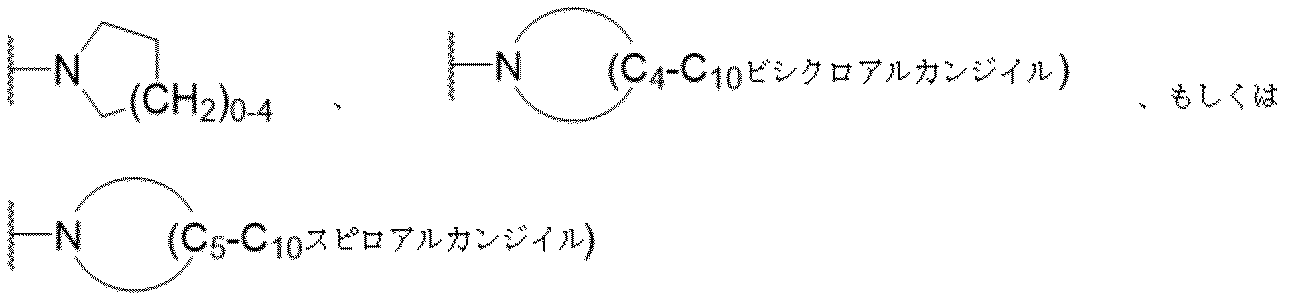
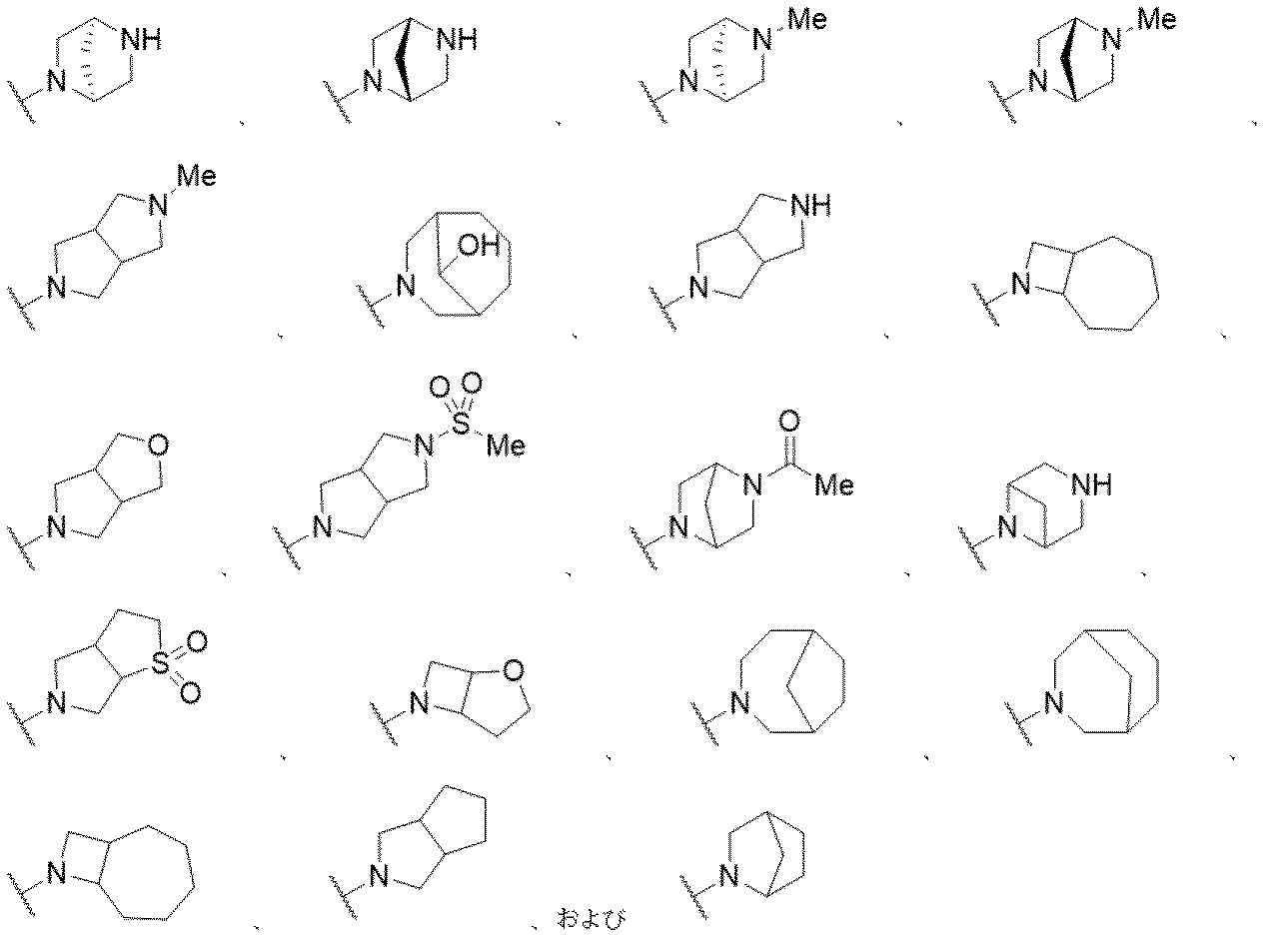
医薬組成物は、一つ以上の添加剤を含むことがある。使用されることがある添加剤には、担体、界面活性剤、増粘または乳化剤、固体結合剤、分散または懸濁助剤、可溶化剤、着色剤、風味剤、コーティング、崩壊剤、滑沢剤、甘味剤、防腐剤、等張化剤、およびそれらの組み合わせが挙げられる。適当な添加剤の選択および使用は、Gennaro編,Remington:The Science and Practice of Pharmacy,第20版(Lippincott Williams & Wilkins 2003)に記載されている。
好ましくは、医薬組成物は、静脈内、筋肉内、皮下、非経口、脊髄または上皮投与(例えば、注射または注入による)に適する。投与経路に応じて、活性化合物は、物質でコーティングされ、化合物を不活化することがある酸および他の自然条件の作用から保護されることがある。「非経口投与」という語句は、通常、注射による、経腸および局所投与以外の投与方法を意味し、例として、静脈内、筋肉内、動脈内、髄腔内、嚢内、眼窩内、心臓内、皮内、腹腔内、経気管、皮下、表皮下、関節内、嚢下、くも膜下、脊髄内、硬膜外および胸骨内注射ならびに注入が挙げられるが、これらに限定されない。あるいは、医薬組成物は、局所、上皮または粘膜投与経路などの非非経口経路(non-parenteral route)で、例えば、鼻腔内、経口的、経膣的、経直腸的、舌下または局所的に投与され得る。
医薬組成物は、滅菌水溶液または滅菌水分散液の形であり得る。それらは、マイクロエマルジョン、リポソーム、または高い薬物濃度を達成するのに適当な他の秩序構造中で製剤化されることもある。組成物は、投与前に水で再調製する凍結乾燥物の形でも提供され得る。
担体物質と結合して単一剤形を生成し得る活性成分の量は、治療を受ける患者および特定の投与方法によって異なり、一般的には治療効果をもたらす組成物の量であろう。一般的に、100パーセントのうち、この量は、活性成分の約0.01パーセントから約99パーセント、好ましくは約0.1パーセントから約70パーセント、最も好ましくは、薬学的に許容される担体との併用で活性成分の約1パーセントから約30パーセントに及ぶであろう。
投与計画は、治療反応を提供するように調整される。例えば、単回のボーラス投与を行ってもよく、用量をいくつかに分けて時間をかけて投与してもよく、状況の緊急性に応じて比例的に用量を増減させてもよい。投与の簡便性および用量の均一性にとって、用量単位形態で非経口組成物を製剤化することは特に有利である。「用量単位形態」は、治療を受ける患者に対する単一の用量として適当な、物理的に別々の単位を指し;各単位には、望ましい治療反応をもたらすように計算された、予め決められた量の活性化合物が、必要な医薬担体とともに含まれる。
用量は、宿主の体重に対して、約0.0001から100mg/kg、より一般的には0.01から5mg/kgに及ぶ。例えば、用量は、0.3mg/kg体重、1mg/kg体重、3mg/kg体重、5mg/kg体重または10mg/kg体重であってもよく、1-10mg/kg、あるいは0.1から5mg/kgの範囲内であってもよい。代表的な治療レジメンは、1週間に1回、2週間に1回、3週間に1回、4週間に1回、1か月に1回、3か月に1回、または3から6か月に1回の投与である。好ましい投与計画には、以下の投薬スケジュール:(i)4週間ごとに6用量を投与し、次に3か月ごとに投与;(ii)3週間ごとに投与;(iii)3mg/kg体重で1回投与し、続いて1mg/kg体重で3週間ごとに投与のうちの一つを用いて、1mg/kg体重または3mg/kg体重で静脈内投与する方法が挙げられる。いくつかの方法において、用量は、約1-1000μg/mLの、いくつかの方法においては約25-300μg/mLの血漿中抗体濃度を達成するように調整される。
本発明の化合物の「治療有効量」は、好ましくは、疾患の症状の重症度の減少、疾患の無症状期間の回数および持続期間の上昇、または疾患の苦痛に起因する機能障害もしくは身体障害の予防をもたらす。例えば、がんを有する患者の治療については、「治療有効量」は、治療を受けていない患者と比較して、好ましくは少なくとも約20%、より好ましくは少なくとも約40%、さらに好ましくは少なくとも約60%、さらに好ましくは少なくとも約80%、腫瘍増殖を阻害する。治療有効量の治療化合物は、腫瘍の大きさを減少させるか、そうでなければ、患者における症状を寛解させることがあり、患者は、一般にはヒトであるが、別の哺乳動物であってもよい。2つ以上の治療剤が併用療法で投与される場合、「治療有効量」は、個々の薬剤としてではなく、全体としての組み合わせの有効性をいう。
医薬組成物は、インプラント、経皮パッチ、およびマイクロカプセル化送達システムなどの放出制御または徐放性製剤であり得る。生分解性の生体適合性ポリマー、例えば、エチレン酢酸ビニル、ポリ酸無水物、ポリグリコール酸、コラーゲン、ポリオルトエステル、およびポリ乳酸などが使用され得る。例えば、Sustained and Controlled Release Drug Delivery Systems,J.R. Robinson編,Marcel Dekker社,ニューヨーク,1978を参照されたい。
治療組成物は、(1)無針皮下注射器具;(2)マイクロ注入ポンプ;(3)経皮デバイス;(4)注入デバイス;および(5)浸透圧装置などの医療機器を用いて投与され得る。
ある実施形態において、医薬組成物は、インビボにおいて適切な分布を確保するように製剤化されることがある。例えば、本発明の治療化合物が血液脳関門を通過することを確実にするために、それらはリポソーム中で製剤化されることがあり、リポソームは、標的化部分をさらに含み、特定の細胞または臓器への選択的輸送を増強することがある。
一つの実施形態において、TLR7アゴニストは、抗がん免疫療法剤-別名を免疫抗がん剤という-と組み合わせて使用される。抗がん免疫療法剤は、がん細胞を攻撃し、破壊する体の免疫系を刺激することにより、特にT細胞の活性化を介して効果を発揮する。免疫系には、それによる正当な標的細胞への攻撃、およびそれによる健康で正常な細胞への攻撃の抑止のバランスの維持を助ける、多数のチェックポイント(調節)分子がある。いくつかは刺激因子(上方調節因子)であり、それらの関与はT細胞活性化を促進し、免疫応答を増強するということを意味する。他は阻害因子(下方制御因子またはブレーキ)であり、それらの関与はT細胞活性化を阻害し、免疫応答を弱めるということを意味する。アゴニスト免疫療法剤の、刺激性チェックポイント分子への結合は、後者の活性化およびがん細胞に対する免疫応答の増強をもたらし得る。交換的に、アンタゴニスト免疫療法剤の、抑制性チェックポイント分子への結合は、後者による免疫系の下方制御を防ぎ、がん細胞に対する活発な応答の維持を助け得る。刺激性チェックポイント分子の例は、B7-1、B7-2、CD28、4-1BB (CD137)、4-1BBL、ICOS、CD40、ICOS-L、OX40、OX40L、GITR、GITRL、CD70、CD27、CD40、DR3およびCD28Hである。抑制性チェックポイント分子の例は、CTLA-4、PD-1、PD-L1、PD-L2、LAG-3、TIM-3、ガレクチン9、CEACAM-1、BTLA、CD69、ガレクチン-1、CD113、GPR56、VISTA、2B4、CD48、GARP、PD1H、LAIR1、TIM-1、CD96およびTIM-4である。
どちらの抗がん免疫療法剤の作用機序においても、その有効性は、TLR7の活性化などの全身的な免疫系の上方制御により上昇し得る。それゆえ、一つの実施形態において、本明細書は、がんを患う患者に、抗がん免疫療法剤および本明細書に開示されるようなTLR7アゴニストの治療的に有効な組み合わせを投与することを特徴とする、がんの治療方法を提供する。投与のタイミングは、同時でも、連続的でも、交互であってもよい。投与方法は、全身的であっても、局所的であってもよい。TLR7アゴニストは、対象を絞った方法で、複合体を用いて送達されることがある。
上記のような併用療法により治療され得るがんには、急性骨髄白血病、副腎皮質癌、カポジ肉腫、リンパ腫、肛門癌、虫垂癌、奇形/ラブドイド腫瘍、基底細胞癌、胆管癌、膀胱癌、骨癌、脳癌、乳癌、気管支腫瘍、カルチノイド腫瘍、心臓腫瘍、子宮頸癌、脊索腫、慢性リンパ性白血病、慢性骨髄増殖性腫瘍、結腸癌、結腸直腸癌、頭蓋咽頭腫、胆管癌、子宮内膜癌、上衣腫、食道癌、感覚神経芽腫、ユーイング肉腫、眼癌、卵管癌、胆嚢癌、消化管カルチノイド腫瘍、消化管間質腫瘍、胚細胞腫瘍、へアリー細胞白血病、頭頸部癌、心臓癌、肝臓癌、下咽頭癌、膵臓癌、腎臓癌、喉頭癌、慢性骨髄性白血病、口唇および口腔癌(lip and oral cavity cancer)、肺癌、黒色腫、メルケル細胞癌、中皮腫、口腔癌(mouth cancer)、口腔癌(oral cancer)、骨肉腫、卵巣癌、陰茎癌、咽頭癌、前立腺癌、直腸癌、唾液腺癌、皮膚癌、小腸癌、軟部組織肉腫、精巣癌、咽喉癌、甲状腺癌、尿道癌、子宮癌、膣癌、および外陰癌が挙げられる。
TLR7アゴニストとの併用療法の一つの実施形態において、抗がん免疫療法剤は、アンタゴニスト抗CTLA-4、抗PD-1、または抗PD-L1抗体である。がんは、肺癌(非小細胞肺癌を含む)、膵臓癌、腎臓癌、頭頸部癌、リンパ腫(ホジキンリンパ腫を含む)、皮膚癌(黒色腫およびメルケル皮膚癌を含む)、尿路上皮癌(膀胱癌を含む)、胃癌、肝細胞癌、または結腸直腸癌であり得る。
TLR7アゴニストとの併用療法のもう一つの実施形態において、抗がん免疫療法剤は、アンタゴニスト抗PD-1抗体、好ましくはニボルマブまたはペムブロリズマブである。
解析手順
NMR
プロトン核磁気共鳴(NMR)スペクトルを得るために以下の条件を用いた:溶媒および内部標準としてDMSO-d6またはCDCl3のいずれかを用いて、400Mzまたは500MhzのBruker装置のいずれかでNMRスペクトルを得た。ADC LabsのACD Spectrusバージョン2015-01またはMestReNovaソフトウェアのいずれかを用いることにより、生のNMRデータを解析した。
化学シフトは、内部のテトラメチルシラン(TMS)から、または重水素化NMR溶媒により推測されるTMSの位置を基準に、低磁場側が百万分率(ppm)で報告される。明らかな多重度は:一重線-s、二重線-d、三重線-t、四重線-q、または多重線-mとして報告する。広幅化を示すピークをbrとしてさらに表す。積分値は近似値である。積分強度、ピーク形状、化学シフトおよび結合定数は、溶媒、濃度、温度、pH、および他の因子に依存し得るということに注意すべきである。さらに、NMRスペクトルにおいて水または溶媒ピークと重複するか、または交換が起こるピークは、信頼できる積分強度を提供しないことがある。場合によっては、NMRスペクトルは、水ピーク抑制を用いて得られることがあるが、重複するピークが目に見えなくなるか、またはその形状および/もしくは積分値が変化することがある。
液体クロマトグラフィー
以下のプレパラティブおよび分析(LC/MS)液体クロマトグラフィー法を用いた:
LCMS手順A:カラム:Waters XBridge C18、2.1 mm x 50 mm、1.7 μm粒子;移動相A:5:95 アセトニトリル:10mM NH4OAc含有水;移動相B:95:5 アセトニトリル:10mM NH4OAc含有水;温度:50℃;グラジエント:3分かけて0%Bから100%B、次いで100%Bで0.50分間保持;流速:1mL/分;検出:MSおよびUV(220nm)
LCMS手順B:カラム:Waters XBridge C18、2.1 mm x 50 mm、1.7 μm粒子;移動相A:5:95 アセトニトリル:0.1%TFA含有水;移動相B:95:5 アセトニトリル:0.1%TFA含有水;温度:50℃;グラジエント:3分かけて0%Bから100%B、次いで100%Bで0.50分間保持;流速:1mL/分;検出:MSおよびUV(220nm)
LCMS手順C:カラム:Waters XBridge BEH C18 XP(50 x 2.1 mm)2.5 μm;移動相A:5:95 アセトニトリル:10mM NH4OAc含有水;移動相B:95:5 アセトニトリル:10mM NH4OAc含有水;温度:50℃;グラジエント:3分かけて0-100%B;流速:1.1mL/分
LCMS手順D:カラム:Ascentis Express C18(50 x 2.1 mm)2.7 μm;移動相A:5:95 アセトニトリル:10mM NH4OAc含有水;移動相B:95:5 アセトニトリル:10mM NH4OAc含有水;温度:50℃;グラジエント:3分かけて0-100%B;流速:1.1mL/分
LCMS手順E:カラム:BEH C18 2.1 x 50 mm;移動相A:0.05% TFA含有水;移動相B:0.05% TFA含有アセトニトリル;温度:50℃;グラジエント:1.7分かけて2-98%B;流速:0.8mL/分
LCMS手順F:カラム:Waters XBridge C18、2.1 mm x 50 mm、1.7 μm粒子;移動相A:5:95 アセトニトリル:10mM NH4OAc含有水;移動相B:95:5 アセトニトリル:10mM NH4OAc含有水;温度:50℃;グラジエント:3分かけて0%Bから100%B、次いで100%Bで0.50分間保持;流速:1mL/分;検出:MSおよびUV(220nm)。この方法は、超高速液体クロマトグラフィー(UPLC(商標)法である。
合成-一般的な手順
一般的に、本明細書に開示される手順は、ピラゾロピリミジン環系の1Hまたは2H位置でアルキル化された位置異性体の混合物をもたらす(それぞれN1およびN2位置異性体とも呼ばれ、アルキル化された窒素に言及している)。簡略化のために、N2位置異性体は、便宜上示されないが、初期に生成される混合物中に存在し、例えばプレパラティブHPLCにより、後で分離されるということが理解されるべきである。
位置異性体の混合物を合成の初期段階に分離し、1H位置異性体を用いて残りの合成段階を実行してもよく、あるいは、必要に応じて、位置異性体の混合物を用いて合成を進め、後期に分離を実行してもよい。
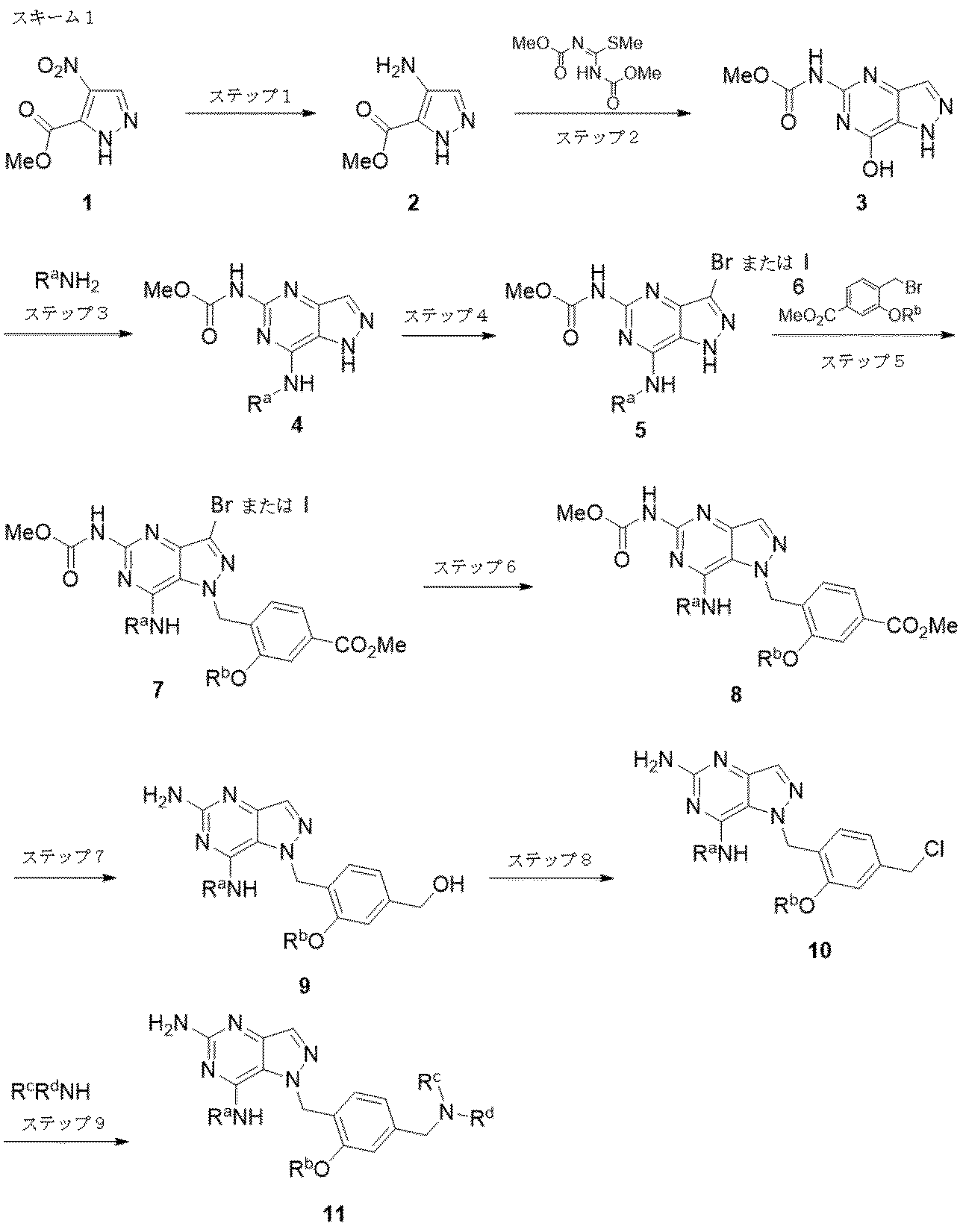
本開示の化合物は、有機合成化学の当業者に周知の多数の方法により調製され得る。これらの方法は、以下に記載される方法、またはそのバリエーションを含む。好ましい方法には、下記のスキームに記載される方法が挙げられるが、これらに限らない。
スキーム1
R
aは、スキーム1およびそれが登場する他の事例において、例えば、
、または他の適当な部分であり得る。R
bは、スキーム1およびそれが登場する他の事例において、例えば、C
1-C
3アルキルである。R
cNHR
dは、スキーム1およびそれが登場する他の事例において、第一級または第二級アミンである。R
a、R
b、R
c、および/またはR
dは、合成過程の間の適切な時点で取り除かれる保護基により覆われた官能基を有してもよい。
化合物11は、上記のスキーム1に図示される合成順序により調製され得る。ニトロピラゾール1の還元により化合物2が得られ、次いで1,3-ビス(メトキシカルボニル)-2-メチル-2-チオシュードウレアとの環化によりヒドロキシピラゾロピリミジン3が得られる。BOP/DBUカップリング条件を用いてアミンR
aNH
2が導入され、次いで、NBSを用いる臭素化またはNISを用いるヨウ素化(ステップ4)によりブロモまたはヨード-ピラゾロピリミジン5が得られる。ベンジルハライド6を用いるアルキル化によりN1およびN2生成物の混合物が得られ、分離することでN1中間体7が得られる。接触水素化(ステップ6)の後、ワンポットでLiAlH
4還元およびカルバメート加水分解を行うことで、中間体アルコール9が得られる。アルコール9を塩化ベンジルに変換し、次いでそれを適当なアミンで置換することで化合物11が得られる(ステップ5における臭素化中間体5のアルキル化の方が、非臭素化中間体4のアルキル化と比較して、より好ましい比率でN1/N2生成物が得られる)。
スキーム2
あるいは、中間体9は、上記のスキーム2に記載される経路を用いて得られることがある。NBSまたはNISを用いて中間体3を臭素化またはヨウ素化し、次いでアルキル化することで中間体エステル12が得られる。次にBOPカップリング条件を用いてアミノ化することで中間体7が得られる。接触水素化の後、アルコールへのLiAlH
4還元およびメチルカルバメート脱保護を行うことで中間体9が得られる。
スキーム3
中間体8への代替経路は、ベンジルハライド6を用いるニトロピラゾール1のアルキル化から始まり、ベンジルピラゾール13が得られる。ニトロ基の還元の後、1,3-ビス(メトキシカルボニル)-2-メチル-2-チオシュードウレアとの環化によりヒドロキシピラゾロピリミジン15が得られ、BOP/DBU条件を用いて適切なアミン誘導体8に変換される。これが上記のスキーム3に図示される。
スキーム4
標的化合物への別の代替経路は、上記のスキーム4に示される。中間体15のエステル基を還元し、NaOHを用いてメチルカルバメートを取り除くとアルコール16が得られる。アルコール16を塩化物に変換し、次いで適当なアミンで置換すると17が得られ、続いてBOP/DBU条件を用いてアミノ化すると標的分子11が得られる。
スキーム5
上記のスキーム5において、7/8または15中のメチルエステルの加水分解、次いでアミド形成により、対応するアミド7a/8aまたは15aを得ることができる。7aの接触水素化、次いでカルバメート脱保護により、化合物7bが生成される。8aのカルバメート脱保護により化合物8bが得られる。最後に、15aのアミン導入、次いでカルバメート脱保護により、化合物15bが得られる。
合成-具体例
上記の内容をさらに説明するために、以下の限定されない代表的な合成スキームが含まれる。請求項の範囲内にあるこれらの実施例のバリエーションは、当業者の範囲内であり、本開示の範囲内にあると見なされる。読者は、本開示を提供された、関連技術に熟練した当業者であれば、網羅的な実施例がなくとも、本明細書に開示される化合物を調製し、使用することができるであろうということを認識するであろう。
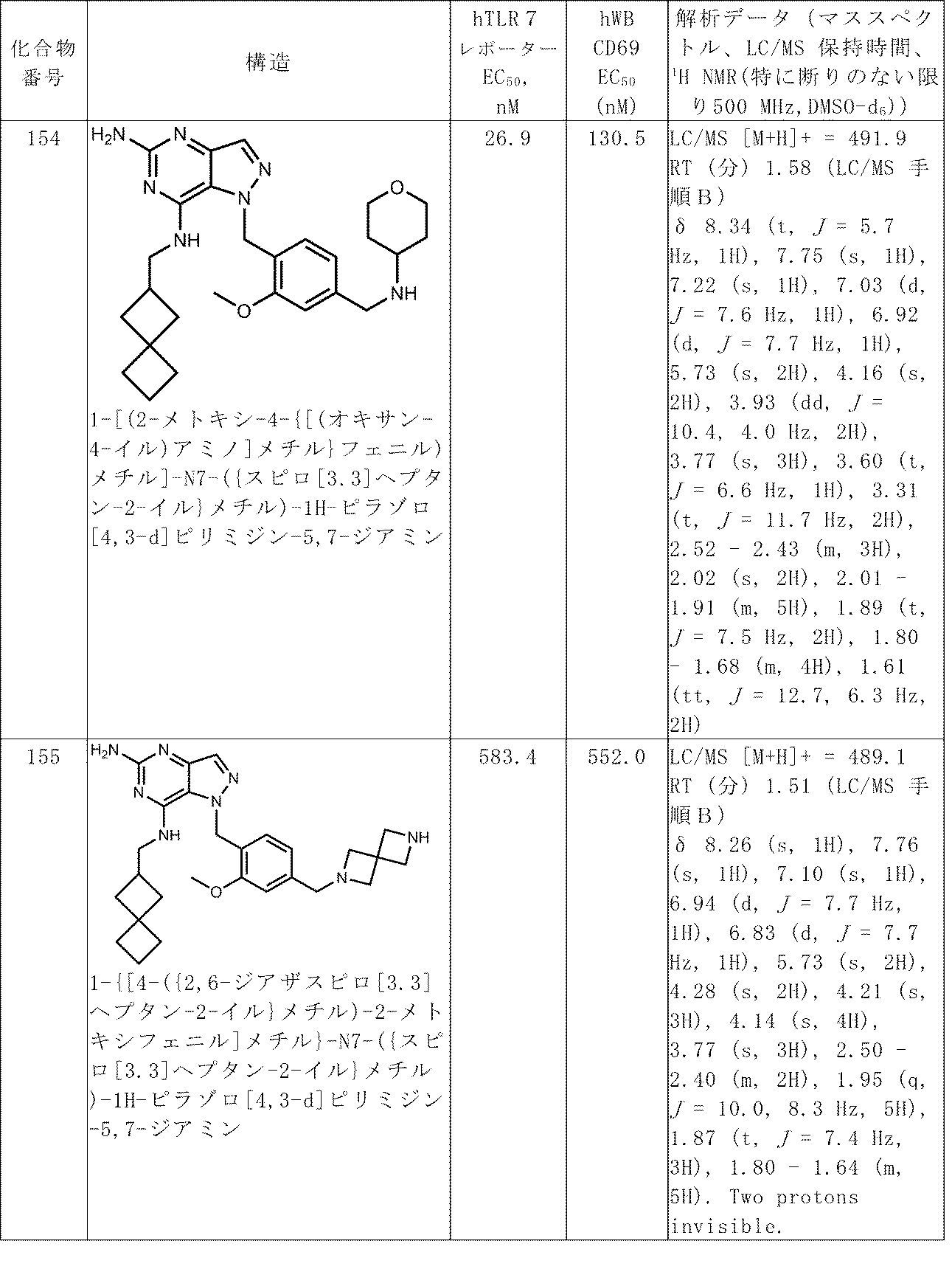
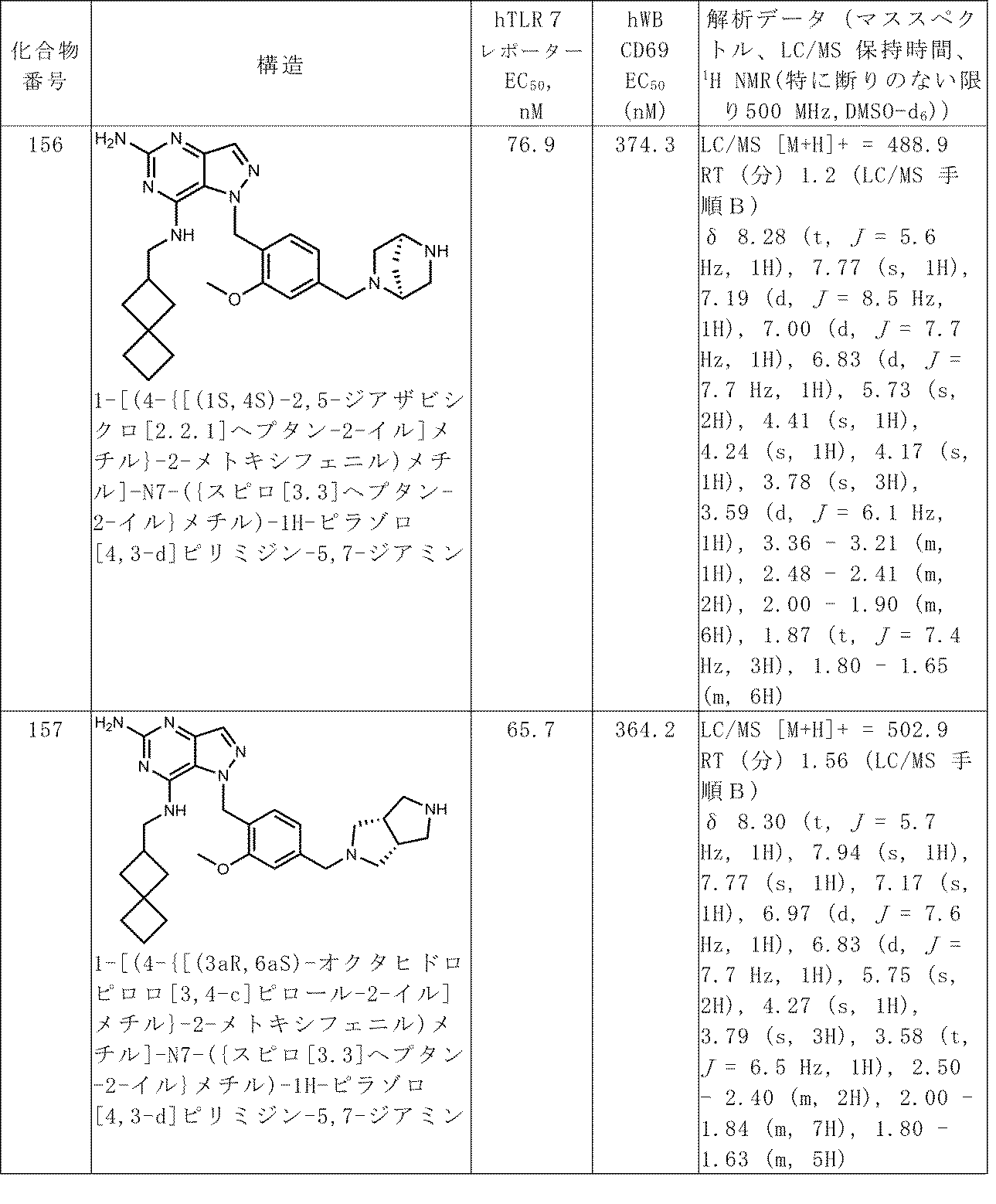
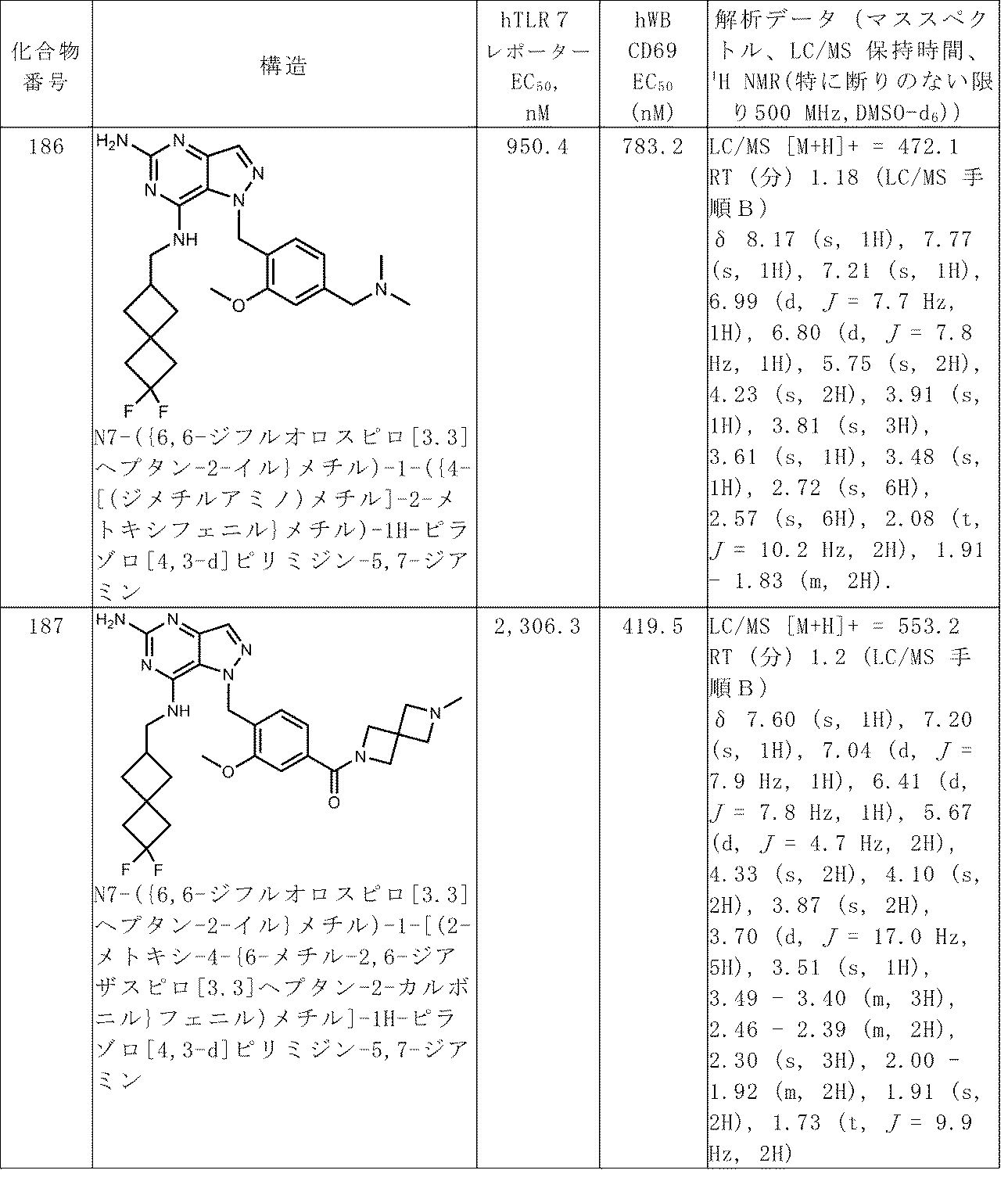
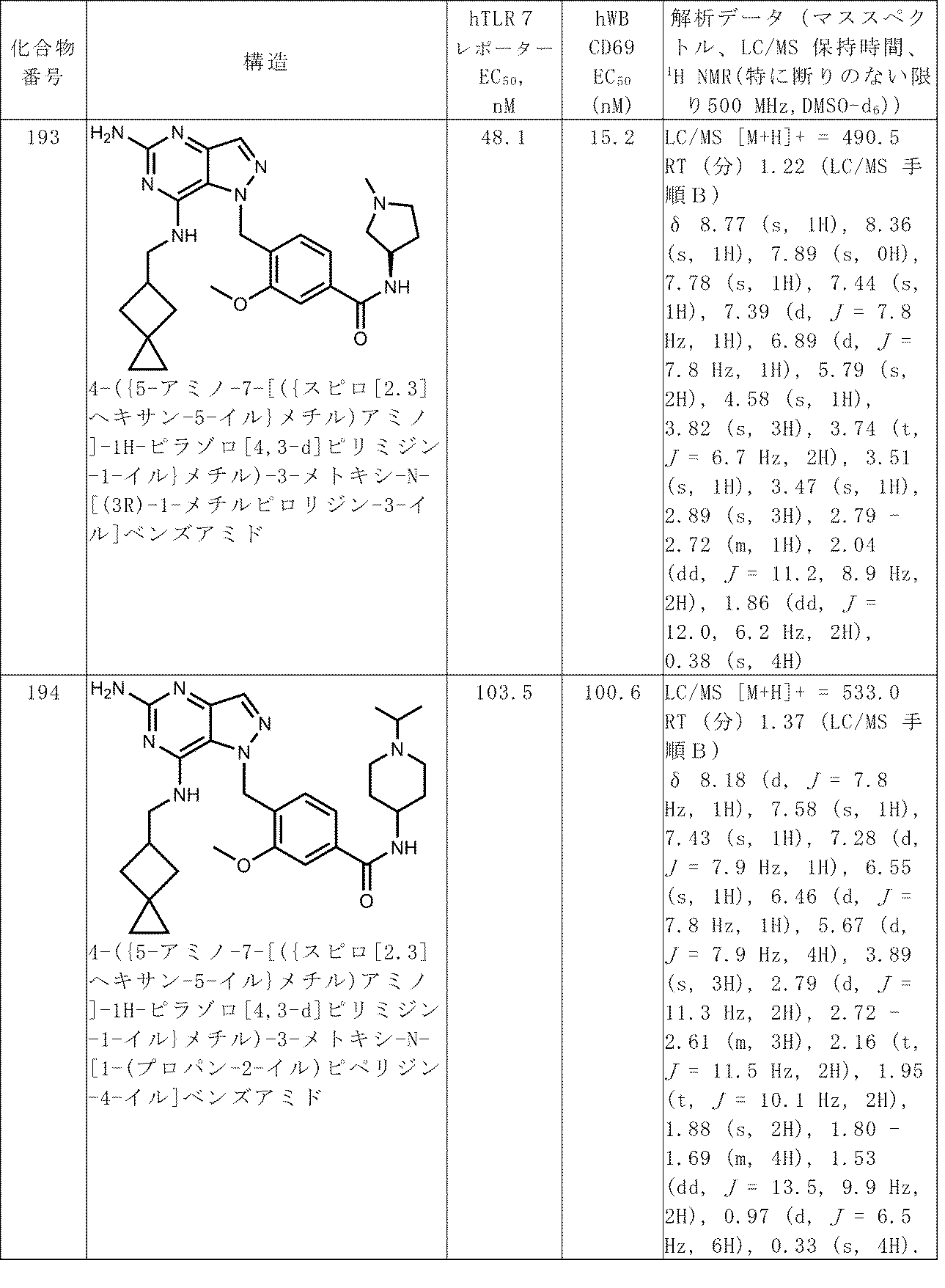
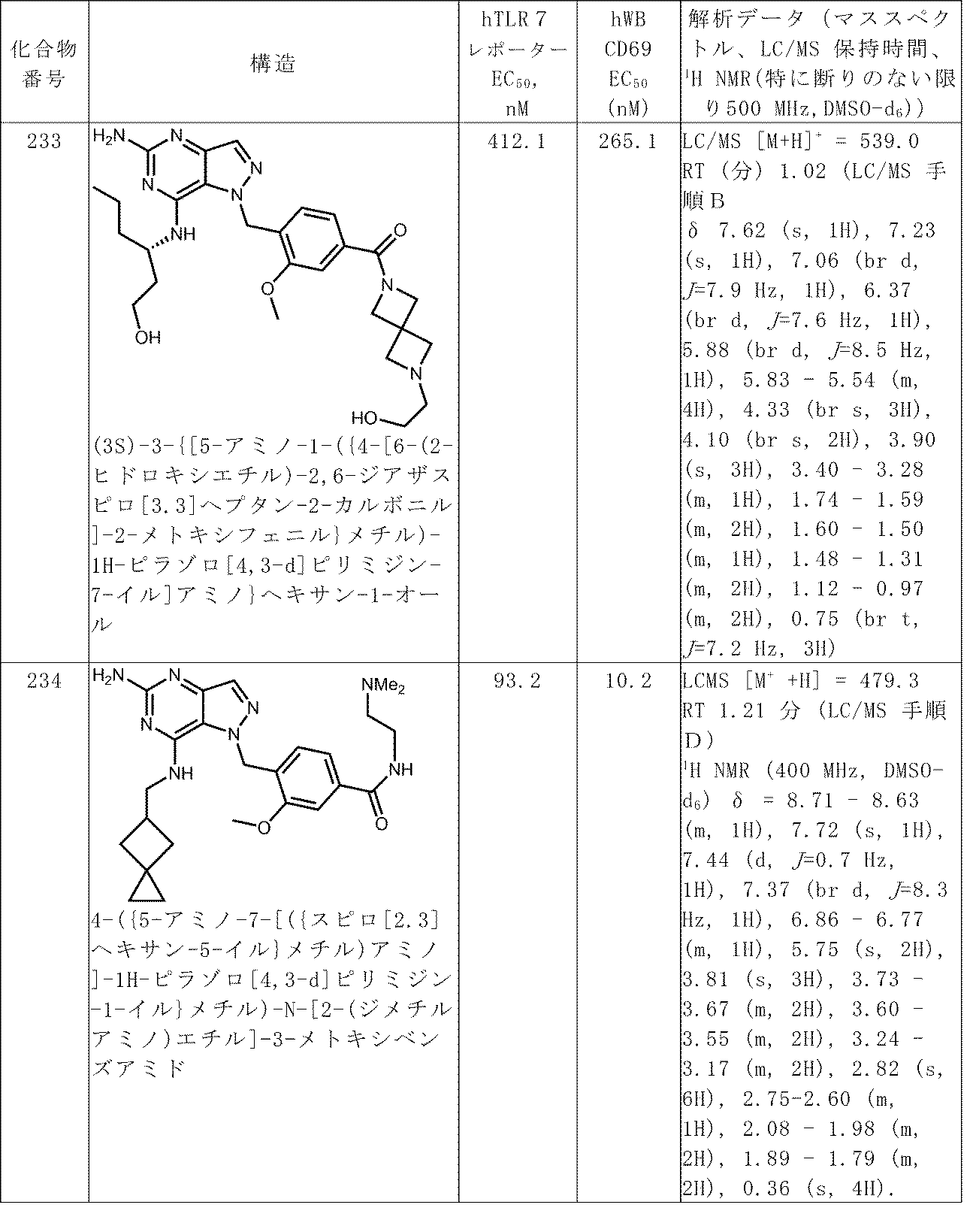
100以上の番号がつけられた化合物についての解析データは、表Aで見つけることができる。
実施例1-化合物101
(S)-3-((1-(4-((2,6-ジアザスピロ[3.3]ヘプタン-2-イル)メチル)-2-メトキシベンジル)-5-アミノ-1H-ピラゾロ[4,3-d]ピリミジン-7-イル)アミノ)ヘキサン-1-オール 1(US 2020/0038403 A1;31 mg、0.065 mmol)のDMF(1 mL)溶液をAc2O(6.09 μL、0.065 mmol)で処理し、RTで1時間攪拌した。溶媒を蒸発させ、残留物をDMF(1 mL)中に溶解した。以下の条件を用いて、粗製残留物をプレパラティブLC/MSで精製した:カラム:XBridge C18、200 mm x 19 mm、5μm 粒子;移動相A:5:95 アセトニトリル:NH4OAc含有水;移動相B:95:5 アセトニトリル:NH4OAc含有水;グラジエント:4%Bで0分保持、20分かけて4-44%B、次いで100%Bで0分保持;流速:20mL/分;カラム温度:25℃。フラクション収集はMSシグナルによりトリガーされた。目的物を含むフラクションを混ぜ合わせ、遠心蒸発で乾燥させ、5mgの化合物101を得た。
以下の化合物を類似的に調製した:化合物106、化合物107、化合物215(ホルムアルデヒドで化合物1を還元的アミノ化することにより作成)、および化合物216(アセトンで化合物1を還元的アミノ化することにより作成)。
実施例2-化合物110
1-(4-((2,6-ジアザスピロ[3.3]ヘプタン-2-イル)メチル)-2-メトキシベンジル)-N7-ブチル-1H-ピラゾロ[4,3-d]ピリミジン-5,7-ジアミン 2(US 2020/0038403 A1;32 mg、0.073 mmol)およびシクロブタンカルボン酸(7.01 μL、0.073 mmol)のDMF(0.5 mL)溶液をヒューニッヒ塩基(0.064 mL、0.366 mmol)およびHATU(33.4 mg、0.088 mmol)で処理し、30分間攪拌した。塩基を蒸発させ、シリンジ濾過した。以下の条件を用いて、粗製物質をプレパラティブLC/MSで精製した:カラム:XBridge C18、200 mm x 19 mm、5μm 粒子;移動相A:5:95 アセトニトリル:10mM NH4OAc含有水;移動相B:95:5 アセトニトリル:10mM NH4OAc含有水;グラジエント:12%Bで0分保持、20分かけて12-52%B、次いで100%Bで0分保持;流速:20mL/分;カラム温度:25℃。フラクション収集はMSおよびUVシグナルによりトリガーされた。目的物を含むフラクションを混ぜ合わせ、遠心蒸発で乾燥させ、化合物110を得た。
以下の化合物を類似的に調製した:化合物104、化合物105、および化合物111。
実施例3-化合物102
N7-ブチル-1-(4-(クロロメチル)-2-メトキシベンジル)-1H-ピラゾロ[4,3-d]ピリミジン-5,7-ジアミン 3(US 2020/0038403 A1;15 mg、0.04 mmol)の2ml DMF溶液を6,6-ジフルオロ-2-アザスピロ[3.3]ヘプタン(10.6 mg、0.08 mml)で処理し、80℃で1時間加熱した。LCMSが反応の完了を示した。反応物をシリンジ濾過した。以下の条件を用いて、粗製物質をプレパラティブLC/MSで精製した:カラム:XBridge C18、200 mm x 19 mm、5μm 粒子;移動相A:5:95 アセトニトリル:10 mM NH4OAc含有水;移動相B:95:5 アセトニトリル:10 mM NH4OAc含有水;グラジエント:21%Bで0分保持、20分かけて21-61%B、次いで100%Bで4分保持;流速:20mL/分;カラム温度:25℃。フラクション収集はMSおよびUVシグナルによりトリガーされた。目的物を含むフラクションを混ぜ合わせ、遠心蒸発で乾燥させ、化合物102を得た。
化合物103を類似的に調製した。
実施例4-化合物112
メチル (S)-(7-((1-((tert-ブチルジフェニルシリル)オキシ)ヘキサン-3-イル)アミノ)-1-(4-(クロロメチル)-2-メトキシベンジル)-1H-ピラゾロ[4,3-d]ピリミジン-5-イル)カルバメート 4(US 2020/0038403;20 mg、0.028 mmol)の1mL DMF溶液を1-オキサ-6-アザスピロ[3.3]ヘプタン(13 mg、0.14 mmol)で処理し、80℃で1時間加熱した。反応混合物をトリエチルアミン-トリヒドロフルオライド(23 μL、0.14 mmol)で処理し、RTで3時間攪拌した。粗生成物をNaOH(112 μL、0.559 mmol)で処理し、80℃で2時間加熱した。6M HCl水溶液で、反応混合物をpH7に中和した。ロータリーエバポレーター中で溶媒を蒸発させた。残留物を1mL DMF中に溶解し、以下の条件を用いて、粗製物質をプレパラティブLC/MSで精製した:カラム:XBridge C18、200 mm x 19 mm、5μm 粒子;移動相A:5:95 アセトニトリル:NH4OAc含有水;移動相B:95:5 アセトニトリル:NH4OAc含有水;グラジエント:9%Bで0分保持、20分かけて9-49%B、次いで100%Bで0分保持;流速:20mL/分;カラム温度:25℃。フラクション収集はMSおよびUVシグナルによりトリガーされた。目的物を含むフラクションを混ぜ合わせ、遠心蒸発で乾燥させ、化合物112を得た。
化合物113および化合物114を類似的に調製した。
実施例5-化合物108
ステップ1.メチル (7-ヒドロキシ-1-(4-(ヒドロキシメチル)-2-メトキシベンジル)-1H-ピラゾロ[4,3-d]ピリミジン-5-イル)カルバメート 5(US 2020/0038403 A1;300 mg、0.835 mmol)、スピロ[2.3]ヘキサン-5-イルメタンアミン ヒドロクロライド(139 mg、1.252 mmol)のDMSO(2 mL)溶液をDBU(0.378 mL、2.505 mmol)で処理した。BOP(554 mg、1.252 mmol)を添加した。反応混合物を40℃で1時間加熱した。反応混合物をNaOH(0.835 mL、4.17 mmol)で処理し、80℃で2時間加熱した。逆相ISCOで、50 g C-18カラムを用いて、0-50% 水/MeCN(0.05% TFA)で溶出させて生成物を直接精製し、フラクションを凍結乾燥させ、化合物166を白色固体として得た。
ステップ2.(4-((5-アミノ-7-((スピロ[2.3]ヘキサン-5-イルメチル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-メトキシフェニル)メタノール 166(300 mg、0.760 mmol)のTHF(2 mL)溶液をSOCl2(0.111 mL、1.521 mmol)で処理し、RTで30分間攪拌した。溶媒をV-10エバポレーター中で蒸発させ、30mgの粗製塩化物をDMSO(0.5 mL)中に溶解し、1-(2,6-ジアザスピロ[3.3]ヘプタン-2-イル)エタン-1-オン(51 mg、0.363 mmol)およびヒューニッヒ塩基(0.127 mL、0.727 mmol)で処理した。反応混合物を80℃で3時間加熱した。過剰な塩基を蒸発させ、以下の条件を用いて、粗生成物をプレパラティブLC/MSで精製した:カラム:XBridge C18、200 mm x 19 mm、5μm 粒子;移動相A:5:95 アセトニトリル:NH4OAc含有水;移動相B:95:5 アセトニトリル:NH4OAc含有水;グラジエント:12%Bで0分保持、20分かけて12-52%B、次いで100%Bで0分保持;流速:20mL/分;カラム温度:25℃。フラクション収集はMSおよびUVシグナルによりトリガーされた。化合物109を含むフラクションを混ぜ合わせ、遠心蒸発で乾燥させ、化合物108を得た。
以下の化合物を類似的に調製した:化合物109(ステップ1において、スピロ[2.2]ペンタン-1-イルメタンアミンをスピロ[2.3]ヘキサン-5-イルメタンアミンの代わりに用いた)、化合物129、化合物130、化合物131、化合物132、化合物133、化合物134、化合物135、化合物145、化合物146、化合物147、化合物148、化合物152(ステップ1において、(3-シクロプロピルシクロブチル)メタンアミンをスピロ[2.3]ヘキサン-5-イルメタンアミンの代わりに用いた)、化合物167、化合物168、化合物169、化合物170、化合物183、および化合物241。
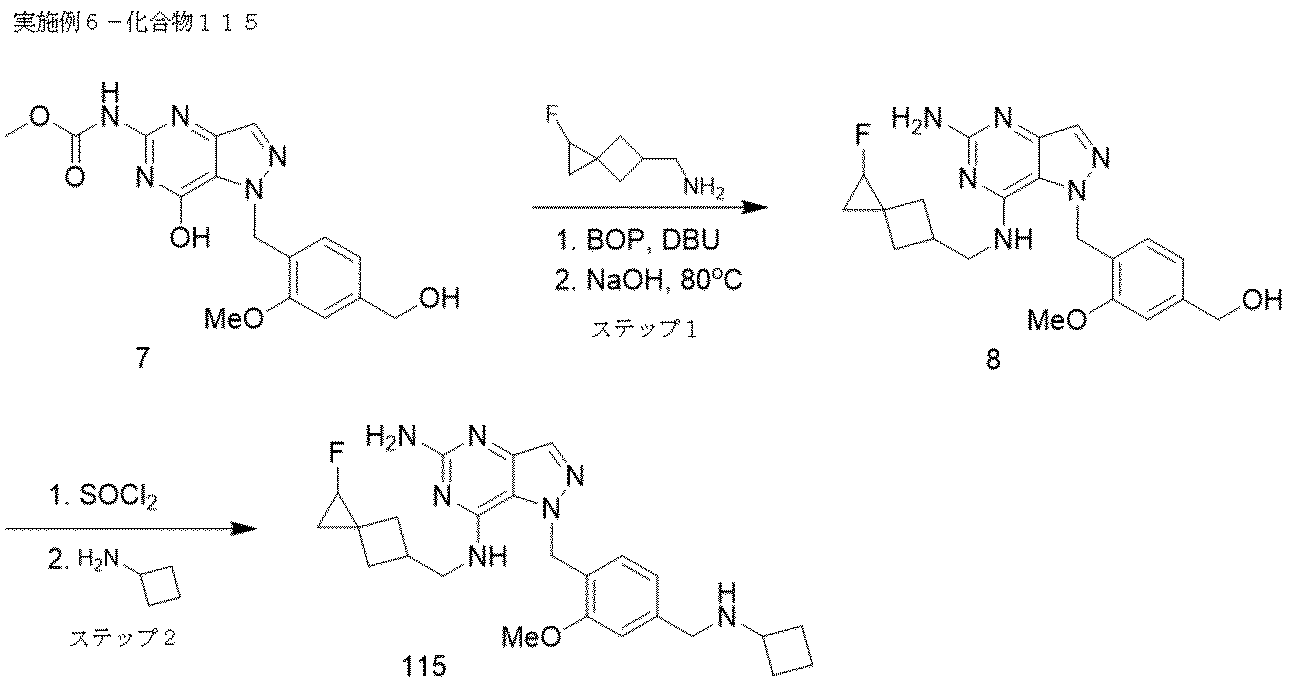
実施例6-化合物115
ステップ1.メチル (7-ヒドロキシ-1-(4-(ヒドロキシメチル)-2-メトキシベンジル)-1H-ピラゾロ[4,3-d]ピリミジン-5-イル)カルバメート 7(US 2020/0038403 A1;100 mg、0.278 mmol)、(1-フルオロスピロ[2.3]ヘキサン-5-イル)メタンアミン(71.9 mg、0.557 mmol)のDMSO(2 mL)溶液をDBU(0.126 mL、0.835 mmol)で処理した。BOP(185 mg、0.417 mmol)を添加した。反応混合物を40℃で1時間加熱した。反応混合物をNaOH(0.278 mL、1.391 mmol)で処理し、80℃で2時間加熱した。逆相ISCで、50 g C-18カラムを用いて、0-50% 水/MeCN(0.05% TFA)で溶出させて生成物を直接精製し、目的のフラクションを凍結乾燥させ、84mgの化合物8を白色固体として、ジアステレオマーの混合物として得た。
LC/MS [M+H]+=469.1
1H NMR(400MHz,DMSO-d6)δ 8.34(s,1H),7.88(s,1H),7.75(d,J = 1.8 Hz,1H),6.98(s,1H),6.86-6.74(m,2H),5.71(s,2H),4.67-4.58(m,1H),4.45(d,J = 3.6 Hz,3H),3.75(d,J = 3.2 Hz,5H),2.80(s,1H),2.18(q,J = 9.1 Hz,1H),2.05-1.90(m,2H),1.85-1.76(m,1H),0.74(ddd,J = 21.0,11.2,5.9 Hz,2H)
ステップ2.SOCl2(0.030 mL、0.407 mmol)を、化合物8(84 mg、0.204 mmol)のTHF(1 mL)溶液に添加した。反応混合物をRTで1時間攪拌した。溶媒をV-10エバポレーター中で蒸発させて粗製塩化物を得て、さらなる精製は行わずに次のステップに進んだ。20mL密封バイアル中の、12mgの塩化物およびシクロブチルアミン(3.96 mg、0.056 mmol)の0.5 mL DMF溶液を70℃で1時間加熱した。過剰の塩基を蒸発させ、以下の条件を用いて、粗製物質をプレパラティブLC/MSで精製した:カラム:XBridge C18、200 mm x 19 mm、5μm 粒子;移動相A:5:95 アセトニトリル:0.05% TFA含有水;移動相B:95:5 アセトニトリル:0.05% TFA含有水;グラジエント:2%Bで0分保持、23分かけて2-42%B、次いで100%Bで0分保持;流速:20mL/分;カラム温度:25℃。フラクション収集はMSシグナルによりトリガーされた。目的物を含むフラクションを混ぜ合わせ、遠心蒸発で乾燥させ、6mgの化合物115をジアステレオマーの混合物として得た。
以下の化合物を類似的に調製した:化合物116、化合物117、化合物118、化合物119、化合物120、化合物121、化合物124、化合物125、化合物126、化合物127、および化合物128。
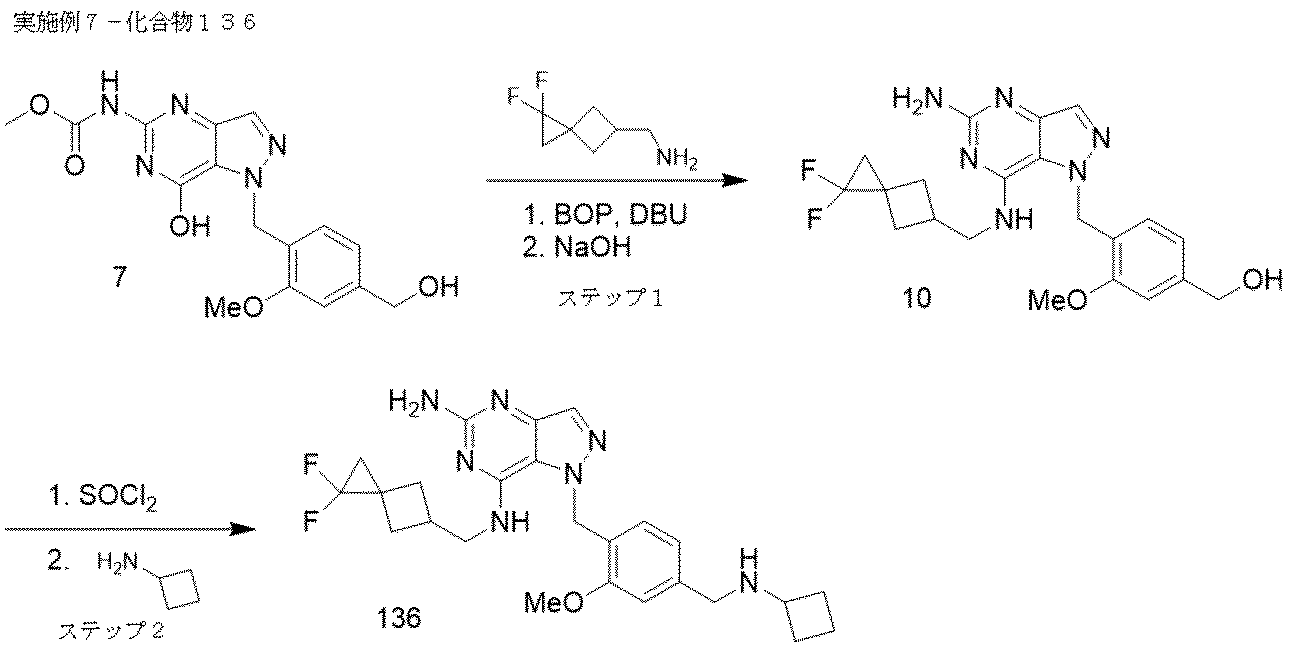
実施例7-化合物136
ステップ1.化合物7(200 mg、0.557 mmol)、(1,1-ジフルオロスピロ[2.3]ヘキサン-5-イル)メタンアミン(164 mg、1.113 mmol)のDMSO(2 mL)溶液をDBU(0.252 mL、1.670 mmol)で処理した。BOP(369 mg、0.835 mmol)を添加した。反応混合物を40℃で1時間加熱した。反応混合物をNaOH(0.557 mL、2.78 mmol)で処理し、80℃で2時間加熱した。逆相ISCで、50 g C-18カラムを用いて、0-50% 水/アセトニトリルで溶出させて反応物を直接精製し、フラクションを凍結乾燥させ、白色固体として目的物を得た。
LC/MS予測 C21H24F2N6O2=431.4 実測[M+H]+=431.2
ステップ2.化合物10(142 mg、0.330 mmol)のテトラヒドロフラン(2 mL)溶液をSOCl2(0.048 mL、0.660 mmol)で処理し、1時間攪拌した。溶媒をV-10エバポレーター中で蒸発させ、粗生成物を次のステップに進めた。0.5 mL DMF中、粗製塩化物およびシクロブチルアミン(11.8 mg、0.167 mmol)の混合物を80℃で1時間加熱した。過剰なアミンを蒸発させ、以下の条件を用いて、粗製物質をプレパラティブLC/MSで精製した:カラム:XBridge C18、200 mm x 19 mm、5μm 粒子;移動相A:5:95 アセトニトリル:NH4OAc含有水;移動相B:95:5 アセトニトリル:NH4OAc含有水;グラジエント:15%Bで0分保持、20分かけて15-55%B、次いで100%Bで0分保持;流速:20mL/分;カラム温度:25℃。フラクション収集はMSシグナルによりトリガーされた。目的物を含むフラクションを混ぜ合わせ、遠心蒸発で乾燥させ、4.2mgの化合物136を得て、ジアステレオマーの混合物として単離した。
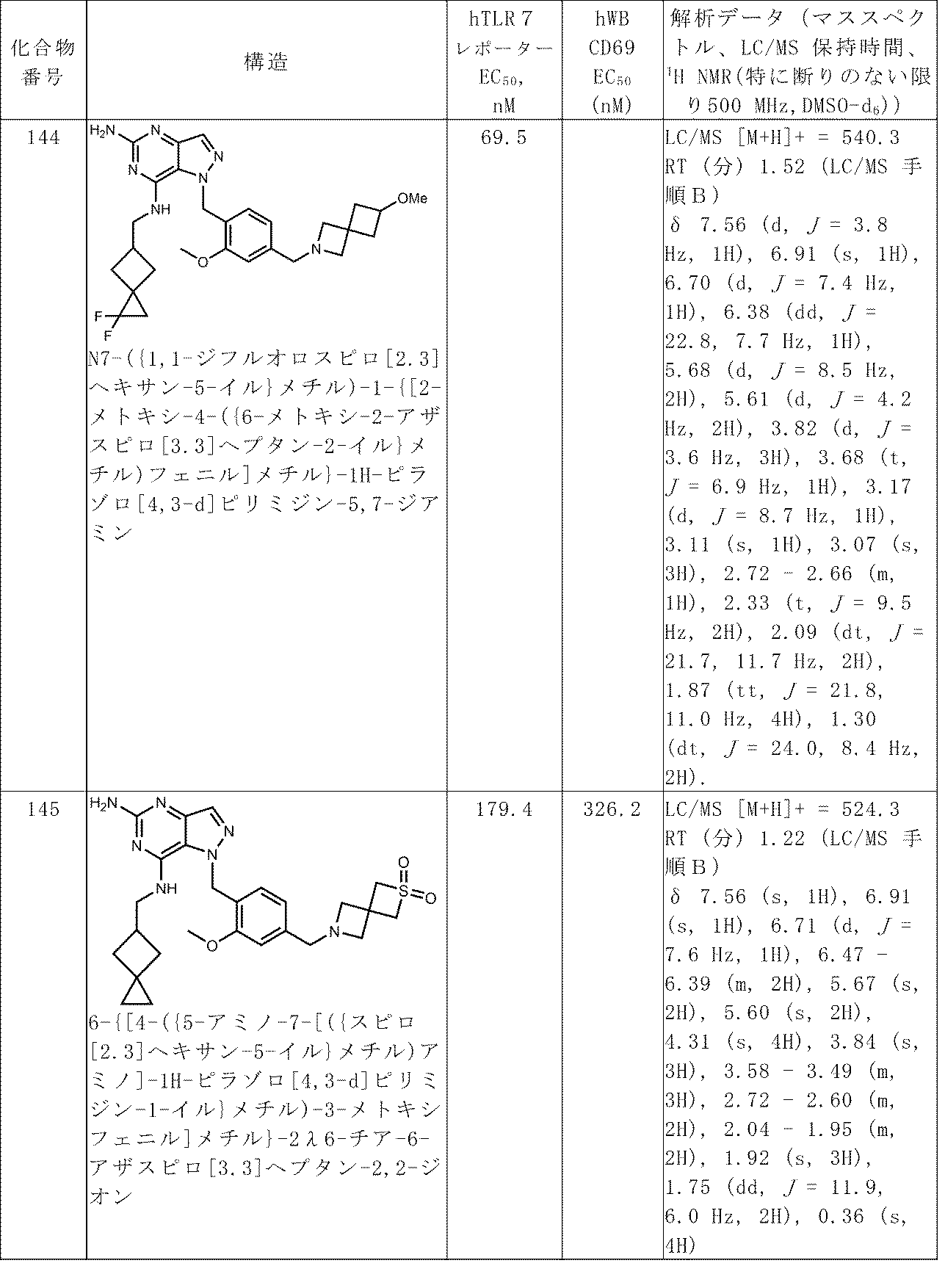
以下の化合物を類似的に調製した:化合物122、化合物123、化合物137、化合物138、化合物139、化合物140、化合物141、化合物142、化合物143、および化合物144。
実施例8-化合物173
ステップ1.化合物11(US 2020/0038403 A1;350 mg、0.904 mmol)、スピロ[2.3]ヘキサン-5-イルメタンアミン ヒドロクロライド(151 mg、1.355 mmol)のDMSO(2 mL)溶液をDBU(0.409 mL、2.71 mmol)で処理した。BOP(599 mg、1.355 mmol)を添加した。反応混合物を40℃で1時間加熱した。反応混合物をNaOH(0.904 mL、4.52 mmol)で処理し、80℃で2時間加熱した。逆相ISCOで、50 g C-18カラムを用いて、0-50% 水/アセトニトリル(0.05% TFA)で溶出させて反応物を直接精製し、フラクションを凍結乾燥させ、白色固体として化合物12を得た。
LC/MS [M+H]+=395.2
1H NMR(400MHz,DMSO-d6) 12.34(s,1H),8.32(t,J = 5.7 Hz,1H),7.86(s,1H),7.80(s,1H),7.53-7.43(m,2H),6.79(d,J = 7.9 Hz,1H),5.81(s,2H),3.84(s,3H),3.72(t,J = 6.5 Hz,3H),2.77-2.65(m,1H),1.82(dd,J = 12.0,6.3 Hz,3H),1.66(s,1H),0.36(s,4H)
ステップ2.化合物12(40 mg、0.098 mmol)および2-メチル-2,6-ジアザスピロ[3.3]ヘプタン(11 mg、0.098 mmol)の0.5 mL DMF溶液をヒューニッヒ塩基(1μL、0.294 mmol)およびHATU(44 mg、0.118 mmol)で処理した。反応混合物をRTで30分間攪拌した。過剰な塩基を蒸発させ、50 g C-18カラムを用いて、0-50% 水/アセトニトリル(0.05% TFA)で溶出させて、粗生成物を逆相ISCOにより精製し、フラクションを凍結乾燥させ、化合物173を白色固体として得た。
以下の化合物を類似的に調製した:化合物171、化合物172、化合物174、化合物175、化合物176、化合物177、化合物178、化合物179、化合物184、化合物190、化合物192、化合物193、化合物194、化合物195、化合物196、化合物197、化合物201、化合物222、化合物225、化合物226、化合物227、化合物228、化合物230、化合物231、化合物234、化合物235、化合物236、化合物237、化合物238、化合物239、および化合物240。
実施例9-化合物149。
ステップ1.化合物11(100 mg、0.258 mmol)、(1,1-ジフルオロスピロ[2.3]ヘキサン-5-イル)メタンアミン(76 mg、0.516 mmol)のDMSO(2 mL)溶液をDBU(0.117 mL、0.774 mmol)で処理した。BOP(171 mg、0.387 mmol)を添加した。反応混合物を40℃で1時間加熱し、NaOH(0.258 mL、1.291 mmol)で処理し、80℃で2時間加熱し、逆相ISCで、50 g C-18カラムを用いて、0-50% 水/アセトニトリル(0.05% TFA)で溶出させて直接精製し、フラクションを凍結乾燥させ、91mgの化合物14を白色固体として得た。
LC/MS [M-H]+=443.2
ステップ2.化合物14(15 mg、0.034 mmol)および2-メチル-2,6-ジアザスピロ[3.3]ヘプタン(3.8 mg、0.034 mmol)の0.5 mL DMF溶液をヒューニッヒ塩基(18μL、0.1 mmol)およびHATU(15.4 mg、0.041 mmol)で処理した。反応物をRTで20分間攪拌した。過剰なアミンを蒸発させ、以下の条件を用いて、粗製物質をプレパラティブLC/MSで精製した:カラム:XBridge C18、200 mm x 19 mm、5μm 粒子;移動相A:5:95 アセトニトリル:NH4OAc含有水;移動相B:95:5 アセトニトリル:NH4OAc含有水;グラジエント:8%Bで0分保持、20分かけて8-48%B、次いで100%Bで0分保持;流速:20mL/分;カラム温度:25℃。フラクション収集はMSシグナルによりトリガーされた。目的物を含むフラクションを混ぜ合わせ、遠心蒸発で乾燥させ、化合物149を白色固体として得た。
以下の化合物を類似的に調製した:化合物150、化合物151、化合物159、化合物160、化合物161、化合物162、化合物163、化合物164、および化合物165。
実施例10-化合物153
ステップ1.化合物7(100 mg、0.278 mmol)、スピロ[3.3]ヘプタン-2-イルメタンアミン(69.7 mg、0.557 mmol)のDMSO(2 mL)溶液をDBU(0.126 mL、0.835 mmol)で処理した。BOP(185 mg、0.417 mmol)を添加した。反応混合物を40℃で1時間加熱し、NaOH(0.278 mL、1.391 mmol)で処理し、80℃で2時間加熱し、逆相ISCで、50 g C-18カラムを用いて、0-50% 水/MeCN(0.05% TFA)で溶出させて直接精製し、フラクションを凍結乾燥させ、化合物16を白色固体として得た。
LC/MS [M+H]+=409.3
ステップ2.化合物16(190 mg、0.465 mmol)のTHF(1 mL)溶液をSOCl2(0.068 mL、0.930 mmol)で処理し、30分間攪拌した。溶媒を蒸発させ、粗製塩化物を次のステップに進めた。前記塩化物(15 mg、0.035 mmol)およびシクロブチルアミン(12 mg、0.176 mmol)の溶液を0.5 mLのDMF中に溶解し、70℃で1時間加熱した。シクロブチルアミンを蒸発させ、以下の条件を用いて、粗製物質をプレパラティブLC/MSで精製した:カラム:XBridge C18、200 mm x 19 mm、5μm 粒子;移動相A:5:95 アセトニトリル:0.05% TFA含有水;移動相B:95:5 アセトニトリル:0.05% TFA含有水;グラジエント:9%Bで0分保持、20分かけて9-49%B、次いで100%Bで0分保持;流速:20mL/分;カラム温度:25℃。フラクション収集はMSシグナルによりトリガーされた。目的物を含むフラクションを混ぜ合わせ、遠心蒸発で乾燥させ、7.9mgの化合物153を得た。
この実施例を通じて、以下の化合物を類似的に調製した:化合物154、化合物155、化合物156、化合物157、化合物158、化合物185、および化合物186。化合物185および化合物186の例において、ステップ1で(6,6-ジフルオロスピロ[3.3]ヘプタン-2-イル)メタンアミンをスピロ[3.3]ヘプタン-2-イルメタンアミンの代わりに使用した。
実施例11-化合物200
ステップ1.化合物7(100 mg、0.258 mmol)、スピロ[3.3]ヘプタン-2-イルメタンアミン(48.5 mg、0.387 mmol)のDMSO(2 mL)溶液をDBU(0.117 mL、0.774 mmol)で処理した。BOP(171 mg、0.387 mmol)を添加した。反応混合物を40℃で1時間加熱し、NaOH(0.258 mL、1.291 mmol)で処理し、80℃で2時間加熱した。逆相ISCで、50 g C-18カラムを用いて、0-50% 水/アセトニトリル(0.05% TFA)で溶出させて、反応生成物を直接精製し、フラクションを凍結乾燥させ、化合物18を白色固体として得た。
LC/MS [M+H]+=422.3
1H NMR(500MHz,DMSO-d6)δ 7.59(s,1H),7.22(s,1H),7.05(d,J = 7.8 Hz,1H),6.46(s,0H),6.37(d,J = 7.8 Hz,1H),5.66(d,J = 12.0 Hz,4H),4.31(s,2H),4.08(s,2H),3.89(s,3H),3.53(s,1H),3.38(t,J = 6.4 Hz,1H),3.31(d,J = 7.6 Hz,1H),3.24(d,J = 7.5 Hz,1H),3.01(d,J = 4.4 Hz,0H),2.31(q,J = 7.7 Hz,1H),2.17(s,3H),1.93-1.86(m,2H),1.85(td,J = 12.7,11.5,3.7 Hz,4H),1.80(d,J = 7.3 Hz,3H),1.72(q,J = 7.7 Hz,2H),1.57-1.50(m,2H)
ステップ2.化合物18(20 mg、0.047 mmol)のDMF(0.5 mL)溶液を2-メチル-2,6-ジアザスピロ[3.3]ヘプタン(5.31 mg、0.047 mmol)で、続いてHATU(21.60 mg、0.057 mmol)およびヒューニッヒ塩基(0.025 mL、0.142 mmol)で処理した。LCMSは、30分後に反応の完了を示した。以下の条件を用いて、粗製物質をプレパラティブLC/MSで精製した:カラム:XBridge C18、200 mm x 19 mm、5μm 粒子;移動相A:5:95 アセトニトリル:NH4OAc含有水;移動相B:95:5 アセトニトリル:NH4OAc含有水;グラジエント:11%Bで0分保持、20分かけて11-51%B、次いで100%Bで0分保持;流速:20mL/分;カラム温度:25℃。フラクション収集はMSおよびUVシグナルによりトリガーされた。目的物を含むフラクションを混ぜ合わせ、遠心蒸発で乾燥させ、化合物200を得た。
以下の化合物を類似的に調製した:化合物180、化合物181、化合物182、化合物187、化合物188、化合物189、化合物202、化合物203、化合物204、および化合物205。化合物187の例において、ステップ1で(6,6-ジフルオロスピロ[3.3]ヘプタン-2-イル)メタンアミンをスピロ[3.3]ヘプタン-2-イルメタンアミンの代わりに使用した。
実施例12-化合物210
ステップ1.化合物7(100 mg、0.278 mmol)および(5-メチルイソキサゾール-3-イル)メタンアミン(62 mg、0.557 mmol)のDMSO(2 mL)溶液をDBU(0.210 mL、1.391 mmol)で処理した。BOP(185 mg、0.417 mmol)を添加した。反応混合物を40℃で1時間加熱し、NaOH(0.278 mL、1.391 mmol)で処理し、80℃で2時間加熱した。逆相ISCで、50 g C-18カラムを用いて、0-50% 水/アセトニトリル(0.05% TFA)で溶出させて、反応混合物を直接精製した。フラクションを凍結乾燥させ、化合物20(白色固体)を得た。
LC/MS [M+H]+=396.1
1H NMR(400MHz,DMSO-d6)δ 8.80(t,J = 5.9 Hz,1H),7.84(s,1H),7.70(s,1H),6.88(s,1H),6.81-6.71(m,2H),6.11(s,1H),5.62(s,2H),4.73(d,J = 5.8 Hz,2H),4.39(s,2H),4.13(d,J = 5.9 Hz,0H),3.61(s,3H),2.29(d,J = 4.0 Hz,3H),1.56-1.43(m,1H),0.59-0.50(m,1H)
ステップ2.化合物20(70 mg、0.177 mmol)のTHF(0.5 mL)溶液をSOCl2(0.026 mL、0.354 mmol)で処理し、RTで30分間攪拌した。溶媒をV-10エバポレーター中で蒸発させ、粗製塩化物を次のステップに進めた。粗製塩化物(18 mg、0.043 mmol)および2,6-ジアザスピロ[3.3]ヘプタン(21 mg、0.217 mmol)を0.5mLのDMSO中で混合し、反応混合物を80℃で1時間加熱した。以下の条件を用いて、粗製物質をプレパラティブLC/MSで精製した:カラム:XBridge C18、200 mm x 19 mm、5μm 粒子;移動相A:5:95 アセトニトリル:NH4OAc含有水;移動相B:95:5 アセトニトリル:NH4OAc含有水;グラジエント:5%Bで0分保持、20分かけて5-45%B、次いで100%Bで0分保持;流速:20mL/分;カラム温度:25℃。フラクション収集はMSシグナルによりトリガーされた。目的物を含むフラクションを混ぜ合わせ、遠心蒸発で乾燥させ、7.4mgの化合物210を白色固体として得た。
以下の化合物を類似的に調製した:化合物211、化合物212、および化合物213。
実施例13-化合物214
ステップ1.メチル 4-((7-ヒドロキシ-5-((メトキシカルボニル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-メトキシベンゾエート(300 mg、0.774 mmol)のDMSO(3.9 mL)溶液を(5-メチルイソキサゾール-3-イル)メタンアミン(174 mg、1.55 mmol)、BOP(411 mg、0.929 mmol)およびDBU(233 μL、1.549 mmol)で処理した。反応混合物をRTで2時間攪拌し、EtOAcで希釈し、H2Oで洗浄した(3回)。有機層をNa2SO4で乾燥させ、濾過し、減圧濃縮して、メチル 3-メトキシ-4-((5-((メトキシカルボニル)アミノ)-7-(((5-メチルイソキサゾール-3-イル)メチル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)ベンゾエート(353 mg、収率95%)を得た。
1H NMR(400MHz,DMSO-d6)δ 9.80(s,1H),7.99-7.93(m,1H),7.77(t,J=5.9 Hz,1H),7.49(d,J=1.5 Hz,1H),7.45(dd,J=7.8,1.5 Hz,1H),6.62(d,J=7.9 Hz,1H),6.10(d,J=0.9 Hz,1H),5.80(s,2H),4.73(d,J=5.9 Hz,2H),3.84(s,3H),3.82(s,3H),3.64(s,3H),2.31(s,3H) LC/MS条件:カラム:Aquity UPLC BEH C18、2.1 mm x 50 mm、1.7 μm 粒子;移動相A:0.05% TFA含有100%水;移動相B:0.05% TFA含有100%アセトニトリル;グラジエント:1分かけて2%Bから98%B、次いで98%Bで0.50分保持;流速:0.8mL/分 LC RT:0.67分 LC/MS(M+H) 482.3
ステップ2.メチル 3-メトキシ-4-((5-((メトキシカルボニル)アミノ)-7-(((5-メチルイソキサゾール-3-イル)メチル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)ベンゾエート(125 mg、0.260 mmol)のジオキサン(1.3 mL)溶液をNaOH(10M水溶液、0.2 mL、2.0 mmol)で処理し、75℃に加熱した。2時間後、反応混合物をRTに冷まし、HCl(4M ジオキサン中、0.52 mL、2.1 mmol)で処理した。得られた溶液を減圧濃縮した。残留物をMeOH/DCM中に再溶解し、再度減圧濃縮して、粗製4-((5-アミノ-7-(((5-メチルイソキサゾール-3-イル)メチル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-メトキシ安息香酸を得た。この粗生成物(40 mg)のDMF(469μL)溶液を2-メチル-2,6-ジアザスピロ[3.3]ヘプタン・2HCl(17 mg、0.094 mmol)、DIEA(57 μl、0.33 mmol)および2,4,6-トリプロピル-1,3,5,2,4,6-トリオキサトリホスホリナン-2,4,6-トリオキシド(50% EtOAc溶液、55.8 μL、0.094 mmol)で処理した。反応混合物をRTで1時間攪拌した。反応混合物をDMF(1 mL)およびH
2O(0.2 mL)で希釈し、PTFEフリットに通して濾過した。以下の条件を用いて、粗製物質をプレパラティブLC/MSで精製した:カラム:XBridge C18、200 mm x 19 mm、5μm 粒子;移動相A:5:95 アセトニトリル:10 mM NH
4OAc含有水;移動相B:95:5 アセトニトリル:10 mM NH
4OAc含有水;グラジエント:5%Bで0分保持、20分かけて5-45%B、次いで100%Bで0分保持;流速:20mL/分;カラム温度:25℃。フラクション収集はMSシグナルによりトリガーされた。目的物を含むフラクションを混ぜ合わせ、遠心蒸発で乾燥させ、化合物214(13.7 mg、収率58%)を得た。
実施例14-化合物198
ステップ1.メチル 4-ニトロ-1H-ピラゾール-5-カルボキシレート(5 g、29.2 mmol)のDMF(30 mL)攪拌溶液にCs2CO3(11.42 g、35.1 mmol)を添加した。氷浴で冷却後、メチル 4-(ブロモメチル)-3-メトキシベンゾエート(7.57 g、29.2 mmol)のDMF(20 mL)溶液を複数回に分けて、5分かけて添加した。ゆっくりと室温に温まるまで反応物をそのままにし、終夜攪拌し、水(150 mL)に注ぎ、EtOAc(3 x 70 mL)で抽出した。合わせた有機相をブライン(4 x 50 mL)で洗浄し、乾燥(MgSO4)させ、濾過し、濃縮した。フラッシュクロマトグラフィー(220 g SiO2カラム、ヘキサン中、0から50%EtOAc)により、メチル 1-(2-メトキシ-4-(メトキシカルボニル)ベンジル)-4-ニトロ-1H-ピラゾール-5-カルボキシレート(1.012 g、2.90 mmol、収率9.92%)を固体として得た。
LC-MS(ES,m/z):[M+H]+ 350.1
1H NMR(400MHz,DMSO-d6)δ 8.40(s,1H),7.57(d,J=7.6 Hz,1H),7.50(s,1H),7.27(d,J=7.9 Hz,1H),5.53(s,2H),3.96(s,3H),3.86(s,3H),3.82(s,3H)
ステップ2.メチル 1-(2-メトキシ-4-(メトキシカルボニル)ベンジル)-4-ニトロ-1H-ピラゾール-5-カルボキシレート(2 g、5.73 mmol)をエタノール(100 mL)中に懸濁した。10%パラジウム炭素(100 mg)を添加し、反応槽を真空にし、水素で6回パージした。水素雰囲気下で反応混合物を終夜攪拌し、EtOH(100 mL)で洗浄しながらCELITE(商標)に通して濾過した。濾液を蒸発乾固させ、メチル 4-アミノ-1-(2-メトキシ-4-(メトキシカルボニル)ベンジル)-1H-ピラゾール-5-カルボキシレート(1.764 g、5.52 mmol、収率96%)を固体として得た。
LC-MS(ES,m/z):[M+H]+ 320.1
1H NMR(400MHz,DMSO-d6)δ 7.50(s,1H),7.46(d,J=7.7 Hz,1H),7.18(s,1H),6.42(d,J=7.9 Hz,1H),5.55(s,2H),5.14(s,2H),3.91(s,3H),3.84(s,3H),3.70(s,3H)
ステップ3.メチル 4-アミノ-1-(2-メトキシ-4-(メトキシカルボニル)ベンジル)-1H-ピラゾール-5-カルボキシレート(1.75 g、5.48 mmol)をMeOH(60 mL)中に懸濁した。1,3-ビス(メトキシカルボニル)-2-メチル-2-チオシュードウレア(1.243 g、6.03 mmol)、続いてHOAc(1.882 mL、32.9 mmol)を添加した。反応混合物をRTで1時間攪拌した。2mLのTFAを添加し、反応混合物を終夜攪拌した。NaOMe(23.69 g、110 mmol、重量25%)を添加し、次にRTで4時間攪拌した。沈殿物を濾過し、MeOH(50 mL)中に懸濁した。NaOMe(3 g、13.88 mmol、重量25%)を添加し、反応物をRTで1時間攪拌した。反応混合物をAcOHで酸性化し、10分間攪拌した後、反応物を濾過し、MeOHで洗浄し、固体メチル 4-((7-ヒドロキシ-5-((メトキシカルボニル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-メトキシベンゾエート(670 mg、1.730 mmol、収率32%)を得た。
LC-MS(ES,m/z):[M+H]+ 388.1 1
H NMR(400MHz,DMSO-d6)δ 7.92(s,1H),7.52(s,1H),7.47(d,J=7.6 Hz,1H),6.70(d,J=7.7 Hz,1H),5.76(s,2H),3.90(s,3H),3.85(s,3H),3.76(s,3H)
ステップ4.20mLシンチレーションバイアルにメチル 4-((7-ヒドロキシ-5-((メトキシカルボニル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-メトキシベンゾエート(180 mg、0.465 mmol)、スピロ[2.3]ヘキサン-5-イルメタンアミン ヒドロクロライド(103 mg、0.697 mmol)、BOP(308 mg、0.697 mmol)およびDMSO(1 mL)を入れた。DBU(0.245 mL、1.626 mmol)を添加した。反応混合物を60℃で2時間攪拌し、冷却し、濾過し、逆相フラッシュクロマトグラフィー(50 g C18カラム、0.05% ギ酸含有水中、0から65% MeCN)を用いて精製し、メチル 3-メトキシ-4-((5-((メトキシカルボニル)アミノ)-7-((スピロ[2.3]ヘキサン-5-イルメチル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)ベンゾエートを得た(165 mg、0.343 mmol、収率73.9%、白色固体)。
LC-MS(ES,m/z):[M+H]+ 481.2
ステップ5.メチル 3-メトキシ-4-((5-((メトキシカルボニル)アミノ)-7-((スピロ[2.3]ヘキサン-5-イルメチル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)ベンゾエート(165 mg、0.343 mmol)をジオキサン(4 mL)中に溶解した。NaOH(1.030 mL、5.15 mmol)を添加し、反応物を80℃で2時間加熱した。冷却後、反応混合物をHClで酸性化し、蒸発乾固させ、生成物を精製せずに次に使用した。
LC-MS(ES,m/z):[M+H]+ 409.3
ステップ6.20mLシンチレーションバイアルに4-((5-アミノ-7-((スピロ[2.3]ヘキサン5-イルメチル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-メトキシ安息香酸(100 mg、0.086 mmol)、HBTU(39.0 mg、0.103 mmol)、1-メチルピペリジン-4-アミン(19.57 mg、0.171 mmol)およびDMF(2 mL)を入れた。DIPEA(0.045 mL、0.257 mmol)を添加した。反応混合物をRTで1時間攪拌し、濾過し、以下の条件を用いて、プレパラティブLC/MSで精製した:カラム:XBridge C18、200 mm x 19 mm、5μm 粒子;移動相A:5:95 アセトニトリル:NH4OAc含有水;移動相B:95:5 アセトニトリル:NH4OAc含有水;グラジエント:0%Bで0分保持、25分かけて0-40%B、次いで100%Bで0分保持;流速:20mL/分;カラム温度:25℃。フラクション収集はMSおよびUVシグナルによりトリガーされた。フラクション化合物198(16.4 mg、0.032 mmol、収率38%)
化合物199を類似的に調製した。
実施例15-化合物207、ジトリフルオロアセテート
ステップ1.メチル 4-ニトロ-1H-ピラゾール-5-カルボキシレート(10 g、58.4 mmol)のEtOH(100 mL)攪拌溶液に10%パラジウム炭素(0.622 g、0.584 mmol)を添加した。反応物を真空にし、水素で6回パージし、次いで水素雰囲気下で2日間攪拌した。反応混合物をCELITE(商標)に通して濾過し、EtOH(100 mL)で洗浄した。濾液を蒸発乾固させ、エーテル/ヘキサンでトリチュレートし、メチル 4-アミノ-1H-ピラゾール-5-カルボキシレート(8.012 g、56.8 mmol、収率97%)を固体として得た。
LC-MS(ES,m/z):[M+H]+ 142.1
ステップ2.メチル 4-アミノ-1H-ピラゾール-5-カルボキシレート(4 g、28.3 mmol)をMeOH(75 mL)中に溶解し、1,3-ビス(メトキシカルボニル)-2-メチル-2-チオシュードウレア(6.43 g、31.2 mmol)、続いて酢酸(6.49 mL、113 mmol)を添加した。反応混合物をRTで5時間攪拌した。NaOMe(36.7 g、170 mmol、重量25%)を添加した。反応混合物をRTで終夜攪拌し、AcOHで酸性化し、濾過し、水(100 mL)、THF(100 mL)およびエーテル(100 mL)で洗浄し、メチル (7-ヒドロキシ-1H-ピラゾロ[4,3-d]ピリミジン-5-イル)カルバメート(5.098 g、24.37 mmol、収率86%)を固体として得た。
LC-MS(ES,m/z):[M+H]+ 210.0
ステップ3.メチル (7-ヒドロキシ-1H-ピラゾロ[4,3-d]ピリミジン-5-イル)カルバメート(5.1 g、24.38 mmol)をDMF(100 mL)中に懸濁した。NBS(4.34 g、24.38 mmol)を添加し、反応物をRTで1時間攪拌した。反応混合物を水(100 mL)でクエンチし、10分間攪拌し、次いで濾過し、水(100 mL)、THF(2 x 50 mL)およびエーテル(2 x 50 mL)で洗浄し、メチル (3-ブロモ-7-ヒドロキシ-1H-ピラゾロ[4,3-d]ピリミジン-5-イル)カルバメート(8.32 g、23.11 mmol、収率95%)を固体として得た。
LC-MS(ES,m/z):[M+H]+ 288.0,290.0
ステップ4.メチル (3-ブロモ-7-ヒドロキシ-1H-ピラゾロ[4,3-d]ピリミジン-5-イル)カルバメート(2.50 g、8.68 mmol)のDMF(35 mL)攪拌溶液にCs2CO3(3.11 g、9.55 mmol)、続いてメチル 4-(ブロモメチル)-3-メトキシベンゾエート(2.249 g、8.68 mmol)のDMF(15 mL)攪拌溶液を添加した。反応混合物をRTで終夜攪拌し、水(400 mL)でクエンチし、EtOAc(3 x 150 mL)で抽出した。合わせた有機相をブライン(4 x 100 mL)で洗浄し、乾燥させ(MgSO4)、濾過し、濃縮した。DCM/エーテル/ヘキサンを用いてトリチュレートし、メチル 4-((3-ブロモ-7-ヒドロキシ-5-((メトキシカルボニル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-メトキシベンゾエート(1.791 g、3.07 mmol、収率35.4%)を得た。LCMSによると生成物は80%純粋であった(残りの20%はN2位置異性体であった)。
LC-MS(ES,m/z):[M+H]+ 466.1,468.1
ステップ5.20mLマイクロウェーブバイアルにメチル 4-((3-ブロモ-7-ヒドロキシ-5-((メトキシカルボニル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-メトキシベンゾエート(500 mg、1.072 mmol)(純度約80%でN2位置異性体が混入している)、2,4,6-トリメチル-1,3,5,2,4,6-トリオキサトリボリナン(TMB、269 mg、2.145 mmol)、[1,1'-ビス(ジフェニルホスフィノ)フェロセン]ジクロロパラジウム(II)(235 mg、0.322 mmol)、K2CO3(296 mg、2.145 mmol)、ジオキサン(12 mL)および水(3 mL)を入れた。反応混合物を電子レンジ内で、120℃で1時間加熱し、蒸発乾固させた。DMSO(3 mL)を残留物に添加した。混合物を濾過し、逆相フラッシュクロマトグラフィー(100 g C18カラム、0.05 TFA含有水中、0から50% アセトニトリル)を用いて精製し、メチル 4-((5-アミノ-7-ヒドロキシ-3-メチル-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-メトキシベンゾエート(117 mg、0.341 mmol、収率31.8%)をオフホワイト固体として得た。
LC-MS(ES,m/z):[M+H]+ 344.1
ステップ6.20mLシンチレーションバイアルにメチル 4-((5-アミノ-7-ヒドロキシ-3-メチル-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-メトキシベンゾエート(130 mg、0.379 mmol)、スピロ[2.3]ヘキサン-5-イルメタンアミン ヒドロクロライド(84 mg、0.568 mmol)、BOP(251 mg、0.568 mmol)およびDMSO(2 mL)を入れた。DBU(0.200 mL、1.325 mmol)を添加した。反応混合物を50℃で1時間攪拌し、冷却し、水(1 mL)で希釈し、濾過し、逆相フラッシュクロマトグラフィー(50 g C18カラム、0.05% TFA含有水中、0から60% アセトニトリル)を用いて精製し、メチル 4-((5-アミノ-3-メチル-7-((スピロ[2.3]ヘキサン-5-イルメチル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-メトキシベンゾエート(80 mg、0.183 mmol、収率48.4%)を固体として得た。
LC-MS(ES,m/z):[M+H]+ 437.3
1H NMR(400MHz,DMSO-d6)δ 8.29(br t,J=5.6 Hz,1H),7.80(br s,2H),7.53-7.46(m,2H),6.79(d,J=7.7 Hz,1H),5.74(s,2H),3.85(s,6H),3.71(br t,J=6.5 Hz,2H),2.78-2.64(m,1H),2.31(s,3H),2.03-1.93(m,2H),1.82(dd,J=12.1,6.4 Hz,2H),0.35(s,4H)
ステップ7.20mLシンチレーションバイアルにメチル 4-((5-アミノ-3-メチル-7-((スピロ[2.3]ヘキサン-5-イルメチル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-メトキシベンゾエート(75 mg、0.172 mmol)、ジオキサン(2 mL)およびNaOH(0.412 mL、2.062 mmol)を入れた。反応混合物を2時間、80℃に加熱し、冷却し、5N HClで中和し、蒸発乾固し、4-((5-アミノ-3-メチル-7-((スピロ[2.3]ヘキサン-5-イルメチル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-メトキシ安息香酸(190 mg、0.157 mmol、収率92%)を固体として得て、精製せずに使用した。
LC-MS(ES,m/z):[M+H]+ 423.3
ステップ8.20mLシンチレーションバイアルに4-((5-アミノ-3-メチル-7-((スピロ[2.3]ヘキサン-5-イルメチル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-メトキシ安息香酸(100 mg、0.083 mmol、純度35%)、HATU(37.8 mg、0.099 mmol)、1-メチルピペリジン-4-アミン(18.92 mg、0.166 mmol)およびDMF(2 mL)を入れた。DIPEA(0.043 mL、0.249 mmol)を添加した。反応混合物をRTで1時間攪拌し、濾過し、以下の条件を用いて、プレパラティブLC/MSで精製した:カラム:XBridge C18、200 mm x 19 mm、5μm 粒子;移動相A:5:95 アセトニトリル:0.05% TFA含有水;移動相B:95:5 アセトニトリル:0.05% TFA含有水;グラジエント:9%Bで0分保持、20分かけて9-49%B、次いで100%Bで0分保持;流速:20mL/分;カラム温度:25℃。フラクション収集はMSおよびUVシグナルによりトリガーされた。目的物を含むフラクションを混ぜ合わせ、遠心蒸発で乾燥させ、化合物207(43.3 mg、0.058 mmol、収率70%)を得た。
化合物208および化合物217を類似的に調製した。
実施例16-化合物218
ステップ1.水酸化カリウム(5N、24.07 mL、120 mmol)の水溶液を、メチル 3-ヒドロキシ-4-メチルベンゾエート(4 g、24.07 mmol)のアセトニトリル(150 mL)冷却(氷浴)溶液に添加した。0℃で5分間攪拌した後、ジエチル (ブロモジフルオロメチル)ホスホネート(12.85 g、48.1 mmol)を添加した。ゆっくりとRTに温まるまで反応混合物をそのままにし、16時間攪拌した。さらにKOH溶液(5N、16 mL、80 mmol)を添加した。反応混合物をRTでさらに30分間攪拌し、水(200 mL)で希釈、EtOAc(3 x 50 mL)で抽出した。合わせた有機相をブライン(2 x 50 mL)で洗浄し、乾燥させ(MgSO4)、濾過し、濃縮した。フラッシュクロマトグラフィー(SiO2カラム、ヘキサン中、0から10% EtOAc)により、メチル 3-(ジフルオロメトキシ)-4-メチルベンゾエート(2.552 g、11.80 mmol、収率49.0%)を油として得た。
LC-MS(ES,m/z):[M+H]+ 217.1
1H NMR(400MHz,DMSO-d6)δ 7.76(dd,J=7.8,1.7 Hz,1H),7.68(br.s,1H),7.51-7.10(m,2H),3.87(s,3H),2.31(s,3H)
ステップ2.NBS(1.811 g、10.18 mmol)およびベンゾイルペルオキシド(0.448 g、1.850 mmol)をメチル 3-(ジフルオロメトキシ)-4-メチルベンゾエート(2 g、9.25 mmol)の四塩化炭素(20 mL)攪拌溶液に添加した。反応物を75℃で4時間、次いでRTで終夜攪拌した。反応混合物を蒸発乾固させ、フラッシュクロマトグラフィー(SiO2カラム、ヘキサン中、0から15% EtOAc)を用いて精製し、メチル 4-(ブロモメチル)-3-(ジフルオロメトキシ)ベンゾエート(1.561 g、5.29 mmol、収率57.2%)を油として得た。
LC-MS(ES,m/z):[M+H]+ 295.0,297.0
1H NMR(400MHz,CDCl3)δ 7.88(dd,J=8.1,1.5 Hz,1H),7.80(s,1H),7.52(d,J=8.1 Hz,1H),6.64(t,J=73.0 Hz,1H),4.57-4.51(m,2H),3.98-3.90(m,3H)
ステップ3.メチル (3-ブロモ-7-ヒドロキシ-1H-ピラゾロ[4,3-d]ピリミジン-5-イル)カルバメート(1.269 g、4.41 mmol)およびCs2CO3(1.579 g、4.85 mmol)のDMF(30 mL)攪拌懸濁液を氷浴で冷却した。メチル 4-(ブロモメチル)-3-(ジフルオロメトキシ)ベンゾエート(1.3 g、4.41 mmol)のDMF(5 mL)溶液を添加した。ゆっくりとRTに温まるまで反応混合物をそのままにし、3時間攪拌した。反応混合物を水(400 mL)に注ぎ、EtOAc(3 x 150 mL)で抽出した。合わせた有機相をブライン(4 x 80 mL)で洗浄し、乾燥させ(MgSO4)、濾過し、濃縮し、メチル 4-((3-ブロモ-7-ヒドロキシ-5-((メトキシカルボニル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-(ジフルオロメトキシ)ベンゾエート(1.69 g、3.37 mmol、収率76%)を固体として得た。
LC-MS(ES,m/z):[M+H]+ 502.1,504.0
1H NMR(400MHz,DMSO-d6)δ 11.72(br s,1H),11.45(br s,1H),7.80(dd,J=7.9,1.3 Hz,1H),7.74(s,1H),7.35(t,J=73.2 Hz,1H),7.26-7.18(m,1H),5.82(s,2H),3.87(s,3H),3.76(s,3H)
ステップ4.メチル 4-((3-ブロモ-7-ヒドロキシ-5-((メトキシカルボニル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-(ジフルオロメトキシ)ベンゾエート(1.6 g、3.19 mmol)のエタノール(150 mL)攪拌懸濁液に10%パラジウム炭素(0.16 g)を添加した。反応混合物を真空にし、水素で6回パージし、水素雰囲気下で24時間攪拌し、CELITE(商標)に通して濾過した。生成物のほとんどが、パラジウムとともにCELITE(商標)に詰まったので、全ての固体物質をCELITE(商標)からこそぎ落として水(100 mL)に入れ、EtOAc(3 x 70 mL)で抽出した。合わせた有機相をブライン(2 x 50 mL)で洗浄し、再度CELITE(商標)に通して濾過した。この濾液を最初の濾液と合わせ、乾燥させ(MgSO4)、濾過、濃縮し、メチル 3-(ジフルオロメトキシ)-4-((7-ヒドロキシ-5-((メトキシカルボニル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)ベンゾエート(1.2 g、2.83 mmol、収率89%)をオフホワイト固体として得た。
LC-MS(ES,m/z):[M+H]+ 424.1
1H NMR(400MHz,DMSO-d6)δ 11.16(br s,1H),7.93(s,1H),7.77(d,J=8.5 Hz,1H),7.73(s,1H),7.36(t,J=73.2 Hz,1H),7.04(d,J=7.9 Hz,1H),5.84(s,2H),3.87(s,3H),3.76(s,3H)
ステップ7.20mLシンチレーションバイアルに、メチル 3-(ジフルオロメトキシ)-4-((7-ヒドロキシ-5-((メトキシカルボニル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)ベンゾエート(1.250 g、2.95 mmol)、スピロ[2.3]ヘキサン-5-イルメタンアミン ヒドロクロライド(0.654 g、4.43 mmol)、BOP(1.959 g、4.43 mmol)およびDMSO(15 mL)を入れた。DBU(1.558 mL、10.33 mmol)を添加し、反応物を50℃で3時間攪拌した。反応混合物を飽和NaHCO3溶液(100 mL)に注ぎ、EtOAc(3 x 50 mL)で抽出した。合わせた有機相をブライン(4 x 50 mL)で洗浄し、乾燥させ(MgSO4)、濾過し、濃縮した。フラッシュクロマトグラフィー(80 g SiO2カラム、CELITE(商標)充填、ヘキサン中、0から100% EtOAc)により、メチル 3-(ジフルオロメトキシ)-4-((5-((メトキシカルボニル)アミノ)-7-((スピロ[2.3]ヘキサン-5-イルメチル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)ベンゾエート(338 mg、0.654 mmol、収率22.16%)を固体として得た。
LC-MS(ES,m/z):[M+H]+ 517.3
ステップ6.メチル 3-(ジフルオロメトキシ)-4-((5-((メトキシカルボニル)アミノ)-7-((スピロ[2.3]ヘキサン-5-イルメチル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)ベンゾエート(330 mg、0.639 mmol)のジオキサン(3600μL)攪拌溶液にNaOH(1278μL、6.39 mmol)を添加した。反応物を2時間、80℃で攪拌した。冷却後、5N HCl(1.28 mL)を用いて反応混合物を中和し、蒸発乾固させた。残留物をDMSO(2 mL)中に懸濁し、水(35 mL)を添加し、生成物を濾過し、水(30 mL)で洗浄して、4-((5-アミノ-7-((スピロ[2.3]ヘキサン-5-イルメチル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-(ジフルオロメトキシ)安息香酸(126 mg、0.284 mmol、収率44.4%)を固体として得た。
LC-MS(ES,m/z):[M+H]+ 445.2
1H NMR(400MHz,DMSO-d6)δ 7.71(s,1H),7.67(d,J=7.9 Hz,1H),7.63(s,1H),7.37(t,J=73.5 Hz,1H),6.80(br t,J=5.4 Hz,1H),6.55(d,J=7.9 Hz,1H),5.92(br s,2H),5.81(s,2H),3.57-3.51(m,2H),2.72-2.57(m,1H),1.98-1.88(m,2H),1.77-1.69(m,2H),0.37-0.25(m,4H)
ステップ7.20mLシンチレーションバイアルに、4-((5-アミノ-7-((スピロ[2.3]ヘキサン-5-イルメチル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-(ジフルオロメトキシ)安息香酸(30 mg、0.068 mmol)、HATU(30.8 mg、0.081 mmol)、(3aR,6aS)-2-メチルオクタヒドロピロロ[3,4-c]ピロール(12.78 mg、0.101 mmol)およびDMF(2 mL)を入れた。DIPEA(0.035 mL、0.203 mmol)を添加した。反応物をRTで終夜攪拌し、濾過し、以下の条件を用いて、プレパラティブLC/MSで精製した:カラム:XBridge C18、200 mm x 19 mm、5μm 粒子;移動相A:5:95 アセトニトリル:NH4OAc含有水;移動相B:95:5 アセトニトリル:NH4OAc含有水;グラジエント:10%Bで0分保持、25分かけて10-50%B、次いで100%Bで0分保持;流速:20mL/分;カラム温度:25℃。フラクション収集はMSおよびUVシグナルによりトリガーされた。目的物を含むフラクションを混ぜ合わせ、遠心蒸発で乾燥させ、化合物218(30.3 mg、収率81%)を得た。
以下の化合物を類似的に調製した:化合物219、化合物220、化合物221、および化合物224。
実施例17-化合物223
ステップ1.Cs2CO3(1329 mg、4.08 mmol)をメチル (3-ブロモ-7-(ブチルアミノ)-1H-ピラゾロ[4,3-d]ピリミジン-5-イル)カルバメート(700 mg、2.040 mmol)のDMF(5 mL)攪拌溶液に添加した。氷浴で冷却後、メチル 4-(ブロモメチル)-3-(ジフルオロメトキシ)ベンゾエート(572 mg、1.938 mmol)のDMF(2 mL)溶液を添加した。RTに温まるまで反応混合物をそのままにし、3時間攪拌した。水(20 mL)を添加し、EtOAc(3 x 5 mL)で反応混合物を抽出した。合わせた有機相をブライン(4 x 10 mL)で洗浄し、乾燥させ(MgSO4)、濾過し、濃縮した。フラッシュクロマトグラフィー(SiO2カラム、DCM充填、ヘキサン中、0から60% EtOAc)により、メチル 4-((3-ブロモ-7-(ブチルアミノ)-5-((メトキシカルボニル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-(ジフルオロメトキシ)ベンゾエート(275 mg、0.493 mmol、収率24.19%)を固体として得た。
LC-MS(ES,m/z):[M+H]+ 557.1,559.1
1H NMR(400MHz,DMSO-d6)δ 9.89(s,1H),7.82-7.69(m,2H),7.61-7.14(m,2H),6.87(d,J=7.9 Hz,1H),5.88(s,2H),3.87(s,3H),3.64(s,3H),3.54-3.45(m,2H),1.58-1.46(m,2H),1.19(dq,J=15.0,7.4 Hz,2H),0.83(t,J=7.3 Hz,3H)
ステップ2.メチル 4-((3-ブロモ-7-(ブチルアミノ)-5-((メトキシカルボニル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-(ジフルオロメトキシ)ベンゾエート(275 mg、0.493 mmol)をエタノール(15 mL)中に溶解した。10% Pd/C(27 mg)を添加した。反応槽を真空にし、水素で6回パージした。反応混合物をH2雰囲気下で2時間攪拌し、濾過し、蒸発乾固させた。残留物をジオキサン(2 mL)中に溶解した。NaOH(0.564 mL、2.82 mmol)を添加した。反応混合物を80℃で2時間攪拌し、冷却し、5N HClで中和し、蒸発乾固させた。残留物をMeOH/水(1:1,8 mL)中に溶解した。メタノールを蒸発により除去した。残った水性懸濁液を濾過し、水で洗浄し、4-((5-アミノ-7-(ブチルアミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-(ジフルオロメトキシ)安息香酸(54 mg、0.133 mmol、収率27%)を固体として得た。
LC-MS(ES,m/z):[M+H]+=407.22
1H NMR(400MHz,DMSO-d6)δ 8.50(br s,1H),7.84(s,2H),7.79-7.68(m,2H),7.63-7.05(t,J=73.2 Hz 1H),6.97(d,J=7.9 Hz,1H),5.94(s,2H),3.54(q,J=6.4 Hz,2H),1.54(quin,J=7.2 Hz,2H),1.19(dq,J=14.9,7.3 Hz,2H),0.84(t,J=7.3 Hz,3H)
ステップ3.20mLシンチレーションバイアルに4-((5-アミノ-7-(ブチルアミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-(ジフルオロメトキシ)安息香酸(50 mg、0.123 mmol)、HATU(56.1 mg、0.148 mmol)、tert-ブチル 2,6-ジアザスピロ[3.3]ヘプタン-2-カルボキシレート(24.39 mg、0.123 mmol)およびDMF(2 mL)を入れた。DIPEA(0.064 mL、0.369 mmol)を添加した。反応混合物をRTで1時間攪拌し、飽和NaHCO
3溶液(10 mL)でクエンチし、EtOAc(3 x 5 mL)で抽出した。合わせた有機相をブライン(4 x 5 mL)で洗浄し、乾燥させ(MgSO
4)、濾過し、濃縮した。残留物をDCM(1.5 mL)中に溶解し、TFA(0.5 mL)を添加した。反応物をRTで30分間攪拌し、次いで蒸発乾固させた。粗製物質をDMF(2 mL)中に溶解し、濾過し、以下の条件を用いて、プレパラティブLC/MSで精製した:カラム:XBridge C18、200 mm x 19 mm、5μm 粒子;移動相A:5:95 アセトニトリル:NH
4OAc含有水;移動相B:95:5 アセトニトリル:NH
4OAc含有水;グラジエント:0%Bで0分保持、20分かけて0-37%B、次いで100%Bで0分保持;流速:20mL/分;カラム温度:25℃。フラクション収集はMSシグナルによりトリガーされた。目的物を含むフラクションを混ぜ合わせ、遠心蒸発で乾燥させ、化合物223(20.9 mg、0.043 mmol、収率35%)を得た。
実施例18-化合物242、トリ-TFA塩
ステップ1.20mLシンチレーションバイアルに、メチル 4-((3-ブロモ-7-ヒドロキシ-5-((メトキシカルボニル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-(ジフルオロメトキシ)ベンゾエート(750 mg、1.493 mmol)、スピロ[2.3]ヘキサン-5-イルメタンアミン ヒドロクロライド(500 mg、2.370 mmol)、BOP(991 mg、2.240 mmol)およびDMSO(7.5 mL)を入れた。DBU(0.788 mL、5.23 mmol)を添加した。反応混合物を50℃で終夜攪拌し、飽和NaHCO3溶液(100 mL)に注ぎ、EtOAc(3 x 50 mL)で抽出した。合わせた有機相をブライン(4 x 50 mL)で洗浄し、乾燥させ(MgSO4)、濾過し、濃縮した。フラッシュクロマトグラフィー(80 g SiO2カラム、ヘキサン中、0から60% EtOAc)により、メチル 4-((3-ブロモ-5-((メトキシカルボニル)アミノ)-7-((スピロ[2.3]ヘキサン-5-イルメチル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-(ジフルオロメトキシ)ベンゾエート(286 mg、0.480 mmol、収率32.2%)を固体として得た。
LC-MS(ES,m/z):[M+H]+ 595.1,597.1
1H NMR(400MHz,DMSO-d6)δ 9.91(s,1H),7.78-7.71(m,2H),7.44(t,J=5.4 Hz,1H),7.38(t,J=73.2 Hz,1H),6.86(d,J=7.9 Hz,1H),5.89(s,2H),3.86(s,3H),3.70-3.59(m,5H),2.76(br t,J=7.2 Hz,1H),2.15-2.03(m,2H),1.80(dd,J=12.1,6.4 Hz,2H),0.32(s,4H)
ステップ2.メチル 4-((3-ブロモ-5-((メトキシカルボニル)アミノ)-7-((スピロ[2.3]ヘキサン-5-イルメチル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-(ジフルオロメトキシ)ベンゾエート(286 mg、0.480 mmol)のエタノール(15 mL)攪拌溶液に10%パラジウム炭素(28 mg)を添加した。反応混合物を真空にし、水素で6回パージし、次いで水素雰囲気下で1時間攪拌した。反応混合物を濾過し、蒸発乾固させて、メチル 3-(ジフルオロメトキシ)-4-((5-((メトキシカルボニル)アミノ)-7-((スピロ[2.3]ヘキサン-5-イルメチル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)ベンゾエート(290 mg、0.477 mmol、収率99%)を白色固体として得た。
LC-MS(ES,m/z):[M+H]+ 517.3
1H NMR(400MHz,DMSO-d6)δ 11.93(br s,1H),8.92(br s,1H),8.17(s,1H),7.80(d,J=7.9 Hz,1H),7.77(s,1H),7.42(t,J=73.0 Hz,1H),7.11(d,J=7.9 Hz,1H),6.04(s,2H),3.92(s,3H),3.90(s,3H),3.82(br t,J=6.5 Hz,2H),2.89-2.75(m,1H),2.03(dd,J=12.1,8.4 Hz,2H),1.92(dd,J=12.2,6.7 Hz,2H),0.40(s,4H)
ステップ3.メチル 3-(ジフルオロメトキシ)-4-((5-((メトキシカルボニル)アミノ)-7-((スピロ[2.3]ヘキサン-5-イルメチル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)ベンゾエート(250 mg、0.484 mmol)のTHF(10 mL)攪拌溶液に、0℃で、LiAlH4(1.065 mL、1.065 mmol)を複数回に分けて、10分かけて添加した。反応混合物を30分間、0℃で攪拌し、次いでロッシェル塩(10 mL、20 w/v)でクエンチした。10分間攪拌した後、反応混合物を50 mLの水が入った分液漏斗に移し、EtOAc(3 x 30 mL)で抽出した。合わせた有機物をブライン(3 x 30 mL)で洗浄し、乾燥させ(MgSO4)、濾過し、濃縮した。フラッシュクロマトグラフィー(24 g SiO2カラム、DCM充填、DCM中、0から10% MeOH)により、メチル (1-(2-(ジフルオロメトキシ)-4-(ヒドロキシメチル)ベンジル)-7-((スピロ[2.3]ヘキサン-5-イルメチル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-5-イル)カルバメート(117 mg、0.240 mmol、収率49.5%)を固体として得た。
LC-MS(ES,m/z):[M+H]+ 489.2
1H NMR(400MHz,DMSO-d6)δ 9.65(s,1H),7.88(s,1H),7.43-6.98(m,4H),6.62(d,J=7.9 Hz,1H),5.79(s,2H),5.29(t,J=5.6 Hz,1H),4.46(d,J=5.5 Hz,2H),3.68-3.59(m,5H),2.78(dt,J=15.0,7.3 Hz,1H),2.00(dd,J=12.0,8.5 Hz,2H),1.83(dd,J=12.2,6.3 Hz,2H),0.35(s,4H)
ステップ4.メチル (1-(2-(ジフルオロメトキシ)-4-(ヒドロキシメチル)ベンジル)-7-((スピロ[2.3]ヘキサン-5-イルメチル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-5-イル)カルバメート(55 mg、0.113 mmol)をDCM(2 mL)中に溶解し、SOCl2(0.025 mL、0.338 mmol)を添加した。反応混合物をRTで30分間攪拌し、次いで蒸発乾固させた。残留物をDMF(2 mL)中に溶解し、tert-ブチル 2,6-ジアザスピロ[3.3]ヘプタン-2-カルボキシレート(33.5 mg、0.169 mmol)、続いてDIPEA(0.059 mL、0.338 mmol)を添加した。反応混合物を50℃で4時間、次いでRTで終夜攪拌し、飽和NaHCO3溶液(10 mL)でクエンチし、EtOAc(3 x 4 mL)で抽出した。合わせた有機相をブライン(3 x 5 mL)で洗浄し、乾燥させ(MgSO4)、濾過し、濃縮した。残留物をDCM(2 mL)中に溶解し、TFA(0.4 mL)を添加した。反応物をRTで1時間攪拌し、次いで蒸発乾固させ、ジオキサン(2 mL)中に再溶解した。NaOH(0.338 mL、1.689 mmol、5N)を添加し、反応物を80℃で1時間攪拌し、冷却、5N HClを用いて中和し、蒸発乾固させた。残留物をDMF(2 mL)中に溶解し、濾過し、以下の条件を用いて、プレパラティブLC/MSで精製した:カラム:XBridge C18、200 mm x 19 mm、5μm 粒子;移動相A:5:95 アセトニトリル:0.05% TFA含有水;移動相B:95:5 アセトニトリル:0.05% TFA含有水;グラジエント:2%Bで0分保持、20分かけて2-42%B、次いで100%Bで0分保持;流速:20mL/分;カラム温度:25℃。フラクション収集はMSおよびUVシグナルによりトリガーされた。目的物を含むフラクションを混ぜ合わせ、遠心蒸発で乾燥させ、化合物242、3TFA塩(21.1 mg、0.025 mmol、収率21.7%)を得た。
化合物243を類似的に調製した。
実施例19-化合物206
ステップ1.DBU(0.856 mL、5.68 mmol)を、メチル 4-((7-ヒドロキシ-5-((メトキシカルボニル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-メトキシベンゾエート(550 mg、1.420 mmol;NaOH処理前の実施例2のステップ6を参照)および(S)-3-アミノヘキサン-1-オール ヒドロクロライド 2(327 mg、2.130 mmol)のDMSO(5 mL)懸濁液に添加した。反応混合物をRTで10分間攪拌すると、混合物は透明な溶液になった。BOP(1256 mg、2.84 mmol)を添加し、反応混合物を70℃で2時間攪拌してからLCMSにかけたところ、出発物質は全く検出されなかった。この溶液を5M NaOH(5 mL、25.00 mmol)溶液で処理し、70℃で0.5時間攪拌した。冷却後、反応混合物をシリンジフィルターディスクに通して濾過した。プレパラティブ逆相C18カラム(150g)で、アセトニトリル:水(0.05% TFA調整剤を含む)=0-50%で溶出させて濾液を精製し、目的のフラクションを凍結および凍結乾燥させて、(S)-4-((5-アミノ-7-((1-ヒドロキシヘキサン-3-イル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-メトキシ安息香酸(860.8 mg、1.246 mmol、収率88%)を得た。
LCMS ESI:計算値 C20H27N6O4=415.2(M+H+)、実測値 415.2(M+H+)
ステップ2.DMF(1 mL)中、(S)-4-((5-アミノ-7-((1-ヒドロキシヘキサン-3-イル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-メトキシ安息香酸(60 mg、0.145 mmol)、2-メチル-2,6-ジアザスピロ[3.3]ヘプタン,2HCl(53.6 mg、0.290 mmol)の混合物をヒューニッヒ塩基(0.126 mL、0.724 mmol)、続いてBOP(96 mg、0.217 mmol)で処理した。反応混合物をRTで3時間攪拌した。以下の条件を用いて、粗製物質をプレパラティブLC/MSで精製した:カラム:XBridge C18、200 mm x 19 mm、5μm 粒子;移動相A:5:95 アセトニトリル:NH4OAc含有水;移動相B:95:5 アセトニトリル:NH4OAc含有水;グラジエント:5%Bで0分保持、25分かけて5-45%B、次いで100%Bで0分保持;流速:20mL/分;カラム温度:25℃。フラクション収集はMSおよびUVシグナルによりトリガーされた。目的物を含むフラクションを混ぜ合わせ、遠心蒸発で乾燥させ、化合物206(15.5 mg、0.030 mmol、収率20.88%)を得た。
以下の化合物を類似的に調製した:化合物209、化合物229、化合物232、および化合物233。
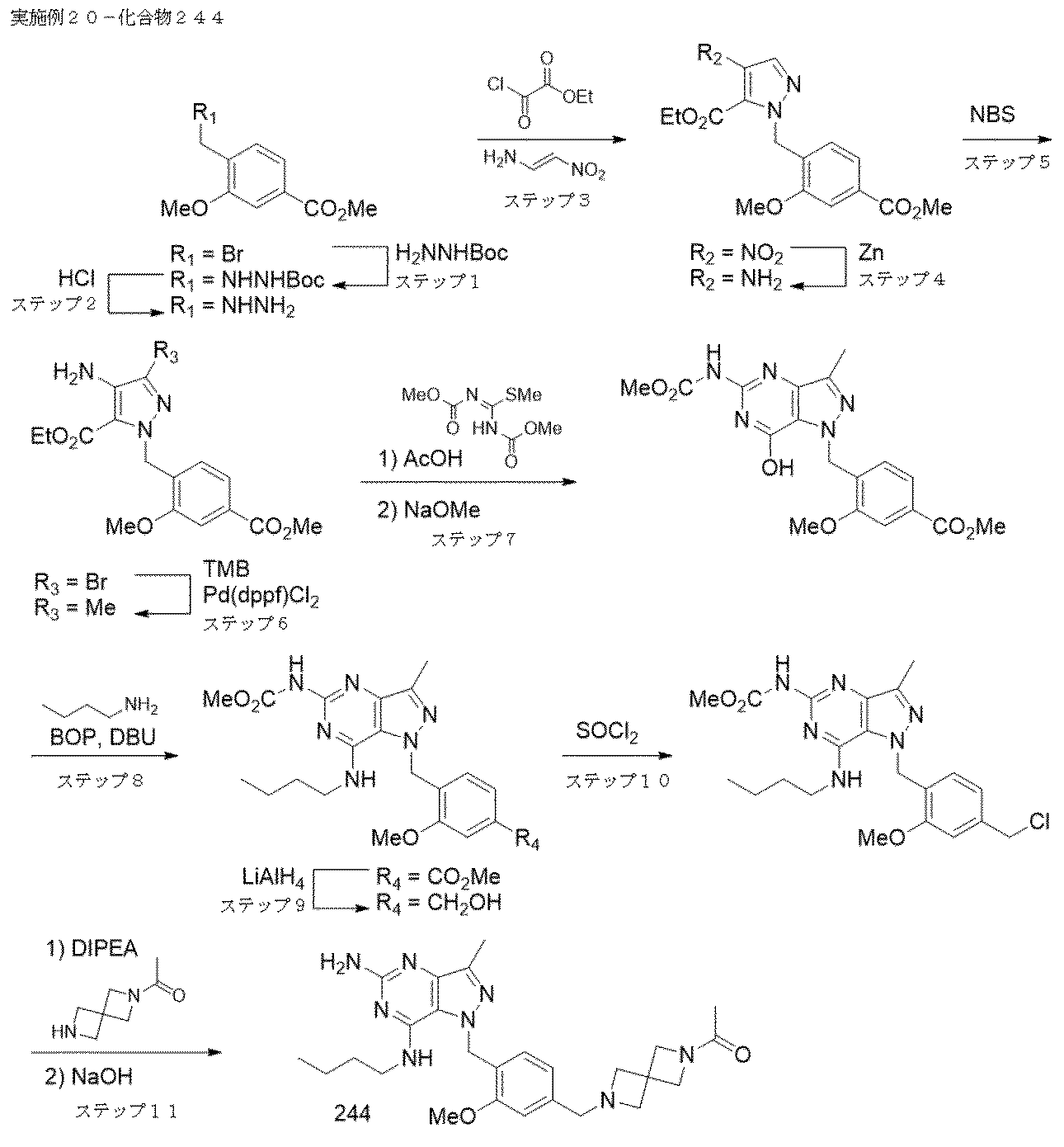
実施例20-化合物244
ステップ1.tert-ブチル ヒドラジンカルボキシレート(12.75 g、96 mmol)およびDIPEAのDMF(24 mL)溶液をRTで、24mLのDMF中、メチル 4-(ブロモメチル)-3-メトキシベンゾエート(5 g、19.30 mmol)を滴下漏斗で1時間かけて滴下することで処理した。反応混合物をRTで終夜攪拌した。EtOAc(135 mL)およびH2O(75 mL)を添加し、二相混合物を30分間攪拌した。反応混合物を分液漏斗に注ぎ、水層を除去した。有機層をH2O(75 mL)でさらに2回、10% LiCl溶液(75 mL)で2回洗浄し、Na2SO4で乾燥させ、濃縮した。カラムクロマトグラフィー(Isco、220 g SiO2、0% CH2Cl2(5分)、次いで15% EtOAc-CH2Cl2)により、tert-ブチル 2-(2-メトキシ-4-(メトキシカルボニル)ベンジル)ヒドラジン-1-カルボキシレートを透明な油(3.85 g)として得た。
LC/MS(M+H) 311.2;LC RT=0.80分(手順E)
1H NMR(400MHz,クロロホルム-d)δ 7.64(dd,J=7.7,1.5 Hz,1H),7.56(d,J=1.5 Hz,1H),7.37(d,J=7.7 Hz,1H),6.08-5.87(m,1H),4.07(s,2H),3.94(d,J=4.6 Hz,6H),1.50-1.40(m,9H)
ステップ2.tert-ブチル 2-(2-メトキシ-4-(メトキシカルボニル)ベンジル)ヒドラジン-1-カルボキシレート(25.4 g、82 mmol)をMeOH(164 mL)中にRTで溶解した。4N HCl-ジオキサン(123 ml、59.5 mmol)を添加し、反応物をRTで終夜攪拌した。白色沈殿を濾過により回収し、乾燥させてメチル 4-(ヒドラジニルメチル)-3-メトキシベンゾエート,ジヒドロクロライド(20 g)を得た。
LC/MS(M+H) 211.1;LC RT=0.51分(手順F)
1H NMR(400MHz,DMSO-d6)δ 9.12(br s),7.62-7.55(m,1H),7.53-7.47(m,2H),4.10(s,2H),3.88(s,3H),3.87(s,3H)
ステップ3.(E)-N,N-ジメチル-2-ニトロエテン-1-アミン(46.4 g、400 mmol)およびピリジン(420 ml、5195 mmol)のCH2Cl2(799 ml)溶液を-10℃に冷却し、エチル 2-クロロ-2-オキソアセテート(51.4 ml、460 mmol)でゆっくりと処理した。25℃に温まるまで2時間の間、反応混合物をそのままにし、終夜攪拌した。CH2Cl2を回転蒸発により除去し、メチル 4-(ヒドラジニルメチル)-3-メトキシベンゾエート ジヒドロクロライド(31.7 g、112 mmol)を一度に添加した。溶液を2時間、RTで攪拌し、溶媒を真空下で除去した。残留物を水、1N HCl水溶液で洗浄し、EtOAcで抽出した。有機層をNa2SO4で乾燥させ、濃縮した。残留物をCH2Cl2中に溶解し、ショートシリカゲルカラムに通し、エタノールから再結晶させて、エチル 1-(2-メトキシ-4-(メトキシカルボニル)ベンジル)-4-ニトロ-1H-ピラゾール-5-カルボキシレート(29.4 g)を得た。
LC/MS(M+Na) 386.0;LC RT=0.98分(手順F)
1H NMR(400MHz,クロロホルム-d)δ 8.06(s,1H),7.64(dd,J=7.9,1.5 Hz,1H),7.56(d,J=1.5 Hz,1H),7.13(d,J=7.8 Hz,1H),5.53(s,2H),4.45(q,J=7.2 Hz,2H),3.94(s,3H),3.88(s,3H),1.37(t,J=7.2 Hz,3H)
ステップ4.ギ酸アンモニウム(1.41 g、22.4 mmol)および亜鉛(0.915 g、14.0 mmol)をエチル 1-(2-メトキシ-4-(メトキシカルボニル)ベンジル)-4-ニトロ-1H-ピラゾール-5-カルボキシレート(2.03 g、5.60 mmol)のTHF(4.67 ml)/MeOH(4.7 ml)溶液に、RTで添加した。反応物をRTで2時間攪拌し、追加量のギ酸アンモニウム(0.353 g、5.60 mmol)および亜鉛(0.229 g、4.67 mmol)を添加した。1時間後、反応混合物をCELITE(商標)のパッドに通して濾過し、濾液を減圧濃縮して白色固体を得た。前記固体をEtOAc中に懸濁し、30分間攪拌し、濾過した。次に、有機濾液を減圧濃縮し、エチル 4-アミノ-1-(2-メトキシ-4-(メトキシカルボニル)ベンジル)-1H-ピラゾール-5-カルボキシレート(1.83 g、98%)を得た。
1H NMR(400MHz,DMSO-d6)δ 7.50-7.49(m,1H),7.48-7.44(m,1H),7.18(s,1H),6.39(d,J=7.8 Hz,1H),5.53(s,2H),5.10(s,2H),4.14(q,J=7.1 Hz,2H),3.90(s,3H),3.83(s,3H),1.13(t,J=7.1 Hz,3H)
LC/MS条件:カラム:Aquity UPLC BEH C18,2.1 mm x 50 mm,1.7 μm 粒子;移動相A:0.05% TFA含有100%水;移動相B:0.05% TFA含有100%アセトニトリル;グラジエント:1分かけて2%Bから98%B、次いで98%Bで0.50分保持;流速:0.8mL/分 LC RT:0.86分 LCMS(M+H)=334.2
ステップ5.エチル 4-アミノ-1-(2-メトキシ-4-(メトキシカルボニル)ベンジル)-1H-ピラゾール-5-カルボキシレート(1.65 g、4.95 mmol)をCHCl3(49.5 ml)中に溶解し、0℃に冷却した。NBS(0.925 g、5.20 mmol)を混合物に、一度に添加した。15分後、反応物をCHCl3で希釈し、10% Na2S2O3水溶液とともに10分間、勢いよく攪拌した。有機相を分離し、H2Oで洗浄し、MgSO4で乾燥させ、濃縮した。粗生成物をカラムクロマトグラフィー(80g SiO2、0から50% EtOAc-ヘキサン グラジエント溶出)により精製し、エチル 4-アミノ-3-ブロモ-1-(2-メトキシ-4-(メトキシカルボニル)ベンジル)-1H-ピラゾール-5-カルボキシレート(1.32 g)を白色固体として得た。
LC/MS(M+H) 412.2/414.2;LC RT=1.02分(手順E)
1H NMR(400MHz,DMSO-d6)δ 7.61-7.41(m,2H),6.55(d,J=8.3 Hz,1H),5.56(s,2H),5.02(s,2H),4.20(q,J=7.1 Hz,2H),3.90(s,3H),3.85(s,3H),1.15(t,J=7.1 Hz,3H)
ステップ6.エチル 4-アミノ-1-(2-メトキシ-4-(メトキシカルボニル)ベンジル)-3-メチル-1H-ピラゾール-5-カルボキシレート(741.2 mg、収率67.1%)、K2CO3(1.098 g、7.94 mmol)および2,4,6-トリメチル-1,3,5,2,4,6-トリオキサトリボリナン(3.5M THF中)(1.816 ml、6.36 mmol)をジオキサン(26.5 ml):水(5.30 ml)(5:1)中に懸濁した。N2気流で反応混合物を5分間通気した後、PdCl2(dppf)-CH2Cl2付加物(0.052 g、0.064 mmol)を添加し、さらに4分間通気し続けた後、反応物を密閉し、90℃に加熱した。3時間後、追加量の2,4,6-トリメチル-1,3,5,2,4,6-トリオキサトリボリナン(3.5M THF中)(0.908 ml、3.18 mmol)およびPdCl2(dppf)-CH2Cl2付加物(0.052 g、0.064 mmol)を添加し、反応物を100℃で16時間攪拌した。冷却した反応混合物を100mLのEtOAcで希釈し、CELITE(商標)に通して濾過し、EtOAcでさらに洗浄した。粗生成物を4 g CELITE(商標)上で濃縮した。カラムクロマトグラフィー(80g SiO2、0から30% EtOAc-CH2Cl2 グラジエント溶出)により、期待される生成物、エチル 4-アミノ-1-(2-メトキシ-4-(メトキシカルボニル)ベンジル)-3-メチル-1H-ピラゾール-5-カルボキシレート(741 mg)をクリーム色の固体として得た。
LC/MS(M+H) 348.2;LC RT=0.89分(手順E)
1H NMR(400MHz,DMSO-d6)δ 7.49(d,J=1.5 Hz,1H),7.46(dd,J=7.9,1.5 Hz,1H),6.40(d,J=7.8 Hz,1H),5.48(s,2H),4.94-4.86(m,2H),4.14(q,J=7.0 Hz,2H),3.90(s,3H),3.84(s,3H),2.10(s,3H),1.15-1.08(m,3H)
ステップ7.エチル 4-アミノ-1-(2-メトキシ-4-(メトキシカルボニル)ベンジル)-3-メチル-1H-ピラゾール-5-カルボキシレート(742 mg、2.136 mmol)をMeOH(10.800 mL)中に懸濁し、勢いよく攪拌しながら穏やかに加熱し、前記物質を可溶化した。1,3-ビス-(メトキシカルボニル)-2-メチル-2-チオシュードウレア(661 mg、3.20 mmol)、続いてAcOH(0.611 mL、10.68 mmol)を添加した。反応混合物をRTで16時間攪拌した。追加量のAcOHを添加し(0.049 mL、0.854 mmol)、次いでRTでさらに72時間攪拌した後、NaOMe(25%wt MeOH中)を添加した(5.69 mL、25.6 mmol)。3時間攪拌した後、反応混合物をAcOHで再び酸性化した。生成物を濾過により回収し、10分間風乾し、実験用乾燥機の中で完全に乾燥させて、メチル 4-((7-ヒドロキシ-5-((メトキシカルボニル)アミノ)-3-メチル-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-メトキシベンゾエート(722.0 mg)をクリーム色の固体として得た。
LC/MS(M+H) 402.3;LC RT=0.86分(手順E)
1H NMR(400MHz,DMSO-d6)δ 11.58-11.17(m,2H),7.51(d,J=1.4 Hz,1H),7.49-7.42(m,1H),6.67(d,J=7.9 Hz,1H),5.67(s,2H),3.90(s,3H),3.84(s,3H),3.71(s,3H),2.31(s,3H)
ステップ8.メチル 4-((7-ヒドロキシ-5-((メトキシカルボニル)アミノ)-3-メチル-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-メトキシベンゾエート(200 mg 、0.498 mmol)およびBOP(331 mg、0.747 mmol)をDMF(2491μl)中にRTで懸濁した。ブタン-1-アミン(64.0μl、0.648 mmol)、続いてDBU(3当量)(225 μl、1.495 mmol)を添加した後、反応混合物は均質になった。反応混合物を40℃で16時間攪拌した。追加量のブタン-1-アミン(64.0 μl、0.648 mmol)、BOP(331 mg、0.747 mmol)およびDBU(3当量)(225 μl、1.495 mmol)を反応物に添加し、40℃で2時間攪拌した後、RTに冷ました。反応混合物をEtOAcおよび飽和NaHCO3の間に分配した。有機層を除去し、水相を3回に分けてEtOAcでさらに抽出した。合わせた有機物を10% LiCl溶液で洗浄し、Na2SO4で乾燥させ、濃縮した。粗生成物をカラムクロマトグラフィー(24g SiO2、0から80% EtOAc-ヘキサン グラジエント溶出)により精製し、メチル 4-((7-(ブチルアミノ)-5-((メトキシカルボニル)アミノ)-3-メチル-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-メトキシベンゾエート(117.4 mg)を得た。
LC/MS(M+H) 457.4;LC RT=0.84分(手順E)
1H NMR(400MHz,クロロホルム-d)δ 7.66(d,J=1.3 Hz,1H),7.64(dd,J=8.0,1.4 Hz,1H),7.11(d,J=7.8 Hz,1H),5.64(s,2H),4.04(s,3H),3.94(s,3H),3.86(s,3H),3.54-3.44(m,2H),2.43(s,3H),1.50(quin,J=7.3 Hz,2H),1.32-1.19(m,2H),0.94-0.87(m,3H)
ステップ9.メチル 4-((7-(ブチルアミノ)-5-((メトキシカルボニル)アミノ)-3-メチル-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-メトキシベンゾエート(117 mg、0.256 mmol)をTHF(854 μl)中にRTで溶解した。LiAlH4(1M THF中)(256 μl、0.256 mmol)を滴下し、反応物をRTで20分間攪拌した。LiAlH4(1M THF中)(256 μl、0.256 mmol)をさらに添加し、反応物をさらに20分間攪拌した。反応混合物をMeOHでクエンチし、ロッシェル塩およびEtOAcで希釈し、16時間攪拌した。有機層を分離し、水相を3回に分けてEtOAcでさらに抽出した。合わせた有機層をNa2SO4で乾燥させ、濃縮し、メチル (7-(ブチルアミノ)-1-(4-(ヒドロキシメチル)-2-メトキシベンジル)-3-メチル-1H-ピラゾロ[4,3-d]ピリミジン-5-イル)カルバメート(86.6 mg)を得た。
LC/MS(M+H) 429.4;LC RT=0.74分(手順E)
1H NMR(400MHz,クロロホルム-d)δ 7.04(s,1H),6.99(d,J=7.8 Hz,1H),6.91-6.86(m,1H),5.58(s,3H),4.70(s,2H),3.97(s,3H),3.81(s,3H),3.50(td,J=6.9,5.6 Hz,2H),2.54(s,3H),1.54-1.43(m,2H),1.31-1.22(m,2H),0.94-0.88(m,3H)
ステップ10.メチル (7-(ブチルアミノ)-1-(4-(ヒドロキシメチル)-2-メトキシベンジル)-3-メチル-1H-ピラゾロ[4,3-d]ピリミジン-5-イル)カルバメート(86 mg、0.201 mmol)をTHF(1004 μl)中にRTで溶解した。SOCl2(73.2 μl、1.004 mmol)を添加した。反応混合物をRTで1時間攪拌し、濃縮してメチル (7-(ブチルアミノ)-1-(4-(クロロメチル)-2-メトキシベンジル)-3-メチル-1H-ピラゾロ[4,3-d]ピリミジン-5-イル)カルバメート(57.1 mg)を得た。
LC/MS(M+H) 447.4;LC RT=0.89分(手順E)
ステップ11.メチル (7-(ブチルアミノ)-1-(4-(クロロメチル)-2-メトキシベンジル)-3-メチル-1H-ピラゾロ[4,3-d]ピリミジン-5-イル)カルバメート(28 mg、0.063 mmol)および1-(2,6-ジアザスピロ[3.3]ヘプタン-2-イル)エタン-1-オン ヒドロクロライド(33.2 mg、0.188 mmol)をアセトニトリル(626 μl)中にRTで溶解した。DIPEA(32.8 μl、0.188 mmol)を添加し、反応混合物を16時間、50℃に加熱した。反応混合物を濃縮し、残留物をジオキサン(0.7 mL)中に再溶解し、NaOH溶液(10 M、125 μl、1.253 mmol)を添加した。反応混合物を3時間、80℃に加熱し、RTに冷まし、濃縮した。残留物をDMF:H
2O:AcOH(6:2:2、1 mL)中に溶解し、PTFEフリットに通して濾過し、以下の条件を用いて、プレパラティブLC/MSで精製した:カラム:XBridge C18、200 mm x 19 mm、5μm 粒子;移動相A:5:95 アセトニトリル:NH
4OAc含有水;移動相B:95:5 アセトニトリル:NH
4OAc含有水;グラジエント:10%Bで0分保持、25分かけて10-50%B、次いで100%Bで0分保持;流速:20mL/分;カラム温度:25℃。フラクション収集はMSおよびUVシグナルによりトリガーされた。目的物を含むフラクションを混ぜ合わせ、遠心蒸発で乾燥させ、0.8当量のAcOHを含む化合物244(4.7 mg)を得た。
実施例21-化合物269
ステップ1.エチル 4-アミノ-1-(2-メトキシ-4-(メトキシカルボニル)ベンジル)-1H-ピラゾール-5-カルボキシレート(1.65 g、4.95 mmol)をCHCl3(49.5 ml)中に溶解し、0℃に冷却した。NBS(0.925 g、5.20 mmol)を一度に添加した。15分後、反応物をCHCl3で希釈し、10% Na2S2O3水溶液とともに10分間勢いよく攪拌した。有機相を分離し、H2Oで洗浄し、MgSO4で乾燥させ、濃縮した。粗生成物をカラムクロマトグラフィー(80g SiO2、0から50% EtOAc-ヘキサン グラジエント溶出)で精製し、エチル 4-アミノ-3-ブロモ-1-(2-メトキシ-4-(メトキシカルボニル)ベンジル)-1H-ピラゾール-5-カルボキシレート(1.32 g)を白色固体として得た。
LC/MS(M+H) 412.2/414.2;LC RT=1.02分(方法A)
1H NMR(400MHz,DMSO-d6)δ 7.61-7.41(m,2H),6.55(d,J=8.3 Hz,1H),5.56(s,2H),5.02(s,2H),4.20(q,J=7.1 Hz,2H),3.90(s,3H),3.85(s,3H),1.15(t,J=7.1 Hz,3H)
ステップ2.エチル 4-アミノ-1-(2-メトキシ-4-(メトキシカルボニル)ベンジル)-3-メチル-1H-ピラゾール-5-カルボキシレート(741.2 mg、収率67.1%)、K2CO3(1.098 g、7.94 mmol)および2,4,6-トリメチル-1,3,5,2,4,6-トリオキサトリボリナン(3.5M THF中)(1.816 ml、6.36 mmol)をジオキサン(26.5 ml):水(5.30 ml)(5:1)中に懸濁した。N2気流で反応混合物を5分間通気した後、PdCl2(dppf)-CH2Cl2付加物(0.052 g、0.064 mmol)を添加し、さらに4分間通気し続けた後、反応槽を密閉し、90℃に加熱した。3時間後、追加量の2,4,6-トリメチル-1,3,5,2,4,6-トリオキサトリボリナン(TMB、3.5M THF中;0.908 ml、3.18 mmol)およびPdCl2(dppf)-CH2Cl2付加物(0.052 g、0.064 mmol)を添加し、反応混合物を100℃で16時間攪拌した。冷却した反応混合物を100mLのEtOAcで希釈し、CELITE(商標)に通して濾過し、EtOAcでさらに洗浄した。粗生成物を4 g CELITE(商標)上で濃縮した。カラムクロマトグラフィー(80g SiO2、0から30% EtOAc-CH2Cl2 グラジエント溶出)により、期待される生成物、エチル 4-アミノ-1-(2-メトキシ-4-(メトキシカルボニル)ベンジル)-3-メチル-1H-ピラゾール-5-カルボキシレート(741 mg)をクリーム色の固体として得た。
LC/MS(M+H) 348.2;LC RT=0.89分(方法A)
1H NMR(400MHz,DMSO-d6)δ 7.49(d,J=1.5 Hz,1H),7.46(dd,J=7.9,1.5 Hz,1H),6.40(d,J=7.8 Hz,1H),5.48(s,2H),4.94-4.86(m,2H),4.14(q,J=7.0 Hz,2H),3.90(s,3H),3.84(s,3H),2.10(s,3H),1.15-1.08(m,3H)
ステップ3.エチル 4-アミノ-1-(2-メトキシ-4-(メトキシカルボニル)ベンジル)-3-メチル-1H-ピラゾール-5-カルボキシレート(742 mg、2.136 mmol)をMeOH(10.800 mL)中に懸濁し、勢いよく攪拌しながら穏やかに加熱し、前記物質を可溶化した。1,3-ビス-(メトキシカルボニル)-2-メチル-2-チオシュードウレア(661 mg、3.20 mmol)、続いてAcOH(0.611 mL、10.68 mmol)を添加した。反応混合物をRTで16時間攪拌した。追加量のAcOHを添加し(0.049 mL、0.854 mmol)、反応物をRTでさらに72時間攪拌した後、NaOMe(25%wt MeOH中)を添加した(5.69 mL、25.6 mmol)。3時間攪拌した後、酸性になるまで反応混合物をAcOHで再び酸性化した。生成物を濾過により回収し、10分間風乾し、実験用乾燥機の中で完全に乾燥させてメチル 4-((7-ヒドロキシ-5-((メトキシカルボニル)アミノ)-3-メチル-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-メトキシベンゾエート(722.0 mg)をクリーム色の固体として得た。
LC/MS(M+H) 402.3;LC RT=0.86分(方法A)
1H NMR(400MHz,DMSO-d6)δ 11.58-11.17(m,2H),7.51(d,J=1.4 Hz,1H),7.49-7.42(m,1H),6.67(d,J=7.9 Hz,1H),5.67(s,2H),3.90(s,3H),3.84(s,3H),3.71(s,3H),2.31(s,3H)
ステップ4.メチル 4-((7-ヒドロキシ-5-((メトキシカルボニル)アミノ)-3-メチル-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-メトキシベンゾエート(300 mg 、0.747 mmol)、(S)-1-((tert-ブチルジフェニルシリル)オキシ)ヘキサン-3-アミン,HCl(381 mg、0.972 mmol)およびBOP(496 mg、1.121 mmol)をDMF(3737 μl)中にRTで懸濁した。DBU(4当量)(451 μl、2.99 mmol)を添加した後、反応混合物は均質になり、これを40℃に加熱した。15分後、追加量のDBU(2当量)(225 μl、1.495 mmol)を添加し、反応物を40℃で16時間攪拌した。(S)-1-((tert-ブチルジフェニルシリル)オキシ)ヘキサン-3-アミン,HCl(381 mg、0.972 mmol)、BOP(496 mg、1.121 mmol)およびDBU(4当量)(451 μl、2.99 mmol)を添加し、反応物をさらに48時間攪拌した。反応混合物をEtOAcで希釈し、H2O(2x)、および10% LiCl溶液(1x)で洗浄した。有機相をNa2SO4で乾燥させ、濃縮した。粗生成物をカラムクロマトグラフィー(24g SiO2、0から80% EtOAc-CH2Cl2 グラジエント溶出)で精製し、次いでさらに精製(12g SiO2、0から70% EtOAc-ヘキサン グラジエント溶出)し、メチル (S)-4-((7-((1-((tert-ブチルジフェニルシリル)オキシ)ヘキサン-3-イル)アミノ)-5-((メトキシカルボニル)アミノ)-3-メチル-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-メトキシベンゾエート(270.6 mg)を得た。
LC/MS(M+H) 739.7;LC RT=1.04分(方法A)
ステップ5.メチル (S)-4-((7-((1-((tert-ブチルジフェニルシリル)オキシ)ヘキサン-3-イル)アミノ)-5-((メトキシカルボニル)アミノ)-3-メチル-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-メトキシベンゾエート(500 mg、0.677 mmol)の乾燥THF(10 mL)およびMeOH(3 mL)溶液に、LiBH4(1.692 mL、3.38 mmol)を窒素雰囲気下で添加した。反応混合物を45℃で24時間加熱した。反応混合物をNH4Cl水溶液およびEtOAcの間に分配した。有機層を水およびブラインで洗浄し、無水Na2SO4で乾燥させ、濃縮した。粗生成物をCombiFlashクロマトグラフィー(60-120シリカゲル;石油エーテル中、10-100% 酢酸エチル)で精製し、(S)-(4-((5-アミノ-7-((1-((tert-ブチルジフェニルシリル)オキシ)ヘキサン-3-イル)アミノ)-3-メチル-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-メトキシフェニル)メタノール(150 mg、0.230 mmol、収率34.0%)を淡黄色固体として得た。
LC/MS(M+H) 653.4
ステップ6:(S)-(4-((5-アミノ-7-((1-((tert-ブチルジフェニルシリル)オキシ)ヘキサン-3-イル)アミノ)-3-メチル-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-メトキシフェニル)メタノール(150 mg、0.230 mmol)のTHF(0.5 mL)攪拌溶液にSOCl2(0.1 ml、1.370 mmol)を添加した。反応混合物を0℃で1時間、窒素雰囲気下で攪拌し、次に減圧濃縮を行い、(S)-N7-(1-((tert-ブチルジフェニルシリル)オキシ)ヘキサン-3-イル)-1-(4-(クロロメチル)-2-メトキシベンジル)-3-メチル-1H-ピラゾロ[4,3-d]ピリミジン-5,7-ジアミンを淡黄色固体として得て、それ以上は精製せずに次のステップに移った。
LC/MS(M+H) 671.4
ステップ7:(S)-N7-(1-((tert-ブチルジフェニルシリル)オキシ)ヘキサン-3-イル)-1-(4-(クロロメチル)-2-メトキシベンジル)-3-メチル-1H-ピラゾロ[4,3-d]ピリミジン-5,7-ジアミン(150 mg、0.223 mmol)のDMF(2 mL)攪拌溶液に、2-メチル-2-アザスピロ[3.3]ヘプタン-6-アミン,HCl(72.7 mg、0.447 mmol)およびK2CO3(61.8 mg、0.447 mmol)を添加した。反応混合物を50℃で3時間攪拌し、次いで濾過した。濾液を減圧濃縮し、(S)-N7-(1-((tert-ブチルジフェニルシリル)オキシ)ヘキサン-3-イル)-1-(2-メトキシ-4-(((2-メチル-2-アザスピロ[3.3]ヘプタン-6-イル)アミノ)メチル)ベンジル)-3-メチル-1H-ピラゾロ[4,3-d]ピリミジン-5,7-ジアミンを薄茶色がかった油として得て、それ以上は精製せずに次のステップに移った。
LC/MS(M+H) 761.5
ステップ8:(S)-N7-(1-((tert-ブチルジフェニルシリル)オキシ)ヘキサン-3-イル)-1-(2-メトキシ-4-(((2-メチル-2-アザスピロ[3.3]ヘプタン-6-イル)アミノ)メチル)ベンジル)-3-メチル-1H-ピラゾロ[4,3-d]ピリミジン-5,7-ジアミン(150 mg、0.197 mmol)のMeOH(3 mL)攪拌溶液にHCl(0.3 mL、9.87 mmol)を添加した。反応混合物を0℃からRTで2時間、窒素雰囲気下で攪拌し、次いで減圧濃縮した。以下の条件を用いて、残留物をプレパラティブHPLCで精製し(カラム:Ascentis Express C18(50 x 2.1 mm),2.7 μm;移動相A:5:95 アセトニトリル:10mM NH
4OAc含有水;移動相B:95:5 アセトニトリル:10mM NH
4OAc含有水;温度:50℃;グラジエント:3分かけて0-100%B;流速:1.1mL/分 インジェクション2条件:カラム:Ascentis Express C18(50 x 2.1 mm),2.7 μm;移動相A:5:95 アセトニトリル:0.1% TFA含有水;移動相B:95:5 アセトニトリル:0.1% TFA含有水;温度:50℃;グラジエント:3分かけて0-100%B;流速:1.1mL/分)、化合物269(14.6 mg、0.027 mmol、収率13.75%)を得た。
実施例22-化合物245
ステップ1.メチル (7-ヒドロキシ-1H-ピラゾロ[4,3-d]ピリミジン-5-イル)カルバメート(2 g、9.56 mmol)およびSelectfluor(商標)(10.16 g、28.7 mmol)をMeCN(20 mL)中に懸濁した。酢酸(2 mL)を添加した。反応混合物を70℃で24時間攪拌し、冷却し、水(100 mL)に注いだ。得られた混合物をフリーザー(-20℃)内に30分間静置した。沈殿した生成物を濾過により回収し、水(40 mL)で洗浄し、メチル (3-フルオロ-7-ヒドロキシ-1H-ピラゾロ[4,3-d]ピリミジン-5-イル)カルバメート(1311 mg、5.77 mmol、収率60.4%)を固体として得た。
LC-MS(ES,m/z):[M+H]+=228.2
1H NMR(400MHz,DMSO-d6) δ 13.69(s,1H),11.63(s,1H),11.26(s,1H),3.76(s,3H)
ステップ2.メチル (3-フルオロ-7-ヒドロキシ-1H-ピラゾロ[4,3-d]ピリミジン-5-イル)カルバメート(1.311 g、5.77 mmol)およびCs2CO3(2.257 g、6.93 mmol)のDMF(5 mL)攪拌懸濁液を氷浴で冷却した。メチル 4-(ブロモメチル)-3-メトキシベンゾエート(1.495 g、5.77 mmol)のDMF(5 mL)溶液を添加した。ゆっくりとRTに温まるまで反応混合物をそのままにし、終夜攪拌し、濾過した。濾液をGenevac装置内で蒸発させた。沈殿物をTHF(100 mL)および水(100 mL)で洗浄し、濾液を別々に回収した。最終沈殿物をDMF濾液およびTHF濾液由来の乾燥物質と混ぜ合わせ、シリカ上で蒸発させ、次いでフラッシュクロマトグラフィー(80 g SiO2カラム、DCM中、0から10% MeOH)を用いて精製し、メチル 4-((3-フルオロ-7-ヒドロキシ-5-((メトキシカルボニル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-メトキシベンゾエート(1.03 g、2.54 mmol、収率44.0%)を固体として得た。
LC-MS(ES,m/z):[M+H]+=406.1
ステップ3.40mLシンチレーションバイアルに、メチル 4-((3-フルオロ-7-ヒドロキシ-5-((メトキシカルボニル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-メトキシベンゾエート(1013 mg、2.499 mmol)、(S)-1-((tert-ブチルジフェニルシリル)オキシ)ヘキサン-3-アミン(1333 mg、3.75 mmol)、BOP(1658 mg、3.75 mmol)、DBU(1.13 mL、7.5 mmol)およびDMSO(10 mL)を入れた。反応混合物を60℃で2時間攪拌し、冷却し、飽和NaHCO3溶液(150 mL)に注ぎ、EtOAc(3 x 60 mL)中に抽出した。合わせた有機相をブライン(4 x 50 mL)で洗浄し、乾燥させ(MgSO4)、濾過し、濃縮した。フラッシュクロマトグラフィー(80 g SiO2カラム、ヘキサン中、0から70% EtOAc)を用いて粗製物質を精製し、メチル (S)-4-((7-((1-((tert-ブチルジフェニルシリル)オキシ)ヘキサン-3-イル)アミノ)-3-フルオロ-5-((メトキシカルボニル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-メトキシベンゾエート(493 mg、0.664 mmol、収率26.6%)を油として得た。
LC-MS(ES,m/z):[M+H]+=743.3
ステップ4.メチル (S)-4-((7-((1-((tert-ブチルジフェニルシリル)オキシ)ヘキサン-3-イル)アミノ)-3-フルオロ-5-((メトキシカルボニル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-メトキシベンゾエート(493 mg、0.664 mmol)のTHF(50 mL)溶液を氷浴で冷却した。LiAlH4(0.697 mL、1.394 mmol)を添加した。反応混合物を0℃で15分間攪拌した。ロッシェル塩(20 mL、20 w/v)を添加し、15分間RTで攪拌した後、反応混合物を水(100 mL)に注ぎ、EtOAc(3 x 50 mL)中に抽出した。合わせた有機相をブライン(3 x 40 mL)で洗浄し、乾燥させ(MgSO4)、濾過し、濃縮した。フラッシュクロマトグラフィー(24 g SiO2カラム、ヘキサン中、0から100% EtOAc)を用いて粗製物質を精製し、メチル (S)-(3-フルオロ-7-((1-ヒドロキシヘキサン-3-イル)アミノ)-1-(4-(ヒドロキシメチル)-2-メトキシベンジル)-1H-ピラゾロ[4,3-d]ピリミジン-5-イル)カルバメート(125 mg、0.262 mmol、収率39.5%)を固体として得た。
LC-MS(ES,m/z):[M+H]+=477.2
ステップ5.メチル (S)-(3-フルオロ-7-((1-ヒドロキシヘキサン-3-イル)アミノ)-1-(4-(ヒドロキシメチル)-2-メトキシベンジル)-1H-ピラゾロ[4,3-d]ピリミジン-5-イル)カルバメート(40 mg、0.084 mmol)のDCM(2 mL)攪拌溶液に、DIPEA(0.044 mL、0.252 mmol)および塩化メタンスルホニル(0.013 mL、0.168 mmol)を添加した。反応混合物をRTで30分間攪拌し、次いで蒸発乾固させた。残留物をDMF(2 mL)中に溶解し、tert-ブチル 2,6-ジアザスピロ[3.3]ヘプタン-2-カルボキシレート(33.3 mg、0.168 mmol)およびDIPEA(0.044 mL、0.252 mmol)を添加した。反応混合物を80℃で2時間攪拌し、冷却し、飽和NaHCO
3溶液(10 mL)でクエンチし、EtOAc(3 x 5 mL)中に抽出した。合わせた有機相をブライン(4 x 5 mL)で洗浄し、乾燥させ(MgSO
4)、濾過し、濃縮した。残留物をDCM(2 mL)中に溶解し、TFA(0.4 mL)を添加した。反応混合物を終夜RTで攪拌し、次いで蒸発乾固させた。次に残留物をジオキサン(2 mL)中に溶解した。NaOH(0.420 mL、2.099 mmol)を添加した。反応混合物を80℃で2時間攪拌し、冷却し、5N HClで酸性化し、蒸発乾固させた。粗製物質をDMF(2 mL)中に溶解し、濾過し、以下の条件を用いて、プレパラティブLC/MSで精製した:カラム:XBridge C18、200 mm x 19 mm、5μm 粒子;移動相A:5:95 アセトニトリル:NH
4OAc含有水;移動相B:95:5 アセトニトリル:NH
4OAc含有水;グラジエント:4%Bで0分保持、20分かけて4-44%B、次いで100%Bで0分保持;流速:20mL/分;カラム温度:25℃。フラクション収集はMSおよびUVシグナルによりトリガーされた。目的物を含むフラクションを混ぜ合わせ、遠心蒸発で乾燥させ、化合物245(6.1 mg、0.012 mmol、収率14.14%)を得た。
実施例23-化合物246
ステップ1.エチル 4-アミノ-1-(2-メトキシ-4-(メトキシカルボニル)ベンジル)-1H-ピラゾール-5-カルボキシレート(1.65 g、4.95 mmol)をCHCl3(49.5 ml)中に溶解し、0℃に冷却した。NBS(0.925 g、5.20 mmol)を反応混合物に、一度に添加した。15分後、反応物をCHCl3で希釈し、10% Na2S2O3水溶液とともに10分間勢いよく攪拌した。有機相を分離し、H2Oで洗浄し、MgSO4で乾燥させ、濃縮した。粗生成物をカラムクロマトグラフィー(80g SiO2、0から50% EtOAc-ヘキサン グラジエント溶出)で精製し、エチル 4-アミノ-3-ブロモ-1-(2-メトキシ-4-(メトキシカルボニル)ベンジル)-1H-ピラゾール-5-カルボキシレート(1.32 g)を白色固体として得た。
LC/MS(M+H) 412.2/414.2;LC RT=1.02分(方法A)
1H NMR(400MHz,DMSO-d6)δ 7.61-7.41(m,2H),6.55(d,J=8.3 Hz,1H),5.56(s,2H),5.02(s,2H),4.20(q,J=7.1 Hz,2H),3.90(s,3H),3.85(s,3H),1.15(t,J=7.1 Hz,3H)
ステップ2.エチル 4-アミノ-1-(2-メトキシ-4-(メトキシカルボニル)ベンジル)-3-メチル-1H-ピラゾール-5-カルボキシレート(741.2 mg、収率67.1%)、K2CO3(1.098 g、7.94 mmol)および2,4,6-トリメチル-1,3,5,2,4,6-トリオキサトリボリナン(3.5M THF中)(1.816 ml、6.36 mmol)をジオキサン(26.5 ml):水(5.30 ml)(5:1)中に懸濁した。N2気流で反応混合物を5分間通気した後、PdCl2(dppf)-CH2Cl2付加物(0.052 g、0.064 mmol)を添加し、さらに4分間通気し続けた後、反応槽を密閉し、90℃に加熱した。3時間後、追加量の2,4,6-トリメチル-1,3,5,2,4,6-トリオキサトリボリナン(3.5M THF中)(0.908 ml、3.18 mmol)およびPdCl2(dppf)-CH2Cl2付加物(0.052 g、0.064 mmol)を添加し、反応混合物を100℃で16時間攪拌した。冷却した反応混合物を100mLのEtOAcで希釈し、CELITE(商標)に通して濾過し、EtOAcでさらに洗浄した。粗生成物を4 g CELITE(商標)上で濃縮した。カラムクロマトグラフィー(80g SiO2、0から30% EtOAc-CH2Cl2 グラジエント溶出)により、期待される生成物、エチル 4-アミノ-1-(2-メトキシ-4-(メトキシカルボニル)ベンジル)-3-メチル-1H-ピラゾール-5-カルボキシレート(741 mg)をクリーム色の固体として得た。
LC/MS(M+H) 348.2;LC RT=0.89分(方法A)
1H NMR(400MHz,DMSO-d6)δ 7.49(d,J=1.5 Hz,1H),7.46(dd,J=7.9,1.5 Hz,1H),6.40(d,J=7.8 Hz,1H),5.48(s,2H),4.94-4.86(m,2H),4.14(q,J=7.0 Hz,2H),3.90(s,3H),3.84(s,3H),2.10(s,3H),1.15-1.08(m,3H)
ステップ3.エチル 4-アミノ-1-(2-メトキシ-4-(メトキシカルボニル)ベンジル)-3-メチル-1H-ピラゾール-5-カルボキシレート(742 mg、2.136 mmol)をMeOH(10.800 mL)中に懸濁し、勢いよく攪拌しながら穏やかに加熱し、前記物質を可溶化した。1,3-ビス-(メトキシカルボニル)-2-メチル-2-チオシュードウレア(661 mg、3.20 mmol)、続いてAcOH(0.611 mL、10.68 mmol)を添加した。反応混合物をRTで16時間攪拌した。追加量のAcOHを添加し(0.049 mL、0.854 mmol)、反応物をRTでさらに72時間攪拌した後、NaOMe(25%wt MeOH中)を添加した(5.69 mL、25.6 mmol)。3時間攪拌した後、酸性になるまで反応混合物をAcOHで再び酸性化した。生成物を濾過により回収し、10分間風乾し、実験用乾燥機の中で完全に乾燥させてメチル 4-((7-ヒドロキシ-5-((メトキシカルボニル)アミノ)-3-メチル-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-メトキシベンゾエート(722.0 mg)をクリーム色の固体として得た。
LC/MS(M+H) 402.3;LC RT=0.86分(方法A)
1H NMR(400MHz,DMSO-d6)δ 11.58-11.17(m,2H),7.51(d,J=1.4 Hz,1H),7.49-7.42(m,1H),6.67(d,J=7.9 Hz,1H),5.67(s,2H),3.90(s,3H),3.84(s,3H),3.71(s,3H),2.31(s,3H)
ステップ4.メチル 4-((7-ヒドロキシ-5-((メトキシカルボニル)アミノ)-3-メチル-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-メトキシベンゾエート(300 mg、0.747 mmol)、(S)-1-((tert-ブチルジフェニルシリル)オキシ)ヘキサン-3-アミン,HCl(381 mg、0.972 mmol)およびBOP(496 mg、1.121 mmol)をDMF(3737 μl)中にRTで懸濁した。DBU(4当量)(451 μl、2.99 mmol)を添加した後、反応混合物は均質になり、これを40℃に加熱した。15分後、追加量のDBU(2当量)(225 μl、1.495 mmol)を添加し、反応物を40℃で16時間攪拌した。(S)-1-((tert-ブチルジフェニルシリル)オキシ)ヘキサン-3-アミン,HCl(381 mg、0.972 mmol)、BOP(496 mg、1.121 mmol)およびDBU(4当量)(451 μl、2.99 mmol)を添加し、反応物をさらに48時間攪拌した。反応混合物をEtOAcで希釈し、H2O(2x)、および10% LiCl溶液(1x)で洗浄した。有機相をNa2SO4で乾燥させ、濃縮した。粗生成物をカラムクロマトグラフィー(24g SiO2、0から80% EtOAc-CH2Cl2 グラジエント溶出)で精製し、次いでさらに精製(12g SiO2、0から70% EtOAc-ヘキサン グラジエント溶出)し、メチル (S)-4-((7-((1-((tert-ブチルジフェニルシリル)オキシ)ヘキサン-3-イル)アミノ)-5-((メトキシカルボニル)アミノ)-3-メチル-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-メトキシベンゾエート(270.6 mg)を得た。
LC/MS(M+H) 739.7;LC RT=1.04分(方法A)
ステップ5.メチル (S)-4-((7-((1-((tert-ブチルジフェニルシリル)オキシ)ヘキサン-3-イル)アミノ)-5-((メトキシカルボニル)アミノ)-3-メチル-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-メトキシベンゾエート(270 mg、0.365 mmol)をTHF(3654 μl)中にRTで溶解した。LiAlH4(731 μl、0.731 mmol)を5分かけて滴下した。反応混合物を15分間RTで攪拌し、MeOHおよびロッシェル塩でクエンチした。EtOAcを添加し、層が透明になるまで混合物を3時間攪拌した。有機相を除去し、水層を3回に分けてEtOAcでさらに抽出した。合わせた有機相をブラインで洗浄し、Na2SO4で乾燥させ、濃縮した。カラムクロマトグラフィー(12g SiO2、0から100% EtOAc-ヘキサン グラジエント溶出、次いで0から20% MeOH-CH2Cl2)により、期待される物質、メチル (S)-(7-((1-((tert-ブチルジフェニルシリル)オキシ)ヘキサン-3-イル)アミノ)-1-(4-(ヒドロキシルメチル)-2-メトキシベンジル)-3-メチル-1H-ピラゾロ[4,3-d]ピリミジン-5-イル)カルバメート(61.7 mg)を得た。
LC/MS(M+H) 711.4;LC RT=1.08分(方法A)
ステップ6.メチル (S)-(7-((1-((tert-ブチルジフェニルシリル)オキシ)ヘキサン-3-イル)アミノ)-1-(4-(ヒドロキシメチル)-2-メトキシベンジル)-3-メチル-1H-ピラゾロ[4,3-d]ピリミジン-5-イル)カルバメート(60 mg、0.084 mmol)をCH2Cl2(844 μl)中にRTで溶解した。SOCl2(30.8 μL、0.422 mmol)を添加し、反応物を 時間攪拌した。濃縮して、期待される生成物、メチル (S)-(7-((1-((tert-ブチルジフェニルシリル)オキシ)ヘキサン-3-イル)アミノ)-1-(4-(クロロメチル)-2-メトキシベンジル)-3-メチル-1H-ピラゾロ[4,3-d]ピリミジン-5-イル)カルバメート(56.6 mg)を得た。
LC/MS(M+H) 729.3;LC RT=1.18分(方法A)
ステップ7.メチル (S)-(7-((1-((tert-ブチルジフェニルシリル)オキシ)ヘキサン-3-イル)アミノ)-1-(4-(クロロメチル)-2-メトキシベンジル)-3-メチル-1H-ピラゾロ[4,3-d]ピリミジン-5-イル)カルバメート(45 mg、0.02 mmol)をアセトニトリル(620 μL)中にRTで溶解した。tert-ブチル 2,6-ジアザスピロ[3.3]ヘプタン-2-カルボキシレート,HCl(29.0 mg、0.123 mmol)、続いてDIPEA(21.55 μl、0.123 mmol)を添加した。反応混合物を80℃で16時間加熱し、濃縮した。残留物をカラムクロマトグラフィー(4g SiO2、0-5% MeOH-CH2Cl2グラジエント)で精製した。いくつかの副生成物をカラムに通し、部分的に精製されたtert-ブチル (S)-6-(4-((7-((1-((tert-ブチルジフェニルシリル)オキシ)ヘキサン-3-イル)アミノ)-5-((メトキシカルボニル)アミノ)-3-メチル-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-メトキシベンジル)-2,6-ジアザスピロ[3.3]ヘプタン-2-カルボキシレート(42 mg)を得た。
LC/MS(M+H) 892.7;LC RT=0.995分(方法A)
ステップ8.tert-ブチル (S)-6-(4-((7-((1-((tert-ブチルジフェニルシリル)オキシ)ヘキサン-3-イル)アミノ)-5-((メトキシカルボニル)アミノ)-3-メチル-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-メトキシベンジル)-2,6-ジアザスピロ[3.3]ヘプタン-2-カルボキシレート(42 mg、0.047 mmol)をCH
2Cl
2(471 μl)中にRTで溶解した。TFA(100 μL)を添加した。2時間後、反応物をN
2気流下で濃縮し、ジオキサン(470 μL)中に再溶解した。トリエチルアミントリヒドロフルオライド(16.79 μL、0.103 mmol)を添加し、反応物を70℃に加熱した。45分後、10M NaOH水溶液(61.3 μl、0.613 mmol)を添加した。反応混合物を70℃で16時間攪拌し、AcOH(54 μL)でクエンチし、N
2気流下で濃縮し、DMF-H
2Oで希釈し、PTFEフリットに通して濾過し、以下の条件を用いて、プレパラティブLC/MSで精製した:カラム:XBridge C18、200 mm x 19 mm、5μm 粒子;移動相A:5:95 アセトニトリル:NH
4OAc含有水;移動相B:95:5 アセトニトリル:NH
4OAc含有水;グラジエント:1%Bで0分保持、20分かけて1-41%B、次いで100%Bで0分保持;流速:20mL/分;カラム温度:25℃。生成物を含むフラクション-収集はMSシグナルによりトリガーされた-を混ぜ合わせ、遠心蒸発で乾燥させ、化合物246(2.9 mg)を得た。
実施例24-化合物247
ステップ1.メチル 4-((7-ヒドロキシ-5-((メトキシカルボニル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-メトキシベンゾエート(510 mg、1.32 mmol;US 2020/0038403 A1、図2A、化合物16)のDMSO(6.6 mL)溶液を(5-メチル-1,2,4-オキサジアゾール-3-イル)メタンアミン・HCl(236 mg、1.58 mmol)、BOP(698 mg、1.58 mmol)およびDBU(595 μL、3.95 mmol)で処理した。反応物をRTで攪拌した。16時間後、追加量の(5-メチル-1,2,4-オキサジアゾール-3-イル)メタンアミン・HCl(50 mg、0.33 mmol)、BOP(50 mg、0.11 mmol)およびDBU(200 μL、1.33 mmol)を添加し、反応物をRTで2時間攪拌した。反応混合物をEtOAcで希釈し、H2O(4x)で洗浄した。有機層をCELITE(商標)上に吸収させ、カラムクロマトグラフィー(100g C18 gold column;移動相A:5:95 アセトニトリル:0.05% TFA含有水;移動相B:95:5 アセトニトリル:0.05% TFA含有水;流速:60mL/分、20-60%グラジエント)で精製した。目的物を含むフラクションを混ぜ合わせ、HCl(1M H2O中、2 mL、2 mmol)で処理し、減圧濃縮して、メチル 3-メトキシ-4-((5-((メトキシカルボニル)アミノ)-7-(((5-メチル-1,2,4-オキサジアゾール-3-イル)メチル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)ベンゾエート(382 mg、収率60%)を得た。
1H NMR(400MHz,DMSO-d6) δ 9.72-9.70(m,1H),7.96-7.94(m,1H),7.83-7.76(m,1H),7.49(d,J=1.4 Hz,1H),7.46(dd,J=7.8,1.5 Hz,1H),6.74(d,J=7.8 Hz,1H),5.79(s,2H),4.86(d,J=5.8 Hz,2H),3.86(s,3H),3.84(s,3H),3.60(s,3H),2.54(s,3H)
LC RT:0.64分 LC/MS[M+H]+ 483.3(方法E)
ステップ2.メチル 3-メトキシ-4-((5-((メトキシカルボニル)アミノ)-7-(((5-メチル-1,2,4-オキサジアゾール-3-イル)メチル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)ベンゾエート(382 mg、0.791 mmol)のジオキサン(9.0 mL)溶液をNaOH(10M 水溶液、0.32 mL、3.2 mmol)で処理し、40℃に加熱した。30分後、温度を60℃に上昇させた。追加量のNaOH(10M水溶液、450 μL、3 mmol)およびMeOH(1 mL)を6時間かけて反応混合物に添加した。反応混合物をRTに冷まし、HOAcで中和し、減圧濃縮した。粗生成物をMeOH中に溶解し、PTFEフリットに通して濾過し、以下の条件を用いて、プレパラティブHPLCで精製した:カラム:Axia C18 100 mm x 30 mm、5μm 粒子;移動相A:10:90 メタノール:0.1% TFA含有水;移動相B:90:10 メタノール:0.1% TFA含有水;グラジエント:15%Bで0分保持、10分かけて15-30%B、次いで30%Bで4分保持;流速:40mL/分;220nmでUV検出;カラム温度:25℃。目的物を含むフラクションを混ぜ合わせ、HCl(1M H2O中、2 mL、2 mmol)で処理し、減圧濃縮して、4-((5-アミノ-7-(((5-メチル-1,2,4-オキサジアゾール-3-イル)メチル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-メトキシ安息香酸・HCl(98.9 mg、収率28%)を得た。
1H NMR(400MHz,DMSO-d6)δ 13.23-12.93(m,1H),12.67-12.43(m,1H),9.06-8.92(m,1H),8.03-7.87(m,2H),7.83(s,1H),7.51-7.46(m,2H),6.98(d,J=8.2 Hz,1H),5.80(s,2H),4.91(d,J=5.7 Hz,2H),3.82(s,3H),2.57(s,3H) LC RT:0.52分
LC/MS[M+H]+ 411.3(方法E)
ステップ3.4-((5-アミノ-7-(((5-メチル-1,2,4-オキサジアゾール-3-イル)メチル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-メトキシ安息香酸・HCl(25 mg、0.056 mmol)のDMF(0.6 mL)溶液を、2-メチル-2,6-ジアザスピロ[3.3]ヘプタン・2HCl(20.7 mg、0.112 mmol)、DIEA(68 μL、0.39 mmol)および2,4,6-トリプロピル-1,3,5,2,4,6-トリオキサトリホスホリナン-2,4,6-トリオキシド(EtOAc中、50%溶液、67 μL、0.11 mmol)で処理した。反応物をRTで攪拌した。16時間後、反応混合物をDMF(1 mL)およびH2O(0.2 mL)で希釈し、PTFEフリットに通して濾過した。以下の条件を用いて、粗製物質をプレパラティブLC/MSで精製した:カラム:XBridge C18、200 mm x 19 mm、5μm 粒子;移動相A:5:95 アセトニトリル:0.05% TFA含有水;移動相B:95:5 アセトニトリル:0.05% TFA含有水;グラジエント:0%Bで0分保持、26分かけて0-40%B、次いで100%Bで0分保持;流速:20mL/分;カラム温度:25℃。フラクション収集はMSシグナルによりトリガーされた。目的物を含むフラクションを混ぜ合わせ、遠心蒸発で乾燥させ、化合物247をビスTFA塩として得た(20.5 mg、収率72%)。
化合物248を類似的に調製した。
実施例25-化合物254
ステップ1:メチル(7-ヒドロキシ-1-(4-(ヒドロキシメチル)-2-メトキシベンジル)-1H-ピラゾロ[4,3-d]ピリミジン-5-イル)カルバメート(700 mg、1.95 mmol;US 2020/0038403 A1;図7、化合物64)のDMSO(9.7 mL)溶液を、(5-メチル-1,2,4-オキサジアゾール-3-イル)メタンアミン・HCl(379 mg、2.53 mmol)、BOP(129 mg、2.92 mmol)およびDBU(1.0 mL、6.8 mmol)で処理した。反応混合物をRTで2時間攪拌し、DCMで希釈し、H2Oで洗浄した。有機層をH2O(6x)で洗浄し、Na2SO4で乾燥させ、濾過し、減圧濃縮した。残留物をDCM/MeOH中に溶解し、CELITE(商標)上に吸収させ、カラムクロマトグラフィー(100g C18 gold column;移動相A:5:95 アセトニトリル:0.05% TFA含有水;移動相B:95:5 アセトニトリル:0.05% TFA含有水;流速:60mL/分、10-50%グラジエント)で精製した。精製した生成物をDCM中に溶解し、飽和NaHCO3水溶液で洗浄した。有機層をNa2SO4で乾燥させ、濾過し、減圧濃縮して、メチル (1-(4-(ヒドロキシメチル)-2-メトキシベンジル)-7-(((5-メチル-1,2,4-オキサジアゾール-3-イル)メチル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-5-イル)カルバメート(372 mg、収率42%)を得た。
1H NMR(400MHz,DMSO-d6)δ 9.69-9.66(m,1H),7.89(s,1H),7.76(t,J=5.8 Hz,1H),6.95(s,1H),6.81-6.77(m,1H),6.76-6.70(m,1H),5.69(s,2H),5.17(t,J=5.7 Hz,1H),4.89(d,J=5.7 Hz,2H),4.45(d,J=5.8 Hz,2H),3.77(s,3H),3.60(s,3H),2.56(s,3H)
LC RT:0.56分 LC/MS[M+H]+ 455.3(方法E)
ステップ2:メチル (1-(4-(ヒドロキシメチル)-2-メトキシベンジル)-7-(((5-メチル-1,2,4-オキサジアゾール-3-イル)メチル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-5-イル)カルバメート(372 mg、0.818 mmol)のDCM(8.2 mL)溶液をSOCl2(179 μL、2.46 mmol)で処理した。反応混合物をRTで10分間攪拌し、減圧濃縮した。残留物をDCM中に再溶解し、減圧濃縮して、メチル (1-(4-(クロロメチル)-2-メトキシベンジル)-7-(((5-メチル-1,2,4-オキサジアゾール-3-イル)メチル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-5-イル)カルバメート(387 mg、100%)を得た。
1H NMR(400MHz,DMSO-d6)δ 11.82-11.60(m,1H),9.40-9.21(m,1H),8.12-8.08(m,1H),7.10(s,1H),7.04-6.95(m,2H),5.81(s,2H),5.02(br d,J=5.3 Hz,2H),4.74(s,2H),3.80(s,3H),3.75(s,3H),2.60(s,3H) LC RT:0.70分
LC/MS[M+H]+=473.3(方法E)
ステップ3:メチル (1-(4-(クロロメチル)-2-メトキシベンジル)-7-(((5-メチル-1,2,4-オキサジアゾール-3-イル)メチル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-5-イル)カルバメート(45 mg、0.095 mmol)のDMF(1.9 mL)溶液を、DIEA(83 μL、0.48 mmol)および2-チア-6-アザスピロ[3.3]ヘプタン 2,2-ジオキシド・HCl(26.2 mg、0.143 mmol)で処理した。反応混合物を60℃で6時間攪拌し、減圧濃縮した。残留物をジオキサン(0.7 mL)中に溶解し、NaOH(10M 水溶液、76 μL、0.76 mmol)で処理し、60℃に5時間加熱した。反応混合物をRTで、HOAcで中和し、減圧濃縮した。粗生成物をDMFおよびH
2O中に溶解し、PTFEフリットに通して濾過し、以下の条件を用いて、プレパラティブLC/MSで精製した:カラム:XBridge C18、200 mm x 19 mm、5μm 粒子;移動相A:5:95 アセトニトリル:10mM NH
4OAc含有水;移動相B:95:5 アセトニトリル:10mM NH
4OAc含有水;グラジエント:1%Bで0分保持、20分かけて1-41%B、次いで100%Bで0分保持;流速:20mL/分;カラム温度:25℃。フラクション収集はMSシグナルによりトリガーされた。目的物を含むフラクションを混ぜ合わせ、遠心蒸発で乾燥させた。以下の条件を用いて、分離した生成物をプレパラティブLC/MSでさらに精製した:カラム:XBridge C18、200 mm x 19 mm、5μm 粒子;移動相A:5:95 アセトニトリル:0.05% TFA含有水;移動相B:95:5 アセトニトリル:0.05% TFA含有水;グラジエント:0%Bで0分保持、20分かけて0-40%B、次いで100%Bで0分保持;流速:20mL/分;カラム温度:25℃。フラクション収集はMSシグナルによりトリガーされた。目的物を含むフラクションを混ぜ合わせ、遠心蒸発で乾燥させ、化合物254(11.7 mg、16%)を得た。
実施例26-化合物263
ステップ1.メチル 4-((5-((tert-ブトキシカルボニル)アミノ)-7-ヒドロキシ-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-メトキシベンゾエート(685 mg、1.59 mmol;US 2020/0038403 A1、図8、化合物71)のTHF(16 mL)溶液を0℃に冷却し、LiAlH4(1M THF中、2.8 mL、2.8 mmol)で処理した。反応混合物を15分間、0℃で攪拌し、H2Oおよびロッシェル塩(飽和水溶液)でクエンチし、RTで3時間攪拌した。有機層をCELITE(商標)上に吸収させ、カラムクロマトグラフィー(24g SiO2;0から20% MeOH-DCM グラジエント溶出)で精製し、tert-ブチル (7-ヒドロキシ-1-(4-(ヒドロキシメチル)-2-メトキシベンジル)-1H-ピラゾロ[4,3-d]ピリミジン-5-イル)カルバメート(460 mg、収率72%)を得た。
1H(400MHz,DMSO-d6)δ 11.69-11.43(m,1H),10.95-10.62(m,1H),7.87-7.79(m,1H),6.97(s,1H),6.77(d,J=7.7 Hz,1H),6.59(d,J=7.8 Hz,1H),5.66(s,2H),5.16(t,J=5.8 Hz,1H),4.45(d,J=5.8 Hz,2H),3.79(s,3H),1.49(s,9H)
LC RT:0.77分 LC/MS[M+H]+=402.2(方法E)
ステップ2.tert-ブチル (7-ヒドロキシ-1-(4-(ヒドロキシメチル)-2-メトキシベンジル)-1H-ピラゾロ[4,3-d]ピリミジン-5-イル)カルバメート(460 mg、1.15 mmol)のDMSO(5.7 mL)溶液を、(5-メチル-1,2,4-オキサジアゾール-3-イル)メタンアミン・HCl(223 mg、1.49 mmol)、BOP(760 mg、1.72 mmol)およびDBU(0.69 mL、4.6 mmol)で処理した。反応混合物をRTで2時間攪拌し、EtOAcで希釈し、H2O(2x)で洗浄した。有機層をCELITE(商標)上に吸収させ、カラムクロマトグラフィー(100g C18 gold column;移動相A:5:95 アセトニトリル:0.05% TFA含有水;移動相B:95:5 アセトニトリル:0.05% TFA含有水;流速:60mL/分、30-50%グラジエント)で精製した。精製した生成物をDCM中に溶解し、飽和NaHCO3水溶液で洗浄した。有機層をNa2SO4で乾燥させ、濾過し、減圧濃縮して、tert-ブチル (1-(4-(ヒドロキシメチル)-2-メトキシベンジル)-7-(((5-メチル-1,2,4-オキサジアゾール-3-イル)メチル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-5-イル)カルバメート(190 mg、収率33%)を得た。
1H NMR(400MHz,DMSO-d6)δ 9.24-9.15(m,1H),7.87(s,1H),7.72(t,J=5.8 Hz,1H),6.95(s,1H),6.82-6.75(m,1H),6.73-6.68(m,1H),5.68(s,2H),5.17(t,J=5.7 Hz,1H),4.87(d,J=5.7 Hz,2H),4.44(d,J=5.7 Hz,2H),3.76(s,3H),2.55(s,3H),1.43(s,9H)
LC RT:0.75分 LC/MS[M+H]+=497.2(方法E)
ステップ3.tert-ブチル (1-(4-(ヒドロキシメチル)-2-メトキシベンジル)-7-(((5-メチル-1,2,4-オキサジアゾール-3-イル)メチル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-5-イル)カルバメート(91.5 mg、0.184 mmol)のジオキサン(0.6 mL)溶液をHCl(4M ジオキサン中、0.69 mL、2.8 mmol)で処理し、40℃で90分間攪拌し、濃縮した。残留物をDCM中に溶解し、減圧濃縮して、(4-((5-アミノ-7-(((5-メチル-1,2,4-オキサジアゾール-3-イル)メチル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-メトキシフェニル)メタノール(73.1 mg、収率100%)を得た。
LC RT:0.65分 LC/MS[M+H]+=397.1(方法E)
ステップ4.1-(4-(クロロメチル)-2-メトキシベンジル)-N7-((5-メチル-1,2,4-オキサジアゾール-3-イル)メチル)-1H-ピラゾロ[4,3-d]ピリミジン-5,7-ジアミン(27 mg、0.065 mmol)のDMSO(1.3 mL)溶液を、DIEA(57 μL、0.33 mmol)および2-イソプロピル-2,6-ジアザスピロ[3.3]ヘプタン(14 mg、0.098 mmol)で処理した。反応混合物を65℃で30分間攪拌し、DMSOで希釈し、PTFEフリットに通して濾過し、以下の条件を用いて、プレパラティブLC/MSで精製した:カラム:XBridge C18、200 mm x 19 mm、5μm 粒子;移動相A:5:95 アセトニトリル:10mM NH4OAc含有水;移動相B:95:5 アセトニトリル:10mM NH4OAc含有水;グラジエント:0%Bで0分保持、20分かけて0-40%B、次いで100%Bで0分保持;流速:20mL/分;カラム温度:25℃。フラクション収集はMSシグナルによりトリガーされた。目的物を含むフラクションを混ぜ合わせ、遠心蒸発で乾燥させ、化合物263(13.7 mg、35%)を酢酸塩として得た。
化合物264を類似的に調製した。
実施例27-化合物249
DMF(1 mL)中、化合物835(20 mg、0.042 mmol)およびアセトアルデヒド(183 mg、0.083 mmol)の混合物を、酢酸(0.024 mL、0.416 mmol)および20mgの4Åモレキュラーシーブ、続いてナトリウムトリアセトキシボロハイドライド(35.3 mg、0.166 mmol)で処理した。反応混合物をRTで1時間攪拌した。酢酸(0.024 mL、0.416 mmol)を蒸発させた。以下の条件を用いて、粗製物質をプレパラティブLC/MSで精製した:カラム:XBridge C18、200 mm x 19 mm、5μm 粒子;移動相A:5:95 アセトニトリル:NH4OAc含有水;移動相B:95:5 アセトニトリル:NH4OAc含有水;グラジエント:3%Bで0分保持、25分かけて3-43%B、次いで100%Bで0分保持;流速:20mL/分;カラム温度:25℃。フラクション収集はMSシグナルによりトリガーされた。目的物を含むフラクションを混ぜ合わせ、遠心蒸発で乾燥させた。以下の条件を用いて、前記物質をプレパラティブLC/MSでさらに精製した:カラム:XBridge C18、200 mm x 19 mm、5μm 粒子;移動相A:5:95 アセトニトリル:0.05% TFA含有水;移動相B:95:5 アセトニトリル:0.05% TFA含有水;グラジエント:0%Bで0分保持、25分かけて0-40%B、次いで100%Bで0分保持;流速:20mL/分;カラム温度:25℃。フラクション収集はMSシグナルによりトリガーされた。目的の化合物249を含むフラクションを混ぜ合わせ、遠心蒸発で乾燥させた。
化合物252および化合物253を類似的に調製した。
実施例28-化合物255
ステップ1.(4-((5-アミノ-7-(ブチルアミノ)-1H-ピラゾロ[4,3-d]ピリミジン-1-イル)メチル)-3-メトキシフェニル)メタノール 818(400 mg、1.122 mmol)のTHF(2 mL)溶液をSOCl2(0.164 mL、2.244 mmol)で処理し、RTで1時間攪拌した。溶媒を蒸発させ、粗製塩化物2をそれ以上は精製せずに次のステップに移った。
ステップ2.塩化物2のDMSO溶液をアミン3(市販されている、CAS:236406-55-6)で処理し、80℃で2時間加熱してからLCMSにかけたところ、反応の完了を示した。反応混合物をTFAで処理し、1時間攪拌した。TFAを蒸発させた。以下の条件を用いて、粗製物質をプレパラティブLC/MSでさらに精製した:カラム:XBridge C18、200 mm x 19 mm、5μm 粒子;移動相A:5:95 アセトニトリル:NH4OAc含有水;移動相B:95:5 アセトニトリル:NH4OAc含有水;グラジエント:4%Bで0分保持、20分かけて4-44%B、次いで100%Bで0分保持;流速:20mL/分;カラム温度:25℃。フラクション収集はMSおよびUVシグナルによりトリガーされた。目的の化合物255を含むフラクションを混ぜ合わせ、遠心蒸発で乾燥させた。
以下の化合物を類似的に調製した:化合物256、化合物257、化合物258、化合物265、および化合物266。
実施例29-化合物259
化合物255(18 mg、0.039 mmol)のDMF(0.5 mL)溶液をK2CO3(16.06 mg、0.116 mmol)/2-ブロモエタン-1-オール(5.49 μl、0.077 mmol)で処理し、50℃で2時間加熱した。以下の条件を用いて、粗製物質をプレパラティブLC/MSで精製した:カラム:XBridge C18、200 mm x 19 mm、5μm 粒子;移動相A:5:95 アセトニトリル:0.05% TFA含有水;移動相B:95:5 アセトニトリル:0.05% TFA含有水;グラジエント:0%Bで0分保持、20分かけて0-40%B、次いで100%Bで0分保持;流速:20mL/分;カラム温度:25℃。フラクション収集はMSおよびUVシグナルによりトリガーされた。目的の化合物259を含むフラクションを混ぜ合わせ、遠心蒸発で乾燥させた。
以下の化合物を類似的に調製した:化合物260、化合物261、化合物262、化合物267、および化合物268。
実施例30-化合物270
ステップ1.(5-ブロモ-3-メトキシピリジン-2-イル)メタノール(Sigma-Aldrich)(2.462 g、11.29 mmol)の、0℃のCH2Cl2(113 ml)溶液にSOCl2(1.235 ml、16.94 mmol)を滴下した。反応物をRTで1時間攪拌し、減圧濃縮した。残留物をCH2Cl2と混合し、減圧濃縮して(2x)、粗製5-ブロモ-2-(クロロメチル)-3-メトキシピリジンを得た。この物質をそれ以上は精製せずに使用した。
LC-MS m/z 236/238[M+H]+
ステップ2.メチル (7-ヒドロキシ-3-ヨード-1H-ピラゾロ[4,3-d]ピリミジン-5-イル)カルバメート(3.44 g、10.26 mmol)の、RTのDMF(45.6 ml)懸濁液にCs2CO3(13.37 g、41.0 mmol)を添加した。混合物を0℃で10分間攪拌し;次いでステップ1由来の粗製物質のDMF(22.80 ml)溶液を添加した。反応混合物を0℃で1時間攪拌した。冷却槽を取り除き、RTで20時間攪拌し続けた。反応混合物をH2O(250 mL)に添加し、得られた混合物をRTで静置した。固体を真空濾過により回収し、H2O(3 x 15 mL)、MeOH(2 x 15 mL)、CH2Cl2(15 mL)、およびヘキサン(15 mL)で洗浄し、メチル (1-((5-ブロモ-3-メトキシピリジン-2-イル)メチル)-7-ヒドロキシ-3-ヨード-1H-ピラゾロ[4,3-d]ピリミジン-5-イル)カルバメート(4.431 g、81%)を得た。
LC-MS m/z 535/537[M+H]+
1H NMR(400MHz,DMSO-d6)δ 13.19-12.96(m,1H),8.95-8.80(m,1H),8.06(s,1H),7.71(s,1H),5.87-5.65(m,2H),3.89(s,3H),3.53(br s,3H)
ステップ3.メチル (1-((5-ブロモ-3-メトキシピリジン-2-イル)メチル)-7-ヒドロキシ-3-ヨード-1H-ピラゾロ[4,3-d]ピリミジン-5-イル)カルバメート(0.990 g、1.850 mmol)の、RTのDMSO(12.33 ml)懸濁液に、(S)-1-((tert-ブチルジフェニルシリル)オキシ)ヘキサン-3-アミン,HCl塩(0.870 g、2.220 mmol)(US 2020/0038403 A1、図8、化合物71a)、続いて1,8-ジアザビシクロ[5.4.0]ウンデカ-7-エン(1.245 ml、8.33 mmol)および(ベンゾトリアゾール-1-イルオキシ)トリス(ジメチルアミノ)ホスホニウム ヘキサフルオロホスフェート(0.982 g、2.220 mmol)を添加した。反応混合物をRTで1時間攪拌し、EtOAc(100 mL)で希釈し、H2O(100 mL)で洗浄した。分液操作を行い、水層をEtOAc(100 mL)で抽出した。合わせた有機層を飽和NaCl水溶液(100 mL)で洗浄し、Na2SO4で乾燥させ、濾過し、減圧濃縮した。粗製物質をフラッシュクロマトグラフィー(80 g シリカゲル;リニアグラジエント 0-100% EtOAc-ヘキサン)により精製し、メチル (S)-(1-((5-ブロモ-3-メトキシピリジン-2-イル)メチル)-7-((1-((tert-ブチルジフェニルシリル)オキシ)ヘキサン-3-イル)アミノ)-3-ヨード-1H-ピラゾロ[4,3-d]ピリミジン-5-イル)カルバメート(810 mg、50%)を黄色の泡沫として得た。
LC-MS m/z 872/874[M+H]+
1H NMR(400MHz,DMSO-d6)δ 9.69(s,1H),7.92(d,J=1.8 Hz,1H),7.79(d,J=1.8 Hz,1H),7.57-7.53(m,2H),7.50-7.46(m,2H),7.42-7.31(m,4H),7.25-7.20(m,2H),7.12(d,J=8.3 Hz,1H),5.78-5.69(m,2H),4.64-4.55(m,1H),3.91(s,3H),3.70-3.64(m,2H),3.58(s,3H),1.90-1.82(m,2H),1.57-1.48(m,2H),1.25-1.13(m,2H),0.92(s,9H),0.81(t,J=7.3 Hz,3H)
ステップ4.MeOH(9.28 ml)およびAcOH(9.28 ml)の混合物中、メチル (S)-(1-((5-ブロモ-3-メトキシピリジン-2-イル)メチル)-7-((1-((tert-ブチルジフェニルシリル)オキシ)ヘキサン-3-イル)アミノ)-3-ヨード-1H-ピラゾロ[4,3-d]ピリミジン-5-イル)カルバメート(0.810 g、0.928 mmol)の、0℃の溶液に亜鉛(0.607 g、9.28 mmol)を添加した。反応混合物を0℃で30分間攪拌し、MeOH(10 mL)およびEtOAc(50 mL)で洗浄しながらCELITE(商標)に通して濾過した。濾液をEtOAc(200 mL)で希釈した。攪拌しながら、飽和NaHCO3水溶液(250 mL)を、この溶液にゆっくりと添加した(添加速度を調節してガス発生速度を制御した)。分液操作を行い、有機層を飽和NaCl水溶液(250 mL)で洗浄し、Na2SO4で乾燥させ、濾過し、減圧濃縮して、粗生成物、メチル (S)-(1-((5-ブロモ-3-メトキシピリジン-2-イル)メチル)-7-((1-((tert-ブチルジフェニルシリル)オキシ)ヘキサン-3-イル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-5-イル)カルバメートを得た。この物質をそれ以上は精製せずに使用した。
LC-MS m/z 746/748[M+H]+
ステップ5.窒素ガスで、化合物5(500 mg、0.670 mmol)、化合物6(304 mg、0.870 mmol、CAS 2240187-78-2)およびK2CO3(370 mg、2.68 mmol)のDMF(2 mL)溶液を2分間通気した。PdCl2(dppf)-CH2Cl2付加物(54.7 mg、0.067 mmol)を添加し、反応混合物をN2で1分間、再度通気した。反応フラスコを密閉し、70℃で5時間加熱した。50 g シリカゲルカラムで、0-50% MeOH/DCMで溶出させて精製し、476mgの化合物7を得た。
LC/MS[M+H]+=889.5
1H NMR(400MHz,クロロホルム-d)δ 8.02(s,5H),7.92(d,J = 19.1 Hz,1H),7.63-7.50(m,3H),7.37-7.23(m,2H),7.22-7.14(m,2H),7.04(s,1H),6.62(d,J = 2.6 Hz,1H),5.64(dd,J = 14.7,1.4 Hz,1H),5.39(d,J = 14.9 Hz,1H),4.63(s,1H),3.95(d,J = 2.0 Hz,2H),3.82(d,J = 7.5 Hz,3H),3.57(s,1H),3.31-3.21(m,1H),2.96(s,7H),2.88(s,7H),2.49(s,1H),1.95(d,J = 17.4 Hz,1H),1.47(s,5H),1.52-1.36(m,2H),1.26(d,J = 13.9 Hz,5H),1.02(s,3H),1.04-0.90(m,2H)
ステップ6-7.固体化合物7(476 mg、0.535 mmol)を、RTで2時間攪拌しながらジオキサン(1.338 mL、5.35 mmol)中、HClで処理し、次いでLC/MSにかけたところ、反応の完了を示した。V-10エバポレーターを用いてHClを蒸発させた。粗生成物8を1mLジオキサン中に溶解し、NaOH水溶液(1.071 mL、10.71 mmol)とともに2時間加熱し、次いでLC/MSにかけたところ、反応の完了を示した。以下の条件を用いて、粗製物質をプレパラティブLC/MSで精製した:カラム:XBridge C18、200 mm x 19 mm、5μm粒子;移動相A:5:95 アセトニトリル:NH4OAc含有水;移動相B:95:5 アセトニトリル:NH4OAc含有水;グラジエント:3%Bで0分保持、20分かけて3-43%B、次いで100%Bで0分保持;流速:20mL/分;カラム温度:25℃。フラクション収集はMSおよびUVシグナルによりトリガーされた。化合物272((3S)-3-({5-アミノ-1-[(5-{7-アザスピロ[3.5]ノン-1-エン-2-イル}-3-メトキシピリジン-2-イル)メチル]-1H-ピラゾロ[4,3-d]ピリミジン-7-イル}アミノ)ヘキサン-1-オール)を含むフラクションを混ぜ合わせ、遠心蒸発で乾燥させた。
ステップ8.化合物272(40 mg、0.081 mmol)、テトラヒドロ-4H-ピラン-4-オン(37.5 μl、0.406 mmol)のDMA(1 mL)溶液を、酢酸(46.5 μL、0.812 mmol)、続いて50mgの顆粒4Åモレキュラーシーブおよびナトリウムトリアセトキシボロハイドライド(86 mg、0.406 mmol)で処理した。反応混合物をRTで終夜攪拌し、シリンジ濾過した。以下の条件を用いて、粗製物質をプレパラティブLC/MSで精製した:カラム:XBridge C18、200 mm x 19 mm、5μm 粒子;移動相A:5:95 アセトニトリル:NH4OAc含有水;移動相B:95:5 アセトニトリル:NH4OAc含有水;グラジエント:7%Bで0分保持、20分かけて7-47%B、次いで100%Bで0分保持;流速:20mL/分;カラム温度:25℃。フラクション収集はMSおよびUVシグナルによりトリガーされた。目的物を含むフラクションを混ぜ合わせ、遠心蒸発で乾燥させ、化合物273 ((3S)-3-{[5-アミノ-1-({3-メトキシ-5-[7-(オキサン-4-イル)-7-アザスピロ[3.5]ノン-1-エン-2-イル]ピリジン-2-イル}メチル)-1H-ピラゾロ[4,3-d]ピリミジン-7-イル]アミノ}ヘキサン-1-オール)を得た。
ステップ9.水素ガスで、化合物273(18 mg、0.026 mmol)のMeOH(1 mL)溶液およびPd/C(2.73 mg、0.026 mmol)を1分間通気した。反応混合物を60℃で、水素バルーンの雰囲気下で2時間加熱した。以下の条件を用いて、粗製物質をプレパラティブLC/MSで精製した:カラム:XBridge C18、200 mm x 19 mm、5μm粒子;移動相A:5:95 アセトニトリル:NH4OAc含有水;移動相B:95:5 アセトニトリル:NH4OAc含有水;グラジエント:7%Bで0分保持、20分かけて7-47%B、次いで100%Bで0分保持;流速:20mL/分;カラム温度:25℃。フラクション収集はMSおよびUVシグナルによりトリガーされた。目的物を含むフラクションを混ぜ合わせ、遠心蒸発で乾燥させ、化合物27を得た。
以下の化合物を類似的に調製した:化合物274、化合物275、および化合物278。
実施例31-化合物271
ステップ1.メチル (S)-(1-((5-ブロモ-3-メトキシピリジン-2-イル)メチル)-7-((1-((tert-ブチルジフェニルシリル)オキシ)ヘキサン-3-イル)アミノ)-1H-ピラゾロ[4,3-d]ピリミジン-5-イル)カルバメート(552 mg、0.739 mmol)、tert-ブチル 7-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-2-アザスピロ[3.5]ノン-6-エン-2-カルボキシレート 1(336 mg、0.961 mmol;CAS 235276-13-4)およびK2CO3(409 mg、2.96 mmol)のDMF(5 mL)溶液をN2で2分間通気した。PdCl2(dppf)-CH2Cl2付加物(60.4 mg、0.074 mmol)を添加し、再度N2で1分間通気した。反応槽を密閉し、70℃で5時間加熱した。50 g シリカゲルカラムで、0-50% MeOH/DCMで溶出させて精製し、477mgの化合物2を得た。
LC/MS[M+H]+=889.5
1H NMR(400MHz,クロロホルム-d)δ 8.70(d,J = 8.1 Hz,1H),8.04-7.95(m,1H),7.87(s,1H),7.65-7.58(m,2H),7.58-7.51(m,2H),7.40-7.31(m,1H),7.33-7.24(m,3H),7.21-7.12(m,3H),7.08(s,1H),6.02(s,1H),5.63(d,J = 14.9 Hz,1H),5.40(d,J = 15.0 Hz,1H),4.58(s,1H),3.94(s,3H),3.85-3.74(m,4H),3.73-3.64(m,2H),3.62(d,J = 8.3 Hz,2H),3.49(s,2H),2.95(s,1H),2.88(s,1H),2.41(d,J = 4.3 Hz,4H),2.02(dd,J = 13.0,6.2 Hz,0H),1.95(s,1H),1.91(d,J = 5.7 Hz,1H),1.45(s,6H),1.45-1.36(m,1H),1.24(s,6H),1.04(d,J = 8.8 Hz,0H),1.03(s,6H),1.02(s,1H),0.94(t,J = 7.3 Hz,3H)
ステップ2.化合物2(90 mg、0.101 mmol)をTFA(0.078 mL、1.012 mmol)で処理した。反応混合物をRTで30分間攪拌した。TFAをV-10エバポレーター中で蒸発させた。残留物をDMA(0.5 mL)中に溶解し、テトラヒドロ-4H-ピラン-4-オン(0.028 mL、0.506 mmol)、酢酸(0.029 mL、0.506 mmol)、50mgの4Åモレキュラーシーブ、および最後にナトリウムトリアセトキシボロハイドライド(107 mg、0.506 mmol)で処理した。RTで1時間撹拌した後、反応混合物をトリエチルアミントリヒドロフルオライド(0.165 mL、1.012 mmol)で処理し、RTで2時間攪拌した。50 g 逆相ISCOで、0-50% MeCN/水(0.05% TFA)で溶出させて、反応混合物を直接精製し、化合物3を白色固体として得た。
LC/MS[M+H]+=635.3
ステップ3、パート1.化合物(58 mg、0.091 mmol)のDMSO(0.5 mL)溶液をNaOH(0.091 mL、0.914 mmol)で処理し、80℃で2時間加熱して、脱カルバモイル化した化合物3を得た。
LC/MS[M+H]+=577.3
ステップ3、パート2.脱カルバモイル化した化合物3(12 mg、0.021 mmol)の、Pd-C(2.214 mg、0.021 mmol)含有MeOH(1 mL)溶液をH2で1分間通気した。反応混合物を水素バルーン雰囲気下で、60℃で2時間加熱した。以下の条件を用いて、粗生成物をプレパラティブLC/MSで精製した:カラム:XBridge C18、200 mm x 19 mm、5μm粒子;移動相A:5:95 アセトニトリル:NH4OAc含有水;移動相B:95:5 アセトニトリル:NH4OAc含有水;グラジエント:9%Bで0分保持、20分かけて9-49%B、次いで100%Bで0分保持;流速:20mL/分;カラム温度:25℃。フラクション収集はMSシグナルによりトリガーされた。目的物を含むフラクションを混ぜ合わせ、遠心蒸発で乾燥させ、4.7mgの化合物271を得た。
化合物277を類似的に調製した。
実施例32-化合物250
ステップ1.ベンジル 6-ヒドロキシ-2-アザスピロ[3.3]ヘプタン-2-カルボキシレート 1(CAS#1363383-32-7;3 g、12.13 mmol)のDCM(20 mL)溶液を、トリエチルアミン(2.029 mL、14.56 mmol)、DMAP(0.296 g、2.426 mmol)およびトシル-Cl(2.54 g、13.34 mmol)で、0℃で処理した。2時間かけて反応を進行させた。反応物を50mLの水でクエンチし、50mLの1M HCl水溶液、ブライン(50 mL)で洗浄し、Na2SO4で乾燥させ、濾過し、濃縮して、粗製トシル化中間体を黄色がかった残留物として得た。これをDMSO(20 mL)中に溶解し、ヨウ化ナトリウム(5.46 g、36.4 mmol)で処理した。120℃で2時間にわたって加熱した後、反応混合物を50mLのEtOAc中に溶解し、飽和Na2S2O3水溶液(50 mL)、水(50 mL)、ブライン(50 mL)で洗浄し、Na2SO4で乾燥させた。濾過、濃縮し、80 g シリカゲルカラムで、0-50% EtOAc/ヘキサンで溶出させて精製し、化合物2を白色固体として得た。
LC/MS[M+H]+=357.9
1H NMR(400MHz,DMSO-d6)δ 7.42-7.27(m,5H),5.01(s,2H),4.43(p,J = 7.9 Hz,1H),3.96(s,4H),2.92(ddd,J = 10.4,7.5,3.1 Hz,2H),2.74-2.61(m,2H)
ステップ2.化合物2(1649 mg、4.62 mmol)の4mL THF溶液を、乾燥機で乾燥させた丸底フラスコ内のTHF(12.08 mL、9.23 mmol)中、Rieke亜鉛に、N2下で添加した。フラスコの温度が上昇し、亜鉛試薬3の生成を示した。反応混合物をRTで1時間攪拌し、今後の使用のためにN2下に保った
ステップ3.5-ブロモ-2-(((tert-ブチルジメチルシリル)オキシ)メチル)-3-メトキシピリジン(1.4 g、4.21 mmol)、1,1'-ビス(ジフェニルホスフィノ)フェロセンジクロロパラジウム(II) ジクロロメタン錯体(0.308 g、0.421 mmol)およびヨウ化銅(I)(0.160 g、0.843 mmol)のDMF(10 mL)溶液を、N2で1分間通気した。(2-((ベンジルオキシ)カルボニル)-2-アザスピロ[3.3]ヘプタン-6-イル)亜鉛(II) アイオダイド 3(17.49 mL、5.06 mmol)を添加した。反応混合物を70℃で 時間加熱した。この溶液をトリエチルアミントリヒドロフルオライド(1.372 mL、8.43 mmol)で処理し、終夜攪拌した。LCMSは化合物5(167 mg、0.453 mmol、収率10.76%)の生成を示した。150 g 逆相C-18カラムで、0-50% MeCN/水(0.05% TFA)で溶出させて反応物を直接精製し、目的のフラクションを収集し、化合物5を淡黄色固体として得た。
LC/MS[M+H]+=369.2
1H NMR(400MHz,DMSO-d6)δ 8.08(s,1H),7.81(s,1H),7.43-7.28(m,5H),5.04(s,2H),4.71(s,2H),4.11(s,2H),3.66-3.56(m,3H),3.61-3.48(m,1H),2.58(ddt,J = 10.6,8.4,2.5 Hz,2H),2.41(td,J = 9.5,2.9 Hz,2H),1.84-1.70(m,3H)
ステップ4-5.化合物5(167 mg、0.453 mmol)のTHF(1 mL)溶液をSOCl2(0.066 mL、0.907 mmol)で処理し、RTで30分間攪拌した。溶媒をV-10エバポレーターで蒸発させた。1mL DMF中の粗生成物6を、メチル (7-ヒドロキシ-3-ヨード-1H-ピラゾロ[4,3-d]ピリミジン-5-イル)カルバメート 7(152 mg、0.453 mmol)およびCs2CO3(295 mg、0.907 mmol)の、1mLのDMF溶液に添加した。60℃で2時間加熱した後、反応物を濾過し、50 g 逆相C-18カラムで、0-50% MeCN/水(0.05% TFA)で溶出させて直接精製した。目的のフラクションを凍結乾燥し、化合物8を淡黄色固体として得た。
LC/MS[M+H]+=886.1
ステップ6-7.化合物8(140 mg、0.204 mmol)および(S)-3-アミノヘキサン-1-オール 9(47.9 mg、0.408 mmol)のDMSO(1 mL)溶液をDBU(0.092 mL、0.613 mmol)、続いてBOP(135 mg、0.306 mmol)で処理した。40℃で1時間加熱した後、LMCSにかけたところ反応の完了を示し、中間体10を得た。反応混合物をNaOH(0.204 mL、2.042 mmol)で処理し、80℃で2時間加熱した。50 g C-18逆相カラムで、0-50% MeCN/水(0.05% TFA)で溶出させて、反応混合物を直接精製した。目的のフラクションを凍結乾燥し、化合物11を淡黄色固体として得た。
LC/MS[M+H]+=593.1
ステップ8.化合物11(30 mg、0.051 mmol)のMeOH(1 mL)溶液を、Pd-C(5.39 mg、0.051 mmol)とともに、H2(10.21 mg、5.06 mmol)で1分間通気した。反応混合物を50℃で、H2バルーン下で2時間加熱した。以下の条件を用いて、粗製物質をプレパラティブLC/MSで精製した:カラム:XBridge C18、200 mm x 19 mm、5μm粒子;移動相A:5:95 アセトニトリル:NH4OAc含有水;移動相B:95:5 アセトニトリル:NH4OAc含有水;グラジエント:6%Bで0分保持、20分かけて6-46%B、次いで100%Bで0分保持;流速:20mL/分;カラム温度:25℃。フラクション収集はMSおよびUVシグナルによりトリガーされた。目的物を含むフラクションを混ぜ合わせ、遠心蒸発で乾燥させ、7.6mgの化合物12を得た。
LC/MS[M+H]+=466.9
1H NMR(500MHz,DMSO-d6)δ 7.93(s,2H),7.90(d,J = 1.6 Hz,1H),7.51(d,J = 7.6 Hz,1H),7.43(d,J = 8.6 Hz,1H),5.96(s,1H),5.71-5.59(m,3H),4.43(s,2H),4.11(s,1H),3.90(d,J = 6.9 Hz,3H),2.68(d,J = 9.9 Hz,0H),2.57(d,J = 22.7 Hz,2H),2.35(d,J = 14.2 Hz,2H),2.24(s,1H),1.92(s,1H),1.78(d,J = 6.3 Hz,2H),1.77-1.69(m,2H),1.58(s,4H),1.29(s,2H),0.91-0.84(m,3H)
ステップ9.化合物12(30 mg、0.064 mmol)およびテトラヒドロ-4H-ピラン-4-オン(0.012 mL、0.129 mmol)のDMF(0.5 mL)溶液を、2滴の酢酸および50mgの4Åモレキュラーシーブ並びにナトリウムトリアセトキシボロハイドライド(54.5 mg、0.257 mmol)で処理した。RTで1時間撹拌した後、以下の条件を用いて、粗製物質をプレパラティブLC/MSで精製した:カラム:XBridge C18、200 mm x 19 mm、5μm粒子;移動相A:5:95 アセトニトリル:NH4OAc含有水;移動相B:95:5 アセトニトリル:NH4OAc含有水;グラジエント:13%Bで0分保持、20分かけて13-53%B、次いで100%Bで0分保持;流速:20mL/分;カラム温度:25℃。フラクション収集はMSおよびUVシグナルによりトリガーされた。目的物を含むフラクションを混ぜ合わせ、遠心蒸発で乾燥させた。以下の条件を用いて、前記物質をプレパラティブLC/MSでさらに精製した:カラム:XBridge Phenyl、200 mm x 19 mm、5μm 粒子;移動相A:5:95 アセトニトリル:NH4OAc含有水;移動相B:95:5 アセトニトリル:NH4OAc含有水;グラジエント:9%Bで0分保持、20分かけて9-49%B、次いで100%Bで0分保持;流速:20mL/分;カラム温度:25℃。フラクション収集はMSシグナルによりトリガーされた。目的物を含むフラクションを混ぜ合わせ、遠心蒸発で乾燥させ、1.9mgの化合物250を得た。
化合物251を類似的に調製した。
実施例33-出発物質および中間体
下記のチャートは、本明細書に開示されるTLR7アゴニストの調製用の出発物質または中間体として有用なことがある化合物を作成するためのスキームを示す。スキームは、出発物質または中間体として使用されることがある他の類似化合物の作成に適用され得る。使用される試薬は当技術分野において周知であり、多くの場合、その使用は前述の実施例に示されている。
チャート1
チャート2
チャート3
生物学的活性
TLR7アゴニストとして本明細書に開示される化合物の生物学的活性は、以下の手順により定量されることがある。
ヒトTLR7アゴニスト活性アッセイ
この手順は、本明細書に開示される化合物のヒトTLR7(hTLR7)アゴニスト活性を定量する方法を説明する。
ヒトTLR7分泌型胚性アルカリホスファターゼ(SEAP)レポータートランスジーンを有する改変ヒト胚性腎臓ブルー細胞(HEK-Blue(商標)TLR細胞;Invivogen)を、非選択培地(10%ウシ胎児血清(Sigma)を添加したDMEM高グルコース(Invitrogen))中に懸濁した。HEK-Blue(商標)TLR7細胞を384ウェル組織培養プレートの各ウェルに添加し(1ウェルあたり15,000細胞)、16-18時間、37℃、5%CO2でインキュベートした。HEK-Blue(商標)TLR細胞が入ったウェルに化合物(100 nl)を添加し、処置した細胞を37℃、5%CO2でインキュベートした。処理から18時間後、10マイクロリットルの新たに調製したQuanti-Blue(商標)試薬(Invivogen)を各ウェルに添加し、30分間インキュベートし(37℃、5%CO2)、Envisionプレートリーダー(OD=620nm)を用いてSEAPレベルを測定した。半数効果濃度値(EC50;アッセイ基準値および最大値の中間の応答を引き起こす化合物濃度)を算出した。
ヒト血液におけるI型インターフェロン遺伝子(MX-1)およびCD69の誘導
I型インターフェロン(IFN)MX-1遺伝子およびB細胞活性化マーカーCD69の誘導は、TLR7経路の活性化で起こる下流のイベントである。以下は、TLR7アゴニストに対する応答におけるそれらの誘導を測定するヒト全血アッセイである。
ヘパリン処置したヒト全血をヒト患者から回収し、1mMで、TLR7アゴニスト試験化合物で処置した。血液をRPMI 1640培地で希釈し、Echoを使用して1ウェルあたり10nLプレドット(predot)し、最終濃度を1μMとした(10μLの血液中に10nL)。30秒間振盪機で混合した後、プレートを覆い、37℃のチャンバー内に終夜=17時間置いた。固定/溶解バッファーを調製し(H20中5x→1x、37℃で温める;Cat# BD 558049)、後で使用するためにパームバッファーを(氷上で)維持した。
表面マーカー染色(CD69)のために表面抗体を調製した:0.045μl hCD14-FITC(ThermoFisher Cat # MHCD1401)+0.6μl hCD19-ef450(ThermoFisher Cat # 48-0198-42)+1.5μl hCD69-PE(cat# BD555531)+0.855μl FACSバッファー。3μl/ウェルで添加し、1000rpmで1分間遠心し、振盪機で30秒間混合し、氷上に30分間置いた。30分後、70μLの予め温めた1x固定/溶解バッファーで刺激を停止させ、Feliex mateを用いて再懸濁し(15回、プレートごとにチップを変えた)、37℃で10分間インキュベートした。
2000rpmで5分間遠心し、HCSプレートウォッシャーで吸引し、振盪機で30秒間混合し、次いで70μLのdPBSで洗浄し、ペレット状にすること2回(2000rpm、5分間)、50μLのFACSバッファーで洗浄し、ペレット状にすること1回(2000rpm、5分間)を行った。振盪機で30秒間混合した。細胞内マーカー染色(MX-1)については:50μlのBD PermバッファーIIIを添加し、振盪機で30秒間混合した。氷上で30分間インキュベートした(遮光)。50μLのFACSバッファーで2回洗浄し(透過処理後2300rpmで5分間遠心)、続いて振盪機で30秒間混合した。MX1抗体((4812)-Alexa 647:Novus Biologicals #NBP2-43704AF647)を含む20μLのFACSバッファーで再懸濁した(20μl FACSバッファー+0.8ul hIgG+0.04μl MX-1)。1000rpmで1分間遠心し、振盪機で30秒間混合し、サンプルをRTで、暗所で45分間インキュベートし、続いて2xFACSバッファーで洗浄した(透過処理後2300rpmで5分間遠心)。20μlのFACSバッファーで再懸濁し(1ウェルあたり合計35μL)、ホイルで覆い、4℃に置き、翌日に読み取った。プレートをiQuePlusで読み取った。結果をツールセットにロードし、カーブマスターでIC50曲線を作成した。y軸の100%は1μMのレシキモドに設定されている。
マウス血液におけるTNF-アルファおよびI型IFN応答遺伝子の誘導
TNF-アルファおよびI型IFN応答遺伝子の誘導は、TLR7経路の活性化で起こる下流のイベントである。以下は、TLR7アゴニストに対する応答における、マウス全血中のそれらの誘導を測定するアッセイである。
ヘパリン処置したマウス全血を、Pen-Strepを含むRPMI 1640培地で、5:4の比率で希釈した(50μLの全血および40μLの培地)。体積90μLの希釈血液をFalcon平底96ウェル組織培養プレートのウェルに移し、プレートを4℃で1時間インキュべートした。100% DMSOストック中の試験化合物を、濃度応答アッセイのために同じ培地で20倍希釈し、次いで10μLの希釈した試験化合物をウェルに添加し、最終DMSO濃度が0.5%となるようにした。コントールウェルに、5% DMSOを含む10μLの培地を添加した。次にプレートを37℃で、5%CO2インキュベーター内で17時間インキュベートした。インキュベート後、100μLの培地を各ウェルに添加した。プレートを遠心し、130μLの上清を除去し、ELISAによるTNFα産生のアッセイに使用した(Invitrogen、カタログ番号 88-7324 Thermo-Fisher Scientificより)。Invitrogen mRNA Catcher Plusキット(Cat# K1570-02)に由来する、DTTを含む体積70μLのmRNAキャッチャー溶解バッファー(1x)を、ウェル中の残りの70μLサンプルに添加し、ピペッティングにより5回混合した。次にプレートをRTで5-10分間振盪し、続いて2μLのプロテイナーゼK(20 mg/mL)を各ウェルに添加した。次にプレートを15-20分間、RTで振盪した。次に、さらに処理するまでの間、プレートを-80℃で保存した。
冷凍サンプルを解凍し、Invitrogen mRNA Catcher Plusキット(Cat# K1570-02)を用いて、製造業者の説明書に従ってmRNAを抽出した。RNA抽出から得られたmRNAの半量を用いて、Invitrogen SuperScript IV VILO Master Mix(Cat# 11756500)を使用して、20μLの逆転写酵素反応でcDNAを合成した。ThermoFisher(Applied Biosystems)のQuantStudio Real-Time PCRシステムを用いて、TaqMan(登録商標)リアルタイムPCRを行った。全てのリアルタイムPCR反応を、市販のマウスIFIT1、IFIT3、MX1およびPPIA遺伝子発現用プレデザインTaqManアッセイ並びにTaqMan Master Mixを用いて、2回繰り返して行った。PPIAは、ハウスキーピング遺伝子として利用した。製造業者からの勧告に従った。全ての生データ(Ct)を平均ハウスキーピング遺伝子(Ct)で正規化し、次に比較Ct(ΔΔCt)法を利用して、実験解析のために、相対的な遺伝子発現量(RQ)を定量化した。
定義
「脂肪族」は、特定の数の炭素原子を有する直鎖または分岐鎖の飽和または不飽和非芳香族炭化水素部分を意味し(例えば、「C3脂肪族」、「C1-5脂肪族」、「C1-C5脂肪族」、または「C1からC5脂肪族」のように。後者3つの表現は1から5個の炭素原子を有する脂肪族部分と同義である)、炭素原子の数が明確に特定されない場合は、1から4個の炭素原子(不飽和脂肪族部分の場合は2から4個の炭素)である。同様の理解が、他の種類における炭素の数、つまりC2-4アルケン、C4-C7シクロ脂肪族などに適用される。同様に、「(CH2)1-3」などの用語は、下付き文字が1、2、または3であることの省略表現として理解されるべきであり、そのため、かかる用語は、CH2、CH2CH2、およびCH2CH2CH2を表すことになる。
「アルキル」は、適用可能な炭素原子の数を指定するための同じ慣習に従う飽和脂肪族部分を意味する。実例として、C
1-C
4アルキル部分には、メチル、エチル、プロピル、イソプロピル、イソブチル、t-ブチル、1-ブチル、2-ブチル、および同類のものが挙げられるが、これらに限らない。「アルカンジイル」(時として「アルキレン」とも呼ばれる)は、アルキル基の2価の対応物を意味し、例えば、
などがある。
「アルケニル」は、適用可能な炭素原子の数を指定するための同じ慣習に従う、少なくとも一つの炭素-炭素二重結合を有する脂肪族部分を意味する。実例として、C2-C4アルケニル部分には、エテニル(ビニル)、2-プロペニル(アリルまたはプロプ-2-エニル)、シス-1-プロペニル、トランス-1-プロペニル、E-(またはZ-)2-ブテニル、3-ブテニル、1,3-ブタジエニル(ブト-1,3-ジエニル)、および同類のものが挙げられるが、これらに限らない。
「アルキニル」は、適用可能な炭素原子の数を指定するための同じ慣習に従う、少なくとも一つの炭素-炭素三重結合を有する脂肪族部分を意味する。実例として、C2-C4アルキニル基には、エチニル(アセチレニル)、プロパルギル(プロプ-2-イニル)、1-プロピニル、ブト-2-イニル、および同類のものが挙げられる。
「シクロ脂肪族」は、1から3個の環を有し、各環が、3から8個(好ましくは3から6個)の炭素原子を有する、飽和または不飽和非芳香族炭化水素部分を意味する。「シクロアルキル」は、各環が飽和であるシクロ脂肪族部分を意味する。「シクロアルケニル」は、少なくとも一つの環が少なくとも一つの炭素-炭素二重結合を有する、シクロ脂肪族部分を意味する。「シクロアルキニル」は、少なくとも一つの環が少なくとも一つの炭素-炭素三重結合を有する、シクロ脂肪族部分を意味する。実例として、シクロ脂肪族部分には、シクロプロピル、シクロブチル、シクロペンチル、シクロペンテニル、シクロヘキシル、シクロヘキセニル、シクロヘプチル、シクロオクチル、およびアダマンチルが挙げられるが、これらに限らない。好ましいシクロ脂肪族部分は、シクロアルキル部分、特にシクロプロピル、シクロブチル、シクロペンチル、およびシクロヘキシルである。「シクロアルカンジイル」(時として「シクロアルキレン」とも呼ばれる)は、シクロアルキル基の2価の対応物を意味する。同様に、「ビシクロアルカンジイル」(または「ビシクロアルキレン」)および「スピロアルカンジイル」(または「スピロアルキレン」)は、ビシクロアルキルおよびスピロアルキル(または「スピロシクロアルキル」)基の2価の対応物を指す。実例として、
部分の例は、
であり、
部分の例は、
である。
「ヘテロシクロ脂肪族」は、少なくとも一つのその環において、最大3個(好ましくは1から2個)の炭素が、N、OまたはSから独立して選択されるヘテロ原子で置換されており、ここでNおよびSは、適宜酸化されてもよく、Nは、適宜四級化されてもよい、シクロ脂肪族部分を意味する。好ましいシクロ脂肪族部分は、5から6員の大きさの1つの環からなる。同様に、「ヘテロシクロアルキル」、「ヘテロシクロアルケニル」、および「ヘテロシクロアルキニル」は、少なくとも一つのその環が、そのように修飾されている、シクロアルキル、シクロアルケニル、またはシクロアルキニル部分をそれぞれ意味する。代表的なヘテロシクロ脂肪族部分には、アジリジニル、アゼチジニル、1,3-ジオキサニル、オキセタニル、テトラヒドロフリル、ピロリジニル、ピペリジニル、ピペラジニル、テトラヒドロピラニル、テトラヒドロチオピラニル、テトラヒドロチオピラニルスルホン、モルホリニル、チオモルホリニル、チオモルホリニルスルホキシド、チオモルホリニルスルホン、1,3-ジオキソラニル、テトラヒドロ-1,1-ジオキソチエニル、1,4-ジオキサニル、チエタニル、および同類のものが挙げられる。「ヘテロシクロアルキレン」は、ヘテロシクロアルキル基の2価の対応物を意味する。
「アルコキシ」、「アリールオキシ」、「アルキルチオ」、および「アリールチオ」は、それぞれ、-O(アルキル)、-O(アリール)、-S(アルキル)、および-S(アリール)を意味する。例は、それぞれ、メトキシ、フェノキシ、メチルチオ、およびフェニルチオである。
「ハロゲン」または「ハロ」は、より狭い意味が指示されない限り、フッ素、塩素、臭素またはヨウ素を意味する。
「アリール」は、各環が3から7個の炭素原子を有し、少なくとも一つの環が芳香族である、単、二、または三環式環系(好ましくは単環式)を有する、炭化水素部分を意味する。環系中の環は、(ナフチルのように)互いに縮合していてもよく、(ビフェニルのように)互いに結合していてもよく、(インダニルまたはシクロヘキシルフェニルのように)非芳香環と縮合または結合していてもよい。さらなる実例として、アリール部分には、フェニル、ナフチル、テトラヒドロナフチル、インダニル、ビフェニル、フェナントリル、アントラセニル、およびアセナフチルが挙げられるが、これらに限らない。「アリーレン」は、アリール基の2価の対応物、例えば1,2-フェニレン、1,3-フェニレン、または1,4-フェニレンを意味する。
「ヘテロアリール」は、各環が3から7個の炭素原子を有し、少なくとも一つの環が、N、O、またはSから独立して選択される1から4個のヘテロ原子を含む芳香環であり、ここでNおよびSは、適宜酸化されてもよく、Nは、適宜四級化されてもよい、単、二、または三環式環系(好ましくは5から7員の単環式)を有する部分を意味する。そのような少なくとも一つのヘテロ原子を含む芳香環は、(ベンゾフラニルまたはテトラヒドロイソキノリルのように)他の種類の環と縮合してもよく、(フェニルピリジルまたは2-シクロペンチルピリジルのように)他の種類の環と直接結合してもよい。さらなる実例として、ヘテロアリール部分には、ピロリル、フラニル、チオフェニル(チエニル)、イミダゾリル、ピラゾリル、オキサゾリル、イソオキサゾリル、チアゾリル、イソチアゾリル、トリアゾリル、テトラゾリル、ピリジル、N-オキソピリジル、ピリダジニル、ピリミジニル、ピラジニル、キノリニル、イソキノリニル、キナゾリニル、シンノリニル、キノザリニル、ナフチリジニル、ベンゾフラニル、インドリル、ベンゾチオフェニル、オキサジアゾリル、チアジアゾリル、フェノチアゾリル、ベンゾイミダゾリル、ベンゾトリアゾリル、ジベンゾフラニル、カルバゾリル、ジベンゾチオフェニル、アクリジニル、および同類のものが挙げられる。「ヘテロアリーレン」は、ヘテロアリール基の2価の対応物を意味する。
例えば「非置換の、または置換された」あるいは「適宜置換されてもよい」を用いる、つまり「非置換の、または置換されたC1-C5アルキル」あるいは「適宜置換されてもよいヘテロアリール」と表現するなどして、部分が置換されてもよいということが示される場合、かかる部分は、一つ以上の独立して選択される置換基、好ましくは数にして1から5個、より好ましくは数にして1から2個の置換基を有してもよい。置換基および置換パターンは、置換基が結合する部分を考慮して当業者により選択されることがあり、化学的に安定で、当技術分野で既知の技術、ならびに本明細書に記載される方法により合成され得る化合物を提供する。部分が、「非置換の、または置換された」あるいは「適宜置換されてもよい」ものとして特定される場合、好ましい実施形態において、かかる部分は非置換である。
「アリールアルキル」、「(ヘテロシクロ脂肪族)アルキル」、「アリールアルケニル」、「アリールアルキニル」、「ビアリールアルキル」、および同類のものは、場合によっては、アリール、ヘテロシクロ脂肪族、ビアリールなどで置換されたアルキル、アルケニル、またはアルキニル部分を、場合によっては、例えば、ベンジル、フェネチル、N-イミダゾイルエチル、N-モルホリノエチル、および同類のもののように、アルキル、アルケニル、またはアルキニル部分で開いた(不満足な)原子価を有する部分を意味する。反対に、「アルキルアリール」、「アルケニルシクロアルキル」、および同類のものは、場合によっては、アルキル、アルケニルなどで置換されたアリール、シクロアルキル、その他の部分、場合によっては、例えば、メチルフェニル(トリル)またはアリルシクロヘキシルのような部分を意味する。「ヒドロキシアルキル」、「ハロアルキル」、「アルキルアリール」、「シアノアリール」、および同類のものは、場合によっては、一つ以上の特定の置換基(場合によっては、ヒドロキシル、ハロなど)で置換されたアルキル、アリール、その他の部分を意味する。
例えば、許容される置換基には、アルキル(特にメチルまたはエチル)、アルケニル(特にアリル)、アルキニル、アリール、ヘテロアリール、シクロ脂肪族、ヘテロシクロ脂肪族、ハロ(特にフルオロ)、ハロアルキル(特にトリフルオロメチル)、ヒドロキシル、ヒドロキシアルキル(特にヒドロキシエチル)、シアノ、ニトロ、アルコキシ、-O(ヒドロキシアルキル)、-O(ハロアルキル)(特に-OCF3)、-O(シクロアルキル)、-O(ヘテロシクロアルキル)、-O(アリール)、アルキルチオ、アリールチオ、=O、=NH、=N(アルキル)、=NOH、=NO(アルキル)、-C(=O)(アルキル)、-C(=O)H、-CO2H、-C(=O)NHOH、-C(=O)O(アルキル)、-C(=O)O(ヒドロキシアルキル)、-C(=O)NH2、-C(=O)NH(アルキル)、-C(=O)N(アルキル)2、-OC(=O)(アルキル)、-OC(=O)(ヒドロキシアルキル)、-OC(=O)O(アルキル)、-OC(=O)O(ヒドロキシアルキル)、-OC(=O)NH2、-OC(=O)NH(アルキル)、-OC(=O)N(アルキル)2、アジド、-NH2、-NH(アルキル)、-N(アルキル)2、-NH(アリール)、-NH(ヒドロキシアルキル)、-NHC(=O)(アルキル)、-NHC(=O)H、-NHC(=O)NH2、-NHC(=O)NH(アルキル)、-NHC(=O)N(アルキル)2、-NHC(=NH)NH2、-OSO2(アルキル)、-SH、-S(アルキル)、-S(アリール)、-S(シクロアルキル)、-S(=O)アルキル、-SO2(アルキル)、-SO2NH2、-SO2NH(アルキル)、-SO2N(アルキル)2、および同類のものが挙げられるが、これらに限らない。
置換される部分が脂肪族部分の場合、好ましい置換基は、アリール、ヘテロアリール、シクロ脂肪族、ヘテロシクロ脂肪族、ハロ、ヒドロキシル、シアノ、ニトロ、アルコキシ、-O(ヒドロキシアルキル)、-O(ハロアルキル)、-O(シクロアルキル)、-O(ヘテロシクロアルキル)、-O(アリール)、アルキルチオ、アリールチオ、=O、=NH、=N(アルキル)、=NOH、=NO(アルキル)、-CO2H、-C(=O)NHOH、-C(=O)O(アルキル)、-C(=O)O(ヒドロキシアルキル)、-C(=O)NH2、-C(=O)NH(アルキル)、-C(=O)N(アルキル)2、-OC(=O)(アルキル)、-OC(=O)(ヒドロキシアルキル)、-OC(=O)O(アルキル)、-OC(=O)O(ヒドロキシアルキル)、-OC(=O)NH2、-OC(=O)NH(アルキル)、-OC(=O)N(アルキル)2、アジド、-NH2、-NH(アルキル)、-N(アルキル)2、-NH(アリール)、-NH(ヒドロキシアルキル)、-NHC(=O)(アルキル)、-NHC(=O)H、-NHC(=O)NH2、-NHC(=O)NH(アルキル)、-NHC(=O)N(アルキル)2、-NHC(=NH)NH2、-OSO2(アルキル)、-SH、-S(アルキル)、-S(アリール)、-S(=O)アルキル、-S(シクロアルキル)、-SO2(アルキル)、-SO2NH2、-SO2NH(アルキル)、および-SO2N(アルキル)2である。より好ましい置換基は、ハロ、ヒドロキシル、シアノ、ニトロ、アルコキシ、-O(アリール)、=O、=NOH、=NO(アルキル)、-OC(=O)(アルキル)、-OC(=O)O(アルキル)、-OC(=O)NH2、-OC(=O)NH(アルキル)、-OC(=O)N(アルキル)2、アジド、-NH2、-NH(アルキル)、-N(アルキル)2、-NH(アリール)、-NHC(=O)(アルキル)、-NHC(=O)H、-NHC(=O)NH2、-NHC(=O)NH(アルキル)、-NHC(=O)N(アルキル)2、および-NHC(=NH)NH2である。特に好ましい置換基は、フェニル、シアノ、ハロ、ヒドロキシル、ニトロ、C1-C4アルコキシ、O(C2-C4アルカンジイル)OH、およびO(C2-C4アルカンジイル)ハロである。
置換される部分がシクロ脂肪族、ヘテロシクロ脂肪族、アリール、またはヘテロアリール部分の場合、好ましい置換基は、アルキル、アルケニル、アルキニル、ハロ、ハロアルキル、ヒドロキシル、ヒドロキシアルキル、シアノ、ニトロ、アルコキシ、-O(ヒドロキシアルキル)、-O(ハロアルキル)、-O(アリール)、-O(シクロアルキル)、-O(ヘテロシクロアルキル)、アルキルチオ、アリールチオ、-C(=O)(アルキル)、-C(=O)H、-CO2H、-C(=O)NHOH、-C(=O)O(アルキル)、-C(=O)O(ヒドロキシアルキル)、-C(=O)NH2、-C(=O)NH(アルキル)、-C(=O)N(アルキル)2、-OC(=O)(アルキル)、-OC(=O)(ヒドロキシアルキル)、-OC(=O)O(アルキル)、-OC(=O)O(ヒドロキシアルキル)、-OC(=O)NH2、-OC(=O)NH(アルキル)、-OC(=O)N(アルキル)2、アジド、-NH2、-NH(アルキル)、-N(アルキル)2、-NH(アリール)、-NH(ヒドロキシアルキル)、-NHC(=O)(アルキル)、-NHC(=O)H、-NHC(=O)NH2、-NHC(=O)NH(アルキル)、-NHC(=O)N(アルキル)2、-NHC(=NH)NH2、-OSO2(アルキル)、-SH、-S(アルキル)、-S(アリール)、-S(シクロアルキル)、-S(=O)アルキル、-SO2(アルキル)、-SO2NH2、-SO2NH(アルキル)、および-SO2N(アルキル)2である。より好ましい置換基は、アルキル、アルケニル、ハロ、ハロアルキル、ヒドロキシル、ヒドロキシアルキル、シアノ、ニトロ、アルコキシ、-O(ヒドロキシアルキル)、-C(=O)(アルキル)、-C(=O)H、-CO2H、-C(=O)NHOH、-C(=O)O(アルキル)、-C(=O)O(ヒドロキシアルキル)、-C(=O)NH2、-C(=O)NH(アルキル)、-C(=O)N(アルキル)2、-OC(=O)(アルキル)、-OC(=O)(ヒドロキシアルキル)、-OC(=O)O(アルキル)、-OC(=O)O(ヒドロキシアルキル)、-OC(=O)NH2、-OC(=O)NH(アルキル)、-OC(=O)N(アルキル)2、-NH2、-NH(アルキル)、-N(アルキル)2、-NH(アリール)、-NHC(=O)(アルキル)、-NHC(=O)H、-NHC(=O)NH2、-NHC(=O)NH(アルキル)、-NHC(=O)N(アルキル)2、および-NHC(=NH)NH2である。特に好ましい置換基は、C1-C4アルキル、シアノ、ニトロ、ハロ、およびC1-C4アルコキシである。
「C1-C5アルキル」または「5から10%」のように範囲が述べられる場合、かかる範囲は、範囲の終点、つまり第一の例においてはC1およびC5、並びに第二の例においては5%および10%を含む。
(例えば、構造式中の関連する立体中心における価標を太線にするか、または破線にすることにより、構造式中で、二重結合をEまたはZ配置を有するものとして描くことにより、あるいは立体化学を指定する命名法または記号を用いることにより)特定の立体異性体が明確に指示されない限り、全ての立体異性体が、純粋化合物ならびにその混合物として本発明の範囲内に含まれる。特に断らない限り、ラセミ体、個々のエナンチオマー(光学的に純粋であろうと部分的に分割されていようと)、ジアステレオマー、幾何異性体、およびそれらの組み合わせ、並びにそれらの混合物は、本発明により全て包含される。
当業者は、化合物が、本明細書で使用される構造式に描かれるものと同等の互変異性体(例えば、ケトおよびエノール形)、共鳴構造、および双性イオン型を有することがあり、構造式は、そのような互変異性体、共鳴構造、双性イオン型を包含するということを認識するであろう。
「薬学的に許容されるエステル」は、インビボで(例えば人体内で)加水分解し、親化合物またはその塩を生成するか、あるいはそれ自体が親化合物の活性と類似のそれを有するエステルを意味する。適当なエステルには、C1-C5アルキル、C2-C5アルケニルまたはC2-C5アルキニルエステル、特にメチル、エチルまたはn-プロピルエステルが挙げられる。
「薬学的に許容される塩」は、医薬製剤に適する化合物の塩を意味する。化合物が一つ以上の塩基性基を有する場合、塩は、酸付加塩、例えば、硫酸塩、臭化水素酸塩、酒石酸塩、メシル酸塩、マレイン酸塩、クエン酸塩、リン酸塩、酢酸塩、パモ酸塩(エンボン酸塩)、ヨウ化水素酸塩、硝酸塩、塩酸塩、乳酸塩、メチル硫酸塩、フマル酸塩、安息香酸塩、コハク酸塩、メシル酸塩、ラクトビオン酸塩、スベリン酸塩、トシル酸塩、および同類のものなどであり得る。化合物が一つ以上の酸性基を有する場合、塩は、カルシウム塩、カリウム塩、マグネシウム塩、メグルミン塩、アンモニウム塩、亜鉛塩、ピペラジン塩、トロメタミン塩、リチウム塩、コリン塩、ジエチルアミン塩、4-フェニルシクロヘキシルアミン塩、ベンザチン塩、ナトリウム塩、テトラメチルアンモニウム塩、および同類のものなどの塩であり得る。多形結晶性形態および溶媒和物も本発明の範囲内に包含される。
「患者(subject)」は動物を指し、霊長類(例えば、ヒト)、サル、ウシ、ブタ、ヒツジ、ヤギ、ウマ、イヌ、ネコ、ウサギ、ラット、またはマウスを含むが、これらに限らない。「患者(subject)」および「患者(patient)」という用語は、例えば、ヒトなどの哺乳動物の患者に関して、本明細書で互換的に使用される。
「治療する(treat)」、「治療する(treating)」、および「治療(treatment)」という用語は、疾患または障害の治療の文脈において、障害、疾患、または病態、あるいは障害、疾患、もしくは病態に関連する症状のうちの一つ以上を軽減するか、または抑制すること;あるいは疾患、障害、または病態の、あるいは一つ以上のそれらの症状の進行、拡大または悪化を遅らせることを含むように意図される。「がんの治療」は、以下の効果のうちの一つ以上を指す:(1)(i)遅延および(ii)完全な増殖停止を含む、ある程度の腫瘍増殖の阻害;(2)腫瘍細胞数の減少;(3)腫瘍の大きさの維持;(4)腫瘍の大きさの減少;(5)末梢臓器への腫瘍細胞浸潤の(i)減少、(ii)遅延または(iii)完全な予防を含む阻害;(6)転移の(i)減少、(ii)遅延または(iii)完全な予防を含む阻害;(7)(i)腫瘍の大きさの維持、(ii)腫瘍の大きさの減少、(iii)腫瘍の増殖の遅延、(iv)浸潤の減少、遅延または予防をもたらすことがある、抗腫瘍免疫応答の増強および/または(8)ある程度の、障害に関連する一つ以上の症状の重症度または数の軽減。
本明細書の式において、価標に対して横方向の波線(
)または価標の末端にあるアスタリスク(*)は、共有結合部位を意味する。例えば、式
において、Rは
である、またはRは
であるという記述は、
を意味する。
本明細書の式において、その2つの炭素の間で芳香環を横切る価標は、その価標に結合する基が、黙示的にそこにある(または、完全に書かれている場合、明示的にそこにある)水素の除去によって空きができる芳香環の位置のうちのどこにあってもよいということを意味する。実例として:
は、
を表し;
は、
を表し;
は、
を表す。
本開示は、本明細書に記載される化合物で生じる原子の全ての同位体を含む。同位体は、原子番号は同じだが異なる質量数を有する原子を含む。一般的な例であり、限定ではないが、水素の同位体には重水素およびトリチウムが挙げられる。炭素の同位体には、13Cおよび14Cが挙げられる。同位体標識した本発明の化合物は、一般的に、他の場合に使用される非標識試薬の代わりに、同位体標識した適切な試薬を用いて、当業者に既知の従来の技術により、または本明細書に記載されるものと類似の工程により調製され得る。例として、C1-C3アルキル基は、重水素化されていなくても、部分的に重水素化されていても、完全に重水素化されていてもよく、「CH3」には、CH3、13CH3、14CH3、CH2T、CH2D、CHD2、CD3などが含まれる。一つの実施形態において、化合物中の様々な元素は、それらの天然の同位体存在度で存在する。
当業者は、特定の構造はどちらの互変異性体-例えば、ケトかエノールか-で描かれてもよく、その2つの形態は等価であるということを認識するであろう。
アクロニムおよび略語
表Cは、本明細書で使用されるアクロニムおよび略語の一覧をその意味とともに提供する。
参考文献
本明細書の初めの方で、筆頭著者(または発明者)および日付により省略された形で引用される以下の参考文献に対する完全な引用を以下に提供する。これらの参考文献のそれぞれは、あらゆる目的のために、参照により本明細書に組み込まれる。
Akinbobuyi et al., Tetrahedron Lett. 2015, 56, 458, “Facile syntheses of functionalized toll-like receptor 7 agonists”.
Akinbobuyi et al., Bioorg. Med. Chem. Lett. 2016, 26, 4246, “Synthesis and immunostimulatory activity of substituted TLR7 agonists.”
Barberis et al., US 2012/0003298 A1 (2012).
Beesu et al., J. Med. Chem. 2017, 60, 2084, “Identification of High-Potency Human TLR8 and Dual TLR7/TLR8 Agonists in Pyrimidine-2,4-diamines.”
Berghoefer et al., J. Immunol. 2007, 178, 4072, “Natural and Synthetic TLR7 Ligands Inhibit CpG-A- and CpG-C-Oligodeoxynucleotide-Induced IFN-α Production.”
Bonfanti et al., US 2014/0323441 A1 (2015) [2015a].
Bonfanti et al., US 2015/0299221 A1 (2015) [2015b].
Bonfanti et al., US 2016/0304531 A1 (2016).
Carson et al., US 2013/0202629 A1 (2013).
Carson et al., US 8,729,088 B2 (2014).
Carson et al., US 9,050,376 B2 (2015).
Carson et al., US 2016/0199499 A1 (2016).
Chan et al., Bioconjugate Chem. 2009, 20, 1194, “Synthesis and Immunological Characterization of Toll-Like Receptor 7 Agonistic Conjugates.”
Chan et al., Bioconjugate Chem. 2011, 22, 445, “Synthesis and Characterization of PEGylated Toll Like Receptor 7 Ligands.”
Chen et al., US 7,919,498 B2 (2011).
Coe et al., US 9,662,336 B2 (2017).
Cortez and Va, Medicinal Chem. Rev. 2018, 53, 481, “Recent Advances in Small-Molecule TLR7 Agonists for Drug Discovery”.
Cortez et al., US 2017/0121421 A1 (2017).
Cortez et al., US 9,944,649 B2 (2018).
Dellaria et al., WO 2007/028129 A1 (2007).
Desai et al., US 9,127,006 B2 (2015).
Ding et al., WO 2016/107536 A1 (2016).
Ding et al., US 2017/0273983 A1 (2017) [2017a].
Ding et al., WO 2017/076346 A1 (2017) [2017b].
Gadd et al., Bioconjugate Chem. 2015, 26, 1743, “Targeted Activation of Toll-Like Receptors: Conjugation of a Toll-Like Receptor 7 Agonist to a Monoclonal Antibody Maintains Antigen Binding and Specificity.”
Graupe et al., US 8,993,755 B2 (2015).
Embrechts et al., J. Med. Chem. 2018, 61, 6236, “2,4-Diaminoquinazolines as Dual Toll Like Receptor (TLR) 7/8 Modulators for the Treatment of Hepatitis B Virus.”
Halcomb et al., US 9,161,934 B2 (2015).
Hashimoto et al., US 2009/0118263 A1 (2009).
He et al., US 10,487,084 B2 (2019) [2019a].
He et al., US 10,508,115 B2 (2019) [2019b].
Hirota et al., US 6,028,076 (2000).
Holldack et al., US 2012/0083473 A1 (2012).
Isobe et al., US 6,376,501 B1 (2002).
Isobe et al., JP 2004137157 (2004).
Isobe et al., J. Med. Chem. 2006, 49 (6), 2088, “Synthesis and Biological Evaluation of Novel 9-Substituted-8-Hydroxyadenine Derivatives as Potent Interferon Inducers.”
Isobe et al., US 7,521,454 B2 (2009) [2009a].
Isobe et al., US 2009/0105212 A1 (2009) [2009b].
Isobe et al., US 2011/0028715 A1 (2011).
Isobe et al., US 8,148,371 B2 (2012).
Jensen et al., WO 2015/036044 A1 (2015).
Jones et al., US 7,691,877 B2 (2010).
Jones et al., US 2012/0302598 A1 (2012).
Kasibhatla et al., US 7,241,890 B2 (2007).
Koga-Yamakawa et al., Int. J. Cancer 2013, 132 (3), 580, “Intratracheal and oral administration of SM-276001: A selective TLR7 agonist, leads to antitumor efficacy in primary and metastatic models of cancer.”
Li et al., US 9,902,730 B2 (2018).
Lioux et al., US 9,295,732 B2 (2016).
Lund et al., Proc. Nat’l Acad. Sci (USA) 2004, 101 (15), 5598, “Recognition of single-stranded RNA viruses by Toll-like receptor 7.”
Maj et al., US 9,173,935 B2 (2015).
McGowan et al., US 2016/0168150 A1 (2016) [2016a].
McGowan et al., US 9,499,549 B2 (2016) [2016b].
McGowan et al., J. Med. Chem. 2017, 60, 6137, “Identification and Optimization of Pyrrolo[3,2-d]pyrimidine Toll-like Receptor 7 (TLR7) Selective Agonists for the Treatment of Hepatitis B.”
Musmuca et al., J. Chem. Information & Modeling 2009, 49 (7), 1777, “Small-Molecule Interferon Inducers. Toward the Comprehension of the Molecular Determinants through Ligand-Based Approaches.”
Nakamura et al., Bioorg. Med. Chem. Lett. 2013, 13, 669, “Synthesis and evaluation of 8-oxoadenine derivatives as potent Toll-like receptor agonists with high water solubility.”
Ogita et al., US 2007/0225303 A1 (2007).
Ota et al., WO 2019/124500 A1 (2019).
Pilatte et al., WO 2017/216293 A1 (2017).
Poudel et al., US 10,472,361 B2 (2019) [2019a].
Poudel et al., US 10,494,370 B2 (2019) [2019b].
Poudel et al., US 2020/0038403 A1 (2020) [2020a].
Poudel et al., US 2020/0039986 A1 (2020) [2020b].
Purandare et al., WO 2019/209811 A1 (2019).
Pryde, US 7,642,350 B2 (2010).
Sato-Kaneko et al., JCI Insight 2017, 2, e93397, “Combination Immunotherapy with TLR Agonists and Checkpoint Inhibitors Suppresses Head and Neck Cancer”.
Smits et al., The Oncologist 2008, 13, 859, “The Use of TLR7 and TLR8 Ligands for the Enhancement of Cancer Immunotherapy”.
Vasilakos and Tomai, Expert Rev. Vaccines 2013, 12, 809, “The Use of Toll-like Receptor 7/8 Agonists as Vaccine Adjuvants”.
Vernejoul et al., US 2014/0141033 A1 (2014).
Young et al., US 10,457,681 B2 (2019).
Yu et al., PLoS One 2013, 8 (3), e56514, “Toll-Like Receptor 7 Agonists: Chemical Feature Based Pharmacophore Identification and Molecular Docking Studies.”
Zhang et al., Immunity 2016, 45, 737, “Structural Analysis Reveals that Toll-like Receptor 7 Is a Dual Receptor for Guanosine and Single-Stranded RNA.”
Zhang et al., WO 2018/095426 A1 (2018)>
Zurawski et al., US 2012/0231023 A1 (2012).
前述の本発明の詳細な説明は、本発明の特定の部分または態様に、主にまたは排他的に関係する節を含む。これは、明確化のため、および便宜のためであり、特定の特徴は、それが開示される節だけでなくその他の節においても関連していることがあり、本明細書における開示は、異なる節に記載される情報の、全ての適切な組み合わせを含むことが理解されるべきである。同様に、本明細書における様々な図および説明は、本発明の特定の実施形態に関するが、具体的な特徴が、特定の図または実施形態の文脈で開示される場合、かかる特徴は、適切な範囲で、別の図または実施形態の文脈で、別の特徴と組み合わせて、または本発明一般においても使用され得るということが理解されるべきである。
さらに、本発明は、特定の好ましい実施形態について特に記載されているが、本発明は、かかる好ましい実施形態に限定されない。それどころか、本発明の範囲は、添付の請求項により定義される。