JP7214925B2 - Pan-RAFキナーゼ阻害剤としてのビアリール化合物 - Google Patents
Pan-RAFキナーゼ阻害剤としてのビアリール化合物 Download PDFInfo
- Publication number
- JP7214925B2 JP7214925B2 JP2022533534A JP2022533534A JP7214925B2 JP 7214925 B2 JP7214925 B2 JP 7214925B2 JP 2022533534 A JP2022533534 A JP 2022533534A JP 2022533534 A JP2022533534 A JP 2022533534A JP 7214925 B2 JP7214925 B2 JP 7214925B2
- Authority
- JP
- Japan
- Prior art keywords
- compound
- μmol
- mmol
- added
- stirred
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- -1 Biaryl compounds Chemical class 0.000 title description 19
- 229940043355 kinase inhibitor Drugs 0.000 title description 2
- 239000003757 phosphotransferase inhibitor Substances 0.000 title description 2
- 150000001875 compounds Chemical class 0.000 claims description 272
- 150000003839 salts Chemical class 0.000 claims description 62
- 125000000217 alkyl group Chemical group 0.000 claims description 48
- 206010028980 Neoplasm Diseases 0.000 claims description 42
- 229910052739 hydrogen Inorganic materials 0.000 claims description 42
- 229910052731 fluorine Inorganic materials 0.000 claims description 41
- 229910052799 carbon Inorganic materials 0.000 claims description 23
- 229910052794 bromium Inorganic materials 0.000 claims description 22
- 229910052801 chlorine Inorganic materials 0.000 claims description 22
- 229910052740 iodine Inorganic materials 0.000 claims description 21
- 239000003814 drug Substances 0.000 claims description 10
- 229910052736 halogen Inorganic materials 0.000 claims description 9
- 150000002367 halogens Chemical class 0.000 claims description 9
- 238000002360 preparation method Methods 0.000 claims description 8
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 5
- 201000011510 cancer Diseases 0.000 claims description 4
- 229940123690 Raf kinase inhibitor Drugs 0.000 claims description 3
- 108700021017 phosphatidylethanolamine binding protein Proteins 0.000 claims description 3
- 102000051624 phosphatidylethanolamine binding protein Human genes 0.000 claims description 3
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical group CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 173
- 239000000243 solution Substances 0.000 description 141
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 81
- 238000006243 chemical reaction Methods 0.000 description 80
- 239000011541 reaction mixture Substances 0.000 description 74
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 67
- 239000012074 organic phase Substances 0.000 description 59
- 235000019439 ethyl acetate Nutrition 0.000 description 58
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 51
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 50
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 44
- 239000000460 chlorine Substances 0.000 description 41
- IJGRMHOSHXDMSA-UHFFFAOYSA-N nitrogen Substances N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 40
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 40
- 229910001873 dinitrogen Inorganic materials 0.000 description 39
- 210000004027 cell Anatomy 0.000 description 38
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 32
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 32
- 239000000741 silica gel Substances 0.000 description 32
- 229910002027 silica gel Inorganic materials 0.000 description 32
- 239000000706 filtrate Substances 0.000 description 28
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 27
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 25
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 24
- 239000000203 mixture Substances 0.000 description 23
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 22
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 20
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 19
- 238000010898 silica gel chromatography Methods 0.000 description 19
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 16
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 16
- 229910000029 sodium carbonate Inorganic materials 0.000 description 16
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 15
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 15
- 229910000024 caesium carbonate Inorganic materials 0.000 description 15
- 238000004440 column chromatography Methods 0.000 description 15
- 125000001424 substituent group Chemical group 0.000 description 15
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 14
- 238000002474 experimental method Methods 0.000 description 14
- 239000012071 phase Substances 0.000 description 14
- 238000000926 separation method Methods 0.000 description 14
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 13
- 241000699670 Mus sp. Species 0.000 description 13
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 13
- 239000011259 mixed solution Substances 0.000 description 12
- 229920006395 saturated elastomer Polymers 0.000 description 12
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 11
- 239000002253 acid Substances 0.000 description 11
- 239000012043 crude product Substances 0.000 description 11
- 238000011282 treatment Methods 0.000 description 11
- 201000005202 lung cancer Diseases 0.000 description 10
- 208000020816 lung neoplasm Diseases 0.000 description 10
- 230000014759 maintenance of location Effects 0.000 description 10
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 9
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 9
- 238000000034 method Methods 0.000 description 9
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 9
- 206010059866 Drug resistance Diseases 0.000 description 8
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical group CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 8
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 8
- 102100033479 RAF proto-oncogene serine/threonine-protein kinase Human genes 0.000 description 8
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical group OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 8
- 229910002092 carbon dioxide Inorganic materials 0.000 description 8
- WDVGNXKCFBOKDF-UHFFFAOYSA-N dicyclohexyl-[3,6-dimethoxy-2-[2,4,6-tri(propan-2-yl)phenyl]phenyl]phosphane Chemical compound COC1=CC=C(OC)C(C=2C(=CC(=CC=2C(C)C)C(C)C)C(C)C)=C1P(C1CCCCC1)C1CCCCC1 WDVGNXKCFBOKDF-UHFFFAOYSA-N 0.000 description 8
- 125000000524 functional group Chemical group 0.000 description 8
- 125000006239 protecting group Chemical group 0.000 description 8
- 102000016914 ras Proteins Human genes 0.000 description 8
- 238000007920 subcutaneous administration Methods 0.000 description 8
- 238000012360 testing method Methods 0.000 description 8
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 7
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical class [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 7
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 7
- 125000004429 atom Chemical group 0.000 description 7
- 230000015572 biosynthetic process Effects 0.000 description 7
- 229940079593 drug Drugs 0.000 description 7
- 239000000284 extract Substances 0.000 description 7
- 239000003112 inhibitor Substances 0.000 description 7
- 230000002401 inhibitory effect Effects 0.000 description 7
- 239000000463 material Substances 0.000 description 7
- OKKJLVBELUTLKV-VMNATFBRSA-N methanol-d1 Chemical compound [2H]OC OKKJLVBELUTLKV-VMNATFBRSA-N 0.000 description 7
- 239000012046 mixed solvent Substances 0.000 description 7
- 238000003786 synthesis reaction Methods 0.000 description 7
- 230000004614 tumor growth Effects 0.000 description 7
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 6
- 102000043136 MAP kinase family Human genes 0.000 description 6
- 108091054455 MAP kinase family Proteins 0.000 description 6
- 102100024193 Mitogen-activated protein kinase 1 Human genes 0.000 description 6
- 230000004913 activation Effects 0.000 description 6
- 230000037396 body weight Effects 0.000 description 6
- 239000001569 carbon dioxide Substances 0.000 description 6
- 239000003153 chemical reaction reagent Substances 0.000 description 6
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 6
- 239000002609 medium Substances 0.000 description 6
- 230000035772 mutation Effects 0.000 description 6
- 230000037361 pathway Effects 0.000 description 6
- 230000026731 phosphorylation Effects 0.000 description 6
- 238000006366 phosphorylation reaction Methods 0.000 description 6
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 6
- 239000002904 solvent Substances 0.000 description 6
- 239000000126 substance Substances 0.000 description 6
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 6
- 239000007821 HATU Substances 0.000 description 5
- 230000001028 anti-proliverative effect Effects 0.000 description 5
- 125000004432 carbon atom Chemical group C* 0.000 description 5
- MXFYYFVVIIWKFE-UHFFFAOYSA-N dicyclohexyl-[2-[2,6-di(propan-2-yloxy)phenyl]phenyl]phosphane Chemical compound CC(C)OC1=CC=CC(OC(C)C)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 MXFYYFVVIIWKFE-UHFFFAOYSA-N 0.000 description 5
- 125000005647 linker group Chemical group 0.000 description 5
- ILMRJRBKQSSXGY-UHFFFAOYSA-N tert-butyl(dimethyl)silicon Chemical group C[Si](C)C(C)(C)C ILMRJRBKQSSXGY-UHFFFAOYSA-N 0.000 description 5
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 5
- 101000984753 Homo sapiens Serine/threonine-protein kinase B-raf Proteins 0.000 description 4
- 241001465754 Metazoa Species 0.000 description 4
- 241000699666 Mus <mouse, genus> Species 0.000 description 4
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 4
- 102100027103 Serine/threonine-protein kinase B-raf Human genes 0.000 description 4
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 4
- 102000004142 Trypsin Human genes 0.000 description 4
- 108090000631 Trypsin Proteins 0.000 description 4
- 125000002252 acyl group Chemical group 0.000 description 4
- 239000013592 cell lysate Substances 0.000 description 4
- 238000012054 celltiter-glo Methods 0.000 description 4
- 238000007796 conventional method Methods 0.000 description 4
- 230000000694 effects Effects 0.000 description 4
- 238000001914 filtration Methods 0.000 description 4
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 4
- 230000005764 inhibitory process Effects 0.000 description 4
- 238000005259 measurement Methods 0.000 description 4
- 239000012312 sodium hydride Substances 0.000 description 4
- 229910000104 sodium hydride Inorganic materials 0.000 description 4
- BCNZYOJHNLTNEZ-UHFFFAOYSA-N tert-butyldimethylsilyl chloride Chemical compound CC(C)(C)[Si](C)(C)Cl BCNZYOJHNLTNEZ-UHFFFAOYSA-N 0.000 description 4
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 4
- 239000012588 trypsin Substances 0.000 description 4
- SZUVGFMDDVSKSI-WIFOCOSTSA-N (1s,2s,3s,5r)-1-(carboxymethyl)-3,5-bis[(4-phenoxyphenyl)methyl-propylcarbamoyl]cyclopentane-1,2-dicarboxylic acid Chemical compound O=C([C@@H]1[C@@H]([C@](CC(O)=O)([C@H](C(=O)N(CCC)CC=2C=CC(OC=3C=CC=CC=3)=CC=2)C1)C(O)=O)C(O)=O)N(CCC)CC(C=C1)=CC=C1OC1=CC=CC=C1 SZUVGFMDDVSKSI-WIFOCOSTSA-N 0.000 description 3
- ONBQEOIKXPHGMB-VBSBHUPXSA-N 1-[2-[(2s,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]oxy-4,6-dihydroxyphenyl]-3-(4-hydroxyphenyl)propan-1-one Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1OC1=CC(O)=CC(O)=C1C(=O)CCC1=CC=C(O)C=C1 ONBQEOIKXPHGMB-VBSBHUPXSA-N 0.000 description 3
- KKVYYGGCHJGEFJ-UHFFFAOYSA-N 1-n-(4-chlorophenyl)-6-methyl-5-n-[3-(7h-purin-6-yl)pyridin-2-yl]isoquinoline-1,5-diamine Chemical compound N=1C=CC2=C(NC=3C(=CC=CN=3)C=3C=4N=CNC=4N=CN=3)C(C)=CC=C2C=1NC1=CC=C(Cl)C=C1 KKVYYGGCHJGEFJ-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- 102000004190 Enzymes Human genes 0.000 description 3
- 108090000790 Enzymes Proteins 0.000 description 3
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 3
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 3
- 101100381978 Mus musculus Braf gene Proteins 0.000 description 3
- 101100523539 Mus musculus Raf1 gene Proteins 0.000 description 3
- 241000209094 Oryza Species 0.000 description 3
- 235000007164 Oryza sativa Nutrition 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- ZEEBGORNQSEQBE-UHFFFAOYSA-N [2-(3-phenylphenoxy)-6-(trifluoromethyl)pyridin-4-yl]methanamine Chemical compound C1(=CC(=CC=C1)OC1=NC(=CC(=C1)CN)C(F)(F)F)C1=CC=CC=C1 ZEEBGORNQSEQBE-UHFFFAOYSA-N 0.000 description 3
- 230000002378 acidificating effect Effects 0.000 description 3
- 150000007513 acids Chemical class 0.000 description 3
- 238000010171 animal model Methods 0.000 description 3
- 239000008346 aqueous phase Substances 0.000 description 3
- 239000006285 cell suspension Substances 0.000 description 3
- 229940125904 compound 1 Drugs 0.000 description 3
- 229940126543 compound 14 Drugs 0.000 description 3
- 229940126142 compound 16 Drugs 0.000 description 3
- 238000011161 development Methods 0.000 description 3
- 230000018109 developmental process Effects 0.000 description 3
- 238000007865 diluting Methods 0.000 description 3
- 239000000539 dimer Substances 0.000 description 3
- 238000011534 incubation Methods 0.000 description 3
- 239000000178 monomer Substances 0.000 description 3
- AICOOMRHRUFYCM-ZRRPKQBOSA-N oxazine, 1 Chemical compound C([C@@H]1[C@H](C(C[C@]2(C)[C@@H]([C@H](C)N(C)C)[C@H](O)C[C@]21C)=O)CC1=CC2)C[C@H]1[C@@]1(C)[C@H]2N=C(C(C)C)OC1 AICOOMRHRUFYCM-ZRRPKQBOSA-N 0.000 description 3
- 235000011056 potassium acetate Nutrition 0.000 description 3
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 3
- 108010077182 raf Kinases Proteins 0.000 description 3
- 102000009929 raf Kinases Human genes 0.000 description 3
- 235000009566 rice Nutrition 0.000 description 3
- 102200124916 rs121434596 Human genes 0.000 description 3
- FVAUCKIRQBBSSJ-UHFFFAOYSA-M sodium iodide Chemical compound [Na+].[I-] FVAUCKIRQBBSSJ-UHFFFAOYSA-M 0.000 description 3
- 238000006467 substitution reaction Methods 0.000 description 3
- 238000010189 synthetic method Methods 0.000 description 3
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 description 2
- QFLWZFQWSBQYPS-AWRAUJHKSA-N (3S)-3-[[(2S)-2-[[(2S)-2-[5-[(3aS,6aR)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoylamino]-3-methylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-[1-bis(4-chlorophenoxy)phosphorylbutylamino]-4-oxobutanoic acid Chemical compound CCCC(NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@@H](NC(=O)CCCCC1SC[C@@H]2NC(=O)N[C@H]12)C(C)C)P(=O)(Oc1ccc(Cl)cc1)Oc1ccc(Cl)cc1 QFLWZFQWSBQYPS-AWRAUJHKSA-N 0.000 description 2
- UNILWMWFPHPYOR-KXEYIPSPSA-M 1-[6-[2-[3-[3-[3-[2-[2-[3-[[2-[2-[[(2r)-1-[[2-[[(2r)-1-[3-[2-[2-[3-[[2-(2-amino-2-oxoethoxy)acetyl]amino]propoxy]ethoxy]ethoxy]propylamino]-3-hydroxy-1-oxopropan-2-yl]amino]-2-oxoethyl]amino]-3-[(2r)-2,3-di(hexadecanoyloxy)propyl]sulfanyl-1-oxopropan-2-yl Chemical compound O=C1C(SCCC(=O)NCCCOCCOCCOCCCNC(=O)COCC(=O)N[C@@H](CSC[C@@H](COC(=O)CCCCCCCCCCCCCCC)OC(=O)CCCCCCCCCCCCCCC)C(=O)NCC(=O)N[C@H](CO)C(=O)NCCCOCCOCCOCCCNC(=O)COCC(N)=O)CC(=O)N1CCNC(=O)CCCCCN\1C2=CC=C(S([O-])(=O)=O)C=C2CC/1=C/C=C/C=C/C1=[N+](CC)C2=CC=C(S([O-])(=O)=O)C=C2C1 UNILWMWFPHPYOR-KXEYIPSPSA-M 0.000 description 2
- HIXDQWDOVZUNNA-UHFFFAOYSA-N 2-(3,4-dimethoxyphenyl)-5-hydroxy-7-methoxychromen-4-one Chemical compound C=1C(OC)=CC(O)=C(C(C=2)=O)C=1OC=2C1=CC=C(OC)C(OC)=C1 HIXDQWDOVZUNNA-UHFFFAOYSA-N 0.000 description 2
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 2
- 238000011729 BALB/c nude mouse Methods 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 2
- 239000006145 Eagle's minimal essential medium Substances 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- 102100039788 GTPase NRas Human genes 0.000 description 2
- 101000744505 Homo sapiens GTPase NRas Proteins 0.000 description 2
- 101000771237 Homo sapiens Serine/threonine-protein kinase A-Raf Proteins 0.000 description 2
- WTDHULULXKLSOZ-UHFFFAOYSA-N Hydroxylamine hydrochloride Chemical compound Cl.ON WTDHULULXKLSOZ-UHFFFAOYSA-N 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- 239000004698 Polyethylene Substances 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- 102100029437 Serine/threonine-protein kinase A-Raf Human genes 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- ABRVLXLNVJHDRQ-UHFFFAOYSA-N [2-pyridin-3-yl-6-(trifluoromethyl)pyridin-4-yl]methanamine Chemical compound FC(C1=CC(=CC(=N1)C=1C=NC=CC=1)CN)(F)F ABRVLXLNVJHDRQ-UHFFFAOYSA-N 0.000 description 2
- WREOTYWODABZMH-DTZQCDIJSA-N [[(2r,3s,4r,5r)-3,4-dihydroxy-5-[2-oxo-4-(2-phenylethoxyamino)pyrimidin-1-yl]oxolan-2-yl]methoxy-hydroxyphosphoryl] phosphono hydrogen phosphate Chemical compound O[C@@H]1[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)O[C@H]1N(C=C\1)C(=O)NC/1=N\OCCC1=CC=CC=C1 WREOTYWODABZMH-DTZQCDIJSA-N 0.000 description 2
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 2
- YRKCREAYFQTBPV-UHFFFAOYSA-N acetylacetone Chemical compound CC(=O)CC(C)=O YRKCREAYFQTBPV-UHFFFAOYSA-N 0.000 description 2
- 150000001412 amines Chemical class 0.000 description 2
- 125000003277 amino group Chemical group 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 125000005002 aryl methyl group Chemical group 0.000 description 2
- 239000012298 atmosphere Substances 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 2
- 230000000903 blocking effect Effects 0.000 description 2
- 238000004113 cell culture Methods 0.000 description 2
- 239000006143 cell culture medium Substances 0.000 description 2
- 238000004587 chromatography analysis Methods 0.000 description 2
- 229940125773 compound 10 Drugs 0.000 description 2
- 229940125797 compound 12 Drugs 0.000 description 2
- 229940125758 compound 15 Drugs 0.000 description 2
- 229940125782 compound 2 Drugs 0.000 description 2
- 229940126214 compound 3 Drugs 0.000 description 2
- 229940125898 compound 5 Drugs 0.000 description 2
- 239000013078 crystal Substances 0.000 description 2
- BFSMGDJOXZAERB-UHFFFAOYSA-N dabrafenib Chemical compound S1C(C(C)(C)C)=NC(C=2C(=C(NS(=O)(=O)C=3C(=CC=CC=3F)F)C=CC=2)F)=C1C1=CC=NC(N)=N1 BFSMGDJOXZAERB-UHFFFAOYSA-N 0.000 description 2
- 229960002465 dabrafenib Drugs 0.000 description 2
- 238000001212 derivatisation Methods 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 125000005982 diphenylmethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])(*)C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- RTZKZFJDLAIYFH-UHFFFAOYSA-N ether Substances CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 2
- 230000012010 growth Effects 0.000 description 2
- 238000002868 homogeneous time resolved fluorescence Methods 0.000 description 2
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 2
- 229940072106 hydroxystearate Drugs 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 239000012442 inert solvent Substances 0.000 description 2
- 238000011081 inoculation Methods 0.000 description 2
- KQNPFQTWMSNSAP-UHFFFAOYSA-N isobutyric acid Chemical compound CC(C)C(O)=O KQNPFQTWMSNSAP-UHFFFAOYSA-N 0.000 description 2
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 201000001441 melanoma Diseases 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- 238000010899 nucleation Methods 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- 239000001301 oxygen Substances 0.000 description 2
- 229910052760 oxygen Inorganic materials 0.000 description 2
- 239000003208 petroleum Substances 0.000 description 2
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 2
- XNGIFLGASWRNHJ-UHFFFAOYSA-N phthalic acid Chemical compound OC(=O)C1=CC=CC=C1C(O)=O XNGIFLGASWRNHJ-UHFFFAOYSA-N 0.000 description 2
- 229920000573 polyethylene Polymers 0.000 description 2
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 2
- 108090000765 processed proteins & peptides Proteins 0.000 description 2
- 102200124923 rs121913254 Human genes 0.000 description 2
- 238000007086 side reaction Methods 0.000 description 2
- 125000003808 silyl group Chemical group [H][Si]([H])([H])[*] 0.000 description 2
- 238000004467 single crystal X-ray diffraction Methods 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- 238000007619 statistical method Methods 0.000 description 2
- 229960005322 streptomycin Drugs 0.000 description 2
- TYFQFVWCELRYAO-UHFFFAOYSA-N suberic acid Chemical compound OC(=O)CCCCCCC(O)=O TYFQFVWCELRYAO-UHFFFAOYSA-N 0.000 description 2
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 210000004881 tumor cell Anatomy 0.000 description 2
- 230000001875 tumorinhibitory effect Effects 0.000 description 2
- GPXBXXGIAQBQNI-UHFFFAOYSA-N vemurafenib Chemical compound CCCS(=O)(=O)NC1=CC=C(F)C(C(=O)C=2C3=CC(=CN=C3NC=2)C=2C=CC(Cl)=CC=2)=C1F GPXBXXGIAQBQNI-UHFFFAOYSA-N 0.000 description 2
- 229960003862 vemurafenib Drugs 0.000 description 2
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- UTQNKKSJPHTPBS-UHFFFAOYSA-N 2,2,2-trichloroethanone Chemical group ClC(Cl)(Cl)[C]=O UTQNKKSJPHTPBS-UHFFFAOYSA-N 0.000 description 1
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 1
- LBLYYCQCTBFVLH-UHFFFAOYSA-M 2-methylbenzenesulfonate Chemical compound CC1=CC=CC=C1S([O-])(=O)=O LBLYYCQCTBFVLH-UHFFFAOYSA-M 0.000 description 1
- NPAXPTHCUCUHPT-UHFFFAOYSA-N 3,4,7,8-tetramethyl-1,10-phenanthroline Chemical compound CC1=CN=C2C3=NC=C(C)C(C)=C3C=CC2=C1C NPAXPTHCUCUHPT-UHFFFAOYSA-N 0.000 description 1
- KVCQTKNUUQOELD-UHFFFAOYSA-N 4-amino-n-[1-(3-chloro-2-fluoroanilino)-6-methylisoquinolin-5-yl]thieno[3,2-d]pyrimidine-7-carboxamide Chemical compound N=1C=CC2=C(NC(=O)C=3C4=NC=NC(N)=C4SC=3)C(C)=CC=C2C=1NC1=CC=CC(Cl)=C1F KVCQTKNUUQOELD-UHFFFAOYSA-N 0.000 description 1
- PXACTUVBBMDKRW-UHFFFAOYSA-N 4-bromobenzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=C(Br)C=C1 PXACTUVBBMDKRW-UHFFFAOYSA-N 0.000 description 1
- POILWHVDKZOXJZ-UHFFFAOYSA-N 4-hydroxypent-3-en-2-one Chemical compound CC(O)=CC(C)=O POILWHVDKZOXJZ-UHFFFAOYSA-N 0.000 description 1
- TXNLQUKVUJITMX-UHFFFAOYSA-N 4-tert-butyl-2-(4-tert-butylpyridin-2-yl)pyridine Chemical compound CC(C)(C)C1=CC=NC(C=2N=CC=C(C=2)C(C)(C)C)=C1 TXNLQUKVUJITMX-UHFFFAOYSA-N 0.000 description 1
- NGFFVZQXSRKHBM-FKBYEOEOSA-N 5-[[(1r,1as,6br)-1-[6-(trifluoromethyl)-1h-benzimidazol-2-yl]-1a,6b-dihydro-1h-cyclopropa[b][1]benzofuran-5-yl]oxy]-3,4-dihydro-1h-1,8-naphthyridin-2-one Chemical compound N1C(=O)CCC2=C1N=CC=C2OC(C=C1[C@@H]23)=CC=C1O[C@@H]3[C@H]2C1=NC2=CC=C(C(F)(F)F)C=C2N1 NGFFVZQXSRKHBM-FKBYEOEOSA-N 0.000 description 1
- OZJPLYNZGCXSJM-UHFFFAOYSA-N 5-valerolactone Chemical compound O=C1CCCCO1 OZJPLYNZGCXSJM-UHFFFAOYSA-N 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- 208000031648 Body Weight Changes Diseases 0.000 description 1
- 101001011741 Bos taurus Insulin Proteins 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- DFCLLLPNKBIWIZ-UHFFFAOYSA-N CO[Ir].C1CCC=CC=CC1 Chemical class CO[Ir].C1CCC=CC=CC1 DFCLLLPNKBIWIZ-UHFFFAOYSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 102100031480 Dual specificity mitogen-activated protein kinase kinase 1 Human genes 0.000 description 1
- 101710146526 Dual specificity mitogen-activated protein kinase kinase 1 Proteins 0.000 description 1
- 102100030013 Endoribonuclease Human genes 0.000 description 1
- 101710199605 Endoribonuclease Proteins 0.000 description 1
- 230000005526 G1 to G0 transition Effects 0.000 description 1
- IAJILQKETJEXLJ-UHFFFAOYSA-N Galacturonsaeure Natural products O=CC(O)C(O)C(O)C(O)C(O)=O IAJILQKETJEXLJ-UHFFFAOYSA-N 0.000 description 1
- 238000010155 Games-Howell test Methods 0.000 description 1
- 206010020751 Hypersensitivity Diseases 0.000 description 1
- HHCBMISMPSAZBF-UHFFFAOYSA-N LY3009120 Chemical compound CC1=NC2=NC(NC)=NC=C2C=C1C1=CC(NC(=O)NCCC(C)(C)C)=C(F)C=C1C HHCBMISMPSAZBF-UHFFFAOYSA-N 0.000 description 1
- 239000012448 Lithium borohydride Substances 0.000 description 1
- 229940124647 MEK inhibitor Drugs 0.000 description 1
- 241000699660 Mus musculus Species 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 108091000080 Phosphotransferase Proteins 0.000 description 1
- 229920002556 Polyethylene Glycol 300 Polymers 0.000 description 1
- 229920002565 Polyethylene Glycol 400 Polymers 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- 102000042888 RAF family Human genes 0.000 description 1
- 108091082327 RAF family Proteins 0.000 description 1
- 101710113029 Serine/threonine-protein kinase Proteins 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 1
- VWMJHAFYPMOMGF-ZCFIWIBFSA-N TAK-580 Chemical compound N([C@H](C)C=1SC(=CN=1)C(=O)NC=1N=CC(Cl)=C(C=1)C(F)(F)F)C(=O)C1=NC=NC(N)=C1Cl VWMJHAFYPMOMGF-ZCFIWIBFSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- DHXVGJBLRPWPCS-UHFFFAOYSA-N Tetrahydropyran Chemical compound C1CCOCC1 DHXVGJBLRPWPCS-UHFFFAOYSA-N 0.000 description 1
- 229920004890 Triton X-100 Polymers 0.000 description 1
- 239000013504 Triton X-100 Substances 0.000 description 1
- DQFAAIWCCHDCLO-UHFFFAOYSA-N acetonitrile;formic acid;hydrate Chemical group O.CC#N.OC=O DQFAAIWCCHDCLO-UHFFFAOYSA-N 0.000 description 1
- 125000003668 acetyloxy group Chemical group [H]C([H])([H])C(=O)O[*] 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 125000004423 acyloxy group Chemical group 0.000 description 1
- IAJILQKETJEXLJ-QTBDOELSSA-N aldehydo-D-glucuronic acid Chemical compound O=C[C@H](O)[C@@H](O)[C@H](O)[C@H](O)C(O)=O IAJILQKETJEXLJ-QTBDOELSSA-N 0.000 description 1
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- 230000000259 anti-tumor effect Effects 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 125000005101 aryl methoxy carbonyl group Chemical group 0.000 description 1
- 238000011914 asymmetric synthesis Methods 0.000 description 1
- 239000002774 b raf kinase inhibitor Substances 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 125000000051 benzyloxy group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])O* 0.000 description 1
- 230000004579 body weight change Effects 0.000 description 1
- IXIBAKNTJSCKJM-BUBXBXGNSA-N bovine insulin Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@H]1CSSC[C@H]2C(=O)N[C@@H](C)C(=O)N[C@@H](CO)C(=O)N[C@H](C(=O)N[C@H](C(N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC=3C=CC(O)=CC=3)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=3C=CC(O)=CC=3)C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=3C=CC(O)=CC=3)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](C)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=3NC=NC=3)NC(=O)[C@H](CO)NC(=O)CNC1=O)C(=O)NCC(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(O)=O)C(=O)N[C@@H](CC(N)=O)C(O)=O)=O)CSSC[C@@H](C(N2)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C(C)C)NC(=O)[C@@H](NC(=O)CN)[C@@H](C)CC)C(C)C)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@@H](NC(=O)[C@@H](N)CC=1C=CC=CC=1)C(C)C)C1=CN=CN1 IXIBAKNTJSCKJM-BUBXBXGNSA-N 0.000 description 1
- 239000012888 bovine serum Substances 0.000 description 1
- 238000009395 breeding Methods 0.000 description 1
- 230000001488 breeding effect Effects 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 238000011095 buffer preparation Methods 0.000 description 1
- 239000007853 buffer solution Substances 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 244000309466 calf Species 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 230000024245 cell differentiation Effects 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 239000012069 chiral reagent Substances 0.000 description 1
- PJGJQVRXEUVAFT-UHFFFAOYSA-N chloroiodomethane Chemical compound ClCI PJGJQVRXEUVAFT-UHFFFAOYSA-N 0.000 description 1
- 238000011281 clinical therapy Methods 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 229910052805 deuterium Inorganic materials 0.000 description 1
- 230000029087 digestion Effects 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 238000001647 drug administration Methods 0.000 description 1
- 229940000406 drug candidate Drugs 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 239000003777 experimental drug Substances 0.000 description 1
- 239000012091 fetal bovine serum Substances 0.000 description 1
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 229940097043 glucuronic acid Drugs 0.000 description 1
- 125000001183 hydrocarbyl group Chemical group 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 230000031146 intracellular signal transduction Effects 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 238000011835 investigation Methods 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 238000006317 isomerization reaction Methods 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 239000012280 lithium aluminium hydride Substances 0.000 description 1
- YNESATAKKCNGOF-UHFFFAOYSA-N lithium bis(trimethylsilyl)amide Chemical compound [Li+].C[Si](C)(C)[N-][Si](C)(C)C YNESATAKKCNGOF-UHFFFAOYSA-N 0.000 description 1
- LZWQNOHZMQIFBX-UHFFFAOYSA-N lithium;2-methylpropan-2-olate Chemical compound [Li+].CC(C)(C)[O-] LZWQNOHZMQIFBX-UHFFFAOYSA-N 0.000 description 1
- 201000005296 lung carcinoma Diseases 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 230000036210 malignancy Effects 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- 108010082117 matrigel Proteins 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 125000005948 methanesulfonyloxy group Chemical group 0.000 description 1
- 125000001434 methanylylidene group Chemical group [H]C#[*] 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 1
- 239000002829 mitogen activated protein kinase inhibitor Substances 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- UEPXBTCUIIGYCY-UHFFFAOYSA-N n-[3-[2-(2-hydroxyethoxy)-6-morpholin-4-ylpyridin-4-yl]-4-methylphenyl]-2-(trifluoromethyl)pyridine-4-carboxamide Chemical compound C1=C(C=2C=C(N=C(OCCO)C=2)N2CCOCC2)C(C)=CC=C1NC(=O)C1=CC=NC(C(F)(F)F)=C1 UEPXBTCUIIGYCY-UHFFFAOYSA-N 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 239000013642 negative control Substances 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 238000010534 nucleophilic substitution reaction Methods 0.000 description 1
- 238000011580 nude mouse model Methods 0.000 description 1
- 238000011275 oncology therapy Methods 0.000 description 1
- 238000001543 one-way ANOVA Methods 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 230000003285 pharmacodynamic effect Effects 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 102000020233 phosphotransferase Human genes 0.000 description 1
- 238000007747 plating Methods 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 239000000047 product Substances 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- VVWRJUBEIPHGQF-MDZDMXLPSA-N propan-2-yl (ne)-n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)\N=N\C(=O)OC(C)C VVWRJUBEIPHGQF-MDZDMXLPSA-N 0.000 description 1
- VVWRJUBEIPHGQF-UHFFFAOYSA-N propan-2-yl n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)N=NC(=O)OC(C)C VVWRJUBEIPHGQF-UHFFFAOYSA-N 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 230000007420 reactivation Effects 0.000 description 1
- 230000008707 rearrangement Effects 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 229930195734 saturated hydrocarbon Natural products 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- 102000034285 signal transducing proteins Human genes 0.000 description 1
- 108091006024 signal transducing proteins Proteins 0.000 description 1
- 230000019491 signal transduction Effects 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- WRIKHQLVHPKCJU-UHFFFAOYSA-N sodium bis(trimethylsilyl)amide Chemical compound C[Si](C)(C)N([Na])[Si](C)(C)C WRIKHQLVHPKCJU-UHFFFAOYSA-N 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 235000009518 sodium iodide Nutrition 0.000 description 1
- GEHJYWRUCIMESM-UHFFFAOYSA-L sodium sulfite Chemical class [Na+].[Na+].[O-]S([O-])=O GEHJYWRUCIMESM-UHFFFAOYSA-L 0.000 description 1
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 1
- 235000019345 sodium thiosulphate Nutrition 0.000 description 1
- 239000007787 solid Substances 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 238000003419 tautomerization reaction Methods 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000001981 tert-butyldimethylsilyl group Chemical group [H]C([H])([H])[Si]([H])(C([H])([H])[H])[*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 1
- 125000004044 trifluoroacetyl group Chemical group FC(C(=O)*)(F)F 0.000 description 1
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 230000003442 weekly effect Effects 0.000 description 1
Images




Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/04—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F5/00—Compounds containing elements of Groups 3 or 13 of the Periodic Table
- C07F5/02—Boron compounds
- C07F5/025—Boronic and borinic acid compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6558—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system
- C07F9/65586—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system at least one of the hetero rings does not contain nitrogen as ring hetero atom
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Description
CN201911242225.7、出願日は2019年12月06日であり;
CN202010367751.2、出願日は2020年04月30日であり;
CN202010652149.3、出願日は2020年07月08日である。

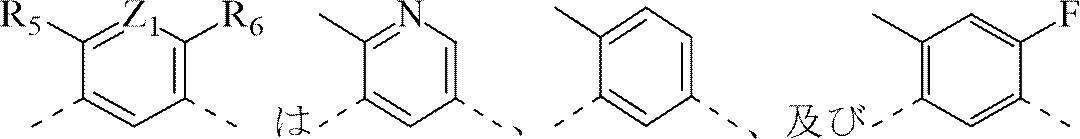
XとYはそれぞれ独立してCH、及びNから選択され;
Lは-O-、-S-、-S(=O)-、及び-S(=O)2-から選択され;
L1は-CH2-、及び単結合から選択され;
Z1とZ2はそれぞれ独立してCH、及びNから選択され;
R1とR2はそれぞれ独立してH、F、及びC1―3アルキルから選択され、前記C1-3アルキルは任意選択で1、2又は3個のRaで置換され;
R3は

R4はH、及びC1-3アルキルから選択され、前記C1-3アルキルは任意選択で1、2又は3個のRbで置換され;
R5はH、及びC1-3アルキルから選択され、前記C1-3アルキルは任意選択で1、2又は3個のRcで置換され;
R6はH、及びFから選択され;
R7はH、及びCNから選択され;
R8はH、及びCH3から選択され;
R9はH、F、及びCH3から選択され;
Raはそれぞれ独立してF、Cl、Br、Iから選択され;
Rbはそれぞれ独立してF、Cl、Br、I、及びCH3から選択され;
Rcはそれぞれ独立してF、Cl、Br、及びIから選択される。

XとYはそれぞれ独立してCH、及びNから選択され;
Lは-O-、-S-、-S(=O)-、及び-S(=O)2-から選択され;
Z1とZ2はそれぞれ独立してCH、及びNから選択され;
R1とR2はそれぞれ独立してH、F、及びC1-3アルキルから選択され、前記C1-3アルキルは任意選択で1、2又は3個のRaで置換され;
R3は

R4はH、及びC1-3アルキルから選択され、前記C1-3アルキルは任意選択で1、2又は3個のRbで置換され;
R5はH、及びC1-3アルキルから選択され、前記C1-3アルキルは任意選択で1、2又は3個のRcで置換され;
R6はH、及びFから選択され;
R7はH、及びCNから選択され;
R8はH、及びCH3から選択され;
R9はH、F、及びCH3から選択され;
Raはそれぞれ独立してF、Cl、Br、Iから選択され;
Rbはそれぞれ独立してF、Cl、Br、I、及びCH3から選択され;
Rcはそれぞれ独立してF、Cl、Br、及びIから選択される。

Lは-O-、-S-、-S(=O)-、及び-S(=O)2-から選択され;
Z1とZ2はそれぞれ独立してCH、及びNから選択され;
R1とR2はそれぞれ独立してH、及びC1-3アルキルから選択され、前記C1-3アルキルは任意選択で1、2又は3個のRaで置換され;
R3は

R4はH、及びC1-3アルキルから選択され、前記C1-3アルキルは任意選択で1、2又は3個のRbで置換され;
R5はH、及びC1-3アルキルから選択され、前記C1-3アルキルは任意選択で1、2又は3個のRcで置換され;
R6はH、及びFから選択され;
Raはそれぞれ独立してF、Cl、Br、Iから選択され;
Rbはそれぞれ独立してF、Cl、Br、I、及びCH3から選択され;
Rcはそれぞれ独立してF、Cl、Br、及びIから選択される。









ただし、R1、R2、R3、R5、R6、R7、R9、Z1、及びZ2は本発明に定義された通りである。

ただし、R1、R2、R3、及びR7は本発明に定義された通りである。




X1はハロゲン、-SO2Me、-OMs、OTf、OTs、

X2はハロゲン、OH、-SO2Me、-OMs、OTf、OTs、及びHから選択され;
R1、R2、R5、R6、R7、R9、X、Y、Z1、及びZ2は本発明に定義された通りである。


本発明の化合物は、良好な薬物様特性を有し、良好なRAF酵素阻害活性と様々な細胞抗増殖活性を有し、同時に良好な生体内有効性を有する。現在のBRAFV600E突然変異癌治療の薬剤耐性問題を解決し、RAS突然変異癌の効果的な治療を提供することが期待されている。
別途に説明しない限り、本明細書で用いられる以下の用語及び連語は以下の意味を含む。一つの特定の用語又は連語は、特別に定義されない場合、不確定又は不明瞭ではなく、普通の定義として理解されるべきである。本明細書で商品名が出た場合、相応の商品又はその活性成分を指す。

別途に説明しない限り、用語「異性体の過剰量」又は「エナンチオマーの過剰量」とは、二つの異性体又は二つのエナンチオマーの間の相対百分率の差の値である。例えば、その一方の異性体又はエナンチオマーの含有量が90%で、もう一方の異性体又はエナンチオマーの含有量が10%である場合、異性体又はエナンチオマーの過剰量(ee値)は80%である。






0℃で、1A-2(192.15mg、1.31mmol)のTHF(1mL)溶液に水素化ナトリウム(43.81mg、1.10mmol、純度:60%)を加え、25℃に昇温させて30分間攪拌した。0℃で、当該混合溶液に1A-1(300mg、1.10mmol)を加え、25℃に昇温させて2時間攪拌した。反応混合物に飽和塩化アンモニウム溶液(10mL)を加え、酢酸エチル(10mL×3)で抽出し、合わせた抽出溶液を濃縮し、カラムクロマトグラフィー(PE/EA=20/1、V/V)で分離して1A-3を得た。
1A-3(200mg、521.36μmol)、1A-4(232.96mg、573.50μmol)、Pd(dppf)Cl2(38.15mg、52.14μmol)、及び炭酸ナトリウム(110.52mg、1.04mmol)をジオキサン(2mL)と水(0.4mL)の混合溶媒に加えた。窒素ガス保護下で、反応混合物を100℃に加熱して2時間攪拌した。反応溶液を水(10mL)で希釈し、ジクロロメタン(10×2mL)で抽出し、合わせた有機相を無水硫酸ナトリウムで乾燥させた後濃縮し、分取シリカゲルプレート(PE/EA=5/1、V/V)で分離して、1A-5を得た。1H NMR (400MHz, DMSO-d6) δ 9.00 (d, J=5.0 Hz, 1H), 8.37 (s, 1H), 8.20 (d, J=4.8 Hz, 1H), 7.77 (dd, J=2.2, 8.2 Hz, 1H), 7.70 (d, J=2.2 Hz, 1H), 7.37 (d, J=8.4 Hz, 1H), 7.12 (d, J=1.0 Hz, 1H), 6.86 (d, J=1.0 Hz, 1H), 4.68 - 4.64 (m, 1H), 4.52 - 4.40 (m, 2H), 3.82 - 3.71 (m, 2H), 3.48 - 3.41 (m, 2H), 2.26 (s, 3H), 1.75 - 1.58 (m, 2H), 1.53 - 1.40 (m, 4H)。

1A-1(1.0g、3.65mmol)、1A-4(1.67g、4.11mmol)、Pd(dppf)Cl2(300mg、367.36μmol)、及び炭酸ナトリウム(774mg、7.30mmol)をジオキサン(10mL)と水(2.5mL)の混合溶媒に加えた。窒素ガス保護下で、反応混合物を70℃に加熱して4時間攪拌した。反応溶液を濾過し、酢酸エチル(10×3mL)で抽出し、合わせた有機相を無水硫酸ナトリウムで乾燥させた後濃縮し、シリカゲルカラムクロマトグラフィー(PE/EA=30/1~3/1、V/V)で分離して1A-6を得た。1HNMR (400MHz, CDCl3) δ 8.98-8.91 (d, J=5.0 Hz, 1H), 8.11 (s, 1H), 8.06 (s, 1H), 7.97-7.92 (d, J=5.0 Hz, 1H), 7.61 - 7.56 (m, 2H), 7.39-7.30 (d, J=9.0 Hz, 1H), 7.28 - 7.26 (m, 2H), 2.37 - 2.22 (m, 3H)。

1A-1(785mg、2.87mmol)、1A-7(1.11g、2.72mmol)、Pd(dppf)Cl2(210mg、287.0μmol)、及び炭酸ナトリウム(609mg、5.75mmol)をDME(10mL)と水(2.5mL)の混合溶媒に加えた。窒素ガス保護下で、反応混合物を90℃に加熱して2時間攪拌した。反応溶液を酢酸エチル(50mL)に加えた後濾過し、水(50×2mL)と飽和食塩水(80×1mL)で洗浄し、合わせた有機相を無水硫酸ナトリウムで乾燥させた後濃縮し、シリカゲルカラムクロマトグラフィー(PE/EA=20/1~3/1、V/V)で分離して1A-8を得た。1HNMR (400 MHz, CDCl3) δ 8.65 (d, J=2.51 Hz, 1 H), 8.26 (d, J=2.51 Hz, 1 H), 8.15 (s, 1 H), 8.10 (d, J=7.91 Hz, 1 H), 8.03 (s, 1 H), 7.87 (d, J=7.78 Hz, 1 H), 7.69 (t, J=7.78 Hz, 1 H), 7.31 (s, 2 H), 2.53 (s, 3 H)。

水素化ナトリウム(163.18mg、4.08mmol、60%)、11-2A-1(897.89mg、4.08mmol)の混合物にDMSO(4mL)を加えた。窒素ガス保護下で、反応混合物を20℃で0.5時間攪拌した。反応溶液に11-2(200mg、1.02mmol)のDMSO(4mL)溶液を加え、反応混合物を80℃に加熱して2.5時間攪拌した。反応混合物に水(2mL)と酢酸エチル(40mL)を加え、水(20×2mL)で洗浄し、有機相を無水硫酸ナトリウムで乾燥させた後濃縮し、分取シリカゲルプレート(PE/EA=5/1、V/V)で分離して化合物2-1Aを得た。1HNMR (400 MHz, CDCl3) δ 7.65 - 7.47 (m, 1H), 7.21 - 7.02 (m, 2H), 4.11 - 3.91 (m, 2H), 3.74 - 3.57 (m, 1H), 3.56 - 3.37 (m, 1H), 2.55 - 2.42 (m, 1H), 2.20 - 2.05 (m, 1H), 1.85 - 1.71 (m, 1H), 1.39 - 1.28 (m, 1H), 1.16 - 1.04 (m, 1H)。
2-1A-1(395.45mg、1.56mmol)、[Ir(COD)OMe]2(23.71mg、35.77μmol)とtmphen(16.91mg、71.54μmol)、及び酢酸カリウム(11.5g、117.18mmol)をMTBE(8mL)に加えた。反応混合物を窒素ガス保護下で、80℃に加熱して5分間攪拌した。次に、2-1A(300mg、1.43mmol)のMTBE(2mL)溶液を加え、80℃で16時間攪拌した。反応溶液を濾過し、濃縮して化合物2-1Bの粗成生物を得た。MS (ESI): m/z 254.1 [M+H-82]+。
2-1B(480mg、1.43mmol)、2-1B-1(320mg、1.72mmol)、Pd(dppf)Cl2(117mg、143μmol)、及び炭酸ナトリウム(454.74mg、4.29mmol)をDME(10mL)と水(2mL)の混合溶媒に加えた。窒素ガス保護下で、反応混合物を80℃に加熱して2時間攪拌した。反応溶液に酢酸エチル(20mL)を加えた後濾過し、濾液を水(20×3mL)で洗浄し、有機相を無水硫酸ナトリウムで乾燥させた後濃縮し、シリカゲルカラムクロマトグラフィー(PE/EA=20/1~10/1)で分離して化合物2-1Cを得た。1H NMR (400 MHz, CDCl3) δ 7.09 - 7.06 (m, 2H), 7.05 (d, J = 1.0 Hz, 1H), 6.70 - 6.64 (m, 1H), 6.53 (d, J = 2.6 Hz, 1H), 4.07 - 4.00 (m, 1H), 3.99 - 3.93 (m, 1H), 3.70 - 3.59 (m, 3H), 3.52 - 3.42 (m, 1H), 2.56 - 2.47 (m, 1H), 2.16 - 2.14 (m, 3H), 2.14 (br s, 1H), 1.75 - 1.60 (m, 1H), 1.40 - 1.33 (m, 1H), 1.11 (dd, J = 4.2, 6.4 Hz, 1H)。
2-1C(100mg、317.65μmol)、2-1C-1(60.71mg、317.65μmol)のDMF(3mL)に、HATU(181.17mg、476.48μmol)とDIPEA(123.16mg、952.96μmol)を加え、20℃で2時間攪拌した。反応混合物を酢酸エチル(20mL)で希釈し、水(10×3mL)で洗浄し、有機相を合わせた後無水硫酸ナトリウムで乾燥させ、濃縮した後、分取シリカゲルプレート(PE/EA=2/1、V/V)で精製して化合物2-1を得た。1HNMR (400 MHz, CDCl3) δ 8.94 (d, J = 5.0 Hz, 1H), 8.16 - 8.06 (m, 1H), 8.00 - 7.85 (m, 2H), 7.61 - 7.56 (m, 1H), 7.55 - 7.51 (m, 1H), 7.36 - 7.31 (m, 1H), 7.11 - 7.06 (m, 2H), 4.07 - 4.01 (m, 1H), 3.99 - 3.93 (m, 1H), 3.68 - 3.59 (m, 1H), 3.53 - 3.43 (m, 1H), 2.57 - 2.47 (m, 1H), 2.30 - 2.25 (m, 3H), 2.18 - 2.08 (m, 1H), 1.88 - 1.79 (m, 1H), 1.41 - 1.35 (m, 1H), 1.20-1.10 (dd, J = 4.2, 6.4 Hz, 1H)。MS (ESI) m/z: 488.1 [M+H] +。

14-2A-1(10.5g、54.97mmol)のDMF(100mL)溶液にヒドロキシルアミン塩酸塩(4.77g、68.71mmol)とトリエチルアミン(16.69g、164.90mmol)を加え、20℃で1時間攪拌した。反応溶液にT3P(34.98g、54.97mmol、50%のDMF溶液)を加え、20℃で4時間攪拌した。反応混合物に飽和炭酸水素ナトリウム溶液(200mL)を加え、EtOAc(50mL×3)で抽出し、合わせた抽出溶液を無水硫酸ナトリウムで乾燥させた後濃縮して、14-2A-2の粗成生物を得た。1H NMR (400MHz, CDCl3) δ 9.24 - 8.56 (m, 1H), 8.08 (s, 1H), 4.35-4.28 (t, J=2.2 Hz, 2H), 3.85-3.76 (t, J=5.6 Hz, 2H), 2.70 - 2.62 (m, 2H)。
14-2A-2(9.5g、46.11mmol)のDCM(100mL)溶液に塩化チオニル(15.67g、131.74mmol)を加え、20℃で1時間攪拌した。0℃で、反応混合物をゆっくりと飽和炭酸水素ナトリウム溶液(100mL)に滴下し、DCM(100mL×2)で抽出し、合わせた抽出溶液を無水硫酸ナトリウムで乾燥させた後濃縮して、14-2Aの粗成生物を得た。1HNMR (400MHz, CDCl3) δ 4.30-4.20 (t, J=2.6 Hz, 2H), 3.92-3.85 (t, J=5.4 Hz, 2H), 2.75-2.64 (tt, J=2.6, 5.4 Hz, 2H)。

1A-5(100mg、186.59μmol)、1-1A(77mg、377.38μmol)、Pd(dppf)Cl2(14mg、19.13μmol)、及び炭酸ナトリウム(40mg、377.40μmol)をDME(2mL)と水(0.2mL)の混合溶媒に加えた。窒素ガス保護下で、反応混合物を100℃に加熱して16時間攪拌した。反応溶液を濾過し、濾液を濃縮し、分取シリカゲルプレート(PE/EA=5/1、V/V)で分離して粗成生物を得、更にカラムクロマトグラフィー(シリカゲル)で分離(HCOOH-MeCN-H2O)して化合物1-1を得た。1H NMR (400 MHz, CDCl3) δ 8.99-8.90 (d, J = 5.0 Hz, 1H), 8.11 (s, 1H), 7.96 - 7.89 (m, 2H), 7.67-7.58 (dd, J = 2.0, 8.6 Hz, 1H), 7.49-7.40 (d, J = 1.8 Hz, 1H), 7.36-7.29 (d, J = 8.4 Hz, 1H), 6.76 (s, 1H), 6.55-6.49 (d, J = 0.8 Hz, 1H), 4.74-4.68 (t, J = 3.6 Hz, 1H), 4.60 - 4.45 (m, 2H), 4.09 - 4.02 (m, 2H), 3.98 - 3.88 (m, 2H), 3.89-3.76 (ddd, J = 4.0, 6.4, 11.2 Hz, 1H), 3.66 - 3.58 (m, 1H), 3.57 - 3.44 (m, 2H), 2.58-2.45 (td, J = 5.2, 13.8 Hz, 1H), 2.27 (s, 3H), 2.14 - 2.07 (m, 1H), 1.89 - 1.81 (m, 2H), 1.77 - 1.71 (m, 1H), 1.68 - 1.61 (m, 2H), 1.45-1.36 (dd, J = 4.0, 9.2 Hz, 1H), 1.10-1.02 (dd, J = 4.0, 6.4 Hz, 1H), 0.99 - 0.73 (m, 2H)。
1-1(54mg、90.36μmol)のジクロロメタン(2mL)にトリフルオロ酢酸(616mg、5.4mmol)を加え、反応溶液を25℃で0.5時間攪拌した。反応溶液をジクロロメタン(20mL)で希釈し、飽和炭酸水素ナトリウム水溶液で中性に調節した。有機相を飽和食塩水(15×1mL)で洗浄し、無水硫酸ナトリウムで乾燥させた後濃縮し、分取シリカゲルプレート(PE/EA=1/1、V/V)で分離して化合物1を得た。 1H NMR (400 MHz, CDCl3) δ 8.99-8.90 (d, J = 5.0 Hz, 1H), 8.11 (s, 1H), 7.96 - 7.85 (m, 2H), 7.67-7.59 (dd, J = 1.8, 8.0 Hz, 1H), 7.51 (s, 1H), 7.37-7.28 (d, J = 8.6 Hz, 1H), 6.84-6.78 (d, J = 1.0 Hz, 1H), 6.57-6.52 (d, J = 1.0 Hz, 1H), 4.53 - 4.46 (m, 2H), 4.08 - 4.03 (m, 1H), 4.00 - 3.94 (m, 3H), 3.69 - 3.58 (m, 1H), 3.54-3.36 (ddd, J = 5.2, 8.6, 11.6 Hz, 1H), 2.53-2.46 (td, J = 5.0, 14.0 Hz, 1H), 2.28 (s, 3H), 2.18-2.09 (ddd, J = 5.4, 8.4, 13.8 Hz, 1H), 1.79 - 1.72 (m, 1H), 1.39-1.28 (dd, J = 4.2, 8.8 Hz, 1H), 1.11 - 1.03 (m, 1H)。MS (ESI) m/z: 514.2 [M+H] +。
化合物1(25mg、48.68μmol)、及びイミダゾール(8mg、117.51μmol)のジクロロメタン(3mL)溶液に、TBSCl(10mg、48.68μmol)を加えた。反応混合物を25℃で12時間攪拌した。反応混合物をジクロロメタン(20mL)で希釈し、水(30×1mL)と飽和食塩水(20×1mL)で洗浄し、有機相を無水硫酸ナトリウムで乾燥させ、濾液を濃縮した後、分取シリカゲルプレート(PE/EA=2/1、V/V)で分離して化合物1-2を得た。MS (ESI) m/z: 628.3 [M+H] +。
化合物1-2をSFCキラル分離(キラルカラム:REGIS(s,s)WHELK-O1 (250mm*50mm,10μm),移動相A:イソプロパノール(0.05%のDIEAを含有);移動相B:二酸化炭素)して化合物1-2A(保持時間:3.846min)と化合物1-2B(保持時間:3.966min)を得た。化合物1-2A:MS (ESI) m/z: 628.3 [M+H] +,99.4% (ee%);化合物1-2B:MS (ESI) m/z: 628.3 [M+H]+ ,94.3% (ee%)。
1-2A(15mg、23.89μmol)のTHF(2mL)にTBAF(1M、0.048mL)を加え、反応溶液を20℃で0.5時間攪拌した。反応溶液濾液をそれぞれ酢酸エチル(30mL)で希釈し、水(20×2mL)で洗浄し、有機相を無水硫酸ナトリウムで乾燥させた後、濃縮し、分取シリカゲルプレート(PE/EA=1/2、V/V)で分離して化合物1Aを得た。 1H NMR (400 MHz, CDCl3) δ 8.96-8.90 (d, J = 5.0 Hz, 1H), 8.15 - 8.11 (m, 1H), 8.08 (s, 1H), 7.98 - 7.94 (m, 1H), 7.65 - 7.58 (m, 1H), 7.55-7.48 (d, J = 2.0 Hz, 1H), 7.36-7.25 (d, J = 8.2 Hz, 1H), 6.80 (s, 1H), 6.58-6.50 (d, J = 1.0 Hz, 1H), 4.54 - 4.48 (m, 2H), 4.09 - 4.01 (m, 1H), 4.00 - 3.94 (m, 3H), 3.67 - 3.62 (m, 1H), 3.52-3.40(ddd, J = 5.0, 8.4, 11.6 Hz, 1H), 2.50-2.40 (td, J = 4.8, 14.2 Hz, 1H), 2.28 (s, 3H), 2.20-2.10 (ddd, J = 5.6, 8.4, 13.8 Hz, 1H), 1.79 - 1.70 (m, 1H), 1.37 - 1.34 (m, 1H), 1.10-1.02 (dd, J = 4.2, 6.0 Hz, 1H)。MS (ESI) m/z:514.2 [M+H] +。
1-2B(11mg、17.52μmol)のTHF(2mL)にTBAF(1M,0.035mL)を加え、反応溶液を20℃で0.5時間攪拌した。反応溶液濾液を、それぞれ酢酸エチル(30mL)で希釈し、水(20×2mL)で洗浄し、有機相を無水硫酸ナトリウムで乾燥させた後、濃縮し、分取シリカゲルプレート(PE/EA=1/2、V/V)で分離して化合物1Bを得た。 1H NMR (400 MHz, CDCl3) δ 8.96-8.90 (d, J = 5.0 Hz, 1H), 8.15 - 8.11 (m, 1H), 7.97 - 7.89 (m, 2H), 7.64-7.55 (dd, J = 1.6, 7.0 Hz, 1H), 7.54-7.48 (br d, J = 0.8 Hz, 1H), 7.38-7.26(d, J = 8.6 Hz, 1H), 6.84-6.78 (d, J = 0.8 Hz, 1H), 6.58-6.50 (d, J = 1.2 Hz, 1H), 4.54 - 4.47 (m, 2H), 4.07 - 4.02 (m, 1H), 4.00 - 3.93 (m, 3H), 3.68 - 3.58 (m, 1H), 3.53 - 3.44 (m, 1H), 2.50-2.40 (td, J = 5.3, 13.6 Hz, 1H), 2.28 (s, 3H), 2.16-2.06 (ddd, J = 5.6, 8.7, 14.1 Hz, 1H), 1.78 - 1.71 (m, 1H), 1.37 - 1.34 (m, 1H), 1.12 - 1.05 (m, 1H)。MS (ESI) m/z: 514.2 [M+H] +。

1A-6(1.0g、2.35mmol)、1-1A(480mg、2.35mmol)、Pd(dppf)Cl2(250mg、341.67μmol)、及び炭酸セシウム(1.6g、4.90mmol)をジオキサン(30mL)と水(5mL)の混合溶媒に加えた。窒素ガス保護下で、反応混合物を110℃に加熱して16時間攪拌した。反応溶液を酢酸エチル(30mL)で希釈した後濾過し、濾液を水(20×2mL)で洗浄し、有機相を無水硫酸ナトリウムで乾燥させた後濃縮し、シリカゲルカラムクロマトグラフィー(PE/EA=20/1~5/1、V/V)で分離して、粗成生物を得、更にカラムクロマトグラフィー(シリカゲル)で分離(HCOOH-MeCN-H2O)して、化合物2-1を得た。1H NMR (400 MHz, CDCl3) δ 8.99-8.92 (d, J = 5.0 Hz, 1H), 8.10 (s, 1H), 7.98-7.91 (d, J = 4.4 Hz, 1H), 7.89 (s, 1H), 7.60-7.52 (br d, J = 8.2 Hz, 1H), 7.54 (s, 1H), 7.38-7.30 (d, J = 8.2 Hz, 1H), 7.14-7.06 (d, J = 3.8 Hz, 2H), 4.09 - 4.01 (m, 1H), 3.99 - 3.92 (m, 1H), 3.69-3.58 (td, J = 5.4, 11.4 Hz, 1H), 3.52-3.45 (ddd, J = 5.4, 8.6, 11.4 Hz, 1H), 2.56-2.48 (td, J = 5.0, 13.8 Hz, 1H), 2.28 (s, 3H), 2.22-2.10 (ddd, J = 5.4, 8.6, 14.0 Hz, 1H), 1.88 - 1.79 (m, 1H), 1.45-1.36 (dd, J = 4.2, 9.2 Hz, 1H), 1.18-1.10 (dd, J = 4.2, 6.4 Hz, 1H), 0.87 - 0.82 (m, 1H)。
2-1(200mg、409.92μmol)、及び2-1A-2(200mg、1.06mmol)のトルエン(5mL)溶液に、Pd2dba3(37.54mg、40.99μmol)、Ruphos(38.26mg、81.98μmol)と炭酸セシウム(248.99mg、764.19μmol)を加えた。反応混合物を窒素ガス保護下で、120℃に加熱して4時間攪拌した。反応混合物を濾過し、濾液を濃縮した後、分取シリカゲルプレート(PE/EA=2/1、V/V)で分離して化合物2-2を得た。MS (ESI) m/z: 641.3 [M+H] +。
2-2(80mg、124.84μmol)のTHF(5mL)にTBAF(1M、0.25mL)を加え、反応溶液を20℃で0.5時間攪拌した。反応溶液を濾過し、濾液をそれぞれクエン酸溶液(1M、20×2mL)と飽和食塩水(15×1mL)で洗浄し、有機相を濃縮した後、分取シリカゲルプレート(PE/EA=1/3、V/V)で分離して化合物2を得た。1H NMR (400 MHz, DMSO-d6) δ 10.67 (s, 1H), 9.07-9.96 (d, J = 5.0 Hz, 1H), 8.36 (s, 1H), 8.24-8.16 (d, J = 4.4 Hz, 1H), 7.78-7.70 (dd, J = 2.2, 8.4 Hz, 1H), 7.66-7.58 (d, J = 2.2 Hz, 1H), 7.36-7.25 (d, J = 8.4 Hz, 1H), 6.44 (s, 1H), 6.29 (s, 1H), 4.68 - 4.62 (m, 1H), 3.98-3.88 (dd, J = 4.8, 11.4 Hz, 1H), 3.85-3.74 (d, J = 11.4 Hz, 1H), 3.60 - 3.53 (m, 3H), 3.52 - 3.45 (m, 1H), 3.40 - 3.36 (m, 1H), 3.30 (s, 2H), 3.04 (s, 3H), 2.23 (s, 3H), 1.98 - 1.89 (m, 1H), 1.76 - 1.68 (m, 1H), 1.30-1.21 (dd, J = 3.2, 9.2 Hz, 1H), 0.99-0.90 (dd, J = 3.6, 5.8 Hz, 1H)。MS (ESI) m/z: 527.2 [M+H] +。

2-1(160mg、329.27μmol)、及び3-1A(150mg、855.46μmol)のトルエン(5mL)溶液に、Pd2dba3(30.15mg、32.93μmol)、Ruphos(30.73mg、65.85μmol)と炭酸セシウム(200mg、613.84μmol)を加えた。反応混合物を窒素ガス保護下で、120℃に加熱して4時間攪拌した。反応混合物を濾過し、濾液を濃縮した後分取シリカゲルプレート(PE/EA=2/1、V/V)で分離して化合物3-1を得た。MS (ESI) m/z: 627.2 [M+H] +。
3-1(50mg、79.77μmol)のTHF(5mL)にTBAF(1M、0.16mL)を加え、反応溶液を20℃で0.5時間攪拌した。反応溶液を濾過し、濾液をそれぞれクエン酸溶液(1M、15×2mL)と飽和食塩水(15×1mL)で洗浄し、有機相を濃縮した後、分取シリカゲルプレート(PE/EA=1/5、V/V)で分離して化合物3を得た。 1H NMR (400 MHz, DMSO-d6) δ 10.66 (s, 1H), 9.05-9.95 (d, J = 5.0 Hz, 1H), 8.36 (s, 1H), 8.24-8.16 (d, J = 4.8 Hz, 1H), 7.80-7.68 (dd, J = 2.2, 8.4 Hz, 1H), 7.65-7.59 (d, J = 2.2 Hz, 1H), 7.36-7.27 (d, J = 8.4 Hz, 1H), 6.52-6.46 (t, J = 5.6 Hz, 1H), 6.38 (s, 1H), 6.25-6.18 (d, J = 0.6 Hz, 1H), 4.73-4.65 (t, J = 5.4 Hz, 1H), 3.98-3.90 (dd, J = 5.0, 11.4 Hz, 1H), 3.86-3.74 (d, J = 11.4 Hz, 1H), 3.58-3.50 (q, J = 5.8 Hz, 2H), 3.50 - 3.43 (m, 1H), 3.40 - 3.36 (m, 1H), 3.30 (s, 2H), 2.47 - 2.42 (m, 1H), 2.22 (s, 3H), 1.99-1.86 (ddd, J = 5.0, 8.2, 13.6 Hz, 1H), 1.74 - 1.67 (m, 1H), 1.30-1.21 (dd, J = 3.2, 9.2 Hz, 1H), 0.99-0.90 (dd, J = 3.6, 6.2 Hz, 1H)。MS (ESI) m/z: 513.2 [M+H] +。

1A-8(460mg、1.08mmol)、1-1A(405mg、1.19mmol)、Pd(dppf)Cl2(79mg、107.97μmol)、及び炭酸セシウム(706mg、2.17mmol)をDME(8mL)と水(2mL)の混合溶媒に加えた。窒素ガス保護下で、反応混合物を100℃に加熱して24時間攪拌した。反応溶液を酢酸エチル(20mL)で希釈した後濾過し、濾液を水(20×2mL)で洗浄し、有機相を無水硫酸ナトリウムで乾燥させた後、濃縮し、シリカゲルカラムクロマトグラフィー(PE/EA=5/1~2/1、V/V)で分離して化合物4-1を得た。1HNMR (400 MHz, CDCl3) δ 8.65 (d, J=2.6 Hz, 1 H), 8.20 (d, J=2.6 Hz, 1 H), 8.16 (s, 1 H), 8.10 (d, J=8.2 Hz, 1 H), 7.87 (d, J=7.6 Hz, 1 H), 7.94 (s, 1 H), 7.69 (s, 1 H), 7.12 (d, J=2.0 Hz, 2 H), 4.08-3.95 (m, 2 H), 3.69 - 3.62 (m, 1 H), 3.49 (ddd, J=11.6, 8. 8, 5.2 Hz, 1 H), 2.51 (s, 3 H), 2.21 - 2.12 (m, 1 H), 1.90 - 1.82 (m, 1 H), 1.40 (dd, J=9.2, 4.0 Hz, 1 H), 1.15 (dd, J=4.4, 2.0 Hz, 1 H), 0.92 (dd, J=12.6, 5.6 Hz, 1 H)。
4-1(280mg、573.89μmol)、及び1A-2(126mg、861.93μmol)のトルエン(5mL)溶液に、Pd2dba3(53mg、57.88μmol)、Brettphos(31mg、57.75μmol)と炭酸セシウム(374mg、1.15mmol)を加えた。反応溶液を酢酸エチル(10mL)で希釈した後濾過し、濾液を水(20×2mL)で洗浄し、有機相を無水硫酸ナトリウムで乾燥させた後濃縮し、シリカゲルカラムクロマトグラフィー(PE/EA=40/1~5/1、V/V)で分離して化合物4-2を得た。1HNMR (400 MHz, CDCl3) δ, 8.66 (d, J=2.6 Hz, 1 H), 8.16 (s, 2 H), 8.10 (d, J=2.6 Hz, 1 H), 7.91 (s, 1 H), 7.86 (br d, J=7.6 Hz, 1 H), 7.66 - 7.70 (m, 1 H), 6.79 (d, J=1.0 Hz, 1 H), 6.55 (d, J=1.0 Hz, 1 H), 4.72 (d, J=3.8 Hz, 1 H), 4.60- 4.54 (m, 2 H), 4.04 - 4.09 (m, 2 H), 3.93 - 3.97 (m, 2 H), 3.82 (s, 1 H), 3.76 (s, 1 H), 3.58 - 3.52 (m, 2 H), 2.80 (br d, J=6.4 Hz, 1 H), 2.51 (s, 3 H), 2.14- 2.08 (m, 1 H), 1.90 - 1.80 (m, 4 H), 1.80 - 1.74 (m, 2 H), 1.62 (br d, J=4.2 Hz, 1 H), 1.39 (d, J=3.8 Hz, 1 H), 1.07 (dd, J=6.2, 4.0 Hz, 1 H)。
4-2(220mg、638.13μmol)のDMF(2mL)にHCl(2M、2.2mL)を加え、反応溶液を15℃で0.5時間攪拌した。反応溶液に飽和NaHCO3溶液を加えてpH=8に調節し、更に水(20mL)で希釈した後酢酸エチル(20×2mL)で抽出し、有機相を無水硫酸ナトリウムで乾燥させた後濃縮し、分取シリカゲルプレート(DCM/MeOH=20/1、V/V)で分離して化合物4を得た。1H NMR (400 MHz, MeOD) δ 8.88-8.80 (d, J=2.4 Hz, 1 H), 8.30 (s, 1 H), 8.26-8.19 (d, J=7.6 Hz, 1 H), 8.15-8.09 (d, J=2.4 Hz, 1 H), 7.99-7.91 (d, J=7.4 Hz, 1 H), 7.72 - 7.78 (m, 1 H), 6.96-6.89 (d, J=1.0 Hz, 1 H), 6.65-6.60 (d, J=1.0 Hz, 1 H), 4.40 - 4.44 (m, 2 H), 4.10-4.02 (dd, J=11.4, 4.6 Hz, 1 H), 3.97-3.90 (d, J=11.4 Hz, 1 H), 3.91 - 3.88 (m, 2 H), 3.68 - 3.60 (m, 1 H), 3.54 - 3.46 (m, 2 H), 2.60-2.51 (br d, J=14.0 Hz, 1 H), 2.47 (s, 3 H), 2.10 (s, 1 H), 1.88 - 1.80 (m, 1 H), 1.46-1.40 (dd, J=9.0, 4.2 Hz, 1 H), 1.34-1.28 (br s, 1 H), 1.10-1.01 (dd, J=6.4, 4.0 Hz, 1 H)。MS (ESI) m/z: 514.2 [M+H] +。

1A-3(2g、5.21mmol)、5-1A(1.51g、5.74mmol)、Pd(dppf)Cl2(190.74mg、260.68μmol)、及び炭酸ナトリウム(1.38g、13.03mmol)をジオキサン(20mL)と水(4mL)の混合溶媒に加えた。窒素ガス保護下で、反応混合物を100℃に加熱して1.5時間攪拌した。反応溶液を酢酸エチル(30mL)で希釈した後濾過し、濾液を水(20×3mL)で洗浄し、有機相を無水硫酸ナトリウムで乾燥させた後濃縮し、シリカゲルカラムクロマトグラフィー(PE/EA=100/1~50/1、V/V)で分離して化合物5-1を得た。1HNMR (400 MHz, CDCl3) δ 8.17 (dd, J = 2.6 8.4 Hz, 1H), 8.07 (d, J = 2.4 Hz, 1H), 7.46 (d, J = 8.4 Hz, 1H), 6.88 (d, J = 1.2 Hz, 1H), 6.67 (d, J = 1.2 Hz, 1H), 4.74 - 4.69 (m, 1H), 4.57 (ddd, J = 3.4, 6.0, 9.2 Hz, 2H), 4.08 (ddd, J = 3.4, 5.8, 11.4 Hz, 1H), 3.96 - 3.78 (m, 2H), 3.60 - 3.50 (m, 1H), 2.38 (s, 3H), 1.94 - 1.80 (m, 1H), 1.79 - 1.70 (m, 1H), 1.69 - 1.53 (m, 4H)。
5-1(200mg、509.12μmol)、1-1A(190mg、558.72μmol)、Pd(dppf)Cl2(38mg、51.93μmol)、及び炭酸セシウム(333mg、1.02mmol)をDME(4mL)と水(1mL)の混合溶媒に加えた。窒素ガス保護下で、反応混合物を100℃に加熱して24時間攪拌した。反応溶液を酢酸エチル(10mL)で希釈した後濾過し、濾液を水(10×2mL)で洗浄し、有機相を無水硫酸ナトリウムで乾燥させた後濃縮し、薄層シリカゲルプレート(PE/EA=3/1、V/V)で分離して化合物5-2を得た。MS (ESI) m/z: 455.2 [M+H] +。
5-2(120mg、264.02μmol)のメタノール(4mL)溶液に、Pd/C(10%、15mg)を加え、水素ガス(15psi)雰囲気下で、25℃で1時間攪拌した。反応溶液を濾過した後、濃縮して化合物5-3の粗成生物を得た。MS (ESI): m/z 447.2 [M+Na+H]+。
5-2A(60mg、346.59μmol)、5-3(120mg、282.66μmol)のDCM(5mL)に、HATU(120mg、315.59μmol)とDIPEA(92mg、711.83μmol)を加え、20℃で16時間攪拌した。反応混合物を水(20mL)で希釈し、DCM(20×2mL)で抽出し、有機相を合わせた後無水硫酸ナトリウムで乾燥させ、濃縮した後、分取シリカゲルプレート(PE/EA=2/1、V/V)で精製して化合物5-4を得た。1HNMR (400 MHz, CDCl3) δ, 8.89-8.80 (d, J=5.0 Hz, 1 H), 8.04 (s, 1 H), 7.96 (s, 1 H), 7.82-7.76 (br d, J=5.4 Hz, 1 H), 7.59 - 7.64 (m, 1 H), 7.50-7.42 (d, J=1.8 Hz, 1 H), 7.38-7.29 (d, J=8.2 Hz, 1 H), 6.58 - 6.88 (m, 2 H), 6.56-6.50 (d, J=0.8 Hz, 1 H), 4.71 (t, J=3.6 Hz, 1 H), 4.45 - 4.58 (m, 2 H), 4.01 - 4.15 (m, 2 H), 3.88 - 3.99 (m, 2 H), 3.82 (ddd, J=11.0, 6.8, 3.8 Hz, 1 H), 3.58 - 3.67 (m, 1 H), 3.43 - 3.56 (m, 2 H), 2.55 (dt, J=13.8, 5.2 Hz, 1 H), 2.03 - 2.14 (m, 1 H), 2.27 (s, 3 H), 1.79 - 1.90 (m, 2 H), 1.70 - 1.77 (m, 1 H), 1.60 - 1.69 (m, 2 H), 1.53 (br d, J=9.2 Hz, 2 H), 1.39 (dd, J=9.0, 3.6 Hz, 1 H), 1.05 (dd, J=6.2, 4.0 Hz, 1 H)。
5-4(100mg、172.52μmol)のDMF(2mL)にHCl(4M、2mL)を加え、反応溶液を15℃で0.5時間攪拌した。反応溶液に飽和NaHCO3溶液を加えてpH=8に調節し、更に水(20mL)で希釈した後酢酸エチル(20×2mL)で抽出し、有機相を無水硫酸ナトリウムで乾燥させた後濃縮して化合物5を得た。1HNMR (400 MHz, MeOD) δ 8.89-8.80 (d, J=5.0 Hz, 1 H), 8.18 (s, 1 H), 8.06-7.95 (d, J=4.6 Hz, 1 H), 7.69-7.60 (dd, J=8.4, 2.2 Hz, 1 H), 7.65-7.58 (d, J=2.2 Hz, 1 H), 7.38-7.30 (d, J=8.4 Hz, 1 H), 6.66 - 7.00 (m, 2 H), 6.58-6.50 (d, J=1.0 Hz, 1 H), 4.36 - 4.44 (m, 2 H), 4.08-4.01 (dd, J=11.4, 4.6 Hz, 1 H), 3.96-3.91 (d, J=1.2 Hz, 1 H), 3.86 - 3.91 (m, 2 H), 3.57 - 3.65 (m, 1 H), 3.52-3.46 (ddd, J=11.60, 8.4, 5.2 Hz, 2 H), 2.26 (s, 3 H), 2.51 - 2.62 (m, 1 H), 2.03 - 2.15 (m, 1 H), 1.76 - 1.90 (m, 1 H), 1.45-1.38 (dd, J=9.2, 3.8 Hz, 1 H), 1.10-1.01 (dd, J=6.2, 4.0 Hz, 1 H)。MS (ESI) m/z: 496.1 [M+H] +。

6-1(250mg、1.34mmol)、6-1A(274mg、1.47mmol)のDCM(5mL)に、HATU(610mg、1.60mmol)とDIPEA(208mg、1.61mmol)を加え、20℃で2時間攪拌した。反応混合物を水(20mL)で希釈し、DCM(15×2mL)で抽出し、有機相を合わせた後無水硫酸ナトリウムで乾燥させ、濃縮した後、分取シリカゲルプレート(PE/EA=2/1、V/V)で精製して化合物6-2を得た。1HNMR (400 MHz, CDCl3) δ 8.83 (d, J = 5.0 Hz, 1H), 8.01 (d, J = 0.8 Hz, 1H), 7.91 (d, J = 2.0 Hz, 1H), 7.87 - 7.79 (m, 2H), 7.49 (dd, J = 2.0, 8.2 Hz, 1H), 7.25 (d, J = 8.2 Hz, 1H), 2.40 (s, 3H), 2.07 (t, J = 18.8 Hz, 3H)。
6-2(370mg、1.04mmol)、2-1A-1(317mg、1.25mmol)とPd(dppf)Cl2(39mg、53.30μmol)、及び酢酸カリウム(205mg、2.09mmol)をDME(6mL)に加えた。反応混合物を窒素ガス保護下で、90℃に加熱して12時間攪拌した。反応混合物を酢酸エチル(20mL)で希釈し、有機相を水(20×1mL)と飽和食塩水(20×1mL)で洗浄し、濃縮した後化合物6-3を得た。MS (ESI): m/z 403.2 [M+H]+。
6-3(600mg、1.49mmol)、1-1A(450mg、1.64mmol)とPd(dppf)Cl2(55mg、75.17μmol)、及び炭酸ナトリウム(317mg、2.99mmol)をDME(8mL)とH2O(2mL)に加えた。反応混合物を窒素ガス保護下で、85℃に加熱して2時間攪拌した。反応混合物を酢酸エチル(50mL)で希釈し、有機相を水(30×1mL)と飽和食塩水(30×1mL)で洗浄し、濃縮した後化合物6-4を得た。1HNMR (400 MHz, CDCl3) δ 8.86-8.81 (d, J = 5.0 Hz, 1H), 8.13 - 7.99 (m, 2H), 7.87-7.79 (d, J = 5.0 Hz, 1H), 7.65 - 7.42 (m, 3H), 7.41-7.38 (d, J = 8.2 Hz, 1H), 2.29 (s, 3H), 2.07 - 1.98 (m, 3H)。
6-4(360mg、852.57μmol)、1-1A(319mg、938.06μmol)、Pd(dppf)Cl2(63mg、86.10μmol)、及び炭酸セシウム(556mg、1.71mmol)をDME(6mL)と水(1mL)の混合溶媒に加えた。窒素ガス保護下で、反応混合物を100℃に加熱して14時間攪拌した。反応溶液を酢酸エチル(50mL)で希釈した後濾過し、濾液を水(30×1mL)で洗浄し、有機相を無水硫酸ナトリウムで乾燥させた後濃縮し、薄層シリカゲルプレート(PE/EA=1/1、V/V)で分離して化合物6-5を得た。1H NMR (400 MHz, CDCl3) δ 8.84 (d, J = 5.0 Hz, 1H), 8.03 (s, 1H), 7.92 (s, 1H), 7.83 (d, J = 4.4 Hz, 1H), 7.62 - 7.53 (m, 2H), 7.33 (d, J = 8.2 Hz, 1H), 7.09 (d, J = 3.6 Hz, 2H), 4.08 - 4.00 (m, 1H), 4.00 - 3.89 (m, 1H), 3.70 - 3.61 (m, 1H), 3.48 (ddd, J = 5.2, 8.8, 11.8 Hz, 1H), 2.52 (td, J = 4.8, 13.8 Hz, 1H), 2.27 (s, 3H), 2.18 - 2.13 (m, 1H), 2.07 - 2.02 (m, 3H), 1.87 - 1.79 (m, 1H), 1.38 (dd, J = 4.2, 9.2 Hz, 1H), 1.13 (dd, J = 4.2, 6.4 Hz, 1H)。
6-5(160mg、330.62μmol)、及び1A-2(73mg、499.37μmol)のトルエン(4mL)溶液に、Pd2dba3(31mg、33.85μmol)、Brettphos(36mg、67.07μmol)と炭酸セシウム(216mg、662.94μmol)を加えた。窒素ガス保護下で、反応混合物を110℃に加熱して2時間攪拌した。反応液を酢酸エチル(30mL)で希釈した後濾過し、濾液を水(20×1mL)で洗浄し、有機相を無水硫酸ナトリウムで乾燥させた後濃縮し、薄層シリカゲルプレート(PE/EA=2/1、V/V)で分離して化合物6-6を得た。1HNMR (400 MHz, CDCl3) δ 8.83 (d, J = 4.6 Hz, 1H), 8.03 (s, 1H), 7.93 (s, 1H), 7.83 (d, J = 4.6 Hz, 1H), 7.62 (dd, J = 2.0, 8.2 Hz, 1H), 7.49 - 7.45 (m, 1H), 7.30 (d, J = 8.4 Hz, 1H), 6.76 (d, J = 1.0 Hz, 1H), 6.51 (d, J = 1.0 Hz, 1H), 4.75 - 4.68 (m, 1H), 4.59 - 4.47 (m, 2H), 4.09 - 4.01 (m, 2H), 3.99 - 3.88 (m, 2H), 3.82 (ddd, J = 4.0, 6.6, 11.0 Hz, 1H), 3.65 - 3.59 (m, 1H), 3.57 - 3.44 (m, 2H), 2.55 (td, J = 5.0, 14.2 Hz, 1H), 2.27 (s, 3H), 2.07 - 2.02 (m, 3H), 1.89 - 1.71 (m, 3H), 1.68 - 1.60 (m, 2H), 1.58 - 1.47 (m, 3H), 1.39 (dd, J = 4.0, 9.0 Hz, 1H), 1.05 (dd, J = 4.0, 6.2 Hz, 1H)。
6-6(150mg、252.67μmol)のDMF(2mL)にHCl(4M、0.5mL)を加え、反応溶液を20℃で2時間攪拌した。反応溶液に飽和NaHCO3溶液を加えてpH=8に調節し、酢酸エチル(20×1mL)で抽出し、有機相を濃縮し、薄層シリカゲルプレート(PE/EA=1/2、V/V)で分離して化合物6を得た。1HNMR (400 MHz, MeOD) δ 8.85 - 8.79 (d, J = 5.0 Hz, 1H), 8.17 (s, 1H), 7.98 - 7.90 (d, J = 4.6 Hz, 1H), 7.69 - 7.60 (dd, J = 2.4, 8.2 Hz, 1H), 7.60 - 7.54 (d, J = 2.2 Hz, 1H), 7.35 - 7.28 (d, J = 8.2 Hz, 1H), 6.89 - 6.84 (m, 1H), 6.58 - 6.51 (d, J = 1.0 Hz, 1H), 4.44 - 4.35 (m, 2H), 4.08 - 3.99 (dd, J = 4.6, 11.2 Hz, 1H), 3.95 - 3.85 (m, 3H), 3.65 - 3.57 (m, 1H), 3.54 - 3.44 (m, 1H), 2.61 - 2.51 (m, 1H), 2.25 (s, 3H), 2.09 - 1.97 (m, 4H), 1.84 - 1.76 (m, 1H), 1.40 - 1.32 (dd, J = 3.4, 9.2 Hz, 1H), 1.06-1.01 (dd, J = 3.8, 6.2 Hz, 1H)。MS (ESI) m/z: 510.3 [M+H] +。
化合物6(150mg、294.38μmol)、及びイミダゾール(41mg、602.23μmol)のDMF(3mL)溶液に、TBSCl(67mg、444.53μmol)を加えた。反応混合物を35℃で4時間攪拌した。反応混合物を酢酸エチル(20mL)に希釈し、水(15×1mL)と飽和食塩水(15×3mL)で洗浄し、有機相を濃縮した後分取シリカゲルプレート(PE/EA=3/1、V/V)で分離して化合物6-7を得た。1HNMR (400 MHz, CDCl3) δ 8.83 (d, J = 5.0 Hz, 1H), 8.03 (s, 1H), 7.90 (s, 1H),7.83 (d, J = 5.0 Hz, 1H), 7.62 (dd, J = 2.0, 8.0 Hz, 1H), 7.46 (d, J = 2.0 Hz, 1H), 7.31 (d, J = 8.0 Hz, 1H), 6.76 (s, 1H), 6.48 (s,1H), 4.41 (t, J = 5.4 Hz, 2H), 4.08 - 4.02 (m, 1H), 3.99 - 3.97 (m, 2H), 3.78 - 3.74 (m, 1H), 3.62 (td, J = 5.4, 11.2 Hz, 1H), 3.48(ddd, J = 5.2, 8.4, 11.6 Hz, 1H), 2.55 (td, J = 5.2, 14.0 Hz, 1H), 2.27 (s, 3H), 2.08 - 2.02 (m, 3H), 1.89 - 1.82 (m, 2H), 1.38 (dd, J = 3.8, 9.0 Hz, 1H), 1.05 (dd, J = 3.8, 6.0 Hz, 1H), 0.91 (s, 9H), 0.09 (s, 6H)。
化合物6-7をSFCキラル分離(キラルカラム:REGIS(R,R)WHELK-O1(250mm*25mm、10μm)、移動相A:イソプロパノール(0.05%のDIEAを含有);移動相B:二酸化炭素)して化合物6-7A(保持時間:2.263min)と化合物6-7B(保持時間:2.325min)を得た。
6-7A(55mg、88.17μmol)のTHF(2mL)にHCl(3M、0.78mL)を加え、反応溶液を25℃で1時間攪拌した。反応溶液を飽和NaHCO3で中和した後、酢酸エチル(15×2mL)で抽出し、有機相を飽和食塩水(30×1mL)で洗浄し、有機相を濃縮し、分取シリカゲルプレート(PE/EA=1/1、V/V)で分離して化合物6Aを得た。1HNMR (400 MHz, MeOD) δ 8.85 - 8.79 (d, J = 5.0 Hz, 1H), 8.17 (s, 1H), 7.98 - 7.90 (d, J = 4.6 Hz, 1H), 7.69 - 7.60 (dd, J = 2.4, 8.2 Hz, 1H), 7.60 - 7.54 (d, J = 2.2 Hz, 1H), 7.35 - 7.28 (d, J = 8.2 Hz, 1H), 6.89 - 6.84 (m, 1H), 6.58 - 6.51 (d, J = 1.0 Hz, 1H), 4.44 - 4.35 (m, 2H), 4.08 - 3.99 (dd, J = 4.6, 11.2 Hz, 1H), 3.95 - 3.85 (m, 3H), 3.65 - 3.57 (m, 1H), 3.54 - 3.44 (m, 1H), 2.61 - 2.51 (m, 1H), 2.25 (s, 3H), 2.09 - 1.97 (m, 4H), 1.84 - 1.76 (m, 1H), 1.40 - 1.32 (dd, J = 3.4, 9.2 Hz, 1H), 1.06-1.01 (dd, J = 3.8, 6.2 Hz, 1H)。MS (ESI) m/z: 510.3 [M+H] +,100% (ee%)。
6-7B(52mg、83.36μmol)のTHF(2mL)にHCl(3M、0.74mL)を加え、反応溶液を25℃で1時間攪拌した。反応溶液を飽和NaHCO3で中和した後、酢酸エチル(10×2mL)で抽出し、有機相を飽和食塩水(20×1mL)で洗浄し、有機相を濃縮し、分取シリカゲルプレート(PE/EA=1/1、V/V)で分離して化合物6Bを得た。1HNMR (400 MHz, MeOD) δ 8.85 - 8.79 (d, J = 5.0 Hz, 1H), 8.17 (s, 1H), 7.98 - 7.90 (d, J = 4.6 Hz, 1H), 7.69 - 7.60 (dd, J = 2.4, 8.2 Hz, 1H), 7.60 - 7.54 (d, J = 2.2 Hz, 1H), 7.35 - 7.28 (d, J = 8.2 Hz, 1H), 6.89 - 6.84 (m, 1H), 6.58 - 6.51 (d, J = 1.0 Hz, 1H), 4.44 - 4.35 (m, 2H), 4.08 - 3.99 (dd, J = 4.6, 11.2 Hz, 1H), 3.95 - 3.85 (m, 3H), 3.65 - 3.57 (m, 1H), 3.54 - 3.44 (m, 1H), 2.61 - 2.51 (m, 1H), 2.25 (s, 3H), 2.09 - 1.97 (m, 4H), 1.84 - 1.76 (m, 1H), 1.40 - 1.32 (dd, J = 3.4, 9.2 Hz, 1H), 1.06-1.01 (dd, J = 3.8, 6.2 Hz, 1H)。MS (ESI) m/z: 510.3 [M+H] +,100% (ee%)。

2-1(80mg、163.97μmol)、及び7-1A(25mg、208.04μmol)のジオキサン(2mL)溶液に、Pd2dba3(16mg、17.47μmol)、Brettphos(10mg、17.28μmol)とDIPEA(44mg、340.45μmol)を加えた。窒素ガス保護下で、反応混合物を100℃に加熱して12時間攪拌した。反応溶液を濃縮し、薄層シリカゲルプレート(PE/EA=1/1、V/V)で分離して化合物7-1を得た。MS (ESI) m/z: 572.3 [M+H] +。
7-1(85mg、148.70μmol)のTHF(3mL)に水素化アルミニウムリチウム(9mg、237.13μmol)を加え、反応溶液を0℃で15分間攪拌した。0℃で、反応溶液に水(10mL)を加え、酢酸エチル(15×2mL)で抽出し、有機相を無水硫酸ナトリウムで乾燥させた後濃縮し、カラムクロマトグラフィーで分離(HCOOH-MeCN-H2O)して化合物7を得た。1HNMR (400 MHz, CDCl3) δ 8.94 - 8.90 (br d, J = 4.8 Hz, 1H), 8.25 (br s, 1H), 8.13 (s, 1H), 7.98 -7.91 (br d, J = 4.0 Hz, 1H), 7.64 - 7.60 (br d, J = 7.8 Hz, 1H), 7.48 (br s, 1H), 7.34 - 7.27 (br d, J = 8.2 Hz, 1H), 7.04 (s, 1H), 6.91 (s, 1H), 4.58 - 4.28 (m, 1H), 4.11 - 3.88 (m, 4H), 3.71 - 3.57 (m, 1H), 3.55 - 3.30 (m, 3H), 2.44 - 2.34 (m, 1H), 2.26 (s, 3H), 2.19 - 2.06 (m, 1H), 1.74-1.69 (br d, J = 4.4 Hz, 1H), 1.37 - 1.24 (m, 1H), 1.10 -1.01 (br t, J = 5.0 Hz, 1H)。MS (ESI) m/z: 530.0 [M+H] +。

1A-3(500mg、1.30mmol)、1-1A(450mg、1.32mmol)とPd(dppf)Cl2(200mg、273.33μmol)及び炭酸セシウム(860mg、2.64mmol)をDME(12mL)とH2O(2mL)に加えた。反応混合物を窒素ガス保護下で、100℃に加熱して12時間攪拌した。反応混合物を酢酸エチル(10mL)と水(15×1mL)で希釈し、酢酸エチル(20×3mL)で抽出し、有機相を無水硫酸ナトリウムで乾燥させた後濃縮し、カラムクロマトグラフィー(シリカゲル)で分離(HCOOH-MeCN-H2O)して化合物8-1を得た。1HNMR (400 MHz, CDCl3) δ 6.68 (d, J=1.2 Hz, 1H), 6.47 (d, J=1.2 Hz, 1H), 4.62 (t, J=3.6 Hz, 1H), 4.40 (ddq, J=3.4, 6.2, 11.6 Hz, 2H), 3.99 - 3.78 (m, 5H), 3.71 (ddd, J=3.4, 6.2, 11.6 Hz, 1H), 3.54 (td, J=5.2, 11.6 Hz, 1H), 3.45 (td, J=5.2, 11.0 Hz, 1H), 3.33 (ddd, J=5.8, 8.4, 11.6 Hz, 1H), 2.05 - 1.97 (m, 2H), 1.82 - 1.61 (m, 2H), 1.60 - 1.50 (m, 2H), 1.45 (br d, J=5.4 Hz, 1H), 1.32 - 1.23 (m, 1H), 1.04 - 0.92 (m, 2H)。
8-1(50mg、141.31μmol)、1A-4(80mg、196.94μmol)、Pd(dppf)Cl2(15mg、20.50μmol)、及び炭酸ナトリウム(30mg、283.05μmol)をジオキサン(2mL)と水(0.4mL)の混合溶媒に加えた。窒素ガス保護下で、反応混合物を100℃に加熱して4時間攪拌した。反応溶液を酢酸エチル(10mL)と水(15mL)で希釈した後濾過し、濾液を酢酸エチル(10×3mL)で抽出し、有機相を無水硫酸ナトリウムで乾燥させた後濃縮し、薄層シリカゲルプレート(PE/EA=1/1、V/V)で分離して化合物8-2を得た。MS (ESI) m/z: 598.4 [M+H] +。
8-2(50mg、83.66μmol)のDMF(2mL)にHCl(4M、1mL)を加え、反応溶液を15℃下で0.5時間攪拌した。反応溶液を酢酸エチル(10mL)と水(15mL)で希釈した後、飽和NaHCO3溶液でpH=8に調節し、酢酸エチル(10×3mL)で抽出し、有機相を無水硫酸ナトリウムで乾燥させた後濃縮し、薄層シリカゲルプレート(PE/EA=1/1、V/V)で分離して化合物8を得た。1HNMR (400 MHz, MeOD) δ 8.97-8.92 (d, J=5.0 Hz, 1H), 8.33 (s, 1H), 8.18-8.11 (d, J=4.0 Hz, 1H), 7.83 - 7.75 (m, 1H), 7.73-7.68 (dd, J=2.2, 8.2 Hz, 1H), 7.36-7.29 (d, J=8.2 Hz, 1H), 7.05-6.95 (d, J=1.4 Hz, 1H), 6.75-6.70 (d, J=1.4 Hz, 1H), 4.43 - 4.38 (m, 2H), 4.10-4.02 (dd, J=4.6, 11.4 Hz, 1H), 3.97-3.90 (d, J=11.4 Hz, 1H), 3.91 - 3.86 (m, 2H), 3.67 - 3.59 (m, 1H), 3.56-3.49 (ddd, J=5.6, 8.2, 11.6 Hz, 1H), 2.39 (s, 3H), 2.25 - 2.14 (m, 2H), 1.56-1.51 (td, J=4.6, 9.2 Hz, 1H), 1.22 - 1.15 (m, 1H), 1.10 - 1.05 (m, 1H), 1.10 - 1.05 (m, 1H)。MS (ESI) m/z: 514.3 [M+H] +。
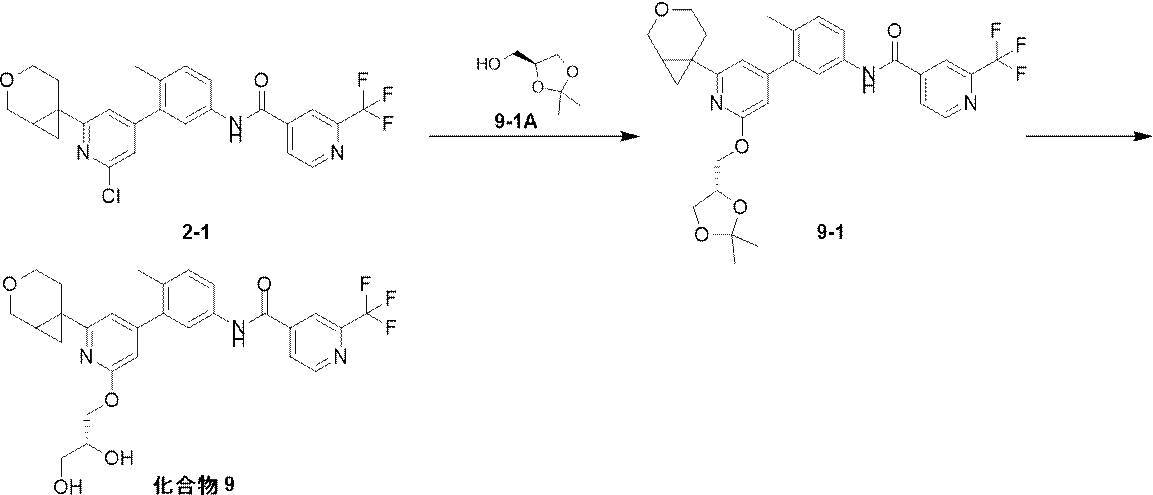
2-1(150mg、307.44μmol)、及び9-1A(82mg、620.47μmol)のトルエン(4mL)溶液に、Pd2dba3(28mg、30.58μmol)、Brettphos(33mg、61.48μmol)と炭酸セシウム(201mg、616.91μmol)を加えた。窒素ガス保護下で、反応混合物を110℃に加熱して4時間攪拌した。反応溶液を酢酸エチル(20mL)で希釈し、水(20×1mL)で洗浄し、有機相を無水硫酸ナトリウムで乾燥させた後濃縮し、薄層シリカゲルプレート(PE/EA=2/1、V/V)で分離して化合物9-1を得た。1HNMR (400 MHz, CDCl3) δ 8.93 (d, J = 5.0 Hz, 1H), 8.11 (s, 1H), 7.94 - 7.85 (m, 2H), 7.59 (dd, J = 2.0, 8.4 Hz, 1H), 7.47 (d, J = 1.8 Hz, 1H), 7.31 (d, J = 8.4 Hz, 1H), 6.78 (s, 1H), 6.52 (d, J = 1.0 Hz, 1H), 4.56 - 4.47 (m, 1H), 4.45 - 4.32 (m, 2H), 4.20 - 4.16 (m, 1H), 4.08 - 4.02 (m, 1H), 3.99 - 3.93 (m, 1H), 3.89 (ddd, J = 1.2, 6.2, 8.4 Hz, 1H), 3.62 (td, J = 5.4, 11.2 Hz, 1H), 3.48 (ddd, J = 5.2, 8.4, 11.6 Hz, 1H), 2.54 (td, J = 5.2, 12.4 Hz, 1H), 2.27 (s, 3H), 2.15 - 2.06 (m, 1H), 1.87 - 1.77 (m, 1H), 1.49 (s, 3H), 1.42 (s, 3H), 1.40 - 1.35 (m, 1H), 1.07 (dd, J = 3.8, 6.2 Hz, 1H)。
9-1(140mg、239.89μmol)のメタノール(3mL)にHCl(3M、0.2mL)を加え、反応溶液を30℃で1時間攪拌した。反応溶液を水(20mL)で希釈した後、飽和NaHCO3溶液でpH=8に調節し、酢酸エチル(20×1mL)で抽出し、有機相を濃縮し、薄層シリカゲルプレート(PE/EA=1/1、V/V)で分離して化合物9を得た。1HNMR (400 MHz, MeOD) δ 8.96-8.89 (d, J = 5.0 Hz, 1H), 8.31 (s, 1H), 8.16-8.11 (dd, J = 1.4, 5.0 Hz, 1H), 7.71-7.65 (dd, J = 2.4, 8.2 Hz, 1H), 7.65-7.59 (d, J = 2.4 Hz, 1H), 7.36-7.30 (d, J = 8.2 Hz, 1H), 6.91-6.87 (d, J = 1.0 Hz, 1H), 6.60-6.54 (d, J = 1.0 Hz, 1H), 4.49 - 4.40 (m, 1H), 4.38 - 4.30 (m, 1H), 4.10 - 3.99 (m, 2H), 3.96-3.90 (dd, J = 1.2, 11.2 Hz, 1H), 3.69 - 3.56 (m, 3H), 3.54-3.38 (ddd, J = 5.2, 8.4, 11.6 Hz, 1H), 2.60-2.53 (td, J = 5.2, 14.0 Hz, 1H), 2.27 (s, 3H), 2.15 - 2.04 (m, 1H), 1.90 - 1.76 (m, 1H), 1.48 - 1.36 (m, 1H), 1.09-1.01 (dd, J = 4.0, 6.4 Hz, 1H)。MS (ESI) m/z: 544.2 [M+H] +。

2-1(140mg、286.94μmol)、及び10-1A(46mg、433.47μmol)のトルエン(4mL)溶液に、Pd2dba3(27mg、29.48μmol)、Brettphos(31mg、57.75μmol)と炭酸セシウム(187mg、573.94μmol)を加えた。窒素ガス保護下で、反応混合物を110℃に加熱して4時間攪拌した。反応溶液を酢酸エチル(20mL)で希釈した後、水(20×1mL)で洗浄し、有機相を無水硫酸ナトリウムで乾燥させた後濃縮し、薄層シリカゲルプレート(PE/EA=2/1、V/V)で分離して化合物10を得た。1HNMR (400 MHz, DMSO-d6) δ 10.69 (s, 1H), 9.01-8.97 (d, J = 5.0 Hz, 1H), 8.36 (s, 1H), 8.23-8.18 (dd, J = 1.0, 5.0 Hz, 1H), 7.77-7.73 (dd, J = 2.4, 8.2 Hz, 1H), 7.69-7.65 (d, J = 2.2 Hz, 1H), 7.37-7.32 (d, J = 8.6 Hz, 1H), 6.89-6.85 (d, J = 1.0 Hz, 1H), 6.55-6.51 (d, J = 1.0 Hz, 1H), 5.08-5.01 (d, J = 5.4 Hz, 1H), 4.32 - 4.23 (m, 1H), 4.22 - 4.14 (m, 1H), 4.00 - 3.92 (m, 2H), 3.83-3.77 (dd, J = 1.2, 11.4 Hz, 1H), 3.54 - 3.46 (m, 1H), 3.45 - 3.35 (m, 3H), 3.28 (s, 3H), 2.55-2.51 (br d, J = 2.0 Hz, 1H), 2.24 (s, 3H), 2.06 - 1.93 (m, 1H), 1.84 - 1.73 (m, 1H), 1.36-1.30 (td, J = 3.0, 9.0 Hz, 1H), 1.05-0.98 (dd, J = 3.6, 6.2 Hz, 1H)。MS (ESI) m/z: 558.0 [M+H] +。
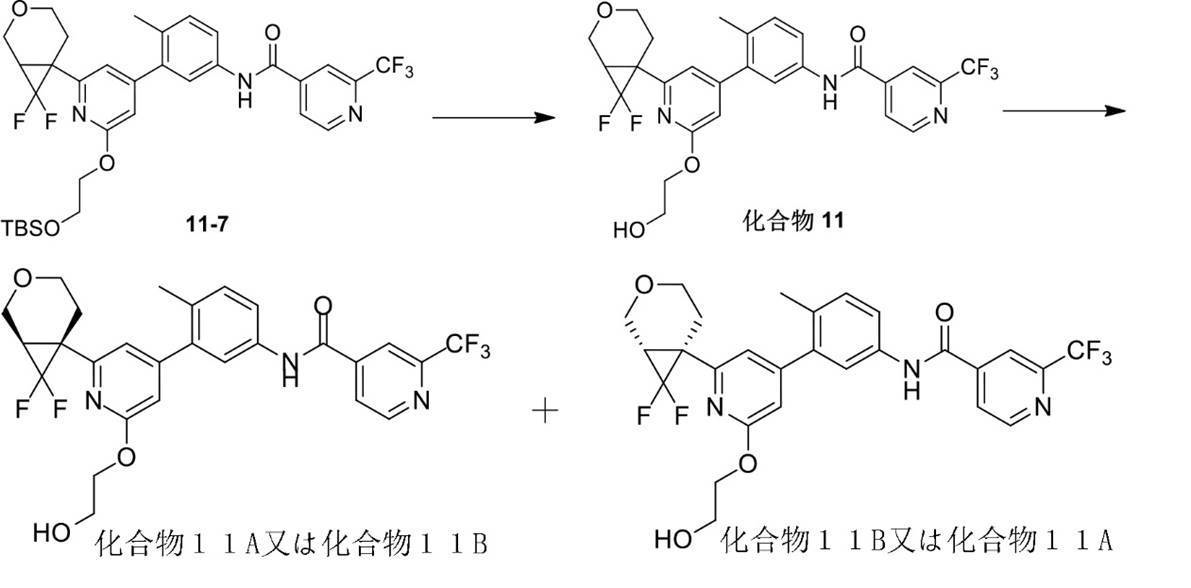
11-1(7.5g、50.68mmol)、11-1A(10.1g、48.15mmol)、Pd(dppf)Cl2(4.1g、5.07mmol)、及び炭酸ナトリウム(10.7g、101.36mmol)をDME(225mL)と水(50mL)の混合溶媒に加えた。窒素ガス保護下で、反応混合物を90℃に加熱して12時間攪拌した。反応溶液を濃縮し、シリカゲルカラムクロマトグラフィー(PE/EA=1/0~0/1、V/V)で分離して化合物11-2を得た。1HNMR (400 MHz, CDCl3) δ 7.56-7.52 (m, 1H), 7.20-7.18 (m, 1H), 7.12-7.03 (m, 1H), 6.72-6.70 (m, 1H), 4.31 - 4.29 (m, 2H), 3.87 - 3.85 (m, 2H), 2.54 - 2.51 (m, 2H) 。
11-2(1.8g、9.20mmol)、11-2A(4.6g、32.11mmol)、ヨウ化ナトリウム(450mg、3.00mmol)をTHF(42mL)溶媒に加えた。窒素ガス保護下で、反応混合物を80℃に加熱して12時間攪拌した。反応溶液にジクロロメタン(20mL)と水(20mL)を加え、分層し、有機相を無水硫酸ナトリウムで乾燥させた後濃縮して化合物11-3を得た。MS (ESI) m/z:246.2[M+H] +。
11-3(2g、8.14mmol)のジクロロメタン(20mL)溶媒にm-クロロペルオキシ安息香酸(4.1g、20.27mmol、85%)を加えた。反応混合物を65℃に加熱して12時間攪拌した。反応溶液にチオ硫酸ナトリウム(20g)、水(200mL)とジクロロメタン(200mL)を加え、30分間攪拌した。炭酸ナトリウム(15g)を加えて20分間攪拌した。分層し、有機相を無水硫酸ナトリウムで乾燥させた後濃縮し、シリカゲルカラムクロマトグラフィー(DCM/MeOH=1/0~1/1、V/V)で分離して化合物11-4を得た。 1HNMR (400MHz, CDCl3) δ 7.51-7.49 (m, 1H), 7.35-7.33 (m, 1H), 7.20-7.16 (m, 1H), 4.23 - 4.21 (m, 1H), 4.13 - 4.08 (m, 1H), 3.82 - 3.80 (m, 2H), 2.30 - 2.25 (m, 1H), 1.77 - 1.67 (m, 2H)。
11-4(100mg、382.19μmol)をオキシ塩化リン(2mL)に加え、90℃に加熱して12時間攪拌した。反応溶液を飽和炭酸水素ナトリウム溶液(20mL)に加え、酢酸エチル(20×1mL)で抽出し、有機相を無水硫酸ナトリウムで乾燥させた後濃縮して化合物11-5を得た。 MS (ESI) m/z: 280.1 [M+H]+。
11-5(100mg、357.02μmol)、及び11-3A(125.9mg、714.04μmol)のTHF(1mL)溶媒にカリウムtert-ブトキシド(1.8mL、1M)を加えた。反応混合物を25℃で3時間攪拌した。反応溶液に水(10mL)と酢酸エチル(10mL)を加え、分層し、有機相を無水硫酸ナトリウムで乾燥させた後濃縮し、薄層シリカゲルプレート(PE/EA=5/1、V/V)で分離して化合物11-6を得た。1HNMR (400MHz, CDCl3) δ 6.89 (d, J=1.4 Hz, 1H), 6.67 (d, J=1.4 Hz, 1H), 4.38 (t, J=5.2 Hz, 2H), 4.18 - 4.01 (m, 2H), 3.94 (t, J=5.2 Hz, 2H), 3.67 - 3.47 (m, 2H), 2.57 - 2.48 (m, 1H), 2.41 - 2.27 (m, 1H), 2.21 - 2.09 (m, 1H), 0.93 - 0.88 (m, 1H), 0.91 (s, 8H), 0.15 - 0.05 (m, 6H)。
11-6(100mg、238.12μmol)、1A-4(116mg、285.74μmol)、Pd(dppf)Cl2(38.9mg、47.62μmol)、及び炭酸ナトリウム(50.5mg、476.23μmol)をジオキサン(1mL)と水(0.2mL)の混合溶媒に加えた。窒素ガス保護下で、反応混合物を90℃に加熱して3時間攪拌した。反応溶液を濃縮し、薄層シリカゲルプレート(PE/EA=3/1、V/V)で分離した後、更にカラムクロマトグラフィー(シリカゲル)で分離(NH4HCO3-MeCN-H2O)して化合物11-7を得た。1HNMR (400MHz, CDCl3) δ 8.94 (d, J=4.8 Hz, 1H), 8.11 (s, 1H), 7.93 (d, J=4.6 Hz, 1H), 7.83 (s, 1H), 7.63 (br d, J=8.6 Hz, 1H), 7.47 (s, 1H), 7.33 (d, J=8.4 Hz, 1H), 6.84 (s, 1H), 6.62 (d, J=1.0 Hz, 1H), 4.45 (t, J=5.2 Hz, 2H), 4.20 - 4.04 (m, 2H), 3.99 (t, J=5.2 Hz, 2H), 3.68 - 3.49 (m, 2H), 2.59 (ddd, J=3.2, 6.4, 9.8 Hz, 1H), 2.43 - 2.32 (m, 1H), 2.27 (s, 3H), 2.26 - 2.17 (m, 1H), 1.56 (s, 4H), 0.91 (s, 9H), 0.10 (s, 6H)。
11-7(120mg、180.79μmol)のTHF(3mL)にHCl(12M、0.03mL)を加え、反応溶液を20℃で3時間攪拌した。反応溶液を水(10mL)で希釈した後、飽和NaHCO3溶液でpH=8に調節し、酢酸エチル(10×1mL)で抽出し、有機相を無水硫酸ナトリウムで乾燥させた後濃縮して化合物11を得た。1HNMR (400MHz, CDCl3) δ 8.97-8.92 (d, J=4.8 Hz, 1H), 8.11 (s, 1H), 7.95-7.90 (br d, J=4.8 Hz, 1H), 7.88 (s, 1H), 7.64-7.58(br d, J=8.0 Hz, 1H), 7.52 (s, 1H), 7.36-7.30 (d, J=8.4 Hz, 1H), 6.90 (s, 1H), 6.68 (s, 1H), 4.59 - 4.50 (m, 2H), 4.17 - 3.97 (m, 4H), 3.67 - 3.49 (m, 2H), 3.27-3.23 (t, J=5.8 Hz, 1H), 2.53 - 2.42 (m, 1H), 2.41 - 2.30 (m, 1H), 2.28 (s, 3H), 2.23 - 2.11 (m, 1H)。MS (ESI) m/z: 550.3 [M+H]+。
化合物11をSFC分離(キラルカラム:DAICEL CHIRALPAK AD-H(250mm*30mm,5μm),移動相A:エタノール(0.05%のDIEAを含有);移動相B:二酸化炭素)して化合物11A(保持時間:1.325min)と化合物11B(保持時間:1.422min)を得た。化合物11A:1HNMR (400MHz, CDCl3) δ 8.97-8.92 (d, J=4.8 Hz, 1H), 8.11 (s, 1H), 7.95-7.90 (br d, J=4.8 Hz, 1H), 7.88 (s, 1H), 7.64-7.58(br d, J=8.0 Hz, 1H), 7.52 (s, 1H), 7.36-7.30 (d, J=8.4 Hz, 1H), 6.90 (s, 1H), 6.68 (s, 1H), 4.59 - 4.50 (m, 2H), 4.17 - 3.97 (m, 4H), 3.67 - 3.49 (m, 2H), 3.27-3.23 (t, J=5.8 Hz, 1H), 2.53 - 2.42 (m, 1H), 2.41 - 2.30 (m, 1H), 2.28 (s, 3H), 2.23 - 2.11 (m, 1H)。MS (ESI) m/z: 550.0 [M+H] +,100% (ee%)。化合物11B:1HNMR (400MHz, CDCl3) δ 8.97-8.92 (d, J=4.8 Hz, 1H), 8.11 (s, 1H), 7.95-7.90 (br d, J=4.8 Hz, 1H), 7.88 (s, 1H), 7.64-7.58(br d, J=8.0 Hz, 1H), 7.52 (s, 1H), 7.36-7.30 (d, J=8.4 Hz, 1H), 6.90 (s, 1H), 6.68 (s, 1H), 4.59 - 4.50 (m, 2H), 4.17 - 3.97 (m, 4H), 3.67 - 3.49 (m, 2H), 3.27-3.23 (t, J=5.8 Hz, 1H), 2.53 - 2.42 (m, 1H), 2.41 - 2.30 (m, 1H), 2.28 (s, 3H), 2.23 - 2.11 (m, 1H)。MS (ESI) m/z: 550.0 [M+H] +,97.5% (ee%)。

12-1(480mg、2.46mmol)、1A-4(1g、2.46mmol)、Pd(dppf)Cl2(400mg、489.81μmol)、及び炭酸ナトリウム(520mg、4.91mmol)をジオキサン(20mL)と水(4mL)の混合溶媒に加えた。窒素ガス保護下で、反応混合物を100℃に加熱して2時間攪拌した。反応溶液を濾過し、濾液を濃縮し、シリカゲルカラムクロマトグラフィー(PE/EA=10/1~5/1、V/V)で分離して化合物12-2を得た。1HNMR (400 MHz, CDCl3) δ 8.92 (d, J=5.0 Hz, 1H), 8.11 (s, 2H), 7.93 (d, J=4.2 Hz, 1H), 7.76 (d, J=2.2 Hz, 1H), 7.66 (dd, J=2.2, 8.2 Hz, 1H), 7.32 (d, J=8.2 Hz, 1H), 7.14 (s, 1H), 2.61 (s, 3H), 2.44 (s, 3H)。
12-2(300mg、683.60μmol)、1-1A(156mg、764.56μmol)とPd(dppf)Cl2(120mg、146.94μmol)及び炭酸セシウム(450mg、1.38mmol)をDME(10mL)とH2O(2mL)に加えた。反応混合物を窒素ガス保護下で、110℃に加熱して16時間攪拌した。反応溶液を濾過し、濾液を濃縮し、シリカゲルカラムクロマトグラフィー(PE/EA=10/1~5/1、V/V)で分離して化合物12-2を得た。 MS (ESI) m/z: 501.2 [M+H] +。
12-3(80mg、159.83μmol)のジクロロメタン(2mL)溶媒にm-クロロペルオキシ安息香酸(65mg、320.17μmol、85%)を加えた。反応混合物を15℃で1時間攪拌した。反応溶液に水(2mL)とジクロロメタン(3mL)を加え、分層し、有機相を飽和亜硫酸ナトリウム(1mL)で洗浄し、有機相を濃縮して化合物12-4を得た。MS (ESI) m/z: 533.3 [M+H] +。
1A-2(16mg、109.45μmol)のジオキサン(2mL)溶液に水素化ナトリウム(16mg、400.04μmol、60%)を加えた。12-4(55mg、103.28μmol)を前記混合物に加え、15℃で1時間攪拌した。反応溶液に飽和塩化アンモニウム溶液(3mL)を加え、酢酸エチル(5×3mL)で抽出し、有機相を無水硫酸ナトリウムで乾燥させた後濃縮して化合物12-5を得た。MS (ESI) m/z: 599.4 [M+H] +。
12-5(50mg、83.53μmol)のDMF(2mL)にHCl(2M、0.15mL)を加え、反応溶液を15℃で1時間攪拌した。反応溶液に飽和NaHCO3溶液(5mL)を加え、酢酸エチル(5×3mL)で抽出し、有機相を無水硫酸ナトリウムで乾燥させた後濃縮し、カラムクロマトグラフィー(シリカゲル)で分離(HCOOH-MeCN-H2O)して化合物12を得た。1HNMR (400 MHz, DMSO-d6) δ 10.80 (s, 1H), 9.02-8.97 (d, J=5.0 Hz, 1H), 8.39 (s, 1H), 8.24-8.20 (d, J=4.4 Hz, 1H), 7.89-7.86 (d, J=2.0 Hz, 1H), 7.87-7.83 (dd, J=2.2, 8.4 Hz, 1H), 7.39-7.34 (d, J=8.4 Hz, 1H), 7.17 (s, 1H), 4.91 (br s, 1H), 4.36-4.31 (t, J=5.2 Hz, 2H), 3.96 - 3.88 (m, 1H), 3.87 - 3.81 (m, 1H), 3.75-3.70 (br t, J=4.8 Hz, 2H), 3.60 - 3.52 (m, 1H), 2.64-2.58 (td, J=4.8, 14.0 Hz, 1H), 2.38 (s, 3H), 1.99-1.89 (ddd, J=5.8, 8.6, 14.2 Hz, 1H), 1.88 - 1.80 (m, 1H), 1.49-1.40 (dd, J=3.8, 9.2 Hz, 1H), 1.16-1.10 (dd, J=3.8, 6.4 Hz, 1H)。MS (ESI) m/z: 515.1 [M+H] +。

2-1(100mg、204.96μmol)、及び13-1A(100mg、533.75μmol)のトルエン(4mL)溶液に、Pd2dba3(30mg、32.76μmol)、Ruphos(30mg、64.29μmol)と炭酸セシウム(150mg、460.38μmol)を加えた。窒素ガス保護下で、反応混合物を120℃に加熱して4時間攪拌した。反応溶液を濾過し、濾液を濃縮した後、薄層シリカゲルプレート(PE/EA=3/1、V/V)で分離して化合物13-1を得た。1HNMR (400 MHz, CDCl3) δ 8.93 (d, J=5.0 Hz, 1H), 8.11 (s, 1H), 7.92 (br d, J=4.6 Hz, 1H), 7.85 (s, 1H), 7.62 (br d, J=8.4 Hz, 1H), 7.42 (s, 1H), 7.30 (d, J=8.4 Hz, 1H), 6.51 (s, 1H), 6.03 (s, 1H), 4.76 (quin, J=5.6 Hz, 1H), 4.22 (t, J=7.6 Hz, 2H), 4.06 - 3.99 (m, 1H), 3.98 - 3.93 (m, 1H), 3.84 - 3.77 (m, 2H), 3.62 (td, J=5.4, 11.2 Hz, 1H), 3.45 (ddd, J=5.4, 8.8, 11.2 Hz, 1H), 2.57 (td, J=4.8, 14.0 Hz, 1H), 2.10 - 2.03 (m, 1H), 1.81 (td, J=4.6, 9.2 Hz, 1H), 1.37 (dd, J=3.8, 9.2 Hz, 1H), 0.99 (dd, J=3.8, 6.4 Hz, 2H), 0.93 - 0.90 (m, 9H), 0.11 - 0.07 (m, 6H)。
13-1(80mg、125.24μmol)のジクロロメタン(4mL)にTFA(1mL)を加え、反応溶液を15℃で13時間攪拌した。反応溶液をジクロロメタン(10mL)で希釈した後、飽和NaHCO3溶液でpH=8に調節し、ジクロロメタン(10×3mL)で抽出し、有機相を無水硫酸ナトリウムで乾燥させた後濃縮し、カラムクロマトグラフィー(シリカゲル)で分離(HCOOH-MeCN-H2O)して化合物13を得た。1HNMR (400 MHz, DMSO-d6) δ 10.69 (s, 1H), 9.02-8.97 (d, J=5.0 Hz, 1H), 8.37 (s, 1H), 8.22-8.18 (d, J=5.0 Hz, 1H), 7.77-7.73 (dd, J=2.0, 8.4 Hz, 1H), 7.64-7.61 (d, J=2.0 Hz, 1H), 7.35-7.30 (d, J=8.4 Hz, 1H), 6.54 (s, 1H), 6.12 (br s, 1H), 5.77 - 5.36 (m, 1H), 4.64 - 4.50 (m, 1H), 4.18-4.14 (br t, J=7.2 Hz, 2H), 3.96-3.92 (dd, J=4.6, 11.2 Hz, 1H), 3.83-3.79 (d, J=10.2 Hz, 1H), 3.75 - 3.61 (m, 2H), 3.51-3.47 (td, J=5.6, 11.4 Hz, 1H), 3.42 - 3.37 (m, 2H), 2.23 (s, 3H), 1.98-1.94 (ddd, J=5.6, 8.2, 13.8 Hz, 1H), 1.77 - 1.70 (m, 1H), 1.29-1.27 (dd, J=3.6, 9.0 Hz, 1H), 0.95-0.90 (dd, J=3.6, 6.0 Hz, 1H)。MS (ESI) m/z: 525.2 [M+H] +。

0℃で、1A-2(11.8g、80.72mmol)のTHF(100 mL)溶液に水素化ナトリウム(5.41g、135.14mmol、純度:60%)を加え、20℃に昇温させて30分間攪拌した。0℃で、当該混合溶液に11-1(10g、67.57mmol)を加え、20℃に昇温させて4時間攪拌した。反応混合物に飽和塩化アンモニウム溶液(30mL)を加え、酢酸エチル(40mL×2)で抽出し、合わせた抽出溶液を濃縮し、カラムクロマトグラフィー(PE/EA=100/0~80/1、V/V)で分離して14-1を得た。1HNMR (400MHz, CDCl3) δ 7.55 - 7.49 (m, 1H), 6.90 (d, J=7.4 Hz, 1H), 6.71 (d, J=8.2 Hz, 1H), 4.71 (t, J=3.6 Hz, 1H), 4.58 - 4.43 (m, 2H), 4.05 (ddd, J=3.6, 5.8, 11.6 Hz, 1H), 3.90 (ddd, J=3.2, 8.0, 11.4 Hz, 1H), 3.81 (ddd, J=3.6, 6.4, 11.4 Hz, 1H), 3.58 - 3.49 (m, 1H), 1.91 - 1.80 (m, 1H), 1.79 - 1.70 (m, 1H), 1.68 - 1.50 (m, 4H)。
14-1(10g、38.80mmol)、2-1A-1(20g、78.76mmol)とPd(dppf)Cl2(2.75g、3.76mmol)、及び酢酸カリウム(11.5g、117.18mmol)をDMF(200mL)に加えた。反応混合物を窒素ガス保護下で、100℃に加熱して8時間攪拌した。反応溶液を濾過して化合物14-2の溶液を得た。MS (ESI): m/z 268.2 [M+H]+。
14-2(9.6g、35.94mmol)、14-2A(6g、31.91mmol)、Pd(dppf)Cl2(2.4g、3.28mmol)、及び炭酸ナトリウム(7.2g、67.93mmol)をDMF(240mL)と水(48mL)の混合溶媒に加えた。窒素ガス保護下で、反応混合物を100℃に加熱して3時間攪拌した。反応溶液に水(300mL)を加え、酢酸エチル(200×2mL)で抽出し、有機相を飽和食塩水(100×2mL)で洗浄し、無水硫酸ナトリウムで乾燥させた後濃縮し、シリカゲルカラムクロマトグラフィー(PE/EA=16/1~10/1、V/V)で分離して化合物14-3を得た。1HNMR (400MHz, CDCl3) δ 7.65 (dd, J=7.4, 8.2 Hz, 1H), 7.17 (d, J=7.4 Hz, 1H), 6.86 (d, J=8.2 Hz, 1H), 4.74 - 4.71 (m, 1H), 4.70 - 4.59 (m, 2H), 4.43 (t, J=2.6 Hz, 2H), 4.10 (ddd, J=3.2, 5.8, 11.6 Hz, 1H), 3.97 (t, J=5.4 Hz, 2H), 3.94 - 3.77 (m, 2H), 3.57 - 3.49 (m, 1H), 2.77 (tt, J=2.8, 5.6 Hz, 2H), 1.91 - 1.82 (m, 1H), 1.80 - 1.71 (m, 1H), 1.70 - 1.62 (m, 2H), 1.58 - 1.52 (m, 2H)。
0℃で、14-3(2g、6.05mmol)とクロロヨードメタン(2g、18.16mmol)のNMP(20mL)溶液にリチウムtert-ブトキシド(1.45g、18.16mmol)のNMP(20mL)溶液を滴下して加え、20℃に昇温させて12時間攪拌した。反応混合物に飽和塩化アンモニウム溶液(50mL)と水(50mL)を加え、酢酸エチル(50mL×2)で抽出し、合わせた抽出溶液を濃縮し、カラムクロマトグラフィー(PE/EA=20/0~8/1)で分離して14-4を得た。1HNMR (400MHz, CDCl3) δ 7.66 - 7.53 (m, 1H), 7.04 (d, J = 7.4 Hz, 1H), 6.71 (d, J = 8.4 Hz, 1H), 6.44 (d, J = 6.2 Hz, 1H), 5.57 (d, J = 6.2 Hz, 1H), 4.74 - 4.67 (m, 1H), 4.62 (ddd, J = 3.8, 6.2, 11.6 Hz, 1H), 4.55 (t, J = 5.0 Hz, 1H), 4.47 (ddd, J = 3.8, 6.4, 11.7 Hz, 1H), 4.29 (d, J = 10.8 Hz, 1H), 4.11 - 4.02 (m, 1H), 3.98 (d, J = 10.8 Hz, 1H), 3.91 (ddd, J = 3.2, 8.0, 11.2 Hz, 1H), 3.87 - 3.77 (m, 1H), 3.57 - 3.48 (m, 1H), 2.78 (ddd, J = 0.8, 4.6, 7.2 Hz, 1H), 1.89 - 1.81 (m, 1H), 1.77 (d, J = 4.0 Hz, 1H), 1.76 - 1.70 (m, 1H), 1.68 - 1.57 (m, 2H), 1.57 - 1.49 (m, 2H)。
14-4(1.5g、4.38mmol)のメタノール(20mL)溶液に、Pd/C(10%、400mg)を加え、水素ガス(15psi)雰囲気下で、20℃で1時間攪拌した。反応溶液を濾過した後、濃縮して化合物14-5の粗成生物を得た。1HNMR (400MHz, CDCl3) δ 7.62 - 7.56 (m, 1H), 6.93 (dd, J=4.2, 7.2 Hz, 1H), 6.71 (d, J=8.4 Hz, 1H), 4.73 (t, J=3.6 Hz, 1H), 4.65 - 4.49 (m, 2H), 4.19 - 4.13 (m, 1H), 4.09 (dddd, J=1.6, 3.8, 5.6, 11.2 Hz, 1H), 4.05 - 4.01 (m, 1H), 3.93 (ddd, J=3.2, 8.0, 11.2 Hz, 1H), 3.89 - 3.79 (m, 2H), 3.58 - 3.48 (m, 2H), 2.59 - 2.49 (m, 1H), 2.28 (dd, J=5.0, 12.4 Hz, 1H), 2.12 - 2.00 (m, 1H), 1.94 - 1.83 (m, 1H), 1.82 - 1.72 (m, 1H), 1.71 - 1.64 (m, 1H), 1.62 (s, 1H), 1.59 - 1.54 (m, 2H), 1.49 (d, J=5.0 Hz, 1H)。
2-1A-1(884.79mg、3.48mmol)、[Ir(COD)OMe]2(200mg、301.72μmol)、及びdtbpy(162.10mg、603.94μmol)をテトラヒドロフラン(10mL)に加えた。反応混合物を窒素ガス保護下で、80℃に加熱して5分間攪拌した。次に、14-5(800mg、2.32mmol)のテトラヒドロフラン(10mL)溶液を加え、80℃で4時間攪拌した。反応溶液を濾過し、濃縮して化合物14-6の粗成生物を得た。MS (ESI): m/z 305.1 [M+H-THP]+。
14-6(500mg、1.39mmol)、14-5A(600mg、1.28mmol)とPd(dppf)Cl2(93.66mg、128.00μmol)、及び炭酸ナトリウム(271.33mg、2.56mmol)をジオキサン(10mL)と水(2mL)に加えた。反応混合物を窒素ガス保護下で、80℃に加熱して2時間攪拌した。反応混合物を濾過し、濾液を濃縮した後酢酸エチル(30mL)を加え、飽和食塩水(20mL×2)で洗浄した後濃縮し、カラムクロマトグラフィー(PE/EA=20/1~5/1)で分離して14-7を得た。MS (ESI): m/z 623.3 [M+H]+。
14-7(50mg、80.30μmol)のジクロロメタン(1mL)にTFA(0.5mL)を加え、反応溶液を20℃で0.5時間攪拌した。反応溶液を飽和炭酸水素ナトリウム溶液でpH=8に調節し、ジクロロメタン(20×3mL)で抽出し、有機相を無水硫酸ナトリウムで乾燥させた後濃縮し、薄層シリカゲルプレート(PE/EA=1/2、V/V)で分離して化合物14を得た。1HNMR (400 MHz, DMSO-d6) δ 10.71 (s, 1H), 9.02-8.95 (d, J = 4.8 Hz, 1H), 8.37 (s, 1H), 8.21-8.18 (d, J = 4.8 Hz, 1H), 7.82-7.74 (dd, J = 2.2, 8.4 Hz, 1H), 7.68-7.60 (d, J = 2.2 Hz, 1H), 7.39-7.30 (d, J = 8.4 Hz, 1H), 7.14 (s, 1H), 6.69 (s, 1H), 4.89-4.83 (t, J = 5.6 Hz, 1H), 4.39-4.32 (t, J = 5.0 Hz, 2H), 4.10-4.05 (d, J = 11.2 Hz, 1H), 3.98-4.86 (d, J = 11.4 Hz, 1H), 3.82 - 3.68 (m, 3H), 3.55 - 3.49 (m, 1H), 2.61 - 2.55 (m, 1H), 2.46-2.38 (d, J = 5.4 Hz, 1H), 2.23 (s, 3H), 2.02 - 1.93 (m, 1H), 1.54-1.47 (d, J = 5.4 Hz, 1H)。MS (ESI): m/z 539.2 [M+H]+。
化合物14をSFCキラル分離(キラルカラム:DAICEL CHIRALPAK AD(250mm*30mm,10μm),移動相A:イソプロパノール(0.05%のDIEAを含有);移動相B:二酸化炭素)して化合物14A(保持時間:1.702min)と化合物14B(保持時間:1.815min)を得た。化合物14A: 1HNMR (400 MHz, DMSO-d6) δ 10.71 (s, 1H), 9.02-8.95 (d, J = 4.8 Hz, 1H), 8.37 (s, 1H), 8.21-8.18 (d, J = 4.8 Hz, 1H), 7.82-7.74 (dd, J = 2.2, 8.4 Hz, 1H), 7.68-7.60 (d, J = 2.2 Hz, 1H), 7.39-7.30 (d, J = 8.4 Hz, 1H), 7.14 (s, 1H), 6.69 (s, 1H), 4.89-4.83 (t, J = 5.6 Hz, 1H), 4.39-4.32 (t, J = 5.0 Hz, 2H), 4.10-4.05 (d, J = 11.2 Hz, 1H), 3.98-4.86 (d, J = 11.4 Hz, 1H), 3.82 - 3.68 (m, 3H), 3.55 - 3.49 (m, 1H), 2.61 - 2.55 (m, 1H), 2.46-2.38 (d, J = 5.4 Hz, 1H), 2.23 (s, 3H), 2.02 - 1.93 (m, 1H), 1.54-1.47 (d, J = 5.4 Hz, 1H)。MS (ESI): m/z 539.2 [M+H]+ ,100% (ee%)。化合物14B:1HNMR (400 MHz, DMSO-d6) δ 10.71 (s, 1H), 9.02-8.95 (d, J = 4.8 Hz, 1H), 8.37 (s, 1H), 8.21-8.18 (d, J = 4.8 Hz, 1H), 7.82-7.74 (dd, J = 2.2, 8.4 Hz, 1H), 7.68-7.60 (d, J = 2.2 Hz, 1H), 7.39-7.30 (d, J = 8.4 Hz, 1H), 7.14 (s, 1H), 6.69 (s, 1H), 4.89-4.83 (t, J = 5.6 Hz, 1H), 4.39-4.32 (t, J = 5.0 Hz, 2H), 4.10-4.05 (d, J = 11.2 Hz, 1H), 3.98-4.86 (d, J = 11.4 Hz, 1H), 3.82 - 3.68 (m, 3H), 3.55 - 3.49 (m, 1H), 2.61 - 2.55 (m, 1H), 2.46-2.38 (d, J = 5.4 Hz, 1H), 2.23 (s, 3H), 2.02 - 1.93 (m, 1H), 1.54-1.47 (d, J = 5.4 Hz, 1H)。MS (ESI): m/z 539.2 [M+H]+ ,100% (ee%)。

2-1(200mg、409.92μmol)、及び15-1A(84.97mg、942.81μmol)のトルエン(5mL)溶液に、Pd2dba3(37.54mg、40.99μmol)、Brettphos(44.01mg、81.98μmol)と炭酸セシウム(267.12mg、819.84μmol)を加えた。窒素ガス保護下で、反応混合物を100℃に加熱して4時間攪拌した。反応溶液を酢酸エチル(10mL)で希釈した後濾過し、濾液を濃縮した後、カラムクロマトグラフィー(シリカゲル)で分離(HCOOH-MeCN-H2O)して化合物15を得た。1HNMR (400 MHz, CD3OD) δ 8.94-8.92 (d, J = 5.0 Hz, 1H), 8.32 (s, 1H), 8.16-8.12 (dd, J = 1.0, 5.0 Hz, 1H), 7.72-7.66 (dd, J = 2.4, 8.2 Hz, 1H), 7.66-7.63 (d, J = 2.2 Hz, 1H), 7.37-7.32 (d, J = 8.6 Hz, 1H), 6.90-6.87 (d, J = 1.0 Hz, 1H), 6.60-6.58 (d, J = 1.0 Hz, 1H), 4.60 (s, 2H), 4.20 (s, 2H), 4.10 - 4.05 (m, 1H), 3.98-3.92 (m, 1H), 3.64 - 3.61 (m, 1H), 3.54 - 3.46 (m, 1H), 2.60-2.58 (m, 1H), 2.29 (s, 3H), 2.06 - 1.98 (m, 1H), 1.85 - 1.76 (m, 1H), 1.43-1.38 (m, 1H), 1.33 (s, 6H), 1.06-1.04 (m, 1H)。MS (ESI) m/z: 542.1 [M+H] +。

-78℃で、16-1(10g、51.96mmol)のジクロロメタン(100mL)溶液にn-ブチルリチウム(23mL、2.5M)を加え、30分間攪拌した。-78℃で、当該混合溶液に16-1A(4.92g、57.16mmol)を加え、25℃に昇温させて0.5時間攪拌した。反応混合物に飽和塩化アンモニウム溶液(40mL)を加え、ジクロロメタン(40mL×2)で抽出し、合わせた抽出溶液を無水硫酸ナトリウムで乾燥させた後濃縮し、カラムクロマトグラフィー(PE/EA=10/1~5/1、V/V)で分離して化合物16-2を得た。1HNMR (400MHz, CDCl3) δ 7.72 (t, J=7.8 Hz, 1H), 7.47 (d, J=7.2 Hz, 1H), 7.28 (d, J=7.2 Hz, 1H), 4.35 (s, 1H), 4.24 - 4.14 (m, 2H), 4.05 - 4.00 (m, 1H), 3.96 - 3.92 (m, 1H), 2.45 (td, J=8.8, 13.0 Hz, 1H), 2.32 - 2.22 (m, 1H)。
16-2(6g、30.06mmol)のトルエン(80mL)溶液にp-トルエンスルホン酸(11.5g、60.46mmol)を加え、110℃に昇温させて16時間攪拌した。反応混合物に飽和炭酸水素ナトリウム溶液(40mL)と酢酸エチル(100mL)を加え、分層し、有機相を飽和炭酸水素ナトリウム溶液(50mL×2)で洗浄し、無水硫酸ナトリウムで乾燥させた後濃縮し、シリカゲルカラムクロマトグラフィー(PE/EA=50/1~20/1、V/V)で分離して化合物16-3を得た。1HNMR (400MHz, CDCl3) δ 7.64 (t, J=7.8 Hz, 1H), 7.23 (dd, J=5.2, 7.8 Hz, 2H), 6.68 (quin, J=2.0 Hz, 1H), 5.08 (dt, J=2.0, 5.0 Hz, 2H), 4.91 (dt, J=2.0, 5.0 Hz, 2H)。
カリウムtert-ブトキシド(3.71g、33.04mmol)、11-2A-1(7.3g、33.17mmol)の混合物にDMSO(50mL)を加えた。窒素ガス保護下で、反応混合物を25℃に加熱して1時間攪拌した。反応溶液に16-3(1g、5.51mmol)のDMSO(5mL)溶液を加え、反応混合物を70℃に加熱して6時間攪拌した。反応混合物に水(180mL)を加え、酢酸エチル(30×3mL)で抽出し、有機相を無水硫酸ナトリウムで乾燥させた後濃縮し、シリカゲルカラムクロマトグラフィー(PE/EA=20/1、V/V)で分離して化合物16-4を得た。1HNMR (400 MHz, CDCl3) δ 7.46 (t, J=7.8 Hz, 1H), 7.05 (d, J=7.8 Hz, 1H), 6.88 (d, J=7.6 Hz, 1H), 4.11 - 4.04 (m, 2H), 3.89 - 3.78 (m, 2H), 2.08 (ddd, J=2.8, 5.2, 8.0 Hz, 1H), 1.32 (dd, J=4.4, 8.0 Hz, 1H), 1.07 (t, J=4.6 Hz, 1H)。
2-1A-1(1.08g、4.25mmol)、[Ir(COD)OMe]2(70mg、105.60μmol)、及びtmphen(50mg、211.59μmol)をメチルtert-ブチルエーテル(20mL)に加えた。反応混合物に窒素ガス保護下で、16-4(770mg、3.94mmol)を加え、80℃に加熱して3時間攪拌した。反応溶液を濾過し、濾液を濃縮して化合物16-5の粗成生物を得た。MS (ESI): m/z 240.3 [M-84+2H]+。
16-5(1.2g、3.73mmol)、14-5A(1.4g、3.90mmol)とPd(dppf)Cl2・DCM(300mg、367.36μmol)、及び炭酸ナトリウム(800mg、7.55mmol)をジオキサン(50mL)と水(10mL)に加えた。反応混合物を窒素ガス保護下で、100℃に加熱して3時間攪拌した。反応混合物を濾過し、濾液を濃縮した後酢酸エチル(20mL)と水(10mL)を加え、分層し、水相を酢酸エチル(10mL×3)で抽出し、合わせた有機相を無水硫酸ナトリウムで乾燥させた後濃縮し、シリカゲルカラムクロマトグラフィー(PE/EA=10/1~5/1)で分離して16-6を得た。1HNMR (400 MHz, CDCl3) δ 8.92 (d, J=5.0 Hz, 1H), 8.20 (s, 1H), 8.11 (s, 1H), 7.94 (d, J=5.0 Hz, 1H), 7.60 - 7.51 (m, 2H), 7.32 (d, J=7.8 Hz, 1H), 7.10 (d, J=1.2 Hz, 1H), 6.90 (d, J=1.2 Hz, 1H), 4.19 - 4.14 (m, 2H), 3.95 - 3.87 (m, 2H), 2.27 (s, 3H), 2.24 - 2.17 (m, 1H), 1.48 - 1.41 (m, 1H), 1.20 - 1.15 (m, 1H)。
16-6(900mg、1.90mmol)、及び11-3A(400mg、2.27mmol)のトルエン(30mL)溶液に、Pd2dba3(180mg、196.57μmol)、Brettphos(200mg、372.60μmol)と炭酸セシウム(1.26g、3.87mmol)を加えた。窒素ガス保護下で、反応混合物を110℃に加熱して4時間攪拌した。反応溶液を濾過し、濾液を濃縮した後、シリカゲルカラムクロマトグラフィー(PE/EA=20/1~6/1)で分離して化合物16-7を得た。1HNMR (400 MHz, DMSO-d6) δ 10.65 (s, 1H), 8.95 (d, J=5.0 Hz, 1H), 8.31 (s, 1H), 8.14 (d, J=4.6 Hz, 1H), 7.70 (dd, J=2.0, 8.2 Hz, 1H), 7.59 (d, J=2.2 Hz, 1H), 7.29 (d, J=8.4 Hz, 1H), 6.68 (s, 1H), 6.49 (s, 1H), 4.31 (br d, J=2.8 Hz, 1H), 4.07 - 3.97 (m, 2H), 3.90 - 3.86 (m, 2H), 3.79 - 3.69 (m, 2H), 2.17 (s, 3H), 2.15 - 2.09 (m, 1H), 1.33 (dd, J=3.8, 7.8 Hz, 1H), 1.13 (t, J=7.2 Hz, 1H), 0.96 - 0.91 (m, 1H), 0.80 (s, 9H), 0.00 (s, 6H)。
16-7(300mg、488.81μmol)のテトラヒドロフラン(10mL)に塩酸(1mL、4M)を加え、反応溶液を25℃で1時間攪拌した。反応溶液を酢酸エチル(20mL)で希釈した後、飽和NaHCO3溶液でpH=8に調節し、酢酸エチル(10×3mL)で抽出し、有機相を無水硫酸ナトリウムで乾燥させた後濃縮し、薄層シリカゲルプレート(PE/EA=1/1、V/V)で分離して化合物16を得た。1HNMR (400 MHz, DMSO-d6) δ 10.70 (s, 1H), 9.06-8.95 (d, J=5.0 Hz, 1H), 8.37 (s, 1H), 8.23-8.18 (d, J=4.2 Hz, 1H), 7.77-7.71 (dd, J=2.2, 8.4 Hz, 1H), 7.68-7.64 (d, J=2.2 Hz, 1H), 7.39-7.31 (d, J=8.4 Hz, 1H), 6.73 (s, 1H), 6.59-6.54 (d, J=1.0 Hz, 1H), 4.86-4.81 (t, J=5.2 Hz, 1H), 4.35-4.30 (t, J=5.2 Hz, 2H), 4.13 - 4.03 (m, 2H), 3.83 - 3.70 (m, 4H), 2.24 (s, 3H), 2.22-2.08 (ddd, J=2.4, 5.0, 7.8 Hz, 1H), 1.40-1.32 (dd, J=3.6, 7.8 Hz, 1H), 1.02-0.96 (t, J=4.4 Hz, 1H)。MS (ESI) m/z: 500.4 [M+H] +。
化合物16をSFCキラル分離(キラルカラム:DAICEL CHIRALCEL OJ-H(250mm*30mm,5μm),移動相A:エタノール(0.05%のDIEAを含有);移動相B:二酸化炭素)して化合物16A(保持時間:1.487min)と化合物16B(保持時間:1.590min)を得た。
化合物16A:1HNMR (400 MHz, DMSO-d6) δ 10.70 (s, 1H), 9.06-8.95 (d, J=5.0 Hz, 1H), 8.37 (s, 1H), 8.23-8.18 (d, J=4.2 Hz, 1H), 7.77-7.71 (dd, J=2.2, 8.4 Hz, 1H), 7.68-7.64 (d, J=2.2 Hz, 1H), 7.39-7.31 (d, J=8.4 Hz, 1H), 6.73 (s, 1H), 6.59-6.54 (d, J=1.0 Hz, 1H), 4.86-4.81 (t, J=5.2 Hz, 1H), 4.35-4.30 (t, J=5.2 Hz, 2H), 4.13 - 4.03 (m, 2H), 3.83 - 3.70 (m, 4H), 2.24 (s, 3H), 2.22-2.08 (ddd, J=2.4, 5.0, 7.8 Hz, 1H), 1.40-1.32 (dd, J=3.6, 7.8 Hz, 1H), 1.02-0.96 (t, J=4.4 Hz, 1H)。MS (ESI) m/z: 500.4 [M+H] +,100% (ee%)。
化合物16B:1HNMR (400 MHz, DMSO-d6) δ 10.70 (s, 1H), 9.06-8.95 (d, J=5.0 Hz, 1H), 8.37 (s, 1H), 8.23-8.18 (d, J=4.2 Hz, 1H), 7.77-7.71 (dd, J=2.2, 8.4 Hz, 1H), 7.68-7.64 (d, J=2.2 Hz, 1H), 7.39-7.31 (d, J=8.4 Hz, 1H), 6.73 (s, 1H), 6.59-6.54 (d, J=1.0 Hz, 1H), 4.86-4.81 (t, J=5.2 Hz, 1H), 4.35-4.30 (t, J=5.2 Hz, 2H), 4.13 - 4.03 (m, 2H), 3.83 - 3.70 (m, 4H), 2.24 (s, 3H), 2.22-2.08 (ddd, J=2.4, 5.0, 7.8 Hz, 1H), 1.40-1.32 (dd, J=3.6, 7.8 Hz, 1H), 1.02-0.96 (t, J=4.4 Hz, 1H)。MS (ESI) m/z: 500.4 [M+H] +,99.7% (ee%)。

-70℃で、アセトニトリル(54.60g、1.33mol)のテトラヒドロフラン(500mL)溶液にn-ブチルリチウム(500mL、2.5M)を加え、1時間攪拌した。-70℃で、当該混合液に11-1(65g、439.22mmol)のテトラヒドロフラン(200mL)溶液を加え、1時間攪拌した後、25℃に昇温させて1時間攪拌した。反応混合物に水(200mL)を加え、酢酸エチル(100mL×2)で抽出し、合わせた抽出溶液を無水硫酸ナトリウムで乾燥させた後濃縮して化合物16B-1の粗成生物を得た。1HNMR (400MHz, CDCl3) δ 7.74 (t, J=7.8 Hz, 1H), 7.43 (d, J=7.4 Hz, 1H), 7.34 (d, J=7.8 Hz, 1H), 3.94 (s, 2H)。
-30℃で、16B-1(130g、852.01mmol)、及び16B-2(80.5g、870.04mmol)のメチルtert-ブチルエーテル(1.2L)溶液にLiHMDS(940.79mL、1M)を滴下し、-10~0℃に昇温させて2時間攪拌した。-30℃で、当該混合液にNaHMDS(855.26mL、1M)を滴下して加え、25℃に昇温させて16時間攪拌した。反応混合物に水(40mL)を加え、反応溶液を濃縮して化合物16B-3の粗成生物を得た。
16B-3(180g、862.71mmol)のエタノール(1L)溶液に水酸化カリウム(900mL、2M)を滴下し、80℃に昇温させて4時間攪拌して化合物16B-4の混合溶液を得た。
50℃で、当該16B-4の混合溶液に塩酸(740mL、6M)を滴下して加え、60℃に昇温させて1時間攪拌した。反応混合物を濃縮した後エタノールを除去し、水相を酢酸エチル(800mL×3)で抽出し、合わせた有機相を飽和食塩水(500mL×1)で洗浄し、無水硫酸ナトリウムで乾燥させた後濃縮し、シリカゲルカラムクロマトグラフィー(PE/EA=20/1~10/1)で分離して化合物16B-5を得た。1HNMR (400 MHz, CDCl3) δ 8.06 (dd, J=0.8, 7.8 Hz, 1H), 7.63 (t, J=7.8 Hz, 1H), 7.16 (dd, J=0.8, 7.8 Hz, 1H), 4.41 (dd, J=4.6, 9.2 Hz, 1H), 4.29 (d, J=9.2 Hz, 1H), 3.02 - 2.84 (m, 1H), 2.12 (dd, J=4.2, 7.8 Hz, 1H), 1.54 - 1.38 (m, 1H)。
-5℃で、16B-5(44g、209.89mmol)のテトラヒドロフラン(400mL)溶液に、水素化ホウ素リチウム(100mL、4M)を加え、20℃に昇温させて2時間攪拌した。反応混合物にゆっくりと飽和塩化アンモニウム水溶液(100mL)を加え、濾過し、濾液を水(60mL×2)で洗浄し、有機相を無水硫酸ナトリウムで乾燥させた後濃縮し、粗成生物をPE/EA(10:1、80mL)でスラリー化させて化合物16B-6を得た。1HNMR (400MHz, CDCl3) δ 7.50 (t, J=7.8 Hz, 1H), 7.07 (dd, J=0.6, 7.8 Hz, 1H), 6.92 (d, J=7.8 Hz, 1H), 4.66 - 4.53 (m, 1H), 4.32 (dd, J=4.6, 8.3 Hz, 1H), 4.10 (ddd, J=5.2, 9.2, 12.4 Hz, 1H), 3.54 - 3.46 (m, 1H), 3.43 - 3.34 (m, 1H), 3.43 - 3.34 (m, 1H), 1.76 - 1.59 (m, 1H), 1.41 (dd, J=5.6, 8.8 Hz, 1H), 0.85 (t, J=5.6 Hz, 1H)。
0℃で、トリフェニルホスフィン(58g、221.13mmol)のテトラヒドロフラン(500mL)溶液にDEAD(45.03g、258.53mmol)を滴下して加え、0℃で30分間攪拌した。0℃で、当該混合溶液に16B-6(40g、187.21mmol)のテトラヒドロフラン(200mL)を加え、25℃に昇温させて1時間攪拌した。反応混合物に水(100mL)と酢酸エチル(300mL)を加え、分層し、有機相を水(100mL×2)で洗浄し、無水硫酸ナトリウムで乾燥させた後濃縮し、カラムクロマトグラフィー(PE/EA=100/1~20/1、V/V)で分離して16B-7を得た。1HNMR (400MHz, CDCl3) δ 7.54 (t, J=7.8 Hz, 1H), 7.12 (dd, J=0.6, 7.8 Hz, 1H), 6.95 (dd, J=0.6, 7.8 Hz, 1H), 4.18 - 4.13 (m, 2H), 3.95 - 3.85 (m, 2H), 2.16 (ddd, J=2.6, 5.2, 8.0 Hz, 1H), 1.40 (dd, J=4.4, 8.0 Hz, 1H), 1.15 (t, J=4.6 Hz, 1H)。
110℃で、カリウムtert-ブトキシド(30.6g、272.70mmol)と1A-2(24g、164.18mmol)のジオキサン(200mL)溶液に16B-7(26.7g、136.47mmol)を加え、110℃で1時間攪拌した。反応溶液を濾過し、濾液を濃縮した後、水(100mL)と酢酸エチル(200mL)を加えた。分層し、有機相を水(100mL×2)で洗浄し、無水硫酸ナトリウムで乾燥させた後濃縮し、カラムクロマトグラフィー(PE/EA=50/1~20/1、V/V)で分離して16B-8を得た。1HNMR (400 MHz, CDCl3) δ 7.50 - 7.42 (m, 1H), 6.59 (t, J=8.0 Hz, 2H), 4.70 (t, J=3.6 Hz, 1H), 4.53 - 4.39 (m, 2H), 4.20 - 4.16 (m, 1H), 4.13 - 4.09 (m, 1H), 4.04 (ddd, J=4.0, 5.8, 11.2 Hz, 1H), 3.92 - 3.84 (m, 3H), 3.80 (ddd, J=4.0, 6.8, 11.2 Hz, 1H), 3.57 - 3.48 (m, 1H), 2.12 - 2.07 (m, 1H), 1.89 - 1.81 (m, 1H), 1.79 - 1.71 (m, 1H), 1.68 - 1.60 (m, 2H), 1.58 - 1.48 (m, 2H), 1.39 (dd, J=4.2, 8.0 Hz, 1H), 1.06 (t, J=4.4 Hz, 1H)。
2-1A-1(36g、141.77mmol)、[Ir(COD)OMe]2(2g、3.02mmol)、及びtmphen(1.6g、6.77mmol)をメチルtert-ブチルエーテル(400mL)に置き、16B-8(40g、130.99mmol)を加えた。反応混合物を窒素ガス保護下で、80℃に加熱して4時間攪拌した。反応溶液を濃縮して化合物16B-9の粗成生物を得た。MS (ESI): m/z 350.4 [M-84+2H]+。
16B-9(52g、120.56mmol)、14-5A(48.53g、135.14mmol)とPd(dppf)Cl2・DCM(10.4g、12.74mmol)、及び炭酸ナトリウム(26g、245.31mmol)をジオキサン(500mL)と水(100mL)に加えた。反応混合物を窒素ガス保護下で、100℃に加熱して3時間攪拌した。反応混合物を濾過し、濾液を濃縮した後、酢酸エチル(400mL)と水(300mL)を加え、分層し、水相を酢酸エチル(100mL×3)で抽出し、合わせた有機相を無水硫酸ナトリウムで乾燥させた後濃縮し、シリカゲルカラムクロマトグラフィー(PE/EA=7/1~3/1)で分離して化合物16B-10を得た。1HNMR (400MHz, CDCl3) δ 8.83 (d, J=5.0 Hz, 1H), 8.11 (br s, 1H), 8.05 (s, 1H), 7.86 (br d, J=4.6 Hz, 1H), 7.53 (br d, J=8.2 Hz, 1H), 7.40 - 7.32 (m, 1H), 7.19 (s, 1H), 6.46 (s, 1H), 6.43 (s, 1H), 4.62 (t, J=3.6 Hz, 1H), 4.51 - 4.35 (m, 2H), 4.13 - 4.06 (m, 1H), 4.05 - 3.97 (m, 2H), 3.86 - 3.71 (m, 4H), 3.51 - 3.40 (m, 1H), 2.18 (s, 3H), 2.05 (ddd, J=2.6, 5.0, 7.8 Hz, 1H), 1.82 - 1.63 (m, 2H), 1.57 - 1.39 (m, 4H), 1.37 - 1.30 (m, 1H), 1.01 (t, J=4.4 Hz, 1H)。
16B-10(60g、102.81mmol)のDMF(60mL)に塩酸(128.51mL、4M)を加え、反応溶液を40℃で2時間攪拌した。反応溶液を水(2.5L)に加え、得られた混合溶液を濾過し、ケーキを乾燥させた後、ジクロロメタンで溶解させ、無水硫酸ナトリウムで乾燥させた後、シリカゲルカラムクロマトグラフィー(PE/EA=3/1~0/1)で分離して、化合物16Bの粗生成物を得、当該粗成生物をPE/EA(4:1、150mL)の混合溶液に置いて攪拌し、濾過して化合物16Bを得た。1HNMR (400 MHz, DMSO-d6) δ 10.70 (s, 1H), 9.06-8.95 (d, J=5.0 Hz, 1H), 8.37 (s, 1H), 8.23-8.18 (d, J=4.2 Hz, 1H), 7.77-7.71 (dd, J=2.2, 8.4 Hz, 1H), 7.68-7.64 (d, J=2.2 Hz, 1H), 7.39-7.31 (d, J=8.4 Hz, 1H), 6.73 (s, 1H), 6.59-6.54 (d, J=1.0 Hz, 1H), 4.86-4.81 (t, J=5.2 Hz, 1H), 4.35-4.30 (t, J=5.2 Hz, 2H), 4.13 - 4.03 (m, 2H), 3.83 - 3.70 (m, 4H), 2.24 (s, 3H), 2.22-2.08 (ddd, J=2.4, 5.0, 7.8 Hz, 1H), 1.40-1.32 (dd, J=3.6, 7.8 Hz, 1H), 1.02-0.96 (t, J=4.4 Hz, 1H)。MS (ESI) m/z: 500.4 [M+H] +,100% ee(キラルカラム:DAICEL CHIRALCEL OJ-H(250mm*30mm,5μm),移動相A:エタノール(0.05%のDIEAを含有);移動相B:二酸化炭素)保持時間:1.592min)。
実験材料:

(1) 化合物の調製:
試験化合物と参照化合物をDMSOで100μMに希釈し、化合物をEchoで3倍に勾配希釈して、11個濃度勾配の標的プレートを得た。
(2) 実験プロセス:
1)緩衝液の調製:50mMのTris-HCl(pH7.4)、3.5mMのMgCl2、150mMのNaCl、1mMのDTT、0.02%のTriton X-100、H2O;
2)緩衝液でMEK1とATPの混合溶液を調製し、384中間プレートに5μLの基質混合溶液を加えた;
3)緩衝液でcRAF酵素を希釈し、384中間プレートに5μLを加えた;
4)Bravoで5μLの反応混合溶液を384実験プレートに移し、15秒間遠心分離した後、23℃の恒温インキュベーターに入れて培養した。
5)1時間後、384プレートに5μLのADP-Gloを加え、振とうし、15秒間遠心分離した後、23℃の恒温インキュベーターに入れて培養した。
6)40分後、384実験プレートに10μLのキナーゼ検出試薬を加え、振とうし、15秒間遠心分離した後、23℃の恒温インキュベーターに入れて培養した。
7)1時間後、Envisionでプレートを読み取った。
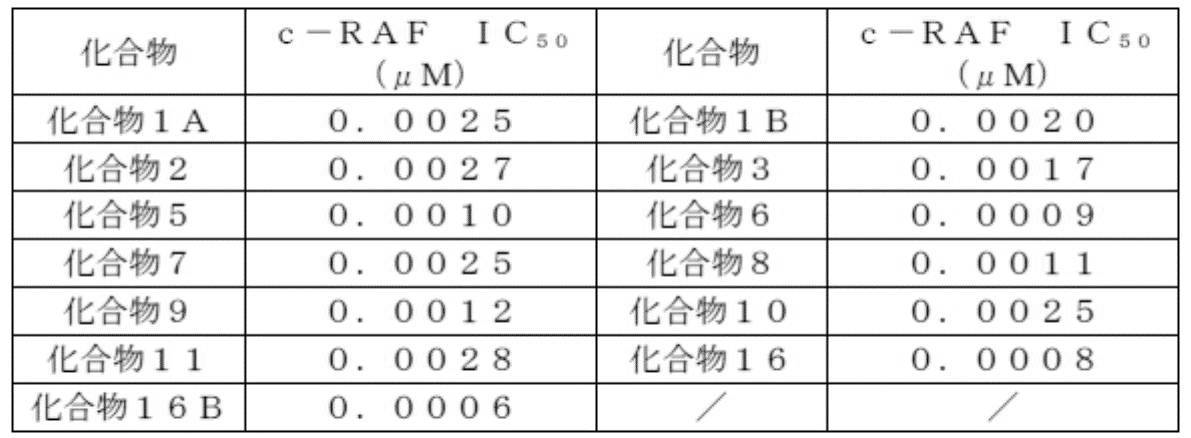
1)実験試薬と消耗品


(1)細胞培地:89%のEMEM、10%のウシ血清、及び1%のペニシリン-ストレプトマイシン;
(2)細胞培養フラスコ内の元の培地を除去し、トリプシンで細胞を消化した後カウントし、細胞懸濁液を培地で、1mlあたり3.75×104細胞のプレーティングに必要な細胞密度に希釈した;
(3)細胞プレートの周りの各ウェルに100μLの培地を加え、他のウェルに80μLの細胞懸濁液を加え、5%のCO2を含む37℃のインキュベーターで一晩培養した。
Echoで化合物を勾配希釈し、薬を投与した後、細胞プレートをインキュベーターに戻させて3日間培養した;
プレートを読み、データを分析:
CTGの添加とプレートの読み取り:細胞プレートの各ウェルに20μLのCellTiterGloを加え、暗所で10分間振とうし、Victor Nivoでプレートを読み取った。

1)実験試薬と消耗品


(1)細胞培地:89%のMc’Coy 5A、10%のウシ血清、及び1%のペニシリン-ストレプトマイシン;
(2)細胞培養フラスコ内の元の培地を除去し、トリプシンで細胞を消化した後カウントし、細胞懸濁液を培地で、1mlあたり2.5×104細胞のプレーティングに必要な細胞密度に希釈した;
(3)細胞プレートの周りの各ウェルに100μLの培地を加え、他のウェルに80μLの細胞懸濁液を加え、5%のCO2を含む37℃のインキュベーターで一晩培養した。
化合物を勾配希釈し、投薬した後、細胞プレートをインキュベーターに戻させて3日間培養した;
プレートを読み、データを分析:
CTGの添加とプレートの読み取り:細胞プレートの各ウェルに20μLのCellTiterGloを加え、暗所で10分間振とうし、Victor Nivoでプレートを読み取った。

1.実験試薬と消耗品



1)細胞を蘇生させて対数増殖期まで培養し、トリプシンで消化して、96ウェルプレートに播種し、インキュベーターで一晩培養した。
2)DMSOで溶解した一連の勾配の化合物を96ウェルプレートに加え、インキュベーターに戻させて、1時間培養した。
3)細胞プレートを取り出し、上清を取り除き、細胞溶解液(1%のブロッキングペプチドを含む)を加え、室温で30分間培養した。
4)ウェルあたり16μLの細胞溶解液をHTRFプレートに移し、次に調製した抗体混合溶液4μLを加えた。
5)一晩の培養した後、Envisionを使用してプレートを読み取り、比率(Ex665/Ex615蛍光強度の比率)にしたがって適合曲線を得、Graphpadの4パラメータフィッティング式Y=Bottom+(Top-Bottom)/(1+10^((LogEC50-X)*HillSlope))によってEC50を計算して得た。

実験試薬と消耗品



1)細胞を蘇生させて対数増殖期まで培養し、トリプシンで消化して、96ウェルプレートに播種し、インキュベーターで一晩培養した。
2)DMSOで溶解した一連の勾配の化合物を96ウェルプレートに加え、インキュベーターに戻させて、1時間培養した。
3)細胞プレートを取り出し、細胞溶解液(1%のブロッキングペプチドを含む)を加え、室温で30分間培養した。
4)ウェルあたり16μLの細胞溶解液をHTRFプレートに移し、次に調製した抗体混合溶液4μLを加えた。
5)一晩の培養した後、Envisionを使用してプレートを読み取り、比率(Ex665/Ex615蛍光強度の比率)にしたがって適合曲線を得、Graphpadの4パラメータフィッティング式Y=Bottom+(Top-Bottom)/(1+10^((LogEC50-X)*HillSlope))によってEC50を計算して得た。

1.1 実験動物と飼育環境
1.1.1 実験動物
種類:マウス
系統:BALB/cヌードマウス
到着年齢:6~8週齢
性別:メス
1.1.2飼育環境
物は、SPF級動物室でIVC(独立した空気供給システム、恒温と一定の湿度)ケージで飼育された(ケージあたり3~5匹)。
温度:20~26℃
湿度:40~70%

細胞:ヒト肺癌Calu-6細胞を体外培養し、EMEM培地に0.2Units/mLのウシインスリン、10%のウシ胎児血清に加え、37℃の5%CO2インキュベーターで培養した。週に2回トリプシン-EDTAで従来の消化処理をし、継代培養した。細胞の飽和度が80%~90%になり、数量が要件に達すると、細胞を収集し、カウントし、播種した。


2.1 腫瘍細胞の接種
細胞の接種:0.2mLのCalu-6細胞(マトリゲルとの比は1:1)を各マウスの右後部に皮下接種し、平均腫瘍体積が173mm3に達した時、群分け投与を開始した。


週に2回ノギスを使用して腫瘍の直径を測定した。腫瘍体積の計算式は、:V=0.5×a×b2であり、aとbは、それぞれ腫瘍の長径と短径を表す。
統計分析は、試験終了時のRTVデータに基づいてSPSSソフトウェアを使用して実行された。群間の比較は、One-way ANOVA分析によって分析され、分散が均一ではない場合(F値に有意差がある場合)、Games-Howell検定を使用して検定した。p<0.05は有意差があると見なされた。
3.1 ヒト肺癌ヌードマウスの皮下移植腫瘍の増殖に対する試験物質の阻害効果
本実験では、ヒト肺癌異種移植腫瘍モデルにおける試験物質の有効性を評価し、溶媒対照群を参照として使用した。異なる時点での各群の腫瘍体積は図1と図3に示された通りである。投与群の化合物1(100mg/kg)は、そのT/Cが18.5%で、TGIが100.9%であり、有意な腫瘍抑制効果があり(P<0.01)、化合物投与後のヒト肺癌Calu-6細胞皮下異種移植腫瘍モデルの担癌マウスにおける腫瘍増殖曲線は図1に示された通りである。投与群の化合物16B(100mg/kg)のT/Cは11.4%で、TGIは99.1%であり、有意な腫瘍抑制効果があった(P<0.01)。化合物投与後のヒト肺癌Calu-6細胞皮下異種移植腫瘍モデルの担癌マウスにおける腫瘍増殖曲線は図3に示された通りである。
マウスの体重と状態は正常であった。マウスの体重に対する試験物の影響は図2と図4に示された通りである。
本発明の化合物は、皮下異種移植腫瘍を有するマウスにおけるヒト肺癌Calu-6細胞の増殖に対して有意な阻害効果を有している。
マウス体内における本発明の化合物の薬物動態パラメータを検出することである。
1)実験医薬品:化合物16B;
2)実験動物:4匹のメスCD-1マウスであり、2つの群に分け、群あたり2匹であった;
3)医薬の調製:適量の薬物を秤量してソルトール(体積の5%)、DMSO(体積5%)とPEG-300(体積の25%)の水溶液に溶解させ、注射投与に使用した。適量の薬物を秤量して5%のソルトール水溶液(体積の80%)に分散させた後、PEG-400(体積の20%)を加え、均一に混合し、胃内投与に使用した。ソルトールはポリエチレングリコール-15-ヒドロキシステアレートである。
第一群の動物には、静脈内注射によって2mg/kgの投与量で1mg/mLの濃度の化合物を投与した。動物は、投与後0.117、0.333、1、2、4、7、及び24時間で血漿サンプルを収集された;第二群の動物には、胃内投与によって100mg/kgの投与量で10mg/mLの濃度の化合物を投与した。動物は、投与後0.0833、0.25、0.5、1、2、4、6、8、及び24時間で血漿サンプルを収集された;各時点での薬物濃度はLC-MS/MS法によって測定され、得られた試験薬物のパラメーターは下記の表に示された通りである:

本発明の化合物は、マウス体内において良好な薬物動態特性を有している。
<付記>
本開示は、以下の態様を含む。
<項1>
式(III)で表される化合物又はその薬学的に許容される塩。

(ただし、
XとYはそれぞれ独立してCH、及びNから選択され;
Lは-O-、-S-、-S(=O)-、及び-S(=O) 2 -から選択され;
L 1 は-CH 2 -、及び単結合から選択され;
Z 1 とZ 2 はそれぞれ独立してCH、及びNから選択され;
R 1 とR 2 はそれぞれ独立してH、F、及びC 1-3 アルキルから選択され、前記C 1-3 アルキルは任意選択で1、2又は3個のR a で置換され;
R 3 は

から選択され;
R 4 はH、及びC 1-3 アルキルから選択され、前記C 1-3 アルキルは任意選択で1、2又は3個のR b で置換され;
R 5 はH、及びC 1-3 アルキルから選択され、前記C 1-3 アルキルは任意選択で1、2又は3個のR c で置換され;
R 6 はH、及びFから選択され;
R 7 はH、及びCNから選択され;
R 8 はH、及びCH 3 から選択され;
R 9 はH、F、及びCH 3 から選択され;
R a はそれぞれ独立してF、Cl、Br、Iから選択され;
R b はそれぞれ独立してF、Cl、Br、I、及びCH 3 から選択され;
R c はそれぞれ独立してF、Cl、Br、及びIから選択される。)
<項2>
化合物が式(I’)から選択される、項1に記載の化合物又はその薬学的に許容される塩。

(ただし、
XとYはそれぞれ独立してCH、及びNから選択され;
Lは-O-、-S-、-S(=O)-、及び-S(=O) 2 -から選択され;
Z 1 とZ 2 はそれぞれ独立してCH、及びNから選択され;
R 1 とR 2 はそれぞれ独立してH、F、及びC 1-3 アルキルから選択され、前記C 1-3 アルキルは任意選択で1、2又は3個のR a で置換され;
R 3 は

から選択され;
R 4 はH、及びC 1-3 アルキルから選択され、前記C 1-3 アルキルは任意選択で1、2又は3個のR b で置換され;
R 5 はH、及びC 1-3 アルキルから選択され、前記C 1-3 アルキルは任意選択で1、2又は3個のR c で置換され;
R 6 はH、及びFから選択され;
R 7 はH、及びCNから選択され;
R 8 はH、及びCH 3 から選択され;
R 9 はH、F、及びCH 3 から選択され;
R a はそれぞれ独立してF、Cl、Br、Iから選択され;
R b はそれぞれ独立してF、Cl、Br、I、及びCH 3 から選択され;
R c はそれぞれ独立してF、Cl、Br、及びIから選択される。)
<項3>
R 1 とR 2 がそれぞれ独立してH、及びFから選択される、項1又は2に記載の化合物又はその薬学的に許容される塩。
<項4>
R 3 が

から選択される、項1又は2に記載の化合物又はその薬学的に許容される塩。
<項5>
R 4 がH、CH 3 、及びCH 2 CH 3 から選択される、項1又は2に記載の化合物又はその薬学的に許容される塩。
<項6>
R 5 がCH 3 から選択される、項1又は2に記載の化合物又はその薬学的に許容される塩。
<項7>
構造単位

から選択される、項1に記載の化合物又はその薬学的に許容される塩。
<項8>
構造単位

から選択される、項1に記載の化合物又はその薬学的に許容される塩。
<項9>
構造単位

から選択される、項8に記載の化合物又はその薬学的に許容される塩。
<項10>
構造単位

から選択される、項1又は2に記載の化合物又はその薬学的に許容される塩。
<項11>
構造単位

から選択される、項1又は2に記載の化合物又はその薬学的に許容される塩。
<項12>
構造単位

から選択される、項1又は2に記載の化合物又はその薬学的に許容される塩。
<項13>
化合物が

から選択される、項1、及び3~6のいずれか1項に記載の化合物又はその薬学的に許容される塩。
(ただし、R 1 、R 2 、R 3 、R 5 、R 6 、R 7 、R 9 、Z 1 とZ 2 は項1~5のいずれか1項に定義された通りである。)
<項14>
化合物が

から選択される、項13に記載の化合物又はその薬学的に許容される塩。
(ただし、R 1 、R 2 、R 3 とR 7 は項13に定義された通りである。)
<項15>
化合物が下記から選択される、化合物又はその薬学的に許容される塩。


<項16>
化合物が下記から選択される、項15に記載の化合物又はその薬学的に許容される塩。

<項17>
式(IV-1)、(IV-2)、(IV-3)、及び(IV-4)で表される化合物又はその薬学的に許容される塩。

(ただし、
X 1 はハロゲン、-SO 2 Me、-OMs、OTf、OTs、

から選択され;
X 2 はハロゲン、OH、-SO 2 Me、-OMs、OTf、OTs、及びHから選択され;
R 1 、R 2 、R 5 、R 6 、R 7 、R 9 、X、Y、Z 1 とZ 2 は項1に定義された通りである。)
<項18>
式(V)で表される化合物又はその薬学的に許容される塩。

(ただし、
X 1 はハロゲン、-SO 2 Me、-OMs、OTf、OTs、

から選択される。)
<項19>
RAFキナーゼ阻害剤の調製における、項1~18のいずれか1項に記載の化合物又はその薬学的に許容される塩の使用。
<項20>
癌を治療する医薬の調製における、項1~18のいずれか1項に記載の化合物又はその薬学的に許容される塩の使用。
Claims (20)
- 式(III)で表される化合物又はその薬学的に許容される塩。

(ただし、
XとYはそれぞれ独立してCH、及びNから選択され;
Lは-O-、-S-、-S(=O)-、及び-S(=O)2-から選択され;
L1は-CH2-、及び単結合から選択され;
Z1とZ2はそれぞれ独立してCH、及びNから選択され;
R1とR2はそれぞれ独立してH、F、及びC1-3アルキルから選択され、前記C1-3アルキルは任意選択で1、2又は3個のRaで置換され;
R3は

から選択され;
R4はH、及びC1-3アルキルから選択され、前記C1-3アルキルは任意選択で1、2又は3個のRbで置換され;
R5はH、及びC1-3アルキルから選択され、前記C1-3アルキルは任意選択で1、2又は3個のRcで置換され;
R6はH、及びFから選択され;
R7はH、及びCNから選択され;
R8はH、及びCH3から選択され;
R9はH、F、及びCH3から選択され;
Raはそれぞれ独立してF、Cl、Br、Iから選択され;
Rbはそれぞれ独立してF、Cl、Br、I、及びCH3から選択され;
Rcはそれぞれ独立してF、Cl、Br、及びIから選択される。) - 化合物が式(I’)から選択される、請求項1に記載の化合物又はその薬学的に許容される塩。

(ただし、
XとYはそれぞれ独立してCH、及びNから選択され;
Lは-O-、-S-、-S(=O)-、及び-S(=O)2-から選択され;
Z1とZ2はそれぞれ独立してCH、及びNから選択され;
R1とR2はそれぞれ独立してH、F、及びC1-3アルキルから選択され、前記C1-3アルキルは任意選択で1、2又は3個のRaで置換され;
R3は
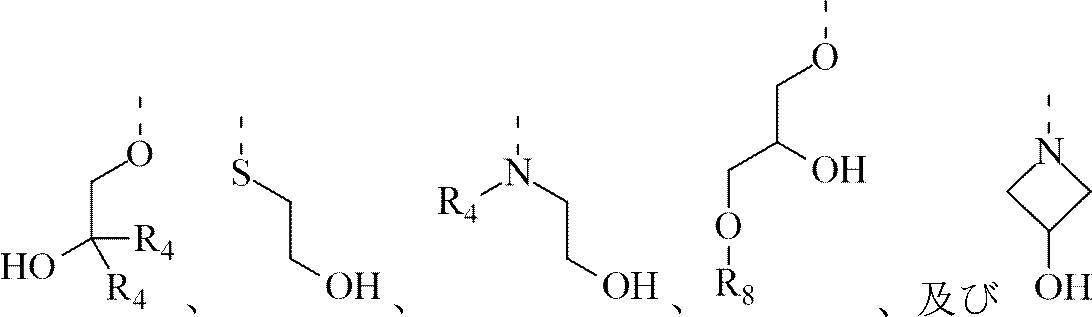
から選択され;
R4はH、及びC1-3アルキルから選択され、前記C1-3アルキルは任意選択で1、2又は3個のRbで置換され;
R5はH、及びC1-3アルキルから選択され、前記C1-3アルキルは任意選択で1、2又は3個のRcで置換され;
R6はH、及びFから選択され;
R7はH、及びCNから選択され;
R8はH、及びCH3から選択され;
R9はH、F、及びCH3から選択され;
Raはそれぞれ独立してF、Cl、Br、Iから選択され;
Rbはそれぞれ独立してF、Cl、Br、I、及びCH3から選択され;
Rcはそれぞれ独立してF、Cl、Br、及びIから選択される。) - R1とR2がそれぞれ独立してH、及びFから選択される、請求項1又は2に記載の化合物又はその薬学的に許容される塩。
- R3が

から選択される、請求項1又は2に記載の化合物又はその薬学的に許容される塩。 - R4がH、CH3、及びCH2CH3から選択される、請求項1又は2に記載の化合物又はその薬学的に許容される塩。
- R5がCH3から選択される、請求項1又は2に記載の化合物又はその薬学的に許容される塩。
- 構造単位

から選択される、請求項1に記載の化合物又はその薬学的に許容される塩。 - 構造単位

から選択される、請求項1に記載の化合物又はその薬学的に許容される塩。 - 構造単位
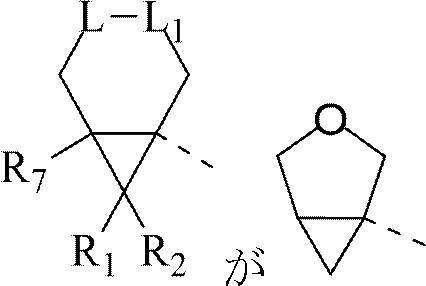
から選択される、請求項8に記載の化合物又はその薬学的に許容される塩。 - 構造単位

から選択される、請求項1又は2に記載の化合物又はその薬学的に許容される塩。 - 構造単位

から選択される、請求項1又は2に記載の化合物又はその薬学的に許容される塩。 - 構造単位

から選択される、請求項1又は2に記載の化合物又はその薬学的に許容される塩。 - 化合物が

から選択される、請求項1、及び3~6のいずれか1項に記載の化合物又はその薬学的に許容される塩。
(ただし、R1、R2、R3、R5、R6、R7、R9、Z1とZ2は請求項1~5のいずれか1項に定義された通りである。) - 化合物が

から選択される、請求項13に記載の化合物又はその薬学的に許容される塩。
(ただし、R1、R2、R3とR7は請求項13に定義された通りである。) - 化合物が下記から選択される、化合物又はその薬学的に許容される塩。


- 化合物が下記から選択される、請求項15に記載の化合物又はその薬学的に許容される塩。

- 式(IV-1)、(IV-2)、(IV-3)、及び(IV-4)で表される化合物又はその薬学的に許容される塩。

(ただし、
X1はハロゲン、-SO2Me、-OMs、OTf、OTs、

から選択され;
X2はハロゲン、OH、-SO2Me、-OMs、OTf、OTs、及びHから選択され;
R1、R2、R5、R6、R7、R9、X、Y、Z1とZ2は請求項1に定義された通りである。) - 式(V)で表される化合物又はその薬学的に許容される塩。

(ただし、
X1はハロゲン、-SO2Me、-OMs、OTf、OTs、

から選択される。) - RAFキナーゼ阻害剤の調製における、請求項1~16のいずれか1項に記載の化合物又はその薬学的に許容される塩の使用。
- 癌を治療する医薬の調製における、請求項1~16のいずれか1項に記載の化合物又はその薬学的に許容される塩の使用。
Applications Claiming Priority (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201911242225 | 2019-12-06 | ||
| CN201911242225.7 | 2019-12-06 | ||
| CN202010367751.2 | 2020-04-30 | ||
| CN202010367751 | 2020-04-30 | ||
| CN202010652149 | 2020-07-08 | ||
| CN202010652149.3 | 2020-07-08 | ||
| PCT/CN2020/133933 WO2021110141A1 (zh) | 2019-12-06 | 2020-12-04 | Pan-RAF激酶抑制剂的联芳基化合物 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2022550219A JP2022550219A (ja) | 2022-11-30 |
| JP7214925B2 true JP7214925B2 (ja) | 2023-01-30 |
Family
ID=76221502
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2022533534A Active JP7214925B2 (ja) | 2019-12-06 | 2020-12-04 | Pan-RAFキナーゼ阻害剤としてのビアリール化合物 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US20230046018A1 (ja) |
| EP (1) | EP4071146A4 (ja) |
| JP (1) | JP7214925B2 (ja) |
| CN (1) | CN114746417B (ja) |
| WO (1) | WO2021110141A1 (ja) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2022253334A1 (zh) * | 2021-06-04 | 2022-12-08 | 南京明德新药研发有限公司 | 一种raf激酶抑制剂的晶型及其制备方法 |
| CN117677398A (zh) | 2021-07-27 | 2024-03-08 | 东丽株式会社 | 用于癌的治疗和/或预防的药品 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2016517417A (ja) | 2013-03-14 | 2016-06-16 | ノバルティス アーゲー | キナーゼ阻害剤としてのビアリールアミド化合物 |
| JP2016523270A (ja) | 2013-06-28 | 2016-08-08 | ベイジーン リミテッド | 複数種類のキナーゼの阻害剤としての縮合三環式アミド系化合物 |
| JP2017527585A (ja) | 2014-09-12 | 2017-09-21 | ノバルティス アーゲー | Rafキナーゼ阻害剤としての化合物および組成物 |
| JP2019517549A (ja) | 2016-06-10 | 2019-06-24 | ノバルティス アーゲー | C−raf阻害薬の治療的使用 |
| WO2019185026A1 (zh) | 2018-03-30 | 2019-10-03 | 南京明德新药研发有限公司 | 作为SGLTs抑制剂的葡糖苷类衍生物及其应用 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104822658B (zh) * | 2013-06-28 | 2017-08-08 | 百济神州有限公司 | 作为多种激酶抑制剂的稠合三环酰胺类化合物 |
| UY36294A (es) * | 2014-09-12 | 2016-04-29 | Novartis Ag | Compuestos y composiciones como inhibidores de quinasa |
| CN109678796B (zh) * | 2017-10-19 | 2023-01-10 | 上海长森药业有限公司 | Pd-1/pd-l1小分子抑制剂及其制备方法和用途 |
| US11098031B1 (en) * | 2019-10-24 | 2021-08-24 | Kinnate Biopharma Inc. | Inhibitors of RAF kinases |
| CN113912591B (zh) * | 2020-07-08 | 2023-10-20 | 齐鲁制药有限公司 | 联芳基化合物 |
-
2020
- 2020-12-04 EP EP20895795.1A patent/EP4071146A4/en not_active Withdrawn
- 2020-12-04 WO PCT/CN2020/133933 patent/WO2021110141A1/zh unknown
- 2020-12-04 JP JP2022533534A patent/JP7214925B2/ja active Active
- 2020-12-04 US US17/781,201 patent/US20230046018A1/en active Pending
- 2020-12-04 CN CN202080084296.3A patent/CN114746417B/zh active Active
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2016517417A (ja) | 2013-03-14 | 2016-06-16 | ノバルティス アーゲー | キナーゼ阻害剤としてのビアリールアミド化合物 |
| JP2016523270A (ja) | 2013-06-28 | 2016-08-08 | ベイジーン リミテッド | 複数種類のキナーゼの阻害剤としての縮合三環式アミド系化合物 |
| JP2017527585A (ja) | 2014-09-12 | 2017-09-21 | ノバルティス アーゲー | Rafキナーゼ阻害剤としての化合物および組成物 |
| JP2019517549A (ja) | 2016-06-10 | 2019-06-24 | ノバルティス アーゲー | C−raf阻害薬の治療的使用 |
| WO2019185026A1 (zh) | 2018-03-30 | 2019-10-03 | 南京明德新药研发有限公司 | 作为SGLTs抑制剂的葡糖苷类衍生物及其应用 |
Also Published As
| Publication number | Publication date |
|---|---|
| US20230046018A1 (en) | 2023-02-16 |
| EP4071146A1 (en) | 2022-10-12 |
| EP4071146A4 (en) | 2023-01-04 |
| CN114746417B (zh) | 2023-12-08 |
| JP2022550219A (ja) | 2022-11-30 |
| WO2021110141A1 (zh) | 2021-06-10 |
| CN114746417A (zh) | 2022-07-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6877407B2 (ja) | Ntrk関連障害の治療に有用な化合物および組成物 | |
| WO2021228161A1 (zh) | 烷氧基烷基取代杂环基类抑制剂及其制备方法和应用 | |
| CN115335372B (zh) | 八氢吡嗪并二氮杂萘啶二酮类化生物 | |
| JP2024505735A (ja) | 置換されたピリダジンフェノール系誘導体 | |
| BR112020001695A2 (pt) | compostos macrocíclicos e usos dos mesmos | |
| AU2020394767B2 (en) | Spiro compound serving as ERK inhibitor, and application thereof | |
| WO2021249563A1 (zh) | 芳基或杂芳基并吡啶酮或嘧啶酮类衍生物及其制备方法和应用 | |
| WO2019165967A1 (zh) | 吡唑并嘧啶衍生物及其用途 | |
| JP2024517965A (ja) | Shp2活性を阻害する複素環化合物、その製造方法および使用 | |
| JP4966186B2 (ja) | 新規ピリミジンヌクレオシド化合物又はその塩 | |
| JP7214925B2 (ja) | Pan-RAFキナーゼ阻害剤としてのビアリール化合物 | |
| CN114829365B (zh) | 作为erk抑制剂的噻唑并内酰胺类化合物及其应用 | |
| JP2017516800A (ja) | 新規な化合物 | |
| CN114787142A (zh) | 作为周期蛋白依赖性激酶9抑制剂的化合物及其应用 | |
| WO2018084321A1 (ja) | Egfr阻害及び腫瘍治療に有用な新規化合物 | |
| CN115403575A (zh) | 杂芳环类衍生物及其制备方法和用途 | |
| CN112955453B (zh) | 作为选择性Trk抑制剂的吡唑并嘧啶衍生物 | |
| CN110066272A (zh) | 取代的苯并[d]咪唑类化合物及其药物组合物 | |
| US11760751B2 (en) | Benzo 2-azaspiro[4.4]nonane compound and use thereof | |
| CN110256408B (zh) | 一种二芳基吡唑化合物及包含该化合物的组合物及其用途 | |
| CN115551842B (zh) | 联苯类化合物 | |
| CN114945576B (zh) | 氘代噻吩并吡啶类化合物 | |
| RU2800042C1 (ru) | Спиросоединения в качестве ингибиторов ERK и их применение |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20220701 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20220701 |
|
| A871 | Explanation of circumstances concerning accelerated examination |
Free format text: JAPANESE INTERMEDIATE CODE: A871 Effective date: 20220701 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20230110 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20230118 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 7214925 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| S111 | Request for change of ownership or part of ownership |
Free format text: JAPANESE INTERMEDIATE CODE: R313113 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |