JP7200928B2 - 熱硬化性樹脂組成物、プリプレグおよび繊維強化複合材料 - Google Patents
熱硬化性樹脂組成物、プリプレグおよび繊維強化複合材料 Download PDFInfo
- Publication number
- JP7200928B2 JP7200928B2 JP2019507971A JP2019507971A JP7200928B2 JP 7200928 B2 JP7200928 B2 JP 7200928B2 JP 2019507971 A JP2019507971 A JP 2019507971A JP 2019507971 A JP2019507971 A JP 2019507971A JP 7200928 B2 JP7200928 B2 JP 7200928B2
- Authority
- JP
- Japan
- Prior art keywords
- thermosetting resin
- resin composition
- groups
- moles
- cyanate
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 229920001187 thermosetting polymer Polymers 0.000 title claims description 124
- 239000011342 resin composition Substances 0.000 title claims description 107
- 239000003733 fiber-reinforced composite Substances 0.000 title claims description 26
- 229920005989 resin Polymers 0.000 claims description 107
- 239000011347 resin Substances 0.000 claims description 107
- 239000003822 epoxy resin Substances 0.000 claims description 72
- 229920000647 polyepoxide Polymers 0.000 claims description 72
- 239000004643 cyanate ester Substances 0.000 claims description 58
- -1 Aromatic amine compound Chemical class 0.000 claims description 43
- XLJMAIOERFSOGZ-UHFFFAOYSA-M cyanate group Chemical group [O-]C#N XLJMAIOERFSOGZ-UHFFFAOYSA-M 0.000 claims description 40
- 125000003055 glycidyl group Chemical group C(C1CO1)* 0.000 claims description 39
- 239000000463 material Substances 0.000 claims description 38
- 239000012783 reinforcing fiber Substances 0.000 claims description 31
- IISBACLAFKSPIT-UHFFFAOYSA-N bisphenol A Chemical compound C=1C=C(O)C=CC=1C(C)(C)C1=CC=C(O)C=C1 IISBACLAFKSPIT-UHFFFAOYSA-N 0.000 claims description 26
- 239000001257 hydrogen Substances 0.000 claims description 26
- 229910052739 hydrogen Inorganic materials 0.000 claims description 26
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 22
- 125000003277 amino group Chemical group 0.000 claims description 20
- 239000004593 Epoxy Substances 0.000 claims description 19
- 238000012360 testing method Methods 0.000 claims description 15
- 239000007787 solid Substances 0.000 claims description 12
- 238000003860 storage Methods 0.000 claims description 12
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 claims description 10
- PXKLMJQFEQBVLD-UHFFFAOYSA-N bisphenol F Chemical compound C1=CC(O)=CC=C1CC1=CC=C(O)C=C1 PXKLMJQFEQBVLD-UHFFFAOYSA-N 0.000 claims description 9
- 229920003986 novolac Polymers 0.000 claims description 9
- AFEQENGXSMURHA-UHFFFAOYSA-N oxiran-2-ylmethanamine Chemical compound NCC1CO1 AFEQENGXSMURHA-UHFFFAOYSA-N 0.000 claims description 8
- 239000004695 Polyether sulfone Substances 0.000 claims description 7
- 229920006393 polyether sulfone Polymers 0.000 claims description 7
- MQJKPEGWNLWLTK-UHFFFAOYSA-N Dapsone Chemical compound C1=CC(N)=CC=C1S(=O)(=O)C1=CC=C(N)C=C1 MQJKPEGWNLWLTK-UHFFFAOYSA-N 0.000 claims description 6
- 150000001412 amines Chemical class 0.000 claims description 6
- 238000010438 heat treatment Methods 0.000 claims description 6
- 238000000113 differential scanning calorimetry Methods 0.000 claims description 3
- 150000001913 cyanates Chemical class 0.000 claims description 2
- 238000010521 absorption reaction Methods 0.000 description 42
- 238000000034 method Methods 0.000 description 32
- 230000000052 comparative effect Effects 0.000 description 29
- 230000009477 glass transition Effects 0.000 description 26
- 238000001723 curing Methods 0.000 description 19
- LNEPOXFFQSENCJ-UHFFFAOYSA-N haloperidol Chemical compound C1CC(O)(C=2C=CC(Cl)=CC=2)CCN1CCCC(=O)C1=CC=C(F)C=C1 LNEPOXFFQSENCJ-UHFFFAOYSA-N 0.000 description 19
- 229920005992 thermoplastic resin Polymers 0.000 description 17
- 239000011159 matrix material Substances 0.000 description 14
- 239000000835 fiber Substances 0.000 description 11
- 238000005259 measurement Methods 0.000 description 11
- 239000000126 substance Substances 0.000 description 10
- 229920000049 Carbon (fiber) Polymers 0.000 description 9
- 239000004917 carbon fiber Substances 0.000 description 9
- 238000006243 chemical reaction Methods 0.000 description 9
- 239000002245 particle Substances 0.000 description 9
- 230000008719 thickening Effects 0.000 description 9
- 238000002156 mixing Methods 0.000 description 8
- 238000000465 moulding Methods 0.000 description 8
- 230000009257 reactivity Effects 0.000 description 8
- 238000004519 manufacturing process Methods 0.000 description 7
- 239000004952 Polyamide Substances 0.000 description 6
- 238000004898 kneading Methods 0.000 description 6
- 239000007788 liquid Substances 0.000 description 6
- 229920002647 polyamide Polymers 0.000 description 6
- 230000008569 process Effects 0.000 description 6
- 125000005843 halogen group Chemical group 0.000 description 5
- 239000000203 mixture Substances 0.000 description 5
- 239000003960 organic solvent Substances 0.000 description 5
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 4
- 125000004432 carbon atom Chemical group C* 0.000 description 4
- 150000001875 compounds Chemical class 0.000 description 4
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical group C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 4
- 125000000524 functional group Chemical group 0.000 description 4
- XSQUKJJJFZCRTK-UHFFFAOYSA-N isourea group Chemical group NC(O)=N XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 4
- 150000002576 ketones Chemical class 0.000 description 4
- 239000010410 layer Substances 0.000 description 4
- IGALFTFNPPBUDN-UHFFFAOYSA-N phenyl-[2,3,4,5-tetrakis(oxiran-2-ylmethyl)phenyl]methanediamine Chemical compound C=1C(CC2OC2)=C(CC2OC2)C(CC2OC2)=C(CC2OC2)C=1C(N)(N)C1=CC=CC=C1 IGALFTFNPPBUDN-UHFFFAOYSA-N 0.000 description 4
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 4
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 3
- LJGHYPLBDBRCRZ-UHFFFAOYSA-N 3-(3-aminophenyl)sulfonylaniline Chemical compound NC1=CC=CC(S(=O)(=O)C=2C=C(N)C=CC=2)=C1 LJGHYPLBDBRCRZ-UHFFFAOYSA-N 0.000 description 3
- AHIPJALLQVEEQF-UHFFFAOYSA-N 4-(oxiran-2-ylmethoxy)-n,n-bis(oxiran-2-ylmethyl)aniline Chemical compound C1OC1COC(C=C1)=CC=C1N(CC1OC1)CC1CO1 AHIPJALLQVEEQF-UHFFFAOYSA-N 0.000 description 3
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- 229920003319 Araldite® Polymers 0.000 description 3
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 3
- 125000002723 alicyclic group Chemical group 0.000 description 3
- 239000000470 constituent Substances 0.000 description 3
- 230000000694 effects Effects 0.000 description 3
- 238000013007 heat curing Methods 0.000 description 3
- 239000012943 hotmelt Substances 0.000 description 3
- 230000000704 physical effect Effects 0.000 description 3
- 229920000090 poly(aryl ether) Polymers 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- PISLZQACAJMAIO-UHFFFAOYSA-N 2,4-diethyl-6-methylbenzene-1,3-diamine Chemical compound CCC1=CC(C)=C(N)C(CC)=C1N PISLZQACAJMAIO-UHFFFAOYSA-N 0.000 description 2
- IWRZKNMUSBNOOD-UHFFFAOYSA-N 2-methyl-4-(oxiran-2-ylmethoxy)-n,n-bis(oxiran-2-ylmethyl)aniline Chemical compound C=1C=C(N(CC2OC2)CC2OC2)C(C)=CC=1OCC1CO1 IWRZKNMUSBNOOD-UHFFFAOYSA-N 0.000 description 2
- QTWJRLJHJPIABL-UHFFFAOYSA-N 2-methylphenol;3-methylphenol;4-methylphenol Chemical compound CC1=CC=C(O)C=C1.CC1=CC=CC(O)=C1.CC1=CC=CC=C1O QTWJRLJHJPIABL-UHFFFAOYSA-N 0.000 description 2
- VAGOJLCWTUPBKD-UHFFFAOYSA-N 3-(oxiran-2-ylmethoxy)-n,n-bis(oxiran-2-ylmethyl)aniline Chemical compound C1OC1COC(C=1)=CC=CC=1N(CC1OC1)CC1CO1 VAGOJLCWTUPBKD-UHFFFAOYSA-N 0.000 description 2
- PMPLQTWAQNSOSY-UHFFFAOYSA-N 4-[[4-[bis(oxiran-2-ylmethyl)amino]-3-ethylphenyl]methyl]-2-ethyl-n,n-bis(oxiran-2-ylmethyl)aniline Chemical compound C=1C=C(N(CC2OC2)CC2OC2)C(CC)=CC=1CC(C=C1CC)=CC=C1N(CC1OC1)CC1CO1 PMPLQTWAQNSOSY-UHFFFAOYSA-N 0.000 description 2
- FAUAZXVRLVIARB-UHFFFAOYSA-N 4-[[4-[bis(oxiran-2-ylmethyl)amino]phenyl]methyl]-n,n-bis(oxiran-2-ylmethyl)aniline Chemical compound C1OC1CN(C=1C=CC(CC=2C=CC(=CC=2)N(CC2OC2)CC2OC2)=CC=1)CC1CO1 FAUAZXVRLVIARB-UHFFFAOYSA-N 0.000 description 2
- 229930185605 Bisphenol Natural products 0.000 description 2
- 239000004697 Polyetherimide Substances 0.000 description 2
- 125000001931 aliphatic group Chemical group 0.000 description 2
- 235000010290 biphenyl Nutrition 0.000 description 2
- 239000004305 biphenyl Substances 0.000 description 2
- 229930003836 cresol Natural products 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- 230000007613 environmental effect Effects 0.000 description 2
- 125000003700 epoxy group Chemical group 0.000 description 2
- 125000001033 ether group Chemical group 0.000 description 2
- 238000011156 evaluation Methods 0.000 description 2
- 230000001747 exhibiting effect Effects 0.000 description 2
- 239000003365 glass fiber Substances 0.000 description 2
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 2
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 2
- 230000001771 impaired effect Effects 0.000 description 2
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- 125000001624 naphthyl group Chemical group 0.000 description 2
- 239000004843 novolac epoxy resin Substances 0.000 description 2
- 238000007344 nucleophilic reaction Methods 0.000 description 2
- 238000013001 point bending Methods 0.000 description 2
- 229920001601 polyetherimide Polymers 0.000 description 2
- 238000002360 preparation method Methods 0.000 description 2
- 239000012798 spherical particle Substances 0.000 description 2
- 229910001220 stainless steel Inorganic materials 0.000 description 2
- 239000010935 stainless steel Substances 0.000 description 2
- 239000002759 woven fabric Substances 0.000 description 2
- HCNHNBLSNVSJTJ-UHFFFAOYSA-N 1,1-Bis(4-hydroxyphenyl)ethane Chemical compound C=1C=C(O)C=CC=1C(C)C1=CC=C(O)C=C1 HCNHNBLSNVSJTJ-UHFFFAOYSA-N 0.000 description 1
- JYEUMXHLPRZUAT-UHFFFAOYSA-N 1,2,3-triazine Chemical group C1=CN=NN=C1 JYEUMXHLPRZUAT-UHFFFAOYSA-N 0.000 description 1
- OUPZKGBUJRBPGC-UHFFFAOYSA-N 1,3,5-tris(oxiran-2-ylmethyl)-1,3,5-triazinane-2,4,6-trione Chemical compound O=C1N(CC2OC2)C(=O)N(CC2OC2)C(=O)N1CC1CO1 OUPZKGBUJRBPGC-UHFFFAOYSA-N 0.000 description 1
- WZCQRUWWHSTZEM-UHFFFAOYSA-N 1,3-phenylenediamine Chemical compound NC1=CC=CC(N)=C1 WZCQRUWWHSTZEM-UHFFFAOYSA-N 0.000 description 1
- HECLRDQVFMWTQS-RGOKHQFPSA-N 1755-01-7 Chemical group C1[C@H]2[C@@H]3CC=C[C@@H]3[C@@H]1C=C2 HECLRDQVFMWTQS-RGOKHQFPSA-N 0.000 description 1
- ZJRAAAWYHORFHN-UHFFFAOYSA-N 2-[[2,6-dibromo-4-[2-[3,5-dibromo-4-(oxiran-2-ylmethoxy)phenyl]propan-2-yl]phenoxy]methyl]oxirane Chemical compound C=1C(Br)=C(OCC2OC2)C(Br)=CC=1C(C)(C)C(C=C1Br)=CC(Br)=C1OCC1CO1 ZJRAAAWYHORFHN-UHFFFAOYSA-N 0.000 description 1
- UUODQIKUTGWMPT-UHFFFAOYSA-N 2-fluoro-5-(trifluoromethyl)pyridine Chemical compound FC1=CC=C(C(F)(F)F)C=N1 UUODQIKUTGWMPT-UHFFFAOYSA-N 0.000 description 1
- OVEUFHOBGCSKSH-UHFFFAOYSA-N 2-methyl-n,n-bis(oxiran-2-ylmethyl)aniline Chemical compound CC1=CC=CC=C1N(CC1OC1)CC1OC1 OVEUFHOBGCSKSH-UHFFFAOYSA-N 0.000 description 1
- WDGCBNTXZHJTHJ-UHFFFAOYSA-N 2h-1,3-oxazol-2-id-4-one Chemical group O=C1CO[C-]=N1 WDGCBNTXZHJTHJ-UHFFFAOYSA-N 0.000 description 1
- CKOFBUUFHALZGK-UHFFFAOYSA-N 3-[(3-aminophenyl)methyl]aniline Chemical compound NC1=CC=CC(CC=2C=C(N)C=CC=2)=C1 CKOFBUUFHALZGK-UHFFFAOYSA-N 0.000 description 1
- RDIGYBZNNOGMHU-UHFFFAOYSA-N 3-amino-2,4,5-tris(oxiran-2-ylmethyl)phenol Chemical compound OC1=CC(CC2OC2)=C(CC2OC2)C(N)=C1CC1CO1 RDIGYBZNNOGMHU-UHFFFAOYSA-N 0.000 description 1
- YBRVSVVVWCFQMG-UHFFFAOYSA-N 4,4'-diaminodiphenylmethane Chemical compound C1=CC(N)=CC=C1CC1=CC=C(N)C=C1 YBRVSVVVWCFQMG-UHFFFAOYSA-N 0.000 description 1
- VPWNQTHUCYMVMZ-UHFFFAOYSA-N 4,4'-sulfonyldiphenol Chemical compound C1=CC(O)=CC=C1S(=O)(=O)C1=CC=C(O)C=C1 VPWNQTHUCYMVMZ-UHFFFAOYSA-N 0.000 description 1
- CBEVWPCAHIAUOD-UHFFFAOYSA-N 4-[(4-amino-3-ethylphenyl)methyl]-2-ethylaniline Chemical compound C1=C(N)C(CC)=CC(CC=2C=C(CC)C(N)=CC=2)=C1 CBEVWPCAHIAUOD-UHFFFAOYSA-N 0.000 description 1
- FVCSARBUZVPSQF-UHFFFAOYSA-N 5-(2,4-dioxooxolan-3-yl)-7-methyl-3a,4,5,7a-tetrahydro-2-benzofuran-1,3-dione Chemical compound C1C(C(OC2=O)=O)C2C(C)=CC1C1C(=O)COC1=O FVCSARBUZVPSQF-UHFFFAOYSA-N 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- KWYHDKDOAIKMQN-UHFFFAOYSA-N N,N,N',N'-tetramethylethylenediamine Chemical compound CN(C)CCN(C)C KWYHDKDOAIKMQN-UHFFFAOYSA-N 0.000 description 1
- 229920000571 Nylon 11 Polymers 0.000 description 1
- 229920000299 Nylon 12 Polymers 0.000 description 1
- 229920002292 Nylon 6 Polymers 0.000 description 1
- 229920000572 Nylon 6/12 Polymers 0.000 description 1
- 240000007594 Oryza sativa Species 0.000 description 1
- 235000007164 Oryza sativa Nutrition 0.000 description 1
- 239000004696 Poly ether ether ketone Substances 0.000 description 1
- 229920000491 Polyphenylsulfone Polymers 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- FDLQZKYLHJJBHD-UHFFFAOYSA-N [3-(aminomethyl)phenyl]methanamine Chemical compound NCC1=CC=CC(CN)=C1 FDLQZKYLHJJBHD-UHFFFAOYSA-N 0.000 description 1
- 150000008065 acid anhydrides Chemical class 0.000 description 1
- 239000000853 adhesive Substances 0.000 description 1
- 230000001070 adhesive effect Effects 0.000 description 1
- 239000004840 adhesive resin Substances 0.000 description 1
- 229920006223 adhesive resin Polymers 0.000 description 1
- 239000002390 adhesive tape Substances 0.000 description 1
- 239000004760 aramid Substances 0.000 description 1
- 229920006231 aramid fiber Polymers 0.000 description 1
- 230000006399 behavior Effects 0.000 description 1
- 238000005452 bending Methods 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 239000006229 carbon black Substances 0.000 description 1
- 229910021393 carbon nanotube Inorganic materials 0.000 description 1
- 239000002041 carbon nanotube Substances 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 238000010538 cationic polymerization reaction Methods 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 239000000460 chlorine Substances 0.000 description 1
- 239000004927 clay Substances 0.000 description 1
- 229910052570 clay Inorganic materials 0.000 description 1
- 239000002131 composite material Substances 0.000 description 1
- 239000000805 composite resin Substances 0.000 description 1
- 238000004883 computer application Methods 0.000 description 1
- 238000010276 construction Methods 0.000 description 1
- 238000005260 corrosion Methods 0.000 description 1
- 230000007797 corrosion Effects 0.000 description 1
- 239000007822 coupling agent Substances 0.000 description 1
- ZSWFCLXCOIISFI-UHFFFAOYSA-N cyclopentadiene Chemical group C1C=CC=C1 ZSWFCLXCOIISFI-UHFFFAOYSA-N 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000002542 deteriorative effect Effects 0.000 description 1
- QGBSISYHAICWAH-UHFFFAOYSA-N dicyandiamide Chemical compound NC(N)=NC#N QGBSISYHAICWAH-UHFFFAOYSA-N 0.000 description 1
- 239000005007 epoxy-phenolic resin Substances 0.000 description 1
- 230000002349 favourable effect Effects 0.000 description 1
- 238000009730 filament winding Methods 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 229910021389 graphene Inorganic materials 0.000 description 1
- 239000010439 graphite Substances 0.000 description 1
- 229910002804 graphite Inorganic materials 0.000 description 1
- 238000009787 hand lay-up Methods 0.000 description 1
- 150000002431 hydrogen Chemical class 0.000 description 1
- 150000002460 imidazoles Chemical class 0.000 description 1
- 238000005470 impregnation Methods 0.000 description 1
- 238000001746 injection moulding Methods 0.000 description 1
- 239000011256 inorganic filler Substances 0.000 description 1
- 229910003475 inorganic filler Inorganic materials 0.000 description 1
- 239000011229 interlayer Substances 0.000 description 1
- 238000010030 laminating Methods 0.000 description 1
- 229940018564 m-phenylenediamine Drugs 0.000 description 1
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Chemical compound C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 1
- JAYXSROKFZAHRQ-UHFFFAOYSA-N n,n-bis(oxiran-2-ylmethyl)aniline Chemical compound C1OC1CN(C=1C=CC=CC=1)CC1CO1 JAYXSROKFZAHRQ-UHFFFAOYSA-N 0.000 description 1
- SJPFBRJHYRBAGV-UHFFFAOYSA-N n-[[3-[[bis(oxiran-2-ylmethyl)amino]methyl]phenyl]methyl]-1-(oxiran-2-yl)-n-(oxiran-2-ylmethyl)methanamine Chemical compound C1OC1CN(CC=1C=C(CN(CC2OC2)CC2OC2)C=CC=1)CC1CO1 SJPFBRJHYRBAGV-UHFFFAOYSA-N 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- 229920001568 phenolic resin Polymers 0.000 description 1
- 229920002492 poly(sulfone) Polymers 0.000 description 1
- 229920002530 polyetherether ketone Polymers 0.000 description 1
- 229920005649 polyetherethersulfone Polymers 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 239000003505 polymerization initiator Substances 0.000 description 1
- 229920001955 polyphenylene ether Polymers 0.000 description 1
- 150000003141 primary amines Chemical class 0.000 description 1
- 239000011208 reinforced composite material Substances 0.000 description 1
- 235000009566 rice Nutrition 0.000 description 1
- 150000003335 secondary amines Chemical class 0.000 description 1
- 238000004904 shortening Methods 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 150000003512 tertiary amines Chemical class 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- 238000001721 transfer moulding Methods 0.000 description 1
- 230000007704 transition Effects 0.000 description 1
- 150000003672 ureas Chemical class 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G59/00—Polycondensates containing more than one epoxy group per molecule; Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups
- C08G59/18—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing
- C08G59/40—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing characterised by the curing agents used
- C08G59/50—Amines
- C08G59/56—Amines together with other curing agents
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G59/00—Polycondensates containing more than one epoxy group per molecule; Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups
- C08G59/18—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing
- C08G59/20—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing characterised by the epoxy compounds used
- C08G59/32—Epoxy compounds containing three or more epoxy groups
- C08G59/3227—Compounds containing acyclic nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L63/00—Compositions of epoxy resins; Compositions of derivatives of epoxy resins
- C08L63/10—Epoxy resins modified by unsaturated compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G59/00—Polycondensates containing more than one epoxy group per molecule; Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups
- C08G59/18—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing
- C08G59/20—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing characterised by the epoxy compounds used
- C08G59/32—Epoxy compounds containing three or more epoxy groups
- C08G59/3218—Carbocyclic compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G59/00—Polycondensates containing more than one epoxy group per molecule; Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups
- C08G59/18—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing
- C08G59/40—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing characterised by the curing agents used
- C08G59/4007—Curing agents not provided for by the groups C08G59/42 - C08G59/66
- C08G59/4014—Nitrogen containing compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G59/00—Polycondensates containing more than one epoxy group per molecule; Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups
- C08G59/18—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing
- C08G59/40—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing characterised by the curing agents used
- C08G59/4007—Curing agents not provided for by the groups C08G59/42 - C08G59/66
- C08G59/4014—Nitrogen containing compounds
- C08G59/4028—Isocyanates; Thioisocyanates
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G59/00—Polycondensates containing more than one epoxy group per molecule; Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups
- C08G59/18—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing
- C08G59/40—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing characterised by the curing agents used
- C08G59/50—Amines
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G59/00—Polycondensates containing more than one epoxy group per molecule; Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups
- C08G59/18—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing
- C08G59/40—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing characterised by the curing agents used
- C08G59/50—Amines
- C08G59/504—Amines containing an atom other than nitrogen belonging to the amine group, carbon and hydrogen
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J5/00—Manufacture of articles or shaped materials containing macromolecular substances
- C08J5/04—Reinforcing macromolecular compounds with loose or coherent fibrous material
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J5/00—Manufacture of articles or shaped materials containing macromolecular substances
- C08J5/24—Impregnating materials with prepolymers which can be polymerised in situ, e.g. manufacture of prepregs
- C08J5/241—Impregnating materials with prepolymers which can be polymerised in situ, e.g. manufacture of prepregs using inorganic fibres
- C08J5/243—Impregnating materials with prepolymers which can be polymerised in situ, e.g. manufacture of prepregs using inorganic fibres using carbon fibres
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/16—Nitrogen-containing compounds
- C08K5/17—Amines; Quaternary ammonium compounds
- C08K5/18—Amines; Quaternary ammonium compounds with aromatically bound amino groups
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L63/00—Compositions of epoxy resins; Compositions of derivatives of epoxy resins
- C08L63/04—Epoxynovolacs
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L79/00—Compositions of macromolecular compounds obtained by reactions forming in the main chain of the macromolecule a linkage containing nitrogen with or without oxygen or carbon only, not provided for in groups C08L61/00 - C08L77/00
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L81/00—Compositions of macromolecular compounds obtained by reactions forming in the main chain of the macromolecule a linkage containing sulfur with or without nitrogen, oxygen or carbon only; Compositions of polysulfones; Compositions of derivatives of such polymers
- C08L81/06—Polysulfones; Polyethersulfones
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2363/00—Characterised by the use of epoxy resins; Derivatives of epoxy resins
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2379/00—Characterised by the use of macromolecular compounds obtained by reactions forming in the main chain of the macromolecule a linkage containing nitrogen with or without oxygen, or carbon only, not provided for in groups C08J2361/00 - C08J2377/00
- C08J2379/04—Polycondensates having nitrogen-containing heterocyclic rings in the main chain; Polyhydrazides; Polyamide acids or similar polyimide precursors
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2463/00—Characterised by the use of epoxy resins; Derivatives of epoxy resins
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2479/00—Characterised by the use of macromolecular compounds obtained by reactions forming in the main chain of the macromolecule a linkage containing nitrogen with or without oxygen, or carbon only, not provided for in groups C08J2461/00 - C08J2477/00
- C08J2479/04—Polycondensates having nitrogen-containing heterocyclic rings in the main chain; Polyhydrazides; Polyamide acids or similar polyimide precursors
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2481/00—Characterised by the use of macromolecular compounds obtained by reactions forming in the main chain of the macromolecule a linkage containing sulfur with or without nitrogen, oxygen, or carbon only; Polysulfones; Derivatives of such polymers
- C08J2481/06—Polysulfones; Polyethersulfones
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Manufacturing & Machinery (AREA)
- Inorganic Chemistry (AREA)
- Reinforced Plastic Materials (AREA)
- Epoxy Resins (AREA)
- Compositions Of Macromolecular Compounds (AREA)
Description
少なくとも次の構成要素[A]~[C]を含み、以下の条件(1)と(2)を満たし、さらにポリエーテルスルホンを含む熱硬化性樹脂組成物、である。
[A]グリシジルアミン型エポキシ、ビスフェノールA型エポキシ、ビスフェノールF型エポキシ、フェノールノボラック型エポキシから選ばれる少なくとも1種のグリシジル基を2つ以上有するエポキシ樹脂
[B]ビスフェノールA型シアネートエステルとフェノールノボラック型シアネートエステルから選ばれる少なくとも1種のシアネート基を2つ以上有するシアネートエステル樹脂
[C]アミノ基を二つ以上有する芳香族アミン化合物
(1)0.25≦熱硬化性樹脂組成物中のグリシジル基のモル数/熱硬化性樹脂組成物中のシアネート基のモル数≦1.5
(2)0.05≦熱硬化性樹脂組成物中のアミノ基に含まれる活性水素のモル数/熱硬化性樹脂組成物中のシアネート基のモル数≦0.5。
上記の熱硬化性樹脂組成物が強化繊維に含浸されてなるプリプレグ、である。
上記のプリプレグが硬化されてなる繊維強化複合材料、
または、
強化繊維と上記の熱硬化性樹脂組成物が硬化されてなる樹脂硬化物を含んでなる繊維強化複合材料、である。

本発明の熱硬化性樹脂組成物は、ポリエーテルスルホンを熱硬化性樹脂組成物中に1~30質量%含むことが好ましい。
少なくとも次の構成要素[A]~[C]を含み、以下の条件(1)と(2)を満たす熱硬化性樹脂組成物。
[A]グリシジル基を2つ以上有するエポキシ樹脂
[B]シアネート基を2つ以上有するシアネートエステル樹脂
[C]アミン化合物
(1)0.25≦熱硬化性樹脂組成物中のエポキシ基のモル数/熱硬化性樹脂組成物中のシアネート基のモル数≦1.5
(2)0.05≦熱硬化性樹脂組成物中のアミノ基に含まれる活性水素のモル数/熱硬化性樹脂組成物中のシアネート基のモル数≦0.5。
本発明における構成要素[C]は、アミン化合物である。25℃で固形のアミン化合物であると、樹脂混練工程や、プリプレグ等の中間基材製造工程等での良好なポットライフが得られるため好ましい。さらに、2個以上のアミノ基を有する芳香族アミン化合物であると、架橋構造を形成することができ、得られる化学構造も剛直となるため、高いガラス転移温度を有する熱硬化性樹脂硬化物が得られ、好ましい。構成要素[C]としては、例えば、3,3’-ジイソプロピル-4,4’-ジアミノジフェニルメタン、3,3’-ジ-t-ブチル-4,4’-ジアミノジフェニルメタン、3,3’,5,5’-テトラエチル-4,4’-ジアミノジフェニルメタン、3,3’-ジアミノジフェニルメタン、4,4’-ジアミノジフェニルメタン、3,3’-ジイソプロピル-4,4’-ジアミノジフェニルケトン、3,3’-ジ-t-ブチル-4,4’-ジアミノジフェニルケトン、3,3’,5,5’-テトラエチル-4,4’-ジアミノジフェニルケトン、4,4’-ジアミノジフェニルケトン、3,3’-ジアミノジフェニルケトン、3,3’-ジイソプロピル-4,4’-ジアミノジフェニルスルホン、3,3’-ジ-t-ブチル-4,4’-ジアミノジフェニルスルホン、3,3’,5,5’-テトラエチル-4,4’-ジアミノジフェニルスルホン、4,4’-ジアミノジフェニルスルホン、3,3’-ジアミノジフェニルスルホン、m-フェニレンジアミン、m-キシリレンジアミン、ジエチルトルエンジアミンなどが挙げられる。
(1)0.25≦熱硬化性樹脂組成物中のグリシジル基のモル数/熱硬化性樹脂組成物中のシアネート基のモル数≦1.5
(2)0.05≦熱硬化性樹脂組成物中のアミノ基に含まれる活性水素のモル数/熱硬化性樹脂組成物中のシアネート基のモル数≦0.5。
構成要素[A]のエポキシ樹脂が有するグリシジル基のモル数=構成要素[A]のエポキシ樹脂の質量部数/構成要素[A]のエポキシ樹脂のエポキシ当量。
構成要素[A]のエポキシ樹脂が有するグリシジル基のモル数=成分1のエポキシ樹脂の質量部数/成分1のエポキシ樹脂のエポキシ当量+成分2のエポキシ樹脂の質量部数/成分2のエポキシ樹脂のエポキシ当量。
構成要素[B]のシアネートエステル樹脂が有するシアネート基のモル数=構成要素[B]のシアネートエステル樹脂の質量部数/構成要素[B]のシアネートエステル樹脂のシアネート当量。
構成要素[C]のアミン化合物の活性水素のモル数=構成要素[C]のアミン化合物の質量部数/構成要素[C]のアミン化合物の活性水素当量。
(1)構成要素[A]:グリシジル基を2つ以上有するエポキシ樹脂
・ビスフェノールA型エポキシ樹脂(“jER”(登録商標)828、三菱ケミカル(株)製)エポキシ当量:189(g/eq.)、グリシジル基数:2)
・ビスフェノールF型エポキシ樹脂(“EPICLON”(登録商標)830、DIC(株)製)エポキシ当量:172(g/eq.)、グリシジル基数:2)
・テトラグリシジルジアミノジフェニルメタン(“アラルダイト”(登録商標)MY721、ハンツマン・アドバンスト・マテリアルズ社製)エポキシ当量:113(g/eq.)、グリシジル基数:4)
・トリグリシジル-m-アミノフェノール(“アラルダイト”(登録商標)MY0600、ハンツマン・アドバンスト・マテリアルズ社製)エポキシ当量:106(g/eq.)、グリシジル基数:3)
・トリグリシジル-p-アミノフェノール(“アラルダイト”(登録商標)MY0500、ハンツマン・アドバンスト・マテリアルズ社製)エポキシ当量:106(g/eq.)、グリシジル基数:3)
・フェノールノボラック型エポキシ樹脂(“jER”(登録商標)154、三菱ケミカル(株)製)エポキシ当量:178(g/eq.)。
・ビスフェノールA型シアネートエステル樹脂(“サイテスタ(CYTESTER)”(登録商標)TA、三菱ガス化学(株)製)シアネート当量:139(g/eq.)
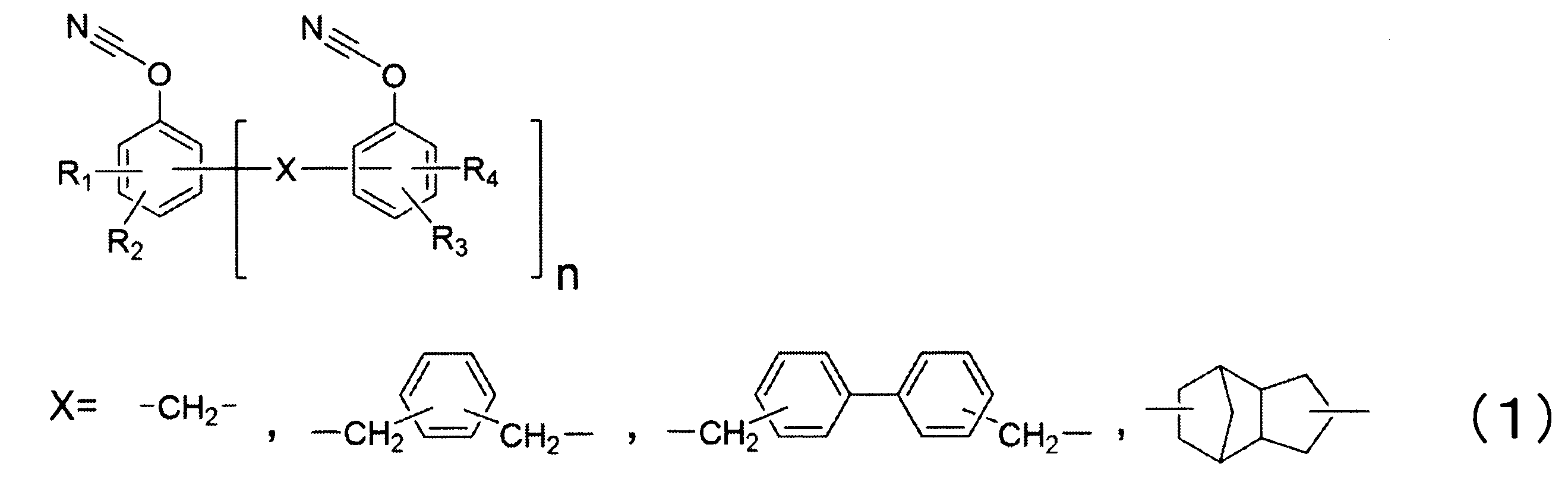
・フェノールノボラック型シアネートエステル樹脂(“AroCy”(登録商標)XU371、ハンツマン・アドバンスト・マテリアルズ社製)シアネート当量:131(g/eq.)、式(1)の構造に含まれるシアネートエステル樹脂。
・4,4’-ジアミノジフェニルスルホン(セイカキュアS、和歌山精化工業(株)製)活性水素当量:62(g/eq.)、25℃で固形、アミノ基を2つ以上有する芳香族アミン化合物
・3,3’-ジアミノジフェニルスルホン(3,3’-DAS、三井化学ファイン(株)製)活性水素当量:62(g/eq.)、25℃で固形、アミノ基を2つ以上有する芳香族アミン化合物
・4,4’-ジアミノジフェニルケトン(和歌山精化工業(株)製)活性水素当量:53(g/eq.)、25℃で固形、アミノ基を2つ以上有する芳香族アミン化合物
・3,3’-ジアミノジフェニルケトン(東京化成工業(株)製)活性水素当量:53(g/eq.)、25℃で固形、アミノ基を2つ以上有する芳香族アミン化合物
・ジエチルトルエンジアミン(“Aradur”(登録商標)5200、ハンツマン・アドバンスト・マテリアルズ社製)活性水素当量:45(g/eq.)、25℃で液状、アミノ基を2つ以上有する芳香族アミン化合物
・4,4’-ジアミノ-3,3’-ジエチルジフェニルメタン(“カヤハード”(登録商標)A-A、日本化薬(株)製)活性水素当量:64(g/eq.)、25℃で液状、アミノ基を2つ以上有する芳香族アミン化合物
・ジシアンジアミド(DICY7、三菱ケミカル(株)製)、25℃で固形、脂肪族アミン化合物。
・ポリエーテルスルホン(“Virangage”(登録商標)VW-10700RP SOLVAY社製)。
・テトラフェニルホスホニウムテトラ-p-トリルボレート(“TPP-MK”(登録商標)北興化学工業(株)製)。
以下の方法にて各実施例および比較例の熱硬化性樹脂組成物を測定した。
混練装置中に、表1~6に記載の構成要素[A]に該当するエポキシ樹脂、および構成要素[B]に該当するシアネートエステル樹脂および熱可塑性樹脂を投入し、加熱混練を行い、熱可塑性樹脂を溶解させた。次いで、混練を続けたまま100℃以下の温度まで降温させ、表1~5に記載の構成要素[C]または硬化促進剤(ただし、比較例においては、構成要素[A]、構成要素[B]、構成要素[C]を加えない場合もある。)を加えて撹拌し、熱硬化性樹脂組成物を得た。
示差走査熱量計(DSC Q2000:TAインスツルメント社製)を用いて、窒素雰囲気中で5℃/分の昇温速度にて、熱硬化性樹脂組成物の発熱曲線を得た。得られた発熱曲線中で、発熱量が100mW/g以上である発熱ピークの頂点の温度を、発熱ピーク温度とした。発熱量が100mW/g以上である発熱ピークが2つ以上ある場合は、低温側のピークの頂点の温度を、発熱ピーク温度とした。速硬化性の評価に関し、表1~6において、発熱ピーク温度が180℃以下をA、180℃超、200℃以下をB、200℃超、220℃以下をC、220℃超をDで表記した。
熱硬化性樹脂組成物の粘度は、動的粘弾性測定装置ARESレオメーター(TAインスツルメント社製)を用いて評価した。上下部測定冶具に直径40mmの平板のパラレルプレートを用い、上部と下部の冶具間距離が1mmとなるように該熱硬化性樹脂組成物をセットし、ねじりモード(測定周波数:0.5Hz)により測定した複素粘性率η*を粘度とした。80℃で1分間保持した時の粘度η* 1、80℃で120分間保持した時の粘度η* 120を測定し、増粘倍率(ポットライフ)をη* 120/η* 1より求めた。ポットライフの評価に関し、表1~6において、増粘倍率が2.0倍以下をA、2.0倍超、2.5倍以下をB、2.5倍超、3.0倍以下をC、3.0倍超をDで表記した。
熱硬化性樹脂組成物の貯蔵弾性率は、動的粘弾性装置ARESレオメーター(TAインスツルメント社製)を用いて評価した。上下部測定冶具に直径40mmの平板のパラレルプレートを用い、上部と下部の冶具間距離が1mmとなるように該熱硬化性樹脂組成物をセットし、ねじりモード(測定周波数:0.5Hz)で測定した。30℃で1分間保持した時の貯蔵弾性率を、30℃貯蔵弾性率として算出した。
上記(1)で調製した熱硬化性樹脂組成物をモールドに注入し、熱風乾燥機中で30℃から速度1.5℃/分で昇温し、210℃で8時間加熱硬化した後、30℃まで速度2.5℃/分で降温して、厚さ2mmの板状の樹脂硬化物を作製した。
上記(5)の方法で作製した樹脂硬化板から、幅12.7mm、長さ45mmの試験片を切り出し、乾燥条件での測定の場合は、試験片を60℃真空オーブン中で24時間乾燥させ、JIS K 7244に従い、動的粘弾性試験によりガラス転移温度を求めた。また、吸湿条件での測定の場合は、試験片を98℃の熱水に48時間浸漬し、同様に動的粘弾性試験によりガラス転移温度を求めた。貯蔵弾性率曲線において、ガラス状態での接線と転移状態での接線との交点の温度の値をガラス転移温度とした。ここでは、昇温速度5℃/分、周波数1Hzで測定した。表1~6において、吸湿後の耐熱性は、吸湿後のガラス転移温度が190℃以上をA、180℃以上190℃未満をB、170℃以上180℃未満をC、170℃未満をDで表記した。
上記(5)の方法で作製した樹脂硬化板から、長さ60mm、幅10mmの試験片を切り出し、材料万能試験機(インストロン・ジャパン(株)製、“インストロン”(登録商標)5565型P8564)を用い、試験速度2.5mm/分、支点間距離32mmで3点曲げ試験を行い、JIS K 7171-1994に従い曲げ弾性率を求めた。乾燥時の室温環境下での測定の場合は、試験片を60℃真空オーブン中で24時間乾燥させ、環境温度25℃で試験した。吸湿後の高温環境下での測定の場合は、試験片を98℃の熱水に48時間浸漬し、環境温度82℃で試験した。表1~6において、吸湿後の高温環境下の力学特性は、吸湿後の82℃での曲げ弾性率が、3.2GPa以上をA、3.0GPa以上3.2GPa未満をB、2.8GPa以上3.0GPa未満をC、2.8GPa未満をDで表記した。
上記(1)で調製した熱硬化性樹脂組成物を離型紙上にコーティングし、所定の樹脂目付の樹脂フィルムを作製した。この樹脂フィルムをプリプレグ作製機にセットし、引き揃えた強化繊維の両面から重ね、加熱加圧して熱硬化性樹脂組成物を含浸させ、繊維目付192g/m2、樹脂含有率が35質量%のプリプレグを作製した。なお、強化繊維は、炭素繊維(“トレカ”(登録商標)T700S-24K、東レ(株)製、繊維数24,000本、引張強度4,900MPa、引張弾性率230MPa)を用いた。
上記(8)で作製したプリプレグを、15cm角および10cm角のシート状に切り出し、15cm角のプリプレグを下側、10cm角のプリプレグを上側にして重ねた。重ねたプリプレグの上側に、粘着性テープを貼り付けた10cm角のステンレス製プレート(400g)を載せ、30秒間保持した。その後、ステンレス製プレートを持ち上げた際に、重ねたプリプレグが2枚に分かれない場合はタック性を「良好」、プリプレグが剥がれて2枚に分かれる場合を、タック性は「不良」と判定した。
実施例1~4では、構成要素[C]として、表1に記載のアミン化合物を用いた結果、表5に記載の比較例1(構成要素[C]非含有)と比べ、80℃2時間保持後の増粘倍率は良好な値を保ちつつ、発熱ピーク温度の大幅な低下がなされ、優れた速硬化性を示した。さらに、実施例1~4は、アミンとシアネートの反応により形成したイソウレア構造の水素結合性により、比較例1対比、吸湿後の82℃環境下の曲げ弾性率が向上し、優れた力学特性を示した。
表1および5に示すように、実施例5~8および比較例2では、実施例1~4および比較例1における構成要素[A]のエポキシ樹脂を、テトラグルシジルジアミノジフェニルメタンから変更し、ビスフェノールA型エポキシ樹脂を用いた。実施例5~8では、比較例2(構成要素[C]非含有)と比べ、80℃2時間保持後の増粘倍率は良好な値を保ちつつ、発熱ピーク温度の大幅な低下がなされ、優れた速硬化性を示した。さらに、実施例5~8は、比較例2対比、吸湿後の82℃環境下での曲げ弾性率が向上し、優れた力学特性を示した。
表1、2および5に示すように、実施例2、9~11および比較例6、7では、構成要素[A]のエポキシ樹脂として、テトラグルシジルジアミノジフェニルメタンを用い、熱硬化性樹脂組成物中のシアネート基のモル数に対する、アミノ基に含まれる活性水素のモル数の比を一定とし、熱硬化性樹脂組成物中のシアネート基のモル数に対するグリシジル基のモル数の比を変更した。グリシジル基のモル数の割合が増加すると、80℃2時間保持後の増粘倍率が低下する傾向が見られ、優れたポットライフを示した。熱硬化性樹脂組成物中のシアネート基のモル数に対するグリシジル基のモル数の比が0.2である比較例6では、吸湿後のガラス転移温度が、実施例2、9~11対比低く、さらに熱硬化性樹脂組成物は30℃で固形であり、プリプレグとしてのタックを示さなかった。また、熱硬化性樹脂組成物中のシアネート基のモル数に対するグリシジル基のモル数の比が1.75である比較例7では、実施例2、9~11対比、吸湿後のガラス転移温度および吸湿後82℃環境下の曲げ弾性率が低い値であった。
表1および2に示すように、実施例6、12~14では、構成要素[A]のエポキシ樹脂として、ビスフェノールA型エポキシ樹脂を用い、熱硬化性樹脂組成物中のシアネート基のモル数に対するアミノ基に含まれる活性水素のモル数の比を一定とし、熱硬化性樹脂組成物中のシアネート基のモル数に対するグリシジル基のモル数の比を変更した。グリシジル基のモル数の割合が増加すると、80℃2時間保持後の増粘倍率が低下する傾向が見られ、優れたポットライフを示した。表5、6に示すとおり、構成要素[C]を含まない比較例2、13~15では、構成要素[C]を含む実施例6、12~14対比、発熱ピーク温度が著しく高く、十分な速硬化性を示さなかった。
表1および2に示すように、実施例2、6、15~18では、熱硬化性樹脂組成物中のシアネート基のモル数に対する、アミノ基に含まれる活性水素のモル数の比および、熱硬化性樹脂組成物中のシアネート基のモル数に対するグリシジル基のモル数の比を一定とし、テトラグルシジルジアミノジフェニルメタンとビスフェノールA型エポキシ樹脂の配合割合を変更した。3官能以上のグリシジルアミン型エポキシ樹脂であるテトラグリシジルジアミノジフェニルメタンの配合量が増加することで、乾燥状態のガラス転移温度および吸湿後のガラス転移温度が大きく向上し、好ましい特性を示した。
表5に示すように、比較例3~5では、構成要素[A]のエポキシ樹脂を含んでいないため、熱硬化性樹脂組成物が30℃で固形状であり、貯蔵弾性率の測定が不可能であった。さらに、作製したプリプレグはタック性を示さなかった。構成要素[A]を含む実施例1~3は、比較例3~5対比、吸湿後の82℃環境下の曲げ弾性率および吸湿後のガラス転移温度が高く、優れた特性を示した。
表2および3に示すように、実施例19~22では、種々の構成要素[A]のエポキシ樹脂を用いた結果、表6に記載の比較例16~19(構成要素[C]非含有)と比べ、80℃2時間保持後の増粘倍率は良好な値を保ちつつ、発熱ピーク温度の大幅な低下が達成され、優れた速硬化性を示した。
表3に示すように、実施例23~26では、実施例2と同じ熱硬化性樹脂組成物中のシアネート基のモル数に対するアミノ基に含まれる活性水素のモル数の比および、熱硬化性樹脂組成物中のシアネート基のモル数に対するグリシジル基のモル数の比とした上で、フェノールノボラック型シアネートエステル樹脂(式(1)の構造に含まれる)とビスフェノールA型シアネートエステル樹脂の配合割合を変更した。フェノールノボラック型シアネートエステル樹脂(式(1)の構造に含まれる)の配合量が増加することで、吸湿後のガラス転移温度が大きく向上し、優れた特性を示した。
表3に示すように、実施例27~30では、フェノールノボラック型シアネートエステル樹脂を全シアネートエステル樹脂に対して20質量部以上含んだ上で、実施例23、24に対して、熱硬化性樹脂組成物中のシアネート基のモル数に対するアミノ基に含まれる活性水素のモル数の比または、熱硬化性樹脂組成物中のシアネート基のモル数に対するグリシジル基のモル数の比を変更した。いずれも優れた吸湿後のガラス転移温度を有しており、優れた特性を示した。
表4に示すように、実施例31~36では、種々の構成要素[C]のアミン化合物を用いた結果、いずれの場合も、表5に記載の比較例1(構成要素[C]非含有)と比べ、発熱ピーク温度の大幅な低下がなされ、優れた速硬化性を示した。構成要素[C]としてジアミノジフェニルホンまたはジアミノジフェニルケトンを用いた実施例31~33では、電子吸引性の官能基を有するためアミンの求核性が適度に抑制されており、特に優れたポットライフと速硬化性のバランスを示した。さらに、ジアミノジフェニルホンまたはジアミノジフェニルケトンの骨格の剛直性に起因し、特に優れた吸湿後のガラス転移温度を示した。
表6に示す通り、構成要素[B]を含まない比較例20は、グリシジル基とアミノ基の反応により形成されるヒドロキシル基の親水性のため吸湿しやすく、構成要素[B]を含む実施例1に対し、吸湿後の82℃環境下での曲げ弾性率が著しく低い特性であった。
表4に示す通り、熱可塑性樹脂を含まない実施例37では、30℃の貯蔵弾性率が低下する傾向を示したが、プリプレグとして良好なタック性を示した。






Claims (10)
- 少なくとも次の構成要素[A]~[C]を含み、以下の条件(1)と(2)を満たし、さらにポリエーテルスルホンを含む熱硬化性樹脂組成物。
[A]グリシジルアミン型エポキシ、ビスフェノールA型エポキシ、ビスフェノールF型エポキシ、フェノールノボラック型エポキシから選ばれる少なくとも1種のグリシジル基を2つ以上有するエポキシ樹脂
[B]ビスフェノールA型シアネートエステルとフェノールノボラック型シアネートエステルから選ばれる少なくとも1種のシアネート基を2つ以上有するシアネートエステル樹脂
[C]アミノ基を二つ以上有する芳香族アミン化合物
(1)0.25≦熱硬化性樹脂組成物中のグリシジル基のモル数/熱硬化性樹脂組成物中のシアネート基のモル数≦1.5
(2)0.05≦熱硬化性樹脂組成物中のアミノ基に含まれる活性水素のモル数/熱硬化性樹脂組成物中のシアネート基のモル数≦0.5 - 構成要素[C]のアミン化合物が25℃において固形である請求項1に記載の熱硬化性樹脂組成物。
- 構成要素[C]のアミン化合物が、ジアミノジフェニルスルホンまたはジアミノジフェニルケトンを含む請求項2に記載の熱硬化性樹脂組成物。
- 構成要素[A]のグリシジル基を2つ以上有するエポキシ樹脂が、グリシジル基を3個以上含むグリシジルアミン型エポキシ樹脂であり、全エポキシ樹脂100質量部に対し、上記グリシジル基を3個以上含むグリシジルアミン型エポキシ樹脂を40~100質量部含む請求項1から3のいずれかに記載の熱硬化性樹脂組成物。
- ポリエーテルスルホンを熱硬化性樹脂組成物中に1~30質量%含む請求項1から4のいずれかに記載の熱硬化性樹脂組成物。
- 昇温速度5℃/分での示差走査熱量測定による発熱曲線において、100mW/g以上の発熱ピークが160℃~200℃に存在する請求項1から5のいずれかに記載の熱硬化性樹脂組成物。
- 周波数0.5Hzの動的粘弾性試験による30℃の貯蔵弾性率が、0.1~100,000Paである請求項1から6のいずれかに記載の熱硬化性樹脂組成物。
- 請求項1から7のいずれかに記載の熱硬化性樹脂組成物が強化繊維に含浸されてなるプリプレグ。
- 請求項8に記載のプリプレグが硬化されてなる繊維強化複合材料。
- 強化繊維と請求項1から7のいずれかに記載の熱硬化性樹脂組成物が硬化されてなる樹脂硬化物を含んでなる繊維強化複合材料。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2018032782 | 2018-02-27 | ||
| JP2018032782 | 2018-02-27 | ||
| PCT/JP2019/004260 WO2019167579A1 (ja) | 2018-02-27 | 2019-02-06 | 熱硬化性樹脂組成物、プリプレグおよび繊維強化複合材料 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JPWO2019167579A1 JPWO2019167579A1 (ja) | 2021-01-07 |
| JP7200928B2 true JP7200928B2 (ja) | 2023-01-10 |
Family
ID=67806031
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2019507971A Active JP7200928B2 (ja) | 2018-02-27 | 2019-02-06 | 熱硬化性樹脂組成物、プリプレグおよび繊維強化複合材料 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US11319435B2 (ja) |
| EP (1) | EP3760661A4 (ja) |
| JP (1) | JP7200928B2 (ja) |
| KR (1) | KR20200125579A (ja) |
| CN (1) | CN111770948A (ja) |
| WO (1) | WO2019167579A1 (ja) |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2021152957A1 (ja) * | 2020-01-31 | 2021-08-05 | 東レ株式会社 | 複合プリプレグ、ならびにそれを用いたプリフォーム、繊維強化複合材料接合体およびその製造方法 |
| US20230122917A1 (en) * | 2020-03-31 | 2023-04-20 | Denka Company Limited | Semicured product complex and method for producing same, cured product complex and method for producing same, and thermosetting composition used to impregnate porous body |
| US12053908B2 (en) | 2021-02-01 | 2024-08-06 | Regen Fiber, Llc | Method and system for recycling wind turbine blades |
| CN113522166B (zh) * | 2021-07-09 | 2024-04-30 | 苏州锦鳞电子科技有限公司 | 造粒粘结剂及其制备方法、粉体造粒方法与应用 |
| CN114106517B (zh) * | 2021-10-25 | 2023-11-21 | 航天特种材料及工艺技术研究所 | 一种高强度高韧性阻燃环氧树脂及其制备方法 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2013181132A (ja) | 2012-03-02 | 2013-09-12 | Sekisui Chem Co Ltd | エポキシ樹脂材料及び多層基板 |
Family Cites Families (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4142034A (en) * | 1978-04-10 | 1979-02-27 | Cincinnati Milacron Inc. | Epoxy resin compositions containing an amine-cyanic acid ester combination curing agent |
| JPH0653790B2 (ja) * | 1986-02-10 | 1994-07-20 | 三菱レイヨン株式会社 | 熱硬化性樹脂組成物 |
| JPH0768379B2 (ja) | 1986-02-12 | 1995-07-26 | 東レ株式会社 | プリプレグ用樹脂組成物の製法 |
| JPH06260536A (ja) * | 1993-03-06 | 1994-09-16 | Kanegafuchi Chem Ind Co Ltd | Tab用テープ |
| JPH10178040A (ja) * | 1996-10-15 | 1998-06-30 | Toray Ind Inc | 半導体集積回路接続用基板およびそれを構成する部品ならびに半導体装置 |
| AU2002228697B2 (en) | 2001-02-27 | 2006-03-02 | Hexcel Corporation | Adhesive prepreg face sheets for sandwich panels |
| JP5369655B2 (ja) | 2008-02-19 | 2013-12-18 | 東レ株式会社 | ポリアミド微粒子、プリプレグ及び炭素繊維強化複合材料 |
| KR20110095384A (ko) * | 2008-11-21 | 2011-08-24 | 헨켈 코포레이션 | 상이 분리된 경화성 조성물 |
| JP5603610B2 (ja) * | 2010-02-12 | 2014-10-08 | 株式会社Adeka | 無溶剤一液型シアン酸エステル−エポキシ複合樹脂組成物 |
| RU2013118705A (ru) * | 2010-09-24 | 2014-10-27 | Торэй Индастриз, Инк. | Композиция эпоксидной смолы для армированного волокнами композиционного материала, препрег и армированный волокнами композиционный материал |
| ES2650268T3 (es) * | 2013-12-04 | 2018-01-17 | Lonza Ltd | Método para preparar piezas reforzadas con fibras a base de mezclas de éster de cianato/epoxi |
| CN106068346B (zh) * | 2014-03-12 | 2018-12-11 | 东丽株式会社 | 涂上浆剂增强纤维、涂上浆剂增强纤维的制造方法、预浸料坯及纤维增强复合材料 |
| JP6397205B2 (ja) * | 2014-04-01 | 2018-09-26 | Jxtgエネルギー株式会社 | プリプレグ、炭素繊維強化複合材料、ロボットハンド部材及びその原料樹脂組成物 |
| WO2017009220A1 (de) * | 2015-07-13 | 2017-01-19 | Basf Se | Verwendung von oligo-n,n-bis-(3-aminopropyl)methylamin als härter für epoxidharze |
| EP3345950B1 (en) | 2015-09-04 | 2021-07-21 | Adeka Corporation | Resin composition for fiber-reinforced plastic, cured product thereof, fiber-reinforced plastic containing said cured product, and method for producing said fiber-reinforced plastic |
| JP2017132896A (ja) | 2016-01-27 | 2017-08-03 | 三菱瓦斯化学株式会社 | シアン酸エステル化合物を含む樹脂組成物及びその硬化物 |
| KR102573168B1 (ko) * | 2017-02-27 | 2023-09-06 | 가부시키가이샤 아데카 | 섬유 강화 플라스틱용 수지 조성물, 그 경화물, 및 상기 경화물로 이루어지는 섬유 강화 플라스틱 |
-
2019
- 2019-02-06 EP EP19760072.9A patent/EP3760661A4/en not_active Withdrawn
- 2019-02-06 KR KR1020207017242A patent/KR20200125579A/ko not_active Withdrawn
- 2019-02-06 WO PCT/JP2019/004260 patent/WO2019167579A1/ja not_active Ceased
- 2019-02-06 US US16/969,650 patent/US11319435B2/en active Active
- 2019-02-06 JP JP2019507971A patent/JP7200928B2/ja active Active
- 2019-02-06 CN CN201980014494.XA patent/CN111770948A/zh not_active Withdrawn
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2013181132A (ja) | 2012-03-02 | 2013-09-12 | Sekisui Chem Co Ltd | エポキシ樹脂材料及び多層基板 |
Also Published As
| Publication number | Publication date |
|---|---|
| US20200399462A1 (en) | 2020-12-24 |
| KR20200125579A (ko) | 2020-11-04 |
| EP3760661A4 (en) | 2021-11-17 |
| US11319435B2 (en) | 2022-05-03 |
| CN111770948A (zh) | 2020-10-13 |
| EP3760661A1 (en) | 2021-01-06 |
| JPWO2019167579A1 (ja) | 2021-01-07 |
| WO2019167579A1 (ja) | 2019-09-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5003827B2 (ja) | 炭素繊維強化複合材料用エポキシ樹脂組成物、プリプレグおよび炭素繊維強化複合材料 | |
| JP7200928B2 (ja) | 熱硬化性樹脂組成物、プリプレグおよび繊維強化複合材料 | |
| JP5800031B2 (ja) | エポキシ樹脂組成物、プリプレグおよび炭素繊維強化複合材料 | |
| JP6812794B2 (ja) | エポキシ樹脂組成物、プリプレグおよび繊維強化複合材料 | |
| JP6710972B2 (ja) | エポキシ樹脂組成物、プリプレグ、樹脂硬化物および繊維強化複合材料 | |
| WO2010109929A1 (ja) | 繊維強化複合材料用エポキシ樹脂組成物、プリプレグおよび繊維強化複合材料 | |
| JP6237951B1 (ja) | エポキシ樹脂組成物、プリプレグおよび繊維強化複合材料 | |
| JPWO2018173716A1 (ja) | エポキシ樹脂組成物、プリプレグおよび炭素繊維強化複合材料 | |
| JP7268355B2 (ja) | エポキシ樹脂組成物、プリプレグおよび繊維強化複合材料 | |
| JP7014153B2 (ja) | エポキシ樹脂組成物、プリプレグおよび繊維強化複合材料 | |
| JP2018012797A (ja) | エポキシ樹脂組成物、プリプレグ、樹脂硬化物および繊維強化複合材料 | |
| JP2019059827A (ja) | エポキシ樹脂組成物、プリプレグ、樹脂硬化物および繊維強化複合材料 | |
| JP2018012798A (ja) | エポキシ樹脂組成物、プリプレグ、樹脂硬化物および繊維強化複合材料 | |
| CN120569423A (zh) | 环氧树脂组合物、预浸料坯及纤维增强复合材料以及环氧树脂组合物的制造方法 | |
| JP2018012796A (ja) | エポキシ樹脂組成物、プリプレグ、樹脂硬化物および繊維強化複合材料 | |
| JP2019183134A (ja) | プリプレグ |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20211130 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20211130 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20221004 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20221107 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20221122 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20221205 |
|
| R151 | Written notification of patent or utility model registration |
Ref document number: 7200928 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R151 |