JP7034496B2 - 芳香族化合物を産生する微生物 - Google Patents
芳香族化合物を産生する微生物 Download PDFInfo
- Publication number
- JP7034496B2 JP7034496B2 JP2019535461A JP2019535461A JP7034496B2 JP 7034496 B2 JP7034496 B2 JP 7034496B2 JP 2019535461 A JP2019535461 A JP 2019535461A JP 2019535461 A JP2019535461 A JP 2019535461A JP 7034496 B2 JP7034496 B2 JP 7034496B2
- Authority
- JP
- Japan
- Prior art keywords
- strain
- gene
- frt
- dna
- fbr
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images


Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P13/00—Preparation of nitrogen-containing organic compounds
- C12P13/04—Alpha- or beta- amino acids
- C12P13/22—Tryptophan; Tyrosine; Phenylalanine; 3,4-Dihydroxyphenylalanine
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P7/00—Preparation of oxygen-containing organic compounds
- C12P7/02—Preparation of oxygen-containing organic compounds containing a hydroxy group
- C12P7/22—Preparation of oxygen-containing organic compounds containing a hydroxy group aromatic
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Zoology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Wood Science & Technology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Microbiology (AREA)
- General Chemical & Material Sciences (AREA)
- Biotechnology (AREA)
- Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
Description
(1)aroA
(2)aroB
(3)aroC
(4)aroGfbr又はaroFfbr
(5)pheAfbr又はtyrAfbr
本開示において、フェニルアラニン及びチロシンは、特に言及のない場合、L-フェニルアラニン及びL-チロシンを意味しうる。
本開示において、「aroGfbr」遺伝子は、aroGのフィードバック阻害の脱感作変異型を意味する。aroGfbrは、一又は複数の実施形態において、特開平05-236947に開示のものを使用できる。
本開示において、「aroFfbr」遺伝子は、aroFのフィードバック阻害の脱感作変異型を意味する。aroFfbrは、一又は複数の実施形態において、特開平05-236947に開示のものを使用できる。
本開示において、「pheAfbr」遺伝子は、pheAのフィードバック阻害の脱感作変異型を意味する。pheAfbrは、一又は複数の実施形態において、特開2006-311833及び特開平05-344881に開示のものを使用できる。
本開示において、「tyrAfbr」遺伝子は、tyrAのフィードバック阻害の脱感作変異型を意味する。tyrAfbrは、一又は複数の実施形態において、特開平05-076352に開示のものを使用できる。
本開示に係る微生物では、一又は複数の実施形態において、フェニルアラニン、チロシン等の芳香族化合物の産生量を向上させる観点から、tyrBは発現誘導可能なプロモーターともに染色体に導入されないことが好ましい。従来、フェニルアラニンやチロシンを産生させる場合、tyrBは過剰発現される場合が多かった。しかし、本発明者らは、tyrBの過剰発現は代謝負荷によるネガティブな影響が大きく、tyrBを過剰発現させないほうが、フェニルアラニンやチロシンの生産を向上できることを見出した。
(1)aroA
(2)aroB
(3)aroC
(4)aroGfbr又はaroFfbr
(5)pheAfbr又はtyrAfbr
本実施形態において、上記(5)の遺伝子がpheAfbrの場合、本開示に係る微生物は、フェニルアラニンの生産能を有し、本開示に係る微生物を用いた発酵法によりフェニルアラニンの製造が可能になる。
本実施形態において、上記(5)の遺伝子がtyrAfbrの場合、本開示に係る微生物は、チロシンの生産能を有し、本開示に係る微生物を用いた発酵法によりチロシンの製造が可能になる。
本実施形態において、上記(4)のaroGfbr又はaroFfbrは同じ酵素をコードしており、相互に代替可能である。なお、aroGfbr及びaroFfbrの代わりにaroH(NCBI GENBANKのGene ID:946229)のフィードバック阻害の脱感作変異型を使用することもできる。
本発明者らは、上記(4)及び(5)に加え、上記(1)~(3)のすべて過剰発現させることで、導入した遺伝子(例えば、上記(4)及び(5))の過剰発現による代謝負荷を低減しうることを見出した。
(6)ppsA
(7)tktA
ここで、発現誘導可能なプロモーターとしてT7プロモーターを使用する場合、目的の芳香族化合物の生産性向上の点から、上記(7)tktAのプロモーターは発現誘導能が低減した変異型T7プロモーターであることが好ましい。
(8)aroD
(9)aroE又はydiB
(10)aroL又はaroK
(8)~(10)の中で、2つ又は全部が導入されていることが好ましい。
本実施形態において、上記(9)のaroE又はydiBは相互に代替可能であるが、上記(9)の遺伝子としては、aroEが好ましい。
本実施形態において、上記(10)のaroL又はaroKは相互に代替可能であるが、上記(10)の遺伝子としては、aroLが好ましい。
例えば、フェニルピルビン酸デカルボキシラーゼをコードする異種起源の遺伝子ipdCが染色体上に発現誘導可能なプロモーターとともに導入されると、本開示に係る微生物は、2-フェニルエタノール又は2-(4-ヒドロキシフェニル)エタノールの生産能を有するから、これらの芳香族化合物の製造に使用できる。
また、例えば、乳酸デヒドロゲナーゼをコードする異種起源の遺伝子ldhAが染色体上に発現誘導可能なプロモーターとともに導入されると、フェニル乳酸又は4-ヒドロキシフェニル乳酸の生産能を有する微生物となり、これらの芳香族化合物の製造に使用できる。
すなわち、フェニルピルビン酸、4-ヒドロキシフェニルピルビン酸、フェニルアラニン、又はチロシンを基質とする酵素をコードする遺伝子をさらに導入すると、その遺伝子に応じて芳香族化合物が製造できる。
また、非特許文献2に記載されているように、例えば、Lactobacillus brevisのチロシンデカルボキシラーゼ遺伝子(NCBI GENBANKのGene ID:4413406)やPseudomonas putida NBRC100650の芳香族アミノ酸デカルボキシラーゼ遺伝子(NCBI GENBANKのGene ID:1045854)又はそれらのオーソログ遺伝子を導入することで、フェニルエチルアミンやチラミンを製造することが可能となる。一方、非特許文献3に記載されているように、例えば、フェニルピルビン酸デカルボキシラーゼ遺伝子と共に、Escherichia coliのフェニルアセトアルデヒドデヒドロゲナーゼ遺伝子(NCBI GENBANKのGene ID:945933)又はそのオーソログ遺伝子を導入することで、フェニル酢酸や4-ヒドロキシフェニル酢酸を製造することが可能となる。
なお、本開示に係る微生物を用いたフェニルアラニン、チロシンを含む芳香族化合物の製造方法では、一又は複数の実施形態において、導入した遺伝子の過剰発現による代謝負荷が低減されているから、前培養にグルコースを添加してカタボライトリプレッションを引き起こさなくても芳香族化合物を産生させることができる。
<1> 形質転換されたエシェリキア属に属する微生物であって、
少なくとも下記5つの遺伝子が染色体上に発現誘導可能なプロモーターとともに導入されている微生物。
(1)aroA
(2)aroB
(3)aroC
(4)aroGfbr又はaroFfbr
(5)pheAfbr又はtyrAfbr
<2> さらに、下記2つの遺伝子が染色体上に発現誘導可能なプロモーターとともに導入されている、<1>に記載の微生物。
(6)ppsA
(7)tktA
<3> さらに、下記3つの遺伝子の少なくとも1つが染色体上に発現誘導可能なプロモーターとともに導入されている、<1>又は<2>に記載の微生物。
(8)aroD
(9)aroE又はydiB
(10)aroL又はaroK
<4> 染色体上に発現誘導可能なプロモーターとともに導入される遺伝子にtyrBが含まれない、<1>から<3>のいずれかに記載の微生物。
<5> 前記(5)の遺伝子が、pheAfbrであり、フェニルアラニン生産能を有する<1>から<4>のいずれかに記載の微生物。
<6> 前記(5)の遺伝子が、tyrAfbrであり、チロシン生産能を有する、<1>から<4>のいずれかに記載の微生物。
<7> さらに、フェニルピルビン酸、4-ヒドロキシフェニルピルビン酸、フェニルアラニン、又はチロシンを基質とする酵素をコードする同種又は異種起源の遺伝子が染色体上に発現誘導可能なプロモーターとともに導入された、<1>から<4>のいずれかに記載の微生物。
<8> さらに、フェニルピルビン酸デカルボキシラーゼをコードする異種起源の遺伝子ipdCが染色体上に発現誘導可能なプロモーターとともに導入され、2-フェニルエタノール又は2-(4-ヒドロキシフェニル)エタノールの生産能を有する、<1>から<4>のいずれかに記載の微生物。
<9> さらに、乳酸デヒドロゲナーゼをコードする異種起源の遺伝子ldhAが染色体上に発現誘導可能なプロモーターとともに導入され、フェニル乳酸又は4-ヒドロキシフェニル乳酸の生産能を有する、<1>から<4>のいずれかに記載の微生物。
<10> <1>から<9>のいずれかに記載の微生物を培地で培養することを含む、芳香族化合物の製造方法。
大腸菌の染色体に部位特異的に複数の遺伝子を導入する方法は、非特許文献2(Koma et al.Appl.Microbiol.Biotechnol.93:815-829,2012)に従った。簡単には、同文献Fig.1(図1)に記載のように、Red相同組み換え、FLP/FRT部位特異的組み換え、及びP1トランスダクションを利用して作製した。
図1は、3つの遺伝子(A,B,C)を染色体に導入する場合のスキームを示す。まず、Red相同組み換えにより、FRT-Km-FRT-A、FRT-Km-FRT-B、FRT-Km-FRT-Cを染色体の所望の位置に有する3つの株(株A,株B,株C)を作製する。株A内でFLP/FRT組み換えによりKmカセットが切り出されたものをアクセプター株Aとして得る。次にドナー株BをP1virファージに感染させて得た溶菌液Bをアクセプター株Aに感染させてP1トランスダクションを行う。得られた株からFLP/FRT組み換えによりKmカセットが切り出されたものをアクセプター株ABとして得る。ドナー株CをP1virファージに感染させて得た溶菌液Cをアクセプター株ABに感染させてP1トランスダクションを行う。最後に、FLP/FRT組み換えによりKmカセットが切り出されたものを選択すれば、3つの遺伝子(A,B,C)が染色体に導入された株を得ることができる。
Red相同組み換えで導入する遺伝子断片を作製するために、FRT-Km-FRTカセットをマルチクローニングサイト(MCS)の上流に備える2つのプラスミドを利用した。pET-21a-FRTは、MCSが1つ、pETDuet-FRTは、MCSが2つである。これらのプラスミドはT7プロモーターによって遺伝子発現を誘導できる。次に、直鎖DNA断片をPCRで増幅する。プライマーには、ターゲット部位の外側の50塩基の配列(H1又はH2)と、プラスミド由来の20塩基の配列(P1又はP2)を有する。そして、増幅断片を、pKD46を有する大腸菌細胞にエレクトロポレートする。得られる組み替え体は、染色体のターゲット部位に所望の遺伝子が導入される。この組み替え体にプラスミドpCP20を導入することで、FLP/FRT組み換えによりKmカセットを除去できる。
[pET21a-FRT-aroGfbrの作製]
大腸菌MG1655のゲノムDNAを鋳型として、EcAroG-F(ATGAATTATCAGAACGACGATTTACG,配列番号01)とAroG-RM-Nde(GCTATTATCCATGTGCGGATCG,配列番号02)プライマーペア及びAroG-FM-Nde(CGATCCGCACATGGATAATAGC,配列番号03)とEcAroG-R(CAAAGCTTTTACCCGCGACGCGCTTTTAC,配列番号04)プライマーペアを用いて、Phusion Hot Start II DNAポリメラーゼ(Thermo Fisher Scientific、以下同じ)により2つのDNA断片を増幅した。なお、PCR反応の条件は、説明書に記載の基本的な条件に基づいて行った。つぎに、2つの増幅断片を鋳型として、EcAroG-FとEcAroG-Rのプライマーペアを用いて、Phusion Hot Start II DNAポリメラーゼによりオーバーラップエクステンションPCRを行い、配列中のNdeIサイトを消失させたaroG遺伝子のDNAを得た。さらに、増幅されたDNA断片を鋳型として、EcAroG-FとEcAroG-D146N-RM(GGGGTGATCATATTGAGAAACTC,配列番号05)及びEcAroG-D146N-FM(GAGTTTCTCAATATGATCACCCC,配列番号06)とEcAroG-R(CAAAGCTTTTACCCGCGACGCGCTTTTAC,配列番号07)のプライマーペアを用いて、Phusion Hot Start II DNAポリメラーゼにより2つのDNA断片を得た。最後に、2つの増幅断片を鋳型として、EcAroG-Nde(CCAACCATATGAATTATCAGAACGACGATTTACG,配列番号08)とEcAroG-Rのプライマーペアを用いて、Phusion Hot Start II DNAポリメラーゼによりオーバーラップエクステンションPCRを行い、146番目のアスパラギン酸がアスパラギンに置換されたAroGをコードするDNA(aroGfbr)を得た。
増幅されたDNAとpET21a-FRTベクター(非特許文献2、Koma et al.Appl.Microbiol.Biotechnol.93:815-829,2012、以下同じ)をそれぞれNdeIとXhoIで消化し、1%アガロースゲル電気泳動でDNAを分離した後、QIAquick Gel Extraction kit(Qiagen、以下同じ)を用いて該当のDNAをゲルから抽出・精製した。精製されたDNAを常法によりLigationし、大腸菌DH5αを形質転換し、20μg/mlのカナマイシンを含むLB寒天培地を用いて37℃で一晩培養した。T7プロモータープライマー(TAATACGACTCACTATAGG,配列番号09、以下同じ)とT7ターミネータープライマー(GCTAGTTATTGCTCAGCGG,配列番号10、以下同じ)を用いたコロニーダイレクトPCRを行い、目的のプラスミドを持つ菌株を選別した。最終的に目的プラスミドを有する菌株を20μg/mlのカナマイシンを含むLB液体培地を用いて37℃で一晩培養し、NucleoSpin Plasmid QuickPure(MACHERY-NAGEL、以下同じ)を用いて目的のプラスミドを抽出し精製した。LB寒天培地は、純水1Lに対して、10gポリペプトン(日本製薬株式会社)、5g乾燥酵母エキス(日本製薬株式会社)、10g塩化ナトリウム、20g寒天、水酸化ナトリウムでpH7.0に調整したもの。LB液体培地は、純水1Lに対して、10gポリペプトン(日本製薬株式会社)、5g乾燥酵母エキス(日本製薬株式会社)、10g塩化ナトリウム、水酸化ナトリウムでpH7.0に調整したもの。以下同じ。
大腸菌MG1655のゲノムDNAを鋳型として、EcPheA-F(CCAACCATATGACATCGGAAAACCCGTTAC,配列番号11)とEcPheA-S330P-RM(GAATCGGGCGTGGTTCCAGAC,配列番号12)プライマーペア及びEcPheA-S330P-FM(GTCTGGAACCACGCCCGATTC,配列番号13)とEcPheA-R(CACTCGAGTCAGGTTGGATCAACAGGCAC,配列番号14)プライマーペアを用いて、Phusion Hot Start II DNAポリメラーゼにより2つのDNA断片を増幅した。なお、PCR反応の条件は、説明書に記載の基本的な条件に基づいて行った。つぎに、2つの増幅断片を鋳型として、EcPheA-FとEcPheA-Rのプライマーペアを用いて、Phusion Hot Start II DNAポリメラーゼによりオーバーラップエクステンションPCRを行い、330番目のセリンがプロリンに置換されたPheAをコードするDNA(PheAfbr)を得た。
増幅されたDNAとpET21a-FRTベクターをそれぞれNdeIとXhoIで消化し、1%アガロースゲル電気泳動でDNAを分離した後、QIAquick Gel Extraction kitを用いて該当のDNAをゲルから抽出・精製した。精製されたDNAを常法によりLigationし、大腸菌DH5αを形質転換し、20μg/mlのカナマイシンを含むLB寒天培地を用いて37℃で一晩培養した。T7プロモータープライマーとT7ターミネータープライマーを用いたコロニーダイレクトPCRを行い、目的のプラスミドを持つ菌株を選別した。最終的に目的プラスミドを有する菌株を20μg/mlのカナマイシンを含むLB液体培地を用いて37℃で一晩培養し、NucleoSpin Plasmid QuickPureを用いて目的のプラスミドを抽出し精製した。
大腸菌MG1655のゲノムDNAを鋳型として、pps-F(CAACCATATGTCCAACAATGGCTCGTC,配列番号15)とpps-RM-Nco(GGATTTTTTTCGACCCCATAGTGCGGCGC,配列番号16)プライマーペア及びpps-FM-Nco(GCGCCGCACTATGGGGTCGAAAAAAATCC,配列番号17)とpps-R(CACTCGAGTTATTTCTTCAGTTCAGCCAGG,配列番号18)プライマーペアを用いて、Phusion Hot Start II DNAポリメラーゼにより2つのDNA断片を増幅した。なお、PCR反応の条件は、説明書に記載の基本的な条件に基づいて行った。つぎに、2つの増幅断片を鋳型としてpps-Fとpps-Rのプライマーペアを用いて、Phusion Hot Start II DNAポリメラーゼによりオーバーラップエクステンションPCRを行い、配列中のNcoIサイトを消失させたppsA遺伝子のDNAを得た。
増幅されたDNAとpET21a-FRTベクターをそれぞれNdeIとXhoIで消化し、1%アガロースゲル電気泳動でDNAを分離した後、QIAquick Gel Extraction kitを用いて該当のDNAをゲルから抽出・精製した。精製されたDNAを常法によりLigationし、大腸菌DH5αを形質転換し、20μg/mlのカナマイシンを含むLB寒天培地を用いて37℃で一晩培養した。T7プロモータープライマーとT7ターミネータープライマーを用いたコロニーダイレクトPCRを行い、目的のプラスミドを持つ菌株を選別した。最終的に目的プラスミドを有する菌株を20μg/mlのカナマイシンを含むLB液体培地を用いて37℃で一晩培養し、NucleoSpin Plasmid QuickPureを用いて目的のプラスミドを抽出し精製した。
大腸菌MG1655のゲノムDNAを鋳型としてtktA-Nde(CCAACCATATGTCCTCACGTAAAGAGCTTGCC,配列番号19)とtktA-Xho(CACTCGAGTTACAGCAGTTCTTTTGCTTTC,配列番号20)プライマーペアを用いて、Phusion Hot Start II DNAポリメラーゼによりtktA遺伝子のDNAを増幅した。なお、PCR反応の条件は、説明書に記載の基本的な条件に基づいて行った。
増幅されたDNAとpET21a-FRTベクターをそれぞれNdeIとXhoIで消化し、1%アガロースゲル電気泳動でDNAを分離した後、QIAquick Gel Extraction kitを用いて該当のDNAをゲルから抽出・精製した。精製されたDNAを常法によりLigationし、大腸菌DH5αを形質転換し、20μg/mlのカナマイシンを含むLB寒天培地を用いて37℃で一晩培養した。T7プロモータープライマーとT7ターミネータープライマーを用いたコロニーダイレクトPCRを行い、目的のプラスミドを持つ菌株を選別した。最終的に目的プラスミドを有する菌株を20μg/mlのカナマイシンを含むLB液体培地を用いて37℃で一晩培養し、NucleoSpin Plasmid QuickPureを用いて目的のプラスミドを抽出し精製した。
pETDuet-FRT(非特許文献2、Koma et al.Appl.Microbiol.Biotechnol.93:815-829,2012)を鋳型としてF-Bgl(CAACAGATCTATTCCGGGGATCCGTCGACC,配列番号21)と-8TC-RM1(CCCCTATAGTGGGTCGTATTAATTTCG,配列番号22)プライマーペアを用いて、Phusion Hot Start II DNAポリメラーゼによりDNA断片を増幅した。なお、PCR反応の条件は、説明書に記載の基本的な条件に基づいて行った。一方、pET21a-FRT-tktAを鋳型として-8TC-FM1(CGAAATTAATACGACCCACTATAGGGG,配列番号23)とT7ターミネータープライマーのペアを用いて、Phusion Hot Start II DNAポリメラーゼによりDNA断片を増幅した。つぎに、2つの増幅断片を鋳型としてF-BglとT7ターミネータープライマーのペアを用いて、Phusion Hot Start II DNAポリメラーゼによりオーバーラップエクステンションPCRを行い、T7プロモーターの転写開始起点から8塩基上流の配列がTからCに置換された変異型T7プロモーター配列を含むDNA断片を得た。
このDNA断片とpET21a-FRTベクターをそれぞれBglIIとXhoIで消化し、1%アガロースゲル電気泳動でDNAを分離した後、QIAquick Gel Extraction kitを用いて該当のDNAをゲルから抽出・精製した。精製されたDNAを常法によりLigationし、大腸菌DH5αを形質転換し、20μg/mlのカナマイシンを含むLB寒天培地を用いて37℃で一晩培養した。T7プロモータープライマーとT7ターミネータープライマーを用いたコロニーダイレクトPCRを行い、目的のプラスミドを持つ菌株を選別した。最終的に目的プラスミドを有する菌株を20μg/mlのカナマイシンを含むLB液体培地を用いて37℃で一晩培養し、NucleoSpin Plasmid QuickPureを用いて目的のプラスミドを抽出し精製した。
大腸菌MG1655のゲノムDNAを鋳型として、EcTyrA-F(CCAACCATATGGTTGCTGAATTGACCGC,配列番号24)とEcTyrA-RM1(CGCGAGGCCAAGATAGATGCCTCGCGC,配列番号25)プライマーペア、EcTyrA-FM1(GCGCGAGGCATCTATCTTGGCCTCGCG,配列番号26)とEcTyrA-RM2(CTCTGAAAACGCTGTACGTAATCGCCGAAC,配列番号27)プライマーペア、及びEcTyrA-FM2(GTTCGGCGATTACGTACAGCGTTTTCAGAG,配列番号28)とEcTyrA-R(CACTCGAGTTACTGGCGATTGTCATTCGCC,配列番号29)プライマーペアを用いて、Phusion Hot Start II DNAポリメラーゼにより3つのDNA断片を増幅した。なお、PCR反応の条件は、説明書に記載の基本的な条件に基づいて行った。つぎに、3つの増幅断片を鋳型としてEcTyrA-FとEcTyrA-Rのプライマーペアを用いて、Phusion Hot Start II DNAポリメラーゼによりオーバーラップエクステンションPCRを行い、53番目のメチオニンがイソロイシンに置換され、かつ354番目のアラニンがバリンに置換されたTyrAをコードするDNA(tyrAfbr)を得た。
増幅されたDNAとpET21a-FRTベクターをそれぞれNdeIとXhoIで消化し、1%アガロースゲル電気泳動でDNAを分離した後、QIAquick Gel Extraction kitを用いて該当のDNAをゲルから抽出・精製した。精製されたDNAを常法によりLigationし、大腸菌DH5αを形質転換し、20μg/mlのカナマイシンを含むLB寒天培地を用いて37℃で一晩培養した。T7プロモータープライマーとT7ターミネータープライマーを用いたコロニーダイレクトPCRを行い、目的のプラスミドを持つ菌株を選別した。最終的に目的プラスミドを有する菌株を20μg/mlのカナマイシンを含むLB液体培地を用いて37℃で一晩培養し、NucleoSpin Plasmid QuickPureを用いて目的のプラスミドを抽出し精製した。
大腸菌MG1655のゲノムDNAを鋳型としてtyrB-Nde(CAACACATATGTTTCAAAAAGTTGACGC,配列番号30)とtyrB-Xho(TACTCGAGTTACATCACCGCAGCAAAC,配列番号31)プライマーペアを用いて、Phusion Hot Start II DNAポリメラーゼによりtyrB遺伝子のDNA断片を増幅した。なお、PCR反応の条件は、説明書に記載の基本的な条件に基づいて行った。
増幅されたDNAとpET21a-FRTベクターをそれぞれNdeIとXhoIで消化し、1%アガロースゲル電気泳動でDNAを分離した後、QIAquick Gel Extraction kitを用いて該当のDNAをゲルから抽出・精製した。精製されたDNAを常法によりLigationし、大腸菌DH5αを形質転換し、20μg/mlのカナマイシンを含むLB寒天培地を用いて37℃で一晩培養した。T7プロモータープライマーとT7ターミネータープライマーを用いたコロニーダイレクトPCRを行い、目的のプラスミドを持つ菌株を選別した。最終的に目的プラスミドを有する菌株を20μg/mlのカナマイシンを含むLB液体培地を用いて37℃で一晩培養し、NucleoSpin Plasmid QuickPureを用いて目的のプラスミドを抽出し精製した。
大腸菌MG1655のゲノムDNAを鋳型としてAroB-opt-gib-F(GTTTAACTTTAAGAAGGAGATATACATATGGAGCGTATTGTCGTTACTC,配列番号32)とAroB-gib-R(GTGGTGGTGGTGCTCGAGTTACGCTGATTGACAATCGGC,配列番号33)プライマーペアを用いて、Phusion Hot Start II DNAポリメラーゼによりaroB遺伝子のDNAを増幅した。なお、PCR反応の条件は、説明書に記載の基本的な条件に基づいて行った。
DpnIで処理したaroB遺伝子のDNAとNdeIとXhoIで消化したpET21a-FRTをギブソン・アッセンブリー・システム(New England Biolabs、以下同じ)を用いて反応させ、大腸菌DH5αを形質転換し、20μg/mlのカナマイシンを含むLB寒天培地を用いて37℃で一晩培養した。T7プロモータープライマーとT7ターミネータープライマーを用いたコロニーダイレクトPCRを行い、目的のプラスミドを持つ菌株を選別した。最終的に目的プラスミドを有する菌株を20μg/mlのカナマイシンを含むLB液体培地を用いて37℃で一晩培養し、NucleoSpin Plasmid QuickPureを用いて目的のプラスミドを抽出し精製した。
大腸菌MG1655のゲノムDNAを鋳型としてEcAroA-F(CAACACCATGGAATCCCTGACGTTACAAC,配列番号34)とEcAroA-R(CAAAGCTTTCAGGCTGCCTGGCTAATCC,配列番号35)プライマーペア、EcAroL-F(CCAACCATATGACACAACCTCTTTTTCTGATC,配列番号36)とEcAroL-R(CACTCGAGTCAACAATTGATCGTCTGTGCC,配列番号37)プライマーペア、及びEcAroC-F(CAACACCATGGCTGGAAACACAATTGGAC,配列番号38)とEcAroC-R(CAAAGCTTTTACCAGCGTGGAATATCAGTC,配列番号39)プライマーペアを用いて、Phusion Hot Start II DNAポリメラーゼによりaroA、aroL、及びaroC遺伝子のDNAを増幅した。なお、PCR反応の条件は、説明書に記載の基本的な条件に基づいて行った。aroA遺伝子とaroC遺伝子のDNAをNcoIとHindIIIで消化し、一方、aroL遺伝子のDNAをNdeIとXhoIで消化し、1%アガロースゲル電気泳動でDNAを分離した後、QIAquick Gel Extraction kitを用いてそれぞれのDNAをゲルから抽出・精製した。pET21a-FRTベクターをNdeIとXhoIで消化し、pET21d-FRTベクター(非特許文献2、Koma et al.Appl.Microbiol.Biotechnol.93:815-829,2012)をNcoIとHindIIIで消化し、1%アガロースゲル電気泳動でDNAを分離した後、QIAquick Gel Extraction kitを用いて該当のDNAをゲルから抽出・精製した。精製されたaroA遺伝子のDNAとpET21d-FRT、精製されたaroC遺伝子のDNAとpET21d-FRT、及び精製されたaroL遺伝子のDNAとpET21a-FRTを常法によりそれぞれLigationし、大腸菌DH5αを形質転換し、20μg/mlのカナマイシンを含むLB寒天培地を用いて37℃で一晩培養した。T7プロモータープライマーとT7ターミネータープライマーを用いたコロニーダイレクトPCRを行い、目的のプラスミドを持つ菌株を選別した。最終的に目的プラスミドを有する菌株を20μg/mlのカナマイシンを含むLB液体培地を用いて37℃で一晩培養し、NucleoSpin Plasmid QuickPureを用いて抽出し精製することで、pET21d-FRT-aroA、pET21d-FRT-aroC、及びpET21a-FRT-aroLを得た。
pET21d-FRT-aroAを鋳型としてALC-F(GAAATTAATACGACTCACTATAGGGGAATTG,配列番号40)とALC-aroA-R(TCAGGCTGCCTGGCTAATC,配列番号41)プライマーペアを用いて、Phusion Hot Start II DNAポリメラーゼによりaroA遺伝子を含むDNA断片を増幅した。また、pET21a-FRT-aroLを鋳型として、ALC-aroL-F(GATTAGCCAGGCAGCCTGACTTTAAGAAGGAGATATACATATGACAC,配列番号42)とALC-aroL-R(GGATGGCCTCCTTTAGATCCTCAACAATTGATCGTCTGTGC,配列番号43)プライマーペアを用いて、Phusion Hot Start II DNAポリメラーゼによりaroL遺伝子を含むDNA断片を増幅した。さらに、pET21d-FRT-aroCを鋳型としてALC-aroC-F(GGATCTAAAGGAGGCCATCCATGGCTGGAAACACAATTGG,配列番号44)とALCorEDB-R(GGATGGCCTCCTTTAGATCCTCAACAATTGATCGTCTGTGC,配列番号45)プライマーペアを用いて、Phusion Hot Start II DNAポリメラーゼによりaroC遺伝子を含むDNA断片を増幅した。つぎに、3つの増幅断片を鋳型としてaroALC-F1(ATGGAATCCCTGACGTTACAAC,配列番号46)とaroALC-R1(TTACCAGCGTGGAATATCAGTCTTC,配列番号47)のプライマーペアを用いて、Phusion Hot Start II DNAポリメラーゼによりオーバーラップエクステンションPCRを行い、aroALCを含むDNA断片を得た。得られたDNA断片をpCRTMII-Blunt-TOPOTM(Thermo Fisher Scientific、以下同じ)を含むTOPOクローニングの試薬と反応させ、大腸菌DH5αを形質転換し、20μg/mlのカナマイシンを含むLB寒天培地を用いて37℃で一晩培養した。aroALC-F1とaroALC-R1を用いたコロニーダイレクトPCRを行い、目的のプラスミドを持つ菌株を選別した。最終的に目的プラスミドを有する菌株を20μg/mlのカナマイシンを含むLB液体培地を用いて37℃で一晩培養し、NucleoSpin Plasmid QuickPureを用いてpCRTMII-Blunt-TOPOTM-aroALCを抽出し精製した。
一方、pET21a-FRTを鋳型として、pET21a-ALC-R(GTAACGTCAGGGATTCCATGGTAATATCTCCTTCTTAAAGTTAAAC,配列番号48)とpET21a-ALC-F(CTGATATTCCACGCTGGTAACTCGAGCACCACCACCACC,配列番号49)プライマーペアを用いて、Phusion Hot Start II DNAポリメラーゼにより、プラスミドバックボーンとなるDNA断片を増幅した。得られたDNA断片をpCRTMII-Blunt-TOPOTMを含むTOPOクローニングの試薬と反応させ、大腸菌DH5αを形質転換し、20μg/mlのカナマイシンを含むLB寒天培地を用いて37℃で一晩培養した。pET21a-ALC-RとpET21a-ALC-Fを用いたコロニーダイレクトPCRを行い、目的のプラスミドを持つ菌株を選別した。最終的に目的プラスミドを有する菌株を20μg/mlのカナマイシンを含むLB液体培地を用いて37℃で一晩培養し、NucleoSpin Plasmid QuickPureを用いてpCRTMII-Blunt-TOPOTM-BB-aroALCを抽出し精製した。
つぎに、pCRTMII-Blunt-TOPOTM-aroALCを鋳型として、aroALC-F1とaroALC-R1プライマーペアを用いて、Phusion Hot Start II DNAポリメラーゼによりaroALC遺伝子を含むDNA断片を増幅した。一方、pCRTMII-Blunt-TOPOTM-BB-aroALCを鋳型として、pET21a-ALC-RとpET21a-ALC-Fプライマーペアを用いて、Phusion Hot Start II DNAポリメラーゼによりプラスミドバックボーンを含むDNA断片を得た。得られた両DNA断片をギブソン・アッセンブリー・システムを用いて反応させ、大腸菌DH5αを形質転換し、50μg/mlのアンピシリンを含むLB寒天培地を用いて37℃で一晩培養した。aroALC-F1とaroALC-R1プライマーペアを用いたコロニーダイレクトPCRを行い、目的のプラスミドを持つ菌株を選別した。最終的に目的プラスミドを有する菌株を50μg/mlのアンピシリンを含むLB液体培地を用いて37℃で一晩培養し、NucleoSpin Plasmid QuickPureを用いて目的のプラスミドを抽出し精製した。
大腸菌MG1655のゲノムDNAを鋳型としてEcAroE-F(CAACCATATGGAAACCTATGCTGTTTTTGG,配列番号50)とEcAroE-R(CACTCGAGTCACGCGGACAATTCCTCC,配列番号51)プライマーペア、EcAroD-F(CCAACCATATGAAAACCGTAACTGTAAAAGATC,配列番号52)とEcAroD-R(CACTCGAGTTATGCCTGGTGTAAAATAGTTAATAC,配列番号53)プライマーペア、及びEcAroB-F(CAACACCATGGAGAGGATTGTCGTTACTC,配列番号54)とEcAroB-R(CAAAGCTTTTACGCTGATTGACAATCGGC,配列番号55)プライマーペアを用いて、Phusion Hot Start II DNAポリメラーゼによりaroE、aroD、及びaroB遺伝子のDNAを増幅した。なお、PCR反応の条件は、説明書に記載の基本的な条件に基づいて行った。aroE遺伝子とaroD遺伝子のDNAをNdeIとXhoIで消化し、一方、aroB遺伝子のDNAをNcoIとHindIIIで消化し、1%アガロースゲル電気泳動でDNAを分離した後、QIAquick Gel Extraction kitを用いてそれぞれのDNAをゲルから抽出・精製した。pET21a-FRTベクターをNdeIとXhoIで消化し、pET21d-FRTベクターをNcoIとHindIIIで消化し、1%アガロースゲル電気泳動でDNAを分離した後、QIAquick Gel Extraction kitを用いて該当のDNAをゲルから抽出・精製した。精製されたaroE遺伝子のDNAとpET21a-FRT、精製されたaroD遺伝子のDNAとpET21a-FRT、及び精製されたaroB遺伝子のDNAとpET21d-FRTを常法によりそれぞれLigationし、大腸菌DH5αを形質転換し、20μg/mlのカナマイシンを含むLB寒天培地を用いて37℃で一晩培養した。T7プロモータープライマーとT7ターミネータープライマーを用いたコロニーダイレクトPCRを行い、目的のプラスミドを持つ菌株を選別した。最終的に目的プラスミドを有する菌株を20μg/mlのカナマイシンを含むLB液体培地を用いて37℃で一晩培養し、NucleoSpin Plasmid QuickPureを用いて抽出し精製することで、pET21d-FRT-aroE、pET21d-FRT-aroD、及びpET21a-FRT-aroBを得た。
pET21d-FRT-aroEを鋳型としてEDB-F(GAAATTAATACGACTCACTATAGGGGAATTG,配列番号56)とEDB-aroE-R(CATATGTATATCTCCTTCTTAAAGTCACGCGGACAATTCCTCC,配列番号57)プライマーペアを用いて、Phusion Hot Start II DNAポリメラーゼによりaroE遺伝子を含むDNA断片を増幅した。また、pET21d-FRT-aroDを鋳型として、EDB-aroD-F(CGTGACTTTAAGAAGGAGATATACATATGAAAACCGTAACTGTAAAAGACC,配列番号58)とEDB-aroD-R(GGATGGCCTCCTTTAGATCCTTATGCCTGGTGTAAAATAGTTAATAC,配列番号59)プライマーペアを用いて、Phusion Hot Start II DNAポリメラーゼによりaroD遺伝子を含むDNA断片を増幅した。さらに、pET21a-FRT-aroBを鋳型としてEDB-aroB-F2(GGATCTAAAGGAGGCCATCCATGGAGCGTATTGTCGTTACTC,配列番号60)とALCorEDB-Rプライマーペアを用いて、Phusion Hot Start II DNAポリメラーゼによりaroB遺伝子を含むDNA断片を増幅した。つぎに、3つの増幅断片を鋳型としてaroEDB-F1(ATGGAAACCTATGCTGTTTTTGG,配列番号61)とaroEDB-R1(TTACGCTGATTGACAATCGGC,配列番号62)のプライマーペアを用いて、Phusion Hot Start II DNAポリメラーゼによりオーバーラップエクステンションPCRを行い、aroEDBを含むDNA断片を得た。得られたDNA断片をpCRTMII-Blunt-TOPOTMを含むTOPOクローニングの試薬と反応させ、大腸菌DH5αを形質転換し、20μg/mlのカナマイシンを含むLB寒天培地を用いて37℃で一晩培養した。aroEDB-F1とaroEDB-R1を用いたコロニーダイレクトPCRを行い、目的のプラスミドを持つ菌株を選別した。最終的に目的プラスミドを有する菌株を20μg/mlのカナマイシンを含むLB液体培地を用いて37℃で一晩培養し、NucleoSpin Plasmid QuickPureを用いてpCRTMII-Blunt-TOPOTM-aroEDBを抽出し精製した。
一方、pET21a-FRTを鋳型として、pET21a-EDB-R(CAAAAACAGCATAGGTTTCCATATGTATATCTCCTTCTTAAAGTTAAAC,配列番号63)とpET21a-EDB-F(CGATTGTCAATCAGCGTAACTCGAGCACCACCACCACC,配列番号64)プライマーペアを用いて、Phusion Hot Start II DNAポリメラーゼにより、プラスミドバックボーンとなるDNA断片を増幅した。得られたDNA断片をpCRTMII-Blunt-TOPOTMを含むTOPOクローニングの試薬と反応させ、大腸菌DH5αを形質転換し、20μg/mlのカナマイシンを含むLB寒天培地を用いて37℃で一晩培養した。pET21a-EDB-RとpET21a-EDB-Fを用いたコロニーダイレクトPCRを行い、目的のプラスミドを持つ菌株を選別した。最終的に目的プラスミドを有する菌株を20μg/mlのカナマイシンを含むLB液体培地を用いて37℃で一晩培養し、NucleoSpin Plasmid QuickPureを用いてpCRTMII-Blunt-TOPOTM-BB-aroEDBを抽出し精製した。
つぎに、pCRTMII-Blunt-TOPOTM-aroEDBを鋳型として、aroEDB-F1とaroEDB-R1プライマーペアを用いて、Phusion Hot Start II DNAポリメラーゼによりaroEDB遺伝子を含むDNA断片を増幅した。一方、pCRTMII-Blunt-TOPOTM-BB-aroEDBを鋳型として、pET21a-EDB-RとpET21a-EDB-Fプライマーペアを用いて、Phusion Hot Start II DNAポリメラーゼによりプラスミドバックボーンを含むDNA断片を得た。得られた両DNA断片をギブソン・アッセンブリー・システムを用いて反応させ、大腸菌DH5αを形質転換し、50μg/mlのアンピシリンを含むLB寒天培地を用いて37℃で一晩培養した。aroEDB-F1とaroEDB-R1プライマーペアを用いたコロニーダイレクトPCRを行い、目的のプラスミドを持つ菌株を選別した。最終的に目的プラスミドを有する菌株を50μg/mlのアンピシリンを含むLB液体培地を用いて37℃で一晩培養し、NucleoSpin Plasmid QuickPureを用いて目的のプラスミドを抽出し精製した。
pET21a-FRT-aroALCを鋳型としてaroLC-DB-F(CTTAAAGTTAAACAAAATTATTTCTAGAGG,配列番号65)とaroLC-R(AAGGAGATATACATATGACACAACCTC,配列番号66)プライマーペアを用いて、Phusion Hot Start II DNAポリメラーゼによりaroLC遺伝子を含むDNA断片を増幅した。得られたDNA断片をQIAquick PCR Purification Kit(Qiagen、以下同じ)で精製した後にDpnIで消化し、再びQIAquick PCR Purification Kitで精製した後に、T4キナーゼ(Takara)を用いてリン酸化した。リン酸化されたDNA断片をQIAquick PCR Purification Kitで精製した後に、DNAライゲーションキット(Takara、以下同じ)を用いてセルフライゲーションさせ、大腸菌DH5αを形質転換して、20μg/mlのカナマイシンを含むLB寒天培地を用いて37℃で一晩培養した。T7プロモータープライマーとT7ターミネータープライマーペアを用いたコロニーダイレクトPCRを行い、目的のプラスミドを持つ菌株を選別した。最終的に目的プラスミドを有する菌株を20μg/mlのカナマイシンを含むLB液体培地を用いて37℃で一晩培養し、NucleoSpin Plasmid QuickPureを用いてプラスミドを抽出し精製することでpET21a-FRT-aroLCを得た。
pET21a-FRT-aroALCを鋳型としてaroAC-F(CTTAAAGTCAGGCTGCCTGGC,配列番号67)とaroAC-R(AAGGAGGCCATCCATGGCTGGAAAC,配列番号68)プライマーペアを用いて、Phusion Hot Start II DNAポリメラーゼによりaroAC遺伝子を含むDNA断片を増幅した。得られたDNA断片をQIAquick PCR Purification Kitで精製した後にDpnIで消化し、再びQIAquick PCR Purification Kitで精製した後に、T4キナーゼ(Takara)を用いてリン酸化した。リン酸化されたDNA断片をQIAquick PCR Purification Kitで精製した後に、DNAライゲーションキットを用いてセルフライゲーションさせ、大腸菌DH5αを形質転換して、20μg/mlのカナマイシンを含むLB寒天培地を用いて37℃で一晩培養した。T7プロモータープライマーとT7ターミネータープライマーペアを用いたコロニーダイレクトPCRを行い、目的のプラスミドを持つ菌株を選別した。最終的に目的プラスミドを有する菌株を20μg/mlのカナマイシンを含むLB液体培地を用いて37℃で一晩培養し、NucleoSpin Plasmid QuickPureを用いてプラスミドを抽出し精製することでpET21a-FRT-aroACを得た。
pET21a-FRT-aroALCを鋳型としてaroAL-F(TCAACAATTGATCGTCTGTGCC,配列番号69)とaroAL-ED-R(CTCGAGCACCACCACCAC,配列番号70)プライマーペアを用いて、Phusion Hot Start II DNAポリメラーゼによりaroAL遺伝子を含むDNA断片を増幅した。得られたDNA断片をQIAquick PCR Purification Kitで精製した後にDpnIで消化し、再びQIAquick PCR Purification Kitで精製した後に、T4キナーゼ(Takara)を用いてリン酸化した。リン酸化されたDNA断片をQIAquick PCR Purification Kitで精製した後に、DNAライゲーションキットを用いてセルフライゲーションさせ、大腸菌DH5αを形質転換して、20μg/mlのカナマイシンを含むLB寒天培地を用いて37℃で一晩培養した。T7プロモータープライマーとT7ターミネータープライマーペアを用いたコロニーダイレクトPCRを行い、目的のプラスミドを持つ菌株を選別した。最終的に目的プラスミドを有する菌株を20μg/mlのカナマイシンを含むLB液体培地を用いて37℃で一晩培養し、NucleoSpin Plasmid QuickPureを用いてプラスミドを抽出し精製することでpET21a-FRT-aroALを得た。
pET21a-FRT-aroEDBを鋳型としてaroLC-DB-FとaroDB-R(AAGGAGATATACATATGAAAACCGTAAC,配列番号71)プライマーペアを用いて、Phusion Hot Start II DNAポリメラーゼによりaroDB遺伝子を含むDNA断片を増幅した。得られたDNA断片をQIAquick PCR Purification Kitで精製した後にDpnIで消化し、再びQIAquick PCR Purification Kitで精製した後に、T4キナーゼ(Takara)を用いてリン酸化した。リン酸化されたDNA断片をQIAquick PCR Purification Kitで精製した後に、DNAライゲーションキットを用いてセルフライゲーションさせ、大腸菌DH5αを形質転換して、20μg/mlのカナマイシンを含むLB寒天培地を用いて37℃で一晩培養した。T7プロモータープライマーとT7ターミネータープライマーペアを用いたコロニーダイレクトPCRを行い、目的のプラスミドを持つ菌株を選別した。最終的に目的プラスミドを有する菌株を20μg/mlのカナマイシンを含むLB液体培地を用いて37℃で一晩培養し、NucleoSpin Plasmid QuickPureを用いてプラスミドを抽出し精製することでpET21a-FRT-aroDBを得た。
pET21a-FRT-aroEDBを鋳型としてaroEB-F(TCACGCGGACAATTCCTCC,配列番号72)とaroEB-R(GGATCTAAAGGAGGCCATCCATG,配列番号73)プライマーペアを用いて、Phusion Hot Start II DNAポリメラーゼによりaroEB遺伝子を含むDNA断片を増幅した。得られたDNA断片をQIAquick PCR Purification Kitで精製した後にDpnIで消化し、再びQIAquick PCR Purification Kitで精製した後に、T4キナーゼ(Takara)を用いてリン酸化した。リン酸化されたDNA断片をQIAquick PCR Purification Kitで精製した後に、DNAライゲーションキットを用いてセルフライゲーションさせ、大腸菌DH5αを形質転換して、20μg/mlのカナマイシンを含むLB寒天培地を用いて37℃で一晩培養した。T7プロモータープライマーとT7ターミネータープライマーペアを用いたコロニーダイレクトPCRを行い、目的のプラスミドを持つ菌株を選別した。最終的に目的プラスミドを有する菌株を20μg/mlのカナマイシンを含むLB液体培地を用いて37℃で一晩培養し、NucleoSpin Plasmid QuickPureを用いてプラスミドを抽出し精製することでpET21a-FRT-aroEBを得た。
pET21a-FRT-aroEDBを鋳型としてaroED-F(TTATGCCTGGTGTAAAATAGTTAATACC,配列番号74)とaroAL-ED-Rプライマーペアを用いて、Phusion Hot Start II DNAポリメラーゼによりaroED遺伝子を含むDNA断片を増幅した。得られたDNA断片をQIAquick PCR Purification Kitで精製した後にDpnIで消化し、再びQIAquick PCR Purification Kitで精製した後に、T4キナーゼ(Takara)を用いてリン酸化した。リン酸化されたDNA断片をQIAquick PCR Purification Kitで精製した後に、DNAライゲーションキットを用いてセルフライゲーションさせ、大腸菌DH5αを形質転換して、20μg/mlのカナマイシンを含むLB寒天培地を用いて37℃で一晩培養した。T7プロモータープライマーとT7ターミネータープライマーペアを用いたコロニーダイレクトPCRを行い、目的のプラスミドを持つ菌株を選別した。最終的に目的プラスミドを有する菌株を20μg/mlのカナマイシンを含むLB液体培地を用いて37℃で一晩培養し、NucleoSpin Plasmid QuickPureを用いてプラスミドを抽出し精製することでpET21a-FRT-aroEDを得た。
表1に示す鋳型DNAとプライマーペアを用いて、PrimeSTAR GXL DNAポリメラーゼ(タカラバイオ)により、表1に示すように、大腸菌の染色体に導入するためのDNAカセットを増幅した。プライマーの配列は表2に示す。



染色体に1遺伝子を導入した菌株を20μg/mlのカナマイシンを含む5mlのLB液体培地に植菌し、37℃でOD660が0.1程度になるまで培養した。1Mの塩化カルシウムを50μl添加し、さらに100μlのP1ファージ溶液(>108pfu/ml)を添加して37℃で3~4時間程度培養した。10,000rpmで5分間遠心分離し、上澄みを0.2μmのポアサイズの滅菌フィルターでろ過することで、P1ファージライセートを得た。
pET21a-FRT-aroGfbrを鋳型としてdelta-tyrR(GTGTCATATCATCATATTAATTGTTCTTTTTTCAGGTGAAGGTTCCCATGATTCCGGGGATCCGTCGACC,配列番号94)とdelta-tyrR-FRT-R(TGGTGTTGCACCATCAGGCATATTCGCGCTTACTCTTCGTTCTTCTTCTGTGTAGGCTGGAGCTGCTTCG,配列番号95)プライマーペアを用いて、PrimeSTAR GXL DNAポリメラーゼ(タカラバイオ)によりFRT-Km-FRT-aroGfbrのDNAを増幅した。得られたDNAをQIAquick PCR Purification Kitで精製した後にDpnIで消化し、再びQIAquick PCR Purification Kitで精製した。
大腸菌MG1655(DE3)/pKD46を10mMのアラビノースと100μg/mlのアンピシリンを含むLB培地でOD660の値が0.5程度になるまで培養した。培養液を氷冷した後、3mlの培養液を10,000rpm、4℃で3分間遠心分離し、上清を捨てた。菌体を氷冷した1mlの滅菌水に懸濁し、10,000rpm、4℃で3分間遠心分離し、上清を捨てた。この操作をもう一度繰り返した後、菌体を50μlの滅菌水に懸濁し、約100ngのFRT-Km-FRT-aroGfbrを含むDNA溶液を加えた後に、エレクトロポレーションを行った。1mlのLB培地を用いてエレクトロポレーションキュベットから菌体を回収し、37℃で2.5時間振とうした後に20μg/mlのカナマイシンを含むLB寒天培地を用いて37℃で一晩培養した。K2とD-tyrR(CGTAAGTTTAACCAACTGGCAACTG,配列番号96)プライマーペアを用いたコロニーダイレクトPCRを行い、tyrR座位にFRT-Km-FRT-aroGfbrが挿入された菌株を選別した。
つぎにこの株を20μg/mlのカナマイシンを含むLB液体培地で培養し、OD660の値が0.5程度になるまで培養した。培養液を氷冷した後、1mlの培養液を10,000rpm、4℃で3分間遠心分離し、上清を捨てた。菌体を100μlのTSS溶液(10%ポリエチレングリコール3350、5%ジメチルスルホキシド、70mM塩化マグネシウム、0.1M塩化カリウム、30mM塩化カルシウム)に懸濁し、50ng/μlのpCP20を1μl添加し、氷中で30分間静置した。42℃で90秒間ヒートショックした後に氷中で2分間静置し、900μlのLB培地を加え、100μlを50μg/mlのアンピシリンを含むLB寒天培地に塗布した。30℃で一晩培養し、出現したコロニーをLB寒天培地にストリークして、42℃で一晩培養した。つぎに、出現したコロニーを新たなLB寒天培地にストリークして、37℃で一晩培養した。U-tyrR(GTTTAATTAATCGCATCGCCACGC,配列番号97)とD-tyrRプライマーペアを用いて、tyrR座位に挿入されたFRT-Km-FRT-aroGfbrからカナマイシン耐性遺伝子が除去されたことを確認し、MG1655(DE3)tyrR::PT7-aroGfbr株を得た。
つぎにこの株を5mlのLB液体培地を用いて37℃で一晩培養した。50μlの1M塩化カルシウムを加えて良く撹拌した後、200μlを1.5mlチューブに移した。そこにS1株のP1ファージライセートを20μl加えて混合し、37℃で20分間恒温した。1Mのクエン酸3ナトリウム溶液を100μlとLB液体培地を700μl加えて混合した後に、37℃でさらに40分間恒温した。20μg/mlのカナマイシンを含むLB寒天培地に菌液を塗布し、37℃で一晩培養した。K2とD-ldhAのプライマーペアを用いたコロニーダイレクトPCRを行い、得られた株のldhA座位にFRT-Km-FRT-PT7-pheAfbrが挿入されていることを確認した。
つぎにこの株を20μg/mlのカナマイシンを含むLB液体培地で培養し、OD660の値が0.5程度になるまで培養し、前述のpCP20を用いた方法によりカナマイシン耐性遺伝子を除去することにより、MG1655(DE3)tyrR::PT7-aroGfbr ldhA::PT7-pheAfbr株を得た。この株をPhe1株と命名した。
Phe1株を5mlのLB液体培地を用いて37℃で一晩培養した。50μlの1M塩化カルシウムを加えて良く撹拌した後、200μlを1.5mlチューブに移した。そこにS2株のP1ファージライセートを20μl加えて混合し、37℃で20分間恒温した。1Mのクエン酸3ナトリウム溶液を100μlとLB液体培地を700μl加えて混合した後に、37℃でさらに40分間恒温した。20μg/mlのカナマイシンを含むLB寒天培地に菌液を塗布し、37℃で一晩培養した。K2とD-adhEのプライマーペアを用いたコロニーダイレクトPCRを行い、得られた株のadhE座位にFRT-Km-FRT-PT7-ppsAが挿入されていることを確認した。つぎに、Phe1株の作製で記載した方法に基づいて、カナマイシン耐性遺伝子を除去し、MG1655(DE3) tyrR::PT7-aroGfbr ldhA::PT7-pheAfbr adhE::PT7-ppsA株を得た。この株をPhe2株と命名した。
Phe2株を5mlのLB液体培地を用いて37℃で一晩培養した。50μlの1M塩化カルシウムを加えて良く撹拌した後、200μlを1.5mlチューブに移した。そこにS3株のP1ファージライセートを20μl加えて混合し、37℃で20分間恒温した。1Mのクエン酸3ナトリウム溶液を100μlとLB液体培地を700μl加えて混合した後に、37℃でさらに40分間恒温した。20μg/mlのカナマイシンを含むLB寒天培地に菌液を塗布し、37℃で一晩培養した。K2とD-pflDCのプライマーペアを用いたコロニーダイレクトPCRを行い、得られた株のpflDC座位にFRT-Km-FRT-PT7-tktAが挿入されていることを確認した。つぎに、Phe1株の作製で記載した方法に基づいて、カナマイシン耐性遺伝子を除去し、MG1655(DE3) tyrR::PT7-aroGfbr ldhA::PT7-pheAfbr adhE::PT7-ppsA pflDC::PT7-tktA株を得た。この株をPhe3株と命名した。
Phe2株を5mlのLB液体培地を用いて37℃で一晩培養した。50μlの1M塩化カルシウムを加えて良く撹拌した後、200μlを1.5mlチューブに移した。そこにS4株のP1ファージライセートを20μl加えて混合し、37℃で20分間恒温した。1Mのクエン酸3ナトリウム溶液を100μlとLB液体培地を700μl加えて混合した後に、37℃でさらに40分間恒温した。20μg/mlのカナマイシンを含むLB寒天培地に菌液を塗布し、37℃で一晩培養した。K2とD-pflDCのプライマーペアを用いたコロニーダイレクトPCRを行い、得られた株のpflDC座位にFRT-Km-FRT-PT7(-8TC)-tktAが挿入されていることを確認した。つぎに、Phe1株の作製で記載した方法に基づいて、カナマイシン耐性遺伝子を除去し、MG1655(DE3) tyrR::PT7-aroGfbr ldhA::PT7-pheAfbr adhE::PT7-ppsA pflDC::PT7(-8TC)-tktA株を得た。この株をPhe4株と命名した。
Phe1株を5mlのLB液体培地を用いて37℃で一晩培養した。50μlの1M塩化カルシウムを加えて良く撹拌した後、200μlを1.5mlチューブに移した。そこにS8株のP1ファージライセートを20μl加えて混合し、37℃で20分間恒温した。1Mのクエン酸3ナトリウム溶液を100μlとLB液体培地を700μl加えて混合した後に、37℃でさらに40分間恒温した。20μg/mlのカナマイシンを含むLB寒天培地に菌液を塗布し、37℃で一晩培養した。K2とD-ascFのプライマーペアを用いたコロニーダイレクトPCRを行い、得られた株のascF座位にFRT-Km-FRT-PT7-aroEDBが挿入されていることを確認した。つぎに、Phe1株の作製で記載した方法に基づいて、カナマイシン耐性遺伝子を除去し、MG1655(DE3) tyrR::PT7-aroGfbr ldhA::PT7-pheAfbr ascF::PT7-aroEDB株を得た。この株をPhe5株と命名した。
Phe3株を5mlのLB液体培地を用いて37℃で一晩培養した。50μlの1M塩化カルシウムを加えて良く撹拌した後、200μlを1.5mlチューブに移した。そこにS8株のP1ファージライセートを20μl加えて混合し、37℃で20分間恒温した。1Mのクエン酸3ナトリウム溶液を100μlとLB液体培地を700μl加えて混合した後に、37℃でさらに40分間恒温した。20μg/mlのカナマイシンを含むLB寒天培地に菌液を塗布し、37℃で一晩培養した。K2とD-ascFのプライマーペアを用いたコロニーダイレクトPCRを行い、得られた株のascF座位にFRT-Km-FRT-PT7-aroEDBが挿入されていることを確認した。つぎに、Phe1株の作製で記載した方法に基づいて、カナマイシン耐性遺伝子を除去し、MG1655(DE3) tyrR::PT7-aroGfbr ldhA::PT7-pheAfbr adhE::PT7-ppsA pflDC::PT7-tktA ascF::PT7-aroEDB株を得た。この株をPhe6株と命名した。
Phe4株を5mlのLB液体培地を用いて37℃で一晩培養した。50μlの1M塩化カルシウムを加えて良く撹拌した後、200μlを1.5mlチューブに移した。そこにS8株のP1ファージライセートを20μl加えて混合し、37℃で20分間恒温した。1Mのクエン酸3ナトリウム溶液を100μlとLB液体培地を700μl加えて混合した後に、37℃でさらに40分間恒温した。20μg/mlのカナマイシンを含むLB寒天培地に菌液を塗布し、37℃で一晩培養した。K2とD-ascFのプライマーペアを用いたコロニーダイレクトPCRを行い、得られた株のascF座位にFRT-Km-FRT-PT7-aroEDBが挿入されていることを確認した。つぎに、Phe1株の作製で記載した方法に基づいて、カナマイシン耐性遺伝子を除去し、MG1655(DE3) tyrR::PT7-aroGfbr ldhA::PT7-pheAfbr adhE::PT7-ppsA pflDC::PT7(-8TC)-tktA ascF::PT7-aroEDB株を得た。この株をPhe7株と命名した。
Phe1株を5mlのLB液体培地を用いて37℃で一晩培養した。50μlの1M塩化カルシウムを加えて良く撹拌した後、200μlを1.5mlチューブに移した。そこにS9株のP1ファージライセートを20μl加えて混合し、37℃で20分間恒温した。1Mのクエン酸3ナトリウム溶液を100μlとLB液体培地を700μl加えて混合した後に、37℃でさらに40分間恒温した。20μg/mlのカナマイシンを含むLB寒天培地に菌液を塗布し、37℃で一晩培養した。K2とD-pykFのプライマーペアを用いたコロニーダイレクトPCRを行い、得られた株のpykF座位にFRT-Km-FRT-PT7-aroALCが挿入されていることを確認した。つぎに、Phe1株の作製で記載した方法に基づいて、カナマイシン耐性遺伝子を除去し、MG1655(DE3) tyrR::PT7-aroGfbr ldhA::PT7-pheAfbr pykF::PT7-aroALC株を得た。この株をPhe8株と命名した。
Phe3株を5mlのLB液体培地を用いて37℃で一晩培養した。50μlの1M塩化カルシウムを加えて良く撹拌した後、200μlを1.5mlチューブに移した。そこにS9株のP1ファージライセートを20μl加えて混合し、37℃で20分間恒温した。1Mのクエン酸3ナトリウム溶液を100μlとLB液体培地を700μl加えて混合した後に、37℃でさらに40分間恒温した。20μg/mlのカナマイシンを含むLB寒天培地に菌液を塗布し、37℃で一晩培養した。K2とD-pykFのプライマーペアを用いたコロニーダイレクトPCRを行い、得られた株のpykF座位にFRT-Km-FRT-PT7-aroALCが挿入されていることを確認した。つぎに、Phe1株の作製で記載した方法に基づいて、カナマイシン耐性遺伝子を除去し、MG1655(DE3) tyrR::PT7-aroGfbr ldhA::PT7-pheAfbr adhE::PT7-ppsA pflDC::PT7-tktA pykF::PT7-aroALC株を得た。この株をPhe9株と命名した。
Phe4株を5mlのLB液体培地を用いて37℃で一晩培養した。50μlの1M塩化カルシウムを加えて良く撹拌した後、200μlを1.5mlチューブに移した。そこにS9株のP1ファージライセートを20μl加えて混合し、37℃で20分間恒温した。1Mのクエン酸3ナトリウム溶液を100μlとLB液体培地を700μl加えて混合した後に、37℃でさらに40分間恒温した。20μg/mlのカナマイシンを含むLB寒天培地に菌液を塗布し、37℃で一晩培養した。K2とD-pykFのプライマーペアを用いたコロニーダイレクトPCRを行い、得られた株のpykF座位にFRT-Km-FRT-PT7-aroALCが挿入されていることを確認した。つぎに、Phe1株の作製で記載した方法に基づいて、カナマイシン耐性遺伝子を除去し、MG1655(DE3) tyrR::PT7-aroGfbr ldhA::PT7-pheAfbr adhE::PT7-ppsA pflDC::PT7(-8TC)-tktA pykF::PT7-aroALC株を得た。この株をPhe10株と命名した。
Phe5株を5mlのLB液体培地を用いて37℃で一晩培養した。50μlの1M塩化カルシウムを加えて良く撹拌した後、200μlを1.5mlチューブに移した。そこにS9株のP1ファージライセートを20μl加えて混合し、37℃で20分間恒温した。1Mのクエン酸3ナトリウム溶液を100μlとLB液体培地を700μl加えて混合した後に、37℃でさらに40分間恒温した。20μg/mlのカナマイシンを含むLB寒天培地に菌液を塗布し、37℃で一晩培養した。K2とD-pykFのプライマーペアを用いたコロニーダイレクトPCRを行い、得られた株のpykF座位にFRT-Km-FRT-PT7-aroALCが挿入されていることを確認した。つぎに、Phe1株の作製で記載した方法に基づいて、カナマイシン耐性遺伝子を除去し、MG1655(DE3) tyrR::PT7-aroGfbr ldhA::PT7-pheAfbr ascF::PT7-aroEDB pykF::PT7-aroALC株を得た。この株をPhe11株と命名した。
Phe6株を5mlのLB液体培地を用いて37℃で一晩培養した。50μlの1M塩化カルシウムを加えて良く撹拌した後、200μlを1.5mlチューブに移した。そこにS9株のP1ファージライセートを20μl加えて混合し、37℃で20分間恒温した。1Mのクエン酸3ナトリウム溶液を100μlとLB液体培地を700μl加えて混合した後に、37℃でさらに40分間恒温した。20μg/mlのカナマイシンを含むLB寒天培地に菌液を塗布し、37℃で一晩培養した。K2とD-pykFのプライマーペアを用いたコロニーダイレクトPCRを行い、得られた株のpykF座位にFRT-Km-FRT-PT7-aroALCが挿入されていることを確認した。つぎに、Phe1株の作製で記載した方法に基づいて、カナマイシン耐性遺伝子を除去し、MG1655(DE3) tyrR::PT7-aroGfbr ldhA::PT7-pheAfbr adhE::PT7-ppsA pflDC::PT7-tktA ascF::PT7-aroEDB pykF::PT7-aroALC株を得た。この株をPhe12株と命名した。
Phe7株を5mlのLB液体培地を用いて37℃で一晩培養した。50μlの1M塩化カルシウムを加えて良く撹拌した後、200μlを1.5mlチューブに移した。そこにS9株のP1ファージライセートを20μl加えて混合し、37℃で20分間恒温した。1Mのクエン酸3ナトリウム溶液を100μlとLB液体培地を700μl加えて混合した後に、37℃でさらに40分間恒温した。20μg/mlのカナマイシンを含むLB寒天培地に菌液を塗布し、37℃で一晩培養した。K2とD-pykFのプライマーペアを用いたコロニーダイレクトPCRを行い、得られた株のpykF座位にFRT-Km-FRT-PT7-aroALCが挿入されていることを確認した。つぎに、Phe1株の作製で記載した方法に基づいて、カナマイシン耐性遺伝子を除去し、MG1655(DE3) tyrR::PT7-aroGfbr ldhA::PT7-pheAfbr adhE::PT7-ppsA pflDC::PT7(-8TC)-tktA ascF::PT7-aroEDB pykF::PT7-aroALC株を得た。この株をPhe13と命名した。Phe13は、独立行政法人製品評価技術基盤機構 特許微生物寄託センター(#122,2-5-8 Kazusakamatari,Kisarazu-shi,Chiba 292-0818,Japan)に寄託した(受託日2017年7月20日、受託番号:NITE BP-02514)。
Phe10株を5mlのLB液体培地を用いて37℃で一晩培養した。50μlの1M塩化カルシウムを加えて良く撹拌した後、200μlを1.5mlチューブに移した。そこにS6株のP1ファージライセートを20μl加えて混合し、37℃で20分間恒温した。1Mのクエン酸3ナトリウム溶液を100μlとLB液体培地を700μl加えて混合した後に、37℃でさらに40分間恒温した。20μg/mlのカナマイシンを含むLB寒天培地に菌液を塗布し、37℃で一晩培養した。K2とD-ascFのプライマーペアを用いたコロニーダイレクトPCRを行い、得られた株のascF座位にFRT-Km-FRT-PT7-aroBが挿入されていることを確認した。作製したMG1655(DE3) tyrR::PT7-aroGfbr ldhA::PT7-pheAfbr adhE::PT7-ppsA pflDC::PT7(-8TC)-tktA pykF::PT7-aroALC ascF::PT7-FRT-Km-FRT-aroB株をPhe14株と命名した。
Phe19株を5mlのLB液体培地を用いて37℃で一晩培養した。50μlの1M塩化カルシウムを加えて良く撹拌した後、200μlを1.5mlチューブに移した。そこにS14株のP1ファージライセートを20μl加えて混合し、37℃で20分間恒温した。1Mのクエン酸3ナトリウム溶液を100μlとLB液体培地を700μl加えて混合した後に、37℃でさらに40分間恒温した。20μg/mlのカナマイシンを含むLB寒天培地に菌液を塗布し、37℃で一晩培養した。K2とD-ascFのプライマーペアを用いたコロニーダイレクトPCRを行い、得られた株のascF座位にFRT-Km-FRT-PT7-aroEBが挿入されていることを確認した。作製したMG1655(DE3) tyrR::PT7-aroGfbr ldhA::PT7-pheAfbr adhE::PT7-ppsA pflDC::PT7(-8TC)-tktA pykF::PT7-aroAC ascF::PT7-FRT-Km-FRT-aroEB株をPhe15株と命名した。
Phe19株を5mlのLB液体培地を用いて37℃で一晩培養した。50μlの1M塩化カルシウムを加えて良く撹拌した後、200μlを1.5mlチューブに移した。そこにS15株のP1ファージライセートを20μl加えて混合し、37℃で20分間恒温した。1Mのクエン酸3ナトリウム溶液を100μlとLB液体培地を700μl加えて混合した後に、37℃でさらに40分間恒温した。20μg/mlのカナマイシンを含むLB寒天培地に菌液を塗布し、37℃で一晩培養した。K2とD-ascFのプライマーペアを用いたコロニーダイレクトPCRを行い、得られた株のascF座位にFRT-Km-FRT-PT7-aroDBが挿入されていることを確認した。作製したMG1655(DE3) tyrR::PT7-aroGfbr ldhA::PT7-pheAfbr adhE::PT7-ppsA pflDC::PT7(-8TC)-tktA pykF::PT7-aroAC ascF::PT7-FRT-Km-FRT-aroDB株をPhe16株と命名した。
Phe19株を5mlのLB液体培地を用いて37℃で一晩培養した。50μlの1M塩化カルシウムを加えて良く撹拌した後、200μlを1.5mlチューブに移した。そこにS6株のP1ファージライセートを20μl加えて混合し、37℃で20分間恒温した。1Mのクエン酸3ナトリウム溶液を100μlとLB液体培地を700μl加えて混合した後に、37℃でさらに40分間恒温した。20μg/mlのカナマイシンを含むLB寒天培地に菌液を塗布し、37℃で一晩培養した。K2とD-ascFのプライマーペアを用いたコロニーダイレクトPCRを行い、得られた株のascF座位にFRT-Km-FRT-PT7-aroBが挿入されていることを確認した。作製したMG1655(DE3) tyrR::PT7-aroGfbr ldhA::PT7-pheAfbr adhE::PT7-ppsA pflDC::PT7(-8TC)-tktA pykF::PT7-aroAC ascF::PT7-FRT-Km-FRT-aroB株をPhe17株と命名した。
Phe13株を5mlのLB液体培地を用いて37℃で一晩培養した。50μlの1M塩化カルシウムを加えて良く撹拌した後、200μlを1.5mlチューブに移した。そこにS10株のP1ファージライセートを20μl加えて混合し、37℃で20分間恒温した。1Mのクエン酸3ナトリウム溶液を100μlとLB液体培地を700μl加えて混合した後に、37℃でさらに40分間恒温した。20μg/mlのカナマイシンを含むLB寒天培地に菌液を塗布し、37℃で一晩培養した。K2とD-pykFのプライマーペアを用いたコロニーダイレクトPCRを行い、得られた株のpykF座位にFRT-Km-FRT-PT7-aroALが挿入されていることを確認した。つぎに、Phe1株の作製で記載した方法に基づいて、カナマイシン耐性遺伝子を除去し、MG1655(DE3) tyrR::PT7-aroGfbr ldhA::PT7-pheAfbr adhE::PT7-ppsA pflDC::PT7(-8TC)-tktA ascF::PT7-aroEDB pykF::PT7-aroAL株を得た。この株をPhe18株と命名した。
Phe13株を5mlのLB液体培地を用いて37℃で一晩培養した。50μlの1M塩化カルシウムを加えて良く撹拌した後、200μlを1.5mlチューブに移した。そこにS11株のP1ファージライセートを20μl加えて混合し、37℃で20分間恒温した。1Mのクエン酸3ナトリウム溶液を100μlとLB液体培地を700μl加えて混合した後に、37℃でさらに40分間恒温した。20μg/mlのカナマイシンを含むLB寒天培地に菌液を塗布し、37℃で一晩培養した。K2とD-pykFのプライマーペアを用いたコロニーダイレクトPCRを行い、得られた株のpykF座位にFRT-Km-FRT-PT7-aroACが挿入されていることを確認した。つぎに、Phe1株の作製で記載した方法に基づいて、カナマイシン耐性遺伝子を除去し、MG1655(DE3) tyrR::PT7-aroGfbr ldhA::PT7-pheAfbr adhE::PT7-ppsA pflDC::PT7(-8TC)-tktA ascF::PT7-aroEDB pykF::PT7-aroAC株を得た。この株をPhe19株と命名した。
Phe13株を5mlのLB液体培地を用いて37℃で一晩培養した。50μlの1M塩化カルシウムを加えて良く撹拌した後、200μlを1.5mlチューブに移した。そこにS12株のP1ファージライセートを20μl加えて混合し、37℃で20分間恒温した。1Mのクエン酸3ナトリウム溶液を100μlとLB液体培地を700μl加えて混合した後に、37℃でさらに40分間恒温した。20μg/mlのカナマイシンを含むLB寒天培地に菌液を塗布し、37℃で一晩培養した。K2とD-pykFのプライマーペアを用いたコロニーダイレクトPCRを行い、得られた株のpykF座位にFRT-Km-FRT-PT7-aroLCが挿入されていることを確認した。つぎに、Phe1株の作製で記載した方法に基づいて、カナマイシン耐性遺伝子を除去し、MG1655(DE3) tyrR::PT7-aroGfbr ldhA::PT7-pheAfbr adhE::PT7-ppsA pflDC::PT7(-8TC)-tktA ascF::PT7-aroEDB pykF::PT7-aroLC株を得た。この株をPhe20株と命名した。
Phe13株を5mlのLB液体培地を用いて37℃で一晩培養した。50μlの1M塩化カルシウムを加えて良く撹拌した後、200μlを1.5mlチューブに移した。そこにS13株のP1ファージライセートを20μl加えて混合し、37℃で20分間恒温した。1Mのクエン酸3ナトリウム溶液を100μlとLB液体培地を700μl加えて混合した後に、37℃でさらに40分間恒温した。20μg/mlのカナマイシンを含むLB寒天培地に菌液を塗布し、37℃で一晩培養した。K2とD-ascFのプライマーペアを用いたコロニーダイレクトPCRを行い、得られた株のascF座位にFRT-Km-FRT-PT7-aroEDが挿入されていることを確認した。つぎに、Phe1株の作製で記載した方法に基づいて、カナマイシン耐性遺伝子を除去し、MG1655(DE3) tyrR::PT7-aroGfbr ldhA::PT7-pheAfbr adhE::PT7-ppsA pflDC::PT7(-8TC)-tktA ascF::PT7-aroED pykF::PT7-aroALC株を得た。この株をPhe21株と命名した。
Phe13株を5mlのLB液体培地を用いて37℃で一晩培養した。50μlの1M塩化カルシウムを加えて良く撹拌した後、200μlを1.5mlチューブに移した。そこにS14株のP1ファージライセートを20μl加えて混合し、37℃で20分間恒温した。1Mのクエン酸3ナトリウム溶液を100μlとLB液体培地を700μl加えて混合した後に、37℃でさらに40分間恒温した。20μg/mlのカナマイシンを含むLB寒天培地に菌液を塗布し、37℃で一晩培養した。K2とD-ascFのプライマーペアを用いたコロニーダイレクトPCRを行い、得られた株のascF座位にFRT-Km-FRT-PT7-aroEBが挿入されていることを確認した。つぎに、Phe1株の作製で記載した方法に基づいて、カナマイシン耐性遺伝子を除去し、MG1655(DE3) tyrR::PT7-aroGfbr ldhA::PT7-pheAfbr adhE::PT7-ppsA pflDC::PT7(-8TC)-tktA ascF::PT7-aroEB pykF::PT7-aroALC株を得た。この株をPhe22株と命名した。
Phe13株を5mlのLB液体培地を用いて37℃で一晩培養した。50μlの1M塩化カルシウムを加えて良く撹拌した後、200μlを1.5mlチューブに移した。そこにS15株のP1ファージライセートを20μl加えて混合し、37℃で20分間恒温した。1Mのクエン酸3ナトリウム溶液を100μlとLB液体培地を700μl加えて混合した後に、37℃でさらに40分間恒温した。20μg/mlのカナマイシンを含むLB寒天培地に菌液を塗布し、37℃で一晩培養した。K2とD-ascFのプライマーペアを用いたコロニーダイレクトPCRを行い、得られた株のascF座位にFRT-Km-FRT-PT7-aroDBが挿入されていることを確認した。つぎに、Phe1株の作製で記載した方法に基づいて、カナマイシン耐性遺伝子を除去し、MG1655(DE3) tyrR::PT7-aroGfbr ldhA::PT7-pheAfbr adhE::PT7-ppsA pflDC::PT7(-8TC)-tktA ascF::PT7-aroDB pykF::PT7-aroALC株を得た。この株をPhe23株と命名した。
Phe13株を5mlのLB液体培地を用いて37℃で一晩培養した。50μlの1M塩化カルシウムを加えて良く撹拌した後、200μlを1.5mlチューブに移した。そこにS7株のP1ファージライセートを20μl加えて混合し、37℃で20分間恒温した。1Mのクエン酸3ナトリウム溶液を100μlとLB液体培地を700μl加えて混合した後に、37℃でさらに40分間恒温した。20μg/mlのカナマイシンを含むLB寒天培地に菌液を塗布し、37℃で一晩培養した。K2とD-ackA-ptaのプライマーペアを用いたコロニーダイレクトPCRを行い、得られた株のackA-pta座位にFRT-Km-FRT-PT7-tyrBが挿入されていることを確認した。作製したMG1655(DE3) tyrR::PT7-aroGfbr ldhA::PT7-pheAfbr adhE::PT7-ppsA pflDC::PT7(-8TC)-tktA pykF::PT7-aroAC ascF::PT7-aroEDB ackA-pta::FRT-Km-FRT-PT7-tyrB株をPhe24株と命名した。
Phe7株を5mlのLB液体培地を用いて37℃で一晩培養した。50μlの1M塩化カルシウムを加えて良く撹拌した後、200μlを1.5mlチューブに移した。そこにS5株のP1ファージライセートを20μl加えて混合し、37℃で20分間恒温した。1Mのクエン酸3ナトリウム溶液を100μlとLB液体培地を700μl加えて混合した後に、37℃でさらに40分間恒温した。20μg/mlのカナマイシンを含むLB寒天培地に菌液を塗布し、37℃で一晩培養した。K2とD-ldhAのプライマーペアを用いたコロニーダイレクトPCRを行い、得られた株のldhA座位にFRT-Km-FRT-PT7-tyrAfbrが挿入されていることを確認した。つぎに、Phe1株の作製で記載した方法に基づいて、カナマイシン耐性遺伝子を除去し、MG1655(DE3) tyrR::PT7-aroGfbr ldhA::PT7-tyrAfbr adhE::PT7-ppsA pflDC::PT7(-8TC)-tktA ascF::PT7-aroEDB株を得た。この株をTyr7と命名した。
Phe10株を5mlのLB液体培地を用いて37℃で一晩培養した。50μlの1M塩化カルシウムを加えて良く撹拌した後、200μlを1.5mlチューブに移した。そこにS5株のP1ファージライセートを20μl加えて混合し、37℃で20分間恒温した。1Mのクエン酸3ナトリウム溶液を100μlとLB液体培地を700μl加えて混合した後に、37℃でさらに40分間恒温した。20μg/mlのカナマイシンを含むLB寒天培地に菌液を塗布し、37℃で一晩培養した。K2とD-ldhAのプライマーペアを用いたコロニーダイレクトPCRを行い、得られた株のldhA座位にFRT-Km-FRT-PT7-tyrAfbrが挿入されていることを確認した。つぎに、Phe1株の作製で記載した方法に基づいて、カナマイシン耐性遺伝子を除去し、MG1655(DE3) tyrR::PT7-aroGfbr ldhA::PT7-tyrAfbr adhE::PT7-ppsA pflDC::PT7(-8TC)-tktA pykF::PT7-aroALC株を得た。この株をTyr10と命名した。
Phe13株を5mlのLB液体培地を用いて37℃で一晩培養した。50μlの1M塩化カルシウムを加えて良く撹拌した後、200μlを1.5mlチューブに移した。そこにS5株のP1ファージライセートを20μl加えて混合し、37℃で20分間恒温した。1Mのクエン酸3ナトリウム溶液を100μlとLB液体培地を700μl加えて混合した後に、37℃でさらに40分間恒温した。20μg/mlのカナマイシンを含むLB寒天培地に菌液を塗布し、37℃で一晩培養した。K2とD-ldhAのプライマーペアを用いたコロニーダイレクトPCRを行い、得られた株のldhA座位にFRT-Km-FRT-PT7-tyrAfbrが挿入されていることを確認した。つぎに、Phe1株の作製で記載した方法に基づいて、カナマイシン耐性遺伝子を除去し、MG1655(DE3) tyrR::PT7-aroGfbr ldhA::PT7-tyrAfbr adhE::PT7-ppsA pflDC::PT7(-8TC)-tktA ascF::PT7-aroEDB pykF::PT7-aroALC株を得た。この株をTyr13と命名した。Tyr13は、独立行政法人製品評価技術基盤機構 特許微生物寄託センター(#122,2-5-8 Kazusakamatari,Kisarazu-shi,Chiba 292-0818,Japan)に寄託した(受託日2017年7月20日、受託番号:NITE BP-02513)。
Phe13株を5mlのLB液体培地を用いて37℃で一晩培養した。50μlの1M塩化カルシウムを加えて良く撹拌した後、200μlを1.5mlチューブに移した。そこにAR-G84のP1ファージライセート(非特許文献3、Koma et al.Appl.Environ.Microbiol.78:6203-6216,2012)を20μl加えて混合し、37℃で20分間恒温した。1Mのクエン酸3ナトリウム溶液を100μlとLB液体培地を700μl加えて混合した後に、37℃でさらに40分間恒温した。20μg/mlのカナマイシンを含むLB寒天培地に菌液を塗布し、37℃で一晩培養した。K2とD-mtlA(GATCAACGACATCATCACCAATGC,配列番号98)のプライマーペアを用い、得られた株のmtlA座位にFRT-Km-FRT-PT7-ipdCが挿入されていることを確認した。作製したMG1655(DE3) tyrR::PT7-aroGfbr ldhA::PT7-pheAfbr adhE::PT7-ppsA pflDC::PT7(-8TC)-tktA pykF::PT7-aroALC ascF::PT7-aroEDB mtlA::FRT-Km-FRT-PT7-ipdC株を2PE2株と命名した。なお、2PE1株は既報(非特許文献3、Koma et al.Appl Environ.Microbiol.78:6203-6216,2012)に記載されているPAR-60株に相当する。
Tyr13株を5mlのLB液体培地を用いて37℃で一晩培養した。50μlの1M塩化カルシウムを加えて良く撹拌した後、200μlを1.5mlチューブに移した。そこにAR-G39のP1ファージライセート(非特許文献3、Koma et al.Appl.Environ.Microbiol.78:6203-6216,2012)を20μl加えて混合し、37℃で20分間恒温した。1Mのクエン酸3ナトリウム溶液を100μlとLB液体培地を700μl加えて混合した後に、37℃でさらに40分間恒温した。20μg/mlのカナマイシンを含むLB寒天培地に菌液を塗布し、37℃で一晩培養した。K2とD-acs(CTCATGCAGGACTTCATTATTAAGACGGTC,配列番号99)のプライマーペアを用い、得られた株のacs座位にFRT-Km-FRT-PT7-ldhAが挿入されていることを確認した。作製したMG1655(DE3) tyrR::PT7-aroGfbr ldhA::PT7-pheAfbr adhE::PT7-ppsA pflDC::PT7(-8TC)-tktA pykF::PT7-aroALC ascF::PT7-aroEDB acs::FRT-Km-FRT-PT7-ldhA株を4HLA2株と命名した。なお、4HLA1株は既報(非特許文献3、Koma et al.Appl.Environ.Microbiol.78:6203-6216,2012)に記載されているPAR-3株に相当する。
菌株を5mlのLB液体培地または0.8%のグルコースを含むLB液体培地を用いて27℃で一晩前培養した。50μlの前培養液をM9M2液体培地に加えて植菌し、37℃でOD660の値が0.3程度になるまで振とう培養した。M9M2液体培地は、900mlの蒸留水に10gのグルコース、6gのリン酸水素2ナトリウム、3gのリン酸2水素カリウム、0.5gの塩化ナトリウム、および4gの塩化アンモニウムを加え、さらに1M 塩化マグネシウムを1ml、10mg/mlチアミン塩酸塩を1ml、100mM硫酸鉄(二価)を0.5ml、1M塩化カルシウムを100μl、微量金属塩溶液を10μl加え、1Lにメスアップしたものを0.2μmのフィルターを用いてろ過除菌したものを用いた。微量金属塩溶液は100mlの蒸留水中に0.371gの(NH4)6Mo7O24・4H2O、2.473gのH3BO3、0.714gのCoCl2・6H2O、0.239gのCuSO4・5H2O、1.583gのMnCl2・4H2O、0.288gのZnSO4・7H2Oを含む。なお、2PE1株と2PE2株を培養する場合にはM9M2培地にフェニルアラニンを2mMとなるように加えた。イソプロピル-β-チオガラクトピラノシド(IPTG)を終濃度が1mMとなるように添加し、27℃で44時間振とう培養した。950μlの蒸留水と200μlのメタノールと50μlの培養液を混合し、10,000rpmで5分間遠心分離し、上清を0.45μmのフィルターを用いてろ過したものをHPLCのサンプルとして用いた。HPLCは既報(非特許文献3、Koma et al.Appl.Environ.Microbiol.78:6203-6216,2012)に記載されているとおりに行い、培養液中のフェニルアラニン、チロシン、2-フェニルエタノール、または4-ヒドロキシフェニル乳酸の生成量を定量した。
その結果を表4~7に示す。これらの表において、Pheはフェニルアラニンを、Tyrはチロシンを、2PEは2-フェニルエタノールを、4HPLAは4-ヒドロキシフェニル乳酸、Glcはグルコースを表す。
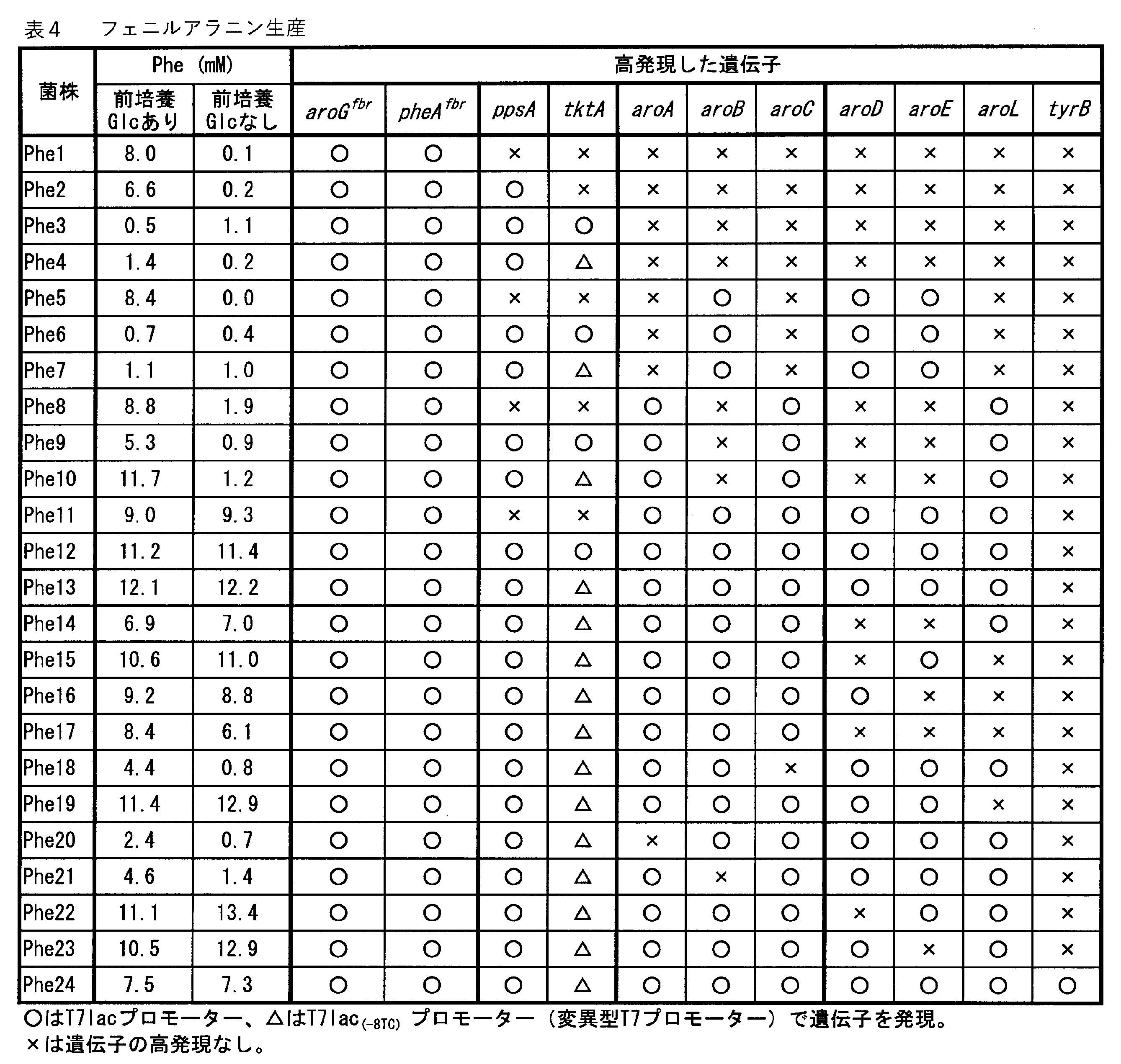
Phe13株とPhe24株との比較から、tyrBの過剰発現は、フェニルアラニン産生にネガティブな影響があることが認められた。
また、Phe11株~Phe17株、Phe19株、Phe22株、及びPhe23株の結果から、良好なフェニルアラニンの産生には、少なくともaroA、aroB、aroC、aroGfbr、及びpheAfbrの過剰発現が必要であると認められる。
Phe11株~Phe13株の比較から、ppsAの過剰発現はフェニルアラニン産生に好ましく、tktAの過剰発現も好ましいが、tktAは誘導が弱まる変異の入ったT7プロモーターによって発現されることがより好ましいことが認められる。
Phe13株~Phe17株、Phe19株、Phe22株、及びPhe23株の結果から、aroD,aroE及びaroLは、これらのうち少なくとも1つが過剰発現することが好ましいと認められた。
表4に示すように、aroGfbr及びpheAfbrを過剰発現し、かつ、aroA、aroB、及びaroCの3つの遺伝子の少なくとも1つを過剰発現していないフェニルアラニン生産株では、導入した遺伝子の過剰発現による代謝負荷がかかっており、構築されたフェニルアラニンの合成経路が円滑に機能していないと考えられる。したがって、このような菌株では前培養にグルコースを添加してカタボライトリプレッションを引き起こすことにより、基底状態での遺伝子発現レベルを低下させる必要がある(例えば、Phe1、2、5、8、9、10、18、20及び21株)。一方、aroGfbr及びpheAfbrに加えてaroA、aroB、及びaroCの3つの遺伝子を過剰発現している菌株では、構築されたフェニルアラニンの合成経路が円滑に機能していると考えられる。そのため、前培養でのグルコース添加の有無にかかわらず、フェニルアラニンの高生産が達成できる(例えば、Phe11~17、19、22、及び23株)。



NITE BP-02514
Claims (10)
- 形質転換されたエシェリキア(Escherichia)属に属する微生物であって、
下記(1)~(7)の7つの遺伝子と、下記(8)~(10)のうち少なくとも2つの遺伝子とが染色体上に発現誘導可能なプロモーターとともに導入されている微生物であって、
(1)aroA
(2)aroB
(3)aroC
(4)aroGfbr又はaroFfbr
(5)pheAfbr又はtyrAfbr
(6)ppsA
(7)tktA
(8)aroD
(9)aroE又はydiB
(10)aroL又はaroK
染色体上に発現誘導可能なプロモーターとともに導入される遺伝子にtyrBが含まれない、微生物。 - 上記(1)~(10)の10つの遺伝子が染色体上に発現誘導可能なプロモーターとともに導入されている、請求項1に記載の微生物。
- 前記(5)の遺伝子が、pheAfbrであり、フェニルアラニン生産能を有する、請求項1又は2に記載の微生物。
- 前記(5)の遺伝子が、tyrAfbrであり、チロシン生産能を有する、請求項1又は2に記載の微生物。
- さらに、フェニルピルビン酸、4-ヒドロキシフェニルピルビン酸、フェニルアラニン、又はチロシンを基質とする酵素をコードする同種又は異種起源の遺伝子が染色体上に発現誘導可能なプロモーターとともに導入された、請求項1から4のいずれかに記載の微生物。
- さらに、フェニルピルビン酸デカルボキシラーゼをコードする異種起源の遺伝子ipdCが染色体上に発現誘導可能なプロモーターとともに導入され、2-フェニルエタノール又は2-(4-ヒドロキシフェニル)エタノールの生産能を有する、請求項1から5のいずれかに記載の微生物。
- さらに、乳酸デヒドロゲナーゼをコードする異種起源の遺伝子ldhAが染色体上に発現誘導可能なプロモーターとともに導入され、フェニル乳酸又は4-ヒドロキシフェニル乳酸の生産能を有する、請求項1から4のいずれかに記載の微生物。
- 芳香族化合物の生産能を有する、請求項1から7のいずれかに記載の微生物。
- 請求項1から8のいずれかに記載の微生物を培地で培養することを含む、芳香族化合物の製造方法。
- 前記芳香族化合物が、フェニルアラニン、チロシン、2-フェニルエタノール、2-(4-ヒドロキシフェニル)エタノール、フェニル酢酸及び4-ヒドロキシフェニル酢酸からなる群から選択される、請求項9に記載の製造方法。
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/JP2017/028650 WO2019030808A1 (ja) | 2017-08-07 | 2017-08-07 | 芳香族化合物を産生する微生物 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JPWO2019030808A1 JPWO2019030808A1 (ja) | 2020-07-02 |
| JP7034496B2 true JP7034496B2 (ja) | 2022-03-14 |
Family
ID=65272020
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2019535461A Active JP7034496B2 (ja) | 2017-08-07 | 2017-08-07 | 芳香族化合物を産生する微生物 |
Country Status (2)
| Country | Link |
|---|---|
| JP (1) | JP7034496B2 (ja) |
| WO (1) | WO2019030808A1 (ja) |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007088977A1 (ja) | 2006-02-02 | 2007-08-09 | Ajinomoto Co., Inc. | L-アミノ酸の製造法 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2148482C (en) * | 1992-12-21 | 2006-10-31 | John W. Frost | Deblocking the common pathway of aromatic amino acid synthesis |
| JP4032441B2 (ja) * | 1995-08-30 | 2008-01-16 | 味の素株式会社 | L−アミノ酸の製造方法 |
| CN106661164B (zh) * | 2014-05-07 | 2019-01-04 | 索尔维公司 | 杂化的氟聚合物复合材料 |
| KR101869869B1 (ko) * | 2015-11-11 | 2018-07-19 | 포항공과대학교 산학협력단 | 타이로신 고생산 균주 및 이를 이용한 타이로신 생산방법 |
-
2017
- 2017-08-07 WO PCT/JP2017/028650 patent/WO2019030808A1/ja active Application Filing
- 2017-08-07 JP JP2019535461A patent/JP7034496B2/ja active Active
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007088977A1 (ja) | 2006-02-02 | 2007-08-09 | Ajinomoto Co., Inc. | L-アミノ酸の製造法 |
Non-Patent Citations (2)
| Title |
|---|
| KOMA, D. et al.,Production of Aromatic Compounds by Metabolically Engineered Escherichia coli with an Expanded Shikimate Pathway,Applied and Environmental Microbiology,2012年,Vol.78, No.17,pp.6203-6216,特にAbstract,Tables 1,4,p.6204右欄第3段落~p.6205右欄第1段落, ISSN: 1098-5336 |
| 駒大輔,遺伝子改変大腸菌を用いた芳香族化成品の生産,分析展と講演・技術発表会講演要旨集,2017年03月,Vol.41st,pp.63-64,特にp.63左欄第4段落,p.64第2段落 |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2019030808A1 (ja) | 2019-02-14 |
| JPWO2019030808A1 (ja) | 2020-07-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8835154B2 (en) | Microorganism having enhanced L-amino acids productivity and process for producing L-amino acids using the same | |
| KR100792095B1 (ko) | L-페닐알라닌 생산능을 갖는 대장균 변이주로부터유전자조작된 l-트립토판 생산능을 갖는 재조합 대장균균주 및 이를 이용한 트립토판 제조방법 | |
| JP2019195330A (ja) | 遺伝子操作された微生物からのムコン酸の生成 | |
| CN104379729B (zh) | 有用微生物和目标物质的制备方法 | |
| JP7437116B2 (ja) | 遺伝子操作された微生物からのムコン酸生成の改善 | |
| US20160304917A1 (en) | Modified Microorganism for Improved Production of Alanine | |
| JP2018161154A (ja) | L−スレオニン産生能を有する組換えエシェリキア属微生物およびこれを用いたl−スレオニンの産生方法 | |
| JP6177995B2 (ja) | L−トリプトファン産生能を有する微生物およびこれを用いてl−トリプトファンを産生する方法 | |
| JP2002512802A6 (ja) | 芳香族代謝/iii物質の微生物による製造 | |
| KR101830001B1 (ko) | Prpp 합성 경로 개선을 통한 l-트립토판이 과발현되는 균주 및 이를 이용하는 l-트립토판의 제조 방법 | |
| US6358714B1 (en) | Materials and methods for the production of D-phenylalanine | |
| KR101483012B1 (ko) | 재조합 대장균을 이용하여 3-히드록시프로피온산을 고수율로 생산하는 방법 | |
| Su et al. | Enhanced production of d-pantothenic acid in Corynebacterium glutamicum using an efficient CRISPR–Cpf1 genome editing method | |
| JP2019013256A (ja) | L−トリプトファン生産能を有するエシェリキア属微生物及びこれを用いたl−トリプトファンの製造方法 | |
| US20230407351A1 (en) | Recombinant host cells to produce anthranilic acid | |
| JP7034496B2 (ja) | 芳香族化合物を産生する微生物 | |
| Turlin et al. | Regulation of the early steps of 3-phenylpropionate catabolism in Escherichia coli | |
| US20080014619A1 (en) | Method of production of para-hydroxycinnamic acid | |
| JP7194950B2 (ja) | ヒドロキシチロソールの製造 | |
| JP5297804B2 (ja) | アミノ酸の製造法 | |
| JP2008522611A (ja) | 染色体上のlysR遺伝子が不活性化されたL−トレオニン生成微生物、これを製造する方法、及び該微生物を利用したL−トレオニンの製造方法 | |
| JP2023144366A (ja) | 3-ヒドロキシアジピン酸および/またはα-ヒドロムコン酸を生産するための遺伝子改変微生物および当該化学品の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A821 Effective date: 20190925 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20200619 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20210720 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20210916 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20220201 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20220222 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 7034496 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |