JP6996443B2 - 4-ボロノフェニルアラニン前駆体、2-[18f]フルオロ-4-ボロノフェニルアラニン前駆体の製造方法、2-[18f]フルオロ-4-ボロノフェニルアラニンの製造方法 - Google Patents
4-ボロノフェニルアラニン前駆体、2-[18f]フルオロ-4-ボロノフェニルアラニン前駆体の製造方法、2-[18f]フルオロ-4-ボロノフェニルアラニンの製造方法 Download PDFInfo
- Publication number
- JP6996443B2 JP6996443B2 JP2018135799A JP2018135799A JP6996443B2 JP 6996443 B2 JP6996443 B2 JP 6996443B2 JP 2018135799 A JP2018135799 A JP 2018135799A JP 2018135799 A JP2018135799 A JP 2018135799A JP 6996443 B2 JP6996443 B2 JP 6996443B2
- Authority
- JP
- Japan
- Prior art keywords
- group
- general formula
- compound
- boronophenylalanine
- carbon atoms
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- DWHXBEAKNROPNE-UHFFFAOYSA-N C[B]1(OC2)OCC2(C)CO1 Chemical compound C[B]1(OC2)OCC2(C)CO1 DWHXBEAKNROPNE-UHFFFAOYSA-N 0.000 description 2
- 0 Cc1c(CC(C(O*)=O)N)ccc(*)c1 Chemical compound Cc1c(CC(C(O*)=O)N)ccc(*)c1 0.000 description 2
- AXFZXOHZQMVFER-UHFFFAOYSA-N C=C(C(OC1(CCCC1)O1)=O)C1=O Chemical compound C=C(C(OC1(CCCC1)O1)=O)C1=O AXFZXOHZQMVFER-UHFFFAOYSA-N 0.000 description 1
- AIEJHDNUAFFGNX-UHFFFAOYSA-N CC(C(C)C(OC1(CCCCC1)O1)=O)C1=O Chemical compound CC(C(C)C(OC1(CCCCC1)O1)=O)C1=O AIEJHDNUAFFGNX-UHFFFAOYSA-N 0.000 description 1
- BASJNTNUQFBSNW-UHFFFAOYSA-N CC(C)(OC(C1=[I]C)=O)OC1=O Chemical compound CC(C)(OC(C1=[I]C)=O)OC1=O BASJNTNUQFBSNW-UHFFFAOYSA-N 0.000 description 1
- ILMMUDFVWXDZNE-UHFFFAOYSA-N CN(C(C(C(N1C)=O)=[I]C)=O)C1=O Chemical compound CN(C(C(C(N1C)=O)=[I]C)=O)C1=O ILMMUDFVWXDZNE-UHFFFAOYSA-N 0.000 description 1
- NSXGXEIATCXWFH-UHFFFAOYSA-N Cc(cccc1C)c1N(C1[ClH]C)C=CN1c1c(C)cccc1C Chemical compound Cc(cccc1C)c1N(C1[ClH]C)C=CN1c1c(C)cccc1C NSXGXEIATCXWFH-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Description
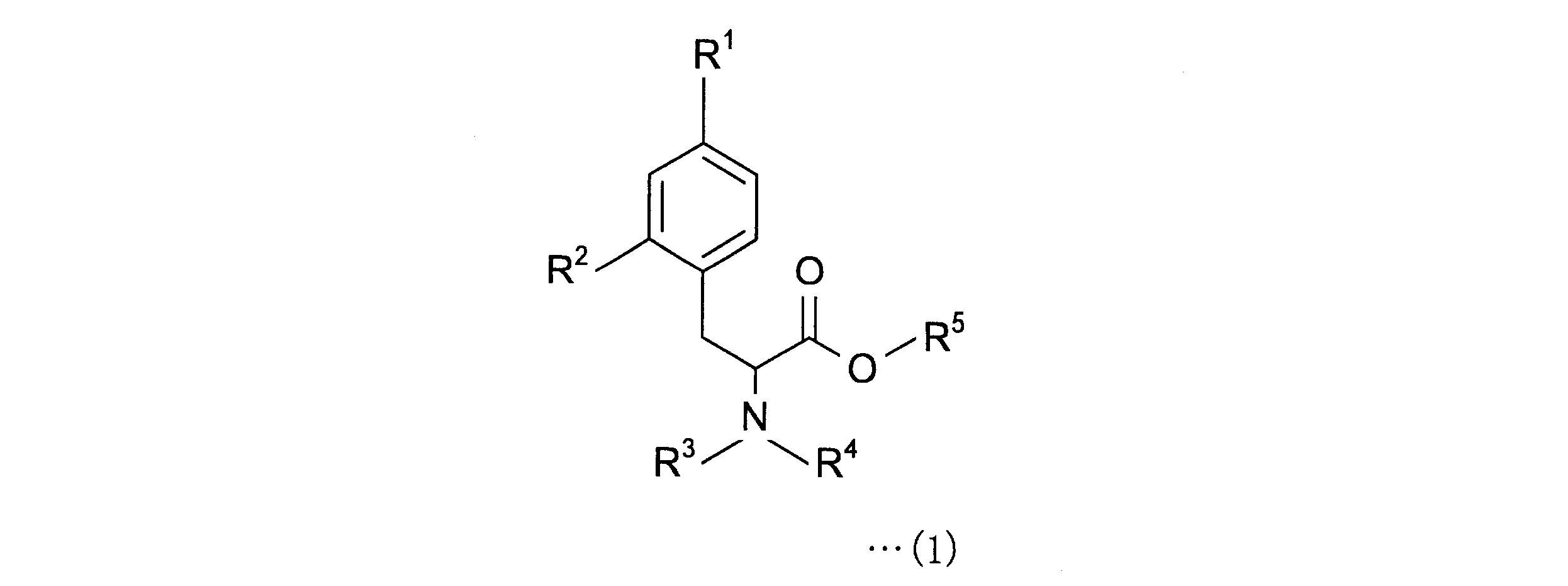
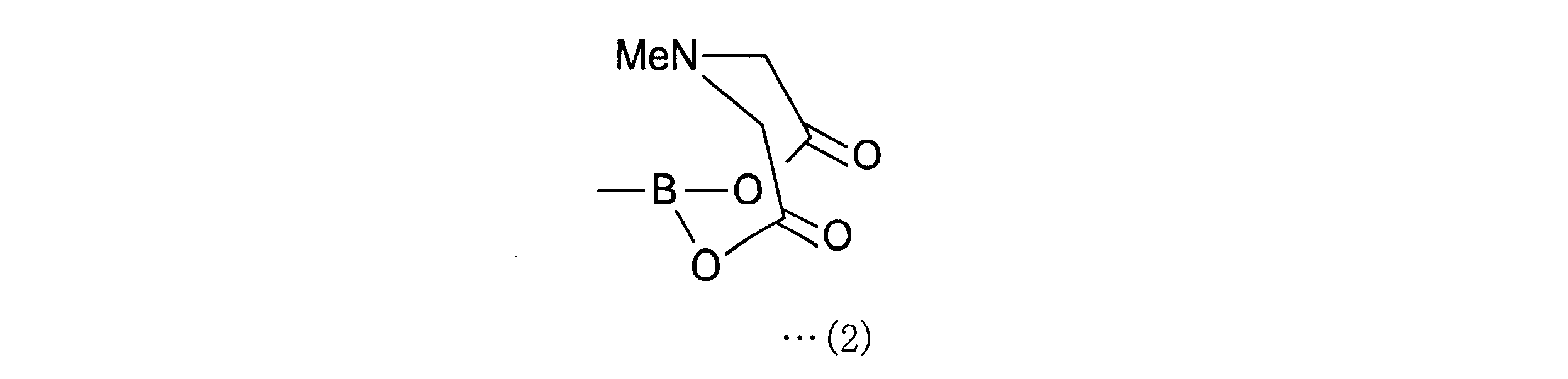
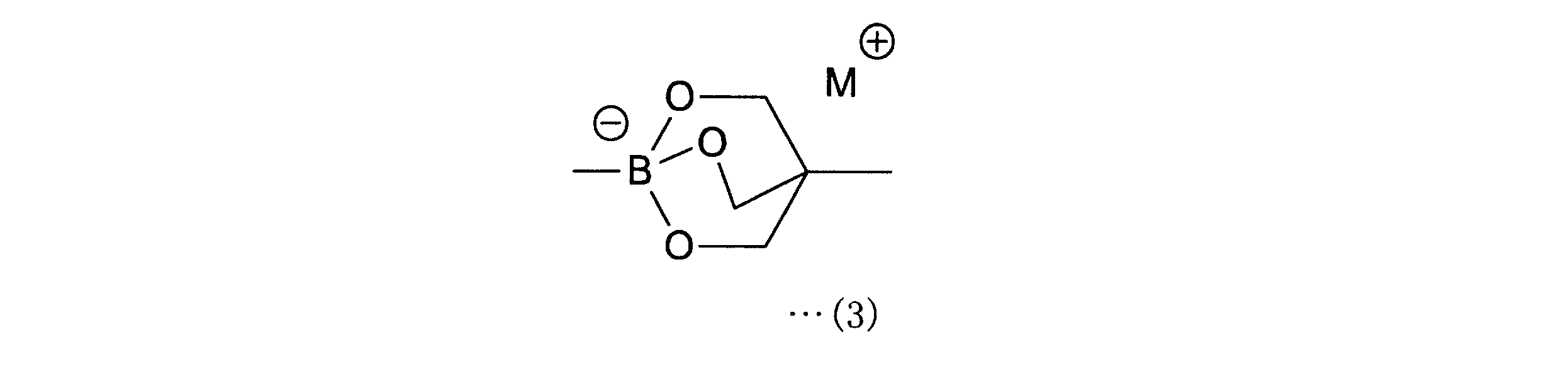
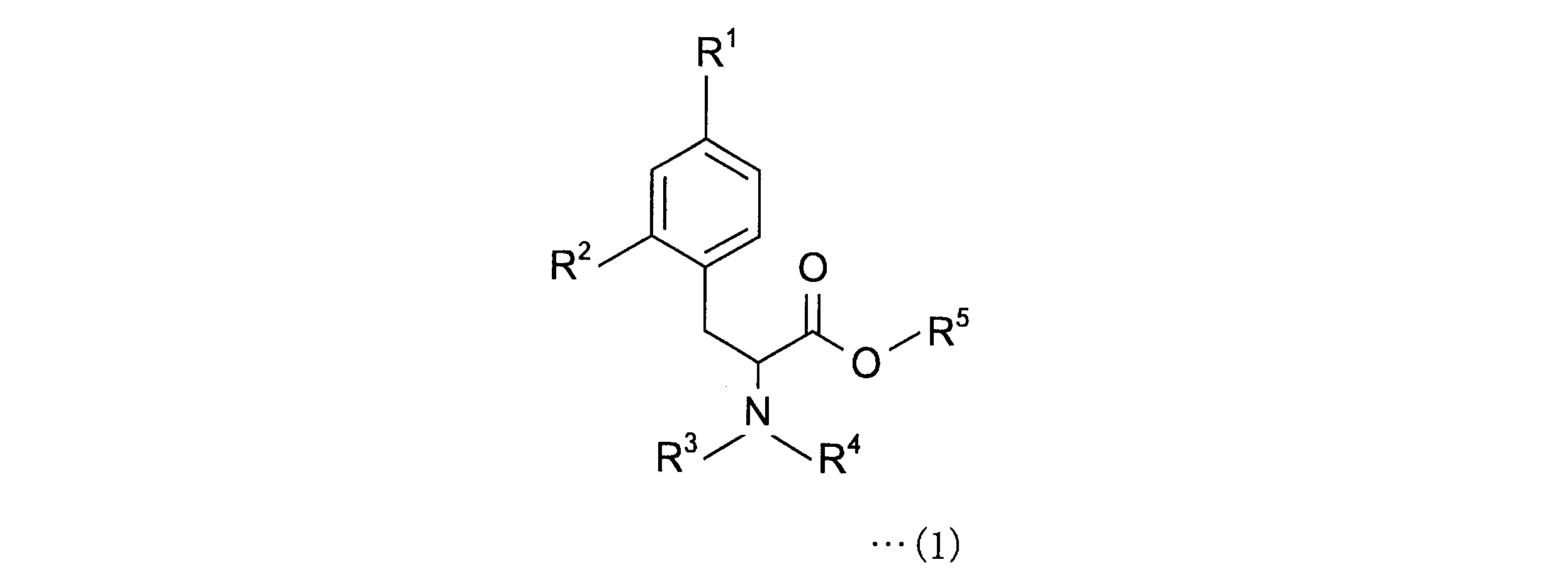
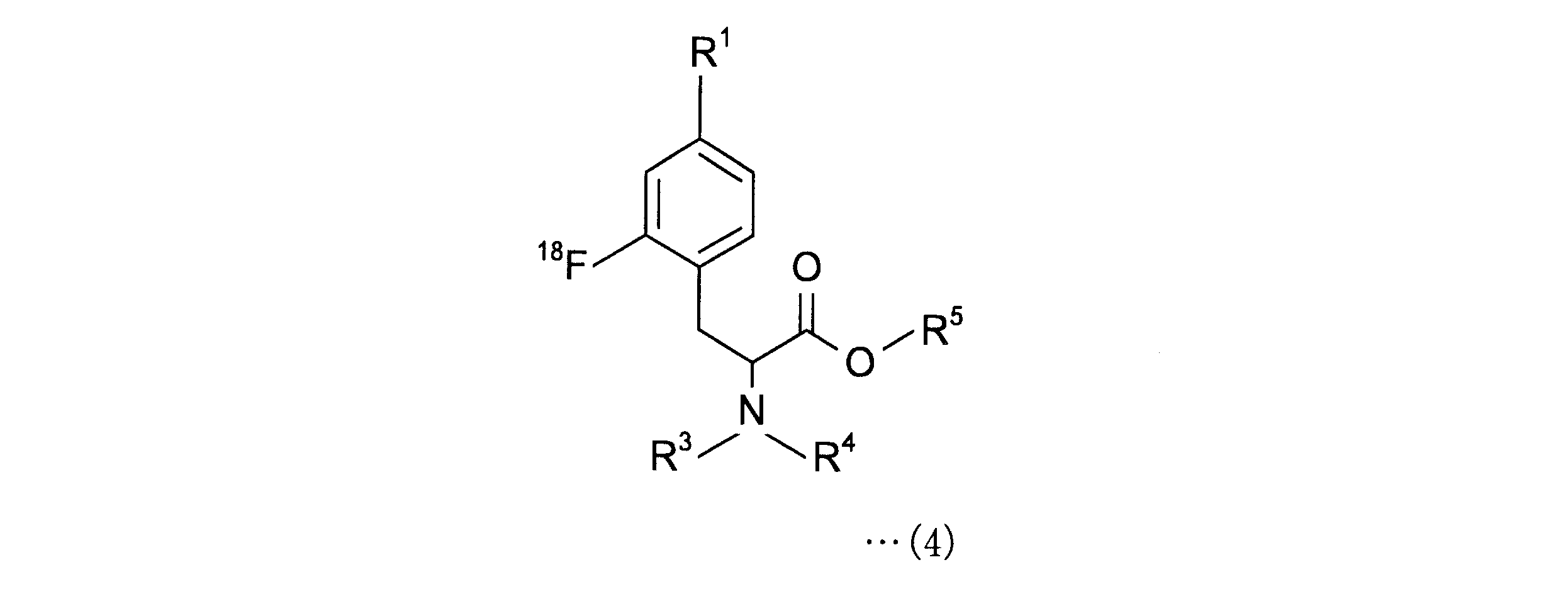

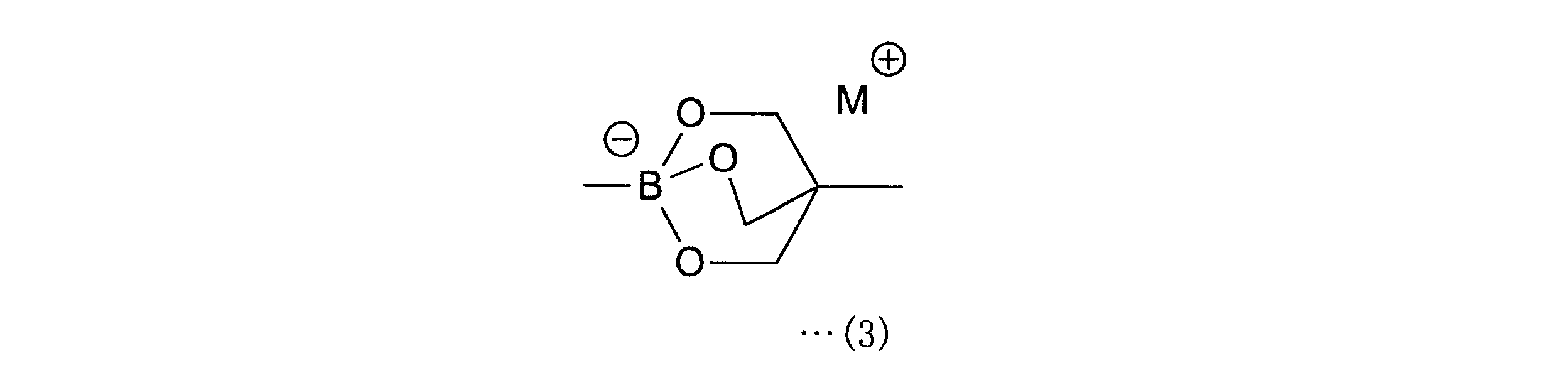
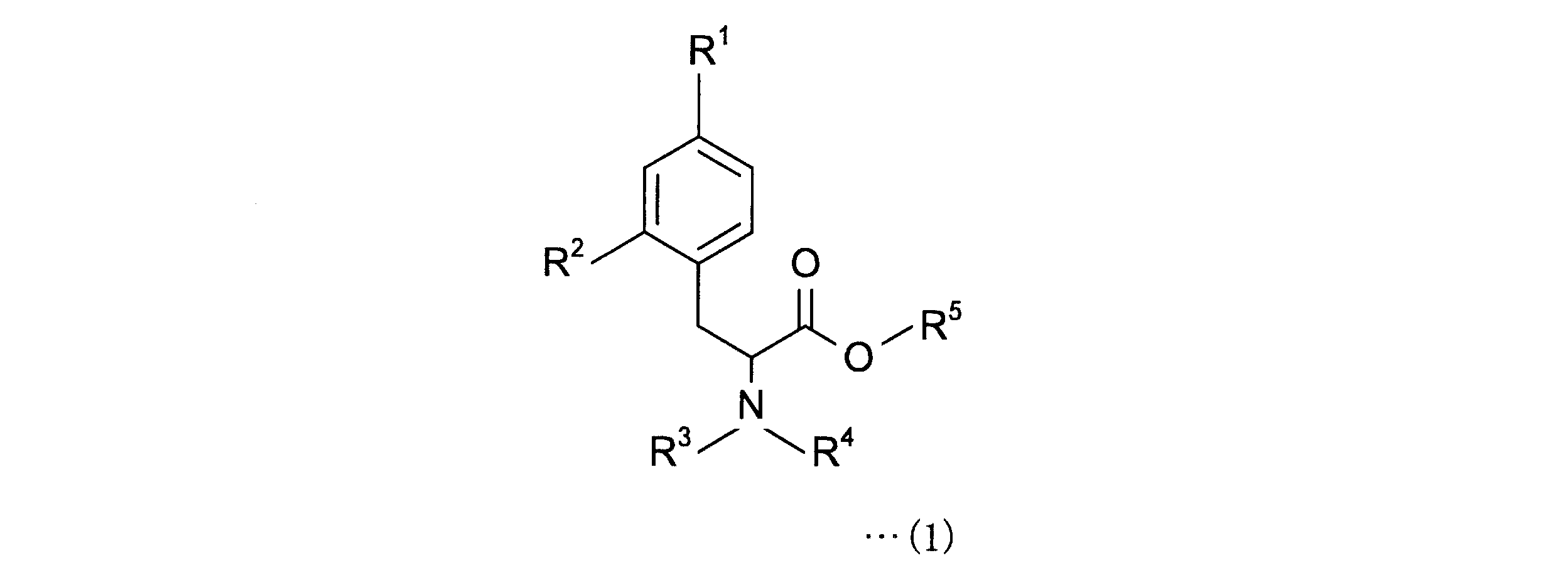
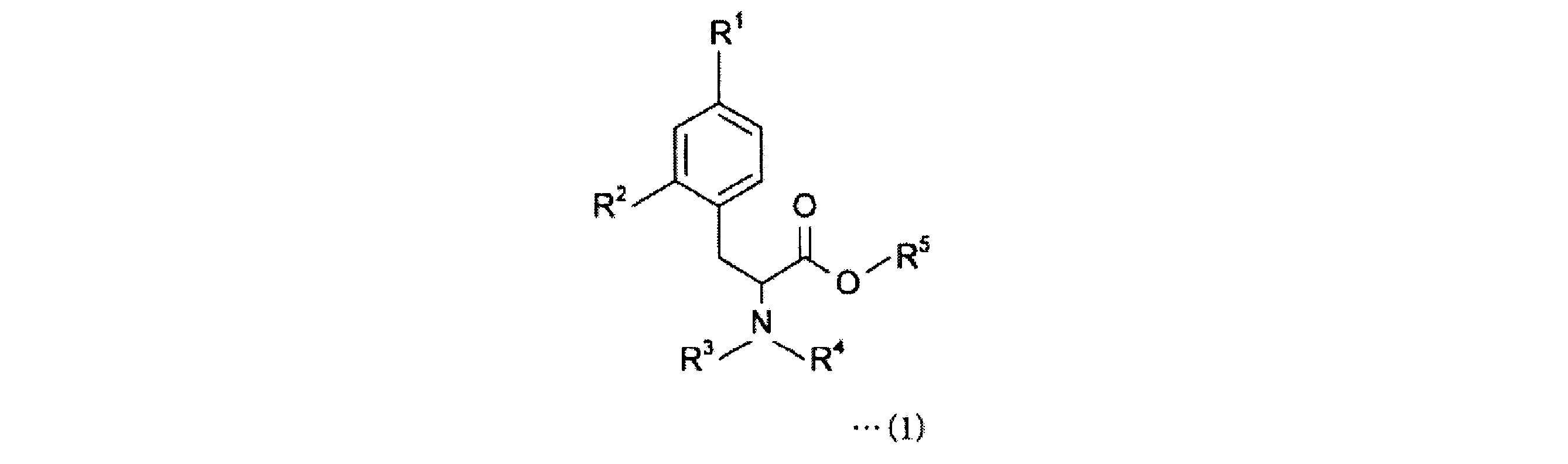
4-ボロノフェニルアラニン前駆体は、下記一般式(1)の化合物である。
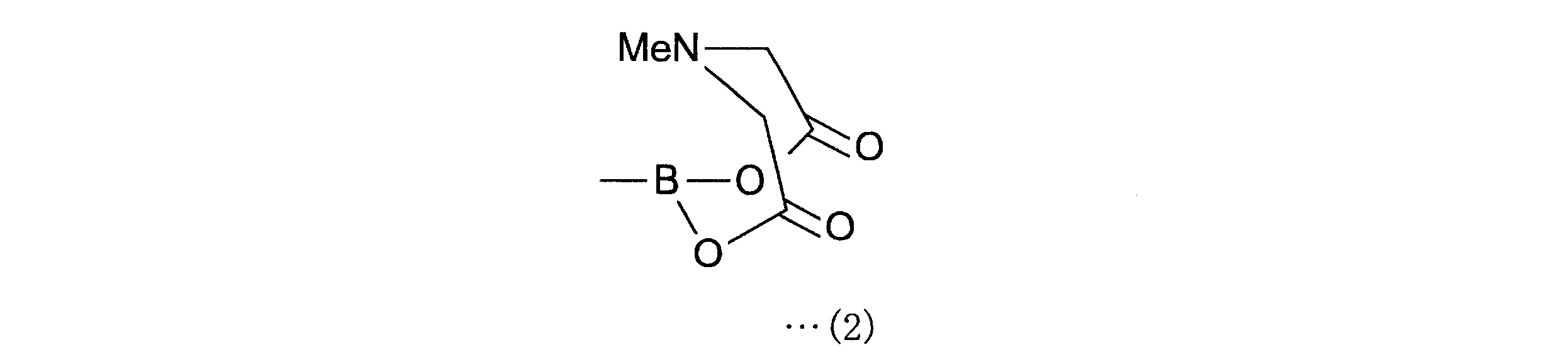
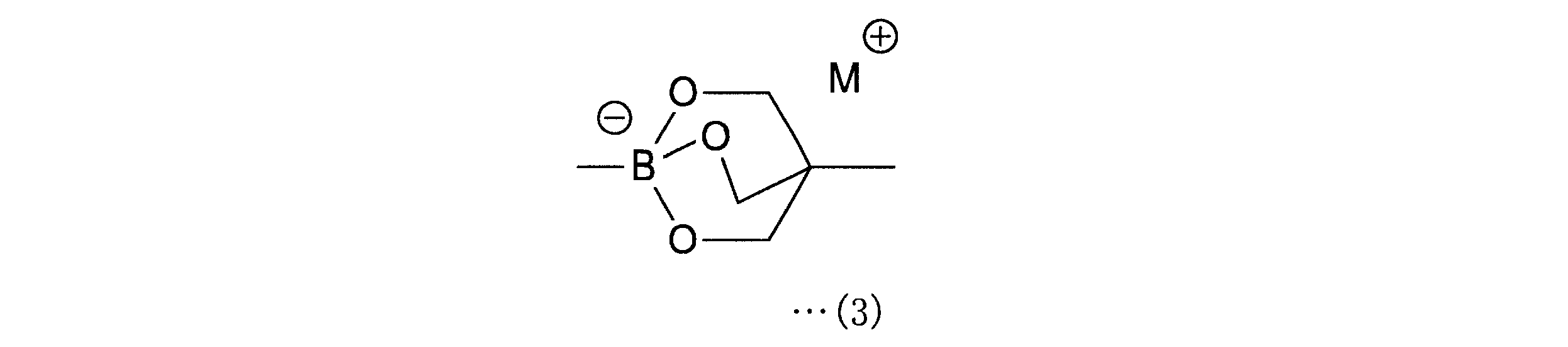

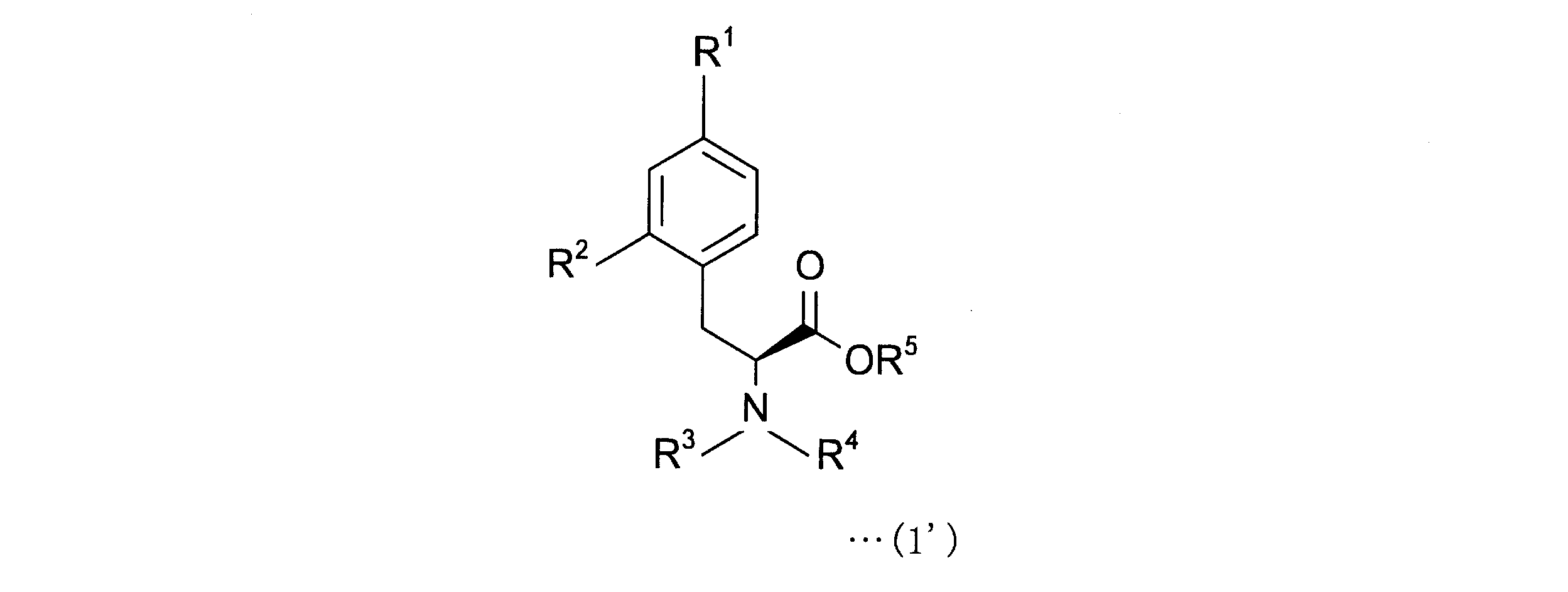
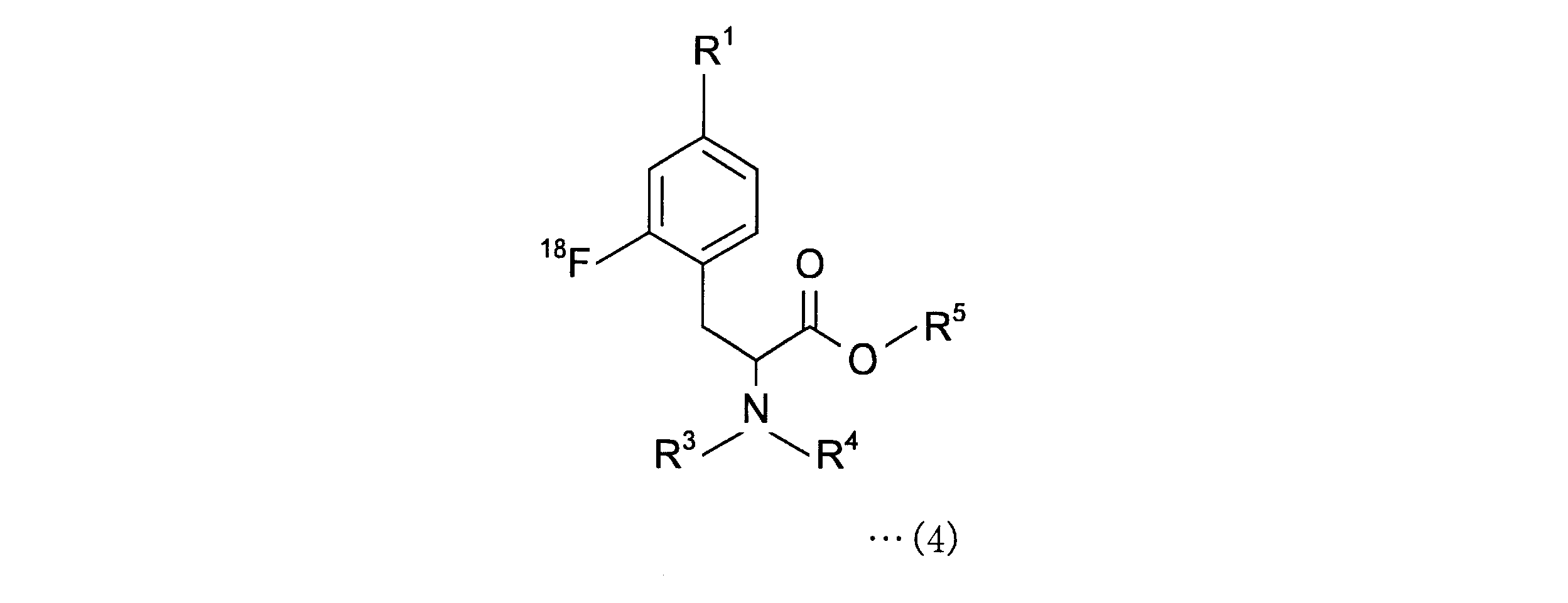
以下、2-[18F]フルオロ-4-ボロノフェニルアラニン前駆体の製造方法について説明する。
本発明の製造方法における各工程において、反応終了後、各工程の目的化合物は、常法にしたがって反応混合物から単離され得る。目的化合物は、例えば、(i)必要に応じて触媒等の不要物を濾去し、(ii)反応混合物に水、および水と混和しない溶媒(例えば、酢酸エチル、クロロホルム等)を加えて目的化合物を抽出し、(iii)有機層を水洗して、必要に応じて無水硫酸マグネシウム等の乾燥剤を用いて乾燥させ、(iv)溶媒を留去することによって得られる。得られた目的化合物は、必要に応じて公知の方法(例えば、シリカゲルカラムクロマトグラフィー等)により、さらに精製することができる。また、各工程の目的化合物は、精製することなく次の反応に提供することも可能である。
反応時間は、1~60分が好ましく、5~30分がより好ましく、10~20分がさらに好ましい。
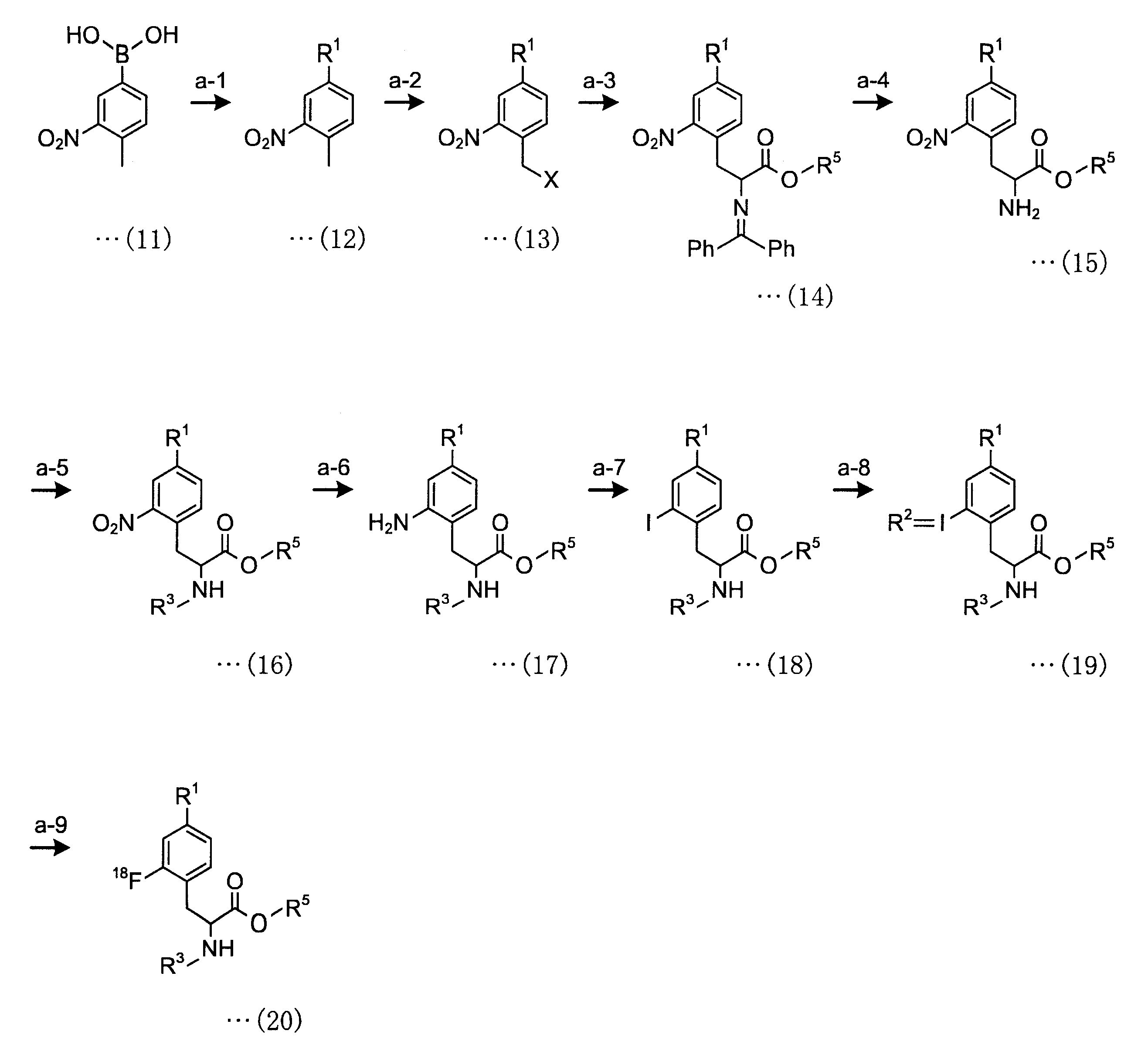
a-1工程は、一般式(11)の化合物をボラン酸の保護基と反応させ、一般式(12)の化合物を得る工程である。a-1工程で用いるホウ酸の保護基としては、ホウ素のsp2空軌道に非共有電子対を供与しうるものが好ましく、R1としては、2,3-ジヒドロ-1H-ナフト[1,8-de]-1,3,2-ジアザボリニル基、トリフルオロボレート基、ボロン酸-N-メチルイミノジアセテート基、環状トリオールボレート基が例示される。
a-2工程は、一般式(12)の化合物をハロゲン化試薬と反応させて、一般式(13)の化合物を得る工程である。a-2工程のXは、フッ素、塩素、ヨウ素または臭素であり、入手性の観点から臭素が好ましい。
a-3工程は、一般式(13)の化合物を修飾アミノ酸と反応させ、一般式(14)の化合物を得る工程である。
反応温度は、-20~100℃が好ましく、-10~50℃がより好ましく、-5~10℃がさらに好ましい。反応時間は、1~60分が好ましく、5~45分がより好ましく、10~30分がさらに好ましい。
a-4工程は、一般式(14)の化合物から、酸性水溶液中にてアミノ酸保護基を脱離させ、一般式(15)の化合物を得る工程である。
a-5工程は、一般式(15)の化合物のアミノ基に、塩基性条件下で保護基を付加させ、一般式(16)の化合物を得る工程である。使用される保護基は、ジ-tert-ブチルジカルボネート(「Boc2O」ともいう)、ベンジルクロロホルメート等が例示される。
好ましい。
a-6工程は、一般式(16)の化合物を、水素還元して一般式(17)の化合物を得る工程である。使用する触媒としては、パラジウム炭素、水酸化パラジウム等が例示される。
a-7工程は、一般式(17)の化合物を、ジアゾニウムを経てヨウ素化して一般式(18)の化合物を得る工程である。ジアゾニウム反応試薬としては、亜硝酸ナトリム、亜硝酸カリウム、亜硝酸イソブチル等が例示される。ヨウ素化試薬としては、ヨウ化ナトリウム、ヨウ化カリウム等が例示される。
a-8工程は、一般式(18)の化合物を、過酸化物等の酸化剤の存在下、ヨウ素を超原子価ヨードニウムイリドとして一般式(19)の化合物を得る工程である。酸化剤としては、3-クロロ過安息香酸、1-クロロメチル-4-フルオロ-1,4-ジアゾニアビシクロ[2.2.2]オクタン-ビス(テトラフルオロボラート)等が例示される。
反応温度は、0~40℃が好ましく、10~40℃がより好ましい。反応時間は、2時間~12時間が好ましく、2分~8時間がより好ましい。
a-9工程は、一般式(19)の化合物を、[18F]フッ化物イオンでフッ素化して一般式(20)の化合物を得る工程である。
反応温度は、40℃~150℃が好ましく、60~140℃がより好ましい。反応時間は、1分~1時間が好ましく、5分~30分がより好ましい。

b-1工程は、一般式(30)の化合物をボラン酸の保護基と反応させ、一般式(31)の化合物を得る工程である。b-1工程で用いるホウ酸の保護基、溶媒、反応時間、反応温度は、a-1工程と同様である。
b-2工程は、一般式(31)の化合物の水酸基に、保護基を付加させ一般式(32)の化合物を得る工程である。保護基R6としては、炭素数6~12のアリール基が好ましい。
反応温度は、0℃~60℃が好ましく、10~40℃がより好ましい。反応時間は、1~24時間が好ましく、3~8時間がより好ましい。
b-3工程は、一般式(32)の化合物をハロゲン化試薬と反応させて、一般式(33)の化合物を得る工程である。b-3工程のXは、フッ素、塩素、ヨウ素または臭素であり、入手性の観点から臭素が好ましい。b-3工程で用いるハロゲン化試薬、溶媒、反応時間、反応温度は、a-2工程と同様である。
b-4工程は、一般式(33)の化合物を修飾アミノ酸と反応させ、一般式(34)の化合物を得る工程である。b-4工程で使用する修飾アミノ酸、塩基、溶媒、反応時間、反応温度は、a-3工程と同様である。
b-5工程は、一般式(34)の化合物から、酸性水溶液中にてアミノ酸保護基を脱離させ、一般式(35)の化合物を得る工程である。b-5工程で使用する溶媒、反応時間、反応温度は、a-4工程と同様である。
b-6工程は、一般式(35)の化合物のアミノ基に、塩基性条件下で保護基を付加させ、一般式(36)の化合物を得る工程である。b-6工程で使用する保護基、塩基、溶媒、反応時間、反応温度は、a-5工程と同様である。
b-7工程は、一般式(36)の化合物を、水素還元して一般式(37)の化合物を得る工程である。b-7工程で使用する溶媒、反応時間、反応温度は、a-6工程と同様である。
b-8工程は、一般式(37)の化合物を、[18F]フッ化物イオンでフッ素化して一般式(38)の化合物を得る工程である。
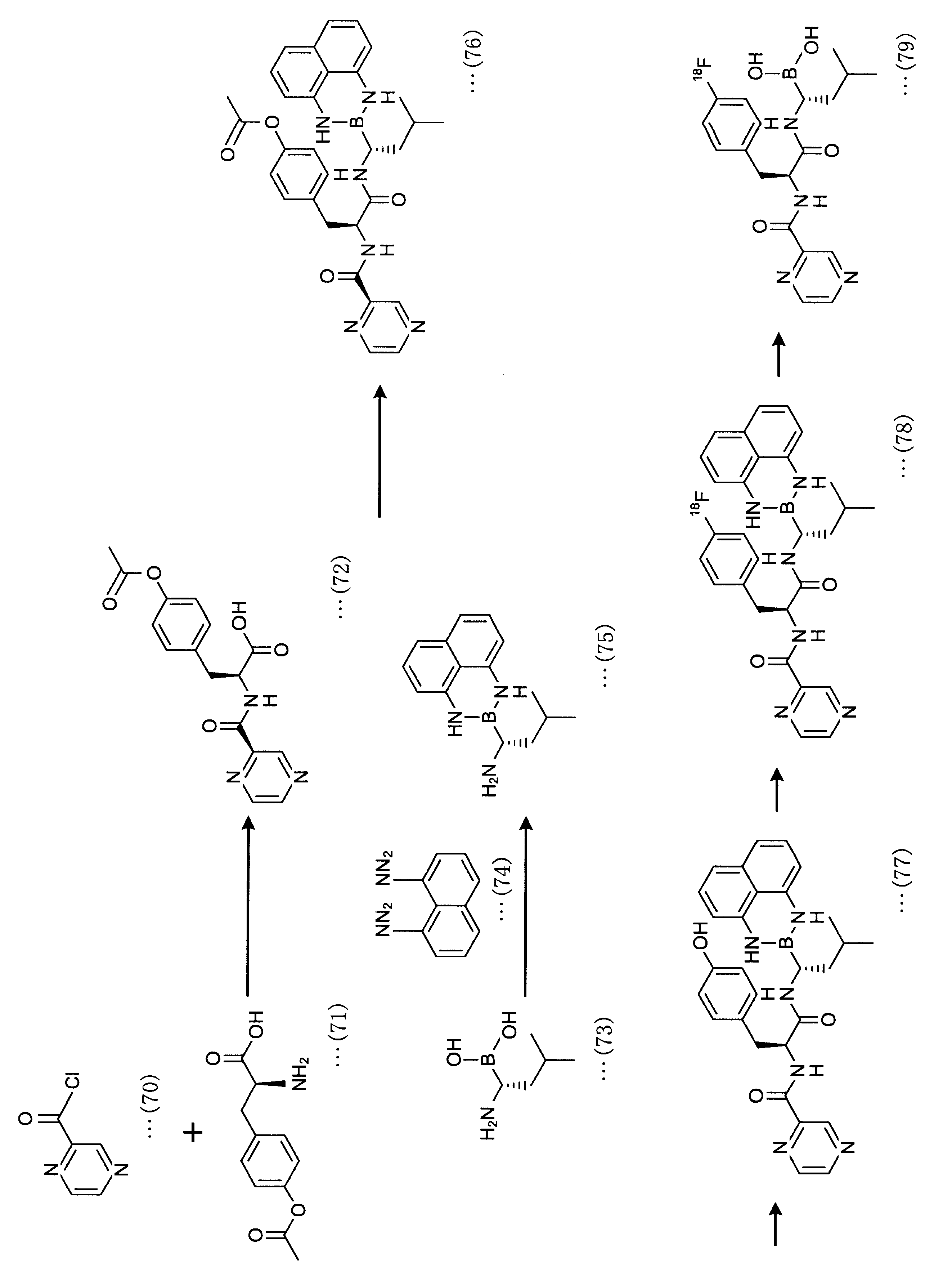
実施例1は、以下の合成スキームで合成を行った。
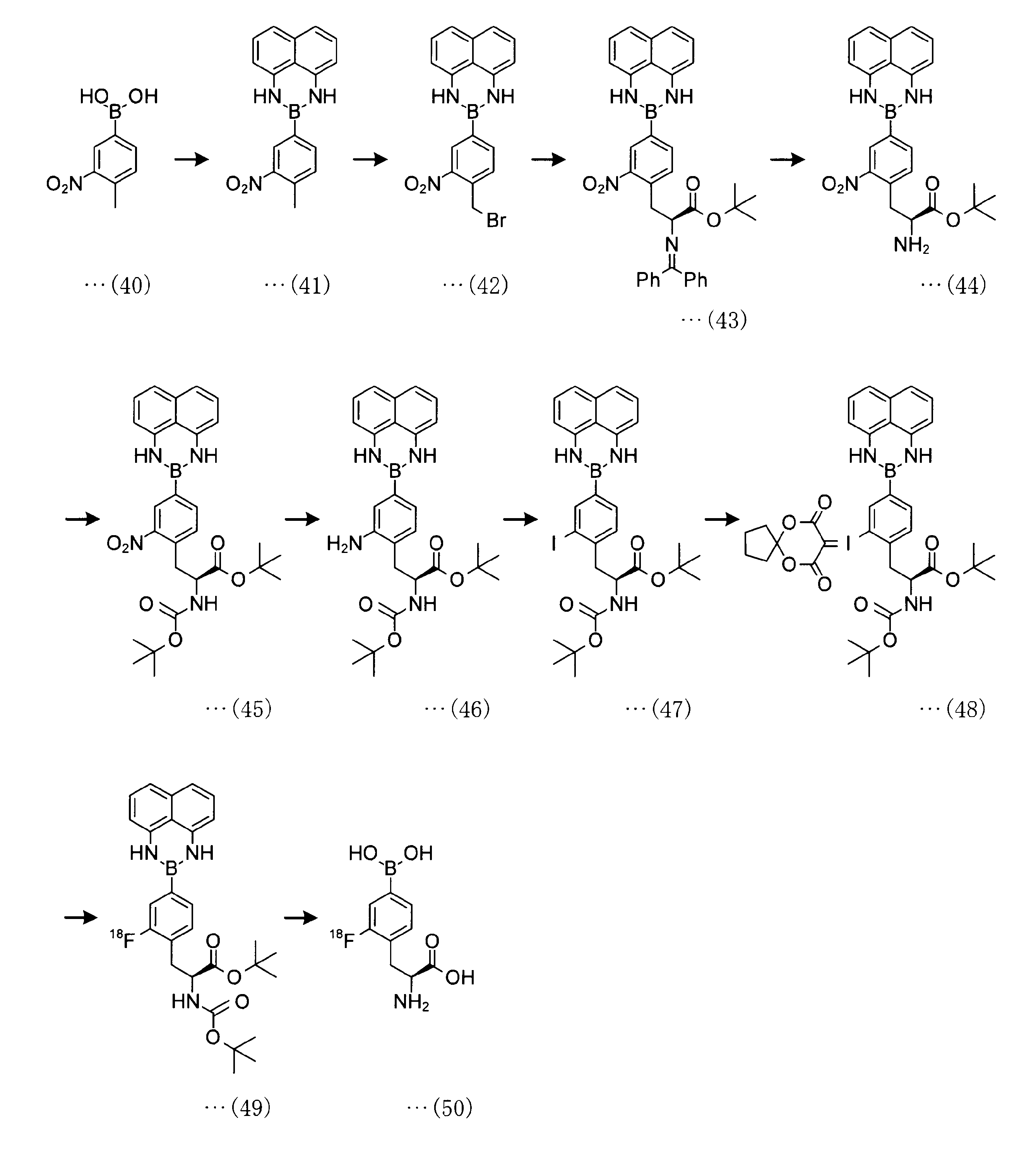
2-(4-メチル-3-ニトロフェニル)-2,3-ジヒドロ-1H-ナフト[1,8-de][1,3,2]ジアザボリニン(化合物(41))の合成
アルゴン雰囲気下、(4-メチル-3-ニトロフェニル)ボロン酸(3.62g、20mmol)、1,8-ジアミノナフタレン(4.8g、15mmol)と4Åモレキュラーシーブス500mgをジメチルスルホキシド、トルエンの混合溶液55mL(1:10(v/v))に溶解させ、12時間Dean-Stark装置中で還流した。反応の終了を薄層クロマトグラフィー(thin-layer chromatography、「TLC」ともいう)で確認したのち、反応溶液を室温付近まで冷却した。反応溶液に30mLの水を加え、30mLの酢酸エチルで3回抽出した。有機層を無水硫酸マグネシウム(「MgSO4」ともいう)で乾燥させ、溶媒を留去した。残渣をn-ヘキサン/酢酸エチル=1/1を溶出溶媒とするシリカゲルカラムクロマトグラフィーにより精製することで化合物(41)を得る(5.55g、91.5%)。
1H-NMR(300MHz,DMSO-d6):8.21(s,1H),8.09(d,1H),7.53(d,2H),7.42(d,1H),7.40(t,2H),6.96(d,2H),2.54(s,3H)
LC-MS(ESI)m/z:383[M+H]+.
2-(4-(ブロモメチル)-3-ニトロフェニル)-2,3-ジヒドロ-1H-ナフト[1,8-de][1,3,2]ジアザボリニン(化合物(42))の合成
化合物(41)(2.8g、9.2mmol)の四塩化炭素溶液(30mL)に、0.05等量のアザビスイソブチルニトリル、1.1等量のN-ブロモスクシンイミド(「NBS」ともいう)を加え、90℃で一晩加熱還流した。溶媒を留去したのち、n-ヘキサン/酢酸エチル=7/1を溶出溶媒とするシリカゲルカラムクロマトグラフィーにより精製することで化合物(42)を得た(3.1g、87%)。
1H-NMR(300MHz,DMSO-d6):8.29(s,1H),8.17(d,1H),7.54(d,1H),7.53(d,2H),7.40(d,2H),6.96(d,2H),4.56(s,2H)
LC-MS(ESI)m/z:304[M+H]+.
tert-ブチル-3-(4-(1H-ナフト[1,8-de][1,3,2]ジアザボリニン-2(3H)-イル)-2-ニトロフェニル)-2-((ジフェニルメチレン)アミノ)プロパノエート(化合物(43))の合成
化合物(42)(3.1g、8mmol)、N-(ジフェニルメチレン)グリシン-tert-ブチルエステル(2.6g、1.1等量)、0.05等量の(R)-4,4-ジブチル-2,6-ビス(3,4,5-トリフルオロフェニル)-4,5-ジヒドロ-3H-ジナフト[2,1-c:1’,2’-e]アゼピニウムブロミドをトルエン30mL、9M水酸化カリウム水溶液(「KOH水溶液」ともいう)30mLに溶解させ、0℃で12時間攪拌した。KOH水溶液を除去したのち、トルエン層を30mLのブラインで3回洗浄し、有機層をMgSO4で乾燥させ、溶媒を減圧留去することで化合物(43)の粗精製物を得る。化合物(43)の粗精製物はシリカゲルカラムクロマトグラフィーにより精製した(3.87g、81%)。
1H-NMR(300MHz,DMSO-d6):8.26(s,1H),8.14(d,1H),7.95(d,2H),7.60-7.35(m,13H,Ar),6.96(d,2H),4.35(s,1H),3.38(dd,1H,CH2-α),3.13(dd,1H,CH2-β),1.42(s,9H)
LC-MS(ESI)m/z:597[M+H]+.
tert-ブチル-3-(4-(1H-ナフト[1,8-de][1,3,2]ジアザボリニン-2(3H)-イル)-2-ニトロフェニル)-2-アミノプロパノエート(化合物(44))の合成
化合物(43)(3.5g、5.9mmol)を20mLのテトラヒドロフラン(「THF」ともいう)と20mLの20%クエン酸水溶液の混液に溶解させ、2時間室温で攪拌させる。THFを減圧留去したのち、20mLのジエチルエーテルに再溶解させ、20mLのブラインで3回洗浄、MgSO4で乾燥、溶媒を留去することで化合物(44)の粗精製物を得る(2.52g)。
1H-NMR(300MHz,DMSO-d6):8.26(s,1H),8.14(d,1H),7.53-7.40(m,5H,Ar(Arは芳香族を表す)),6.96(d,2H),4.14(s,1H),3.54(dd,1H,CH2-α),3.29(dd,1H,CH2-β),1.42(s,9H)
LC-MS(ESI)m/z:433[M+H]+.
tert-ブチル-3-(4-(1H-ナフト[1,8-de][1,3,2]ジアザボリニン-2(3H)-イル)-2-ニトロフェニル)-2-((tert-ブロキシカルボニル)アミノ)プロパノエート(化合物(45))の合成
化合物(44)(2.52g)の粗精製物を20mLのアセトンに溶解し、10mLの炭酸カリウム水溶液(「K2CO3水溶液」ともいう)を加える。1.2等量のジ-tert-ブチルジカルボネートを反応溶液に加え室温で17時間撹拌する。反応終了をTLCで確認後、アセトンを減圧留去し、酢酸エチルに再溶解し、有機層をブラインで洗浄する。有機層をMgSO4で乾燥させたのち、n-ヘキサン/酢酸エチル=7/1を溶出溶媒とするシリカゲルカラムクロマトグラフィーにより精製し、化合物(45)(2.61g、83%、2steps)を得る。
1H-NMR(300MHz,DMSO-d6):8.26(s,1H),8.14(d,1H),7.53-7.40(m,5H,Ar),6.96(d,2H),4.14(s,1H),3.54(dd,1H,CH2-α),3.29(dd,1H,CH2-β),1.42(s,18H)
LC-MS(ESI)m/z:533[M+H]+.
tert-ブチル-3-(2-アミノ-4-(1H-ナフト[1,8-de][1,3,2]ジアザボリニン-2(3H)-イル)フェニル)-2-((tert-ブトキシカルボニル)アミノ)プロパノエート(化合物(46))の合成
アルゴン雰囲気下、化合物(45)(2.5g、4.7mmol)の15mLエタノール溶液を氷冷し、ゆっくりとパラジウム炭素(100mg、21μmol)を加えた。アルゴンを水素に置換後、室温にて30時間攪拌した。アルゴン置換、セライトろ過を行い、ろ液を減圧濃縮した。残渣をn-ヘキサン/酢酸エチル=4/1を溶出溶媒とするシリカゲルカラムクロマトグラフィーで精製することによりオイル状の化合物(46)(1.98g、84%)を得た。
1H-NMR(300MHz,DMSO-d6):7.53(d,2H),7.40(t,2H),7.21(d,1H),7.05(d,1H),6.96(d,2H),6.73(s,1H),4.68(s,1H),3.54(dd,1H,CH2-α),3.29(dd,1H,CH2-β),1.42(s,18H)
LC-MS(ESI)m/z:503[M+H]+.
tert-ブチル-2-((tert-ブトキシカルボニル)アミノ)-3-(2-ヨード-4-(1H-ナフト[1,8-de][1,3,2]ジアザボリニン-2(3H)-イル)フェニル)プロパノエート(化合物(47))の合成
アルゴン雰囲気下、化合物(46)(1.8g、3.6mmol)の2-プロパノール溶液(5mL)を0℃で撹拌し、3.6Mのテトラフルオロホウ酸水溶液を1.33mL(4.8mmol)加える。反応溶液を0℃で30分撹拌したのち、亜硝酸ナトリウム(3.04g、44mmol)をゆっくりと加え、さらに30分撹拌した。生成した結晶をろ過し、冷メタノールで洗浄後、減圧下乾燥した。結晶をアセトニトリルに再溶解させ、ヨウ化カリウムの水溶液を加え、0℃で30分、続いて室温で30分撹拌した。アセトニトリルを減圧留去後、20mLの酢酸エチルに再溶解させ、有機層を10mLのチオ硫酸ナトリウム水溶液で洗浄後、20mLのブラインでさらに2回洗浄した。有機層をMgSO4で乾燥させたのち、ろ液を減圧濃縮した。残渣をn-ヘキサン/酢酸エチル=10/1を溶出溶媒とするシリカゲルカラムクロマトグラフィーで精製することによりオイル状の化合物(47)(674mg、31%)を得た。
1H-NMR(300MHz,DMSO-d6):7.74(d,1H),7.71(s,1H),7.53(d,2H),7.40(t,2H),6.97(d,1H),6.96(d,2H),4.68(s,1H),3.54(dd,1H,CH2-α),3.29(dd,1H,CH2-β),1.42(s,18H)
LC-MS(ESI)m/z:614[M+H]+.
tert-ブチル-2-((tert-ブトキシカルボニル)アミノ)-3-(2-((7,9-ジオキソ-6,10-ジオキサスピロ[4.5]デカン-8-イリデン)-3-ヨーダニル)-4-(1H-ナフト[1,8-de][1,3,2]ジアザボリニン-2(3H)-イル)フェニル))プロパノエート(化合物(48))の合成
化合物(47)(613mg、1mmol)のクロロホルム溶液にメタクロロ過安息香酸(「mCPBA」ともいう)を加え、室温で3時間撹拌した。化合物(47)が全て反応したことをTLCにより確認したのち、6,10-ジオキサスピロ[4.5]デカン-7,9-ジオンの10%炭酸ナトリウム溶液(「Na2CO3溶液」ともいう)(w/v、2mL,0.33M溶液)を反応溶液に加える。反応溶液をさらに4時間撹拌し、反応の完結をTLCで確認する。反応溶液に水を加え、クロロホルムで3回抽出した。有機層を集め、MgSO4で乾燥、溶媒を留去した。残渣にヘキサンと酢酸エチルを適量加え、冷蔵庫中で再結晶させた。結晶を集めることで化合物(48)(266mg、34%)を得た。
1H-NMR(300MHz,DMSO-d6):7.74(d,1H),7.71(s,1H),7.53(d,2H),7.40(t,H),6.97(d,1H),6.96(d,2H),4.68(s,1H),3.54(dd,1H,CH2-α),3.29(dd,1H,CH2-β),2.16(m,4H),1.80(m,4H),1.42(s,18H)
LC-MS(ESI)m/z:782[M+H]+.
tert-ブチル-2-((tert-ブトキシカルボニル)アミノ)-3-(2-[18F]フルオロ-4-(1H-ナフト[1,8-de][1,3,2]ジアザボリニン-2(3H)-イル)フェニル)プロパノエート(化合物(49))の合成
サイクロトロンより生成した[18F]フッ化物イオンをSep-Pak Light Accell Plus QMA Carbonate(46mg)に通しトラップする。トラップした[18F]フッ化物イオンをクリプトフィックス222(3.77mg、10μmol)、K2CO3(691μg、5μmol)のアセトニトリル(960μL)、水(40μL)混液を用いて溶出し、反応バイアルに移送する。この反応バイアルを140℃、3分加熱し乾固した。さらに少量のアセトニトリルを加え3回共沸を行なった。
続いて化合物(48)(10mg、15.5μmol)のDMF(400μL)溶液で溶解し、反応バイアルに加え、80℃で5分反応を行う。
冷却後、反応バイアルに緩衝液2mL、蒸留水16mLを加え希釈し、Sep-Pak C18を通し、未反応の[18F]フッ化物イオンを除去する。さらに蒸留水2mLで洗浄し、エタノール1mLで溶離し、化合物(49)を得た。
2-[18F]フルオロ-4-ボロノフェニルアラニン(化合物(50))の合成
化合物(49)の反応溶液に臭化水素(1mL)を加え、150℃で15分加熱した。反応溶液を水で希釈し、フィルターろ過し、HPLCおよびラジオTLCを用いて目的物の生成を確認した。
実施例2は、以下の合成スキームで合成を行った。
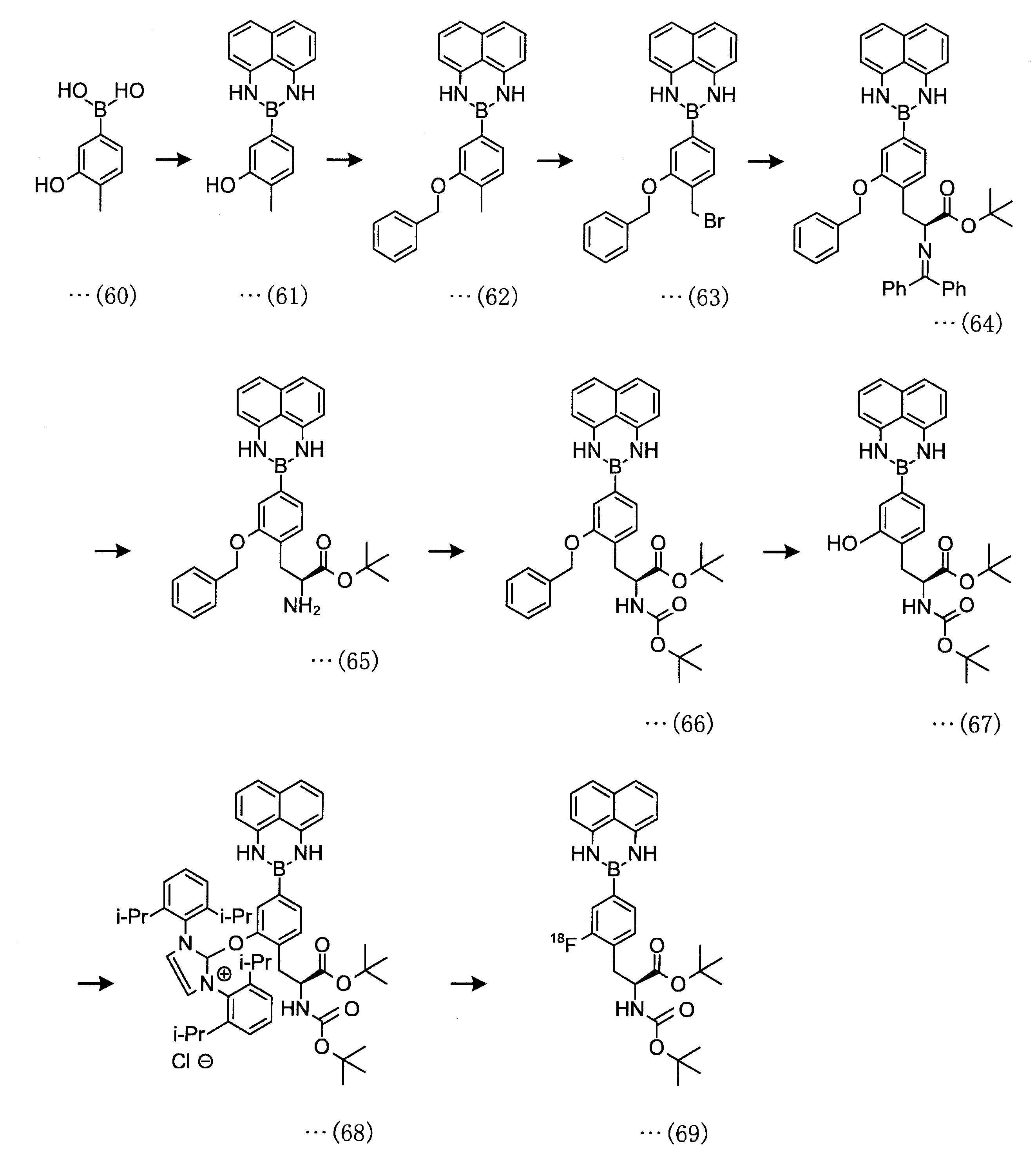
2-メチル-5-(1H-ナフト[1,8-de][1,3,2]ジアザボリニン-2(3H)-イル)フェノ-ル(化合物(61))の合成
アルゴン雰囲気下、化合物(60)(3.04g、20mmol)、1,8-ジアミノナフタレン(3.84g、24mmol)と4Åモレキュラーシーブス500mgをDMSO、トルエンの混合溶液44mL(1:10(v/v))に溶解させ、12時間Dean-Stark装置中で還流した。反応の終了をTLCで確認したのち、反応溶液を室温付近まで冷却した。反応溶液に30mLの水を加え、30mLの酢酸エチルで3回抽出した。有機層をMgSO4で乾燥させ、溶媒を留去した。残渣をn-ヘキサン/酢酸エチル=1/1を溶出溶媒とするシリカゲルカラムクロマトグラフィーにより精製することで化合物(61)を得る(4.99g、91%)。
1H-NMR(300MHz,DMSO-d6):9.68(s,1H),7.53(d,2H),7.40(t,2H),7.26(d,1H),6.99(d,1H),6.96(d,2H),6.75(s,1H),2.15(s,3H)
LC-MS(ESI)m/z:275[M+H]+.
2-(3-(ベンジルオキシ)-4-メチルフェニル)-2,3-ジヒドロ-1H-ナフト[1,8-de][1,3,2]ジアザボリニン(化合物(62))の合成
化合物(61)(4.66g、17mmol)をDMF(50mL)に溶解し、ベンジルブロミド(3.55mL、19mmol)と水素化ナトリウム(456mg、19mmol)を加え、反応溶液を室温で6時間撹拌した。TLCを用いて反応の進行を確認したのち、20mLの炭酸カリウムとメタノール溶液を加え、30分撹拌したのち、有機溶媒を減圧留去後、残渣を50mLのクロロホルムに溶解した。有機層を40mLのブラインで3回洗浄し、有機層をMgSO4で乾燥した。ろ液を減圧留去することで化合物(62)を得た(5.94g、96%)。
1H-NMR(300MHz,DMSO-d6):7.53-7.32(m,10H),7.16(d,1H),6.96(d,2H),6.90(s,1H),5.16(s,2H),2.15(s,3H)
LC-MS(ESI)m/z:365[M+H]+.
2-(3-(ベンジルオキシ)-4-(ブロモメチル)フェニル)-2,3-ジヒドロ-1H-ナフト[1,8-de][1,3,2]ジアザボリニン(化合物(63))の合成
化合物(62)(5.83g、16mmol)の四塩化炭素溶液(40mL)に、0.05等量のアゾビスイソブチロニトリル、1.1等量のNBSを加え、100℃で一晩加熱還流した。溶媒を留去したのち、n-ヘキサン/酢酸エチル=6/1を溶出溶媒とするシリカゲルカラムクロマトグラフィーにより精製することで化合物(63)を得る(5.81g、82%)。
1H-NMR(300MHz,DMSO-d6):7.53-7.28(m,11H),6.98(s,1H),6.96(d,2H),5.16(s,2H),4.56(s,2H)
LC-MS(ESI)m/z:444[M+H]+.
tert-ブチル-3-(2-(ベンジルオキシ)-4-(1H-ナフト[1,8-de][1,3,2]ジアザボリニン-2(3H)-イル)フェニル)-2-((ジフェニルメチレン)アミノ)プロパノエート(化合物(64))の合成
4.43gの化合物(63)(10mmol)、3.25gのN-(ジフェニルメチレン)グリシン-tert-ブチルエステル、0.05等量の(R)-4,4-ジブチル-2,6-ビス(3,4,5-トリフルオロフェニル)-4,5-ジヒドロ-3H-ジナフト[2,1-c:1’,2’-e]アゼピニウムブロミドをトルエン30mL、9MのKOH水溶液30mLに溶解させ、0℃で12時間攪拌する。KOH層を除去したのち、トルエン層を30mLのブラインで3回洗浄し、有機層をMgSO4で乾燥させたのち、溶媒を減圧留去することで化合物(64)の粗精製物を得る。化合物(64)の粗精製物はシリカゲルカラムクロマトグラフィーにより精製した(5.33g、81%)。
1H-NMR(300MHz,DMSO-d6):7.95(d,2H),7.63-7.32(m,18H,Ar),7.20(d,1H),6.96(d,2H),6.95(s,1H),5.16(s,2H),4.35(s,1H),3.38(dd,1H,CH2-α),3.13(dd,1H,CH2-β),1.42(s,9H)
LC-MS(ESI)m/z:659[M+H]+.
tert-ブチル-2-アミノ-3-(2-(ベンジルオキシ)-4-(1H-ナフト[1,8-de][1,3,2]ジアザボリニン-2(3H)-イル)フェニル)プロパノエート(化合物(65))の合成
化合物(64)(5.0g、7.6mmol)を50mLのTHFと50mLの20%クエン酸水溶液の混液に溶解させ、2時間室温で攪拌させる。THFを減圧留去したのち、50mLのジエチルエーテルに再溶解させ、50mLのブラインで3回洗浄、MgSO4で乾燥、溶媒を留去することで化合物(65)の粗精製物を得る(3.66g)。
1H-NMR(300MHz,DMSO-d6):7.53-7.32(m,10H,Ar),7.20(d,1H),6.96(d,2H),6.95(s,1H),5.16(s,2H),4.14(s,1H),3.54(dd,1H,CH2-α),3.29(dd,1H,CH2-β),1.42(s,9H)
LC-MS(ESI)m/z:494[M+H]+.
tert-ブチル-3-(2-(ベンジルオキシ)-4-(1H-ナフト[1,8-de][1,3,2]ジアザボリニン-2(3H)-イル)フェニル)-2-((tert-ブトキシカルボニル)アミノ)プロパノエート(化合物(66))の合成
化合物(65)(3.66g)の粗精製物を30mLのアセトンに溶解し、10mLのK2CO3水溶液を加える。1.2等量のBoc2Oを反応溶液に加え室温で17時間撹拌する。反応終了をTLCで確認後、アセトンを減圧留去し、酢酸エチルに再溶解し、有機層をブラインで洗浄する。有機層をMgSO4で乾燥させたのち、n-ヘキサン/酢酸エチル=7/1を溶出溶媒とするシリカゲルカラムクロマトグラフィーにより精製し、化合物(66)(3.74g、83%,2steps)を得る。
1H-NMR(300MHz,DMSO-d6):7.53-7.32(m,10H,Ar),7.20(d,1H),6.96(d,2H),6.95(s,1H),5.16(s,2H),4.68(s,1H),3.54(dd,1H,CH2-α),3.29(dd,1H,CH2-β),1.42(s,18H)
LC-MS(ESI)m/z:595[M+H]+.
tert-ブチル-2-((tert-ブトキシカルボニル)アミノ)-3-(2-ヒドロキシ-4-(1H-ナフト[1,8-de][1,3,2]ジアザボリニン-2(3H)-イル)フェニル)プロパノエート(化合物(67))の合成
アルゴン雰囲気下、化合物(66)(3.5g、5.9mmol)の15mLエタノール溶液を氷冷し、ゆっくりとパラジウム炭素(100mg、21μmol)を加えた。アルゴンを水素に置換後、室温にて30時間攪拌した。アルゴン置換、セライトろ過を行い、ろ液を減圧濃縮した。残渣をn-ヘキサン/酢酸エチル=7/1を溶出溶媒とするシリカゲルカラムクロマトグラフィーで精製することによりオイル状の化合物(67)(2.49g、84%)を得た。
1H-NMR(300MHz,DMSO-d6):9.68(s,1H),7.53(d,2H),7.40(t,2H),7.31(d,1H),7.03(d,1H),6.96(d,2H),6.80(s,1H),4.68(s,1H),3.54(dd,1H,CH2-α),3.29(dd,1H,CH2-β),1.42(s,18H)
LC-MS(ESI)m/z:504[M+H]+.
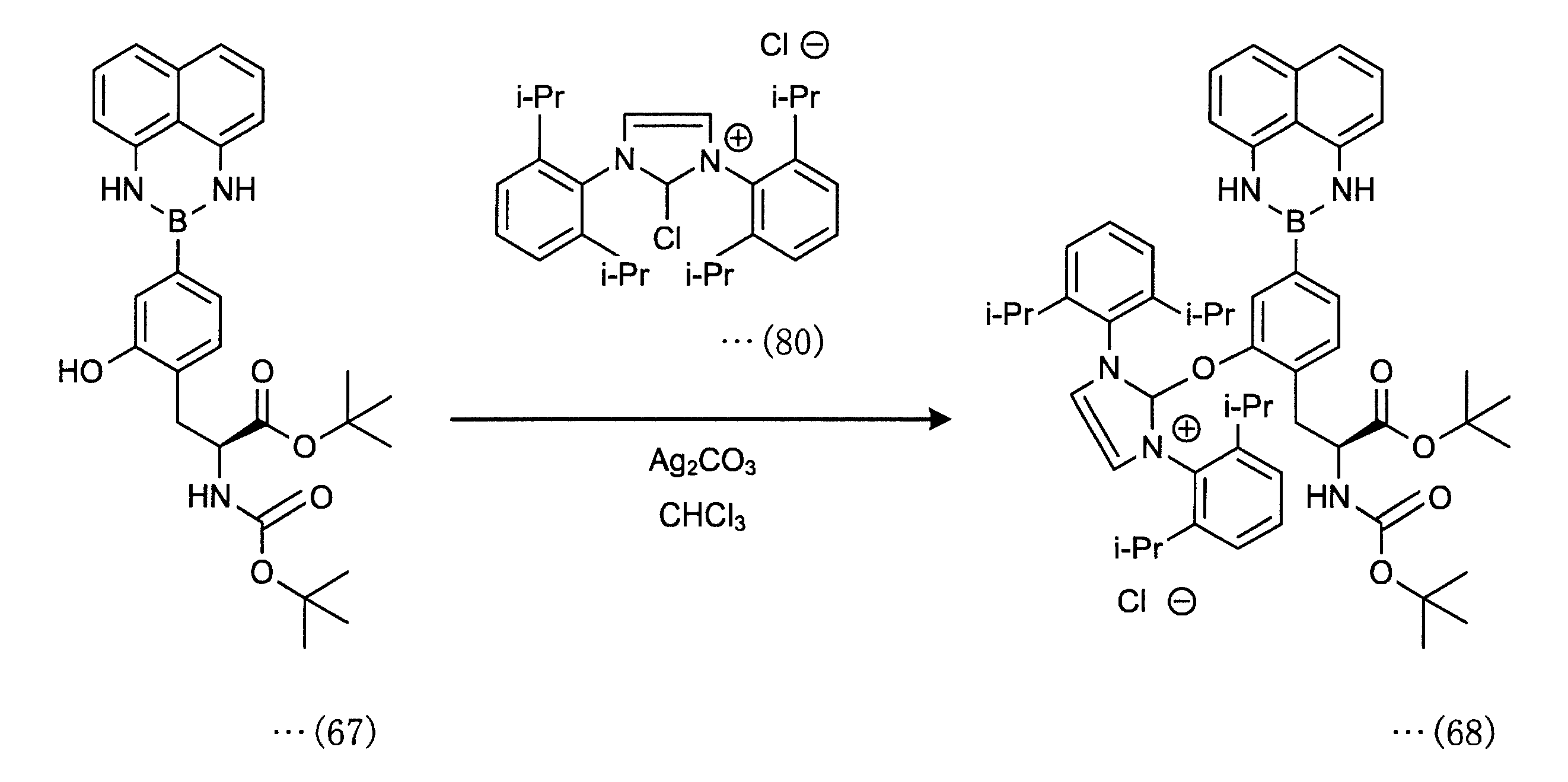
脱酸素的フッ素化剤(化合物(80))を使用した化合物(68)の合成
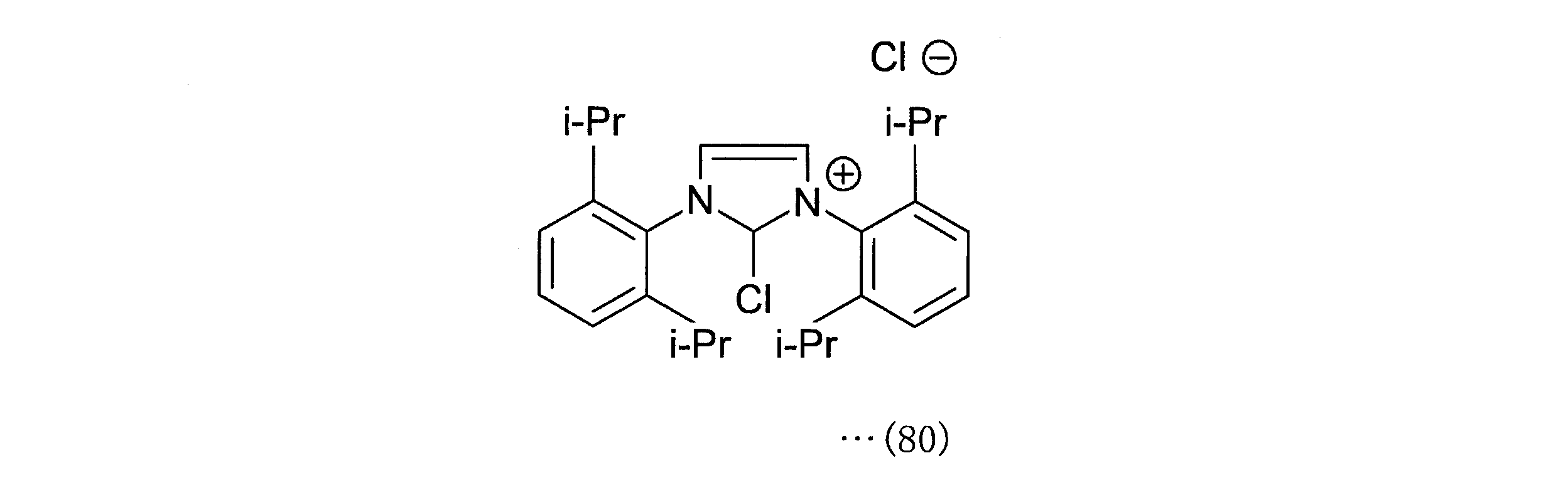
クロロイミダゾリウムクロライド(化合物(80))(500mg、1.07mmol、1.00equiv)と炭酸銀(「Ag2CO3」ともいう)(147mg、0.530mmol、0.500equiv)が混合され、脱酸素的フッ素化剤(PhenoFluor、化22の化合物(81))は褐色バイアルに保存された。
脱酸素的フッ素化剤(化合物(81))(1.00equiv)と化合物(67)(1.00equiv)、クロロホルム(2mL/mmol)をバイアルに入れ、縣濁液を60℃、4時間攪拌した。沈殿物をろ過により除去し、ろ液は濃縮され、化合物(68)が得られた。脱酸素的フッ素化剤(化合物(81))は下記スキームで製造される。

脱酸素的フッ素化剤(化合物(80))を使用した化合物(68)の合成
化合物(68)は、下記スキームのように、脱酸素的フッ素化剤(化合物(80))から直接合成してもよい。クロロイミダゾリウムクロライド(化合物(80))(500mg、1.07mmol、1.00equiv)、Ag2CO3(147mg、0.530mmol、0.500equiv、0.50equiv)、および化合物(67)(1.00equiv)、クロロホルム(2mL/mmol)をバイアルに入れ、縣濁液を60℃、4時間攪拌した。沈殿物をろ過により除去し、ろ液は濃縮され、化合物(68)が得られる。
tert-ブチル-2-((tert-ブトキシカルボニル)アミノ)-3-(2-[18F]フルオロ-4-(1H-ナフト[1,8-de][1,3,2]ジアザボリニン-2(3H)-イル)フェニル)プロパノエート(化合物(69))の合成
サイクロトロンより生成した[18F]フッ化物イオンをChromafix 30-PS-HCO3 に通しトラップする。2-ブタノン:エタノール=10:1(1mL)により洗浄する。トラップした[18F]フッ化物イオンを化合物(68)(8.0mg)の2-ブタノン:エタノール:NBu3(10:1:0.1)溶液1.0mLを用いて溶離する。溶離液を130℃、15分攪拌し、化合物(69)を得る。化合物(69)(化合物(49)と同一)を、実施例1、合成例10と同様の工程により加水分解することにより、化合物(50)を得ることができる。
Claims (5)
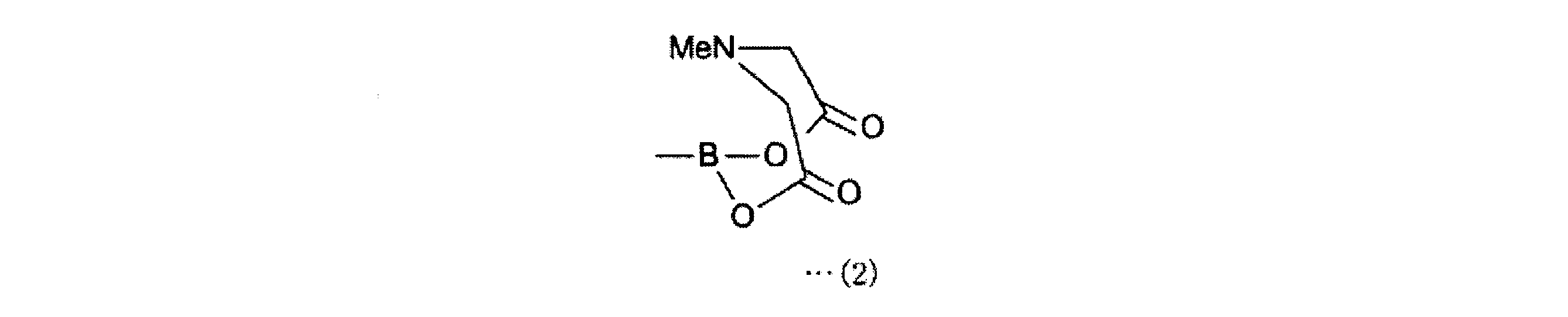
- 下記一般式(1)で表される4-ボロノフェニルアラニン前駆体
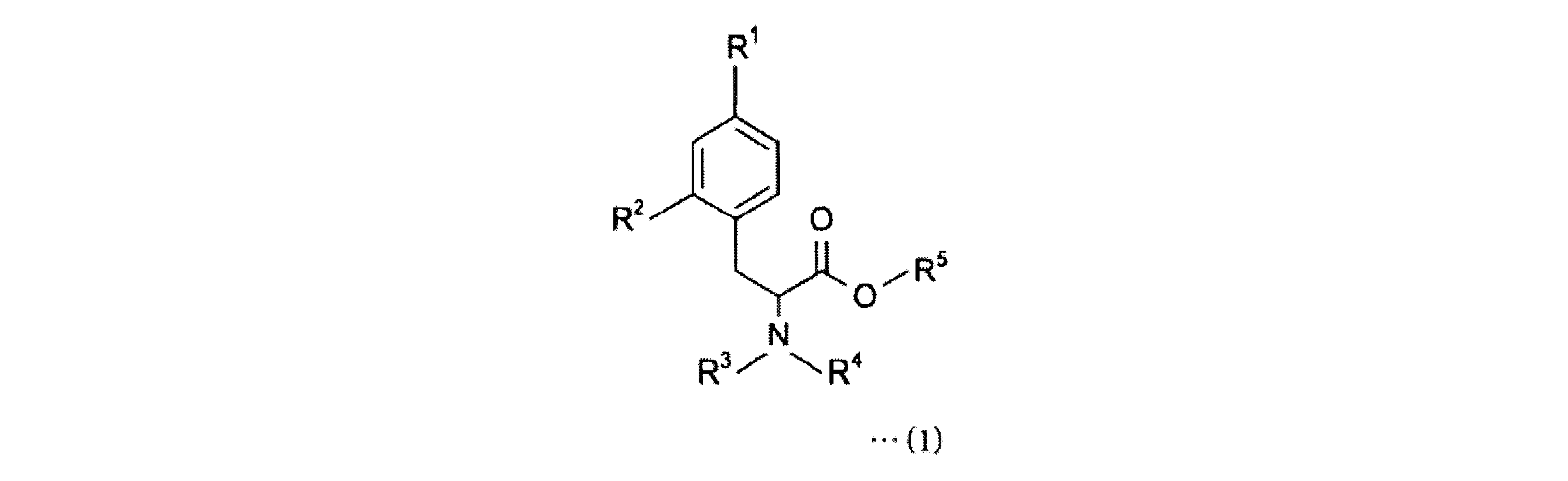
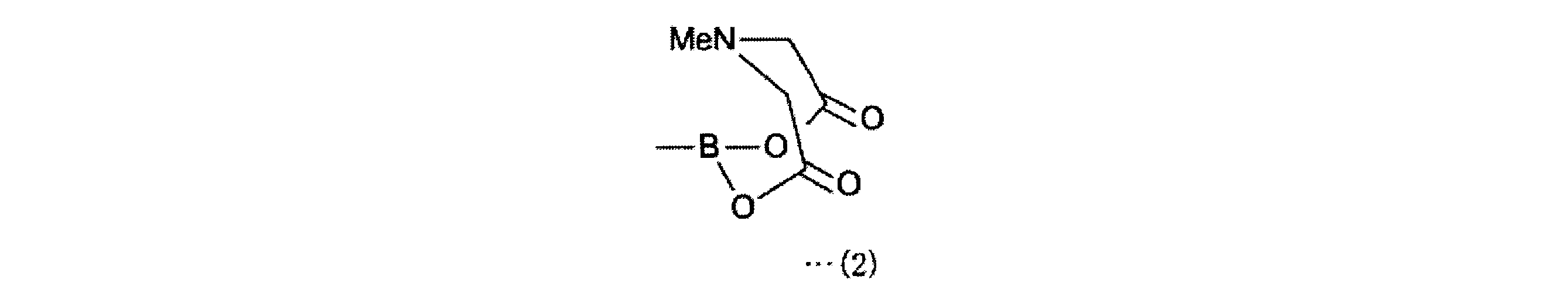
- 上記一般式(1)で表される化合物中、ヨードニウムイリド基はスピロ環を有するものであることを特徴とする請求項1に記載の4-ボロノフェニルアラニン前駆体。
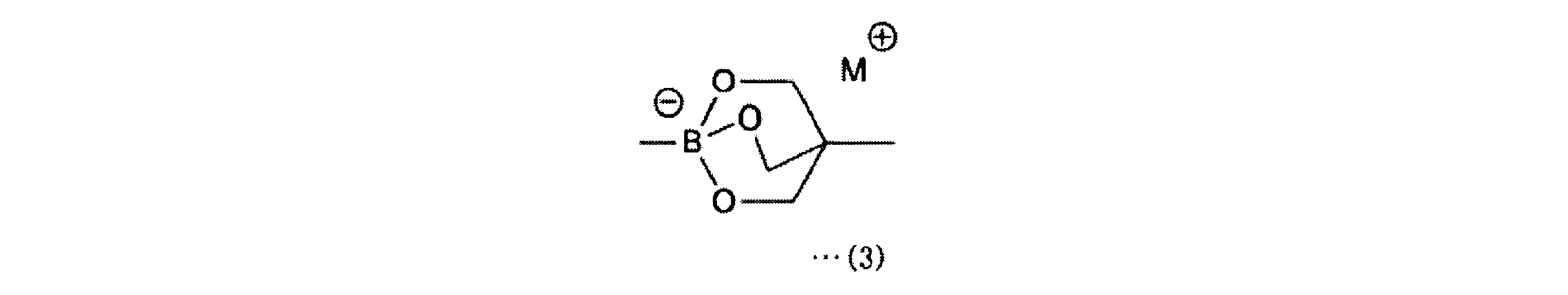
- 下記一般式(1)で表される4-ボロノフェニルアラニン前駆体
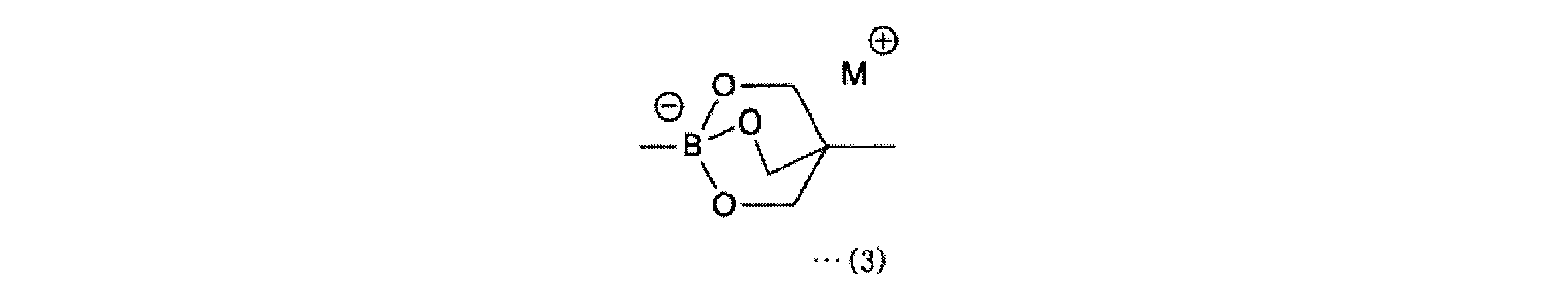

- 上記一般式(1)で表される化合物中、ヨードニウムイリド基はスピロ環を有するものであることを特徴とする請求項3に記載の2-[18F]フルオロ-4-ボロノフェニルアラニン前駆体の製造方法。
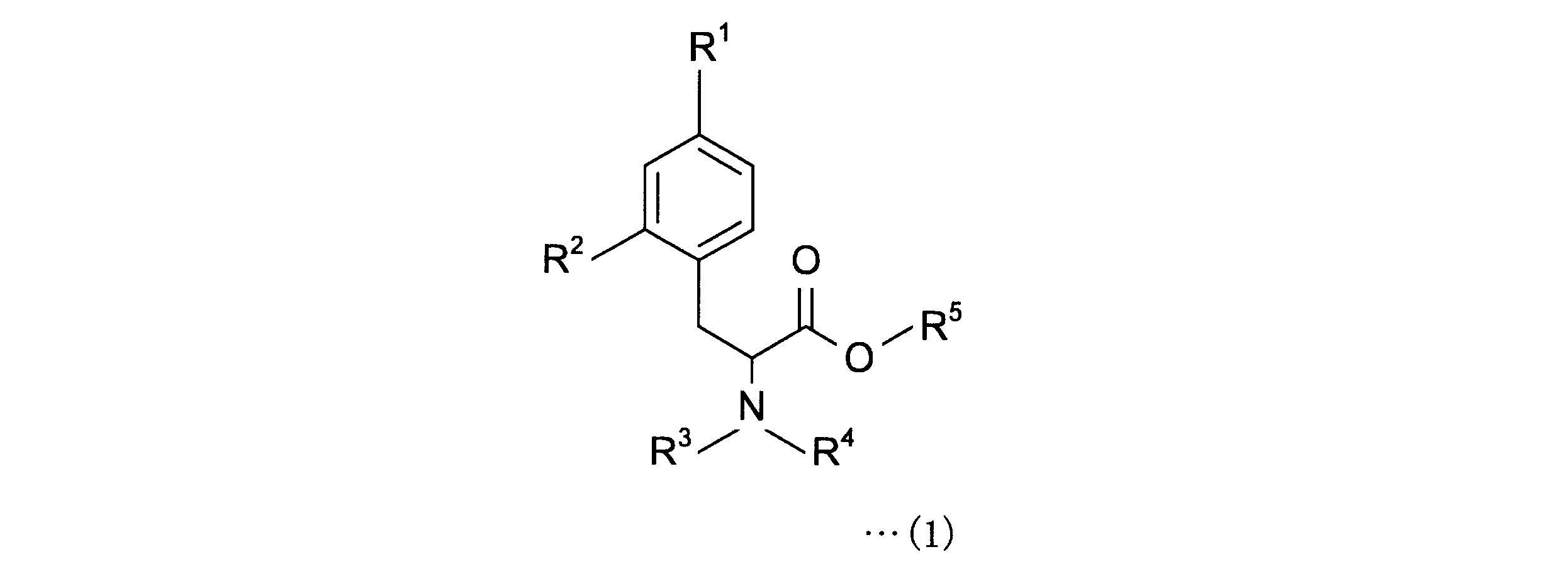
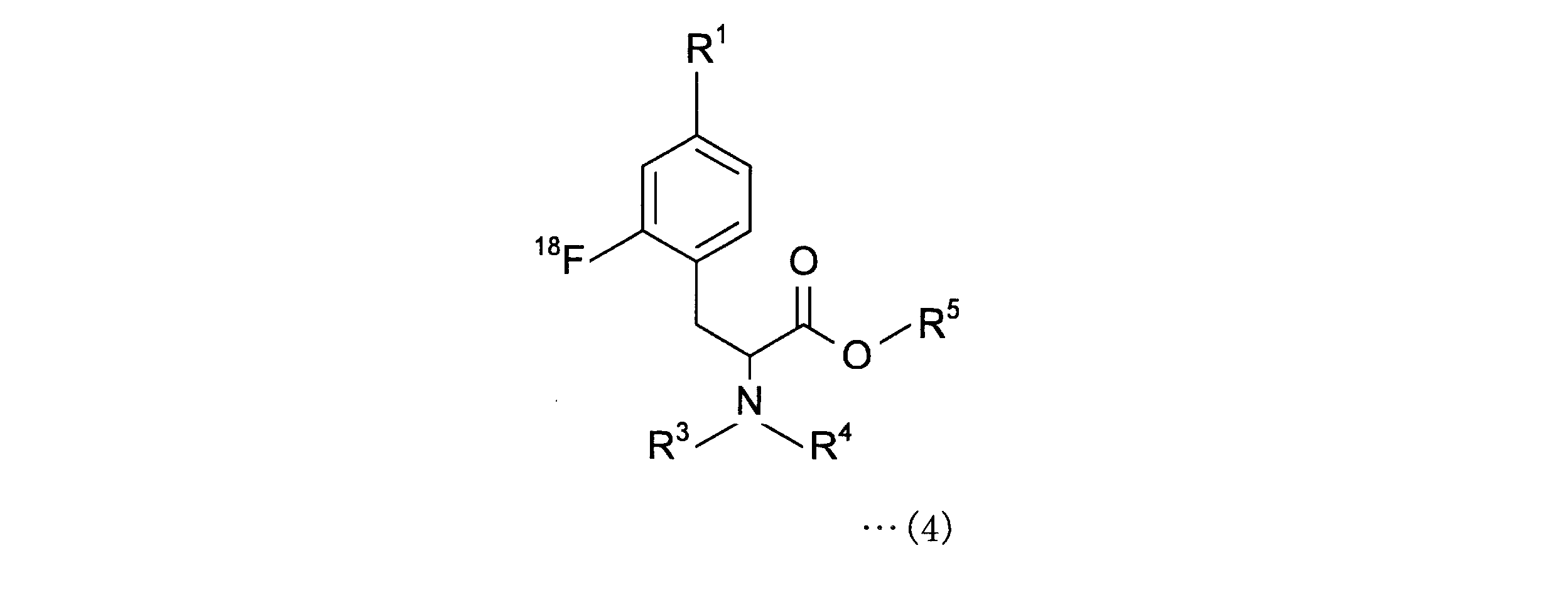
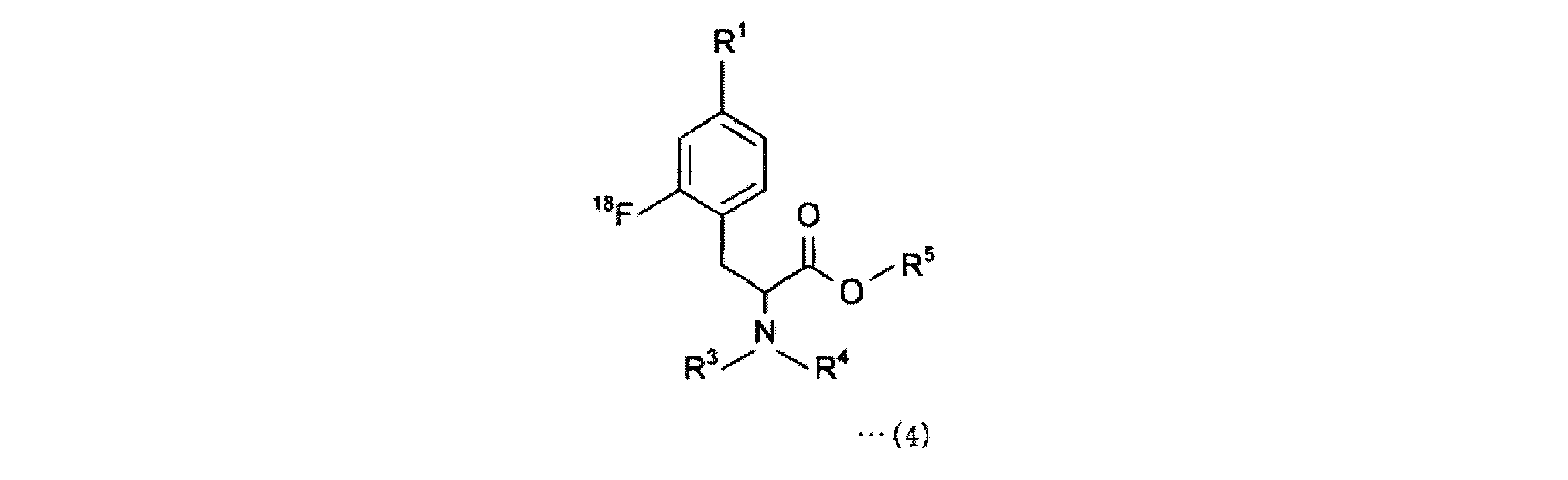
- 下記一般式(4)で表される2-[18F]フルオロ-4-ボロノフェニルアラニン前駆体
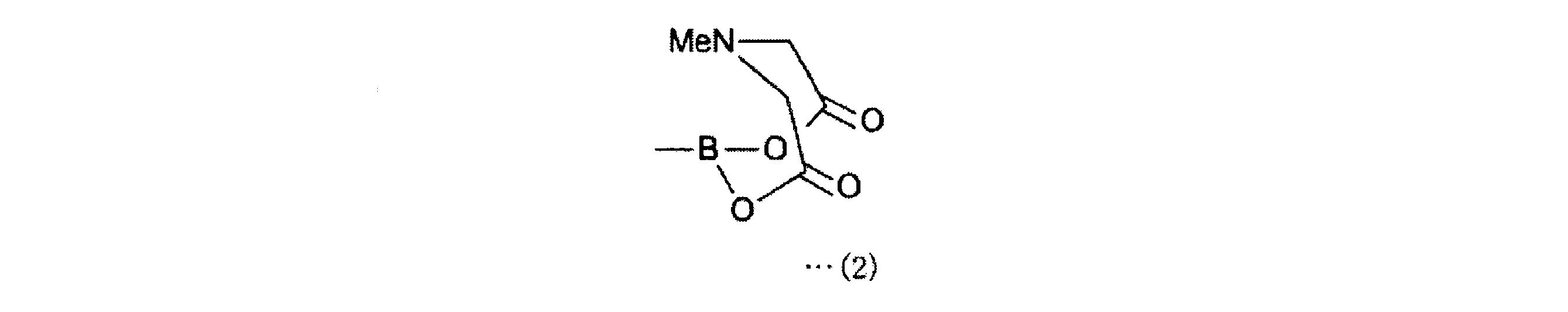
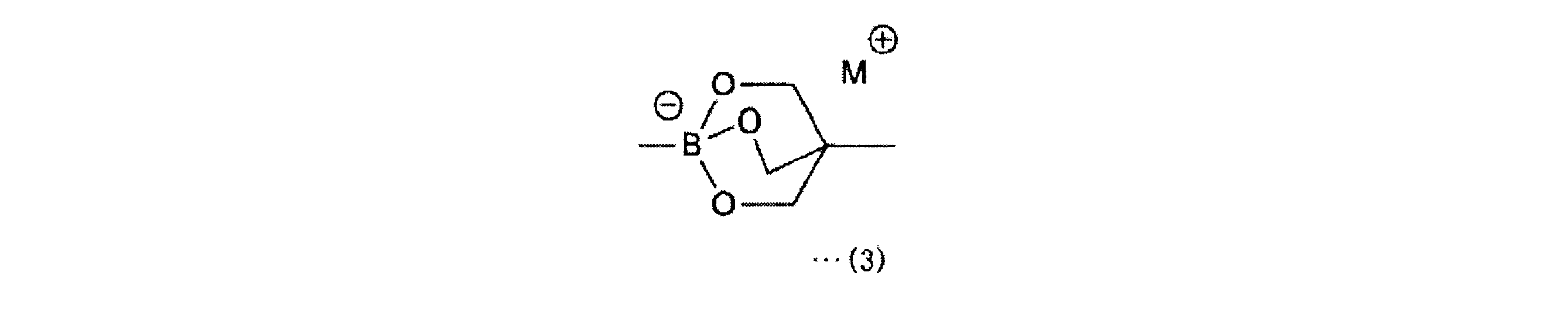
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2018135799A JP6996443B2 (ja) | 2018-07-19 | 2018-07-19 | 4-ボロノフェニルアラニン前駆体、2-[18f]フルオロ-4-ボロノフェニルアラニン前駆体の製造方法、2-[18f]フルオロ-4-ボロノフェニルアラニンの製造方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2018135799A JP6996443B2 (ja) | 2018-07-19 | 2018-07-19 | 4-ボロノフェニルアラニン前駆体、2-[18f]フルオロ-4-ボロノフェニルアラニン前駆体の製造方法、2-[18f]フルオロ-4-ボロノフェニルアラニンの製造方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2020011928A JP2020011928A (ja) | 2020-01-23 |
| JP6996443B2 true JP6996443B2 (ja) | 2022-02-04 |
Family
ID=69168856
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2018135799A Active JP6996443B2 (ja) | 2018-07-19 | 2018-07-19 | 4-ボロノフェニルアラニン前駆体、2-[18f]フルオロ-4-ボロノフェニルアラニン前駆体の製造方法、2-[18f]フルオロ-4-ボロノフェニルアラニンの製造方法 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP6996443B2 (ja) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20220156001A (ko) * | 2020-02-21 | 2022-11-24 | 국립대학법인 홋가이도 다이가쿠 | 방향족 아스타틴 화합물의 제조 방법 |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014061508A1 (ja) | 2012-10-15 | 2014-04-24 | ステラファーマ株式会社 | キラルな4-ボロノフェニルアラニン(bpa)誘導体と製造方法、およびその誘導体を用いた18f標識化bpaの製造方法 |
| WO2015093469A1 (ja) | 2013-12-17 | 2015-06-25 | ステラファーマ株式会社 | 2-フルオロ-4-ボロノ-l-フェニルアラニンの製造方法および2-フルオロ-4-ボロノ-l-フェニルアラニンの前駆体 |
| WO2015129374A1 (ja) | 2014-02-28 | 2015-09-03 | ステラファーマ株式会社 | 18f原子が導入された4‐ボロノ-l-フェニルアラニンの製造方法および18f原子が導入された4‐ボロノ-l-フェニルアラニンの前駆体 |
| JP2016204314A (ja) | 2015-04-23 | 2016-12-08 | 国立研究開発法人理化学研究所 | 化合物及び4−ボロノフェニルアラニン誘導体の製造方法 |
| JP2017513930A (ja) | 2014-03-20 | 2017-06-01 | オックスフォード ユニバーシティ イノベーション リミテッドOxford University Innovation Limited | フッ素化方法 |
| JP2017529312A (ja) | 2014-03-07 | 2017-10-05 | ザ ジェネラル ホスピタル コーポレイション | ヨウ素(iii)を媒介とする放射性フッ素化 |
-
2018
- 2018-07-19 JP JP2018135799A patent/JP6996443B2/ja active Active
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014061508A1 (ja) | 2012-10-15 | 2014-04-24 | ステラファーマ株式会社 | キラルな4-ボロノフェニルアラニン(bpa)誘導体と製造方法、およびその誘導体を用いた18f標識化bpaの製造方法 |
| WO2015093469A1 (ja) | 2013-12-17 | 2015-06-25 | ステラファーマ株式会社 | 2-フルオロ-4-ボロノ-l-フェニルアラニンの製造方法および2-フルオロ-4-ボロノ-l-フェニルアラニンの前駆体 |
| WO2015129374A1 (ja) | 2014-02-28 | 2015-09-03 | ステラファーマ株式会社 | 18f原子が導入された4‐ボロノ-l-フェニルアラニンの製造方法および18f原子が導入された4‐ボロノ-l-フェニルアラニンの前駆体 |
| JP2017529312A (ja) | 2014-03-07 | 2017-10-05 | ザ ジェネラル ホスピタル コーポレイション | ヨウ素(iii)を媒介とする放射性フッ素化 |
| JP2017513930A (ja) | 2014-03-20 | 2017-06-01 | オックスフォード ユニバーシティ イノベーション リミテッドOxford University Innovation Limited | フッ素化方法 |
| JP2016204314A (ja) | 2015-04-23 | 2016-12-08 | 国立研究開発法人理化学研究所 | 化合物及び4−ボロノフェニルアラニン誘導体の製造方法 |
Non-Patent Citations (1)
| Title |
|---|
| Neumann, Constanze N.; Hooker, Jacob M.; Ritter, Tobias,Concerted nucleophilic aromatic substitution with 19F- and 18F-,Nature (London, United Kingdom) ,2016年,534(7607),369-373 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2020011928A (ja) | 2020-01-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4981683B2 (ja) | アルコール溶媒中での有機フルオロ化合物の調製方法 | |
| EP2537826B1 (en) | Fluorination of aromatic ring systems | |
| EP2417119B1 (en) | No-carrier-added nucleophilic ýf-18¨fluorination of aromatic compounds | |
| EP3838890B1 (en) | Method for preparing 18f-bpa and intermediate | |
| JP5732198B2 (ja) | 放射性フッ素標識有機化合物の製造方法 | |
| JP2016204314A (ja) | 化合物及び4−ボロノフェニルアラニン誘導体の製造方法 | |
| JP6563401B2 (ja) | 放射性ヨウ素化化合物 | |
| JP6996443B2 (ja) | 4-ボロノフェニルアラニン前駆体、2-[18f]フルオロ-4-ボロノフェニルアラニン前駆体の製造方法、2-[18f]フルオロ-4-ボロノフェニルアラニンの製造方法 | |
| Riss et al. | Direct, nucleophilic radiosynthesis of [18 F] trifluoroalkyl tosylates: improved labelling procedures | |
| CN101316812A (zh) | 放射性卤素标记的有机化合物的前体化合物 | |
| Kniess et al. | “Hydrous 18F-fluoroethylation”–Leaving off the azeotropic drying | |
| CN119431194B (zh) | 一种18f-dopa的制备方法及其f-18标记前体 | |
| JP2008214319A (ja) | フェニルホウ素誘導体およびそれを用いたp−ボロノフェニルアラニンの製造方法 | |
| Kovac et al. | 3D QSAR study, synthesis, and in vitro evaluation of (+)-5-FBVM as potential PET radioligand for the vesicular acetylcholine transporter (VAChT) | |
| WO2014095739A1 (en) | Precursors and process for the production of 18f-labelled amino acids | |
| KR101879181B1 (ko) | 18f-표지 pet 방사성의약품의 전구체 및 그 제조방법 | |
| CN113307758B (zh) | 一种医用放射性同位素标记的p2x7受体靶向探针前体 | |
| WO2024019014A1 (ja) | 放射性標識活性化エステル及びその前駆体 | |
| JPWO2018164043A1 (ja) | 放射性フッ素標識前駆体化合物及びそれを用いた放射性フッ素標識化合物の製造方法 | |
| KR101619564B1 (ko) | F-18 표지 pet 추적자에 관한 신규 전구체 분자 | |
| US20210205482A1 (en) | Method for preparing fluorine-18-labeled fluoromethyl-substituted radiopharmaceuticals using selective azide substitution reaction and precursor scavenging | |
| JP7159157B2 (ja) | 放射性フッ素標識化合物の製造方法および放射性医薬の製造方法 | |
| WO2017106340A1 (en) | Isotopic fluorination and applications thereof | |
| US20130190529A1 (en) | Fluorine Radiolabelling Process | |
| HK1180299B (en) | Fluorination of aromatic ring systems |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20200910 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20210624 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20210713 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20210910 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20211116 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20211129 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6996443 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| S111 | Request for change of ownership or part of ownership |
Free format text: JAPANESE INTERMEDIATE CODE: R313111 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |