JP6969848B2 - プリナブリン組成物 - Google Patents
プリナブリン組成物 Download PDFInfo
- Publication number
- JP6969848B2 JP6969848B2 JP2018501232A JP2018501232A JP6969848B2 JP 6969848 B2 JP6969848 B2 JP 6969848B2 JP 2018501232 A JP2018501232 A JP 2018501232A JP 2018501232 A JP2018501232 A JP 2018501232A JP 6969848 B2 JP6969848 B2 JP 6969848B2
- Authority
- JP
- Japan
- Prior art keywords
- mixture
- prinabrin
- monohydrate
- composition
- purinabrin
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images

























Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/50—Pyridazines; Hydrogenated pyridazines
- A61K31/501—Pyridazines; Hydrogenated pyridazines not condensed and containing further heterocyclic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4965—Non-condensed pyrazines
- A61K31/497—Non-condensed pyrazines containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D9/00—Crystallisation
- B01D9/004—Fractional crystallisation; Fractionating or rectifying columns
- B01D9/0045—Washing of crystals, e.g. in wash columns
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D9/00—Crystallisation
- B01D9/005—Selection of auxiliary, e.g. for control of crystallisation nuclei, of crystal growth, of adherence to walls; Arrangements for introduction thereof
- B01D9/0054—Use of anti-solvent
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D9/00—Crystallisation
- B01D9/0063—Control or regulation
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D9/00—Crystallisation
- B01D2009/0086—Processes or apparatus therefor
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
Description
本出願は、2015年7月13日に出願された「プリナブリン組成物(PlinabulinCompositions)」と題する米国特許仮出願第62/191990号に基づく優先権を主張し、該仮出願の開示は参照によりその全体が本明細書に組み込まれる。
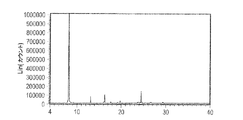
プリナブリン一水和物(形態1)は、プリナブリンの安定した結晶形態である。プリナブリン一水和物(形態1)のX線粉末回折(PXRD)パターンは、図1に示されるのと実質的に同じであり、対応する表形式ピークデータは表1に示される。

内で標準物質の角度設定と一致し、ピークの相対強度が20%を超えて異ならない場合には同一であることが確定されると述べられている。したがって、本明細書に記載の位置の0.2°範囲内のピーク位置は同一と見なされる。別段の指示がない限り、本明細書に記載のX線回析角はすべてCuKα源に基づく。
いくつかの実施形態は、組成物の総重量に対して、約50重量%を超える本明細書に記載のプリナブリン一水和物(形態1)を含むプリナブリン組成物に関する。いくつかの実施形態では、プリナブリン組成物は約75%を超える本明細書に記載のプリナブリン一水和物(形態1)を含む。いくつかの実施形態では、プリナブリン組成物は約90%を超える本明細書に記載のプリナブリン一水和物を含む。いくつかの実施形態では、プリナブリン組成物は約95%を超える本明細書に記載のプリナブリン一水和物を含む。いくつかの実施形態では、プリナブリン組成物は約98%を超える本明細書に記載のプリナブリン一水和物を含む。いくつかの実施形態では、プリナブリン組成物は約99%を超える本明細書に記載のプリナブリン一水和物を含む。いくつかの実施形態では、プリナブリン組成物は、組成物の総重量に対して、約50%〜約99%、約60%〜約99%、約70%〜約99%、約80%〜約99%、約90%〜約99%、約95%〜約99%、又は約97.5%〜約99%の範囲で、本明細書に記載のプリナブリン一水和物を含む。プリナブリン組成物の残りの部分は、他の形のプリナブリン及び/又は他の化学物質であってよい。
いくつかの実施形態では、組成物中のプリナブリンは、上記のように少なくとも部分的にプリナブリン一水和物で存在する。
いくつかの実施形態は、本明細書に記載のプリナブリン一水和物又はプリナブリン組成物を調製する方法に関し、その方法は、プリナブリン及び第1の溶媒系を混合して第1の混合物を形成すること、第1の混合物を約50℃〜90℃の範囲の温度に加熱すること、並びに第1の混合物を冷却して第1の沈殿物を形成することを含む。
いくつかの実施形態では、第1の溶媒系は、水、アルコール、又は水とアルコールの混合物であることができる。
いくつかの実施形態では、第1の混合物を加熱することは、第1の混合物を還流することを含む。
いくつかの実施形態では、第1の混合物を熱することは、第1の混合物を少なくとも65℃に熱することを含み、第1の混合物を冷却することは、第1の混合物を約50℃〜60℃に冷却することを含む。
いくつかの実施形態では、本明細書に記載の方法は、第1の沈殿物を分析して第1の沈殿物中のプリナブリン組成物を決定することをさらに含む。
合して第2の混合物を形成すること;第2の混合物を約50℃〜90℃の範囲の温度に加熱すること;第2の混合物を冷却して第2の沈殿物を形成すること;並びに第2の沈殿物を濾過すること及び第2の沈殿物を洗うことをさらに含む。
いくつかの実施形態は、プリナブリンの結晶形態2及びその調製方法に関する。いずれの特定の理論に縛られるものではないが、形態2は、プリナブリンイソプロピルアルコール(IPA)溶媒和物であると考えられている。
いくつかの実施形態は、プリナブリンの結晶形態3及びその調製方法に関する。いずれの特定の理論に縛られるものではないが、形態3はプリナブリンの無水形であると考えられている。
いくつかの実施形態は、プリナブリンの結晶形態4及びその調製方法に関する。いずれの特定の理論に縛られるものではないが、形態4はプリナブリンメタノール溶媒和物であると考えられている。
いくつかの実施形態は、プリナブリンの結晶形態5及びその調製方法に関する。
いくつかの実施形態は、プリナブリンの結晶形態6及びその調製方法に関する。
いくつかの実施形態は、プリナブリンの結晶形態7及びその調製方法に関する。
いくつかの実施形態は、プリナブリンの結晶形態8及びその調製方法に関する。
いくつかの実施形態は、プリナブリンの結晶形態9及びその調製方法に関する。
プリナブリン一水和物(形態1)は、同定された9つの多形のなかで最も安定した多形である。プリナブリン一水和物(形態1)は乾燥過程でも湿度ベース安定性試験下でも安定した状態を保つ(週末をかけた50℃での真空下における乾燥に対して安定、相対湿度
95%を超える湿度への13日間の曝露時に固体形の変化なし)。
いくつかの実施形態は、本明細書に記載のプリナブリン多形及び薬学的に許容される担体を含む医薬組成物を含む。そのような組成物は、治療療法の一部として対象に投与することができる。
腸、局所(経皮及び皮内を含む)、眼、脳内、頭蓋内、髄腔内、動脈内、静脈内、筋肉内、又は投与の他の非経口経路のための多様な適切な形態のいずれかでありうる。経口及び経鼻組成物を吸入によって投与され、利用可能な方法を用いて作られる組成物を含むことを当業者なら理解するであろう。所望の特定の投与経路に応じて、多様な当該技術分野において周知である薬学的に許容される担体が使用されうる。薬学的に許容される担体としては、例えば、固体又は液体充填剤、希釈剤、ハイドロトロピー剤、界面活性剤、及び被包物質が挙げられる。化合物又は組成物の活性を実質的に干渉しない随意の薬学的に活性のある物質が含まれてもよい。化合物又は組成物と併せて用いられる担体の量は、化合物の単位投与あたりの投与のための材料の実際の量を提供するのに十分である。本明細書に記載の方法において有用な剤形を作る技術及び組成物は、以下の参考文献に記載され、該文献はすべて、参照により本明細書に組み込まれる。Modern Pharmaceutics, 4th Ed., Chapters 9 and 10 (Banker & Rhodes, editors, 2002); Lieberman et al., Pharmaceutical Dosage Forms: Tablets (1989); 及びAnsel, Introduction to Pharmaceutical Dosage Forms 8th Edition (2004)。
り、所望の作用を延長するためにさまざまな時点で放出されたりするように、従来の方法、典型的にはpH依存性コーティング又は時間依存性コーティングでコーティングされてもよい。そのような剤形は典型的に、限定されないが、1つ以上の酢酸フタル酸セルロース、ポリ酢酸フタル酸ビニル、フタル酸ヒドロキシプロピルメチルセルロース、エチルセルロース、オイドラギットコーティング、ロウ、及びシェラックを含む。
0mg/m2、約40mg/m2、約50mg/m2、約60mg/m2、約70mg/m2、約80mg/m2、約90mg/m2、又は約100mg/m2でありうる。
プリナブリン化合物の試料をエタノール中で撹拌し、熱して還流した。全試料が溶けるまでエタノールを分けて添加し還流を維持し、透明な黄色溶液を得た。還流において試料を完全に溶かすために合計124.7gのエタノールを要した。次いで溶液を冷まし、沈殿をモニターした。溶液が49℃のとき、沈殿が見られた。混合物を再び熱して還流し、透明な黄色溶液を得た。熱い溶液をより大きい三角フラスコに9.45gのエタノールリンスとともに移した(熱濾過を擬するため)。この還流溶液に6.6gの水(エタノールの約5%の水)を加えた。この溶液を撹拌しながらゆっくりと冷ました。溶液が70℃まで冷えたとき、沈殿が見られた。この時点で、追加の水(128.6g)をゆっくりと加え、大量の固体を沈殿させた。溶液を撹拌しながら室温にまで冷やした。固体を17℃で濾過し、20gの水で3回洗った。合計134gの水を濾液に加え、濁った溶液、濾過に
不十分な量の固体が生じた。さらなる生成物を沈殿させようとして137gの追加の水を加えたが、さらなる固体は回収できなかった。固体を40〜45℃で3日間乾燥させ、4.73gを得た。97.5%回収。XRPDによる分析から、生成物がプリナブリン一水和物(形態1)であることが示された。カール・フィッシャーによる分析は水分レベルが4.9%であることを示した。
プリナブリン化合物の試料(4.92g)をエタノール(147.6g)中で撹拌し、熱して還流した(75℃で完全に可溶)。次いで溶液を冷まし、沈殿をモニターした。溶液が48℃のとき、沈殿が見られた。混合物を再び熱して還流し、透明な黄色溶液を得た。添加中に混合物を冷ましながら、熱い溶液に295gの水(エタノールの質量の約2倍)を加えた。約150mLの温度48℃の水を入れた後、沈殿が見られた。この溶液を室温にまで冷やした。固体を濾過し、20gの水で3回洗った。固体を40〜45℃で2.5日間乾燥させ、4.82gを得た。98.0%回収。XRPDによる分析は、生成物が、プリナブリン一水和物(形態1)(主生成物)及び無水プリナブリン(形態3)(少量生成物)の混合物であることを示した。カール・フィッシャーによる分析は水分レベルが5.0%であることを示した。45℃にて真空下でさらに70時間乾燥させた後のKFによる分析は、水分レベルが4.4%に減少したことを示した。この試料のXRPDは、本質的に不変であり、さらなるピーク(12.26°、15.19°、及び28.79°2θ度)をもつ形態1と形態3の混合物を示す。この試料を蓋のない容器中のグローブバッグに、水の入った蓋のない容器とともに置き、水分レベルをモニターした。4時間後に水分レベルが測定され、5.0%であった。18.5時間後に水分レベルが測定され、4.9%であり、51時間後にKF結果は5.1%であった。
プリナブリン化合物の試料を、激しく撹拌するか又は穏やかに熱して(50℃)、1,2−プロパンジオールに溶かした。4時間後、試料は形態3に関連するピークを示した。週末にかけて撹拌した後、試料は水でスラリー化され、XRPDスキャンで見られる結晶形態3に関連するピークをもたないプリナブリン一水和物(形態1)に完全に変換された。エタノール/水での実験についても同様の結果が見られ、非常に小さいピークが形態3について1時間及び4時間で見られ、形態1については66時間後のみに観察された。
再加工手順では、プリナブリン化合物(形態3)を、還流時に(重量で)1:25の比でエタノールに溶かした。この溶液を50℃より高い温度で濾過し、濾液を等しい質量の水と混ぜ合わせて生成物を得た。全量の水を加える前にポリッシュ濾過した(polish-filtered)エタノール溶液を再び熱し、(エタノールに対して)約5%の水を加え、この溶液を約70℃で撹拌して形態1への変換を確実にすることが望ましいことがある。次いで、追加の水を加え、混合物を冷却して生成物を濾過によって分離した。試料を長期間乾燥させて、カール・フィッシャー分析で測定した水分含量を下げることができる。1つの試料を3日間乾燥させ、4.9%水分のKF分析となった。さらに3日間乾燥させることにより、KF結果は4.1%に下がった。
含水量に関してKF分析が約3.1%であるプリナブリンのバッチをクレモフォール(kolliphor)(40重量%)とプロピレングリコール(60重量%)の混合物に加えた。不溶性粒子が溶液中に形成され、不溶性粒子は無水プリナブリン(形態3)であると判断した。バッチを図29に記載のステップに従って再加工してプリナブリン一水和物(形態1)を形成した。KFによる分析は、再加工したプリナブリンの含水量が約5.1%であることを示した。それは一水和物の理論含水量と一致する。プリナブリン一水和物(形態1)はクレモフォール(kolliphor)(40重量%)とプロピレングリコール(60重量%)の混合物に完全に溶け、不溶性粒子は溶液中に形成されなかった。したがって、無水プリナブリン(形態3)を含有するプリナブリン組成物と比べて、プリナブリン一水和物(形態1)のほうが良好な溶解度を示した。
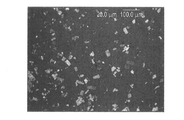
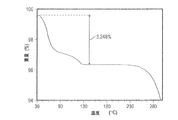
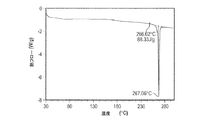
プリナブリン一水和物(形態1の結晶)を、XRPD(結晶、図1)、光学顕微鏡法(図2)、DSC(図3A)、TGA(図3B)、及びKFによって特徴付けた。
。
重量法による溶解度を測定するために、プリナブリン化合物を表9に示されるような15の異なる溶媒/溶媒混合物に15℃及び45℃で3日間スラリー化した。各実験について約70mgの固体をバイアルに加えた後に、0.7mLのそれぞれの溶媒を加えた。次に、スラリーを遠心分離し、上清を予め重さを量ったバイアルに入れ、真空下で蒸発させて乾燥させた。残留固体が入ったバイアルの重さを再び量って、溶解度を計算した。化合物は、THFに溶解性が高く、試験した他の溶媒には中程度から低い溶解度を示した。溶解度データを表10に示す。
さまざまな結晶形態を、200mgスケールでの他の形への変換について試験した。
Claims (46)
- 少なくとも8.1°±0.2°、13.1°±0.2°、16.3°±0.2°、23.9°±0.2°、24.2°±0.2°、24.5°±0.2°、及び26.6°±0.2°2θにおけるピークを含む粉末X線回折パターンを示す、プリナブリン一水和物の結晶。
- 少なくとも8.1°±0.2°、13.1°±0.2°、16.3°±0.2°、22.9°±0.2°、23.9°±0.2°、24.2°±0.2°、24.5°±0.2°、及び26.6°±0.2°2θにおけるピークを含む粉末X線回折パターンを示す、請求項1に記載のプリナブリン一水和物の結晶。
- 少なくとも8.1°±0.2°、13.1°±0.2°、14.8°±0.2°、16.1°±0.2°、16.3°±0.2°、17.6°±0.2°、19.8°±0.2°、22.4°±0.2°、22.9°±0.2°、23.9°±0.2°、24.2°±0.2°、24.5°±0.2°、26.6°±0.2°、及び29.3°±0.2°2θにおけるピークを含む粉末X線回折パターンを示す、請求項1に記載のプリナブリン一水和物の結晶。
- 前記結晶が267℃の融点をもつ、請求項1に記載のプリナブリン一水和物の結晶。
- 吸熱ピークが141℃及び267℃にある示差走査熱量測定サーモグラムをもつ、請求項1に記載のプリナブリン一水和物の結晶。
- プリナブリン組成物であって、前記組成物の総重量に対して50重量%を超える、請求項1〜5のいずれか一項に記載の前記プリナブリン一水和物の結晶を含むプリナブリン組成物。
- 前記組成物の総重量に対して75重量%を超えるプリナブリン一水和物の結晶を含む、請求項6の組成物。
- 前記組成物の総重量に対して90重量%を超えるプリナブリン一水和物の結晶を含む、請求項6の組成物。
- 前記組成物の総重量に対して95重量%を超えるプリナブリン一水和物の結晶を含む、請求項6の組成物。
- 前記組成物の総重量に対して98重量%を超えるプリナブリン一水和物の結晶を含む、請求項6の組成物。
- 前記組成物の総重量に対して99重量%を超えるプリナブリン一水和物の結晶を含む、請求項6の組成物。
- 請求項1〜5のいずれか一項に記載のプリナブリン一水和物の結晶を固体形態で含む無菌容器。
- 請求項1〜5のいずれか一項に記載のプリナブリン一水和物の結晶を調製する方法であって、
プリナブリン及び第1の溶媒を混合して第1の混合物を形成すること;
前記第1の混合物を50℃〜90℃の範囲の温度に加熱すること;並びに
前記第1の混合物を冷却して第1の沈殿物を形成すること
を含む方法。 - 請求項6〜11のいずれか一項に記載のプリナブリン組成物を調製する方法であって、
プリナブリン及び第1の溶媒を混合して第1の混合物を形成すること;
前記第1の混合物を50℃〜90℃の範囲の温度に加熱すること;並びに
前記第1の混合物を冷却して第1の沈殿物を形成すること
を含む方法。 - 前記第1の混合物を冷却する前に濾過することをさらに含む、請求項13又は14に記載の方法。
- 加熱前に水を前記第1の混合物に加えることをさらに含む、請求項13〜15のいずれか一項に記載の方法。
- 前記第1の溶媒が、水、アルコール、又は水とアルコールの混合物である、請求項13〜16のいずれか一項に記載の方法。
- 前記アルコールが、メタノール、エタノール、イソプロピルアルコール、tert−ブチルアルコール、及びn−ブチルアルコール、又はそれらの混合物から選択される、請求項17に記載の方法。
- 前記アルコールがエタノールである、請求項18に記載の方法。
- 前記第1の沈殿物を濾過することをさらに含む、請求項13〜19のいずれか一項に記載の方法。
- 前記第1の沈殿物を洗うことをさらに含む、請求項13〜20のいずれか一項に記載の方法。
- 前記第1の混合物が70℃〜78℃に加熱される、請求項13〜21のいずれか一項に
記載の方法。 - 前記第1の混合物を冷却する前に前記第1の混合物を70℃〜78℃の範囲の温度に1時間維持することをさらに含む、請求項13〜22のいずれか一項に記載の方法。
- 前記第1の混合物を加熱することが、前記第1の混合物を少なくとも65℃に加熱することを含み、且つ前記第1の混合物を冷却することが、前記第1の混合物を50℃〜60℃に冷却することを含む、請求項13〜23のいずれか一項に記載の方法。
- 前記第1の混合物を前記冷却することが、水を前記第1の混合物に加えて前記第1の沈殿物を生じさせることを含む、請求項13〜24のいずれか一項に記載の方法。
- 前記第1の混合物を前記冷却することが、前記第1の混合物を少なくとも4時間撹拌することを含む、請求項13〜25のいずれか一項に記載の方法。
- X線粉末回折分析を用いて前記第1の沈殿物を分析することをさらに含む、請求項13〜26のいずれか一項に記載の方法。
- 前記第1の沈殿物及び第2の溶媒を混合して第2の混合物を形成すること;
前記第2の混合物を50℃〜90℃の範囲の温度に加熱すること;
前記第2の混合物を冷却して第2の沈殿物を形成すること;並びに
前記第2の沈殿物を濾過すること及び前記第2の沈殿物を洗うこと
をさらに含む、請求項13〜27のいずれか一項に記載の方法。 - 前記第2の溶媒が、水、アルコール、又は水とアルコールの混合物である、請求項28に記載の方法。
- 前記第2の溶媒が、メタノール、エタノール、イソプロピルアルコール、tert−ブチルアルコール、及びn−ブチルアルコール、又はそれらの混合物から選択される、請求項29に記載の方法。
- 前記第2の溶媒がエタノールである、請求項28に記載の方法。
- 前記第2の混合物を加熱することが、前記第2の混合物を還流することを含む、請求項28〜31のいずれか一項に記載の方法。
- 前記第2の混合物が70℃〜78℃に加熱される、請求項28〜32のいずれか一項に記載の方法。
- 前記第2の混合物を冷却する前に前記第2の混合物を還流温度で1時間維持することをさらに含む、請求項28〜33のいずれか一項に記載の方法。
- 前記第2の混合物を前記冷却することが、15℃〜30℃に冷却することを含む、請求項28〜34のいずれか一項に記載の方法。
- 前記第2の混合物を前記冷却することが、水を前記第2の混合物に加えて前記第2の沈殿物を生じさせることを含む、請求項28〜35のいずれか一項に記載の方法。
- 前記第2の混合物を前記冷却することが、前記第2の混合物を少なくとも4時間撹拌することを含む、請求項28〜36のいずれか一項に記載の方法。
- 前記第1の沈殿物がアルコールで洗浄され、前記洗浄したアルコールが回収されて、加熱前に第2の混合物に加えられる、請求項28〜37のいずれか一項に記載の方法。
- 前記第2の沈殿物を乾燥することをさらに含む、請求項28〜38のいずれか一項に記載の方法。
- 前記混合、冷却、及び濾過ステップが、前記第2の沈殿物中のプリナブリンの量に基づいて1回以上繰り返される、請求項28〜39のいずれか一項に記載の方法。
- プリナブリン、エタノール、及び水を混ぜて混合物を形成すること
を含む、請求項1〜5のいずれか一項に記載のプリナブリン一水和物の結晶を調製する方法。 - プリナブリン、エタノール、及び水を混ぜて混合物を形成すること
を含む、請求項6〜11のいずれか一項に記載のプリナブリン組成物を調製する方法。 - 前記混合物を撹拌することをさらに含む、請求項41又は42に記載の方法。
- 前記エタノールの水に対する比が体積で15:1〜20:1の範囲にある、請求項41〜43のいずれか一項に記載の方法。
- 前記混合物が少なくとも2時間撹拌される、請求項41〜44のいずれか一項に記載の方法。
- 前記混ぜることが20℃〜40℃の範囲の温度で行われる、請求項41〜45のいずれか一項に記載の方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2021138150A JP2021181497A (ja) | 2015-07-13 | 2021-08-26 | プリナブリン組成物 |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201562191990P | 2015-07-13 | 2015-07-13 | |
| US62/191,990 | 2015-07-13 | ||
| PCT/US2016/041773 WO2017011399A1 (en) | 2015-07-13 | 2016-07-11 | Plinabulin compositions |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2021138150A Division JP2021181497A (ja) | 2015-07-13 | 2021-08-26 | プリナブリン組成物 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2018520178A JP2018520178A (ja) | 2018-07-26 |
| JP2018520178A5 JP2018520178A5 (ja) | 2019-08-29 |
| JP6969848B2 true JP6969848B2 (ja) | 2021-11-24 |
Family
ID=57757489
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2018501232A Active JP6969848B2 (ja) | 2015-07-13 | 2016-07-11 | プリナブリン組成物 |
| JP2021138150A Pending JP2021181497A (ja) | 2015-07-13 | 2021-08-26 | プリナブリン組成物 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2021138150A Pending JP2021181497A (ja) | 2015-07-13 | 2021-08-26 | プリナブリン組成物 |
Country Status (21)
| Country | Link |
|---|---|
| US (4) | US10155748B2 (ja) |
| EP (2) | EP4011878A1 (ja) |
| JP (2) | JP6969848B2 (ja) |
| KR (1) | KR20180027563A (ja) |
| CN (3) | CN108026075B (ja) |
| AU (2) | AU2016291708B2 (ja) |
| BR (1) | BR112018000229A2 (ja) |
| CA (1) | CA2991059C (ja) |
| CL (1) | CL2018000098A1 (ja) |
| CO (1) | CO2018000350A2 (ja) |
| DK (1) | DK3334726T3 (ja) |
| EC (1) | ECSP18010481A (ja) |
| ES (1) | ES2910035T3 (ja) |
| HK (1) | HK1251559A1 (ja) |
| IL (1) | IL256559B2 (ja) |
| MX (1) | MX2018000451A (ja) |
| MY (1) | MY181892A (ja) |
| PE (1) | PE20180528A1 (ja) |
| PH (1) | PH12018500071A1 (ja) |
| SG (1) | SG10202108194XA (ja) |
| WO (1) | WO2017011399A1 (ja) |
Families Citing this family (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| MX2017011375A (es) | 2015-03-06 | 2018-01-23 | Beyondspring Pharmaceuticals Inc | Método para tratar un tumor cerebral. |
| MX2017011374A (es) | 2015-03-06 | 2018-01-23 | Beyondspring Pharmaceuticals Inc | Método de tratamiento de cáncer asociado con una mutación de ras. |
| CA2991059C (en) | 2015-07-13 | 2024-01-16 | Beyondspring Pharmaceuticals, Inc. | Plinabulin monohydrate polymorphs |
| WO2017139231A1 (en) | 2016-02-08 | 2017-08-17 | Beyondspring Pharmaceuticals, Inc. | Compositions containing tucaresol or its analogs |
| RU2760348C2 (ru) | 2016-06-06 | 2021-11-24 | Бейондспринг Фармасьютикалс, Инк. | Способ уменьшения нейтропении |
| CN107011331A (zh) * | 2016-08-12 | 2017-08-04 | 青岛海洋生物医药研究院股份有限公司 | 脱氢苯基阿夕斯丁类化合物的多晶型及其制备方法 |
| EP4089085B1 (en) | 2016-08-12 | 2024-04-03 | Shenzhen Huahong Marine Biomedicine Co., Ltd | Manufacturing and purification method of polycrystalline form of dehydrophenylahistin-like compound |
| CN110431135A (zh) | 2017-01-06 | 2019-11-08 | 大连万春布林医药有限公司 | 微管蛋白结合化合物及其治疗用途 |
| CA3052190A1 (en) | 2017-02-01 | 2018-08-09 | Beyondspring Pharmaceuticals, Inc. | Method of reducing neutropenia |
| EP3661927A1 (en) | 2017-08-03 | 2020-06-10 | Pliva Hrvatska D.O.O. | Salts and solid state forms of plinabulin |
| US11786523B2 (en) | 2018-01-24 | 2023-10-17 | Beyondspring Pharmaceuticals, Inc. | Composition and method for reducing thrombocytopenia |
| WO2023154755A1 (en) * | 2022-02-10 | 2023-08-17 | Beyondspring Pharmaceuticals, Inc. | Stable plinabulin formulations and methods of their preparation and use |
Family Cites Families (98)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS509164A (ja) | 1973-05-11 | 1975-01-30 | ||
| HU190371B (en) | 1980-12-18 | 1986-08-28 | The Wellcome Foundation Ltd,Gb | Process for producing ether-type compounds and pharmaceutical compositions containing them as active agents |
| AU576322B2 (en) | 1983-07-22 | 1988-08-25 | Ici Australia Limited | Alpha-substituted-alpha-cyanomethyl alcohols |
| US4783443A (en) | 1986-03-03 | 1988-11-08 | The University Of Chicago | Amino acyl cephalosporin derivatives |
| JPH05255106A (ja) | 1990-10-31 | 1993-10-05 | Toray Ind Inc | 血小板減少症治療剤 |
| JPH059164A (ja) | 1991-07-02 | 1993-01-19 | Sumitomo Chem Co Ltd | 光学活性マンデロニトリル誘導体の製造方法 |
| GB9217331D0 (en) | 1992-08-14 | 1992-09-30 | Xenova Ltd | Pharmaceutical compounds |
| EP0678298A3 (en) | 1992-10-01 | 1996-05-29 | Wellcome Found | Use of 4- (2-formyl-3-hydroxyphenoxymethyl) benzoic acid as an immunopotentiating agent. |
| US6096786A (en) | 1992-10-01 | 2000-08-01 | Glaxo Wellcome Inc. | Immunopotentiatory agent and physiologically acceptable salts thereof |
| US5872151A (en) | 1992-10-01 | 1999-02-16 | Glaxo Wellcome Inc. | Immunopotentiatory agents and physiologically acceptable salts thereof |
| PT835657E (pt) | 1992-11-27 | 2004-11-30 | Mayne Pharma Usa Inc | Composicao injectavel estavel de paclitaxel |
| JPH0725858A (ja) | 1993-07-13 | 1995-01-27 | Otsuka Pharmaceut Co Ltd | ピペラジン誘導体 |
| US5958980A (en) | 1993-08-26 | 1999-09-28 | Glaxo Wellcome, Inc. | Immunopotentiatory agent and physiologically acceptable salts thereof |
| IL110787A0 (en) | 1993-08-27 | 1994-11-11 | Sandoz Ag | Biodegradable polymer, its preparation and pharmaceutical composition containing it |
| GB9402809D0 (en) | 1994-02-14 | 1994-04-06 | Xenova Ltd | Pharmaceutical compounds |
| GB9402807D0 (en) | 1994-02-14 | 1994-04-06 | Xenova Ltd | Pharmaceutical compounds |
| GB9426224D0 (en) | 1994-12-23 | 1995-02-22 | Xenova Ltd | Pharmaceutical compounds |
| US5891877A (en) | 1995-02-14 | 1999-04-06 | Xenova Limited | Pharmaceutical compounds |
| US5874443A (en) | 1995-10-19 | 1999-02-23 | Trega Biosciences, Inc. | Isoquinoline derivatives and isoquinoline combinatorial libraries |
| US5886210A (en) | 1996-08-22 | 1999-03-23 | Rohm And Haas Company | Method for preparing aromatic compounds |
| JP3131574B2 (ja) | 1996-09-04 | 2001-02-05 | 新日本製鐵株式会社 | 新規抗腫瘍物質、該物質を製造するための微生物及び方法、並びに該物質を有効成分とする細胞周期阻害剤及び抗腫瘍剤 |
| US5939098A (en) | 1996-09-19 | 1999-08-17 | Schering Corporation | Cancer treatment with temozolomide |
| US5922683A (en) | 1997-05-29 | 1999-07-13 | Abbott Laboratories | Multicyclic erythromycin derivatives |
| IL136044A (en) | 1997-11-21 | 2005-08-31 | Euro Celtique Sa | 2-aminoacetamide derivatives and pharmaceutical compositions containing the same |
| JP2002501944A (ja) | 1998-01-29 | 2002-01-22 | アヴェンティス ファーマシューティカルズ プロダクツ インコーポレイテッド | N−[(脂肪族又は芳香族)カルボニル]−2−アミノアセトアミド化合物及び環化化合物の製造方法 |
| US7141603B2 (en) | 1999-02-19 | 2006-11-28 | The Regents Of The University California | Antitumor agents |
| US6069146A (en) | 1998-03-25 | 2000-05-30 | The Regents Of The University Of California | Halimide, a cytotoxic marine natural product, and derivatives thereof |
| IL137974A0 (en) | 1998-03-26 | 2001-10-31 | Shionogi & Co | Indole derivatives having antiviral activity |
| US6509331B1 (en) | 1998-06-22 | 2003-01-21 | Elan Pharmaceuticals, Inc. | Deoxyamino acid compounds, pharmaceutical compositions comprising same, and methods for inhibiting β-amyloid peptide release and/or its synthesis by use of such compounds |
| FR2784988B1 (fr) | 1998-10-23 | 2002-09-20 | Adir | Nouveaux composes dihydro et tetrahydroquinoleiniques, leur procede de preparation et les compositions pharmaceutiques qui les contiennent |
| US6358957B1 (en) | 1998-11-12 | 2002-03-19 | Nereus Pharmaceuticals, Inc. | Phenylahistin and the phenylahistin analogs, a new class of anti-tumor compounds |
| US7026322B2 (en) | 1998-11-12 | 2006-04-11 | Nereus Pharmaceuticals, Inc. | Phenylahistin and the phenylahistin analogs, a new class of anti-tumor compounds |
| JP4896327B2 (ja) | 1999-08-23 | 2012-03-14 | ダナ−ファーバー キャンサー インスティテュート,インコーポレイテッド | Pd−1、b7−4の受容体、およびその使用 |
| WO2001014556A1 (en) | 1999-08-23 | 2001-03-01 | Dana-Farber Cancer Institute, Inc. | Novel b7-4 molecules and uses therefor |
| WO2001039722A2 (en) | 1999-11-30 | 2001-06-07 | Mayo Foundation For Medical Education And Research | B7-h1, a novel immunoregulatory molecule |
| WO2001053290A1 (fr) | 2000-01-18 | 2001-07-26 | Nippon Steel Corporation | Inhibiteurs de la division cellulaire et procede de production de ces inhibiteurs |
| AU2001245823A1 (en) | 2000-03-17 | 2001-10-03 | Corixa Corporation | Novel amphipathic aldehydes and their use as adjuvants and immunoeffectors |
| ATE417614T1 (de) | 2000-05-09 | 2009-01-15 | Angiorx Corp | Piperazindion-verbindungen |
| JP2004518731A (ja) | 2000-12-28 | 2004-06-24 | ニューロクライン バイオサイエンシーズ, インコーポレイテッド | 三環式crfレセプターアンタゴニスト |
| US20030082140A1 (en) | 2001-08-20 | 2003-05-01 | Fisher Paul B. | Combinatorial methods for inducing cancer cell death |
| US7012100B1 (en) | 2002-06-04 | 2006-03-14 | Avolix Pharmaceuticals, Inc. | Cell migration inhibiting compositions and methods and compositions for treating cancer |
| ATE374767T1 (de) | 2002-08-02 | 2007-10-15 | Nereus Pharmaceuticals Inc | Dehydrophenylahistine und analoge davon sowie ein verfahren zur herstellung von dehydrophenylahistinen und analogen davon |
| US7919497B2 (en) | 2002-08-02 | 2011-04-05 | Nereus Pharmaceuticals, Inc. | Analogs of dehydrophenylahistins and their therapeutic use |
| US7935704B2 (en) | 2003-08-01 | 2011-05-03 | Nereus Pharmaceuticals, Inc. | Dehydrophenylahistins and analogs thereof and the synthesis of dehydrophenylahistins and analogs thereof |
| CN101633655B (zh) * | 2002-08-02 | 2014-04-30 | 大连万春药业有限公司 | 脱氢苯基阿夕斯丁及其类似物以及脱氢苯基阿夕斯丁及其类似物的合成 |
| CN1753912B (zh) | 2002-12-23 | 2011-11-02 | 惠氏公司 | 抗pd-1抗体及其用途 |
| US20040197312A1 (en) | 2003-04-02 | 2004-10-07 | Marina Moskalenko | Cytokine-expressing cellular vaccine combinations |
| NZ548659A (en) * | 2004-02-04 | 2011-01-28 | Nereus Pharmaceuticals Inc | Dehydrophenylahistins and analogs thereof and the synthesis of dehydrophenylahistins and analogs thereof |
| CN1934101B (zh) * | 2004-02-04 | 2011-10-12 | 尼瑞斯药品公司 | 脱氢苯基阿夕斯丁及其类似物以及脱氢苯基阿夕斯丁及其类似物的合成 |
| DE202004018940U1 (de) | 2004-12-07 | 2006-04-13 | Asf Verwaltungs Gmbh | Druckverschluß, Druckverschlußband und wiederverschließbarer Beutel |
| CN105315373B (zh) | 2005-05-09 | 2018-11-09 | 小野药品工业株式会社 | 程序性死亡-1(pd-1)的人单克隆抗体及使用抗pd-1抗体来治疗癌症的方法 |
| KR101607288B1 (ko) | 2005-07-01 | 2016-04-05 | 이. 알. 스퀴부 앤드 선즈, 엘.엘.씨. | 예정 사멸 리간드 1 (피디-엘1)에 대한 인간 모노클로날 항체 |
| ES2521679T3 (es) | 2006-01-18 | 2014-11-13 | Merck Patent Gmbh | Terapia específica usando ligandos de integrinas para el tratamiento del cáncer |
| US8129527B2 (en) | 2006-11-03 | 2012-03-06 | Nereus Pharmacuticals, Inc. | Analogs of dehydrophenylahistins and their therapeutic use |
| WO2008100598A2 (en) | 2007-02-15 | 2008-08-21 | Mannkind Corporation | A method for enhancing t cell response |
| EP2146735B1 (en) | 2007-04-13 | 2012-06-20 | Abraxis BioScience, Inc. | Compositions comprising sparc polypeptides |
| US20090170837A1 (en) | 2007-08-17 | 2009-07-02 | Thallion Pharmaceuticals Inc. | Methods for treating ras driven cancer in a subject |
| US8569262B2 (en) | 2007-11-02 | 2013-10-29 | Momenta Pharmaceuticals, Inc. | Polysaccharide compositions and methods of use for the treatment and prevention of disorders associated with progenitor cell mobilization |
| AU2009204194A1 (en) | 2008-01-08 | 2009-07-16 | Bristol-Myers Squibb Company | Combination of anti-CTLA4 antibody with tubulin modulating agents for the treatment of proliferative diseases |
| WO2009114335A2 (en) | 2008-03-12 | 2009-09-17 | Merck & Co., Inc. | Pd-1 binding proteins |
| TWI461423B (zh) | 2008-07-02 | 2014-11-21 | Astrazeneca Ab | 用於治療Pim激酶相關病狀及疾病之噻唑啶二酮化合物 |
| SG196798A1 (en) | 2008-12-09 | 2014-02-13 | Genentech Inc | Anti-pd-l1 antibodies and their use to enhance t-cell function |
| WO2010114922A1 (en) | 2009-03-31 | 2010-10-07 | Agios Pharmaceuticals, Inc. | Methods of treating cancer having an aberrant egfr or kras genotype |
| GB0907551D0 (en) | 2009-05-01 | 2009-06-10 | Univ Dundee | Treatment or prophylaxis of proliferative conditions |
| AU2010295646B2 (en) | 2009-09-15 | 2016-02-11 | Ellipses Pharma Limited | Treatment of cancer |
| CA2778707A1 (en) | 2009-10-23 | 2011-04-28 | Mannkind Corporation | Cancer immunotherapy and method of treatment |
| CA2778714C (en) | 2009-11-24 | 2018-02-27 | Medimmune Limited | Targeted binding agents against b7-h1 |
| CN101766815B (zh) | 2009-12-31 | 2012-04-25 | 胡松华 | 紫杉醇及多西紫杉醇的用途 |
| WO2011109625A1 (en) | 2010-03-03 | 2011-09-09 | Targeted Molecular Diagnostics, Llc | Methods for determining responsiveness to a drug based upon determination of ras mutation and/or ras amplification |
| EP2571577A1 (en) | 2010-05-17 | 2013-03-27 | Bristol-Myers Squibb Company | Improved immunotherapeutic dosing regimens and combinations thereof |
| WO2011151423A1 (en) | 2010-06-04 | 2011-12-08 | Exonhit S.A. | Substituted isoquinolines and their use as tubulin polymerization inhibitors |
| JP2012033526A (ja) | 2010-07-28 | 2012-02-16 | Fuji Electric Co Ltd | 薄膜太陽電池およびその製造方法 |
| WO2012035436A1 (en) * | 2010-09-15 | 2012-03-22 | Tokyo University Of Pharmacy And Life Sciences | Plinabulin prodrug analogs and therapeutic uses thereof |
| WO2012074904A2 (en) | 2010-11-29 | 2012-06-07 | Precision Therapeutics, Inc. | Methods and systems for evaluating the sensitivity or resistance of tumor specimens to chemotherapeutic agents |
| ES2669310T3 (es) | 2011-04-20 | 2018-05-24 | Medimmune, Llc | Anticuerpos y otras moléculas que se unen con B7-H1 y PD-1 |
| JP2014533955A (ja) | 2011-11-28 | 2014-12-18 | ナショナル リサーチ カウンシル オブ カナダ | パクリタキセル応答性がんマーカー |
| US20150004175A1 (en) | 2011-12-13 | 2015-01-01 | Yale University | Compositions and Methods for Reducing CTL Exhaustion |
| PL2846809T3 (pl) | 2012-05-09 | 2021-07-26 | Cantex Pharmaceuticals, Inc. | Leczenie mielosupresji |
| AU2013204313C1 (en) | 2012-06-01 | 2016-04-07 | Bionomics Limited | Combination Therapy |
| WO2014066834A1 (en) | 2012-10-26 | 2014-05-01 | The University Of Chicago | Synergistic combination of immunologic inhibitors for the treatment of cancer |
| EP3626741A1 (en) | 2013-02-20 | 2020-03-25 | The Trustees Of The University Of Pennsylvania | Treatment of cancer using humanized anti-egfrviii chimeric antigen receptor |
| US11285169B2 (en) | 2013-03-13 | 2022-03-29 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Methods for modulating chemotherapeutic cytotoxicity |
| WO2014195852A1 (en) | 2013-06-03 | 2014-12-11 | Glaxosmithkline Intellectual Property (No.2) Limited | Combinations of an anti-pd-l1 antibody and a mek inhibitor and/or a braf inhibitor |
| ES2902665T3 (es) | 2013-10-11 | 2022-03-29 | Beyondspring Inc | Tratamiento del cáncer con combinación de plinabulina y taxano |
| WO2015069770A1 (en) | 2013-11-05 | 2015-05-14 | Cognate Bioservices, Inc. | Combinations of checkpoint inhibitors and therapeutics to treat cancer |
| KR102357699B1 (ko) | 2013-11-06 | 2022-02-04 | 더 유나이티드 스테이츠 오브 어메리카, 애즈 리프리젠티드 바이 더 세크러테리, 디파트먼트 오브 헬쓰 앤드 휴먼 서비씨즈 | 발현 프로파일링에 의한 림프종 유형의 하위유형 분류 방법 |
| US9850225B2 (en) | 2014-04-14 | 2017-12-26 | Bristol-Myers Squibb Company | Compounds useful as immunomodulators |
| CN106794246B (zh) | 2014-08-08 | 2021-10-15 | OncoQuest制药有限公司 | 肿瘤抗原特异性抗体和tlr3刺激以增强检查点干扰癌症疗法的性能 |
| JP7243021B2 (ja) | 2015-02-12 | 2023-03-22 | ビヨンドスプリング ファーマシューティカルズ,インコーポレイテッド | 免疫チェックポイント阻害薬と併用するプリナブリンの使用 |
| MX2017011374A (es) | 2015-03-06 | 2018-01-23 | Beyondspring Pharmaceuticals Inc | Método de tratamiento de cáncer asociado con una mutación de ras. |
| MX2017011375A (es) | 2015-03-06 | 2018-01-23 | Beyondspring Pharmaceuticals Inc | Método para tratar un tumor cerebral. |
| CA2991059C (en) | 2015-07-13 | 2024-01-16 | Beyondspring Pharmaceuticals, Inc. | Plinabulin monohydrate polymorphs |
| EP3359692A4 (en) | 2015-10-05 | 2019-05-01 | Cedars-Sinai Medical Center | METHOD OF CLASSIFYING AND DIAGNOSING CANCER |
| WO2017139231A1 (en) | 2016-02-08 | 2017-08-17 | Beyondspring Pharmaceuticals, Inc. | Compositions containing tucaresol or its analogs |
| RU2760348C2 (ru) | 2016-06-06 | 2021-11-24 | Бейондспринг Фармасьютикалс, Инк. | Способ уменьшения нейтропении |
| CN110431135A (zh) | 2017-01-06 | 2019-11-08 | 大连万春布林医药有限公司 | 微管蛋白结合化合物及其治疗用途 |
| CA3052190A1 (en) | 2017-02-01 | 2018-08-09 | Beyondspring Pharmaceuticals, Inc. | Method of reducing neutropenia |
| BR112019018880A2 (pt) | 2017-03-13 | 2020-04-14 | Beyondspring Pharmaceuticals Inc | composições de plinabulina e seu uso |
-
2016
- 2016-07-11 CA CA2991059A patent/CA2991059C/en active Active
- 2016-07-11 PE PE2018000054A patent/PE20180528A1/es unknown
- 2016-07-11 CN CN201680051286.3A patent/CN108026075B/zh active Active
- 2016-07-11 US US15/741,635 patent/US10155748B2/en active Active
- 2016-07-11 AU AU2016291708A patent/AU2016291708B2/en active Active
- 2016-07-11 WO PCT/US2016/041773 patent/WO2017011399A1/en active Application Filing
- 2016-07-11 MY MYPI2017705031A patent/MY181892A/en unknown
- 2016-07-11 CN CN202110926846.8A patent/CN113735834B/zh active Active
- 2016-07-11 DK DK16824996.9T patent/DK3334726T3/da active
- 2016-07-11 JP JP2018501232A patent/JP6969848B2/ja active Active
- 2016-07-11 CN CN201910023460.9A patent/CN109516981B/zh active Active
- 2016-07-11 ES ES16824996T patent/ES2910035T3/es active Active
- 2016-07-11 BR BR112018000229A patent/BR112018000229A2/pt not_active Application Discontinuation
- 2016-07-11 EP EP22151146.2A patent/EP4011878A1/en active Pending
- 2016-07-11 EP EP16824996.9A patent/EP3334726B1/en active Active
- 2016-07-11 SG SG10202108194XA patent/SG10202108194XA/en unknown
- 2016-07-11 MX MX2018000451A patent/MX2018000451A/es active IP Right Grant
- 2016-07-11 KR KR1020187003671A patent/KR20180027563A/ko not_active Application Discontinuation
-
2017
- 2017-12-25 IL IL256559A patent/IL256559B2/en unknown
-
2018
- 2018-01-08 PH PH12018500071A patent/PH12018500071A1/en unknown
- 2018-01-12 CL CL2018000098A patent/CL2018000098A1/es unknown
- 2018-01-15 CO CONC2018/0000350A patent/CO2018000350A2/es unknown
- 2018-02-09 EC ECIEPI201810481A patent/ECSP18010481A/es unknown
- 2018-08-24 HK HK18110939.3A patent/HK1251559A1/zh unknown
- 2018-12-06 US US16/212,386 patent/US10550104B2/en active Active
-
2020
- 2020-02-03 US US16/780,780 patent/US11254657B2/en active Active
-
2021
- 2021-03-22 AU AU2021201784A patent/AU2021201784B2/en active Active
- 2021-08-26 JP JP2021138150A patent/JP2021181497A/ja active Pending
-
2022
- 2022-02-18 US US17/675,453 patent/US20220169635A1/en active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6969848B2 (ja) | プリナブリン組成物 | |
| US11690836B2 (en) | Solid forms of {[5-(3-chlorophenyl)-3-hydroxypyridine-2-carbonyl]amino}acetic acid, compositions, and uses thereof | |
| US20120035142A1 (en) | Polymorphs of darunavir | |
| US20220356173A1 (en) | Polymorphic forms of kinase inhibitor compound, pharmaceutical composition containing same, preparation method therefor and use thereof | |
| KR20200118098A (ko) | 약제학적 화합물, 이의 염, 이의 제형, 그리고 이의 제조 방법 및 사용 방법 | |
| RU2773843C1 (ru) | Композиции плинабулина | |
| EP3526217B1 (en) | Crystalline forms of 4-(2-((1r,2r)-2-hydroxycyclohexylamino) benzothiazol-6-yloxy)-n-methylpicolinamide | |
| JP2019089822A (ja) | トピロキソスタットの新規結晶形及びその製造方法 | |
| WO2020187674A1 (en) | Crystalline (s)-[3,4-difluoro-2-(2-fluoro-4-iodophenylamino)phenyl][3-hydroxy-3-(piperidin-2-yl)azetidin-1-yl]methanone hemisuccinate | |
| NZ719970B2 (en) | Solid forms of {[5-(3-chlorophenyl)-3-hydroxypyridine-2-carbonyl]amino}acetic acid, compositions, and uses thereof | |
| NZ759132B2 (en) | Solid forms of {[5-(3-chlorophenyl)-3-hydroxypyridine-2-carbonyl]amino}acetic acid, compositions, and uses thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20190709 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20190709 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20200528 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20200616 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20200916 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20201116 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20210427 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20210826 |
|
| C60 | Trial request (containing other claim documents, opposition documents) |
Free format text: JAPANESE INTERMEDIATE CODE: C60 Effective date: 20210826 |
|
| A911 | Transfer to examiner for re-examination before appeal (zenchi) |
Free format text: JAPANESE INTERMEDIATE CODE: A911 Effective date: 20210903 |
|
| C21 | Notice of transfer of a case for reconsideration by examiners before appeal proceedings |
Free format text: JAPANESE INTERMEDIATE CODE: C21 Effective date: 20210907 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20210928 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20211026 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6969848 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |