JP5941607B2 - 血液脳関門輸送用融合タンパク質 - Google Patents
血液脳関門輸送用融合タンパク質 Download PDFInfo
- Publication number
- JP5941607B2 JP5941607B2 JP2008534618A JP2008534618A JP5941607B2 JP 5941607 B2 JP5941607 B2 JP 5941607B2 JP 2008534618 A JP2008534618 A JP 2008534618A JP 2008534618 A JP2008534618 A JP 2008534618A JP 5941607 B2 JP5941607 B2 JP 5941607B2
- Authority
- JP
- Japan
- Prior art keywords
- bdnf
- composition
- fusion protein
- bbb
- sequence
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 230000008499 blood brain barrier function Effects 0.000 title claims description 295
- 210000001218 blood-brain barrier Anatomy 0.000 title claims description 294
- 108020001507 fusion proteins Proteins 0.000 title claims description 284
- 102000037865 fusion proteins Human genes 0.000 title claims description 280
- 108090000715 Brain-derived neurotrophic factor Proteins 0.000 claims description 315
- 102000004219 Brain-derived neurotrophic factor Human genes 0.000 claims description 308
- 229940077737 brain-derived neurotrophic factor Drugs 0.000 claims description 307
- 239000000203 mixture Substances 0.000 claims description 180
- 108090000765 processed proteins & peptides Proteins 0.000 claims description 152
- 210000004556 brain Anatomy 0.000 claims description 121
- 150000001413 amino acids Chemical class 0.000 claims description 112
- 239000003814 drug Substances 0.000 claims description 102
- 241000282414 Homo sapiens Species 0.000 claims description 97
- 239000003795 chemical substances by application Substances 0.000 claims description 94
- 102000005962 receptors Human genes 0.000 claims description 89
- 108020003175 receptors Proteins 0.000 claims description 89
- 230000000694 effects Effects 0.000 claims description 68
- 230000027455 binding Effects 0.000 claims description 51
- 101000852815 Homo sapiens Insulin receptor Proteins 0.000 claims description 44
- 102000047882 human INSR Human genes 0.000 claims description 44
- 239000000126 substance Substances 0.000 claims description 44
- 230000000324 neuroprotective effect Effects 0.000 claims description 42
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 39
- 108060003951 Immunoglobulin Proteins 0.000 claims description 37
- 102000018358 immunoglobulin Human genes 0.000 claims description 37
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 33
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 27
- 230000002093 peripheral effect Effects 0.000 claims description 26
- 210000002966 serum Anatomy 0.000 claims description 26
- 102000003746 Insulin Receptor Human genes 0.000 claims description 24
- 108010001127 Insulin Receptor Proteins 0.000 claims description 24
- 238000011282 treatment Methods 0.000 claims description 24
- 208000015114 central nervous system disease Diseases 0.000 claims description 23
- 208000035475 disorder Diseases 0.000 claims description 23
- 230000001684 chronic effect Effects 0.000 claims description 22
- 229940077456 human brain-derived neurotrophic factor Drugs 0.000 claims description 21
- 238000001990 intravenous administration Methods 0.000 claims description 21
- 241000282412 Homo Species 0.000 claims description 18
- 101000739876 Homo sapiens Brain-derived neurotrophic factor Proteins 0.000 claims description 18
- 102000051542 human BDNF Human genes 0.000 claims description 18
- 238000004519 manufacturing process Methods 0.000 claims description 18
- 230000001154 acute effect Effects 0.000 claims description 17
- 201000006474 Brain Ischemia Diseases 0.000 claims description 16
- 206010008120 Cerebral ischaemia Diseases 0.000 claims description 16
- 108010019476 Immunoglobulin Heavy Chains Proteins 0.000 claims description 16
- 206010008118 cerebral infarction Diseases 0.000 claims description 16
- 238000007920 subcutaneous administration Methods 0.000 claims description 14
- 206010002026 amyotrophic lateral sclerosis Diseases 0.000 claims description 13
- 208000015122 neurodegenerative disease Diseases 0.000 claims description 13
- 208000024827 Alzheimer disease Diseases 0.000 claims description 12
- 230000004770 neurodegeneration Effects 0.000 claims description 12
- 208000024891 symptom Diseases 0.000 claims description 12
- 208000023105 Huntington disease Diseases 0.000 claims description 11
- 208000018737 Parkinson disease Diseases 0.000 claims description 11
- 201000007737 Retinal degeneration Diseases 0.000 claims description 11
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 11
- 230000004258 retinal degeneration Effects 0.000 claims description 11
- 102000015534 trkB Receptor Human genes 0.000 claims description 11
- 108010064880 trkB Receptor Proteins 0.000 claims description 11
- GOJUJUVQIVIZAV-UHFFFAOYSA-N 2-amino-4,6-dichloropyrimidine-5-carbaldehyde Chemical group NC1=NC(Cl)=C(C=O)C(Cl)=N1 GOJUJUVQIVIZAV-UHFFFAOYSA-N 0.000 claims description 10
- 102000006496 Immunoglobulin Heavy Chains Human genes 0.000 claims description 10
- 238000007918 intramuscular administration Methods 0.000 claims description 10
- 208000012902 Nervous system disease Diseases 0.000 claims description 9
- 239000008194 pharmaceutical composition Substances 0.000 claims description 9
- 208000020431 spinal cord injury Diseases 0.000 claims description 8
- 208000017667 Chronic Disease Diseases 0.000 claims description 7
- 208000029028 brain injury Diseases 0.000 claims description 7
- 238000007912 intraperitoneal administration Methods 0.000 claims description 7
- 108010078286 Ataxins Proteins 0.000 claims description 6
- 102000014461 Ataxins Human genes 0.000 claims description 6
- 208000022526 Canavan disease Diseases 0.000 claims description 6
- 206010008025 Cerebellar ataxia Diseases 0.000 claims description 6
- 208000024412 Friedreich ataxia Diseases 0.000 claims description 6
- 201000011240 Frontotemporal dementia Diseases 0.000 claims description 6
- 208000026072 Motor neurone disease Diseases 0.000 claims description 6
- 208000000609 Pick Disease of the Brain Diseases 0.000 claims description 6
- 208000024777 Prion disease Diseases 0.000 claims description 6
- 208000006289 Rett Syndrome Diseases 0.000 claims description 6
- 208000009415 Spinocerebellar Ataxias Diseases 0.000 claims description 6
- 208000026911 Tuberous sclerosis complex Diseases 0.000 claims description 6
- 201000004562 autosomal dominant cerebellar ataxia Diseases 0.000 claims description 6
- 239000000463 material Substances 0.000 claims description 6
- 208000005264 motor neuron disease Diseases 0.000 claims description 6
- 201000006417 multiple sclerosis Diseases 0.000 claims description 6
- 208000001749 optic atrophy Diseases 0.000 claims description 6
- 230000003836 peripheral circulation Effects 0.000 claims description 6
- 208000009174 transverse myelitis Diseases 0.000 claims description 6
- 208000009999 tuberous sclerosis Diseases 0.000 claims description 6
- 210000005036 nerve Anatomy 0.000 claims description 5
- 230000009471 action Effects 0.000 claims description 2
- 210000003712 lysosome Anatomy 0.000 claims description 2
- 230000001868 lysosomic effect Effects 0.000 claims description 2
- 208000032253 retinal ischemia Diseases 0.000 claims description 2
- 238000009825 accumulation Methods 0.000 claims 1
- 108010025020 Nerve Growth Factor Proteins 0.000 description 158
- 102000007072 Nerve Growth Factors Human genes 0.000 description 139
- 235000001014 amino acid Nutrition 0.000 description 114
- 229940024606 amino acid Drugs 0.000 description 111
- 108090000623 proteins and genes Proteins 0.000 description 84
- 230000032258 transport Effects 0.000 description 84
- 210000003169 central nervous system Anatomy 0.000 description 72
- 150000007523 nucleic acids Chemical group 0.000 description 72
- 238000000034 method Methods 0.000 description 67
- 230000001225 therapeutic effect Effects 0.000 description 66
- 210000004027 cell Anatomy 0.000 description 61
- 229940079593 drug Drugs 0.000 description 59
- 239000013598 vector Substances 0.000 description 58
- 230000001404 mediated effect Effects 0.000 description 53
- 102000039446 nucleic acids Human genes 0.000 description 50
- 108020004707 nucleic acids Proteins 0.000 description 50
- 125000003275 alpha amino acid group Chemical group 0.000 description 48
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 48
- 239000002299 complementary DNA Substances 0.000 description 43
- 125000002091 cationic group Chemical group 0.000 description 39
- 235000018102 proteins Nutrition 0.000 description 39
- 102000004169 proteins and genes Human genes 0.000 description 39
- 102000004196 processed proteins & peptides Human genes 0.000 description 38
- 229940124597 therapeutic agent Drugs 0.000 description 38
- 239000002773 nucleotide Substances 0.000 description 37
- 125000003729 nucleotide group Chemical group 0.000 description 37
- 229940027941 immunoglobulin g Drugs 0.000 description 35
- 230000004927 fusion Effects 0.000 description 28
- 125000005647 linker group Chemical group 0.000 description 28
- 101100127166 Escherichia coli (strain K12) kefB gene Proteins 0.000 description 24
- 108090001061 Insulin Proteins 0.000 description 24
- 102000004877 Insulin Human genes 0.000 description 24
- 108010076504 Protein Sorting Signals Proteins 0.000 description 24
- 229940125396 insulin Drugs 0.000 description 24
- 108010078791 Carrier Proteins Proteins 0.000 description 23
- -1 Percefin Proteins 0.000 description 22
- 108090000099 Neurotrophin-4 Proteins 0.000 description 20
- 210000004369 blood Anatomy 0.000 description 20
- 230000014509 gene expression Effects 0.000 description 20
- 238000003752 polymerase chain reaction Methods 0.000 description 20
- 238000006467 substitution reaction Methods 0.000 description 20
- 108020004414 DNA Proteins 0.000 description 19
- 102000015336 Nerve Growth Factor Human genes 0.000 description 19
- 108091028043 Nucleic acid sequence Proteins 0.000 description 19
- 239000008280 blood Substances 0.000 description 19
- 210000004899 c-terminal region Anatomy 0.000 description 19
- 229940053128 nerve growth factor Drugs 0.000 description 19
- 102000018233 Fibroblast Growth Factor Human genes 0.000 description 18
- 108050007372 Fibroblast Growth Factor Proteins 0.000 description 18
- 108010022394 Threonine synthase Proteins 0.000 description 18
- 229940126864 fibroblast growth factor Drugs 0.000 description 18
- 239000012634 fragment Substances 0.000 description 18
- 241000699666 Mus <mouse, genus> Species 0.000 description 17
- 108010009583 Transforming Growth Factors Proteins 0.000 description 17
- 102000009618 Transforming Growth Factors Human genes 0.000 description 17
- 102000004419 dihydrofolate reductase Human genes 0.000 description 17
- JVJUWEFOGFCHKR-UHFFFAOYSA-N 2-(diethylamino)ethyl 1-(3,4-dimethylphenyl)cyclopentane-1-carboxylate;hydrochloride Chemical compound Cl.C=1C=C(C)C(C)=CC=1C1(C(=O)OCCN(CC)CC)CCCC1 JVJUWEFOGFCHKR-UHFFFAOYSA-N 0.000 description 16
- CJLHTKGWEUGORV-UHFFFAOYSA-N Artemin Chemical compound C1CC2(C)C(O)CCC(=C)C2(O)C2C1C(C)C(=O)O2 CJLHTKGWEUGORV-UHFFFAOYSA-N 0.000 description 16
- 108010005939 Ciliary Neurotrophic Factor Proteins 0.000 description 16
- 102100031614 Ciliary neurotrophic factor Human genes 0.000 description 16
- 102000003951 Erythropoietin Human genes 0.000 description 16
- 108090000394 Erythropoietin Proteins 0.000 description 16
- 102000034615 Glial cell line-derived neurotrophic factor Human genes 0.000 description 16
- 108091010837 Glial cell line-derived neurotrophic factor Proteins 0.000 description 16
- 102000003745 Hepatocyte Growth Factor Human genes 0.000 description 16
- 108090000100 Hepatocyte Growth Factor Proteins 0.000 description 16
- 102000004058 Leukemia inhibitory factor Human genes 0.000 description 16
- 108090000581 Leukemia inhibitory factor Proteins 0.000 description 16
- 102000010803 Netrins Human genes 0.000 description 16
- 108010063605 Netrins Proteins 0.000 description 16
- 108010038512 Platelet-Derived Growth Factor Proteins 0.000 description 16
- 102000010780 Platelet-Derived Growth Factor Human genes 0.000 description 16
- 108010073929 Vascular Endothelial Growth Factor A Proteins 0.000 description 16
- 102000005789 Vascular Endothelial Growth Factors Human genes 0.000 description 16
- 108010019530 Vascular Endothelial Growth Factors Proteins 0.000 description 16
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 16
- 229940105423 erythropoietin Drugs 0.000 description 16
- 238000009472 formulation Methods 0.000 description 16
- 239000013612 plasmid Substances 0.000 description 16
- OXCMYAYHXIHQOA-UHFFFAOYSA-N potassium;[2-butyl-5-chloro-3-[[4-[2-(1,2,4-triaza-3-azanidacyclopenta-1,4-dien-5-yl)phenyl]phenyl]methyl]imidazol-4-yl]methanol Chemical compound [K+].CCCCC1=NC(Cl)=C(CO)N1CC1=CC=C(C=2C(=CC=CC=2)C2=N[N-]N=N2)C=C1 OXCMYAYHXIHQOA-UHFFFAOYSA-N 0.000 description 16
- 102000007644 Colony-Stimulating Factors Human genes 0.000 description 15
- 108010071942 Colony-Stimulating Factors Proteins 0.000 description 15
- 101001076407 Homo sapiens Interleukin-1 receptor antagonist protein Proteins 0.000 description 15
- 229940119178 Interleukin 1 receptor antagonist Drugs 0.000 description 15
- 102000051628 Interleukin-1 receptor antagonist Human genes 0.000 description 15
- 206010035226 Plasma cell myeloma Diseases 0.000 description 15
- 238000004422 calculation algorithm Methods 0.000 description 15
- 239000013604 expression vector Substances 0.000 description 15
- 239000003407 interleukin 1 receptor blocking agent Substances 0.000 description 15
- 201000000050 myeloid neoplasm Diseases 0.000 description 15
- 241000282560 Macaca mulatta Species 0.000 description 14
- 210000004155 blood-retinal barrier Anatomy 0.000 description 14
- 230000004378 blood-retinal barrier Effects 0.000 description 14
- ZXQYGBMAQZUVMI-GCMPRSNUSA-N gamma-cyhalothrin Chemical compound CC1(C)[C@@H](\C=C(/Cl)C(F)(F)F)[C@H]1C(=O)O[C@H](C#N)C1=CC=CC(OC=2C=CC=CC=2)=C1 ZXQYGBMAQZUVMI-GCMPRSNUSA-N 0.000 description 14
- 238000002347 injection Methods 0.000 description 14
- 239000007924 injection Substances 0.000 description 14
- 230000009871 nonspecific binding Effects 0.000 description 14
- 108020004705 Codon Proteins 0.000 description 13
- 108010065825 Immunoglobulin Light Chains Proteins 0.000 description 13
- 230000006870 function Effects 0.000 description 13
- 230000002441 reversible effect Effects 0.000 description 13
- 102000007350 Bone Morphogenetic Proteins Human genes 0.000 description 12
- 108010007726 Bone Morphogenetic Proteins Proteins 0.000 description 12
- 108090000723 Insulin-Like Growth Factor I Proteins 0.000 description 12
- 102000007238 Transferrin Receptors Human genes 0.000 description 12
- 230000008901 benefit Effects 0.000 description 12
- 229940112869 bone morphogenetic protein Drugs 0.000 description 12
- 238000005259 measurement Methods 0.000 description 12
- 239000000243 solution Substances 0.000 description 12
- 102000014429 Insulin-like growth factor Human genes 0.000 description 11
- 102000003683 Neurotrophin-4 Human genes 0.000 description 11
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 11
- 239000013613 expression plasmid Substances 0.000 description 11
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 11
- 102000007056 Recombinant Fusion Proteins Human genes 0.000 description 10
- 108010008281 Recombinant Fusion Proteins Proteins 0.000 description 10
- 208000006011 Stroke Diseases 0.000 description 10
- 108010033576 Transferrin Receptors Proteins 0.000 description 10
- 210000003527 eukaryotic cell Anatomy 0.000 description 10
- 238000002741 site-directed mutagenesis Methods 0.000 description 10
- 102000015696 Interleukins Human genes 0.000 description 9
- 108010063738 Interleukins Proteins 0.000 description 9
- 102100033857 Neurotrophin-4 Human genes 0.000 description 9
- 241000288906 Primates Species 0.000 description 9
- 150000001768 cations Chemical class 0.000 description 9
- 150000001875 compounds Chemical class 0.000 description 9
- 230000029087 digestion Effects 0.000 description 9
- 229940047122 interleukins Drugs 0.000 description 9
- 239000007788 liquid Substances 0.000 description 9
- 239000003550 marker Substances 0.000 description 9
- 229940097998 neurotrophin 4 Drugs 0.000 description 9
- 238000002360 preparation method Methods 0.000 description 9
- VBEQCZHXXJYVRD-GACYYNSASA-N uroanthelone Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CS)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CS)C(=O)N[C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C(C)C)[C@@H](C)O)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CCSC)NC(=O)[C@H](CS)NC(=O)[C@@H](NC(=O)CNC(=O)CNC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CS)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CS)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC(N)=O)C(C)C)[C@@H](C)CC)C1=CC=C(O)C=C1 VBEQCZHXXJYVRD-GACYYNSASA-N 0.000 description 9
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 8
- 102100026376 Artemin Human genes 0.000 description 8
- 101710205806 Artemin Proteins 0.000 description 8
- 108090000368 Fibroblast growth factor 8 Proteins 0.000 description 8
- 244000060234 Gmelina philippensis Species 0.000 description 8
- 101150027568 LC gene Proteins 0.000 description 8
- 108010092801 Midkine Proteins 0.000 description 8
- 102000016776 Midkine Human genes 0.000 description 8
- 102000014413 Neuregulin Human genes 0.000 description 8
- 108050003475 Neuregulin Proteins 0.000 description 8
- 108090000556 Neuregulin-1 Proteins 0.000 description 8
- 102000048238 Neuregulin-1 Human genes 0.000 description 8
- 102100021584 Neurturin Human genes 0.000 description 8
- 108010015406 Neurturin Proteins 0.000 description 8
- 241000700159 Rattus Species 0.000 description 8
- 102000017852 Saposin Human genes 0.000 description 8
- 108050007079 Saposin Proteins 0.000 description 8
- 229960002685 biotin Drugs 0.000 description 8
- 239000011616 biotin Substances 0.000 description 8
- 238000011161 development Methods 0.000 description 8
- 230000018109 developmental process Effects 0.000 description 8
- 210000003714 granulocyte Anatomy 0.000 description 8
- 102000005162 pleiotrophin Human genes 0.000 description 8
- 150000003839 salts Chemical class 0.000 description 8
- 210000000130 stem cell Anatomy 0.000 description 8
- YNJBWRMUSHSURL-UHFFFAOYSA-N trichloroacetic acid Chemical compound OC(=O)C(Cl)(Cl)Cl YNJBWRMUSHSURL-UHFFFAOYSA-N 0.000 description 8
- 102100028892 Cardiotrophin-1 Human genes 0.000 description 7
- 238000002965 ELISA Methods 0.000 description 7
- 108010017080 Granulocyte Colony-Stimulating Factor Proteins 0.000 description 7
- 102000004269 Granulocyte Colony-Stimulating Factor Human genes 0.000 description 7
- 102000013463 Immunoglobulin Light Chains Human genes 0.000 description 7
- 102000003960 Ligases Human genes 0.000 description 7
- 108090000364 Ligases Proteins 0.000 description 7
- 241001465754 Metazoa Species 0.000 description 7
- 241000699670 Mus sp. Species 0.000 description 7
- 108090001012 Transforming Growth Factor beta Proteins 0.000 description 7
- 102000004887 Transforming Growth Factor beta Human genes 0.000 description 7
- 238000004458 analytical method Methods 0.000 description 7
- 239000000427 antigen Substances 0.000 description 7
- 108091007433 antigens Proteins 0.000 description 7
- 102000036639 antigens Human genes 0.000 description 7
- 230000015572 biosynthetic process Effects 0.000 description 7
- 239000000872 buffer Substances 0.000 description 7
- 108010041776 cardiotrophin 1 Proteins 0.000 description 7
- 239000003937 drug carrier Substances 0.000 description 7
- 238000003780 insertion Methods 0.000 description 7
- 230000037431 insertion Effects 0.000 description 7
- 238000000185 intracerebroventricular administration Methods 0.000 description 7
- 210000002540 macrophage Anatomy 0.000 description 7
- 239000000178 monomer Substances 0.000 description 7
- 210000002569 neuron Anatomy 0.000 description 7
- 230000004112 neuroprotection Effects 0.000 description 7
- 229940046166 oligodeoxynucleotide Drugs 0.000 description 7
- 229920001184 polypeptide Polymers 0.000 description 7
- 239000011780 sodium chloride Substances 0.000 description 7
- 210000000278 spinal cord Anatomy 0.000 description 7
- ZRKFYGHZFMAOKI-QMGMOQQFSA-N tgfbeta Chemical compound C([C@H](NC(=O)[C@H](C(C)C)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CC(C)C)NC(=O)CNC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CCSC)C(C)C)[C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(O)=O)C1=CC=C(O)C=C1 ZRKFYGHZFMAOKI-QMGMOQQFSA-N 0.000 description 7
- 208000014644 Brain disease Diseases 0.000 description 6
- 108010047041 Complementarity Determining Regions Proteins 0.000 description 6
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 6
- 101150021185 FGF gene Proteins 0.000 description 6
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 6
- 208000015439 Lysosomal storage disease Diseases 0.000 description 6
- 239000002202 Polyethylene glycol Substances 0.000 description 6
- 102000014105 Semaphorin Human genes 0.000 description 6
- 108050003978 Semaphorin Proteins 0.000 description 6
- 108090000901 Transferrin Proteins 0.000 description 6
- 239000004480 active ingredient Substances 0.000 description 6
- 238000013459 approach Methods 0.000 description 6
- 238000003556 assay Methods 0.000 description 6
- 239000002775 capsule Substances 0.000 description 6
- 238000012377 drug delivery Methods 0.000 description 6
- 230000013595 glycosylation Effects 0.000 description 6
- 238000006206 glycosylation reaction Methods 0.000 description 6
- 238000001727 in vivo Methods 0.000 description 6
- 238000010253 intravenous injection Methods 0.000 description 6
- 238000001155 isoelectric focusing Methods 0.000 description 6
- 102000005861 leptin receptors Human genes 0.000 description 6
- 108010019813 leptin receptors Proteins 0.000 description 6
- 229960000485 methotrexate Drugs 0.000 description 6
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 6
- 229920001223 polyethylene glycol Polymers 0.000 description 6
- 238000011160 research Methods 0.000 description 6
- 210000001525 retina Anatomy 0.000 description 6
- 239000012679 serum free medium Substances 0.000 description 6
- 150000003384 small molecules Chemical class 0.000 description 6
- 239000000829 suppository Substances 0.000 description 6
- 238000012360 testing method Methods 0.000 description 6
- 238000002560 therapeutic procedure Methods 0.000 description 6
- 108090001008 Avidin Proteins 0.000 description 5
- 101000976075 Homo sapiens Insulin Proteins 0.000 description 5
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 5
- 102000007330 LDL Lipoproteins Human genes 0.000 description 5
- 108010007622 LDL Lipoproteins Proteins 0.000 description 5
- 102000011965 Lipoprotein Receptors Human genes 0.000 description 5
- 108010061306 Lipoprotein Receptors Proteins 0.000 description 5
- 229910019142 PO4 Inorganic materials 0.000 description 5
- 239000002253 acid Substances 0.000 description 5
- 239000003242 anti bacterial agent Substances 0.000 description 5
- 230000004888 barrier function Effects 0.000 description 5
- 230000001588 bifunctional effect Effects 0.000 description 5
- 150000001720 carbohydrates Chemical class 0.000 description 5
- 235000014633 carbohydrates Nutrition 0.000 description 5
- 230000002860 competitive effect Effects 0.000 description 5
- 238000009295 crossflow filtration Methods 0.000 description 5
- 230000002068 genetic effect Effects 0.000 description 5
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 5
- PBGKTOXHQIOBKM-FHFVDXKLSA-N insulin (human) Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@H]1CSSC[C@H]2C(=O)N[C@H](C(=O)N[C@@H](CO)C(=O)N[C@H](C(=O)N[C@H](C(N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC=3C=CC(O)=CC=3)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=3C=CC(O)=CC=3)C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=3C=CC(O)=CC=3)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](C)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=3NC=NC=3)NC(=O)[C@H](CO)NC(=O)CNC1=O)C(=O)NCC(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H]([C@@H](C)O)C(O)=O)C(=O)N[C@@H](CC(N)=O)C(O)=O)=O)CSSC[C@@H](C(N2)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C(C)C)NC(=O)[C@@H](NC(=O)CN)[C@@H](C)CC)[C@@H](C)CC)[C@@H](C)O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@@H](NC(=O)[C@@H](N)CC=1C=CC=CC=1)C(C)C)C1=CN=CN1 PBGKTOXHQIOBKM-FHFVDXKLSA-N 0.000 description 5
- 238000000670 ligand binding assay Methods 0.000 description 5
- 239000011159 matrix material Substances 0.000 description 5
- 239000002609 medium Substances 0.000 description 5
- 201000001119 neuropathy Diseases 0.000 description 5
- 230000007823 neuropathy Effects 0.000 description 5
- 239000003900 neurotrophic factor Substances 0.000 description 5
- 208000033808 peripheral neuropathy Diseases 0.000 description 5
- 230000000144 pharmacologic effect Effects 0.000 description 5
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 5
- 239000010452 phosphate Substances 0.000 description 5
- 229920000642 polymer Polymers 0.000 description 5
- 125000006239 protecting group Chemical group 0.000 description 5
- 229920005989 resin Polymers 0.000 description 5
- 239000011347 resin Substances 0.000 description 5
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 5
- 239000003826 tablet Substances 0.000 description 5
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 4
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 4
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 4
- 241000699802 Cricetulus griseus Species 0.000 description 4
- 241000701022 Cytomegalovirus Species 0.000 description 4
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 4
- 241000283086 Equidae Species 0.000 description 4
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 4
- 208000010496 Heart Arrest Diseases 0.000 description 4
- 206010021143 Hypoxia Diseases 0.000 description 4
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 4
- 241000124008 Mammalia Species 0.000 description 4
- 229930195725 Mannitol Natural products 0.000 description 4
- 108090000742 Neurotrophin 3 Proteins 0.000 description 4
- 102100029268 Neurotrophin-3 Human genes 0.000 description 4
- 238000012300 Sequence Analysis Methods 0.000 description 4
- 239000007983 Tris buffer Substances 0.000 description 4
- 238000010521 absorption reaction Methods 0.000 description 4
- 239000013543 active substance Substances 0.000 description 4
- 239000011543 agarose gel Substances 0.000 description 4
- 125000000539 amino acid group Chemical group 0.000 description 4
- 230000003321 amplification Effects 0.000 description 4
- 239000007864 aqueous solution Substances 0.000 description 4
- 230000009286 beneficial effect Effects 0.000 description 4
- 230000036765 blood level Effects 0.000 description 4
- 210000000988 bone and bone Anatomy 0.000 description 4
- 210000004781 brain capillary Anatomy 0.000 description 4
- 238000013461 design Methods 0.000 description 4
- 238000010586 diagram Methods 0.000 description 4
- 239000000539 dimer Substances 0.000 description 4
- 201000010099 disease Diseases 0.000 description 4
- 238000002474 experimental method Methods 0.000 description 4
- 239000000499 gel Substances 0.000 description 4
- 238000003365 immunocytochemistry Methods 0.000 description 4
- 230000006872 improvement Effects 0.000 description 4
- 230000001965 increasing effect Effects 0.000 description 4
- 239000004615 ingredient Substances 0.000 description 4
- 239000000543 intermediate Substances 0.000 description 4
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 4
- 239000000594 mannitol Substances 0.000 description 4
- 235000010355 mannitol Nutrition 0.000 description 4
- 230000000921 morphogenic effect Effects 0.000 description 4
- 238000003199 nucleic acid amplification method Methods 0.000 description 4
- 210000001672 ovary Anatomy 0.000 description 4
- 230000036961 partial effect Effects 0.000 description 4
- 210000005259 peripheral blood Anatomy 0.000 description 4
- 239000011886 peripheral blood Substances 0.000 description 4
- 239000000843 powder Substances 0.000 description 4
- 239000003755 preservative agent Substances 0.000 description 4
- 239000000047 product Substances 0.000 description 4
- 230000000069 prophylactic effect Effects 0.000 description 4
- 238000000746 purification Methods 0.000 description 4
- 108091008146 restriction endonucleases Proteins 0.000 description 4
- 210000003625 skull Anatomy 0.000 description 4
- 238000003860 storage Methods 0.000 description 4
- 239000000725 suspension Substances 0.000 description 4
- 238000003786 synthesis reaction Methods 0.000 description 4
- 230000009885 systemic effect Effects 0.000 description 4
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 4
- HDTRYLNUVZCQOY-UHFFFAOYSA-N α-D-glucopyranosyl-α-D-glucopyranoside Natural products OC1C(O)C(O)C(CO)OC1OC1C(O)C(O)C(O)C(CO)O1 HDTRYLNUVZCQOY-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- 201000004569 Blindness Diseases 0.000 description 3
- 125000001433 C-terminal amino-acid group Chemical group 0.000 description 3
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 3
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 3
- QMMFVYPAHWMCMS-UHFFFAOYSA-N Dimethyl sulfide Chemical compound CSC QMMFVYPAHWMCMS-UHFFFAOYSA-N 0.000 description 3
- 102100031780 Endonuclease Human genes 0.000 description 3
- 102000004190 Enzymes Human genes 0.000 description 3
- 108090000790 Enzymes Proteins 0.000 description 3
- 108700039691 Genetic Promoter Regions Proteins 0.000 description 3
- 241000725303 Human immunodeficiency virus Species 0.000 description 3
- 108010067060 Immunoglobulin Variable Region Proteins 0.000 description 3
- 102000017727 Immunoglobulin Variable Region Human genes 0.000 description 3
- AGPKZVBTJJNPAG-WHFBIAKZSA-N L-isoleucine Chemical compound CC[C@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-WHFBIAKZSA-N 0.000 description 3
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 3
- 102000003992 Peroxidases Human genes 0.000 description 3
- 238000012228 RNA interference-mediated gene silencing Methods 0.000 description 3
- 108010092799 RNA-directed DNA polymerase Proteins 0.000 description 3
- AABIBDJHSKIMJK-FXQIFTODSA-N Ser-Ser-Met Chemical compound [H]N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCSC)C(O)=O AABIBDJHSKIMJK-FXQIFTODSA-N 0.000 description 3
- HDTRYLNUVZCQOY-WSWWMNSNSA-N Trehalose Natural products O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-WSWWMNSNSA-N 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- 239000000654 additive Substances 0.000 description 3
- 230000032683 aging Effects 0.000 description 3
- HDTRYLNUVZCQOY-LIZSDCNHSA-N alpha,alpha-trehalose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-LIZSDCNHSA-N 0.000 description 3
- 229940088710 antibiotic agent Drugs 0.000 description 3
- 210000004958 brain cell Anatomy 0.000 description 3
- 210000000170 cell membrane Anatomy 0.000 description 3
- 238000006243 chemical reaction Methods 0.000 description 3
- 230000002759 chromosomal effect Effects 0.000 description 3
- 230000019771 cognition Effects 0.000 description 3
- 230000000295 complement effect Effects 0.000 description 3
- 239000003636 conditioned culture medium Substances 0.000 description 3
- 230000006378 damage Effects 0.000 description 3
- 230000003111 delayed effect Effects 0.000 description 3
- 238000012217 deletion Methods 0.000 description 3
- 230000037430 deletion Effects 0.000 description 3
- 238000009826 distribution Methods 0.000 description 3
- 229940088598 enzyme Drugs 0.000 description 3
- 231100000318 excitotoxic Toxicity 0.000 description 3
- 230000003492 excitotoxic effect Effects 0.000 description 3
- 125000000524 functional group Chemical group 0.000 description 3
- 230000009368 gene silencing by RNA Effects 0.000 description 3
- 238000010353 genetic engineering Methods 0.000 description 3
- 230000009851 immunogenic response Effects 0.000 description 3
- 208000015181 infectious disease Diseases 0.000 description 3
- 208000028867 ischemia Diseases 0.000 description 3
- 239000003446 ligand Substances 0.000 description 3
- 238000012417 linear regression Methods 0.000 description 3
- 210000004185 liver Anatomy 0.000 description 3
- 229920002521 macromolecule Polymers 0.000 description 3
- 230000015654 memory Effects 0.000 description 3
- 239000004090 neuroprotective agent Substances 0.000 description 3
- 238000010647 peptide synthesis reaction Methods 0.000 description 3
- 108040007629 peroxidase activity proteins Proteins 0.000 description 3
- 239000006187 pill Substances 0.000 description 3
- 230000036470 plasma concentration Effects 0.000 description 3
- 239000000244 polyoxyethylene sorbitan monooleate Substances 0.000 description 3
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 3
- 229940068968 polysorbate 80 Drugs 0.000 description 3
- 229920000053 polysorbate 80 Polymers 0.000 description 3
- 239000002243 precursor Substances 0.000 description 3
- 210000004129 prosencephalon Anatomy 0.000 description 3
- 239000012217 radiopharmaceutical Substances 0.000 description 3
- 229940121896 radiopharmaceutical Drugs 0.000 description 3
- 230000002799 radiopharmaceutical effect Effects 0.000 description 3
- 230000004044 response Effects 0.000 description 3
- 239000000523 sample Substances 0.000 description 3
- 238000010532 solid phase synthesis reaction Methods 0.000 description 3
- 239000002904 solvent Substances 0.000 description 3
- 239000000600 sorbitol Substances 0.000 description 3
- 238000001890 transfection Methods 0.000 description 3
- 238000001262 western blot Methods 0.000 description 3
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 description 2
- BDNKZNFMNDZQMI-UHFFFAOYSA-N 1,3-diisopropylcarbodiimide Chemical compound CC(C)N=C=NC(C)C BDNKZNFMNDZQMI-UHFFFAOYSA-N 0.000 description 2
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide Chemical compound CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 2
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 2
- 239000004475 Arginine Substances 0.000 description 2
- DCXYFEDJOCDNAF-UHFFFAOYSA-N Asparagine Natural products OC(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-N 0.000 description 2
- 206010003805 Autism Diseases 0.000 description 2
- 208000020706 Autistic disease Diseases 0.000 description 2
- 208000003174 Brain Neoplasms Diseases 0.000 description 2
- 229920002261 Corn starch Polymers 0.000 description 2
- 101150074155 DHFR gene Proteins 0.000 description 2
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 2
- 208000027534 Emotional disease Diseases 0.000 description 2
- 108010010803 Gelatin Proteins 0.000 description 2
- 208000032612 Glial tumor Diseases 0.000 description 2
- 206010018338 Glioma Diseases 0.000 description 2
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 2
- 239000004471 Glycine Substances 0.000 description 2
- 206010019196 Head injury Diseases 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 2
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 2
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 2
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 description 2
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 2
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 2
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 2
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 2
- LRQKBLKVPFOOQJ-YFKPBYRVSA-N L-norleucine Chemical compound CCCC[C@H]([NH3+])C([O-])=O LRQKBLKVPFOOQJ-YFKPBYRVSA-N 0.000 description 2
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 2
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 2
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 2
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- 108090001090 Lectins Proteins 0.000 description 2
- 102000004856 Lectins Human genes 0.000 description 2
- 102000016267 Leptin Human genes 0.000 description 2
- 108010092277 Leptin Proteins 0.000 description 2
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 2
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 2
- 239000004472 Lysine Substances 0.000 description 2
- 241001529936 Murinae Species 0.000 description 2
- 241000699660 Mus musculus Species 0.000 description 2
- 101100278853 Mus musculus Dhfr gene Proteins 0.000 description 2
- 230000004988 N-glycosylation Effects 0.000 description 2
- 108700026244 Open Reading Frames Proteins 0.000 description 2
- 108091093037 Peptide nucleic acid Proteins 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 2
- 208000024571 Pick disease Diseases 0.000 description 2
- 239000004365 Protease Substances 0.000 description 2
- 101000798098 Rattus norvegicus Serotransferrin Proteins 0.000 description 2
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 2
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 2
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 2
- 239000004473 Threonine Substances 0.000 description 2
- 102000004338 Transferrin Human genes 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 2
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 description 2
- 208000027418 Wounds and injury Diseases 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- 235000004279 alanine Nutrition 0.000 description 2
- 125000003282 alkyl amino group Chemical group 0.000 description 2
- SHGAZHPCJJPHSC-YCNIQYBTSA-N all-trans-retinoic acid Chemical compound OC(=O)\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-YCNIQYBTSA-N 0.000 description 2
- RDOXTESZEPMUJZ-UHFFFAOYSA-N anisole Chemical compound COC1=CC=CC=C1 RDOXTESZEPMUJZ-UHFFFAOYSA-N 0.000 description 2
- 239000005557 antagonist Substances 0.000 description 2
- 230000000844 anti-bacterial effect Effects 0.000 description 2
- 230000000692 anti-sense effect Effects 0.000 description 2
- 229940121375 antifungal agent Drugs 0.000 description 2
- 239000003429 antifungal agent Substances 0.000 description 2
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 2
- 235000009697 arginine Nutrition 0.000 description 2
- 235000009582 asparagine Nutrition 0.000 description 2
- 229960001230 asparagine Drugs 0.000 description 2
- 235000003704 aspartic acid Nutrition 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- 230000001580 bacterial effect Effects 0.000 description 2
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 235000020958 biotin Nutrition 0.000 description 2
- 108010006025 bovine growth hormone Proteins 0.000 description 2
- 230000006931 brain damage Effects 0.000 description 2
- 231100000874 brain damage Toxicity 0.000 description 2
- 210000005252 bulbus oculi Anatomy 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 238000012512 characterization method Methods 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 239000007958 cherry flavor Substances 0.000 description 2
- OSASVXMJTNOKOY-UHFFFAOYSA-N chlorobutanol Chemical compound CC(C)(O)C(Cl)(Cl)Cl OSASVXMJTNOKOY-UHFFFAOYSA-N 0.000 description 2
- 230000006726 chronic neurodegeneration Effects 0.000 description 2
- 238000003776 cleavage reaction Methods 0.000 description 2
- 238000002648 combination therapy Methods 0.000 description 2
- 238000010276 construction Methods 0.000 description 2
- 239000008120 corn starch Substances 0.000 description 2
- 230000008878 coupling Effects 0.000 description 2
- 238000010168 coupling process Methods 0.000 description 2
- 238000005859 coupling reaction Methods 0.000 description 2
- 230000007812 deficiency Effects 0.000 description 2
- 230000001934 delay Effects 0.000 description 2
- 239000000032 diagnostic agent Substances 0.000 description 2
- 229940039227 diagnostic agent Drugs 0.000 description 2
- 238000009792 diffusion process Methods 0.000 description 2
- BGRWYRAHAFMIBJ-UHFFFAOYSA-N diisopropylcarbodiimide Natural products CC(C)NC(=O)NC(C)C BGRWYRAHAFMIBJ-UHFFFAOYSA-N 0.000 description 2
- 239000002612 dispersion medium Substances 0.000 description 2
- VYFYYTLLBUKUHU-UHFFFAOYSA-N dopamine Chemical compound NCCC1=CC=C(O)C(O)=C1 VYFYYTLLBUKUHU-UHFFFAOYSA-N 0.000 description 2
- 238000009509 drug development Methods 0.000 description 2
- 238000010828 elution Methods 0.000 description 2
- 239000006274 endogenous ligand Substances 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- 230000008029 eradication Effects 0.000 description 2
- ZMMJGEGLRURXTF-UHFFFAOYSA-N ethidium bromide Chemical compound [Br-].C12=CC(N)=CC=C2C2=CC=C(N)C=C2[N+](CC)=C1C1=CC=CC=C1 ZMMJGEGLRURXTF-UHFFFAOYSA-N 0.000 description 2
- 229960005542 ethidium bromide Drugs 0.000 description 2
- 239000000796 flavoring agent Substances 0.000 description 2
- 235000013355 food flavoring agent Nutrition 0.000 description 2
- 235000003599 food sweetener Nutrition 0.000 description 2
- 239000008273 gelatin Substances 0.000 description 2
- 229920000159 gelatin Polymers 0.000 description 2
- 235000019322 gelatine Nutrition 0.000 description 2
- 235000011852 gelatine desserts Nutrition 0.000 description 2
- 235000013922 glutamic acid Nutrition 0.000 description 2
- 239000004220 glutamic acid Substances 0.000 description 2
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 2
- 235000004554 glutamine Nutrition 0.000 description 2
- 239000003102 growth factor Substances 0.000 description 2
- 125000004356 hydroxy functional group Chemical class O* 0.000 description 2
- 238000003384 imaging method Methods 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 238000010874 in vitro model Methods 0.000 description 2
- 239000007972 injectable composition Substances 0.000 description 2
- 208000014674 injury Diseases 0.000 description 2
- 230000010354 integration Effects 0.000 description 2
- 230000000302 ischemic effect Effects 0.000 description 2
- AGPKZVBTJJNPAG-UHFFFAOYSA-N isoleucine Natural products CCC(C)C(N)C(O)=O AGPKZVBTJJNPAG-UHFFFAOYSA-N 0.000 description 2
- 229960000310 isoleucine Drugs 0.000 description 2
- 239000007951 isotonicity adjuster Substances 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 239000002523 lectin Substances 0.000 description 2
- NRYBAZVQPHGZNS-ZSOCWYAHSA-N leptin Chemical compound O=C([C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)CNC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](N)CC(C)C)CCSC)N1CCC[C@H]1C(=O)NCC(=O)N[C@@H](CS)C(O)=O NRYBAZVQPHGZNS-ZSOCWYAHSA-N 0.000 description 2
- 229940039781 leptin Drugs 0.000 description 2
- 235000019359 magnesium stearate Nutrition 0.000 description 2
- 239000012528 membrane Substances 0.000 description 2
- 230000002503 metabolic effect Effects 0.000 description 2
- MYWUZJCMWCOHBA-VIFPVBQESA-N methamphetamine Chemical compound CN[C@@H](C)CC1=CC=CC=C1 MYWUZJCMWCOHBA-VIFPVBQESA-N 0.000 description 2
- 229930182817 methionine Natural products 0.000 description 2
- 230000003278 mimic effect Effects 0.000 description 2
- 230000036651 mood Effects 0.000 description 2
- 230000000626 neurodegenerative effect Effects 0.000 description 2
- 230000000926 neurological effect Effects 0.000 description 2
- 238000006386 neutralization reaction Methods 0.000 description 2
- 239000008203 oral pharmaceutical composition Substances 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- 229910052760 oxygen Inorganic materials 0.000 description 2
- 239000001301 oxygen Substances 0.000 description 2
- 238000007911 parenteral administration Methods 0.000 description 2
- 230000035515 penetration Effects 0.000 description 2
- 230000010412 perfusion Effects 0.000 description 2
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 2
- 229910052698 phosphorus Inorganic materials 0.000 description 2
- 239000011574 phosphorus Substances 0.000 description 2
- 230000002265 prevention Effects 0.000 description 2
- 229940002612 prodrug Drugs 0.000 description 2
- 239000000651 prodrug Substances 0.000 description 2
- 210000001176 projection neuron Anatomy 0.000 description 2
- 238000011321 prophylaxis Methods 0.000 description 2
- 230000004224 protection Effects 0.000 description 2
- 238000010188 recombinant method Methods 0.000 description 2
- 238000011084 recovery Methods 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 238000000611 regression analysis Methods 0.000 description 2
- 210000001116 retinal neuron Anatomy 0.000 description 2
- 229930002330 retinoic acid Natural products 0.000 description 2
- 238000012552 review Methods 0.000 description 2
- CVHZOJJKTDOEJC-UHFFFAOYSA-N saccharin Chemical compound C1=CC=C2C(=O)NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 description 2
- 238000013391 scatchard analysis Methods 0.000 description 2
- 230000007017 scission Effects 0.000 description 2
- 230000028327 secretion Effects 0.000 description 2
- 230000035945 sensitivity Effects 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 239000007790 solid phase Substances 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 239000007921 spray Substances 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- 235000000346 sugar Nutrition 0.000 description 2
- 229910052717 sulfur Inorganic materials 0.000 description 2
- 239000011593 sulfur Substances 0.000 description 2
- 239000006228 supernatant Substances 0.000 description 2
- 239000004094 surface-active agent Substances 0.000 description 2
- 239000003765 sweetening agent Substances 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- 231100000331 toxic Toxicity 0.000 description 2
- 230000002588 toxic effect Effects 0.000 description 2
- 239000012581 transferrin Substances 0.000 description 2
- 238000011830 transgenic mouse model Methods 0.000 description 2
- 230000001052 transient effect Effects 0.000 description 2
- 229960001727 tretinoin Drugs 0.000 description 2
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 2
- 239000004474 valine Substances 0.000 description 2
- 210000005166 vasculature Anatomy 0.000 description 2
- 239000003981 vehicle Substances 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- FDKWRPBBCBCIGA-REOHCLBHSA-N (2r)-2-azaniumyl-3-$l^{1}-selanylpropanoate Chemical compound [Se]C[C@H](N)C(O)=O FDKWRPBBCBCIGA-REOHCLBHSA-N 0.000 description 1
- AOFUBOWZWQFQJU-SNOJBQEQSA-N (2r,3s,4s,5r)-2,5-bis(hydroxymethyl)oxolane-2,3,4-triol;(2s,3r,4s,5s,6r)-6-(hydroxymethyl)oxane-2,3,4,5-tetrol Chemical compound OC[C@H]1O[C@](O)(CO)[C@@H](O)[C@@H]1O.OC[C@H]1O[C@H](O)[C@H](O)[C@@H](O)[C@@H]1O AOFUBOWZWQFQJU-SNOJBQEQSA-N 0.000 description 1
- UKAUYVFTDYCKQA-UHFFFAOYSA-N -2-Amino-4-hydroxybutanoic acid Natural products OC(=O)C(N)CCO UKAUYVFTDYCKQA-UHFFFAOYSA-N 0.000 description 1
- VGONTNSXDCQUGY-RRKCRQDMSA-N 2'-deoxyinosine Chemical group C1[C@H](O)[C@@H](CO)O[C@H]1N1C(N=CNC2=O)=C2N=C1 VGONTNSXDCQUGY-RRKCRQDMSA-N 0.000 description 1
- NBWRJAOOMGASJP-UHFFFAOYSA-N 2-(3,5-diphenyl-1h-tetrazol-1-ium-2-yl)-4,5-dimethyl-1,3-thiazole;bromide Chemical compound [Br-].S1C(C)=C(C)N=C1N1N(C=2C=CC=CC=2)N=C(C=2C=CC=CC=2)[NH2+]1 NBWRJAOOMGASJP-UHFFFAOYSA-N 0.000 description 1
- YEDUAINPPJYDJZ-UHFFFAOYSA-N 2-hydroxybenzothiazole Chemical compound C1=CC=C2SC(O)=NC2=C1 YEDUAINPPJYDJZ-UHFFFAOYSA-N 0.000 description 1
- 206010001022 Acute psychosis Diseases 0.000 description 1
- 208000007848 Alcoholism Diseases 0.000 description 1
- 208000019901 Anxiety disease Diseases 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 206010003571 Astrocytoma Diseases 0.000 description 1
- 238000012935 Averaging Methods 0.000 description 1
- 241000167854 Bourreria succulenta Species 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- 108091035707 Consensus sequence Proteins 0.000 description 1
- 102000004127 Cytokines Human genes 0.000 description 1
- 108090000695 Cytokines Proteins 0.000 description 1
- FDKWRPBBCBCIGA-UWTATZPHSA-N D-Selenocysteine Natural products [Se]C[C@@H](N)C(O)=O FDKWRPBBCBCIGA-UWTATZPHSA-N 0.000 description 1
- 206010012289 Dementia Diseases 0.000 description 1
- 208000016192 Demyelinating disease Diseases 0.000 description 1
- 206010012305 Demyelination Diseases 0.000 description 1
- 235000019739 Dicalciumphosphate Nutrition 0.000 description 1
- BWGNESOTFCXPMA-UHFFFAOYSA-N Dihydrogen disulfide Chemical compound SS BWGNESOTFCXPMA-UHFFFAOYSA-N 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- JOYRKODLDBILNP-UHFFFAOYSA-N Ethyl urethane Chemical compound CCOC(N)=O JOYRKODLDBILNP-UHFFFAOYSA-N 0.000 description 1
- 108090000379 Fibroblast growth factor 2 Proteins 0.000 description 1
- 102000003974 Fibroblast growth factor 2 Human genes 0.000 description 1
- KRHYYFGTRYWZRS-UHFFFAOYSA-N Fluorane Chemical compound F KRHYYFGTRYWZRS-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 241000711549 Hepacivirus C Species 0.000 description 1
- 101000599951 Homo sapiens Insulin-like growth factor I Proteins 0.000 description 1
- 101001076292 Homo sapiens Insulin-like growth factor II Proteins 0.000 description 1
- 108010021625 Immunoglobulin Fragments Proteins 0.000 description 1
- 102000008394 Immunoglobulin Fragments Human genes 0.000 description 1
- 108700029228 Immunoglobulin Heavy Chain Genes Proteins 0.000 description 1
- 108700029227 Immunoglobulin Light Chain Genes Proteins 0.000 description 1
- 208000001019 Inborn Errors Metabolism Diseases 0.000 description 1
- 208000026350 Inborn Genetic disease Diseases 0.000 description 1
- 102100037852 Insulin-like growth factor I Human genes 0.000 description 1
- 102100025947 Insulin-like growth factor II Human genes 0.000 description 1
- 108010050904 Interferons Proteins 0.000 description 1
- 102000014150 Interferons Human genes 0.000 description 1
- 102000019223 Interleukin-1 receptor Human genes 0.000 description 1
- 108050006617 Interleukin-1 receptor Proteins 0.000 description 1
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 1
- ONIBWKKTOPOVIA-BYPYZUCNSA-N L-Proline Chemical compound OC(=O)[C@@H]1CCCN1 ONIBWKKTOPOVIA-BYPYZUCNSA-N 0.000 description 1
- UKAUYVFTDYCKQA-VKHMYHEASA-N L-homoserine Chemical compound OC(=O)[C@@H](N)CCO UKAUYVFTDYCKQA-VKHMYHEASA-N 0.000 description 1
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 1
- QEFRNWWLZKMPFJ-ZXPFJRLXSA-N L-methionine (R)-S-oxide Chemical compound C[S@@](=O)CC[C@H]([NH3+])C([O-])=O QEFRNWWLZKMPFJ-ZXPFJRLXSA-N 0.000 description 1
- QEFRNWWLZKMPFJ-UHFFFAOYSA-N L-methionine sulphoxide Natural products CS(=O)CCC(N)C(O)=O QEFRNWWLZKMPFJ-UHFFFAOYSA-N 0.000 description 1
- ZFOMKMMPBOQKMC-KXUCPTDWSA-N L-pyrrolysine Chemical compound C[C@@H]1CC=N[C@H]1C(=O)NCCCC[C@H]([NH3+])C([O-])=O ZFOMKMMPBOQKMC-KXUCPTDWSA-N 0.000 description 1
- 102000000853 LDL receptors Human genes 0.000 description 1
- 108010001831 LDL receptors Proteins 0.000 description 1
- 102000004895 Lipoproteins Human genes 0.000 description 1
- 108090001030 Lipoproteins Proteins 0.000 description 1
- 240000005265 Lupinus mutabilis Species 0.000 description 1
- 235000008755 Lupinus mutabilis Nutrition 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 description 1
- 208000036626 Mental retardation Diseases 0.000 description 1
- 244000246386 Mentha pulegium Species 0.000 description 1
- 235000016257 Mentha pulegium Nutrition 0.000 description 1
- 235000004357 Mentha x piperita Nutrition 0.000 description 1
- 208000019695 Migraine disease Diseases 0.000 description 1
- 208000019022 Mood disease Diseases 0.000 description 1
- BKAYIFDRRZZKNF-VIFPVBQESA-N N-acetylcarnosine Chemical compound CC(=O)NCCC(=O)N[C@H](C(O)=O)CC1=CN=CN1 BKAYIFDRRZZKNF-VIFPVBQESA-N 0.000 description 1
- 241001045988 Neogene Species 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- 208000028389 Nerve injury Diseases 0.000 description 1
- 208000025966 Neurological disease Diseases 0.000 description 1
- 108090000189 Neuropeptides Proteins 0.000 description 1
- 101150056950 Ntrk2 gene Proteins 0.000 description 1
- WLHBRQYVZLNQFE-UHFFFAOYSA-N O[S+]1C2=CC=CC=C2N=C1 Chemical compound O[S+]1C2=CC=CC=C2N=C1 WLHBRQYVZLNQFE-UHFFFAOYSA-N 0.000 description 1
- 108091034117 Oligonucleotide Proteins 0.000 description 1
- 108010093625 Opioid Peptides Proteins 0.000 description 1
- 102000001490 Opioid Peptides Human genes 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 108090000854 Oxidoreductases Proteins 0.000 description 1
- 108090000526 Papain Proteins 0.000 description 1
- 102000057297 Pepsin A Human genes 0.000 description 1
- 108090000284 Pepsin A Proteins 0.000 description 1
- 108091005804 Peptidases Proteins 0.000 description 1
- 102000035195 Peptidases Human genes 0.000 description 1
- 102100036660 Persephin Human genes 0.000 description 1
- 108010002747 Pfu DNA polymerase Proteins 0.000 description 1
- 206010034912 Phobia Diseases 0.000 description 1
- 108010039918 Polylysine Proteins 0.000 description 1
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-N Propionic acid Chemical class CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 1
- 108010007568 Protamines Proteins 0.000 description 1
- 102000007327 Protamines Human genes 0.000 description 1
- 108020004511 Recombinant DNA Proteins 0.000 description 1
- 102000006382 Ribonucleases Human genes 0.000 description 1
- 108010083644 Ribonucleases Proteins 0.000 description 1
- 108091028664 Ribonucleotide Proteins 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- 235000019095 Sechium edule Nutrition 0.000 description 1
- 108010086019 Secretin Proteins 0.000 description 1
- 102100037505 Secretin Human genes 0.000 description 1
- 229920002684 Sepharose Polymers 0.000 description 1
- 229920001800 Shellac Polymers 0.000 description 1
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 1
- 102000013275 Somatomedins Human genes 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 108091081024 Start codon Proteins 0.000 description 1
- RYYWUUFWQRZTIU-UHFFFAOYSA-N Thiophosphoric acid Chemical class OP(O)(S)=O RYYWUUFWQRZTIU-UHFFFAOYSA-N 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- 108700019146 Transgenes Proteins 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- 229920001807 Urea-formaldehyde Polymers 0.000 description 1
- 108010046516 Wheat Germ Agglutinins Proteins 0.000 description 1
- 239000003070 absorption delaying agent Substances 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 239000000853 adhesive Substances 0.000 description 1
- 230000001070 adhesive effect Effects 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 239000000556 agonist Substances 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 230000000172 allergic effect Effects 0.000 description 1
- 150000001370 alpha-amino acid derivatives Chemical class 0.000 description 1
- 235000008206 alpha-amino acids Nutrition 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 230000001773 anti-convulsant effect Effects 0.000 description 1
- 230000001430 anti-depressive effect Effects 0.000 description 1
- 230000002942 anti-growth Effects 0.000 description 1
- 230000000845 anti-microbial effect Effects 0.000 description 1
- 229940125644 antibody drug Drugs 0.000 description 1
- 239000003146 anticoagulant agent Substances 0.000 description 1
- 239000001961 anticonvulsive agent Substances 0.000 description 1
- 229960003965 antiepileptics Drugs 0.000 description 1
- 239000000739 antihistaminic agent Substances 0.000 description 1
- 229940125715 antihistaminic agent Drugs 0.000 description 1
- 230000036506 anxiety Effects 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- 235000021407 appetite control Nutrition 0.000 description 1
- 239000012736 aqueous medium Substances 0.000 description 1
- 208000006673 asthma Diseases 0.000 description 1
- 208000010668 atopic eczema Diseases 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 150000001558 benzoic acid derivatives Chemical class 0.000 description 1
- 230000003115 biocidal effect Effects 0.000 description 1
- 229920002988 biodegradable polymer Polymers 0.000 description 1
- 239000004621 biodegradable polymer Substances 0.000 description 1
- 230000017531 blood circulation Effects 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 238000006664 bond formation reaction Methods 0.000 description 1
- 210000000133 brain stem Anatomy 0.000 description 1
- 239000006189 buccal tablet Substances 0.000 description 1
- 244000309464 bull Species 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 230000004858 capillary barrier Effects 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 238000005341 cation exchange Methods 0.000 description 1
- 150000001767 cationic compounds Chemical class 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 230000002490 cerebral effect Effects 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 235000019693 cherries Nutrition 0.000 description 1
- 229960004926 chlorobutanol Drugs 0.000 description 1
- 238000010367 cloning Methods 0.000 description 1
- 238000012761 co-transfection Methods 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 238000011284 combination treatment Methods 0.000 description 1
- 238000006482 condensation reaction Methods 0.000 description 1
- 230000001268 conjugating effect Effects 0.000 description 1
- 230000021615 conjugation Effects 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 230000009146 cooperative binding Effects 0.000 description 1
- 235000018417 cysteine Nutrition 0.000 description 1
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 1
- 125000000151 cysteine group Chemical group N[C@@H](CS)C(=O)* 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 231100000517 death Toxicity 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- 230000008260 defense mechanism Effects 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 230000002939 deleterious effect Effects 0.000 description 1
- 238000002716 delivery method Methods 0.000 description 1
- 239000005549 deoxyribonucleoside Substances 0.000 description 1
- 239000005547 deoxyribonucleotide Substances 0.000 description 1
- 125000002637 deoxyribonucleotide group Chemical group 0.000 description 1
- 230000000994 depressogenic effect Effects 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 239000000645 desinfectant Substances 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 238000011026 diafiltration Methods 0.000 description 1
- 238000003745 diagnosis Methods 0.000 description 1
- 238000002405 diagnostic procedure Methods 0.000 description 1
- NEFBYIFKOOEVPA-UHFFFAOYSA-K dicalcium phosphate Chemical compound [Ca+2].[Ca+2].[O-]P([O-])([O-])=O NEFBYIFKOOEVPA-UHFFFAOYSA-K 0.000 description 1
- 229940038472 dicalcium phosphate Drugs 0.000 description 1
- 229910000390 dicalcium phosphate Inorganic materials 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000000378 dietary effect Effects 0.000 description 1
- 238000002050 diffraction method Methods 0.000 description 1
- OZRNSSUDZOLUSN-LBPRGKRZSA-N dihydrofolic acid Chemical compound N=1C=2C(=O)NC(N)=NC=2NCC=1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 OZRNSSUDZOLUSN-LBPRGKRZSA-N 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 229940042399 direct acting antivirals protease inhibitors Drugs 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 238000010494 dissociation reaction Methods 0.000 description 1
- 230000005593 dissociations Effects 0.000 description 1
- VHJLVAABSRFDPM-QWWZWVQMSA-N dithiothreitol Chemical compound SC[C@@H](O)[C@H](O)CS VHJLVAABSRFDPM-QWWZWVQMSA-N 0.000 description 1
- 229960003638 dopamine Drugs 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 239000012154 double-distilled water Substances 0.000 description 1
- 206010013663 drug dependence Diseases 0.000 description 1
- 238000004520 electroporation Methods 0.000 description 1
- 210000001671 embryonic stem cell Anatomy 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 230000012202 endocytosis Effects 0.000 description 1
- 230000003511 endothelial effect Effects 0.000 description 1
- 239000002702 enteric coating Substances 0.000 description 1
- 238000009505 enteric coating Methods 0.000 description 1
- 206010015037 epilepsy Diseases 0.000 description 1
- 210000000981 epithelium Anatomy 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 239000003205 fragrance Substances 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 238000001502 gel electrophoresis Methods 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 238000001415 gene therapy Methods 0.000 description 1
- 208000016361 genetic disease Diseases 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 230000001279 glycosylating effect Effects 0.000 description 1
- 230000012010 growth Effects 0.000 description 1
- 230000035876 healing Effects 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 210000001320 hippocampus Anatomy 0.000 description 1
- 239000000710 homodimer Substances 0.000 description 1
- 235000001050 hortel pimenta Nutrition 0.000 description 1
- 230000008348 humoral response Effects 0.000 description 1
- 210000004408 hybridoma Anatomy 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- 150000002431 hydrogen Chemical class 0.000 description 1
- 230000007954 hypoxia Effects 0.000 description 1
- 230000001146 hypoxic effect Effects 0.000 description 1
- 230000008105 immune reaction Effects 0.000 description 1
- 238000003018 immunoassay Methods 0.000 description 1
- 230000005847 immunogenicity Effects 0.000 description 1
- 229940072221 immunoglobulins Drugs 0.000 description 1
- 230000008676 import Effects 0.000 description 1
- 208000016245 inborn errors of metabolism Diseases 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 239000003701 inert diluent Substances 0.000 description 1
- 230000036512 infertility Effects 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 208000015978 inherited metabolic disease Diseases 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 229940047124 interferons Drugs 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 239000007928 intraperitoneal injection Substances 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 210000003140 lateral ventricle Anatomy 0.000 description 1
- 238000007834 ligase chain reaction Methods 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- XMGQYMWWDOXHJM-UHFFFAOYSA-N limonene Chemical compound CC(=C)C1CCC(C)=CC1 XMGQYMWWDOXHJM-UHFFFAOYSA-N 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 239000006193 liquid solution Substances 0.000 description 1
- 239000006194 liquid suspension Substances 0.000 description 1
- 230000004807 localization Effects 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- ZLNQQNXFFQJAID-UHFFFAOYSA-L magnesium carbonate Chemical compound [Mg+2].[O-]C([O-])=O ZLNQQNXFFQJAID-UHFFFAOYSA-L 0.000 description 1
- 239000001095 magnesium carbonate Substances 0.000 description 1
- 229910000021 magnesium carbonate Inorganic materials 0.000 description 1
- 150000002690 malonic acid derivatives Chemical class 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 239000000155 melt Substances 0.000 description 1
- 230000008384 membrane barrier Effects 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 239000002207 metabolite Substances 0.000 description 1
- UZKWTJUDCOPSNM-UHFFFAOYSA-N methoxybenzene Substances CCCCOC=C UZKWTJUDCOPSNM-UHFFFAOYSA-N 0.000 description 1
- OSWPMRLSEDHDFF-UHFFFAOYSA-N methyl salicylate Chemical compound COC(=O)C1=CC=CC=C1O OSWPMRLSEDHDFF-UHFFFAOYSA-N 0.000 description 1
- 230000000813 microbial effect Effects 0.000 description 1
- 244000005700 microbiome Species 0.000 description 1
- 210000001724 microfibril Anatomy 0.000 description 1
- 206010027599 migraine Diseases 0.000 description 1
- 230000006540 mitochondrial respiration Effects 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 108091005601 modified peptides Proteins 0.000 description 1
- 238000002703 mutagenesis Methods 0.000 description 1
- 231100000350 mutagenesis Toxicity 0.000 description 1
- 210000003928 nasal cavity Anatomy 0.000 description 1
- 229940037525 nasal preparations Drugs 0.000 description 1
- 239000005445 natural material Substances 0.000 description 1
- 101150091879 neo gene Proteins 0.000 description 1
- 230000008764 nerve damage Effects 0.000 description 1
- 210000000653 nervous system Anatomy 0.000 description 1
- 230000001537 neural effect Effects 0.000 description 1
- 230000004766 neurogenesis Effects 0.000 description 1
- 230000002981 neuropathic effect Effects 0.000 description 1
- 231100000189 neurotoxic Toxicity 0.000 description 1
- 230000002887 neurotoxic effect Effects 0.000 description 1
- 230000000269 nucleophilic effect Effects 0.000 description 1
- 235000003715 nutritional status Nutrition 0.000 description 1
- OIPZNTLJVJGRCI-UHFFFAOYSA-M octadecanoyloxyaluminum;dihydrate Chemical compound O.O.CCCCCCCCCCCCCCCCCC(=O)O[Al] OIPZNTLJVJGRCI-UHFFFAOYSA-M 0.000 description 1
- 239000003921 oil Substances 0.000 description 1
- 239000003399 opiate peptide Substances 0.000 description 1
- 238000000879 optical micrograph Methods 0.000 description 1
- 239000007968 orange flavor Substances 0.000 description 1
- 238000006213 oxygenation reaction Methods 0.000 description 1
- 238000004806 packaging method and process Methods 0.000 description 1
- 229940055729 papain Drugs 0.000 description 1
- 235000019834 papain Nutrition 0.000 description 1
- 230000001575 pathological effect Effects 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 229940111202 pepsin Drugs 0.000 description 1
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 1
- 108010070453 persephin Proteins 0.000 description 1
- 230000002085 persistent effect Effects 0.000 description 1
- 239000008177 pharmaceutical agent Substances 0.000 description 1
- 238000009522 phase III clinical trial Methods 0.000 description 1
- 229960003742 phenol Drugs 0.000 description 1
- 208000019899 phobic disease Diseases 0.000 description 1
- 150000008298 phosphoramidates Chemical class 0.000 description 1
- 230000001766 physiological effect Effects 0.000 description 1
- 239000002504 physiological saline solution Substances 0.000 description 1
- 239000000049 pigment Substances 0.000 description 1
- 229920000724 poly(L-arginine) polymer Polymers 0.000 description 1
- 229920001515 polyalkylene glycol Polymers 0.000 description 1
- 108010011110 polyarginine Proteins 0.000 description 1
- 229920000656 polylysine Polymers 0.000 description 1
- 108091033319 polynucleotide Proteins 0.000 description 1
- 239000002157 polynucleotide Substances 0.000 description 1
- 102000040430 polynucleotide Human genes 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 150000004804 polysaccharides Chemical class 0.000 description 1
- 230000001323 posttranslational effect Effects 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 230000003449 preventive effect Effects 0.000 description 1
- 238000004393 prognosis Methods 0.000 description 1
- 210000001236 prokaryotic cell Anatomy 0.000 description 1
- 230000009465 prokaryotic expression Effects 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- 230000000644 propagated effect Effects 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- 229940048914 protamine Drugs 0.000 description 1
- 230000004952 protein activity Effects 0.000 description 1
- 238000000159 protein binding assay Methods 0.000 description 1
- 238000001243 protein synthesis Methods 0.000 description 1
- 230000012743 protein tagging Effects 0.000 description 1
- 230000002685 pulmonary effect Effects 0.000 description 1
- 239000002510 pyrogen Substances 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- 230000002285 radioactive effect Effects 0.000 description 1
- 238000000163 radioactive labelling Methods 0.000 description 1
- 239000000700 radioactive tracer Substances 0.000 description 1
- 229960000160 recombinant therapeutic protein Drugs 0.000 description 1
- 239000006215 rectal suppository Substances 0.000 description 1
- 210000000664 rectum Anatomy 0.000 description 1
- 230000002829 reductive effect Effects 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 230000008886 response to retinoic acid Effects 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 230000004241 retinal aging Effects 0.000 description 1
- 238000010839 reverse transcription Methods 0.000 description 1
- 239000002342 ribonucleoside Substances 0.000 description 1
- 239000002336 ribonucleotide Substances 0.000 description 1
- 125000002652 ribonucleotide group Chemical group 0.000 description 1
- 238000011808 rodent model Methods 0.000 description 1
- 229940081974 saccharin Drugs 0.000 description 1
- 235000019204 saccharin Nutrition 0.000 description 1
- 239000000901 saccharin and its Na,K and Ca salt Substances 0.000 description 1
- 238000010845 search algorithm Methods 0.000 description 1
- 229960002101 secretin Drugs 0.000 description 1
- OWMZNFCDEHGFEP-NFBCVYDUSA-N secretin human Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CO)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(O)=O)C(=O)NCC(=O)N[C@@H](C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(N)=O)[C@@H](C)O)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)C1=CC=CC=C1 OWMZNFCDEHGFEP-NFBCVYDUSA-N 0.000 description 1
- 230000003248 secreting effect Effects 0.000 description 1
- 235000016491 selenocysteine Nutrition 0.000 description 1
- ZKZBPNGNEQAJSX-UHFFFAOYSA-N selenocysteine Natural products [SeH]CC(N)C(O)=O ZKZBPNGNEQAJSX-UHFFFAOYSA-N 0.000 description 1
- 229940055619 selenocysteine Drugs 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 230000009919 sequestration Effects 0.000 description 1
- 238000007493 shaping process Methods 0.000 description 1
- 239000004208 shellac Substances 0.000 description 1
- ZLGIYFNHBLSMPS-ATJNOEHPSA-N shellac Chemical compound OCCCCCC(O)C(O)CCCCCCCC(O)=O.C1C23[C@H](C(O)=O)CCC2[C@](C)(CO)[C@@H]1C(C(O)=O)=C[C@@H]3O ZLGIYFNHBLSMPS-ATJNOEHPSA-N 0.000 description 1
- 229940113147 shellac Drugs 0.000 description 1
- 235000013874 shellac Nutrition 0.000 description 1
- 238000007086 side reaction Methods 0.000 description 1
- 229940126586 small molecule drug Drugs 0.000 description 1
- 239000007909 solid dosage form Substances 0.000 description 1
- 239000012453 solvate Substances 0.000 description 1
- 239000004334 sorbic acid Substances 0.000 description 1
- 235000010199 sorbic acid Nutrition 0.000 description 1
- 229940075582 sorbic acid Drugs 0.000 description 1
- 210000000952 spleen Anatomy 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 238000010186 staining Methods 0.000 description 1
- 230000005477 standard model Effects 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 238000007619 statistical method Methods 0.000 description 1
- 208000005809 status epilepticus Diseases 0.000 description 1
- 238000009168 stem cell therapy Methods 0.000 description 1
- 238000009580 stem-cell therapy Methods 0.000 description 1
- 239000008223 sterile water Substances 0.000 description 1
- 230000001954 sterilising effect Effects 0.000 description 1
- 208000011117 substance-related disease Diseases 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- RWSOTUBLDIXVET-UHFFFAOYSA-O sulfonium group Chemical group [SH3+] RWSOTUBLDIXVET-UHFFFAOYSA-O 0.000 description 1
- 239000013589 supplement Substances 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 238000007910 systemic administration Methods 0.000 description 1
- RTKIYNMVFMVABJ-UHFFFAOYSA-L thimerosal Chemical compound [Na+].CC[Hg]SC1=CC=CC=C1C([O-])=O RTKIYNMVFMVABJ-UHFFFAOYSA-L 0.000 description 1
- 229940033663 thimerosal Drugs 0.000 description 1
- 230000002537 thrombolytic effect Effects 0.000 description 1
- 210000001578 tight junction Anatomy 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 230000005030 transcription termination Effects 0.000 description 1
- 230000002103 transcriptional effect Effects 0.000 description 1
- 238000010361 transduction Methods 0.000 description 1
- 230000026683 transduction Effects 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 230000014616 translation Effects 0.000 description 1
- 150000003626 triacylglycerols Chemical class 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- 230000003827 upregulation Effects 0.000 description 1
- XSQUKJJJFZCRTK-UHFFFAOYSA-N urea group Chemical group NC(=O)N XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 1
- 210000003708 urethra Anatomy 0.000 description 1
- 238000001291 vacuum drying Methods 0.000 description 1
- 239000006216 vaginal suppository Substances 0.000 description 1
- 210000003462 vein Anatomy 0.000 description 1
- 230000002861 ventricular Effects 0.000 description 1
- 238000012795 verification Methods 0.000 description 1
- 230000000007 visual effect Effects 0.000 description 1
- 239000002699 waste material Substances 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Chemical compound O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 1
- 230000003442 weekly effect Effects 0.000 description 1
- 238000005303 weighing Methods 0.000 description 1
- 210000004885 white matter Anatomy 0.000 description 1
- 239000009637 wintergreen oil Substances 0.000 description 1
- DGVVWUTYPXICAM-UHFFFAOYSA-N β‐Mercaptoethanol Chemical compound OCCS DGVVWUTYPXICAM-UHFFFAOYSA-N 0.000 description 1
Images







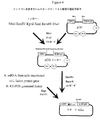

























Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2869—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against hormone receptors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
- A61P21/04—Drugs for disorders of the muscular or neuromuscular system for myasthenia gravis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/475—Growth factors; Growth regulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/20—Immunoglobulins specific features characterized by taxonomic origin
- C07K2317/24—Immunoglobulins specific features characterized by taxonomic origin containing regions, domains or residues from different species, e.g. chimeric, humanized or veneered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/30—Non-immunoglobulin-derived peptide or protein having an immunoglobulin constant or Fc region, or a fragment thereof, attached thereto
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Neurosurgery (AREA)
- Immunology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Molecular Biology (AREA)
- Genetics & Genomics (AREA)
- Biophysics (AREA)
- Biochemistry (AREA)
- Psychiatry (AREA)
- Psychology (AREA)
- Gastroenterology & Hepatology (AREA)
- Endocrinology (AREA)
- Zoology (AREA)
- Toxicology (AREA)
- Physical Education & Sports Medicine (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Ophthalmology & Optometry (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Urology & Nephrology (AREA)
- Pain & Pain Management (AREA)
- Vascular Medicine (AREA)
- Hospice & Palliative Care (AREA)
- Peptides Or Proteins (AREA)
Description
本発明は、国立衛生研究所の助成金番号R44−NS−44654によって合衆国政府の資金提供を受けた。合衆国政府は、本発明に一定の権利を有し得る。
本発明の新たな特性は、添付した特許請求の範囲に詳細にわたって記載されている。本発明の特性および利点のよりよい理解は、本発明の原理を利用した、例示的実施形態を説明した以下の詳細な説明および添付した図面を参考にすることによって得られるだろう。
図2は、修飾された5’−および3’−リンカーを備えたvBDNFcDNAをコードする細菌発現プラスミドの遺伝子操作を示した図である。
図3は、融合タンパク質を生成するタンデムベクター構築における中間体である様々な構造物の大きさを示した臭化エチジウム染色アガロースゲルを示した図である。(A)列1〜2:NruIで消化した図2のプラスミドで、0.4kbのvBDNFおよび3.5kbのベクター主鎖を示している。列3:1.4〜0.1kbの範囲の大きさのMW標準物。列4:23〜0.6kbの範囲の大きさのMW標準物。(B)列1:0.4kbのvBDNFcDNAは、ポリA+ヒトU87神経膠腫細胞から単離されたRNAから逆転写されたcDNAを使用した、ポリメラーゼ連鎖反応(PCR)によって生成される。PCRプライマー配列を表2に示す。列2および3:パネルAに示したのと同じ大きさのMW標準物。(C)列1:NheIおよびBamHIで消化した後のクローン416。列2:陰性クローン。列3:NheIおよびBamHIで消化した後のクローン400。列4および5:パネルAで示したのと同じ大きさのMW標準物。(D)融合タンパク質HC(列1)およびLC(列2)をコードするDNAの特性のPCR断片。列3〜4:パネルAで示したのと同じ大きさのMW標準物。(E)列1〜4:NheIで消化した後のクローン422aの4種の、異なるが一致しているコピーで、0.4kbの融合タンパク質HC可変領域(VH)cDNAの遊離が示されている。列5〜6:パネルAで示したのと同じ大きさのMW標準物。(F)列1〜4:EcoRVおよびBamHIで消化した後のクローン423aの5種の、異なるが一致しているコピーで、0.7kbの完全なLCcDNAの遊離が示されている。列5〜6:パネルAで示したのと同じ大きさのMW標準物。(G)PvuI(列1)およびEcoRI−HindIII(列2)によるタンデムベクター(図12)の制限エンドヌクレアーゼ地図。PvuI(切断1回)によって、約11kbの予測直線状DNAバンドが生成した。EcoRIおよびHindIIIによる消化によって、融合タンパク質軽鎖(すなわち、1.8kb)およびDHFR(すなわち、1.5kb)発現カセットの両方が遊離する。約8kbのバンドは、融合タンパク質重鎖発現カセットを含む主鎖ベクターを表す。列3〜4:順番は逆であるが、パネルAで示したのと同じ大きさのMW標準物。
図4は、融合タンパク質HCのカルボキシル末端とvBDNFのアミノ末端との間の融合部位のヌクレオチド(配列番号21)およびアミノ酸(配列番号22)を示した図である。HIRMAb HCとvBDNFとの間の3−アミノ酸リンカーならびにvBDNFのカルボキシル末端の新たな終止コドンを示す。
図5は、プラスミド416にクローニングされた融合タンパク質HC遺伝子のヌクレオチド配列(配列番号23)を示した図である。イタリック体:ヒトIgG1定常領域イントロン。太字:ヒトIgG1エキソン配列。下線部:vBDNF。
図6は、融合タンパク質HCのアミノ酸配列(配列番号24)を示した図である。CH3領域とvBDNFとの間の3−アミノ酸リンカーのように、アミノ酸19個のシグナルペプチドに下線を引いてある。CH2内のN−結合グリコシル化コンセンサス配列に下線を引いてある。
図7は、融合タンパク質HCの様々なドメインのアミノ酸配列(配列番号25)を示した図である。
図8は、融合タンパク質HCをコードする、イントロンのない真核発現ベクター、クローン422aの生成を示した図である。融合タンパク質HC cDNAは、クローン416を形質移入した骨髄腫細胞から単離されたRNAの逆転写酵素によって生じたcDNAをPCRすることによって生成した。
図9は、(A)クローン422aに挿入された融合タンパク質HC cDNAのヌクレオチド配列(配列番号26)を示した図である。(B)(配列番号27および28)図Aに示したヌクレオチド配列から推定される融合タンパク質HCのアミノ酸配列を示した図である。シグナルペプチドの配列には下線が引いてある。
図10は、融合タンパク質LCをコードする、イントロンのない真核発現ベクター、クローン423aの生成を示した図である。融合タンパク質LC cDNAは、キメラHIRMAb LCのVLを含むヒトカッパLC遺伝子のイントロン/エキソン配列をコードする染色体断片由来のLC遺伝子を生成する発現ベクターを形質移入された骨髄腫細胞から単離されたRNAの逆転写酵素によって生成したcDNAをPCRすることによって生成した。
図11は、(A)クローン423aに挿入された融合タンパク質LC cDNAのヌクレオチド配列(配列番号29)を示した図である。(B)(配列番号30および31)図Aに示したヌクレオチド配列から推定される融合タンパク質LCのアミノ酸配列を示した図である。シグナルペプチドの配列には下線が引いてある。
図12は、融合タンパク質のHCおよびLC遺伝子をコードするタンデムベクターの構造物を示した図である。TVは、cDNA発現ベクター、HCについてはクローン422aおよびLCについては423a、ならびにマウスDHFRの発現カセットをコードする細菌発現プラスミドから操作された。
図13Aおよび図13Bは、融合タンパク質HC遺伝子およびLC遺伝子ならびにタンデムベクターに組み込まれたDHFR遺伝子のヌクレオチド配列(配列番号32)を示した図である。
図14は、タンデムベクターヌクレオチド配列分析に基づいて融合タンパク質HCの推定アミノ酸配列を示した図である(配列番号33および34)。シグナルペプチド配列には下線が引いてある。
図15は、タンデムベクターヌクレオチド配列分析に基づいて融合タンパク質LCの推定アミノ酸配列を示した図である(配列番号35および36)。シグナルペプチド配列には下線が引いてある。
図16は、タンデムベクターヌクレオチド配列分析に基づいてDHFRの推定アミノ酸配列を示した図である(配列番号37および38)。
図17は、バイオリアクターで50日間一定に維持したCHO細胞の生細胞および全細胞密度を示した図で、CHO細胞は、融合タンパク質をコードするタンデムベクターを永久的に形質移入された。
図18は、融合タンパク質、(a)血液からBBBを通過する輸送を可能にするヒトBBBヒトインシュリン受容体(HIR)に結合し、(b)神経保護を導くニューロン上のtrkBに結合する2機能性分子の構造を示した図である。
図19は、2次抗体がヒトIgG1のFc領域(x軸)またはヒトBDNF(y軸)のいずれかに特異的である、2種類の異なる「サンドイッチ」免疫測定法の相関を示した図である。どちらの測定法でも1次抗体はヒトカッパ軽鎖に特異的である。CHO細胞調整培地中の融合タンパク質の測定レベルは、抗Fcを使用しても、抗BDNF抗体を使用しても同一である。
図20は、キメラHIRMAbおよび融合タンパク質の還元(左)および非還元(右)ドデシル硫酸ナトリウムポリアクリルアミドゲル電気泳動(SDS−PAGE)を示した図である。還元条件下では、軽鎖の大きさ、30kDaは、キメラHIRMAbおよび融合タンパク質と同一であり、融合タンパク質の重鎖の大きさは、BDNFが存在するため、キメラHIRMAb重鎖より約15kDa大きい。非還元条件下では、キメラHIRMAbおよび融合タンパク質はそれぞれ、分子量180および200kDaの単一な異種四量体種として移動する。
図21は、(左図)抗ヒトIgG1次抗体のウェスタンブロットを示した図である。融合タンパク質およびHIRMAbの重鎖の大きさはそれぞれ、64kDaおよび50kDaであり、融合タンパク質またはキメラHIRMAbの軽鎖の大きさは25kDaである。(右図)融合タンパク質またはBDNFには反応するが、キメラHIRMAbには反応しない抗ヒトBDNF抗体によるウェスタンブロットを示した図である。MW標準物(STDS)は、右側に示してある。
図22は、等電点(pI)標準物(列1)、キメラHIRMAb(列2および4)、BDNF(列3)および融合タンパク質(列5)の等電点電気泳動(IEF)を示した図である。BDNFは、pI>10で強い陽イオンである一方、融合タンパク質のpIはキメラHIRMAbのpIに近似しており、約8.5で、融合タンパク質の理論的pIに近い。
図23は、(A)ヒトインシュリン受容体(HIR)競合リガンド結合測定法(CLBA)の概略を示した図である。HIR細胞外ドメイン(ECD)に[125I]標識マウスHIRMAbを結合させ、図Bに示したように、この結合をキメラHIRMAbまたは融合タンパク質のいずれかと競合的に置換させる。(B)キメラIHRMAbまたは融合タンパク質のいずれかによる、HIR ECDに対する[125I]標識マウスHIRMAbの結合の置換を示した図である。HIR ECDに対するキメラHIRMAbの親和性は高く、HIR ECDに対する融合タンパク質の親和性は、キメラHIRMAbの親和性と有意差があるとはいえない。これらの結果は、キメラHIRMAb重鎖のカルボキシル末端にvBDNFを融合しても、HIRに対する融合タンパク質の結合を損なわないことを示している。
図24は、(A)trkB競合リガンド結合測定(CLBA)の設計。PEGリンカーの利点は、それによる修飾でELISAウェルに対する陽イオンBDNFの非特異的結合(NSB)が排除され、それによってシグナル/ノイズ比の高い測定が得られることである。trkB ECDへのBDNF−PEG2000−ビオチンの結合は、アビジンおよびビオチンペルオキシダーゼを使用したペルオキシダーゼ系によって検出された。(B)trkB ECDへのBDNF−PEG2000−ビオチンの結合は、組換えBDNFによって競合的に置換される。この結合データは、非線形回帰分析によって分析したところ、BDNF結合のKI、3.5±1.3pmol/ウェルおよびNSBパラメータが得られた。(C)trkB ECDへのBDNF−PEG2000−ビオチンの結合は、融合タンパク質によって競合的に置換される。この結合データは、非線形回帰分析によって分析したところ、融合タンパク質結合のKIは2.2±1.2pmol/ウェルで、天然BDNFのKIと有意差があるとはいえない。これらのデータは、trkB受容体に対する融合タンパク質の親和性が天然BDNFの親和性と等しいことを示している。
図25は、(A)ヒト神経SH−SY5Y細胞における低酸素症−再酸素負荷神経保護測定法の設計を示した図である。細胞をレチノイン酸に7日間曝露して、trkB、BDNF受容体の遺伝子発現の上方制御を引き起こす。(B)3−(4,5−ジメチルチアゾール−2−イル)−2,5−ジフェニル−2H−テトラゾリウムブロミド(MTT)によるミトコンドリア呼吸の測定に基づいた神経保護測定法。最大の神経保護は、BDNF4nMによって確立され、融合タンパク質4nMは、ヒト神経細胞において、同程度の神経保護レベルを生じる。MTTレベルは、細胞の約50%のみがレチノイン酸に応答してtrkBを誘導するので、低酸素症ではない細胞のレベルにはもどらない。
図26は、(A)ヒトBBBのin vitroモデル系として使用した、ヒト脳から単離された毛細血管の光学顕微鏡写真である。(B)ヒトBBB上のHIRに対する[3H]−融合タンパク質の結合の放射線−受容体測定法。結合は非標識融合タンパク質によって自己阻害される。非線形回帰分析によって飽和データをスキャッチャードプロットに当てはめると、結合パラメータ、KD=0.55±0.07nM、Bmax=1.35±0.10pmol/mgpが得られる。
図27は、成体アカゲザルにおける融合タンパク質の薬物動態および脳への取り込み。(A)麻酔した成体アカゲザルに[3H]−融合タンパク質または[125I]マウスHIRMAbのいずれかのタンパク質を1回静脈注射した後、[3H]−融合タンパク質または[125I]マウスHIRMAbの血清濃度を時間に対してプロットする。(B)麻酔した成体アカゲザルに[3H]融合タンパク質を、または麻酔した成体ラットの[3H]−BDNFを1回静脈注射した後、トリクロロ酢酸(TCA)によって沈殿可能な血清放射活性を時間に対してプロットする。(C)麻酔した成体アカゲザルに、[3H]融合タンパク質または[3H]マウスIgG2aのいずれかを1回静脈注射してから180分後の脳分布の毛細血管欠乏分析。(D)融合タンパク質373μgを静脈注射してから180分後の融合タンパク質の霊長類脳における濃度を、霊長類脳の内在性BDNF濃度と比較して示した図である。
I.導入
II.定義
III.血液脳関門
A.輸送系
B.血液脳関門受容体媒介輸送系に結合する構造
IV.血液脳関門を通過して輸送するための薬剤
A.ニューロトロフィン
B.脳由来神経栄養因子
V.組成物
VI.核酸、ベクター、細胞および製造
A.核酸
B.ベクター
C.細胞
D.製造
VII.方法
VIII.キット
略語
ALS 筋萎縮性側索硬化症
BBB 脳血液関門
BDNF 脳由来神経栄養因子
BRB 脳網膜関門
CDR 相補性決定領域
CED 対流増強送達
CH1 IgG定常領域の第1部
CH2 IgG定常領域の第2部
CH3 IgG定常領域の第3部
CHO チャイニーズハムスター卵巣細胞
CHOP CHO細胞宿主タンパク質
CLBA 競合リガンド結合測定法
CMV サイトメガロウイルス
CNS 中枢神経系
DHFR ジヒドロ葉酸レダクターゼ
ECD 細胞外ドメイン
FR フレームワーク領域
FWD フォワード
HIR ヒトインシュリン受容体
HIRMAb ヒトインシュリン受容体に対するモノクローナル抗体
HIV ヒト免疫不全ウイルス
IC 脳内
ICC 免疫細胞化学
ICV 側脳室内
ID 注射用量
IEF 等電点電気泳動
IGF インシュリン様成長因子
IgG イムノグロブリンG
LDL 低密度リポタンパク質
MAb モノクローナル抗体
MRT 平均滞留時間
MTH 分子版トロイの木馬
MTX メトトレキセート
MW 分子量
NSB 非特異的結合
NT−3 ニューロトロフィン−3
NT−4/5 ニューロトロフィン−4/5
ODN オリゴデオキシヌクレオチド
PEG ポリエチレングリコール
PRO プロモーター
REV リバース
RMT 受容体媒介輸送
SDM 部位特異的変異誘発
SDS−PAGE ドデシル硫酸ナトリウムポリアクリル
アミドゲル電気泳動
SFM 無血清培地
SNP 一塩基多形
TCA トリクロロ酢酸
TFF タンジェンシャルフロー濾過
TFI 一過性前脳虚血
TfR トランスフェリン受容体
TrkB BDNF受容体
TV タンデムベクター
vBDNF BDNF変種
VD 分布量
VH 重鎖の可変領域
VL 軽鎖の可変領域
血液脳関門は、多くの末梢投与薬を中枢神経系に輸送するのを制限する因子である。本発明は、BBBを通過してCNSに薬剤を輸送するのに重要な3つの要素、1)薬剤がBBBと十分に接触してBBBを横断するために、十分な時間末梢循環中にあることが可能な薬剤の薬物動態プロファイル、2)BBBを通過させるための薬剤の修飾、および3)一旦BBBを通過した薬剤の活性の保持に取り組む。本発明の様々な態様は、BBBを通過して輸送されるために、および/または構造に結合していながら、脳においてその活性のいくらか、もしくは全てを保持するために、薬剤の血清半減期の延長をもたらす構造に共有結合した薬剤(例えば、治療薬)の融合構造(例えば、融合タンパク質)を提供することによって、これらの要素に取り組む。
本明細書で使用したように、「薬剤」には、生物における生理学的または生化学的効果を含む効果を生じるのに有用ないかなる物質も含まれる。「治療薬」とは、治療効果、すなわち、障害の改善、予防、および/または完全な、もしくは部分的な治癒を導く効果を生じさせる、または生じさせるようにする物質である。「治療効果」はまた、本明細書で使用したように、障害に罹患していない個体における平均的または正常な状態よりも良好な状態を作り出すこと、すなわち、正常または平均的な状態と比較して、認知、記憶、気分またはCNSの機能の少なくとも一部に起因すると考えられるその他の特性の改善などの平均以上の効果を含む。「神経治療薬」は、CNSにおいて治療効果をもたらす薬剤である。「治療用ペプチド」には、ペプチドからなる治療薬が含まれる。「陽イオン治療用ペプチド」には、等電点が約7.4を上回る、いくつかの実施形態では、約8、8.5、9、9.5、10、10.5、11、11.5、12、または約12.5を上回る治療用ペプチドが包含される。陽イオン治療用ペプチドの下位区分は陽イオン神経治療用ペプチドである。
一態様では、本発明は、血液脳関門(BBB)を通過できる構造に共有結合した薬剤を利用する組成物および方法を提供する。この組成物および方法は、薬剤、例えば、神経治療薬などの治療薬を末梢血から、血液脳関門を通過してCNSに輸送するのに有用である。本明細書で使用したように、「血液脳関門」とは、脳毛細血管内皮原形質膜内で緊密な結合によって形成される末梢循環と脳および脊髄との間の関門のことであり、分子量60Daの尿素と同じくらい小さい分子であっても、脳内への分子の輸送を制限する極めて厳重な関門を形成する。脳内の血液脳関門、脊髄内の血液脊髄関門、網膜内の血液網膜関門は、中枢神経系(CNS)内の近接した毛細血管関門であり、集合的に血液脳関門またはBBBと称される。
いくつかの実施形態では、本発明は、BBBを通過して輸送することが望ましい薬剤、例えば、神経治療薬を結合させた、BBB受容体媒介輸送系に結合する構造を含む組成物を提供する。本発明の組成物および方法は、選択された内在性BBB受容体媒介輸送系によって輸送でき、所望する薬剤に結合させることもできる任意の適切な構造を利用することができる。いくつかの実施形態では、この構造は、抗体である。いくつかの実施形態では、この抗体はモノクローナル抗体(MAb)、例えば、キメラMAbである。
CNSに対する薬剤を送達するための非侵襲的な1取り組みは、BBB上の受容体と結合する構造、例えば、分子に関心のある薬剤を結合させることである。次に、この構造は、BBBを通過して薬剤を輸送するための媒介物として役立つ。このような構造は、本明細書では「分子版トロイの木馬(MTH)」と称する。一般的に、必ずしも必要ではないけれども、MTHは、内在性BBB受容体媒介輸送系でBBBを横断する内在性BBB受容体媒介輸送系に結合することができる外来ペプチドまたはペプチド様部分(例えば、MAb)である。ある実施形態では、MTHは、輸送系の受容体、例えば、インシュリン受容体に対する抗体であることができる。いくつかの実施形態では、この抗体はモノクローナル抗体(MAb)である。いくつかの実施形態では、このMAbはキメラMAbである。したがって、Abは通常脳から排除されるという事実があるにもかかわらず、それらがBBB上の受容体に特異性を有するならば、分子を脳実質に送達するための効果的な媒体であることができる。
BBBを通過して輸送することが所望される薬剤は、CNSに導入するための任意の適切な物質であってよい。一般的に、この薬剤は、BBBを通過して輸送することが所望され、天然の形態では、有意な量でBBBを通過しない物質である。この薬剤は、例えば、治療薬、診断薬、または研究薬であってよい。診断薬には、ペプチド放射線医薬品、例えば、脳癌を画像診断するための上皮成長因子(EGF)(Kurihara and Pardridge(1999)Canc.Res.54:6159〜6163)、およびアルツハイマーなど脳アミロイドを画像診断するためのアミロイドペプチド(Lee他(2002)J.Cereb.Blood Flow Metabol.22:223〜231)が含まれる。いくつかの実施形態では、この薬剤は、治療薬、例えば神経治療薬である。ニューロトロフィンの他に、潜在的に有用な治療用タンパク質剤には、リソソーム蓄積症のための組換え酵素(例えば、本明細書に全体を参考として援用した2005年2月17日出願の米国特許出願公開第20050142141号)、内在性ペプチドを模倣しているか、または脳障害のために内在性ペプチド、ポリペプチド、例えば、自閉症のためにセクレチンの作用を遮断するモノクローナル抗体(Ratliff−Schaub他(2005)Autism9:256〜265)、薬物またはアルコール中毒のためのオピオイドペプチド(Cowen他(2004)、J.Neurochem.89:273〜285)、あるいは食欲制御のための神経ペプチド(Jethwa他(2005)Am.J.Physiol.289:E301〜305)が含まれる。いくつかの実施形態では、この薬剤は、本明細書では「ニューロトロフィン」とも呼ばれる神経栄養因子である。したがって、いくつかの実施形態では、本発明は、ニューロトロフィンを利用する組成物および方法を提供する。いくつかの実施形態では、単一のニューロトロフィンを使用できる。その他では、ニューロトロフィンの組み合わせを使用する。いくつかの実施形態では、本発明は、脳由来神経栄養因子(BDNF)を利用する。
多くの神経栄養因子は、脳において神経を保護するが、血液脳関門を通過できない。これらの因子は、本発明の組成物および方法で使用するために適しており、脳由来神経栄養因子(BDNF)、神経成長因子(NGF)、ニューロトロフィン−4/5、繊維芽細胞増殖因子(FGF)−2およびその他のFGF、ニューロトロフィン(NT)−3、エリスロポエチン(EPO)、肝細胞増殖因子(HGF)、上皮成長因子(EGF)、トランスフォーミング成長因子(TGF)−α、TGF−β、血管内皮増殖因子(VEGF)、インターロイキン−1受容体アンタゴニスト(IL−1ra)、繊毛様神経栄養因子(CNTF)、グリア細胞由来神経栄養因子(GDNF)、ニュールツリン、血小板由来成長因子(PDGF)、ヘレグリン、ニューレグリン、アルテミン、ペルセフィン、インターロイキン類、顆粒球コロニー刺激因子(CSF)、顆粒球マクロファージCSF、ネトリン、カルジオトロフィン−1、ヘッジホッグ、白血病抑制因子(LIF)、ミッドカイン、プレイオトロフィン、骨形態形成蛋白質(BMP)、ネトリン、サポシン、セマフォリンおよび幹細胞因子(SCF)が含まれる。BBBを通過する融合タンパク質の前駆体として使用されるニューロトロフィンを利用した本発明のいくつかの実施形態で特に有用なのは、BDNFと同様に自然に二量体構造を形成するものである。BDNFまたはNT−3などのある種のニューロトロフィンは、異種二量体構造を形成することができ、いくつかの実施形態では、本発明は、抗体、例えば、HIRMAbの1本目の鎖(例えば、軽鎖または重鎖)に融合した1個のニューロトロフィン単量体および抗体の2本目の鎖(例えば、軽鎖または重鎖)に融合した別のニューロトロフィン単量体から構成された融合タンパク質を提供する。一般的に、分子版トロイの木馬に融合させることができる組換えタンパク質の分子量範囲は、1000ダルトンから500000ダルトンである。
本発明の実施形態で特に有用な1ニューロトロフィンは、脳由来神経栄養因子(BDNF)である。BDNFは、多くの急性および慢性脳疾患において神経保護薬として使用できる強力な神経治療薬である。しかし、BBBを通過してBDNFを輸送できないことは、脳および脊髄のための神経治療薬としてこの分子を開発することの妨げとなってきた。
本発明の組成物は、陽イオン化合物の血清半減期の延長、BBBを通過する薬剤の輸送、および/またはBBBを通過して一旦輸送された薬剤の活性の保持の1種または複数において有用である。したがって、いくつかの実施形態では、本発明は血液脳関門(BBB)を通過できる構造に共有結合した神経治療薬を含有する組成物であって、末梢投与後、脳内の神経治療薬の濃度を平均して少なくとも約1、2、3、4、5、10、20、30、40または50ng/グラム脳上昇させることができる組成物を提供する。本発明はまた、ヒトBBBインシュリン受容体に対するキメラMAbに共有結合した薬剤を含有する組成物を提供する。本発明はさらに、中枢神経系(CNS)で活性のあるペプチドに共有結合した、BBBを通過することができる構造を含有する融合タンパク質であって、血液脳関門を通過できる構造および中枢神経系で活性のあるペプチドはそれぞれ、別々の実体としての活性と比較して、平均してそれらの活性の少なくとも約10、20、30、40、50、60、70、80、90、95、99または100%を保持している融合タンパク質を提供する。ある種の実施形態では、本発明はさらに、陽イオン物質の血清半減期を延長する組成物を提供する。本発明はまた、本発明の1個または複数の組成物および薬学的に許容される賦形剤を含有する医薬組成物を提供する。
本発明はまた、核酸、ベクター、細胞および製造法を提供する。
いくつかの実施形態では、本発明は本発明のタンパク質またはペプチドをコードする核酸を提供する。ある種の実施形態では、本発明はイムノグロブリンの軽鎖をコードする第1の配列およびイムノグロブリンの重鎖をコードする第2の配列を含有する単一の核酸配列であって、前記第1の配列はまた、軽鎖に共有結合したペプチドの融合タンパク質として発現されるペプチドをコードするか、または前記第2の配列はまた、重鎖に共有結合したペプチドの融合タンパク質として発現されるペプチドをコードする核酸配列を提供する。いくつかの実施形態では、本発明は、核酸配列を提供し、いくつかの実施形態では、本発明は、特定のヌクレオチド配列と少なくとも約60、70、80、90、95、99または100%同一な核酸配列を提供する。例えば、いくつかの実施形態では、本発明は、配列番号33のヌクレオチド58〜1386と少なくとも約60、70、80、90、95、99または100%同一である第1の配列および配列番号33のヌクレオチド1396〜1746と少なくとも約60、70、80、90、95、99または100%同一である第2の配列を含有する核酸を提供する。
本発明はまた、ベクターを提供する。このベクターは、本明細書で記載した核酸配列のいずれかを含有することができる。いくつかの実施形態では、本発明は、ペプチド、例えば、ニューロトロフィンなどの治療用ペプチドに融合した抗体重鎖をコードする核酸、および抗体の軽鎖をコードする核酸を含有する、いずれもが核酸の1片、例えば、DNAの1片に組み込まれている1本のタンデム発現ベクターを提供する。この1本のタンデムベクターは、1種または複数の選択および/または増幅遺伝子も含むことができる。本発明の例示的ベクターの作製方法は、実施例に挙げてある。しかし、当業界で知られているような任意の適切な技術をベクターの構築に使用することができる。
本発明はさらに、本発明の1種または複数のベクターを組み込んだ細胞を提供する。この細胞は、原核細胞または真核細胞であってよい。いくつかの実施形態では、この細胞は真核細胞である。いくつかの実施形態では、この細胞はマウス骨髄腫細胞である。いくつかの実施形態では、この細胞はチャイニーズハムスター卵巣(CHO)細胞である。ベクターを細胞に組み込む例示的方法を実施例に挙げる。しかし、当業界で知られているように、任意の適切な技術を、ベクターの細胞への組み込みに使用することができる。いくつかの実施形態では、本発明はイムノグロブリン融合タンパク質を発現できる細胞であって、永久的に1本のタンデム発現ベクターを導入した細胞であり、イムノグロブリン軽鎖遺伝子および治療薬に融合させたイムノグロブリン重鎖の遺伝子の両方が、核酸、例えば、DNAの1片に組み込まれている細胞を提供する。いくつかの実施形態では、本発明はイムノグロブリン融合タンパク質を発現できる細胞であって、永久的に1本のタンデム発現ベクターを導入した細胞であり、イムノグロブリン重鎖遺伝子および治療薬に融合させたイムノグロブリン軽鎖の遺伝子の両方が、核酸、例えば、DNAの1片に組み込まれている細胞を提供する。タンデムベクターの導入は、例えば、染色体核酸への永久的組み込みによって、または、例えば、エピソーム遺伝子因子の導入によることができる。
さらに、本発明は製造方法を提供する。いくつかの実施形態では、本発明はイムノグロブリン融合タンパク質の製造方法であって、真核細胞に1本のタンデム発現ベクターを永久的に導入することによって、前記融合タンパク質が、治療薬と融合したイムノグロブリン重鎖を含有しており、イムノグロブリン軽鎖遺伝子および治療薬に融合したイムノグロブリン重鎖の遺伝子の両方が核酸、例えば、DNAの1片に永久的に組み込まれている製造方法を提供する。いくつかの実施形態では、本発明はイムノグロブリン融合タンパク質の製造方法であって、真核細胞に1本のタンデム発現ベクターを永久的に導入することによって、前記融合タンパク質が、治療薬と融合したイムノグロブリン軽鎖を含有しており、イムノグロブリン重鎖遺伝子および治療薬に融合したイムノグロブリン軽鎖の遺伝子の両方が核酸、例えば、DNAの1片に永久的に組み込まれている製造方法を提供する。いくつかの実施形態では、ベクターの導入は、宿主細胞ゲノムに永久的に統合することによって実現される。いくつかの実施形態では、ベクターの導入は、エピソームの遺伝的因子を含有するベクターを宿主細胞に導入することによって実現される。エピソームの遺伝的因子は当業界では周知である。いくつかの実施形態では、この治療薬は神経治療薬である。いくつかの実施形態では、この核酸の一片はさらに、1個または複数の選択可能なマーカーの遺伝子を含む。いくつかの実施形態では、この核酸の一片はさらに、1個または複数の増幅遺伝子を含む。いくつかの実施形態では、イムノグロブリンはIgG,例えば、キメラMAbなどのMAbである。この方法にはさらに、イムノグロブリン融合タンパク質を発現し、および/またはイムノグロブリン融合タンパク質を精製することを含んでよい。発現および精製を含む例示的製造方法を実施例に挙げる。
本発明はまた、方法を提供する。いくつかの実施形態では、本発明はCNSで活性のある薬剤を有効量でBBBを通過して輸送する方法を提供する。いくつかの実施形態では、本発明は治療法、診断法または研究法を提供する。診断法には、BBBを通過して輸送することができるペプチド放射性医薬品、例えば、ペプチドリガンドの融合物または脳内の内在性受容体のペプチド様MAbの開発、その後の融合タンパク質の放射標識、その後の全身投与およびペプチド放射性医薬品の脳内における局在の外部画像診断が含まれる。
本発明の組成物、例えば、融合タンパク質は、容器または適切な包装内に製剤、例えば、融合タンパク質を含むキットとして提供することができる。組成物は、乾燥粉末形態、固形(すなわち、凍結乾燥)、溶液、または懸濁液で提供することができる。組成物がタンパク質の場合、このタンパク質に、乳化剤、塩、保存剤、その他のタンパク質、核酸、プロテアーゼ阻害剤、抗生物質、香料、多糖類、接着剤、ポリマー、ミクロフィブリル、油などを添加することができる。輸送、保存および/または消費者が使用するために組成物を包装する。輸送、保存および使用のための治療用組成物のこのような包装は、当業界で周知である。包装された組成物には、組成物を分配および保存するための成分をさらに含めることができ、例えば、製剤、例えば、融合タンパク質を患者に投与する前に溶解するための滅菌水または適切な緩衝液からなる別々に包装された希釈剤も含めることができる。本発明のキットには、使用指示書、臨床研究の結果、治療の所望される予後および予測経過、予防措置および副作用の情報などの文書を含めることもできる。このキットは、所望によりさらに、その他の成分、例えば、手袋、はさみ、テープ、使用済みバイアルおよびその他の廃棄物を処分するための器具、マスク、殺菌剤、抗生物質などを含有することができる。
IgG−神経治療薬融合物の完全な遺伝子を含有する1本のタンデムベクターの構築
融合タンパク質の重鎖(HC)をコードする真核発現ベクターの遺伝子操作を図1に概略する。最終的な融合タンパク質HC発現ベクターは、pHIRMAb−BDNF、またはクローン416と示される。このベクターは、HIRMAbのHCに融合したBDNF変種から構成される融合タンパク質を産生するように設計された。BDNFまたはBDNFの変種(vBDNF)のいずれかをHIRMAbに融合させることができる。vBDNFは、ある種のアミノ酸が置換していることによって天然のヒトBDNFとは異なっており、例えば、vBDNFではBDNFのカルボキシル末端の2アミノ酸が存在しない。クローン416プラスミドは、HIRMAbのキメラ型のHCを産生するクローン400およびは図2に記載した通りに作製された成熟ヒトvBDNFをコードするcDNAから得られた。クローン400は、ヒトIgG1定常領域をコードする染色体断片から得られ、イントロンおよびエキソン配列の両方から構成されるキメラヒトIgG1をコードする。クローン400におけるキメラHIRMAbのHC遺伝子を、部位特異的変異誘発(SDM)によるCH3領域の3’末端に位置する終止コドンの操作を容易にするために、pCRIIプラスミドのBamHI部位にサブクローニングした。CH3領域の末端に位置する終止コドンの操作は、SspI部位を創出する部位特異的変異誘発によって実施した。SspI部位は、クローン415を作製するために、クローン400への平滑末端連結によるvBDNFcDNAの挿入を可能にする(図3)。SDMは、QuickChange SDMキット(Stratagene、CA)を使用して実施した。センスおよび相補的変異誘発プライマーは、CH3終止コドン(aaTGAg)がSspI部位(aaTATt)に変異するように設計した。さらに、プライマーは、終止コドン5’−および3’−周囲領域の15個のヌクレオチドを含有しており、SDM−SspIフォワード(FWD)およびリバース(REV)と称されるこれらのプライマーの配列を表1に挙げる。

融合タンパク質タンデムベクターによるCHO細胞のエレクトロポレーションおよびバイオリアクターでの培養。融合タンパク質タンデムベクター(図12)をPvuIで直線化して、CHO−K1細胞にエレクトロポレーションし、その後G418(375μg/ml)で3週間選択した。96ウェルプレートにおいて、ヒトIgG1 HCおよびヒトカッパLCの両方に対する2種類の1次抗体を使用するヒトIgG ELISAによって、陽性クローンを検出した。導入遺伝子のコピー数が多い細胞系を、MTXを600nMまで段階的に増加させることによって選択した。MTX選択細胞系をT175フラスコ中で増殖させ、次にCHO細胞無血清培地(SFM)の10L量を有する20Lバイオリアクターに移した。図17に示したように、CHO細胞をバイオリアクター中において1000万生細胞/mLを超える高密度で潅流様式によって約50日間維持した。融合タンパク質のこれらの細胞による分泌は、ヒトIgGまたはヒトBDNFのいずれかに対する抗体を使用したELISAによって検出した。図18に示したように、融合タンパク質は、HIRMAb重鎖のカルボキシル末端に対してvBDNFが1:1で融合したものであり、融合タンパク質重鎖が形成される。図18に示したように、重鎖は軽鎖と結合する。したがって、融合タンパク質は、(i)ヒトIgG1 HC、(ii)ヒトカッパLC、または(iii)ヒトBDNFの3種類の抗体と同等によく反応するはずである。図19に示したように、抗ヒトIgG抗体または抗ヒトBDNF抗体のいずれかをELISAで使用することによって、CHO細胞培地における融合タンパク質の測定値は比例している。これらのELISAの結果は、免疫細胞化学(ICC)によって確かめられており、TV−120で形質移入したCHO細胞がヒトIgGまたはヒトBDNFに対する抗体と免疫応答し、BDNF免疫シグナルは組換えBDNFによる抗BDNF抗体の吸収によって除去されることが示された。
バイオリアクターによって生成した融合タンパク質の精製および特徴付け。潅流様式でバイオリアクターから得られた調整培地を1μmフィルターに通過させて、培地を滅菌条件下で200LBioprocess容器に収集し、バイオリアクターに隣接したガラス製扉の冷蔵庫内で4℃に維持した。次に、細胞残渣を除去するために、調整培地200Lのバッチを1μmおよび0.4μmの予備フィルターに通過させた。次いで、この培地をタンジェンシャルフロー濾過(TFF)で濃縮した。TFFシステムは、Millipore社製Pellicon2モデルで、分子量のカットオフ値が30kDaで、総表面積が2.5cm2である0.5m2濾過カセットから構成される。膜間に15PSIの勾配が生じ、2時間以内に200Lから2Lに体積が減少する。濃縮した培地を、ProsepA(Millipore)組換えプロテインAアフィニティーカラム100mLで溶出する前に、0.22μフィルターに通過させた。試料添加後、カラムを緩衝液A(NaCl 0.025M、Tris 0.025M、pH=7.4、EDTA 3mM)で洗浄した。CHO細胞宿主タンパク質(CHOP)の溶出は、Shimadzu検出器でA280でモニターした。融合タンパク質は、クエン酸0.1M(pH3)で溶出し、すぐにpH7まで中和するためにTris塩基を含有した試験管に入れた。中和した酸溶出収集物を、電気伝導率が<7mSになるまで、2回蒸留水で希釈し、この物質をTris、0.02M、pH=7.5で平衡化したセファロースSP陽イオン交換カラム(Amersham)50mLに添加した。Tris緩衝液で洗浄後、残存するCHOPを、NaCl、0から1MのNaCl直線勾配で融合タンパク質から分離した。融合タンパク質ピークを収集し、緩衝液を交換し、分子量カットオフ値30kDaのMilliporeダイアフィルトレーションで濃縮した。最終濃度の抗体溶液を滅菌濾過(0.22μm)し、4℃で保存した。融合タンパク質は、図20に示したように、ドデシル硫酸ナトリウムポリアクリルアミドゲル電気泳動(SDS−PAGE)で均一になるまで精製した。融合タンパク質重鎖の大きさは、54kDaであるHIRMAb重鎖の大きさと比較して68kDaであった。融合タンパク質とHIRMAb重鎖の大きさの違いは、融合タンパク質の各重鎖に融合した付加vBDNF単量体(14kDa)を反映している。この融合タンパク質は、ウェスタンブロッティング上で抗ヒトIgG抗体と抗ヒトBDNF抗体の両方と、予測した免疫反応バンドの分子量の大きさで反応する(図21)。等電点電気泳動(IEF)は、組換えBDNFの導電点(pI)がPI>10で強い陽イオンであることを示している(図22)。融合タンパク質で観察されたpIは8.5で、HIRMAbのpIと近似している(図22)。融合タンパク質で観察されたpI、8.5は、融合タンパク質HCが9.04で、LCが5.27である算出されたpIと矛盾しなかった(http:scansite.mit.edu/)。
融合タンパク質は、2機能性であり、ヒトインシュリン受容体およびヒトtrkB受容体の両方に高い親和性で結合する。融合タンパク質のHIR細胞外ドメイン(ECD)への親和性は、レクチンアフィニティー精製HIR ECDを使用した競合的リガンド結合測定法(CLBA)で測定した。HIR ECDを永久的に形質移入したCHO細胞を無血清培地(SFM)中で増殖させ、HIR ECDをコムギ胚芽凝集素アフィニティーカラムで精製した。HIR ECDをNunc−Maxisorb96ウェル皿に播種し、マウスHIRMAbのHIR ECDに対する結合は、結合測定法のリガンドとして[125I]マウスHIRMAbを添加した後、放射活性測定法によって検出した(図23A)。[125I]マウスHIRMAbのHIR ECDへの結合は、図23Bに示したように、非標識融合タンパク質またはHIRMAbを添加することによって置換された。CLBAは、HIRMAbまたは融合タンパク質と同程度の結合を示す。高親和性および低親和性結合部位モデルを使用したスキャッチャード分析および非線形回帰分析を実施して、融合タンパク質のHIRへの結合の親和定数を決定した。融合タンパク質およびHIRMAbは両方とも、高い親和結合定数、Ki=0.63±0.07nMでHIRに等しく十分に結合する(図23B)。
低酸素症に罹ったヒト神経細胞は、組換えBDNFと同等の活性を備えた融合タンパク質によって神経保護される。ヒトSH−SY5Y神経細胞をレチノイン酸10μMに7日間曝露して、trkB、BDNF受容体の遺伝子発現を誘導した。次に、この細胞を、酸素センサーを備えた密封した容器内で16時間酸素遮断した。その後、興奮毒性の神経損傷を4時間再度酸素添加することによって誘導した(図25A)。この4時間の再酸素添加の間に、細胞は何も処理しないか、等モル濃度のヒト組換えBDNFまたは融合タンパク質に曝露した。図25Bに示したように、この融合タンパク質は、興奮毒性虚血再酸素添加に曝露したヒト神経細胞における神経保護の誘導に関して、天然のヒトBDNFと効力が等しかった。
融合タンパク質の、単離されたヒト脳毛細血管内のヒト血液脳関門インシュリン受容体に対する高い結合親和性。単離されたヒト脳毛細血管は、ヒトBBBのin vitroモデル系として使用されている(図26A)。この融合タンパク質は、3H−N−スクシニミジルプロピオネートで放射標識され、ヒトBBBのHIRに結合する融合タンパク質の放射性受容体測定法(RRA)を確立するためにヒト脳に添加された。[3H]−融合タンパク質は、非標識融合タンパク質によって結合が自己阻害されるので、BBBに特異的に結合している(図26B)。マウスHIRMAb(mHIRMAb)はまた、ヒトBBBに対する[3H]−融合タンパク質の結合を阻害するので、この融合タンパク質は、ヒトBBBのインシュリン受容体に結合される。図26Bの結合データを非線形回折分析でスキャッチャードプロットに当てはめると、結合定数:KD=0.55±0.07nM、Bmax=1.35×0.10pmol/mgp、およびNSB=0.39±0.02pmol/mgpが得られ、KDは解離定数であり、Bmaxは最大結合であり、NSBは不飽和結合である。KDは<1nMで、融合タンパク質がヒトBBB上のHIRに非常に高い親和性で結合することを示している。
成体アカゲザルによる融合タンパク質の薬物動態および脳への取り込み。融合タンパク質を、比活性が2.0μCi/μgになるまで[3H]−N−スクシニミジルプロピオネートでトリチウム標識した。5歳のメスアカゲザル、体重5.2kgに、746μCi(373μg)の用量を1回静脈注射することによって投与し、180分間かけて複数の時点で血清を採取した。麻酔し、一晩絶食させた霊長類の血清グルコースは、実験期間180分の間ずっと一定で、平均72±2mg%で、このことは、HIRMAbをベースにした融合タンパク質の投与が内在性インシュリン受容体の妨害を引き起こさず、糖血症の制御に影響を及ぼさないことを示している。
BNDFおよびBBB分子版トロイの木馬の結合による局所的脳虚血の神経保護。急性脳卒中を治療する神経保護薬を開発するために、数多くの試みが開発されてきた。神経保護薬は、特定の小分子の場合、毒性が強いか、または、薬剤がBBBを通過できないために効力がないので、今までのところ成功していない。BDNFは、齧歯類の実験的脳卒中および局所的脳虚血と同時に脳に直接注射すると、神経保護的である。高分子はBBBを通過しないので、BDNFは、頭蓋骨を貫通して脳に直接注射しなければならない。BBBは、局所脳虚血後早い時間に元通りになり、BDNFはBBBを通過しないので、BDNFのみを静脈内投与しても、虚血した脳において神経保護効果は表れない。BBBを通過してBDNFを輸送するために、ニューロトロフィンをラットトランスフェリン受容体(TfR)に対するマウスMAbに結合させた。このペプチド様MAbはBBBを通過してBDNFを運搬し、結合したBDNF−MAb結合体は、このBDNFはBBBを通過し、脳から血液に入ることができるので、実験的脳卒中に遅延的に静脈内投与した後でも神経保護効果が高い。一旦、脳の内側、BBBの向こう側に入ると、BDNFはそのコグネイト受容体、trkBを活性化し、その後虚血したニューロンにおいて神経保護効果を誘発し、一連のアポトーシス死を停止させる。BDNF−MAb結合体の神経保護効果は、用量応答効果、時間応答効果を示し、7日目の神経保護効果は、BDNF−MAb結合体を1回静脈内投与して1日目の神経保護効果と同一であるので、長期間持続する。例えば、本明細書に全体を参考として援用した、Zhang and Pardridge(2001)Brain Res.889:49〜56、およびZhang and Pardridge(2001)Stroke 32:1378〜1374を参照のこと。BDNFはHIRに対するMAbに融合され、in vitroで両方のヒトBBBに迅速に結合し、in vivoにおいて霊長類BBBを通過して迅速に輸送されるので、この融合タンパク質は、ヒト脳卒中においても神経保護的である。
BNDFおよびBBB分子版トロイの木馬の結合体の全脳虚血における神経保護効果。脳にBDNFを直接注射するとまた、心停止後に生じ得るような一過性前脳虚血(TFI)において神経保護的である。しかし、BDNFはBBBを通過せず、TFI後の早い時間ではBBBは完全であるので、神経保護が可能である場合でも、静脈内BDNFはTFIに神経保護的ではない。反対に、BDNFをラットトランスフェリン受容体(TfR)に対するマウスMAbに結合させると、BBBを通過してBDNFを脳内まで運ぶ分子版トロイの木馬として作用し、静脈内BDNFはTFIにおいて神経保護的であった。成体ラットを脳電図(EEG)が約10分間平坦線を生じるTFIに罹患させた。動物を蘇生し、その後、4種類の異なる治療薬、(a)緩衝液、(b)未結合BDNF、(c)BDNFが結合していない受容体特異的MAb、および(d)BDNF−MAb結合体のうち1種類を静脈内投与した。生理食塩水、未結合BDNFまたはMAb単独で治療した動物の場合、海馬のCA1部分の錐体ニューロンは神経保護されなかった。しかし、BDNF−MAb結合体の場合、遅延型静脈内投与後、CA1錐体ニューロン密度は完全に正常化される。本明細書に全体を参考として援用したWu and Pardridge(199)、PNAS(USA)96:254〜259を参照のこと。これは、BDNFをBBB分子版トロイの木馬に結合させるならば、BDNFを遅延して静脈内投与した後、全脳虚血で強力な神経保護効果があることを示している。BDNFの組換え融合タンパク質および受容体特異的MAbは、脳障害を永久的に防御するために心停止後投与することができた。
ニューロトロフィンが脳細胞を通過できるならば、BDNFは脳および脊髄損傷において神経保護的である。BDNFはBBBを通過しないので、頭蓋骨を貫通してニューロトロフィンを直接注射するならば、BDNFは脳損傷に神経保護的である。BDNFはまた、神経毒による興奮毒性損傷に罹患した脳において神経保護的であり、ヒト免疫不全ウイルス(HIV)−1に感染した脳において神経保護的である。BDNFは急性脊髄損傷においても神経保護的であるが、前脳のBBBと同じように、BDNFは血液脊髄関門を通過しないので、BDNFは脊椎管に直接注入することによって投与しなければならない。これらの場合全てにおいて、BDNFはBBBを通過せず、BBBは損傷後早い時間では脳損傷内において完全であるので、神経保護効果がまだある場合でも、BDNFの静脈投与は神経保護的ではない。反対に、BDNF融合タンパク質は、BDNFをBBB分子版トロイの木馬に融合させ、末梢投与後に血液から脳および脊髄に浸透できるので、静脈投与後のこれらの症状において神経保護的である。
ニューロトロフィンが脳細胞を通過できるならば、BDNFは慢性神経変性症状において神経保護的である。ニューロトロフィンがBBBを通過できないならば、BDNFなどのニューロトロフィンは末梢投与経路のための薬剤として開発することができる。BDNFのキメラHIRMAbへの融合は、プリオン病、アルツハイマー病(AD)、パーキンソン病(PD)、ハンチントン病(HD)、ALS、横断性脊髄炎、運動ニューロン疾患、ピック病、結節性硬化症、リソソーム蓄積症、カナバン病、レット症候群、脊髄小脳失調症、フリードライヒ失調症、視神経萎縮および網膜変性症を含む神経変性疾患ならびに脳加齢の慢性的治療のためにヒトの脳にBDNFの非侵襲的に輸送する新たな取り組みを提案する。
網膜変性および失明における治療薬としてのBDNF。網膜は、脳のように、血液網膜関門(BRB)によって血液から保護されている。インシュリン受容体は、BBBおよびBRBの両方の上で発現し、HIRMAbは、RMTを介してBRBを通過して網膜まで治療薬を輸送することが示された(Zhang他、(2003)Mol.Ther.7:11〜18)。BDNFはBRBを通過しないので、BDNFは、網膜変性において神経保護的であるが、眼球に直接ニューロトロフィンを注射する必要がある。HIRMAbはBRBを通過してBDNFを輸送し、したがって、ニューロトロフィンは血液区画から網膜神経細胞に曝露されるので、融合タンパク質は、静脈注射または皮下注射と同程度の非侵襲的な投与経路で、網膜変性および失明を治療するために使用することができた。
うつ病の治療薬としてのBDNF。うつ病患者の小集団は、BDNFが脳で欠乏している可能性があり、一塩基多形(SNP)と感情障害との関連が報告されている。BDNFを脳に直接注射すると、齧歯類モデルでは持続性のある抗うつ効果がある。ニューロトロフィンはBBBを通過しないので、BDNFは、脳に直接注射しなければならない。融合タンパク質を慢性的に投与することによって、BDNFの脳レベルを上昇させる手段を提供し、うつ病患者および脳BDNFの産生が減少した患者において治療効果があり得る。
IgG融合タンパク質の製造方法。イムノグロブリンG(IgG)遺伝子の真核細胞系への形質移入には一般的に、IgGを構成する重鎖(HC)および軽鎖(LC)をコードする別々のプラスミドを細胞系に同時形質移入することが必要である。IgG融合タンパク質の場合、組換え治療用タンパク質をコードする遺伝子は、HCまたはLC遺伝子のいずれかに融合させることができる。しかし、この同時形質移入の取り組みでは、HCおよびLC両方の融合遺伝子またはHC融合物およびLC遺伝子を平等に高く取り込む細胞系を選択することが困難である。融合タンパク質の製造の好ましい取り組みは、DNAの1本鎖上に、HC融合タンパク質遺伝子、LC遺伝子、選択遺伝子、例えば、ネオ、および増幅遺伝子、例えば、ジヒドロ葉酸レダクターゼ遺伝子を含む必要な遺伝子全てを含有する1本のプラスミドDNAを永久的に形質移入された細胞系を作製することである。図12の融合タンパク質タンデムベクターの図に示したように、HC融合遺伝子、LC遺伝子、ネオ遺伝子およびDHFR遺伝子は全て、別々の、しかしタンデムなプロモーターおよび別々の、しかしタンデムな転写終止配列の制御下にある。したがって、治療用タンパク質およびHCまたはLC IgG遺伝子のいずれかの融合遺伝子を含む遺伝子は全て、宿主細胞ゲノムに平等に組み入れられる。
Claims (58)
- そのアミノ末端で、内在性血液脳関門(BBB)インシュリン受容体を介してBBBを通過し得るヒトインシュリン受容体に対するモノクローナル抗体(HIRMAb)の重鎖カルボキシル末端と共有結合している物質を含む組成物であって、該物質が、配列番号24のアミノ酸466〜582と少なくとも80%同一であるアミノ酸配列を含む脳由来神経栄養因子(BDNF)であり、該組成物が末梢投与後、脳内の該物質の濃度を平均して少なくとも5ng/グラム上昇させ、該物質が受容体と結合して神経保護作用を誘導し、該抗体および該物質がそれぞれ、別々の実体としての活性と比較して、それらの活性の少なくとも30%を保持している、組成物。
- 該物質の分子量が400ダルトンを超える、請求項1記載の組成物。
- 該物質が単独では、末梢投与後の治療有効量でBBBを通過しない、請求項1記載の組成物。
- 該HIRMAbがキメラHIRMAbである、請求項1記載の組成物。
- 該BDNFがヒトBDNFである、請求項1記載の組成物。
- 該HIRMAbの重鎖が、配列番号24のアミノ酸20〜462と少なくとも80%同一である配列を含む、請求項1記載の組成物。
- 該HIRMAbの重鎖と該BDNFとの間にリンカーをさらに含む、請求項6記載の組成物。
- 該リンカーがS−S−Mである、請求項7記載の組成物。
- 該軽鎖が、配列番号36のアミノ酸21〜234と少なくとも80%同一である配列を含む、請求項8記載の組成物。
- 該HIRMAbがグリコシル化されている、請求項9記載の組成物。
- 該物質が配列番号24のアミノ酸466〜582を含む、請求項1記載の組成物。
- 該BDNFが2アミノ酸カルボキシル切断型変種である、請求項1記載の組成物。
- 該BDNFが、配列番号24のアミノ酸466〜582と少なくとも95%同一である配列を含む、請求項1記載の組成物。
- 請求項1記載の組成物および血液脳関門(BBB)を通過する第2の抗体と共有結合している第2の物質を含む第2の組成物を含む組成物。
- 該第1および第2の物質が異なっており、血液脳関門(BBB)を通過する該第1および第2の抗体が同一の抗体である、請求項14記載の組成物。
- 該第2の物質が第2の抗体の重鎖と共有結合している、請求項15記載の組成物。
- 請求項1〜16のいずれか1項に記載の組成物および薬学的に許容される賦形剤を含む医薬組成物。
- そのアミノ末端で、内在性血液脳関門(BBB)インシュリン受容体を介してBBBを通過し得るヒトインシュリン受容体に対するモノクローナル抗体(HIRMAb)の重鎖カルボキシル末端と共有結合している物質を含む組成物であって、該物質が、配列番号24のアミノ酸466〜582の配列と少なくとも80%同一であるアミノ酸配列を含むBDNFであり、該HIRMAbが重鎖および軽鎖を含み、該物質が受容体と結合して神経保護作用を誘導する、組成物。
- 該BDNFが、配列番号24のアミノ酸466〜582と少なくとも95%同一であるアミノ酸配列を含む、請求項18記載の組成物。
- 該BDNFが、配列番号24のアミノ酸466〜582を含む、請求項19記載の組成物。
- 該BDNFが、2アミノ酸カルボキシル末端切断型BDNFである、請求項19記載の組成物。
- 該HIRMAbの重鎖およびBDNFが、融合タンパク質を形成し、該融合タンパク質の配列が、配列番号24のアミノ酸20〜462と少なくとも80%同一である第1の配列および配列番号24のアミノ酸466〜582と少なくとも80%同一である第2の配列を含む、請求項18記載の組成物。
- 該HIRMAbがグリコシル化されている、請求項22記載の組成物。
- 該HIRMAbの軽鎖が、配列番号36のアミノ酸21〜234と少なくとも80%同一である配列を含む、請求項22記載の組成物。
- 該第1の配列のカルボキシル末端と該第2の配列のアミノ末端との間にペプチドリンカーをさらに含む、請求項24記載の組成物。
- 該リンカーがS−S−Mを含む、請求項25記載の組成物。
- 該HIRMAbがグリコシル化されている、請求項26記載の組成物。
- 該抗体および該物質がそれぞれ、平均してそれらの活性の少なくとも40%を保持している、請求項18記載の組成物。
- そのアミノ末端で、内在性血液脳関門(BBB)インシュリン受容体を介してBBBを通過し得るヒトインシュリン受容体に対するモノクローナル抗体(HIRMAb)の重鎖カルボキシル末端と共有結合しているBDNFを含む、神経障害を治療するための組成物であって、該BDNFがTrkB受容体と結合して神経保護作用を誘導するのに有効であり、該共有結合しているBDNFが神経障害を治療するのに有効な量で内在性ヒトインシュリン受容体を介してBBBを通過する、組成物。
- 該抗体および該物質がそれぞれ、平均してそれらの活性の少なくとも40%を保持している、請求項29記載の組成物。
- そのアミノ末端でイムノグロブリンの重鎖カルボキシル末端と共有結合している物質を含む組成物であって、該組成物中の該物質が、該物質単独の血清半減期よりも平均して少なくとも5倍長い血清半減期を有し、該物質が、配列番号24のアミノ酸466〜582と少なくとも80%同一であるアミノ酸配列を含むBDNFであり、該物質が受容体と結合して神経保護作用を誘導し、該イムノグロブリンが内在性血液脳関門(BBB)インシュリン受容体を介してBBBを通過し得るヒトインシュリン受容体に対するモノクローナル抗体(HIRMAb)である、組成物。
- 該抗体および該物質がそれぞれ、平均してそれらの活性の少なくとも40%を保持している、請求項31記載の組成物。
- 該物質が、受容体と結合してヒトにおいて神経保護作用を誘導する、請求項1または31記載の組成物。
- そのアミノ末端で、内在性血液脳関門(BBB)インシュリン受容体を介してBBBを通過し得るヒトインシュリン受容体に対するモノクローナル抗体(HIRMAb)の重鎖カルボキシル末端と共有結合している物質を含む、神経障害を治療するための組成物であって、該物質が、配列番号24のアミノ酸466〜582と少なくとも80%同一であるアミノ酸配列を含むBDNFであり、該物質が受容体と結合して神経保護作用を誘導し、該共有結合している物質が神経障害を治療するのに有効な量でBBBを通過し、該物質および該HIRMAbがそれぞれ、別々の実体としての活性と比較して、平均してそれらの活性の少なくとも30%を保持している、組成物。
- 該HIRMAbが重鎖および軽鎖を含む、請求項34記載の組成物。
- 該組成物中のBDNFが、BDNF単独の血清半減期よりも平均して少なくとも5倍長い血清半減期を有する、請求項35記載の組成物。
- 配列番号24のアミノ酸466〜582と少なくとも80%同一であるアミノ酸配列を含む脳由来神経栄養因子(BDNF)である物質のアミノ末端とその重鎖カルボキシル末端で共有結合している内在性血液脳関門(BBB)インシュリン受容体を介してBBBを通過し得るヒトインシュリン受容体に対するモノクローナル抗体(HIRMAb)を含む融合タンパク質であって、該抗体および該物質がそれぞれ、別々の実体としての活性と比較して、平均してそれらの活性の少なくとも30%を保持しており、該物質が受容体と結合して神経保護作用を誘導する、融合タンパク質。
- 該HIRMAbがキメラHIRMAbである、請求項37記載の融合タンパク質。
- 該BDNFが切断型BDNFである、請求項37記載の融合タンパク質。
- 該切断型BDNFがカルボキシル切断型BDNFである、請求項39記載の融合タンパク質。
- 該カルボキシル切断型BDNFが、2個のカルボキシル末端アミノ酸を欠如している、請求項40記載の融合タンパク質。
- 該抗体および該物質が、ペプチドリンカーによって共有結合している、請求項37記載の融合タンパク質。
- 該HIRMAbが重鎖および軽鎖を含む、請求項37記載の融合タンパク質。
- 該融合タンパク質中のBDNFが、該BDNF単独の血清半減期よりも平均して少なくとも5倍長い血清半減期を有する、請求項37記載の融合タンパク質。
- 該物質が、受容体と結合してヒトにおいて神経保護作用を誘導する、請求項37記載の融合タンパク質。
- BDNFを末梢循環からBBBを通過させて輸送するための医薬の製造のための請求項1記載の組成物の使用。
- 個体におけるCNS障害の治療のための医薬の製造のための請求項1記載の組成物の使用。
- 経口、静脈内、筋肉内、皮下、腹腔内、直腸、頬膜貫通(transbuccal)、鼻腔内、経皮および吸入投与からなる群から選択される投与が適用される、請求項47記載の使用。
- 該投与が、静脈内、筋肉内または皮下投与である、請求項48記載の使用。
- 該CNS障害が急性CNS障害である、請求項47記載の使用。
- 該急性CNS障害が、脊髄損傷、脳損傷局所脳虚血および全脳虚血からなる群から選択される、請求項50記載の使用。
- 該組成物が1回投与される、請求項50記載の使用。
- 該組成物が1週間に約1回以下の頻度で投与される、請求項50記載の使用。
- 該CNS障害が慢性障害である、請求項47記載の使用。
- 該慢性障害が、慢性神経変性疾患、網膜虚血およびうつ病からなる群から選択される、請求項54記載の使用。
- 前記慢性神経変性疾患が、プリオン病、アルツハイマー病、パーキンソン病、筋萎縮性側索硬化症、ハンチントン病、多発硬化症、横断性脊髄炎、運動ニューロン疾患、ピック病、結節性硬化症、リソソーム蓄積症、カナバン病、レット症候群、脊髄小脳失調症、フリードライヒ失調症、視神経萎縮および網膜変性症からなる群から選択される、請求項55記載の使用。
- 該個体がヒトである、請求項47記載の使用。
- 該組成物が、1から100mgの用量で投与される、請求項57記載の使用。
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US11/245,710 US8053569B2 (en) | 2005-10-07 | 2005-10-07 | Nucleic acids encoding and methods of producing fusion proteins |
| US11/245,546 | 2005-10-07 | ||
| US11/245,546 US8142781B2 (en) | 2005-10-07 | 2005-10-07 | Fusion proteins for blood-brain barrier delivery |
| US11/245,710 | 2005-10-07 | ||
| PCT/US2006/038587 WO2007044323A2 (en) | 2005-10-07 | 2006-09-28 | Fusion proteins for blood-brain barrier delivery |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2014116085A Division JP6320179B2 (ja) | 2005-10-07 | 2014-06-04 | 血液脳関門輸送用融合タンパク質 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2009515819A JP2009515819A (ja) | 2009-04-16 |
| JP2009515819A5 JP2009515819A5 (ja) | 2009-11-12 |
| JP5941607B2 true JP5941607B2 (ja) | 2016-06-29 |
Family
ID=37943316
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2008534618A Active JP5941607B2 (ja) | 2005-10-07 | 2006-09-28 | 血液脳関門輸送用融合タンパク質 |
| JP2014116085A Active JP6320179B2 (ja) | 2005-10-07 | 2014-06-04 | 血液脳関門輸送用融合タンパク質 |
| JP2016100640A Active JP6283711B2 (ja) | 2005-10-07 | 2016-05-19 | 血液脳関門輸送用融合タンパク質 |
Family Applications After (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2014116085A Active JP6320179B2 (ja) | 2005-10-07 | 2014-06-04 | 血液脳関門輸送用融合タンパク質 |
| JP2016100640A Active JP6283711B2 (ja) | 2005-10-07 | 2016-05-19 | 血液脳関門輸送用融合タンパク質 |
Country Status (6)
| Country | Link |
|---|---|
| US (2) | US8053569B2 (ja) |
| EP (2) | EP3118218A3 (ja) |
| JP (3) | JP5941607B2 (ja) |
| AU (1) | AU2006302589A1 (ja) |
| CA (1) | CA2625293C (ja) |
| WO (1) | WO2007044323A2 (ja) |
Families Citing this family (95)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7388079B2 (en) * | 2002-11-27 | 2008-06-17 | The Regents Of The University Of California | Delivery of pharmaceutical agents via the human insulin receptor |
| ES2368941T3 (es) | 2003-01-06 | 2011-11-23 | Angiochem Inc. | Angiopep-1, compuestos relacionados y utilizaciones correspondientes. |
| US7485706B2 (en) * | 2003-07-30 | 2009-02-03 | The Board Of Trustees Of The Leland Stanford Junior University | Neurodegenerative protein aggregation inhibition methods and compounds |
| BRPI0613005A8 (pt) | 2005-07-15 | 2017-12-26 | Angiochem Inc | uso de polipeptídeos de aprotinina como veículos em conjugados farmacêuticos. |
| US8741260B2 (en) * | 2005-10-07 | 2014-06-03 | Armagen Technologies, Inc. | Fusion proteins for delivery of GDNF to the CNS |
| US8053569B2 (en) * | 2005-10-07 | 2011-11-08 | Armagen Technologies, Inc. | Nucleic acids encoding and methods of producing fusion proteins |
| US8124095B2 (en) * | 2005-10-07 | 2012-02-28 | Armagen Technologies, Inc. | Fusion proteins for delivery of erythropoietin to the CNS |
| US7981417B2 (en) * | 2006-06-07 | 2011-07-19 | Wisconsin Alumni Research Foundation | Blood-brain barrier targeting anti-bodies |
| US8497246B2 (en) * | 2006-08-18 | 2013-07-30 | Armagen Technologies, Inc. | Methods for diagnosing and treating CNS disorders by trans-blood-brain barrier delivery of protein compositions |
| US20080200441A1 (en) * | 2007-02-14 | 2008-08-21 | University Of Southern California | Estrogen receptor modulators associated pharmaceutical compositions and methods of use |
| US20090011040A1 (en) * | 2007-05-02 | 2009-01-08 | Naash Muna I | Use of compacted nucleic acid nanoparticles in non-viral treatments of ocular diseases |
| US9365634B2 (en) | 2007-05-29 | 2016-06-14 | Angiochem Inc. | Aprotinin-like polypeptides for delivering agents conjugated thereto to tissues |
| US20100197012A1 (en) * | 2007-06-05 | 2010-08-05 | National Tsing Hua University | Application of RNA Interference Targeting dhfr Gene, to Cell for Producing Secretory Protein |
| JP5901877B2 (ja) * | 2007-07-27 | 2016-04-13 | アーメイゲン・テクノロジーズ・インコーポレイテッドArmagen Technologies, Inc. | 中枢神経系のα−L−イデュロニダーゼ活性を増加させるための方法および組成物 |
| MX355683B (es) | 2008-04-18 | 2018-04-26 | Angiochem Inc | Composiciones farmacéuticas de paclitaxel, análogos de paclitaxel o conjugados de paclitaxel, y métodos relacionados de preparación y uso. |
| JP5705118B2 (ja) | 2008-10-15 | 2015-04-22 | アンジオケム インコーポレーテッド | 薬物送達のためのエトポシドおよびドキソルビシン複合体 |
| AU2009304560A1 (en) | 2008-10-15 | 2010-04-22 | Angiochem Inc. | Conjugates of GLP-1 agonists and uses thereof |
| AU2009322043A1 (en) | 2008-12-05 | 2011-07-07 | Angiochem Inc. | Conjugates of neurotensin or neurotensin analogs and uses thereof |
| CA2747220A1 (en) | 2008-12-17 | 2010-06-24 | Richard Beliveau | Membrane type-1 matrix metalloprotein inhibitors and uses thereof |
| WO2010108048A2 (en) * | 2009-03-18 | 2010-09-23 | Armagen Technologies, Inc. | Compositions and methods for blood-brain barrier delivery of igg-decoy receptor fusion proteins |
| KR101456326B1 (ko) | 2009-04-07 | 2014-11-12 | 로슈 글리카트 아게 | 3가, 이중특이적 항체 |
| WO2010121379A1 (en) | 2009-04-20 | 2010-10-28 | Angiochem Inc | Treatment of ovarian cancer using an anticancer agent conjugated to an angiopep-2 analog |
| CN102459348A (zh) * | 2009-06-11 | 2012-05-16 | 安吉奥开米公司 | 用于将gdnf和bdnf递送至中枢神经系统的融合蛋白 |
| US9676845B2 (en) | 2009-06-16 | 2017-06-13 | Hoffmann-La Roche, Inc. | Bispecific antigen binding proteins |
| JP5932642B2 (ja) | 2009-07-02 | 2016-06-08 | アンジオケム インコーポレーテッド | 多量体ペプチドコンジュゲートおよびその使用 |
| US20120244147A1 (en) | 2009-08-17 | 2012-09-27 | Theuer Charles P | Combination therapy of cancer with anti-endoglin antibodies and anti-vegf agents |
| US8221753B2 (en) | 2009-09-30 | 2012-07-17 | Tracon Pharmaceuticals, Inc. | Endoglin antibodies |
| PT2485761T (pt) | 2009-10-09 | 2019-05-30 | Armagen Inc | Métodos e composições para aumentar a atividade da iduronato 2-sulfatase no snc |
| US20130079382A1 (en) | 2009-10-12 | 2013-03-28 | Larry J. Smith | Methods and Compositions for Modulating Gene Expression Using Oligonucleotide Based Drugs Administered in vivo or in vitro |
| AU2011258301B2 (en) * | 2010-05-27 | 2013-11-21 | Janssen Biotech Inc. | Insulin-like growth factor 1 receptor binding peptides |
| EP2603221A4 (en) * | 2010-08-13 | 2014-02-26 | Univ Georgetown | GGF2 AND METHOD FOR ITS APPLICATION |
| PL2646470T3 (pl) | 2010-11-30 | 2017-07-31 | F.Hoffmann-La Roche Ag | Przeciwciała przeciw receptorowi transferyny o niskim powinowactwie oraz ich zastosowanie do przenoszenia terapeutycznego scfv przez barierę krew-mózg |
| US9539324B2 (en) | 2010-12-01 | 2017-01-10 | Alderbio Holdings, Llc | Methods of preventing inflammation and treating pain using anti-NGF compositions |
| US9067988B2 (en) | 2010-12-01 | 2015-06-30 | Alderbio Holdings Llc | Methods of preventing or treating pain using anti-NGF antibodies |
| US8728473B2 (en) | 2010-12-01 | 2014-05-20 | Alderbio Holdings Llc | Methods of preventing or treating pain using anti-NGF antibodies |
| US11214610B2 (en) | 2010-12-01 | 2022-01-04 | H. Lundbeck A/S | High-purity production of multi-subunit proteins such as antibodies in transformed microbes such as Pichia pastoris |
| US9078878B2 (en) | 2010-12-01 | 2015-07-14 | Alderbio Holdings Llc | Anti-NGF antibodies that selectively inhibit the association of NGF with TrkA, without affecting the association of NGF with p75 |
| US9884909B2 (en) | 2010-12-01 | 2018-02-06 | Alderbio Holdings Llc | Anti-NGF compositions and use thereof |
| KR101572338B1 (ko) | 2011-02-28 | 2015-11-26 | 에프. 호프만-라 로슈 아게 | 1가 항원 결합 단백질 |
| ES2549638T3 (es) | 2011-02-28 | 2015-10-30 | F. Hoffmann-La Roche Ag | Proteínas de unión a antígeno |
| US10227387B2 (en) * | 2011-05-18 | 2019-03-12 | Children's Hospital Medical Center | Targeted delivery of proteins across the blood-brain barrier |
| ES2714999T3 (es) * | 2011-09-24 | 2019-05-31 | Csl Behring Gmbh | Terapia de combinación que usa inmunoglobulina e inhibidor de C1 |
| AU2012346448B2 (en) | 2011-12-02 | 2017-09-14 | Armagen, Inc. | Methods and compositions for increasing arylsulfatase A activity in the CNS |
| WO2013177062A2 (en) | 2012-05-21 | 2013-11-28 | Genentech, Inc. | Methods for improving safety of blood-brain barrier transport |
| US9790264B2 (en) | 2012-06-25 | 2017-10-17 | The Brigham And Women's Hospital, Inc. | Compounds and methods for modulating pharmacokinetics |
| HUE035607T2 (en) | 2012-06-25 | 2018-05-28 | Brigham & Womens Hospital Inc | Targeted therapeutic agents |
| JP2015526434A (ja) | 2012-08-14 | 2015-09-10 | アンジオケム インコーポレーテッド | ペプチド−デンドリマーコンジュゲート及びその使用 |
| JO3462B1 (ar) | 2012-08-22 | 2020-07-05 | Regeneron Pharma | أجسام مضادة بشرية تجاه gfr?3 وطرق لاستخدامها |
| UA115789C2 (uk) | 2012-09-05 | 2017-12-26 | Трейкон Фармасутікалз, Інк. | Композиція антитіла до cd105 та її застосування |
| CN105263514B (zh) | 2013-03-15 | 2019-04-26 | 本质生命科学有限公司 | 抗铁调素抗体及其用途 |
| PE20151926A1 (es) | 2013-05-20 | 2016-01-07 | Genentech Inc | Anticuerpos de receptores de antitransferrina y metodos de uso |
| US10906981B2 (en) * | 2013-07-19 | 2021-02-02 | The Regents Of The University Of California | Compositions and methods related to structures that cross the blood brain barrier |
| WO2015031673A2 (en) | 2013-08-28 | 2015-03-05 | Bioasis Technologies Inc. | Cns-targeted conjugates having modified fc regions and methods of use thereof |
| WO2015091130A1 (en) * | 2013-12-20 | 2015-06-25 | F. Hoffmann-La Roche Ag | Method for improving the recombinant production of soluble fusion polypeptides |
| KR20160098277A (ko) * | 2013-12-20 | 2016-08-18 | 에프. 호프만-라 로슈 아게 | 개선된 재조합 폴리펩티드 제조 방법 |
| BR112016015589A2 (pt) * | 2014-01-06 | 2017-10-31 | Hoffmann La Roche | módulos de trânsito monovalentes para a barreira hematoencefálica |
| EP3197915A4 (en) | 2014-09-22 | 2018-12-19 | Intrinsic Lifesciences LLC | Humanized anti-hepcidin antibodies and uses thereof |
| US9926375B2 (en) | 2014-11-12 | 2018-03-27 | Tracon Pharmaceuticals, Inc. | Anti-endoglin antibodies and uses thereof |
| WO2016077451A1 (en) | 2014-11-12 | 2016-05-19 | Tracon Pharmaceuticals, Inc. | Anti-endoglin antibodies and uses thereof |
| JP6993228B2 (ja) | 2014-11-19 | 2022-03-03 | ジェネンテック, インコーポレイテッド | 抗トランスフェリン受容体/抗bace1多重特異性抗体および使用方法 |
| US10508151B2 (en) | 2014-11-19 | 2019-12-17 | Genentech, Inc. | Anti-transferrin receptor antibodies and methods of use |
| CN107001482B (zh) | 2014-12-03 | 2021-06-15 | 豪夫迈·罗氏有限公司 | 多特异性抗体 |
| US10538589B2 (en) | 2015-01-14 | 2020-01-21 | Armagen Inc. | Methods and compositions for increasing N-acetylglucosaminidase (NAGLU) activity in the CNS using a fusion antibody comprising an anti-human insulin receptor antibody and NAGLU |
| DK3307326T3 (da) | 2015-06-15 | 2020-10-19 | Angiochem Inc | Fremgangsmåder til behandling af leptomeningeal karcinomatose |
| EA039366B1 (ru) | 2015-06-24 | 2022-01-19 | Джей Си Ар ФАРМАСЬЮТИКАЛЗ КО., ЛТД. | Антитело к рецептору трансферрина человека, проникающее через гематоэнцефалический барьер |
| RS62986B1 (sr) | 2015-06-24 | 2022-03-31 | Hoffmann La Roche | Antitela na transferinski receptor sa prilagođenim afinitetom |
| CN107849150B (zh) | 2015-06-24 | 2021-12-14 | Jcr制药股份有限公司 | 含有bdnf的融合蛋白 |
| AR106189A1 (es) | 2015-10-02 | 2017-12-20 | Hoffmann La Roche | ANTICUERPOS BIESPECÍFICOS CONTRA EL A-b HUMANO Y EL RECEPTOR DE TRANSFERRINA HUMANO Y MÉTODOS DE USO |
| MA43023A (fr) | 2015-10-02 | 2018-08-08 | Hoffmann La Roche | Anticorps de récepteur de la transferrine humaine/anti-humaine cd20 bispécifique et leurs procédés d'utilisation |
| US11752315B1 (en) | 2016-10-07 | 2023-09-12 | Carlos A. Hakim | Method of treating normal pressure hydrocephalus |
| CN110621774A (zh) * | 2016-10-14 | 2019-12-27 | 儿童医疗中心有限公司 | 用于治疗中枢神经系统疾病和病症的组合物和方法 |
| CA3052936A1 (en) | 2016-12-26 | 2018-07-05 | Jcr Pharmaceuticals Co., Ltd. | Fusion protein of bdnf and anti-transferrin receptor antibody |
| NZ752703A (en) | 2016-12-26 | 2022-09-30 | Japan Chem Res | Novel anti-human transferrin receptor antibody capable of penetrating blood-brain barrier |
| US12000843B2 (en) | 2017-01-17 | 2024-06-04 | Children's Medical Center Corporation | Compositions and methods for diagnosing and treating peroxisomal diseases |
| CN108467428A (zh) * | 2018-03-26 | 2018-08-31 | 江苏中新医药有限公司 | 一种清除rhNGF中N端截短及异常变异体的方法 |
| MX2020010740A (es) | 2018-04-11 | 2021-01-20 | Salubris Biotherapeutics Inc | Composiciones de proteina de fusion recombinante neuregulina-1 (nrg-1) humana y metodos para su uso. |
| JP7532338B2 (ja) * | 2018-08-07 | 2024-08-13 | アーマジェン・インコーポレイテッド | ヘキソサミニダーゼa、酸スフィンゴミエリナーゼおよびパルミトイル-タンパク質チオエステラーゼ1のcnsにおける活性を増加させるための方法および組成物 |
| US20220041673A1 (en) * | 2019-01-03 | 2022-02-10 | The Board Of Trustees Of The University Of Illinois | Method and kit for treating a neurodegenerative disease |
| KR20220035452A (ko) | 2019-07-19 | 2022-03-22 | 온코리스폰스 인크. | 면역조절 항체 및 이의 사용 방법 |
| EP4013877A1 (en) | 2019-08-14 | 2022-06-22 | Codiak BioSciences, Inc. | Extracellular vesicle-aso constructs targeting stat6 |
| CA3147366A1 (en) | 2019-08-14 | 2021-02-18 | Adam T. BOUTIN | Extracellular vesicles with stat3-antisense oligonucleotides |
| CA3147365A1 (en) | 2019-08-14 | 2021-02-18 | Joanne LIM | Extracellular vesicle-nlrp3 antagonist |
| BR112022002690A2 (pt) | 2019-08-14 | 2022-08-23 | Codiak Biosciences Inc | Construtos de vesícula-aso extracelular tendo como alvo cebp/beta |
| JP2022544290A (ja) | 2019-08-14 | 2022-10-17 | コディアック バイオサイエンシーズ, インコーポレイテッド | Krasを標的とするアンチセンスオリゴヌクレオチドを有する細胞外小胞 |
| WO2021062058A1 (en) | 2019-09-25 | 2021-04-01 | Codiak Biosciences, Inc. | Sting agonist comprising exosomes for treating neuroimmunological disorders |
| WO2021184021A1 (en) | 2020-03-13 | 2021-09-16 | Codiak Biosciences, Inc. | Extracellular vesicle-aso constructs targeting pmp22 |
| WO2021184020A1 (en) | 2020-03-13 | 2021-09-16 | Codiak Biosciences, Inc. | Methods of treating neuroinflammation |
| WO2021248133A1 (en) | 2020-06-05 | 2021-12-09 | Codiak Biosciences, Inc. | Anti-transferrin extracellular vesicles |
| CA3128035A1 (en) | 2020-08-13 | 2022-02-13 | Bioasis Technologies, Inc. | Combination therapies for delivery across the blood brain barrier |
| WO2022153957A1 (ja) | 2021-01-12 | 2022-07-21 | Jcrファーマ株式会社 | リガンドと生理活性を有する蛋白質の融合蛋白質をコードする遺伝子が組み込まれた核酸分子 |
| IL305171A (en) | 2021-02-17 | 2023-10-01 | Lonza Sales Ag | EXTRACELLULAR VESILE-NLRP3 antagonist |
| CN112851759B (zh) * | 2021-02-23 | 2022-12-27 | 李婧炜 | 噬菌体展示技术筛选可穿越血-脑脊液屏障的多肽 |
| IL312864A (en) | 2021-11-19 | 2024-07-01 | Japan Chem Res | Peptide having affinity for human transferrin receptor |
| EP4455290A1 (en) | 2021-12-28 | 2024-10-30 | JCR Pharmaceuticals Co., Ltd. | Fusion protein of anti-transferrin receptor antibody and bioactive protein for safe gene therapy |
| WO2024080305A1 (ja) * | 2022-10-11 | 2024-04-18 | Jcrファーマ株式会社 | 血清アルブミンと生理活性を有する蛋白質との融合蛋白質 |
Family Cites Families (51)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5618920A (en) * | 1985-11-01 | 1997-04-08 | Xoma Corporation | Modular assembly of antibody genes, antibodies prepared thereby and use |
| US4902505A (en) * | 1986-07-30 | 1990-02-20 | Alkermes | Chimeric peptides for neuropeptide delivery through the blood-brain barrier |
| US4801575A (en) | 1986-07-30 | 1989-01-31 | The Regents Of The University Of California | Chimeric peptides for neuropeptide delivery through the blood-brain barrier |
| US5530101A (en) * | 1988-12-28 | 1996-06-25 | Protein Design Labs, Inc. | Humanized immunoglobulins |
| US5229500A (en) | 1989-08-30 | 1993-07-20 | Regeneron Pharmaceuticals, Inc. | Brain derived neurotrophic factor |
| US5180820A (en) * | 1989-08-30 | 1993-01-19 | Barde Yves Alain | Brain-derived neurotrophic factor |
| US5672683A (en) * | 1989-09-07 | 1997-09-30 | Alkermes, Inc. | Transferrin neuropharmaceutical agent fusion protein |
| US5977307A (en) | 1989-09-07 | 1999-11-02 | Alkermes, Inc. | Transferrin receptor specific ligand-neuropharmaceutical agent fusion proteins |
| US6329508B1 (en) * | 1989-09-07 | 2001-12-11 | Alkermes, Inc. | Transferrin receptor reactive chimeric antibodies |
| US5154924A (en) * | 1989-09-07 | 1992-10-13 | Alkermes, Inc. | Transferrin receptor specific antibody-neuropharmaceutical agent conjugates |
| US5527288A (en) * | 1990-12-13 | 1996-06-18 | Elan Medical Technologies Limited | Intradermal drug delivery device and method for intradermal delivery of drugs |
| TW279133B (ja) | 1990-12-13 | 1996-06-21 | Elan Med Tech | |
| US6060069A (en) * | 1991-05-20 | 2000-05-09 | Dura Pharmaceuticals, Inc. | Pulmonary delivery of pharmaceuticals |
| US6287792B1 (en) * | 1991-06-17 | 2001-09-11 | The Regents Of The University Of California | Drug delivery of antisense oligonucleotides and peptides to tissues in vivo and to cells using avidin-biotin technology |
| GEP20002243B (en) | 1991-09-20 | 2000-09-25 | Amgen Inc | Glial Derived Neurotrophic Factor |
| JPH06228199A (ja) * | 1992-11-27 | 1994-08-16 | Takeda Chem Ind Ltd | 血液脳関門通過可能なペプチド結合体 |
| US5997501A (en) | 1993-11-18 | 1999-12-07 | Elan Corporation, Plc | Intradermal drug delivery device |
| EP0706799B1 (en) * | 1994-09-16 | 2001-11-14 | MERCK PATENT GmbH | Immunoconjugates II |
| US5656284A (en) * | 1995-04-24 | 1997-08-12 | Balkin; Michael S. | Oral transmucosal delivery tablet and method of making it |
| WO1997010806A1 (en) | 1995-09-19 | 1997-03-27 | Fujisawa Pharmaceutical Co., Ltd. | Aerosol compositions |
| US5837231A (en) | 1996-06-27 | 1998-11-17 | Regents Of The University Of Minnesota | GM-CSF administration for the treatment and prevention of recurrence of brain tumors |
| WO1999000150A2 (en) * | 1997-06-27 | 1999-01-07 | Regents Of The University Of California | Drug targeting of a peptide radiopharmaceutical through the primate blood-brain barrier in vivo with a monoclonal antibody to the human insulin receptor |
| US6482923B1 (en) | 1997-09-17 | 2002-11-19 | Human Genome Sciences, Inc. | Interleukin 17-like receptor protein |
| US6016800A (en) * | 1997-10-24 | 2000-01-25 | Century; Theodore J. | Intrapulmonary aerosolizer |
| US6743427B1 (en) | 1997-12-02 | 2004-06-01 | Neuralab Limited | Prevention and treatment of amyloidogenic disease |
| ES2314995T3 (es) | 1997-12-11 | 2009-03-16 | Alza Corporation | Dispositivo para mejorar el flujo de agentes tansdermicos. |
| US6348210B1 (en) * | 1998-11-13 | 2002-02-19 | Alza Corporation | Methods for transdermal drug administration |
| US6375975B1 (en) | 1998-12-21 | 2002-04-23 | Generex Pharmaceuticals Incorporated | Pharmaceutical compositions for buccal and pulmonary application |
| US6284262B1 (en) | 1999-01-26 | 2001-09-04 | Virgil A. Place | Compact dosage unit for buccal administration of a pharmacologically active agent |
| US7309687B1 (en) * | 1999-04-13 | 2007-12-18 | The Kenneth S. Warren Institute, Inc. | Methods for treatment and prevention of neuromuscular and muscular conditions by peripherally administered erythropoietin |
| CZ299516B6 (cs) * | 1999-07-02 | 2008-08-20 | F. Hoffmann-La Roche Ag | Konjugát erythropoetinového glykoproteinu, zpusobjeho výroby a použití a farmaceutická kompozice sjeho obsahem |
| US6372250B1 (en) * | 2000-04-25 | 2002-04-16 | The Regents Of The University Of California | Non-invasive gene targeting to the brain |
| US6696274B2 (en) | 2000-05-03 | 2004-02-24 | Supratek Pharma, Inc. | Ligand for enhancing oral and CNS delivery of biological agents |
| US7078376B1 (en) * | 2000-08-11 | 2006-07-18 | Baxter Healthcare S.A. | Therapeutic methods for treating subjects with a recombinant erythropoietin having high activity and reduced side effects |
| WO2002055736A2 (en) * | 2000-12-04 | 2002-07-18 | Univ California | Antisense imaging of gene expression of the brain in vivo |
| BR0207016A (pt) * | 2001-02-06 | 2004-02-03 | Merck Patent Gmbh | Fator neurotrófico derivado de cérebro humano modificado (bdnf) com imunogenicidade reduzida |
| EP1554572B1 (en) * | 2001-07-25 | 2009-10-14 | Raptor Pharmaceutical Inc. | Compositions and methods for modulating blood-brain barrier transport |
| US7053202B2 (en) * | 2001-10-19 | 2006-05-30 | Millennium Pharmaceuticals, Inc. | Immunoglobulin DNA cassette molecules, monobody constructs, methods of production, and methods of use therefor |
| JP4219152B2 (ja) * | 2002-10-31 | 2009-02-04 | ヤマハマリン株式会社 | 船舶推進機におけるオイル交換時期警報装置 |
| US6850492B2 (en) * | 2002-11-22 | 2005-02-01 | Nokia Corporation | Method and system for enabling a route and flow table update in a distributed routing platform |
| US20040102369A1 (en) | 2002-11-27 | 2004-05-27 | The Regents Of The University Of California | Transport of basic fibroblast growth factor across the blood brain barrier |
| US20050142141A1 (en) * | 2002-11-27 | 2005-06-30 | Pardridge William M. | Delivery of enzymes to the brain |
| US7388079B2 (en) * | 2002-11-27 | 2008-06-17 | The Regents Of The University Of California | Delivery of pharmaceutical agents via the human insulin receptor |
| US7294704B2 (en) * | 2003-08-15 | 2007-11-13 | Diadexus, Inc. | Pro108 antibody compositions and methods of use and use of Pro108 to assess cancer risk |
| EP1548031A1 (en) | 2003-12-22 | 2005-06-29 | Dubai Genetics FZ-LLC | Nature-identical erythropoietin |
| US20060222668A1 (en) * | 2005-04-01 | 2006-10-05 | Wellspring Pharmaceutical Corporation | Stannsoporfin compositions, drug products and methods of manufacture |
| US8053569B2 (en) | 2005-10-07 | 2011-11-08 | Armagen Technologies, Inc. | Nucleic acids encoding and methods of producing fusion proteins |
| US8124095B2 (en) | 2005-10-07 | 2012-02-28 | Armagen Technologies, Inc. | Fusion proteins for delivery of erythropoietin to the CNS |
| US8741260B2 (en) * | 2005-10-07 | 2014-06-03 | Armagen Technologies, Inc. | Fusion proteins for delivery of GDNF to the CNS |
| KR101248252B1 (ko) * | 2006-11-28 | 2013-03-27 | 한올바이오파마주식회사 | 변형된 에리스로포이에틴 폴리펩티드와 이의 치료용 용도 |
| JP6083219B2 (ja) * | 2012-12-06 | 2017-02-22 | 富士通セミコンダクター株式会社 | プラズマフラッドガン及びイオン注入装置 |
-
2005
- 2005-10-07 US US11/245,710 patent/US8053569B2/en active Active
- 2005-10-07 US US11/245,546 patent/US8142781B2/en active Active
-
2006
- 2006-09-28 EP EP16171496.9A patent/EP3118218A3/en not_active Withdrawn
- 2006-09-28 WO PCT/US2006/038587 patent/WO2007044323A2/en active Application Filing
- 2006-09-28 AU AU2006302589A patent/AU2006302589A1/en not_active Abandoned
- 2006-09-28 EP EP06825389A patent/EP1945259A4/en not_active Ceased
- 2006-09-28 CA CA2625293A patent/CA2625293C/en active Active
- 2006-09-28 JP JP2008534618A patent/JP5941607B2/ja active Active
-
2014
- 2014-06-04 JP JP2014116085A patent/JP6320179B2/ja active Active
-
2016
- 2016-05-19 JP JP2016100640A patent/JP6283711B2/ja active Active
Also Published As
| Publication number | Publication date |
|---|---|
| EP1945259A2 (en) | 2008-07-23 |
| AU2006302589A1 (en) | 2007-04-19 |
| CA2625293C (en) | 2016-10-18 |
| JP2009515819A (ja) | 2009-04-16 |
| US8142781B2 (en) | 2012-03-27 |
| EP1945259A4 (en) | 2010-03-24 |
| JP2014221762A (ja) | 2014-11-27 |
| WO2007044323A2 (en) | 2007-04-19 |
| EP3118218A2 (en) | 2017-01-18 |
| US20070082380A1 (en) | 2007-04-12 |
| CA2625293A1 (en) | 2007-04-19 |
| JP6283711B2 (ja) | 2018-02-21 |
| US8053569B2 (en) | 2011-11-08 |
| US20070081992A1 (en) | 2007-04-12 |
| EP3118218A3 (en) | 2017-03-15 |
| JP2016196469A (ja) | 2016-11-24 |
| JP6320179B2 (ja) | 2018-05-09 |
| WO2007044323A3 (en) | 2009-05-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6283711B2 (ja) | 血液脳関門輸送用融合タンパク質 | |
| US20200254113A1 (en) | Fusion proteins for delivery of gdnf to the cns | |
| US8124095B2 (en) | Fusion proteins for delivery of erythropoietin to the CNS | |
| US20230279156A1 (en) | Methods and compositions for increasing alpha-l-iduronidase activity in the cns | |
| US20100077498A1 (en) | Compositions and methods for blood-brain barrier delivery in the mouse | |
| JP5873003B2 (ja) | IgGデコイ受容体融合タンパク質の血液脳関門送達のための組成物および方法 | |
| AU2011205186B2 (en) | Fusion proteins for blood-brain barrier delivery | |
| AU2013206657A1 (en) | Methods and compositions for increasing alpha-iduronidase activity in the CNS |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20090925 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20090925 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20120313 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20120613 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20130129 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20130426 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20130508 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20130528 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20130604 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20130619 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20130626 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20130729 |
|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20140120 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20140204 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20140604 |
|
| A911 | Transfer to examiner for re-examination before appeal (zenchi) |
Free format text: JAPANESE INTERMEDIATE CODE: A911 Effective date: 20140730 |
|
| A912 | Re-examination (zenchi) completed and case transferred to appeal board |
Free format text: JAPANESE INTERMEDIATE CODE: A912 Effective date: 20140822 |
|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20141215 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20150417 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20150519 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20150619 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20151228 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20160125 |
|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20160219 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20160229 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20160328 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20160523 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5941607 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |