JP5919192B2 - βアミノ酸の調製 - Google Patents
βアミノ酸の調製 Download PDFInfo
- Publication number
- JP5919192B2 JP5919192B2 JP2012528394A JP2012528394A JP5919192B2 JP 5919192 B2 JP5919192 B2 JP 5919192B2 JP 2012528394 A JP2012528394 A JP 2012528394A JP 2012528394 A JP2012528394 A JP 2012528394A JP 5919192 B2 JP5919192 B2 JP 5919192B2
- Authority
- JP
- Japan
- Prior art keywords
- group
- reaction
- hydantoinase
- enzyme
- amino acid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 0 *C(C(*)N)C(O)=O Chemical compound *C(C(*)N)C(O)=O 0.000 description 1
Images












Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/78—Hydrolases (3) acting on carbon to nitrogen bonds other than peptide bonds (3.5)
- C12N9/86—Hydrolases (3) acting on carbon to nitrogen bonds other than peptide bonds (3.5) acting on amide bonds in cyclic amides, e.g. penicillinase (3.5.2)
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/10—Antimycotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/78—Hydrolases (3) acting on carbon to nitrogen bonds other than peptide bonds (3.5)
- C12N9/80—Hydrolases (3) acting on carbon to nitrogen bonds other than peptide bonds (3.5) acting on amide bonds in linear amides (3.5.1)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P13/00—Preparation of nitrogen-containing organic compounds
- C12P13/001—Amines; Imines
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P13/00—Preparation of nitrogen-containing organic compounds
- C12P13/005—Amino acids other than alpha- or beta amino acids, e.g. gamma amino acids
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P13/00—Preparation of nitrogen-containing organic compounds
- C12P13/02—Amides, e.g. chloramphenicol or polyamides; Imides or polyimides; Urethanes, i.e. compounds comprising N-C=O structural element or polyurethanes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P13/00—Preparation of nitrogen-containing organic compounds
- C12P13/04—Alpha- or beta- amino acids
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y305/00—Hydrolases acting on carbon-nitrogen bonds, other than peptide bonds (3.5)
- C12Y305/01—Hydrolases acting on carbon-nitrogen bonds, other than peptide bonds (3.5) in linear amides (3.5.1)
- C12Y305/01087—N-Carbamoyl-L-amino-acid hydrolase (3.5.1.87)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y305/00—Hydrolases acting on carbon-nitrogen bonds, other than peptide bonds (3.5)
- C12Y305/02—Hydrolases acting on carbon-nitrogen bonds, other than peptide bonds (3.5) in cyclic amides (3.5.2)
- C12Y305/02002—Dihydropyrimidinase (3.5.2.2), i.e. hydantoinase
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y305/00—Hydrolases acting on carbon-nitrogen bonds, other than peptide bonds (3.5)
- C12Y305/02—Hydrolases acting on carbon-nitrogen bonds, other than peptide bonds (3.5) in cyclic amides (3.5.2)
- C12Y305/02014—N-Methylhydantoinase (ATP-hydrolyzing) (3.5.2.14)
Description

第1の態様において、本発明の目的は、βアミノ酸前駆体を、生体触媒によって立体特異的に、より詳細にはエナンチオ選択的に生産するためのプロセスによって解決されたが、該プロセスは、少なくとも1つの一般式(I)
置換されていてもよい、直鎖もしくは分岐鎖低級アルキル基;
置換されていてもよい、直鎖もしくは分岐鎖低級アルケニル基;
置換されていてもよい、環状アルキル基;
置換されていてもよい、単環式もしくは多環式アリール基;
置換されていてもよい、単環式もしくは多環式ヘテロアリール基;
置換されていてもよい、直鎖もしくは分岐鎖アルコキシ基;
アミノ基;
置換されていてもよい、直鎖もしくは分岐鎖アルキルアミノ基;
置換されていてもよい、直鎖もしくは分岐鎖アルキルチオ基;
置換されていてもよい、直鎖もしくは分岐鎖アシル基;
カルボキシル基または
アルデヒド基
から選択される]
で表される基質であって、
立体異性として純粋な形態、たとえば(R)-もしくは(S)-異性体、または立体異性体の混合物、たとえばラセミ混合物、
または前記化合物の塩、たとえば酸付加塩;
の形態をとる前記基質を、
ヒダントインおよび/またはジヒドロピリミジン環の加水分解を触媒する、少なくとも1つの酵素、具体的にはヒダントイナーゼおよびジヒドロピリミジナーゼから選択されるが、より詳細にはヒダントイナーゼであって、特に、変換すべき化合物の特定の異性体に優先傾向を有する酵素の存在下で;ならびに、必要に応じて、一般式(I)の前記化合物の立体異性体を相互転換する能力をもつ少なくとも1つの酵素の存在下で、反応させ、
その結果、一般式(II)のβアミノ酸前駆体
を生成すること;
を含んでなるプロセスであって、
前記プロセスは、ヒダントインおよび/またはジヒドロピリミジン環の加水分解を触媒する少なくとも1つの酵素、具体的にはヒダントイナーゼおよび/またはジヒドロピリミジナーゼが、アズキ(Vigna angularis)から得られること、および/または、下記の部分配列の少なくとも1つに対して約60から100%の同一性を有する、少なくとも1つの部分配列を含有すること、
ITGPEGQRLAGP (配列番号7)
IELGITGPEGQRLAGPTVL (配列番号1)
IELGITGPEGQRLAGPVL (配列番号2)
IELITGPEGQRLAGPTVL (配列番号3)
IELITGPEGQRLAGPVL (配列番号4)
EEIARARKSGQRVIGEPVAS (配列番号5)、
たとえば、配列番号5および7の部分配列の少なくとも1つに対して60%から100%までの同一性を有する、少なくとも1つの部分配列を含有することを特徴とする。
ITGPEGQRLAGP (配列番号7)
IELGITGPEGQRLAGPTVL (配列番号1)
IELGITGPEGQRLAGPVL (配列番号2)
IELITGPEGQRLAGPTVL (配列番号3)
IELITGPEGQRLAGPVL (配列番号4)
EEIARARKSGQRVIGEPVAS (配列番号5)、
たとえば、配列番号5および7の部分配列の少なくとも1つ、
に対して60パーセントから100パーセントの同一性を有する、少なくとも1つの部分配列を含有するヒダントイナーゼおよび/またはジヒドロピリミジナーゼを、本発明のプロセスのために使用することに関する。
ITGPEGQRLAGP (配列番号7)
IELGITGPEGQRLAGPTVL (配列番号1)
IELGITGPEGQRLAGPVL (配列番号2)
IELITGPEGQRLAGPTVL (配列番号3)
IELITGPEGQRLAGPVL (配列番号4)
EEIARARKSGQRVIGEPVAS (配列番号5)、
たとえば、配列番号5および7の部分配列の少なくとも1つ、
に対して60パーセントから100パーセント、たとえば100パーセント、の同一性を有する少なくとも1つの部分配列を含めて、上記部分配列の少なくとも1つを含有する、実質的に純粋なヒダントイナーゼに関する。
a) イオン交換クロマトグラフィー
b) 疎水性クロマトグラフィー
c) ゲル濾過
d) アフィニティクロマトグラフィー
e) 陰イオン交換
f) ゲル濾過。
A. 定義
本発明の文脈において、置換されたヒダントインおよび/または置換されたジヒドロピリミジン環、たとえば5-フェニルヒダントインおよび6-フェニルジヒドロウラシルのような「ヒダントインおよび/またはジヒドロピリミジン環の加水分解を触媒する酵素」は、少なくとも1つの式(I)の化合物の加水分解を触媒して式(II)の化合物を生成する能力を示す必要がある。具体的には、このような酵素は、ヒダントイナーゼおよび/またはジヒドロピリミジナーゼと呼ばれることもあるが、天然基質がヒダントイン環である酵素はヒダントイナーゼと呼ばれるのが好ましく、天然基質がジヒドロウラシル環である酵素は、ジヒドロピリミジナーゼと称されるのが好ましいであろう。
ee (%) = [(Sエナンチオマー - Rエナンチオマー) / (Sエナンチオマー + Rエナンチオマー)] x 100。
一般式(I)の基質の残基R1および/またはR2は、直鎖もしくは分岐鎖低級アルキル基から選択することができるが、これは一箇所もしくは複数箇所で置換されていてもよい。残基R1およびR2は、具体的には、1から10炭素原子、特に2から6炭素原子を含有する直鎖もしくは分岐アルキル基、たとえば、メチル基、エチル基、i-もしくはn-プロピル基、sec-もしくはtert-ブチル基、n-ペンチル基、2-メチルブチル基、n-ヘキシル基、ヘプチル基、オクチル基、ノニル基、デシル基からなる基とすることができる。分岐アルキル基の例には、イソプロピル、イソブチル、イソペンチル、2,2-ジメチルプロピル、イソヘキシル、3-メチルペンチル、2,2-ジメチルブチル、2,3-ジメチルブチル、イソヘプチル、2-エチルブチル、3-メチルヘキシル、2,2-ジメチルペンチル、2,3-ジメチルペンチル、2,4-ジメチルペンチル、2,2,3-トリメチルブチル、イソオクチル、3-メチルヘプチル、4-メチルヘプチル、2,2-ジメチルヘキシル、2,4-ジメチルヘキシル、2,5-ジメチルヘキシル、2,2,3-トリメチルペンチル、2,2,4-トリメチルペンチル、2,2,5-トリメチルペンチル、およびイソノニル基がある。
βアミノ酸前駆体を生産するための方法に際して存在する、ヒダントイナーゼおよびジヒドロピリミジナーゼから選択された、少なくとも1つの酵素は、その酵素(1つもしくは複数)を自然に産生する、または組換え生産する、生きた細胞中に存在していてもよく、収集された細胞、死んだ細胞、透過性細胞、細胞粗抽出物、精製抽出物中に、または基本的に純粋な形態で、もしくは完全に純粋な形態で存在することもできる。少なくとも1つの酵素は、溶液中に存在していてもよく、担体上に固定化された酵素であってもよい。1つもしくは複数の酵素は、同時に可溶型および固定化型として存在してもよい。
βアミノ酸前駆体を生体触媒により生産する、特に、βアミノ酸前駆体を生体触媒によりエナンチオ選択的に生産するためのプロセスに使用される少なくとも1つの酵素は、ヒダントイナーゼおよびジヒドロピリミジナーゼから選択されるが、好ましくは、アズキ(Vigna angularis)から得られる任意のヒダントイナーゼもしくは任意のジヒドロピリミジナーゼから選択される。βアミノ酸前駆体の生体触媒による生産中に、1つもしくは複数のヒダントイナーゼ、1つもしくは複数のジヒドロピリミジナーゼ、またはそれらの任意の組み合わせが存在していてもよい。少なくとも1つのヒダントイナーゼが存在することが好ましい。少なくとも1つの酵素は、好ましくはエナンチオ選択性酵素であり、D-ジヒドロピリミジナーゼおよび/またはD-ヒダントイナーゼが好ましい。特にその酵素は、D-ヒダントイナーゼである。少なくとも1つの酵素は、(±)-6-モノ置換ジヒドロウラシルを加水分解することによって、対応する(S)-N-カルバモイル-βアミノ酸を生成する能力、たとえば(±)-6-フェニルジヒドロウラシルから(S)-N-カルバモイル-βフェニルアラニンを生成する能力、を有することが好ましい。
a) イオン交換クロマトグラフィー(たとえば、Q-Sepharose FF-クロマトグラフィーカラムを使用)
b) 疎水性クロマトグラフィー(たとえば、フェニルセファロースカラムを使用)
c) ゲル濾過(たとえば、Superdex Prep Grade 200ゲル濾過カラムを使用)
d) アフィニティクロマトグラフィー(たとえば、Blue HiTrap 5 mlアフィニティクロマトグラフィーカラム使用)
e) 陰イオン交換クロマトグラフィー(たとえば、Mono Q HR 5/5 陰イオン交換カラムを使用)および
f) ゲル濾過(たとえばSuperose 6 prep gradeゲル濾過カラムを使用)。
IELGITGPEGQRLAGPTVL (配列番号1)
IELGITGPEGQRLAGPVL (配列番号2)
IELITGPEGQRLAGPTVL (配列番号3)
IELITGPEGQRLAGPVL (配列番号4)
EEIARARKSGQRVIGEPVAS (配列番号5)
ITGPEGQRLAGP (配列番号7)。
βアミノ酸前駆体もしくはβアミノ酸を得るための方法の際に、1つもしくは複数の追加の酵素が、必要に応じて反応中に存在していてもよい。こうした酵素は一般式(I)のジヒドロウラシル基質のD-およびL-エナンチオマー間の変換を触媒する。適当な酵素の例は、ジヒドロウラシルデヒドロゲナーゼ(たとえば、哺乳動物起源)、ジヒドロウラシルオキシダーゼ(Rhodotorula glutinis)およびエノエートレダクターゼ(旧黄色酵素)などである。
一般式(I)の基質を一般式(II)のβアミノ酸前駆体に変換するための本発明の方法は、好ましくは約7.0から約11.0までのpHで実施されるが、約7.5から10.0までのpHが好ましい。具体的には、pHは7.5から8.5までの範囲にあり、より詳細には約7.5から8.0までの範囲にある。特に好ましいpH値は約7.5、7.6、7.7、7.8、7.9および8.0である。
一般式(I)の基質の生体触媒による変換のためのプロセスは、使用するヒダントイナーゼおよび/またはジヒドロピリミジナーゼに耐えられる、任意の温度で行うことができる。通常、温度は、酵素を内包する、または酵素抽出の起源として使用される、生物または微生物の増殖至適温度と相関しているが、当業者は容易に確定することができる。概して、プロセスは30℃〜60℃、特に40℃〜50℃の温度で実施することができる。反応温度の例は、約30℃、約35℃、約37℃、約40℃、約45℃、約50℃、約55℃、および約60℃である。
基本的に疎水性のR1および/またはR2を有する基質の溶解性を高めるために、1つもしくは複数の有機共溶媒が反応媒体中に含まれていてもよい。
反応媒体からアミノ酸およびN-カルバモイルアミノ酸を単離する方法は、当技術分野で知られており、たとえば、ゲル濾過、HPLC、逆相クロマトグラフィーおよびイオン交換クロマトグラフィーなどの技術が含まれる。イオン交換クロマトグラフィー、特に陰イオン交換クロマトグラフィーは、好ましい方法である。適当なイオン交換マトリックスには、強イオン交換体(たとえば、四級アンモニウムに基づくQ Sepharose)および弱陰イオン交換体(たとえば、ジエチルアミノエチルに基づくDEAE Sepharose)がある。好ましい方法の例は、DEAEカラムを用いた陰イオン交換クロマトグラフィー、または有機溶媒による抽出であるが、これは、生物学的変換系から基質を抽出するために酢酸エチルを、次に生成物を抽出するために2-ブタノールを使用する。
化学的実験
厚さ0.2 mmのシリカゲル60F254 (Mackerey-Nagel)でコーティングしたプラスチック支持プレートを用いてTLCを行った。プレートをUV光(254 nm)または過マンガン酸塩浸漬によって可視化した。化学物質は、特に指示しない限りSigma-Aldrich、AcrosまたはFlukaから購入した。特に明記しない限り、試薬はすべて標準的な研究室グレードとし、溶媒は無水であり、供給時に使用される。1H-NMRおよび13C-NMRは、Bruker AC300 またはAC400スペクトロメータで記録した。次の略号を使用した:δ、化学シフト; bs、ブロード・シングレット(broad singlet);d、ダブレット(doublet);dd、ダブル・ダブレット(doublet of doublets);J、カップリング定数;m、マルチプレット(multiplet);q、カルテット(quartet);qui、クインテット(quintet);s、シングレット(singlet);sep、セプテット(septet)。化学シフト(δ)は、100万分の1の単位(ppm)で記録され、カップリング定数(J)はHz単位で記録される。残留プロトン性溶媒、DMSO-d6 (δ H 2.50, qui)は、1H NMRスペクトルの内部標準として使用し、13C NMRシフトは、ブロードなバンドをデカップリングしたDMSO-d6 (δ C 39.5, sep)を標準として用いた。
特に明記しない限り、すべての実験は、15 ml Falconチューブ内で、5 mL Trisバッファー(0.1M pH 7.5)中5 mM 6-PDHU(4.75 mg)のスケールで、1Uの市販のアズキ(Vigna angularis)由来ヒダントイナーゼ(35 mg、Sigma, St.Louis, USAより購入)を用いて行われ、サーモミキサー(Falcon)内で、750 rpm、温度50℃にて、インキュベートした。
ヒダントイナーゼ活性を測定するために、ホウ酸バッファー(1.5 mL、0.1M、pH 9.0)に溶解した5-フェニルヒダントイン(1.32 mg)にD-ヒダントイナーゼ(10.5 mg)を添加した。生物学的変換は、Eppendorf “Themomixer comfort”, (40℃、1400 rpm)においてインキュベートした。定められた時点で、混合物の一定分量(100μL)を採り、TCA(12%w/v、175μL)の添加により酵素を変性させて可溶性タンパク質を沈殿させることによって、反応を終了させた。遠心分離後、上清を集め、HPLCで分析したが、そこで下記のパラメーターを使用した:
カラム:Agilent Zorbax XBD-C18 4.6 mm x 50 mm、3.5 μm。
条件:25℃、H2O:アセトニトリル、85:15 (+0.1% TFA) を1 ml/分。
c-18 rpHPLC: 均一濃度移動相、25 ℃、H2O: ACN, 90:10 + 0.1 % TFA
保持時間:6-フェニルジヒドロウラシル (12.6)、N-カルバモイルβフェニルアラニン(NCBPA, 4, 9.6) TCA (4.3) DMSO (1.6)
Astec Chirobiotic T カラム、25 cm x 4.6 mm、5 μm:均一濃度移動相、5℃、20 mM 酢酸アンモニウム(pH 6.5):MeOH、70:30
保持時間:6PDHU (15.9, 27.5)、NCBPA (7.8, 6.8)。
ヒダントイナーゼ活性を有するタンパク質を次のように、アズキ(Vigna angularis)から単離した。
885 gのアズキ(健康食品店より購入)を、2分割量として通常のミキサーを用いてドライアイスとともに粉砕した。アズキ粉末を得るために、4Lの抽出バッファー(20 mM Tris、10 mM アスコルビン酸、10 mM リジン、pH 7.5)を加え、懸濁液を4℃にて一晩撹拌し、ガーゼで濾過して遠心分離した。タンパク質含量9.5 mg/mlの粗抽出液2500 mLが得られ、次のクロマトグラフィーステップ(Amersham PharmaciaまたはGE Healthare製クロマトグラフィーカラムを使用)を行うまで-20℃で保存した。
Q-Sepharose FFクロマトグラフィーカラム(直径5 cm;長さ21 cm)を200 mM Tris/HCL(pH 7.5)で洗浄し、ランニングバッファーA(20 mM Tris/HCl、1 mM アスコルビン酸、1 mM L-リジン塩酸塩、pH 7.5)で平衡化して、粗抽出物(総タンパク質5 g)をロードした。バッファーAからバッファーB(20 mM Tris/HCl、750 mM NaCl、1 mMアスコルビン酸、1 mM L-リジン塩酸塩、pH 7.5)への直線濃度勾配を適用し、活性画分を集めた。
直径5 cm、長さ21 cmのPhenyl Sepharoseカラムを使用した。水を加えて、プールしたイオン交換クロマトグラフィーの活性画分(280 ml)を500 mlに希釈し、67 g (NH4)2SO4を添加して25%飽和として、その結果得られた混合物をカラムにかけた。ランニングバッファーA(20 mM Tris、(NH4)2SO4で25%飽和, 1 mM リジン、1 mM アスコルビン酸、pH 7.5;洗浄バッファーとしても使用)を15 mL/分の低流速で加えた。カラム体積の2倍量のランニングバッファーB(20 mM Tris、1 mM リジン、1 mM アスコルビン酸、pH 7.5)の添加によりカラム環境をランニングバッファーBへと直線的に変化させた後、カラム体積の2倍量のバッファーBを溶出のために添加した。その後洗浄のためにバッファーC(10 mM Tris/HCl、pH 7.5、10% 2-propanol)を使用した。Ehrlich試薬を用いたヒダントイナーゼアッセイによる測定で最大の吸収を示す活性画分をプールした。
次の精製ステップのために、Superdex 200カラム(直径2.6 cm、長さ60 cm)を4 mL/分の流速で稼働させた。疎水性クロマトグラフィーから得られた、プールされた活性画分に、70.2 gの(NH4)2SO4を添加して80%飽和とし、得られた混合物を遠心分離(20分、12,000 rpm)してペレットを得た。得られたペレットを10 mLの均一濃度ランニングバッファー(20 mM Tris/HCl、pH 7.5)に溶解し、カラムに加えた。
Blue HiTrap 5 mLアフィニティクロマトグラフィーカラムを、バッファーA(20 mM Tris/HCl, pH 7.5)で平衡化した。分子ふるいクロマトグラフィーから、Ehrlich試薬を用いたヒダントイナーゼアッセイにより測定され(確認されたい)、プールされた活性画分をロードした後、カラムを、均一濃度で5カラム容のバッファーB(20 mM Tris/HCl、500 mM NaCl、pH 7.5)を用いて作動させ、次に2カラム容のバッファーC(20 mM Tris/HCl、pH 7.5、500 mM NaCl、1 mM NAD、1 mM NADP)で作動させた。このアフィニティクロマトグラフィーステップを用いて、カラムへの結合によりグルコースデヒドロゲナーゼ(グルコースおよびNAD/NADPに関して活性を示さない)を除去した。
前のアフィニティクロマトグラフィーから得られた素通り画分をMono Qカラム(直径0.5 cm)に、流速1 mL/分でロードした。ランニングバッファーA(20 mM Tris/HCl、pH 7.5)からランニングバッファーB(20 mM Tris/HCl、750 mM NaCl、pH 7.5)への直線濃度勾配を適用した。画分のプールは、それぞれの画分の活性およびタンパク質ゲル中のバンドの存在に基づいて行った。
先行の陰イオン交換クロマトグラフィーから得られた、プールされた活性画分を1 mLの容積まで濃縮し(分子量カットオフ10 kDのCentriprepデバイスを使用)、1 mL/分の流速でSuperoseカラムにロードした。ランニングバッファーとして20 mM Tris/HCl (pH 7.5)を使用した。
1) 分子量標準(Precision Plus Protein Marker from BioRad、それぞれ10、15、20、25、37、50、75、100、150および250 kDに対応する)
2) 精製前の無細胞抽出液
3) Q-Sepharose FF陰イオン交換画分
4) Phenylsepharose FF HIC画分
5) Superdex Prep Grade 200ゲル濾過画分
6) Blue HiTrap 5 ml アフィニティクロマトグラフィー画分
7) Mono Q HR 5/5 陰イオン交換画分
8) Superose 6 prep grade 125 mlゲル濾過画分
9) 分子量標準(レーン1と同様)
10) 別個の標品(Mono Q HR 陰イオン交換画分に相当)。
IELGITGPEGQRLAGPTVL (配列番号1)
IELGITGPEGQRLAGPVL (配列番号2)
IELITGPEGQRLAGPTVL (配列番号3)
IELITGPEGQRLAGPVL (配列番号4)
EEIARARKSGQRVIGEPVAS (配列番号5)
ITGPEGQRLAGP (配列番号7)
得られた部分配列は、Genbankアクセッション番号ACU20291(配列番号6)のダイズ(Glycine max)由来配列に対して高い相同性を有するが、このダイズ由来配列はD-ヒダントイナーゼに帰せられるので、単離されたタンパク質のヒダントイナーゼ性を裏付ける。配列番号6を下記に示し、相同性を有する領域に下線を付す。
Sigmaから購入したヒダントイナーゼ、および実施例2で単離されたヒダントイナーゼの活性を比較した。
必要とされる6-置換ジヒドロウラシル4a-i(図2)は、2つの選択可能な経路の1つによって調製した。方法A(Cabaleiro, M. C., Journal of Chemical Research 7:318-320, 2000)では、尿素を適当なケイ皮酸誘導体7とともに、190℃で2-4時間加熱した後、生成物を再結晶し、4a-iを45%の収率で得た。方法Bは、対応するラセミ体βアミノ酸をシアン酸カリウムとともに処理し、続いて加熱して環化を生じさせ、高い収率で4a-iを与えた。方法Bはラセミ化なしで進行することが知られており、エナンチオマーとして純粋な(S)-6aを出発物質として使用することによりエナンチオマーとして純粋な(S)-4aおよび(S)-5aを調製するためにも用いられた。
a) 下記の式の(±)-6-フェニルジヒドロウラシルの合成
6-PDHUおよびNCBPAの、Q SepharoseおよびDEAE Sepharoseへの結合はマイクロキャップ(Eppendorf)内でテストした。1 mL Q SepharoseまたはDEAE Sepharose(市販のエタノール懸濁液)を水(3 x 1 mL)で洗浄した後、2.5 mM NCBPAおよび2.5 mM 6-PDHUを含有する溶液(1 mL)を添加し、マイクロキャップをボルテックスしてから上清の一定分量を取り出した。上清中のNCBPAおよび6PDHUのパーセンテージをrpHPLCで測定し、それを表2に示す。
キラルrpHPLCのために、下記のカラム、バッファーおよびランニング条件を使用した:
Astec ChirobioticTM T 25 cm x 4.6 mm、5μm (Sigma-Aldrich, St. Louis, USA)
酢酸アンモニウムバッファー(pH 5.5):メタノール、70:30、25℃、0.5 mL/分。
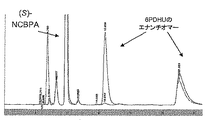
カラムは、逆さにしたシリンジから調製したが、グラスウールで塞いで、10カラム容のdH2Oで洗浄済みのDEAE(4 mL)を充填した。200 mM酢酸アンモニウムバッファー(pH 4)で平衡化後、H2O中NCBPA(2.5 mM)、6PDHU(2.5 mM)および酵素(7 mg/mL)の合成混合物をカラムにかけ、素通り画分をrpHPLC(6PDHU/NCBPA検出)およびBradfordアッセイ(酵素検出)で分析した。6PDHUをカラムからすべて洗い流した後、200 mM酢酸アンモニウムバッファー(pH 8.17)を用いてNCBPAをカラムから溶出した。結果を図3に示す(四角は6-PDHUを表す;菱形はNCBPAを表し、三角は酵素を表す;6-PDHUおよびNCBPAはmM単位、酵素はmg/Lの単位で示す)。pH 4で、NCBPAと6PDHUとの明確な分離がみられ、NCBPAの収率90%が可能となった。
はじめに、ヒダントイナーゼ(Sigma, St. Louis, USAより購入)により触媒される(±)-6-PDHU(ラセミ体6-フェニルジヒドロウラシル)の加水分解(図2の4a:R = Ph)をモデル系として検討した。反応はTrisバッファー[pH 7.5]中5 mMの基質濃度で行い、実施例7に記載の逆相キラルHPLCでモニターしたが、これにより、反応における変換の程度、ならびに未反応ジヒドロウラシル4aおよびN-カルバモイル誘導体5の鏡像体過剰率の両者の同時測定が可能となった(図4、スキーム2も参照されたい)。
(i) ヒダントイナーゼ酵素は、6-PDHU (4a)の(S)-エナンチオマーに対して高度に選択的であって、E値>100であることが判明した。生成物(S)-N-カルバモイル-β-フェニルアラニン[(S)-NCBPA]および未反応基質(R)-6-DPHUの絶対配置は標準品と比較して決定した;
(ii) 高い(S)エナンチオ選択性が観察されたにもかかわらず、反応時間を延長した後でも、変換は50%まで進行しなかった。すべての反応は、6-PDHU:(S)-NCBPA = 51:49の平衡濃度を与えた。(S)-NCBPAの鏡像体過剰率は全観察期間の間、96%を越えた。図5は小さい反応器内で25 mL反応から得られた結果を示すが、ここで菱形はNCBPAの濃度[mM]、四角は6-DPHUの濃度[mM]を表し、×印は鏡像体過剰率[%]を表す。
6-PDHUは、さまざまなpH値のTrisバッファー中で、50℃にて24時間インキュベートした。6-PDHUの相当なバックグラウンド加水分解は、pHおよびバッファーに強く依存して生じた。pH 7.0のTrisバッファー中で、0.5%のバックグラウンド加水分解が認められた。pH 9では、バックグラウンド加水分解は約20%に達した(図7)。
6-PDHUをリン酸バッファー(0.1M、リン酸二水素カリウムおよびリン酸水素二カリウム、適当なpHに調整された量)またはTrisバッファー中で、さまざまなpH値にてインキュベートした(8時間まで、Sigma製D-ヒダントイナーゼ7 g/Lを使用)。約pH 7.5から約pH 9.0までの範囲で、もっとも高い変換率が観察された(図8、四角はリン酸バッファーを、菱形はTrisバッファーを表す)。
実施例4に記載のように、さまざまな基質を合成し、ヒダントイナーゼが触媒する変換反応に供した[Trisバッファー (pH 7.5, 0.1M, 1.0 mL)、D-ヒダントイナーゼ (Sigma, 7g/L)、基質 (5mM溶液、芳香族置換基(4a, 4b, 4c)でなければバッファー中にあらかじめ溶解、芳香族置換基の場合、基質は100μL DMSOに溶解して900μLバッファーに加えた)]。反応は図10に示し、結果は表4に示すが、このE値は下記のように定義される:
E値 = (ln(1-[生成物]*(1+ee)))/(ln(1-[生成物]*(1-ee)))
5 mM 6-PDHUの生物学的変換は、2% DMSO含有バッファー、およびMTBE(約5容量%)またはブタン-2-オール(約15容量%)で飽和したバッファー中で、5 mLスケールで実施した。結果を図11に示すが、そこで菱形はバッファーを表し、四角は2% DMSO含有バッファー、三角はMTBE含有バッファー、そして×印はブタン-2-オール含有バッファーを表す。DMSOもMTBEも変換にあまり影響を及ぼさなかったが、ブタン-2-オールは使用した濃度でNCBPAの量を減少させた。
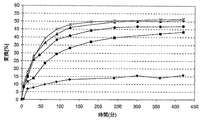
5 mM 6-PDHUを含有するそれぞれ5 mL Trisバッファー、pH 7.5の全反応容量で、温度50℃にて、3つの反応設定を並行して実施した。1つの反応では、通常の酵素量(7 g/L)を用いたが、他の設定は0.25倍相当量(1.75 g/L)および4倍相当量(28 g/L)をそれぞれ含有した。N-カルバモイル-β-フェニルアラニン(NCBPA)の生成、およびS-エナンチオマーの鏡像体過剰率をさまざまな時点で測定した。結果を図12に示すが、この縦線、斜線、または横線で網掛けされた柱状図はそれぞれ、0.25倍、1倍、または4倍相当量の酵素の存在下で得られたNCBPAの濃度を表し、菱形、四角および三角はそれぞれ、0.25倍、1倍、または4倍相当量の酵素の存在下での対応する鏡像体過剰率を表す。
3つの独立して行われた生物学的変換において、20 mM 6-PDHUを、25 mMバッファー(TrisバッファーpH 7.5)の全反応容量で、175 mgヒダントイナーゼの存在下50℃にてインキュベートした。バッファーのpHは6-PDHUの添加後に調整し、NaOHの自動滴定により反応過程のあいだ一定に保った。NCBPAの濃度、およびS-エナンチオマーの鏡像体過剰率を測定するために、さまざまな時点で一定量を分け取った。結果をそれぞれ図13および図14に示す。図13において、菱形、四角および三角はそれぞれリアクターラン1、リアクターラン2、およびリアクターラン3を表す。図14において、菱形、四角および三角はそれぞれリアクターラン1、リアクターラン2、およびリアクターラン3で観察された鏡像体過剰率を表し、実線、破線、および点線はそれぞれリアクターラン1、リアクターラン2、およびリアクターラン3で観察された鏡像体過剰率の、もっとも当てはまる曲線を表す。
配列番号1 - 単離されたヒダントイナーゼから得られたペプチド配列
配列番号2 - 単離されたヒダントイナーゼから得られたペプチド配列
配列番号3 - 単離されたヒダントイナーゼから得られたペプチド配列
配列番号4 - 単離されたヒダントイナーゼから得られたペプチド配列
配列番号5 - 単離されたヒダントイナーゼから得られたペプチド配列
配列番号6 - 配列基準としてダイズ(Glycine max)由来のヒダントイナーゼ
配列番号7 - 単離されたヒダントイナーゼから得られた部分的ペプチド配列
略号
NCBPA - N-カルバモイル-β-フェニルアラニン
6PDHU - 6-フェニルジヒドロウラシル
Claims (17)
- βアミノ酸前駆体を生体触媒により生産するための方法であって、該方法は、少なくとも1つの一般式(I)、
水素、
置換されていてもよい、直鎖もしくは分岐鎖低級アルキル基、
置換されていてもよい、直鎖もしくは分岐鎖低級アルケニル基、
置換されていてもよい、環状アルキル基、
置換されていてもよい、単環式もしくは多環式アリール基、
置換されていてもよい、単環式もしくは多環式ヘテロアリール基、
置換されていてもよい、直鎖もしくは分岐鎖アルコキシ基、
アミノ基、
置換されていてもよい、直鎖もしくは分岐鎖アルキルアミノ基、
置換されていてもよい、直鎖もしくは分岐鎖アルキルチオ基、
置換されていてもよい、直鎖もしくは分岐鎖アシル基、
カルボキシル基または
アルデヒド基
から選択される]
で表される基質、
ここで該基質は立体異性として純粋な形態で、または立体異性体の混合物として存在しており、該立体異性として純粋な形態または立体異性体の混合物は塩であってもよい、
をヒダントインおよび/またはジヒドロピリミジン環の加水分解を触媒する、少なくとも1つの酵素の存在下で反応させ、
それにより、一般式(II)のβアミノ酸前駆体、
を生成すること、
を含んでなる方法であり、
前記方法に使用される少なくとも1つの酵素が、
(a)ヒダントイナーゼおよび/もしくはジヒドロピリミジナーゼ活性を有し、アズキ(Vigna angularis)から得られ、配列番号5および配列番号7のアミノ酸配列の少なくとも1つを含み、SDS-PAGEによる測定で約55kDの分子量を示す酵素であって、「約」という用語は、示された値の±5%の変動を示す、酵素であるか、
または
(b)(a)の酵素に対して1〜10アミノ酸の付加、置換、欠失および/もしくは逆位を有し、配列番号5もしくは配列番号7に対して少なくとも95%の配列同一性を有するアミノ酸配列を含み、ヒダントイナーゼおよび/もしくはジヒドロピリミジナーゼ活性を有する酵素である
ことを特徴とする、前記方法。 - 式(II)の前記βアミノ酸前駆体を、対応する式(III)のβアミノ酸
に変換することをさらに含んでなる、請求項1に記載の方法。 - βアミノ酸前駆体の変換が、酸性pHで、またはカルバモイラーゼの存在下で起こる、請求項2に記載の方法。
- R2がHであり、R1が置換されていてもよいアリール基である、請求項1〜3のいずれか1つに記載の方法。
- 反応がTris緩衝、またはホウ酸緩衝反応混合物中で行われる、請求項1〜4のいずれか1つに記載の方法。
- 反応が約7.0から約11.0までのpHで実施され、ここで、「約」という用語は、示された値の±5%の変動を示す、請求項1〜5のいずれか1つに記載の方法。
- 反応が、約1%から約20%までのジメチルスルホキシド存在下で実施され、ここで、「約」という用語は、示された値の±5%の変動を示す、請求項1〜6のいずれか1つに記載の方法。
- 反応が、約30℃〜約60℃の温度で実施され、ここで、「約」という用語は、示された値の±5%の変動を示す、請求項1〜7のいずれか1つに記載の方法。
- 反応が、約1時間から約25時間まで行われ、ここで、「約」という用語は、示された値の±5%の変動を示す、請求項1〜8のいずれか1つに記載の方法。
- 少なくとも1つの基質が、5位または6位で一置換されたジヒドロウラシルである、請求項1〜9のいずれか1つに記載の方法。
- 少なくとも1つの酵素が、配列番号5のアミノ酸配列および配列番号7のアミノ酸配列を含む、請求項1〜10のいずれか1つに記載の方法。
- 反応がTris緩衝反応混合物中で行われる、請求項1〜11のいずれか1つに記載の方法。
- 反応が約7.5から約8.0までのpHで実施され、ここで、「約」という用語は、示された値の±5%の変動を示す、請求項1〜12のいずれか1つに記載の方法。
- 反応が、約10%のジメチルスルホキシド存在下で実施され、ここで、「約」という用語は、示された値の±5%の変動を示す、請求項1〜13のいずれか1つに記載の方法。
- 反応が、約40℃〜約50℃の温度で実施され、ここで、「約」という用語は、示された値の±5%の変動を示す、請求項1〜14のいずれか1つに記載の方法。
- 反応が、約4時間から約5時間まで行われ、ここで、「約」という用語は、示された値の±5%の変動を示す、請求項1〜15のいずれか1つに記載の方法。
- 少なくとも1つの基質が、6-フェニルジヒドロウラシル、4-フルオロ-6-フェニルジヒドロウラシル、3-ブロモ-6-フェニルジヒドロウラシル、5-メチルジヒドロウラシル、および6-メチルジヒドロウラシルからなる群より選択される、請求項1〜16のいずれか1つに記載の方法。
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP09170364.5 | 2009-09-15 | ||
| EP09170364 | 2009-09-15 | ||
| US24530309P | 2009-09-24 | 2009-09-24 | |
| US61/245,303 | 2009-09-24 | ||
| PCT/EP2010/063558 WO2011032990A1 (en) | 2009-09-15 | 2010-09-15 | Preparation of beta-amino acids |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2013504313A JP2013504313A (ja) | 2013-02-07 |
| JP2013504313A5 JP2013504313A5 (ja) | 2015-08-13 |
| JP5919192B2 true JP5919192B2 (ja) | 2016-05-18 |
Family
ID=43259766
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2012528394A Expired - Fee Related JP5919192B2 (ja) | 2009-09-15 | 2010-09-15 | βアミノ酸の調製 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US9096841B2 (ja) |
| EP (1) | EP2478098B1 (ja) |
| JP (1) | JP5919192B2 (ja) |
| CN (1) | CN102782128A (ja) |
| ES (1) | ES2628225T3 (ja) |
| WO (1) | WO2011032990A1 (ja) |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20120070867A1 (en) | 2009-06-04 | 2012-03-22 | Basf Se | process for the enzymatic reduction of enoates |
| US20130273619A1 (en) | 2012-04-16 | 2013-10-17 | Basf Se | Process for the Preparation of (3E, 7E)-Homofarnesol |
| CN106191147B (zh) * | 2016-07-08 | 2020-03-27 | 凯莱英医药集团(天津)股份有限公司 | 手性β-氨基酸的制备方法 |
| CN106831606B (zh) * | 2017-01-20 | 2019-02-22 | 湖南有色郴州氟化学有限公司 | 一种5-三氟甲基-5,6-二氢尿嘧啶的制备方法 |
| CN109896980B (zh) * | 2017-12-07 | 2022-04-08 | 浙江九洲药业股份有限公司 | 一种西格列汀中间体的生物合成方法 |
Family Cites Families (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH06261787A (ja) * | 1993-03-10 | 1994-09-20 | Kanegafuchi Chem Ind Co Ltd | 光学活性β−アミノ酸の製造法 |
| DE19931847A1 (de) | 1999-07-09 | 2001-01-11 | Basf Ag | Immobilisierte Lipase |
| DE10019377A1 (de) | 2000-04-19 | 2001-10-25 | Basf Ag | Verfahren zur Immobilisierung von biologisch aktiven Stoffen auf Trägermaterialien und Verwendung der mit biologisch aktiven Stoffen geträgerten Materialien für chirale Synthesen |
| DE10019380A1 (de) | 2000-04-19 | 2001-10-25 | Basf Ag | Verfahren zur Herstellung von kovalent gebundenen biologisch aktiven Stoffen an Polyurethanschaumstoffen sowie Verwendung der geträgerten Polyurethanschaumstoffe für chirale Synthesen |
| CN1260347C (zh) * | 2002-05-13 | 2006-06-21 | 中国人民解放军军事医学科学院生物工程研究所 | 一株产乙内酰脲酶系的节杆菌 |
| CN1908159B (zh) * | 2005-02-07 | 2011-04-20 | 中国科学院上海生命科学研究院 | D-氨基酸生产菌株及其构建方法 |
| EP2065470A1 (en) | 2007-11-28 | 2009-06-03 | Basf Se | New malonate decarboxylases for industrial applications |
| EP2229449A2 (de) | 2007-12-10 | 2010-09-22 | Basf Se | Verfahren zur enzymatischen reduktion von alpha- und beta-dehydroaminosäuren |
| JP5361904B2 (ja) | 2007-12-20 | 2013-12-04 | ビーエーエスエフ ソシエタス・ヨーロピア | (s)−1−メトキシ−2−プロピルアミンからの(s)−2−アミノ−1−プロパノール(l−アラニノール)の製造方法 |
| EP2145904A1 (de) | 2008-07-18 | 2010-01-20 | Basf Se | Verfahren zur enzymkatalysierten Hydrolyse von Polyacrylsäureestern sowie dafür zu verwendende Esterasen |
| WO2010079068A1 (de) | 2008-12-17 | 2010-07-15 | Basf Se | Verbesserte biokatalysatoren zur herstellung von duloxetinalkohol |
| CN102282122B (zh) | 2009-01-16 | 2014-10-29 | 巴斯夫欧洲公司 | 分离(r)-和(s)-3-氨基-1-丁醇的对映体混合物 |
| DE102009003035A1 (de) | 2009-05-12 | 2010-11-18 | Basf Se | Verfahren zur Herstellung von urethangruppenhaltigen (Meth)acrylsäureestern |
| US20120070867A1 (en) | 2009-06-04 | 2012-03-22 | Basf Se | process for the enzymatic reduction of enoates |
| CN102449158B (zh) | 2009-06-05 | 2017-08-25 | 巴斯夫欧洲公司 | 龙涎呋喃的生物催化产生 |
| US20120123155A1 (en) | 2009-07-30 | 2012-05-17 | Basf Se | Biocatalyst for catalytic hydroamination |
-
2010
- 2010-09-15 ES ES10752833.3T patent/ES2628225T3/es active Active
- 2010-09-15 CN CN2010800513638A patent/CN102782128A/zh active Pending
- 2010-09-15 WO PCT/EP2010/063558 patent/WO2011032990A1/en active Application Filing
- 2010-09-15 JP JP2012528394A patent/JP5919192B2/ja not_active Expired - Fee Related
- 2010-09-15 EP EP10752833.3A patent/EP2478098B1/en not_active Not-in-force
- 2010-09-15 US US13/496,086 patent/US9096841B2/en not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| CN102782128A (zh) | 2012-11-14 |
| EP2478098A1 (en) | 2012-07-25 |
| US9096841B2 (en) | 2015-08-04 |
| WO2011032990A1 (en) | 2011-03-24 |
| US20120270280A1 (en) | 2012-10-25 |
| JP2013504313A (ja) | 2013-02-07 |
| ES2628225T3 (es) | 2017-08-02 |
| EP2478098B1 (en) | 2017-03-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5735493B2 (ja) | アンブロキサンの生物触媒的生産 | |
| US11447800B2 (en) | Method for producing vanillin | |
| CZ20021449A3 (cs) | L-Pantolakton hydroláza a způsob přípravy D-pantolaktonu | |
| JP5919192B2 (ja) | βアミノ酸の調製 | |
| JP2024038047A (ja) | ホモファルネシル酸の酵素的環化 | |
| WO2009009117A2 (en) | Chemoenzymatic processes for preparation of levetiracetam | |
| US7399624B2 (en) | Cephalosporin C acylases | |
| Zheng et al. | Hydroxynitrile lyase isozymes from Prunus communis: identification, characterization and synthetic applications | |
| US11345907B2 (en) | Method for producing albicanol compounds | |
| US20100196970A1 (en) | Butynol I esterase | |
| WO2023088077A1 (en) | Biocatalysts and methods for the synthesis of pregabalin intermediates | |
| JP5453238B2 (ja) | ブタ肝臓エステラーゼのイソ型 | |
| WO2011078667A2 (en) | Method of finding a biocatalyst having ammonia lyase activity | |
| JP5619400B2 (ja) | 酸性ホスファターゼ及びそれをコードする核酸、並びにこれらを利用したリボフラビン−5’−リン酸の製造方法 | |
| Liang et al. | Biocatalytic synthesis of chiral five-membered carbasugars intermediates utilizing CV2025 ω-transaminase from Chromobacterium violaceum | |
| Hu et al. | Engineered ketoreductase-catalyzed stereoselective reduction of ethyl 2′-ketopantothenate and its analogues: chemoenzymatic synthesis of d-pantothenic acid | |
| Yamamura et al. | A novel method of producing the key intermediate ASI-2 of ranirestat using a porcine liver esterase (PLE) substitute enzyme | |
| WO2024041972A1 (en) | Nucleic acids encoding improved lipase proteins | |
| JP5291367B2 (ja) | 新規加水分解酵素 | |
| KR20060113697A (ko) | 신규 아세토아세틸-CoA 환원 효소 및 광학 활성알코올의 제조 방법 | |
| JP2010187573A (ja) | (r)−3−ヒドロキシイソ酪酸の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20130912 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20141216 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20150310 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20150616 |
|
| A524 | Written submission of copy of amendment under article 19 pct |
Free format text: JAPANESE INTERMEDIATE CODE: A524 Effective date: 20150616 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20151027 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20160226 |
|
| A911 | Transfer to examiner for re-examination before appeal (zenchi) |
Free format text: JAPANESE INTERMEDIATE CODE: A911 Effective date: 20160307 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20160329 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20160411 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5919192 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| LAPS | Cancellation because of no payment of annual fees |