JP5699152B2 - リジン特異的デメチラーゼ−1阻害剤およびその使用 - Google Patents
リジン特異的デメチラーゼ−1阻害剤およびその使用 Download PDFInfo
- Publication number
- JP5699152B2 JP5699152B2 JP2012530181A JP2012530181A JP5699152B2 JP 5699152 B2 JP5699152 B2 JP 5699152B2 JP 2012530181 A JP2012530181 A JP 2012530181A JP 2012530181 A JP2012530181 A JP 2012530181A JP 5699152 B2 JP5699152 B2 JP 5699152B2
- Authority
- JP
- Japan
- Prior art keywords
- trans
- alkyl
- ethyl
- pharmaceutically acceptable
- cancer
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- KJRNXFVJPCQLFL-GGYWPGCISA-N CC(C)(C)OC(N[C@@H](C1)C1c(cc1)ccc1OCc1ccccc1)=O Chemical compound CC(C)(C)OC(N[C@@H](C1)C1c(cc1)ccc1OCc1ccccc1)=O KJRNXFVJPCQLFL-GGYWPGCISA-N 0.000 description 1
- QYBLNMULDUNRBR-VIFPVBQESA-N CN(C)C[C@H]1CN(CCN)CC1 Chemical compound CN(C)C[C@H]1CN(CCN)CC1 QYBLNMULDUNRBR-VIFPVBQESA-N 0.000 description 1
- RODXJVOOUYCESN-KEKNWZKVSA-N CN1CCN(CCN[C@@H](C2)C2c(cc2)ccc2-c2cc(Cl)ccc2)CC1 Chemical compound CN1CCN(CCN[C@@H](C2)C2c(cc2)ccc2-c2cc(Cl)ccc2)CC1 RODXJVOOUYCESN-KEKNWZKVSA-N 0.000 description 1
- UXILYVCUBVSGOI-DTIOYNMSSA-N ClCCN[C@@H](C1)C1c1ccccc1 Chemical compound ClCCN[C@@H](C1)C1c1ccccc1 UXILYVCUBVSGOI-DTIOYNMSSA-N 0.000 description 1
- YFSDVCYHUDZZKO-LBAQZLPGSA-N FC(c1cccc(-c2ccc(C(C3)[C@H]3NCCN3CCNCC3)cc2)c1)(F)F Chemical compound FC(c1cccc(-c2ccc(C(C3)[C@H]3NCCN3CCNCC3)cc2)c1)(F)F YFSDVCYHUDZZKO-LBAQZLPGSA-N 0.000 description 1
- IMUDHTPIFIBORV-UHFFFAOYSA-N NCCN1CCNCC1 Chemical compound NCCN1CCNCC1 IMUDHTPIFIBORV-UHFFFAOYSA-N 0.000 description 1
Images





Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/075—Ethers or acetals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/095—Sulfur, selenium, or tellurium compounds, e.g. thiols
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4965—Non-condensed pyrazines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C211/00—Compounds containing amino groups bound to a carbon skeleton
- C07C211/33—Compounds containing amino groups bound to a carbon skeleton having amino groups bound to carbon atoms of rings other than six-membered aromatic rings
- C07C211/39—Compounds containing amino groups bound to a carbon skeleton having amino groups bound to carbon atoms of rings other than six-membered aromatic rings of an unsaturated carbon skeleton
- C07C211/40—Compounds containing amino groups bound to a carbon skeleton having amino groups bound to carbon atoms of rings other than six-membered aromatic rings of an unsaturated carbon skeleton containing only non-condensed rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C217/00—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton
- C07C217/02—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups and amino groups bound to acyclic carbon atoms of the same carbon skeleton
- C07C217/04—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups and amino groups bound to acyclic carbon atoms of the same carbon skeleton the carbon skeleton being acyclic and saturated
- C07C217/06—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups and amino groups bound to acyclic carbon atoms of the same carbon skeleton the carbon skeleton being acyclic and saturated having only one etherified hydroxy group and one amino group bound to the carbon skeleton, which is not further substituted
- C07C217/08—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups and amino groups bound to acyclic carbon atoms of the same carbon skeleton the carbon skeleton being acyclic and saturated having only one etherified hydroxy group and one amino group bound to the carbon skeleton, which is not further substituted the oxygen atom of the etherified hydroxy group being further bound to an acyclic carbon atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C217/00—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton
- C07C217/54—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton
- C07C217/74—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton with rings other than six-membered aromatic rings being part of the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C323/00—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups
- C07C323/23—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton
- C07C323/24—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton having the sulfur atoms of the thio groups bound to acyclic carbon atoms of the carbon skeleton
- C07C323/25—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton having the sulfur atoms of the thio groups bound to acyclic carbon atoms of the carbon skeleton the carbon skeleton being acyclic and saturated
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/04—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D207/10—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D207/14—Nitrogen atoms not forming part of a nitro radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/08—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms
- C07D211/10—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with radicals containing only carbon and hydrogen atoms attached to ring carbon atoms
- C07D211/14—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with radicals containing only carbon and hydrogen atoms attached to ring carbon atoms with hydrocarbon or substituted hydrocarbon radicals attached to the ring nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D241/00—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings
- C07D241/02—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings
- C07D241/04—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/02—Systems containing only non-condensed rings with a three-membered ring
Description
(A’)x−(A)−(B)−(Z)−(L)−(D)
I
式中:
(A)は(B)に、存在するならば、(A’)に共有結合するヘテロアリールまたはアリールであり;
(A’)は、各々、存在するならば、アリール、アリールアルコキシ、アリールアルキル、ヘテロシクリル、アリールオキシ、ハロ、アルコキシ、ハロアルキル、シクロアルキル、ハロアルコキシおよびシアノから独立して選択され、ここで(A’)は、各々、ハロ、ハロアルキル、アリール、アリールアルコキシ、アルキル、アルコキシ、シアノ、スルホニル、アミドおよびスルフィニルから独立して選択される0、1、2または3個の置換基を有し;
xは0、1、2または3であり;
(B)は(A)および(Z)に共有結合するシクロプロピル環であり、ここで(A)および(Z)は(B)の異なる炭素原子に共有結合しており;
(Z)は−NH−であり;したがって、(Z)は(B)に、(L)に、および水素原子に共有結合する窒素原子であり;
(L)は(Z)に、および(D)に共有結合するリンカーであり、ここで該リンカーは−CH2CH2−、−CH2CH2CH2−および−CH2CH2CH2CH2−から選択され;および
(D)は−N(−R1)−R2、−O−R3および−S−R3から選択され、ここで:
R1およびR2は相互に連結して、R1およびR2が結合する窒素原子と一緒になってヘテロ環式環を形成し、ここで該ヘテロ環式環は−NH2、−NH(C1−C6アルキル)、−N(C1−C6アルキル)(C1−C6アルキル)、アルキル、ハロ、シアノ、アルコキシ、ハロアルキルおよびハロアルコキシから独立して選択される0、1、2または3個の置換基を有するか、または
R1およびR2は−H、アルキル、シクロアルキル、ハロアルキルおよびヘテロシクリルから独立して選択され、ここでR1およびR2上の置換基の合計は0、1、2または3であり、該置換基は−NH2、−NH(C1−C6アルキル)、−N(C1−C6アルキル)(C1−C6アルキル)およびフルオロより独立して選択され;および
R3は−H、アルキル、シクロアルキル、ハロアルキルおよびヘテロシクリルより選択され、ここでR3は−NH2、−NH(C1−C6アルキル)、−N(C1−C6アルキル)(C1−C6アルキル)およびフルオロより独立して選択される、0、1、2または3個の置換基を有する;
で示される化合物またはその医薬上許容される塩または溶媒和物、あるいはそのエナンチオマー、ジアステレオマーまたは混合物を提供する。
R1およびR2は、(D)の窒素と一緒になって、−NH2、−N(C1−C6アルキル)(C1−C6アルキル)、−NH(C1−C6アルキル)、アルキル、ハロ、シアノ、アルコキシ、ハロアルキルおよびハロアルコキシから独立して選択される0、1、2または3個の置換基を有するヘテロ環式環を形成するか、または
R1およびR2は−H、アルキル、シクロアルキル、ハロアルキルおよびヘテロシクリルから独立して選択され、ここで、R1およびR2上の置換基の合計は0、1、2または3であり、該置換基は−NH2、−N(C1−C6アルキル)(C1−C6アルキル)、−NH(C1−C6アルキル)およびフルオロより独立して選択されるか、あるいは
(D)は(L)に共有結合する酸素または硫黄原子であり、−H、アルキル、シクロアルキル、ハロアルキルおよびヘテロシクリルより選択される一の置換基R3を有し、ここで一の該置換基R3は−NH2、−N(C1−C6アルキル)(C1−C6アルキル)、−NH(C1−C6アルキル)およびフルオロより独立して選択される0、1、2または3個の置換基を有する。
N1−[(トランス)−2−フェニルシクロプロピル]−N2−ウンデシル−rel−1,2−エタンジアミン;
N1−[(トランス)−2−フェニルシクロプロピル]−N2−トリシクロ[3.3.1.13,7]デカ−2−イル−rel−1,2−エタンジアミン;
N1−シクロオクチル−N2−[(トランス)−2−フェニルシクロプロピル]−rel−1,2−エタンジアミン;
N1,N1−ジメチル−N2−(2−フェニルシクロプロピル)−1,3−プロパンジアミン;
N1,N1−ジメチル−N2−(2−フェニルシクロプロピル)−1,2−エタンジアミン;および
トランス−1−フェニル−2−[(2−ヒドロキシエチル)アミノ]シクロプロパン。
(A’)x−(A)−(B)−(Z)−(L)−(D)
I
式中:
(A)は(B)に、存在するならば(A’)に共有結合するヘテロアリールまたはアリールであり;
(A’)は、各々、存在するならば、アリール、アリールアルコキシ、アリールアルキル、ヘテロシクリル、アリールオキシ、ハロ、アルコキシ、ハロアルキル、シクロアルキル、ハロアルコキシおよびシアノから独立して選択され、ここで(A’)は、各々、ハロ、ハロアルキル、アリール、アリールアルコキシ、アルキル、アルコキシ、シアノ、スルホニルおよびスルフィニルから独立して選択される0、1または2個の置換基で置換されており;
xは0、1、2または3であり;
(B)は(A)および(Z)に共有結合するシクロプロピル環であり、ここで(A)および(Z)は(B)の異なる炭素原子に共有結合しており;
(Z)は−NH−であり;したがって、(Z)は(B)に、(L)に、および水素原子に共有結合する窒素原子であり;
(L)は(Z)に、および(D)に共有結合するリンカーであり、ここで該リンカーは−CH2CH2−、−CH2CH2CH2−および−CH2CH2CH2CH2−より選択され;
(D)は−N(−R1)−R2、−O−R3および−S−R3より選択され、ここで:
R1およびR2は相互に連結して、R1およびR2が結合する窒素原子と一緒になってヘテロ環式環を形成し、ここで該ヘテロ環式環は−NH2、−N(C1−C6アルキル)(C1−C6アルキル)、アルキル、ハロ、シアノ、アルコキシ、ハロアルキルおよびハロアルコキシより独立して選択される0、1、2または3個の置換基を有するか、または
R1およびR2は−H、アルキル、シクロアルキル、ハロアルキルおよびヘテロシクリルより独立して選択され、ここで、R1およびR2上の置換基の合計は0、1、2または3であり、該置換基は−NH2、−N(C1−C6アルキル)(C1−C6アルキル)およびフルオロより独立して選択され;および
R3は−H、アルキル、シクロアルキル、ハロアルキルおよびヘテロシクリルより選択され、ここでR3は−NH2、−N(C1−C6アルキル)(C1−C6アルキル)およびフルオロより独立して選択される0、1、2または3個の置換基を有する;
で示される化合物またはその医薬上許容される塩または溶媒和物、あるいはそのエナンチオマー、ジアステレオマーまたは混合物を提供する;
ただし、式Iの化合物は:
N1−[(トランス)−2−フェニルシクロプロピル]−N2−ウンデシル−rel−1,2−エタンジアミン;
N1−[(トランス)−2−フェニルシクロプロピル]−N2−トリシクロ[3.3.1.13,7]デカ−2−イル−rel−1,2−エタンジアミン;
N1−シクロオクチル−N2−[(トランス)−2−フェニルシクロプロピル]−rel−1,2−エタンジアミン;
N1,N1−ジメチル−N2−(2−フェニルシクロプロピル)−1,2−エタンジアミン;
N1,N1−ジメチル−N2−(2−フェニルシクロプロピル)−1,3−プロパンジアミン;
トランス−1−フェニル−2−[(2−ヒドロキシエチル)アミン]シクロプロパン;
あるいは該化合物のエナンチオマー、ジアステレオマーまたはその混合物
以外の化合物である。
ここでR1およびR2は(D)の窒素と一緒になって−NH2、−N(C1−C6アルキル)(C1−C6アルキル)、アルキル、ハロ、シアノ、アルコキシ、ハロアルキルおよびハロアルコキシから独立して選択される0、1、2または3個の置換基を有するヘテロ環式環を形成するか、または
R1およびR2は、−H、アルキル、シクロアルキル、ハロアルキルおよびヘテロシクリルから独立して選択され、ここで、R1およびR2上の置換基の合計は0、1、2または3であり、その置換基は−NH2、−N(C1−C6アルキル)(C1−C6アルキル)およびフルオロから独立して選択されるか、あるいは
(D)は(L)に共有結合する酸素または硫黄原子であり、−H、アルキル、シクロアルキル、ハロアルキルおよびヘテロシクリルから選択される一の置換基R3を有し、該R3置換基は−NH2、−N(C1−C6アルキル)(C1−C6アルキル)およびフルオロから独立して選択される0、1、2または3個の置換基を有する。
(A’)がアリールおよびアリールアルコキシから選択され、該アリールまたはアリールアルコキシがハロおよびハロアルキルから選択される0または1個の置換基を有し、他の可変基が本願発明の第一の態様の最も広い定義にて上記したとおりである、化合物を提供する。
(A’)x−(A)−(B)−(Z)−(L)−(D)
I
式中:
(A)は(B)に、存在するならば、(A’)に共有結合するヘテロアリールまたはアリールであり;
(A’)は、各々、存在するならば、アリール、アリールアルコキシ、アリールアルキル、ヘテロシクリル、アリールオキシ、ハロ、アルコキシ、ハロアルキル、シクロアルキル、ハロアルコキシおよびシアノより独立して選択され、ここで(A’)は、各々、ハロ、ハロアルキル、アリール、アリールアルコキシ、アルキル、アルコキシ、シアノ、スルホニルおよびスルフィニルから独立して選択される0、1または2個の置換基で置換されており;
xは0、1、2または3であり;
(B)は(A)および(Z)に共有結合するシクロプロピル環であり、ここで(A)および(Z)は(B)の異なる炭素原子に共有結合しており;
(Z)は−NH−であり;したがって、(Z)は(B)に、(L)に、および水素原子に共有結合する窒素原子であり;
(L)は(Z)に、および(D)に共有結合するリンカーであり、ここで該リンカーは−CH2CH2−、−CH2CH2CH2−および−CH2CH2CH2CH2−より選択され;
(D)はR1に、R2に、および(L)に共有結合する窒素原子であり、
ここでR1およびR2は(D)の窒素と一緒になって−NH2、−N(C1−C6アルキル)(C1−C6アルキル)、アルキル、ハロ、シアノ、アルコキシ、ハロアルキルおよびハロアルコキシから独立して選択される0、1、2または3個の置換基を有するヘテロ環式環を形成するか、または
R1およびR2は−H、アルキル、シクロアルキル、ハロアルキルおよびヘテロシクリルより独立して選択され、ここで、R1およびR2上の置換基の合計は0、1、2または3であり、その置換基は−NH2、−N(C1−C6アルキル)(C1−C6アルキル)およびフルオロより独立して選択されるか、あるいは
(D)は(L)に共有結合する酸素または硫黄原子であり、アルキル、シクロアルキル、ハロアルキルおよびヘテロシクリルより選択される一の置換基R3を有し、ここで該一の置換基は−NH2、−N(C1−C6アルキル)(C1−C6アルキル)およびフルオロより独立して選択される0、1、2または3個の置換基を有する;
で示される化合物またはその医薬上許容される塩または溶媒和物、あるいはエナンチオマー、ジアステレオマーまたはその混合物:ただし、式(I)の化合物は、
N1−[(トランス)−2−フェニルシクロプロピル]−N2−ウンデシル−rel−1,2−エタンジアミン;
N1−[(トランス)−2−フェニルシクロプロピル]−N2−トリシクロ[3.3.1.13,7]デカ−2−イル−rel−1,2−エタンジアミン;
N1−シクロオクチル−N2−[(トランス)−2−フェニルシクロプロピル]−rel−1,2−エタンジアミン;
N1,N1−ジメチル−N2−(2−フェニルシクロプロピル)−1,2−エタンジアミン;
N,N−ジメチル−N’−(2−フェニルシクロプロピル)−1,3−プロパンジアミン;
ではなく、あるいは該化合物のエナンチオマー、ジアステレオマーまたはその混合物ではない。
(A’)x−(A)−(B)−(Z)−(L)−(D)
I’
式中:
(A)は(B)に、存在するならば、(A’)に共有結合するヘテロアリールまたはアリールであり;
(A’)は、各々、存在するならば、アリール、アリールアルコキシ、アリールアルキル、ヘテロシクリル、アリールオキシ、ハロ、アルコキシ、ハロアルキル、シクロアルキル、ハロアルコキシおよびシアノより独立して選択され、ここで(A’)は、各々、ハロ、ハロアルキル、アリール、アリールアルコキシ、アルキル、アルコキシ、シアノ、スルホニルおよびスルフィニルから独立して選択される0、1または2個の置換基を有しており;
xは0、1、2または3であり;
(B)は(A)および(Z)に共有結合するシクロプロピル環であり、ここで(A)および(Z)は(B)の異なる炭素原子に共有結合しており;
(Z)は−NH−であり;従って、Zは(B)に、(L)に、および水素原子に共有結合する窒素原子であり;
(L)は(Z)に、および(D)に共有結合するリンカーであり、ここで該リンカーは−CH2CH2−、−CH2CH2CH2−および−CH2CH2CH2CH2−より選択され;
(D)はR1に、R2に、および(L)に共有結合する窒素原子であり;
ここでR1およびR2は(D)の窒素と一緒になって−NH2、−N(C1−C6アルキル)(C1−C6アルキル)、アルキル、ハロ、シアノ、アルコキシ、ハロアルキルおよびハロアルコキシから独立して選択される0、1、2または3個の置換基を有するヘテロ環式環を形成するか、または
R1およびR2は−H、アルキル、シクロアルキル、ハロアルキルおよびヘテロシクリルより独立して選択され、ここで、R1およびR2上の置換基の合計は0、1、2または3であり、該置換基は−NH2、−N(C1−C6アルキル)(C1−C6アルキル)およびフルオロより独立して選択されるか、あるいは
(D)は(L)に共有結合する酸素または硫黄原子であり、−H、アルキル、シクロアルキル、ハロアルキルおよびヘテロシクリルより選択される一の置換基R3を有し、ここで該一の置換基R3は−NH2、−N(C1−C6アルキル)(C1−C6アルキル)およびフルオロより独立して選択される0、1、2または3個の置換基を有する;
で示される化合物またはその医薬上許容される塩と、医薬上許容される担体とを含む医薬組成物を、LSD1活性を阻害するのに十分な量にて投与することを含む、LSD1活性の阻害方法を提供する。
R1およびR2は相互に連結して、R1およびR2が結合する窒素原子と一緒になってヘテロ環式環を形成し、ここで該ヘテロ環式環は−NH2、−N(C1−C6アルキル)(C1−C6アルキル)、アルキル、ハロ、シアノ、アルコキシ、ハロアルキルおよびハロアルコキシより独立して選択される0、1、2または3個の置換基を有するか、または
R1およびR2は−H、アルキル、シクロアルキル、ハロアルキルおよびヘテロシクリルより独立して選択され、ここで、R1およびR2上の置換基の合計は0、1、2または3であり、該置換基は−NH2、−N(C1−C6アルキル)(C1−C6アルキル)およびフルオロより独立して選択され;
R3は−H、アルキル、シクロアルキル、ハロアルキルおよびヘテロシクリルより選択され、ここでR3は−NH2、−N(C1−C6アルキル)(C1−C6アルキル)およびフルオロより独立して選択される0、1、2または3個の置換基を有する。
本願発明は、化合物の同定およびその疾患の治療または予防における使用に関する。本願発明は、式Iの化合物、式Iの化合物またはまたはその医薬上許容される塩または溶媒和物と、医薬上許容される担体とを含む医薬組成物、および疾患を治療または予防するための該化合物および組成物の使用を提供する。式Iの化合物の一の使用は癌を治療または予防するためである。式Iの化合物はMAO−AおよびMAO−BよりもLSD1をより大きく阻害するLSD1選択的阻害剤として使用され得る。特に、式Iの(ヘテロ)アリールシクロプロピルアミン誘導体、とりわけ式Iに包含されるフェニルシクロプロピルアミン誘導体が、予期せぬ強力なLSD1阻害を有する化合物を生成することが判明した。本願明細書に記載の実施例は、式Iの化合物が、LSD1阻害について、500ナノモルより低いKi値を有し(表1を参照)、そのことが該化合物をトラニルシプロミンよりもLSD1阻害について少なくとも約40〜50倍またはそれ以上強力にすることを示す。これらの化合物はMAO−AおよびMAO−BよりもLSD1をより大きく阻害する点でLSD1選択的である。
(A’)x−(A)−(B)−(Z)−(L)−(D)
I
式中:
(A)は(B)に、存在するならば、(A’)に共有結合するヘテロアリールまたはアリールであり;
(A’)は、各々、存在するならば、アリール、アリールアルコキシ、アリールアルキル、ヘテロシクリル、アリールオキシ、ハロ、アルコキシ、ハロアルキル、シクロアルキル、ハロアルコキシおよびシアノから独立して選択され、ここで(A’)は、各々、ハロ、ハロアルキル、アリール、アリールアルコキシ、アルキル、アルコキシ、シアノ、スルホニル、アミドおよびスルフィニルから独立して選択される0、1、2または3個の置換基を有し;
xは0、1、2または3であり;
(B)は(A)および(Z)に共有結合するシクロプロピル環であり、ここで(A)および(Z)は(B)の異なる炭素原子に共有結合しており;
(Z)は−NH−であり;したがって、(Z)は(B)に、(L)に、および水素原子に共有結合する窒素原子であり;
(L)は(Z)に、および(D)に共有結合するリンカーであり、ここで該リンカーは−CH2CH2−、−CH2CH2CH2−および−CH2CH2CH2CH2−から選択され;および
(D)はR1およびR2に共有結合する窒素原子であり、ここで:
R1およびR2は(D)の窒素と一緒になって、−NH2、−N(C1−C6アルキル)(C1−C6アルキル)、−NH(C1−C6アルキル)、アルキル、ハロ、シアノ、アルコキシ、ハロアルキルおよびハロアルコキシから独立して選択される0、1、2または3個の置換基を有するヘテロ環式環を形成するか、または
R1およびR2は−H、アルキル、シクロアルキル、ハロアルキルおよびヘテロシクリルから独立して選択され、ここでR1およびR2上の置換基の合計は0、1、2または3であり、該置換基は−NH2、−N(C1−C6アルキル)(C1−C6アルキル)、−NH(C1−C6アルキル)およびフルオロより独立して選択されるか;または
(D)は(L)に共有結合する酸素または硫黄原子であり、−H、アルキル、シクロアルキル、ハロアルキルおよびヘテロシクリルから選択される一の置換基R3を有し、ここで一の該置換基R3は−NH2、−N(C1−C6アルキル)(C1−C6アルキル)、−NH(C1−C6アルキル)およびフルオロより独立して選択される0、1、2または3個の置換基を有する
で示される化合物またはその医薬上許容される塩もしくは溶媒和物、あるいはエナンチオマー、ジアステレオマーまたはその混合物を提供する:
ここで、xが0であり、(D)が酸素である場合、その場合、R3はアルキル、シクロアルキル、ハロアルキル、ヘテロシクリルより選択されるか、または
xが0であり、(L)が−CH2CH2−または−CH2CH2CH2−であって、(D)が窒素である場合、その場合、R1およびR2は共にメチル以外の基であるか、または
(L)が−CH2CH2−であり、R1およびR2の一方が−Hである場合、R1およびR2の他方はウンデシルまたはトリシクロ[3.3.1.13,7]デカ−2−イル以外の基である。
(A’)x−(A)−(B)−(Z)−(L)−(D)
I
式中:
(A)は(B)に、存在するならば、(A’)に共有結合するヘテロアリールまたはアリールであり;
(A’)は、各々、存在するならば、アリール、アリールアルコキシ、アリールアルキル、ヘテロシクリル、アリールオキシ、ハロ、アルコキシ、ハロアルキル、シクロアルキル、ハロアルコキシおよびシアノより独立して選択され、ここで(A’)は、各々、ハロ、ハロアルキル、アリール、アリールアルコキシ、アルキル、アルコキシ、シアノ、スルホニルおよびスルフィニルから独立して選択される0、1または2個の置換基を有しており;
xは0、1、2または3であり;
(B)は(A)および(Z)に共有結合するシクロプロピル環であり、ここで、
(A)および(Z)は(B)の異なる炭素原子に共有結合しており;
(Z)は−NH−であり;したがって、(Z)は(B)に、(L)に、および水素原子に共有結合する窒素原子であり;
(L)は(Z)に、および(D)に共有結合するリンカーであり、ここで該リンカーは−CH2CH2−、−CH2CH2CH2−および−CH2CH2CH2CH2−より選択され;
(D)はR1に、およびR2に共有結合する窒素原子であり、
ここで、R1およびR2は(D)の窒素と一緒になって、−NH2、−N(C1−C6アルキル)(C1−C6アルキル)、アルキル、ハロ、シアノ、アルコキシ、ハロアルキルおよびハロアルコキシから独立して選択される0、1、2または3個の置換基を有するヘテロ環式環を形成するか、または
R1およびR2は、−H、アルキル、シクロアルキル、ハロアルキルおよびヘテロシクリルより独立して選択され、ここで、R1およびR2上の置換基の合計は0、1、2または3であり、該置換基は−NH2、−N(C1−C6アルキル)(C1−C6アルキル)およびフルオロより独立して選択されるか、あるいは
(D)は(L)に共有結合する酸素または硫黄原子であり、−H、アルキル、シクロアルキル、ハロアルキルおよびヘテロシクリルより選択される一の置換基R3を有し、ここで一の該置換基R3は−NH2、−N(C1−C6アルキル)(C1−C6アルキル)およびフルオロより独立して選択される0、1、2または3個の置換基を有する;
で示される化合物またはその医薬上許容される塩または溶媒和物、あるいはエナンチオマー、ジアステレオマーまたはその混合物を提供する:
ただし、式(I)の化合物は、
N1−[(トランス)−2−フェニルシクロプロピル]−N2−ウンデシル−rel−1,2−エタンジアミン;
N1−[(トランス)−2−フェニルシクロプロピル]−N2−トリシクロ[3.3.1.13,7]デカ−2−イル−rel−1,2−エタンジアミン;
N1−シクロオクチル−N2−[(トランス)−2−フェニルシクロプロピル]−rel−1,2−エタンジアミン;
N1,N1−ジメチル−N2−(2−フェニルシクロプロピル)−1,2−エタンジアミン;
N1,N1−ジメチル−N2−(2−フェニルシクロプロピル)−1,3−プロパンジアミン;
トランス−1−フェニル−2−[(2−ヒドロキシエチル)アミノ]シクロプロパン;
あるいはエナンチオマー、ジアステレオマーまたはその混合物以外の化合物である。

で示される。
で示される。
で示される。
N−[2−(4−メチルピペラジン−1−イル)エチル]−N−[(トランス)−2−フェニルシクロプロピル] アミン;
N−シクロプロピル−N’−[(トランス)−2−フェニルシクロプロピル]エタン−1,2−ジアミン;
N,N−ジメチル−N’−(2−{[(トランス)−2−フェニルシクロプロピル]アミノ}エチル)エタン−1,2−ジアミン;
(3R)−1−(2−{[(トランス)−2−フェニルシクロプロピル]アミノ}エチル)ピロリジン−3−アミン;
(3S)−N,N−ジメチル−1−(2−{[(トランス)−2−フェニルシクロプロピル]アミノ}エチル)ピロリジン−3−アミン;
(3R)−N,N−ジメチル−1−(2−{[(トランス)−2−フェニルシクロプロピル]アミノ}エチル)ピロリジン−3−アミン;
N−[(トランス)−2−フェニルシクロプロピル]−N−(2−ピペラジン−1−イルエチル)アミン;
N1,N1−ジエチル−N2−((トランス)−2−フェニルシクロプロピル)エタン−1,2−ジアミン;
N−[(トランス)−2−フェニルシクロプロピル]−N−(2−ピペリジン−1−イルエチル)アミン;
(トランス)−2−(4−(ベンジルオキシ)フェニル)−N−(2−(4−メチルピペラジン−1−イル)エチル)シクロプロパンアミン;
(トランス)−N−(2−(4−メチルピペラジン−1−イル)エチル)−2−(3’−(トリフルオロメチル)ビフェニル−4−イル)シクロプロパンアミン;
(トランス)−2−(3’−クロロビフェニル−4−イル)−N−(2−(4−メチルピペラジン−1−イル)エチル)シクロプロパンアミン;
(R)−1−(2−((トランス)−2−(3’−(トリフルオロメチル)ビフェニル−4−イル)シクロプロピルアミノ)エチル)ピロリジン−3−アミン;および
N1−シクロプロピル−N2−((トランス)−2−(3’−(トリフルオロメチル)ビフェニル−4−イル)シクロプロピル)エタン−1,2−ジアミン;
またはその医薬上許容される塩または溶媒和物
より選択される式Iの化合物を提供する。
N−(トランス)−2−(イソブチルチオ)−エチル−2−フェニルシクロプロパンアミン、
N−トランス−(2−エトキシエチル)−2−フェニルシクロプロパンアミン、および
N−トランス−(2−メトキシエチル)−2−フェニルシクロプロパンアミン;
またはその医薬上許容される塩または溶媒和物
より選択される式Iの化合物を提供する。
N1−((トランス)−2−(4−(3−ブロモベンジルオキシ)フェニル)シクロプロピル)−N2−シクロプロピルエタン−1,2−ジアミン;
N1−((トランス)−2−(3’−クロロビフェニル−4−イル)シクロプロピル)−N2−シクロプロピルエタン−1,2−ジアミン;
N1−シクロプロピル−N2−((トランス)−2−(4−フェネトキシフェニル)シクロプロピル)エタン−1,2−ジアミン;
N1,N1−ジエチル−N2−((トランス)−2−(4−(3−フルオロベンジルオキシ)フェニル)シクロプロピル)エタン−1,2−ジアミン;
(トランス)−2−(4−ブロモフェニル)−N−(2−(4−メチルピペラジン−1−イル)エチル)シクロプロパンアミン;
N1−((トランス)−2−(テルフェニル−4−イル)シクロプロピル)−N2−シクロプロピルエタン−1,2−ジアミン;
(トランス)−N−(2−(ピペリジン−1−イル)エチル)−2−(3’−(トリフルオロメチル)ビフェニル−4−イル)シクロプロパンアミン;
N1,N1−ジエチル−N2−((トランス)−2−(3’−(トリフルオロメチル)ビフェニル−4−イル)シクロプロピル)エタン−1,2−ジアミン;
(トランス)−N−(2−(ピペラジン−1−イル)エチル)−2−(3’−(トリフルオロメチル)ビフェニル−4−イル)シクロプロパンアミン;
(S)−1−(2−((トランス)−2−(3’−(トリフルオロメチル)ビフェニル−4−イル)シクロプロピルアミノ)エチル)ピロリジン−3−アミン;
(R)−1−(2−((トランス)−2−(3’−クロロビフェニル−4−イル)シクロプロピルアミノ)エチル)ピロリジン−3−アミン;
(R)−1−(2−((トランス)−2−(4’−クロロビフェニル−4−イル)シクロプロピルアミノ)エチル)ピロリジン−3−アミン;
(R)−1−(2−((トランス)−2−(3’−メトキシビフェニル−4−イル)シクロプロピルアミノ)エチル)ピロリジン−3−アミン;
(R)−1−(2−((トランス)−2−(4−(3−ブロモベンジルオキシ)フェニル)シクロプロピルアミノ)エチル)ピロリジン−3−アミン;および
(R)−1−(2−((トランス)−2−(6−(3−(トリフルオロメチル)フェニル)ピリジン−3−イル)シクロプロピルアミノ)エチル)ピロリジン−3−アミン;
またはその医薬上許容される塩または溶媒和物
より選択される式Iの化合物を提供する。
(R)−1−(2−((トランス)−2−(4−(4−ブロモベンジルオキシ)フェニル)シクロプロピルアミノ)エチル)ピロリジン−3−アミン;
(R)−1−(2−((トランス)−2−(4−(4−クロロベンジルオキシ)フェニル)シクロプロピルアミノ)エチル)ピロリジン−3−アミン;
(R)−1−(2−((トランス)−2−(4−(ビフェニル−4−イルメトキシ)フェニル)シクロプロピルアミノ)エチル)ピロリジン−3−アミン;
(R)−1−(2−((トランス)−2−(3’,5’−ジクロロビフェニル−4−イル)シクロプロピルアミノ)エチル)ピロリジン−3−アミン;
N1−((トランス)2−(2−[1,1’;4’,1’’]テルフェニル−4’’−イル−シクロプロピル)−N2−シクロプロピルエタン−1,2−ジアミン;
(R)−1−(2−((トランス)−2−(6−(ベンジルオキシ)−4’−(トリフルオロメチル)ビフェニル−3−イル)シクロプロピルアミノ)エチル)ピロリジン−3−アミン;および
(R)−1−(2−((トランス)−2−(6−(ベンジルオキシ)ビフェニル−3−イル)シクロプロピルアミノ)エチル)ピロリジン−3−アミン;
またはその医薬上許容される塩または溶媒和物
より選択される式Iの化合物を提供する。
(R)−1−(2−((トランス)−2−(4−フェネトキシフェニル)シクロプロピルアミノ)エチル)ピロリジン−3−アミン;
(R)−1−(2−((トランス)−2−(6−(3−メトキシフェニル)ピリジン−3−イル)シクロプロピルアミノ)エチル)ピロリジン−3−アミン;
(R)−1−(2−((トランス)−2−(6−(4−クロロフェニル)ピリジン−3−イル)シクロプロピルアミノ)エチル)ピロリジン−3−アミン;および
4−((4−((トランス)−2−(2−((R)−3−アミノピロリジン−1−イル)エチルアミノ)シクロプロピル)フェノキシ)メチル)ベンゾニトリル;
またはその医薬上許容される塩または溶媒和物
より選択される式Iの化合物を提供する。
(A’)x−(A)−(B)−(Z)−(L)−(D)
I
式中:
(A)は(B)に、存在するならば、(A’)に共有結合するヘテロアリールまたはアリールであり;
(A’)は、各々、存在するならば、アリール、アリールアルコキシ、アリールアルキル、ヘテロシクリル、アリールオキシ、ハロ、アルコキシ、ハロアルキル、シクロアルキル、ハロアルコキシおよびシアノから独立して選択され、ここで(A’)は、各々、ハロ、ハロアルキル、アリール、アリールアルコキシ、アルキル、アルコキシ、シアノ、スルホニルおよびスルフィニルから独立して選択される0、1または2個の置換基を有し;
xは0、1、2または3であり;
(B)は(A)および(Z)に共有結合するシクロプロピル環であり、ここで(A)および(Z)は(B)の異なる炭素原子に共有結合しており;
(Z)は−NH−であり;したがって(Z)は(B)に、(L)に、および水素原子に共有結合している窒素原子であり;
(L)は(Z)に、および(D)に共有結合するリンカーであり、ここで該リンカーは−CH2CH2−、−CH2CH2CH2−および−CH2CH2CH2CH2−から選択され;
(D)はR1に、R2に、および(L)に共有結合する窒素原子であり、ここで
R1およびR2は(D)の窒素と一緒になって、−NH2、−N(C1−C6アルキル)(C1−C6アルキル)、アルキル、ハロ、シアノ、アルコキシ、ハロアルキルおよびハロアルコキシから独立して選択される0、1、2または3個の置換基を有する4、5または6員のヘテロ環式環を形成する
で示される化合物またはその医薬上許容される塩または溶媒和物を提供する。
(A’)x−(A)−(B)−(Z)−(L)−(D)
I
式中:
(A)は(B)に、存在するならば、(A’)に共有結合するヘテロアリールまたはアリールであり;
(A’)は、各々、存在するならば、アリール、アリールアルコキシ、アリールアルキル、ヘテロシクリル、アリールオキシ、ハロ、アルコキシ、ハロアルキル、シクロアルキル、ハロアルコキシおよびシアノより独立して選択され、ここで(A’)の各々は、ハロ、ハロアルキル、アリール、アリールアルコキシ、アルキル、アルコキシ、シアノ、スルホニルおよびスルフィニルより選択される0、1または2個の置換基を有し;
xは0、1、2または3であり;
(B)は(A)および(Z)に共有結合するシクロプロピル環であり、ここで(A)および(Z)は(B)の異なる炭素原子に共有結合しており;
(Z)は−NH−であり;したがって、(Z)は(B)に、(L)に、および水素原子に共有結合する窒素原子であり;
(L)は(Z)に、および(D)に共有結合するリンカーであり、ここで該リンカーは−CH2CH2−、−CH2CH2CH2−および−CH2CH2CH2CH2−より選択され;
(D)はR1に、R2に、および(L)に共有結合する窒素原子であり、ここで
R1およびR2は−H、アルキルおよびシクロアルキルより独立して選択され、ここで該アルキルまたはシクロアルキル基は−NH2および−N(C1−C6アルキル)(C1−C6アルキル)から独立して選択される0、1または2個の置換基を有する
で示される化合物またはその医薬上許容される塩または溶媒和物を提供する。
(A’)x−(A)−(B)−(Z)−(L)−(D)
I
式中:
(A)は(B)に、および(A’)に共有結合するヘテロアリールまたはアリールであり;
(A’)はアリールおよびアリールアルコキシより選択され、ここで(A’)はハロおよびハロアルキルから独立して選択される0、1または2個の置換基で置換されており;
xは1であり;
(B)は(A)および(Z)に共有結合するシクロプロピル環であり、ここで(A)および(Z)は(B)の異なる炭素原子に共有結合しており;
(Z)は−NH−であり;したがって、(Z)は(B)に、(L)に、および水素原子に共有結合する窒素原子であり;
(L)は(Z)に、および(D)に共有結合するリンカーであり、ここで該リンカーは−CH2CH2−、−CH2CH2CH2−および−CH2CH2CH2CH2−より選択され;
(D)はR1に、R2に、および(L)に共有結合する窒素原子であり、ここで
R1およびR2は(D)の窒素と一緒になって−NH2、−N(C1−C6アルキル)(C1−C6アルキル)、アルキル、ハロ、シアノ、アルコキシ、ハロアルキルおよびハロアルコキシから独立して選択される0、1、2または3個の置換基を有する3、4、5、6または7員のヘテロ環式環を形成するか、または
R1およびR2は−H、アルキル、シクロアルキル、ハロアルキルおよびヘテロシクリルより独立して選択され、ここで、R1およびR2上の置換基の合計は0、1、2または3であり、該置換基は−NH2、−N(C1−C6アルキル)(C1−C6アルキル)およびフルオロより独立して選択されるか、または
(D)は(L)に共有結合する酸素または硫黄原子であり、−H、アルキル、シクロアルキル、ハロアルキルおよびヘテロシクリルより選択される一の置換基R3を有し、ここで一の該置換基R3は−NH2、−N(C1−C6アルキル)(C1−C6アルキル)およびフルオロより独立して選択される0、1、2または3個の置換基を有する
で示される化合物またはその医薬上許容される塩または溶媒和物を提供する。
(A’)x−(A)−(B)−(Z)−(L)−(D)
I
式中:
(A)は(B)に、そして(A’)に共有結合するフェニルであり;
(A’)はアリールおよびアリールアルコキシより選択される0または1個の置換基を有するフェニルであり、ここで該アリールおよびアリールアルコキシ基は、ハロおよびハロアルキルより独立して選択される0、1または2個の基で置換されており;
xは1であり;
(B)は(A)および(Z)に共有結合するシクロプロピル環であり、ここで(A)および(Z)は(B)の異なる炭素原子に共有結合しており、シクロプロピル環について(A)および(Z)基の立体化学はトランス位にあり;
(Z)は−NH−であり;したがって、(Z)は(B)に、(L)に、そして水素原子に共有結合している窒素原子であり;
(L)は(Z)に、および(D)に共有結合するリンカーであり、ここで該リンカーは−CH2CH2−および−CH2CH2CH2−より選択され;
(D)はR1に、R2に、および(L)に共有結合する窒素原子であり、ここで
R1およびR2は一緒になって−NH2、−N(C1−C6アルキル)(C1−C6アルキル)およびアルキルから独立して選択される0、1、2または3個の置換基を有する4、5または6員のヘテロ環式環を形成するか、または
R1およびR2は−H、アルキル、ハロアルキルおよびシクロアルキルから独立して選択され、
ここで、R1およびR2上の置換基の合計は0、1、2または3であり、該置換基は−NH2、−N(C1−C6アルキル)(C1−C6アルキル)およびフルオロより独立して選択される
で示される化合物またはその医薬上許容される塩または溶媒和物を提供する。
(A’)x−(A)−(B)−(Z)−(L)−(D)
I
式中:
(A)は、(B)に、そして(A’)に共有結合するフェニルであり;
(A’)はアリールおよびアリールアルコキシより選択され、ここで該アリールまたはアリールアルコキシは、ハロおよびハロアルキルから独立して選択される0、1または2個の置換基を有し;
xは1であり;
(B)は(A)および(Z)に共有結合するシクロプロピル環であり、ここで(A)および(Z)は(B)の異なる炭素原子に共有結合しており、シクロプロピル環について(A)および(Z)基の立体化学はトランス位にあり;
(Z)は−NH−であり;したがって、(Z)は、(B)に、(L)に、そして水素原子に共有結合する窒素原子であり;
(L)は(Z)に、および(D)に共有結合するリンカーであり、ここで該リンカーは−CH2CH2−および−CH2CH2CH2−より選択され;
(D)はR1に、R2に、および(L)に共有結合する窒素原子であり、ここで
R1およびR2は一緒になって−NH2、−N(C1−C6アルキル)(C1−C6アルキル)およびアルキルから独立して選択される0、1、2または3個の置換基を有する4、5または6員のヘテロ環式環を形成するか;または
R1およびR2は−H、アルキル、ハロアルキルおよびシクロアルキルより独立して選択され、
ここで、R1およびR2上の置換基の合計は0、1、2または3であり、該置換基は−NH2、−N(C1−C6アルキル)(C1−C6アルキル)およびフルオロより独立して選択される
で示される化合物またはその医薬上許容される塩または溶媒和物を提供する。
(A’)x−(A)−(B)−(Z)−(L)−(D)
I
式中:
(A) is (B)に、存在するならば、(A’)に共有結合するヘテロアリールであり;
(A’)は、各々、存在するならば、(A)に共有結合し、アリール、アリールアルコキシ、アリールアルキル、ヘテロシクリル、アリールオキシ、ハロ、アルコキシ、ハロアルキル、シクロアルキル、ハロアルコキシおよびシアノから独立して選択され、ここで(A’)は、ハロ、ハロアルキル、アリール、アリールアルコキシ、アルキル、アルコキシ、シアノ、スルホニルおよびスルフィニルから独立して選択される0、1または2個の置換基で置換されており;
xは0、1、2または3であり;
(B)は(A)および(Z)に共有結合するシクロプロピル環であり、ここで(A)および(Z)は(B)の異なる炭素原子に共有結合しており;
(Z)は−NH−であり;したがって、(Z)は(B)に、(L)に、および水素原子に共有結合する窒素原子であり;
(L)は(Z)に、および(D)に共有結合するリンカーであり、ここで該リンカーは−CH2CH2−、−CH2CH2CH2−および−CH2CH2CH2CH2−から選択され;
(D)はR1に、R2に、および(L)に共有結合する窒素原子であり、ここで
R1およびR2は(D)の窒素と一緒になって−NH2、−N(C1−C6アルキル)(C1−C6アルキル)、アルキル、ハロ、シアノ、アルコキシ、ハロアルキルおよびハロアルコキシから独立して選択される0、1、2または3個の置換基を有するヘテロ環式環を形成するか、または
R1およびR2は−H、アルキル、シクロアルキル、ハロアルキルおよびヘテロシクリルより独立して選択され、ここで、R1およびR2上の置換基の合計は0、1、2または3であり、該置換基は−NH2、−N(C1−C6アルキル)(C1−C6アルキル)およびフルオロより独立して選択されるか、または
(D)は−H、アルキル、シクロアルキル、ハロアルキルおよびヘテロシクリルより選択される1個の置換基R3を有する酸素または硫黄原子であり、ここで1個の該置換基R3は−NH2、−N(C1−C6アルキル)(C1−C6アルキル)およびフルオロより独立して選択される0、1、2または3個の置換基を有する
で示される化合物またはその医薬上許容される塩または溶媒和物を提供する。
(A’)x−(A)−(B)−(Z)−(L)−(D)
I
式中:
(A)は(B)に、存在するならば、(A’)に共有結合するヘテロアリールまたはアリールであり;
(A’)は、各々、存在するならば、(A)に共有結合し、アリール、アリールアルコキシ、アリールアルキル、ヘテロシクリル、アリールオキシ、ハロ、アルコキシ、ハロアルキル、シクロアルキル、ハロアルコキシおよびシアノより独立して選択され、ここで(A’)は、ハロ、ハロアルキル、アリール、アリールアルコキシ、アルキル、アルコキシ、シアノ、スルホニルおよびスルフィニルから独立して選択される0、1または2個の置換基で置換されており;
xは0、1、2または3であり;
(B)は(A)および(Z)に共有結合するシクロプロピル環であり、ここで(A)および(Z)は(B)の異なる炭素原子に共有結合しており;
(Z)は−NH−であり;したがって、(Z)は(B)に、(L)に、および水素原子に共有結合する窒素原子であり;
(L)は(Z)に、および(D)に共有結合するリンカーであり、ここで該リンカーは−CH2CH2−、−CH2CH2CH2−および−CH2CH2CH2CH2−より選択され;
(D)はR1に、R2に、および(L)に共有結合する窒素原子であり、ここで
R1およびR2は(D)の窒素と一緒になって−NH2、−N(C1−C6アルキル)(C1−C6アルキル)、アルキル、ハロ、シアノ、アルコキシ、ハロアルキルおよびハロアルコキシから独立して選択される0、1、2または3個の置換基を有するヘテロ環式環を形成するか、または
R1およびR2は、−H、アルキル、シクロアルキル、ハロアルキルおよびヘテロシクリルより独立して選択され、ここでR1およびR2上の置換基の合計は0、1、2または3であり、該置換基は−NH2、−N(C1−C6アルキル)(C1−C6アルキル)およびフルオロより独立して選択されるか、または
(D)は、アルキル、シクロアルキル、ハロアルキルおよびヘテロシクリルより独立して選択される一の置換基R3を有する酸素または硫黄原子であり、ここで一の該置換基R3は−NH2、−N(C1−C6アルキル)(C1−C6アルキル)およびフルオロより独立して選択される0、1、2または3個の置換基を有する
で示される化合物またはその医薬上許容される塩または溶媒和物を提供する:
ただし、式(I)の化合物は、
N1−[(トランス)−2−フェニルシクロプロピル]−N2−ウンデシル−rel−1,2−エタンジアミン;
N1−[(トランス)−2−フェニルシクロプロピル]−N2−トリシクロ[3.3.1.13,7]デカ−2−イル−rel−1,2−エタンジアミン;
N1−シクロオクチル−N2−[(トランス)−2−フェニルシクロプロピル]−rel−1,2−エタンジアミン;
N1,N1−ジメチル−N2−(2−フェニルシクロプロピル)−1,2−エタンジアミン;
N,N−ジメチル−N’−(2−フェニルシクロプロピル)−1,3−プロパンジアミン;
またはそのエナンチオマー、ジアステレオーもしくはその混合物以外の化合物である。
(A’)x−(A)−(B)−(Z)−(L)−(D)
I’
式中:
(A)は(B)に、存在するならば、(A’)に共有結合するヘテロアリールまたはアリールであり;
(A’)は、各々、存在するならば、アリール、アリールアルコキシ、アリールアルキル、ヘテロシクリル、アリールオキシ、ハロ、アルコキシ、ハロアルキル、シクロアルキル、ハロアルコキシおよびシアノより独立して選択され、ここで(A’)は、各々、ハロ、ハロアルキル、アリール、アリールアルコキシ、アルキル、アルコキシ、シアノ、スルホニルおよびスルフィニルから独立して選択される0、1または2個の置換基で置換されており;
xは0、1、2または3であり;
(B)は(A)および(Z)に共有結合するシクロプロピル環であり、ここで(A)および(Z)は(B)の異なる炭素原子に共有結合しており;
(Z)は−NH−であり;したがって、(Z)は(B)に、(L)に、および水素原子に共有結合する窒素原子であり;
(L)は(Z)に、および(D)に共有結合するリンカーであり、ここで該リンカーは−CH2CH2−、−CH2CH2CH2−および−CH2CH2CH2CH2−より選択され;
(D)はR1に、R2に、および(L)に共有結合する窒素原子であり、ここで;
R1およびR2は(D)の窒素と一緒になって−NH2、−N(C1−C6アルキル)(C1−C6アルキル)、アルキル、ハロ、シアノ、アルコキシ、ハロアルキルおよびハロアルコキシから独立して選択される0、1、2または3個の置換基を有するヘテロ環式環を形成するか、または
R1およびR2は、−H、アルキル、シクロアルキル、ハロアルキルおよびヘテロシクリルより独立して選択され、ここで、R1およびR2上の置換基の合計は0、1、2または3であり、該置換基は−NH2、−N(C1−C6アルキル)(C1−C6アルキル)およびフルオロより独立して選択されるか、または
(D)は(L)に共有結合する酸素または硫黄原子であり、ここで該(D)基は−H、アルキル、シクロアルキル、ハロアルキルおよびヘテロシクリルより選択される一の置換基R3を有し、一の該置換基R3は−NH2、−N(C1−C6アルキル)(C1−C6アルキル)およびフルオロより独立して選択される0、1、2または3個の置換基を有する
で示される化合物またはその医薬上許容される塩または溶媒和物と、医薬上許容される担体とを含む組成物を、LSD1活性を阻害するのに十分な量にて投与することを含む、方法を提供する。この態様はLSD1阻害剤として用いるために本願明細書にて上記した式I’の化合物またはその医薬上許容される塩または溶媒和物として再構成され得る。この態様はまた、LSD1と関連する疾患の治療または予防にて用いるための、式I’の化合物またはその医薬上許容される塩または溶媒和物として再構成され得る。
N1−[(トランス)−2−フェニルシクロプロピル]−N2−ウンデシル−rel−1,2−エタンジアミン(CAS登録番号627525−03−5に相当する);
N1−[(トランス)−2−フェニルシクロプロピル]−N2−トリシクロ[3.3.1.13,7]デカ−2−イル−rel−1,2−エタンジアミン(CAS登録番号627519−38−4に相当する);
N1−シクロオクチル−N2−[(トランス)−2−フェニルシクロプロピル]−rel−1,2−エタンジアミン(CAS登録番号627519−36−2に相当する);
N1,N1−ジメチル−N2−(2−フェニルシクロプロピル)−1,2−エタンジアミン(CAS登録番号106572−06−9に相当する);
N,N−ジメチル−N’−(2−フェニルシクロプロピル)−1,3−プロパンジアミン(CAS登録番号100407−49−6に相当する);および
トランス−1−フェニル−2−[(2−ヒドロキシエチル)アミノ]シクロプロパン(CAS登録番号32752−04−8に相当する);
またはそのエナンチオマー、ジアステレオマー、またはその混合物。
(A’)x−(A)−(B)−(Z)−(L)−(D)
I
で示される化合物またはそのエナンチオマー、ジアステレオマー、またはその混合物、あるいはその医薬上許容される塩または溶媒和物であって、ここで(A’)、x、(A)、(B)、(Z)、(L)および(D)は以下に示される意義または好ましい意義を有する。
N1−[(トランス)−2−フェニルシクロプロピル]−N2−ウンデシル−rel−1,2−エタンジアミン;
N1−[(トランス)−2−フェニルシクロプロピル]−N2−トリシクロ[3.3.1.13,7]デカ−2−イル−rel−1,2−エタンジアミン;
N1−シクロオクチル−N2−[(トランス)−2−フェニルシクロプロピル]−rel−1,2−エタンジアミン;
N1,N1−ジメチル−N2−(2−フェニルシクロプロピル)−1,3−プロパンジアミン;
N1,N1−ジメチル−N2−(2−フェニルシクロプロピル)−1,2−エタンジアミン;および
トランス−1−フェニル−2−[(2−ヒドロキシエチル)アミノ]シクロプロパン。
本願明細書で使用される場合、「アルキル」なる語は、直鎖および/または分岐鎖基を含め、1〜20個の炭素原子を有する飽和脂肪族炭化水素をいう(本願明細書にて明らかな場合はいつでも、「1〜20」などの数値範囲は所定の範囲内にある各整数をいい:例えば、「1〜20個の炭素原子」はアルキル基が1個の炭素原子、2個の炭素原子、3個の炭素原子等、20個までの炭素原子からなってもよいことを意味する)。好ましくは、「アルキル」は1〜10個の炭素原子を有する。より好ましくは、「アルキル」または「低級アルキル」は1〜6個の炭素原子を有し、その上さらに好ましくは1〜4個の炭素原子を有する。
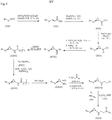
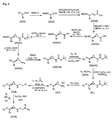
本願発明の化合物は、スキーム1、2 、3、4、5、6および7に記載の一般的経路により合成され得る。
以下の実施例の化合物の構造に対応する名称を付すのに用いたプログラムはMDL ISIS Draw 2.5(ISIS Draw add-in用のACD/Nameを用いる)またはCHEMDRAW(CambridgeSoft製のChemBioDraw Ultra version 11.0.1)であった。このプログラムは該分子をその入力構造の配置により(1S,2R)配置を命名し、該プログラムにより特定される(1S,2R)なる語の代わりに「トランス」なる語が用いられる。以下の実施例に記載される構造はフェニルシクロプロピルアミン核のシクロプロピル炭素原子について一の特定の立体化学配置(1S,2R)を有するものとして示される。実施例で合成される化合物はすべて配置(1R,2S)と(1S,2R)の両方を有する混合物であり、いわゆる、シクロプロピル環系のシクロプロピル環について「トランス」である。これは使用する出発物質のフェニルシクロプロピルアミンが「トランス」であることによるものである。シス配置の出発物質または個々のジアステレオマーを用いることができ、それらは市販されているか、合成可能であることが理解されよう。かくして、本願発明は、シクロプロピル環について特定の立体化学配置を有する化合物、例えば、トランス((1R,2S)および(1S,2R))およびシス((1R,2R)および(1S,2S))または個々のそのジアステレオマーに関する。フェニルシクロプロピルアミンのシクロプロピル環について好ましい立体化学配置はトランスである。
1H−NMR(CDCl3、250MHz、δ):1.10−1.02(m,2H)、1.46(s,9H)、1.99−1.94(m,1H)、2.66(br,1H)、4.90(br,1H)、6.46(br,1H)、6.69(d,2H)、6.93(d,2H)
中間体E(300mg、1当量)の乾燥DMF(2倍容量)中溶液を、1.5当量のNaHの乾燥DMF(10倍容量)中懸濁液に0℃で添加した。30分間攪拌した後、1.1当量の1−(2−ブロモエチル)−4−メチルピペラジンを加え、0℃ないし室温で16時間攪拌した。反応の進行をTLCでモニター観察し、反応終了後、該反応混合物を水上に注ぎ、EtOAc(2x20mL)で抽出した。合した抽出液を水(10mL)、ブライン(20mL)で洗浄し、無水Na2SO4で乾燥させ、濾過し、蒸発させて220mgの粗生成物を得た。それを分取用HPLCで精製し、90mgの(トランス)−2−(4−(ベンジルオキシ)フェニル)−N−(2−(4−メチルピペラジン−1−イル)エチル)シクロプロパンアミンを得た。收率:28%
0℃のHCl/1,4−ジオキサン(4mL)を(トランス)−2−(4−(ベンジルオキシ)フェニル)−N−(2−(4−メチルピペラジン−1−イル)エチル)シクロプロパンアミン(120mg、1当量)のジオキサン(5mL)中冷却溶液に添加し、16時間攪拌した。反応の進行をTLCでモニター観察した。反応終了後、濾過して62mgの(トランス)−2−(4−(ベンジルオキシ)フェニル)−N−(2−(4−メチルピペラジン−1−イル)エチル)シクロプロパンアミン塩酸塩を得た。收率:43%
1H−NMR(DMSO−d6)δ(ppm):1.21(q,1H)、1.54(quin,1H)、2.76(s,3H)、2.91(br,2H)、3.09(br,2H)、3.28(br,2H)、3.44(br,2H)、5.08(s,2H)、6.94(d,2H)、7.11(d,2H)、7.33(m,1H)、7.40(m,4H)、9.50(br,1H)、10.92(br,1H) MS(M+H):366.2
1H−NMR(CDCl3)δ(ppm):1.21(m,1H)、1.44(m,1H)、2.41(m,1H)、2.92(m,1H)、3.20(m,2H)、3.61(m,2H)、7.19−7.25(m,5H)、9.2(bs,2H) MS(M+H):192.
(トランス)−2−(4−ヒドロキシフェニル)シクロプロピルカルバミン酸tert−ブチル(中間体N、10g、40.16ミリモル)のDMF中溶液に、K2CO3(13.75g、100.40ミリモル)および4−ブロモベンジルブロミド(10.03、40.16ミリモル)を添加し、室温で18時間攪拌した。反応の終了をTLCでモニター観察し、それを水(100mL)中に注ぎ、EtOAc(2x100mL)で抽出した。合わせた有機抽出液を水(50mL)、ブライン(50mL)で洗浄し、無水Na2SO4で乾燥させ、濾過し、蒸発させた。粗残渣をEtOAc:石油エーテル(2:8)を用いるカラムクロマトグラフィー(SiO2)に付して精製し、(トランス)−2−(4−(4−ブロモベンジルオキシ) フェニル)シクロプロピルカルバミン酸tert−ブチル(11g、654.86%)を白色固体として得た。
(トランス)−2−(4−(4−ブロモベンジルオキシ)フェニル)シクロプロピルカルバミン酸tert−ブチル(11g、26.37ミリモル)のジオキサン(110mL)中溶液に、0℃で、HCl/ジオキサン(110mL)を添加し、1時間攪拌した。反応完了後、溶媒を蒸発させ、残渣をEt2O(15mL)でトリチュレートし、粗塩を得た、該粗塩を水(150mL)に溶かし、Na2CO3溶液で塩基性にし、EtOAc(3x100mL)で抽出した。合わせた抽出液を水(50mL)、ブライン(50mL)で洗浄し、無水Na2SO4で乾燥させ、濾過し、蒸発させて(トランス)−2−(4−(4−ブロモベンジルオキシ)フェニル)シクロプロパンアミン(8.2g、98%)を得た。
(トランス)−2−(4−(4−ブロモベンジルオキシ)フェニル)シクロプロパンアミン(4.2g、13.24ミリモル)のDCM(42mL)中溶液に、4Åモレキュラーシーブを加え、つづいてクロロアセトアルデヒド(1.0g、13.24ミリモル)を添加し、室温で1時間攪拌した。反応終了後、TLCでモニター観察し、反応混合物を−10℃に冷却し、Na(CN)BH3(0.99mg、15.88ミリモル)を加え、室温で2時間攪拌した。反応終了後、反応混合物をNH4Cl(5%)でクエンチし、セライトパッドを介して濾過した。濾液をDCM(2x150mL)で抽出し、合わせた抽出液を水(50mL)で洗浄し、無水Na2SO4で乾燥させ、濾過して蒸発させた。残渣をEtOAc:石油エーテルを用いるフラッシュカラムクロマトグラフィーに付して精製し、(トランス)−2−(4−(4−ブロモベンジルオキシ)フェニル)−N−(2−クロロエチル)シクロプロパンアミン(4g、80%)を得た。
(トランス)−2−(4−(4−ブロモベンジルオキシ)フェニル)−N−(2−クロロエチル)シクロプロパンアミン(5g、13.15ミリモル)の乾燥DMF(25mL)中溶液に、N−Bocピロリジン(5.13g、27.6ミリモル)を加え、ついで室温で18時間攪拌した。添加終了後、反応混合物を氷水(50mL)中に注ぎ、EtOAc(2x50mL)で抽出した。合わせた抽出液を水(50mL)、ブラインで洗浄し、無水Na2SO4で乾燥させ、濾過し、蒸発させた。粗残渣をMeOH:CHCl3(4:96)を用いるカラムクロマトグラフィー(SiO2)に付して精製し、(R)−1−(2−((トランス)−2−(4−(4−ブロモベンジルオキシ)フェニル)シクロプロピルアミノ)エチル)ピロリジン−3−イルカルバミン酸tert−ブチル(1.7g、24.63 %)を白色固体として得た。
(R)−1−(2−((トランス)−2−(4−(4−ブロモベンジルオキシ)フェニル)シクロプロピルアミノ)エチル)ピロリジン−3−イルカルバミン酸tert−ブチル(0.6g、1.132ミリモル)のジオキサン(6mL)中冷却溶液に、0℃にて、HCl/1,4−ジオキサン(6mL)を添加し、ついで室温で16時間攪拌した。反応の進行をTLCでモニター観察した。反応終了後、溶媒を蒸発させ、残渣をEt2Oでトリチュレートし、(R)−1−(2−((トランス)−2−(4−(4−ブロモベンジルオキシ)フェニル)シクロプロピルアミノ)エチル)ピロリジン−3−アミン塩酸塩(500mg、82%)を白色固体として得た。
1H−NMR(D2O)δ(ppm):1.38(q,1H)、1.50(quin,1H)、2.17(m,1H)、2.52(m,1H)、2.61(m,1H)、2.95(m,1H)、3.43(m,2H)、3.58(m,5H)、3.78(m,1H)、4.17(m,1H)、5.11(s,2H)、6.99(d,2H)、7.14(d,2H)、7.37(d,2H)、7.56(d,2H) MS(M+H):430.1
(トランス)−2−(4−ブロモフェニル) シクロプロピルカルバミン酸tert−ブチル(中間体H、5g、16.02ミリモル)、3−トリフルオロメチル安息香酸(3.6g、19.23ミリモル)およびK2CO3(7.9g、57.69ミリモル)のCH3CN:H2O(4:1)中溶液を20分間脱気した。Pd(PPh3)4(0.185g、0.160ミリモル)を添加し、4時間加熱還流した。反応終了後、該反応混合物を水(100mL)中に注ぎ、EtOAc(2x50mL)で抽出した。合わせた有機抽出液を水(50mL)、ブライン(50mL)で洗浄し、無水Na2SO4で乾燥させ、濾過し、蒸発させた。粗残渣をEtOAc:石油エーテル(2:8)を用いるカラムクロマトグラフィー(SiO2)に付して精製し、(トランス)−2−(3’−(トリフルオロメチル)ビフェニル−4−イル)シクロプロピルカルバミン酸tert−ブチル(5g、83%)を得た。
(トランス)−2−(3’−(トリフルオロメチル)ビフェニル−4−イル)シクロプロピルカルバミン酸tert−ブチル(5g)のジエチルエーテル(50mL)中溶液に、0℃にて、HCl/ジエチルエーテル(20mL)を添加し、1時間攪拌した。完了後、溶媒を蒸発させ、残渣をEt2O(20mL)でトリチュレートし、粗塩を得た。該粗塩を水(50mL)に溶かし、Na2CO3溶液で塩基性にし、EtOAc(3x50mL)で抽出した。合わせた抽出液を水(50mL)、ブライン(50mL)で洗浄し、無水Na2SO4で乾燥させ、濾過し、蒸発させて粗(トランス)−2−(3’−(トリフルオロメチル)ビフェニル−4−イル)シクロプロパンアミン(3.6g、98.09%)を白色固体として得た。
(トランス)−2−(3’−(トリフルオロメチル)ビフェニル−4−イル)シクロプロパンアミン(3.6g、38.26ミリモル)のDCM(36mL)中溶液に、4Åモレキュラーシーブを加え、つづいてクロロアセトアルデヒド(5.8mL、38.26ミリモル)を添加し、ついで室温で1時間攪拌した。終了後、TLCでモニター観察し、反応混合物を−10℃で冷却し、Na(CN)BH3(2.88g、45.92ミリモル)を加え、室温で2時間攪拌した。反応終了後に、該反応混合物をNH4Cl(5%)でクエンチし、セライトパッドを介して濾過した。濾液をDCM(2x50mL)で抽出し、合わせた抽出液を水(40mL)で洗浄し、無水Na2SO4で乾燥させ、濾過して蒸発させた。残渣をEtOAc:石油エーテルを用いるフラッシュカラムクロマトグラフィーに付して精製し、(トランス)−N−(2−クロロエチル)−2−(3’−(トリフルオロメチル)ビフェニル−4−イル)シクロプロパンアミン(3.2g、72.72%)を液体として得た。
(トランス)−N−(2−クロロエチル)−2−(3’−(トリフルオロメチル)ビフェニル−4−イル)シクロプロパンアミン(750mg、2.21ミリモル)の乾燥DMF(7.5mL)中溶液に、(S)−ピロリジン−3−イルカルバミン酸tert−ブチル(864mg、4.64ミリモル)を添加し、室温で48時間攪拌した。反応終了後、該反応混合物を氷水(25mL)中に注ぎ、EtOAc(2x20mL)で抽出した。合わせた抽出液を水(25mL)、ブライン(20mL)で洗浄し、無水Na2SO4で乾燥させ、濾過し、蒸発させた。粗残渣をMeOH:CHCl3(4:96)を用いるカラムクロマトグラフィー(SiO2)に付して精製し、(S)−1−(2−((トランス)−2−(3’−(トリフルオロメチル)ビフェニル−4−イル)シクロプロピルアミノ)エチル)ピロリジン−3−イルカルバミン酸tert−ブチル(400mg、37%)を白色固体として得た。
(S)−1−(2−((トランス)−2−(3’−(トリフルオロメチル)ビフェニル−4−イル)シクロプロピルアミノ)エチル)ピロリジン−3−イルカルバミン酸tert−ブチル(400mg)のジオキサン(5mL)中の冷却溶液に、0℃にて、HCl/1,4−ジオキサン(5mL)を添加し、室温で16時間攪拌した。反応の進行をTLCでモニター観察した。反応終了後、溶媒を蒸発させ、残渣をEt2Oでトリチュレートし、(S)−1−(2−((トランス)−2−(3’−(トリフルオロメチル)ビフェニル−4−イル)シクロプロピルアミノ)エチル)ピロリジン−3−アミン塩酸塩(250mg、62%)を白色固体として得た。
1H−NMR(D2O)δ(ppm):1.36(q,1H)、1.47(quin,1H)、2.09(m,1H)、2.49(m,2H)、2.97(m,1H)、3.42(m,3H)、3.52(s,4H)、3.74(m,1H)、4.07(m,1H)、7.16(s,2H)、7.52(m,4H)、7.71(m,1H)、7.80(s,1H) MS(M+H):390.2
(トランス)−2−(4−(ベンジルオキシ)−3−ブロモフェニル)シクロプロピルカルバミン酸tert−ブチル(1g、2.4ミリモル)、4−トリフルオロメチルボロン酸(545mg、2.86ミリモル)およびK2CO3(1.17g、8.58ミリモル)のACN:H2O(4:1)中溶液を20分間脱気した。Pd(PPh3)4(27mg、0.02ミリモル)を添加し、4時間加熱還流した。反応終了後、反応混合物を水(20mL)中に注ぎ、EtOAc(2x20mL)で抽出した。合わせた有機抽出液を水(20mL)、ブライン(20mL)で洗浄し、無水Na2SO4で乾燥させ、濾過して蒸発させた。粗残渣をEtOAc:石油エーテル(2:8)を用いるカラムクロマトグラフィー(SiO2)に付して精製し、(トランス)−2−(6−(ベンジルオキシ)−4’−(トリフルオロメチル)ビフェニル−3−イル)シクロプロピルカルバミン酸tert−ブチル(800mg、69.56 %)を得た。
(トランス)−2−(6−(ベンジルオキシ)−4’−(トリフルオロメチル)ビフェニル−3−イル)シクロプロピルカルバミン酸tert−ブチル(800mg、1.65ミリモル)のジエチルエーテル(8mL)中溶液に、0℃で、HCl/ジエチルエーテル(8mL)を加え、1時間攪拌した。反応終了後、溶媒を蒸発させ、残渣をEt2O(5mL)でトリチュレートし、粗塩を得た。該粗塩を水(20mL)に溶かし、Na2CO3溶液で塩基性にし、EtOAc(3x20mL)で抽出した。合わせた抽出液を水(20mL)、ブライン(20mL)で洗浄し、無水Na2SO4で乾燥させ、濾過し、蒸発させて粗(トランス)−2−(6−(ベンジルオキシ)−4’−(トリフルオロメチル)ビフェニル−3−イル)シクロプロパンアミン(520mg、82.5%)を白色固体として得た。
(トランス)−2−(6−(ベンジルオキシ)−4’−(トリフルオロメチル)ビフェニル−3−イル)シクロプロパンアミン(520mg、2.08ミリモル)のDCM(6mL)中溶液に、4Åモレキュラーシーブを加え、つづいてクロロアセトアルデヒド(0.33mL、2.5ミリモル)を添加し、室温で1時間攪拌した。反応終了後、TLCでモニター観察し、反応混合物を−10℃に冷却し、Na(CN)BH3(157mg、2.5ミリモル)を加え、室温で2時間攪拌した。反応終了後、該反応混合物をNH4Cl(5%)でクエンチし、セライトパッドを介して濾過した。濾液をDCM(2x20mL)で抽出し、合わせた抽出液を水(20mL)で洗浄し、無水Na2SO4で乾燥させ、濾過して蒸発させた。該残渣をEtOAc:石油エーテルを用いるフラッシュカラムクロマトグラフィーに付して精製し、(トランス)−2−(6−(ベンジルオキシ)−4’−(トリフルオロメチル)ビフェニル−3−イル)−N−(2−クロロエチル)シクロプロパンアミン(500mg、83.3%)を液体として得た。
(トランス)−2−(6−(ベンジルオキシ)−4’−(トリフルオロメチル)ビフェニル−3−イル)−N−(2−クロロエチル)シクロプロパンアミン(500mg、1.12ミリモル)の乾燥DMF(5mL)中溶液、(R)−ピロリジン−3−イルカルバミン酸tert−ブチル(438mg、2.3ミリモル)を添加し、室温で48時間攪拌した。反応終了後、該反応混合物を氷水(20mL)中に注ぎ、EtOAc(2x20mL)で抽出した。合わせた抽出液を水(25mL)、ブラインで洗浄し、無水Na2SO4で乾燥させ、濾過して蒸発させた。粗残渣をMeOH:CHCl3(4:96)を用いるカラムクロマトグラフィー(SiO2)に付して精製し、(R)−1−(2−((トランス)−2−(6−(ベンジルオキシ)−4’−(トリフルオロメチル)ビフェニル−3−イル)シクロプロピルアミノ)エチル)ピロリジン−3−イルカルバミン酸tert−ブチル(400mg、60.6%)を白色固体として得た。
(R)−1−(2−((トランス)−2−(6−(ベンジルオキシ)−4’−(トリフルオロメチル)ビフェニル−3−イル)シクロプロピルアミノ)エチル)ピロリジン−3−イルカルバミン酸tert−ブチル(400mg、0.67ミリモル)のジオキサン(8mL)中の冷却した溶液に、0℃で、HCl/1,4−ジオキサン(4mL)を添加し、室温で16時間攪拌した。反応の進行をTLCでモニター観察した。反応終了後、溶媒を蒸発させ、残渣をEt2Oでトリチュレートし、(R)−1−(2−((トランス)−2−(6−(ベンジルオキシ)−4’−(トリフルオロメチル)ビフェニル−3−イル)シクロプロピルアミノ)エチル)ピロリジン−3−アミン二塩酸塩(380mg、95%)を白色固体として得た。
1H−NMR(DMSO−d6)δ(ppm):1.32(q,1H)、1.59(quin,1H)、2.25(br,2H)、2.62(m,1H)、3.08(m,1H)、3.40 − 4.00(br,12H)、5.15(s,2H)、7.18(d,2H)、7.23(d,1H)、7.30(m,1H)、7.35(s,4H)、7.77(s,4H)、8.60(br,3H)、10.10(br,2H) MS(M+H):496.2
本願発明の化合物は、そのLSD1を阻害する能力について試験され得る。本願発明の化合物のLSD1阻害能は次のように試験され得る。ヒト組換えLSD1蛋白をBPS Bioscience Inc.より入手した。目的とする発明者らの阻害剤によるLSD1酵素活性およびその阻害割合をモニター観察するために、ジメチル化H3−K4ペプチド(Millipore)を基質として選択した。デメチラーゼ活性を、好気条件下、触媒工程の間に産生されるH2O2の放出をAmplex(登録商標)Redペルオキシド/ペルオキシダーゼ連結アッセイキット(Invitrogen)を用いて測定することで評価した。
表1にて報告されるKi値の範囲は、MAO−Aについて、I=40μMより大きい、およびII=1μMと40μMの間;MAO−Bについて、I=40μMより大きい、II=1μMと40μMの間、およびIII=0.1μMと1μMの間;LSD1について、I=40μMより大きい、II=1μMと40μMの間、III=0.1μMと1μMの間、およびIV=0.001μMと0.1μMの間である。実施例7はMAO−Aで約40μMのKi値を有する。
(R)−1−(2−((トランス)−2−(4−フェネトキシフェニル)シクロプロピルアミノ)エチル)ピロリジン−3−アミン
(R)−1−(2−((トランス)−2−(6−(3−(トリフルオロメチル)フェニル)ピリジン−3−イル)シクロプロピルアミノ)エチル)ピロリジン−3−アミン
(R)−1−(2−((トランス)−2−(6−(3−メトキシフェニル)ピリジン−3−イル)シクロプロピルアミノ)エチル)ピロリジン−3−アミン
(R)−1−(2−((トランス)−2−(6−(4−クロロフェニル)ピリジン−3−イル)シクロプロピルアミノ)エチル)ピロリジン−3−アミン
4−((4−((トランス)−2−(2−((R)−3−アミノピロリジン−1−イル)エチルアミノ)シクロプロピル)フェノキシ)メチル)ベンゾニトリル
ヒト結腸癌細胞系HCT116をAmerican Type Culture Collection(ATCC;CCL−247)より入手した。HCT116細胞系は10%ウシ胎仔血清を捕捉したDMEM GlutaMAX(Invitrogen)中に維持された。
薬物を添加する24時間前に、100μlの培地中、6000細胞/ウェルの密度で、細胞を96−ウェルプレートに置いた。ついで、100μM〜0.45nM(各濃度を3回重複して試験する)の薬物を投与した。そのために、2倍のスクリーニング濃度の薬物希釈プレートを調製した。72時間後に、アラマーブルー(Biosource, Invitrogen)生存能力アッセイを製造業者のプロトコルに従って行った。簡単には、培地で希釈したアラマーブルーを細胞に加え、5%溶液を得た。細胞を37℃で3時間および室温で30分間インキュベートした。薬物不含細胞、および薬物を含まず、トリトンX−100で溶解させた細胞を対照として用いた。530nmの励起および590nmの発光波長で蛍光をモニター観察した。Infinite F200 Microplate Reader(Tecan Group, Ltd.)を用いて結果を定量化した。EC50を、細胞増殖を50%まで阻害するのに必要な薬物の用量として、Origin 7.0コンピュータプログラムを用いて計算した。
Claims (17)
- 式I:
(A’)x−(A)−(B)−(Z)−(L)−(D)
I
[式中:
(A)はヘテロアリールまたはアリールであり;
(A’)は、存在する場合、各々独立して、アリール、アリールアルコキシ、アリールアルキル、ヘテロシクリル、アリールオキシ、ハロ、アルコキシ、ハロアルキル、シクロアルキル、ハロアルコキシおよびシアノより選択され、ここで(A’)は、各々、ハロ、ハロアルキル、アリール、アリールアルコキシ、アルキル、アルコキシ、シアノ、スルホニル、アミドおよびスルフィニルより独立して選択される0、1、2または3個の置換基で置換されており;
xは0、1、2または3であり;
(B)はシクロプロピル環であり、ここで(A)および(Z)は(B)の異なる炭素原子に共有結合しており;
(Z)は−NH−であり;
(L)は−CH2CH2−、−CH2CH2CH2−および−CH2CH2CH2CH2−より選択され;および
(D)は−N(−R1)−R2、−O−R3および−S−R3より選択され、ここで:
R1およびR2は相互に連結して、R1およびR2が結合する窒素原子と一緒になってヘテロ環式環を形成し、ここで該ヘテロ環式環は−NH2、−NH(C1−C6アルキル)、−N(C1−C6アルキル)(C1−C6アルキル)、アルキル、ハロ、シアノ、アルコキシ、ハロアルキルおよびハロアルコキシより独立して選択される0、1、2または3個の置換基を有するか、または
R1およびR2は−H、アルキル、C3−7シクロアルキル、ハロアルキルおよびヘテロシクリルより独立して選択され、ここで、R1およびR2上の置換基の合計は0、1、2または3であり、該置換基は−NH2、−NH(C1−C6アルキル)、−N(C1−C6アルキル)(C1−C6アルキル)およびフルオロより独立して選択され;および
R3は−H、アルキル、シクロアルキル、ハロアルキルおよびヘテロシクリルより選択され、ここでR3は−NH2、−NH(C1−C6アルキル)、−N(C1−C6アルキル)(C1−C6アルキル)およびフルオロより独立して選択される0、1、2または3個の置換基を有する]
で示される化合物またはそのエナンチオマー、ジアステレオマーまたはその混合物、あるいはその医薬上許容される塩または溶媒和物;
ただし、以下の化合物:
N1−[(トランス)−2−フェニルシクロプロピル]−N2−ウンデシル−rel−1,2−エタンジアミン;
N1,N1−ジメチル−N2−(2−フェニルシクロプロピル)−1,3−プロパンジアミン;
N1,N1−ジメチル−N2−(2−フェニルシクロプロピル)−1,2−エタンジアミン;
トランス−1−フェニル−2−[(2−ヒドロキシエチル)アミン]シクロプロパン;
は除かれる。 - (A)がフェニルである、請求項1記載の化合物あるいはその医薬上許容される塩または溶媒和物。
- xが1であり、(A’)がアリールおよびアリールアルコキシから選択され、ここで該アリールまたは該アリールアルコキシがハロおよびハロアルキルから選択される0または1個の置換基を有する、請求項1または2記載の化合物あるいはその医薬上許容される塩または溶媒和物。
- (D)が−N(−R1)−R2であり、さらにはR1およびR2が相互に連結して、R1およびR2が結合する窒素原子と一緒になってヘテロ環式環を形成し、ここで該ヘテロ環式環が−NH2、−NH(C1−C6アルキル)、−N(C1−C6アルキル)(C1−C6アルキル)、アルキル、ハロ、シアノ、アルコキシ、ハロアルキルおよびハロアルコキシより独立して選択される0、1、2または3個の置換基を有する、請求項1−3のいずれか一項に記載の化合物あるいはその医薬上許容される塩または溶媒和物。
- 該ヘテロ環式環が、飽和状態の4、5または6員のヘテロ環式環である、請求項4記載の化合物あるいはその医薬上許容される塩または溶媒和物。
- 該ヘテロ環式環がアゼチジニル、ピロリジニル、ピペリジニルおよびモルホリノより選択される、請求項4−5のいずれか一項に記載の化合物あるいはその医薬上許容される塩または溶媒和物。
- 該ヘテロ環式環が、−NH2、−N(C1−C6アルキル)(C1−C6アルキル)およびアルキルから選択される0または1個の置換基を有する、請求項4−6のいずれか一項に記載の化合物あるいはその医薬上許容される塩または溶媒和物。
- (D)が−N(−R1)−R2であり、さらにはR1およびR2が−H、アルキル、C3−7シクロアルキルおよびハロアルキルより独立して選択され、ここで、R1およびR2上の置換基の合計が0、1、2または3であり、該置換基が−NH2、−NH(C1−C6アルキル)、−N(C1−C6アルキル)(C1−C6アルキル)およびフルオロより独立して選択される、請求項1−3のいずれか一項に記載の化合物あるいはその医薬上許容される塩または溶媒和物。
- (L)が−CH2CH2−である、請求項1−8のいずれか一項に記載の化合物あるいはその医薬上許容される塩または溶媒和物。
- (A)および(Z)がシクロプロピル環(B)に対してトランス配置にある、請求項1−9のいずれか一項に記載の化合物あるいはその医薬上許容される塩または溶媒和物。
- N−[2−(4−メチルピペラジン−1−イル)エチル]−N−[(トランス)−2−フェニルシクロプロピル]アミン;
N−シクロプロピル−N’−[(トランス)−2−フェニルシクロプロピル]エタン−1,2−ジアミン;
N,N−ジメチル−N’−(2−{[(トランス)−2−フェニルシクロプロピル]アミノ}エチル)エタン−1,2−ジアミン;
(3R)−1−(2−{[(トランス)−2−フェニルシクロプロピル]アミン}エチル)ピロリジン−3−アミン;
(3S)−N,N−ジメチル−1−(2−{[(トランス)−2−フェニルシクロプロピル]アミノ}エチル) ピロリジン−3−アミン;
(3R)−N,N−ジメチル−1−(2−{[(トランス)−2−フェニルシクロプロピル]アミノ}エチル)ピロリジン−3−アミン;
N−[(トランス)−2−フェニルシクロプロピル]−N−(2−ピペラジン−1−イルエチル)アミン;
N1,N1−ジエチル−N2−((トランス)−2−フェニルシクロプロピル)エタン−1,2−ジアミン;
N−[(トランス)−2−フェニルシクロプロピル]−N−(2−ピペリジン−1−イルエチル)アミン;
(トランス)−2−(4−(ベンジルオキシ)フェニル)−N−(2−(4−メチルピペラジン−1−イル)エチル)シクロプロパンアミン;
(トランス)−N−(2−(4−メチルピペラジン−1−イル)エチル)−2−(3’−(トリフルオロメチル)ビフェニル−4−イル)シクロプロパンアミン;
(トランス)−2−(3’−クロロビフェニル−4−イル)−N−(2−(4−メチルピペラジン−1−イル)エチル)シクロプロパンアミン;
(R)−1−(2−((トランス)−2−(3’−(トリフルオロメチル)ビフェニル−4−イル)シクロプロピルアミノ)エチル)ピロリジン−3−アミン;
N1−シクロプロピル−N2−((トランス)−2−(3’−(トリフルオロメチル)ビフェニル−4−イル)シクロプロピル)エタン−1,2−ジアミン;
N1−((トランス)−2−(4−(3−ブロモベンジルオキシ)フェニル)シクロプロピル)−N2−シクロプロピルエタン−1,2−ジアミン;
N1−((トランス)−2−(3’−クロロビフェニル−4−イル)シクロプロピル)−N2−シクロプロピルエタン−1,2−ジアミン;
N1−シクロプロピル−N2−((トランス)−2−(4−フェネトキシフェニル)シクロプロピル)エタン−1,2−ジアミン;
N1,N1−ジエチル−N2−((トランス)−2−(4−(3−フルオロベンジルオキシ)フェニル)シクロプロピル)エタン−1,2−ジアミン;
(トランス)−2−(4−ブロモフェニル)−N−(2−(4−メチルピペラジン−1−イル)エチル)シクロプロパンアミン;
(トランス)−N−(2−(ピペリジン−1−イル)エチル)−2−(3’−(トリフルオロメチル)ビフェニル−4−イル)シクロプロパンアミン;
N1,N1−ジエチル−N2−((トランス)−2−(3’−(トリフルオロメチル)ビフェニル−4−イル)シクロプロピル)エタン−1,2−ジアミン;
(トランス)−N−(2−(ピペラジン−1−イル)エチル)−2−(3’−(トリフルオロメチル)ビフェニル−4−イル)シクロプロパンアミン;
(S)−1−(2−((トランス)−2−(3’−(トリフルオロメチル)ビフェニル−4−イル)シクロプロピルアミノ)エチル)ピロリジン−3−アミン;
(R)−1−(2−((トランス)−2−(3’−クロロビフェニル−4−イル)シクロプロピルアミノ)エチル)ピロリジン−3−アミン;
(R)−1−(2−((トランス)−2−(4’−クロロビフェニル−4−イル)シクロプロピルアミノ)エチル)ピロリジン−3−アミン;
(R)−1−(2−((トランス)−2−(3’−メトキシビフェニル−4−イル)シクロプロピルアミノ)エチル)ピロリジン−3−アミン;
(R)−1−(2−((トランス)−2−(4−(3−ブロモベンジルオキシ)フェニル)シクロプロピルアミノ)エチル)ピロリジン−3−アミン;
(R)−1−(2−((トランス)−2−(6−(3−(トリフルオロメチル)フェニル)ピリジン−3−イル)シクロプロピルアミノ)エチル)ピロリジン−3−アミン;
N−(トランス)−2−(イソブチルチオ)−エチル−2−フェニルシクロプロパンアミン;
N−トランス−(2−エトキシエチル)−2−フェニルシクロプロパンアミン;
N−トランス−(2−メトキシエチル)−2−フェニルシクロプロパンアミン;
(R)−1−(2−((トランス)−2−(4−(4−ブロモベンジルオキシ)フェニル)シクロプロピルアミノ)エチル)ピロリジン−3−アミン;
(R)−1−(2−((トランス)−2−(4−(4−クロロベンジルオキシ)フェニル)シクロプロピルアミノ)エチル)ピロリジン−3−アミン;
(R)−1−(2−((トランス)−2−(4−(ビフェニル−4−イルメトキシ)フェニル)シクロプロピルアミノ)エチル)ピロリジン−3−アミン;
(R)−1−(2−((トランス)−2−(3’,5’−ジクロロビフェニル−4−イル)シクロプロピルアミノ)エチル)ピロリジン−3−アミン;
N1−((トランス)−2−([1,1’;4’,1’’]−テルフェニル−4−イル)シクロプロピル)−N2−シクロプロピルエタン−1,2−ジアミン;
(R)−1−(2−((トランス)−2−(6−(ベンジルオキシ)−4’−(トリフルオロメチル)ビフェニル−3−イル)シクロプロピルアミノ)エチル)ピロリジン−3−アミン;
(R)−1−(2−((トランス)−2−(6−(ベンジルオキシ)ビフェニル−3−イル)シクロプロピルアミノ)エチル)ピロリジン−3−アミン;
(R)−1−(2−((トランス)−2−(4−フェネトキシフェニル)シクロプロピルアミノ)エチル)ピロリジン−3−アミン;
(R)−1−(2−((トランス)−2−(6−(3−メトキシフェニル)ピリジン−3−イル)シクロプロピルアミノ)エチル)ピロリジン−3−アミン;
(R)−1−(2−((トランス)−2−(6−(4−クロロフェニル)ピリジン−3−イル)シクロプロピルアミノ)エチル)ピロリジン−3−アミン;および
4−((4−((トランス)−2−(2−((R)−3−アミノピロリジン−1−イル)エチルアミノ)シクロプロピル)フェノキシ)メチル)ベンゾニトリル
より選択される請求項1記載の化合物またはその医薬上許容される塩または溶媒和物。 - 医薬として用いるための請求項1−11のいずれか一項に記載の式Iの化合物あるいはその医薬上許容される塩または溶媒和物。
- 請求項1−11のいずれか一項に記載の化合物あるいはその医薬上許容される塩または溶媒和物と、医薬上許容される担体とを含む、医薬組成物。
- 癌の治療または予防に用いるための、請求項1−11のいずれか一項に記載の化合物またはその医薬上許容される塩もしくは溶媒和物。
- 癌の治療または予防に用いるための請求項13記載の医薬組成物。
- 該癌が脳腫瘍、結腸直腸癌、乳癌、肺癌、前立腺癌、皮膚癌、精巣癌および血液癌より選択される、請求項14記載の化合物またはその医薬上許容される塩もしくは溶媒和物。
- 該癌が脳腫瘍、結腸直腸癌、乳癌、肺癌、前立腺癌、皮膚癌、精巣癌および血液癌より選択される、請求項15記載の医薬組成物。
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP09171425 | 2009-09-25 | ||
| EP09171425.3 | 2009-09-25 | ||
| EP10150866.1 | 2010-01-15 | ||
| EP10150866 | 2010-01-15 | ||
| PCT/EP2010/055131 WO2011035941A1 (en) | 2009-09-25 | 2010-04-19 | Lysine specific demethylase-1 inhibitors and their use |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2013505903A JP2013505903A (ja) | 2013-02-21 |
| JP2013505903A5 JP2013505903A5 (ja) | 2013-06-06 |
| JP5699152B2 true JP5699152B2 (ja) | 2015-04-08 |
Family
ID=42829238
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2012530181A Expired - Fee Related JP5699152B2 (ja) | 2009-09-25 | 2010-04-19 | リジン特異的デメチラーゼ−1阻害剤およびその使用 |
Country Status (12)
| Country | Link |
|---|---|
| US (1) | US8859555B2 (ja) |
| EP (1) | EP2480528B1 (ja) |
| JP (1) | JP5699152B2 (ja) |
| KR (1) | KR101736218B1 (ja) |
| CN (1) | CN102639496B (ja) |
| AU (1) | AU2010297557C1 (ja) |
| BR (1) | BR112012006572A2 (ja) |
| CA (1) | CA2812683C (ja) |
| IL (1) | IL218778A (ja) |
| MX (1) | MX338041B (ja) |
| RU (1) | RU2602814C2 (ja) |
| WO (1) | WO2011035941A1 (ja) |
Families Citing this family (66)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010043721A1 (en) | 2008-10-17 | 2010-04-22 | Oryzon Genomics, S.A. | Oxidase inhibitors and their use |
| EP2389362B1 (en) | 2009-01-21 | 2019-12-11 | Oryzon Genomics, S.A. | Phenylcyclopropylamine derivatives and their medical use |
| WO2011035941A1 (en) | 2009-09-25 | 2011-03-31 | Oryzon Genomics S.A. | Lysine specific demethylase-1 inhibitors and their use |
| EP2486002B1 (en) | 2009-10-09 | 2019-03-27 | Oryzon Genomics, S.A. | Substituted heteroaryl- and aryl- cyclopropylamine acetamides and their use |
| US9616058B2 (en) * | 2010-02-24 | 2017-04-11 | Oryzon Genomics, S.A. | Potent selective LSD1 inhibitors and dual LSD1/MAO-B inhibitors for antiviral use |
| WO2011106106A2 (en) | 2010-02-24 | 2011-09-01 | Oryzon Genomics, S.A. | Lysine demethylase inhibitors for diseases and disorders associated with hepadnaviridae |
| CN102947265B (zh) | 2010-04-19 | 2015-07-29 | 奥瑞泽恩基因组学股份有限公司 | 赖氨酸特异性脱甲基酶-1抑制剂及其应用 |
| ES2564352T3 (es) * | 2010-04-20 | 2016-03-22 | Università Degli Studi Di Roma "La Sapienza" | Derivados de tranilcipromina como inhibidores de la histona-desmetilasa LSD1 y/o LSD2 |
| US9006449B2 (en) | 2010-07-29 | 2015-04-14 | Oryzon Genomics, S.A. | Cyclopropylamine derivatives useful as LSD1 inhibitors |
| CN103124724B (zh) * | 2010-07-29 | 2015-05-20 | 奥瑞泽恩基因组学股份有限公司 | Lsd1的基于芳基环丙胺的脱甲基酶抑制剂及其医疗用途 |
| US20130303545A1 (en) | 2010-09-30 | 2013-11-14 | Tamara Maes | Cyclopropylamine derivatives useful as lsd1 inhibitors |
| WO2012045883A1 (en) * | 2010-10-08 | 2012-04-12 | Oryzon Genomics S.A. | Cyclopropylamine inhibitors of oxidases |
| WO2012072713A2 (en) | 2010-11-30 | 2012-06-07 | Oryzon Genomics, S.A. | Lysine demethylase inhibitors for diseases and disorders associated with flaviviridae |
| WO2012107498A1 (en) | 2011-02-08 | 2012-08-16 | Oryzon Genomics S.A. | Lysine demethylase inhibitors for myeloproliferative disorders |
| EP2712316A1 (en) | 2011-02-08 | 2014-04-02 | Oryzon Genomics, S.A. | Lysine demethylase inhibitors for myeloproliferative or lymphoproliferative diseases or disorders |
| CA2831143C (en) | 2011-03-25 | 2019-05-21 | Glaxosmithkline Intellectual Property Development Limited | Cyclopropylamines as lsd1 inhibitors |
| EP2750671A2 (en) | 2011-05-19 | 2014-07-09 | Oryzon Genomics, S.A. | Lysine demethylase inhibitors for thrombosis and cardiovascular diseases |
| WO2012156531A2 (en) | 2011-05-19 | 2012-11-22 | Oryzon Genomics, S.A. | Lysine demethylase inhibitors for inflammatory diseases or conditions |
| KR20140052018A (ko) | 2011-08-09 | 2014-05-02 | 다케다 야쿠힌 고교 가부시키가이샤 | 시클로프로판아민 화합물 |
| WO2013033688A1 (en) * | 2011-09-01 | 2013-03-07 | The Brigham And Women's Hospital, Inc. | Treatment of cancer |
| EP4074695A1 (en) | 2011-10-20 | 2022-10-19 | Oryzon Genomics, S.A. | (hetero)aryl cyclopropylamine compounds as lsd1 inhibitors |
| RU2681211C2 (ru) | 2011-10-20 | 2019-03-05 | Оризон Дженомикс С.А. | (гетеро)арилциклопропиламины в качестве ингибиторов lsd1 |
| JP6325449B2 (ja) * | 2012-10-12 | 2018-05-16 | 武田薬品工業株式会社 | シクロプロパンアミン化合物およびその用途 |
| EP2740474A1 (en) | 2012-12-05 | 2014-06-11 | Instituto Europeo di Oncologia S.r.l. | Cyclopropylamine derivatives useful as inhibitors of histone demethylases kdm1a |
| EP3003301B1 (en) * | 2013-05-30 | 2021-02-24 | Board Of Regents Of The Nevada System Of Higher Education On Behalf Of The University Of Nevada, Las Vegas | Novel suicidal lsd1 inhibitors targeting sox2-expressing cancer cells |
| CN105592888A (zh) | 2013-08-06 | 2016-05-18 | 伊美格生物科学公司 | 用于治疗疾病的kdm1a抑制剂 |
| US9186391B2 (en) | 2013-08-29 | 2015-11-17 | Musc Foundation For Research Development | Cyclic peptide inhibitors of lysine-specific demethylase 1 |
| LT3105218T (lt) | 2014-02-13 | 2019-12-10 | Incyte Corp | Ciklopropilaminai kaip lsd1 inhibitoriai |
| HUE045725T2 (hu) | 2014-02-13 | 2020-01-28 | Incyte Corp | Ciklopropilaminok mint LSD1 inhibitorok |
| WO2015123437A1 (en) | 2014-02-13 | 2015-08-20 | Incyte Corporation | Cyclopropylamines as lsd1 inhibitors |
| EP3392244A1 (en) | 2014-02-13 | 2018-10-24 | Incyte Corporation | Cyclopropylamines as lsd1 inhibitors |
| CN106459024B (zh) | 2014-04-11 | 2019-11-05 | 武田药品工业株式会社 | 环丙胺化合物及其用途 |
| TW201613925A (en) | 2014-07-10 | 2016-04-16 | Incyte Corp | Imidazopyrazines as LSD1 inhibitors |
| US9758523B2 (en) | 2014-07-10 | 2017-09-12 | Incyte Corporation | Triazolopyridines and triazolopyrazines as LSD1 inhibitors |
| US9695167B2 (en) | 2014-07-10 | 2017-07-04 | Incyte Corporation | Substituted triazolo[1,5-a]pyridines and triazolo[1,5-a]pyrazines as LSD1 inhibitors |
| WO2016007731A1 (en) | 2014-07-10 | 2016-01-14 | Incyte Corporation | Imidazopyridines and imidazopyrazines as lsd1 inhibitors |
| KR102626978B1 (ko) | 2015-02-12 | 2024-01-18 | 이마고 바이오사이언시즈 인코포레이티드 | 질환 치료를 위한 kdm1a 저해제 |
| MA51438A (fr) | 2015-04-03 | 2021-04-14 | Incyte Corp | Composés hétérocycliques utilisés en tant qu'inhibiteurs de lsd1 |
| EP3090998A1 (en) | 2015-05-06 | 2016-11-09 | F. Hoffmann-La Roche AG | Solid forms |
| SG10201911989SA (en) | 2015-06-12 | 2020-02-27 | Oryzon Genomics Sa | Biomarkers associated with lsd1 inhibitors and uses thereof |
| WO2017013061A1 (en) | 2015-07-17 | 2017-01-26 | Oryzon Genomics, S.A. | Biomarkers associated with lsd1 inhibitors and uses thereof |
| MX2018001706A (es) | 2015-08-12 | 2018-09-06 | Incyte Corp | Sales de un inhibidor de dimetilasa 1 especifica para lisina (lsd1). |
| US10059668B2 (en) | 2015-11-05 | 2018-08-28 | Mirati Therapeutics, Inc. | LSD1 inhibitors |
| CA3009805C (en) | 2015-12-29 | 2023-10-17 | Mirati Therapeutics, Inc. | Lsd1 inhibitors |
| RU2018127657A (ru) | 2015-12-30 | 2020-01-31 | Новартис Аг | Виды терапии на основе иммуноэффекторных клеток с улучшенной эффективностью |
| SG11201807965YA (en) | 2016-03-15 | 2018-10-30 | Oryzon Genomics Sa | Combinations of lsd1 inhibitors for use in the treatment of solid tumors |
| WO2017157813A1 (en) | 2016-03-15 | 2017-09-21 | F. Hoffmann-La Roche Ag | Combinations of lsd1 inhibitors for the treatment of hematological malignancies |
| JP2019512546A (ja) * | 2016-03-16 | 2019-05-16 | オリゾン・ゲノミクス・ソシエダッド・アノニマ | Kdm1a標的会合を決定するための方法、およびそれに有用な化学プローブ |
| CN109414410B (zh) | 2016-04-22 | 2022-08-12 | 因赛特公司 | Lsd1抑制剂的制剂 |
| PL3307267T3 (pl) | 2016-06-10 | 2019-10-31 | Oryzon Genomics Sa | Leczenie stwardnienia rozsianego |
| US11390590B2 (en) | 2016-08-16 | 2022-07-19 | Imago Biosciences, Inc. | Methods and processes for the preparation of KDM1A inhibitors |
| US20190256930A1 (en) | 2016-11-03 | 2019-08-22 | Oryzon Genomics, S.A. | Biomarkers for determining responsiveness to lsd1 inhibitors |
| MA50466A (fr) * | 2017-05-26 | 2020-09-02 | Taiho Pharmaceutical Co Ltd | Nouveau composé de biphényle ou sel de celui-ci |
| WO2019025588A1 (en) | 2017-08-03 | 2019-02-07 | Oryzon Genomics, S.A. | METHODS OF TREATING ALTERATIONS IN BEHAVIOR |
| WO2019068326A1 (en) | 2017-10-05 | 2019-04-11 | Université D'aix-Marseille | INHIBITORS OF LSD1 FOR THE TREATMENT AND PREVENTION OF CARDIOMYOPATHIES |
| JP7450559B2 (ja) | 2018-05-11 | 2024-03-15 | イマーゴ バイオサイエンシーズ インコーポレイテッド | 疾患の処置のためのkdm1a阻害剤 |
| WO2020047198A1 (en) | 2018-08-31 | 2020-03-05 | Incyte Corporation | Salts of an lsd1 inhibitor and processes for preparing the same |
| SG11202109159VA (en) | 2019-03-20 | 2021-10-28 | Oryzon Genomics Sa | Methods of treating borderline personality disorder |
| EP3941465A1 (en) | 2019-03-20 | 2022-01-26 | Oryzon Genomics, S.A. | Methods of treating attention deficit hyperactivity disorder using kdm1a inhibitors such as the compound vafidemstat |
| JP2022546908A (ja) | 2019-07-05 | 2022-11-10 | オリゾン・ゲノミクス・ソシエダッド・アノニマ | Kdm1a阻害剤を使用した小細胞肺がんの個別化された処置のためのバイオマーカーおよび方法 |
| CN115667190A (zh) * | 2020-06-17 | 2023-01-31 | 贝达药业股份有限公司 | 双环类化合物及其应用 |
| EP3964204A1 (en) | 2020-09-08 | 2022-03-09 | Université d'Aix-Marseille | Lsd1 inhibitors for use in the treatment and prevention of fibrosis of tissues |
| EP4319732A1 (en) | 2021-04-08 | 2024-02-14 | Oryzon Genomics, S.A. | Combinations of lsd1 inhibitors for treating myeloid cancers |
| WO2023217758A1 (en) | 2022-05-09 | 2023-11-16 | Oryzon Genomics, S.A. | Methods of treating malignant peripheral nerve sheath tumor (mpnst) using lsd1 inhibitors |
| WO2023217784A1 (en) | 2022-05-09 | 2023-11-16 | Oryzon Genomics, S.A. | Methods of treating nf1-mutant tumors using lsd1 inhibitors |
| CN114853680A (zh) * | 2022-06-06 | 2022-08-05 | 沈阳药科大学 | 芳香六元环并咪唑类衍生物及其制备方法和应用 |
Family Cites Families (125)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3106578A (en) | 1960-09-16 | 1963-10-08 | Smith Kline French Lab | Nu-phenethyl-2-phenylcyclopropylamine derivatives |
| US3365458A (en) | 1964-06-23 | 1968-01-23 | Aldrich Chem Co Inc | N-aryl-n'-cyclopropyl-ethylene diamine derivatives |
| US3532749A (en) | 1965-05-11 | 1970-10-06 | Aldrich Chem Co Inc | N'-propargyl-n**2-cyclopropyl-ethylenediamines and the salts thereof |
| US3532712A (en) | 1967-09-29 | 1970-10-06 | Aldrich Chem Co Inc | N'-cyclopropyl ethylenediamine derivatives |
| US3471522A (en) | 1967-09-29 | 1969-10-07 | Aldrich Chem Co Inc | N-cyclopropyl-n'-furfuryl-n'-methyl ethylene diamines |
| US3654306A (en) | 1970-01-26 | 1972-04-04 | Robins Co Inc A H | 5-azaspiro(2.4)heptane-4 6-diones |
| US3758684A (en) | 1971-09-07 | 1973-09-11 | Burroughs Wellcome Co | Treating dna virus infections with amino purine derivatives |
| US4530901A (en) | 1980-01-08 | 1985-07-23 | Biogen N.V. | Recombinant DNA molecules and their use in producing human interferon-like polypeptides |
| US4409243A (en) | 1981-11-09 | 1983-10-11 | Julian Lieb | Treatment of auto-immune and inflammatory diseases |
| US4522811A (en) | 1982-07-08 | 1985-06-11 | Syntex (U.S.A.) Inc. | Serial injection of muramyldipeptides and liposomes enhances the anti-infective activity of muramyldipeptides |
| GB9311282D0 (en) | 1993-06-01 | 1993-07-21 | Rhone Poulenc Rorer Ltd | New compositions of matter |
| ES2234768T3 (es) | 1994-10-21 | 2005-07-01 | Nps Pharmaceuticals, Inc. | Compuestos capaces de modular la actividad del receptor de calcio. |
| US5652258A (en) | 1995-05-30 | 1997-07-29 | Gliatech, Inc. | 2-(4-imidazoyl) cyclopropyl derivatives |
| US20040132820A1 (en) | 1996-02-15 | 2004-07-08 | Jean Gosselin | Agents with leukotriene B4-like antiviral (DNA) and anti-neoplastic activities |
| GB9615730D0 (en) | 1996-07-26 | 1996-09-04 | Medical Res Council | Anti-viral agent 1 |
| US5961987A (en) | 1996-10-31 | 1999-10-05 | University Of Iowa Research Foundation | Ocular protein stimulants |
| DE19647615A1 (de) | 1996-11-18 | 1998-05-20 | Bayer Ag | Verfahren zur Herstellung von Cyclopropylaminen |
| AR017014A1 (es) | 1997-07-22 | 2001-08-22 | Astrazeneca Ab | Compuestos de triazolo [4,5-d]pirimidina, composiciones farmaceuticas, uso de los mismos para preparar medicamentos y procesos para la preparacionde dichos compuestos |
| SE9702772D0 (sv) | 1997-07-22 | 1997-07-22 | Astra Pharma Prod | Novel compounds |
| AU1631699A (en) | 1997-12-18 | 1999-07-05 | E.I. Du Pont De Nemours And Company | Cyclohexylamine arthropodicides and fungicides |
| US6809120B1 (en) | 1998-01-13 | 2004-10-26 | University Of Saskatchewan Technologies Inc. | Composition containing propargylamine for enhancing cancer therapy |
| ES2203141T3 (es) | 1998-04-21 | 2004-04-01 | Micromet Ag | Polipeptidos cd19 x cd3 especificos y su utilizacion. |
| SE9802206D0 (sv) | 1998-06-22 | 1998-06-22 | Astra Pharma Inc | Novel compounds |
| TWI229674B (en) | 1998-12-04 | 2005-03-21 | Astra Pharma Prod | Novel triazolo[4,5-d]pyrimidine compounds, pharmaceutical composition containing the same, their process for preparation and uses |
| JP4574112B2 (ja) | 2000-05-26 | 2010-11-04 | シェーリング コーポレイション | アデノシンa2aレセプターアンタゴニスト |
| JP2001354563A (ja) | 2000-06-09 | 2001-12-25 | Sankyo Co Ltd | 置換ベンジルアミン類を含有する医薬 |
| US8519005B2 (en) | 2000-07-27 | 2013-08-27 | Thomas N. Thomas | Compositions and methods to prevent toxicity of antiinflammatory agents and enhance their efficacy |
| EP1193268A1 (en) | 2000-09-27 | 2002-04-03 | Applied Research Systems ARS Holding N.V. | Pharmaceutically active sulfonamide derivatives bearing both lipophilic and ionisable moieties as inhibitors of protein Junkinases |
| WO2002079152A1 (en) | 2001-03-29 | 2002-10-10 | Bristol-Myers Squibb Company | Cyclopropylindole derivatives as selective serotonin reuptake inhibitors |
| DE10123163A1 (de) | 2001-05-09 | 2003-01-16 | Gruenenthal Gmbh | Substituierte Cyclohexan-1,4-diaminderivate |
| US20030008844A1 (en) | 2001-05-17 | 2003-01-09 | Keryx Biopharmaceuticals, Inc. | Use of sulodexide for the treatment of inflammatory bowel disease |
| US7544675B2 (en) | 2002-04-18 | 2009-06-09 | Ucb, S.A. | Chemical compounds with dual activity, processes for their preparation and pharmaceutical compositions |
| US7704995B2 (en) | 2002-05-03 | 2010-04-27 | Exelixis, Inc. | Protein kinase modulators and methods of use |
| EP1501514B1 (en) | 2002-05-03 | 2012-12-19 | Exelixis, Inc. | Protein kinase modulators and methods of use |
| AU2003273179A1 (en) | 2002-05-10 | 2003-12-12 | Bristol-Myers Squibb Company | 1,1-disubstituted cycloalkyl derivatives as factor xa inhibitors |
| US20040033986A1 (en) | 2002-05-17 | 2004-02-19 | Protopopova Marina Nikolaevna | Anti tubercular drug: compositions and methods |
| US6951961B2 (en) | 2002-05-17 | 2005-10-04 | Marina Nikolaevna Protopopova | Methods of use and compositions for the diagnosis and treatment of infectious disease |
| US7456222B2 (en) | 2002-05-17 | 2008-11-25 | Sequella, Inc. | Anti tubercular drug: compositions and methods |
| AU2003299519A1 (en) | 2002-05-20 | 2004-05-04 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| US7611704B2 (en) | 2002-07-15 | 2009-11-03 | Board Of Regents, The University Of Texas System | Compositions and methods for treating viral infections using antibodies and immunoconjugates to aminophospholipids |
| SE0202539D0 (sv) | 2002-08-27 | 2002-08-27 | Astrazeneca Ab | Compounds |
| EP1569934B1 (en) | 2002-12-13 | 2008-01-23 | Smithkline Beecham Corporation | Cyclopropyl compounds as ccr5 antagonists |
| CA2851462A1 (en) | 2003-01-08 | 2004-07-29 | University Of Washington | Antibacterial agents |
| US7223785B2 (en) | 2003-01-22 | 2007-05-29 | Boehringer Ingelheim International Gmbh | Viral polymerase inhibitors |
| GB0303439D0 (en) | 2003-02-14 | 2003-03-19 | Pfizer Ltd | Antiparasitic terpene alkaloids |
| US7186832B2 (en) | 2003-02-20 | 2007-03-06 | Sugen Inc. | Use of 8-amino-aryl-substituted imidazopyrazines as kinase inhibitors |
| US7135575B2 (en) | 2003-03-03 | 2006-11-14 | Array Biopharma, Inc. | P38 inhibitors and methods of use thereof |
| EP1631548B1 (en) | 2003-04-24 | 2009-10-28 | Merck & Co., Inc. | Inhibitors of akt activity |
| JP2007520440A (ja) | 2003-07-03 | 2007-07-26 | イーライ リリー アンド カンパニー | ムスカリン性受容体アゴニストとしてのインダン誘導体 |
| JP4895811B2 (ja) | 2003-09-11 | 2012-03-14 | ケミア,インコーポレイテッド | サイトカイン阻害剤 |
| WO2005025558A1 (en) | 2003-09-12 | 2005-03-24 | Applied Reserach Systems Ars Holding N.V. | Sulfonamide derivatives for the treatment of diabetes |
| CN1897950A (zh) | 2003-10-14 | 2007-01-17 | 惠氏公司 | 稠合芳基和杂芳基衍生物及其使用方法 |
| CA2542536A1 (en) | 2003-10-21 | 2005-05-12 | Merck & Co., Inc. | Triazolo-pyridazine compounds and derivatives thereof useful in the treatment of neuropathic pain |
| US7026339B2 (en) | 2003-11-07 | 2006-04-11 | Fan Yang | Inhibitors of HCV NS5B polymerase |
| WO2005058808A1 (en) | 2003-12-15 | 2005-06-30 | Japan Tobacco Inc. | N-substituted-n-sulfonylaminocyclopropane compounds and pharmaceutical use thereof |
| AU2004299455A1 (en) | 2003-12-15 | 2005-06-30 | Japan Tobacco Inc. | Cyclopropane compounds and pharmaceutical use thereof |
| UA83101C2 (en) | 2003-12-15 | 2008-06-10 | Алмирал Аг | 2,6-bisheteroaryl-4-aminopyrimidines as adenosine receptor antagonists |
| US7399825B2 (en) | 2003-12-24 | 2008-07-15 | Lipps Binie V | Synthetic peptide, inhibitor to DNA viruses |
| EP1756103A2 (en) | 2004-04-26 | 2007-02-28 | Pfizer, Inc. | Pyrrolopyridine derivatives and their use as hiv-integrase inhibitors |
| US20090275099A1 (en) | 2004-04-27 | 2009-11-05 | Regents Of The University Of Michigan | Methods and compositions for treating diseases and conditions associated with mitochondrial function |
| DE102004057594A1 (de) | 2004-11-29 | 2006-06-08 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Substitueirte Pteridine zur Behandlung von entzündlichen Erkrankungen |
| CA2591717A1 (en) | 2004-12-16 | 2006-07-06 | President And Fellows Of Harvard College | Histone demethylation mediated by the nuclear amine oxidase homolog lsd1 |
| DK1704859T3 (da) | 2005-02-18 | 2010-11-22 | Universitaetsklinikum Freiburg | Kontrol af androgen receptor-afhængig genenkspression ved inhibering af amin-oxidase-aktiviteten af lysin-specifik demetylase (LSD1) |
| US20060275366A1 (en) | 2005-06-02 | 2006-12-07 | Schering Corporation | Controlled-release formulation |
| WO2007002220A2 (en) | 2005-06-21 | 2007-01-04 | Bristol-Myers Squibb Company | Aminoacetamide acyl guanidines as beta-secretase inhibitors |
| EP1741708A1 (en) | 2005-06-28 | 2007-01-10 | Sanofi-Aventis Deutschland GmbH | Heteroaryl-substituted amides comprising an unsaturated or cyclic linker group, and their use as pharmaceuticals |
| AU2006276246B2 (en) | 2005-07-25 | 2012-09-27 | Intermune, Inc. | Novel macrocyclic inhibitors of hepatitis C virus replication |
| EP1940773B1 (en) | 2005-08-10 | 2015-03-18 | Johns Hopkins University | Polyamines useful as anti-parasitic and anti-cancer therapeutics and as lysine-specific demethylase inhibitors |
| US20090203750A1 (en) | 2005-08-24 | 2009-08-13 | Alan Kozikowski | 5-HT2C Receptor Agonists as Anorectic Agents |
| GB0517740D0 (en) * | 2005-08-31 | 2005-10-12 | Novartis Ag | Organic compounds |
| TW200745067A (en) | 2006-03-14 | 2007-12-16 | Astrazeneca Ab | Novel compounds |
| ATE449068T1 (de) | 2006-05-18 | 2009-12-15 | Syngenta Participations Ag | Neue mikrobiozide |
| US8198301B2 (en) | 2006-07-05 | 2012-06-12 | Hesheng Zhang | Quinazoline and quinoline derivatives as irreversibe protein tyrosine kinase inhibitors |
| EP2521786B1 (en) | 2006-07-20 | 2015-06-24 | Vical Incorporated | Compositions for vaccinating against hsv-2 |
| WO2008033466A2 (en) | 2006-09-14 | 2008-03-20 | Combinatorx (Singapore) Pre. Ltd. | Compositions and methods for treatment of viral diseases |
| US20110092601A1 (en) | 2007-04-13 | 2011-04-21 | The Johns Hopkins University | Lysine-specific demethylase inhibitors |
| US7906513B2 (en) | 2007-04-26 | 2011-03-15 | Enanta Pharmaceuticals, Inc. | Hydrazide-containing hepatitis C serine protease inhibitors |
| CA2691215C (en) | 2007-06-27 | 2016-03-29 | Astrazeneca Ab | Pyrazinone derivatives and their use in the treatment of lung diseases |
| JP2010535773A (ja) | 2007-08-10 | 2010-11-25 | グラクソスミスクライン エルエルシー | ウイルス感染を治療するための窒素含有二環式化学物質 |
| SI2203431T1 (sl) | 2007-09-17 | 2011-12-30 | Abbott Lab | Antiinfekcijski pririmidini in njihove uporabe |
| US20100016262A1 (en) | 2007-10-18 | 2010-01-21 | Yale University | Compositions and methods for reducing hepatotoxicity associated with drug administration and treating non-alcoholic fatty liver disease, non-alcoholic steatohepatitis and associated cirrhosis |
| JP5497650B2 (ja) | 2007-10-19 | 2014-05-21 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | Ccr10アンタゴニスト |
| JP5517153B2 (ja) | 2007-12-26 | 2014-06-11 | 塩野義製薬株式会社 | グリコペプチド抗生物質配糖化誘導体 |
| WO2009109991A2 (en) | 2008-01-23 | 2009-09-11 | Sun Pharma Advanced Research Company Ltd., | Novel hydrazide containing tyrosine kinase inhibitors |
| NZ586831A (en) | 2008-01-28 | 2012-02-24 | Janssen Pharmaceutica Nv | 6-substituted-thio-2-amino-quinoline derivatives useful as inhibitors of beta-secretase (bace) |
| US9206143B2 (en) | 2008-03-19 | 2015-12-08 | Aurimmed Pharma, Inc. | Compounds advantageous in the treatment of central nervous system diseases and disorders |
| RU2525236C2 (ru) | 2008-03-27 | 2014-08-10 | Грюненталь Гмбх | Замещенные производные 4-аминоциклогексана |
| CA2723205C (en) | 2008-04-16 | 2017-03-14 | Portola Pharmaceuticals, Inc. | 2,6-diamino-pyrimidin-5-yl-carboxamides as syk or jak kinase inhibitors |
| WO2009153197A1 (en) | 2008-06-18 | 2009-12-23 | F. Hoffmann-La Roche Ag | Halo-substituted pyrimidodiazepines as plkl inhibitors |
| EP2317992A2 (en) | 2008-07-24 | 2011-05-11 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Preventing or treating viral infection using an inhibitor of the lsd1 protein, a mao inhibitor or an inhibitor of lsd1 and a mao inhibitor |
| EP2331542B1 (en) | 2008-08-01 | 2016-07-27 | The United States of America, as Represented by The Secretary, Department of Health and Human Services | A3 adenosine receptor antagonists and partial agonists |
| US8048887B2 (en) | 2008-09-11 | 2011-11-01 | Bristol-Myers Squibb Company | Compounds for the treatment of hepatitis C |
| WO2010043721A1 (en) | 2008-10-17 | 2010-04-22 | Oryzon Genomics, S.A. | Oxidase inhibitors and their use |
| EP2389362B1 (en) | 2009-01-21 | 2019-12-11 | Oryzon Genomics, S.A. | Phenylcyclopropylamine derivatives and their medical use |
| WO2010085749A2 (en) | 2009-01-23 | 2010-07-29 | Northwestern University | Potent and selective neuronal nitric oxide synthase inhibitors with improved membrane permeability |
| MY160130A (en) | 2009-02-27 | 2017-02-28 | Enanta Pharm Inc | Hepatitis c virus inhibitors |
| AU2010247414B2 (en) | 2009-05-15 | 2013-08-01 | Novartis Ag | Aryl pyridine as aldosterone synthase inhibitors |
| US8389580B2 (en) | 2009-06-02 | 2013-03-05 | Duke University | Arylcyclopropylamines and methods of use |
| EP2258865A1 (en) | 2009-06-05 | 2010-12-08 | Universitätsklinikum Freiburg | Lysine-specific demethylase 1 (LSD1) is a biomarker for breast cancer |
| WO2010143582A1 (ja) | 2009-06-11 | 2010-12-16 | 公立大学法人名古屋市立大学 | フェニルシクロプロピルアミン誘導体及びlsd1阻害剤 |
| US9708255B2 (en) | 2009-08-18 | 2017-07-18 | Robert A. Casero | (bis)urea and (bis)thiourea compounds as epigenic modulators of lysine-specific demethylase 1 and methods of treating disorders |
| WO2011031934A1 (en) | 2009-09-11 | 2011-03-17 | Enanta Pharmaceuticals, Inc. | Hepatitis c virus inhibitors |
| WO2011035941A1 (en) | 2009-09-25 | 2011-03-31 | Oryzon Genomics S.A. | Lysine specific demethylase-1 inhibitors and their use |
| EP2486002B1 (en) | 2009-10-09 | 2019-03-27 | Oryzon Genomics, S.A. | Substituted heteroaryl- and aryl- cyclopropylamine acetamides and their use |
| WO2011057262A2 (en) | 2009-11-09 | 2011-05-12 | Evolva Inc. | Treatment of infections with tp receptor antagonists |
| US9616058B2 (en) | 2010-02-24 | 2017-04-11 | Oryzon Genomics, S.A. | Potent selective LSD1 inhibitors and dual LSD1/MAO-B inhibitors for antiviral use |
| WO2011106106A2 (en) | 2010-02-24 | 2011-09-01 | Oryzon Genomics, S.A. | Lysine demethylase inhibitors for diseases and disorders associated with hepadnaviridae |
| US20130197088A1 (en) | 2010-03-12 | 2013-08-01 | Robert A. Casero, JR. | Compositions and Methods for Combinations of Oligoamines with 2-Difluoromethylornithine (DFMO) |
| CN102947265B (zh) | 2010-04-19 | 2015-07-29 | 奥瑞泽恩基因组学股份有限公司 | 赖氨酸特异性脱甲基酶-1抑制剂及其应用 |
| ES2564352T3 (es) | 2010-04-20 | 2016-03-22 | Università Degli Studi Di Roma "La Sapienza" | Derivados de tranilcipromina como inhibidores de la histona-desmetilasa LSD1 y/o LSD2 |
| EP2560939A2 (en) | 2010-04-20 | 2013-02-27 | Actavis Group Ptc Ehf | Novel process for preparing phenylcyclopropylamine derivatives using novel intermediates |
| RU2013103794A (ru) | 2010-06-30 | 2014-08-10 | Актавис Груп Птс Ехф | Новые способы получения производных фенилциклопропиламина и их применение для получения тикагрелора |
| CN103124724B (zh) | 2010-07-29 | 2015-05-20 | 奥瑞泽恩基因组学股份有限公司 | Lsd1的基于芳基环丙胺的脱甲基酶抑制剂及其医疗用途 |
| US9006449B2 (en) | 2010-07-29 | 2015-04-14 | Oryzon Genomics, S.A. | Cyclopropylamine derivatives useful as LSD1 inhibitors |
| WO2012034116A2 (en) | 2010-09-10 | 2012-03-15 | The Johns Hopkins University | Small molecules as epigenetic modulators of lysine-specific demethylase 1 and methods of treating disorders |
| US20130303545A1 (en) | 2010-09-30 | 2013-11-14 | Tamara Maes | Cyclopropylamine derivatives useful as lsd1 inhibitors |
| WO2012045883A1 (en) | 2010-10-08 | 2012-04-12 | Oryzon Genomics S.A. | Cyclopropylamine inhibitors of oxidases |
| WO2012072713A2 (en) | 2010-11-30 | 2012-06-07 | Oryzon Genomics, S.A. | Lysine demethylase inhibitors for diseases and disorders associated with flaviviridae |
| EP2712316A1 (en) | 2011-02-08 | 2014-04-02 | Oryzon Genomics, S.A. | Lysine demethylase inhibitors for myeloproliferative or lymphoproliferative diseases or disorders |
| WO2012107498A1 (en) | 2011-02-08 | 2012-08-16 | Oryzon Genomics S.A. | Lysine demethylase inhibitors for myeloproliferative disorders |
| CA2831143C (en) | 2011-03-25 | 2019-05-21 | Glaxosmithkline Intellectual Property Development Limited | Cyclopropylamines as lsd1 inhibitors |
| EP2750671A2 (en) | 2011-05-19 | 2014-07-09 | Oryzon Genomics, S.A. | Lysine demethylase inhibitors for thrombosis and cardiovascular diseases |
| WO2012156531A2 (en) | 2011-05-19 | 2012-11-22 | Oryzon Genomics, S.A. | Lysine demethylase inhibitors for inflammatory diseases or conditions |
| RU2681211C2 (ru) | 2011-10-20 | 2019-03-05 | Оризон Дженомикс С.А. | (гетеро)арилциклопропиламины в качестве ингибиторов lsd1 |
| EP4074695A1 (en) | 2011-10-20 | 2022-10-19 | Oryzon Genomics, S.A. | (hetero)aryl cyclopropylamine compounds as lsd1 inhibitors |
-
2010
- 2010-04-19 WO PCT/EP2010/055131 patent/WO2011035941A1/en active Application Filing
- 2010-04-19 BR BR112012006572A patent/BR112012006572A2/pt not_active IP Right Cessation
- 2010-04-19 CN CN201080053636.2A patent/CN102639496B/zh not_active Expired - Fee Related
- 2010-04-19 CA CA2812683A patent/CA2812683C/en not_active Expired - Fee Related
- 2010-04-19 RU RU2012116584/04A patent/RU2602814C2/ru not_active IP Right Cessation
- 2010-04-19 EP EP10720278.0A patent/EP2480528B1/en not_active Not-in-force
- 2010-04-19 KR KR1020127010672A patent/KR101736218B1/ko active IP Right Grant
- 2010-04-19 JP JP2012530181A patent/JP5699152B2/ja not_active Expired - Fee Related
- 2010-04-19 US US13/497,994 patent/US8859555B2/en not_active Expired - Fee Related
- 2010-04-19 MX MX2012003499A patent/MX338041B/es active IP Right Grant
- 2010-04-19 AU AU2010297557A patent/AU2010297557C1/en not_active Ceased
-
2012
- 2012-03-22 IL IL218778A patent/IL218778A/en not_active IP Right Cessation
Also Published As
| Publication number | Publication date |
|---|---|
| AU2010297557A1 (en) | 2012-05-17 |
| CN102639496A (zh) | 2012-08-15 |
| IL218778A0 (en) | 2012-06-28 |
| CN102639496B (zh) | 2014-10-29 |
| US8859555B2 (en) | 2014-10-14 |
| MX2012003499A (es) | 2012-08-03 |
| KR20120104525A (ko) | 2012-09-21 |
| CA2812683A1 (en) | 2011-03-31 |
| RU2012116584A (ru) | 2013-10-27 |
| IL218778A (en) | 2015-06-30 |
| EP2480528A1 (en) | 2012-08-01 |
| BR112012006572A2 (pt) | 2016-04-26 |
| EP2480528B1 (en) | 2018-08-29 |
| CA2812683C (en) | 2017-10-10 |
| JP2013505903A (ja) | 2013-02-21 |
| US20120283266A1 (en) | 2012-11-08 |
| MX338041B (es) | 2016-03-30 |
| KR101736218B1 (ko) | 2017-05-16 |
| AU2010297557B2 (en) | 2016-10-06 |
| RU2602814C2 (ru) | 2016-11-20 |
| AU2010297557C1 (en) | 2017-04-06 |
| WO2011035941A1 (en) | 2011-03-31 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5699152B2 (ja) | リジン特異的デメチラーゼ−1阻害剤およびその使用 | |
| JP5868948B2 (ja) | リジン特異的脱メチル化酵素1阻害薬およびその使用 | |
| US8524717B2 (en) | Oxidase inhibitors and their use | |
| EP2486002B1 (en) | Substituted heteroaryl- and aryl- cyclopropylamine acetamides and their use | |
| JP6054868B2 (ja) | Lsd1のアリールシクロプロピルアミンをベースとしたデメチラーゼ阻害剤およびそれらの医学的使用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20130417 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20130417 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20140228 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20140311 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20140610 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20150120 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20150216 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5699152 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| LAPS | Cancellation because of no payment of annual fees |