JP5536070B2 - ペメトレキセド二酸の新規結晶型及びその製造方法 - Google Patents
ペメトレキセド二酸の新規結晶型及びその製造方法 Download PDFInfo
- Publication number
- JP5536070B2 JP5536070B2 JP2011527193A JP2011527193A JP5536070B2 JP 5536070 B2 JP5536070 B2 JP 5536070B2 JP 2011527193 A JP2011527193 A JP 2011527193A JP 2011527193 A JP2011527193 A JP 2011527193A JP 5536070 B2 JP5536070 B2 JP 5536070B2
- Authority
- JP
- Japan
- Prior art keywords
- pemetrexed
- pemetrexed diacid
- water
- diacid
- crystal form
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- QOFFJEBXNKRSPX-ZDUSSCGKSA-N pemetrexed Chemical compound C1=N[C]2NC(N)=NC(=O)C2=C1CCC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 QOFFJEBXNKRSPX-ZDUSSCGKSA-N 0.000 title claims description 146
- 229960005079 pemetrexed Drugs 0.000 title claims description 142
- 239000013078 crystal Substances 0.000 title claims description 137
- 238000000034 method Methods 0.000 title description 17
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 89
- 238000004519 manufacturing process Methods 0.000 claims description 38
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 claims description 33
- NYDXNILOWQXUOF-UHFFFAOYSA-L disodium;2-[[4-[2-(2-amino-4-oxo-1,7-dihydropyrrolo[2,3-d]pyrimidin-5-yl)ethyl]benzoyl]amino]pentanedioate Chemical compound [Na+].[Na+].C=1NC=2NC(N)=NC(=O)C=2C=1CCC1=CC=C(C(=O)NC(CCC([O-])=O)C([O-])=O)C=C1 NYDXNILOWQXUOF-UHFFFAOYSA-L 0.000 claims description 23
- 229960003349 pemetrexed disodium Drugs 0.000 claims description 21
- 239000002904 solvent Substances 0.000 claims description 19
- 238000000634 powder X-ray diffraction Methods 0.000 claims description 18
- 239000003513 alkali Substances 0.000 claims description 11
- 150000003839 salts Chemical class 0.000 claims description 11
- 239000011259 mixed solution Substances 0.000 claims description 10
- 239000002244 precipitate Substances 0.000 claims description 9
- 239000003960 organic solvent Substances 0.000 claims description 7
- 239000012046 mixed solvent Substances 0.000 claims description 3
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 33
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 30
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 30
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 24
- -1 2-amino-4,7-dihydro-4-oxo-1H-pyrrolo [2,3-d] pyrimidin-5-yl Chemical group 0.000 description 23
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 21
- 239000007864 aqueous solution Substances 0.000 description 20
- 239000000203 mixture Substances 0.000 description 17
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 15
- 238000001035 drying Methods 0.000 description 15
- 239000000047 product Substances 0.000 description 15
- 238000003756 stirring Methods 0.000 description 14
- 239000000243 solution Substances 0.000 description 12
- 239000002253 acid Substances 0.000 description 10
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 9
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 9
- 238000002441 X-ray diffraction Methods 0.000 description 9
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 8
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 8
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 8
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 8
- 235000011054 acetic acid Nutrition 0.000 description 8
- 239000012065 filter cake Substances 0.000 description 8
- 238000002360 preparation method Methods 0.000 description 8
- 230000001376 precipitating effect Effects 0.000 description 7
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 6
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 6
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 6
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 6
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 6
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 6
- 238000009835 boiling Methods 0.000 description 6
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 6
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 6
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 6
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 6
- QXNVGIXVLWOKEQ-UHFFFAOYSA-N Disodium Chemical class [Na][Na] QXNVGIXVLWOKEQ-UHFFFAOYSA-N 0.000 description 5
- 238000001556 precipitation Methods 0.000 description 5
- 238000012360 testing method Methods 0.000 description 5
- 108010022394 Threonine synthase Proteins 0.000 description 4
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 4
- 238000010438 heat treatment Methods 0.000 description 4
- 229910003002 lithium salt Inorganic materials 0.000 description 4
- XAEFZNCEHLXOMS-UHFFFAOYSA-M potassium benzoate Chemical compound [K+].[O-]C(=O)C1=CC=CC=C1 XAEFZNCEHLXOMS-UHFFFAOYSA-M 0.000 description 4
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 4
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 3
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 3
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 3
- 239000005711 Benzoic acid Substances 0.000 description 3
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- 150000007513 acids Chemical class 0.000 description 3
- 239000000908 ammonium hydroxide Substances 0.000 description 3
- 235000010233 benzoic acid Nutrition 0.000 description 3
- AXCZMVOFGPJBDE-UHFFFAOYSA-L calcium dihydroxide Chemical compound [OH-].[OH-].[Ca+2] AXCZMVOFGPJBDE-UHFFFAOYSA-L 0.000 description 3
- 239000000920 calcium hydroxide Substances 0.000 description 3
- 229910001861 calcium hydroxide Inorganic materials 0.000 description 3
- ZPWVASYFFYYZEW-UHFFFAOYSA-L dipotassium hydrogen phosphate Chemical compound [K+].[K+].OP([O-])([O-])=O ZPWVASYFFYYZEW-UHFFFAOYSA-L 0.000 description 3
- 238000004090 dissolution Methods 0.000 description 3
- 238000001914 filtration Methods 0.000 description 3
- 235000019253 formic acid Nutrition 0.000 description 3
- 238000004108 freeze drying Methods 0.000 description 3
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 3
- 229940071870 hydroiodic acid Drugs 0.000 description 3
- 229940098779 methanesulfonic acid Drugs 0.000 description 3
- 229910000403 monosodium phosphate Inorganic materials 0.000 description 3
- 235000019799 monosodium phosphate Nutrition 0.000 description 3
- 208000002154 non-small cell lung carcinoma Diseases 0.000 description 3
- NYDXNILOWQXUOF-GXKRWWSZSA-L pemetrexed disodium Chemical compound [Na+].[Na+].C=1NC=2NC(N)=NC(=O)C=2C=1CCC1=CC=C(C(=O)N[C@@H](CCC([O-])=O)C([O-])=O)C=C1 NYDXNILOWQXUOF-GXKRWWSZSA-L 0.000 description 3
- 229910000027 potassium carbonate Inorganic materials 0.000 description 3
- 229910000160 potassium phosphate Inorganic materials 0.000 description 3
- 235000011009 potassium phosphates Nutrition 0.000 description 3
- WBHQBSYUUJJSRZ-UHFFFAOYSA-M sodium bisulfate Chemical compound [Na+].OS([O-])(=O)=O WBHQBSYUUJJSRZ-UHFFFAOYSA-M 0.000 description 3
- 229910000342 sodium bisulfate Inorganic materials 0.000 description 3
- 229910000029 sodium carbonate Inorganic materials 0.000 description 3
- AJPJDKMHJJGVTQ-UHFFFAOYSA-M sodium dihydrogen phosphate Chemical compound [Na+].OP(O)([O-])=O AJPJDKMHJJGVTQ-UHFFFAOYSA-M 0.000 description 3
- 239000001488 sodium phosphate Substances 0.000 description 3
- 229910000162 sodium phosphate Inorganic materials 0.000 description 3
- 235000011008 sodium phosphates Nutrition 0.000 description 3
- 239000012453 solvate Substances 0.000 description 3
- 238000011282 treatment Methods 0.000 description 3
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 3
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 description 3
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 2
- ZHNUHDYFZUAESO-UHFFFAOYSA-N Formamide Chemical compound NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 description 2
- 101000606741 Homo sapiens Phosphoribosylglycinamide formyltransferase Proteins 0.000 description 2
- 229930195725 Mannitol Natural products 0.000 description 2
- 102100039654 Phosphoribosylglycinamide formyltransferase Human genes 0.000 description 2
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 2
- 102000005497 Thymidylate Synthase Human genes 0.000 description 2
- 239000012670 alkaline solution Substances 0.000 description 2
- 150000003863 ammonium salts Chemical class 0.000 description 2
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 2
- 238000006243 chemical reaction Methods 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- 238000002425 crystallisation Methods 0.000 description 2
- 230000008025 crystallization Effects 0.000 description 2
- 102000004419 dihydrofolate reductase Human genes 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 2
- 239000012467 final product Substances 0.000 description 2
- 239000004052 folic acid antagonist Substances 0.000 description 2
- 229960002989 glutamic acid Drugs 0.000 description 2
- 159000000002 lithium salts Chemical class 0.000 description 2
- 208000006178 malignant mesothelioma Diseases 0.000 description 2
- 201000005282 malignant pleural mesothelioma Diseases 0.000 description 2
- 239000000594 mannitol Substances 0.000 description 2
- 235000010355 mannitol Nutrition 0.000 description 2
- 238000011519 second-line treatment Methods 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 206010005003 Bladder cancer Diseases 0.000 description 1
- 206010006187 Breast cancer Diseases 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- 229930091371 Fructose Natural products 0.000 description 1
- 239000005715 Fructose Substances 0.000 description 1
- RFSUNEUAIZKAJO-ARQDHWQXSA-N Fructose Chemical compound OC[C@H]1O[C@](O)(CO)[C@@H](O)[C@@H]1O RFSUNEUAIZKAJO-ARQDHWQXSA-N 0.000 description 1
- 208000005016 Intestinal Neoplasms Diseases 0.000 description 1
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 1
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 1
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 1
- 208000005718 Stomach Neoplasms Diseases 0.000 description 1
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 150000008064 anhydrides Chemical class 0.000 description 1
- 230000000259 anti-tumor effect Effects 0.000 description 1
- 239000002246 antineoplastic agent Substances 0.000 description 1
- 239000011260 aqueous acid Substances 0.000 description 1
- 235000011116 calcium hydroxide Nutrition 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- 150000001875 compounds Chemical class 0.000 description 1
- 229940127089 cytotoxic agent Drugs 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- BNIILDVGGAEEIG-UHFFFAOYSA-L disodium hydrogen phosphate Chemical compound [Na+].[Na+].OP([O-])([O-])=O BNIILDVGGAEEIG-UHFFFAOYSA-L 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 229960003668 docetaxel Drugs 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 239000003814 drug Substances 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- 238000009093 first-line therapy Methods 0.000 description 1
- 206010017758 gastric cancer Diseases 0.000 description 1
- 201000010536 head and neck cancer Diseases 0.000 description 1
- 208000014829 head and neck neoplasm Diseases 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- 201000002313 intestinal cancer Diseases 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 201000002528 pancreatic cancer Diseases 0.000 description 1
- 208000008443 pancreatic carcinoma Diseases 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 235000011181 potassium carbonates Nutrition 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 238000003672 processing method Methods 0.000 description 1
- 230000002250 progressing effect Effects 0.000 description 1
- 125000004528 pyrimidin-5-yl group Chemical group N1=CN=CC(=C1)* 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 238000009094 second-line therapy Methods 0.000 description 1
- 235000017550 sodium carbonate Nutrition 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 239000007787 solid Substances 0.000 description 1
- 201000011549 stomach cancer Diseases 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 201000005112 urinary bladder cancer Diseases 0.000 description 1
- 239000008215 water for injection Substances 0.000 description 1
Images



Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Description
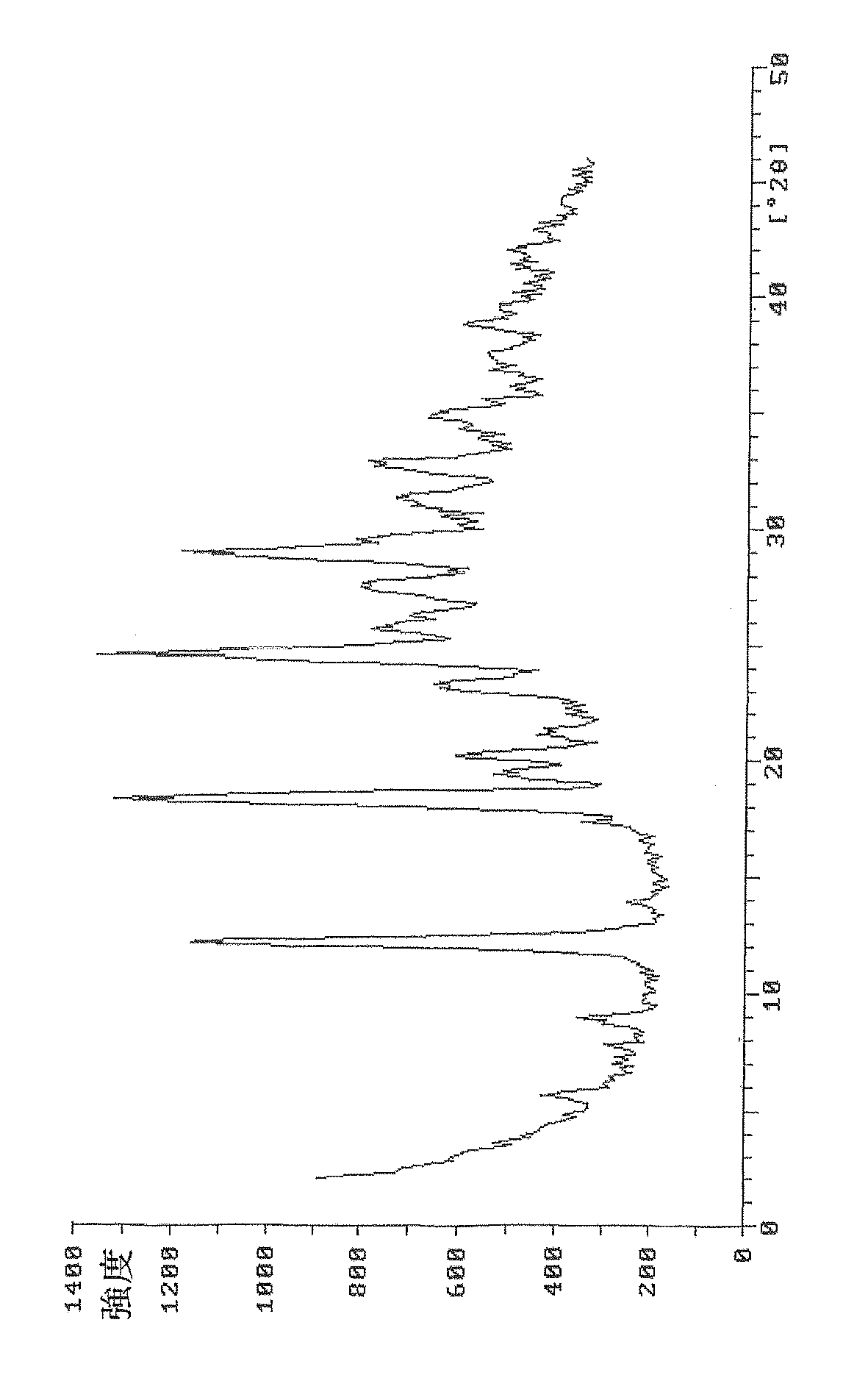
−PW1710 BASED X線回折装置
-CuKα源(λ=1.5406A)
−管電圧:30kV
−管電流:30mA
−受光スリット:0.05
-走査ステップ幅:0.1
-ステップ毎の時間:2s
ペメトレキセド二ナトリウム(乾燥品)5gを水50mlに溶解させ、エタノール60mlを加え、2mol/Lの塩酸水溶液にてpHを1.5〜2.0まで調整し、加熱して清澄させ、室温で1.5時間かけて攪拌しながら結晶を析出させ、ろ過し、適当な量のpH4〜5の水で洗浄し、45〜50℃、減圧(真空度、0.085〜0.090MPa)で30時間乾燥することで、含水量が7.1%であるペメトレキセド二酸のH結晶型2.9gを得た。
ペメトレキセド二ナトリウム(湿品)8gを水24mlに溶解させ、エタノール24mlを加え、1mol/Lの塩酸水溶液にてpHを2.0〜2.5まで調整し、加熱して清澄させ、更に水36mlを加え、攪拌しながら0.5時間で結晶を析出させ、ろ過し、濾過ケーキを適当な量の水で洗浄することで、含水量が約75%であるペメトレキセド二酸のH結晶型を得た。
ペメトレキセド二酸(乾燥品)5gを水75mlとアセトン70mlとの混合液に加え、1.5mol/Lの塩酸水溶液にてpHを1.5〜2.0まで調整し、加熱して清澄させ、攪拌しながら1時間で結晶を析出させ、ろ過し、濾過ケーキを適当な量の水で洗浄し、55〜60℃、減圧(真空度、0.090〜0.095MPa)で15時間乾燥することで、含水量が8.5%であるペメトレキセド二酸のH結晶型3.2gを得た。
ペメトレキセド二ナトリウム(乾燥品)10gを水500mlに溶解させ、0〜5℃まで冷却し、酢酸にて溶液のpHを4〜5まで調整し、更に2mol/Lの塩酸水溶液にてpHを2〜3まで調整し、続いて0.5時間攪拌し、ろ過し、濾過ケーキを適当な量の水で洗浄することで、含水量が約65%であるペメトレキセド二酸のI結晶型を得た。
ペメトレキセド二酸(湿品)10gを水400mlとエタノール100mlの混合溶媒に加え、4mol/Lの水酸化ナトリウム溶液にてpHを11〜12まで調整し、攪拌して溶解させ、2mol/Lの塩酸溶液にてpHを2〜3まで調整し、続いて1時間攪拌し、ろ過し、濾過ケーキを適当な量の水で洗浄し、45〜50℃、減圧(真空度、0.085〜0.090MPa)で35時間乾燥することで、含水量が6.7%であるペメトレキセド二酸のI結晶型3.7gを得た。
ペメトレキセド二ナトリウム(乾燥品)15gを水375mlに攪拌して溶解させ、0〜5℃まで冷却し、酢酸にて溶液pHを3〜4まで調整し、続いて10分攪拌し、ろ過し、濾過ケーキを適当な量の水で洗浄し、J結晶型のペメトレキセド二酸を得、60〜65℃、減圧(真空度、0.090〜0.095MPa)で24時間乾燥することで、含水量が7.7%であるペメトレキセド二酸のJ結晶型12.1gを得た。
ペメトレキセド二ナトリウム(湿品)15gを水100mlに攪拌して溶解させ、エタノール40mlを加え、1mol/Lの塩酸溶液にてpHを2〜3まで調整し、続いて約0.5時間攪拌し、ろ過し、濾過ケーキを適当な量の水で洗浄することで、含水量が約50%であるペメトレキセド二酸のJ結晶型を得た。
ペメトレキセド二ナトリウム(乾燥品)10gを水100mlに攪拌して溶解させ、アセトン100mlを加え、2mol/Lの塩酸にて溶液pHを3〜4まで調整し、系に固体が多量出現した後、60〜65℃まで加熱して約10分攪拌し、約1.5時間にかけて攪拌して冷却し、ろ過し、濾過ケーキを適当な量の水で洗浄することで、ペメトレキセド二酸のJ結晶型を得た。
上記実施例で得られたペメトレキセド二酸の新規結晶型(乾燥品)5gを水35mlに加え、5mol/Lの水酸化ナトリウム水溶液にてpHを11〜12まで調整し、攪拌して溶解させ、更に2mol/Lの塩酸にてpHを8〜9まで調整し、混合液を40〜45℃まで加熱し、アセトン170mlを加え、約1.5時間にかけて攪拌しながら冷却して結晶を析出させ、ろ過し、濾過ケーキを適当な量のアセトンで洗浄し、50℃、減圧(真空度、0.090〜0.095MPa)で24時間乾燥することで、ペメトレキセド二ナトリウム5.3gを得た。
上記実施例で得られたペメトレキセド二酸の新規結晶型(湿品)7gを水10mlに加え、5mol/Lの水酸化ナトリウム溶液にてpHを10〜11まで調整し、攪拌して清澄させ、アセトニトリル44mlを加え、室温で約2時間にかけて攪拌しながら結晶を析出させ、ろ過し、適当な量のアセトニトリル/水の混合液、及びアセトンで洗浄することで、ペメトレキセド二ナトリウムを得た。
上記実施例で得られたペメトレキセド二酸の新規結晶型(乾燥品)10.8gを注射用水180mlに加え、2mol/Lの水酸化ナトリウム溶液にてpHを11〜12まで調整し、攪拌して溶解させ、更に2mol/Lの塩酸にてpHを7.0〜8.5まで調整し、次に250mlに定容した。マンニトール10.0g、0.05%活性炭を加え、10分間攪拌し、ろ過し、更にろ液を滅菌ろ過した。凍結乾燥ボトルに12.5ml/ボトルで装入し、凍結乾燥することで、凍結乾燥したペメトレキセド二ナトリウムを得た。
Claims (7)
- H結晶型の粉末X線回折パターンにおいて、2θ値が9.9°、12.2°、16.1°、18.9°、19.8°、22.6°、25.1°である位置に対応する回折ピークがあるペメトレキセド二酸のH結晶型。
- 含水量が5wt%〜80wt%の範囲であることを特徴する請求項1に記載のペメトレキセド二酸のH結晶型。
- ペメトレキセド二酸をペメトレキセド二酸、水および水と相溶する溶媒を含む混合溶液から結晶させることを特徴する、請求項1または2に記載のペメトレキセド二酸のH結晶型の製造方法。
- ペメトレキセド二酸をペメトレキセド二酸、水および水と相溶する溶媒を含む混合溶液から結晶させることが、具体的に、水及び水と相溶する溶媒からなる混合溶液にペメトレキセド塩を溶解させ、pHを1〜2.5に調整して、ペメトレキセド二酸の結晶を析出させることであり;または水及び水と相溶する溶媒からなる混合溶液にペメトレキセド二酸をそのまま溶解させ、更に結晶を析出させることであることを特徴する請求項3に記載の製造方法。
- 対応するアルカリと作用させることによってペメトレキセド二酸の薬学的に許容される塩を得るための請求項1に記載のペメトレキセド二酸の結晶型の使用。
- 対応するアルカリと作用させることによってペメトレキセド二酸の薬学的に許容される塩を得るためのペメトレキセド二酸の結晶型の使用は、
ペメトレキセド二酸を水含有溶媒に添加して対応するアルカリにてペメトレキセド二酸を溶解させ、溶解した後、適宜な水と相溶する有機溶媒を添加してペメトレキセド二酸の薬学的に許容される塩を析出させ、或いはそのまま凍結乾燥して凍結乾燥した状態でのペメトレキセド二酸の薬学的に許容される塩を得るものを含み、上記の水含有溶媒は、
水や、水及び水と相溶する有機溶媒からなる混合溶媒を含有することを特徴する請求項5に記載の使用。 - 前記の対応するアルカリは水酸化ナトリウムであり、前記のペメトレキセド二酸の薬学的に許容される塩はペメトレキセド二ナトリウムであることを特徴する請求項5または請求項6に記載の使用。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2008100703459A CN101684121B (zh) | 2008-09-22 | 2008-09-22 | 培美曲塞二酸的新晶型及其制备方法 |
| CN200810070345.9 | 2008-09-22 | ||
| PCT/CN2009/074059 WO2010031357A1 (zh) | 2008-09-22 | 2009-09-21 | 培美曲塞二酸的新晶型及其制备方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2012502933A JP2012502933A (ja) | 2012-02-02 |
| JP5536070B2 true JP5536070B2 (ja) | 2014-07-02 |
Family
ID=42039100
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2011527193A Expired - Fee Related JP5536070B2 (ja) | 2008-09-22 | 2009-09-21 | ペメトレキセド二酸の新規結晶型及びその製造方法 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US8324382B2 (ja) |
| EP (1) | EP2351755B1 (ja) |
| JP (1) | JP5536070B2 (ja) |
| CN (1) | CN101684121B (ja) |
| AU (1) | AU2009295094B2 (ja) |
| CA (1) | CA2737967C (ja) |
| WO (1) | WO2010031357A1 (ja) |
Families Citing this family (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6754487B1 (en) | 2000-02-28 | 2004-06-22 | Telecom Network Optimization, Inc. | Radio network test analysis system |
| EP2311838A1 (en) | 2006-08-14 | 2011-04-20 | Sicor, Inc. | Crystalline form of pemetrexed diacid and process for the preparation thereof |
| JP2008543975A (ja) | 2006-08-14 | 2008-12-04 | シコール インコーポレイティド | 高度に純粋なペメトレキセド二酸およびその調製方法 |
| ES2639639T3 (es) * | 2011-03-25 | 2017-10-27 | Scinopharm Taiwan, Ltd. | Proceso para la producción de pemetrexed disódico |
| US20150259348A1 (en) * | 2012-10-17 | 2015-09-17 | Shilpa Medicare Limited | Process for preparing pemetrexed di potassium and its hydrates |
| WO2014060962A1 (en) * | 2012-10-17 | 2014-04-24 | Shilpa Medicare Limited | Pemetrexed dipotassium formulations |
| WO2014060959A1 (en) * | 2012-10-17 | 2014-04-24 | Shilpa Medicare Limited | Crystalline pemetrexed dipotassium process |
| CN103040834B (zh) * | 2012-12-25 | 2014-07-02 | 山西普德药业股份有限公司 | 一种注射用培美曲塞二钠及其制备方法 |
| CN104098573A (zh) * | 2013-04-10 | 2014-10-15 | 重庆医药工业研究院有限责任公司 | 培美曲塞盐及其制备方法 |
| WO2014185797A1 (en) | 2013-05-17 | 2014-11-20 | Instytut Farmaceutyczny | Process for the preparation of high purity amorphous pemetrexed disodium and crystalline forms of n-[4-[2-(2-amino-4,7-dihydro-4-oxo-3h-pyrrolo[2,3- d] pyrimidin-5-yl)ethyl] benzoyl]-l-glutamic acid |
| EP3021849B1 (en) | 2013-07-16 | 2019-10-09 | Dr. Reddy's Laboratories Ltd. | Novel crystalline forms of pemetrexed tromethamine salts |
| NZ630292A (en) * | 2013-11-25 | 2015-02-27 | Shilpa Medicare Ltd | Process for crystalline pemetrexed dipotassium salt |
| CN104119345B (zh) * | 2014-06-18 | 2016-04-27 | 威海昊同医药科技有限公司 | 一种注射级培美曲塞二钠的纯化方法 |
| NZ630299A (en) | 2014-06-30 | 2014-11-28 | Shilpa Medicare Ltd | Pemetrexed dipotassium formulations |
| CA3015436C (en) | 2014-10-30 | 2020-07-21 | Scinopharm Taiwan, Ltd. | Crystalline forms of pemetrexed diacid and manufacturing processes therefor |
| WO2017168442A1 (en) * | 2016-03-26 | 2017-10-05 | Dharmesh Mahendrabhai Shah | Novel stable salts of pemetrexed |
| CN107641124A (zh) * | 2016-07-22 | 2018-01-30 | 上海创诺制药有限公司 | 一种培美曲塞二酸新晶型及其制备方法 |
| CN114262332A (zh) * | 2020-09-16 | 2022-04-01 | 齐鲁制药有限公司 | 培美曲塞二酸新晶型及其制备方法 |
| CN115197224A (zh) * | 2021-04-12 | 2022-10-18 | 齐鲁制药有限公司 | 培美曲塞二酸新晶型及其制备方法 |
Family Cites Families (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| NO169490C (no) | 1988-03-24 | 1992-07-01 | Takeda Chemical Industries Ltd | Analogifremgangsmaate for fremstilling av terapeutisk aktive pyrrolopyrimidinderivater |
| IL96531A (en) * | 1989-12-11 | 1995-08-31 | Univ Princeton | History of Acid N- (Diomeric-H1-Pyrolo] D-2,3 [Pyrimidine-3-Ilacyl (-glutamic, preparation and pharmaceutical preparations containing them) |
| KR0162654B1 (ko) | 1989-12-11 | 1998-11-16 | 알렌 제이. 시니스갤리 | N-(피롤로[2,3-d]피리미딘-3-일아크릴)-글루타민산 유도체 |
| US5416211A (en) | 1992-09-25 | 1995-05-16 | Eli Lilly And Company | Process for preparing 5-substituted pyrrolo-[2,3-d]pyrimidines |
| AU740087B2 (en) * | 1997-09-26 | 2001-11-01 | Eli Lilly And Company | Processes and intermediates useful to make antifolates |
| US6262262B1 (en) | 1997-09-26 | 2001-07-17 | Eli Lilly And Company | Processes and intermediates useful to make antifolates |
| EP1212325A2 (en) | 1999-08-23 | 2002-06-12 | Eli Lilly And Company | A novel crystalline form of disodium n-[4-[2-(2-amino-4,7-dihydro-4-oxo-3h-pyrrolo[2,3-d]-pyrimidin-5-yl)ethyl]benzoyl]-l-glutamic acid salt and processes therefor |
| KR100744917B1 (ko) | 2000-02-25 | 2007-08-01 | 일라이 릴리 앤드 캄파니 | 신규한 결정형의N-[4-[2-(2-아미노-4,7-디히드로-4-옥소-3H-피롤로[2,3-d]피리미딘-5-일)에틸]벤조일]-L-글루탐산 및 이의 제조방법 |
| CN1536999B (zh) * | 2001-08-03 | 2012-08-08 | 西巴特殊化学品控股有限公司 | 氟伐他汀钠的晶形 |
| CN100344615C (zh) * | 2004-11-25 | 2007-10-24 | 重庆医药工业研究院有限责任公司 | 制备N-(吡咯并[2,3-d]嘧啶-5-基)酰基谷氨酸衍生物的方法及中间体 |
| CN1840530B (zh) * | 2005-03-28 | 2010-06-02 | 齐鲁制药有限公司 | 培美曲塞的制备方法 |
| CN100379729C (zh) * | 2006-04-06 | 2008-04-09 | 海南天源康泽医药科技有限公司 | 硝基化合物及其在培美曲塞制备中的应用 |
| JP2008543975A (ja) | 2006-08-14 | 2008-12-04 | シコール インコーポレイティド | 高度に純粋なペメトレキセド二酸およびその調製方法 |
| JP2008543976A (ja) * | 2006-08-14 | 2008-12-04 | シコール インコーポレイティド | ペメトレキセド二酸の医薬上許容される凍結乾燥塩の調製方法 |
| US7994180B2 (en) * | 2006-08-14 | 2011-08-09 | Sicor Inc. | Processes for preparing intermediates of pemetrexed |
| EP2311838A1 (en) * | 2006-08-14 | 2011-04-20 | Sicor, Inc. | Crystalline form of pemetrexed diacid and process for the preparation thereof |
| JP2008059088A (ja) | 2006-08-29 | 2008-03-13 | Toshiba Corp | 画像表示システム、及び画像表示装置 |
| CN101687872A (zh) * | 2007-04-03 | 2010-03-31 | 雷迪博士实验室有限公司 | 培美曲塞的固体形式 |
| EP2334685A4 (en) | 2008-09-08 | 2011-10-26 | Reddys Lab Ltd Dr | AMORPHOUS PEMETREXED DISODIUM |
-
2008
- 2008-09-22 CN CN2008100703459A patent/CN101684121B/zh active Active
-
2009
- 2009-09-21 CA CA2737967A patent/CA2737967C/en not_active Expired - Fee Related
- 2009-09-21 WO PCT/CN2009/074059 patent/WO2010031357A1/zh not_active Ceased
- 2009-09-21 JP JP2011527193A patent/JP5536070B2/ja not_active Expired - Fee Related
- 2009-09-21 AU AU2009295094A patent/AU2009295094B2/en not_active Ceased
- 2009-09-21 US US13/120,080 patent/US8324382B2/en active Active
- 2009-09-21 EP EP09814077.5A patent/EP2351755B1/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| CN101684121A (zh) | 2010-03-31 |
| US8324382B2 (en) | 2012-12-04 |
| JP2012502933A (ja) | 2012-02-02 |
| AU2009295094A1 (en) | 2010-03-25 |
| EP2351755B1 (en) | 2014-11-12 |
| EP2351755A1 (en) | 2011-08-03 |
| CA2737967A1 (en) | 2010-03-25 |
| WO2010031357A1 (zh) | 2010-03-25 |
| AU2009295094B2 (en) | 2012-11-01 |
| CA2737967C (en) | 2017-04-04 |
| US20110172424A1 (en) | 2011-07-14 |
| CN101684121B (zh) | 2013-04-03 |
| EP2351755A4 (en) | 2012-06-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5536070B2 (ja) | ペメトレキセド二酸の新規結晶型及びその製造方法 | |
| KR101045616B1 (ko) | 페메트렉세드 이산의 결정형 및 이의 제조 방법 | |
| JP5215505B2 (ja) | フェブキソスタットの結晶形 | |
| US9718829B2 (en) | Crystalline forms of pemetrexed diacid and processes for the preparation thereof | |
| EP2213674B1 (en) | Purification method of pemetrexed salts,sodium salts and disodium salts | |
| JP5744017B2 (ja) | チエノピリミジン誘導体の結晶 | |
| WO2025167975A1 (en) | Crystalline cyclin-dependent kinase (cdk) 12 and/or cdk13 inhibitor and uses thereof | |
| CN108864063A (zh) | 一种治疗癌症的药物溶剂合物及其制备方法 | |
| WO2021043200A1 (zh) | 一种喹唑啉衍生物的制备方法及其结晶 | |
| HK1259175A1 (zh) | 一种tlr7激动剂的马来酸盐、其晶型c、晶型d、晶型e及其制备方法和用途 | |
| HK1259175B (zh) | 一种tlr7激动剂的马来酸盐、其晶型c、晶型d、晶型e及其制备方法和用途 | |
| HK1259182A1 (zh) | 一种tlr7激动剂的三氟乙酸盐、晶型b、制备方法和用途 | |
| HK1259182B (zh) | 一种tlr7激动剂的三氟乙酸盐、晶型b、制备方法和用途 | |
| HK1259170B (zh) | 一种tlr7激动剂的晶型a、其制备方法和用途 | |
| CN102675183A (zh) | 一种药物中间体的大规模制备方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20130702 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20131002 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20140408 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20140423 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5536070 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |