JP4758525B2 - 生理活性ポリペプチドの徐放性製剤およびその製造法 - Google Patents
生理活性ポリペプチドの徐放性製剤およびその製造法 Download PDFInfo
- Publication number
- JP4758525B2 JP4758525B2 JP07479399A JP7479399A JP4758525B2 JP 4758525 B2 JP4758525 B2 JP 4758525B2 JP 07479399 A JP07479399 A JP 07479399A JP 7479399 A JP7479399 A JP 7479399A JP 4758525 B2 JP4758525 B2 JP 4758525B2
- Authority
- JP
- Japan
- Prior art keywords
- hgh
- physiologically active
- sustained
- active polypeptide
- release preparation
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images

Description
【発明の属する技術分野】
本発明は、生理活性ポリペプチドを含有する徐放性製剤およびその製造法に関する。
【0002】
【従来の技術】
生理活性ポリペプチドは、生体において種々の薬理作用を示すことが知られており、このうちいくつかについては遺伝子工学、細胞工学の手法の発達により大腸菌、酵母、動物細胞あるいはヤギ、ハムスター等の生体を用いて大量に生産され、医薬品としての応用が図られている。しかしながら、これらの生理活性ポリペプチドは一般的に生体内での半減期が短いために、頻回投与が必要であり、注射に伴う患者の肉体的負担は無視できないものがある。
例えば、成長ホルモン(以下、GHと略記することがある)は、元来下垂体前葉で産生・分泌される代表的なホルモンで、身体の成長促進に働くほか、糖・脂質代謝、蛋白同化、細胞増殖や分化に関与する等、幅広く多彩な生理作用を有する生理活性ポリペプチドであり、現在では遺伝子組換え技術を用いて大腸菌により大量生産され、医薬品として全世界で広く臨床応用されているが、GHは生体内半減期が短く、有効血中濃度を維持するためには頻回投与が必要であり、特に下垂体性小人症の場合には、乳幼児あるいは若年患者に対して数カ月から10年以上の長期に亘る連日皮下投与がなされているのが実情である。
【0003】
特開平8−3055号公報(EP−A−633020)には、水溶性ポリペプチドを生体内分解性ポリマーと脂肪酸金属塩とからなる生体内分解性マトリックスに水中で浸透させる徐放性製剤の製造法及び該製造法で調製された徐放性マイクロカプセルが開示されている。
特開平8−217691号公報(WO 96/07399)には、水溶性ペプチド性生理活性物質を塩化亜鉛水溶液等により水不溶性ないし水難溶性多価金属塩とし、これと生体内分解性ポリマーとを含有してなる徐放性製剤の製造法が開示されている。
WO 94/12158には、ヒトGH(以下、hGHと略記することがある)と生体分解性ポリマーとの徐放性製剤の製造法として、ポリマーに対して0.1ないし30%(W/W)の水酸化亜鉛等のポリマー分解促進剤をポリマー溶液に加えることができるとの記載がある。また、hGHとポリマーとを含有する有機溶媒溶液を液体窒素中に噴霧し多孔性粒子として生物活性を保持した形で徐放性マイクロカプセルを調製する方法が開示されている。
また、WO 92/17200及びネイチャー メディシン(Nature Medicine), 第2巻, 795頁(1996)には、hGHの亜鉛塩を用いる徐放性製剤の製造法が開示されている。
更に、WO 95/29664には、炭酸亜鉛等の金属塩を固体状でポリマー溶液に分散させた後、生理活性物質(ホルモン等)を添加し、生理活性物質と金属カチオンとを別々に生体分解性ポリマーに分散させてなる徐放性マイクロカプセルの製造法が開示されている。
【0004】
【発明が解決しようとする課題】
前記のように生理活性ポリペプチドの生理活性を保持しながら徐放性製剤を製造する試みは種々なされているものの、生理活性ポリペプチドによっては、製剤内への生理活性ポリペプチドの取り込み率が低い、投与初期の過剰放出がある、長期にわたる安定した放出性が達成されない、十分な血中濃度が長期にわたって保持できない等の点で、未だ臨床上満足すべき製剤は得られていない。また、製造法においても大量生産を前提とする工業化に合致しないのが現状である。
生理活性ポリペプチド粉体(固相:以下、S相と略記することがある)を、ポリマーを溶解した有機溶媒(O相)に直接添加し分散させた、いわゆるS/Oエマルションを用いて徐放性製剤を製造する方法においては、比較的安定に生理活性ポリペプチドを封入することが可能であるが、一方で製剤工程において大量の生理活性ポリペプチド粉体を固体として効率的に取り扱う必要があり、更にはS/O分散液中の粉体粒径が、得られる徐放性製剤の品質に大きく影響するため、その制御が必須である等、製造上の問題が数多くある。
従って、製剤化工程中での生理活性ポリペプチドの安定性の保持、操作性のよい粉体の作成、粉体の微粒化等を全て満たし、しかも収率の良い、安定した品質の徐放性製剤および該徐放性製剤を大量生産できる製造法が望まれている。
【0005】
【課題を解決するための手段】
本発明者らは前記の問題点を解決するため鋭意研究を進め、生理活性ポリペプチド水溶液に水混和性の有機溶媒及び/又は揮発性の塩類を添加後、凍結乾燥することにより、予想外にも当該ポリペプチドの生理活性を保持したまま、製剤化工程において操作性がよく、しかも粒子径の小さな生理活性ポリペプチド粉体が得られることを初めて見出した。更に、このようにして得られた生理活性ポリペプチド粉体を、生体内分解性ポリマーの有機溶媒液に分散させた後、有機溶媒を除去して徐放性製剤を製造すると、意外にも徐放性製剤における生理活性ポリペプチドの封入率及び徐放性が改善されることを見出し、これらに基づいて本発明を完成した。
【0006】
すなわち、本発明は、
(1)生理活性ポリペプチド水溶液に、水混和性の有機溶媒及び/又は揮発性の塩類を添加後、凍結乾燥して得られる生理活性ポリペプチド粉体を、生体内分解性ポリマーの有機溶媒液に分散させた後、有機溶媒を除去することを特徴とする徐放性製剤の製造法、
(2)生理活性ポリペプチド粉体が平均粒子径が10μm以下のものである前記(1)記載の製造法、
(3)生理活性ポリペプチドが成長ホルモンである前記(1)記載の製造法、
(4)成長ホルモンがヒト成長ホルモンである前記(3)記載の製造法、
(5)水混和性の有機溶媒がアルコールである前記(1)記載の製造法、
(6)アルコールがメタノール又はエタノールである前記(5)記載の製造法、
(7)アルコールがエタノールである前記(5)記載の製造法、
(8)揮発性の塩類がアンモニウム塩である前記(1)記載の製造法、
(9)アンモニウム塩が酢酸アンモニウム、重炭酸アンモニウム又は炭酸アンモニウムである前記(8)記載の製造法、
(10)アンモニウム塩が酢酸アンモニウムである前記(8)記載の製造法、
(11)生理活性ポリペプチド水溶液に対する濃度が0.03%(V/V)ないし0.5%(V/V)になる様にアルコールが添加される前記(5)記載の製造法、
(12)生理活性ポリペプチドに対するモル比が10倍ないし80倍のアンモニウム塩が添加される前記(8)記載の製造法、
(13)生体内分解性ポリマーが乳酸−グリコール酸重合体である前記(1)記載の製造法、
(14)乳酸−グリコール酸重合体の乳酸/グリコール酸組成比(モル%)が100/0ないし40/60である前記(13)記載の製造法、
(15)乳酸−グリコール酸重合体の重量平均分子量が3,000ないし50,000である前記(13)記載の製造法、
(16)乳酸−グリコール酸重合体が多価金属塩である前記(13)記載の製造法、
(17)多価金属が亜鉛である前記(16)記載の製造法、
(18)徐放性製剤がマイクロカプセルである前記(1)記載の製造法、
(19)マイクロカプセルの平均粒子径が0.1μmないし300μmである前記(18)記載の製造法、
(20)徐放性製剤が注射用である前記(1)記載の製造法、
(21)前記(1)記載の製造法により製造される徐放性製剤、
(22)生理活性ポリペプチド水溶液に、水混和性の有機溶媒及び/又は揮発性塩類を添加後、凍結乾燥することを特徴とする生理活性ポリペプチド粉体の製造法、
(23)前記(22)記載の製造法により得られる生理活性ポリペプチド粉体、
(24)生理活性ポリペプチド粉体が平均粒子径10μm以下である前記(23)記載の生理活性ポリペプチド粉体、
(25)医薬製剤を製造するための、前記(23)記載の生理活性ポリペプチド粉体の使用、
(26)粒径の小さな生理活性ポリペプチド粉体を製造するための水混和性の有機溶媒及び/又は揮発性の塩類の使用
(27)生理活性ポリペプチド粉体が平均粒子径が10μm以下である前記(26)記載の使用。
(28)請求項1記載の製造法により得られうる徐放性製剤。
(29)生理活性ポリペプチドの初期放出率が40%以下である、生理活性ポリペプチドと生体内分解性ポリマーの多価金属塩を含有する徐放性製剤、
(30)生理活性ポリペプチドが平均粒子径が10μm以下の粉体である前記(29)記載の徐放性製剤、
(31)生理活性ポリペプチドが成長ホルモンである前記(29)記載の徐放性製剤、
(32)生体内分解性ポリマーが乳酸−グリコール酸重合体である前記(29)記載の徐放性製剤、
(33)徐放性製剤がマイクロカプセルである前記(29)記載の徐放性製剤、
(34)マイクロカプセルの平均粒子径が0.1ないし300μmである前記(33)記載の徐放性製剤、
(35)1週間ないし1ヶ月型徐放性製剤である前記(29)記載の徐放性製剤、
および
(36)0.1ないし30%(W/W)の生理活性ポリペプチドを徐放性製剤中に含有する前記(29)記載の徐放性製剤等に関する。
【0007】
本発明に用いられる生体内分解性ポリマーとしては、例えばα-ヒドロキシカルボン酸類(例えば、グリコール酸、乳酸等)、ヒドロキシジカルボン酸類(例えば、リンゴ酸等)、ヒドロキシトリカルボン酸(例えば、クエン酸等)等の1種以上から無触媒脱水重縮合で合成され、遊離のカルボキシル基を有する重合体あるいはこれらの混合物、ポリ-α-シアノアクリル酸エステル、ポリアミノ酸(例えば、ポリ-γ-ベンジル-L-グルタミン酸等)、無水マレイン酸系重合体(例えば、スチレン-マレイン酸重合体等)等が挙げられる。これらはホモポリマーまたはコポリマーのいずれであってもよい。重合の形式は、ランダム、ブロック、グラフトのいずれでもよい。また、上記のα-ヒドロキシカルボン酸類、ヒドロキシジカルボン酸類、ヒドロキシトリカルボン酸類が分子内に光学活性中心を有する場合、D−,L−,DL−体のいずれも用いることができる。
これらの中では、末端に遊離のカルボキシル基を有する生体内分解性ポリマー、例えばα-ヒドロキシカルボン酸類(例えば、グリコール酸、乳酸等)から合成された重合体(例えば、乳酸重合体、乳酸−グリコール酸共重合体等)、ポリ-α-シアノアクリル酸エステル等が好ましい。
生体内分解性ポリマーとしては、更に好ましくはα-ヒドロキシカルボン酸類から合成された重合体等、特に好ましくは乳酸-グリコール酸重合体等である。
【0008】
本明細書においては、ポリ乳酸、ポリグリコール酸等の単重合体のみならず乳酸-グリコール酸共重合体も含めて、単に乳酸-グリコール酸重合体と称することがある。
生体内分解性ポリマーとして乳酸-グリコール酸重合体(乳酸-グリコール酸共重合体又は単重合体)を用いる場合、その組成比(モル%)は約100/0ないし約40/60が好ましく、約85/15ないし約50/50が更に好ましい。
乳酸-グリコール酸重合体の重量平均分子量は、約3,000ないし約50,000が好ましく、約3,000ないし約25,000がより好ましく、約5,000から約20,000が更に好ましい。
乳酸-グリコール酸重合体の分散度(重量平均分子量/数平均分子量)は約1.2ないし約4.0が好ましく、約1.5ないし約3.5が更に好ましい。
【0009】
なお、本明細書での重量平均分子量及び分散度に関し、前者は重量平均分子量が120,000、52,000、22,000、9,200、5,050、2,950、1,050、580、162の9種類のポリスチレンを基準物質としてゲルパーミエーションクロマトグラフィー(GPC)で測定したポリスチレン換算の値、後者はこの値から算出した値である。測定は、GPCカラムKF804L x 2(昭和電工製)、RIモニター L-3300(日立製作所製)を使用し、移動相としてクロロホルムを用いて行う。
【0010】
また、末端に遊離のカルボキシル基を有する生体内分解性ポリマーとは、末端基定量による数平均分子量と上記のGPC測定による数平均分子量がほぼ一致するポリマーであり、末端基定量による数平均分子量は以下のようにして算出される。
約1gないし約3gの生体内分解性ポリマーをアセトン(25ml)とメタノール(5ml)との混合溶媒に溶解し、フェノールフタレインを指示薬としてこの溶液中のカルボキシル基を0.05Nアルコール性水酸化カリウム溶液で、室温(20℃)で撹拌下、速やかに滴定して末端基定量による数平均分子量を次式で算出した。
末端基定量による数平均分子量=20000×A/B
A:生体内分解性ポリマーの質量(g)
B:滴定終点までに添加した0.05Nアルコール性水酸化カリウム溶液 (ml)
末端基定量による数平均分子量が絶対値であるのに対して、GPC測定による数平均分子量は、各種分析・解析条件(例えば、移動相の種類、カラムの種類、基準物質、スライス幅の選択、ベースラインの選択等)によって変動する相対値であるため、一義的な数値化は困難であるが、両測定による数平均分子量がほぼ一致するとは、例えば、α−ヒドロキシカルボン酸類から合成された重合体において、末端基定量による数平均分子量がGPC測定による数平均分子量の約0.5倍から約2倍の範囲内であること、好ましくは約0.7倍から約1.5倍の範囲内であることをいう。
例えば、1種類以上のα-ヒドロキシカルボン酸類から無触媒脱水重縮合法で合成され、末端に遊離のカルボキシル基を有する重合体では、GPC測定による数平均分子量と末端基定量による数平均分子量とがほぼ一致する。これに対し、環状二量体から触媒を用いて開環重合法で合成され、末端に遊離のカルボキシル基を本質的には有しない重合体では、末端基定量による数平均分子量がGPC測定による数平均分子量の約2倍以上に大きく上回る。この相違によって末端に遊離のカルボキシル基を有する重合体は、末端に遊離のカルボキシル基を有しない重合体と明確に区別することができる。
【0011】
末端に遊離のカルボキシル基を有する乳酸-グリコール酸重合体は、自体公知の製造法、例えば特開昭61−28521号公報に記載の方法(例えば無触媒下の脱水重縮合反応又は無機固体酸触媒下での脱水重縮合反応による製造方法等)に従って製造することができる。
乳酸-グリコール酸重合体の分解・消失速度は、組成比あるいは重量平均分子量によって大きく変化するが、一般的にはグリコール酸分率が低いほど分解・消失が遅いため、グリコール酸分率を低くするかあるいは分子量を大きくすることによって放出期間を長くすること(例えば、約6ヶ月)ができる。逆に、グリコール酸分率を高くするあるいは分子量を小さくすることによって放出期間を短くこと(例えば、約1週間)もできる。例えば、1週間ないし2ヶ月型徐放性製剤とするには、前記組成比及び重量平均分子量の範囲の乳酸-グリコール酸重合体を用いるのが好ましい。
【0012】
従って、本発明において用いる生体内分解性ポリマーの組成は、目的とする生理活性ポリペプチドの種類、所望の徐放期間等に応じて、適宜選択されることが好ましい。その具体例としては、例えば、生理活性ポリペプチドとしてGHを用いる場合、乳酸−グリコール酸重合体を用いることが好ましく、該乳酸−グリコール酸重合体としては、その乳酸/グリコール酸組成比(モル%)が約85/15ないし約50/50の乳酸−グリコール酸共重合体が好ましく、更に好ましくは約75/25ないし約50/50の乳酸−グリコール酸共重合体である。またその重量平均分子量は約8,000ないし約20,000が好ましく、更に好ましくは約10,000ないし約20,000である。また、乳酸−グリコール酸重合体の分散度(重量平均分子量/数平均分子量)は約1.2ないし約4.0が好ましく、更に好ましくは約1.5ないし約3.5である。
用いる乳酸−グリコール酸重合体は、前記公報記載の方法等、公知の方法に従い製造できる。該重合体は無触媒脱水重縮合で製造されたものが好ましい。前記GPC測定法による数平均分子量と末端基定量法による数平均分子量とが、ほぼ一致する乳酸−グリコール酸重合体(PLGA)を用いることが好ましい。
また、該重合体は組成比及び/または重量平均分子量の異なる2種の乳酸−グリコール酸重合体を任意の割合で混合して用いてもよい。このような例としては、組成比(乳酸/グリコール酸)(モル%)が約75/25で重量平均分子量が約10,000の乳酸−グリコール酸共重合体と、組成比(乳酸/グリコール酸)(モル%)が約50/50で重量平均分子量が約12,000の乳酸−グリコール酸共重合体との混合物等が用いられる。混合する際の重量比は、好ましくは約25/75ないし約75/25である。
【0013】
また、本発明で用いる生体内分解性ポリマーは、前記した生体内分解性ポリマーの金属塩であってもよく、例えば、WO97/01331号公報に記載の各種生体内分解性ポリマーの多価金属塩等が用いられる。好ましくは乳酸−グリコール酸重合体の多価金属塩(さらに好ましくは亜鉛塩,カルシウム塩,マグネシウム塩等、より好ましくは亜鉛塩等)等が用いられる。該多価金属塩の金属種としては、生体に悪影響を及ぼさない化合物であれば特に限定されず、例えば2価(例、鉄、亜鉛、銅、カルシウム、マグネシウム、アルミニウム、スズ、マンガン等)、3価(例、鉄、アルミニウム、マンガン等)、4価(例、スズ等)などの多価金属も用いることができる。
本明細書においては、生体内分解性ポリマーが金属塩の場合も含めて生体内分解性ポリマーと称することがあり、例えば乳酸−グリコール酸重合体が多価金属塩の場合も乳酸-グリコール酸重合体と称することがある。
これらの生体内分解性ポリマーの多価金属塩はWO97/01331号公報に記載の方法及びこれに準じる方法により製造することができる。
また、生体内分解性ポリマーの多価金属塩が亜鉛塩の場合には、生体内分解性ポリマーと酸化亜鉛とを有機溶媒中で反応させることによって製造することもできる。
該製造法においては、まず生体内分解性ポリマーと酸化亜鉛とを有機溶媒中に共存させて、生体内分解性ポリマー・酸化亜鉛体の有機溶媒溶液を製造する。この際、生体内分解性ポリマーの溶液中濃度は分子量、有機溶媒等の種類によって異なるが、例えば約0.1ないし約80%(W/W)、好ましくは約1ないし約70%(W/W)、更に好ましくは約2ないし約60%(W/W)である。また、添加する酸化亜鉛量は、特開平10−231252号公報に記載されたように、有機溶媒の種類によって異なるが、例えば生体内分解性ポリマー量の約0.001ないし約2%(W/W)、好ましくは約0.01ないし約1.5%(W/W)、更に好ましくは約0.1ないし約1%(W/W)である。
有機溶媒への生体内分解性ポリマー及び酸化亜鉛の添加順序は、生体内分解性ポリマーの有機溶媒溶液に酸化亜鉛を粉末状であるいは該有機溶媒に懸濁した状態で添加してもよく、逆に酸化亜鉛の有機溶媒懸濁液中に生体内分解性ポリマーの有機溶媒溶液を添加してもよい。また、両者を粉末状で混和後、有機溶媒を添加してもよい。
【0014】
本発明における生理活性ポリペプチドとしては、好ましくは分子量約1,000ないし約50,000、更に好ましくは分子量約5,000ないし約40,000の生理活性ポリペプチドが用いられる。
生理活性ポリペプチドの活性として代表的なものとしては、ホルモン作用が挙げられる。該生理活性ポリペプチドは天然物、合成物、半合成物のいずれでもよく、更にそれらの誘導体ないし類縁体でもよい。該生理活性ポリペプチドの作用機作は、作動性あるいは拮抗性のいずれでもよい。
本発明における生理活性ポリペプチドとしては、例えばペプチドホルモン、サイトカイン、ペプチド性神経伝達物質、造血因子、各種増殖因子、酵素、ポリペプチド系抗生物質、鎮痛性ペプチド等が用いられる。
ペプチドホルモンとしては、例えばインスリン、ソマトスタチン、ソマトスタチン誘導体(サンドスタチン,米国特許第4,087,390号,同第4,093,574号,同第4,100,117号,同第4,253,998号参照)、成長ホルモン(GH)、ナトリウム利尿ペプチド、ガストリン、プロラクチン、副腎皮質刺激ホルモン(ACTH)、ACTH誘導体(エビラタイド等)、メラノサイト刺激ホルモン(MSH)、甲状腺ホルモン放出ホルモン(TRH)、その塩及びその誘導体(特開昭50−121273号、特開昭52−116465号公報参照)、甲状腺刺激ホルモン(TSH)、黄体形成ホルモン(LH)、卵胞刺激ホルモン(FSH)、ヒト絨毛ゴナドトロピン(HCG)、サイモシン(チモシン)、モチリン、バソプレシン、バソプレシン誘導体{デスモプレシン〔日本内分泌学会雑誌,第54巻 第5号 第676ないし691頁(1978)〕参照}、オキシトシン、カルシトニン、副甲状腺ホルモン(PTH)、グルカゴン、セクレチン、パンクレオザイミン、コレシストキニン、アンジオテンシン、ヒト胎盤ラクトーゲン等が用いられる。ペプチドホルモンとしては、好ましくはインスリン及び成長ホルモン等である。
サイトカインとしては、例えばリンホカイン、モノカイン等が用いられる。リンホカインとしては、例えばインターフェロン類(アルファ型、ベータ型、ガンマ型等)、インターロイキン類(例えば、IL−2,3,4,5,6,7,8,9,10,11,12等)等が用いられる。モノカインとしては、例えばインターロイキン−1(IL−1)、腫瘍壊死因子(TNF)等が用いられる。サイトカインとしては、好ましくはリンホカイン等、更に好ましくはインターフェロン等、特に好ましくはインターフェロンアルファ等である。
ペプチド性神経伝達物質としては、例えばサブスタンスP、セロトニン、GABA等が用いられる。
【0015】
造血因子としては、例えばエリスロポエチン(EPO)、コロニー刺激因子(G−CSF,GM−CSF,M−CSF等)、トロンボポエチン(TPO)、血小板増殖刺激因子、メガカリオサイトポテンシエーター等が用いられる。
各種増殖因子としては、例えば塩基性あるいは酸性の繊維芽細胞増殖因子(FGF)あるいはこれらのファミリー(例えば、EGF、TGF−α、TGF−β、PDGF,酸性FGF,塩基性FGF、FGF−9等)、神経細胞増殖因子(NGF)あるいはこれらのファミリー(例えば、BDNF、NT−3、NT−4、CNTF、GDNF等)、インスリン様成長因子(例えば、IGF−1,IGF−2等)、骨増殖に関与する因子(BMP)あるいはこれらのファミリー等が用いられる。
酵素としては、例えばスーパーオキシドディスミュターゼ(SOD)、ウロキナーゼ、ティシュープラスミノーゲンアクティベーター(TPA)、アスパラギナーゼ、カリクレイン等が用いられる。
ポリペプチド系抗生物質としては、例えばポリミキシンB、コリスチン、グラミシジン、バシトラシン等が用いられる。
鎮痛性ペプチドとしては、例えばエンケファリン、エンケファリン誘導体〔米国特許第4,277,394号,ヨーロッパ特許出願公開第31567号公報参照〕,エンドルフィン、キョウトルフィン等が用いられる。
その他、生理活性ポリペプチドとしては、サイモポエチン、ダイノルフィン、ボムベシン、セルレイン、サイモスチムリン、胸腺液性因子(THF)、血中胸腺因子(FTS)及びその誘導体(米国特許第4,229,438号参照)、及びその他の胸腺因子〔医学のあゆみ、第125巻,第10号,835−843頁(1983年)〕、ニューロテンシン、ブラジキニン及びエンドセリン拮抗作用を有するペプチド類(ヨーロッパ特許公開第436189号,同第457195号,同第496452号,特開平3−94692号,同3−130299号公報参照)等が挙げられる。
本発明に特に好ましく適用される生理活性ポリペプチドとしては、成長ホルモン、インスリン等が挙げられ、中でも成長ホルモン、とりわけヒト成長ホルモンが好ましい。
【0016】
本発明において、生理活性ポリペプチドが金属を含有する場合、その金属含有量は0.1%(w/w)以下が好ましく、更に好ましくは0.01%(w/w)以下、特に好ましくは0.001%(w/w)以下であって実質的に金属を含まない生理活性ポリペプチドが最適である。例えば結晶性インスリンは、通常亜鉛、ニッケル、コバルト、カドミウム等の少量の重金属を含んでいる。0.4%(w/w)亜鉛を含んでいるインスリンは6量体で存在し、それ自身で安定に存在し、生体内分解性高分子重合物の金属塩との相互作用が弱められると考えられる。
必要な場合には、生理活性ポリペプチドに含有されている金属を前もって除去しておいてもよく、金属を除去する方法としては公知の方法が用いられる。例えばインスリンの塩酸酸性水溶液を、水あるいは酢酸アンモニウム塩溶液に対して透析したのち凍結乾燥することによりアモルファス状態で金属が最小限のインスリンが得られる。
成長ホルモンとしては、いずれの種由来のものでも良いが、好ましくはヒト成長ホルモンである。また、脳下垂体等から抽出される天然由来も本発明に用いられるが、好ましくは遺伝子組換え型GH(特公平6−12996号公報、特公平6−48987号公報参照)であり、更に好ましくはN末端にメチオニンを有さない天然型と同じ構造を有する組換え型hGHである。該GHとしては金属塩であってもよいが、実質的に金属を含有しないGHも用いられる。hGHとしては、分子量約22Kダルトンのみならず、分子量約20Kダルトンのもの(特開平7−101877号公報、特開平10−265404号公報参照)を用いてもよい。また、hGHの誘導体あるいはその関連タンパク質(WO99/03887号公報参照)を用いてもよい。
【0017】
生理活性ポリペプチドとしては、生体より抽出・精製されたもの、化学的に合成されたもの、あるいは遺伝子組換え法により作製されたもの等、いずれの方法によって作製されたものでも用いられるが、高純度の製品を大量に合成するためには遺伝子組換え法の利用が好ましい。また、いずれの方法を採用する場合においても、生理活性ポリペプチドを含有する組織・体液、化学合成粗調製物、あるいは組換え細胞・菌体より、それぞれ当該ポリペプチドを純化・精製する必要がある。この場合、一般的なペプチドあるいは蛋白の分離精製法が使用できるが(「タンパク質」、佐竹一夫著、朝倉書店;「生理活性ペプチド」、ファルマシアレビュー、No.3、日本薬学会)、特に幾つかの液体クロマトグラフィーを組み合わせることにより(「タンパク質・ペプチドの高速液体クロマトグラフィー」、宇井信生ら共編、化学同人)、高純度の生理活性ポリペプチドをその生理活性を損なうことなく収率よく得られる。また、所望により精製法の最終工程において、脱塩操作に供されることが好ましい。水溶液中での生理活性ポリペプチド濃度は、特に限定されないが、例えば0.01%(W/V)ないし30%(W/V)、好ましくは0.03%(W/V)ないし10%(W/V)、特に好ましくは0.05%(W/V)ないし3%(W/V)等である。生理活性ポリペプチドがhGHである場合、水溶液中でのhGH濃度は、好ましくは約0.01ないし約5%(W/V)、より好ましくは約0.05ないし約0.5%(W/V)である。
【0018】
本発明において生理活性ポリペプチド水溶液に添加される水混和性の有機溶媒としては、例えばアルコール類(例えば、メタノール、エタノール、イソプロパノール等、好ましくはメタノール、エタノール等)、アセトン等が挙げられる。これらは適宜の割合で混合して用いてもよいが、好ましくはアルコール類、特にエタノールを単独で用いることが望ましい。また、生理活性ポリペプチド水溶液への添加量(濃度)は、体積比において約0.03ないし0.5%(V/V)であり、好ましくは約0.06ないし0.25%(V/V)、更に好ましくは約0.1ないし0.15%(V/V)である。このような水混和性の有機溶媒の添加により得られる生理活性ポリペプチド水溶液を、更に凍結乾燥することにより、取り扱いが容易で(操作性のよい)、かつ微細な(粒子径の小さな)生理活性ポリペプチド粉体が作成できる。
本発明において生理活性ポリペプチド水溶液に添加される揮発性の塩類としては、例えばアンモニウム塩(例えば酢酸アンモニウム、重炭酸アンモニウム、炭酸アンモニウム、塩化アンモニウム等、好ましくは酢酸アンモニウム等)が挙げられる。これらは適宜の割合で混合して用いてもよい。揮発性の塩類の生理活性ポリペプチド水溶液への添加量は、モル比において約10倍ないし約80倍モルであり、好ましくは約10倍ないし約70倍モルであり、さらに好ましくは約15倍ないし約70倍モルであり、より好ましくは約20倍ないし約70倍モルであり、最も好ましくは約20倍ないし約50倍モルである。水混和性の有機溶媒を添加する場合と同様に、揮発性塩類の添加により得られる生理活性ポリペプチド水溶液を、更に凍結乾燥することにより、取り扱いが容易で(操作性のよい)、かつ微細な(粒子径の小さな)微細生理活性ポリペプチド粉体が作成できる。
本発明において、生理活性ポリペプチド水溶液に添加される水混和性の有機溶媒及び/又は揮発性の塩類は、単独で用いてもよいし、適宜組み合わせて用いてもよい。水混和性の有機溶媒及び揮発性の塩類を組み合せて用いる時は、上記のそれぞれの添加量に従って、生理活性ポリペプチド水溶液に添加することができる
上記の方法により得られた水混和性の有機溶媒及び/又は揮発性の塩類を含有する生理活性ポリペプチド水溶液は、本発明の凍結乾燥操作に供されるが、本操作としては医薬品・食品関係のみならず、水に関わるあらゆる分野で利用されている方法が使用される(「医薬品の開発」第11巻、仲井由宣編集、広川書店)。例えば生理活性ポリペプチド水溶液をバイアル又はアンプル内等に充填し、次いで水溶液を凍結後、乾燥処理に供する。この場合、機外で凍結した後、乾燥機に供してもよいが、通常は機内で十分な温度制御のもとに凍結するのが好ましい。また、乾燥操作においても段階的に実施することができる。近年の凍結乾燥機は、これらの温度制御又は真空制御が可能であり、対象とする生理活性ポリペプチドに最適の条件が選ばれる。凍結乾燥操作に供する水溶液中での生理活性ポリペプチド濃度は、特に限定されないが、例えば0.01%(W/V)ないし30%(W/V)、好ましくは0.03%(W/V)ないし10%(W/V)、特に好ましくは0.05%(W/V)ないし3%(W/V)等である。生理活性ポリペプチドがhGHである場合、水溶液中でのhGH濃度は、好ましくは約0.01ないし約5%(W/V)、より好ましくは約0.05ないし約0.5%(W/V)である。
【0019】
本発明における生体内分解性ポリマーの溶解に用いる有機溶媒は、沸点120℃以下であることが好ましい。該有機溶媒としては、例えばハロゲン化炭化水素(例えば、ジクロロメタン、クロロホルム、四塩化炭素等)、アルコール類(例えば、エタノール、メタノール、1,4−ブタンジオール、1,5−ペンタンジオール等)、酢酸エチル、アセトニトリル等が挙げられる。これらは適宜の割合で混合して用いてもよい。有機溶媒を単独で用いる場合、例えばジクロロメタン、アセトニトリル等が好ましい。有機溶媒を混合溶媒として用いる場合、例えばハロゲン化炭化水素(例えば、ジクロロメタン、クロロホルム等)と、アルコール類(例えば、エタノール、メタノール、1,4−ブタンジオール、1,5−ペンタンジオール等)あるいはアセトニトリルとの組み合わせが好ましい。特に、ジクロロメタンとアセトニトリルとの組み合わせが汎用される。ハロゲン化炭化水素と、アルコール類あるいはアセトニトリルとの混合比(体積比)は約40:1ないし約1:1であり、好ましくは約20:1ないし約1:1である。特に、ハロゲン化炭化水素(ジクロロメタン等)を単独、あるいはハロゲン化炭化水素とアセトニトリルとの9:1ないし1:1の混合溶媒を用いることが望ましい。また、生体内分解性ポリマーの溶液中濃度は分子量、有機溶媒等の種類によって異なるが、例えば約0.01ないし約80%(W/W)、好ましくは約0.1ないし約70%(W/W)、更に好ましくは約1ないし約60%(W/W)である。
【0020】
本発明の徐放性製剤は、水混和性の有機溶媒及び/又は揮発性塩類を添加した生理活性ポリペプチド溶液を凍結乾燥して得られる粉体(S相)を、生体内分解性ポリマーを溶解した有機溶媒液(O相)に分散させた、S/O型分散液から溶媒を除去することにより製造される。その製造法としては、例えば(a)水中乾燥法(S/O/W法)、(b)相分離法(コアセルベーション法)及び(c)噴霧乾燥法、あるいはこれらに準じた方法等が挙げられる。以下に徐放性製剤として、例えばマイクロカプセルを製造する場合の製造方法について記述する。
(a)水中乾燥法(S/O/W法)
本法によれば、まず生理活性ポリペプチド水溶液に水混和性の有機溶媒及び/又は揮発性塩類を添加した後、凍結乾燥により生理活性ポリペプチド粉体(S相)を作成する。次に生体内分解性ポリマーを有機溶媒に溶解し、この有機溶媒液中に上記の生理活性ポリペプチド粉体を添加し分散させる。この際、生理活性ポリペプチドと生体内分解性ポリマーとの比率(重量比)は、例えば約1:1000ないし約1:1、好ましくは約1:200ないし約1:5、更に好ましくは約1:100ないし約1:5である。また、生理活性ポリペプチド粉体を有機溶媒液中に均一に分散させるため、外部物理的エネルギーを加えることが好ましい。その方法としては例えば、超音波照射、タービン型撹拌器、ホモジナイザー等が用いられる。この時の有機溶媒液中での生理活性ポリペプチドの平均粒子径としては約10μm以下、さらに好ましくは約0.1μmないし約10μm、より好ましくは約0.5μmないし5μmであることが望ましく、本発明の製造法により得られた生理活性ポリペプチド粉体を用いることにより容易に達成される。本発明における生理活性ポリペプチドの平均粒子径は、ホモジナイザーを用いて該生理活性ポリペプチドをジクロロメタン等の有機溶媒中で分散した後に、レーザー解析式粒度分布測定装置(SALD2000A:島津)により得られる値を示す。その際、生理活性ポリペプチドはジクロロメタン等の有機溶媒に、例えば約20ないし100mg/mlの濃度で添加後、ホモジナイザー(例えば、ポリトロン(キネマチカ社))を用いて約20,000rpmで約30秒ないし1分間攪拌することにより分散液とされ、さらに上記粒度分布測定装置の測定可能な範囲となるように適宜、該有機溶媒で希釈し、供試される。
次いでこのようにして調製された有機溶媒分散液(S/O型分散液)を、更に水性溶媒(W相)中に添加して、上記と同様の外部物理的エネルギー、例えば超音波照射、タービン型撹拌器、あるいはホモジナイザー等によりS/O/W型エマルションを形成させる。以後、油相溶媒を蒸発させマイクロカプセルを製造する。この際の水相体積は、一般的には油相体積の約1倍ないし約10,000倍から選ばれる。更に好ましくは約2倍ないし約5,000倍から選ばれる。特に好ましくは約5倍ないし約2,000倍から選ばれる。
上記外水相中には、乳化剤を加えてもよい。該乳化剤としては、一般的に安定なS/O/Wエマルションを形成できるものであれば何れでもよい。乳化剤としては、例えばアニオン性界面活性剤、非イオン性界面活性剤、ポリオキシエチレンヒマシ油誘導体、ポリビニルピロリドン、ポリビニルアルコール、カルボキシメチルセルロース、レシチン、ゼラチン、ヒアルロン酸等が挙げられる。これらは適宜組み合わせて使用してもよい。外水相中の乳化剤の濃度は、好ましくは約0.001%ないし20%(w/w)である。更に好ましくは約0.01%ないし10%(w/w)、特に好ましくは約0.05%ないし5%(w/w)である。
【0021】
このようにして得られたマイクロカプセルは、遠心分離あるいは濾過操作により分取した後、マイクロカプセルの表面に付着している乳化剤等を蒸留水による洗浄で除去し、再び蒸留水等に分散して凍結乾燥する。その後必要であれば、加温してマイクロカプセル中の水分及び有機溶媒を更に除去する。減圧下に加温してもよい。加温条件としては、用いた生体内分解性ポリマーのガラス転移温度以上で、マイクロカプセルの各粒子が互いに付着しない程度の温度で加熱乾燥する。好ましくは、生体内分解性ポリマーのガラス転移温度からガラス転移温度より約30℃高い温度の範囲で加熱乾燥する。ここでガラス転移温度とは、示差走査熱量計を用い、加温速度毎分10ないし20℃で昇温した際に得られる中間点を云う。
【0022】
(b)相分離法(コアセルベーション法)
本法においては、前記(a)のS/O型分散液にコアセルベーション剤を撹拌下徐々に加えマイクロカプセルを析出、固化させる。該コアセルベーション剤は、上記分散液の約0.01倍ないし約1,000倍の体積量が加えられる。更に好ましくは、約0.05倍ないし約500倍、特に好ましくは約0.1倍ないし約200倍の体積量である。コアセルベーション剤としては、生体内分解性ポリマーを溶解する有機溶媒と混和する高分子系、鉱物油系又は植物油系の化合物で使用した生体内分解性ポリマーを溶解しないものであればよい。具体的には、例えばシリコン油、ゴマ油、大豆油、コーン油、綿実油、ココナッツ油、アマニ油、鉱物油、n−ヘキサン、n−ヘプタン等が用いられる。これらは2種以上混合して用いてもよい。このようにして得られたマイクロカプセルを濾過分取した後、ヘプタン等により繰り返し洗浄してコアセルベーション剤を除去する。更に、前記(a)と同様に洗浄し、次いで凍結乾燥する。
水中乾燥法及びコアセルベーション法でのマイクロカプセルの製造では、粒子同士の凝集を防ぐために凝集防止剤を加えてもよい。該凝集防止剤としては、例えばマンニトール、ラクトース、ブドウ糖、デンプン類(例えば、コーンスターチ等)、ヒアルロン酸あるいはこのアルカリ金属塩等の水溶性多糖、グリシン、フィブリン、コラーゲン等の蛋白質、塩化ナトリウム、リン酸水素ナトリウム等の無機塩類等が適宜用いられる。
【0023】
(c)噴霧乾燥法
本法においては、前記(a)のS/O型分散液を、ノズルを用いてスプレードライヤー(噴霧乾燥器)の乾燥室内へ噴霧し、極めて短時間に微粒化液滴内の有機溶媒を揮発させ、マイクロカプセルを製造する。該ノズルとしては、例えば二流体ノズル型、圧力ノズル型、回転ディスク型等がある。この際所望により、上記の分散液と同時に、マイクロカプセル粒子同志の凝集防止を目的として前記凝集防止剤の水溶液を別ノズルより噴霧することも有効である。このようにして得られたマイクロカプセルは、前記(a)と同様にして洗浄し、必要であれば加温(要すれば減圧下)により、水分及び有機溶媒を更に除去する。
【0024】
本発明の徐放性製剤は微粒子状であることが好ましい。なぜならば徐放性製剤は、皮下あるいは筋肉内注射に通常使用される注射針を通して投与される方が、患者に対し過度の苦痛を与えることがないからである。該徐放性製剤の粒子径は、例えば平均粒子径として約0.1ないし300μm、好ましくは約1ないし150μm、特に好ましくは約2ないし100μmである。
本発明の徐放性製剤に含まれる生理活性ポリペプチドの含量は、例えば約0.1ないし30%(W/W)、好ましくは約0.2ないし20%(W/W)、更に好ましくは約0.5ないし10%(W/W)である。該生理活性ポリペプチドの平均粒子径は好ましくは約10μm以下、さらに好ましくは約0.1μmないし10μm、より好ましくは約0.5μmないし5μmである。
また、本発明の徐放性製剤に含まれる生体内分解性ポリマーの含量は、例えば約30ないし99.9%(W/W)、好ましくは約60ないし97%(W/W)、さらに好ましくは約70ないし90%(W/W)である。
本発明の徐放性製剤の生理活性ポリペプチドの初期放出率[投与後1日(24時間)までの放出率]は、好ましくは約40%以下、さらに好ましくは約1ないし40%、より好ましくは約3ないし35%である。なお、該初期放出率は本発明の徐放性製剤皮下投与後24時間までの血中濃度のAUC(Area Under the Concentration)を、生理活性ポリペプチド溶液皮下投与後24時間までのAUCから得られる投与量−AUC直線に適用させることにより初期放出量が得られ、さらに初期放出率が算出される。
【0025】
本発明の徐放性製剤は、例えばマイクロカプセルとして、あるいはマイクロカプセルを原料物質として種々の剤形に製剤化し、非経口剤(例えば、筋肉内、皮下、臓器等への注射剤又は埋め込み剤、鼻腔、直腸、子宮等への経粘膜剤等)、経口剤(例えば、カプセル剤(例えば、硬カプセル剤、軟カプセル剤等)、顆粒剤、散剤等の固形製剤、懸濁剤等の液剤等)等として投与することができる。
本発明の徐放性製剤は特に注射用であることが好ましい。例えば、徐放性製剤がマイクロカプセルである場合、マイクロカプセルを分散剤(例えば、Tween 80、HCO-60 等の界面活性剤、カルボキシメチルセルロース、アルギン酸ナトリウム、ヒアルロン酸等の多糖類等)、保存剤(例えば、メチルパラベン、プロピルパラベン等)、等張化剤(例えば、塩化ナトリウム、マンニトール、ソルビトール、ブドウ糖等)等と共に水性懸濁剤とすることにより実用的な注射用徐放製剤が得られる。また、ゴマ油、コーン油等の植物油あるいはこれにレシチン等のりん脂質を混合したもの、あるいは中鎖脂肪酸トリグリセリド(例えば、ミグリオール812)と共に分散して油性懸濁剤として実際に使用できる徐放性注射剤とする。
【0026】
徐放性製剤が例えばマイクロカプセルである場合、マイクロカプセルの粒子径は、懸濁注射剤として使用する場合には、その分散度、通針性を満足する範囲であればよく、例えば平均粒子径として約0.1ないし約300μmの範囲が挙げられる。平均粒子径は、好ましくは約1ないし約150μm、特に好ましくは約2ないし約100μmの範囲である。
上記したマイクロカプセルを無菌製剤にするには、製造全工程を無菌にする方法、ガンマ線で滅菌する方法、防腐剤を添加する方法等が挙げられるが、特に限定されない。
【0027】
本発明の徐放性製剤は、低毒性で、哺乳動物(例えば、ヒト、牛、豚、犬、ネコ、マウス、ラット、ウサギ等)に対して安全に用いることができる。
徐放性製剤の適応は、使用する生理活性ポリペプチドにより種々異なる。生理活性ポリペプチドが、例えばインスリンである場合には、糖尿病等、インターフェロン−αである場合には、ウイルス性肝炎(例えば、C型肝炎、HBe 抗原陽性活動性肝炎等)、癌(例えば、腎癌、多発性骨髄腫等)等、エリスロポエチンの場合には貧血(例えば、腎透析時貧血等)等、G−CSFの場合には好中球減少症(例えば、制ガン剤治療時)、感染症等、IL−2の場合には癌(例えば、血管内皮腫等)等、FGFの場合には骨折、創傷(床ずれ等)、歯周病、消化管潰瘍等、FGF−9の場合には血小板減少症等、NGFの場合には老人性痴呆、神経病(ニューロパシー)等、TPAの場合には血栓症等、腫瘍壊死因子の場合には癌等の治療又は予防に有効である。また、GH含有徐放性製剤では、GHの成長ホルモン作用に基づき、下垂体性小人症の他、ターナー症候群、慢性腎疾患、軟骨異栄養症、更には成人性下垂体不全症に適応できる。また、GHはダウン症候群、シルバー症候群、骨形成不全症、あるいは若年性慢性関節症等の疾患にも適応され、有効な治療効果を得たとの報告もあり、GH含有徐放性製剤はこれらの疾患にも適応可能である。更にはうっ血性心不全症等の治療又は予防にも有効である。
【0028】
徐放性製剤の投与量は、生理活性ポリペプチドの種類と含量、放出の持続時間、対象疾病、対象動物等によって種々異なるが、該生理活性ポリペプチドの有効濃度が体内で保持される量であればよい。該生理活性ポリペプチドの投与量としては、例えば徐放性製剤が2週間型製剤である場合、好ましくは成人1人当たり約0.0001ないし約10mg/kg体重の範囲から適宜選ぶことができる。更に好ましくは約0.05ないし約1mg/kg体重の範囲から適宜選ぶことができる。投与回数は、1週間に1回、2週間に1回、1ヶ月に1回、2ヶ月に1回等、該生理活性ポリペプチドの種類と含量、剤型、放出の持続時間、対象疾病、対象動物等によって適宜選ぶことができる。好ましくは1週間ないし1ヶ月型徐放性製剤、さらに好ましくは1ないし3週間型徐放性製剤、より好ましくは10ないし20日型徐放性製剤、とりわけ好ましくは2週間型徐放性製剤が挙げられる。
徐放性製剤の有効成分である生理活性ポリペプチドが、例えばインスリンである場合には、糖尿病の成人に対する投与量は、有効成分として通常、約0.001ないし約1mg/kg体重、好ましくは約0.01ないし約0.2mg/kg体重の範囲から適宜選び、1週間に1回投与するのがよい。
【0029】
徐放性製剤の有効成分である生理活性ポリペプチドが、GHの場合には、投与量は、GHの種類と含量、放出の持続時間、対象疾病、対象動物等によって種々異なるが、該GHの有効濃度が体内で保持される量であればよい。前記した疾患の治療において、例えば徐放性製剤が2週間型製剤である場合、GHの投与量は有効成分として、好ましくは、小児あるいは成人1人当たり約0.01ないし約5mg/kg体重(約0.03ないし約15 IU/kg体重)の範囲から適宜選択して安全に投与することができる。更に好ましくは約0.05ないし約1mg/kg体重(約0.15ないし約3 IU/kg体重)の範囲から適宜選ぶことができる。投与回数は、1週間に1回、2週間に1回あるいは1ケ月に1回等、GH含量、剤型、放出の持続時間、対象疾病、対象動物等によって適宜選ぶことができる。好ましくは1週間ないし1ヶ月型徐放性製剤、さらに好ましくは1ないし3週間型徐放性製剤、より好ましくは10ないし20日型徐放性製剤、とりわけ好ましくは2週間型徐放性製剤が挙げられる。
徐放性製剤は、常温あるいは冷所に保存することが好ましい。徐放性製剤は、冷所に保存することが更に好ましい。ここでいう常温あるいは冷所とは、日本薬局方において定義されるものである。すなわち、常温とは15ないし25℃を、冷所とは15℃以下を意味する。冷所のうち、とりわけ2ないし8℃が好ましい。
【0030】
【発明の実施の形態】
以下に参考例、実施例、比較例及び試験例を挙げて、更に具体的に説明するが、これらは本発明を限定するものではない。また、本発明の明細書においてアミノ酸等の略号で表示する場合には、IUPAC−IUB Commission on Biochemical Nomenclature による略号あるいは当該分野における慣用略号に基づくものであり、その例を下に示す。またアミノ酸に関して光学異性がある場合は、特に明示しなければL−体を示す。
SDS : ドデシル硫酸ナトリウム
Gly : グリシン
Ala : アラニン
Val : バリン
Leu : ロイシン
Ile : イソロイシン
Ser : セリン
Thr : スレオニン
Cys : システイン
Met : メチオニン
Glu : グルタミン酸
Gln : グルタミン
Asp : アスパラギン酸
Asn : アスパラギン
Lys : リジン
Arg : アルギニン
His : ヒスチジン
Phe : フェニルアラニン
Tyr : チロシン
Trp : トリプトファン
Pro : プロリン
Asx : Asp + Asn
Glx : Glu + Gln
【0031】
【実施例】
参考例1 T7プロモーターを用いたヒト成長ホルモン(hGH)発現ベクターの構築
hGHの構造遺伝子はプラスミド pHGH107(特公平6−12996号公報に記載、寄託番号ATCC31538及びATCC40011)からEcoRI及びEcoRVで切断し約0.75kbの断片を単離した。一方、pET−3C〔Rosenberg et al.,ジーン(Gene)、56巻、125頁(1987年)〕をNdeI及びBamHIで切断し、T7プロモーター及びアンピシリン耐性遺伝子を有する約4.6kbの断片を単離した。
両断片をT4DNAポリメラーゼ(DNA Blunting kit、宝酒造株式会社製)で平滑末端とし、T4DNAリガーゼで連結したのち、大腸菌JM109に導入し、アンピシリン耐性を指標として形質転換体を選択した。出現したコロニーの中から12個を拾い、これらからプラスミドを調製し制限酵素PstIによる切断パターンを調べたところ、6個のコロニーからのプラスミドにおいてhGH遺伝子が正しい方向で挿入されていることがわかった。この中の1株から得られたプラスミドをpTGA201と命名した。
【0032】
参考例2 大腸菌でのMet−hGHの発現
大腸菌JM109を、T7ファージのRNAポリメラーゼ遺伝子で組み換えられているラムダファージ(スチュディエ、スプラ)で溶原化した。その後、参考例1で得られたhGH発現ベクターpTGA201をこの大腸菌JM109(DE3)へ導入し、大腸菌JM109(DE3)/pTGA201を得た。
この形質転換細胞を、50μg/mlのアンピシリンを含むLB培地(1%ペプトン、0.5%酵母エキス、0.5%塩化ナトリウム)1リットルを含む2リットル容フラスコ中で30℃、16時間振とう培養した。得られた培養液を、0.02%ニューポールLB−625(消泡剤;三洋化成工業製)及び50μg/mlのアンピシリンを含む20リットルのLB培地を仕込んだ50リットル容発酵槽へ移植して、37℃、6時間通気撹拌培養した。この培養液を360リットルの主発酵培地(1.68%リン酸一水素ナトリウム、0.3%リン酸二水素カリウム、0.1%塩化アンモニウム、0.05%塩化ナトリウム、0.0246%硫酸マグネシウム、0.02%ニューポールLB−625、0.0005%塩酸チアミン、1.5%ブドウ糖、1.5%カザミノ酸)を仕込んだ500リットル容発酵槽に移植して、37℃で通気撹拌培養を開始した。培養液の濁度が約500クレット単位になった時点で、5.95mg/リットル分のイソプロピル−β−D−チオガラクトピラノシド(IPTG)を添加し、更に培養を続け、4時間後に培養液を遠心分離して、約4.5kgの湿菌体を得、−80℃に凍結保存した。
上記の形質転換大腸菌JM109(DE3)/pTGA201は、受託番号FERM BP−5632として通商産業省工業技術院生命工学工業技術研究所(NIBH)に寄託され、また受託番号IFO 16001として財団法人発酵研究所(IFO)に寄託されている。
【0033】
参考例3 Met−hGHの活性化
参考例2で得られた菌体2kgに50mMトリス/HCl、8Mグアニジン塩酸塩溶液(pH8.0)6リットルを加えて菌体を溶解後、遠心分離(10000rpm、120分間)を行った。得られた上澄液6リットルに50mMトリス/HCl、0.28mMGSSG、1.4mMGSH、0.7Mアルギニン(pH8.0)18リットルを加えて pH8.0に調整した後、4℃で5日間活性化を行った。
【0034】
参考例4 Met−hGHの精製
参考例3で活性化の終了した再生液をペリコンカセットシステム(PTGC膜、ミリポア社)で、20mMトリス/HCl、2.5M尿素(pH8.0)を加えながら電気伝導度が10mS以下になるまで脱塩、濃縮を行った後、得られた濃縮液を遠心分離(10000rpm、60分間)し、濃縮液の上清5リットルを得た。ついでこの液を20mMトリス/HCl、2.5M尿素(pH8.0)で平衡化したDEAE−トヨパール650Mカラム(20cmφ×84cm、東ソー社)に吸着させ、十分に洗浄した後、0ないし25%B(B=20mMトリス/HCl、2.5M尿素、1M塩化ナトリウム、pH8.0)の濃度勾配で100分間、300ml/分の流速で溶出を行い、Met−hGH画分として10リットルの溶出液を得た。更に、この溶出液をペリコンカセットシステム(PTGC膜、ミリポア社)で濃縮、脱塩した。更に、高速液体クロマトグラフ法(ギルソンHPLCシステム、ギルソン社)により、この溶液をDEAE−5PWカラム(21cm×30cm、東ソー社)に通液吸着させた後、A=50mMトリス/HCl+2.5M尿素(pH8.0)、B=50mMMES[2−(N−モルホリノ)エタンスルホン酸]+2.5M尿素(pH4.0)とによる70ないし85%Bの pH勾配で、70分間、320ml/分の流速で溶出を行い、Met−hGH画分6リットルを得た。この溶出液に2Mトリス/HCl(pH7.8)溶液300ml加えて pH7.2に調整し、ついでペリコンカセットシステム(PTGC膜、ミリポア社)で濃縮、脱塩し、9,979ミリグラムのMet−hGHを得た。
【0035】
参考例5 N末端Metの除去
参考例4で得たMet−hGH溶液1650mlに2.5Mグリオキシル酸、35mM硫酸銅、6Mピリジン溶液413mlを加え、よく撹拌した後、25℃で60分間反応させた。次いで、20mMトリス/HCl、2.5M尿素(pH8.0)で平衡化したセファデックスG−25カラム(11.3cmφ×125cm、ファルマシア社)に3リットル/hの流速で通液し、平衡化と同じ緩衝液を用いて展開し、溶出してきたMet−hGHのジケトン体画分を、よく撹拌しながら直接4M酢酸、4M酢酸ナトリウム、80mMo−フェニレンジアミン、3M尿素溶液4リットル中に加えた。溶出終了後、この反応溶液8リットルを4℃に3日間静置した。静置後、ペリコンカセットシステム(PTGC膜、ミリポア社)で4リットルに濃縮し、20mMトリス/HCl、2.5M尿素(pH8.0)で平衡化したセファデックスG−25カラム(11.3cmφ×140cm、ファルマシア社)に3リットル/hの流速で通液し、hGH画分4.7リットルを集めた。更に、高速液体クロマトグラフ法(ギルソンHPLCシステム、ギルソン社)により、この溶液をDEAE−5PWカラム(21cm×30cm、東ソー社)に通液吸着させた後、A=50mMトリス/HCl+2.5M尿素(pH8.0)、B=50mMMES[2−(N−モルホリノ)エタンスルホン酸]+2.5M尿素(pH4.0)とによる70ないし85%Bの pH勾配で、70分間、320ml/分の流速で溶出を行い、hGH画分10リットルを得た。このhGH画分に2Mトリス/HCl(pH7.8)溶液を500ml加えて pH7.2に調整後、ミニタンII(PTGC膜、ミリポア社)で濃縮し、この濃縮液500mlを蒸留水で平衡化したセファクリルS−100カラム(113.cmφ×50cm、ファルマシア社)に2リットル/hの流速で通液、展開しhGH画分1651mlを得た。更に、この溶液をミリパック60(ミリポア社)でろ過し、hGH溶液1487ml(3309mgのhGH)を得た。
【0036】
参考例6 hGHの性状確認
(a)SDSポリアクリルアミドゲル電気泳動を用いた分析
参考例5で得られたhGHに100mMDTTを含むサンプルバッファー[Laemmli, Nature, 227, 680(1970)]を等量加えてよく撹拌し、95℃で2分間加熱後、マルチゲル10/20(第一化学薬品)で電気泳動を行った。泳動後のゲルをクーマシー・ブリリアント・ブルー(Coomassie brilliant blue)で染色した結果、約22kdに単一バンドが認められたことから、精製hGHはほぼ単一であることが確認された。
【0037】
(b)アミノ酸組成分析
アミノ酸組成をアミノ酸分析計(L−8500A,日立)を用いて決定した。その結果、得られたhGHのアミノ酸組成はcDNAの塩基配列から推定されるアミノ酸組成と一致した(表1)。
【表1】

1)0時間に外挿した値
2)未検出
約20μgを用いて分析を行った。
【0038】
(c)N末端アミノ酸配列分析
N末端アミノ酸配列を気相プロテインシーケンサー(アプライドバイオシステムズ社、モデル477A)を用いて決定した。その結果、得られたhGHのN末端アミノ酸配列はcDNAの塩基配列から推定されたhGHのN末端アミノ酸配列と一致した(表2)。
【表2】
1nmolを用いて分析を行った。
【0039】
(d)C末端アミノ酸分析
C末端アミノ酸をアミノ酸分析計(L−8500A,日立)を用いて決定した。得られたhGHのC末端アミノ酸はcDNAの塩基配列から推定されたC末端アミノ酸と一致した(表3)。
【表3】
18nmolを用いて分析を行った。
(e)hGHの活性測定
参考例5で得られた精製hGHのNb2細胞〔ジャーナル・オブ・クリニカル・エンドクリノロジー・アンド・メタボリズム、51巻、1058頁(1980)〕に対する増殖促進効果は、標準品と同等であった。
【0040】
実施例1
(1)hGH粉体の製造
参考例5に準じて得られた遺伝子組換え型hGH水溶液(最終hGH濃度=2mg/ml)に、エタノール[最終濃度(V/V%)=0.03%,0.06%,0.1%,0.15%,0.25%,0.5%)]を添加し、凍結乾燥することにより6種のhGH粉体を得た。
(2)hGH含有マイクロカプセルの製造
乳酸−グリコール酸共重合体(乳酸/グリコール=50/50,ポリスチレン換算平均分子量=12,000,粘度=0.145dl/g)1.85gと酸化亜鉛10mgとをジクロロメタン3.0mlに溶解した。この有機溶媒液に上記(1)のhGH粉体140mgを添加し、ポリトロン(キネマチカ社)を用いて微粒化した。このS/O分散液を0.1%ポリビニルアルコール水溶液800mlに添加し、ホモミキサーを用いて撹拌・乳化した。室温で3時間撹拌してジクロロメタンを揮散させた後、遠心分離(約1,500rpm)することによりマイクロカプセルを分取した。次いで蒸留水400mlを用いて2回洗浄後、D−マンニトール0.2gを添加し凍結乾燥した。更に残留溶媒除去のため、46℃で3日間真空乾燥して6種のhGH含有マイクロカプセルを得た。
【0041】
実施例2
(1)hGH粉体の製造
参考例5に準じて得られた遺伝子組換え型hGH水溶液(最終hGH濃度=2mg/ml)に、酢酸アンモニウムの10,20,25,40,50,70及び100倍モル等量を添加し、凍結乾燥することにより7種のhGH粉体を得た。
(2)hGH含有マイクロカプセルの製造
実施例1(2)と同様の操作により、上記(1)記載のhGH粉体を用いて7種のhGH含有マイクロカプセルを得た。
【0042】
比較例1
(1)hGH粉体の製造
参考例5に準じて得られた遺伝子組換え型hGH水溶液(最終hGH濃度=2mg/ml)に、エタノール及び/又は酢酸アンモニウムを添加せずに、凍結乾燥することによりhGH粉体を得た。
(2)hGH含有マイクロカプセルの製造
実施例1(2)と同様の操作により、上記(1)記載のhGH粉体を用いてhGH含有マイクロカプセルを得た。
【0043】
試験例1
実施例1(1)及び比較例1(1)において得られるhGH粉体について、その取扱い易さを、粉体容積(嵩高さ)及び静電気への帯電性の観点から判定した。結果を〔表4〕に示す。
〔表4〕の結果から明らかなように、エタノール無添加のhGH粉体に比べ、エタノールを添加したhGH粉体では取り扱い易さが格段に向上した。
【0044】
試験例2
実施例1(2)及び比較例1(2)の中間工程において得られるhGH粉体/PLGA有機溶媒のS/O分散液50μl に、更にジクロロメタン2mlを添加した後、hGH粉体の平均粒子径をレーザー回折式粒度分布測定装置(SALD2000A:島津)を用いて測定した。結果を〔表4〕に示す。
〔表4〕の結果から明らかなように、エタノール無添加のhGH粉体は平均粒子径が10μm以上であるのに対し、エタノールを添加したhGH粉体はいずれも平均粒子径が10μm以下の微細粉末であった。
【表4】
試験例3
実施例1(2)及び比較例1(2)で製造したhGH含有マイクロカプセルを用いて以下の試験を実施した。
(1)hGH含量
マイクロカプセル5mgに0.3mlのアセトニトリルを添加後、超音波処理に付した。得られたアセトニトリル溶液に0.7mlの10mMリン酸食塩緩衝液(pH8.0)を添加し、主薬であるhGHを抽出した。次いでこのhGH抽出液を下記条件のサイズ排除高速液体クロマトグラフィーに供してhGH含量を測定し、封入率を算定した。結果を〔表5〕に示す。
カラム:TSKgelG 3000SWXL(Tosoh)
抽出液:0.2Mリン酸緩衝液(pH6.8)−0.2M NaCl
流 速:0.6ml/min
〔表5〕の結果から明らかなように、エタノール無添加のhGH粉体を用いて製造したhGH含有マイクロカプセルに比べ、エタノールを添加したhGH粉体を用いて製造したhGH含有マイクロカプセルでは高いhGH封入率が得られた。
【表5】
(2)in vivo 放出性
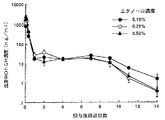
免疫抑制SDラット(雄性,6週齢)にマイクロカプセルを皮下投与し(hGH量として6mg/ラット)、経時的に採血後、その血清中のhGH濃度をラジオイムノアッセイ(Ab ビーズHGH:栄研化学)により測定し、hGH徐放性を評価した。免疫抑制SDラットは、プログラフ(藤沢薬品)を、マイクロカプセル投与3日前に0.4mg/ラット、投与時、4日後、7日後、11日後及び14日後にそれぞれ0.2mg/ラットを皮下投与して作成した。結果を〔図1〕に示す。
〔図1〕の結果から明らかなように、エタノールを添加したhGH粉体を用いて得られたhGH含有マイクロカプセルでは、初期放出[投与後1日(24時間)までの放出量]が小さく、その後の持続濃度[投与後1日(24時間)を超え2週間までの放出量]が高かった。すなわち、エタノールを添加したhGH粉体を用いることにより、初期放出の抑制と、2週に亘る高い血中hGH濃度が達成された。
【0047】
試験例4
実施例2(1)及び比較例1(1)において得られるhGH粉体について、その取り扱い易さを、粉体容積(嵩高さ)及び静電気への帯電性の観点から判定した。結果を〔表6〕に示す。
〔表6〕の結果から明らかなように、酢酸アンモニウム無添加のhGH粉体に比べ、酢酸アンモニウムを添加したhGH粉体では取扱い易さが格段に向上した。
【0048】
試験例5
実施例2(2)及び比較例1(2)の中間工程において得られるhGH粉体/PLGA有機溶媒のS/O分散液を、試験例2と同様の操作によりレーザー回折式粒度分布測定装置(SALD2000A:島津)に供して、hGHの平均粒子径を測定した。結果を〔表6〕に示す。
【表6】
試験例6
実施例2(2)及び比較例1(2)で製造したhGH含有マイクロカプセルを用いて以下の実験を実施した。
(1)hGH含量
試験例1(1)と同様の操作により、各種マイクロカプセルのhGH封入率を測定した。結果を〔表7〕に示す。
〔表7〕の結果から明らかなように、酢酸アンモニウム無添加のhGH粉体を用いて製造したhGH含有マイクロカプセルに比べ、酢酸アンモニウムを添加したhGH粉体を用いて製造したhGH含有マイクロカプセルでは高いhGH封入率が得られた。
(2)初期放出率
以下に示す方法により、マイクロカプセルの初期放出率を算出した。結果を〔表7〕に示す。
免疫抑制SDラット(雄性,6週齢)にhGH水溶液(濃度5,10,20mg/Kg)を皮下投与し、経時的に24時間まで採血後、その血清中のhGH濃度をラジオイムノアッセイ(Ab ビーズHGH:栄研化学)により測定し、各濃度におけるAUCを求め、投与量−AUC(Area Under the Concentration)直線を得た。
また、免疫抑制SDラット(雄性,6週齢)にマイクロカプセル(hGH量として6mg)を皮下投与し、経時的に24時間まで採血後、その血清中のhGH濃度をラジオイムノアッセイ(Ab ビーズHGH:栄研化学)により測定し、各マイクロカプセルにおけるAUCを求めた。得られたAUCを投与量−AUC直線に適用することにより初期放出量を得、初期放出率を算出した。
【表7】
試験例1(2)と同様の操作により、マイクロカプセルの免疫抑制SDラットにおけるhGH徐放性を評価した。結果を〔図2〕に示す。
〔図2〕の結果から明らかなように、酢酸アンモニウムを添加したhGH粉体を用いて得られたhGH含有マイクロカプセルでは、初期放出[投与後1日(24時間)までの放出量]が小さく、その後の持続濃度[投与後1日(24時間)を超え2週間までの放出量]が高かった。すなわち、酢酸アンモニウムを添加したhGH粉体を用いることにより、初期放出の抑制と、2週に亘る高い血中hGH濃度が達成された。
【0050】
【発明の効果】
本発明によれば、製剤工程における生理活性ポリペプチド粉体の取り扱い易さを改善し、徐放性製剤の工業的大量生産を可能にする。また、徐放性製剤への生理活性ポリペプチドの封入率を高め、初期放出率が低く長期に亘り安定した高い血中維持濃度を示す徐放性製剤を提供できる。
【0051】
【図面の簡単な説明】
【図1】免疫抑制SDラットにおける、エタノールを添加したhGH粉体を用いて得られたhGH含有マイクロカプセル投与後の血清中のhGH濃度推移を示すグラフである。
【図2】免疫抑制SDラットにおける、酢酸アンモニウムを添加したhGH粉体を用いて得られたhGH含有マイクロカプセル投与後の血清中のhGH濃度推移を示すグラフである。
Claims (14)
- 生理活性ポリペプチド水溶液に、(1)生理活性ポリペプチド水溶液に対する濃度が0.03%(V/V)ないし0.5%(V/V)のエタノール及び/又は(2)生理活性ポリペプチドに対するモル比が10倍ないし80倍の酢酸アンモニウムを添加後、凍結乾燥して得られる生理活性ポリペプチド粉体を、生体内分解性ポリマーの有機溶媒液に分散させた後、有機溶媒を除去することを特徴とする徐放性製剤の製造法。
- 生理活性ポリペプチド粉体が平均粒子径10μm以下のものである請求項1記載の製造法。
- 生理活性ポリペプチドが成長ホルモンである請求項1記載の製造法。
- 成長ホルモンがヒト成長ホルモンである請求項3記載の製造法。
- 生体内分解性ポリマーが乳酸−グリコール酸重合体である請求項1記載の製造法。
- 乳酸−グリコール酸重合体の乳酸/グリコール酸組成比(モル%)が100/0ないし40/60である請求項5記載の製造法。
- 乳酸−グリコール酸重合体の重量平均分子量が3,000ないし50,000である請求項5記載の製造法。
- 乳酸−グリコール酸重合体が多価金属塩である請求項5記載の製造法。
- 多価金属が亜鉛である請求項8記載の製造法。
- 徐放性製剤がマイクロカプセルである請求項1記載の製造法。
- マイクロカプセルの平均粒子径が0.1ないし300μmである請求項10記載の製造法。
- 徐放性製剤が注射用である請求項1記載の製造法。
- 生理活性ポリペプチド水溶液に、(1)生理活性ポリペプチド水溶液に対する濃度が0.03%(v/v)ないし0.5%(v/v)のエタノール及び/又は(2)生理活性ポリペプチドに対するモル比が10倍ないし80倍の酢酸アンモニウムを添加後、凍結乾燥することを特徴とする生理活性ポリペプチド粉体の製造法。
- 平均粒子径が10μm以下である生理活性ポリペプチド粉体を製造するための(1)生理活性ポリペプチド水溶液に対する濃度が0.03%(v/v)ないし0.5%(v/v)のエタノール及び/又は(2)生理活性ポリペプチドに対するモル比が10倍ないし80倍の酢酸アンモニウムの使用。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP07479399A JP4758525B2 (ja) | 1998-03-20 | 1999-03-19 | 生理活性ポリペプチドの徐放性製剤およびその製造法 |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP10-71853 | 1998-03-20 | ||
| JP7185398 | 1998-03-20 | ||
| JP1998071853 | 1998-03-20 | ||
| JP07479399A JP4758525B2 (ja) | 1998-03-20 | 1999-03-19 | 生理活性ポリペプチドの徐放性製剤およびその製造法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JPH11322631A JPH11322631A (ja) | 1999-11-24 |
| JP4758525B2 true JP4758525B2 (ja) | 2011-08-31 |
Family
ID=26412966
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP07479399A Expired - Fee Related JP4758525B2 (ja) | 1998-03-20 | 1999-03-19 | 生理活性ポリペプチドの徐放性製剤およびその製造法 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP4758525B2 (ja) |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| NZ516083A (en) * | 1999-05-27 | 2003-08-29 | Acusphere Inc | Porous drug matrices and methods of manufacture thereof |
| EP1346722B1 (en) | 2000-12-01 | 2008-12-10 | Takeda Pharmaceutical Company Limited | Method for producing preparation containing bioactive substance |
| JP5160005B2 (ja) * | 2000-12-28 | 2013-03-13 | 武田薬品工業株式会社 | 徐放性製剤 |
| KR100700869B1 (ko) * | 2005-06-03 | 2007-03-29 | 재단법인 목암생명공학연구소 | Pth, 완충제 및 안정제를 포함하는 안정한 pth조성물 |
| KR101424163B1 (ko) * | 2010-12-24 | 2014-08-01 | 주식회사 삼양바이오팜 | 수난용성 약물 함유 서방성 마이크로입자 및 그 제조방법 |
| JP5938762B1 (ja) * | 2015-09-01 | 2016-06-22 | 日揮株式会社 | マイクロカプセル製剤及びその製造方法 |
| JP6587942B2 (ja) * | 2016-01-19 | 2019-10-09 | 日揮株式会社 | マイクロカプセル製剤及びその製造方法 |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH107583A (ja) * | 1995-06-27 | 1998-01-13 | Takeda Chem Ind Ltd | 徐放性製剤の製造法 |
| JP3913316B2 (ja) * | 1996-06-14 | 2007-05-09 | 武田薬品工業株式会社 | N末端メチオニンの除去方法 |
-
1999
- 1999-03-19 JP JP07479399A patent/JP4758525B2/ja not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| JPH11322631A (ja) | 1999-11-24 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6197350B1 (en) | Method of producing a sustained-release preparation | |
| CA2433037C (en) | Sustained-release preparation | |
| JP3249147B2 (ja) | 生理活性蛋白含有経口製剤 | |
| RU2098121C1 (ru) | Микрокапсула для длительного высвобождения физиологически активного пептида | |
| JP5160005B2 (ja) | 徐放性製剤 | |
| CA2127317C (en) | Method of producing sustained-release preparation | |
| TW555569B (en) | Biodegradable microparticles for the sustained delivery of therapeutic drugs | |
| US6482864B1 (en) | Sustained-release preparation of physiologically active polypeptide and production thereof | |
| US20090142399A1 (en) | Dispersant agent for sustained-release preparations | |
| JP4683319B2 (ja) | 徐放性製剤用の分散剤 | |
| US6429296B2 (en) | Complex of human growth hormone and zinc and use | |
| JP4758525B2 (ja) | 生理活性ポリペプチドの徐放性製剤およびその製造法 | |
| JP4117922B2 (ja) | 徐放性製剤およびその製造法 | |
| JP4459315B2 (ja) | 徐放性製剤の製造法 | |
| JPH11158200A (ja) | ヒト成長ホルモン・亜鉛複合体及びその用途 | |
| JPH083055A (ja) | 徐放性製剤の製造法 | |
| JP2001151694A (ja) | タンパク質粉体の製造法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20051020 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20051020 |
|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20061225 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20090210 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20090326 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20100209 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20100407 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20110510 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20110603 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20140610 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20140610 Year of fee payment: 3 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |