JP4371694B2 - 標的成分の分離方法、検出方法、スクリーニング方法 - Google Patents
標的成分の分離方法、検出方法、スクリーニング方法 Download PDFInfo
- Publication number
- JP4371694B2 JP4371694B2 JP2003127363A JP2003127363A JP4371694B2 JP 4371694 B2 JP4371694 B2 JP 4371694B2 JP 2003127363 A JP2003127363 A JP 2003127363A JP 2003127363 A JP2003127363 A JP 2003127363A JP 4371694 B2 JP4371694 B2 JP 4371694B2
- Authority
- JP
- Japan
- Prior art keywords
- pha
- target component
- carrier
- group
- magnetic
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 0 CC(CC(C1)C(C)CCCC1N(C=[*+])N(C(NC(OC)=C)=C)OC(C)(C)C)O Chemical compound CC(CC(C1)C(C)CCCC1N(C=[*+])N(C(NC(OC)=C)=C)OC(C)(C)C)O 0.000 description 3
Images


Landscapes
- Investigating Or Analysing Materials By The Use Of Chemical Reactions (AREA)
- Investigating Or Analysing Materials By Optical Means (AREA)
- Other Investigation Or Analysis Of Materials By Electrical Means (AREA)
Description
【発明の属する技術分野】
本発明は、検体中に含有される、特定の標的成分を分離する方法、該標的成分を検出する方法、あるいは、特定の標的成分をスクリーニングする方法に関し、より具体的には、特定の標的成分に対する特異的な親和結合性を有する分子、例えば、天然由来の、あるいは人工的な改変を施さた核酸分子、タンパク質およびペプチド、糖鎖、脂質、低分子化合物、ならびにこれらの複合体を利用し、担体の表面上に前記特異的な親和結合性を有する分子を担持した構造体とした上で、該構造体と特定の標的成分との結合を行わせることによって、検体中に含有される特定の標的成分を選択的に分離する方法、検出する方法、または、スクリーニングする方法、さらには、前記方法の実施に専ら利用可能な装置に関する。
【0002】
【背景技術】
検体試料中に含まれる標的成分、特には、医学的治療・診断に有効な、あるいは産業的に価値のある標的成分に関して、その分離・回収、検出、スクリーニングを行う際、例えば、プローブ分子等、標的成分に対して結合親和力を有する分子を担持する担体として、マイクロメートルサイズからナノメートルサイズの微小な微粒子を用いる分離・回収、検出、スクリーニング方法が様々に開発、利用されている。特に、前記担体として、磁性を有する微粒子(以下、磁性微粒子と称する)を利用する方法は、検体試料から担体を分離、回収する際、磁性微粒子は、磁力による分離、回収が容易に行える利点を有している。そのため、磁性微粒子を利用する方法における、多くの開発例を挙げることができる。
【0003】
上記のプローブ分子等を担持する担体として利用される、磁性微粒子の多くは、その安定性の向上、磁性の制御等の目的で、表面を有機高分子により被覆された形で用いられる。
【0004】
有機高分子により被覆された磁性微粒子の、担体への応用の一領域として、従来から、抗原抗体反応を利用した免疫検出・診断方法が開発されてきた。
【0005】
その一例として、特開平07−151755号公報には、担体として、ローヌプーラン社製 平均粒径0.7μmの磁性体含有ポリスチレンラテックス粒子を用いて、免疫検出を行う方法が開示されている。
【0006】
特開平10−221341号公報には、日本ダイナル社製 トシル化磁性微粒子(Dynabead M−280、平均粒径2.8μm)を用いた免疫測定方法が開示されている。同じく、特開平09−229936号公報には、磁性微粒子 Dynabeads M−450 uncoated、ダイナル社製、粒径:4.5 μm、3%(w/v)を用いた免疫測定方法および装置が開示されている。
【0007】
また、有機高分子により被覆された磁性微粒子の、担体への応用の更なる一領域として、DNA等の核酸分子の検査・診断方法が挙げられる。
【0008】
特開平05−281230号公報には、担体用の磁性微粒子として、ディノ・インダストリア社(Dyno Industrier A. S.,Norway)製XP−6006を用い、抗原抗体反応に加え、DNA等の核酸分子の検査診断方法が開示されている。
【0009】
加えて、有機高分子により被覆された磁性微粒子の応用の更なる一領域として、標的成分の分離・回収方法も開発されてきている。
【0010】
特開平09−304385号公報には、ダイナビーズ M−450 アンコーテッド (ダイナル社:Dynal)(磁性微粒子自体の直径:45μm)を用いた好塩基球の分離・回収方法および装置が開示されている。
【0011】
特開平10−068731号公報には、ローヌ−プーラン(Rhone−Poulenc)社製磁性微粒子を、免疫学的に活性な物質、または核酸との共有結合を形成させる磁性微粒子として用いて、液体中の被検成分を磁気的に分離する方法が開示されている。
【0012】
また、米国特許第4,230,685号、同第3,970,518号、同第5,508,164号、同第5,567,326号および同第4,018,886号の米国特許公報においても、磁性微粒子を標的成分に結合せしめ、磁性微粒子と結合した標的成分を処理および分離するために用いる方法が開示されている。
【0013】
米国特許第5,900,481号には、コーティングを施した磁性微粒子を用いて、DNAを結合することにより、そのDNAに操作を施すことが開示されている。
【0014】
米国特許第5,834,197号には、コーティングを施した磁性微粒子を用いて、液体から一定種の菌株を捕捉する方法が開示されている。このビーズには、抗原に対する選択的親和力を備えている、標識抗体を添加して、対象の抗原を挟持させ、標識抗体と反応する抗原を容易に検出および回収できるように、磁性微粒子に検知可能な標識を結合させる方法が開示されている。
【0015】
以上に述べた特許文献以外に、磁性微粒子に結合される様々な分子の操作に関する開示を含む非特許文献として、Analytical Chemistry68(13):2122〜6(1996)およびNucleic AcidsResearch 23(16):3126〜31(1995年)を挙げることができる。
【0016】
加えて、上で述べた磁性微粒子以外にも、標的成分を検出・回収・スクリーニングする方法に用いる目的で既に市販されている磁性微粒子の例として、スフェロテック(Spherotech)社のフェロマグネティック・パーティクル(Ferromagnetic Particle)、セラディン(Seradyn)社のセラ−マグ(Sera−Mag)、バングス・ラボラトリー(Bangs Laboratory)社のエスタポール(Esteapor)を挙げることができる。
【0017】
【特許文献1】
特開平07−151755号公報
【特許文献2】
特開平10−221341号公報
【特許文献3】
特開平09−229936号公報
【特許文献4】
特開平05−281230号公報
【特許文献5】
特開平09−304385号公報
【特許文献6】
特開平10−068731号公報
【特許文献7】
米国特許第4,230,685号明細書
【特許文献8】
米国特許第3,970,518号明細書
【特許文献9】
米国特許第5,508,164号明細書
【特許文献10】
米国特許第5,567,326号明細書
【特許文献11】
米国特許第4,018,886号明細書
【特許文献12】
米国特許第5,900,481号明細書
【特許文献13】
米国特許第5,834,197号明細書
【非特許文献1】
Analytical Chemistry 68(13):2122〜6(1996)
【非特許文献2】
Nucleic Acids Research 23(16):3126〜31(1995年)
【0018】
【発明が解決しようとする課題】
標的成分の分離・回収方法、検出方法、スクリーニング方法において、対象とする標的成分は、生理的活性を有するか、あるいは医療・診断に対し有効である場合が多く、そのためできるだけ生体条件に近く、生理条件を損なわないような環境中で操作を行うことが望ましい。しかし、上記する従来の磁性微粒子を含む担体に用いた方法では、該磁性微粒子の表面を被覆あるいはコーティングしている被覆層成分は、スチレン系、アクリル系、ビニル系等の合成高分子であるため、非特異吸着の課題や、合成高分子中の残留モノマー成分の漏出による機能の低下といった課題が依然として生じている。さらには、標的成分のみならず、磁性微粒子を含む担体上に担持・固定化される標的成分結合分子も、生体由来の成分かあるいは、その派生物質である場合が多く、該磁性微粒子を含む担体の被覆層に用いている、非天然の高分子表面がこれら標的成分結合分子の標的成分に対する結合機能に悪影響を及ぼしていないとは断言できない。
【0019】
本発明は、前記の課題を解決するものであり、担体上に標的成分結合分子を担持・固定化してなる構造体を利用して、標的成分の分離・回収、検出、スクリーニングを行う際、対象とする標的成分、ならびに、担体上に担持・固定化されている標的成分結合分子を、より生体条件に近い条件に維持して、標的成分の分離・回収、検出、スクリーニングを行うことを可能とする方法を提供するものである。
【0020】
【課題を解決するための手段】
本発明者らは、前記の課題を解決すべく、鋭意研究を進めたところ、標的成分の分離・回収、検出、スクリーニングを行う際、利用する担体上に標的成分結合分子を担持・固定化してなる構造体について、その担体の表面に設ける高分子被覆層として、従前の非天然の高分子に代えて、生体親和性の高い高分子材料を用いることで、より生体条件に近い条件を維持可能であることを見出した。さらには、担体の表面に設ける高分子被覆層に利用する生体親和性の高い高分子材料として、微生物等の生物細胞内で酵素的に重合されるポリマーであるポリヒドロキシアルカノエート(以下、PHAと略記する場合もある)を採用すると、より好ましい結果が得られることも見出した。
【0021】
一方、本発明者らは既に、微生物等の生物細胞内で酵素的に重合されるポリマーであるポリヒドロキシアルカノエートについて、人工構造体上に固定・担持したPHA合成酵素に該PHAの前駆体モノマーである3−ヒドロキシアシルCoAを接触させて酵素的に重合反応を行わせしむことにより、該人工構造体表面を良好に生成するPHAで被覆せしめる方法を見出している(特開2002−327046号公報、および特開2003−11312号公報参照)。
【0022】
本発明者らは、この手法を、例えば、磁性体を含む担体に応用することにより、該磁性体を含む担体の表面を該PHAで被覆する構造体を調製することが可能であり、さらには、被覆層に用いるPHA自体は、生分解性および生体適合性に優れた材料であることから、この様なPHA被覆を施した磁性体を含む担体上に、所望の標的成分結合分子を担持・固定化してなる構造体は、生化学分野、診断分野、医療分野、創薬分野に利用価値の大きい標的成分を、良好に分離・回収、検出、スクリーニングし得ることを検証して、本発明を完成するに至った。
【0023】
すなわち、本発明にかかる標的成分の分離方法は、
検体中に含まれる標的成分を分離する方法であって、
前記標的成分に対して結合親和力を有する分子が、その表面に固定された担体を用意する工程と、
前記担体と前記検体とを混合する工程と、
前記混合工程において混合される、前記検体中に含まれる標的成分と、前記担体表面に固定されている前記結合親和力を有する分子とを結合させる工程と、
前記結合工程により、前記結合親和力を有する分子との結合を介して、前記担体に固定化された前記標的成分を、前記担体とともに前記検体中から分離する工程とを有し、
前記担体は、ポリヒドロキシアルカノエートで少なくとも一部を被覆されていることを特徴とする標的成分の分離方法である。
【0024】
その際、前記担体として、磁性体を含んでなる担体を用いることができる。例えば、前記ポリヒドロキシアルカノエートとして、3−ヒドロキシアルカン酸ユニットを含有するポリヒドロキシアルカノエートを好適に利用できる。磁性体を含んでなる担体を用いる際には、前記結合親和力を有する分子との結合を介して、前記担体に固定化された前記標的成分を、前記担体とともに前記検体中から分離する工程において、
前記磁性体を含んでなる担体に対して、磁界を作用させて、前記検体より磁気的に分離する工程を採用することが好ましい。
【0025】
一方、本発明にかかる標的成分の分離方法に利用する、前記標的成分に対して結合親和力を有する分子の、前記担体表面への固定は、
前記担体表面の少なくとも一部を被覆されている、前記ポリヒドロキシアルカノエートとして、エポキシ基、アミノ基、カルボキシ基、およびハロゲン分子からなる群から選択される官能基を一つ以上有するポリヒドロキシアルカノエートを用い、
前記ポリヒドロキシアルカノエートの有する官能基の少なくとも一つ以上を利用して、前記標的成分に対して結合親和力を有する分子を固定化することが好ましい。
【0026】
また、本発明にかかる標的成分の分離方法は、前記担体表面へに固定される、前記標的成分に対して結合親和力を有する分子は、
核酸、タンパク質、ペプチド、糖鎖、脂質、低分子化合物、およびこれらの複合体からなる群より選択される一種類以上を含んでいる分子である場合に、好適に適用できる。加えて、前記検体中から分離される前記標的成分は、
核酸、タンパク質、ペプチド、糖鎖、脂質、低分子化合物、およびこれらの複合体からなる群より選択される一種類以上を含んでいる成分である場合に、本発明にかかる標的成分の分離方法は、より好適な方法となる。
【0027】
なお、本発明にかかる標的成分の分離方法では、利用する担体の形状は、任意に選択することが可能であり、例えば、前記磁性体を含んでなる担体の形状は、粒状であることが、一般に望ましいが、用途に応じて、前記磁性体を含んでなる担体の形状として、平板状またはフィルム状を選択することも可能である。なお、前記磁性体を含んでなる担体中の磁性体は、磁性を有する金属または金属化合物からなる磁性体であることが望ましい。
【0028】
また、本発明にかかる標的成分の検出方法は、
検体中に含まれる標的成分を検出する方法であって、
前記標的成分に対して結合親和力を有する分子が、その表面に固定された担体を用意する工程と、
前記担体と前記検体とを混合する工程と、
前記混合工程において混合される、前記検体中に含まれる標的成分と、前記担体表面に固定されている前記結合親和力を有する分子とを結合させる工程と、
前記結合工程により、前記結合親和力を有する分子との結合を介して、前記担体に固定化された前記標的成分について、選択的に検出する工程とを有し、
前記担体は、ポリヒドロキシアルカノエートで少なくとも一部を被覆されていることを特徴とする標的成分の検出方法である。
【0029】
その際、前記結合工程により、前記結合親和力を有する分子との結合を介して、前記担体に固定化された前記標的成分について、選択的に検出する工程は、
前記結合工程により、前記結合親和力を有する分子との結合を介して、前記担体に固定化された前記標的成分を、前記担体とともに前記検体中から分離する工程と、
前記担体とともに前記検体中から分離される、前記結合親和力を有する分子との結合を介して、前記担体に固定化された前記標的成分について、選択的に検出する工程とを含む構成を採用することが好ましい。
【0030】
本発明にかかる標的成分の検出方法でも、前記担体として、磁性体を含んでなる担体を用いることができる。例えば、前記ポリヒドロキシアルカノエートとして、3−ヒドロキシアルカン酸ユニットを含有するポリヒドロキシアルカノエートを好適に利用できる。磁性体を含んでなる担体を用いる際には、前記結合親和力を有する分子との結合を介して、前記担体に固定化された前記標的成分を、前記担体とともに前記検体中から分離する工程において、
前記磁性体を含んでなる担体に対して、磁界を作用させて、前記検体より磁気的に分離する工程を採用することが好ましい。
【0031】
一方、本発明にかかる標的成分の検出方法では、標的成分の検出手段として、種々の分光学的な手法を採用することができ、例えば、前記担体に固定化された前記標的成分について、選択的に検出する工程において、
検出手法として、比色法、蛍光法、化学発光法、ラジオアイソトープ法からなる群から選択される一つの検出手法または、二つ以上の検出手法の組み合わせを用いることができる。あるいは、前記担体に固定化された前記標的成分について、選択的に検出する工程において、
検出手法として、核磁気共鳴スペクトロスコピー、マススペクトロスコピー、赤外吸収スペクトロスコピー、および紫外吸収スペクトロスコピーからなる群から選択される一つの検出手法または、二つ以上の検出手法の組み合わせを用いることもできる。
【0032】
加えて、本発明にかかる標的成分のスクリーニング方法は、
媒体中に含まれる標的成分のスクリーニング方法であって、
前記スクリーニングは、前記標的成分を含んだ混合物が前記媒体中に含まれる混合試料を対象とし、
前記標的成分に対して結合親和力を有する分子が、その表面に固定された担体を用意する工程と、
前記担体と前記混合試料とを混合する工程と、
前記混合工程において混合される、前記混合試料中に含まれる標的成分と、前記担体表面に固定されている前記結合親和力を有する分子とを結合させる工程と、
前記結合工程により、前記結合親和力を有する分子との結合を介して、前記担体に固定化された前記標的成分を、前記担体とともに前記混合試料中から分離する工程とを有し、
前記担体は、ポリヒドロキシアルカノエートで少なくとも一部を被覆されていることを特徴とする標的成分のスクリーニング方法である。
【0033】
その際、前記担体として、磁性体を含んでなる担体を用いることができる。例えば、前記ポリヒドロキシアルカノエートとして、3−ヒドロキシアルカン酸ユニットを含有するポリヒドロキシアルカノエートを好適に利用できる。磁性体を含んでなる担体を用いる際には、前記結合親和力を有する分子との結合を介して、前記担体に固定化された前記標的成分を、前記担体とともに前記混合試料中から分離する工程において、
前記磁性体を含んでなる担体に対して、磁界を作用させて、前記混合試料より磁気的に分離する工程を採用することが好ましい。
【0034】
なお、本発明において、ポリヒドロキシアルカノエートとは、構成ユニットとして、ヒドロキシアルカン酸ユニットを含むポリヒドロキシアルカノエートを意味する。加えて、下記する本発明の実施の形態を説明する際、ポリヒドロキシアルカノエートとして、3−ヒドロキシアルカン酸ユニットを含むポリヒドロキシアルカノエートを用いている形態を具体例に採用するが、本発明は、3−ヒドロキシアルカン酸ユニットを含むポリヒドロキシアルカノエートを用いる形態に限定されることなく、一般に、前記の定義を満たすポリヒドロキシアルカノエートに包含される限り、いずれのポリヒドロキシアルカノエートも制限なく使用することができる。
【0035】
【発明の実施の形態】
まず、図1〜図3を参照して、本発明の特徴を概説する。
【0036】
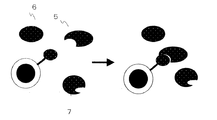
本発明にかかる標的成分の分離・回収方法、検出方法、スクリーニング方法は、標的成分を選択的に採取する手段として、標的成分に対して結合親和力を有する分子を用いるが、この標的成分に対して結合親和力を有する分子(標的成分結合分子)4は予め担体表面に担持・固定化して、標的成分結合分子担持構造体の形態で利用する。より具体的には、用いる担体は、担体基材の表面に、有機高分子材料からなる被覆層を設ける構造を選択し、その際、担体基材の表面に設ける有機高分子材料からなる被覆層として、図1に示すように、例えば、磁性体からなる基体を、担体基材1として用い、担体基材1の表面の少なくとも一部は、生体親和性の高い高分子材料である、ポリヒドロキシアルカノエート2によって被覆する形態としている。担体基材が、磁性体を含む際には、ポリヒドロキシアルカノエート(以下、PHAと記載する場合もある)の被覆を設けた磁性担体を、「PHA磁性構造体」と表記することもある。このPHA磁性構造体のような、ポリヒドロキシアルカノエート2による被覆を有する担体上に、標的成分結合分子4を担持する際には、ポリヒドロキシアルカノエート2による被覆中の、標的成分に対して結合親和力を有する分子4に対する、選択的な担持可能な部位3を利用する。磁性体からなる基体を、担体基材1として用いると、このPHA磁性構造体上への標的成分結合分子4を担持する工程によって、標的成分結合分子担持PHA磁性構造体が調製される。
【0037】
次いで、標的成分結合分子担持PHA被覆担体と、媒体中に溶解、または、分散されている標的成分5を含む液、具体的には、検体、あるいは、混合試料とを、混合する工程を設け、図2に示すように、担体上に担持されている標的成分結合分子4に、検体、あるいは、混合試料中に含まれる標的成分5を接触させ、標的成分結合分子4と標的成分5との結合をさせる工程を終えると、担体上に標的成分5を固定化することがなされる。一方、検体、あるいは、混合試料中に含まれる標的成分以外の成分6、7は、標的成分結合分子4との結合を起こさず、また、担体表面を被覆するPHAに対する、標的成分以外の成分6、7の非選択的な付着も少なく、また、標的成分以外の成分6、7の非選択的な付着が生じたとしても、簡単な清浄操作を施すことで、非選択的な付着した標的成分以外の成分6、7を容易に除去することができる。
【0038】
担体上に標的成分5を固定化した後、この担体とともに、例えば、固・液分離手段を利用して、担体を回収、分離すると、この回収、分離される担体群中には、標的成分5を固定化した担体も同様に回収、分離される。また、図3に例示するように、磁性体からなる基体を、担体基材1として用いる担体では、磁力を発揮する構造体8(永久磁石や電磁石等)により磁界を印加すると、磁性体からなる基体と磁力を発揮する構造体8との間の磁気的引力によって、担体は、磁力を発揮する構造体8(永久磁石や電磁石等)上に集積される。
【0039】
本発明にかかる標的成分の分離・回収方法、検出方法、スクリーニング方法では、以上に説明するように、標的成分結合分子の担持は、生体親和性の高い高分子材料である、ポリヒドロキシアルカノエートで被覆される部分になされ、標的成分と標的成分結合分子との結合も、ポリヒドロキシアルカノエートで被覆される表面部分で起こるため、より生体条件に近い条件に維持して、標的成分の分離・回収、検出、スクリーニングを実施することができる。
【0040】
なお、本発明にかかる標的成分の分離・回収方法、および標的成分の検出方法においては、検体中に標的成分以外の成分は存在しない場合もある。つまり、前記媒体中に標的成分が単独で存在しているものの、該標的成分が低濃度で広範囲にわたって存在している場合、担体上に標的成分を固定化した後、この担体とともに、回収すると、分離・回収される担体群中にで、該標的成分の存在比率は高く、濃縮がなされ、その検出をより容易なものとしている。
【0041】
なお、ポリヒドロキシアルカノエート:1およびその担持部分:3については<ポリヒドロキシアルカノエート>および<標的成分結合分子のPHA磁性構造体への担持>の項で、磁性体:2については<磁性体>の項で、標的成分に対して結合親和力を有する分子:4および標的成分:5については<標的成分および標的成分に対して結合親和力を有する分子>の項で、磁気を発揮する構造体:8については<標的成分結合磁性構造体の磁気分離および洗浄>の項でそれぞれ詳細に後述する。
【0042】
以下に、本発明をさらに詳細に説明する。
【0043】
<ポリヒドロキシアルカノエート>
本発明の方法で利用することができるポリヒドロキシアルカノエートは、そのモノマー・ユニットである3−ヒドロキシアルカン酸ユニットの炭素数が4または5である短鎖長ポリヒドロキシアルカノエート(short−chain−length PHA:以下scl−PHAと略記する場合もある);炭素数が4であるポリヒドロキシブチレート(PHB)、あるいは炭素数が5であるポリヒドロキシバレレート(PHV)のみならず、炭素数が6から12程度までの中鎖長(medium−chain−length:以下mclと記載する場合もある)の3−ヒドロキシアルカン酸ユニットからなるポリヒドロキシアルカノエート(以下、mcl−PHAと略記する場合がある)が含まれる。さらには、上記の側鎖にアルキル基を有するモノマーユニットからなるポリヒドロキシアルカノエート(以下、usual−PHAと略記する場合がある)以外にも、より広範囲な応用、例えば、機能性ポリマーとしての応用をも考慮して、アルキル基以外の、各種置換基(例えば、フェニル基、不飽和炭化水素基、エステル基、アリール基、シアノ基、ハロゲン化炭化水素基、エポキシ基(−O−)など)を側鎖に導入したモノマーユニットを含むポリヒドロキシアルカノエート(以下、unusual−PHAと略記する場合がある)、あるいはこれらのモノマーユニット複数を、任意のユニット比で含んでいるコポリマーをも、本発明の方法に用いることができる。
【0044】
本発明の方法に用いるPHAは、PHA合成酵素によって合成可能なPHA(例えば、各種のmcl−PHAやunusual−PHAなど)であれば、特に限定はされない。前述の通り、PHA合成酵素は、生物体内でのPHA合成反応系における最終段階を触媒する酵素であり、従って、生物体内において合成されることが判明しているPHAは、いずれも、該合成酵素による触媒作用を受けて、生合成されていることになる。さらには、本発明においては、生物体内において生合成されることが確認されているPHAの全て種類に関して、所望のPHAに含まれるモノマーユニットに対応する3−ヒドロキシアシルCoAを、磁性体に固定化された該PHA合成酵素に作用させることによって、所望のPHAを生成させて、磁性体の表面を被覆した構造体を作製することが可能である。
【0045】
本発明における、所望のPHAを生成させて、磁性体の表面を被覆した構造体を作製する工程で利用するPHA合成酵素は、合成するPHAに応じて、該合成酵素を生産する微生物群から適宜選択された微生物、あるいは、それら微生物由来のPHA合成酵素遺伝子を導入した形質転換体により生産されたものを用いることができる。
【0046】
scl−PHAの生合成は、scl−PHA生産菌の体内で、種々の炭素源から、生体内の様々な代謝経路を経て生成された(R)−3−ヒドロキシプロピオニルCoA、(R)−3−ヒドロキシブチリルCoAおよび(R)−3−ヒドロキシバレリルCoAの少なくとも一種を基質とした、PHA合成酵素による重合反応によって行われる。このscl−PHAの重合反応を触媒する酵素を、本発明では、特に、scl−PHA合成酵素と呼ぶことにする。scl−PHA合成酵素中でも、例えば、PHBの生合成を行うPHA合成酵素であれば、通常、PHB合成酵素(PHBポリメラーゼ、PHBシンターゼとも呼ばれる)と呼ばれている。
【0047】
本発明に用いるscl−PHA合成酵素は、該合成酵素を生産する微生物から適宜選択された微生物、あるいは、それら微生物由来のscl−PHA合成酵素遺伝子を導入した形質転換体により生産された、遺伝子組換え型scl−PHA合成酵素を用いることができる。かかるscl−PHA合成酵素を生産する微生物としては、例えば、PHBやPHB/V生産菌として知られている微生物を用いることができる。前記PHBやPHB/V生産性を有する微生物として、アエロモナス属(Aeromonas sp.)、アルカリゲネス属(Alcaligenes sp.)、クロマチウム属(Chromatium sp.)、コマモナス属(Comamonas sp.)、メチロバクテリウム属(Methylobacterium sp.)、パラコッカス属(Paracoccussp.)、シュードモナス属(Pseudomonas sp.)などが知られている。また、本発明者らにより、別途分離された菌株、バルクホルデリア・セパシア・KK01株(Burkholderia cepacia KK01)、ラルストーニャ・ユートロファ・TB64株(Ralstonia eutroSCL−PHA TB64)、アルカリゲネス属・TL2株(Alcaligenes sp. TL2)などを用いることができる。なお、KK01株は、寄託番号:FERM BP−4235として、TB64株は、寄託番号:FERM BP−6933として、TL2株は、寄託番号:FERM BP−6913として、それぞれ、「特許手続上の微生物の寄託の国際的承認に関するブタペスト条約」に基づき、国際寄託機関である、独立行政法人 産業技術総合研究所特許生物寄託センター(旧 経済産業省 産業技術総合研究所 生命工学工業技術研究所(NIBH)特許微生物寄託センター)に国際寄託されている。
【0048】
また、上述するscl−PHA合成酵素を生産する野生株以外に、scl−PHA合成酵素を生産するために、形質転換体を用いることもできる。野生株由来のscl−PHA合成酵素遺伝子のクローニング、発現ベクターの作製、および、遺伝子組換えによる形質転換体の創製は、常法に従って行うことができる。scl−PHA合成酵素遺伝子のクローニングに関しては、既に、アルカリゲネス・ユートロファスのPHB合成酵素遺伝子(phbC)がクローニングされ、報告されている。また、本発明者らは、バークホルデリア・セパシア KK01株のphbCについて、クローニングを完了しており、また、ラルストーニャ・ユートロファ TB64株のphbCについても、クローニングを完了している。scl−PHA合成酵素を産生する形質転換体は、宿主微生物に、このphbC遺伝子を含むベクターを導入することによって創製できる。形質転換に利用する、phbC遺伝子を含むベクターは、例えば、プラスミド・ベクター、ファージ・ベクター等にphbC遺伝子を挿入することによって調製される。宿主としては、例えば、大腸菌(エシェリチア・コリ:Escherichia coli)が広く利用されている。
【0049】
scl−PHA合成酵素の生産には、以上に挙げたようなscl−PHA生産菌は、単独で、あるいは必要に応じて2種以上を組み合わせて用いることができる。
【0050】
また、前述のscl−PHA生産菌由来のscl−PHA合成酵素遺伝子を導入した形質転換体を用いて、所望のscl−PHA合成酵素を生産することも可能である。宿主として、大腸菌等の細菌を用いている形質転換体に付いては、その培養に用いる培地として、天然培地あるいは合成培地、例えば、LB培地,M9培地等が挙げられる。また、培養温度を25〜37℃の範囲に選択し、好気的に8〜27時間培養することにより、微生物の増殖を図る。その後、集菌して、菌体内に蓄積されたscl−PHA合成酵素の回収を行うことができる。必要に応じて、培地には、発現ベクターに含まれるマーカー遺伝子、各種耐性遺伝子に応じて、カナマイシン、アンピシリン、テトラサイクリン、クロラムフェニコール、ストレプトマイシン等の抗生物質を添加してもよい。また、発現ベクターにおいて、挿入されるscl−PHA合成酵素遺伝子の転写用に、誘導性のプロモーターを採用している場合は、形質転換体を培養する際に、該プロモーターに対応する誘導物質を培地に添加して、発現を促してもよい。利用可能な誘導物質の例として、イソプロピル−β−D−チオガラクトピラノシド(IPTG)、テトラサイクリン,インドールアクリル酸(IAA)等が挙げられる。
【0051】
本発明では、scl−PHA合成酵素として、微生物の菌体破砕液や、菌体破砕液から、タンパク質成分を硫酸アンモニウム等により沈澱・回収した硫安塩析物などの粗酵素を用いてもよい。さらには、各種方法で精製した精製酵素を用いてもよい。該scl−PHA合成酵素は、必要に応じて、金属塩、グリセリン、ジチオトレイトール、EDTA、ウシ血清アルブミン(BSA)などの安定化剤、付活剤を適宜添加して、用いることができる。
【0052】
培養された微生物菌体から、scl−PHA合成酵素を分離・精製する工程では、scl−PHA合成酵素の酵素活性が保持される方法であれば、いかなる分離・精製方法をも用いることができる。例えば、培養後、集菌して得られた微生物菌体を、フレンチプレス,超音波破砕機,リゾチームや各種界面活性剤等を用いて破砕する。微生物の菌体破砕液を遠心分離して、上清として得られる粗酵素液、あるいは、粗酵素液から調製した硫安塩析物に対して、アフィニティ・クロマトグラフィー、陽イオンまたは陰イオン交換クロマトグラフィー、ゲル濾過等の精製手段を、単独または適宜組み合わせて、適用することによって、精製酵素を得ることができる。特に、遺伝子組換えタンパク質では、N末端やC末端にヒスチジン残基等の「タグ」を結合した融合タンパク質の形で発現させ、目的の融合タンパク質を選択的に、このタグを介して親和性樹脂に結合させる手法を利用することによって、より簡便に精製することができる。融合タンパク質を選択的に結合している親和性樹脂から、目的の酵素タンパク質を分離するには、トロンビン、血液凝固因子 Xa等のプロテアーゼで「タグ」部分を切断する、pHを低下せしめる、結合競合剤として、高濃度のイミダゾールを添加する等の方法を用いるとよい。また、pTYB1(New Englan Biolab社製)などの発現ベクターを用いる場合など、タグを介した親和性樹脂との結合に「インテイン」を含む場合には、ジチオトレイトールなどの還元性物質を添加して還元条件とすることで、切断する。アフィニティ・クロマトグラフィーによる精製を可能とする、融合タンパク質の融合パートナーの例は、ヒスチジン・タグの他に、グルタチオン S−トランスフェラーゼ(GST)、キチン結合ドメイン(CBD)、マルトース結合タンパク質(MBP)、あるいはチオレドキシン(TRX)等も公知である。例えば、GST融合タンパク質は、GST親和性レジンによって精製することができる。
【0053】
また、PHA合成酵素を生産する微生物としては、例えば、mcl−PHAやunusual−PHAの生産菌を用いることができる。このようなmcl−PHAやunusual−PHA産生能を有する微生物として、前述のシュードモナス・オレオボランス、シュードモナス・レジノボランス、シュードモナス属 61−3株,シュードモナス・プチダ・KT2442株,シュードモナス・アエルギノーサなど、さらには、本発明者らにより分離された、シュードモナス・プチダ・P91株(Pseudomonas putida P91),シュードモナス・チコリアイ・H45株(Pseudomonas cichorii H45),シュードモナス・チコリアイ・YN2株(Pseudomonas cichorii YN2),シュードモナス・ジェッセニイ・P161株(Pseudomonas jessenii P161)等のシュードモナス属微生物や、特開2001−78753号公報に記載のバークホルデリア属・OK3株(Burkholderia sp.OK3、寄託番号:FERM P−17370),特開2001−69968号公報に記載のバークホルデリア属・OK4株(Burkholderia sp.OK4、寄託番号:FERM P−17371)などのバークホルデリア属微生物を用いることができる。加えて、アエロモナス属(Aeromonas sp.)、コマモナス属(Comamonas sp.)などに属し、mcl−PHAやunusual−PHAを生産する微生物を用いることも可能である。
【0054】
なお、P91株は、寄託番号:FERM BP−7373として、H45株は、寄託番号:FERM BP−7374として、YN2株は、寄託番号:FERM BP−7375として、P161株は、寄託番号:FERM BP−7376として、それぞれ、「特許手続上の微生物の寄託の国際的承認に関するブタペスト条約」に基づき、国際寄託機関である、独立行政法人 産業技術総合研究所特許生物寄託センター(旧 経済産業省 産業技術総合研究所 生命工学工業技術研究所(NIBH)特許微生物寄託センター)に国際寄託されている。
【0055】
また、これらPHA生産微生物、ならびに、該PHA生産微生物由来のPHA合成酵素に関しては、特開2002−327046号公報、および特開2003−11312号公報に詳細に記載されているので参考にされたい。
【0056】
本発明の方法で利用する、磁性複合体において、その表面を被覆しているPHAの具体的な例として、下記化学式[1]〜化学式[10]で表されるモノマーユニットを少なくとも含むPHAを例示することができる。
【0057】
化学式[1]:
【0058】
【化1】

(式中、R1およびaは、下記するR1とaとの組み合わせからなる群より選択されるいずれか一つの組み合わせで表される:
R1は、水素原子(H)であり、aは、3〜10の整数のいずれかである;
R1は、ハロゲン原子であり、aは、1〜10の整数のいずれかである;
R1は、発色団であり、aは、1〜10の整数のいずれかである;
R1は、カルボキシ基あるいはその塩であり、aは、1〜10の整数のいずれかである;あるいは、
R1は、下記:
【0060】
【化2】
1,2−エポキシエチル基であり、aは、1〜7の整数のいずれかである。)
化学式[1]:
【0062】
【化3】
(式中、
bは、0〜7の整数のいずれかを表し、
R2は、水素原子(H)、ハロゲン原子、−CN、−NO2、−CF3、−C2F5、−C3F7からなる群から選択されるいずれか一つを表す。)
化学式[3]:
【0064】
【化4】
(式中、
cは、1〜8の整数のいずれかを表し、
R3は、水素原子(H)、ハロゲン原子、−CN、−NO2、−CF3、−C2F5、−C3F7からなる群から選択されるいずれか一つを表す。)
化学式[4]:
【0066】
【化5】
(式中、
dは、0〜7の整数のいずれかを表し、
R4は、水素原子(H)、ハロゲン原子、−CN、−NO2、−CF3、−C2F5、−C3F7からなる群から選択されるいずれか一つを表す。)
化学式[5]:
【0068】
【化6】
(式中、
eは、1〜8の整数のいずれかを表し、
R5は、
水素原子(H)、ハロゲン原子、−CN、−NO2、−CF3、−C2F5、−C3F7、−CH3、−C2H5、−C3H7からなる群から選択されるいずれか一つを表す。)
化学式[6]:
【0070】
【化7】
(式中、fは、0〜7の整数のいずれかを表す。)
化学式[7]:
【0072】
【化8】
(式中、gは、1〜8の整数のいずれかを表す。)
化学式[8]:
【0074】
【化9】
(式中、
hは、1〜7の整数のいずれかを表し、
R6は、水素原子(H)、ハロゲン原子、−CN、−NO2、−COOR’、−SO2R”、−CH3、−C2H5、−C3H7、−CH(CH3)2、−C(CH3)3からなる群から選択されるいずれか一つを表し、
ここで、
R’は、水素原子(H)、Na、K、−CH3、−C2H5からなる群から選択されるいずれか一つであり、
R”は、−OH、−ONa、−OK、ハロゲン原子、−OCH3、−OC2H5からなる群から選択されるいずれか一つである。)
化学式[9]:
【0076】
【化10】
(式中、
iは、1〜7の整数のいずれかを表し、
R7は、水素原子(H)、ハロゲン原子、−CN、−NO2、−COOR’、−SO2R”からなる群から選択されるいずれか一つを表し、
ここで、
R’は、水素原子(H)、Na、K、−CH3,−C2H5からなる群から選択されるいずれか一つであり、
R”は、−OH、−ONa、−OK、ハロゲン原子、−OCH3、−OC2H5からなる群から選択されるいずれか一つである。)
化学式[10]:
【0078】
【化11】
(式中、jは、1〜9の整数のいずれかを表す。)
なお、前記ハロゲン原子の具体例として、フッ素、塩素、臭素などを挙げることができる。また、前記発色団は、化学式[1]で示されるモノマーユニットに対応する、3−ヒドロキシアシルCoA体が、PHA合成酵素の触媒作用を受け得るものである限り、特に限定はされないが、PHA合成酵素による高分子合成時における立体障害などを考慮すると、3−ヒドロキシアシルCoA 分子内において、CoAと結合するカルボキシ基と発色団との間の離反を保つ上では、側鎖として、炭素数1〜5のアルキレン鎖、例えば、直鎖のアルキレン鎖を有する方が望ましい。一方、該発色団の光吸収波長が可視域にあれば、かかるPHAを被覆してなる磁性複合体は、着色したものが得られ、また、該発色団の光吸収波長が可視域以外にあっても、かかるPHAは、種々の電子材料としての利用に供することができる。上記化学式[1]で示されるモノマーユニットにおける発色団の例として、ニトロソ、ニトロ、アゾ、ジアリールメタン、トリアリールメタン、キサンテン、アクリジン、キノリン、メチン、チアゾール、インダミン、インドフェノール、ラクトン、アミノケトン、ヒドロキシケトン、スチルベン、アジン、オカサジン、チアジン、アントラキノン、フタロシアニン、インジゴイドなどを挙げることができる。
【0080】
本発明の方法で利用する、磁性複合体において、その表面を被覆しているPHAの具体的な例として、さらに、下記化学式[11]〜化学式[18]で表されるモノマーユニットを少なくとも含むPHAを例示することができる。
【0081】
化学式[11]:
【0082】
【化12】
(式中、
R1は、ビニル基であり、
aは、1〜10の整数のいずれかである。)
化学式[12]:
【0084】
【化13】
(式中、
bは、0〜7の整数のいずれかを表し、
R2は、CH3基、C2H5基、C3H7基、ビニル基、1,2−エポキシエチル基、COOR21(ここで、R21は、H原子、Na原子、K原子のいずれか一つを表す)からなる群から選択されるいずれか一つを表す。
なお、PHA中に、化学式[12]で示されるモノマーユニット複数種が存在する場合、各モノマーユニット毎に、そのbとR2とは、独立して上記の意味を表す。)
化学式[13]:
【0086】
【化14】
(式中、
cは、1〜8の整数のいずれかを表し、
R3は、CH3基、C2H5基、C3H7基、SCH3基からなる群から選択されるいずれか一つを表す。
なお、PHA中に、化学式[13]で示されるモノマーユニット複数種が存在する場合、各モノマーユニット毎に、そのcとR3とは、独立して上記の意味を表す。)
化学式[14]:
【0088】
【化15】
(式中、
dは、0〜8の整数のいずれかを表し、
R4は、
dが0の場合、H原子、CN基、NO2基、ハロゲン原子、CH3基、C2H5基、C3H7基、CF3基、C2F5基またはC3F7基からなる群から選択されるいずれか一つを表し、
dが1〜8の場合、CH3基、C2H5基、C3H7基からなる群から選択されるいずれか一つを表す。
なお、PHA中に、化学式[14]で示されるモノマーユニット複数種が存在する場合、各モノマーユニット毎に、そのdとR4とは、独立して上記の意味を表す。)
化学式[15]:
【0090】
【化16】
(式中、eは、1〜8の整数のいずれかを表す。)
化学式[16]:
【0092】
【化17】
(式中、
fは、1〜8の整数のいずれかを表し、
R6は、CH3基、C2H5基、C3H7基、(CH3)2−CH基または(CH3)3−C基からなる群から選択されるいずれか一つを表す。
なお、PHA中に、化学式[16]で示されるモノマーユニット複数種が存在する場合、各モノマーユニット毎に、そのfとR6とは、独立して上記の意味を表す。)
化学式[17]:
【0094】
【化18】
(式中、
gは、1〜8の整数のいずれかを表し、
R7は、H原子、ハロゲン原子、CN基、NO2基、COOR71(ここで、R71は、H、Na、K、CH3、C2H5のいずれかを表す)、SO2R72(ここで、R72は、−OH、−ONa、−OK、ハロゲン原子、−OCH3、−OC2H5のいずれかを表す)、CH3基、C2H5基、C3H7基、(CH3)2−CH基または(CH3)3−C基からなる群から選択されるいずれか一つを表す。
なお、PHA中に、化学式[17]で示されるモノマーユニット複数種が存在する場合、各モノマーユニット毎に、そのgとR7とは、独立して上記の意味を表す。)
化学式[18]:
【0096】
【化19】
(式中、
gは、1〜8の整数のいずれかを表し、
R7は、H原子、ハロゲン原子、CN基、NO2基、COOR71(ここで、R71は、H、Na、K、CH3、C2H5のいずれかを表す)、SO2R72(ここで、R72は、−OH、−ONa、−OK、ハロゲン原子、−OCH3、−OC2H5のいずれかを表す)、CH3基、C2H5基、C3H7基、(CH3)2−CH基または(CH3)3−C基からなる群から選択されるいずれか一つを表す。
なお、PHA中に、化学式[18]で示されるモノマーユニット複数種が存在する場合、各モノマーユニット毎に、そのgとR7とは、独立して上記の意味を表す。)
本発明において利用されるPHAとして、上記する化学式[1]〜化学式[18]で示されるモノマーユニットを複数種含むランダム共重合体やブロック共重合体を用いることも可能であり、かかる複数種のモノマーユニットを含むPHAでは、各モノマーユニットや含まれる官能基の特性を利用したPHAの物性制御や複数の機能の付与、官能基間の相互作用を利用した新たな機能の発現等が可能となる。
【0098】
さらには、例えば、磁性微粒子の表面にPHAの被覆を行う際、PHA合成酵素の基質である3−ヒドロキシアシルCoAの種類や濃度などを経時的に変化させることによって、作製されるPHAの組成も経時的に変化させることができる。従って、構造体の形状が粒状であれば、表面のPHA被覆は、内側から外側に向かう方向に、また、構造体の形状が平面状であれば、表面のPHA被覆は、その平面と垂直な方向に、被覆されているPHAのモノマーユニット組成を変化させることも可能である。
【0099】
例えば、最終的に作製される構造体の形態を、核となる磁性体に対して、その構造体の最表面には、該磁性体と親和性の低いPHAによる被覆がなされているものにする必要がある際には、先ず、核となる磁性体の表面を、該磁性体と親和性の高いPHAで被覆して、次いで、その磁性体と親和性の高いPHAのモノマーユニット組成から、目的とするPHAのモノマーユニット組成へと、内側から外側に向かう方向、あるいは、垂直方向に、組成が変化する構造、例えば、多層構造あるいはグラディエント構造とすることで、核となる磁性体と、その表面を被覆するPHA層との結合を強固にしたPHA被膜を形成することが可能となる。
【0100】
また、3−ヒドロキシプロピオン酸ユニット、3−ヒドロキシ−n−酪酸ユニット、3−ヒドロキシ−n−吉草酸ユニット、4−ヒドロキシ−n−酪酸ユニット、さらには、炭素数6〜14のヒドロキシアルカン酸ユニットからなるホモポリマー、あるいはこれらの複数のユニットからなるコポリマーなども、本発明の方法において利用可能である。さらには、必要に応じて、一旦PHAを酵素合成した後、あるいは、酵素合成中に、さらに化学修飾等を施してもよい。
【0101】
また、被覆に用いるPHAの分子量制御、および得られるPHA被覆膜の親水性向上という観点から、反応溶液中に水酸基を有する化合物を適宜添加してもよい。
【0102】
本発明の方法において、前記の目的で反応溶液中に添加する水酸基を有する化合物は、アルコール類、ジオール類、トリオール類、アルキレングリコール類、ポリエチレングリコール類、ポリエチレンオキサイド類、アルキレングリコールモノエステル類、ポリエチレングリコールモノエステル類、ポリエチレンオキサイドモノエステル類化合物から選ばれる少なくとも1種類であるが、さらに詳しく述べると以下の通りである。すなわち、好適に利用できるアルコール類、ジオール類およびトリオール類化合物は、炭素数3〜14の直鎖状または分岐のアルコール、ジオール、トリオールである。好適に利用できるアルキレングリコール類およびアルキレングリコールモノエステル類化合物は、そのアルキレングリコールの炭素鎖は、炭素数2〜10の直鎖状または分岐状構造を有している化合物である。また、好適に利用できるポリエチレングリコール類、ポリエチレンオキサイド類、ポリエチレングリコールモノエステル類、ポリエチレンオキサイドモノエステル類化合物は、その数平均分子量が100〜20,000の範囲であるものである。
【0103】
上記目的で反応溶液中に添加する、水酸基を有する化合物の添加濃度は、PHA合成酵素による3−ヒドロキシアシルCoAの重合反応が阻害されない濃度であれば、特に制限はないが、好ましくは、PHA合成酵素と3−ヒドロキシアシルCoAとを含む反応溶液に対して、0.01%〜10%(w/v)の範囲、さらに好ましくは、0.02%〜5%(w/v)の範囲で添加することがよく、その添加方法は、反応初期に一括して添加する方法、反応時間内に、数回に分けて反応溶液中に添加する方法のいずれでもよい。
【0104】
なお、化学式[12]で示されるモノマーユニットにおいて、R2として、カルボキシ基(COOR21)を含むモノマーユニットは、この化学式[12]で示され、R2がビニル基である、すなわち、側鎖末端にビニルフェニル基を有するモノマーユニットから、そのビニル基の二重結合部分を選択的に酸化開裂することで、カルボキシ基へ変換することで製造することができ、結果として、化学式[12]で示されるモノマーユニットの側鎖末端にカルボキシフェニル基を有するユニットを含むPHAが得られる。
【0105】
前記のビニル基をカルボキシ基へと変換する方法、すなわち、炭素−炭素の二重結合を酸化剤を用いて、酸化開裂してカルボン酸を得る方法としては、例えば、過マンガン酸塩を用いる方法(J. Chem. Soc., Perkin. Trans. 1, 806 (1973))、重クロム酸塩を用いる方法(Org. Synth., 4, 698 (1963))、過ヨウ素酸塩を用いる方法(J. Org. Chem., 46, 19 (1981))、硝酸を用いる方法(特開昭59−190945号広報)、オゾンを用いる方法(J. Am. Chem. Soc., 81, 4273 (1959))等が知られており、さらに、PHAに関しては、前述のMacromolecular Chemistry, 4, 289−293 (2001)に、PHAの側鎖末端の炭素−炭素二重結合を、酸化剤として過マンガン酸カリウムを用いて、反応を酸性条件下で行うことで、カルボン酸を得る方法が報告されており、本発明において、前記ビニル基をカルボキシ基へと変換する反応を行う際、これらの酸化開裂方法を参考とすることができる。
【0106】
前記ビニル基をカルボキシ基へと変換する反応に際して、酸化剤として用いる前記過マンガン酸塩としては、過マンガン酸カリウムが一般的である。過マンガン酸塩の使用量は、酸化開裂反応が化学量論的反応であるため、R2としてビニル基を有する化学式[12]で示されるモノマーユニット1モルに対して、通常1モル当量以上、好ましくは、2〜4モル当量使用するのがよい。
【0107】
酸化剤として過マンガン酸カリウムを用いる、酸化開裂反応において、反応系を酸性条件下にするためには、通常、硫酸、塩酸、酢酸、硝酸などの各種の無機酸や有機酸が用いられる。しかしながら、硫酸、硝酸、塩酸などの酸を用いた場合、PHAの主鎖のエステル結合が切断され、分子量低下を引き起こす恐れがある。そのため、酸性条件下にする際、酢酸を用いることが好ましい。反応系に添加される、酸の添加量は、R2としてビニル基を有する化学式[12]で示されるモノマーユニット1モルに対して、通常、0.2〜200モル当量、好ましくは、0.4〜100モル当量の範囲に選択する。反応系に添加される、酸の添加量が0.2モル当量に満たない場合には、酸化開裂反応は低収率となり、200モル当量を超える場合には、添加した酸による分解物が副生するため、いずれの場合も好ましくない。また、酸化開裂反応を促進する目的で、クラウン−エーテルを用いることができる。この場合、クラウン−エーテルと過マンガン酸塩とが、錯体を形成する結果、その反応活性が増大する効果が得られる。前記目的で利用可能なクラウン−エーテルとして、ジベンゾ−18−クラウン−6−エーテル、ジシクロ−18−クラウン−6−エーテル、18−クラウン−6−エーテルが、一般的に用いられる。反応系へのクラウン−エーテルの添加量は、過マンガン塩1モルに対して、通常、1.0〜2.0モル当量の範囲、好ましくは、1.0〜1.5モル当量の範囲に選択することが望ましい。
【0108】
本発明において、前記酸化反応を行う際、R2としてビニル基を有する化学式[12]で示すユニットを含むPHAで被覆された構造体、および過マンガン酸塩と酸を、一括して最初からともに仕込んで反応させてもよく、あるいは、それぞれ、連続的に、もしくは、断続的に反応系内に加えながら、反応させてもよい。あるいは、過マンガン酸塩のみを先に反応系に溶解、もしくは懸濁させておき、続いて、PHAで被覆された構造体と酸とを、連続的に、もしくは、断続的に反応系内に加えて反応させてもよく、PHAで被覆された構造体のみを先に反応系に懸濁させておき、続いて、過マンガン酸塩と酸とを、連続的に、もしくは断続的に反応系内に加えて反応させてもよい。さらには、PHAで被覆された構造体と酸を先に仕込んでおき、続いて、過マンガン酸塩を連続的に、もしくは断続的に反応系内に加えて反応させてもよく、過マンガン酸塩と酸を先に仕込んでおき、続いて、PHAで被覆された構造体を連続的に、もしくは断続的に反応系内に加えて反応させてもよく、PHAで被覆された構造体と過マンガン酸塩を先に仕込んでおき、続いて、酸を連続的に、もしくは断続的に反応系内に加えて反応させてもよい。
【0109】
R2としてビニル基を有する化学式[12]で示すユニットを含むPHAで被覆された構造体を対象とする、酸化剤として過マンガン酸カリウムを用いる、酸化開裂反応では、反応温度は、通常、−20〜40℃の範囲、好ましくは、0〜30℃の範囲に選択するのがよい。反応速度は、R2としてビニル基を有する化学式[12]で示すユニットと過マンガン酸塩との量論比、ならびに反応温度に依存し、加えて、前記ビニル基をカルボキシ基へと変換する目標比率に応じて、反応時間は選択されるものであるが、かかる目標比率を概ね100%と選択する際には、反応時間を、通常、2〜48時間とするのがよい。
【0110】
同様の酸化開裂反応を、R1がビニル基である化学式[11]で示すユニットに適用すると、側鎖末端のビニル基をカルボキシ基へと変換することも可能である。
【0111】
また、化学式[17]で示されるモノマーユニット、あるいは化学式[18]で示されるモノマーユニットを含むPHAは、一旦化学式[8]で示されるモノマーユニットを含むPHAを作製した後、そのスルファニル基(−S−)を選択的に酸化して、スルフィニル基(−SO−)、あるいはスルホニル基(−SO2−)へと変換することで製造することができる。かかるスルファニル基(−S−)の選択的な酸化は、例えば、過酸化化合物による酸化処理を施すことで達成でき、その際、スルファニル基(−S−)の酸化に寄与し得るものであれば、いかなる種類の過酸化化合物をも用いることが可能である。なお、酸化効率、PHA主鎖骨格ならびにPHAに含まれる他のモノマーユニットへの影響、処理の簡便さ等を考慮した場合、特に、過酸化水素、過炭酸ナトリウム、メタクロロ過安息香酸、過蟻酸、過酢酸からなる群から選択される過酸化化合物を用いることが好ましい。
【0112】
例えば、スルファニル基(−S−)の酸化に利用する過酸化化合物として、メタクロロ過安息香酸(MCPBA)を用いると、化学式[8]で示されるモノマーユニット中のスルファニル基(−S−)に対する酸化は、化学量論的に進行するため、化学式[17]で示されるモノマーユニット、あるいは化学式[18]で示されるモノマーユニットの含有比率の制御がし易い。また、その反応条件が温和であるため、PHA主鎖骨格の切断や活性部位の架橋反応等、不要な副次反応が起こり難い。
【0113】
一般的な反応条件として、スルファニル基(−S−)をスルフィニル基(−SO−)まで選択的に酸化するためには、PHA中に含有されるスルファニル基(−S−)を含む化学式[8]で示されるモノマーユニットのユニットモル量、1モルに対して、MCPBAを若干過剰量、具体的には、1.1〜1.4モル量の範囲に選択し、クロロホルム中、温度を0℃〜30℃の範囲に選択して、反応を行う。前記の反応条件範囲においては、反応時間を10時間程度とすると、理論値のほぼ90%、20時間程度とすると、理論値のほぼ100%まで、スルフィニル基(−SO−)へと酸化を進行させることができる。また、スルファニル基(−S−)を全てスルホニル基(−SO2−)まで酸化するためには、PHA中に含有されるスルファニル基(−S−)を含む化学式[8]で示されるモノマーユニットのユニットモル量、1モルに対して、MCPBAを2モルより若干過剰量、具体的には、2.1〜2.4モル量の範囲に選択し、前記と同様の溶媒、温度、時間条件を選択して、反応を行えばよい。かかる過酸化化合物として、MCPBAを用いる酸化処理では、スルファニル基(−S−)にMCPBA1分子が作用して、スルフィニル基(−SO−)へと変換し、さらに、MCPBA1分子が作用して、スルホニル基(−SO2−)へと段階的に酸化が進行するが、スルファニル基(−S−)からスルフィニル基(−SO−)への変換は、スルフィニル基(−SO−)からスルホニル基(−SO2−)への変換より、より高い反応性を示す。
【0114】
また、R2として1,2−エポキシエチル基を有する化学式[12]で示すユニットは、R2としてビニル基を有する化学式[12]で示すユニットに対して、側鎖末端ビニルフェニル基中のビニル基の二重結合部分を選択的に部分酸化して、エポキシ基の導入を行うことで製造することができる。すなわち、R2としてビニル基を有する化学式[12]で示すユニットを含むPHAに対して、ビニル基に選択的な酸化処理を施すことで、R2として1,2−エポキシエチル基を有する化学式[12]で示すユニットを含むPHAが得られる。
【0115】
前記ビニル基から1,2−エポキシエチル基へとエポキシ化を行う酸化処理についても、例えば、過酸化化合物を利用することができ、ビニル基の選択的な部分酸化に寄与し得るものであれば、いかなる種類の過酸化化合物をも用いることが可能である。その際、酸化効率、PHA主鎖骨格ならびにPHAに含まれる他のモノマーユニットへの影響、処理の簡便さ等を考慮した場合、特に、過酸化水素、過炭酸ナトリウム、メタクロロ過安息香酸、過蟻酸、過酢酸からなる群から選択される過酸化化合物を用いることが好ましい。ビニル基から1,2−エポキシエチル基へとエポキシ化を行う酸化処理に過酸化化合物を用いる場合、その反応条件は、過酸化化合物による前記スルファニル基の選択的な部分酸化処理における反応条件を参考とすることができる。
【0116】
本発明の方法で利用されるポリヒドロキシアルカノエートの分子量は、数平均分子量として、1,000から10,000,000程度とするのが望ましい。
【0117】
なお、本発明において、PHA被覆された構造体の作製工程に利用される、PHA合成酵素により合成されるPHAは、一般に、R体のみから構成されるアイソタクチックなポリマーである。
【0118】
本発明の方法で利用される、磁性体を核とする構造体に含有される磁性体の量は、1〜80重量%の範囲、好ましくは5〜70重量%の範囲、さらに好ましくは10〜60重量%の範囲に選択する。構造体中の磁性体含有量が1重量%より少ないと、磁気性能が不足して磁性体含有構造体としての性能が不十分となる恐れがあり、また、磁性体含量が80重量%を超えると、核とする磁性体に対して、その表面を被覆するPHAの含有比率が相対的に低下するため、表面に十分なPHA被覆層を設けることで達成される、かかる構造体本来の機能が損なわれ、実用性能の面で満足できなくなる恐れがある。
【0119】
本発明の方法に利用される、粒状構造体の粒子径は、その個別的な用途等に応じて適宜選定されるが、通常、0.02〜100μmの範囲、好ましくは0.05〜20μmの範囲に選択する。
【0120】
<標的成分、ならびに標的成分に対して結合親和力を有する分子>
本発明の方法が対象とする「標的成分」は、生理学的に有用であり、多くの場合、検体試料中では、その他の物質と混在しているか、あるいは、他の物質の混在がない、単一体であっても、広範囲に低濃度で存在しているものである。従って、検体試料中に含有されている、「標的成分」のみを、分離・回収、検出、およびスクリーニングする手段・方法が求められている。
【0121】
本発明の方法では、前記目的のために、「標的成分」のみを捕捉する上で好適に利用可能な、「標的成分に対して結合親和力を有する分子(以下、「標的成分結合分子」と記載する場合もある)」を用いる。
【0122】
本発明の方法が対象とする「標的成分」ならびに「標的成分に対して結合親和力を有する分子」の具体的な例として、核酸、タンパク質、ペプチド、糖鎖、脂質、および低分子化合物、あるいはこれらの複合体、さらには、これらの分子を部分的に含んでなる物質を挙げることができる。
【0123】
ここで、「核酸」は、デオキシリボ核酸、リボ核酸、オリゴヌクレオチド、ポリヌクレオチド、アプタマー、リボザイム等を包括する。
【0124】
ここで、「タンパク質」は、糖タンパク質、リポタンパク質、膜タンパク質、および標識タンパク質、ならびに低分子量ペプチドなどの天然および人工的に誘導された異形分子を包括する。
【0125】
当該タンパク質が免疫反応体であれば、例えば、抗体、抗原、ハプテン、あるいはこれらの錯体が可能であり、特に、「抗体」の場合、モノクローナル抗体またはポリクローナル抗体、組換えタンパク質型抗体または天然型抗体、キメラ抗体、ハイブリッド抗体混合物(単数あるいは複数)、単鎖抗体提示ファージ抗体(単鎖抗体を提示しているファージ全体を含む)、ならびに抗体およびタンパク質の融合体、あるいはこれらのハイブリッド混合物も包含される。当該タンパク質が触媒反応体の場合は、天然酵素、遺伝子改変により作製された変異酵素、ポリエチレングリコール等の合成分子と複合化された半人工酵素、非天然アミノ酸が導入された半人工酵素等を包括する。
【0126】
前記「抗体」として、通常、IgG(免疫グロブリンG、あるいはイムノグロブリンG)が挙げられるが、ペプシン、パパインなどの消化酵素、あるいはジチオトレイトール、2−スルファニルエタノール(2−メルカプトエタノール)などの還元剤を用いて、F(ab’)2、Fab’、Fab、Fvなどの低分子化処理が施されたものであってもよい。また、IgGだけでなく、IgM、あるいは前記IgGと同様の処理で低分子化したIgM由来のフラグメントであってもよい。また、モノクローナル抗体、ポリクローナル抗体のいずれも、本発明における「標的成分結合分子」として適用できる。「標的成分結合分子」として、モノクロール抗体を用いる際には、B型肝炎ウイルス表面抗原のように繰り返し構造を有するタンパク質や、CA19−9抗原のように分子内に複数のエピトープが存在する抗原に対しては、各エピトープに対する反応性を示すモノクローナル抗体複数種を併用することもできる。また、認識エピトープの異なるものを2種類以上組み合わせても使用できる。
【0127】
一方、「抗原」としては、例えば、タンパク質、ポリペプチド、ステロイド、多糖類、脂質、花粉、遺伝子工学的に産生された組換えタンパク質、薬物など種々のものが挙げられる。すなわち、本発明において対象とする「抗原」は、人、あるいは動物に対して、抗体産生を惹起する能力のある全ての物質のうち、例えば、診断等特別の目的の下に選択された単一あるいは複数の物質、ないしはそれらを含有する混合物である。
【0128】
「ペプチド」は、主に、本発明においては、その分子量とは無関係にタンパク質の断片を指す。
【0129】
「低分子化合物」は、受容体に認知可能である低分子量の、好ましくは有機分子である。通常、低分子化合物は、タンパク質に対する特異結合化合物であり、その多くは、生理活性物質あるいは薬剤候補物質あって、特に、抗原性の低分子化合物は、「ハプテン」と称される場合もある。
【0130】
「糖鎖」としては、グルコース、マンノース、N−アセチルグルコサミン、フコース、ガラクトース、グルクロン酸、N−アセチルグルコサミン、シアル酸から選択される単糖ユニットが複数(数個から数十個)連なった、直鎖および分岐のオリゴマーおよびポリマーを挙げることができる。また、本発明の方法では、これらの糖鎖とタンパク質の複合体である糖タンパク質、脂質との複合体である糖脂質、さらには、これら糖鎖が生体細胞表面に提示されている場合は、細胞膜表面に糖鎖を提示している細胞そのもの、あるいは細胞膜断片も含まれる。
【0131】
「脂質」としては、生物の神経組織、形質膜、ミトコンドリア、ミクロソーム、細胞核等細胞内オルガネラの膜として存在する複合脂質、組織境界脂質膜等の天然脂質(アシルグリセロール)や、タンパク質との複合体であるリポタンパク質、レシチン(ホスファジチルコリン)のようなリン脂質やその脂質二重膜カプセルとしてのリポソーム等を挙げることができる。
【0132】
<磁性体>
本発明の方法において、構造体の核として用いる磁性体としては、その表面にPHA合成酵素を固定化することのできるものであれば、磁性材料の種類、形状、大きさに関しては、その個別的な用途に応じて、適宜選択して用いることができる。すなわち、PHA合成酵素の固定化方法や、作製した構造体の応用の形態等に応じて、磁性体の種類や構造を適宜選択して用いることができる。
【0133】
本発明の方法で利用する、構造体を構成する磁性体の例として、例えば、磁性を有する金属または金属化合物が挙げられ、さらに具体的には、四三酸化鉄(Fe3O4)、γ−重三二酸化鉄(γ−Fe2O3)、MnZnフェライト、NiZnフェライト、YFeガーネット、GaFeガーネット、Baフェライト、Srフェライト等各種フェライト、鉄、マンガン、コバルト、ニッケル、クロムなどの金属、鉄、マンガン、コバルト、ニッケルなどの合金を挙げることができるが、これらに限定されるものではない。ここで、例えば、磁性複合体上に生体物質を固定する場合、あるいは、磁性複合体を生体内に投与する場合には、生体に対する適合性の良好なマグネタイト(Fe3O4)のほか、必要に応じて、マグネタイトの金属元素の一部を少なくとも1種類の他の金属元素で置換した各種フェライト組成などが好適に適用可能である。これら磁性体の形状は、生成条件によって変化し、多面体、8面体、6面体、球状、棒状、鱗片状等などがあるが、異方性の少ない構造が、構造体とした際、その表面を被覆するPHA層が示す機能の安定発現のためにはより好ましい。本発明の構造体を構成する磁性体の一次粒子の粒子径は、その用途に応じて適宜選択可能であるが、例えば、0.001〜10μmの範囲内の粒径を有する粒子を用いるとよい。
【0134】
また、本発明の方法で利用する、構造体を構成する磁性体として、超常磁性を有するものについても好ましく用いることができる。例えば、フェライトの粒子径が20nm程度以下と小さい場合には、フェライトは熱擾乱影響を受け超常磁性を示すようになり、残留磁化や保磁力を持たなくなる。超常磁性であっても、外部磁界を印加することにより、磁気的操作が可能であり、また、超常磁性であれば、残留磁化や保磁力を持たないので、外部磁界が印加されていない際に、残留磁化に起因する磁気的な凝集の生じる恐れもない。
【0135】
さらには、本発明の方法で利用する磁性体は、金属または金属化合物を含むマトリックスなどのような複合材料であってもよく、そのマトリックスは、有機または無機の各種材料から構成されるものである。フェライトめっきのような手法で、有機高分子表面を磁性体により被覆した材料、有機高分子中に磁性体が分散した材料等も、その表面の一部に磁性体が露出した形態であれば、利用可能である。
【0136】
なお、本発明の方法に用いる磁性体は、粒子状、平板状、フィルム状として、用いることができるが、本発明の方法における、磁気的操作による分離の具体的な手法を考慮する場合、粒子状が好ましく、さらには、液体試料中への分散を想定すると、その粒子径は、0.001〜10μm程度の微粒子であることがより好ましい。
【0137】
<PHA磁性構造体の作製>
本発明の方法で利用するPHA磁性構造体の製造方法は、磁性体にPHA合成酵素を固定化する工程と、該固定化PHA合成酵素を用いて、3−ヒドロキシアシルCoAに対する酵素反応により、PHAを合成する工程とを含むものである。
【0138】
磁性体にPHA合成酵素を固定化する方法としては、該PHA合成酵素の活性が保持され得るものであり、かつ、目的とする磁性体に対して適用可能なものであれば、通常行われている酵素固定化方法の中から、任意に選択される方法を用いることができる。例えば、PHA合成酵素を固定化する方法として、共有結合法、イオン吸着法、疎水吸着法、物理的吸着法、アフィニティ吸着法、架橋法、格子型包括法などを例示することができるが、特に、イオン吸着や疎水吸着を利用した固定化方法を用いると、簡便である。
【0139】
一般に、PHA合成酵素などの酵素タンパク質は、アミノ酸が多数結合したポリペプチドであり、リシン、ヒスチジン、アルギニン、アスパラギン酸、グルタミン酸などの側鎖上にイオン性基を有するアミノ酸残基によって、イオン吸着体としての性質を示し、また、アラニン、バリン、ロイシン、イソロイシン、メチオニン、トリプトファン、フェニルアラニン、プロリンなどの側鎖として疎水性基を有するアミノ酸残基によって、あるいは、全体として、有機高分子であるという点で、疎水吸着体としての性質を有している。従って、程度の差はあるが、イオン性や疎水性、もしくはイオン性と疎水性の両方の性質を有する固体表面に吸着させることが可能である。
【0140】
主にイオン吸着法によってPHA合成酵素を固定化する方法を適用する際、その核には、イオン性官能基を表面に発現しているコアを用いればよく、例えば、カオリナイト,ベントナイト、タルク、雲母等の粘土鉱物、アルミナ、二酸化チタン等の金属酸化物、シリカゲル、ヒドロキシアパタイト、リン酸カルシウムゲル等の不溶性無機塩などをコアとして用いることができる。また、これらを主要な成分とする無機顔料、イオン交換樹脂、キトサン、ポリアミノポリスチレン等の、イオン性官能基を有する重合体も、イオン吸着性コアとして用いることができる。
【0141】
本発明に利用するPHA磁性構造体において、基材が磁性体の場合、例えば、フェライトなどの金属酸化物においては、その表面に水酸基が存在しており、PHA合成酵素表面のカルボキシ基との水素結合による固定化法などが好適に用いうる。
【0142】
一方、主に疎水吸着によってPHA合成酵素を固定化する方法を適用する際、その核には、表面が非極性であるコアを用いればよく、例えば、スチレン系ポリマー、アクリル系ポリマー、メタクリル系ポリマー、ビニルエステル類、ビニル系ポリマーなど、イオン性官能基を表面に発現していない、もしくは疎水性官能基を表面に発現している多くの重合体をコアとして用いることができる。具体的には、芳香環を複数有するアゾ顔料や縮合多環のフタロシアニン系顔料、アントラキノン系顔料等の有機顔料、カーボンブラックなどは疎水吸着性である。また、疎水吸着法は、親油化処理を施した磁性体にも適用することが可能である。
【0143】
イオン吸着法または疎水吸着法によるPHA合成酵素のコアへの固定化は、該PHA合成酵素とコアとを所定の反応液中で混合することによって達成される。このとき、該PHA合成酵素がコアの表面に均等に吸着されるよう、反応容器を適当な強度で振盪あるいは攪拌することが望ましい。
【0144】
反応液のpHや塩濃度、温度によって、コアおよびPHA合成酵素の表面電荷の正負や電荷量、疎水性が変化するので、用いるコアの性質に合わせて、酵素活性上許容される範囲内で溶液の調整を行うことが望ましい。例えば、コアが主にイオン吸着性である場合には、塩濃度を下げることにより、コアとPHA合成酵素との吸着に寄与する電荷量を増やすことができる。また、pHを調整する事により、両者の反対電荷を増やすことができる。コアが主に疎水吸着性である場合には、塩濃度を上げることによって、両者の疎水性を増やすことができる。また、予め電気泳動やぬれ角等を測定し、コアやPHA合成酵素の荷電状態や疎水性を調べることで、吸着に適した溶液条件の設定をすることもできる。さらに、コアとPHA合成酵素との吸着量を直接測定して条件を求めることもできる。吸着量の測定は、例えば、コアが分散された溶液に濃度既知のPHA合成酵素溶液を添加し、吸着処理を行った後、溶液中のPHA合成酵素濃度を測定し、差し引き法により吸着酵素量を求める等の方法を用いればよい。
【0145】
イオン吸着法や疎水吸着法では酵素を固定化し難いコア材質の場合には、操作の煩雑さや酵素の失活の可能性を考慮した上で、共有結合法によってもかまわない。例えば、芳香族アミノ基を有するコア材質(固体粒子)をジアゾ化し、これに酵素をジアゾカップリングする方法や、カルボキシ基、アミノ基を有するコア材質(固体粒子)とPHA合成酵素の間にペプチド結合を形成させる方法、ハロゲン基(ハロゲノアルキル基)を有するコア材質(固体粒子)とPHA合成酵素のアミノ基等との間でアルキル化する方法、臭化シアンで活性化した多糖類コア粒子とPHA合成酵素のアミノ基を反応させる方法、コア材質(固体粒子)のアミノ基と酵素のアミノ基との間を架橋する方法、アルデヒド基またはケトン基を有する化合物とイソシアニド化合物の存在下、カルボキシ基,アミノ基を有するコア材質(固体粒子)とPHA合成酵素とを反応させる方法、ジスルフィド基(−S−S−)を有するコア材質(固体粒子)とPHA合成酵素のスルファニル基(−SH)との間で交換反応させる方法などがある。
【0146】
また、アフィニティ吸着によって、コア材質(固体粒子)の表面にPHA合成酵素を吸着してもよい。アフィニティ吸着とは、生体高分子とそれに特異的な親和力を示す、リガンドと呼ばれる特定物質との間の生物学的吸着で、例えば、酵素と基質、抗体と抗原、レセプターとアセチルコリン等の情報物質、mRNAとtRNAなどがある。一般に、アフィニティ吸着を利用して、酵素タンパク質を固定化する方法では、酵素の基質や反応生成物、拮抗阻害剤、補酵素、アロステリックエフェクター等をリガンドとして固体表面に結合させ、このリガンドと添加した酵素タンパク質との結合を介して、固体表面にアフィニティ吸着させる方法を採用する。しかしながら、PHA合成酵素に対しては、例えば、基質である3−ヒドロキシアシルCoAをリガンドとして用いた場合、該PHA合成酵素中のPHA合成を触媒する活性部位がリガンドとの結合により塞がれてしまうため、PHAを合成できなくなるという問題を生じる。しかしながら、他の生体高分子をPHA合成酵素に予め融合させ、該生体高分子のリガンドを利用して、アフィニティ吸着法を適用することによって、アフィニティ吸着による固定化後も、PHA合成酵素自体のPHA合成活性を維持することができる。なお、PHA合成酵素と生体高分子との融合は遺伝子工学的手法によって行ってもよく、また、生体高分子をPHA合成酵素に化学的に結合させてもよい。用いる生体高分子としては、生体高分子に対するリガンドの入手容易で、そのリガンドがコアに容易に結合できるものであればいかなるものでも構わないが、遺伝子組換えによって融合物を発現させる場合には、融合させる生体高分子はタンパク質であることが好ましい。具体的には、形質転換によって、融合パートナーのGSTを発現する遺伝子配列にPHA合成酵素の遺伝子配列を繋げた遺伝子を導入した大腸菌を用いて、GSTとPHA合成酵素との融合タンパク質を生産し、この融合タンパク質をGSTのリガンドであるグルタチオンを結合したSepharoseに添加することで、融合タンパク質型のPHA合成酵素をSepharose上にアフィニティ吸着させることができる。
【0147】
また、磁性体に対して結合能を有するアミノ酸配列を含むペプチドをPHA合成酵素に融合して提示させ、該磁性体に対して結合能を有するアミノ酸配列のペプチド部分と、磁性体との結合性に基づいて、該磁性体表面にPHA合成酵素を固定化することもできる。
【0148】
磁性体に対する結合能を有するアミノ酸配列は、例えば、ランダム・ペプチド・ライブラリーのスクリーニングによって決定することができる。特に、例えば、M13系ファージの表面蛋白質(例えば。geneIII蛋白質)のN末端側遺伝子にランダム合成遺伝子を連結して調製されたファージ・ディスプレイ・ペプチド・ライブラリーを好適に用いることができるが、その場合、磁性体に対する結合能を有するアミノ酸配列を決定するには、次のような手順をとる。すなわち、磁性体あるいは該磁性体を構成する少なくとも一成分に対して、ファージ・ディスプレイ・ペプチド・ライブラリーを添加することによって接触させ、その後、洗浄により結合ファージと非結合ファージを分離する。磁性体結合ファージを酸などにより溶出し、緩衝液で中和した後、大腸菌に感染させ、磁性体結合ファージを増幅する。この選別を複数回繰り返すと、目的の磁性体に高い結合能を示す複数のクローンが濃縮される。ここで、単一なクローンを得るため、再度大腸菌に感染させた状態で培地プレート上にコロニーを作らせる。それぞれの単一コロニーを液体培地で培養した後、培地上清中に存在するファージをポリエチレングリコール等で沈澱精製し、そのクローンが有する合成遺伝子の塩基配列を解析すれば、磁性体に対する結合能を有するペプチドの構造(アミノ酸配列)を知ることができる。
【0149】
上記スクリーニング方法により選別される、磁性体に対する結合能を有するペプチドのアミノ酸配列をコードする合成遺伝子は、通常の遺伝子工学的手法を用いて、PHA合成酵素の遺伝子に融合して利用される。磁性体に対する結合能を有するペプチドは、PHA合成酵素のN末端あるいはC末端に連結して発現することができる。また、適当なスペーサー配列を挿入して発現することもできる。スペーサー配列としては、約3〜約400アミノ酸が好ましく、また、スペーサー配列はいかなるアミノ酸を含んでもよい。最も好ましくは、スペーサー配列は、PHA合成酵素の機能を妨害せず、また、前記磁性体に対する結合能を有するペプチドを介して、融合タンパク質型PHA合成酵素が磁性体に結合する際、結合能を有するペプチドによる結合を妨害しないものである。
【0150】
上記方法により作製された固定化PHA合成酵素は、そのままでも用いることができるが、さらに、凍結乾燥等を施した上で保存し、その都度、酵素反応液用媒体に添加して、使用することもできる。
【0151】
固定化PHA合成酵素による3−ヒドロキシアシルCoAの重合によって、PHAが合成される反応において放出されるCoA量が、1分間に1μmolとなるPHA合成酵素量を1単位(U)としたとき、磁性体に固定するPHA合成酵素の量は、例えば、磁性体がカプセル構造体のコアである場合、磁性体1 g当たり、10 単位(U)〜1,000単位(U)の範囲、望ましくは、50 単位(U)〜500単位(U)の範囲内に設定するとよい。
【0152】
前記の固定化PHA合成酵素は、所望のPHAの原料となる3−ヒドロキシアシルCoAを含む水系反応液中に添加され、磁性体表面に固定化したPHA合成酵素によってPHAを合成することにより、磁性体の表面が合成されるPHAにより被覆された構造体を形成する。その際、水系反応液は、固定化PHA合成酵素の活性を発揮させ得る条件に調整された反応系に構成するべきであり、例えば、通常、pH5.5〜pH9.0の範囲、好ましくは、pH7.0〜pH 8.5の範囲となるよう、緩衝液によりpH調整を行う。ただし、使用する固定化PHA合成酵素の至適pHやpH安定性によっては、上記のpH範囲以外に条件を設定することも除外されない。緩衝液の種類は、使用する固定化PHA合成酵素の活性を発揮させ得るものであれば、設定するpH領域等に応じて、適宜選択して用いることができるが、例えば、一般の生化学反応に用いられる緩衝液、具体的には、酢酸バッファー、リン酸バッファー、リン酸カリウムバッファー、3−(N−モルフォリノ)プロパンスルフォン酸(MOPS)バッファー、N−トリス(ヒドロキシメチル)メチル−3−アミノプロパンスルフォン酸(TAPS)バッファー、トリス塩酸バッファー、グリシンバッファー、2−(シクロヘキシルアミノ)エタンスルフォン酸(CHES)バッファーなどを用いるとよい。緩衝液の濃度も、使用する固定化PHA合成酵素の活性を発揮させ得るものであれば、特に限定はされないが、通常、5.0 mM〜1.0 Mの範囲、好ましくは0.1 M〜0.2 Mの濃度のものを使用するとよい。反応温度は、使用するPHA合成酵素の特性に応じて、適宜設定するものであるが、通常、4℃〜50℃の範囲、好ましくは、20℃〜40℃の範囲に設定するとよい。ただし、使用するPHA合成酵素の至適温度や耐熱性によっては、上記の範囲以外に条件を設定することも除外されない。反応時間は、合成すべきPHAの被覆層の厚さに応じて、選択すべきものであり、また、使用するPHA合成酵素の安定性等にもよるが、通常、1分間〜24時間の範囲、好ましくは、30分間〜3時間の範囲内で適宜選択して設定する。反応液中の基質3−ヒドロキシアシルCoA濃度は、合成すべきPHAの被覆層の組成、厚さに応じて、適宜選択するべきものであり、また、使用する固定化PHA合成酵素の活性を発揮させ得る範囲内で、適宜設定するものであるが、通常、0.1 mM〜1.0 M、好ましくは0.2 mM〜0.2 Mの範囲内で設定するとよい。なお、反応液中における基質3−ヒドロキシアシルCoA濃度が高い場合、一般に、反応系のpHが低下する傾向にあるため、基質3−ヒドロキシアシルCoA濃度を高く設定する場合は、pHの変動を抑制するため、前記の緩衝液濃度も高めに設定することが好ましい。
【0153】
また、上記工程において、水系反応液中の基質3−ヒドロキシアシルCoAの種類や濃度などの組成を経時的に変化させることによって、形成される構造体の形状が粒状であれば内側から外側に向かう方向に、構造体の形状が平面状であれば垂直方向に、磁性体表面を被覆するPHAの膜厚方向におけるモノマーユニット組成を変化させることができる。
【0154】
このPHAのモノマーユニット組成の変化した構造体の形態として、例えば、PHA被膜の組成変化が連続的で、内側から外側に向かう方向、もしくは垂直方向に組成の勾配を形成した1層のPHAが磁性体を被覆した形態を挙げることができる。組成の勾配を形成した1層のPHAの製造方法としては、例えば、PHAを合成しながら、反応液中に別組成の基質3−ヒドロキシアシルCoAを添加するなどの方法によればよい。
【0155】
また別の形態として、PHA被膜の組成変化が段階的で、組成の異なるPHAが磁性体を多層に被覆した形態を挙げることができる。この組成の異なるPHA多層被覆を製造方法としては、ある3−ヒドロキシアシルCoAの組成でPHAを合成した後、遠心分離などによって調製中の構造体を反応液から一旦回収し、回収された調製中の構造体表面に、異なる3−ヒドロキシアシルCoAの組成からなる反応液を再度添加して、組成の異なるPHAを積層するなどの方法を適用よればよい。
【0156】
<標的成分結合分子のPHA磁性構造体への担持>
PHA磁性構造体の表面に、標的成分に対して結合親和力を有する分子(以下、標的成分結合分子と記載する。)を担持する方法としては、該表面を被覆するPHAと標的成分結合分子との疎水性、イオン性、ファンデルワールス力等の物理的親和力による物理吸着によることもできるが、再現性や安定性を考慮すると、該PHA側鎖の官能基と、標的成分結合分子の有する官能基とを、そのまま、あるいは変換・修飾・活性化試薬の存在下に結合させて、不可逆的な共有結合を介在させることがより望ましい。
【0157】
本発明の方法に用いるPHA磁性構造体の一形態として、該表面を被覆するPHAの側鎖上にエポキシ基を有するPHA磁性構造体を用いることができる。該エポキシ基は、標的成分結合分子が有するアミノ基(−NH2)あるいはスルファニル基(−SH)と直接的に共有結合を形成することができる。結合形成に試薬を必要とせず、共有結合の形成が可能であることから、用いる標的成分結合分子として、酵素タンパク質等の変性を起こし易いタンパク質や、Aタンパク質、Gタンパク質といった抗体(Fc)受容体タンパク質等の担持に有効である。さらには、該表面を被覆するPHA上にイミノ二酢酸(IDA)を担持し、Ni2+等の金属イオンを添加することにより、ヒスタグ化された標的タンパク質の効率的回収に利用することが可能である。本エポキシ基との反応を利用して、共有結合を形成する方法は、標的成分結合分子が薬剤候補物質のような低分子化学物質である場合にも、該低分子化学物質がアミノ基(−NH2)あるいはスルファニル基(−SH)を有していれば、勿論応用することができる。
【0158】
該エポキシ基含有PHA磁性構造体は、エポキシ基に対して、10〜100倍モル量の水酸化アンモニウム、あるいはヘキサメチレンジアミン塩酸塩等を用いて、アルカリ条件下で反応させることにより、アミノ基を有するPHA磁性構造体に変換することができる。該アミノ基は、標的成分結合分子がタンパク質やペプチドの場合は、その主鎖カルボキシ末端やアスパラギン酸、グルタミン酸といった、該タンパク質やペプチドが有するアミノ酸残基の側鎖カルボキシ基との間で、NHS(N−ヒドロキシスクシンイミド)等の架橋化剤によりアミド結合を形成させることができる。その場合、標的成分結合分子側のカルボキシ基をNHS処理により活性化エステルとする必要があるので、NHS処理を施した際、標的成分結合分子の機能が十分保持されるか否かを、予め確認しておく必要がある。本アミド結合形成を利用する担持方法は、標的成分結合分子が薬剤候補物質のような低分子化学物質である場合にも、該低分子化学物質がカルボキシ基を有していれば、勿論応用することができる。
【0159】
さらに、該アミノ基は、標的成分結合分子が、糖鎖やレクチン等の糖タンパク質、リポ多糖等の糖脂質の場合には、その糖鎖中のアルデヒド構造(ホルミル基:−CHO)との間で、シッフ塩基(−CH=N−)形成および還元的アミネーションによる安定な結合を形成することができる。本糖鎖とアミノ基との反応を利用する共有結合の形成方法は、糖鎖中にアルデヒド構造を導入する、糖鎖部分の部分的酸化により促進され、また、そのFc部分に糖鎖を有するIgG等の抗体分子の担持にも応用可能である。本アミノ基とアルデヒド構造(ホルミル基:−CHO)との方法は標的成分結合分子が薬剤候補物質のような低分子化学物質である場合にも、該低分子化学物質がアルデヒド構造(ホルミル基:−CHO)を有しているか、部分的酸化によりアルデヒド構造(ホルミル基:−CHO)を導入することが可能であれば、勿論応用することができる。
【0160】
さらに、該アミノ基は、マレイミド誘導体、ピリジルジチオ化合物、ヨウ素/臭素アセチル化合物の存在下で、スルファニル基(−SH)を有する標的成分結合分子と結合させることができる。このアミノ基とスルファニル基(−SH)との反応の条件は、マレイミド誘導体を用いる場合は、0.1Mリン酸ナトリウム(pH6.5−7.5)中で、4℃から室温で2−4時間、ピリジルジチオ化合物を用いる場合は、PBS緩衝液(pH7.5)中で、室温にて15−20時間程度、ヨウ素/臭素アセチル化合物を用いる場合には、0.05Mホウ酸ナトリウム溶液(pH8.3)中で、遮光下に室温にて、1時間の反応が基本的な反応条件となるが、該反応条件は、標的成分結合分子の種類、その後の目的に応じて適宜変更することも可能である。
【0161】
本発明の方法に用いるPHA磁性構造体への担持の別の一形態として、該PHAの側鎖にカルボキシ基を有するPHA磁性構造体を利用するものを挙げることができる。該カルボキシ基は、標的成分結合分子が、タンパク質やペプチドの場合は、その主鎖アミノ末端や、リジン、アルギニンといった該タンパク質やペプチドが有するアミノ酸残基側鎖上のアミノ基との間で、NHS(N−ヒドロキシスクシンイミド)等の架橋化剤によって、アミド結合を形成することができる。このアミド結合形成反応は、PHAのカルボキシ基をNHS処理により予め活性化エステルとすることにより、アミド結合の形成速度・頻度を向上したものである。また、本PHA側鎖のカルボキシ基を利用するアミド結合の形成方法は、標的成分結合分子がDNAやオリゴヌクレオチドである場合には、通常の既知の方法で、その末端をアミノ化したDNA、オリゴヌクレオチドを用いることで、上記の反応方法によって、PHA上に担持することができる。本PHA側鎖のカルボキシ基を利用するアミド結合の形成方法は、標的成分結合分子が薬剤候補物質のような低分子化学物質である場合にも、該低分子化学物質がアミノ基を有していれば、勿論応用することができる。
【0162】
本発明の方法に用いるPHA磁性構造体への担持の別の一形態として、該PHAの側鎖にクロロ基(−Cl)、ブロモ基(−Br)、フルオロ基(−F)といったハロゲンを有するPHA磁性構造体を利用するものを挙げることができる。該ハロゲンがクロロ基やブロモ基の場合、スルファニル基(−SH)を有する標的成分結合分子との間で、温和な条件により、スルフィド結合(−S−)を形成することができる。
【0163】
その他、上記の活性官能基を用いて、表面にPHAを被覆した構造体に担持したい「標的成分結合分子」と特異的に結合する物質(例えば、「標的成分結合分子」が抗体の場合には、プロテインAやプロテインGなど)を、その表面に結合させた後、「標的成分結合分子」と特異的に結合する物質に対して、目的の「標的成分結合分子」を特異的に結合させることにより、構造体上に担持する方法も挙げられるし、「標的成分結合分子」を、さらに修飾した後、同様の方法で担持することも可能である。後者の例としては、カルボキシ型PHA磁性構造体の表面に、試薬NHS−iminobiotin(Pierce製)を用いて、ビオチンを担持し、一方、アビジンあるいはストレプトアビジンで修飾した標的成分結合分子をビオチンに特異的に結合させることで、構造体上に担持する方法が挙げられる。利用可能な親和性結合対の例として、他に、レクチンと糖、ハプテンと抗体、タンパク質AあるいはGと抗体Fc、他の特異結合タンパク質対、フェニルボロン酸とサリチルヒドロキサム酸、あるいは、互いに反応はするが、一般に、タンパク質とは反応しないその他の化学的部分対が挙げられる。
【0164】
構造体上への非特異吸着を防止するため、担持される標的成分結合分子の活性を損失しないような「ブロッキング剤」で、担持がなされていない構造体表面部分をコーティングすると好ましい。この「ブロッキング処理」に適したブロッキング剤として、コラーゲン、ゼラチン(特に、冷水魚皮ゼラチン)、スキムミルク、BSA等の血清タンパク質および、タンパク質とは反応しない疎水性部分およびも親水性部分を含む数多くの化合物が挙げられる。
【0165】
<標的成分結合分子担持PHA磁性構造体と標的成分との接触および結合>
本発明の方法における、PHA磁性構造体上に担持された標的成分結合分子と標的成分との接触は、通常、水性媒体中で行われるが、標的成分がある種の薬剤候補物質のような難水溶性分子である場合には、アルコールやアセトン、DMSO(ジメチルスルホキシド)やDMF(ジメチルホルムアミド)といった極性溶媒や、TweenやTriton、SDSといった界面活性剤を添加し、さらには、トルエンやキシレン、ヘキサンのような非極性溶媒を加えて、エマルジョン系で接触を行い、結合反応を促進してもよい。但し、これらの溶媒、界面活性剤を用いる場合には、その添加濃度は、担持されている標的成分結合分子の親和結合機能が損なわれないような濃度範囲に選択することが必要である。
【0166】
本発明の方法における、PHA磁性構造体上に担持された標的成分結合分子と標的成分との接触、結合を促進するために、標的成分結合分子の親和結合機能が損なわれない程度において、加熱手段や攪拌手段を用いることも可能であり、その際、超音波等の手段を用いることも可能である。
【0167】
また、本発明の方法で利用する構造体は、特には、磁性構造体であるので、その表面に担持された標的成分結合分子と標的成分との接触、結合を促進する手段として、磁力による操作を利用することも可能である。磁力による操作を施す場合、磁力を発揮する構造体(永久磁石や電磁石等:以下まとめて磁石と記載する場合もある)を用い、磁力の発揮、解放を繰り返す操作を施すことができる。電磁石を用いる場合、スイッチ切換えにより、電磁石に対する通電と遮断を反復して、磁性構造体の捕捉および解放を行う。磁石を用いた操作の一例としては、プローブ状の電磁石を反応容器内に挿入し、スイッチの切換え、電源のオン・オフ等により電磁石に対する通電と遮断を反復して、磁性構造体の捕捉・解放を繰り返す方法がある。また、別の一例として、磁石を容器側部の外側に配置して、スイッチの切換え、電源のオン・オフ、あるいは磁石自身の反応容器との距離の調節により、印加される磁場強度を反復的に変動させ、磁性構造体の捕捉・解放を繰り返す方法がある。
【0168】
なお、本発明の方法でいう標的成分と標的成分結合分子との「結合」は、結合対の要素、すなわち、化学的あるいは物理的作用により一方の分子が他方の分子に特異的に結合することである。周知の抗原・抗体反応による、抗原と抗体間の結合はもとより、ビオチンとアビジン、炭水化物とレクチン、核酸・ヌクレオチドの相補配列間、作動体と受容体分子、酵素補助因子と酵素、酵素抑制剤と酵素、ペプチド配列とその配列あるいはタンパク質全体に特異な抗体、ポリマーの酸と塩基、色素とタンパク質バインダー、ペプチドと特異タンパク質バインダー(リボヌクレアーゼ、S−ペプチドおよびリボヌクレアーゼS−タンパク質)、糖とホウ酸、および結合アッセイにおける分子会合を可能とする親和力を備えた同様の分子対の間における結合が挙げられるが、これに限定するものではない。さらに、結合対として、組換え技術あるいは分子工学により製造される分析物類似体あるいは結合要素など、元の結合要素の類似体である要素を挙げることができる。結合要素が免疫反応体であれば、例えば、抗体、抗原、ハプテンあるいはこれらの錯体が可能であり、「抗体」を用いる場合、単クローン抗体あるいはポリクローナル抗体、組換えタンパク質型抗体あるいは天然型抗体、キメラ抗体、混合物(単数あるいは複数)、単鎖抗体提示ファージ抗体(ファージ全体を含む)、単鎖抗体を提示するその断片(単数あるいは複数)、ならびに抗体およびタンパク質の結合要素の混合物でよい。
【0169】
近年、進化分子工学の発達により、ランダムなオリゴヌクレオチド・ライブラリーから蛋白質等の標的分子に対して高いアフィニティーを有する核酸分子、すなわちアプタマー(核酸抗体と称される場合もある)をスクリーニングする技術(“systematic evolution of ligands byexponential enrichment”; SELEX or in vitro selection)が開発された。このアプタマーを用いるスクリーニング方法を用いて、抗体よりも迅速且つ容易な高アフィニティー・リガンドの調製が数多く報告されている(例えば、Nature, 355: 564 (1992) 、国際特許出願WO92/14843号公報、特開平8−252100号公報、特開平9−216895号公報等)。
【0170】
あるいは、タンパク質である核酸の転写因子と、ある特定の塩基配列を含む核酸との結合に関しても、疾病発生の原因究明や、さらには、効果的な診断・治療への応用も期待されている。
【0171】
本発明の方法が対象とする「結合」には、勿論、このような核酸−タンパク質間の親和性結合も包含される。
【0172】
また、本発明の方法が対象とする「結合」には、永久的あるいは一次的を問わず、あらゆる物理的付着あるいは化学的付着、あるいは密接な特異的・選択的な会合を含む。一般に、イオン結合の相互作用、水素結合、疎水力、ファンデルワールス力などにより、対象のリガンド分子と受容体との間を物理的に付着させることができる。「結合」の相互作用は、結合により化学変化が起こる場合のように短い可能性がある。これは、結合成分が酵素であり、その分析物「標的成分結合分子」が酵素用基質である場合に一般的である。さらには、化学的連結が、永久的あるいは可逆的結合である可能性もある。結合は、特に異なる条件下において、特異的となる可能性がある。
【0173】
実際に、本発明の方法における、PHA磁性構造体上に担持された標的成分結合分子と標的成分との接触および結合工程の一例としては、標的成分結合分子を担持するPHA磁性構造体を、組織や細胞ホモジェネートあるいは血清などの流体といった天然タンパク質を含む生物学的試料に接触させて、その天然タンパク質をPHA磁性構造体表面に担持する標的成分結合分子に特異的に吸着・結合させる工程である。
【0174】
また、該工程の別の一例としては、周知のファージ・ディスプレイ抗体選択法で用いられているように、標的成分結合分子であるタンパク質を担持する磁性構造体を用いて、適したバクテリオファージの表面に提示される抗体部分を選択的に結合することができる。
【0175】
また、該工程の別の一例としては、ハイブリドーマ上澄み液、ファージ・ディスプレイなどの流体など、受容体を含有した生物学的試料に接触させて、生物学的試料中に含まれる受容体を、特異的にPHA磁性構造体上に担持された標的成分結合分子に吸着させる工程も考え得る。
【0176】
<標的成分結合磁性複合体の磁気分離および洗浄>
磁性複合体(標的成分結合分子が担持されている磁性構造体)に標的成分を結合させた後、標的成分結合磁性複合体を分離および洗浄する方法においては、磁力を発揮する構造体(永久磁石や電磁石等)を用い、磁力の発揮、解放を繰り返すことにより、磁気分離の操作を施す。電磁石を用いる場合、スイッチ切換えにより、電磁石に対する通電と遮断を行って、標的成分結合磁性複合体の捕捉および解放を行う。
【0177】
実施形態の一例としては、プローブ型磁石を用いて、容器内で標的成分結合磁性複合体を捕捉した後、残余する液体を容器から除去する。洗浄液をその容器内に投入して、プローブ上にまだ捕捉されている標的成分結合磁性複合体を洗浄する。あるいは、標的成分結合磁性複合体が捕捉されたプローブ型磁石を反応溶液から取り出し、洗浄液へと移動させ、洗浄操作を行うこともできる。
【0178】
別の方法として、磁石を容器側部の外側に配置して、液体の交換時に標的成分結合磁性複合体を容器内側部に付着させておくこともできる。この外部磁石を取り除くと、標的成分結合磁性複合体は解放され、交換された液体(洗浄液)と混ざり合うことで洗浄が行われる。
【0179】
なお、本磁気分離の操作には、磁性微粒子を操作する用途で市販されている、数多くの磁選機を利用することも可能である。市販されている磁選機の例としては、DYNAL社製 DYNAL MCP、 Serono Diagnostics社製 MAIA Magnetic Separator、 宝酒造社製Magnetight Saparation Stand、 Advanced Magnetics社製 BioMag Separator、等が挙げられる。
【0180】
<標的成分の磁性構造体からの溶離・遊離>
本発明の方法においては、その必要に応じて、標的成分結合磁性複合体を単離した後、結合している標的成分を標的成分結合分子から溶離・遊離することもできる。標的成分結合分子と標的成分とが、タンパク質−タンパク質の組み合わせである場合は、通常の解離条件(pH2、4Mグアニジン、2Mチオシアン酸アンモニウム、あるいは1%SDSなど)で遊離することができるが、その際、遊離する標的成分タンパク質は変性を受けていることが多い。遊離した標的成分タンパク質が仮に変性していても、電気泳動やHPLCで精製を施す場合は、特に問題はないが、標的成分タンパク質本来の立体構造等を検証したい場合には、透析等の復元工程が必要な場合がある。
【0181】
<標的成分の検出>
本発明の方法で用いる、標的成分の検出方法として、通常の比色法、蛍光法、化学発光法、ラジオアイソトープ法等、イムノアッセイ、ハイブリダイゼーション・アッセイで用いることのできる如何なる方法も、採用することが可能である。さらには、標的成分結合分子から上記の方法で溶離・遊離された標的成分は、同様の方法を用いて分析することもできるし、さらに、標的成分がDNAである場合は、PCR等の手法で増幅した上で、シーケンサー等で塩基配列を検証することも可能であるし、標的成分がタンパク質である場合は、酵素分解した後、その酵素消化断片を、二次元電気泳動やHPLCにより分離した後、エレクトロスプレイ・イオン化法−MS分析を行うことも可能であるし、また、標的成分タンパク質について、直接あるいは酵素分解した後、マトリックス支援レーザ脱離イオン化法−飛行時間型質量分析法(MALDI ToF−MS)で分析することも可能である。
【0182】
勿論、核磁気共鳴スペクトロスコピー(NMR)や赤外吸収スペクトロスコピー(IR)、紫外吸収スペクトロスコピー(UV)のような、その他のスペクトル分析を単独で、あるいは組み合わせて、標的成分の検出を行うことも可能である。
【0183】
【実施例】
以下、実施例により、本発明をさらに具体的に説明する。ただし、以下に述べる実施例は、本発明にかかる最良の実施形態の一例ではあるが、本発明の技術的範囲はこれら実施例に限定されるものではない。
【0184】
参考例1. PHA合成酵素生産能を有する形質転換体の作製
YN2株を100 mlのLB培地(1%ポリペプトン(日本製薬(株)製)、0.5%酵母エキス(Difco社製)、0.5%塩化ナトリウム、pH7.4)中で30℃、一晩培養した後、マーマーらの方法により染色体DNAを分離回収した。得られたYN2株の染色体DNAを制限酵素Hind IIIで完全分解した。クローニング・ベクターには、pUC18を使用し、制限酵素Hind IIIで切断した。末端の脱リン酸処理(Molecular Cloning、 1、 572、 (1989); Cold Spring HarborLaboratory 出版)の後、DNAライゲーション・キット Ver. II(宝酒造(株)製)を用いて、ベクターの切断部位(クローニング・サイト)と染色体DNAのHind III完全分解断片とを連結した。この染色体DNA断片を組み込んだプラスミド・ベクターを用いて、大腸菌(Escherichia coli)HB101株を形質転換し、YN2株のDNAライブラリーを作製した。
【0185】
次に、YN2株のPHA合成酵素遺伝子を含むDNA断片を選択するため、コロニー・ハイブリダイズ用のプローブ調製を行った。配列番号:2および配列番号:3の塩基配列からなるオリゴヌクレオチドを合成し(アマシャム・ファルマシア・バイオテク(株))、このオリゴヌクレオチドをプライマーに用いて、染色体DNAをテンプレートとしてPCRを行った。PCR増幅されてきたDNA断片を、コロニー・ハイブリダイズ用のプローブとして用いた。プローブの標識化は、市販の標識酵素系AlkPhos・Direct(アマシャム・ファルマシア・バイオテク(株)製)を利用して行った。得られた標識化プローブを用いて、YN2株の染色体DNAライブラリーからコロニー・ハイブリダイゼーション法によって、PHA合成酵素遺伝子を含む組換えプラスミドを有する大腸菌菌株を選抜した。
【0186】
選抜した菌株から、アルカリ法によってプラスミドを回収することで、YN2株由来のPHA合成酵素遺伝子を含むDNA断片を得ることができた。ここで取得した遺伝子DNA断片を、不和合性グループであるIncP、IncQ、あるいはIncWの何れにも属さない広宿主域複製領域を含むベクターpBBR122(Mo BiTec)に組み換えた。この組み換えプラスミドをシュードモナス・チコリアイYN2ml株(PHA合成能欠損株)にエレクトロポレーション法により挿入して、形質転換したところ、YN2ml株のPHA合成能が復帰し、相補性を示した。従って、選抜された遺伝子DNA断片は、シュードモナス・チコリアイYN2ml株内において、PHA合成酵素に翻訳可能な、PHA合成酵素遺伝子領域を含むことが確認される。
【0187】
この遺伝子DNA断片について、サンガー法により塩基配列を決定した。その結果、決定された塩基配列中には、それぞれペプチド鎖をコードする、配列番号:4および配列番号:5で示される塩基配列が存在することが確認された。これらのPHA合成酵素遺伝子について、染色体DNAをテンプレートとしてPCRを行い、PHA合成酵素遺伝子の完全長を再調製した。すなわち、配列番号:4で示される塩基配列のPHA合成酵素遺伝子に対する、上流側プライマー(配列番号:6)および下流側プライマー(配列番号:7)と、配列番号:5で示される塩基配列のPHA合成酵素遺伝子に対する、上流側プライマー(配列番号:8)および下流側プライマー(配列番号:9)とを、それぞれ合成した(アマシャム・ファルマシア・バイオテク(株))。
【0188】
これらのプライマー対を用いて、配列番号:4ならびに配列番号:5で示される塩基配列のそれぞれについて、PCR増幅を行い、PHA合成酵素遺伝子の完全長を増幅した(LA−PCRキット;宝酒造(株)製)。次に、得られたPCR増幅断片および発現ベクターpTrc99Aを制限酵素Hind IIIで切断し、脱リン酸化処理(Molecular Cloning, 1巻, 572頁, 1989年; Cold Spring Harbor Laboratory出版)した後、この発現ベクターpTrc99Aの切断部位に、両末端の不用な塩基配列を除いたPHA合成酵素遺伝子の完全長を含むDNA断片を、DNAライゲーション・キット Ver. II(宝酒造(株)製)を用いて連結した。
【0189】
得られた組換えプラスミドで、大腸菌(Escherichia coli HB101:宝酒造)を塩化カルシウム法により形質転換した。得られた組換え体を培養し、組換えプラスミドの増幅を行い、組換えプラスミドをそれぞれ回収した。配列番号:4の遺伝子DNAを保持する組換えプラスミドを、pYN2−C1、配列番号:5の遺伝子DNAを保持する組換えプラスミドを、pYN2−C2とした。pYN2−C1、pYN2−C2で、大腸菌(Escherichia coli HB101fB fadB欠損株)を塩化カルシウム法により形質転換し、それぞれの組換えプラスミドを保持する組換え大腸菌株、pYN2−C1組換え株、pYN2−C2組換え株を得た。
【0190】
参考例2. PHA合成酵素の生産1
pYN2−C1に対して、上流側プライマーとなる、オリゴヌクレオチド(配列番号:10)および下流側プライマーとなる、オリゴヌクレオチド(配列番号:11)をそれぞれ設計・合成した(アマシャム・ファルマシア・バイオテク(株))。このオリゴヌクレオチドをプライマーとして、pYN2−C1をテンプレートとしてPCRを行い、上流にBamHI制限部位、下流にXhoI制限部位を有するPHA合成酵素遺伝子の完全長を増幅した(LA−PCRキット;宝酒造(株)製)。
【0191】
同様に、pYN2−C2に対して、上流側プライマーとなる、オリゴヌクレオチド(配列番号:12)および下流側プライマーとなる、オリゴヌクレオチド(配列番号:13)をそれぞれ設計・合成した(アマシャム・ファルマシア・バイオテク(株))。このオリゴヌクレオチドをプライマーとして、pYN2−C2をテンプレートとしてPCRを行い、上流にBamHI制限部位、下流にXhoI制限部位を有するPHA合成酵素遺伝子の完全長を増幅した(LA−PCRキット;宝酒造(株)製)。
【0192】
精製したそれぞれのPCR増幅産物をBamHIおよびXhoIにより消化し、プラスミドpGEX−6P−1(アマシャム・ファルマシア・バイオテク(株)製)の対応する部位に挿入した。これらのベクターを用いて、大腸菌(JM109)を形質転換し、発現用菌株を得た。発現用菌株の確認は、Miniprep(Wizard Minipreps DNA Purification Systems、PROMEGA社製)を用いて、大量に調製したプラスミドDNAをBamHI、XhoIで処理して得られるDNA断片によって行った。得られた菌株を、LB−Amp培地10 mlで一晩プレ・カルチャーした後、その培養物0.1 mlを、10 mlのLB−Amp培地に添加し、37℃、170 rpmで3時間振とう培養した。その後、IPTGを添加(終濃度 1 mM)し、37℃で4〜12時間培養を続けた。
【0193】
IPTG誘導した大腸菌を集菌(8,000×g, 2分、4℃)し、1/10量の4℃リン酸緩衝生理食塩水(PBS;8 g NaCl,1.44 g Na2HPO4,0.24 g KH2PO4,0.2 g KCl,1,000 ml精製水)に再懸濁した。凍結融解およびソニケーションにより菌体を破砕し、遠心(8,000×g, 10分、4℃)して固形夾雑物を取り除いた。目的の発現タンパク質が上清に存在することを、SDS−PAGEで確認した後、誘導され発現されたGST融合タンパク質をグルタチオン・セファロース4B(アマシャム・ファルマシア・バイオテク(株)製)で精製した。使用するグルタチオン・セファロースは、予め非特異的吸着を抑える処理を行った。すなわち、グルタチオン・セファロースを同量のPBSで3回洗浄(8,000×g、 1分、4℃)した後、4%ウシ血清アルブミン含有PBSを同量加えて、4℃で1時間処理した。処理後、同量のPBSで2回洗浄し、1/2量のPBSに再懸濁した。前処理したグルタチオン・セファロース 40μlを、無細胞抽出液1 mlに添加し、4℃で静かに攪拌した。前記の手順で、融合タンパク質GST−YN2−C1および融合タンパク質GST−YN2−C2をグルタチオン・セファロースに吸着させた。吸着後、遠心(8,000×g、1分、4℃)してグルタチオン・セファロースを回収し、400μlのPBSで3回洗浄した。その後、10 mMグルタチオン40μlを添加し、4℃で1時間攪拌して、吸着した融合タンパク質を溶出した。遠心(8,000×g、2分、4℃)して上清を回収した後、PBSに対して透析し、GST融合タンパク質を精製した。SDS−PAGEにより、GST融合タンパク質由来のシングル・バンドを確認した。
【0194】
各GST融合タンパク質500μgをPreScissionプロテアーゼ(アマシャム・ファルマシア・バイオテク(株)製、5U)で消化した後、グルタチオン・セファロースに通してプロテアーゼとGSTとを除去した。フロー・スルー分画をさらに、PBSで平衡化したセファデックスG200カラムにかけ、発現タンパク質YN2−C1およびYN2−C2の最終精製物を得た。SDS−PAGEにより、それぞれ60.8kDa、および61.5kDaのシングル・バンドを確認した。
【0195】
該組換え型酵素を生体溶液試料濃縮剤(みずぶとりくん AB−1100、アトー(株)製)を用いて濃縮し、10 U/mlの精製酵素溶液を得た。なお、各精製酵素活性は、前述の方法で測定した。また、試料中のタンパク質濃度は、マイクロBCAタンパク質定量試薬キット(ピアスケミカル社製)によって測定した。表1に、各精製酵素の活性測定の結果を示す。
【0196】
【表1】
参考例3.P HA合成酵素の生産2
P91株、H45株、YN2株またはP161株を、酵母エキス(Difco社製)0.5%、オクタン酸0.1%とを含むM9培地200mlに植菌して、30℃、125ストローク/分で振盪培養した。24時間後、菌体を遠心分離(10,000×g、4℃、10分間)によって回収し、0.1M トリス塩酸バッファー(pH8.0) 200 mlに再懸濁して、再度遠心分離することによって洗浄した。洗浄後の菌体を、0.1M トリス塩酸バッファー(pH8.0)2.0 mlに再懸濁し、超音波破砕機にて破砕した後、遠心分離(12,000×g、4℃、10分間)して上清を回収して粗酵素を得た。
各精製酵素活性は前述の方法で測定し、表2に測定結果を示す。
【0198】
【表2】
参考例4. 磁性体の調製
硫酸第一鉄水溶液中に、鉄イオンに対してl.0〜1.1当量の苛性ソーダ溶液を混合し、水酸化第一鉄を含む水溶液を調製した。この水溶液のpHを8前後に維持しながら、空気を吹き込み、80〜90℃で酸化反応を行い、種晶を生成させるスラリー液を調製した。
【0200】
次いで、このスラリー液に、当初のアルカリ量(苛性ソーダのナトリウム成分)に対して、0.9〜1.2当量となるよう硫酸第一鉄水溶液を加えた後、スラリー液をpH8前後に維持して、空気を吹き込みながら酸化反応を進める。酸化反応後に、生成した磁性酸化鉄粒子を洗浄、濾過、乾燥し、凝集している粒子を解砕し、平均粒径が0.1μmの粒状の磁性体1を得た。
【0201】
参考例5. エポキシ型PHA磁性構造体微粒子の作製1
pYN2−C1組換え株由来のPHA合成酵素溶液(10 U/ml)10質量部に、参考例4で作製した磁性体を1質量部、PBS 39質量部を添加し、30℃にて30分間緩やかに振盪して、PHA合成酵素を磁性体表面に吸着させた。これを遠心分離(10,000×g、4℃、10分間)し、沈澱をPBS溶液に懸濁し、再度遠心分離(10,000×g、4℃、10分間)して固定化PHA合成酵素を得た。
【0202】
前記固定化PHA合成酵素を0.1 Mリン酸バッファー(pH7.0)48質量部に懸濁し、(R,S)−3−ヒドロキシ−5−フェノキシバレリルCoA(3−フェノキシプロパナールとブロモ酢酸エチルとのReformatsky反応で得られた3−ヒドロキシ−5−フェノキシ吉草酸エステルを加水分解して3−ヒドロキシ−5−フェノキシ吉草酸を得た後、Eur. J. Biochem., 250, 432−439 (1997)に記載の方法で調製)0.8質量部、(R,S)−3−ヒドロキシ−7,8−エポキシオクタノイルCoA(Int. J. Biol. Macromol., 12, 85−91(1990)に記載の方法で合成した3−ヒドロキシ−7−オクテン酸の不飽和部分を3−クロロ安息香酸でエポキシ化したのち、 Eur. J. Biochem., 250, 432−439 (1997)に記載の方法で調製)0.2質量部、ウシ血清アルブミン(Sigma社製)0.1質量部を添加し、30℃で2時間緩やかに振盪して、PHA磁性構造体微粒子を得た。
【0203】
上記の試料10μlをスライドグラス上に採取し、1%ナイルブルーA水溶液10μlを添加し、スライドグラス上で混合した後、カバーグラスを載せ、蛍光顕微鏡(330〜380 nm励起フィルタ、420 nmロングパス吸収フィルタ、(株)ニコン製)観察を行った。その結果、いずれの試料においても、PHA磁性構造体微粒子表面が蛍光を発していることが確認された。従って、該PHA磁性構造体微粒子は、確かにPHAにより表面を被覆されていることが示された。
【0204】
さらに、試料の一部を遠心分離(10,000×g、4℃、10分間)により回収し、真空乾燥した後、クロロホルムに懸濁し、60℃で20時間攪拌して、外被を成すPHAを抽出した。この抽出液について、1H−NMR分析を行った(使用機器:FT−NMR:Bruker DPX400、測定核種:1H,使用溶媒:重クロロホルム(TMS入り))。その結果、該PHA磁性構造体微粒子の外被PHAの組成およびユニット%は、3−ヒドロキシ−5−フェノキシ吉草酸ユニット:75%,3−ヒドロキシ−7,8−エポキシオクタン酸ユニット:25%であることが確認された。
【0205】
参考例6. ビニルフェニル型PHA磁性構造体微粒子の作製
pYN2−C1組換え株由来のPHA合成酵素溶液(10 U/ml)10質量部に湿式法で合成した粒子径0.3μmのマグネタイト微粒子「マグネタイトEPT500」(戸田工業(株)製)を1質量部、PBS 39質量部を添加し、30℃にて30分間緩やかに振盪して、PHA合成酵素を微粒子表面に吸着させた。これを遠心分離(10,000×g、4℃、10分間)し、沈澱をPBS溶液に懸濁し、再度遠心分離(10,000×g、4℃、10分間)して、固定化PHA合成酵素を得た。
【0206】
上記固定化PHA合成酵素を0.1 Mリン酸バッファー(pH7.0)48質量部に懸濁し、(R)−3−ヒドロキシ−5−フェニルバレリルCoA(Eur. J. Biochem., 250, 432−439 (1997)に記載の方法で調製)0.8質量部、(R)−3−ヒドロキシ−5−(4−ビニルフェニル)バレリルCoA(Eur. J. Biochem., 250, 432−439 (1997)に記載の方法で調製)0.2質量部、ウシ血清アルブミン(Sigma社製)0.1質量部を添加し、30℃で2時間緩やかに振盪して、PHA磁性構造体微粒子を得た。
【0207】
上記の試料10μlをスライドグラス上に採取し、1%ナイルブルーA水溶液10μlを添加し、スライドグラス上で混合した後、カバーグラスを載せ、蛍光顕微鏡(330〜380 nm励起フィルタ、420 nmロングパス吸収フィルタ、(株)ニコン製)観察を行った。その結果、PHA磁性構造体微粒子の表面が蛍光を発していることが確認され、該PHA磁性構造体微粒子は、確かにPHAにより表面を被覆されていることが判った。
【0208】
さらに、試料の一部を遠心分離(10,000×g、4℃、10分間)により回収し、真空乾燥した後、クロロホルムに懸濁し、60℃で20時間攪拌して外被を成すPHAを抽出した。この抽出液について、1H−NMR分析を行った(使用機器:FT−NMR:Bruker DPX400、測定核種:1H,使用溶媒:重クロロホルム(TMS入り))。その結果、該PHA磁性構造体微粒子の外被PHAの組成およびユニット%は、3−ヒドロキシ−5−フェニル吉草酸ユニット:83%,3−ヒドロキシ−5−(4−ビニルフェニル)吉草酸ユニット:17%であることが確認された。
【0209】
参考例7. エポキシ型PHA磁性構造体微粒子の作製2
参考例6で製造したビニルフェニル型PHA磁性構造体微粒子について、そのビニル基のエポキシ化反応を行った。ビニルフェニル型PHA磁性構造体微粒子の1質量部を四つ口丸底フラスコに加え、蒸留水6質量部を加えて攪拌した。フラスコ内を40℃に加温し、これに過酢酸30%のヘキサン溶液1質量部を連続的に滴下し、攪拌下40℃で5時間反応させた。反応は、PHA磁性構造体微粒子同士が凝集することなく進行した。反応終了後、室温まで冷却し、反応液をろ過することで、PHA磁性構造体微粒子を回収した。この回収したPHA磁性構造体微粒子は蒸留水に再懸濁した後、遠心分離(3000×g、4℃、30分間)した。分離した後、PHA磁性構造体微粒子を蒸留水に再懸濁し、再度遠心分離を行って洗浄した。さらに、この洗浄操作を3回繰り返した。その後、真空乾燥することで、下記組成のPHA磁性構造体微粒子を得た。
【0210】
さらに、試料の一部を遠心分離(10,000×g、4℃、10分間)により回収し、真空乾燥した後、クロロホルムに懸濁し、60℃で20時間攪拌して、外被を成すPHAを抽出した。この抽出液について、1H−NMR分析を行った(使用機器:FT−NMR:Bruker DPX400、測定核種:1H,使用溶媒:重クロロホルム(TMS入り))。この測定結果から計算した、外被PHAに含まれる各側鎖ユニットのユニット%は、3−ヒドロキシ−5−フェニル吉草酸ユニット:85%、3−ヒドロキシ−5−(4−ビニルフェニル)吉草酸ユニット:11%、3−ヒドロキシ−5−(4−(1,2−エポキシエチル)フェニル)吉草酸ユニット:4%であった。この反応系が不均一反応であるため、微粒子表面のPHAはエポキシ化され、被覆内部では、未反応PHAのままに残されていると考えられる。
【0211】
参考例8. カルボキシ型PHA磁性構造体微粒子の作製
参考例6で製造したビニルフェニル型PHA磁性構造体微粒子について、そのビニル基の酸化開裂反応を行った。ビニルフェニル型PHA磁性構造体微粒子の10質量部を三つ口フラスコに移して、過酸化水素50ppmを添加した300重量部の蒸留水を加えた。オゾンを1重量部/時間の割りで吹き込み、3時間室温で攪拌した。反応は、磁性構造体微粒子同士が凝集することなく進行した。反応終了後、反応液をろ過することで、PHA磁性構造体微粒子を回収した。PHA磁性構造体微粒子は、蒸留水に再懸濁した後、遠心分離(3000×g、4℃、30分間)を行って、残余する過酸化水素水を洗浄した。さらに、この洗浄操作を2回繰り返した。その後、真空乾燥することで、下記する組成のPHA磁性構造体微粒子を得た。
【0212】
さらに、試料の一部を遠心分離(10,000×g、4℃、10分間)により回収し、真空乾燥した後、クロロホルムに懸濁し、60℃で20時間攪拌して外被を成すPHAを抽出した。この抽出液について、1H−NMR分析を行った(使用機器:FT−NMR:Bruker DPX400、測定核種:1H,使用溶媒:重クロロホルム(TMS入り))。この測定結果から計算した外被PHAに含まれる各側鎖ユニットのユニット%は、3−ヒドロキシ−5−フェニル吉草酸ユニット:84%、3−ヒドロキシ−5−(4−ビニルフェニル)吉草酸ユニット:11%、3−ヒドロキシ−5−(4−カルボキシフェニル)吉草酸ユニット:5%であった。この反応系が不均一反応であるため、微粒子表面のPHAはエポキシ化され、被覆内部では、未反応PHAのままに残されていると考えられる。
【0213】
参考例9. ブロモ型PHA磁性構造体微粒子の作製
YN2−C1組換え株由来のPHA合成酵素溶液(10 U/ml)10質量部に、参考例4で作製した磁性体を1質量部、PBS 39質量部を添加し、30℃にて30分間緩やかに振盪してPHA合成酵素を磁性体表面に吸着させた。これを遠心分離(10,000×g、4℃、10分間)し、沈澱をPBS溶液に懸濁し、再度遠心分離(10,000×g、4℃、10分間)して、固定化PHA合成酵素を得た。
【0214】
上記固定化PHA合成酵素を0.1 Mリン酸バッファー(pH7.0)48質量部に懸濁し、(R,S)−3−ヒドロキシ−5−フェノキシバレリルCoA(3−フェノキシプロパナールとブロモ酢酸エチルとのReformatsky反応で得られた3−ヒドロキシ−5−フェノキシ吉草酸エステルを加水分解して3−ヒドロキシ−5−フェノキシ吉草酸を得た後、Eur. J. Biochem., 250, 432−439 (1997)に記載の方法で調製)0.8質量部、(R)−3−ヒドロキシ−8−ブロモオクタノイルCoA(Eur .J. Biochem., 250, 432−439( 1997) に記載の方法で調製)0.2質量部、ウシ血清アルブミン(Sigma社製)0.1質量部を添加し、30℃で2時間緩やかに振盪して、PHA磁性構造体微粒子を得た。
【0215】
上記のPHA磁性構造体微粒子の10μlをスライドグラス上に採取し、1%ナイルブルーA水溶液10μlを添加し、スライドグラス上で混合した後、カバーグラスを載せ、蛍光顕微鏡(330〜380 nm励起フィルタ、420 nmロングパス吸収フィルタ、(株)ニコン製)観察を行った。その結果、磁性構造体の粒子表面が蛍光を発していることが確認された。従って、該PHA磁性構造体微粒子は、確かにPHAにより表面を被覆されていること示された。
【0216】
さらに、該PHA磁性構造体微粒子の一部を遠心分離(10,000×g、4℃、10分間)により回収し、真空乾燥した後、クロロホルムに懸濁し、60℃で20時間攪拌して、外被を成すPHAを抽出した。この抽出液について、1H−NMR分析を行った(使用機器:FT−NMR:Bruker DPX400、測定核種:1H,使用溶媒:重クロロホルム(TMS入り))。この測定結果から計算した外被PHAに含まれる各側鎖ユニットのユニット%は、3−ヒドロキシ−5−フェノキシ吉草酸ユニット:89%、3−ヒドロキシ−8−ブロモオクタン酸ユニット:11%であった。
【0217】
実施例1. エポキシ型PHA磁性構造体微粒子上に担持された抗AFP抗体によるAFPの免疫検出
1. エポキシ型PHA磁性構造体微粒子への抗体の担持
リン酸緩衝液(0.1M;pH7.4)中に、参考例5において作製されたエポキシ型PHA磁性構造体微粒子を均一に分散させ(理論濃度5−10×108個/mL)、同様のリン酸緩衝液に溶解した抗AFP(α−フェトプロテイン)抗体を、前記PHA磁性構造体微粒子と抗体の最終比率が5−10μg/107となるよう加え、ピペッティングにより穏やかに攪拌した。
【0218】
ロータリー・シェーカーで30℃1時間反応させた後、ブロッキングのためにウシ血清アルブミン(BSA)を最終濃度0.3%になるように加え、さらに、15時間反応を行うことにより、エポキシ型PHA磁性構造体微粒子表面に担持された抗体を得た。
【0219】
2.標的成分(抗原:AFP)との反応
5mL容エッペンドルフチューブ(BSA処理)中、1で作製した抗AFP抗体担持エポキシ型PHA磁性構造体微粒子の分散液500μLに、濃度1μg/mLのAFP溶液20μLを混合し、37℃で30分間反応させた。このチューブを磁石に接して、磁性構造体微粒子を捕捉し、上清をデカンテーションにより除去した。その後、チューブ中に0.04%NaCl溶液2mLを加え攪拌した。前記と同様に、磁石により磁性構造体微粒子を捕捉し、上清をデカンテーションにより除去した。この洗浄操作を3回繰り返した。
【0220】
3.酵素標識二次抗体との反応
J. Immunoassay, 4, 209(1983)およびBiochemistry, 11(12), 2291 (1972)に記載の方法に従って、アルカリ・ホスファターゼ標識抗AFP抗体Fab’を作製し、最終濃度濃度0.1μg/mLになるように、0.1Mトリス−塩酸緩衝液(2% BSA、1mM MgCl2 、0.1mM ZnCl2 含有;pH7.5)と混合・溶解し、その500μLを3.で得られた抗原AFP結合磁性複合体微粒子に加え、37℃10分間、反応させた。その後、2.と同様の方法により、酵素標識二次抗体を反応させた抗原AFP結合磁性複合体微粒子の洗浄を行った。
【0221】
4.検出
3.で得られた酵素標識二次抗体を反応させた抗原AFP結合磁性複合体微粒子を含むチューブに、500μLのグリシン−NaOH緩衝液(0.1M:pH10.3:MgCl2(1mM)および卵白アルブミン(250mg/L)を含む)を加え、37℃で5分間反応させた後、4−ニトロフェニルリン酸(最終濃度5.5mM)を含む前記緩衝液500μlを加え、37℃で60分間反応させた。反応終了後、1M NaOH水溶液500μLを加えて、反応を停止し、紫外・可視吸光度を測定した。その結果、標識酵素アルカリ・ホスファターゼによる反応産物に由来する405nmの吸収が検出され、実際に、抗原AFPが回収されていることが確認された。
【0222】
実施例2. セロペンタオース担持アミノ化PHA磁性構造体微粒子を用いたコンカナバリンAの回収
1.エポキシ型PHA磁性構造体微粒子のアミノ化
参考例7で作製されたエポキシ型PHA磁性構造体微粒子に、2,2’−(エチレンジオキシ)−ジメチルアミンを加え、30℃で15時間反応を行いアミノ化を行った。反応終了後、ビス−2−メトキシエチルエーテルで5回洗浄し、残留アミンを除去した後、蒸留水でさらに3回洗浄することにより、アミノ化PHA磁性構造体微粒子を得た。
【0223】
2.アミノ化PHA磁性構造体微粒子へのセロペンタオースの担持
工程1.で得られたアミノ化PHA磁性構造体微粒子を、実施例1と同様にリン酸緩衝液中に均一に分散させ、セロペンタオース(D−(+)−Cellopentaose:Sigma社製)を予め過ヨウ素酸ナトリウムで非還元末端を酸化することにより、アルデヒド構造(−CHO:ホルミル基)を生成させたものと30℃で30時間攪拌し、セロペンタオースをアミノ化PHA磁性構造体微粒子上への担持を行った。その後、微粒子表面に残留する余剰のアミノ基を保護するため、無水ブタンジエン酸を加えて反応させ、蒸留水で3回洗浄することにより、セロペンタオース担持アミノ化PHA磁性構造体微粒子を得た。
【0224】
3.コンカナバリンAの回収
前記リン酸緩衝液にコンカナバリンA(Concanavalin A:Sigma社製)とBSAを溶解させ、2.で得られたセロペンタオース担持アミノ化PHA磁性構造体微粒子を加えて、30℃で15時間反応した後、磁力により微粒子を回収した。回収した微粒子を、ドデシル硫酸ナトリウム(SDS)で処理し、溶出液のSDS−PAGE分析を行ったところ、104kDaにほぼ単一のバンドが見られ、コンカナバリンAが回収されたことが示された。
【0225】
実施例3.カルボキシ型PHA磁性構造体微粒子を用いたDNA断片のスクリーニング
1.モデル核酸M13p18ssDNAの合成とアミノ化
配列番号:14に示す、大腸菌M13ファージmp18一本鎖DNAに相補的な塩基配列を有する20量体オリゴヌクレオチドをDNA自動合成機(ABI社製381A)で合成した。
配列番号:14: 5’−GTTGTAAAACGACGGCCAGT−3’
その後、通常のアミダイド試薬に変わり、アミノ基を導入したデオキシウリジル酸誘導体モノマー(化合物[19])を用いて、上記20量体オリゴヌクレオチドの5’側にアミノ基(−NH2)を導入した(化合物[20])。常法により、CPGサポートからの切り出し、脱保護基、高速液体クロマトグラフィー(HPLC)による精製を行った。
【0226】
【化20】
【化21】
2.アミノ化プローブ・オリゴヌクレオチドのカルボキシ型PHA磁性構造体微粒子への担持
参考例8で得られたカルボキシ型PHA磁性構造体微粒子を、予め0.01M水酸化ナトリウム溶液で洗浄し、得られた微粒子に対し、理論上カルボキシ基の2モル倍程度のEDC(1−エチル−3−(3−ジエチルアミノプロピル)−カルボジイミド−塩酸)および式[20]に示すアミノ化オリゴヌクレオチドを加えて、4℃で18時間反応を行い、プローブ・オリゴヌクレオチドを担持したPHA磁性構造体微粒子を得た。
【0229】
3.ターゲット・モデルオリゴヌクレオチドの合成と標識化
上記配列番号:14のオリゴヌクレオチドに対して、完全相補的な20量体オリゴヌクレオチド(配列番号:15)、1塩基ミスマッチの(配列番号:16)、2塩基ミスマッチの(配列番号:17)オリゴヌクレオチドを、それぞれ自動合成機で合成し、常法により精製した。
配列番号15: 5’−ACTGGCCGTCGTTTTACAAC−3’
配列番号16: 5’−ACTGGCCGTCCTTTTACAAC−3’
配列番号17: 5’−ACTGGCGGTCGTTATACAAC−3’
精製したモデルオリゴヌクレオチド三種を、それぞれ工程1と同様の方法で、デオキシウリジル酸誘導体モノマー(化合物[19])を用いて、5’末端をアミノ化した。
【0230】
これとは別に、特許第03368011号公報に開示の方法に従って、シアニン色素(化合物[21])170mgを5mlの乾燥DMFに溶解し、これに乾燥ピリジン50μlを加えた。さらに、DSC(ジスクシイミジルカーボネート)128mgを加えた後、暗所、室温で20時間攪拌した。反応混液にジエチルエーテル150mlを加え、析出した沈澱を集め、ジエチルエーテルで洗った後乾燥させた。得られた活性エステル体(化合物[22])を、そのままターゲット・モデルオリゴヌクレオチド(完全相補:配列番号15)の標識に用いた。
【0231】
【化22】
【化23】
また、同様の方法でアズレン色素活性エステルである化合物[23]で、上記の1塩基ミスマッチの(配列番号:16)、2塩基ミスマッチの(配列番号:17)オリゴヌクレオチドのアミノ化物を標識した(化合物[24])。
【0234】
【化24】
【化25】
4.完全相補オリゴヌクレオチドのスクリーニング
工程2で得られたプローブ・オリゴヌクレオチド担持PHA磁性構造体微粒子と、工程3で得られた標識化ターゲット・オリゴヌクレオチドを、プローブ/ターゲット量比が理論上1/10になるよう調整し、80℃で2分間保持した後、時間をかけて室温に戻した(ハイブリダイゼーション条件)。
【0237】
磁力により微粒子を分離し、780nmのレーザーで励起して、蛍光光度計で測定したところ、化学式[21]の化合物に由来する、820nm付近にピークを持つ蛍光が見られ、配列番号:15のターゲットDNAが、選択的に回収されていることが確認された。
【0238】
実施例4.抗アルキルフェノール抗体担持カルボキシ型PHA磁性構造体微粒子を用いたノニルフェノールの回収
1.抗アルキルフェノール抗体のカルボキシ型PHA磁性構造体微粒子への担持
参考例8で作製したカルボキシ型PHA磁性構造体微粒子のPHA側鎖上のカルボキシ基を、常法に従って、N−エチル−N’−(ジメチルアミノプロピル)カルボジイミド(EDC)およびN−ヒドロキシスクシンイミド(NHS)で活性化し、理論上2倍量の抗アルキルフェノール抗体(和光純薬製)を反応させて、抗アルキルフェノール抗体を担持したPHA磁性構造体微粒子を得た。
【0239】
2.ノニルフェノールの回収、検出
和光純薬製「アルキルフェノールELISAキット」の手法に準じ、ノニルフェノール標準原液と抗原酵素複合体溶解液を混合し、工程1で得られた抗アルキルフェノール抗体担持PHA磁性構造体微粒子を加えて、容器の外側から密着させた電磁石のスイッチ切換えにより、反応を促進した。本操作を30℃で30分間行った後、電磁石のスイッチをオンにした状態で微粒子を容器内壁面に捕捉し、反応液を除去した。次いで、容器に洗浄液を加え、電磁石のスイッチ切換えにより、洗浄を促進した。本洗浄工程を、洗浄液を置換して5分ずつ3回行った後、発色基質溶液を加えて、吸光度を測定したところ450nmに強い吸収が見られ、本方法により、ノニルフェノールが回収され、その検出が行えることが示された。
【0240】
実施例5.長鎖アルカン担持ブロモ型PHA磁性構造体微粒子を用いたリポソームの分離・回収
1.ドデカンチオールを用いたブロモ型PHA磁性構造体微粒子へのドデカン担持
1−ドデカンチオールをヘキサンに溶解し、ヨウ化ナトリウム、炭酸カリウムおよびジエチルアミンを加えて、参考例9で作製したブロモ型PHA磁性構造体微粒子と混合し、室温で20時間攪拌することで、スルフィド結合(−S−)を介したドデカン担持PHA磁性構造体微粒子を得た。
【0241】
2.蛍光リポソームの回収・検出
Sigma社が製造販売している「Liopsome Kit」を用い、そのプロトコルに従って、内部にFITCを含んだリポソームを作製し、工程1で得られたドデカン担持PHA磁性構造体微粒子と攪拌・混合した。混合15分後、プローブ型電磁石を容器内に装入し、プローブ型電磁石用電源スイッチをオンにして、微粒子をプローブ型電磁石上に捕捉し、次いで、反応容器より洗浄液の入った容器へ移し、電源スイッチをオフにして微粒子を解放し、15分攪拌した。この洗浄操作を3回繰り返した後、蛍光光度計で測定したところ、FITCに基づく520nmに極大を有する蛍光が確認され、本方法により、リポソームが効率よく回収され、検出できることが示された。
【0242】
実施例6.コンセンサス結合配列遺伝子断片担持カルボキシ型PHA磁性構造体微粒子を用いた転写因子タンパク質の分離・回収
塩基性のロイシンジッパーを持った転写因子であるATF−2とそのコンセンサス結合配列との親和性を利用して、ATF−2の分離・回収を試みた。
【0243】
1.末端チオール型ATF−2コンセンサス結合配列DNA断片の合成
DNA自動合成機を用いて、配列番号:18の一本鎖核酸を合成した。なお、配列番号:18の一本鎖DNAの5’末端には、DNA自動合成機での合成時にチオール・モディファイア(Thiol−Modifier)(グレンリサーチ(GlenResearch)社製)を用いることによって、スルファニル基(−SH)を導入した(化合物[25])。続いて、通常の脱保護によりDNAを回収し、高速液体クロマトグラフィーにて精製し、以下の実験に用いた。
配列番号:18: 5’−TGACATCA−3’
【0244】
【化26】
2.末端チオール化DNA断片のカルボキシ型PHA磁性構造体微粒子への担持
参考例8で作製したカルボキシ型PHA磁性構造体微粒子のPHA側鎖上のカルボキシ基を、常法に従って、N−エチル−N’−(ジメチルアミノプロピル)カルボジイミド(EDC)およびN−ヒドロキシスクシンイミド(NHS)で活性化し、ホウ酸緩衝液(pH8.5:NaOHにて調整)に溶解した2−(2−ピリジニルジチオ)エタンアミン(PDEA)により、チオールに対して活性化した後、工程1で得られた末端チオール化DNA断片を加えて、担持を行った。なお、反応終了後、PHA磁性構造体微粒子上に残留する活性基は、システイン−NaClで不活性化した。
【0246】
3.結合反応、標識化
クロンテック社のプロトコルに従い、AFT−2のPBS溶液中に工程2で得られたDNA断片担持PHA磁性構造体微粒子を導入し、実施例4と同様の方法で、磁石によりDNA断片とAFT−2の親和性に起因する結合反応を促進した。反応終了後、実施例4と同様の方法で洗浄し、常法によりFITC標識された抗AFT−2抗体(ポリクローナル)溶液中で、抗原・抗体反応を行った。
【0247】
4.洗浄および検出
抗原・抗体反応終了後、実施例4の方法で、磁石を用いた操作により洗浄を行い、蛍光検出を行ったところ、標識FITCに基づく520nmに極大を有する蛍光が確認され、本方法により、転写因子タンパク質−コンセンサス配列DNA断片の親和性結合に基づいた回収がなされ、検出できることが示された。
【0248】
【発明の効果】
本発明では、担体表面を被覆する高分子材料層として、生体親和性の高いポリマー材料である、ポリヒドロキシアルカノエート、例えば、3−ヒドロキシアルカン酸ユニットを含有するポリヒドロキシアルカノエートを用いることにより、より生体条件に近い条件で、検体中の標的物質の分離、検出を効率よく行うことができる。
【0249】
【配列表】
【図面の簡単な説明】
【図1】本発明において利用される、担体(磁性体)表面を被覆するPHA上に標的成分に対して結合親和力を有する分子を担持した、標的成分結合分子担持PHA構造体の構成を模式的に示す図である。
【図2】本発明における、標的成分結合分子担持PHA構造体に対する、標的成分と標的成分結合分子との選択的な結合形成過程を模式的に示す図である。
【図3】本発明における、標的成分との選択的な結合を形成した標的成分結合分子担持PHA磁性構造体の、磁力を発揮する構造体を利用した磁気的分離過程を模式的に示す図である。
【符号の説明】
1 担体基体(磁性体)
2 ポリヒドロキシアルカノエート被覆層
3 ポリヒドロキシアルカノエート中の標的成分に対して結合親和力を有する分子を選択的に担持する部位
4 標的成分に対して結合親和力を有する分子
5 標的成分
6、7 標的成分以外の混在成分
8 磁力を発揮する構造体(永久磁石や電磁石等)
Claims (11)
- 検体中に含まれる標的成分を分離する方法であって、
前記標的成分に対して結合親和力を有する分子が、その表面に固定された担体を用意する工程と、
前記担体と前記検体とを混合する工程と、
前記混合工程において混合される、前記検体中に含まれる標的成分と、前記担体表面に固定されている前記結合親和力を有する分子とを結合させる工程と、
前記結合工程により、前記結合親和力を有する分子との結合を介して、前記担体に固定化された前記標的成分を、前記担体とともに前記検体中から分離する工程とを有し、
前記担体は、磁性体を含んでなる担体であり、
前記担体は、ポリヒドロキシアルカノエートで少なくとも一部を被覆されており、
前記標的成分に対して結合親和力を有する分子の、前記担体表面への固定は、
前記担体表面の少なくとも一部を被覆されている、前記ポリヒドロキシアルカノエートとして、エポキシ基、アミノ基、カルボキシ基、およびハロゲン分子からなる群から選択される官能基を一つ以上有するポリヒドロキシアルカノエートを用い、
前記ポリヒドロキシアルカノエートの有する官能基の少なくとも一つ以上を利用して、前記標的成分に対して結合親和力を有する分子を固定化する
ことを特徴とする標的成分の分離方法。 - 前記ポリヒドロキシアルカノエートは、少なくとも、その側鎖上に前記官能基を有している3−ヒドロキシアルカン酸ユニットを含有するポリヒドロキシアルカノエートである
ことを特徴とする請求項1に記載の方法。 - 前記結合親和力を有する分子との結合を介して、前記担体に固定化された前記標的成分を、前記担体とともに前記検体中から分離する工程において、
前記磁性体を含んでなる担体に対して、磁界を作用させて、前記検体より磁気的に分離する工程を有する
ことを特徴とする請求項1または2に記載の方法。 - 前記担体表面に固定される、前記標的成分に対して結合親和力を有する分子は、
核酸、タンパク質、ペプチド、糖鎖、脂質、低分子化合物、およびこれらの複合体からなる群より選択される一種類以上を含んでいる分子である
ことを特徴とする請求項1〜3のいずれか一項に記載の方法。 - 前記検体中から分離される前記標的成分は、
核酸、タンパク質、ペプチド、糖鎖、脂質、低分子化合物、およびこれらの複合体からなる群より選択される一種類以上を含んでいる成分である
ことを特徴とする請求項1〜4のいずれか一項に記載の方法。 - 前記磁性体を含んでなる担体の形状は、粒状である
ことを特徴とする請求項1〜5のいずれか一項に記載の方法。 - 前記磁性体を含んでなる担体の形状は、平板状またはフィルム状である
ことを特徴とする請求項1〜5のいずれか一項に記載の方法。 - 前記磁性体を含んでなる担体中の磁性体は、磁性を有する金属または金属化合物からなる磁性体である
ことを特徴とする請求項1〜7のいずれか一項に記載の方法。 - 検体中に含まれる標的成分を検出する方法であって、
前記標的成分に対して結合親和力を有する分子が、その表面に固定された担体を用意する工程と、
前記担体と前記検体とを混合する工程と、
前記混合工程において混合される、前記検体中に含まれる標的成分と、前記担体表面に固定されている前記結合親和力を有する分子とを結合させる工程と、
前記結合工程により、前記結合親和力を有する分子との結合を介して、前記担体に固定化された前記標的成分について、選択的に検出する工程とを有し、
前記担体は、磁性体を含んでなる担体であり、
前記担体は、ポリヒドロキシアルカノエートで少なくとも一部を被覆されており、
前記標的成分に対して結合親和力を有する分子の、前記担体表面への固定は、
前記担体表面の少なくとも一部を被覆されている、前記ポリヒドロキシアルカノエートとして、エポキシ基、アミノ基、カルボキシ基、およびハロゲン分子からなる群から選択される官能基を一つ以上有するポリヒドロキシアルカノエートを用い、
前記ポリヒドロキシアルカノエートの有する官能基の少なくとも一つ以上を利用して、前記標的成分に対して結合親和力を有する分子を固定化する
ことを特徴とする標的成分の検出方法。 - 前記ポリヒドロキシアルカノエートは、少なくとも、その側鎖上に前記官能基を有している3−ヒドロキシアルカン酸ユニットを含有するポリヒドロキシアルカノエートである
ことを特徴とする請求項9に記載の方法。 - 前記磁性体を含んでなる担体中の磁性体は、磁性を有する金属または金属化合物からなる磁性体である
ことを特徴とする請求項9または10に記載の方法。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2003127363A JP4371694B2 (ja) | 2003-05-02 | 2003-05-02 | 標的成分の分離方法、検出方法、スクリーニング方法 |
| US10/544,942 US20070003975A1 (en) | 2003-05-02 | 2004-05-06 | Structured construct and producing method therefor |
| PCT/JP2004/006420 WO2004097417A1 (en) | 2003-05-02 | 2004-05-06 | Structured construct and producing method therefor |
| US12/201,338 US20090029423A1 (en) | 2003-05-02 | 2008-08-29 | Method for separating target component |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2003127363A JP4371694B2 (ja) | 2003-05-02 | 2003-05-02 | 標的成分の分離方法、検出方法、スクリーニング方法 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2004333232A JP2004333232A (ja) | 2004-11-25 |
| JP2004333232A5 JP2004333232A5 (ja) | 2006-06-22 |
| JP4371694B2 true JP4371694B2 (ja) | 2009-11-25 |
Family
ID=33503936
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2003127363A Expired - Fee Related JP4371694B2 (ja) | 2003-05-02 | 2003-05-02 | 標的成分の分離方法、検出方法、スクリーニング方法 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP4371694B2 (ja) |
Families Citing this family (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006084461A1 (en) * | 2005-02-11 | 2006-08-17 | Merck Patent Gmbh | Solid-phase oligosaccharide tagging: a technique for manipulation of immobilized carbohydrates |
| JP4883640B2 (ja) | 2005-05-26 | 2012-02-22 | 独立行政法人科学技術振興機構 | 新規糖固定化金属ナノ粒子およびこれを用いて糖−タンパク質相互作用を測定する方法、並びに糖−タンパク質相互作用体からタンパク質を回収する方法 |
| JP4897581B2 (ja) * | 2007-06-12 | 2012-03-14 | 積水化学工業株式会社 | 親水化磁性体内包粒子の製造方法、親水化磁性体内包粒子及び免疫測定用粒子 |
| JP5504587B2 (ja) * | 2008-05-27 | 2014-05-28 | 東レ株式会社 | 分析用チップ |
| JP6111569B2 (ja) * | 2012-09-04 | 2017-04-12 | Jsr株式会社 | 無機材料で構成される表面用の表面処理剤、該表面処理剤でコーティングされた器具、その製造方法、及び新規重合体 |
| JP6085983B2 (ja) * | 2013-02-07 | 2017-03-01 | Jsr株式会社 | ブロッキング剤、標的物質に対する抗原または抗体が固定化された担体、これを含む体外診断用試薬およびキット、並びに標的物質の検出方法 |
| JPWO2014126111A1 (ja) * | 2013-02-13 | 2017-02-02 | Jsr株式会社 | 担体、当該担体の製造方法、生体物質の免疫学的測定方法 |
| JP2013226149A (ja) * | 2013-06-10 | 2013-11-07 | Universal Bio Research Co Ltd | 生体関連物質の分離回収方法 |
| JP6269773B2 (ja) * | 2016-10-06 | 2018-01-31 | Jsr株式会社 | 無機材料で構成される表面用の表面処理剤、該表面処理剤でコーティングされた器具、その製造方法、及び新規重合体 |
| JP2017044705A (ja) * | 2016-11-21 | 2017-03-02 | 国立大学法人北海道大学 | 糖鎖アレイ |
| JP7324998B2 (ja) * | 2018-04-25 | 2023-08-14 | パナソニックIpマネジメント株式会社 | センサ基板、センサ基板の製造方法および検出装置 |
| CN112305154B (zh) * | 2020-10-16 | 2022-09-23 | 中石化石油工程技术服务有限公司 | 一种膨润土吸蓝量的自动分析检测仪及其检测方法 |
| CN114958748B (zh) * | 2022-04-24 | 2024-02-13 | 东北大学 | 高效捕获和无损释放循环肿瘤细胞的纳米磁性亲和材料 |
| CN114990095B (zh) * | 2022-06-28 | 2024-05-24 | 天津天隆江大生物科技有限公司 | 一种生产抗冻蛋白多肽的复合酶制剂及其应用 |
-
2003
- 2003-05-02 JP JP2003127363A patent/JP4371694B2/ja not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| JP2004333232A (ja) | 2004-11-25 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4078247B2 (ja) | 磁性体−生体物質複合体型構造体、磁性体に対して結合能を有するアミノ酸配列を有するペプチド断片及びその遺伝子、ならびに磁性体−生体物質複合体型構造体の製造方法 | |
| JP4371694B2 (ja) | 標的成分の分離方法、検出方法、スクリーニング方法 | |
| US20090029423A1 (en) | Method for separating target component | |
| KR20030013244A (ko) | 폴리히드록시알카노에이트 함유 구조체 및 그 제조방법 | |
| KR101273964B1 (ko) | 단백질 g다합체가 고정화된 자성실리카 나노입자 및 이의 제조방법과 이를 이용한 검출용 면역센서 및 이를 이용한 면역검출방법 | |
| Urusov et al. | Application of magnetic nanoparticles in immunoassay | |
| EP2089714B1 (en) | Magnetic recognition system | |
| US7919263B2 (en) | Organic material-immobiling structure and method for production of the same, and peptide and DNA therefor | |
| JP2006327962A (ja) | 目的物質の分離方法および分子コンプレックス | |
| KR20020083519A (ko) | 구조체 및 그 제조방법 | |
| US6960457B1 (en) | Reversible immobilization of arginine-tagged moieties on a silicate surface | |
| JP4073034B2 (ja) | 磁性体−生体物質複合体型構造体、磁性体に対して結合能を有するアミノ酸配列を有するペプチド断片及びその遺伝子、ならびに磁性体−生体物質複合体型構造体の製造方法 | |
| Kunc et al. | Polycarboxylated Dextran as a Multivalent Linker: Synthesis and Target Recognition of the Antibody–Nanoparticle Bioconjugates in PBS and Serum | |
| JP4579502B2 (ja) | 構造体及びその製造方法、該構造体を含むトナー並びにそれを用いた画像形成方法及び装置 | |
| KR20150101067A (ko) | 암세포 바이오마커 검출용 패턴화된 탄소 나노튜브 바이오센서 | |
| WO2009133202A1 (en) | Set of magnetic labels | |
| Elaissari et al. | Biomedical application for magnetic latexes | |
| JP2007228909A (ja) | 標的成分の分離方法およびキット | |
| US20100075394A1 (en) | Method of Crosslinking Two Objects of Interest | |
| WO2010060209A1 (en) | Single domain antibody - targeted nanoparticle architectures for increased pathogen detection specificity and sensitivity | |
| Mary et al. | Phage-based Biosensors | |
| EP2474612A1 (en) | Affinity separation means, and uses thereof to separate, purify or concentrate a target molecule | |
| Elaissari et al. | Latexes: Magnetic | |
| WO1999012036A1 (en) | Reversible immobilization of arginine-tagged moieties on a silicate surface | |
| JP2004335622A (ja) | 構造体及びその製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20060501 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20060501 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20090527 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20090727 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20090826 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20090901 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120911 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120911 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130911 Year of fee payment: 4 |
|
| LAPS | Cancellation because of no payment of annual fees |