JP2019123770A - ブテン系樹脂組成物および成形体 - Google Patents
ブテン系樹脂組成物および成形体 Download PDFInfo
- Publication number
- JP2019123770A JP2019123770A JP2018003486A JP2018003486A JP2019123770A JP 2019123770 A JP2019123770 A JP 2019123770A JP 2018003486 A JP2018003486 A JP 2018003486A JP 2018003486 A JP2018003486 A JP 2018003486A JP 2019123770 A JP2019123770 A JP 2019123770A
- Authority
- JP
- Japan
- Prior art keywords
- butene
- mass
- resin composition
- polybutene
- olefin copolymer
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Images


Classifications
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A30/00—Adapting or protecting infrastructure or their operation
Abstract
Description
[1]
下記要件(A1)〜(A2)を満たすブテン・α−オレフィン共重合体(A)を70〜99.9質量部、および下記要件(B1)〜(B4)を満たすポリブテン−1(B)を0.1〜30質量部(ただし、ブテン・α−オレフィン共重合体(A)およびポリブテン−1(B)の合計を100質量部とする。)を含むブテン系樹脂組成物。
(A1):1−ブテン由来の構成単位の含有量が80.0〜99.9モル%であり、エチレンおよび炭素数3〜20の1−ブテン以外のα−オレフィンから選ばれる少なくとも1種のオレフィン由来の構成単位の総含有量(K)が0.1〜20.0モル%である。(ただし、1−ブテン由来の構成単位の含有量と前記総含有量(K)との合計を100モル%とする)
(A2):135℃のデカリン中で測定された極限粘度[η]が0.5〜5.5dl/gである。
(B1):13C−NMRにより測定されたアイソタクティックペンタッド分率が94.0%以上である。
(B2):13C−NMRにより測定されたシンジオタクティックトライアッド分率が1.5%以下である。
(B3):135℃のデカリン中で測定された極限粘度[η]が0.5〜5.5dl/gである。
(B4):ゲルパーミエーションクロマトグラフィー(GPC)で測定した、分子量が10,000以下の成分の割合が、1.3質量%未満である。
前記[1]のブテン系樹脂組成物からなる成形体。
[3]
パイプまたはパイプ継手である前記[2]の成形体。
[ブテン系樹脂組成物]
本発明に係るブテン系樹脂組成物は、後述する要件(A1)〜(A2)を満たすブテン・α−オレフィン共重合体(A)を70〜99.9質量部、および後述する要件(B1)〜(B4)を満たすポリブテン−1(B)を0.1〜30質量部(ただし、ブテン・α−オレフィン共重合体(A)およびポリブテン−1(B)の合計を100質量部とする)を含んでいる。
〔(A)ブテン・α−オレフィン共重合体〕
ブテン・α−オレフィン共重合体(A)は、以下に説明する要件(A1)〜(A2)を充足し、好ましくは、以下に説明する要件(A3)〜(A4)からなる群から選ばれる少なくとも1つの要件をさらに充足し、より好ましくは以下に説明する要件(A3)および(A4)を充足する。
要件(A1)は、1−ブテン由来の構成単位の含有量が80.0〜99.9モル%であり、エチレンおよび炭素数3〜20の1−ブテン以外のα−オレフィンから選ばれる少なくとも1種のオレフィン由来の構成単位の総含有量(K)が0.1〜20.0モル%である(ただし、1−ブテン由来の構成単位の含有量と前記総含有量(K)との合計を100モル%とする。)というものである。
前記ブテン・α−オレフィン共重合体(A)中の各構成単位の含有量は、例えば、重合反応中に添加するそれぞれのオレフィン(例:1−ブテン、エチレン、炭素数3〜20の1−ブテン以外のα−オレフィン)の量によって調整することができる。
前記ブテン・α−オレフィン共重合体(A)は、本発明の効果が損なわれない限り、エチレンおよび炭素数3〜20のα−オレフィン以外の、他のモノマーに由来する構成単位を有してもよい。他のモノマーに由来する構成単位の含有量の上限値は、1−ブテン由来の構成単位の含有量と前記総含有量(K)との合計100質量%に対して、例えば5質量以下である。
要件(A2)は、135℃のデカリン中で測定された極限粘度[η]が0.5〜5.5dl/gであるというものである。
要件(A3)は、後述する実施例で採用した条件下で13C−NMRにより測定されたアイソタクティックペンタッド分率(以下「mmmm分率」とも記載する。)が90.0%以上94.0%以下であるというものである。
要件(A4)は、後述する実施例で採用した条件下で13C−NMRにより測定されたシンジオタクティックトライアッド分率(以下「rr分率」とも記載する。)が1.5%以上3.0%以下であるというものである。
前記rr分率は、ブテン・α−オレフィン共重合体(A)を重合する際の重合用触媒の選択、重合温度等の重合条件の調整などによって、調整することができる。
前記ブテン・α−オレフィン共重合体(A)は、従来公知の触媒、例えば、バナジウム系触媒、チタン系触媒、マグネシウム担持型チタン触媒、または国際公開第01/53369号、国際公開第01/27124号、もしくは特開平3−193796号公報中に記載のメタロセン触媒などを用いて、従来公知の重合法により製造することができる。
本発明に係るブテン系樹脂組成物は、ポリブテン−1(1−ブテン単独重合体)(B)を含有する。ポリブテン−1(B)は、以下に説明する要件(B1)〜(B4)を充足する。
要件(B1)は、後述する実施例で採用した条件下で13C−NMRにより測定されたアイソタクティックペンタッド分率(mmmm分率)が94.0%以上であるというものである。
前記mmmm分率は、ポリブテン−1(B)を重合する際の重合用触媒を適切に選択し、重合温度等の重合条件を設定することにより上記範囲内に調整できる。
要件(B2)は、後述する実施例で採用した条件下で13C−NMRにより測定されたシンジオタクティックトライアッド分率(rr分率)が1.5%以下であるというものである。
前記rr分率は、ポリブテン−1(B)を重合する際の重合用触媒を適切に選択し、重合温度等の重合条件を適切に設定することにより上記範囲内に調整できる。
要件(B3)は、135℃のデカリン中で測定された極限粘度[η]が0.5〜5.5dl/gであるというものである。
前記極限粘度[η]は、好ましくは0.7〜5.0dl/g、より好ましくは0.7〜4.5dl/g、さらに好ましくは0.7〜4.0dl/g、特に好ましくは0.7〜2.8dl/gである。
要件(B4)は、ポリブテン−1(B)の、ゲルパーミエーションクロマトグラフィー(GPC;標準ポリスチレン換算)で測定した、分子量が10,000以下の成分の割合が、1.3質量%未満であるというものである。この割合は好ましくは0.9質量%以下である。下限値は、たとえば0.1質量%であってもよい。
分子量が10,000以下の成分の割合は、ポリブテン−1(B)を重合する際の重合用触媒を適切に選択することで上記範囲内に調整できる。
前記ポリブテン−1(B)は、従来公知の方法、たとえば国際公開第2017/130895号の[0059]〜[0144]に開示された方法において、原料モノマーとして1−ブテンのみを使用することで製造することができる。
本発明に係るブテン系樹脂組成物は、前記ブテン・α−オレフィン共重合体(A)を70〜99.9質量部、および前記ポリブテン−1(B)を0.1〜30質量部(ただし、ブテン・α−オレフィン共重合体(A)およびポリブテン−1(B)の合計を100質量部とする)を含んでいる。
本発明のブテン系樹脂組成物の結晶化度を評価する手法として、例えばサンプル間の融解熱量ΔHを比較する方法がある。ΔHは示差走査型熱量測定(昇温速度:10℃/分)によって決定される。
本発明のブテン系樹脂組成物は、上述したブテン・α−オレフィン共重合体(A)およびポリブテン−1(B)を含む。
ブテン・α−オレフィン共重合体(A)およびポリブテン−1(B)の合計の含有量は、本発明のブテン系樹脂組成物の総質量に対して、0.1〜99.999質量%であることが好ましく、下限値は好ましくは20質量%であり、一実施態様として、ブテン・α−オレフィン共重合体(A)の含有量の下限値は、例えば40質量%、さらに好ましくは60質量%、特に好ましくは80質量%、最も好ましくは85質量%である。
他の重合体(C)としては、熱可塑性樹脂を広く用いることができる。他の重合体(C)の含有量は、本発明のブテン系樹脂組成物の総質量に対して、0.001〜99.9質量%であることが好ましく、上限値は好ましくは80質量%であり、一実施態様として、他の重合体(C)の含有量の上限値は、より好ましくは60質量%、さらに好ましくは40質量%、特に好ましくは20質量%、最も好ましくは15質量%である。
熱可塑性ポリオレフィン系樹脂:例えば、低密度、中密度、高密度ポリエチレン、高圧法低密度ポリエチレン等のポリエチレン、アイソタクティックポリプロピレン、シンジオタクティックポリプロピレン等のポリプロピレン、ポリブテン−1(B)以外のポリ1−ブテン、ポリ4−メチル−1−ペンテン、ポリ3−メチル−1−ペンテン、ポリ3−メチル−1−ブテン、エチレン・α−オレフィン共重合体、プロピレン・α−オレフィン共重合体、1−ブテン・α−オレフィン共重合体、4−メチル−1−ペンテン・α−オレフィン共重合体、環状オレフィン共重合体、塩素化ポリオレフィン、およびこれらのオレフィン系樹脂を変性した変性ポリオレフィン樹脂(上述した熱可塑性ポリオレフィン系樹脂のうち、例えばポリエチレン、ポリプロピレンは結晶核剤として用いることもでき、その場合の好ましい含有量はブテン系樹脂組成物の総質量に対して、0.001〜5質量%である。)、
熱可塑性ポリアミド系樹脂:例えば、脂肪族ポリアミド(ナイロン6、ナイロン11、ナイロン12、ナイロン66、ナイロン610、ナイロン612)、
熱可塑性ポリエステル系樹脂:例えば、ポリエチレンテレフタレート、ポリブチレンテレフタレート、ポリエステル系エラストマー、
熱可塑性ビニル芳香族系樹脂:例えば、ポリスチレン、ABS樹脂、AS樹脂、スチレン系エラストマー(スチレン・ブタジエン・スチレンブロックポリマー、スチレン・イソプレン・スチレンブロックポリマー、スチレン・イソブチレン・スチレンブロックポリマー、これらの水素添加物)、
熱可塑性ポリウレタン;塩化ビニル樹脂;塩化ビニリデン樹脂;アクリル樹脂;エチレン・酢酸ビニル共重合体等の酢酸ビニル共重合体;エチレン・メタクリル酸アクリレート共重合体;アイオノマー;エチレン・ビニルアルコール共重合体;ポリビニルアルコール;フッ素系樹脂;ポリカーボネート;ポリアセタール;ポリフェニレンオキシド;ポリフェニレンサルファイドポリイミド;ポリアリレート;ポリスルホン;ポリエーテルスルホン;ロジン系樹脂;テルペン系樹脂および石油樹脂;
共重合体ゴム:例えば、エチレン・α−オレフィン・ジエン共重合体、プロピレン・α−オレフィン・ジエン共重合体、1−ブテン・α−オレフィン・ジエン共重合体、ポリブタジエンゴム、ポリイソプレンゴム、ネオプレンゴム、ニトリルゴム、ブチルゴム、ポリイソブチレンゴム、天然ゴム、シリコーンゴム;
等が例示される。
他の重合体(C)は1種単独で用いてもよく、2種以上を併用してもよい。
添加剤(D)としては、例えば、核剤、アンチブロッキング剤、顔料、染料、充填剤、滑剤、可塑剤、離型剤、酸化防止剤、難燃剤、紫外線吸収剤、抗菌剤、界面活性剤、帯電防止剤、耐候安定剤、耐熱安定剤、スリップ防止剤、発泡剤、結晶化助剤、防曇剤、老化防止剤、塩酸吸収剤、衝撃改良剤、架橋剤、共架橋剤、架橋助剤、粘着剤、軟化剤、加工助剤が挙げられる。
添加剤(D)の含有量は、本発明の目的を損なわない範囲内で用途に応じて、特に限定されないが、本発明のブテン系樹脂組成物の総質量に対して、配合される添加剤それぞれについて0.001〜30質量%であることが好ましい。
抗菌剤としては、例えば、4級アンモニウム塩、ピリジン系化合物、有機酸、有機酸エステル、ハロゲン化フェノール、有機ヨウ素が挙げられる。
有機ペルオキシドとしては、例えば、ジクミル有機ペルオキシド、ジ−tert−ブチル有機ペルオキシド、2,5−ジメチル−2,5−ジ−(tert−ブチルペルオキシ)ヘキサン、2,5−ジメチル−2,5−ジ−(tert−ブチルペルオキシ)ヘキシン−3、1,3−ビス(tert−ブチルペルオキシイソプロピル)ベンゼン、1,1−ビス(tert−ブチルペルオキシ)−3,3,5−トリメチルシクロヘキサン、n−ブチル−4,4−ビス(tert−ブチルペルオキシ)バレレート、ベンゾイル有機ペルオキシド、p−クロロベンゾイルペルオキシド、2,4−ジクロロベンゾイル有機ペルオキシド、tert−ブチルペルオキシベンゾエート、tert−ブチルペルベンゾエート、tert−ブチルペルオキシイソプロピルカーボネート、ジアセチル有機ペルオキシド、ラウロイル有機ペルオキシド、tert−ブチルクミル有機ペルオキシドが挙げられる。
グラフト変性に用いられる極性モノマーとしては、例えば、水酸基含有エチレン性不飽和化合物、アミノ基含有エチレン性不飽和化合物、エポキシ基含有エチレン性不飽和化合物、芳香族ビニル化合物、不飽和カルボン酸またはその誘導体、ビニルエステル化合物、塩化ビニル、カルボジイミド化合物が挙げられる。特に、不飽和カルボン酸またはその誘導体が好ましい。不飽和カルボン酸またはその誘導体としては、カルボン酸基を1以上有する不飽和化合物、カルボン酸基を有する化合物とアルキルアルコールとのエステル、無水カルボン酸基を1以上有する不飽和化合物が挙げられる。不飽和基としては、例えば、ビニル基、ビニレン基、不飽和環状炭化水素基が挙げられる。
被変性体の極性モノマーによるグラフト変性は、従来公知の方法で行うことができ、例えば被変性体を有機溶媒に溶解し、次いで極性モノマーおよびラジカル開始剤などを溶液に加え、通常70〜200℃、好ましくは80〜190℃の温度で、通常0.5〜15時間、好ましくは1〜10時間反応させることにより行うことができる。
本発明のブテン系樹脂組成物の製造方法は特に限定されないが、例えば、前記ブテン・α−オレフィン共重合体(A)と、前記ポリブテン−1(B)と、必要に応じて任意成分とを上述の添加割合で混合したのち、溶融混練するか、または良溶媒中で溶解させ十分攪拌したのちに溶媒を乾燥させる方法があげられる。
本発明のブテン系樹脂組成物から形成された各種成形体は、従来公知のポリオレフィン用途に広く用いることができる。成形体は、例えば、押出成形、射出成形、インフレーション成形、ブロー成形、押出ブロー成形、射出ブロー成形、プレス成形、真空成形、パウダースラッシュ成形、カレンダー成形、発泡成形等の公知の熱成形方法により得られる。
[遷移金属錯体の合成]
国際公開第2014/050817号の合成例4に従い、(8‐オクタメチルフルオレン-12'-イル-(2-(アダマンタン-1-イル)-8-メチル-3,3b,4,5,6,7,7a,8-オクタヒドロシクロペンタ[a]インデン))ジルコニウムジクロライドを合成した。この化合物を「触媒(a)」とも記載する。
全圧を0.5MPaGとしたこと以外は特開2002−241553号公報の[製造例1]に記載のTi系触媒を用いた従来公知のブテン系重合体の重合方法により、評価用ポリマー(1−ブテン/プロピレン2元共重合体)を合成した。
全圧を0.6MPaGに変更したこと以外は比較例1と同様の操作を行い、評価用ポリマー(1−ブテン/プロピレン2元共重合体)を合成した。
充分に乾燥し窒素置換したシュレンク管に磁気攪拌子を入れ、メタロセン化合物として触媒(a)2.0μmolを入れ、修飾メチルアルミノキサンの懸濁液を触媒(a)に対して300当量分(n−ヘキサン溶媒、アルミニウム原子換算で0.60mmol)、攪拌しながら室温で加え、次いで触媒(a)が1μmol/mLとなる量のヘプタンを加えて、触媒液(a)を調製した。
冷却/脱圧したオートクレーブから取り出した重合液を、メタノール中に投入し、ポリマーを析出させて濾過回収した。その後回収したポリマーを80℃で12時間減圧乾燥して、評価用ポリマー(1−ブテン単独重合体)を得た。
充分に乾燥し窒素置換した内容積1,500mlのSUS製オートクレーブに、重合溶媒としてヘプタン500mLとトリイソブチルアルミニウムのヘキサン溶液(Al=0.5M、0.75mmol)1.5mLとを装入し、次いで850回転/分で撹拌しながら1−ブテン180gを加えた後に、重合温度60℃に昇温した。その温度で71.0NmLの水素を加えた後にオートクレーブ内圧が0.7MPaGになるまで窒素を加えた。
冷却/脱圧したオートクレーブから取り出した重合液を、メタノール中に投入し、ポリマーを析出させて濾過回収した。その後回収したポリマーを80℃で10時間減圧乾燥して、評価用ポリマー(1−ブテン単独重合体)を得た。
プロピレンを用いなかったこと以外は比較例1と同様の操作を行い、評価用ポリマー(1−ブテン単独重合体)を合成した。
比較例1で得られた1−ブテン/プロピレン共重合体と製造例3で得られた1−ブテン単独重合体とを質量比で75対25となるように磁気攪拌子を入れたサンプル瓶に秤量した。p−キシレン溶液を加え、140℃に設定したホットスターラーに置き溶解させ攪拌した。1時時間後、未溶解物がないことを確認し溶液をガラスシャーレに展開し室温で一晩放置した。翌日溶媒が蒸発した後の固形物をさらに120℃、窒素雰囲気において24時間乾燥させ、樹脂組成物を得た。
原料である樹脂およびその割合を、比較例1で得られた1−ブテン/プロピレン共重合体を95質量%および製造例1で得られた1−ブテン単独重合体を5質量%に変更したこと以外は比較例3と同様の操作を行い、樹脂組成物を得た。
原料である樹脂およびその割合を、比較例1で得られた1−ブテン/プロピレン共重合体を75質量%および製造例1で得られた1−ブテン単独重合体を25質量%に変更したこと以外は比較例3と同様の操作を行い、樹脂組成物を得た。
原料である樹脂およびその割合を、比較例1で得られた1−ブテン/プロピレン共重合体を75質量%および製造例2で得られた1−ブテン単独重合体を25質量%に変更したこと以外は比較例3と同様の操作を行い、樹脂組成物を得た。
原料である樹脂およびその割合を、比較例2で得られた1−ブテン/プロピレン共重合体を75質量%および製造例1で得られた1−ブテン単独重合体を25質量%に変更したこと以外は比較例3と同様の操作を行い、樹脂組成物を得た。
実施例等で得られたポリマーないし樹脂組成物の各種物性を、下記方法により測定した。結果を表1に示す。
ブテン・α−オレフィン共重合体中のプロピレン由来の構成単位(コモノマー単位)の含有量は、以下の装置および条件により、13C−NMRスペクトルより算出した。
コモノマー単位の含有量(モル%)=[P/(P+M)]×100
ここで、Pはコモノマー主鎖メチレンシグナルの全ピーク面積を示し、Mは1−ブテン主鎖メチレンシグナルの全ピーク面積を示す。
mmmm分率(%)は、下記式で表される。
mmmm分率(%)=Smmmm/S×100
ここで、13C−NMRスペクトルにおいて、Smmmmは27.50ppmをピークトップとするmmmm由来のブテン側鎖メチレンシグナルのピーク面積を示し、Sは29.0ppmから25.0ppmの範囲に現れる、ブテン側鎖メチレンシグナルの全ピーク面積を示す。
rr分率(%)は、下記式で表される。
rr分率(%)=Srr/S×100
ここで、13C−NMRスペクトルにおいて、Srrは26.6ppmから25.0ppmの範囲に現れるrr由来のブテン側鎖メチレンシグナルのピーク面積を示し、Sは29.0ppmから25.0ppmの範囲に現れる、ブテン側鎖メチレンシグナルの全ピーク面積を示す。
ブテン系重合体の極限粘度[η]は、デカリンを用いて、135℃で測定した。具体的には、前記重合で得られたブテン系重合体約20mgをデカリン15mlに溶解し、135℃のオイルバス中で比粘度ηspを測定した。このデカリン溶液にデカリンを5ml追加して希釈後、同様にして比粘度ηspを測定した。この希釈操作をさらに2回繰り返し、濃度(C)を0に外挿した時のηsp/Cの値を極限粘度として求めた(下式参照)。
[η]=lim(ηsp/C) (C→0)
ブテン系重合体の分子量10,000以下の成分の割合は、ゲルパーミエーションクロマトグラフィー(GPC)により、カラムとして東ソー株式会社製TSKgelGMH6−HT×2本およびTSKgelGMH6−HTL×2本(カラムサイズはいずれも直径7.5mm、長さ300mm)を直列接続した、液体クロマトグラフ(Waters製Alliance/GPC2000型)を用いて、測定した。移動相媒体は、o−ジクロロベンゼンおよび酸化防止剤としてBHT(武田薬品)0.025質量%を用い、試料濃度は0.15%(V/W)、流速1.0ml/分、140℃で測定を行った。標準ポリスチレンは、分子量が500〜20,600,000については東ソー社製を用いた。得られたクロマトグラムはWaters製データ処理ソフトEmpower2を用いて、公知の方法によって、標準ポリスチレンサンプルを使用した検量線を用いて解析することで、分子量10,000以下の成分の割合を算出した。
パーキンエルマ社製DSC測定装置(Diamond DSC)により、発熱・吸熱曲線を求め、昇温時の最大融解ピークの積算値から融解熱量ΔHを算出した。
パーキンエルマ社製DSC測定装置(Diamond DSC)を用いて、半結晶化時間τ1/2を測定した。30℃から320℃/分の設定速度で200℃まで昇温し、10分間保持した後、85または80℃まで降温速度320℃/分の設定速度で降温し、等温下での結晶化ピークを測定した。測定した結晶化ピークから、結晶化が始まった時点から、結晶化が半分促進する時点(全体の結晶化ピーク面積に対して、面積が1/2になる時点)での時間をτ1/2とした。
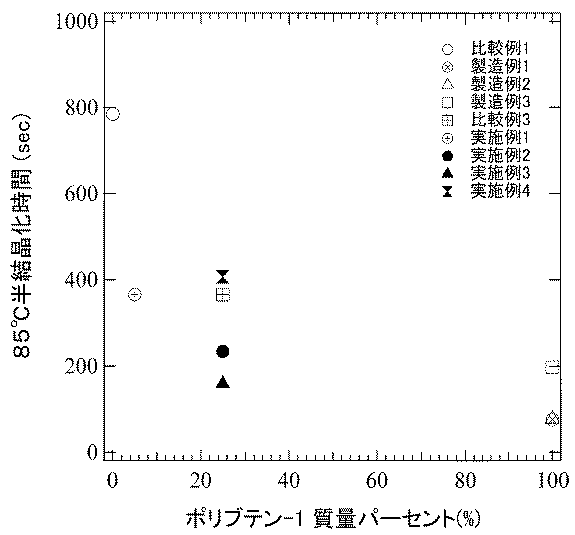
図1は、実施例等において、ポリブテン−1の割合(質量パーセント)に対して融解熱量ΔHをプロットしたものである。図中点線はブレンド物を形成する単身のブテン・プロピレン共重合体およびポリブテン−1の融解熱量ΔH値を結んだものである。それぞれのブレンド物で点線よりも大きな融解熱量ΔHを示すことは、単純な加成性が成り立たないことを示す。したがって、ポリブテン−1の添加には結晶化促進効果があると言え、特に、実施例で示すようなmmmm分率の値が大きく、rr分率の小さなポリブテン−1添加した際の効果が顕著である。
Claims (3)
- 下記要件(A1)〜(A2)を満たすブテン・α−オレフィン共重合体(A)を70〜99.9質量部、および下記要件(B1)〜(B4)を満たすポリブテン−1(B)を0.1〜30質量部(ただし、ブテン・α−オレフィン共重合体(A)およびポリブテン−1(B)の合計を100質量部とする)を含むブテン系樹脂組成物。
(A1):1−ブテン由来の構成単位の含有量が80.0〜99.9モル%であり、エチレンおよび炭素数3〜20の1−ブテン以外のα−オレフィンから選ばれる少なくとも1種のオレフィン由来の構成単位の総含有量(K)が0.1〜20.0モル%である。(ただし、1−ブテン由来の構成単位の含有量と前記総含有量(K)との合計を100モル%とする。)
(A2):135℃のデカリン中で測定された極限粘度[η]が0.5〜5.5dl/gである。
(B1):13C−NMRにより測定されたアイソタクティックペンタッド分率が94.0%以上である。
(B2):13C−NMRにより測定されたシンジオタクティックトライアッド分率が1.5%以下である。
(B3):135℃のデカリン中で測定された極限粘度[η]が0.5〜5.5dl/gである。
(B4):ゲルパーミエーションクロマトグラフィー(GPC)で測定した、分子量が10,000以下の成分の割合が、1.3質量%未満である。 - 請求項1に記載のブテン系樹脂組成物からなる成形体。
- パイプまたはパイプ継手である請求項2に記載の成形体。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2018003486A JP7044557B2 (ja) | 2018-01-12 | 2018-01-12 | ブテン系樹脂組成物および成形体 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2018003486A JP7044557B2 (ja) | 2018-01-12 | 2018-01-12 | ブテン系樹脂組成物および成形体 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2019123770A true JP2019123770A (ja) | 2019-07-25 |
| JP7044557B2 JP7044557B2 (ja) | 2022-03-30 |
Family
ID=67397950
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2018003486A Active JP7044557B2 (ja) | 2018-01-12 | 2018-01-12 | ブテン系樹脂組成物および成形体 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP7044557B2 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP4312191A1 (en) | 2022-07-29 | 2024-01-31 | FUJI-FILM Corporation | Image processing device, glasses-type information display device, image processing method, and image processing program |
| EP4312205A1 (en) | 2022-07-29 | 2024-01-31 | FUJIFILM Corporation | Control device, glasses-type information display device, control method, and control program |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002002659A1 (fr) * | 2000-07-03 | 2002-01-10 | Mitsui Chemicals, Inc. | Copolymere de butene, composition de resine renfermant ce copolymere et produits moules de cette composition, et catalyseur solide au titane pour la production du copolymere, ainsi que procede de preparation du catalyseur |
| WO2017130895A1 (ja) * | 2016-01-28 | 2017-08-03 | 三井化学株式会社 | ブテン系重合体、樹脂組成物および成形体 |
-
2018
- 2018-01-12 JP JP2018003486A patent/JP7044557B2/ja active Active
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002002659A1 (fr) * | 2000-07-03 | 2002-01-10 | Mitsui Chemicals, Inc. | Copolymere de butene, composition de resine renfermant ce copolymere et produits moules de cette composition, et catalyseur solide au titane pour la production du copolymere, ainsi que procede de preparation du catalyseur |
| WO2017130895A1 (ja) * | 2016-01-28 | 2017-08-03 | 三井化学株式会社 | ブテン系重合体、樹脂組成物および成形体 |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP4312191A1 (en) | 2022-07-29 | 2024-01-31 | FUJI-FILM Corporation | Image processing device, glasses-type information display device, image processing method, and image processing program |
| EP4312205A1 (en) | 2022-07-29 | 2024-01-31 | FUJIFILM Corporation | Control device, glasses-type information display device, control method, and control program |
Also Published As
| Publication number | Publication date |
|---|---|
| JP7044557B2 (ja) | 2022-03-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5020524B2 (ja) | プロピレン系重合体組成物、該組成物からなる成形体、プロピレン系重合体組成物からなるペレット、熱可塑性重合体用改質剤、熱可塑性重合体組成物の製造方法 | |
| JP7118137B2 (ja) | 4-メチル-1-ペンテン系重合体粒子および4-メチル-1-ペンテン系樹脂の製造方法 | |
| KR19990013979A (ko) | 불포화 공중합체, 이 공중합체의 제조방법 및 이 공중합체를 함유한 조성물 | |
| JP2020180279A (ja) | ブテン系重合体、樹脂組成物および成形体 | |
| JP2015193710A (ja) | 熱可塑性エラストマー組成物 | |
| JP5330637B2 (ja) | プロピレン系重合体組成物、該組成物からなる成形体、プロピレン系重合体組成物の製造方法 | |
| JP6560342B2 (ja) | ポリ−1−ブテン樹脂組成物およびそれから得られる成形体 | |
| JP7044557B2 (ja) | ブテン系樹脂組成物および成形体 | |
| JP2016147983A (ja) | ブテン系重合体、樹脂組成物および成形体 | |
| JP6514682B2 (ja) | グラフト変性プロピレン・α−オレフィン共重合体およびその製造方法 | |
| JP2017222850A (ja) | プロピレン系樹脂組成物およびその製造方法、ならびに該プロピレン系樹脂組成物を用いた成形体 | |
| JP5631696B2 (ja) | 4−メチル−1−ペンテン(共)重合体および該重合体から得られるブロー成形体 | |
| JP2019178254A (ja) | 1−ブテン共重合体、当該1−ブテン共重合体を含む重合体組成物および当該1−ブテン共重合体からなる成形体 | |
| JP2018115280A (ja) | 樹脂組成物および成形体 | |
| KR102649475B1 (ko) | 프로필렌·에틸렌·α-올레핀 공중합체, 프로필렌계 중합체 조성물 및 이들의 용도 | |
| JP7449705B2 (ja) | 4-メチル-1-ペンテン系樹脂組成物およびその用途 | |
| JP6890474B2 (ja) | プロピレン系樹脂組成物およびその製造方法、ならびに該プロピレン系樹脂組成物を用いた成形体 | |
| JP5858803B2 (ja) | インフレーションフィルム、インフレーションフィルムの製造方法、ブロー成形体、ブロー成形体の製造方法、熱成形体及び熱成形体の製造方法 | |
| JP7088689B2 (ja) | 4-メチル-1-ペンテン系樹脂組成物およびその成形体 | |
| JP2007063395A (ja) | ポリプロピレン系樹脂組成物、およびその組成物からなるフィルムまたはシート | |
| JP2009079110A (ja) | ポリプロピレン樹脂組成物およびその射出成形体 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20201023 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20210812 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20210817 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20211018 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20220301 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20220317 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 7044557 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |