JP2015532307A - 凝固因子viiポリペプチド - Google Patents
凝固因子viiポリペプチド Download PDFInfo
- Publication number
- JP2015532307A JP2015532307A JP2015536187A JP2015536187A JP2015532307A JP 2015532307 A JP2015532307 A JP 2015532307A JP 2015536187 A JP2015536187 A JP 2015536187A JP 2015536187 A JP2015536187 A JP 2015536187A JP 2015532307 A JP2015532307 A JP 2015532307A
- Authority
- JP
- Japan
- Prior art keywords
- factor vii
- polypeptide
- fviia
- factor
- antithrombin
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Withdrawn
Links
Images









Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/48—Hydrolases (3) acting on peptide bonds (3.4)
- C12N9/50—Proteinases, e.g. Endopeptidases (3.4.21-3.4.25)
- C12N9/64—Proteinases, e.g. Endopeptidases (3.4.21-3.4.25) derived from animal tissue
- C12N9/6421—Proteinases, e.g. Endopeptidases (3.4.21-3.4.25) derived from animal tissue from mammals
- C12N9/6424—Serine endopeptidases (3.4.21)
- C12N9/6437—Coagulation factor VIIa (3.4.21.21)
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/43—Enzymes; Proenzymes; Derivatives thereof
- A61K38/46—Hydrolases (3)
- A61K38/48—Hydrolases (3) acting on peptide bonds (3.4)
- A61K38/482—Serine endopeptidases (3.4.21)
- A61K38/4846—Factor VII (3.4.21.21); Factor IX (3.4.21.22); Factor Xa (3.4.21.6); Factor XI (3.4.21.27); Factor XII (3.4.21.38)
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/56—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule
- A61K47/59—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyureas or polyurethanes
- A61K47/60—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyureas or polyurethanes the organic macromolecular compound being a polyoxyalkylene oligomer, polymer or dendrimer, e.g. PEG, PPG, PEO or polyglycerol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/56—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule
- A61K47/61—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule the organic macromolecular compound being a polysaccharide or a derivative thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/62—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being a protein, peptide or polyamino acid
- A61K47/64—Drug-peptide, drug-protein or drug-polyamino acid conjugates, i.e. the modifying agent being a peptide, protein or polyamino acid which is covalently bonded or complexed to a therapeutically active agent
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/62—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being a protein, peptide or polyamino acid
- A61K47/64—Drug-peptide, drug-protein or drug-polyamino acid conjugates, i.e. the modifying agent being a peptide, protein or polyamino acid which is covalently bonded or complexed to a therapeutically active agent
- A61K47/643—Albumins, e.g. HSA, BSA, ovalbumin or a Keyhole Limpet Hemocyanin [KHL]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/62—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being a protein, peptide or polyamino acid
- A61K47/64—Drug-peptide, drug-protein or drug-polyamino acid conjugates, i.e. the modifying agent being a peptide, protein or polyamino acid which is covalently bonded or complexed to a therapeutically active agent
- A61K47/644—Transferrin, e.g. a lactoferrin or ovotransferrin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/04—Antihaemorrhagics; Procoagulants; Haemostatic agents; Antifibrinolytic agents
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y304/00—Hydrolases acting on peptide bonds, i.e. peptidases (3.4)
- C12Y304/21—Serine endopeptidases (3.4.21)
- C12Y304/21021—Coagulation factor VIIa (3.4.21.21)
Abstract
本発明は、凝固活性を有する修飾凝固因子VII(第VII因子)ポリペプチド、ならびにこのようなポリペプチドをコードするポリヌクレオチド構築物、このようなポリヌクレオチドを含むおよび発現するベクターおよび宿主細胞、医薬組成物、治療の使用および方法に関する。
Description
本発明は、プロコアグラント活性を有する修飾された凝固因子VII(第VII因子)ポリペプチドに関する。本発明はまた、このようなポリペプチドをコードするポリヌクレオチド構築物、このようなポリヌクレオチドを含むおよび発現するベクターおよび宿主細胞、このようなポリペプチドを含む医薬組成物、ならびにこのようなポリペプチドの治療の使用および方法にも関する。
配列リストの参照による組み込み
配列番号1:野生型ヒト凝固因子VII
配列番号1:野生型ヒト凝固因子VII
血管への損傷は、細胞成分と分子成分の間の複雑な相互作用を伴う止血系を活性化する。出血を最終的に止めさせる過程は、止血として知られている。止血の重要な部分は、血液の凝固および損傷部位での血餅の形成である。凝固過程は、幾つかのタンパク質分子の機能に高度に依存する。これらは凝固因子として知られている。凝固因子の幾つかは、不活性のチモーゲンまたは酵素的に活性な形態で存在し得るプロテアーゼである。チモーゲン形態は、別のタンパク質分解的に活性な凝固因子により触媒されたポリペプチド鎖の特異的切断により、酵素的に活性な形態に変換され得る。第VII因子は、肝臓で合成され、1本鎖糖タンパク質として血液に分泌されるビタミンK依存性血漿タンパク質である。第VII因子チモーゲンは、特異的タンパク質分解切断により単一の部位、すなわち配列番号1のR152とI153の間で活性形態(第VIIa因子)に変換され、単一のジスルフィド結合により連結された2つの鎖分子をもたらす。第VIIa因子における2つのポリペプチド鎖は、それぞれ配列番号1(野生型ヒト凝固因子VII)の残基1〜152および153〜406に対応する軽鎖および重鎖と呼ばれる。第VII因子は主にチモーゲンとして循環するが、わずかな画分は活性形態(第VIIa因子)にある。
血液凝固過程は3期、すなわち、開始、増幅および伝播に分けることができる。開始期および伝播期は、止血において多くの重要な機能を有する凝固因子であるトロンビンの形成に寄与する。凝固カスケードは、血管の内部表面を裏打ちする内皮細胞の単層バリアが損傷を受けると開始する。これは、内皮下細胞および血管外マトリックスタンパク質を露出し、これに血中の血小板が張り付く。これが起こると、内皮下細胞の表面に存在する組織因子(TF)が、血中を循環する第VIIa因子に曝されるようになる。TFは膜結合タンパク質であり、第VIIa因子の受容体として機能する。第VIIa因子は、本質的に活性が低い酵素、セリンプロテアーゼである。しかし、第VIIa因子がTFに結合されると、この活性は大幅に増加する。TFとの第VIIa因子相互作用はまた、TF担持細胞のリン脂質表面に第VIIa因子を局在化させ、第Xa因子へ第X因子を活性化するために第VIIa因子を最適に配置する。これが起こると、第Xa因子は、第Va因子と結合してTF担持細胞の表面でいわゆる「プロトロンビナーゼ」複合体を形成することができる。プロトロンビナーゼ複合体は、次いで、プロトロンビンの切断によりトロンビンを生成する。循環第VIIa因子へのTFの曝露により活性化されトロンビンの最初の生成をもたらす経路は、TF経路として知られる。TF:第VIIa因子複合体はまた、第IXa因子への第IX因子の活性化も触媒する。次いで、活性化された第IXa因子は、損傷部位に張り付き活性化した血小板の表面に拡散し得る。これは、第IXa因子がFVIIIaと結合して、活性化血小板の表面で「テナーゼ」複合体を形成できるようにする。この複合体は、第Xa因子への第X因子の活性化における著しい効率性により、伝播期において重要な役割を果たす。第Xa因子活性の効率的テナーゼ触媒生成は、今度は、プロトロンビナーゼ複合体により触媒されたトロンビンへのプロトロンビンの効率的切断をもたらす。
第IX因子または第VIII因子のどちらかに何らかの欠損があれば、これは重要なテナーゼ活性を損ない、凝固に必要なトロンビンの産生を低下させる。TF経路により最初に形成されるトロンビンは、損傷部位で血小板の動員、活性化、および凝集を促すプロコアグラントシグナルとして機能する。これは、血小板の緩い一次血栓の形成をもたらす。しかし、この血小板の一次血栓は不安定であり、止血を持続するのに補強を必要とする。血栓の安定化は、フィブリン線維の網に血小板を固定し、絡ませることを必要とする。
強い安定な血餅の形成は、局所トロンビン活性の頑強なバーストの生成に依存する。故に、血管損傷後のトロンビン生成に至る過程における欠損は、出血障害、例えば血友病AおよびBをもたらす可能性がある。血友病AおよびBを有する人は、それぞれ機能的第VIIIa因子または第IXa因子を欠く。伝播期でのトロンビン生成は、テナーゼ活性に非常に依存し、すなわち第VIIIa因子および第FIXa因子の両方を必要とする。したがって、血友病AまたはBを有する人では、一次血小板血栓の適切な凝固がなく、出血が持続する。
補充療法は血友病AおよびBに対する伝統的な治療であり、第VIII因子または第IX因子の静脈内投与を必要とする。しかし、多くの場合、患者は、注入されたタンパク質に対する抗体(インヒビターとしても知られる)を発生させ、該抗体は治療の有効性を低下させ、または無効にする。組換え第VIIa因子[Novoseven(登録商標)]は、インヒビターを有する血友病AまたはB患者の治療に対して認可されており、また、出血エピソードを停止させ、または外傷および/もしくは手術に伴う出血を防ぐのにも使用される。組換え第VIIa因子はまた、先天性第VII因子欠乏症患者の治療に対しても認可されている。組換えFVIIaは、TF非依存的機構を通じて作用することが提案されている。このモデルによれば、組換えFVIIaは、このGlaドメインにより活性化血小板の表面に向けられ、該表面で次いでタンパク質分解的に第X因子を第Xa因子に活性化し、故に機能的テナーゼ複合体の必要性を回避する。TFの非存在下のFVIIaの低酵素活性および膜に対するGlaドメインの低親和性は、血友病を有する人において止血を達成するのに必要とされる循環FVIIaの超生理学的レベルの必要性を説明し得る。
組換え第VIIa因子は、2〜3時間のインビボでの機能的半減期(functional half-life)を有し、患者における出血を解決するために頻回投与を必要とする場合がある。さらに、患者は多くの場合、予防策としてではなく出血が始まった後に第VIIa因子療法を受けるのみであり、これは多くの場合、患者の全般的な生活の質に影響を与える。インビボでの機能的半減期がより長い組換え第VIIa因子バリアントは、必要な投与回数を減少させ、より頻度の少ない投与を支持することになり、故に患者および介護者の利益に対する第VIIa因子療法の大幅な改善が期待できる。
WO2007031559(7012)は、アンチトロンビンによる阻害に対する感受性が低下した第VII因子バリアントを開示している。
WO2009126307(触媒)は、プロコアグラント活性が変化した修飾第VII因子ポリペプチドを開示している。
「Summary Basis for Approval for NovoSeven(著作権)」、FDA参照番号96 0597
Agersoら(2011)J Thromb Haemost、9、333〜338頁
ZollerおよびSmith(DNA 3:479〜488頁、1984)
「Splicing by extension overlap」、Hortonら、Gene 77、1989、61〜68頁
PCR Protocols、1990、Academic Press、San Diego、California、USA
Sambrookら、Molecular Cloning: A Laboratory Manual、第2版 Cold Spring Harbor Laboratory、Cold Spring Harbor、New York、1989
BeaucageおよびCaruthers、Tetrahedron Letters 22(1981)、1859〜1869頁
Matthesら、EMBO Journal 3(1984)、801〜805頁
Saikiら、Science 239(1988)、487〜491頁
Subramaniら、Mol. Cell Biol. 1(1981)、854〜864頁
Palmiterら、Science 222(1983)、809〜814頁
Boshartら、Cell 41:521〜530頁、1985
KaufmanおよびSharp、Mol. Cell. Biol、2:1304〜1319頁、1982
Vasuvedanら、FEBS Lett. 311,(1992)7〜11頁
J.M. Vlakら、J. Gen. Virology 69、1988、765〜776頁
Hitzemanら、J. Biol. Chem. 255(1980)、12073〜12080頁
AlberおよびKawasaki、J. Mol. Appl. Gen. 1(1982)、419〜434頁
Youngら、Genetic Engineering of Microorganisms for Chemicals(Hollaenderら編)、Plenum Press、New York、1982
Russellら、Nature 304(1983)、652〜654頁
McKnightら、The EMBO J. 4(1985)、2093〜2099頁
AlberおよびKawasaki、J. Mol. Appl. Gen. 1、1982、419〜434頁
DeNotoら Nucl. Acids Res. 9:3719〜3730頁、1981
O. Hagenbuchleら、Nature 289、1981、643〜646頁
L.A. Vallsら、Cell 48、1987、887〜897頁
M. Egel-Mitaniら、Yeast 6、1990、127〜137頁
KaufmanおよびSharp、J. Mol. Biol. 159(1982)、601〜621頁
SouthernおよびBerg、J. Mol. Appl.Genet. 1(1982)、327〜341頁
Loyterら、Proc. Natl. Acad. Sci. USA 79(1982)、422〜426頁
Wiglerら、Cell 14(1978)、725頁
CorsaroおよびPearson、Somatic Cell Genetics 7(1981)、603頁
Grahamおよびvan der Eb、Virology 52(1973)、456頁
Neumannら、EMBO J. 1(1982)、841〜845頁
Mammalian Cell Technology、Butterworth Publishers、Stoneham、MA
Grahamら、J. Gen. Virol. 36:59〜72頁、1977
WaechterおよびBaserga、Proc. Natl. Acad. Sci. USA 79:1106〜1110頁、1982
UrlaubおよびChasin、Proc. Natl. Acad. Sci. USA 77:4216〜4220、1980
Gleesonら、J. Gen. Microbiol. 132、1986、3459〜3465頁
Malardierら、1989、Gene 78: 147〜156頁
Whitelawら、Biochem. J. 286: 31〜39頁(1992)
Brinsterら、Proc. Natl. Acad. Sci. USA 85: 836〜840頁(1988)
Palmiterら、Proc. Natl. Acad. Sci. USA 88: 478〜482頁(1991)
Whitelawら、Transgenic Res. 1: 3〜13頁(1991)
von Heijne、Nucl. Acids Res. 14: 4683〜4690頁(1986)
Jaenisch、Science 240: 1468〜1474頁(1988)
Bradleyら、Bio/Technology 10: 534〜539頁(1992)
Hoganら、Manipulating the Mouse Embryo: A Laboratory Manual、Cold Spring Harbor Laboratory、1986
Simonsら、Bio/Technology 6: 179〜183頁(1988)
Wallら、Biol. Reprod. 32: 645〜651頁(1985)
Buhlerら、Bio/Technology 8: 140〜143頁(1990)
Ebertら、Bio/Technology 9: 835〜838頁(1991)
Krimpenfortら、Bio/Technology 9: 844〜847頁(1991)
Wallら、J. Cell. Biochem. 49: 113〜120頁(1992)
Gordonら、Proc. Natl. Acad. Sci. USA 77: 7380〜7384頁(1980)
GordonおよびRuddle、Science 214: 1244〜1246頁(1981)
PalmiterおよびBrinster、Cell 41: 343〜345頁(1985)
Brinsterら、Proc. Natl. Acad. Sci. USA 82: 4438〜4442頁(1985)
Hiatt、Nature 344:469〜479頁(1990)
Edelbaumら、J. Interferon Res. 12:449〜453頁(1992)
Sijmonsら、Bio/Technology 8:217〜221頁(1990)
Protein Purification、J.-C. JansonおよびLars Ryden編者、VCH Publishers、New York、1989
Wakabayashiら、J. Biol. Chem. 261:11097〜11108頁、(1986)
Thimら、Biochemistry 27: 7785〜7793頁、(1988)
Scopes、R.、Protein Purification、Springer-Verlag、N.Y.、1982
Osterudら、Biochemistry 11:2853〜2857頁(1972)
HednerおよびKisiel、J. Clin. Invest. 71:1836〜1841頁(1983)
KisielおよびFujikawa、Behring Inst. Mitt. 73:29〜42頁(1983)
Bjoernら(Research Disclosure、269 September 1986、564〜565頁)
Monroeら(1997)Brit. J. Haematol. 99、542〜547頁の543頁
Remington: The Science and Practice of Pharmacy、第19版、1995
Durocherら(2002)Nucleic Acid Res. 30(2):e9
Busbyら(1991)J.Biol.Chem.、266:15286〜15292頁
Arteltら(1988) Gene 62:213〜219頁
Perssonら(1996)FEBS Lett.、385:241〜243頁
J. H. Morrisseyら、Blood. 81:734〜744頁(1993)
Viuffら Thrombosis Research、2010;126, 144〜149頁
一般に、凝固障害を有する人には多くの満たされていない医学的必要性がある。血餅形成を促進する組換え第VIIa因子の使用は、治療剤としての増大する重要性を明確に示す。しかし、組換え第VIIa因子療法は、重大な満たされていない医学的必要性を依然として残しており、改善された医薬的特性(例えばインビボでの増大した機能的半減期および改善された活性)を有する組換え第VIIa因子ポリペプチドが必要である。
本発明は、改善された医薬的特性を有するように設計された修飾第VII因子ポリペプチドを提供する。広範な態様において、本発明は、ヒト野生型第VIIa因子と比較してインビボでの機能的半減期の増大を示す第VII因子ポリペプチドに関する。別の広範な態様において、本発明は、内因性血漿インヒビター、特にアンチトロンビンによる不活化に対する耐性の増加を示す第VII因子ポリペプチドに関する。さらなる広範な態様において、本発明は、活性が増強または実質的に保存された第VII因子ポリペプチドに関する。
本明細書で提供されるのは、アンチトロンビン不活化に対する耐性の増加をもたらし、タンパク質分解活性の喪失をほとんどまたは全くもたらさない変異の組み合わせを含む、インビボでの機能的半減期が増大した第VII因子ポリペプチドである。本発明の特に興味深い態様において第VII因子ポリペプチドは、1つまたは複数の「半減期延長基」にカップリングされてインビボでの機能的半減期を増大する。
1つの態様において、本発明は、ヒト第VII因子(配列番号1)のアミノ酸配列と比べて2つ以上の置換を含む第VII因子(a)ポリペプチドであって、置換の少なくとも1つが、T293がLys(K)、Tyr(Y)、Arg(R)もしくはPhe(F)により置換され、Q176がLys(K)、Arg(R)、Asn(N)により置換され、および/またはQ286がAsn(N)により置換されている所にあり、ならびに置換の少なくとも1つが、M298がGln(Q)、Lys(K)、Arg(R)、Asn(N)、Gly(G)、Pro(P)、Ala(A)、Val(V)、Leu(L)、Ile(I)、Phe(F)、Trp(W)、Tyr(Y)、Asp(D)、Glu(E)、His(H)、Cys(C)、Ser(S)、またはThr(T)により置換されている所にある、第VII因子(a)ポリペプチドに関する。
興味深い実施形態において本発明は、少なくとも1つの半減期延長部分とカップリングされている第VII因子(a)ポリペプチドに関する。
別の態様において、本発明は、本発明の第VII因子(a)ポリペプチドを産生する方法に関する。
さらなる態様において、本発明は、本発明の第VII因子(a)ポリペプチドを含む医薬組成物に関する。
本発明の一般的な目的は、凝固障害を有する人における現在利用可能な治療選択肢を改善し、治療的有用性が改善された第VII因子ポリペプチドを得ることである。本発明が有する1つの目的は、医薬的に許容されるタンパク質分解活性を維持しながら、インビボでの機能的半減期が延長された第VII因子ポリペプチドを得ることである。これを達成するために、本発明の第VII因子ポリペプチドは、タンパク質分解活性を実質的に保存しながら、血漿インヒビターアンチトロンビンによる不活化に対する感受性の低下をもたらす変異の組み合わせを含む。本発明の特に興味深い実施形態において、第VII因子ポリペプチドはまた、1つまたは複数の「半減期延長基」にカップリングされる。
本発明の修飾第VII因子ポリペプチドによる医学的治療は、注射と注射の間のより長い期間、より低い投与量、より便利な投与、および注射と注射の間の潜在的に改善された止血防御など、現在利用可能な治療計画に対して幾つかの利点を提供する。
本発明は、インビボでの機能的半減期の増大、血漿インヒビターアンチトロンビンによる不活化に対する感受性の低下、および保存されたタンパク質分解活性を示す第VII因子ポリペプチドの設計および使用に関する。半減期延長部分へのコンジュゲーションと組み合わせたヒト第VII因子における変異の特定の組み合わせは、上述の特性をもたらすことが本発明の発明者らにより見出された。本発明の第VII因子ポリペプチドは、より長く続くプロコアグラント活性が望まれる状況において治療的に有用な血中での延長された機能的半減期を有する。
本発明は、ヒト第VII因子(配列番号1)のアミノ酸配列と比べて2つ以上の置換を含む第VII因子(a)ポリペプチドであって、置換の少なくとも1つが、T293がLys(K)、Tyr(Y)、Arg(R)もしくはPhe(F)により置換され、Q176がLys(K)、Arg(R)、Asn(N)により置換され、および/またはQ286がAsn(N)により置換されている所にあり、ならびに置換の少なくとも1つが、M298がGln(Q)、Lys(K)、Arg(R)、Asn(N)、Gly(G)、Pro(P)、Ala(A)、Val(V)、Leu(L)、Ile(I)、Phe(F)、Trp(W)、Tyr(Y)、Asp(D)、Glu(E)、His(H)、Cys(C)、Ser(S)、またはThr(T)により置換されている所にある、第VII因子(a)ポリペプチドに関する。
第VII因子
凝固因子VII(第VII因子)は、肝臓で主に産生される糖タンパク質である。成熟タンパク質は、配列番号1により定義された406個のアミノ酸残基から成り、4つのドメインから構成される。N末端ガンマカルボキシグルタミン酸(Gla)リッチドメインと、これに続く2つの表皮成長因子(EGF)様ドメイン、およびC末端トリプシン様セリンプロテアーゼドメインがある。第VII因子は、主に1本鎖分子として血漿中に循環する。第VII因子は、残基Arg152とIle153の間の切断により第VIIa因子に活性化されて、ジスルフィド結合により結合された2本鎖タンパク質をもたらす。軽鎖はGlaおよびEGF様ドメインを含有するが、重鎖はプロテアーゼドメインである。第VII因子における配列番号1による特異的Glu(E)残基、すなわちE6、E7、E14、E16、E19、E20、E25、E26、E29、およびE35は、翻訳後にガンマカルボキシル化され得る。Glaドメイン中のガンマカルボキシグルタミン酸残基は、リン脂質膜との相互作用を仲介するコンフォメーションにおいてGlaドメインを維持する幾つかのカルシウムイオンの調整を必要とされる。
凝固因子VII(第VII因子)は、肝臓で主に産生される糖タンパク質である。成熟タンパク質は、配列番号1により定義された406個のアミノ酸残基から成り、4つのドメインから構成される。N末端ガンマカルボキシグルタミン酸(Gla)リッチドメインと、これに続く2つの表皮成長因子(EGF)様ドメイン、およびC末端トリプシン様セリンプロテアーゼドメインがある。第VII因子は、主に1本鎖分子として血漿中に循環する。第VII因子は、残基Arg152とIle153の間の切断により第VIIa因子に活性化されて、ジスルフィド結合により結合された2本鎖タンパク質をもたらす。軽鎖はGlaおよびEGF様ドメインを含有するが、重鎖はプロテアーゼドメインである。第VII因子における配列番号1による特異的Glu(E)残基、すなわちE6、E7、E14、E16、E19、E20、E25、E26、E29、およびE35は、翻訳後にガンマカルボキシル化され得る。Glaドメイン中のガンマカルボキシグルタミン酸残基は、リン脂質膜との相互作用を仲介するコンフォメーションにおいてGlaドメインを維持する幾つかのカルシウムイオンの調整を必要とされる。
用語「第VII因子(a)」は、未切断1本鎖チモーゲン、第VII因子、および切断された2本鎖の、故に活性化されたプロテアーゼ、第VIIa因子を包含する。「第VII因子(a)」には、個々に存在し得、個々に異なり得る第VII因子(a)の天然対立遺伝子バリアントが含まれる。野生型ヒト第VII因子配列は、配列番号1に示されている。
第VII因子(a)は、血漿由来であってもよく、または産生および精製のよく知られた方法を用いて組換え的に産生されてもよい。グリコシル化、ガンマカルボキシル化、および他の翻訳後修飾の程度および位置は、選択された宿主細胞およびこの増殖条件によって異なり得る。
第VII因子ポリペプチド
用語「第VII因子(a)ポリペプチド」は本明細書において、野生型第VII因子(a)分子のほか、第VII因子(a)バリアントおよび第VII因子(a)コンジュゲートを指す。このようなバリアントおよびコンジュゲートは、野生型ヒト第VIIa因子と比べて実質的に同じ、または改善された活性を示すことができる。
用語「第VII因子(a)ポリペプチド」は本明細書において、野生型第VII因子(a)分子のほか、第VII因子(a)バリアントおよび第VII因子(a)コンジュゲートを指す。このようなバリアントおよびコンジュゲートは、野生型ヒト第VIIa因子と比べて実質的に同じ、または改善された活性を示すことができる。
用語、第VII因子ポリペプチドの「活性」は本明細書で使用されるとき、野生型ヒト第VII因子(a)により示される任意の活性を指し、凝固(coagulation)または凝固(coagulant)活性、プロコアグラント活性、第X因子活性化または第IX因子活性化に影響を与えるようなタンパク質分解活性または触媒活性; TF、第X因子または第IX因子を結合する能力;および/またはリン脂質に結合する能力を含むが、これらに限定されない。これらの活性は、一般に認められているアッセイを用いて、例えば、インビトロまたはインビボで凝固を測定して、インビトロまたはインビボで評価することができる。このようなアッセイの結果は、ポリペプチドがインビボでの該ポリペプチドの活性と相関している可能性がある活性を示すことを示し、インビボ活性は生物活性と呼ばれてもよい。第VII因子ポリペプチドの活性を決定するためのアッセイは、当業者に知られている。FVIIポリペプチドの活性を評価するための例示的アッセイには、以下の実施例に記載されているようなインビトロタンパク質分解アッセイが含まれる。
用語「増加したまたは保存された活性」は本明細書で使用されるとき、野生型ヒト第VIIa因子と比較して、実質的に同じまたは増加した活性、例えばi)組換え野生型ヒト第VIIa因子と比較して、TFの存在下および/または非存在下で実質的に同じまたは増加したタンパク質分解活性を示す第VIIa因子ポリペプチド; ii)組換え野生型ヒト第VIIa因子と比較して、TF親和性が実質的に同じまたは増加した第VII因子(a)ポリペプチド; iii)活性化血小板に対する親和性が実質的に同じまたは増加した第VII因子(a)ポリペプチド;またはiv)組換え野生型ヒト第VIIa因子と比較して、第X因子もしくは第IX因子に結合する親和性/能力が実質的に同じもしくは増加した第VII因子(a)ポリペプチドを指す。例えば、保存された活性は、保持された活性量が、野生型ヒト第VIIa因子と比較して活性の10%、20%、30%、40%、50%、60%、70%、80%、90%、100%、200%、300%、400%、500%以上、または約10%、約20%、約30%、約40%、約50%、約60%、約70%、約80%、約90%、約100%、約200%、約300%、約400%、約500%以上であることを意味する。
用語「第VII因子(a)バリアント」は本明細書で使用されるとき、親タンパク質の1つもしくは複数のアミノ酸が別の自然発生のアミノ酸で置換されている、および/または親タンパク質の1つもしくは複数のアミノ酸が欠失されている、および/または1つもしくは複数のアミノ酸が該タンパク質に挿入されている、および/または1つもしくは複数のアミノ酸が親タンパク質に付加されている、配列番号1の配列を有する第VII因子を表すことが意図される。このような付加は、親タンパク質のN末端もしくはC末端のどちらか、または両方で生じ得る。1つの実施形態においてバリアントは、配列番号1の配列と少なくとも95%同一である。別の実施形態においてバリアントは、配列番号1の配列と少なくとも99%同一である。本明細書で使用されるとき、特定の位置への任意の言及は、配列番号1の対応する位置を指す。
本明細書で使用されるアミノ酸置換に対する用語は、次の通りである。最初の文字は、配列番号1の位置に自然に存在するアミノ酸を表す。次の数字は、配列番号1における位置を表す。2番目の文字は、天然のアミノ酸を置換する異なるアミノ酸を表す。一例は、配列番号1の197位のリジンがアラニンにより置換されているK197A第VII因子である。
本文脈においてアミノ酸の3文字または1文字の略号は、Table 1(表1)に示された従来の意味で使用されている。明確に示されない限り、本明細書で言及されるアミノ酸はL-アミノ酸である。

用語「第VII因子(a)コンジュゲート」は本明細書で使用されるとき、野生型第VII因子(a)と比べて、実質的に同じまたは改善された生物活性を示し、1つもしくは複数のアミノ酸または1つもしくは複数の付加されたグリカン鎖が、アルキル化、グリコシル化、アシル化、エステル形成、ジスルフィド結合形成、またはアミド形成によるなど化学的および/または酵素的に修飾されている第VII因子ポリペプチドを表すことが意図される。
本発明の第VII因子ポリペプチドは、ヒト第VII因子のアミノ酸配列(配列番号1)と比べて2つ以上の置換を含み、置換の少なくとも1つが、T293がLys(K)、Tyr(Y)、Arg(R)もしくはPhe(F)により置換され、Q176がLys(K)、Arg(R)、Asn(N)により置換され、および/またはQ286がAsn(N)により置換されている所にあり、ならびに置換の少なくとも1つが、M298がGln(Q)、Lys(K)、Arg(R)、Asn(N)、Gly(G)、Pro(P)、Ala(A)、Val(V)、Leu(L)、Ile(I)、Phe(F)、Trp(W)、Tyr(Y)、Asp(D)、Glu(E)、His(H)、Cys(C)、Ser(S)、またはThr(T)により置換されている所にある。
一連の興味深い実施形態において本発明は、ポリペプチドが、以下の置換T293K/M298Q、T293Y/M298Q、T293R/M298Q、T293F/M298Q、Q176K/M298Q、Q176R/M298Q、Q176N/M298Q、Q286N/M298Q、T293Y/V158D/E296V/M298Q、T293R/V158D/E296V/M298Q、T293K/V158D/E296V/M298Q、Q176K/V158D/E296V/M298Q、およびQ176R/V158D/E296V/M298Qの1つを有する、第VII因子ポリペプチドに関する。
半減期-血漿インヒビターによる不活化に対する耐性
インビボでのクリアランスに加えて、インビボでの機能的半減期もまた、化合物が体内で「治療的に利用可能」である期間にとって重要である。組換えヒト野生型第VIIa因子の循環半減期は、約2.3時間である(「Summary Basis for Approval for NovoSeven(著作権)」、FDA参照番号96 0597)。
インビボでのクリアランスに加えて、インビボでの機能的半減期もまた、化合物が体内で「治療的に利用可能」である期間にとって重要である。組換えヒト野生型第VIIa因子の循環半減期は、約2.3時間である(「Summary Basis for Approval for NovoSeven(著作権)」、FDA参照番号96 0597)。
用語「インビボでの機能的半減期」は、この通常の意味で使用され、すなわち、体内/標的器官に残っている第VII因子ポリペプチドの生物活性を低下させる(終末相で50%)のに必要とされる時間、または第VII因子ポリペプチドの活性が初期値の50%となる時間である。インビボ半減期に対する代替用語には、終末半減期、血漿半減期、循環(circulating)半減期、循環(circulatory)半減期、およびクリアランス半減期が含まれる。インビボでの機能的半減期は、以下(実施例12)でさらに論じられるように、当技術分野で知られた任意の適切な方法により決定することができる。
用語「増大した」はインビボでの機能的半減期または血漿半減期について使用されるとき、同等の条件下で決定された野生型ヒト第VIIa因子などの参照分子の半減期と比べて、ポリペプチドの関連半減期が増大されることを示すのに使用される。
1つの態様において、本発明の第VIIa因子ポリペプチドは、野生型ヒト第VIIa因子と比べて増大したインビボでの機能的半減期を示す。例えば関連半減期は、少なくとも約50%(例えば少なくとも約100%、150%、200%、250%、または500%)など少なくとも約25%増大され得る。
凝固カスケードの生化学および病態生理学に関する詳細な理解にもかかわらず、循環からの個々の凝固因子のクリアランスに関する機構原理は、大部分が不明のままである。他のビタミンK依存性タンパク質のチモーゲンおよび酵素形態と比較した、第VII因子およびこの活性形態第VIIa因子の循環半減期における著しい違いは、第VIIa因子の特異的および異なるクリアランス機構の存在を示唆している。2つのタイプの経路が、第VIIa因子のクリアランスにおいて作動可能であるらしい - 一方はインタクトなタンパク質の排除をもたらし、他方は血漿インヒビターにより仲介され、タンパク質分解不活化をもたらす。
アンチトロンビンIII(アンチトロンビン、AT)は豊富な血漿インヒビターであり、第VIIa因子を含む凝固系のほとんどのプロテアーゼを標的にする。アンチトロンビンIIIは、血漿中にマイクロモル濃度で存在し、自殺基質機構により標的プロテアーゼを不可逆的に結合し不活化するセリンプロテアーゼインヒビターのセルピンファミリーに属する。アンチトロンビンによる阻害は、静脈内投与後、インビボで組換え第VIIa因子の優勢なクリアランス経路となるらしい。血友病患者における組換え第VIIa因子薬物動態に関する最近の研究において、総クリアランスの約60%が、この経路に起因している可能性があった(Agersoら(2011)J Thromb Haemost、9、333〜338頁)。
幾つかの実施形態において、本発明の第VII因子(a)ポリペプチドは、野生型ヒト第VIIa因子と比べて、内因性血漿インヒビター、特にアンチトロンビンによる不活化に対する耐性の増加を示す。
本発明の1つの実施形態において、第VIIa因子ポリペプチドは、アンチトロンビンなどの内因性血漿インヒビターなどのインヒビターによる不活化に対する耐性のため、増大した半減期を示す。本発明の第VII因子(a)ポリペプチドの、天然第VIIa因子と比較して阻害に対するより高い耐性のため、作用部位で機能的に適切な濃度を得るのにより低用量が適切となり得、故に、出血エピソードを有するまたは正常な止血系の増強を必要とする対象に対し、より低用量をおよび/またはより低頻度で投与することが可能となろう。
以下の変異T293Y、T293R、T293K、Q176K、Q176R、Q286Nを有する第VII因子ポリペプチドは、アンチトロンビン不活化に対する耐性の増加をもたらすことが本発明の発明者らにより見出された。理論に縛られることなく、これらの第VIIa因子ポリペプチドバリアントの、インヒビター不活化に対するこの耐性は、TF依存性活性を減少させることと引き換えに達成され得、活性の点からこれらの第VIIa因子ポリペプチドバリアントの欠点となる可能性がある。
以下の変異M298Q、およびV158D/E296V/M298Qを有する第VIIa因子ポリペプチドは、タンパク質分解活性の増加をもたらすことが知られている。しかし、これらの第VIIa因子ポリペプチドバリアントもまた、インヒビター不活化に対する感受性の増加を示し、インビボでの機能的半減期の点からこれらの第VIIa因子ポリペプチドバリアントの欠点となる可能性がある。
上述の変異のこれら2つの群を組み合わせることにより、増加したまたは保存された活性は、インヒビター不活化に対する高い耐性を維持しながら達成されることが本発明の発明者らにより見出された。すなわち、変異の組み合わせを含む本発明の第VII因子ポリペプチドは、アンチトロンビン不活化に対する耐性の増加、および実質的に保存されたタンパク質分解活性を示す。本発明の第VII因子ポリペプチドが、1つまたは複数の半減期延長部分とコンジュゲートされる場合、半減期延長に対する驚くほど改善された効果が得られる。これらの特性を考慮すると、本発明のこのようなコンジュゲートされた第VII因子ポリペプチドは、医薬的に許容されるタンパク質分解活性を維持しながら改善された循環半減期を示す。
追加の修飾
本発明の第VII因子ポリペプチドは、さらなる修飾、特に、追加の有利な特性を第VII因子ポリペプチドにもたらすさらなる修飾を含んでもよい。故に、上述の少なくとも2つの置換に加えて、本発明の第VII因子ポリペプチドは、例えば、さらなるアミノ酸修飾、例えば1つのさらなるアミノ酸置換を含んでもよい。1つのこのような実施形態において、本発明の第VII因子ポリペプチドは、例えばWO2002077218に記載された基R396C、Q250C、および407Cから選択される追加の変異または付加を有する。
本発明の第VII因子ポリペプチドは、さらなる修飾、特に、追加の有利な特性を第VII因子ポリペプチドにもたらすさらなる修飾を含んでもよい。故に、上述の少なくとも2つの置換に加えて、本発明の第VII因子ポリペプチドは、例えば、さらなるアミノ酸修飾、例えば1つのさらなるアミノ酸置換を含んでもよい。1つのこのような実施形態において、本発明の第VII因子ポリペプチドは、例えばWO2002077218に記載された基R396C、Q250C、および407Cから選択される追加の変異または付加を有する。
本発明の第VII因子ポリペプチドは、第VII因子ポリペプチドの一次配列中にあるまたはない追加の修飾を含んでもよい。追加の修飾には、糖質部分の付加、半減期延長部分の付加、例えばPEG部分、Fcドメインの付加等が含まれるが、これらに限定されない。例えば、このような追加の修飾は、第VII因子ポリペプチドの安定性または半減期を増大させるように行うことができる。
半減期延長部分または基
用語「半減期延長部分」および「半減期延長基」は、本明細書において互換的に使用され、-SH、-OH、-COOH、-CONH2、-NH2などの1つもしくは複数のアミノ酸側鎖官能基、または1つもしくは複数のN-および/もしくはO-グリカン構造に付加された1つまたは複数の化学基、ならびにタンパク質/ペプチドのインビボ循環半減期を、これらのタンパク質/ペプチドにコンジュゲートされた場合に増大することができる1つまたは複数の化学基を指すと理解される。半減期延長部分の例には、生体適合性脂肪酸およびこの誘導体、ヒドロキシアルキルデンプン(HAS)、例えばヒドロキシエチルデンプン(HES)、ポリエチレングリコール(PEG)、Poly(Glyx-Sery)n(HAP)、ヒアルロン酸(HA)、ヘパロサンポリマー(HEP)、ホスホリルコリンベースのポリマー(PCポリマー)、フレキシマー(Fleximer)、デキストラン、ポリシアル酸(PSA)、Fcドメイン、トランスフェリン、アルブミン、エラスチン様(ELP)ペプチド、XTENポリマー、PASポリマー、PAポリマー、アルブミン結合ペプチド、CTPペプチド、FcRn結合ペプチド、およびこれらの任意の組み合わせが含まれる。
用語「半減期延長部分」および「半減期延長基」は、本明細書において互換的に使用され、-SH、-OH、-COOH、-CONH2、-NH2などの1つもしくは複数のアミノ酸側鎖官能基、または1つもしくは複数のN-および/もしくはO-グリカン構造に付加された1つまたは複数の化学基、ならびにタンパク質/ペプチドのインビボ循環半減期を、これらのタンパク質/ペプチドにコンジュゲートされた場合に増大することができる1つまたは複数の化学基を指すと理解される。半減期延長部分の例には、生体適合性脂肪酸およびこの誘導体、ヒドロキシアルキルデンプン(HAS)、例えばヒドロキシエチルデンプン(HES)、ポリエチレングリコール(PEG)、Poly(Glyx-Sery)n(HAP)、ヒアルロン酸(HA)、ヘパロサンポリマー(HEP)、ホスホリルコリンベースのポリマー(PCポリマー)、フレキシマー(Fleximer)、デキストラン、ポリシアル酸(PSA)、Fcドメイン、トランスフェリン、アルブミン、エラスチン様(ELP)ペプチド、XTENポリマー、PASポリマー、PAポリマー、アルブミン結合ペプチド、CTPペプチド、FcRn結合ペプチド、およびこれらの任意の組み合わせが含まれる。
特に興味深い実施形態において、本発明の第VII因子ポリペプチドは、1つまたは複数の延長基(protracting group)/半減期延長部分とカップリングされる。
1つの実施形態において本発明のシステインコンジュゲート第VII因子ポリペプチドは、第VII因子ポリペプチドに導入されたシステインのスルフヒドリル基にコンジュゲートされた1つまたは複数の疎水性半減期延長部分を有する。本発明のシステインコンジュゲート第VII因子ポリペプチドは、さらに、遅延性(protractive)半減期延長部分を他のアミノ酸残基に連結させることが可能である。
1つの実施形態において本発明の第VII因子ポリペプチドは、例えばWO2007115953に記載されているように、組織因子にジスルフィド連結される。
別の実施形態において本発明の第VII因子ポリペプチドは、血小板親和性が増加した第VIIa因子バリアントである。
ペグ化誘導体
本発明による「ペグ化第VII因子ポリペプチドバリアント/誘導体」は、第VII因子ポリペプチドの任意のアミノ酸残基または糖質部分を含むFVIIポリペプチドの任意の部分に付加された、1つまたは複数のポリエチレングリコール(PEG)分子を有することができる。化学的および/または酵素的方法が、PEG基または他の遅延基を第VII因子ポリペプチド上のグリカンにコンジュゲートするのに使用され得る。酵素的コンジュゲーション過程の例は、例えばWO03031464に記載されている。グリカンは自然発生的であってもよく、または例えば、当技術分野でよく知られた方法を用いて、N-グリコシル化モチーフ(NXT/S、Xは任意の自然発生アミノ酸)を第VII因子のアミノ酸配列に導入して改変されてもよい。本発明による「システインペグ化第VII因子ポリペプチドバリアント」は、FVIIポリペプチドに存在するまたはFVIIポリペプチドに導入されたシステイン残基のスルフヒドリル基にコンジュゲートされた1つまたは複数のPEG分子を有する。
本発明による「ペグ化第VII因子ポリペプチドバリアント/誘導体」は、第VII因子ポリペプチドの任意のアミノ酸残基または糖質部分を含むFVIIポリペプチドの任意の部分に付加された、1つまたは複数のポリエチレングリコール(PEG)分子を有することができる。化学的および/または酵素的方法が、PEG基または他の遅延基を第VII因子ポリペプチド上のグリカンにコンジュゲートするのに使用され得る。酵素的コンジュゲーション過程の例は、例えばWO03031464に記載されている。グリカンは自然発生的であってもよく、または例えば、当技術分野でよく知られた方法を用いて、N-グリコシル化モチーフ(NXT/S、Xは任意の自然発生アミノ酸)を第VII因子のアミノ酸配列に導入して改変されてもよい。本発明による「システインペグ化第VII因子ポリペプチドバリアント」は、FVIIポリペプチドに存在するまたはFVIIポリペプチドに導入されたシステイン残基のスルフヒドリル基にコンジュゲートされた1つまたは複数のPEG分子を有する。
ヘパロサンコンジュゲート
本発明による第VII因子ポリペプチドヘパロサンコンジュゲートは、第VII因子ポリペプチドの任意のアミノ酸残基または糖質部分を含むFVIIポリペプチドの任意の部分に付加された、1つまたは複数のヘパロサンポリマー(HEP)分子を有することができる。化学的および/または酵素的方法が、HEPを第VII因子ポリペプチド上のグリカンにコンジュゲートするのに使用され得る。酵素的コンジュゲーション過程の例は、例えばWO03031464に記載されている。グリカンは自然発生的であってもよく、または例えば、当技術分野でよく知られた方法を用いて、N-グリコシル化モチーフ(NXT/S、Xは任意の自然発生のアミノ酸)を第VII因子のアミノ酸配列に導入して改変されてもよい。
本発明による第VII因子ポリペプチドヘパロサンコンジュゲートは、第VII因子ポリペプチドの任意のアミノ酸残基または糖質部分を含むFVIIポリペプチドの任意の部分に付加された、1つまたは複数のヘパロサンポリマー(HEP)分子を有することができる。化学的および/または酵素的方法が、HEPを第VII因子ポリペプチド上のグリカンにコンジュゲートするのに使用され得る。酵素的コンジュゲーション過程の例は、例えばWO03031464に記載されている。グリカンは自然発生的であってもよく、または例えば、当技術分野でよく知られた方法を用いて、N-グリコシル化モチーフ(NXT/S、Xは任意の自然発生のアミノ酸)を第VII因子のアミノ酸配列に導入して改変されてもよい。
本発明による「システイン-HEP第VII因子ポリペプチドコンジュゲート」は、FVIIポリペプチドに存在するまたはFVIIポリペプチドに導入されたシステイン残基のスルフヒドリル基にコンジュゲートされた1つまたは複数のHEP分子を有する。
本発明の1つの興味深い実施形態において、第VII因子ポリペプチドはHEPポリマーにカップリングされる。
融合タンパク質
融合タンパク質は、もともと別個のタンパク質もしくはペプチドまたはこの断片をコードする2つ以上のDNA配列のインフレーム結合を通じて作り出されたタンパク質である。融合タンパク質DNA配列の翻訳は、もともとの各タンパク質またはペプチドのそれぞれに由来する機能的特性を有し得る、単一のタンパク質配列をもたらすであろう。融合タンパク質をコードするDNA配列は、オーバーラップPCRまたはDNAライゲーションなどの標準的な分子生物学方法により人工的に作り出すことができ、アセンブリは、最初の5'末端DNA配列中の終止コドンを排除する一方、3'末端DNA配列中の終止コドンを保持して行われる。得られた融合タンパク質DNA配列は、細菌、酵母、真菌、昆虫細胞または哺乳動物細胞などの標準的宿主生物における異種融合タンパク質発現を支持する適切な発現ベクターに挿入することができる。
融合タンパク質は、もともと別個のタンパク質もしくはペプチドまたはこの断片をコードする2つ以上のDNA配列のインフレーム結合を通じて作り出されたタンパク質である。融合タンパク質DNA配列の翻訳は、もともとの各タンパク質またはペプチドのそれぞれに由来する機能的特性を有し得る、単一のタンパク質配列をもたらすであろう。融合タンパク質をコードするDNA配列は、オーバーラップPCRまたはDNAライゲーションなどの標準的な分子生物学方法により人工的に作り出すことができ、アセンブリは、最初の5'末端DNA配列中の終止コドンを排除する一方、3'末端DNA配列中の終止コドンを保持して行われる。得られた融合タンパク質DNA配列は、細菌、酵母、真菌、昆虫細胞または哺乳動物細胞などの標準的宿主生物における異種融合タンパク質発現を支持する適切な発現ベクターに挿入することができる。
融合タンパク質は、リンカー、または融合タンパク質を規定するタンパク質もしくはペプチド部分を分離するスペーサーペプチド配列を含有することができる。リンカーまたはスペーサーペプチド配列は、個々のタンパク質またはペプチド部分の正確な折り畳みを促進し得、個々のタンパク質またはペプチド部分がこの個々の機能的特性を保持する可能性を高め得る。リンカーまたはスペーサーペプチド配列は、完全な融合タンパク質DNA配列を構成する個々のDNA断片のインフレームアセンブリ中に、すなわちオーバーラップPCRまたはDNAライゲーション中に、融合タンパク質DNA配列に挿入することができる。
Fc融合タンパク質
用語「Fc融合タンパク質」は、本明細書において、任意の抗体アイソタイプに由来し得るFcドメインに融合された本発明の第VII因子ポリペプチドを包含することが意味される。IgG Fcドメインは、IgG抗体の比較的長い循環半減期のため、多くの場合好ましいであろう。Fcドメインは、例えば補体結合および/または特定のFc受容体への結合などの特定のエフェクター機能を調節するために、さらに修飾されてもよい。FcRn受容体に結合する能力を有するFcドメインとのFVIIポリペプチドの融合は、一般的に、wt FVIIポリペプチドの半減期と比較して、融合タンパク質の循環半減期の延長をもたらすであろう。IgG Fcドメインにおける234、235および237位の変異は、一般的に、FcγRI受容体、ならびに恐らく同様にFcγRIIaおよびFcγRIII受容体への結合の減少をもたらすであろう。これらの変異は、エンドサイトーシスリサイクリング経路により長い循環半減期を促進するFcRn受容体への結合を変化させない。好ましくは、本発明による融合タンパク質の修飾IgG Fcドメインは、1つまたは複数の以下の変異を含み、該変異はそれぞれ、特定のFc受容体に対する親和性の減少(L234A、L235E、およびG237A)、およびC1q媒介補体固定の低下(A330SおよびP331S)をもたらすであろう。あるいは、Fcドメインは、好ましくはS241P/S228P変異を含む、IgG4 Fcドメインであってもよい。
用語「Fc融合タンパク質」は、本明細書において、任意の抗体アイソタイプに由来し得るFcドメインに融合された本発明の第VII因子ポリペプチドを包含することが意味される。IgG Fcドメインは、IgG抗体の比較的長い循環半減期のため、多くの場合好ましいであろう。Fcドメインは、例えば補体結合および/または特定のFc受容体への結合などの特定のエフェクター機能を調節するために、さらに修飾されてもよい。FcRn受容体に結合する能力を有するFcドメインとのFVIIポリペプチドの融合は、一般的に、wt FVIIポリペプチドの半減期と比較して、融合タンパク質の循環半減期の延長をもたらすであろう。IgG Fcドメインにおける234、235および237位の変異は、一般的に、FcγRI受容体、ならびに恐らく同様にFcγRIIaおよびFcγRIII受容体への結合の減少をもたらすであろう。これらの変異は、エンドサイトーシスリサイクリング経路により長い循環半減期を促進するFcRn受容体への結合を変化させない。好ましくは、本発明による融合タンパク質の修飾IgG Fcドメインは、1つまたは複数の以下の変異を含み、該変異はそれぞれ、特定のFc受容体に対する親和性の減少(L234A、L235E、およびG237A)、およびC1q媒介補体固定の低下(A330SおよびP331S)をもたらすであろう。あるいは、Fcドメインは、好ましくはS241P/S228P変異を含む、IgG4 Fcドメインであってもよい。
第VII因子ポリペプチドの産生
1つの態様において、本発明は、第VII因子ポリペプチドを産生する方法に関する。本明細書に記載された第VII因子ポリペプチドは、組換え核酸法により産生することができる。一般に、クローン化ヒト野生型第VII因子核酸配列は、所望のタンパク質をコードするように修飾される。この修飾配列は、次いで発現ベクターに挿入され、今度は宿主細胞に形質転換またはトランスフェクトされる。より高等な真核細胞、特に培養哺乳動物細胞が、宿主細胞として好ましい。
1つの態様において、本発明は、第VII因子ポリペプチドを産生する方法に関する。本明細書に記載された第VII因子ポリペプチドは、組換え核酸法により産生することができる。一般に、クローン化ヒト野生型第VII因子核酸配列は、所望のタンパク質をコードするように修飾される。この修飾配列は、次いで発現ベクターに挿入され、今度は宿主細胞に形質転換またはトランスフェクトされる。より高等な真核細胞、特に培養哺乳動物細胞が、宿主細胞として好ましい。
さらなる態様において、本発明は、該ポリヌクレオチド構築物を含有および発現するトランスジェニック動物に関する。
さらなる態様において、本発明は、該ポリヌクレオチド構築物を含有および発現するトランスジェニック植物に関する。
ヒト野生型第VII因子に関する完全なヌクレオチド配列およびアミノ酸配列が知られている(組換えヒト第VII因子のクローニングおよび発現が記載されているU.S. 4,784,950参照)。
アミノ酸配列変異は、様々な手法により達成され得る。核酸配列の修飾は、部位特異的変異誘発によってもよい。部位特異的変異誘発の手法は、当技術分野でよく知られており、例えば、ZollerおよびSmith(DNA 3:479〜488頁、1984)または「Splicing by extension overlap」、Hortonら、Gene 77、1989、61〜68頁に記載されている。故に、第VII因子のヌクレオチドおよびアミノ酸配列を用いて、選択した変異を導入することができる。同様に、特定のプライマーを用いたポリメラーゼ連鎖反応を用いてDNA構築物を調製する手順は、当業者によく知られている(PCR Protocols、1990、Academic Press、San Diego、California、USA参照)。
本発明の第VII因子ポリペプチドをコードする核酸構築物は適切には、標準的な手法(Sambrookら、Molecular Cloning: A Laboratory Manual、第2版 Cold Spring Harbor Laboratory、Cold Spring Harbor、New York、1989参照)に従って、ゲノムライブラリーまたはcDNAライブラリーを調製すること、および合成オリゴヌクレオチドプローブを用いてハイブリダイゼーションにより該ポリペプチドの全部または一部をコードするDNA配列をスクリーニングすることにより例えば得られた、ゲノム起源またはcDNA起源であり得る。
第VII因子ポリペプチドをコードする核酸構築物はまた、確立された標準的な方法、例えばBeaucageおよびCaruthers、Tetrahedron Letters 22(1981)、1859〜1869頁により記載されたホスホラミダイト方法、またはMatthesら、EMBO Journal 3(1984)、801〜805頁により記載された方法により合成的に調製することもできる。ホスホラミダイト方法により、オリゴヌクレオチドは、例えば自動DNA合成器において合成され、精製され、アニールされ、ライゲートされ、適切なベクターにクローン化される。ヒト第VII因子ポリペプチドをコードするDNA配列はまた、例えばUS 4,683,202、Saikiら、Science 239(1988)、487〜491頁、またはSambrookら(上記参照)に記載されているように、特異的プライマーを用いてポリメラーゼ連鎖反応により調製することもできる。
さらに、核酸構築物は、標準的な手法に従って、合成起源、ゲノム起源またはcDNA起源(適宜)の断片(全核酸構築物の様々な部分に対応する断片)をライゲートして調製された、合成起源およびゲノム起源の混合、合成起源およびcDNA起源の混合、またはゲノム起源およびcDNA起源の混合であってもよい。
核酸構築物は、好ましくはDNA構築物である。本発明による第VII因子ポリペプチドの産生において使用するDNA配列は、適切な翻訳後プロセシング(例えばグルタミン酸残基のガンマカルボキシル化)および宿主細胞からの分泌を得るために、第VII因子のアミノ末端にプレプロポリペプチドを典型的にはコードするであろう。プレプロポリペプチドは、第VII因子、または第IX因子、第X因子、プロトロンビン、プロテインCもしくはプロテインSなどの別のビタミンK依存性血漿タンパク質のプレプロポリペプチドであってもよい。当業者により理解されるように、追加の修飾は、これらの修飾が、凝固剤として作用するタンパク質の能力を大幅に損なわない第VII因子ポリペプチドのアミノ酸配列において行われ得る。
ヒト第VII因子ポリペプチドをコードするDNA配列は、好都合には組換えDNA手順に供することができる任意のベクターであり得る組換えベクターに通常は挿入され、ベクターの選択は、ベクターが導入される宿主細胞に多くの場合依存するであろう。故に、ベクターは、自己複製ベクター、すなわち、複製が染色体複製から独立している染色体外実体、例えばプラスミドとして存在するベクターであってもよい。あるいは、ベクターは、宿主細胞に導入される場合、宿主細胞ゲノムに組み込まれ、ベクターが組み込まれた染色体と一緒に複製されるものであってもよい。
ベクターは好ましくは、ヒト第VII因子ポリペプチドをコードするDNA配列が、DNAの転写に必要とされる追加のセグメントに作動可能に連結される発現ベクターである。一般に、発現ベクターは、プラスミドもしくはウイルスDNAに由来し、または両方のエレメントを含有してもよい。用語「作動可能に連結される」は、セグメントが、意図された目的に対して協調して機能する(例えば転写がプロモーターにおいて開始し、ポリペプチドをコードするDNA配列を通って進む)ように整列されることを示す。
第VIIa因子ポリペプチドバリアントの発現に使用する発現ベクターは、クローン化遺伝子またはcDNAの転写を指示することができるプロモーターを含むであろう。プロモーターは、選択した宿主細胞において転写活性を示し、および宿主細胞と同種または異種のタンパク質をコードする遺伝子に由来し得る、任意のDNA配列であってもよい。
哺乳動物細胞においてヒト第VII因子ポリペプチドをコードするDNAの転写を指示する適切なプロモーターの例は、SV40プロモーター(Subramaniら、Mol. Cell Biol. 1(1981)、854〜864頁)、MT-1(メタロチオネイン遺伝子)プロモーター[Palmiterら、Science 222(1983)、809〜814頁]、CMVプロモーター(Boshartら、Cell 41:521〜530頁、1985)、またはアデノウイルス2型主要後期プロモーター(KaufmanおよびSharp、Mol. Cell. Biol、2:1304〜1319頁、1982)である。
昆虫細胞において使用する適切なプロモーターの例は、ポリへドリンプロモーター(US 4,745,051; Vasuvedanら、FEBS Lett. 311,(1992)7〜11頁)、P10プロモーター(J.M. Vlakら、J. Gen. Virology 69、1988、765〜776頁)、オートグラファカリフォルニカ多角体病ウイルス(Autographa californica polyhedrosis virus)塩基性タンパク質プロモーター(EP 397 485)、バキュロウイルス最初期遺伝子1プロモーター(US 5,155,037; US 5,162,222)、またはバキュロウイルス39K遅延型初期遺伝子プロモーター(US 5,155,037; US 5,162,222)である。
酵母宿主細胞において使用する適切なプロモーターの例には、酵母解糖遺伝子(Hitzemanら、J. Biol. Chem. 255(1980)、12073〜12080頁; AlberおよびKawasaki、J. Mol. Appl. Gen. 1(1982)、419〜434頁)、もしくはアルコールデヒドロゲナーゼ遺伝子[Youngら、Genetic Engineering of Microorganisms for Chemicals(Hollaenderら編)、Plenum Press、New York、1982において]由来のプロモーター、またはTPI1(US 4,599,311)もしくはADH2-4c[Russellら、Nature 304(1983)、652〜654頁]プロモーターが含まれる。
糸状菌宿主細胞において使用する適切なプロモーターの例は、例えば、ADH3プロモーター[McKnightら、The EMBO J. 4(1985)、2093〜2099頁]またはtpiAプロモーターである。他の有用なプロモーターの例は、アスペルギルス・オリゼ(A. oryzae) TAKAアミラーゼ、リゾムコール・ミエヘイ(Rhizomucor miehei)アスパラギン酸プロテイナーゼ、アスペルギルス・ニガー(A. niger)中性α-アミラーゼ、アスペルギルス・ニガー酸安定性α-アミラーゼ、アスペルギルス・ニガーもしくはアスペルギルス・アワモリ(A. awamori)グルコアミラーゼ(gluA)、リゾムコール・ミエヘイリパーゼ、アスペルギルス・オリゼアルカリプロテアーゼ、アスペルギルス・オリゼトリオースリン酸イソメラーゼ、またはアスペルギルス・ニデュランス(A. nidulans)アセトアミダーゼをコードする遺伝子に由来するものである。好ましいのは、TAKAアミラーゼおよびgluAプロモーターである。適切なプロモーターは、例えばEP 238 023およびEP 383 779に述べられている。
第VII因子ポリペプチドをコードするDNA配列はまた、必要であれば、ヒト成長ホルモンターミネーター(Palmiterら、Science 222、1983、809〜814頁)、またはTPI1(AlberおよびKawasaki、J. Mol. Appl. Gen. 1、1982、419〜434頁)もしくはADH3(McKnightら、The EMBO J. 4、1985、2093〜2099頁)ターミネーターなどの適切なターミネーターに作動可能に結合することもできる。発現ベクターはまた、プロモーターから下流、および第VII因子配列自体の挿入部位から上流に位置する一連のRNAスプライス部位を含有することもできる。好ましいRNAスプライス部位は、アデノウイルス遺伝子および/または免疫グロブリン遺伝子から得ることができる。同様に発現ベクターに含有されるのは、挿入部位の下流に位置するポリアデニル化シグナルである。特に好ましいポリアデニル化シグナルには、SV40由来の初期または後期ポリアデニル化シグナル(KaufmanおよびSharp、同上)、アデノウイルス5 Elb領域由来のポリアデニル化シグナル、ヒト成長ホルモン遺伝子ターミネーター(DeNotoら Nucl. Acids Res. 9:3719〜3730頁、1981)、またはヒト第VII因子遺伝子もしくはウシ第VII因子遺伝子由来のポリアデニル化シグナルが含まれる。発現ベクターは、プロモーターとRNAスプライス部の間に位置するアデノウイルス2型三連リーダー(tripartite leader)などの非コードウイルスリーダー配列、およびSV40エンハンサーなどのエンハンサー配列も含むことができる。
本発明の第VII因子ポリペプチドを宿主細胞の分泌経路に向けるために、分泌シグナル配列(リーダー配列、プレプロ配列またはプレ配列としても知られる)が組換えベクター中に提供されてもよい。分泌シグナル配列は、正しいリーディングフレームにヒト第VII因子ポリペプチドをコードするDNA配列に結合される。分泌シグナル配列は通常、ペプチドをコードするDNA配列の5'に位置する。分泌シグナル配列は普通、タンパク質と関連するものであってもよく、または別の分泌タンパク質をコードする遺伝子由来であってもよい。
酵母細胞からの分泌に関して、分泌シグナル配列は、発現されたヒト第VII因子ポリペプチドを細胞の分泌経路に効率的に向けることを確保する任意のシグナルペプチドをコードしてもよい。シグナルペプチドは、自然発生のシグナルペプチド、もしくはこの機能的部分であってもよく、またはシグナルペプチドは、合成ペプチドであってもよい。適切なシグナルペプチドは、α因子シグナルペプチド(US 4,870,008参照)、マウス唾液アミラーゼのシグナルペプチド(O. Hagenbuchleら、Nature 289、1981、643〜646頁参照)、修飾カルボキシペプチダーゼシグナルペプチド(L.A. Vallsら、Cell 48、1987、887〜897頁)、酵母BAR1シグナルペプチド(WO 87/02670参照)、または酵母アスパラギン酸プロテアーゼ3(YAP3)シグナルペプチド(M. Egel-Mitaniら、Yeast 6、1990、127〜137頁参照)であることが見出されている。
酵母における効率的な分泌のために、リーダーペプチドをコードする配列はまた、シグナル配列の下流、およびヒト第VII因子ポリペプチドをコードするDNA配列の上流に挿入することもできる。リーダーペプチドの機能は、培養培地への分泌のために、発現されたペプチドが小胞体からゴルジ装置へ、さらには分泌小胞へ向けられることを可能にすることである(すなわち酵母細胞のペリプラズム空間への、細胞壁を越えたまたは少なくとも細胞膜を通じたヒト第VII因子ポリペプチドの輸出)。リーダーペプチドは、酵母アルファ因子リーダーであってもよい(例えばUS 4,546,082、US 4,870,008、EP 16 201、EP 123 294、EP 123 544、およびEP 163 529に記載されている使用)。あるいは、リーダーペプチドは、合成リーダーペプチド、すなわち天然に見出されないリーダーペプチドであってもよい。合成リーダーペプチドは、例えば、WO 89/02463またはWO 92/11378に記載されているように構築することができる。
糸状菌において使用するために、シグナルペプチドは好都合には、アスペルギルス属種アミラーゼもしくはグルコアミラーゼをコードする遺伝子、リゾムコール・ミエヘイリパーゼもしくはプロテアーゼまたはフミコラ・ラヌギノサ(Humicola lanuginosa)リパーゼをコードする遺伝子に由来してもよい。シグナルペプチドは、好ましくは、アスペルギルス・オリゼTAKAアミラーゼ、アスペルギルス・ニガー中性α-アミラーゼ、アスペルギルス・ニガー酸安定性アミラーゼ、またはアスペルギルス・ニガーグルコアミラーゼをコードする遺伝子に由来する。適切なシグナルペプチドは、例えばEP 238 023およびEP 215 594に開示されている。
昆虫細胞において使用するために、シグナルペプチドは好都合には、鱗翅目マンジュカ・セクスタ(Manduca sexta)脂質動員ホルモン前駆物質シグナルペプチド(US 5,023,328参照)などの昆虫遺伝子に由来してもよい(WO 90/05783参照)。
第VII因子ポリペプチドをコードするDNA配列、プロモーター配列、ならびに場合によりターミネーターおよび/または分泌シグナル配列をそれぞれライゲートし、複製に必要な情報を含有する適切なベクターにこれらを挿入するのに使用される手順は、当業者によく知られている(例えば、Sambrookら、Molecular Cloning: A Laboratory Manual、Cold Spring Harbor、New York、1989参照)。
哺乳動物細胞をトランスフェクトし、細胞に導入されたDNA配列を発現させる方法は、例えば、KaufmanおよびSharp、J. Mol. Biol. 159(1982)、601〜621頁; SouthernおよびBerg、J. Mol. Appl.Genet. 1(1982)、327〜341頁; Loyterら、Proc. Natl. Acad. Sci. USA 79(1982)、422〜426頁; Wiglerら、Cell 14(1978)、725頁; CorsaroおよびPearson、Somatic Cell Genetics 7(1981)、603頁、Grahamおよびvan der Eb、Virology 52(1973)、456頁;およびNeumannら、EMBO J. 1(1982)、841〜845頁に記載されている。
クローン化DNA配列は、例えば、リン酸カルシウムトランスフェクション(Wiglerら、Cell 14:725〜732頁、1978; CorsaroおよびPearson、Somatic Cell Genetics 7:603〜616頁、1981; GrahamおよびVan der Eb、Virology 52d:456〜467頁、1973)、またはエレクトロポレーション(Neumannら、EMBO J. 1:841〜845頁、1982)により培養哺乳動物細胞に導入される。外因性DNAを発現する細胞を同定および選択するために、選択可能な表現型をもたらす遺伝子(選択可能なマーカー)が、一般的に、目的とする遺伝子またはcDNAに沿って細胞に導入される。好ましい選択可能なマーカーは、ネオマイシン、ハイグロマイシン、およびメトトレキサートなどの薬剤に対する耐性をもたらす遺伝子を含む。選択可能なマーカーは、増幅可能な選択可能なマーカーであってもよい。好ましい増幅可能な選択可能なマーカーは、ジヒドロ葉酸還元酵素(DHFR)配列である。選択可能なマーカーは、Thillyによりレビューされている(参照により本明細書に組み込まれているMammalian Cell Technology、Butterworth Publishers、Stoneham、MA)。当業者は、適切な選択可能なマーカーを容易に選択できよう。
選択可能なマーカーは、目的とする遺伝子と同時に別個のプラスミド上で細胞に導入されてもよく、または選択可能なマーカーは、同じプラスミド上で導入されてもよい。同じプラスミド上であるならば、選択可能なマーカーおよび目的とする遺伝子は、異なるプロモーターまたは同じプロモーターの制御下にあってもよく、後者の配置はジシストロニックなメッセージを生成する。このタイプの構築物は、当技術分野で知られている(例えば、LevinsonおよびSimonsen、U.S. 4,713,339)。細胞に導入された混合物に、「担体DNA」として知られる追加のDNAを付加することもまた有利であり得る。
細胞が該DNAを取り込んだ後、細胞は、適切な増殖培地で典型的には1〜2日増殖されて、目的とする遺伝子を発現し始める。本明細書で使用されるとき、用語「適切な増殖培地」は、細胞の増殖および目的とする第VII因子ポリペプチドの発現に必要とされる栄養素および他の成分を含有する培地を意味する。培地は一般的に、炭素源、窒素源、必須アミノ酸、必須糖、ビタミン、塩、リン脂質、タンパク質、および増殖因子を含む。ガンマカルボキシル化タンパク質を産生するために、培地は、ビタミンKを好ましくは約0.1μg/mlから約5μg/mlの濃度で含有するであろう。薬剤選択は、次いで、選択可能なマーカーを安定な様式で発現している細胞の増殖を選択するために適用される。増幅可能な選択可能なマーカーをトランスフェクトされた細胞に関して、増加したコピー数のクローン化配列を選択するために薬剤濃度が増加され得、これにより発現レベルを増加させることができる。安定にトランスフェクトされた細胞のクローンは、次いで目的とするヒト第VII因子ポリペプチドの発現についてスクリーニングされる。
第VII因子ポリペプチドをコードするDNA配列が導入される宿主細胞は、翻訳後修飾されたヒト第VII因子ポリペプチドを産生することができ、ならびに酵母、真菌、およびより高等な真核細胞を含むいずれの細胞であってもよい。
本発明において使用するための哺乳動物細胞系の例は、COS-1(ATCC CRL 1650)、ベビーハムスター腎臓(BHK)、および293(ATCC CRL 1573; Grahamら、J. Gen. Virol. 36:59〜72頁、1977)細胞系である。好ましいBHK細胞系は、tk-ts13 BHK細胞系(参照により本明細書に組み込まれているWaechterおよびBaserga、Proc. Natl. Acad. Sci. USA 79:1106〜1110頁、1982)であり、以下BHK 570細胞と呼ばれる。BHK 570細胞系は、ATCCアクセッション番号CRL 10314のもと、アメリカンタイプカルチャーコレクション、12301 Parklawn Dr., Rockville、Md. 20852に寄託されている。tk-ts13 BHK細胞系はまた、アクセッション番号CRL 1632のもとATCCから入手することもできる。さらに、Rat Hep I(ラット肝細胞癌; ATCC CRL 1600)、Rat Hep II(ラット肝細胞癌; ATCC CRL 1548)、TCMK(ATCC CCL 139)、ヒト肺(ATCC HB 8065)、NCTC 1469(ATCC CCL 9.1)、CHO(ATCC CCL 61)、およびDUKX細胞(UrlaubおよびChasin、Proc. Natl. Acad. Sci. USA 77:4216〜4220、1980)を含む幾つかの他の細胞系が、本発明内で使用され得る。
適切な酵母細胞の例には、サッカロミセス属種またはシゾサッカロミセス属種、特にサッカロミセス・セレビシエ(Saccharomyces cerevisiae)またはサッカロミセス・クルイベリ(Saccharomyces kluyveri)の株の細胞が含まれる。酵母細胞を異種DNAで形質転換し、そこから異種ポリペプチドを産生する方法は、例えばUS 4,599,311、US 4,931,373、US 4,870,008、5,037,743、およびUS 4,845,075に記載されており、これらの全ては参照により本明細書に組み込まれている。形質転換細胞は、選択可能なマーカーにより決定された表現型、通常は薬剤耐性、または特定の栄養素、例えばロイシンの非存在下で増殖する能力により選択される。酵母において使用するための好ましいベクターは、US 4,931,373に開示されたPOT1ベクターである。ヒト第VII因子ポリペプチドをコードするDNA配列は、例えば上記に記載されているように、シグナル配列および場合によりリーダー配列が前にあってもよい。適切な酵母細胞のさらなる例は、クルイベロミセス・ラクティス(K. lactis)などのクルイベロミセス属、ハンゼヌラ属、例えばハンゼヌラ・ポリモルファ(H. polymorpha)、またはピキア属、例えばピキア・パストリス(P. pastoris)の株である(Gleesonら、J. Gen. Microbiol. 132、1986、3459〜3465頁; US 4,882,279参照)。
他の真菌細胞の例は、糸状菌、例えばアスペルギルス属種、ニューロスポラ属種、フザリウム属種、またはトリコデルマ属種、特にアスペルギルス・オリザ、アスペルギルス・ニデュランスまたはアスペルギルス・ニガーの株の細胞である。タンパク質の発現のためのアスペルギルス属種の使用は、例えば、EP 272 277、EP 238 023、EP 184 438に記載されている。フザリウム・オキシスポラム(F. oxysporum)の形質転換は、例えば、Malardierら、1989、Gene 78: 147〜156頁により記載されているように実施されてもよい。トリコデルマ属種の形質転換は、例えばEP 244 234に記載されているように行われてもよい。
糸状菌が宿主細胞として使用される場合、好都合には本発明のDNA構築物を宿主染色体に組み込んで組換え宿主細胞を得ることにより、糸状菌は本発明のDNA構築物で形質転換され得る。DNA配列は、該細胞で安定に維持される可能性が高いことから、この組み込みは一般的に利点であると考えられる。宿主染色体へのDNA構築物の組み込みは、従来の方法により、例えば同種または異種組換えにより行うことができる。
昆虫細胞の形質転換およびその中での異種ポリペプチドの産生は、US 4,745,051、US 4,879,236、US 5,155,037、5,162,222、EP 397,485に記載されているように行うことができ、これらの全ては参照により本明細書に組み込まれている。宿主として使用される昆虫細胞系は適切には、スポドプテラ・フルギペルダ(Spodoptera frugiperda)細胞またはトリコプルシア・ニ(Trichoplusia ni)細胞などの鱗翅目細胞系であってもよい(US 5,077,214参照)。培養条件は適切には、例えば、WO 89/01029もしくはWO 89/01028、または上述の参考文献のいずれかに記載されている通りであってもよい。
上記の形質転換またはトランスフェクトされた宿主細胞は、次いで、第VII因子ポリペプチドの発現を可能にする条件下、適切な栄養培地で培養され、この後、得られたペプチドの全部または一部が培養から回収され得る。細胞を培養するのに使用される培地は、適切なサプリメントを含有する最小または複合培地などの、宿主細胞を増殖させるのに適切な任意の従来の培地であってもよい。適切な培地は、商業的供給者から入手可能であり、または公開されたレシピ(例えばアメリカンタイプカルチャーコレクションのカタログ中の)により調製することができる。細胞により産生された第VII因子ポリペプチドは、次いで、遠心分離または濾過による培地からの宿主細胞の分離、塩(例えば硫酸アンモニウム)による上清または濾液のタンパク質性成分の沈殿、様々なクロマトグラフィー手順(例えば当該ポリペプチドのタイプに依存したイオン交換クロマトグラフィー、ゲル濾過クロマトグラフィー、親和性クロマトグラフィー等)による精製を含む従来の手順により、培養培地から回収することができる。
トランスジェニック動物技術が、本発明の第VII因子ポリペプチドを産生するのに使用されてもよい。宿主メス哺乳動物の乳腺内でタンパク質を産生することが好ましい。乳腺での発現、およびこの後の乳汁への目的とするタンパク質の分泌は、他の供給源からのタンパク質の単離において遭遇する多くの問題を克服する。乳汁は容易に採取され、大量に入手可能であり、生化学的に十分に特徴付けられている。さらに、主な乳タンパク質は、高濃度(典型的には約1〜15g/l)で乳汁に存在する。
商業的観点から、大きな乳収量を有する種を宿主として使用することが明らかに好ましい。マウスおよびラットなどのより小型の動物が使用され得る(原理段階の証明では好ましい)が、ブタ、ヤギ、ヒツジおよびウシを含むがこれらに限定されない家畜哺乳動物を使用することが好ましい。ヒツジは、この種における遺伝子導入の前歴、乳収量、コスト、およびヒツジ乳を採取する装置の入手しやすさといった要因のため、特に好ましい(例えば、宿主種の選択に影響を与える要因の比較にはWO 88/00239参照)。イーストフリースランドシープ(East Friesland sheep)などの酪農用に育種された宿主動物の品種を選択すること、または後日、トランスジェニック系の交配により酪農家畜を導入することが、一般的に望ましい。いずれの場合も、既知の良好な健康状態の動物が使用されるべきである。
乳腺での発現を得るために、乳タンパク質遺伝子由来の転写プロモーターが使用される。乳タンパク質遺伝子には、カゼイン(U.S. 5,304,489参照)、ベータ-ラクトグロブリン、a-ラクトアルブミン、および乳清酸性タンパク質をコードする遺伝子が含まれる。ベータ-ラクトグロブリン(BLG)プロモーターが好ましい。ヒツジベータ-ラクトグロブリン遺伝子の場合では、遺伝子の5'フランキング配列の少なくとも近位406bpの領域が一般的に使用されるであろうが、ベータ-ラクトグロブリン遺伝子の5'フランキングプロモーターおよび非コード部分を包含する約4.25kbp DNAセグメントなどの、5'フランキング配列のより大きい部分(最大約5kbp)が好ましい[Whitelawら、Biochem. J. 286: 31〜39頁(1992)参照]。他の種由来のプロモーターDNAの類似の断片もまた適切である。
ベータ-ラクトグロブリン遺伝子の他の領域もまた、発現される遺伝子のゲノムの領域と同様に構築物に組み込まれてよい。イントロンを欠く構築物は、例えば、このようなDNA配列を含有する構築物と比較して、十分に発現されないことが当技術分野で一般的に認められている[Brinsterら、Proc. Natl. Acad. Sci. USA 85: 836〜840頁(1988); Palmiterら、Proc. Natl. Acad. Sci. USA 88: 478〜482頁(1991); Whitelawら、Transgenic Res. 1: 3〜13頁(1991); WO 89/01343;およびWO 91/02318参照。これらの各々は、参照により本明細書に組み込まれている]。この点に関して、可能であれば、目的とするタンパク質またはポリペプチドをコードする遺伝子の天然イントロンの全てまたは幾つかを含有するゲノムの配列を使用することが一般的に好ましく、故に、例えばベータ-ラクトグロブリン遺伝子由来の、少なくとも幾つかのイントロンをさらに含むことが好ましい。1つのこのような領域は、ヒツジベータ-ラクトグロブリン遺伝子の3'非コード領域由来の、イントロンスプライシングおよびRNAポリアデニル化をもたらすDNAセグメントである。遺伝子の天然3'非コード配列が置換される場合、このヒツジベータ-ラクトグロブリンセグメントは、目的とするタンパク質またはポリペプチドの発現レベルを増大させることも安定化させることもできる。他の実施形態において、バリアント第VII因子配列の開始ATGを囲む領域は、乳特異的タンパク質遺伝子由来の対応する配列と置換される。このような置換は、発現を増大させる推定上の組織特異的開始環境をもたらす。バリアント第VII因子プレプロ配列全体および5'非コード配列を、例えばBLG遺伝子のものと置換することが好都合であるが、より小さい領域が置換されてもよい。
トランスジェニック動物における第VII因子ポリペプチドの発現のために、バリアント第VII因子をコードするDNAセグメントが、この発現に必要とされる追加のDNAセグメントに作動可能に連結されて、発現ユニットを生成する。このような追加のセグメントは、上述のプロモーター、ならびにmRNAの転写およびポリアデニル化の停止をもたらす配列を含む。発現ユニットは、修飾第VII因子をコードするセグメントに作動可能に連結された分泌シグナル配列をコードするDNAセグメントをさらに含むであろう。分泌シグナル配列は、天然第VII因子分泌シグナル配列であってもよく、または乳タンパク質などの別のタンパク質のものであってもよい(例えば、参照により本明細書に組み込まれているvon Heijne、Nucl. Acids Res. 14: 4683〜4690頁(1986);およびMeadeら、U.S. 4,873,316参照)。
トランスジェニック動物において使用するための発現ユニットの構築は、好都合には、追加のDNAセグメントを含有するプラスミドまたはファージベクターにバリアント第VII因子配列を挿入して実施されるが、発現ユニットは、ライゲーションの本質的に任意の配列により構築することができる。乳タンパク質をコードするDNAセグメントを含有するベクターを提供すること、および乳タンパク質のコード配列を第VII因子バリアントのものと置換し、これにより乳タンパク質遺伝子の発現制御配列を含む遺伝子融合を作り出すことが特に好都合である。いずれの場合も、プラスミドまたは他のベクターへの発現ユニットのクローニングは、バリアント第VII因子配列の増幅を促進する。増幅は、好都合には細菌[例えば大腸菌(E. coli)]宿主細胞で実施され、故にベクターは典型的には、複製起源および細菌宿主細胞において機能的な選択可能なマーカーを含むであろう。発現ユニットは次いで、選択された宿主種の受精卵(初期の胚を含む)に導入される。異種DNAの導入は、マイクロインジェクション[例えば米国特許第4,873,191号)、レトロウイルス感染(Jaenisch、Science 240: 1468〜1474頁(1988)]、または胚性幹(ES)細胞を用いる部位特異的組み込み(Bradleyら、Bio/Technology 10: 534〜539頁(1992)によりレビューされている)を含む、幾つかの経路の1つにより達成することができる。受精卵は次いで、偽妊娠したメスの卵管または子宮に移植され、出産まで発育させられる。導入されたDNAを生殖細胞系に担持する子は、この子孫に正常なメンデル様式でDNAを伝えることができ、トランスジェニック集団の発生を可能にする。トランスジェニック動物を作製する一般的な手順は、当技術分野で知られている(例えば、Hoganら、Manipulating the Mouse Embryo: A Laboratory Manual、Cold Spring Harbor Laboratory、1986; Simonsら、Bio/Technology 6: 179〜183頁(1988); Wallら、Biol. Reprod. 32: 645〜651頁(1985); Buhlerら、Bio/Technology 8: 140〜143頁(1990); Ebertら、Bio/Technology 9: 835〜838頁(1991); Krimpenfortら、Bio/Technology 9: 844〜847頁(1991); Wallら、J. Cell. Biochem. 49: 113〜120頁(1992); U.S. 4,873,191; U.S. 4,873,316; WO 88/00239、WO 90/05188、WO 92/11757;およびGB 87/00458参照)。外来DNA配列を哺乳動物およびこの生殖細胞に導入する手法は、もともとマウスにおいて開発された(例えば、Gordonら、Proc. Natl. Acad. Sci. USA 77: 7380〜7384頁(1980); GordonおよびRuddle、Science 214: 1244〜1246頁(1981); PalmiterおよびBrinster、Cell 41: 343〜345頁(1985); Brinsterら、Proc. Natl. Acad. Sci. USA 82: 4438〜4442頁(1985);ならびにHoganら(同上)参照)。これらの手法は、この後、家畜種を含むより大型の動物で使用するために適合された[例えば、WO 88/00239、WO 90/05188、およびWO 92/11757;ならびにSimonsら、Bio/Technology 6: 179〜183頁(1988)参照]。要約すると、トランスジェニックマウスまたは家畜の発生において今日までに使用された最も効率的な経路において、確立された手法により、目的とするDNAの数百の直鎖状分子が受精卵の前核の1つに注入される。接合子の細胞質へのDNAの注入も使用することができる。
トランスジェニック植物における産生もまた使用され得る。発現は一般化されてもよく、または塊茎などの特定の器官に向けられてもよい[Hiatt、Nature 344:469〜479頁(1990); Edelbaumら、J. Interferon Res. 12:449〜453頁(1992); Sijmonsら、Bio/Technology 8:217〜221頁(1990);およびEP 0 255 378参照]。
精製
本発明の第VII因子ポリペプチドは、細胞培養培地から回収される。本発明の第VII因子ポリペプチドは、クロマトグラフィー(例えば、イオン交換、親和性、疎水性、クロマトフォーカシング、およびサイズ排除)、電気泳動手順[例えば、分取等電点電気泳動(IEF)]、示差溶解度(例えば、硫安塩析)、または抽出を含むがこれらに限定されない、当技術分野で知られた様々な手順により精製することができる(例えば、Protein Purification、J.-C. JansonおよびLars Ryden編者、VCH Publishers、New York、1989参照)。好ましくは、本発明の第VII因子ポリペプチドは、抗第VII因子抗体カラム上で親和性クロマトグラフィーにより精製され得る。Wakabayashiら、J. Biol. Chem. 261:11097〜11108頁、(1986)、およびThimら、Biochemistry 27: 7785〜7793頁、(1988)に記載されているようなカルシウム依存性モノクローナル抗体の使用が特に好ましい。さらなる精製は、高速液体クロマトグラフィーなどの従来の化学的精製手段により達成することができる。クエン酸バリウム沈殿を含む精製の他の方法が当技術分野で知られており、本明細書に記載された新規の第VII因子ポリペプチドの精製に適用されてもよい(例えば、Scopes、R.、Protein Purification、Springer-Verlag、N.Y.、1982参照)。
本発明の第VII因子ポリペプチドは、細胞培養培地から回収される。本発明の第VII因子ポリペプチドは、クロマトグラフィー(例えば、イオン交換、親和性、疎水性、クロマトフォーカシング、およびサイズ排除)、電気泳動手順[例えば、分取等電点電気泳動(IEF)]、示差溶解度(例えば、硫安塩析)、または抽出を含むがこれらに限定されない、当技術分野で知られた様々な手順により精製することができる(例えば、Protein Purification、J.-C. JansonおよびLars Ryden編者、VCH Publishers、New York、1989参照)。好ましくは、本発明の第VII因子ポリペプチドは、抗第VII因子抗体カラム上で親和性クロマトグラフィーにより精製され得る。Wakabayashiら、J. Biol. Chem. 261:11097〜11108頁、(1986)、およびThimら、Biochemistry 27: 7785〜7793頁、(1988)に記載されているようなカルシウム依存性モノクローナル抗体の使用が特に好ましい。さらなる精製は、高速液体クロマトグラフィーなどの従来の化学的精製手段により達成することができる。クエン酸バリウム沈殿を含む精製の他の方法が当技術分野で知られており、本明細書に記載された新規の第VII因子ポリペプチドの精製に適用されてもよい(例えば、Scopes、R.、Protein Purification、Springer-Verlag、N.Y.、1982参照)。
治療目的には、本発明の第VII因子ポリペプチドは、実質的に純粋であることが好ましい。故に、本発明の好ましい実施形態において、本発明の第VII因子ポリペプチドは、少なくとも約90から95%均一性、好ましくは少なくとも約98%均一性まで精製される。純度は、例えばゲル電気泳動およびアミノ末端アミノ酸配列決定により評価することができる。
第VII因子ポリペプチドは、この2本鎖形態に変換するためにこの活性化部位で切断される。活性化は、Osterudら、Biochemistry 11:2853〜2857頁(1972); Thomas、米国特許第4,456,591号; HednerおよびKisiel、J. Clin. Invest. 71:1836〜1841頁(1983);またはKisielおよびFujikawa、Behring Inst. Mitt. 73:29〜42頁(1983)により開示されたものなど、当技術分野で知られた手順により実施することができる。あるいは、Bjoernら(Research Disclosure、269 September 1986、564〜565頁)により記載されているように、第VII因子は、Mono Q(登録商標)(Pharmacia fine Chemicals社)等などのイオン交換クロマトグラフィーカラムに通過させて活性化されてもよい。得られた活性化第VII因子バリアントは次いで、以下に記載されているように製剤化および投与することができる。
アッセイ
本発明はまた、本発明による好ましい第VII因子ポリペプチドを選択するための適切なアッセイも提供する。これらのアッセイは、単純な予備的インビトロ試験として行うことができる。
本発明はまた、本発明による好ましい第VII因子ポリペプチドを選択するための適切なアッセイも提供する。これらのアッセイは、単純な予備的インビトロ試験として行うことができる。
第VIIa因子ポリペプチドの活性も、適切には5〜1000nM(30〜40nMなどの)濃度で、第X因子などの生理学的基質を用いて測定することができ(「インビトロタンパク質分解アッセイ」)(実施例5参照)、生成された第Xa因子が、適切な色素産生基質(例えばS-2765)の添加後に測定される。さらに、活性アッセイは生理学的温度で行うことができる。
トロンビンを生成する第VIIa因子ポリペプチドの能力も、生理学的濃度で全ての関連凝固因子およびインヒビター[マイナス第VIII因子(血友病A状態を模倣する場合)]、ならびに活性化血小板を含むアッセイで測定することができる[参照により本明細書に組み込まれているMonroeら(1997)Brit. J. Haematol. 99、542〜547頁の543頁に記載されているように]。実施例8参照。
医薬組成物
1つの態様において、本発明は、本発明の第VII因子ポリペプチドを含む組成物および製剤に関する。例えば、本発明は、医薬的に許容される担体と一緒に製剤化される本発明の第VII因子ポリペプチドを含む医薬組成物を提供する。
1つの態様において、本発明は、本発明の第VII因子ポリペプチドを含む組成物および製剤に関する。例えば、本発明は、医薬的に許容される担体と一緒に製剤化される本発明の第VII因子ポリペプチドを含む医薬組成物を提供する。
したがって、本発明の1つの目的は、0.25mg/mlから100mg/mlの濃度で存在する第VII因子ポリペプチドを含む医薬製剤であって、pH2.0から10.0を有する製剤を提供することである。製剤はさらに、1つまたは複数の緩衝系、保存料、等張化剤、キレート剤、安定剤、抗酸化剤、または界面活性剤、および様々なこれらの組み合わせを含んでもよい。医薬組成物における保存料、等張剤、キレート剤、安定剤、抗酸化剤、および界面活性剤の使用は、当業者に十分に知られている。Remington: The Science and Practice of Pharmacy、第19版、1995を参照することができる。
1つの実施形態において、医薬製剤は水性製剤である。このような製剤は、典型的には溶液または懸濁液であるが、コロイド、分散剤、エマルジョン、および多相材料を含むこともできる。用語「水性製剤」は、少なくとも50%w/w水を含む製剤として定義される。同様に、用語「水性溶液」は、少なくとも50%w/w水を含む溶液として定義され、用語「水性懸濁液」は、少なくとも50%w/w水を含む懸濁液として定義される。
別の実施形態において、医薬製剤は、医師または患者が使用前に溶媒および/または希釈剤を添加する凍結乾燥製剤である。
さらなる態様において、医薬製剤は、第VII因子ポリペプチドの水性溶液および、該ポリペプチドが1mg/ml以上からの濃度で存在し、前記製剤が約2.0から約10.0のpHを有する緩衝液を含む。
本発明の第VII因子ポリペプチドは、静脈内、筋肉内、皮下など非経口的に投与することができる。あるいは、本発明のFVIIポリペプチドは、経口的または局所的になど非経口でない経路を介して投与することができる。本発明のポリペプチドは予防的に投与することができる。本発明のポリペプチドは、(要求に応じて)治療的に投与することができる。
治療的使用
広範な態様において、本発明の第VII因子ポリペプチドまたは前記ポリペプチドを含む医薬製剤は、医薬品として使用することができる。
広範な態様において、本発明の第VII因子ポリペプチドまたは前記ポリペプチドを含む医薬製剤は、医薬品として使用することができる。
1つの態様において、本発明の第VII因子ポリペプチドまたは前記ポリペプチドを含む医薬製剤は、凝固障害を有する対象を治療するのに使用することができる。
別の態様において、本発明の第VII因子ポリペプチドまたは前記ポリペプチドを含む医薬製剤は、出血障害もしくは出血エピソードを治療するための医薬品の調製、または正常な止血系の増強に使用することができる。
さらなる態様において、本発明の第VII因子ポリペプチドまたは前記ポリペプチドを含む医薬製剤は、血友病A、血友病B、または後天性インヒビターを有する血友病AもしくはBの治療に使用することができる。
別の態様において、本発明の第VII因子ポリペプチドまたは前記ポリペプチドを含む医薬製剤は、対象における出血障害もしくは出血エピソードを治療するための、または正常な止血系の増強のための方法であって、本発明の第VII因子ポリペプチドの治療的または予防的に有効な量をこれを必要とする対象に投与する工程を含む方法において使用することができる。
本明細書で使用されるとき、用語「対象」には、任意のヒト患者、またはヒト以外の脊椎動物が含まれる。
用語「治療」は本明細書で使用されるとき、これを必要とする任意のヒトまたは他の脊椎動物対象の薬物療法を指す。前記対象は、前記特定の治療の使用が、前記ヒトまたは他の脊椎動物の健康に有益であることを示す暫定または確定診断を与えた医師、または獣医師による身体検査を受けていることが期待される。前記治療のタイミングおよび目的は、対象の健康の現状により1人1人異なる可能性がある。故に、前記治療は、予防的、緩和的、対症的および/または根治的であり得る。本発明に関して、予防的、緩和的、対症的および/または根治的治療は、本発明の別の態様となり得る。
用語「凝固障害」は本明細書で使用されるとき、正常な凝固カスケードのいずれかのプロコアグラント成分のいずれかの質的もしくは量的欠乏、または線維素溶解のいずれかの上方制御に起因し得る出血傾向の増加を指す。このような凝固障害は、先天性および/または後天性および/または医原性であり得、当業者により同定される。先天性凝固低下障害(hypocoagulopathy)の非限定例は、血友病A、血友病B、第VII因子欠乏症、第X因子欠乏症、第XI因子欠乏症、フォンヴィルブランド病、ならびにグランツマン血小板無力症およびベルナールスーリエ症候群などの血小板減少症である。血友病AまたはBの臨床的重症度は、血中のFIX/第VIII因子の機能的ユニットの濃度により決定され、軽度、中等度、または重度に分類される。重度の血友病は、正常レベルの<1%に相当する<0.01U/mlの凝固因子レベルにより定義されるが、中等度および軽度血友病を有する人は、それぞれ1〜5%および>5%のレベルを有する。「インヒビター」(すなわち、第VIII因子に対する同種抗体)を有する血友病A、および「インヒビター」(すなわち、第IX因子に対する同種抗体)を有する血友病Bは、一部は先天性であり一部は後天性である凝固障害の非限定例である。
後天性凝固障害の非限定例は、ビタミンK欠乏症に起因するセリンプロテアーゼ欠乏症である。このようなビタミンK欠乏症は、ワルファリンなどのビタミンKアンタゴニストの投与に起因し得る。後天性凝固障害はまた、広範な外傷後にも生じ得る。別名「外傷死の3徴」として知られるこの場合において、後天性凝固障害は、血液希釈(希釈性血小板減少症および凝固因子の希釈)、低体温、凝固因子の消費、および代謝異常(アシドーシス)を特徴とする。輸液療法および線維素溶解の増加は、この状況を悪化させ得る。前記出血は、身体のいずれの部分からもあり得る。
医原性凝固障害の非限定例は、血栓塞栓症を治療するために処方され得るヘパリン、アスピリン、ワルファリン、および他の血小板凝集阻害剤などの抗凝固剤の過剰投与である。医原性凝固障害の第2の非限定例は、輸血により誘発され得るものなど、過剰なおよび/または不適切な輸液療法により誘発されるものである。
本発明の1つの実施形態において、出血は血友病AまたはBと関連する。別の実施形態において、出血は後天性インヒビターを有する血友病AまたはBと関連する。別の実施形態において、出血は血小板減少症と関連する。別の実施形態において、出血はフォンヴィルブランド病と関連する。別の実施形態において、出血は重度組織損傷と関連する。別の実施形態において、出血は重度外傷と関連する。別の実施形態において、出血は手術と関連する。別の実施形態において、出血は出血性胃炎および/または腸炎と関連する。別の実施形態において、出血は胎盤早期剥離におけるなどの大量子宮出血である。別の実施形態において、出血は、頭蓋内、耳内(intraaurally)、または眼内などの機械的止血の可能性が限られた器官で生じる。別の実施形態において、出血は抗凝固剤療法と関連する。
本発明は、以下の例によりさらに例証されるが、これらは保護の範囲を制限すると見なされるべきではない。前述の記載および以下の実施例に開示された特徴は、個別におよびこれらの任意の組み合わせの両方において、本発明をこの多様な形態で実現する材料となり得る。
タンパク質
ヒト血漿由来第X因子(FX)および第Xa因子(FXa)は、Enzyme Research Laboratories Inc.社(サウスベンド、IN)から入手した。可溶性組織因子1-219(sTF)または1-209は、公開されている手順(Freskgardら、1996)により調製した。組換え野生型FVIIaの発現および精製は、以前に記載されているように行った(Thimら、1988; PerssonおよびNielsen、1996)。ヒト血漿由来アンチトロンビン(Baxter社)は、公開されている手順(Olsonら、1993)によりヘパリンセファロースクロマトグラフィー(GE Healthcare社)により再精製した。
ヒト血漿由来第X因子(FX)および第Xa因子(FXa)は、Enzyme Research Laboratories Inc.社(サウスベンド、IN)から入手した。可溶性組織因子1-219(sTF)または1-209は、公開されている手順(Freskgardら、1996)により調製した。組換え野生型FVIIaの発現および精製は、以前に記載されているように行った(Thimら、1988; PerssonおよびNielsen、1996)。ヒト血漿由来アンチトロンビン(Baxter社)は、公開されている手順(Olsonら、1993)によりヘパリンセファロースクロマトグラフィー(GE Healthcare社)により再精製した。
(実施例1)
FVIIa-アンチトロンビン相互作用をマッピングするために、FVIIaバリアントライブラリーを、FVIIa-アンチトロンビン複合体の複合体の構造的モデルに基づきインシリコで設計した。図1に示されたモデルは、テンプレートとしてFXa-アンチトロンビンミカエリス複合体の公開されているX線構造(Johnsonら 2006)を用いて構築した。該モデルにおいてアンチトロンビンに近接するFVIIa残基(FVIIaとアンチトロンビン側鎖の間の最大距離は12Åであった)を、変異誘発に供した。第1ライブラリーを設計して、アンチトロンビンへのヒトFVIIa結合に対する保存的変化の影響を調べた。様々な種(チンパンジー、イヌ、ブタ、ウシ、マウス、ラットおよびウサギ)由来のFVII配列のアライメントを図2に示す。他の種において異なる側鎖を有する、アンチトロンビンに近接するヒトFVIIaにおける側鎖を、対応する種の側鎖に変異させた。1つの例は、ヒトFVIIにおけるGlnおよびブタFVIIにおけるArgである286位の残基である。第1ライブラリーをスクリーニング後、第2の集中ライブラリーを次いで設計し、選択した位置における可能性のあるアミノ酸置換(CysおよびProとは別に)の全てまたは幾つかを試験した。例には、配列番号1による176、286、および293位が含まれる。
FVIIa-アンチトロンビン相互作用をマッピングするために、FVIIaバリアントライブラリーを、FVIIa-アンチトロンビン複合体の複合体の構造的モデルに基づきインシリコで設計した。図1に示されたモデルは、テンプレートとしてFXa-アンチトロンビンミカエリス複合体の公開されているX線構造(Johnsonら 2006)を用いて構築した。該モデルにおいてアンチトロンビンに近接するFVIIa残基(FVIIaとアンチトロンビン側鎖の間の最大距離は12Åであった)を、変異誘発に供した。第1ライブラリーを設計して、アンチトロンビンへのヒトFVIIa結合に対する保存的変化の影響を調べた。様々な種(チンパンジー、イヌ、ブタ、ウシ、マウス、ラットおよびウサギ)由来のFVII配列のアライメントを図2に示す。他の種において異なる側鎖を有する、アンチトロンビンに近接するヒトFVIIaにおける側鎖を、対応する種の側鎖に変異させた。1つの例は、ヒトFVIIにおけるGlnおよびブタFVIIにおけるArgである286位の残基である。第1ライブラリーをスクリーニング後、第2の集中ライブラリーを次いで設計し、選択した位置における可能性のあるアミノ酸置換(CysおよびProとは別に)の全てまたは幾つかを試験した。例には、配列番号1による176、286、および293位が含まれる。
(実施例2)
Novagen社製KOD Xtreme(商標)Hot Start DNAポリメラーゼ、またはStratagene社製QuickChange(登録商標)部位特異的変異誘発キットを用いる部位特異的変異誘発PCRベースの方法を用いて、FVII cDNAをコードする哺乳動物発現ベクターに変異を導入した。以下の発現ベクターを使用した: HEK293FおよびHKB11細胞のトランスフェクションにはpTT5[Durocherら(2002)Nucleic Acid Res. 30(2):e9]; CHOEBNALT85のトランスフェクションにはIcosagen社(エストにエア)製pQMCF; CHO-K1のトランスフェクションにはpZEM219b[Busbyら(1991)J.Biol.Chem.、266:15286〜15292頁]およびpMPSVHE[Arteltら(1988) Gene 62:213〜219頁]; BHK細胞のトランスフェクションにはpLN174[Perssonら(1996)FEBS Lett.、385:241〜243頁]。プライマーは、製造者の推奨に従って設計した。所望の変異の導入は、DNA配列決定(MWG Biotech社、ドイツ)により検証した。
Novagen社製KOD Xtreme(商標)Hot Start DNAポリメラーゼ、またはStratagene社製QuickChange(登録商標)部位特異的変異誘発キットを用いる部位特異的変異誘発PCRベースの方法を用いて、FVII cDNAをコードする哺乳動物発現ベクターに変異を導入した。以下の発現ベクターを使用した: HEK293FおよびHKB11細胞のトランスフェクションにはpTT5[Durocherら(2002)Nucleic Acid Res. 30(2):e9]; CHOEBNALT85のトランスフェクションにはIcosagen社(エストにエア)製pQMCF; CHO-K1のトランスフェクションにはpZEM219b[Busbyら(1991)J.Biol.Chem.、266:15286〜15292頁]およびpMPSVHE[Arteltら(1988) Gene 62:213〜219頁]; BHK細胞のトランスフェクションにはpLN174[Perssonら(1996)FEBS Lett.、385:241〜243頁]。プライマーは、製造者の推奨に従って設計した。所望の変異の導入は、DNA配列決定(MWG Biotech社、ドイツ)により検証した。
(実施例3)
FVIIバリアントは、ベビーハムスター腎臓(BHK)細胞、Freestyle(商標)293-Fヒト胚性腎臓細胞[HEK293F; Gibco社(Life Technologies社、ナエウム、DKによる]、HKB11(HEK293およびヒトB細胞系のハイブリッド細胞系)細胞(ATCC、LGC Standards AB社、ボロース、スウェーデン)、チャイニーズハムスター卵巣(CHOK1)細胞、またはIcosagen Cell Factory社(エストニア)製CHO-EBNALT85細胞のいずれかで発現させた。
FVIIバリアントは、ベビーハムスター腎臓(BHK)細胞、Freestyle(商標)293-Fヒト胚性腎臓細胞[HEK293F; Gibco社(Life Technologies社、ナエウム、DKによる]、HKB11(HEK293およびヒトB細胞系のハイブリッド細胞系)細胞(ATCC、LGC Standards AB社、ボロース、スウェーデン)、チャイニーズハムスター卵巣(CHOK1)細胞、またはIcosagen Cell Factory社(エストニア)製CHO-EBNALT85細胞のいずれかで発現させた。
BHK接着細胞は、安定な細胞系を産生するために製造者の指示に従って、Merck Millipore社(ヘレルプ、デンマーク)製GeneJuice(登録商標)を用いてFVIIバリアント構築物でトランスフェクトした。メトトレキサート(Sigma-Aldrich社)を選択試薬として使用した。安定な細胞系を培地で大規模に培養し、合計5から10リットルの細胞上清を得た。2%(V/V)ウシ胎仔血清[Gibco社(Life Technologies社、ナエウム、DKによる)]、1%(v/v)ペニシリン/ストレプトマイシン[Gibco社(Life Technologies社、ナエウム、DKによる)]、および5mg/lビタミンK1(Sigma-Aldrich社)を補充したDMEM[Gibco社(Life Technologies社、ナエウム、DKによる)]中、37℃、5または8% CO2でインキュベーター中で細胞を培養した。
HEK293FおよびHKB11懸濁細胞は、製造者の指示に従って293Fectin[(商標)(Invitrogen社(Life Technologies社、ナエウム、DKによる)]を用いて一過性トランスフェクトした。細胞を37℃、5または8% CO2および85から125rpmで、振盪インキュベーター中で培養した。トランスフェクト細胞を中規模から大規模発現に増大させ、合計250ml〜1リットルの細胞上清を得た。上清を遠心分離により回収し、この後、0.22μM PESフィルター(Corning社; Fischer Scientific Biotech社、スランゲルプ、DK)により濾過した。HEK293FおよびHKB11細胞を、1%(v/v)ペニシリン/ストレプトマイシン[Gibco社(Life Technologies社、ナエウム、DKによる)]およびビタミンK1(Sigma-Aldrich社)を補充したFreestyle 293発現培地[Gibco社(Life Technologies社、ナエウム、DKによる)]中で培養した。
CHOEBNALT85懸濁細胞は、エレクトロポレーション(Gene Pulse Xcell、Biorad社、コペンハーゲン、DK)により一過性トランスフェクトした。トランスフェクト細胞を700μg/l Geneticin(登録商標)[Gibco社(Life Technologies社による)]により選択し、中規模/大規模に発現させ、合計500mlから10リットルの上清を得た。細胞を、5mg/lビタミンK1 (Sigma-Aldrich社)を補充した培地で製造者の指示に従って培養した。細胞を37℃、5または8% CO2および85または125rpmで、振盪インキュベーター中で培養した。上清を遠心分離により回収し、この後、0.22μM PESフィルター(Corning社; Fischer Scientific Biotech社、スランゲルプ、DK)により濾過した。
懸濁細胞に適合させたCHOK1細胞は、製造者の推奨に従ってエレクトロポレーション(Gene Pulse Xcell、Biorad社、コペンハーゲン、DK)によりトランスフェクトした。700μg/l Geneticin(登録商標)[Gibco社(Life Technologies社による)]を選択試薬として使用した。安定な細胞系を大規模発現に使用した。細胞を37℃、5または8% CO2、および85または125rpmでインキュベーター中で培養した。Thermo Scientific Hyclone CDM4CHOTM培地を補充した1%(v/v)ペニシリン/ストレプトマイシン[Gibco社(Life Technologies社、ナエウム、DKによる)]および5mg/lビタミンK1(Sigma-Aldrich社)を発現に使用した。上清を0.22μM PESフィルター[Corning社(Fischer Scientific社、スランゲルプ、DK)]により濾過した。
大規模発現(BHK)- 接着BKH細胞系を、5mg/lビタミンK1および2%胎仔ウシ血清[Invitrogen社(Life Technologies、ナエウム、DKによる)]を補充したDMEM/F12培地[Invitrogen社(Life Technologies、ナエウム、DKによる)]中で培養した。バイオリアクター用の種培養の増殖中は、10%胎仔ウシ血清を使用し、培地に1μMメトトレキサート(Sigma-Aldrich社、コペンハーゲン、DK)を補充した。簡単には、細胞をベントT-175フラスコ中で増殖させ、2層および10層の細胞ファクトリーを37℃および5% CO2でインキュベートした。次の工程に移る前に、TrypLE(商標)Express[Gibco社(Life Technologies社、ナエウム、DKによる)]を用いて集密で細胞を解離させた。産生期は、マイクロキャリア(5g/L、Cytodex 3、GE Life Sciences社、ウプサラ、SE)によりバイオリアクター中で反復バッチ培養として行った。pHは、CO2およびNa2CO3の添加により約7に制御した。溶存酸素濃度は、酸素を注入して大気中飽和の50%超を保った。温度は36.5℃で維持した。取り出した回収物は、精製前に濾過(3μm Clarigard、Opticap XL10; 0.22μm Durapore、Opticap XL10、Merck Millipore社、ヘレルプ、DK)により清澄化させた。
大規模発現(CHOK1)- 懸濁液適合CHOK1細胞系を、5mg/LビタミンK1を補充した既知組成培地(CDM4CHO、Thermo Scientific HyClone、Fisher Scientific社、スランゲルプ、DK)中で培養した。バイオリアクター用の種培養の増殖中は、培地に600μg/ml Geneticin(登録商標)[Invitrogen社(Life Technologies、ナエウム、DKによる)]を補充した。簡単には、細胞をベント振盪フラスコ中で増殖させ、37℃および5% CO2でオービタルシェーカー中でインキュベートした。産生期は、バイオリアクター中で反復バッチ培養として行った。pHは、CO2およびNa2CO3の添加により約7に制御した。溶存酸素濃度は、酸素を注入して大気中飽和の50%超を保った。温度は36.5℃で維持した。取り出した回収物は、精製前に濾過(3μm Clarigard、Opticap XL10; 0.22μm Durapore、Opticap XL10、Merck Millipore社、ヘレルプ、DK)により清澄化させた。
(実施例4)
FVIIバリアントを、他で本質的に記載されているように(Thimら 1988)、Glaドメイン特異的抗体親和性クロマトグラフィーにより精製した。簡単には、プロトコルは1から3工程を含んだ。工程1では、5m MCaCl2を馴化培地に添加し、試料を親和性カラムに充填した。20mM HEPES、2M NaCl、10mM CaCl2、0.005% Tween80、pH8.0による広範囲な洗浄後、結合タンパク質を20mM HEPES、20mM NaCl、20mM EDTA、0.005% Tween80、pH8.0で、(工程2)アニオン交換カラム(Source 15Q、GE Healthcare社)に溶出した。20mM HEPES、20mM NaCl、0.005% Tween80、pH8.0による洗浄後、結合タンパク質を、20mM HEPES、135mM NaCl、10mM CaCl2、0.005% Tween80、pH8.0で、(工程3)ヒト血漿由来FXaが製造者の指示に従って1mg/mlの密度でカップリングされたCNBr-Sepharose Fast Flowカラム(GE Healthcare社)に溶出した。流速を最適化して、活性形態への精製チモーゲンバリアントの完全活性化を本質的に確保した。馴化培地またはアニオン交換カラムにおいて自己活性化することができる増強した活性を有するFVIIaバリアントについては、タンパク質分解変性を防ぐために工程2および/または工程3を省略た。精製タンパク質は-80℃で保管した。タンパク質の品質をSDS-PAGE分析により評価し、活性部位滴定または以下に記載されたrpHPLCによる軽鎖含量の定量化により、機能的分子の濃度を測定した。
FVIIバリアントを、他で本質的に記載されているように(Thimら 1988)、Glaドメイン特異的抗体親和性クロマトグラフィーにより精製した。簡単には、プロトコルは1から3工程を含んだ。工程1では、5m MCaCl2を馴化培地に添加し、試料を親和性カラムに充填した。20mM HEPES、2M NaCl、10mM CaCl2、0.005% Tween80、pH8.0による広範囲な洗浄後、結合タンパク質を20mM HEPES、20mM NaCl、20mM EDTA、0.005% Tween80、pH8.0で、(工程2)アニオン交換カラム(Source 15Q、GE Healthcare社)に溶出した。20mM HEPES、20mM NaCl、0.005% Tween80、pH8.0による洗浄後、結合タンパク質を、20mM HEPES、135mM NaCl、10mM CaCl2、0.005% Tween80、pH8.0で、(工程3)ヒト血漿由来FXaが製造者の指示に従って1mg/mlの密度でカップリングされたCNBr-Sepharose Fast Flowカラム(GE Healthcare社)に溶出した。流速を最適化して、活性形態への精製チモーゲンバリアントの完全活性化を本質的に確保した。馴化培地またはアニオン交換カラムにおいて自己活性化することができる増強した活性を有するFVIIaバリアントについては、タンパク質分解変性を防ぐために工程2および/または工程3を省略た。精製タンパク質は-80℃で保管した。タンパク質の品質をSDS-PAGE分析により評価し、活性部位滴定または以下に記載されたrpHPLCによる軽鎖含量の定量化により、機能的分子の濃度を測定した。
活性部位滴定によるFVIIaバリアント濃度の測定- 精製調製物中の機能的分子の濃度は、他で本質的に記載されているように(Bock, 1992)、準化学量論的レベルのd-Phe-Phe-Argクロロメチルケトン(FFR-cmk; Bachem社)で滴定した時のアミド分解活性の不可逆的喪失から、活性部位滴定により決定した。簡単には、全てのタンパク質をアッセイ緩衝液[50mM HEPES(pH7.4)、100mM NaCl、10mM CaCl2、1mg/mL BSA、および0.1%(w/v)PEG8000]に希釈した。最終濃度の150nM FVIIaバリアントを、500nM可溶性組織因子(sTF)と10分間プレインキュベートし、この後、96ウェルプレートにおいて100μLの総反応容量中0〜300nMの最終濃度でFFR-cmkを添加した(n=2)。反応物を室温で一晩インキュベートした。別の96ウェルプレートにおいて、20μLの各反応物を、1mM S-2288(Chromogenix社、ミラノ、イタリア)を含有するアッセイ緩衝液に10倍希釈した。吸光度増加は、SOFTmax PROソフトウェアを備えたSpectramax 190マイクロプレート分光光度計において、405nMで連続的に10分間測定した。アミド分解活性は、ブランク減算後の線形プログレス曲線の傾斜として報告した。活性部位濃度は、アミド分解活性を完全に消失させるのに必要とされるFFR-cmkの最小濃度として外挿により決定した。
逆相HPLCを用いた軽鎖含量からのFVIIaバリアント濃度の測定- 代替のアプローチにおいて、精製調製物中の機能的FVIIa分子の濃度は、逆相HPLC(rpHPLC)によるFVIIa軽鎖(LC)含量の定量化により決定した。野生型FVIIaによる検量線を、0から3μMの範囲のFVIIa濃度を用いて作成する一方、未知濃度の試料は、1.5μMの推定濃度で調製した(n=2)。全ての試料を、20%(v/v)の濃度まで試料に添加した0.5Mトリス(2-カルボキシエチル)ホスフィン(TCEP; Calbiochem/Merck KGaA社、ダルムシュタット、ドイツ)およびギ酸の1:1混合物を用いて還元し、この後、試料を70℃で10分間加熱した。還元FVIIaバリアントを、30℃に維持したC4カラム(Vydac社、300Å、粒径5μM、4.6mm、250mm)に充填した。移動相は、0.09% TFA水溶液(溶媒A)および0.085% TFAアセトニトリル溶液(溶媒B)から成った。80μL試料を注入後、システムを4分間、25%溶媒Bで均一濃度、この後、10分にわたり25〜46% Bの直線勾配で実行した。ピークは、それぞれ280および348nmの励起および発光波長を用いて、蛍光により検出した。軽鎖定量化はピーク積分により行い、FVIIaバリアントの相対量を野生型FVIIa標準曲線に基づき計算した。
(実施例5)
実質的に保存された活性を有するが、血漿インヒビターアンチトロンビンとの反応性が低下したFVIIaバリアントを同定するために、生成したバリアントライブラリーを、手動および自動フォーマットの両方で確立された以下に詳述されたスクリーニングアッセイに供した。簡単には、活性を、リン脂質ベシクルの存在下、巨大分子基質第X因子をタンパク質分解的に活性化する各バリアントの能力として測定した(インビトロタンパク質分解アッセイ)。組換えFVIIaの、可能性のあるTF依存性および非依存性作用機構を模倣するために、各反応は、補因子である組織因子(sTF)の存在下または非存在下で行った。アンチトロンビンによる阻害に対する各バリアントの感受性は、インビボでの反応を加速させる内因性ヘパリン様グリコサミノグリカン(GAG)の能力を模倣するための低分子量ヘパリンの存在下、擬一次条件(pseudo-first order condition)下で定量化した。図4に示されている通り、測定したインビトロでのアンチトロンビン反応性は、FVIIa-アンチトロンビン複合体のインビボ蓄積と相関することが見出され、故にインビトロスクリーニング手順の予測性を確証した。
実質的に保存された活性を有するが、血漿インヒビターアンチトロンビンとの反応性が低下したFVIIaバリアントを同定するために、生成したバリアントライブラリーを、手動および自動フォーマットの両方で確立された以下に詳述されたスクリーニングアッセイに供した。簡単には、活性を、リン脂質ベシクルの存在下、巨大分子基質第X因子をタンパク質分解的に活性化する各バリアントの能力として測定した(インビトロタンパク質分解アッセイ)。組換えFVIIaの、可能性のあるTF依存性および非依存性作用機構を模倣するために、各反応は、補因子である組織因子(sTF)の存在下または非存在下で行った。アンチトロンビンによる阻害に対する各バリアントの感受性は、インビボでの反応を加速させる内因性ヘパリン様グリコサミノグリカン(GAG)の能力を模倣するための低分子量ヘパリンの存在下、擬一次条件(pseudo-first order condition)下で定量化した。図4に示されている通り、測定したインビトロでのアンチトロンビン反応性は、FVIIa-アンチトロンビン複合体のインビボ蓄積と相関することが見出され、故にインビトロスクリーニング手順の予測性を確証した。
バリアントスクリーニングからの結果をTable 2(表2)に示す。バリアントのうち、Lys(K)、Arg(R)、Tyr(Y)、またはPhe(F)とのT293の置換は、アンチトロンビン反応性を野生型FVIIaの10%以下のレベルまで低下させたが、sTFの非存在下、タンパク質分解活性は野生型レベルをわずかに上回って維持された。T293Yバリアントについては、野生型FVIIaの>200%の活性レベルが観察された。同様に、Q176のLys(K)、Arg(R)、およびAsn(N)置換は、アンチトロンビン反応性を劇的に低下させる一方、実質的にタンパク質分解活性を野生型レベルで保存した。著しくは、1%未満のアンチトロンビン反応性がQ176Rバリアントについて観察された。
第X因子を基質として用いるタンパク質分解活性の測定(手動インビトロタンパク質分解アッセイ)- FVIIaバリアントのタンパク質分解活性を、血漿由来第X因子(FX)を基質として用いて推定した。全てのタンパク質を、アッセイ緩衝液[50mM HEPES(pH7.4)、100mM NaCl、10mM CaCl2、1mg/mL BSA、および0.1%(w/v)PEG8000]に希釈した。FX活性化の速度論的パラメータを、96ウェルプレートにおいて100μlの総反応容量中、25μM 75:25ホスファチジルコリン:ホスファチジルセリンリン脂質ベシクル(PS:PC; Haematologic Technologies社、バーモント、USA)の存在下、10nMの各FVIIaコンジュゲートを40nM FXと室温で30分間インキュベートして決定した(n=2)。sTFの存在下のFX活性化は、100μlの総反応容量中、25μM PC:PSリン脂質の存在下、5pMの各FVIIaコンジュゲートを30nM FXと室温で20分間インキュベートして決定した(n=2)。インキュベーション後、50μl停止緩衝液[50mM HEPES(pH7.4)、100mM NaCl、80mM EDTA]の添加と、これに続く50μl 2mM S-2765色素産生基質(Chromogenix社、ミラノ、イタリア)の添加により反応を停止させた。最後に、吸光度増加を、Spectramax 190マイクロプレートリーダーにおいて405nMで連続的に測定した。見かけの触媒速度値(kcat/Km)を、線形回帰を用いてミカエリスメンテンの式(v=kcat*[S]*[E]/Km)の単純化形式にデータをフィットさせて推定した。この理由は、FX基質濃度([S])が活性化反応に関してKmを下回るためである。生成されたFXaの量を、同一条件下のヒト血漿由来FXaで作成した標準曲線から推定した。推定kcat/km値は、野生型FVIIaのものと比べて報告した。結果をTable 2(表2)およびTable 3(表3)に示す。
第X因子を基質として用いるタンパク質分解活性の測定(自動インビトロタンパク質分解アッセイ)- FVIIaバリアントのタンパク質分解活性を、血漿由来第X因子(FX)を基質として用いて推定した。全てのタンパク質を、50mM HEPES(pH7.4)、100mM NaCl、10mM CaCl2、1mg/mL BSA、および0.1%(w/v)PEG8000に希釈した。相対的タンパク質分解活性を、96ウェルプレートにおいて100μLの総反応容量中、25μmM 75:25ホスファチジルコリン:ホスファチジルセリン(PC:PS)リン脂質(Haematologic technologies社、バーモント、USA)の存在下、10nMの各FVIIaコンジュゲートを40nM FXと室温で30分間インキュベートして決定した(n=2)。sTFの存在下のFX活性化は、100μLの総反応容量中、25μM PC:PSリン脂質の存在下、5pMの各FVIIaコンジュゲートを30nM FXと室温で20分間インキュベートして決定した(n=2)。インキュベーション後、100μL 1mM S-2765を停止緩衝液[50mM HEPES(pH7.4)、100mM NaCl、80mM EDTA]に添加して反応を停止させた。停止直後に、吸光度増加を、Envisionマイクロプレートリーダー(PerkinElmer社、ウォルサム、MA)において405nMで連続的に測定した。全ての添加、インキュベーション、およびプレート移動は、Envisionマイクロプレートリーダーにカップリングしたライン上のHamilton Microlab Starロボット(Hamilton社、ボナドゥウ、スイス)により行った。タンパク質分解活性は、野生型FVIIaと比べて計算した。結果をTable 2(表2)およびTable 3(表3)に示す。
アンチトロンビンによるFVIIa阻害の測定(手動アッセイ)- 不連続方法を使用して、低分子量(LMW)ヘパリン(Calbiochem/Merck KGaA社、ダルムシュタット、ドイツ)の存在下、擬一次条件下でヒト血漿由来アンチトロンビン(AT)による阻害のインビトロ速度を測定した。アッセイは、200μlの総反応容量中、50mM HEPES(pH7.4)、100mM NaCl、10mM CaCl2、1mg/mL BSA、および0.1%(w/v)PEG8000を含有するアッセイ緩衝液を用いて、96ウェルプレートで行った。200nM FVIIaおよび12μM LMWヘパリンの混合物に対し、100μlの最終反応容量中5μMアンチトロンビンを添加した。種々の時間で、180μLのsTF(200nM)、反応を停止させるためのポリブレン(0.5mg/ml;臭化ヘキサジメトリン、Sigma-Aldrich社)、およびS-2288(1mM)を含有する別のマイクロタイタープレートに、20μlの反応混合物を移して反応を停止させた。種々の時間で移した直後に、基質切断をSpectramax 190マイクロプレートリーダーにおいて405nmで10分間モニタリングした。擬一次速度定数(kobs)は、指数関数的減衰関数へのデータの非線形最小二乗フィッティングにより得、および二次速度定数(k)は、以下の関係k=kobs/[AT]から得た。阻害の速度は、野生型FVIIaのものと比べて報告した。結果をTable 2(表2)およびTable 3(表3)に示す。
アンチトロンビンによるFVIIa阻害の測定(自動アッセイ)- 不連続方法を使用して、低分子量(LMW)ヘパリン(Calbiochem/Merck KGaA社、ダルムシュタット、ドイツ)の存在下、擬一次条件下でヒト血漿由来アンチトロンビン(AT)による阻害のインビトロ速度を測定した。アッセイは、200μLの総反応容量中、50mM HEPES(pH7.4)、100mM NaCl、10mM CaCl2、1mg/mL BSA、および0.1%(w/v)PEG8000を含有する緩衝液を用いて、96ウェルプレートで行った。200nM FVIIaおよび12μM LMWヘパリンの混合物に対し、100μLの最終反応容量中5μMアンチトロンビンを添加した。種々の時間で、180μLのsTF(200nM)、ポリブレン(0.5mg/mL;臭化ヘキサジメトリン、Sigma-Aldrich社)、およびS-2288(1mM)を含有する別のマイクロタイタープレートに、20μLの反応混合物を移して反応を停止させた。種々の時間で移した直後に、基質切断をEnvisionマイクロプレートリーダーにおいて405nmで10分間モニタリングした。擬一次速度定数(kobs)は、指数関数的減衰関数へのデータの非線形最小二乗フィッティングにより得、二次速度定数(k)は、以下の関係k=kobs/[AT]から得た。全ての添加、インキュベーション、およびプレート移動は、Envisionマイクロプレートリーダー(PerkinElmer社、ウォルサム、MA)にカップリングしたライン上のHamilton Microlab Starロボット(Hamilton社、ボナドゥウ、スイス)により行った。阻害の速度は、野生型FVIIaのものと比べて報告した。結果をTable 2(表2)およびTable 3(表3)に示す。
(実施例6)
同定したアンチトロンビン耐性FVIIaバリアントのセレクションを、活性増強置換M298Q、V158D/E296V/M298Q、またはL305V/S314E/K337A/F374Yと組み合わせてさらに評価した。
同定したアンチトロンビン耐性FVIIaバリアントのセレクションを、活性増強置換M298Q、V158D/E296V/M298Q、またはL305V/S314E/K337A/F374Yと組み合わせてさらに評価した。
実施例5に記載されたタンパク質分解アッセイを用いる精製タンパク質調製物の特徴付けは、該バリアントがスーパー活性を保持することを示した。これは、実施例7に記載されたSTACLOT(登録商標)VIIa-rTF血漿ベースのアッセイを用いる効力(potency)推定により確認され、幾つかのバリアントについては、実施例8に記載されたFVIII欠乏血漿におけるトロンビン生成およびトロンボエラストグラフィーによっても確認された[Table 2(表2)参照]。さらに、T293KおよびQ176K変異は、M298Qのアンチトロンビン反応性を野生型FVIIaの10%未満まで効果的に低下させたが、あまり顕著でない低下が、より活性な(およびアンチトロンビン反応性)バリアントV158D/E296V/M298QまたはL305V/S314E/K337A/F374Yとの組み合わせにおいて観察された[Table 4(表4)参照]。V158D/E296V/M298Qバックグラウンドでは、T293K変異は、アンチトロンビン反応性を野生型レベルの約20%まで低下させた。反対に、T293AまたはT293Lはいずれも、M298Qバックグラウンドでアンチトロンビン反応性を100%未満に維持することができなかった。これらのデータは、Q176KおよびT293K変異は活性の維持に関して優れている一方、アンチトロンビン反応性を実質的に低下させることを示している。
(実施例7)
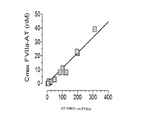
効力は、市販のFVIIa特異的凝固アッセイ、Diagnostica Stago社製STACLOT(登録商標)VIIa-rTFを用いて推定した。アッセイは、J. H. Morrisseyら、Blood. 81:734〜744頁(1993)により公開されている方法に基づいた。これは、リン脂質の存在下、FVII欠乏血漿におけるフィブリン血栓形成までのsTF開始FVIIa活性依存的時間(sTF initiated FVIIa activity-dependent time)を測定するものである。凝固時間は、ACL9000(ILS社)凝固計器で測定し、結果は、FVIIa検量線に基づく両対数関数的(bilogarithmic)スケールで線形回帰を用いて計算した。同じアッセイを、動物PK試験からの血漿試料におけるFVIIa凝固活性の測定に使用した。血漿中の定量下限(LLOQ)は0.25U/mlと推定した。血漿活性レベルを特異的活性を用いてnMに変換した。結果をTable 2(表2)および図3に示す。
効力は、市販のFVIIa特異的凝固アッセイ、Diagnostica Stago社製STACLOT(登録商標)VIIa-rTFを用いて推定した。アッセイは、J. H. Morrisseyら、Blood. 81:734〜744頁(1993)により公開されている方法に基づいた。これは、リン脂質の存在下、FVII欠乏血漿におけるフィブリン血栓形成までのsTF開始FVIIa活性依存的時間(sTF initiated FVIIa activity-dependent time)を測定するものである。凝固時間は、ACL9000(ILS社)凝固計器で測定し、結果は、FVIIa検量線に基づく両対数関数的(bilogarithmic)スケールで線形回帰を用いて計算した。同じアッセイを、動物PK試験からの血漿試料におけるFVIIa凝固活性の測定に使用した。血漿中の定量下限(LLOQ)は0.25U/mlと推定した。血漿活性レベルを特異的活性を用いてnMに変換した。結果をTable 2(表2)および図3に示す。
(実施例8)
非修飾および40kグリコペグ化形態でのアンチトロンビン耐性バリアントのセレクションを、FVIII欠乏血漿におけるトロンビン生成および血栓形成に対する該バリアントの影響について以下に記載されているように試験した。結果をTable 2(表2)に示す。
非修飾および40kグリコペグ化形態でのアンチトロンビン耐性バリアントのセレクションを、FVIII欠乏血漿におけるトロンビン生成および血栓形成に対する該バリアントの影響について以下に記載されているように試験した。結果をTable 2(表2)に示す。
ヒトドナー血液のトロンビン生成アッセイ(TGA)- トロンビン生成は、健康なドナー由来の血小板リッチ血漿(PRP)において測定した(最終血小板濃度は150×109/lであった)。PRPを阻害性抗ヒト第VIII因子IgGで処理して、血友病A様状態を誘発した。アッセイを開始する約5分前に、100μM最終濃度のPAR-1アゴニストSFFLRN(Bachem社、ブーベンドルフ、スイス)、および100ng/ml最終濃度のコラーゲン受容体(GPVI)アゴニストコンバルキシン(Pentapharm社、バーゼル、スイス)を添加して血小板を活性化した。FVIIaおよびFVIIaバリアントを、血小板アゴニストを含有する80μl PRPと一緒に20μlの容量でマイクロタイタープレートに添加した。16.7mMの最終濃度でCaCl2を含有する20μl蛍光発生トロンビン基質(FluCaキット、Thrombinoscope bv社、マーストリヒト、オランダ)を添加して、反応を開始した。トロンビン生成は、Fluoroscan Ascent(登録商標)蛍光光度計(Thermo Fisher Scientific社、ヘルシンキ、フィンランド)を用いて120分間連続的に測定した。蛍光シグナルは、記載されているように(Hemkerら 2003)、390nm(励起)および460nm(発光)の波長で検出し、α2-マクログロブリン結合トロンビンについて補正し、較正器(Thrombinoscope社)およびThrombinoscopeソフトウェア(Synapse BV社、マーストリヒト、オランダ)により生成されたモル(nM)トロンビンに変換した。トロンビン生成の速度は、トロンビンピーク/(ピークまでの時間-ラグタイム)として計算した。トップのプラトーレベルを得ることができなかったため、EC50値を生成することはできなかった。代わりに、グラフ上の最も急勾配の部分に表された特定の速度を得るのに必要とされる化合物の濃度を比較して、野生型FVIIaと比べたバリアントの活性を推定した。
ヒト全血のトロンボエラストグラフィー(TEG)- TEG分析は、健康なドナー由来の全血で行った(Viuffら Thrombosis Research、2010;126, 144〜149頁に本質的に記載されているように)。血液を阻害性抗ヒト第VIII因子IgGで処理して、血友病A様状態を誘発した。FVIIa、FVIIaバリアントまたは緩衝液(HEPES 20mM、NaCl 150mM、BSA 2%)を、カオリン(Haemoscope社、ナイルズ、IL、USA)を含有する試験管に添加し、試験管を反転させて全血と慎重に混合した。試料をTEGカップに移し、再石灰化して凝固を開始させた。止血過程は、TEG凝固分析器(5000 series TEG分析器、Haemoscope Corporation社、シカゴ、USA)により記録した。TEG凝固時間[R、試料カップに血液を入れてから血栓が形成し始めるまでの待ち時間を表す(2mm振幅)]、および血栓形成(MTG、最大血栓生成)の速度を分析に使用した。試料は単一試料として分析し、実験は2回行った(毎回異なるドナー)。データ分析は、Haemoscopeソフトウェア、バージョン4により行った。EC50値は、パラメータごとにフィットした4パラメータロジスティック濃度反応曲線に基づき計算した。
(実施例9)
同定した置換がアンチトロンビン認識に影響を与える機構を調べるために、代表的なバリアント(FVIIa Q176K)の結晶構造を決定した。
同定した置換がアンチトロンビン認識に影響を与える機構を調べるために、代表的なバリアント(FVIIa Q176K)の結晶構造を決定した。
可溶性組織因子(断片1-209)との複合体における精製H-D-Phe-Phe-Argクロロメチルケトン(FFR-cmk; Bachem社、スイス)活性部位阻害FVIIa Q176Kは、(Bjelkeら 2008)に従って懸滴方法を用いて結晶化させた。タンパク質緩衝液溶液は、10mMトリス-HCl、100mM NaCl、15mM CaCl2、pH7.5の混合物であり、タンパク質濃度は5.8mg/mlであった。沈殿ウェル溶液(precipitation, well, solution)は、100mMクエン酸ナトリウム、pH5.6、16.6% PEG 3350、および12% 1-プロパノールであった。懸滴は、1.5μlのプロテイン溶液および0.5μlのウェル溶液の混合物により、1mlのウェル溶液を用いて24ウェルVDXプレートでセットした。結晶は、最大0.3×0.15×0.05mmの寸法を有する薄いプレートとして成長した。
結晶を、80容量%結晶化ウェル溶液および20%グリセロール(99%純度)を含有する溶液に移した。結晶を約30秒間浸したままにし、この後、結晶を液体窒素に移し、液体窒素中で急速冷凍した。X線回析データは、ビームライン911-3、MAX-lab synchrotron社、ルンド、スウェーデン(Mammenら、2002)で収集した。結晶の一部は単一であったが、他の部分は双晶形成を示した。結晶の非双晶部分からの完全なデータセットを得た。データをXDSデータ整理ソフトウェア(Kabsch、2010)により処理し、1.95Åの最終分解能カットオフを得た。結晶学的データ、精密化統計値およびモデル統計値をTable 5(表5)に示す。
タンパク質データバンク(Bermanら、2000)からの3ELAエントリー(Bjelkeら 2008)の結晶座標を、CCP4ソフトウェアパッケージ(Collaborative Computational Project、1994)のREFMAC5(Murshudovら、2011)における剛体精密化の開始モデルとして使用した。精密化に続いて、コンピュータグラフィックソフトウェアCOOT(Emsleyら、2010)で双方向モデル補正を行った。座標精密化およびモデル構築は、この後、PHENIXソフトウェアパッケージ(Adamsら、2010)およびPHENIX.REFINEソフトウェア(Afonineら、2012)に移した。得られた最終R-freeおよびR-freeは、それぞれ0.183および0.216であった。結晶学的データ、精密化およびモデル統計値をTable 5(表5)に示す。
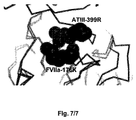
構造解析- 重鎖FVIIa Lys 176残基は、ベータストランドA1とB1の間のループ/ベータストランドB1の極めて始めに位置する。重鎖Lys 176残基の電子密度は、尤度重み付け2mFo-DFcマップで1.0カットオフを用いる場合、主鎖および側鎖C原子までに対して明白に示される。Lys 176側鎖の配向は、重鎖293 Thr残基の方向におよそ3.5Å離れている。FVIIIa Q176K構造とタンパク質データバンク(Berman、Westbrook、Feng、Gilliland、Bhat、Weissig、Shindyalov、& Bourne、2000) 1DAN構造(Bannerら、1996)との比較は、構造間の高い類似性を示す。FVIIa FFR-cmk阻害およびGlaドメイン含有1DAN構造は、別の空間群、P41212で結晶化し、ドメイン間配向に小さな違いがある。Cα原子間の平均二乗偏差(RMSD)を、GESAMT(Krissinel、2012)およびCCP4ソフトウェアプログラムパッケージ(Collaborative Computational Project、1994)のLSQKABにより計算した。2つの複合体の3つの共通する鎖、FVIIa重鎖(H)、FVIIa軽鎖(L)、および組織因子(T)の全体的なRMSDは、0.796Å(529 Cα原子対で)であるが、触媒ドメイン、FVIIa重鎖のみのRMSDは0.347Åである。図6は、LSQKAB重ね合わせからの個々のCα-Cα距離が、FVIIa変異体Q176Kの触媒ドメインと1DAN構造からの触媒ドメインの間に走っていることを示している。
アンチトロンビンとFVIIa変異体Q176Kの間の可能性のある相互作用を試験するために、PDBコード2GD4を有する座標の第Xa因子分子(Johnsonら、2006)においてFVIIa変異体の重ね合わせを行った。FXa分子とFVIIa分子間の識別位置(identity position)は34.1%であるが、コンセンサス位置(consensus position)は49.4%である。GESAMTソフトウェアを用いた重ね合わせは、1.05ÅのRMSD、Q=0.783、および229残基のアライメント長を示した。ライディング(riding)アンチトロンビン分子から、もしアンチトロンビンがFVIIa(Q176K)と相互作用していれば、2つの正に帯電したアミノ酸、アンチトロンビンのArg399およびFVIIa重鎖のK176は、極めて近くなるが[Table 6(表6)および図7参照]、両方の残基が正に帯電していることから、結果は2つの残基間の静電反発力であることが次いで明らかである。さらに、アンチトロンビンArg399は、アンチトロンビン分子の反応性中心ループ(RCL)の一部であり(Johnsonら 2006)、記載された反発力は、アンチトロンビンがこのRCLをFVIIaの活性部位に置く可能性に対してほぼ確実にマイナスに影響するであろう。これによりQ176K変異FVIIa分子は、アンチトロンビンによる阻害を受けにくくなろうであろう。これは、アンチトロンビンによる不活化に対する耐性の増加、半減期の延長とこれに続く、活性のほんのわずかな低下を示す、Table 2(表2)、Table 3(表3)および図3で観察されるものと一致しており、これを説明するものである。
(実施例10)
活性増強置換および化学的修飾と組み合わせたアンチトロンビン耐性の効果を評価するために、FVIIaバリアントのセレクション[Table 2(表2)参照]を、以下のセクションに記載された酵素的糖コンジュゲーションにより40kDa PEGにコンジュゲートした。
活性増強置換および化学的修飾と組み合わせたアンチトロンビン耐性の効果を評価するために、FVIIaバリアントのセレクション[Table 2(表2)参照]を、以下のセクションに記載された酵素的糖コンジュゲーションにより40kDa PEGにコンジュゲートした。
N-グリカン特異的ペグ化は、他で本質的に公開されているように(Stennickeら 2008)実施した。簡単には、4-アミノベンズアミジン(Sigma社)を、10mMヒスチジン、50mM NaCl、10mM CaCl2、pH5.8中、タンパク質(約1.55mg/ml)溶液に10mMの最終濃度まで添加した。ウレアファシエンスシアリダーゼ(Urefaciens sialidase)を次いで4μg/mlの最終濃度まで添加して、N-グリカンの末端シアル酸を除去した。脱シアリル化反応を室温で1時間実施した。この後、アシアロタンパク質を、他で記載されているように(Thimら 1988)、50mM Hepes、100mM NaCl、10mM CaCl2、pH7.4を用いてGlaドメイン特異的モノクローナル抗体親和性クロマトグラフィーにより精製して、溶出緩衝液として余分なベンズアミジンおよび50mM Hepes、100mM NaCl、10mM EDTA pH7.4を洗い流した。塩化カルシウムおよびベンズアミジンを、収集した画分に10mMの最終濃度まで直ちに添加した。
得られた産物を、製造者の指示に従って4〜12% Bis-Trisゲル(Invitrogen社)を用いて、還元および非還元SDS-PAGEにより分析した。タンパク質濃度は、軽鎖rpHPLC分析により決定した。得られたアシアロタンパク質は、SDS-PAGEおよびRP-HPLC分析の両方に基づき均一であった。アシアロタンパク質(最終濃度約26μM)に、40kDa-PEG-GSC[N-((2,3-ジ(20kDa mPEGyl)プロポキシカルボニルアミノ)アセチル)-O2-[5']シチジリル-ζ-ノイラミン酸;10等量]およびST3Gal-III(最終濃度0.22U/ml)を添加した。反応は32℃で3時間実施した。NAN-CMP(シチジン-5'-一リン酸-N-アセチルノイラミン酸;3mM)により32℃で1時間、グリコペグ化産物をキャッピングした後、上記に記載されているように抗体親和性クロマトグラフィーにより産物を単離した。グリコペグ化産物を、Superdex 200pg 26/600カラム(GE Healthcare社)を用いるサイズ排除クロマトグラフィーによりさらに精製した。モノ-グリコペグ化産物に対応する画分をプールし、上記に記載されているようにSDS-PAGEにより分析した。この後、Amicon 10kDaカットオフ超遠心分離装置(Millipore社)を用いて、産物を約1mg/mlに濃縮した。
ジ-グリコペグ化FVIIaの含量を、TSK-Gel G300SWXLカラム、ならびに蛍光(励起280nm、354nmの発光)および吸光度(280nm)による検出を用いて分析的SEC HPLCにより評価した。カラム温度は30℃であり、流速は200mMリン酸ナトリウム、300mM NaCl、10%イソプロパノール、pH6.9中、1ml/分で維持した。
試験した全てのバリアント[Table 4(表4)のリスト参照]がグリコPEG化に適しており、主にモノ-PEG化(>85%)産物を生じた。インビトロ特徴付けは、FVIIa野生型におけるQ286N、Q176KおよびT293Kバリアント、M298QおよびV158D/E296V/M298Qバックグラウンドは、グリコPEG化時にアンチトロンビン耐性のわずかな増加を示すことを示した。さらに、タンパク質分解活性の低下が観察された。しかし、Q176KおよびT293Kベースのバリアントは、M298QおよびV158D/E296V/M298Qとの組み合わせで>100% TF非依存性タンパク質分解活性を依然として保持し、これらの変異の優位性を示した。
(実施例11)
定量化方法:
Hep化(Hepylated)FVIIaコンジュゲートを、HPLCにより純度について分析した。HPLCは、FVIIa参照分子に基づく単離したコンジュゲートの量の定量化にも使用した。試料は、非還元または還元形態のいずれかで分析した。Zorbax 300SB-C3カラム(4.6×50mm; 3.5um Agilent社、カタログ番号:865973-909)を使用した。カラムは30℃で操作した。5ug試料を注入し、0.1%トリフルオロ酢酸を含有する水(A)-アセトニトリル(B)溶媒系でカラムを溶出した。勾配プログラムは次の通りであった: 0分(25% B); 4分(25% B); 14分(46% B); 35分(52% B); 40分(90% B); 40.1分(25% B)。10ul TCEP/ギ酸溶液(70mM トリス(2-カルボキシエチル)ホスフィンおよび10%ギ酸水溶液)を25ul/30ug FVIIa(またはコンジュゲート)に添加して、還元試料を調製した。反応物は、HPLC(5ul注入)での分析前に70℃で10分間放置した。
定量化方法:
Hep化(Hepylated)FVIIaコンジュゲートを、HPLCにより純度について分析した。HPLCは、FVIIa参照分子に基づく単離したコンジュゲートの量の定量化にも使用した。試料は、非還元または還元形態のいずれかで分析した。Zorbax 300SB-C3カラム(4.6×50mm; 3.5um Agilent社、カタログ番号:865973-909)を使用した。カラムは30℃で操作した。5ug試料を注入し、0.1%トリフルオロ酢酸を含有する水(A)-アセトニトリル(B)溶媒系でカラムを溶出した。勾配プログラムは次の通りであった: 0分(25% B); 4分(25% B); 14分(46% B); 35分(52% B); 40分(90% B); 40.1分(25% B)。10ul TCEP/ギ酸溶液(70mM トリス(2-カルボキシエチル)ホスフィンおよび10%ギ酸水溶液)を25ul/30ug FVIIa(またはコンジュゲート)に添加して、還元試料を調製した。反応物は、HPLC(5ul注入)での分析前に70℃で10分間放置した。
HEP-マレイミドポリマーの調製
規定サイズのマレイミド官能化ヘパロサンポリマーを、2つの糖ヌクレオチドUDP-GlcNAcおよびUDP-GlcUAを用いて酵素(PmHS1)重合反応により調製した。反応を開始させるのにプライミング三糖(GlcUA-GlcNAc-GlcUA)NH2を使用し、重合は糖ヌクレオチド構成単位が枯渇するまで行う。末端アミン(プライマーに由来)を次いで、遊離システインにコンジュゲーションするために設計された適切な反応基(この場合、マレイミド官能基)で官能化する。N-(g-マレイミドブチリルオキシ)スルホスクシンイミドエステル(スルホ-GMBS、Pierce社)などの試薬を、アミンからマレイミドへの変換に使用することができる。ヘパロサンポリマーのサイズは、糖ヌクレオチド:プライマー化学量論の変動により予め決定することができる。該手法は、US 2010/0036001に詳細に記載されている。
規定サイズのマレイミド官能化ヘパロサンポリマーを、2つの糖ヌクレオチドUDP-GlcNAcおよびUDP-GlcUAを用いて酵素(PmHS1)重合反応により調製した。反応を開始させるのにプライミング三糖(GlcUA-GlcNAc-GlcUA)NH2を使用し、重合は糖ヌクレオチド構成単位が枯渇するまで行う。末端アミン(プライマーに由来)を次いで、遊離システインにコンジュゲーションするために設計された適切な反応基(この場合、マレイミド官能基)で官能化する。N-(g-マレイミドブチリルオキシ)スルホスクシンイミドエステル(スルホ-GMBS、Pierce社)などの試薬を、アミンからマレイミドへの変換に使用することができる。ヘパロサンポリマーのサイズは、糖ヌクレオチド:プライマー化学量論の変動により予め決定することができる。該手法は、US 2010/0036001に詳細に記載されている。
FVIIa Q286N 407Cの選択的還元:
FVIIa Q286N 407Cを、グルタチオンベースのレドックス緩衝系を用いてUS 20090041744に記載されているように還元した。0.5mM GSH、15uM GSSG、25mM p-アミノベンズアミジンおよび2μM Grx2を含有する18.2 ml 10mM Hepes、10mM CaCl2、50mM NaCl、0.01% Tween80、pH6.0の総容量中、非還元FVIIa Q286N 407C(20mg)を5℃で18時間インキュベートした。溶液を20ml 50mM Hepes、100mM NaCl、pH7.0で希釈し、氷上で冷却した。pHを中性に保ちながら、4.0ml 100mM EDTA溶液を次いで添加した。緩衝液A(50mM HEPES、100mMNaCl、1mM EDTA、pH7.0)で平衡化した2つの連結した5ml HiTrap Q FFカラム[Amersham Biosciences社(GE Healthcare社)]に、全含量を充填してFVIIa Q286N 407Cを捕捉した。緩衝液Aで洗浄して非結合グルタチオン緩衝液およびGrx2pを除去した後、FVIIa Q286N 407Cを緩衝液B(50mM HEPES、100mM NaCl、10mM CaCl2、pH7.0)により一工程で溶出した。溶出液中のFVIIa Q286N 407Cの濃度をHPLCにより決定した。12.2ml 50mM Hepes、100mM NaCl、10mM CaCl2、pH7.0において、17mg FVIIa Q286N 407Cが単離された。反応を2度繰り返した場合、8ml 50mM Hepes、100mM NaCl、10mM CaCl2、pH7.0において、定量的収量(20mg)の50mM Hepes、100mM NaCl、10mM CaCl2、pH7.0が単離された。
FVIIa Q286N 407Cを、グルタチオンベースのレドックス緩衝系を用いてUS 20090041744に記載されているように還元した。0.5mM GSH、15uM GSSG、25mM p-アミノベンズアミジンおよび2μM Grx2を含有する18.2 ml 10mM Hepes、10mM CaCl2、50mM NaCl、0.01% Tween80、pH6.0の総容量中、非還元FVIIa Q286N 407C(20mg)を5℃で18時間インキュベートした。溶液を20ml 50mM Hepes、100mM NaCl、pH7.0で希釈し、氷上で冷却した。pHを中性に保ちながら、4.0ml 100mM EDTA溶液を次いで添加した。緩衝液A(50mM HEPES、100mMNaCl、1mM EDTA、pH7.0)で平衡化した2つの連結した5ml HiTrap Q FFカラム[Amersham Biosciences社(GE Healthcare社)]に、全含量を充填してFVIIa Q286N 407Cを捕捉した。緩衝液Aで洗浄して非結合グルタチオン緩衝液およびGrx2pを除去した後、FVIIa Q286N 407Cを緩衝液B(50mM HEPES、100mM NaCl、10mM CaCl2、pH7.0)により一工程で溶出した。溶出液中のFVIIa Q286N 407Cの濃度をHPLCにより決定した。12.2ml 50mM Hepes、100mM NaCl、10mM CaCl2、pH7.0において、17mg FVIIa Q286N 407Cが単離された。反応を2度繰り返した場合、8ml 50mM Hepes、100mM NaCl、10mM CaCl2、pH7.0において、定量的収量(20mg)の50mM Hepes、100mM NaCl、10mM CaCl2、pH7.0が単離された。
60k HEP-[C]-FVIIa Q286N 407Cの合成:
単一のシステイン還元FVIIa Q286N 407C(20mg)を、50mM Hepes、100mM NaCl、10mM CaCl2、pH7.0緩衝液(8.0ml)中、室温で14時間、60K HEP-マレイミド(32mg)と反応させた。反応混合物を次いで、Glaドメイン特異的抗体で修飾したFVIIa特異的親和性カラム(CV=25ml)に充填し、最初は2カラム容量の緩衝液A(50mM Hepes、100mM NaCl、10mM CaCl2、pH7.4)、次いで2カラム容量の緩衝液B(50mM Hepes、100mM NaCl、10m MEDTA、pH7.4)でステップ溶出した。方法は本質的に、Thim、Lら Biochemistry(1988)27、7785〜779頁により記載された原理に従っている。折り畳まれていないGlaドメインを含む産物を収集し、10mM His、100mM NaCl、pH7.5で平衡化した2つの連結した5ml HiTrap Q FFイオン交換カラム[Amersham Biosciences社(GE Healthcare社)、CV=10ml]に直接適用した。カラムを4カラム容量の10mM His、100mM NaCl、pH7.5、および5カラム容量の10mM His、100mM NaCl、10mM CaCl2、pH=7.5で洗浄して非修飾FVIIa Q286N 407Cを溶出した。次いで10mM His、100mM NaCl、10mM CaCl2、pH=6.0(12カラム容量)によりpHを6.0まで低下させた。10カラム容量の40%緩衝液A(10mM His、100mM NaCl、10mM CaCl2、pH=6.0)および60%緩衝液B(10mM His、1M NaCl、10mM CaCl2、pH=6.0)溶媒混合物で、60k-HEP-[C]-FVIIa Q286N 407Cを溶出した。コンジュゲートを含有する画分を合わせ、10kDのカットオフによるSlide-A-Lyzerカセット(Thermo Scientific社)を用いて、10mM His、100mM NaCl、10mM CaCl2、pH=6.0に対して透析した。収量(2.61mg、13%)は、逆相HPLCを用いたトリス(2-カルボキシエチル)ホスフィン還元後に、FVIIa標準と比較したFVIIa軽鎖含量の定量化により決定した。12.6mg非修飾FVIIa Q286N 407Cも単離された。
単一のシステイン還元FVIIa Q286N 407C(20mg)を、50mM Hepes、100mM NaCl、10mM CaCl2、pH7.0緩衝液(8.0ml)中、室温で14時間、60K HEP-マレイミド(32mg)と反応させた。反応混合物を次いで、Glaドメイン特異的抗体で修飾したFVIIa特異的親和性カラム(CV=25ml)に充填し、最初は2カラム容量の緩衝液A(50mM Hepes、100mM NaCl、10mM CaCl2、pH7.4)、次いで2カラム容量の緩衝液B(50mM Hepes、100mM NaCl、10m MEDTA、pH7.4)でステップ溶出した。方法は本質的に、Thim、Lら Biochemistry(1988)27、7785〜779頁により記載された原理に従っている。折り畳まれていないGlaドメインを含む産物を収集し、10mM His、100mM NaCl、pH7.5で平衡化した2つの連結した5ml HiTrap Q FFイオン交換カラム[Amersham Biosciences社(GE Healthcare社)、CV=10ml]に直接適用した。カラムを4カラム容量の10mM His、100mM NaCl、pH7.5、および5カラム容量の10mM His、100mM NaCl、10mM CaCl2、pH=7.5で洗浄して非修飾FVIIa Q286N 407Cを溶出した。次いで10mM His、100mM NaCl、10mM CaCl2、pH=6.0(12カラム容量)によりpHを6.0まで低下させた。10カラム容量の40%緩衝液A(10mM His、100mM NaCl、10mM CaCl2、pH=6.0)および60%緩衝液B(10mM His、1M NaCl、10mM CaCl2、pH=6.0)溶媒混合物で、60k-HEP-[C]-FVIIa Q286N 407Cを溶出した。コンジュゲートを含有する画分を合わせ、10kDのカットオフによるSlide-A-Lyzerカセット(Thermo Scientific社)を用いて、10mM His、100mM NaCl、10mM CaCl2、pH=6.0に対して透析した。収量(2.61mg、13%)は、逆相HPLCを用いたトリス(2-カルボキシエチル)ホスフィン還元後に、FVIIa標準と比較したFVIIa軽鎖含量の定量化により決定した。12.6mg非修飾FVIIa Q286N 407Cも単離された。
FVIIa T293K 407Cの選択的還元:
FVIIa T293K 407Cを、グルタチオンベースのレドックス緩衝系を用いて、FVIIa Q286N 407Cについて上記に記載されているように還元した。12ml 50mM Hepes、100mM NaCl、10mM CaCl2、pH7.0において、合計22.8mg FVIIa T293K 407Cが単離された。
FVIIa T293K 407Cを、グルタチオンベースのレドックス緩衝系を用いて、FVIIa Q286N 407Cについて上記に記載されているように還元した。12ml 50mM Hepes、100mM NaCl、10mM CaCl2、pH7.0において、合計22.8mg FVIIa T293K 407Cが単離された。
40k HEP-[C]-FVIIa T293K 407Cの合成:
単一のシステイン還元FVIIa T293K 407C(22mg)および40K HEP-マレイミド(24mg)を、0.5mM p-アミノベンズアミジンを含有する8ml 50mM Hepes、100mM NaCl、10mM CaCl2、pH7.0緩衝液中、室温で18時間インキュベートした。反応混合物を次いで、20ml 50mM Hepes、100mM NaCl、10mM CaCl2、pH7.4で希釈し、Glaドメイン特異的抗体で修飾したFVIIa特異的親和性カラム(CV=64ml)に充填した。カラムを、最初は2つのカラム容量の緩衝液A(50mM Hepes、100mM NaCl、10mM CaCl2、pH7.4)、次いで2つのカラム容量の緩衝液B(50mM Hepes、100mM NaCl、10mM EDTA、pH7.4)でステップ溶出した。折り畳まれていないGlaドメインを含む産物を収集し、10mM His、100mM NaCl、pH7.5で平衡化した3つの連結した5ml HiTrap Q FFイオン交換カラム[Amersham Biosciences社(GE Healthcare社)、CV=15 ml]に直接適用した。カラムを4カラム容量の10mM His、100mM NaCl、pH7.5、および15カラム容量の10mM His、100mM NaCl、10mM CaCl2、pH=7.5で洗浄して非修飾FVIIa T293K 407Cを溶出した。次いで10mM His、100mM NaCl、10mM CaCl2、pH=6.0(12カラム容量)によりpHを6.0まで低下させた。15カラム容量の40%緩衝液A(10mM His、100mM NaCl、10mM CaCl2、pH=6.0)および60%緩衝液B(10mM His、1M NaCl、10mM CaCl2、pH=6.0)溶媒混合物で、40k-HEP-[C]-FVIIa T293K 407Cを溶出した。コンジュゲートを含有する画分を合わせ、10kDのカットオフによるSlide-A-Lyzerカセット(Thermo Scientific社)を用いて、10mM His、100mM NaCl、10mM CaCl2、pH=6.0に対して透析した。収量(15.3mg、69%)は、逆相HPLCを用いたトリス(2-カルボキシエチル)ホスフィン還元後に、FVIIa標準と比較したFVIIa軽鎖含量の定量化により決定した。
単一のシステイン還元FVIIa T293K 407C(22mg)および40K HEP-マレイミド(24mg)を、0.5mM p-アミノベンズアミジンを含有する8ml 50mM Hepes、100mM NaCl、10mM CaCl2、pH7.0緩衝液中、室温で18時間インキュベートした。反応混合物を次いで、20ml 50mM Hepes、100mM NaCl、10mM CaCl2、pH7.4で希釈し、Glaドメイン特異的抗体で修飾したFVIIa特異的親和性カラム(CV=64ml)に充填した。カラムを、最初は2つのカラム容量の緩衝液A(50mM Hepes、100mM NaCl、10mM CaCl2、pH7.4)、次いで2つのカラム容量の緩衝液B(50mM Hepes、100mM NaCl、10mM EDTA、pH7.4)でステップ溶出した。折り畳まれていないGlaドメインを含む産物を収集し、10mM His、100mM NaCl、pH7.5で平衡化した3つの連結した5ml HiTrap Q FFイオン交換カラム[Amersham Biosciences社(GE Healthcare社)、CV=15 ml]に直接適用した。カラムを4カラム容量の10mM His、100mM NaCl、pH7.5、および15カラム容量の10mM His、100mM NaCl、10mM CaCl2、pH=7.5で洗浄して非修飾FVIIa T293K 407Cを溶出した。次いで10mM His、100mM NaCl、10mM CaCl2、pH=6.0(12カラム容量)によりpHを6.0まで低下させた。15カラム容量の40%緩衝液A(10mM His、100mM NaCl、10mM CaCl2、pH=6.0)および60%緩衝液B(10mM His、1M NaCl、10mM CaCl2、pH=6.0)溶媒混合物で、40k-HEP-[C]-FVIIa T293K 407Cを溶出した。コンジュゲートを含有する画分を合わせ、10kDのカットオフによるSlide-A-Lyzerカセット(Thermo Scientific社)を用いて、10mM His、100mM NaCl、10mM CaCl2、pH=6.0に対して透析した。収量(15.3mg、69%)は、逆相HPLCを用いたトリス(2-カルボキシエチル)ホスフィン還元後に、FVIIa標準と比較したFVIIa軽鎖含量の定量化により決定した。
(実施例12)
同定したアンチトロンビン耐性変異単独、またはM298QおよびV158D/E296V/M298Qおよび40k-グリコペグ化との組み合わせでの薬物動態分析をラットおよびイヌで行って、FVIIaのインビボ生存率に対するこの影響を評価した。Sprague Dawleyラット(1群あたり3匹)またはビーグル犬(1群あたり2匹)を静脈内投与した。Stabylite(商標)(TriniLize Stabylite試験管; Tcoag Ireland Ltd社、アイルランド)安定化血漿試料を適切な時点で完全なプロファイルとして収集し、さらなる分析まで凍結させた。凝固活性(実施例7に記載されている)について、およびFVIIa-アンチトロンビン複合体を定量化するELISAにより、血漿試料を分析した。薬物動態分析は、WinNonlin(Pharsight Corporation社)を用いた非コンパートメント方法により実施した。以下のパラメータを推定した: FVIIa-アンチトロンビン複合体のCmax(最大濃度)、ならびに凝固活性のT1/2(機能的終末半減期)およびMRT(機能的平均滞留時間)。
同定したアンチトロンビン耐性変異単独、またはM298QおよびV158D/E296V/M298Qおよび40k-グリコペグ化との組み合わせでの薬物動態分析をラットおよびイヌで行って、FVIIaのインビボ生存率に対するこの影響を評価した。Sprague Dawleyラット(1群あたり3匹)またはビーグル犬(1群あたり2匹)を静脈内投与した。Stabylite(商標)(TriniLize Stabylite試験管; Tcoag Ireland Ltd社、アイルランド)安定化血漿試料を適切な時点で完全なプロファイルとして収集し、さらなる分析まで凍結させた。凝固活性(実施例7に記載されている)について、およびFVIIa-アンチトロンビン複合体を定量化するELISAにより、血漿試料を分析した。薬物動態分析は、WinNonlin(Pharsight Corporation社)を用いた非コンパートメント方法により実施した。以下のパラメータを推定した: FVIIa-アンチトロンビン複合体のCmax(最大濃度)、ならびに凝固活性のT1/2(機能的終末半減期)およびMRT(機能的平均滞留時間)。
簡単には、FVIIa-アンチトロンビン複合体は、酵素免疫アッセイ(EIA)を使用して測定した。EGFドメインのN末端に結合し、アンチトロンビン結合をブロックしないモノクローナル抗FVIIa抗体を、複合体の捕捉に使用した(Dako Denmark A/S社、グロストルップ;製品コードO9572)。ポリクローナル抗ヒトAT抗体ペルオキシダーゼコンジュゲートを検出に使用した(Siemens Healthcare Diagnostics ApS社、バレルプ/デンマーク;製品コードOWMG15)。ヒト野生型またはバリアントFVIIaおよび血漿由来ヒトアンチトロンビンの予め形成した精製複合体を、EIA検量線を構築するための基準として使用した。血漿試料を希釈し、分析し、二重の測定の平均濃度を計算した。EIAのアッセイ内精度は、1〜8%の間であった。
薬物動態プロファイルを図3に示し、推定パラメータをTable 3(表3)にリストする。FVIIa野生型および40k-グリコペグ化バックグラウンドにおけるアンチトロンビン耐性バリアントについて、FVIIa-アンチトロンビン複合体の蓄積は検出レベル未満まで低下した。さらに、これは、グリコペグ化FVIIaと比較して(ラットで7.4時間、イヌで8時間)、グリコペグ化FVIIa Q286Nの著しく延長した機能的半減期に反映された(ラットで16時間、イヌで20時間)。同様に、バリアントがM298QおよびV158D/E296V/M298Qとさらに組み合わされた場合、アンチトロンビンによるインビボ不活化は大いに低減した。著しくは、FVIIa M298Q T293K 40k-PEGの半減期は、FVIIa 40k-PEGの7.4時間と比較してラットにおいて13時間であった一方、222%のタンパク質分解TF非依存性活性を保持した[Table 3(表3)参照]。
(実施例13)
野生型FVIIaと比較したQ176K FVIIaのインビボ有効性を、FVIIIノックアウト(Biら 1996)マウスの尾出血モデルにおいて、0.5、2、5および10mg/kgの用量で調べた。尾出血は、イソフルラン麻酔マウスにおいて、尾静脈にFVIIaまたはビヒクルを静脈内投与した5分後に尾の先端4mmを切断して開始した。出血時間および失血は、他で記載されているように(Elmら 2012)、37℃生理食塩水中で30分間測定した。結果を図5に示す。失血ED50は、FVIIaについては1.8mg/kg、Q176K FVIIaについては2.6mg/kg、p=0.50とそれぞれ計算された。出血時間対用量および失血、ならびに出血時間対野生型FVIIaおよびFVIIa Q176Kの曝露も、極めて類似した用量反応曲線を示す。結論として、FVIII欠乏マウスでの急性尾出血において、アンチトロンビン耐性FVIIa Q176Kバリアントと野生型FVIIaの間に用量反応の有意な差はなかった。
(参考文献)
野生型FVIIaと比較したQ176K FVIIaのインビボ有効性を、FVIIIノックアウト(Biら 1996)マウスの尾出血モデルにおいて、0.5、2、5および10mg/kgの用量で調べた。尾出血は、イソフルラン麻酔マウスにおいて、尾静脈にFVIIaまたはビヒクルを静脈内投与した5分後に尾の先端4mmを切断して開始した。出血時間および失血は、他で記載されているように(Elmら 2012)、37℃生理食塩水中で30分間測定した。結果を図5に示す。失血ED50は、FVIIaについては1.8mg/kg、Q176K FVIIaについては2.6mg/kg、p=0.50とそれぞれ計算された。出血時間対用量および失血、ならびに出血時間対野生型FVIIaおよびFVIIa Q176Kの曝露も、極めて類似した用量反応曲線を示す。結論として、FVIII欠乏マウスでの急性尾出血において、アンチトロンビン耐性FVIIa Q176Kバリアントと野生型FVIIaの間に用量反応の有意な差はなかった。
(参考文献)
本発明は、以下の非限定的実施形態によりさらに記載される。
実施形態1:ヒト第VII因子のアミノ酸配列(配列番号1)と比べて2つ以上の置換を含む第VII因子(a)ポリペプチドであって、置換の少なくとも1つが、T293がLys(K)、Tyr(Y)、Arg(R)もしくはPhe(F)により置換され、Q176がLys(K)、Arg(R)、Asn(N)により置換され、および/またはQ286がAsn(N)により置換されている所にあり、ならびに置換の少なくとも1つが、M298が、Gln(Q)、Lys(K)、Arg(R)、Asn(N)、Gly(G)、Pro(P)、Ala(A)、Val(V)、Leu(L)、Ile(I)、Phe(F)、Trp(W)、Tyr(Y)、Asp(D)、Glu(E)、His(H)、Cys(C)、Ser(S)、またはThr(T)により置換されている所にある、第VII因子(a)ポリペプチド。
実施形態2: T293がLys(K)、Tyr(Y)、Arg(R)、またはPhe(F)により置換されている、実施形態1による第VII因子(a)ポリペプチド。
実施形態3: Q176がLys(K)、Arg(R)、またはAsn(N)により置換されている、実施形態1による第VII因子(a)ポリペプチド。
実施形態4: Q286がAsn(N)により置換されている、実施形態1による第VII因子(a)ポリペプチド。
実施形態5: M298がQにより置換されている、実施形態1〜4による第VII因子(a)ポリペプチド。
実施形態6:追加の置換としてV158がAsp(D)により置換され、E296がVal(V)により置換されている、実施形態5による第VII因子(a)ポリペプチド。
実施形態7:さらなる追加の置換としてK337がAla(A)により置換されている、実施形態6による第VII因子(a)ポリペプチド。
実施形態8:ポリペプチドが、以下の置換の群T293K/M298Q、T293Y/M298Q、T293R/M298Q、T293F/M298Q、Q176K/M298Q、Q176R/M298Q、Q176N/M298Q、Q286N/M298Q、T293Y/V158D/E296V/M298Q、T293R/V158D/E296V/M298Q、T293K/V158D/E296V/M298Q、Q176K/V158D/E296V/M298Q、またはQ176R/V158D/E296V/M298Qの1つを有する、実施形態1による第VII因子(a)ポリペプチド。
実施形態9:第VII因子(a)ポリペプチドが少なくとも1つの半減期延長部分とカップリングされている、実施形態1〜8による第VII因子(a)ポリペプチド。
実施形態10:「半減期延長部分」が、生体適合性脂肪酸およびこの誘導体、ヒドロキシアルキルデンプン(HAS)、例えばヒドロキシエチルデンプン(HES)、ポリエチレングリコール(PEG)、Poly(Glyx-Sery)n(HAP)、ヒアルロン酸(HA)、ヘパロサンポリマー(HEP)、ホスホリルコリンベースのポリマー(PCポリマー)、フレキシマー、デキストラン、ポリシアル酸(PSA)、Fcドメイン、トランスフェリン、アルブミン、エラスチン様(ELP)ペプチド、XTENポリマー、PASポリマー、PAポリマー、アルブミン結合ペプチド、CTPペプチド、およびFcRn結合ペプチドから選択される、実施形態9による第VII因子(a)ポリペプチド。
実施形態11:半減期延長部分がHEPである、実施形態10による第VII因子(a)ポリペプチド。
実施形態12:第VII因子(a)ポリペプチドが追加の変異R396C、Q250C、または+407Cを有する、実施形態9〜11のいずれか1つによる第VII因子(a)ポリペプチド。
実施形態13:前記第VII因子(a)ポリペプチドが組織因子にジスルフィド結合されている、実施形態1〜12のいずれか1つによる第VII因子(a)ポリペプチド。
実施形態14:前記第VII因子(a)ポリペプチドが、血小板親和性が増加した第VIIa因子バリアントである、実施形態1〜13のいずれか1つによる第VII因子(a)ポリペプチド。
実施形態15:実施形態1〜14のいずれか1つによる第VII因子(a)ポリペプチドをコードするポリヌクレオチド構築物。
実施形態16:実施形態15によるポリヌクレオチド構築物を含む宿主細胞。
実施形態17:実施形態1〜14のいずれか1つに定義された第VII因子(a)ポリペプチドを産生する方法。
実施形態18:実施形態1〜14のいずれか1つに定義された第VII因子(a)ポリペプチドを含む医薬組成物。
実施形態19:血友病AまたはBの治療において医薬品として使用するための、実施形態18による医薬組成物。
実施形態20:出血障害もしくは出血エピソードの治療、または正常な止血系の増強のための医薬品を調製するための、実施形態1〜14のいずれか1つに定義された第VII因子(a)ポリペプチドの使用。
実施形態21:血友病AまたはBを治療するための、実施形態20による使用。
実施形態22:対象における出血障害もしくは出血エピソードの治療、または正常な止血系の増強のため方法であって、実施形態1〜14のいずれか1つに定義された第VII因子(a)ポリペプチドの治療的または予防的に有効な量を、これを必要とする対象に投与する工程を含む方法。
実施形態23:医薬品として使用するための、実施形態1〜14のいずれか1つに定義された第VII因子(a)ポリペプチド。
実施形態24:血友病AまたはBの治療において医薬品として使用するための、実施形態23による第VII因子(a)ポリペプチド。
Claims (15)
- ヒト第VII因子のアミノ酸配列(配列番号1)と比べて2つ以上の置換を含む第VII因子(a)ポリペプチドであって、
置換の少なくとも1つが、T293がLys(K)、Tyr(Y)、Arg(R)もしくはPhe(F)により置換され、Q176がLys(K)、Arg(R)、Asn(N)により置換され、および/またはQ286がAsn(N)により置換されている所にあり、
置換の少なくとも1つが、M298がGln(Q)、Lys(K)、Arg(R)、Asn(N)、Gly(G)、Pro(P)、Ala(A)、Val(V)、Leu(L)、Ile(I)、Phe(F)、Trp(W)、Tyr(Y)、Asp(D)、Glu(E)、His(H)、Cys(C)、Ser(S)、またはThr(T)により置換されている所にある、
第VII因子(a)ポリペプチド。 - T293がLys(K)、Tyr(Y)、Arg(R)、またはPhe(F)により置換されている、請求項1に記載の第VII因子(a)ポリペプチド。
- Q176がLys(K)、Arg(R)、またはAsn(N)により置換されている、請求項1に記載の第VII因子(a)ポリペプチド。
- Q286がAsn(N)により置換されている、請求項1に記載の第VII因子(a)ポリペプチド。
- M298がQにより置換されている、請求項1から4のいずれか一項に記載の第VII因子(a)ポリペプチド。
- ポリペプチドが、以下の置換の群T293K/M298Q、T293Y/M298Q、T293R/M298Q、T293F/M298Q、Q176K/M298Q、Q176R/M298Q、Q176N/M298Q、Q286N/M298Q、T293Y/V158D/E296V/M298Q、T293R/V158D/E296V/M298Q、T293K/V158D/E296V/M298Q、Q176K/V158D/E296V/M298Q、またはQ176R/V158D/E296V/M298Qの1つを有する、請求項1に記載の第VII因子(a)ポリペプチド。
- 第VII因子(a)ポリペプチドが少なくとも1つの半減期延長部分とカップリングされている、請求項1から6のいずれか一項に記載の第VII因子(a)ポリペプチド。
- 半減期延長部分が、生体適合性脂肪酸およびこの誘導体、ヒドロキシアルキルデンプン(HAS)、例えばヒドロキシエチルデンプン(HES)、ポリエチレングリコール(PEG)、Poly(Glyx-Sery)n(HAP)、ヒアルロン酸(HA)、ヘパロサンポリマー(HEP)、ホスホリルコリンベースのポリマー(PCポリマー)、フレキシマー、デキストラン、ポリシアル酸(PSA)、Fcドメイン、トランスフェリン、アルブミン、エラスチン様(ELP)ペプチド、XTENポリマー、PASポリマー、PAポリマー、アルブミン結合ペプチド、CTPペプチド、およびFcRn結合ペプチドから選択される、請求項7に記載の第VII因子(a)ポリペプチド。
- 半減期延長部分がヘパロサンポリマーである、請求項8に記載の第VII因子(a)ポリペプチド。
- 第VII因子(a)ポリペプチドが追加の変異R396C、Q250C、または407Cを有する、請求項7から8のいずれか一項に記載の第VII因子(a)ポリペプチド。
- 請求項1から10のいずれか一項に記載の第VII因子(a)ポリペプチドを産生する方法。
- 請求項1から10のいずれか一項に記載の第VII因子(a)ポリペプチドを含む医薬組成物。
- 対象における出血障害もしくは出血エピソードの治療、または正常な止血系の増強のための方法であって、請求項1から10のいずれか一項に記載の第VII因子(a)ポリペプチドの治療的または予防的に有効な量を、これを必要とする対象に投与する工程を含む、方法。
- 医薬品として使用するための、請求項1から10のいずれか一項に記載の第VII因子(a)ポリペプチド。
- 血友病AまたはBの治療において医薬品として使用するための、請求項14に記載の第VII因子(a)ポリペプチド。
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP12188473.8 | 2012-10-15 | ||
| EP12188473 | 2012-10-15 | ||
| US201261715920P | 2012-10-19 | 2012-10-19 | |
| US61/715,920 | 2012-10-19 | ||
| PCT/EP2013/071510 WO2014060401A1 (en) | 2012-10-15 | 2013-10-15 | Coagulation factor vii polypeptides |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2015532307A true JP2015532307A (ja) | 2015-11-09 |
| JP2015532307A5 JP2015532307A5 (ja) | 2016-12-01 |
Family
ID=47022566
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2015536187A Withdrawn JP2015532307A (ja) | 2012-10-15 | 2013-10-15 | 凝固因子viiポリペプチド |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US20150307865A1 (ja) |
| EP (1) | EP2906693A1 (ja) |
| JP (1) | JP2015532307A (ja) |
| CN (1) | CN104704118A (ja) |
| WO (1) | WO2014060401A1 (ja) |
Families Citing this family (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI538916B (zh) | 2008-04-11 | 2016-06-21 | 介控生化科技公司 | 經修飾的因子vii多肽和其用途 |
| WO2011075185A1 (en) | 2009-12-18 | 2011-06-23 | Oligasis | Targeted drug phosphorylcholine polymer conjugates |
| EP2906247A1 (en) | 2012-10-15 | 2015-08-19 | Novo Nordisk Health Care AG | Factor vii conjugates |
| WO2015035342A2 (en) | 2013-09-08 | 2015-03-12 | Oligasis Llc | Factor viii zwitterionic polymer conjugates |
| JP2016537321A (ja) | 2013-10-15 | 2016-12-01 | ノヴォ・ノルディスク・ヘルス・ケア・アーゲー | 凝固因子viiポリペプチド |
| US9840553B2 (en) | 2014-06-28 | 2017-12-12 | Kodiak Sciences Inc. | Dual PDGF/VEGF antagonists |
| CN107208076A (zh) | 2014-10-17 | 2017-09-26 | 科达制药 | 丁酰胆碱酯酶两性离子聚合物缀合物 |
| EP3397276A4 (en) | 2015-12-30 | 2019-12-18 | Kodiak Sciences Inc. | ANTIBODIES AND CONJUGATES THEREOF |
| BR112019000610A2 (pt) * | 2016-07-11 | 2019-07-02 | Opko Biologics Ltd | fator vii de coagulação de longa ação e métodos de produção do mesmo |
| CN106279436B (zh) * | 2016-08-19 | 2017-10-31 | 安源医药科技(上海)有限公司 | 活化的人凝血因子vii融合蛋白及其制备方法与用途 |
| CN107102125B (zh) * | 2017-04-17 | 2019-07-09 | 中国科学院苏州生物医学工程技术研究所 | 血栓弹力图仪的校准方法 |
| CN110218258A (zh) * | 2019-06-12 | 2019-09-10 | 福建农林大学 | 融合三肽囊素的克隆表达及应用 |
| US20210069306A1 (en) * | 2019-08-15 | 2021-03-11 | Catalyst Biosciences, Inc. | Modified factor vii polypeptides for subcutaneous administration and on-demand treatment |
| CN114786731A (zh) | 2019-10-10 | 2022-07-22 | 科达制药股份有限公司 | 治疗眼部病症的方法 |
Family Cites Families (53)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4203570A (en) | 1978-08-23 | 1980-05-20 | The Western States Machine Company | Power-operated loading gate for centrifugal machines incorporating an auxiliary drive device |
| US4873191A (en) | 1981-06-12 | 1989-10-10 | Ohio University | Genetic transformation of zygotes |
| US4456591A (en) | 1981-06-25 | 1984-06-26 | Baxter Travenol Laboratories, Inc. | Therapeutic method for activating factor VII |
| US4546082A (en) | 1982-06-17 | 1985-10-08 | Regents Of The Univ. Of California | E. coli/Saccharomyces cerevisiae plasmid cloning vector containing the alpha-factor gene for secretion and processing of hybrid proteins |
| US4599311A (en) | 1982-08-13 | 1986-07-08 | Kawasaki Glenn H | Glycolytic promotersfor regulated protein expression: protease inhibitor |
| US4713339A (en) | 1983-01-19 | 1987-12-15 | Genentech, Inc. | Polycistronic expression vector construction |
| JPS60501140A (ja) | 1983-04-22 | 1985-07-25 | アムジエン | 酵母による外因性ポリペプチドの分泌 |
| NZ207926A (en) | 1983-04-25 | 1988-04-29 | Genentech Inc | Use of yeast #a#-factor to assist in expression of proteins heterologus to yeast |
| US4745051A (en) | 1983-05-27 | 1988-05-17 | The Texas A&M University System | Method for producing a recombinant baculovirus expression vector |
| US4870008A (en) | 1983-08-12 | 1989-09-26 | Chiron Corporation | Secretory expression in eukaryotes |
| US4879236A (en) | 1984-05-16 | 1989-11-07 | The Texas A&M University System | Method for producing a recombinant baculovirus expression vector |
| US4931373A (en) | 1984-05-25 | 1990-06-05 | Zymogenetics, Inc. | Stable DNA constructs for expression of α-1 antitrypsin |
| DK58285D0 (da) | 1984-05-30 | 1985-02-08 | Novo Industri As | Peptider samt fremstilling og anvendelse deraf |
| US4766073A (en) | 1985-02-25 | 1988-08-23 | Zymogenetics Inc. | Expression of biologically active PDGF analogs in eucaryotic cells |
| US4885249A (en) | 1984-12-05 | 1989-12-05 | Allelix, Inc. | Aspergillus niger transformation system |
| US4683202A (en) | 1985-03-28 | 1987-07-28 | Cetus Corporation | Process for amplifying nucleic acid sequences |
| GR860984B (en) | 1985-04-17 | 1986-08-18 | Zymogenetics Inc | Expression of factor vii and ix activities in mammalian cells |
| DE3650202T3 (de) | 1985-08-29 | 2004-06-03 | Genencor International, Inc., South San Francisco | Expression von heterologen Polypeptiden in filamentösen Pilzen, Verfahren zu deren Herstellung und Vektoren zu deren Herstellung. |
| US4882279A (en) | 1985-10-25 | 1989-11-21 | Phillips Petroleum Company | Site selective genomic modification of yeast of the genus pichia |
| AU6543286A (en) | 1985-10-25 | 1987-05-19 | Zymogenetics Inc. | Method of using bar1 for secreting foreign proteins |
| US4935349A (en) | 1986-01-17 | 1990-06-19 | Zymogenetics, Inc. | Expression of higher eucaryotic genes in aspergillus |
| DK122686D0 (da) | 1986-03-17 | 1986-03-17 | Novo Industri As | Fremstilling af proteiner |
| GB8610600D0 (en) | 1986-04-30 | 1986-06-04 | Novo Industri As | Transformation of trichoderma |
| GB8615942D0 (en) | 1986-06-30 | 1986-08-06 | Animal & Food Research Council | Peptide production |
| NZ221259A (en) | 1986-07-31 | 1990-05-28 | Calgene Inc | Seed specific transcriptional regulation |
| EP0279582A3 (en) | 1987-02-17 | 1989-10-18 | Pharming B.V. | Dna sequences to target proteins to the mammary gland for efficient secretion |
| US4873316A (en) | 1987-06-23 | 1989-10-10 | Biogen, Inc. | Isolation of exogenous recombinant proteins from the milk of transgenic mammals |
| CA1309680C (en) | 1987-07-24 | 1992-11-03 | David Harano | Airlift insect cell culture |
| US5024947A (en) | 1987-07-24 | 1991-06-18 | Cetus Corporation | Serum free media for the growth on insect cells and expression of products thereby |
| US5004697A (en) | 1987-08-17 | 1991-04-02 | Univ. Of Ca | Cationized antibodies for delivery through the blood-brain barrier |
| JP2703598B2 (ja) | 1987-09-04 | 1998-01-26 | ノボ―ノルディスク アクティーゼルスカブ | アスペルギルス菌中でのタンパク質生産物の生産方法およびアスペルギルス菌中で使用するためのプロモーター |
| DK463887D0 (da) | 1987-09-07 | 1987-09-07 | Novo Industri As | Gaerleader |
| US5037743A (en) | 1988-08-05 | 1991-08-06 | Zymogenetics, Inc. | BAR1 secretion signal |
| GB8826446D0 (en) | 1988-11-11 | 1988-12-14 | Agricultural & Food Res | Peptide production |
| AU4647889A (en) | 1988-11-18 | 1990-06-12 | Cetus Corporation | Insect signal peptide mediated secretion of recombinant proteins |
| GB8910962D0 (en) | 1989-05-12 | 1989-06-28 | Natural Environment Res | Novel baculovirus expression vectors and use thereof in the expression of foreign proteins in insects or insect cells |
| US5077214A (en) | 1989-07-07 | 1991-12-31 | The Texas A&M University System | Use of baculovirus early promoters for expression of foreign genes in stably transformed insect cells |
| US5162222A (en) | 1989-07-07 | 1992-11-10 | Guarino Linda A | Use of baculovirus early promoters for expression of foreign genes in stably transformed insect cells or recombinant baculoviruses |
| US5073964A (en) | 1989-08-04 | 1991-12-17 | Aware, Inc. | Signal processing device and method |
| US5155037A (en) | 1989-08-04 | 1992-10-13 | The Texas A&M University System | Insect signal sequences useful to improve the efficiency of processing and secretion of foreign genes in insect systems |
| US5023328A (en) | 1989-08-04 | 1991-06-11 | The Texas A&M University System | Lepidopteran AKH signal sequence |
| DK300090D0 (da) | 1990-12-19 | 1990-12-19 | Novo Nordisk As | Fremgangsmaade til fremstilling af leadersekvenser |
| WO1992011757A1 (en) | 1991-01-11 | 1992-07-23 | American Red Cross | Expression of active human protein c in mammary tissue of transgenic animals |
| RU2278123C2 (ru) * | 2000-02-11 | 2006-06-20 | Максиджен Холдингз Лтд. | Молекулы, подобные фактору vii или viia |
| CZ2003611A3 (cs) * | 2000-09-13 | 2003-08-13 | Novo Nordisk A/S | Varianty lidského koagulačního faktoru VII |
| HUP0401124A3 (en) | 2001-03-22 | 2006-01-30 | Novo Nordisk Healthcare Ag | Coagulation factor vii derivatives |
| PT2279753E (pt) | 2001-10-10 | 2016-01-15 | Novo Nordisk As | Remodelação e glicoconjugação de péptidos |
| WO2006134174A2 (en) * | 2005-06-17 | 2006-12-21 | Novo Nordisk Health Care Ag | Dimeric and multimeric fviia compound |
| EP1926817A2 (en) | 2005-09-14 | 2008-06-04 | Novo Nordisk Health Care AG | Human coagulation factor vii polypeptides |
| DE602007009956D1 (de) | 2006-04-07 | 2010-12-02 | Novo Nordisk Healthcare Ag | Kovalenter faktor vii-gewebefaktor-komplex |
| US9687559B2 (en) | 2008-03-19 | 2017-06-27 | The Board Of Regents Of The University Of Oklahoma | Heparosan polymers and methods of making and using same for the enhancement of therapeutics |
| TWI538916B (zh) | 2008-04-11 | 2016-06-21 | 介控生化科技公司 | 經修飾的因子vii多肽和其用途 |
| CA2773755C (en) * | 2008-09-09 | 2017-04-25 | The Board Of Regents Of The University Of Oklahoma | Heparosan polymers and methods of making and using same for the enhancement of therapeutics |
-
2013
- 2013-10-15 JP JP2015536187A patent/JP2015532307A/ja not_active Withdrawn
- 2013-10-15 US US14/675,887 patent/US20150307865A1/en not_active Abandoned
- 2013-10-15 WO PCT/EP2013/071510 patent/WO2014060401A1/en active Application Filing
- 2013-10-15 EP EP13777046.7A patent/EP2906693A1/en not_active Withdrawn
- 2013-10-15 CN CN201380053832.3A patent/CN104704118A/zh not_active Withdrawn
Also Published As
| Publication number | Publication date |
|---|---|
| CN104704118A (zh) | 2015-06-10 |
| WO2014060401A1 (en) | 2014-04-24 |
| US20150307865A1 (en) | 2015-10-29 |
| EP2906693A1 (en) | 2015-08-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2015532307A (ja) | 凝固因子viiポリペプチド | |
| US20220073895A1 (en) | Human coagulation factor vii polypeptides | |
| JP4537059B2 (ja) | ヒト凝固第vii因子ポリペプチド | |
| EP1908782B1 (en) | Human coagulation factor VII polypeptides | |
| US7416861B2 (en) | Human coagulation factor VII polypeptides | |
| EP1451315B1 (en) | Human coagulation factor vii polypeptides | |
| US20090104661A1 (en) | Human Coagulation Factor VII Polypeptides | |
| MX2010011170A (es) | Polipeptidos del factor vii que se modifican y usos de los mismos. | |
| US20080010693A1 (en) | Human coagulation factor VII polypeptides | |
| US20210002624A1 (en) | Short-acting factor vii polypeptides | |
| US9370583B2 (en) | Coagulation factor VII polypeptides | |
| US20090011992A1 (en) | Human Coagulation Factor VII Polypeptides |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20161012 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20161012 |
|
| A761 | Written withdrawal of application |
Free format text: JAPANESE INTERMEDIATE CODE: A761 Effective date: 20170106 |