JP2015114197A - 乾燥分離材を用いた分離方法及び測定方法 - Google Patents
乾燥分離材を用いた分離方法及び測定方法 Download PDFInfo
- Publication number
- JP2015114197A JP2015114197A JP2013256153A JP2013256153A JP2015114197A JP 2015114197 A JP2015114197 A JP 2015114197A JP 2013256153 A JP2013256153 A JP 2013256153A JP 2013256153 A JP2013256153 A JP 2013256153A JP 2015114197 A JP2015114197 A JP 2015114197A
- Authority
- JP
- Japan
- Prior art keywords
- component
- eluent
- separation material
- sample
- separation
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images












Landscapes
- Solid-Sorbent Or Filter-Aiding Compositions (AREA)
- Investigating Or Analysing Biological Materials (AREA)
- Sampling And Sample Adjustment (AREA)
- Investigating Or Analysing Materials By Optical Means (AREA)
- Treatment Of Liquids With Adsorbents In General (AREA)
Abstract
【課題】小スケールにも適用でき、かつ定量性にも優れる分離方法を提供する。
【解決手段】試料中の成分S1と成分S2を分離する方法であって、
溶離液Aにより成分S2を特異的に分離材に結合させる第1の工程と、
溶離液Bにより成分S2を分離材から解離させる第2の工程を有し、
前記第1の工程は、乾燥状態の分離材に試料及び溶離液Aを接触させ、分離材を分散又は攪拌することによって行われ、その後、濾過操作により成分S2を含む濾過残渣を残して濾液中に成分S1を分画し、
前記第2の工程は、前記濾過残渣の分離材に溶離液Bを接触させ、分離材に結合していた成分S2を解離させ、次いで濾過操作により濾液中に成分S2を分画する。またこの方法により、成分S1とS2の量の総和に対するS1又はS2の量比を求める。
【選択図】図8
【解決手段】試料中の成分S1と成分S2を分離する方法であって、
溶離液Aにより成分S2を特異的に分離材に結合させる第1の工程と、
溶離液Bにより成分S2を分離材から解離させる第2の工程を有し、
前記第1の工程は、乾燥状態の分離材に試料及び溶離液Aを接触させ、分離材を分散又は攪拌することによって行われ、その後、濾過操作により成分S2を含む濾過残渣を残して濾液中に成分S1を分画し、
前記第2の工程は、前記濾過残渣の分離材に溶離液Bを接触させ、分離材に結合していた成分S2を解離させ、次いで濾過操作により濾液中に成分S2を分画する。またこの方法により、成分S1とS2の量の総和に対するS1又はS2の量比を求める。
【選択図】図8
Description
本発明は乾燥分離材を用いた分離方法及び測定方法に関するものであり、特にヘモグロビンA1cの簡易測定に好適に使用できる方法に関する。
血液中のヘモグロビンは、α鎖N末端のアミノ酸のバリンのアミノ基とグルコースのアルデヒド基の間の非酵素的な反応によりグルコースと結合する。この結合の第一段階は、可逆的なシッフ塩基反応であるが、さらにアマドリ転移反応を経て不可逆的なケトアミンを形成する。このようにして生成するヘモグロビンとグルコースの結合物は、ヘモグロビンA1c(以下、HbA1c又はA1cともいう)として知られている。ヘモグロビンの血液中での寿命は約3ヵ月であり、その間グルコースと結合して生成するHbA1cが徐々に蓄積するが、その一方で寿命が尽きたHbA1cは逐次分解されていく。すなわち、ある時点で血液中に存在しているHbA1cの濃度は、その時点からヘモグロビンの寿命のおよそ半分の1〜2ヵ月程遡った過去の血液中のグルコース濃度、すなわち血糖濃度を平均的に反映するものである。このような特徴を有するHbA1cは、一般的な糖尿病の指標である血糖濃度等のように一時的な変動が無く過去の血糖状態を正確に把握できる。
HbA1cの測定方法としては、高速液体クロマトグラフィ法、免疫学的方法、酵素法などが知られている。
免疫学的方法は、ラテックスを用いた免疫反応を利用してHbA1cを測定する方法である。前処理が必要ではあるが、生化学自動分析装置で測定可能な試薬もあり、大量処理に向いている(特許文献1参照)。酵素法は、酵素により、HbA1cの糖化ジペプジドを切り出し、それをオキシダーゼ、ペルオキシダーゼによる発色へ導き測定する方法である。生化学自動分析装置で測定可能であり、大量処理に向いている(特許文献2参照)。
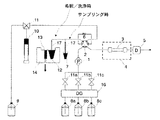
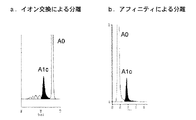
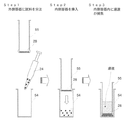
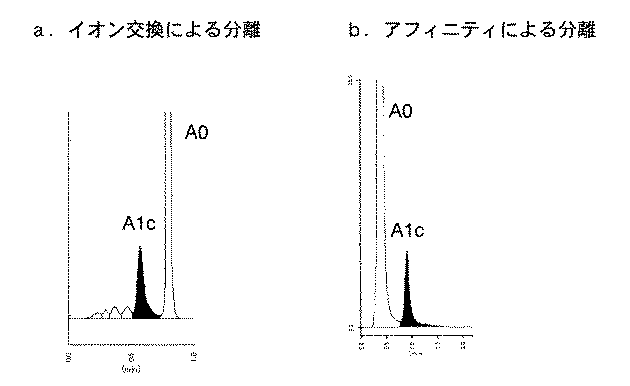
高速液体クロマトグラフィ法はカラムによりHbA1cを分離してその面積比率から定量を行うものである。大量処理には向かないとされてきたが、近年分析時間が大幅に短縮され、現在のところ採用施設も多く実質的な標準的測定方法である。図1にその一般的な流路系を示す。高速液体クロマトグラフィ法は、分離モードによりイオン交換モード法とアフィニティモード法に大別される。イオン交換モード法はHbA1cの電荷の違いにより分離する方法(特許文献3、4参照)、アフィニティモード法は、ホウ酸がHbA1cのシスジオール結合にアフィニティ結合する性質を利用した方法(特許文献5参照)である。図2はイオン交換モード、および、アフィニティモードでの代表的なクロマトグラムである。
分離分析の分野で広く使用される「固相抽出法」は試料の前処理の用途で用いられることが多い手法である(図3参照)。一般的な固相抽出法は、シリンジ内に目的の検体の成分と相互作用する分離材を充填しておき、検体を負荷し、複数の溶離液にて溶出操作を行い処理するものである。溶出操作は、目的に応じて加圧または減圧で行われている(図5参照)。この固相抽出法は、試料の精製目的で行われることが多く、定量分析には用いられることは少ない。
これは、測定毎の、固相抽出によって得られた溶質の量が一定でも、溶液の量が異なってしまうと濾液の濃度が変化してしまうため、定量的に測定するには、溶出操作に使用する溶液量を常に一定にするか、濾液を全て分析に供するか、あるいは、濾液を一度乾固させ一定量の別の溶液を添加し分析に供する必要があるからである。
また、固相抽出に使用されるカラムは、分離性能を一定に保つため/分離性能を向上させるために、何らかの溶液で湿潤させておく必要もあり、その液量も考慮しないと定量分析の場合は誤差を生じてしまう。
固相抽出に使用されるカラムは、分離材の充填のし易さ、操作性、品質の均一性のため、微小スケールは扱いにくい。一般的には分離材量は1mL以上必要とすることが多く、小さなスケールでの操作には不向きであるという欠点がある。
従来の固相抽出法では、分離床となる分離材をシリンジ内に充填して使用するものであり、分離を確実に行うため、それ相応の分離床の高さが必要である。
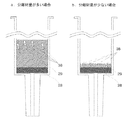
分離材の充填容量を極端に少なくすると、充填の均一性が乏しくなり、ゲル床に隙間やひび割れが生じやすくなる。このような不適切な固相抽出カラムで分離を行った場合、試料の一部が分離材粒子に全く接触することなくゲル床を通過してしまい、分離材との相互作用が全く無いまま溶出し、分離精度を落とす原因になる(図4参照)。そのため、従来の固相抽出法では、少なくとも1mL程度の分離材量が必要とされている。このように、従来の固相抽出法では、処理スケールを小さくすることは困難である。
また、固相抽出法で使用される分離材は保存液等の溶液で満たされていることから、抽出のために添加する溶媒の容量を正確に把握することはできず、そのため、溶出物の定量を行うには手間も掛かり、精度も劣るという難点がある。
本発明は、前記課題を解決するために成されたものであり、小スケールにも適用しうる分離方法であり、かつ定量性にも優れるため測定方法に適用でき、その結果、グリコヘモグロビン(A1c)の定量をも可能とすることを目的とするものである。
本発明者らは上記課題について鋭意検討した結果、本発明に到達した。即ち本発明は以下の通りである。
[1]試料中の成分S1と成分S2を分離する方法であって、
溶離液Aにより成分S2を特異的に分離材に結合させる第1の工程と、
溶離液Bにより成分S2を分離材から解離させる第2の工程を有し、
前記第1の工程は、乾燥状態の分離材に試料及び溶離液Aを接触させ、分離材を分散又は攪拌することによって行われ、その後、濾過操作により成分S2を含む濾過残渣を残して濾液中に成分S1を分画し、
前記第2の工程は、前記濾過残渣の分離材に溶離液Bを接触させ、分離材に結合していた成分S2を解離させ、次いで濾過操作により濾液中に成分S2を分画する、
ことを特徴とする方法。
[2]前記試料が血液であり、前記成分S2がヘモグロビンA1cである、[1]に記載の方法。
[3]前記分離材が、ポリマーを基材とし、m−アミノフェニルボロン酸を固定化したアフィニティ分離材であり、その粒径が10μmから200μmであり、1回の分離に使用する分離材の量が乾燥状態で20mg以下である、[1]又は[2]に記載の方法。
[4]前記溶離液Aが、前記分離材のボロネートアニオンとヘモグロビンA1cの1,2−シスジオールとが結合できる塩基性緩衝液であり、
前記溶離液Bが、前記分離材のボロネートアニオンとヘモグロビンA1cの1,2−シスジオールとが解離できる糖類またはアミンを含有する緩衝液である、
[3]に記載の方法。
[5]上述の[1]〜[4]いずれかに記載の方法で試料中の成分S1と成分S2を分離し、成分S1の量と成分S2の量の総和に対する成分S1または成分S2の量の比を求めることを特徴とする、成分S1または成分S2の測定方法。
[6]成分S1または成分S2の量を吸光度により測定する、[5]に記載の測定方法。
以下に本発明を更に詳細に説明する。
溶離液Aにより成分S2を特異的に分離材に結合させる第1の工程と、
溶離液Bにより成分S2を分離材から解離させる第2の工程を有し、
前記第1の工程は、乾燥状態の分離材に試料及び溶離液Aを接触させ、分離材を分散又は攪拌することによって行われ、その後、濾過操作により成分S2を含む濾過残渣を残して濾液中に成分S1を分画し、
前記第2の工程は、前記濾過残渣の分離材に溶離液Bを接触させ、分離材に結合していた成分S2を解離させ、次いで濾過操作により濾液中に成分S2を分画する、
ことを特徴とする方法。
[2]前記試料が血液であり、前記成分S2がヘモグロビンA1cである、[1]に記載の方法。
[3]前記分離材が、ポリマーを基材とし、m−アミノフェニルボロン酸を固定化したアフィニティ分離材であり、その粒径が10μmから200μmであり、1回の分離に使用する分離材の量が乾燥状態で20mg以下である、[1]又は[2]に記載の方法。
[4]前記溶離液Aが、前記分離材のボロネートアニオンとヘモグロビンA1cの1,2−シスジオールとが結合できる塩基性緩衝液であり、
前記溶離液Bが、前記分離材のボロネートアニオンとヘモグロビンA1cの1,2−シスジオールとが解離できる糖類またはアミンを含有する緩衝液である、
[3]に記載の方法。
[5]上述の[1]〜[4]いずれかに記載の方法で試料中の成分S1と成分S2を分離し、成分S1の量と成分S2の量の総和に対する成分S1または成分S2の量の比を求めることを特徴とする、成分S1または成分S2の測定方法。
[6]成分S1または成分S2の量を吸光度により測定する、[5]に記載の測定方法。
以下に本発明を更に詳細に説明する。
初めに従来の固相抽出法での難点である、使用する分離材量が多いという点を解決する手段について説明する。
従来の固相抽出法では、(1)固相抽出カラムに試料を添加し、(2)溶離液Aを添加し、成分S2を分離材に結合させ、(3)成分S1をカラムから溶出させ分画する。続いて、(4)溶離液Bを添加し、分離材に結合した成分S2を解離させ、(5)成分2をカラムから溶出させ分画する。固相抽出法では、分離は全てカラム内で行われる(図7参照)。
これに対し本発明では、乾燥状態の分離材に、試料及び溶離液Aを接触させ、分離材を分散又は攪拌することにより、成分S2を分離材に結合させることが大きな特徴である。次いで濾過することにより、分離材に結合しなかった成分S1を濾液中に分画する工程を行うことも大きな特徴である。次いで、濾過残渣(分離材)に溶離液Bを接触させ、分離材に結合していた成分S2を解離させ、次いで濾過操作により濾液中に成分2を分画する(図8参照)。
ここで、乾燥分離材への試料と溶離液Aの接触順序には特に限定はなく、同時に接触させてもよく、また順次接触させてもよい。また事前に試料と溶離液Aを混合してから、乾燥分離材に接触させてもよい。
このように乾燥状態の分離材に、試料及び溶離液を接触させ、分離材を分散または撹拌することにより、従来の固相抽出法のように、分離材の充填状態による性能への影響が少なくなる。そのため、使用する分離材の絶対量を大幅に少なくすることが可能であり、これにより、処理全体のスケールも大幅に少なくすることが可能となる。
また、従来の固相抽出法では、洗浄液/保存液/溶離液Aなどの溶液により湿潤状態の分離材を使用することから、添加した溶離液の容量を正確に把握することができなかったが、本発明の方法では、乾燥状態の分離材を使用することで、添加した溶離液の容量を正確に把握することが可能となる。これにより、分画された成分の定量分析を正確に行うことが可能となる。
本発明に用いる濾過方式は、分離材を含む溶液を分離材と溶液に効率よく分離できる方式であれば良く、特に濾過方式を限定するものではない。
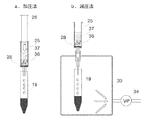
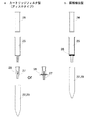
濾過方式の第1の例として、シリンジに液体クロマトグラフィの前処理用に市販されているカートリッジフィルタ(ディスクタイプ)を装着した方式が挙げられる(図6a)。シリンジ内に分離材を含む溶液を入れ、ピストンで押し出し濾過する方法である。この方法は、専用の真空チャンバ等の器材を必要としないことから簡便に使用することができる。シリンジの容量は、検体の処理容量に応じて選択すれば良いが、処理量の1.5から2倍程度の容量が好適である。
濾過方式の第2の例として、一般的な固相抽出で用いられる底面部にフィルタが圧入されている、空のカートリッジを使用することができる(図6b)。カートリッジ内に分離材を含む溶液を入れ、減圧により濾過する方法である。この方法は、専用の真空チャンバ等の器材を必要とするが、複数の検体を同時に処理できる利点がある。
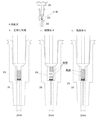
濾過方式の第3の例として、フィルタを底面に内蔵した内側容器と気密性のある外容器からなる濾過方式でも同様な効果が得られる。一例として、GEヘルスケア・ジャパン株式会社から発売されている「ミニユニ(Mini−UniPrep)」を使用することができる。
これは、外側容器(54)に微粒子を含む溶液を入れ、フィルタ部(28)を底面に内蔵した内側容器(55)を前記の外側容器に挿入する。内側容器(55)と外側容器(54)は気密性が保たれているため、前記溶液はフィルタ部(28)を通して内側の容器に濾過され、微粒子は外側容器(54)と内側容器(55)の隙間に残ることになる。つまり、内側の容器に捕集された溶液が「濾液」となる。
このような濾過方式を本発明に適用すると、使用する部材が少なく、操作も簡便化することが可能となる(図24参照)。
本発明において、試料には特に限定はないが、例えば血液等をあげることができ、こととき成分S2としてHbA1c、成分S1としてヘモグロビンA0(以下、ヘモグロビンA0を単にA0と記載する場合もある)をあげることができる。
本発明で使用する溶血液は、血球の細胞膜を破壊する組成であれば良く、組成、濃度、pH等を限定するものではない。
本発明で使用する分離材は、試料中の成分S2を溶離液Aにより結合し、かつ溶離液Bにより解離するものであり、その結果、試料中の成分S1とS2を分離することができるものであれば特に限定はない。ここで結合とは、化学的結合でもよく、また吸着であってもよい。例えばS1がヘモグロビンA0、S2がヘモグロビンA1cの場合、分離材はポリマーを基材とし、m−アミノフェニルボロン酸を固定化したものが好ましい。
また本発明で用いられる分離材は、粒子状が好ましく、その粒径には特に限定はないが、吸引濾過、加圧濾過が簡便な機構または用手法で容易に行えるよう、粒径が数10μm〜数100μmが望ましく、10μm〜200μmがさらに好ましい。
一般的にクロマト用分離材は湿潤状態又はスラリー状態で供給されるが、本発明に使用する分離材は、乾燥状態で用いることが必要である。そのため、例えば、事前に湿潤状態又はスラリー状態の分離材を吸引濾過し、純水を分離材量の3倍程度用いて洗浄した後、可能な限り純水を除去する。このような分離材を、シリカゲル等の乾燥剤を入れたデシケータ中で、水分を完全に除去した後、本発明の分離材として使用することができる。本発明では小スケールで分離を行うこともできるため、1回の分離での分離材の使用量が乾燥状態で20mg以下とすることもできる。
本発明で使用する溶離液については、試料中の成分S2を溶離液Aにより分離材に結合し、かつ溶離液Bにより分離材から解離するものであり、その結果、試料中の成分S1とS2を分離することができるものであれば特に限定はない。ここで結合とは、化学的結合でもよく、また吸着であってもよい。例えばS1がヘモグロビンA0、S2がヘモグロビンA1cの場合、分離材として上述のポリマーを基材としたm−アミノフェニルボロン酸を固定化したものを用いた場合は、溶離液Aは分離材のボロネートアニオンとA1cの1,2−シスジオールが結合できる塩基性緩衝液であればよく、組成、濃度、pH等を限定するものではない。また溶離液Bは、分離材のボロネートアニオンとA1cの1,2−シスジオールが解離できる糖類またはアミンを含有する緩衝液であればよく、組成、濃度、pH等を限定するものではない。溶離液Aの一例として、100mmol/Lグリシン+150mmol/L 硫酸ナトリウム(pH9.2)、溶離液Bの一例として、100mmol/L トリスヒドロキシメチルアミノメタン+50mmol/L硫酸ナトリウム+100mmol/L D−ソルビトール(pH8.0)をあげることができる。
本発明では、このような方法で試料中の成分S1と成分S2を分離し、成分S1の量と成分S2の量の総和に対する成分S1または成分S2の量の比を求めることができる。このとき、成分S1または成分S2の量の測定法については特に限定はないが、例えば吸光度により測定することができる。
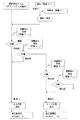
以下に、前記の方式を血液中に含まれるA1cを分離し測定に適応するプロトコールを説明する。プロトコールは一例として、以下の概ね11ステップから構成される(図12参照)。
(I)検体溶血工程(検体容量:Vs、溶血液容量:Vd)
(II)溶離液A添加工程(容量:V11)
(III)アフィニティ分離材との反応工程(分離材重量:G)
(IV)溶離液AによるA0相当成分溶出工程
(V)溶離液Aによる洗浄工程(容量:V12)
(VI)空気圧送工程
(VII)A0相当成分の吸光度測定工程(Aa)
(VIII)溶離液BによるA1c相当成分溶出工程(容量:V21)
(IX)溶離液Bによる洗浄工程(容量:V22)
(X)空気圧送工程
(XI)A1c相当成分の吸光度測定工程(Ab)
事前に十分に乾燥させたアフィニティ分離材を、容器Bに秤量する(分離材重量:G)。
(I)検体溶血工程(検体容量:Vs、溶血液容量:Vd)
(II)溶離液A添加工程(容量:V11)
(III)アフィニティ分離材との反応工程(分離材重量:G)
(IV)溶離液AによるA0相当成分溶出工程
(V)溶離液Aによる洗浄工程(容量:V12)
(VI)空気圧送工程
(VII)A0相当成分の吸光度測定工程(Aa)
(VIII)溶離液BによるA1c相当成分溶出工程(容量:V21)
(IX)溶離液Bによる洗浄工程(容量:V22)
(X)空気圧送工程
(XI)A1c相当成分の吸光度測定工程(Ab)
事前に十分に乾燥させたアフィニティ分離材を、容器Bに秤量する(分離材重量:G)。
容器Aに検体(容量:Vs)と溶血液(容量:Vd)を添加し、攪拌により検体を溶血させる(I)。前記容器Aに溶離液A(容量:V11)を添加し攪拌する(II)。
分離材の入った容器Bに前記検体溶液を全量移送し、十分に攪拌し、A1c相当成分を分離材に結合させる(III)。
フィルタを付けたシリンジに前記の分離材を含んだ溶液を全量移送し、加圧または減圧により濾過し、濾液を容器Cに捕集する(IV)。シリンジに溶離液A(容量:V12)を添加し、加圧または減圧により濾過し、濾液を容器Cに捕集する(V)。空気圧送により溶液を完全に押し出す(VI)。以上の操作によりA0相当成分が濾液に捕集される。濾液の吸光度(Aa)を測定する(VII)。
前記操作の残渣の分離材に溶離液B(容量:V21)を添加し、加圧または減圧により濾過し、濾液を容器Dに捕集する(VIII)。シリンジに溶離液B(容量:V22)を添加し、加圧または減圧により濾過し、濾液を容器Dに捕集する(IX)。空気圧送により溶液を完全に押し出す(X)。
以上の操作によりA1c相当成分が分離材から解離し濾液に捕集される。濾液の吸光度(Ab)を測定する(XI)。
A0相当成分の吸光度(Aa)とA1c相当成分の吸光度(Ab)から、下記式1により
A1c相当成分の比率を求めA1c比率(%)を算出する。
A1c比率(%)=[Ab/(Aa+Ab)]×100 (式1)
工程(I)から(VI)で使用する溶液容量、即ちA0相当成分の溶出に供した溶液量(Vs+Vd+V11+V12)と溶離液Bの容量、即ちA1c相当成分の溶出に供した溶液量(V21+V22)は式2のようにできるだけ同じにすることが望ましい。
(Vs+Vd+V11+V12)=(V21+V22) (式2)
容量が異なる場合、A0相当成分の吸光度(Aa)とA1c相当成分の吸光度(Ab)を添加した容量で補正する必要がある。
A1c相当成分の比率を求めA1c比率(%)を算出する。
A1c比率(%)=[Ab/(Aa+Ab)]×100 (式1)
工程(I)から(VI)で使用する溶液容量、即ちA0相当成分の溶出に供した溶液量(Vs+Vd+V11+V12)と溶離液Bの容量、即ちA1c相当成分の溶出に供した溶液量(V21+V22)は式2のようにできるだけ同じにすることが望ましい。
(Vs+Vd+V11+V12)=(V21+V22) (式2)
容量が異なる場合、A0相当成分の吸光度(Aa)とA1c相当成分の吸光度(Ab)を添加した容量で補正する必要がある。
吸光度を測定する波長は、A1cを特異的に検出できる波長であり、溶離液Aおよび溶離液Bに対して吸収ができるだけ無い波長であれば良く、特に限定するものではない。一般的には405〜420nm程度が使用可能であるが、415nmが好適である。
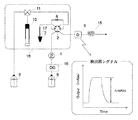
吸光度を測定する方法は、目的とする測定精度で決めれば良く特に限定するものではない。図23は吸光度測定方法の例を幾つか示したものである。図23aは、シングルビームにより簡易測定を行う例である。測定精度を必要としない場合や、検体の濃度が比較的高く溶離液のバックグラウンドの影響が少ない場合などは、この方法で測定を行ってもよい。
図23bはデュアルビーム分光光度計により測定を行う例である。A0相当成分の吸光度(Aa)、A1c相当成分の吸光度(Ab)の吸光度を測定する波長(一例、415nm)であっても、溶離液A、溶離液Bの吸収が僅かに存在するため、この影響を補正することが可能な方法で、精度良く測定を行うことができる。A0相当成分の吸光度(Aa)を測定する際はリファレンスとして溶離液A(吸光度:Ara)を、A1c相当成分の吸光度(Ab)を測定する際はリファレンスとして溶離液B(吸光度:Arb)を使用することで、溶離液の吸光度を補正することができる。検体の濃度が低い場合などには適した方法である。
図23cは2つの波長により測定を行う例である。A0相当成分、A1c相当成分に対して吸収がある波長を第1波長とし、A0相当成分、A1c相当成分に対して吸収がなく、溶離液A、Bにのみ前記第1波長と同程度の吸収がある波長を第2波長とし、2つの波長で同時に測定を行い、第1波長の吸光度から第2波長の吸光度を差し引くことで、溶離液A、溶離液Bの吸収の影響を補正することが可能な方法である。一例として、第1波長として415nm、第2波長として450nmを使用することで、溶離液の吸収の影響を補正することができる。
図23dは、フローインジェクション法による測定を行う例である。この方法は、溶液の流れに測定試料を導入し、吸光度測定する方法であり、効率よく吸光度測定を行うことが可能な方法である。
何れの例でも、試料を測定するセルには、専用のセルを使用しても良いが、本発明の操作で使用する容器C、容器Dに直接光を照射して測定することも可能であり、操作がより簡便に行うことができる。この場合、容器C、Dには可視光透過樹脂(ポリメチルメタクリレート、ポリスチレン樹脂等)のものを使用する必要がある。
ここでは、測定の手順として、(I)検体溶血工程、(II)溶離液A添加工程、(III)アフィニティ分離材との反応工程の順番で記載したが、(I)検体溶血工程、(III)アフィニティ分離材との反応工程、(II)溶離液A添加工程、の順番に行っても同様な効果が得られ、特に順番を限定するものではない。また、溶離液A,溶離液Bの添加工程を2回で記載したが、更に回数を増やしても同様な効果が得られ、特に回数を限定するものではない。逆に、精度は低下するものの、溶離液Aによる洗浄工程(V)、溶離液Bによる洗浄工程(IX)を省略したり、(VI)(X)の空気圧送工程を省略したりして工程の短縮を行ってもよい。
図25に前記の濾過方式を本発明に適用した場合の操作手順を示す。
事前に十分に乾燥させたアフィニティ分離材を、外側容器(54)に秤量する(分離材重量:G)。
(I)容器Aに検体(容量:Vs)と溶血液(容量:Vd)を分注し、攪拌により検体を溶血させる。
(II)前記容器Aに溶離液A(容量:V11)を添加し攪拌する。
(III)分離材の入った外側容器(54)に前記検体溶液を全量移送し、十分に攪拌し、A1c相当成分を分離材に結合させる。
(IV)フィルタの付いた内側容器(55)を挿入し、内側容器内に濾液を捕集する。
(V)一度、内側容器(55)を取り外し、外側容器に溶離液A(容量:V12)を添加する。
(VI)再び内側容器を挿入し濾液を捕集する。
(VII)内側容器に捕集されたA0相当成分の吸光度(Aa)を測定する。
(VIII)内側容器(55)を取り外し、外側容器(54)に溶離液B(容量:V21)を添加し、内側容器を挿入し、内側容器に濾液を捕集する。
(IX)一度、内側容器(55)を取り外し、外側容器(54)に溶離液B(容量:V22)を添加する。
(X)再び内側容器を挿入し濾液を捕集する。
(XI)内側容器に捕集されたA1c相当成分の吸光度(Ab)を測定する。
A0相当成分の吸光度(Aa)とA1c相当成分の吸光度(Ab)からA1cの存在比率を式1により算出する。
(I)容器Aに検体(容量:Vs)と溶血液(容量:Vd)を分注し、攪拌により検体を溶血させる。
(II)前記容器Aに溶離液A(容量:V11)を添加し攪拌する。
(III)分離材の入った外側容器(54)に前記検体溶液を全量移送し、十分に攪拌し、A1c相当成分を分離材に結合させる。
(IV)フィルタの付いた内側容器(55)を挿入し、内側容器内に濾液を捕集する。
(V)一度、内側容器(55)を取り外し、外側容器に溶離液A(容量:V12)を添加する。
(VI)再び内側容器を挿入し濾液を捕集する。
(VII)内側容器に捕集されたA0相当成分の吸光度(Aa)を測定する。
(VIII)内側容器(55)を取り外し、外側容器(54)に溶離液B(容量:V21)を添加し、内側容器を挿入し、内側容器に濾液を捕集する。
(IX)一度、内側容器(55)を取り外し、外側容器(54)に溶離液B(容量:V22)を添加する。
(X)再び内側容器を挿入し濾液を捕集する。
(XI)内側容器に捕集されたA1c相当成分の吸光度(Ab)を測定する。
A0相当成分の吸光度(Aa)とA1c相当成分の吸光度(Ab)からA1cの存在比率を式1により算出する。
溶血液、溶離液Aおよび溶離液Bの使用容量は、処理する試料の量によって最適化すればよく、限定されるものではない。一例として、試料2μLとした場合、乾燥アフィニティ分離材20mg、溶血液98μL、初段の溶離液A2.9mL(V11)、次段の溶離液A1.0mL(V12)、初段の溶離液B2.0mL(V21)、次段の溶離液B2.0mL(V22)で処理を行うことができる。
本発明の分離方法は、小スケールにも適用しうるものであり、かつ定量性にも優れるため、試料中の成分の量比を求める方法にも適用できる。その結果、試料中のグリコヘモグロビン(A1c)等の量比を測定することも可能である。
以下、実施例により本発明をさらに詳細に説明するが、これら実施例は本発明を限定するものではない。
[実施例1]
本発明の効果を検証するため、従来の固相抽出法との比較を実施した。
本発明の効果を検証するため、従来の固相抽出法との比較を実施した。
まず、吸光度測定方法について説明する。本発明の方法および従来の固相抽出法のいずれの吸光度測定も、フローインジェクション法によりオンラインで実施した。図9はその装置構成および流路を示した図である。装置は主に送液ポンプ(1)、試料注入装置(18)、紫外可視検出器(5)で構成される。一定流速で溶離液を送液しながら、試料注入装置により処理された試料を大量に注入し、検出器出力が平衡(台形状出力)になった値から吸光度を算出した。送液ポンプ(1)は東ソー製DP−8020を毎分0.35ミリリッタの流速で、試料注入装置(18)は東ソー製AS−8020(注入量800マイクロリッタ)を、紫外可視検出器(5)は東ソー製UV−8020を415nmの波長で使用した。
次に処理に使用した部材について説明する。検体はA1cが4.8%程度の全血、A1cが7.3%程度の全血、およびキャリブレータ(基準値:5.59%、10.33% 東ソー(株)製)を使用した。
溶血液は、グリコヘモグロビン分析計HLC−723GHbV(東ソー(株)製)専用溶血液をそのまま使用した。A1c分離の溶離液としては、下記の組成のものを調整し使用した。
溶離液A:酢酸アンモニウム 150mmol/L、塩化マグネシウム 50nmoL/L、5%エタノール、pH8.7
溶離液B:トリスヒドロキシメチルアミノメタン 50nmoL/L、EDTA・2Na 50mmoL/L、ソルビトール 100mmoL/L、5%エタノール、pH8.0 なお、前記のフローインジェクション用の溶離液にも前記溶離液Aを使用した。フィルタはクロマト用の前処理用に使用されるディスク型カートリッジフィルタを使用した(孔径:0.45μm、材質:セルロースアセテート、外径:3mm、「マイショリディスク」東ソー(株)製)。シリンジは2.5mLのディスポプラスティック注射器を使用し、加圧法で使用した。分離材は、メタクリレートポリマーを基材としm−アミノフェニルボロン酸を固定化した充てん剤であるTSKgel Boronate−5PW(東ソー製)を用いた。従来の固相抽出法で使用する場合は、1g/mLの濃度に調整された前記分離材スラリー200μL(分離材量20mg相当)を、ディスクフィルタを装着した2.5mLのプラスティックシリンジに分注し、固相抽出カラムを作製し測定に使用した。本発明の測定方法で使用する場合は、前記分離材スラリーを吸引濾過し、ほぼ完全に水分を除去した後、シリカゲル存在下のデシケータ中で完全に乾燥させ測定に使用した。
溶離液A:酢酸アンモニウム 150mmol/L、塩化マグネシウム 50nmoL/L、5%エタノール、pH8.7
溶離液B:トリスヒドロキシメチルアミノメタン 50nmoL/L、EDTA・2Na 50mmoL/L、ソルビトール 100mmoL/L、5%エタノール、pH8.0 なお、前記のフローインジェクション用の溶離液にも前記溶離液Aを使用した。フィルタはクロマト用の前処理用に使用されるディスク型カートリッジフィルタを使用した(孔径:0.45μm、材質:セルロースアセテート、外径:3mm、「マイショリディスク」東ソー(株)製)。シリンジは2.5mLのディスポプラスティック注射器を使用し、加圧法で使用した。分離材は、メタクリレートポリマーを基材としm−アミノフェニルボロン酸を固定化した充てん剤であるTSKgel Boronate−5PW(東ソー製)を用いた。従来の固相抽出法で使用する場合は、1g/mLの濃度に調整された前記分離材スラリー200μL(分離材量20mg相当)を、ディスクフィルタを装着した2.5mLのプラスティックシリンジに分注し、固相抽出カラムを作製し測定に使用した。本発明の測定方法で使用する場合は、前記分離材スラリーを吸引濾過し、ほぼ完全に水分を除去した後、シリカゲル存在下のデシケータ中で完全に乾燥させ測定に使用した。
次に、比較のための、従来の固相抽出法について説明する。図10は従来の固相抽出法での処理のプロトコール、図11は実際の測定手順を示した図である。処理のプロトコールは次の通りである。容器A(20)に検体2μLを分注し、溶血液98μLを添加し攪拌し溶血した後、全量を前記手順で作製した固相抽出カラム(39)に負荷する。次に溶離液Aを2.9mL添加し、ピストンで加圧し容器C(22)に溶出液を捕集する(A1c相当成分を分離材に吸着)。
次に溶離液Aを1mL添加し洗浄を行う。ピストンで加圧した後、最後に空気を圧送し完全に溶液を排出させる。容器C(22)に捕集された成分を図9の装置にてA0相当成分の吸光度(Aa)を測定する。続いて、溶離液Bを2mL添加し、ピストンで加圧し容器D(23)に溶出液を捕集する。溶離液Bを2mL添加し、ピストンで加圧し洗浄を行う。最後に空気を圧送し完全に溶液を排出させる。容器D(23)に捕集された成分を図9の装置にてA1c相当成分の吸光度(Ab)を測定した。前記で得られた吸光度(Aa、Ab)から前述の式1によりA1c比率(%)を算出した。
次に、本発明の方法による測定法を説明する。図12は処理のプロトコール、図13は実際の測定手順を示した図である。処理のプロトコールは以下の通りである。事前に、容器B(21)に前記の乾燥させた分離材を20mg秤量しておく。別の容器A(20)に検体2μLを分注し、溶血液98μLを添加し攪拌し溶血を行う。次に溶離液Aを2.9mL添加し攪拌する。この試料混合液(容器A)を、前記分離材を入れた容器B(21)に全量添加し、1分以上攪拌し、A1c相当成分を分離材に吸着させる。2.5mLのプラスティックシリンジに前記フィルタを装着し、前記分離材を含む混合液を入れ、ピストンで加圧し容器C(22)に濾過する。次に溶離液Aを1mL添加し洗浄を行う。ピストンで加圧した後、最後に空気を圧送し完全に溶液を排出させる。容器C(22)に捕集された濾液成分を図9の装置にてA0相当成分の吸光度(Aa)を測定する。続いて、溶離液Bを2mL添加し、ピストンで加圧し容器D(23)に濾過する。溶離液Bを2mL添加し、ピストンで加圧し洗浄を行う。最後に空気を圧送し完全に溶液を排出させる。容器D(23)に捕集された濾液成分を図9の装置にてA1c相当成分の吸光度(Ab)を測定した。
ここでは、検体容量(Vs)、溶血液容量(Vd)、溶離液Aの総容量(V11+V12)及び溶離液Bの総容量(V21+V22)との間に前述の式2の関係が成立していることから、前述の式1によりA1c比率(%)を算出した。
従来の固相抽出法および本発明の方法とも、A1cの値が異なる2つの検体で各20回の測定を実施した。検量線は、高濃度に調整したキャリブレータ(通常4mLに溶解するところを、100μLに溶解)を使用し、同様な手法でそれぞれ測定を行った。
図14、15、16および表1、2は従来の固相抽出法で測定結果を、図17、18、19および表3、4は本発明の方法で測定結果を示した図である。
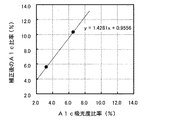
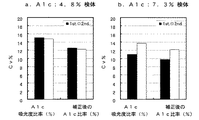
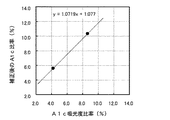
キャリブレータ(基準値:5.59%、10.33%)による検量線について説明する(図14、図17)。横軸が式1で計算したA1c比率(%)、縦軸がキャリブレータの基準値、即ち補正後のA1c比率(%)を示している。従来の固相抽出法、本発明の方法の何れでも、正の傾きの検量線が得られることから、何れの方法でもA1c比率(%)の測定が可能であることがうかがえる。
次に実検体(A1c約4.8%、約7.3%の検体)の測定値の変動について説明する(図15、図18参照)。横軸が測定回数、縦軸がA1c比率(%)である。凡例の白丸は式1で計算したA1c比率(%)、凡例の黒丸は図14又は図17の検量線を用いて補正した後の補正後のA1c比率(%)である。従来の固相抽出法(図15)では、20回の測定で、低値検体(A1c:4.8%)で2.3%、中値検体(A1c:7.3%)で3.6%の変動に対して、本発明の方法(図18)では、低値検体(A1c:4.8%)で0.6%、中値検体(A1c:7.3%)で0.6%の変動に収まっており、本発明の精度の良さが分かる。最終結果である補正後のA1c比率のCv%(n=10)で比較すると、より差異が明らかになる。従来の固相抽出法では、Cvが低値検体で12.52%、12.23%、中値検体で9.71%、12.21%であるのに対して(図16、表1、2参照)、本発明の方法では、Cvが低値検体で4.36%、1.68%、中値検体で1.79%、2.61%と大幅に改善されている(図19、表3、4参照)。
このように、本発明は、簡易測定法でありながら、僅かな分離材量であっても精度良くA1cを定量することができる。
[実施例2]
本発明の方法の精度を確認するため、アフィニティモードの液体クロマトグラフィ法との相関性試験を実施した。検体は全血(27検体)を使用した。比較のため、実施例1と同じように、従来の固相抽出法での測定も実施した。
本発明の方法の精度を確認するため、アフィニティモードの液体クロマトグラフィ法との相関性試験を実施した。検体は全血(27検体)を使用した。比較のため、実施例1と同じように、従来の固相抽出法での測定も実施した。
アフィニティクロマト法での測定は下記の条件で実施した。分析カラムは内径1.5mm、長さ10mmのカラム管にTSKgelBorate−5PW(10μm)を充填したものを、45℃で温調されたカラムオーブン内に設置し使用した。溶離液A、Bは前記実施例1で使用したものを使用した。溶離液は毎分250マイクロリッタで送液し、ステップグラジエントは、試料注入後、0分から0.6分まで溶離液A、0.6分から1.4分まで溶離液Bが流れる条件で実施した。本方法の測定手順および従来の固相抽出法での測定手順は、実施例1と同じ条件で実施した。
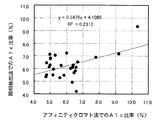
図21は従来の固相抽出法で相関性試験の結果を、図22および表5は本発明の方法で相関性試験の結果を示した図である。横軸がアフィニティモードの液体クロマトグラフィ法での測定値、縦軸が固相抽出法または本発明の方法による測定値である。表5は各々の結果を集計した表である(表中の丸数字の2は前述の検量線による補正後のA1c比率(%)である)。従来の固相抽出法(図21)では、相関係数が0.4809(R2:0.2313)であるのに対して、本発明の方法(図22)では相関係数が0.9720(R2:0.9447)と非常に良好な相関性が得られた。これは、実施例1で明らかなように、固相抽出法での再現性が悪いことから、相関性にも悪影響を及ぼしている。
この結果からも、本発明は、簡易測定法でありながら、従来の測定法との相関性を良好で、A1cを定量することが可能であることが明らかとなった。
1:送液ポンプ
2:試料導入バルブ
3:分析カラム
4:カラム恒温槽
5:紫外可視検出器
6:ループ
7:試料(検体)
8,8a,8b,8c:溶離液
9:洗浄液
10:シリンジ
11:電磁弁
12:希釈ポート
13:ドレンポート
14:希釈部
15:抵抗管
16:脱気装置
17:ニードル
18:試料注入装置
19:容器
20:容器A
21:容器B
22:容器C
23:容器D
24:分注器
25:シリンジ
26:ピストン
27:カートリッジフィルタ
28:フィルタ部
29:アフィニティ分離材
30:溶離液A
31:溶離液B
32:溶血液
33.真空チャンバ
34.真空ポンプ
35.充填溶媒
36.分離材
37.溶離液
38.亀裂/空洞
39.固相抽出カラム
40.光源
41.ハーフミラー
42.全反射ミラー
43.フォトセンサ(サンプル側)
44.フォトセンサ(リファレンス側)
45.フォトセンサ
46.セル(サンプル側)
47.セル(リファレンス側)
48.測定容器
49.フローセル
50.測定試料
51.試料導入機構
52.送液機構
53.溶液
54.外側容器
55.内側容器
56.ダイクロイックミラー
2:試料導入バルブ
3:分析カラム
4:カラム恒温槽
5:紫外可視検出器
6:ループ
7:試料(検体)
8,8a,8b,8c:溶離液
9:洗浄液
10:シリンジ
11:電磁弁
12:希釈ポート
13:ドレンポート
14:希釈部
15:抵抗管
16:脱気装置
17:ニードル
18:試料注入装置
19:容器
20:容器A
21:容器B
22:容器C
23:容器D
24:分注器
25:シリンジ
26:ピストン
27:カートリッジフィルタ
28:フィルタ部
29:アフィニティ分離材
30:溶離液A
31:溶離液B
32:溶血液
33.真空チャンバ
34.真空ポンプ
35.充填溶媒
36.分離材
37.溶離液
38.亀裂/空洞
39.固相抽出カラム
40.光源
41.ハーフミラー
42.全反射ミラー
43.フォトセンサ(サンプル側)
44.フォトセンサ(リファレンス側)
45.フォトセンサ
46.セル(サンプル側)
47.セル(リファレンス側)
48.測定容器
49.フローセル
50.測定試料
51.試料導入機構
52.送液機構
53.溶液
54.外側容器
55.内側容器
56.ダイクロイックミラー
Claims (6)
- 試料中の成分S1と成分S2を分離する方法であって、
溶離液Aにより成分S2を特異的に分離材に結合させる第1の工程と、
溶離液Bにより成分S2を分離材から解離させる第2の工程を有し、
前記第1の工程は、乾燥状態の分離材に試料及び溶離液Aを接触させ、分離材を分散又は攪拌することによって行われ、その後、濾過操作により成分S2を含む濾過残渣を残して濾液中に成分S1を分画し、
前記第2の工程は、前記濾過残渣の分離材に溶離液Bを接触させ、分離材に結合していた成分S2を解離させ、次いで濾過操作により濾液中に成分S2を分画する、
ことを特徴とする方法。 - 前記試料が血液であり、前記成分S2がヘモグロビンA1cである、請求項1に記載の方法。
- 前記分離材が、ポリマーを基材とし、m−アミノフェニルボロン酸を固定化したアフィニティ分離材であり、その粒径が10μmから200μmであり、1回の分離に使用する分離材の量が乾燥状態で20mg以下である、請求項1又は2に記載の方法。
- 前記溶離液Aが、前記分離材のボロネートアニオンとヘモグロビンA1cの1,2−シスジオールとが結合できる塩基性緩衝液であり、
前記溶離液Bが、前記分離材のボロネートアニオンとヘモグロビンA1cの1,2−シスジオールとが解離できる糖類またはアミンを含有する緩衝液である、
請求項3に記載の方法。 - 請求項1〜4いずれかに記載の方法で試料中の成分S1と成分S2を分離し、成分S1の量と成分S2の量の総和に対する成分S1または成分S2の量の比を求めることを特徴とする、成分S1または成分S2の測定方法。
- 成分S1または成分S2の量を吸光度により測定する、請求項5に記載の測定方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013256153A JP2015114197A (ja) | 2013-12-11 | 2013-12-11 | 乾燥分離材を用いた分離方法及び測定方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013256153A JP2015114197A (ja) | 2013-12-11 | 2013-12-11 | 乾燥分離材を用いた分離方法及び測定方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2015114197A true JP2015114197A (ja) | 2015-06-22 |
Family
ID=53528122
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2013256153A Pending JP2015114197A (ja) | 2013-12-11 | 2013-12-11 | 乾燥分離材を用いた分離方法及び測定方法 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2015114197A (ja) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN107966498A (zh) * | 2016-10-18 | 2018-04-27 | 湖北生物医药产业技术研究院有限公司 | 一种检测艾代拉里斯中溶剂残留的方法 |
| JP2018128394A (ja) * | 2017-02-09 | 2018-08-16 | 東ソー株式会社 | 分離材を用いた測定方法 |
| JP2023516808A (ja) * | 2020-03-09 | 2023-04-20 | アイデックス ラボラトリーズ インコーポレイテッド | 液体サンプルの干渉成分を化学試薬検査スライドへの適用前に除去するための方法 |
| WO2023199678A1 (ja) * | 2022-04-14 | 2023-10-19 | 国立研究開発法人産業技術総合研究所 | 分離対象物質の純度が高められた精製液を製造する方法及び分離対象物質を精製するための精製キット |
| US12392795B2 (en) | 2020-03-09 | 2025-08-19 | Idexx Laboratories, Inc. | Matrix and associated sample or mixing cup used for removing components of a liquid sample |
Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS63500002A (ja) * | 1985-06-10 | 1988-01-07 | バテル メモリアル インステイチユ−ト | 生物学的活性物質を生物学的特異的吸着により精製する方法及びそのための吸着剤 |
| JPH0346566A (ja) * | 1989-07-11 | 1991-02-27 | Miles Inc | 非遠心性及び非毛細管性の操作による逐次的分析試験実施用の反応カセット |
| JPH09304382A (ja) * | 1996-03-14 | 1997-11-28 | Tosoh Corp | ヘモグロビンA1cの測定法及びキット |
| JP2001524681A (ja) * | 1997-11-28 | 2001-12-04 | プロヴァリス・ダイアグノスティクス・リミテッド | アッセイを案内するための設備および装置 |
| JP2002139481A (ja) * | 2000-11-02 | 2002-05-17 | Tosoh Corp | 糖化ヘモグロビン測定装置 |
| JP2007255912A (ja) * | 2006-03-20 | 2007-10-04 | Nokodai Tlo Kk | 糖化タンパク質分離・検出用デバイス |
| JP2008519986A (ja) * | 2004-11-12 | 2008-06-12 | プロメガ コーポレイション | 生物学的物質の精製のための器具及び方法 |
| JP2009092643A (ja) * | 2007-10-08 | 2009-04-30 | Infopia Co Ltd | 糖化ヘモグロビン測定カセット及びそれを利用した糖化ヘモグロビン測定方法 |
| JP2012002593A (ja) * | 2010-06-15 | 2012-01-05 | Hitachi High-Technologies Corp | 生体試料前処理方法及び装置 |
| JP2012215470A (ja) * | 2011-03-31 | 2012-11-08 | Tosoh Corp | 糖化ヘモグロビンの測定方法及び糖化ヘモグロビン測定装置 |
-
2013
- 2013-12-11 JP JP2013256153A patent/JP2015114197A/ja active Pending
Patent Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS63500002A (ja) * | 1985-06-10 | 1988-01-07 | バテル メモリアル インステイチユ−ト | 生物学的活性物質を生物学的特異的吸着により精製する方法及びそのための吸着剤 |
| JPH0346566A (ja) * | 1989-07-11 | 1991-02-27 | Miles Inc | 非遠心性及び非毛細管性の操作による逐次的分析試験実施用の反応カセット |
| JPH09304382A (ja) * | 1996-03-14 | 1997-11-28 | Tosoh Corp | ヘモグロビンA1cの測定法及びキット |
| JP2001524681A (ja) * | 1997-11-28 | 2001-12-04 | プロヴァリス・ダイアグノスティクス・リミテッド | アッセイを案内するための設備および装置 |
| JP2002139481A (ja) * | 2000-11-02 | 2002-05-17 | Tosoh Corp | 糖化ヘモグロビン測定装置 |
| JP2008519986A (ja) * | 2004-11-12 | 2008-06-12 | プロメガ コーポレイション | 生物学的物質の精製のための器具及び方法 |
| JP2007255912A (ja) * | 2006-03-20 | 2007-10-04 | Nokodai Tlo Kk | 糖化タンパク質分離・検出用デバイス |
| JP2009092643A (ja) * | 2007-10-08 | 2009-04-30 | Infopia Co Ltd | 糖化ヘモグロビン測定カセット及びそれを利用した糖化ヘモグロビン測定方法 |
| JP2012002593A (ja) * | 2010-06-15 | 2012-01-05 | Hitachi High-Technologies Corp | 生体試料前処理方法及び装置 |
| JP2012215470A (ja) * | 2011-03-31 | 2012-11-08 | Tosoh Corp | 糖化ヘモグロビンの測定方法及び糖化ヘモグロビン測定装置 |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN107966498A (zh) * | 2016-10-18 | 2018-04-27 | 湖北生物医药产业技术研究院有限公司 | 一种检测艾代拉里斯中溶剂残留的方法 |
| CN107966498B (zh) * | 2016-10-18 | 2021-06-22 | 湖北生物医药产业技术研究院有限公司 | 一种检测艾代拉里斯中溶剂残留的方法 |
| JP2018128394A (ja) * | 2017-02-09 | 2018-08-16 | 東ソー株式会社 | 分離材を用いた測定方法 |
| JP2023516808A (ja) * | 2020-03-09 | 2023-04-20 | アイデックス ラボラトリーズ インコーポレイテッド | 液体サンプルの干渉成分を化学試薬検査スライドへの適用前に除去するための方法 |
| JP7679396B2 (ja) | 2020-03-09 | 2025-05-19 | アイデックス ラボラトリーズ インコーポレイテッド | 液体サンプルの干渉成分を化学試薬検査スライドへの適用前に除去するための方法 |
| US12366506B2 (en) | 2020-03-09 | 2025-07-22 | Idexx Laboratories, Inc. | Method for removing interfering components of a liquid sample prior to dispensing same on a chemical reagent test slide |
| US12392795B2 (en) | 2020-03-09 | 2025-08-19 | Idexx Laboratories, Inc. | Matrix and associated sample or mixing cup used for removing components of a liquid sample |
| WO2023199678A1 (ja) * | 2022-04-14 | 2023-10-19 | 国立研究開発法人産業技術総合研究所 | 分離対象物質の純度が高められた精製液を製造する方法及び分離対象物質を精製するための精製キット |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Hage | Affinity chromatography: a review of clinical applications | |
| Reuhs et al. | High-performance liquid chromatography | |
| Kema et al. | Profiling of tryptophan-related plasma indoles in patients with carcinoid tumors by automated, on-line, solid-phase extraction and HPLC with fluorescence detection | |
| Baghdady et al. | Online comprehensive high pH reversed phase× low pH reversed phase approach for two-dimensional separations of intact proteins in top-down proteomics | |
| JP2015114197A (ja) | 乾燥分離材を用いた分離方法及び測定方法 | |
| WO2000008460A1 (en) | Method for determining hemoglobins | |
| CN113933403B (zh) | 一种丙氨酰谷氨酰胺有关物质的测定方法 | |
| JPH03102259A (ja) | 液体クロマトグラフの方法,その装置,そのシステム,及びその分離カラム | |
| Emara et al. | An eco-friendly direct injection HPLC method for methyldopa determination in serum by mixed-mode chromatography using a single protein-coated column | |
| CN116626191A (zh) | 检测游离睾酮的超滤-液相色谱串联质谱法 | |
| JP2017198666A (ja) | 液体クロマトグラフィ測定方法、液体クロマトグラフィ測定装置、及び液体クロマトグラフィ測定プログラム | |
| WO1997041771A1 (en) | Chromatographic method for the identification and characterization of hemoglobin variants in blood | |
| JP2017187319A (ja) | 区間分割によるバックグラウンド補正方法 | |
| CA2088360A1 (en) | Quantitative analysis and monitoring of protein structure by substractive chromatography | |
| JP2001228133A (ja) | ヘモグロビン類の測定方法 | |
| JP6885089B2 (ja) | 分離材を用いた測定方法 | |
| Clarke et al. | Clinical applications of affinity chromatography | |
| CN115774110A (zh) | 生物安全的糖化血红蛋白解离液及糖化血红蛋白解离方法 | |
| US4914036A (en) | Method and apparatus for simultaneously analyzing vanillylmandelic acid, homovanillic acid and creatinine | |
| CN209102674U (zh) | 一种二维色谱质谱联用分析烟草中农残的装置 | |
| JPH08105875A (ja) | リポタンパク質分離用充填剤、その充填剤を充填してなる抗体カラム及びそのカラムを用いたレムナント様リポタンパク質の分析方法 | |
| Wajcman | Analysis of hemoglobins and globin chains by high-performance liquid chromatography | |
| JPH10227780A (ja) | 糖化ヘモグロビンの測定方法および装置 | |
| JP5471287B2 (ja) | セロトニンの測定方法 | |
| Tutar | High Performance Liquid Chromatography (HPLC) Theoretical and Practical Aspects |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20161122 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20170906 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20171031 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20180522 |