JP2010516750A - オリーブからの抽出物又はオリーブ・オイルの抽出後の残留物を含む固体成分からヒドロキティロソルを生成するプロセスと装置 - Google Patents
オリーブからの抽出物又はオリーブ・オイルの抽出後の残留物を含む固体成分からヒドロキティロソルを生成するプロセスと装置 Download PDFInfo
- Publication number
- JP2010516750A JP2010516750A JP2009546832A JP2009546832A JP2010516750A JP 2010516750 A JP2010516750 A JP 2010516750A JP 2009546832 A JP2009546832 A JP 2009546832A JP 2009546832 A JP2009546832 A JP 2009546832A JP 2010516750 A JP2010516750 A JP 2010516750A
- Authority
- JP
- Japan
- Prior art keywords
- hydroxytyrosol
- resin
- hydrolysis
- chromatography column
- starting material
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- JUUBCHWRXWPFFH-UHFFFAOYSA-N Hydroxytyrosol Chemical compound OCCC1=CC=C(O)C(O)=C1 JUUBCHWRXWPFFH-UHFFFAOYSA-N 0.000 title claims abstract description 304
- 235000003248 hydroxytyrosol Nutrition 0.000 title claims abstract description 152
- 229940095066 hydroxytyrosol Drugs 0.000 title claims abstract description 152
- 238000000034 method Methods 0.000 title claims abstract description 64
- 239000000284 extract Substances 0.000 title claims abstract description 50
- 239000007787 solid Substances 0.000 title claims abstract description 38
- 241000207836 Olea <angiosperm> Species 0.000 title claims abstract description 9
- 238000000605 extraction Methods 0.000 title claims description 23
- 239000004006 olive oil Substances 0.000 title claims description 16
- 235000008390 olive oil Nutrition 0.000 title claims description 16
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims abstract description 55
- 239000011347 resin Substances 0.000 claims abstract description 54
- 229920005989 resin Polymers 0.000 claims abstract description 54
- 238000004587 chromatography analysis Methods 0.000 claims abstract description 44
- 238000006460 hydrolysis reaction Methods 0.000 claims abstract description 36
- 230000007062 hydrolysis Effects 0.000 claims abstract description 35
- 240000007817 Olea europaea Species 0.000 claims abstract description 27
- 239000003957 anion exchange resin Substances 0.000 claims abstract description 26
- 239000007858 starting material Substances 0.000 claims abstract description 26
- 239000002253 acid Substances 0.000 claims abstract description 18
- 238000005903 acid hydrolysis reaction Methods 0.000 claims abstract description 12
- 239000007864 aqueous solution Substances 0.000 claims abstract description 11
- 238000004519 manufacturing process Methods 0.000 claims abstract description 6
- 239000012539 chromatography resin Substances 0.000 claims abstract description 3
- 239000012530 fluid Substances 0.000 claims description 41
- 238000004128 high performance liquid chromatography Methods 0.000 claims description 25
- 239000000243 solution Substances 0.000 claims description 16
- 238000004659 sterilization and disinfection Methods 0.000 claims description 16
- 230000001954 sterilising effect Effects 0.000 claims description 14
- 230000003204 osmotic effect Effects 0.000 claims description 12
- 239000002250 absorbent Substances 0.000 claims description 11
- 230000002745 absorbent Effects 0.000 claims description 11
- 230000002441 reversible effect Effects 0.000 claims description 10
- 239000003921 oil Substances 0.000 claims description 9
- 235000019198 oils Nutrition 0.000 claims description 9
- 125000003118 aryl group Chemical group 0.000 claims description 5
- 229920000642 polymer Polymers 0.000 claims description 4
- 230000003301 hydrolyzing effect Effects 0.000 claims description 3
- 230000026676 system process Effects 0.000 claims 1
- 239000002798 polar solvent Substances 0.000 abstract description 7
- 239000000047 product Substances 0.000 description 41
- 238000000746 purification Methods 0.000 description 25
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 21
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 18
- 229920002774 Maltodextrin Polymers 0.000 description 17
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 16
- 239000005913 Maltodextrin Substances 0.000 description 16
- 229940035034 maltodextrin Drugs 0.000 description 16
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 15
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 15
- 239000006286 aqueous extract Substances 0.000 description 15
- 239000008346 aqueous phase Substances 0.000 description 14
- 238000006243 chemical reaction Methods 0.000 description 14
- 239000006227 byproduct Substances 0.000 description 13
- YCCILVSKPBXVIP-UHFFFAOYSA-N 2-(4-hydroxyphenyl)ethanol Chemical compound OCCC1=CC=C(O)C=C1 YCCILVSKPBXVIP-UHFFFAOYSA-N 0.000 description 12
- 235000013305 food Nutrition 0.000 description 12
- 239000007921 spray Substances 0.000 description 11
- 239000007791 liquid phase Substances 0.000 description 10
- 239000000843 powder Substances 0.000 description 10
- RFWGABANNQMHMZ-HYYSZPHDSA-N Oleuropein Chemical compound O([C@@H]1OC=C([C@H](C1=CC)CC(=O)OCCC=1C=C(O)C(O)=CC=1)C(=O)OC)[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O RFWGABANNQMHMZ-HYYSZPHDSA-N 0.000 description 9
- 238000005119 centrifugation Methods 0.000 description 9
- 238000010828 elution Methods 0.000 description 9
- 239000012467 final product Substances 0.000 description 9
- 239000003960 organic solvent Substances 0.000 description 9
- 239000012071 phase Substances 0.000 description 9
- RFWGABANNQMHMZ-UHFFFAOYSA-N 8-acetoxy-7-acetyl-6,7,7a,8-tetrahydro-5H-benzo[g][1,3]dioxolo[4',5':4,5]benzo[1,2,3-de]quinoline Natural products CC=C1C(CC(=O)OCCC=2C=C(O)C(O)=CC=2)C(C(=O)OC)=COC1OC1OC(CO)C(O)C(O)C1O RFWGABANNQMHMZ-UHFFFAOYSA-N 0.000 description 8
- HKVGJQVJNQRJPO-UHFFFAOYSA-N Demethyloleuropein Natural products O1C=C(C(O)=O)C(CC(=O)OCCC=2C=C(O)C(O)=CC=2)C(=CC)C1OC1OC(CO)C(O)C(O)C1O HKVGJQVJNQRJPO-UHFFFAOYSA-N 0.000 description 8
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 8
- 235000011576 oleuropein Nutrition 0.000 description 8
- RFWGABANNQMHMZ-CARRXEGNSA-N oleuropein Natural products COC(=O)C1=CO[C@@H](O[C@H]2O[C@@H](CO)[C@H](O)[C@@H](O)[C@@H]2O)C(=CC)[C@H]1CC(=O)OCCc3ccc(O)c(O)c3 RFWGABANNQMHMZ-CARRXEGNSA-N 0.000 description 8
- 241000196324 Embryophyta Species 0.000 description 7
- 150000001875 compounds Chemical class 0.000 description 7
- 238000001914 filtration Methods 0.000 description 7
- 238000010438 heat treatment Methods 0.000 description 7
- 239000000463 material Substances 0.000 description 7
- 239000000203 mixture Substances 0.000 description 7
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 6
- DBLDQZASZZMNSL-QMMMGPOBSA-N L-tyrosinol Natural products OC[C@@H](N)CC1=CC=C(O)C=C1 DBLDQZASZZMNSL-QMMMGPOBSA-N 0.000 description 6
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 6
- 239000012141 concentrate Substances 0.000 description 6
- 239000002537 cosmetic Substances 0.000 description 6
- 238000001035 drying Methods 0.000 description 6
- 239000000126 substance Substances 0.000 description 6
- 235000004330 tyrosol Nutrition 0.000 description 6
- 239000007795 chemical reaction product Substances 0.000 description 5
- 238000001704 evaporation Methods 0.000 description 5
- 230000008020 evaporation Effects 0.000 description 5
- 238000005342 ion exchange Methods 0.000 description 5
- 239000007788 liquid Substances 0.000 description 5
- 238000001223 reverse osmosis Methods 0.000 description 5
- 150000003839 salts Chemical class 0.000 description 5
- 238000000926 separation method Methods 0.000 description 5
- 238000001694 spray drying Methods 0.000 description 5
- 238000003756 stirring Methods 0.000 description 5
- 244000061456 Solanum tuberosum Species 0.000 description 4
- 235000002595 Solanum tuberosum Nutrition 0.000 description 4
- 238000010521 absorption reaction Methods 0.000 description 4
- YBCVMFKXIKNREZ-UHFFFAOYSA-N acoh acetic acid Chemical compound CC(O)=O.CC(O)=O YBCVMFKXIKNREZ-UHFFFAOYSA-N 0.000 description 4
- 238000009295 crossflow filtration Methods 0.000 description 4
- 239000003814 drug Substances 0.000 description 4
- 239000002245 particle Substances 0.000 description 4
- 230000002572 peristaltic effect Effects 0.000 description 4
- 238000003825 pressing Methods 0.000 description 4
- 238000010992 reflux Methods 0.000 description 4
- MYRTYDVEIRVNKP-UHFFFAOYSA-N 1,2-Divinylbenzene Chemical compound C=CC1=CC=CC=C1C=C MYRTYDVEIRVNKP-UHFFFAOYSA-N 0.000 description 3
- 102100027324 2-hydroxyacyl-CoA lyase 1 Human genes 0.000 description 3
- NOEGNKMFWQHSLB-UHFFFAOYSA-N 5-hydroxymethylfurfural Chemical compound OCC1=CC=C(C=O)O1 NOEGNKMFWQHSLB-UHFFFAOYSA-N 0.000 description 3
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 3
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 3
- 101001009252 Homo sapiens 2-hydroxyacyl-CoA lyase 1 Proteins 0.000 description 3
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 3
- 230000002378 acidificating effect Effects 0.000 description 3
- -1 amide compound Chemical class 0.000 description 3
- 229920001429 chelating resin Polymers 0.000 description 3
- 229920001577 copolymer Polymers 0.000 description 3
- 239000008121 dextrose Substances 0.000 description 3
- 235000013399 edible fruits Nutrition 0.000 description 3
- 150000002148 esters Chemical group 0.000 description 3
- 239000002778 food additive Substances 0.000 description 3
- RJGBSYZFOCAGQY-UHFFFAOYSA-N hydroxymethylfurfural Natural products COC1=CC=C(C=O)O1 RJGBSYZFOCAGQY-UHFFFAOYSA-N 0.000 description 3
- 239000012528 membrane Substances 0.000 description 3
- 230000007935 neutral effect Effects 0.000 description 3
- 150000002989 phenols Chemical class 0.000 description 3
- 229920005597 polymer membrane Polymers 0.000 description 3
- 230000000717 retained effect Effects 0.000 description 3
- 239000012265 solid product Substances 0.000 description 3
- 239000002904 solvent Substances 0.000 description 3
- 239000002699 waste material Substances 0.000 description 3
- OWEGMIWEEQEYGQ-UHFFFAOYSA-N 100676-05-9 Natural products OC1C(O)C(O)C(CO)OC1OCC1C(O)C(O)C(O)C(OC2C(OC(O)C(O)C2O)CO)O1 OWEGMIWEEQEYGQ-UHFFFAOYSA-N 0.000 description 2
- VVJKKWFAADXIJK-UHFFFAOYSA-N Allylamine Chemical compound NCC=C VVJKKWFAADXIJK-UHFFFAOYSA-N 0.000 description 2
- 101100313763 Arabidopsis thaliana TIM22-2 gene Proteins 0.000 description 2
- 239000004375 Dextrin Substances 0.000 description 2
- 229920001353 Dextrin Polymers 0.000 description 2
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 2
- 239000005909 Kieselgur Substances 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- GUBGYTABKSRVRQ-PICCSMPSSA-N Maltose Natural products O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@@H](CO)OC(O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-PICCSMPSSA-N 0.000 description 2
- PPBRXRYQALVLMV-UHFFFAOYSA-N Styrene Chemical compound C=CC1=CC=CC=C1 PPBRXRYQALVLMV-UHFFFAOYSA-N 0.000 description 2
- 230000004913 activation Effects 0.000 description 2
- 238000007792 addition Methods 0.000 description 2
- 239000000654 additive Substances 0.000 description 2
- 230000000274 adsorptive effect Effects 0.000 description 2
- 230000009286 beneficial effect Effects 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- 230000015556 catabolic process Effects 0.000 description 2
- 238000006731 degradation reaction Methods 0.000 description 2
- 235000019425 dextrin Nutrition 0.000 description 2
- 239000008157 edible vegetable oil Substances 0.000 description 2
- 239000003480 eluent Substances 0.000 description 2
- 235000013373 food additive Nutrition 0.000 description 2
- 238000004108 freeze drying Methods 0.000 description 2
- 239000012535 impurity Substances 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 239000000787 lecithin Substances 0.000 description 2
- 235000010445 lecithin Nutrition 0.000 description 2
- 239000011259 mixed solution Substances 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- WTJKGGKOPKCXLL-RRHRGVEJSA-N phosphatidylcholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCC=CCCCCCCCC WTJKGGKOPKCXLL-RRHRGVEJSA-N 0.000 description 2
- 238000006116 polymerization reaction Methods 0.000 description 2
- 150000008442 polyphenolic compounds Chemical class 0.000 description 2
- 235000013824 polyphenols Nutrition 0.000 description 2
- 238000002360 preparation method Methods 0.000 description 2
- 239000011541 reaction mixture Substances 0.000 description 2
- 238000011084 recovery Methods 0.000 description 2
- 239000007790 solid phase Substances 0.000 description 2
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 2
- 235000013311 vegetables Nutrition 0.000 description 2
- QFRYQWYZSQDFOS-UHFFFAOYSA-N verbascoside Natural products CC1OC(COC2C(O)C(COC3OC(C(O)C(O)C3O)C(=O)O)OC(Oc4cc(O)cc5OC(=CC(=O)c45)c6ccc(O)c(O)c6)C2O)C(O)C(O)C1O QFRYQWYZSQDFOS-UHFFFAOYSA-N 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- GMQXOLRKJQWPNB-UHFFFAOYSA-N (8E)-ligstroside Natural products CC=C1C(CC(=O)OCCC=2C=CC(O)=CC=2)C(C(=O)OC)=COC1OC1OC(CO)C(O)C(O)C1O GMQXOLRKJQWPNB-UHFFFAOYSA-N 0.000 description 1
- NWUYHJFMYQTDRP-UHFFFAOYSA-N 1,2-bis(ethenyl)benzene;1-ethenyl-2-ethylbenzene;styrene Chemical class C=CC1=CC=CC=C1.CCC1=CC=CC=C1C=C.C=CC1=CC=CC=C1C=C NWUYHJFMYQTDRP-UHFFFAOYSA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- SMZOUWXMTYCWNB-UHFFFAOYSA-N 2-(2-methoxy-5-methylphenyl)ethanamine Chemical compound COC1=CC=C(C)C=C1CCN SMZOUWXMTYCWNB-UHFFFAOYSA-N 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-N 2-Propenoic acid Natural products OC(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 1
- CETXOEGRUBXUAL-UHFFFAOYSA-N 3-(hydroxymethyl)furan-2-carbaldehyde Chemical class OCC=1C=COC=1C=O CETXOEGRUBXUAL-UHFFFAOYSA-N 0.000 description 1
- 239000004925 Acrylic resin Substances 0.000 description 1
- 229920000178 Acrylic resin Polymers 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- RPNUMPOLZDHAAY-UHFFFAOYSA-N Diethylenetriamine Chemical compound NCCNCCN RPNUMPOLZDHAAY-UHFFFAOYSA-N 0.000 description 1
- 101001055216 Homo sapiens Interleukin-9 Proteins 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 102100026871 Interleukin-9 Human genes 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- CERQOIWHTDAKMF-UHFFFAOYSA-N Methacrylic acid Chemical compound CC(=C)C(O)=O CERQOIWHTDAKMF-UHFFFAOYSA-N 0.000 description 1
- UEEJHVSXFDXPFK-UHFFFAOYSA-N N-dimethylaminoethanol Chemical compound CN(C)CCO UEEJHVSXFDXPFK-UHFFFAOYSA-N 0.000 description 1
- 241000047703 Nonion Species 0.000 description 1
- 235000002725 Olea europaea Nutrition 0.000 description 1
- 239000004743 Polypropylene Substances 0.000 description 1
- 230000002292 Radical scavenging effect Effects 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 239000003463 adsorbent Substances 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 230000000845 anti-microbial effect Effects 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- 238000010923 batch production Methods 0.000 description 1
- 230000000975 bioactive effect Effects 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 238000011097 chromatography purification Methods 0.000 description 1
- 238000005352 clarification Methods 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 238000010924 continuous production Methods 0.000 description 1
- 239000000490 cosmetic additive Substances 0.000 description 1
- 238000012258 culturing Methods 0.000 description 1
- 229960002887 deanol Drugs 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- IUNMPGNGSSIWFP-UHFFFAOYSA-N dimethylaminopropylamine Chemical compound CN(C)CCCN IUNMPGNGSSIWFP-UHFFFAOYSA-N 0.000 description 1
- 239000012972 dimethylethanolamine Substances 0.000 description 1
- 150000002016 disaccharides Chemical class 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- UYMKPFRHYYNDTL-UHFFFAOYSA-N ethenamine Chemical compound NC=C UYMKPFRHYYNDTL-UHFFFAOYSA-N 0.000 description 1
- 238000004880 explosion Methods 0.000 description 1
- 239000010462 extra virgin olive oil Substances 0.000 description 1
- 235000021010 extra-virgin olive oil Nutrition 0.000 description 1
- 229930003935 flavonoid Natural products 0.000 description 1
- 235000017173 flavonoids Nutrition 0.000 description 1
- 150000002215 flavonoids Chemical class 0.000 description 1
- 229930182478 glucoside Natural products 0.000 description 1
- 150000008131 glucosides Chemical class 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 239000013067 intermediate product Substances 0.000 description 1
- GMQXOLRKJQWPNB-MVVLSVRYSA-N ligstroside Chemical compound O([C@@H]\1OC=C([C@H](C/1=C\C)CC(=O)OCCC=1C=CC(O)=CC=1)C(=O)OC)[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O GMQXOLRKJQWPNB-MVVLSVRYSA-N 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 150000002739 metals Chemical class 0.000 description 1
- 235000013336 milk Nutrition 0.000 description 1
- 239000008267 milk Substances 0.000 description 1
- 210000004080 milk Anatomy 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 239000000178 monomer Substances 0.000 description 1
- 238000001728 nano-filtration Methods 0.000 description 1
- 229930014626 natural product Natural products 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 239000012466 permeate Substances 0.000 description 1
- 150000003904 phospholipids Chemical class 0.000 description 1
- 238000000053 physical method Methods 0.000 description 1
- 239000002952 polymeric resin Substances 0.000 description 1
- 230000000379 polymerizing effect Effects 0.000 description 1
- 229920001155 polypropylene Polymers 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 238000002203 pretreatment Methods 0.000 description 1
- 239000003586 protic polar solvent Substances 0.000 description 1
- 239000002994 raw material Substances 0.000 description 1
- 230000001172 regenerating effect Effects 0.000 description 1
- 238000002390 rotary evaporation Methods 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 238000001179 sorption measurement Methods 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 239000012609 strong anion exchange resin Substances 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 229920003002 synthetic resin Polymers 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- KDSWDGKIENPKLB-QJDQKFITSA-N verbascoside Chemical compound O[C@@H]1[C@H](O)[C@@H](O)[C@H](C)O[C@H]1O[C@H]1[C@H](OC(=O)CCC=2C=C(O)C(O)=CC=2)[C@@H](CO)O[C@@H](OCCC=2C=C(O)C(O)=CC=2)[C@@H]1O KDSWDGKIENPKLB-QJDQKFITSA-N 0.000 description 1
- 239000012610 weak anion exchange resin Substances 0.000 description 1
Images

Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J19/00—Chemical, physical or physico-chemical processes in general; Their relevant apparatus
- B01J19/18—Stationary reactors having moving elements inside
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/18—Antioxidants, e.g. antiradicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P39/00—General protective or antinoxious agents
- A61P39/06—Free radical scavengers or antioxidants
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C37/00—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom of a six-membered aromatic ring
- C07C37/004—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom of a six-membered aromatic ring by obtaining phenols from plant material or from animal material
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2219/00—Chemical, physical or physico-chemical processes in general; Their relevant apparatus
- B01J2219/00002—Chemical plants
- B01J2219/00004—Scale aspects
- B01J2219/00006—Large-scale industrial plants
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2219/00—Chemical, physical or physico-chemical processes in general; Their relevant apparatus
- B01J2219/00049—Controlling or regulating processes
- B01J2219/00051—Controlling the temperature
- B01J2219/00074—Controlling the temperature by indirect heating or cooling employing heat exchange fluids
- B01J2219/00087—Controlling the temperature by indirect heating or cooling employing heat exchange fluids with heat exchange elements outside the reactor
- B01J2219/00094—Jackets
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Health & Medical Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Biochemistry (AREA)
- Botany (AREA)
- Toxicology (AREA)
- Communicable Diseases (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Dermatology (AREA)
- Oncology (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Cosmetics (AREA)
- Medicines Containing Plant Substances (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Solid-Sorbent Or Filter-Aiding Compositions (AREA)
Abstract
【課題】 有機極性溶剤を使用せずに、オリーブからヒドロキティロソルを含む抽出物を溶離する方法を提供する。
【解決手段】 本発明のオリーブからヒドロキティロソルを含有する抽出物を生成する方法は、(a)水中で前記出発材料を酸性加水分解するステップと、前記加水分解ステップは、温度は、70℃−140℃であり、ゲージ圧は、20psiまであり、pHは、1.0−6.0であり、(b)前記(a)ステップの加水分解水溶液から懸濁した固体を除去するステップと、前記ステップにより、分離した水溶液を確保し、(c)前記(b)ステップで得た水溶液(A)を、第1のクロマトグラフィ・カラムに入れるステップと、前記第1のクロマトグラフィ・カラム内の樹脂は、酸で活性化した陰イオン交換樹脂から選択され、(d)前記クロマトグラフィ・樹脂上に残った生成物を、水で溶離するステップとを有する。
【選択図】 図1
Description
ポマス(pomaces:搾りかす)、即ち、圧搾後の残留物、又は三相プロセスの圧搾後の残留物、あるいは二相プロセスの圧搾後の残留物を含む固体である。この固体状態においては、遠心分離のステップの間、オリーブの小片には水は添加されない。orujo(搾りかす)とalperujo(遠心分離後の残渣)とdefatted orujo(脱脂した搾りかす)は、相当量(45−70%)の水を含有する。抽出残留物は、絞りかすと、オリーブの乾燥固体とを含む。これらは、圧搾後のオイルの抽出後、15%未満の水しか含まず、残留オイルを含まない。
(a)水中で前記出発材料を酸性加水分解するステップと、
前記加水分解ステップは、温度は、70℃−140℃であり、ゲージ圧は、20psiまであり、pHは、1.0−6.0であり、
(b)前記(a)ステップの加水分解水溶液から、懸濁した固体を除去するステップと、
前記ステップにより、分離した水溶液を確保し、
(c)前記(b)ステップで得た水溶液(A)を、第1クロマトグラフィ・カラムに入れるステップと、
前記第1クロマトグラフィ・カラム内の樹脂は、酸で活性化した陰イオン交換樹脂から選択され、
(d)前記第1クロマトグラフィ・カラムの樹脂上に残った生成物を、水で溶離するステップと
を有する。
本発明の好まし実施例によれば、本発明は、更に純化ステップを実行する。この純化ステップは、前記の第1のクロマトグラフィ・カラム(陰イオン交換樹脂を含む)からの水溶離溶液を、第2のクロマトグラフィ・カラム(吸着性の非イオン性樹脂を含む)の内に入れるステップと、前記第2のクロマトグラフィ・カラム内に残った生成物を水で溶離するステップである。
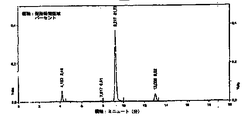
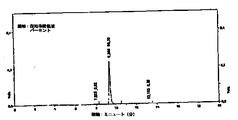
本発明のより好ましい態様によれば、本発明の方法により得られたヒドロキティロソルを含む固体生成物は、ヒドロキティロソルを20%(w/w)以上含む。その純度は、90%以上(HPLC 280nm)であり、フェノールの全量は、20%以上である。
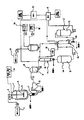
本発明の装置又はプラントは、反応容器と、第1のクロマトグラフィ・カラムと、第2クロマトグラフィ・カラムとを含む。前記反応容器は、圧力のかかった状態で酸性の加水分解を実行する。前記クロマトグラフィ・カラムは、樹脂と、酸で活性化した陰イオン交換樹脂(弱塩基性(weakly basic))と、水溶離性の陰イオン交換樹脂を含む。第2クロマトグラフィ・カラムは、吸着性の非イオン樹脂を含む。この吸着性の非イオン樹脂は、マクロ網状の架橋された芳香族ポリマ・樹脂(macroreticular cross-link aromatic polymer resin)である
実験例1
オリーブ廃棄物(orujillo)からヒドロキティロソルの抽出、水相の純化
250gの乾燥したオリーブ廃棄物のサンプルを、838mlの脱塩(脱イオン)水と、16.7gの硫酸(98%)で混合した。かくして得られた混合液を、高圧釜内で30分間121℃で保持した。その後、水相を、固体残留物からフィルタで濾過することにより分離した。この固相(個体物)は、フィルタ上に残ったものであるが、310mlの脱塩水で洗浄した。この洗浄ステップから得られた水を、前に回収された水相と共に収集した。水相は、約860mlあるが、その後、遠心分離で精製し、フィルタを通過した固体粒子を除去した。この固体除去の後、835mlの原水性抽出物(crude aqueous extract)を得た。この原水性抽出物は、HPLC純度が47.5%の1.41gのヒドロキティロソルを含む。
イオン交換によるヒドロキティロソルの純化
835mlの原水性抽出物(実験例1により得られた1.41gのヒドロキティロソルを含む)のサンプルを、陰イオン交換樹脂を含むクロマトグラフィ・カラムに入れた。陰イオン交換樹脂は、その前にアセテート(酢酸塩)・サイクル手段で活性化しておいた。これには、ダイヤイオン(Diaion)WA10を使用した。カラムの出口で回収され液相は、ヒドロキティロソルを含まないが、樹脂を脱塩水で連続的に溶離した。この溶離作業は、最初に入れたヒドロキティロソルの90%が回収されるまで、行われた。この溶離された相は、1.27gのヒドロキティロソルを含み、そのHPLC純度は80.85%であった。
イオン交換と吸収によるヒドロキティロソルの純化
835mlの原水性溶離物(実験例1により得られた1.41gのヒドロキティロソルを含む)のサンプルを、陰イオン交換樹脂を含むクロマトグラフィ・カラムに入れた。陰イオン交換樹脂は、その前にアセテート(酢酸塩)・サイクル手段で活性化しておいた。これには、IRA−67を使用した。カラムの出口で回収され液相は、ヒドロキティロソルを含まないが、樹脂を脱塩水で連続的に溶離した。この溶離作業は、最初に入れたヒドロキティロソルの90%以上が回収されるまで、行われた。
蒸発によるエンリッチにされたヒドロキティロソル原抽出物の濃縮
遠心分離するステップの前の実験例1で得られた水相(物)約860mlを蒸発法で濃縮して、最終的に193mlの容積を得た。その後、この水相を遠心分離で精製して、フィルタを通して、固体粒子を除去した。この固体除去の後、160mlの原水性抽出物は、1.41gのヒドロキティロソルを含み、そのHPLC純度は47.5%であった。
エンリッチにしたイオン交換により純化したヒドロキティロソル抽出物の、逆浸透圧方法による濃縮
80リットルの原水溶抽出液(実験例2によりパイロット・プラントで得られた150gのヒドロキティロソルを含む)のサンプルを、逆浸透圧のパイロット・プラントを用いて濃縮し、10リットルの濃縮生成物にした。この逆浸透圧装置は、2.5m2の高分子膜を具備する。同一材料から形成した0.3m2の膜をその後用いて、ヒドロキティロソルを10.8%含むヒドロキティロソル濃縮物を得た。そのHPLC純度は80.53%であった。
イオン交換と吸収により純化したヒドロキティロソル溶離物の、逆浸透圧方法による濃縮
546リットルの原水溶抽出液(実験例3によりパイロット・プラントで得られた135gのヒドロキティロソルを含む)のサンプルを、逆浸透圧のパイロット・プラントを用いて濃縮し、10リットルの濃縮生成物にした。このパイロット・プラントは、2.5m2の高分子膜を具備する。同一材料から形成した0.3m2の膜をその後用いて、ヒドロキティロソルを12.20%含むヒドロキティロソル濃縮物を得た。そのHPLC純度は95.27%であった。
純化ステップなしで、ヒドロキティロソル・リッチの原抽出物の噴霧乾燥
442mlの原水性抽出物(実験例1により得られた1.02gのヒドロキティロソルを含む)のサンプルを、100gのマルトデキストリンと混合した。この混合は、マルトデキストリンが完全に溶解するまで行った。100gのマルトデキストリンの代わりに、例えば、等価の10デキストロース・ポテト・マルトデキストリン(dextrose potato maltodextrin)を用いてもよい。蠕動ポンプ(peristaltic pump)を用いて噴霧乾燥機にこれを供給した。噴霧乾燥機は、入口空気温度が150℃で予め平衡状態にしてある。この供給速度は、出口空気温度が100℃未満となるよう調整した。95gの褐色の粉末が得られた。その湿度は6.85%(karl Fischer:カール・フィッシャー滴定;各種物質の広範囲な水分測定に世界中で使用されている方法)であり、ヒドロキティロソルのリッチネスが0.98%であった。
ヒドロキティロソル・リッチの一部純化した水性抽出物の噴霧乾燥
290mlの水性抽出物(実験例2により得られた0.38gのヒドロキティロソルを含む)のサンプルを、その後逆浸透圧法で濃縮し、50gのマルトデキストリンと混合した。この混合は、マルトデキストリンが完全に溶解するまで行った。マルトデキストリンの代わりに、例えば、等価の10デキストロース・ポテト・マルトデキストリンを用いてもよい。蠕動ポンプを用いて、噴霧乾燥機にこれを供給した。噴霧乾燥機は、入口空気温度が150℃で予め平衡状態にしてある。この供給速度は、出口空気温度が100℃未満となるよう調整した。48.25gの灰色の粉末が得らた。その湿度は6.72%(karl Fischer)であり、ヒドロキティロソルのリッチネスが0.71%であった。
ヒドロキティロソル・リッチの純化した水性抽出物の噴霧乾燥
188mlの純化した水性抽出物(実験例3により得られた0.29gのヒドロキティロソルを含む)のサンプルを、その後逆浸透圧法で濃縮し、28.5gのマルトデキストリンとゆっくりと撹拌した。マルトデキストリンの代わりに、例えば、等価の10デキストロース・ポテト・マルトデキストリンを用いてもよい。蠕動ポンプを用いて、噴霧乾燥機にこれを供給した。噴霧乾燥機は、入口空気温度が175℃で予め平衡状態にしてある。この供給速度は、出口空気温度が100℃未満となるよう調整した。27.1gの白色の粉末が得られた。その湿度は5.45%(karl Fischer)であり、ヒドロキティロソルのリッチネスは0.97%であった。
ヒドロキティロソルが豊富に含まれる粉末の用意
1750lの水性抽出物(実験例3により得られた432gのヒドロキティロソルを含む)サンプルを、パイロットプラントで、実験例6により濃縮した。39.04%のヒドロキティロソルを含む濃縮物を得た。そのHPLC純度は95.60%であった。この濃縮溶液を、噴霧乾燥機に供給した。噴霧乾燥機は、入口空気温度が150℃で予め平衡状態にしてある。この供給速度は、出口空気温度が100℃未満となるよう調整した。375.84gの褐色の粉末が得られた。その湿度は4.35%(karl Fischer)であり、ヒドロキティロソルのリッチネスは94.74%であった。
オリーブの実からのオリーブの抽出物
25Kgのオリーブの実のサンプルを、50Lの脱塩(脱イオン)水と混合した。かくして得られた混合液を、数分間ブレンドした。その後636gの硫酸(98%)を添加した。この混合液を、高圧釜内で30分間121℃で保持した。その後、水相を固体残留物からフィルタで濾過することにより分離した。この固相は、フィルタ上に残ったものであるが、12.5lの脱塩水で洗浄した。この洗浄ステップから得られた水を、前に回収された水相と共に収集した。水相は、約63Lあるが、その後、Kieselguhlar フィルタを通して濾過し、抽出されたオイルを除去した。このフィルタは、Celite 500珪藻土で予めコーティングしておいて。これにより、オイルを含有しない水相物を、56Lあるが、遠心分離で精製し、Kieselguhlar フィルタを通過した固体粒子を除去した。この固体除去の後、52Lの原水性抽出物(crude aqueous extract)を得た。これは、HPLC純度が50.5%の141gのヒドロキティロソルを含む。
噴霧乾燥によるオリーブの実の抽出粉末の準備
260mlの純化した液状のオリーブ抽出物(実験例11により得られた19.5gのヒドロキティロソルを含む)のサンプルを、58gのマルトデキストリンとゆっくりと撹拌した。このマルトデキストリンは、260mlの脱塩水で予め溶解しておいた、ポテト・マルトデキストリンを用いてもよい。蠕動ポンプを用いて、噴霧乾燥機にこれを供給した。噴霧乾燥機は、入口空気温度が150℃で予め平衡状態にしてある。この供給速度は、出口空気温度が100℃未満となるよう調整した。76gの白色の粉末が得られた。その湿度は5.4%(karl Fischer)であり、ヒドロキティロソルのリッチネスは、21.9%であった。
3相のポマス(脱脂搾りかす)からヒドロキティロソルの抽出、水相の純化
湿度が60.55%の475.5gの3相ポマスのサンプルを、800mlの脱塩(脱イオン)水と、26.36gの硫酸(98%)で混合した。かくして得られた混合液を、高圧釜内で30分間121℃で保持した。その後、水相を固体残留物から、600ミクロンのポリプロピレン製フィルタで濾過することにより分離した。濾過された水相約795mlを、蒸発させることにより濃縮して、最終的に343.8mlの容量を得た。その後、この水相を遠心分離で精製して、フィルタを通過した固体粒子を除去した。この固体除去の後、275mlの原水性抽出物(crude aqueous extract)を得た。これは、0.97gのヒドロキティロソルを含む。そのHPLC純度は47.5%であった。
3相のポマス(脱脂搾りかす)から得られたヒドロキティロソルのイオン交換と吸着による純化
275mlの原水性抽出物(実験例13により得られた0.97gのヒドロキティロソルを含む)のサンプルを、陰イオン交換樹脂を含むカラムに入れた。陰イオン交換樹脂は、その前にアセテート(酢酸塩)・サイクル手段で予め活性化しておいた。これには、Diaion WA10を使用した。カラムの出口で回収され液相は、ヒドロキティロソルを含まないが、樹脂を、脱塩水で連続的に溶離した。この溶離作業は、最初に入れたヒドロキティロソルの90%が回収されるまで、行われた。
2 供給手段
3 加熱手段
4 脱塩(脱イオン)水
5 酸タンク
6 攪拌手段
7 フィルタ
8 遠心分離機
9 貯蔵器
10 濃縮手段
11 カラム
12 陰イオン交換樹脂
13 水源
14 貯蔵器
15 カラム
16 非イオン性吸収樹脂
17 貯蔵器
18 酢酸ソース
19 NaOHソース
20 乾燥手段
21 硫酸ソース
図2−4の翻訳
横軸:ミニュート(分)、
縦軸:保持時間領域パーセント
Claims (16)
- ある出発材料から、ヒドロキティロソルを含有する抽出物を生成する方法において、
前記出発材料は、オリーブ・オイルを抽出した後のオリーブ残留物、又はオリーブから選択され、
前記方法は、前記出発材料を酸性加水分解し、得られた溶液を純化し、
(a)水中で前記出発材料を酸性加水分解するステップと、
前記加水分解ステップは、温度は、70℃−140℃であり、ゲージ圧は、20psi(137.8KPa)以下あり、pHは、1.0−6.0であり、
(b)前記(a)ステップの加水分解水溶液から、懸濁した固体を除去するステップと、
前記ステップにより、分離した水溶液を確保し、
(c)前記(b)ステップで得た水溶液(A)を、第1クロマトグラフィ・カラムに入れるステップと、
前記第1クロマトグラフィ・カラム内の樹脂は、酸で活性化した陰イオン交換樹脂から選択され、
(d)前記第1クロマトグラフィ・カラムの樹脂上に残った生成物を、水で溶離するステップと
を有する
ことを特徴とするヒドロキティロソルを含有する抽出物を生成する方法。 - (e)前記ステップ(d)で得られた溶液(B)を、第2クロマトグラフィ・カラムに入れるステップと、
前記第2クロマトグラフィ・カラムの樹脂は、吸収性の非イオン性樹脂から選択され、
(f)前記第2クロマトグラフィ・樹脂上に残った生成物を、水で溶離するステップと
をさらに有する
ことを特徴とする請求項1記載の方法。 - 前記加水分解は、連続する殺菌システムで行われ、
前記加水分解の温度は、110℃−130℃であり、
前記加水分解のゲージ圧は、10−20psi(68.94−137.8KPa)である
ことを特徴とする請求項1又は2記載の方法。 - 前記ステップ(a)は、15分−45分間行われる
ことを特徴とする請求項3記載の方法。 - 前記ステップ(a)の温度は、118℃−126℃の範囲である
ことを特徴とする請求項3又は4記載の方法。 - (g)前記ステップ(d)で得られた溶液(B)、又は前記ステップ(f)で得られた溶液(C)を、逆浸透圧で濃縮ステップ、
を更に有する
ことを特徴とする請求項1−5の何れかに記載の方法。 - (h)前記生成物が固体状態となるまで、脱水するステップ、
を更に有する
ことを特徴とする請求項1−6の何れかに記載の方法。 - オイルを、前記出発材料から除去する、あるいは前記ステップ(b)で得られた溶液から除去する
ことを特徴とする請求項1−7の何れかに記載の方法。 - 請求項1−6の何れかに記載された方法により得られたヒドロキティロソルを含む流体抽出物において、
前記流体抽出物は、ヒドロキティロソルを10%以上を含み、
その純度は、80%(HPLC280nmによる)以上である
ことを特徴とするヒドロキティロソルを含む流体抽出物。 - 請求項1−6の何れかに記載された方法により得られたヒドロキティロソルを含む流体抽出物において、
前記流体抽出物は、ヒドロキティロソルを30%以上を含み、
その純度は、90%(HPLC280nmによる)以上である
ことを特徴とするヒドロキティロソルを含む流体抽出物。 - 請求項1−7の何れかに記載された方法により得られたヒドロキティロソルを含む固体抽出物において、
前記固体抽出物は、ヒドロキティロソルを10%以上を含み、
その純度は、40%(HPLC280nmによる)以上である
ことを特徴とするヒドロキティロソルを含む固体抽出物。 - 請求項10に記載された方法により得られたヒドロキティロソルを含む固体抽出物において、
前記固体抽出物は、ヒドロキティロソルを40%以上を含み、
その純度は、90%(HPLC280nmによる)以上である
ことを特徴とするヒドロキティロソルを含む固体抽出物。 - ある出発材料からヒドロキティロソルを含有する抽出物を生成する装置において、
前記出発材料は、オリーブ・オイルを抽出した後のオリーブ残留物、又はオリーブから選択され、
前記装置は、前記出発材料を加水分解する手段を有し、
(a)水中で前記出発材料を酸性加水分解する手段と、
前記加水分解は、温度は70℃−140℃で、pHは1.0−6.0で、実行され、
(b)分離した水溶液を得る為、前記加水分解による生成物を処理する手段と、
(c)第1のクロマトグラフィ・カラムと、
前記第1のクロマトグラフィ・カラム内の樹脂は、酸で活性化した陰イオン交換樹脂から選択され、
を有する
ことを特徴とするヒドロキティロソルを含有する抽出物を生成する装置。 - (d)第2クロマトグラフィ・カラムと、
前記第2クロマトグラフィ・カラムの樹脂は、吸収性の非イオン性樹脂から選択され、
を有する
ことを特徴とする請求項13記載の装置。 - 前記加水分解する手段は、連続する殺菌システムを有し、
前記連続する殺菌システムは、前記出発材料の連続する流れを圧力をかけながら処理する
ことを特徴とする請求項13又は14記載の装置。 - 前記の酸で活性化した陰イオン交換樹脂は、弱塩基性の水溶離陰イオン交換樹脂であり、
前記吸収性の非イオン性樹脂は、マクロ網状の架橋された芳香族ポリマである
ことを特徴とする請求項14又は15記載の装置。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP07001791A EP1953133A1 (en) | 2007-01-26 | 2007-01-26 | Process and apparatus for the production of hydroxytyrosol from olive oil extraction residues |
| EP07014390 | 2007-07-23 | ||
| PCT/IB2008/000173 WO2008090460A1 (en) | 2007-01-26 | 2008-01-25 | Process and apparatus for the production of hydroxytyrosol containing extract from olives and solids containing residues of olive oil extraction |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2010516750A true JP2010516750A (ja) | 2010-05-20 |
| JP2010516750A5 JP2010516750A5 (ja) | 2011-01-27 |
Family
ID=39339229
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2009546832A Pending JP2010516750A (ja) | 2007-01-26 | 2008-01-25 | オリーブからの抽出物又はオリーブ・オイルの抽出後の残留物を含む固体成分からヒドロキティロソルを生成するプロセスと装置 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US8236993B2 (ja) |
| EP (2) | EP2489651A1 (ja) |
| JP (1) | JP2010516750A (ja) |
| ES (1) | ES2451011T3 (ja) |
| WO (1) | WO2008090460A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2017537119A (ja) * | 2014-12-12 | 2017-12-14 | ヴァーディア, インコーポレイテッド | セルロースをフラン生成物に変換するための方法 |
| JP2020516315A (ja) * | 2017-04-14 | 2020-06-11 | レヴィウス ヴィタ フーズ エス.アール.エル.Levius Vita Foods S.R.L. | 高ポリフェノールオリーブオイルを調製するための方法および装置 |
Families Citing this family (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ATE508642T1 (de) * | 2007-07-23 | 2011-05-15 | Probelte Pharma S A | Hydroxytyrosol-verstärkte öle und deren verwendung |
| EP2108692A1 (fr) * | 2008-04-11 | 2009-10-14 | Brasseries Kronenbourg | Procédé d'obtention d'extraits concentrés en polyphénols issus du procédé de brassage |
| ES2341526B1 (es) * | 2008-12-19 | 2011-06-08 | Consejo Superior De Investigaciones Cientificas (Csic) | Procedimiento de purificacion de 3,4-dihidroxifenilglicol (dhfg) a partir de productos vegetales. |
| MY161839A (en) | 2009-05-18 | 2017-05-15 | Malaysian Palm Oil Board | A composition for use in the prevention and treatment of cardiovascular diseases |
| EP2338500A1 (en) * | 2009-12-23 | 2011-06-29 | Phenofarm S.r.l. | Process for producing concentrated and refined actives from tissues and byproducts of Olea europaea with membrane technologies |
| ITMI20110941A1 (it) | 2011-05-25 | 2012-11-26 | Phenofarm S R L | Processo di produzione di un fito-estratto da acque di vegetazione esanse olearie |
| ES2395317B1 (es) * | 2011-07-08 | 2014-04-16 | Consejo Superior De Investigaciones Cientificas (Csic) | Procedimiento para la obtención de extracto de hidroxitirosol, extracto mezcla de hidroxitirosol y 3,4-dihidroxifenilglicol, y extracto de acetato de hidroxitirosilo, a partir de subproductos del olivo y su purificación. |
| DE102011053527A1 (de) * | 2011-09-12 | 2013-03-14 | Gea Mechanical Equipment Gmbh | Verfahren und Anlage zur Aufarbeitung von Alpeorujo |
| US10532022B2 (en) * | 2014-07-21 | 2020-01-14 | The United States Of America, As Represented By The Secretary Of Agriculture | Whole stablized olive mill process water, production thereof and uses thereof |
| ITUA20162673A1 (it) * | 2016-04-18 | 2017-10-18 | Laboratori Archa Srl | Procedimento di trattamento di acque di vegetazione olearia |
| ES2953687T3 (es) | 2016-07-21 | 2023-11-15 | Board Of Supervisors For The Univ Of Louisiana System | Aislamiento de oleocantal y tratamiento del cáncer |
| CL2021001885A1 (es) * | 2021-07-15 | 2021-12-17 | American Bioprocess Ltda | Proceso para la obtención de extractos concentrados en hidroxitirosol (ht) |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20040102657A1 (en) * | 2001-02-15 | 2004-05-27 | Juan Fernandez-Bolanos Guzman | Method for obtaining purified hydroxytyrosol from products and by-products derived from the olive tree |
| JP2005532398A (ja) * | 2002-07-05 | 2005-10-27 | クリアグリ, インコーポレイテッド | オリーブ植物水由来のヒドロキシチロソール富化組成物およびその使用方法 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6358542B2 (en) | 1999-12-20 | 2002-03-19 | Usana, Inc. | Antioxidant compositions extracted from olives and olive by-products |
| ES2172429B1 (es) | 2000-10-06 | 2003-12-16 | Consejo Superior Investigacion | Procedimiento de obtencion de hidroxitirosol purificado a partir de productos y subproductos derivados del olivo. |
| ES2199069B1 (es) | 2002-07-17 | 2005-02-01 | Centro De Investigaciones Energeticas, Medioambientales Y Tecnologicas, (C.I.E.M.A.T) | Procedimiento para la extraccion de compuestos fenolicos a partir de un material vegetal residual mediante un tratamiento hidrotermico. |
| EP1582512A1 (en) | 2004-03-31 | 2005-10-05 | Cognis IP Management GmbH | Process for obtaining hydroxytyrosol from olive leaves extracts |
| ITMI20041627A1 (it) | 2004-08-06 | 2004-11-06 | Lachifarma S R L Lab Chimi Co | Processo per il recupero di tirosolo idrossitirosolo e altri componenti fenolici da acque di vegetazione e metodo di ossidazione catalitica di tirosolo a idrossitirosolo |
-
2008
- 2008-01-25 US US12/524,603 patent/US8236993B2/en active Active
- 2008-01-25 EP EP12003813A patent/EP2489651A1/en not_active Withdrawn
- 2008-01-25 WO PCT/IB2008/000173 patent/WO2008090460A1/en active Application Filing
- 2008-01-25 JP JP2009546832A patent/JP2010516750A/ja active Pending
- 2008-01-25 EP EP08702319.8A patent/EP2049458B1/en active Active
- 2008-01-25 ES ES08702319.8T patent/ES2451011T3/es active Active
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20040102657A1 (en) * | 2001-02-15 | 2004-05-27 | Juan Fernandez-Bolanos Guzman | Method for obtaining purified hydroxytyrosol from products and by-products derived from the olive tree |
| JP2005532398A (ja) * | 2002-07-05 | 2005-10-27 | クリアグリ, インコーポレイテッド | オリーブ植物水由来のヒドロキシチロソール富化組成物およびその使用方法 |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2017537119A (ja) * | 2014-12-12 | 2017-12-14 | ヴァーディア, インコーポレイテッド | セルロースをフラン生成物に変換するための方法 |
| US10532990B2 (en) | 2014-12-12 | 2020-01-14 | Virdia, Inc. | Methods for converting cellulose to furanic products |
| JP2020516315A (ja) * | 2017-04-14 | 2020-06-11 | レヴィウス ヴィタ フーズ エス.アール.エル.Levius Vita Foods S.R.L. | 高ポリフェノールオリーブオイルを調製するための方法および装置 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2489651A1 (en) | 2012-08-22 |
| WO2008090460A1 (en) | 2008-07-31 |
| EP2049458B1 (en) | 2013-12-25 |
| ES2451011T3 (es) | 2014-03-26 |
| EP2049458A1 (en) | 2009-04-22 |
| US8236993B2 (en) | 2012-08-07 |
| US20100160690A1 (en) | 2010-06-24 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2010516750A (ja) | オリーブからの抽出物又はオリーブ・オイルの抽出後の残留物を含む固体成分からヒドロキティロソルを生成するプロセスと装置 | |
| Takaç et al. | Recovery of phenolic antioxidants from olive mill wastewater | |
| Suwal et al. | Technologies for the Extraction, Separation and Purification of polyphenols–A Review | |
| EP1967078A1 (en) | Process and apparatus for preparing pomegranate extracts | |
| EP0817824B1 (en) | Process for extracting and purifying lignans and cinnamic acid derivatives from flaxseed | |
| JP5734208B2 (ja) | オリーブ果実水からのフェノール性化合物の抽出方法ならびにオリーブおよびブドウのポリフェノールを有する濃縮された抽出物の製造方法 | |
| AU2008310307B2 (en) | Method to recover bioactive compounds | |
| US20040102657A1 (en) | Method for obtaining purified hydroxytyrosol from products and by-products derived from the olive tree | |
| EP1953133A1 (en) | Process and apparatus for the production of hydroxytyrosol from olive oil extraction residues | |
| Pinto et al. | Separation and recovery of polyphenols and carbohydrates from Eucalyptus bark extract by ultrafiltration/diafiltration and adsorption processes | |
| Conde et al. | Recovery and concentration of antioxidants from industrial effluents and from processing streams of underutilized vegetal biomass. | |
| CA2614360C (en) | Salix extract, its use and formulations containing it | |
| EP2743248A1 (en) | Method for obtaining hydroxytyrosol extract, mixture of hydroxytyrosol and 3,4-dihydroxyphenylglycol extract, and hydroxytyrosyl acetate extract, from by-products of the olive tree, and the purification thereof | |
| US20040019226A1 (en) | Process for producing high purity isoflavones | |
| CN111454242A (zh) | 一种从花生衣中分离多种活性成分的方法 | |
| RU2180566C1 (ru) | Способ выделения дигидрокверцетина | |
| JP4738788B2 (ja) | アルブチンの分離精製方法 | |
| CA3139363A1 (en) | Anthocyanin containing extract powder and method of production | |
| JP2007290971A (ja) | グリセロールガラクトシドの抽出方法 | |
| CN113816933B (zh) | 一种羟基红花黄色素a的制备方法 | |
| CN109824738A (zh) | 一种肉苁蓉总寡糖的脱盐脱色方法 | |
| RU2727130C1 (ru) | Способ хроматографического выделения и концентрирования антоцианов | |
| RU2317093C1 (ru) | Способ выделения биологически активных изомеров дигидрокверцетина | |
| WO2025038360A1 (en) | Processes for separating products from plant biomass | |
| Cassano et al. | from Agro-Food By-products via |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20101203 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20110120 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20110517 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20130215 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20130220 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20130520 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20130527 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20130618 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20130625 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20130701 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20130731 |