JP2005290301A - ポリベンゾオキサゾール樹脂前駆体の製造方法およびポリベンゾオキサゾール樹脂の製造方法 - Google Patents
ポリベンゾオキサゾール樹脂前駆体の製造方法およびポリベンゾオキサゾール樹脂の製造方法 Download PDFInfo
- Publication number
- JP2005290301A JP2005290301A JP2004110276A JP2004110276A JP2005290301A JP 2005290301 A JP2005290301 A JP 2005290301A JP 2004110276 A JP2004110276 A JP 2004110276A JP 2004110276 A JP2004110276 A JP 2004110276A JP 2005290301 A JP2005290301 A JP 2005290301A
- Authority
- JP
- Japan
- Prior art keywords
- polybenzoxazole resin
- reaction
- group
- resin precursor
- temperature
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 239000011347 resin Substances 0.000 title claims abstract description 81
- 229920005989 resin Polymers 0.000 title claims abstract description 81
- 229920002577 polybenzoxazole Polymers 0.000 title claims abstract description 78
- 239000002243 precursor Substances 0.000 title claims abstract description 77
- 238000004519 manufacturing process Methods 0.000 title claims abstract description 29
- 238000006243 chemical reaction Methods 0.000 claims abstract description 78
- 239000007788 liquid Substances 0.000 claims abstract description 30
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 claims abstract description 29
- -1 diamine compound Chemical class 0.000 claims abstract description 27
- 238000002156 mixing Methods 0.000 claims abstract description 25
- 239000002904 solvent Substances 0.000 claims abstract description 16
- 238000007363 ring formation reaction Methods 0.000 claims abstract description 13
- 239000012295 chemical reaction liquid Substances 0.000 claims abstract description 12
- 230000018044 dehydration Effects 0.000 claims abstract description 10
- 238000006297 dehydration reaction Methods 0.000 claims abstract description 10
- 125000000962 organic group Chemical group 0.000 claims description 22
- 125000001153 fluoro group Chemical group F* 0.000 claims description 13
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 11
- 238000000034 method Methods 0.000 claims description 4
- 125000005843 halogen group Chemical group 0.000 claims description 3
- 238000002844 melting Methods 0.000 claims description 3
- 230000008018 melting Effects 0.000 claims description 3
- ZCQWOFVYLHDMMC-UHFFFAOYSA-N Oxazole Chemical compound C1=COC=N1 ZCQWOFVYLHDMMC-UHFFFAOYSA-N 0.000 claims description 2
- 125000001183 hydrocarbyl group Chemical group 0.000 claims 2
- 239000000126 substance Substances 0.000 abstract description 8
- 230000003287 optical effect Effects 0.000 abstract description 7
- 239000000243 solution Substances 0.000 description 35
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 12
- 125000003118 aryl group Chemical group 0.000 description 10
- AMXOYNBUYSYVKV-UHFFFAOYSA-M lithium bromide Chemical compound [Li+].[Br-] AMXOYNBUYSYVKV-UHFFFAOYSA-M 0.000 description 10
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 8
- 239000004793 Polystyrene Substances 0.000 description 7
- 125000004122 cyclic group Chemical group 0.000 description 7
- 229920002223 polystyrene Polymers 0.000 description 7
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 6
- 125000003545 alkoxy group Chemical group 0.000 description 6
- 125000000217 alkyl group Chemical group 0.000 description 6
- 229910052731 fluorine Inorganic materials 0.000 description 6
- 125000003709 fluoroalkyl group Chemical group 0.000 description 6
- 125000000951 phenoxy group Chemical group [H]C1=C([H])C([H])=C(O*)C([H])=C1[H] 0.000 description 6
- 230000015572 biosynthetic process Effects 0.000 description 5
- 238000005227 gel permeation chromatography Methods 0.000 description 5
- 238000003756 stirring Methods 0.000 description 5
- 238000003786 synthesis reaction Methods 0.000 description 5
- 150000002430 hydrocarbons Chemical group 0.000 description 4
- 238000006116 polymerization reaction Methods 0.000 description 4
- 238000001577 simple distillation Methods 0.000 description 4
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 4
- HDGLPTVARHLGMV-UHFFFAOYSA-N 2-amino-4-(1,1,1,3,3,3-hexafluoropropan-2-yl)phenol Chemical compound NC1=CC(C(C(F)(F)F)C(F)(F)F)=CC=C1O HDGLPTVARHLGMV-UHFFFAOYSA-N 0.000 description 3
- UGWGABORRHXAFP-UHFFFAOYSA-N 4-(4-carbonochloridoyl-2,3,5,6-tetrafluorophenyl)-2,3,5,6-tetrafluorobenzoyl chloride Chemical compound FC1=C(C(Cl)=O)C(F)=C(F)C(C=2C(=C(F)C(C(Cl)=O)=C(F)C=2F)F)=C1F UGWGABORRHXAFP-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- 239000007864 aqueous solution Substances 0.000 description 3
- 230000000052 comparative effect Effects 0.000 description 3
- 238000000354 decomposition reaction Methods 0.000 description 3
- 125000001142 dicarboxylic acid group Chemical group 0.000 description 3
- 239000012153 distilled water Substances 0.000 description 3
- 238000001035 drying Methods 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 239000012299 nitrogen atmosphere Substances 0.000 description 3
- 239000003960 organic solvent Substances 0.000 description 3
- 239000004810 polytetrafluoroethylene Substances 0.000 description 3
- 229920001343 polytetrafluoroethylene Polymers 0.000 description 3
- 150000003222 pyridines Chemical class 0.000 description 3
- 238000000926 separation method Methods 0.000 description 3
- 239000007787 solid Substances 0.000 description 3
- 238000005406 washing Methods 0.000 description 3
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Chemical compound O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 3
- NAWXUBYGYWOOIX-SFHVURJKSA-N (2s)-2-[[4-[2-(2,4-diaminoquinazolin-6-yl)ethyl]benzoyl]amino]-4-methylidenepentanedioic acid Chemical compound C1=CC2=NC(N)=NC(N)=C2C=C1CCC1=CC=C(C(=O)N[C@@H](CC(=C)C(O)=O)C(O)=O)C=C1 NAWXUBYGYWOOIX-SFHVURJKSA-N 0.000 description 2
- HPYNZHMRTTWQTB-UHFFFAOYSA-N 2,3-dimethylpyridine Chemical compound CC1=CC=CN=C1C HPYNZHMRTTWQTB-UHFFFAOYSA-N 0.000 description 2
- KKENYPMBRJYGTH-UHFFFAOYSA-N 2,4,5,6-tetrafluorobenzene-1,3-dicarbonyl chloride Chemical compound FC1=C(F)C(C(Cl)=O)=C(F)C(C(Cl)=O)=C1F KKENYPMBRJYGTH-UHFFFAOYSA-N 0.000 description 2
- YEJRWHAVMIAJKC-UHFFFAOYSA-N 4-Butyrolactone Chemical compound O=C1CCCO1 YEJRWHAVMIAJKC-UHFFFAOYSA-N 0.000 description 2
- DLFVBJFMPXGRIB-UHFFFAOYSA-N Acetamide Chemical compound CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 description 2
- 0 CC*(C)*CN(C)C Chemical compound CC*(C)*CN(C)C 0.000 description 2
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 2
- 239000002253 acid Substances 0.000 description 2
- 125000002947 alkylene group Chemical group 0.000 description 2
- 125000000732 arylene group Chemical group 0.000 description 2
- 238000011088 calibration curve Methods 0.000 description 2
- 150000001875 compounds Chemical class 0.000 description 2
- LZCLXQDLBQLTDK-UHFFFAOYSA-N ethyl 2-hydroxypropanoate Chemical compound CCOC(=O)C(C)O LZCLXQDLBQLTDK-UHFFFAOYSA-N 0.000 description 2
- 238000000465 moulding Methods 0.000 description 2
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 2
- 239000002966 varnish Substances 0.000 description 2
- 229940058015 1,3-butylene glycol Drugs 0.000 description 1
- RRQYJINTUHWNHW-UHFFFAOYSA-N 1-ethoxy-2-(2-ethoxyethoxy)ethane Chemical compound CCOCCOCCOCC RRQYJINTUHWNHW-UHFFFAOYSA-N 0.000 description 1
- ARXJGSRGQADJSQ-UHFFFAOYSA-N 1-methoxypropan-2-ol Chemical compound COCC(C)O ARXJGSRGQADJSQ-UHFFFAOYSA-N 0.000 description 1
- PGRIMKUYGUHAKH-UHFFFAOYSA-N 2,4,5,6-tetrafluorobenzene-1,3-dicarboxylic acid Chemical compound OC(=O)C1=C(F)C(F)=C(F)C(C(O)=O)=C1F PGRIMKUYGUHAKH-UHFFFAOYSA-N 0.000 description 1
- RLXBOUUYEFOFSW-UHFFFAOYSA-N 2,5-diaminobenzene-1,4-diol Chemical compound NC1=CC(O)=C(N)C=C1O RLXBOUUYEFOFSW-UHFFFAOYSA-N 0.000 description 1
- RFWAJLMIKBTJAY-UHFFFAOYSA-N 2-amino-4-(1,1,1,3,3,3-hexafluoropropan-2-yl)-6-(trifluoromethyl)phenol Chemical compound NC=1C=C(C=C(C1O)C(F)(F)F)C(C(F)(F)F)C(F)(F)F RFWAJLMIKBTJAY-UHFFFAOYSA-N 0.000 description 1
- KZLDGFZCFRXUIB-UHFFFAOYSA-N 2-amino-4-(3-amino-4-hydroxyphenyl)phenol Chemical group C1=C(O)C(N)=CC(C=2C=C(N)C(O)=CC=2)=C1 KZLDGFZCFRXUIB-UHFFFAOYSA-N 0.000 description 1
- ZGDMDBHLKNQPSD-UHFFFAOYSA-N 2-amino-5-(4-amino-3-hydroxyphenyl)phenol Chemical group C1=C(O)C(N)=CC=C1C1=CC=C(N)C(O)=C1 ZGDMDBHLKNQPSD-UHFFFAOYSA-N 0.000 description 1
- CDAWCLOXVUBKRW-UHFFFAOYSA-N 2-aminophenol Chemical compound NC1=CC=CC=C1O CDAWCLOXVUBKRW-UHFFFAOYSA-N 0.000 description 1
- CRWNQZTZTZWPOF-UHFFFAOYSA-N 2-methyl-4-phenylpyridine Chemical compound C1=NC(C)=CC(C=2C=CC=CC=2)=C1 CRWNQZTZTZWPOF-UHFFFAOYSA-N 0.000 description 1
- QCAHUFWKIQLBNB-UHFFFAOYSA-N 3-(3-methoxypropoxy)propan-1-ol Chemical compound COCCCOCCCO QCAHUFWKIQLBNB-UHFFFAOYSA-N 0.000 description 1
- JSGVZVOGOQILFM-UHFFFAOYSA-N 3-methoxy-1-butanol Chemical compound COC(C)CCO JSGVZVOGOQILFM-UHFFFAOYSA-N 0.000 description 1
- CEBWCJILGIWCOV-UHFFFAOYSA-N 4-(4-carboxy-2,3,5,6-tetrafluorophenyl)-2,3,5,6-tetrafluorobenzoic acid Chemical group FC1=C(F)C(C(=O)O)=C(F)C(F)=C1C1=C(F)C(F)=C(C(O)=O)C(F)=C1F CEBWCJILGIWCOV-UHFFFAOYSA-N 0.000 description 1
- LPEKGGXMPWTOCB-UHFFFAOYSA-N 8beta-(2,3-epoxy-2-methylbutyryloxy)-14-acetoxytithifolin Natural products COC(=O)C(C)O LPEKGGXMPWTOCB-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- MRABAEUHTLLEML-UHFFFAOYSA-N Butyl lactate Chemical compound CCCCOC(=O)C(C)O MRABAEUHTLLEML-UHFFFAOYSA-N 0.000 description 1
- XXRCUYVCPSWGCC-UHFFFAOYSA-N Ethyl pyruvate Chemical compound CCOC(=O)C(C)=O XXRCUYVCPSWGCC-UHFFFAOYSA-N 0.000 description 1
- JLTDJTHDQAWBAV-UHFFFAOYSA-N N,N-dimethylaniline Chemical compound CN(C)C1=CC=CC=C1 JLTDJTHDQAWBAV-UHFFFAOYSA-N 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 239000001191 butyl (2R)-2-hydroxypropanoate Substances 0.000 description 1
- 229940019778 diethylene glycol diethyl ether Drugs 0.000 description 1
- SBZXBUIDTXKZTM-UHFFFAOYSA-N diglyme Chemical compound COCCOCCOC SBZXBUIDTXKZTM-UHFFFAOYSA-N 0.000 description 1
- ODQWQRRAPPTVAG-GZTJUZNOSA-N doxepin Chemical compound C1OC2=CC=CC=C2C(=C/CCN(C)C)/C2=CC=CC=C21 ODQWQRRAPPTVAG-GZTJUZNOSA-N 0.000 description 1
- 239000004210 ether based solvent Substances 0.000 description 1
- 229940116333 ethyl lactate Drugs 0.000 description 1
- 229940117360 ethyl pyruvate Drugs 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 239000012433 hydrogen halide Substances 0.000 description 1
- 229910000039 hydrogen halide Inorganic materials 0.000 description 1
- 238000011835 investigation Methods 0.000 description 1
- BDJSOPWXYLFTNW-UHFFFAOYSA-N methyl 3-methoxypropanoate Chemical compound COCCC(=O)OC BDJSOPWXYLFTNW-UHFFFAOYSA-N 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 229940057867 methyl lactate Drugs 0.000 description 1
- CWKLZLBVOJRSOM-UHFFFAOYSA-N methyl pyruvate Chemical compound COC(=O)C(C)=O CWKLZLBVOJRSOM-UHFFFAOYSA-N 0.000 description 1
- 239000012778 molding material Substances 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 125000006551 perfluoro alkylene group Chemical group 0.000 description 1
- 238000006068 polycondensation reaction Methods 0.000 description 1
- LLHKCFNBLRBOGN-UHFFFAOYSA-N propylene glycol methyl ether acetate Chemical compound COCC(C)OC(C)=O LLHKCFNBLRBOGN-UHFFFAOYSA-N 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 239000002994 raw material Substances 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 238000010792 warming Methods 0.000 description 1
Images
Landscapes
- Macromolecular Compounds Obtained By Forming Nitrogen-Containing Linkages In General (AREA)
Abstract
【課題】 これまでよりも分子量の大きいポリベンゾオキサゾール樹脂前駆体と、耐熱性、機械的強度、耐薬品性、光学的特性、電気的特性等にすぐれたポリベンゾオキサゾール樹脂とを製造するための製造方法を提供する。
【解決手段】 ポリベンゾオキサゾール樹脂前駆体の製造方法は、ジアミン化合物と、ジカルボン酸ジハライドと、溶媒とを、液温を0℃以下に維持しつつ混合して反応液とし、次いで、この反応液の液温を0℃以下に維持して一定時間、反応を続ける。ポリベンゾオキサゾール樹脂は、上記の前駆体を、脱水環化反応または脱シラノール環化反応させる。
【選択図】 なし
【解決手段】 ポリベンゾオキサゾール樹脂前駆体の製造方法は、ジアミン化合物と、ジカルボン酸ジハライドと、溶媒とを、液温を0℃以下に維持しつつ混合して反応液とし、次いで、この反応液の液温を0℃以下に維持して一定時間、反応を続ける。ポリベンゾオキサゾール樹脂は、上記の前駆体を、脱水環化反応または脱シラノール環化反応させる。
【選択図】 なし
Description
本発明は、ポリベンゾオキサゾール樹脂前駆体の製造方法と、それによって製造された前駆体を用いる、ポリベンゾオキサゾール樹脂の製造方法に関するものである。
耐熱性、機械的強度、耐薬品性、光学的特性、電気的特性等にすぐれた、剛直な分子構造を有する樹脂として、一般式(5):
〔式中R1は4価の有機基、R2は2価の有機基を示す。〕
で表される繰り返し単位を有するポリベンゾオキサゾール樹脂が知られている。
で表される繰り返し単位を有するポリベンゾオキサゾール樹脂が知られている。
上記ポリベンゾオキサゾール樹脂は、一般に、ビス(アミノフェノール)化合物と呼ばれる、一般式(2):
〔式中R1は4価の有機基を示す。〕
で表されるジアミン化合物と、一般式(3):
〔式中R2は2価の有機基、Xはハロゲン原子を示す。〕
で表されるジカルボン酸ジハライドと反応させて、一般式(1):
〔式中R1は4価の有機基、R2は2価の有機基を示す。〕
で表される繰り返し単位を有するポリベンゾオキサゾール樹脂前駆体を製造したのち、この前駆体を、脱水環化反応させることによって合成される。
(特許文献1〜3参照)。
特開2000−128985号公報(第0007欄〜第0008欄、第0012欄以降の実施例)
特開2001−114893号公報(第0016欄、第0022欄〜第0023欄、第0028欄以降の実施例)
特開2002−173532号公報(第0026欄、第0037欄、第0045欄以降の実施例)
で表されるジアミン化合物と、一般式(3):
で表されるジカルボン酸ジハライドと反応させて、一般式(1):
で表される繰り返し単位を有するポリベンゾオキサゾール樹脂前駆体を製造したのち、この前駆体を、脱水環化反応させることによって合成される。
(特許文献1〜3参照)。
ところが、特許文献1〜3に記載の製造方法では、前記式(1)で表される繰り返し単位の繰り返し数で表される平均重合度が、およそ15を超えるか、または、ポリスチレン換算の重量平均分子量Mwが、およそ10000を超えるような分子量の大きいポリベンゾオキサゾール樹脂前駆体を製造するのが難しいという問題がある。また、そのため、かかるポリベンゾオキサゾール樹脂前駆体を脱水環化反応させても、耐熱性、機械的強度、耐薬品性、電気的特性、光学的特性等にすぐれたポリベンゾオキサゾール樹脂を製造できないという問題がある。
本発明の目的は、これまでよりも分子量の大きいポリベンゾオキサゾール樹脂前駆体と、耐熱性、機械的強度、耐薬品性、光学的特性、電気的特性等にすぐれたポリベンゾオキサゾール樹脂とを製造するための製造方法を提供することにある。
請求項1記載の発明は、一般式(1):
〔式中R1は4価の有機基、R2は2価の有機基を示す。〕
で表される繰り返し単位を有するポリベンゾオキサゾール樹脂前駆体を製造する方法であって、一般式(2):
〔式中R1は4価の有機基を示す。〕
で表されるジアミン化合物と、一般式(3):
〔式中R2は2価の有機基、Xはハロゲン原子を示す。〕
で表されるジカルボン酸ジハライドと、溶媒とを、液温を0℃以下に維持しつつ混合して反応液とする混合工程と、この混合工程に続いて、反応液の液温を0℃以下に維持して一定時間、反応を続ける低温反応工程とを含むことを特徴とするポリベンゾオキサゾール樹脂前駆体の製造方法である。
で表される繰り返し単位を有するポリベンゾオキサゾール樹脂前駆体を製造する方法であって、一般式(2):
で表されるジアミン化合物と、一般式(3):
で表されるジカルボン酸ジハライドと、溶媒とを、液温を0℃以下に維持しつつ混合して反応液とする混合工程と、この混合工程に続いて、反応液の液温を0℃以下に維持して一定時間、反応を続ける低温反応工程とを含むことを特徴とするポリベンゾオキサゾール樹脂前駆体の製造方法である。
請求項2記載の発明は、混合工程および低温反応工程における液温を、液の融点以上で、かつ0℃以下の範囲内とする請求項1記載のポリベンゾオキサゾール樹脂前駆体の製造方法である。
請求項3記載の発明は、反応液の液温を、0℃を超える温度に昇温した際に、生成したポリベンゾオキサゾール樹脂前駆体中に占める、分子量5000以下のオリゴマーの割合が、35重量%以下になるまで、低温反応工程を続ける請求項1記載のポリベンゾオキサゾール樹脂前駆体の製造方法である。
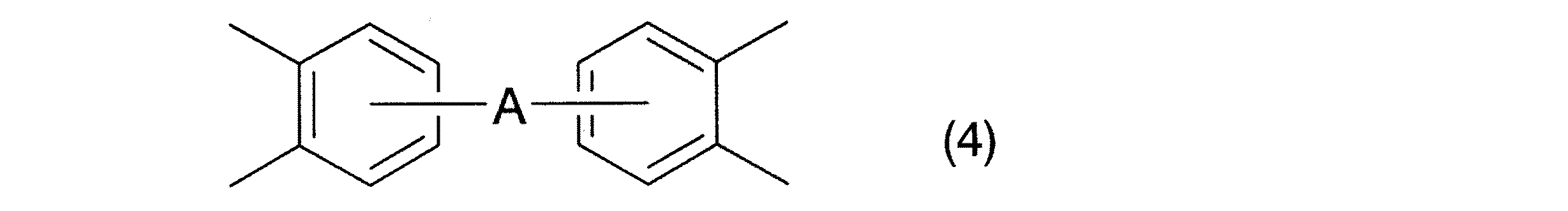
請求項4記載の発明は、基R1が、一般式(4):
〔式中Aは2価の有機基を示す。〕
で表される4価の基である請求項1記載のポリベンゾオキサゾール樹脂前駆体の製造方法である。
で表される4価の基である請求項1記載のポリベンゾオキサゾール樹脂前駆体の製造方法である。
請求項5記載の発明は、基R2が、2価の炭化水素基の全ての水素原子を、フッ素原子に置換したパーフルオロ炭化水素基である請求項1記載のポリベンゾオキサゾール樹脂前駆体の製造方法である。
請求項6記載の発明は、一般式(5):
〔式中R1は4価の有機基、R2は2価の有機基を示す。〕
で表される繰り返し単位を有するポリベンゾオキサゾール樹脂を製造する方法であって、請求項1記載の製造方法で製造したポリベンゾオキサゾール樹脂前駆体を、脱水環化反応させることを特徴とするポリベンゾオキサゾール樹脂の製造方法である。
で表される繰り返し単位を有するポリベンゾオキサゾール樹脂を製造する方法であって、請求項1記載の製造方法で製造したポリベンゾオキサゾール樹脂前駆体を、脱水環化反応させることを特徴とするポリベンゾオキサゾール樹脂の製造方法である。
特許文献1〜3に記載された従来の製造方法では、重合反応中にジカルボン酸ジハライドが分解して、それ以上、反応が進まなくなることがある。また、前記一般式(1)の繰り返し単位からなるポリベンゾオキサゾール樹脂前駆体が、ポリスチレン換算の重量平均分子量Mw=5000以下程度まで成長したオリゴマーの段階で、主鎖の両末端が結合して環状になりやすい。そして、生成した環状のオリゴマーは、それ以上、主鎖が成長しない。そのため、ポリベンゾオキサゾール樹脂前駆体は、全体として見たときの平均重合度および重量平均分子量が、前記のように小さくなる。
従来の製造方法において、ジカルボン酸ジハライドが分解したり、環状のオリゴマーが生成したりするのは、ジアミン化合物とジカルボン酸ジハライドと溶媒とを、液温を0℃以下に維持して混合した後、反応速度を高めるため、直ちに、液温を、室温程度まで高めているためである。つまり、0℃を超える温度条件下では、ジカルボン酸ジハライドが分解してジカルボン酸になる反応が起こったり、ポリベンゾオキサゾール樹脂前駆体の主鎖の自由度が大きくなり、主鎖の両末端が結合可能な距離まで近接する機会が増加して、両末端が結合して環状になったりしやすい。
そこで発明者は、反応液の液温についてさらに検討した結果、液温を0℃以下に維持してジアミン化合物とジカルボン酸ジハライドと溶媒とを混合後、直ちに昇温せず、この混合工程に続いて、反応液の液温を0℃以下に維持して一定時間、反応を続ける低温反応工程を設けて、ジカルボン酸ジハライドの分解を抑制するとともに、成長途上の主鎖の自由度をある程度、抑制してやればよいことを見出した。低温反応工程を設けて、ジカルボン酸ジハライドの分解を抑制し、かつ主鎖の自由度を抑制した状態で主鎖を成長させて、環状のオリゴマーの生成を抑制すれば、前駆体の分子量を、これまでよりも大きくできることを見出したのである。したがって、請求項1記載の発明によれば、これまでよりも分子量の大きいポリベンゾオキサゾール樹脂前駆体を製造することが可能となる。
また、請求項2記載の発明によれば、ジアミン化合物とジカルボン酸ジハライドとを、混合工程においてより均一に混合し、低温反応工程においてより均一に反応させることができるため、分子量の大きいポリベンゾオキサゾール樹脂前駆体を、より効率よく製造することができる。
また、請求項3記載の発明によれば、ジカルボン酸ジハライドの分解と、環状のオリゴマーの生成を確実に抑制できるため、分子量の大きいポリベンゾオキサゾール樹脂前駆体を、より効率よく製造することができる。
請求項4記載の発明によれば、一般式(1)中の基R1が一般式(4)で表される4価の基である、高分子量のポリベンゾオキサゾール樹脂前駆体を製造することができる。そして、かかる前駆体を脱水環化反応させることによって、主鎖に上記の基が導入された、とくに剛直で、耐熱性、機械的強度、耐薬品性、電気的特性等にすぐれたポリベンゾオキサゾール樹脂を製造することが可能となる。
請求項5記載の発明によれば、一般式(1)中の基R2が、2価の炭化水素基の全ての水素原子をフッ素原子に置換したパーフルオロ炭化水素基である、高分子量のポリベンゾオキサゾール樹脂前駆体を製造することができる。そして、かかる前駆体を脱水環化反応させることによって、主鎖に上記の基が導入された、とくに剛直で、耐熱性、機械的強度、耐薬品性、光学的特性、高周波特性、電気的特性等にすぐれたポリベンゾオキサゾール樹脂を製造することが可能となる。
請求項6記載の発明によれば、上記製造方法で製造した前駆体を、さらに、脱水環化反応させることによって、従来のものと同等の、耐熱性、機械的強度、耐薬品性、電気的特性等にすぐれた剛直な分子構造を有するポリベンゾオキサゾール樹脂を、従来に比べてより効率よく製造することができる。
以下に、本発明を説明する。
前記のように、本発明の前駆体の製造方法は、一般式(2)で表されるジアミン化合物と、一般式(3)で表されるジカルボン酸ジハライドと、溶媒とを、混合工程において、液温を0℃以下に維持しつつ混合して反応液とし、この反応液を、低温反応工程において、液温を0℃以下に維持して一定時間、反応を続けることを特徴とする。これにより、環状のオリゴマーが生成するのを防止して、これまでよりも分子量の大きいポリベンゾオキサゾール樹脂前駆体を製造することが可能となる。
前記のように、本発明の前駆体の製造方法は、一般式(2)で表されるジアミン化合物と、一般式(3)で表されるジカルボン酸ジハライドと、溶媒とを、混合工程において、液温を0℃以下に維持しつつ混合して反応液とし、この反応液を、低温反応工程において、液温を0℃以下に維持して一定時間、反応を続けることを特徴とする。これにより、環状のオリゴマーが生成するのを防止して、これまでよりも分子量の大きいポリベンゾオキサゾール樹脂前駆体を製造することが可能となる。
前駆体の製造原料である、一般式(2)で表されるジアミン化合物、および一般式(3)で表されるジカルボン酸ジハライドとしては、それぞれの式中の、基R1、R2のうちの少なくとも一方が芳香族基である種々の化合物を用いることができ、とくに、基R1、R2がともに芳香族基である、ジアミン化合物とジカルボン酸ジハライドとを組み合わせて用いるのが、分子の剛直性を高めて、耐熱性、機械的強度、耐薬品性、光学的特性、電気的特性等にすぐれたポリベンゾオキサゾール樹脂を製造する上で好ましい。また、上記ジアミン化合物、およびジカルボン酸ジハライドとしては、それぞれ、基R1、R2が異なる2種以上を、任意に併用してもよい。
一般式(2)中の基R1に好適な4価の芳香族基としては、これに限定されないが、例えば、一般式(6):
〔式中R5、R6は同一または異なって水素原子、フッ素原子、アルキル基、アルコキシ基、フルオロアルキル基、アリール基、フェノキシ基、またはフルオロフェノキシ基を示す。〕
で表される基、一般式(7):
〔式中R7、R8、R9、R10、R11、R12は同一または異なって水素原子、フッ素原子、アルキル基、アルコキシ基、フルオロアルキル基、アリール基、フェノキシ基、またはフルオロフェノキシ基を示す。Aは2価の有機基を示す。〕
で表される基、または一般式(8):
〔式中R7、R8、R9、R10、R11、R12は同一または異なって水素原子、フッ素原子、アルキル基、アルコキシ基、フルオロアルキル基、アリール基、フェノキシ基、またはフルオロフェノキシ基を示す。〕
で表される基が挙げられる。
で表される基、一般式(7):
で表される基、または一般式(8):
で表される基が挙げられる。
とくに、一般式(7)中のR7、R8、R9、R10、R11、R12がいずれも水素原子である、一般式(4):
〔式中Aは2価の有機基を示す。〕
で表される基が好ましい。また、一般式(4)中のAとしては、−O−、−SO2−、−S−、−R13−、−(OR13)n−、−(R13O)n−、または−(OR13O)n−が挙げられる。これらの基においてR13はアルキレン基、フルオロアルキレン基、アリーレン基、フルオロアリーレン基を示し、nは1以上の整数を示す。
で表される基が好ましい。また、一般式(4)中のAとしては、−O−、−SO2−、−S−、−R13−、−(OR13)n−、−(R13O)n−、または−(OR13O)n−が挙げられる。これらの基においてR13はアルキレン基、フルオロアルキレン基、アリーレン基、フルオロアリーレン基を示し、nは1以上の整数を示す。
これらの条件を満たす、一般式(2)で表されるジアミン化合物の具体例としては、例えば、式(9):
で表される2,2′−ビス(3−アミノ−4−ヒドロキシフェニル)ヘキサフルオロプロパンが挙げられる。また、一般式(2)で表されるジアミン化合物の他の例としては、2,2′−ビス(3−アミノ−4−ヒドロキシ−5−トリフルオロメチルフェニル)ヘキサフルオロプロパン、4,4′−ジアミノ−3,3′−ジヒドロキシビフェニル、3,3′−ジアミノ−4,4′−ジヒドロキシビフェニル、2,5−ジアミノ−1,4−ジヒドロキシベンゼン、4,6−ジアミノ−1,3−ジヒドロキシベンゼン等が挙げられる。
一般式(6)で表されるジカルボン酸ジハライドにおいて、基R2に好適な2価の芳香族基としては、これに限定されないが、例えば、一般式(10):
〔式中R14、R15、R16、R17は同一または異なって水素原子、フッ素原子、アルキル基、アルコキシ基、フルオロアルキル基、アリール基、フェノキシ基、またはフルオロフェノキシ基を示す。〕
で表される基、一般式(11):
〔式中R18、R19、R20、R21、R22、R23、R24、R25は同一または異なって水素原子、フッ素原子、アルキル基、アルコキシ基、フルオロアルキル基、アリール基、フェノキシ基、またはフルオロフェノキシ基を示す。Eは−O−、−SO2−、−S−、−R26−、−(OR26)n−、−(R26O)n−、または−(OR26O)n−を示す。またこれらの基においてR26はアルキレン基、フルオロアルキレン基、アリーレン基、フルオロアリーレン基を示し、nは1以上の整数を示す。〕
で表される基、または一般式(12):
〔式中R18、R19、R20、R21、R22、R23、R24、R25は同一または異なって水素原子、フッ素原子、アルキル基、アルコキシ基、フルオロアルキル基、アリール基、フェノキシ基、またはフルオロフェノキシ基を示す。〕
で表される基が挙げられる。
で表される基、一般式(11):
で表される基、または一般式(12):
で表される基が挙げられる。
とくに、ポリベンゾオキサゾール樹脂を光学的用途や高周波用途に使用する場合は、基R2としては、2価の炭化水素基の全ての水素原子をフッ素原子に置換したパーフルオロ炭化水素基が好ましい。すなわち、前記一般式(10)中の基R14、R15、R16、R17がいずれもフッ素原子である基や、一般式(11)中の基R18、R19、R20、R21、R22、R23、R24、R25がいずれもフッ素原子で、なおかつEがパーフルオロアルキレン基、またはパーフルオロアリーレン基である基、あるいは一般式(12)中の基R18、R19、R20、R21、R22、R23、R24、R25がいずれもフッ素原子である基が好ましい。
これらの条件を満たす、一般式(3)で表されるジカルボン酸ジハライドの具体例としては、例えば、式(13):
で表される2,4,5,6−テトラフルオロイソフタル酸ジクロリドや、式(14):
で表される2,3,5,6,2′,3′,5′,6′−オクタフルオロビフェニル−4,4′−ジカルボン酸ジクロリド等が挙げられる。
上記ジアミン化合物と、ジカルボン酸ジハライドとを反応させてポリベンゾオキサゾール樹脂前駆体を製造するために、本発明では、まず混合工程において、この両成分と、溶媒とを、液温を0℃以下に維持しつつ混合して反応液を調製する。具体的には、ジアミン化合物およびジカルボン酸ジハライドを、ともに、良好に溶解しうる共通の溶媒に、ジアミン化合物を溶解した溶液と、同じ溶媒に、ジカルボン酸ジハライドを溶解した溶液とを用意する。そして、例えば、ジアミン化合物を溶解した溶液の液温を0℃以下に維持しつつ、液をかく拌しながら、ジカルボン酸ジハライドを溶解した溶液を滴下することで、反応液が調製される。
溶媒としては、例えば、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N−メチル−2−ピロリドン等のアミド系溶媒、ジメチルスルホキシド、テトラメチルスルホン等のイオウ系溶媒、テトラヒドロフラン等のエーテル系溶媒などが挙げられる。これらは、それぞれ1種単独で使用してもよいし、2種以上を併用してもよい。
また、反応液には、ジアミン化合物とジカルボン酸ジハライドとの反応によって生成するハロゲン化水素を受容するための酸受容剤を加えておくのが好ましい。酸受容剤としては、例えば、トリメチルアミン、トリエチルアミン、ピリジン、ジメチルアニリン、ジメチルピリジン、イミダゾール等が挙げられる。これらは、それぞれ1種単独で使用してもよいし、2種以上を併用してもよい。
次に、調製した反応液を、低温反応工程において、反応液の液温を0℃以下に維持しつつ、液をかく拌しながら一定時間、反応を続ける。この低温反応工程において、低温で反応を続ける時間は、ジアミン化合物、ジカルボン酸ジハライドおよび溶媒の種類や組み合わせ等によって異なり、特に制限されない。しかし、反応液の液温を、0℃を超える温度に昇温した際に、生成したポリベンゾオキサゾール樹脂前駆体中に占める、分子量5000以下のオリゴマーの割合が、35重量%以下になるまで、低温反応工程を続けるのが、環状のオリゴマーの生成をできるだけ抑制して、前駆体の分子量を高めるために、好ましい。
具体的には、特定のジアミン化合物、ジカルボン酸ジハライドおよび溶媒の種類、組み合わせについて、混合工程および低温反応工程の温度を所定値に設定して合成反応を行い、反応開始から所定時間の経過後に、反応液の液温を0℃を超える温度に昇温した際に、生成したポリベンゾオキサゾール樹脂前駆体中に占める、分子量5000以下のオリゴマーの割合を測定する操作を、低温反応工程の時間を違えながら、繰り返し行って検量線を作成し、この検量線に基づいて、低温反応工程の時間を設定して反応を行う。あるいは、反応開始から一定時間が経過するごとに反応液をサンプリングし、サンプリングした反応液を、0℃を超える温度に昇温して、生成したポリベンゾオキサゾール樹脂前駆体中に占める、分子量5000以下のオリゴマーの割合を測定しながら、反応を行ってもよい。
そして、低温反応工程の終了後、反応液を、0℃を超える温度に昇温して反応を完結させれば、分子量5000以下のオリゴマーの割合が前記の範囲に抑制されて、平均重合度が、およそ15を超えるか、または、ポリスチレン換算の平均分子量Mwが、およそ10000を超える、分子量の大きいポリベンゾオキサゾール樹脂前駆体を製造することができる。
なお、前記混合工程および低温反応工程での液温は、0℃以下であればよい。しかし、ジアミン化合物とジカルボン酸ジハライドとを均一に混合し、また均一に反応させることを考慮すると、液温は、0℃以下で、かつ、溶液および反応液の融点以上であるのが好ましい。かかる反応により、前記一般式(2)で表されるジアミン化合物と、一般式(3)で表されるジカルボン酸ジハライドとが縮重合して、一般式(1)で表される繰り返し単位を有するポリベンゾオキサゾール樹脂前駆体が合成される。
次に、合成された前駆体を、脱水環化反応させることによって、一般式(5)で表される繰り返し単位を有するポリベンゾオキサゾール樹脂が製造される。例えば、反応液から分離した上記の前駆体を、適当な有機溶媒に溶解してワニス化し、これを、基材上に塗布した状態で、一定時間、加熱して脱水環化反応させることによって、ポリベンゾオキサゾール樹脂からなるフィルムが製造される。
また、合成反応後の、前駆体を含む反応液を、そのままワニスとして使用することもできる。その場合は、必要に応じて有機溶媒を加えるなどして、粘度を調整しても良い。また、前駆体を成形材料として使用して所定の形状に成形するのと同時に、もしくは、成形後に、さらに加熱して脱水環化反応させることによって、ポリベンゾオキサゾール樹脂からなる成形体が製造される。
前駆体をワニス化するために用いる有機溶媒としては、当該前駆体の合成反応に用いるのと同じ溶媒が使用される。また、その他の溶媒としては、γ−ブチロラクトン、ジエチレングリコールジメチルエーテル、ジエチレングリコールジエチルエーテル、ジエチレングリコールジブチルエーテル、プロピレングリコールモノメチルエーテル、ジプロピレングリコールモノメチルエーテル、プロピレングリコールモノメチルエーテルアセテート、乳酸メチル、乳酸エチル、乳酸ブチル、メチル−1,3−ブチレングリコールアセテート、1,3−ブチレングリコール−3−モノメチルエーテル、ピルビン酸メチル、ピルビン酸エチル、メチル−3−メトキシプロピオネート等が挙げられる。これらは、それぞれ1種単独で使用してもよいし、2種以上を併用してもよい。脱水環化反応の反応温度は、100〜500℃の範囲で適宜、設定することができる。また、反応時間は、数秒から数十時間程度の範囲で適宜、設定することができる。
以下に、本発明を実施例、比較例に基づいて説明する。
実施例1:
工程1(ジカルボン酸ジクロリドの合成)
4,4′−ジカルボキシ−2,3,5,6,2′,3′,5′,6′−オクタフルオロビフェニル100g(0.26モル)を、塩化チオニル300g、およびN,N−ジメチルアセトアミド2gとともに反応容器に入れ、30分かけて60℃に昇温後、60℃で2時間、反応させた。
実施例1:
工程1(ジカルボン酸ジクロリドの合成)
4,4′−ジカルボキシ−2,3,5,6,2′,3′,5′,6′−オクタフルオロビフェニル100g(0.26モル)を、塩化チオニル300g、およびN,N−ジメチルアセトアミド2gとともに反応容器に入れ、30分かけて60℃に昇温後、60℃で2時間、反応させた。
反応終了後、反応液を、60℃、約1.3×104Paで1時間、減圧濃縮した。次いで、濃縮液を、100℃、約4.00×102Paで単蒸留し、さらに110℃、1.33×102Paで単蒸留を繰り返して、前記式(14)で表される2,3,5,6,2′,3′,5′,6′−オクタフルオロビフェニル−4,4′−ジカルボン酸ジクロリド92gを得た。
工程2(ポリベンゾオキサゾール樹脂前駆体の製造)
上記工程1で合成した2,3,5,6,2′,3′,5′,6′−オクタフルオロビフェニル−4,4′−ジカルボン酸ジクロリド12.7g(0.03モル)を、脱水N,N−ジメチルアセトアミド44mlに溶解して溶液を調製した。
上記工程1で合成した2,3,5,6,2′,3′,5′,6′−オクタフルオロビフェニル−4,4′−ジカルボン酸ジクロリド12.7g(0.03モル)を、脱水N,N−ジメチルアセトアミド44mlに溶解して溶液を調製した。
次に、前記式(9)で表される2,2′−ビス(3−アミノ−4−ヒドロキシフェニル)ヘキサフルオロプロパン11g(0.03モル)を、脱水N,N−ジメチルアセトアミド88mlに溶解し、脱水ピリジン6.4g(0.08モル)を添加後、乾燥窒素雰囲気下、液温を−30℃に維持しつつ、かく拌しながら、先の溶液を、30分かけて徐々に滴下した(混合工程)。そして、滴下終了後、反応液の液温を−30℃に維持して反応を続け(低温反応工程)、一定時間が経過した時点で、液温を、1時間かけて23℃まで昇温して反応を終了した。
反応後の液を、1Nの塩酸水溶液に滴下し、PTFEフィルタを通して固形分をロ別し、蒸留水による洗浄とロ別とを2回、繰り返した後、60℃で12時間、減圧乾燥してポリベンゾオキサゾール樹脂前駆体を製造した。
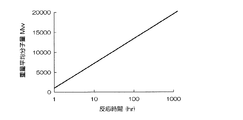
得られたポリベンゾオキサゾール樹脂前駆体の、ポリスチレン換算の重量平均分子量Mwを、臭素化リチウムを溶解させたN−メチル−2−ピロリドンを展開溶液とするゲルパーミェーションクロマトグラムによって測定した。低温反応工程の時間と、得られたポリベンゾオキサゾール樹脂前駆体の、ポリスチレン換算の重量平均分子量Mwとの関係を、表1および図1に示す。
また、得られたポリベンゾオキサゾール樹脂前駆体中に占める、分子量5000以下のオリゴマーの割合(重量%)をゲルパーミェーションクロマトグラフ法によって測定した。低温反応工程の時間と、分子量5000以下のオリゴマーの割合(重量%)との関係を、表1に示す。表より、低温反応工程の時間が長いほど、オリゴマーの割合を少なくして、ポリベンゾオキサゾール樹脂前駆体の分子量を大きくできることが確認された。
比較例1:
混合工程の終了後、直ちに、液温を23℃まで上昇させて5時間、反応を行ったこと以外は、実施例1と同様にして、ポリベンゾオキサゾール樹脂前駆体を製造した。得られたポリベンゾオキサゾール樹脂前駆体の、ポリスチレン換算の重量平均分子量Mwを、臭素化リチウムを溶解させたN−メチル−2−ピロリドンを展開溶液とするゲルパーミェーションクロマトグラムによって測定したところ、4100であった。また、得られたポリベンゾオキサゾール樹脂前駆体中に占める、分子量5000以下のオリゴマーの割合(重量%)をゲルパーミェーションクロマトグラフ法によって測定したところ、45重量%であった。
混合工程の終了後、直ちに、液温を23℃まで上昇させて5時間、反応を行ったこと以外は、実施例1と同様にして、ポリベンゾオキサゾール樹脂前駆体を製造した。得られたポリベンゾオキサゾール樹脂前駆体の、ポリスチレン換算の重量平均分子量Mwを、臭素化リチウムを溶解させたN−メチル−2−ピロリドンを展開溶液とするゲルパーミェーションクロマトグラムによって測定したところ、4100であった。また、得られたポリベンゾオキサゾール樹脂前駆体中に占める、分子量5000以下のオリゴマーの割合(重量%)をゲルパーミェーションクロマトグラフ法によって測定したところ、45重量%であった。
実施例2:
工程1(ジカルボン酸ジクロリドの合成)
2,4,5,6−テトラフルオロイソフタル酸63g(0.26モル)を、塩化チオニル300g、およびN,N−ジメチルアセトアミド2gとともに反応容器に入れ、30分かけて60℃に昇温後、60℃で2時間、反応させた。
工程1(ジカルボン酸ジクロリドの合成)
2,4,5,6−テトラフルオロイソフタル酸63g(0.26モル)を、塩化チオニル300g、およびN,N−ジメチルアセトアミド2gとともに反応容器に入れ、30分かけて60℃に昇温後、60℃で2時間、反応させた。
反応終了後、反応液を、60℃、約1.3×104Paで1時間、減圧濃縮した。次いで、濃縮液を、100℃、約4.00×102Paで単蒸留し、さらに110℃、1.33×102Paで単蒸留を繰り返して、前記式(13)で表される2,4,5,6−テトラフルオロイソフタル酸ジクロリド61gを得た。
工程2(ポリベンゾオキサゾール樹脂前駆体の製造)
上記工程1で合成した2,4,5,6−テトラフルオロイソフタル酸ジクロリド8.25g(0.03モル)を、脱水N,N−ジメチルアセトアミド44mlに溶解して溶液を調製した。
上記工程1で合成した2,4,5,6−テトラフルオロイソフタル酸ジクロリド8.25g(0.03モル)を、脱水N,N−ジメチルアセトアミド44mlに溶解して溶液を調製した。
次に、前記式(9)で表される2,2′−ビス(3−アミノ−4−ヒドロキシフェニル)ヘキサフルオロプロパン11g(0.03モル)を、脱水N,N−ジメチルアセトアミド88mlに溶解し、脱水ピリジン6.4g(0.08モル)を添加後、乾燥窒素雰囲気下、液温を−25℃に維持しつつ、かく拌しながら、先の溶液を、30分かけて徐々に滴下した(混合工程)。そして、滴下終了後、反応液の液温を−25℃に維持して反応を続け(低温反応工程)、168.5時間が経過した時点で、液温を、1時間かけて23℃まで昇温して反応を終了した。
反応後の液を、1Nの塩酸水溶液に滴下し、PTFEフィルタを通して固形分をロ別し、蒸留水による洗浄とロ別とを2回、繰り返した後、60℃で12時間、減圧乾燥してポリベンゾオキサゾール樹脂前駆体を製造した。
得られたポリベンゾオキサゾール樹脂前駆体の、ポリスチレン換算の重量平均分子量Mwを、臭素化リチウムを溶解させたN−メチル−2−ピロリドンを展開溶液とするゲルパーミェーションクロマトグラムによって測定したところ、10100であった。また、得られたポリベンゾオキサゾール樹脂前駆体中に占める、分子量5000以下のオリゴマーの割合(重量%)をゲルパーミェーションクロマトグラフ法によって測定したところ、35重量%であった。
比較例2:
混合工程での液温を−15℃に設定し、かつ、混合工程の終了後、直ちに、液温を23℃まで上昇させて5時間、反応を行ったこと以外は、実施例2と同様にして、ポリベンゾオキサゾール樹脂前駆体を製造した。
混合工程での液温を−15℃に設定し、かつ、混合工程の終了後、直ちに、液温を23℃まで上昇させて5時間、反応を行ったこと以外は、実施例2と同様にして、ポリベンゾオキサゾール樹脂前駆体を製造した。
得られたポリベンゾオキサゾール樹脂前駆体の、ポリスチレン換算の重量平均分子量Mwを、臭素化リチウムを溶解させたN−メチル−2−ピロリドンを展開溶液とするゲルパーミェーションクロマトグラムによって測定したところ、7000であった。また、得られたポリベンゾオキサゾール樹脂前駆体中に占める、分子量5000以下のオリゴマーの割合(重量%)をゲルパーミェーションクロマトグラフ法によって測定したところ、50重量%であった。
実施例3:
工程2(ポリベンゾオキサゾール樹脂前駆体の製造)
実施例1の工程1で合成した2,3,5,6,2′,3′,5′,6′−オクタフルオロビフェニル−4,4′−ジカルボン酸ジクロリド12.7g(0.03モル)を、脱水N,N−ジメチルアセトアミド44mlに溶解して溶液を調製した。
工程2(ポリベンゾオキサゾール樹脂前駆体の製造)
実施例1の工程1で合成した2,3,5,6,2′,3′,5′,6′−オクタフルオロビフェニル−4,4′−ジカルボン酸ジクロリド12.7g(0.03モル)を、脱水N,N−ジメチルアセトアミド44mlに溶解して溶液を調製した。
次に、2,5′−ジアミノ−1,4−ジハイドロベンゼン6.49g(0.03モル)を、脱水N,N−ジメチルアセトアミド88mlに溶解し、脱水ピリジン6.4g(0.08モル)を添加後、乾燥窒素雰囲気下、液温を−25℃に維持しつつ、かく拌しながら、先の溶液を、30分かけて徐々に滴下した(混合工程)。そして、滴下終了後、反応液の液温を−25℃に維持して反応を続け(低温反応工程)、24時間が経過した時点で、液温を、1時間かけて23℃まで昇温して反応を終了した。
反応後の液を、1Nの塩酸水溶液に滴下し、PTFEフィルタを通して固形分をロ別し、蒸留水による洗浄とロ別とを2回、繰り返した後、60℃で12時間、減圧乾燥してポリベンゾオキサゾール樹脂前駆体を製造した。
得られたポリベンゾオキサゾール樹脂前駆体の、ポリスチレン換算の重量平均分子量Mwを、臭素化リチウムを溶解させたN−メチル−2−ピロリドンを展開溶液とするゲルパーミェーションクロマトグラムによって測定したところ、34000であった。また、得られたポリベンゾオキサゾール樹脂前駆体中に占める、分子量5000以下のオリゴマーの割合(重量%)をゲルパーミェーションクロマトグラフ法によって測定したところ、3重量%であった。
Claims (6)
- 一般式(1):
で表される繰り返し単位を有するポリベンゾオキサゾール樹脂前駆体を製造する方法であって、一般式(2):
で表されるジアミン化合物と、一般式(3):
で表されるジカルボン酸ジハライドと、溶媒とを、液温を0℃以下に維持しつつ混合して反応液とする混合工程と、この混合工程に続いて、反応液の液温を0℃以下に維持して一定時間、反応を続ける低温反応工程とを含むことを特徴とするポリベンゾオキサゾール樹脂前駆体の製造方法。 - 混合工程および低温反応工程における液温を、液の融点以上で、かつ0℃以下の範囲内とする請求項1記載のポリベンゾオキサゾール樹脂前駆体の製造方法。
- 反応液の液温を、0℃を超える温度に昇温した際に、生成したポリベンゾオキサゾール樹脂前駆体中に占める、分子量5000以下のオリゴマーの割合が、35重量%以下になるまで、低温反応工程を続ける請求項1記載のポリベンゾオキサゾール樹脂前駆体の製造方法。
- 基R1が、一般式(4):
で表される4価の基である請求項1記載のポリベンゾオキサゾール樹脂前駆体の製造方法。 - 基R2が、2価の炭化水素基の全ての水素原子を、フッ素原子に置換したパーフルオロ炭化水素基である請求項1記載のポリベンゾオキサゾール樹脂前駆体の製造方法。
- 一般式(5):
で表される繰り返し単位を有するポリベンゾオキサゾール樹脂を製造する方法であって、請求項1記載の製造方法で製造したポリベンゾオキサゾール樹脂前駆体を、脱水環化反応させることを特徴とするポリベンゾオキサゾール樹脂の製造方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2004110276A JP2005290301A (ja) | 2004-04-02 | 2004-04-02 | ポリベンゾオキサゾール樹脂前駆体の製造方法およびポリベンゾオキサゾール樹脂の製造方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2004110276A JP2005290301A (ja) | 2004-04-02 | 2004-04-02 | ポリベンゾオキサゾール樹脂前駆体の製造方法およびポリベンゾオキサゾール樹脂の製造方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2005290301A true JP2005290301A (ja) | 2005-10-20 |
Family
ID=35323593
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2004110276A Pending JP2005290301A (ja) | 2004-04-02 | 2004-04-02 | ポリベンゾオキサゾール樹脂前駆体の製造方法およびポリベンゾオキサゾール樹脂の製造方法 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2005290301A (ja) |
Cited By (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10308644B2 (en) | 2016-12-22 | 2019-06-04 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US10618916B2 (en) | 2018-05-11 | 2020-04-14 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US10669271B2 (en) | 2018-03-30 | 2020-06-02 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US10793565B2 (en) | 2016-12-22 | 2020-10-06 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US10806785B2 (en) | 2016-12-22 | 2020-10-20 | Incyte Corporation | Immunomodulator compounds and methods of use |
| US11401279B2 (en) | 2019-09-30 | 2022-08-02 | Incyte Corporation | Pyrido[3,2-d]pyrimidine compounds as immunomodulators |
| US11407749B2 (en) | 2015-10-19 | 2022-08-09 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11465981B2 (en) | 2016-12-22 | 2022-10-11 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11535615B2 (en) | 2015-12-22 | 2022-12-27 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| CN115636846A (zh) * | 2022-11-02 | 2023-01-24 | 洛阳中硅高科技有限公司 | 高纯三甲硅烷基胺的制备方法及装置 |
| US11572366B2 (en) | 2015-11-19 | 2023-02-07 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11608337B2 (en) | 2016-05-06 | 2023-03-21 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11613536B2 (en) | 2016-08-29 | 2023-03-28 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11673883B2 (en) | 2016-05-26 | 2023-06-13 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11718605B2 (en) | 2016-07-14 | 2023-08-08 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11753406B2 (en) | 2019-08-09 | 2023-09-12 | Incyte Corporation | Salts of a PD-1/PD-L1 inhibitor |
| US11760756B2 (en) | 2020-11-06 | 2023-09-19 | Incyte Corporation | Crystalline form of a PD-1/PD-L1 inhibitor |
| US11780836B2 (en) | 2020-11-06 | 2023-10-10 | Incyte Corporation | Process of preparing a PD-1/PD-L1 inhibitor |
| US11866451B2 (en) | 2019-11-11 | 2024-01-09 | Incyte Corporation | Salts and crystalline forms of a PD-1/PD-L1 inhibitor |
| US11866434B2 (en) | 2020-11-06 | 2024-01-09 | Incyte Corporation | Process for making a PD-1/PD-L1 inhibitor and salts and crystalline forms thereof |
| US11873309B2 (en) | 2016-06-20 | 2024-01-16 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US12466822B2 (en) | 2016-12-22 | 2025-11-11 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US12516044B2 (en) | 2015-12-17 | 2026-01-06 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
-
2004
- 2004-04-02 JP JP2004110276A patent/JP2005290301A/ja active Pending
Cited By (37)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11407749B2 (en) | 2015-10-19 | 2022-08-09 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11572366B2 (en) | 2015-11-19 | 2023-02-07 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US12516044B2 (en) | 2015-12-17 | 2026-01-06 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11866435B2 (en) | 2015-12-22 | 2024-01-09 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11535615B2 (en) | 2015-12-22 | 2022-12-27 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11608337B2 (en) | 2016-05-06 | 2023-03-21 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11673883B2 (en) | 2016-05-26 | 2023-06-13 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11873309B2 (en) | 2016-06-20 | 2024-01-16 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11718605B2 (en) | 2016-07-14 | 2023-08-08 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11613536B2 (en) | 2016-08-29 | 2023-03-28 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11465981B2 (en) | 2016-12-22 | 2022-10-11 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US10793565B2 (en) | 2016-12-22 | 2020-10-06 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US10308644B2 (en) | 2016-12-22 | 2019-06-04 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11339149B2 (en) | 2016-12-22 | 2022-05-24 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11787793B2 (en) | 2016-12-22 | 2023-10-17 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11566026B2 (en) | 2016-12-22 | 2023-01-31 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US12466822B2 (en) | 2016-12-22 | 2025-11-11 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US10806785B2 (en) | 2016-12-22 | 2020-10-20 | Incyte Corporation | Immunomodulator compounds and methods of use |
| US10800768B2 (en) | 2016-12-22 | 2020-10-13 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US10669271B2 (en) | 2018-03-30 | 2020-06-02 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US12247026B2 (en) | 2018-03-30 | 2025-03-11 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11124511B2 (en) | 2018-03-30 | 2021-09-21 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11414433B2 (en) | 2018-05-11 | 2022-08-16 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US10906920B2 (en) | 2018-05-11 | 2021-02-02 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US12187743B2 (en) | 2018-05-11 | 2025-01-07 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US10618916B2 (en) | 2018-05-11 | 2020-04-14 | Incyte Corporation | Heterocyclic compounds as immunomodulators |
| US11753406B2 (en) | 2019-08-09 | 2023-09-12 | Incyte Corporation | Salts of a PD-1/PD-L1 inhibitor |
| US11401279B2 (en) | 2019-09-30 | 2022-08-02 | Incyte Corporation | Pyrido[3,2-d]pyrimidine compounds as immunomodulators |
| US12247038B2 (en) | 2019-09-30 | 2025-03-11 | Incyte Corporation | Pyrido[3,2-d]pyrimidine compounds as immunomodulators |
| US11866451B2 (en) | 2019-11-11 | 2024-01-09 | Incyte Corporation | Salts and crystalline forms of a PD-1/PD-L1 inhibitor |
| US11866434B2 (en) | 2020-11-06 | 2024-01-09 | Incyte Corporation | Process for making a PD-1/PD-L1 inhibitor and salts and crystalline forms thereof |
| US12084443B2 (en) | 2020-11-06 | 2024-09-10 | Incyte Corporation | Process of preparing a PD-1/PD-L1 inhibitor |
| US11780836B2 (en) | 2020-11-06 | 2023-10-10 | Incyte Corporation | Process of preparing a PD-1/PD-L1 inhibitor |
| US12404272B2 (en) | 2020-11-06 | 2025-09-02 | Incyte Corporation | Process for making a PD-1/PD-L1 inhibitor and salts and crystalline forms thereof |
| US11760756B2 (en) | 2020-11-06 | 2023-09-19 | Incyte Corporation | Crystalline form of a PD-1/PD-L1 inhibitor |
| CN115636846B (zh) * | 2022-11-02 | 2024-07-23 | 洛阳中硅高科技有限公司 | 高纯三甲硅烷基胺的制备方法及装置 |
| CN115636846A (zh) * | 2022-11-02 | 2023-01-24 | 洛阳中硅高科技有限公司 | 高纯三甲硅烷基胺的制备方法及装置 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2005290301A (ja) | ポリベンゾオキサゾール樹脂前駆体の製造方法およびポリベンゾオキサゾール樹脂の製造方法 | |
| Zhang et al. | A smart latent catalyst containing o-trifluoroacetamide functional benzoxazine: precursor for low temperature formation of very high performance polybenzoxazole with low dielectric constant and high thermal stability | |
| US6291635B1 (en) | Fluorine-containing polybenzoxazole | |
| CN102604156B (zh) | 碱产生剂 | |
| Watanabe et al. | Synthesis of wholly alicyclic polyimides from N-silylated alicyclic diamines and alicyclic dianhydrides | |
| US9410055B2 (en) | Polybenzoxazole resin and precursor thereof | |
| US9255186B2 (en) | Thermosetting resins with enhanced cure characteristics containing organofunctional silane moieties | |
| JP2006001968A (ja) | トリプチセン骨格を有するポリアミド酸、ポリイミド樹脂及びこれを用いた光学部品 | |
| JP2759796B2 (ja) | 新規なビス(マレイミド)シロキサンおよびその製造方法 | |
| JP7734616B2 (ja) | アミノ化合物、当該アミノ化合物を用いたポリアミド酸及びポリイミド、並びにこれらの製造方法 | |
| JP2002327060A (ja) | ポリベンズオキサゾールおよびその製造方法 | |
| JP2005097365A (ja) | 低線熱膨張係数を有するポリベンゾオキサゾール及びその製造方法 | |
| JP6224154B2 (ja) | 環状オリゴマーを用いたポリ(エーテルスルホン−イミドまたは−アミド)共重合体の製造方法 | |
| CN1041603A (zh) | 具有热稳定性含有二有机多分子硅醚基团的共聚酰亚胺-酰胺的制备方法 | |
| KR20100115994A (ko) | 고리형 올리고머를 이용한 폴리〔이미드-아미드〕공중합체의 제조방법 | |
| JP2714661B2 (ja) | エチレン性不飽和基を末端基とするポリ(ベンズヒドロールイミド)、熱重合によって得られる網状ポリイミド及びこれらの用途 | |
| Mallakpour et al. | Thermoplastic nonvinyl polymers: From macro to nanostructure | |
| US6384182B2 (en) | Fluorine-containing polybenzoxazole | |
| KR102140776B1 (ko) | 사다리 구조의 폴리실세스퀴옥산을 포함하는 유무기 복합 폴리이미드 및 이의 제조방법 | |
| JP2841682B2 (ja) | 含フッ素ポリベンゾオキサゾール及びその前駆体である含フッ素ポリヒドロキシアミド | |
| JP2011174020A (ja) | ポリアミド酸エステルの製造方法 | |
| JP3546971B2 (ja) | ポリベンザゾールの製造方法 | |
| JP2890646B2 (ja) | 含フッ素ベンゾオキサゾール系重合体の製造法 | |
| JP2016153445A (ja) | ポリアミド樹脂の製造方法 | |
| JPH07228837A (ja) | 含フッ素コーティング剤 |