ES2936864T3 - Derivado de pirimidinosulfamida y método de preparación y aplicación médica del mismo - Google Patents
Derivado de pirimidinosulfamida y método de preparación y aplicación médica del mismo Download PDFInfo
- Publication number
- ES2936864T3 ES2936864T3 ES18881280T ES18881280T ES2936864T3 ES 2936864 T3 ES2936864 T3 ES 2936864T3 ES 18881280 T ES18881280 T ES 18881280T ES 18881280 T ES18881280 T ES 18881280T ES 2936864 T3 ES2936864 T3 ES 2936864T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- mmol
- added
- room temperature
- alkyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 238000002360 preparation method Methods 0.000 title abstract description 5
- 238000000034 method Methods 0.000 title description 14
- 150000001875 compounds Chemical class 0.000 claims abstract description 403
- 239000000203 mixture Substances 0.000 claims abstract description 132
- 229940044551 receptor antagonist Drugs 0.000 claims abstract description 9
- 239000002464 receptor antagonist Substances 0.000 claims abstract description 9
- -1 C1-4alkylO-C1-4alkyl Chemical group 0.000 claims description 89
- 125000000217 alkyl group Chemical group 0.000 claims description 34
- 150000003839 salts Chemical class 0.000 claims description 32
- 125000005842 heteroatom Chemical group 0.000 claims description 26
- 125000000592 heterocycloalkyl group Chemical group 0.000 claims description 20
- 229910052739 hydrogen Inorganic materials 0.000 claims description 19
- 229910052794 bromium Inorganic materials 0.000 claims description 14
- 229910052801 chlorine Inorganic materials 0.000 claims description 14
- 229910052740 iodine Inorganic materials 0.000 claims description 14
- 229910052731 fluorine Inorganic materials 0.000 claims description 13
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 claims description 7
- 125000006716 (C1-C6) heteroalkyl group Chemical group 0.000 claims description 6
- 125000006570 (C5-C6) heteroaryl group Chemical group 0.000 claims description 6
- 125000002883 imidazolyl group Chemical group 0.000 claims description 6
- 125000003226 pyrazolyl group Chemical group 0.000 claims description 6
- 125000000719 pyrrolidinyl group Chemical group 0.000 claims description 6
- 125000000335 thiazolyl group Chemical group 0.000 claims description 6
- 125000005958 tetrahydrothienyl group Chemical group 0.000 claims description 5
- JPRPJUMQRZTTED-UHFFFAOYSA-N 1,3-dioxolanyl Chemical group [CH]1OCCO1 JPRPJUMQRZTTED-UHFFFAOYSA-N 0.000 claims description 4
- 206010020772 Hypertension Diseases 0.000 claims description 4
- 206010064911 Pulmonary arterial hypertension Diseases 0.000 claims description 4
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims description 4
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 claims description 3
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims description 3
- 208000007342 Diabetic Nephropathies Diseases 0.000 claims description 3
- 208000007530 Essential hypertension Diseases 0.000 claims description 3
- 239000004480 active ingredient Substances 0.000 claims description 3
- 125000006430 alkyl cyclopropyl group Chemical group 0.000 claims description 3
- 208000033679 diabetic kidney disease Diseases 0.000 claims description 3
- 208000001286 intracranial vasospasm Diseases 0.000 claims description 3
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 3
- JNCMHMUGTWEVOZ-UHFFFAOYSA-N F[CH]F Chemical compound F[CH]F JNCMHMUGTWEVOZ-UHFFFAOYSA-N 0.000 claims description 2
- 108010081348 HRT1 protein Hairy Proteins 0.000 claims description 2
- 102100021881 Hairy/enhancer-of-split related with YRPW motif protein 1 Human genes 0.000 claims description 2
- VUWZPRWSIVNGKG-UHFFFAOYSA-N fluoromethane Chemical compound F[CH2] VUWZPRWSIVNGKG-UHFFFAOYSA-N 0.000 claims description 2
- 239000008194 pharmaceutical composition Substances 0.000 claims description 2
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 claims 4
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims 3
- 239000003814 drug Substances 0.000 abstract description 17
- 229940079593 drug Drugs 0.000 abstract description 12
- 201000010099 disease Diseases 0.000 abstract description 6
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 abstract description 6
- GZRPWFCHDLJSDX-UHFFFAOYSA-N pyrimidine;sulfamide Chemical class NS(N)(=O)=O.C1=CN=CN=C1 GZRPWFCHDLJSDX-UHFFFAOYSA-N 0.000 abstract 1
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 395
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 203
- 239000011541 reaction mixture Substances 0.000 description 153
- 235000019439 ethyl acetate Nutrition 0.000 description 132
- 239000000243 solution Substances 0.000 description 131
- 238000006243 chemical reaction Methods 0.000 description 123
- 239000002904 solvent Substances 0.000 description 118
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 104
- 239000012074 organic phase Substances 0.000 description 98
- 238000003786 synthesis reaction Methods 0.000 description 93
- 230000015572 biosynthetic process Effects 0.000 description 92
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 91
- 239000012299 nitrogen atmosphere Substances 0.000 description 88
- 239000000706 filtrate Substances 0.000 description 83
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 83
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 80
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 78
- 238000005160 1H NMR spectroscopy Methods 0.000 description 72
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 69
- 239000007787 solid Substances 0.000 description 61
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 57
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 54
- 239000003480 eluent Substances 0.000 description 54
- 239000003208 petroleum Substances 0.000 description 52
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 42
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 42
- 238000004440 column chromatography Methods 0.000 description 41
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 38
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 30
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 29
- 238000012360 testing method Methods 0.000 description 29
- 239000000872 buffer Substances 0.000 description 24
- 229910052757 nitrogen Inorganic materials 0.000 description 24
- 239000012634 fragment Substances 0.000 description 22
- 108050009340 Endothelin Proteins 0.000 description 21
- 102000002045 Endothelin Human genes 0.000 description 21
- 230000000694 effects Effects 0.000 description 21
- 125000001424 substituent group Chemical group 0.000 description 21
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 20
- 238000005481 NMR spectroscopy Methods 0.000 description 20
- 229910052799 carbon Inorganic materials 0.000 description 20
- 210000004027 cell Anatomy 0.000 description 19
- BKLHHTTZTSYHBV-UHFFFAOYSA-N 5-bromo-4,6-dichloropyrimidine Chemical compound ClC1=NC=NC(Cl)=C1Br BKLHHTTZTSYHBV-UHFFFAOYSA-N 0.000 description 18
- NXQGGXCHGDYOHB-UHFFFAOYSA-L cyclopenta-1,4-dien-1-yl(diphenyl)phosphane;dichloropalladium;iron(2+) Chemical compound [Fe+2].Cl[Pd]Cl.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1 NXQGGXCHGDYOHB-UHFFFAOYSA-L 0.000 description 18
- ZUBDGKVDJUIMQQ-UBFCDGJISA-N endothelin-1 Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(O)=O)NC(=O)[C@H]1NC(=O)[C@H](CC=2C=CC=CC=2)NC(=O)[C@@H](CC=2C=CC(O)=CC=2)NC(=O)[C@H](C(C)C)NC(=O)[C@H]2CSSC[C@@H](C(N[C@H](CO)C(=O)N[C@@H](CO)C(=O)N[C@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)N[C@H](CC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N2)=O)NC(=O)[C@@H](CO)NC(=O)[C@H](N)CSSC1)C1=CNC=N1 ZUBDGKVDJUIMQQ-UBFCDGJISA-N 0.000 description 17
- 125000003118 aryl group Chemical group 0.000 description 16
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 15
- 239000012043 crude product Substances 0.000 description 15
- 102000005962 receptors Human genes 0.000 description 15
- 108020003175 receptors Proteins 0.000 description 15
- 125000004429 atom Chemical group 0.000 description 14
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 14
- 239000007788 liquid Substances 0.000 description 13
- 239000003921 oil Substances 0.000 description 13
- 101800004490 Endothelin-1 Proteins 0.000 description 12
- 102400000686 Endothelin-1 Human genes 0.000 description 12
- 230000002401 inhibitory effect Effects 0.000 description 11
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 10
- MWUXSHHQAYIFBG-UHFFFAOYSA-N Nitric oxide Chemical compound O=[N] MWUXSHHQAYIFBG-UHFFFAOYSA-N 0.000 description 10
- 239000008346 aqueous phase Substances 0.000 description 10
- 125000004432 carbon atom Chemical group C* 0.000 description 10
- 230000006698 induction Effects 0.000 description 10
- JGCMEBMXRHSZKX-UHFFFAOYSA-N macitentan Chemical compound C=1C=C(Br)C=CC=1C=1C(NS(=O)(=O)NCCC)=NC=NC=1OCCOC1=NC=C(Br)C=N1 JGCMEBMXRHSZKX-UHFFFAOYSA-N 0.000 description 10
- 229960001039 macitentan Drugs 0.000 description 10
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 10
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 10
- 125000006239 protecting group Chemical group 0.000 description 10
- 241000282472 Canis lupus familiaris Species 0.000 description 9
- 108010044467 Isoenzymes Proteins 0.000 description 9
- 239000002253 acid Substances 0.000 description 9
- 238000002474 experimental method Methods 0.000 description 9
- 125000000623 heterocyclic group Chemical group 0.000 description 9
- 125000001183 hydrocarbyl group Chemical group 0.000 description 9
- 239000005457 ice water Substances 0.000 description 9
- 229910052760 oxygen Inorganic materials 0.000 description 9
- 239000013641 positive control Substances 0.000 description 9
- 238000004237 preparative chromatography Methods 0.000 description 9
- 229910052717 sulfur Inorganic materials 0.000 description 9
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 9
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 8
- 108010081668 Cytochrome P-450 CYP3A Proteins 0.000 description 8
- 102000000804 Pregnane X Receptor Human genes 0.000 description 8
- 108010001511 Pregnane X Receptor Proteins 0.000 description 8
- 241000700159 Rattus Species 0.000 description 8
- 230000003042 antagnostic effect Effects 0.000 description 8
- 239000002585 base Substances 0.000 description 8
- 239000003153 chemical reaction reagent Substances 0.000 description 8
- 239000000460 chlorine Substances 0.000 description 8
- 125000001072 heteroaryl group Chemical group 0.000 description 8
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 8
- 125000004433 nitrogen atom Chemical group N* 0.000 description 8
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 7
- 239000012981 Hank's balanced salt solution Substances 0.000 description 7
- 230000010100 anticoagulation Effects 0.000 description 7
- 239000003833 bile salt Substances 0.000 description 7
- 239000012230 colorless oil Substances 0.000 description 7
- 238000001514 detection method Methods 0.000 description 7
- 125000000524 functional group Chemical group 0.000 description 7
- 238000001294 liquid chromatography-tandem mass spectrometry Methods 0.000 description 7
- 239000000523 sample Substances 0.000 description 7
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 7
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical class [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 6
- 102000004328 Cytochrome P-450 CYP3A Human genes 0.000 description 6
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 6
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 6
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 6
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 6
- 239000012298 atmosphere Substances 0.000 description 6
- 239000001257 hydrogen Substances 0.000 description 6
- 230000005764 inhibitory process Effects 0.000 description 6
- 230000001404 mediated effect Effects 0.000 description 6
- 229920006395 saturated elastomer Polymers 0.000 description 6
- 239000012312 sodium hydride Substances 0.000 description 6
- 229910000104 sodium hydride Inorganic materials 0.000 description 6
- 125000004434 sulfur atom Chemical group 0.000 description 6
- XPGIBDJXEVAVTO-UHFFFAOYSA-N 5-bromo-2-chloropyrimidine Chemical compound ClC1=NC=C(Br)C=N1 XPGIBDJXEVAVTO-UHFFFAOYSA-N 0.000 description 5
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 5
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 5
- IPWKHHSGDUIRAH-UHFFFAOYSA-N bis(pinacolato)diboron Chemical compound O1C(C)(C)C(C)(C)OB1B1OC(C)(C)C(C)(C)O1 IPWKHHSGDUIRAH-UHFFFAOYSA-N 0.000 description 5
- 150000001721 carbon Chemical group 0.000 description 5
- 238000004587 chromatography analysis Methods 0.000 description 5
- 125000000753 cycloalkyl group Chemical group 0.000 description 5
- 238000000338 in vitro Methods 0.000 description 5
- 239000000463 material Substances 0.000 description 5
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 5
- 230000003228 microsomal effect Effects 0.000 description 5
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 5
- 235000011056 potassium acetate Nutrition 0.000 description 5
- 229910000027 potassium carbonate Inorganic materials 0.000 description 5
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 5
- 125000004076 pyridyl group Chemical group 0.000 description 5
- 238000001308 synthesis method Methods 0.000 description 5
- 210000001519 tissue Anatomy 0.000 description 5
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 4
- 102000002004 Cytochrome P-450 Enzyme System Human genes 0.000 description 4
- 108010015742 Cytochrome P-450 Enzyme System Proteins 0.000 description 4
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 4
- JRNVZBWKYDBUCA-UHFFFAOYSA-N N-chlorosuccinimide Chemical compound ClN1C(=O)CCC1=O JRNVZBWKYDBUCA-UHFFFAOYSA-N 0.000 description 4
- 230000002378 acidificating effect Effects 0.000 description 4
- 125000003545 alkoxy group Chemical group 0.000 description 4
- 239000008280 blood Substances 0.000 description 4
- 210000004369 blood Anatomy 0.000 description 4
- 125000004122 cyclic group Chemical group 0.000 description 4
- 231100000673 dose–response relationship Toxicity 0.000 description 4
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 4
- 125000004404 heteroalkyl group Chemical group 0.000 description 4
- 125000004356 hydroxy functional group Chemical group O* 0.000 description 4
- 239000001301 oxygen Substances 0.000 description 4
- 125000004430 oxygen atom Chemical group O* 0.000 description 4
- 239000012071 phase Substances 0.000 description 4
- 125000003386 piperidinyl group Chemical group 0.000 description 4
- 238000002953 preparative HPLC Methods 0.000 description 4
- 239000011734 sodium Substances 0.000 description 4
- 238000012453 sprague-dawley rat model Methods 0.000 description 4
- 239000000758 substrate Substances 0.000 description 4
- 238000012546 transfer Methods 0.000 description 4
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 3
- QGJOPFRUJISHPQ-UHFFFAOYSA-N Carbon disulfide Chemical compound S=C=S QGJOPFRUJISHPQ-UHFFFAOYSA-N 0.000 description 3
- IGXWBGJHJZYPQS-SSDOTTSWSA-N D-Luciferin Chemical compound OC(=O)[C@H]1CSC(C=2SC3=CC=C(O)C=C3N=2)=N1 IGXWBGJHJZYPQS-SSDOTTSWSA-N 0.000 description 3
- CYCGRDQQIOGCKX-UHFFFAOYSA-N Dehydro-luciferin Natural products OC(=O)C1=CSC(C=2SC3=CC(O)=CC=C3N=2)=N1 CYCGRDQQIOGCKX-UHFFFAOYSA-N 0.000 description 3
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 3
- 102000004190 Enzymes Human genes 0.000 description 3
- 108090000790 Enzymes Proteins 0.000 description 3
- BJGNCJDXODQBOB-UHFFFAOYSA-N Fivefly Luciferin Natural products OC(=O)C1CSC(C=2SC3=CC(O)=CC=C3N=2)=N1 BJGNCJDXODQBOB-UHFFFAOYSA-N 0.000 description 3
- 108060001084 Luciferase Proteins 0.000 description 3
- 239000005089 Luciferase Substances 0.000 description 3
- DDWFXDSYGUXRAY-UHFFFAOYSA-N Luciferin Natural products CCc1c(C)c(CC2NC(=O)C(=C2C=C)C)[nH]c1Cc3[nH]c4C(=C5/NC(CC(=O)O)C(C)C5CC(=O)O)CC(=O)c4c3C DDWFXDSYGUXRAY-UHFFFAOYSA-N 0.000 description 3
- 241001465754 Metazoa Species 0.000 description 3
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 3
- WBWWGRHZICKQGZ-UHFFFAOYSA-N Taurocholic acid Natural products OC1CC2CC(O)CCC2(C)C2C1C1CCC(C(CCC(=O)NCCS(O)(=O)=O)C)C1(C)C(O)C2 WBWWGRHZICKQGZ-UHFFFAOYSA-N 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 3
- 125000003342 alkenyl group Chemical group 0.000 description 3
- 125000000304 alkynyl group Chemical group 0.000 description 3
- 239000005557 antagonist Substances 0.000 description 3
- 238000003556 assay Methods 0.000 description 3
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 3
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 3
- 125000002619 bicyclic group Chemical group 0.000 description 3
- 229910052796 boron Inorganic materials 0.000 description 3
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Chemical compound BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 3
- 238000010790 dilution Methods 0.000 description 3
- 239000012895 dilution Substances 0.000 description 3
- 230000002496 gastric effect Effects 0.000 description 3
- 230000002440 hepatic effect Effects 0.000 description 3
- 238000011534 incubation Methods 0.000 description 3
- 125000005647 linker group Chemical group 0.000 description 3
- ZCSHNCUQKCANBX-UHFFFAOYSA-N lithium diisopropylamide Chemical compound [Li+].CC(C)[N-]C(C)C ZCSHNCUQKCANBX-UHFFFAOYSA-N 0.000 description 3
- 239000002609 medium Substances 0.000 description 3
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 3
- 239000011259 mixed solution Substances 0.000 description 3
- 125000002950 monocyclic group Chemical group 0.000 description 3
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 3
- 230000007935 neutral effect Effects 0.000 description 3
- 229910052763 palladium Inorganic materials 0.000 description 3
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 3
- 125000003373 pyrazinyl group Chemical group 0.000 description 3
- 230000004044 response Effects 0.000 description 3
- 238000000926 separation method Methods 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 238000006467 substitution reaction Methods 0.000 description 3
- WBWWGRHZICKQGZ-GIHLXUJPSA-N taurocholic acid Chemical compound C([C@@H]1C[C@H]2O)[C@@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@@H]([C@@H](CCC(=O)NCCS(O)(=O)=O)C)[C@@]2(C)[C@H](O)C1 WBWWGRHZICKQGZ-GIHLXUJPSA-N 0.000 description 3
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 3
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 3
- 238000004293 19F NMR spectroscopy Methods 0.000 description 2
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 2
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 2
- 239000004215 Carbon black (E152) Substances 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- 206010007559 Cardiac failure congestive Diseases 0.000 description 2
- 108010000543 Cytochrome P-450 CYP2C9 Proteins 0.000 description 2
- 102100029358 Cytochrome P450 2C9 Human genes 0.000 description 2
- 102100039205 Cytochrome P450 3A4 Human genes 0.000 description 2
- LCGLNKUTAGEVQW-UHFFFAOYSA-N Dimethyl ether Chemical compound COC LCGLNKUTAGEVQW-UHFFFAOYSA-N 0.000 description 2
- 108050001739 Endothelin receptor Proteins 0.000 description 2
- 102000010180 Endothelin receptor Human genes 0.000 description 2
- 229940118365 Endothelin receptor antagonist Drugs 0.000 description 2
- 102100040611 Endothelin receptor type B Human genes 0.000 description 2
- 102100029110 Endothelin-2 Human genes 0.000 description 2
- 108090000387 Endothelin-2 Proteins 0.000 description 2
- 102100029109 Endothelin-3 Human genes 0.000 description 2
- 108010072844 Endothelin-3 Proteins 0.000 description 2
- OUVXYXNWSVIOSJ-UHFFFAOYSA-N Fluo-4 Chemical compound CC1=CC=C(N(CC(O)=O)CC(O)=O)C(OCCOC=2C(=CC=C(C=2)C2=C3C=C(F)C(=O)C=C3OC3=CC(O)=C(F)C=C32)N(CC(O)=O)CC(O)=O)=C1 OUVXYXNWSVIOSJ-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- 206010019280 Heart failures Diseases 0.000 description 2
- 206010019851 Hepatotoxicity Diseases 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- 208000007920 Neurogenic Inflammation Diseases 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 108010029485 Protein Isoforms Proteins 0.000 description 2
- 102000001708 Protein Isoforms Human genes 0.000 description 2
- 208000001647 Renal Insufficiency Diseases 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 2
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 2
- YRKCREAYFQTBPV-UHFFFAOYSA-N acetylacetone Chemical compound CC(=O)CC(C)=O YRKCREAYFQTBPV-UHFFFAOYSA-N 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 125000002252 acyl group Chemical group 0.000 description 2
- 125000006242 amine protecting group Chemical group 0.000 description 2
- 150000001412 amines Chemical class 0.000 description 2
- 235000012538 ammonium bicarbonate Nutrition 0.000 description 2
- 125000003710 aryl alkyl group Chemical group 0.000 description 2
- 125000005002 aryl methyl group Chemical group 0.000 description 2
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 2
- 230000000903 blocking effect Effects 0.000 description 2
- 125000001589 carboacyl group Chemical group 0.000 description 2
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- JQXXHWHPUNPDRT-YOPQJBRCSA-N chembl1332716 Chemical compound O([C@](C1=O)(C)O\C=C/[C@@H]([C@H]([C@@H](OC(C)=O)[C@H](C)[C@H](O)[C@H](C)[C@@H](O)[C@@H](C)/C=C\C=C(C)/C(=O)NC=2C(O)=C3C(O)=C4C)C)OC)C4=C1C3=C(O)C=2\C=N\N1CCN(C)CC1 JQXXHWHPUNPDRT-YOPQJBRCSA-N 0.000 description 2
- 125000000392 cycloalkenyl group Chemical group 0.000 description 2
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 2
- 230000001086 cytosolic effect Effects 0.000 description 2
- IEJIGPNLZYLLBP-UHFFFAOYSA-N dimethyl carbonate Chemical compound COC(=O)OC IEJIGPNLZYLLBP-UHFFFAOYSA-N 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical compound C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 2
- 230000009977 dual effect Effects 0.000 description 2
- 239000002308 endothelin receptor antagonist Substances 0.000 description 2
- 239000000066 endothelium dependent relaxing factor Substances 0.000 description 2
- 238000011156 evaluation Methods 0.000 description 2
- 239000012526 feed medium Substances 0.000 description 2
- 239000012065 filter cake Substances 0.000 description 2
- 239000007850 fluorescent dye Substances 0.000 description 2
- 230000005714 functional activity Effects 0.000 description 2
- 125000002541 furyl group Chemical group 0.000 description 2
- 238000003304 gavage Methods 0.000 description 2
- 125000001188 haloalkyl group Chemical group 0.000 description 2
- 230000007686 hepatotoxicity Effects 0.000 description 2
- 231100000304 hepatotoxicity Toxicity 0.000 description 2
- 125000004366 heterocycloalkenyl group Chemical group 0.000 description 2
- 229930195733 hydrocarbon Natural products 0.000 description 2
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 2
- 125000002632 imidazolidinyl group Chemical group 0.000 description 2
- 125000001041 indolyl group Chemical group 0.000 description 2
- 239000012442 inert solvent Substances 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- 238000010253 intravenous injection Methods 0.000 description 2
- 150000002500 ions Chemical group 0.000 description 2
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 2
- KQNPFQTWMSNSAP-UHFFFAOYSA-N isobutyric acid Chemical compound CC(C)C(O)=O KQNPFQTWMSNSAP-UHFFFAOYSA-N 0.000 description 2
- 238000006317 isomerization reaction Methods 0.000 description 2
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- 125000000842 isoxazolyl group Chemical group 0.000 description 2
- 201000006370 kidney failure Diseases 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 210000004185 liver Anatomy 0.000 description 2
- 210000001853 liver microsome Anatomy 0.000 description 2
- 210000003712 lysosome Anatomy 0.000 description 2
- 239000011777 magnesium Substances 0.000 description 2
- 230000014759 maintenance of location Effects 0.000 description 2
- 238000004519 manufacturing process Methods 0.000 description 2
- 239000012528 membrane Substances 0.000 description 2
- 125000002757 morpholinyl group Chemical group 0.000 description 2
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 125000001624 naphthyl group Chemical group 0.000 description 2
- 210000000056 organ Anatomy 0.000 description 2
- 125000001181 organosilyl group Chemical group [SiH3]* 0.000 description 2
- 125000002971 oxazolyl group Chemical group 0.000 description 2
- 230000002093 peripheral effect Effects 0.000 description 2
- XNGIFLGASWRNHJ-UHFFFAOYSA-N phthalic acid Chemical compound OC(=O)C1=CC=CC=C1C(O)=O XNGIFLGASWRNHJ-UHFFFAOYSA-N 0.000 description 2
- 125000004193 piperazinyl group Chemical group 0.000 description 2
- DBABZHXKTCFAPX-UHFFFAOYSA-N probenecid Chemical compound CCCN(CCC)S(=O)(=O)C1=CC=C(C(O)=O)C=C1 DBABZHXKTCFAPX-UHFFFAOYSA-N 0.000 description 2
- 229960003081 probenecid Drugs 0.000 description 2
- WGYKZJWCGVVSQN-UHFFFAOYSA-N propylamine Chemical compound CCCN WGYKZJWCGVVSQN-UHFFFAOYSA-N 0.000 description 2
- KAQKFAOMNZTLHT-OZUDYXHBSA-N prostaglandin I2 Chemical compound O1\C(=C/CCCC(O)=O)C[C@@H]2[C@@H](/C=C/[C@@H](O)CCCCC)[C@H](O)C[C@@H]21 KAQKFAOMNZTLHT-OZUDYXHBSA-N 0.000 description 2
- 125000003072 pyrazolidinyl group Chemical group 0.000 description 2
- 125000000714 pyrimidinyl group Chemical group 0.000 description 2
- 125000000168 pyrrolyl group Chemical group 0.000 description 2
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 2
- 230000002285 radioactive effect Effects 0.000 description 2
- 229960001225 rifampicin Drugs 0.000 description 2
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- 238000007086 side reaction Methods 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- 238000010561 standard procedure Methods 0.000 description 2
- TYFQFVWCELRYAO-UHFFFAOYSA-N suberic acid Chemical compound OC(=O)CCCCCCC(O)=O TYFQFVWCELRYAO-UHFFFAOYSA-N 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- XOAAWQZATWQOTB-UHFFFAOYSA-N taurine Chemical compound NCCS(O)(=O)=O XOAAWQZATWQOTB-UHFFFAOYSA-N 0.000 description 2
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 2
- ILMRJRBKQSSXGY-UHFFFAOYSA-N tert-butyl(dimethyl)silicon Chemical compound C[Si](C)C(C)(C)C ILMRJRBKQSSXGY-UHFFFAOYSA-N 0.000 description 2
- 125000001981 tert-butyldimethylsilyl group Chemical group [H]C([H])([H])[Si]([H])(C([H])([H])[H])[*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- VEPTXBCIDSFGBF-UHFFFAOYSA-M tetrabutylazanium;fluoride;trihydrate Chemical compound O.O.O.[F-].CCCC[N+](CCCC)(CCCC)CCCC VEPTXBCIDSFGBF-UHFFFAOYSA-M 0.000 description 2
- 125000001544 thienyl group Chemical group 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 2
- 239000005526 vasoconstrictor agent Substances 0.000 description 2
- 210000003462 vein Anatomy 0.000 description 2
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- DYLIWHYUXAJDOJ-OWOJBTEDSA-N (e)-4-(6-aminopurin-9-yl)but-2-en-1-ol Chemical compound NC1=NC=NC2=C1N=CN2C\C=C\CO DYLIWHYUXAJDOJ-OWOJBTEDSA-N 0.000 description 1
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 1
- AVQQQNCBBIEMEU-UHFFFAOYSA-N 1,1,3,3-tetramethylurea Chemical compound CN(C)C(=O)N(C)C AVQQQNCBBIEMEU-UHFFFAOYSA-N 0.000 description 1
- 125000004502 1,2,3-oxadiazolyl group Chemical group 0.000 description 1
- 125000004511 1,2,3-thiadiazolyl group Chemical group 0.000 description 1
- 125000004504 1,2,4-oxadiazolyl group Chemical group 0.000 description 1
- 125000004514 1,2,4-thiadiazolyl group Chemical group 0.000 description 1
- 125000004506 1,2,5-oxadiazolyl group Chemical group 0.000 description 1
- 125000004517 1,2,5-thiadiazolyl group Chemical group 0.000 description 1
- 125000001781 1,3,4-oxadiazolyl group Chemical group 0.000 description 1
- 125000004520 1,3,4-thiadiazolyl group Chemical group 0.000 description 1
- 125000001637 1-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C(*)=C([H])C([H])=C([H])C2=C1[H] 0.000 description 1
- 125000001462 1-pyrrolyl group Chemical group [*]N1C([H])=C([H])C([H])=C1[H] 0.000 description 1
- WJFKNYWRSNBZNX-UHFFFAOYSA-N 10H-phenothiazine Chemical compound C1=CC=C2NC3=CC=CC=C3SC2=C1 WJFKNYWRSNBZNX-UHFFFAOYSA-N 0.000 description 1
- 125000005955 1H-indazolyl group Chemical group 0.000 description 1
- UTQNKKSJPHTPBS-UHFFFAOYSA-N 2,2,2-trichloroethanone Chemical group ClC(Cl)(Cl)[C]=O UTQNKKSJPHTPBS-UHFFFAOYSA-N 0.000 description 1
- KIPSRYDSZQRPEA-UHFFFAOYSA-N 2,2,2-trifluoroethanamine Chemical compound NCC(F)(F)F KIPSRYDSZQRPEA-UHFFFAOYSA-N 0.000 description 1
- 125000004206 2,2,2-trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 description 1
- VEPOHXYIFQMVHW-XOZOLZJESA-N 2,3-dihydroxybutanedioic acid (2S,3S)-3,4-dimethyl-2-phenylmorpholine Chemical compound OC(C(O)C(O)=O)C(O)=O.C[C@H]1[C@@H](OCCN1C)c1ccccc1 VEPOHXYIFQMVHW-XOZOLZJESA-N 0.000 description 1
- IZOVVFPEDIYGKF-UHFFFAOYSA-N 2-(5-bromopyrimidin-2-yl)oxyethanol Chemical compound OCCOC1=NC=C(Br)C=N1 IZOVVFPEDIYGKF-UHFFFAOYSA-N 0.000 description 1
- WZIMSXIXZTUBSO-UHFFFAOYSA-N 2-[[bis(carboxymethyl)amino]methyl-(carboxymethyl)amino]acetic acid Chemical compound OC(=O)CN(CC(O)=O)CN(CC(O)=O)CC(O)=O WZIMSXIXZTUBSO-UHFFFAOYSA-N 0.000 description 1
- 125000004174 2-benzimidazolyl group Chemical group [H]N1C(*)=NC2=C([H])C([H])=C([H])C([H])=C12 0.000 description 1
- BPGIOCZAQDIBPI-UHFFFAOYSA-N 2-ethoxyethanamine Chemical compound CCOCCN BPGIOCZAQDIBPI-UHFFFAOYSA-N 0.000 description 1
- GRCLBOGXTPXNPL-UHFFFAOYSA-N 2-fluoropyridine-3-carboxamide Chemical compound NC(=O)C1=CC=CN=C1F GRCLBOGXTPXNPL-UHFFFAOYSA-N 0.000 description 1
- 125000002941 2-furyl group Chemical group O1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- ASUDFOJKTJLAIK-UHFFFAOYSA-N 2-methoxyethanamine Chemical compound COCCN ASUDFOJKTJLAIK-UHFFFAOYSA-N 0.000 description 1
- 125000001622 2-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C(*)C([H])=C([H])C2=C1[H] 0.000 description 1
- HPJALMWOZYIZGE-UHFFFAOYSA-N 2-oxa-6-azaspiro[3.3]heptane Chemical compound C1NCC11COC1 HPJALMWOZYIZGE-UHFFFAOYSA-N 0.000 description 1
- 125000000094 2-phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- HMWXCSCBUXKXSA-UHFFFAOYSA-N 2-propoxyethanamine Chemical compound CCCOCCN HMWXCSCBUXKXSA-UHFFFAOYSA-N 0.000 description 1
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 125000000389 2-pyrrolyl group Chemical group [H]N1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- 125000000175 2-thienyl group Chemical group S1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- HGWUUOXXAIISDB-UHFFFAOYSA-N 3-azabicyclo[3.1.0]hexane Chemical compound C1NCC2CC21 HGWUUOXXAIISDB-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- 125000000474 3-butynyl group Chemical group [H]C#CC([H])([H])C([H])([H])* 0.000 description 1
- 125000003682 3-furyl group Chemical group O1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- FAXDZWQIWUSWJH-UHFFFAOYSA-N 3-methoxypropan-1-amine Chemical compound COCCCN FAXDZWQIWUSWJH-UHFFFAOYSA-N 0.000 description 1
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 1
- 125000001397 3-pyrrolyl group Chemical group [H]N1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- 125000001541 3-thienyl group Chemical group S1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- PXACTUVBBMDKRW-UHFFFAOYSA-N 4-bromobenzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=C(Br)C=C1 PXACTUVBBMDKRW-UHFFFAOYSA-N 0.000 description 1
- POILWHVDKZOXJZ-UHFFFAOYSA-N 4-hydroxypent-3-en-2-one Chemical compound CC(O)=CC(C)=O POILWHVDKZOXJZ-UHFFFAOYSA-N 0.000 description 1
- 125000005986 4-piperidonyl group Chemical group 0.000 description 1
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 1
- KDDQRKBRJSGMQE-UHFFFAOYSA-N 4-thiazolyl Chemical group [C]1=CSC=N1 KDDQRKBRJSGMQE-UHFFFAOYSA-N 0.000 description 1
- 125000002471 4H-quinolizinyl group Chemical group C=1(C=CCN2C=CC=CC12)* 0.000 description 1
- CWDWFSXUQODZGW-UHFFFAOYSA-N 5-thiazolyl Chemical group [C]1=CN=CS1 CWDWFSXUQODZGW-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 201000001320 Atherosclerosis Diseases 0.000 description 1
- KHBQMWCZKVMBLN-UHFFFAOYSA-N Benzenesulfonamide Chemical compound NS(=O)(=O)C1=CC=CC=C1 KHBQMWCZKVMBLN-UHFFFAOYSA-N 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 1
- KZBUYRJDOAKODT-UHFFFAOYSA-N Chlorine Chemical compound ClCl KZBUYRJDOAKODT-UHFFFAOYSA-N 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- 101000862089 Clarkia lewisii Glucose-6-phosphate isomerase, cytosolic 1A Proteins 0.000 description 1
- 108010074922 Cytochrome P-450 CYP1A2 Proteins 0.000 description 1
- 108010026925 Cytochrome P-450 CYP2C19 Proteins 0.000 description 1
- 108010001237 Cytochrome P-450 CYP2D6 Proteins 0.000 description 1
- 102100026533 Cytochrome P450 1A2 Human genes 0.000 description 1
- 102100029363 Cytochrome P450 2C19 Human genes 0.000 description 1
- 102100021704 Cytochrome P450 2D6 Human genes 0.000 description 1
- 108090000695 Cytokines Proteins 0.000 description 1
- 102000004127 Cytokines Human genes 0.000 description 1
- AEMOLEFTQBMNLQ-AQKNRBDQSA-N D-glucopyranuronic acid Chemical compound OC1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-AQKNRBDQSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- UFHFLCQGNIYNRP-VVKOMZTBSA-N Dideuterium Chemical compound [2H][2H] UFHFLCQGNIYNRP-VVKOMZTBSA-N 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 206010016654 Fibrosis Diseases 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- 108090000045 G-Protein-Coupled Receptors Proteins 0.000 description 1
- 102000003688 G-Protein-Coupled Receptors Human genes 0.000 description 1
- 230000005526 G1 to G0 transition Effects 0.000 description 1
- IAJILQKETJEXLJ-UHFFFAOYSA-N Galacturonsaeure Natural products O=CC(O)C(O)C(O)C(O)C(O)=O IAJILQKETJEXLJ-UHFFFAOYSA-N 0.000 description 1
- 239000007821 HATU Substances 0.000 description 1
- 239000007995 HEPES buffer Substances 0.000 description 1
- 229910004373 HOAc Inorganic materials 0.000 description 1
- 101000603877 Homo sapiens Nuclear receptor subfamily 1 group I member 2 Proteins 0.000 description 1
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 1
- 206010020751 Hypersensitivity Diseases 0.000 description 1
- 206010020880 Hypertrophy Diseases 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- 239000005909 Kieselgur Substances 0.000 description 1
- 208000019695 Migraine disease Diseases 0.000 description 1
- 229910017906 NH3H2O Inorganic materials 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- PCNDJXKNXGMECE-UHFFFAOYSA-N Phenazine Natural products C1=CC=CC2=NC3=CC=CC=C3N=C21 PCNDJXKNXGMECE-UHFFFAOYSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-L Phosphate ion(2-) Chemical compound OP([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-L 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical group C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- 206010040047 Sepsis Diseases 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- 208000032851 Subarachnoid Hemorrhage Diseases 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- PNDPGZBMCMUPRI-XXSWNUTMSA-N [125I][125I] Chemical compound [125I][125I] PNDPGZBMCMUPRI-XXSWNUTMSA-N 0.000 description 1
- QUWBSOKSBWAQER-UHFFFAOYSA-N [C].O=C=O Chemical compound [C].O=C=O QUWBSOKSBWAQER-UHFFFAOYSA-N 0.000 description 1
- CKUAXEQHGKSLHN-UHFFFAOYSA-N [C].[N] Chemical group [C].[N] CKUAXEQHGKSLHN-UHFFFAOYSA-N 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- XPOLVIIHTDKJRY-UHFFFAOYSA-N acetic acid;methanimidamide Chemical compound NC=N.CC(O)=O XPOLVIIHTDKJRY-UHFFFAOYSA-N 0.000 description 1
- 125000003668 acetyloxy group Chemical group [H]C([H])([H])C(=O)O[*] 0.000 description 1
- 125000000641 acridinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3C=C12)* 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 206010000891 acute myocardial infarction Diseases 0.000 description 1
- 125000004423 acyloxy group Chemical group 0.000 description 1
- 210000004100 adrenal gland Anatomy 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 1
- 125000003282 alkyl amino group Chemical group 0.000 description 1
- 125000004414 alkyl thio group Chemical group 0.000 description 1
- 208000030961 allergic reaction Diseases 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 125000000539 amino acid group Chemical group 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 125000006615 aromatic heterocyclic group Chemical group 0.000 description 1
- 210000001367 artery Anatomy 0.000 description 1
- 125000005101 aryl methoxy carbonyl group Chemical group 0.000 description 1
- 125000005110 aryl thio group Chemical group 0.000 description 1
- 125000004104 aryloxy group Chemical group 0.000 description 1
- 208000006673 asthma Diseases 0.000 description 1
- 238000011914 asymmetric synthesis Methods 0.000 description 1
- 125000002393 azetidinyl group Chemical group 0.000 description 1
- 125000004931 azocinyl group Chemical group N1=C(C=CC=CC=C1)* 0.000 description 1
- 229910052788 barium Inorganic materials 0.000 description 1
- DSAJWYNOEDNPEQ-UHFFFAOYSA-N barium atom Chemical compound [Ba] DSAJWYNOEDNPEQ-UHFFFAOYSA-N 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 125000004604 benzisothiazolyl group Chemical group S1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000004603 benzisoxazolyl group Chemical group O1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 125000003354 benzotriazolyl group Chemical group N1N=NC2=C1C=CC=C2* 0.000 description 1
- 125000004935 benzoxazolinyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 1
- 125000002618 bicyclic heterocycle group Chemical group 0.000 description 1
- 239000004305 biphenyl Substances 0.000 description 1
- 235000010290 biphenyl Nutrition 0.000 description 1
- 125000000319 biphenyl-4-yl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 1
- 230000036772 blood pressure Effects 0.000 description 1
- 210000004204 blood vessel Anatomy 0.000 description 1
- 239000012267 brine Substances 0.000 description 1
- HQABUPZFAYXKJW-UHFFFAOYSA-N butan-1-amine Chemical compound CCCCN HQABUPZFAYXKJW-UHFFFAOYSA-N 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 125000004369 butenyl group Chemical group C(=CCC)* 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- 125000000609 carbazolyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3NC12)* 0.000 description 1
- 229910002092 carbon dioxide Inorganic materials 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical compound OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- 239000007810 chemical reaction solvent Substances 0.000 description 1
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 1
- 239000012069 chiral reagent Substances 0.000 description 1
- QZHPTGXQGDFGEN-UHFFFAOYSA-N chromene Chemical compound C1=CC=C2C=C[CH]OC2=C1 QZHPTGXQGDFGEN-UHFFFAOYSA-N 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 230000007012 clinical effect Effects 0.000 description 1
- 230000008602 contraction Effects 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- KZZKOVLJUKWSKX-UHFFFAOYSA-N cyclobutanamine Chemical compound NC1CCC1 KZZKOVLJUKWSKX-UHFFFAOYSA-N 0.000 description 1
- JVZOVVGVPJVLSL-UHFFFAOYSA-N cyclobutylmethanamine;hydron;chloride Chemical compound Cl.NCC1CCC1 JVZOVVGVPJVLSL-UHFFFAOYSA-N 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- NLUNLVTVUDIHFE-UHFFFAOYSA-N cyclooctylcyclooctane Chemical compound C1CCCCCCC1C1CCCCCCC1 NLUNLVTVUDIHFE-UHFFFAOYSA-N 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- IGSKHXTUVXSOMB-UHFFFAOYSA-N cyclopropylmethanamine Chemical compound NCC1CC1 IGSKHXTUVXSOMB-UHFFFAOYSA-N 0.000 description 1
- 125000004186 cyclopropylmethyl group Chemical group [H]C([H])(*)C1([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000004856 decahydroquinolinyl group Chemical group N1(CCCC2CCCCC12)* 0.000 description 1
- HQWPLXHWEZZGKY-UHFFFAOYSA-N diethylzinc Chemical compound CC[Zn]CC HQWPLXHWEZZGKY-UHFFFAOYSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-M dihydrogenphosphate Chemical compound OP(O)([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-M 0.000 description 1
- NZZFYRREKKOMAT-UHFFFAOYSA-N diiodomethane Chemical compound ICI NZZFYRREKKOMAT-UHFFFAOYSA-N 0.000 description 1
- 125000000532 dioxanyl group Chemical group 0.000 description 1
- ZZVUWRFHKOJYTH-UHFFFAOYSA-N diphenhydramine Chemical group C=1C=CC=CC=1C(OCCN(C)C)C1=CC=CC=C1 ZZVUWRFHKOJYTH-UHFFFAOYSA-N 0.000 description 1
- 125000005982 diphenylmethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])(*)C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 125000005883 dithianyl group Chemical group 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 239000003937 drug carrier Substances 0.000 description 1
- 210000002889 endothelial cell Anatomy 0.000 description 1
- 108010074630 endothelin-3 receptor Proteins 0.000 description 1
- 210000003989 endothelium vascular Anatomy 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 229960001123 epoprostenol Drugs 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- XWBDWHCCBGMXKG-UHFFFAOYSA-N ethanamine;hydron;chloride Chemical compound Cl.CCN XWBDWHCCBGMXKG-UHFFFAOYSA-N 0.000 description 1
- VFRSADQPWYCXDG-LEUCUCNGSA-N ethyl (2s,5s)-5-methylpyrrolidine-2-carboxylate;2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.CCOC(=O)[C@@H]1CC[C@H](C)N1 VFRSADQPWYCXDG-LEUCUCNGSA-N 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 230000005284 excitation Effects 0.000 description 1
- 230000004761 fibrosis Effects 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 201000005206 focal segmental glomerulosclerosis Diseases 0.000 description 1
- 231100000854 focal segmental glomerulosclerosis Toxicity 0.000 description 1
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 125000003838 furazanyl group Chemical group 0.000 description 1
- 229910052732 germanium Inorganic materials 0.000 description 1
- GNPVGFCGXDBREM-UHFFFAOYSA-N germanium atom Chemical compound [Ge] GNPVGFCGXDBREM-UHFFFAOYSA-N 0.000 description 1
- 229960004580 glibenclamide Drugs 0.000 description 1
- 229940097043 glucuronic acid Drugs 0.000 description 1
- ZNNLBTZKUZBEKO-UHFFFAOYSA-N glyburide Chemical compound COC1=CC=C(Cl)C=C1C(=O)NCCC1=CC=C(S(=O)(=O)NC(=O)NC2CCCCC2)C=C1 ZNNLBTZKUZBEKO-UHFFFAOYSA-N 0.000 description 1
- 229910052736 halogen Inorganic materials 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- 150000002367 halogens Chemical class 0.000 description 1
- 210000002216 heart Anatomy 0.000 description 1
- 150000002391 heterocyclic compounds Chemical class 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 102000046617 human NR1I2 Human genes 0.000 description 1
- 150000002431 hydrogen Chemical class 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 229940071870 hydroiodic acid Drugs 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- 125000002636 imidazolinyl group Chemical group 0.000 description 1
- 125000004926 indolenyl group Chemical group 0.000 description 1
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000003406 indolizinyl group Chemical group C=1(C=CN2C=CC=CC12)* 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 229940044173 iodine-125 Drugs 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 125000001977 isobenzofuranyl group Chemical group C=1(OC=C2C=CC=CC12)* 0.000 description 1
- 125000004594 isoindolinyl group Chemical group C1(NCC2=CC=CC=C12)* 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 1
- 125000005956 isoquinolyl group Chemical group 0.000 description 1
- 125000004628 isothiazolidinyl group Chemical group S1N(CCC1)* 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 230000000155 isotopic effect Effects 0.000 description 1
- 125000003965 isoxazolidinyl group Chemical group 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 238000012417 linear regression Methods 0.000 description 1
- 238000004020 luminiscence type Methods 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 239000002207 metabolite Substances 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- OKKJLVBELUTLKV-VMNATFBRSA-N methanol-d1 Chemical compound [2H]OC OKKJLVBELUTLKV-VMNATFBRSA-N 0.000 description 1
- HRDXJKGNWSUIBT-UHFFFAOYSA-N methoxybenzene Chemical group [CH2]OC1=CC=CC=C1 HRDXJKGNWSUIBT-UHFFFAOYSA-N 0.000 description 1
- LSEFCHWGJNHZNT-UHFFFAOYSA-M methyl(triphenyl)phosphanium;bromide Chemical compound [Br-].C=1C=CC=CC=1[P+](C=1C=CC=CC=1)(C)C1=CC=CC=C1 LSEFCHWGJNHZNT-UHFFFAOYSA-M 0.000 description 1
- 206010027599 migraine Diseases 0.000 description 1
- 238000013508 migration Methods 0.000 description 1
- 230000005012 migration Effects 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 125000006682 monohaloalkyl group Chemical group 0.000 description 1
- 125000004572 morpholin-3-yl group Chemical group N1C(COCC1)* 0.000 description 1
- 125000004573 morpholin-4-yl group Chemical group N1(CCOCC1)* 0.000 description 1
- 210000002464 muscle smooth vascular Anatomy 0.000 description 1
- 208000010125 myocardial infarction Diseases 0.000 description 1
- RLKHFSNWQCZBDC-UHFFFAOYSA-N n-(benzenesulfonyl)-n-fluorobenzenesulfonamide Chemical compound C=1C=CC=CC=1S(=O)(=O)N(F)S(=O)(=O)C1=CC=CC=C1 RLKHFSNWQCZBDC-UHFFFAOYSA-N 0.000 description 1
- 125000003136 n-heptyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 210000002569 neuron Anatomy 0.000 description 1
- 239000002858 neurotransmitter agent Substances 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 125000005593 norbornanyl group Chemical group 0.000 description 1
- 125000004930 octahydroisoquinolinyl group Chemical group C1(NCCC2CCCC=C12)* 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 238000003305 oral gavage Methods 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- IPBPLHNLRKRLPJ-UHFFFAOYSA-N oxan-4-ylmethanamine Chemical compound NCC1CCOCC1 IPBPLHNLRKRLPJ-UHFFFAOYSA-N 0.000 description 1
- 125000000160 oxazolidinyl group Chemical group 0.000 description 1
- 125000003551 oxepanyl group Chemical group 0.000 description 1
- 125000003566 oxetanyl group Chemical group 0.000 description 1
- YNOGYQAEJGADFJ-UHFFFAOYSA-N oxolan-2-ylmethanamine Chemical compound NCC1CCCO1 YNOGYQAEJGADFJ-UHFFFAOYSA-N 0.000 description 1
- PIBWKRNGBLPSSY-UHFFFAOYSA-L palladium(II) chloride Chemical compound Cl[Pd]Cl PIBWKRNGBLPSSY-UHFFFAOYSA-L 0.000 description 1
- 125000006340 pentafluoro ethyl group Chemical group FC(F)(F)C(F)(F)* 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 125000004934 phenanthridinyl group Chemical group C1(=CC=CC2=NC=C3C=CC=CC3=C12)* 0.000 description 1
- 125000004625 phenanthrolinyl group Chemical group N1=C(C=CC2=CC=C3C=CC=NC3=C12)* 0.000 description 1
- 229950000688 phenothiazine Drugs 0.000 description 1
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 1
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 1
- 125000004592 phthalazinyl group Chemical group C1(=NN=CC2=CC=CC=C12)* 0.000 description 1
- 230000001766 physiological effect Effects 0.000 description 1
- 125000000587 piperidin-1-yl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000004483 piperidin-3-yl group Chemical group N1CC(CCC1)* 0.000 description 1
- 125000004928 piperidonyl group Chemical group 0.000 description 1
- 229960005235 piperonyl butoxide Drugs 0.000 description 1
- 125000004591 piperonyl group Chemical group C(C1=CC=2OCOC2C=C1)* 0.000 description 1
- 125000006684 polyhaloalkyl group Polymers 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 229910000160 potassium phosphate Inorganic materials 0.000 description 1
- 235000011009 potassium phosphates Nutrition 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 230000007015 preclinical effect Effects 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 108090000765 processed proteins & peptides Proteins 0.000 description 1
- 239000000047 product Substances 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- VVWRJUBEIPHGQF-MDZDMXLPSA-N propan-2-yl (ne)-n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)\N=N\C(=O)OC(C)C VVWRJUBEIPHGQF-MDZDMXLPSA-N 0.000 description 1
- VVWRJUBEIPHGQF-UHFFFAOYSA-N propan-2-yl n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)N=NC(=O)OC(C)C VVWRJUBEIPHGQF-UHFFFAOYSA-N 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 125000001042 pteridinyl group Chemical group N1=C(N=CC2=NC=CN=C12)* 0.000 description 1
- 125000004309 pyranyl group Chemical group O1C(C=CC=C1)* 0.000 description 1
- 125000002755 pyrazolinyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- 125000005344 pyridylmethyl group Chemical group [H]C1=C([H])C([H])=C([H])C(=N1)C([H])([H])* 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000005493 quinolyl group Chemical group 0.000 description 1
- 125000004621 quinuclidinyl group Chemical group N12C(CC(CC1)CC2)* 0.000 description 1
- 230000006340 racemization Effects 0.000 description 1
- 238000007634 remodeling Methods 0.000 description 1
- 229930195734 saturated hydrocarbon Natural products 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- 239000010703 silicon Substances 0.000 description 1
- 238000004088 simulation Methods 0.000 description 1
- 210000000329 smooth muscle myocyte Anatomy 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 1
- 239000012086 standard solution Substances 0.000 description 1
- 125000000446 sulfanediyl group Chemical group *S* 0.000 description 1
- 125000001273 sulfonato group Chemical group [O-]S(*)(=O)=O 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 238000004808 supercritical fluid chromatography Methods 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 229960003080 taurine Drugs 0.000 description 1
- 238000003419 tautomerization reaction Methods 0.000 description 1
- ISIJQEHRDSCQIU-UHFFFAOYSA-N tert-butyl 2,7-diazaspiro[4.5]decane-7-carboxylate Chemical compound C1N(C(=O)OC(C)(C)C)CCCC11CNCC1 ISIJQEHRDSCQIU-UHFFFAOYSA-N 0.000 description 1
- DZLFLBLQUQXARW-UHFFFAOYSA-N tetrabutylammonium Chemical compound CCCC[N+](CCCC)(CCCC)CCCC DZLFLBLQUQXARW-UHFFFAOYSA-N 0.000 description 1
- 125000004192 tetrahydrofuran-2-yl group Chemical group [H]C1([H])OC([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000003039 tetrahydroisoquinolinyl group Chemical group C1(NCCC2=CC=CC=C12)* 0.000 description 1
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 1
- 125000000147 tetrahydroquinolinyl group Chemical group N1(CCCC2=CC=CC=C12)* 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 125000004627 thianthrenyl group Chemical group C1(=CC=CC=2SC3=CC=CC=C3SC12)* 0.000 description 1
- 125000002053 thietanyl group Chemical group 0.000 description 1
- 125000004001 thioalkyl group Chemical group 0.000 description 1
- 238000004448 titration Methods 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- 125000003866 trichloromethyl group Chemical group ClC(Cl)(Cl)* 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 1
- 125000004044 trifluoroacetyl group Chemical group FC(C(=O)*)(F)F 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 125000004417 unsaturated alkyl group Chemical group 0.000 description 1
- 230000002792 vascular Effects 0.000 description 1
- 108010080213 vascular factor Proteins 0.000 description 1
- 230000025102 vascular smooth muscle contraction Effects 0.000 description 1
- 230000024883 vasodilation Effects 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 239000012224 working solution Substances 0.000 description 1
- 125000001834 xanthenyl group Chemical group C1=CC=CC=2OC3=CC=CC=C3C(C12)* 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D407/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00
- C07D407/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/10—Spiro-condensed systems
- C07D491/107—Spiro-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Cardiology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Heart & Thoracic Surgery (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Se describen una serie de compuestos de pirimidina sulfamida y aplicaciones de los mismos en la preparación de un fármaco para una enfermedad relacionada con un antagonista del receptor ETA. En particular, se describe un compuesto derivado representado por la fórmula (I) o un tautómero o una composición farmacéuticamente aceptable del mismo. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Derivado de pirimidinosulfamida y método de preparación y aplicación médica del mismo
Referencia a la solicitud relacionada
La presente solicitud reivindica la siguiente prioridad:
Documento CN201711168111.3, fecha de presentación 21 de noviembre de 2017.
Campo de la invención
La presente divulgación se relaciona con una clase de compuestos derivados de pirimidinosulfamida y su uso en la fabricación de un medicamento para una enfermedad relacionada con el antagonista de receptores ETa. Específicamente, se divulga un compuesto derivado de fórmula (I), uno de sus tautómeros o una de sus composiciones farmacéuticamente aceptables.
Técnica anterior
La endotelina (ET) es una familia de péptidos isómeros que contienen 21 aminoácidos, todos los cuales tienen un extremo C hidrófobo que consiste en 6 residuos de aminoácido idénticos y 2 enlaces disulfuro intracatenarios. Hay tres isoformas de endotelina codificadas por diferentes genes en el cuerpo humano, a saber, ET-1, ET-2 y ET-3. Entre ellas, ET-1 tiene la actividad vasoconstrictora más fuerte, siendo las venas de 3 a 10 veces más altas que las arterias, que es la principal isoforma que causa la enfermedad. ET-1 es el tipo más abundante de la familia de las endotelinas y también tiene la función más importante. Se expresa principalmente en el endotelio vascular y también se distribuye en tejidos no vasculares como el corazón, riñón, pulmón, glándula suprarrenal y otros órganos.
La ET no solo funciona como un factor vascular que modula la presión arterial, sino también como una hormona que induce muchas progresiones (como la proliferación, la apoptosis y la migración) celulares que conducen a la hipertrofia, remodelación, fibrosis e inflamación de los tejidos. Los niveles de ET-1 en plasma y tejido aumentarán en diversas enfermedades tales como hipertensión arterial pulmonar, hipertensión, septicemia, aterosclerosis, infarto agudo de miocardio, insuficiencia cardiaca congestiva, migraña, asma y similares. Por lo tanto, los antagonistas de los receptores de endotelina se investigan ampliamente como agentes terapéuticos con mucho potencial.
Los receptores de endotelina pertenecen a los receptores acoplados a proteína G, que principalmente tienen tres tipos: ETa, ETb y ETc . Tienen diferentes distribuciones en diferentes tejidos y órganos, tienen diferentes afinidades por los tres subtipos de endotelina y tienen una gran diferencia en los efectos fisiológicos. Los receptores ETA de endotelina se distribuyen principalmente en las células del músculo liso y se unen selectivamente a ET-1 y median en la contracción del músculo liso vascular. Los receptores ETb de endotelina se dividen en dos subtipos, a saber, ETb1 y ETb2, donde el primero se distribuye en las células endoteliales y media en la liberación del factor relajante derivado del endotelio (EDRF), la prostaciclina (PGI2) y el óxido nítrico (NO), provocando así vasodilatación, mientras que el segundo se localiza en el músculo liso vascular, el efecto es el mismo que el del receptor ETa para mediar directamente en la contracción de vasos sanguíneos venosos y la afinidad del receptor ETb de endotelina para ET-1, ET-2 y ET-3 es similar. El receptor ETc es un receptor selectivo de ET-3, distribuido principalmente en las células neuronales, y funciona como un neurotransmisor. ET-1 actúa principalmente a través de los receptores ETa y ETb. El antagonista del receptor de la endotelina se puede dividir en el antagonista de receptores ETa, el antagonista de receptores ETb y el antagonista doble de ETa/ETb, de los cuales se han demostrado los efectos preclínicos y/o clínicos en una variedad de enfermedades tales como hemorragia subaracnoidea, insuficiencia cardíaca, hipertensión arterial pulmonar, hipertensión primaria, hipertensión refractaria, inflamación neurogénica, nefropatía diabética, glomeruloesclerosis focal y segmentaria, insuficiencia renal, inflamación neurogénica, vasoespasmo cerebral después de insuficiencia renal e infarto de miocardio y similares. Los antagonistas de receptores ETA altamente selectivos inhiben el fuerte efecto vasoconstrictor de la ET-1, al tiempo que evitan alguna respuesta adversa de los antagonistas dobles de receptores ETa/ETb no selectivos, lo que reduce los efectos secundarios clínicos.
La patente WO02/053557 divulga un compuesto, macitentán, que puede usarse para tratar enfermedades asociadas con la acción de la endotelina.

Contenido de la presente invención
La presente divulgación proporciona un compuesto de fórmula (I), uno de sus isómeros geométricos o estereoisómeros o una de sus sales farmacéuticamente aceptables,

donde
R1 se selecciona de H, F, Cl, Br, I, OH y NH2 ;
R2 se selecciona de H y alquilo C1-3, donde el alquilo C1-3 está opcionalmente sustituido con uno, dos o tres R;
R3 se selecciona de H, alquilo C1-6, heteroalquilo C1-6, -alquil(C1-3)-cicloalquilo(C3-6), cicloalquilo C3-6 y -alquil(C1-3)-heterocicloalquilo(de 3-7 miembros), donde el alquilo C1-6, heteroalquilo C1-6, -alquil(C1-3)-cicloalquilo(C3-6), cicloalquilo C3-6 o -alquil(C1-3)-heterocicloalquilo(de 3-7 miembros) está opcionalmente sustituido con uno, dos o tres R;
o R2 y R3 están conectados para formar un anillo de 3-8 miembros opcionalmente sustituido con uno, dos o tres R; el anillo B se selecciona de heterocicloalquilo de 3-7 miembros y heteroarilo de 5-6 miembros, el heterocicloalquilo de 3-7 miembros o el heteroarilo de 5-6 miembros está opcionalmente sustituido con uno, dos o tres R;
R se selecciona independientemente de H, F, Cl, Br, I, OH, NH2 , CN, alquilo C1-6 y heteroalquilo C1-6, donde el alquilo C1-6 o heteroalquilo C1-6 está opcionalmente sustituido con uno, dos o tres R';
R' se selecciona independientemente de F, Cl, Br, I, OH, NH2 , CN, Me, CH2F, CHF2 , CF3 y Et;
cada uno del heteroalquilo C1-6, heterocicloalquilo de 3-7 miembros y heteroarilo de 5-6 miembros contiene uno, dos, tres o cuatro heteroátomos o grupos de heteroátomos seleccionados independientemente de N, -O-, -S-, -NH-, -S(=O)2- y -S(=O)-.
En algunas realizaciones de la presente divulgación, R se selecciona de H, F, Cl, Br, I, OH, NH2 , CN, alquilo C1-3, alquil(C1-3)-S(=O)2- y alquil(C1-3)-O-, donde el alquilo C1-3, alquil(C1-3)-S(=O)2- o alquil(C1-3)-O- está opcionalmente sustituido con uno, dos o tres R', y otras variables son como se definen en la presente divulgación.
En algunas realizaciones de la presente divulgación, R se selecciona de H, F, Cl, Br, I, OH, NH2, CN, Me, Et,
OII o w
,.S1>
y , , o o está opcionalmente sustituido con uno, dos o tres R', y otras variables son como se definen en la presente divulgación.
En algunas realizaciones de la presente divulgación, R se selecciona de H, F, Cl, Br, I, OH, NH2, CN, Me, CH2F, CHF2,
O
' 1
C F 3 , Et, y O y otras variables son como se definen en la presente divulgación. En algunas realizaciones de la presente divulgación, R2 se selecciona de H y Me, y otras variables son como se definen en la presente divulgación.
En algunas realizaciones de la presente divulgación, R3 se selecciona de H, alquilo C1-4, alquil(C1-4)-O-alquilo(C1-4), ciclobutilo, -alquil(C1-3)-ciclobutilo, -alquil(C1-3)-ciclopropilo, -alquil(C1-3)-tetrahidrofuranilo y -alquil(C1-3)-tetrahidropiranilo, donde el alquilo C1-4, alquil(C1-4)-O-alquilo(C1-4), ciclobutilo, -alquil(C1-3)-ciclobutilo, -alquil(C1-3)-ciclopropilo, -alquil(C1-3)-tetrahidrofuranilo o -alquil(C1-3)-tetrahidropiranilo está opcionalmente sustituido con uno, dos o tres R, y otras variables son como se definen en la presente divulgación.
En algunas realizaciones de la presente divulgación, R3 se selecciona de H, Me, Et,


opcionalmente sustituido con uno, dos o tres R, y otras variables son como se definen en la presente divulgación. En al unas realizaciones de la presente divul ación, R3 se selecciona de H, Me, Et,


divulgación.
En algunas realizaciones de la presente divulgación, R2 y R3 están conectados para formar un heterocicloalquilo de 6 8 miembros opcionalmente sustituido con uno, dos o tres R.
R 2 v n . R 3
En al unas realizaciones de la presente divul ación, la unidad estructural se selecciona de

otras variables son como se definen en la presente divulgación.
FVN' R3
En al unas realizaciones de la presente divulgación, la unidad estructural se selecciona de

En algunas realizaciones de la presente divulgación, el anillo B se selecciona de tetrahidrofuranilo, tetrahidrotienilo, 1,3-dioxolanilo, pirrolidinilo, tiazolilo, pirazolilo e imidazolilo, donde el tetrahidrofuranilo, tetrahidrotienilo, 1,3-dioxolanilo, pirrolidinilo, tiazolilo, pirazolilo o imidazolilo está opcionalmente sustituido con uno, dos o tres R, y otras variables son como se definen en la presente divulgación.
En al unas realizaciones de la presente divulgación, la unidad estructural \ se selecciona de

Se pueden obtener otras realizaciones de la presente divulgación mediante la combinación arbitraria de variables. En algunas realizaciones de la presente divulgación, el compuesto, su geometría o estereoisómero o su sal farmacéuticamente aceptable se selecciona de

donde, R, Ri o R2 es como se define en la presente divulgación.
La presente divulgación también proporciona un compuesto, su isómero geométrico o estereoisómero o su sal farmacéuticamente aceptable, que se selecciona de



En algunas realizaciones de la presente divulgación, el compuesto, su isómero geométrico o estereoisómero o su sal farmacéuticamente aceptable se selecciona de

La divulgación también proporciona una composición farmacéutica, que comprende una cantidad terapéuticamente eficaz del compuesto o su sal farmacéuticamente aceptable como ingrediente activo, y un vehículo farmacéuticamente
aceptable.
La presente divulgación también proporciona un uso del compuesto o sus sales farmacéuticamente aceptables o las composiciones en la fabricación de un medicamento relacionado con antagonistas de receptores ETa .
En algunas realizaciones de la presente divulgación, el medicamento relacionado con el antagonista de receptores ETa es un medicamento para indicaciones tales como hipertensión arterial pulmonar, hipertensión primaria, hipertensión refractaria, nefropatía diabética y vasoespasmo intracraneal.
Definición y descripción
A menos que se indique lo contrario, los siguientes términos, cuando se usan en las descripciones y las reivindicaciones de la presente divulgación, tienen los siguientes significados. Un término o expresión específicos no deben considerarse indefinidos o poco claros en ausencia de una definición particular, sino que debe entenderse en el sentido ordinario. Cuando aparece un nombre comercial en este documento, se pretende hacer referencia a su correspondiente producto o ingrediente activo del mismo. El término "farmacéuticamente aceptable" se usa en este documento en relación con aquellos compuestos, materiales, composiciones y/o formas de dosificación que son adecuados para usar en contacto con tejidos humanos y animales dentro del alcance del juicio médico confiable, sin toxicidad, irritación, reacción alérgica u otros problemas o complicaciones excesivos, de acuerdo con una relación riesgo/beneficio razonable.
El término "sal farmacéuticamente aceptable" se refiere a una sal del compuesto de la presente divulgación que se prepara haciendo reaccionar el compuesto que tiene un sustituyente específico de la presente divulgación con un ácido o base relativamente atóxicos. Cuando el compuesto de la presente divulgación contiene un grupo funcional relativamente ácido, se puede obtener una sal por adición de base poniendo en contacto la forma neutra del compuesto con una cantidad suficiente de base en una solución pura o un disolvente inerte adecuado. La sal por adición de base farmacéuticamente aceptable incluye una sal de sodio, potasio, calcio, amonio, amina orgánica o magnesio o sales similares. Cuando el compuesto de la presente divulgación contiene un grupo funcional relativamente básico, se puede obtener una sal por adición de ácido poniendo en contacto la forma neutra del compuesto con una cantidad suficiente de ácido en una solución pura o un disolvente inerte adecuado. Ejemplos de la sal por adición de ácido farmacéuticamente aceptable incluyen una sal de ácido inorgánico, en la que el ácido inorgánico incluye, por ejemplo, ácido clorhídrico, ácido bromhídrico, ácido nítrico, ácido carbónico, bicarbonato, ácido fosfórico, monohidrogenofosfato, dihidrogenofosfato, ácido sulfúrico, hidrogenosulfato, ácido yodhídrico, ácido fosforoso y similares; y una sal de ácido orgánico, en la que el ácido orgánico incluye, por ejemplo, ácido acético, ácido propiónico, ácido isobutírico, ácido maleico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido láctico, ácido mandélico, ácido ftálico, ácido bencenosulfónico, ácido p-toluenosulfónico, ácido cítrico, ácido tartárico y ácido metanosulfónico, y similares; y una sal de aminoácido (como arginina y similares), y una sal de un ácido orgánico como ácido glucurónico y similares. Ciertos compuestos específicos de la presente divulgación que contienen grupos funcionales tanto básicos como ácidos pueden convertirse en cualquier sal por adición de base o ácido.
La sal farmacéuticamente aceptable de la presente divulgación se puede preparar a partir del compuesto original que contiene un resto ácido o básico mediante un método químico convencional. Generalmente, esta sal se puede preparar haciendo reaccionar la forma de ácido o base libre del compuesto con una cantidad estequiométrica de una base o ácido apropiados en agua o un disolvente orgánico o una de sus mezclas.
El compuesto de la presente divulgación puede tener una forma geométrica o estereoisómera específica. La presente divulgación contempla todos estos compuestos, incluidos el isómero cis y trans, el enantiómero (-) y (+), el enantiómero (R) y (S), el diastereoisómero, el isómero (D), el isómero (L) y la mezcla racémica y otras mezclas, por ejemplo, una mezcla enriquecida en enantiómero o diastereoisómero, todos los cuales están incluidos dentro del alcance de la presente divulgación. El sustituyente, como alquilo, puede tener un átomo de carbono asimétrico adicional. Todos estos isómeros y sus mezclas están incluidos dentro del alcance de la presente divulgación.
A menos que se especifique lo contrario, el término "enantiómero" o "isómero óptico" se refiere a estereoisómeros que son imágenes especulares entre sí.
A menos que se especifique lo contrario, el término "isómero cis-trans" o "isómero geométrico" está provocado por la incapacidad para girar libremente de un enlace doble o un enlace simple de átomos de carbono en el anillo.
A menos que se especifique lo contrario, el término "diastereoisómero" se refiere a estereoisómeros en los que las moléculas tienen dos o más centros quirales y no son imágenes especulares entre sí.
A menos que se especifique lo contrario, "(D)"o "(+)" significa dextrorrotación, "(L)" o "(-)" significa levorrotación, "(DL)" o "(±)" significa racemización.
A menos que se especifique lo contrario, la configuración absoluta de un centro estereogénico está representada por
un enlace sólido en cuña( y un enlace discontinuo en cuña ( X y la configuración relativa de un centro
estereogénico está representada por un enlace sólido lineal K ) V un enlace discontinuo lineal )• Una línea
ondulada r " ) ) representa un enlace sólido en cuña ( ' ) o un enlace discontinuo en cuña ( O representa un
O representa un
enlace sólido lineal ( - O o un enlace discontinuo lineal (*' )•
A menos que se indique lo contrario, los términos "tautómero" o "forma tautómera" se refieren al hecho de que los diferentes isómeros funcionales están en equilibrio dinámico a temperatura ambiente y pueden convertirse rápidamente entre sí. Si los tautómeros son posibles (como en solución), se puede lograr el equilibrio químico de los isómeros. Por ejemplo, los tautómeros protónicos (también conocidos como tautómeros prototrópicos) incluyen interconversiones por transferencia de protones, como la isomerización ceto-enol y la isomerización imina-enamina. El tautómero de valencia incluye la transformación mutua de algunos electrones de enlace. Un ejemplo específico de tautomerización ceto-enol es la interconversión entre dos tautómeros de pentano-2,4-diona y 4-hidroxipent-3-en-2-ona.
A menos que se especifique lo contrario, los términos "enriquecido en un isómero", "enriquecido isómeramente", "enriquecido en un enantiómero" o "enriquecido enantiómeramente" se refieren a que el contenido de uno de los isómeros o enantiómeros es inferior al 100%, y el contenido del isómero o enantiómero es 60% o más, o 70% o más, u 80% o más, o 90% o más, o 95% o más, o 96% o más, o 97% o más, o 98% o más más, o 99% o más, o 99,5% o más, o 99,6% o más, o 99,7% o más, o 99,8% o más, o 99,9% o más.
A menos que se especifique lo contrario, los términos "exceso de isómero" o "exceso de enantiómero" se refieren a la diferencia entre los porcentajes relativos de los dos isómeros o enantiómeros. Por ejemplo, cuando el contenido de uno de los isómeros o enantiómeros es del 90% y el otro es del 10%, entonces el exceso de isómero o enantiómero (valor de ee) es del 80%.
Se puede preparar un isómero (R) y (S) o D y L ópticamente activo usando síntesis quiral o reactivos quirales u otras técnicas convencionales. Si se va a obtener un tipo de enantiómero de cierto compuesto de la presente divulgación, el enantiómero deseado puro se puede obtener mediante síntesis asimétrica o acción derivada del auxiliar quiral seguido de la separación de la mezcla diastereoisómera resultante y la escisión del grupo auxiliar. Alternativamente, cuando la molécula contiene un grupo funcional básico (como amino) o un grupo funcional ácido (como carboxilo). El compuesto reacciona con un ácido o una base ópticamente activos apropiados para formar una sal del isómero diastereoisómero que a continuación se somete a resolución diastereoisómera a través del método convencional en la técnica para dar el enantiómero puro. Además, el enantiómero y el diastereoisómero generalmente se aíslan mediante cromatografía que usa una fase estacionaria quiral y, opcionalmente, se combina con un método derivado químico (como carbamato generado a partir de amina). El compuesto de la presente divulgación puede contener una proporción no natural de isótopo atómico en uno o más de uno de los átomos que constituyen el compuesto. Por ejemplo, el compuesto se puede radiomarcar con un isótopo radiactivo, como tritio (3H), yodo-125 (125I) o C-14 (14C). Para otro ejemplo, el hidrógeno puede ser reemplazado por hidrógeno pesado para formar un fármaco deuterado, y el enlace compuesto por bario y carbono es más fuerte que el enlace compuesto por hidrógeno y carbono comunes. En comparación con los fármacos no deuterados, los fármacos deuterados tienen efectos secundarios reducidos y mayor estabilidad del fármaco, mejoran la eficacia y prolongan la semivida biológica del fármaco. Todas las variaciones isotópicas del compuesto de la presente divulgación, radiactivas o no, están incluidas dentro del alcance de la presente divulgación.
"Opcional" u "opcionalmente" significa que el evento o condición posterior puede producirse pero no es un requisito, que el término incluye el caso en el que el evento o la condición se produce y el caso en el que el evento o la condición no se produce.
El término "sustituido" significa que uno o más átomos de hidrógeno en un átomo específico están sustituidos con el sustituyente, incluidas las variantes deuteradas e hidrogenadas, siempre que la valencia del átomo específico sea normal y el compuesto sustituido sea estable. Cuando el sustituyente es un oxígeno (es decir, =O), significa que se sustituyen dos átomos de hidrógeno. Las posiciones en un anillo aromático no se pueden sustituir con una cetona. El término "opcionalmente sustituido" significa que un átomo puede estar sustituido o no con un sustituyente, a menos que se especifique lo contrario. El tipo y el número del sustituyente pueden ser arbitrarios siempre que se puedan lograr químicamente.
Cuando cualquier variable (como R) aparece en la constitución o estructura del compuesto más de una vez, la definición de la variable en cada aparición es independiente. Así, por ejemplo, si un grupo está sustituido con 0-2 R, el grupo puede estar opcionalmente sustituido con hasta dos R, donde la definición de R en cada aparición es independiente. Por otra parte, solo se permite una combinación del sustituyente y/o su variante cuando la combinación dé como resultado un compuesto estable.
Cuando el número de un grupo de ligazón es 0, como -(CRR)ü-, significa que el grupo de ligazón es un enlace simple.
Cuando una de las variables se selecciona de un enlace simple, significa que los dos grupos ligados por el enlace simple están conectados directamente. Por ejemplo, cuando L en A-L-Z representa un enlace simple, la estructura de A-L-Z es en realidad A-Z.
Cuando un sustituyente está vacante, significa que el sustituyente no existe. Por ejemplo, cuando X está vacante en A-X, la estructura de A-X es en realidad A. Cuando no se indica por qué átomo están unidos los sustituyentes
enumerados al grupo sustituido, dicho sustituyente puede estar enlazado a través de cualquiera de sus átomos, por ejemplo, el grupo piridilo como sustituyente puede estar enlazado al grupo sustituido a través de uno cualquiera de los átomos de carbono del anillo de piridina. Cuando el grupo de ligazón enumerado no indica la dirección de ligazón, la
dirección de ligazón es arbitraria, por ejemplo, el grupo de ligazón L contenido es -MW-,
es -MW-,
entonces -MW- puede ligar el anillo A y el anillo B para formar en la misma dirección que
en la misma dirección que
el orden de lectura de izquierda a derecha, y formar la dirección contraria al orden de lectura de izquierda a derecha. Las combinaciones de los grupos de ligazón, sus sustituyentes y/o variantes solo se permiten si dichas combinaciones dan como resultado compuestos estables.
la dirección contraria al orden de lectura de izquierda a derecha. Las combinaciones de los grupos de ligazón, sus sustituyentes y/o variantes solo se permiten si dichas combinaciones dan como resultado compuestos estables.
A menos que se especifique lo contrario, el término "hetero" representa un heteroátomo o un grupo heteroatómico (por ejemplo, un grupo atómico que contiene un heteroátomo), incluido el átomo excepto el carbono (C) y el hidrógeno (H) y el grupo atómico que contiene el heteroátomo, por ejemplo, incluido oxígeno (O), nitrógeno (N), azufre (S), silicio (Si), germanio (Ge), aluminio (Al), boro (B), -O-, -S-, =O, =S, -C(=O)O-, -C(=O)-, -C(=S)-, -S(=O), -S(=O)2-, y el grupo que consiste en -C(=O)N(H)-, -N(H)-, -C(=NH)-, -S(=O)2N(H)- y -S(=O)N(H)-, cada uno de los cuales está opcionalmente sustituido.
A menos que se especifique lo contrario, el término "anillo" se refiere a un cicloalquilo, heterocicloalquilo, cicloalquenilo, heterocicloalquenilo, cicloalquinilo, heterocicloalquinilo, arilo o heteroarilo sustituido o no sustituido. El llamado anillo incluye un anillo simple, un anillo doble, un anillo en espiral, un anillo condensado o un anillo con puente. El número de átomos en el anillo generalmente se define como el número de miembros del anillo, por ejemplo, un "anillo de 5 a 7 miembros" significa que de 5 a 7 átomos están dispuestos en un anillo. A menos que se especifique lo contrario, el anillo contiene opcionalmente de 1 a 3 heteroátomos. Por lo tanto, un "anillo de 5 a 7 miembros" incluye, por ejemplo, fenilo, piridinilo y piperidinilo; por otra parte, el término "anillo de heterocicloalquilo de 5 a 7 miembros" incluye piridilo y piperidinilo, pero excluye fenilo. El término "anillo" también incluye un sistema de anillos que contiene al menos un anillo, en el que cada anillo cumple independientemente la definición.
A menos que se especifique lo contrario, el término "heterociclo" se refiere a un anillo monocíclico, bicíclico o tricíclico estable que contiene un heteroátomo o un grupo de heteroátomos, que puede ser saturado, parcialmente insaturado o insaturado (aromático) y puede contener átomos de carbono y uno, dos, tres o cuatro heteroátomos de anillo seleccionados independientemente de N, O y S, donde cualquiera de los heterociclos puede estar condensado a un anillo de benceno para formar un anillo bicíclico. Los heteroátomos nitrógeno y azufre opcionalmente pueden estar oxidados (es decir, NO y S(O)p, p es 1 o 2). El átomo de nitrógeno puede estar sustituido o no sustituido (es decir, N o NR, donde R es H u otros sustituyentes ya definidos en este documento). El heterociclo puede estar enlazado al grupo colgante de cualquier heteroátomo o átomo de carbono para formar una estructura estable. Si el compuesto resultante es estable, el heterociclo descrito en este documento puede tener una sustitución en una posición de carbono o nitrógeno. El átomo de nitrógeno en el heterociclo está opcionalmente cuaternizado. En una realización preferida, cuando el número total de átomos de S y O del heterociclo es superior a 1, los heteroátomos no son adyacentes entre sí. En otra realización preferida. El número total de átomos de S y O del heterociclo no es más de 1. Como se usa en el presente documento, el término "grupo heterocíclico aromático" o "heteroarilo" se refiere a un anillo aromático heterocíclico monocíclico o bicíclico de 5, 6 o 7 miembros o bicíclico de 7, 8, 9 o 10 miembros estable que contiene átomos de carbono y uno, dos, tres o cuatro heteroátomos en el anillo seleccionados independientemente de N, O y S. El átomo de nitrógeno puede estar sustituido o no sustituido (es decir, N o NR, donde R es H u otros sustituyentes ya definidos en este documento). Los heteroátomos nitrógeno y azufre pueden opcionalmente estar oxidados (es decir, NO y S(O)p , p es 1 o 2). Vale la pena señalar que el número total de átomos de S y O de un heterociclo aromático no es más de uno. El anillo con puente también se incluye en la definición del heterociclo. Un anillo con puente se forma cuando uno o más de un átomo (es decir, C, O, N o S) liga dos átomos de carbono o nitrógeno no adyacentes. Un anillo con puente preferido incluye, pero no se limita a, un átomo de carbono. Dos átomos de carbono, un átomo de nitrógeno, dos átomos de nitrógeno y un grupo carbono-nitrógeno. Vale la pena señalar que un puente siempre convierte un anillo monocíclico en un anillo tricíclico. En un anillo con puente, el sustituyente en el anillo también puede estar presente en el puente.
Ejemplos del compuesto heterocíclico incluyen, pero no se limitan a: acridinilo, azocinilo, bencimidazolilo, benzofuranilo, benzomercaptofuranilo, benzomercaptofenilo, benzoxazolilo, benzoxazolinilo, benzotiazolilo, benzotriazolilo, benzotetrazolilo, benzoisoxazolilo, benzoisotiazolilo, benzoimidazolinilo, carbazolilo, 4aH-carbazolilo, carbolinilo, cromanilo, cromeno, cinolinilo decahidroquinolinilo, 2H,6H-1,5,2-ditiazinilo, dihidrofuro[2,3-£>]tetrahidrofuranilo, furanilo, furazanilo, imidazolidinilo, imidazolinilo, imidazolilo, 1 H-indazolilo, indolenilo, indolinilo, indolizinilo, indolilo, 3H-indolilo, isobenzofuranilo, isoindolilo, isoindolinilo, isoquinolinilo, isotiazolilo, isoxazolilo, metilendioxifenilo, morfolinilo, naftiridinilo, octahidroisoquinolinilo, oxadiazolilo, 1,2,3-oxadiazolilo, 1,2,4-oxadiazolilo, 1,2,5-oxadiazolilo, 1,3,4-oxadiazolilo, oxazolidinilo, oxazolilo, hidroxindolilo, pirimidinilo, fenantridinilo, fenantrolinilo,
fenazina, fenotiazina, benzoxantinilo, fenoloxazinilo, ftalazinilo, piperazinilo, piperidinilo, piperidonilo, 4-piperidonilo, piperonilo, pteridinilo, purinilo, piranilo, pirazinilo, pirazolidinilo, pirazolinilo, pirazolilo, piridazinilo, piridoxazolilo, piridoimidazolilo, piridotiazolilo, piridinilo, pirrolidinilo, pirrolinilo, 2H-pirrolilo, pirrolilo, quinazolinilo, quinolinilo, 4H-quinolizinilo, quinoxalinilo, quinuclidinilo, tetrahidrofuranilo, tetrahidroisoquinolinilo, tetrahidroquinolinilo, tetrazolilo, 6H-1,2,5-tiadiazinilo, 1,2,3-tiadiazolilo, 1,2,4-tiadiazolilo, 1,2,5-tiadiazolilo, 1,3,4-tiadiazolilo, tiantrenilo, tiazolilo, isotiazoliltienilo, tienoxazolilo , tienotiazolilo, tienoimidazolilo, tienilo, triazinilo, 1H-1,2,3-triazolilo, 2H-1,2,4-triazolilo, 1H-1,2,4-triazolilo, 4H-1,2,4-triazolilo y xantenilo. También se incluyen compuestos de anillos condensados y compuestos tipo espiro.
A menos que se especifique lo contrario, el término "hidrocarbilo" o sus hipónimos (p. ej., alquilo, alquenilo, alquinilo y arilo, etc.), por sí mismo o como parte de otro sustituyente, se refiere a un radical hidrocarbonado lineal, de cadena ramificada o cíclico o cualquiera de sus combinaciones, pueden estar completamente saturados (p. ej., alquilo), mono o poliinsaturados (p. ej., alquenilo, alquinilo y arilo), pueden estar mono, di o polisustituidos, pueden ser monovalentes (p. ej., metilo), divalentes (por ejemplo, metileno) o multivalentes (por ejemplo, metenilo), también pueden incluir un grupo divalente o multivalente, tiener un número específico de átomos de carbono (por ejemplo, C1-C12 indica de 1 a 12 átomos de carbono, C1-12 se selecciona de C1, C2 , C3 , C4 , C5, C6 , C7 , C8 , C9 , C10, C11 y C12; C3-12 se selecciona de C3 , C4 , C5 , C6 , C7 , C8 , C9 , C10, C11 y C12). El término "hidrocarbilo" incluye, pero no se limita a, hidrocarbilo alifático e hidrocarbilo aromático, el hidrocarbilo alifático incluye hidrocarbilo lineal y cíclico, incluye específicamente, pero no se limita a, alquilo, alquenilo y alquinilo. El hidrocarbilo aromático incluye, pero no se limita a, hidrocarbilo aromático de 6-12 miembros como fenilo, naftilo y similares. En algunas realizaciones, el término "hidrocarbilo" se refiere a un grupo lineal o ramificado o una de sus combinaciones que puede estar completamente saturado, mono o poliinsaturado y puede incluir un grupo divalente o multivalente. Ejemplos del grupo hidrocarbilo saturado incluyen, pero no se limitan a, metilo, etilo, n-propilo, isopropilo, n-butilo, terc-butilo, isobutilo, sec-butilo, ciclohexilo, (ciclohexil)metilo, ciclopropilmetilo y el homólogo o isómero de n-amilo, n-hexilo, n-heptilo, n-octilo y otros grupos atómicos. El hidrocarbilo insaturado tiene uno o más de un doble o triple enlace. Ejemplos del alquilo insaturado incluyen, pero no se limitan a, vinilo, 2-propenilo, butenilo, crotilo, 2-isopentenilo, 2-(butadienilo), 2,4-pentadienilo, 3-(1,4-pentadienilo), etinilo, 1- y 3-propinilo, 3-butinilo y más homólogos e isómeros superiores.
A menos que se especifique lo contrario, el término "heterohidrocarbilo" o sus hipónimos (tales como heteroalquilo, heteroalquenilo, heteroalquinilo y heteroarilo, etc.), por sí mismo o como parte de otro sustituyente, se refiere a un grupo hidrocarbonado lineal, ramificado o cíclico estable o cualquiera de sus combinaciones, que tiene un número específico de átomos de carbono y al menos un heteroátomo. En algunas realizaciones, el término "heteroalquilo" por sí mismo o en combinación con otro término se refiere a un radical hidrocarbonado de cadena lineal o ramificado estable o una de sus combinaciones que tiene un número específico de átomos de carbono y al menos un heteroátomo. En una realización específica, un heteroátomo se selecciona de B, O, N y S, donde los átomos de nitrógeno y azufre están opcionalmente oxidados y el átomo de nitrógeno está opcionalmente cuaternizado. El heteroátomo o grupo de heteroátomos puede ubicarse en cualquier posición interior de un heterohidrocarbilo, incluida la posición en la que el hidrocarbilo se une a la parte restante de la molécula. Pero los términos "alcoxi", "alquilamino" y "alquiltio" (o tioalquilo) se utilizan en el sentido convencional y se refieren a un grupo alquilo conectado a la ICtante de la molécula a través de un átomo de oxígeno, un amino o un átomo de azufre, respectivamente. Ejemplos incluyen, pero no se limitan a, -CH2-CH2-O-CH3 , -CH2-CH2-NH-CH3 , -CH2-CH2-N(CH3)-CH3 , -CH2-S-CH2-CH3 , - CH2-CH2 , -S(O)-CH3 , -CH2-CH2-S(O)2-CH3 , -CH=CH-O-CH3 , -CH2-CH=N-OCH3 y -CH=CH-N(CH3)-CH3. Pueden estar presentes hasta dos heteroátomos consecutivos, como -CH2-NH-OCH3.
A menos que se especifique lo contrario, el término "ciclohidrocarbilo", "heterociclohidrocarbilo" o sus hipónimos (tales como arilo, heteroarilo, cicloalquilo, heterocicloalquilo, cicloalquenilo, heterocicloalquenilo, cicloalquinilo, heterocicloalquinilo, etc.) por sí mismos o en combinación con otro término se refiere a "hidrocarbilo” o "heterohidrocarbilo" ciclados. Además, para heterohidrocarbilo o heterociclohidrocarbilo (p. ej., heteroalquilo y heterocicloalquilo), un heteroátomo puede ocupar la posición en la que el heterociclo se une a la posición restante de la molécula. Ejemplos del cicloalquilo incluyen, pero no se limitan a, ciclopentilo, ciclohexilo, 1-ciclohexenilo, 3-ciclohexenilo, cicloheptilo y similares. Ejemplos no limitativos de heterocicloalquilo incluyen 1 -(1,2,5,6-tetrahidropiridilo), 1 -piperidinilo, 2-piperidinilo, 3-piperidinilo, 4-morfolinilo, 3-morfolinilo, tetrahidrofuran-2-ilo, tetrahidrofurano-3-ilo, tetrahidrotiofen-2-ilo, tetrahidrotiofen-3-ilo, 1 -piperazinilo y 2-piperazinilo.
A menos que se especifique lo contrario, el término "heterocicloalquilo" por sí mismo o en combinación con otros términos indica un "heteroalquilo" ciclado y, además, con respecto al "heterocicloalquilo", el heteroátomo puede ocupar la posición de unión del grupo heterocicloalquilo al resto de la molécula. En algunas realizaciones, el heterocicloalquilo es heterocicloalquilo de 4-6 miembros; en otras realizaciones, el heterocicloalquilo es un heterocicloalcano de 5-6 miembros. Ejemplos de grupos heterocicloalquilo incluyen, pero no se limitan a, azetidinilo, oxetanilo, tietanilo, pirrolidinilo, pirazolidinilo, imidazolidinilo, tetrahidrotienilo, tetrahidrofuranilo, tetrahidropiranilo, piperidinilo, piperazinilo, morfolinilo, dioxanilo, ditianilo, isoxazolidinilo, isotiazolidinilo, 1,2-oxazinilo, 1,2-tiazinilo, hexahidropiridazinilo, homopiperazinilo, homopiperidinilo u oxepanilo.
A menos que se especifique lo contrario, el término "alquilo" se refiere a un grupo hidrocarbonado saturado de cadena lineal o ramificado, que puede estar monosustituido (p. ej., -CH2 F) o polisustituido (p. ej., -CF3), puede ser monovalente (por ejemplo, metilo), divalente (por ejemplo, metileno) o multivalente (por ejemplo, metenilo). Ejemplos de alquilo incluyen metilo (Me), etilo (Et), propilo (como n-propilo e isopropilo), butilo (como n-butilo, isobutilo, s-butilo, t-butilo),
pentilo (como n-pentilo, isopentilo, neopentilo) y similares.
A menos que se especifique lo contrario, cicloalquilo incluye cualquier hidrocarbilo cíclico o policíclico estable, y cualquier átomo de carbono está saturado, puede estar monosustituido o polisustituido y puede ser monovalente, divalente o multivalente. Ejemplos de cicloalquilo incluyen, pero no se limitan a, ciclopropilo, norbornanilo, [2.2.2]biciclooctano, [4.4.0]biciclodecanilo y similares.
A menos que se especifique lo contrario, el término "halo" o "halógeno" por sí mismo o como parte de otro sustituyente se refiere a un átomo de flúor, cloro, bromo o yodo. Por otra parte, el término "haloalquilo" pretende incluir monohaloalquilo y polihaloalquilo. Por ejemplo, el término "halo-alquilo(C1-C4)" incluye, pero no se limita a, trifluorometilo, 2,2,2-trifluoroetilo, 4-clorobutilo, 3-bromopropilo y similares. Ejemplos de haloalquilo incluyen, pero no se limitan a, trifluorometilo, triclorometilo, pentafluoroetilo y pentacloroetilo. .
El término "alcoxi" representa cualquier alquilo definido anteriormente que tiene un número específico de átomos de carbono unidos por un puente de oxígeno. A menos que se especifique lo contrario, alcoxi C1-6 incluye alcoxi C1, C2 , C3 , C4 , C5 y C6. Ejemplos de alcoxi incluyen, pero no se limitan a, metoxi, etoxi, n-propoxi, isopropoxi, n-butoxi, sec-butoxi, terc-butoxi, n-pentiloxi y s-pentoxi.
A menos que se especifique lo contrario, el término "arilo" se refiere a un sustituyente aromático poliinsaturado, puede estar mono-, di- o polisustituido, puede ser monovalente, divalente o multivalente, puede ser un solo anillo o un anillo múltiple (por ejemplo, de uno a tres anillos, en los que al menos un anillo es aromático), que están condensados entre sí o conectados covalentemente. El término "heteroarilo" se refiere a un arilo (o anillo) que contiene de uno a cuatro heteroátomos. En un ejemplo ilustrativo, el heteroátomo se selecciona de B, O, N y S, donde los átomos de nitrógeno y azufre están opcionalmente oxidados y el átomo de nitrógeno está opcionalmente cuaternizado. Un heteroarilo puede unirse a la parte restante de una molécula a través de un heteroátomo. Ejemplos no limitativos de arilo o heteroarilo incluyen fenilo, naftilo, bifenilo, pirrolilo, pirazolilo, imidazolilo, pirazinilo, oxazolilo, feniloxazolilo, isoxazolilo, tiazolilo, furanilo, tienilo, piridilo, pirimidinilo, benzotiazolilo, purinilo, bencimidazolilo, indolilo, isoquinolilo, quinoxalinilo, quinolilo, 1 -naftilo, 2-naftilo, 4-bifenilo, 1 -pirrolilo, 2-pirrolilo, 3-pirrolilo, 3-pirazolilo, 2-imidazolilo, 4-imidazolilo, pirazinilo, 2-oxazolilo, 4-oxazolilo, 2-fenil-4-oxazolilo, 5-oxazolilo, 3-isoxazolilo, 4-isoxazolilo, 5-isoxazolilo, 2-tiazolilo, 4-tiazolilo, 5-tiazolilo, 2-furilo, 3-furilo, 2-tienilo, 3-tienilo, 2-piridilo, 3-piridilo, 4-piridilo, 2-pirimidilo, 4-pirimidilo, 5-benzotiazolilo, purinilo, 2-bencimidazolilo, 5-indolilo, 1 -isoquinolilo, 5-isoquinolilo, 2-quinoxalinilo, 5-quinoxalinilo, 3-quinolilo y 6-quinolilo. El sustituyente de cualquiera de los sistemas de anillo arílico y heteroarílico se selecciona del sustituyente aceptable descrito a continuación.
A menos que se especifique lo contrario, cuando el arilo se combina con otros términos (como ariloxi, ariltio, arilalquilo), el arilo incluye el anillo arílico y heteroarílico como se definió anteriormente. Así, el término "aralquilo" pretende incluir el grupo (p. ej., bencilo, fenetilo, piridilmetilo, etc.) en el que un arilo está unido a un alquilo, incluido un alquilo en el que el átomo de carbono (p. ej., metileno) ha sido reemplazado por un átomo tal como oxígeno, por ejemplo, fenoximetilo, 2-piridiloxi, 3-(1-naftiloxi)propilo y similares.
El término "grupo saliente" se refiere a un grupo funcional o átomo que puede ser reemplazado por otro grupo funcional o átomo a través de una reacción de sustitución (tal como una reacción de sustitución por afinidad). Por ejemplo, grupos salientes representativos incluyen triflato; cloro, bromo y yodo; grupo sulfonato, como mesilato, tosilato, p-bromobencenosulfonato, p-toluenosulfonatos y similares; aciloxi, tal como acetoxi, trifluoroacetoxi y similares.
El término "grupo protector" incluye, pero no se limita a, "grupo protector de amino", "grupo protector de hidroxi" o "grupo protector de tio". El término "grupo protector de amino" se refiere a un grupo protector adecuado para bloquear la reacción secundaria en el nitrógeno de un amino. Grupos protectores de amino representativos incluyen, pero no se limitan a: formilo; acilo, tal como alcanoílo (por ejemplo, acetilo, tricloroacetilo o trifluoroacetilo); alcoxicarbonilo, como terc-butoxicarbonilo (Boc); arilmetoxicarbonilo como benciloxicarbonilo (Cbz) y 9-fluorenilmetoxicarbonilo (Fmoc); arilmetilo como bencilo (Bn), tritilo (Tr), 1,1-bis-(4'-metoxifenil)metilo; sililo como trimetilsililo (TMS) y terc-butildimetilsililo (TBS) y similares. El término "grupo protector de hidroxi" se refiere a un grupo protector adecuado para bloquear la reacción secundaria en un hidroxi. Grupos protectores de hidroxi representativos incluyen, pero no se limitan a: alquilo como metilo, etilo y terc-butilo; acilo como alcanoílo (p. ej., acetilo); arilmetilo como bencilo (Bn), p-metoxibencilo (PMB), 9-fluorenilmetilo (Fm) y difenilmetilo (benzhidrilo, DPM); sililo como trimetilsililo (TMS) y terc-butildimetilsililo (TBS) y similares.
El compuesto de la presente divulgación se puede preparar mediante una variedad de métodos sintéticos bien conocidos por los expertos en la técnica, incluida la siguiente realización enumerativa, la realización formada por la siguiente realización enumerativa en combinación con otros métodos de síntesis química y el reemplazo equivalente bien conocido por los expertos en la técnica. La realización preferida incluye, pero no se limita a, la realización de la presente divulgación.
Los compuestos de la presente divulgación pueden tener diversos usos o indicaciones, incluidos, pero no limitados a, los usos o indicaciones específicos enumerados en la presente solicitud.
El disolvente utilizado en la presente divulgación está disponible comercialmente. La presente divulgación emplea las siguientes abreviaturas: ac significa agua; HATU significa 0-(7-azabenzotriazol-1-il)-W,W,W',W-hexafluorofosfato de
tetrametiluronio; EDC significa hicrocloruro de W-(3-dimetilaminopropil)-W-etilcarbodiimida; m-CPBA significa ácido 3-cloroperoxibenzoico; eq significa equivalente; CDI significa carbonildiimidazol; DCM significa diclorometano; PE significa éter de petróleo; DIAD significa azodicarboxilato de diisopropilo; DMF significa W,W-dimetilformamida; DMSO significa dimetilsulfóxido; EtOAc significa ésteres de ácido acético; EtOH significa etanol; MeOH significa metanol; CBz significa benciloxicarbonilo, que es un grupo protector de amina; BOC significa terc-butoxicarbonilo, que es un grupo protector de amina; HOAc significa ácido acético; NaCNBH3 significa cianoborohidruro de sodio; Ta representa la temperatura ambiente; O/N significa durante la noche; THF significa tetrahidrofurano; Boc2O representa dicarbonato de di-terc-butilo; TFA significa ácido trifluoroacético; DIPEA significa diisopropiletilamina; SOCl2 significa cloruro de tionilo; CS2 significa disulfuro de carbono; TsOH significa ácido p-toluenosulfónico; NFSI significa W-fluoro-W-(fenilsulfonil)bencenosulfonamida; NCS significa 1-cloropirrolidina-2,5-diona; n-Bu4NF significa tetrabutilamonio; iPrOH representa 2-propanol; pf significa punto de fusión; LDA significa diisopropilaminolitio; DEA significa dietilamina; ACN significa acetonitrilo.
Los compuestos se nombran manualmente o mediante el programa ChemDraw® , los compuestos disponibles comercialmente utilizan sus nombres del directorio de proveedores.
Efectos técnicos: todos los compuestos de la presente divulgación exhiben una actividad antagonista muy alta contra receptores ETa humanos in vitro, y la selectividad para ETa/ETb es más de 10000 veces; los compuestos de la divulgación son superiores al compuesto de control macitentán en experimentos de caracterización de la inducción mediada por PXR de la expresión de CYP3A. Al caracterizar los experimentos del efecto inhibidor sobre 5 isoenzimas principales del citocromo P450 microsómico hepático humano, los compuestos de la presente divulgación son superiores al macitentán; el efecto inhibidor de los compuestos de la presente divulgación sobre las bombas de exportación de sales biliares es mucho más débil que el macitentán, lo que reduce significativamente el riesgo de desarrollar hepatotoxicidad. Los compuestos de la presente divulgación tienen buenas propiedades farmacocinéticas tanto en ratas SD como en perros beagle.
Descripción detallada de la realización preferida
Los siguientes ejemplos ilustran adicionalmente la presente divulgación, pero la presente divulgación no se limita a ellos. La presente divulgación se ha descrito en detalle en el texto, y también se han divulgado sus realizaciones específicas, para un experto en la técnica, es obvio modificar y mejorar las realizaciones de la presente divulgación dentro del alcance de la presente divulgación.
Realización de referencia 1: fragmento BB-1

Etapa 1: Síntesis del compuesto BB-1-2
A temperatura ambiente, se disolvió el compuesto BB-1-1 (30,00 g, 211,97 mmol, 18,40 ml) en diclorometano (200 ml), a continuación la mezcla se enfrió hasta 0°C, se añadió lentamente gota a gota (el tiempo de goteo fue de aproximadamente 1 hora) una solución de terc-butanol (15,71 g, 211,97 mmol, 20,40 ml) en diclorometano (100 ml) y la mezcla de reacción se calentó hasta temperatura ambiente y se agitó durante 1 hora. El compuesto BB-1-2 buscado (producto bruto) se retuvo en el disolvente de reacción diclorometano y se utilizó directamente en la siguiente reacción.
Etapa 2: Síntesis del compuesto BB-1-3
A temperatura ambiente, se disolvieron el compuesto 2,2,2-trifluoroetilamina (8,00 g, 80,77 mmol, 6,35 ml) y trietilamina (24,52 g, 242,30 mmol, 33,59 ml) en diclorometano (100,00 ml), a continuación la mezcla se enfrió hasta 0°C, y se añadió gota a gota lentamente (el tiempo de goteo fue de aproximadamente 1 hora) una solución del compuesto BB-1-2 (80,77 mmol, producto bruto) en diclorometano, y la mezcla de reacción se calentó hasta temperatura ambiente y se agitó durante 14 horas. Una vez completada la reacción, se eliminó el disolvente a presión reducida, se añadió agua (150 ml) al residuo, se extrajo con diclorometano (100 ml) y se descartó la fase orgánica. La fase acuosa se ajustó hasta un pH de 5-6 con ácido clorhídrico diluido 1 M y se extrajo con acetato de etilo (100 ml x 3). Las fases orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente se eliminó a presión reducida para obtener el compuesto BB-1-3 buscado (sólido blanco, 15,00 g, producto bruto). RMN 1H (400 MHz, DMSO_afe) 5: 3,55 (q, j=9,8 Hz, 2H), 1,37 (s, 9H).
Etapa 3: Síntesis del compuesto BB-1-4
A temperatura ambiente, se añadió el compuesto BB-1-3 (15,00 g, 53,91 mmol) a agua (150,00 ml) y la mezcla de reacción se calentó hasta 110°C y se agitó durante 1 hora. Una vez completada la reacción, la mezcla de reacción se enfrió hasta temperatura ambiente y se extrajo con acetato de etilo (100 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera (100 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida para obtener el compuesto BB-1-4 buscado (sólido amarillo, 7,50 g, producto bruto). RMN 1H (400 MHz, DMSO_afe) 5: 7,51 (t, j=7,0 Hz, 1H), 6,83 (s, 2H), 3,69-3,54 (m, 2H). RMN 19F (400 MHz, DMSO_de) 5: -70,81 (s, 3F).
Etapa 4: Síntesis del compuesto BB-1-5
A temperatura ambiente, se disolviero el compuesto BB-1-4 (1,56 g, 8,78 mmol) y terc-butóxido de potasio (1,97 g, 17,55 mmol) en dimetilsulfóxido (80,00 ml) y la mezcla de reacción se agitó durante 1 hora en atmósfera de nitrógeno a temperatura ambiente. A continuación, se añadió 5-bromo-4,6-dicloropirimidina (2,00 g, 8,78 mmol) a la mezcla de reacción y la mezcla de reacción se agitó adicionalmente a temperatura ambiente durante 10 horas. Una vez completada la reacción, se añadió agua (100 ml), el pH se ajustó hasta 5-6 con ácido clorhídrico diluido 1 M y la mezcla se extrajo con acetato de etilo (50 ml x 3). Las fases orgánicas se combinaron, se lavaron con agua (50 ml x 2), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante cromatografía en columna (eluyente: éter de petróleo/acetato de etilo = 10/1-4/1, relación en volumen) para obtener el compuesto BB-1-5 buscado (sólido amarillo, 1,90 g, rendimiento: 58,56%). RMN 1H (400 MHz, DMSO_afe) 5: 8,60 (s, 1H), 7,51 (t, j=7,0 Hz, 1H), 6,83 (s, 1H), 3,84 (q, j=9,6 Hz, 2H).
Etapa 5: Síntesis del compuesto BB-1
A temperatura ambiente, se añadió terc-butóxido de potasio (1,73 g, 15,42 mmol) a etilenglicol (52,68 g, 848,46 mmol, 47,46 ml) y éter dimetílico de etilenglicol (10 ml) y la mezcla de reacción se calentó hasta 40°C en atmósfera de nitrógeno y se agitó durante 0,5 horas, se añadió una solución de compuesto BB-1-5 (1,90 g, 5,14 mmol) en éter dimetílico de etilenglicol (20 ml) a la solución y la mezcla de reacción se calentó hasta 100°C y se agitó durante 16 horas en atmósfera de nitrógeno. Una vez completada la reacción, la solución de reacción se enfrió hasta temperatura ambiente, se añadió agua (100 ml), el pH se ajustó hasta 5-6 con ácido clorhídrico diluido 2 M, a continuación la mezcla se extrajo con acetato de etilo (60 ml x 3). Las fases orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante cromatografía en columna (eluyente: éter de petróleo/acetato de etilo = 8/1 -3/1, relación en volumen) para obtener el compuesto BB-1 buscado (sólido amarillo, 1,55 g, rendimiento: 76,31%). MS-ESI m/z: 394,7 [M+H]+, 396,7 [M+H+2]+. RMN 1H (400 MHz, CDCL) 5: 8,33 (s, 1H), 6,04 (s, 1H), 4,53 (t, j=4,4 Hz, 2H), 3,93 (t, j=4,4 Hz, 2H), 3,67 (q, j=8,6 Hz, 2H). RMN 19F (400 MHz, CDCh) 5: -71,87 (s, 3F).
Realización de referencia 2: fragmento BB-2

Ruta sintética:

Etapa 1: Síntesis del compuesto BB-2-1
A temperatura ambiente, se añadieron hidrocloruro de etilamina (5,00 g, 61,32 mmol) y trietilamina (18,61 g, 183,96 mmol, 25,49 ml) a diclorometano (100,00 ml), a continuación la mezcla de reacción se enfrió hasta 0°C y se añadió gota a gota lentamente (el tiempo de goteo fue de aproximadamente 1 hora) una solución del compuesto BB-1-2 (61,32 mmol, producto bruto) en diclorometano y la mezcla de reacción se calentó hasta temperatura ambiente y se agitó durante 16 horas. Una vez completada la reacción, se eliminó el disolvente a presión reducida. Se añadió agua (150 ml) al residuo, se extrajo con diclorometano (100 ml) y se descartó la fase orgánica. La fase acuosa se ajustó hasta un pH de 5-6 con ácido clorhídrico diluido 1 M y se extrajo con acetato de etilo (100 ml x 3). Las fases orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, se filtraron y a continuación se eliminó el disolvente a presión reducida para obtener el compuesto BB-2-1 buscado (sólido blanco, 6,00 g, producto bruto). RMN 1H (400 MHz, CDCh) 5: 5,07 (t, j=5,6 Hz, 1H), 3,13-3,01 (m, 2H), 1,43 (s, 9H), 1,16 (t, j=7,3 Hz, 3H).
Etapa 2: Síntesis del compuesto BB-2-2
A temperatura ambiente, se añadió el compuesto BB-2-1 (7,02 g, 31,30 mmol) a agua (200,00 ml) y la mezcla de reacción se calentó hasta 110°C y se agitó durante 1 hora. Una vez completada la reacción, la mezcla de reacción se enfrió hasta temperatura ambiente y se extrajo con acetato de etilo (100 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (50 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida para obtener el compuesto BB-2-2 buscado (aceite amarillo, 2,87 g, producto bruto). RMN 1H (400 MHz, CDCh) 5: 4,80 (s, 2H), 4,57 (s, 1H), 3,23-3,14 (m, 2H), 1,24 (t, j=7,3 Hz, 3H).
Etapa 3: Síntesis del compuesto BB-2-3
A temperatura ambiente, se añadieron el compuesto BB-2-2 (2,87 g, 23,12 mmol) y terc-butóxido de potasio (5,19 g, 46,24 mmol) a dimetilsulfóxido (80,00 ml), a continuación se añadió 5-bromo-4,6-dicloropirimidina (5,27 g, 23,12 mmol) a la mezcla de reacción y la mezcla de reacción se agitó a temperatura ambiente durante 10 horas en atmósfera de nitrógeno. Una vez completada la reacción, se añadió agua (150 ml), el pH se ajustó hasta 5-6 con ácido clorhídrico diluido 1 M y la solución se extrajo con acetato de etilo (100 ml x 3). Las fases orgánicas se combinaron, se lavaron con agua (50 ml x 2), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante cromatografía en columna (eluyente: éter de petróleo/acetato de etilo = 10/1-4/1, relación en volumen) para obtener el compuesto BB-2-3 buscado (sólido amarillo, 2,40 g, rendimiento: 32,89%). RMN 1H (400 MHz, DMSO_afe) 5: 8,59 (s, 1H), 2,96 (q, j=7,1 Hz, 2H), 1,02 (t, j=7,0 Hz, 3H).
Etapa 4: Síntesis del compuesto BB-2
A temperatura ambiente, se añadió terc-butóxido de potasio (1,50 g, 13,41 mmol) a una solución mixta de etilenglicol (33,30 g, 536,49 mmol, 30,00 ml) y éter dimetílico de etilenglicol (10 ml), la mezcla de reacción se calentó hasta 40°C y se agitó durante 0,5 horas en atmósfera de nitrógeno, a continuación se añadió una solución del compuesto BB-2-3 (1,41 g, 4,47 mmol) en éter dimetílico de etilenglicol (20 ml) a la solución en una porción, y la mezcla de reacción se calentó hasta 100°C y se agitó durante 16 horas. Una vez completada la reacción, la mezcla de reacción se enfrió hasta temperatura ambiente, se añadió agua (100 ml), el pH se ajustó hasta 5-6 con ácido clorhídrico diluido 2 M y a continuación la mezcla se extrajo con acetato de etilo (60 ml x 3). Las fases orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante cromatografía en columna (eluyente: éter de petróleo/acetato de etilo = 8/1-3/1, relación en volumen) para obtener el compuesto BB-2 buscado (sólido amarillo, 1,36 g, rendimiento: 87,21%). MS-ESI m/z: 340,7 [M+H]+, 342,7 [M+H+2]+. RMN 1H (400 MHz, CDCh) 5: 8,38 (s, 1H), 7,66 (s, 1H), 5,54 (t, j=5,9 Hz, 1H), 4,60 (t, j=4,8 Hz, 2H), 4,00 (t, j=4,0 Hz, 2H), 3,19-3,03 (m, 2H), 2,45 (s ancho, 1H), 1,21 (t, j=7,2 Hz, 3H).
Realización de referencia 3: fragmento BB-3

Etapa 1: Síntesis del compuesto BB-3-1
A temperatura ambiente, se disolvieron n-propilamina (7,61 g, 128,70 mmol, 10,57 ml) y trietilamina (14,21 g, 140,40 mmol, 19,47 ml) en diclorometano (100,00 ml), a continuación la mezcla se enfrió hasta 0°C, a continuación se añadió lentamente una solución del compuesto BB-1-2 (117,00 mmol, producto bruto) en diclorometano a la solución de reacción (el tiempo de goteo fue de aproximadamente 0,5 horas) y la mezcla de reacción se agitó a temperatura ambiente durante 18 horas en atmósfera de nitrógeno. Una vez completada la reacción, se añadió agua (200 ml) y la mezcla se extrajo con diclorometano (200 ml x 2). Las fases orgánicas se combinaron, se lavaron con ácido clorhídrico diluido 1 M (50 ml) y salmuera saturada (200 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida para obtener el compuesto BB-3-1 buscado (sólido blanco, 21,00 g, rendimiento: 75,32%). RMN 1H (400 MHz, CDCh) 5: 2,93 (t, j=7,0 Hz, 2H), 1,58-1,48 (m, 2H), 1,46-1,37 (s, 9H), 0,88 (t, j=7,4 Hz, 3H).
Etapa 2: Síntesis del compuesto BB-3-2
A temperatura ambiente, se añadió el compuesto BB-3-1 (20,00 g, 83,93 mmol) a agua (100,00 ml) y la mezcla de reacción se calentó hasta 100°C y se agitó durante 1 hora en atmósfera de nitrógeno. Una vez completada la reacción, la solución de reacción se enfrió hasta temperatura ambiente y se extrajo con acetato de etilo (100 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (100 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente se eliminó a presión reducida para obtener el compuesto BB-3-2 buscado (aceite incoloro, 10,00 g, rendimiento: 86,22%). RMN 1H (400 MHz, DMSO_afe) 5: 6,44 (s, 2H), 2,88-2,78 (m, 2H), 1,52-1,43 (m, 2H), 0,87 (t, j=7,5 Hz, 3H).
Etapa 3: Síntesis del compuesto BB-3-3
A temperatura ambiente, se disolvió el compuesto BB-3-2 (18,19 g, 131,66 mmol) en dimetilsulfóxido (300,00 ml), a continuación se añadió terc-butóxido de potasio (19,70 g, 175,54 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 0,5 horas. A continuación, se añadió 5-bromo-4,6-dicloropirimidina (20,00 g, 87,77 mmol) a la solución de reacción y la mezcla de reacción se agitó adicionalmente a temperatura ambiente durante 48 horas. Una vez completada la reacción, se añadió salmuera saturada (1000 ml), el pH se ajustó hasta 4-5 con ácido clorhídrico diluido al 10% y la mezcla se extrajo con acetato de etilo (500 ml x 3). Las fases orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante cromatografía en columna (eluyente: éter de petróleo/acetato de etilo = 10/1-1/1, relación en volumen) para obtener el compuesto BB-3-3 buscado (sólido blanco, 15,00 g, rendimiento: 51,85%). RMN 1H (400 MHz, CDCh) 5: 8,58 (s, 1H), 7,84 (s, 1H), 5,52-5,54 (m, 1 H), 3,07 (q, j=6,8 Hz, 2H), 1,59-1,64 (m, 2H), 0,96 (t, j=7,2 Hz, 3H).
Etapa 4: Síntesis del compuesto BB-3
A temperatura ambiente, se añadió terc-butóxido de potasio (10,21 g, 91,02 mmol) a etilenglicol (56,50 g, 910,19 mmol) y la mezcla de reacción se calentó hasta 40°C y se agitó durante 0,5 horas en atmósfera de nitrógeno. A continuación, se añadió una solución del compuesto BB-3-3 (15,00 g, 45,51 mmol) en éter dimetílico de etilenglicol
(50,00 ml) a la solución y la mezcla de reacción se calentó hasta 100°C y se agitó durante 48 horas en atmósfera de nitrógeno. Una vez completada la reacción, la mezcla se enfrió hasta temperatura ambiente, se añadió agua (200 ml), el pH se ajustó hasta 4 con ácido clorhídrico diluido 1 M y la mezcla se extrajo con acetato de etilo (200 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (200 ml), se secaron sobre sulfato de sodio anhidro y se filtraron, el disolvente del filtrado se eliminó a presión reducida y el residuo se separó mediante cromatografía en columna (eluyente: éter de petróleo/acetato de etilo) = 10/1-1/1, relación de volumen) para obtener el compuesto BB-3 buscado (sólido amarillo, 7,10 g, rendimiento: 40,13%). MS-ESI m/z: 354,8 [M+H]+, 356,8 [M+H+2]+ . RMN 1H (400 MHz, CDCl3) 5: 8,39 (s, 1H), 7,68 (s, 1H), 5,59-5,62 (m, 1H), 4,83-4,75 (m, 2H), 4,02-4,00 (m, 2H), 3,04 (q, j=6,8 Hz, 2H), 2,05 (s ancho, 1 H) 1,63-1,57 (m, 2H), 0,95 (t, j=7,2 Hz, 3H).
Realización de referencia 4: fragmento BB-4

A temperatura ambiente, se disolvieron el compuesto 2-metoxietilamina (2,00 g, 26,63 mmol, 2,33 ml) y trietilamina (5,39 g, 53,26 mmol, 7,38 ml) en diclorometano (100,00 ml) y a continuación la mezcla de reacción se enfrió hasta 0°C, se añadió lentamente una solución del compuesto BB-1-2 (26,63 mmol, producto bruto) en diclorometano a la solución de reacción (el tiempo de goteo fue de aproximadamente 0,5 horas) y la mezcla de reacción se calentó hasta temperatura ambiente y se agitó durante 15 horas. Una vez completada la reacción, se eliminó el disolvente a presión reducida y se añadió agua (100 ml) al residuo, se ajustó el pH hasta 5 con ácido clorhídrico 1 M y se extrajo la mezcla con acetato de etilo (100 ml x 3 ). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (100 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida para obtener el compuesto BB-4-1 buscado (sólido blanco, 6,00 g, rendimiento: 88,59%). RMN 1H (400 MHz, CDCh) 5: 7,37 (s, 1H), 5,50 (s ancho, 1H), 3,53 (t, j=5,0 Hz, 2H), 3,40 (s, 3H), 3,26 (d, j=4,8 Hz, 2H), 1,51 (s, 9H).
Etapa 2: Síntesis del compuesto BB-4-2
A temperatura ambiente, se añadió el compuesto BB-4-1 (6,00 g, 23,59 mmol) a agua (100,00 ml) y la mezcla de reacción se calentó hasta 100°C y se agitó durante 1 hora. Una vez completada la reacción, la mezcla se enfrió hasta temperatura ambiente y se extrajo con acetato de etilo (100 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (100 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida para obtener el compuesto BB-4-2 buscado (sólido amarillo, 2,00 g, rendimiento: 54,99%). RMN 1H (400 MHz, CDCh) 5: 5,52 (s ancho, 2H), 3,58-3,48 (m, 2H), 3,41-3,19 (m, 5H).
Etapa 3: Síntesis del compuesto BB-4-3
A temperatura ambiente, se añadieron el compuesto BB-4-2 (1,12 g, 7,24 mmol) y terc-butóxido de potasio (2,22 g, 19,75 mmol) a dimetilsulfóxido (20,00 ml) y la mezcla de reacción se agitó a temperatura ambiente durante 0,5 horas, a continuación se añadió 5-bromo-4,6-dicloropirimidina (1,50 g, 6,58 mmol) a la solución de reacción y la mezcla de reacción se agitó adicionalmente a temperatura ambiente durante 6 horas. Una vez completada la reacción, se añadió agua (100 ml), el pH se ajustó hasta 6 con ácido clorhídrico diluido 1 M y la mezcla se extrajo con acetato de etilo (100
ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (100 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante cromatografía en columna (eluyente: diclorometano/metanol = 30/1, relación en volumen) para obtener el compuesto BB-4-3 buscado (sólido amarillo, 1,40 g, rendimiento: 61,56%). RMN 1H (400 MHz, CDCL) 5: 8,57 (s, 1H), 7,89 (s ancho, 1 H), 5,99 (s ancho, 1 H), 3,36 (br d, j=2,3 Hz, 2H), 3,32-3,20 (m, 5H).
Etapa 4: Síntesis del compuesto BB-4
A temperatura ambiente, se añadió terc-butóxido de potasio (1,36 g, 12,15 mmol) a etilenglicol (22,20 g, 357,66 mmol, 20,00 ml), la mezcla de reacción se calentó hasta 40°C y se agitó durante 0,5 horas, y a continuación se añadió la solución del compuesto BB-43 (1,40 g, 4,05 mmol) en éter dimetílico de etilenglicol (10,00 ml) a la solución y la mezcla de reacción se calentó hasta 110°C y se agitó durante 12 horas. Una vez completada la reacción, la mezcla se enfrió hasta temperatura ambiente, se añadió agua (50 ml), el pH se ajustó hasta 3 con ácido clorhídrico diluido 1 M y la mezcla se extrajo con acetato de etilo (50 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (50 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante cromatografía en columna (eluyente: cloruro de metileno/metanol = 20/1, relación en volumen) para obtener el compuesto BB-4 buscado (sólido amarillo, 1,20 g, rendimiento: 76,63%). MS-ESI m/z: 370,8 [M+H]+, 372,8 [M+H]+. RMN 1H (400 MHz, CDCL) 5: 8,39 (s, 1H), 7,64 (s ancho, 1H), 6,03-5,94 (m, 1H), 4,65-4,54 (m, 2H), 3,99 (d, j=3,0 Hz, 2H), 3,49 (t, j=5,0 Hz, 2H), 3,33-3,19 (m, 5H), 2,39 (t, j=5,3 Hz, 1H).
Realización de referencia 5: fragmento BB-5

Etapa 1: Síntesis del compuesto BB-5-1
A temperatura ambiente, se disolvieron el compuesto 2-etoxietilamina (5,00 g, 56,09 mmol) y trietilamina (11,35 g, 112,18 mmol, 15,55 ml) en diclorometano (50,00 ml) en atmósfera de nitrógeno, la mezcla se enfrió hasta 0°C y a continuación se añadió gota a gota una solución del compuesto BB-1-2 (56,09 mmol, producto bruto) en diclorometano a la solución de reacción y la mezcla de reacción se agitó a temperatura ambiente durante 12 horas en atmósfera de nitrógeno. Una vez completada la reacción, se añadió agua (80 ml) y la mezcla se extrajo con diclorometano (80 ml x 2). Las fases orgánicas se combinaron, se lavaron con ácido clorhídrico diluido 1 M (50 ml) y salmuera saturada (200 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente se eliminó a presión reducida para obtener el compuesto BB-5-1 buscado (sólido blanco, 11,00 g, rendimiento: 73,09%). RMN 1H (400 MHz, CDCL) 5: 7,41 (s, 1H), 5,43 (t, j=5,7 Hz, 1H), 3,50 (t, j=5,0 Hz, 2H), 3,46-3,40 (m, 2H), 3,19 (q, J=5,5 Hz, 2H), 1,44 (s, 9H), 1,14 (t, j=7,0 Hz, 3H).
Etapa 2: Síntesis del compuesto BB-5-2
A temperatura ambiente, se añadió el compuesto BB-5-1 (10,00 g, 37,27 mmol) a agua (100,00 ml) y la mezcla de reacción se calentó hasta 100°C y se agitó durante 12 horas. Una vez completada la reacción, la mezcla se enfrió hasta temperatura ambiente y se extrajo con acetato de etilo (80 ml x 3). Las fases orgánicas se combinaron, se
lavaron con salmuera saturada (100 ml x 2), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente se eliminó a presión reducida para obtener el compuesto BB-5-2 buscado (sólido blanco, 5,20 g, rendimiento: 82,95%). RMN 1H (400 MHz, CDCh) ó: 5,02 (t, j=5,8 Hz, 1H), 5,00-4,88 (m, 2H), 3,63-3,57 (m, 2H), 3,55 (d, j=7,0 Hz, 2H), 3,33 (d, j=5,0 Hz, 2H), 1,22 (t, j=6,2 Hz, 3H).
Etapa 3: Síntesis del compuesto BB-5-3
A temperatura ambiente, se añadieron el compuesto BB-5-2 (5,00 g, 29,72 mmol) y terc-butóxido de potasio (10,01 g, 89,17 mmol) a dimetilsulfóxido (50,00 ml) y la mezcla de reacción se calentó hasta 35°C y se agitó durante 0,5 horas, a continuación se añadió 5-bromo-4,6-dicloropirimidina (6,77 g, 29,72 mmol) a la solución de reacción y la mezcla de reacción se agitó adicionalmente a 35°C durante 12 horas. Una vez completada la reacción, se añadió ácido clorhídrico (0,5 M, 50 ml) y la mezcla se extrajo con acetato de etilo (50 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (100 ml x 2), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante cromatografía en columna (eluyente: éter de petróleo/acetato de etilo = 10/1-3/1, relación en volumen) para obtener el compuesto BB-5-3 buscado (sólido amarillo claro, 2,10 g, rendimiento: 16,76%). MS-ESI m/z: 358,9 [M+H]+, 360,8 [M+H+2]+. RMN 1H (400 MHz, CDCL) ó: 8,49 (s, 1H), 7,82 (s, 1H), 5,99 (t, J=5,5 Hz, 1H), 3,47-3,43 (m, 2H), 3,34 (d, j=7,0 Hz, 2H), 3,18 (d, j=4,7 Hz, 2H), 1,05 (t, j=6,9 Hz, 3H).
Etapa 4: Síntesis del compuesto BB-5
A temperatura ambiente, se añadió terc-butóxido de potasio (1,87 g, 16,68 mmol) a etilenglicol (33,30 g, 536,49 mmol, 30,00 ml), la mezcla de reacción se calentó hasta 40°C y se agitó durante 0,5 horas en atmósfera de nitrógeno, a continuación se añadió una solución del compuesto BB-5-3 (2,00 g, 5,56 mmol) en éter dimetílico de etilenglicol (20,00 ml) a la solución y la mezcla de reacción se calentó hasta 110°C y se agitó durante 12 horas en atmósfera de nitrógeno. Una vez completada la reacción, la mezcla se enfrió hasta temperatura ambiente, se añadió ácido clorhídrico (0,5 M, 50 ml) y la mezcla se extrajo con acetato de etilo (50 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (100 ml x 2), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante cromatografía en columna (eluyente: éter de petróleo/acetato de etilo = 10/1-1/1, relación en volumen) para obtener el compuesto BB-5 buscado (sólido amarillo claro, 1,30 g, rendimiento: 60,69%). RMN 1H (400 MHz, CDCh) ó: 8,38 (s, 1H), 7,67 (s ancho, 1H), 6,09 (d, j=5,0 Hz, 1H), 4,72-4,52 (m, 2H), 4,00 (s ancho, 2H), 3,62-3,50 (m, 2H), 3,47-3,36 (m, 2H), 3,31-3,20 (m, 2H), 2,46 (s ancho, 1H), 1,21-1,05 (m, 3H).
Realización de referencia 6: fragmento BB-6

A temperatura ambiente, se disolvieron el compuesto 2-n-propoxietilamina (5,00 g, 48,47 mmol) y trietilamina (9,81 g, 96,94 mmol, 13,44 ml) en diclorometano (50,00 ml), la mezcla de reacción se enfrió hasta 0°C en atmósfera de
nitrógeno y a continuación se añadió gota a gota lentamente una solución del compuesto BB-1-2 (48,47 mmol, producto bruto) en diclorometano a la solución de reacción y la mezcla de reacción se calentó hasta temperatura ambiente y se agitó durante 12 horas. Una vez completada la reacción, se añadió agua (80 ml) y la mezcla se extrajo con diclorometano (80 ml x 2). Las fases orgánicas se combinaron, se lavaron con ácido clorhídrico diluido 1 M (50 ml) y salmuera saturada (200 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente se eliminó a presión reducida para obtener el compuesto BB-6-1 buscado (sólido blanco, 11,00 g, rendimiento: 80,37%).
Etapa 2: Síntesis del compuesto BB-6-2
A temperatura ambiente, se añadió el compuesto BB-6-1 (11,00 g, 38,96 mmol) a agua (100,00 ml) y la mezcla de reacción se calentó hasta 100°C y se agitó durante 2 horas. Una vez completada la reacción, la mezcla se enfrió hasta temperatura ambiente y se extrajo con acetato de etilo (80 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (100 ml x 2), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente se eliminó a presión reducida para obtener el compuesto BB-6-2 buscado (sólido blanco, 5,60 g, rendimiento: 78,87%). RMN 1H (400 MHz, CDCta) 5: 5,01-4,96 (m, 1H), 4,90 (s ancho, 2H), 3,58-3,66 (m, 2H), 3,41-3,47 (m, 2H), 3,34 (d, J=4,5 Hz, 2H), 1,55-1,68 (m, 2H), 0,90-0,96 (m, 3H).
Etapa 3: Síntesis del compuesto BB-6-3
A temperatura ambiente, se añadieron el compuesto BB-6-2 (5,00 g, 27,44 mmol) y terc-butóxido de potasio (9,24 g, 82,32 mmol) a dimetilsulfóxido (50,00 ml) y la mezcla de reacción se calentó hasta 35°C y se agitó durante 0,5 horas, a continuación se añadió 5-bromo-4,6-dicloropirimidina (6,25 g, 27,44 mmol) a la solución de reacción y la mezcla de reacción se agitó adicionalmente a 35°C durante 12 horas. Una vez completada la reacción, se añadió ácido clorhídrico (0,5 M, 50 ml) y la mezcla se extrajo con acetato de etilo (50 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (100 ml x 2), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante cromatografía en columna (eluyente: éter de petróleo/acetato de etilo = 10/1 -3/1, relación en volumen) para obtener el compuesto BB-6-3 buscado (sólido amarillo claro, 2,00 g, rendimiento: 18,15%). MS-ESI m/z: 372,8 [M+H]+, 374,8 [M+H+2]+. RMN 1H (400 MHz, CDCh) 5: 8,48 (s, 1 H), 7,77 (s, 1H), 5,96 (t, j=5,6 Hz, 1H), 3,43-3,47 (m, 2H), 3,24 (t, j=6,6 Hz, 2H), 3,18 (d, j=4,7 Hz, 2H), 1,43 (d, j=7,2 Hz, 2H), 0,81 (t, j=7,4 Hz, 3H).
Etapa 4: Síntesis del compuesto BB-6
A temperatura ambiente, se añadió terc-butóxido de potasio (1,80 g, 16,06 mmol) a etilenglicol (33,30 g, 536,49 mmol, 30,00 ml), la mezcla de reacción se calentó hasta 40°C y se agitó durante 0,5 horas en atmósfera de nitrógeno, a continuación se añadió una solución del compuesto BB-6-3 (2,00 g, 5,35 mmol) en éter dimetílico de etilenglicol (20,00 ml) a la solución y la mezcla de reacción se calentó hasta 110°C y se agitó durante 12 horas en atmósfera de nitrógeno. Una vez completada la reacción, la mezcla se enfrió hasta temperatura ambiente, se añadió ácido clorhídrico (0,5 M, 30 ml) y la mezcla se extrajo con acetato de etilo (50 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (100 ml x 2), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante cromatografía en columna (eluyente: éter de petróleo/acetato de etilo = 10/1-1/1, relación en volumen) para obtener el compuesto BB-6 buscado (sólido amarillo claro, 1,20 g, rendimiento: 56,18%). RMN 1H (400 MHz, CDCta) 5: 8,39 (s, 1H), 7,67 (s, 1H), 6,08 (t, j=5,7 Hz, 1H), 4,56-4,65 (m, 2H), 3,97-4,03 (m, 2H), 3,52-3,57 (m, 2H), 3,33 (t, J=6,6 Hz, 2H), 3,24 (q, J=5,5 Hz, 2H), 2,44 (s ancho, 1H), 1,44-1,59 (m, 2H), 0,90 (t, j=7,4Hz, 3H).
Realización de referencia 7: fragmento BB-7

Ruta sintética:

Etapa 1: Síntesis del compuesto BB-7-1
A temperatura ambiente, se disolvieron n-butilamina (2,83 g, 38,64 mmol, 3,82 ml) y trietilamina (3,91 g, 38,64 mmol, 5,36 ml) en diclorometano (100 ml) y la mezcla de reacción se enfrió hasta 0°C, a continuación una solución del compuesto BB-1-2 (46,37 mmol, producto bruto) en diclorometano se añadió gota a gota lentamente a la solución de reacción (el tiempo de goteo fue de aproximadamente 0,5 horas) y la mezcla de reacción se calentó hasta temperatura ambiente y se agitó durante 16 horas. Una vez completada la reacción, se eliminó el disolvente a presión reducida. Se añadió diclorometano (200 ml) al residuo y se lavó con ácido clorhídrico diluido 1 M (80 ml) y agua (100 ml x 2), respectivamente. La fase orgánica se secó sobre sulfato de sodio anhidro, se filtró y el disolvente se eliminó a presión reducida para obtener el compuesto BB-7-1 buscado (sólido blanco, 3,00 g, rendimiento: 30,77%). RMN 1H (400 MHz, CDCl3) 5: 2,98 (q, j=8,0 Hz, 2H), 1,47 (t, j=4,0 Hz 2H), 1,24-1,38 (m, 11 H), 0,86 (t, j=4,0 Hz, 3H).
Etapa 2: Síntesis del compuesto BB-7-2
A temperatura ambiente, se añadió el compuesto BB-7-1 (3,00 g, 11,89 mmol) a agua (150,00 ml) y la mezcla de reacción se calentó hasta 110°C y se agitó durante 0,5 horas. Una vez completada la reacción, la mezcla se enfrió hasta temperatura ambiente y se extrajo con diclorometano (50 ml). La fase orgánica se descartó y la fase acuosa se extrajo con acetato de etilo (100 ml x 3). A continuación, las fases orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente se eliminó a presión reducida para obtener el compuesto BB-7-2 buscado (aceite incoloro, 1,10 g, rendimiento: 60,78%). RMN 1H (400 MHz, CDCh) 5: 3,06 (q, j=8,0 Hz, 2H), 1,46-1,54 (m, 2H), 1,32 (s, 2H), 0,87 (t, j=8,0 Hz, 3H).
Etapa 3: Síntesis del compuesto BB-7-3
A temperatura ambiente, se disolvió el compuesto BB-7-2 (1,10 g, 7,23 mmol) en dimetilsulfóxido (50,00 ml), a continuación se disolvió terc-butóxido de potasio (1,22 g, 10,85 mmol), la mezcla de reacción se agitó a temperatura ambiente durante 0,5 horas en atmósfera de nitrógeno. A continuación, se añadió 5-bromo-4,6-dicloropirimidina (1,98 g, 8,68 mmol) a la solución de reacción y la mezcla de reacción se agitó adicionalmente durante 3 horas a temperatura ambiente bajo protección con nitrógeno. Una vez completada la reacción, se añadió salmuera saturada (50 ml), el pH se ajustó hasta 4-5 con ácido clorhídrico diluido al 10% y la mezcla se extrajo con acetato de etilo (80 ml). Las fases orgánicas se combinaron, se lavaron con agua (50 ml x 2), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante cromatografía en columna (eluyente: éter de petróleo/acetato de etilo = 10/1-1/1, relación en volumen) para obtener el compuesto BB-7-3 buscado (sólido blanco, 350,00 mg, rendimiento: 9,58%). MS-ESI m/z: 342,7 [M+H]+, 344,7 [M+H+2]+ . RMN 1H (400 MHz, CDCh) 5: 8,49 (s, 1H), 7,74 (s, 1H), 5,41 (t, j=6,0 Hz, 1H), 3,00 (q, j=7,2 Hz, 2H), 1,47 (q, j=7,6 Hz, 2H), 1,28 -1,32 (m, 2H), 0,84 (t, j=7,2 Hz, 3H).
Etapa 4: Síntesis del compuesto BB-7
A temperatura ambiente, se añadió terc-butóxido potásico (343,36 mg, 3,06 mmol) a etilenglicol (3,17 g, 51,00 mmol, 2,85 ml), la mezcla de reacción se calentó hasta 40°C y se agitó durante 0,5 horas en atmósfera de nitrógeno, a continuación se añadió una solución del compuesto BB-7-3 (350,00 mg, 1,02 mmol) en éter dimetílico de etilenglicol (20,00 ml) a la solución en una porción, y la mezcla de reacción se calentó hasta 110°C y se agitó durante 15 horas en atmósfera de nitrógeno. Una vez completada la reacción, la mezcla se enfrió hasta temperatura ambiente, se añadió agua helada (50 ml), se ajustó hasta pH 4 con ácido clorhídrico diluido 1 M y se extrajo con acetato de etilo (20 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (50 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante cromatografía preparativa (eluyente: diclorometano/metanol = 20/1, relación en volumen) para obtener el compuesto BB-7 buscado (sólido amarillo, 300,00 mg, rendimiento: 79,66%). RMN 1H (400 MHz, CDCh) 5: 8,30 (s, 1H), 7,54 (s,
1H), 5,44 (t, j=6,0 Hz, 1H), 4,52 (t, j=4,8 Hz, 2H), 3,92 (q, j=3,2 Hz, 2H), 2,98 (q, j=6,8 Hz, 2H), 2,31 (t, j=6,0 Hz, 1 H), 1,45 (q, j=8,0 Hz, 2H), 1,26-1,32 (m, 2H), 0,83 (t, j=7,2 Hz, 3H).
Realización de referencia 8: fragmento BB-8

A temperatura ambiente, se disolvieron ciclobutilamina (5,00 g, 70,30 mmol, 6,02 ml) y trietilamina (8,54 g, 84,36 mmol, 11,70 ml) en diclorometano (100,00 ml) y la mezcla de reacción se enfrió hasta 0°C, a continuación se añadió gota a gota una solución del compuesto BB-1-2 (84,36 mmol, producto bruto) en diclorometano a la solución de reacción (el tiempo de goteo fue de aproximadamente 0,5 horas) y la mezcla de reacción se calentó hasta temperatura ambiente y se agitó durante 15 horas. Una vez completada la reacción, la mezcla se extrajo con agua (100 ml x 3). Las fases acuosas se combinaron, se ajustaron hasta pH 5 con ácido clorhídrico diluido 1 M y se extrajeron con acetato de etilo (100 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (100 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida para obtener el compuesto BB-8-1 buscado (sólido blanco, 12,00 g, rendimiento: 68,19%). RMN 1H (400 MHz, CDCh) 5: 5,35 (d, j=9,8 Hz, 1H), 3,94 3,84 (m, 1H), 3,15 (d, j=7,3 Hz, 1 H), 2,38-2,30 (m, 2H), 2,03-1,90 (m, 2H), 1,77-1,61 (m, 2H), 1,50 (s, 9H).
Etapa 2: Síntesis del compuesto BB-8-2
A temperatura ambiente, se añadió el compuesto BB-8-1 (5,00 g, 19,98 mmol) a agua (100,00 ml) y la mezcla de reacción se calentó hasta 100°C y se agitó durante 1 hora. Una vez completada la reacción, la mezcla se enfrió hasta temperatura ambiente y se extrajo con acetato de etilo (100 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (100 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida para obtener el compuesto BB-8-2 buscado (sólido blanco, 2,90 g, rendimiento: 96,63%). RMN 1H (400 MHz, CDCl3) 5: 4,72-4,48 (m, 2H), 4,07-3,81 (m, 1H), 2,47-2,25 (m, 2H), 2,04-1,90 (m, 2H), 1,83-1,65 (m, 2H).
Etapa 3: Síntesis del compuesto BB-8-3
A temperatura ambiente, se añadieron el compuesto BB-8-2 (2,90 g, 19,31 mmol) y terc-butóxido de potasio (4,33 g, 38,62 mmol) a dimetilsulfóxido (80,00 ml) y la mezcla de reacción se agitó a temperatura ambiente durante 0,5 horas, a continuación se añadió 5-bromo-4,6-dicloropirimidina (3,52 g, 15,45 mmol) a la solución de reacción y la mezcla de reacción se agitó adicionalmente a temperatura ambiente durante 15 horas. Una vez completada la reacción, se añadió agua (150 ml), el pH se ajustó hasta 6 con ácido clorhídrico diluido 1 M y la mezcla se extrajo con acetato de etilo (200 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (200 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante cromatografía en columna (eluyente: éter de petróleo/acetato de etilo = 10/1 -3/1, relación en volumen) para obtener el compuesto BB-8-3 buscado (sólido amarillo, 2,50 g, rendimiento: 37,90%). RMN 1H (400 MHz, CDCh) 5: 8,59 (s, 1H), 7,82 (s, 1H), 5,71 (d, j=8,5 Hz, 1H), 4,10-3,74 (m, 1H), 2,30-2,17 (m, 2H), 1,94-1,79 (m, 2H), 1,74-1,58 (m, 2H). Etapa 4: Síntesis del compuesto BB-8
A temperatura ambiente, se añadió terc-butóxido de potasio (2,46 g, 21,96 mmol) a etilenglicol (22,20 g, 357,66 mmol, 20,00 ml), la mezcla de reacción se calentó hasta 40°C y se agitó durante 0,5 horas en atmósfera de nitrógeno, a
continuación se añadió una solución del compuesto BB-8-3 (2,50 g, 7,32 mmol) en éter dimetílico de etilenglicol (80,00 ml) a la solución y la mezcla de reacción se calentó hasta 110°C y se agitó durante 15 horas en atmósfera de nitrógeno. Una vez completada la reacción, la mezcla se enfrió hasta temperatura ambiente, se añadió agua (200 ml), se ajustó hasta pH 4 con ácido clorhídrico diluido 1 M y se extrajo con acetato de etilo (200 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (200 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante cromatografía en columna (eluyente: éter de petróleo/acetato de etilo = 5/1-1/1, relación en volumen) para obtener el compuesto BB-8 buscado (sólido amarillo, 1,1 g, rendimiento: 40,92%). RMN 1H (400 MHz, CDCh) 5: 8,41 (s, 1H), 7,62 (s, 1H), 5,72 (br d, y=8,8 Hz, 1H), 4,81 -4,42 (m, 2H), 4,03-3,96 (m, 2H), 3,96-3,87 (m, 1H), 2,31 -2,16 (m, 2H), 1,93-1,79 (m, 2H) ), 1,73-1,61 (m, 2H).
Realización de referencia 9: fragmento BB-9

A temperatura ambiente, se disolvieron ciclopropilmetilamina (5,00 g, 70,30 mmol) y trietilamina (14,23 g, 140,60 mmol, 19,49 ml) en diclorometano (100,00 ml), la mezcla de reacción se enfrió hasta 0°C y a continuación se añadió una solución del compuesto BB-1-2 (70,30 mmol, producto bruto) en diclorometano (tiempo de goteo de aproximadamente 0,5 horas), la mezcla de reacción se calentó hasta temperatura ambiente y se agitó durante 15 horas en atmósfera de nitrógeno. Una vez completada la reacción, se eliminó el disolvente a presión reducida y se añadió agua (100 ml) al residuo, se ajustó hasta pH 5 con ácido clorhídrico diluido 1 M y se extrajo con acetato de etilo (100 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (100 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida para obtener el compuesto BB-9-1 buscado (sólido blanco, 11,00 g, rendimiento: 62,51%). RMN 1H (400 MHz, CDCh) 5: 2,94 (dd, y=4,0, 6,8 Hz, 2H), 1,53-1,44 (m, 9H), 1,11-0,94 (m, 1H), 0,64-0,52 (m, 2H), 0,30-0,12 (m, 2H).
Etapa 2: Síntesis del compuesto BB-9-2
A temperatura ambiente, se añadió el compuesto BB-9-1 (10,00 g, 39,95 mmol) a agua (100,00 ml) y la mezcla de reacción se calentó hasta 100°C y se agitó durante 1 hora. Una vez completada la reacción, la mezcla se enfrió hasta temperatura ambiente y se extrajo con acetato de etilo (100 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (100 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida para obtener el compuesto BB-9-2 buscado (sólido blanco, 5,00 g, rendimiento: 83,33%). RMN 1H (400 MHz, CDCh) 5: 4,64-4,54 (m, 2H), 3,64 (s ancho, 1H), 3,03-2,86 (m, 2H), 1,16-0,98 (m, 1H), 0,63-0,42 (m, 2H), 0,29-0,10 (m, 2H).
Etapa 3: Síntesis del compuesto BB-9-3
A temperatura ambiente, se añadieron el compuesto BB-9-2 (4,94 g, 32,91 mmol) y terc-butóxido de potasio (4,92 g, 43,88 mmol) a dimetilsulfóxido (80,00 ml), la mezcla de reacción se agitó a temperatura ambiente durante 0,5 horas en atmósfera de nitrógeno, a continuación se añadió 5-bromo-4,6-dicloropirimidina (5,00 g, 21,94 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 15 horas en atmósfera de nitrógeno. Una vez completada la reacción, se añadió agua (100 ml), el pH se ajustó hasta 6 con ácido clorhídrico diluido 1 M y la mezcla se extrajo con acetato de etilo (200 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (200 ml), se
secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante cromatografía en columna (eluyente: éter de petróleo/acetato de etilo = 10/1-3/1, relación en volumen) para obtener el compuesto BB-9-3 buscado (sólido blanco, 5,00 g, rendimiento: 66,71%). RMN 1H (400 MHz, CDCl3) ó: 8,57 (s, 1H), 7,80 (s ancho, 1 H), 5,63 (t, j=5,4 Hz, 1H), 2,96 (t, j=6,7 Hz, 2H), 1,09-0,86 (m, 1 H), 0,62-0,39 (m, 2H), 0,26-0,03 (m, 2H).
Etapa 4: Síntesis del compuesto BB-9
A temperatura ambiente, se añadió terc-butóxido de potasio (4,93 g, 43,91 mmol) a etilenglicol (22,20 g, 357,66 mmol, 20,00 ml), la mezcla de reacción se calentó hasta 40°C y se agitó durante 0,5 horas, y a continuación se añadió una solución del compuesto BB-9-3 (5,00 g, 14,64 mmol) en éter dimetílico de etilenglicol (80,00 ml) a la mezcla y la mezcla de reacción se calentó hasta 110°C y se agitó durante 15 horas. Una vez completada la reacción, la mezcla se enfrió hasta temperatura ambiente, se añadió agua (200 ml), se ajustó el pH hasta 3 con ácido clorhídrico diluido 1 M y se extrajo con acetato de etilo (200 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (50 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante cromatografía en columna (eluyente: éter de petróleo/acetato de etilo = 5/1-1/1, relación en volumen) para obtener el compuesto BB-9 buscado (aceite amarillo, 3,50 g, rendimiento: 65,10%). RMN 1H (400 MHz, CDCh): 8,45-8,29 (m, 1H), 7,68 (s ancho, 1 H), 5,74 (t, j=5,5 Hz, 1H), 4,73-4,52 (m, 2H), 4,04-3,93 (m, 2H), 2,93 (t, j=6,5 Hz, 2H), 2,04 (s, 1H), 1,11-0,78 (m, 1H), 0,62-0,41 (m, 2H), 0,14 (q, j=5,0 Hz, 2H).
Realización de referencia 10: fragmento BB-10

Etapa 1: Síntesis del compuesto BB-10-1
A 0°C, se añadieron hidrocloruro de ciclobutilmetilamina (5,00 g, 41,12 mmol), trietilamina (10,40 g, 102,80 mmol, 14,25 ml) y diclorometano (50,00 ml) a una solución del compuesto BB-1-2 (41,12 mmol, producto bruto) en diclorometano, y la mezcla de reacción se calentó hasta temperatura ambiente y se agitó durante 18 horas en atmósfera de nitrógeno. Una vez completada la reacción, se añadió agua (60 ml) y la mezcla se extrajo con diclorometano (60 ml x 2). Las fases orgánicas se combinaron, se lavaron con ácido clorhídrico diluido 1 M (50 ml) y salmuera saturada (100 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida para obtener el compuesto BB-10-1 buscado (sólido amarillo, 7,20 g, rendimiento: 66,24%).
Etapa 2: Síntesis del compuesto BB-10-2
A temperatura ambiente, se añadió el compuesto BB-10-1 (7,00 g, 26,48 mmol) a agua (100,00 ml) y la mezcla de reacción se calentó hasta 110°C y se agitó durante 2 horas. Una vez completada la reacción, la mezcla se enfrió hasta temperatura ambiente y se extrajo con acetato de etilo (100 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (200 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida para obtener el compuesto BB-10-2 buscado (aceite incoloro, 3,80 g, rendimiento: 87,38%). RMN 1H (400 MHz, CDCh) 5: 5,10-4,92 (m, 2H), 3,15-3,10 (m, 2H), 2,54-2,50 (m, 1H), 2,07-2,04 (m, 2H), 1,90-1,88 (m, 2H), 1,88-1,69 (m, 2H).
Etapa 3: Síntesis del compuesto BB-10-3
A temperatura ambiente, se añadió terc-butóxido de potasio (3,14 g, 28,00 mmol) a una solución del compuesto BB-10-2 (2,30 g, 14,00 mmol) en dimetilsulfóxido (40,00 ml), la mezcla de reacción se agitó a temperatura ambiente durante 0,5 horas en atmósfera de nitrógeno, a continuación se añadió 5-bromo-4,6-dicloropirimidina (3,19 g, 14,00 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 15 horas en atmósfera de nitrógeno. Una vez completada la reacción, se añadió agua (80 ml), el pH se ajustó hasta 4 con ácido clorhídrico diluido 1 M y la mezcla se extrajo con acetato de etilo (40 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (40 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente se eliminó a presión reducida. El residuo se separó mediante cromatografía en columna (eluyente: éter de petróleo/acetato de etilo = 1/1, relación en volumen) para obtener el compuesto BB-10-3 buscado (sólido amarillo, 3,10 g, rendimiento: 62,29%). RMN 1H (400 MHz, CDCta) 5: 8,49 (s, 1H), 7,79 (s, 1 H), 5,45 (t, j=6,0 Hz, 1H), 3,03-2,98 (m, 2H), 2,53-2,33 (m, 1 H), 2,04-1,98 (m, 2H), 1,84-1,78 (m, 2H), 1,63-1,57 (m, 2H) ).
Etapa 4: Síntesis del compuesto BB-10
A temperatura ambiente, se añadió terc-butóxido de potasio (1,89 g, 16,88 mmol) a etilenglicol (15,79 g, 254,55 mmol, 14,23 ml), la mezcla de reacción se calentó hasta 40°C y se agitó durante 0,5 horas en atmósfera de nitrógeno, a continuación se añadió una solución del compuesto BB-10-3 (3,00 g, 8,44 mmol) en éter dimetílico de etilenglicol (30,00 ml) a la mezcla y la mezcla de reacción se calentó hasta 120°C y se agitó durante 15 horas en atmósfera de nitrógeno. Una vez completada la reacción, se añadió agua (60 ml), el pH se ajustó hasta 4 con ácido clorhídrico diluido 1 M y la mezcla se extrajo con acetato de etilo (50 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (30 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante cromatografía en columna (eluyente: éter de petróleo/acetato de etilo = 1/2, relación en volumen) para obtener el compuesto BB-10 buscado (aceite amarillo, 2,50 g, rendimiento: 77,73%). RMN 1H (400 MHz, CDCh) 5: 8,39 (s, 1H), 7,65 (s ancho, 1H), 5,52 (t, j=6,0 Hz, 1H), 4,71-4,49 (m, 2H), 4,02 (br d, j=3,8 Hz, 2H), 3,12-3,01 (m, 2H), 2,60-2,47 (m, 1 H), 2,43 (s ancho, 1 H), 2,09-2,01 (m, 2H), 1,98-1,77 (m, 2H) , 1,74-1,64 (m, 2H).
Realización de referencia 11: fragmento BB-11

Etapa 1: Síntesis del compuesto BB-11-1
A 0°C, se añadió lentamente gota a gota una solución del compuesto BB-1-2 (78,00 milimoles, producto bruto) en diclorometano a una solución de 3-metoxipropilamina (6,95 g, 78,00 milimoles, 7,99 ml) y trietilamina (15,79 g, 156,00 milimoles, 21,63 ml) en diclorometano (50,00 ml) (el tiempo de goteo fue aproximadamente 0,5 horas) y la mezcla de reacción se calentó hasta temperatura ambiente y se agitó durante 18 horas. Una vez completada la reacción, se añadió agua (200 ml) y la mezcla se extrajo con diclorometano (150 ml x 2). Las fases orgánicas se combinaron, se lavaron con ácido clorhídrico diluido 1 M (50 ml) y salmuera saturada (200 ml), respectivamente, se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida para obtener el compuesto
BB-11-1 buscado (sólido blanco, 16,00 g, rendimiento: 76,45%). RMN 1H (400 MHz, CDCI3) ó: 3,42 (t, j=5,8 Hz, 2H), 3,28 (s, 3H), 3,15-3,04 (m, 2H), 1,91-1,64 (m, 2H), 1,43 (s, 9H).
Etapa 2: Síntesis del compuesto BB-11-2
A temperatura ambiente, se añadió el compuesto BB-11-1 (16,00 g, 59,63 mmol) a agua (100,00 ml) y la mezcla de reacción se calentó hasta 100°C y se agitó durante 1 hora. Una vez completada la reacción, la mezcla se enfrió hasta temperatura ambiente y se extrajo con acetato de etilo (100 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (100 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida para obtener el compuesto BB-11-2 buscado (aceite incoloro, 8,50 g, rendimiento: 84,74%). RMN 1H (400 MHz, DMSO _o6) 5: 6,49-6,38 (m, 3H), 3,37-3,32 (m, 2H), 3,23-3,19 (m, 3H), 2,96-2,82 (m, 2H), 1,73-1,63 (m, 2H).
Etapa 3: Síntesis del compuesto BB-11-3
A temperatura ambiente, se añadió terc-butóxido de potasio (2,67 g, 23,78 mmol) a una solución del compuesto BB-11-2 (2,00 g, 11,89 mmol) en dimetilsulfóxido (10,00 ml), la mezcla de reacción se agitó a temperatura ambiente durante 0,5 horas en atmósfera de nitrógeno, a continuación se añadió 5-bromo-4,6-dicloropirimidina (2,71 g, 11,89 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 15 horas en atmósfera de nitrógeno. Una vez completada la reacción, se añadió agua (60 ml), el pH se ajustó hasta 4 con ácido clorhídrico diluido 0,5 M y la mezcla se extrajo con acetato de etilo (30 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (30 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante cromatografía en columna (eluyente: éter de petróleo/acetato de etilo = 1/1, relación en volumen) para obtener el compuesto BB-11-3 buscado (sólido blanco, 3,30 g, rendimiento: 77,21%). RMN 1H (400 MHz, DMSO _de) 5: 8,59 (s, 1H), 3,29-3,25 (m, 2H), 3,16 (s, 3H), 2,96 (t, j=6,9 Hz, 2H), 1,70-1,62 (m, 2H).
Etapa 4: Síntesis del compuesto BB-11
A temperatura ambiente, se añadió terc-butóxido de potasio (2,06 g, 18,35 mmol) a etilenglicol (30,24 g, 487,25 mmol, 27,25 ml), la mezcla de reacción se calentó hasta 40°C y se agitó durante 0,5 horas en atmósfera de nitrógeno, a continuación se añadió una solución del compuesto BB-11-3 (3,30 g, 9,18 mmol) en éter dimetílico de etilenglicol (10,00 ml) a la mezcla y la mezcla de reacción se calentó hasta 110°C y se agitó durante 24 horas en atmósfera de nitrógeno. Una vez completada la reacción, la mezcla se enfrió hasta temperatura ambiente, se añadió agua (60 ml), se ajustó el pH hasta 4 con ácido clorhídrico diluido 1 M y se extrajo con acetato de etilo (30 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (30 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante cromatografía en columna (eluyente: éter de petróleo/acetato de etilo = 1/2, relación en volumen) para obtener el compuesto BB-11 buscado (aceite amarillo, 2,20 g, rendimiento: 61,34%). RMN 1H (400 MHz, CDCl3) 5: 8,51-8,08 (m, 1H), 7,65 (s, 1H), 6,10 (t, j=5,9 Hz, 1H), 4,67-4,45 (m, 2H), 4,01 (d, j=3,8 Hz, 2H), 3,53-3,39 (m, 2H), 3,34 (s, 3H), 3,26-3,13 (m, 2H), 2,46 (s ancho, 1 H), 1,85 (q, j=6,0 Hz, 2H).
Realización de referencia 12: fragmento BB-12

Etapa 1: Síntesis del compuesto BB-12-1
A 0°C, se añadió lentamente una solución del compuesto BB-1-2 (74,19 mmol, producto bruto) en diclorometano a una solución de 3-etoxipropil-1 -amina (7,65 g, 74,19 mmol, 8,90 ml) y trietilamina (22,52 g, 222,58 mmol, 30,85 ml) en diclorometano (40,00 ml) (el tiempo de goteo fue de aproximadamente 1 hora) y la mezcla de reacción se calentó hasta temperatura ambiente y se agitó durante 14 horas en atmósfera de nitrógeno. Una vez completada la reacción, se eliminó el disolvente a presión reducida y se añadió agua (200 ml) al residuo y se extrajo con diclorometano (100 ml). La fase orgánica se descartó y la fase acuosa se ajustó hasta un pH de 5-6 con ácido clorhídrico diluido 1 M, a continuación la fase acuosa se extrajo con acetato de etilo (100 ml x 3). Las fases orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida para obtener el compuesto BB-12-1 buscado (sólido amarillo, 17,00 g, producto bruto). RMN 1H (400 MHz, DMSO_afe) ó: 10,80 (s, 1H), 7,51 (t, 7=5,8 Hz, 1 H), 3,40-3,37 (m, 2H), 2,93 (q, 7=6,4 Hz, 2H), 2,51 (s, 2H), 1,74-1,61 (m, 2H), 1,43 (s, 9H), 1,10 (t, 7=6,8 Hz, 3H).
Etapa 2: Síntesis del compuesto BB-12-2
A temperatura ambiente, se añadió el compuesto BB-12-1 (17,00 g, 60,21 mmol) a agua (100,00 ml) y la mezcla de reacción se calentó hasta 110°C y se agitó durante 1 hora. Una vez completada la reacción, la mezcla se enfrió hasta temperatura ambiente y se extrajo con acetato de etilo (100 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (100 ml), se secaron sobre sulfato de sodio anhidro y se filtraron, y el disolvente del filtrado se eliminó a presión reducida para obtener el compuesto BB-12-2 buscado (aceite amarillo, 9,00 g, producto bruto). RMN 1H (400 MHz, DMSO _d6) ó: 6,46 (s, 2H), 6,41 (t, 7=6,2 Hz, 1H), 3,43-3,37 (m, 4H), 2,90 (q, 7=6,4 Hz, 2H), 1,75-1,60 (m, 2H), 1,10 (t, 7=7,0 Hz, 3H).
Etapa 3: Síntesis del compuesto BB-12-3
A temperatura ambiente, se añadieron el compuesto BB-12-2 (1,60 g, 8,78 mmol) y terc-butóxido de potasio (1,97 g, 17,55 mmol) a dimetilsulfóxido (20,00 ml), la mezcla de reacción se agitó a temperatura ambiente durante 1 hora en atmósfera de nitrógeno, se añadió 5-bromo-4,6-dicloropirimidina (2,00 g, 8,78 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 11 horas en atmósfera de nitrógeno. Una vez completada la reacción, se añadió agua (100 ml), el pH se ajustó hasta 5-6 con ácido clorhídrico diluido 1 M y la mezcla se extrajo con acetato de etilo (50 ml x 3). Las fases orgánicas se combinaron, se lavaron con agua (50 ml x 2), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante cromatografía en columna (eluyente: éter de petróleo/acetato de etilo = 10/1 -4/1, relación en volumen) para obtener el compuesto BB-12-3 buscado (sólido amarillo, 1,30 g, rendimiento: 39,64%). RMN 1H (400 MHz, Dm So _afe) ó: 8,59 (s, 1 h ), 3,34-3,29 (m, 4H), 2,98 (t, 7=6,8 Hz, 2H), 1,69-1,61 (m, 2H), 1,06 (t, 7=6,8 Hz, 3H).
Etapa 4: Síntesis del compuesto BB-12
A temperatura ambiente, se añadió terc-butóxido de potasio (1,17 g, 10,44 mmol) a una solución mixta de etilenglicol (35,63 g, 574,20 mmol, 32,10 ml) y éter dimetílico de etilenglicol (10,00 ml), la mezcla de reacción se calentó hasta 40°C y se agitó durante 0,5 horas en atmósfera de nitrógeno, a continuación se añadió una solución del compuesto BB-12-3 (1,30 g, 3,48 mmol) en éter dimetílico de etilenglicol (20,00 ml) a la mezcla en una porción, y la mezcla de reacción se calentó hasta 100°C y se agitó durante 15 horas en atmósfera de nitrógeno. Una vez completada la reacción, la mezcla se enfrió hasta temperatura ambiente, se añadió agua (100 ml), se ajustó hasta un pH de 5-6 con ácido clorhídrico diluido 2 M y se extrajo con acetato de etilo (60 ml x 3). Las fases orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante cromatografía en columna (eluyente: éter de petróleo/acetato de etilo = 3/1-1/3, relación en volumen) para obtener el compuesto BB-12 buscado (sólido blanco, 1,10 g, rendimiento: 79,17%). MS-ESI m/z: 398,9 [M+H]+, 400,9 [M+H+2]+. RMN 1H (400 MHz, CDCh) ó: 8,29 (s, 1H), 7,56 (s, 1H), 6,02 (t, 7=6,0 Hz, 1H), 4,52 (t, 7=4,6 Hz, 2H), 3,99-3,87 (m, 2H), 3,46-3,31 (m, 4H), 3,11 (q, 7=6,4 Hz, 2H), 2,38 (t, 7=6,0 Hz, 1H), 1,80-1,71 (m, 2H), 1,14 (t, 7=7,0 Hz, 3H).
Realización de referencia 13: fragmento BB-13

Ruta sintética:

Etapa 1: Síntesis del compuesto BB-13-1
A 0°C, se añadió lentamente una solución del compuesto BB-1-2 (49,43 milimoles, producto bruto) en diclorometano a una solución de 2-tetrahidrofurfurilamina (5,00 g, 49,43 milimoles, 5,10 ml) y trietilamina (10,00 g, 98,86 milimoles, 13,70 ml) en diclorometano (50,00 ml) (el tiempo de goteo fue de aproximadamente 0,5 horas) y la mezcla de reacción se calentó hasta temperatura ambiente y se agitó durante 18 horas en atmósfera de nitrógeno. Una vez completada la reacción, se añadió agua (100 ml) y la mezcla se extrajo con diclorometano (90 ml x 2). Las fases orgánicas se combinaron, se lavaron con ácido clorhídrico diluido 1 M (50 ml) y salmuera saturada (200 ml), respectivamente, se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida para obtener el compuesto BB-13-1 buscado (sólido blanco, 8,30 g, rendimiento: 59,90%). RMN 1H (400 MHz, CDCh) 5: 5,75 (d, j=4,8Hz, 1H), 4,02 (dd, j=3,9, 6,7 Hz, 1 H), 3,88-3,64 (m, 2H), 3,30-2,83 (m, 2H), 2,06-1,76 (m, 3H), 1,71-1,18 (m, 10H).
Etapa 2: Síntesis del compuesto BB-13-2
A temperatura ambiente, se añadió el compuesto BB-13-1 (8,00 g, 28,54 mmol) a agua (100,00 ml) y la mezcla de reacción se calentó hasta 110°C y se agitó durante 2 horas en atmósfera de nitrógeno. Una vez completada la reacción, la mezcla se enfrió hasta temperatura ambiente y se extrajo con acetato de etilo (100 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (200 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida para obtener el compuesto BB-13-2 buscado (aceite incoloro, 4,90 g, rendimiento: 95,27%). RMN 1H (400 MHz, CDCh) 5: 5,18-5,01 (m, 1H), 4,07-3,91 (m, 1H), 3,86-3,61 (m, 2H), 3,27 2,90 (m, 2H), 1,96-1,74 (m, 3H), 1,62-1,41 (m, 1 H).
Etapa 3: Síntesis del compuesto BB-13-3
A temperatura ambiente, se añadió terc-butóxido de potasio (3,86 g, 34,40 mmol) a una solución del compuesto BB-13-2 (3,10 g, 17,20 mmol) en dimetilsulfóxido (20,00 ml), la mezcla de reacción se agitó a temperatura ambiente durante 0,5 horas en atmósfera de nitrógeno, a continuación se añadió 5-bromo-4,6-dicloropirimidina (3,92 g, 17,20 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 15 horas en atmósfera de nitrógeno. Una vez completada la reacción, se añadió agua (60 ml), el pH se ajustó hasta 4 con ácido clorhídrico diluido 1 M y la mezcla se extrajo con acetato de etilo (30 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (30 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante cromatografía en columna (eluyente: éter de petróleo/acetato de etilo = 1/1, relación en volumen) para obtener el compuesto BB-13-3 buscado (sólido amarillo, 2,10 g, rendimiento: 32,85%). RMN 1H (400 MHz, CDCh) 5: 8,65-8,36 (m, 1H), 8,08-7,70 (m, 1H), 5,99-5,80 (m, 1H), 4,08-3,90 (m, 1H), 3,82-3,55 (m, 2H), 3,25 3,13 (m, 1H), 3,04-2,89 (m, 1H), 1,98-1,72 (m, 3H), 1,60-1,44 (m, 1H).
Etapa 4: Síntesis del compuesto BB-13
A temperatura ambiente, se añadió terc-butóxido de potasio (1,27 g, 11,30 mmol) a etilenglicol (10,58 g, 170,40 mmol, 9,53 ml), la mezcla de reacción se calentó hasta 40°C y se agitó durante 0,5 horas en atmósfera de nitrógeno, a continuación se añadió una solución de compuesto BB-13-3 (2,10 g, 5,65 mmol) en éter dimetílico de etilenglicol (30,00 ml) a la mezcla y la mezcla de reacción se calentó hasta 120°C y se agitó durante 15 horas en atmósfera de nitrógeno. Una vez completada la reacción, la mezcla se enfrió hasta temperatura ambiente, se añadió agua (60 ml), se ajustó hasta pH 4 con ácido clorhídrico diluido 1 M y se extrajo con acetato de etilo (50 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (50 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante cromatografía en columna (eluyente: éter de petróleo/acetato de etilo = 1/2, relación en volumen) para obtener el compuesto BB-13 buscado (aceite amarillo, 1,80 g, rendimiento: 78,35%). MS-ESI m/z: 396,8 [M+H]+, 398,8 [M+H+2]+.
Realización de referencia 14: fragmento BB-14

A 0°C, se añadió una solución del compuesto BB-1-2 (43,41 mmol, producto bruto) en diclorometano a una solución de 4-(aminometil)tetrahidro-2H-pirano (5,00 g, 43,41 mmol) y trietilamina (8,79 g, 86,82 mmol, 12,04 ml) en diclorometano (50,00 ml), la mezcla de reacción se calentó hasta temperatura ambiente y se agitó durante 12 horas en atmósfera de nitrógeno. Una vez completada la reacción, se añadió agua (80 ml) y la mezcla se extrajo con diclorometano (80 ml x 2). Las fases orgánicas se combinaron, se lavaron con ácido clorhídrico diluido 1 M (50 ml) y salmuera saturada (200 ml), respectivamente, se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida para obtener el compuesto BB-14-1 buscado (aceite blanquecino, 5,20 g, rendimiento: 40,69%).
Etapa 2: Síntesis del compuesto BB-14-2
A temperatura ambiente, se añadió el compuesto BB-14-1 (5,00 g, 16,99 mmol) a agua (100,00 ml) y la mezcla de reacción se calentó hasta 100°C y se agitó durante 12 horas. Una vez completada la reacción, la mezcla se enfrió hasta temperatura ambiente y se extrajo con acetato de etilo (80 ml x 2). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (100 ml x 2), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida para obtener el compuesto BB-14-2 buscado (aceite amarillo claro, 1,70 g, rendimiento: 51,51%).
Etapa 3: Síntesis del compuesto BB-14-3
A 35°C, una mezcla del compuesto BB-14-2 (1,70 g, 8,75 mmol) y terc-butóxido de potasio (2,95 g, 26,25 mmol) en dimetilsulfóxido (50,00 ml) se agitó durante 0,5 horas, a continuación se añadió 5-bromo-4,6-dicloropirimidina (1,99 g, 8,75 mmol), la mezcla de reacción se agitó a 35°C durante 12 horas. Una vez completada la reacción, se añadió a la mezcla ácido clorhídrico diluido 0,5 M (50 ml) y se extrajo con acetato de etilo (50 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (100 ml x 2), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante cromatografía en columna (eluyente: éter de petróleo/acetato de etilo = 10/1-1/1, relación en volumen) para obtener el compuesto BB-14-3 buscado (sólido amarillo, 1,20 g, rendimiento: 12,80%). MS-ESI m/z: 384,8 [M+H]+, 386,8 [M+H+2]+.
Etapa 4: Síntesis del compuesto BB-14
A temperatura ambiente, se añadió terc-butóxido de potasio (1,05 g, 9,33 mmol) a etilenglicol (33,30 g, 536,49 mmol, 30,00 ml), la mezcla de reacción se calentó hasta 40°C y se agitó durante 0,5 horas en atmósfera de nitrógeno, a continuación se añadió una solución del compuesto BB-14-3 (1,20 g, 3,11 mmol) en éter dimetílico de etilenglicol (20,00 ml) a la mezcla y la mezcla de reacción se calentó hasta 110°C y se agitó durante 12 horas en atmósfera de
nitrógeno. Una vez completada la reacción, la mezcla se enfrió hasta temperatura ambiente, se añadió ácido clorhídrico diluido 0,5 M (30 ml) y se extrajo con acetato de etilo (50 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (100 ml x 2), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante cromatografía en columna (eluyente: éter de petróleo/acetato de etilo = 10/1-0/1, relación en volumen) para obtener el compuesto BB-14 buscado (sólido amarillo claro, 200,00 mg, rendimiento: 14,59%). MS-ESI m/z: 410,9 [M+H]+, 412,9 [M+H+2]+. RMN 1H (400 MHz, CDCh) 5: 8,38 (s, 1H), 7,62 (s ancho, 1H), 5,65 (t, j=6,5 Hz, 1H), 4,60 (t, j=4,8 Hz, 2H), 4,05-3,89 (m, 4H), 3,38 (t, j=10,8 Hz, 2H), 2,93 (t, j=6,5 Hz, 2H), 1,86-1,75 (m, 1H), 1,71 -1,61 (m, 2H), 1,31 -1,22 (m, 2H).
Realización de referencia 15: fragmento BB-15

Etapa 1: Síntesis del compuesto BB-15-1
A 0°C, se añadió lentamente gota a gota una solución del compuesto BB-1-2 (31,32 milimoles, producto bruto) en diclorometano a una solución mixta de hidrocloruro de 2-aminoetilmetilsulfona (5,00 g, 31,32 milimoles) y trietilamina (6,34 g, 62,64 milimoles, 8,68 ml) en diclorometano (50,00 ml) (el tiempo de goteo fue de aproximadamente 0,5 horas) y la mezcla de reacción se calentó hasta temperatura ambiente y se agitó durante 18 horas en atmósfera de nitrógeno. Una vez completada la reacción, se añadió agua (100 ml) y la mezcla se extrajo con diclorometano (100 ml x 2). Las fases orgánicas se combinaron, se lavaron con ácido clorhídrico diluido 1 M (50 ml) y salmuera saturada (80 ml), respectivamente, se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida para obtener el compuesto BB-15-1 buscado (sólido blanco 5,00 g, rendimiento: 52,81%). RMN 1H (400 MHz, DMSO_afe) 5: 11,04 (s, 1H), 7,83 (s ancho, 1 H), 3,44 (s ancho, 2H), 3,35-3,30 (m, 2H), 3,02 (s, 3H), 1,44 (s, 9H).
Etapa 2: Síntesis del compuesto BB-15-2
A temperatura ambiente, se añadió el compuesto BB-15-1 (4,80 g, 15,87 mmol) a agua (100,00 ml) y la mezcla de reacción se calentó hasta 110°C y se agitó durante 2 horas en atmósfera de nitrógeno. Una vez completada la reacción, la mezcla se enfrió hasta temperatura ambiente, se extrajo con acetato de etilo (100 ml), la fase orgánica se descartó y la fase acuosa se concentró a presión reducida para obtener el compuesto BB-15-2 buscado (aceite incoloro, 2,10 g, rendimiento: 65,43%). RMN 1H (400 MHz, MeOD) 5: 3,55-3,47 (m, 2H), 3,41-3,35 (m, 2H), 3,06-3,02 (s, 3H).
Etapa 3: Síntesis del compuesto BB-15-3
A temperatura ambiente, se añadió terc-butóxido de potasio (2,22 g, 19,78 mmol) a una solución del compuesto BB-15-2 (2,00 g, 9,89 mmol) en dimetilsulfóxido (20,00 ml), la mezcla de reacción se agitó a temperatura ambiente durante 0,5 horas en atmósfera de nitrógeno, a continuación se añadió 5-bromo-4,6-dicloropirimidina (2,25 g, 9,89 mmol) y la mezcla de reacción se agitó durante 15 horas a temperatura ambiente en atmósfera de nitrógeno. Una vez completada la reacción, se añadió agua (60 ml), el pH se ajustó hasta 4 con ácido clorhídrico diluido 1 M y la mezcla se extrajo con acetato de etilo (30 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (30 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante cromatografía en columna (eluyente: éter de petróleo/acetato de etilo = 1/1, relación en volumen) para obtener el compuesto BB-15-3 buscado (sólido amarillo, 1,80 g, rendimiento: 43,98%). MS-ESI m/z: 392,8 [M+H]+,
394,8 [M+H+2]+ . RMN 1H (400 MHz, CDCI3) 5: 8,60 (s, 1H), 6,36 (s ancho, 1H), 3,66 (q, j=6,0 Hz, 2H), 3,46-3,25 (m, 2H), 3,09-2,82 (m, 3H).
Etapa 4: Síntesis del compuesto BB-15
En atmósfera de nitrógeno y a 40°C, una mezcla de etilenglicol (4,28 g, 68,95 mmol, 3,86 ml) y terc-butóxido de potasio (513,07 mg, 4,57 mmol) se agitó durante 0,5 horas y a continuación se añadió una solución del compuesto BB-15-3 (0,9 g, 2,29 mmol) en éter dimetílico de etilenglicol (30,00 ml) a la mezcla y la mezcla de reacción se calentó hasta 60°C y se agitó durante 3 horas en atmósfera de nitrógeno. Una vez completada la reacción, la mezcla se enfrió hasta temperatura ambiente, se añadió agua (60 ml), se ajustó hasta pH 4 con ácido clorhídrico diluido 1 M y se extrajo con acetato de etilo (50 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (50 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante cromatografía en columna (eluyente: éter de petróleo/acetato de etilo = 1/2, relación en volumen) para obtener el compuesto BB-15 buscado (aceite amarillo claro, 327,27 mg, rendimiento: 33,40%). MS-ESI m/z: 440,9 [M+Na]+ , 442,9 [M+Na+2]+ .
Realización de referencia 16: fragmento BB-16

A 0-5°C, se añadió lentamente gota a gota una solución del compuesto BB-1-2 (9,19 milimoles, producto bruto) en diclorometano a una solución de 3-azabiciclo[3.1.0]hexano en hidrocloruro (900,00 mg, 7,53 milimoles) y trietilamina (2,28 g, 22,58 milimoles) en diclorometano (10 ml) (el tiempo de goteo fue de aproximadamente 1 hora) y la mezcla de reacción se calentó hasta temperatura ambiente y se agitó durante 16 horas en atmósfera de nitrógeno. Una vez completada la reacción, se eliminó el disolvente a presión reducida y se añadió agua (20 ml) al residuo, se ajustó hasta un pH de 4-5 con ácido clorhídrico diluido 1 M y se extrajo con acetato de etilo (25 ml x 4 ). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (100 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida para obtener el compuesto BB-16-1 buscado (sólido blanco, 1,90 g, rendimiento: 96,19%). RMN 1H (400 MHz, CDCta) 5: 3,56-3,68 (m, 4H), 1,55-1,59 (m, 2H), 1,51 (s, 9H), 0,69-0,74 (m, 1 H), 0,41-0,45 (m, 1H).
Etapa 2: Síntesis del compuesto BB-16-2
A temperatura ambiente, se añadió ácido trifluoroacético (3,30 g, 28,96 mmol) a una solución del compuesto BB-16-1 (1,90 g, 7,24 mmol) en diclorometano (10,00 ml) en una porción, la mezcla de reacción se agitó a temperatura ambiente durante 16 horas en atmósfera de nitrógeno. Una vez completada la reacción, el disolvente de la mezcla de reacción se eliminó a presión reducida para obtener el compuesto BB-16-2 buscado (sólido blanquecino, 1,44 g, rendimiento: 72,00%, sal de trifluoroacetato). RMN 1H (400 MHz, CDCta) 5: 3,28-3,36 (m, 4H), 1,54-1,59 (m, 2H), 0,61-0,69 (m, 1 H), 0,44-0,58 (m, 1H).
Etapa 3: Síntesis del compuesto BB-16-3
A temperatura ambiente, se añadió terc-butóxido de potasio (1,75 g, 15,63 mmol) a una mezcla de la sal de
trifluoroacetato del compuesto BB-16-2 (1,44 g, 5,21 mmol) en dimetilsulfóxido (30,00 ml) en una porción, la mezcla de reacción se agitó a temperatura ambiente durante 0,5 horas en atmósfera de nitrógeno, a continuación se añadió 5-bromo-4,6-dicloropirimidina (1,42 g, 6,25 mmol) a la mezcla, la mezcla de reacción se agitó durante 16 horas a temperatura ambiente en atmósfera de nitrógeno. Una vez completada la reacción, se añadió agua helada (60 ml), el pH se ajustó hasta 4-5 con ácido clorhídrico diluido 4 M y la mezcla se extrajo con acetato de etilo (100 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (200 ml), se secaron sobre sulfato de sodio anhidro y se filtraron, el disolvente del filtrado se eliminó a presión reducida y el residuo se separó mediante cromatografía en columna (eluyente: éter de petróleo/acetato de etilo). = 10/1-1/1, relación de volumen) para obtener el compuesto BB-16-3 buscado (sólido marrón, 1,40 g, rendimiento: 74,14%). RMN 1H (400 MHz, CDCh) ó: 8,59 (s, 1H), 7,80 (s ancho, 1H), 3,73-3,79 (m, 2H), 3,66-3,72 (m, 2H), 1,58-1,62 (m, 2H), 0,69-0,76 (m , 1 H), 0,33 (q, y=4,3 Hz, 1H).
Etapa 4: Síntesis del compuesto BB-16
A temperatura ambiente, se añadió terc-butóxido de potasio (760,78 mg, 6,78 mmol) a etilenglicol (22,20 g, 357,67 mmol), la mezcla de reacción se calentó hasta 40°C y se agitó durante 0,5 horas en atmósfera de nitrógeno, y a continuación se añadió una solución del compuesto BB-16-3 (799,18 mg, 2,26 mmol) en éter dimetílico de etilenglicol (10,00 ml) a la mezcla y la mezcla de reacción se calentó hasta 110°C y se agitó durante 39 horas en atmósfera de nitrógeno. Una vez completada la reacción, la mezcla se enfrió hasta temperatura ambiente y el disolvente se eliminó a presión reducida. Se añadió agua helada (60 ml) al residuo, se ajustó hasta un pH de 4-5 con ácido clorhídrico diluido 1 M y se extrajo con acetato de etilo (60 ml x 2). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (120 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante cromatografía preparativa (eluyente: éter de petróleo/acetato de etilo = 1/1, relación en volumen) para obtener el compuesto BB-16 buscado (sólido amarillo, 520,00 mg, rendimiento: 59,45%). RMN 1H (400 MHz, CDCl3) 5: 8,38 (s, 1H), 7,63 (s ancho, 1H), 4,55-4,61 (m, 2H), 3,94-4,02 (m, 2H), 3,69-3,74 (m, 2H), 3,64-3,69 (m , 2H), 2,48 (s ancho, 1 H), 1,49-1,58 (m, 2H), 0,62-0,71 (m, 1H), 0,30-0,37 (m, 1 H).
Realización de referencia 17: fragmento BB-17

Etapa 1: Síntesis del compuesto BB-17-1
A 0°C, se añadió gota a gota una solución del compuesto BB-1-2 (12,39 mmol, producto bruto) en diclorometano a una solución de 2-oxa-6-azaespiro[3,3]heptano (1,17 g, 11,80 mmol) y trietilamina (3,58 g, 35,40 mmol) en diclorometano (10 ml) (tiempo de goteo aproximadamente 1 hora), la mezcla de reacción se calentó hasta temperatura ambiente y se agitó durante 16 horas. Una vez completada la reacción, se eliminó el disolvente a presión reducida, se añadió agua al residuo (20 ml), se ajustó el pH hasta 4-5 con ácido clorhídrico diluido 5 M y se extrajo con acetato de etilo (25 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (100 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente se eliminó a presión reducida para obtener el compuesto BB-17-1 buscado (sólido blanco, 1,70 g, rendimiento: 51,76%). RMN 1H (400 MHz, CDCh) 5: 7,07 (s ancho, 1H), 4,79 (s, 4H); 4,34 (s, 4H), 1,52 (s, 9H).
E ta p a 2 : S ín te s is d e l c o m p u e s to
BB-17-2
A temperatura ambiente, se añadió el compuesto BB-17-1 (1,70 g, 6,11 mmol) a agua (10,00 ml) y la mezcla de reacción se calentó hasta 100°C y se agitó durante 1 hora. Una vez completada la reacción, la mezcla se enfrió hasta temperatura ambiente y se extrajo con acetato de etilo (10 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (30 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida para obtener el compuesto BB-17-2 buscado (sólido blanco, 920,00 mg, rendimiento: 84,49%). RMN 1H (400 MHz, CDCta) 5: 4,78 (s, 4H), 4,39 (s ancho, 2H), 4,04 (s, 4H).
Etapa 3: Síntesis del compuesto BB-17-3
A temperatura ambiente, el compuesto BB-17-2 (920,00 mg, 5,16 mmol) y terc-butóxido de potasio (1,50 g, 13,37 mmol) se añadieron a dimetilsulfóxido (15,00 ml) y la mezcla de reacción se agitó a temperatura ambiente durante 0,5 horas. A continuación, se añadió a la mezcla 5-bromo-4,6-dicloropirimidina (1,41 g, 6,19 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 18 horas. Una vez completada la reacción, se añadió agua helada (40 ml), el pH se ajustó hasta 4-5 con ácido clorhídrico diluido 4 M y la mezcla se extrajo con acetato de etilo (40 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (120 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante una placa de cromatografía preparativa (eluyente: diclorometano/metanol = 10/1, relación en volumen) para obtener el compuesto BB-17-3 buscado (sólido marrón, 430,00 mg, rendimiento: 16,43%). MS-ESI m/z: 368,8 [M+H]+, 370,8 [M+H+2]+. Etapa 4: Síntesis del compuesto BB-17
A temperatura ambiente, se añadió terc-butóxido de potasio (390,49 mg, 3,48 mmol) a etilenglicol (15,23 g, 245,36 mmol), la mezcla de reacción se calentó hasta 40°C y se agitó durante 0,5 horas en atmósfera de nitrógeno, y a continuación, se añadió lentamente gota a gota una solución del compuesto BB-17-3 (430,00 mg, 1,16 mmol) en éter dimetílico de etilenglicol (10,00 ml) a la mezcla y la mezcla de reacción se calentó hasta 110°C y se agitó durante 48 horas en atmósfera de nitrógeno. Una vez completada la reacción, la mezcla se enfrió hasta temperatura ambiente y el disolvente se eliminó a presión reducida. Se añadió agua helada (30 ml) al residuo, se ajustó el pH hasta 4-5 con ácido clorhídrico diluido 1 M y se extrajo con acetato de etilo (30 ml x 4). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (120 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante cromatografía preparativa (eluyente: diclorometano/metanol = 10/1, relación de volumen) para obtener el compuesto BB-17 buscado (sólido amarillo, 150,00 mg, rendimiento: 30,86%). MS-ESI m/z: 417,0 [M+Na]+, 417,0 [M+Na+2]+.
Realización de referencia 18: fragmento BB-18

Ruta sintética:

A 0°C, se añadió gota a gota una solución de terc-butóxido de potasio (7,22 g, 64,31 mmol) en tetrahidrofurano (50 ml) a una suspensión de bromuro de metiltrifenilfosfonio (22,97 g, 64,31 mmol) en tetrahidrofurano (100 ml), la mezcla de reacción se calentó hasta temperatura ambiente y se agitó durante 1 hora en atmósfera de nitrógeno, a continuación la mezcla de reacción se enfrió hasta 0°C, y se añadió una solución del compuesto BB-18-1 (10,00 g, 42,87 mmol) en tetrahidrofurano (50 ml) en una porción. La mezcla de reacción se calentó hasta temperatura ambiente y se agitó durante 64 horas en atmósfera de nitrógeno. Una vez completada la reacción, se añadieron secuencialmente agua (50 ml) y éter de petróleo (50 ml), y se separó el líquido. La fase acuosa se extrajo con éter de petróleo (50 ml x 2). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (150 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante cromatografía en columna (eluyente: éter de petróleo/acetato de etilo = 1/0-10/1, relación en volumen) para obtener el compuesto BB-18-2 buscado (aceite incoloro, 5,20 g, rendimiento: 48,56%). MS-ESI m/z: 232,0 [M+H]+. RMN 1H (400 MHz, CDCh) 5: 7,40-7,29 (m, 5H), 5,15 (s, 2H), 4,76 (s, 2H), 3,51 (t, j=5,8 Hz, 4H), 2,19 (t, j=5,8 Hz, 4H).
Etapa 2: Síntesis del compuesto BB-18-3
A -40°C, se añadió gota a gota una solución de ácido trifluoroacético (10,25 g, 89,92 mmol) en diclorometano (10 ml) a una solución de dietilcinc (1 M, 89,92 ml) en diclorometano (50 ml), la mezcla de reacción se agitó a -40°C durante 0,5 horas en atmósfera de nitrógeno y a continuación se añadió una solución de diyodometano (24,08 g, 89,92 mmol) en diclorometano (10 ml) a la mezcla, la mezcla de reacción se agitó a -40°C durante 0,5 horas en atmósfera de nitrógeno. A continuación, se añadió una solución del compuesto BB-18-2 (5,20 g, 22,48 mmol) en diclorometano (10 ml), la mezcla de reacción se calentó hasta temperatura ambiente y se agitó durante 16 horas en atmósfera de nitrógeno. Una vez completada la reacción, se añadieron diclorometano (30 ml) y solución acuosa saturada de bicarbonato de sodio (80 ml) y se agitó durante 5 minutos, seguido de precipitación, filtración y separación del líquido. La fase orgánica se lavó con salmuera saturada (100 ml), se secó sobre sulfato de sodio anhidro, se filtró, el disolvente del filtrado se eliminó a presión reducida y el residuo se separó mediante cromatografía en columna (eluyente: éter de petróleo/acetato de etilo = 100/1-10/1, relación de volumen) para obtener un aceite amarillo, que se separó nuevamente por HPLC preparativa para obtener el compuesto BB-18-3 buscado (aceite amarillo claro, 4,00 g, rendimiento: 47,46%). MS-ESI m/z: 246,0 [M+H]+. RMN 1H (400 MHz, CDCh) 5: 7,40-7,29 (m, 5H), 5,15 (s, 2H), 3,57 3,47 (m, 4H), 1,35 (s ancho, 4H), 0,34 (s, 4H).
Etapa 3: Síntesis del compuesto BB-18-4
A temperatura ambiente, se añadió paladio sobre carbono húmedo (150,00 mg, pureza: 10%) a una solución del compuesto BB-18-3 (1,50 g, 6,11 mmol) en tetrahidrofurano (15,00 ml) y la mezcla de reacción se hizo reaccionar en una atmósfera de hidrógeno (3,5 MPa) y se agitó a temperatura ambiente durante 40 horas. Una vez completada la reacción, la mezcla de reacción se filtró, seguido de la adición de ácido clorhídrico/acetato de etilo (4 M, 10 ml), se agitó durante 15 minutos y se concentró a presión reducida para obtener el compuesto BB-18-4 (sólido amarillo, 900,00 mg, rendimiento: 99,76%, hidrocloruro). RMN 1H (400 MHz, DMSO_de) 5: 9,13 (s ancho, 2H), 3,02 (s ancho, 4H), 1,54 (t, j=5,0 Hz, 4H), 0,37 (s, 4H).
E ta p a 4 : S ín te s is d e l c o m p u e s to
BB-18-5
A 0°C, se añadió gota a gota una solución del compuesto BB-18-4 (1,20 g, 8,13 mmol, hidrocloruro) en diclorometano (20 ml) a una solución del compuesto BB-1-2 (8,54 mmol, producto bruto) en diclorometano y una mezcla de diclorometano (10 ml) y trietilamina (3,29 g, 32,52 mmol) (el tiempo de goteo fue de aproximadamente 1 hora). La mezcla de reacción se calentó hasta temperatura ambiente y se agitó durante 16 horas en atmósfera de nitrógeno. Una vez completada la reacción, el disolvente se eliminó a presión reducida y se añadió agua (20 ml) al residuo, se ajustó hasta un pH de 4-5 con ácido clorhídrico diluido 4 M (10 ml) y se extrajo con acetato de etilo ( 25 ml x 4). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (100 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida para obtener el compuesto BB-18-5 (sólido amarillo claro, 1,32 g, rendimiento: 55,91%). RMN 1H (400 MHz, CDCta) 5: 7,05 (s ancho, 1H), 3,47-3,40 (m, 4H), 1,52 (s, 9H), 1,50-1,47 (m, 4H), 0,38 (s, 4H).
Etapa 5: Síntesis del compuesto BB-18-6
A temperatura ambiente, se añadió ácido trifluoroacético (1,53 g, 13,42 mmol) a una solución del compuesto BB-19-5 (580,00 mg, 2,00 mmol) en diclorometano (3,00 ml) en una porción, la mezcla de reacción se agitó a temperatura ambiente durante 16 horas en atmósfera de nitrógeno. Una vez completada la reacción, se eliminó el disolvente a presión reducida para obtener el compuesto BB-18-6 (sólido amarillo claro, 600,00 mg, rendimiento: 98,50%, trifluoroacetato). RMN 1H (400 MHz, CDCta)5: 3,23 (t, j=5,5 Hz, 4H), 1,50 (t, j=5,5 Hz, 4H), 0,36 (s, 4H).
Etapa 6: Síntesis del compuesto BB-18-7
A temperatura ambiente, se añadió terc-butóxido de potasio (1,39 g, 12,42 mmol) a una solución del compuesto BB-18-6 (1,26 g, 4,14 mmol, trifluoroacetato) en dimetilsulfóxido (10,00 ml) en una porción, la mezcla de reacción se agitó a temperatura ambiente durante 0,5 horas en atmósfera de nitrógeno, a continuación se añadió 5-bromo-4,6-dicloropirimidina (1,04 g, 4,55 mmol) a la mezcla de reacción, la mezcla de reacción se agitó durante 20 horas a temperatura ambiente en atmósfera de nitrógeno. Una vez completada la reacción, se añadió agua helada (20 ml), el pH se ajustó hasta 4-5 con ácido clorhídrico diluido 1 M y la mezcla se extrajo con acetato de etilo (30 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (120 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente se eliminó a presión reducida. El residuo se separó mediante una placa de cromatografía (eluyente: éter de petróleo/acetato de etilo = 1/1, relación en volumen) para obtener el compuesto BB-18-7 (aceite amarillo, 600,00 mg, rendimiento: 35,86%). RMN 1H (400 MHz, CDCh) 5: 8,53 (s, 1H), 4,40 (s ancho, 1H), 3,20-3,28 (m, 4H), 1,49-1,55 (m, 4H), 0,35 (s, 4H).
Etapa 7: Síntesis del compuesto BB-18
A temperatura ambiente, se añadió terc-butóxido de potasio (353,46 mg, 3,15 mmol) a una solución de etilenglicol (16,03 g, 258,27 mmol) en éter dimetílico de etilenglicol (3,00 ml) y la mezcla de reacción se calentó hasta 40°C y se agitó durante 0,5 horas en atmósfera de nitrógeno, a continuación se añadió una solución del compuesto BB-18-7 (400,00 mg, 1,05 mmol) en éter dimetílico de etilenglicol (5,00 ml) a la solución de reacción y la mezcla de reacción se calentó hasta 110°C y se agitó durante 40 horas en atmósfera de nitrógeno. Una vez completada la reacción, la mezcla se enfrió hasta temperatura ambiente y el disolvente se eliminó a presión reducida. Se añadió agua helada (30 ml) al residuo, se ajustó hasta un pH de 5-6 con ácido clorhídrico diluido 1 M y se extrajo con acetato de etilo (30 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (100 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante cromatografía (eluyente: acetato de etilo/éter de petróleo = relación en volumen 1/1) para obtener el compuesto BB-18 (aceite amarillo, 180,00 mg, rendimiento: 42,09%). RMN 1H (400 MHz, CDCh) 5: 8,37 (s, 1H), 4,61-4,56 (m, 2H), 4,01 -3,97 (m, 2H), 3,54-3,47 (m, 4H), 1,52-1,45 (m, 4H), 0,36-0,31 ( m, 4H).
Realización 1: WX001
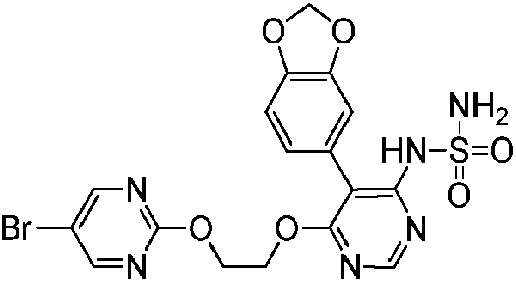
Ruta sintética:

Etapa 1: Síntesis del compuesto WX001-2
A 0°C, se añadió lentamente gota a gota diclorosulfóxido (58,11 g, 488,46 mmol, 35,43 ml) a una solución del compuesto WX001-1 (80 g, 444,06 mmol) en metanol (400 ml) (el tiempo de goteo fue de aproximadamente 0,5 horas) y la mezcla de reacción se calentó hasta temperatura ambiente y se agitó durante 10 horas en atmósfera de nitrógeno. Una vez completada la reacción, se eliminó el disolvente a presión reducida y se añadió agua (300 ml) al residuo y se extrajo con acetato de etilo (300 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (300 ml), se secaron sobre sulfato de sodio anhidro y se filtraron, y el disolvente del filtrado se eliminó a presión reducida para obtener el compuesto WX001-2. RMN 1H (400 MHz, CDCh) 5: 6,80-6,71 (m, 3H), 5,94 (s, 2H), 3,70 (s, 3H), 3,55 (s, 2H).
Etapa 2: Síntesis del compuesto WX001-3
A 0°C, se añadió gota a gota lentamente una solución del compuesto WX001-2 (35 g, 180,24 mmol) en carbonato de dimetilo (118,51 g, 1,32 mol, 110,76 ml) a una solución mixta de hidruro de sodio (10,81 g, 270,36 mmol, pureza: 60%) en carbonato de dimetilo (118,51 g, 1,32 mol , 110,76 ml) (el tiempo de goteo es de aproximadamente 1 hora), la mezcla de reacción se calentó hasta temperatura ambiente y se agitó durante 15 horas en atmósfera de nitrógeno. Una vez completada la reacción, se añadió agua helada (500 ml) y la mezcla se extrajo con acetato de etilo (300 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (300 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante cromatografía en columna (eluyente: éter de petróleo/acetato de etilo = 10/1 -5/1, relación en volumen) para obtener el compuesto WX001-3. RMN 1H (400 MHz, CDCh) 5: 7,00-6,93 (m, 1H), 6,85-6,77 (m, 2H), 5,98 (s, 2H), 4,58 (s, 1H), 3,78 (s, 6H).
Etapa 3: Síntesis del compuesto WX001-4
A 0°C, se añadió en porciones un bloque de sodio (10,94 g, 475,78 mmol) a metanol anhidro (150 ml), la mezcla de reacción se agitó a temperatura ambiente durante 0,5 horas en atmósfera de nitrógeno y a continuación se añadió una solución del compuesto WX001-3 (40 g, 158,59 mmol) en metanol (100 ml) a la solución de reacción. La mezcla de reacción se calentó hasta temperatura ambiente y se agitó durante 15 horas en atmósfera de nitrógeno, a continuación se añadió acetato de formamidina (19,81 g, 190,31 mmol) a la solución de reacción en una porción y la mezcla de reacción se agitó adicionalmente a temperatura ambiente durante 15 horas en atmósfera de nitrógeno. Una vez completada la reacción, se eliminó el disolvente a presión reducida y se añadió al residuo ácido clorhídrico diluido 2 M (200 ml) y se agitó a temperatura ambiente durante 30 minutos, durante los cuales precipitó una gran cantidad de sólido blanco. La mezcla de reacción se filtró, la torta de filtración se lavó con metanol (50 ml x 2) y la torta de filtración se recogió y se secó al vacío para obtener el compuesto WX001-4.
Etapa 4: Síntesis del compuesto WX001-5
A temperatura ambiente, se añadió el compuesto WX001-4 (24 g, 103,36 mmol) a oxicloruro de fósforo (594,00 g, 3,87 mol, 360,00 ml) y la mezcla de reacción se calentó hasta 90°C y se agitó durante 5 horas en atmósfera de nitrógeno. Una vez completada la reacción, la mezcla se enfrió hasta temperatura ambiente, la solución de reacción se vertió lentamente en agua helada (1000 ml) y se agitó a temperatura ambiente durante 0,5 horas y a continuación se extrajo con acetato de etilo (1000 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (1000 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante cromatografía en columna (eluyente: éter de petróleo/acetato de etilo = 10/1, relación en volumen) para obtener el compuesto WX001-5. RMN 1H (400 MHz, CDCh) 5: 8,78 (s, 1H), 6,96 (d, j=8,3 Hz, 1H), 6,78 (dd, j=2,5, 4,0 Hz, 2H), 6,09 (s, 2H).
E ta p a 5 : S ín te s is d e l c o m p u e s to
WX001-6
A temperatura ambiente, se disolvieron el compuesto 2-(5-bromopirimidin-2-il)oxietanol (8,06 g, 36,79 mmol) y el compuesto WX001-5 (11 g, 40,88 mmol) en tolueno (100 ml), a continuación la mezcla se enfrió hasta 0°C, se añadió terc-butóxido de potasio (9,17 g, 81,76 mmol) en porciones y la mezcla de reacción se agitó a 0°C durante 0,5 horas en atmósfera de nitrógeno. Una vez completada la reacción, se añadió ácido clorhídrico diluido 0,5 M (100 ml) a la solución de reacción y la mezcla se extrajo con acetato de etilo (100 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (100 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante cromatografía en columna (eluyente: éter de petróleo/acetato de etilo = 5/1, relación en volumen) para obtener el compuesto WX001-6. RMN 1H (400 MHz, CDCh) 5: 8,54-8,48 (m, 3H), 6,86-6,74 (m, 3H), 6,02 (s, 2H), 4,80-4,74 (m, 2H), 4,73-4,64 (m, 2H).
Etapa 6: Síntesis del compuesto WX001
A temperatura ambiente, se disolvieron el compuesto sulfamida (1,52 g, 15,83 mmol) y el compuesto WX001-6 (6,5 g, 14,39 mmol) en dimetilsulfóxido (100 ml) y a continuación se añadieron en una porción carbonato de potasio (5,97 g, 43,17 mmol) y trihidrato de fluoruro de tetrabutilamonio (9,08 g, 28,78 mmol). La mezcla de reacción se calentó hasta 70°C y se agitó durante 5 horas en atmósfera de nitrógeno. Una vez completada la reacción, la mezcla se enfrió hasta temperatura ambiente, se añadieron ácido clorhídrico diluido 0,5 M (100 ml) y agua (500 ml) y se extrajo con acetato de etilo (400 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (500 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante cromatografía en columna (eluyente: éter de petróleo/acetato de etilo = 3/1, a continuación diclorometano/acetato de etilo = 5/1, relación en volumen) para obtener el compuesto WX001 buscado. MS-ESI m/z: 510,8 [M+H]+, 512,8 [M+H+2]+. RMN 1H (400 MHz, DMSO_de) 5: 9,29 (s, 1H), 8,72 (s, 2H), 8,46 (s, 1H), 7,21 (s, 2H), 6,91 (d, J=7,8 Hz, 1 H), 6,76-6,61 (m, 2H ), 6,05 (s, 2H), 4,69-4,62 (m, 2H), 4,62-4,54 (m, 2H).
Realización 2: WX002

Etapa 1: Síntesis del compuesto WX002-2
A temperatura ambiente, se añadieron el compuesto WX002-1 (8,00 g, 37,37 mmol), bis(pinacolato)diboro (11,39 g, 44,84 mmol) y acetato de potasio (7,33 g, 74,74 mmol) a 1,4-dioxano (100,00 ml), a continuación se añadió [1,1'-bis(difenilfosfino)ferroceno]dicloropaladio (II) (5,47 g, 7,47 mmol). La mezcla de reacción se calentó hasta 100°C y se agitó durante 10 horas en atmósfera de nitrógeno. Una vez completada la reacción, la mezcla se enfrió hasta temperatura ambiente, se filtró y el disolvente del filtrado se eliminó a presión reducida. Se añadió agua (100 ml) al residuo y se extrajo con acetato de etilo (100 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (50 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante cromatografía en columna (eluyente: éter de petróleo/acetato de etilo = 20/1, relación en volumen) para obtener el compuesto WX002-2 buscado. RMN 1H (400 MHz, CDCh) 5: 8,92 (s, 1H), 8,51 (s, 1H), 7,89 (d, j=8,0 Hz, 1 H), 7,78 (d, j=8,0 Hz, 1H), 1,30 (s, 12H).
E ta p a 2 : S ín te s is d e l c o m p u e s to
WX002-3
A temperatura ambiente, se añadieron el compuesto BB-2 (250,00 mg, 732,75 pmol) y el compuesto WX002-2 (287,04 mg, 1,10 mmol) a 1,4-dioxano (15,00 ml), a continuación se añadió una solución de carbonato de potasio (303,82 mg, 2,20 mmol) en agua (5,00 ml). La mezcla de reacción se agitó a temperatura ambiente durante 0,5 horas en atmósfera de nitrógeno y a continuación se añadió [1,1'-bis(difenilfosfino)ferroceno]dicloropaladio(II) (160,85 mg, 219,83 pmol) a la mezcla. La mezcla de reacción se calentó hasta 80°C y se agitó durante 11 horas en atmósfera de nitrógeno. Una vez completada la reacción, la mezcla se enfrió hasta temperatura ambiente, se añadió agua (100 ml), se extrajo con acetato de etilo (20 ml x 1) y se descartó la fase orgánica. La fase acuosa se ajustó hasta un pH de 5-6 con ácido clorhídrico diluido 3 M y se extrajo con acetato de etilo (20 ml x 3). Las fases orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante cromatografía en columna (eluyente: éter de petróleo/acetato de etilo = 10/1-1/1, relación en volumen) para obtener el compuesto WX002-3 buscado. MS-ESI m/z: 395,9 [M+H]+. RMN 1H (400 MHz, CDCh) ó: 9,01 (s, 1H), 8,47 (s, 1H), 8,05 (s, 1 H), 8,02 (s, 1H), 7,31 (d, j=8,3 Hz, 1 H), 7,06 (s, 1H), 5,52 (t, j=6,0 Hz, 1H), 4,44 (t, j=4,4 Hz, 2H), 3,77 (s, 2H), 3,08-2,98 (m, 2H), 2,58 (s ancho, 1H), 1,15 (t, j=7,2 Hz, 3 H).
Etapa 3: Síntesis del compuesto WX002
A temperatura ambiente, se añadió hidruro de sodio (194,20 mg, 4,86 mmol, pureza: 60%) a tetrahidrofurano anhidro (20,00 ml), a continuación se añadió una solución del compuesto WX002-3 (240,00 mg, 606,89 pmol) en N,N-dimetilformamida anhidra (1 ml) y se añadió una solución de 5-bromo-2-cloropirimidina (234,78 mg, 1,21 mmol) en tetrahidrofurano anhidro (1 ml), la mezcla de reacción se calentó hasta 70°C y se agitó durante 2 horas en atmósfera de nitrógeno. Una vez completada la reacción, la mezcla se enfrió hasta temperatura ambiente, se añadió una solución saturada de cloruro de amonio (30 ml), se ajustó hasta un pH de 4-5 con ácido clorhídrico diluido 1 M y se extrajo con acetato de etilo (20 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (50 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante HPLC preparativa (fase móvil: acetonitrilo/agua; sistema básico: NH4HCO3 y NH3-H2Ü) para obtener el compuesto WX002 buscado. MS-ESI m/z: 552,0 [M+H]+, 554,0 [M+H+2]+. RMN 1H (400 MHz, CDCh) 5: 9,08 (s, 1H), 8,52 (s, 1H), 8,43 (s, 2H), 8,04 (d, j=2,0 Hz, 2H), 7,34 (dd, j=8,5, 1,3 Hz, 1H), 6,87 (s ancho, 1H), 5,58 (s, 1H), 4,74 (t, j=4,4 Hz, 2H), 4,62 (t, j=4,4 Hz, 2H), 3,15-3,02 (m, 2H), 1,21 (t, j=7,3 Hz, 3H).
Refiriéndose al método de síntesis de la realización 2 (BB-2 en la etapa 2 se reemplazó por la estructura correspondiente del fragmento 2), se sintetizaron las realizaciones de la tabla 1.
Tabla 1: Fórmula estructural de las realizaciones 3-7

Los datos de LCMS y RMN H de cada realización se muestran en la tabla 2.
T a b la 2 : D a to s d e R M N y L C M S d e la s
realizaciones 3-7

Realización 8: WX008

Etapa 1: Síntesis del compuesto WX008-2
A temperatura ambiente, se añadieron el compuesto WX008-1 (3,00 g, 14,92 mmol), bis(pinacolato)diboro (7,58 g, 29,84 mmol) y acetato de potasio (4,39 g, 44,76 mmol) a 1,4-dioxano (30,00 ml), a continuación se añadió [1,1'-bis(difenilfosfino)ferroceno]dicloropaladio (II) (3,28 g, 4,48 mmol). La mezcla de reacción se calentó hasta 80°C y se agitó durante 16 horas en atmósfera de nitrógeno. Una vez completada la reacción, la mezcla se enfrió hasta temperatura ambiente, se filtró y el disolvente del filtrado se eliminó a presión reducida. Se añadió agua (30 ml) al residuo y se extrajo con acetato de etilo (20 ml x 3). Las fases orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, se filtraron, el disolvente del filtrado se eliminó a presión reducida y el residuo se separó mediante cromatografía en columna (eluyente: éter de petróleo/acetato de etilo = 1/0-100/1, relación en volumen) para obtener
el compuesto WX008-2 buscado. RMN 1H (400 MHz, CDCis) 5:7,38 (dd, j=7,8, 0,8 Hz, 1H), 7,26 (s, 1H), 6,85 (d, j=7,8 Hz, 1H), 5,97 (s, 2H), 1,35 (s, 12H).
Etapa 2: Síntesis del compuesto WX008-3
A temperatura ambiente, se añadieron el compuesto BB-3 (300,00 mg, 844,57 pmol), el compuesto WX008-2 (419,04 mg, 1,69 mmol) y fosfato de potasio (537,83 mg, 2,53 mmol) a N,N-dimetilformamida (20,00 ml), a continuación se añadió [1,1'-bis(difenilfosfino)ferroceno]dicloropaladio (II) (185,39 mg, 253,37 pmol). La mezcla de reacción se calentó hasta 80°C y se agitó durante 16 horas en atmósfera de nitrógeno. Una vez completada la reacción, la mezcla se enfrió hasta temperatura ambiente, se añadió agua (100 ml), se extrajo con acetato de etilo (20 ml x 1) y se descartó la fase orgánica. La fase acuosa se ajustó hasta un pH de 5-6 con ácido clorhídrico diluido 3 M y se extrajo con acetato de etilo (20 ml x 3). Las fases orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante una placa de cromatografía preparativa (eluyente: éter de petróleo/acetato de etilo = 1/2, relación en volumen) para obtener el compuesto WX008-3 buscado. MS-ESI m/z: 397,0 [M+H]+.
Etapa 3: Síntesis del compuesto WX008
A temperatura ambiente, se añadió hidruro de sodio (145,30 mg, 3,63 mmol, pureza: 60%) a tetrahidrofurano anhidro (20 ml), a continuación se añadió una solución del compuesto WX008-3 (180,00 mg, 454,06 pmol) en N,N-dimetilformamida anhidra (1 ml) y se añadió a esto una solución de 5-bromo-2-cloropirimidina (175,66 mg, 908,13 pmol) en tetrahidrofurano anhidro (1 ml). La mezcla de reacción se calentó hasta 70°C y se agitó durante 2 horas en atmósfera de nitrógeno. Una vez completada la reacción, la mezcla se enfrió hasta temperatura ambiente, se añadió una solución saturada de cloruro de amonio (30 ml), se ajustó hasta un pH de 4-5 con ácido clorhídrico diluido 1 M y se extrajo con acetato de etilo (20 ml x 3). . Las fases orgánicas se combinaron, se lavaron con salmuera saturada (50 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante HPLC preparativa (fase móvil: acetonitrilo/agua; sistema neutro) para obtener el compuesto WX008 buscado. MS-ESI m/z: 552,8 [M+H]+, 554,8 [M+H+2]+. RMN 1H (400 MHz, CDCb) 5: 8,49 (s, 2H), 8,43 (s, 1H), 6,87 (d, j=8,3 Hz, 1 H), 6,73-6,68 (m, 2H), 6,03 (s, 2H), 5,61 (t, j=6,2 Hz, 1 H), 4,73 (q, j=5,0 Hz, 2H), 4,64 (t, j=4,8 Hz, 2H), 2,96 (q, j=6,8 Hz, 2H), 1,64-1,57 (m, 2H), 0,94 (t, j=7,4 Hz, 3H).
Refiriéndose al método de síntesis de la realización 8 (BB-3 en la etapa 2 se reemplazó por la estructura correspondiente del fragmento 2), se sintetizaron las realizaciones de la tabla 3.
Tabla 3: Fórmula estructural de las realizaciones 9-22


Los datos de LCMS y RMN H de cada realización se muestran en la tabla 4.
T a b la 4 : D a to s d e R M N y L C M S d e la s
realizaciones 9-22


Realización 23 y Realización 24: WX023 y WX024

El compuesto WX021 (500,00 mg, 839,74 pmol) se resolvió mediante cromatografía de fluidos supercríticos (condiciones de separación, columna: chiralpak AD-350 * 4,6 mm de DI, 3 pm; fase móvil: A: dióxido de carbono; B: isopropanol (dietilamina al 0,05%), 40%; temperatura de la columna: 40°C; longitud de onda: 220 nm) para obtener la muestra con un tiempo de retención de 1,149 min como WX023 (% ee: 100%) y la muestra con un tiempo de retención de 3,199 min como WX024 (% ee: 100%).
Realización 25: WX025

Ruta sintética:

Etapa 1: Síntesis del compuesto WX025-2
A temperatura ambiente, se añadieron el compuesto WX025-1 (400,00 mg, 2,03 mmol), bis(pinacolato)diboro (618,60 mg, 2,44 mmol), acetato de potasio (597,72 mg, 6,09 mmol) y [1,1'-bis(difenilfosfino)ferroceno]dicloropaladio(II) (297,09 mg, 406,03 pmol) a dioxano (20,00 ml), la mezcla de reacción se calentó hasta 100°C y se agitó durante 15 horas en atmósfera de nitrógeno. Una vez completada la reacción, la mezcla se enfrió hasta temperatura ambiente, el disolvente se eliminó a presión reducida y se añadió agua (100 ml) al residuo y se extrajo con acetato de etilo (100 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (50 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante cromatografía en columna (eluyente: éter de petróleo/acetato de etilo = relación de volumen 1/1) para obtener el compuesto WX025-2 buscado. RMN 1H (400 MHz, CDCh) 5: 8,70 (s, 1H), 7,99-7,98 (m, 1H), 7,51 (d, j=8,8 Hz, 1H), 7,37 (d, j=8,8 Hz, 1H), 6,48 (s, 1H), 1,36 (s, 12H).
Etapa 2: Síntesis del compuesto WX025-3
A temperatura ambiente, se añadieron el compuesto BB-3 (300,00 mg, 844,57 pmol), el compuesto WX025-2 (309,24 mg, 1,27 mmol), [1,1'-bis(difenilfosfino)ferroceno] dicloropaladio(II) (123,59 mg, 168,91 pmol) y carbonato de potasio (350,18 mg, 2,53 mmol) a una mezcla de dioxano (20,00 ml) y agua (2,00 ml) y la mezcla de reacción se calentó hasta 80°C y se agitó durante 10 horas en atmósfera de nitrógeno. Una vez completada la reacción, la mezcla se enfrió hasta temperatura ambiente, se añadió agua (100 ml), se ajustó hasta pH 5 con ácido clorhídrico diluido 1 M y se extrajo con acetato de etilo (50 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (50 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se sometió a cromatografía preparativa (eluyente: éter de petróleo/acetato de etilo = 1/2, relación en volumen) para obtener el compuesto WX025-3 buscado. MS-ESI m/z: 393,0 [M+H]+.
Etapa 3: Síntesis del compuesto WX025
A temperatura ambiente, se añadieron una solución del compuesto WX025-3 (180,00 mg, 346,76 pmol) en N,N-dimetilformamida (2 ml) y una solución de 5-bromo-2-cloropirimidina (67,07 mg, 346,76 pmol) en tetrahidrofurano (1 ml) a una mezcla de hidruro de sodio (83,08 mg, 2,08 mmol, pureza: 60%) en tetrahidrofurano anhidro (20 ml) en una porción. La mezcla de reacción se calentó hasta 70°C y se agitó durante 2 horas en atmósfera de nitrógeno. Una vez completada la reacción, la mezcla se enfrió hasta temperatura ambiente, se añadió una solución saturada de cloruro de amonio (50 ml), se ajustó hasta un pH de 4-5 con ácido clorhídrico diluido 1 M y se extrajo con acetato de etilo (50 ml x 3). . Las fases orgánicas se combinaron, se lavaron con salmuera saturada (50 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante HPLC preparativa (fase móvil: acetonitrilo/agua; sistema básico: NH4HCO3 y NH3H2O) para obtener el compuesto WX025 buscado. MS-ESI m/z: 549,0 [M+H]+, 551,0 [M+H+2]+. RMN 1H (400 MHz, CDCh) 5: 8,53 (s, 1H), 8,46-8,44 (m, 3H), 8,03 (s, 1H), 7,62 (d, j=9,2 Hz, 1H), 6,98 (d, j=9,2 Hz, 1H), 6,62 (s, 1H), 5,64 (s, 1 H), 4,78-4,76 (m, 2H), 4,67 4,65 (m, 2H), 3,02-3,00 (m, 2H), 1,65-1,63 (m, 2H), 0,99 (t,j=7,2 Hz, 3H).
Refiriéndose al método de síntesis de la realización 25 (BB-3 en la etapa 2 se reemplazó por la estructura correspondiente del fragmento 2), se sintetizaron las realizaciones de la tabla 5.
T a b la 5 : F ó rm u la e s tru c tu ra s d e la s
realizaciones 26-27

Los datos de LCMS y RMN H de cada realización se muestran en la tabla 6.
Tabla 6 : Datos de RMN y LCMS de las realizaciones 26-27

Realización 28: WX028

Etapa 1: Síntesis del compuesto WX028-2
A temperatura ambiente, se añadieron el compuesto WX028-1 (2,00 g, 10,15 mmol), bis(pinacolato)diboro (3,87 g, 15,23 mmol) y acetato de potasio (2,99 g, 30,45 mmol) a acetonitrilo (30,00 ml), a continuación se añadió [1,1'-bis(difenilfosfino)ferroceno]dicloropaladio (II) (1,11 g, 1,52 mmol). La mezcla de reacción se calentó hasta 60°C y se agitó durante 16 horas en atmósfera de nitrógeno. Una vez completada la reacción, la mezcla se enfrió hasta temperatura ambiente y el disolvente se eliminó a presión reducida. Se añadió agua (20 ml) al residuo y se extrajo con acetato de etilo (20 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (80 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante cromatografía preparativa (eluyente: éter de petróleo) para obtener el compuesto WX028-2 buscado. RMN 1H (400 MHz, CDCla) 5: 7,99 (s, 1H), 7,72-7,69 (m, 1H), 7,69 (d, j=2,0 Hz, 1H), 7,64-7,61 (m, 1H), 6,80 (d, j=1,2 Hz, 1H), 1,39 (s, 12H).
Etapa 2: Síntesis del compuesto WX028-3
A temperatura ambiente, se añadió paladio sobre carbono húmedo (50,00 mg, pureza: 10%) a una solución del compuesto WX028-2 (1,50 g, 6,15 mmol) en metanol (30,00 ml) y la mezcla de reacción se agitó durante 16 horas a temperatura ambiente en atmósfera de hidrógeno (1,05 kg/cm2 [15 psi]). Una vez completada la reacción, la mezcla de reacción se filtró y el disolvente del filtrado se eliminó a presión reducida para obtener el compuesto WX028-3 buscado. RMN 1H (400 MHz, CDCla) 5: 7,32 (d, j=7,0 Hz, 1H), 7,21 (m, 2H), 4,55 (t, j=8,5 Hz, 2H), 3,22 (t, j=8,5 Hz, 2H), 1,34 (s, 12H).
Etapa 3: Síntesis del compuesto WX028-4
A temperatura ambiente, se añadieron una solución de carbonato de potasio (254,51 mg, 1,84 mmol) en agua (2,00 ml) y [(bis(1-adamantil)-N-n-butilfosfina]-2-(2-aminobifenil)]dicloropaladio (40,00 mg) a una solución del compuesto BB-18 (250,00 mg, 613,83 pmol) y el compuesto WX028-3 (226,60 mg, 920,74 pmol) en dioxano (20,00 ml). La mezcla de reacción se calentó hasta 60°C y se agitó durante 16 horas en atmósfera de nitrógeno. Una vez completada la reacción, la mezcla se enfrió hasta temperatura ambiente y el disolvente se eliminó a presión reducida. A continuación, se añadió agua (15 ml) a la mezcla, se ajustó hasta un pH de 4-5 con ácido clorhídrico diluido 1 M y se extrajo con acetato de etilo (20 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (60 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante cromatografía preparativa (eluyente: éter de petróleo/acetato de etilo = 1/1, relación en volumen) para obtener el compuesto WX028-4 buscado. MS-ESI m/z: 447,2 [M+H]+.
Etapa 4: Síntesis del compuesto WX028
A temperatura ambiente, se añadieron una solución del compuesto WX028-4 (220,00 mg, 492,70 pmol) en N,N-dimetilformamida y 5-bromo-2-cloropirimidina (190,61 mg, 985,40 pmol) a una mezcla de hidruro de sodio (300,00 mg, 7,50 mmol, pureza: 60%) en tetrahidrofurano anhidro (15,00 ml) en una porción. La mezcla de reacción se calentó hasta 75°C y se agitó durante 1,5 horas en atmósfera de nitrógeno. Una vez completada la reacción, la solución se enfrió hasta temperatura ambiente, se añadió solución saturada de cloruro de amonio (20 ml), se ajustó hasta un pH de 4-5 con ácido clorhídrico diluido 1 M y se extrajo con acetato de etilo (30 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (100 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante cromatografía preparativa (eluyente: éter de petróleo/acetato de etilo = 1/ 1, relación en volumen) para obtener el compuesto WX028 buscado. MS-ESI m/z: 603,1 [M+H]+, 605,1 [M+H+2]+. RMN 1H (400 MHz, CDCh) 5: 8,48 (s, 2H), 8,46 (s, 1H), 7,23 (d, j=7,5 Hz, 1H), 6,99 (s, 1H), 6,69 (d, j=7,5 Hz, 1H), 6,63 (s, 1H), 4,74-4,70 (m, 2H), 4,66-4,64 (m, 2H), 4,63-4,59 (m, 2H), 3,49-3,44 (m, 4H), 3,25 (t, j=8,5 Hz, 2H), 1,48-1,44 (m, 4H), 0,34 (s, 4H).
Refiriéndose al método de síntesis de la realización 28 (BB-18 en el etapa 3 se reemplazó por la estructura correspondiente del fragmento 2), se sintetizaron las realizaciones de la tabla 7.
T a b la 7 : F ó rm u la e s tru c tu ra l d e la s
realizaciones 29-32

Los datos de LCMS y RMN H de cada realización se muestran en la tabla 8.
Tabla 8 : Datos de RMN y LCMS de las realizaciones 29-32

Realización 33: WX033

Ruta sintética:

A temperatura ambiente, se añadieron el compuesto WX033-1 (2,50 g, 12,69 mmol), bis(pinacolato)diboro (6,44 g, 25,38 mmol) y acetato de potasio (3,74 g, 38,07 mmol) a W,W-dimetilformamida (20,00 ml), a continuación se añadió [1,1'-bis(difenilfosfino)ferroceno]dicloropaladio(II) (1,86 g, 2,54 mmol). La mezcla de reacción se calentó hasta 90°C y se agitó durante 12 horas en atmósfera de nitrógeno. Una vez completada la reacción, la mezcla se enfrió hasta temperatura ambiente, se añadió agua (100 ml) y se extrajo con acetato de etilo (100 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (100 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante cromatografía en columna (eluyente: éter de petróleo/acetato de etilo = 30/1-20/1, relación en volumen) para obtener el compuesto WX033-2 buscado. RMN 1H (400 MHz, CDCh) 5:8,14 (s, 1H), 7,79 (d, j=8,3 Hz, 1H), 7,64 (d, j=2,0 Hz, 1H), 7,53 (d, j=8,3 Hz, 1H), 6,79 (d, j=1 ,5 Hz, 1 H), 1,39 (s, 12H).
Etapa 2: Síntesis del compuesto WX033-3
A temperatura ambiente, el compuesto WX033-2 (500,00 mg, 2,05 mmol) se disolvió en metanol (30,00 ml), a continuación se añadió paladio sobre carbono húmedo (200,00 mg, pureza: 10%) y la mezcla de reacción se calentó hasta 40°C y se agitó durante 48 horas bajo atmósfera de hidrógeno (1,05 kg/cm2 [15 psi]). Una vez completada la reacción, la mezcla se enfrió hasta temperatura ambiente, se filtró a través de tierra de diatomeas y el disolvente del filtrado se eliminó a presión reducida para obtener el compuesto WX033-3 buscado. RMN 1H (400 MHz, CDCh) 5: 7,62 (s, 1H), 7,60-7,50 (m, 1H), 6,78-6,77 (m,1 H), 4,60-4,56 (t, j=8,8 Hz, 2H), 3,22-3,17 (t, j=8,8 Hz, 2H), 1,33(s, 12H).
Etapa 3: Síntesis del compuesto WX033-4
A temperatura ambiente, se añadieron el compuesto BB-3 (500,00 mg, 703,81 pmol), el compuesto WX033-3 (259,82 mg, producto bruto), carbonato de potasio (291,82 mg, 2,11 mmol) y [1,1'-bis(difenilfosfino)ferroceno]dicloropaladio(II) (103,00 mg, 140,76 pmol) a una mezcla de dioxano (20,00 ml) y agua (2,00 ml). La mezcla de reacción se calentó hasta 80°C y se agitó durante 10 horas en atmósfera de nitrógeno. Una vez completada la reacción, la mezcla se enfrió hasta temperatura ambiente y el disolvente se eliminó a presión reducida. Se añadió agua (100 ml) al residuo, se ajustó hasta pH 5 con ácido clorhídrico diluido 1 M y se extrajo con acetato de etilo (50 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (50 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante cromatografía preparativa (eluyente: éter de petróleo/acetato de etilo = 1/1, relación en volumen) para obtener el compuesto WX033-4 buscado. MS-ESI m/z: 395,0 [M+H]+.
Etapa 4: Síntesis del compuesto WX033
A temperatura ambiente, se añadieron secuencialmente una solución del compuesto WX033-4 (140,00 mg, 314,28 pmol) en W,W-dimetilformamida (2,00 ml) y una solución de 5-bromo-2-cloropirimidina (121,58 mg, 628,56 pmol) en tetrahidrofurano (1 ml) a una solución de hidruro de sodio (75,43 mg, 1,89 mmol, pureza: 60%) en tetrahidrofurano anhidro (20,00 ml) en una porción. La mezcla de reacción se calentó hasta 70°C y se agitó durante 2 horas en atmósfera de nitrógeno. Una vez completada la reacción, la mezcla se enfrió hasta temperatura ambiente, se añadió una solución acuosa saturada de cloruro de amonio (50 ml), se ajustó hasta un pH de 4-5 con ácido clorhídrico diluido 1 M y se extrajo con acetato de etilo (50 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (50 ml), se secaron sobre sulfato de sodio anhidro, se filtraron y el disolvente del filtrado se eliminó a presión reducida. El residuo se separó mediante cromatografía preparativa (eluyente: éter de petróleo/acetato de etilo = 1/1, relación en volumen) para obtener el compuesto WX033 buscado. MS-ESI m/z: 551,1 [M+H]+, 553,1 [M+H+2]+. RMN 1H (400 MHz, CDCl3) 5: 8,50 (s, 2H), 8,43 (s, 1H), 7,07 (s, 1 H), 7,01 -6,97 (m, 2H), 6,83-6,78 (m, 1 H), 5,65 (s, 1 H), 4,73-4,71 (m, 2H), 4,65-4,61 (m, 4H), 3,25-3,20 (m, 2H), 2,98-2,93 (m, 2H), 1,60-1,56 (m, 2H), 0,96 (t, j=7,6Hz, 3H).
Refiriéndose al método de síntesis de la realización 33 (BB-3 en la etapa 3 se reemplazó por el fragmento 2), se sintetizó la realización de la tabla 9.
Tabla 9: Fórmula estructural de la realización 34

Los datos de LCMS y RMN H de la realización se muestran en la tabla 10.
Tabla 10: Datos de RMN y LCMS de la realización 34

Realización experimental 1: Prueba in vitro del efecto antagonista contra receptor ETa humano Propósito experimental:
La actividad antagonista de los compuestos contra receptores ETa humanos expresados endógenamente en células SK-N-MC se evaluó midiendo el efecto de los compuestos sobre los cambios en la señal de iones Ca2+ citoplásmicos inducida por antagonistas de receptores ETA humanos usando métodos de detección de fluorescencia. La actividad funcional del efecto antagonista sobre receptores ETa se probó en Eurofins-Cerep SA según procedimientos estándar actuales. Protocolo experimental:
1. Las células se suspendieron en solución de medio de Eagle con modificación de Dulbecco (DMEM, Invitrogen) complementado con FCSd al 1%, a continuación se distribuyeron en una placa de 384 pocillos (100 pl/pocillo) a una densidad de 5x10345células/pocillo;
2. Se mezcló probenecid en solución salina equilibrada de Hank (HBSS, Invitrogen) complementada con ácido 4-(2-hidroxietil)piperazin-1 -etanosulfónico 20 mM (Hepes, Invitrogen) (pH 7,4) con sonda fluorescente (Fluo4 NW, Invitrogen), la mezcla se añadió a cada pocillo, a continuación se equilibró con células a 37°C durante 60 minutos y a continuación se equilibró con células a 22°C durante 15 minutos;
3. La placa de prueba se colocó en un lector de microplacas (CellLux, PerkinElmer), se añadió una solución de DMSO o tampón de HBSS con una concentración apropiada del compuesto de prueba y el control positivo, y se añadió endotelina-1 1 nM o tampón de HBSS (control de base) después de 5 minutos, y a continuación se midió el cambio en la intensidad de fluorescencia proporcional a la concentración de iones Ca2+ de lisosomas celulares libres;
4. Los resultados se mostraron como el porcentaje de inhibición de la respuesta de control a endotelina-1 1 nM; 5. El control positivo estándar fue BQ-123, se probaron varias concentraciones en cada experimento. Los datos se analizaron usando Prism para generar una curva de respuesta a la concentración, y se calcularon los valores de IC50 de los compuestos.
Realización experimental 2: Prueba in vitro de efectos antagonistas contra receptor ETa humano Propósito experimental:
La actividad antagonista del compuesto frente a receptores ETb humanos expresados en células CHO transfectadas se evaluó midiendo el efecto del compuesto sobre los cambios en la señal de iones Ca2+ citoplásmicos inducida por antagonistas de receptores ETb humanos. La actividad funcional del efecto sobre receptores ETB se probó en Eurofins-Cerep SA según los procedimientos estándar actuales.
Protocolo experimental:
1. Las células se suspendieron en tampón de DMEM (Invitrogen) y a continuación se distribuyeron en una placa de 384 pocillos (100 pl/pocillo) a una densidad de 3x104 células/pocillo;
2. Se mezcló probenecid en tampón de HBSS (Invitrogen) complementado con Hepes 20 mM (Invitrogen) (pH 7,4) con sonda fluorescente (Fluo4 NW, Invitrogen), la mezcla se añadió a cada pocillo y a continuación se equilibró con células a 37°C durante 60 minutos y a continuación se equilibró con células a 22°C durante 15 minutos;
3. La placa de prueba se colocó en un lector de microplacas (CellLux, PerkinElmer), se añadió una solución de DMSO o tampón de HBSS con una concentración apropiada del compuesto de prueba y el control positivo, y se añadió endotelina-1 0,3 nM o tampón de HBSS (control de base) después de 5 minutos, y a continuación se midió el cambio en la intensidad de fluorescencia proporcional a la concentración de iones Ca2+ de lisosomas celulares libres;
4. Los resultados se mostraron como el porcentaje de inhibición de la respuesta de control a endotelina-1 0,3 nM; 5. El control positivo estándar fue BQ-788, se probaron varias concentraciones en cada experimento. Los datos se analizaron usando Prism para generar una curva de respuesta a la concentración, y se calcularon los valores de IC50 de los compuestos.
Tabla 11: Actividad antagonista in vitro de los compuestos de la presente divulgación frente a receptores ETa y ETb humanos y su selectividad para ETB

Conclusión:
Todos los compuestos de la presente divulgación exhiben una actividad antagonista muy alta contra receptores ETa humanos in vitro. La selectividad de los compuestos de la presente divulgación para ETa y ETb es de más de 10000 veces.
Realización experimental 3: Ensayo del receptor X de pregnano (PXR) humano
Propósito experimental:
Se evaluó el efecto de los compuestos sobre la inducción de la expresión de CYP3A mediada por PXR.
Materiales y dispositivos experimentales:

Protocolo experimental:
1. Las células DPX2 se descongelaron en condiciones estériles.
2. La solución de células DPX2 se distribuyó en una placa de 96 pocillos (100 pl/pocillo) y la placa se colocó en una incubadora de CO2 al 5%/37°C durante la noche.
3. El medio de alimentación cuantitativo se descongeló en un baño de agua a 37°C. El control positivo rifampicina se descongeló a temperatura ambiente. Se prepararon una serie de compuestos de prueba y diluciones de control positivo en medio de alimentación cuantitativo. Las células se aspiraron cuidadosamente de cada pocillo sin alterar las células durante la aspiración y se descartó el medio. Se transfirieron 100 pl de cada concentración de compuesto de prueba a pocillos premarcados. El funcionamiento del grupo de control positivo y del grupo en blanco fue el mismo. La placa se volvió a colocar en la incubadora durante 24 horas de exposición.
4. Prueba de actividad enzimática:
(1) Se añadieron 7 pl de Luciferin-IPA a 7 pl de medio de alimentación cuantitativo, la mezcla se mezcló por inversión y se vertió en un depósito de reactivo Luciferin-IPA.
(2) Se sacó la placa de 96 pocillos de la incubadora y se aspiró cuidadosamente el medio de cada pocillo. Se añadieron 50 pl del reactivo Luciferin-IPA a cada pocillo y la placa celular se volvió a colocar en la incubadora durante 60 minutos.
(3) Durante la incubación, el tampón P450-Glo se vertió en el reactivo de detección de luciferina y la mezcla se mezcló por inversión.
(4) Se sacó la placa de 96 pocillos de la incubadora y se transfirieron 40 pl de solución de cada pocillo a la placa blanca de 96 pocillos correspondiente, y la posición correspondiente de cada pocillo coincidía con la placa celular original.
(5) Después de transferir la solución P450-Glo™ a la placa de réplica, se transfirieron 10 ml de tampón de titulación celular (tampón de CTF) a un tubo cónico estéril de 15 ml, seguido de la adición de 5 pl de reactivo CellTiter-Fluor™, y la mezcla se mezcló por inversión.
(6) Se utilizó un pipeteador de líquidos de múltiples canales, se añadieron suavemente 100 pl de CellTiter-Fluor™ a la placa de 96 pocillos donde se cultivaron originalmente las células y, a continuación, se volvió a colocar la placa celular en la incubadora durante 60 minutos.
(7) A cada pocillo de la placa de réplica se le añadieron 40 pl de reactivo de detección de luciferina/tampón P450-Glo, se agitó y se incubó durante 20 minutos a temperatura ambiente.
(8) Después de la incubación con el reactivo de detección de luciferina durante 20 minutos, se usó un fotómetro (ajustado de 1 a 5 segundos. Tiempo de lectura) para medir la luminiscencia de cada pocillo de la placa blanca de 96 pocillos. Debe usarse un ajuste de ganancia relativamente alto.
(9) Se añadió tampón de detección de luciferasa ONE-Glo™ al reactivo de detección ONE-Glo™ y la mezcla se mezcló por inversión.
(10) Después de incubar durante 60 minutos a 37°C, se sacó de la incubadora la placa original de 96 pocillos. El lector se ajustó al modo de fluorescencia, la longitud de onda de excitación se ajustó a 380-400 nm, la longitud de onda de emisión se ajustó a 505 nm y se midió la intensidad de fluorescencia de cada pocillo.
(11) Se sacó la placa celular del lector enzimático y se añadieron 100 pl de reactivo de detección ONE-Glo™ a cada pocillo. La mezcla se mezcló agitando las placas y se incubó a temperatura ambiente durante 5 minutos. (12) El lector enzimático se ajustó a 5 segundos para la preagitación y 5 segundos para la lectura de pocillos, y
se midió la intensidad de fluorescencia en cada orificio. Deben utilizarse ajustes altos de ganancia (sensibilidad) del instrumento.
5. El efecto de activación del fármaco sobre PXR se reflejó por la inducción en número de veces, es decir, la inducción en número de veces de cada grupo = valor de actividad de luciferasa del grupo de tratamiento con fármaco/valor de actividad de luciferasa del grupo de control de disolvente, y la inducción en número de veces se usó para predecir el efecto de inducción sobre CYP3A4.
El control positivo fue rifampicina, se probaron seis concentraciones en cada experimento. Los datos se analizaron usando Prism para generar una curva de respuesta a la concentración y se calculó el valor de EC50 del compuesto.
Resultados experimentales:
Los resultados de la prueba se muestran en la tabla 12.
Tabla 12: Resultados del efecto de inducción del compuesto de la presente divulgación sobre la expresión de CYP3A mediada por PXR

* Error de cálculo de la curva de simulación
Conclusión:
El compuesto WX013 de la presente divulgación tiene un efecto de inducción relativamente débil sobre la expresión de CYP3A mediada por PXR, y el compuesto macitentán tiene un efecto de inducción relativamente fuerte sobre la expresión de CYP3A mediada por PXR. Por lo tanto, en el experimento de caracterización de la inducción de la expresión de CYP3A mediada por PXR, WX013 es superior a macitentán.
Realización experimental 4: Ensayo de inhibición de isoenzimas del citocromo P450 microsómico hepático humano
Propósito experimental:
El propósito del ensayo fue evaluar la actividad inhibidora de las muestras contra isoenzima del citocromo P450 microsómico hepático humano (CYP1A2, CYP2C9, CYP2C19, CYP2D6 y CYP3A4) utilizando el sustrato de sonda de 5 en 1 de la isoenzima CYP.
Protocolo experimental:
Se adquirieron microsomas hepáticos humanos (HLM) mixtos de Corning Inc. (Steuben, Nueva York, EE. UU.) y se almacenaron por debajo de -80°C antes de su uso. La solución de trabajo del compuesto de prueba que se ha diluido en concentraciones en serie se añadió a un sistema de incubación que contenía microsomas hepáticos humanos, sustratos de sonda y cofactores del sistema circulatorio, y se usó un control que contenía disolvente sin el compuesto de prueba como control de la actividad enzimática (100%). La concentración del metabolito producido por el sustrato de sonda en la muestra se determinó usando un método de cromatografía de líquidos-espectrometría de masas en tándem (LC-MS/MS). Se realizó un análisis de regresión no lineal sobre el porcentaje medio de actividad frente a la concentración de los sujetos usando SigmaPlot (V.11). El valor de IC50 se calculó mediante una ecuación logarítmica inversa de tres parámetros o cuatro parámetros.
Resultados experimentales:
Los resultados de la prueba se mostraban en la tabla 13.
Tabla 13: Resultados de la inhibición de los compuestos de la presente divulgación sobre la isoenzima del citocromo P450 microsómico hepático humano

Conclusión:
Los compuestos WX005, WX013 y WX025 de la presente divulgación tienen efectos inhibidores muy débiles sobre cinco isoenzimas principales de CYP. El macitentán tiene efectos inhibidores débiles sobre cuatro isoenzimas principales de CYP, y los efectos inhibidores sobre las isoenzimas CYP2C9 son moderados. Por lo tanto, WX005, WX013 y WX025 eran superiores al macitentán en el experimento de caracterización sobre la inhibición de cinco isoenzimas principales de la citocina microsómica hepática humana P450.
Realización experimental 5: Prueba de inhibición del compuesto sobre la bomba de transferencia de sales biliares (BSEP)
Propósito experimental:
Este experimento evalúa si el compuesto de prueba tiene un efecto inhibidor durante el proceso de transporte de la bomba de transferencia de sales biliares (BSEP) usando LC/MS/MS para detectar la capacidad de absorción de la bomba de transferencia de sales biliares (BSEP) al sustrato taurina TCA.
Materiales experimentales:

Preparación de la solución:
1. Tampón A:
HEPES 50 mM pH 7,4, KNO3100 mM, Mg(NO3)210 mM, sacarosa 50 mM.
2. Tampón B:
TRIS 10 mM pH 7,4, KNO3100 mM, Mg(NO3)210 mM, sacarosa 50 mM.
3. Tampón de ATP:
Preparado con tampón A, ATP 8,16 mM y ácido taurocólico 4,08 pM estaban contenidos en 12 ml de tampón A.
4. Búfer de AMP:
Preparado con tampón A, AMP 8,16 mM, ácido taurocólico 4,08 pM, 12 ml de tampón A estaban contenidos en el tampón A.
5. Solución vesicular de BSEP-Hi5-VT:
Se preparó una solución que contenía 5 pg/pl de BSEP-Hi5-VT con tampón A.
Preparación de la muestra:
1. El compuesto se diluyó hasta 100 mM con DMSO; a continuación se diluyó en serie 3 veces para una dilución de 11 puntos; la concentración mínima fue de 0,169 pM.
2. La referencia positiva gliburida se diluyó hasta 20 mM con DMSO; a continuación se diluyó dos veces en serie para una dilución de 11 puntos; la concentración mínima fue de 19,5 pM.
Protocolo experimental:
1. Se transfirieron 0,3 pl de una solución del compuesto en DMSO o una solución de DMSO diluida a los pocillos correspondientes de la placa del compuesto usando ECHO, respectivamente.
2. Se añadieron 14,7 pl de tampón de ATP al compuesto y a los pocillos correspondientes de cero por ciento de efecto (ZPE), respectivamente.
3. Se añadieron 14,7 pl de tampón de AMP a los pocillos correspondientes de 100% de efecto (HPE), respectivamente.
4. La placa se agitó durante 10 minutos a 25°C.
5. Se añadieron 15 pl de solución vesicular de BSEP-Hl5-VT a todos los pocillos, respectivamente, y la placa se agitó durante otros 40 minutos a 25°C.
6. Se añadieron inmediatamente 5 pl de una solución de ácido etilendiaminotetraacético (EDTA) 0,5 M a todos los pocillos, seguido de la adición de 65 pl de tampón B y se completó toda la reacción.
7. Se transfirieron 95 pl de líquido a la placa de filtración desde la placa de compuestos al final de la reacción usando un instrumento.
8. Después de colocar la placa de recepción de líquido debajo de la placa de filtración, se eliminó el líquido usando una centrífuga y se descartó el líquido de la placa de recepción.
9. Se añadieron 90 pl de tampón B a la placa de filtración. Después de colocar la placa de recepción de líquido debajo de la placa de filtración, se eliminó el líquido usando una centrífuga y la placa de recepción de líquido se descartó y la placa de filtración se lavó tres veces en total.
10. La placa de filtración se secó durante la noche.
11. Al día siguiente, se añadieron a la placa de filtración 80 pl de solución de metanol/agua (80/20, relación en volumen).
12. La placa se agitó durante 15 minutos después de unir la placa de filtración a la membrana.
13. Se colocó una nueva placa de recepción de líquido debajo de la placa de filtración y la placa de filtración se centrifugó durante 5 minutos y todos los líquidos de la placa de filtración se filtraron a la placa de recepción. 14. Se añadieron 15 pl de solución de estándar interno a cada pocillo en la placa de recepción de líquido y la placa se selló con una membrana.
15. El contenido de ácido taurocólico en la placa de recepción se detectó usando LC/MS/MS.
Se probaron varias concentraciones en cada experimento. Los datos se analizaron usando Prism para generar una curva de respuesta a la concentración, y se calcularon los valores de IC50 de los compuestos.
Resultados experimentales:
Los resultados experimentales se muestran en la tabla 14.
Tabla 14: Efecto inhibidor del compuesto de la presente divulgación sobre la bomba de transporte de sales biliares
(BSEP)

Conclusión:
El efecto inhibidor del compuesto WX013 de la presente divulgación sobre la bomba de transporte de sales biliares (BSEP) fue extremadamente débil, pero el efecto inhibidor del macitentán fue fuerte. Por lo tanto, el efecto inhibidor de WX013 sobre la bomba de transporte de sales biliares fue mucho más débil que el del macitentán, lo que reducía significativamente el riesgo de desarrollar hepatotoxicidad.
Realización experimental 6: Evaluación de la farmacocinética de los compuestos en ratas
Propósito experimental:
Se seleccionaron ratas SD macho como animales de prueba en este estudio, y se midió cuantitativamente la concentración de fármaco en el plasma de las ratas en diferentes momentos mediante inyección intravenosa o administración gástrica oral del compuesto de prueba usando el método de LC/MS/MS para evaluar las características farmacocinéticas de los compuestos de prueba en ratas.
Materiales experimentales:
Ratas Sprague Dawley (SD) (macho, 200-300 g, 7-10 semanas de edad, Beijing Viton Lihua o Shanghai Slake).
Protocolo experimental:
Se inyectó una solución transparente del compuesto de prueba en ratas SD a través de la vena caudal (ayuno durante la noche), o se administró por vía oral por sonda (ayuno durante la noche). Para la administración intravenosa, se recogieron 200 pl de sangre yugular mediante venopunción a las 0 horas (antes de la dosificación) y a las 0,0833, 0,25, 0,5, 1, 2, 4, 6, 8 y 24 horas después de la dosificación, que a continuación se colocaron en un tubo de anticoagulación complementado con EDTA-K2 (Suzuki Healthcare Medical Co., Ltd.). La mezcla del tubo de anticoagulación se sometió a turbulencia para mezclar a fondo a 4°C y a continuación se centrifugó a 13000 rpm durante 10 minutos para recoger el plasma. Para la administración por sonda oral, se recogieron 200 pl de sangre yugular mediante venopunción a las 0 horas (antes de la dosificación) y a las 0,5, 1, 2, 4, 6, 8 y 24 horas después de la dosificación, que a continuación se colocaron en un tubo de anticoagulación complementado con EDTA-K2 (Suzuki Healthcare Medical Co., Ltd.). La mezcla del tubo de anticoagulación se sometió a turbulencia para mezclar a fondo y a continuación se centrifugó a 13000 rpm durante 10 minutos para recoger el plasma. La concentración de fármaco en plasma se midió mediante el método de LC/MS/MS, y los parámetros farmacocinéticos relacionados se calcularon usando el programa de farmacocinética WinNonlin™ Versión 6.3 (Pharsight, Paul View, CA) en el método trapezoidal logarítmico lineal de modelo no compartimentado.
Resultados experimentales:
Los resultados experimentales se muestran en la tabla 15.
Tabla 15: Parámetros farmacocinéticos de los compuestos de la divulgación en ratas

Conclusión:
Los compuestos WX001 y WX013 de la presente divulgación tienen una velocidad de depuración plasmática baja (<
5 ml/min/kg) y una alta biodisponibilidad oral (> 70%) en ratas.
Realización experimental 7: Evaluación de la farmacocinética de compuestos en perros beagle
Propósito experimental:
Se seleccionaron perros beagle macho como los animales probados en este estudio, y se midió cuantitativamente la concentración de fármaco en plasma de los perros beagle en diferentes momentos a través de inyección intravenosa o administración gástrica por perfusión oral de los compuestos de prueba usando el método de LC/Ms /MS para evaluar las características farmacocinéticas de los compuestos de prueba en los perros beagle.
Materiales experimentales:
Perro beagle (macho, 6-15 kg, más de 6 meses de edad, Beijing Marshall Biotechnology Co., Ltd.).
Protocolo experimental:
Una solución transparente del compuesto de prueba se inyectó por vía intravenosa en perros beagle (ayuno durante la noche), o se administró por vía oral a perros beagle por sonda (ayuno durante la noche). Para la administración intravenosa, se recogieron 500 gl de sangre de los vasos periféricos a las 0 horas (antes de la dosificación) y a las 0. 0833, 0,25, 0,5, 1, 2, 4, 6, 8 y 24 horas después de la dosificación, que a continuación se introdujeron en un tubo de anticoagulación complementado con EDTA-K2 (Suzuki Healthcare Medical Co., Ltd.). Para la administración gástrica oral, se recogieron 500 gl de sangre de los vasos periféricos a las 0 horas (antes de la dosificación) y a las 0,25, 0,5, 1, 2, 4, 6, 8 y 24 horas después de la dosificación, que a continuación se introdujeron en un tubo de anticoagulación complementado con EDTA-K2. La mezcla en el tubo de anticoagulación se sometió a turbulencia para mezclar a fondo y a continuación se centrifugó a 13000 rpm durante 10 minutos para recoger el plasma. La concentración de fármaco en plasma se midió mediante el método de LC/MS/MS, y los parámetros farmacocinéticos relacionados se calcularon usando el programa de farmacocinética WinNonlin™ Versión 6.3 (Pharsight, Paul View, CA) en el método trapezoidal logarítmico lineal con modelo no compartimentado.
Resultados experimentales:
Los resultados experimentales se muestran en la tabla 16.
Tabla 16: Parámetros farmacocinéticos de los compuestos de la presente divulgación en perros beagle

Conclusión:
Los compuestos WX001 y WX013 de la presente divulgación tienen una baja velocidad de depuración plasmática (< 5 ml/min/kg) y una alta biodisponibilidad oral (> 50%) en perros beagle.
Claims (15)
1. Un compuesto de fórmula (I), uno de sus isómeros geométricos o estereoisómeros o una de sus sales farmacéuticamente aceptables,

donde,
R1 se selecciona de H, F, Cl, Br, I, OH y NH2;
R2 se selecciona de H y alquilo C1-3, donde el alquilo C1-3 está opcionalmente sustituido con uno, dos o tres R;
R3 se selecciona de H, alquilo C1-6, heteroalquilo C1-6, -alquil(C1-3)-cicloalquilo(C3-6), cicloalquilo C3-6 y -alquil(C1-3)-heterocicloalquilo(de 3-7 miembros), donde el heteroalquilo C1-6, -alquil(C1-3)-cicloalquilo(C3-6), cicloalquilo C3-6 y -alquil(C1-3)-heterocicloalquilo(de 3-7 miembros) está opcionalmente sustituido con uno, dos o tres R;
o R2 y R3 están conectados para formar un anillo de 3 a 8 miembros opcionalmente sustituido con uno, dos o tres R; el anillo B se selecciona de heterocicloalquilo de 3-7 miembros y heteroarilo de 5-6 miembros, donde el heterocicloalquilo de 3-7 miembros o el heteroarilo de 5-6 miembros está opcionalmente sustituido con uno, dos o tres R;
R se selecciona independientemente de H, F, Cl, Br, I, OH, NH2, CN, alquilo C1-6 y heteroalquilo C1-6, donde el alquilo C1-6 o el heteroalquilo C1-6 está opcionalmente sustituido con uno, dos o tres R';
R' se selecciona independientemente de F, Cl, Br, I, OH, NH2, CN, Me, CH2F, CHF2, CF3 y Et;
cada uno del heteroalquilo C1-6, heterocicloalquilo de 3-7 miembros y heteroarilo de 5-6 miembros contiene uno, dos, tres o cuatro heteroátomos o grupos de heteroátomos seleccionados independientemente de N, -O-, -S-, -NH-, -S(=O)2- y -S(=O)-.
2. El compuesto, su isómero geométrico o estereoisómero o su sal farmacéuticamente aceptable según la reivindicación 1, donde R se selecciona de H, F, Cl, Br, I, OH, NH2, CN, alquilo C1-3, alquil(C1-3)-S(=O)2- y alquil(C1-3)-O-, donde el alquilo C1-3, alquil(C1-3)-S(=O)2- o alquil(C1-3)-O- está opcionalmente sustituido con uno, dos o tres R'; opcionalmente, R2 se selecciona de H y Me;
opcionalmente, R3 se selecciona de H, alquilo C1-4, alquil(C1-4)-O-alquilo(C1-4), ciclobutilo, -alquil(C1-3)-ciclobutilo, -alquil(C1-3)-ciclopropilo, -alquil(C1-3)-tetrahidrofuranilo y -alquil(C1-3)-tetrahidropiranilo, donde el alquilo C1-4, alquil(C1-4)-O-alquilo(C1-4), ciclobutilo, -alquil(C1-3)-ciclobutilo, -alquil(C1-3)-ciclopropilo, -alquil(C1-3)-tetrahidrofuranilo o -alquil(C1-3)-tetrahidropiranilo está opcionalmente sustituido con uno, dos o tres R.
3. El compuesto, su isómero geométrico o estereoisómero o su sal farmacéuticamente aceptable según la o

,0 .  II
II
el Me, Et, ' o O está opcionalmente sustituido con uno, dos o tres R'; opcionalmente, R3 se

, .
4. El compuesto, su isómero geométrico o estereoisómero o su sal farmacéuticamente aceptable según la reivindicación 3, donde R se selecciona de H, F, Cl, Br, I, OH, N H 2 , C N , Me, C H 2 F , C H F 2 , F C 3 , Et,

5. El compuesto, su isómero geométrico o estereoisómero o su sal farmacéuticamente aceptable según una cualquiera de las reivindicaciones 1-4, donde R2 y R3 están conectados para formar un heterocicloalquilo de 6-8 miembros opcionalmente sustituido con uno, dos o tres R.
6. El compuesto, su isómero geométrico o estereoisómero o su sal farmacéuticamente aceptable según la
Rz' r R3 
reivindicación 5, donde la unidad estructural se selecciona ’ donde el

7. El compuesto, su isómero geométrico o estereoisómero o su sal farmacéuticamente aceptable según la
R2' r Rs 
reivindicación 6, donde la unidad estructural ' se selecciona
8. El compuesto, su isómero geométrico o estereoisómero o su sal farmacéuticamente aceptable según una cualquiera de las reivindicaciones 1-4, donde el anillo B se selecciona de tetrahidrofuranilo, tetrahidrotienilo, 1,3-dioxolanilo, pirrolidinilo, tiazolilo, pirazolilo e imidazolilo, donde el tetrahidrofuranilo, tetrahidrotienilo, 1,3-dioxolanilo, pirrolidinilo, tiazolilo, pirazolilo o imidazolilo está opcionalmente sustituido con uno, dos o tres R.
9. El compuesto, su isómero geométrico o estereoisómero o su sal farmacéuticamente aceptable según la

reivindicación 8, donde la unidad estructural \ se selecciona de

10. El compuesto, su isómero geométrico o estereoisómero o su sal farmacéuticamente aceptable según una cualquiera de las reivindicaciones 1-7, que se selecciona de

donde R, Ri o R2 es como se define en una cualquiera de las reivindicaciones 1-7.
11. Un compuesto según la reivindicación 1, uno de sus isómeros geométricos o estereoisómeros o una de sus sales farmacéuticamente aceptables, donde el compuesto se selecciona de
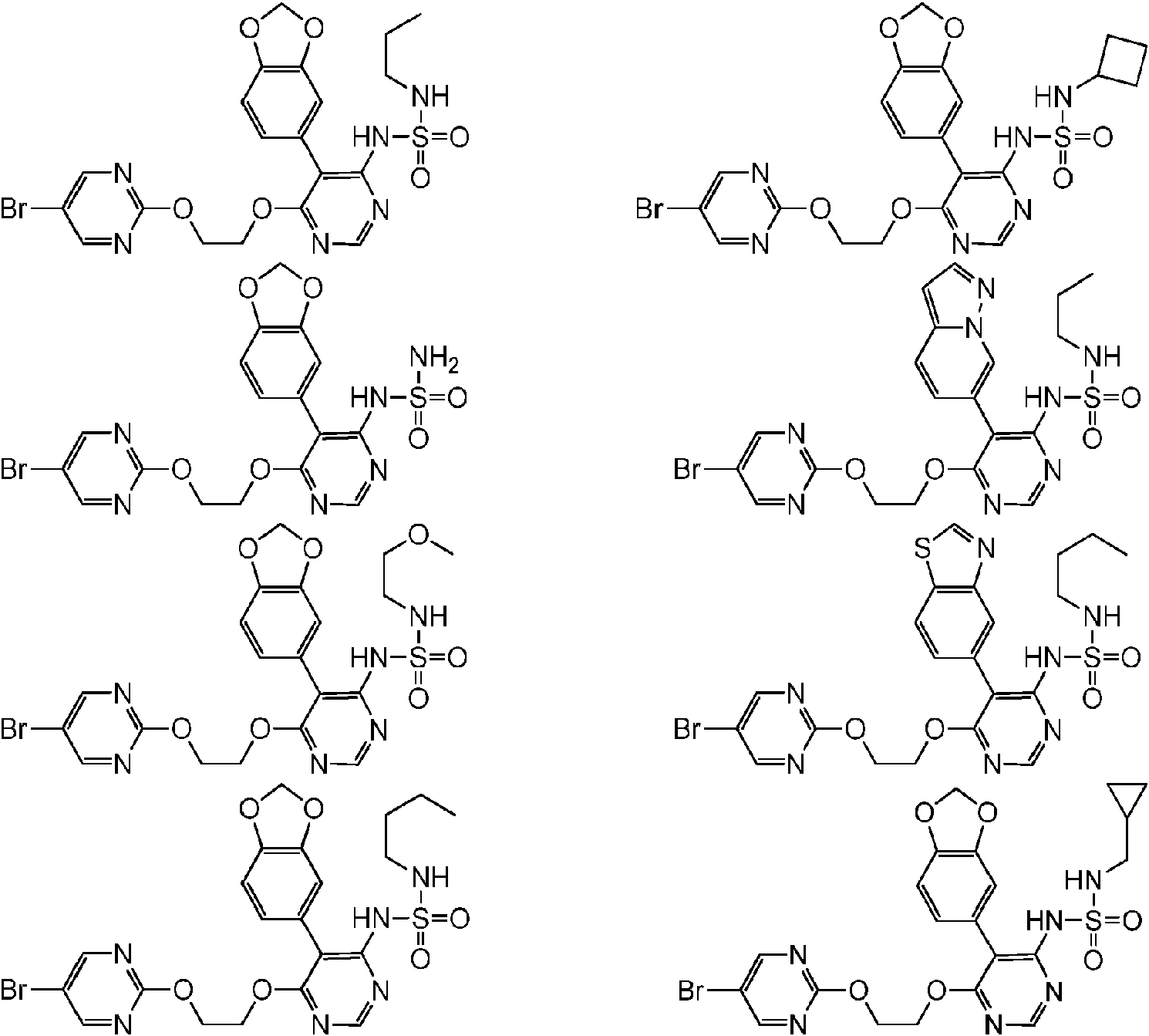


12. El compuesto, su isómero geométrico o estereoisómero o su sal farmacéuticamente aceptable según la reivindicación 11, que se selecciona de

13. Una composición farmacéutica, que comprende una cantidad terapéuticamente eficaz del compuesto o la sal farmacéuticamente aceptable según una cualquiera de las reivindicaciones 1-12 como ingrediente activo y un vehículo
farmacéuticamente aceptable.
14. El compuesto o la sal farmacéuticamente aceptable según una cualquiera de las reivindicaciones 1-12 o la composición según la reivindicación 13, para el uso como antagonista de los receptores ETa.
15. El compuesto o la sal farmacéuticamente aceptable según una cualquiera de las reivindicaciones 1-12 o la composición según la reivindicación 13, para el uso en el tratamiento de la hipertensión arterial pulmonar, la hipertensión primaria, la hipertensión refractaria, la nefropatía diabética y el vasoespasmo intracraneal.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201711168111 | 2017-11-21 | ||
| PCT/CN2018/116196 WO2019101039A1 (zh) | 2017-11-21 | 2018-11-19 | 嘧啶磺酰胺类衍生物、其制备方法及其在医药上的应用 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2936864T3 true ES2936864T3 (es) | 2023-03-22 |
Family
ID=66630436
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES18881280T Active ES2936864T3 (es) | 2017-11-21 | 2018-11-19 | Derivado de pirimidinosulfamida y método de preparación y aplicación médica del mismo |
Country Status (19)
| Country | Link |
|---|---|
| US (1) | US11319305B2 (es) |
| EP (1) | EP3719013B1 (es) |
| JP (1) | JP7245832B2 (es) |
| CN (1) | CN111479808B (es) |
| AU (1) | AU2018372752B2 (es) |
| BR (1) | BR112020010149A8 (es) |
| CA (1) | CA3083019A1 (es) |
| DK (1) | DK3719013T3 (es) |
| ES (1) | ES2936864T3 (es) |
| FI (1) | FI3719013T3 (es) |
| HR (1) | HRP20230318T1 (es) |
| HU (1) | HUE061515T2 (es) |
| IL (1) | IL274773B2 (es) |
| LT (1) | LT3719013T (es) |
| PL (1) | PL3719013T3 (es) |
| PT (1) | PT3719013T (es) |
| RS (1) | RS64040B1 (es) |
| SI (1) | SI3719013T1 (es) |
| WO (1) | WO2019101039A1 (es) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN109232546B (zh) * | 2018-09-25 | 2020-09-04 | 中国人民解放军总医院 | 一种嘧啶磺酰胺类衍生物的医药用途 |
| BR112021023328A8 (pt) * | 2019-05-22 | 2023-04-25 | Shijiazhuang Sagacity New Drug Dev Co Ltd | Forma de cristal de composto de pirimidina sulfonamida e método de preparação para a mesma |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2315614C (en) * | 1999-07-29 | 2004-11-02 | Pfizer Inc. | Pyrazoles |
| EP1160248A1 (en) * | 2000-05-31 | 2001-12-05 | Pfizer Inc. | N-(Isoxazol-5-yl)-sulfonamide derivatives and their use as endothelin antagonists |
| JP3490959B2 (ja) | 2000-07-11 | 2004-01-26 | 三洋電機株式会社 | 受光素子及び受光モジュール |
| IL155805A0 (en) * | 2000-12-18 | 2003-12-23 | Actelion Pharmaceuticals Ltd | Novel sulfamides and their use as endothelin receptor antagonists |
| ATE423103T1 (de) | 2002-12-02 | 2009-03-15 | Actelion Pharmaceuticals Ltd | Pyrimidin-sulfonamide und ihre verwendung als endothelin-rezeptor-antagonisten |
| TW200628467A (en) | 2004-11-11 | 2006-08-16 | Actelion Pharmaceuticals Ltd | Novel sulfamides |
-
2018
- 2018-11-19 DK DK18881280.4T patent/DK3719013T3/da active
- 2018-11-19 PL PL18881280.4T patent/PL3719013T3/pl unknown
- 2018-11-19 US US16/764,654 patent/US11319305B2/en active Active
- 2018-11-19 LT LTEPPCT/CN2018/116196T patent/LT3719013T/lt unknown
- 2018-11-19 RS RS20230197A patent/RS64040B1/sr unknown
- 2018-11-19 HU HUE18881280A patent/HUE061515T2/hu unknown
- 2018-11-19 EP EP18881280.4A patent/EP3719013B1/en active Active
- 2018-11-19 PT PT188812804T patent/PT3719013T/pt unknown
- 2018-11-19 WO PCT/CN2018/116196 patent/WO2019101039A1/zh unknown
- 2018-11-19 AU AU2018372752A patent/AU2018372752B2/en active Active
- 2018-11-19 SI SI201830884T patent/SI3719013T1/sl unknown
- 2018-11-19 FI FIEP18881280.4T patent/FI3719013T3/fi active
- 2018-11-19 BR BR112020010149A patent/BR112020010149A8/pt unknown
- 2018-11-19 CN CN201880074739.3A patent/CN111479808B/zh active Active
- 2018-11-19 ES ES18881280T patent/ES2936864T3/es active Active
- 2018-11-19 HR HRP20230318TT patent/HRP20230318T1/hr unknown
- 2018-11-19 CA CA3083019A patent/CA3083019A1/en active Pending
- 2018-11-19 JP JP2020528076A patent/JP7245832B2/ja active Active
-
2020
- 2020-05-19 IL IL274773A patent/IL274773B2/en unknown
Also Published As
| Publication number | Publication date |
|---|---|
| CN111479808A (zh) | 2020-07-31 |
| HUE061515T2 (hu) | 2023-07-28 |
| HRP20230318T1 (hr) | 2023-05-12 |
| EP3719013A1 (en) | 2020-10-07 |
| PT3719013T (pt) | 2023-02-01 |
| BR112020010149A8 (pt) | 2023-01-17 |
| IL274773B2 (en) | 2023-08-01 |
| CN111479808B (zh) | 2023-02-24 |
| EP3719013B1 (en) | 2023-01-04 |
| IL274773B1 (en) | 2023-04-01 |
| WO2019101039A1 (zh) | 2019-05-31 |
| JP2021504330A (ja) | 2021-02-15 |
| RU2020119900A3 (es) | 2021-12-23 |
| EP3719013A4 (en) | 2021-04-21 |
| US20200283425A1 (en) | 2020-09-10 |
| LT3719013T (lt) | 2023-05-10 |
| AU2018372752B2 (en) | 2023-02-02 |
| RU2020119900A (ru) | 2021-12-23 |
| US11319305B2 (en) | 2022-05-03 |
| DK3719013T3 (da) | 2023-04-03 |
| FI3719013T3 (fi) | 2023-04-03 |
| PL3719013T3 (pl) | 2023-05-08 |
| CA3083019A1 (en) | 2019-05-31 |
| BR112020010149A2 (pt) | 2020-11-10 |
| IL274773A (en) | 2020-07-30 |
| RS64040B1 (sr) | 2023-04-28 |
| JP7245832B2 (ja) | 2023-03-24 |
| SI3719013T1 (sl) | 2023-05-31 |
| AU2018372752A1 (en) | 2020-07-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2870524T3 (es) | Derivado de piridina como inhibidor de ASK1, y método de preparación y uso del mismo | |
| ES2953512T3 (es) | Compuestos de arilo o heteroarilo sustituidos con amina como inhibidores de EHMT1 y EHMT2 | |
| ES2688868T3 (es) | Piridopirazinas anticáncer a través de la inhibición de FGFR quinasas | |
| AU2015225745B2 (en) | Heterocyclic compounds | |
| ES2739348T3 (es) | Pteridinas como inhibidores del FGFR | |
| ES2621461T3 (es) | Inhibidores de quinasa de pirazolilquinoxalina | |
| CN104011052B (zh) | 化合物 | |
| ES2666155T3 (es) | Moduladores de P2X7 | |
| PT2552909E (pt) | Derivados de 4-aminopirimidina e sua utilização como antagonistas de recetor de adenosina a2a | |
| AU2014250836A1 (en) | Quinazolines and azaquinazolines as dual inhibitors of RAS/RAF/MEK/ERK and PI3K/AKT/PTEN/mTOR pathways | |
| ES2936864T3 (es) | Derivado de pirimidinosulfamida y método de preparación y aplicación médica del mismo | |
| US20220185811A1 (en) | Fgfr4 kinase inhibitor and preparation method therefor and use thereof | |
| AU2024200781A1 (en) | Substituted fused bi- or tri-heterocyclic compounds as ehmt2 inhibitors | |
| EP3533787B1 (en) | Pyridone compound as c-met inhibitor | |
| ES2954453T3 (es) | Compuestos de pirimidina y métodos que utilizan los mismos | |
| CN114436976B (zh) | 一种新型喹唑啉类衍生物及其制备和应用 | |
| CN111655689B (zh) | 吡唑并吡啶酮化合物 | |
| ES2926518T3 (es) | Compuestos de pirazolopiridinona | |
| RU2773844C2 (ru) | Производное пиримидинсульфамида и способ его получения, и медицинское применение | |
| RU2773844C9 (ru) | Производное пиримидинсульфамида и способ его получения, и медицинское применение | |
| WO2023004438A2 (en) | Fret-based assays | |
| EP3006438A1 (en) | Cyclic aminomethyl pyrimidine derivative | |
| CN111362928A (zh) | 2,4-二氨基嘧啶衍生物 |