ES2922158T3 - Compuestos cristalinos - Google Patents
Compuestos cristalinos Download PDFInfo
- Publication number
- ES2922158T3 ES2922158T3 ES19196556T ES19196556T ES2922158T3 ES 2922158 T3 ES2922158 T3 ES 2922158T3 ES 19196556 T ES19196556 T ES 19196556T ES 19196556 T ES19196556 T ES 19196556T ES 2922158 T3 ES2922158 T3 ES 2922158T3
- Authority
- ES
- Spain
- Prior art keywords
- crystalline form
- xrpd
- radiation
- pattern
- crystal
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 150000001875 compounds Chemical class 0.000 title description 30
- ACVMJAJGCQUPKX-LIOBNPLQSA-N (1r,5s)-1-naphthalen-2-yl-3-azabicyclo[3.1.0]hexane;hydrochloride Chemical compound Cl.C1=CC=CC2=CC([C@@]34CNC[C@H]4C3)=CC=C21 ACVMJAJGCQUPKX-LIOBNPLQSA-N 0.000 claims abstract description 24
- 238000000634 powder X-ray diffraction Methods 0.000 claims description 354
- 230000005855 radiation Effects 0.000 claims description 163
- 229910002483 Cu Ka Inorganic materials 0.000 claims description 75
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 claims description 46
- 239000008194 pharmaceutical composition Substances 0.000 claims description 11
- 208000036864 Attention deficit/hyperactivity disease Diseases 0.000 claims description 10
- 208000015802 attention deficit-hyperactivity disease Diseases 0.000 claims description 10
- 238000011282 treatment Methods 0.000 claims description 10
- 239000003085 diluting agent Substances 0.000 claims description 9
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 claims description 9
- 208000006096 Attention Deficit Disorder with Hyperactivity Diseases 0.000 claims description 7
- 238000011321 prophylaxis Methods 0.000 claims description 6
- 239000012730 sustained-release form Substances 0.000 claims description 6
- 238000013268 sustained release Methods 0.000 claims description 5
- 208000019901 Anxiety disease Diseases 0.000 claims description 3
- 208000011117 substance-related disease Diseases 0.000 claims description 3
- 230000036506 anxiety Effects 0.000 claims description 2
- 201000009032 substance abuse Diseases 0.000 claims 1
- 231100000736 substance abuse Toxicity 0.000 claims 1
- 239000000203 mixture Substances 0.000 abstract description 96
- 238000000034 method Methods 0.000 abstract description 30
- 238000013519 translation Methods 0.000 abstract description 2
- 238000004519 manufacturing process Methods 0.000 abstract 1
- 239000013078 crystal Substances 0.000 description 209
- 239000007787 solid Substances 0.000 description 94
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 88
- 239000000523 sample Substances 0.000 description 81
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 75
- 239000000243 solution Substances 0.000 description 73
- 238000007792 addition Methods 0.000 description 47
- JMMWKPVZQRWMSS-UHFFFAOYSA-N isopropanol acetate Natural products CC(C)OC(C)=O JMMWKPVZQRWMSS-UHFFFAOYSA-N 0.000 description 43
- 229940011051 isopropyl acetate Drugs 0.000 description 43
- GWYFCOCPABKNJV-UHFFFAOYSA-N isovaleric acid Chemical compound CC(C)CC(O)=O GWYFCOCPABKNJV-UHFFFAOYSA-N 0.000 description 43
- 239000007858 starting material Substances 0.000 description 37
- 239000000725 suspension Substances 0.000 description 36
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 34
- 239000011541 reaction mixture Substances 0.000 description 32
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 31
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 30
- 230000005540 biological transmission Effects 0.000 description 30
- 239000012071 phase Substances 0.000 description 30
- 239000000047 product Substances 0.000 description 29
- 238000003756 stirring Methods 0.000 description 28
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 26
- 239000012065 filter cake Substances 0.000 description 26
- 239000002904 solvent Substances 0.000 description 26
- 238000004458 analytical method Methods 0.000 description 25
- 150000003839 salts Chemical class 0.000 description 25
- 125000004429 atom Chemical group 0.000 description 23
- 238000006243 chemical reaction Methods 0.000 description 23
- 238000001816 cooling Methods 0.000 description 23
- 238000012856 packing Methods 0.000 description 22
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 22
- 238000000113 differential scanning calorimetry Methods 0.000 description 21
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 21
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 21
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 20
- 208000035475 disorder Diseases 0.000 description 20
- 239000012074 organic phase Substances 0.000 description 20
- 238000001757 thermogravimetry curve Methods 0.000 description 20
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 18
- 239000008186 active pharmaceutical agent Substances 0.000 description 18
- 238000010586 diagram Methods 0.000 description 18
- 238000010438 heat treatment Methods 0.000 description 18
- 238000002474 experimental method Methods 0.000 description 17
- 239000000463 material Substances 0.000 description 17
- 229910052757 nitrogen Inorganic materials 0.000 description 17
- 239000008346 aqueous phase Substances 0.000 description 16
- 239000001257 hydrogen Substances 0.000 description 16
- 229910052739 hydrogen Inorganic materials 0.000 description 16
- HKHCSWPSUSWGLI-CABCVRRESA-N (1r,5s)-1-naphthalen-2-yl-3-azabicyclo[3.1.0]hexane Chemical compound C1=CC=CC2=CC([C@@]34CNC[C@H]4C3)=CC=C21 HKHCSWPSUSWGLI-CABCVRRESA-N 0.000 description 15
- 238000004090 dissolution Methods 0.000 description 15
- VYFYYTLLBUKUHU-UHFFFAOYSA-N dopamine Chemical compound NCCC1=CC=C(O)C(O)=C1 VYFYYTLLBUKUHU-UHFFFAOYSA-N 0.000 description 14
- 238000001035 drying Methods 0.000 description 14
- 239000000284 extract Substances 0.000 description 14
- 238000002360 preparation method Methods 0.000 description 14
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 13
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 13
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 13
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 13
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 13
- 239000000843 powder Substances 0.000 description 13
- 229910052710 silicon Inorganic materials 0.000 description 13
- 239000010703 silicon Substances 0.000 description 13
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 13
- 238000001914 filtration Methods 0.000 description 12
- 238000012546 transfer Methods 0.000 description 12
- 238000010583 slow cooling Methods 0.000 description 11
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 10
- 239000004677 Nylon Substances 0.000 description 10
- 238000010899 nucleation Methods 0.000 description 10
- 229920001778 nylon Polymers 0.000 description 10
- 239000002245 particle Substances 0.000 description 10
- 239000004926 polymethyl methacrylate Substances 0.000 description 10
- QZAYGJVTTNCVMB-UHFFFAOYSA-N serotonin Chemical compound C1=C(O)C=C2C(CCN)=CNC2=C1 QZAYGJVTTNCVMB-UHFFFAOYSA-N 0.000 description 10
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 10
- 238000013480 data collection Methods 0.000 description 9
- 239000000706 filtrate Substances 0.000 description 9
- 230000004580 weight loss Effects 0.000 description 9
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 8
- FNYLWPVRPXGIIP-UHFFFAOYSA-N Triamterene Chemical compound NC1=NC2=NC(N)=NC(N)=C2N=C1C1=CC=CC=C1 FNYLWPVRPXGIIP-UHFFFAOYSA-N 0.000 description 8
- 238000010521 absorption reaction Methods 0.000 description 8
- QGZKDVFQNNGYKY-UHFFFAOYSA-N ammonia Natural products N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 8
- 230000008859 change Effects 0.000 description 8
- 238000001704 evaporation Methods 0.000 description 8
- 230000008020 evaporation Effects 0.000 description 8
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 8
- 238000001556 precipitation Methods 0.000 description 8
- 238000010992 reflux Methods 0.000 description 8
- 230000007704 transition Effects 0.000 description 8
- 238000003828 vacuum filtration Methods 0.000 description 8
- WSVLPVUVIUVCRA-KPKNDVKVSA-N Alpha-lactose monohydrate Chemical compound O.O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O WSVLPVUVIUVCRA-KPKNDVKVSA-N 0.000 description 7
- 239000012296 anti-solvent Substances 0.000 description 7
- 230000008033 biological extinction Effects 0.000 description 7
- 229910052799 carbon Inorganic materials 0.000 description 7
- 239000012141 concentrate Substances 0.000 description 7
- 229960003638 dopamine Drugs 0.000 description 7
- 238000002844 melting Methods 0.000 description 7
- 230000008018 melting Effects 0.000 description 7
- 230000006911 nucleation Effects 0.000 description 7
- 238000012360 testing method Methods 0.000 description 7
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 6
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 description 6
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 6
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 6
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 6
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 6
- UORVGPXVDQYIDP-UHFFFAOYSA-N borane Chemical compound B UORVGPXVDQYIDP-UHFFFAOYSA-N 0.000 description 6
- 239000003795 chemical substances by application Substances 0.000 description 6
- 238000012937 correction Methods 0.000 description 6
- 230000001404 mediated effect Effects 0.000 description 6
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 6
- 230000035939 shock Effects 0.000 description 6
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 6
- 239000007790 solid phase Substances 0.000 description 6
- SFLSHLFXELFNJZ-QMMMGPOBSA-N (-)-norepinephrine Chemical compound NC[C@H](O)C1=CC=C(O)C(O)=C1 SFLSHLFXELFNJZ-QMMMGPOBSA-N 0.000 description 5
- LPCWDVLDJVZIHA-UHFFFAOYSA-N 2-naphthalen-2-ylacetonitrile Chemical compound C1=CC=CC2=CC(CC#N)=CC=C21 LPCWDVLDJVZIHA-UHFFFAOYSA-N 0.000 description 5
- 102220470359 Thymosin beta-10_H14A_mutation Human genes 0.000 description 5
- 102220531684 WD repeat and coiled-coil-containing protein_H12A_mutation Human genes 0.000 description 5
- 102220540101 WD repeat and coiled-coil-containing protein_H16A_mutation Human genes 0.000 description 5
- 150000001412 amines Chemical class 0.000 description 5
- 230000006399 behavior Effects 0.000 description 5
- 230000015572 biosynthetic process Effects 0.000 description 5
- 238000002425 crystallisation Methods 0.000 description 5
- 230000008025 crystallization Effects 0.000 description 5
- 238000000354 decomposition reaction Methods 0.000 description 5
- 238000002050 diffraction method Methods 0.000 description 5
- 230000000694 effects Effects 0.000 description 5
- 238000000605 extraction Methods 0.000 description 5
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 5
- 235000019341 magnesium sulphate Nutrition 0.000 description 5
- 239000011159 matrix material Substances 0.000 description 5
- 229960002748 norepinephrine Drugs 0.000 description 5
- SFLSHLFXELFNJZ-UHFFFAOYSA-N norepinephrine Natural products NCC(O)C1=CC=C(O)C(O)=C1 SFLSHLFXELFNJZ-UHFFFAOYSA-N 0.000 description 5
- 230000003389 potentiating effect Effects 0.000 description 5
- 238000003786 synthesis reaction Methods 0.000 description 5
- HKHCSWPSUSWGLI-UHFFFAOYSA-N 1-naphthalen-2-yl-3-azabicyclo[3.1.0]hexane Chemical compound C1=CC=CC2=CC(C34CNCC4C3)=CC=C21 HKHCSWPSUSWGLI-UHFFFAOYSA-N 0.000 description 4
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 4
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 4
- 238000010268 HPLC based assay Methods 0.000 description 4
- 229910052782 aluminium Inorganic materials 0.000 description 4
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 4
- MCQRPQCQMGVWIQ-UHFFFAOYSA-N boron;methylsulfanylmethane Chemical compound [B].CSC MCQRPQCQMGVWIQ-UHFFFAOYSA-N 0.000 description 4
- 239000012267 brine Substances 0.000 description 4
- 238000004364 calculation method Methods 0.000 description 4
- 238000007707 calorimetry Methods 0.000 description 4
- 235000011089 carbon dioxide Nutrition 0.000 description 4
- 238000009792 diffusion process Methods 0.000 description 4
- 238000006073 displacement reaction Methods 0.000 description 4
- 230000006870 function Effects 0.000 description 4
- 239000011521 glass Substances 0.000 description 4
- 238000000227 grinding Methods 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- 235000019359 magnesium stearate Nutrition 0.000 description 4
- KJIFKLIQANRMOU-UHFFFAOYSA-N oxidanium;4-methylbenzenesulfonate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1 KJIFKLIQANRMOU-UHFFFAOYSA-N 0.000 description 4
- 239000002244 precipitate Substances 0.000 description 4
- 229920006395 saturated elastomer Polymers 0.000 description 4
- WRIKHQLVHPKCJU-UHFFFAOYSA-N sodium bis(trimethylsilyl)amide Chemical compound C[Si](C)(C)N([Na])[Si](C)(C)C WRIKHQLVHPKCJU-UHFFFAOYSA-N 0.000 description 4
- 239000012453 solvate Substances 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- 208000024891 symptom Diseases 0.000 description 4
- 239000003174 triple reuptake inhibitor Substances 0.000 description 4
- 238000005406 washing Methods 0.000 description 4
- BRLQWZUYTZBJKN-GSVOUGTGSA-N (+)-Epichlorohydrin Chemical compound ClC[C@@H]1CO1 BRLQWZUYTZBJKN-GSVOUGTGSA-N 0.000 description 3
- BYEAHWXPCBROCE-UHFFFAOYSA-N 1,1,1,3,3,3-hexafluoropropan-2-ol Chemical compound FC(F)(F)C(O)C(F)(F)F BYEAHWXPCBROCE-UHFFFAOYSA-N 0.000 description 3
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 3
- RHQDFWAXVIIEBN-UHFFFAOYSA-N Trifluoroethanol Chemical compound OCC(F)(F)F RHQDFWAXVIIEBN-UHFFFAOYSA-N 0.000 description 3
- 230000004888 barrier function Effects 0.000 description 3
- 239000012455 biphasic mixture Substances 0.000 description 3
- 229910000085 borane Inorganic materials 0.000 description 3
- 238000012512 characterization method Methods 0.000 description 3
- 230000002860 competitive effect Effects 0.000 description 3
- 239000012043 crude product Substances 0.000 description 3
- BNIILDVGGAEEIG-UHFFFAOYSA-L disodium hydrogen phosphate Chemical compound [Na+].[Na+].OP([O-])([O-])=O BNIILDVGGAEEIG-UHFFFAOYSA-L 0.000 description 3
- 235000019439 ethyl acetate Nutrition 0.000 description 3
- 238000011156 evaluation Methods 0.000 description 3
- 238000004108 freeze drying Methods 0.000 description 3
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 3
- 239000000314 lubricant Substances 0.000 description 3
- 238000005259 measurement Methods 0.000 description 3
- 238000010907 mechanical stirring Methods 0.000 description 3
- 238000005191 phase separation Methods 0.000 description 3
- 238000002390 rotary evaporation Methods 0.000 description 3
- 229940076279 serotonin Drugs 0.000 description 3
- 238000004467 single crystal X-ray diffraction Methods 0.000 description 3
- 239000011734 sodium Substances 0.000 description 3
- 229910052708 sodium Inorganic materials 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- 238000002336 sorption--desorption measurement Methods 0.000 description 3
- 238000003860 storage Methods 0.000 description 3
- 238000002076 thermal analysis method Methods 0.000 description 3
- 238000002411 thermogravimetry Methods 0.000 description 3
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical class CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 3
- 230000009466 transformation Effects 0.000 description 3
- 241000669003 Aspidiotus destructor Species 0.000 description 2
- 208000027448 Attention Deficit and Disruptive Behavior disease Diseases 0.000 description 2
- 208000027691 Conduct disease Diseases 0.000 description 2
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 2
- 208000020401 Depressive disease Diseases 0.000 description 2
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 2
- WHNWPMSKXPGLAX-UHFFFAOYSA-N N-Vinyl-2-pyrrolidone Chemical compound C=CN1CCCC1=O WHNWPMSKXPGLAX-UHFFFAOYSA-N 0.000 description 2
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 2
- 208000008589 Obesity Diseases 0.000 description 2
- 230000002547 anomalous effect Effects 0.000 description 2
- 238000013459 approach Methods 0.000 description 2
- 238000003556 assay Methods 0.000 description 2
- 208000029560 autism spectrum disease Diseases 0.000 description 2
- 238000013477 bayesian statistics method Methods 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 230000000035 biogenic effect Effects 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 238000004296 chiral HPLC Methods 0.000 description 2
- 208000010877 cognitive disease Diseases 0.000 description 2
- 239000002178 crystalline material Substances 0.000 description 2
- 238000002447 crystallographic data Methods 0.000 description 2
- 239000002274 desiccant Substances 0.000 description 2
- 229940061607 dibasic sodium phosphate Drugs 0.000 description 2
- 238000001938 differential scanning calorimetry curve Methods 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- 239000006185 dispersion Substances 0.000 description 2
- 238000009826 distribution Methods 0.000 description 2
- 206010013663 drug dependence Diseases 0.000 description 2
- 230000001747 exhibiting effect Effects 0.000 description 2
- 239000010439 graphite Substances 0.000 description 2
- 229910002804 graphite Inorganic materials 0.000 description 2
- 239000012729 immediate-release (IR) formulation Substances 0.000 description 2
- 208000024714 major depressive disease Diseases 0.000 description 2
- QSHDDOUJBYECFT-UHFFFAOYSA-N mercury Chemical compound [Hg] QSHDDOUJBYECFT-UHFFFAOYSA-N 0.000 description 2
- 229910052753 mercury Inorganic materials 0.000 description 2
- 238000000386 microscopy Methods 0.000 description 2
- 239000012452 mother liquor Substances 0.000 description 2
- 150000002825 nitriles Chemical class 0.000 description 2
- 235000020824 obesity Nutrition 0.000 description 2
- 239000008203 oral pharmaceutical composition Substances 0.000 description 2
- 230000036961 partial effect Effects 0.000 description 2
- 230000037361 pathway Effects 0.000 description 2
- 238000001907 polarising light microscopy Methods 0.000 description 2
- 230000010287 polarization Effects 0.000 description 2
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 2
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 2
- 230000002265 prevention Effects 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- 238000010791 quenching Methods 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 239000012047 saturated solution Substances 0.000 description 2
- 201000000980 schizophrenia Diseases 0.000 description 2
- JHJLBTNAGRQEKS-UHFFFAOYSA-M sodium bromide Chemical compound [Na+].[Br-] JHJLBTNAGRQEKS-UHFFFAOYSA-M 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- 230000009897 systematic effect Effects 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- 208000016686 tic disease Diseases 0.000 description 2
- 230000000007 visual effect Effects 0.000 description 2
- 239000003039 volatile agent Substances 0.000 description 2
- SNICXCGAKADSCV-JTQLQIEISA-N (-)-Nicotine Chemical compound CN1CCC[C@H]1C1=CC=CN=C1 SNICXCGAKADSCV-JTQLQIEISA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- OKMWKBLSFKFYGZ-UHFFFAOYSA-N 1-behenoylglycerol Chemical compound CCCCCCCCCCCCCCCCCCCCCC(=O)OCC(O)CO OKMWKBLSFKFYGZ-UHFFFAOYSA-N 0.000 description 1
- 208000007848 Alcoholism Diseases 0.000 description 1
- 206010003805 Autism Diseases 0.000 description 1
- 208000020706 Autistic disease Diseases 0.000 description 1
- 229920003084 Avicel® PH-102 Polymers 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- 208000000094 Chronic Pain Diseases 0.000 description 1
- 208000022497 Cocaine-Related disease Diseases 0.000 description 1
- 208000028698 Cognitive impairment Diseases 0.000 description 1
- 229910017488 Cu K Inorganic materials 0.000 description 1
- 229910017541 Cu-K Inorganic materials 0.000 description 1
- HTJDQJBWANPRPF-UHFFFAOYSA-N Cyclopropylamine Chemical compound NC1CC1 HTJDQJBWANPRPF-UHFFFAOYSA-N 0.000 description 1
- 208000026331 Disruptive, Impulse Control, and Conduct disease Diseases 0.000 description 1
- 208000030814 Eating disease Diseases 0.000 description 1
- 208000019454 Feeding and Eating disease Diseases 0.000 description 1
- 238000005033 Fourier transform infrared spectroscopy Methods 0.000 description 1
- 208000001613 Gambling Diseases 0.000 description 1
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 1
- 208000030990 Impulse-control disease Diseases 0.000 description 1
- 208000020358 Learning disease Diseases 0.000 description 1
- 206010026749 Mania Diseases 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 208000003863 Marijuana Abuse Diseases 0.000 description 1
- 229920003096 Methocel™ K100M Polymers 0.000 description 1
- 229920003095 Methocel™ K15M Polymers 0.000 description 1
- 229920003094 Methocel™ K4M Polymers 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 229920000881 Modified starch Polymers 0.000 description 1
- 208000021384 Obsessive-Compulsive disease Diseases 0.000 description 1
- 208000026251 Opioid-Related disease Diseases 0.000 description 1
- 208000002193 Pain Diseases 0.000 description 1
- 208000018737 Parkinson disease Diseases 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 208000003028 Stuttering Diseases 0.000 description 1
- 208000030886 Traumatic Brain injury Diseases 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 238000013019 agitation Methods 0.000 description 1
- 201000007930 alcohol dependence Diseases 0.000 description 1
- 208000028505 alcohol-related disease Diseases 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 238000004164 analytical calibration Methods 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 230000003542 behavioural effect Effects 0.000 description 1
- GGNALUCSASGNCK-UHFFFAOYSA-N carbon dioxide;propan-2-ol Chemical compound O=C=O.CC(C)O GGNALUCSASGNCK-UHFFFAOYSA-N 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 208000015114 central nervous system disease Diseases 0.000 description 1
- 239000003610 charcoal Substances 0.000 description 1
- 239000000460 chlorine Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 229940069078 citric acid / sodium citrate Drugs 0.000 description 1
- 238000010549 co-Evaporation Methods 0.000 description 1
- 230000019771 cognition Effects 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 229960000913 crospovidone Drugs 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 238000003795 desorption Methods 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 238000007416 differential thermogravimetric analysis Methods 0.000 description 1
- 238000002845 discoloration Methods 0.000 description 1
- 229910000397 disodium phosphate Inorganic materials 0.000 description 1
- 235000019800 disodium phosphate Nutrition 0.000 description 1
- 235000014632 disordered eating Nutrition 0.000 description 1
- 208000035548 disruptive behavior disease Diseases 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 239000003814 drug Substances 0.000 description 1
- 208000024732 dysthymic disease Diseases 0.000 description 1
- 238000011067 equilibration Methods 0.000 description 1
- MVPICKVDHDWCJQ-UHFFFAOYSA-N ethyl 3-pyrrolidin-1-ylpropanoate Chemical compound CCOC(=O)CCN1CCCC1 MVPICKVDHDWCJQ-UHFFFAOYSA-N 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 239000000835 fiber Substances 0.000 description 1
- 239000011888 foil Substances 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- 239000012520 frozen sample Substances 0.000 description 1
- 230000004927 fusion Effects 0.000 description 1
- 208000037870 generalized anxiety Diseases 0.000 description 1
- 229940049654 glyceryl behenate Drugs 0.000 description 1
- 239000000380 hallucinogen Substances 0.000 description 1
- 239000001307 helium Substances 0.000 description 1
- 229910052734 helium Inorganic materials 0.000 description 1
- SWQJXJOGLNCZEY-UHFFFAOYSA-N helium atom Chemical compound [He] SWQJXJOGLNCZEY-UHFFFAOYSA-N 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- -1 hydroxypropoxyl Chemical group 0.000 description 1
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 1
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 1
- 229920003130 hypromellose 2208 Polymers 0.000 description 1
- 229940031707 hypromellose 2208 Drugs 0.000 description 1
- 229910052738 indium Inorganic materials 0.000 description 1
- APFVFJFRJDLVQX-UHFFFAOYSA-N indium atom Chemical compound [In] APFVFJFRJDLVQX-UHFFFAOYSA-N 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 230000001788 irregular Effects 0.000 description 1
- 230000002427 irreversible effect Effects 0.000 description 1
- 239000010410 layer Substances 0.000 description 1
- 201000003723 learning disability Diseases 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- 239000007791 liquid phase Substances 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229940057948 magnesium stearate Drugs 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 239000000155 melt Substances 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- GRVDJDISBSALJP-UHFFFAOYSA-N methyloxidanyl Chemical group [O]C GRVDJDISBSALJP-UHFFFAOYSA-N 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 208000027061 mild cognitive impairment Diseases 0.000 description 1
- 229910052759 nickel Inorganic materials 0.000 description 1
- 229960002715 nicotine Drugs 0.000 description 1
- SNICXCGAKADSCV-UHFFFAOYSA-N nicotine Natural products CN1CCCC1C1=CC=CN=C1 SNICXCGAKADSCV-UHFFFAOYSA-N 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- 208000024196 oppositional defiant disease Diseases 0.000 description 1
- 239000012044 organic layer Substances 0.000 description 1
- 238000004806 packaging method and process Methods 0.000 description 1
- 208000019906 panic disease Diseases 0.000 description 1
- 208000022821 personality disease Diseases 0.000 description 1
- 238000011170 pharmaceutical development Methods 0.000 description 1
- 208000019899 phobic disease Diseases 0.000 description 1
- 238000010935 polish filtration Methods 0.000 description 1
- 235000013809 polyvinylpolypyrrolidone Nutrition 0.000 description 1
- 229920000523 polyvinylpolypyrrolidone Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 208000028173 post-traumatic stress disease Diseases 0.000 description 1
- 229940069328 povidone Drugs 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 208000020016 psychiatric disease Diseases 0.000 description 1
- 238000010926 purge Methods 0.000 description 1
- 230000000171 quenching effect Effects 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 230000000306 recurrent effect Effects 0.000 description 1
- 239000012925 reference material Substances 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 239000012266 salt solution Substances 0.000 description 1
- 238000013341 scale-up Methods 0.000 description 1
- 230000009329 sexual behaviour Effects 0.000 description 1
- 229920002379 silicone rubber Polymers 0.000 description 1
- 239000004945 silicone rubber Substances 0.000 description 1
- 208000019116 sleep disease Diseases 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 230000000391 smoking effect Effects 0.000 description 1
- 229920003109 sodium starch glycolate Polymers 0.000 description 1
- 229940079832 sodium starch glycolate Drugs 0.000 description 1
- 239000008109 sodium starch glycolate Substances 0.000 description 1
- 229940045902 sodium stearyl fumarate Drugs 0.000 description 1
- 230000002269 spontaneous effect Effects 0.000 description 1
- 229910001220 stainless steel Inorganic materials 0.000 description 1
- 239000010935 stainless steel Substances 0.000 description 1
- 229940032147 starch Drugs 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 229940033134 talc Drugs 0.000 description 1
- 230000008685 targeting Effects 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 230000009529 traumatic brain injury Effects 0.000 description 1
- 238000011179 visual inspection Methods 0.000 description 1
- 238000005303 weighing Methods 0.000 description 1
- 230000004584 weight gain Effects 0.000 description 1
- 235000019786 weight gain Nutrition 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/52—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring condensed with a ring other than six-membered
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/403—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with carbocyclic rings, e.g. carbazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C211/00—Compounds containing amino groups bound to a carbon skeleton
- C07C211/01—Compounds containing amino groups bound to a carbon skeleton having amino groups bound to acyclic carbon atoms
- C07C211/16—Compounds containing amino groups bound to a carbon skeleton having amino groups bound to acyclic carbon atoms of a saturated carbon skeleton containing rings other than six-membered aromatic rings
- C07C211/17—Compounds containing amino groups bound to a carbon skeleton having amino groups bound to acyclic carbon atoms of a saturated carbon skeleton containing rings other than six-membered aromatic rings containing only non-condensed rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Neurosurgery (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Psychiatry (AREA)
- Addiction (AREA)
- Epidemiology (AREA)
- Psychology (AREA)
- Pain & Pain Management (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Indole Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Crystals, And After-Treatments Of Crystals (AREA)
Abstract
La presente invención se relaciona con las formas cristalinas de (1R, 5S) -1- (naftalen-2-il) -3-azabiciclo [3.1.0] clorhidrato de hexano y composiciones que comprenden lo mismo y métodos de fabricación y uso de lo mismo. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Compuestos cristalinos
Campo de la invención
La presente invención se refiere a la forma cristalina B de clorhidrato de (1R,5S)-1-(naftalen-2-il)-3-azabiciclo[3.1.0]hexano y a composiciones que la comprenden y a métodos para elaborarla y usarla.
Antecedentes de la invención
(1R,5S)-1-(Naftalen-2-il)-3-azabiciclo[3.1.0]hexano, también conocido como (+)-1-(naftalen-2-il)-3-azabiciclo[3.1.0]hexano, es un compuesto útil como inhibidor de la recaptación triple (TRI) desequilibrado, el más potente en la recaptación de norepinefrina (NE), una sexta parte tan potente en la recaptación de dopamina (DA) y una catorceava parte en la recaptación de serotonina (5-HT). Este compuesto y su utilidad se divulgan con más detalle en el documento US 2007/0082940.
Pueden existir principios activos farmacéuticos en diferentes formas físicas (por ejemplo, líquidas o sólidas en diferentes formas cristalinas, amorfas, hidratadas o solvatadas), que pueden variar la procesabilidad, estabilidad, solubilidad, biodisponibilidad, farmacocinética (absorción, distribución, metabolismo, excreción, o similares), y/o bioequivalencia del principio activo farmacéutico y de las composiciones farmacéuticas que lo comprenden. No puede predecirse si un compuesto existirá en una forma de polimorfo particular. Es importante en el desarrollo farmacéutico generar e identificar formas físicas ventajosas (por ejemplo, base libre o sal en formas sólidas, líquidas, cristalinas, hidratadas, solvatadas o amorfas) de principios activos farmacéuticos. Por tanto, sigue habiendo la necesidad de formas de polimorfo particulares de (1R,5S)-1-(naftalen-2-il)-3-azabiciclo[3.1.0]hexano.
Sumario de la invención
(1R,5S)-1-(Naftalen-2-il)-3-azabiciclo[3.1.0]hexano, también conocido como (+)-1-(naftalen-2-il)-3-azabiciclo[3.1.0]hexano (“el compuesto”) se muestra como la fórmula I a continuación:

Los inventores han encontrado polimorfos particulares del compuesto en forma de sal de adición de ácido clorhídrico, designados formas cristalinas A, B y C. Estos polimorfos particulares tienen diferentes perfiles de estabilidad y disolución y son especialmente ventajosos en la preparación de formulaciones galénicas de tipos diversos y variados, especialmente la forma cristalina B tal como se describe a continuación. Por tanto, en el primer aspecto, la invención proporciona la forma cristalina B de clorhidrato de (1R,5S)-1-(naftalen-2-il)-3-azabiciclo[3.1.0]hexano, por ejemplo: 1.1 Forma cristalina B del compuesto en forma de sal de adición de ácido clorhídrico (clorhidrato de (1R,5S)-1-(naftalen-2-il)-3-azabiciclo[3.1.0]hexano) (“forma cristalina B”), en la que la forma cristalina B comprende menos del 20% en peso de cualquier otra forma cristalina de clorhidrato de (1R,5S)-1-(naftalen-2-il)-3-azabiciclo[3.1.0]hexano.
1.2 Fórmula 1.1 en la que la forma cristalina B pertenece al grupo espacial P2i2i2i y tiene los siguientes parámetros de celda unitaria:
a = 5.9055(2) k,b = 7.4645(3) Á, c = 29.1139(13) Á, ce = j3 = / = 90°
1.3 Fórmula 1.1 en la que la forma cristalina B pertenece al grupo espacial P 2^21 y tiene cualquier combinación de los siguientes parámetros de celda unitaria:
a = 5-7 Á, por ejemplo, 6 Á, por ejemplo, 5,7-6,1 Á, por ejemplo, 5,8-6,0 Á, por ejemplo, 5,9 Á, por ejemplo, 5,91, por ejemplo, 5,906 Á;
b = 6-8 Á, por ejemplo, 7 Á, por ejemplo, 7,3-7,7 Á, por ejemplo, 7,4-7,6 Á, por ejemplo, 7,5 Á, por ejemplo, 7,46 Á, por ejemplo, 7,465 Á;
c = 28-30 A, por ejemplo, 29 A, por ejemplo, 28,9-29,3 A, por ejemplo, 29,0-29,2 A, por ejemplo, 29,1 A, por ejemplo, 29,11 A, por ejemplo, 29,114 A; y
a = f í = y= 90°.
1.4 Cualquiera de las fórmulas 1.1-1.3 en las que la forma cristalina B tiene un volumen calculado de V = 1283,39(9) A3.
1.5 Cualquiera de las fórmulas 1.1-1.4 en las que la estructura cristalina de la forma cristalina B se obtiene con un cristal que tiene unas dimensiones aproximadas de 0,31 mm * 0,21 mm * 0,09 mm, por ejemplo, una placa incolora que tiene unas dimensiones aproximadas de 0,31 mm * 0,21 mm * 0,09 mm.
1.6 Cualquiera de las fórmulas 1.1-1.5 en las que la estructura cristalina de la forma cristalina B se obtiene con radiación Cu Ka, por ejemplo, Cu Ka que tiene X = 1,54178 A.
1.7 Cualquiera de las fórmulas 1.1-1.6 en las que la estructura cristalina de la forma cristalina B se obtiene a 100(2) K.
1.8 Cualquiera de las fórmulas 1.1-1.7 en las que la forma cristalina B tiene una estructura monocristalina representada por el dibujo elipsoide de desplazamiento atómico de la figura 24.
1.9 Cualquiera de las fórmulas 1.1-1.8 en las que la forma cristalina B tiene un patrón de XRPD calculado tal como se muestra en la figura 32.
1.10 Cualquiera de las fórmulas 1.1-1.9 en las que la forma cristalina B presenta un patrón de XRPD que comprende al menos tres valores 2-theta (°) seleccionados del grupo que consiste en 6,0, 17,4, 18,9, 19,2 y 24,4, en las que la XRPD se mide usando un haz incidente de radiación Cu, por ejemplo, radiación Cu Ka, por ejemplo, en las que la XRPD se mide usando radiación de longitud de onda 1,54059 A.
1.11 Cualquiera de las fórmulas 1.1-1.10 en las que la forma cristalina B presenta un patrón de XRPD que comprende valores 2-theta (°) de 6,0, 17,4, 18,9, 19,2 y 24,4, en las que la XRPD se mide usando un haz incidente de radiación Cu, por ejemplo, radiación Cu Ka, por ejemplo, en las que la XRPD se mide usando radiación de longitud de onda 1,54059 A.
1.12 Cualquiera de las fórmulas 1.1-1.11 en las que la forma cristalina B presenta un patrón de XRPD que tiene valores 2-theta (°) característicos de 6,0, 17,4, 18,9, 19,2 y 24,4, en las que la XRPD se mide usando un haz incidente de radiación Cu, por ejemplo, radiación Cu Ka, por ejemplo, en las que la XRPD se mide usando radiación de longitud de onda 1,54059 A.
1.13 Cualquiera de las fórmulas 1.1-1.12 en las que la forma cristalina B presenta un patrón de XRPD que comprende al menos tres valores 2-theta (°) seleccionados del grupo que consiste en 6,04, 17,41, 18,94, 19,19 y 24,39, en las que la XRPD se mide usando un haz incidente de radiación Cu, por ejemplo, radiación Cu Ka, por ejemplo, en las que la XRPD se mide usando radiación de longitud de onda 1,54059 A.
1.14 Cualquiera de las fórmulas 1.1-1.13 en las que la forma cristalina B presenta un patrón de XRPD que comprende valores 2-theta (°) de 6,04, 17,41, 18,94, 19,19 y 24,39, en las que la XRPD se mide usando un haz incidente de radiación Cu, por ejemplo, radiación Cu Ka, por ejemplo, en las que la XRPD se mide usando radiación de longitud de onda 1,54059 A.
1.15 Cualquiera de las fórmulas 1.1-1.14 en las que la forma cristalina B presenta un patrón de XRPD que tiene valores 2-theta (°) característicos de 6,04, 17,41, 18,94, 19,19 y 24,39, en las que la XRPD se mide usando un haz incidente de radiación Cu, por ejemplo, radiación Cu Ka, por ejemplo, en las que la XRPD se mide usando radiación de longitud de onda 1,54059 A.
1.16 Cualquiera de las fórmulas 1.1-1.15 en las que la forma cristalina B presenta un patrón de XRPD que comprende al menos tres valores 2-theta (°) seleccionados de los expuestos en la tabla D a continuación:
Tabla D.
°20 espacio d (A) Intensidad (%)
6,04 ± 0,20 14,620 ± 0,484 13
17,41 ± 0,20 5,089 ± 0,058 14
18,94 ± 0,20 4,681 ± 0,049 79
19,19 ± 0,20 4,622 ± 0,048 100
24,39 ± 0,20 3,646 ± 0,029 23
en las que la XRPD se mide usando un haz incidente de radiación Cu, por ejemplo, radiación Cu Ka, por ejemplo, en las que la XRPD se mide usando radiación de longitud de onda 1,54059 A.
1.17 Cualquiera de las fórmulas 1.1-1.16 en las que la forma cristalina B presenta un patrón de XRPD que comprende los valores 2-theta (°) expuestos en la tabla D de la fórmula 1.93, en las que la XRPD se mide usando un haz incidente de radiación Cu, por ejemplo, radiación Cu Ka, por ejemplo, en las que la XRPD se mide usando radiación de longitud de onda 1,54059 A.
1.18 Cualquiera de las fórmulas 1.1-1.17 en las que la forma cristalina B presenta un patrón de XRPD que tiene valores 2-theta (°) característicos tal como se expone en la tabla D de la fórmula 1.93, en las que la XRPD se mide usando un haz incidente de radiación Cu, por ejemplo, radiación Cu Ka, por ejemplo, en las que la XRPD se mide usando radiación de longitud de onda 1,54059 A.
1.19 Cualquiera de las fórmulas 1.1-1.18 en las que la forma cristalina B presenta un patrón de XRPD que comprende al menos tres, por ejemplo, al menos cinco, valores 2-theta (°) seleccionados del grupo que consiste en 6,0, 13,2, 17,4, 18,9, 19,2, 23,6, 23,8, 24,4 y 28,2, en las que la XRPD se mide usando un haz incidente de radiación Cu, por ejemplo, radiación Cu Ka, por ejemplo, en las que la XRPD se mide usando radiación de longitud de onda 1,54059 A.
1.20 Cualquiera de las fórmulas 1.1-1.19 en las que la forma cristalina B presenta un patrón de XRPD que comprende valores 2-theta (°) de 6,0, 13,2, 17,4, 18,9, 19,2, 23,6, 23,8, 24,4 y 28,2, en las que la XRPD se mide usando un haz incidente de radiación Cu, por ejemplo, radiación Cu Ka, por ejemplo, en las que la XRPD se mide usando radiación de longitud de onda 1,54059 A.
1.21 Cualquiera de las fórmulas 1.1-1.20 en las que la forma cristalina B presenta un patrón de XRPD que tiene valores 2-theta (°) representativos de 6,0, 13,2, 17,4, 18,9, 19,2, 23,6, 23,8, 24,4 y 28,2, en las que la XRPD se mide usando un haz incidente de radiación Cu, por ejemplo, radiación Cu Ka, por ejemplo, en las que la XRPD se mide usando radiación de longitud de onda 1,54059 A.
1.22 Cualquiera de las fórmulas 1.1-1.21 en las que la forma cristalina B presenta un patrón de XRPD que comprende al menos tres, por ejemplo, al menos cinco, valores 2-theta (°) seleccionados del grupo que consiste en 6,04, 13,21, 17,41, 18,94, 19,19, 23,59, 23,79, 24,39 y 28,15, en las que la XRPD se mide usando un haz incidente de radiación Cu, por ejemplo, radiación Cu Ka, por ejemplo, en las que la XRPD se mide usando radiación de longitud de onda 1,54059 A.
1.23 Cualquiera de las fórmulas 1.1-1.22 en las que la forma cristalina B presenta un patrón de XRPD que comprende valores 2-theta (°) de 6,04, 13,21, 17,41, 18,94, 19,19, 23,59, 23,79, 24,39 y 28,15, en las que la XRPD se mide usando un haz incidente de radiación Cu, por ejemplo, radiación Cu Ka, por ejemplo, en las que la XRPD se mide usando radiación de longitud de onda 1,54059 A.
1.24 Cualquiera de las fórmulas 1.1-1.23 en las que la forma cristalina B presenta un patrón de XRPD que tiene valores 2-theta (°) representativos de 6,04, 13,21, 17,41, 18,94, 19,19, 23,59, 23,79, 24,39 y 28,15, en las que la XRPD se mide usando un haz incidente de radiación Cu, por ejemplo, radiación Cu Ka, por ejemplo, en las que la XRPD se mide usando radiación de longitud de onda 1,54059 A.
1.25 Cualquiera de las fórmulas 1.1-1.24 en las que la forma cristalina B presenta un patrón de XRPD que comprende al menos tres, por ejemplo, al menos cinco, valores 2-theta (°) seleccionados de los expuestos en la tabla E a continuación:
Tabla E.
°20 espacio d (A) Intensidad (%)
6,04 ± 0,20 14,620 ± 0,484 13
13,21 ± 0,20 6,699 ± 0,101 21
17,41 ± 0,20 5,089 ± 0,058 14
18,94 ± 0,20 4,681 ± 0,049 79
19,19 ± 0,20 4,622 ± 0,048 100
23,59 ± 0,20 3,769 ± 0,032 16
23,79 ± 0,20 3,737 ± 0,031 43
24,39 ± 0,20 3,646 ± 0,029 23
28,15 ± 0,20 3,168 0,022 24
en las que la XRPD se mide usando un haz incidente de radiación Cu, por ejemplo, radiación Cu Ka, por ejemplo, en las que la XRPD se mide usando radiación de longitud de onda 1,54059 A.
1.26 Cualquiera de las fórmulas 1.1-1.25 en las que la forma cristalina B presenta un patrón de XRPD que comprende los valores 2-theta (°) expuestos en la tabla E de la fórmula 1.102, en las que la XRPD se mide usando un haz incidente de radiación Cu, por ejemplo, radiación Cu Ka, por ejemplo, en las que la XRPD se mide usando radiación de longitud de onda 1,54059 A.
1.27 Cualquiera de las fórmulas 1.1-1.26 en las que la forma cristalina B presenta un patrón de XRPD que tiene valores 2-theta (°) representativos tal como se expone en la tabla E de la fórmula 1.102, en las que la XRPD se mide usando un haz incidente de radiación Cu, por ejemplo, radiación Cu Ka, por ejemplo, en las que la XRPD se mide usando radiación de longitud de onda 1,54059 A.
1.28 Cualquiera de las fórmulas 1.1-1.27 en las que la forma cristalina B presenta un patrón de XRPD que comprende al menos tres, por ejemplo, al menos cinco, por ejemplo, al menos nueve, por ejemplo, al menos diez, por ejemplo, al menos quince, por ejemplo, al menos veinte, por ejemplo, al menos veinticinco, valores 2-theta (°) seleccionados del grupo que consiste en 6,0, 12.1, 13,2, 14,9, 15,1, 16,0, 16,9, 17,4, 18,2, 18,9, 19,2, 19,9, 21,1, 21,3, 21,7, 22,6, 23,6, 23,8, 24,4, 25,3, 26,1,26,6, 27,2, 28,2, 28,7 y 29,5, en las que la XRPD se mide usando un haz incidente de radiación Cu, por ejemplo, radiación Cu Ka, por ejemplo, en las que la XRPD se mide usando radiación de longitud de onda 1,54059 A.
1.29 Cualquiera de las fórmulas 1.1-1.28 en las que la forma cristalina B presenta un patrón de XRPD que comprende los siguientes valores 2-theta (°):
6,0, 12.1, 13,2, 14,9, 15,1, 16,0, 16,9, 17,4, 18,2, 18,9, 19,2, 19,9, 21,1, 21,3, 21,7, 22,6, 23,6, 23,8, 24,4, 25,3, 26,1, 26,6, 27,2, 28,2, 28,7 y 29,5,
en las que la XRPD se mide usando un haz incidente de radiación Cu, por ejemplo, radiación Cu Ka, por ejemplo, en las que la XRPD se mide usando radiación de longitud de onda 1,54059 A.
1.30 Cualquiera de las fórmulas 1.1-1.29 en las que la forma cristalina B presenta un patrón de XRPD que comprende al menos tres, por ejemplo, al menos cinco, por ejemplo, al menos nueve, por ejemplo, al menos diez, por ejemplo, al menos quince, por ejemplo, al menos veinte, por ejemplo, al menos veinticinco, valores 2-theta (°) seleccionados del grupo que consiste en 6,04, 12,12, 13,21, 14,86, 15,13, 16,02, 16,90, 17,41, 18,23, 18,94, 19,19, 19,91, 21,05, 21,27, 21,74, 22,55, 23,59, 23,79, 24,39, 25,34, 26,06, 26,61, 27,15, 28,15, 28,66 y 29,47, en las que la XRPD se mide usando un haz incidente de radiación Cu, por ejemplo, radiación Cu Ka, por ejemplo, en las que la XRPD se mide usando radiación de longitud de onda 1,54059 A.
1.31 Cualquiera de las fórmulas 1.1-1.30 en las que la forma cristalina B presenta un patrón de XRPD que comprende los siguientes valores 2-theta (°):
6,04, 12,12, 13,21, 14,86, 15,13, 16,02, 16,90, 17,41, 18,23, 18,94, 19,19, 19,91, 21,05, 21,27, 21,74, 22,55, 23,59, 23,79, 24,39, 25,34, 26,06, 26,61,27,15, 28,15, 28,66 y 29,47,
en las que la XRPD se mide usando un haz incidente de radiación Cu, por ejemplo, radiación Cu Ka, por ejemplo, en las que la XRPD se mide usando radiación de longitud de onda 1,54059 A.
1.32 Cualquiera de las fórmulas 1.1-1.31 en las que la forma cristalina B presenta un patrón de XRPD que comprende al menos tres, por ejemplo, al menos cinco, por ejemplo, al menos nueve, por ejemplo, al menos diez, por ejemplo, al menos quince, por ejemplo, al menos veinte, por ejemplo, al menos veinticinco, por ejemplo, al menos cinco, valores 2-theta (°) seleccionados de los expuestos en la tabla F a continuación:
Tabla F.
°20 espacio d (A) Intensidad (%)
6,04 ± 0,20 14,620 ± 0,484 13
12,12 ± 0,20 7,296 ± 0,120 6
13,21 ± 0,20 6,699 ± 0,101 21
14,86 ± 0,20 5,958 ± 0,080 8
15,13 ± 0,20 5,853 ± 0,077 5
16,02 ± 0,20 5,529 ± 0,069 1
16,90 ± 0,20 5,242 ± 0,062 4
17,41 ± 0,20 5,089 ± 0,058 14
18,23 ± 0,20 4,861 ± 0,053 10
18,94 ± 0,20 4,681 ± 0,049 79
19,19 ± 0,20 4,622 ± 0,048 100
19,91 ± 0,20 4,457 ± 0,044 4
21.05 ± 0,20 4,217 ± 0,040 11
21,27 ± 0,20 4,173 ± 0,039 2
21,74 ± 0,20 4,085 ± 0,037 4
22,55 ± 0,20 3,939 ± 0,034 6
23,59 ± 0,20 3,769 ± 0,032 16
23,79 ± 0,20 3,737 ± 0,031 43
24,39 ± 0,20 3,646 ± 0,029 23
25,34 ± 0,20 3,512 ± 0,027 1
26.06 ± 0,20 3,416 ± 0,026 2
26,61 ± 0,20 3,347 ± 0,025 1
27.15 ± 0,20 3,282 ± 0,024 2
28.15 ± 0,20 3,168 ± 0,022 24
28,66 ± 0,20 3,112 ± 0,021 13
29,47 ± 0,20 3,028 ± 0,020 13
en las que la XRPD se mide usando un haz incidente de radiación Cu, por ejemplo, radiación Cu Ka, por ejemplo, en las que la XRPD se mide usando radiación de longitud de onda 1,54059 A.
1.33 Cualquiera de las fórmulas 1.1-1.32 en las que la forma cristalina B presenta un patrón de XRPD que comprende los valores 2-theta (°) expuestos en la tabla F de la fórmula 1.109, en las que la XRPD se mide usando un haz incidente de radiación Cu, por ejemplo, radiación Cu Ka, por ejemplo, en las que la XRPD se mide usando radiación de longitud de onda 1,54059 A.
1.34 Cualquiera de las fórmulas 1.1-1.33 en las que la forma cristalina B presenta un patrón de XRPD que comprende al menos tres valores de espaciamiento d (A) seleccionados del grupo que consiste en 14,6, 5,1,4,7, 4,6 y 3,6.
1.35 Cualquiera de las fórmulas 1.1-1.34 en las que la forma cristalina B presenta un patrón de XRPD que comprende valores de espaciamiento d (A) de 14,6, 5,1,4,7, 4,6 y 3,6.
1.36 Cualquiera de las fórmulas 1.1-1.35 en las que la forma cristalina B presenta un patrón de XRPD que comprende al menos tres valores de espaciamiento d (A) seleccionados del grupo que consiste en 14,62, 5,09, 4,68, 4,62 y 3,65.
1.37 Cualquiera de las fórmulas 1.1-1.36 en las que la forma cristalina B presenta un patrón de XRPD que comprende valores de espaciamiento d (A) de 14,62, 5,09, 4,68, 4,62 y 3,65.
1.38 Cualquiera de las fórmulas 1.1-1.37 en las que la forma cristalina B presenta un patrón de XRPD que comprende al menos tres valores de espaciamiento d (A) seleccionados del grupo que consiste en 14,620, 5,089, 4,681,4,622 y 3,646.
1.39 Cualquiera de las fórmulas 1.1-1.38 en las que la forma cristalina B presenta un patrón de XRPD que comprende valores de espaciamiento d (A) de 14,620, 5,089, 4,681, 4,622 y 3,646.
1.40 Cualquiera de las fórmulas 1.1-1.39 en las que la forma cristalina B presenta un patrón de XRPD que comprende al menos tres valores de espaciamiento d (A) seleccionados de los expuestos en la tabla D de la fórmula 1.93.
1.41 Cualquiera de las fórmulas 1.1-1.40 en las que la forma cristalina B presenta un patrón de XRPD que comprende los valores de espaciamiento d (A) expuestos en la tabla D de la fórmula 1.93.
1.42 Cualquiera de las fórmulas 1.1-1.41 en las que la forma cristalina B presenta un patrón de XRPD que comprende al menos tres, por ejemplo, al menos cinco, valores de espaciamiento d (A) seleccionados del grupo que consiste en 14,6, 6,7, 5,1, 4,7, 4,6, 3,8, 3,7, 3,6 y 3,2.
1.43 Cualquiera de las fórmulas 1.1-1.42 en las que la forma cristalina B presenta un patrón de XRPD que comprende valores de espaciamiento d (A) de 14,6, 6,7, 5,1, 4,7, 4,6, 3,8, 3,7, 3,6 y 3,2.
1.44 Cualquiera de las fórmulas 1.1-1.43 en las que la forma cristalina B presenta un patrón de XRPD que comprende al menos tres, por ejemplo, al menos cinco, valores de espaciamiento d (A) seleccionados del grupo que consiste en 14,62, 6,70, 5,09, 4,68, 4,62, 3,77, 3,74, 3,65 y 3,17.
1.45 Cualquiera de las fórmulas 1.1-1.44 en las que la forma cristalina B presenta un patrón de XRPD que comprende valores de espaciamiento d (A) de 14,62, 6,70, 5,09, 4,68, 4,62, 3,77, 3,74, 3,65 y 3,17.
1.46 Cualquiera de las fórmulas 1.1-1.45 en las que la forma cristalina B presenta un patrón de XRPD que comprende al menos tres, por ejemplo, al menos cinco, valores de espaciamiento d (A) seleccionados del grupo que consiste en 14,620, 6,699, 5,089, 4,681, 4,622, 3,769, 3,737, 3,646 y 3,168.
1.47 Cualquiera de las fórmulas 1.1-1.46 en las que la forma cristalina B presenta un patrón de XRPD que comprende valores de espaciamiento d (A) de 14,620, 6,699, 5,089, 4,681, 4,622, 3,769, 3,737, 3,646 y 3,168.
1.48 Cualquiera de las fórmulas 1.1-1.47 en las que la forma cristalina B presenta un patrón de XRPD que comprende al menos tres, por ejemplo, al menos cinco, valores de espaciamiento d (A) seleccionados de los expuestos en la tabla E de la fórmula 1.102.
1.49 Cualquiera de las fórmulas 1.1-1.48 en las que la forma cristalina B presenta un patrón de XRPD que comprende los valores de espaciamiento d (A) expuestos en la tabla E de la fórmula 1.102.
1.50 Cualquiera de las fórmulas 1.1-1.49 en las que la forma cristalina B presenta un patrón de XRPD que comprende al menos tres, por ejemplo, al menos cinco, por ejemplo, al menos nueve, por ejemplo, al menos diez, por ejemplo, al menos quince, por ejemplo, al menos veinte, valores de espaciamiento d (A) seleccionados del grupo que consiste en 14,6, 7,3, 6,7, 6,0, 5,9, 5,5, 5,2, 5,1, 4,9, 4,7, 4,6, 4,5, 4,2, 4,1, 3,9, 3,8, 3,7, 3,6, 3,5, 3,4, 3,3, 3,2, 3,1 y 3,0.
1.51 Cualquiera de las fórmulas 1.1-1.50 en las que la forma cristalina B presenta un patrón de XRPD que comprende valores de espaciamiento d (A) de 14,6, 7,3, 6,7, 6,0, 5,9, 5,5, 5,2, 5,1, 4,9, 4,7, 4,6, 4,5, 4,2, 4,1, 3,9, 3,8, 3,7, 3,6, 3,5, 3,4, 3,3, 3,2, 3,1 y 3,0.
1.52 Cualquiera de las fórmulas 1.1-1.51 en las que la forma cristalina B presenta un patrón de XRPD que comprende al menos tres, por ejemplo, al menos cinco, por ejemplo, al menos nueve, por ejemplo, al menos diez, por ejemplo, al menos quince, por ejemplo, al menos veinte, por ejemplo, al menos veinticinco, valores de espaciamiento d (A) seleccionados del grupo que consiste en 14,62, 7,30, 6,70, 5,96, 5,85, 5,53, 5,24, 5,09, 4,86, 4,68, 4,62, 4,46, 4,22, 4,17, 4,09, 3,94, 3,77, 3,74, 3,65, 3,51, 3,42, 3,35, 3,28, 3,17, 3,11 y 3,03.
1.53 Cualquiera de las fórmulas 1.1-1.52 en las que la forma cristalina B presenta un patrón de XRPD que comprende valores de espaciamiento d (A) de 14,62, 7,30, 6,70, 5,96, 5,85, 5,53, 5,24, 5,09, 4,86, 4,68, 4,62, 4,46, 4,22, 4,17, 4,09, 3,94, 3,77, 3,74, 3,65, 3,51, 3,42, 3,35, 3,28, 3,17, 3,11 y 3,03.
1.54 Cualquiera de las fórmulas 1.1-1.53 en las que la forma cristalina B presenta un patrón de XRPD que comprende al menos tres, por ejemplo, al menos cinco, por ejemplo, al menos nueve, por ejemplo, al menos diez, por ejemplo, al menos quince, por ejemplo, al menos veinte, por ejemplo, al menos veinticinco, valores de espaciamiento d (A) seleccionados del grupo que consiste en 14,620, 7,296, 6,699, 5,958, 5,853, 5,529, 5,242, 5,089, 4,861,4,681,4,622, 4,457, 4,217, 4,173, 4,085, 3,939, 3,769, 3,737, 3,646, 3,512, 3,416, 3,347, 3,282, 3,168, 3,112 y 3,028.
1.55 Cualquiera de las fórmulas 1.1-1.54 en las que la forma cristalina B presenta un patrón de XRPD que comprende valores de espaciamiento d (A) de 14,620, 7,296, 6,699, 5,958, 5,853, 5,529, 5,242, 5,089, 4,861, 4,681,4,622, 4,457, 4,217, 4,173, 4,085, 3,939, 3,769, 3,737, 3,646, 3,512, 3,416, 3,347, 3,282, 3,168, 3,112 y 3,028.
1.56 Cualquiera de las fórmulas 1.1-1.55 en las que la forma cristalina B presenta un patrón de XRPD que comprende al menos tres, por ejemplo, al menos cinco, por ejemplo, al menos nueve, por ejemplo, al menos diez, por ejemplo, al menos quince, por ejemplo, al menos veinte, por ejemplo, al menos veinticinco, valores de espaciamiento d (A) seleccionados de los expuestos en la tabla F de la fórmula 1.109.
1.57 Cualquiera de las fórmulas 1.1-1.56 en las que la forma cristalina B presenta un patrón de XRPD que comprende los valores de espaciamiento d (A) expuestos en la tabla F de la fórmula 1.109.
1.58 Cualquiera de las fórmulas 1.1-1.57 en las que la forma cristalina B presenta un patrón de difracción de rayos X de polvo que comprende picos característicos del patrón de XRPD mostrado en la figura 5, en las que la XRPd se mide usando radiación Cu, por ejemplo, radiación Cu Ka, por ejemplo, en las que la XRPD se mide usando radiación de longitud de onda 1,541871 A.
1.59 Cualquiera de las fórmulas 1.1-1.58 en las que la forma cristalina B presenta un patrón de difracción de rayos X de polvo que comprende picos representativos del patrón de XRPD mostrados en la figura 5, en las que la XRPD se
mide usando radiación Cu, por ejemplo, radiación Cu Ka, por ejemplo, en las que la XRPD se mide usando radiación de longitud de onda 1,541871 A.
1.60 Cualquiera de las fórmulas 1.1-1.59 en las que la forma cristalina B presenta un patrón de difracción de rayos X de polvo, por ejemplo, un patrón de difracción de rayos X de polvo medido usando un haz incidente de radiación Cu, por ejemplo, radiación Cu Ka, por ejemplo, en las que la XRPD se mide usando radiación de longitud de onda 1,541871 A, que comprende tres picos, en algunas realizaciones, cinco picos, seleccionados de los mostrados en la figura 5.
1.61 Cualquiera de las fórmulas 1.1-1.60 en las que la forma cristalina B presenta un patrón de difracción de rayos X de polvo, por ejemplo, un patrón de difracción de rayos X de polvo medido usando un haz incidente de radiación Cu, por ejemplo, radiación Cu Ka, por ejemplo, en las que la XRPD se mide usando radiación de longitud de onda 1,541871 A, que comprende al menos cinco picos, por ejemplo, al menos nueve picos, por ejemplo, al menos diez picos, por ejemplo, al menos quince picos, por ejemplo, al menos veinte picos, por ejemplo, al menos veinticinco picos, seleccionados de los mostrados en la figura 5.
1.62 Cualquiera de las fórmulas 1.1-1.61 en las que la forma cristalina B presenta un patrón de difracción de rayos X de polvo, por ejemplo, un patrón de difracción de rayos X de polvo medido usando un haz incidente de radiación Cu, por ejemplo, radiación Cu Ka, por ejemplo, en las que la XRPD se mide usando radiación de longitud de onda 1,541871 A, sustancialmente tal como se muestra en la figura 5.
1.63 Cualquiera de las fórmulas 1.1-1.62 en las que la forma cristalina B presenta un patrón de difracción de rayos X de polvo, por ejemplo, un patrón de difracción de rayos X de polvo medido usando un haz incidente de radiación Cu, por ejemplo, radiación Cu Ka, por ejemplo, en las que la XRPD se mide usando radiación de longitud de onda 1,541871 A, tal como se muestra en la figura 5.
1.64 Cualquiera de las fórmulas 1.1-1.63 en las que la forma cristalina B presenta un patrón de difracción de rayos X de polvo que comprende picos característicos del patrón de XRPD mostrado en la figura 7, en las que la XRPd se mide usando radiación Cu, por ejemplo, radiación Cu Ka, por ejemplo, en las que la XRPD se mide usando radiación de longitud de onda 1,54059 A, por ejemplo, en las que el patrón de XRPD también comprende picos de la forma cristalina A (por ejemplo, una mezcla de formas cristalinas A y B).
1.65 Cualquiera de las fórmulas 1.1-1.64 en las que la forma cristalina B presenta un patrón de difracción de rayos X de polvo que comprende picos representativos del patrón de XRPD mostrados en la figura 7, en las que la XRPd se mide usando radiación Cu, por ejemplo, radiación Cu Ka, por ejemplo, en las que la XRPD se mide usando radiación de longitud de onda 1,54059 A, por ejemplo, en las que el patrón de XRPD también comprende picos de la forma cristalina A (por ejemplo, una mezcla de formas cristalinas A y B).
1.66 Cualquiera de las fórmulas 1.1-1.65 en las que la forma cristalina B presenta un patrón de difracción de rayos X de polvo, por ejemplo, un patrón de difracción de rayos X de polvo medido usando un haz incidente de radiación Cu, por ejemplo, radiación Cu Ka, por ejemplo, un patrón de difracción de rayos X de polvo de alta resolución medido usando un haz incidente de radiación Cu Ka, por ejemplo, en las que la XRPD se mide usando radiación de longitud de onda 1,54059 A, que comprende tres picos, en algunas realizaciones, cinco picos, seleccionados de los mostrados en la figura 7, por ejemplo, en las que el patrón de XRPD también comprende picos de la forma cristalina A (por ejemplo, una mezcla de formas cristalinas A y B).
1.67 Cualquiera de las fórmulas 1.1-1.66 en las que la forma cristalina B presenta un patrón de difracción de rayos X de polvo, por ejemplo, un patrón de difracción de rayos X de polvo medido usando un haz incidente de radiación Cu, por ejemplo, radiación Cu Ka, por ejemplo, un patrón de difracción de rayos X de polvo de alta resolución medido usando un haz incidente de radiación Cu Ka, por ejemplo, en las que la XRPD se mide usando radiación de longitud de onda 1,54059 A, que comprende al menos cinco picos, por ejemplo, al menos nueve picos, por ejemplo, al menos diez picos, por ejemplo, al menos quince picos, por ejemplo, al menos veinte picos, por ejemplo, al menos veinticinco picos, seleccionados de los mostrados en la figura 7, por ejemplo, en las que el patrón de XRPD comprende picos de la forma cristalina A (por ejemplo, una mezcla de formas cristalinas A y B).
1.68 Cualquiera de las fórmulas 1.1-1.67 en las que la forma cristalina B presenta un patrón de difracción de rayos X de polvo, por ejemplo, un patrón de difracción de rayos X de polvo medido usando un haz incidente de radiación Cu, por ejemplo, radiación Cu Ka, por ejemplo, un patrón de difracción de rayos X de polvo de alta resolución medido usando un haz incidente de radiación Cu Ka, por ejemplo, en las que la XRPD se mide usando radiación de longitud de onda 1,54059 A, sustancialmente tal como se muestra en la figura 7, por ejemplo, en las que el patrón de XRPD comprende picos de la forma cristalina A (por ejemplo, una mezcla de formas cristalinas A y B).
1.69 Cualquiera de las fórmulas 1.1-1.68 en las que la forma cristalina B presenta un patrón de difracción de rayos X de polvo, por ejemplo, un patrón de difracción de rayos X de polvo medido usando un haz incidente de radiación Cu, por ejemplo, radiación Cu Ka, por ejemplo, un patrón de difracción de rayos X de polvo de alta resolución medido usando un haz incidente de radiación Cu Ka, por ejemplo, en las que la XRPD se mide usando radiación de longitud
de onda 1,54059 A, tal como se muestra en la figura 7, por ejemplo, en las que el patrón de XRPD comprende picos de la forma cristalina A (por ejemplo, una mezcla de formas cristalinas A y B).
1.70 Cualquiera de las fórmulas 1.1-1.69 en las que la forma cristalina B presenta un patrón de XRPD que comprende picos característicos del patrón de XRPD mostrado en cualquiera de las figuras 7, 40 y 48, por ejemplo, la figura 7, por ejemplo, la figura 40, por ejemplo, la figura 48, en las que la XRPD se mide usando radiación Cu, por ejemplo, radiación Cu Ka, por ejemplo, en las que la XRPD se mide usando radiación de longitud de onda 1,54059 A.
1.71 Cualquiera de las fórmulas 1.1-1.70 en las que la forma cristalina B presenta un patrón de XRPD que comprende picos representativos del patrón de XRPD mostrados en cualquiera de las figuras 7, 40 y 48, por ejemplo, la figura 7, por ejemplo, la figura 40, por ejemplo, la figura 48, en las que la XRPD se mide usando radiación Cu, por ejemplo, radiación Cu Ka, por ejemplo, en las que la XRPD se mide usando radiación de longitud de onda 1,54059 A.
1.72 Cualquiera de las fórmulas 1.1-1.71 en las que la forma cristalina B presenta un patrón de XRPD, por ejemplo, un patrón de difracción de rayos X de polvo medido usando un haz incidente de radiación Cu, por ejemplo, radiación Cu Ka, por ejemplo, un patrón de difracción de rayos X de polvo de alta resolución medido usando un haz incidente de radiación Cu Ka, por ejemplo, en las que la XRPD se mide usando radiación de longitud de onda 1,54059 A, que comprende tres picos, en algunas realizaciones, cinco picos, seleccionados de los mostrados en cualquiera de las figuras 7, 40 y 48, por ejemplo, la figura 7, por ejemplo, la figura 40, por ejemplo, la figura 48.
1.73 Cualquiera de las fórmulas 1.1-1.72 en las que la forma cristalina B presenta un patrón de XRPD, por ejemplo, un patrón de difracción de rayos X de polvo medido usando un haz incidente de radiación Cu, por ejemplo, radiación Cu Ka, por ejemplo, un patrón de difracción de rayos X de polvo de alta resolución medido usando un haz incidente de radiación Cu Ka, por ejemplo, en las que la XRPD se mide usando radiación de longitud de onda 1,54059 A, que comprende al menos cinco, por ejemplo, al menos nueve, por ejemplo, al menos diez, por ejemplo, al menos quince, por ejemplo, al menos veinte, por ejemplo, al menos veinticinco, seleccionados de los mostrados en cualquiera de las figuras 7, 40 y 48, por ejemplo, la figura 7, por ejemplo, la figura 40, por ejemplo, la figura 48.
1.74 Cualquiera de las fórmulas 1.1-1.73 en las que la forma cristalina B presenta un patrón de XRPD, por ejemplo, un patrón de difracción de rayos X de polvo medido usando un haz incidente de radiación Cu, por ejemplo, radiación Cu Ka, por ejemplo, en las que la XRPD se mide usando radiación de longitud de onda 1,54059 A, sustancialmente tal como se muestra en cualquiera de las figuras 7, 40 y 48, por ejemplo, la figura 7, por ejemplo, la figura 40, por ejemplo, la figura 48.
1.75 Cualquiera de las fórmulas 1.1-1.74 en las que la forma cristalina B presenta un patrón de difracción de rayos X de polvo (XRPD), por ejemplo, un patrón de difracción de rayos X de polvo medido usando un haz incidente de radiación Cu, por ejemplo, radiación Cu Ka, por ejemplo, en las que la XRPD se mide usando radiación de longitud de onda 1,54059 A, tal como se muestra en cualquiera de las figuras 7, 40 y 48, por ejemplo, la figura 7, por ejemplo, la figura 40, por ejemplo, la figura 48.
1.76 Cualquiera de las fórmulas 1.1-1.75 en las que la forma cristalina B presenta un termograma de calorimetría diferencial de barrido (DSC) que comprende un pico endotérmico entre 247°C y 248°C.
1.77 Cualquiera de las fórmulas 1.1-1.76 en las que la forma cristalina B presenta un termograma de calorimetría diferencial de barrido (DSC) que comprende un pico endotérmico a 247°C.
1.78 Cualquiera de las fórmulas 1.1-1.77 en las que la forma cristalina B presenta un termograma de calorimetría diferencial de barrido (DSC) que comprende un pico endotérmico a 248°C, por ejemplo, un pico endotérmico a 248°C con una aparición a 246°C.
1.79 Cualquiera de las fórmulas 1.1-1.78 en las que la forma cristalina B presenta un termograma de calorimetría diferencial de barrido (DSC) que comprende un pico endotérmico a 251°C.
1.80 Cualquiera de las fórmulas 1.1-1.79 en las que la forma cristalina B presenta un termograma de calorimetría diferencial de barrido (DSC) que comprende un pico endotérmico a 264°C.
1.81 Cualquiera de las fórmulas 1.1-1.80 en las que la forma cristalina B presenta un termograma de calorimetría diferencial de barrido (DSC) que comprende un pico endotérmico a 141°C, por ejemplo, un pico endotérmico a 141°C con una aparición entre 137°C y 138°C, por ejemplo, un pico endotérmico a 141°C con una aparición a 137°C, por ejemplo, un pico endotérmico a 141°C con una aparición a 138°C.
1.82 Cualquiera de las fórmulas 1.1-1.81 en las que la forma cristalina B presenta un termograma de calorimetría diferencial de barrido (DSC) tal como se muestra en la figura 8.
1.83 Cualquiera de las fórmulas 1.1-1.82 en las que la forma cristalina B presenta un termograma de análisis
termogravimétrico (TGA) que comprende el 0,2% de pérdida de peso hasta 200°C.
1.84 Cualquiera de las fórmulas 1.1-1.83 en las que la forma cristalina B presenta un termograma de análisis termogravimétrico (TGA) que comprende una temperatura de inicio de descomposición a 281°C.
1.85 Cualquiera de las fórmulas 1.1-1.84 en las que la forma cristalina B presenta un termograma de análisis termogravimétrico (TGA) tal como se muestra en la figura 8.
1.86 La forma cristalina B de cualquiera de las fórmulas 1.1-1.85, en la que dicha forma cristalina B contiene menos del 20% en peso, por ejemplo, menos del 15% en peso, por ejemplo, menos del 10% en peso, preferiblemente menos del 5% en peso, preferiblemente menos del 3% en peso, más preferiblemente menos del 2% en peso, todavía preferiblemente menos del 1% en peso, todavía preferiblemente menos del 0,1% en peso, lo más preferiblemente menos del 0,01% en peso, de la forma amorfa.
1.87 La forma cristalina B de cualquiera de las fórmulas 1.1-1.86, en la que dicha forma cristalina B está libre o sustancialmente libre de cualquier otra forma, por ejemplo, contiene menos del 10% en peso, preferiblemente menos del 5% en peso, preferiblemente menos del 3% en peso, más preferiblemente menos del 2% en peso, todavía preferiblemente menos del 1% en peso, todavía preferiblemente menos del 0,1% en peso, lo más preferiblemente menos del 0,01% en peso, de cualquier otra forma cristalina de clorhidrato de (1R,5S)-1-(naftalen-2-il)-3-azabiciclo[3.1.0]hexano.
1.88 La forma cristalina B según cualquiera de las fórmulas 1.1-1.87 cuando se elabora mediante cualquiera de los procedimientos descritos a continuación en el presente documento o descritos de manera similar en cualquiera de los ejemplos o que tiene una difracción de rayos X de polvo o estructura cristalina de rayos X tal como se representa en cualquiera de las figuras.
Las transiciones de fase de los sólidos pueden ser termodinámicamente reversibles o irreversibles. Las formas cristalinas que se transforman de manera reversible a una temperatura de transición (Tt) específica son polimorfos enantiotrópicos. Si las formas cristalinas no son interconvertibles en estas condiciones, el sistema es monotrópico (una forma termodinámicamente estable).
Las formas cristalinas A, B y C son enantiótropos anhidros de clorhidrato de (1R,5S)-1-(naftalen-2-il)-3-azabiciclo[3.1.0]hexano. La forma cristalina C es la fase sólida estable por debajo de la temperatura de transición Tt,ô B, la forma cristalina B es la fase sólida estable entre Tt,ô B y Tíb^ a y la forma cristalina A es la fase sólida estable por encima de Tt,B^ A. Se espera que Tt,ô B está por debajo de 2°C. Tt,ô A estará entre 2°C y temperatura ambiental y Tt.B^A está entre 37 y 54°C.
Debido a las limitaciones cinéticas, la transformación termodinámica de la forma cristalina A a la forma cristalina B está impedida. Por tanto, sorprendentemente, la forma cristalina A parece ser lo suficientemente estable cinéticamente como para persistir en estado sólido en condiciones de temperatura en las que es termodinámicamente metaestable.
Agitar la forma cristalina A como suspensión durante 16 días en diclorometano a temperatura ambiental (véase el ejemplo 6a) no provoca una conversión de forma mediada por disolvente a la forma cristalina B, la forma más estable a esa temperatura. Esto indica que la barrera de energía libre crítica para la nucleación no se supera en ausencia de semillas del polimorfo más estable dentro del intervalo de tiempo evaluado.
Bajo exposición a condiciones de estrés acelerado durante dos semanas, las formas cristalinas A y B permanecen sin cambios a 30°C/el 56% de HR o 40°C/el 75% de HR (ejemplo 11). En cambio, la forma cristalina C se convierte en una mezcla de las formas cristalinas A y B en el plazo de dos semanas a 40°C/el 75% de HR (ejemplo 11). Por tanto, a diferencia de la forma cristalina A, la forma cristalina C se convierte en condiciones en las que es metaestable.
Para la forma cristalina A, en ausencia de semillas del polimorfo más estable, la barrera de energía libre crítica para la nucleación de la forma cristalina B no se supera en el estado sólido o en experimentos de conversión mediada por disolventes dentro del tiempo evaluado.
Por tanto, la forma cristalina A puede sintetizarse fácilmente a gran escala, pero también, sorprendentemente, persiste en estado sólido incluso en condiciones en las que es termodinámicamente metaestable.
En el segundo aspecto, la invención proporciona lo siguiente:
2.1. Una composición farmacéutica que comprende la forma cristalina B de cualquiera de las fórmulas 1.1-1.85 y un diluyente o portador farmacéuticamente aceptable.
2.2. La composición farmacéutica según la fórmula 2.1, en la que la composición es de liberación sostenida.
2.3. La composición farmacéutica según la fórmula 2.1 ó 2.2, que comprende de 1 mg a 1800 mg, por ejemplo, de 10
mg a 1800 mg, por ejemplo, de 25 mg a 1800 mg, por ejemplo, de 10 mg a 1600 mg, por ejemplo, de 10 mg a 1200 mg, por ejemplo, de 50 mg a 1200 mg, por ejemplo, de 50 mg a 1000 mg, por ejemplo, de 75 mg a 1000 mg, por ejemplo, de 75 mg a 800 mg, por ejemplo, de 75 mg a 500 mg, por ejemplo, de 100 mg a 750 mg, por ejemplo, de 100 mg a 500 mg, por ejemplo, de 100 mg a 400 mg, por ejemplo, de 100 mg a 300 mg, por ejemplo, de 100 mg a 200 mg, de la forma cristalina B de cualquiera de las fórmulas 1.1-1.85.
2.4 La composición según una cualquiera de las fórmulas 2.1-2.3 que comprende de 75 mg a 1000 mg, por ejemplo, de 100 mg a 600 mg, por ejemplo, de 100 mg a 400 mg, por ejemplo, de 100 mg a 200 mg, de la forma cristalina B de cualquiera de las fórmulas 1.1-1.85.
2.5 La composición según una cualquiera de las fórmulas 2.1-2.3 que comprende de 50 mg a 600 mg, por ejemplo, de 100 mg a 600 mg, por ejemplo, de 100 mg a 400 mg, por ejemplo, de 100 mg a 200 mg, de la forma cristalina B de cualquiera de las fórmulas 1.1-1.85.
2.6 La composición según una cualquiera de las fórmulas 2.1-2.3 que comprende de 5 mg a 500 mg, por ejemplo, de 5 mg a 10 mg, por ejemplo, de 10 mg a 25 mg, por ejemplo, de 30 mg a 50 mg, por ejemplo, de 10 mg a 300 mg, por ejemplo, de 25 mg a 300 mg, por ejemplo, de 50 mg a 100 mg, por ejemplo, a 100 mg a 250 mg, por ejemplo, de 250 mg a 500 mg, de una cualquiera de la forma cristalina B de cualquiera de las fórmulas 1.1-1.85.
2.7 La composición según una cualquiera de las fórmulas 2.1-2.3 para la administración de 0,5 mg/kg a 20 mg/kg al día, por ejemplo, de 1 mg/kg a 15 mg/kg al día, por ejemplo, de 1 mg/kg a 10 mg/kg al día, por ejemplo, de 2 mg/kg a 20 mg/kg al día, por ejemplo, de 2 mg/kg a 10 mg/kg al día, por ejemplo, de 3 mg/kg a 15 mg/kg al día, de la forma cristalina B de cualquiera de las fórmulas 1.1-1.85.
2.8 La composición según una cualquiera de las fórmulas 2.1-2.7 que comprende menos del 50% p/p de la forma cristalina B de la invención, por ejemplo, menos del 40% p/p, por ejemplo, menos del 30% p/p, menos del 20% p/p, por ejemplo, el 1-40% p/p, por ejemplo, el 5-40% p/p, por ejemplo, el 10-30% p/p, por ejemplo, el 15-25% p/p, por ejemplo, el 15-20% p/p, por ejemplo, el 17% p/p, por ejemplo, el 25% p/p, por ejemplo, de la forma cristalina B de cualquiera de las fórmulas 1.1-1.85.
2.9 La composición según una cualquiera de las fórmulas 2.1-2.8 en la que el diluyente o portador farmacéuticamente aceptable comprende hidroxipropilmetilcelulosa.
2.10 La composición según la fórmula 2.9, en la que la composición comprende al menos el 10% p/p de la hidroxipropilmetilcelulosa, por ejemplo, el 10-50% p/p, por ejemplo, el 10-40% p/p, por ejemplo, el 20-50% p/p, por ejemplo, el 20-40% p/p, por ejemplo, el 30-40% p/p, por ejemplo, el 37% p/p.
2.11 La composición según la fórmula 2.9 ó 2.10, en la que el grado de sustitución con metoxilo de la hidroxipropilmetilcelulosa es del19-24%.
2.12 La composición según una cualquiera de las fórmulas 2.9-2,11, en la que el grado de sustitución con hidroxipropoxilo de la hidroxipropilmetilcelulosa es del 4-12%.
2.13 La composición según una cualquiera de las fórmulas 2.9-2.12, en la que la hidroxipropilmetilcelulosa es Hypromellose 2208.
2.14 La composición según una cualquiera de las fórmulas 2.9-2.13, en la que la hidroxipropilmetilcelulosa tiene una viscosidad nominal de 4.000 mPA-a.
2.15 La composición según una cualquiera de las fórmulas 2.9-2.13, en la que la hidroxipropilmetilcelulosa tiene una viscosidad de 2.000-6.000 mPA-a, por ejemplo, de 2.600 a 5.000 mPA-a, por ejemplo, de 2.663 a 4.970 mPA-a. 2.16 La composición según una cualquiera de las fórmulas 2.9-2.15, en la que el diluyente o portador farmacéuticamente aceptable comprende alfa-lactosa monohidratada.
2.17 La composición según la fórmula 2.16, en la que la composición comprende al menos el 10% p/p de alfa-lactosa monohidratada, por ejemplo, el 10-80% p/p, por ejemplo, el 20-70% p/p, por ejemplo, el 20-60% p/p, por ejemplo, el 20-50% p/p, por ejemplo, el 20-40% p/p, por ejemplo, el 20-30% p/p, por ejemplo, el 30-70% p/p, por ejemplo, el 30-60% p/p, por ejemplo, el 30-50% p/p, por ejemplo, el 30%-40% p/p, por ejemplo, el 37% p/p.
2.18 La composición según la fórmula 2.16 ó 2.17, en la que la composición comprende alfa-lactosa monohidratada molida.
2.19 La composición según una cualquiera de las fórmulas 2.1-2.18, en la que la composición comprende una mezcla coprocesada de hidroxipropilmetilcelulosa y alfa-lactosa monohidratada (por ejemplo, Retalac®).
2.20 La composición según la fórmula 2.19, en la que la mezcla comprende partes iguales de hidroxipropilmetilcelulosa y alfa-lactosa monohidratada.
2.21 La composición según la fórmula 2.19 ó 2.20, en la que la mezcla comprende partículas de hidroxipropilmetilcelulosa y alfa-lactosa monohidratada con d50 (diámetro mediano) en el intervalo de 100 |im a 200 |im, por ejemplo, 125 |im.
2.22 La composición según una cualquiera de las fórmulas 2.19-2.21, en la que la mezcla comprende partículas de hidroxipropilmetilcelulosa y alfa-lactosa monohidratada, en la que la distribución del tamaño de partícula es tal como sigue:
< 63 |im < 25%
< 100 |im: 35%
< 250 |im > 80%.
2.23 La composición según una cualquiera de las fórmulas 2.19-2.22, en la que la composición comprende al menos el 20% p/p de la mezcla, por ejemplo, al menos el 30% p/p, por ejemplo, al menos el 40% p/p, por ejemplo, al menos el 50% p/p, por ejemplo, al menos el 60% p/p, por ejemplo, al menos el 70% p/p, por ejemplo, al menos el 80% p/p, por ejemplo, el 20-90% p/p, por ejemplo, el 30-80% p/p, por ejemplo, el 40-80% p/p, por ejemplo, el 50-80% p/p, por ejemplo, el 60-80% p/p, por ejemplo, el 70-80% p/p, por ejemplo, el 75% p/p.
2.24 La composición según una cualquiera de las fórmulas 2.1-2.23, en la que el diluyente o portador farmacéuticamente aceptable comprende un lubricante, por ejemplo, estearato de magnesio.
2.25 La composición según la fórmula 3.24, en la que el lubricante es uno o más de behenato de glicerilo, estearato de magnesio, talco y fumarato de estearilo de sodio, por ejemplo, estearato de magnesio.
2.26 La composición según la fórmula 23.24 ó 2.25, en la que la composición comprende menos del 10% p/p del lubricante, por ejemplo, menos del 5% p/p, menos del 3% p/p, menos del 1% p/p, por ejemplo, del 0,1 al 1% p/p, por ejemplo, del 0,1 al 0,8% p/p, por ejemplo, el 0,5% p/p.
2.27 La composición según una cualquiera de las fórmulas 2.24-2.26, en la que la composición comprende menos del 10% p/p de estearato de magnesio, por ejemplo, menos del 5% p/p, menos del 3% p/p, menos del 1% p/p, por ejemplo, del 0,1 al 1% p/p, por ejemplo, del 0,1 al 0,8% p/p, por ejemplo, el 0,5% p/p.
2.28 La composición según una cualquiera de las fórmulas 2.1-2.27, en la que el diluyente o portador farmacéuticamente aceptable comprende uno o más de un diluyente, agente disgregante, aglutinante y agente de liberación modificada.
2.29 La composición según la fórmula 2.28, en la que el diluyente es uno o más de manitol (por ejemplo, Pearlitol 300 DC), celulosa microcristalina (por ejemplo, Avicel pH 102) y almidón pregelatinizado (por ejemplo, Starch 1500).
2.30 La composición según la fórmula 2.29, en la que el agente disgregante es uno o ambos de crospovidona (por ejemplo, Polyplasdone XL-10) y glicolato de almidón de sodio (por ejemplo, Explotab).
2.31 La composición según la fórmula 2.28, en la que el aglutinante es polivinilpirrolidona (por ejemplo, povidona K29/32).
2-32 La composición según la fórmula 2.28, en la que el agente de liberación modificada es uno o más de hidroxipropilcelulosa (por ejemplo, Klucel EXF, Klucel MXF y/o Klucel HXF) e hidroxipropilmetilcelulosa (por ejemplo, Methocel K100M, Methocel K4M PREM, Methocel K15M PREM CR).
2.33 La composición según la fórmula 2.28 ó 23.32, en la que la composición comprende al menos el 5% p/p del agente de liberación modificada, por ejemplo, el 5-60% p/p, por ejemplo, el 10-50% p/p, por ejemplo, el 10-40% p/p.
2.34 La composición según la fórmula 2.32 ó 2.33, en la que el agente de liberación modificada es hidroxipropilmetilcelulosa.
2.35 Forma cristalina B según cualquiera de las fórmulas 1.1-1.85 para su uso en la profilaxis o el tratamiento de los trastornos descritos en la fórmula 2.35, o para su uso en el tratamiento o la profilaxis de cualquiera de los trastornos descritos en la fórmula 2.35 trastornos por déficit de atención.
Breve descripción de los dibujos
La figura 1 muestra un patrón de difracción de rayos X de polvo (XRPD) de alta resolución de la forma cristalina A. La figura 2 muestra los termogramas de DSC y TGA de la forma cristalina A.
La figura 3 muestra la isoterma de sorción/desorción dinámica de vapor de la forma cristalina A.
La figura 4 muestra una superposición de patrones de difracción de rayos X de polvo (XRPD) de la forma A, forma B y forma C cristalinas (de arriba a abajo):
la figura 4A representa un patrón de difracción de rayos X de polvo de alta resolución de la forma cristalina A; la figura 4B representa un patrón de difracción de rayos X de polvo de la forma cristalina B; y
La figura 4C representa un patrón de difracción de rayos X de polvo de la forma cristalina C.
La figura 5 representa un patrón de difracción de rayos X de polvo (XRPD) de la forma cristalina B.
La figura 6 representa una solución de indexación para la forma cristalina B.
La figura 7 representa un patrón de difracción de rayos X de polvo (XRPD) de alta resolución de la forma cristalina B. La figura 8 representa los termogramas de DSC y TGA de la forma cristalina B.
La figura 9 representa un patrón de difracción de rayos X de polvo (XRPD) de la forma cristalina C.
La figura 10 representa una solución de indexación para la forma cristalina C.
La figura 11 representa un patrón de difracción de rayos X de polvo (XRPD) de alta resolución de la forma cristalina C.
La figura 12 representa los termogramas de DSC y TGA de la forma cristalina C.
La figura 13 representa una superposición de patrones de difracción de rayos X de polvo (XRPD) de la forma A, forma B y forma C cristalinas (de arriba a abajo):
la figura 13A representa un patrón de difracción de rayos X de polvo de la forma B cristalina (enfriamiento lento en IPA, los sólidos precipitan en el refrigerador);
la figura 13B representa un patrón de difracción de rayos X de polvo de la forma cristalina C la forma cristalina B (enfriamiento cristalino lento en IPA, con semillas, los sólidos precipitan en el congelador);
la figura 13C representa un patrón de difracción de rayos X de polvo de la forma cristalina C la forma cristalina A (enfriamiento lento en IPA, precipitación de sólidos en el congelador);
la figura 13D representa un patrón de difracción de rayos X de polvo de la forma B cristalina (enfriamiento lento en IPA, los sólidos precipitan en el congelador);
la figura 13E representa un patrón de difracción de rayos X de polvo de la forma cristalina B la forma cristalina A (enfriamiento de choque en IPA, precipitación de sólidos en hielo seco/IPA);
La figura 13F representa un patrón de difracción de rayos X de polvo de la forma cristalina A la forma cristalina C (enfriamiento lento en IPA, precipitación de sólidos en el congelador); y
La figura 13G representa un patrón de difracción de rayos X de polvo de la forma cristalina C, enfriamiento lento en IPA.
La figura 14 representa una superposición de patrones de difracción de rayos X de polvo (XRPD) de la forma D, forma E y forma F cristalinas (de arriba a abajo):
la figura 14D representa un patrón de difracción de rayos X de polvo de la forma cristalina D (agitación de 30 min a 70°C en tampón de pH 4,4);
la figura 14E representa un patrón de difracción de rayos X de polvo de la forma cristalina E (contiene picos de la forma cristalina F, suspensión a 50°C en tampón de pH 6,0); y
la figura 14F representa un patrón de difracción de rayos X de polvo de la forma cristalina F (agitación de 30 min a 70°C en tampón de pH 8,1).
La figura 15 representa un patrón de difracción de rayos X de polvo (XRPD) de la forma cristalina D.
La figura 16 representa un patrón de difracción de rayos X de polvo (XRPD) de la forma cristalina E (contiene picos de la forma cristalina F).
La figura 17 representa un patrón de difracción de rayos X de polvo (XRPD) de la forma cristalina F.
La figura 18 representa un dibujo ORTEP de la forma cristalina A. Los átomos están representados por elipsoides térmicos anisotrópicos al 50% de probabilidad.
La figura 19 representa un diagrama de empaquetamiento de la forma cristalina A vista en el eje cristalográfico a. La figura 20 representa un diagrama de empaquetamiento de la forma cristalina A vista en el eje cristalográfico b . La figura 21 representa un diagrama de empaquetamiento de la forma cristalina A vista en el eje cristalográfico c. La figura 22 representa enlace de hidrógeno en la forma cristalina A.
La figura 23 representa un patrón de difracción de rayos X de polvo (XRPD) calculado de la forma cristalina A. La figura 24 representa un dibujo de elipsoide de desplazamiento atómico para la forma cristalina B (los átomos están representados por elipsoides térmicos anisotrópicos al 50% de probabilidad).
La figura 25 representa un diagrama de empaquetamiento de la forma cristalina B vista a lo largo del eje cristalográfico a.
La figura 26 representa un diagrama de empaquetamiento de la forma cristalina B vista a lo largo del eje cristalográfico b.
La figura 27 representa un diagrama de empaquetamiento de la forma cristalina B vista a lo largo del eje cristalográfico c.
La figura 28 representa el enlace de hidrógeno en la estructura de la forma cristalina B.
La figura 29 representa las conformaciones moleculares del (1R,5S)-1-(naftalen-2-il)-3-azabiciclo[3.1.0]hexano en las estructuras de las formas cristalinas A y B (izquierda: (1R,5S)-1-(naftalen-2-il)-3-azabiciclo[3.1.0]hexano en la estructura de la forma cristalina A; derecha: (1R,5S)-1-(naftalen-2-il)-3-azabiciclo[3.1.0]hexano en la estructura de la forma cristalina B).
La figura 30 representa un diagrama de empaquetamiento de las formas cristalinas A y B vistas a lo largo del eje cristalográfico a (izquierda: empaquetamiento de la forma cristalina A; derecha: empaquetamiento de la forma cristalina B).
La figura 31 representa el enlace de hidrógeno en las estructuras de las formas cristalinas A y B (izquierda: enlace de hidrógeno en la estructura de la forma cristalina A; derecha: enlace de hidrógeno en la estructura de la forma B). La figura 32 representa un patrón de polvo de rayos X calculado de la forma cristalina B.
La figura 33 representa patrones de XRPD experimentales y calculados de la forma cristalina B (arriba: patrón de XRPD experimental a temperatura ambiente; centro: patrón de XRPD calculado ajustado a temperatura ambiente; abajo: patrón de XRPD calculado a 100 K).
La figura 34 representa los patrones de XRPD experimentales y calculados de la forma cristalina A (arriba: patrón de XRPD calculado; abajo: patrón de XRPD experimental a temperatura ambiente).
La figura 35 representa un patrón de XRPD de la forma cristalina A.
La figura 36 representa una comparación de patrones de XRPD de clorhidrato de (1R,5S)-1-(naftalen-2-il)-3-azabiciclo[3.1.0]hexano de los ejemplos 1 y 5 (arriba: ejemplo 5; abajo: ejemplo 1) (los patrones están desplazados a lo largo del eje y para la comparación).
La figura 37 representa un patrón de XRPD de la forma cristalina A recogido con radiación Cu Ka.
La figura 38 representa un resultado de indexación para el patrón de XRPD representado en la figura 37 recogido con radiación Cu Ka.
La figura 39 representa los picos observados para el patrón de XRPD representado en la figura 37 recogido con radiación Cu Ka.
La figura 40 representa un patrón de XRPD de la forma cristalina B.
La figura 41 representa un resultado de indexación para el patrón de XRPD representado en la figura 40 recogido con radiación Cu Ka.
La figura 42 representa los picos observados en el patrón de XRPD representado en la figura 40 recogido con radiación Cu Ka.
La figura 43 representa un patrón de XRPD de la forma cristalina C.
La figura 44 representa un resultado de indexación para el patrón de XRPD representado en la figura 43 recogido con radiación Cu Ka.
La figura 45 representa los picos observados en el patrón de XRPD representado en la figura 43 recogido con radiación Cu Ka.
La figura 46 representa los gráficos de energía y temperatura propuestos para las formas cristalinas A, B y C.
La figura 47 representa un patrón de XRPD de la forma cristalina A.
La figura 48 representa un patrón de XRPD de la forma cristalina B.
La figura 49 representa un patrón de XRPD de una mezcla de la forma cristalina A y una pequeña cantidad de la forma cristalina B.
La figura 50 representa los patrones de XRPD de la forma cristalina A antes y después del análisis de DVS (arriba: antes, abajo: después).
Las figuras 51-54 representan los patrones de XRPD de la forma cristalina A desordenada.
La figura 55 representa un termograma de DSC de la forma cristalina B.
La figura 56 representa un patrón de XRPD de una mezcla de las formas cristalinas A y B.
Descripción detallada de la invención
Tal como se usa en el presente documento, el término “el compuesto” se refiere a (1R,5S)-1-(naftalen-2-il)-3-azabiciclo[3.1.0]hexano, también conocido como (+)-1-(naftalen-2-il)-3-azabiciclo[3.1.0]hexano. El término “el compuesto en forma de sal de adición de ácido clorhídrico” se refiere a clorhidrato de (1R,5S)-1-(naftalen-2-il)-3-azabiciclo[3.1.0]hexano o clorhidrato de (+)-1-(naftalen-2-il)-3-azabiciclo[3.1.0]hexano que tiene la siguiente estructura:

Este compuesto está libre o sustancialmente libre del correspondiente (-)-enantiómero, por ejemplo, que contiene no más del 20% p/p (peso/peso) del correspondiente (-) enantiómero, en forma de sal libre o farmacéuticamente aceptable, por ejemplo, no más del 10% p/p del correspondiente (-) enantiómero, en forma de sal libre o farmacéuticamente aceptable, por ejemplo, no más del 5% p/p del correspondiente (-) enantiómero, en forma de sal libre o farmacéuticamente aceptable, por ejemplo, no más del 2% p/p del correspondiente (-) enantiómero, en forma de sal libre o farmacéuticamente aceptable, por ejemplo, no más del 1% p/p del correspondiente (-) enantiómero, en forma de sal libre o farmacéuticamente aceptable.
“Forma cristalina A” se refiere a una forma cristalina del compuesto en forma de sal de adición de ácido clorhídrico tal
como se caracteriza en secciones relevantes de los ejemplos a continuación.
“Forma cristalina B” se refiere a una forma cristalina del compuesto en forma de sal de adición de ácido clorhídrico tal como se describe en cualquiera de las fórmulas 1.1-1.85 o tal como se caracteriza en secciones relevantes de los ejemplos a continuación.
“Forma cristalina C” se refiere a una forma cristalina del compuesto en forma de sal de adición de ácido clorhídrico tal como se caracteriza en secciones relevantes de los ejemplos a continuación.
“Forma cristalina D” se refiere a una forma cristalina tal como se caracteriza en secciones relevantes de los ejemplos a continuación.
“Forma cristalina E” se refiere a una forma cristalina tal como se caracteriza en secciones relevantes de los ejemplos a continuación.
“Forma cristalina F” se refiere a una forma cristalina tal como se caracteriza en secciones relevantes de los ejemplos a continuación.
La invención proporciona la forma cristalina B tal como se describe en el presente documento, por ejemplo en cualquiera de las fórmulas 1.1-1.89 o tal como se caracteriza en la sección de ejemplos a continuación.
El término “sustancialmente libre” de otras formas cristalinas se refiere a menos del 10% en peso, en algunas realizaciones menos del 5% en peso, en algunas realizaciones menos del 2% en peso, todavía en algunas realizaciones menos del 1% en peso, todavía en algunas realizaciones menos del 0,1% en peso, aún en algunas realizaciones menos del 0,01% en peso de otras formas u otras formas cristalinas, por ejemplo, formas amorfas u otras formas cristalinas.
El término “solvato” se refiere a aductos sólidos cristalinos que contienen cantidades o bien estequiométricas o bien no estequiométricas de un disolvente incorporadas dentro de la estructura cristalina. Por tanto, el término forma de “no solvato” en el presente documento se refiere a formas cristalinas que están libres o sustancialmente libres de moléculas de disolvente dentro de las estructuras cristalinas de la invención. De manera similar, el término forma de no “hidrato” en el presente documento se refiere a cristales de sal que están libres o sustancialmente libres de moléculas de agua dentro de las estructuras cristalinas de la invención.
El término forma “amorfa” se refiere a sólidos de disposiciones desordenadas de moléculas y no presenta una red cristalina distinguible.
El término “paciente” incluye humano y no humano.
El término “antidisolvente” significa un disolvente en el que el compuesto y/o el compuesto en forma de sal de adición de ácido clorhídrico (clorhidrato de (1R,5S)-1-(naftalen-2-il)-3-azabiciclo[3.1.0]hexano) tiene baja solubilidad o es insoluble. Por ejemplo, un antidisolvente incluye un disolvente en el que el compuesto y/o el compuesto en forma de sal de adición de ácido clorhídrico tiene una solubilidad de menos del 35 mg/ml, por ejemplo, una solubilidad de 10 30 mg/ml, por ejemplo, una solubilidad de 1-10 mg/ml, por ejemplo, una solubilidad de menos de 1 mg/ml.
El término “XRPD” significa difracción de rayos X de polvo.
Debe entenderse que un patrón de difracción de rayos X de polvo de una muestra dada puede variar (desviación estándar) dependiendo del instrumento usado, del tiempo y de la temperatura de la muestra en el momento de la medición, y de los errores estándar experimentales. Por tanto, los valores 2-theta, los valores de espaciamiento d, las alturas y la intensidad relativa de los picos tendrán un nivel aceptable de desviación. Por ejemplo, los valores pueden tener una desviación aceptable de, por ejemplo, aproximadamente el 20%, el 15%, el 10%, el 5%, el 3%, el 2% o el 1%. En una realización particular, los valores 2-theta (°) o los valores de espaciamiento d (A) del patrón de XRPD de las formas cristalinas de la presente invención pueden tener una desviación aceptable de ± 0,2 grados y/o ± 0,2 A. Además, el patrón de XRPD de las formas cristalinas de la invención puede identificarse por los picos característicos tal como los reconoce un experto en la técnica. Por ejemplo, las formas cristalinas de la invención pueden identificarse por, por ejemplo, dos picos característicos, en algunos casos, tres picos característicos, en otro caso, cinco picos característicos. Por tanto, el término “sustancialmente como” se establece en una tabla particular o se representa o se muestra en una figura particular se refiere a cualquier cristal que tiene una XRPD que tiene los picos principales o característicos tal como se establece en las tablas/figuras tal como reconoce un experto en la técnica.
También debe entenderse que los termogramas de calorimetría diferencial de barrido o de análisis termogravimétrico de una muestra determinada pueden variar (desviación estándar) en función del instrumento usado, del tiempo y de la temperatura de la muestra en el momento de la medición y de los errores estándar experimentales. El valor de la temperatura en sí puede desviarse en ± 10°C, preferiblemente ± 5°C, preferiblemente ± 3°C de la temperatura de referencia.
En la mayoría de las circunstancias para los XRPD, se seleccionan los picos dentro del intervalo de hasta aproximadamente 30° 20. Se usan algoritmos de redondeo para redondear cada pico al 0,1° o al 0,01° 20 más cercano, dependiendo del instrumento usado para recoger los datos y/o de la resolución inherente del pico. Las variabilidades de posición de los picos se dan con una precisión de ±0,2° 20.
La longitud de onda usada para calcular los valores de espaciamiento d (A) en el presente documento es de 1,5405929Á, la longitud de onda Cu-K«i (Phys. Rev., A56 (6), 4554-4568 (1997)).
Según las directrices de la USP, los hidratos y solvatos variables pueden presentar variaciones de pico mayores de ±0,2° 20.
Los “picos prominentes” son un subconjunto de toda la lista de picos observados y se seleccionan a partir de los picos observados mediante la identificación de picos preferiblemente no superpuestos, de ángulo bajo y de fuerte intensidad.
Si están disponibles múltiples patrones de difracción, es posible evaluar entonces las estadísticas de las partículas (PS) y/o la orientación preferida (PO). La reproducibilidad entre los patrones de XRPD de múltiples muestras analizadas en un solo difractómetro indica que las estadísticas de las partículas son adecuadas. La coherencia de la intensidad relativa entre los patrones de XRPD de múltiples difractómetros indica que las estadísticas de orientación son buenas. Alternativamente, el patrón de XRPD observado puede compararse con un patrón de XRPD calculado basado en una estructura monocristalina, si está disponible. Los patrones de dispersión bidimensional que usan detectores de área también pueden usarse para evaluar PS/PO. Si se determina que los efectos de PS y PO son insignificantes, entonces el patrón de XRPD es representativo de la intensidad promedio del polvo para la muestra y los picos prominentes pueden identificarse como “picos representativos”. En general, cuantos más datos se recojan para determinar los picos representativos, más se puede confiar en la clasificación de esos picos.
Los “picos característicos”, en la medida en que existen, son un subconjunto de los picos representativos y se usan para diferenciar un polimorfo cristalino de otro polimorfo cristalino (los polimorfos son formas cristalinas que tienen la misma composición química). Los picos característicos se determinan evaluando qué picos representativos, si los hay, están presentes en un polimorfo cristalino de un compuesto frente a todos los demás polimorfos cristalinos conocidos de ese compuesto con una precisión de ±0,2° 20. No todos los polimorfos cristalinos de un compuesto tienen necesariamente al menos un pico característico.
Se ha observado que en las reacciones para elaborar la forma cristalina A, también puede formarse la forma cristalina B. Sin embargo, la síntesis de los productos puede controlarse, por ejemplo, sembrando con la forma cristalina A.
La forma cristalina B, por ejemplo, de las fórmulas 1.1-1.89, tal como se describe en el presente documento, es útil como inhibidor de la recaptación triple (TRI), más potente en la recaptación de norepinefrina (NE), una sexta parte tan potente en la recaptación de dopamina (DA) y una cuarta parte tan potente en la recaptación de serotonina (5-HT). Por tanto, la forma cristalina B de las fórmulas 1.1-1.89 tal como se describe en el presente documento es útil para la profilaxis o el tratamiento de un trastorno y/o el alivio de los síntomas asociados de cualquier trastorno tratable mediante la inhibición de la recaptación de múltiples aminas biógenas relacionadas causalmente con el trastorno del SNC objetivo, en la que las aminas biógenas seleccionadas como diana para la inhibición de la recaptación se seleccionan de norepinefrina, y/o serotonina, y/o dopamina. Por tanto, la forma cristalina B es para su uso en la profilaxis o el tratamiento de cualquiera de los siguientes trastornos:
• trastorno por déficit de atención con hiperactividad (TDAH) y trastornos conductuales relacionados, así como formas y síntomas de toxicomanía (alcoholismo, drogadicción), comportamientos obsesivos compulsivos, trastornos del aprendizaje, problemas de lectura, ludomanía, síntomas maníacos, fobias, ataques de pánico, comportamiento de oposición desafiante, trastorno de conducta, problemas académicos en la escuela, tabaquismo, comportamientos sexuales anómalos, comportamientos esquizoides, somatización, depresión, trastornos del sueño, ansiedad generalizada, tartamudez, trastornos por tics, otros trastornos tales como los que se divulgan en el documento US 2007/0082940 ;
• depresión, trastornos de ansiedad, autismo, lesiones cerebrales traumáticas, deterioro cognitivo y esquizofrenia (en particular para la cognición), obesidad, trastornos de dolor crónico, trastorno de la personalidad y deterioro cognitivo leve;
• trastorno de pánico, trastorno de estrés postraumático, trastorno obsesivo compulsivo, esquizofrenia y trastornos afines, obesidad, trastornos por tics, enfermedad de Parkinson;
• trastorno asociado al cromosoma X frágil;
• trastorno asociado al cromosoma X frágil en el que el paciente era resistente a un plan de tratamiento previo para el trastorno asociado al cromosoma X frágil;
• trastorno por déficit de atención/hiperactividad (TDAH) en el que el TDAH es comórbido con uno o ambos de ansiedad y depresión (por ejemplo, depresión), por ejemplo, en un paciente con un trastorno asociado al cromosoma X frágil;
• trastorno del espectro autista (TEA),
administrando a un paciente que lo necesita una cantidad terapéuticamente eficaz de la forma cristalina B según cualquiera de las fórmulas 1.1-1.89.
Los trastornos contemplados para el tratamiento empleando la forma cristalina B de la invención, tal como se describe en el presente documento, incluyen los trastornos de la Quick Reference to the Diagnostic Criteria From DSM-IV (Diagnostic and Statistical Manual of Mental Disorders, cuarta edición), The American Psychiatric Association, Washington, D.C., 1994. Estos trastornos objetivo incluyen, pero no se limitan a, el trastorno por déficit de atención/hiperactividad, tipo predominantemente inatento; el trastorno por déficit de atención/hiperactividad, tipo predominantemente hiperactivo-impulsivo; el trastorno por déficit de atención/hiperactividad, tipo combinado; el trastorno por déficit de atención/hiperactividad no especificado (NOS); el trastorno de conducta; el trastorno de oposición desafiante; y el trastorno de conducta perturbadora no especificado (NOS).
Los trastornos depresivos susceptibles de tratamiento y/o prevención según la invención incluyen el trastorno depresivo mayor, recurrente; el trastorno distímico; el trastorno depresivo no especificado (NOS); y el trastorno depresivo mayor, episodio único.
Los trastornos adictivos susceptibles de tratamiento y/o prevención empleando los métodos y composiciones de la invención incluyen los trastornos alimentarios, los trastornos del control de los impulsos, los trastornos relacionados con el alcohol, los trastornos relacionados con la nicotina, los trastornos relacionados con las anfetaminas, los trastornos relacionados con el cannabis, los trastornos relacionados con la cocaína, los trastornos relacionados con el consumo de alucinógenos, los trastornos relacionados con los medicamentos inhalatorios y los trastornos relacionados con los opioides.
Tal como se usa en el presente documento, “cantidad terapéuticamente eficaz” se refiere a una cantidad eficaz, cuando se administra a un paciente humano o no humano, para proporcionar un beneficio terapéutico, tal como la mejora de los síntomas. La dosis específica de la sustancia administrada para obtener un beneficio terapéutico se determinará, por supuesto, en función de las circunstancias particulares que rodean el caso, incluyendo, por ejemplo, la sustancia específica administrada, la vía de administración, el estado que está tratándose y el individuo que está tratándose.
La dosis o el método de administración de la dosis de la presente divulgación no está particularmente limitado. Las dosis empleadas en la práctica de la presente divulgación variarán, por supuesto, dependiendo, por ejemplo, del modo de administración y de la terapia deseada. En general, se indica que se obtienen resultados satisfactorios en la administración oral a dosis del orden de desde aproximadamente 0,01 hasta 2,0 mg/kg. Una dosis diaria indicada para la administración oral puede estar en el intervalo de desde aproximadamente 0,75 mg hasta 200 mg, convenientemente administrada una vez, o en dosis divididas de 2 a 4 veces, diariamente o en forma de liberación sostenida. Las formas de dosificación unitarias para la administración oral pueden comprender, por ejemplo, desde aproximadamente 0,2 mg hasta 75 mg o 150 mg, por ejemplo, desde aproximadamente 0,2 mg o 2,0 mg o 50 mg o 75 mg o 100 mg hasta 200 mg o 500 mg de la forma cristalina B de cualquiera de las fórmulas 1.1-1.85, junto con un diluyente o portador farmacéuticamente aceptable para la misma.
La forma cristalina B de la invención puede administrarse por cualquier vía adecuada, incluyendo por vía oral, parenteral, transdérmica o por inhalación, incluyendo por liberación sostenida, aunque también pueden emplearse otras vías, dispositivos y métodos de administración conocidos. En algunas realizaciones, se proporciona una composición farmacéutica de liberación sostenida, por ejemplo, una composición farmacéutica oral de liberación sostenida, que comprende cualquiera de las formas cristalinas B de la invención de las fórmulas 1.1-1.85, durante un periodo de administración sostenida de aproximadamente 6 h o más, por ejemplo, 8 h o más, por ejemplo, 12 h o más, por ejemplo, 18 h o más, por ejemplo, 24 h o más. En algunas realizaciones, se proporciona una composición farmacéutica de liberación inmediata, por ejemplo, una composición farmacéutica oral de liberación inmediata, que comprende la forma cristalina B de la invención de las fórmulas 1.1-1.85.
El (1R,5S)-1-(naftalen-2-il)-3-azabiciclo[3.1.0]hexano en forma de sal de adición de ácido clorhídrico puede prepararse tal como se describe en los documentos US 2007/0082940 o WO 2013/019271.
Aunque tanto el documento US 2007/0082940 como el documento WO 2013/019271 describen la síntesis de clorhidrato de (1R,5S)-1-(naftalen-2-il)-3-azabiciclo[3.1.0]hexano, ninguno de ellos comenta ninguna forma cristalina particular de clorhidrato de (1R,5S)-1-(naftalen-2-il)-3-azabiciclo[3.1.0]hexano.
La siguiente sección ilustra los métodos de elaboración y caracterización de la forma cristalina B de la invención. Se
emplean técnicas de cristalización tanto termodinámicas como cinéticas. Estas técnicas se describen con más detalle a continuación.
Precipitación con antidisolvente: se preparan disoluciones en diversos disolventes y se filtran a través de un filtro de nailon de 0,2 |im en un vial. Luego se añade el antidisolvente hasta que se observa la precipitación. Los sólidos resultantes se aíslan por filtración a vacío y se analizan.
Enfriamiento de choque (CC): se preparan disoluciones en diversos disolventes a una temperatura elevada y se filtran en caliente a través de un filtro de nailon de 0,2 |im en un vial enfriado previamente. El vial se coloca en un baño de enfriamiento (hielo seco isopropanol). Las muestras se colocan en un congelador si no se observa que los sólidos precipiten inmediatamente. Los sólidos resultantes se aíslan por filtración a vacío y se analizan.
Evaporación rápida (FE): se prepararán las disoluciones en diversos disolventes y se someten a sonicación entre las adiciones de alícuotas para ayudar a la disolución. Una vez que la mezcla alcanza la disolución completa, tal como se juzga mediante observación visual, la disolución se filtra a través de un filtro de nailon de 0,2 |im. La disolución filtrada se deja evaporar en condiciones ambientales en un vial sin tapa. Las disoluciones se evaporan hasta sequedad, a menos que se designen como evaporaciones parciales. Los sólidos formados se aíslan y se analizan.
Secado por congelación (liofilización): se prepararán las disoluciones en dioxano:agua 1:1 o agua, se filtran a través de un filtro de nailon de 0,2 |im y se congelan en un vial o matraz sumergido en un baño de hielo seco e isopropanol. El vial o matraz que contiene la muestra congelada se acopla a un liofilizador Flexi-Dry y se seca durante un periodo de tiempo medido. Después del secado, los sólidos se aíslan y se almacenan en el congelador sobre desecante hasta su uso.
Molienda: se coloca una muestra sólida en un frasco de molienda de acero inoxidable con una bola de molienda. La muestra se muele a 30 Hz en un molino de bolas (Retsch Mixer Mill modelo MM200) durante una cantidad de tiempo determinada. Los sólidos se recogen y se analizan.
Estrés de humedad relativa: los sólidos se almacenan a una condición de aproximadamente 40°C/75% de HR durante un periodo de tiempo medido, colocando los sólidos en un vial dentro de una cámara sellada de temperatura/humedad en la condición controlada. Las muestras se analizan después de sacarlas del entorno de estrés.
Evaporación rotatoria: se preparan disoluciones del compuesto en forma de sal de adición de ácido clorhídrico de ((1R,5S)-1-(naftalen-2-il)-3-azabiciclo[3.1.0]hexano) en HFlPA. Los sólidos se obtienen por evaporación rotatoria del disolvente a vacío, con el vial de muestra sumergido en un baño de agua caliente a aproximadamente 40°C. Luego los sólidos se secan durante aproximadamente 10 minutos adicionales a vacío a temperatura ambiental. Después de la evaporación, los sólidos se almacenan en el congelador sobre desecante hasta su uso.
Enfriamiento lento (SC): se prepararán las disoluciones en diversos disolventes a una temperatura elevada. Las disoluciones se filtran en caliente a través de un filtro de nailon de 0,2 |im en un vial caliente. El vial se tapa y se deja en la placa caliente, y se apaga la placa caliente para permitir que la muestra se enfríe lentamente hasta temperatura ambiental. Si no hay sólidos presentes después del enfriamiento hasta temperatura ambiental, la muestra se coloca en un refrigerador y/o congelador para que se siga enfriando. Los sólidos se recogen por filtración a vacío y se analizan.
Evaporación lenta (SE): se prepararán disoluciones en diversos disolventes y se someten a sonicación para ayudar a la disolución. Una vez que la mezcla alcanza la disolución completa, tal como se juzga mediante observación visual, la disolución se filtra a través de un filtro de nailon de 0,2 |im. La disolución filtrada se deja evaporar en condiciones ambientales en un vial cubierto con una lámina de aluminio perforada con orificios. Las disoluciones se evaporan hasta sequedad, a menos que se designen como evaporaciones parciales. Los sólidos formados se aíslan y se analizan.
Experimentos con suspensiones: se preparan suspensiones añadiendo una cantidad suficiente de sólidos a un disolvente determinado para que haya un exceso de sólidos. Luego la mezcla se agita en un vial sellado a temperatura ambiental o a una temperatura elevada. Después de un periodo de tiempo determinado, los sólidos se aíslan por filtración a vacío y se analizan.
Difusión de vapor (VD): se preparan disoluciones en diversos disolventes y se filtran a través de un filtro de nailon de 0,2 |im. La disolución filtrada se dispensa en un vial de 1 dracma, que se coloca entonces dentro de un vial de 20 ml que contiene antidisolvente. El vial de 1 dracma se deja sin tapar y el vial de 20 ml se tapa para permitir se produzca la difusión de vapor. Los sólidos resultantes se aíslan y se analizan.
Estrés de vapor (VS): se coloca una muestra sólida en un vial de 1 dracma. Luego el vial de 1 dracma se coloca en un vial de 20 ml que contiene disolvente. El vial de 20 ml se tapa y se deja en condiciones ambientales durante un periodo de tiempo medido. Las muestras se analizan después de sacarlas del entorno de estrés.
Superposiciones de XRPD: las superposiciones de patrones de XRPD se generan usando Pattern Match 2.3.6.
Indexación de XRPD: los patrones de XRPD de alta resolución de las formas cristalinas de la invención se indexan usando X'Pert High Score Plus (X'Pert High Score Plus 2.2a (2.2.1)) o un software propio. La indexación y el refinamiento de la estructura son estudios computacionales.
Técnicas instrumentales: los materiales de prueba de este estudio se analizan usando las técnicas instrumentales descritas a continuación.
Calorimetría diferencial de barrido (DSC): la DSC se realiza usando un calorímetro diferencial de barrido de TA Instruments. La calibración de la temperatura se realiza usando metal indio rastreable por NIST. La muestra se coloca en una bandeja de DSC de aluminio, se cubre con una tapa y se registra el peso con precisión. En el lado de referencia de la celda se coloca una bandeja de aluminio pesada configurada como bandeja de muestra. Los parámetros de adquisición de datos y la configuración de la bandeja se muestran en la imagen de cada termograma. El código del método en el termograma es una abreviatura de la temperatura inicial y final, así como de la velocidad de calentamiento; por ejemplo, -30-250-10 significa “desde -30°C hasta 250°C, a 10°C/min”. La siguiente tabla resume las abreviaturas usadas en cada imagen para las configuraciones de la bandeja:
Abreviatura Significado
T0C Bandeja doblada Tcero
HS Tapa sellada herméticamente
HSLP Tapa sellada herméticamente y perforada con un orificio láser
C Tapa doblada
NC Tapa no doblada
Análisis termogravimétrico (TGA): los análisis TG se realizan usando un analizador termogravimétrico de TA Instruments. La calibración de la temperatura se realiza usando níquel y Alumel™. Cada muestra se coloca en una bandeja de aluminio. La muestra se cierra herméticamente, se perfora la tapa y se introduce en el horno TG. El horno se calienta bajo nitrógeno. Los parámetros de adquisición de datos se muestran en la imagen de cada termograma. El código del método en el termograma es una abreviatura de la temperatura inicial y final, así como de la velocidad de calentamiento; por ejemplo, 25-350-10 significa “desde 25°C hasta 350°C, a 10°C/min”.
Difracción de rayos X de polvo (XRPD): Inel XRG-300. Los análisis de difracción de rayos X de polvo se realizan en un difractómetro Inel XRG-3000, equipado con un detector curvo sensible a la posición con un intervalo de 20 de 120°. Los datos en tiempo real se recogen usando radiación Cu Ka con una resolución de 0,03° 20. El voltaje y el amperaje del tubo se ajustan a 40 kV y 30 mA, respectivamente. Los patrones se muestran desde 2,5 hasta 40° 20 para facilitar comparaciones de patrones directas. Las muestras se preparan para el análisis empaquetándolas en capilares de vidrio de paredes finas. Cada capilar se monta en un cabezal de goniómetro que está motorizado para permitir el giro del capilar durante la adquisición de datos. La calibración del instrumento se realiza diariamente usando un patrón de referencia de silicio. Los parámetros de adquisición y procesamiento de datos se muestran en cada patrón que se encuentra en la sección de datos.
Difracción de rayos X de polvo (XRPD): difractómetro Bruker D-8 Discover. Los patrones de XRPD se recogen con un difractómetro Bruker D-8 Discover y el sistema de detección de difracción de área general de Bruker (GADDS, v.
4.1.20). Se produce un haz incidente de radiación Cu Ka usando un tubo de enfoque fino (40 kV, 40 mA), un espejo Gobel y un colimador de doble orificio de 0,5 mm. La muestra se empaqueta entre láminas de 3 |im de grosor para formar una probeta portátil en forma de disco. La probeta preparada se carga en un soporte fijado a una platina de traslación y se analiza en geometría de transmisión. El haz incidente se escanea y se rastrea para optimizar las estadísticas de orientación. Se usa una trampa de haz para minimizar la dispersión en el aire del haz incidente en ángulos bajos. Los patrones de difracción se recogen con un detector de área Hi-Star situado a 15 cm de la muestra y se procesan con GADDS. Antes del análisis, se analiza un patrón de silicio para verificar la posición del pico de Si 111. Los parámetros de adquisición y procesamiento de datos se muestran en cada patrón que se encuentra en la sección de datos.
Difracción de rayos X de polvo (XRPD): difractómetro PANalytical X'Pert Pro. Los patrones de XRPD se recogen usando un difractómetro PANalytical X'Pert Pro. La probeta se analiza con radiación Cu producida usando una fuente de enfoque fino largo Optix. Se usa un espejo multicapa elípticamente graduado para enfocar los rayos X de Cu Ka de la fuente a través de la probeta y sobre el detector. La probeta se intercala entre películas de 3 micrómetros de grosor, se analiza en geometría de transmisión, y se gira en paralelo al vector de difracción para optimizar las estadísticas de orientación. Para minimizar el fondo generado por la dispersión en el aire, se usa una trampa de haz, una extensión corta de antidispersión, un filo de cuchilla de antidispersión y una purga de helio. Se usan ranuras de Soller para los haces incidentes y difractados para minimizar la divergencia axial. Los patrones de difracción se recogen usando un detector sensible a la posición de barrido (X'Celerator) situado a 240 mm de la probeta. Los parámetros de adquisición de datos de cada patrón de difracción se muestran por encima de la imagen de cada patrón
en la sección de datos. Antes del análisis, se analiza una probeta de silicio (material de referencia convencional 640d de NIST) para verificar la posición del pico de silicio 111.
Para la indexación, la concordancia entre las posiciones de los picos permitidos, marcadas con barras, y los picos observados indica una determinación coherente de la celda unitaria. La indexación exitosa del patrón indica que la muestra se compone principalmente de una sola fase cristalina. Los grupos espaciales coherentes con el símbolo de extinción asignado, los parámetros de celda unitaria y las cantidades derivadas se tabulan debajo de la figura. Para confirmar la solución de indexación tentativa, deben determinarse los motivos de empaquetamiento molecular dentro de las celdas unitarias cristalográficas. No se realiza ningún intento de empaquetamiento molecular.
Abreviaturas
acetonitrilo (ACN)
birrefringencia (B)
salmuera (disolución acuosa satura de cloruro de sodio)
densidad (d)
diclorometano (DCM)
equivalentes (eq)
etanol (EtOH)
acetato de etilo (EtOAc)
extinción (E)
peso de fórmula (FW)
gramo (g)
hora u horas (h)
hexafluoroisopropanol (HFIPA)
cromatografía de líquidos de alta resolución (presión) (HPLC)
isopropanol (IPA)
acetato de isopropilo (IPAc)
isopropil éter (IPE)
kilogramo (kg)
litros (l)
metanol (MeOH)
metil etil cetona (MEK)
minuto(s) (min)
mililitros(ml)
molaridad de una disolución (mol/l) (M)
peso molecular (PM)
moles (mol)
temperatura ambiente (TA)
saturado (sat)
hexametildisililazano de sodio (NaHMDS)
material de partida (SM)
tetrahidrofurano (THF)
2,2,2,-trifluoroetanol (TFE)
frente a (vs.)
peso (p)
Aquellas partes de los siguientes ejemplos no cubiertas por el alcance de las reivindicaciones adjuntas son con fines ilustrativos.
Ejemplo 1 - Preparación de la forma cristalina A

Cargar 2-naftilacetonitrilo (1500 g, 8,97 mol, SM) a un matraz de fondo redondo de 3 bocas de 50 l equipado con un agitador superior, embudo de adición, termopar, baño de enfriamiento, entrada de nitrógeno y tubo de secado. Cargar tetrahidrofurano (6,0 l, 4 ml/g, SM) al recipiente de reacción. Agitar a temperatura ambiente hasta que se disuelva todo el 2-naftilacetonitrilo. Cargar (S)-(+)-epiclorhidrina (1081 g, 11,67 mol, 1,30 eq) al recipiente de reacción. Enfriar la mezcla de reacción hasta una temperatura interna de -28°C. Usar baño de hielo seco/acetona para enfriar. Se añade hielo seco de manera intermitente para mantener el baño de enfriamiento entre -35 y -25°C durante la adición de bis(trimetilsilil)amida de sodio. Cargar una disolución de bis(trimetilsilil)amida de sodio en THF (9,0 l, 18,0 mol, 2 eq. en mol) al embudo de adición y añadir lentamente a la mezcla de reacción enfriada a una velocidad tal que la temperatura interna permanece a menos de -14°C. La adición requiere 1 h 40 min. Durante la adición, la temperatura interna está generalmente entre -20 y -17°C. Después de la finalización de la adición, la disolución resultante se agita a entre -21 y -16°C durante 2 h 30 minutos. Monitorizar la reacción mediante HPLC. Mantener una temperatura de -20 a -15°C de la mezcla de reacción mientras se analiza la muestra mediante HPLC.
Ensayo de HPLC a 2 h 30 min muestra que la reacción no es completa. Añadir bis(trimetilsilil)amida de sodio adicional en t Hf (0,30 l, 0,60 mol, 0,067 eq. en mol) durante 10 min mediante el embudo de adición, mantener la temperatura interna de la mezcla de reacción a menos de -15°C. Agitar 15 min en cuyo momento el ensayo de HPLC muestra que la reacción es completa. Cargar complejo de dimetilsulfuro de borano (2,25 l, 22,5 mol, 2,5 eq. en mol) mediante el embudo de adición a una velocidad tal que la temperatura interna de la mezcla de reacción permanece por debajo de 0°C. La adición requiere 40 min. Después de la finalización de la adición de borano, calentar lentamente la mezcla de reacción hasta 40°C. Una vez se obtiene una temperatura interna de 40°C, interrumpir el calentamiento. Se observa una exoterma estable lenta durante aproximadamente dos horas, lo que da como resultado una temperatura interna máxima de 49°C. Tras la finalización de la exoterma, aumentar la temperatura interna hasta 60°C. Agitar la mezcla de reacción durante la noche a 60°C. Monitorizar la reacción mediante HPLC. Mantener una temperatura de 60°C de la mezcla de reacción mientras se analiza la muestra mediante HPLC.
Cargar dimetilsulfuro de borano adicional (0,35 l, 0,70 mol, 0,39 eq. en mol) a la mezcla de reacción mediante el embudo de adición. Agitar la mezcla de reacción durante 3 h 30 min a 60°C. Enfriar la mezcla de reacción hasta temperatura ambiente.
Cargar HCl 2 M en agua (17,3 l, 11,5 ml/g SM, preparado a partir de 2,9 l de HCl concentrado y 14,4 l de agua) a un segundo matraz de fondo redondo de 3 bocas de 50 l equipado con un agitador superior, termopar, baño de enfriamiento y entrada de nitrógeno. Enfriar la disolución de HCl/agua hasta 3°C. Transferir lentamente la mezcla de reacción a temperatura ambiente que contiene la ciclopropilamina a la disolución de HCl enfriada a una velocidad tal que la temperatura interna máxima de la mezcla de extinción es de 23°C. La extinción requiere 2 h 50 min. Cuando la extinción de la reacción es completa, calentar la mezcla bifásica hasta 50°C. Agitar durante una hora a 50°C. Enfriar hasta temperatura ambiente. Añadir acetato de isopropilo (6,0 l, 4 ml/g SM). Añadir agua (7,5 l, 5 ml/g SM). Agitar la mezcla durante al menos 15 min. Interrumpir la agitación y dejar que las fases sedimenten durante al menos 30 min. Desechar la fase orgánica (superior). Añadir amoniaco acuoso (2,25 l, 1,5 ml/g SM) a la fase acuosa. Añadir acetato de isopropilo (7,5 l, 5 ml/g). Agitar la mezcla durante al menos 15 min. Interrumpir la agitación y dejar que las fases sedimenten durante al menos 30 min. Separar las fases. El producto está en la fase orgánica (superior). Añadir acetato de isopropilo (7,5 l, 5 ml/g SM) a la fase acuosa. Agitar la mezcla durante al menos 15 min. Interrumpir la agitación y dejar que las fases sedimenten durante al menos 30 min. Separar las fases. El producto está en la fase orgánica (superior). Combinar los dos extractos de acetato de isopropilo. Añadir fosfato de sodio dibásico al 5% en agua (6,0 l, 4 ml/g SM) a los extractos combinados. Agitar la mezcla durante al menos 15 min. Interrumpir la agitación y dejar que las fases sedimenten durante al menos 30 min. Separar las fases y desechar la fase acuosa (inferior). Añadir salmuera saturada (6,0 l, 4 ml/g SM) a los extractos combinados. Agitar la mezcla durante al menos 15 min. Interrumpir la agitación y dejar que las fases sedimenten durante al menos 30 min. Separar las fases y desechar la fase acuosa (inferior). Concentrar la fase orgánica final en un matraz de Buchi de 20 l tarado a vacío. Obtener un total de 1967,6 g de un sólido ceroso naranja claro. Transferir los sólidos a un matraz de fondo redondo de 3 bocas de 50 l equipado con un agitador superior, termopar, manto calefactor, entrada de nitrógeno y tubo de secado. Añadir acetato de isopropilo (15 l, 10 ml/g SM). Calentar la mezcla hasta 50°C. Añadir ácido p-toluenosulfónico monohidratado (1586 g, 8,34 mol, 0,93 eq. en mol) en porciones durante 30 min manteniendo la temperatura a menos de 60°C. Tras la finalización de la adición, interrumpir el calentamiento y dejar que la mezcla se enfríe hasta temperatura ambiente. Recoger los sólidos mediante filtración. Lavar la torta de filtro con acetato de isopropilo (3 l, 2 ml/g SM). Lavar la torta de filtro una segunda vez con acetato de isopropilo (3 l, 2 ml/g SM). Secar la torta de filtro hasta un peso constante en el embudo de filtro arrastrando aire a través de la torta usando vacío. Después de un periodo de secado inicial, la torta de filtro se rompe con una espátula y se agita la torta a intervalos para promover el secado. Obtener 2049 g de un sólido blanco. Ensayo de HPLC: 98,2% para el pico principal y una razón cis:trans de 98,5:1,5.

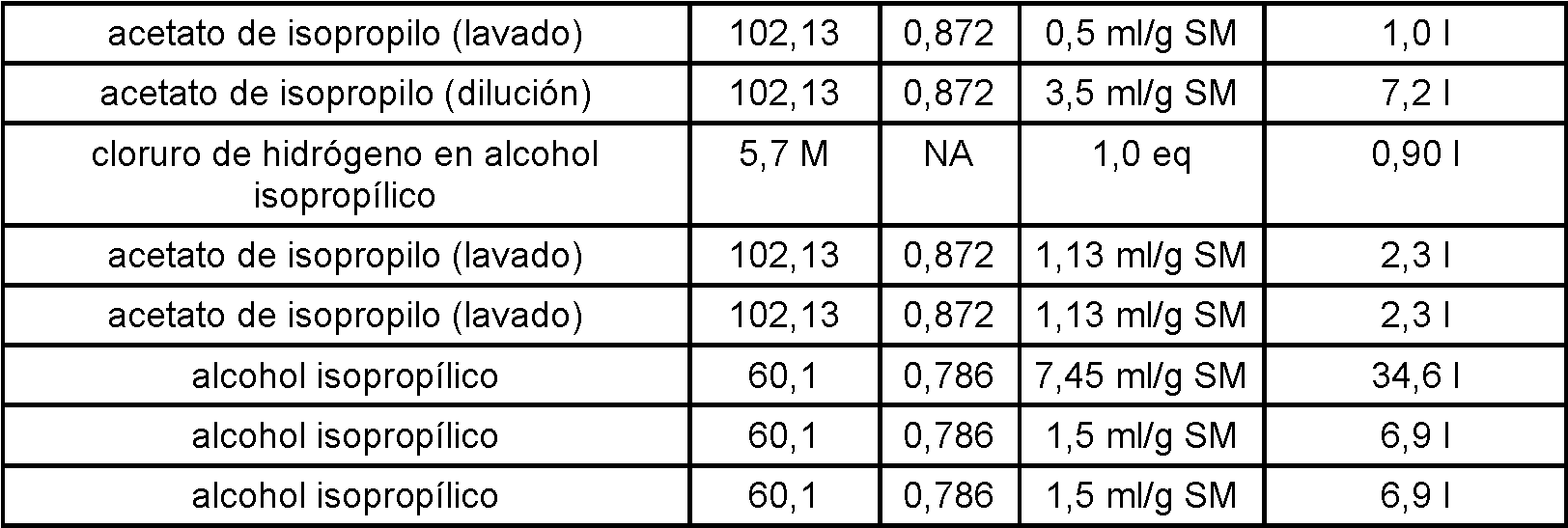
Nota: la adición de NaOH 5 M a la mezcla de reacción es exotérmica y requiere un enfriamiento activo.
Cargar 2039,7 g (5,10 mol, 1,0 eq. en mol) de la sal de tosilato de naftilciclopropilamina obtenida anteriormente a un matraz de fondo redondo de 3 bocas de 50 ml equipado con un agitador superior, termopar, embudo de adición, entrada de nitrógeno, tubo de secado y baño de agua a temperatura ambiente. Cargar 13,2 l de acetato de isopropilo (IPAc, 13,2 1, 6,5 ml/g SM) al matraz de reacción y agitar a temperatura ambiente para dar una suspensión blanca. Añadir 445 ml de cloruro de tionilo (6,13 mol, 1,2 eq. en mol) mediante el embudo de adición manteniendo la temperatura a menos de 25°C. La adición requiere 1 h 5 min. Agitar la suspensión espesa a temperatura ambiental durante al menos dos h. Monitorizar la reacción mediante HPLC. Mantener la mezcla de reacción a temperatura ambiental mientras se analiza la muestra mediante HPLC. Añadir NaOH 5 M (6,1 l, 30,5 mol, 6,0 eq. en mol) mediante el embudo de adición usando un baño de hielo/agua para mantener a menos de 30°C. La adición requiere 1 h 40 min. Monitorizar la reacción mediante HPLC. Mantener la mezcla de reacción a temperatura ambiental mientras se analiza la muestra mediante HPLC. Agitar la mezcla de reacción a 25°C durante 1 h 5 min luego dejar que las fases sedimenten. Separar las fases. Lavar la fase orgánica (superior) con NaOH 1 M (2.1 l, 1 ml/g SM). Combinar las dos fases acuosas. Someter a retroextracción las fases acuosas combinadas con acetato de isopropilo (7,6 l, 3,75 ml/g SM). Combinar la fase orgánica lavada y el retroextracto. Lavar las fases orgánicas combinadas con salmuera saturada (4,1 l, 2 ml/g SM). Secar las fases orgánicas sobre sulfato de magnesio granular. Filtrar para retirar los sólidos. Lavar la torta de filtro con acetato de isopropilo (1 l, 0,5 ml/g SM). Concentrar el filtrado combinado y lavar en un matraz evaporador rotativo de Buchi de 20 l hasta un volumen total de 4,2 l. Transferir a un matraz de fondo redondo de 3 bocas de 22 l equipado con un agitador superior, embudo de adición, termopar, baño de enfriamiento, entrada de nitrógeno y tubo de secado. Diluir con acetato de isopropilo (7,2 l, volumen de disolución total = 11,4 l, 5,6 ml/g SM). Añadir cloruro de hidrógeno en alcohol isopropílico (5,7 M, 0,90 l, 5,13 mol, 1,0 eq. en mol) mediante el embudo de adición durante 50 min a una velocidad tal que la temperatura interna permanece por debajo de 30°C. Agitar la suspensión durante 45 min a temperatura ambiente. Filtrar para recoger los sólidos. Lavar la torta de filtro con acetato de isopropilo (2,3 l, 1,13 ml/g SM). Lavar la torta de filtro una segunda vez con acetato de isopropilo (2,3 l, 1,13 ml/g SM). Secar parcialmente la torta de filtro arrastrando aire a través de la torta con vacío. El ensayo de HPLC de la torta húmeda muestra una pureza del 96,3% de área y un EE de 89,5%.
Combinar las tortas húmedas de este experimento y de otro lote en un matraz de fondo redondo de 3 bocas de 50 ml equipado con un agitador superior, manto calefactor, termopar, condensador de reflujo, entrada de nitrógeno y tubo de secado. Añadir alcohol isopropílico (34,6 l, 7,45 ml/g SM). Calentar la suspensión a reflujo. Mantener el reflujo durante tres horas. Interrumpir el calentamiento y dejar enfriar hasta temperatura ambiente. Filtrar para recoger los sólidos. Lavar la torta de filtro con alcohol isopropílico (6,9 l, 1,5 ml/g SM). Lavar la torta de filtro una segunda vez con alcohol isopropílico (6,9 l, 1,5 ml/g SM). Secar la torta de filtro hasta un peso constante arrastrando aire a través de la torta usando vacío. Obtener 2009 g de producto como un sólido marrón. HPLC: > 99,5%. HPLC quiral: 95,4%.

Nota: debe usarse una cantidad mínima de etanol necesaria para disolver completamente el material de partida.
Cargar clorhidrato de (1R,5S)-1-(naftalen-2-il)-3-azabiciclo[3.1.0]hexano a un matraz de fondo redondo de 3 bocas de 50 ml equipado con un agitador superior, termopar, condensador de reflujo, manto calefactor, entrada de nitrógeno y tubo de secado. Añadir etanol (20 l, ml/g SM). Calentar la suspensión con agitación hasta 77°C. Añadir etanol adicional en alícuotas de 0,5 l y devolver la mezcla a reflujo hasta que todos los sólidos se disuelvan. Completar la disolución después de la adición de 1,5 l de etanol adicional, 21,5 l total. Interrumpir el calentamiento y dejar que la disolución se
enfríe hasta temperatura ambiente. Filtrar para recoger los sólidos. Lavar la torta de filtro con etanol (4,3 1, 2,14 ml/g SM). Secar la torta de filtro hasta un peso constante arrastrando aire a través de la torta de filtro usando vacío. Obtener 1435 g de sólidos marrón claro. Rendimiento = 74%. HPLC: 99,5%. HPLC quiral: 99,9%.

Cargar el compuesto en forma de sal de adición de ácido clorhídrico (clorhidrato de (1R,5S)-1-(naftalen-2-il)-3-azabiciclo[3.1.0]hexano) (1406 g, 5,72 mol, 1,0 eq. en mol) (el compuesto obtenido de la etapa anterior y de otro lote) a un matraz de fondo redondo de 3 bocas de 22 l equipado con un agitador superior, manto calefactor, termopar y entrada de nitrógeno. Añadir agua (14 l, 10 ml/g SM). Calentar la suspensión hasta una temperatura interna de 34°C para disolver todos los sólidos. Transferir a un gran embudo separador. Añadir tetrahidrofurano (2,8 l, 2 ml/g SM). Añadir acetato de isopropilo (2,8 l, 2 ml/g SM). Interrumpir la agitación y dejar que las fases se separen. Desechar la fase orgánica (superior). El producto está en la fase inferior (acuosa). A la fase acuosa (inferior) añadir amoniaco acuoso (1,14 l, 17,1 mol, 3,0 eq. en mol). Añadir acetato de isopropilo (14,0 l, 10 ml/g SM). Agitar la mezcla durante al menos 15 min. Interrumpir la agitación y dejar que las fases sedimenten durante al menos 30 min. Separar las fases. El producto está en una fase orgánica (superior). Añadir sulfato de magnesio granular a la fase orgánica. Filtrar para retirar los sólidos. Lavar la torta de filtro con acetato de isopropilo (1 l). Lavar la torta de filtro una segunda vez con acetato de isopropilo (1 l). Concentrar el filtrado combinado y lavar en un matraz evaporador rotativo de Buchi de 20 l para dar un sólido blanquecino. Cargar el sólido a un matraz de fondo redondo de 22 l equipado con un agitador superior, termopar, embudo de adición, entrada de nitrógeno y tubo de secado. Añadir alcohol isopropílico (14 l, 10 ml/g SM). Agitar a temperatura ambiente para disolver todos los sólidos. Cargar HCl 5,7 N en IPA (175 ml, 1,0 mol, 0,17 eq. en mol) mediante el embudo de adición durante 10 min para formar sólidos blancos. Agitar la suspensión fina a temperatura ambiente durante 30 min. Cargar HCl 5,7 N en IPA (670 ml, 3,82 mol, 0,67 eq. en mol) seguido de HCl 5,6 N en IPA (110 ml, 0,62 mol, 0,11 eq. en mol) mediante el embudo de adición durante 55 min. Agitar la suspensión durante 35 min luego someter a ensayo las aguas madre para determinar la pérdida. Añadir HCl 5,6 N en IPA (60 ml, 0,34 mol, 0,06 eq. en mol) durante 10 min mediante el embudo de adición. Agitar la suspensión durante 30 min, luego someter a ensayo las aguas madre para determinar la pérdida. Filtrar para recoger los sólidos. Lavar la torta de filtro con alcohol isopropílico (2,8 l, 2 ml/g SM). Lavar la torta de filtro una segunda vez con alcohol isopropílico (2,8 l, 2 ml/g SM). Secar la torta de filtro hasta un peso constante arrastrando aire a través de la torta de filtro usando vacío. Obtener 1277 g de producto como sólido blanquecino. HPLC: 99,9%.
El compuesto resultante presenta un patrón de XRPD cristalino (figura 1) y se designa como forma cristalina A. Se recoge el patrón de XRPD con un difractómetro PANalytical X'Pert PRO MPD usando un haz incidente de radiación Cu producido usando una fuente de enfoque fino largo Optix. Se usa un espejo multicapa elípticamente graduado para enfocar los rayos X de Cu Ka a través de la probeta y sobre el detector. Antes del análisis, una probeta de silicio (NIST SRM 640d) se analiza para verificar la posición del pico de Si 111. Se intercala una probeta de la muestra entre películas de 3 |im de grosor y se analiza en geometría de transmisión. Se usan una trampa de haz, una extensión corta antidispersión y un filo de cuchilla antidispersión para minimizar el fondo generado por el aire. Se usan ranuras de Soller para los haces incidentes y difractados para minimizar el ensanchamiento a partir de la divergencia axial. El
patrón de difracción se recoge usando un detector sensible a la posición de barrido (X'Celerator) ubicado 240 mm de la probeta y software Data Collector v. 2.2b. El patrón de XRPD experimental se recoge según especificaciones de BPFa. El patrón de XRPD recogido se muestra en la figura 1 (Panalytical X-Pert Pro MPD PW3040 Pro, tubo de rayos X: Cu (1,54059 A), voltaje: 45 kV, amperaje: 40 mA, intervalo de barrido: 1,01-40,00 °20, tamaño de paso: 0,017 o20, tiempo de recogida: 1939 s, velocidad de barrido: 1,2°/min, ranura: DS: 1/2°, SS: nulo, tiempo de revolución: 1,0 s, modo: transmisión).
Se muestran resultados de análisis térmicos en la figura 2 (DSC, tamaño: 1,7800 mg, método: (-30)-300-10, T0C; TGA, tamaño: 6,8320 mg, método: 00-350-10). Mediante TGA, la forma cristalina A presenta aproximadamente el 0,4% de pérdida de peso hasta 200°C. El drástico cambio de peso en el TGA a aproximadamente 276°C es coherente con la descomposición. El termograma de DSC (figura 2) presenta múltiples endotermas entre aproximadamente 245 y 248°C de manera simultánea con el drástico cambio de peso mediante TGA, lo que sugiere que se producen acontecimientos solapados durante el calentamiento.
Los datos de caracterización para la forma cristalina 1 se resumen en la tabla 1 a continuación:
Tabla 1.

a. Las temperaturas se redondean hasta el °C más cercano; los valores de pérdida de peso se redondean a un decimal. Basándose en los datos de sorción/desorción de vapor dinámico recogidos (figura 3), la forma cristalina A obtenida es un material no higroscópico. Tras el equilibrio inicial al 5% de HR, la forma cristalina A muestra una pérdida de peso del 0,03%; un aumento de peso del 0,10% se observa desde el 5% hasta el 95% de HR. Durante la etapa de desorción desde el 95% hasta el 5% de HR, la forma cristalina A presenta aproximadamente una pérdida de peso del 0,10%. El material tras el equilibrio de humedad es similar al material de partida mediante XRPD (figura 50).
Parámetros de adquisición de datos para isoterma de sorción/desorción de vapor dinámico:


Ejemplo 2 - Preparación de cristales de la forma A
Se prepara una disolución del compuesto en forma de sal de adición de ácido clorhídrico (clorhidrato de (1R,5S)-1-(naftalen-2-il)-3-azabiciclo[3.1.0]hexano) usando 98,5 mg del compuesto del ejemplo 1 en 2 ml de metanol y se filtra a través de un filtro de nailon de 0,2 |am. Una alícuota de 0,5 ml de la disolución filtrada se dispensa en un vial abierto de 1 dracma, que luego se coloca dentro de un vial de 20 ml que contiene 3 ml de antidisolvente acetato de etilo. Se deja el vial de 1 dracma sin tapar y se tapa el vial de 20 ml para dejar que se produzca la difusión de vapor. Se hacen crecer cristales individuales en el vial de 1 dracma después de aproximadamente 7 días.
Recogida de datos: una placa incolora de C i5Hi6ClN [C1, Ci 5Hi 6N] que tiene unas dimensiones aproximadas de 0,38 x 0,30 x 0,18 mm, se monta sobre una fibra en orientación aleatoria. Se realizan examen preliminar y recogida de datos con radiación Mo Ka (X = 0,71073 Á) en un difractómetro Nonius Kappa CCD equipado con un cristal de grafito, monocromador de haz incidente. Se realizan refinamientos usando SHELX97 (Sheldrick, G. M. Acta Cryst., 2008, A64, 112). Se obtienen las constantes de celdas y una matriz de orientación para recogida de datos a partir del refinamiento por mínimos cuadrados usando los ángulos de ajuste de 5812 reflexiones en el intervalo 1° < 0 < 27°. La mosaicidad refinada de DENZO/SCALEPACK es de 0,38°, lo que indica una buena calidad del cristal (Otwinowski, Z.; Minor, W. Methods Enzymol. 1997, 276, 307). El grupo espacial se determina mediante el programa XPREP (Bruker, XPREP en SHELXTL v. 6,12., Bruker AXS Inc., Madison, Wl, EE:UU., 2002). A partir de la presencia sistemática de las siguientes condiciones: h00 h = 2n; 0k0 k = 2n; 00l l = 2n y de posterior refinamiento por mínimos cuadrados, se determina que el grupo espacial es P212121 (n.° 19). Los datos se recogen hasta un valor máximo de 20 de 55,71°, a una temperatura de 150 ± 1 K.
Reducción de datos: se integran las tramas con DENZO-SMN (Otwinowski, Z.; Minor, W. Methods Enzymol. 1997, 276, 307). Se recoge un total de 5812 reflexiones, de las que 2930 son únicas. Se aplican a los datos correcciones de Lorentz y polarización. El coeficiente de absorción lineal es de 0,273 mm-1 para radiación Mo Ka. Se aplica una corrección de absorción empírica usando SCALEPACK (Otwinowski, Z.; Minor, W. Methods Enzymol. 1997, 276, 307). Los coeficientes de transmisión oscilan desde 0,953 hasta 0,953. Se promedian las intensidades de reflexiones equivalentes. El factor de coincidencia para el promedio es del 2,9% basándose en la intensidad.
Resolución de la estructura y refinamiento: la estructura se resuelve mediante métodos directos usando SIR2004 (Burla, M.C., Caliandro, R., Camalli, M,. Carrozzini, B., Cascarano, G.l., De Caro, l., Giacovazzo, C., Polidori, G. y Spagna, R., J. Appl. Cryst. 2005, 38, 381). Los átomos restantes se ubican en las posteriores síntesis de Fourier de diferencia. Se incluyen átomos de hidrógeno en el refinamiento pero se limitan a montarse en el átomo al que se unen. La estructura se refina en mínimos cuadrados de matriz completa minimizando la función:

El peso w se define como 1/[ct2(F o 2) (0,0384P)2 (0,2436P)], donde P = (Fo2 +2F c 2)/3.
Se toman factores de dispersión de las “Tablas internacionales de cristalografía” (International Tables for Crystallography, Vol. C, Kluwer Academic Publishers: Dordrecht, Países Bajos, 1992, tablas 4.2.6.8 y 6.1.1,4). De las 2930 reflexiones usadas en los refinamientos, sólo las reflexiones con F o 2 > 2a(F o 2) se usan para calcular R. Se usa un total de 2678 reflexiones en el cálculo. El ciclo final de refinamiento incluye 162 parámetros variables y converge (el desplazamiento más grande de parámetro es < 0,01 veces su desviación estándar estimada) con factores de coincidencias no ponderados y ponderados:
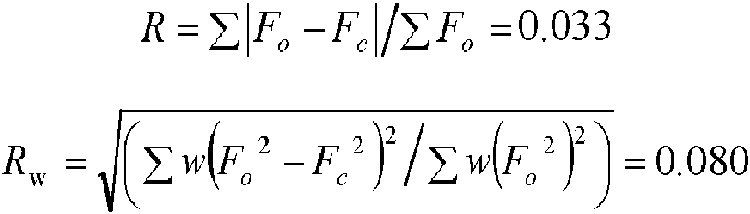
La desviación estándar de una observación de peso unitario (bondad del ajuste) es de 1,066. El pico más alto en la Fourier de diferencia final tiene una altura de 0,19 e/Á3. El pico mínimo negativo tiene una altura de -0,24 e/Á3. El factor de Flack para la determinación de la estructura absoluta (Flack, H. D. Acta Cryst. 1983, A39, 876) se refina a -0,02(6). Patrón de difracción de rayos X de polvo (XRPD) calculado: se genera un patrón de XRPD calculado para la radiación Cu usando PowderCell 2.3 (PowderCell para Windows versión 2.3 Kraus, W.; Nolze, G. Federal Institute for Materials Research and Testing, Berlín, Alemania, UE, 1999) y las coordenadas atómicas, el grupo espacial y los parámetros de celda unitaria a partir de los datos del monocristal. Debido a que los datos del monocristal se recogen a bajas temperaturas (150 K), los desplazamientos de picos pueden ser evidentes entre el patrón calculado a partir de los datos de baja temperatura y el patrón de difracción de polvo experimental a temperatura ambiente, particularmente en ángulos de difracción altos.
Diagramas ORTEP y de empaquetamiento: el diagrama ORTEP se prepara usando el programa ORTEP III (Johnson, C. K. ORTEPIII, Report ORNL-6895, Oak Ridge National Laboratory, TN, EE.UU. 1996. OPTEP-3 para Windows VI.05, Farrugia, L.J., J. Appl. Cryst. 1997, 30, 565) dentro del software p La t On (Spek, A. L. PLATON. Molecular Graphics Program. Universidad de Utrecht, Utrecht, Países Bajos, 2008. Spek, A. L, J. Appl. Cryst. 2003, 36, 7). Los átomos se representan mediante elipsoides térmicos anisotrópicos al 50% de probabilidad. Los diagramas de empaquetamiento se preparan usando el software de modelado cA m ERON (Watkin, D. J.; Prout, C. K.; Pearce, L. J. Ca MERON, Chemical Crystallography Laboratory, Universidad de Oxford, Oxford, 1996). La evaluación de los centros quirales se realiza con el paquete de software PLATON (Spek, A. L. PLATON. Molecular Graphics Program. Universidad de Utrecht, Utrecht, Países Bajos, 2008. Spek, A. L, J. Appl. Cryst. 2003, 36, 7). La configuración absoluta se evalúa usando la especificación de reglas de quiralidad molecular (Cahn, R.S.; Ingold, C; Prelog, V. Angew. Chem. Intern. Ed. Eng., 1966, 5, 385; Prelog, V. G. Helmchen Angew. Chem. Intern. Ed. Eng., 1982, 21,567). Las figuras adicionales se generan con el paquete de visualización Mercury 2.4 (Macrae, C. F. Edgington, P. R. McCabe, P. Pidcock, E. Shields, G. P. Taylor, R. Towler M. and van de Streek, J.; J. Appl. Cryst., 2006, 39, 453-457). El enlace de hidrógeno se representa con líneas discontinuas.
Resultados: los parámetros de la celda ortorrómbica y el volumen calculado son: a = 5,7779(2) Á, b = 8,6633(2) Á, c = 25,7280(8) Á, a = p = y = 90°, V = 1287,83(7) Á3. El peso de fórmula de la unidad asimétrica en la estructura cristalina es de 245,75 g mol-1 con Z = 4, lo que da como resultado una densidad calculada de 1,267 g cm-3. Se ha determinado que el grupo espacial es P2^21. En la tabla 2 a continuación se proporciona un resumen de los datos del cristal y de los parámetros de recogida de datos cristalográficos.
El valor R es de 0,033 (3,3%).
En la figura 18 se muestra un dibujo ORTEP de la forma cristalina A.
La unidad asimétrica, mostrada en la figura 18, contiene una molécula protonada de (1R,5S)-1-(naftalen-2-il)-3-azabiciclo[3.1.0]hexano y un contraión cloruro. El protón se encuentra en el mapa de diferencias y se permite que refine libremente en el nitrógeno, lo que indica la formación de la sal.
Los diagramas de empaquetamiento vistos a lo largo de los ejes cristalográficos a, b y c se muestran en las figuras 19-21, respectivamente. El enlace de hidrógeno se produce entre los átomos de cloro y nitrógeno, y la estructura consiste en infinitas cadenas unidimensionales unidas con enlaces de hidrógeno a lo largo del eje cristalográfico a , mostradas en la figura 22.
La estructura absoluta puede determinarse mediante un análisis de la dispersión anómala de rayos X por el cristal. Un parámetro refinado x, conocido como parámetro de Flack (Flack, H.D.; Bernardinelli, G., Acta Cryst., 1999, A55, 908; Flack, H.D.; Bernardinelli, G., J. Appl. Cryst., 2000, 33, 1143), codifica la abundancia relativa de los dos componentes en un gemelo de inversión. La estructura contiene una fracción 1-x del modelo que se está refinando, y x de su inverso. Siempre que se obtenga una incertidumbre estándar baja, el parámetro de Flack debería estar cerca de 0 si la estructura resuelta es correcta, y cerca de 1 si el modelo inverso es correcto. El parámetro de Flack medido para la estructura de la forma cristalina A mostrada en la figura 18 es de -0,02 con una incertidumbre estándar de 0,06. Después de que se resuelve una estructura, la calidad de los datos puede evaluarse para determinar su poder de distinción de la inversión, lo que se realiza mediante un examen de la incertidumbre estándar del parámetro de Flack. Para la forma cristalina A, la incertidumbre estándar, (u), es igual a 0,06, lo que indica un fuerte poder de distinción de la inversión. El compuesto es enantiopuro y la estructura absoluta puede asignarse directamente a partir de la estructura cristalina.
El refinamiento del parámetro de Flack (x) (Flack, H.D. Acta Cryst. 1983, A39, 876) no da lugar a una declaración cuantitativa sobre la asignación de la estructura absoluta. Sin embargo, un enfoque que aplica la estadística bayesiana a las diferencias de Bijvoet puede proporcionar una serie de probabilidades para diferentes hipótesis de la estructura absoluta (Hooft, R.W., J. Appl. Cryst., 2008, 41, 96-103; Bijvoet, J.M.; Peederman, A.F.; van Bommel, A.J., Nature 1951, 168, 271). Este análisis proporciona un parámetro equivalente a Flack (Hooft) además de las probabilidades de que la estructura absoluta sea correcta, incorrecta o un gemelo racémico. Para el conjunto de datos actual, se determina que el parámetro equivalente a Flack (Hooft) es de -0,01(3), la probabilidad de que la estructura sea correcta es de 1.000, la probabilidad de que la estructura sea incorrecta es de 0,000 y la probabilidad de que el material sea un gemelo racémico es de 0,4'59.
La estructura contiene dos centros quirales situados en C11 y C15 (véase la figura 18, dibujo ORTEP), que se asignan como configuración R y S, respectivamente.
La figura 23 muestra un patrón de difracción de rayos X de polvo calculado de la forma cristalina A, generado a partir de los datos del monocristal.
En la figura 1 se muestra el patrón de difracción de rayos X de polvo experimental de la forma cristalina A.
La XRPD experimental de la forma cristalina A de la figura 1 se superpone con el patrón calculado en la figura 34.
Las diferencias de intensidad entre los patrones de difracción de rayos X de polvo calculados y experimentales se deben a menudo a la orientación preferida. La orientación preferida es la tendencia de los cristales a alinearse con cierto grado de orden. Esta orientación preferida de la muestra puede afectar significativamente a las intensidades de los picos, pero no a las posiciones de los picos, en el patrón de difracción de polvo experimental. Además, es de esperar que se produzca algún cambio en la posición de los picos entre los patrones de difracción de polvo calculados y experimentales, ya que el patrón de polvo experimental se recoge a temperatura ambiental y los datos del monocristal se recogen a 150 K. Las bajas temperaturas se usan en el análisis del monocristal para mejorar la calidad de la estructura, pero pueden contraer el cristal, lo que da como resultado un cambio en los parámetros de celda unitaria, que se refleja en el patrón de difracción de polvo calculado. Estos desplazamientos son especialmente evidentes en los ángulos de difracción elevados.
A continuación se proporcionan las tablas de los parámetros posicionales y sus desviaciones estándar estimadas (tabla 3), los coeficientes del factor de temperatura anisotrópico (tabla 4), las distancias de enlaces (tabla 5), los ángulos de enlace (tabla 6), los enlaces de hidrógeno y ángulos (tabla 7) y los ángulos de torsión (tabla 8).
Tabla 2. Datos de cristales y parámetros de recogida de datos para la forma A de clorhidrato de (1R,5S)-1-(naftalen-2-il)-3-azabiciclo[3.1.0]hexano (forma cristalina A)
fórmula C15H16ClN
peso de fórmula 245,75
grupo espacial P2-|2-|21 (n.° 19)
a, A 5,7779(2)
b, A 8,6633(2)
c, A 25,7280(8)
V, A3 1287,83(7)
Z 4
dcalc, g cm- -33 1,267
dimensiones del cristal, mm 0,38 x 0,30 x 0,18
temperatura, K 150
radiación (longitud de onda, A) Mo Kx (0,71073)
monocromador grafito
coef. de abs. lineal, mm- -11 0,273
corrección de absorción aplicada empíricaa
factores de transmisión: mín, máx 0,953, 0,953
difractómetro Nonius Kappa CCD
Intervalo h, k, l de -7 a 7 de -11 a 11 de -33 a 33 intervalo 20, grados 1,58-55,71
mosaicidad, grados 0,38
programas usados SHELXTL F000 520,0
ponderación
1/[ü2(Fo2) (0,0384P)2 (0,2436P)], donde P=(F02 +2Fc2)/3
datos recogidos 5812
datos únicos 2930
Pint 0,029
datos usados en el refinamiento 2930
Límite usados en cálculos del factor R F c 2>2,0o(F c 2)
datos con -£>2.00(7) 2678
número de variables 162
desplazamiento más grande/est. en ciclo final 0,00
P(Fo) 0,033
Pw(Fo 2) 0,080
bondad del ajuste 1,066
determinación de la estructura absoluta parámetro de Flackb (-0,02(6)) parámetro de Hooftc (-0,01(3)) Cobertura de Friedel 90% a Otwinowski, Z.; Minor, W. Methods Enzymol. 1997, 276, 307.
b Flack, H. D. Acta Cryst., 1983 A39, 876.
c Hooft, R.W.W., Straver, L.H. y Spek, A.L. J. Appl. Cryst., 2008, 41, 96-103.
Tabla 3. Parámetros posicionales y sus desviaciones estándar estimadas para la forma cristalina A
Átomo x y z U(A2)
C11 -0,21843(7) 1,09587(4) 0,483829(15) 0,02856(9)
N13 0,2878(3) 1,04618(14) 0,53004(5) 0,0234(3)
C1 0,4183(3) 0,93704(19) 0,70605(6) 0,0294(4)
C2 0,2847(3) 0,88296(17) 0,66572(6) 0,0268(4)
C3 0,0828(3) 0,7983(2) 0,67700(7) 0,0380(5)
C4 0,0151(3) 0,7719(3) 0,72723(8) 0,0426(6)
C5 0,1497(3) 0,8274(2) 0,76923(7) 0,0340(5)
C6 0,0855(4) 0,8007(3) 0,82173(8) 0,0465(6)
C7 0,2208(4) 0,8543(2) 0,86149(7) 0,0483(6)
C8 0,4249(4) 0,9340(2) 0,85125(7) 0,0447(6)
C9 0,4915(4) 0,9627(2) 0,80087(7) 0,0391(5)
C10 0,3549(3) 0,9099(2) 0,75855(6) 0,0294(4)
C11 0,3521(3) 0,91598(19) 0,61066(6) 0,0261(4)
C12 0,2704(3) 1,06743(16) 0,58785(5) 0,0270(4)
C14 0,2577(3) 0,87808(16) 0,51906(6) 0,0282(4)
C15 0,3409(3) 0,7984(2) 0,56741(7) 0,0314(5)
C16 0,5712(3) 0,8497(2) 0,58846(7) 0,0352(5)
H131 0,436(4) 1,082(2) 0,5177(8) 0,036(5)*
H132 0,168(4) 1,105(2) 0,5138(7) 0,039(5)*
H1 0,555 0,993 0,699 0,035 H3 -0,008 0,759 0,649 0,046 H4 -0,123 0,716 0,734 0,051 H6 -0,052 0,745 0,829 0,056 H7 0,175 0,837 0,896 0,058
H8 0,519 0,969 0,879 0,054
H9 0,630 1,018 0,794 0,047
H15 0,285 0,692 0,575 0,038
H12A 0,109 1,089 0,598 0,032
H12B 0,370 1,154 0,600 0,032
H14A 0,351 0,847 0,489 0,034
H14B 0,093 0,853 0,512 0,034
H16A 0,659 0,776 0,610 0,042
H16B 0,667 0,918 0,566 0,042
Los átomos con asterisco están refinados isotópicamente
Ueq _ (1/3)XiXj Uija*ia*jai.aj
Se incluyen átomos de hidrógeno en el cálculo de factores de estructura pero no están refinados.
Tabla 4. Coeficientes de factor de temperatura anisotrópico - U para la forma cristalina A
Nombre U(1,1) U(2,2) U(3,3) U(1,2) U(1,3) U(2,3) C11 0,02543(19) 0,02561(17) 0,03463(19) 0,00075(15) 0,00262(16) 0,00196(16) N13 0,0268(7) 0,0213(6) 0,0222(6) 0,0008(6) -0,0013(6) -0,0002(5) C1 0,0292(9) 0,0301(9) 0,0290(8) -0,0056(7) 0,0005(7) 0,0014(7) C2 0,0258(8) 0,0290(8) 0,0256(7) 0,0017(7) -0,0019(6) 0,0053(6) C3 0,0278(9) 0,0550(12) 0,0313(9) -0,0099(9) -0,0063(8) 0,0089(8) C4 0,0286(10) 0,0605(13) 0,0388(11) -0,0118(10) -0,0015(8) 0,0154(10) C5 0,0326(10) 0,0394(10) 0,0301(8) 0,0019(8) 0,0016(7) 0,0094(8) C6 0,0458(12) 0,0584(13) 0,0354(10) -0,0020(11) 0,0068(10) 0,0160(9) C7 0,0664(14) 0,0518(11) 0,0266(8) 0,0055(12) 0,0037(10) 0,0084(8) C8 0,0628(14) 0,0437(12) 0,0276(9) 0,0012(10) -0,0062(9) -0,0020(8) C9 0,0479(12) 0,0386(10) 0,0309(10) -0,0053(9) -0,0015(8) -0,0037(8) C10 0,0334(9) 0,0282(8) 0,0265(8) 0,0020(7) -0,0002(6) 0,0017(7) C11 0,0252(8) 0,0282(8) 0,0249(7) -0,0008(7) -0,0014(6) 0,0018(7) C12 0,0352(9) 0,0244(7) 0,0215(7) -0,0015(7) 0,0001(7) -0,0019(5) C14 0,0343(8) 0,0221(7) 0,0283(7) 0,0013(6) -0,0041(7) -0,0040(6) C15 0,0393(11) 0,0245(8) 0,0303(8) 0,0047(7) -0,0011(7) 0,0004(7) C16 0,0308(9) 0,0452(10) 0,0297(8) 0,0105(8) 0,0006(7) 0,0081(8)
La forma del factor de temperatura anisotrópico es:
exp[-2^h2a*2U(1,1) k2b*2U(2,2) 12C*2 U(3,3) 2hka*b*U(1,2) 2hla*c*U(1,3)+ 2klb*c*U(2,3)] donde a*, b* y c* son constantes de red cristalina recíprocas.
Tabla 5. Distancias de enlaces en Angstroms para la forma cristalina A
Átomo 1 Átomo 2 Distancia Átomo 1 Átomo 2 Distancia
N13 C14 1,4936(18) C7 H7 0,950
N13 C12 1,5023(18) C8 C9 1,375(3)
N13 H131 0,96(2) C8 H8 0,950
N13 H132 0,96(2) C9 C10 1,420(3)
C1 C2 1,376(2) C9 H9 0,950
C1 C10 1,419(2) C11 C16 1,503(2)
C1 H1 0,950 C11 C15 1,510(2)
C2 C3 1,408(2) C11 C12 1,513(2)
C2 C11 1,497(2) C12 H12A 0,990
C3 C4 1,370(3) C12 H12B 0,990
C3 H3 0,950 C14 C15 1,501(2)
C4 C5 1,415(3) C14 H14A 0,990
C4 H4 0,950 C14 H14B 0,990
C5 C10 1,412(3) C15 C16 1,504(3)
C5 C6 1,420(3) C15 H15 1,000
C6 C7 1,369(3) C16 H16A 0,990
C6 H6 0,950 C16 H16B 0,990
C7 C8 1,391(3)
Los números entre paréntesis son desviaciones estándar estimadas en los dígitos menos significativos.
Tabla 6. Ángulos de enlaces en grados para la forma cristalina A
Átomo 1 Átomo 2 Átomo 3 Ángulo Átomo 1 Átomo 2 Átomo 3 Ángulo
C14 N13 C12 107,39(11) C5 C10 C1 119,08(16)
C14 N13 H131 110,6(12) C5 C10 C9 118,71(16)
C12 N13 H131 110,3(12) C1 C10 C9 122,21(17)
C14 N13 H132 110,8(13) C2 C11 C16 120,40(14)
C12 N13 H132 108,7(12) C2 C11 C15 123,87(14)
H131 N13 H132 109,2(16) C16 C11 C15 59,90(12)
C2 C1 C10 121,10(16) C2 C11 C12 116,85(14)
C2 C1 H1 119,50 C16 C11 C12 116,53(15)
C10 C1 H1 119,50 C15 C11 C12 106,60(13)
C1 C2 C3 119,14(15) N13 C12 C11 104,89(12)
C1 C2 C11 120,17(15) N13 C12 H12A 110,80
C3 C2 C11 120,69(15) C11 C12 H12A 110,80
C4 C3 C2 121,22(17) N13 C12 H12B 110,80
C4 C3 H3 119,40 C11 C12 H12B 110,80
C2 C3 H3 119,40 H12A C12 H12B 108,80
C3 C4 C5 120,43(18) N13 C14 C15 104,74(12)
C3 C4 H4 119,80 N13 C14 H14A 110,80
C5 C4 H4 119,80 C15 C14 H14A 110,80
C10 C5 C4 119,01(16) N13 C14 H14B 110,80
C10 C5 C6 119,16(17) C15 C14 H14B 110,80
C4 C5 C6 121,82(18) H14A C14 H14B 108,90
C7 C6 C5 120,4(2) C14 C15 C16 116,45(15)
C7 C6 H6 119,80 C14 C15 C11 108,31(14)
C5 C6 H6 119,80 C16 C15 C11 59,81(11)
C6 C7 C8 120,71(18) C14 C15 H15 119,20
C6 C7 H7 119,60 C16 C15 H15 119,20
C8 C7 H7 119,60 C11 C15 H15 119,20
C9 C8 C7 120,36(19) C11 C16 C15 60,29(12)
C9 C8 H8 119,80 C11 C16 H16A 117,70
C7 C8 H8 119,80 C15 C16 H16A 117,70
C8 C9 C10 120,6(2) C11 C16 H16B 117,70
C8 C9 H9 119,70 C15 C16 H16B 117,70
C10 C9 H9 119,70 H16A C16 H16B 114,90
Los números entre paréntesis son desviaciones estándar estimadas en los dígitos menos significativos.
Tabla 7. Distancias de enlaces de hidrógeno en Angstroms y ángulos en grados para la forma cristalina A
D H A D-H A-H D-A D-H-A
N13 H131 C11 0,96(2) 2.18(2) 3,121(2) 164,1(15)
N13 H132 C11 0,96(2) 2,36(2) 3,187(2) 144,0(15)
N13 H132 C11 0,96(2) 2,674(18) 3,1217(19) 109,2(14)
Los números entre paréntesis son desviaciones estándar estimadas en los dígitos menos significativos.
Tabla 8. Ángulos de torsión en grados para la forma cristalina A
Átomo 1 Átomo 2 Átomo 3 Átomo 4 Ángulo
C14 N13 C12 C11 28,20(0,18)
C12 N13 C14 C15 -27,51 (0,18)
C10 C1 C2 C3 -0,50 (0,25)
C10 C1 C2 C11 178,63 (0,15)
C2 C1 C10 C5 -0,71 (0,25)
C2 C1 C10 C9 179,13 (0,16)
C1 C2 C3 C4 1,39 (0,26)
C11 C2 C3 C4 -177,73 (0,18)
C1 C2 C11 C12 -85,92 (0,20)
C1 C2 C11 C15 137,54 (0,17)
C1 C2 C11 C16 65,41 (0,21)
C3 C2 C11 C12 93,19 (0,19)
C3 C2 C11 C15 -43,34(0,24)
C3 C2 C11 C16 -115,47 (0,18)
C2 C3 C4 C5 -1,05 (0,30)
C3 C4 C5 C6 -179,38 (0,20)
C3 C4 C5 C10 -0,18 (0,30)
C4 C5 C6 C7 179,21 (0,21)
C10 C5 C6 C7 0,02 (0,46)
C4 C5 C10 C1 1,04 (0,26)
C4 C5 C10 C9 -178,80 (0,18)
C6 C5 C10 C1 -179,74 (0,18)
C6 C5 C10 C9 0,42 (0,27)
C5 C6 C7 C8 -0,85 (0,33)
C6 C7 C8 C9 1,25 (0,30)
C7 C8 C9 C10 -0,80 (0,29)
C8 C9 C10 C1 -179,87 (0,17)
C8 C9 C10 C5 -0,03 (0,25)
C2 C11 C12 N13 -160,97 (0,14) C15 C11 C12 N13 -17,56 (0,17)
C16 C11 C12 N13 46,58 (0,18)
C2 C11 C15 C14 141,11 (0,16)
C2 C11 C15 C16 -108,36 (0,18) C12 C11 C15 C14 0,94(0,18)
C12 C11 C15 C16 111,47 (0,15)
C16 C11 C15 C14 -110,53 (0,16)
C2 C11 C16 C15 114,01 (0,17)
C12 C11 C16 C15 -94,57(0,15)
N13 C14 C15 C11 16,15 (0,18)
N13 C14 C15 C16 -48,59(0,19)
C14 C15 C16 C11 96,68(0,16)
Los números entre paréntesis son desviaciones estándar estimadas en los dígitos menos significativos.
Ejemplo 3 - Preparación de las formas cristalinas A a F
Las formas cristalinas A a F se preparan tal como sigue usando la forma cristalina A obtenida en el ejemplo 1 anterior. Se usa una variedad de técnicas de cristalización, incluyendo evaporación, enfriamiento, precipitación con disolvente/antidisolvente, suspensión, estrés de vapor y difusión de vapor, tal como se describió anteriormente. Los resultados se presentan en la tabla 9 a continuación:
Tabla 9.



a. Los valores de temperaturas, tiempos y HR informados son aproximados.
b. Escala a aproximadamente 25 mg. Concentración de disolución de IPA: 10 mg/ml.
c. Escala a aproximadamente 27 mg. Concentración de disolución de IPA: 10 mg/ml.
Forma cristalina B - Tal como se resumió anteriormente, la forma cristalina B se obtiene a partir de evaporación y suspensión en agua, suspensión, enfriamiento lento y de choque en DCM, así como el enfriamiento lento en 1-propanol. Además, los materiales que presentan patrones de XRPD de la forma cristalina A con picos de la forma cristalina B resultan de la evaporación en DCM, etanol, HFIPA y TFE. El material que presenta un patrón de XRPD de la forma cristalina B con picos débiles de la forma cristalina A y de la forma cristalina C se observa a partir de un experimento de enfriamiento de choque en 1-propanol.
La forma cristalina B se indexa a partir de un patrón de XRPD de alta resolución usando X'Pert High Score Plus (X'Pert High Score Plus 2.2a (2.2.1)) (figura 6, el patrón de XRPD de alta resolución también se muestra en la figura 7). El patrón parece representar una mezcla de las formas cristalinas B y A. La coincidencia entre las posiciones de los picos permitidos, marcadas con barras para la forma actual, y los picos observados indica una determinación coherente de la celda unitaria. Los picos a 18,5°, 20,7°, 25,7° y 27,5° dos-theta no son coherentes con la solución de indexación de la forma cristalina B y son probablemente de la forma cristalina A. Los grupos espaciales coherentes con el símbolo de extinción asignado, los parámetros de celda unitaria y las cantidades derivadas se tabulan debajo de la figura. Para confirmar la solución de indexación tentativa, deben determinarse los motivos de empaquetamiento molecular dentro de la celda unitaria cristalográfica. No se realiza ningún intento de empaquetamiento molecular. La forma cristalina B se indexa con un volumen similar por unidad de fórmula en comparación con la forma cristalina A, lo que sugiere que la forma cristalina B es una forma cristalina no solvatada.
Parámetros de adquisición de datos de XRPD para las figuras 4B y 5: INEL XRG-3000, tubo de rayos X: 1,54187100 A, voltaje: 40 (kV), amperaje: 30 (mA), tiempo de adquisición: 300 s, capilar giratorio, tamaño de paso: aproximadamente 0,03° 20.
Parámetros de adquisición de datos de XRPD para las figuras 6 y 7: Panalytical X-Pert Pro MPD PW3040 Pro, tubo de rayos X: Cu (1,54059 A), voltaje: 45 kV, amperaje: 40 mA, intervalo de barrido: 1,00-39,99° 20, tamaño de paso: 0,017° 20, tiempo de recogida: 1939 s, velocidad de barrido: 1,2°/min, ranura: DS: 1/2°, SS: nulo, tiempo de revolución: 1,0 s, modo: transmisión.
Los datos de caracterización de la forma Cristalina B se resumen en la tabla 10 a continuación:
Tabla 10.

a. Las temperaturas se redondean hasta el °C más cercano; los valores de pérdida de peso se redondean a un decimal.
b. XRPD de alta resolución.
Los resultados del análisis térmico de la forma cristalina B se muestran en la figura 8 (DSC, tamaño: 1,2600 mg, método: (-30)-300-10, T0C; TGA, tamaño: 9,4320 mg, método: 00-350-10). Mediante TGA, la forma cristalina B muestra una pequeña pérdida de peso de aproximadamente el 0,2% desde temperatura ambiental hasta 200°C, posiblemente debido a cantidades traza de disolvente. El cambio drástico en la pendiente del termograma de TGA a aproximadamente 281°C es coherente con la descomposición. Mediante DSC, se sospecha que una amplia endoterma observada a aproximadamente 141°C (pico) se atribuye a un cambio de forma sólida o posiblemente a una pérdida de compuestos volátiles al calentarse. La forma cristalina B presenta una endoterma a aproximadamente 248°C (pico), similar al comportamiento térmico observado para la forma cristalina A, seguida de dos amplias endotermas a aproximadamente 251 y 264°C. Basándose en los datos obtenidos, la forma cristalina B es un material cristalino no solvatado.
Forma cristalina C - La forma cristalina C puede elaborarse mediante enfriamiento lento en isopropanol. El material que presenta un patrón de XRPD de la forma cristalina A con picos débiles de la forma cristalina C resulta de un experimento de enfriamiento lento en etanol; mientras que los experimentos de enfriamiento de choque en etanol e isopropanol ofrecen un patrón de XRPD de la forma cristalina C con picos débiles de la forma cristalina A.
Se realizan seis intentos de escalado para preparar la forma cristalina C mediante enfriamiento en isopropanol en una escala de aproximadamente 50-150 mg (tabla 11) y los sólidos se someten a prueba mediante XRPD. A la temperatura del refrigerador, los sólidos precipitados producen la forma B. La siembra con la forma C después del enfriamiento en el refrigerador (no se observan sólidos) y antes de colocarla en el congelador produce el patrón de XRPD de la forma C con picos B. La precipitación a la temperatura del congelador da como resultado sólidos con un patrón de XRPD de la forma C con picos A. Una disolución colocada en el congelador después de enfriar hasta temperatura ambiente con una concentración más baja (7 mg/ml en comparación con 10 mg/ml) produce la forma B. Mediante el enfriamiento de choque (disolución a temperatura ambiental colocada en hielo seco/isopropanol), los sólidos generados son una mezcla de las formas B y A. El último intento a escala aproximada de 50 mg genera una mezcla de las formas A y C. Los diferentes resultados de estos experimentos sugieren posibles factores que afectan a la cristalización de la forma C a mayor escala (por ejemplo, concentración, temperatura, tiempo de enfriamiento y siembra), y la cristalización competitiva de las formas A y B que posiblemente sean más estables en las condiciones experimentales usadas. Obsérvese que la forma C permanece inalterada por XRPD después de 22 días de almacenamiento a temperatura ambiental.
Parámetros de adquisición de datos de XRPD para las figuras 13A, C y F: Panalytical X-Pert Pro MPD PW3040 Pro, tubo de rayos X: Cu (1,54059 A), voltaje: 45 kV, amperaje: 40 mA, intervalo de barrido: 1,00-39,99° 20, tamaño de paso: 0,017° 20, tiempo de recogida: 717 s, velocidad de barrido: 3,3°/min, ranura: DS: 1/2°, SS: nulo, tiempo de revolución: 1,0 s, modo: transmisión.
Parámetros de adquisición de datos de XRPD para la figura 13B: Panalytical X-Pert Pro MPD PW3040 Pro, tubo de rayos X: Cu (1,54059 A), voltaje: 45 kV, amperaje: 40 mA, intervalo de barrido: 1,00-39,99° 20, tamaño de paso: 0,017° 20, tiempo de recogida: 720 s, velocidad de barrido: 3,2°/min, ranura: DS: 1/2°, SS: nulo, tiempo de revolución: 1,0 s, modo: transmisión.
Parámetros de adquisición de datos de XRPD para la figura 13D: Panalytical X-Pert Pro MPD PW3040 Pro, tubo de rayos X: Cu (1,54059 A), voltaje: 45 kV, amperaje: 40 mA, intervalo de barrido: 1,00-39,99° 20, tamaño de paso: 0,017° 20, tiempo de recogida: 718 s, velocidad de barrido: 3,3°/min, ranura: DS: 1/2°, SS: nulo, tiempo de revolución: 1,0 s, modo: transmisión.
Parámetros de adquisición de datos de XRPD para la figura 13E: Panalytical X-Pert Pro MPD PW3040 Pro, tubo de rayos X: Cu (1,54060 A), voltaje: 45 kV, amperaje: 40 mA, intervalo de barrido: 1,00-39,99° 20, tamaño de paso: 0,017° 20, tiempo de recogida: 720 s, velocidad de barrido: 3,2°/min, ranura: DS: 1/2°, SS: nulo, tiempo de revolución: 1,0 s, modo: transmisión.
Tabla 11.


a. Las temperaturas y tiempos informados son aproximados.
b. Concentración de disolución de IPA: 11 mg/ml.
c. Concentración de disolución de IPA: 10 mg/ml.
d. Sembrado con la forma cristalina C (para XRPD de semillas véanse las figuras 4C y 9) antes del traslado al congelador.
e. Concentración de disolución de IPA: 7 mg/ml.
La forma C se indexa a partir de un patrón de XRPD de alta resolución (figura 10) usando un software propio. El patrón parece representar una mezcla de las formas C y A. La concordancia entre las posiciones de los picos permitidos, marcadas con barras para la forma actual, y los picos observados indica una determinación coherente de la celda unitaria. Los picos a 12,3°, 15,4°, 16,6°, 20,7° y 25,7° dos-theta no son coherentes con la solución de indexación de la forma C y son probablemente de la forma A. Los grupos espaciales coherentes con el símbolo de extinción asignado, los parámetros de celda unitaria y las cantidades derivadas se tabulan debajo de la figura. Para confirmar la solución de indexación tentativa, deben determinarse los motivos de empaquetamiento molecular dentro de la celda unitaria cristalográfica. No se realiza ningún intento de empaquetamiento molecular. La forma C se indexa con un volumen por unidad de fórmula similar en comparación con la forma A, lo que sugiere que la forma C es una forma cristalina no solvatada.
Parámetros de adquisición de datos de XRPD para las figuras 4C, 9 y 13G: INEL XRG-3000, tubo de rayos X: 1,54187100 A, voltaje: 40 (kV), amperaje: 30 (mA), tiempo de adquisición: 300 s, capilar giratorio, tamaño de paso: aproximadamente 0,03° 20.
Parámetros de adquisición de datos de XRPD para las figuras 10 y 11: Panalytical X-Pert Pro MPD PW3040 Pro, tubo de rayos X: Cu (1,54059 A), voltaje: 45 kV, amperaje: 40 mA, intervalo de barrido: 1,00-39,99° 20, tamaño de paso: 0,017° 20, tiempo de recogida: 720 s, velocidad de barrido: 3,2°/min, ranura: DS: 1/2°, SS: nulo, tiempo de revolución: 1,0 s, modo: transmisión.
Los datos de caracterización de la forma C se resumen en la tabla 12 a continuación:
Tabla 12.

a. Las temperaturas se redondean hasta el °C más cercano; los valores de pérdida de peso se redondean a un decimal; los valores de AH informados se redondean hasta el número entero más cercano.
b. XRPD de alta resolución, reanalizado después de 22 días de almacenamiento a temperatura ambiente.
Los resultados del análisis térmico de la forma C se muestran en la figura 12 (DSC, tamaño: 1,0100 mg, método: (-30)-300-10, T0C; TGA, tamaño: 2,2300 mg, método: 00-350-10). Mediante TGA, la forma C presenta una pérdida de peso de aproximadamente el 1,3% desde temperatura ambiental hasta 200°C, posiblemente debido a la pérdida de
compuestos volátiles al calentarse. El cambio drástico en la pendiente del termograma de TGA a aproximadamente 266°C es coherente con la descomposición. Mediante DSC, se sospecha que una pequeña y amplia endoterma observada a aproximadamente 122°C (pico) se atribuye a un cambio de forma sólida o posiblemente a una pérdida de compuestos volátiles al calentarse. La forma C presenta una endoterma a aproximadamente 248°C (pico), similar al comportamiento térmico observado para la forma A, seguida de una amplia endoterma a aproximadamente 271°C.
Según los datos obtenidos, la forma C es un material cristalino no solvatada.
Formas cristalinas D, E y F - La forma cristalina A se disuelve en un medio con pH ajustado. El sólido no disuelto o el precipitado observado se analiza mediante XRPD. Algunos experimentos se realizan a temperatura elevada para aumentar la solubilidad, los sólidos no disueltos también se analizan mediante XRPD. Las formas cristalinas D, E y F resultantes se generan durante estos experimentos tal como se resume en la tabla 13 a continuación.
Parámetros de adquisición de datos de XRPD para las figuras 14D-F: INEL XRG-3000, tubo de rayos X: 1,54187100 A, voltaje: 40 (kV), amperaje: 30 (mA), tiempo de adquisición: 300 s, capilar giratorio, tamaño de paso: aproximadamente 0,03° 20.
Tabla 13.
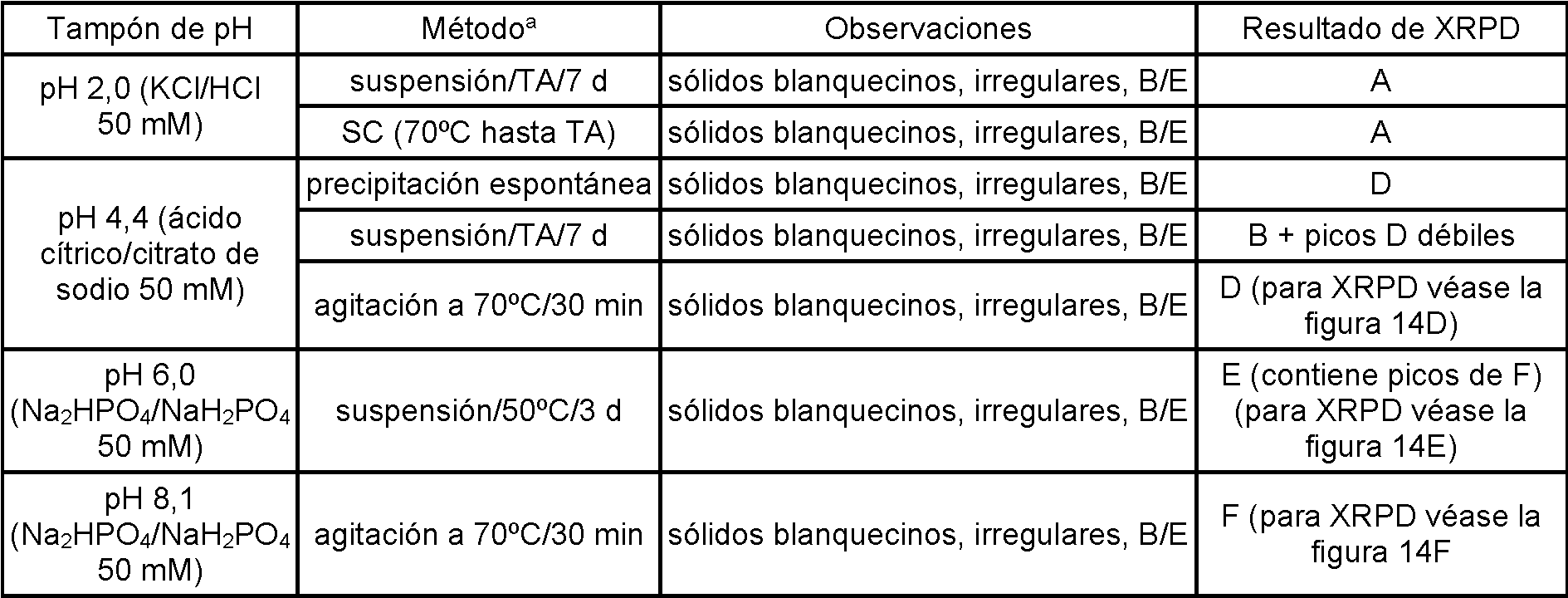
a. Los tiempos y las temperaturas informados son aproximados.
- tampón pH 2,0 (KCl/HCl 50 mM): la forma cristalina A se recupera a partir de un enfriamiento lento (aproximadamente de 70°C a temperatura ambiental) y suspensión a temperatura ambiente.
- tampón pH 4,4 (ácido cítrico/citrato de sodio 50 mM): la forma cristalina D resulta de la precipitación espontánea a temperatura ambiente y después de agitar una suspensión a aproximadamente 70°C; una suspensión a temperatura ambiente produce la forma cristalina B con picos débiles de la forma cristalina D mediante XRPD.
- tampón pH 6,0 (Na2HPO^NaH2PO450 mM): la forma cristalina E con picos también encontrados en la forma cristalina F mediante XRPD se observa a partir de la suspensión a aproximadamente 50°C.
- tampón pH 8,1 (Na2HPO4/NaH2PO450 mM): la forma cristalina F resulta de la agitación de una suspensión a aproximadamente 70°C.
Las formas cristalinas D, E y F se caracterizan mediante XRPD tal como se muestra en la figura 14.
Ejemplo 4 - Amorfo
Los intentos por preparar clorhidrato de (1R,5S)-1-(naftalen-2-il)-3-azabiciclo[3.1.0]hexano amorfo se realizan mediante molienda, liofilización y evaporación rotatoria (tabla 14). Los posibles materiales de la forma cristalina A desordenada se recuperan en todos los intentos usados en este estudio.
Parámetros de adquisición de datos de XRPD para las figuras 52-55: Bruker Discovery D8, tubo de rayos X: Cu (1,54059 A), intervalo de barrido: 2,14-37,02° 20, tamaño de paso: 0,04° 20, tiempo de adquisición: 900 s.
.

a. Los tiempos informados son aproximados.
Ejemplo 5 - Preparación de la forma cristalina A

Se usan reactivos disponibles comercialmente tal como se reciben a menos que se indique lo contrario. Las reacciones que requieren una atmósfera inerte se ejecutan bajo nitrógeno a menos que se indique lo contrario.
Etapas 1 y 2:


Se disuelve 2-naftilacetonitrilo (4500 g) en THF (321), se añaden 3,2 kg de (S)-(+)-epiclorhidrina y se enfría la disolución hasta -16°C. Luego se añade una disolución hexametildisililazano de sodio 2,0 M en tetrahidrofurano (THF) (24,7 kg), manteniendo la temperatura interna por debajo de -10°C. Esta adición requiere 2 h 45 min hasta finalización. Luego se agita la mezcla de reacción unas seis horas adicionales a aproximadamente -15°C después de que se analiza una muestra mediante HPLC. Mientras se mantiene la temperatura interna a menos de 0°C, se añade dimetilsulfuro de borano (6,5 kg) durante 36 min. Después de la finalización de la adición de borano, se calienta la mezcla de reacción lentamente hasta 60°C para reducir el nitrilo a la amina. Durante este calentamiento, se observa que se inicia una exoterma a 45°C. Después de calentar a 60°C durante dos horas, se analiza una muestra de la mezcla de reacción mediante HPLC. Se enfría la mezcla de reacción a 24°C y se transfiere a una disolución de HCl 2 M durante 1 h. Se calienta la mezcla bifásica hasta 50°C y se agita durante 1 h a esta temperatura, seguida de un enfriamiento hasta 29°C. Se mide el pH de la mezcla de reacción extinguida y se comprueba que es 5. Se añade HCl 2 M adicional, se calienta la mezcla hasta 50°C y se agita durante una hora, enfriándose después a 25°C. Se mide el pH y se comprueba que es 1. La reacción continúa con la adición de acetato de isopropilo (IPAc), la agitación, la separación de fases y el descarte de la fase orgánica. Se añade amoníaco acuoso a la fase acuosa y se mide el pH, que muestra un pH de 8. Se añade amoníaco adicional y se vuelve a medir el pH, que es de 8,5. A continuación se continúa con la extracción con dos extracciones de la fase acuosa con IPAc. Luego se lavan los extractos orgánicos combinados con fosfato de sodio dibásico al 5% en agua, seguido de un lavado con salmuera. Se concentra parcialmente la fase orgánica resultante para el secado azeotrópico, seguida de una dilución con IPAc. Luego se añaden porciones de ácido p-toluenosulfónico hidratado (4,9 kg) para precipitar el producto deseado como su sal pTsOH, que se aísla mediante filtración. Se lava la torta de filtración con IPAc y se seca hasta peso constante para dar 5785 g del producto deseado como sólido blanco. Rendimiento: 54%. HPLC: 98,2%.
Etapas 3 y 4:

Etapa 3:
Se suspende la sal de amina-pTsOH (5785 g) obtenida de la etapa 2 en IPAc (176 l) para dar una suspensión. Luego se añade cloruro de tionilo (2,1 kg) durante una hora. Tras la finalización de la adición de cloruro de tionilo, se agita la mezcla de reacción una hora adicional y se analiza una muestra mediante HPLC. Se añade hidróxido de sodio acuoso (5 M, 6 equivalentes en mol) durante una hora seguido de cuatro horas de agitación adicional. Se dejan sedimentar las fases y se comprueba que el pH de la fase acuosa es de 9. Se separan las fases y se lava la fase orgánica con NaOH 1 M en agua. Se combinan las fases acuosas y se retroextraen con IPAc y se combinan la fase orgánica inicial y el retroextracto. Se lavan estas fases orgánicas combinadas con HCl 0,5 M para extraer (1R,5S)-1-(naftalen-2-il)-3-azabiciclo[3.1.0]hexano en la fase acuosa. Se lava la fase acuosa ácida con una mezcla 1:1 de IPAc y THF para eliminar color. Se basifica la fase acuosa con amoniaco acuoso seguido de extracción con IPAc. Después de la separación de fases, se lava la fase orgánica con salmuera, se seca sobre sulfato de magnesio y se concentra parcialmente. Después de la concentración, se añade cloruro de hidrógeno en alcohol isopropílico (IPA) (1,0 equivalente en mol de HCl, 0,90 l) para formar la sal en bruto, que se aísla mediante filtración, se lava con IPAc y luego se seca parcialmente. Se somete a reflujo la torta húmeda en IPAc. Se somete a reflujo la sal en bruto en IPA y
se aíslan los sólidos mediante filtración, se lavan con IPA y luego se secan. > 99,5 porcentaje de área de HPLC y 97,7% de pureza en porcentaje de área quiral. 1759 g del producto deseado.
Etapa 4:
Se disuelve el clorhidrato de (1R,5S)-1-(naftalen-2-il)-3-azabiciclo[3.1.0]hexano en bruto (1753 g) obtenido de la etapa 3 en 20 volúmenes de etanol caliente (70°C) y luego se filtra mediante un filtro en línea como filtración de pulido. Luego se enjuagan el recipiente de disolución y el filtro en línea y la línea de transferencia con etanol caliente adicional (61°C) y se combina el enjuague con el filtrado. Se concentran parcialmente el filtrado y los lavados combinados a vacío hasta aproximadamente 11,5 volúmenes en total (en relación con la entrada de clorhidrato de (1R,5S)-1-(naftalen-2-il)-3-azabiciclo[3.1.0]hexano en bruto) y luego se recalientan para volver a disolver los sólidos. Se enfría la disolución hasta 65°C y se añaden cristales de semilla como suspensión en etanol. Después de agitar a aproximadamente 65°C para desarrollar el lecho de semilla, se enfría la suspensión hasta temperatura ambiente. Se aíslan los sólidos resultantes mediante filtración, se lava la torta de filtro con etanol y se secan los sólidos lavados. Se obtiene un total de 1064 g de producto marrón. >99,5% tanto para HPLC quiral como aquiral.
Etapa 5:
Se disuelve el clorhidrato de (1R,5S)-1-(naftalen-2-il)-3-azabiciclo[3.1.0]hexano (1064 g) obtenido de la etapa 4 en 10,7 l de agua mientras se calienta hasta 35°C. Una vez se disuelven todos los sólidos, se lava la disolución acuosa con THF:IPAc 1:1 para eliminar la mayor parte del color. Después del lavado, se añade amoniaco acuoso a la fase acuosa y se extrae (1R,5S)-1-(naftalen-2-il)-3-azabiciclo[3.1.0]hexano en IPAc. Se seca la fase orgánica sobre sulfato de magnesio y luego se concentra a vacío para dar un sólido blanquecino. Se disuelve el sólido en IPA y se transfiere a un matraz de fondo redondo de 3 bocas de 22 l mediante filtración en línea. Luego se añade cloruro de hidrógeno filtrado en IPA para formar de nuevo la sal, que se aísla mediante filtración. Se lava la torta de filtro con IPA y luego se seca para dar 926 g de clorhidrato de (1R,5S)-1-(naftalen-2-il)-3-azabiciclo[3.1.0]hexano como un sólido ligeramente blanquecino.
Se muestra una XRPD del producto en la figura 35 y es coherente con la forma cristalina A. Se recoge el patrón de XRPD con un difractómetro PANalytical X'Pert PRO MPD usando un haz incidente de radiación Cu producido usando una fuente de enfoque fino largo Optix. Se usa un espejo multicapa elípticamente graduado para enfocar los rayos X de Cu Ka a través de la probeta y sobre el detector. Antes del análisis, se analiza una probeta de silicio (NIST SRM 640d) para verificar si la posición observada del pico de Si 111 es coherente con la posición certificada por NIST. Se intercala una probeta de la muestra entre películas de 3 |im de grosor y se analiza en geometría de transmisión. Se usan una trampa de haz, una extensión corta antidispersión y un filo de cuchilla antidispersión para minimizar el fondo generado por el aire. Se usan ranuras de Soller para los haces incidentes y difractados para minimizar el ensanchamiento a partir de la divergencia axial. El patrón de difracción se recoge usando un detector sensible a la posición de barrido (X'Celerator) ubicado 240 mm de la probeta y software Data Collector v. 2.2b. Los parámetros de adquisición de datos son: Panalytical X-Pert Pro MPD PW3040 Pro, tubo de rayos X: Cu (1,54059 A), voltaje: 45 kV, amperaje: 40 mA, intervalo de barrido: 1,00-39,99° 20, tamaño de paso: 0,017° 20, tiempo de recogida: 717 s, velocidad de barrido: 3,3°/min, ranura: DS: 1/2°, SS: nulo, tiempo de revolución: 1,0 s, modo: transmisión.
La figura 36 superpone los patrones de XRPD de la figura 1 y la figura 35. Hay algunas diferencias en intensidades de pico relativas que se deben probablemente a la orientación preferida (PO). PO es la tendencia de los cristales, habitualmente placas o agujas, de empaquetarse entre sí con cierto grado de orden. PO puede afectar a las intensidades de pico, pero no a las posiciones de pico, en patrones de XRPD.
Se muestra una XRPD del producto después del almacenamiento a largo plazo en la figura 37 y es coherente con la forma cristalina A. Se recoge el patrón de XRPD con un difractómetro PANalytical X'Pert PRO MPD usando un haz incidente de radiación Cu producido usando una fuente de enfoque fino largo Optix. Se usa un espejo multicapa elípticamente graduado para enfocar los rayos X de Cu Ka a través de la probeta y sobre el detector. Antes del análisis, se analiza una probeta de silicio (NIST SRM 640e) para verificar si la posición observada del pico de Si 111 es coherente con la posición certificada por NIST. Se intercala una probeta de la muestra entre películas de 3 |im de grosor y se analiza en geometría de transmisión. Se usan una trampa de haz, una extensión antidispersión corta y u filo de cuchilla antidispersión para minimizar el fondo generado por el aire. Se usan ranuras de Soller para los haces incidentes y difractados para minimizar el ensanchamiento a partir de la divergencia axial. El patrón de difracción se recoge usando un detector sensible a la posición de barrido (X'Celerator) ubicado 240 mm de la probeta y software Data Collector v. 2.2b. Los parámetros de adquisición de datos son: Panalytical X-Pert Pro MPD PW3040 Pro, tubo de rayos X: Cu (1,54059 A), voltaje: 45 kV, amperaje: 40 mA, intervalo de barrido: 1,00-39,99° 20, tamaño de paso: 0,017°20, tiempo de recogida: 719 s, velocidad de barrido: 3,3°/min, ranura: DS: 1/2°, SS: nulo, tiempo de revolución: 1,0 s, modo: transmisión.
Se analiza un patrón PANalytical para detectar la forma cristalina A y se evalúan los efectos de la orientación preferida y de las estadísticas de las partículas mediante la comparación con patrones de XRPD adicionales analizados usando una geometría alternativa, además de un patrón de XRPD calculado a partir del análisis de monocristal. En la figura
38 se muestra un resultado de indexación para la XRPD mostrada en la figura 37 recogido con radiación Cu Ka. El patrón de XRPD se indexa usando X’Pert High Score Plus 2.2a (2.2.1). Los picos observados se muestran en la figura 39 y se enumeran en la tabla C en la fórmula 1.32 anterior, los picos representativos se enumeran en la tabla B en la fórmula 1.25 anterior, y los picos característicos se enumeran en la tabla A en la fórmula 1.16 anterior.
Ejemplo 6 - Preparación de cristales de la forma B
Ejemplo 6a
Se suspenden 558,9 mg de la forma cristalina A del ejemplo 5 anterior en 5 ml de diclorometano. Se agita la preparación (300 rpm) en un vial sellado a temperatura ambiental durante 16 días. Se aíslan sólidos blancos mediante filtración a vacío, se enjugan con 1 ml de diclorometano y se secan brevemente bajo nitrógeno. El producto es la forma cristalina A. Un patrón de XRPD del producto está en la figura 47. Se recoge el patrón de XRPD con un difractómetro PANalytical X ’Pert PRO MPD usando un haz incidente de radiación Cu producido usando una fuente de enfoque fino largo Optix. Se usa un espejo multicapa elípticamente graduado para enfocar los rayos X de Cu Ka a través de la probeta y sobre el detector. Antes del análisis, se analiza una probeta de silicio (NIST SRM 640e) para verificar si la posición observada del pico de Si 111 es coherente con la posición certificada por NIST. Se intercala una probeta de la muestra entre películas de 3 ^m de grosor y se analiza en geometría de transmisión. Se usan una trampa de haz, una extensión antidispersión corta y un filo de cuchilla antidispersión para minimizar el fondo generado por el aire. Se usan ranuras de Soller para los haces incidentes y difractados para minimizar el ensanchamiento a partir de la divergencia axial. Se recogen patrones de difracción usando un detector sensible a la posición de barrido (X’Celerator) ubicado 240 mm de la probeta y software Data Collector v. 2.2b. Los parámetros de adquisición de datos son: Panalytical X-Pert Pro m Pd PW3040 Pro, tubo de rayos X: Cu (1,54059 Á), voltaje: 45 kV, amperaje: 40 mA, intervalo de barrido: 1,00-39,99° 20, tamaño de paso: 0,017° 20, tiempo de recogida: 720 s, velocidad de barrido: 3,2°/min, ranura: DS: 1/2°, SS: nulo, tiempo de revolución: 1,0 s, modo: transmisión.
Ejemplo 6b
Se ponen en contacto 34,3 mg de la forma cristalina A del ejemplo 6a con 1 ml de agua. Se somete a sonicación la muestra hasta que se disuelven los sólidos. Se tapa la muestra y se deja a temperatura ambiental hasta que se observa nucleación, en el plazo de un día. Se aíslan partes individuales de la muestra a granel para análisis.
Recogida de datos: se monta una placa incolora de C i5Hi6ClN [Ci5Hi 6N, C1], que tiene unas dimensiones aproximadas de 0,31 x 0,21 x 0,09 mm, sobre un bucle de nailon en orientación aleatoria. Se realizan examen preliminar y recogida de datos con radiación Cu Ka (^ = 1,54178 Á) en un difractómetro Rigaku Rapid II equipado con óptica confocal. Se realizan refinamientos usando SHELX2014 (Sheldrick, G. M. Acta Cryst. 2015, C 71,3-8). Se obtienen las constantes de celdas y una matriz de orientación para recogida de datos a partir del refinamiento de mínimos cuadrados usando los ángulos de ajuste de 22958 reflexiones en el intervalo 2° < 0 < 26°. A partir de la presencia sistemática de las siguientes condiciones: h00 h = 2n; 0k0 k = 2n; 00l l = 2n y del posterior refinamiento por mínimos cuadrados, se determina que el grupo espacial es P212121 (n.° 19). Se recogen los datos hasta un ángulo de difracción máximo (20) de 144,79°, a una temperatura de 100 K.
Reducción de datos: se integran las tramas con HKL3000 (Otwinowski, Z.; Minor, W. Methods Enzymol. 1997, 276, 307). Se recoge un total de 22958 reflexiones, de las que 2415 son únicas. Se aplican a los datos correcciones de Lorentz y polarización. El coeficiente de absorción lineal es de 2,422 mm-1 para radiación Cu Ka. Se aplica una corrección de absorción empírica usando SCALEPACK (Otwinowski, Z.; Minor, W. Methods Enzymol. 1997, 276, 307). Los coeficientes de transmisión oscilan desde 0,753 hasta 0,976. Se aplica una corrección de extinción secundaria (Sheldrick, G. M. Acta Cryst. 2015, C71, 3-8). El coeficiente final, refinado en mínimos cuadrados, es 0,0055(8) (en unidades absolutas). Se promedian las intensidades de reflexiones equivalentes. El factor de coincidencia para el promedio es del 4,95% basándose en la intensidad.
Resolución de la estructura y refinamiento: la estructura se resuelve mediante métodos directos usando SHELXS-97 (Sheldrick, G. M. Acta Cryst. 2015, C71, 3-8). Los átomos restantes se ubican en las posteriores síntesis de Fourier de diferencia. Se incluyen átomos de hidrógeno en el refinamiento pero se limitan a montarse en el átomo al que se unen. La estructura se refina en mínimos cuadrados de matriz completa minimizando la función:

El peso w se define como 1/[ct2(F02) (0,0437P)2 (2.1802P)], donde P = (P02 2Fc2)/3. Se toman factores de dispersión de las “Tablas internacionales de cristalografía” (International Tables for Crystallography, vol. C, Kluwer Academic Publishers: Dordrecht, Países Bajos, 1992, tablas 4.2.6.8 y 6.1.1.4). De las 2415 reflexiones usadas en los refinamientos, sólo las reflexiones con Fo2 > 2u(Fo2) se usan para calcular el ajuste residual, R. Se usa un total de 2372 reflexiones en el cálculo. El ciclo final de refinamiento incluye 155 parámetros variables y converge con factores de coincidencias no ponderados y ponderados:

La desviación estándar de una observación de peso unitario (bondad del ajuste) es de 1,150. El pico más alto en la Fourier de diferencia final tiene una altura de 0,318 e/Á3. El pico mínimo negativo tiene una altura de -0,313 e/Á3. Patrón de difracción de rayos X de polvo (XRPD) calculado: se genera un patrón de XRPD calculado para la radiación Cu usando Mercury (Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Tailor, R.; Towler, M.; y van de Streek, J., J. Appl. Cryst., 2006, 39, 453-457) y las coordenadas atómicas, el grupo espacial y los parámetros de celda unitaria a partir de la estructura del monocristal. Debido a que los datos del monocristal se recogen a bajas temperaturas (100 K), los desplazamientos de picos pueden ser evidentes entre el patrón calculado a partir de los datos de baja temperatura y el patrón de difracción de polvo experimental a temperatura ambiente, particularmente en los ángulos de difracción altos. El patrón de XRPD calculado se ajusta a temperatura ambiente usando los parámetros de celda unitaria obtenidos previamente a partir de la indexación de XRPD.
Diagramas de elipsoide de desplazamiento atómico y de empaquetamiento: el diagrama de elipsoide de desplazamiento atómico se prepara usando Mercury (Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; y van de Streek, J. Appl. Cryst., 2006, 39, 453-457). Los átomos se representan mediante elipsoides térmicos anisotrópicos al 50% de probabilidad. Los diagramas de empaquetamiento y las figuras adicionales se preparan usando Mercury. El enlace de hidrógeno se representa con líneas discontinuas. La evaluación de los centros quirales se realiza con PLATON (Spek, A.L. PLATON. Molecular Graphics Program. Universidad de Utrecht, Utrecht, Países Bajos, 2008. Spek, A.L., J. Appl. Cryst. 2003, 36, 7). La configuración absoluta se evalúa usando la especificación de reglas de quiralidad molecular (Cahn, R.S.; Ingold, C; Prelog, V. Angew. Chem. Intern. Ed. Eng., 1966, 5, 385 y Prelog, V., Helmchen, G. Angew. Chem. Intern. Ed. Eng., 1982, 21,567).
Resultados: los parámetros de la celda ortorrómbica y el volumen calculado son: a = 5,9055(2) Á, b = 7,4645(3) Á, c = 29,1139(13) Á (a =p = y = 90°), V= 1283,39(9) Á3. El peso de fórmula de la unidad asimétrica en la forma cristalina B es de 245,74 g mol-1 con Z = 4, lo que da como resultado una densidad calculada de 1,272 g cm-3. Se ha determinado que el grupo espacial es P2\2{2\ (n.° 19). En la tabla 15 se ofrece un resumen de los datos del cristal y de los parámetros de recogida de datos cristalográficos. Los parámetros de grupo espacial y de celda unitaria son coherentes con los obtenidos para la forma B por indexación de XRPD.
El valor R es de 0,0453 (4,53%).
En la figura 24 se muestra un dibujo de elipsoide de desplazamiento atómico de la forma cristalina B.
La unidad asimétrica mostrada en la figura 24 contiene una molécula protonada de (1R,5S)-1-(naftalen-2-il)-3-azabiciclo[3.1.0]hexano y un contraión cloruro.
Los diagramas de empaquetamiento vistos a lo largo de los ejes cristalográficos a, b y c se muestran en las figuras 25-27, respectivamente. El enlace de hidrógeno se produce desde la amina hasta el cloruro, formando cadenas helicoidales unidimensionales unidas con enlaces de hidrógeno a lo largo del eje a, que se muestra en la figura 28. La conformación molecular de las moléculas de (1R,5S)-1-(naftalen-2-il)-3-azabiciclo[3.1.0]hexano en la estructura de la forma cristalina B se compara con la conformación molecular observada en la estructura de la forma cristalina A en la figura 29, y el empaquetamiento de las dos formas visto a lo largo del eje a se compara en la figura 30. El enlace de hidrógeno en las estructuras de las formas cristalinas A y B se muestra en la figura 31. Las moléculas adyacentes están unidas a través de los iones cloruro en el enlace de hidrógeno de la forma cristalina A, formando cadenas rectas a lo largo del eje a. Los grupos amino de las moléculas adyacentes están demasiado separados en el empaquetamiento de la forma cristalina B para estar unidos de manera similar, y en su lugar el enlace de hidrógeno en la forma cristalina B forma una cadena helicoidal.
La estructura absoluta puede determinarse mediante un análisis de la dispersión anómala de rayos X por el cristal. Un parámetro refinado x, conocido como el parámetro de Flack (Flack, H.D.; Bernardinelli, G., Acta Cryst. 1999, A55, 908; Flack, H.D., Bernardinelli, G., J. Appl. Cryst. 2000, 33, 1143, Flack, H.D., Acta Cryst. 1983, A39, 876; Parsons, S.; Flack, H.D.; Wagner, T., Acta Cryst. 2013, B69, 249-259), codifica la abundancia relativa de los dos componentes en un gemelo de inversión. La estructura contiene una fracción 1-x del modelo que se está refinando, y x de su inverso. Siempre que se obtenga una incertidumbre estándar baja, el parámetro de Flack debería estar cerca de 0 si la estructura resuelta es correcta, y cerca de 1 si el modelo inverso es correcto. El parámetro de Flack medido para la estructura de la forma cristalina B mostrada en la figura 24 es de 0,010 con una incertidumbre estándar de 0,010, lo que indica un fuerte poder de distinción de la inversión. El compuesto es enantiopuro y la configuración absoluta puede
asignarse directamente a partir de la estructura cristalina.
El refinamiento del parámetro de Flack (x) no da lugar a una declaración cuantitativa sobre la asignación de la estructura absoluta. Sin embargo, un enfoque que aplica la estadística bayesiana a las diferencias de Bijvoet puede proporcionar una serie de probabilidades para diferentes hipótesis de la estructura absoluta (Hooft, R.W.W.; Straver, L.H.; y Spek, A.L., J. Appl. Cryst., 2008, 41, 96-103 y Bijvoet, J.M.; Peerdeman, A.F.; van Bommel, A.J., Nature, 1951, 168, 271). Este análisis proporciona un parámetro equivalente a Flack (Hooft) además de las probabilidades de que la estructura absoluta sea correcta, incorrecta o un gemelo racémico. Para el conjunto de datos actual, se determina que el parámetro equivalente a Flack (Hooft) es de -0,001(7), la probabilidad de que la estructura sea correcta es de 1,000, la probabilidad de que la estructura sea incorrecta es de 0,000 y la probabilidad de que el material sea un gemelo racémico es de 0,000.
Esta estructura contiene dos centros quirales situados en C2 y C3 (véase la figura 24), que se unen en la configuración S y R, respectivamente.
La figura 32 muestra un patrón de XRPD calculado de la forma cristalina B, generado a partir de la estructura monocristalina.
En la figura 33 se muestra un patrón de XRPD experimental de la forma cristalina B (el mismo que el patrón de XRPD de la figura 40, ejemplo 8), superpuesto al patrón calculado y a un patrón calculado que se ha ajustado a temperatura ambiente. Todos los picos de los patrones experimentales están representados en el patrón de XRPD calculado, lo que indica una única fase.
Las diferencias de intensidad entre los patrones de difracción de polvo calculados y experimentales se deben a menudo a la orientación preferida. La orientación preferida es la tendencia de los cristales a alinearse con cierto grado de orden. Esta orientación preferida de la muestra puede afectar significativamente a las intensidades de los picos, pero no a las posiciones de los picos, en el patrón de difracción de polvos experimental. Además, es de esperar que se produzca algún cambio en la posición de los picos entre los patrones de difracción de polvo calculados y experimentales, ya que el patrón de polvo experimental se recoge a temperatura ambiental y los datos del monocristal se recogen a 100 K. Las bajas temperaturas se usan en el análisis del monocristal para mejorar la calidad de la estructura, pero pueden contraer el cristal, lo que da como resultado un cambio en los parámetros de celda unitaria, que se refleja en el patrón de difracción de polvo calculado. Estos cambios son especialmente evidentes en los ángulos de difracción elevados. El patrón de XRPD calculado se ha ajustado a la temperatura ambiente usando la celda unitaria obtenida previamente a partir de la indexación de XRPD.
Tabla 15. Parámetros de recogida de datos y datos del cristal para clorhidrato de (1R,5S)-1-(naftalen-2-il)-3-azabiciclo[3.1.0]hexano Forma B (forma cristalina B)
Fórmula empírica C15H16ClN
Peso de fórmula 245,74
Temperatura 100(2) K
Longitud de onda 1,54178 A
Sistema cristalino Ortorrómbico
Grupo espacial P212121
Dimensiones de la celda unitaria a = 5,9055(2) A
b = 7,4645(3) A P= 90°.
c = 29,1139(13) A
Volumen 1283,39(9) A3
Z 4
Densidad (calculada) 1,272 Mg/m3
Coeficiente de absorción 2,422 mm-1
F(000) 520
Tamaño del cristal 0,310 x 0,210 x 0,090 mm3
Intervalo theta para recogida de datos De 6,080 a 72,393°.
Intervalos de índice -7<=h<=7, -8<=k<=8, -35<=l<=35
Reflexiones recogidas 22958
Reflexiones independientes 2415 [R(int) = 0,0495]
Completitud hasta theta = 67,679° 98,5%
Corrección de la absorción Semiempíricas a partir de equivalentes
Transmisión máx. y mín. 0,976 y 0,753
Método de refinamiento Mínimos cuadrados de matriz completa en F2 Datos/restricciones/parámetros 2415/0/155
Bondad del ajuste en F2 1,150
Índices R finales [I>2sigma(I)] R1 = 0,0453, wR2 = 0,1224
Índices R (todos los datos) R1 = 0,0464, wR2 = 0,1240
Parámetro de estructura absoluta Parámetro de Flack: 0,010(10)
Parámetro de Hooft: -0,001(7)
Coeficiente de extinción 0,0055(8)
Mayor dif. de pico y agujero 0,318 y -0,313 e.A-3
Ejemplo 7 - Preparación de la forma cristalina B
Se mezclan 470,9 mg de la forma cristalina A del ejemplo 5 anterior con 5 ml de agua en un vial de vidrio de 20 ml. Se agita la suspensión a temperatura ambiental durante 16 días con una barra de agitación para permitir que se produzca la conversión. Se recogen los sólidos mediante filtración a vacío y se secan brevemente bajo nitrógeno.
Ejemplo 8 -Preparación de la forma cristalina B
Se agita 1 g del producto del ejemplo 16 a continuación en 5 ml de etanol 200 especial para uso industrial (desnaturalizado) a lo largo del fin de semana a temperatura ambiental. Se filtra la mezcla y se aclara con 2 ml de etanol 200 especial para uso industrial (desnaturalizado) y seguido de acetato de isopropilo (2 * 3 ml). Extraer en seco los sólidos durante 2 h y luego secar a 40°C durante 6 h para dar 0,81 g de producto.
Una XRPD muestra que el producto es la forma cristalina B (figura 40 y también se muestra como el patrón de XRPD superior en la figura 33). Se recoge el patrón de XRPD con un difractómetro PANalytical X'Pert PRO MPD usando un haz incidente de radiación Cu producido usando una fuente de enfoque fino largo Optix. Se usa un espejo multicapa elípticamente graduado para enfocar los rayos X de Cu Ka a través de la probeta y sobre el detector. Antes del análisis, se analiza una probeta de silicio (NIST SRM 640d) para verificar si la posición observada del pico de Si 111 es coherente con la posición certificada por NIST. Se intercala una probeta de la muestra entre películas de 3 |im de grosor y se analiza en geometría de transmisión. Se usan una trampa de haz, una extensión corta antidispersión y un filo de cuchilla antidispersión para minimizar el fondo generado por el aire. Se usan ranuras de Soller para los haces incidentes y difractados para minimizar el ensanchamiento a partir de la divergencia axial. El patrón de difracción se recoge usando un detector sensible a la posición de barrido (X'Celerator) ubicado 240 mm de la probeta y software Data Collector v. 2.2b. Los parámetros de adquisición de datos son: Panalytical X-Pert Pro MPD PW3040 Pro, tubo de rayos X: Cu (1,54059 A), voltaje: 45 kV, amperaje: 40 mA, intervalo de barrido: 1,01-39,98° 20, tamaño de paso: 0,017° 20, tiempo de recogida: 720 s, velocidad de barrido: 3,2°/min, ranura: DS: 1/2°, SS: nulo, tiempo de revolución: 1,0 s, modo: transmisión.
Se analiza un patrón PANalytical para este material, y se evalúan los efectos de la orientación preferida y de las estadísticas de las partículas mediante la comparación con patrones de XRPD adicionales analizados usando una geometría alternativa, además de un patrón de XRPD calculado a partir del análisis del monocristal. En la figura 41 se muestra un resultado de indexación para la XRPD mostrada en la figura 40 recogido con radiación Cu Ka. El patrón de XRPD se indexa usando X'Pert High Score Plus 2.2a (2.2.1). Los picos observados se muestran en la figura 42 y se enumeran en la tabla F en la fórmula 1.109, los picos representativos se enumeran en la tabla E en la fórmula 1.102, y los picos característicos se enumeran en la tabla D en la fórmula 1.93.
Ejemplo 9 - Forma cristalina C
Se genera una disolución turbia que contiene 458,2 mg de la forma cristalina A del ejemplo 5 y 40 ml de IPA a temperatura elevada. Se filtra la disolución caliente con un filtro de nailon de 0,2 |im en un vial limpio y se coloca en un congelador. Después de dos días, se recuperan los sólidos por filtración a vacío y se secan brevemente bajo nitrógeno. Los sólidos se identifican como una mezcla de las formas cristalinas A y C. Se genera una suspensión con 42,2 mg de la mezcla y 0,8 ml de una disolución saturada de DCM. (La disolución saturada se genera con 65,4 mg de la forma cristalina A del ejemplo 5 en 5 ml de DCM a temperatura ambiente. El exceso de sólidos se filtra de la disolución al día siguiente con un filtro de nailon de 0,2 |im). Se agita la suspensión a 100 rpm con una bola de ágata a 2°C durante 3 semanas para permitir que se produzca la conversión. Se almacenan los sólidos aislados de la suspensión resultante mediante filtración a vacío a una temperatura entre - 25 y - 10°C.
Se muestra una XRPD del producto en la figura 43. Se recoge el patrón de XRPD con un difractómetro PANalytical
X'Pert PRO MPD usando un haz incidente de radiación Cu producido usando una fuente de enfoque fino largo Optix. Se usa un espejo multicapa elípticamente graduado para enfocar los rayos X de Cu Ka a través de la probeta y sobre el detector. Antes del análisis, se analiza una probeta de silicio (NIST SRM 640d) para verificar si la posición observada del pico de Si 111 es coherente con la posición certificada por NIST. Se intercala una probeta de la muestra entre películas de 3 |im de grosor y se analiza en geometría de transmisión. Se usan una trampa de haz, una extensión corta antidispersión y un filo de cuchilla antidispersión para minimizar el fondo generado por el aire. Se usan ranuras de Soller para los haces incidentes y difractados para minimizar el ensanchamiento a partir de la divergencia axial. El patrón de difracción se recoge usando un detector sensible a la posición de barrido (X'Celerator) ubicado 240 mm de la probeta y software Data Collector v. 2.2b. Los parámetros de adquisición de datos son: Panalytical X-Pert Pro MPD PW3040 Pro, tubo de rayos X: Cu (1,54059 A), voltaje: 45 kV, amperaje: 40 mA, intervalo de barrido: 1,00-39,99° 20, tamaño de paso: 0,017° 20, tiempo de recogida: 720 s, velocidad de barrido: 3,2°/min, ranura: DS: 1/2°, SS: nulo, tiempo de revolución: 1,0 s, modo: transmisión.
Se analiza un patrón PANalytical para detectar este material, y se evalúan los efectos de la orientación preferida y de las estadísticas de las partículas mediante la comparación con otros patrones de XRPD analizados usando una geometría alternativa. En la figura 44 se muestra un resultado de indexación para el patrón de XRPD mostrado en la figura 43, recogido con radiación Cu Ka. El patrón de XRPD se indexa usando un software propio (patente estadounidense n.° 8.576.985). Los picos observados se muestran en la figura 45 y se enumeran en la tabla I en la fórmula 1.183, los picos representativos se enumeran en la tabla H en la fórmula 1.176, y los picos característicos se enumeran en la tabla G en la fórmula 1.168.
Ejemplo 10 - Experimentos con suspensiones de interconversión
Un diagrama de energía-temperatura es una disolución gráfica semicuantitativa de la ecuación de Gibbs-Helmholtz, en la que se representan las isobaras de entalpía (H) y energía libre (G) de cada forma en función de la temperatura. El gráfico supone que las isobaras de energía libre se cruzan como máximo una vez y, en segundo lugar, que las isobaras de entalpía de los polimorfos no se cruzan. El punto de fusión de un polimorfo se define como la temperatura a la que la isobara de energía libre del polimorfo se cruza con la isobara de energía libre del líquido. La temperatura de transición se define como la temperatura a la que la isobara de energía libre de un polimorfo se cruza con la isobara de energía libre del segundo. Por tanto, a Tt ambos polimorfos tienen igual energía libre y, por tanto, están en equilibrio entre sí.
En la figura 46 se muestra el diagrama de energía-temperatura propuesto para las formas cristalinas A, B y C. En el diagrama se representan las isobaras de entalpía (H) y energía libre (G) para cada forma en función de la temperatura (T). Los subíndices A, B, C y L se refieren a las formas cristalinas A, B, C y a la fase líquida, respectivamente. Los subíndices f, t y m se refieren a fusión, punto de transición y punto de fusión, respectivamente. El gráfico supone que las isobaras de energía libre se cruzan como máximo una vez y, en segundo lugar, que las isobaras de entalpía de los polimorfos no se cruzan. El punto de fusión de un polimorfo se define como la temperatura a la que la isobara de energía libre del polimorfo se cruza con la isobara de energía libre del líquido. La temperatura de transición se define como la temperatura a la que la isobara de energía libre de un polimorfo se cruza con la isobara de energía libre del segundo. Por tanto, a Tt ambas formas polimórficas tienen igual energía libre y, por tanto, están en equilibrio entre sí. La forma cristalina C es la fase sólida estable por debajo de Tí,c^ b (porque la energía libre de la forma cristalina C es menor que la de la forma cristalina B), la forma cristalina B es la fase sólida estable entre Tí,c^ b y Tí,b^ a, y la forma cristalina A es la fase sólida estable por encima de Tí,b^ a. El polimorfo de baja energía tendrá una fugacidad, una presión de vapor, una actividad termodinámica, una solubilidad, una velocidad de disolución por unidad de superficie y una velocidad de reacción menores en relación con los otros polimorfos.
Se realizan experimentos de interconversión para someter a prueba la relación termodinámica hipotética entre materiales ilustrada por el diagrama de energía-temperatura anterior. Los experimentos de interconversión o con suspensiones competitivos son un procedimiento mediado por la disolución que proporciona una ruta para que el cristal menos soluble (más estable) crezca a expensas de la forma cristalina más soluble (Bernstein, J. Polymorphism in Molecular Crystals. Clarendon Press, Oxford, 2006; Brittain, H.G., Polymorphism in Pharmaceutical Solids. Marcel Dekker, Inc., Nueva York, 1999). Fuera de la formación de un solvato o una degradación, el polimorfo resultante más estable de un experimento de interconversión es independiente del disolvente usado porque el polimorfo más estable termodinámicamente tiene una energía más baja y, por tanto, una solubilidad menor. La elección del disolvente afecta a la cinética de la conversión del polimorfo y no a la relación termodinámica entre las formas polimórficas (Gu, C.H., Young, V. Jr., Grant, D.J., J. Pharm. Sci. 2001, 90 (11), 1878-1890).
Los experimentos con suspensiones de interconversión binarias entre las formas cristalinas A, B y C en diferentes sistemas de disolventes a temperaturas que abarcan aproximadamente de 2 a 67°C se resumen en la tabla 16 a continuación. Se generan disoluciones saturadas y luego se añaden a mezclas que se componen de cantidades aproximadamente equivalentes de dos de los polimorfos. Las muestras se suspenden desde durante la noche hasta tres semanas y se recogen los sólidos y se analizan mediante XRPD. Los resultados de los estudios de interconversión indican que la estabilidad termodinámica relativa de los enantiótropos, es decir, las formas cristalinas A, B y C, está correctamente representada por el diagrama de energía-temperatura propuesto. Además, se espera que la Tí,c^ b esté
por debajo de 2°C (no se ha determinado), la Tt,c^A estará entre 2°C y la temperatura ambiental, y la Tt,B^A estará entre 37 y 54°C.
Tabla 16. Suspensiones de interconversión binarias entre formas cristalinas A, B y C


1 La duración y las temperaturas son aproximadas.
2 La flecha hacia abajo indica que las intensidades de los picos de la fase cristalina asociada han disminuido en relación con las de la mezcla inicial. La duración del experimento no es suficiente para alcanzar el equilibrio; sin embargo, pueden sacarse conclusiones de la forma predominante basándose en la mezcla resultante.
3 El procedimiento de interconversión mediado por la disolución proporciona una ruta para que el cristal menos soluble (más estable en relación con el otro) crezca a expensas de la forma cristalina más soluble. Sin embargo, cuando ninguna de las formas implicadas en la mezcla competitiva binaria es la forma más estable termodinámicamente, también puede darse la posibilidad de que el cristal más estable crezca a expensas de las otras dos formas cristalinas más solubles. Esta transformación polimórfica mediada por el disolvente está controlada por su tasa de nucleación, que generalmente es mayor en un disolvente que proporciona mayor solubilidad. Además de la solubilidad, también es importante la fuerza de las interacciones disolvente-soluto. El grado de agitación y la temperatura también modifican la tasa de transformación polimórfica al influir en la cinética de cristalización del polimorfo más estable.
La forma cristalina B presenta una solubilidad aparente más baja que la forma cristalina A tanto en metanol como en agua (tabla 17 a continuación). También se realizan análisis de calorimetría en disolución (SolCal) para determinar los calores de disolución en metanol a 25°C y confirmar la forma estable a esta temperatura (véase el ejemplo 15). Basándose en los datos de SolCal, las disoluciones de ambas formas cristalinas A y B en metanol son acontecimientos endotérmicos con calores de disolución promedia de 48,618 y 64,567 J/g, respectivamente, lo que indica que la forma cristalina B es más estable que la cristalina A a 25°C.
Sección experimental: solubilidad aproximada
Se trata una muestra pesada con alícuotas del disolvente de prueba a temperatura ambiente. Se somete a sonicación la mezcla entre las adiciones para facilitar la disolución. Se determina la disolución completa del material de prueba
por inspección visual. La solubilidad se estima basándose en el total de disolvente usado para proporcionar una disolución completa. La solubilidad real puede ser mayor que el valor calculado debido al uso de alícuotas de disolvente demasiado grandes o a una velocidad de disolución lenta.
Tabla 17. Solubilidad aproximada de las formas cristalinas A y B

1 Nucleación observada después de un día. Se aísla un monocristal de la forma cristalina B.
2 Nucleación de finos irregulares sin birrefringencia observada después de 7 días.
Ejemplo 11 - Condiciones aceleradas de estrés
Se exponen las formas cristalinas A, B y C a condiciones aceleradas de estrés durante dos semanas (tabla 18). Basándose en la XRPD, las formas cristalinas A y B permanecen sin cambios a 30°C/56% de HR o 40°C/75% de HR dentro del periodo de tiempo evaluado. Sin embargo, la forma cristalina C se convierte en una mezcla de las formas cristalinas A y B en dos semanas a 40°C/75% de HR. La forma cristalina C es metaestable en estas condiciones. En el caso de la forma cristalina A, en ausencia de semillas del polimorfo más estable, la barrera de energía libre crítica para la nucleación de la forma cristalina B no se supera en el estado sólido ni en los experimentos de conversión de forma mediada por disolvente dentro del marco temporal evaluado.
Tabla 18. Evaluación de la estabilidad acelerada de la forma cristalina

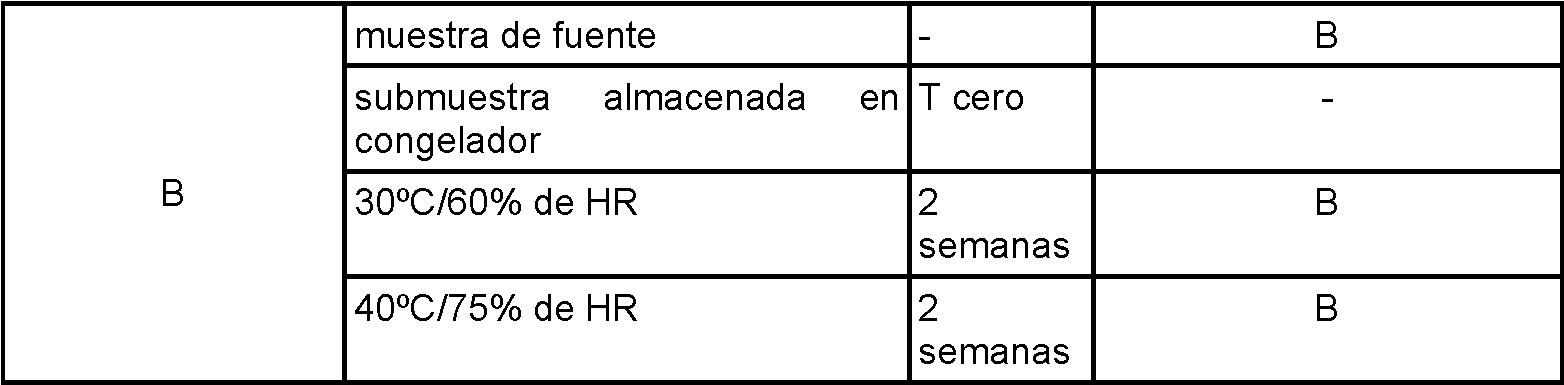

Tt,B^A está entre 37 y 54°C. Una mezcla de las formas A y B (combinación de porciones 1 y 2 del ejemplo 17), se convierte completamente en la forma A tras exposición a 23o°C (tabla 19 a continuación).
Sección experimental: estrés de humedad relativa
Se utilizan los siguientes frascos de humedad relativa (se usan disoluciones salinas saturadas para generar una humedad relativa deseada): el 75% de HR (NaCl) y el 56% de HR (NaBr) (Nyqvist, H., Int. J. Pharm. Tech. & Prod. Mfr. 1983, 4 (2), 47-48).
Tabla 19. Estabilidad física de la mezcla de las formas A y B
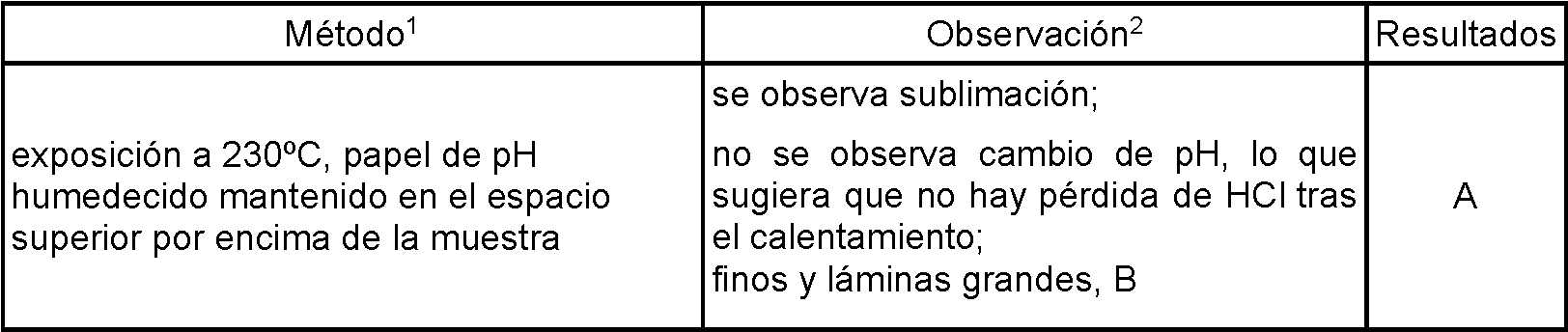
1 El tiempo y la temperatura son aproximados.
2 B = birrefringente cuando se observa mediante microscopía de luz polarizada
3 La flecha hacia arriba indica que las intensidades de los picos de la fase cristalina asociada han aumentado en relación con las de la mezcla inicial.
Ejemplo 12 - Preparación de la forma cristalina B
Se suspende una porción de la forma cristalina A del ejemplo 5 anterior con agua a temperatura ambiental durante 16 días. Se aísla la forma cristalina B. Una XRPD del producto está en la figura 48. Se recoge el patrón de XRPD con un difractómetro PANalytical X'Pert PRO MPD usando un haz incidente de radiación Cu producido usando una fuente de enfoque fino largo Optix. Se usa un espejo multicapa elípticamente graduado para enfocar los rayos X de Cu Ka a través de la probeta y sobre el detector. Antes del análisis, se analiza una probeta de silicio (NIST SRM 640e) para verificar si la posición observada del pico de Si 111 es coherente con la posición certificada por NIST. Se intercala una probeta de la muestra entre películas de 3 |im de grosor y se analiza en geometría de transmisión. Se usan una trampa de haz, una extensión antidispersión corta y un filo de cuchilla antidispersión para minimizar el fondo generado por el aire. Se usan ranuras de Soller para los haces incidentes y difractados para minimizar el ensanchamiento a partir de la divergencia axial. Se recogen patrones de difracción usando un detector sensible a la posición de barrido (X'Celerator) ubicado 240 mm de la probeta y software Data Collector v. 2.2b. Los parámetros de adquisición de datos son: Panalytical X-Pert Pro MPD PW3040 Pro, tubo de rayos X: Cu (1,54059 A), voltaje: 45 kV, amperaje: 40 mA, intervalo de barrido: 1,00-39,99° 20, tamaño de paso: 0,017° 20, tiempo de recogida: 716 s, velocidad de barrido: 3,3°/min, ranura: DS: 1/2°, SS: nulo, tiempo de revolución: 1,0 s, modo: transmisión.
Ejemplo 13 - XRPD de una mezcla de la forma cristalina A y una pequeña cantidad de la cristalina B
Un patrón de XRPD de una mezcla de la forma cristalina A y una pequeña cantidad del producto de la forma cristalina B está en la figura 49 (ejemplo 17 para síntesis). Se recoge el patrón de XRPD con un difractómetro PANalytical X'Pert PRO MPD usando un haz incidente de radiación Cu producido usando una fuente de enfoque fino largo Optix. Se usa un espejo multicapa elípticamente graduado para enfocar los rayos X de Cu Ka a través de la probeta y sobre el detector. Antes del análisis, se analiza una probeta de silicio (NIST SRM 640e) para verificar si la posición observada del pico de Si 111 es coherente con la posición certificada por NIST. Se intercala una probeta de la muestra entre películas de 3 |im de grosor y se analiza en geometría de transmisión. Se usan una trampa de haz, una extensión antidispersión corta y un filo de cuchilla antidispersión para minimizar el fondo generado por el aire. Se usan ranuras de Soller para los haces incidentes y difractados para minimizar el ensanchamiento a partir de la divergencia axial. Se recogen patrones de difracción usando un detector sensible a la posición de barrido (X'Celerator) ubicado 240 mm de la probeta y software Data Collector v. 2.2b. Los parámetros de adquisición de datos son: Panalytical X-Pert Pro MPD PW3040 Pro, tubo de rayos X: Cu (1,54059 A), voltaje: 45 kV, amperaje: 40 mA, intervalo de barrido: 1,00-39,99° 20, tamaño de paso: 0,017° 20, tiempo de recogida: 720 s, velocidad de barrido: 3,2°/min, ranura: DS: 1/2°, SS: nulo, tiempo de revolución: 1,0 s, modo: transmisión.
Ejemplo 14 - Análisis de calorimetría en disolución (SolCal) de las formas cristalinas A y B
Se mide el análisis de calorimetría en disolución para cada forma por triplicado en metanol y se resumen los datos en la tabla 21. Para cada prueba, se obtienen dos calores de disolución, uno calculado usando una calibración que precede al análisis de la muestra y uno calculado usando una calibración tras el análisis de la muestra. Los valores medios de las dos calibraciones también se proporcionan en la tabla. Se observan disoluciones transparentes después de cada prueba.
Las disoluciones de las formas cristalinas tanto A como B en metanol son acontecimientos endotérmicos con calores de disolución promedio de 48,618 y 64,567 J/g, respectivamente. La desviación estándar para cada conjunto es de 0,457 y 0,344 J/g, respectivamente.
La forma cristalina B tiene un mayor valor de calor de disolución que la forma A, lo que indica que la forma cristalina B es más estable que A a 25°C. La entalpía de la transición calculada a partir de los datos de SolCal de la forma B a
la forma A es de aproximadamente 15,9 J/g. La diferencia en el calor de fusión en la transición en estado sólido en la DSC de la forma cristalina B es de 15,9 J/g (véanse las figuras 8 y 55), que coincide bien con los resultados de SolCal. Se realiza calorimetría en disolución usando un calorímetro de disolución de precisión Thermometric 2225, un calorímetro semiadiabático. Se usa el software Solution Calorimeter System v. 1.2. Se pesan muestras en ampollas de aplastamiento de vidrio y se sellan usando tapones de caucho de silicona y cera caliente. Se llevan a cabo los experimentos en 100 ml de metanol a 25°C. La medición de los calores de disolución de las muestras es tanto precedida como seguida de calibraciones usando un calentador interno. Los calores de disolución se calculan usando un modelo dinámico de calibración.
Tabla 21. Calores de disolución de las formas cristalinas A y B en metanol

(a): Calculado usando la calibración antes de romper el vial de muestra.
(b) : Calculado usando la calibración después de romper el vial de muestra.
(c) : Se hacen observaciones en el momento en el que se completan las pruebas.
Ejemplo 15 - Microscopía de platina caliente (HSM) de la forma cristalina A del ejemplo 1
Se realiza microscopía de platina caliente usando una platina caliente Linkam (modelo FTIR 600) montada en un microscopio Leica DM LP. Se observan las muestras con un objetivo de 20x (obj.). Se colocan las muestras en un cubreobjetos, y luego se coloca un segundo cubreobjetos sobre la muestra. Se observa visualmente cada muestra mientras se calienta la platina. Se capturan imágenes usando una cámara digital en color SPOT Insight™ con software SPOT v. 4.5.9. Se calibra la platina caliente usando patrones de punto de fusión de la USP.
Mediante HSM de la forma cristalina A, entre 182 y 239°C, las partículas más pequeñas se evaporan y el vapor resultante recristaliza en cristales más grandes. La condensación y la fusión se observan entre 239 y 247°C; las agujas parecen fundirse en último lugar, coherente con las múltiples endotermas observadas mediante DSC. Se usan dos preparaciones para el análisis. En la primera, se observa una decoloración (descomposición) después de la fusión. En la segunda, el enfriamiento rápido provoca la recristalización de la masa fundida.
Ejemplo 16 - Preparación de una mezcla de las formas cristalinas A y B
Se usan reactivos disponibles comercialmente tal como se reciben a menos que se indique lo contrario. Las reacciones que requieren atmósferas inertes se realizan bajo nitrógeno a menos que se indique lo contrario.

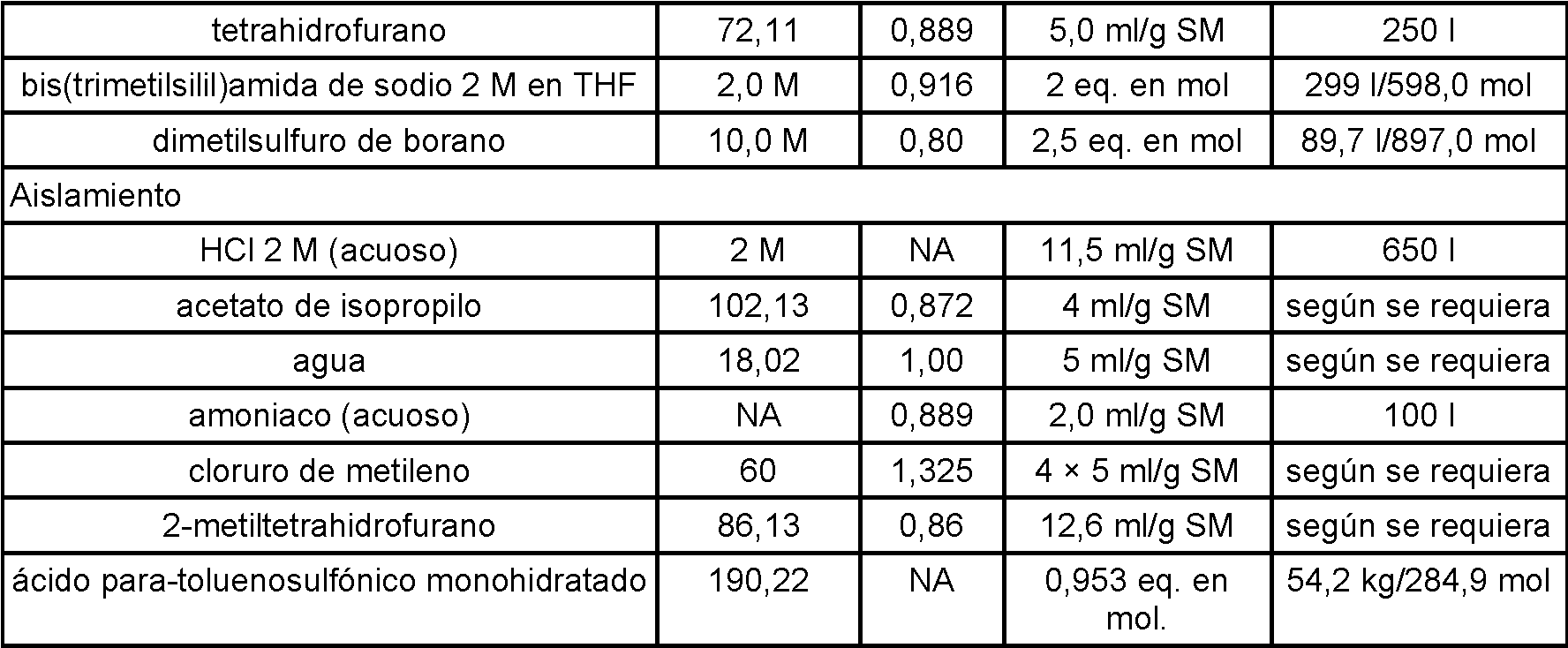
Etapas 1 y 2
Se disuelve 2-naftilacetonitrilo (50 kg) en THF (250 l), se añaden 32 kg de (S)-(+)-epiclorhidrina y se enfría la disolución hasta -10°C. Luego se añade una disolución 2,0 M de hexametildisililazano de sodio en THF (299 l) manteniendo la temperatura interna por debajo de -10°C. Esta adición requiere 14 h, 14 min hasta finalización. Luego se agita la mezcla de reacción cuatro horas adicionales a aproximadamente -10°C, después de lo cual se analiza una muestra de la mezcla de reacción mediante HPLC. Mientras se mantiene la temperatura interna a menos de 0°C, se añade dimetilsulfuro de borano (71 kg) durante cuatro horas y 33 min. Después de la finalización de la adición de borano, se calienta la mezcla de reacción lentamente hasta 60°C para reducir el nitrilo a la amina. Durante este calentamiento, se observa una exoterma, que se inicia a 45°C. Después de calentar a 60°C durante 14 h y 46 min, se analiza una muestra de la mezcla de reacción mediante HPLC.
Luego se enfría la mezcla de reacción hasta 24°C y se transfiere a una disolución de HCl 2 M durante 2 h y 28 min y se enjuaga el reactor con THF (22,3 kg) y se transfiere a la mezcla de reacción que contiene HCl. Se calienta la mezcla bifásica hasta de 45°C a 55°C y se agita durante 1 h 48 min a esta temperatura seguido de enfriamiento hasta 30°C. Se mide el pH de la mezcla de reacción extinguida y se comprueba que es 1. La reacción continúa mediante la adición de IPAc, agitación y separación de las fases. Cargar disolución de HCl 1 M a la fase orgánica, agitar, separar las fases y desechar la fase orgánica. Se añade amoniaco acuoso a la fase acuosa combinada y se mide el pH que muestra un pH de 9. A continuación se continúa con la extracción con dos extracciones de la fase acuosa con IPAc. Luego se lavan los extractos orgánicos combinados con disolución de cloruro de sodio al 5%. Se concentra parcialmente la fase orgánica resultante para el secado azeotrópico y coevaporación con cloruro de metileno cuatro veces y seguido de dilución con cloruro de metileno y transferencia de la mezcla de reacción mediante filtro en línea para limpiar, secar el reactor y diluir con IPAc. Luego se añaden porciones de ácido p-toluenosulfónico hidratado (54 kg) para precipitar el producto deseado como su sal pTsOH y se agita la suspensión de reacción durante tres horas a de 10°C a 15°C y se aísla el producto mediante filtración. Se lava la torta de filtro con 2-metiltetrahidrofurano y seguido de IPAc, luego se extrae en seco durante dos horas. Se purifica el producto en bruto mediante agitación con 2-metiltetrahidrofurano durante 11 h 36 min a de 10°C a 15°C y se aísla el producto mediante filtración. Se lava el sólido filtrado con 2-metiltetrahidrofurano y luego se seca hasta un peso constante para dar 73,8 kg del producto deseado como un sólido blanco. Rendimiento = 73,8 kg (62%). HPLC = 96,8%.
Etapas 3 y 4
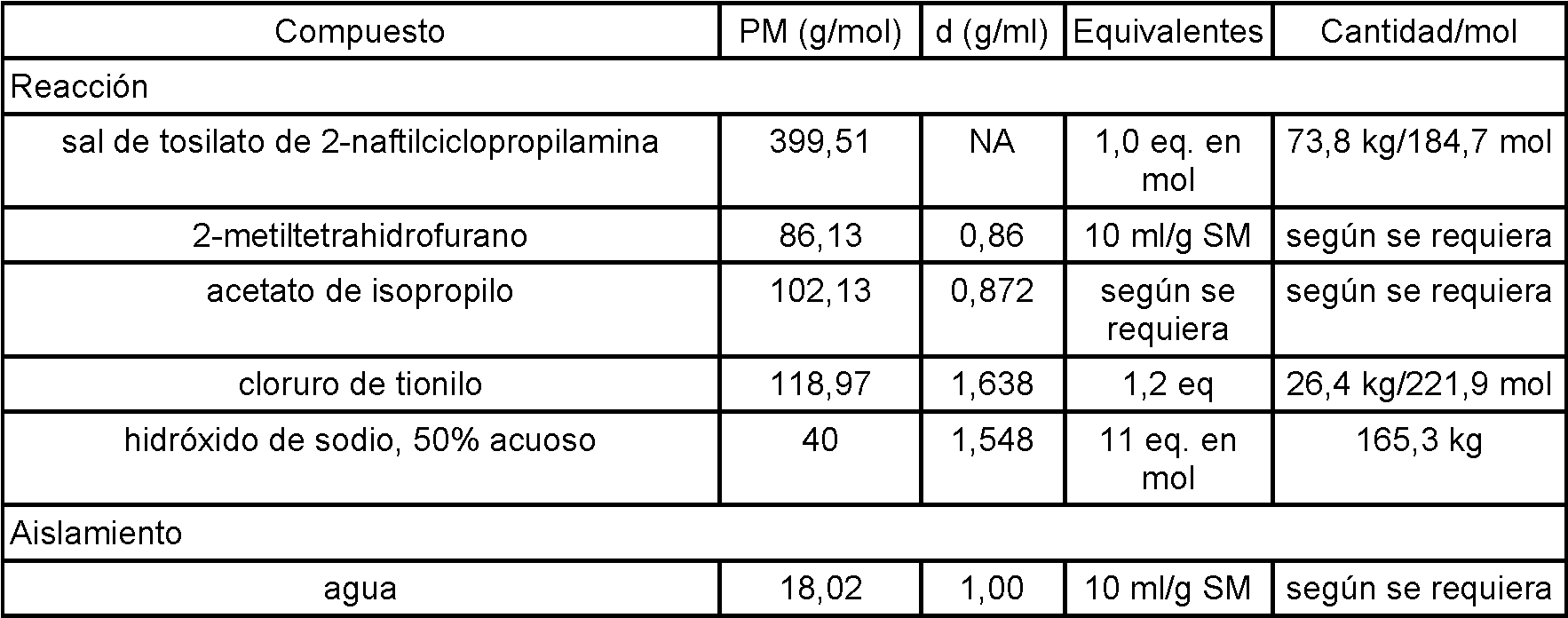

Se suspende la sal de amina-pTsOH (73,8 kg) obtenida de la etapa 2 anterior en 2-metiltetrahidrofurano (738 l) para dar una suspensión. Luego se añade cloruro de tionilo (26,4 kg) durante tres horas. Tras la finalización de la adición de cloruro de tionilo, se agita la mezcla de reacción tres horas adicionales. Se añade hidróxido de sodio acuoso (5 M, 10 equivalentes en mol) durante tres horas seguido de dos horas de agitación adicional. Se dejaron sedimentar las fases y se comprueba el pH de la fase acuosa y se encuentra que es de 9. Se añade agua (2 ml/g, SM), se agita la mezcla de reacción 15 más min a temperatura ambiente y se separan las fases y se lava la fase orgánica dos veces con agua. Se combinan las fases acuosas y se retroextraen con 2-metiltetrahidrofurano y se combinan la fase orgánica inicial y el retroextracto. Se lavan estas fases orgánicas combinadas con salmuera, se seca sobre sulfato de magnesio y se concentra parcialmente. Después de la concentración, se añade cloruro de hidrógeno en IPA (1,0 equivalente en mol de HCl en IPA) y se agita 2 h para formar la sal en bruto que se aísla mediante filtración, se lava con 2-metiltetrahidrofurano y seguido de IPAc y luego se extrae en seco durante 2 h a vacío.
Se disuelve el producto en bruto (82,6 kg) obtenido de lo anterior en 14 volúmenes de etanol caliente (70°C) y luego se filtra mediante un filtro de carbono encapsulado para mejorar el color. Luego se enjuagan el recipiente de disolución y el filtro de carbono encapsulado y la línea de transferencia con etanol caliente adicional (70°C) y se combina el enjuague con el filtrado. Se concentran parcialmente el filtrado y los lavados combinados a vacío hasta aproximadamente 5 volúmenes en total (en relación con la entrada de producto en bruto) y luego se agitan durante dos horas a 0°C. Se aíslan los sólidos resultantes mediante filtración, se lava la torta de filtro con etanol enfriado (de 0°C a 5°C) y seguido de IPAc y luego se secan los sólidos lavados para dar 33,6 kg del producto como un sólido ligeramente blanquecino. Rendimiento = 33,6 kg (73% rendimiento). HPLC aquiral = 98%.
Luego se seca el material mediante secado en cono. Después del secado, se tamiza el material.
Luego se disuelve una porción del material (14 kg) en 15 volúmenes de etanol caliente (70°C) y se filtra mediante un filtro de carbono encapsulado para mejorar el color. Luego se enjuagan el recipiente de disolución y el filtro de carbono encapsulado y la línea de transferencia con etanol caliente adicional (70°C) y se combina el enjuague con el filtrado. Se concentran parcialmente el filtrado y los lavados combinados a vacío hasta aproximadamente 8 volúmenes en total (en relación con la entrada de 14 kg de clorhidrato de (1R,5S)-1-(naftalen-2-il)-3-azabiciclo[3.1.0]hexano de partida) y luego se agita durante dos horas a 18°C. Se aíslan los sólidos resultantes mediante filtración, se lava la torta de filtro con etanol enfriado (de 5°C a 10°C) y seguido de IPAc y luego se secan los sólidos lavados para dar 9,4 kg (67,1% de rendimiento) de clorhidrato de (1R,5S)-1-(naftalen-2-il)-3-azabiciclo[3.1.0]hexano como un sólido blanco. HPLC aquiral = 98%.
Se muestra una XRPD del producto en la figura 56. La XRPD es coherente con la forma cristalina A con evidencia de picos de menor intensidad a 18,9°, 19,2°, 23,6°, 23,8°, 28,2° y 28,7° 20 atribuidos a la forma cristalina B. Se recoge el patrón de XRPD con un difractómetro PANalytical X'Pert PRO MPD usando un haz incidente de radiación Cu producido usando una fuente de enfoque fino largo Optix. Se usa un espejo multicapa elípticamente graduado para enfocar los rayos X de Cu Ka a través de la probeta y sobre el detector. Antes del análisis, se analiza una probeta de silicio (NIST s Rm 640e) para verificar si la posición observada del pico de Si 111 es coherente con la posición certificada por NIST. Se intercala una probeta de la muestra entre películas de 3 |im de grosor y se analiza en geometría de transmisión. Se usan una trampa de haz, una extensión de antidispersión corta, filo de cuchilla antidispersión, para minimizar el fondo generado por el aire. Se usan ranuras de Soller para los haces incidentes y difractados para minimizar el ensanchamiento a partir de la divergencia axial. Se recogen patrones de difracción usando un detector sensible a la posición de barrido (X'Celerator) ubicado 240 mm de la probeta y software Data Collector v. 2.2b.
Los parámetros de adquisición de datos XRPD son: Panalytical X-Pert Pro MPD PW3040 Pro, tubo de rayos X: Cu (1,54059 A), voltaje: 45 kV, amperaje: 40 mA, intervalo de barrido: 1,00-39,99° 20, tamaño de paso: 0,017° 20, tiempo de recogida: 721 s, velocidad de barrido: 3,2°/min, ranura: DS: 1/2°, SS: nulo, tiempo de revolución: 1,0 s, modo: transmisión.
Ejemplo 17 - Preparación de una mezcla de las formas cristalinas A y B
Se añaden 50 g del producto del ejemplo 16 anterior y EtOH especial para uso industrial (750 ml, 15 vol) a un matraz de fondo redondo de 3 bocas de 2 l con agitación mecánica, condensador de reflujo, entrada de nitrógeno, termopar y manto calefactor. Se calienta la mezcla hasta reflujo (77°C). Se disuelven los sólidos formando una disolución transparente a 72°C. Se añade suspensión de carbón vegetal suelto (5 g, 0,1 eq en 100 ml de EtOH) y se agita la mezcla durante 1 h. Filtrar y enjuagar con EtOH caliente (150 ml). Dividir el filtrado en dos porciones iguales.
Porción 1
Concentrar hasta 10 vol (250 ml) a 50°C. Comienza a precipitar una pequeña cantidad de sólidos durante la concentración. Transferir a un matraz de fondo redondo de 3 bocas de 500 ml con agitación mecánica y dejar enfriar hasta temperatura ambiente. Agitar durante 2 h a temperatura ambiente. Se forma una suspensión. Filtrar y enjuagar con EtOH (50 ml, 2 vol) seguido de IPAc (50 ml). Extraer en seco con filtro. Rendimiento = 20,5 g (82%).
Porción 2
Concentrar hasta 7 vol (175 ml) a 50°C. Comienzan a precipitar pequeñas cantidades de sólidos durante la concentración. Transferir a un matraz de fondo redondo de 3 bocas de 500 ml con agitación mecánica y dejar enfriar hasta temp temperatura ambiente. Agitar durante 2 h a temperatura ambiente. Se forma una suspensión. Filtrar y enjugar con EtOH (50 ml, 2 vol) seguido de IPAc (50 ml). Extraer en seco con filtro. Rendimiento = 19,8 g (79,2%).
Se combina el producto de las dos porciones y un patrón de XRPD de las porciones combinadas está en la figura 49 (Ejemplo 13).
Ejemplo 18 - Preparación de las formas cristalinas
Se usa la forma cristalina A del ejemplo 5 para elaborar las siguientes formas cristalinas.
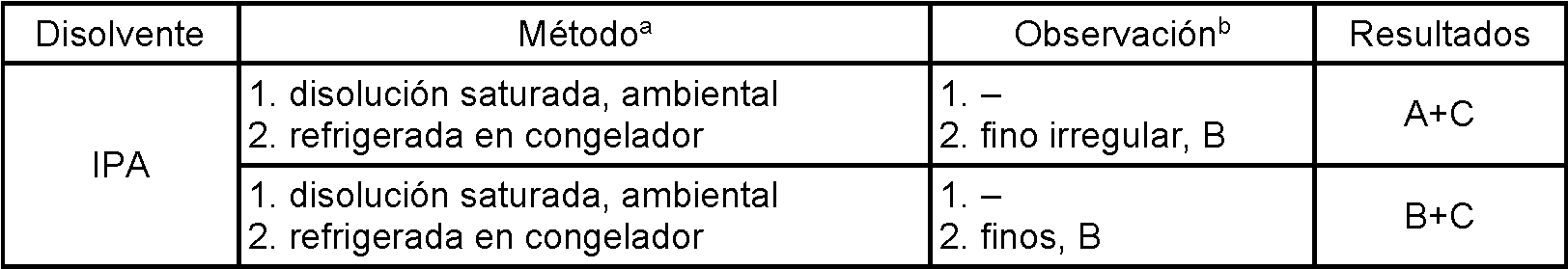
a. El tiempo y la temperatura son aproximados.
b. B = birrefringente cuando se observa mediante microscopio de luz polarizada.
Claims (11)
- REIVINDICACIONESi. Forma cristalina B de clorhidrato de (1R,5S)-1-(naftalen-2-il)-3-azabicido[3.1.0]hexano, en la que la forma cristalina B presenta un patrón de difracción de rayos X de polvo (XRPD) que comprende al menos tres valores 2-theta (°) seleccionados del grupo que consiste en 6,0±0,2, 17,4±0,2, 18,9±0,2, 19,2±0,2 y 24,4±0,2, en la que la XRPD se mide usando un haz incidente de radiación Cu Ka que tiene una longitud de onda de 1,54059 A y en la que la forma cristalina B comprende menos del 20% en peso de cualquier otra forma cristalina de clorhidrato de (1R,5S)-1-(naftalen-2-il)-3-azabiciclo[3.1.0]hexano.
- 2. Forma cristalina B según la reivindicación 1, en la que la forma cristalina B presenta un patrón de difracción de rayos X de polvo (XRPD) que comprende valores 2-theta (°) de 6,0±0,2, 17,4±0,2, 18,9±0,2, 19,2±0,2 y 24,4±0,2.
- 3. Forma cristalina B según la reivindicación 1 ó 2, en la que el patrón de XRPD de la forma cristalina B comprende además al menos uno, dos o tres valores 2-theta (°) seleccionados del grupo que consiste en 12,1±0,2, 13,2±0,2, 14,9±0,2, 15,1±0,2, 16,0±0,2, 16,9±0,2, 18,2±0,2, 19,9±0,2, 21,1±0,2, 21,3±0,2, 21,7±0,2, 22,6±0,2, 23,6±0,2, 23,8±0,2, 25,3±0,2, 26,1±0,2, 26,6±0,2, 27,2±0,2, 28,2±0,2, 28,7±0,2 y 29,5±0,2.
- 4. Forma cristalina B según una cualquiera de las reivindicaciones 1-3, en la que la forma cristalina B presenta un patrón de difracción de rayos X de polvo (XRPD) que comprende al menos tres valores de espaciamiento d (A) seleccionados del grupo que consiste en 14,6, 5,1,4,7, 4,6 y 3,6.
- 5. Forma cristalina B según la reivindicación 4, en la que la forma cristalina B presenta un patrón de difracción de rayos X de polvo (XRPD) que comprende valores de espaciamiento d (A) de 14,6, 5,1,4,7, 4,6 y 3,6. 6. Forma cristalina B según la reivindicación 4 ó 5, en la que el patrón de XRPD de la forma cristalina B comprende además al menos uno, dos o tres valores de espaciamiento d (A) seleccionados del grupo que consiste en 7,3, 6,7,
- 6,0, 5,9, 5,5, 5,2, 4,9, 4,5, 4,2, 4,1, 3,9, 3,8, 3,7, 3,5, 3,4, 3,3, 3,2, 3,1 y 3,0.
- 7. Forma cristalina B según una cualquiera de las reivindicaciones 1-6, en la que la forma cristalina B pertenece al grupo espacial P2-|2-|21 y tiene los siguientes parámetros de celda unitaria: a = 5,9055(2) A, b = 7,4645(3) A, c = 29,1139(13) A, a = p = y = 90°.
- 8. Composición farmacéutica que comprende la forma cristalina B según una cualquiera de las reivindicaciones 1-7 y un diluyente o portador farmacéuticamente aceptable.
- 9. Composición farmacéutica según la reivindicación 8, que es una composición farmacéutica de liberación sostenida oral.
- 10. Forma cristalina B según una cualquiera de las reivindicaciones 1-7, o composición farmacéutica según una cualquiera de las reivindicaciones 8-9, para su uso en la profilaxis o el tratamiento del trastorno por déficit de atención con hiperactividad.
- 11. Forma cristalina B o composición farmacéutica para su uso en la profilaxis o el tratamiento del trastorno por déficit de atención con hiperactividad según la reivindicación 10, en la que el trastorno por déficit de atención con hiperactividad es comorbilidad con depresión, toxicomanía o ansiedad.
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201562181174P | 2015-06-17 | 2015-06-17 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2922158T3 true ES2922158T3 (es) | 2022-09-09 |
Family
ID=57546597
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES19196556T Active ES2922158T3 (es) | 2015-06-17 | 2016-06-17 | Compuestos cristalinos |
Country Status (25)
| Country | Link |
|---|---|
| US (5) | US9708261B2 (es) |
| EP (3) | EP4049997A1 (es) |
| JP (4) | JP6896651B2 (es) |
| KR (1) | KR102593783B1 (es) |
| CN (3) | CN117466800A (es) |
| AU (2) | AU2016279075C1 (es) |
| CA (2) | CA2989431C (es) |
| CY (1) | CY1125352T1 (es) |
| DK (1) | DK3597189T3 (es) |
| ES (1) | ES2922158T3 (es) |
| HK (1) | HK1247125A1 (es) |
| HR (1) | HRP20220829T1 (es) |
| HU (1) | HUE059348T2 (es) |
| LT (1) | LT3597189T (es) |
| MX (2) | MX387591B (es) |
| MY (1) | MY194868A (es) |
| NZ (2) | NZ739044A (es) |
| PH (1) | PH12017502324A1 (es) |
| PL (1) | PL3597189T3 (es) |
| PT (1) | PT3597189T (es) |
| SA (1) | SA517390552B1 (es) |
| SG (1) | SG10201911417PA (es) |
| SI (1) | SI3597189T1 (es) |
| TW (1) | TWI751998B (es) |
| WO (1) | WO2016205762A1 (es) |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2006275870B2 (en) | 2005-07-27 | 2013-01-10 | Otsuka America Pharmaceutical, Inc. | Novel 1-aryl-3-azabicyclo[3.1.0]hexanes: preparation and use to treat neuropsychiatric disorders |
| US20140206740A1 (en) | 2011-07-30 | 2014-07-24 | Neurovance, Inc. | Use Of (1R,5S)-(+)-(Napthalen-2-yl)-3-Azabicyclo[3.1.0]Hexane In The Treatment Of Conditions Affected By Monoamine Neurotransmitters |
| WO2015102826A1 (en) * | 2013-12-09 | 2015-07-09 | Neurovance, Inc. | Novel compositions |
| NZ739044A (en) | 2015-06-17 | 2022-08-26 | Otsuka America Pharmaceutical Inc | Crystalline compounds |
| CN111417624B (zh) * | 2017-12-11 | 2022-03-25 | 苏州科睿思制药有限公司 | Eb-1020的晶型及其制备方法和用途 |
| CN114555557A (zh) | 2019-10-16 | 2022-05-27 | 大塚制药株式会社 | 森塔纳法啶的制备方法 |
| WO2022256215A1 (en) | 2021-05-31 | 2022-12-08 | Teva Pharmaceuticals International Gmbh | Solid state form of centanafadine hcl and process for preparation thereof |
| US12502358B1 (en) | 2024-08-13 | 2025-12-23 | Innovate Therapeutics Llc | Centanafadine multiphasic controlled-release pharmaceutical formulation |
Family Cites Families (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2005117630A (ru) | 2002-11-08 | 2006-01-20 | Дов Фармасьютикал, Инк. (Us) | Полиморфы гидрохлорида бицифадина |
| HRP20160757T1 (hr) | 2004-04-19 | 2016-08-12 | Krka, Tovarna Zdravil, D.D., Novo Mesto | Postupak za proizvodnju polimorfnog oblika i od klopidogrel-vodik-sulfata |
| US20070043100A1 (en) | 2005-08-16 | 2007-02-22 | Hagen Eric J | Novel polymorphs of azabicyclohexane |
| JP2008510715A (ja) | 2004-08-18 | 2008-04-10 | ディーオーヴィ ファーマスーティカル インコーポレイテッド | アザビシクロヘキサンの新規多型 |
| US20070082939A1 (en) | 2005-07-26 | 2007-04-12 | Lippa Arnold S | Methods and compositions for the treatment of neuropathies and related disorders |
| AU2006275870B2 (en) | 2005-07-27 | 2013-01-10 | Otsuka America Pharmaceutical, Inc. | Novel 1-aryl-3-azabicyclo[3.1.0]hexanes: preparation and use to treat neuropsychiatric disorders |
| US7919598B2 (en) | 2006-06-28 | 2011-04-05 | Bristol-Myers Squibb Company | Crystal structures of SGLT2 inhibitors and processes for preparing same |
| US20080058535A1 (en) * | 2006-07-25 | 2008-03-06 | Zhengming Chen | Methods and compositions for production, formulation and use of 1 aryl-3-azabicyclo[3.1.0]hexanes |
| US8576985B2 (en) | 2009-09-01 | 2013-11-05 | Aptuit (West Lafayette) Llc | Methods for indexing solid forms of compounds |
| EA024026B1 (ru) | 2010-11-25 | 2016-08-31 | Рациофарм Гмбх | Новые соли и полиморфные формы афатиниба |
| US20140206740A1 (en) * | 2011-07-30 | 2014-07-24 | Neurovance, Inc. | Use Of (1R,5S)-(+)-(Napthalen-2-yl)-3-Azabicyclo[3.1.0]Hexane In The Treatment Of Conditions Affected By Monoamine Neurotransmitters |
| SI2819516T1 (sl) | 2011-07-30 | 2020-07-31 | Otsuka America Pharmaceutical, Inc. | Uporaba (1R,5S)-(+)-1-(naftalen-2-IL)-3-azabiciklo(3.1.0)heksana pri zdravljenju stanj na katere vplivajo monoamin nevrotransmiterji |
| US20140228421A1 (en) | 2011-09-07 | 2014-08-14 | Anthony McKinney | Methods For Inhibiting Native And Promiscuous Uptake Of Monoamine Neurotransmitters |
| WO2015102826A1 (en) * | 2013-12-09 | 2015-07-09 | Neurovance, Inc. | Novel compositions |
| NZ739044A (en) | 2015-06-17 | 2022-08-26 | Otsuka America Pharmaceutical Inc | Crystalline compounds |
-
2016
- 2016-06-17 NZ NZ739044A patent/NZ739044A/en unknown
- 2016-06-17 SG SG10201911417PA patent/SG10201911417PA/en unknown
- 2016-06-17 CA CA2989431A patent/CA2989431C/en active Active
- 2016-06-17 AU AU2016279075A patent/AU2016279075C1/en active Active
- 2016-06-17 CN CN202310843667.7A patent/CN117466800A/zh active Pending
- 2016-06-17 EP EP22164478.4A patent/EP4049997A1/en active Pending
- 2016-06-17 MY MYPI2017704811A patent/MY194868A/en unknown
- 2016-06-17 SI SI201631552T patent/SI3597189T1/sl unknown
- 2016-06-17 CN CN201680035027.1A patent/CN107921021A/zh active Pending
- 2016-06-17 HK HK18106861.3A patent/HK1247125A1/zh unknown
- 2016-06-17 NZ NZ776973A patent/NZ776973A/en unknown
- 2016-06-17 CA CA3200692A patent/CA3200692A1/en active Pending
- 2016-06-17 PL PL19196556.5T patent/PL3597189T3/pl unknown
- 2016-06-17 DK DK19196556.5T patent/DK3597189T3/da active
- 2016-06-17 EP EP16812592.0A patent/EP3310352A4/en not_active Ceased
- 2016-06-17 ES ES19196556T patent/ES2922158T3/es active Active
- 2016-06-17 MX MX2017016430A patent/MX387591B/es unknown
- 2016-06-17 LT LTEP19196556.5T patent/LT3597189T/lt unknown
- 2016-06-17 CN CN202310848120.6A patent/CN117088802A/zh active Pending
- 2016-06-17 JP JP2017565236A patent/JP6896651B2/ja active Active
- 2016-06-17 HU HUE19196556A patent/HUE059348T2/hu unknown
- 2016-06-17 US US15/186,415 patent/US9708261B2/en active Active
- 2016-06-17 HR HRP20220829TT patent/HRP20220829T1/hr unknown
- 2016-06-17 KR KR1020187001083A patent/KR102593783B1/ko active Active
- 2016-06-17 PT PT191965565T patent/PT3597189T/pt unknown
- 2016-06-17 WO PCT/US2016/038256 patent/WO2016205762A1/en not_active Ceased
- 2016-06-17 EP EP19196556.5A patent/EP3597189B1/en active Active
- 2016-12-21 TW TW105142467A patent/TWI751998B/zh active
-
2017
- 2017-06-01 US US15/611,580 patent/US9856217B2/en active Active
- 2017-11-21 US US15/820,241 patent/US10280141B2/en active Active
- 2017-12-14 SA SA517390552A patent/SA517390552B1/ar unknown
- 2017-12-15 PH PH12017502324A patent/PH12017502324A1/en unknown
- 2017-12-15 MX MX2020009949A patent/MX2020009949A/es unknown
-
2019
- 2019-03-11 US US16/298,179 patent/US10800740B2/en active Active
-
2020
- 2020-08-14 US US16/993,648 patent/US11299458B2/en active Active
-
2021
- 2021-01-19 AU AU2021200314A patent/AU2021200314B2/en active Active
- 2021-06-09 JP JP2021096354A patent/JP7244575B2/ja active Active
-
2022
- 2022-07-11 CY CY20221100470T patent/CY1125352T1/el unknown
-
2023
- 2023-03-09 JP JP2023036730A patent/JP7583851B2/ja active Active
-
2024
- 2024-11-01 JP JP2024192608A patent/JP2025022896A/ja active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2922158T3 (es) | Compuestos cristalinos | |
| TR201816159T4 (tr) | Bir pürin türevinin kristal formları. | |
| JP2018517742A5 (es) | ||
| CN102917694A (zh) | 依泽替米贝的片剂制剂 | |
| RU2789672C2 (ru) | Кристаллические соединения | |
| HK40020220B (en) | Crystalline compounds | |
| HK40020220A (en) | Crystalline compounds | |
| HK40077679A (en) | Crystalline compounds | |
| HK40102359A (zh) | 结晶化合物 | |
| HK40099983A (zh) | 结晶化合物 | |
| WO2021028678A1 (en) | Polymorph of venetoclax and method for preparing the polymorph | |
| NZ623344B2 (en) | Polymorphic form of pridopidine hydrochloride |