ES2917549T3 - Un derivado de carboxamida y sus diastereoisómeros en forma cristalina estable - Google Patents
Un derivado de carboxamida y sus diastereoisómeros en forma cristalina estable Download PDFInfo
- Publication number
- ES2917549T3 ES2917549T3 ES16704029T ES16704029T ES2917549T3 ES 2917549 T3 ES2917549 T3 ES 2917549T3 ES 16704029 T ES16704029 T ES 16704029T ES 16704029 T ES16704029 T ES 16704029T ES 2917549 T3 ES2917549 T3 ES 2917549T3
- Authority
- ES
- Spain
- Prior art keywords
- pyrazole
- carboxamide
- hydroxyethyl
- pyrazol
- propan
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/415—1,2-Diazoles
- A61K31/4155—1,2-Diazoles non condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/02—Halogenated hydrocarbons
- A61K31/025—Halogenated hydrocarbons carbocyclic
- A61K31/03—Halogenated hydrocarbons carbocyclic aromatic
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
- A61K31/165—Amides, e.g. hydroxamic acids having aromatic rings, e.g. colchicine, atenolol, progabide
- A61K31/166—Amides, e.g. hydroxamic acids having aromatic rings, e.g. colchicine, atenolol, progabide having the carbon of a carboxamide group directly attached to the aromatic ring, e.g. procainamide, procarbazine, metoclopramide, labetalol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/275—Nitriles; Isonitriles
- A61K31/277—Nitriles; Isonitriles having a ring, e.g. verapamil
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/14—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Epidemiology (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
La presente divulgación se relaciona con formas cristalinas sólidas de N-((S) -1- (3- (3-cloro-4-cianofenil) -1h-pirazol-1-il) -propan-2-il) -5- (1-hidroxietil) -1h-pirazol-3- carboxamida (i) y los diastereómeros de los mismos, y a los métodos para preparar tales formas cristalinas. El compuesto (i) y los diastereómeros de los mismos son potentes moduladores de receptor de andrógenos (AR) útiles como medicamento. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Un derivado de carboxamida y sus diastereoisómeros en forma cristalina estable
Campo de la invención
La presente descripción se refiere a formas cristalinas sólidas de los diastereoisómeros del compuesto farmacéutico N-((s)-1-(3-(3-doro-4-cianofenil)-1H-pirazol-1-il)-propan-2-il)-5-(1-hidroxietil)-1H-pirazol-3-carboxamida (I) y a métodos para la preparación de tales formas cristalinas.
Antecedentes de la invención
El compuesto N-((S)-1-(3-(3-cloro-4-cianofenil)-1H-pirazol-1-il)-propan-2-il)-5-(1-hidroxietil)-1H-pirazol-3-carboxamida (I) y la fabricación del mismo se han divulgado en el documento WO 2011/051540. El compuesto (I) es un potente modulador del receptor de andrógenos (RA) útil en el tratamiento del cáncer, particularmente el cáncer dependiente del RA, tal como el cáncer de próstata, y otras enfermedades donde se desea el antagonismo del RA. El compuesto (I) se representa mediante la estructura:

Como el átomo de hidrógeno del anillo de pirazol puede existir en equilibrio tautomérico entre las posiciones 1 y 2, la persona experta reconoce que la estructura anterior y el nombre químico "N-((S)-1-(3-(3-cloro-4-cianofenil)-1H-pirazol-1-il)-propan-2-il)-5-(1-hidroxietil)-1H-pirazol-3-carboxamida (I)", tal como se hace referencia en el presente documento, incluyen el tautómero del compuesto (I), en concreto, N-((S)-1-(3-(3-cloro-4-cianofenil)-1H-pirazol-1-il)-propan-2-il)-3-(1-hidroxietil)-1H-pirazol-5-carboxamida.
Además del átomo de carbono quiral que se muestra en la estructura química anterior, el compuesto (I) tiene otro átomo de carbono quiral con un grupo hidroxi unido al mismo. Por lo tanto, el compuesto (I) tiene dos diastereómeros, en concreto, N-((S)-1-(3-(3-cloro-4-cianofenil)-1 H-pirazol-1-il)propan-2-il)-5-((S)-1-hidroxietil)-1H-pirazol-3-carboxamida (la)

y N-((S)-1-(3-(3-cloro-4-cianofenil)-1H-pirazol-1-il)propan-2-il)-5-((R)-1-hidroxietil)-1H-pirazol-3-carboxamida (lb).
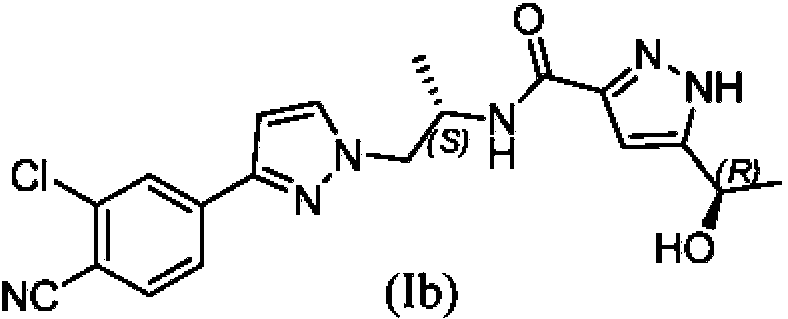
Debido al equilibrio tautomérico del átomo de hidrógeno entre las posiciones 1 y 2 en el anillo de pirazol, el nombre químico de los diastereómeros (la) y (lb) incluye los tautómeros de (la) y (lb), de manera similar al compuesto (I), tal como se ha explicado anteriormente.
Los compuestos (la) y (lb) también son potentes moduladores del receptor de andrógenos (RA) útiles en el tratamiento del cáncer, particularmente el cáncer dependiente del RA, tal como el cáncer de próstata, y otras enfermedades donde se desea el antagonismo del RA.
Sumario de la invención
Actualmente, se ha hallado que los diastereoisómeros (la) y (Ib) se pueden obtener en una forma cristalina estable y sustancialmente pura mediante la cristalización en determinadas condiciones.
Por tanto, la presente invención proporciona la forma cristalina I' de N-((S)-1-(3-(3-cloro-4-cianofenil)-1H-pirazol-1-il)propan-2-il)-5-((S)-1-hidroxietil)-1H-pirazol-3-carboxamida (la), que tiene un patrón de difracción de rayos X en polvo que comprende picos característicos a 9,3, 15,7, 17,0, 24,1 y 25,1 ± 0,15 grados 2-theta medidos usando un tubo de rayos X llenado de Cu.
La presente invención proporciona, además, la forma cristalina I' de N-((S)-1-(3-(3-cloro-4-cianofenil)-1H-pirazol-1-il)propan-2-il)-5-((S)-1-hidroxietil)-1H-pirazol-3-carboxamida (la), que tiene un patrón de difracción de rayos X en polvo que comprende picos característicos a 9,3, 11,4, 11,5, 13,6, 14,7, 14,9, 15,7, 16,1, 17,0, 17,7, 18,5, 19,1, 20,5, 21,5, 22,1, 22,6, 23,2, 23,6, 24,1, 25,1, 26,2 y 27,2 ± 0,15 grados 2-theta medidos usando un tubo de rayos X llenado de Cu.
La presente invención proporciona, además, la forma cristalina I" de N-((S)-1-(3-(3-cloro-4-cianofenil)-1H-pirazol-1-il)propan-2-il)-5-((R)-1-hidroxietil)-1H-pirazol-3-carboxamida (lb), que tiene un patrón de difracción de rayos X en polvo que comprende picos característicos a 9,2, 10,9, 15,1, 15,8 y 22,1 ± 0,15 grados 2-theta medidos usando un tubo de rayos X llenado de Cu.
La presente invención proporciona, además, la forma cristalina I" de N-((S)-1-(3-(3-cloro-4-cianofenil)-1H-pirazol-1-il)propan-2-il)-5-((R)-1-hidroxietil)-1H-pirazol-3-carboxamida (lb), que tiene un patrón de difracción de rayos X en polvo que comprende picos característicos a 7,9, 9,2, 10,9, 13,2, 14,8, 15,1, 15,5, 15,8, 16,9, 18,4, 20,2, 20,5, 21,8, 22,1 y 24,3 ± 0,15 grados 2-theta medidos usando un tubo de rayos X llenado de Cu.
La presente invención proporciona, además, un proceso para la preparación de la forma cristalina I' o I" de N-((S)-1-(3-(3-cloro-4-cianofenil)-1H-pirazol-1-il)propan-2-il)-5-((S)-1-hidroxietil)-1H-pirazol-3-carboxamida (la) o N-((S)-1-(3-(3-cloro-4-cianofenil)-1H-pirazol-1-il)propan-2-il)-5-((R)-1-hidroxietil)-1H-pirazol-3-carboxamida (lb), respectivamente, que comprende
a) mezclar N-((S)-1-(3-(3-cloro-4-cianofenil)-1H-pirazol-1-il)propan-2-il)-5-((S)-1-hidroxietil)-1H-pirazol-3-carboxamida (la) o N-((s)-1-(3-(3-cloro-4-cianofenil)-1H-pirazol-1-il)propan-2-il)-5-((R)-1-hidroxietil)-1H-pirazol-3-carboxamida (lb) con una mezcla de acetonitrilo y agua;
b) calentar la mezcla de la Etapa a) para formar una solución;
c) enfriar la solución de la Etapa b) hasta aproximadamente 0-50 °C; y
d) aislar la forma cristalina.
Breve descripción de los dibujos
La Figura 1 muestra el patrón de difracción de rayos X en polvo de la forma cristalina I del compuesto (I) obtenido en el Ejemplo de referencia 1.
La Figura 2 muestra el patrón de difracción de rayos X en polvo de la forma cristalina I' del compuesto (la) obtenido en el Ejemplo 4.
La Figura 3 muestra el patrón de difracción de rayos X en polvo de la forma cristalina I" del compuesto (lb) obtenido en el Ejemplo 7.
Descripción detallada de la invención
La forma cristalina I del compuesto (I), la forma cristalina I' del compuesto (la) y la forma cristalina I" del compuesto (lb) se han caracterizado mediante estudios de difracción de rayos X en polvo (XRPD por sus siglas en inglés). Por consiguiente, en un aspecto, la presente divulgación proporciona la forma cristalina I' de N-((S)-1-(3-(3-cloro-4-cianofenil)-1H-pirazol-1-il)propan-2-il)-5-((S)-1-hidroxietil)-1H-pirazol-3-carboxamida (la), que tiene un patrón de difracción de rayos X en polvo que comprende picos característicos a 9,3, 15,7, 17,0, 24,1 y 25,1 ± 0,15 grados 2-theta.
En otro aspecto, la presente divulgación proporciona la forma cristalina I" de N-((S)-1-(3-(3-cloro-4-cianofenil)-1H-pirazol-1-il)propan-2-il)-5-((R)-1-hidroxietil)-1H-pirazol-3-carboxamida (lb), que tiene un patrón de difracción de rayos X en polvo que comprende picos característicos a 9,2, 10,9, 15,1, 15,8 y 22,1 ± 0,15 grados 2-theta.
En otro aspecto más, la presente divulgación proporciona la forma cristalina I' de N-((S)-1-(3-(3-cloro-4-cianofenil)-1H-pirazol-1-il)propan-2-il)-5-((S)-1-hidroxietil)-1H-pirazol-3-carboxamida (la), que tiene un patrón de difracción de rayos X en polvo que comprende picos característicos a 9,3, 11,4, 11,5, 13,6, 14,7, 14,9, 15,7, 16,1, 17,0, 17,7, 18,5, 19,1, 20,5, 21,5, 22,1, 22,6, 23,2, 23,6, 24,1, 25,1, 26,2 y 27,2 ± 0,15 grados 2-theta.
En otro aspecto más, la presente divulgación proporciona la forma cristalina I" de N-((S)-1-(3-(3-cloro-4-cianofenil)-1H-pirazol-1-il)propan-2-il)-5-((R)-1-hidroxietil)-1H-pirazol-3-carboxamida (lb), que tiene un patrón de difracción de rayos X en polvo que comprende picos característicos a aproximadamente 7,9, 9,2, 10,9, 13,2, 14,8, 15,1, 15,5, 15,8, 16,9, 18,4, 20,2, 20,5, 21,8, 22,1 y 24,3 ± 0,15 grados 2-theta.
De acuerdo con otro aspecto más, la presente divulgación proporciona la forma cristalina I' de N-((S)-1-(3-(3-cloro-4-cianofenil)-1H-pirazol-1-il)propan-2-il)-5-((S)-1-hidroxietil)-1H-pirazol-3-carboxamida (la), que tiene un patrón de difracción de rayos X en polvo sustancialmente tal como se ilustra en la Figura 2.
De acuerdo con otro aspecto más, la presente divulgación proporciona la forma cristalina I" de N-((S)-1-(3-(3-cloro-4-cianofenil)-1H-pirazol-1-il)propan-2-il)-5-((R)-1-hidroxietil)-1H-pirazol-3-carboxamida (lb), que tiene un patrón de difracción de rayos X en polvo sustancialmente tal como se ilustra en la Figura 3.
La persona experta reconoce que las posiciones de los picos del patrón de difracción de rayos X en polvo a las que se hace referencia en el presente documento se pueden someter a variaciones de /- 0,15 grados 2-theta de acuerdo con diversos factores, tales como la temperatura, la concentración y la instrumentación usada.
De acuerdo con otro aspecto más, la presente divulgación proporciona un proceso para la preparación de la forma cristalina I' o I" de N-((S)-1-(3-(3-cloro-4-cianofenil)-1H-pirazol-1-il)propan-2-il)-5-((S)-1-hidroxietil)-1H-pirazol-3-carboxamida (la) o N-((S)-1-(3-(3-cloro-4-cianofenil)-1H-pirazol-1-il)propan-2-il)-5-((R)-1-hidroxietil)-1H-pirazol-3-carboxamida (lb), respectivamente, que comprende
a) mezclar N-((S)-1-(3-(3-cloro-4-cianofenil)-1H-pirazol-1-il)propan-2-il)-5-((S)-1-hidroxietil)-1H-pirazol-3-carboxamida (la) o N-((S)-1-(3-(3-cloro-4-cianofenil)-1H-pirazol-1-il)propan-2-il)-5-((R)-1-hidroxietil)-1H-pirazol-3-carboxamida (lb) con una mezcla de acetonitrilo y agua;
b) calentar la mezcla de la Etapa a) para formar una solución;
c) enfriar la solución de la Etapa b) hasta aproximadamente 0-50 °C; y
d) aislar la forma cristalina.
También se describe en el presente documento un proceso para la fabricación del diastereómero (la) o (lb), que comprende
a) reducir el compuesto de la Fórmula (1)

en donde R es H o alquilo C1-6, con una enzima cetorreductasa para obtener el compuesto de la Fórmula (2) en forma ópticamente activa, en donde R es tal como se ha definido anteriormente;

b) opcionalmente, proteger el grupo hidroxilo del compuesto de la Fórmula (2);
c) en caso de que R sea un alquilo C1-6, someter el compuesto de la Fórmula (2), en donde el grupo hidroxilo está opcionalmente protegido, a la escisión del enlace de éster para obtener el compuesto (3);

en donde el grupo hidroxilo está opcionalmente protegido; y
d) tratar el compuesto (3), en donde el grupo hidroxilo está opcionalmente protegido, con (S)-4-(1-(2-aminopropil)-1H-pirazol-3-il)-2-dorobenzonitrilo y, en caso de que el grupo hidroxilo esté protegido, desproteger el grupo hidroxilo, a fin de obtener el compuesto (la) o (lb).
El compuesto (I) se puede sintetizar usando los procedimientos descritos en el documento WO 2011/051540.
Los diastereómeros puros (la) y (lb) se pueden sintetizar de manera adecuada, por ejemplo, usando enzimas cetorreductasa (KRED por sus siglas en inglés) para la reducción selectiva de tanto S como R del compuesto 1 en el compuesto 2, tal como se muestra en el Esquema 1, en donde R es H o alquilo C m
Protección

Esquema 1.
Por ejemplo, las enzimas Codexis KRED-130 y KRED-NADH-110 son útiles para la obtención de una excelente estereoselectividad, incluso estereoespecificidad. En el Esquema 1, el material de partida 1 es preferentemente un éster (R=alquilo C1-6), por ejemplo, éster de etilo (R=etilo), tal como para facilitar la extracción del producto en la fase orgánica como compuesto, donde R=H tiene la tendencia a permanecer en la fase acuosa. El producto intermedio 2 se puede proteger, preferentemente con derivados de sililo, tales como terc-butildifenilsililo, con el fin de evitar la esterificación en la etapa de amidación. En el caso de R=alquilo C1-6, la hidrólisis del éster se realiza típicamente antes de la etapa de amidación, preferentemente en presencia de LiOH, NaOH o KOH. La amidación del compuesto 3 en el compuesto 5 se lleva a cabo de manera adecuada usando el sistema de EDCI HBTU, DIPEA, pero también resulta posible usar otros métodos de amidación típicos. La desprotección de 5 proporciona los diastereoisómeros puros (la) y (lb).
El anillo de pirazol sin sustitución de NH es de una funcionalidad tautomerizable conocida y se describe en el presente documento únicamente como tautómero individual, pero en el presente documento todos los productos intermedios y finales pueden existir en ambas formas tautoméricas al mismo tiempo.
La estereoquímica de los compuestos se puede confirmar mediante el uso de materiales de partida ópticamente puros con una configuración absoluta conocida, tal como se demuestra en el Esquema 2, en donde R=H o alquilo C1-6, preferentemente alquilo, por ejemplo, etilo. Los productos finales del Esquema 2 se obtienen típicamente como mezcla de tautómeros en análisis de Rm N en 1H en DMSO a aprox. 27 °C (+300 °K).


Esquema 2. Vía de síntesis en estereoisómeros mediante el uso de materiales de partida con una configuración absoluta conocida
Las formas cristalinas I, I' y I" de los compuestos (I), (la) y (lb), respectivamente, se pueden preparar, por ejemplo, mediante la disolución del compuesto en cuestión en una mezcla de acetonitrilo:agua que tiene una relación en volumen de 85:15 a 99:1, tal como de 90:10 a 98:2, por ejemplo, de 95:5, con el calentamiento y el enfriamiento lento de la solución hasta que la forma cristalina se precipita de la solución. La concentración del compuesto en la mezcla de disolvente de acetonitrilo:agua es, de manera adecuada, de aproximadamente 1 kg del compuesto en 5-25 litros de la mezcla de disolvente de acetonitrilo:agua, por ejemplo, 1 kg del compuesto en 10-20 litros de la mezcla de disolvente de acetonitrilo:agua. El compuesto se disuelve de manera adecuada en la mezcla de disolvente de acetonitrilo:agua mediante el calentamiento de la solución, por ejemplo, cerca de la temperatura de reflujo, por ejemplo, hasta 60-80 °C, por ejemplo, hasta 75 °C, con agitación y filtración, si resulta necesario. A continuación, la solución se enfría de manera adecuada hasta 0-50 °C, por ejemplo, hasta 5-35 °C, por ejemplo, hasta TA, durante 5 a 24 horas, por ejemplo, durante 6 a 12 horas, y se agita a esta temperatura durante 3 a 72 horas, por ejemplo, durante 5 a 12 horas. A continuación, el producto cristalino obtenido se puede filtrar, lavar y secar. El secado se lleva a cabo de manera adecuada al vacío a una temperatura de 40 a 60 °C, por ejemplo, a 55 °C, durante 1 a 24 horas, tal como durante 2 a 12 horas, por ejemplo, durante 2 a 6 horas.
Las formas cristalinas I, I' y I" de los compuestos (I), (la) y (lb), respectivamente, son útiles como medicamentos y se pueden formular en formas de dosificación farmacéuticas, tales como comprimidos y cápsulas para administración oral, mediante el mezclado con excipientes farmacéuticos conocidos en la técnica.
La divulgación se ilustra adicionalmente mediante los siguientes ejemplos.
Ejemplo de referencia 1. Cristalización de N-((S)-1-(3-(3-cloro-4-cianofenil)-1H-pirazol-1-il)-propan-2-il)-5-(1-hidroxietil)-1H-pirazol-3-carboxamida (I)
Se cargaron N-((S)-1-(3-(3-cloro-4-cianofenil)-1H-pirazol-1-il)-propan-2-il)-5-(1-hidroxietil)-1H-pirazol-3-carboxamida (I) (5 g), 71,25 ml de acetonitrilo y 3,75 ml de agua destilada se cargaron en un matraz y la mezcla se calentó hasta 75 °C. La mezcla se enfrió lentamente hasta TA y se agitó a TA durante 3 días. El sólido obtenido se filtró y se lavó dos veces con acetonitrilo:agua (9,5 ml:0,5 ml). El producto se secó al vacío a 40 °C y, finalmente, a 60 °C para obtener 4,42 g del compuesto del título cristalino (rendimiento del 88 %), que se usó en el estudio de difracción de rayos X.
Ejemplo de referencia 2. Estudio de difracción de rayos X del compuesto cristalino (I)
La forma cristalina del compuesto (I) obtenido en el Ejemplo 1 se analizó mediante el método de difracción de rayos X en polvo. Las mediciones se realizaron con el difractómetro de rayos X en polvo PANalytical X'Pert PRO a temperatura ambiente usando un tubo de rayos X llenado de Cu (45 kV * 40 mA) como fuente de rayos X, una rendija de antidispersión fija de 1°, una rendija de divergencia programable con una longitud irradiada de 5,0 mm y el detector de tiras múltiples en tiempo real X'Celerator. La recopilación de datos se realizó en etapas de 0,008° a una velocidad de barrido de 1°/min en el intervalo de 3-80° 20. La forma cristalina se caracterizó mediante un patrón de difracción de rayos X en polvo, tal como se muestra en la Figura 1, y presentado picos característicos a aproximadamente los siguientes valores 2-theta:

Ejemplo 3. Síntesis de N-((S)-1-(3-(3-cloro-4-cianofenil)-1H-pirazol-1-il)-propan-2-il)-5-((S)-1-hidroxietil)-1H-pirazol-3-carboxamida (Ia)
a) Etil-5-((S)1-hidroxietil)-1H-pirazol-3-carboxilato
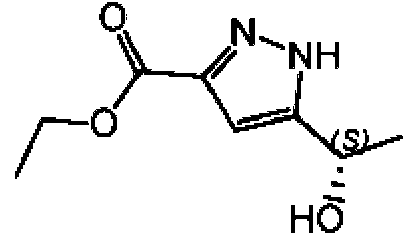
Se añadieron MgSO4 x 7 H2O (341 mg), sal monosódica de NADP (596 mg), D(+)-glucosa (9,26 g) y polvo liofilizado de enzima optimizada CDX-901 (142 mg) a un tampón de 0,2 mM de KH2PO4 (pH 7,0, 709 ml) para preparar la solución I. A esta solución I se añadió la solución II que contenía etil-5-acetil-1H-pirazol-3-carboxilato (8,509 g; 46,70 mmol), EtOH (28 ml) y KRED-130 (cetorreductasa de NADPH, 474 mg). La mezcla se agitó a 30-32 °C durante 5,5 h (seguimiento mediante HPLC) y se dejó enfriar hasta TA. La mezcla se evaporó hasta un volumen menor y el residuo se agitó con tierra de diatomeas y se filtró. El líquido madre se extrajo con 3 * 210 ml de EtOAc y se secó. La solución se filtró a través de sílice (83 g) y se evaporó hasta sequedad para dar 7,40 g del compuesto del título. La pureza óptica fue del 100 % ee.
b) Etil 5-((S)-1-((ferc-butildifenilsilil)oxi)etil)-1 H-pirazol-3-carboxilato
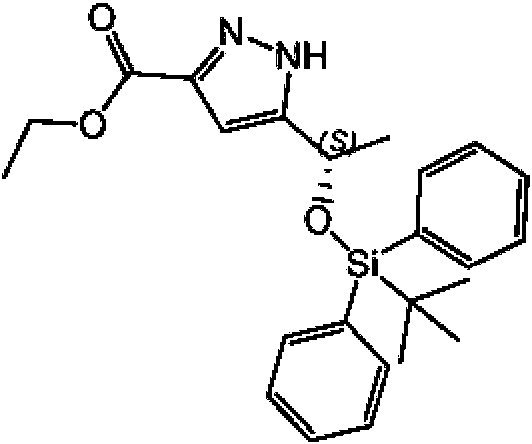
Se añadió difenil-ferc-butilclorosilano (7,48 g, 27,21 mmol) en 26 ml de DMF a una mezcla del compuesto del Ejemplo 3(a) (5,00 g, 27,15 mmol) e imidazol (2,81 g, 41,27 mmol) en DMF (50 ml) a TA. La mezcla se agitó a TA durante 24 h. Se añadieron NaHCO3 acuoso saturado (56 ml) y agua (56 ml) y la mezcla se agitó a TA durante 20 min. La mezcla se extrajo con 2 * 100 ml de EtOAc. Las fases orgánicas combinadas se lavaron con agua (1 * 100 ml, 1 * 50 ml), se secaron (Na2SO4), se filtraron y se concentraron para dar 10,92 g del compuesto del título en bruto.
c) Ácido 5-((S)-1-((ferc-butildifenilsilil)oxi)etil)-1H-pirazol-3-carboxílico

Se añadió NaOH 2 M (ac.) (38,8 ml; 77,5 mmol) a una solución del compuesto del Ejemplo 3(b) (10,9 g, 25,8 mmol) en 66 ml de THF. La mezcla se calentó hasta la temperatura de reflujo. Se continuó el calentamiento durante 2,5 horas y se retiró el THF al vacío. Se añadieron agua (40 ml) y EtOAc (110 ml). Se obtuvo una solución transparente después de la adición de más agua (10 ml). Las capas se separaron y la fase acuosa se extrajo con 100 ml de EtOAc. Las fases orgánicas combinadas se secaron (Na2SO4), se filtraron y se concentraron para dar 9,8 g del compuesto del título.
d) 5-((S)-1-((íerc-butildifenilsilil)oxi)etil)-N-((S)-1-(3-(3-cloro-4-cianofenil)-1 H-pirazol-1-il)propan-2-il)-1 H-pirazol-3-carboxamida

En una atmósfera de nitrógeno, se añadieron HBTU (0,84 g; 2,22 mmol), EDCI * HCl (3,26 g; 17,02 mmol) y (S)-4-(1-(2-aminopropil)-1H-pirazol-3-il)-2-clorobenzonitrilo (3,86 g; 14,80 mmol) a una mezcla del compuesto en bruto del Ejemplo 3(c) (8,68 g; pureza del 77,4 % del área) y DlPEA (2,20 g; 17,02 mmol) en DCM (50 ml). La mezcla se agitó a TA durante 46 h (se añadieron 6 ml de DCM después de 20 h). La mezcla se lavó con 3 * 20 ml de agua, se secó (Na2SO4), se filtró y se concentró para dar 13,7 g del compuesto del título en bruto.
e) N-((S)-1-(3-(3-cloro-4-cianofenil)-1H-pirazol-1-il)propan-2-il)-5-((S)-1-hidroxietil)-1H-pirazol-3-carboxamida (la) Se añadió hidrato de TBAF (Bu4NF * 3 H2O; 2,34 g; 7,40 mmol) en 10 ml de THF a la solución del compuesto del Ejemplo 3(d) (9,43 g; 14,79 mmol) en THF (94 ml) a 0 °C en una atmósfera de nitrógeno. Se continuó la agitación a TA durante 21,5 h y la mezcla se concentró. Se añadió DCM (94 ml) al residuo y la solución se lavó con 3 * 50 ml de agua, se secó (Na2SO4), se filtró y se concentró. El producto en bruto se purificó mediante cromatografía ultrarrápida (EtOAc/n-heptano) para dar 2,1 g del compuesto del título. RMN en 1H (400 MHz; d6-DMSO; aprox.
27 °C (300 °K)): tautómero mayor (~85 %): 81,11 (d, 3H), 1,39 (d, 3H), 4,24-4,40 (m, 2H), 4,40-4,50 (m, 1H), 6,41 (s, 1H), 6,93 (d, 1H), 7,77-7,82 (m, 1H), 7,88-8,01 (m, 2H), 8,08 (s, 1H), 8,19 (d, 1H), 13,02 (s ancho, 1H). Tautómero menor (~15 %): 81,07-1,19 (m, 3H), 1,32-1,41 (m, 3H), 4,24-4,40 (m, 2H), 4,40-4,50 (m, 1H), 6,80 (s ancho, 1H), 6,91-6,94 (m, 1H), 7,77-7,82 (m, 1H), 7,88-8,01 (m, 2H), 8,05-8,09 (m, 1H), 8,31 (d, 1H), 13,10 (s ancho, 1H).
Ejemplo 4. Cristalización de N-((S)-1-(3-(3-cloro-4-cianofenil)-1H-pirazol-1-il)propan-2-il)-5-((S)-1-hidroxietil)-1H-pirazol-3-carboxamida (la)
Se mezcló N-((S)-1-(3-(3-cloro-4-cianofenil)-1H-pirazol-1-il)propan-2-il)-5-((S)-1-hidroxietil)-1H-pirazol-3-carboxamida (la) (5,00 g; 12,54 mmol) con 47,5 ml de ACN y 2,5 ml de agua. La mezcla se calentó hasta que el compuesto (la) se disolvió por completo. La solución se dejó enfriar lentamente hasta TA para formar un precipitado. A continuación, la mezcla se enfrió nuevamente hasta 0 °C y se mantuvo a esta temperatura durante 30 min. La mezcla se filtró y el precipitado se secó al vacío para obtener 4,50 g del compuesto del título cristalino, que se usó en el estudio de difracción de rayos X.
Ejemplo 5. Estudio de difracción de rayos X del compuesto cristalino (la)
La forma cristalina del compuesto (la) obtenido en el Ejemplo 4 se analizó mediante el método de difracción de rayos X en polvo, tal como se describe en el Ejemplo 2. La forma cristalina se caracterizó mediante un patrón de difracción
de rayos X en polvo, tal como se muestra en la Figura 2, y presentado picos característicos a aproximadamente los siguientes valores 2-theta:

Ejemplo 6. Síntesis de N-((S)-1-(3-(3-doro-4-cianofenil)-1H-pirazol-1-il)-propan-2-il)-5-((R)-1-hidroxietil)-1H-pirazol-3-carboxamida (Ib)
a) Etil-5-((R)-1-hidroxietil)-1H-pirazol-3-carboxilato

Se preparó un tampón de dihidrógeno fosfato de potasio (Solución I) mediante la disolución de dihidrógeno fosfato de potasio (11,350 g, 54,89 mmol) en agua (333 ml) y el ajuste del pH de la solución a 7,0 mediante la adición de una solución de NaOH 5 M. Se añadieron MgSO4 x 7 H2O (1,650 g), sal monosódica de NAD (0,500 g), D(+)-glucosa (10,880 g) y polvo liofilizado de enzima optimizada CDX-901 (0,200 g) a la Solución I. A esta solución (Solución II) se añadieron KRED-NADH-110 (0,467 g), etil-5-acetil-1H-pirazol-3-carboxilato (10,00 g; 54,89 mmol) y 2-metiltetrahidrofurao (16 ml). La mezcla se agitó a 30 °C durante 11 h y se dejó enfriar hasta TA durante la noche. El pH de la mezcla se mantuvo en 7 mediante la adición de una solución de NaOH 5 M. La mezcla se evaporó a un volumen más pequeño. El residuo de la evaporación se agitó durante 10 min con tierra de diatomeas (40 g) y carbón activado (0,54 g) y se filtró. El material del filtro se lavó con agua (40 ml) y los lavados se combinaron con el filtrado. Las capas se separaron y la fase acuosa se extrajo con EtOAc (450 ml y 2 * 270 ml). Las fases orgánicas combinadas se secaron sobre Na2SO4, se filtraron y se evaporaron hasta sequedad para dar 9,85 g del compuesto del título (100 % ee).
b) Etil-5-((R)-1-((ferc-butildifenilsilil)oxi)etil)-1 H-pirazol-3-carboxilato

Se añadió imidazol (5,32 g; 78,08 mmol) a una solución de DCM (67 ml) del compuesto del Ejemplo 6(a) (9,85 g;
53,48). La mezcla se agitó hasta que todo el reactivo se disolvió y se añadió íerc-butildifenil clorosilano (13,21 ml;
50,80 mmol) a la mezcla. La mezcla se agitó durante 1,5 h, se añadieron 70 ml de agua y se continuó la agitación durante 15 min. Las capas se separaron y la fase orgánica se lavó con 2 * 70 ml de agua y se secó sobre Na2SO4, se filtró y se concentró para dar 22,07 g del compuesto del título en bruto.
c) Ácido 5-((R)-1-((ferc-butildifenilsilil)oxi)etil)-1H-pirazol-3-carboxílico

El compuesto del Ejemplo 6(b) (11,3 g; 26,74 mmol; rendimiento teórico de la etapa anterior) se disolvió en 34 ml de THF y se añadieron 50 ml de NaOH 2 M (ac.). La mezcla se calentó a temperatura de reflujo durante 70 min.
La mezcla se extrajo con 2 * 55 ml de EtOAc y las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron. El residuo de la evaporación se trituró en 250 ml de nheptano, se filtró y se secó para dar 17,58 g del compuesto del título en bruto.
d) 5-((R)-1-((íerc-butildifenilsilil)oxi)etil)-N-((S)-1-(3-(3-cloro-4-cianofenil)-1 H-pirazol-1-il)propan-2-il)-1 H-pirazol-3-carboxamida

Una mezcla del compuesto del Ejemplo 6(c) (11,14 g; 26,75 mmol; rendimiento teórico de la etapa anterior), 91 ml de DCM, HBTU (1,52 g; 4,01 mmol), EDCI * HCl (5,90 g; 30,76 mmol), (S)-4-(1-(2-aminopropil)-1H-pirazol-3-il)-2-clorobenzonitrilo (6,97 g; 26,75 mmol) y DIPEA (3,98 g; 30,76 mmol) se agitó a Ta durante 3 h y a 30 °C durante 22 h. La mezcla se lavó con 2 * 90 ml de HCl 0,5 M y 4 x 90 ml de agua, se secó sobre Na2SO4, se filtró y se concentró. El producto en bruto se purificó mediante cromatografía en columna ultrarrápida (n-heptano-EtOAc) para dar 16,97 g del compuesto del título.
e) N-((S)-1-(3-(3-cloro-4-cianofenil)-1H-pirazol-1-il)propan-2-il)-5-((R)-1-hidroxietil)-1H-pirazol-3-carboxamida (Ib) Una mezcla del compuesto del Ejemplo 6(d) (6,09 g; 9,56 mmol), 61 ml de THF y TBAf se agitó a 40 °C durante 6,5 h. La mezcla se concentró y se añadieron 61 ml de EtOAc al residuo de la evaporación. La solución se lavó con 2 * 50 ml de HCl 0,5 M y 4 * 50 ml de agua, se secó sobre Na2SO4, se filtró y se concentró. El producto en bruto se purificó mediante cromatografía en columna ultrarrápida (n-heptano-EtOAc) para dar 1,71 g del compuesto del título. RMN en 1H (400 MHz; d6-DMSO; aprox. 27 °C (300 °K)): tautómero mayor (~85 %): 81,10 (d, 3H), 1,38 (d, 3H), 4,14-4,57 (m, 2H), 5,42 (d, 1H), 6,39 (s, 1H), 6,86-6,98 (m, 1H), 7,74-7,84 (m, 1H), 7,86-8,02 (m, 2H), 8,08 (s, 1H), 8,21 (d, 1H), 13,04 (s ancho, 1H). Tautómero menor (~15 %): 80,95-1,24 (m, 3H), 1,25-1,50 (m, 3H), 4,14
4,57 (m, 2H), 4,60-4,90 (m, 1H), 5,08 (d, 1H), 6,78 (s ancho, 1H), 6,86-6,98 (m, 1H), 7,77-7,84 (m, 1H), 7,86-8,02 (m, 2H), 8,02-8,12 (m, 1H), 8,32 (d, 1H), 13,11 (s ancho, 1H).
Ejemplo 7. Cristalización de N-((S)-1-(3-(3-cloro-4-cianofenil)-1H-pirazol-1-il)propan-2-il)-5-((R)-1-hidroxietil)-1H-pirazol-3-carboxamida (Ib)
Se mezcló N-((S)-1-(3-(3-doro-4-danofenil)-1H-pirazol-1-il)propan-2-il)-5-((R)-1-hidroxietil)-1H-pirazol-3-carboxamida (lb) (3,7 g; 9,28 mmol) con 70 ml de ACN y 3,5 ml de agua. La mezcla se calentó hasta la temperatura de reflujo hasta que el compuesto (lb) se disolvió por completo. La solución se dejó enfriar lentamente. La mezcla se filtró a 50 °C para obtener 6,3 mg del precipitado. El líquido madre se enfrió hasta 41 °C y se filtró nuevamente para obtener 20,7 mg del precipitado. A continuación, el líquido madre obtenido se enfrió hasta 36 °C y se filtró para obtener 173 mg del precipitado. El líquido madre final se enfrió hasta TA, se agitó durante la noche, se enfrió hasta 0 °C, se filtró, se lavó con ACN:agua en frío (1:1) y se secó para obtener 2,71 g del precipitado. Se comprobó la pureza óptica de los precipitados y se usó el último precipitado del compuesto del título cristalino (pureza óptica del 100 %) en el estudio de difracción de rayos X.
Ejemplo 8. Estudio de difracción de rayos X del compuesto cristalino (lb)
La forma cristalina del compuesto (lb) obtenido en el Ejemplo 7 se analizó mediante el método de difracción de rayos X en polvo, tal como se describe en el Ejemplo 2. La forma cristalina se caracterizó mediante un patrón de difracción de rayos X en polvo, tal como se muestra en la Figura 3, y presentado picos característicos a aproximadamente los siguientes valores 2-theta:

Ejemplo 9. Síntesis de etil-5-((S)1-hidroxietil)-1 H-pirazol-3-carboxilato

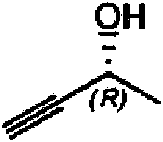
Se añadieron trifluorometanosulfonato de zinc (0,259 g; 0,713 mmol) y (S)-(-)-3-butin-2-ol (0,25 g; 3,57 mmol) a 0,75 ml (5,35 mmol) de Et3N en una atmósfera de nitrógeno. Se añadió lentamente diazoacetato de etilo (0,45 ml; 4,28 mmol) y la mezcla se calentó a 100 °C durante 2 h. La mezcla se enfrió hasta TA y se añadieron 5 ml de agua. La mezcla se lavó con 15 ml de DCM, se añadieron 5 ml de agua y se separaron las fases. La fase acuosa se lavó dos veces con DCM, todas las capas orgánicas se combinaron, se secaron con filtración por separador de fases y se evaporaron hasta sequedad para dar 0,523 g del material en bruto. El producto se purificó mediante cromatografía en columna de fase normal (MeOH:DCM al 0-5 %) para dar 0,165 mg del compuesto del título. RMN en 1H (400 MHz; d6-DMSO; temperatura a aprox. 27 °C (+300 °K)): tautómero 1 (mayor al 77 %): 81,28 (t, 3H), 1,39 (d, 3H), 4,20-4,28 (m, 2H), (d, 1H), 4,75-4,85 (m, 1H), 5,43 (d ancho, 1H), 6,54 (s ancho, 1H), 13,28 (s ancho, 1H). Tautómero 2 (menor al 23 %): 81,28 (t, 3H), 1,39 (d, 3H), 4,20-4,28 (m, 2H), 4,66-4,85 (m, 1H), 5,04-5,15 (s ancho, 1H), 6,71 (s ancho, 1H), 13,60 (s ancho, 1H).
Ejemplo 10. Etil-5-((R)-1-hidroxietil)-1H-pirazol-3-carboxilato

Se añadieron trifluorometanosulfonato de zinc (1,037 g; 2,85 mmol) y (R)-(+)-3-butin-2-ol (1,00 g; 14,27 mmol) a 2,98 ml (21,40 mmol) de Et3N en una atmósfera de nitrógeno. Se añadió lentamente diazoacetato de etilo (1,80 ml; 21,40 mmol) y, a continuación, se sometió a reflujo durante 3 h. La mezcla se enfrió hasta TA y se añadieron 45 ml de agua. La mezcla se extrajo con 3 * 50 ml de DCM, las capas orgánicas se combinaron, se secaron con filtración por separador de fases y se evaporaron hasta sequedad para dar 2,503 g del material en bruto, que se purificó mediante cromatografía en columna de fase normal (MeOH:DCM al 0-10 %) para dar 0,671 mg del compuesto del título. RMN en 1H (400 MHz; d6-DMSO; temperatura a aprox. 27 °C (+300 °K)): tautómero 1 (mayor al 78 %): 81,28 (t, 3H), 1,39 (d, 3H), 4,18-4,35 (m, 2H), (d, 1H), 4,75-4,85 (m, 1H), 5,42 (d ancho, 1H), 6,54 (s, 1H), 13,29 (s ancho, 1H). Tautómero 2 (menor al 22 %): 81,28 (t, 3H), 1,39 (d, 3H), 4,18-4,35 (m, 2H), 4,66-4,85 (m, 1H), 5,09 (s ancho, 1H), 6,71 (s ancho, 1H), 13,61 (s ancho, 1H).
Abreviaturas:
ACN: acetonitrilo
DCM: diclorometano
DIPEA: N,N-diisopropil-etil amina
DMF: dimetilformamida
DMSO: dimetilsulfóxido
EDCI * HCI: clorhidrato de N-(3-dimetilaminopropil)-N-etilcarbodiimida
EtOAc: acetato de etilo
EtOH: etanol
HBTU: o-benzotriazol-N,N,N',N'-tetrametil-uroniohexafluoro-fosfato
KRED: cetorreductasa (enzima)
TA: temperatura ambiente
TFA: ácido trifluoroacético.
Claims (11)
1. Forma cristalina I' de N-((S)-1-(3-(3-cloro-4-cianofenil)-1H-pirazol-1-il)propan-2-il)-5-((S)-1-hidroxietil)-1H-pirazol-3-carboxamida (la), que tiene un patrón de difracción de rayos X en polvo que comprende picos característicos a 9,3, 15,7, 17,0, 24,1 y 25,1 ± 0,15 grados 2-theta medidos usando un tubo de rayos X llenado de Cu.
2. Forma cristalina I' de N-((S)-1-(3-(3-cloro-4-cianofenil)-1H-pirazol-1-il)propan-2-il)-5-((S)-1-hidroxietil)-1H-pirazol-3-carboxamida (la) de acuerdo con la reivindicación 1, que tiene un patrón de difracción de rayos X en polvo que comprende picos característicos a 9,3, 11,4, 11,5, 13,6, 14,7, 14,9, 15,7, 16,1, 17,0, 17,7, 18,5, 19,1,20,5, 21,5, 22,1, 22,6, 23,2, 23,6, 24,1, 25,1,26,2 y 27,2 ± 0,15 grados 2-theta medidos usando un tubo de rayos X llenado de Cu.
3. Forma cristalina I" de N-((S)-1-(3-(3-cloro-4-cianofenil)-1H-pirazol-1-il)propan-2-il)-5-((R)-1-hidroxietil)-1H-pirazol-3-carboxamida (lb), que tiene un patrón de difracción de rayos X en polvo que comprende picos característicos a 9,2, 10,9, 15,1, 15,8 y 22,1 ± 0,15 grados 2-theta medidos usando un tubo de rayos X llenado de Cu.
4. Forma cristalina I" de N-((S)-1-(3-(3-cloro-4-cianofenil)-1H-pirazol-1-il)propan-2-il)-5-((R)-1-hidroxietil)-1H-pirazol-3-carboxamida (lb) de acuerdo con la reivindicación 3, que tiene un patrón de difracción de rayos X en polvo que comprende picos característicos a 7,9, 9,2, 10,9, 13,2, 14,8, 15,1, 15,5, 15,8, 16,9, 18,4, 20,2, 20,5, 21,8, 22,1 y 24,3 ± 0,15 grados 2-theta medidos usando un tubo de rayos X llenado de Cu.
5. Un proceso para la preparación de la forma cristalina I' o I" de N-((S)-1-(3-(3-cloro-4-cianofenil)-1H-pirazol-1-il)propan-2-il)-5-((S)-1-hidroxietil)-1H-pirazol-3-carboxamida (la) o N-((S)-1-(3-(3-cloro-4-danofenil)-1H-pirazol-1-il)propan-2-il)-5-((R)-1-hidroxietil)-1H-pirazol-3-carboxamida (lb), respectivamente, que comprende
a) mezclar N-((S)-1-(3-(3-cloro-4-cianofenil)-1H-pirazol-1-il)propan-2-il)-5-((S)-1-hidroxietil)-1H-pirazol-3-carboxamida (la) o N-((S)-1-(3-(3-cloro-4-cianofenil)-1H-pirazol-1-il)propan-2-il)-5-((R)-1-hidroxietil)-1H-pirazol-3-carboxamida (lb) con una mezcla de acetonitrilo y agua;
b) calentar la mezcla de la Etapa a) para formar una solución;
c) enfriar la solución de la Etapa b) hasta aproximadamente 0-50 °C; y
d) aislar la forma cristalina.
6. El proceso de acuerdo con la reivindicación 5, en donde la mezcla de acetonitrilo:agua tiene una relación en volumen de 85:15 a 99:1.
7. El proceso de acuerdo con la reivindicación 6, en donde la mezcla de acetonitrilo:agua tiene una relación en volumen de 90:10 a 98:2.
8. El proceso de acuerdo con la reivindicación 7, en donde la mezcla de acetonitrilo:agua tiene una relación en volumen de aproximadamente 95:5.
9. El proceso de acuerdo con cualquiera de las reivindicaciones 5 a 8, en donde la Etapa c) de enfriamiento se produce durante 5 a 24 horas.
10. El proceso de acuerdo con la reivindicación 9, en donde la Etapa c) de enfriamiento se produce durante 6 a 12 horas.
11. El proceso de acuerdo con cualquiera de las reivindicaciones 5 a 10, en donde la forma cristalina aislada se seca al vacío a una temperatura de 40 °C a 60 °C.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FI20150033 | 2015-01-30 | ||
| PCT/FI2016/050054 WO2016120530A1 (en) | 2015-01-30 | 2016-01-28 | A carboxamide derivative and its diastereomers in stable crystalline form |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2917549T3 true ES2917549T3 (es) | 2022-07-08 |
Family
ID=55349869
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16704029T Active ES2917549T3 (es) | 2015-01-30 | 2016-01-28 | Un derivado de carboxamida y sus diastereoisómeros en forma cristalina estable |
Country Status (13)
| Country | Link |
|---|---|
| US (4) | US10010530B2 (es) |
| EP (1) | EP3250554B1 (es) |
| JP (2) | JP7283861B2 (es) |
| DK (1) | DK3250554T3 (es) |
| ES (1) | ES2917549T3 (es) |
| HR (1) | HRP20220998T1 (es) |
| HU (1) | HUE058986T2 (es) |
| LT (1) | LT3250554T (es) |
| PL (1) | PL3250554T3 (es) |
| PT (1) | PT3250554T (es) |
| RS (1) | RS63477B1 (es) |
| SI (1) | SI3250554T1 (es) |
| WO (1) | WO2016120530A1 (es) |
Families Citing this family (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018036558A1 (zh) * | 2016-08-26 | 2018-03-01 | 苏州科睿思制药有限公司 | 一种雄激素受体拮抗剂药物的晶型及其制备方法和用途 |
| CN108218908A (zh) * | 2016-12-15 | 2018-06-29 | 连云港润众制药有限公司 | 一种新型雄激素受体拮抗剂的制备方法 |
| WO2018162793A1 (en) | 2017-03-07 | 2018-09-13 | Orion Corporation | Manufacture of a crystalline pharmaceutical product |
| CN116396220A (zh) * | 2017-08-09 | 2023-07-07 | 杭州领业医药科技有限公司 | Odm-201晶型及其制备方法和药物组合物 |
| CN107602471B (zh) * | 2017-09-22 | 2021-04-27 | 成都恒汇化成医药科技有限公司 | 一种达罗鲁胺的晶型制备方法 |
| US20210087175A1 (en) * | 2018-02-27 | 2021-03-25 | Sandoz Ag | Crystalline Form II of Darolutamide |
| WO2021001603A1 (en) | 2019-07-02 | 2021-01-07 | Orion Corporation | Pharmaceutical composition of darolutamide |
| CN110590668A (zh) * | 2019-07-17 | 2019-12-20 | 江苏君若医药有限公司 | N-[(1s)-2-[3-(3-氯-4-氰基苯基)-1h-吡唑-1-基]-1-甲基乙基]-5-(1-羟基乙基)- 1h-吡唑-3-甲酰胺的制备方法 |
| CN110452123B (zh) * | 2019-08-30 | 2021-10-29 | 南京师范大学 | 一步构建c-c、c-n、c-s键的合成方法 |
| CN111116476A (zh) * | 2019-12-27 | 2020-05-08 | 武汉九州钰民医药科技有限公司 | 制备抗肿瘤药物多拉米胺的方法 |
| WO2022036782A1 (zh) * | 2020-08-18 | 2022-02-24 | 拜耳消费者护理股份有限公司 | 一种雄激素受体拮抗剂药物的晶型csvi及其制备方法和用途 |
| WO2022049075A1 (en) | 2020-09-02 | 2022-03-10 | Bend Research, Inc. | Amorphous solid dispersion of darolutamide |
| WO2022144926A1 (en) * | 2020-12-31 | 2022-07-07 | Msn Laboratories Private Limited, R&D Center | Process for the preparation of amorphous n-{(2s)-1-[3-(3-chloro-4-cyanophenyl)-1h-pyrazol-1-yl]propan-2-yl}-5-(1-hydroxyethyl)-1h-pyrazole-3-carboxamide |
| MX2024009980A (es) | 2022-02-28 | 2024-08-26 | Quim Sintetica S A | Forma cristalina de darolutamida. |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AR078793A1 (es) * | 2009-10-27 | 2011-12-07 | Orion Corp | Derivados de carboxamidas no esteroidales y acil hidrazona moduladores de receptores androgenicos de tejido selectivo (sarm), composiciones farmaceuticas que los contienen y uso de los mismos en el tratamiento del cancer de prostata entre otros |
| MX360611B (es) | 2011-04-21 | 2018-11-09 | Orion Corp | Carboxamidas que modulan el receptor de andrógeno. |
-
2016
- 2016-01-28 PT PT167040294T patent/PT3250554T/pt unknown
- 2016-01-28 HU HUE16704029A patent/HUE058986T2/hu unknown
- 2016-01-28 JP JP2017540225A patent/JP7283861B2/ja active Active
- 2016-01-28 US US15/547,193 patent/US10010530B2/en active Active
- 2016-01-28 ES ES16704029T patent/ES2917549T3/es active Active
- 2016-01-28 SI SI201631571T patent/SI3250554T1/sl unknown
- 2016-01-28 DK DK16704029.4T patent/DK3250554T3/da active
- 2016-01-28 HR HRP20220998TT patent/HRP20220998T1/hr unknown
- 2016-01-28 LT LTEPPCT/FI2016/050054T patent/LT3250554T/lt unknown
- 2016-01-28 PL PL16704029.4T patent/PL3250554T3/pl unknown
- 2016-01-28 RS RS20220737A patent/RS63477B1/sr unknown
- 2016-01-28 EP EP16704029.4A patent/EP3250554B1/en active Active
- 2016-01-28 WO PCT/FI2016/050054 patent/WO2016120530A1/en active Application Filing
-
2018
- 2018-06-04 US US15/997,040 patent/US10376494B2/en active Active
- 2018-06-04 US US15/997,028 patent/US10383853B2/en active Active
-
2019
- 2019-07-01 US US16/458,435 patent/US10835515B2/en active Active
-
2020
- 2020-10-22 JP JP2020177607A patent/JP2021020935A/ja active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| HRP20220998T1 (hr) | 2022-11-11 |
| EP3250554B1 (en) | 2022-05-18 |
| US20180008577A1 (en) | 2018-01-11 |
| US20190321333A1 (en) | 2019-10-24 |
| WO2016120530A1 (en) | 2016-08-04 |
| US10010530B2 (en) | 2018-07-03 |
| US20180280354A1 (en) | 2018-10-04 |
| EP3250554A1 (en) | 2017-12-06 |
| JP2021020935A (ja) | 2021-02-18 |
| SI3250554T1 (sl) | 2022-09-30 |
| JP2018503662A (ja) | 2018-02-08 |
| LT3250554T (lt) | 2022-06-10 |
| JP7283861B2 (ja) | 2023-05-30 |
| US10376494B2 (en) | 2019-08-13 |
| US20180280355A1 (en) | 2018-10-04 |
| PT3250554T (pt) | 2022-06-02 |
| PL3250554T3 (pl) | 2022-09-19 |
| US10835515B2 (en) | 2020-11-17 |
| RS63477B1 (sr) | 2022-08-31 |
| HUE058986T2 (hu) | 2022-09-28 |
| DK3250554T3 (da) | 2022-06-27 |
| US10383853B2 (en) | 2019-08-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2917549T3 (es) | Un derivado de carboxamida y sus diastereoisómeros en forma cristalina estable | |
| US8846896B2 (en) | Methods of preparing substituted nucleotide analogs | |
| CA2863772C (en) | Process for preparing tiotropium bromide | |
| ES2738211T3 (es) | Procedimiento mejorado de preparación de profármacos a base de duocarmicina | |
| US9233963B2 (en) | Method for preparing meropenem using zinc powder | |
| CA3030555A1 (en) | Intermediates in processes for the preparation of 4-alkoxy-3-(acyl or alkyl)oxypicolinamides_____________ | |
| CA3082499A1 (en) | Quinazolinone compound and application thereof | |
| EP2151442A2 (en) | Process for preparing temozolomide | |
| EP3507282B1 (en) | Process for preparing indole carboxamide compounds | |
| ES2919330T3 (es) | Proceso para preparar temozolomida y un compuesto intermedio | |
| JP4294121B2 (ja) | ピリドンカルボン酸誘導体の製造方法およびその中間体 | |
| JPWO2016121777A1 (ja) | ピラジンカルボキサミド化合物の製造方法及びその合成中間体 | |
| EP2643308B1 (en) | Process for the preparation of taurolidine and its intermediates thereof | |
| EP3986400B1 (en) | Processes and intermediates for producing diazaspiro lactam compounds | |
| JP2009525964A (ja) | 6,7−ジヒドロ−5H−イミダゾ[1,2−a]イミダゾール−3−スルホン酸アミドの合成 | |
| EP3154942A1 (en) | Preparation of piperidine-4-carbothioamide | |
| AU2011311847A1 (en) | Method of preparation of antiviral compounds and useful intermediates thereof | |
| KR101184433B1 (ko) | 결정형 올메사탄 실렉세틸의 제조방법 | |
| IT201900009777A1 (it) | Processo per la sintesi di lofexidina | |
| CN117659032A (zh) | 海鞘素类衍生物的中间体及其的合成方法 | |
| JP2009508922A (ja) | 4−アミノピラゾール誘導体の製造方法 | |
| KR20080107545A (ko) | 프란루카스트 또는 이의 약학적으로 허용가능한 염의제조방법 | |
| WO2016071380A1 (en) | Synthesis of pi3k inhibitor and salts thereof |