ES2848122T3 - Fosforamidita de agrupación de GalNAc - Google Patents
Fosforamidita de agrupación de GalNAc Download PDFInfo
- Publication number
- ES2848122T3 ES2848122T3 ES16797831T ES16797831T ES2848122T3 ES 2848122 T3 ES2848122 T3 ES 2848122T3 ES 16797831 T ES16797831 T ES 16797831T ES 16797831 T ES16797831 T ES 16797831T ES 2848122 T3 ES2848122 T3 ES 2848122T3
- Authority
- ES
- Spain
- Prior art keywords
- galnac
- formula
- ethoxy
- comp
- cluster
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 CC(C)(CC(C)(C)N)CO* Chemical compound CC(C)(CC(C)(C)N)CO* 0.000 description 5
- JQDDWLPYLNOSNU-YLXYFGKXSA-N CC(O)OC[C@H]([C@@H]1OC(C)=O)OC(C)CC1OC(C)=[O]=[IH]=CCC(C)=O Chemical compound CC(O)OC[C@H]([C@@H]1OC(C)=O)OC(C)CC1OC(C)=[O]=[IH]=CCC(C)=O JQDDWLPYLNOSNU-YLXYFGKXSA-N 0.000 description 1
- KAQNWSPSALZVGB-UHFFFAOYSA-N FC(CCCCCCOCc1ccccc1)(F)F Chemical compound FC(CCCCCCOCc1ccccc1)(F)F KAQNWSPSALZVGB-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H15/00—Compounds containing hydrocarbon or substituted hydrocarbon radicals directly attached to hetero atoms of saccharide radicals
- C07H15/02—Acyclic radicals, not substituted by cyclic structures
- C07H15/04—Acyclic radicals, not substituted by cyclic structures attached to an oxygen atom of the saccharide radical
- C07H15/08—Polyoxyalkylene derivatives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H1/00—Processes for the preparation of sugar derivatives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
- C07H21/04—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids with deoxyribosyl as saccharide radical
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Molecular Biology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- Engineering & Computer Science (AREA)
- Genetics & Genomics (AREA)
- General Health & Medical Sciences (AREA)
- Biotechnology (AREA)
- Crystallography & Structural Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Saccharide Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Derivados de fosforamidita de GalNAc de fórmula I **(Ver fórmula)** en la que R1 es un grupo acilo, n es un número entero de 0 a 10 y m es un número entero de 0 a 20, enantiómeros y/o isómeros ópticos correspondientes de los mismos.
Description
DESCRIPCIÓN
Fosforamidita de agrupación de GalNAc
La invención se refiere a derivados de fosforamidita de GalNAc de fórmula I

en la que R1 es un grupo protector de hidroxi como se define a continuación, n es un número entero de 0 a 10 y m es un número entero de 0 a 20, enantiómeros y/o isómeros ópticos correspondientes de los mismos.
Las fosforamiditas de GalNAc de fórmula I portan el resto GalNAc que es el resto de direccionamiento de los conjugados que comprenden el resto GalNAc. El resto GalNAc, debido a su afinidad por el receptor de asialoglucoproteína que está localizado en los hepatocitos, permite el suministro funcional de conjugados oligonucleotídicos a los hepatocitos. Dichos conjugados con agrupaciones de GalNAc tienen el potencial de actuar como moduladores farmacocinéticos y, por lo tanto, de ser compuestos terapéuticamente valiosos como se describe, por ejemplo, en la publicación PCT Wo 2012/083046 o en la publicación de solicitud de patente de EE. UU. US 2011/0207799.
Debido a la combinación única de resto GalNAc y fosforamidita, la fosforamidita de GalNAc de fórmula I se puede introducir directamente como componente básico conjuntamente con los componentes básicos nucleotídicos en la síntesis de oligonucleótidos en fase sólida. Por lo tanto, se puede evitar una etapa de conjugación separada para introducir el resto GalNAc.
Por lo tanto, el objetivo de la invención fue proporcionar dicho componente básico. Otro objetivo de la invención fue el uso de derivados de fosforamidita de GalNAc de fórmula I para la preparación de conjugados de oligonucleótidos con agrupaciones de GalNAc.
El objetivo se podría conseguir con los nuevos derivados de fosforamidita de GalNAc de fórmula I como se describe anteriormente.
Las siguientes definiciones se exponen para ilustrar y definir el significado y alcance de los diversos términos usados para describir la invención en el presente documento.
Siempre que un carbono quiral esté presente en una estructura química, se entiende que todos los estereoisómeros asociados con ese carbono quiral están englobados por la estructura como estereoisómeros puros, así como mezclas
de los mismos.
El término "alquilo C1-12" indica un grupo hidrocarburo saturado lineal o ramificado monovalente de 1 a 12 átomos de carbono y, en modos de realización más particulares, de 1 a 6 átomos de carbono. Los ejemplos de alquilo incluyen metilo, etilo, propilo, isopropilo, n-butilo, isobutilo, sec-butilo, tere-butilo y pentilo y hexilo con sus isómeros.
El término "acilo" indica un grupo carbonilo que está unido a un grupo alquilo. El término representa en particular un grupo alquilcarbonilo C1-12, más en particular un grupo alquilcarbonilo C1-6 que está sustituido opcionalmente con alquilo C1-6 o sustituido opcionalmente con fenilo. Ejemplos de grupos acilo son acetilo, pivaloílo o benzoílo. Sustituciones opcionales para el grupo fenilo son halógeno, tal como cloro, bromo o yodo, o un grupo alquilo C1-6 como se define anteriormente.
El término "grupo protector de hidroxi" indica grupos que están destinados a proteger un grupo hidroxi e incluyen grupos formadores de éster y éter, en particular grupos tetrahidropiranilo, acilo (por ejemplo, benzoílo, acetilo, carbamoílo), bencilo y éteres silílicos (por ejemplo, TBS, TBDPS). Otros ejemplos de estos grupos se encuentran en T. W. Greene y P. G. M. Wuts, "Protective Groups in Organic Synthesis", 2.a ed., John Wiley & Sons, Inc., Nueva York, NY, 1991, capítulos 2-3; E. Haslam, "Protective Groups in Organic Chemistry", J. G. W. McOmie, Ed., Plenum Press, Nueva York, NY, 1973, capítulo 5, y T.W. Greene, "Protective Groups in Organic Synthesis", John Wiley and Sons, Nueva York, NY, 1981.
El término "halógeno" representa flúor, cloro, bromo o yodo.
El grupo protector de hidroxi R1 es un grupo acilo, en particular un grupo alquilcarbonilo C 1-12, más en particular un grupo alquilcarbonilo C1-6 que está sustituido opcionalmente con alquilo C1-6 o fenilo. El grupo protector de hidroxi R1 preferente se puede seleccionar de acetilo, pivaloílo o benzoílo, donde el acetilo es el más preferente.
n es preferentemente un número entero de 0 a 5, más preferentemente de 1 a 3, pero lo más preferente es 2. m es preferentemente un número entero de 0 a 10, más preferentemente de 3 a 7, pero lo más preferente es 5. En un modo de realización preferente, los derivados de fosforamidita de GalNAc tienen la fórmula la

en la que R1, n y m son como anteriormente.
Preferentemente, n es 0, 1,2, 3, 4 o 5 y m es 0, 1,2, 3, 4, 5, 6, 7, 8, 8 o 10, más preferentemente R1 es acetilo, pivaloílo o benzoílo; n es 1, 2 o 3 y m es 3, 4, 5, 6 o 7.
En un modo de realización incluso más preferente, el derivado de fosforamidita de GalNAc tiene la fórmula Ib

El procedimiento para la preparación de un derivado de fosforamidita de GalNAc de fórmula I comprende a) la reacción de un derivado de ácido de GalNAc de fórmula III

en la que R1 y n son como anteriormente
con una amina de fórmula IV

IV
en la que R3 es un grupo protector de hidroxi y m es como anteriormente, para formar una amida de fórmula V

en la que R1 y R3, n y m son como anteriormente;
5 b) la eliminación del grupo protector de hidroxi R3 para formar la amida de ácido de GalNAc de fórmula VI

en la que R1, n y m son como anteriormente y
c) la reacción de la amida de ácido de GalNAc de fórmula VI con un agente fosforamidante para formar el derivado de fosforamidita de GalNAc de fórmula I.
Etapa a)
La etapa a) comprende la reacción de un derivado de ácido de GalNAc de fórmula III con la amina de fórmula V. El precursor de éster bencílico del derivado de GalNAc de fórmula III se puede preparar de acuerdo con el esquema 1 a continuación. La hidrogenólisis posterior del éster bencílico por medio de una hidrogenación catalítica con hidrógeno en presencia de un catalizador de hidrogenación tal como, por ejemplo, con paladio sobre carbón vegetal proporciona el derivado de GalNAc de fórmula III.

La amina de fórmula V se puede preparar como se ejemplifica para la 6-benciloxihexan-1-amina en el esquema 2 a continuación. La reacción se describe, por ejemplo, en Saneyoshi, Hisao et al., Organic Letters, 16(1), 30-33; 2014 o en Álvarez, M. et al., Anales de Química, Serie C: Química Orgánica y Bioquímica, 83(2), 155-61; 1987 o Álvarez, M. et al., Journal of Medicinal Chemistry, 30(7), 1186-93; 1987. Comenzando a partir de un halogenohexanoximetilbenceno disponible comercialmente, se forma la isoindolin-1,3-diona respectiva con una sal de 1.3- dioxoisoindolina. Las sales de 1,3-dioxoisoindolina adecuadas son las sales alcalinas como la sal de sodio o potasio o las sales de tetraalquilamonio, tal como la sal de tetrabutilamonio. En una etapa posterior, el grupo isoindolin-1.3- diona se puede escindir con hidrazina o con una amina primaria, preferentemente metilamina, para formar la amina libre.

La formación de amida en la etapa a) se realiza en presencia de un agente de acoplamiento peptídico, una base amínica y un disolvente orgánico.
El acoplamiento puede seguir los procedimientos clásicos conocidos por los expertos en la técnica usando un agente de acoplamiento carbodiimídico como DCC (W,W'-diciclohexilcarbodiimida) y un aditivo como HOBt (1-hidroxibenzotriazol), HOSu (W-hidroxisuccinimida), TBTU (tetrafluoroborato de W,W,W',W'-tetrametil-O-[benzotriazol-1-il]uronio), HBTU (hexafluorofosfato de 2-[1H-benzotriazol-1-il]-1,1,3,3-tetrametiluronio) o HOAt (1 -hidroxi-7-azabenzotriazol) y combinaciones comunes de los mismos tales como TBTU/HOBt o HBTU/HOAt.
En un modo de realización preferente, el anhídrido del ácido n-propilfosfónico (T3P) se selecciona como agente de acoplamiento conjuntamente con una base amínica terciaria, como trietilamina o W-etildiisopropilamina, pero preferentemente con W-etildiisopropilamina.
La reacción de acoplamiento tiene lugar normalmente en un disolvente aprótico polar como acetonitrilo o tetrahidrofurano o mezclas de los mismos a temperaturas de reacción en el intervalo de 20 °C a 70 °C, preferentemente en el intervalo de 20 °C a 40 °C.
El producto en bruto de amida de fórmula V se puede aislar por evaporación del disolvente. La purificación adicional del producto se puede conseguir mediante cromatografía de fase inversa preparativa usando una fase estacionaria apolar y una fase móvil polar, tal como acetonitrilo/agua o por SFC (cromatografía de fluidos supercríticos) usando CO2/metanol/etanol/2-propanol.
Etapa b)
La etapa b) requiere la eliminación del grupo protector de hidroxi R3 para formar la amida de ácido de GalNAc de fórmula VI.
Es importante que el grupo protector de hidroxi R3 sea químicamente diferente del grupo protector de hidroxi R1, de modo que las condiciones de eliminación se puedan seleccionar de manera que el grupo protector de hidroxi R3 se escinda mientras que el grupo protector de hidroxi R1 permanezca inalterado.
Un grupo protector de hidroxi R3 adecuado es bencilo, que está sustituido opcionalmente con halógeno o alquilo C 1-6 o es benzhidrilo o tritilo, es decir, un grupo que se puede escindir por hidrogenólisis. De forma alternativa, R3 es sililo como ferc-butildimetilsililo. Los grupos sililo se pueden escindir en presencia de iones fluoruro.
En un modo de realización preferente, R3 es bencilo y la hidrogenólisis es una hidrogenación catalítica con hidrógeno en presencia de un catalizador de hidrogenación adecuado.
El catalizador de hidrogenación adecuado para la eliminación del grupo bencilo es paladio sobre carbono (Pd/C). La reacción se realiza normalmente en presencia de un disolvente aprótico polar como acetonitrilo o tetrahidrofurano o mezclas de los mismos a temperaturas de reacción de entre 0 °C y 40 °C, preferentemente 10 °C y 30 °C y a una presión de hidrógeno de 1 bar a 5 bar.
La amida de ácido de GalNAc de fórmula VI se puede separar retirando por filtración el catalizador y eliminando el disolvente por evaporación.
Etapa c)
La etapa c) requiere la reacción de la amida de ácido de GalNAc de fórmula VI con un agente fosforamidante para formar el derivado de fosforamidita de GalNAc de fórmula I
El agente fosforamidante se puede seleccionar de 2-cianoetil-W,W-di-(2-propil)clorofosforamidita o de 2-cianometil-W,W,W,W-tetra(2-propil)fosforodiamidita.
En el caso de que se use 2-cianometil-W,W,W,W-tetra(2-propil)fosforodiamidita, la reacción se realiza en presencia de un compuesto activador como tetrazol, 5-(etiltio)-1H-tetrazol, 5-(benciltio)-1H-tetrazol o 4,5-dicianoimidazol.
En un modo de realización preferente se elige 2-cianoetil-W,W-di-(2-propil)clorofosforamidita. A continuación, la reacción se realiza en presencia de una amina terciaria tal como trietilamina y un disolvente aprótico polar como acetonitrilo o tetrahidrofurano o mezclas de los mismos a una temperatura de reacción de entre -20 °C y 50 °C, preferentemente en acetonitrilo a una temperatura de reacción de entre 0 °C y 20 °C.
El derivado de fosforamidita de GalNAc de fórmula I se puede separar, por regla general, del clorhidrato de trietilamina por filtración. El filtrado con el producto en bruto se puede usar directamente para la síntesis de nucleótidos en fase sólida sin purificación adicional.
Uso del derivado de fosforamidita de GalNAc de fórmula I para la preparación de conjugados de oligonucleótidos con agrupaciones de GalNAc
En otro modo de realización de la presente invención, los derivados de fosforamidita de GalNAc de fórmula I se pueden usar para la preparación de conjugados de oligonucleótidos con agrupaciones de GalNAc terapéuticamente valiosos. La preparación de conjugados de oligonucleótidos con agrupaciones de GalNAc comprende
a2) la preparación del derivado de fosforamidita de GalNAc de fórmula I como se describe anteriormente;
b2) su empleo en la síntesis de oligonucleótidos en fase sólida conjuntamente con los componentes básicos nucleosídicos deseados en la secuencia deseada para formar un conjugado de oligonucleótidos con agrupaciones de GalNAc deseado unido al soporte sólido y, finalmente, la
c2) escisión del conjugado de oligonucleótidos con agrupaciones de GalNAc del soporte en fase sólida y desprotección
completa.
En un modo de realización preferente se usa el derivado de fosforamidita de GalNAc de fórmula la, preferentemente de fórmula Ib.
El término "oligonucleótido", como se usa en el presente documento, se define como lo entiende en general el experto en la técnica, como una molécula que comprende dos o más nucleósidos unidos covalentemente. Para su uso como un oligonucleótido terapéuticamente valioso, los oligonucleótidos se sintetizan típicamente con una longitud de 7 a 30 nucleótidos. Los oligonucleótidos se preparan normalmente en el laboratorio mediante síntesis química en fase sólida seguida de purificación. Cuando se hace referencia a una secuencia del oligonucleótido, se hace referencia a la secuencia u orden de los restos de nucleobases, o modificaciones de los mismos, de los nucleótidos o nucleósidos unidos covalentemente. El oligonucleótido de la invención es artificial, y se sintetiza químicamente, y típicamente se purifica o se aísla. El oligonucleótido de la invención puede comprender uno o más nucleósidos o nucleótidos modificados. En algunos modos de realización, el oligonucleótido es un oligonucleótido de antisentido.
Los oligonucleótidos pueden consistir en ADN, ARN, ARN modificado o monómeros de nucleósidos de LNA o combinaciones de los mismos. Los monómeros de nucleósidos de LNA son nucleósidos modificados que comprenden un grupo conector (denominado birradical o puente) entre C2' y C4' del anillo glucídico de ribosa de un nucleótido. Estos nucleósidos también se denominan ácido nucleico con puente o ácido nucleico bicíclico (BNA) en la literatura. En un modo de realización no limitante, el oligonucleótido se puede seleccionar del grupo que consiste en:
5'-caGsmCsGstsasasasgsasgsasGsG-3'; (comp. n.° 10)
5'-caGsmCsGstsasasasgsasgsasGsGsT-3'; (comp. n.° 11)
5'-caGsmCsGstsasasasgsasgsAsGsG-3'; (comp. n.° 12)
5'-caGsGsmCsGstsasasasgsasgsasGsG-3'; (comp. n.° 13)
5'-caAsGsmCsgsasasgstsgscsascsAsmCsG-3'; (comp. n.° 14)
5'-caAsGsmCsgsasasgstsgscsascsasCsGsG-3'; (comp. n.° 15)
5'-(5'Br)caGsmCsGstsasasasgsasgsasGsG-3'; (comp. n.° 16)
5'-cagcgtaaagagagg-3'; (comp. n.° 17)
5 -AsAsTsgscstsascsasasasascsCsCsA-3, (comp. n.° 18)
en el que las letras mayúsculas indican unidades beta-D-oxi-LNA; las letras minúsculas indican unidades de ADN; el subíndice "s" indica un enlace fosforotioato; el superíndice m indica una unidad de ADN o beta-D-oxi-LNA que contiene una base 5-metilcitosina, el superíndice 5-Br indica una unidad de ADN que contiene una base 5-bromocitosina. El uso del derivado de fosforamidita de GalNAc de fórmula I como componente básico en la síntesis de oligonucleótidos en fase sólida permite introducir el resto GalNAc conjuntamente con un conector amino adecuado en el extremo 5' del oligonucleótido y formar conjugados de oligonucleótidos con agrupaciones de GalNAc deseados. En un modo de realización no limitante, los conjugados de oligonucleótidos con agrupaciones de GalNAc se pueden seleccionar del grupo que consiste en:
Agrupación de GalNAc-AM-C6-5'-caGsmCsGstsasasasgsasgsasGsG-3'; (comp. n.° 1)
Agrupación de GalNAc-AM-C6-5'-caGsmCsGstsasasasgsasgsasGsGsT-3'; (comp. n.° 2)
Agrupación de GalNAc-AM-C6-5'-caGsmCsGstsasasasgsasgsAsGsG-3'; (comp. n.° 3)
Agrupación de GalNAc-AM-C6-5'-caGsGsmCsGstsasasasgsasgsasGsG-3'; (comp. n.° 4)
Agrupación de GalNAc-AM-C6-5'-caAsGsmCsgsasasgstsgscsascsAsmCsG-3'; (comp. n.° 5)
Agrupación de GalNAc-AM-C6-5'-caAsGsmCsgsasasgstsgscsascsasCsGsG-3'; (comp. n.° 6)
Agrupación de GalNAc-AM-C6-5'-(5'Br)caGsmCsGstsasasasgsasgsasGsG-3'; (comp. n.° 7)
Agrupación de GalNAc-AM-C6-5'-cagcgtaaagagagg-3'; (comp. n.° 8)
Agrupación de GalNAc-AM-C6s-5'-AsAsTsgsCstsasCsasasasasCsCsCsA-3'; (comp. n.° 9)
en el que AM-C6 indica un enlace 6-aminohexil-1-fosfato o 1-tiofosfato de la fórmula.

Después de la síntesis en fase sólida, el conjugado de oligonucleótido con agrupaciones de GalNAc todavía está unido al soporte sólido y todavía porta grupos de protección como el grupo protector de hidroxi R1.
La escisión del soporte y la desprotección pueden tener lugar usando procedimientos conocidos por los expertos en la técnica y descritos en la literatura, tal como en Wincott et al.; Nucl. Acids Res. (1995) 23 (14): 2677-2684. Normalmente, el conjugado de oligonucleótido con agrupaciones de GalNAc se obtiene en forma de una sal adecuada, tal como la sal de amonio o la sal de metal alcalino, como la sal de sodio o potasio.
Los compuestos divulgados en el presente documento tienen una secuencia de nucleobases seleccionada del grupo que consiste en SEQ ID NO: 1, 2, 3, 4 y 5:
SEQ ID NO: 1: cagcgtaaag agagg (comp. 1, 3, 4, 7, 8, 10, 12, 13, 16 y 17)
SEQ ID NO: 2: cagcgtaaag agaggt (comp. 2 y 11)
SEQ ID NO: 3: caagcgaagt gcacacg (comp. 5 y 14)
SEQ ID NO: 4: caagcgaagt gcacacgg (comp. 6 y 15)
SEQ ID NO: 5: aatgctacaa aaccca (comp. 9 y 18)
Ejemplos
Abreviaturas:
AcOH ácido acético
DMAP 4-(dimetilamino)-piridina
DMF W,W-dimetilformamida
EtOH etanol
MeOH metanol
t.a. temperatura ambiente (20-25 °C)
THF tetrahidrofurano
MTBE éter metil-terc-butílico
Síntesis del precursor ácido de GalNAc
Componente básico A:
Ejemplo 1
(2S)-6-(terc-Butoxicarbonilamino)-2-(9H-fluoren-9-ilmetoxicarbonilamino)hexanoato de bencilo
Se suspendieron 234,0 g (500,0 mmol) de ácido (2S)-6-(terc-butoxicarbonilamino)-2-(9H-fluoren-9-ilmetoxicarbonilamino)hexanoico en 500 ml de diclorometano, se añadieron 62,0 ml (600 mmol, 1,2 equiv.) de alcohol bencílico y 3,05 g de DMAP (25,0 mmol, 0,05 equiv.). La solución se enfrió hasta 0-5 °C en el transcurso de 40 min, se añadió gota a gota una solución de 108,0 g (525,0 mmol, 1,05 equiv.) de W,W-diciclohexilcarbodiimida en 500 ml de diclorometano. La suspensión blanca se agitó durante 1 h a 0-5 °C y, a continuación, durante 15 h a temperatura ambiente. La suspensión se filtró sobre un filtro de vidrio G3, la torta de filtración blanca se lavó por partes con un total de 250 ml de diclorometano. El filtrado se evaporó a 650-10 mbar/1 h para obtener un aceite amarillo, que se disolvió en 2,0 l de acetato de etilo, se extrajo con 2,0 l de ácido clorhídrico 0,5 M, 2,0 l de NaHCÜ3 1 M y 1,0 l de salmuera, se evaporó la fase orgánica a sequedad a 40 °C/150-10 mbar/5 h para obtener 291,1 g de (2S)-6-(tercbutoxicarbonilamino)-2-(9H-fluoren-9-ilmetoxicarbonilamino)hexanoato de bencilo en bruto como un sólido blanco con un rendimiento del 104 % y una pureza del 96,4 % (% del área por HPLC; contiene aprox. 5 % de alcohol bencílico). El material se usó en la siguiente etapa sin purificación adicional. EM: m/z = 459,22735 (M-Boc+H)+.
Ejemplo 2
Sal ácida metanosulfónica de (2S)-2-amino-6-(terc-butoxicarbonilamino)hexanoato de bencilo

Se disolvieron 291,1 g (500,0 mmol) de (2S)-6-(terc-butoxicarbonilamino)-2-(9H-fluoren-9-ilmetoxicarbonilamino)hexanoato de bencilo (pureza por HPLC: 95,8 %; contiene aprox. 5 % de alcohol bencílico) en 1,4 l de THF a temperatura ambiente. En 10 minutos, se añadieron 1,04 l de dietilamina (10,0 mol, 20 equiv.), la solución de color amarillo claro se agitó durante 2 h a temperatura ambiente y, a continuación, se evaporó a 40 °C/200-10 mbar, se añadieron 200 ml de acetonitrilo y se evaporó nuevamente para eliminar eficazmente la dietilamina a 40 °C/100-10 mbar/1 h. Finalmente, se obtuvieron 268,1 g de un aceite amarillo, que se disolvió en 2,5 l de acetonitrilo, agitando durante 10 min a temperatura ambiente. Las partículas insolubles se filtraron sobre un filtro de fibra de vidrio y se lavaron con 500 ml de acetonitrilo. El filtrado se trató gota a gota en el transcurso de 10 min con 34,0 ml de ácido metanosulfónico a 20 °C-25 °C. La suspensión blanca formada se agitó durante 17 h a temperatura ambiente y se filtró sobre un filtro de vidrio G3. Los residuos de filtración se lavaron por partes con 500 ml de acetonitrilo. Los cristales blancos se secaron a 40 °C/15 mbar/4 h para obtener 195,8 g de sal ácida metanosulfónica de (2S)-2-amino-6-(tercbutoxicarbonilamino)hexanoato de bencilo en forma de cristales blancos con un rendimiento del 91 % (2 etapas) y un 99,3 % de pureza (% del área por HPLC). EM: m/z = 337,2149 (M+H)+.
Ejemplo 3
(2S)-2-[[(2S)-2,6-bis(terc-Butoxicarbonilamino)hexanoil]amino]-6-(terc-butoxicarbonilamino)hexanoato de bencilo

Se suspendieron 190,0 g (439,0 mmol) de sal acida metanosulfonica de (2S)-2-amino-6-(fercbutoxicarbonilamino)hexanoato de bencilo en 1,9 l de THF a temperatura ambiente. Se añadieron 335 ml (1,98 mol, 4,5 equiv.) de W-etildiisopropilamina, con lo que la temperatura disminuyó ligeramente hasta 15 °C. A continuación, se añadieron 213 g (615 mmol, 1,4 equiv.) de ácido (S)-2,6-bis((ferc-butoxicarbonil)amino)hexanoico y la suspensión blanca se agitó a temperatura ambiente durante 20 min. Se añadieron gota a gota 390 ml de anhídrido del ácido npropilfosfónico (T3P como trímero cíclico al 50 % en acetato de etilo, 659 mmol, 1,5 equiv.) en el transcurso de 20 min a 20-25 °C (enfriado en un baño de agua fría). La solución turbia de color amarillo claro resultante se agitó a temperatura ambiente durante 1,5 h, se transfirió a un embudo de separación, se diluyó con 1,9 l de MTBE y se extrajo con 1,9 l de agua, 1,9 l de ácido clorhídrico 0,5 M, 1,9 l de NaOH 0,5 M, 1,9 l de agua y 1,9 l de salmuera. La fase orgánica separada, aún turbia, se filtró sobre un filtro de fibra de vidrio, el filtro se lavó con 100 ml de MTBE y los filtrados combinados se evaporaron a 40 °C/100 mbar/1 h, se añadió de nuevo 1,0 l de MTBE (para eliminar el agua de forma azeotrópica) y se evaporó a 40 °C/250-10 mbar/1 h para obtener 296,4 g en bruto como un residuo sólido blanco.
El sólido en bruto se trató con 500 ml de acetonitrilo y la solución turbia se calentó a 60-65 °C durante 10 min. La mezcla se enfrió a 20-25 °C, se agitó durante 10 min, se filtró sobre un filtro de fibra de vidrio y se lavó con 50 ml de acetonitrilo. La solución de color amarillo claro se evaporó a 40 °C/100-10 mbar/4 h para obtener 295 g de (2S)-2-[[(2S)-2,6-bis(ferc-butoxicarbonilamino)hexanoil]amino]-6-(ferc-butoxicarbonilamino)hexanoato de bencilo como un sólido blanquecino con un rendimiento del 101 % (pureza por HPLC: 100 %, pureza del diastereómero (SS): 98,6 %) que se usó sin purificación adicional en la siguiente etapa. EM: m/z = 565,3741 (M-Boc+H)+.
Ejemplo 4
Sal ácida trimetanosulfónica de (2S)-6-amino-2-[[(2S)-2,6-diaminohexanoil]amino]hexanoato de bencilo

Se suspendieron 124,0 g (187 mmol) de (2S)-2-[[(2S)-2,6-bis(ferc-butoxicarbonilamino)hexanoil]amino]-6-(fercbutoxicarbonilamino)hexanoato de bencilo en 1,25 l de acetonitrilo. Se añadieron 61,0 ml (935,0 mmol, 5,0 equiv.) de ácido metanosulfónico a 20-25 °C en el transcurso de 10 min (producción de gas). La suspensión anaranjada resultante se calentó en 40 min hasta 55-60 °C y se agitó durante 1 h adicional a 55-60 °C. La emulsión de color anaranjado rojizo se enfrió a temperatura ambiente (la eliminación de Boc se controló mediante 1H-RMN) y se usó sin
purificación adicional en la etapa de ensamblaje A+B, ejemplo 8. EM: m/z = 365,2558 (M+H)+.
Componente básico B:
Ejemplo 5a
2-[2-[2-(2-Hidroxietoxi)etoxi]etoxi]acetato de bencilo

-
Se disolvieron 30,0 g (200,0 mmol) de 2-[2-(2-hidroxietoxi)etoxi]etanol en 50 ml de DMF a 20-25 °C y, a continuación, se añadieron 46,0 ml de metóxido de sodio al 25 % (200,0 mmol, 1,0 equiv.) en metanol. La solución formada se evaporó a 40 °C/50 mbar/0,5 h (eliminación de 40 ml de disolvente), se añadieron nuevamente 50 ml de DMF y se evaporó a 40 °C/20 mbar/0,5 h (eliminación de 15 ml de disolvente). A la suspensión ligeramente gelatinosa se le añadió una solución de 13,9 g de ácido bromoacético (100 mmol, 0,5 equiv.) en 50 ml de DMF a 20-25 °C y la mezcla se agitó durante 6 h. Se añadieron 11,9 ml de bromuro de bencilo (100 mmol, 0,5 equiv.) y la mezcla se agitó durante otras 16 h a 20-25 °C. La mezcla de reacción se trató, a continuación, con 200 ml de salmuera y se extrajo con 200 ml de MTBE. La fase de MTBE separada se extrajo con 200 ml de salmuera, la fase de MTBE separada se secó a continuación con sulfato de sodio anhidro, se filtró y se evaporó a 40 °C/300-10 mbar/1 h para obtener 23,9 g en bruto de 2-[2-[2-(2-hidroxietoxi)etoxi]etoxi]acetato de bencilo.
Después de la cromatografía (600 g de sílice 60 [0,063-0,2 mm], fase móvil: acetato de etilo), se aislaron un total de 7,85 g de 2-[2-[2-(2-hidroxietoxi)etoxi]etoxi]acetato de bencilo como un aceite incoloro con un rendimiento del 13 % y un 99,0 % de pureza (% del área por HPLC). EM: m/z = 299,1517 (M+H)+.
Ejemplo 5b
2-[2-[2-(2-Hidroxietoxi)etoxi]etoxi]acetato de bencilo

Se suspendieron 11,2 g de ferc-butilato de potasio (100,0 mmol, 0,5 equiv.) en 70 ml de 2-metil-2-butanol (ligeramente exotérmica a 35 °C) y, a continuación, se añadieron gota a gota 30,0 g (200,0 mmol) de 2-[2-(2-hidroxietoxi)etoxi]etanol en el transcurso de 5 min. El embudo de decantación se enjuagó con 10 ml de 2-metil-2-butanol (aumento de temperatura hasta 45 °C), la solución se calentó a 60-65 °C, se añadieron 11,6 g (100 mmol, 0,5 equiv.) de cloroacetato de sodio y se agitó durante 16 h a 60-65 °C. A continuación, se añadieron 11,9 ml de bromuro de bencilo (100 mmol, 0,5 equiv.) y la mezcla se agitó durante otras 16 h a 60-65 °C. La mezcla de reacción se enfrió a t.a. y, a continuación, se trató con 50 ml de agua y se extrajo con 80 ml de MTBE y 40 ml de MTBE. La fase combinada de MTBE se lavó con 50 ml de salmuera semisaturada, la fase orgánica se evaporó a 40 °C/300-10 mbar/1 h para obtener 27,0 g en bruto de 2-[2-[2-(2-hidroxietoxi)etoxi]etoxi]acetato de bencilo.
Después de la cromatografía (270 g de sílice 60 [0,063-0,2 mm], fase móvil: comenzar con acetato de etilo/n-heptano 1/1, cuando el producto puro fue visible, la fase móvil se cambió a acetato de etilo al 100 %), se aislaron un total de 11,4 g de 2-[2-[2-(2-hidroxietoxi)etoxi]etoxi]acetato de bencilo como un aceite casi incoloro con un rendimiento del 19 % (38 % a partir del cloroacetato de sodio) y un 99,0 % de pureza (% del área por HPLC)
Ejemplo 6
2-[2-[2-[2-[(2R,3R,4R,5R,6R)-3-Acetamido-4,5-diacetoxi-6-(acetoximetil)tetrahidropiran-2-il]oxietoxi]etoxi]etoxi]acetato de bencilo

Se disolvieron 268,0 g de 2-(2-(2-(2-hidroxietoxi)etoxi)etoxi)acetato de bencilo (900 mol) en 2,4 l de diclorometano. Se añadieron 385,0 g de triacetato de (2S,3R,4R,5R,6R)-3-acetamido-6-(acetoximetil)tetrahidro-2H-piran-2,4,5-triilo (990 mmol, 1,1 equiv.) y 12,0 ml de ácido trifluorometanosulfónico (135 mmol, 0,15 equiv.). La suspensión se calentó a reflujo con un separador Dean-Stark (50 ml, para eliminar el AcOH). Después de 1 h, se añadieron 4,50 ml de ácido trifluorometanosulfónico (50,7 mmol, 0,05 equiv.) y 50 ml de diclorometano a la suspensión anaranjada y se descargó el disolvente (50 ml) del separador Dean-Stark. Cada media hora se repitió este procedimiento, hasta un total de 6 veces (3 h). Después de un total de 4,5 h, la solución roja se enfrió a 10-15 °C y se añadió en 30 min a 20-25 °C a una solución de 1,8 l de hidrogenocarbonato de sodio 1 M (1,8 mol, 2,0 equiv.; producción de CO2, pH 7-8). La capa orgánica amarilla se separó y se evaporó a 40 °C/600-10 mbar/3 h para obtener 585,4 g en bruto de 2-[2-[2-[2-[(2R,3R,4R,5R,6R)-3-acetamido-4,5-diacetoxi-6-(acetoximetil)tetrahidropiran-2-il]oxietoxi]etoxi]etoxi]acetato de bencilo como un aceite amarillo (pureza por HPLC: 87 %). El producto en bruto se disolvió en 700 ml de acetona y se cargó en una columna de sílice precargada (3,0 kg de sílice 60; 0,063-0,2 mm). La cromatografía se realizó usando nheptano/acetona como fase móvil (gradiente de 5:1 a 1:2). Las fracciones recogidas combinadas se evaporaron a 40 °C/600-10 mbar y se secaron a 20-25 °C/0,3 mbar/3 h para obtener 465,0 g de 2-[2-[2-[2-[(2R,3R,4R,5R,6R)-3-acetamido-4,5-diacetoxi-6-(acetoximetil)tetrahidropiran-2-il]oxietoxi]etoxi]etoxi]acetato de bencilo como un aceite amarillo con un rendimiento del 83 % y una pureza del 100 % (% del área por HPLC). EM: m/z = 628,2627 (M+H)+.
Ejemplo 7
Ácido 2-[2-[2-[2-[(2R,3R,4R,5R,6R)-3-acetamido-4,5-diacetoxi-6-(acetoximetil)tetrahidropiran-2-il]oxietoxi]etoxi]etoxi]acético

Bajo atmósfera de argón, se disolvieron 456,0 g de 2-[2-[2-[2-[(2R,3R,4R,5R,6R)-3-acetamido-4,5-diacetoxi-6-(acetoximetil)tetrahidropiran-2-il]oxietoxi]etoxi]etoxi]acetato de bencilo (727 mmol) en 1,4 l de THF. Se añadieron 4,56 g de Pd/C al 10 % y la atmósfera de argón se reemplazó por hidrógeno (1 bar). La suspensión negra se hidrogenó a 20-25 °C durante 2 h. La atmósfera de hidrógeno se reemplazó por argón, la suspensión negra se filtró y la torta de filtración se lavó por partes con un total de 400 ml de THF. El filtrado incoloro (pureza por HPLC: 71 % y 27 % de tolueno) se usó sin ninguna purificación en la etapa de ensamblaje A+B, ejemplo 8. EM: m/z = 538,2191 (M+H)+.
Ensamblaje de los componentes básicos A y B
Ejemplo 8
(2S)-6-[[2-[2-[2-[2-[(2R,3R,4R,5R,6R)-3-Acetamido-4,5-diacetoxi-6-(acetoximetil)tetrahidropiran-2-il]oxietoxi]etoxi]etoxi]acetil]amino]-2-[[(2S)-2,6-bis[[2-[2-[2-[2-[(2R,3R,4R,5R,6R)-3-acetamido-4,5-diacetoxi-6-(acetoximetil)tetrahidropiran-2-il]oxietoxi]etoxi]etoxi]acetil]amino]hexanoil]amino]hexanoato de bencilo

La solución de color rojo anaranjado (~1,4 l) de trimetanosulfonato de (2S)-6-amino-2-[[(2S)-2,6-diaminohexanoil]amino]hexanoato de bencilo (180,0 mmol) del ejemplo 4 se diluyó con 3,60 l de acetonitrilo. A 20 25 °C, se añadieron 365,0 ml de /V-etildiisopropilamina (2,16 mol, 12,0 equiv.) en 5 min. A la suspensión pegajosa formada se le añadió una solución (~2,25 l) de ácido 2-[2-[2-[2-[(2R,3R,4R,5R,6R)-3-acetamido-4,5-diacetoxi-6-(acetoximetil)tetrahidropiran-2-il]oxietoxi]etoxi]etoxi]acético (720 mmol, 4,0 equiv.) del ejemplo 7 a 20-25 °C en 10 min, con lo que la temperatura se incrementó ligeramente hasta 40 °C. A 45-50 °C se añadió una solución de 425 ml de anhídrido del ácido n-propilfosfónico (T3P, trímero al 50 % en acetato de etilo, 720 mmol, 4,0 equiv.) en 10 min. La solución de reacción se agitó durante 1 h a 45-50 °C. La solución de color amarillo claro se enfrió a 20-25 °C y se evaporó a 40 °C/10 mbar/6 h para obtener 1,06 kg en bruto de (2S)-6-[[2-[2-[2-[2-[(2R,3R,4R,5R,6R)-3-acetamido-4,5-diacetoxi-6-(acetoximetil)tetrahidropiran-2-il]oxietoxi]etoxi]etoxi]acetil]amino]-2-[[(2S)-2,6-bis[[2-[2-[2-[2-[(2R,3R,4R,5R,6R)-3-acetamido-4,5-diacetoxi-6-(acetoximetil)tetrahidropiran-2-il]oxietoxi]etoxi]etoxi]acetil]amino]hexanoil]amino]hexanoato de bencilo (pureza por HPLC: 24,1 %). El producto en bruto se precipitó en tres partes para eliminar el ácido metanosulfónico, la V-etildiisopropilamina y el T3P residual. Se disolvieron 353 g del producto en bruto en 7,0 l de 2-propanol, se enfriaron en 1 h a -25 °C, se agitó durante 1 h a -25 °C, se filtró sobre un filtro de vidrio G3 enfriado previamente (-25 °C; sin enjuague), con una parte del producto precipitado depositado sobre la pared de vidrio del reactor. Todos los precipitados se disolvieron por partes del filtro y de la pared de vidrio con un total de 1,0 l de THF. Las soluciones combinadas se evaporaron a 40 °C/20 mbar/6 h para obtener 390,0 g de (2S)-6-[[2-[2-[2-[2-[(2R,3R,4R,5R,6R)-3-acetamido-4,5-diacetoxi-6-(acetoximetil)tetrahidropiran-2-il]oxietoxi]etoxi]etoxi]acetil]amino]-2-[[(2S)-2,6-bis[[2-[2-[2-[2-[(2R,3R,4R,5R,6R)-3-acetamido-4,5-diacetoxi-6-(acetoximetil)tetrahidropiran-2-il]oxietoxi]etoxi]etoxi]acetil]amino]hexanoil]amino]hexanoato de bencilo (pureza por HPLC: 71,9 %), que se usó sin purificación adicional en la siguiente etapa. EM: m/z = 1923,8438 (M+H)+
Síntesis de la fosforamidita de GalNAc
Ejemplo 9
Ácido (2S)-6-[[2-[2-[2-[2-[(2R,3R,4R,5R,6R)-3-acetamido-4,5-diacetoxi-6-(acetoximetil)tetrahidropiran-2-il]oxietoxi]etoxi]etox¡]acetil]amino]-2-[[(2S)-2,6-bis[[2-[2-[2-[2-[(2R,3R,4R,5R,6R)-3-acetamido-4,5-diacetoxi-6-(acetoximetil)tetrahidropiran-2-il]oxietoxi]etoxi]etoxi]acetil]amino]hexanoil]amino]hexanoico

Se disolvieron 5,0 g (2,6 mmol) de ((2S)-6-[[2-[2-[2-[2-[(2R,3R,4R,5R,6R)-3-acetamido-4,5-diacetoxi-6-(acetoximetil)tetrahidropiran-2-il]oxietoxi]etoxi]etoxi]acetil]amino]-2-[[(2S)-2,6-bis[[2-[2-[2-[2-[(2R,3R,4R,5R,6R)-3-acetamido-4,5-diacetoxi-6-(acetoximetil)tetrahidropiran-2-il]oxietoxi]etoxi]etoxi]acetil]amino]hexanoil]amino]hexanoato de bencilo en 25,0 ml de THF, se añadieron 100 mg de Pd/C al 10 %, la mezcla se purgó tres veces con argón y a continuación tres veces con gas hidrógeno. La suspensión negra se hidrogenó a 20-25 °C durante 3,0 h en atmósfera de hidrógeno. La suspensión se filtró sobre un filtro de fibra de vidrio y el filtrado se evaporó a 20 °C/100-10 mbar/1 h para obtener 4,5 g de ácido (2S)-6-[[2-[2-[2-[2-[(2R,3R,4R,5R,6R)-3-acetamido-4,5-d¡acetox¡-6-(acetoximetil)tetrahidropiran-2-il]oxietoxi]etoxi]etoxi]acetil]amino]-2-[[(2S)-2,6-bis[[2-[2-[2-[2-[(2R,3R,4R,5R,6R)-3-acetamido-4,5-diacetoxi-6-(acetoximetil)tetrahidropiran-2-il]oxietoxi]etoxi]etoxi]acetil]amino]hexanoil]amino]hexanoico, como un aceite amarillo claro. CL-EM, (IEN) MH+ 1832,7941
Ejemplo 10a
2-(6-Benciloxihexil)isoindolin-1,3-diona

Se disolvieron 10,8 g (40 mmol) de 6-bromohexoximetilbenceno en 55,0 ml de THF, se añadieron 17,1 g (44,0 mmol, 1,1 equiv.) de 1,3-dioxoisoindolin-2-ida de tetrabutilamonio (ftalimida/hidróxido de tetrabutilamonio al 25 % en metanol 1/1, evaporación azeotrópica con tolueno y secado a 40 °C) a la solución incolora (ligera exotermia). La mezcla de reacción se agitó durante 2 h a 20-25 °C, la suspensión formada se filtró y la torta de filtración se lavó con 20,0 ml de THF, el filtrado combinado se diluyó con 50 ml de MTBE y se extrajo con 50 ml de agua, la fase orgánica se separó y se secó con sulfato de sodio anhidro, se filtró, se lavó con 30 ml de MTBE y se evaporó a 40 °C/400-15 mbar/3 h para obtener 12,9 g de 2-(6-benciloxihexil)isoindolin-1,3-diona en bruto como un sólido amarillo claro, que se usó sin purificación adicional en la etapa 10c. EM: m/z = (M+H)+ 338,1752
Ejemplo 10b
2-(6-Benciloxihexil)isoindolin-1,3-diona

Se disolvieron 12,7 g (47 mmol) de 6-bromohexoximetilbenceno en 64,0 ml de THF, se añadieron 9,6 g (51,7 mmol, 1,1 equiv.) de isoindolin-2-ida-1,3-diona de potasio y 0,32 g (0,94 mmol, 0,02 equiv.) de hidrogenosulfato de
tetrabutilamonio a la solución incolora. La mezcla de reacción se calentó a reflujo durante 18 h, se diluyó con 64 ml de MTBE, se extrajo dos veces con 64 ml de agua, la fase orgánica se separó y se secó con sulfato de sodio anhidro, se filtró, se lavó con 30 ml de MTBE y se evaporó a 40 °C/400-15 mbar/3 h para obtener 14,7 g de 2-(6-benciloxihexil)isoindolin-1,3-diona en bruto como un sólido blanquecino. Se trataron 10,6 g de producto en bruto con una mezcla de 37 ml de metanol y 5,5 ml de agua, se calentó a reflujo, después de 5 min la solución turbia se enfrió a 20-25 °C y se agitó durante 1 hora a 20-25 °C, la suspensión blanca se filtró y la torta de filtración se lavó con una mezcla de 7,0 ml de metanol y 1,0 ml de agua. Los cristales blancos se secaron a 40 °C/15 mbar/1 h para obtener 9,6 g de 2-(6-(benciloxi)hexil)isoindolin-1,3-diona como cristales blancos. EM: m/z = (M+H)+ 338,1752
Ejemplo 10c
6-Benciloxihexan-1-amina

Se disolvieron 12,8 g (37,9 mmol) de 2-(6-(benciloxi)hexil)isoindolin-1,3-diona en 40,0 ml de THF, a temperatura ambiente se añadieron 5,5 ml (113 mmol, 3,0 equiv.) de hidrazina monohidrato al 64 % en agua. La solución incolora se calentó a 50-55 °C durante 2 horas, la suspensión blanca formada se enfrió a temperatura ambiente y se filtró y la torta de filtración blanca se lavó con un total de 30,0 ml de THF. El filtrado se diluyó con 60,0 ml de MTBE y se extrajo con 50 ml de agua. La fase orgánica se separó, se secó con sulfato de sodio anhidro, se filtró y se evaporó a 40 °C/350-15 mbar/2 h para obtener 6,44 g de 6-benciloxihexan-1-amina en bruto como un aceite incoloro.
Formación de la sal clorhidrato: se disolvieron 6,44 g (31,1 mmol) en 32,0 ml de metanol, a temperatura ambiente se añadieron 7,80 ml de HCl 4 M en metanol, la solución de color amarillo claro se evaporó a 40 °C/150-15 mbar/15 min para obtener 8,1 g en bruto como un aceite amarillo. A este aceite se le añadieron 65,0 ml de acetato de etilo, con lo que se formó una suspensión blanca, que se agitó durante 16 horas a temperatura ambiente. La suspensión blanca se filtró, la torta de filtración se lavó con un total de 40,0 ml de acetato de etilo y se secó a 40 °C/15 mbar/3 h para obtener 4,95 g de clorhidrato de 6-benciloxihexan-1-amina como cristales blancos.
Formación de la base libre: se trataron 2,0 g de clorhidrato de 6-benciloxihexan-1-amina con 15,0 ml de NaOH 1 M/NaCl al 13 % y se extrajeron con 15,0 ml de MTBE, la fase orgánica se separó, se secó con sulfato de sodio anhidro, se filtró, se lavó con 5 ml de MTBE y se evaporó a 40 °C/350-15 mbar/2 h para obtener 1,67 g de 6-benciloxihexan-1-amina como un aceite incoloro
EM: m/z = (M+H)+ 208,17006
Ejemplo 10d
Acetato de [(2R,3R,4R,5R,6R)-5-acetamido-6-[2-[2-[2-[2-[[(5S)-5-[[2-[2-[2-[2-[(2R,3R,4R,5R,6R)-3-acetamido-4,5-diacetoxi-6-(acetoximetil)tetrahidropiran-2-il]oxietoxi]etoxi]etoxi]acetil]aminoj-6-[[(1S)-5-[[2-[2-[2-[2-[(2R,3R,4R,5R,6R)-3-acetamido-4,5-diacetoxi-6-(acetoximetil)tetrahidropiran-2-il]oxietoxi]etoxi]etoxi]acetil]amino]-1-(6-benciloxihexilcarbamoil)pentil]amino]-6-oxo-hexil]amino]-2-oxo-etoxi]etoxi]etoxi]etoxi]-3,4-diacetoxi-tetrahidropiran-2-iljmetilo
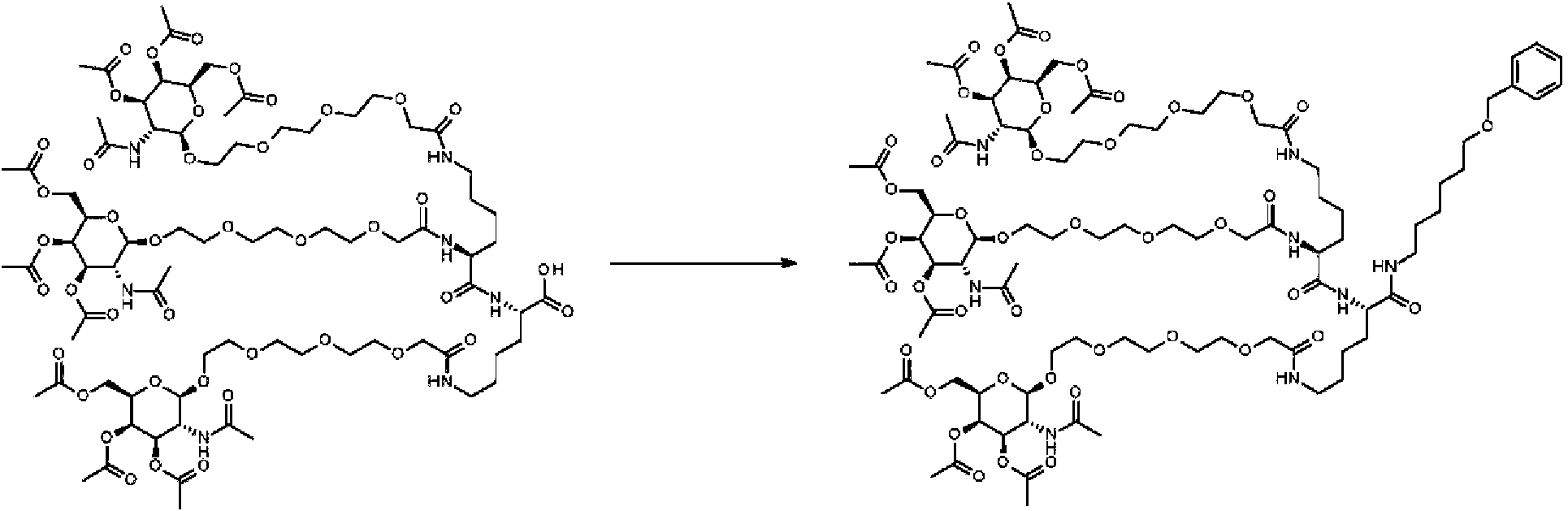
Se disolvieron 4,4 g de ácido (2S)-6-[[2-[2-[2-[2-[(2R,3R,4R,5R,6R)-3-acetamido-4,5-diacetoxi-6-(acetoximetil)tetrahidropiran-2-il]oxietoxi]etoxi]etoxi]acetil]amino]-2-[[(2S)-2,6-bis[[2-[2-[2-[2-[(2R,3R,4R,5R,6R)-3-acetamido-4,5-diacetoxi-6-(acetoximetil)tetrahidropiran-2-il]oxietoxi]etoxi]etoxi]acetil]amino]hexanoil]amino]hexanoico en 45,0 ml de THF, a 20-25 °C se añadieron 0,55 g (2,64 mmol, 1,1 equiv.) de 6-benciloxihexan-l-amina y 1,22 ml (7,2 mmol, 3,0 equiv.) de /V-etildiisopropilamina, la solución se calentó a 35-40 °C y se añadieron 1,84 ml de anhídrido de ácido n-propilfosfónico (T3P como trímero cíclico al 50 % en acetato de etilo, 3,12 mmol, 1,3 equiv.) a la solución de color amarillo claro. Se agitó la mezcla durante 4,0 h a 35-40 °C, se enfrió a 20-25 °C y se evaporó a 20-25 °C/100-10 mbar/2 h para obtener 6,5 g de producto en bruto. El producto se purificó con SFC/RP-C18/2-propanol para obtener 2,91 g de acetato de [(2R,3R,4R,5R,6R)-5-acetamido-6-[2-[2-[2-[2-[[(5S)-5-[[2-[2-[2-[2-[(2R,3R,4R,5R,6R)-3-acetamido-4,5-diacetoxi-6-(acetoximetil)tetrahidropiran-2-il]oxietoxi]etoxi]etoxi]acetil]amino]-6-[[(1S)-5-[[2-[2-[2-[2-[(2R,3R,4R,5R,6R)-3-acetamido-4,5-diacetoxi-6-(acetoximetil)tetrahidropiran-2-il]oxietoxi]etoxi]etoxi]acetil]amino]-1-(6-benciloxihexilcarbamoil)pentil]amino]-6-oxo-hexil]amino]-2-oxo-etoxi]etoxi]etoxi]etoxi]-3,4-diacetoxi-tetrahidropiran-2-il]metilo, como una espuma blanca. CL-EM, (IEN) MH+ 2021,945.
Ejemplo 11
Acetato de [(2R,3R,4R,5R,6R)-5-acetamido-6-[2-[2-[2-[2-[[(5S)-5-[[2-[2-[2-[2-[(2R,3R,4R,5R,6R)-3-acetamido-4,5-diacetoxi-6-(acetoximetil)tetrahidropiran-2-il]oxietoxi]etoxi]etoxi]acetil]amino]-6-[[(1 S)-5-[[2-[2-[2-[2-[(2R,3R,4R,5R,6R)-3-acetamido-4,5-diacetoxi-6-(acetoximetil)tetrahidropiran-2-il]oxietoxi]etoxi]etoxi]acetil]amino]-1-(6-hidroxihexilcarbamoil)pentil]amino]-6-oxo-hexil]amino]-2-oxo-etoxi]etoxi]etoxi]etoxi]-3,4-diacetoxi-tetrahidropiran-2-il]metilo

Se disolvieron 1,62 g (0,80 mmol) de acetato de [(2R,3R,4R,5R,6R)-5-acetamido-6-[2-[2-[2-[2-[[(5S)-5-[[2-[2-[2-[2-[(2R,3R,4R,5R,6R)-3-acetamido-4,5-diacetoxi-6-(acetoximetil)tetrahidropiran-2-il]oxietoxi]etoxi]etoxi]acetil]amino]-6-[[(1S)-5-[[2-[2-[2-[2-[(2R,3R,4R,5R,6R)-3-acetamido-4,5-diacetoxi-6-(acetoximetil)tetrahidropiran-2-il]oxietoxi]etoxi]etoxi]acetil]amino]-1-(6-benciloxihexilcarbamoil)pentil]amino]-6-oxo-hexil]amino]-2-oxoetoxi]etoxi]etoxi]etoxi]-3,4-diacetoxi-tetrahidropiran-2-il]metilo en 8,0 ml de metanol, se añadieron 162 mg de Pd/C al 10 %, la mezcla se purgó tres veces con argón en primer lugar y a continuación tres veces con gas hidrógeno. La suspensión negra se hidrogenó a 20-25 °C durante 1,5 horas. La suspensión negra se filtró sobre un filtro de fibra de vidrio, el filtrado incoloro se evaporó a 20 °C/100-10 mbar/1 h. El aceite incoloro se disolvió en 10 ml de acetonitrilo y se evaporó a 100-10 mbar/1 h y se trató de nuevo con 10 ml de acetonitrilo y se evaporó a 100-10 mbar/1 h, a continuación se secó el aceite incoloro a 20-25 °C a 1 mbar durante 2 h para obtener 1,50 g de acetato de
[(2R,3R,4R,5R,6R)-5-acetamido-6-[2-[2-[2-[2-[[(5S)-5-[[2-[2-[2-[2-[(2R,3R,4R,5R,6R)-3-acetamido-4,5-diacetoxi-6-(acetoximetil)tetrahidropiran-2-il]oxietoxi]etoxi]etoxi]acetil]amino]-6-[[(1S)-5-[[2-[2-[2-[2-[(2R,3R,4R,5R,6R)-3-acetamido-4,5-diacetoxi-6-(acetoximetil)tetrahidropiran-2-il]oxietoxi]etoxi]etoxi]acetil]amino]-1-(6-hidroxihexilcarbamoil)pentil]amino]-6-oxo-hexil]amino]-2-oxo-etoxi]etoxi]etoxi]etoxi]-3,4-diacetoxi-tetrahidropiran-2-il]metilo, como un aceite incoloro. CL-EM MH+ 1931,90142
Ejemplo 12
Acetato de [(2R,3R,4R,5R,6R)-5-acetamido-6-[2-[2-[2-[2-[[(5S)-5-[[2-[2-[2-[2-[(2R,3R,4R,5R,6R)-3-acetamido-4,5-diacetoxi-6-(acetoximetil)tetrahidropiran-2-il]oxietoxi]etoxi]etoxi]acetil]amino]-6-[[(1 S)-5-[[2-[2-[2-[2-[(2R,3R,4R,5R,6R)-3-acetamido-4,5-diacetoxi-6-(acetoximetil)tetrahidropiran-2-il]oxietoxi]etoxi]etoxi]acetil]amino]-1-[6-[2-cianoetoxi-(diisopropilamino)fosfanil]oxihexilcarbamoil]pentil]amino]-6-oxo-hexil]amino]-2-oxoetoxi]etoxi]etoxi]etoxi]-3,4-diacetoxi-tetrahidropiran-2-il]metilo

Se disolvieron 1,50 g (0,77 mmol) de acetato de [(2R,3R,4R,5R,6R)-5-acetamido-6-[2-[2-[2-[2-[[(5S)-5-[[2-[2-[2-[2-[(2R,3R,4R,5R,6R)-3-acetamido-4,5-diacetoxi-6-(acetoximetil)tetrahidropiran-2-il]oxietoxi]etoxi]etoxi]acetil]amino]-6-[[(1S)-5-[[2-[2-[2-[2-[(2R,3R,4R,5R,6R)-3-acetamido-4,5-diacetoxi-6-(acetoximetil)tetrahidropiran-2-il]oxietoxi]etoxi]etoxi]acetil]amino]-1-(6-hidroxihexilcarbamoil)pentil]amino]-6-oxo-hexil]amino]-2-oxoetoxi]etoxi]etoxi]etoxi]-3,4-diacetoxi-tetrahidropiran-2-il]metilo en 5,0 ml de acetonitrilo, la solución de color amarillo claro se enfrió a 0-5 °C, se añadieron 0,184 ml de trietilamina y 2-cianoetil-W,W-diisopropilclorofosforamidita a 0-5 °C. Después de 15 min, se añadieron 1,50 g de tamices moleculares de 3A en microesferas de 4-8 mesh y se agitó a 0 5 °C durante 10 min, la suspensión se filtró sobre un filtro de fibra de vidrio y la solución de color amarillo claro se evaporó a 20-25 °C/100-10 mbar/0,5 h para obtener 1,50 g de acetato de [(2R,3R,4R,5R,6R)-5-acetamido-6-[2-[2-[2-[2-[[(5S)-5-[[2-[2-[2-[2-[(2R,3R,4R,5R,6R)-3-acetamido-4,5-diacetoxi-6-(acetoximetil)tetrahidropiran-2-il]oxietoxi]etoxi]etoxi]acetil]amino]-6-[[(1S)-5-[[2-[2-[2-[2-[(2R,3R,4R,5R,6R)-3-acetamido-4,5-diacetoxi-6-(acetoximetil)tetrahidropiran-2-il]oxietoxi]etoxi]etoxi]acetil]amino]-1-[6-[2-cianoetoxi-(diisopropilamino)fosfanil]oxihexilcarbamoil]pentil]amino]-6-oxo-hexil]amino]-2-oxo-etoxi]etoxi]etoxi]etoxi]-3,4-diacetoxi-tetrahidropiran-2-il]metilo, como un aceite amarillo, que se usó en la siguiente etapa sin purificación adicional.
31P-RMN DMSO-d6 146,33 ppm (s 1P). CL-EM, M(NH4)+ 2149,0.
Ejemplo 13
Hexadecaamonio; fosfato de [(3S)-2-[[6-[[6-[[2-[2-[2-[2-[(2R)-3-acetamido-4,5-dihidroxi-6-(hidroximetilo)tetrahidropiran-2-il]oxietoxi]etoxi]etoxi]acetil]amino]-2-[2,6-bis[[2-[2-[2-[2-[(2R)-3-acetamido-4,5-dihidroxi-6-(hidroximetil)tetrahidropiran-2-il]oxietoxi]etoxi]etoxi]acetil]amino]hexanoilamino]hexanoil]amino]hexoxioxido-fosforil]oximetil]-5-(4-amino-2-oxo-pirimidin-1-il)tetrahidrofuran-3-il] [(2R,3s ,5R)-3-[[(1R,4R,6R,7S)-7-[[(1R,4R,6R,7s )-6-(4-amino-5-metil-2-oxo-pirimidin-1-il)-7-[[(1R,4R,6R,7S)-6-(2-amino-6-oxo-1H-purin-9-il)-7-[[(2R,3S,5R)-3-[[(2R,3S,5R)-3-[[(2R,3S,5R)-3-[[(2R,3S,5R)-3-[[(2R,3S,5R)-5-(2-amino-6-oxo-1H-purin-9-il)-3-[[(2R,3S,5R)-3-[[(2R,3S,5R)-5-(2-amino-6-oxo-1H-purin-9-il)-3-[[(2R,3S,5R)-3-[[(4R)-6-(2-amino-6-oxo-1H-purin-9-il)-7-[[6-(2-amino-6-oxo-1H-purin-9-il)-7-[[7-hidroxi-6-(5-metil-2,4-dioxo-pirimidin-1-il)-2,5-dioxabiciclo[2.2.1]heptan-4-il]metoxi-oxido-fosfinotioil]oxi-2,5-dioxabiciclo[2.2.1]heptan-4-il]metoxi-oxido-fosfinotioil]oxi-2,5-dioxabiciclo[2.2.1]heptan-4-il]metoxi-oxido-fosfinotioil]oxi-5-(6-aminopurin-9-il)tetrahidrofuran-2-il]metoxi-oxidofosfinotioil]oxi-tetrahidrofuran-2-il]metoxi-oxido-fosfinotioil]oxi-5-(6-aminopurin-9-il)tetrahidrofuran-2-il]metoxi-oxidofosfinotioil]oxi-tetrahidrofuran-2-il]metoxi-oxido-fosfinotioil]oxi-5-(6-aminopurin-9-il)tetrahidrofuran-2-il]metoxi-oxidofosfinotioil]oxi-5-(6-aminopurin-9-il)tetrahidrofuran-2-il]metoxi-oxido-fosfinotioil]oxi-5-(6-aminopurin-9-il)tetrahidrofuran-2-il]metoxi-oxido-fosfinotioil]oxi-5-(5-metil-2,4-dioxo-pirimidin-1-il)tetrahidrofuran-2-il]metoxi-oxidofosfinotioil]oxi-2,5-dioxabiciclo[2.2.1]heptan-4-il]metoxi-oxido-fosfinotioil]oxi-2,5-dioxabiciclo[2.2.1]heptan-4-il]metoxioxido-fosfinotioil]oxi-6-(2-amino-6-oxo-1H-purin-9-il)-2,5-dioxabiciclo[2.2.1]heptan-4-il]metoxi-oxido-fosforil]oxi-5-(6aminopurin-9-il)tetrahidrofuran-2-il]metilo

El LNA/ADN modificado con agrupaciones de GalNAc se produjo mediante química estándar de fosforamidita en fase sólida a una escala de 0,200 mmol usando un AKTA Oligopilot 100 (GE Healthcare, Friburgo, Alemania) y Primer Support 5G Unylinker 350 (GE Healthcare, Friburgo, Alemania). El LNA que contiene nucleótidos unidos mediante puente 2'-OCH2-4' (Sigma-Aldrich, SAFC, Hamburgo, Alemania) y el ADN (Sigma-Aldrich, SAFC, Hamburgo, Alemania) se generaron empleando la fosforamidita y fosforamidita con agrupaciones de GalNAc correspondientes. La escisión y la desprotección se consiguieron mediante procedimientos conocidos en el campo (Wincott F. et al. Nucleic Acid Research, 1995, 23,14, 2677-84). Se caracterizó el LNA modificado de agrupaciones de GalNAc en bruto desprotegido y secado como sal de amonio (1,6 g) y se confirmó la identidad por HPLC-EM de pares iónicos como agrupación de GalNAc-AM-C6-5'-caGsmCsGstsasasasgsasgsasGsGsT-3'; (comp. n.° 2) C231H316N78O118P16S13, CL-EM, M (IEN) 6981,3.
(Las letras mayúsculas indican unidades de beta-D-oxi-LNA; las letras minúsculas indican unidades de ADN; el subíndice "s" indica un enlace fosforotioato; el superíndice m indica una unidad de ADN o beta-D-oxi-LNA que contiene una base 5-metilcitosina y AM-C6 indica un enlace 6-aminohexil-1-fosfato)
Claims (17)
1. Derivados de fosforamidita de GalNAc de fórmula I

en la que R1 es un grupo acilo, n es un número entero de 0 a 10 y m es un número entero de 0 a 20, enantiómeros y/o isómeros ópticos correspondientes de los mismos.
2. Derivado de fosforamidita de GalNAc de la reivindicación 1, en el que R1 es un grupo alquilcarbonilo C1-6 que está sustituido opcionalmente con alquilo C1-6 o fenilo.
3. Derivado de fosforamidita de GalNAc de una cualquiera de las reivindicaciones 1 o 2, en el que n es un número entero de 0 a 5 y m es un número entero de 0 a 10.
4. Derivado de fosforamidita de GalNAc de una cualquiera de las reivindicaciones 1 a 3, de fórmula Ia

en la que R1, m y n son como anteriormente.
5. Derivado de fosforamidita de GalNAc de una cualquiera de las reivindicaciones 1 a 4, en el que R1 es acetilo, n es 2 y m es 5.
6. Derivado de fosforamidita de GalNAc de una cualquiera de las reivindicaciones 1 a 5, de fórmula Ib

7. Procedimiento para la preparación de un derivado de fosforamidita de GalNAc de fórmula I, que comprende a) hacer reaccionar un derivado de ácido de GalNAc de fórmula III

en la que R1 y n son como se definen en la reivindicación 1
con una amina de fórmula IV

en la que R3 es un grupo protector de hidroxi y m es como anteriormente, para formar una amida de fórmula V

en la que R1, R3, n y m son como anteriormente;
5 b) la eliminación del grupo protector de hidroxi R3 para formar la amida de ácido de GalNAc de fórmula VI
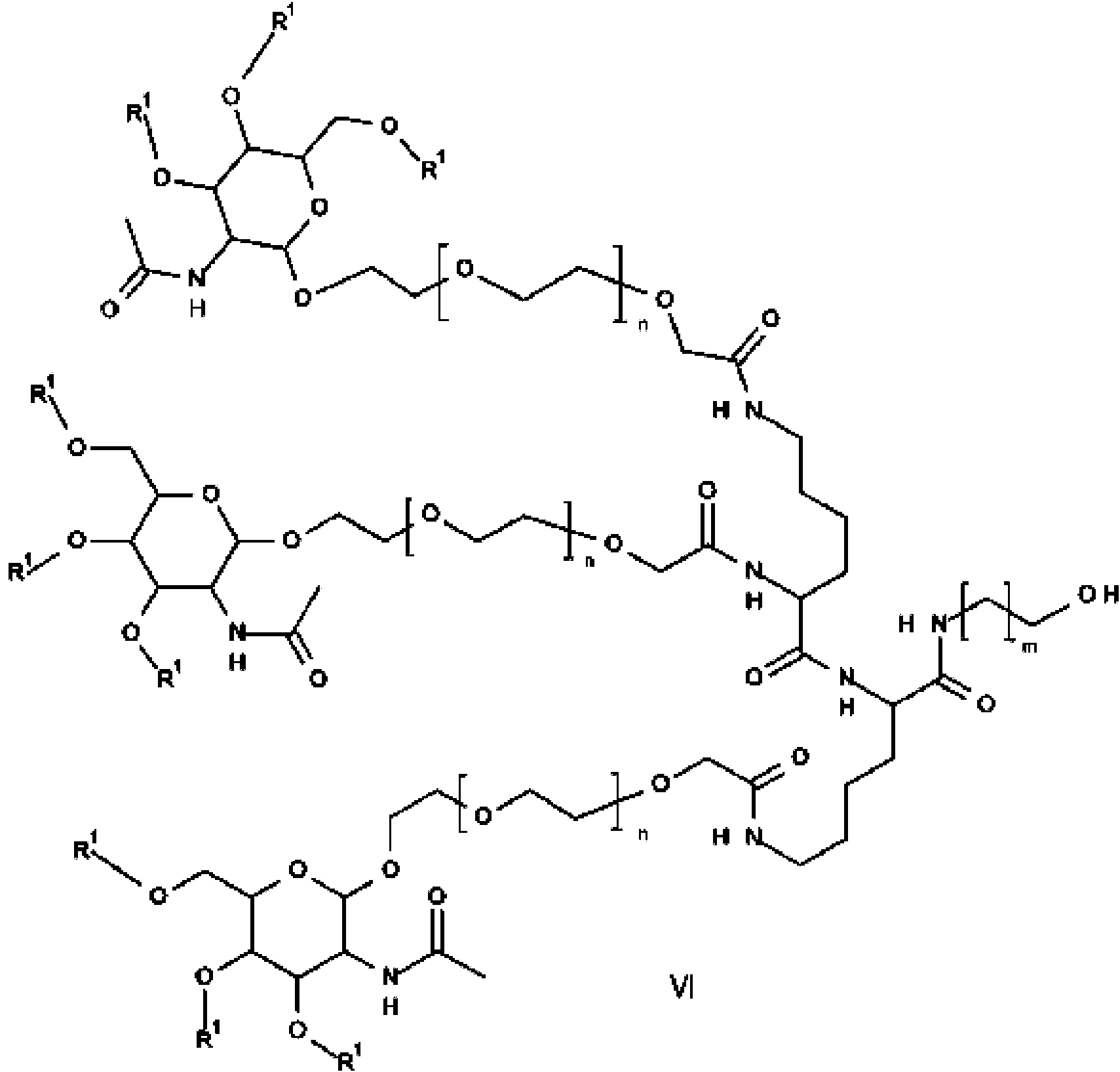
en la que R1, n y m son como anteriormente y
c) hacer reaccionar la amida de ácido de GalNAc de fórmula VI con un agente fosforamidante para formar el derivado de fosforamidita de GalNAc de fórmula I.
8. Procedimiento de la reivindicación 7, en el que la formación de amida en la etapa a) se realiza en presencia de un agente de acoplamiento peptídico, una base amínica y un disolvente orgánico.
9. Procedimiento de las reivindicaciones 7 u 8, en el que el agente de acoplamiento peptídico es anhídrido de ácido npropilfosfónico, la base amínica es una amina terciaria, el disolvente orgánico es un disolvente aprótico polar y la temperatura de reacción se selecciona de 20 °C a 70 °C.
10. Procedimiento de la reivindicación 9, en el que la eliminación en la etapa b) es una hidrogenólisis y se realiza por medio de una hidrogenación catalítica con hidrógeno en presencia de un catalizador de hidrogenación.
11. Procedimiento de una cualquiera de las reivindicaciones 7 a 10, en el que el grupo protector de hidroxi R3 es bencilo.
12. Procedimiento de la reivindicación 7, en el que el agente fosforamidante en la etapa c) se selecciona de 2-cianoetil-W,W-di-(2-propil)clorofosforamidita o de 2-cianoetil-W,W,W,W-tetra(2-propil)fosforodiamidita.
13. Procedimiento de las reivindicaciones 7 o 12, en el que la reacción en la etapa c) se realiza con 2-cianoetil-W,W-diisopropilclorofosforamidita en presencia de una amina terciaria y un disolvente aprótico polar a una temperatura de reacción de entre -20 °C y 50 °C.
14. Uso de los derivados de fosforamidita de GalNAc de fórmula I, como se definen en la reivindicación 1, para la preparación de conjugados de oligonucleótidos con agrupaciones de GalNAc.
15. Uso de la reivindicación 14, en el que la preparación de conjugados de oligonucleótidos con agrupaciones de GalNAc comprende
a2) la preparación del derivado de fosforamidita de GalNAc de fórmula I de acuerdo con las reivindicaciones 7 a 13; b2) el empleo del derivado de fosforamidita de GalNAc de fórmula I en la síntesis de oligonucleótidos en fase sólida conjuntamente con los componentes básicos nucleosídicos deseados en la secuencia deseada para formar un conjugado de oligonucleótidos con agrupaciones de GalNAc deseado unido al soporte sólido y, finalmente, la c2) escisión del conjugado de oligonucleótidos con agrupaciones de GalNAc del soporte en fase sólida y desprotección completa.
16. Uso de la reivindicación 14 o 15, en el que se usa el derivado de fosforamidita de GalNAc de fórmula Ia, preferentemente de fórmula Ib.
17. Uso de una cualquiera de las reivindicaciones 14 a 16, en el que el conjugado de oligonucleótidos con agrupaciones de GalNAc se selecciona de
Agrupación de GalNAc-AM-C6-5'-caGsmCsGstsasasasgsasgsasGsG-3'; (comp. n.° 1)
Agrupación de GalNAc-AM-C6-5'-caGsmCsGstsasasasgsasgsasGsGsT-3'; (comp. n.° 2)
Agrupación de GalNAc-AM-C6-5'-caGsmCsGstsasasasgsasgsAsGsG-3'; (comp. n.° 3)
Agrupación de GalNAc-AM-C6-5'-caGsGsmCsGstsasasasgsasgsasGsG-3'; (comp. n.° 4)
Agrupación de GalNAc-AM-C6-5'-caAsGsmCsgsasasgstsgscsascsAsmCsG-3'; (comp. n.° 5)
Agrupación de GalNAc-AM-C6-5'-caAsGsmCsgsasasgstsgscsascsasCsGsG-3'; (comp. n.° 6)
Agrupación de GalNAc-AM-C6-5'-(5'Br)caGsmCsGstsasasasgsasgsasGsG-3'; (comp. n.° 7)
Agrupación de GalNAc-AM-C6-5'-cagcgtaaagagagg-3'; (comp. n.° 8)
Agrupación de GalNAc-AM-C6s-5'-AsAsTsgscstsascsasasasascsCsCsA-3'; (comp. n.° 9)
en el que AM-C6 indica un enlace 6-aminohexil-1-fosfato o 1-tiofosfato, las letras mayúsculas indican unidades beta-D-oxi-LNA; las letras minúsculas indican unidades de ADN; el subíndice "s" indica un enlace fosforotioato; el superíndice m indica una unidad de ADN o beta-D-oxi-LNA que contiene una base 5-metilcitosina y el superíndice 5-Br indica una unidad de ADN que contiene una base 5-bromocitosina.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP15194811 | 2015-11-16 | ||
| PCT/EP2016/077516 WO2017084987A1 (en) | 2015-11-16 | 2016-11-14 | GalNAc CLUSTER PHOSPHORAMIDITE |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2848122T3 true ES2848122T3 (es) | 2021-08-05 |
Family
ID=54695483
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16797831T Active ES2848122T3 (es) | 2015-11-16 | 2016-11-14 | Fosforamidita de agrupación de GalNAc |
Country Status (14)
| Country | Link |
|---|---|
| US (1) | US10590156B2 (es) |
| EP (1) | EP3377510B1 (es) |
| JP (1) | JP6649482B2 (es) |
| KR (1) | KR102117172B1 (es) |
| CN (1) | CN108738321B (es) |
| AR (1) | AR106683A1 (es) |
| AU (1) | AU2016357980B2 (es) |
| BR (1) | BR112018003910B1 (es) |
| CA (1) | CA2994787A1 (es) |
| ES (1) | ES2848122T3 (es) |
| IL (1) | IL258149B (es) |
| MX (1) | MX2018005733A (es) |
| PL (1) | PL3377510T3 (es) |
| WO (1) | WO2017084987A1 (es) |
Families Citing this family (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20230136689A (ko) | 2016-03-14 | 2023-09-26 | 에프. 호프만-라 로슈 아게 | Pd-l1 발현의 감소를 위한 올리고뉴클레오티드 |
| JP7048574B2 (ja) | 2017-03-10 | 2022-04-05 | 国立研究開発法人国立成育医療研究センター | アンチセンスオリゴヌクレオチドおよび糖原病Ia型予防または治療用組成物 |
| US11447773B2 (en) | 2017-09-08 | 2022-09-20 | Mina Therapeutics Limited | Stabilized HNF4A saRNA compositions and methods of use |
| SG11202002149XA (en) * | 2017-09-14 | 2020-04-29 | Janssen Biopharma Inc | Galnac derivatives |
| CN111511915A (zh) | 2018-03-09 | 2020-08-07 | 第一三共株式会社 | 糖原病Ia型治疗药 |
| CN113840828B (zh) | 2019-05-20 | 2024-01-16 | 豪夫迈·罗氏有限公司 | 用于制备GalNAc亚磷酰胺差向异构体的方法 |
| US20220378920A1 (en) | 2019-09-10 | 2022-12-01 | Daiichi Sankyo Company, Limited | CONJUGATE OF GalNAc-OLIGONUCLEOTIDE FOR DELIVERY TO LIVER AND MANUFACTURING METHOD THEREOF |
| CN110668933B (zh) * | 2019-09-30 | 2024-09-24 | 兰州交通大学 | 一种合成烯丙基脂肪醇聚氧乙烯醚羧酸(apea-7)的方法 |
| CN110759824B (zh) * | 2019-09-30 | 2024-09-24 | 兰州交通大学 | 一种合成烯丙基脂肪醇聚氧乙烯醚羧酸(apea-3)的方法 |
| AU2022291742A1 (en) | 2021-06-14 | 2023-12-21 | Generation Bio Co. | Cationic lipids and compositions thereof |
| WO2023143571A1 (zh) * | 2022-01-30 | 2023-08-03 | 大睿生物医药科技(上海)有限公司 | 含有n-乙酰半乳糖胺的靶向配体 |
| WO2023176862A1 (ja) | 2022-03-16 | 2023-09-21 | 第一三共株式会社 | トランスフェリン受容体2の発現を抑制するsiRNA |
| WO2023176863A1 (ja) | 2022-03-16 | 2023-09-21 | 第一三共株式会社 | RNAi活性を有する化学修飾オリゴヌクレオチド |
| WO2024040222A1 (en) | 2022-08-19 | 2024-02-22 | Generation Bio Co. | Cleavable closed-ended dna (cedna) and methods of use thereof |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| LT3964502T (lt) | 2009-03-19 | 2024-08-26 | The Johns Hopkins University | Junginiai nukreipti į psma ir jų panaudojimas |
| MX2012009178A (es) | 2010-02-24 | 2012-11-30 | Arrowhead Res Corp | Composiciones para liberacion dirigida de arnsi. |
| US8501930B2 (en) * | 2010-12-17 | 2013-08-06 | Arrowhead Madison Inc. | Peptide-based in vivo siRNA delivery system |
| CA2816155C (en) * | 2010-12-17 | 2020-10-27 | Arrowhead Research Corporation | Galactose cluster-pharmacokinetic modulator targeting moiety for sirna |
| PL2992098T3 (pl) * | 2013-05-01 | 2019-09-30 | Ionis Pharmaceuticals, Inc. | Kompozycje i sposoby modulowania ekspresji hbv i ttr |
| EP3591054A1 (en) * | 2013-06-27 | 2020-01-08 | Roche Innovation Center Copenhagen A/S | Antisense oligomers and conjugates targeting pcsk9 |
| RU2016122168A (ru) | 2013-11-14 | 2017-12-19 | Рош Инновейшен Сентер Копенгаген А/С | Антисмысловые конъюгаты, направленные на аполипопротеин b |
| ES2844593T3 (es) * | 2014-05-01 | 2021-07-22 | Ionis Pharmaceuticals Inc | Composiciones y procedimientos para modular la expresión de la angiopoyetina de tipo 3 |
-
2016
- 2016-11-14 BR BR112018003910-1A patent/BR112018003910B1/pt active IP Right Grant
- 2016-11-14 EP EP16797831.1A patent/EP3377510B1/en active Active
- 2016-11-14 CN CN201680065443.6A patent/CN108738321B/zh active Active
- 2016-11-14 JP JP2018523484A patent/JP6649482B2/ja active Active
- 2016-11-14 CA CA2994787A patent/CA2994787A1/en active Pending
- 2016-11-14 WO PCT/EP2016/077516 patent/WO2017084987A1/en active Application Filing
- 2016-11-14 MX MX2018005733A patent/MX2018005733A/es unknown
- 2016-11-14 AU AU2016357980A patent/AU2016357980B2/en active Active
- 2016-11-14 KR KR1020187008037A patent/KR102117172B1/ko active IP Right Grant
- 2016-11-14 PL PL16797831T patent/PL3377510T3/pl unknown
- 2016-11-14 ES ES16797831T patent/ES2848122T3/es active Active
- 2016-11-14 AR ARP160103468A patent/AR106683A1/es unknown
-
2018
- 2018-03-15 IL IL258149A patent/IL258149B/en active IP Right Grant
- 2018-05-14 US US15/978,815 patent/US10590156B2/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| PL3377510T3 (pl) | 2021-05-04 |
| US10590156B2 (en) | 2020-03-17 |
| IL258149A (en) | 2018-05-31 |
| AU2016357980A1 (en) | 2018-02-22 |
| KR20180041225A (ko) | 2018-04-23 |
| EP3377510A1 (en) | 2018-09-26 |
| WO2017084987A1 (en) | 2017-05-26 |
| CN108738321A (zh) | 2018-11-02 |
| MX2018005733A (es) | 2018-08-09 |
| JP2018532764A (ja) | 2018-11-08 |
| BR112018003910B1 (pt) | 2022-03-22 |
| US20180258122A1 (en) | 2018-09-13 |
| JP6649482B2 (ja) | 2020-02-19 |
| KR102117172B1 (ko) | 2020-06-01 |
| EP3377510B1 (en) | 2020-12-02 |
| CN108738321A9 (zh) | 2021-01-29 |
| BR112018003910A2 (pt) | 2018-09-25 |
| IL258149B (en) | 2020-06-30 |
| AU2016357980B2 (en) | 2020-06-25 |
| CN108738321B (zh) | 2021-06-11 |
| CA2994787A1 (en) | 2017-05-26 |
| AR106683A1 (es) | 2018-02-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2848122T3 (es) | Fosforamidita de agrupación de GalNAc | |
| ES2744437T3 (es) | Procedimientos para la preparación de derivados ácidos de GalNAc | |
| US11021503B2 (en) | Process for GalNAc oligonucleotide conjugates | |
| CN106661074A (zh) | 磷酰胺的合成 | |
| JP7558201B2 (ja) | GalNAcホスホラミダイトエピマーを調製するための方法 | |
| BR112017021926B1 (pt) | Processo para a preparação de derivados de ácido galnac, sal de amina e processo para a preparação de conjugados de oligonucleotídeos galnac |