ES2836255T3 - Derivados de indol y su uso como inhibidores de proteína cinasa - Google Patents
Derivados de indol y su uso como inhibidores de proteína cinasa Download PDFInfo
- Publication number
- ES2836255T3 ES2836255T3 ES17711736T ES17711736T ES2836255T3 ES 2836255 T3 ES2836255 T3 ES 2836255T3 ES 17711736 T ES17711736 T ES 17711736T ES 17711736 T ES17711736 T ES 17711736T ES 2836255 T3 ES2836255 T3 ES 2836255T3
- Authority
- ES
- Spain
- Prior art keywords
- methyl
- phenyl
- amino
- carboxylate
- methylene
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 CC(C)(C)OC(Nc1ccc(*)c(*)c1)=O Chemical compound CC(C)(C)OC(Nc1ccc(*)c(*)c1)=O 0.000 description 4
- XEVLOESWJMCIIZ-DCMSUDLNSA-N CC/C(/[N+]([O-])=O)=C\C[C@H](C)C(O)OC Chemical compound CC/C(/[N+]([O-])=O)=C\C[C@H](C)C(O)OC XEVLOESWJMCIIZ-DCMSUDLNSA-N 0.000 description 1
- UDEOEHAYSKOZAQ-UHFFFAOYSA-N COC(c(cc(c(C1)c2)NC1=O)c2I)=O Chemical compound COC(c(cc(c(C1)c2)NC1=O)c2I)=O UDEOEHAYSKOZAQ-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/30—Indoles; Hydrogenated indoles with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to carbon atoms of the hetero ring
- C07D209/32—Oxygen atoms
- C07D209/34—Oxygen atoms in position 2
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/403—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with carbocyclic rings, e.g. carbazole
- A61K31/404—Indoles, e.g. pindolol
- A61K31/4045—Indole-alkylamines; Amides thereof, e.g. serotonin, melatonin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4412—Non condensed pyridines; Hydrogenated derivatives thereof having oxo groups directly attached to the heterocyclic ring
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/454—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. pimozide, domperidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/527—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim spiro-condensed
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/54—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one sulfur as the ring hetero atoms, e.g. sulthiame
- A61K31/541—Non-condensed thiazines containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/551—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having two nitrogen atoms, e.g. dilazep
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/63—Compounds containing para-N-benzenesulfonyl-N-groups, e.g. sulfanilamide, p-nitrobenzenesulfonyl hydrazide
- A61K31/635—Compounds containing para-N-benzenesulfonyl-N-groups, e.g. sulfanilamide, p-nitrobenzenesulfonyl hydrazide having a heterocyclic ring, e.g. sulfadiazine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C205/00—Compounds containing nitro groups bound to a carbon skeleton
- C07C205/49—Compounds containing nitro groups bound to a carbon skeleton the carbon skeleton being further substituted by carboxyl groups
- C07C205/57—Compounds containing nitro groups bound to a carbon skeleton the carbon skeleton being further substituted by carboxyl groups having nitro groups and carboxyl groups bound to carbon atoms of six-membered aromatic rings of the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/04—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms
- C07D295/14—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D295/145—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals with the ring nitrogen atoms and the carbon atoms with three bonds to hetero atoms attached to the same carbon chain, which is not interrupted by carbocyclic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/10—Spiro-condensed systems
Abstract
Un compuesto de fórmula (I): **(Ver fórmula)** en donde R1 representa Me, Et, CH=CH2, C≡C-H o C≡C-Me; uno de R2 y R3 representa un grupo seleccionado de H, alquilo-C1-C6, alcoxi-C1-C6, cicloalquilo-C3-C8, CH2- (cicloalquilo-C3-C8), halógeno y ciano y el otro representa el grupo -Z-Rx; Z representa CO o SO2; Rx representa la fórmula (iv): **(Ver fórmula)** en donde V representa CO; en donde v representa 0 o 1; en donde n representa 0, 1 o 2, excepto que cuando v representa 1, n representa 1 o 2; m representa 1 o 2; X representa CH o N, excepto que cuando n representa 0 o 1, X representa CH; Y representa CH o N; W representa un grupo seleccionado de alquilo-C1-C4, hidroxialquilo-C1-C4, alcoxi-C1-C4-alquilo(C1-C4), alquileno-C1-C4CONR20R21, alquileno-C1-C4NR20COR21, SO2alquilo(C1-C4), CO-alquilo-(C1-C4), halógeno, CN, OH y NR22R23 excepto que cuando W representa NR22R23, alquileno-C1NR20COR21 o halógeno, Y representa CH; R20, R21, R22, R23, R24 y R25 representan independientemente H o-alquilo-C1-C4; o un compuesto seleccionado del grupo que consiste en: del grupo que consiste en: **(Ver fórmula)** o una sal farmacéuticamente aceptable del mismo.
Description
DESCRIPCIÓN
Derivados de indol y su uso como inhibidores de proteína cinasa
Campo de la invención
La invención se refiere, inter alia, a nuevos compuestos que inhiben las proteínas cinasas y a su uso en terapia, particularmente para el tratamiento de enfermedades fibróticas o enfermedades pulmonares intersticiales, especialmente Fibrosis Pulmonar Idiopática y enfermedades respiratorias. En un aspecto, la invención se refiere a composiciones farmacéuticas que comprenden los compuestos.
Antecedentes de la invención
Las enfermedades pulmonares intersticiales (ILD) se caracterizan por la cicatrización del pulmón que conduce a una disfunción pulmonar, que eventualmente puede conducir a insuficiencia respiratoria. Hay muchas ILD sin causa conocida, que se denominan idiopáticas. La Fibrosis Pulmonar Idiopática (IPF) es el tipo más común de ILD. La IPF afecta a aproximadamente 170.000 personas en Europa y 130.000 personas en los Estados Unidos, con aproximadamente 48.000 nuevos casos diagnosticados cada año solo en EE.UU. y con 40.000 personas muertas al año en EE.UU. Las tasas de mortalidad por IPF son muy altas, con una mediana de supervivencia de 3-5 años desde el diagnóstico y tasas de supervivencia informadas a 5 años inferiores a 30%, a la par con los cánceres más letales. Hasta hace poco, pocas opciones de tratamiento distintas del trasplante de pulmón han demostrado ser efectivas y el tratamiento para la mayoría de los pacientes ha sido el control de los síntomas y los cuidados paliativos.
La IPF es una enfermedad crónica y mortal caracterizada principalmente por una disminución progresiva de la función pulmonar causada por la cicatrización del tejido pulmonar que da como resultado un empeoramiento de la disnea. El factor de crecimiento endotelial vascular (VEGF), el factor de crecimiento de fibroblastos (FGF) y el factor de crecimiento derivado de plaquetas (PDGF) son mitógenos potentes conocidos para las células de fibroblastos, que luego reemplazan el tejido normal en los pulmones cuando se produce la fibrosis. En las ILD, se ha demostrado clínicamente la evidencia de un papel patógeno para PDGF, VEGF y FGF. El sitio principal afectado es el intersticio, el tejido entre los sacos de aire en el pulmón, pero también afecta a los espacios aéreos, las rutas respiratorias periféricas y los vasos. Se cree que el proceso de la enfermedad se inicia por una serie de microlesiones en el epitelio alveolar del pulmón. Después de la lesión, la permeabilidad vascular aumentada conduce a la formación de coágulos y las células epiteliales residentes proliferan en un intento de reemplazar aquellas células que murieron como resultado de la lesión. Este proceso desencadena la liberación de una variedad de factores de crecimiento (p. ej., PDGF, VEGF, FGF y factor p de crecimiento transformante (TGFp)), lo que conduce a la activación aberrante de las células epiteliales, la remodelación vascular anormal y, lo más notablemente, la proliferación y migración de fibroblastos al pulmón. Los factores de crecimiento también inducen a las células residentes a transformarse en miofibroblastos, que junto con los fibroblastos se organizan en focos (King TE Jr, et al., Lancet, 2011,3;378(9807):1949-61; Selman M, et al., Ann Intern Med., 2001, 16;134(2):136-51). Estos cambios celulares dan como resultado la ruptura de la membrana basal y la acumulación excesiva de proteínas de la matriz extracelular en el espacio intersticial. El resultado es la eventual destrucción de la arquitectura normal de la unidad capilar alveolar y la cicatrización pulmonar. Las patologías que definen el patrón intersticial habitual (UIP) de fibrosis característico de la IPF son un patrón heterogéneo de áreas alternas de pulmón normal, inflamación intersticial, fibrosis densa, focos fibroblásticos y panalización, especialmente en el área subpleural del pulmón (Du Bois RM., Nat Rev Drug Discov., 2010, 9(2):129-40; Selman M, et al., Ann Intern Med., 2001, 16;134(2):136-51; King TE Jr, et al., Lancet, 2011, 3;378(9807):1949-61). La pérdida de la arquitectura normal y la cicatrización del espacio intersticial conduce a una disminución significativa en la capacidad de intercambio de gases que conduce al desarrollo de los síntomas clásicos de la enfermedad, a saber, disnea, tos crónica, crepitaciones inspiratorias en la auscultación y espirometría anormal (Castriotta RJ, et al., Chest, 2010, 138(3):693-703). Aunque el curso de la enfermedad es heterogéneo, la mediana de supervivencia es de aproximadamente 3-5 años y la causa más común de muerte es la insuficiencia respiratoria debido a patologías progresivas que alteran el funcionamiento normal de los pulmones y el intercambio de gases.
Para lograr una mejor tolerabilidad y también una mejor eficacia en el tratamiento de los trastornos pulmonares, puede ser ventajoso administrar un fármaco directamente en el lugar de acción en el pulmón. Esto permite que se alcancen concentraciones más altas de fármaco en el sitio de acción, lo que da como resultado una dosis total más baja que, en consecuencia, reduce los efectos secundarios sistémicos.
El nintedanib, un inhibidor de la proteína quinasa, fue aprobado por la FDA para el tratamiento de la IPF en 2014 mediante administración oral. Sin embargo, se asocia con eventos adversos sistémicos importantes, que incluyen dolor abdominal, vómitos y diarrea. El documento WO2006/067165 enseña que se puede esperar que los inhibidores de VEGFR, FGFR y PDGFR, tales como nintedanib, sean útiles en el tratamiento de enfermedades fibróticas, tales como IPF. Fehrenbach. H., et al., Virchows Arch., 1999, 435(1):20-31 divulga que el VEGFR está relacionado con la causa de la fibrosis pulmonar. Lindroos. P., Am J Physio Lung Cell Mol Physiol., 2001,280:L354-L362 enseña que la regulación positiva del receptor de PDGF es un mecanismo de hiperplasia de miofibroblastos durante la fibrosis pulmonar. El documento WO01/27081 muestra que los compuestos que tienen efectos inhibidores sobre cinasas, incluyendo VEGFR, PDGFR y FGFR, son adecuados para tratar enfermedades fibróticas y divulga una serie de indolinonas sustituidas en posición 6. Del mismo modo, los documentos WO2006/067165 y WO2006/067168 también
describen indolinonas sustituidas en posición 6 para su uso como medicamentos para el tratamiento o prevención de enfermedades fibróticas.
Sigue existiendo una necesidad en la técnica de desarrollar compuestos adicionales, especialmente compuestos que se toleren mejor que nintedanib, p°ara tratar enfermedades fibróticas y enfermedades pulmonares intersticiales, tales como IPF. Deseablemente, tales compuestos tendrían una dosis baja, una acción de larga duración adecuada para una dosificación de una, dos o tres veces al día y una buena eficacia y tolerabilidad, especialmente cuando se administran tópicamente al pulmón. Los compuestos de fórmula (I) descritos en este documento abordan este problema.
Compendio de la invención
Según la invención, se proporciona un compuesto de fórmula (I):

en la que
Ri representa Me, Et, CH=CH2 , C=CH o C=C-Me;
uno de R2 y R3 representa un grupo seleccionado de H, alquilo-Ci-C6, alcoxi-Ci-C6, cicloalquilo-C3-Cs, -CH2-(cicloalquilo-C3-C8), halógeno y ciano y el otro representa el grupo-Z-Rx;
Z representa CO o SO2 ;
Rx representa la fórmula (iv):

en donde V representa CO;
en donde v representa 0 o 1;
en donde n representa 0, 1 o 2, excepto que cuando v representa 1, n representa 1 o 2;
m representa 1 o 2;
X representa CH o N, excepto que cuando n representa 0 o 1, X representa CH;
Y representa CH o N;
W representa un grupo seleccionado de alquilo-C1-C4, hidroxialquilo-C1-C4, alcoxi-C1-C4-alquilo(C1-C4), alquileno-C1-C4-CONR20R21, alquileno-C1-C4NR20COR21, SO2-alquilo(C1-C4), CO-alquilo-(C1-C4), halógeno, CN, OH y NR22R23 excepto que cuando W representa NR22R23, alquileno-C1NR20COR21 o halógeno, Y representa CH;
R20, R21, R22, R23, R24 y R25 representan independientemente H o alquilo-C1-C4;
o un compuesto seleccionado del grupo que consiste en:

o una sal farmacéuticamente aceptable del mismo.
(en adelante en este documento "compuestos de la invención" o "un compuesto de la invención").
Breve descripción de las figuras
Figura 1: muestra la permeabilidad de la membrana artificial de los ejemplos representativos (3-12, 17-22, 25-27, 29 40, 43-49) de la invención y nintedanib (véanse los resultados del ensayo de permeabilidad PAMPA y la Tabla 7: se utilizaron valores medios para los compuestos en los que se repitió el experimento)
Figura 2: muestra la exposición pulmonar total después de la administración intravenosa e intratraqueal de ejemplos de la invención o nintedanib en ratas (véanse los resultados de mediciones farmacocinéticas en roedores)
Figura 3: muestra la exposición pulmonar total después de la administración intravenosa e intratraqueal de ejemplos de la invención o nintedanib en ratas (véanse los resultados de mediciones farmacocinéticas en roedores)
Descripción detallada de la invención
Los grupos alquilo pueden ser de cadena lineal o ramificada. Los grupos alquilo-C1-8 pueden representar, por ejemplo, alquilo-C1-6, alquilo-C1-4 o alquilo-C1-3. Los grupos alquilo ilustrativos incluyen metilo, etilo, n-propilo, i-propilo, n-butilo, t-butilo y CH2CHMe2. En una realización, alquilo se refiere a alquilo de cadena lineal. El alquileno debe interpretarse de la misma forma que el alquilo, excepto que es un grupo divalente.
Alcoxi, como se usa en este documento, significa -Oalquilo e incluye alcoxi de cadena lineal o ramificada, por ejemplo, metoxi, etoxi, propoxi, butoxi.
Hidroxialquilo significa alquilo con un sustituyente hidroxilo en cualquier posición. Los ejemplos incluyen hidroximetilo, 2-hidroxietilo, 3-hidroxi-n-propilo y 4-hidroxi-n-butilo.
Los halógenos pueden ser adecuadamente Br, Cl o F, especialmente Cl o F, particularmente F.
Los ejemplos de anillos heterocíclicos alifáticos de 4-8 miembros que contienen uno o más heteroátomos seleccionados independientemente entre N, O y S incluyen azetidina, pirrolidina, piperidina, piperazina, morfolina, dioxano, tetrahidrofurano, tiomorfolina, tetrahidropirano y diazepan. De forma adecuada, el anillo heterocíclico incluye 1 o 2, especialmente 1 heteroátomo. Dichos anillos pueden contener un carbonilo y los ejemplos incluyen pirrolidinona, piperazinona, diazepanona y piperidinona. Tales anillos que pueden representar R4 y R5 pueden contener un grupo sulfona tal como 1,1-dioxo-1 -tiomorfolin-1 -ilo.
Cicloalquilo-C3-C8 se refiere a un anillo carbocíclico alifático que contiene típicamente de 3 a 8 miembros en el anillo con ramificación opcional y que contiene de 3 a 8 átomos de carbono en total. Los ejemplos incluyen ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, metilciclohexilo, cicloheptilo y ciclooctilo. En una realización, los anillos no están sustituidos. En otra realización, el anillo lleva un sustituyente, por ejemplo, dimetilamino. Un ejemplo de un anillo de cicloalquilo sustituido es 1-(dimetilamino)ciclobutilo.
Los anillos heterocíclicos alifáticos de 4-8 miembros pueden estar opcionalmente sustituidos. En una realización, los anillos no están sustituidos. En otra forma de realización, el anillo tiene un sustituyente. En otra forma de realización, el anillo tiene dos sustituyentes. Un sustituyente puede estar en un átomo de carbono o nitrógeno. Dichos anillos que Het puede representar o estar dentro de la definición de espiro-C pueden estar sustituidos con metilo. Los ejemplos incluyen morfolinometilo, pirrolidin-1 -ilmetilo, 4-metil-1,4-diazepan-2-ona y 4-metilpiperazin-2-ona.
Los ejemplos de anillos heterocíclicos sustituidos que pueden representar R4 y R5 incluyen 1-metil-piperazina, 1-metilpiperidina, 1-metil-1,4-diazepano, 4-dimetilamino-piperidina, 4-hidroxi-piperidina , 1-(2-hidroxietil)piperidina, 4-(hidroximetil)piperidina, 1-(2-metoxietil)piperidina, 4,4-difluoropiperidina, 4-fluoropiperidina, 1-acetilpiperazina, 4-metil-1,4-diazepan-2-ona, 1 -acetil-1,4-diazepano, 4-(dimetilamino)tetrahidro-2H-pirano, 4-(metilsulfonil)piperazina y 3-hidroxi-1 -metilpirrolidina.
Espiro-C se refiere a un átomo de carbono cuaternario que conecta dos anillos heterocíclicos alifáticos juntos según la definición anterior para formar un anillo espiro bicíclico. Ejemplos de anillos espiro bicíclicos incluyen espiro[5.5]undecano y espiro[5.6]dodecano. Ejemplos de anillos espiro bicíclicos sustituidos incluyen 1-metil-5-oxo-1,4,9-triazaspiro[5.5]undecano y 7-metil-12-oxo-3,7,11 -triazaspiro[5.6]dodecano.
En una realización, se proporciona una sal farmacéuticamente aceptable del compuesto de la invención.
Los compuestos de la divulgación incluyen aquellos en los que el átomo especificado es un isótopo de origen natural o no natural. En una realización, el isótopo es un isótopo estable. Por tanto, los compuestos de la divulgación incluyen, por ejemplo, aquellos que contienen uno o más átomos de deuterio en lugar de átomos de hidrógeno y similares.
La divulgación también se extiende a todas las formas polimórficas de los compuestos definidos en este documento, incluidas las sales de los mismos.
La divulgación también se extiende a todos los solvatos de los compuestos definidos en este documento. Los ejemplos de solvatos incluyen hidratos.
Adecuadamente, R1 representa Me.
En una realización preferida R2 representa un grupo seleccionado entre H, alquilo-C1-C6 , alcoxi-C1-C6 , cicloalquilo-C3-C8, -CH2-(cicloalquilo-C3-C8), halógeno y ciano y R3 representa el grupo -Z-Rx. En una realización alternativa R2 representa el grupo -Z-Rx y R3 representa un grupo seleccionado entre H, -alquilo-C1-C6 , alcoxi-C1-C6 , cicloalquilo-C3-C8,-CH2-(cicloalquilo-C3-C8), halógeno y ciano.
Adecuadamente, cuando R2 o R3 no representan -Z-Rx, representan un grupo seleccionado de H, alquilo-Ci-C6, alcoxi-C1-C6 , cicloalquilo-C3-C6, halógeno y ciano, más adecuadamente H, alquilo-C1-C4 o halógeno, aún más adecuadamente H, Me o halógeno, más adecuadamente H, Me o F, especialmente H.
En una realización preferida, Z representa CO. En una realización alternativa, Z es SO2.
Adecuadamente, R1 representa Me.
Adecuadamente, Z representa CO.
Adecuadamente, R3 representa -Z-Rx.
Cuando v es 0, adecuadamente n representa 0 o 2, especialmente 0.
Cuando v es 1, adecuadamente n representa 1.
Adecuadamente, v representa 0.
Adecuadamente, m representa 1.
De forma adecuada, X representa CH.
Adecuadamente, Y representa N.
De forma adecuada, W representa alquilo-C1-C4 (especialmente Me) o NR22R23 (especialmente NMe2).
Los compuestos de fórmula (I) se pueden preparar convenientemente mediante un proceso que comprende hacer reaccionar un compuesto de fórmula (II), en el que L es un grupo saliente, tal como -O-alquilo-C1-C4, p. ej., -Oetilo:

Normalmente, los compuestos de fórmulas (II) y (III) se pueden hacer reaccionar en presencia de un disolvente, como DMF, y calentarse a aproximadamente 80°C durante aproximadamente 18 horas. Después de esta etapa, se realiza una etapa de desprotección para eliminar el grupo protector, acetilo. Para lograr esto, la mezcla de reacción se puede enfriar a temperatura ambiente y se puede añadir un nucleófilo, tal como piperidina, y agitar durante 1 a 24 horas. Los compuestos de fórmula (II) en la que L representa -Oetilo se pueden preparar haciendo reaccionar un compuesto de fórmula (IV):

o un derivado protegido del mismo con un compuesto de fórmula (V):
Normalmente, los compuestos de fórmulas (IV) y (V) se pueden hacer reaccionar en presencia de anhídrido acético a una temperatura de aproximadamente 110°C durante aproximadamente 4 horas. Se pueden preparar otros compuestos de fórmula (II) de manera análoga.
Los compuestos de fórmula (IV) se pueden preparar haciendo reaccionar un compuesto de fórmula (VI):

con anhídrido acético. Normalmente, la reacción se realiza a aproximadamente 110 ° C. Alternativamente, los compuestos de fórmula (II) en la que L representa-Oetilo se pueden preparar directamente a partir de compuestos de fórmula (VI) mediante tratamiento con un compuesto de fórmula (V) en presencia de anhídrido acético a una temperatura de aproximadamente 110 °C durante aproximadamente 4 horas.
Los compuestos de fórmula (VI) se pueden preparar reduciendo el grupo -NO2 de un compuesto de fórmula (VII):

a un grupo-NH2 seguido de una ciclación formadora de amida, que es un procedimiento bien conocido en el campo. Las condiciones de reducción y ciclación de amida pueden incluir típicamente el uso de H2-Pd/C a temperatura ambiente e hidrogenando a una presión de 5 bares durante aproximadamente 36 horas en un disolvente, tal como ácido acético, que es un procedimiento bien conocido en la técnica.
Los compuestos de fórmula (VII) se pueden preparar haciendo reaccionar un compuesto de fórmula (VIII):

con cloroacetato de metilo. Normalmente, la reacción se produce en presencia de un disolvente orgánico polar, como DMF, y una base, como KOBu, bajo una atmósfera de nitrógeno entre aproximadamente-20 y -10 °C.
Alternativamente, los compuestos de fórmula (Ia), que son compuestos de fórmula (I) en la que Z es CO, pueden prepararse haciendo reaccionar un compuesto de fórmula (IXa) o (IXb):

o derivados protegidos de los mismos con un compuesto de fórmula H-Rx o un derivado protegido del mismo mediante una reacción de condensación entre el grupo NH-amino presente en H-Rx y el COOH en (IXa) y (IXb). Los compuestos se pueden hacer reaccionar normalmente durante aproximadamente 2 a 18 horas a temperatura ambiente en presencia de un agente de acoplamiento, como HATU y una base, como la base de Hünig (DIPEA), en un disolvente orgánico polar como DMF, aunque se pueden utilizar otros disolventes orgánicos. Este proceso puede ir seguido, en su caso, de desprotección.
Los compuestos de fórmulas (IXa) y (IXb) se pueden preparar mediante desprotección de compuestos de fórmulas (Xa) y (Xb) respectivamente:

La desprotección se puede lograr usando reactivos estándar en la técnica, tales como TFA, y los compuestos se agitan típicamente a temperatura ambiente durante aproximadamente 16 horas en un disolvente, como DCM.
Los compuestos de fórmulas (Xa) y (Xb) pueden prepararse haciendo reaccionar un compuesto de fórmula (II) con un compuesto de fórmula (XIa) y (XIb) respectivamente:

Los compuestos se pueden hacer reaccionar en presencia de DMF durante aproximadamente 18 horas a 100°C.
La síntesis de compuestos de fórmula (111) se puede preparar haciendo reaccionar un compuesto de fórmula H-Rx con un compuesto de fórmula (XIla) o (XIIb) respectivamente:

Los compuestos se pueden hacer reaccionar típicamente en presencia de base de Hünig (DIPEA) y DMF durante aproximadamente 16 horas a temperatura ambiente, seguido de una etapa de desprotección usando reactivos estándar en la técnica, tales como TFA.
Nuevos compuestos intermedios de fórmula (IXb) y (Xb) en donde R1 es Me, Et, CH=CH2 , C=C-H o C=C-Me y sus sales se reivindican como un aspecto de la invención.
Los compuestos de fórmulas (V), (VIII), (XIa), (XIb), (Xlla), (XIIb), (XIV), NHR14R15 y HRx pueden prepararse mediante métodos conocidos o métodos análogos a aquellos descritos en este documento.
Los compuestos de fórmula (I) pueden prepararse o emplearse en forma de una sal farmacéuticamente aceptable, incluidas las sales de adición de ácido no tóxicas y terapéuticamente activas que los compuestos de fórmula (I) pueden formar. Estas sales de adición a un ácido farmacéuticamente aceptables se pueden obtener de forma conveniente tratando la forma de base con tales ácidos apropiados en un disolvente o mezcla de disolventes adecuados. Los ácidos apropiados comprenden, por ejemplo, etanosulfónico, maleico, malónico, L-tartárico, fumárico, cítrico, succínico, acético, trifenilacético, clorhídrico, sulfúrico, fosfórico, 1-hidroxi-2-naftoico, bromhídrico, metanosulfónico, tartárico, palmítico, isetiónico, pamoico, fórmico, cinámico benzoico, ascórbico, galáctico, láctico, málico, oxálico, paratoluenosulfónico, bencenosulfónico, propiónico, furoico, fosfónico y glutárico. Por el contrario, dichas formas de sales se pueden convertir por tratamiento con una base apropiadas en la forma de base libre.
La invención proporciona un compuesto de la invención para uso como un producto farmacéutico.
En consecuencia, la presente invención también proporciona una composición farmacéutica que comprende un compuesto según la invención opcionalmente en combinación con uno o más diluyentes o portadores farmacéuticamente aceptables.
Los diluyentes y portadores pueden incluir aquellos adecuados para administración parenteral, oral, tópica, que incluye por inhalación a través de la boca a los pulmones o inhalación a través de la nariz, administración mucosal y rectal, y pueden ser diferentes dependiendo de la ruta de administración.
En una realización, las composiciones pueden prepararse, por ejemplo, para administración parenteral, p. ej., administración subcutánea, intramuscular, intravenosa, intradérmica, intraarticular o periarticular, particularmente en forma de disoluciones o suspensiones líquidas; para administración oral, particularmente en forma de comprimidos, cápsulas, polvos, gránulos, dispersiones sólidas o en forma de soluciones o suspensiones líquidas que incluyen nanosuspensiones; para inhalación en los pulmones o la nariz, p. ej., administración pulmonar o intranasal, particularmente en forma de polvos secos, disoluciones, suspensiones que incluyen nanosuspensiones para nebulización, aerosoles nasales o gotas que comprenden disoluciones o suspensiones o suspensión o aerosoles de disolución o suspensión presurizada o no presurizada; para administración tópica o transdérmica, p. ej., como cremas, aerosoles, espumas, geles, ungüentos, líquidos, parches; para la administración mucosal, p. ej., a la mucosa bucal, sublingual o vaginal, y para la administración rectal, p. ej., en forma de espuma o supositorio.
Las composiciones pueden administrarse convenientemente en forma de dosificación unitaria y pueden prepararse mediante cualquiera de los métodos bien conocidos en la técnica farmacéutica, por ejemplo, como se describe en Remington’s Pharmaceutical Sciences, 17a ed., Mack Publishing Company, Easton, PA., (1985 ). Las composiciones también pueden administrarse convenientemente en forma de dosificación unitaria múltiple.
Las formulaciones para administración parenteral pueden contener como excipientes agua estéril o disolución salina, tampones, agentes ajustadores de tonicidad, conservantes, antioxidantes, agentes ajustadores de viscosidad, alquilenglicoles como propilenglicol, polialquilenglicoles como polietilenglicol, aceites de origen vegetal, naftalenos hidrogenados y similares.
Las composiciones adecuadas para la administración oral pueden comprender uno o más portadores fisiológicamente compatibles y/o excipientes y pueden estar en forma sólida o líquida. Los comprimidos y cápsulas se pueden preparar con agentes aglutinantes, por ejemplo, jarabe, goma arábiga, gelatina, sorbitol, tragacanto, celulosas o polivinilpirrolidona; cargas, tales como lactosa, sacarosa, almidón de maíz, fosfato cálcico, sorbitol o glicina; lubricantes, tales como estearato de magnesio, talco, polietilenglicol o sílice; y tensioactivos, como lauril sulfato de sodio. Las composiciones líquidas pueden contener aditivos convencionales tales como agentes de suspensión, por ejemplo, jarabe de sorbitol, metilcelulosa, jarabe de azúcar, gelatina, carboximetilcelulosa o grasas comestibles; agentes emulsionantes y tensioactivos tales como lecitina o goma arábiga; aceites vegetales tales como aceite de almendras, aceite de coco, aceite de hígado de bacalao o aceite de cacahuete; conservantes como el hidroxianisol butilado (BHA) e hidroxitolueno butilado (BHT). Las composiciones líquidas pueden encapsularse, por ejemplo, en gelatina para proporcionar una forma de dosificación unitaria.
Las formas de dosificación oral sólidas incluyen comprimidos, cápsulas de cubierta dura de dos piezas y cápsulas de gelatina elástica blanda (SEG). Tales cápsulas de cubierta dura de dos piezas pueden estar hechas de, por ejemplo, gelatina o hidroxipropilmetilcelulosa (HPMC).
Una formulación de cubierta seca generalmente comprende aproximadamente una concentración de 40%-60% de gelatina, aproximadamente una concentración de 20%-30% de plastificante (como glicerina, sorbitol o propilenglicol) y aproximadamente una concentración de 30%-40% de agua. También pueden estar presentes otros materiales tales como conservantes, tintes, opacificantes y aromatizantes. El material de relleno líquido comprende un fármaco sólido que se ha disuelto, solubilizado o dispersado (con agentes de suspensión como cera de abejas, aceite de ricino hidrogenado o polietilenglicol 4000) o un fármaco líquido en vehículos o combinaciones de vehículos como aceite mineral, aceites vegetales, triglicéridos, glicoles, polioles y agentes tensioactivos.
Las formulaciones para administración nasal pueden ser polvos y pueden contener excipientes, por ejemplo, lactosa o dextrano, o pueden ser disoluciones acuosas o aceitosas para su uso en forma de gotas nasales o aerosol dosificado. Las formulaciones para administración nasal también pueden estar en forma de suspensiones acuosas o disoluciones o suspensiones no acuosas presurizadas. Para la administración bucal, los excipientes típicos incluyen azúcares, estearato de calcio, estearato de magnesio, almidón pregelatinizado y similares.
De manera adecuada, el compuesto de fórmula (I) se administra tópicamente al pulmón. Por tanto, en una realización se proporciona una composición farmacéutica que comprende un compuesto de la divulgación opcionalmente en combinación con uno o más diluyentes o portadores tópicamente aceptables.
La administración tópica al pulmón se puede lograr mediante el uso de una formulación no presurizada, como una disolución o suspensión acuosa. Estas formulaciones pueden administrarse por medio de un nebulizador, p. ej., uno que puede ser de mano y portátil o para uso doméstico u hospitalario (es decir, no portátil). La formulación puede comprender excipientes tales como agua, tampones, agentes de ajuste de la tonicidad, agentes de ajuste del pH, tensioactivos, conservantes, agentes de suspensión, agentes de carga y codisolventes. Las formulaciones de aerosol y líquido en suspensión (persurizadas o sin presurizar) contendrán generalmente el compuesto de la invención en forma finamente dividida adecuada para depósito en el pulmón, por ejemplo, con un D50 de 0,5-10 pm p. ej., alrededor de 1-5 pm. Los polvos en forma finamente dividida se pueden preparar mediante un proceso de micronización o
molienda, mediante secado por pulverización, por congelación por pulverización o por molienda en húmedo seguida de secado por pulverización. La micronización se puede realizar utilizando un molino de chorro como los fabricados por Hosokawa Alpine. La distribución de tamaño de partícula resultante puede medirse usando difracción láser (por ejemplo, con un instrumento Malvern Mastersizer 2000S o Mastersizer 3000). Las distribuciones de tamaño de partículas se pueden representar usando valores D10, D50 y D90. El valor de la mediana de D50 de las distribuciones de tamaño de partícula se define como el tamaño de partícula que divide la distribución a la mitad. La medición derivada de la difracción láser se describe con mayor precisión como una distribución de volumen y, en consecuencia, el valor D50 obtenido usando este procedimiento se denomina más significativamente como un valor Dv50 (mediana para una distribución de volumen). Como se usa en este documento, los valores Dv se refieren a distribuciones de tamaño de partículas medidas usando difracción láser. De manera similar, los valores D10 y D90, utilizados en el contexto de la difracción láser, se toman como valores de Dv10 y Dv90 y se refieren al tamaño de partícula en el que el 10% de la distribución se encuentra por debajo del valor D10, y el 90% de la distribución se encuentra por debajo del valor D90, respectivamente. En otra realización, las partículas de material compuesto del compuesto de la divulgación y los excipientes para su uso en la nebulización de una formulación en suspensión pueden formarse mediante co-molienda y/o co-secado por pulverización del compuesto y excipientes juntos, en donde las partículas de material compuesto que comprenden tanto el agente activo como excipientes tienen un D50 de 1-10 m. Las formulaciones de suspensión acuosa para administración al pulmón también podrían comprender nanosuspensiones o suspensiones de partículas de material compuesto que contienen nanopartículas.
La administración tópica al pulmón también se puede lograr mediante el uso de una formulación en aerosol presurizada. Las formulaciones en aerosol comprenden típicamente el ingrediente activo suspendido o disuelto en un propelente de aerosol adecuado, tal como un clorofluorocarbono (CFC) o un hidrofluorocarbono (HFC). Los propelentes de CFC adecuados incluyen tricloromonofluorometano (propelente 11), diclorotetrafluorometano (propelente 114) y diclorodifluorometano (propelente 12). Los propelente HFC adecuados incluyen tetrafluoroetano (HFC-134a) y heptafluoropropano (HFC-227). El propelente normalmente comprende 40%-99,5% p. ej., 40%-90% en peso de la composición de inhalación total. La formulación puede comprender excipientes que incluyen codisolventes (p. ej., etanol) y tensioactivos o estabilizadores (por ejemplo, lecitina, trioleato de sorbitán y similares). Otros posibles excipientes incluyen polietilenglicol, polivinilpirrolidona, glicerina y similares. Las formulaciones en aerosol se envasan en botes y se administra una dosis adecuada por medio de una válvula dosificadora (p. ej., la suministrada por Bespak, Aptar o 3M o alternativamente por Coster o Vari). Las formulaciones de suspensiones presurizadas para administración al pulmón también podrían comprender nanosuspensiones o suspensiones de partículas de material compuesto que contienen nanopartículas.
La administración tópica al pulmón también se puede lograr mediante el uso de una formulación de polvo seco. Una formulación de polvo seco contendrá el compuesto de la divulgación en forma finamente dividida, típicamente con un D50 de 0,5-10 pm p. ej., alrededor de 1-5 pm. Los polvos en forma finamente dividida se pueden preparar mediante un proceso de micronización o molienda, mediante secado por pulverización, por congelación por pulverización o por molienda en húmedo seguida de secado por pulverización. La micronización se puede realizar utilizando un molino de chorro como los fabricados por Hosokawa Alpine. La distribución de tamaño de partícula resultante puede medirse usando difracción láser (por ejemplo, con un instrumento Malvern Mastersizer 2000S o Mastersizer 3000). La formulación contendrá típicamente uno o más diluyentes tópicamente aceptables tales como lactosa, glucosa, trehalosa o manitol (preferiblemente lactosa), por lo general de comparativamente tamaño de partícula, p. ej., un D50 de 15-250 m. En otra realización, las partículas de material compuesto del compuesto de la divulgación y los excipientes también pueden formarse mediante co-molienda y/o co-secado por pulverización del compuesto y excipientes juntos, en donde las partículas de material compuesto que comprenden tanto agente activo y excipientes tienen un D50 de 1-10 m. Como se usa en este documento, el término "lactosa" se refiere a un componente que contiene lactosa, que incluye monohidrato de a-lactosa, monohidrato de p-lactosa, a-lactosa anhidra, p-lactosa anhidra y lactosa amorfa. Los componentes de lactosa pueden procesarse mediante micronización, tamizado, molienda, compresión, aglomeración o secado por pulverización. También se incluyen las formas disponibles comercialmente de lactosa en diversas formas, por ejemplo, productos Lactohale® (DFE Pharma), InhaLac® (Meggle), Pharmatose® (DFE Pharma) y Respitose® (DFE Pharma). En una realización, el componente de lactosa se selecciona del grupo que consiste en monohidrato de a-lactosa, a-lactosa anhidra y lactosa amorfa. Preferiblemente, la lactosa es monohidrato de a-lactosa.
Las formulaciones de polvo seco también pueden contener otros excipientes como leucina, estearato de sodio, estearato de calcio o estearato de magnesio. Las partículas de polvo seco podrían ser partículas de material compuesto y podrían estar comprendidas por nanopartículas en una matriz compuesta.
Una formulación de polvo seco generalmente se administra mediante un dispositivo inhalador de polvo seco (DPI). Puede ser un dispositivo de dosis unitaria donde la formulación se presenta en unidades individuales, ya sea como cápsulas o ampollas, o en un dispositivo multidosis donde más de una dosis de formulación está contenida en un dispositivo, ya sea en un depósito a granel o como múltiples recipientes dentro de un dispositivo (p. ej., múltiples ampollas o bolsillos). Ejemplos de inhaladores de polvo seco incluyen SPINHALER, DISKHALER, Tu RBOHALER, DISKUS, ELLIPTA,CLICKHALER, ECLIPSE, ROTAHALER, HANDIHALER, AEROLISER, CYCLOHALER, MONODOSE, BREEZHALER/NEOHALER, FLOWCAPS, TWINCAPS, X-CAPS, TWISTER, TURBOSPIN, ELPENHALER, TURBUHALER, MIATHALER, NEXTHaler, TWISTHALER, NOVOLIZER, GENUAIR, SKYEHALER,
inhalador polvo seco ORIEL, MICRODOSE, ACCUHALER, PULVINAL, EASYHALER, ULTRAHALER, TAIFUN, PULMOJET, OMNIHALER, GYROHALER, TAPER, CONIX, XCELOVAIR y PROHALER.
Se espera que los compuestos de la invención sean útiles en el tratamiento de enfermedades fibróticas tales como fibrosis pulmonar y enfermedades pulmonares con un componente fibrótico, p. ej., seleccionado de IPF, neumonía intersticial celular gigante, sarcoidosis, fibrosis quística, síndrome de distrés respiratorio, fibrosis pulmonar inducida por fármacos, granulomatosis, silicosis, asbestosis, esclerodermia sistémica, cirrosis hepática inducida por virus seleccionada de cirrosis hepática inducida por hepatitis C, o enfermedades de la piel con un componente fibrótico, p. ej., seleccionado de esclerodermia, sarcoidosis y lupus eritematoso sistémico, y especialmente IPF. Más generalmente, se espera que los compuestos de la invención sean útiles en el tratamiento de enfermedades pulmonares intersticiales. Además, se espera que los compuestos de la invención sean útiles en el tratamiento de enfermedades caracterizadas por hiperproliferación de células, por ejemplo, cáncer y en donde los compuestos se administran por inhalación, particularmente cáncer de pulmón. Además, los compuestos de la invención también pueden ser útiles en el tratamiento de trastornos respiratorios que incluyen COPD (incluyendo bronquitis crónica y enfisema), asma, asma pediátrica, rinitis alérgica, rinitis, sinusitis, especialmente asma, bronquitis crónica y COPD.
También se espera que los compuestos de la invención sean útiles en el tratamiento de otras enfermedades fibróticas tales como fibrosis pulmonar asociada con artritis reumatoide, síndrome de distrés respiratorio que incluye síndrome de distrés respiratorio agudo, lesión pulmonar aguda, fibrosis o neumonitis pulmonar inducida por radiación, neumonitis por hipersensibilidad crónica, esclerosis sistémica, síndrome de Sjogren, enfermedades pulmonares intersticiales, hipertensión arterial pulmonar (PAH), incluido el componente vascular de la PAH, o enfermedades de la piel con un componente fibrótico, p. ej., seleccionadas de cicatrices hipertróficas y queloides, o enfermedades oculares en las que la fibrosis es un componente incluyendo glaucoma, degeneración macular relacionada con la edad, edema macular diabético, enfermedad del ojo seco y retinopatía diabética, o fibrosis en el intestino, p. ej., asociada con enfermedad inflamatoria intestinal.
Además, se espera que los compuestos de la invención sean útiles en la prevención de enfermedades caracterizadas por la hiperproliferación de células, por ejemplo, cáncer, p. ej., cuando los compuestos se administran por inhalación, particularmente cáncer de pulmón.
La invención proporciona un compuesto de la invención para su uso en el tratamiento de una o más de las enfermedades mencionadas anteriormente. La invención también proporciona el uso de un compuesto de la invención en la fabricación de un medicamento para el tratamiento de una o más de las enfermedades mencionadas anteriormente.
La palabra "tratamiento" pretende abarcar tanto la profilaxis como el tratamiento terapéutico.
Los compuestos de la invención se pueden administrar una, dos o tres veces al día, especialmente una o dos veces al día. Se puede determinar una cantidad de dosificación adecuada con referencia a la gravedad de la enfermedad y el tamaño del sujeto. Las cantidades de dosificación típicas están en el intervalo de 0,01 mg a 100 mg, p. ej., 0,1 mg a 10 mg p. ej., 0,25 mg a 5 mg por dosis a ser humano para ser suministrada una vez, dos veces o tres veces por día, especialmente una o dos veces al día.
El compuesto de la divulgación también puede administrarse en combinación con uno o más de otros ingredientes activos, p. ej., ingredientes activos adecuados para tratar las afecciones mencionadas anteriormente. Por ejemplo, los posibles ingredientes activos incluyen nintedanib o pirfenidona (estos son conocidos para el tratamiento de la IPF). Otros ingredientes activos que se van a usar en combinación incluyen sustancias con actividad secretorolítica, broncolítica y/o antiinflamatoria, como agentes anticolinérgicos, miméticos beta-2, esteroides, inhibidores de PDE-IV, inhibidores de la cinasa p38 MAP, inhibidores de MK2, inhibidores de galectina, antagonistas de NK1, antagonistas de LTD4, inhibidores de EGFR, inhibidores de VEGF, inhibidores de PDGF, inhibidores de FGF, inhibidores de TGFbeta, antagonistas de LPA1, inhibidores de LOXL2, inhibidores de CTGF, pentoxifilina, N-acetilcisteína, agentes anti-IL13, agentes anti IL4, inhibidores de la integrina alfavp6, inhibidores de IGF, inhibidores de PI3K, inhibidores de mTOR, inhibidores de JNK, antagonistas de pentraxina2 y endotelina.
Otros ingredientes activos que se pueden usar en combinación incluyen sustancias con actividad antifibrótica, como inhibidores de PDE-III, agentes anti-IL4/13 combinados, inhibidores PI3k/mTOR combinados, inhibidores de autotaxina, antagonistas de P2X3, antagonistas de CTGF, antagonistas de 5-LO, antagonistas de leucotrienos e inhibidores de ROCK.
En una realización, se co-formula la combinación de ingredientes activos.
En una realización, la combinación de ingredientes activos se coadministra de forma secuencial o simultánea.
En una realización, los compuestos de la invención se administran por inhalación y los otros posibles ingredientes activos se administran por ruta oral o parenteral.
En una realización se proporciona un producto de combinación que comprende:
(A) un compuesto de la invención; y
(B) un ingrediente activo adicional (como se mencionó anteriormente)
en donde cada uno de los componentes (A) y (B) se formula en mezcla con diluyente(s) o portador(es) farmacéuticamente aceptables. La combinación puede comprender opcionalmente excipientes relevantes adicionales. En una realización, se proporciona un compuesto de la invención para uso como un medicamento para administrar en combinación con uno o más ingredientes activos adicionales (como se ha mencionado anteriormente).
Se espera que los compuestos de la invención tengan una o más de las siguientes propiedades ventajosas:
• Buena actividad inhibidora de quinasas seleccionadas de VEGFR (por ejemplo, VEGFR1 y VEGFR2), FGFR y PDGFR;
• Buena actividad anti-fibrosis, por ejemplo, según se determina en modelos in vivo (p. ej., modelo de fibrosis con bleomicina) cuando se administra tópicamente al pulmón;
• Propiedades físicas y químicas adecuadas y dosis baja para un medicamento, particularmente uno destinado a administrarse tópicamente al pulmón;
• Buen tiempo de permanencia en el pulmón o duración de la acción cuando se administra tópicamente al pulmón;
• Baja permeabilidad en el ensayo de permeabilidad PAMPA;
• Buena duración de la acción, por ejemplo, medida por inhibición de la fosforilación inducida por PDGF-BB de BDGFRp en células de fibroblastos de pulmón fetal humano;
• Buena seguridad y tolerabilidad cuando se administra tópicamente al pulmón;
• Biodisponibilidad oral baja.
Sección experimental
Las abreviaturas utilizadas en el presente documento se definen a continuación (Tabla 1). Cualquier abreviatura no definida pretende transmitir su significado generalmente aceptado.
Tabla 1: Abreviaturas:


Ejemplos de Química
Procedimientos Generales
Todos los materiales de partida y disolventes se obtuvieron de fuentes comerciales o se prepararon de acuerdo con la cita de la bibliografía. A menos que se indique lo contrario, todas las reacciones se agitaron. Las disoluciones
orgánicas se secaron de forma rutinaria sobre sulfato de magnesio anhidro. Las hidrogenaciones se realizaron en un reactor de flujo Thales H-cube en las condiciones indicadas o a presión en un autoclave de gas (bomba).
La cromatografía en columna se realizó en cartuchos de sílice preempaquetados (malla 230-400, 40-63 gm) usando la cantidad indicada. Se adquirió SCX de Supelco y se trató con ácido clorhídrico 1 M antes de su uso. A menos que se indique lo contrario, la mezcla de reacción que se iba a purificar se diluyó primero con MeOH y se acidificó con unas pocas gotas de AcOH. Esta disolución se cargó directamente en el SCX y se lavó con MeOH. El material deseado se eluyó luego lavando con NH3 al 1% en MeOH.
Cromatografía Líquida Preparativa de Fase Inversa de Alto Rendimiento
Se realizó usando detección UV a 215 y 254 nm con o bien una columna Waters X-Select Prep-C18, 5 gm, 19x50 mm que eluye con un gradiente de H2O-MeCN que contiene ácido fórmico al 0,1% v/v durante 10 min (Método A), o una columna Waters X-Bridge Prep-C18, 5 gm, 19x50 mm que eluye con un gradiente H2O-MeCN que contiene bicarbonato de amoniaco al 0,1% durante 10 min (Método B).
Métodos Analíticos
Cromatografía líquida de fase inversa de alto rendimiento
Método 1: Waters XSelect CSH C182,5 gm (4,6 x 30 mm) a 40°C; caudal 2,5-4,5 mL min-1 eluido con un gradiente de H2O-MeCN que contiene ácido fórmico al 0,1% v/v durante 4 min empleando detección UV a 254 y 215 nm. Información de gradiente: 0-3,00 min, subida en rampa desde H2O al 95%-MeCN al 5% a H2O al 5%-MeCN al 95%; 3.00- 3,01 min, mantenido a H2O-al 5%-MeCN al 95%, caudal aumentado a 4,5 mL min-1; 3,01 -3,50 min, mantenido a H2O al 5%-MeCN al 95%; 3,50-3,60 min, se hace volver a H2O al 95%-MeCN al 5%, caudal reducido a 3,50 mL min-1; 3,60-3,90 min, mantenido a H2O al 95%-MeCN al 5%; 3,90-4,00 min, mantenido a H2O al 95%-MeCN al 5%, caudal reducido a 2,5 mL min-1.
Método 2: Waters XBridge BEH C18, 2,5 gm (4,6 x 30 mm) a 40°C; caudal 2,5-4,5 mL min-1 eludo con un gradiente de H2O-MeCN que contiene bicarbonato de sodio 10 mM durante 4 min empleando detección UV a 254 nm. Información de gradiente: 0-3,00 min, elevado en rampa desde H2O al 95%-MeCN al 5% a H2O al 5%-MeCN al 95%; 3.00- 3,01 min, mantenido a H2O al 5%-MeCN al 95%, caudal aumentado a 4,5 mL min-1; 3,01-3,50 min, mantenido a H2O al 5%-MeCN al 95%; 3,50-3,60 min, se hace volver a H2O al 95%-MeCN al 5%, caudal reducido a 3,50 mL min-1; 3,60-3,90 min, mantenido a H2O al 95%-MeCN al 5%; 3,90-4,00 min, mantenido a H2O al 95%-MeCN al 5%, caudal reducido a 2,5 mL min-1.
Espectroscopia 1H RMN
Los espectros de 1H RMN se adquirieron en un espectrómetro Bruker Avance III a 400 MHz usando disolvente sin deuterar residual como referencia y a menos que se especifique otra cosa se realizaron en DMSO-d6.
Todos los nombres químicos se han generado utilizando CambridgeSoft ENotebook 12.0.
Ruta 1A
Ejemplo 1: 5-metil-3-(((4-(N-(2-(4-metilpiperazin-1 -iletil)sulfamoil)fenil)amino)(fenil)metilen)-2-oxoindolin-6-carboxilato, 0.8 form iato de (Z)-metilo
Intermedio A: 4-(2-metoxi-2-oxoetil)-2-metil-5-nitrobenzoato de metilo

A una disolución agitada de terc-butóxido de potasio (35,9 g, 320 mmol) en DMF (350 mL), en una atmósfera de nitrógeno a-20 °C, se añadió una disolución de 2-metil-5-nitrobenzoato de metilo (25,0 g, 128 mmol) y 2-cloroacetato de metilo (16,9 mL, 192 mmol) en DMF (300 mL) gota a gota durante 40 minutos. La mezcla de reacción se calentó a-10°C durante 2 h y luego se vertió sobre una suspensión de hielo-HCl (900 g de hielo, 500 mL de HCl acuoso al 35% en peso). La mezcla resultante se extrajo con DCM (2 x 600 mL) y las capas orgánicas combinadas se lavaron con salmuera (2 x 400 mL) y después se evaporaron a presión reducida. El producto bruto así obtenido se purificó mediante cromatografía en columna ultrarrápida (SiO2 , 330 g, EtOAc 0-10% en DCM, gradiente de elución) para proporcionar el compuesto de subtítulo 4-(2-metoxi-2-oxoetil)-2-metil-5-nitrobenzoato de metilo como un jarabe naranja (31,0 g, 89%); Rt 2,06 min (Método 1); m/z 266 (M-H)-(ES-); 1H RMN 5: 2,61 (3H, s), 3,62 (3H, s), 3,88 (3H, s), 4,12 (2H, s), 7,59 (1 H, s), 8,51 (1H, s).
Intermedio B: 5-metil-2-oxoindolin-6-carboxilato de metilo

A una disolución de 4-(2-metoxi-2-oxoetil)-2-metil-5-nitrobenzoato de metilo (Intermedio A) (23,0 g, 86,0 mmol) en ácido acético (301 mL, 5,25 mol) se añadió paladio sobre carbono [5 % en peso, 58% de agua, tipo 87L] (3,30 g, 1,55 mmol). La mezcla se hidrogenó a ta en una atmósfera de H2 (5 bar) durante 36 h y después se filtró a través de una almohadilla de celite. La torta filtrada se lavó con EtOAc (500 mL) y se concentró el filtrado a presión reducida. El residuo bruto se disolvió en MeOH caliente a reflujo (200 mL) y la mezcla se enfrió a ta. El sólido resultante se filtró, se enjuagó con MeOH (200 mL) y se secó in vacuo para proporcionar el compuesto del subtítulo 5-metil-2-oxoindolin-6-carboxilato de metilo como un polvo marrón (7,00 g, 39%); Rt 1,48 min (Método 1); m/z 206 (M+H)+ (ES+); 1H RMN 5: 2,45 (3H, s), 3,32 (2H, s), 3,81 (3H, s), 7,17 (1H, s), 7,22 (1 H, s), 10,43 (1 H, s).
Intermedio C: 1-acetil-3-(etoxi(fenil)metileno)-5-metil-2-oxoindolin-6-carboxilato de (£)-metilo

A una disolución agitada de 5-metil-2-oxoindolin-6-carboxilato de metilo (Intermedio B) (5,00 g, 24,4 mmol) en anhídrido acético (50,6 mL, 536 mmol) se añadió (trietoximetil)benceno (22,1 mL, 97,0 mmol) y la mezcla se agitó a 110 °C durante 3 h. A continuación, se continuó agitando a ta durante 18 h. Se concentró la mezcla de reacción a presión reducida y se diluyó el residuo con MeOH (50 mL). El sólido resultante se filtró, se enjuagó con MeOH (50 mL) y se secó in vacuo para proporcionar el compuesto del subtítulo 1-acetil-3-(etoxi(fenil)metilen)-5-metil-2-oxoindolin-6-carboxilato de (E)-metilo como un polvo amarillo (4,50 g, 48%); Rt 2,70 min (Método 1); m/z 380 (M+H)+ (ES+); 1H RMN 5: 1,35 (3H, t), 2,42 (3H, s), 2,58 (3H, s), 3,84 (3H, s), 4,01 (2H, q), 7,45-7,62 (5H, solapando a m), 7,90 (1H, s), 8,64 (1 H, s).
5-metil-3-(((4-(W-(2-(4-metilpiperazin-1-il)etil)sulfamoil)fenil)amino)(fenil)metilen)-2-oxoindolin-6-carboxilato de (Z)-metilo, 0.8 form iato

Una mezcla de 1 -acetil-3-(etoxi(fenil)metilen)-5-metil-2-oxoindolin-6-carboxilato de (E)-metilo (Intermedio C) (100 mg, 0,264 mmol) y 4-amino-W-(2-(4-metilpiperazin-1-il)etil)bencenosulfonamida, sal de di-trifluoroacetato (387 mg, 0,316 mmol) en DMF (3 mL) se calentó a 80 °C durante 16 h. Se añadió piperidina (261 gl, 2,64 mmol) y la mezcla se agitó a ta durante 2 h. El disolvente se eliminó a presión reducida y el residuo se disolvió en una disolución de MeOH al 10% en DCM (20 mL) y se lavó con agua (20 mL). Las capas se separaron usando un cartucho separador de fases y la capa orgánica se concentró a presión reducida. El producto bruto se purificó mediante HPLC preparativa (Método A, 20-50% MeCN en agua) para proporcionar el compuesto del título 5-metil-3-(((4-(W-(2-(4-metilpiperazin-1
il)etil)sulfamoil)fenil)amino)(fenil)metilen)-2-oxoindolin-6-carboxilato , 0.8 formiato de (Z)-metilo como un sólido amarillo claro (25 mg, 14%); Rt 1,58 min (Método 1); m/z 590 (M+H)+ (ES+); 1H RMN 5: 2,14 (6H, s), 2,17-2,35 (8H, solapando a m), 2,74-2,82 (2H, m), 3,76 (3H, s), 5,64 (1H, s), 6,96 (2H, m), 7,37 (1H, s), 7,41 (1 H, m), 7,50-7,59 (4H, solapando a m), 7,60-7,73 (3H, solapando a m), 8,19 (0,8H, s), 10,92 (1H, s), 12,24 (1H, s). (Falta 2H, supuestamente oscurecido por el disolvente).
Ruta 1B
Ejemplo de referencia 2: 3-(((4-((2-(dimetilamino)etil)carbamoil)fenil)amino)(fenil)metilen)-5-metil-2-oxoindolin-6-carboxilato de (Z)-metilo, form iato
Intermedio D: 1-acetil-3-(((4-(fórc-butoxicarbonil)fenil)amino)(fenil) metilen)-5-metil-2-oxoindolin-6-carboxilato de (Z)-metilo

Una mezcla de 1-acetil-3-(etoxi(fenil)metilen)-5-metil-2-oxoindolin-6-carboxilato de (E)-metilo (Intermedio C) (1,00 g, 2,64 mmol) y 4-aminobenzoato de terc-butilo (509 mg, 2,64 mmol) en DMF (9 mL) se calentó a 100 °C durante 18 h. Después de enfriar a ta, el precipitado se recogió mediante filtración, se lavó con Et2O (10 mL) y se secó in vacuo para proporcionar el compuesto del subtítulo 1-acetil-3-(((4-(te/-c-butoxicarbonil)fenil)amino)(fenil)metilen)-5-metil-2-oxoindolin-6-carboxilato de (Z)-metilo como un sólido amarillo (1,20 g, 86%); Rt 3,23 min (Método 1); m/z 527 (M+H)+ (ES+); 1H RMN 5: 1,50 (9H, s), 2,13 (3H, s), 2,73 (3H, s), 3,78 (3H, s), 5,54 (1H, s), 7,04 (2H, m), 7,51 (2H, m), 7,58 7,72 (5H, solapando a m), 8,68 (1H, s), 11,89 (1H, s).
Intermedio E: Ácido (Z)-4-(((1-acetil-6-(metoxicarbonil)-5-metil-2-oxoindolin-3-iliden)(fenil)metil)amino)benzoico, aducto de trifluoroacetato

A una disolución de 1-acetil-3-(((4-(te/"c-butoxicarbonil)fenil)amino)(fenil)metilen)-5-metil-2-oxoindolin-6-carboxilato de (Z)-metilo (Intermedio D) (1,20 g, 2,28 mmol) en Dc M (14 mL) se añadió TFA (1,76 mL, 22,8 mmol) y la mezcla se agitó a ta durante 72 h. El disolvente se eliminó a presión reducida para proporcionar el compuesto del subtítulo ácido (Z)-4-(((1 -acetil-6-(metoxicarbonil)-5-metil-2-oxoindolin-3-iliden)(fenil)metil)amino)benzoico, aducto de trifluoroacetato como un sólido amarillo (1,00 g, 72%); Rt 2,72 min (Método 1); m/z 471 (M+H)+ (ES+); 1H RMN 5: 2,13 (3H, s), 2,73 (3H, s), 3,78 (3H, s), 5,54 (1 H, s), 7,04 (2H, m), 7,50 (2H, m), 7,57-7,76 (5H, solapando a m), 8,68 (1H, s), 11,89 (1H, s), 12,89 (1H, s).
3-(((4-((2-(dimetilamino)etil)carbamoil)fenil)amino)(fenil)metilen)-5-metil-2-oxoindolin-6-carboxilato de (Z)-metilo, formiato

Acido (Z)-4-(((1 -acetil-6-(metoxicarbonil)-5-metil-2-oxoindolin-3-iliden)(fenil)metil)amino)benzoico, aducto de trifluoroacetato (Intermedio E) (75 mg, 0,128 mmol) y HATU (73,2 mg, 0,192 mmol) en DMF (2 mL) se agitaron a ta durante 10 min. A continuación, se añadió la base de Hünig (179 pl, 1,027 mmol) y W1,W1-dimetiletano-1,2-diamina (37,8 pl, 0,346 mmol). Se agitó la mezcla a ta durante 16 h. Se añadió piperidina (127 pl, 1,28 mmol). La mezcla se agitó a ta durante 4 h y la mezcla de reacción se repartió entre DCM (25 mL) y disolución acuosa saturada de NaHCÜ3(10 mL). Se lavó la fase orgánica con salmuera (10 mL) y el disolvente se evaporó a presión reducida. El producto bruto se purificó mediante HPLC preparativa (Método A, 20-50% MeCN en agua) para proporcionar el compuesto del título (Z)-metil-3-(((4-((2-(dimetilamino)etil)carbamoil)fenil)amino)(fenil)metilen)-5-metil-2-oxoindolin-6-carboxilato, formiato como un sólido amarillo claro (3,7 mg, 5%); Rt 1,53 min (Método 1); m/z 499 (M+H)+ (ES+); 1H RMN 5: 2,14 (3H, s), 2,80 (6H, s), 3,17 3,23 (2H, solapando a m), 3,50-3,56 (2H, solapando a m), 3,75 (3H, s), 5,63 (1H, s), 6,90 (2H, m), 7,37 (1 H, s), 7,52 (2H, m), 7,56-7,72 (5H, solapando a m), 8,56 (1H, m), 10,89 (1H, s), 12,22 (1H, s).
Ejemplo 3: 5-metil-3-(((4-(((1-metilpiperidin-4-il)metil)carbamoil)fenil)amino)(fenil)metilen)-2-oxoindolin-6-carboxilato de (Z)-metilo, form iato

Ácido (Z)-4-(((1 -acetil-6-(metoxicarbonil)-5-metil-2-oxoindolin-3-iliden)(fenil)metil)amino)benzoico, aducto de trifluoroacetato (Intermedio E) (75 mg, 0,128 mmol) y HATU (73,2 mg, 0,192 mmol) en DMF (2 mL) se agitaron a ta durante 10 min. A continuación, se añadió base de Hünig (179 pl, 1,02 mmol) y (1 -metilpiperidin-4-il)metanamina (32,9 mg, 0,257 mmol) en DMF (0,2 mL). Se agitó la mezcla a ta durante 16 h. Se añadió piperidina (127 pl, 1,28 mmol). La mezcla se agitó a TA durante 4 h y la mezcla de reacción se repartió entre DCM (25 mL) y disolución acuosa saturada de NAHCÜ3(10 mL). Se lavó la fase orgánica con salmuera, (10 mL) y se evaporó el disolvente a presión reducida. El producto bruto se purificó mediante HPLC preparativa (Método A, 20-50% MeCN en agua) para proporcionar el compuesto del título 5-metil-3-(((4-(((1-metilpiperidin-4-il)metil)carbamoil)fenil)amino)(fenill)methilen)-2-oxoindolin-6-carboxilato de (Z)-metilo , formiato como un sólido amarillo claro (19 mg, 25%); Rt 1,55 min (Método 1); m/z 539 (M+H)+ (ES+);1 H RMN 5: 1,11-1,28 (2H, solapando a m), 1,53 (1H, m), 1,60-1,73 (2H, solapando a m), 2,13 (3H, s), 2.15-2.24 (2H, solapando a m), 2.34 (3H, s), 2,89-3,00 (2H, solapando a m), 3,09 (2H, t), 3,75 (3H, s), 5,61 (1H, s), 6,87 (2H, m), 7,36 (1 H, s), 7,52 (2H, m), 7,58-7,69 (5H, solapando a m), 8.17 (1H, s), 8,36 (1 H, t), 10,88 (1H, s), 12,23 (1H, s).
Ejemplo 4: 5-metil-3-(((4-((2-(4-metilpiperazin-1-il)etil)carbamoil)fenil)amino)(fenil)metilen)-2-oxoindolin-6-carboxilato de (Z)-metilo

Ácido (Z)-4-(((1 -acetil-6-(metoxicarbonil)-5-metil-2-oxoindolin-3-iliden)(fenil)metil)amino)benzoico, aducto de trifluoroacetato (Intermedio E) (75 mg, 0,128 mmol), y HATU (73 mg, 0,192 mmol) en DMF (2 mL) se agitaron a ta durante 10 min , después se añadió base de Hünig (179 pl, 1,03 mmol) y 2-(4-metilpiperazin-1 -il)etanamina (50 mg, 0,346 mmol) en DMF (0,5 mL). La mezcla se agitó a ta durante 3 h y se añadió piperidina (127 pl, 1,28 mmol). Se agitó la mezcla a ta durante 18 h. La mezcla de reacción se repartió entre DCM (25 mL) y disolución acuosa saturada de NaHCÜ3 (10 mL). Se lavó la fase orgánica con salmuera (10 mL) y se evaporó el disolvente a presión reducida. El producto bruto se purificó mediante HPLC preparativa (Método A, 20-50% MeCN en agua) para proporcionar el compuesto del título 5-metil-3-(((4-((2-(4-metilpiperazin-1-il)etil)carbamoil)fenil)amino)(fenil)metilen)-2-oxoindoline-6-carboxilato de (Z)-metilo como un sólido amarillo claro (19 mg, 25%); Rt 1,46 min (Método 1); m/z 554 (M+H)+ (ES+); 1H RMN 5: 2,13 (3H, s), 2,21 (3H, s), 2,32-2,46 (8H, solapando a m), 3,27-3,34 (4H, solapando a m), 3,75 (3H, s), 5,62 (1H, s), 6,87 (2H, m), 7,36 (1H, s), 7,52 (2H, m), 7,56-7,69 (5H, solapando a m), 8,26 (1H, t), 10,87 (1H, s), 12,22 (1H, s).
Ejemplo 5: 5-metil-3-(((4-((2-(4-metil-1,4-diazepan-1-il)etil)carbamoil)fenil)amino)(fenil)metileno)-2-oxoindolin-6-carboxilato de (Z)-metilo, form iato

Ácido (Z)-4-(((1 -acetil-6-(metoxicarbonil)-5-metil-2-oxoindolin-3-iliden)(fenil)metil)amino)benzoico, aducto de trifluoroacetato (Intermedio E) (75 mg, 0,128 mmol), y HATU (73 mg, 0,192 mmol) en DMF (2 mL) se agitaron a ta durante 10 min, después se añadió base de Hünig (179 pl, 1,03 mmol) y 2-(4-metil-1,4-diazepan-1 -il)etanamina (54,5 mg, 0,346 mmol) en DMF (0,5 mL). La mezcla se agitó a ta durante 3 h y se añadió piperidina (127 pl, 1,28 mmol). Se agitó la mezcla a ta durante 18 h. La mezcla de reacción se repartió entre DCM (25 mL) y disolución acuosa saturada de NaHCÜ3 (10 mL). Se lavó la fase orgánica con salmuera (10 mL) y se evaporó el disolvente a presión reducida. El producto bruto se purificó mediante HPLC preparativa (Método A, 10-40% MeCN en agua) para proporcionar el compuesto del título 5-metil-3-(((4-((2-(4-metil-1,4-diazepan-1 -il)etil)carbamoil)fenil)amino)(fenil)metilen)-2-oxoindolin-6-carboxilato de (Z)-metilo, formiato como un sólido amarillo claro (27 mg, 34%); Rt 1,29 min (Método 1); m/z 568 (M+H)+ (ES+); 1H RMN 5: 1,68-1,77 (2H, solapando a m), 2,13 (3H, s), 2,34 (3H, s), 2,58 (2H, t), 2,61-2,74 (8H, solapando a m), 3,23-3,32 (2H, solapando a m), 3,75 (3H, s), 5,62 (1H, s), 6,87 (2H, m), 7,36 (1H, s), 7,52 (2H, m), 7,56-7,71 (5H, solapando a m), 8,20 (1H, s), 8,24 (1H, t), 10,88 (1H, s), 12,22 (1H, s).
Ejemplo 6: 3-(((4-((2-(4-(dimetilamino)piperidin-1-il)etil)carbamoil)fenil)amino)(fenil)metilen)-5-metil-2-oxoindolin-6-carboxilato de (Z)-metilo, formiato

Ácido (Z)-4-(((1 -acetil-6-(metoxicarbonil)-5-metil-2-oxoindolin-3-iliden)(fenil)metil)amino)benzoico, aducto de trifluoroacetato (Intermedio E) (75 mg, 0,128 mmol), y HATU (73 mg, 0,192 mmol) en DMF (2 mL) se agitaron a ta durante 10 min, después se añadió base de Hünig (179 pl, 1,03 mmol) y 1-(2-aminoetil)-W,W-dimetilpiperidin-4-amina (59,3 mg, 0,346 mmol) en DMF (0,2 mL). La mezcla se agitó a ta durante 3 h y se añadió piperidina (127 pl, 1,28 mmol). Se agitó la mezcla a temperatura ambiente durante 1 h. La mezcla de reacción se repartió entre DCM (25 mL) y disolución acuosa saturada de NaHCÜ3 (10 mL). Se lavó la fase orgánica con salmuera (10 mL) y se evaporó el disolvente a presión reducida. El producto bruto se purificó mediante HPLC preparativa (Método A, 10-40% MeCN en agua) para proporcionar el compuesto del título 3-(((4-((2-(4-(dimetilamino)piperidin-1-il)etil)carbamoil)fenil)amino)(fenil)metilen)-5-metil-2-oxoindolin-6-carboxilato de (Z)-metilo, formiate como un sólido amarillo claro (20 mg, 24%); Rt 1,26 min (Método 1); m/z 582 (M+H)+ (ES+); 1H RMN 5: 1,29-1,44 (2H, solapando a m), 1,65-1,77 (2H, solapando a m), 1,86-1,98 (2H, solapando a m), 2,14 (3H, s), 2,24 (6H, s), 2,40 (2H, t), 2,85-2,95 (2H, solapando a m), 3,76 (3H, s), 5,62 (1H, s), 6,88 (2H, m), 7,37 (1H, s), 7,53 (2H, m), 7,57-7,70 (5H, solapando a m), 8,22 (1H, s), 8,28 (1H, t), 10,89 (1H, s), 12,23 (1H, s). (Falta 3H, supuestamente oscurecido por el disolvente).
Ejemplo 7: 3-(((4-((2-(4-(2-hidroxietil)piperazin-1-il)-2-oxoetil)carbamoil)fenil)amino)(fenil)metilen)-5-metil-2-oxoindolin-6-carboxilato de (Z)-metilo, form iato

Ácido (Z)-4-(((1 -acetil-6-(metoxicarbonil)-5-metil-2-oxoindolin-3-iliden)(fenil)metil)amino)benzoico, aducto de trifluoroacetato (Intermedio E) (75 mg, 0,128 mmol), y HATU (73 mg, 0,192 mmol) en DMF (2 mL) se agitaron a ta durante 10 min, a continuación se añadió base de Hünig (179 pl, 1,03 mmol) y 2-amino-1-(4-(2-hidroxietil)piperazin-1-il)etanona, 2HCl (90 mg, 0,346 mmol). La mezcla se agitó a ta durante 3 h y se añadió piperidina (127 pl, 1,28 mmol). Se agitó la mezcla a ta durante 18 h. La mezcla de reacción se repartió entre DCM (25 mL) y disolución acuosa saturada de NaHCÜ3 (10 mL). Se lavó la fase orgánica con salmuera (10 mL) y se evaporó el disolvente a presión reducida. El producto bruto se purificó mediante HPLC preparativa (Método A, 20-50% MeCN en agua) para proporcionar el compuesto del título 3-(((4-((2-(4-(2-hidroxietil)piperazin-1-il)-2-oxoetil)carbamoil)fenil)amino)(fenil)metilen)-5-metil-2-oxoindolin-6-carboxilatode (Z)-metilo, formiato como un sólido amarillo claro (24 mg, 27%); Rt 1,51 min (Método 1); m/z 598 (M+H)+ (ES+); 1H RMN 5: 2,14 (3H, s), 2,90-3,58 (8H, solapando a m), 3,68-3,79 (5H, solapando a m), 4,00-4,45 (4H, solapando a m), 5,40 (1H, br s), 5,63 (1H, s), 6,90 (2H, m), 7,37 (1H, s), 7,53 (2H, m), 7,57-7,70 (5H, solapando a m), 8,52 (1H, s), 9,63 (1H, br s), 10,89 (1H, s), 12,23 (1H, s).
Ejemplo 8: 5-metil-3-(((4-((2-(4-metilpiperazin-1-il)-2-oxoetil)carbamoil)fenil)amino)(fenil)metilen)-2-oxoindolin-6-carboxilato de (Z)-metilo, 0.5 formiato

Ácido (Z)-4-(((1 -acetil-6-(metoxicarbonil)-5-metil-2-oxoindolin-3-iliden)(fenil)metil)amino)benzoico, aducto de trifluoroacetato (Intermedio E) (75 mg, 0,128 mmol), y HATU (73 mg, 0,192 mmol) en d Mf (2 mL) se agitaron a ta durante 10 min, después se añade la base de Hünig (179 pl, 1,03 mmol) y 2-amino-1-(4-metilpiperazin-1-il)etanona, 2HCl (80 mg, 0,346 mmol). La mezcla se agitó a ta durante 3 h y se añadió piperidina (127 pl, 1,28 mmol). Se agitó la mezcla a ta durante 18 h. La mezcla de reacción se repartió entre DCM (25 mL) y disolución acuosa saturada de NaHCÜ3(10 mL). Se lavó la fase orgánica con salmuera (10 mL) y se evaporó el disolvente a presión reducida. El producto bruto se purificó mediante HPLC preparativa (Método A, 20-50% MeCN en agua) para proporcionar el compuesto del título 5-metil-3-(((4-((2-(4-metilpiperazin-1 -il)-2-oxoetil)carbamoil)fenil)amino)(fenil)metilen)-2-oxoindoline-6-carboxolato de (Z)-metilo, 0,5 formiato como un sólido amarillo claro (28 mg, 36%); Rt 1,52 min (Método 1); m/z 568 (M+H)+ (ES+); 1H RMN 5: 2,14 (3H, s), 2,26 (3H, s), 2,31-2,46 (4H, solapando a m), 3,41-3,54 (4H, solapando a m), 3,76 (3H, s), 4,06 (2H, d), 5,64 (1H, s), 6,89 (2H, m), 7,37 (1H, s), 7,53 (2H, m), 7,57-7,70 (5H, solapando a m), 8,13 (0,5H, s), 8,46 (1H, t), 10,89 (1H, s), 12,23 (1H, s).
Ejemplo 9: 5-metil-2-oxo-3-(((4-((2-(3-oxopiperazin-1-il)etil)carbamoil)fenil)amino)(fenil)metilen)indolin-6-carboxilato de (Z)-metilo

Ácido (Z)-4-(((1 -acetil-6-(metoxicarbonil)-5-metil-2-oxoindolin-3-iliden)(fenil)metil)amino)benzoico, aducto de trifluoroacetato (Intermedio E) (75 mg, 0,128 mmol), y HATU (73 mg, 0,192 mmol) en d Mf (2 mL) se agitaron a ta durante 10 min a continuación se añadió base de Hünig (179 pl, 1,03 mmol) y 4-(2-aminoetil)piperazin-2-ona (49,6 mg, 0,346 mmol). La mezcla se agitó a ta durante 3 h y se añadió piperidina (127 pl, 1,28 mmol). Se agitó la mezcla a ta durante 18 h. La mezcla de reacción se repartió entre DCM (25 mL) y NaHCÜ3 (10 mL) acuoso saturado. Se lavó la fase orgánica con salmuera (10 mL) y se evaporó el disolvente a presión reducida. El producto bruto se purificó mediante HPLC preparativa (Método A, 20-50% MeCN en agua) para proporcionar el compuesto del título 5-metil-2-oxo-3-(((4-((2-(3-oxopiperazin-1-il)etil)carbamoil)fenil)amino)(fenil)metilen)indolin-6-carboxilato de (Z)-metilo como un sólido amarillo claro (43 mg, 58%); Rt 1,59 min (Método 1); m/z 554 (M+H)+ (ES+); 1H RMN 5: 2,14 (3H, s), 2,40-2,63 (2H, solapando a m), 2,96 (1H, m), 3,13 (1H, m), 3,20-3,48 (4H, solapando a m), 3,58 (1H, m), 3,75 (3H, s), 3,99 (1H, m), 5,62 (1H, s), 6,89 (2H, m), 7,36 (1H, s), 7,53 (2H, m), 7,57-7,69 (5H, solapando a m), 8,27-8,63 (2H, solapando a m), 10,89 (1H, s), 12,23 (1H, s).
Ejemplo 10: 5-metil-3-(((4-((3-(4-metilpiperazin-1-il)-3-oxopropil)carbamoil)fenil)amino)(fenil)metilen)-2-oxoindolin-6-carboxilato de (Z)-metilo

Ácido (Z)-4-(((1 -acetil-6-(metoxicarbonil)-5-metil-2-oxoindolin-3-iliden)(fenil)metil)amino)benzoico, aducto de trifluoroacetato (Intermedio E) (800 mg, 1,37 mmol), y HATU (781 mg, 2,05 mmol) en DMF (6 mL) se agitaron a ta durante 10 min a continuación se añadió base de Hünig (1,43 mL, 8,21 mmol) y 3-amino-1-(4-metilpiperazin-1-il)propan-1-ona (223 mg, 1,30 mmol). La mezcla se agitó a ta durante 3 h y se añadió piperidina (1,35 mL, 13,7 mmol). Se agitó la mezcla a ta durante 18 h. La mezcla de reacción se repartió entre MeOH al 10% en DCM (25 mL) y disolución acuosa saturada de NaHCO3 (10 mL). Se lavó la fase orgánica con salmuera (10 mL) y se evaporó el disolvente a presión reducida. El material se cargó en un cartucho SCX en MeOH (10 mL). La columna se lavó con MeOH (30 mL) y el filtrado se desechó. El producto se eluyó con amoniaco al 1% en MeOH. El disolvente se evaporó y el residuo se purificó mediante cromatografía en columna ultrarrápida (SiO2 , 80 g, 0-30% amoniaco al 1% en MeOH en DCM, gradiente de elución) para proporcionar 5--metil-3-(((4-((3-(4-metilpiperazin-1-il)-3-oxopropil)carbamoil)fenil)amino)(fenil)metilen)-2-oxoindolin-6-carboxilato de (Z)-metilo como un sólido amarillo claro (386 mg, 48%); Rt 1,60 min (Método 1); m/z 582 (M+H)+ (ES+); 1H RMN 5: 2,13 (3H, s), 2,15 (3H, s), 2,21 (2H, t), 2,24 (2H, t), 2,53 (2H, t), 3,35-3,45 (6H, solapando a m), 3,75 (3H, s), 5,62 (1H, s), 6,87 (2H, m), 7,36 (1 H, s), 7,51 (2H, m), 7,57-7,68 (5H, solapando a m), 8,37 (1H, t), 10,88 (1H, s), 12,21 (1H, s).
Ejemplo 11: 5-metil-3-(((4-(4-(2-(metilamino)-2-oxoetil)piperazin-1 -carbonil)fenil)amino)(fenil)metilen)-2-oxoindolin-6-carboxilato de (Z)-metilo

Ácido (Z)-4-(((1 -acetil-6-(metoxicarbonil)-5-metil-2-oxoindolin-3-iliden)(fenil)metil)amino)benzoico, aducto de trifluoroacetato (Intermedio E) (75 mg, 0,128 mmol), y HATU (73 mg, 0,192 mmol) en DMF (2 mL) se agitaron a ta durante 10 min a continuación se añadió base de Hünig (179 pl, 1,03 mmol) y N-metil-2-(piperazin-1 -il)acetamida (54,5 mg, 0,346 mmol). La mezcla se agitó a ta durante 3 h y se añadió piperidina (127 pl, 1,28 mmol). Se agitó la mezcla a ta durante 18 h. La mezcla de reacción se repartió entre DCM (25 mL) y disolución acuosa saturada de NaHCO3 (10 mL). Se lavó la fase orgánica con salmuera (10 mL) y se evaporó el disolvente a presión reducida. El producto bruto se purificó mediante HPLC preparativa (Método A, 20-50% MeCN en agua) para proporcionar el compuesto del título 5-metil-3-(((4-(4-(2-(metilamino)-2-oxoetil)-piperazin-1-carbonil)fenil)amino)(fenil)metilen)-2-oxoindolin-6-carboxilato de (Z)-metilo como un sólido amarillo claro (8,8 mg, 12%); Rt 1,59 min (Método 1); m/z 568 (M+H)+ (ES+); 1H RMN 5: 2,13 (3H, s), 2,61-2,70 (4H, solapando a m), 3,75 (3H, s), 5,60 (1H, s), 6,88 (2H, m), 7,24 (2H, m), 7,37 (1H, s), 7,53 (2H, m), 7,57-7,71 (3H, solapando a m), 8,26 (1H, br s), 10,87 (1H, s), 12,23 (1H, s). (Falta 9H, supuestamente oscurecido por disolvente).
Ejemplo 12: 3-(((4-((2-(4-hidroxipiperidin-1-il)etil)carbamoil)fenil)amino)(fenil)metilen)-5-metil-2-oxoindolin-6-carboxilato de (2)-metilo, 0.8 formiato

Ácido (Z)-4-(((1 -acetil-6-(metoxicarbonil)-5-metil-2-oxoindolin-3-iliden)(fenil)metil)amino)benzoico, aducto de trifluoroacetato (Intermedio E) (75 mg, 0,128 mmol), y HATU (73 mg, 0,192 mmol) en DMF (2 mL) se agitaron a ta durante 10 min a continuación se añadió base de Hünig (179 pl, 1,03 mmol) y 1-(2-aminoetil)piperidin-4-ol (50 mg, 0,346 mmol). La mezcla se agitó a ta durante 3 h y se añadió piperidina (127 pl, 1,28 mmol). Se agitó la mezcla a ta durante 18 h. La mezcla de reacción se repartió entre DCM (25 mL) y disolución acuosa saturada de NaHCÜ3 (10 mL). Se lavó la fase orgánica con salmuera (10 mL) y el disolvente se evaporó a presión reducida. El producto bruto se purificó mediante HPLC preparativa (Método A, 20-50% MeCN en agua) para proporcionar el compuesto del título 3-(((4-((2-(4-hidroxipiperidin-1-il)etil)carbamoil)fenil)amino)(fenil)metilen)-5-metil-2-oxoindoline-6-carboxilato de (Z)-metilo, 0.8 formiato como un sólido amarillo claro (9 mg, 11%); Rt 1,54 min (Método 1); m/z 555 (M+H)+ (ES+); 1H RMN 5: 1,28-1,42 (2H, solapando a m), 1,64-1,72 (2H, solapando a m), 2,02-2,11 (2H, solapando a m), 2,13 (3H, s), 2,42 (2H, t), 2,69-2,78 (2H, solapando a m), 3,75 (3H, s), 4,56 (1H, br s), 5,62 (1 H, s), 6,87 (2H, m), 7,36 (1H, s), 7,52 (2H, m), 7,57-7,69 (5H, solapando a m), 8,16 (0,8H, s), 8,28 (1H, t), 10,88 (1 H, s), 12,22 (1H, s). (Falta 3H, supuestamente oscurecido por el disolvente).
Ejemplo 13: 3-(((4-((1 -(2-hidroxietil) piperidin-4-il) carbamoil)fenil)amino)(fenil)(metilen)-5-metil-2-oxoindolin-6-carboxilato de ( z )-metilo

Ácido (Z)-4-(((1 -acetil-6-(metoxicarbonil)-5-metil-2-oxoindolin-3-iliden)(fenil)metil)amino)benzoico, aducto de trifluoroacetato (Intermedio E) (75 mg, 0,128 mmol), y HATU (73 mg, 0,192 mmol) en DMF (2 mL) se agitaron a ta durante 10 min a continuación se añadió base de Hünig (179 pl, 1,03 mmol) y 2-(4-aminopiperidin-1-il)etanol, 2HCl (100 mg, 0,462 mmol) en DMF (1 mL). La mezcla se agitó a ta durante 2 h y se añadió piperidina (127 pl, 1,28 mmol). Se agitó la mezcla a ta durante 18 h. La mezcla de reacción se repartió entre DCM (25 mL) y disolución saturada acuosa de NaHCÜ3 (10 mL). Se lavó la fase orgánica con salmuera (10 mL) y se evaporó el disolvente a presión reducida. El producto bruto se cargó en una columna de SCX (2 g) en AcÜH al 5% en MeÜH (10 mL). La columna se lavó con MeÜH (10 mL) y el filtrado se desechó. El producto se eluyó con amoniaco al 1% en MeÜH (25 mL). El disolvente se evaporó a presión reducida y el producto se purificó mediante cromatografía en columna ultrarrápida (SiÜ2 , 12 g, 0-30% MeÜH en DCM, gradiente de elución) para proporcionar 3-(((4-((1-(2-hidroxietil)piperidin-4-il)carbamoil)fenil)amino)(fenil)metilen)-5-metil-2-oxoindolin-6-carboxilato de (Z)-metilo como un sólido amarillo claro (33 mg, 34%); Rt 1,56 min (Método 1); m/z 555 (M+H)+ (ES+); 1H RMN 5: 1,43-1,56 (2H, solapando a m), 1,65-1,74 (2H, solapando a m), 2,00 (2H, t), 2,13 (3H, s), 2,36 (2H, m), 2,85 (2H, m), 3.47 (2H, m), 3,66 (1H, m), 3,75 (3H, s), 4,37 (1H, m), 5,61 (1H, s), 6,86 (2H, m), 7,36 (1 H, s), 7,52 (2H, m), 7,56-7,76 (5H, solapando a m), 8,09 (1H, d), 10,88 (1 H, s), 12,23 (1 H, s).
Ejemplo 14: 3-(((4-(((1-(2-hidroxietil)piperidin-4-il)metil)carbamoil)fenil)amino)(fenil)metilen)-5-metil-2-oxoindolin-6-carboxilato de ( z )-metilo

Ácido (Z)-4-(((1 -acetil-6-(metoxicarbonil)-5-metil-2-oxoindolin-3-iliden)(fenil)metil)amino)benzoico, aducto de trifluoroacetato (Intermedio E) (75 mg, 0,128 mmol), y HATU (73 mg, 0,192 mmol) en DMF (2 mL) se agitaron a ta durante10 min a continuación se añadió base de Hünig (179 pl, 1,03 mmol) y 2-(4-(aminometil)piperidin-1-il)etanol (73,1 mg, 0,462 mmol). La mezcla se agitó a ta durante 3 h y se añadió piperidina (127 pl, 1,28 mmol). Se agitó la mezcla a ta durante 18 h. La mezcla de reacción se repartió entre DCM (25 mL) y disolución acuosa saturada de NaHCÜ3 (10 mL). Se lavó la fase orgánica con salmuera (10 mL) y se evaporó el disolvente a presión reducida. El producto bruto se cargó en una columna de SCX (2 g) en AcÜH al 5% en MeÜH (10 mL). La columna se lavó con MeÜH (10 mL) y el filtrado se desechó. Luego, el producto se eluyó con amoniaco al 1% en MeÜH (25 mL). El disolvente se evaporó a presión reducida y el producto se purificó mediante cromatografía en columna ultrarrápida (SiÜ2, 12 g, 0-30% MeÜH en DCM, gradiente de elución) para proporcionar 3-(((4-((1-(2-hidroxietil)piperidin-4-il)metil)carbamoil)fenil)amino)(fenil)metilen)-5-metil-2-oxoindolin-6-carboxilato de (Z)-metilo como un sólido amarillo claro (63 mg, 63%); Rt 1,56 min (Método 1); m/z 569 (M+H)+ (ES+); 1H RMN 5: 1,05-1,19 (2H, solapando a m), 1,47 (1H, m), 1,58 (2H, m), 1,81-1,94 (2H, solapando a m), 2,13 (3H, s), 2,28-2,40 (2H, solapando a m), 2,82 (2H, m), 3,07 (2H, t), 3,46 (2H, m), 3,75 (3H, s), 4,34 (1H, s), 5,61 (1H, s), 6,86 (2H, m), 7,36 (1H, s), 7,52 (2H, m), 7,57-7,70 (5H, m), 8,32 (1 H, t), 10,88 (1H, s), 12,23 (1H, s).
Ruta 1C:
Ejemplo 15: 3-(((4-(4-(2-((2-hidroxietil)amino)-2-oxoetil)piperazin-1 -carbonil)fenil)amino)(fenil)metilen)-5-metil-2-oxoindolin-6-carboxilato de (Z)-metilo
Intermedio F: 1-acetil-3-(((4-(4-(2-(terc-butoxi)-2-oxoetil)piperazin-1-carbonil)fenil)amino)(fenil)metilen)-5-metil-2-oxoindolin-6-carboxilato de (z)-metilo

Ácido (Z)-4-(((1 -acetil-6-(metoxicarbonil)-5-metil-2-oxoindolin-3-iliden)(fenil)metil)amino)benzoico, aducto de trifluoroacetato (Intermedio E) (500 mg, 0,855 mmol), y HATU (488 mg, 1,28 mmol) en DMF (5 mL) se agitaron a ta durante 10 min a continuación se añadió base de Hünig (448 pl, 2,57 mmol) y 2-(piperazin-1-il)acetato de tere-butilo (171 mg, 0,855 mmol) en DMF (1 mL). Se agitó la mezcla a temperatura ambiente durante 3 h. La mezcla de reacción se repartió entre DCM (100 mL) y disolución acuosa saturada de NaHCÜ3 (40 mL). Se lavó la fase orgánica con salmuera (40 mL) y se evaporó el disolvente a presión reducida. el producto bruto se purificó mediante cromatografía en columna ultrarrápida (SiÜ2 , 12 g, 0-30% amoniaco al 1% en MeÜH en DCM, gradiente de elución) para proporcionar el compuesto del subtítulo 1-acetil-3-(((4-(4-(2-(terc-butoxi)-2-oxoetil)piperazin-1-carbonil)fenil)amino)(fenil)metilen)-5-metil-2-oxoindolin-6-carboxilato de (Z)-metilo como un sólido amarillo claro (483 mg, 83%); Rt 2,31 min (Método 1); m/z 653 (M+H)+ (ES+); 1H RMN 5: 1,40 (9H, s), 2,13 (3H, s), 2,73 (3H, s), 3,13 (2H, s), 3,17-3,30 (2H, solapando a m), 3,43-3,62 (2H, solapando a m), 3,77 (3H, s), 5,49 (1H, s), 7,03 (2H, m), 7,21 (2H, m), 7,50 (2H, m), 7,57-7,68 (3H, solapando a m), 8,68 (1H, s), 11,88 (1H, s). (Falta 4H, supuestamente oscurecido por disolvente).
Intermedio G: Ácido (Z)-2-(4-(4-(((1-acetil-6-(metoxicarbonil)-5-metil-2-oxoindolin-3-iliden)(fenil)metil)amino)benzoil)piperazin-1 -il) acético

A una disolución de 1-acetil-3-(((4-(4-(2-(terc-butoxi)-2-oxoetil)piperazin-1-carbonil)fenil)amino)(fenil)metilen)-5-metil-2-oxoindolin-6-carboxilato de (Z)-metilo (Intermedio F) (481 mg, 0,737 mmol) en DCM (4,8 mL) se añadió TFA (568 pl, 7,37 mmol) y la mezcla se agitó a ta durante 16 h. Se añadió otra porción de TFA (1 mL, 13,0 mmol) y la reacción se agitó a ta durante otras 3 h después de las cuales se añadió otra porción de TFA (0,5 mL, 7,50 mmol) y la reacción se agitó a ta durante otra hora. La mezcla de reacción se añadió gota a gota a una disolución saturada acuosa de NaHCÜ3 (50 mL). Las capas orgánicas combinadas se concentraron a presión reducida. El sólido formado en la fase acuosa se recogió mediante filtración y se secó a presión reducida. Ambos sólidos se combinaron para proporcionar ácido (Z)-2-(4-(4-(((1 -acetil-6-(metoxicarbonil)-5-metil-2-oxoindolin-3-iliden)(fenil)metil)amino)benzoil)piperazin-1 -il)acético (410 mg, 89%) como un sólido amarillo claro; Rt 1,94 min (Método 1); m/z 597 (m H)+ (ES+); 1H RMN 5: 2,13 (3H, s), 2,73 (3H, s), 3,17 (2H, s), 3,47-3,65 (2H, solapando a m), 3,77 (3H, s), 5,50 (1H, s), 7,03 (2H, m), 7,22 (2H, m), 7,50 (2H, m), 7,55-7,68 (4H, solapando a m), 8,68 (1H, s), 11,89 (1H, s). (Falta 6H, presuntamente oscurecido por el disolvente).
3-(((4-(4-(2-((2-hidroxietil) amino)-2-oxoetil)piperazin-1-carbonil)fenil)amino)(fenil) metilen)-5-metil-2-oxoindolin-6-carboxilato de (ZJ-metilo.

Ácido (Z)-2-(4-(4-(((1-acetil-6-(metoxicarbonil)-5-metil-2-oxoindolin-3-iliden)(fenil)metil)amino)benzoil)piperazin-1-il)acético (Intermedio G) (200 mg, 0,335 mmol), y HATU (191 mg, 0,503 mmol) en DMF (2 mL) se agitaron a ta durante 10 min a continuación se añadió base de Hünig (176 pl, 1,00 mmol) y 2-aminoetanol (22,52 mg, 0,369 mmol) en DMF (1 mL). La mezcla se agitó a ta durante 4 h y se añadió piperidina (332 pl, 3,35 mmol). Se agitó la mezcla a ta durante 18 h. La mezcla de reacción se repartió entre DCM (25 mL) y disolución acuosa saturada NaHCÜ3 (10 mL). Se lavó la fase orgánica con salmuera (10 mL) y se evaporó el disolvente a presión reducida. El producto crudo se cargó en un cartucho SCX en AcÜH al 5% en MeOH:MeÜH:DCM (2:1:1,20 mL). La columna se lavó con MeÜH (30 mL) y el filtrado se desechó. El producto se eluyó con amoniaco al 1% en MeÜH (25 mL). El disolvente se evaporó a presión reducida y el producto se purificó mediante cromatografía en columna ultrarrápida (SiÜ2 , 12 g, 0-30% amoniaco al 1% en MeÜH en DCM, gradiente de elución) para proporcionar el compuesto del subtítulo 3-(((4-(4-(2-((2-hidroxietil)amino)-2-oxoetil)piperazin-1-carbonil)fenil)amino)(fenil)metilen)-5-metil-2-oxoindolin-6-carboxilato de (Z)-metilo (81 mg, 39%) se obtuvo como un sólido amarillo claro; Rt 1,57 min (Método 1); m/z 598 (M+H)+ (ES+); 1H RMN 5: 2,13 (3H, s), 2,33-2,47 (4H, solapando a m), 2,93 (2H, s), 3,16 (2H, q), 3,39 (2H, q), 3,75 (3H, s), 4,68 (1H, t), 5,60 (1H, s), 6,87 (2H, m), 7,19 (2H, m), 7,36 (1 H, s), 7,52 (2H, m), 7,57-7,68 (3H, solapando a m), 7,72 (1H, s), 10,85 (1H, s), 12,21 (1H, s). (Falta 4H, supuestamente oscurecido por el disolvente).
Los siguientes ejemplos de compuestos (Tabla 2) se pueden preparar mediante métodos sintéticos similares a los ejemplos mencionados anteriormente o mediante métodos descritos en otra parte de este documento:
Tabla 2: Ejemplos de Compuestos Adicionales de la Invención





1H, s),













Un método de síntesis adicional para el Ejemplo 18:
Ejemplo 18: 5-metil-3-(((4-((1-metilpiperidin-4-il)carbamoil)fenil)amino)(fenil)metilen)-2-oxoindolin-6-carboxilato de (Z)-metilo

Ácido Z)-4-(((1-acetil-6-(metoxicarbonil)-5-metil-2-oxoindolin-3-iliden)(fenil)metil)amino)benzoico (Intermedio E) (10 g, 17,54 mmol) y HATU (10,01 g, 26,3 mmol) se agitaron a ta durante 10 min en DMF (200 mL) a continuación se añadió base de Hünig (18,38 mL, 105 mmol) y 1-metilpiperidin-4-amina (2,204 g, 19,30 mmol) en DMF (10mL). La mezcla se agitó a TA durante 3 horas. A continuación, se añadió piperidina (17,37 mL, 175 mmol) y la mezcla resultante se agitó a TA durante 3 h. La mezcla de reacción se repartió entre MeOH al 10% en DCM (750 mL) y disolución saturada de hidrogenocarbonato de sodio (500 mL). Las capas orgánicas combinadas se lavaron con salmuera (300 mL) y se concentró el disolvente. El sólido resultante se trituró con CH3CN, se filtró y se secó para dar 7 g de producto bruto.
La reacción se repitió a la misma escala y los productos brutos se combinaron y disolvieron en MeOH al 10% (amoniaco al 1%) en DCM y se cargaron en una columna de sílice (300 g). El producto se eluyó con MeOH al 30%
(NH3 al 1%) en disolución de DCM y las fracciones que contenían el producto se recogieron y el disolvente se evaporó a continuación. El material se volvió a disolver en MeOH al 30% (NH3 al 1%) en disolución de DCM (500 mL) y se lavó con agua (200 mL). La capa orgánica se secó sobre MgSO4, se filtró y el disolvente se evaporó. El material se secó a presión reducida, a 40 °C, toda la noche, para dar 5-metil-3-(((4-((1 -metilpiperidin-4-il)carbamoil)fenil)amino)(fenil)metilen)-2-oxoindolin-6-carboxilato de (Z)-metilo 1478-45-2 (12,8 g, 23,91 mmol, rendimiento 68,1 % ).
El producto se analizó mediante LCMS (método Agilent, X-Select, Waters X-Select C18, 2,5 gm, 4,6x30 mm, Ácido (ácido fórmico al 0,1%) 4 min, 5-95% MeCN/agua): 1478-45-2-Fin, m/z 525(M+H)+ (ES+); a 1,57 min, 98% pureza @ 254 nm. 1H RMN 5: Rt 1,57 min (Método 1); m/z 525 (M+H)+ (ES+); 1H RMN 5: 1,44-1,57 (2H, solapando a m), 1,65 1,72 (2H, solapando a m), 1,85-1,93 (2H, solapando a m), 2,13 (6H, s), 2,70-2,76 (2H, solapando a m), 3,64 (1h , m), 3,75 (3H, s), 5,61 (1H, s), 6,86 (2H, m), 7,36 (1H, s), 7,52 (2H, m), 7,57-7,68 (5H, solapando a m), 8,09 (1H, d), 10,88 (1H, s), 12,23 (1H, s).
Ensayos B iológicos: Métodos Experimentales
Ensayos de Inhibición de Enzimas
Las actividades inhibidoras de enzimas de los compuestos descritos en este documento se determinaron usando el ensayo ADP-Glo™ (Promega, Reino Unido). Los ensayos para FGFR1, PDGFRa, PDGFRp y VEGFR2 se realizaron en tampón que contenía Tris 40 mM pH 7,5, MgCh 20 mM, BSA 0,1 mg/mL y DTT 1 mM; mientras que los ensayos para FGFR3 y VEGFR1 se realizaron en el tampón anterior suplementado con MnCl22 mM.
Inhibición de la Enzima FGFR1
Las actividades inhibidoras de los compuestos de la invención frente a FGFR1 (Sistema de Enzima Cinasa FGFR1: Promega), se evaluaron mezclando la proteína FGFR1 (3,12 ng/mL, 2 gL), sustrato (Poly (4:1 Glu4, Tyn), 100 ng/mL, 2 gL) con el compuesto de ensayo (2 gL a 3 gM, 0,67 gM, 0,15 gM, 0,033 gM, 0,0073 gM, 0,0016 gM, 0,0036 gM o 0,00008 gM) durante 90 min a 25°C. La reacción de la cinasa se inició después añadiendo ATP (50 gM, 2 gL) y la mezcla se incubó durante 1 h a 25°C. Se añadió reactivo ADP-Glo™ durante 40 min (8 gL), a continuación, se añadió reactivo de desarrollo (16 gL) durante 40 min antes de la detección en un lector de microplacas (EnVision® Multilabel Reader, Perkin Elmer).
Inhibición de Enzima FGFR3
Las actividades inhibidoras de los compuestos de la invención frente a FGFR3 (Sistema de Enzima cinasa FGFR3: Promega), se evaluaron mezclando la proteína FGFR3 (12,5 ng/mL, 2 gL), sustrato (Poly (Ala6, Glu2 , Lyss, Tyn), 100 ng/mL, 2 gL) con el compuesto de ensayo (2 gL a 3 gM, 0,67 gM, 0,15 gM, 0,033 gM, 0,0073 gM, 0,0016 gM, 0,0036 gM o 0,00008 gM) durante 90 min a 25°C. La reacción de la cinasa se inició añadiendo ATP (50 gM, 2 gL) y la mezcla se incubó durante 90 min a 25°C. Se añadió reactivo ADP-Glo™ durante 40 min (8 gL), a continuación, se añadió reactivo de desarrollo (16 gL) durante 40 min antes de la detección en un lector de microplacas (EnVision® Multilabel Reader, Perkin Elmer).
Inhibición de la Enzima PDGFRa
Las capacidades inhibidoras de los compuestos de la invención frente a PDGFRa (Sistema de Enzima Cinasa PDGFRa: Promega), se evaluaron mezclando la proteína PDGFRa (12,5 ng/mL, 2 gL), sustrato (Poly (4:1 Glu4, Tyn), 100 ng/mL, 2 gL) con el compuesto de ensayo (2 gL a 3 gM, 0,67 gM, 0,15 gM, 0,033 gM, 0,0073 gM, 0,0016 gM, 0,0036 gM o 0,00008 gM) durante 90 min a 25°C. La reacción de la cinasa se inició añadiendo ATP (25 gM, 2 gL) y la mezcla se incubó durante 1 h a 25°C. Se añadió reactivo ADP-Glo™ durante 40 min (8 gL), a continuación, se añadió reactivo de desarrollo (16 gL) durante 40 min antes de la detección en un lector de microplacas (EnVision® Multilabel Reader, Perkin Elmer).
Inhibición de la Enzima PDGFRp
Las actividades inhibidoras de los compuestos de la invención frente a PDGFRp (Sistema de Enzima Cinasa PDGFRp : Promega), se evaluaron mezclando la proteína PDGFRp (6,25 ng/mL, 2 gL), sustrato (Poly (4:1 Glu4, Tyn), 100 ng/mL, 2 gL) con el compuesto de ensayo (2 gL a 3 gM, 0,67 gM, 0,15 gM, 0,033 gM, 0,0073 gM, 0,0016 gM, 0,0036 gM o 0,00008 gM) durante 90 min a 25°C. La reacción de la cinasa se inició añadiendo ATP (25 gM, 2 gL) y la mezcla se incubó durante 1 h a 25°C. Se añadió reactivo ADP-Glo™ durante 40 min (8 gL), a continuación, se añadió reactivo de desarrollo (16 gL) durante 40 min antes de la detección en un lector de microplacas (EnVision® Multilabel Reader, Perkin Elmer).
Inhibición de la Enzima VEGFR1
Las actividades inhibidoras de los compuestos de la invención frente a VEGFR1 (Sistema de Enzima Cinasa VEGFR1 : Promega), se evaluaron mezclando la proteína VEGFR1 (12,5 ng/mL, 2 gL), sustrato (IGFR1 Rtide, 100 ng/mL, 2 gL) con el compuesto de ensayo (2 gL a 3 gM, 0,67 gM, 0,15 gM, 0,033 gM, 0,0073 gM, 0,0016 gM, 0,0036 gM o 0,00008
pM) durante 90 min a 25°C. La reacción de la cinasa se inició añadiendo ATP (50 pM, 2 pL) y la mezcla se incubó durante 90 min a 25°C. Se añadió reactivo ADP-Glo™ durante 40 min (8 pL), a continuación, se añadió reactivo de desarrollo (16 pL) durante 40 min antes de la detección en un lector de microplacas (EnVision® Multilabel Reader, Perkin Elmer).
Inhibición de la Enzima VEGFR2
Las actividades inhibidoras de los compuestos de la invención frente a VEGFR2 (VEGFR2 Sistema de Enzima Cinasa: Promega), se evaluaron mezclando la proteína VEGFR2 (1,56 ng/mL, 2 pL), sustrato (Poly (4:1 Glu4, Tyri), 100 ng/mL, 2 pL) con el compuesto de ensayo (2 pL a 3 pM, 0,67 pM, 0,15 pM, 0,033 pM, 0,0073 pM, 0,0016 pM, 0,0036 pM o 0,00008 pM) durante 90 min a 25°C. La reacción de la quinasa se inició añadiendo ATP (50 pM, 2 pL) y la mezcla se incubó durante 1 h a 25 ° C. Se añadió reactivo ADP-Glo™ durante 40 min (8 pL), a continuación, se añadió reactivo de desarrollo (16 pL) durante 40 min antes de la detección en un lector de microplacas (EnVision® Multilabel Reader, Perkin Elmer).
En todos los casos, la cinasa convierte el ATP en ADP y el reactivo ADP-Glo™, después agota cualquier ATP restante. El reactivo de detección convierte el ADP que se ha producido de nuevo en ATP y genera luciferasa que puede detectarse como luminiscencia. Por tanto, la señal luminiscente es directamente proporcional a la cantidad de ADP producida por la reacción enzimática y una reducción de esta señal después del tratamiento del compuesto demuestra inhibición. El porcentaje de inhibición producido por cada concentración de compuesto se calculó mediante la ecuación que se muestra a continuación:
% de Inhibición  100
100
A continuación, se representó gráficamente el porcentaje de inhibición frente a la concentración del compuesto y se determinó la concentración inhibitoria relativa del 50% (RIC50) a partir de la curva de concentración-respuesta resultante. Una vez que se determinó, el Ki se calculó mediante la siguiente ecuación:

Ensayos Celulares y Otros In Vitro
Proliferación de Fibroblastos de Pulmón Humano Normal (NHLF) inducida por PDGF-BB
Los NHLF (grupo Lonza Ltd) se expanden hasta un 90% de confluencia en medios de crecimiento FGM-2 complementados con FBS al 2% (más factores de crecimiento SingleQuot™; Lonza). Se recolectan los fibroblastos (Tripsina/EDTA), suspendido a 25x103 por mL en medio de crecimiento y se añaden 200 pl por pocillo (5x103 células/pocillo) de una placa de cultivo de tejidos de 96 pocillos. Después de 24 h de incubación (37 C/ CO2 al 5%/Ü2 al 95%), las células se les priva de suero (24 h) reduciendo la concentración de FBS a 0,1% en el medio de cultivo. Las células se pre-incubaron con el compuesto de ensayo durante 1 h, después se estimularon con rhuPDGF-BB (100 ng/ml, R&D Systems) durante 48 h. La proliferación celular se evalúa mediante la incorporación de BrdU (ELISA Colorimétrico de Proliferación Celular, Roche). El porcentaje de inhibición de la proliferación de NHLF inducida por rhuPDGF-BB por el compuesto de ensayo en cada concentración se calcula como un porcentaje del alcanzado por rhuPDGF-BB a cada concentración de compuesto de ensayo en comparación con el control del vehículo (proliferación basal). La concentración inhibidora relativa del 50% (R1C50) se determina a partir de la curva de concentraciónrespuesta resultante.
Proliferación de fibroblastos de pulmón humano fetal MRC-5 inducida por PDGF-BB /FGF-Básico
Los fibroblastos MRC-5 (Estándares LGC) se expanden hasta un 90% de confluencia en medio DMEM suplementado con FBS al 10%. Las células se recolectan (Tripsina/EDTA), suspendidas a 25x103por mL en medio de crecimiento y se añaden 200 pl por pocillo (5x103 células/pocillo) de una placa de cultivo de tejidos de 96 pocillos. Después de 24 h de incubaron (37 C/ CÜ2 al 5%/Ü2 al 95%), las células se les priva de suero (3 h) reemplazando el medio de crecimiento que contiene FBS al 0,1%. A continuación, las células se preincuban con el compuesto de ensayo durante 1 h seguido de estimulación con rhuPDGF-BB (100ng/ml, R&D Systems) o rhuFGF-básico (5ng/ml; R&D Systems) durante 48 h. La proliferación celular se evalúa mediante la incorporación de BrdU (ELISA Colorimétrico de Proliferación Celular, Roche). El porcentaje de inhibición de la proliferación de MRC-5 inducida por rhuPDGF-BB/rhuFGF por el compuesto de ensayo a cada concentración se calcula como un porcentaje del logrado por rhuPDGF-BB/FGF-básico en cada concentración de compuesto de ensayo por comparación con el control del vehículo (proliferación basal). La concentración inhibidora relativa del 50% (RIC50) se determina a partir de la curva de concentración-respuesta resultante.
Proliferación de células endoteliales inducida por FGF-Basic/VEGFi
65
TeloHAEC (Células Endoteliales Aórticas Humanas Inmortalizadas con telomerasa; ATCC) se siembran en placas de cultivo de 96 pocillos a una densidad celular de 4000 células por pocillo (100gl) en un medio de privación de células endoteliales (FBS al 0,5%, sin factores de crecimiento FGF y VEGF) y cultivadas durante 3 h (37 C/ CO2 al 5%/O2 al 95%). Las células se preincuban con el compuesto de ensayo durante 1 hora seguido de estimulación con rhuVEGFi65(10 ng/ml, R & D Systems) o rhuFGF-básico (5ng/ml, R & D Systems) durante 48 h. La proliferación celular se evalúa mediante la incorporación de BrdU (ELISA Colorimétrico de Proliferación Celular, Roche). El porcentaje de inhibición de proliferación de TeloHAEC inducida por rhuVEGFWrhuFGF-básico por el compuesto de ensayo a cada concentración se calcula como un porcentaje del alcanzado por rhuVEGFWFGF-básico a cada concentración de compuesto de ensayo por comparación frente al control del vehículo (proliferación basal). La concentración inhibidora relativa del 50% (RIC50) se determina a partir de la curva de concentración-respuesta resultante.
Fosforilación inducida por PDGF-BB de fibroblastos PDGFRp
Se usaron MRC-5/NHLF/NIH-3T3 (fibroblastos embrionarios de ratón, estándares LGC) para evaluar el efecto inhibidor del compuesto de ensayo sobre la fosforilación de PDGFRp usando el kit de ensayo celular de fosfo-PDGFRp (Tyr751) de HTRF (Fluorescencia Resuelta en Tiempo Homogéneo) (Cisbio). MRC-5/NHLF se sembraron en placas de cultivo de tejidos de 96 pocillos a una densidad celular de 10000 células por pocillo en medio de crecimiento DMEM (FBS al 10%) o medio de crecimiento FGM-2 (FBS al 2%) respectivamente y se cultivaron durante 48 h (37 C/CO2 al 5%/O2 al 95%). NIH-3T3 se sembró en placas de cultivo de tejidos de 96 pocillos a una densidad celular de 7000 células por pocillo en medio de crecimiento DMEM (FBS al 10%) y se cultivaron durante 48 h (37 C/CO2 al 5%/O2 al 95%). El medio celular se reemplazó con el medio de privación respectivo que contenía FBS al 0,1% y las placas se incubaron adicionalmente durante 24 h (37 C/CO2 al 5%/O2 al 95%) para MRC-5/NHLF y durante 3 h para NIH-3T3. Las células se preincubaron con el compuesto de ensayo durante 1 h y luego se estimularon con rhuPDGF-BB (25-50 ng/ml, R & D Systems) durante 5 min y rmPDGF-BB (25 ng/ml, Life Technologies) para NIH-3T3s. Se aspiró el medio y las células se lisaron inmediatamente mediante la adición de 50 gl de tampón de lisis proporcionado en el kit de ensayo de HTRF. Se transfirieron 16 gl de lisado celular de cada pocillo a una placa blanca de 384 pocillos de bajo volumen a la que se añadieron los reactivos apropiados del kit según las instrucciones del kit. La fosforilación del PDGFRp se cuantificó calculando la proporción de fluorescencia leída a 665 nm y 620 nm. El porcentaje de inhibición de la fosforilación de PDGFRp inducida por rPDGF-BB por el compuesto de ensayo se calculó como un porcentaje del alcanzado por rPDGF-BB en cada concentración de compuesto de ensayo en comparación con el control del vehículo. La concentración inhibidora relativa del 50% (RIC50) se determinó a partir de la curva de concentración-respuesta resultante.
VEGF
165
indujo la fosforilación de VEGFR2 en células endoteliales
Se usaron TeloHAEC (Células Endoteliales Aórticas Humanas inmortalizadas con telomerasa; ATCC) para evaluar el efecto inhibidor del compuesto de ensayo sobre la fosforilación de VEGFR2 usando el kit de ensayo celular de fosfo-VEGFR2 (Tyr1175) de HTRF (fluorescencia resuelta en tiempo homogéneo) (Cisbio). Se sembraron TeloHAEC en placas de cultivo de tejidos de 96 pocillos a una densidad celular de 12000 células por pocillo en medio de crecimiento endotelial (ATCC; FBS al 2%) y se cultivaron durante 48 horas (37 C/CO2 al 5%/O2 al 95%). El medio celular se reemplazó con medio de privación (sin factores de crecimiento VEGF y FGF) que contenía FBS al 0,5 % y las placas se incubaron adicionalmente durante 24 h (37 C/CO2 al 5%/O2 al 95%). Las células se preincubaron con el compuesto de ensayo durante 1 h seguido de estimulación con rhuVEGF165(50 ng/ml, R&D Systems) durante 5 min. Se aspiró el medio y las células se lisaron inmediatamente mediante la adición de 50 gl de tampón de lisis proporcionado en el kit de ensayo de HTRF. Se transfirieron 16 gl de lisado celular de cada pocillo a una placa blanca de 384 pocillos de bajo volumen a la que se añadieron los reactivos apropiados del kit según las instrucciones del kit. La fosforilación del VEGFR2 se cuantificó calculando la relación de fluorescencia leída a 665 nm y 620 nm. El porcentaje de inhibición de la fosforilación de VEGFR2 inducida por rhuVEGF165 por el compuesto de ensayo se calculó como un porcentaje del alcanzado por rhuVEGF165 a cada concentración de compuesto de ensayo en comparación con el control del vehículo. La concentración inhibidora relativa del 50% (RIC50) se determinó a partir de la curva de concentración-respuesta resultante.
Ensayo de contracción en gel de fibroblastos
Los NHLF se expanden hasta un 90% de confluencia en medio de crecimiento FGM-2 (Lonza) complementado con FBS al 2% (más factores de crecimiento SingleQuot™). Se recolectan los fibroblastos (Tripsina/EDTA) y se suspendieron a 1 x106por mL en medio libre de suero. Basado en el kit de ensayo de concentración celular (Cell Biolabs) ase prepara una celdilla celular mezclando 1 parte de suspensión celular 4 partes de disolución de gel de colágeno según las instrucciones del kit. Se añade 0,9 mL de alícuotas de la celdilla de colágeno a tubos centrífugos de 1,5 mL y se trataron con concentraciones de ensayo finales del compuesto de ensayo. A continuación, se pipetearon 250 gl de la celdilla tratada con compuesto en cada pocillo de una placa de cultivo de tejido de 48 pocillos (triplicado por concentración de ensayo). La placa se incuba durante 90 min (37 C/ CO2 al 5%/O2 al 95%) para permitir que los geles se polimericen. A continuación, se añaden 250 gl de medio sin suero que contiene las concentraciones de ensayo finales del compuesto de ensayo a cada gel correspondiente. Después de 30 minutos más de incubación, los geles se estimularon con TGFp1. (10ng/ml; R&D Systems). Después de un periodo de incubación de 24 h (37 C/CO2 al
5%/o2 al 95%), cada gel individual se retira y se pesa en una balanza de precisión. El efecto del compuesto de ensayo a cada concentración se expresa como el porcentaje de inversión de la contracción inducida por TGFp1 en relación con la contracción basal tratada con vehículo.
Ensayo de liberación de fibroblastos de IL-6
Los NHLF se expanden hasta un 90% de confluencia en medios de crecimiento FGM-2 (Lonza) complementados con FBS al 2% (más factores de crecimiento SingleQuot™). Se recolectaron los fibroblastos (Tripsina/EDTA), suspendidos a 50 x 103 por mL en medio de crecimiento y se añadieron 200 pl por pocillo (10 x 103 células/pocillo) de una placa de cultivo de tejidos de 96 pocillos. Después de 24 h de incubación (37 C/CO2 al 5%/O2 al 95%), las células se privaron de suero (24 h) reduciendo la concentración de FBS del medio a 0,1%. Las células se preincuban con el compuesto de ensayo durante 1 h, luego se estimulan con TGFp1 (5ng/ml, R&D Systems) durante 24 h. Los sobrenadantes libres de células se recuperan para la determinación de las concentraciones de IL-6 mediante ELISA sándwich (Duo-set, R&D Systems). La inhibición de la producción de IL-6 se calcula como un porcentaje del logrado por 5ng/ml de TGFp1 a cada concentración de compuesto de ensayo por comparación con el control del vehículo. La concentración inhibidora relativa del 50% (RIC50) se determina a partir de la curva de concentración-respuesta resultante.
Apoptosis de mastocitos
Los mastocitos se diferencian de las células CD34 de sangre del cordón umbilical Lonza) durante 8 semanas en un medio de crecimiento suplementado con 100 ng/ml de SCF y 10 ng/ml de IL-6. Los mastocitos se siembran en placas de fondo blanco transparente de 384 pocillos entre 2500 y 10000 células/pocillo en medios de crecimiento que contenía SCF (100 ng/ml). Como control positivo para la apoptosis, se incubaron 8 pocillos en medio de crecimiento sin SCF. Las células se incubaron con compuestos de ensayo o vehículo durante 24 horas (37 C/CO2 al 5%/O2 al 95%). Se añade sustrato lumonógeno caspasa-3/7 (Ensayo Caspasa-Glo 3/7, Promega) a las células y se incuban a temperatura ambiente durante 30 min, antes de leer la señal de luminiscencia. La inducción de apoptosis por los compuestos de ensayo se calcula como un porcentaje del logrado por las células incubadas en ausencia de SCF (respuesta apoptótica máxima) para cada concentración de compuesto de ensayo en comparación con el vehículo (apoptosis inicial). La concentración inhibidora relativa del 50% (RIC50) se determina a partir de la curva de concentración-respuesta resultante.
El Efecto de los Compuestos de Ensayo sobre la Viabilidad Celular
Células MRC-5 se sembraron en placas de cultivo de tejido de 96 pocillos de fondo transparente blanco (para lecturas de fluorescencia/luminiscencia) o transparente (para lecturas colorimétricas) a una densidad celular de 12 x 10 3 células por pocillo en medio de crecimiento DMEM (FBS al 10%) . Después de 24 h de incubación (37 C/CO2 al 5%/O2 al 95%), the growth media was replaced with media containing 0,1% FBS plus test compound/ vehicle y incubated for a further 48 hrs. Para el ensayo MTT colorimétrico (evaluación de actividad metabólica celular, los sobrenadantes de cada pocillo se aspiraron, se reemplazaron con 100 pl/pocillo de medio recién preparado (FBS al 0,1%) y 10 pl/pocillo de 5mg/ml de MTT. Después de un periodo de incubación de 1 h (37 C/CO2 al 5%/O2 al 95%), se aspiró el medio y se añadió DMSO al 100% (100 pl) a cada pocillo. Las placas se agitaron ligeramente durante 15 minutos antes de leer la absorbancia a 550 nm. Se calculó el porcentaje de pérdida de viabilidad celular (representado por una reducción en valores de absorbancia) para cada concentración de compuesto con relación a las células tratadas de vehículo (0,5% DMSO). El ensayo de citotoxicidad MultiTox-Fluor Multiplex junto con el ensayo Caspasa-Glo 3/7 (Promega) se utilizaron para medir la citotoxicidad/viabilidad celular y la apoptosis. El ensayo de citotoxicidad MultiTox-Fluor Multiplex es un ensayo fluorescente de adición de reactivo único que mide simultáneamente el número relativo de células vivas y muertas en poblaciones celulares. El ensayo proporciona medidas ratiométricas, inversamente correlacionadas, de viabilidad celular y citotoxicidad. La proporción de células viables a células muertas es independiente del número de células y, por lo tanto, se puede utilizar para normalizar los datos. La adición del reactivo único Caspase-Glo® 3/7 en un formato de "añadir-mezclar-medir" da como resultado la lisis celular, seguida de la escisión de caspasa de un sustrato y la generación de una señal luminiscente de "tipo resplandor". Para este ensayo de citotoxicidad multiplex, se retiraron cuidadosamente de cada pocillo 100 pl de sobrenadantes celulares de las células tratadas con compuesto de 48 h y luego se añadieron 50 pl de reactivo MultiTox (se preparó una disolución de trabajo de reactivos MultiTox patentados diluyendo 10 pl de GF-AFC y bis AAF-R110 en 10 mL de tampón de ensayo según las instrucciones del kit). Las células se incubaron en la oscuridad durante 30 minutos antes de tomar dos lecturas de fluorescencia separadas a; 400Ej/505Em (lectura de célula en vivo) y 485Ej/ Em (lectura de célula muerta). A continuación, se retiraron cuidadosamente 100 pl de sobrenadante de cada pocillo y se añadió 50 pl de reactivo caspasa 3/7 Glo a la placa celular y se incubó durante 30 minutos en la oscuridad. La actividad de la caspasa 3/7 se cuantificó leyendo la señal de luminiscencia. Un aumento de la señal por encima de las células de control tratadas con vehículo representó un aumento de la apoptosis celular.
Ensayo de transición de fibroblastos a miofibroblastos
Para evaluar la actividad antifibrótica de los compuestos de ensayo, se utilizaron dos protocolos alternativos. En el primero, se siembran fibroblastos pulmonares aislados en placas de 96 pocillos a 3000 células/pocillo. Cinco (5) días después de la siembra, las células se refrescan y se añaden a las células los compuestos de ensayo o el vehículo. Después de una (1) hora, se añade TGF-p1 (1,25 ng/mL) para inducir la transición fibroblasto-a-miofibroblasta. La
expresión de aSMA, un marcador de la transición de miofibroblastos, se mide después de 72 horas mediante inmunotinción, se evalúa mediante imágenes de alto contenido en el IN Cell Analyzer 2200 (GE Healthcare) y se cuantifica utilizando un algoritmo patentado (BioFocus) con el software de desarrollo IN Cell (GE Cuidado de la salud). La salida del algoritmo representa la intensidad de la tinción multiplicada por el área teñida (niveles de DxA). Se realiza una tinción conjunta de núcleos celulares con 4',6-diamidino-2-fenilindol (DAPI) para cuantificar el número de células, como medida de toxicidad potencial y/o para normalizar aSMA para diferencias en la densidad celular.
En el protocolo alternativo, los fibroblastos pulmonares aislados se siembran en placas de 96 pocillos a 5100 células/pocillo. Después de 24 horas, las células se refrescan con medio desprovisto durante 24 horas más. Los compuestos de ensayo o el vehículo se añaden a las células. Después de una (1) hora, se añade TGF-p1 (0,1 ng/mL) para inducir la transición fibroblasto-a-miofibroblasto. La expresión de aSMA, un marcador de transición de miofibroblastos, se mide después de 48 horas mediante inmunotinción, se evalúa mediante imágenes de alto contenido en ImageXpres micro (dispositivos moleculares) y se cuantifica usando el software MetaXpress (Dispositivos Moleculares). El porcentaje de células positivas se usa para evaluar la tinción de aSMA y, por lo tanto, el grado de transición de fibroblasto a miofibroblastos. Se realiza una tinción conjunta de núcleos celulares con 4',6-diamidino-2-fenilindol (DAPI) para cuantificar el número de células, como medida de toxicidad potencial y/o para normalizar aSMA para diferencias en la densidad celular.
Evaluación de la duración de la acción de los compuestos de ensayo frente a la fosforilación inducida por PDGF-BB de PDGFRp en células de fibroblastos de pulmón fetal humano
Para determinar la persistencia relativa de los efectos de la exposición al fármaco en la fosforilación de PDGFRp inducida por PDGF-BB, se utiliza un experimento de lavado que utiliza células de fibroblastos de pulmón fetal humano MRC-5 y un kit de ensayo celular fosfo-PDGFRp (Tyr751) HTRF homogénea (Fluorescencia de Resolución de Tiempo Homogénea) (Cisbio), que detecta la modulación del receptor de la fosforilación inducida por PDGF-BB de PDGFRp.
Protocolo de lavado
Las células MRC-5 se retiran con tripsima y se neutralizan con medio completo (medio de crecimiento DMEM, FBS al 10%). Se colocan 1,2x106 células en tubos de microfuga, se centrifugan a 10.000 rpm durante 30 s utilizando una centrífuga de mesa superior, el sobrenadante se retira y las células se lavan en 1ml de tampón de lavado (HBSS pH7,4, 0,1% BSA y 0,04% ácido plurónico) precalentado (37°C) para eliminar cualquier FBS residual. A continuación, las células se centrifugan de nuevo, se retira el sobrenadante y se incuban con 500 pl de vehículo, compuesto de control de ensayo y compuesto de ensayo (a una concentración que proporciona una inhibición del 70%) durante 1 h con agitación suave a 37°C. Después de la incubación, las células se dispersan uniformemente y se extrae una alícuota de 100 pl de cada tubo, para un control sin lavado. A continuación, las células restantes se someten a 5 etapas de lavado repetidas en las que las células se centrifugan, se retira el sobrenadante y se resuspende en 1 mL de tampón de lavado recién preparado precalentado (37°C). Para evitar los efectos del arrastre del compuesto debido a la adherencia al plástico, las células se transfieren a tubos nuevos después de cada etapa de lavado y se colocan en un agitador a 37°C (900 rpm) durante 10 minutos de incubación entre lavados para permitir el reequilibrio entre las células y el tampón. Después de la etapa de lavado final, las células se resuspenden en 360 ul de tampón de lavado precalentado. Se colocan 5 pl de los tubos lavados y sin lavar en una placa de microtitulación de polipropileno blanco 384 (Greiner) y se incuban a 37°C durante 15 minutos. A continuación, las células se estimulan con 5 pL de rhuPDGF-BB (3ng/ml, R&D Systems) durante 5 min, después de lo cual las células se lisan inmediatamente mediante la adición de 10 pl de tampón de lisis proporcionado en el kit de ensayo HTRF. Las células se colocan en un agitador de placas (rpm 1450) durante 1 hora, después de lo cual la placa se centrifuga brevemente durante 30 segundos (3000 rpm) y se añaden los reactivos del kit HTRF patentados según las instrucciones del kit. La fosforilación del PDGFRp se cuantifica calculando la relación de fluorescencia leída a 665 nm y 620 nm. El porcentaje de inhibición de la fosforilación de PDGFRp inducida por rhuPDGF-BB por el compuesto de ensayo se calcula como un porcentaje del alcanzado a 3 pg/ml rhuPDGF-BB frente al control del vehículo de ensayo.
Ensayo de permeabilidad PAMPA
Este ensayo mide la permeabilidad a través de una membrana artificial y fue realizado por Cyprotex usando un ensayo de permeabilidad de membrana artificial paralelo. Es un modelo in vitro de permeación transcelular pasiva a través de una membrana de hexadecano artificial, (Wohnsland F et al., Med. Chem., 2001 44; 923-930). Los compuestos se pueden clasificar en baja y alta permeabilidad. Generalmente, los compuestos que tienen una Pap P< 10 x 10-6cm/s se clasifican como compuestos de baja permeabilidad y con una Paap > 10 x 10-6 cm/s se clasifican como de alta permeabilidad. Se cree que los compuestos que tienen baja permeabilidad probablemente tengan un tiempo de permanencia prolongado en el pulmón debido a su absorción más lenta (Tronde Ann et al., J Pharm. Sci., 2003, 92 (6), 1216-33).
Resumen del protocolo
El compuesto de ensayo se añade al lado donante de una membrana de PAMPA artificial recubierta con filtro, y la permeabilidad se mide monitorizando la apariencia del compuesto de ensayo en el lado aceptor de la membrana celular usando LC-MS/MS.
Procedimiento experimental
Se preparó una disolución de hexadecano en hexano (5 % v/v) y se añadió una alícuota a la membrana de cada pocilio en la placa de filtro (donante) (placa de filtro Multiscreen para permeabilidad, Millipore). A continuación, se dejaron secar las placas donantes para asegurar la evaporación del hexano. Se añadió a cada pocillo de las placas aceptoras tampón (pH 7) que contenía DMSO (5 %). Las disoluciones del compuesto de ensayo se prepararon diluyendo concentrados de DMSO 10 mM en tampón que dio una concentración final del compuesto de ensayo de 10 pM (concentración final de DMSO 5 %). El marcador de integridad fluorescente amarillo lucifer también se incluyó en la disolución del compuesto de ensayo. La placa donante se insertó en la placa aceptora y luego las placas se incubaron a temperatura ambiente durante 5 h. Se prepararon patrones analíticos a partir de disoluciones de compuesto de ensayo. La permeabilidad del compuesto de ensayo se evaluó por cuadriplicado. En cada placa se procesaron compuestos de características de permeabilidad conocidas como controles.
Al final del período de incubación, la placa donante se retiró de la placa aceptora. Las muestras de donantes y aceptores para los compuestos de ensayo y control se cuantificaron mediante análisis de casete LC-MS/MS utilizando una calibración de 5 puntos con la dilución adecuada de las muestras. La recuperación experimental se calculó a partir de las concentraciones del compartimento donante y aceptor.
Si la permeabilidad del amarillo lucifer estaba por encima de los límites de QC para uno, dos o tres pocillos individuales del compuesto de ensayo, entonces se reportaron n = 3, 2 o 1 resultados.
Análisis de Datos
El coeficiente de permeabilidad aparente (Pap) para cada compuesto se calculó a partir de la siguiente ecuación:
[fármacojaceptor
Papipap C x - ln( 1 -[fármacO]equilibrio
donde
c _ vD * v A
(y + VA)Á rea x tiempo
donde V d y V a son los volúmenes de los compartimentos donante y aceptor, respectivamente, área es el área específica de la membrana multiplicada por la porosidad y la concentración de equilibrio del fármaco es la concentración del compuesto de ensayo en el volumen total de los compartimentos donante y aceptor.
Cribado in vivo: Farmacodinámica y Farmacocinética
Fibrosis inducida por bleomicina en ratones (un modelo de ratón de fibrosis pulmonar)
A ratones C57BL6/J se les administra por ruta intratraqueal vehículo o sulfato de bleomicina (MP Biomedicals, 2 U/kg) el día 0. Los compuestos se administran por ruta intranasal (como disoluciones o suspensiones acuosas, 25-50 ul) una vez al día desde el día 5 hasta el día 20. Después de otras 24 h, los animales se anestesian, se canulan sus tráqueas y se ventilan mecánicamente utilizando un ventilador de pistón controlado por ordenador (flexiVent, SCIREQ Inc., Montreal, Canadá). Después de las mediciones de la función pulmonar, los ratones se desangran y se extrae el líquido de lavado broncoalveolar (BALF). Los recuentos de glóbulos blancos totales y diferenciales en las muestras de BALF se miden usando un hemocitómetro Neubaur. Los frotis de citospina de las muestras de BALF se preparan mediante centrifugación y se tiñen utilizando un sistema de tinción DiffQuik (Dade Behring). Las células se cuentan usando microscopía de inmersión en aceite. Los sobrenadantes de BALF sobrantes se guardan para el análisis de citocinas.
Se inflan pulmones enteros a una presión de 25 cm H2O con formalina tamponada neutra al 10% a través de la cánula traqueal y se sumergen en formalina durante al menos 24 h. Después de procesarse en bloques de parafina, los pulmones se seccionan (5 pm) y se tiñen con hematoxilina y eosina (H&E), rojo picro-sirius (PSR) o tricrómico de Masson. Se toman aleatoriamente un número constante de fotografías de 4 secciones transversales. Las imágenes se puntúan de 0 (sin PF) a 8 (PF máximo) por 2 investigadores ciegos para evaluar los cambios fibróticos en los pulmones. Además, se realiza la tinción para alfa-actina de músculo liso (Thermo Scientific, Freemont, CA) utilizando metodologías estándar. El colágeno soluble en homogeneizado de pulmón completo se evalúa mediante el ensayo de colágeno Sircol (Biocolor Ltd, Carrickfergus, Reino Unido).
Fosforilación de POGFRfi inducida por PDGFBB en ratones
Ratones de 7 a 12 semanas C57BL/6 (Charles River) se alojan bajo un ciclo luz/oscuridad de 12 horas y reciben comida y agua ad libitum. Se preparan disoluciones o suspensiones acuosas de compuestos para administrar las dosis indicadas. (mg/kg) basadas en el peso promedio de los ratones en los grupos. Los animales se anestesian y los compuestos o vehículos se administran por ruta intranasal (50 ul). Después de la recuperación en los puntos de tiempo
indicados, los animales se anestesian nuevamente y se administra PDGF-BB de ratón recombinante (50 ug/animal, Cambridge Biosciences) o el vehículo por ruta intratraqueal, y luego, de 5 a 30 minutos después de la instilación de PDGF-BB, se toman muestras de sangre terminal y se extirpan los pulmones completos. El plasma se aísla de la sangre mediante centrifugación. Aproximadamente la mitad de los lóbulos izquierdos se homogeneizan utilizando un instrumento FastPrep-245G en tubos de lisis Matrix D (MP Biomedicals). Los niveles de fosforilación de PDGFRp se miden mediante transferencia Western usando anti-phospho-PDGFRa(Y849)/p(Y857), anti-total PDGFRp/a y anticuerpos anti GAPDH (señalización celular). Los niveles de fosforilación de PDGFRp se informan como relaciones con los niveles de PDGFRp totales o con GAPDH. El porcentaje de inhibición de la fosforilación de PDGFRp se calcula para cada tratamiento en relación con el tratamiento con vehículo. Las mediciones de los niveles de compuestos en el plasma o los homogeneizados de pulmón se determinan mediante LC-MS/MS.
Medidas farmacocinéticas en roedores
Se utilizaron ratas CD macho o ratones C57BL/6J para estudios farmacocinéticos en los que los animales se dosificaron por ruta intratraqueal, oral o intravenosa para ratas e intranasal, oral o intravenosa para ratones. animales fueron dosificados por ruta intravenosa a través de la vena lateral de la cola. Los animales que recibieron dosis por ruta intratraqueal o intranasal fueron anestesiados antes de la dosificación usando isoflurano y óxido nitroso en estudios con ratas u oxígeno en estudios con ratones.
Los animales se canularon en la vena lateral de la cola y se colocaron en una caja caliente (37-40°C) aproximadamente 5 minutos antes de cada momento de muestreo para dilatar las venas de la cola. Los compuestos se formularon como disoluciones para administración intravenosa y como disoluciones o suspensiones para todos los demás métodos. Los animales se pesaron el día de la dosificación y recibieron una sola administración de la formulación con el volumen ajustado de acuerdo con el peso corporal del individuo (Tabla 3). Para muestras seriadas, se tomaron muestras de sangre de rata de 150-200 gl o muestras de sangre de ratón de 20 gl (con la misma adición de volumen de agua) del puerto de la cánula en microtainers recubiertos con K2 EDTA en puntos de tiempo predeterminados durante 24 horas. El plasma se aisló por centrifugación (8200rcf durante 5 minutos), solo en estudios en ratas.
Al final, se recogieron muestras de sangre a través de la vena cava descendente en microtainers recubiertos con K2EDTA y fluido BAL descargando los pulmones a través de la tráquea con 3 x 4ml de instilaciones de BSA al 4% en PBS para ratas y 3 x 0,4ml instilaciones de BSA al 4% en PBS para ratones. Se extirparon los pulmones, se pesaron y se registraron los pesos, y se congelaron rápidamente en nitrógeno líquido.
Las mediciones de los niveles de compuesto en el plasma o la sangre, los homogeneizados pulmonares y el líquido BAL se determinaron mediante LC-MS/MS. El fármaco se extrajo mediante precipitación de proteínas usando un exceso de disolvente que contenía un patrón interno apropiado. Las concentraciones de fármaco se determinaron frente a una curva estándar comparada con la matriz externa.
Las concentraciones de fármaco se representaron frente al tiempo en un gráfico semilogarítmico (ver Figuras 2 y 3). Los parámetros farmacocinéticos se calcularon usando análisis no compartimental. Las concentraciones en el pulmón y el fluido BAL en cada momento se expresaron como el porcentaje de dosis administrada.

Resultados
Los resultados de los ensayos en los ensayos de inhibición enzimática y en los ensayos celulares y otros ensayos in vitro se muestran en las Tablas 4 a 8 siguientes y en las Figuras 1 a 3.
Tabla 4 Las actividades inhibidoras de los compuestos de la invención y nintedanib frente a FGFR1, FGFR3, PDGFRa, PDGFRp, VEGFR1 y VEGFR2.


Tabla 5 Los efectos inhibidores de los compuestos de la invención y nintedanib sobre la fosforilación de PDGFRp en fibroblastos inducida por PDGFBB en células MRC5 y sobre la fosforilación de VEGFR2 en células endoteliales inducida por VEGF165.



Tabla 6 El efecto de los compuestos de la invención y nintedanib sobre la viabilidad celular de las células MRC5 (ensayo MTT).


Tabla 7 Determinación de Pap para compuestos de la invención en el ensayo de permeabilidad PAMPA


Tabla 8 Mediciones farmacocinéticas durante un período de 24 horas en ratas que recibieron dosis intravenosa (i.v.) e intratraqueal (i.t.) con compuestos de la invención o nintedanib.



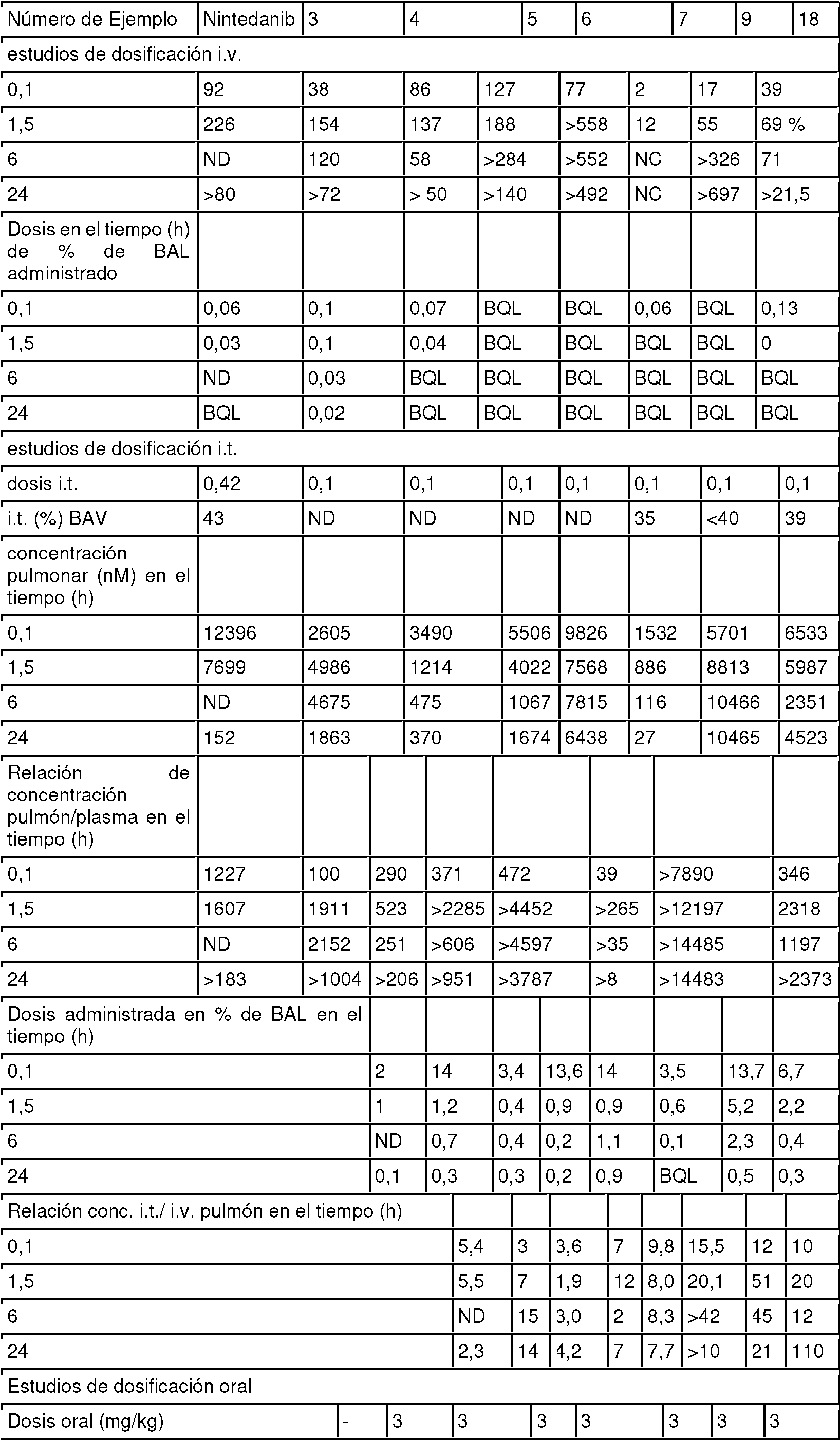

Resumen de los resultados
Los compuestos de la invención, como se describen en el presente documento, demuestran actividad inhibidora frente a FGFR1, FGFR3, PDGFRa, PDGFRp, VEGFR1 y VEGFR2 (Tabla 4 y Tabla 5) y potencia nanomolar. Una gran mayoría de los compuestos ensayados no muestran efectos adversos en los ensayos de viabilidad celular (Tabla 6). Además, los compuestos generalmente tienen baja permeabilidad a través de las membranas (Tabla 7 y Figura 1). Las mediciones farmacocinéticas muestran que generalmente los compuestos de la invención tienen un contenido relativo más alto que permanece en el pulmón 24 horas después de la administración tópica después de que el sujeto recibe la dosis que nintedanib (Tabla 8 y Figuras 2 y 3). Los parámetros farmacocinéticos también indican que los compuestos de la invención mantienen niveles pulmonares más altos cuando se administran por ruta tópica en comparación con las dosis equivalentes administradas por ruta i.v. y, en general, estos niveles relativos más altos son más altos que los niveles correspondientes para nintedanib, particularmente en el punto de tiempo de 24 h. Los niveles bajos de BAL observados junto con las cantidades reductoras durante un período de 24 h indican que se está produciendo disolución y absorción en el tejido pulmonar con los compuestos de la invención.
Este perfil indica que se espera que los compuestos de la invención sean adecuados como medicamentos, entre otros, para el tratamiento de enfermedades fibróticas o enfermedades pulmonares intersticiales, como la IPF, o trastornos respiratorios, especialmente cuando se administran tópicamente al pulmón.
Referencias
Castriotta RJ, et al., Chest, 2010, 138(3):693-703
Du Bois RM., Nat Rev Drug Discov., 2010, 9(2):129-40
Fehrenbach. H., et al., Virchows Arch., 1999, 435(1):20-31
King TE Jr, et al., Lancet, 2011,3;378(9807):1949-61
Lindroos. P., Am J Physio Lung Cell Mol Physiol., 2001,280:L354-L362
Tronde Ann et al., J Pharm. Sci., 2003, 92 (6), 1216-33
Selman M, et al., Ann Intern Med., 2001, 16;134(2):136-51
Wohnsland F et al., Med Chem., 2001,44;923-930
A través de la memoria descriptiva y las reivindicaciones que siguen, a menos que el contexto lo exija de otro modo, el término "comprende", y variaciones tales como "que comprende", deben entenderse como que implican la inclusión de un número entero, etapa, grupo de números enteros o grupo de etapas indicados, pero no la exclusión de cualquier otro número entero, etapa, grupo de números enteros o grupo de etapas.
Claims (15)
1. Un compuesto de fórmula (I):

en donde
R1 representa Me, Et, CH=CH2 , C=C-H o C=C-Me;
uno de R2 y R3 representa un grupo seleccionado de H, alquilo-C1-C6 , alcoxi-C1-C6 , cicloalquilo-C3-C8, CH2-(cicloalquilo-C3-C8), halógeno y ciano y el otro representa el grupo -Z-Rx;
Z representa CO o SO2 ;
Rx representa la fórmula (iv):

en donde V representa CO;
en donde v representa 0 o 1;
en donde n representa 0, 1 o 2, excepto que cuando v representa 1, n representa 1 o 2;
m representa 1 o 2;
X representa CH o N, excepto que cuando n representa 0 o 1, X representa CH;
Y representa CH o N;
W representa un grupo seleccionado de alquilo-C1-C4, hidroxialquilo-C1-C4, alcoxi-C1-C4-alquilo(C1-C4), alquileno-C1-C4CONR20R21, alquileno-C1-C4NR20COR21, SO2alquilo(C1-C4), CO-alquilo-(C1-C4), halógeno, CN, OH y NR22R23 excepto que cuando W representa NR22R23, alquileno-C1NR20COR21 o halógeno, Y representa CH;
R20, R21, R22, R23, R24 y R25 representan independientemente H o-alquilo-C1-C4;
o un compuesto seleccionado del grupo que consiste en: del grupo que consiste en:


o una sal farmacéuticamente aceptable del mismo.
2. El compuesto de la reivindicación 1 en donde R1 representa Me.
3. El compuesto de la reivindicación 1 o 2, en el que Z representa CO.
4. El compuesto de una cualquiera de las reivindicaciones 1 a 3, en donde R3 representa -Z-Rx.
5. El compuesto de una cualquiera de las reivindicaciones 1 a 4, en donde v representa 1 y n representa 1.
6. El compuesto de una cualquiera de las reivindicaciones 1 a 5, en donde m representa 1.
7. El compuesto de una cualquiera de las reivindicaciones 1 a 6, en donde X representa CH.
8. El compuesto de una cualquiera de las reivindicaciones 1 a 7, en donde Y representa N.
9. El compuesto de una cualquiera de las reivindicaciones 1 a 8, en donde W representa-alquilo C1-C4 o NR22R23.
10. Un compuesto de fórmula (I) según la reivindicación 1, que se selecciona de:
5-metil-3-(((4-(W-(2-(4-metilpiperazin-1-il)etil)sulfamoil)fenil)amino)(fenil)metilen)-2-oxoindolin-6-carboxilato de (Z)-metilo; 5-metil-3-(((4-(((1-metilpiperidin-4-il)metil)carbamoil)fenil)amino)(fenil)metilen)-2-oxoindolin-6-carboxilato de (Z)-metilo;
5-metil-3-(((4-((2-(4-metilpiperazin-1-il)etil)carbamoil)fenil)amino)(fenil)metilen)-2-oxoindolin-6-carboxilato de (Z)-metilo; 5-metil-3-(((4-((2-(4-metil-1,4-diazepan-1-il)etil)carbamoil)fenil)amino)(fenil)metileno)-2-oxoindolin-6-carboxilato de (Z)-metilo;
3-(((4-((2-(4-(dimetilamino)piperidin-1-il)etil)carbamoil)fenil)amino)(fenil)metilen)-5-metil-2-oxoindolin-6-carboxilato de (Z)-metilo;
3-(((4-((2-(4-(2-hidroxietil)piperazin-1-il)-2-oxoetil)carbamoil)fenil)amino)(fenil)metilen)-5-metil-2-oxoindolin-6-carboxilatode (Z)-metilo;
5-metil-3-(((4-((2-(4-metilpiperazin-1-il)-2-oxoetil)carbamoil)fenil)amino)(fenil)metilen)-2-oxoindolin-6-carboxilato de (Z)-metilo;
5-metil-2-oxo-3-(((4-((2-(3-oxopiperazin-1 -il)etil)carbamoil)fenil) amino)(fenil) metilen)indolin-6-carboxilato de (Z)-metilo; 5-metil-3-(((4-((3-(4-metilpiperazin-1-il)-3-oxopropil)carbamoil)fenil)amino)(fenil)metilen)-2-oxoindolin-6-carboxilato de (Z)-metilo;
3-(((4-((2-(4-hidroxipiperidin-1-il)etil)carbamoil)fenil)amino)(fenil)metilen)-5-metil-2-oxoindolin-6-carboxilato de (Z)-metilo; 3-(((4-((1-(2-hidroxietil) piperidin-4-il)carbamoil)fenil)amino)(fenil)metilen)-5-metil-2-oxoindolin-6-carboxilato de (Z)-metilo; 3-(((4-(((1 -(2-hidroxietil) piperidin-4-il)metil)carbamoil)fenil)amino)(fenil) metilen)-5-metil-2-oxoindolin-6-carboxilato de (Z)-metilo;
5-metil-3-(((4-((1-metilpiperidin-4-il)carbamoil)fenil)amino)(fenil)metilen)-2-oxoindolin-6-carboxilato de (Z)-metilo;
3-(((4-((1-(2-metoxietil)piperidin-4-il)carbamoil)fenil)amino)(fenil)metilen)-5-metil-2-oxoindolin-6-carboxilato de (Z)-metilo; 5-metil-3-(((4-((3-(4-metilpiperazin-1-il)propil)carbamoil)fenil)amino)(fenil)metilen)-2-oxoindolin-6-carboxilato de (Z)-metilo;
3-(((4-((2-(4,4-difluoropiperidin-1-il)etil)carbamoil)fenil)amino)(fenil)metilen)-5-metil-2-oxoindolin-6-carboxilato de (Z)-metilo;
3-(((4-((2-(4-acetilpiperazin-1-il)etil)carbamoil)fenil)amino)(fenil)metilen)-5-metil-2-oxoindolin-6-carboxilato de (Z)-metilo; 3-(((4-((2-(4-fluoropiperidin-1-il)etil)carbamoil)fenil)amino)(fenil)metilen)-5-metil-2-oxoindolin-6-carboxilato de (Z)-metilo; 5-metil-3-(((4-((3-morfolinopropil)carbamoil)fenil)amino)(fenil)metilen)-2-oxoindolin-6-carboxilato de (Z)-metilo;
3-(((4-((2-(4-acetil-1,4-diazepan-1-il)etil)carbamoil)fenil)amino)(fenil)metilen)-5-metil-2-oxoindolin-6-carboxilato de (Z)-metilo;
3-(((4-((2-(4-(hidroximetil)piperidin-1-il)etil)carbamoil)fenil)amino)(fenil)metilen)-5-metil-2-oxoindolin-6-carboxilato de (Z)-metilo;
5-metil-3-(((4-((2-(4-(metilsulfonil)piperazin-1-il)etil)carbamoil)fenil)amino)(fenil)metilen)-2-oxoindolin-6-carboxilato de (Z)-metilo;
3-(((4-((2-(1,1-dioxidotiomorfolino)etil)carbamoil)fenil)amino)(fenil)metilen)-5-metil-2-oxoindolin-6-carboxilato; de (Z)-metilo;
5-metil-2-oxo-3-(((4-((2-(5-oxo-1,4-diazepan-1 -il)etil)carbamoil)fenil)amino)(fenil)metilen)indolin-6-carboxilato de (Z)-metilo;
3-(((3-fluoro-4-((2-(3-oxopiperazin-1-il)etil)carbamoil)fenil)amino)(fenil)metilen)-5-metil-2-oxoindolin-6-carboxilato de (Z)-metilo;
5-metil-2-oxo-3-(((4-((2-(3-oxo-1,4-diazepan-1 -il)etil)carbamoil)fenil)amino)(fenil)metilen)indolin-6-carboxilato de (Z)-metilo;
y sales farmacéuticamente aceptables de uno cualquiera de los mismos.
11. Una composición farmacéutica que comprende un compuesto según una cualquiera de las reivindicaciones 1 a 10 en combinación con uno o más diluyentes o portadores farmacéuticamente aceptables.
12. Un compuesto de fórmula (I) según una cualquiera de las reivindicaciones 1 a 10 para uso como un medicamento.
13. Un compuesto de fórmula (I) según una cualquiera de las reivindicaciones 1 a 10 o una composición según la reivindicación 11 para uso en el tratamiento de enfermedades fibróticas o enfermedades pulmonares intersticiales, p. ej., seleccionadas de IPF, neumonía intersticial celular gigante, sarcoidosis, fibrosis cística, síndrome de distrés respiratorio, fibrosis pulmonar inducida por fármaco, granulomatosis, silicosis, asbestosis, escleroderma sistémico, la cirrosis hepática inducida por virus seleccionada de cirrosis hepática inducida por hepatitis C o enfermedades de la piel con un componente fibrótico, p. ej., seleccionada de escleroderma, sarcoidosis y lupus eritematoso sistémico, e IPF o para uso en el tratamiento de enfermedades caracterizado por la hiperproliferación de células, por ejemplo, cáncer, particularmente cáncer de pulmón o para uso en el tratamiento de trastornos respiratorios que incluyen COPD (incluyendo bronquitis crónica y enfisema), asma, asma pediátrico, rinitis alérgica, rinitis, sinusitis, o un compuesto de fórmula (I) según una cualquiera de las reivindicaciones 1 a 10 o una composición según la reivindicación 11 para uso en el tratamiento de enfermedades fibróticas tales como fibrosis pulmonar asociada con artritis reumatoide, síndrome de distrés respiratorio que incluye síndrome de distrés respiratorio agudo, lesión pulmonar aguda, fibrosis o neumonitis pulmonar inducida por radiación, neumonitis de hipersensibilidad crónica, esclerosis sistémica, síndrome de Sjogren, enfermedades pulmonares intersticiales, hipertensión pulmonar arterial (PAH), que incluye el componente vascular de PAH, o enfermedades de la piel con un componente fibrótico p. ej., seleccionadas de cicatrización hipertrófica y queloides, o enfermedades oculares donde la fibrosis es un componente que incluye glaucoma, degeneración macular asociada a la edad, edema macular diabético, enfermedad del ojo seco y retinopatía diabética, o fibrosis en el intestino, p. ej., asociada con enfermedad inflamatoria de bowel; o
un compuesto de fórmula (I) según una cualquiera de las reivindicaciones 1 a 10 o una composición según la reivindicación 11 en combinación con nintedanib o pirfenidona para uso en el tratamiento de enfermedades fibróticas o enfermedades pulmonares intersticiales seleccionadas de IPF, neumonía intersticial celular gigante, sarcoidosis, fibrosis cística, síndrome de distrés respiratorio, fibrosis pulmonar inducida por fármaco, granulomatosis, silicosis, asbestosis, escleroderma sistémica, la cirrosis hepática inducida viralmente, seleccionada de cirrosis hepática inducida por hepatitis C, o enfermedades de la piel con un componente fibrótico, p. ej., seleccionadas de escleroderma, sarcoidosis y lupus eritematoso sistémico, e IPF, o para uso en el tratamiento de enfermedades caracterizado por la hiperproliferación de células, por ejemplo, cáncer, particularmente cáncer de pulmón o para el tratamiento de trastornos respiratorios que incluyen COPD (que incluye bronquitis crónica y enfisema), asma, asma pediátrica, rinitis alérgica, rinitis, sinusitis; o
un compuesto de fórmula (I) según una cualquiera de las reivindicaciones 1 a 10 o una composición según la reivindicación 11 en combinación con nintedanib o pirfenidona para uso en el tratamiento de enfermedades fibróticas tales como fibrosis pulmonar asociada con artritis reumatoide, síndrome de distrés respiratorio que incluye 'síndrome de distrés respiratorio agudo, lesión de pulmón aguda, fibrosis o neumonitis de pulmón inducida por radiación, neumonitis con hipersensibilidad crónica, esclerosis sistémica, síndrome de Sjogren, enfermedades de pulmón intersticiales, hipertensión arterial pulmonar (PAH), que incluye el componente vascular de PAH, o enfermedades de la piel con un componente fibrótico, p. ej., seleccionadas de cicatrización hipertrófica y queloides, o enfermedades oculares donde la fibrosis es un componente que incluye glaucoma, degeneración macular asociada a la edad, edema macular diabético, enfermedad del ojo seco y retinopatía diabética, o fibrosis en el intestino, p. ej., asociada con la enfermedad inflamatoria de bowel.
14. Un compuesto de fórmula (IXb):

en donde
R1 representa Me, Et, CH=CH2 , C=C-H o C=C-Me; y
R3 representa independientemente H, alquilo-C1-C6, alcoxi-C1-C6 , cicloalquilo-C3-Cs, -CH2-(cicloalquilo-C3-Cs), halógeno o ciano;
o sales de los mismos; o
un compuesto de fórmula (Xb):

en donde
Ri representa Me, Et, CH=CH2 , C=C-H o C=C-Me; y
R3 representa independientemente H, alquilo-C1-C6 , alcoxi-C1-C6 , cicloalquilo-C3-C8, -CH2-(cicloalquilo-C3-C8), halógeno o ciano;
o sales de estos.
15. Un proceso para preparar un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo según

R1 es como se ha definido en una cualquiera de las reivindicaciones 1 a 10 y L representa un grupo saliente; o una sal del mismo;
con un compuesto de fórmula (III):

en donde
R2 y R3 son como se definen en una cualquiera de las reivindicaciones 1 a 10;
o una sal del mismo.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP16159231 | 2016-03-08 | ||
| PCT/GB2017/050619 WO2017153748A1 (en) | 2016-03-08 | 2017-03-08 | Indole derivatives and their use as protein kinase inhibitors |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2836255T3 true ES2836255T3 (es) | 2021-06-24 |
Family
ID=55521584
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES17711736T Active ES2836255T3 (es) | 2016-03-08 | 2017-03-08 | Derivados de indol y su uso como inhibidores de proteína cinasa |
Country Status (10)
| Country | Link |
|---|---|
| US (2) | US10669235B2 (es) |
| EP (1) | EP3426649B1 (es) |
| JP (1) | JP7039480B2 (es) |
| CN (1) | CN109153666B (es) |
| AU (1) | AU2017231860C1 (es) |
| BR (1) | BR112018068213A2 (es) |
| CA (1) | CA3017308A1 (es) |
| EA (1) | EA038773B1 (es) |
| ES (1) | ES2836255T3 (es) |
| WO (1) | WO2017153748A1 (es) |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20180128390A (ko) | 2015-12-24 | 2018-12-03 | 레스피버트 리미티드 | 인돌리논 화합물과 섬유성 질환의 치료에서 이의 용도 |
| CA3017308A1 (en) | 2016-03-08 | 2017-09-14 | Respivert Limited | Indole derivatives and their use as protein kinase inhibitors |
| CN112574094B (zh) * | 2020-12-14 | 2022-07-01 | 成都大学 | 吲哚酮衍生物及其制药用途 |
| WO2023043473A1 (en) * | 2021-09-14 | 2023-03-23 | R-Pharm Overseas, Inc. | TGF-β INHIBITOR COMPOSITION AND USE THEREOF |
Family Cites Families (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| UA75054C2 (uk) | 1999-10-13 | 2006-03-15 | Бьорінгер Інгельхайм Фарма Гмбх & Ко. Кг | Заміщені в положенні 6 індолінони, їх одержання та їх застосування як лікарського засобу |
| DE10117204A1 (de) * | 2001-04-06 | 2002-10-10 | Boehringer Ingelheim Pharma | In 6-Stellung substituierte Indolinone, ihre Herstellung und ihre Verwendung als Arzneimittel |
| JP2004182726A (ja) | 2002-11-22 | 2004-07-02 | Yamanouchi Pharmaceut Co Ltd | 2−オキソインドリン誘導体 |
| JP2005145928A (ja) | 2003-11-19 | 2005-06-09 | Yamanouchi Pharmaceut Co Ltd | 3−キノリン−2(1h)−イリデンインドリン−2−オン誘導体を有効成分とする医薬組成物 |
| PE20060777A1 (es) | 2004-12-24 | 2006-10-06 | Boehringer Ingelheim Int | Derivados de indolinona para el tratamiento o la prevencion de enfermedades fibroticas |
| US20060154939A1 (en) | 2004-12-24 | 2006-07-13 | Boehringer Ingelheim International Gmbh | Medicaments for the Treatment or Prevention of Fibrotic Diseases |
| WO2007008895A1 (en) | 2005-07-13 | 2007-01-18 | Allergan, Inc. | Kinase inhibitors |
| CA2728228A1 (en) | 2008-07-14 | 2010-01-21 | Gilead Sciences, Inc. | Oxindolyl inhibitor compounds |
| IN2014DN06869A (es) | 2012-01-26 | 2015-05-22 | Angion Biomedica Corp | |
| WO2015179436A1 (en) | 2014-05-19 | 2015-11-26 | Sanford-Burnham Medical Research Institute | Inflammation therapy using mekk3 inhibitors or blocking peptides |
| KR20180128390A (ko) | 2015-12-24 | 2018-12-03 | 레스피버트 리미티드 | 인돌리논 화합물과 섬유성 질환의 치료에서 이의 용도 |
| CA3017308A1 (en) | 2016-03-08 | 2017-09-14 | Respivert Limited | Indole derivatives and their use as protein kinase inhibitors |
-
2017
- 2017-03-08 CA CA3017308A patent/CA3017308A1/en not_active Abandoned
- 2017-03-08 ES ES17711736T patent/ES2836255T3/es active Active
- 2017-03-08 CN CN201780023315.XA patent/CN109153666B/zh active Active
- 2017-03-08 EP EP17711736.3A patent/EP3426649B1/en active Active
- 2017-03-08 EA EA201892020A patent/EA038773B1/ru unknown
- 2017-03-08 JP JP2018547928A patent/JP7039480B2/ja active Active
- 2017-03-08 BR BR112018068213A patent/BR112018068213A2/pt not_active IP Right Cessation
- 2017-03-08 US US16/083,886 patent/US10669235B2/en active Active
- 2017-03-08 WO PCT/GB2017/050619 patent/WO2017153748A1/en active Application Filing
- 2017-03-08 AU AU2017231860A patent/AU2017231860C1/en active Active
-
2020
- 2020-06-01 US US16/889,780 patent/US11208381B2/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| CA3017308A1 (en) | 2017-09-14 |
| EP3426649A1 (en) | 2019-01-16 |
| EP3426649B1 (en) | 2020-07-22 |
| US20190071399A1 (en) | 2019-03-07 |
| BR112018068213A2 (pt) | 2019-01-29 |
| AU2017231860C1 (en) | 2022-01-20 |
| US20210114982A1 (en) | 2021-04-22 |
| EA201892020A1 (ru) | 2019-02-28 |
| AU2017231860B2 (en) | 2021-06-10 |
| JP7039480B2 (ja) | 2022-03-22 |
| EA038773B1 (ru) | 2021-10-18 |
| US10669235B2 (en) | 2020-06-02 |
| CN109153666B (zh) | 2021-07-16 |
| WO2017153748A1 (en) | 2017-09-14 |
| JP2019509294A (ja) | 2019-04-04 |
| AU2017231860A1 (en) | 2018-09-27 |
| US11208381B2 (en) | 2021-12-28 |
| CN109153666A (zh) | 2019-01-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2796358T3 (es) | Compuestos de indolinonas y su uso en el tratamiento de enfermedades fibróticas | |
| CN111433200B (zh) | 用于治疗疾病的化合物及其药物组合物 | |
| ES2836255T3 (es) | Derivados de indol y su uso como inhibidores de proteína cinasa | |
| CN106573914B (zh) | 作为激酶抑制剂的吡唑基-脲 | |
| ES2864035T3 (es) | Derivados de 4,5,6,7-tetrahidro-1h-imidazo[4,5-c]piridina y 1,4,5,6,7,8-hexahidroimidazo[4,5-d]azepina como inhibidores de la quinasa janus | |
| EP2010185B1 (de) | Dihydrothienopyrimidine zur behandlung von entzündlichen erkrankungen | |
| ES2796235T3 (es) | Derivados de naftiridina como antagonistas de la integrina alfa V beta 6 para el tratamiento de enfermedades fibróticas | |
| ES2890226T3 (es) | Compuestos de benzodiazolio como inhibidores del ENaC | |
| PT2215092E (pt) | Piperidino-di-hidrotienopirimidinas substituídas | |
| JP2011510010A (ja) | 3H−[1,2,3]トリアゾロ[4,5−d]ピリミジン化合物、mTORキナーゼおよびPI3キナーゼ阻害剤としてのそれらの使用、ならびにそれらの合成 | |
| JP2010526801A (ja) | プロスタグランジンe2拮抗薬としてのアゼチジン誘導体およびその使用 | |
| WO2019220147A1 (en) | Compounds | |
| CN107108557A (zh) | 用于治疗和预防呼吸道合胞体病毒(rsv)感染的螺环二氢吲哚类 | |
| AU2019207140A1 (en) | Compound, preparation method therefor and use thereof | |
| ES2371450T3 (es) | Dihidropirimidinas heterocíclicas como inhibidores de los canales de potasio. |