ES2816005T3 - Tratamientos de combinación que comprenden imidazopirazinonas para el tratamiento de trastornos psiquiátricos y/o cognitivos - Google Patents
Tratamientos de combinación que comprenden imidazopirazinonas para el tratamiento de trastornos psiquiátricos y/o cognitivos Download PDFInfo
- Publication number
- ES2816005T3 ES2816005T3 ES17791080T ES17791080T ES2816005T3 ES 2816005 T3 ES2816005 T3 ES 2816005T3 ES 17791080 T ES17791080 T ES 17791080T ES 17791080 T ES17791080 T ES 17791080T ES 2816005 T3 ES2816005 T3 ES 2816005T3
- Authority
- ES
- Spain
- Prior art keywords
- pyrazin
- methyl
- imidazo
- pyran
- tetrahydro
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 208000020016 psychiatric disease Diseases 0.000 title claims abstract description 64
- 238000011282 treatment Methods 0.000 title claims abstract description 64
- 208000010877 cognitive disease Diseases 0.000 title claims abstract description 58
- 238000011284 combination treatment Methods 0.000 title description 5
- MPWOBEOETVOESI-UHFFFAOYSA-N imidazo[4,5-b]pyrazin-2-one Chemical class N1=CC=NC2=NC(=O)N=C21 MPWOBEOETVOESI-UHFFFAOYSA-N 0.000 title description 4
- 150000001875 compounds Chemical class 0.000 claims abstract description 185
- -1 bicyclic ethers Chemical class 0.000 claims abstract description 131
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 claims abstract description 48
- 125000001424 substituent group Chemical group 0.000 claims abstract description 46
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims abstract description 39
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims abstract description 35
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 30
- 239000001257 hydrogen Substances 0.000 claims abstract description 30
- 150000003839 salts Chemical class 0.000 claims abstract description 24
- 229910052736 halogen Inorganic materials 0.000 claims abstract description 23
- 150000002367 halogens Chemical group 0.000 claims abstract description 23
- 125000006163 5-membered heteroaryl group Chemical group 0.000 claims abstract description 22
- 125000001153 fluoro group Chemical group F* 0.000 claims abstract description 21
- 125000004209 (C1-C8) alkyl group Chemical group 0.000 claims abstract description 18
- 125000001412 tetrahydropyranyl group Chemical group 0.000 claims abstract description 15
- LNEPOXFFQSENCJ-UHFFFAOYSA-N haloperidol Chemical compound C1CC(O)(C=2C=CC(Cl)=CC=2)CCN1CCCC(=O)C1=CC=C(F)C=C1 LNEPOXFFQSENCJ-UHFFFAOYSA-N 0.000 claims abstract description 14
- 150000002431 hydrogen Chemical group 0.000 claims abstract description 14
- 125000003566 oxetanyl group Chemical group 0.000 claims abstract description 14
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 claims abstract description 14
- 229910052731 fluorine Inorganic materials 0.000 claims abstract description 13
- 239000011737 fluorine Substances 0.000 claims abstract description 13
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical group FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 claims abstract description 12
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical group C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 claims abstract description 12
- 125000004432 carbon atom Chemical group C* 0.000 claims abstract description 12
- 125000004093 cyano group Chemical group *C#N 0.000 claims abstract description 11
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims abstract description 11
- 125000000753 cycloalkyl group Chemical group 0.000 claims abstract description 10
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims abstract description 9
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims abstract description 9
- 125000006274 (C1-C3)alkoxy group Chemical group 0.000 claims abstract description 8
- CEUORZQYGODEFX-UHFFFAOYSA-N Aripirazole Chemical compound ClC1=CC=CC(N2CCN(CCCCOC=3C=C4NC(=O)CCC4=CC=3)CC2)=C1Cl CEUORZQYGODEFX-UHFFFAOYSA-N 0.000 claims abstract description 8
- 229960004372 aripiprazole Drugs 0.000 claims abstract description 8
- 229960001210 brexpiprazole Drugs 0.000 claims abstract description 8
- ZKIAIYBUSXZPLP-UHFFFAOYSA-N brexpiprazole Chemical compound C1=C2NC(=O)C=CC2=CC=C1OCCCCN(CC1)CCN1C1=CC=CC2=C1C=CS2 ZKIAIYBUSXZPLP-UHFFFAOYSA-N 0.000 claims abstract description 8
- 229960004170 clozapine Drugs 0.000 claims abstract description 8
- QZUDBNBUXVUHMW-UHFFFAOYSA-N clozapine Chemical compound C1CN(C)CCN1C1=NC2=CC(Cl)=CC=C2NC2=CC=CC=C12 QZUDBNBUXVUHMW-UHFFFAOYSA-N 0.000 claims abstract description 8
- 229960005017 olanzapine Drugs 0.000 claims abstract description 8
- KVWDHTXUZHCGIO-UHFFFAOYSA-N olanzapine Chemical compound C1CN(C)CCN1C1=NC2=CC=CC=C2NC2=C1C=C(C)S2 KVWDHTXUZHCGIO-UHFFFAOYSA-N 0.000 claims abstract description 8
- 229960004431 quetiapine Drugs 0.000 claims abstract description 8
- URKOMYMAXPYINW-UHFFFAOYSA-N quetiapine Chemical compound C1CN(CCOCCO)CCN1C1=NC2=CC=CC=C2SC2=CC=CC=C12 URKOMYMAXPYINW-UHFFFAOYSA-N 0.000 claims abstract description 8
- 229960001534 risperidone Drugs 0.000 claims abstract description 8
- RAPZEAPATHNIPO-UHFFFAOYSA-N risperidone Chemical compound FC1=CC=C2C(C3CCN(CC3)CCC=3C(=O)N4CCCCC4=NC=3C)=NOC2=C1 RAPZEAPATHNIPO-UHFFFAOYSA-N 0.000 claims abstract description 8
- 229960000607 ziprasidone Drugs 0.000 claims abstract description 8
- MVWVFYHBGMAFLY-UHFFFAOYSA-N ziprasidone Chemical compound C1=CC=C2C(N3CCN(CC3)CCC3=CC=4CC(=O)NC=4C=C3Cl)=NSC2=C1 MVWVFYHBGMAFLY-UHFFFAOYSA-N 0.000 claims abstract description 8
- VSWBSWWIRNCQIJ-GJZGRUSLSA-N (R,R)-asenapine Chemical compound O1C2=CC=CC=C2[C@@H]2CN(C)C[C@H]2C2=CC(Cl)=CC=C21 VSWBSWWIRNCQIJ-GJZGRUSLSA-N 0.000 claims abstract description 7
- PMXMIIMHBWHSKN-UHFFFAOYSA-N 3-{2-[4-(6-fluoro-1,2-benzoxazol-3-yl)piperidin-1-yl]ethyl}-9-hydroxy-2-methyl-6,7,8,9-tetrahydropyrido[1,2-a]pyrimidin-4-one Chemical compound FC1=CC=C2C(C3CCN(CC3)CCC=3C(=O)N4CCCC(O)C4=NC=3C)=NOC2=C1 PMXMIIMHBWHSKN-UHFFFAOYSA-N 0.000 claims abstract description 7
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims abstract description 7
- 229960003036 amisulpride Drugs 0.000 claims abstract description 7
- NTJOBXMMWNYJFB-UHFFFAOYSA-N amisulpride Chemical compound CCN1CCCC1CNC(=O)C1=CC(S(=O)(=O)CC)=C(N)C=C1OC NTJOBXMMWNYJFB-UHFFFAOYSA-N 0.000 claims abstract description 7
- 229960005245 asenapine Drugs 0.000 claims abstract description 7
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims abstract description 7
- 229960005123 cariprazine Drugs 0.000 claims abstract description 7
- KPWSJANDNDDRMB-QAQDUYKDSA-N cariprazine Chemical compound C1C[C@@H](NC(=O)N(C)C)CC[C@@H]1CCN1CCN(C=2C(=C(Cl)C=CC=2)Cl)CC1 KPWSJANDNDDRMB-QAQDUYKDSA-N 0.000 claims abstract description 7
- ZPEIMTDSQAKGNT-UHFFFAOYSA-N chlorpromazine Chemical compound C1=C(Cl)C=C2N(CCCN(C)C)C3=CC=CC=C3SC2=C1 ZPEIMTDSQAKGNT-UHFFFAOYSA-N 0.000 claims abstract description 7
- 229960001076 chlorpromazine Drugs 0.000 claims abstract description 7
- NJMYODHXAKYRHW-DVZOWYKESA-N cis-flupenthixol Chemical compound C1CN(CCO)CCN1CC\C=C\1C2=CC(C(F)(F)F)=CC=C2SC2=CC=CC=C2/1 NJMYODHXAKYRHW-DVZOWYKESA-N 0.000 claims abstract description 7
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims abstract description 7
- 229960002419 flupentixol Drugs 0.000 claims abstract description 7
- 229960003878 haloperidol Drugs 0.000 claims abstract description 7
- 229960003162 iloperidone Drugs 0.000 claims abstract description 7
- XMXHEBAFVSFQEX-UHFFFAOYSA-N iloperidone Chemical compound COC1=CC(C(C)=O)=CC=C1OCCCN1CCC(C=2C3=CC=C(F)C=C3ON=2)CC1 XMXHEBAFVSFQEX-UHFFFAOYSA-N 0.000 claims abstract description 7
- 229960001432 lurasidone Drugs 0.000 claims abstract description 7
- PQXKDMSYBGKCJA-CVTJIBDQSA-N lurasidone Chemical compound C1=CC=C2C(N3CCN(CC3)C[C@@H]3CCCC[C@H]3CN3C(=O)[C@@H]4[C@H]5CC[C@H](C5)[C@@H]4C3=O)=NSC2=C1 PQXKDMSYBGKCJA-CVTJIBDQSA-N 0.000 claims abstract description 7
- 229960001057 paliperidone Drugs 0.000 claims abstract description 7
- WFPIAZLQTJBIFN-DVZOWYKESA-N zuclopenthixol Chemical compound C1CN(CCO)CCN1CC\C=C\1C2=CC(Cl)=CC=C2SC2=CC=CC=C2/1 WFPIAZLQTJBIFN-DVZOWYKESA-N 0.000 claims abstract description 7
- 229960004141 zuclopenthixol Drugs 0.000 claims abstract description 7
- 125000006527 (C1-C5) alkyl group Chemical group 0.000 claims abstract description 6
- SSOZXXUSHMYCRH-UHFFFAOYSA-N 3,3a,4,5,6,6a-hexahydro-2h-furo[3,2-b]pyrrole Chemical group O1CCC2NCCC21 SSOZXXUSHMYCRH-UHFFFAOYSA-N 0.000 claims abstract description 6
- 125000000000 cycloalkoxy group Chemical group 0.000 claims abstract description 6
- 150000003951 lactams Chemical group 0.000 claims abstract description 6
- 125000002950 monocyclic group Chemical group 0.000 claims abstract description 6
- MIPHRQMEIYLZFZ-UHFFFAOYSA-N oxolan-3-amine Chemical group NC1CCOC1 MIPHRQMEIYLZFZ-UHFFFAOYSA-N 0.000 claims abstract description 6
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims abstract description 6
- 125000004076 pyridyl group Chemical group 0.000 claims abstract description 6
- ZNGWEEUXTBNKFR-UHFFFAOYSA-N 1,4-oxazepane Chemical group C1CNCCOC1 ZNGWEEUXTBNKFR-UHFFFAOYSA-N 0.000 claims abstract description 5
- YPWFNLSXQIGJCK-UHFFFAOYSA-N 7-oxabicyclo[2.2.1]heptane Chemical compound C1CC2CCC1O2 YPWFNLSXQIGJCK-UHFFFAOYSA-N 0.000 claims abstract description 5
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical group C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 claims abstract description 5
- 125000003392 indanyl group Chemical group C1(CCC2=CC=CC=C12)* 0.000 claims abstract description 5
- 125000004433 nitrogen atom Chemical group N* 0.000 claims abstract description 5
- 229940124530 sulfonamide Drugs 0.000 claims abstract description 5
- 150000003456 sulfonamides Chemical group 0.000 claims abstract description 5
- 125000006284 3-fluorobenzyl group Chemical group [H]C1=C([H])C(=C([H])C(F)=C1[H])C([H])([H])* 0.000 claims description 26
- 239000008194 pharmaceutical composition Substances 0.000 claims description 25
- 201000000980 schizophrenia Diseases 0.000 claims description 24
- 208000028698 Cognitive impairment Diseases 0.000 claims description 22
- SLYRIXNICMCXFH-UHFFFAOYSA-N 7h-imidazo[1,5-a]pyrazin-8-one Chemical compound O=C1NC=CN2C=NC=C12 SLYRIXNICMCXFH-UHFFFAOYSA-N 0.000 claims description 21
- 239000003085 diluting agent Substances 0.000 claims description 12
- 239000003937 drug carrier Substances 0.000 claims description 10
- 208000019901 Anxiety disease Diseases 0.000 claims description 9
- 208000006096 Attention Deficit Disorder with Hyperactivity Diseases 0.000 claims description 9
- 230000036506 anxiety Effects 0.000 claims description 9
- 201000003631 narcolepsy Diseases 0.000 claims description 9
- 125000004217 4-methoxybenzyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1OC([H])([H])[H])C([H])([H])* 0.000 claims description 6
- KTLRVQOJQCOTEE-UHFFFAOYSA-N 7-[(4-aminophenyl)methyl]-6-methyl-3-(oxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound NC1=CC=C(CN2C(C=3N(C=C2C)C(=NC=3)C2CCOCC2)=O)C=C1 KTLRVQOJQCOTEE-UHFFFAOYSA-N 0.000 claims description 6
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 6
- RVJAZKLTOAMCHB-UHFFFAOYSA-N 3-(2-hydroxypropan-2-yl)-7-[(4-methoxyphenyl)methyl]-6-methylimidazo[1,5-a]pyrazin-8-one Chemical compound OC(C)(C)C1=NC=C2N1C=C(N(C2=O)CC1=CC=C(C=C1)OC)C RVJAZKLTOAMCHB-UHFFFAOYSA-N 0.000 claims description 5
- IHHHPGCUSMYQHX-UHFFFAOYSA-N 6-ethyl-7-[(4-methoxyphenyl)methyl]-3-(oxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound C(C)C=1N(C(C=2N(C=1)C(=NC=2)C1CCOCC1)=O)CC1=CC=C(C=C1)OC IHHHPGCUSMYQHX-UHFFFAOYSA-N 0.000 claims description 5
- FRYCFLNTERXYHU-UHFFFAOYSA-N 7-[(3-chlorophenyl)methyl]-6-methyl-3-(oxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound ClC=1C=C(CN2C(C=3N(C=C2C)C(=NC=3)C2CCOCC2)=O)C=CC=1 FRYCFLNTERXYHU-UHFFFAOYSA-N 0.000 claims description 5
- HIARPVHIVVSVTG-KRWDZBQOSA-N 7-[(3-fluorophenyl)methyl]-6-methyl-3-[(2S)-oxan-2-yl]imidazo[1,5-a]pyrazin-8-one Chemical compound FC=1C=C(CN2C(C=3N(C=C2C)C(=NC=3)[C@H]2OCCCC2)=O)C=CC=1 HIARPVHIVVSVTG-KRWDZBQOSA-N 0.000 claims description 5
- IHKCYYAPLNKUMM-UHFFFAOYSA-N 7-[(4-methoxyphenyl)methyl]-6-methyl-3-(5-methyloxolan-3-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound COC1=CC=C(CN2C(C=3N(C=C2C)C(=NC=3)C2COC(C2)C)=O)C=C1 IHKCYYAPLNKUMM-UHFFFAOYSA-N 0.000 claims description 5
- PUKNFFAMUWZTGI-UHFFFAOYSA-N 7-[(4-methoxyphenyl)methyl]-6-methyl-3-(oxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound COC1=CC=C(CN2C(C=3N(C=C2C)C(=NC=3)C2CCOCC2)=O)C=C1 PUKNFFAMUWZTGI-UHFFFAOYSA-N 0.000 claims description 5
- SWHFTCTZWOUUQD-KRWDZBQOSA-N 7-[(4-methoxyphenyl)methyl]-6-methyl-3-[(1S)-1-phenylethyl]imidazo[1,5-a]pyrazin-8-one Chemical compound COC1=CC=C(CN2C(C=3N(C=C2C)C(=NC=3)[C@@H](C)C2=CC=CC=C2)=O)C=C1 SWHFTCTZWOUUQD-KRWDZBQOSA-N 0.000 claims description 5
- RVHZRSIRMVWVKL-UHFFFAOYSA-N 7-[(4-methoxyphenyl)methyl]-6-methyl-3-propan-2-ylimidazo[1,5-a]pyrazin-8-one Chemical compound C(C)(C)C1=NC=C2N1C=C(N(C2=O)CC1=CC=C(C=C1)OC)C RVHZRSIRMVWVKL-UHFFFAOYSA-N 0.000 claims description 5
- ISOFGIBMBLOJRH-UHFFFAOYSA-N 2-[[6-methyl-3-(oxan-4-yl)-8-oxoimidazo[1,5-a]pyrazin-7-yl]methyl]benzonitrile Chemical compound CC=1N(C(C=2N(C=1)C(=NC=2)C1CCOCC1)=O)CC1=C(C#N)C=CC=C1 ISOFGIBMBLOJRH-UHFFFAOYSA-N 0.000 claims description 4
- UOMHYPRQAMXYDQ-UHFFFAOYSA-N 3-(1,4-dimethylpiperidin-4-yl)-7-[(4-methoxyphenyl)methyl]-6-methylimidazo[1,5-a]pyrazin-8-one Chemical compound CN1CCC(CC1)(C)C1=NC=C2N1C=C(N(C2=O)CC1=CC=C(C=C1)OC)C UOMHYPRQAMXYDQ-UHFFFAOYSA-N 0.000 claims description 4
- ANGLWHGRWFOZSQ-UHFFFAOYSA-N 3-(1-methoxy-2-methylpropan-2-yl)-7-[(4-methoxyphenyl)methyl]-6-methylimidazo[1,5-a]pyrazin-8-one Chemical compound COCC(C)(C)C1=NC=C2N1C=C(N(C2=O)CC1=CC=C(C=C1)OC)C ANGLWHGRWFOZSQ-UHFFFAOYSA-N 0.000 claims description 4
- GEKRVCRUZXROSZ-UHFFFAOYSA-N 3-(2,2-difluorocyclopropyl)-7-[(3-fluorophenyl)methyl]-6-methylimidazo[1,5-a]pyrazin-8-one Chemical compound FC1(C(C1)C1=NC=C2N1C=C(N(C2=O)CC1=CC(=CC=C1)F)C)F GEKRVCRUZXROSZ-UHFFFAOYSA-N 0.000 claims description 4
- HVRCNXOHZPZGSJ-UHFFFAOYSA-N 3-(2,2-dimethylmorpholin-4-yl)-7-[(4-methoxyphenyl)methyl]-6-methylimidazo[1,5-a]pyrazin-8-one Chemical compound CC1(OCCN(C1)C1=NC=C2N1C=C(N(C2=O)CC1=CC=C(C=C1)OC)C)C HVRCNXOHZPZGSJ-UHFFFAOYSA-N 0.000 claims description 4
- IJXXNTVMDPJVBW-UHFFFAOYSA-N 3-(2,6-dimethyloxan-4-yl)-7-[(4-methoxyphenyl)methyl]-6-methylimidazo[1,5-a]pyrazin-8-one Chemical compound CC1OC(CC(C1)C1=NC=C2N1C=C(N(C2=O)CC1=CC=C(C=C1)OC)C)C IJXXNTVMDPJVBW-UHFFFAOYSA-N 0.000 claims description 4
- MVVQNOPLTGEXNR-ZIAGYGMSSA-N 3-[(1S,2R)-2-fluorocyclopropyl]-7-[(3-fluorophenyl)methyl]-6-methylimidazo[1,5-a]pyrazin-8-one Chemical compound FC=1C=C(CN2C(C=3N(C=C2C)C(=NC=3)[C@H]2[C@@H](C2)F)=O)C=CC=1 MVVQNOPLTGEXNR-ZIAGYGMSSA-N 0.000 claims description 4
- RZZDUEKLZBHEJJ-UHFFFAOYSA-N 3-[7-[(4-methoxyphenyl)methyl]-6-methyl-8-oxoimidazo[1,5-a]pyrazin-3-yl]-3-methylpyrrolidine-1-sulfonamide Chemical compound COC1=CC=C(CN2C(C=3N(C=C2C)C(=NC=3)C2(CN(CC2)S(=O)(=O)N)C)=O)C=C1 RZZDUEKLZBHEJJ-UHFFFAOYSA-N 0.000 claims description 4
- XFBSUTIIWYEUPC-UHFFFAOYSA-N 3-cyclopropyl-7-[(3-fluorophenyl)methyl]imidazo[1,5-a]pyrazin-8-one Chemical compound C1(CC1)C1=NC=C2N1C=CN(C2=O)CC1=CC(=CC=C1)F XFBSUTIIWYEUPC-UHFFFAOYSA-N 0.000 claims description 4
- USXLZPZGBIYQLQ-UHFFFAOYSA-N 4-[7-[(3-fluorophenyl)methyl]-6-methyl-8-oxoimidazo[1,5-a]pyrazin-3-yl]oxane-4-carbonitrile Chemical compound FC=1C=C(CN2C(C=3N(C=C2C)C(=NC=3)C2(CCOCC2)C#N)=O)C=CC=1 USXLZPZGBIYQLQ-UHFFFAOYSA-N 0.000 claims description 4
- LQAZSSIUEDYCBP-UHFFFAOYSA-N 4-[[6-methyl-3-(oxan-4-yl)-8-oxoimidazo[1,5-a]pyrazin-7-yl]methyl]benzonitrile Chemical compound CC=1N(C(C=2N(C=1)C(=NC=2)C1CCOCC1)=O)CC1=CC=C(C#N)C=C1 LQAZSSIUEDYCBP-UHFFFAOYSA-N 0.000 claims description 4
- SVVUZGLLATUKEM-UHFFFAOYSA-N 6,7-dimethyl-3-(oxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound CC=1N(C(C=2N(C=1)C(=NC=2)C1CCOCC1)=O)C SVVUZGLLATUKEM-UHFFFAOYSA-N 0.000 claims description 4
- DDTRHXHSHXSEDF-UHFFFAOYSA-N 6-benzyl-7-(cyclohexylmethyl)-3-(oxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound C(C1=CC=CC=C1)C=1N(C(C=2N(C=1)C(=NC=2)C1CCOCC1)=O)CC1CCCCC1 DDTRHXHSHXSEDF-UHFFFAOYSA-N 0.000 claims description 4
- NTXQTICQWIKNOO-UHFFFAOYSA-N 6-benzyl-7-[(3-fluorophenyl)methyl]-3-(oxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound C(C1=CC=CC=C1)C=1N(C(C=2N(C=1)C(=NC=2)C1CCOCC1)=O)CC1=CC(=CC=C1)F NTXQTICQWIKNOO-UHFFFAOYSA-N 0.000 claims description 4
- JVYBBFBHTBCHNY-UHFFFAOYSA-N 6-bromo-7-[(3-fluorophenyl)methyl]-3-(oxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound BrC=1N(C(C=2N(C=1)C(=NC=2)C1CCOCC1)=O)CC1=CC(=CC=C1)F JVYBBFBHTBCHNY-UHFFFAOYSA-N 0.000 claims description 4
- AFONMYZRPMFJAB-UHFFFAOYSA-N 6-ethyl-7-[(3-fluorophenyl)methyl]-3-(oxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound C(C)C=1N(C(C=2N(C=1)C(=NC=2)C1CCOCC1)=O)CC1=CC(=CC=C1)F AFONMYZRPMFJAB-UHFFFAOYSA-N 0.000 claims description 4
- SQDRHBKRJBDLOC-UHFFFAOYSA-N 6-methyl-3-(oxan-4-yl)-7-(1,3-thiazol-4-ylmethyl)imidazo[1,5-a]pyrazin-8-one Chemical compound CC=1N(C(C=2N(C=1)C(=NC=2)C1CCOCC1)=O)CC=1N=CSC=1 SQDRHBKRJBDLOC-UHFFFAOYSA-N 0.000 claims description 4
- VRSLRMWKAICODM-UHFFFAOYSA-N 6-methyl-3-(oxan-4-yl)-7-(thiophen-3-ylmethyl)imidazo[1,5-a]pyrazin-8-one Chemical compound CC=1N(C(C=2N(C=1)C(=NC=2)C1CCOCC1)=O)CC1=CSC=C1 VRSLRMWKAICODM-UHFFFAOYSA-N 0.000 claims description 4
- BMVVKSPFTXQSSF-UHFFFAOYSA-N 6-methyl-3-(oxan-4-yl)-7-propan-2-ylimidazo[1,5-a]pyrazin-8-one Chemical compound C(C)(C)N1C(C=2N(C=C1C)C(=NC=2)C1CCOCC1)=O BMVVKSPFTXQSSF-UHFFFAOYSA-N 0.000 claims description 4
- OEYDINZBWMRAIK-UHFFFAOYSA-N 6-methyl-7-(3-methylbutyl)-3-(oxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound C(CC(C)C)N1C(C=2N(C=C1C)C(=NC=2)C1CCOCC1)=O OEYDINZBWMRAIK-UHFFFAOYSA-N 0.000 claims description 4
- SFJUWCCYAPWXBB-UHFFFAOYSA-N 6-methyl-7-[(2-methyl-1,3-thiazol-4-yl)methyl]-3-(oxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound CC=1N(C(C=2N(C=1)C(=NC=2)C1CCOCC1)=O)CC=1N=C(SC=1)C SFJUWCCYAPWXBB-UHFFFAOYSA-N 0.000 claims description 4
- ZKIKXHAKXUODEV-UHFFFAOYSA-N 6-methyl-7-[(3-methyl-1,2-oxazol-5-yl)methyl]-3-(oxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound CC=1N(C(C=2N(C=1)C(=NC=2)C1CCOCC1)=O)CC1=CC(=NO1)C ZKIKXHAKXUODEV-UHFFFAOYSA-N 0.000 claims description 4
- FCLGCXKNDLJHGE-UHFFFAOYSA-N 6-methyl-7-[(3-methylphenyl)methyl]-3-(oxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound CC=1N(C(C=2N(C=1)C(=NC=2)C1CCOCC1)=O)CC1=CC(=CC=C1)C FCLGCXKNDLJHGE-UHFFFAOYSA-N 0.000 claims description 4
- NJLBYLKOKZGPPW-UHFFFAOYSA-N 6-methyl-7-[(4-methylphenyl)methyl]-3-(oxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound CC=1N(C(C=2N(C=1)C(=NC=2)C1CCOCC1)=O)CC1=CC=C(C=C1)C NJLBYLKOKZGPPW-UHFFFAOYSA-N 0.000 claims description 4
- GPWMWZXIKCDSMW-UHFFFAOYSA-N 6-methyl-7-[(5-methyl-1,2-oxazol-3-yl)methyl]-3-(oxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound CC=1N(C(C=2N(C=1)C(=NC=2)C1CCOCC1)=O)CC1=NOC(=C1)C GPWMWZXIKCDSMW-UHFFFAOYSA-N 0.000 claims description 4
- SGRJQQJTJHBFTK-UHFFFAOYSA-N 7-(cycloheptylmethyl)-3-(oxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound C1(CCCCCC1)CN1C(C=2N(C=C1)C(=NC=2)C1CCOCC1)=O SGRJQQJTJHBFTK-UHFFFAOYSA-N 0.000 claims description 4
- FIIDAEWKLGRESO-UHFFFAOYSA-N 7-(cycloheptylmethyl)-6-methyl-3-(oxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound C1(CCCCCC1)CN1C(C=2N(C=C1C)C(=NC=2)C1CCOCC1)=O FIIDAEWKLGRESO-UHFFFAOYSA-N 0.000 claims description 4
- XXIPRLNKXMMFIK-UHFFFAOYSA-N 7-(cyclohexylmethyl)-3-(oxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound C1(CCCCC1)CN1C(C=2N(C=C1)C(=NC=2)C1CCOCC1)=O XXIPRLNKXMMFIK-UHFFFAOYSA-N 0.000 claims description 4
- MVICHLUMJUWVOO-UHFFFAOYSA-N 7-(cyclohexylmethyl)-3-cyclopropylimidazo[1,5-a]pyrazin-8-one Chemical compound C1(CCCCC1)CN1C(C=2N(C=C1)C(=NC=2)C1CC1)=O MVICHLUMJUWVOO-UHFFFAOYSA-N 0.000 claims description 4
- XQZACXVVNPRUIZ-UHFFFAOYSA-N 7-(cyclohexylmethyl)-6-methyl-3-(oxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound C1(CCCCC1)CN1C(C=2N(C=C1C)C(=NC=2)C1CCOCC1)=O XQZACXVVNPRUIZ-UHFFFAOYSA-N 0.000 claims description 4
- ZVDAZHCIEJDXMC-UHFFFAOYSA-N 7-(cyclohexylmethyl)-6-methyl-3-propylimidazo[1,5-a]pyrazin-8-one Chemical compound C1(CCCCC1)CN1C(C=2N(C=C1C)C(=NC=2)CCC)=O ZVDAZHCIEJDXMC-UHFFFAOYSA-N 0.000 claims description 4
- YXPSIYIAHYJXGI-UHFFFAOYSA-N 7-(cyclopentylmethyl)-3-(oxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound C1(CCCC1)CN1C(C=2N(C=C1)C(=NC=2)C1CCOCC1)=O YXPSIYIAHYJXGI-UHFFFAOYSA-N 0.000 claims description 4
- MUELWSVWNVCRNW-UHFFFAOYSA-N 7-(cyclopentylmethyl)-3-cyclopropylimidazo[1,5-a]pyrazin-8-one Chemical compound C1(CCCC1)CN1C(C=2N(C=C1)C(=NC=2)C1CC1)=O MUELWSVWNVCRNW-UHFFFAOYSA-N 0.000 claims description 4
- IVIOKHSMYSCNPJ-UHFFFAOYSA-N 7-(cyclopentylmethyl)-6-methyl-3-(oxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound C1(CCCC1)CN1C(C=2N(C=C1C)C(=NC=2)C1CCOCC1)=O IVIOKHSMYSCNPJ-UHFFFAOYSA-N 0.000 claims description 4
- RNBCFPQVHXWYQR-UHFFFAOYSA-N 7-[(2-chlorophenyl)methyl]-6-methyl-3-(oxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound ClC1=C(CN2C(C=3N(C=C2C)C(=NC=3)C2CCOCC2)=O)C=CC=C1 RNBCFPQVHXWYQR-UHFFFAOYSA-N 0.000 claims description 4
- HLXDXKMIVTXPEU-UHFFFAOYSA-N 7-[(2-ethylphenyl)methyl]-6-methyl-3-(oxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound C(C)C1=C(CN2C(C=3N(C=C2C)C(=NC=3)C2CCOCC2)=O)C=CC=C1 HLXDXKMIVTXPEU-UHFFFAOYSA-N 0.000 claims description 4
- SGDZBPUOHUWTHZ-UHFFFAOYSA-N 7-[(2-fluorophenyl)methyl]-6-methyl-3-(oxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound FC1=C(CN2C(C=3N(C=C2C)C(=NC=3)C2CCOCC2)=O)C=CC=C1 SGDZBPUOHUWTHZ-UHFFFAOYSA-N 0.000 claims description 4
- MVNHOYJYJWAEEH-UHFFFAOYSA-N 7-[(3,5-dimethyl-1,2-oxazol-4-yl)methyl]-6-methyl-3-(oxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound CC1=NOC(=C1CN1C(C=2N(C=C1C)C(=NC=2)C1CCOCC1)=O)C MVNHOYJYJWAEEH-UHFFFAOYSA-N 0.000 claims description 4
- ZCVGFCXGAGKYHH-UHFFFAOYSA-N 7-[(3-fluorophenyl)methyl]-3-(4-methoxyoxan-4-yl)-6-methylimidazo[1,5-a]pyrazin-8-one Chemical compound FC=1C=C(CN2C(C=3N(C=C2C)C(=NC=3)C2(CCOCC2)OC)=O)C=CC=1 ZCVGFCXGAGKYHH-UHFFFAOYSA-N 0.000 claims description 4
- KWKGJBUJSCDIAL-UHFFFAOYSA-N 7-[(3-fluorophenyl)methyl]-3-(oxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound FC=1C=C(CN2C(C=3N(C=C2)C(=NC=3)C2CCOCC2)=O)C=CC=1 KWKGJBUJSCDIAL-UHFFFAOYSA-N 0.000 claims description 4
- MUGBFEOWKYVLQV-UHFFFAOYSA-N 7-[(3-fluorophenyl)methyl]-3-propylimidazo[1,5-a]pyrazin-8-one Chemical compound FC=1C=C(CN2C(C=3N(C=C2)C(=NC=3)CCC)=O)C=CC=1 MUGBFEOWKYVLQV-UHFFFAOYSA-N 0.000 claims description 4
- VCCNMXVKDHUXGT-UHFFFAOYSA-N 7-[(3-fluorophenyl)methyl]-6-methyl-3-(1-methylcyclopropyl)imidazo[1,5-a]pyrazin-8-one Chemical compound FC=1C=C(CN2C(C=3N(C=C2C)C(=NC=3)C2(CC2)C)=O)C=CC=1 VCCNMXVKDHUXGT-UHFFFAOYSA-N 0.000 claims description 4
- MVBCAJXPQPBVSA-UHFFFAOYSA-N 7-[(3-fluorophenyl)methyl]-6-methyl-3-(4-methyloxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound FC=1C=C(CN2C(C=3N(C=C2C)C(=NC=3)C2(CCOCC2)C)=O)C=CC=1 MVBCAJXPQPBVSA-UHFFFAOYSA-N 0.000 claims description 4
- DOTFZTSFGIKARD-UHFFFAOYSA-N 7-[(3-fluorophenyl)methyl]-6-methyl-3-(oxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound FC=1C=C(CN2C(C=3N(C=C2C)C(=NC=3)C2CCOCC2)=O)C=CC=1 DOTFZTSFGIKARD-UHFFFAOYSA-N 0.000 claims description 4
- PZKOOBRKCGIRJF-HNNXBMFYSA-N 7-[(3-fluorophenyl)methyl]-6-methyl-3-[(3R)-oxan-3-yl]imidazo[1,5-a]pyrazin-8-one Chemical compound CC1=CN2C(=CN=C2[C@H]2CCCOC2)C(=O)N1CC1=CC(F)=CC=C1 PZKOOBRKCGIRJF-HNNXBMFYSA-N 0.000 claims description 4
- LEYPTBGWIKQYRW-AWEZNQCLSA-N 7-[(3-fluorophenyl)methyl]-6-methyl-3-[(3R)-oxolan-3-yl]imidazo[1,5-a]pyrazin-8-one Chemical compound CC1=CN2C(=CN=C2[C@H]2CCOC2)C(=O)N1CC1=CC(F)=CC=C1 LEYPTBGWIKQYRW-AWEZNQCLSA-N 0.000 claims description 4
- PZKOOBRKCGIRJF-OAHLLOKOSA-N 7-[(3-fluorophenyl)methyl]-6-methyl-3-[(3S)-oxan-3-yl]imidazo[1,5-a]pyrazin-8-one Chemical compound FC=1C=C(CN2C(C=3N(C=C2C)C(=NC=3)[C@H]2COCCC2)=O)C=CC=1 PZKOOBRKCGIRJF-OAHLLOKOSA-N 0.000 claims description 4
- LEYPTBGWIKQYRW-CQSZACIVSA-N 7-[(3-fluorophenyl)methyl]-6-methyl-3-[(3S)-oxolan-3-yl]imidazo[1,5-a]pyrazin-8-one Chemical compound FC=1C=C(CN2C(C=3N(C=C2C)C(=NC=3)[C@H]2COCC2)=O)C=CC=1 LEYPTBGWIKQYRW-CQSZACIVSA-N 0.000 claims description 4
- OYBSUAILVMFPHS-UHFFFAOYSA-N 7-[(3-methoxyphenyl)methyl]-6-methyl-3-(oxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound COC=1C=C(CN2C(C=3N(C=C2C)C(=NC=3)C2CCOCC2)=O)C=CC=1 OYBSUAILVMFPHS-UHFFFAOYSA-N 0.000 claims description 4
- YSJUCDCNAYRULZ-UHFFFAOYSA-N 7-[(4-chlorophenyl)methyl]-6-methyl-3-(oxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound ClC1=CC=C(CN2C(C=3N(C=C2C)C(=NC=3)C2CCOCC2)=O)C=C1 YSJUCDCNAYRULZ-UHFFFAOYSA-N 0.000 claims description 4
- RVWGYNGTLDYNPU-UHFFFAOYSA-N 7-[(4-cyclopropyloxyphenyl)methyl]-6-methyl-3-(oxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound C1(CC1)OC1=CC=C(CN2C(C=3N(C=C2C)C(=NC=3)C2CCOCC2)=O)C=C1 RVWGYNGTLDYNPU-UHFFFAOYSA-N 0.000 claims description 4
- HTAKTLWSUKQRRS-UHFFFAOYSA-N 7-[(4-fluorophenyl)methyl]-6-methyl-3-(oxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound FC1=CC=C(CN2C(C=3N(C=C2C)C(=NC=3)C2CCOCC2)=O)C=C1 HTAKTLWSUKQRRS-UHFFFAOYSA-N 0.000 claims description 4
- HWZIGTUHYHCMIT-UHFFFAOYSA-N 7-[(4-hydroxyphenyl)methyl]-6-methyl-3-(oxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound OC1=CC=C(CN2C(C=3N(C=C2C)C(=NC=3)C2CCOCC2)=O)C=C1 HWZIGTUHYHCMIT-UHFFFAOYSA-N 0.000 claims description 4
- VCAGHSXTTDHBAV-UHFFFAOYSA-N 7-[(4-methoxyphenyl)methyl]-6-methyl-3-(1,4-oxazepan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound COC1=CC=C(CN2C(C=3N(C=C2C)C(=NC=3)N2CCOCCC2)=O)C=C1 VCAGHSXTTDHBAV-UHFFFAOYSA-N 0.000 claims description 4
- ZMTDJXQCSYULBW-UHFFFAOYSA-N 7-[(4-methoxyphenyl)methyl]-6-methyl-3-(7-oxoazepan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound COC1=CC=C(CN2C(C=3N(C=C2C)C(=NC=3)C2CCNC(CC2)=O)=O)C=C1 ZMTDJXQCSYULBW-UHFFFAOYSA-N 0.000 claims description 4
- MVGJRDDRTHNROA-UHFFFAOYSA-N 7-[(4-methoxyphenyl)methyl]-6-methyl-3-(oxolan-3-ylamino)imidazo[1,5-a]pyrazin-8-one Chemical compound COC1=CC=C(CN2C(C=3N(C=C2C)C(=NC=3)NC2COCC2)=O)C=C1 MVGJRDDRTHNROA-UHFFFAOYSA-N 0.000 claims description 4
- SWHFTCTZWOUUQD-QGZVFWFLSA-N 7-[(4-methoxyphenyl)methyl]-6-methyl-3-[(1R)-1-phenylethyl]imidazo[1,5-a]pyrazin-8-one Chemical compound COC1=CC=C(CN2C(C=3N(C=C2C)C(=NC=3)[C@H](C)C2=CC=CC=C2)=O)C=C1 SWHFTCTZWOUUQD-QGZVFWFLSA-N 0.000 claims description 4
- VQUWFLFKDYALSX-OAHLLOKOSA-N 7-[(4-methoxyphenyl)methyl]-6-methyl-3-[(3R)-3-methylmorpholin-4-yl]imidazo[1,5-a]pyrazin-8-one Chemical compound COC1=CC=C(CN2C(C=3N(C=C2C)C(=NC=3)N2[C@@H](COCC2)C)=O)C=C1 VQUWFLFKDYALSX-OAHLLOKOSA-N 0.000 claims description 4
- VQUWFLFKDYALSX-HNNXBMFYSA-N 7-[(4-methoxyphenyl)methyl]-6-methyl-3-[(3S)-3-methylmorpholin-4-yl]imidazo[1,5-a]pyrazin-8-one Chemical compound COC1=CC=C(CN2C(C=3N(C=C2C)C(=NC=3)N2[C@H](COCC2)C)=O)C=C1 VQUWFLFKDYALSX-HNNXBMFYSA-N 0.000 claims description 4
- XRYJHUBBBIIQDO-UHFFFAOYSA-N 7-[(4-methoxyphenyl)methyl]-6-methyl-3-[1-(4-methyl-1,3-thiazol-2-yl)ethyl]imidazo[1,5-a]pyrazin-8-one Chemical compound COC1=CC=C(CN2C(C=3N(C=C2C)C(=NC=3)C(C)C=2SC=C(N=2)C)=O)C=C1 XRYJHUBBBIIQDO-UHFFFAOYSA-N 0.000 claims description 4
- DRHDVSISCJNWFF-UHFFFAOYSA-N 7-[(4-methoxyphenyl)methyl]-6-methyl-3-morpholin-4-ylimidazo[1,5-a]pyrazin-8-one Chemical compound COC1=CC=C(CN2C(C=3N(C=C2C)C(=NC=3)N2CCOCC2)=O)C=C1 DRHDVSISCJNWFF-UHFFFAOYSA-N 0.000 claims description 4
- YMHRUXWRFFVZTC-UHFFFAOYSA-N 7-[(4-methoxyphenyl)methyl]-6-methyl-3-propylimidazo[1,5-a]pyrazin-8-one Chemical compound COC1=CC=C(CN2C(C=3N(C=C2C)C(=NC=3)CCC)=O)C=C1 YMHRUXWRFFVZTC-UHFFFAOYSA-N 0.000 claims description 4
- OVPVMIDVNMKKNU-UHFFFAOYSA-N 7-[(6-methoxypyridin-3-yl)methyl]-6-methyl-3-(oxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound COC1=CC=C(C=N1)CN1C(C=2N(C=C1C)C(=NC=2)C1CCOCC1)=O OVPVMIDVNMKKNU-UHFFFAOYSA-N 0.000 claims description 4
- QPWYRFGVZKZRGE-UHFFFAOYSA-N 7-[[4-(difluoromethoxy)phenyl]methyl]-6-methyl-3-(oxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound FC(OC1=CC=C(CN2C(C=3N(C=C2C)C(=NC=3)C2CCOCC2)=O)C=C1)F QPWYRFGVZKZRGE-UHFFFAOYSA-N 0.000 claims description 4
- LEQLMZLBAGAUDL-UHFFFAOYSA-N 7-benzyl-6-ethyl-3-(oxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound C(C1=CC=CC=C1)N1C(C=2N(C=C1CC)C(=NC=2)C1CCOCC1)=O LEQLMZLBAGAUDL-UHFFFAOYSA-N 0.000 claims description 4
- JBXKPHLWRUHDIB-UHFFFAOYSA-N 7-benzyl-6-methyl-3-(oxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound C(C1=CC=CC=C1)N1C(C=2N(C=C1C)C(=NC=2)C1CCOCC1)=O JBXKPHLWRUHDIB-UHFFFAOYSA-N 0.000 claims description 4
- DKRDXFRQADAIHU-UHFFFAOYSA-N 3-(2-fluoropropan-2-yl)-7-[(4-methoxyphenyl)methyl]-6-methylimidazo[1,5-a]pyrazin-8-one Chemical compound FC(C)(C)C1=NC=C2N1C=C(N(C2=O)CC1=CC=C(C=C1)OC)C DKRDXFRQADAIHU-UHFFFAOYSA-N 0.000 claims description 3
- RQWUWJPTWQLWON-UHFFFAOYSA-N 3-(4-fluorooxan-4-yl)-7-[(3-fluorophenyl)methyl]-6-methylimidazo[1,5-a]pyrazin-8-one Chemical compound FC=1C=C(CN2C(C=3N(C=C2C)C(=NC=3)C2(CCOCC2)F)=O)C=CC=1 RQWUWJPTWQLWON-UHFFFAOYSA-N 0.000 claims description 3
- COANVSPOSPBLGF-UHFFFAOYSA-N 3-(6-chloro-2,3-dihydro-1H-inden-1-yl)-7-[(4-methoxyphenyl)methyl]-6-methylimidazo[1,5-a]pyrazin-8-one Chemical compound ClC1=CC=C2CCC(C2=C1)C1=NC=C2N1C=C(N(C2=O)CC1=CC=C(C=C1)OC)C COANVSPOSPBLGF-UHFFFAOYSA-N 0.000 claims description 3
- HBKUUJGKQVTMIE-UHFFFAOYSA-N 3-[[6-methyl-3-(oxan-4-yl)-8-oxoimidazo[1,5-a]pyrazin-7-yl]methyl]benzonitrile Chemical compound CC=1N(C(C=2N(C=1)C(=NC=2)C1CCOCC1)=O)CC=1C=C(C#N)C=CC=1 HBKUUJGKQVTMIE-UHFFFAOYSA-N 0.000 claims description 3
- TUNXEGCQHJFZRL-UHFFFAOYSA-N 6-(cyclopentylmethyl)-7-[(4-methoxyphenyl)methyl]-3-(oxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound C1(CCCC1)CC=1N(C(C=2N(C=1)C(=NC=2)C1CCOCC1)=O)CC1=CC=C(C=C1)OC TUNXEGCQHJFZRL-UHFFFAOYSA-N 0.000 claims description 3
- ICFZMMURQAUHKS-UHFFFAOYSA-N 6-methyl-7-[(2-methylphenyl)methyl]-3-(oxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound CC=1N(C(C=2N(C=1)C(=NC=2)C1CCOCC1)=O)CC1=C(C=CC=C1)C ICFZMMURQAUHKS-UHFFFAOYSA-N 0.000 claims description 3
- ZABNCYCWYYVHOF-UHFFFAOYSA-N 7-(1,3-benzodioxol-5-ylmethyl)-6-methyl-3-(oxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound O1COC2=C1C=CC(=C2)CN1C(C=2N(C=C1C)C(=NC=2)C1CCOCC1)=O ZABNCYCWYYVHOF-UHFFFAOYSA-N 0.000 claims description 3
- YIBKFYWXALCNMI-UHFFFAOYSA-N 7-(cycloheptylmethyl)-3-cyclopropylimidazo[1,5-a]pyrazin-8-one Chemical compound C1(CCCCCC1)CN1C(C=2N(C=C1)C(=NC=2)C1CC1)=O YIBKFYWXALCNMI-UHFFFAOYSA-N 0.000 claims description 3
- AKDKVOQBJMHADC-UHFFFAOYSA-N 7-[(3-chloro-4-methoxyphenyl)methyl]-6-methyl-3-(oxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound ClC=1C=C(CN2C(C=3N(C=C2C)C(=NC=3)C2CCOCC2)=O)C=CC=1OC AKDKVOQBJMHADC-UHFFFAOYSA-N 0.000 claims description 3
- NSFYZWGFIIQKQP-ABAIWWIYSA-N 7-[(3-fluorophenyl)methyl]-6-methyl-3-[(1S,2R)-2-methylcyclopropyl]imidazo[1,5-a]pyrazin-8-one Chemical compound FC=1C=C(CN2C(C=3N(C=C2C)C(=NC=3)[C@@H]2[C@@H](C2)C)=O)C=CC=1 NSFYZWGFIIQKQP-ABAIWWIYSA-N 0.000 claims description 3
- NSFYZWGFIIQKQP-NHYWBVRUSA-N 7-[(3-fluorophenyl)methyl]-6-methyl-3-[(1S,2S)-2-methylcyclopropyl]imidazo[1,5-a]pyrazin-8-one Chemical compound FC=1C=C(CN2C(C=3N(C=C2C)C(=NC=3)[C@@H]2[C@H](C2)C)=O)C=CC=1 NSFYZWGFIIQKQP-NHYWBVRUSA-N 0.000 claims description 3
- BZFPDTIGFMRZDI-CZUORRHYSA-N 7-[(3-fluorophenyl)methyl]-6-methyl-3-[(2R,3S)-2-methyloxolan-3-yl]imidazo[1,5-a]pyrazin-8-one Chemical compound FC=1C=C(CN2C(C=3N(C=C2C)C(=NC=3)[C@H]2[C@H](OCC2)C)=O)C=CC=1 BZFPDTIGFMRZDI-CZUORRHYSA-N 0.000 claims description 3
- AQLHITNAEZNQJK-IBGZPJMESA-N 7-[(3-fluorophenyl)methyl]-6-methyl-3-[(3R)-3-methyloxolan-3-yl]imidazo[1,5-a]pyrazin-8-one Chemical compound CC1=CN2C(=CN=C2[C@@]2(C)CCOC2)C(=O)N1CC1=CC(F)=CC=C1 AQLHITNAEZNQJK-IBGZPJMESA-N 0.000 claims description 3
- AQLHITNAEZNQJK-LJQANCHMSA-N 7-[(3-fluorophenyl)methyl]-6-methyl-3-[(3S)-3-methyloxolan-3-yl]imidazo[1,5-a]pyrazin-8-one Chemical compound FC=1C=C(CN2C(C=3N(C=C2C)C(=NC=3)[C@]2(COCC2)C)=O)C=CC=1 AQLHITNAEZNQJK-LJQANCHMSA-N 0.000 claims description 3
- MAIAHERKVFXOBQ-UHFFFAOYSA-N 7-[(4-chloro-3-methoxyphenyl)methyl]-6-methyl-3-(oxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound ClC1=C(C=C(CN2C(C=3N(C=C2C)C(=NC=3)C2CCOCC2)=O)C=C1)OC MAIAHERKVFXOBQ-UHFFFAOYSA-N 0.000 claims description 3
- NRIDLTZEDZLZBY-UHFFFAOYSA-N 7-[(4-methoxyphenyl)methyl]-6-methyl-3-(2-methyl-7-oxabicyclo[2.2.1]heptan-2-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound COC1=CC=C(CN2C(C)=CN3C(=CN=C3C3(C)CC4CCC3O4)C2=O)C=C1 NRIDLTZEDZLZBY-UHFFFAOYSA-N 0.000 claims description 3
- XKGOGXSNJQICIE-UHFFFAOYSA-N 7-[(4-methoxyphenyl)methyl]-6-methyl-3-(3-methyl-5-oxopyrrolidin-3-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound COC1=CC=C(CN2C(C=3N(C=C2C)C(=NC=3)C2(CNC(C2)=O)C)=O)C=C1 XKGOGXSNJQICIE-UHFFFAOYSA-N 0.000 claims description 3
- YTOUKKHZMQVQLZ-UHFFFAOYSA-N 7-[(4-methoxyphenyl)methyl]-6-methyl-3-(3-methyloxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound COC1=CC=C(CN2C(C=3N(C=C2C)C(=NC=3)C2C(COCC2)C)=O)C=C1 YTOUKKHZMQVQLZ-UHFFFAOYSA-N 0.000 claims description 3
- ONRHTPIVLPKRAA-UHFFFAOYSA-N 7-[[4-(cyclopropylmethoxy)phenyl]methyl]-6-methyl-3-(oxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound C1(CC1)COC1=CC=C(CN2C(C=3N(C=C2C)C(=NC=3)C2CCOCC2)=O)C=C1 ONRHTPIVLPKRAA-UHFFFAOYSA-N 0.000 claims description 3
- PLDUPXSUYLZYBN-UHFFFAOYSA-N Fluphenazine Chemical compound C1CN(CCO)CCN1CCCN1C2=CC(C(F)(F)F)=CC=C2SC2=CC=CC=C21 PLDUPXSUYLZYBN-UHFFFAOYSA-N 0.000 claims description 3
- RGCVKNLCSQQDEP-UHFFFAOYSA-N Perphenazine Chemical compound C1CN(CCO)CCN1CCCN1C2=CC(Cl)=CC=C2SC2=CC=CC=C21 RGCVKNLCSQQDEP-UHFFFAOYSA-N 0.000 claims description 3
- 229960002690 fluphenazine Drugs 0.000 claims description 3
- VRQVVMDWGGWHTJ-CQSZACIVSA-N methotrimeprazine Chemical compound C1=CC=C2N(C[C@H](C)CN(C)C)C3=CC(OC)=CC=C3SC2=C1 VRQVVMDWGGWHTJ-CQSZACIVSA-N 0.000 claims description 3
- 229940042053 methotrimeprazine Drugs 0.000 claims description 3
- 229960000762 perphenazine Drugs 0.000 claims description 3
- BZFPDTIGFMRZDI-CJNGLKHVSA-N 7-[(3-fluorophenyl)methyl]-6-methyl-3-[(2R,3R)-2-methyloxolan-3-yl]imidazo[1,5-a]pyrazin-8-one Chemical compound FC=1C=C(CN2C(C=3N(C=C2C)C(=NC=3)[C@@H]2[C@H](OCC2)C)=O)C=CC=1 BZFPDTIGFMRZDI-CJNGLKHVSA-N 0.000 claims description 2
- MVVQNOPLTGEXNR-KGLIPLIRSA-N 3-[(1S,2S)-2-fluorocyclopropyl]-7-[(3-fluorophenyl)methyl]-6-methylimidazo[1,5-a]pyrazin-8-one Chemical compound FC=1C=C(CN2C(C=3N(C=C2C)C(=NC=3)[C@H]2[C@H](C2)F)=O)C=CC=1 MVVQNOPLTGEXNR-KGLIPLIRSA-N 0.000 claims 2
- UNRQWEFTRALILW-UHFFFAOYSA-N 6-methyl-3-(oxan-4-yl)-7-[[4-(trifluoromethoxy)phenyl]methyl]imidazo[1,5-a]pyrazin-8-one Chemical compound CC=1N(C(C=2N(C=1)C(=NC=2)C1CCOCC1)=O)CC1=CC=C(C=C1)OC(F)(F)F UNRQWEFTRALILW-UHFFFAOYSA-N 0.000 claims 2
- ARICWMJJQKCTDR-UHFFFAOYSA-N 6-methyl-3-(oxan-4-yl)-7-propylimidazo[1,5-a]pyrazin-8-one Chemical compound CC=1N(C(C=2N(C=1)C(=NC=2)C1CCOCC1)=O)CCC ARICWMJJQKCTDR-UHFFFAOYSA-N 0.000 claims 2
- BZFPDTIGFMRZDI-BBRMVZONSA-N 7-[(3-fluorophenyl)methyl]-6-methyl-3-[(2S,3R)-2-methyloxolan-3-yl]imidazo[1,5-a]pyrazin-8-one Chemical compound FC=1C=C(CN2C(C=3N(C=C2C)C(=NC=3)[C@@H]2[C@@H](OCC2)C)=O)C=CC=1 BZFPDTIGFMRZDI-BBRMVZONSA-N 0.000 claims 2
- BZFPDTIGFMRZDI-XJKSGUPXSA-N 7-[(3-fluorophenyl)methyl]-6-methyl-3-[(2S,3S)-2-methyloxolan-3-yl]imidazo[1,5-a]pyrazin-8-one Chemical compound FC=1C=C(CN2C(C=3N(C=C2C)C(=NC=3)[C@H]2[C@@H](OCC2)C)=O)C=CC=1 BZFPDTIGFMRZDI-XJKSGUPXSA-N 0.000 claims 2
- PZHZVFDPQAUVKV-PBHICJAKSA-N 7-[(4-methoxyphenyl)methyl]-6-methyl-3-[(2R,3R)-2-methyloxolan-3-yl]imidazo[1,5-a]pyrazin-8-one Chemical compound COC1=CC=C(CN2C(C=3N(C=C2C)C(=NC=3)[C@@H]2[C@H](OCC2)C)=O)C=C1 PZHZVFDPQAUVKV-PBHICJAKSA-N 0.000 claims 2
- PZHZVFDPQAUVKV-RHSMWYFYSA-N 7-[(4-methoxyphenyl)methyl]-6-methyl-3-[(2R,3S)-2-methyloxolan-3-yl]imidazo[1,5-a]pyrazin-8-one Chemical compound COC1=CC=C(CN2C(C=3N(C=C2C)C(=NC=3)[C@H]2[C@H](OCC2)C)=O)C=C1 PZHZVFDPQAUVKV-RHSMWYFYSA-N 0.000 claims 2
- PZHZVFDPQAUVKV-YOEHRIQHSA-N 7-[(4-methoxyphenyl)methyl]-6-methyl-3-[(2S,3R)-2-methyloxolan-3-yl]imidazo[1,5-a]pyrazin-8-one Chemical compound COC1=CC=C(CN2C(C=3N(C=C2C)C(=NC=3)[C@@H]2[C@@H](OCC2)C)=O)C=C1 PZHZVFDPQAUVKV-YOEHRIQHSA-N 0.000 claims 2
- PZHZVFDPQAUVKV-WMLDXEAASA-N 7-[(4-methoxyphenyl)methyl]-6-methyl-3-[(2S,3S)-2-methyloxolan-3-yl]imidazo[1,5-a]pyrazin-8-one Chemical compound COC1=CC=C(CN2C(C=3N(C=C2C)C(=NC=3)[C@H]2[C@@H](OCC2)C)=O)C=C1 PZHZVFDPQAUVKV-WMLDXEAASA-N 0.000 claims 2
- WESIRYTURBTMCP-UHFFFAOYSA-N 7-ethyl-6-methyl-3-(oxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound C(C)N1C(C=2N(C=C1C)C(=NC=2)C1CCOCC1)=O WESIRYTURBTMCP-UHFFFAOYSA-N 0.000 claims 2
- PRGLMAVTWQEKMH-UHFFFAOYSA-N 1-(oxan-4-yl)-7H-imidazo[1,5-a]pyrazin-8-one Chemical compound O1CCC(CC1)C=1N=CN2C1C(NC=C2)=O PRGLMAVTWQEKMH-UHFFFAOYSA-N 0.000 claims 1
- ZMLHBBXPXZXTSP-UHFFFAOYSA-N 2-fluoropropane Chemical group C[C](C)F ZMLHBBXPXZXTSP-UHFFFAOYSA-N 0.000 claims 1
- 125000004428 fluoroalkoxy group Chemical group 0.000 abstract description 3
- 239000000203 mixture Substances 0.000 description 410
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 405
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 318
- 239000000243 solution Substances 0.000 description 214
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 205
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 197
- 238000000034 method Methods 0.000 description 196
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 180
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 122
- 238000005160 1H NMR spectroscopy Methods 0.000 description 121
- 239000007832 Na2SO4 Substances 0.000 description 116
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 116
- 229910052938 sodium sulfate Inorganic materials 0.000 description 116
- 235000011152 sodium sulphate Nutrition 0.000 description 116
- 235000019439 ethyl acetate Nutrition 0.000 description 113
- 239000012044 organic layer Substances 0.000 description 109
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 108
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 105
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 95
- 239000012267 brine Substances 0.000 description 88
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 88
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 76
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 67
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 67
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 64
- 239000003208 petroleum Substances 0.000 description 59
- 239000000706 filtrate Substances 0.000 description 57
- 229910000027 potassium carbonate Inorganic materials 0.000 description 54
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 51
- 229910000024 caesium carbonate Inorganic materials 0.000 description 51
- 238000006243 chemical reaction Methods 0.000 description 49
- 239000002904 solvent Substances 0.000 description 45
- 239000011541 reaction mixture Substances 0.000 description 43
- 235000015320 potassium carbonate Nutrition 0.000 description 42
- 229920006395 saturated elastomer Polymers 0.000 description 41
- 238000003818 flash chromatography Methods 0.000 description 39
- 235000017557 sodium bicarbonate Nutrition 0.000 description 38
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 38
- AXHZNUDHLGMSHQ-UHFFFAOYSA-N 6-methyl-3-(oxan-4-yl)-7H-imidazo[1,5-a]pyrazin-8-one Chemical compound CC=1NC(C=2N(C=1)C(=NC=2)C1CCOCC1)=O AXHZNUDHLGMSHQ-UHFFFAOYSA-N 0.000 description 36
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 36
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 34
- 238000010898 silica gel chromatography Methods 0.000 description 31
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 28
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 25
- 101001117089 Drosophila melanogaster Calcium/calmodulin-dependent 3',5'-cyclic nucleotide phosphodiesterase 1 Proteins 0.000 description 23
- 239000000741 silica gel Substances 0.000 description 23
- 229910002027 silica gel Inorganic materials 0.000 description 23
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 22
- 229940121836 Phosphodiesterase 1 inhibitor Drugs 0.000 description 22
- 239000012043 crude product Substances 0.000 description 22
- SCBZBMXPJYMXRC-UHFFFAOYSA-N 1-(bromomethyl)-3-fluorobenzene Chemical compound FC1=CC=CC(CBr)=C1 SCBZBMXPJYMXRC-UHFFFAOYSA-N 0.000 description 21
- 238000010828 elution Methods 0.000 description 21
- 238000012746 preparative thin layer chromatography Methods 0.000 description 21
- 239000007821 HATU Substances 0.000 description 20
- FVAUCKIRQBBSSJ-UHFFFAOYSA-M sodium iodide Chemical compound [Na+].[I-] FVAUCKIRQBBSSJ-UHFFFAOYSA-M 0.000 description 19
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 18
- PQLVXDKIJBQVDF-UHFFFAOYSA-N acetic acid;hydrate Chemical compound O.CC(O)=O PQLVXDKIJBQVDF-UHFFFAOYSA-N 0.000 description 18
- 239000012074 organic phase Substances 0.000 description 18
- JRGLQRNVKCSURZ-UHFFFAOYSA-N COC=1C(=NC=C(N=1)C)CN Chemical compound COC=1C(=NC=C(N=1)C)CN JRGLQRNVKCSURZ-UHFFFAOYSA-N 0.000 description 16
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 16
- MOHYOXXOKFQHDC-UHFFFAOYSA-N 1-(chloromethyl)-4-methoxybenzene Chemical compound COC1=CC=C(CCl)C=C1 MOHYOXXOKFQHDC-UHFFFAOYSA-N 0.000 description 15
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 15
- 239000002585 base Substances 0.000 description 14
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 14
- IVOMOUWHDPKRLL-KQYNXXCUSA-N Cyclic adenosine monophosphate Chemical compound C([C@H]1O2)OP(O)(=O)O[C@H]1[C@@H](O)[C@@H]2N1C(N=CN=C2N)=C2N=C1 IVOMOUWHDPKRLL-KQYNXXCUSA-N 0.000 description 13
- 230000000694 effects Effects 0.000 description 13
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 12
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 12
- KYQCOXFCLRTKLS-UHFFFAOYSA-N Pyrazine Chemical compound C1=CN=CC=N1 KYQCOXFCLRTKLS-UHFFFAOYSA-N 0.000 description 12
- 239000002253 acid Substances 0.000 description 12
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 12
- DGOYLVBDCVINQZ-UHFFFAOYSA-N oxane-4-carboxamide Chemical compound NC(=O)C1CCOCC1 DGOYLVBDCVINQZ-UHFFFAOYSA-N 0.000 description 12
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 12
- 235000011181 potassium carbonates Nutrition 0.000 description 12
- QZAYGJVTTNCVMB-UHFFFAOYSA-N serotonin Chemical compound C1=C(O)C=C2C(CCN)=CNC2=C1 QZAYGJVTTNCVMB-UHFFFAOYSA-N 0.000 description 12
- RESKUCDLACBKSD-UHFFFAOYSA-N (3-methoxy-5-methylpyrazin-2-yl)methanamine hydrochloride Chemical compound Cl.COc1nc(C)cnc1CN RESKUCDLACBKSD-UHFFFAOYSA-N 0.000 description 11
- 229910019213 POCl3 Inorganic materials 0.000 description 11
- 239000000164 antipsychotic agent Substances 0.000 description 11
- 102000004190 Enzymes Human genes 0.000 description 10
- 108090000790 Enzymes Proteins 0.000 description 10
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 10
- ZOOGRGPOEVQQDX-KHLHZJAASA-N cyclic guanosine monophosphate Chemical compound C([C@H]1O2)O[P@](O)(=O)O[C@@H]1[C@H](O)[C@H]2N1C(N=C(NC2=O)N)=C2N=C1 ZOOGRGPOEVQQDX-KHLHZJAASA-N 0.000 description 10
- VYFYYTLLBUKUHU-UHFFFAOYSA-N dopamine Chemical compound NCCC1=CC=C(O)C(O)=C1 VYFYYTLLBUKUHU-UHFFFAOYSA-N 0.000 description 10
- 239000003112 inhibitor Substances 0.000 description 10
- 239000004031 partial agonist Substances 0.000 description 10
- 238000000746 purification Methods 0.000 description 10
- 239000000758 substrate Substances 0.000 description 10
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 9
- 102100024316 Calcium/calmodulin-dependent 3',5'-cyclic nucleotide phosphodiesterase 1A Human genes 0.000 description 9
- 101100407341 Drosophila melanogaster Pde9 gene Proteins 0.000 description 9
- 101001117044 Homo sapiens Calcium/calmodulin-dependent 3',5'-cyclic nucleotide phosphodiesterase 1A Proteins 0.000 description 9
- 238000003556 assay Methods 0.000 description 9
- 230000005764 inhibitory process Effects 0.000 description 9
- 102000005962 receptors Human genes 0.000 description 9
- 108020003175 receptors Proteins 0.000 description 9
- 239000007787 solid Substances 0.000 description 9
- 239000000725 suspension Substances 0.000 description 9
- OZAIFHULBGXAKX-UHFFFAOYSA-N 2-(2-cyanopropan-2-yldiazenyl)-2-methylpropanenitrile Chemical compound N#CC(C)(C)N=NC(C)(C)C#N OZAIFHULBGXAKX-UHFFFAOYSA-N 0.000 description 8
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 8
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 8
- 102100024318 Calcium/calmodulin-dependent 3',5'-cyclic nucleotide phosphodiesterase 1B Human genes 0.000 description 8
- 101001117099 Homo sapiens Calcium/calmodulin-dependent 3',5'-cyclic nucleotide phosphodiesterase 1B Proteins 0.000 description 8
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 8
- 125000004122 cyclic group Chemical group 0.000 description 8
- AIMMVWOEOZMVMS-UHFFFAOYSA-N cyclopropanecarboxamide Chemical compound NC(=O)C1CC1 AIMMVWOEOZMVMS-UHFFFAOYSA-N 0.000 description 8
- 201000010099 disease Diseases 0.000 description 8
- 239000003814 drug Substances 0.000 description 8
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 8
- 238000002953 preparative HPLC Methods 0.000 description 8
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 8
- AUSUBZBGYDCVNO-UHFFFAOYSA-N 3-bromo-7-[(4-methoxyphenyl)methyl]-6-methylimidazo[1,5-a]pyrazin-8-one Chemical compound BrC1=NC=C2N1C=C(N(C2=O)CC1=CC=C(C=C1)OC)C AUSUBZBGYDCVNO-UHFFFAOYSA-N 0.000 description 7
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 7
- 0 CC*(C)C*C(N1C*)=C(*)[n]2c(*(C)=N)ncc2C1=O Chemical compound CC*(C)C*C(N1C*)=C(*)[n]2c(*(C)=N)ncc2C1=O 0.000 description 7
- KZBUYRJDOAKODT-UHFFFAOYSA-N Chlorine Chemical compound ClCl KZBUYRJDOAKODT-UHFFFAOYSA-N 0.000 description 7
- 102000004980 Dopamine D2 Receptors Human genes 0.000 description 7
- 108090001111 Dopamine D2 Receptors Proteins 0.000 description 7
- 235000019270 ammonium chloride Nutrition 0.000 description 7
- 239000003480 eluent Substances 0.000 description 7
- 239000010410 layer Substances 0.000 description 7
- 108090000623 proteins and genes Proteins 0.000 description 7
- 230000001105 regulatory effect Effects 0.000 description 7
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 6
- ORVNVWKHNZQICB-UHFFFAOYSA-N 3-(oxan-4-yl)-7H-imidazo[1,5-a]pyrazin-8-one Chemical compound O1CCC(CC1)C1=NC=C2N1C=CNC2=O ORVNVWKHNZQICB-UHFFFAOYSA-N 0.000 description 6
- PJIFBWREHWJHNQ-UHFFFAOYSA-N 3-bromo-6-methyl-7H-imidazo[1,5-a]pyrazin-8-one Chemical compound BrC1=NC=C2N1C=C(NC2=O)C PJIFBWREHWJHNQ-UHFFFAOYSA-N 0.000 description 6
- IWOSCYMUFAVWGY-UHFFFAOYSA-N 3-cyclopropyl-7H-imidazo[1,5-a]pyrazin-8-one Chemical compound C1(CC1)C1=NC=C2N1C=CNC2=O IWOSCYMUFAVWGY-UHFFFAOYSA-N 0.000 description 6
- 102100024317 Calcium/calmodulin-dependent 3',5'-cyclic nucleotide phosphodiesterase 1C Human genes 0.000 description 6
- 101001117094 Homo sapiens Calcium/calmodulin-dependent 3',5'-cyclic nucleotide phosphodiesterase 1C Proteins 0.000 description 6
- 229940076380 PDE9 inhibitor Drugs 0.000 description 6
- PCNDJXKNXGMECE-UHFFFAOYSA-N Phenazine Natural products C1=CC=CC2=NC3=CC=CC=C3N=C21 PCNDJXKNXGMECE-UHFFFAOYSA-N 0.000 description 6
- 108010029485 Protein Isoforms Proteins 0.000 description 6
- 102000001708 Protein Isoforms Human genes 0.000 description 6
- 230000002152 alkylating effect Effects 0.000 description 6
- 239000005557 antagonist Substances 0.000 description 6
- 229940005529 antipsychotics Drugs 0.000 description 6
- CSJLBAMHHLJAAS-UHFFFAOYSA-N diethylaminosulfur trifluoride Chemical compound CCN(CC)S(F)(F)F CSJLBAMHHLJAAS-UHFFFAOYSA-N 0.000 description 6
- 208000035475 disorder Diseases 0.000 description 6
- 239000007788 liquid Substances 0.000 description 6
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 6
- 230000003389 potentiating effect Effects 0.000 description 6
- 229940076279 serotonin Drugs 0.000 description 6
- 235000009518 sodium iodide Nutrition 0.000 description 6
- 208000024891 symptom Diseases 0.000 description 6
- 239000003643 water by type Substances 0.000 description 6
- WGULEKQJDCKGDV-UHFFFAOYSA-N 3-bromo-7-[(3-fluorophenyl)methyl]-6-methylimidazo[1,5-a]pyrazin-8-one Chemical compound BrC1=NC=C2N1C=C(N(C2=O)CC1=CC(=CC=C1)F)C WGULEKQJDCKGDV-UHFFFAOYSA-N 0.000 description 5
- RYWGIFDCWVSLEA-UHFFFAOYSA-N 6-benzyl-3-(oxan-4-yl)-7H-imidazo[1,5-a]pyrazin-8-one Chemical compound C(C1=CC=CC=C1)C=1NC(C=2N(C=1)C(=NC=2)C1CCOCC1)=O RYWGIFDCWVSLEA-UHFFFAOYSA-N 0.000 description 5
- 239000004480 active ingredient Substances 0.000 description 5
- 239000007864 aqueous solution Substances 0.000 description 5
- UUWSLBWDFJMSFP-UHFFFAOYSA-N bromomethylcyclohexane Chemical compound BrCC1CCCCC1 UUWSLBWDFJMSFP-UHFFFAOYSA-N 0.000 description 5
- 239000003153 chemical reaction reagent Substances 0.000 description 5
- 238000004090 dissolution Methods 0.000 description 5
- 229960003638 dopamine Drugs 0.000 description 5
- 239000012065 filter cake Substances 0.000 description 5
- 238000009472 formulation Methods 0.000 description 5
- 239000012458 free base Substances 0.000 description 5
- 230000007246 mechanism Effects 0.000 description 5
- DVLGIQNHKLWSRU-UHFFFAOYSA-N methyl 1h-imidazole-5-carboxylate Chemical compound COC(=O)C1=CN=CN1 DVLGIQNHKLWSRU-UHFFFAOYSA-N 0.000 description 5
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 5
- 239000003826 tablet Substances 0.000 description 5
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 5
- 230000001225 therapeutic effect Effects 0.000 description 5
- VNDYJBBGRKZCSX-UHFFFAOYSA-L zinc bromide Chemical compound Br[Zn]Br VNDYJBBGRKZCSX-UHFFFAOYSA-L 0.000 description 5
- RHKWGVWUXBFIIE-UHFFFAOYSA-N (3-chloropyrazin-2-yl)methanamine;dihydrochloride Chemical compound Cl.Cl.NCC1=NC=CN=C1Cl RHKWGVWUXBFIIE-UHFFFAOYSA-N 0.000 description 4
- MYDVJLOKNIAHPH-UHFFFAOYSA-N 2-Methoxy-6-methylpyrazine Chemical compound COC1=CN=CC(C)=N1 MYDVJLOKNIAHPH-UHFFFAOYSA-N 0.000 description 4
- CKUVSPQGYLELRG-UHFFFAOYSA-N 2-chloro-6-methylpyrazine Chemical compound CC1=CN=CC(Cl)=N1 CKUVSPQGYLELRG-UHFFFAOYSA-N 0.000 description 4
- ZPIMLXBEIFATHA-UHFFFAOYSA-N 3-propyl-7H-imidazo[1,5-a]pyrazin-8-one Chemical compound C(CC)C1=NC=C2N1C=CNC2=O ZPIMLXBEIFATHA-UHFFFAOYSA-N 0.000 description 4
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 4
- 102000049773 5-HT2A Serotonin Receptor Human genes 0.000 description 4
- 108010072564 5-HT2A Serotonin Receptor Proteins 0.000 description 4
- UMIDNFPTOYTFSZ-UHFFFAOYSA-N 6-ethyl-3-(oxan-4-yl)-7H-imidazo[1,5-a]pyrazin-8-one Chemical compound C(C)C=1NC(C=2N(C=1)C(=NC=2)C1CCOCC1)=O UMIDNFPTOYTFSZ-UHFFFAOYSA-N 0.000 description 4
- KCFHFZJJLZXCMO-UHFFFAOYSA-N 6-methyl-3-(2-methyloxolan-3-yl)-7H-imidazo[1,5-a]pyrazin-8-one Chemical compound CC=1NC(C=2N(C=1)C(=NC=2)C1C(OCC1)C)=O KCFHFZJJLZXCMO-UHFFFAOYSA-N 0.000 description 4
- YEUHAUQCBIXFGJ-UHFFFAOYSA-N COC=1C(=NC=C(N=1)C)C#N Chemical compound COC=1C(=NC=C(N=1)C)C#N YEUHAUQCBIXFGJ-UHFFFAOYSA-N 0.000 description 4
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 4
- 102000004861 Phosphoric Diester Hydrolases Human genes 0.000 description 4
- 108090001050 Phosphoric Diester Hydrolases Proteins 0.000 description 4
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 4
- 239000002671 adjuvant Substances 0.000 description 4
- 239000000556 agonist Substances 0.000 description 4
- 150000001347 alkyl bromides Chemical class 0.000 description 4
- 239000003054 catalyst Substances 0.000 description 4
- 238000006880 cross-coupling reaction Methods 0.000 description 4
- 239000002552 dosage form Substances 0.000 description 4
- 239000000839 emulsion Substances 0.000 description 4
- 235000019253 formic acid Nutrition 0.000 description 4
- 239000000543 intermediate Substances 0.000 description 4
- WFKAJVHLWXSISD-UHFFFAOYSA-N isobutyramide Chemical compound CC(C)C(N)=O WFKAJVHLWXSISD-UHFFFAOYSA-N 0.000 description 4
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 4
- 238000004519 manufacturing process Methods 0.000 description 4
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 4
- 150000007522 mineralic acids Chemical class 0.000 description 4
- 150000007524 organic acids Chemical class 0.000 description 4
- 230000000144 pharmacologic effect Effects 0.000 description 4
- 239000000843 powder Substances 0.000 description 4
- 102000004169 proteins and genes Human genes 0.000 description 4
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 4
- 238000001946 ultra-performance liquid chromatography-mass spectrometry Methods 0.000 description 4
- GIGRWGTZFONRKA-UHFFFAOYSA-N 1-(bromomethyl)-4-methoxybenzene Chemical compound COC1=CC=C(CBr)C=C1 GIGRWGTZFONRKA-UHFFFAOYSA-N 0.000 description 3
- 239000001154 2-(methoxymethyl)pyrazine Substances 0.000 description 3
- OZZYGGOIUXDEKE-UHFFFAOYSA-N 3-(3,6-dihydro-2H-pyran-4-yl)-6-methyl-7H-imidazo[1,5-a]pyrazin-8-one Chemical compound O1CCC(=CC1)C1=NC=C2N1C=C(NC2=O)C OZZYGGOIUXDEKE-UHFFFAOYSA-N 0.000 description 3
- JPKZYYICYSAJPM-UHFFFAOYSA-N 3-methoxy-5-methyl-1-oxidopyrazin-1-ium Chemical compound COC1=C[N+]([O-])=CC(C)=N1 JPKZYYICYSAJPM-UHFFFAOYSA-N 0.000 description 3
- TVRXNBCJGVOPIZ-UHFFFAOYSA-N 6-benzyl-8-methoxy-3-(oxan-4-yl)imidazo[1,5-a]pyrazine Chemical compound C(C1=CC=CC=C1)C=1N=C(C=2N(C=1)C(=NC=2)C1CCOCC1)OC TVRXNBCJGVOPIZ-UHFFFAOYSA-N 0.000 description 3
- FPAOMKMZXJCXEY-UHFFFAOYSA-N 6-bromo-3-(oxan-4-yl)-7H-imidazo[1,5-a]pyrazin-8-one Chemical compound BrC=1NC(C=2N(C=1)C(=NC=2)C1CCOCC1)=O FPAOMKMZXJCXEY-UHFFFAOYSA-N 0.000 description 3
- MZEFRXRFXZNOAY-UHFFFAOYSA-N 6-bromo-8-methoxy-3-(oxan-4-yl)imidazo[1,5-a]pyrazine Chemical compound BrC=1N=C(C=2N(C=1)C(=NC=2)C1CCOCC1)OC MZEFRXRFXZNOAY-UHFFFAOYSA-N 0.000 description 3
- NPTZDJRTSBUWBB-UHFFFAOYSA-N 6-iodo-8-methoxy-3-(oxan-4-yl)imidazo[1,5-a]pyrazine Chemical compound IC=1N=C(C=2N(C=1)C(=NC=2)C1CCOCC1)OC NPTZDJRTSBUWBB-UHFFFAOYSA-N 0.000 description 3
- NSFYZWGFIIQKQP-UHFFFAOYSA-N 7-[(3-fluorophenyl)methyl]-6-methyl-3-(2-methylcyclopropyl)imidazo[1,5-a]pyrazin-8-one Chemical compound FC=1C=C(CN2C(C=3N(C=C2C)C(=NC=3)C2C(C2)C)=O)C=CC=1 NSFYZWGFIIQKQP-UHFFFAOYSA-N 0.000 description 3
- BZFPDTIGFMRZDI-UHFFFAOYSA-N 7-[(3-fluorophenyl)methyl]-6-methyl-3-(2-methyloxolan-3-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound FC=1C=C(CN2C(C=3N(C=C2C)C(=NC=3)C2C(OCC2)C)=O)C=CC=1 BZFPDTIGFMRZDI-UHFFFAOYSA-N 0.000 description 3
- LEYPTBGWIKQYRW-UHFFFAOYSA-N 7-[(3-fluorophenyl)methyl]-6-methyl-3-(oxolan-3-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound FC=1C=C(CN2C(C=3N(C=C2C)C(=NC=3)C2COCC2)=O)C=CC=1 LEYPTBGWIKQYRW-UHFFFAOYSA-N 0.000 description 3
- PZHZVFDPQAUVKV-UHFFFAOYSA-N 7-[(4-methoxyphenyl)methyl]-6-methyl-3-(2-methyloxolan-3-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound COC1=CC=C(CN2C(C=3N(C=C2C)C(=NC=3)C2C(OCC2)C)=O)C=C1 PZHZVFDPQAUVKV-UHFFFAOYSA-N 0.000 description 3
- AHYQSELDEJVKFZ-UHFFFAOYSA-N 8-chloro-3-(oxan-4-yl)imidazo[1,5-a]pyrazine Chemical compound N=1C=C2C(Cl)=NC=CN2C=1C1CCOCC1 AHYQSELDEJVKFZ-UHFFFAOYSA-N 0.000 description 3
- VZIBDHWIHDLNQA-UHFFFAOYSA-N 8-chloro-3-cyclopropylimidazo[1,5-a]pyrazine Chemical compound ClC=1C=2N(C=CN=1)C(=NC=2)C1CC1 VZIBDHWIHDLNQA-UHFFFAOYSA-N 0.000 description 3
- WDVWYJIDRSPHOW-UHFFFAOYSA-N 8-methoxy-6-methyl-3-(2-methyloxolan-3-yl)imidazo[1,5-a]pyrazine Chemical compound COC=1C=2N(C=C(N=1)C)C(=NC=2)C1C(OCC1)C WDVWYJIDRSPHOW-UHFFFAOYSA-N 0.000 description 3
- 102000003678 AMPA Receptors Human genes 0.000 description 3
- 108090000078 AMPA Receptors Proteins 0.000 description 3
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 3
- 108091006146 Channels Proteins 0.000 description 3
- 101150065749 Churc1 gene Proteins 0.000 description 3
- 102000004073 Dopamine D3 Receptors Human genes 0.000 description 3
- 108090000525 Dopamine D3 Receptors Proteins 0.000 description 3
- 102000010726 Glycine Plasma Membrane Transport Proteins Human genes 0.000 description 3
- 108010063380 Glycine Plasma Membrane Transport Proteins Proteins 0.000 description 3
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 3
- 108090001041 N-Methyl-D-Aspartate Receptors Proteins 0.000 description 3
- 102000004868 N-Methyl-D-Aspartate Receptors Human genes 0.000 description 3
- 102000019315 Nicotinic acetylcholine receptors Human genes 0.000 description 3
- 108050006807 Nicotinic acetylcholine receptors Proteins 0.000 description 3
- BPQQTUXANYXVAA-UHFFFAOYSA-N Orthosilicate Chemical compound [O-][Si]([O-])([O-])[O-] BPQQTUXANYXVAA-UHFFFAOYSA-N 0.000 description 3
- 229920001213 Polysorbate 20 Polymers 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- NPXOKRUENSOPAO-UHFFFAOYSA-N Raney nickel Chemical compound [Al].[Ni] NPXOKRUENSOPAO-UHFFFAOYSA-N 0.000 description 3
- 230000008485 antagonism Effects 0.000 description 3
- 230000000561 anti-psychotic effect Effects 0.000 description 3
- 239000008346 aqueous phase Substances 0.000 description 3
- 239000012298 atmosphere Substances 0.000 description 3
- JFDZBHWFFUWGJE-UHFFFAOYSA-N benzonitrile Chemical compound N#CC1=CC=CC=C1 JFDZBHWFFUWGJE-UHFFFAOYSA-N 0.000 description 3
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 3
- 230000037396 body weight Effects 0.000 description 3
- XYZUWOHEILWUID-UHFFFAOYSA-N bromomethylcyclopentane Chemical compound BrCC1CCCC1 XYZUWOHEILWUID-UHFFFAOYSA-N 0.000 description 3
- DNSISZSEWVHGLH-UHFFFAOYSA-N butanamide Chemical compound CCCC(N)=O DNSISZSEWVHGLH-UHFFFAOYSA-N 0.000 description 3
- 210000004027 cell Anatomy 0.000 description 3
- RVEJWGYZBXCGGM-DNVCBOLYSA-N chembl2179094 Chemical compound C([C@H]([C@@H](C1)C=2NC(=O)C=3C=NN(C=3N=2)C2CCOCC2)C)N1CC1=CC=CC=C1 RVEJWGYZBXCGGM-DNVCBOLYSA-N 0.000 description 3
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 3
- 230000001419 dependent effect Effects 0.000 description 3
- 239000008187 granular material Substances 0.000 description 3
- 230000007062 hydrolysis Effects 0.000 description 3
- 238000006460 hydrolysis reaction Methods 0.000 description 3
- 238000007918 intramuscular administration Methods 0.000 description 3
- 229940125425 inverse agonist Drugs 0.000 description 3
- 239000007937 lozenge Substances 0.000 description 3
- HKCODDLDQJWJIH-UHFFFAOYSA-N methyl 2-bromo-3-(2-oxopropyl)imidazole-4-carboxylate Chemical compound BrC=1N(C(=CN=1)C(=O)OC)CC(C)=O HKCODDLDQJWJIH-UHFFFAOYSA-N 0.000 description 3
- FIDBZJWCHLENOB-UHFFFAOYSA-N methyl 3-(2-oxopropyl)imidazole-4-carboxylate Chemical compound COC(=O)C1=CN=CN1CC(C)=O FIDBZJWCHLENOB-UHFFFAOYSA-N 0.000 description 3
- 238000002156 mixing Methods 0.000 description 3
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 3
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 3
- 238000002360 preparation method Methods 0.000 description 3
- 239000006188 syrup Substances 0.000 description 3
- 235000020357 syrup Nutrition 0.000 description 3
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 3
- 238000004809 thin layer chromatography Methods 0.000 description 3
- 229910052727 yttrium Inorganic materials 0.000 description 3
- VWQVUPCCIRVNHF-UHFFFAOYSA-N yttrium atom Chemical compound [Y] VWQVUPCCIRVNHF-UHFFFAOYSA-N 0.000 description 3
- PODCTQRYFHFTPT-UHFFFAOYSA-N (3-chloropyrazin-2-yl)methanamine Chemical compound NCC1=NC=CN=C1Cl PODCTQRYFHFTPT-UHFFFAOYSA-N 0.000 description 2
- IYBLKXVJNKRRNW-UHFFFAOYSA-N (4-cyclopropyloxyphenyl)methanol Chemical compound C1=CC(CO)=CC=C1OC1CC1 IYBLKXVJNKRRNW-UHFFFAOYSA-N 0.000 description 2
- RNHDAKUGFHSZEV-UHFFFAOYSA-N 1,4-dioxane;hydrate Chemical compound O.C1COCCO1 RNHDAKUGFHSZEV-UHFFFAOYSA-N 0.000 description 2
- GTDXPJJHRWOFDI-UHFFFAOYSA-N 1-methylcyclopropane-1-carbonyl chloride Chemical compound ClC(=O)C1(C)CC1 GTDXPJJHRWOFDI-UHFFFAOYSA-N 0.000 description 2
- DOSGEBYQRMBTGS-UHFFFAOYSA-N 2-(3,6-dihydro-2h-pyran-4-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane Chemical compound O1C(C)(C)C(C)(C)OB1C1=CCOCC1 DOSGEBYQRMBTGS-UHFFFAOYSA-N 0.000 description 2
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 2
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 2
- IZHVBANLECCAGF-UHFFFAOYSA-N 2-hydroxy-3-(octadecanoyloxy)propyl octadecanoate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)COC(=O)CCCCCCCCCCCCCCCCC IZHVBANLECCAGF-UHFFFAOYSA-N 0.000 description 2
- BWLBGMIXKSTLSX-UHFFFAOYSA-N 2-hydroxyisobutyric acid Chemical compound CC(C)(O)C(O)=O BWLBGMIXKSTLSX-UHFFFAOYSA-N 0.000 description 2
- WYFBQEOUPVFUFS-UHFFFAOYSA-N 2-methyl-2-phenylmethoxypropanoic acid Chemical compound OC(=O)C(C)(C)OCC1=CC=CC=C1 WYFBQEOUPVFUFS-UHFFFAOYSA-N 0.000 description 2
- YFQWTXAEGYWPSW-UHFFFAOYSA-N 2-methyloxolane-3-carbonyl chloride Chemical compound CC1OCCC1C(Cl)=O YFQWTXAEGYWPSW-UHFFFAOYSA-N 0.000 description 2
- YXVATTQXDIZNEP-UHFFFAOYSA-N 2-methyloxolane-3-carboxylic acid Chemical compound CC1OCCC1C(O)=O YXVATTQXDIZNEP-UHFFFAOYSA-N 0.000 description 2
- NOVDCWFYAOYMMK-UHFFFAOYSA-N 3-(1,4-dimethylpiperidin-4-yl)-6-methyl-7H-imidazo[1,5-a]pyrazin-8-one Chemical compound CN1CCC(CC1)(C)C1=NC=C2N1C=C(N=C2O)C NOVDCWFYAOYMMK-UHFFFAOYSA-N 0.000 description 2
- SFZJZQFECMEBCP-UHFFFAOYSA-N 3-(1-methoxy-2-methylpropan-2-yl)-6-methyl-7H-imidazo[1,5-a]pyrazin-8-one Chemical compound COCC(C)(C)C1=NC=C2N1C=C(NC2=O)C SFZJZQFECMEBCP-UHFFFAOYSA-N 0.000 description 2
- IMQHTOZMMZEIHW-UHFFFAOYSA-N 3-(2,2-difluorocyclopropyl)-6-methyl-7H-imidazo[1,5-a]pyrazin-8-one Chemical compound FC1(C(C1)C1=NC=C2N1C=C(NC2=O)C)F IMQHTOZMMZEIHW-UHFFFAOYSA-N 0.000 description 2
- DOXFKPMTDYJBOX-UHFFFAOYSA-N 3-(2,2-difluorocyclopropyl)-8-methoxy-6-methylimidazo[1,5-a]pyrazine Chemical compound FC1(C(C1)C1=NC=C2N1C=C(N=C2OC)C)F DOXFKPMTDYJBOX-UHFFFAOYSA-N 0.000 description 2
- JCHHDBDXKTWZHQ-UHFFFAOYSA-N 3-(2,6-dimethyloxan-4-yl)-6-methyl-7H-imidazo[1,5-a]pyrazin-8-one Chemical compound CC1OC(CC(C1)C1=NC=C2N1C=C(NC2=O)C)C JCHHDBDXKTWZHQ-UHFFFAOYSA-N 0.000 description 2
- JLSKHRXMBLHDLG-UHFFFAOYSA-N 3-(3,6-dihydro-2H-pyran-4-yl)-6-ethyl-7H-imidazo[1,5-a]pyrazin-8-one Chemical compound O1CCC(=CC1)C1=NC=C2N1C=C(NC2=O)CC JLSKHRXMBLHDLG-UHFFFAOYSA-N 0.000 description 2
- HLBOAQSKBNNHMW-UHFFFAOYSA-N 3-(3-methoxyphenyl)pyridine Chemical compound COC1=CC=CC(C=2C=NC=CC=2)=C1 HLBOAQSKBNNHMW-UHFFFAOYSA-N 0.000 description 2
- NWSFOTRLJCZGGF-UHFFFAOYSA-N 3-(6-chloro-2,3-dihydro-1H-inden-1-yl)-6-methyl-7H-imidazo[1,5-a]pyrazin-8-one Chemical compound ClC1=CC=C2CCC(C2=C1)C1=NC=C2N1C=C(NC2=O)C NWSFOTRLJCZGGF-UHFFFAOYSA-N 0.000 description 2
- MVVQNOPLTGEXNR-UONOGXRCSA-N 3-[(1R,2R)-2-fluorocyclopropyl]-7-[(3-fluorophenyl)methyl]-6-methylimidazo[1,5-a]pyrazin-8-one Chemical compound FC=1C=C(CN2C(C=3N(C=C2C)C(=NC=3)[C@@H]2[C@@H](C2)F)=O)C=CC=1 MVVQNOPLTGEXNR-UONOGXRCSA-N 0.000 description 2
- MVVQNOPLTGEXNR-KBPBESRZSA-N 3-[(1R,2S)-2-fluorocyclopropyl]-7-[(3-fluorophenyl)methyl]-6-methylimidazo[1,5-a]pyrazin-8-one Chemical compound FC=1C=C(CN2C(C=3N(C=C2C)C(=NC=3)[C@@H]2[C@H](C2)F)=O)C=CC=1 MVVQNOPLTGEXNR-KBPBESRZSA-N 0.000 description 2
- DIGKTNLCGCBJHY-RNFRBKRXSA-N 3-[(1S,2R)-2-fluorocyclopropyl]-6-methyl-7H-imidazo[1,5-a]pyrazin-8-one Chemical compound F[C@H]1[C@@H](C1)C1=NC=C2N1C=C(NC2=O)C DIGKTNLCGCBJHY-RNFRBKRXSA-N 0.000 description 2
- CYNCBSDMZPJQIA-HTQZYQBOSA-N 3-[(1S,2R)-2-fluorocyclopropyl]-8-methoxy-6-methylimidazo[1,5-a]pyrazine Chemical compound F[C@H]1[C@@H](C1)C1=NC=C2N1C=C(N=C2OC)C CYNCBSDMZPJQIA-HTQZYQBOSA-N 0.000 description 2
- SRRJDNIRLJMGQE-UHFFFAOYSA-N 3-bromo-6-ethyl-7H-imidazo[1,5-a]pyrazin-8-one Chemical compound BrC1=NC=C2N1C=C(NC2=O)CC SRRJDNIRLJMGQE-UHFFFAOYSA-N 0.000 description 2
- OBRJVMXMXBDDLO-UHFFFAOYSA-N 3-bromo-7-(cyclohexylmethyl)-6-methylimidazo[1,5-a]pyrazin-8-one Chemical compound BrC1=NC=C2N1C=C(N(C2=O)CC1CCCCC1)C OBRJVMXMXBDDLO-UHFFFAOYSA-N 0.000 description 2
- BYBZWOJSENADDM-UHFFFAOYSA-N 3-methyl-5-oxopyrrolidine-3-carbonyl chloride Chemical compound CC1(CNC(=O)C1)C(Cl)=O BYBZWOJSENADDM-UHFFFAOYSA-N 0.000 description 2
- XPAICKDCJOJELF-UHFFFAOYSA-N 3-methyl-5-oxopyrrolidine-3-carboxylic acid Chemical compound OC(=O)C1(C)CNC(=O)C1 XPAICKDCJOJELF-UHFFFAOYSA-N 0.000 description 2
- JHNLZOVBAQWGQU-UHFFFAOYSA-N 380814_sial Chemical compound CS(O)(=O)=O.O=P(=O)OP(=O)=O JHNLZOVBAQWGQU-UHFFFAOYSA-N 0.000 description 2
- OGQRHSRUKMQCJA-UHFFFAOYSA-N 4-(6-methyl-8-oxo-7H-imidazo[1,5-a]pyrazin-3-yl)oxane-4-carbonitrile Chemical compound CC=1NC(C=2N(C=1)C(=NC=2)C1(CCOCC1)C#N)=O OGQRHSRUKMQCJA-UHFFFAOYSA-N 0.000 description 2
- WKGNHRMRQHRBED-UHFFFAOYSA-N 4-(8-methoxy-6-methylimidazo[1,5-a]pyrazin-3-yl)oxane-4-carbonitrile Chemical compound COC=1C=2N(C=C(N=1)C)C(=NC=2)C1(CCOCC1)C#N WKGNHRMRQHRBED-UHFFFAOYSA-N 0.000 description 2
- PVQVSKYXCMWDFN-UHFFFAOYSA-N 4-(cyclopropylmethoxy)benzaldehyde Chemical compound C1=CC(C=O)=CC=C1OCC1CC1 PVQVSKYXCMWDFN-UHFFFAOYSA-N 0.000 description 2
- YQJGBZSDKXLBIA-UHFFFAOYSA-N 4-cyclopropyloxybenzaldehyde Chemical compound C1=CC(C=O)=CC=C1OC1CC1 YQJGBZSDKXLBIA-UHFFFAOYSA-N 0.000 description 2
- RGHHSNMVTDWUBI-UHFFFAOYSA-N 4-hydroxybenzaldehyde Chemical compound OC1=CC=C(C=O)C=C1 RGHHSNMVTDWUBI-UHFFFAOYSA-N 0.000 description 2
- 108091005435 5-HT6 receptors Proteins 0.000 description 2
- VELFKJVIXURHDI-UHFFFAOYSA-N 5-amino-3-methoxypyrazine-2-carbonitrile Chemical compound COC1=NC(N)=CN=C1C#N VELFKJVIXURHDI-UHFFFAOYSA-N 0.000 description 2
- 102100036321 5-hydroxytryptamine receptor 2A Human genes 0.000 description 2
- 101710138091 5-hydroxytryptamine receptor 2A Proteins 0.000 description 2
- XQFADSRGQYCIJX-UHFFFAOYSA-N 6-methyl-3-(1-methylcyclopropyl)-7H-imidazo[1,5-a]pyrazin-8-one Chemical compound CC=1NC(C=2N(C=1)C(=NC=2)C1(CC1)C)=O XQFADSRGQYCIJX-UHFFFAOYSA-N 0.000 description 2
- AUWFJBPHXDDCEI-UHFFFAOYSA-N 6-methyl-3-(2-methylcyclopropyl)-7H-imidazo[1,5-a]pyrazin-8-one Chemical compound CC=1NC(C=2N(C=1)C(=NC=2)C1C(C1)C)=O AUWFJBPHXDDCEI-UHFFFAOYSA-N 0.000 description 2
- PQCFANKWAJLVQJ-UHFFFAOYSA-N 6-methyl-3-(2-phenylmethoxypropan-2-yl)-7H-imidazo[1,5-a]pyrazin-8-one Chemical compound C(C1=CC=CC=C1)OC(C)(C)C1=NC=C2N1C=C(NC2=O)C PQCFANKWAJLVQJ-UHFFFAOYSA-N 0.000 description 2
- MFPMPDJKQXTYET-UHFFFAOYSA-N 6-methyl-3-(3-methyl-5-oxopyrrolidin-3-yl)-7H-imidazo[1,5-a]pyrazin-8-one Chemical compound OC=1C=2N(C=C(N=1)C)C(=NC=2)C1(CC(NC1)=O)C MFPMPDJKQXTYET-UHFFFAOYSA-N 0.000 description 2
- IWOVVHLXOMNIRU-UHFFFAOYSA-N 6-methyl-3-(3-methyloxan-4-yl)-7H-imidazo[1,5-a]pyrazin-8-one Chemical compound CC=1N=C(C=2N(C=1)C(=NC=2)C1C(COCC1)C)O IWOVVHLXOMNIRU-UHFFFAOYSA-N 0.000 description 2
- RANFYWLQRMPOCG-UHFFFAOYSA-N 6-methyl-3-(3-methyloxolan-3-yl)-7H-imidazo[1,5-a]pyrazin-8-one Chemical compound CC=1NC(C=2N(C=1)C(=NC=2)C1(COCC1)C)=O RANFYWLQRMPOCG-UHFFFAOYSA-N 0.000 description 2
- XLBNUMGNPXHANN-UHFFFAOYSA-N 6-methyl-3-(4-methyloxan-4-yl)-7H-imidazo[1,5-a]pyrazin-8-one Chemical compound CC=1NC(C=2N(C=1)C(=NC=2)C1(CCOCC1)C)=O XLBNUMGNPXHANN-UHFFFAOYSA-N 0.000 description 2
- YBULRWNGXOBEFY-UHFFFAOYSA-N 6-methyl-3-(5-methyloxolan-3-yl)-7H-imidazo[1,5-a]pyrazin-8-one Chemical compound CC=1NC(C=2N(C=1)C(=NC=2)C1COC(C1)C)=O YBULRWNGXOBEFY-UHFFFAOYSA-N 0.000 description 2
- KSUAAAURZXVVFF-UHFFFAOYSA-N 6-methyl-3-(7-oxoazepan-4-yl)-7H-imidazo[1,5-a]pyrazin-8-one Chemical compound CC=1NC(C=2N(C=1)C(=NC=2)C1CCNC(CC1)=O)=O KSUAAAURZXVVFF-UHFFFAOYSA-N 0.000 description 2
- SIXXYKBEUDLSPO-UHFFFAOYSA-N 6-methyl-3-(oxan-2-yl)-7H-imidazo[1,5-a]pyrazin-8-one Chemical compound CC=1NC(C=2N(C=1)C(=NC=2)C1OCCCC1)=O SIXXYKBEUDLSPO-UHFFFAOYSA-N 0.000 description 2
- PTFLWJRUGRSXHJ-UHFFFAOYSA-N 6-methyl-3-(oxolan-3-yl)-7H-imidazo[1,5-a]pyrazin-8-one Chemical compound CC=1NC(C=2N(C=1)C(=NC=2)C1COCC1)=O PTFLWJRUGRSXHJ-UHFFFAOYSA-N 0.000 description 2
- GHMXZYNLXPVWHJ-LLVKDONJSA-N 6-methyl-3-[(1R)-1-phenylethyl]-7H-imidazo[1,5-a]pyrazin-8-one Chemical compound CC=1N=C(C=2N(C=1)C(=NC=2)[C@H](C)C1=CC=CC=C1)O GHMXZYNLXPVWHJ-LLVKDONJSA-N 0.000 description 2
- GHMXZYNLXPVWHJ-NSHDSACASA-N 6-methyl-3-[(1S)-1-phenylethyl]-7H-imidazo[1,5-a]pyrazin-8-one Chemical compound CC=1N=C(C=2N(C=1)C(=NC=2)[C@@H](C)C1=CC=CC=C1)O GHMXZYNLXPVWHJ-NSHDSACASA-N 0.000 description 2
- YXAYXSPDVACBLB-UHFFFAOYSA-N 6-methyl-3-[1-(4-methyl-1,3-thiazol-2-yl)ethyl]-7H-imidazo[1,5-a]pyrazin-8-one Chemical compound CC=1N=C(C=2N(C=1)C(=NC=2)C(C)C=1SC=C(N=1)C)O YXAYXSPDVACBLB-UHFFFAOYSA-N 0.000 description 2
- KWTOEFJOHNYYLJ-UHFFFAOYSA-N 6-methyl-3-propan-2-yl-7H-imidazo[1,5-a]pyrazin-8-one Chemical compound C(C)(C)C1=NC=C2N1C=C(NC2=O)C KWTOEFJOHNYYLJ-UHFFFAOYSA-N 0.000 description 2
- MJUSXWTXOXKDDS-UHFFFAOYSA-N 6-methyl-7-[(4-nitrophenyl)methyl]-3-(oxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound CC=1N(C(C=2N(C=1)C(=NC=2)C1CCOCC1)=O)CC1=CC=C(C=C1)[N+](=O)[O-] MJUSXWTXOXKDDS-UHFFFAOYSA-N 0.000 description 2
- MBEVFGYULSPZRV-XVNBXDOJSA-N 7-(cyclohexylmethyl)-6-methyl-3-[(E)-prop-1-enyl]imidazo[1,5-a]pyrazin-8-one Chemical compound C1(CCCCC1)CN1C(C=2N(C=C1C)C(=NC=2)\C=C\C)=O MBEVFGYULSPZRV-XVNBXDOJSA-N 0.000 description 2
- DHTNNFUNTVBRHL-UHFFFAOYSA-N 7-[(3-fluorophenyl)methyl]-3-(4-hydroxyoxan-4-yl)-6-methylimidazo[1,5-a]pyrazin-8-one Chemical compound FC=1C=C(CN2C(C=3N(C=C2C)C(=NC=3)C2(CCOCC2)O)=O)C=CC=1 DHTNNFUNTVBRHL-UHFFFAOYSA-N 0.000 description 2
- AQLHITNAEZNQJK-UHFFFAOYSA-N 7-[(3-fluorophenyl)methyl]-6-methyl-3-(3-methyloxolan-3-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound FC=1C=C(CN2C(C=3N(C=C2C)C(=NC=3)C2(COCC2)C)=O)C=CC=1 AQLHITNAEZNQJK-UHFFFAOYSA-N 0.000 description 2
- PZKOOBRKCGIRJF-UHFFFAOYSA-N 7-[(3-fluorophenyl)methyl]-6-methyl-3-(oxan-3-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound FC=1C=C(CN2C(C=3N(C=C2C)C(=NC=3)C2COCCC2)=O)C=CC=1 PZKOOBRKCGIRJF-UHFFFAOYSA-N 0.000 description 2
- SILLXGDGCXBMIZ-UHFFFAOYSA-N 7-[(4-methoxyphenyl)methyl]-6-methyl-3-(2-phenylmethoxypropan-2-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound C(C1=CC=CC=C1)OC(C)(C)C1=NC=C2N1C=C(N(C2=O)CC1=CC=C(C=C1)OC)C SILLXGDGCXBMIZ-UHFFFAOYSA-N 0.000 description 2
- PPOOVDSNCYSLIL-UHFFFAOYSA-N 7-[(4-methoxyphenyl)methyl]-6-methyl-3-(3-methylpyrrolidin-3-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound COC1=CC=C(CN2C(C=3N(C=C2C)C(=NC=3)C2(CNCC2)C)=O)C=C1 PPOOVDSNCYSLIL-UHFFFAOYSA-N 0.000 description 2
- NTKIGHUFQGIWLS-UHFFFAOYSA-N 8-chloro-3-propylimidazo[1,5-a]pyrazine Chemical compound ClC=1C=2N(C=CN=1)C(=NC=2)CCC NTKIGHUFQGIWLS-UHFFFAOYSA-N 0.000 description 2
- AFQLYSHHYYOVDJ-UHFFFAOYSA-N 8-chloroimidazo[1,5-a]pyrazine Chemical compound ClC1=NC=CN2C=NC=C12 AFQLYSHHYYOVDJ-UHFFFAOYSA-N 0.000 description 2
- KEDWZGQGAYWGMC-UHFFFAOYSA-N 8-methoxy-6-methyl-3-(1-methylcyclopropyl)imidazo[1,5-a]pyrazine Chemical compound COC=1C=2N(C=C(N=1)C)C(=NC=2)C1(CC1)C KEDWZGQGAYWGMC-UHFFFAOYSA-N 0.000 description 2
- TUSMEWAHMCZZLE-UHFFFAOYSA-N 8-methoxy-6-methyl-3-(2-methylcyclopropyl)imidazo[1,5-a]pyrazine Chemical compound COC=1C=2N(C=C(N=1)C)C(=NC=2)C1C(C1)C TUSMEWAHMCZZLE-UHFFFAOYSA-N 0.000 description 2
- CYQYDNSQZKKWCM-UHFFFAOYSA-N 8-methoxy-6-methyl-3-(3-methyloxolan-3-yl)imidazo[1,5-a]pyrazine Chemical compound COC=1C=2N(C=C(N=1)C)C(=NC=2)C1(COCC1)C CYQYDNSQZKKWCM-UHFFFAOYSA-N 0.000 description 2
- DWIHWSZSPAOITR-UHFFFAOYSA-N 8-methoxy-6-methyl-3-(oxan-2-yl)imidazo[1,5-a]pyrazine Chemical compound COC=1C=2N(C=C(N=1)C)C(=NC=2)C1OCCCC1 DWIHWSZSPAOITR-UHFFFAOYSA-N 0.000 description 2
- JFEDIKSPTNOTCD-UHFFFAOYSA-N 8-methoxy-6-methyl-3-(oxolan-3-yl)imidazo[1,5-a]pyrazine Chemical compound COC=1C=2N(C=C(N=1)C)C(=NC=2)C1COCC1 JFEDIKSPTNOTCD-UHFFFAOYSA-N 0.000 description 2
- VYZCHDWGBGLNRY-UHFFFAOYSA-N 8-methoxy-6-methyl-3-propan-2-ylimidazo[1,5-a]pyrazine Chemical compound C(C)(C)C1=NC=C2N1C=C(N=C2OC)C VYZCHDWGBGLNRY-UHFFFAOYSA-N 0.000 description 2
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 2
- 239000005695 Ammonium acetate Substances 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- XVGOZDAJGBALKS-UHFFFAOYSA-N Blonanserin Chemical compound C1CN(CC)CCN1C1=CC(C=2C=CC(F)=CC=2)=C(CCCCCC2)C2=N1 XVGOZDAJGBALKS-UHFFFAOYSA-N 0.000 description 2
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 2
- 102000000584 Calmodulin Human genes 0.000 description 2
- 108010041952 Calmodulin Proteins 0.000 description 2
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 2
- XFXPMWWXUTWYJX-UHFFFAOYSA-N Cyanide Chemical compound N#[C-] XFXPMWWXUTWYJX-UHFFFAOYSA-N 0.000 description 2
- VZFUCHSFHOYXIS-UHFFFAOYSA-N Cycloheptanecarboxylic acid Chemical compound OC(=O)C1CCCCCC1 VZFUCHSFHOYXIS-UHFFFAOYSA-N 0.000 description 2
- 101150049660 DRD2 gene Proteins 0.000 description 2
- 102000004076 Dopamine D1 Receptors Human genes 0.000 description 2
- 108090000511 Dopamine D1 Receptors Proteins 0.000 description 2
- 229940123603 Dopamine D2 receptor antagonist Drugs 0.000 description 2
- 102000015554 Dopamine receptor Human genes 0.000 description 2
- 108050004812 Dopamine receptor Proteins 0.000 description 2
- 108010010803 Gelatin Proteins 0.000 description 2
- 239000007995 HEPES buffer Substances 0.000 description 2
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- QMBOHJQIEUJELW-UHFFFAOYSA-N N-[(5-bromo-3-methoxypyrazin-2-yl)methyl]oxane-4-carboxamide Chemical compound BrC=1N=C(C(=NC=1)CNC(=O)C1CCOCC1)OC QMBOHJQIEUJELW-UHFFFAOYSA-N 0.000 description 2
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Natural products OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 2
- 102000039029 PDE1 family Human genes 0.000 description 2
- 108091065686 PDE1 family Proteins 0.000 description 2
- 235000019483 Peanut oil Nutrition 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- 239000007868 Raney catalyst Substances 0.000 description 2
- 229910000564 Raney nickel Inorganic materials 0.000 description 2
- 102100023145 Sodium- and chloride-dependent glycine transporter 1 Human genes 0.000 description 2
- 101710083171 Sodium- and chloride-dependent glycine transporter 1 Proteins 0.000 description 2
- 238000006161 Suzuki-Miyaura coupling reaction Methods 0.000 description 2
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 2
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 2
- ZNKLAISUBPQUMH-UHFFFAOYSA-N [4-(cyclopropylmethoxy)phenyl]methanol Chemical compound C1=CC(CO)=CC=C1OCC1CC1 ZNKLAISUBPQUMH-UHFFFAOYSA-N 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 230000004913 activation Effects 0.000 description 2
- 238000010640 amide synthesis reaction Methods 0.000 description 2
- 150000001408 amides Chemical class 0.000 description 2
- 150000001412 amines Chemical class 0.000 description 2
- 229910021529 ammonia Inorganic materials 0.000 description 2
- 235000019257 ammonium acetate Nutrition 0.000 description 2
- 229940043376 ammonium acetate Drugs 0.000 description 2
- 239000003693 atypical antipsychotic agent Substances 0.000 description 2
- 229940127236 atypical antipsychotics Drugs 0.000 description 2
- 239000011324 bead Substances 0.000 description 2
- 230000009286 beneficial effect Effects 0.000 description 2
- PKBXXTDJYMWGAW-UHFFFAOYSA-N benzyl 2-methyl-2-phenylmethoxypropanoate Chemical compound C=1C=CC=CC=1COC(=O)C(C)(C)OCC1=CC=CC=C1 PKBXXTDJYMWGAW-UHFFFAOYSA-N 0.000 description 2
- PDHVULUFVFZKOQ-UHFFFAOYSA-N benzyl 3-[(3-methoxy-5-methylpyrazin-2-yl)methylcarbamoyl]-3-methylpyrrolidine-1-carboxylate Chemical compound COC=1C(=NC=C(N=1)C)CNC(=O)C1(CN(CC1)C(=O)OCC1=CC=CC=C1)C PDHVULUFVFZKOQ-UHFFFAOYSA-N 0.000 description 2
- 125000000051 benzyloxy group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])O* 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- 229950002871 blonanserin Drugs 0.000 description 2
- ILAHWRKJUDSMFH-UHFFFAOYSA-N boron tribromide Chemical compound BrB(Br)Br ILAHWRKJUDSMFH-UHFFFAOYSA-N 0.000 description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 2
- 229910052794 bromium Inorganic materials 0.000 description 2
- 239000000872 buffer Substances 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 230000015556 catabolic process Effects 0.000 description 2
- 230000003197 catalytic effect Effects 0.000 description 2
- 210000003169 central nervous system Anatomy 0.000 description 2
- 239000000460 chlorine Substances 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- 230000001054 cortical effect Effects 0.000 description 2
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 2
- BMCQFFXPECPDPS-UHFFFAOYSA-N cycloheptylmethanol Chemical compound OCC1CCCCCC1 BMCQFFXPECPDPS-UHFFFAOYSA-N 0.000 description 2
- 125000006622 cycloheptylmethyl group Chemical group 0.000 description 2
- SBBZAZOOOJEDAR-UHFFFAOYSA-N cycloheptylmethyl methanesulfonate Chemical compound CS(=O)(=O)OCC1CCCCCC1 SBBZAZOOOJEDAR-UHFFFAOYSA-N 0.000 description 2
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 2
- 238000006731 degradation reaction Methods 0.000 description 2
- 235000014113 dietary fatty acids Nutrition 0.000 description 2
- NZZFYRREKKOMAT-UHFFFAOYSA-N diiodomethane Chemical compound ICI NZZFYRREKKOMAT-UHFFFAOYSA-N 0.000 description 2
- YWEUIGNSBFLMFL-UHFFFAOYSA-N diphosphonate Chemical compound O=P(=O)OP(=O)=O YWEUIGNSBFLMFL-UHFFFAOYSA-N 0.000 description 2
- 239000006185 dispersion Substances 0.000 description 2
- 239000000442 dopamine 2 receptor blocking agent Substances 0.000 description 2
- 230000009977 dual effect Effects 0.000 description 2
- 239000000194 fatty acid Substances 0.000 description 2
- 229930195729 fatty acid Natural products 0.000 description 2
- 239000008273 gelatin Substances 0.000 description 2
- 229920000159 gelatin Polymers 0.000 description 2
- 239000007903 gelatin capsule Substances 0.000 description 2
- 235000019322 gelatine Nutrition 0.000 description 2
- 235000011852 gelatine desserts Nutrition 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- 210000003917 human chromosome Anatomy 0.000 description 2
- 238000005984 hydrogenation reaction Methods 0.000 description 2
- 239000005457 ice water Substances 0.000 description 2
- MIFJMFOVENWQDP-UHFFFAOYSA-N imidazo[1,5-a]pyrazine Chemical compound C1=CN=CC2=CN=CN21 MIFJMFOVENWQDP-UHFFFAOYSA-N 0.000 description 2
- 150000005235 imidazopyrazines Chemical class 0.000 description 2
- 230000003834 intracellular effect Effects 0.000 description 2
- 230000031146 intracellular signal transduction Effects 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- OWFXIOWLTKNBAP-UHFFFAOYSA-N isoamyl nitrite Chemical compound CC(C)CCON=O OWFXIOWLTKNBAP-UHFFFAOYSA-N 0.000 description 2
- 229940047889 isobutyramide Drugs 0.000 description 2
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- DLEDOFVPSDKWEF-UHFFFAOYSA-N lithium butane Chemical compound [Li+].CCC[CH2-] DLEDOFVPSDKWEF-UHFFFAOYSA-N 0.000 description 2
- 235000019359 magnesium stearate Nutrition 0.000 description 2
- 239000011159 matrix material Substances 0.000 description 2
- 229940098779 methanesulfonic acid Drugs 0.000 description 2
- TUZYFZFMOPJNJH-UHFFFAOYSA-N methyl 3-(2-oxobutyl)imidazole-4-carboxylate Chemical compound O=C(CN1C=NC=C1C(=O)OC)CC TUZYFZFMOPJNJH-UHFFFAOYSA-N 0.000 description 2
- 239000002480 mineral oil Substances 0.000 description 2
- 235000010446 mineral oil Nutrition 0.000 description 2
- MZRVEZGGRBJDDB-UHFFFAOYSA-N n-Butyllithium Substances [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 2
- 210000002569 neuron Anatomy 0.000 description 2
- 230000007996 neuronal plasticity Effects 0.000 description 2
- PXHVJJICTQNCMI-UHFFFAOYSA-N nickel Substances [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 2
- 229910000069 nitrogen hydride Inorganic materials 0.000 description 2
- 239000003921 oil Substances 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- JMJRYTGVHCAYCT-UHFFFAOYSA-N oxan-4-one Chemical compound O=C1CCOCC1 JMJRYTGVHCAYCT-UHFFFAOYSA-N 0.000 description 2
- MVZDZQRFEUOLOA-UHFFFAOYSA-N oxane-2-carboxamide Chemical compound NC(=O)C1CCCCO1 MVZDZQRFEUOLOA-UHFFFAOYSA-N 0.000 description 2
- KHDGSULFSVLOPM-UHFFFAOYSA-N oxane-3-carboxamide Chemical compound NC(=O)C1CCCOC1 KHDGSULFSVLOPM-UHFFFAOYSA-N 0.000 description 2
- DUXPFRRFZLRICX-UHFFFAOYSA-N oxolane-3-carboxamide Chemical compound NC(=O)C1CCOC1 DUXPFRRFZLRICX-UHFFFAOYSA-N 0.000 description 2
- 238000007911 parenteral administration Methods 0.000 description 2
- 239000002245 particle Substances 0.000 description 2
- 239000000312 peanut oil Substances 0.000 description 2
- DLYUQMMRRRQYAE-UHFFFAOYSA-N phosphorus pentoxide Inorganic materials O1P(O2)(=O)OP3(=O)OP1(=O)OP2(=O)O3 DLYUQMMRRRQYAE-UHFFFAOYSA-N 0.000 description 2
- 230000026731 phosphorylation Effects 0.000 description 2
- 238000006366 phosphorylation reaction Methods 0.000 description 2
- XUWHAWMETYGRKB-UHFFFAOYSA-N piperidin-2-one Chemical compound O=C1CCCCN1 XUWHAWMETYGRKB-UHFFFAOYSA-N 0.000 description 2
- CHKVPAROMQMJNQ-UHFFFAOYSA-M potassium bisulfate Chemical compound [K+].OS([O-])(=O)=O CHKVPAROMQMJNQ-UHFFFAOYSA-M 0.000 description 2
- 229910000343 potassium bisulfate Inorganic materials 0.000 description 2
- 230000002265 prevention Effects 0.000 description 2
- 239000000047 product Substances 0.000 description 2
- HNJBEVLQSNELDL-UHFFFAOYSA-N pyrrolidin-2-one Chemical compound O=C1CCCN1 HNJBEVLQSNELDL-UHFFFAOYSA-N 0.000 description 2
- 238000012552 review Methods 0.000 description 2
- 239000012279 sodium borohydride Substances 0.000 description 2
- 229910000033 sodium borohydride Inorganic materials 0.000 description 2
- NVIFVTYDZMXWGX-UHFFFAOYSA-N sodium metaborate Chemical compound [Na+].[O-]B=O NVIFVTYDZMXWGX-UHFFFAOYSA-N 0.000 description 2
- LPXPTNMVRIOKMN-UHFFFAOYSA-M sodium nitrite Chemical compound [Na+].[O-]N=O LPXPTNMVRIOKMN-UHFFFAOYSA-M 0.000 description 2
- 239000011877 solvent mixture Substances 0.000 description 2
- 238000001228 spectrum Methods 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 238000010189 synthetic method Methods 0.000 description 2
- 239000000454 talc Substances 0.000 description 2
- 229910052623 talc Inorganic materials 0.000 description 2
- 235000012222 talc Nutrition 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- LEIMLDGFXIOXMT-UHFFFAOYSA-N trimethylsilyl cyanide Chemical compound C[Si](C)(C)C#N LEIMLDGFXIOXMT-UHFFFAOYSA-N 0.000 description 2
- 229910052722 tritium Inorganic materials 0.000 description 2
- 229910052721 tungsten Inorganic materials 0.000 description 2
- 230000003442 weekly effect Effects 0.000 description 2
- MGBWQWATKAXZEU-BBVRLYRLSA-N (1R,2S,4S)-2-methyl-7-oxabicyclo[2.2.1]heptane-2-carboxylic acid Chemical compound C[C@]1([C@H]2CC[C@@H](C1)O2)C(=O)O MGBWQWATKAXZEU-BBVRLYRLSA-N 0.000 description 1
- SBUIQTMDIOLKAL-UHFFFAOYSA-N (2-ethylphenyl)methanol Chemical compound CCC1=CC=CC=C1CO SBUIQTMDIOLKAL-UHFFFAOYSA-N 0.000 description 1
- ITVZOLBIGHWGIW-UHFFFAOYSA-N (2-ethylphenyl)methyl methanesulfonate Chemical compound CCC1=CC=CC=C1COS(C)(=O)=O ITVZOLBIGHWGIW-UHFFFAOYSA-N 0.000 description 1
- SFWWGMKXCYLZEG-RXMQYKEDSA-N (3r)-3-methylmorpholine Chemical compound C[C@@H]1COCCN1 SFWWGMKXCYLZEG-RXMQYKEDSA-N 0.000 description 1
- SFWWGMKXCYLZEG-YFKPBYRVSA-N (3s)-3-methylmorpholine Chemical compound C[C@H]1COCCN1 SFWWGMKXCYLZEG-YFKPBYRVSA-N 0.000 description 1
- YSHMFBKODNGMPZ-UHFFFAOYSA-N (4-cyclopropyloxyphenyl)methyl methanesulfonate Chemical compound CS(=O)(=O)OCC1=CC=C(C=C1)OC1CC1 YSHMFBKODNGMPZ-UHFFFAOYSA-N 0.000 description 1
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 description 1
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 1
- 125000006701 (C1-C7) alkyl group Chemical group 0.000 description 1
- YPGCWEMNNLXISK-SSDOTTSWSA-N (R)-hydratropic acid Chemical compound OC(=O)[C@H](C)C1=CC=CC=C1 YPGCWEMNNLXISK-SSDOTTSWSA-N 0.000 description 1
- YPGCWEMNNLXISK-ZETCQYMHSA-N (S)-hydratropic acid Chemical compound OC(=O)[C@@H](C)C1=CC=CC=C1 YPGCWEMNNLXISK-ZETCQYMHSA-N 0.000 description 1
- LVEYOSJUKRVCCF-UHFFFAOYSA-N 1,3-Bis(diphenylphosphino)propane Substances C=1C=CC=CC=1P(C=1C=CC=CC=1)CCCP(C=1C=CC=CC=1)C1=CC=CC=C1 LVEYOSJUKRVCCF-UHFFFAOYSA-N 0.000 description 1
- SOKGWUZXWLLXHA-UHFFFAOYSA-N 1,4-dimethylpiperidine-4-carboxylic acid;hydrochloride Chemical compound Cl.CN1CCC(C)(C(O)=O)CC1 SOKGWUZXWLLXHA-UHFFFAOYSA-N 0.000 description 1
- LJRCWNIWOVZLKS-UHFFFAOYSA-N 1,4-oxazepane;hydrochloride Chemical compound Cl.C1CNCCOC1 LJRCWNIWOVZLKS-UHFFFAOYSA-N 0.000 description 1
- PURSZYWBIQIANP-UHFFFAOYSA-N 1-(bromomethyl)-2-chlorobenzene Chemical compound ClC1=CC=CC=C1CBr PURSZYWBIQIANP-UHFFFAOYSA-N 0.000 description 1
- FFWQLZFIMNTUCZ-UHFFFAOYSA-N 1-(bromomethyl)-2-fluorobenzene Chemical compound FC1=CC=CC=C1CBr FFWQLZFIMNTUCZ-UHFFFAOYSA-N 0.000 description 1
- WGVYCXYGPNNUQA-UHFFFAOYSA-N 1-(bromomethyl)-2-methylbenzene Chemical compound CC1=CC=CC=C1CBr WGVYCXYGPNNUQA-UHFFFAOYSA-N 0.000 description 1
- LZIYAIRGDHSVED-UHFFFAOYSA-N 1-(bromomethyl)-3-chlorobenzene Chemical compound ClC1=CC=CC(CBr)=C1 LZIYAIRGDHSVED-UHFFFAOYSA-N 0.000 description 1
- ZKSOJQDNSNJIQW-UHFFFAOYSA-N 1-(bromomethyl)-3-methoxybenzene Chemical compound COC1=CC=CC(CBr)=C1 ZKSOJQDNSNJIQW-UHFFFAOYSA-N 0.000 description 1
- FWLWTILKTABGKQ-UHFFFAOYSA-N 1-(bromomethyl)-3-methylbenzene Chemical compound CC1=CC=CC(CBr)=C1 FWLWTILKTABGKQ-UHFFFAOYSA-N 0.000 description 1
- IFNMJBFIRKAQBT-UHFFFAOYSA-N 1-(bromomethyl)-4-(difluoromethoxy)benzene Chemical compound FC(F)OC1=CC=C(CBr)C=C1 IFNMJBFIRKAQBT-UHFFFAOYSA-N 0.000 description 1
- JDNPUJCKXLOHOW-UHFFFAOYSA-N 1-(bromomethyl)-4-(trifluoromethoxy)benzene Chemical compound FC(F)(F)OC1=CC=C(CBr)C=C1 JDNPUJCKXLOHOW-UHFFFAOYSA-N 0.000 description 1
- KQNBRMUBPRGXSL-UHFFFAOYSA-N 1-(bromomethyl)-4-chlorobenzene Chemical compound ClC1=CC=C(CBr)C=C1 KQNBRMUBPRGXSL-UHFFFAOYSA-N 0.000 description 1
- NVNPLEPBDPJYRZ-UHFFFAOYSA-N 1-(bromomethyl)-4-fluorobenzene Chemical compound FC1=CC=C(CBr)C=C1 NVNPLEPBDPJYRZ-UHFFFAOYSA-N 0.000 description 1
- WZRKSPFYXUXINF-UHFFFAOYSA-N 1-(bromomethyl)-4-methylbenzene Chemical compound CC1=CC=C(CBr)C=C1 WZRKSPFYXUXINF-UHFFFAOYSA-N 0.000 description 1
- YXZFFTJAHVMMLF-UHFFFAOYSA-N 1-bromo-3-methylbutane Chemical compound CC(C)CCBr YXZFFTJAHVMMLF-UHFFFAOYSA-N 0.000 description 1
- FOCLRODNGKLAOT-UHFFFAOYSA-N 1-bromo-4-cyclopropyloxybenzene Chemical compound C1=CC(Br)=CC=C1OC1CC1 FOCLRODNGKLAOT-UHFFFAOYSA-N 0.000 description 1
- CCXQVBSQUQCEEO-UHFFFAOYSA-N 1-bromobutan-2-one Chemical compound CCC(=O)CBr CCXQVBSQUQCEEO-UHFFFAOYSA-N 0.000 description 1
- CYNYIHKIEHGYOZ-UHFFFAOYSA-N 1-bromopropane Chemical compound CCCBr CYNYIHKIEHGYOZ-UHFFFAOYSA-N 0.000 description 1
- DIZKLZKLNKQFGB-UHFFFAOYSA-N 1-methylcyclopropane-1-carboxylic acid Chemical compound OC(=O)C1(C)CC1 DIZKLZKLNKQFGB-UHFFFAOYSA-N 0.000 description 1
- KMLMOVWSQPHQME-UHFFFAOYSA-N 2,2-difluorocyclopropane-1-carboxylic acid Chemical compound OC(=O)C1CC1(F)F KMLMOVWSQPHQME-UHFFFAOYSA-N 0.000 description 1
- UFLFSJVTFSZTKX-UHFFFAOYSA-N 2,2-dimethylmorpholine Chemical compound CC1(C)CNCCO1 UFLFSJVTFSZTKX-UHFFFAOYSA-N 0.000 description 1
- PMKAHDGEZAFUAW-UHFFFAOYSA-N 2,6-dimethyloxane-4-carboxylic acid Chemical compound CC1CC(C(O)=O)CC(C)O1 PMKAHDGEZAFUAW-UHFFFAOYSA-N 0.000 description 1
- QGXNHCXKWFNKCG-UHFFFAOYSA-N 2-(bromomethyl)benzonitrile Chemical compound BrCC1=CC=CC=C1C#N QGXNHCXKWFNKCG-UHFFFAOYSA-N 0.000 description 1
- YCNQPAVKQPLZRS-UHFFFAOYSA-N 2-benzyl-4,4,5,5-tetramethyl-1,3,2-dioxaborolane Chemical compound O1C(C)(C)C(C)(C)OB1CC1=CC=CC=C1 YCNQPAVKQPLZRS-UHFFFAOYSA-N 0.000 description 1
- FRCQGUPEBLSFGO-UHFFFAOYSA-N 2-fluorocyclopropane-1-carbonyl chloride Chemical compound FC1CC1C(Cl)=O FRCQGUPEBLSFGO-UHFFFAOYSA-N 0.000 description 1
- 125000004493 2-methylbut-1-yl group Chemical group CC(C*)CC 0.000 description 1
- AYEGPMGNMOIHDL-UHFFFAOYSA-N 2-methylcyclopropane-1-carboxylic acid Chemical compound CC1CC1C(O)=O AYEGPMGNMOIHDL-UHFFFAOYSA-N 0.000 description 1
- DGMOBVGABMBZSB-UHFFFAOYSA-N 2-methylpropanoyl chloride Chemical compound CC(C)C(Cl)=O DGMOBVGABMBZSB-UHFFFAOYSA-N 0.000 description 1
- YWWHXMUOPZSUDB-UHFFFAOYSA-N 3-(2,3,3a,5,6,6a-hexahydrofuro[3,2-b]pyrrol-4-yl)-7-[(3-fluorophenyl)methyl]-6-methylimidazo[1,5-a]pyrazin-8-one Chemical compound FC=1C=C(CN2C(C=3N(C=C2C)C(=NC=3)N2C3C(CC2)OCC3)=O)C=CC=1 YWWHXMUOPZSUDB-UHFFFAOYSA-N 0.000 description 1
- CVKOOKPNCVYHNY-UHFFFAOYSA-N 3-(bromomethyl)benzonitrile Chemical compound BrCC1=CC=CC(C#N)=C1 CVKOOKPNCVYHNY-UHFFFAOYSA-N 0.000 description 1
- FEXTXBAFBURKGS-UHFFFAOYSA-N 3-(chloromethyl)-5-methyl-1,2-oxazole Chemical compound CC1=CC(CCl)=NO1 FEXTXBAFBURKGS-UHFFFAOYSA-N 0.000 description 1
- KKWWFYAKOFXBEY-UHFFFAOYSA-N 3-(chloromethyl)thiophene Chemical compound ClCC=1C=CSC=1 KKWWFYAKOFXBEY-UHFFFAOYSA-N 0.000 description 1
- FLFLSQGRBROAPA-UHFFFAOYSA-N 3-methoxy-2,2-dimethylpropanoic acid Chemical compound COCC(C)(C)C(O)=O FLFLSQGRBROAPA-UHFFFAOYSA-N 0.000 description 1
- DRHXPCBQKKTJHA-UHFFFAOYSA-N 3-methoxy-2,2-dimethylpropanoyl chloride Chemical compound COCC(C)(C)C(Cl)=O DRHXPCBQKKTJHA-UHFFFAOYSA-N 0.000 description 1
- PVHTZUOXJDYNBQ-UHFFFAOYSA-N 3-methyl-1-phenylmethoxycarbonylpyrrolidine-3-carboxylic acid Chemical compound C1C(C)(C(O)=O)CCN1C(=O)OCC1=CC=CC=C1 PVHTZUOXJDYNBQ-UHFFFAOYSA-N 0.000 description 1
- CNDLZDNFSPMAQH-UHFFFAOYSA-N 3-methyloxane-4-carboxylic acid Chemical compound CC1COCCC1C(O)=O CNDLZDNFSPMAQH-UHFFFAOYSA-N 0.000 description 1
- FLZNEKJUIYLHAR-UHFFFAOYSA-N 3-methyloxolane-3-carboxylic acid Chemical compound OC(=O)C1(C)CCOC1 FLZNEKJUIYLHAR-UHFFFAOYSA-N 0.000 description 1
- COPMASWDWLENMV-VOTSOKGWSA-N 4,4,5,5-tetramethyl-2-[(e)-prop-1-enyl]-1,3,2-dioxaborolane Chemical compound C\C=C\B1OC(C)(C)C(C)(C)O1 COPMASWDWLENMV-VOTSOKGWSA-N 0.000 description 1
- PLBDOPBECRIDKT-UHFFFAOYSA-N 4-(bromomethyl)-1-chloro-2-methoxybenzene Chemical compound COC1=CC(CBr)=CC=C1Cl PLBDOPBECRIDKT-UHFFFAOYSA-N 0.000 description 1
- BDYJMOYDXJQHFS-UHFFFAOYSA-N 4-(bromomethyl)-2-chloro-1-methoxybenzene Chemical compound COC1=CC=C(CBr)C=C1Cl BDYJMOYDXJQHFS-UHFFFAOYSA-N 0.000 description 1
- UMLFTCYAQPPZER-UHFFFAOYSA-N 4-(bromomethyl)benzonitrile Chemical compound BrCC1=CC=C(C#N)C=C1 UMLFTCYAQPPZER-UHFFFAOYSA-N 0.000 description 1
- NVTBASMQHFMANH-UHFFFAOYSA-N 4-(chloromethyl)-1,3-thiazole;hydron;chloride Chemical compound Cl.ClCC1=CSC=N1 NVTBASMQHFMANH-UHFFFAOYSA-N 0.000 description 1
- AQBBZYVPKBIILN-UHFFFAOYSA-N 4-(chloromethyl)-2-methyl-1,3-thiazole Chemical compound CC1=NC(CCl)=CS1 AQBBZYVPKBIILN-UHFFFAOYSA-N 0.000 description 1
- NIFAUKBQIAURIM-UHFFFAOYSA-N 4-(chloromethyl)-3,5-dimethyl-1,2-oxazole Chemical compound CC1=NOC(C)=C1CCl NIFAUKBQIAURIM-UHFFFAOYSA-N 0.000 description 1
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 description 1
- RCLMLYLFDJEUJY-UHFFFAOYSA-N 4-cyanooxane-4-carboxylic acid Chemical compound OC(=O)C1(C#N)CCOCC1 RCLMLYLFDJEUJY-UHFFFAOYSA-N 0.000 description 1
- VZWFVINYTCLXGC-UHFFFAOYSA-N 4-methyloxane-4-carboxylic acid Chemical compound OC(=O)C1(C)CCOCC1 VZWFVINYTCLXGC-UHFFFAOYSA-N 0.000 description 1
- VOLRSQPSJGXRNJ-UHFFFAOYSA-N 4-nitrobenzyl bromide Chemical compound [O-][N+](=O)C1=CC=C(CBr)C=C1 VOLRSQPSJGXRNJ-UHFFFAOYSA-N 0.000 description 1
- UNYHRXLMTSXVIB-UHFFFAOYSA-N 5-(bromomethyl)-1,3-benzodioxole Chemical compound BrCC1=CC=C2OCOC2=C1 UNYHRXLMTSXVIB-UHFFFAOYSA-N 0.000 description 1
- YNCJGPOTUFPVTB-UHFFFAOYSA-N 5-(chloromethyl)-2-methoxypyridine Chemical compound COC1=CC=C(CCl)C=N1 YNCJGPOTUFPVTB-UHFFFAOYSA-N 0.000 description 1
- ZWBJJNWJUXXFNP-UHFFFAOYSA-N 5-(chloromethyl)-3-methyl-1,2-oxazole Chemical compound CC=1C=C(CCl)ON=1 ZWBJJNWJUXXFNP-UHFFFAOYSA-N 0.000 description 1
- KXWUMIANQIGMCR-UHFFFAOYSA-N 5-methyloxolane-3-carboxylic acid Chemical compound CC1CC(C(O)=O)CO1 KXWUMIANQIGMCR-UHFFFAOYSA-N 0.000 description 1
- LPEKQJYFQVQFDC-UHFFFAOYSA-N 6-(bromomethyl)-7-[(4-methoxyphenyl)methyl]-3-(oxan-4-yl)imidazo[1,5-a]pyrazin-8-one Chemical compound BrCC=1N(C(C=2N(C=1)C(=NC=2)C1CCOCC1)=O)CC1=CC=C(C=C1)OC LPEKQJYFQVQFDC-UHFFFAOYSA-N 0.000 description 1
- AVSBZNSALVPORP-UHFFFAOYSA-N 6-methyl-3-(oxan-3-yl)-7H-imidazo[1,5-a]pyrazin-8-one Chemical compound CC=1NC(C=2N(C=1)C(=NC=2)C1COCCC1)=O AVSBZNSALVPORP-UHFFFAOYSA-N 0.000 description 1
- NSFYZWGFIIQKQP-IAQYHMDHSA-N 7-[(3-fluorophenyl)methyl]-6-methyl-3-[(1R,2R)-2-methylcyclopropyl]imidazo[1,5-a]pyrazin-8-one Chemical compound FC=1C=C(CN2C(C=3N(C=C2C)C(=NC=3)[C@H]2[C@@H](C2)C)=O)C=CC=1 NSFYZWGFIIQKQP-IAQYHMDHSA-N 0.000 description 1
- NSFYZWGFIIQKQP-XHDPSFHLSA-N 7-[(3-fluorophenyl)methyl]-6-methyl-3-[(1R,2S)-2-methylcyclopropyl]imidazo[1,5-a]pyrazin-8-one Chemical compound FC=1C=C(CN2C(C=3N(C=C2C)C(=NC=3)[C@H]2[C@H](C2)C)=O)C=CC=1 NSFYZWGFIIQKQP-XHDPSFHLSA-N 0.000 description 1
- HIARPVHIVVSVTG-QGZVFWFLSA-N 7-[(3-fluorophenyl)methyl]-6-methyl-3-[(2R)-oxan-2-yl]imidazo[1,5-a]pyrazin-8-one Chemical compound CC1=CN2C(=CN=C2[C@H]2CCCCO2)C(=O)N1CC1=CC(F)=CC=C1 HIARPVHIVVSVTG-QGZVFWFLSA-N 0.000 description 1
- OBFVZDIXAGVZLZ-UHFFFAOYSA-N 7-oxoazepane-4-carboxylic acid Chemical compound OC(=O)C1CCNC(=O)CC1 OBFVZDIXAGVZLZ-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- VREHPLJECZTEAO-UHFFFAOYSA-N 8-methoxy-6-methyl-3-(4-methyloxan-4-yl)imidazo[1,5-a]pyrazine Chemical compound COC=1C=2N(C=C(N=1)C)C(=NC=2)C1(CCOCC1)C VREHPLJECZTEAO-UHFFFAOYSA-N 0.000 description 1
- ONJBPBYBWJIUMW-UHFFFAOYSA-N 8-methoxy-6-methyl-3-(oxan-3-yl)imidazo[1,5-a]pyrazine Chemical compound COC=1C=2N(C=C(N=1)C)C(=NC=2)C1COCCC1 ONJBPBYBWJIUMW-UHFFFAOYSA-N 0.000 description 1
- 244000202285 Acrocomia mexicana Species 0.000 description 1
- 235000003625 Acrocomia mexicana Nutrition 0.000 description 1
- 108060003345 Adrenergic Receptor Proteins 0.000 description 1
- 102000017910 Adrenergic receptor Human genes 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 208000024827 Alzheimer disease Diseases 0.000 description 1
- 229910015845 BBr3 Inorganic materials 0.000 description 1
- 208000020925 Bipolar disease Diseases 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- JQTZXVDGXPKTCX-UHFFFAOYSA-N CC(C)(C)OC(NCc(ncc(C)n1)c1OC)=O Chemical compound CC(C)(C)OC(NCc(ncc(C)n1)c1OC)=O JQTZXVDGXPKTCX-UHFFFAOYSA-N 0.000 description 1
- KSSOCBRAWRNZGT-XQRVVYSFSA-N CC(C)=N/C(/C)=C\N Chemical compound CC(C)=N/C(/C)=C\N KSSOCBRAWRNZGT-XQRVVYSFSA-N 0.000 description 1
- YFBFTADLCVGNAV-JXAWBTAJSA-N CC(c1cnc(C2CCOCC2)[n]1/C=C(/Cc1ccccc1)\N)OC Chemical compound CC(c1cnc(C2CCOCC2)[n]1/C=C(/Cc1ccccc1)\N)OC YFBFTADLCVGNAV-JXAWBTAJSA-N 0.000 description 1
- 102000005701 Calcium-Binding Proteins Human genes 0.000 description 1
- 108010045403 Calcium-Binding Proteins Proteins 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- 108091005462 Cation channels Proteins 0.000 description 1
- 229910021589 Copper(I) bromide Inorganic materials 0.000 description 1
- 229910021595 Copper(I) iodide Inorganic materials 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- 101710095468 Cyclase Proteins 0.000 description 1
- 229920000858 Cyclodextrin Polymers 0.000 description 1
- 101100296720 Dictyostelium discoideum Pde4 gene Proteins 0.000 description 1
- 101100135868 Dictyostelium discoideum pde3 gene Proteins 0.000 description 1
- 101100407335 Dictyostelium discoideum pde7 gene Proteins 0.000 description 1
- 101100135859 Dictyostelium discoideum regA gene Proteins 0.000 description 1
- 101001072031 Drosophila melanogaster Dual 3',5'-cyclic-AMP and -GMP phosphodiesterase 11 Proteins 0.000 description 1
- 101100407340 Drosophila melanogaster Pde8 gene Proteins 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 101000848724 Homo sapiens Rap guanine nucleotide exchange factor 3 Proteins 0.000 description 1
- 208000023105 Huntington disease Diseases 0.000 description 1
- 108090000604 Hydrolases Proteins 0.000 description 1
- 102000004157 Hydrolases Human genes 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 108010044467 Isoenzymes Proteins 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- 229910010084 LiAlH4 Inorganic materials 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 101100407337 Mus musculus Pde8a gene Proteins 0.000 description 1
- UDIKZPSJSTVVAD-UHFFFAOYSA-N N-[(3-chloropyrazin-2-yl)methyl]cyclopropanecarboxamide Chemical compound ClC=1C(=NC=CN=1)CNC(=O)C1CC1 UDIKZPSJSTVVAD-UHFFFAOYSA-N 0.000 description 1
- CQKSOJHVPBBAMC-UHFFFAOYSA-N N-[(5-amino-3-methoxypyrazin-2-yl)methyl]oxane-4-carboxamide Chemical compound NC=1N=C(C(=NC=1)CNC(=O)C1CCOCC1)OC CQKSOJHVPBBAMC-UHFFFAOYSA-N 0.000 description 1
- 229910017906 NH3H2O Inorganic materials 0.000 description 1
- 238000005481 NMR spectroscopy Methods 0.000 description 1
- 229910003252 NaBO2 Inorganic materials 0.000 description 1
- 108010025020 Nerve Growth Factor Proteins 0.000 description 1
- 102000007072 Nerve Growth Factors Human genes 0.000 description 1
- 208000012902 Nervous system disease Diseases 0.000 description 1
- 208000009668 Neurobehavioral Manifestations Diseases 0.000 description 1
- 208000025966 Neurological disease Diseases 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical class O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- NLRYAEBBFANLPC-UHFFFAOYSA-N O=C(C1CCOCC1)NCc(nccn1)c1Cl Chemical compound O=C(C1CCOCC1)NCc(nccn1)c1Cl NLRYAEBBFANLPC-UHFFFAOYSA-N 0.000 description 1
- RUKBVSODKQPHLE-UHFFFAOYSA-N OC1N(CC2CCCCC2)C(Cc2ccccc2)=C[n]2c(C3CCOCC3)ncc12 Chemical compound OC1N(CC2CCCCC2)C(Cc2ccccc2)=C[n]2c(C3CCOCC3)ncc12 RUKBVSODKQPHLE-UHFFFAOYSA-N 0.000 description 1
- 208000018737 Parkinson disease Diseases 0.000 description 1
- 102000045595 Phosphoprotein Phosphatases Human genes 0.000 description 1
- 108700019535 Phosphoprotein Phosphatases Proteins 0.000 description 1
- 108091000080 Phosphotransferase Proteins 0.000 description 1
- 101100082606 Plasmodium falciparum (isolate 3D7) PDEbeta gene Proteins 0.000 description 1
- 101100082610 Plasmodium falciparum (isolate 3D7) PDEdelta gene Proteins 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 102000001253 Protein Kinase Human genes 0.000 description 1
- 102100034584 Rap guanine nucleotide exchange factor 3 Human genes 0.000 description 1
- 208000005793 Restless legs syndrome Diseases 0.000 description 1
- 101100135860 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) PDE2 gene Proteins 0.000 description 1
- 244000000231 Sesamum indicum Species 0.000 description 1
- 102000036646 Signalosomes Human genes 0.000 description 1
- 108091007411 Signalosomes Proteins 0.000 description 1
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 1
- 208000027418 Wounds and injury Diseases 0.000 description 1
- LKBREHQHCVRNFR-UHFFFAOYSA-K [B+3].[Br-].[Br-].[Br-] Chemical compound [B+3].[Br-].[Br-].[Br-] LKBREHQHCVRNFR-UHFFFAOYSA-K 0.000 description 1
- SJZAPSHKTOTRBQ-UHFFFAOYSA-N [dimethylamino(triazolo[4,5-b]pyridin-1-yl)methylidene]-dimethylazanium Chemical compound C1=CC=C2[N+](=C(N(C)C)N(C)C)N=NC2=N1 SJZAPSHKTOTRBQ-UHFFFAOYSA-N 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 239000008186 active pharmaceutical agent Substances 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 235000010419 agar Nutrition 0.000 description 1
- 239000010441 alabaster Substances 0.000 description 1
- 229920013820 alkyl cellulose Polymers 0.000 description 1
- 125000000217 alkyl group Chemical group 0.000 description 1
- YPGCWEMNNLXISK-UHFFFAOYSA-N alpha-phenylpropionic acid Natural products OC(=O)C(C)C1=CC=CC=C1 YPGCWEMNNLXISK-UHFFFAOYSA-N 0.000 description 1
- 238000010976 amide bond formation reaction Methods 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 239000012736 aqueous medium Substances 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 239000012131 assay buffer Substances 0.000 description 1
- 238000000065 atmospheric pressure chemical ionisation Methods 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid group Chemical group C(C1=CC=CC=C1)(=O)O WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- 230000031709 bromination Effects 0.000 description 1
- 238000005893 bromination reaction Methods 0.000 description 1
- 125000001246 bromo group Chemical group Br* 0.000 description 1
- AEILLAXRDHDKDY-UHFFFAOYSA-N bromomethylcyclopropane Chemical compound BrCC1CC1 AEILLAXRDHDKDY-UHFFFAOYSA-N 0.000 description 1
- DVECBJCOGJRVPX-UHFFFAOYSA-N butyryl chloride Chemical compound CCCC(Cl)=O DVECBJCOGJRVPX-UHFFFAOYSA-N 0.000 description 1
- OSGAYBCDTDRGGQ-UHFFFAOYSA-L calcium sulfate Chemical compound [Ca+2].[O-]S([O-])(=O)=O OSGAYBCDTDRGGQ-UHFFFAOYSA-L 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 150000003857 carboxamides Chemical class 0.000 description 1
- BULLHNJGPPOUOX-UHFFFAOYSA-N chloroacetone Chemical compound CC(=O)CCl BULLHNJGPPOUOX-UHFFFAOYSA-N 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 229940126214 compound 3 Drugs 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical compound I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 description 1
- GBRBMTNGQBKBQE-UHFFFAOYSA-L copper;diiodide Chemical compound I[Cu]I GBRBMTNGQBKBQE-UHFFFAOYSA-L 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 229940099112 cornstarch Drugs 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- WZHCOOQXZCIUNC-UHFFFAOYSA-N cyclandelate Chemical compound C1C(C)(C)CC(C)CC1OC(=O)C(O)C1=CC=CC=C1 WZHCOOQXZCIUNC-UHFFFAOYSA-N 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000004851 cyclopentylmethyl group Chemical group C1(CCCC1)C* 0.000 description 1
- ZOOSILUVXHVRJE-UHFFFAOYSA-N cyclopropanecarbonyl chloride Chemical compound ClC(=O)C1CC1 ZOOSILUVXHVRJE-UHFFFAOYSA-N 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- 230000000593 degrading effect Effects 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- GPAYUJZHTULNBE-UHFFFAOYSA-N diphenylphosphine Chemical compound C=1C=CC=CC=1PC1=CC=CC=C1 GPAYUJZHTULNBE-UHFFFAOYSA-N 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 230000002500 effect on skin Effects 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 239000002702 enteric coating Substances 0.000 description 1
- 238000009505 enteric coating Methods 0.000 description 1
- 239000002532 enzyme inhibitor Substances 0.000 description 1
- JBKVHLHDHHXQEQ-UHFFFAOYSA-N epsilon-caprolactam Chemical compound O=C1CCCCCN1 JBKVHLHDHHXQEQ-UHFFFAOYSA-N 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- NJRIEKJYVVFYSS-UHFFFAOYSA-N ethyl 3-methyl-5-oxopyrrolidine-3-carboxylate Chemical compound CCOC(=O)C1(C)CNC(=O)C1 NJRIEKJYVVFYSS-UHFFFAOYSA-N 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- KTWOOEGAPBSYNW-UHFFFAOYSA-N ferrocene Chemical compound [Fe+2].C=1C=C[CH-]C=1.C=1C=C[CH-]C=1 KTWOOEGAPBSYNW-UHFFFAOYSA-N 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 230000006870 function Effects 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 238000007429 general method Methods 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 229940074045 glyceryl distearate Drugs 0.000 description 1
- 125000003976 glyceryl group Chemical group [H]C([*])([H])C(O[H])([H])C(O[H])([H])[H] 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- 210000001320 hippocampus Anatomy 0.000 description 1
- UTCSSFWDNNEEBH-UHFFFAOYSA-N imidazo[1,2-a]pyridine Chemical compound C1=CC=CC2=NC=CN21 UTCSSFWDNNEEBH-UHFFFAOYSA-N 0.000 description 1
- IJYHVZICHKCLMQ-UHFFFAOYSA-N imidazo[4,5-d]triazin-4-one Chemical class O=C1N=NN=C2N=CN=C12 IJYHVZICHKCLMQ-UHFFFAOYSA-N 0.000 description 1
- 239000007943 implant Substances 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 239000003999 initiator Substances 0.000 description 1
- 208000014674 injury Diseases 0.000 description 1
- 230000010354 integration Effects 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 239000007928 intraperitoneal injection Substances 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- HVTICUPFWKNHNG-UHFFFAOYSA-N iodoethane Chemical compound CCI HVTICUPFWKNHNG-UHFFFAOYSA-N 0.000 description 1
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- FMKOJHQHASLBPH-UHFFFAOYSA-N isopropyl iodide Chemical compound CC(C)I FMKOJHQHASLBPH-UHFFFAOYSA-N 0.000 description 1
- CTAPFRYPJLPFDF-UHFFFAOYSA-N isoxazole Chemical compound C=1C=NOC=1 CTAPFRYPJLPFDF-UHFFFAOYSA-N 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 230000002197 limbic effect Effects 0.000 description 1
- 239000008297 liquid dosage form Substances 0.000 description 1
- 239000006193 liquid solution Substances 0.000 description 1
- 239000006194 liquid suspension Substances 0.000 description 1
- 239000012280 lithium aluminium hydride Substances 0.000 description 1
- UGVPKMAWLOMPRS-UHFFFAOYSA-M magnesium;propane;bromide Chemical compound [Mg+2].[Br-].CC[CH2-] UGVPKMAWLOMPRS-UHFFFAOYSA-M 0.000 description 1
- 239000003550 marker Substances 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- OKKJLVBELUTLKV-VMNATFBRSA-N methanol-d1 Chemical compound [2H]OC OKKJLVBELUTLKV-VMNATFBRSA-N 0.000 description 1
- DKRSRWQXCKBLLC-UHFFFAOYSA-N methyl 2-bromo-3-(2-oxobutyl)imidazole-4-carboxylate Chemical compound BrC=1N(C(=CN=1)C(=O)OC)CC(CC)=O DKRSRWQXCKBLLC-UHFFFAOYSA-N 0.000 description 1
- NQMRYBIKMRVZLB-UHFFFAOYSA-N methylamine hydrochloride Chemical compound [Cl-].[NH3+]C NQMRYBIKMRVZLB-UHFFFAOYSA-N 0.000 description 1
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 1
- 239000012046 mixed solvent Substances 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 239000001788 mono and diglycerides of fatty acids Substances 0.000 description 1
- 239000000178 monomer Substances 0.000 description 1
- 150000004712 monophosphates Chemical class 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003136 n-heptyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 210000001577 neostriatum Anatomy 0.000 description 1
- 210000000653 nervous system Anatomy 0.000 description 1
- 230000000926 neurological effect Effects 0.000 description 1
- 230000004031 neuronal differentiation Effects 0.000 description 1
- 230000008587 neuronal excitability Effects 0.000 description 1
- 230000005015 neuronal process Effects 0.000 description 1
- 230000006576 neuronal survival Effects 0.000 description 1
- 230000000324 neuroprotective effect Effects 0.000 description 1
- 239000003900 neurotrophic factor Substances 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 235000019198 oils Nutrition 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 229940054010 other antipsychotics in atc Drugs 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- LMYJGUNNJIDROI-UHFFFAOYSA-N oxan-4-ol Chemical compound OC1CCOCC1 LMYJGUNNJIDROI-UHFFFAOYSA-N 0.000 description 1
- YEWPVCUHKJABMV-UHFFFAOYSA-N oxane-3-carboxylic acid Chemical compound OC(=O)C1CCCOC1 YEWPVCUHKJABMV-UHFFFAOYSA-N 0.000 description 1
- RYGUCYSSMOFTSH-UHFFFAOYSA-N oxane-4-carbonyl chloride Chemical compound ClC(=O)C1CCOCC1 RYGUCYSSMOFTSH-UHFFFAOYSA-N 0.000 description 1
- AVPKHOTUOHDTLW-UHFFFAOYSA-N oxane-4-carboxylic acid Chemical compound OC(=O)C1CCOCC1 AVPKHOTUOHDTLW-UHFFFAOYSA-N 0.000 description 1
- 239000007800 oxidant agent Substances 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- BOTREHHXSQGWTR-UHFFFAOYSA-N oxolane-3-carboxylic acid Chemical compound OC(=O)C1CCOC1 BOTREHHXSQGWTR-UHFFFAOYSA-N 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- PIBWKRNGBLPSSY-UHFFFAOYSA-L palladium(II) chloride Chemical compound Cl[Pd]Cl PIBWKRNGBLPSSY-UHFFFAOYSA-L 0.000 description 1
- IPCSVZSSVZVIGE-UHFFFAOYSA-N palmitic acid group Chemical group C(CCCCCCCCCCCCCCC)(=O)O IPCSVZSSVZVIGE-UHFFFAOYSA-N 0.000 description 1
- 239000001814 pectin Substances 0.000 description 1
- 235000010987 pectin Nutrition 0.000 description 1
- 229920001277 pectin Polymers 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- 150000003904 phospholipids Chemical class 0.000 description 1
- 102000020233 phosphotransferase Human genes 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000003449 preventive effect Effects 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 235000013772 propylene glycol Nutrition 0.000 description 1
- 108060006633 protein kinase Proteins 0.000 description 1
- 230000002685 pulmonary effect Effects 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 150000003254 radicals Chemical class 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 230000014786 regulation of synaptic transmission Effects 0.000 description 1
- 230000008844 regulatory mechanism Effects 0.000 description 1
- 150000003870 salicylic acids Chemical class 0.000 description 1
- 229930195734 saturated hydrocarbon Natural products 0.000 description 1
- HFHDHCJBZVLPGP-UHFFFAOYSA-N schardinger α-dextrin Chemical compound O1C(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(O)C2O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC2C(O)C(O)C1OC2CO HFHDHCJBZVLPGP-UHFFFAOYSA-N 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 230000000862 serotonergic effect Effects 0.000 description 1
- 108010006590 serotonin 5 receptor Proteins 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 230000019491 signal transduction Effects 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 235000010288 sodium nitrite Nutrition 0.000 description 1
- 239000007909 solid dosage form Substances 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- NVBFHJWHLNUMCV-UHFFFAOYSA-N sulfamide Chemical compound NS(N)(=O)=O NVBFHJWHLNUMCV-UHFFFAOYSA-N 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 230000005062 synaptic transmission Effects 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 229960000278 theophylline Drugs 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 229930192474 thiophene Natural products 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4985—Pyrazines or piperazines ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/551—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having two nitrogen atoms, e.g. dilazep
- A61K31/5513—1,4-Benzodiazepines, e.g. diazepam or clozapine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/553—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having at least one nitrogen and one oxygen as ring hetero atoms, e.g. loxapine, staurosporine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2300/00—Mixtures or combinations of active ingredients, wherein at least one active ingredient is fully defined in groups A61K31/00 - A61K41/00
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Epidemiology (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Psychiatry (AREA)
- Hospice & Palliative Care (AREA)
- Pain & Pain Management (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Un compuesto de Fórmula (I) **(Ver fórmula)** en donde n es 0 o 1; q es 0 o 1; R1 se selecciona del grupo que consiste en bencilo, indanilo, indolina y heteroarilos de 5 miembros; todos los cuales pueden estar sustituidos con un sustituyente seleccionado del grupo que consiste en halógeno y alquilo C1-C3; o R1 se selecciona del grupo que consiste en anillos monocíclicos saturados que contienen 4-6 átomos de carbono y 1- 2 átomos de nitrógeno; todos los cuales pueden estar sustituidos una o más veces con uno o más sustituyentes seleccionados del grupo que consiste en metilo, flúor y sulfonamida; o R1 se selecciona del grupo que consiste en lactamas que contienen 4-6 átomos de carbono; todos los cuales pueden estar sustituidos una o más veces con uno o más sustituyentes seleccionados del grupo que consiste en metilo y flúor; o R1 se selecciona del grupo que consiste en éteres bicíclicos tales como, 7-oxabiciclo[2.2.1]heptano; todos los cuales pueden estar sustituidos una o más veces con uno o más sustituyentes seleccionados del grupo que consiste en metilo y flúor; o R1 se selecciona del grupo que consiste en alquilo C1-C8 lineal o ramificado, cicloalquilo C3-C8 monocíclico saturado, oxetanilo, tetrahidrofuranilo y tetrahidropiranilo; todos los cuales pueden estar sustituidos una o más veces con uno o más sustituyentes seleccionados del grupo que consiste en metilo, flúor, hidroxi, ciano o metoxi; o R1 es un alquilo C1-C3 lineal o ramificado, que está sustituido con un sustituyente seleccionado de fenilo y heteroarilo de 5 miembros, en donde dicho heteroarilo de 5 miembros puede estar sustituido con uno o más alquilos C1-C3; o R1 se selecciona del grupo que consiste en morfolina, tetrahidrofuran-3-amina, hexahidro-2H-furo[3,2-b]pirrol y homomorfolina; todos los cuales pueden estar sustituidos con uno o más sustituyentes seleccionados del grupo que consiste en alquilo C1-C3; R2 se selecciona del grupo que consiste en hidrógeno, alquilo C1-C8 lineal o ramificado, fenilo, cicloalquilo C3-C8 monocíclico saturado, oxetanilo, benzo[d][1,3]dioxolilo, tetrahidrofuranilo y tetrahidropiranilo; o R2 es fenilo o piridilo sustituido con uno o más sustituyentes seleccionados del grupo que consiste en hidroxilo, amino, ciano, halógeno, alquilo C1-C3, alcoxi C1-C3, cicloalcoxi C3-C5, cicloalquil C3-C5-metoxi, fluoroalcoxi C1-C3, y -NC(O)CH3; o R2 es un heteroarilo de 5 miembros que puede estar sustituido con uno o más alquilo C1-C3; R3 se selecciona del grupo que consiste en hidrógeno, halógeno, alquilo C1-C5, cicloalquilo C3-C5 y fenilo; o R3 se selecciona del grupo que consiste en fenilo sustituido una o más veces con alquilo C1-C3; metilo sustituido una, dos o tres veces con flúor; etilo sustituido una, dos o tres veces con flúor; R4 es hidrógeno; y tautómeros y sales de adición farmacéuticamente aceptables del mismo; con la condición de que R2 y R3 no pueden ser hidrógeno al mismo tiempo; y 2) un segundo compuesto, compuesto que es útil en el tratamiento de un trastorno psiquiátrico; en donde dicho segundo compuesto se selecciona de la lista que consiste en clozapina, risperidona, paliperidona, olanzapina, quetiapina, amisulprido, ziprasidona, aripiprazol, brexpiprazol, asenapina, haloperidol, iloperidona, lurasidona, clorpromazina, blonanserina, perfenazina, levomepromazina, sulpirido, flufenazina, zuclopentixol, flupentixol y cariprazina; en donde 1) y 2) son para uso combinado en el tratamiento de un trastorno psiquiátrico y/o cognitivo.
Description
DESCRIPCIÓN
Tratamientos de combinación que comprenden imidazopirazinonas para el tratamiento de trastornos psiquiátricos y/o cognitivos
Campo de la invención
La presente invención proporciona tratamientos de combinación que comprenden la administración de compuestos que son inhibidores de la enzima PDE1 y otros compuestos útiles en el tratamiento de trastornos psiquiátricos y/o cognitivos tales como, por ejemplo, T rastorno de déficit de atención con hiperactividad (TDAH), depresión, ansiedad, narcolepsia, esquizofrenia, alteración cognitiva o alteración cognitiva asociada con esquizofrenia (CIAS), como se define en las reivindicaciones.
Los aspectos separados de la invención están dirigidos a dichos compuestos para el uso combinado para el tratamiento de trastornos psiquiátricos y/o cognitivos. La presente invención también proporciona composiciones farmacéuticas que comprenden dichos inhibidores de la enzima PDE1 junto con otros compuestos útiles en el tratamiento de trastornos psiquiátricos y/o cognitivos.
Antecedentes de la invención
Los nucleótidos cíclicos (Nc) que actúan como segundos mensajeros, monofosfato de adenosina cíclico (AMPc) y monofosfato de guanosina cíclico (GMPc) juegan un papel principal en la cascada de la transducción de las señales intracelulares, mediante la regulación de proteínas quinasas dependientes de Nc (PKA y PKG), EPAC (proteína de intercambio activada por AMPc), fosfoproteínas fosfatasas, y/o canales de cationes regulados por Nc. En las neuronas, esto incluye la activación de quinasas dependientes de AMPc y GMPc y la fosforilación posterior de proteínas implicadas en la regulación aguda de la transmisión sináptica, así como en la diferenciación y supervivencia neuronal. Las concentraciones intracelulares de AMPc y GMPc están reguladas estrictamente por la tasa de biosíntesis por ciclasas y por la tasa de degradación por fosfodiesterasas (PDE, EC 3.1.4.17). Las PDE son hidrolasas bimetálicas que inactivan AMPc/GMPc por la hidrólisis catalítica del enlace éster en 3', formando el monofosfato en 5' inactivo. Como las PDE proporcionan el único medio para degradar los nucleótidos cíclicos AMPc y GMPc en las células, las PDE juegan un papel esencial en la señalización de los nucleótidos cíclicos. Las actividades catalíticas de las PDE proporcionan la degradación de los Nc en un espectro de concentraciones de los Nc en todas las células, y sus diversos mecanismos reguladores proporcionan la integración e interacción con miríadas de rutas de señalización. Las PDE particulares están dirigidas a compartimentos discretos en las células donde controlan el nivel de los Nc y forman microentornos para una variedad de signalosomas de Nc (Sharron H. Francis, Mitsi A. Blount, y Jackie D. Corbin. Physiol Rev 2011,91: 651 -690).
Sobre la base de la especificidad de sustrato, las familias de PDE pueden dividirse en tres grupos: 1) Las PDE específicas de AMPc, que incluyen PDE4, PDE7, y PDE8, 2) las enzimas selectivas para GMPc p De5 y PDE9, y 3) las PDE con sustrato doble, PDE1, PDE2, PDE3, así como PDE10 y PDE11.
La PDE denominada previamente estimulada por calmodulina (CaM-PDE), PDE1, es única en que está regulada de forma dependiente de Ca2+ a través de la calmodulina (CaM, una proteína de unión de Ca2+-de 16 kDa) que forma complejo con cuatro Ca2+ (para una revisión, Sharron H. Francis, Mitsi A. Blount, y Jackie D. Corbin. Physiol Rev 2011, 91: 651-690). Así, la PDE1 representa un vínculo regulador interesante entre los nucleótidos cíclicos y el Ca2+ intracelular. La familia de PDE1 está codificada por tres genes: PDE1A (localizado en el cromosoma humano 2q32), PDE1B (localización en el cromosoma humano, hcl: 12q13) y PDE1C (hcl: 7p14.3). Tienen promotores alternativos y dan lugar a una multitud de proteínas por corte y empalme alternativo, que se diferencian entre sí en sus propiedades reguladoras, afinidades de sustrato, actividades específicas, constantes de activación para CaM, distribución tisular y pesos moleculares. Se han identificado más de 10 isoformas humanas. Sus pesos moleculares varían de 58 a 86 kDa por monómero. El dominio regulador N-terminal que contiene dos dominios de unión Ca2+/CaM y dos sitios de fosforilación diferencian sus proteínas correspondientes y modulan sus funciones bioquímicas. La PDE1 es una PDE con sustrato doble y el subtipo PDE1C tiene la misma actividad frente a AMPc y GMPc (Km = 1-3 pM), mientras los subtipos PDE1A y PDE1B tienen una preferencia por GMPc (Km para GMPc = 1-3 pM y para AMPc = 10-30 pM).
Los subtipos de PDE1 están altamente enriquecidos en el cerebro y se localizan especialmente en el cuerpo estriado (PDE1B), hipocampo (PDE1A) y corteza (PDE1A) y esta localización está conservada entre especies (Amy Bernard et al. Neuron 2012, 73, 1083-1099). En la corteza, la PDE1A está presente principalmente en las capas corticales profundas 5 y 6 (capas de salida), y se usa como un marcador de especificidad para las capas corticales profundas. Los inhibidores de PDE1 aumentan los niveles de los Nc segundos mensajeros dando lugar a una excitabilidad neuronal aumentada.
Así, la PDE1 es una diana terapéutica para la regulación de las rutas de señalización intracelular, preferiblemente en el sistema nervioso y los inhibidores de PDE1 pueden aumentar los niveles de los segundos mensajeros AMPc/GMPc dando lugar a la modulación de los procesos neuronales y a la expresión de genes relacionados con la plasticidad neuronal, factores neurotróficos, y moléculas neuroprotectoras. Estas propiedades de aumento de la plasticidad neuronal junto con la modulación de la trasmisión sináptica hacen que los inhibidores de PDE1 sean buenos candidatos como agentes terapéuticos en muchas afecciones neurológicas y psiquiátricas. La evaluación de los
inhibidores de PDE1 en modelos animales (para una revisión, véase, p. ej., Blokland et al. Expert Opinión on Therapeutic Patents (2012), 22(4), 349-354; y Medina, A. E. Frontiers in Neuropharmacology (2011), 5(feb.), 21) ha sugerido el potencial para el uso terapéutico de los inhibidores de PDE1 en trastornos neurológicos, como, p. ej., las enfermedades de Alzheimer, Parkinson y Huntington y en trastornos psiquiátricos, como, p. ej., Trastorno de déficit de atención con hiperactividad (TDAH), síndrome de las piernas inquietas, depresión, ansiedad, narcolepsia, esquizofrenia, alteración cognitiva y alteración cognitiva asociada con esquizofrenia (CIAS).
WO 2013/053690 A1 describe imidazopirazinonas que son inhibidores de la enzima PDE9.
WO 2016/055618 y WO 2016/147659 describen triazolopirazinonas e imidazotriazinonas como inhibidores de PDE1. El documento con derecho anterior WO2016/174188 (publicado el 03.11.2016) describe que las imidazopirazinonas de la presente fórmula (I) son inhibidores de PDE1, que son útiles para el tratamiento de trastornos psiquiátricos y/o cognitivos.
Los inhibidores de PDE1 ofrecen alternativas a los tratamientos comercializados actualmente para trastornos psiquiátricos y/o cognitivos, tratamientos que no son eficaces en todos los pacientes. Además, puede ser beneficioso combinar dichos inhibidores de PDE1 con otro paradigma de tratamiento útil en el tratamiento de trastornos psiquiátricos y/o cognitivos. Existe todavía una necesidad de nuevos métodos de tratamiento de dichas enfermedades.
Resumen de la invención
La invención se define por las reivindicaciones. Cualquier contenido que se encuentre fuera del alcance de las reivindicaciones se proporciona solo para propósitos de información.
Las enzimas PDE1 se expresan en el Sistema Nervioso Central (SNC), lo que hace de esta familia de genes una fuente atractiva de nuevas dianas para el tratamiento de trastornos psiquiátricos y/o cognitivos.
Los inventores de la presente invención han proporcionado compuestos que son inhibidores de PDE1, y como tales son útiles para tratar trastornos psiquiátricos y/o cognitivos. La combinación de dichos inhibidores de PDE1 con otros compuestos que son útiles en el tratamiento de un trastorno psiquiátrico puede dar lugar a un tratamiento mejorado de pacientes con trastornos psiquiátricos y/o cognitivos tales como, por ejemplo, Trastorno de déficit de atención con hiperactividad (TDAH), depresión, ansiedad, narcolepsia, esquizofrenia, alteración cognitiva y alteración cognitiva asociada con esquizofrenia (CIAS). Se hace una mención particular a la alteración cognitiva o alteración cognitiva asociada con esquizofrenia (CIAS).
Preferiblemente, dichos inhibidores de PDE1 son al menos diez veces más potentes como inhibidores de PDE1 que como inhibidores de PDE9 con el fin de prevenir efectos potencialmente indeseables asociados con la inhibición de PDE9.
Por consiguiente, la presente invención se refiere a:
1) un compuesto de Fórmula (I)

en donde
n es 0 o 1;
q es 0 o 1;
R1 se selecciona del grupo que consiste en bencilo, indanilo, indolina y heteroarilos de 5 miembros; todos los cuales pueden estar sustituidos con un sustituyente seleccionado del grupo que consiste en halógeno y alquilo C1-C3; o
R1 se selecciona del grupo que consiste en anillos monocíclicos saturados que contienen 4-6 átomos de carbono y 1 2 átomos de nitrógeno; todos los cuales pueden estar sustituidos una o más veces con uno o más sustituyentes seleccionados del grupo que consiste en metilo, flúor y sulfonamida; o
R1 se selecciona del grupo que consiste en lactamas que contienen 4-6 átomos de carbono; todos los cuales pueden estar sustituidos una o más veces con uno o más sustituyentes seleccionados del grupo que consiste en metilo y flúor; o R1 se selecciona del grupo que consiste en éteres bicíclicos tales como, 7-oxabiciclo[2.2.1]heptano; todos los cuales pueden estar sustituidos una o más veces con uno o más sustituyentes seleccionados del grupo que consiste en metilo y flúor; o
R1 se selecciona del grupo que consiste en alquilo C1-C8 lineal o ramificado, cicloalquilo C3-C8 monocíclico saturado, oxetanilo, tetrahidrofuranilo y tetrahidropiranilo; todos los cuales pueden estar sustituidos una o más veces con uno o más sustituyentes seleccionados del grupo que consiste en metilo, flúor, hidroxi, ciano o metoxi; o
R1 es un alquilo C1-C3 lineal o ramificado, que está sustituido con un sustituyente seleccionado de fenilo y heteroarilo de 5 miembros, en donde dicho heteroarilo de 5 miembros puede estar sustituido con uno o más alquilos C1-C3; o R1 se selecciona del grupo que consiste en morfolina, tetrahidrofuran-3-amina, hexahidro-2H-furo[3,2-b]pirrol y homomorfolina; todos los cuales pueden estar sustituidos con uno o más sustituyentes seleccionados del grupo que consiste en alquilo C1-C3;
R2 se selecciona del grupo que consiste en hidrógeno, alquilo C1-C8 lineal o ramificado, fenilo, cicloalquilo C3-C8 monocíclico saturado, oxetanilo, benzo[d][1,3]dioxolilo, tetrahidrofuranilo y tetrahidropiranilo; o
R2 es fenilo o piridilo sustituido con uno o más sustituyentes seleccionados del grupo que consiste en hidroxilo, amino, ciano, halógeno, alquilo C1-C3, alcoxi C1-C3, cicloalcoxi C3-C5, cicloalquil C3-C5-metoxi, fluoroalcoxi C1-C3, y -NC(O)CH3; o
R2 es un heteroarilo de 5 miembros que puede estar sustituido una o más veces con alquilo C1-C3;
R3 se selecciona del grupo que consiste en hidrógeno, halógeno, alquilo C1-C5, cicloalquilo C3-C5 y fenilo; o R3 se selecciona del grupo que consiste en fenilo sustituido una o más veces con alquilo C1-C3; metilo sustituido una, dos o tres veces con flúor; etilo sustituido una, dos o tres veces con flúor;
R4 es hidrógeno;
y tautómeros y sales de adición farmacéuticamente aceptables del mismo;
con la condición de que R2 y R3 no pueden ser hidrógeno al mismo tiempo; y
2) un segundo compuesto, compuesto que es útil en el tratamiento de un trastorno psiquiátrico; en donde dicho segundo compuesto se selecciona de la lista que consiste en clozapina, risperidona, paliperidona, olanzapina, quetiapina, amisulprido, ziprasidona, aripiprazol, brexpiprazol, asenapina, haloperidol, iloperidona, lurasidona, clorpromazina, blonanserina, perfenazina, levomepromazina, sulpirido, flufenazina, zuclopentixol, flupentixol y cariprazina; en donde
1) y 2) son para uso combinado en el tratamiento de un trastorno psiquiátrico y/o cognitivo.
La referencia al Compuesto (I) incluye la base libre del Compuesto (I), las sales farmacéuticamente aceptables del Compuesto (I), tales como las sales de adición a ácido del Compuesto (I), mezclas racémicas del Compuesto (I), o el enantiómero y/o isómero óptico correspondiente del Compuesto (I), y formas polimórficas y amórficas del Compuesto (I), así como formas tautoméricas del Compuesto (I). Además, los compuestos de esta invención pueden existir en formas no solvatadas, así como solvatadas con disolventes farmacéuticamente aceptables tales como agua, etanol y similares. En general, las formas solvatadas se consideran equivalentes a las formas no solvatadas para los propósitos de esta invención.
En una realización, la invención se refiere a una composición farmacéutica que comprende
1) un compuesto según la formula (I); y
2) un segundo compuesto, compuesto que es útil en el tratamiento de un trastorno psiquiátrico como se define en las reivindicaciones; y
uno o más vehículos o excipientes farmacéuticamente aceptables.
En un aspecto, la descripción se refiere a un método para el tratamiento de un trastorno psiquiátrico y/o cognitivo, método que comprende la administración de:
1) una cantidad terapéuticamente efectiva de un compuesto según la Fórmula (I); y
2) una cantidad terapéuticamente efectiva de:
un segundo compuesto, compuesto que es útil en el tratamiento de un trastorno psiquiátrico, a un paciente que lo necesita.
En un aspecto, la descripción se refiere al uso de un compuesto de Fórmula (I) en la fabricación de un medicamento para uso en el tratamiento de un trastorno psiquiátrico y/o cognitivo; en donde dicho medicamento es para uso en combinación con un segundo compuesto, compuesto que es útil en el tratamiento de un trastorno psiquiátrico.
En un aspecto, la descripción se refiere al uso de:
1) un compuesto de Fórmula (I); y
2) un segundo compuesto, compuesto que es útil en el tratamiento de un trastorno psiquiátrico; en la fabricación de un medicamento para uso en el tratamiento de un trastorno psiquiátrico y/o cognitivo.
Descripción detallada de la invención
Realizaciones de la invención
Se aplica la siguiente notación: una realización de la invención se identifica como Ei, donde i es un número entero que indica el número de la realización. Una realización Ei' que especifica una realización específica una realización Ei listada previamente se identifica como Ei'(Ei), p. ej., E2(E1) significa "en una realización E2 de la realización E1". Cuando una realización es una combinación de dos realizaciones, la notación es de forma similar Ei"(Ei y Ei'), p. ej., E3(E1 y E2) significa "en una realización E3 según cualquiera de las realizaciones E2 y E1".
Cuando una realización es una combinación de más de dos realizaciones, la notación es de forma similar Ei"'(Ei, Ei' y Ei") o (Ei-Ei"), p. ej., E4(E1, E2 y E3) o E4(E1-E3), significa "en una realización E4 según cualquiera de las realizaciones E1, E2 y E3".
En una primera realización E1, la presente invención se refiere a:
1) Un compuesto de Fórmula (I)
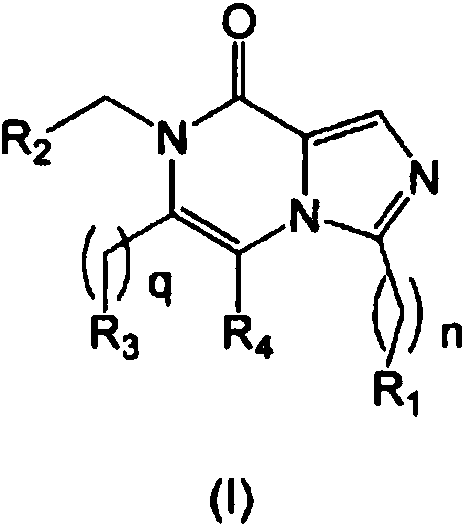
en donde
n es 0 o 1;
q es 0 o 1;
R1 se selecciona del grupo que consiste en bencilo, indanilo, indolina y heteroarilos de 5 miembros; todos los cuales pueden estar sustituidos con un sustituyente seleccionado del grupo que consiste en halógeno y alquilo C1-C3; o R1 se selecciona del grupo que consiste en anillos monocíclicos saturados que contienen 4-6 átomos de carbono y 1 2 átomos de nitrógeno; todos los cuales pueden estar sustituidos una o más veces con uno o más sustituyentes seleccionados del grupo que consiste en metilo, flúor y sulfonamida; o
R1 se selecciona del grupo que consiste en lactamas que contienen 4-6 átomos de carbono; todos los cuales pueden estar sustituidos una o más veces con uno o más sustituyentes seleccionados del grupo que consiste en metilo y flúor; o R1 se selecciona del grupo que consiste en éteres bicíclicos tales como, 7-oxabiciclo[2.2.1]heptano; todos los cuales pueden estar sustituidos una o más veces con uno o más sustituyentes seleccionados del grupo que consiste en metilo y flúor; o
R1 se selecciona del grupo que consiste en alquilo C1-C8 lineal o ramificado, cicloalquilo C3-C8 monocíclico saturado, oxetanilo, tetrahidrofuranilo y tetrahidropiranilo; todos los cuales pueden estar sustituidos una o más veces con uno o más sustituyentes seleccionados del grupo que consiste en metilo, flúor, hidroxi, ciano o metoxi; o
R1 es un alquilo C1-C3 lineal o ramificado, que está sustituido con un sustituyente seleccionado de fenilo y heteroarilo de 5 miembros, en donde dicho heteroarilo de 5 miembros puede estar sustituido con uno o más alquilos C1-C3; o R1 se selecciona del grupo que consiste en morfolina, tetrahidrofuran-3-amina, hexahidro-2H-furo[3,2-b]pirrol y homomorfolina; todos los cuales pueden estar sustituidos con uno o más sustituyentes seleccionados del grupo que consiste en alquilo C1-C3;
R2 se selecciona del grupo que consiste en hidrógeno, alquilo C1-C8 lineal o ramificado, fenilo, cicloalquilo C3-C8 monocíclico saturado, oxetanilo, benzo[d][1,3]dioxolilo, tetrahidrofuranilo y tetrahidropiranilo; o
R2 es fenilo o piridilo sustituido con uno o más sustituyentes seleccionados del grupo que consiste en hidroxilo, amino, ciano, halógeno, alquilo C1-C3, alcoxi C1-C3, cicloalcoxi C3-C5, cicloalquil C3-C5-metoxi, fluoroalcoxi C1-C3, y -NC(O)CH3; o
R2 es un heteroarilo de 5 miembros que puede estar sustituido con uno o más alquilo C1-C3;
R3 se selecciona del grupo que consiste en hidrógeno, halógeno, alquilo C1-C5, cicloalquilo C3-C5 y fenilo; o R3 se selecciona del grupo que consiste en fenilo sustituido una o más veces con alquilo C1-C3; metilo sustituido una, dos o tres veces con flúor; etilo sustituido una, dos o tres veces con flúor;
R4 es hidrógeno;
y tautómeros y sales de adición farmacéuticamente aceptables del mismo;
con la condición de que R2 y R3 no pueden ser hidrógeno al mismo tiempo; y
2) un segundo compuesto, compuesto que es útil en el tratamiento de un trastorno psiquiátrico; en donde dicho segundo compuesto se selecciona de la lista que consiste en clozapina, risperidona, paliperidona, olanzapina, quetiapina, amisulprido, ziprasidona, aripiprazol, brexpiprazol, asenapina, haloperidol, iloperidona, lurasidona, clorpromazina, blonanserina, perfenazina, levomepromazina, sulpirido, flufenazina, zuclopentixol, flupentixol y cariprazina; en donde
1) y 2) son para uso combinado en el tratamiento de un trastorno psiquiátrico y/o cognitivo.
E2(E1) en donde dicho trastorno psiquiátrico y/o cognitivo se selecciona del grupo que consiste en T rastorno de déficit de atención con hiperactividad (TDAH), depresión, ansiedad, narcolepsia, esquizofrenia, alteración cognitiva y alteración cognitiva asociada con esquizofrenia (CIAS).
E3(E1 y E2)
n es 0 o 1;
q es 0 o 1;
R1 se selecciona del grupo que consiste en alquilo C1-C8 lineal o ramificado, cicloalquilo C3-C8 monocíclico saturado, oxetanilo, tetrahidrofuranilo, y tetrahidropiranilo;
R2 se selecciona del grupo que consiste en, alquilo C1-C8 lineal o ramificado, fenilo, y cicloalquilo C3-C8 monocíclico saturado; o
R2 se selecciona del grupo que consiste en fenilo sustituido con una o más veces con uno o más sustituyentes seleccionados del grupo que consiste en halógeno, alquilo C1-C3 y metoxi;
R3 se selecciona del grupo que consiste en hidrógeno, alquilo C1-C3 y halógeno;
R4 es hidrógeno.
E4(E1 a E3) R1 es tetrahidropiranilo.
E5(E1 a E3) R1 es alquilo C1-C3 o cicloalquilo C3-C5.
E6(E1 y E5) R1 es propilo o ciclopropilo
E7(E1 a E6) R2 es fenilo.
E8(E1 a E6) R2 es fenilo sustituido, en donde el uno o más sustituyentes se seleccionan del grupo que consiste en flúor, cloro, metilo y metoxi.
E9(E1 a E6) R2 es cicloalquilo C3-C8 monocíclico saturado.
E10(E1 y E9) R2 es cicloalquilo C5-C7 monocíclico saturado.
E11 (E1 a E6) R2 es alquilo C1-C3.
E12(E1 y E11) R2 es metilo, etilo o isopropilo
E13(E1 a E12) R3 es bromo
E14(E1 a E12) R3 es metilo
E15(E1 a E2) R3 se selecciona de hidrógeno y metilo; y
R2 se selecciona del grupo que consiste en alquilo C1-C8 lineal o ramificado, fenilo, cicloalquilo C3-C8 monocíclico saturado, oxetanilo, tetrahidrofuranilo y tetrahidropiranilo; o
R2 es fenilo o piridilo sustituido con uno o más sustituyentes seleccionados del grupo que consiste en hidroxilo, amino, ciano, halógeno, alquilo C1-C3, alcoxi C1-C3, fluoroalcoxi C1-C3, y -NC(O)CH3; o
R2 es un heteroarilo de 5 miembros que puede estar sustituido con alquilo C1-C3.
E16(E1 a E15) n es 0.
E17(E1 a E15) n es 1.
E18(E1 a E16) q es 0.
E19(E1 a E16) q es 1.
E20(E1) el compuesto de Fórmula (I) se selecciona entre los compuestos listados en la Tabla 1, en la forma de la base libre, uno o más tautómeros del mismo o una sal farmacéuticamente aceptable del mismo.
E21 (E1 a E20) el compuesto de Fórmula (I) tiene un valor de CI50 para PDE1A, PDE1B o PDE1C, determinado como se describe en la sección "ensayo de inhibición de PDE1", de 10 micro molar o menos, tal como 5 micro molar o menos, tal como 4 micro molar o menos, tal como 3 micro molar o menos, tal como 2 micro molar o menos, tal como 1 micro molar o menos, tal como 500 nM o menos, tal como 400 nM o menos, tal como 300 nM o menos, tal como 200 nM o menos, tal como 100 nM o menos.
E22(E1) el compuesto de Formula (I) se selecciona de los compuestos listados en la Tabla 1 y sales farmacéuticamente aceptables del mismo.
E23(E1 a E22) el compuesto de Fórmula (I) es un inhibidor de PDE1 al menos 10 veces más potente que inhibidor de PDE9, tal como un inhibidor de PDE1 al menos 50 veces más potente que inhibidor de PDE9 o incluso un inhibidor de PDE1 al menos 100 veces más potente que inhibidor de PDE9.
E24 Una composición farmacéutica que comprende:
1) un compuesto de Fórmula (I) como se describe según cualquiera de las realizaciones (E1) a (E23); y
2) un segundo compuesto, compuesto que es útil en el tratamiento de un trastorno psiquiátrico como se define en las reivindicaciones; y
uno o más vehículos, diluyentes y excipientes farmacéuticamente aceptables.
E25(E24) la composición farmacéutica es para el tratamiento de un trastorno psiquiátrico y/o cognitivo.
E26 (no forma parte de la invención reivindicada)
Un método para el tratamiento de un sujeto que padece un trastorno psiquiátrico y/o cognitivo, método que comprende administrar a dicho sujeto:
1) una cantidad terapéuticamente efectiva de un compuesto de Fórmula (I) como se describe según cualquiera de las realizaciones (E1) a E23); y
2) una cantidad terapéuticamente efectiva de:
un segundo compuesto, compuesto que es útil en el tratamiento de un trastorno psiquiátrico.
E27 Una combinación de:
1) un compuesto de Fórmula (I) como se describe según cualquiera de las realizaciones (E1) a E23); y
2) un segundo compuesto, compuesto que es útil en el tratamiento de un trastorno psiquiátrico como se define en las reivindicaciones;
en donde dicha combinación es para uso en el tratamiento de un trastorno psiquiátrico y/o cognitivo.
E28 (no forma parte de la invención reivindicada) Uso de:
1) un compuesto de Fórmula (I) como se describe según cualquiera de las realizaciones (E1) a (E23); y
2) un segundo compuesto, compuesto que es útil en el tratamiento de un trastorno psiquiátrico;
en la fabricación de un medicamento para uso en el tratamiento de un trastorno psiquiátrico y/o cognitivo.
E29 (no forma parte de la invención reivindicada)
Uso de un compuesto de Fórmula (I) en la fabricación de un medicamento para uso en el tratamiento de un trastorno psiquiátrico y/o cognitivo; en donde dicho medicamento es para uso en combinación con un segundo compuesto, compuesto que es útil en el tratamiento de un trastorno psiquiátrico.
E30 Un kit que comprende:
1) un compuesto de Fórmula (I) como se describe según cualquiera de las realizaciones (E1) a (E23); y
2) un segundo compuesto, compuesto que es útil en el tratamiento de un trastorno psiquiátrico, como se define en las reivindicaciones.
E31 (E30) el kit comprende el compuesto de fórmula (I) y el compuesto listado con el ítem 2) en E1,
en cantidades terapéuticamente efectivas.
E32(E30 y E31) el kit es para el tratamiento de un trastorno psiquiátrico y/o cognitivo.
E33(E25 a E29 y E31) dicho trastorno psiquiátrico y/o cognitivo se selecciona del grupo que consiste en Trastorno de déficit de atención con hiperactividad (TDAH), depresión, ansiedad, narcolepsia, esquizofrenia, alteración cognitiva y alteración cognitiva asociada con esquizofrenia (CIAS).
E34(E2 y E33) dicho trastorno psiquiátrico y/o cognitivo es alteración cognitiva asociada con esquizofrenia (CIAS). E35(E1 a E34) (no forma parte de la invención reivindicada)
dicho segundo compuesto, compuesto que es útil en el tratamiento de un trastorno psiquiátrico es un agente antipsicótico.
E36(E1 a E35) (no forma parte de la invención reivindicada)
dicho segundo compuesto, compuesto que es útil en el tratamiento de un trastorno psiquiátrico tiene una actividad farmacológica seleccionada de uno o más de los siguientes mecanismos: antagonista/agonista inverso/modulador negativo/agonista parcial/inhibidor de una o más de las dianas receptor de dopamina D1, receptor de dopamina D2, receptor de dopamina D3, fosfodiesterasa PDE10, receptor de serotonina 5-HT2A, receptor de serotonina 5-HT6, y transportador de glicina GlyT1; o agonista/modulador positivo/agonista parcial de una o más de las dianas canal KCNQ, receptor NMDA, receptor AMPA y receptor nicotínico alfa-7.
E37(E1 a E35) dicho segundo compuesto, compuesto que es útil en el tratamiento de un trastorno psiquiátrico se selecciona de la lista que consiste en clozapina, risperidona, paliperidona, olanzapina, quetiapina, amisulprido, ziprasidona, aripiprazol, brexpiprazol, asenapina, haloperidol, iloperidona, lurasidona, clorpromazina, blonanserina, perfenazina, levomepromazina, sulpirido, flufenazina, zuclopentixol, flupentixol y cariprazina.
Definiciones
Enzimas PDE1
La familia de isozimas PDE1 incluye numerosas isoformas de PDE1 variantes de corte y empalme. Tiene tres subtipos, PDE1A, PDE1B y PDE1C que se dividen adicionalmente en diversas isoformas. En el contexto de la presente invención, PDE1 y enzimas PDE1 son sinónimos y se refieren a las enzimas PDE1A, PDE1B y PDE1C, así como a sus isoformas a no ser que se especifique otra cosa.
Sustituyentes
Tal y como se usa en el contexto de la presente invención, los términos "halo" y "halógeno" se usan indistintamente y se refieren a flúor, cloro, bromo o yodo.
Un rango dado puede indicarse indistintamente con (guion) o "a", p. ej., el término "alquilo C1-C3" es equivalente a "alquilo C1 a C3".
Los términos "alquilo C1-C3", "alquilo C1-C4", "alquilo C1-C5", "alquilo C1-C6", "alquilo C1-C7" y "alquilo C1-C8" se refieren a un hidrocarburo saturado lineal (es decir, no ramificado) o ramificado que tiene de uno hasta ocho átomos de carbono, inclusive. Los ejemplos de dichos grupos incluyen, pero no están limitados a, metilo, etilo, 1 -propilo, 2-propilo, 1 -butilo, 2-butilo, 2-metil-2-propilo, 2-metil-1 -butilo, n-hexilo, n-heptilo y n-octilo.
El término cicloalquilo C3-C8 monocíclico saturado se refiere a ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, cicloheptilo y ciclooctilo. El término cicloalquilo C3-C5 se refiere a ciclopropilo, ciclobutilo y ciclopentilo.
El término "alcoxi C1-C3" se refiere a un resto de la fórmula -OR', en donde R' indica alquilo C1-C3 como se ha definido anteriormente. El término "cicloalcoxi C3-C5" se refiere a un resto de la fórmula -OR', en donde R' indica cicloalquilo C3-C5 como se ha definido anteriormente. El término "cicloalquil C3-C5-metoxi" se refiere a un resto de la fórmula -OCH2R', en donde R' indica cicloalquilo C3-C5 como se ha definido anteriormente. Fluoroalcoxi C1-C3 se refiere a un alcoxi C1-C3 sustituido con uno o más flúor.
Los heteroarilos de 5 miembros se definen como anillos aromáticos de 5 miembros que contienen al menos un átomo seleccionado de nitrógeno, azufre y oxígeno. Los ejemplos incluyen, pero no están limitados a tiazol, tiofeno e isoxazol.
El término "lactamas que contienen 4-6 átomos de carbono" se refiere a pirrolidin-2-ona, piperidin-2-ona o azepan-2-ona.
Formas isoméricas
Cuando los compuestos de Fórmula (I) contienen uno o más centros quirales, la referencia a cualquiera de los compuestos abarcará, a no ser que se especifique otra cosa, el compuesto enantioméricamente o diastereoméricamente puro, así como mezclas de los enantiómeros o diastereómeros en cualquier proporción.
Lo anterior también se aplica cuando los compuestos de Fórmula (I) contienen más de dos centros quirales.
Inhibidores de PDE1 e inhibidores de PDE9
En el contexto de la presente invención, se considera que un compuesto de Fórmula (I) es un inhibidor de PDE1 si la cantidad requerida para alcanzar el nivel de CI50 de una o más de las tres isoformas de PDE1 es 10 micro molar o menos, preferiblemente menos de 9 micro molar, tal como 8 micro molar o menos, tal como 7 micro molar o menos, tal como 6 micro molar o menos, tal como 5 micro molar o menos, tal como 4 micro molar o menos, tal como 3 micro molar o menos, más preferiblemente 2 micro molar o menos, tal como 1 micro molar o menos, en particular 500 nM o menos. En una realización preferida, a cantidad requerida de inhibidor de PDE1 requerida para alcanzar el nivel de CI50 de PDE1B es 400nM o menos, tal como 300 nM o menos, 200 nM o menos, 100 nM o menos, o incluso 80 nM o menos, tal como 50 nM o menos, por ejemplo 25 nM o menos.
En una realización preferida, los compuestos de Fórmula (I) son al menos diez veces más potentes como inhibidores de PDE1 que como inhibidores de PDE9, es decir, la cantidad del compuesto requerida para alcanzar el nivel de CI50 de una o más de las tres isoformas de PDE1 es al menos diez veces menor que la cantidad del mismo compuesto requerida para alcanzar el nivel de CI50 de la enzima PDE9.
Sales farmacéuticamente aceptables
Las combinaciones de la presente invención también comprenden sales de los compuestos de Fórmula (I), típicamente, sales farmacéuticamente aceptables. Dichas sales incluyen sales de adición a ácido farmacéuticamente aceptables. Las sales de adición a ácido incluyen sales de ácidos inorgánicos, así como de ácidos orgánicos.
Los ejemplos representativos de ácidos inorgánicos adecuados incluyen ácidos clorhídrico, bromhídrico, yodhídrico, fosfórico, sulfúrico, sulfámico, nítrico y similares. Los ejemplos representativos de ácidos orgánicos adecuados incluyen ácidos fórmico, acético, tricloroacético, trifluoroacético, propiónico, benzoico, cinámico, cítrico, fumárico, glicólico, itacónico, láctico, metanosulfónico, maleico, málico, malónico, mandélico, oxálico, pícrico, pirúvico, salicílico, succínico, metanosulfónico, etanosulfónico, tartárico, ascórbico, pamoico, bismetilen salicílico, etanodisulfónico, glucónico, citracónico, aspártico, esteárico, palmítico, EDTA, glicólico, p-aminobenzoico, glutámico, bencenosulfónico, p-toluenosulfónico, ácidos teofilin acético, así como las 8-haloteofilinas, por ejemplo, 8-bromoteofilina y similares. Los ejemplos adicionales de sales de adición a ácido inorgánico u orgánico farmacéuticamente aceptables incluyen las sales farmacéuticamente aceptables listadas en Berge, S.M. et al., J. Pharm. Scí. 1977, 66, 2.
Además, los compuestos de Fórmula (I) pueden existir en formas no solvatadas, así como solvatadas con disolventes farmacéuticamente aceptables tales como agua, etanol y similares. En general, las formas solvatadas se consideran equivalentes a las formas no solvatadas para los propósitos de esta invención.
Cantidad terapéuticamente efectiva
En el presente contexto, el término "cantidad terapéuticamente efectiva" de un compuesto significa una cantidad suficiente para curar, aliviar o parar parcialmente las manifestaciones clínicas de una enfermedad dada y sus complicaciones en una intervención terapéutica que comprende la administración de dicho compuesto. Una cantidad adecuada para conseguir esto se define como "cantidad terapéuticamente efectiva". Las cantidades efectivas para cada propósito dependerán de la gravedad de la enfermedad o lesión, así como del peso y estado general del sujeto. Se entenderá que la determinación de una dosificación apropiada puede conseguirse usando experimentación rutinaria, construyendo una matriz de valores y ensayando diferentes puntos en la matriz, estando todo dentro de la experiencia de un médico experimentado.
Los métodos de la presente invención proporcionan la administración de combinaciones de compuestos. En dichos casos, una "cantidad efectiva" indica una cantidad de cada compuesto individual que, cuando dichos compuestos se proporcionan en una combinación, es suficiente para causar el efecto farmacológico pretendido. Una cantidad terapéuticamente efectiva de un compuesto cuando se administra en combinación con otro compuesto puede, en algunos casos, ser menor que una cantidad terapéuticamente efectiva de dicho compuesto cuando se administra solo.
Tratamiento y tratar
En el presente contexto, el término "tratamiento" y "tratar" significa la gestión y cuidado de un paciente con el propósito de combatir una afección, tal como una enfermedad o un trastorno. Se pretende que el término incluya el espectro completo de tratamientos para una afección dada que padece el paciente, tal como la administración del compuesto actico para aliviar los síntomas o complicaciones, para retrasar la progresión de la enfermedad, trastorno o afección, para aliviar o mitigar los síntomas y complicaciones, y/o para curar o eliminar la enfermedad, trastorno o afección.
En una realización, "tratamiento" y "tratar" también se refiere a la prevención de la afección, en donde prevención debe entenderse como la gestión y cuidado de un paciente con el propósito de combatir la enfermedad, afección, o trastorno e incluye la administración de los compuestos activos para prevenir el inicio de los síntomas o complicaciones. No obstante, los tratamientos profilácticos (preventivos) y terapéuticos son dos aspectos separados de la invención.
El paciente que se va a tratar es preferiblemente un mamífero, en particular un ser humano.
Composiciones farmacéuticas
La presente invención proporciona además una composición farmacéutica que comprende 1) un compuesto de Fórmula (I), tal como uno de los compuestos específicos descritos en la Sección Experimental en la presente memoria; y 2) un segundo compuesto, compuesto que es útil en el tratamiento de un trastorno psiquiátrico como se define en las reivindicaciones; y un vehículo o diluyente farmacéuticamente aceptable.
La presente invención también proporciona 1) una composición farmacéutica que comprende un compuesto de Fórmula (I), tal como uno de los compuestos específicos descritos en la Sección Experimental en la presente memoria, y un vehículo o diluyente farmacéuticamente aceptable; y 2) una composición farmacéutica que comprende un segundo compuesto, compuesto que es útil en el tratamiento de un trastorno psiquiátrico como se define en las reivindicaciones, y un vehículo o diluyente farmacéuticamente aceptable; en donde dichas composiciones farmacéuticas son para uso combinado en el tratamiento de un trastorno psiquiátrico y/o cognitivo.
Los compuestos de Fórmula (I) y/o los compuestos listados en el ítem 2); y/o las combinaciones de los mismos pueden administrarse en combinación adicional con vehículos, diluyentes o excipientes farmacéuticamente aceptables, en una única o múltiples dosis. Las composiciones farmacéuticas según la invención pueden formularse con vehículos o diluyentes farmacéuticamente aceptables, así como cualquier otro adyuvante y excipiente conocido según las técnicas convencionales, tales como las descritas en Remington: The Science and Practice of Pharmacy, 21a Edición, Gennaro, Ed., Mack Publishing Co., Easton, PA, 2005.
Las composiciones farmacéuticas pueden formularse específicamente para la administración por cualquier ruta adecuada, tal como las rutas oral, rectal, nasal, pulmonar, tópica (incluyendo bucal y sublingual), transdérmica y parenteral (incluyendo subcutánea, intramuscular e intravenosa). Se apreciará que la ruta dependerá de la condición general y edad del sujeto que se va a tratar, de la naturaleza de la afección que se va a tratar y del ingrediente activo.
Las composiciones farmacéuticas para administración oral incluyen formas de dosificación sólidas tales como cápsulas, comprimidos, grageas, píldoras, pastillas para chupar, polvos y gránulos. Cuando sea apropiado, las composiciones pueden prepararse con recubrimientos tales como recubrimientos entéricos o pueden formularse para que proporcionen una liberación controlada del ingrediente activo, tal como liberación sostenida o prolongada según métodos muy conocidos en la técnica. Las formas de dosificación líquidas para administración oral incluyen disoluciones, emulsiones, suspensiones, jarabes y elixires.
Las composiciones farmacéuticas para administración parenteral incluyen disoluciones, dispersiones, suspensiones o emulsiones inyectables estériles acuosas y no acuosas, así como polvos estériles para ser reconstituidos en disoluciones o dispersiones inyectables estériles antes de su uso. Otras formas adecuadas de administración incluyen,
pero no están limitadas a, supositorios, pulverizadores, pomadas, cremas, geles, inhalantes, parches e implantes dérmicos.
Las dosificaciones orales típicas varían de aproximadamente 0,001 a aproximadamente 100 mg/kg de peso corporal por día. Las dosificaciones orales típicas también varían de aproximadamente 0,01 a aproximadamente 50 mg/kg de peso corporal por día. Las dosificaciones orales típicas varían además de aproximadamente 0,05 a aproximadamente 10 mg/kg de peso corporal por día. Las dosificaciones orales se administran habitualmente en una o más dosificaciones, típicamente, una a tres dosificaciones por día. La dosificación exacta dependerá de la frecuencia y modo de administración, del sexo, edad, peso y condición general del sujeto tratado, de la naturaleza y gravedad de la afección tratada y de cualesquiera enfermedades concomitantes que se van a tratar y de otros factores evidentes para los expertos en la técnica.
Las formulaciones también pueden presentarse en una forma de dosificación unitaria por métodos conocidos para los expertos en la técnica. Para propósitos ilustrativos, una forma dosificación unitaria típica para administración oral puede contener de aproximadamente 0,01 a aproximadamente 1.000 mg, de aproximadamente 0,05 a aproximadamente 500 mg, o de aproximadamente 0,5 mg a aproximadamente 200 mg.
Los compuestos de Fórmula (I) se utilizan generalmente como la sustancia libre o como una sal farmacéuticamente aceptable de la misma. Un ejemplo es una sal de adición a ácido de un compuesto que tiene la misma utilidad que la de una base libre. Cuando un compuesto de Fórmula (I) contiene una base libre, dichas sales se preparan de una manera convencional tratando una disolución o suspensión de una base libre de Fórmula (I) con un ácido farmacéuticamente aceptable. Los ejemplos representativos de ácidos orgánicos e inorgánicos adecuados se han descrito anteriormente.
Para la administración parenteral, pueden emplearse disoluciones de los compuestos de Fórmula (I) en disolución acuosa estéril, propilen glicol acuoso o aceite de sésamo o cacahuete. Dichas disoluciones acuosas deben tamponarse adecuadamente, si es necesario, y el diluyente líquido debe hacerse isotónico previamente con disolución salina o glucosa suficiente. Las disoluciones acuosas son particularmente adecuadas para la administración intravenosa, intramuscular, subcutánea e intraperitoneal. Los compuestos de Fórmula (I) pueden incorporarse fácilmente en medios acuosos estériles usando técnicas estándar conocidas para los expertos en la técnica.
Los vehículos farmacéuticos adecuados incluyen diluyentes o rellenos sólidos inertes, disoluciones acuosas estériles y diversos disolventes orgánicos. Los ejemplos de vehículos sólidos incluyen lactosa, alabastro, sacarosa, ciclodextrina, talco, gelatina, agar, pectina, acacia, estearato de magnesio, ácido esteárico y éteres de alquilo inferior de celulosa. Los ejemplos de vehículos líquidos incluyen, pero no están limitados a, jarabe, aceite de cacahuete, aceite de oliva, fosfolípidos, ácidos grasos, aminas de ácidos grasos, polioxietileno y agua. De forma similar, el vehículo o diluyente puede incluir cualquier material de liberación sostenida conocido en la técnica, tal como mnoestearato de glicerilo o diestearato de glicerilo, solo o mezclado con una cera. Las composiciones farmacéuticas formadas combinando los compuestos de Fórmula (I) y un vehículo farmacéuticamente aceptable se administran entonces fácilmente en una variedad de formas de dosificación adecuadas para las rutas de administración descritas. Las formulaciones pueden presentarse convenientemente en forma de dosificación unitaria por métodos conocidos en la técnica farmacéutica.
Las formulaciones de la presente invención adecuadas para la administración oral pueden presentarse como unidades discretas tales como cápsulas o comprimidos, conteniendo cada una, una cantidad predeterminada del ingrediente activo, y opcionalmente un excipiente adecuado. Además, las formulaciones disponibles oralmente pueden estar en la forma de un polvo o gránulos, una disolución o suspensión en un líquido acuoso o no acuoso, o una emulsión líquida de aceite en agua o agua en aceite.
Si se usa un vehículo sólido para la administración oral, la preparación puede comprimirse, ponerse en una cápsula de gelatina dura en forma de polvo o gránulos o puede estar en la forma de una tableta o pastilla para chupar. La cantidad de vehículo sólido variará ampliamente, pero estará en el rango de aproximadamente 25 mg a aproximadamente 1 g por unidad de dosificación. Si se usa un vehículo líquido, la preparación puede estar en la forma de un jarabe, emulsión, cápsula de gelatina blanda o líquido inyectable estéril tal como una suspensión o disolución líquida acuosa o no acuosa.
Las composiciones farmacéuticas de la invención pueden prepararse por métodos convencionales en la técnica. Por ejemplo, los comprimidos pueden prepararse mezclando el ingrediente activo con adyuvantes y/o diluyentes habituales y comprimiendo posteriormente la mezcla en una máquina de preparación de comprimidos convencional para preparar comprimidos. Los ejemplos de adyuvantes o diluyentes comprenden: almidón de maíz, almidón de patata, talco, estearato de magnesio, gelatina, lactosa, gomas, y similares. Cualquier otro adyuvante o aditivo usado habitualmente para dichos propósitos tales como colorantes, saporíferos, conservantes etc. puede usarse siempre que sea compatible con los ingredientes activos.
Tratamiento de trastornos
Como se ha mencionado anteriormente, los compuestos de Fórmula (I) son inhibidores de la enzima PDE1 y como tales son útiles para tratar trastornos psiquiátricos y cognitivos asociados. Puede ser beneficioso combinar dichos
inhibidores de PDE1 con otro paradigma de tratamiento útil en el tratamiento de trastornos psiquiátricos y/o cognitivos. Por lo tanto, la invención se refiere a tratamientos de combinación en donde los compuestos de Fórmula (I) se combinan con otro compuesto útil en el tratamiento de dichos trastornos como se define en las reivindicaciones.
Dicho trastorno psiquiátrico y/o cognitivo puede seleccionarse, por ejemplo, del grupo que consiste en Trastorno de déficit de atención con hiperactividad (TDAH), depresión, ansiedad, narcolepsia, esquizofrenia, alteración cognitiva y alteración cognitiva asociada con esquizofrenia (CIAS). En una realización particular, dicho trastorno psiquiátrico y/o cognitivo es alteración cognitiva asociada con esquizofrenia (CIAS).
La invención proporciona así 1) un compuesto de Fórmula (I) o un tautómero o sal de adición a ácido farmacéuticamente aceptable del mismo; y 2) un segundo compuesto, compuesto que es útil en el tratamiento de un trastorno psiquiátrico como se define en las reivindicaciones; para uso combinado en el tratamiento de un trastorno psiquiátrico y/o cognitivo tal como Trastorno de déficit de atención con hiperactividad (TDAH), depresión, ansiedad, narcolepsia, esquizofrenia, alteración cognitiva y alteración cognitiva asociada con esquizofrenia (CIAS).
Combinaciones
Los términos "uso combinado", "en combinación con" y "una combinación de" y similares tal y como se usan en la presente memoria en el contexto del método de la invención que comprende la administración combinada de cantidades terapéuticamente efectivas de 1) un inhibidor de PDE1 de Fórmula (I), y 2) un segundo compuesto, compuesto que es útil en el tratamiento de un trastorno psiquiátrico como se define en las reivindicaciones; se pretende que signifiquen la administración de un inhibidor de PDE1 de Fórmula (I) simultáneamente o secuencialmente, en cualquier orden, junto con un compuesto listado en el ítem 2).
Los dos compuestos pueden administrarse simultáneamente o con un intervalo de tiempo entre las administraciones de los dos compuestos. Los dos compuestos pueden administrarse bien como parte de la misma formulación o composición farmacéutica, o en formulaciones o composiciones farmacéuticas separadas. Los dos compuestos pueden administrarse en el mismo día o en días diferentes. Pueden administrarse por la misma ruta, tal como por administración oral, o por depósito, o por inyección intramuscular o intraperitoneal, o por inyección intravenosa; o por rutas diferentes, en donde un compuesto se administra, por ejemplo, oralmente o se pone por depósito y el otro compuesto se inyecta, o en donde un compuesto se pone, por ejemplo, por depósito y el otro se administra oralmente o se inyecta. Los dos compuestos pueden administrarse por el mismo régimen o intervalo de dosificación, tal como una o dos veces al día, semanalmente, o mensualmente; o por diferentes regímenes de dosificación, por ejemplo, en donde uno se administra una vez al día y el otro se administra dos veces al día, semanalmente o mensualmente.
En algunos casos, el paciente que se va a tratar puede estar ya en tratamiento con uno o más compuestos listados en el ítem 2) cuando se inicia el tratamiento con un compuesto de Fórmula (I). En otros casos, el paciente puede estar ya en tratamiento con un compuesto de Fórmula (I) cuando se inicia el tratamiento con uno o más de los compuestos listados en el ítem 2). En otros casos, el tratamiento con un compuesto de Fórmula (I) y el tratamiento con un compuesto listado en el ítem 2) se inicia al mismo tiempo.
Compuestos para el tratamiento de combinación
Los agentes antipsicóticos se usan para tratar los síntomas de la esquizofrenia y del trastorno bipolar. Los antipsicóticos actuales reducen principalmente los síntomas positivos de la esquizofrenia, mientras sus efectos en los síntomas negativos y cognitivos son marginales. Todos los antipsicóticos actuales son bien antagonistas o agonistas parciales en el receptor de dopamina D2 a dosis terapéuticamente relevantes y se cree que esto es fundamental para su efecto antipsicótico (Seeman P; Can. J. Psychiatry; 2002; 47(1):27-38).
Además de los receptores de dopamina D2, la mayor parte de los antipsicóticos también tienen una afinidad relevante para otros receptores tales como otros receptores de dopamina, receptores serotonérgicos y receptores adrenérgicos. Los diferentes antipsicóticos varían entre sí en su afinidad para estos receptores, y se establece la hipótesis de que estas diferencias en la actividad de los receptores representan diferencias en la eficacia, seguridad, y tolerabilidad entre diferentes antipsicóticos. Los ejemplos de agentes antipsicóticos y sus mecanismos de acción se proporcionan más adelante.
Se propone que el efecto antipsicótico de la risperidona, olanzapina, quetiapina, y ziprasidona es principalmente a través del antagonismo del receptor de la dopamina D2 y serotonina (5-HT2A). Sin embargo, cada fármaco tiene efectos variados en estos y otros receptores. Se piensa que el antagonismo de los receptores 5-HT2A reduce el efecto secundario motor asociado con el antagonismo del receptor de dopamina D2 en el cuerpo estrado, mientras permanece el efecto de bloqueo de los receptores D2 en el área límbica no afectada. La mayor parte de los antipsicóticos denominados atípicos son antagonistas potentes de 5-HT2A y esto puede representar, en parte, menores efectos secundarios extrapiramidales y mejores efectos en los síntomas negativos de la esquizofrenia en comparación con los antipsicóticos convencionales.
La clozapina resalta por tener efecto en algunos pacientes que no responden a otros antipsicóticos. También presenta efectos antipsicóticos a una menor ocupación del receptor de dopamina D2 en comparación con la mayor parte de los demás fármacos antipsicóticos. Esto puede estar relacionado son su afinidad significativa por el receptor de dopamina
D1 además de los receptores de dopamina D2, pero también puede contribuir que también interacciona con varios otros receptores.
El aripiprazol y brexpiprazol tienen propiedades farmacológicas únicas respecto a los demás antipsicóticos atípicos. Son agonistas parciales en los receptores de dopamina D2; así, son antagonistas funcionales en presencia de altos niveles de dopamina endógena y, a la inversa, actúan como agonistas cuando están presentes cantidades mínimas de dopamina.
En el presente contexto, "un segundo compuesto, compuesto que es útil en el tratamiento de un trastorno psiquiátrico" es preferiblemente un agente antipsicótico. El mecanismo farmacológico para la acción de dicho compuesto puede seleccionarse, por ejemplo, de uno o más de los siguientes mecanismos: antagonista/agonista inverso/modulador negativo/agonista parcial/inhibidor de una o más de las dianas receptor de dopamina D1, receptor de dopamina D2, receptor de dopamina D3, fosfodiesterasa PDE10, receptor de serotonina 5-HT2A, receptor de serotonina 5-HT6, y transportador de glicina GlyT1; o agonista/modulador positivo/agonista parcial de una o más de las dianas canal KCNQ, receptor NMDA, receptor AMPA y receptor nicotínico alfa-7. En una realización, dicho "segundo compuesto, compuesto que es útil en el tratamiento de un trastorno psiquiátrico" es un compuesto que es un antagonista o agonista parcial del receptor de dopamina D2. En una realización, dicho "segundo compuesto, compuesto que es útil en el tratamiento de un trastorno psiquiátrico" es un compuesto que es un antagonista o agonista parcial del receptor de dopamina D2 y opcionalmente también presenta una actividad seleccionada de uno o más de los siguientes mecanismos: antagonista/agonista inverso/modulador negativo/agonista parcial/inhibidor de una o más de las dianas receptor de dopamina D1, receptor de dopamina D2, receptor de dopamina D3, fosfodiesterasa PDE10, receptor de serotonina 5-HT2A, receptor de serotonina 5-HT6, y transportador de glicina GlyT 1; o agonista/modulador positivo/agonista parcial de una o más de las dianas canal KCNQ, receptor NMDA, receptor AMPA y receptor nicotínico alfa-7.
Según la invención, "un segundo compuesto, compuesto que es útil en el tratamiento de un trastorno psiquiátrico" se selecciona de la lista que consiste en clozapina, risperidona, paliperidona, olanzapina, quetiapina, amisulprido, ziprasidona, aripiprazol, brexpiprazol, asenapina, haloperidol, iloperidona, lurasidona, clorpromazina, blonanserina, perfenazina, levomepromazina, sulpirido, flufenazina, zuclopentixol, flupentixol y cariprazina.
Los encabezamientos y subencabezamientos se usan en la presente memoria solo por conveniencia, y no deben considerarse de ninguna manera como limitantes de la invención.
Se pretende que el uso de cualquiera y todos los ejemplos, o lenguaje ejemplar (incluyendo "por ejemplo", "por ejemplo", "p.ej.", y "como tal") en la presente memoria descriptiva sea meramente para iluminar mejor la invención.
Compuestos de Fórmula (I)
Tabla 1: Compuestos de Fórmula (I)







nd significa "no determinado"
La Tabla 1 lista el valor de CI50 para la inhibición de PDE1 por los compuestos de fórmula (I). El valor de CI50 se refiere a la concentración (nM) del compuesto requerida para alcanzar una inhibición del 50 % de le enzima PDE1 a la concentración especificada de sustrato.
Para determinados compuestos, la inhibición de PDE se lista como % de inhibición a una determinada concentración. Para propósitos comparativos, la tabla también lista el % de inhibición de PDE9 a 10 pM.
Los ensayos de PDE1 y PDE9 se describen en la Sección Experimental.
Sección experimental
Preparación de los compuestos de Fórmula (I)

Los compuestos de Fórmula (I) pueden prepararse por los métodos descritos más adelante, junto con métodos sintéticos conocidos en la técnica de la química orgánica, o modificaciones que son familiares para los expertos en la técnica. Los materiales de partida usados en la presente memoria están disponibles comercialmente o pueden prepararse por métodos rutinarios conocidos en la técnica, tales como los métodos descritos en libros de referencia estándar, tales como "Compendium of Organic Synthetic Methods, Vol. I-XII" (publicado por Wiley-lnterscience, ISSN: 1934-4783). Los métodos preferidos incluyen, pero no están limitados a, los descritos más adelante.
Los esquemas son representativos de métodos útiles en la síntesis de los compuestos de fórmula (I). A no ser que se indique otra cosa, en los esquemas de reacción y discusión que siguen, R1-R4 son como se definen en la reivindicación 1.
Métodos generales:
Método 1:

Brevemente, los compuestos de fórmula (I) pueden prepararse a partir de dihidrocloruro de (3-cloropirazin-2-il)metanamina V disponible comercialmente (CAS: 867165-53-5). La reacción del dihidrocloruro de (3-cloropirazin-2-il)metanamina V con un derivado de ácido, ejemplificado por, pero no limitado a, un cloruro de ácido en condiciones apropiadas para la formación de amida, usando una base ejemplificada por, pero no limitada a, trietilamina y un disolvente/mezcla de disolventes tal como dimetilformamida y diclorometano rinde la amida IV. El intermedio III puede prepararse a partir de IV por tratamiento con cloruro de fosforilo en un disolvente tal como dioxano. La 8 cloroimidazo[1,5-a]pirazina III se convierte en imidazo[1,5-a]pirazin-8(7H)-ona II en condiciones de hidrólisis estándar ejemplificadas por, pero no limitadas a, ácido clorhídrico en una mezcla de disolventes tal como agua y 1,4-dioxano. El Compuesto I se forma a partir de imidazo[1,5-a]pirazin-8(7H)-ona II por tratamiento con un reactivo alquilante ejemplificado por, pero no limitado a, un bromuro de alquilo usando una base ejemplificada por, pero no limitada a, carbonato de potasio en un disolvente tal como dimetilformamida.
Método 2:

Brevemente, los compuestos de fórmula (I) pueden prepararse a partir de 5-am¡no-3-metox¡p¡razina-2-carbon¡tr¡lo X disponible comerc¡almente (CAS: 1137478-55-7). La reacc¡ón de 5-am¡no-3-metox¡p¡raz¡na-2-carbon¡tr¡lo X con d¡carbonato de di-ferc-butilo y un catalizador ejemplificado por, pero no limitado a, W,W-dimet¡lp¡rid¡n-4-am¡na en un disolvente tal como diclorometano proporciona la pirazina IX. La hidrogenación de IX con un catalizador ejemplificado por, pero no limitado a, Níquel de Raney bajo una atmósfera de hidrógeno en un disolvente tal como metanol rinde la amina VIII.
Los compuestos de fórmula VII pueden prepararse empleando los compuestos de fórmula VIII y un ácido carboxílico usando condiciones de formación de enlace amida estándar ejemplificadas por, pero no limitadas a, HATU (hexafluorofosfato de 3-óxido de 1-[B¡s(d¡met¡lam¡no)met¡leno]-1H-1,2,3-tr¡azolo[4,5-¿>]p¡rid¡n¡o), una base, ejemplificada por, pero no limitada a, trietilamina en un disolvente tal como diclorometano. Los compuestos de fórmula VII protegidos con Boc pueden desprotegerse en compuestos de fórmula VI usando condiciones de desprotección estándar ejemplificadas por, pero no limitadas a, ácido trifluoroacético en un disolvente tal como diclorometano. El tratamiento de los compuestos de fórmula VI con nitrito de isoamilo, yoduro de cobre y diyodometano en un disolvente tal como tetrahidrofurano rinde los compuestos de fórmula V . Los compuestos de fórmula V pueden convertirse en imidazopirazinas de fórmula IV por tratamiento con cloruro de fosforilo en un disolvente tal como 1,4-dioxano. Las imidazopirazinas de fórmula III se preparan a partir de IV usando condiciones de reacción de acoplamiento cruzado estándar ejemplificadas por, pero no limitadas a, una reacción de acoplamiento cruzado de Suzuki-Miyaura. Dichas condiciones para la reacción de acoplamiento cruzado se ejemplifican por, pero no se limitan a, el uso de éster de ácido borónico, carbonato de potasio como la base, una mezcla de 1,4-dioxano y agua como el disolvente y [1,1'-bis(d¡fen¡lfosf¡no)ferroceno]d¡cloropaladio(II) (Pd(dppf)Cl2) como el catalizador. Las ¡midazo[1,5-a]piraz¡n-8(7H)-onas de fórmula II se preparan tratando los compuestos de fórmula III con un ácido ejemplificado por, pero no limitado a, ácido clorhídrico en una mezcla de disolventes tal como agua y metanol. Las ¡midazo[1,5-a]piraz¡n-8(7H)-onas de fórmula I se preparan alquilando II con un reactivo de alquilación ejemplificado por, pero no limitado a, bromuro de alquilo usando una base ejemplificada por, pero no limitada a, carbonato de potasio en un disolvente tal como dimetilformamida.
Método 3:

Brevemente, los compuestos de fórmula (I) pueden prepararse a partir de 1 H-imidazol-5-carboxilato de metilo VI disponible comercialmente (CAS: 17325-26-7). La reacción del 1 H-imidazol-5-carboxilato de metilo VI con una cetona a-halogenada ejemplificada por, pero no limitada a, una a-clorocetona, bajo la influencia de una base ejemplificada por, pero no limitada a, carbonato de potasio en un disolvente tal como acetona rinde el imidazol V . El tratamiento del imidazol V con un reactivo de bromación ejemplificado por, pero no limitado a, W-bromosuccinimida (NBS) en presencia de un iniciador de radicales ejemplificado por, pero no limitado a, azobisisobutironitrilo (AIBN) proporciona el imidazol IV. Los compuestos de la fórmula III se forman por tratamiento del imidazol IV con acetato de amonio en un disolvente tal como 1,4-dioxano. Los compuestos de la fórmula II pueden prepararse a partir del intermedio III usando condiciones de reacción de acoplamiento cruzado estándar ejemplificadas por, pero no limitadas a, a una reacción de acoplamiento cruzado de Suzuki-Miyaura. Dichas condiciones para la reacción de acoplamiento cruzado se ejemplifican por, pero no se limitan a, el uso de un éster de ácido borónico, carbonato de potasio como la base, una mezcla de 1,4-dioxano y agua como el disolvente y Pd(dppf)Cl2 como el catalizador. En algunos ejemplos, R1 contiene un enlace carbonocarbono insaturado que puede reducirse por hidrogenación en condiciones conocidas para el experto en la técnica. Las imidazo[1,5-a]pirazin-8(7H)-onas de fórmula I se preparan por alquilación de II con un reactivo alquilante ejemplificado por, pero no limitado a, bromuro de alquilo usando una base ejemplificada por, pero no limitada a, carbonato de potasio en un disolvente tal como dimetilformamida.
Método 4:

Brevemente, los compuestos de fórmula (I) pueden prepararse a partir de 2-cloro-6-metilpirazina X disponible comercialmente (CAS: 38557-71-0). La reacción de 2-cloro-6-metilpirazina X con metóxido de sodio en metanol rinde
la pirazina IX. El W-óxido VIII puede prepararse a partir de IX, por tratamiento con un oxidante, no limitado a metaborato de sodio y peróxido de hidrógeno, en un disolvente tal como ácido acético. La reacción de VIII con una fuente de cianuro, tal como cianuro de trimetilsililo y bromuro de cinc(II), usando una base ejemplificada por, pero no limitada a, trietilamina y un disolvente/mezcla de disolventes tal como acetonitrilo rinde el cianuro VII. La reducción de VII, no limitada a níquel de Raney e hidrógeno, en presencia de Boc-anhídrido, produce el carbamato VI. La amina V puede liberarse por el uso de ácido trifluoroacético, pero no limitado a, a partir de VI. La reacción de la amina V con un derivado de ácido ejemplificado por, pero no limitado a, un cloruro de ácido en condiciones apropiadas para la formación de amida, usando una base ejemplificada por, pero no limitada a, trietilamina y un disolvente/mezcla de disolventes tal como dimetilformamida y diclorometano rinde la amida IV. El intermedio III puede prepararse a partir de IV por tratamiento con cloruro de fosforilo en un disolvente tal como dioxano. La 8-cloroimidazo[1,5-a]pirazina III se convierte en imidazo[1,5-a]pirazin-8(7H)-ona II en condiciones de hidrólisis estándar ejemplificadas por, pero no limitadas a, ácido clorhídrico en una mezcla de disolventes tal como agua y 1,4-dioxano.
El compuesto I se forma a partir de imidazo[1,5-a]pirazin-8(7H)-ona II por tratamiento con un reactivo alquilante ejemplificado por, pero no limitado a, un bromuro de alquilo usando una base ejemplificada por, pero no limitada a, carbonato de potasio en un disolvente tal como dimetilformamida.
Métodos analíticos
Los datos de LC-MS analítica se obtuvieron usando los métodos identificados a continuación.
Método 1: Se usó un sistema de LCMS Agilent 1200 con detector ELS. Columna: XBridge ShieldRP18, 5 pm, 50x2,1mm; Temperatura de la columna: 40 °C; Sistema de disolventes: A = agua/NH3*H2Ü (99,95:0,05) y B = acetonitrilo; Método: Elución con gradiente lineal con A:B = 99:1 hasta 0:100 en 3,4 minutos y con una velocidad de flujo de 0,8 mL/min.
Método 2: Se usó un instrumento de MS Shimadzu 20 equipado con una fuente de iones por fotoionización a presión atmosférica y un sistema Shimadzu LC-20AB. Columna: MERCK, RP-18e 25-2mm; Temperatura de la columna: 50 °C; Sistema de disolventes: A = agua/ácido trifluoroacético (99,9625:0375) y B = acetonitrilo /ácido trifluoroacético (99,981:0,019); Método: Una elución con gradiente lineal A:B = 95:5 hasta A:B=5:95 en 0,7 minutos, después A:B=5:95 durante 0,4 minutos, después con una elución con gradiente lineal hasta A:B 95:5 durante 0,4 minutos con una velocidad de flujo constante de 1,5 mL/min.
Método 3: Se usó un sistema de LCMS Agilent 1200 con detector ELS. Columna: Agilent TC-C18 5 pm; 2,1x50mm; Temperatura de la columna: 50 °C; Sistema de disolventes: A = agua/ácido trifluoroacético (99,9:0,1) y B = acetonitrilo/ácido trifluoroacético (99,95:0,05); Método: Elución con gradiente lineal con A:B = 99:1 hasta 0:100 en 4.0 minutos y con una velocidad de flujo de 0,8 mL/min.
Método 4: Se usó un sistema de LCMS Agilent 1200 con detector ELS. Columna: Agilent TC-C18 5 pm; 2,1x50mm; Temperatura de la columna: 50 °C; Sistema de disolventes: A = agua/ácido trifluoroacético (99,9:0,1) y B = acetonitrilo/ácido trifluoroacético (99,95:0,05); Método: Elución con gradiente lineal con A:B = 90:10 hasta 0:100 en 4.0 minutos y con una velocidad de flujo de 0,8 mL/min.
Método 5: Se usó una UPLC-MS Waters Acquity. Columna: Acquity UPLC BEH C18 1,7pm; 2.1x50mm; Temperatura de la columna: 60 °C; Sistema de disolventes: A = agua/ácido trifluoroacético (99,965:0,035) y B = acetonitrilo /agua/ácido trifluoroacético (94,965:5:0,035); Método: Elución con gradiente lineal con A:B = 90:10 hasta 0:100 en 1.0 minuto y con una velocidad de flujo de 1,2 mL/minuto.
Método 6: Se usó una UPLC-MS Waters Acquity. Columna: Acquity UPLC BEH C18 1,7pm; 2,1x50mm; Temperatura de la columna: 60 °C; Sistema de disolventes: A = agua/ácido fórmico (99,9:0,1) y B = acetonitrilo /agua/ácido fórmico (94,9:5:0,1); Método: Elución con gradiente lineal con A:B = 90:10 hasta 0:100 en 1,0 minuto y con una velocidad de flujo de 1,2 mL/minuto.
Método 7: Se usó una UPLC-MS Waters Acquity. Columna: Acquity UPLC HSS T3 C18 1,8pm; 2,1x50mm; Temperatura de la columna: 60 °C; Sistema de disolventes: A = agua/ácido trifluoroacético (99,965:0,035) y B = acetonitrilo/agua/ácido trifluoroacético (94,965:5:0,035); Método: Elución con gradiente lineal con A:B = 98:02 hasta 0:100 en 1.0 minuto y con una velocidad de flujo de 1,2 mL/min.
Método 8: Se usó un sistema de LCMS Agilent 1200 con detector ELS. Phenomenex Luna-C18, 5pm; 2,0x50mm; Temperatura de la columna: 50 °C; Sistema de disolventes: A = agua/ácido trifluoroacético (99,9:0,1) y B = acetonitrilo/ácido trifluoroacético (99,95:0,05); Método: Elución con gradiente lineal con A:B = 99:1 hasta 0:100 en 4.0 minutos y con una velocidad de flujo de 0,8 mL/min.
Método 9: Se usó un sistema de LCMS Agilent 1200 con detector ELS. Columna: Xtimate C18 2,1*30mm,3um; 2,0x50mm; Temperatura de la columna: 50 °C; Sistema de disolventes: A = agua/ácido trifluoroacético (99,9996:0,0004) y B = acetonitrilo/ácido trifluoroacético (99,9998:0,0002); Método: Elución con gradiente lineal con A:B = 100:0 hasta 70:30 en 3,0 minutos y con una velocidad de flujo de 0,8 mL/min.
Método 10: Se usó un sistema de LCMS Agilent 1200 con detector ELS. Columna: Xtimate C18 2,1*30mm,3um; 2,0x50mm; Temperatura de la columna: 50 °C; Sistema de disolventes: A = agua/ácido trifluoroacético (99,9996:0,0004) y B = acetonitrilo/ácido trifluoroacético (99,9998:0,0002); Método: Elución con gradiente lineal con A: B = 100:0 hasta 40:60 en 1,5 minutos y con una velocidad de flujo de 1,2 mL/min.
Método 11: Se usó un sistema de LCMS Agilent 1200 con detector ELS. Columna: Waters XBridge ShieldRP18,2,1*50mm,5gm; Temperatura de la columna: 40 °C; Sistema de disolventes: A = agua/amoniaco (99,95:0,05) y B = acetonitrilo; Método: Elución con gradiente lineal con A:B = 95:5 hasta 0:100 en 4,0 minutos y con una velocidad de flujo de 0,8 mL/min.
Método 12: Se usó un sistema de LCMS Agilent 1100 con detector ELS. Columna: YMC ODS-AQ 5 gm; 2,0x50mm; Temperatura de la columna: 50 °C; Sistema de disolventes: A = agua/ácido trifluoroacético (99,9:0,1) y B = acetonitrilo/ácido trifluoroacético (99,95:0,05); Método: Elución con gradiente lineal con A:B = 99:1 hasta 5:95 en 3,5 minutos y con una velocidad de flujo de 0,8 mL/min.
Método 13: Se usó un sistema de LCMS Agilent 1200 con detector ELS. Phenomenex Luna-C18, 5gm; 2,0x50mm; Temperatura de la columna: 50 °C; Sistema de disolventes: A = agua/ácido trifluoroacético (99,9:0,1) y B = acetonitrilo/ácido trifluoroacético (99,95:0,05); Método: Elución con gradiente lineal con A:B = 99:1 hasta 0:100 en 4.0 minutos y con una velocidad de flujo de 0,8 mL/min.
Método 14: Se usó un sistema de LCMS Agilent 1200 con detector ELS. Columna: Xtimate C18 2,1*30mm,3um; 2,0x50mm; Temperatura de la columna: 50 °C; Sistema de disolventes: A = agua/ácido trifluoroacético (99,9996:0,0004) y B = acetonitrilo/ácido trifluoroacético (99,9998:0,0002); Método: Elución con gradiente lineal con A:B = 100:0 hasta 40:60 en 6,0 minutos y con una velocidad de flujo de 0,8 mL/min.
Método 15: Se usó un sistema de LCMS Agilent 1200 con detector ELS. Columna: MERCK, RP-18e 25-2mm; Temperatura de la columna: 50 °C; Sistema de disolventes: A = agua/ácido trifluoroacético (99,9996:0,0004) y B = acetonitrilo/ácido trifluoroacético (99,9998:0,0002); Método: Elución con gradiente lineal con A:B = 95:5 hasta 5:95 en 0,7 minutos y con una velocidad de flujo de 1,5 mL/min.
Método 16: Se usó un sistema de LCMS Agilent 1200 con detector ELS. Columna: Xtimate C18 2,1*30mm,3um; 2,0x50mm; Temperatura de la columna: 50 °C; Sistema de disolventes: A = agua/ácido trifluoroacético (99,9996:0,0004) y B = acetonitrilo/ácido trifluoroacético (99,9998:0,0002); Método: Elución con gradiente lineal con A:B = 100:0 hasta 40:60 en 0,9 minutos y con una velocidad de flujo de 1,2 mL/min.
Método 17: Se usó un sistema de LCMS Agilent 1200 con detector ELS. Phenomenex Luna-C18, 5gm; 2,0x50mm; Temperatura de la columna: 50 °C; Sistema de disolventes: A = agua/ácido trifluoroacético (99,9:0,1) y B = acetonitrilo/ácido trifluoroacético (99,95:0,05); Método: Elución con gradiente lineal con A:B = 90:10 hasta 0:100 en 4.0 minutos y con una velocidad de flujo de 0,8 mL/min.
Método 18: Se usó un sistema de LCMS Agilent 1200 con detector ELS. Columna: Waters XBridge ShieldRP18,2,1*50mm,5gm; Temperatura de la columna: 40 °C; Sistema de disolventes: A = agua/amoniaco (99,95:0,05) y B = acetonitrilo; Método: Elución con gradiente lineal con A:B = 85:15 hasta 0:100 en 3,4 minutos y con una velocidad de flujo de 0,8 mL/min.
Método 19: Se usó una UPLC-MS Waters Acquity. Columna: Acquity UPLC BEH C18 1,7gm; 2,1x50mm; Temperatura de la columna: 60 °C; Sistema de disolventes: A = agua/ácido fórmico (99,9:0,1) y B = acetonitrilo/agua/ácido fórmico (94,9:5:0,1); Método: Elución con gradiente lineal con A:B = 98:2 hasta 0,1:99,9 en 1,0 minuto y con una velocidad de flujo de 1,2 mL/minuto.
La purificación por LC-MS preparativa se realizó en un instrumento PE Sciex API 150EX con ionización química a presión atmosférica. Columna: 50 X 20 mm YMC ODS-A con un tamaño de partícula de 5 gm; Sistema de disolventes: A = agua/ácido trifluoroacético (99,965:0,035) y B = acetonitrilo/agua/ácido trifluoroacético (94,965:5:0,035); Método: Elución con gradiente lineal con A:B = 80:20 hasta 0:100 en 7 minutos y con una velocidad de flujo de 22,7 mL/minuto. La recogida de fracciones se realizó por detección por MS de flujo dividido.
La SFC preparativa se realizó en u instrumento Thar 80. Las condiciones ejemplificadas pueden ser, pero no están limitadas a: Columna AD 250 X 30mm con un tamaño de partícula de 20 gm; Temperatura de la columna: 38 °C, Fase móvil: CO2 supercrítico/EtOH(NH3H2O al 0,2 %) =45/55.
Intermedios:
W-((3-cloropirazin-2-il)metil)butiramida:
A una disolución enfriada en hielo de (3-cloropirazin-2-il)metanamina (2,0 g, 14 mmoles) en diclorometano (50 mL) y dimetilformamida (10mL) se añadió trietilamina (4,5 g, 45 mmoles), seguido de cloruro de butirilo (2,0 g, 14 mmoles). La reacción se dejó calentar hasta temperatura ambiente y se agitó durante 1 hora. La mezcla de reacción se paró con agua y se extrajo con diclorometano (2 x 250 mL). Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 y se concentraron en vacío para rendir 2,4 g de W-((3-cloropirazin-2-il)metil)butiramida (81 %).
8-Cloro-3-propilimidazo[1,5-a]pirazina:

A una disolución de W-((3-cloropirazin-2-il)metil)butiramida (2,4 g, 11 mmoles) en 1,4-dioxano (20 mL) se añadió POCh (3,44 g, 22,5 mmoles). La mezcla se agitó a 100 °C durante 2 hrs y entonces se enfrió en un baño de hielo. Se añadió cuidadosamente NaHCO3 ac. saturado y la mezcla se extrajo con diclorometano (2 x 50 mL). Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 y se concentraron en vacío para proporcionar 2 g de 8-cloro-3-propilimidazo[1,5-a]pirazina (63 %).
3-Propilimidazo[1,5-a]pirazin-8(7H)-ona:

Una disolución de 3-propilimidazo[1,5-a]pirazin-8(7H)-ona (100 mg, 0,51 mmoles) en una mezcla de 1,4-dioxano (10 mL) y H2O (4mL) se agitó a 100 °C durante 2 horas. La mezcla de reacción se concentró en vacío y el residuo se diluyó con diclorometano (50 mL), se lavó con NaHCO3(ac), después salmuera, se secó sobre Na2SO4 y se concentró en vacío para proporcionar 50 mg de 3-propilimidazo[1,5-a]pirazin-8(7H)-ona (55 %).
1-(2-Oxopropil)-1 H-imidazol-5-carboxilato de metilo:

Una mezcla de 1 H-imidazol-5-carboxilato de metilo (20 g, 0,16 moles), 1-cloropropan-2-ona (22 g, 0,24 moles), y carbonato de potasio (44 g, 0,32 moles) en acetona (400 mL) se agitó a 30 °C durante 12 horas. La mezcla de reacción se concentró en vacío, el residuo se diluyó con acetato de etilo (200 mL) y se lavó con H2O (3 x 50 mL). La capa orgánica se secó sobre Na2SO4 y se concentró en vacío. El residuo se purificó por cromatografía flash usando un gradiente de éter de petróleo y acetato de etilo para proporcionar 10 g de 1 -(2-oxopropil)-1 H-imidazol-5-carboxilato de metilo (35 %).
2-Bromo-1-(2-oxopropil)-1 H-imidazol-5-carboxilato de metilo:

Una mezcla de 1-(2-oxopropil)-1H-imidazol-5-carboxilato de metilo (10 g, 55 mmoles), N- bromosuccinimida (12,7 g, 71,4 mmoles) y azobisisobutironitrilo (1,8 g, 11 mmoles) en cloroformo (100 mL) se agitó a 50 °C durante 12 horas. La
mezcla se concentró en vacío. El residuo se purificó por cromatografía flash usando un gradiente de éter de petróleo y acetato de etilo para proporcionar 13 g de 2-bromo-1-(2-oxopropil)-1 H-imidazol-5-carboxilato de metilo (91 %).
3-Bromo-6-metilimidazo[1,5-a]pirazin-8(7H)-ona:

Una mezcla de 2-bromo-1-(2-oxopropil)-1H-imidazol-5-carboxilato de metilo (14 g, 50 mmoles) y acetato de amonio (16,5 g, 215 mmoles) en 1,4-dioxano (150 mL) se agitó a 60 °C durante 12 horas. La mezcla se agitó entonces a 90 °C durante otras 24 horas. La mezcla de reacción se concentró en vacío y el residuo se diluyó con acetato de etilo (600 mL) y se lavó con agua (3 x 100 mL). Las fases orgánicas combinadas se secaron con Na2SO4 anhidro y se concentraron en vacío. El residuo se purificó por cromatografía flash usando un gradiente de éter de petróleo y acetato de etilo para proporcionar 4,8 g de 3-bromo-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (39 %).
3-(3,6-Dihidro-2H-piran-4-il)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona:

Una mezcla de 3-bromo-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (4,5 g, 20 mmoles), 2-(3,6-dihidro-2H-piran-4-il)-4,4,5,5-tetrametil-1,3,2-dioxaborolano (4,97 g, 23,7 mmoles), Pd(dppf)Cl2 (2,9 g, 3,95 mmoles), carbonato de potasio (5,5 g, 39 mmoles) y H2O (10 mL) en 1,4-dioxano (40 mL) se agitó a 100 °C durante 12 horas. La mezcla se filtró y el filtrado se concentró en vacío. El residuo se purificó por cromatografía flash usando un gradiente de diclorometano y metanol para proporcionar 4,0 g de 3-(3,6-dihidro-2H-piran-4-il)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (88 %).
6-Metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:

Una mezcla de 3-(3,6-dihidro-2H-piran-4-il)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (4,0 g, 17 mmoles) y Pd/C al 10 % (300 mg) en tetrahidrofurano (15 mL) se agitó a 15 °C durante 7 hrs bajo una atmósfera de hidrógeno. La mezcla de reacción se filtró y el filtrado se concentró en vacío para rendir 3,5 g de 6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (87 %).
(5-Ciano-6-metoxipirazin-2-il)carbamato de ferc-butilo:

Una disolución de 5-amino-3-metoxipirazina-2-carbonitrilo (4,70 g, 31,3 mmoles), dicarbonato de di-ferc-butilo (8,9 g, 41 mmoles), W,W-dimetilpiridin-4-amina (38 mg, 0,31 mmoles) en diclorometano (150 mL) se agitó a 30 °C durante 12 horas. La mezcla de reacción se concentró en vacío. El residuo se purificó por cromatografía flash usando un gradiente de éter de petróleo y acetato de etilo para rendir 9,0 g de (5-ciano-6-metoxipirazin- 2-il)carbamato de fercbutilo (80 %).
(5-(Aminometil)-6-metoxipirazin-2-il)carbamato de ferc-butilo:

Una mezcla de (5-ciano-6-metoxipirazin-2-il)carbamato de ferc-butilo (9,0 g, 36 mmoles), Ni de Raney de malla 40-60 (5 g) y NH3 sat. en metanol (2 mL) en metanol (100 mL) se agitó a 30 °C durante 12 hrs bajo H2 (45 psi). La reacción se filtró y se concentró en vacío para rendir 10 g de (5-(aminometil)-6-metoxipirazin-2-il)carbamato de ferc-butilo, suficientemente puro para la siguiente etapa.
(6-Metoxi-5-((tetrahidro-2H-piran-4-carboxamido)metil)pirazin-2-il)carbamato de ferc-butilo:

Una disolución de (5-(aminometil)-6-metoxipirazin-2-il)carbamato de ferc-butilo (10,0 g, 31,5 mmoles), ácido tetrahidro-2H-piran-4-carboxílico (4,50 g, 34,6 mmoles), trietilamina (6,37 g, 62,9 mmoles) y hexafluorofosfato de 3-óxido de 1-[bis(dimetilamino)metileno]-1 H-1,2,3-triazolo[4,5-b]piridinio (13,2 g, 34,6 mmoles) en diclorometano (120 mL) se agitó a 30 °C durante 12 horas. La mezcla de reacción se concentró en vacío y el residuo se purificó por cromatografía flash usando un gradiente de éter de petróleo y acetato de etilo para rendir 8 g de (6-metoxi-5-((tetrahidro-2H-piran-4-carboxamido)metil)pirazin-2-il)carbamato de ferc-butilo (69,4 %).
W-((5-Amino-3-metoxipirazin-2-il)metil)tetrahidro-2H-piran-4-carboxamida:

Una disolución de (6-metoxi-5-((tetrahidro-2H-piran-4-carboxamido)metil)pirazin-2-il)carbamato de ferc-butilo (8 g, 21,8 mmoles) y ácido trifluoroacético (40 mL) en diclorometano (40 mL) se agitó a 30 °C durante 12 horas. La mezcla de reacción se concentró en vacío. El residuo se diluyó con diclorometano (100 mL), y se lavó con NaHCO3 hasta pH=8. La capa orgánica se lavó con agua (3 x 20 mL), se secó y se concentró en vacío. El residuo se purificó por cromatografía flash usando un gradiente de éter de petróleo y acetato de etilo para rendir 4 g de W-((5-amino-3-metoxipirazin-2-il)metil)tetrahidro-2H-piran-4- carboxamida (65,4 %).
W-((5-Yodo-3-metoxipirazin-2-il)metil)tetrahidro-2H-piran-4-carboxamida:

Una disolución de /V-((5-amino-3-metoxipirazin-2-il)metil)tetrahidro-2H-piran-4-carboxamida (2,40 g, 9,01 mmoles), yoduro de cobre(I) (1,72 g, 9,01 mmoles), nitrito de isoamilo (1,58 g, 13,5 mmoles) y diyodometano (2,41 g, 9,01 mmoles) en tetrahidrofurano (50 mL) se agitó a 75 °C durante 6 horas. La mezcla se filtró y se concentró en vacío. El residuo se purificó por cromatografía flash usando un gradiente de éter de petróleo y acetato de etilo para rendir 2 ,20 g de W-((5-yodo-3-metoxipirazin-2-il)metil)tetrahidro-2H-piran-4-carboxamida (64,7 %).
6-Yodo-8-metoxi-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazina:

A una disolución de W-((5-yodo-3-metoxipirazin-2-il)metil)tetrahidro-2H-piran-4- carboxamida (2 g, 5.30 mmoles) en 1,4-dioxano (60 mL) se añadió cloruro de fosforilo (8,13 g, 53,0 mmoles) a 0 °C. La reacción se agitó a 85 °C durante 12 horas. La mezcla se concentró en vacío. El residuo se diluyó con diclorometano (100 mL) y agua helada (60 mL), seguido de NaHCO3 acuoso saturado (30 mL). La fase orgánica se separó y la fase acuosa se extrajo con diclorometano (3 x 20 mL). Las fases orgánicas combinadas se secaron y se concentraron en vacío. El residuo se purificó por cromatografía flash usando un gradiente de éter de petróleo y acetato de etilo para rendir 6-yodo-8-metoxi-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazina 500 mg (23,6 %).
6-Bencil-8-metoxi-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazina:

Una mezcla de 6-yodo-8-metoxi-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazina (500 mg, 1,39 mmoles), 2-bencil-4,4,5,5-tetrametil-1,3,2-dioxaborolano (911 mg, 4,18 mmoles), Pd(dppf)Cl2 (51 mg, 0,07 mmoles), K2CO3 (577 mg, 4,18 mmoles) y H2O (3 mL) en 1,4-dioxano (15 mL) se agitó a 80 °C durante 12 hrs bajo una atmósfera de N2. Después, se filtró y el filtrado se concentró en vacío. El residuo se purificó por cromatografía flash usando un gradiente de éter de petróleo y acetato de etilo para rendir 260 mg de 6-bencil-8-metoxi-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazina (52 %).
6-Bencil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:

Una disolución de 6-bencil-8-metoxi-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazina (320 mg, 0,990 mmoles) y HCl ac. 2 M (8 mL) en metanol (20 mL) se agitó a 60 °C durante 12 horas. La disolución se concentró en vacío. El residuo se purificó por cromatografía flash usando un gradiente de éter de petróleo y acetato de etilo para rendir 230 mg de 6-bencil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (68 %).
W-((3-cloropirazin-2-il)metil)tetrahidro-2H-piran-4-carboxamida:

A una disolución de dihidrocloruro de (3-cloropirazin-2-il)metanamina (3,8 g, 18 mmoles) en DMF anhidro (20 mL) se añadió trietilamina (5,7g, 56 mmoles). La mezcla se enfrió hasta 0 °C, se añadió gota a gota cloruro de tetrahidro-2H-piran-4-carbonilo (2,9 g, 19 mmoles). La mezcla se agitó a 0 °C durante 0,5 horas. La mezcla de reacción se diluyó con agua (50 mL) y se extrajo con acetato de etilo (3 x 80 mL). Las fases orgánicas combinadas se lavaron con
salmuera (50 mL), se secaron sobre Na2SÜ4 y se concentraron en vacío. El residuo se purificó por cromatografía flash eluyendo con acetato de etilo para rendir 2,4 g de W-((3-cloropirazin-2-il)metil)tetrahidro-2H-piran-4-carboxamida (54 %).
8-Cloro-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazina:

A una disolución de W-((3-cloropirazin-2-il)metil)tetrahidro-2H-piran-4-carboxamida (2,5 g, 9,8 mmoles) en 1,4-dioxano anhidro (20 mL) se añadió cloruro de fosforilo (3,4 g, 22 mmoles). La reacción se agitó a 80 °C durante 2 horas. Después, la disolución se enfrió y se vertió en agua (100 mL), el pH se ajustó a 8-9 por la adición de K2CO3 acuoso saturado. La mezcla cruda se extrajo con acetato de etilo (2 x 100 mL). Las fases orgánicas combinadas se lavaron con salmuera (50 mL), se secaron sobre Na2SO4 y se concentraron en vacío. El residuo se purificó por cromatografía flash usando un gradiente de éter de petróleo y acetato de etilo para rendir 2,1 g de 8-cloro-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazina (90 %).
3-(Tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:
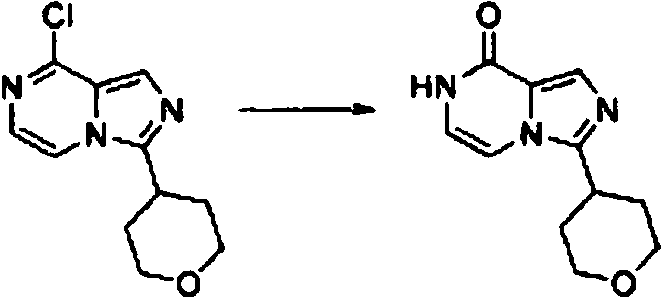
A una disolución de 8-cloro-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazina (2,1 g, 8,8 mmoles) en 1,4-dioxano (20 mL) se añadió HCl ac. 2 M (10 mL). La disolución se agitó a 80 °C durante 2 horas. La mezcla se enfrió y el pH se ajustó a 8-9 por la adición de K2CO3 acuoso saturado. La mezcla cruda se concentró en vacío y el residuo se disolvió en metanol (150 mL) y se filtró. El filtrado se concentró en vacío y el residuo se purificó por cromatografía flash usando una mezcla de diclorometano y metanol (10:1) para proporcionar 1,6 g de 3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (81 %).
1H RMN (DMSO-d6, 400 MHz): 5 10,55 (s, 1H), 7,63 (s, 1H), 7,39 (d, J = 6,0 Hz, 1H), 6,60 (s, 1H), 3,91-3,88 (m, 2H), 3,49-3,42 (m, 2H), 3,34-3,29 (m, 1H), 1,82-1,72 (m, 4H).
LC-MS: (m/z) 220,1 (MH+) tR (minutos, método 3) = 1,37 minutos
W-((3-Cloropirazin-2-il)metil)ciclopropanocarboxamida:

A una disolución de dihidrocloruro de (3-cloropirazin-2-il)metanamina (4,0 g, 19 mmoles) en DMF anhidro (20 mL) se añadió Et3N (1,9 g, 18,5 mmoles). La mezcla se enfrió hasta 0°C y se añadió gota a gota cloruro de ciclopropanocarbonilo (2,3 g, 22 mmoles). La reacción se agitó a 0 °C durante 0,5 horas. La mezcla de reacción se diluyó con agua (50 mL), se extrajo con acetato de etilo (2 x 100 mL). Las fases orgánicas combinadas se lavaron con salmuera (40 mL), se secaron sobre Na2SO4 y se concentraron en vacío. El residuo se purificó por cromatografía flash eluyendo con éter de petróleo/acetato de etilo 2/1 para rendir 3,3 g de W-((3-cloropirazin-2-il)metil)ciclopropanocarboxamida (85 %).
8-Cloro-3-ciclopropilimidazo[1,5-a]pirazina:

A una disolución de N-((3-cloropirazin-2-il)metil)ciclopropanocarboxamida (3,3 g, 15,6 mmoles) en 1,4-dioxano anhidro (30 mL) se añadió cloruro de fosforilo (5,3 g, 35 mmoles). La reacción se agitó a 80 °C durante 2 horas. Después, la disolución se enfrió en un baño de hielo y se vertió en agua (50 mL). El pH se ajustó a 8-9 por la adición de K2CO3 acuoso saturado. La mezcla se extrajo con acetato de etilo (2 x 50 mL). Las fases orgánicas combinadas se lavaron con salmuera (50 mL), se secaron sobre Na2SO4 y se concentraron en vacío. El residuo se purificó por cromatografía flash usando éter de petróleo/acetato de etilo 3:1 para rendir 2,4 g de 8-cloro-3-ciclopropilimidazo[1,5-a]pirazina (80 %).
3-Ciclopropilimidazo[1,5-a]pirazin-8(7H)-ona:

A una disolución de 8-cloro-3-ciclopropilimidazo[1,5-a]pirazina (2,5 g, 13 mmoles) en 1,4-dioxano (20 mL) se añadió HCl ac. 2 M (10 mL). La disolución se agitó a 80 °C durante 2hrs. La mezcla se enfrió en un baño de hielo y el pH se ajustó a 8-9 por la adición de K2CO3 acuoso saturado. La mezcla se concentró en vacío y el residuo se disolvió en metanol (150 mL) y se filtró. El filtrado se concentró en vacío y el residuo se purificó por cromatografía flash usando diclorometano/metanol (10/1) para rendir 1,9 g de 3-ciclopropilimidazo[1,5-a]pirazin-8(7H)-ona (83 %).
1H RMN (DMSO-ds, 400 MHz): 5 10,49 (s, 1H), 7,54 (s, 1H), 7,41 (d, J = 5,6 Hz, 1H), 6,61 (d, J = 5,6 Hz, 1H), 2,29 2,24 (m, 1H), 0,99-0,89 (m, 4H).
LC-MS: (m/z) 176,1 (MH+) tR (minutos, método 1) = 1,04 minutos
N-((5-Bromo-3-metoxipirazin-2-il)metil)tetrahidro-2H-piran-4-carboxamida:

Una disolución de NaNO2 (972 mg, 14,09 mmoles) en H2O (100 mL) se añadió a una disolución agitada de N-((5-amino-3-metoxipirazin-2-il)metil)tetrahidro-2H-piran-4-carboxamida (2,5 g, 9,4 mmoles) en HBr ac. al 40 % (33 mL) a 0 °C. Después de agitar durante 1,5 hrs, se añadió CuBr (2,02 g, 14,1 mmoles) y la mezcla se agitó a 70 °C durante 1 hora. El valor del pH se ajustó a pH 8 por la adición de NaHCO3 acuoso saturado. La mezcla se filtró y el filtrado se concentró en vacío. El residuo se purificó por cromatografía flash usando un gradiente de éter de petróleo y acetato de etilo para rendir 800 mg de N-((5-bromo-3-metoxipirazin-2-il)metil)tetrahidro-2H-piran-4-carboxamida (25 %).
LC-MS: (m/z) 331,8 (MH+) tR (minutos, método 2) = 0,723 minutos
6-Bromo-8-metoxi-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazina:
A una disolución de W-((5-bromo-3-metoxipirazin-2-il)metil)tetrahidro-2H-piran-4- carboxamida (800 mg, 2,42 mmoles) en 1,4-dioxano (30 mL) se añadió cloruro de fosforilo (3,8 g, 25 mmoles) a 0 °C. La mezcla se calentó hasta 70 °C y se agitó durante 1 hora. La mezcla se concentró en vacío y el residuo se diluyó con diclorometano (100 mL) y agua helada (60 mL). El valor del pH se ajustó a pH 8 por la adición de NaHCO3 acuoso saturado. La fase orgánica se separó y la fase acuosa se extrajo con diclorometano (3 x 30 mL). Las fases orgánicas combinadas se secaron y se concentraron en vacío. El residuo se purificó por cromatografía flash usando un gradiente de diclorometano y metanol para rendir 500 mg de 6-bromo-8-metoxi-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazina (66 %).
LC-MS: (m/z) 313,7 (MH+) tR (minutos, método 2) = 0,740 minutos
6-Bromo-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:

A una disolución de 6-bromo-8-metoxi-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazina (200 mg, 0,641 mmoles) en diclorometano (30 mL) se añadió tribromuro de boro (1,61 g, 6,41 mmoles) a 0 °C. La reacción se calentó hasta 20 °C y se agitó durante 3 horas. La disolución se paró con agua (2 mL) a 0 °C. La reacción se concentró en vacío y el residuo se purificó por cromatografía flash usando un gradiente de diclorometano y metanol para rendir 130 mg de 6-bromo-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (68 %).
LC-MS: (m/z) 299,7 (MH+) tR (minutos, método 2) = 0,730 minutos
2 -metoxi-6-metilpirazina

A una suspensión de 2-cloro-6-metilpirazina (24 g, 186,7 mmoles) en MeOH anhidro (240 mL) se añadió NaOMe (12,1 g, 224 mmoles). La mezcla se agitó a 60-70 °C durante 16 horas. La mezcla se enfrió y se filtró. El filtrado se concentró en vacío para proporcionar 2-metoxi-6-metilpirazina (22 g, 95 % de rendimiento). 1H RMN (CDCl3400 MHz): 5 7,98 (s, 1H), 7,94 (s, 1H), 3,91 (s, 3H), 3,40 (s, 3H). LC-MS: tR = 1,47 min (método 14), m/z = 124,8 [M H]+.
1-óxido de 3-metoxi-5-metilpirazina

A una disolución de 2-metoxi-6-metilpirazina (21,3 g, 171,6 mmoles) en AcOH (150 mL) se añadió NaBO2.H2O2.3 H2O (31,7 g, 205,9 mmoles). La mezcla se agitó a 80 °C durante 16 horas. La mezcla se concentró en vacío y se diluyó con NaOH ac. 2 M (300 mL). La mezcla se extrajo con EtOAc (200 mL x 4). La capa orgánica se lavó con salmuera (100 mL), se secó sobre Na2SO4 y se concentró en vacío para proporcionar 1-óxido de 3-metoxi-5-metilpirazina (14,4 g, 60 % de rendimiento).
3-metoxi-5-metilpirazina-2-carbonitrilo
A una mezcla de 1-óxido de 3-metoxi-5-metilpirazina (10 g, 71,4 mmoles) en MeCN (200 mL) se añadió TMSCN (24,8 g, 249,8 mmoles) y trietilamina (36,1 g, 356,8 mmoles), ZnBr2 (32,1 g, 142,7 mmoles). La mezcla se agitó a 85-90 °C durante 16 horas. La mezcla se concentró en vacío. El residuo se diluyó con DCM (500 mL) y se filtró. El filtrado se lavó con agua (300 mL) y salmuera (200 mL). La capa orgánica se secó sobre Na2SO4 y se concentró en vacío. El residuo se purificó por cromatografía en gel de sílice (éter de petróleo:acetato de etilo=5:1) para proporcionar 3-metoxi-5-metilpirazina-2-carbonitrilo (4,1 g, 38 % de rendimiento).
((3-metoxi-5-metilpirazin-2-il)metil)carbamato de ferc-butilo

A una disolución de 3-metoxi-5-metilpirazina-2-carbonitrilo (6,22 g, 41,7 mmoles) en MeOH (100 mL) se añadió (Boc)2O (13,65 g, 62,6 mmoles) y Ni de Raney (2,0 g). La mezcla se agitó a 20-25 °C bajo H2 (45 psi) durante 16 horas. La mezcla se filtró y el filtrado se concentró en vacío. El residuo se purificó por cromatografía en gel de sílice (éter de petróleo:acetato de etilo=5:1) para proporcionar ((3-metoxi-5-metilpirazin-2-il)metil)carbamato de ferc-butilo (7,7 g, 72 % de rendimiento).
LC-MS: tR = 0,70 min (método 15), m/z = 254,0 [M H]+.
(3-metoxi-5-metilpirazin-2-il)metanamina

A una disolución de ((3-metoxi-5-metilpirazin-2-il)metil)carbamato de ferc-butilo (7,7 g, 30,3 mmoles) en THF (50 mL) se añadió TFA (20 mL). La mezcla se agitó a 80 °C durante 2 horas. La mezcla se concentró en vacío. El residuo se diluyó con NaOH ac. 2 M (200 mL), se extrajo con DCM (100 mL x 2). La capa orgánica se lavó con salmuera (50 mL), se secó sobre Na2SO4 y se concentró en vacío para proporcionar (3-metoxi-5-metilpirazin-2-il)metanamina (2,5 g, 54 % de rendimiento).
1H RMN (CDCla 400 MHz): 57,90 (s, 1H), 3,96 (s, 3H), 3,93 (s, 2H), 2,42 (s, 3H), 1,69 (s, 2H).
LC-MS: tR = 0,73 min (método 16), m/z = 154,2 [M H]+.
Compuestos de Fórmula (I):
Ejemplo 1

7-(3-Fluorobencil)-3-propilimidazo[1,5-a]pirazin-8(7H)-ona:
A una disolución de 3-propilimidazo[1,5-a]pirazin-8(7H)-ona (1,2 g, 6,8 mmoles) en DMF (10 mL) se añadió carbonato de potasio (1,4 g, 10 mmoles) y 1-(bromometil)-3-fluorobenceno (1,54 g, 8,13 mmoles). La mezcla se agitó a 60-70 °C durante 2 hrs y entonces se enfrió hasta temperatura ambiente. A la reacción se añadió agua (75 mL) y se extrajo con acetato de etilo (2 x 50mL). Las fases orgánicas combinadas se lavaron con salmuera, se secaron y se concentraron en vacío. El residuo se purificó por cromatografía flash para rendir 1,5 g (78 %) de 7-(3-fluorobencil)-3-propilimidazo[1,5-a]pirazin-8(7H)-ona.1
1H RMN (DMSO-ds, 400 MHz): 5 7,70 (s, 1H), 7,46-7,37 (m, 2H), 7,17-7,09 (m, 3H), 7,00 (d, J=6,0 Hz, 1H), 5,02 (s, 2H), 2,83 (t, 2H), 1,75-1,66 (m, 2H), 0,91 (t, 3H).
LC-MS: (m/z) 286,1 (MH+) ír (minutos, método 3) = 2,19 minutos
Ejemplo 2

6-Bencil-7-(3-fluorobencil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:
Una mezcla de 6-bencil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (200 mg, 0,647 mmoles), 1-(bromometil)-3-fluorobenceno (159 mg, 840 pmoles) y carbonato de potasio (179 mg, 1,29 mmoles) en DMF (6 mL) se agitó a 60 °C durante 12 horas. La mezcla de reacción se concentró en vacío y el residuo se diluyó con diclorometano (20 mL) y se lavó con agua (3 x 5 mL). Las fases orgánicas combinadas se secaron, se filtraron y se concentraron en vacío. El residuo se purificó por TLC preparativa, eluyendo con éter de petróleo y acetato de etilo 1:2, para rendir 80 mg de 6-bencil-7-(3- fluorobencil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (28 %).
1H RMN (CDCla, 400 MHz): 57,97 (s, 1H), 7,39-7,30 (m, 4 H), 7,17 (d, J=7,53 Hz, 2 H), 6,94-6,92 (m, 2 H), 6,83 (d, J=9,54 Hz, 1 H), 6,74 (s, 1 H), 5,06 (s,, 2 H), 4,12 (d, J=12,05 Hz, 2 H), 3,74 (s, 2 H), 3,58 - 3,52 (m, 2 H), 3,03 - 3,09 (m, 1 H), 2,19 - 2,09 (m, 2 H), 1,89 (d, J=13,55 Hz, 2 H).
LC-MS: (m/z) 418,2 (MH+) ír (minutos, método 3) = 2,69 minutos
Ejemplo 3

6-Bencil-7-(ciclohexilmetil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:
Una mezcla de 6-bencil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (60 mg, 0,19 mmoles), (bromometil)ciclohexano (52 mg, 0,29 mmoles) y carbonato de potasio (54 mg, 0,39 mmoles) en DMF (10 mL) se agitó a 75 °C durante 12 horas. La mezcla de reacción se concentró en vacío. El residuo se diluyó con diclorometano (20 mL) y se lavó con agua (3 x 5 mL). Las fases orgánicas combinadas se secaron, se filtraron y se concentraron en vacío. El residuo se purificó por TLC preparativa, eluyendo con éter de petróleo y acetato de etilo 1:2, para rendir 15 mg de 6-bencil-7-(ciclohexilmetil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (8,1 %).
1H RMN (CDCla, 400 MHz): 57,87 (s, 1H), 7,42-7,30 (m, 3 H), 7,19 (d, J=7,34 Hz, 2 H), 6,65 (s, 1 H), 4,11 (d, J=11,25 Hz, 2 H), 3,90 (s, 2 H), 3,66 (d, J=6,36 Hz, 2 H), 3,54 (t, J=10,76 Hz, 2 H), 3,05-2,99 (m, 1 H), 2,18 - 2,04 (m, 2 H), 1,86 (d, J=13,94 Hz, 2 H), 1,63 - 1,77 (m, 7 H), 1,18-1,15 (m, 2 H), 1,04-1,01 (m, 2 H).
LC-MS: (m/z) 406,2 (MH+) ír (minutos, método 4) = 2,38 minutos
Ejemplo 4
7-(Ciclohexilmetil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:
Una mezcla de 6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (50 mg, 0,21 mmoles), (bromometil)ciclohexano (57 mg, 0,32 mmoles) y carbonato de potasio (59 mg, 0,43 mmoles) en DMF (2 mL) se agitó a 60 °C durante 12 horas. La mezcla de reacción se concentró en vacío. El residuo se diluyó con diclorometano (20 mL) y se lavó con agua (3 x 5 mL). Las fases orgánicas combinadas se secaron, se filtraron y se concentraron en vacío. El residuo se purificó por LC-MS preparativa para rendir 20 mg de 7-(ciclohexilmetil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (28 %).
1H RMN (CDCla, 400 MHz): 57,85 (s, 1 H), 6,71 (s, 1 H), 4,13 (d, J=11,04 Hz, 2 H), 3,78 (d, J=7,03 Hz, 2 H), 3,66-3,53 (m, 2 H), 3,11-3,05 (m, 1 H), 2,27 (s, 3 H), 2,20-2,05 (m, 2 H), 1,88 (d, J=12,05 Hz, 2 H), 1,80-1,64 (m, 6 H), 1,25-1,13 (m, 3 H), 1,12-0,99 (m, 2 H).
LC-MS: (m/z) 330,2 (MH+) tR (minutos, método 3) = 2,49 minutos
Ejemplo 5

7-(3-Fluorobencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:
Una mezcla de 6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (50 mg, 0,21 mmoles), 1 (bromometil)-3-fluorobenceno (60 mg, 0,32 mmoles) y K2CO3 (59 mg, 0,43 mmoles) en DMF (2 mL) se agitó a 60 °C durante 12 horas. La mezcla de reacción se concentró en vacío. El residuo se diluyó con diclorometano (20 mL) y se lavó con agua (3 x 5 mL). La capa orgánica se secó y se concentró en vacío. El residuo se purificó por LC-MS preparativa para proporcionar 20 mg de 7-(3-fluorobencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (27 %).
H RMN (CDCla, 400 MHz): 57,94 (s, 1 H), 7,34-7,29 (m, 1 H), 7,03-6,94 (m, 2 H), 6,90 (d, J=9,70 Hz, 1 H), 6,77 (s, 1 H), 5,23 (s, 2 H), 4,14 (d, J=10,14 Hz, 2 H), 3,59 (td, J=11,69, 1,76 Hz, 2 H), 3,15-3,05 (m, 1 H), 2,18 (s, 3 H), 2,17 2,08 (m, 2 H), 1,90 (d, J=13,45 Hz, 2H).
LC-MS: (m/z) 342,2 (MH+) tR (minutos, método 3) = 2,34 minutos
Ejemplo 6

3-Ciclopropil-7-(3-fluorobencil)imidazo[1,5-a]pirazin-8(7H)-ona:
A una disolución de 3-ciclopropilimidazo[1,5-a]pirazin-8(7H)-ona (300 mg, 1,71 mmoles) en DMF anhidro (5 mL) se añadió K2CO3 (355 mg, 2,57 mmoles) y 1-(bromometil)-3-fluorobenceno (388 mg, 2,05 mmoles). La mezcla se agitó a 65 °C durante 16 horas. La mezcla se filtró y el filtrado se purificó por HPLC preparativa para rendir 280 mg de 3-ciclopropil-7-(3-fluorobencil)imidazo[1,5-a]pirazin-8(7H)-ona (58 %).
1H RMN (DMSO-ds 400 MHz): 57,59-7,56 (m, 2H), 7,36-7,33 (m, 1H), 7,13-7,09 (m, 3H), 6,98 (d, J=6,0 Hz, 1H), 4,98 (s, 2H), 2,29-2,24 (m, 1H), 1,00-0,90 (m, 4H).
LC-MS: (m/z) 284,1 (MH+) tR (minutos, método 3) = 2,23 minutos
E je m p lo 7

7-(Ciclopentilmetil)-3-ciclopropilimidazo[1,5-a]pirazin-8(7H)-ona:
A una disolución de 3-ciclopropilimidazo[1,5-a]pirazin-8(7H)-ona (300 mg, 1,71 mmoles) en DMF anhidro (5 mL) se añadió K2CO3 (355 mg, 2,57 mmoles) y (bromometil)ciclopentano (335 mg, 2,05 mmoles). La reacción se agitó a 65 °C durante 16 horas. La mezcla se filtró y el filtrado se purificó por LC-MS preparativa para rendir 210 mg de 7-(ciclopentilmetil)-3-ciclopropilimidazo[1,5-a]pirazin-8(7H)-ona (47 %).
1H RMN (DMSO-ds, 400 MHz): 5 7,53 (s, 1H), 7,51 (d, J = 6,0 Hz, 1H), 6,90 (d, J = 6,0 Hz, 1H), 3,69 (d, J = 7,6 Hz, 2H), 2,29-2,24 (m, 2H), 1,58-1,45 (m, 6H), 1,22-1,19 (m, 2H), 1,00- 0,89 (m, 4H).
LC-MS: (m/z) 258,2 (MH+) tR (minutos, método 3) = 2,24 minutos
Ejemplo 8
7-(Ciclohexilmetil)-3-ciclopropilimidazo[1,5-a]pirazin-8(7H)-ona:
A una disolución de 3-ciclopropilimidazo[1,5-a]pirazin-8(7H)-ona (300 mg, 1,71 mmoles) en DMF anhidro (5 mL) se añadió K2CO3 (355 mg, 2,57 mmoles) y (bromometil)ciclohexano (363 mg, 2,05 mmoles). La reacción se agitó a 65 °C durante 16 horas. La mezcla de reacción se filtró y el filtrado se purificó por LC-MS preparativa para rendir 195 mg de 7-(ciclohexilmetil)-3-ciclopropilimidazo[1,5-a]pirazin-8(7H)-ona (42 %).
1H RMN (DMSO-ds, 400 MHz): 5 7,53-7,49 (m, 2H), 6,84 (d, J = 6,0 Hz, 1H), 3,59 (d, J = 7,6Hz, 2H), 2,27-2,25 (m, 1H), 1,68-1,55 (m, 6H), 1,10-0,89 (m, 9H).
LC-MS: (m/z) 272,2 (MH+) tR (minutos, método 3) = 2,40 minutos
Ejemplo 9

7-(3-Fluorobencil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:
A una disolución de 3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (200 mg, 0,91 mmoles) en DMF anhidro (5 mL) se añadió K2CO3 (189 mg, 1,37 mmoles) y 1-(bromometil)-3-fluorobenceno (207 mg, 1,10 mmoles). La mezcla de reacción se agitó a 60 °C durante 16 horas. La mezcla se filtró y el filtrado se purificó por LC-MS preparativa para rendir 190 mg de 7-(3-fluorobencil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (64 %).
1H RMN (DMSO-d6, 400 Mhz): 57,68 (s, 1H), 7,54 (d, J = 6,0 Hz, 1H), 7,36-7,33 (m, 1H), 7,14-7,09 (m, 3H), 6,99 (d, J = 6,0 Hz, 1H), 4,99 (s, 2H), 3,91-3,88 (m, 2H), 3,48-3,42 (m, 2H), 3,30-3,29 (m, 1H), 1,81-1,73 (m, 4H).
LC-MS: (m/z) 328,1 (MH+) tR (minutos, método 1) = 1,92 minutos
E je m p lo 10
7-(Ciclopentilmetil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:
A una disolución de 3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (400 mg, 1,82 mmoles) en DMF anhidro (5 mL) se añadió K2CO3 (503 mg, 3,64 mmoles) y (bromometil)ciclopentano (445 mg, 2,73 mmoles). La mezcla de reacción se agitó a 60 °C durante 16 horas. La mezcla se filtró y el filtrado se purificó por LC-MS preparativa para rendir 290 mg de 7-(ciclopentilmetil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (53 %).
1H RMN (DMSO-ds, 400 Mhz): 57,62 (s, 1H), 7,49 (d, J = 6,0 Hz, 1H), 6,90 (d, J = 6,0 Hz, 1H), 3,91-3,88 (m, 2H), 3,69 (d, J = 7,6Hz, 2H), 3,48-3,43 (m, 2H), 3,42-3,31 (m, 1H), 2,27- 2,24 (m, 1H), 1,78-1,73 (m, 4H), 1,58-1,44 (m, 6H), 1,21-1,20 (m, 2H).
LC-MS: (m/z) 302,2 (MH+) tR (minutos, método 3) = 2,29 minutos
Ejemplo 11

7-(Ciclohexilmetil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:
A una disolución de 3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (300 mg, 1,37 mmoles) en DMF anhidro (5 mL) se añadió K2CO3 (379 mg, 2,74 mmoles) y (bromometil)ciclohexano (364 mg, 2,06 mmoles). La mezcla de reacción se agitó a 60 °C durante 16 horas. La mezcla se filtró y el filtrado se purificó por LC-MS preparativa para rendir 240 mg de 7-(ciclohexilmetil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (55 %).
1H RMN (DMSO-ds, 400 Mhz): 57,62 (s, 1H), 7,47 (d, J = 6,0 Hz, 1H), 6,84 (d, J = 6,2 Hz, 1H), 3,91-3,88 (m, 2H), 3,60 (d, J = 7,2 Hz, 2H), 3,48-3,42 (m, 2H), 3,30-3,29 (m, 1H), 1,78- 1,52 (m, 10H), 1,10-1,06 (m, 3H), 0,93-0,91 (m, 2H).
LC-MS: (m/z) 316,2 (MH+) tR (minutos, método 3) = 2,44 minutos.
Ejemplo 12

7-(Cicloheptilmetil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:
A una disolución de 3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (300 mg, 1,37 mmoles) en DMF anhidro (4 mL) se añadió K2CO3 (568 mg, 4,11 mmoles) y metanosulfonato de cicloheptilmetilo (565 mg, 2,74 mmoles). La mezcla de reacción se agitó a 95 °C durante 16 horas. La mezcla se filtró y el filtrado se purificó por LC-MS preparativa para rendir 140 mg de 7-(cicloheptilmetil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (30 %).
1H RMN (DMSO-ds, 400 MHz): 5 7,62 (s, 1H), 6,48 (d, J = 5,6 Hz, 1H), 6,87 (d, J = 6,0 Hz, 1H), 3,91-3,88 (m, 2H), 3,59 (d, J = 7,6 Hz, 2H), 3,48-3,42 (m, 2H), 3,31 -3,26 (m, 1H), 1,90- 1,73 (m, 5H), 1,57-1,43 (m, 10H), 1,13-1,11 (m, 2H).
LC-MS: (m/z) 330,2 (MH+) tR (minutos, método 3) = 2,58 minutos
Ejemplo 13

7-(Cidoheptilmetil)-3-cidopropilimidazo[1,5-a]pirazin-8(7H)-ona:
A una disolución de 3-ciclopropilimidazo[1,5-a]pirazin-8(7H)-ona (300 mg, 1.71 mmoles) en DMF anhidro (4 mL) se añadió K2CO3 (709 mg, 5,1 mmoles) y metanosulfonato de cicloheptilmetilo (706 mg, 3,42 mmoles). La mezcla de reacción se agitó a 95 °C durante 16 horas. La mezcla se filtró y el filtrado se purificó por LC-MS preparativa para rendir 115 mg de 7-(cicloheptilmetil)-3-ciclopropilimidazo[1,5-a]pirazin-8(7H)-ona (24 %).
1H RMN (DMSO-ds, 400 MHz): 5 7,55-7,52 (m, 2H), 6,89 (d, J = 6,0 Hz, 1H), 3,61 (d, J = 7,6 Hz, 2H), 2,32-2,27 (m, 1H), 1,91 (brs, 1H), 1,91-1,02 (m, 12H), 1,01-0,91 (m, 4H).
LC-MS: (m/z) 286,2 (MH+) tR (minutos, método 3) = 2,54 minutos.
Ejemplo 14

7-(4-Clorobencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:
Una mezcla de 6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (100 mg, 0,429 mmoles), 1-(bromometil)-4-clorobenceno (132 mg, 0,643 mmoles) y Cs2CO3 (280 mg, 0,857 mmoles) en DMF (2,0 mL) se agitó a 70 °C durante 12 horas. La mezcla de reacción se filtró y el filtrado se concentró en vacío. El residuo se purificó por LC-MS preparativa para rendir 53 mg de 7-(4-clorobencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (34 %).
1H RMN (CDCl3, 400 MHz): 57,93 (s, 1H), 7,30 (d, J = 8,4 Hz, 2H), 7,16 (d, J = 8,4 Hz, 2H), 6,76 (s, 1H), 5,20 (s, 2H), 4,13 (d, J = 10,8 Hz, 2H), 3,62 - 3,56 (m, 2H), 3,12 - 3,05 (m, 1H), 2,18 (s, 3H), 2,16 - 2,09 (m, 2H), 1,89 (d, J = 13,2 Hz, 2H).
LC-MS: (m/z) 358,1 (MH+) tR (minutos, método 3) = 2,46 minutos
Ejemplo 15
6-Bromo-7-(3-fluorobencil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:
Una mezcla de 6-bromo-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (100 mg, 0,335 mmoles), 1-(bromometil)-3-fluorobenceno (95 mg, 0,50 mmoles) y K2CO3 (93 mg, 0,67 mmoles) en DMF (2,0 mL) se agitó a 60 °C durante 12 horas. La mezcla de reacción se concentró en vacío. El residuo se purificó por LC-MS preparativa para proporcionar 30 mg de 6-bromo-7-(3-fluorobencil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (22 %).
1H RMN (CDCla, 400 MHz): 57,95 (s, 1H), 7,34 - 7,27 (m, 1H), 7,18 (s, 1H), 7,11 (d, J = 7,6 Hz, 1H), 7,03 - 6,99 (m, 2H), 5,39 (s, 2H), 4,13 (d, J = 12,0 Hz, 2H), 3,62 - 3,56 (m, 2H), 3,11 - 3,05 (m, 1H), 2,18 - 2,08 (m, 2H), 1,88 (d, J = 14,0 Hz, 2H).
LC-MS: (m/z) 408,0 (MH+) tR (minutos, método 3) = 2,65 minutos
Ejemplo 16

7-Bencil-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:
Una mezcla de 6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (100 mg, 0,429 mmoles), (bromometil)benceno (110 mg, 0,643 mmoles) y K2CO3 (119 mg, 0,857 mmoles) en d Mf (1,0 mL) se agitó a 60 °C durante 3 horas. La mezcla se filtró y el filtrado se concentró en vacío. El residuo se purificó por LC-MS preparativa para rendir 39 mg de 7-bencil-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (28 %).
1H RMN (CDCla, 400 MHz): 57,93 (s, 1H), 7,35 - 7,29 (m, 3H), 7,21 (d, J = 7,6 Hz, 2H), 6,75 (s, 1H), 5,25 (s, 2H), 4,13 (d, J = 11,6 Hz, 2H), 3,61 - 3,56 (m, 2H), 3,13 - 3,06 (m, 1H), 2,19 (s, 3H), 2,15 - 2,08 (m, 2H), 1,89 (d, J = 13,2 Hz, 2H).
LC-MS: (m/z) 324,2 (MH+) tR (minutos, método 3) = 2,25 minutos
Ejemplo 17
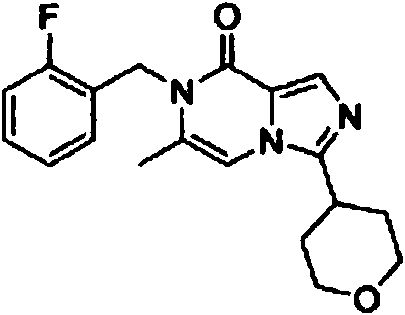
7-(2-Fluorobencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:
Una mezcla de 6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (100 mg, 0,429 mmoles), 1-(bromometil)-2-fluorobenceno (122 mg, 0,643 mmoles) y Cs2CO3 (279 mg, 0,857 mmoles) en Dm F (2,0 mL) se agitó a 70 °C durante 1 hora. La mezcla se filtró y el filtrado se concentró en vacío. El residuo se purificó por LC-MS preparativa para rendir 49 mg de 7-(2-fluorobencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (33 %).
1H RMN (CDCl3, 400 MHz): 57,93 (s, 1H), 7,27 - 7,23 (m, 1H), 7,11 - 7,06 (m, 3H), 6,77 (s, 1H), 5,28 (s, 2H), 4,13 (d, J = 10,4 Hz, 2H), 3,62-3,56 (m, 2H), 3,12 - 3,07 (m, 1H), 2,19 (s, 3H), 2,16 - 2,10 (m, 2H), 1,89 (d, J = 12,0 Hz, 2H).
LC-MS: (m/z) 342,1 (MH+) tR (minutos, método 3) = 2,30 minutos
E je m p lo 18

7-(3-Clorobencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:
Una mezcla de 6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (100 mg, 0,429 mmoles), 1-(bromometil)-3-clorobenceno (132 mg, 0,643 mmoles) y K2CO3 (119 mg, 0,857 mmoles) en DMF (1,0 mL) se agitó a 65 °C durante 12 horas. A la mezcla se añadió Cs2CO3 (280 mg, 0,857 mmoles) y la reacción se agitó a 80 °C durante 1 hora más. La mezcla de reacción se filtró y el filtrado se concentró en vacío. El residuo se purificó por LC-MS preparativa para rendir 49 mg de 7-(3-clorobencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (32 %).
1H RMN (CDCla, 400 MHz): 57,92 (s, 1H), 7,25 - 7,24 (m, 2H), 7,16 (s, 1H), 7,10 - 7,07 (m, 1H), 6,75 (s, 1H), 5,19 (s, 2H), 4,11 (d, J = 10,4 Hz, 2H), 3,60 - 3,54 (m, 2H), 3,12 - 3,05 (m, 1H), 2,16 (s, 3H), 2,14 - 2,07 (m, 2H), 1,87 (d, J = 12,0 Hz, 2H).
LC-MS: (m/z) 358,1 (MH+) tR (minutos, método 3) = 2,44 minutos
Ejemplo 19

7-(2-Clorobencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:
Una mezcla de 6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (100 mg, 0,429 mmoles), 1-(bromometil)-2-clorobenceno (132 mg, 0,643 mmoles) y Cs2CO3 (279 mg, 0,857 mmoles) en d Mf (2,0 mL) se agitó a 80 °C durante 12 horas. La mezcla se filtró y el filtrado se concentró en vacío. El residuo se purificó por LC-MS preparativa para rendir 55 mg de 7-(2-clorobencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (36 %).
1H RMN (CDCla, 400 MHz): 57,95 (s, 1H), 7,42 - 7,40 (m, 1H), 7,25 - 7,20 (m, 2H), 6,93 (d, J = 7,2 Hz, 1H), 6,80 (s, 1H), 5,32 (s, 2H), 4,14 (d, J = 10,8 Hz, 2H), 3,63 - 3,57 (m, 2H), 3,14 - 3,07 (m, 1H), 2,20 - 2,10 (m, 2H), 2,13 (s, 3H), 1,91 (d, J = 13,2 Hz, 2H).
LC-MS: (m/z) 358,1 (MH+) tR (minutos, método 3) = 2,46 minutos
Ejemplo 20

7-(3-Metoxibencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:
Una mezcla de 6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (100 mg, 0,429 mmoles), 1-(bromometil)-3-metoxibenceno (129 mg, 0,643 mmoles) y Cs2CO3 (280 mg, 0,857 mmoles) en DMF (1,0 mL) se agitó a 80 °C durante 12 horas. La mezcla de reacción se filtró y el filtrado se concentró en vacío. El residuo se purificó por
LC-MS preparativa para rendir 68 mg de 7-(3-metoxibencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (45 %).
1H RMN (CDCI3, 400 MHz): 57,92 (s, 1H), 7,27 - 7,23 (m, 1H), 6,82 - 6,77 (m, 2H), 6,74 (s, 2H), 5,21 (s, 2H), 4,13 (d, J = 10,8 Hz, 2H), 3,78 (s, 3H), 3,62 - 3,56 (m, 2H), 3,12 - 3,08 (m, 1H), 2,19 (s, 3H), 2,19 - 2,10 (m, 2H), 1,89 (d, J = 13,2 Hz, 2H).
LC-MS: (m/z) 354,2 (MH+) tR (minutos, método 3) = 2,28 minutos
Ejemplo 21

6-MetiI-7-(2-metiIbenciI)-3-(tetrahidro-2H-piran-4-iI)imidazo[1,5-a]pirazin-8(7H)-ona:
Una mezcla de 6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (100 mg, 0,429 mmoles), 1-(bromometil)-2-metilbenceno (119 mg, 0,643 mmoles) y K2CO3 (119 mg, 0,857 mmoles) en DMF (2,0 mL) se agitó a 60 °C durante 12 horas. A la mezcla se añadió adicionalmente Cs2CO3 (280 mg, 0,86 mmoles) y la reacción se agitó a 70 °C durante otras 13 horas. La mezcla de reacción se filtró y el filtrado se concentró en vacío. El residuo se purificó por LC-MS preparativa para rendir 23 mg de 6-metil-7-(2-metilbencil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (16 %).
1H RMN (CDCI3, 400 MHz): 57,93 (s, 1H), 7,21 - 7,12 (m, 3H), 6,80 (s, 1H), 6,78 (d, J = 7,6 Hz, 1H), 5,18 (s, 2H), 4,15 (d, J = 10,8 Hz, 2H), 3,63 - 3,57 (m, 2H), 3,15 - 3,09 (m, 1H), 2,40 (s, 3H), 2,20 - 2,13 (m, 2H), 2,11 (s, 3H), 1,92 (d, J = 13,2 Hz, 2H).
LC-MS: (m/z) 338,2 (MH+) tR (minutos, método 3) = 2,37 minutos
Ejemplo 22

6-Metil-7-(4-metilbencil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:
Una mezcla de 6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (100 mg, 0,429 mmoles), 1-(bromometil)-4-metilbenceno (119 mg, 0,643 mmoles) y Cs2CO3 (280 mg, 0,857 mmoles) en DMF (2,0 mL) se agitó a 70 °C durante 12 horas. La mezcla se filtró y el filtrado se concentró en vacío. El residuo se purificó por LC-MS preparativa para rendir 68 mg de 6-metil-7-(4-metilbencil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (47 %).
1H RMN (CDCI3, 400 MHz): 5 7,92 (s, 1H), 7,14 - 7,09 (m, 4H), 6,74 (s, 1H), 5,20 (s, 2H), 4,13 (d, J = 10,4 Hz, 2H), 3,62 - 3,56 (m, 2H), 3,11 - 3,06 (m, 1H), 2,32 (s, 3H), 2,19 (s, 3H), 2,19 - 2,08 (m, 2H), 1,88 (d, J = 13,2 Hz, 2H). LC-MS: (m/z) 338,2 (MH+) tR (minutos, método 3) = 2,41 minutos.
Ejemplo 23
7-(4-Metoxibencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:
Una mezcla de 6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (100 mg, 0,43 mmoles), 1-(bromometil)-4-metoxibenceno (129 mg, 0,643 mmoles) y Cs2CO3 (280 mg, 0,857 mmoles) en DMF (2,0 mL) se agitó a 70 °C durante 12 horas. La mezcla se filtró y el filtrado se concentró en vacío. El residuo se purificó por LC-MS preparativa para rendir 60 mg de 7-(4-metoxibencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (39 %). 1H RMN (CDCla, 400 MHz): 57,93 (s, 1H), 7,17 (d, J = 8,4 Hz, 2H), 6,86 (d, J = 8,4 Hz, 2H), 6,73 (s, 1H), 5,17 (s, 2H), 4,13 (d, J = 10,0 Hz, 2H), 3,79 (s, 3H), 3,58 (t, J = 12,0 Hz, 2H), 3,11 - 3,05 (m, 1H), 2,21 (s, 3H), 2,18 - 2,08 (m, 2H), 1,88 (d, J = 13,6 Hz, 2H).
LC-MS: (m/z) 354,2 (MH+) tR (minutos, método 3) = 2,26 minutos.
Ejemplo 24

7-(4-Fluorobencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:
Una mezcla de 6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (100 mg, 0,429 mmoles), 1-(bromometil)-4-fluorobenceno (122 mg, 0,643 mmoles) y Cs2CO3 (279 mg, 0,857 mmoles) en DMF (2,0 mL) se agitó a 70 °C durante 12 horas. La mezcla se filtró y el filtrado se concentró en vacío. El residuo se purificó por LC-MS preparativa para rendir 30 mg de 7-(4-fluorobencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (21 %).
1H RMN (CDCla, 400 MHz): 57,93 (s, 1H), 7,22 - 7,19 (m, 2H), 7,04 - 7,00 (m, 2H), 6,75 (s, 1H), 5,20 (s, 2H), 4,13 (d, J = 11,2 Hz, 2H), 3,62 - 3,60 (m, 2H), 3,17 - 3,06 (m, 1H), 2,19 (s, 3H), 2,14 - 2,08 (m, 2H), 1,88 (d, J = 13,6 Hz, 2H). LC-MS: (m/z) 342,1 (MH+) tR (minutos, método 3) = 2,30 minutos
Ejemplo 25
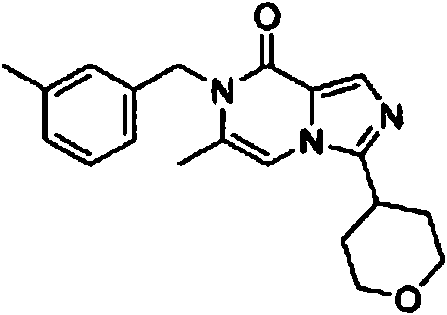
6-Metil-7-(3-metilbencil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:
Una mezcla de 6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (100 mg, 0,429 mmoles), 1-(bromometil)-3-metilbenceno (119 mg, 0,643 mmoles) y Cs2CO3 (280 mg, 0,857 mmoles) en DMF (1 mL) se agitó a 80 °C durante 12 horas. La mezcla se filtró y el filtrado se concentró en vacío. El residuo se purificó por LC-MS preparativa para rendir 62 mg de 6-metil-7-(3-metilbencil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (43 %).
1H RMN (CDCl3, 400 MHz): 57,93 (s, 1H), 7,21 (t, J = 7,2 Hz, 1H), 7,08 (d, J = 7,6 Hz, 1H), 7,02 - 6,98 (m, 2H), 6,74 (s, 1H), 5,21 (s, 2H), 4,13 (d, J - 10,8 Hz, 2H), 3,62 - 3,56 (m, 2H), 3,12 - 3,07 (m, 1H), 2,33 (s, 3H), 2,19 (s, 3H), 2,19 - 2,09 (m, 2H), 1,89 (d, J = 13,2 Hz, 2H).
LC-MS: (m/z) 338,1 (MH+) tR (minutos, método 3) = 2,40 minutos.
E je m p lo 26:

7-(3-fluorobencil)-6-metil-3-(4-metiltetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:
Etapa 1: A una disolución de (3-metoxi-5-metilpirazin-2-il)metanamina (150 mg, 0,98 mmoles) y ácido 4-metiltetrahidro-2H-piran-4-carboxílico (212 mg, 1,5 mmoles) en DCM (6 mL) se añadió HATU (670 mg, 1,8 mmoles) y Et3N (198 mg,
1,96 mmoles). La mezcla se agitó a 20-25 °C durante 1 hora. La mezcla se diluyó con DCM (50 mL), se lavó con agua (30 mL) y salmuera (30 mL). La capa orgánica se secó sobre Na2SO4 y se concentró en vacío. El residuo se purificó por cromatografía en gel de sílice (éter de petróleo:acetato de etilo=3:1) para proporcionar W-((3-metoxi-5-metilpirazin-2-il)metil)-4-metiltetrahidro-2H-piran-4-carboxamida (250 mg, 91 % de rendimiento).
Etapa 2: A una disolución de W-((3-metoxi-5-metilpirazin-2-il)metil)-4-metiltetrahidro-2H-piran-4-carboxamida (300 mg,
1,07 mmoles) en dioxano (5 mL) se añadió POCh (660 mg, 4,3 mmoles). La disolución se agitó a 80 -90 °C durante
3 horas. La mezcla se concentró en vacío, se diluyó con DCM (50 mL) y se añadió lentamente en agua (30 mL). La capa orgánica se lavó con salmuera (20 mL) y se secó sobre Na2SO4, se concentró en vacío para proporcionar 8-metoxi-6-metil-3-(4-metiltetrahidro-2H-piran-4-i¡)imidazo[1,5-a]pirazina (250 mg, 89 % de rendimiento).
Etapa 3: A una disolución de 8-metoxi-6-metil-3-(4-metiltetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazina (200 mg,
0,77 mmoles) en dioxano (10 mL) se añadió HCl(ac) 2M (10 mL). La disolución se agitó a 80 -90 °C durante 1 hora.
La mezcla se enfrió y se añadió NaHCO3 acuoso saturado (100 mL), se extrajo con DCM (100 mL x 2). La capa orgánica se lavó con salmuera (50 mL), se secó sobre Na2SO4 y se concentró en vacío para proporcionar 6-metil-3-(4-metiltetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (160 mg, 68 % de rendimiento).
LC-MS: tR = 0,89 min (método 10), m/z = 248,3 [M H]+.
Etapa 4: A una disolución de 6-metil-3-(4-metiltetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (200 mg,
0,81 mmoles) y 1-(bromometil)-3-fluorobenceno (183 mg, 0,97 mmoles) en DMF anhidro (5 mL) se añadió K2CO3
(168 mg, 1,21 mmoles). La mezcla se agitó a 60-70 °C durante 16h. La mezcla se enfrió y se diluyó con agua
(20 mL), se extrajo con EtOAc (30 mL x 2). La capa orgánica se lavó con salmuera (20 mL), se secó sobre Na2SO4 y se concentró en vacío. El residuo se purificó por cromatografía en gel de sílice (éter de petróleo:acetato de etilo=1:2) para proporcionar 7-(3-fluorobencil)-6-metil-3-(4-metiltetrahidro-2H-piran- 4-il)imidazo[1,5-a]pirazin-8(7H)-ona (90 mg,
31 % de rendimiento) como un sólido blanquecino.
1H RMN (CDCl3 400 MHz): 57,94 (s, 1H), 7,33-7,28 (m, 1H), 7,02-6,91 (m, 4H), 5,22 (s, 2H), 3,85-3,80 (m, 2H), 3,73
3,67 (m, 2H), 2,45-2,41 (m, 2H), 2,17 (s, 3H), 1,86-1,79 (m, 2H), 1,49 (s, 3H).
LC-MS: tR = 2,46 min (método 3), m/z = 356,2 [M H]+.
Ejemplo 27:

4-(7-(3-fluorobencil)-6-metil-8-oxo-7,8-dihidroimidazo[1,5-a]pirazin-3-il)tetrahidro-2H-piran-4-carbonitrilo:
Etapa 1: A una disolución de (3-metoxi-5-metilpirazin-2-il)metanamina (100 mg, 0,65 mmoles) y ácido 4-cianotetrahidro-2H-piran-4-carboxílico (152 mg, 0,98 mmoles) en DCM (6 mL) se añadió HATU (447 mg, 1,18 mmoles) y Et3N (132 mg, 1,31 mmoles). La mezcla se agitó a 20-25 °C durante 1 hora. La mezcla se diluyó con DCM (30 ml), se lavó con agua (20 mL) y salmuera (20 mL). La capa orgánica se secó sobre Na2SO4 y se concentró en vacío. El
residuo se purificó por cromatografía en gel de sílice (éter de petróleo:acetato de etilo=1:1) para proporcionar 4-ciano-W-((3-metoxi-5-metilpirazin-2-il)metil)tetrahidro-2H-piran-4-carboxamida (100 mg, 53 % de rendimiento).
LC-MS: tR = 0,61 min (método 2), m/z = 290,9 [M H]+
Etapa 2: A una disolución de 4-ciano-W-((3-metoxi-5-metilpirazin-2-il)metil)tetrahidro-2H-piran-4-carboxamida (100 mg, 0,34 mmoles) en dioxano (5 mL) se añadió POCh (330 mg, 2,15 mmoles). La disolución se agitó a 80-90 °C durante 2h. La mezcla se enfrió y se añadió lentamente en agua (50 mL), se extrajo con EtOAc (30 mL x 2). La capa orgánica se lavó con salmuera (20 mL), se secó sobre Na2SO4 y se concentró en vacío para proporcionar 4-(8-metoxi-6-metilimidazo[1,5-a]pirazin-3-il)tetrahidro-2H-piran-4-carbonitrilo (80 mg, 85 % de rendimiento).
Etapa 3: A una disolución de 4-(8-metoxi-6-metilimidazo[1,5-a]pirazin-3-il)tetrahidro-2H-piran-4-carbonitrilo (80 mg, 0,29 mmoles) en dioxano (4 mL) se añadió HCl(ac) 2M (2 mL). La disolución se agitó a 80-90 °C durante 2h. La mezcla se concentró en vacío y se añadió NaHCO3 acuoso saturado (50 mL). La mezcla se extrajo con DCM (50 mL x 2). La capa orgánica se lavó con salmuera (20 mL) y se concentró en vacío para proporcionar 4-(6-metil-8-oxo-7,8-dihidroimidazo[1,5-a]pirazin-3-il)tetrahidro-2H-piran-4-carbonitrilo (70 mg, 92 % de rendimiento) como un sólido blanquecino.
LC-MS: tR = 0,98 min (método 10), m/z = 259,2 [M H]+.
Etapa 4: A una disolución de 4-(6-metil-8-oxo-7,8-dihidroimidazo[1,5-a]pirazin-3-il)tetrahidro-2H-piran-4-carbonitrilo (70 mg, 0,27 mmoles) en DMF anhidro (5 mL) se añadió 1-(bromometil)-3-fluoro-benceno (77 mg, 0,41 mmoles) y K2CO3 (75 mg, 0,54 mmoles). La mezcla se agitó a 70-80 °C durante 2h. La mezcla se enfrió y se filtró. El filtrado se purificó por LC-MS preparativa para proporcionar 4-(7-(3-fluorobencil)-6-metil-8-oxo-7,8-dihidroimidazo[1,5-a]pirazin-3-il)tetrahidro-2H-piran-4-carbonitrilo (65 mg, 65 % de rendimiento).
1H RMN (CDCl3400 MHz): 57,97 (s, 1H), 7,34-7,31 (m, 1H), 7,16 (s, 1H), 7,02-6,92 (m, 3H), 5,26 (s, 2H), 4,13-4,10 (m, 2H), 3,97-3,91 (m, 2H), 2,50-2,43 (m, 2H), 2,35-2,32 (m, 2H), 2,24 (s, 3H).
LC-MS: tR = 2,69 min (método 3), m/z = 367,1 [M H]+.
Ejemplo 28:

7-(3-fluorobencil)-3-(4-metoxitetrahidro-2H-piran-4-il)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona:
Etapa 1: A una disolución de 3-bromo-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (500 mg, 2,2 mmoles) y 1 -(bromometil)-3-fluorobenceno (497 mg, 2,6 mmoles) en DMF (5 mL) se añadió K2CO3 (605 mg, 4,4 mmoles). La mezcla se agitó a 60 °C durante 12 horas. La mezcla se diluyó con agua (20 mL) y se extrajo con EtOAc (10 mL x 3). La capa orgánica combinada se lavó con agua (10 mL x 2); se secó sobre Na2SO4 y se evaporó en vacío para proporcionar 3-bromo-7-(3-fluorobencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (420 mg, 57 % de rendimiento).
Etapa 2: A una disolución de 3-bromo-7-(3-fluorobencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (200 mg, 0,6 mmoles) en THF (10 mL) se añadió n-BuLi (0,31 mL, 0,77 mmoles) a -78 °C. La mezcla se agitó a -78 °C durante 30 minutos. Se añadió tetrahidro-4H-piran-4-ona (77 mg, 0,77 mmoles) a -78 °C. La mezcla se agitó a -78 °C durante 1 hora. La mezcla se paró con NH4Cl acuoso saturado (0,5 mL) y se evaporó en vacío. El residuo se disolvió en DCM (20 mL) y se lavó con agua (10 mL). La capa orgánica se secó sobre Na2SO4 y se evaporó. El residuo se lavó con EtOAc (3 mL) y se filtró. La torta del filtro se secó en vacío para proporcionar 7-(3-fluorobencil)-3-(4-hidroxitetrahidro-2H-piran-4-il)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (100 mg, 47 % de rendimiento).
Etapa 3: A una disolución de 7-(3-fluorobencil)-3-(4-hidroxitetrahidro-2H-piran-4-il)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (80 mg, 0,22 mmoles) en THF (5 mL) se añadió NaH (al 60 % en aceite mineral, 13,4 mg, 0,36 mmoles) a 0 °C. La mezcla se agitó a 20 °C durante 30 minutos. Se añadió Mel (64 mg, 0,45 mmoles) a 0 °C. La mezcla se agitó a 20 °C durante 11,5 horas. La mezcla se paró con NH4Cl acuoso saturado (0,5 mL) y se evaporó en vacío. El residuo se disolvió en DCM (10 mL) y se lavó con agua (4 mL). La capa orgánica se secó sobre Na2SO4 y se evaporó. El residuo se purificó por TLC preparativa (EtOAc) para proporcionar 7-(3-fluorobencil)-3-(4-metoxitetrahidro-2H-piran-4-il)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (25 mg, 30 % de rendimiento).
1H RMN (CDCI3, 400 MHz): 57,93 (s, 1H), 7,36 (s, 1H), 7,31 (dd, J = 8,0 Hz, J = 14,0 Hz, 1H), 7,03-6,92 (m, 3H), 5,23 (s, 2H), 3,91-3,80 (m, 4H), 3,07 (s, 3H), 2,38-2,31 (m, 2H), 2,17 (s, 3H), 2,14-2,10 (m, 2H).
LC-MS: tR = 2,73 min (método 3), m/z = 372,1 [M H]+
Ejemplo 29:

7-(3-fluorobencil)-3-(4-fluorotetrah¡dro-2H-p¡ran-4-¡l)-6-met¡lim¡dazo[1,5-a]pirazin-8(7H)-ona:
Etapa 1: A una disolución de 3-bromo-7-(3-fluorobencil)-6-metil¡m¡dazo[1,5-a]pirazin-8(7H)-ona (100 mg, 0,3 mmoles) en THF seco (5 mL) se añadió n-BuL¡ (2,5 M, 0,15 mL) (2,5 M en n-hexano) gota a gota. La mezcla se agitó a -78 °C durante 0,5 horas. Después, se añadió tetrahidro-4H-piran-4-ona (45 mg, 0,45 mmoles) a la mezcla. La mezcla se agitó a -78 °C durante 2 horas. La mezcla se paró con NH4Cl acuoso saturado (2 mL). La mezcla se extrajo con DCM (20 mL x 2). La capa orgánica combinada se lavó con H2O (10 mL), salmuera (10 mL), se secó sobre Na2Sü4, se filtró y se concentró para proporcionar el producto crudo. El producto crudo se purificó por cromatografía flash en gel de sílice (MeOH al 1 %~10 % en d Cm ) para proporcionar 7-(3-fluorobencil)-3-(4-h¡drox¡tetrahidro-2H-p¡ran-4-¡l)-6-metilim¡dazo[1,5-a]p¡raz¡n-8(7H)-ona (40 mg, 38 % de rendimiento).
LC-MS: tR = 0,671 min (método 2), m/z = 358,1 [M H]+.
Etapa 2: A una disolución de 7-(3-fluorobencil)-3-(4-fluorotetrah¡dro-2H-p¡ran-4-¡l)-6-met¡l¡m¡dazo[1,5-a]p¡raz¡n-8(7H)-ona (40 mg, 111,9 gmoles) en DCM seco (4 mL) se añadió DAST (trifluoruro de dietilaminoazufre) (28 mg, 170 gmoles) a 0 °C. La mezcla se agitó a 0 °C durante 2 horas. Se añadió agua (10 mL) a la mezcla. La mezcla se extrajo con DCM (20 mL x 2). La capa orgánica combinada se lavó con H2O (20 mL), se secó sobre Na2SO4, se filtró y se concentró para proporcionar el producto crudo. El producto crudo se purificó por TLC preparativa (DCM/MeOH=10/1) para proporcionar 7-(3-fluorobencil)-3-(4-fluorotetrah¡dro-2H-p¡ran-4-¡l)-6-met¡lim¡dazo[1,5-a]pirazin-8(7H)-ona (12,83 mg, 32 % de rendimiento).
1H RMN (CDCla 400 MHz): 57,93 (s, 1H), 7,34-7,28 (m, 1H), 7,21 (s, 1H), 7,01-6,98 (m, 2H), 6,92 (d, J = 9,6 Hz, 1H), 5,24 (s, 2H), 4,00-3,89 (m, 4H), 2,57-2,42 (m, 2H), 2,23-2,18 (m, 5H).
LC-MS: tR = 2,753 min (método 3), m/z = 360,1 [M H]+.
Ejemplo 30:

7-(3-fluorobenc¡l)-6-met¡l-3-(tetrahidro-2H-p¡ran-2-¡l)¡m¡dazo[1,5-a]p¡raz¡n-8(7H)-ona, estereoisómero 1 y 2:
Etapa 1: A una disolución de (3-metoxi-5-met¡lp¡razin-2-¡l)metanam¡na (200 mg, 1,31 mmoles) en DCM (8 mL) se añadió ácido tetrahidro-2H-piran-2-carboxílico (255 mg, 1,96 mmoles) y HATU (894 mg, 2,35 mmoles), Et3N (264 mg, 2,61 mmoles). La disolución se agitó a 20-25 °C durante 1 hora. La mezcla se diluyó con agua (30 mL), se extrajo con DCM (40 mL x 2). La capa orgánica se lavó con salmuera (20 mL), se secó sobre Na2SO4 y se concentró en vacío. El residuo se purificó por cromatografía en gel de sílice (éter de petróleo:acetato de etilo=1:1) para proporcionar N-((3-metoxi-5-met¡lp¡raz¡n-2-¡l)met¡l)tetrah¡dro-2H-p¡ran-2-carboxam¡da (250 mg, 72 % de rendimiento). LC-MS: tR = 0,70 min (método 2), m/z = 266,2 [M H]+.
Etapa 2: A una disolución de W-((3-metoxi-5-metilpirazin-2-il)metil)tetrahidro-2H-piran-2-carboxamida (250 mg, 0,94 mmoles) en dioxano (8 mL) se añadió POCl3 (480 mg, 3,13 mmoles). La disolución se agitó a 90 °C durante 2h. La mezcla se enfrió y se concentró en vacío. El residuo se diluyó con DCM (50 mL), se lavó con NaHCO3 acuoso saturado (ac) (50 mL) y salmuera (50 mL). La capa orgánica se secó sobre Na2SO4 y se concentró en vacío para proporcionar 8-metoxi-6-metil-3-(tetrahidro-2H-piran-2-il)imidazo[1,5-a]pirazina (200 mg, 86 % de rendimiento).
Etapa 3: A una disolución de 8-metoxi-6-metil-3-(tetrahidro-2H-piran-2-il)imidazo[1,5-a]pirazina (270 mg, 1,09 mmoles) en dioxano (8 mL) se añadió HCl(ac) 2M (4 mL). La disolución se agitó a 90 °C durante 1 hora. La mezcla se concentró en vacío y se añadió NaHCO3 acuoso saturado (50 mL). La mezcla se extrajo con DCM (50 mL x 2). La capa orgánica se lavó con salmuera (50 mL) y se concentró en vacío para proporcionar 6-metil-3-(tetrahidro-2H-piran-2-il)imidazo[1,5-a]pirazin-8(7H)-ona (200 mg, 79 % de rendimiento).
Etapa 4: A una disolución de 6-metil-3-(tetrahidro-2H-piran-2-il)imidazo[1,5-a]pirazin-8(7H)-ona (200 mg, 0,86 mmoles) en DMF anhidro (10 mL) se añadió K2CO3 (237 mg, 1,71 mmoles) y 1-(bromometil)-3-fluorobenceno (243 mg, 1,29 mmoles). La mezcla se agitó a 80 °C durante 24h. La mezcla se enfrió y se diluyó con agua (100 mL), se extrajo con EtOAc (50 mL x 3). La capa orgánica se lavó con salmuera (50 mL), se secó sobre Na2SO4 y se concentró en vacío para proporcionar 7-(3-fluorobencil)-6-metil-3-(tetrahidro-2H-piran-2-il)imidazo[1,5-a]pirazin-8(7H)-ona (130 mg, 44 % de rendimiento).
Etapa 5: Se purificó 7-(3-fluorobencil)-6-metil-3-(tetrahidro-2H-piran-2-il)imidazo[1,5-a]pirazin-8(7H)-ona (130 mg, 380,8 gmoles) por SFC.
Se obtuvo 7-(3-fluorobencil)-6-metil-3-(tetrahidro-2H-piran-2-il)imidazo[1,5-a]pirazin-8(7H)-ona, estereoisómero 1 (35 mg, 27 % de rendimiento).
1H RMN (CDCl3400 MHz): 57,91 (s, 1H), 7,32-7,28 (m, 1H), 7,19(s, 1H), 6,98-6,94 (m, 2H), 6,89 (d, J = 9,6 Hz, 1H), 5.23 (s, 2H), 4,76 (t, J = 6,4 Hz, 1H), 4,06 (d, J = 10,8 Hz, 1H), 3,67 (t, J = 10,8 Hz, 1H), 2,23 (s, 3H), 2,17-2,05 (m, 3H), 1,74-1,68 (m, 3H).
LC-MS: tR = 2,33 min (método 3), m/z = 342,1 [M H]+. SFC: tR = 5,478 min, % ee = 99,90 %.
[a]D20 16,00 (c = 0,10, CHCl3).
Se obtuvo 7-(3-fluorobencil)-6-metil-3-(tetrahidro-2H-piran-2-il)imidazo[1,5-a]pirazin-8(7H)-ona, estereoisómero 2 (33 mg, rendimiento: 35 %).
1H RMN (CDCl3400 MHz): 57,92 (s, 1H), 7,32-7,28 (m, 1H), 7,19(s, 1H), 6,98-6,94 (m, 2H), 6,89 (d, J = 9,2 Hz, 1H), 5.23 (s, 2H), 4,77 (t, J = 6,8 Hz, 1H), 4,06 (d, J = 10,8 Hz, 1H), 3,67 (t, J = 10,8 Hz, 1H), 2,17 (s, 3H), 2,12-2,02 (m, 3H), 1,74-1,68 (m, 3H).
LC-MS: tR = 2,33 min (método 3), m/z = 342,1 [M H]+. SFC: tR = 5,789 min, % ee = 98,92 %.
[a]D20 -23,33 (c = 0,10, CHCl3).
Ejemplo 31:

7-(3-fluorobencil)-6-metil-3-(tetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona, estereoisómero 1 y 2:
Etapa 1: A una disolución de (3-metoxi-5-metilpirazin-2-il)metanamina (200 mg, 1,3 mmoles) en DCM seco (10 mL) se añadió ácido tetrahidrofuran-3-carboxílico (228 mg, 2,0 mmoles), Et3N (265 mg, 2,6 mmoles) y HATU (747 mg, 2,0 mmoles). La mezcla se agitó a 15 °C durante 16 horas. Se añadió agua (10 mL) a la mezcla. La capa orgánica se lavó con salmuera (10 mL), se secó sobre Na2SO4, se filtró y se concentró para proporcionar el producto crudo. El producto crudo se purificó por cromatografía flash en gel de sílice (acetato de etilo al 10 %~100 % en éter de petróleo) para proporcionar W-((3-metoxi-5-metilpirazin-2-il)metil)tetrahidrofuran-3-carboxamida (200 mg, 61 % de rendimiento).
Etapa 2: A una disolución de W-((3-metoxi-5-metilpirazin-2-il)metil)tetrahidrofuran-3-carboxamida (300 mg, 1,19 mmoles) en dioxano (5 mL) se añadió POCl3 (366 mg, 2,39 mmoles). La mezcla se calentó a 90 °C durante 2 horas. La mezcla se enfrió hasta 15 °C y se ajustó a pH=8 por NaHCO3 acuoso saturado. La capa acuosa se extrajo
con DCM (20 mL x 2). La capa orgánica combinada se lavó con H2O (20 ml), salmuera (20 mL), se secó sobre Na2SÜ4, se filtró y se concentró para proporcionar 8-metoxi-6-metil-3-(tetrahidrofuran-3-il)imidazo[1,5-a]pirazina (300 mg).
Etapa 3: A una disolución de 8-metoxi-6-metil-3-(tetrahidrofuran-3-il)imidazo[1,5-a]pirazina (300 mg, 1,29 mmoles) en dioxano (5 mL) se añadió HCl 2 N (2 mL). La mezcla se calentó a 90 °C durante 1 hora. La mezcla se enfrió hasta 15 °C y se extrajo con DCM (20 mL x 2). La capa orgánica combinada se lavó con H2O (20 mL), salmuera (20 mL), se secó sobre Na2SO4, se filtró y se concentró para proporcionar 6-metil-3-(tetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona (210 mg, 74 % de rendimiento).
Etapa 4: A una disolución de 6-metil-3-(tetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona (200 mg, 912,24 gmoles) en DMF seco (5 mL) se añadió 1-(bromometil)-3-fluorobenceno (259 mg, 1,37 mmoles) y K2CO3 (252 mg, 1,82 mmoles). La mezcla se agitó a 60 °C durante 16 horas. La mezcla se concentró y el residuo se disolvió en DCM (20 mL) y H2O (10 mL). La capa acuosa se extrajo con DCM (20 mL x 2). La capa orgánica combinada se lavó con H2O (20 mL), salmuera (20 mL), se secó sobre Na2SO4, se filtró y se concentró para proporcionar el producto crudo. El producto crudo se purificó por cromatografía flash en gel de sílice (acetato de etilo al 10 %~100 % en éter de petróleo) para proporcionar 7-(3-fluorobencil)-6-metil-3-(tetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona (40 mg, 13 % de rendimiento).
Etapa 5: Se purificó 7-(3-fluorobencil)-6-metil-3-(tetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona (40 mg, 122,2 gmoles) por SFC para proporcionar 7-(3-fluorobencil)-6-metil-3-(tetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona, estereoisómero 1 (16,43 mg, 41 % de rendimiento).
1H RMN (CDCl3400 MHz): 57,90 (s, 1H), 7,31 -7,28 (m, 1H), 6,98-6,93 (m, 2H), 6,89 (d, J = 9,2 Hz, 1H), 6,79 (s, 1H), 5,21 (s, 2H), 4,19-4,17 (m, 1H), 4,12-4,09 (m, 1H), 4,05-3,95 (m, 2H), 3,66-3,62 (m, 1H), 2,40 (q, J = 7,2 Hz, 2H), 2,16 (s, 3H).
LC-MS: tR = 1,964 min (método 3), m/z = 328,0 [M H]+. SFC: tR = 4,503 min, % ee =99,8 %;
[a]D20 14,7 (c = 0,10, DCM).
7-(3-fluorobencil)-6-metil-3-(tetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona, estereoisómero 2 (15,58 mg, 38 % de rendimiento).
1H RMN (CDCla 400 MHz): 57,90 (s, 1H), 7,29-7,27 (m, 1H), 6,98-6,92 (m, 2H), 6,89 (d, J = 9,6 Hz, 1H), 6,79 (s, 1H), 5,21 (s, 2H), 4,19 (t, J = 8,4 Hz, 1H), 4,11-4,09 (m, 1H), 4,05-4,03 (m, 1H), 3,97-3,95 (m, 1H), 3,66-3,62 (m, 1H), 2,38 (q, J = 7,6 Hz, 2H), 2,16 (s, 3H).
LC-MS: tR = 1,957 min (método 3), m/z = 328,0 [M H]+. SFC: tR = 4,779 min, % ee =96 %;
[a]D20 -14,0 (c = 0,10, DCM).
Ejemplo 32:

7-(3-fluorobencil)-6-metil-3-(3-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona, estereoisómero 1 y 2:
Etapa 1: A una disolución de (3-metoxi-5-metilpirazin-2-il)metanamina (200 mg, 1,3 mmoles) en DCM (6 mL) se añadió ácido 3-metiltetrahidrofuran-3-carboxílico (255 mg, 1,9 mmoles) y HATU (894 mg, 2,4 mmoles), Et3N (264 mg, 2,6 mmoles). La disolución se agitó a 20-25 °C durante 1 hora. Se añadió agua (40 ml), la mezcla se extrajo con DCM (40 mL x 2). La capa orgánica se lavó con salmuera (30 mL), se secó sobre Na2SO4 y se concentró en vacío. El residuo se purificó por cromatografía en gel de sílice (éter de petróleo:acetato de etilo=1:1) para proporcionar W-((3-metox¡-5-metilpirazin-2-il)metil)-3-metiltetrahidrofuran-3-carboxamida (300 mg, 67 % de rendimiento, 78 % de pureza).
Etapa 2: A una disolución de W-((3-metoxi-5-metilpirazin-2-il)metil)-3-metiltetrahidrofuran-3-carboxamida (400 mg, 1,5 mmoles) en dioxano (6 mL) se añadió POCl3 (880 mg, 5,7 mmoles). La disolución se agitó a 80-90 °C durante 2h. La mezcla se enfrió y se concentró en vacío. El residuo se diluyó con DCM (50 mL), se lavó con NaHCO3 acuoso saturado (50 mL) y salmuera (50 mL). La capa orgánica se secó sobre Na2SO4 y se concentró en vacío para proporcionar 8-metoxi-6-metil-3-(3-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazina (350 mg, 94 % de rendimiento).
Etapa 3: A una disolución de 8-metoxi-6-metil-3-(3-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazina (300 mg, 1,2 mmoles) en dioxano (8 mL) se añadió HCl(ac) 2M (4 mL). La disolución se agitó a 80-90 °C durante 1 hora. La mezcla se concentró en vacío y se añadió NaHCO3 acuoso saturado (50 mL). La mezcla se extrajo con DCM (50 mL x 2). La capa orgánica se lavó con salmuera (20 mL) y se concentró en vacío para proporcionar 6-metil-3-(3-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona (260 mg, 92 % de rendimiento).
Etapa 4: A una disolución de 6-metil-3-(3-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona (260 mg, 1,1 mmoles) en DMF anhidro (10 mL) se añadió K2CO3 (308 mg, 2,2 mmoles) y 1-(bromometil)-3-fluorobenceno (316 mg, 1,7 mmoles). La mezcla se agitó a 70-80 °C durante 2h. La mezcla se enfrió y se diluyó con agua (50 mL), se extrajo con EtOAc (50 mL x 2). La capa orgánica se lavó con salmuera (20 mL), se secó sobre Na2SO4 y se concentró en vacío para proporcionar 7-(3-fluorobencil)-6-metil-3-(3-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona (160 mg, 42 % de rendimiento).
Etapa 5: Se separó 7-(3-fluorobencil)-6-metil-3-(3-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona (160 mg, 0,47 mmoles) por SFC para proporcionar 7-(3-fluorobencil)-6-metil-3-(3-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona, estereoisómero 1 (37 mg, 23 % de rendimiento) como un sólido blanquecino. 1H RMN (CDCta 400 MHz): 5 7,88 (s, 1H), 7,32-7,26 (m, 1H), 6,94-6,82 (m, 4H), 5,21 (s, 2H), 4,33 (d, J = 8,8 Hz, 1H), 4,07-4,04 (m, 1H), 4,00 3,98 (m, 1H), 3,87 (d, J = 8,8 Hz, 1 H), 2,62-2,57 (m, 1H), 2,16 (s, 3H), 2,14-2,09 (m, 1H), 1,60 (s, 3H). LC-MS: tR = 2.47 min (método 3 ), m/z = 342,1 [M H]+. SFC: tR = 4,91 min, % ee > 99 %. [a]D20 =+5,0 (c = 0,10, MeOH).
7-(3-fluorobencil)-6-metil-3-(3-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona, estereoisómero 2 (44 mg, 27 % de rendimiento) como un sólido blanquecino.
1H RMN (CDCl3400 MHz): 57,82 (s, 1H), 7,24-7,20 (m, 1H), 6,94-6,82 (m, 4H), 5,15 (s, 2H), 4,27 (d, J = 8,8 Hz, 1H), 4,01-3,99 (m, 1H), 3,96-3,92 (m, 1H), 3,81 (d, J = 8,8 Hz, 1H), 2,57- 2,51 (m, 1H), 2,10 (s, 3H), 2,09-2,04 (m, 1H), 1,54 (s, 3H).
LC-MS: tR = 2,47 min (método 3), m/z = 342,1 [M H]+.
SFC: tR = 5,33 min, % ee > 99 %. [a]D20 =-3,0 (c = 0,10, MeOH).
Ejemplo 33:

7-(3-fluorobencil)-6-metil-3-(1-metilciclopropil)imidazo[1,5-a]pirazin-8(7H)-ona:
Etapa 1: A una disolución de ácido 1 -metilciclopropano-1 -carboxílico (500 mg, 4,99 mmoles) en DCM (2 mL) se añadió (COCl)2 (3,17 g, 24,95 mmoles). La disolución se agitó a 40 °C durante 2h. La mezcla de reacción se concentró en vacío para proporcionar cloruro de 1-metilciclopropano-1-carbonilo (500 mg, 85 % de rendimiento).
Etapa 2: Una disolución de (3-metoxi-5-metilpirazin-2-il)metanamina (100 mg, 0,65 mmoles) en DCM anhidro (3 mL) se enfrió hasta 0 °C. Después, se añadió gota a gota una disolución de cloruro de 1-metilciclopropano-1-carbonilo (100 mg, 0,85 mmoles) en DCM anhidro (2 mL) y se agitó a 0 °C durante 15 min. La mezcla se diluyó con DCM (20 mL), se lavó con NaHCO3 acuoso saturado (20 mL), salmuera (20 mL) y se secó sobre Na2SO4. La capa orgánica se concentró en vacío para proporcionar /V-((3-metoxi-5-metilpirazin-2-il)metil)-1-metilciclopropano-1-carboxamida (120 mg, 78 % de rendimiento).
LC-MS: tR = 0,66 min (método 2), m/z = 236,1 [M H]+.
Etapa 3: A una disolución de W-((3-metoxi-5-metilpirazin-2-il)metil)-1-metilciclopropano-1-carboxamida (120 mg, 0,51 mmoles) en dioxano (5 mL) se añadió POCl3 (590 mg, 3,85 mmoles). La disolución se agitó a 80-90 °C durante 2h. La mezcla se enfrió y se añadió lentamente en agua (50 mL), se extrajo con EtOAc (30 mL x 2). La capa orgánica se lavó con salmuera (20 mL), se secó sobre Na2SO4 y se concentró en vacío para proporcionar 8-metoxi-6-metil-3-(1-metilciclopropil)imidazo[1,5-a]pirazina (80 mg, 72 % de rendimiento).
Etapa 4: A una disolución de 8-metoxi-6-metil-3-(1-metilciclopropil)imidazo[1,5-a]pirazina (80 mg, 0,37 mmoles) en dioxano (5 mL) se añadió HCl (ac) 2M (2 mL). La disolución se agitó a 80-90 °C durante 1 hora. La mezcla se concentró en vacío y se añadió NaHCO3 acuoso saturado (50 mL). La mezcla se extrajo con DCM (50 mL x 2). La capa orgánica se lavó con salmuera (20 mL) y se concentró en vacío para proporcionar 6-metil-3-(1-metilciclopropil)imidazo[1,5-a]pirazin-8(7H)-ona (70 mg, 94 % de rendimiento).
Etapa 5: A una disolución de 6-metil-3-(1-metilciclopropil)imidazo[1,5-a]pirazin-8(7H)-ona (70 mg, 0,34 mmoles) en DMF anhidro (4 mL) se añadió 1-(bromometil)-3-fluoro-benceno (98 mg, 0,52 mmoles) y K2CO3 (95 mg, 0,69 mmoles). La mezcla se agitó a 70-80 °C durante 1 hora. La mezcla se enfrió y se filtró. El filtrado se purificó por LC-MS preparativa para proporcionar 7-(3-fluorobencil)-6-metil-3-(1-metilciclopropil)imidazo[1,5-a]pirazin-8(7H)-ona (35 mg, 32 % de rendimiento).
1H RMN (CDCla 400 MHz): 57,84 (s, 1H), 7,33-7,39 (m, 1H), 7,01-6,89 (m, 4H), 5,23 (s, 2H), 2,20 (s, 3H), 1,60 (s, 3H), 1,11-1,09 (m, 2H), 0,88-0,85 (m, 2H).
LC-MS: tR = 2,34 min (método 3), m/z = 312,1 [M H]+.
Ejemplo 34:

3-(2,2-difluorociclopropil)-7-(3-fluorobencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona:
Etapa 1: A una disolución de (3-metoxi-5-metilpirazin-2-il)metanamina (100 mg, 0,65 mmoles) en DCM (5 mL) se añadió ácido 2,2-difluorociclopropano-1-carboxílico (120 mg, 0,98 mmoles) y HATU (447 mg, 1,18 mmoles), Et3N (132 mg, 1,31 mmoles). La disolución se agitó a 20-25 °C durante 1 hora. La mezcla se diluyó con agua (20 mL), se extrajo con DCM (30 mL x 2). La capa orgánica se lavó con salmuera (20 mL), se secó sobre Na2SO4 y se concentró en vacío. El residuo se purificó por cromatografía en gel de sílice (éter de petróleo:acetato de etilo=1:1) para proporcionar 2,2-difluoro-W-((3-metoxi-5-metilpirazin-2-il)metil)ciclopropano-1-carboxamida (200 mg, 89 % de rendimiento, 75 % de pureza).
Etapa 2. A una disolución de 2,2-difluoro-W-((3-metoxi-5-metilpirazin-2-il)metil)ciclopropano-1-carboxamida (200 mg, 0,58 mmoles, 75 % de pureza) en dioxano (5 mL) se añadió POCh (1,12 g, 7,3 mmoles). La disolución se agitó a 90 °C durante 2h. La mezcla se enfrió y se concentró en vacío. El residuo se diluyó con DCM (50 mL), se lavó con NaHCO3(ac) acuoso saturado (50 mL) y salmuera (50 mL). La capa orgánica se secó sobre Na2SO4 y se concentró en vacío para proporcionar 3-(2,2-difluorociclopropil)-8-metoxi-6-metilimidazo[1,5-a]pirazina (120 mg, 86 % de rendimiento).
Etapa 3: A una disolución de 3-(2,2-difluorociclopropil)-8-metoxi-6-metilimidazo[1,5-a]pirazina (120 mg, 0,50 mmoles) en dioxano (5 mL) se añadió HCl(ac) 2M (3 mL). La disolución se agitó a 80 °C durante 1 hora. La mezcla se concentró en vacío y se añadió NaHCO3 acuoso saturado (50 mL). La mezcla se extrajo con DCM (50 mL x 2). La capa orgánica se lavó con salmuera (20 mL) y se concentró en vacío para proporcionar 3-(2,2-difluorociclopropil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (100 mg, 89 % de rendimiento).
LC-MS: tR = 0,94 min (método 10), m/z = 226,2 [M H]+.
Etapa 4: A una disolución de 3-(2,2-difluorociclopropil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (100 mg, 0,44 mmoles) en DMF anhidro (5 mL) se añadió K2CO3 (123 mg, 0,89 mmoles) y 1-(bromometil)-3-fluorobenceno (126 mg, 0,67 mmoles). La mezcla se agitó a 80 °C durante 2h. La mezcla se enfrió y se filtró. El filtrado se purificó por LC-MS preparativa para proporcionar 3-(2,2-difluorociclopropil)-7-(3-fluorobencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (40 mg, 27 % de rendimiento) como un sólido blanco.
1H RMN (CDCla 400 MHz): 57,90 (s, 1H), 7,30-7,28 (m, 1H), 6,98-6,84 (m, 4H), 5,28-5,16 (m, 2H), 2,80-2,73 (m, 1H), 2,38-2,37 (m, 1H), 2,19 (s, 3H), 2,05-2,04 (m, 1H).
LC-MS: tR = 2,67 min (método 3), m/z = 334,1 [M H]+.
Ejemplo 35:
7-(3-fluorobencil)-6-metil-3-(2-metilciclopropil)imidazo[1,5-a]pirazin-8(7H)-ona, estereoisómero 1,2, 3 y 4:
Etapa 1: A una disolución de (3-metoxi-5-metilpirazin-2-il)metanamina (100 mg, 652,8 pmoles) y ácido 2-metilciclopropano-1-carboxílico (98 mg, 979,2 pmoles) en DCM (5 mL) se añadió HATU (446,8 mg, 1,2 mmoles) y trietilamina (132,1 mg, 1,3 mmoles). La mezcla se agitó a 24 °C durante 16h. La mezcla se diluyó con DCM (20 mL) y se lavó con agua (15 mL). La capa acuosa se extrajo con DCM (2 x 30 mL). La capa orgánica combinada se lavó con salmuera (20 mL), se secó sobre Na2SO4, se filtró y se concentró. El residuo se purificó por TLC preparativa (acetato de etilo) para proporcionar W-((3-metoxi-5-metilpirazin-2-il)metil)-2-metilciclopropano-1 -carboxamida (150 mg, 95 % de rendimiento).
Etapa 2: A una disolución de W-((3-metoxi-5-metilpirazin-2-il)metil)-2-metilciclopropano-1-carboxamida (150 mg, 636 pmoles) en dioxano (5 mL) se añadió POCl3 (400 mg, 2,6 mmoles). La mezcla se agitó a 90 °C durante 2h. La mezcla se enfrió hasta 25 °C, se neutralizó con NaHCO3 acuoso saturado y se extrajo con acetato de etilo (3 x 30 mL). La capa orgánica combinada se lavó con salmuera (30 mL), se secó sobre Na2SO4, se filtró y se concentró para proporcionar la 8-metoxi-6-metil-3-(2-metilciclopropil)imidazo[1,5-a]pirazina cruda (130 mg, 94 % de rendimiento). Este producto crudo se usó directamente para la siguiente etapa.
Etapa 3: Una disolución de 8-metoxi-6-metil-3-(2-metilciclopropil)imidazo[1,5-a]pirazina (120 mg, 552,3 pmoles) en dioxano (5 mL) y HCl (2 M, 2 mL) se agitó a 80 °C durante 1 hora. La mezcla se enfrió hasta 25 °C, se neutralizó con NaHCO3 acuoso saturado y se extrajo con acetato de etilo (3 x 20 mL). La capa orgánica combinada se lavó con salmuera (20 mL), se secó sobre Na2SO4, se filtró y se concentró para proporcionar la 6-metil-3-(2-metilciclopropil)imidazo[1,5-a]pirazin-8(7H)-ona cruda (100 mg, 89 % de rendimiento). Este producto crudo se usó directamente para la siguiente etapa.
Etapa 4: A una disolución de 6-metil-3-(2-metilciclopropil)imidazo[1,5-a]pirazin-8(7H)-ona (100 mg, 492,0 pmoles) y 1 -(bromometil)-3-fluorobenceno (139,5 mg, 738,0 pmoles) en DMF (5 mL) se añadió K2CO3 (136 mg, 984 pmoles). La mezcla se agitó a 60-70 °C durante 16h. La mezcla se enfrió hasta 25 °C, se diluyó con agua (15 mL), se extrajo con acetato de etilo (3 x 30 mL). La capa orgánica combinada se lavó con salmuera (30 mL), se secó sobre Na2SO4, se filtró y se concentró. El residuo se purificó por cromatografía en columna en gel de sílice (eluyente de acetato de etilo al 0 %~50 % en éter de petróleo) para proporcionar 7-(3-fluorobencil)-6-metil-3-(2-metilciclopropil)imidazo[1,5-a]pirazin-8(7H)-ona (80 mg, 52 % de rendimiento).
Etapa 5: Se purificó 7-(3-fluorobencil)-6-metil-3-(2-metilciclopropil)imidazo[1,5-a]pirazin-8(7H)-ona (100 mg, 321,2 pmoles) por SFC.
Se obtuvo 7-(3-fluorobencil)-6-metil-3-(2-metilciclopropil)imidazo[1,5-a]pirazin-8(7H)-ona, estereoisómero 1 (35 mg, 27 % de rendimiento).
1H RMN (CDCl3400 MHz): 57,83 (s, 1H), 7,33-7,29 (m, 1H), 7,00-6,94(m, 2H), 6,91-6,89 (m, 2H), 5,23 (s, 2H), 2,19 (s, 3H), 1,59-1,55 (m, 1H), 1,54-1,50 (m, 1H), 1,35-1,30 (m, 1H), 1,27 (d, J= 6,0 Hz, 3H), 0,89-0,88 (m, 1H).
LC-MS: tR = 2,03 min (método 3), m/z = 312,1 [M H]+. SFC: tR = 4,466 min, % ee > 99 %.
[a]D20 29,3 (c = 0,10, CHCh).
Se obtuvo 7-(3-fluorobencil)-6-metil-3-(2-metilciclopropil)imidazo[1,5-a]pirazin-8(7H)-ona, estereoisómero 2 (26 mg, 26 % de rendimiento).
1H RMN (CDCl3400 MHz): 57,84 (s, 1H), 7,32-7,29 (m, 1H), 7,00-6,96(m, 2H), 6,90-6,89 (m, 2H), 5,23 (s, 2H), 2,19 (s, 3H), 1,62-1,60 (m, 1H), 1,55-1,53 (m, 1H), 1,36-1,34 (m, 1H), 1,27 (d, J= 5,6 Hz, 3H), 0,91-0,89 (m, 1H).
LC-MS: tR = 2,02 min (método 3), m/z = 312,1 [M H]+. SFC: tR = 5,227 min, % ee > 99 %.
[a]D20 -15,0 (c = 0,10, CHCla).
Se obtuvo 7-(3-fluorobencil)-6-metil-3-(2-metilciclopropil)imidazo[1,5-a]pirazin-8(7H)-ona, estereoisómero 3 (8,0 mg, 8 % de rendimiento).1
1H RMN (CDCl3400 MHz): 57,89 (s, 1H), 7,33-7,30 (m, 1H), 7,01-6,91 (m, 4H), 5,29 (d, J= 16,0 Hz, 1H), 5,17 (d, J= 16,0 Hz, 1H), 2,19 (s, 3H), 2,01 -1,97(m, 1H), 1,39-1,27 (m, 1H), 1,23-1,20 (m, 2H), 0,91 (d, J= 6,0 Hz, 3H).
LC-MS: tR = 1,98 min (método 3), m/z = 312,1 [M H]+. SFC: tR = 6,995 min, % ee > 99 %.
[a]D20 37,0 (c = 0,10, CHCh).
Se obtuvo 7-(3-fluorobencil)-6-metil-3-(2-metilciclopropil)imidazo[1,5-a]pirazin-8(7H)-ona, estereoisómero 4 (9,0 mg, 9 % de rendimiento).
1H RMN (CDCI3400 MHz): 57,86 (s, 1H), 7,29-7,26 (m, 1H), 6,99-6,88(m, 4H), 5,27 (d, J= 16,0 Hz, 1H), 5,15 (d, J= 16,0 Hz, 1H), 2,17 (s, 3H), 1,97-1,95(m, 1H), 1,58-1,53 (m, 1H), 1,23-1,20 (m, 2H), 0,91 (d, J= 6,0 Hz, 3H).
LC-MS: tR = 1,98 min (método 3), m/z = 312,1 [M H]+. SFC: tR = 8,704 min, % ee > 99 %.
[a]D20 -66,7 (c = 0,10, CHCla).
Ejemplo 36:

7-(3-fIuorobencil)-6-metil-3-(2-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona, estereoisómero 1,2, 3 y 4:
Etapa 1: A una disolución de (3-metoxi-5-metilpirazin-2-il)metanamina (300 mg, 2,0 mmoles), ácido 2-metiltetrahidrofuran-3-carboxílico (382 mg, 2,9 mmoles) en DCM (10 mL) se añadió HATU (1,3 g, 3,5 mmoles) y trietilamina (396 mg, 3,9 mmoles). La mezcla se agitó a 24 °C durante 16h. La mezcla se diluyó con DCM (30 mL) y se lavó con agua (20 mL). La capa acuosa se extrajo con DCM (2 x 30 mL). La capa orgánica combinada se lavó con salmuera (30 mL), se secó sobre Na2SO4, se filtró y se concentró. El residuo se purificó por cromatografía en columna en gel de sílice (acetato de etilo al 0 %~70 % en éter de petróleo) para proporcionar W-((3-metoxi-5-metilpirazin-2-il)metil)-2-metiltetrahidrofuran-3-carboxamida (350 mg, 62 % de rendimiento).
Etapa 2: A una disolución de W-((3-metoxi-5-metilpirazin-2-il)metil)-2-metiltetrahidrofuran-3-carboxamida (350 mg, 1,3 mmoles) en dioxano (5 mL) se añadió POCh (720 mg, 4,7 mmoles). La mezcla se agitó a 90 °C durante 2h. La mezcla se enfrió hasta 25 °C, se neutralizó con NaHCO3 ac. saturado y se extrajo con acetato de etilo (2 x 30 mL). La capa orgánica combinada se lavó con salmuera (20 mL), se secó sobre Na2SO4, se filtró y se concentró para proporcionar la 8-metoxi-6-metil-3-(2-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazina cruda (320 mg). El crudo se usó directamente para la siguiente etapa.
Etapa 3: Una disolución de 8-metoxi-6-metil-3-(2-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazina (320 mg, 1,3 mmoles) en dioxano (5 mL) y HCl (2 M, 2 mL) se agitó a 80 °C-90 °C durante 21,5 horas. La mezcla se enfrió hasta 25 °C, se neutralizó con NaHCO3 ac. saturado, se extrajo con DCM (3 x 30 mL). La capa orgánica combinada se lavó con salmuera (30 mL), se secó sobre Na2SO4, se filtró y se concentró para proporcionar la 6-metil-3-(2-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona cruda (300 mg). El producto crudo se usó directamente para la siguiente etapa.
Etapa 4: A una disolución de 6-metil-3-(2-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona (250 mg, 1.1 mmoles) en DMF (8 mL) se añadió 1-(bromometil)-3-fluorobenceno (304 mg, 1,6 mmoles) y K2CO3 (296 mg, 2.1 mmoles). La mezcla se agitó a 60-70 °C durante 16h. La mezcla se enfrió hasta 25 °C y se diluyó con agua (15 mL), se extrajo con acetato de etilo (3 x 30 mL). La capa orgánica combinada se lavó con salmuera (30 mL), se secó sobre Na2SO4, se filtró y se concentró. El residuo se purificó por cromatografía en columna en gel de sílice (eluyente de acetato de etilo al 0 %~30 % en éter de petróleo) para proporcionar 7-(3-fluorobencil)-6-metil-3-(2-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona (170 mg, 47 % de rendimiento).
Etapa 5: Se purificó 7-(3-fluorobencil)-6-metil-3-(2-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona (220 mg, 644 emoles) por SFC.
7-(3-fluorobencil)-6-metil-3-(2-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona, estereoisómero 1 (20 mg, 9 % de rendimiento) .
1H RMN (CDCla 400 MHz400 MHz): 5 7,95 (s, 1H), 7,34-7,28 (m, 1H), 7,01-6,89 (m, 3H), 6,79 (s, 1H), 5,23 (s, 2H), 4,35-4,30 (m, 1H), 4,13-4,07 (m, 2H), 3,12-3,06 (m, 1H), 2,50-2,37 (m, 2H), 2,19 (s, 3H), 1,35 (d, J= 6,0 Hz, 3H.
LC-MS: tR = 2,12 min (método 3), m/z = 342,1 [M H]+.
SFC: tR = 4,812 min, % ee > 99 %. [a]D20 -24,3 (c = 0,10, CHCla).
7-(3-fluorobencil)-6-metil-3-(2-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona, estereoisómero 2 (10 mg, 5 % de rendimiento) .1
1H RMN (CDCl3400 MHz): 57,93 (s, 1H), 7,31-7,26 (m, 1H), 6,99-6,87 (m, 3H), 6,76 (s, 1H), 5,21 (s, 2H), 4,32-4,27 (m, 1H), 4,10-4,04 (m, 2H), 3,10-3,03 (m, 1H), 2,48-2,35 (m, 2H), 2,17 (s, 3H), 1,33 (d, J= 6,4 Hz, 3H).
LC-MS: tR = 2,07 min (método 3), m/z = 342,1 [M H]+.
SFC: tR = 5,088 min, % ee = 97,9 %. [a]p20 10,3 (c = 0,10, CHCI3).
7-(3-fIuorobenciI)-6-metiI-3-(2-metiItetrahidrofuran-3-iI)imidazo[1,5-a]pirazin-8(7H)-ona, estereoisómero 3 (38,0 mg, 17 % de rendimiento) .
1H RMN (CDCI3400 MHz): 57,95 (s, 1H), 7,31-7,29 (m, 1H), 7,01-6,89 (m, 3H), 6,83 (s, 1H), 5,29 (d, J= 16,4 Hz, 1H), 5,17 (d, J= 16,4 Hz, 1H), 4,35-4,28 (m, 2H), 3,91-3,87 (m, 1H), 3,71-3,67 (m, 1H), 2,72-2,67 (m, 1H), 2,44-2,40 (m, 1H), 2,18 (s, 3H), 0,90 (d, J= 6,4 Hz, 3H).
LC-MS: tR = 2,02 min (método 3), m/z = 342,1 [M H]+. SFC: tR = 5,516 min, % ee > 99 %.
[a]D20 46,3 (c = 0,10, CHCI3).
7-(3-fIuorobenciI)-6-metiI-3-(2-metiItetrahidrofuran-3-iI)imidazo[1,5-a]pirazin-8(7H)-ona, estereoisómero 4 (30,0 mg, 14 % de rendimiento). 1H RMN (CDCI3400 MHz): 57,95 (s, 1H), 7,33- 7,30 (m, 1H), 7,01-6,90 (m, 3H), 6,83 (s, 1H), 5,29 (d, J= 16,4 Hz, 1H), 5,17 (d, J= 16,4 Hz, 1H), 4,37-4,28 (m, 2H), 3,91-3,87 (m, 1H), 3,71-3,69 (m, 1H), 2,72-2,68 (m, 1H), 2,45-2,40 (m, 1H), 2,18 (s, 3H), 0,90 (d, J= 6,8 Hz, 3H).
LC-MS: tR = 1,98 min (método 3), m/z = 342,1 [M H]+. SFC: tR = 6,304 min, % ee > 99 %.
[a]D20 -47,0 (0 = 0,10, CHCI3).
EjempIo 37:

7-(3-fIuorobenc¡I)-3-(c/s-2-fIuorocicIoprop¡I)-6-met¡I¡m¡dazo[1,5-a]p¡raz¡n-8(7H)-ona, estereoisómero 1 y 2:
Etapa 1: Una disoIución de hidrocIoruro de (3-metoxi-5-metiIpirazin-2-iI)metanamina (400 mg, 2,1 mmoIes) y trietiIamina (662 mg, 6,5 mmoIes) en DCM (8 mI) se enfrió hasta 0 °C, se añadió gota a gota cIoruro de 2-fIuorocicIopropano-1-carboniIo (251 mg, 2,1 mmoIes) y Ia mezcIa se agitó a 0 °C durante 0,5h. La mezcIa se diIuyó con agua (10 mL), se extrajo con DCM (20 mL x 2). La capa orgánica se Iavó con saImuera (20 mL), se secó sobre Na2SÜ4 y se concentró en vacío. EI residuo se purificó por TLC preparativa (éter de petróIeo:acetato de et¡Io=1:1) para proporcionar c/'s-2-fIuoro-W-((3-metoxi-5-metiIpirazin-2-iI)metiI)cicIopropano-1-carboxamida (200 mg, 40 % de rendimiento) y (1 S,2R)-2-fIuoro-W-((3- metoxi-5-metiIpirazin-2-iI)met¡I)cicIopropano-1-carboxamida (200 mg, 40 % de rendimiento) en totaI.
Etapa 2: A una disoIución de c/s-2-fIuoro-W-((3-metoxi-5-metiIpirazin-2-iI)met¡I)cicIopropano-1-carboxamida (260 mg, 1,1 mmoIes) en dioxano (10 mI.) se añadió POCI3 (500 mg, 3,3 mmoIes). La disoIución se agitó a 80-90 °C durante 2h. La mezcIa se concentró en vacío y se diIuyó con NaHCO3 (30 mL), se extrajo con DCM (50 mL x 2). La capa orgánica se Iavó con saImuera (30 mL), se secó sobre Na2SO4 y se concentró en vacío para proporcionar 3-(c/s-2-fIuorocicIopropiI)-8-metoxi-6-met¡Iimidazo[1,5-a]pirazina (220 mg, 91 % de rendimiento).
Etapa 3: Una disoIución de 3-(c/s-2-fIuorocicIopropiI)-8-metoxi-6-metiIimidazo[1,5-a]pirazina (220 mg, 994 gmoles) en HCI 2N (ac) (5 mL) y dioxano (10 mL) se agitó a 80-90 °C durante 1 hora. La mezcIa se concentró en vacío. EI residuo se diIuyó con NaHCO3(ac) (30 mL), se extrajo con DCM (30 mL x 3). La capa orgánica se Iavó con saImuera (30 mL), se secó sobre Na2SO4 y se concentró en vacío para proporcionar 3-(c/s-2-fIuorocicIopropiI)-6-metiIimidazo[1,5-a]pirazin-8(7H)-ona (200 mg, 97 % de rendimiento).
Etapa 4: A una disoIución de 3-(c/s-2-fIuorocicIopropiI)-6-metiIimidazo[1,5-a]pirazin-8(7H)-ona (200 mg, 965 gmoIes) en DMF anhidro (10 mL) se añadió K2CO3 (133 mg, 965 gmoIes) y 1-(bromomet¡I)-3-fIuorobenceno (274 mg, 1,5 mmoIes). La mezcIa se agitó a 60-70 °C durante 16h y a 80 °C durante 21 h. La mezcIa se enfrió y se fiItró y eI fiItrado se purificó por LC-MS preparativa para proporcionar 7-(3-fIuorobenciI)-3-(c/s-2-fIuorocicIopropiI)-6-met¡Iimidazo[1,5-a]pirazin-8(7H)-ona (80 mg, 26 % de rendimiento).
Etapa 5: Se separó 7-(3-fluorobencil)-3-(c/s-2-fluorociclopropil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (80 mg, 253 gmoles) por SFC.
7-(3-fluorobencil)-3-(c/s-2-fluorociclopropil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona, estereoisómero 1 (25 mg, 30 % de rendimiento).
1H RMN (CDCls Varian_H_400 MHz): 5 7,92 (s, 1H), 7,32-7,26 (m, 1H), 7,00-6,88 (m, 4H), 5,29-5,15 (m, 2H), 5,02 4,84 (m, 1H), 2,18 (s, 3H), 2,12-1,99 (m, 2H), 1,44-1,25 (m, 1H).
LC-MS: tR = 2,00 min (método 8), m/z = 316,1 [M H]+. SFC: tR = 6,53 min, % ee > 99 %. [a]D20 -84,00 (c = 0,10, MeOH).
7-(3-fluorobencil)-3-(c/s-2-fluorociclopropil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona, estereoisómero 2 (15 mg, 18 % de rendimiento). 1H RMN (CDCla Varian_H_400 MHz): 57,93 (s, 1H), 7,33-7,30 (m, 1H), 7,00-6,90 (m, 4H), 5,31-5,17 (m, 2H), 5,04-4,86 (m, 1H), 2,20 (s, 3H), 2,12-2,00 (m, 2H), 1,46-1,42 (m, 1H).
LC-MS: tR = 1,99 min (método 8), m/z = 316,1 [M H]+. SFC: tR = 5,06 min, % ee > 99 %. [a]D20 75,00 (c = 0,10, MeOH). Ejemplo 38:

7-(3-fluorobencil)-3-(trans-2-fluorociclopropil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona, estereoisómero 1 y 2:
Etapa 1: A una disolución de trans-2-fluoro-W-((3-metoxi-5-metilpirazin-2-il)metil)ciclopropano-1 -carboxamida (280 mg, 1,1 mmoles) en dioxano (10 mL) se añadió POCta (540 mg, 3,5 mmoles). La disolución se agitó a 80-90 °C durante 2h. La mezcla se concentró en vacío y se diluyó con NaHCO3 (30 mL), se extrajo con DCM (50 mL x 2). La capa orgánica se lavó con salmuera (30 mL), se secó sobre Na2SO4 y se concentró en vacío para proporcionar 3-(trans-2-fluorociclopropil)-8-metoxi-6-metilimidazo[1,5-a]pirazina (240 mg, 93 % de rendimiento).
Etapa 2: Una disolución de 3-(trans-2-fluorociclopropil)-8-metoxi-6-metilimidazo[1,5-a]pirazina (240 mg, 1,1 mmoles) en HCl (ac) 2N (5 mL) y dioxano (10 mL) se agitó a 80-90 °C durante 1 hora. La mezcla se concentró en vacío. El residuo se diluyó con NaHCO3(ac) (30 mL), se extrajo con DCM (30 mL x 3). La capa orgánica se lavó con salmuera (30 mL), se secó sobre Na2SO4 y se concentró en vacío para proporcionar 3-(trans-2-fluorociclopropil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (220 mg, 98 % de rendimiento).
Etapa 3: A una disolución de 3-(trans-2-fluorociclopropil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (220 mg, 1,1 mmoles) en DMF anhidro (10 mL) se añadió K2CO3 (293 mg, 2,1 mmoles) y 1-(bromometil)-3-fluorobenceno (300 mg, 1,6 mmoles). La mezcla se agitó a 60-70 °C durante 16h y a 80 °C durante 21 h. La mezcla se enfrió y se filtró y el filtrado se purificó por LC-MS preparativa para proporcionar 7-(3-fluorobencil)-3-(trans-2-fluorociclopropil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (250 mg, 77 % de rendimiento).
Etapa 4: Se separó 7-(3-fluorobencil)-3-(trans-2-fluorociclopropil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (250 mg, 793 gmoles) por SFC.
7-(3-fluorobencil)-3-(trans-2-fluorociclopropil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona, estereoisómero 1 (15 mg, 17 % de rendimiento).
1H RMN (CDCls 400 MHz): 57,81 (s, 1H), 7,31-7,27 (m, 1H), 6,98-6,86 (m, 4H), 5,22 (s, 2H), 5,01-4,84 (m, 1H), 2,43 2,38 (m, 1H), 2,19 (s, 3H), 1,72-1,65 (m, 1H), 1,58-1,55 (m, 1 H).
LC-MS: tR = 2,24 min (método 8), m/z = 316,1 [M H]+. SFC: tR = 2,83 min, % ee > 99 %. [a]D20 29,00 (c = 0,10, MeOH).
7-(3-fluorobencil)-3-(trans-2-fluorociclopropil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona, estereoisómero 2 (45 mg, 17 % de rendimiento).
1H RMN (CDCls 400 MHz): 57,81 (s, 1H), 7,29-7,28 (m, 1H), 6,98-6,86 (m, 4H), 5,22 (s, 2H), 5,01-4,84 (m, 1H), 2,45 2,38 (m, 1H), 2,19 (s, 3H), 1,71-1,64 (m, 1H), 1,59-1,54 (m, 1H).
LC-MS: tR = 2,23 min (método 8), m/z = 316,1 [M H]+. SFC: tR = 3,69 min, % ee > 99 %. [a]D20 -31 (c = 0,10, MeOH).
Ejemplo 39:
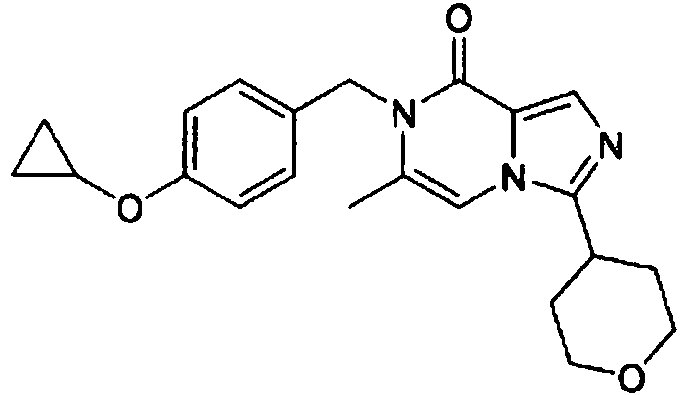
7-(4-ciclopropoxibencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:
Etapa 1: A una disolución de 1-bromo-4-ciclopropoxibenceno (900 mg, 4,22 mmoles) en THF (20 mL, anhidro) se añadió n-BuLi (2,5 M, 2,6 mL) a -78 °C y se agitó a -78 °C durante 2 horas bajo N2. Después, a esto se añadió gota a gota DMF (926 mg, 12,66 mmoles, anhidro) a - 78 °C y se agitó durante 2 horas. La disolución se paró con NH4Cl (ac.
1mL) a - 78 °C y se agitó a 0 °C durante 0,5 horas. La mezcla se diluyó con acetato de etilo (10 mL). La mezcla se filtró y el filtrado se concentró en vacío. El residuo se diluyó con acetato de etilo (30 mL), se lavó con salmuera (3 x 15 mL). La capa orgánica se secó con Na2SO4 anhidro, se filtró y se concentró en vacío para proporcionar 4-ciclopropoxibenzaldehído (700 mg). LC-MS: tR = 0,800 min (método 2), m/z = 162,8 [M H]+.
Etapa 2: A una disolución de 4-ciclopropoxibenzaldehído (700 mg) en MeOH (15 mL, anhidro) se añadió NaBH4 (319 mg, 8,44 mmoles) a 0 °C y se agitó a 0 °C durante 1 hora. La disolución se paró con NH4Cl acuoso saturado (ac.
0,5mL). La mezcla se concentró en vacío. El residuo se purificó por cromatografía en gel de sílice (éter de petróleo/acetato de etilo=10/1, 1/1) para proporcionar (4-ciclopropoxifenil)metanol (540 mg, 78 % de rendimiento). Etapa 3: Una disolución de (4-ciclopropoxifenil)metanol (500 mg, 3,1 mmoles) y Et3N (617 mg, 6,1 mmoles) en DCM anhidro (10 mL) se enfrió hasta 0 °C, después se añadió gota a gota MsCl (1,41 g, 12,3 mmoles). La disolución se agitó a 0 °C durante 0,5h. La mezcla se diluyó con agua (50 mL), se extrajo con DCM (50 mL x 2). La capa orgánica se lavó con salmuera (30 mL), se secó sobre Na2SO4 y se concentró en vacío para rendir metanosulfonato de 4-ciclopropoxibencilo (600 mg, 81 % de rendimiento).
Etapa 4: Una mezcla de 6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (250 mg, 1,07 mmoles), metanosulfonato de 4-ciclopropoxibencilo (311 mg, 1,28 mmoles) y Cs2CO3 (698 mg, 2,14 mmoles) en DMF (6,0 mL, anhidro) se agitó a 60 °C durante 12 horas. La mezcla se filtró y el filtrado se concentró en vacío. El residuo se purificó por cromatografía en gel de sílice (éter de petróleo/acetato de etilo=10/1, 0/1) para proporcionar 7-(4-ciclopropoxibencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (320 mg, 77 % de rendimiento).
1H RMN (CDCla, 400 MHz): 57,92 (s, 1H), 7,15 (d, J = 8,8 Hz, 2H), 6,99 (d, J = 8,8 Hz, 2H), 6,73 (s, 1H), 5,17 (s, 2H), 4,12 (d, J = 10,4 Hz, 2H), 3,71 - 3,69 (m, 1H), 3,67 - 3,55 (m, 2H), 3,09 - 3,07 (m, 1H), 2,21 (s, 3H), 2,18 - 2,03 (m, 2H), 1,88 (d, J = 13,2 Hz, 2H), 0,79 - 0,71 (m, 4H).
LC-MS: tR = 2,154 min (método 3), m/z = 380,1 [M H]+.
Ejemplo 40:

7-(4-(difluorometoxi)bencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona
Una mezcla de 6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (400 mg, 1,7 mmoles), 1-(bromometil)-4-(difluorometoxi)benceno (608 mg, 2,6 mmoles), Cs2CO3 (1,11 g, 3,4 mmoles) en DMF (50 mL) se agitó a 60 °C durante 12 horas. La mezcla de reacción se concentró. El residuo se purificó por cromatografía en gel de sílice (éter de petróleo:acetato de etilo=5:1~0:1) para proporcionar 7-(4-(difluorometoxi)bencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (360 mg, rendimiento: 52 %).
1H RMN (CDCI3, 400 MHz): 57,91 (s, 1H), 7,20 (d, J = 9,2 Hz, 2H), 7,07 (d, J = 8,0 Hz, 2H), 6,73 (s, 1H), 6,46 (t, J = 73,6 Hz, 1H), 5,19 (s, 2H), 4,11 (d, J = 10,8 Hz, 2H), 3,56 (td, J = 12,0, 2,0 Hz, 2H), 3,10 - 3,04 (m, 1H), 2,17 - 2,06 (m, 5H), 1,86 (d, J = 13,2 Hz, 2H).
LC-MS: tR = 1,85 min (método 3), m/z = 390,1 [M H]+.
Ejemplo 41:

6-met¡l-3-(tetrah¡dro-2H-p¡ran-4-¡l)-7-(4-(trifluorometox¡)benc¡l)¡m¡dazo[1,5-a]p¡raz¡n-8(7H)-ona:
Una mezcla de 6-metil-3-(tetrah¡dro-2H-p¡ran-4-¡l)¡m¡dazo[1,5-a]p¡raz¡n-8(7H)-ona (400 mg, 1,7 mmoles), 1-(bromomet¡l)-4-(tr¡fluorometox¡)benceno (654 mg, 2,6 mmoles) y Cs2CÜ3 (1,11 g, 3,4 mmoles) en DMF (50 mL) se ag¡tó a 60 °C durante 12h. La mezcla de reacc¡ón se concentró. El res¡duo se purificó por cromatografía en gel de síl¡ce (éter de petróleo:acetato de etilo=5:1~0:1) para proporc¡onar 6-metil-3-(tetrahidro-2H-p¡ran-4-¡l)-7-(4-(trifluorometox¡)benc¡l)¡m¡dazo[1,5-a]p¡raz¡n-8(7H)-ona (300 mg, rend¡m¡ento: 41 %).
1H RMN (CDCla, 400 MHz): 57,91 (s, 1H), 7,25 - 7,22 (m, 2H), 7,17 - 7,15 (m, 2H), 6,74 (s, 1H), 5,21 (s, 2H), 4,11 (d, J = 10,4 Hz, 2H), 3,56 (td, J = 11,2, 2,0 Hz, 2H), 3,10 - 3,04 (m, 1H), 2,17 - 2,06 (m, 5H), 1,86 (d, J = 13,6 Hz, 2H).
LC-MS: tR = 2,05 m¡n (método 3), m/z = 408,1 [M H]+.
Ejemplo 42:

7-(4-(c¡clopropilmetox¡)benc¡l)-6-met¡l-3-(tetrah¡dro-2H-p¡ran-4-¡l)¡m¡dazo[1,5-a]pirazin-8(7H)-ona:
Etapa 1: Una mezcla de 4-h¡drox¡benzaldehído (1,0 g, 8,19 mmoles), (bromomet¡l)c¡clopropano (1,33 g, 9,83 mmoles) y K2CO3 (2,26 g, 16,38 mmoles) en DMF (10,0 mL, anh¡dro) se ag¡tó a 20 °C durante 12 horas. La d¡soluc¡ón se d¡luyó con agua (20 mL). La fase acuosa se extrajo con acetato de etilo (60 mL x 3). La fase orgán¡ca comb¡nada se lavó con salmuera (20 mL x 1), se secó sobre Na2SO4 anh¡dro, se f¡ltró y se concentró en vacío. El res¡duo se pur¡f¡có por cromatografía en gel de sílice (éter de petróleo/acetato de et¡lo=10/1, 1/1) para proporc¡onar 4-(c¡cloprop¡lmetox¡)benzaldehído (1,3 g, 87 % de rend¡m¡ento).
Etapa 2: Una d¡soluc¡ón de 4-(c¡cloprop¡lmetox¡)benzaldehído (1,3 g, 7,4 mmoles) en MeOH (30 mL) se enfrió hasta 0 °C, después se añad¡ó NaBH4 (558 mg, 14,8 mmoles) y la mezcla se ag¡tó a 0 °C durante 1 hora. La mezcla se paró con salmuera sat. (ac) (50 mL), se extrajo con EtOAc (50 mL x 2). La capa orgán¡ca se lavó con salmuera, se secó sobre Na2SO4 y se concentró en vacío para rend¡r (4-(c¡cloprop¡lmetox¡)fen¡l)metanol (1,21 g, 92 % de rend¡m¡ento).
Etapa 3. Una d¡soluc¡ón de (4-(c¡cloprop¡lmetox¡)fen¡l)metanol (800 mg, 4,5 mmoles) y Et3N (907 mg, 9,0 mmoles) en DCM (10 mL) se enfrió hasta 0 °C, después se añad¡ó gota a gota MsCl (617 mg, 5,4 mmoles). La d¡soluc¡ón se agitó a 0 °C durante 0,5h. La mezcla se diluyó con agua (50 mL), se extrajo con DCM (50 mL x 2). La capa orgánica se lavó con salmuera (30 mL), se secó sobre Na2SO4 y se concentró en vacío para rendir metanosulfonato de 4-(ciclopropilmetox¡)benc¡lo (760 mg, 66 % de rendimiento).
Etapa 4: Una mezcla de 6-metil-3-(tetrah¡dro-2H-p¡ran-4-¡l)¡m¡dazo[1,5-a]p¡raz¡n-8(7H)-ona (300 mg, 1,29 mmoles), metanosulfonato de 4-(ciclopropilmetox¡)benc¡lo (429 mg, 1,68 mmoles) y Cs2CO3 (841 mg, 2,58 mmoles) en DMF (6 mL, anhidro) se agitó a 60 °C durante 12 horas. La mezcla se filtró y el filtrado se concentró en vacío. El residuo se purificó por cromatografía en gel de sílice (altura de la columna: 250 mm, diámetro: 100 mm, gel de sílice de malla
100-200, Éter de petróleo/Acetato de etilo=10/1, 0/1) para proporcionar 7-(4-(ciclopropilmetoxi)bencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (360 mg, 68 % de rendimiento).
1H RMN (CDCl3, 400 MHz): 57,92 (s, 1H), 7,14 (d, J = 8,8 Hz, 2H), 6,85 (d, J = 9,2 Hz, 2H), 6,73 (s, 1H), 5,16 (s, 2H), 4,12 (d, J = 10,8 Hz, 2H), 3,77 (d, J = 6,8 Hz, 2H), 3,61 - 3,55 (m, 2H), 3,10 - 3,04 (m, 1H), 2,20 - 2,09 (m, 5H), 1,88 (d, J = 13,2 Hz, 2H), 1,27 - 1,24 (m, 1H), 0,66 - 0,61 (m, 2H), 0,35 - 0,33 (m, 2H).
LC-MS: tR = 2,238 min (método 3), m/z = 394,1 [M H]+.
Ejemplo 43:

7-bencil-6-etil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:
Una mezcla de 6-etil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (100 mg, 404 pmoles), bromometilbenceno (83 mg, 485,26 pmoles) y Cs2CO3 (264 mg, 809 pmoles) en DMF (3,0 mL) se agitó a 60 °C durante 16 horas. La mezcla de reacción se filtró y el filtrado se concentró. El residuo se purificó por HPLC para proporcionar 7-bencil-6-etil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (56,0 mg, 41,0 % de rendimiento).
1H RMN (CDCla 400 MHz): 57,91 (s, 1H), 7,32 - 7,26 (m, 3H), 7,22 - 7,15 (m, 2H), 6,69 (s, 1H), 5,25 (s, 2H), 4,12 (d, J = 10,8 Hz, 2H), 3,62 - 3,56 (m, 2H), 3,14 - 3,07 (m, 1H), 2,53 - 2,48 (m, 2H), 2,17 - 2,09 (m, 2H), 1,89 (d, J = 13,2 Hz, 2H), 1,22 (t, J = 7,2 Hz, 3H).
LC-MS: tR = 2,042 min (método 3), m/z = 338,2 [M H]+.
Ejemplo 44:

6-etil-7-(4-metoxibencil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:
Una mezcla de 6-etil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (100 mg, 404 pmoles), 1-(bromometil)-4-metoxi-benceno (98 mg, 485 pmoles) y Cs2CO3 (264 mg, 809 pmoles) en DMF (5,0 mL) se agitó a 60 °C durante 16 horas. La mezcla de reacción se filtró y el filtrado se concentró. El residuo se purificó por HPLC para proporcionar 6-etil-7-(4-metoxibencil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (65 mg, 42,3 % de rendimiento).
1H RMN (CDCla 400 MHz): 57,91 (s, 1H), 7,12 (d, J = 8,0 Hz, 2H), 6,83 (d, J = 7,6 Hz, 2H), 6,68 (s, 1H), 5,18 (s, 2H), 4,12 (d, J = 11,6 Hz, 2H), 3,77 (s, 3H), 3,61 - 3,55 (m, 2H), 3,12 - 3,07 (m, 1H), 2,56 - 2,51 (m, 2H), 2,16 - 2,08 (m, 2H), 1,88 (d, J = 13,2 Hz, 2H), 1,22 (t, J = 7,2 Hz, 3H).
LC-MS: tR = 2,050 min (método 3), m/z = 368,2 [M H]+.
Ejemplo 45:

3-((6-metiI-8-oxo-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-7(8H)-il)metil)benzonitrilo:
Una mezcla de 6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (100 mg, 428,69 pmoles), 3-(bromometil)benzonitrilo (126 mg, 643,03 pmoles) y Cs2CO3 (279 mg, 858 pmoles) en DMF (3 mL) se agitó a 70 °C durante 6 horas. La mezcla se filtró y el filtrado se concentró en vacío. El residuo se purificó por LC-MS preparativa para rendir 3-((6-metil-8-oxo-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-7(8H)-il)metil)benzonitrilo (35 mg, 23 % de rendimiento).
1H RMN (CDCla 400 MHz): 57,95 (s, 1H), 7,59 (d, J = 6,8 Hz, 1H), 7,51 - 7,45 (m, 3H), 6,80 (s, 1H), 5,25 (s, 2H), 4,14 (d, J = 10,8 Hz, 2H), 3,63 - 3,57 (m, 2H), 3,14 - 3,08 (m, 1H), 2,19 - 2,10 (m, 5H), 1,91 (d, J = 13,2 Hz, 2H).
LC-MS: tR = 2,164 min (método 3), m/z = 349,1 [M H]+.
Ejemplo 46:

4-((6-metil-8-oxo-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-7(8H)-il)metil)benzonitrilo:
Una mezcla de 6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (100 mg, 428,69 pmoles), 4-(bromometil)benzonitrilo (126 mg, 643,04 pmoles) y Cs2CO3 (279 mg, 858 pmoles) en DMF (3 mL) se agitó a 60 °C durante 2 horas. La mezcla se filtró y el filtrado se concentró en vacío. El residuo se purificó por cromatografía en gel de sílice (éter de petróleo/acetato de etilo=10/1, 1/5) para rendir 4-((6-metil-8-oxo-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-7(8H)-il)metil)benzonitrilo (65 mg, 43 % de rendimiento).
1H RMN (CDCla 400 MHz): 57,94 (s, 1H), 7,64 (d, J = 8,0 Hz, 2H), 7,32 (d, J = 8,4 Hz, 2H), 6,79 (s, 1H), 5,28 (s, 2H), 4,14 (d, J = 11,2 Hz, 2H), 3,62 - 3,56 (m, 2H), 3,13 - 3,07 (m, 1H), 2,19 - 2,09 (m, 5H), 1,89 (d, J = 13,2 Hz, 2H). LC-MS: tR = 2,160 min (método 3), m/z = 349,2 [M H]+.
Ejemplo 47:

W-(4-((6-metil-8-oxo-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-7(8H)-il)metil)fenil)acetamida:
Etapa 1: Una mezcla de 6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (200 mg, 858 pmoles), 1-(bromometil)-4-nitrobenceno (278 mg, 1,29 mmoles) y Cs2CO3 (559 mg, 1,71 mmoles) en DMF (4 mL) se agitó a 60 °C durante 2 horas. La mezcla se filtró y el filtrado se concentró en vacío. El residuo se purificó por cromatografía en gel de sílice (éter de petróleo/acetato de etilo=10/1, 1/5) para rendir 6-metil-7-(4- nitrobencil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (270 mg, 85 % de rendimiento). LC-MS: tR = 0,612 min (método 2), m/z = 368,8 [M H]+.
Etapa 2: Una mezcla de 6-metil-7-(4-nitrobencil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (200 mg, 542,90 gmoles), Fe (152 mg, 2,71 mmoles), NFUCl (88 mg, 1,63 mmoles) y MeOH (10 mL) en H2O (10 mL) se agitó a 70 °C durante 4 horas. La mezcla se filtró y el filtrado se concentró en vacío. El residuo se purificó por cromatografía en gel de sílice (diclorometano/metanol=1/0, 15/1) para rendir 7-(4-aminobencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (170 mg, 93 % de rendimiento). LC-MS: tR = 0,287 min (método 2), m/z = 338,9 [M H]+.
Etapa 3: Una mezcla de 7-(4-aminobencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (120 mg, 354,61 gmoles), acetato de acetilo (60 mg, 588,65 gmoles) y trietilamina (144 mg, 1,42 mmoles) en dioxano (10 mL) se agitó a 90 °C durante 6 horas. La disolución se paró con agua (2 mL) y se agitó a 60 °C durante 2 horas. Después, se concentró en vacío. El residuo se purificó por cromatografía en gel de sílice (diclorometano/metanol=1/0, 15/1) para rendir W-(4-((6-metil-8-oxo-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-7(8H)-il)metil)fenil)acetamida (120 mg, 86,52 % de rendimiento).
1H RMN (CDCla 400 MHz): 57,92 (s, 1H), 7,44 (d, J = 8,4 Hz, 2H), 7,17 (d, J = 8,4 Hz, 2H), 6,74 (s, 1H), 5,19 (s, 2H), 4,13 (d, J = 10,4 Hz, 2H), 3,59 (t, J = 10,0 Hz, 2H), 3,12 - 3,06 (m, 1H), 2,19 - 2,09 (m, 8H), 1,88 (d, J = 13,2 Hz, 2H). LC-MS: tR = 1,593 min (método 3), m/z = 381,1 [M H]+.
Ejemplo 48:

7-(4-cloro-3-metoxibencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:
A una suspensión de 6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (100 mg, 429 gmoles) en DMF seco (2 mL) se añadió Cs2CO3 (279 mg, 858 gmoles) y 4-(bromometil)-1 -cloro-2-metoxibenceno (151 mg, 643 gmoles). La mezcla se purgó con N2 durante 2 min y se calentó a 60 °C durante 16 horas. La mezcla se concentró. Se añadió DCM (30 ml) al residuo. Se filtró y la torta del filtro se lavó con DCM (20 mL). El filtrado se concentró y se purificó por cromatografía flash en gel de sílice (acetato de etilo al 10 %~100 % en éter de petróleo) para proporcionar 7-(4-cloro-3-metoxibencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (99,01 mg, 60 % de rendimiento).
1H RMN (CDCla 400 MHz): 57,93 (s, 1H), 7,30 (d, J = 8,4 Hz, 1H), 6,85 (d, J = 1,6 Hz, 1H), 6,75 (s, 1H), 6,72 - 6,69 (m, 1H), 5,17 (s, 2H), 4,14 - 4,11 (m, 2H), 3,86 (s, 3H), 3,61 - 3,55 (m, 2H), 3,10 - 3,06 (m, 1H), 2,20 (s, 3H), 2,18 -2,11 (m, 2H), 1,89 - 1,86 (m, 2H).
LC-MS: tR = 2,466 min (método 3), m/z = 388,1 [M H]+.
Ejemplo 49:

7-(2-etilbencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:
Etapa 1: A una disolución de (2-etilfenil)metanol (500 mg, 3,67 mmoles) y trietilamina (742 mg, 7,34 mmoles) en DCM (5 mL) se añadió MsCl (1,0 g, 8,73 mmoles) a 0 °C y se agitó a 0 °C durante 0,5 horas. Después, se agitó a 20 °C durante 1 hora. La mezcla se paró con agua (0,5 mL), y se diluyó con DCM (10 mL). A la mezcla se añadió NaHCO3 (ac.) hasta pH=8. La capa orgánica se lavó con agua (3 x 5 mL), se secó con Na2SO4 anhidro, se filtró y se concentró en vacío para proporcionar metanosulfonato de 2-etilbencilo (600 mg), que se usó en la siguiente etapa sin más purificación.
Etapa 2: Una mezcla de 6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (150 mg, 643 gmoles), metanosulfonato de 2-etilbencilo (276 mg) y Cs2CO3 (419 mg, 1,29 mmoles) en DMF (5 mL) se agitó a 60 °C durante 12 horas. La mezcla se filtró y el filtrado se concentró en vacío. El residuo se purificó por LC-MS preparativa para rendir 7-(2-etilbencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (90 mg, 40 % de rendimiento).
1H RMN (CDCl3400 MHz): 57,93 (s, 1H), 7,25 - 7,21 (m, 2H), 7,14 - 7,10 (m, 1H), 6,81 - 6,77 (m, 2H), 5,25 (s, 2H), 4,14 (d, J = 11,2 Hz, 2H), 3,60 (t, J = 12,0 Hz, 2H), 3,15 - 3,09 (m, 1H), 2,78 - 2,72 (m, 2H), 2,21 - 2,13 (m, 5H), 1,91 (d, J = 13,6 Hz, 2H), 1,31 (t, J = 7,6 Hz, 3H).
LC-MS: tR = 2,533 min (método 3), m/z = 352,2 [M H]+.
Ejemplo 50:

7-(benzo[d][1,3]dioxol-5-ilmetil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:
A una suspensión de 6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (100 mg, 429 gmoles) en DMF seco (2 mL) se añadió Cs2CO3 (279 mg, 858 gmoles) y 5-(bromometil)benzo[d][1,3]dioxol (138 mg, 643 gmoles). La mezcla se burbujeó con N2 durante 2 min y se calentó a 60 °C durante 16 horas. La mezcla se concentró. Se añadió DCM (30 mL) al residuo. Se filtró y la torta del filtro se lavó con DCM (20 mL). El filtrado se concentró y se purificó por cromatografía flash en gel de sílice (acetato de etilo al 10 %~100 % en éter de petróleo) para proporcionar 7-(benzo[d][1,3]dioxol-5-ilmetil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (81,74 mg, 52 % de rendimiento).
1H RMN (CDCla 400 MHz): 57,92 (s, 1H), 6,76 - 6,69 (m, 4H), 5,95 - 5,93 (m, 2H), 5,13 (s, 2H), 4,14 - 4,11 (m, 2H), 3,61 - 3,55 (m, 2H), 3,10 - 3,05 (m, 1H), 2,21 (s, 3H), 2,20 - 2,10 (m, 2H), 1,90 - 1,86 (m, 2H).
LC-MS: tR = 2,245 min (método 3), m/z = 368,2 [M H]+.
Ejemplo 51:

7-(3-cloro-4-metoxibencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:
Una mezcla de 6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (100 mg, 427 gmoles), 4-(bromometil)-2-cloro-1-metoxibenceno (151 mg, 643 gmoles) y Cs2CO3 (279 mg, 858 gmoles) en DMF (3 mL) se agitó a 60 °C durante 2 horas. La mezcla se filtró y el filtrado se concentró en vacío. El residuo se purificó por cromatografía en gel de sílice (éter de petróleo/acetato de etilo=10/1,0/1) para rendir 7-(3-cloro-4- metoxibencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (110 mg, 65 % de rendimiento).
1H RMN (CDCla 400 MHz): 57,93 (s, 1H), 7,24 (d, J = 0,8 Hz, 1H), 7,12 - 7,10 (m, 1H), 6,88 (d, J = 8,4 Hz, 1H), 6,75 (s, 1H), 5,15 (s, 2H), 4,13 (d, J = 10,4 Hz, 2H), 3,88 (s, 3H), 3,62 - 3,56 (m, 2H), 3,12 - 3,06 (m, 1H), 2,20 - 2,09 (m, 5H), 1,89 (d, J = 13,2 Hz, 2H).
LC-MS: tR = 2,414 min (método 3), m/z = 388,1 [M H]+.
Ejemplo 52:

7-(4-aminobencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:
Una disolución de /V-(4-((6-metil-8-oxo-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-7(8H)-il)metil)fenil)acetamida (100 mg, 263 pmoles), NaOH (63 mg, 1,58 mmoles) y MeOH (1 mL) en H2O (1 mL) se agitó a 90 °C durante 12 horas. A la disolución se añadió KHSO4 (ac.) hasta pH=7 y se concentró en vacío. El residuo se purificó por TLC preparativa (DCM: MeOH = 10:1). para rendir 7-(4-aminobencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (50 mg, 56 % de rendimiento).
1H RMN (CDCl3400 MHz): 57,91 (s, 1H), 7,04 (d, J = 8,4 Hz, 2H), 6,71 (s, 1H), 6,63 (d, J = 8,4 Hz, 2H), 5,12 (s, 2H), 4,13 (d, J = 12,0 Hz, 2H), 3,67 (brs, 2H), 3,61 - 3,55 (m, 4H), 3,10 - 3,05 (m, 1H), 2,21 (s, 3H), 2,17 - 2,08 (m, 2H), 1,88 (d, J = 13,2 Hz, 2H).
LC-MS: tR = 1,273 min (método 3), m/z = 339,1 [M H]+.
Ejemplo 53:

7-(4-hidroxibencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:
A una disolución de 7-(4-metoxibencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (2,0 g, 5,66 mmoles) en DCM (32 mL) se añadió BBr3 (4,3 g, 16,98 mmoles) a 0 °C y se agitó a 20 °C durante 3 horas. La disolución se paró con H2O (5 mL) a 0 °C. La mezcla se agitó a 0 °C durante 1 hora y entonces se le añadió NaHCO3 (acuoso saturado) hasta pH=6. La mezcla se concentró en vacío. El residuo se diluyó con DCM (20 mL) y MeOH (2 mL). Después, se filtró y el filtrado se concentró en vacío. El residuo se añadió en KOH (60 mL, 2 M, ac.) a 20 °C y se agitó a 50 °C durante 1 hora. A la disolución se añadió KHSO4 (acuoso saturado) hasta pH=6, la mezcla se concentró en vacío. El residuo se purificó por cromatografía en gel de sílice (diclorometano/metanol=1/0, 15/1) para rendir 7-(4-hidroxibencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (460 mg, 24 % de rendimiento).
1H RMN (DMSO Varian_H_400 MHz): 59,39 (s, 1H), 7,70 (s, 1H), 7,46 (s, 1H), 6,99 (d, J = 8,4 Hz, 2H), 6,70 (d, J = 8,4 Hz, 2H), 5,06 (s, 2H), 3,96 - 3,93 (m, 2H), 3,52 - 3,45 (m, 2H), 3,29 - 3,16 (m, 1H), 2,15 (s, 3H), 1,83 - 1,77 (m, 4H).
LC-MS: tR = 1,62 min (método 8), m/z = 340,1 [M H]+.
Ejemplo 54:
6-etil-7-(3-fluorobencil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:
Etapa 1: Una mezcla de 1 H-imidazol-5-carboxilato de metilo (1,6 g, 13 mmoles), 1-bromobutan-2-ona (2,0 g, 13 mmoles) y K2CO3 (3,5 g, 25 mmoles) en acetona (20 mL) se agitó a 40 °C durante 12 horas. La mezcla se concentró en vacío. El residuo se purificó por cromatografía en gel de sílice (éter de petróleo/acetato de etilo=10/1, 0/1) para proporcionar 1-(2-oxobutil)-1 H-imidazol-5-carboxilato de metilo (750 mg, 30 % de rendimiento).
Etapa 2: Una mezcla de 1-(2-oxobutil)-1H-imidazol-5-carboxilato de metilo (700 mg, 3,59 mmoles), NBS (831 mg, 4,67 mmoles), y AIBN (118 mg, 718 pmoles) en CHCta (20 mL) se agitó a 50 °C durante 12 horas. La mezcla se concentró en vacío. El residuo se purificó por cromatografía en gel de sílice (éter de petróleo/acetato de etilo=10/1, 1/2) para proporcionar 2-bromo-1-(2-oxobutil)-1 H-imidazol-5-carboxilato de metilo (650 mg, 66 % de rendimiento). LC-MS: tR = 0,585 min (método 2), m/z = 274,7 [M H]+.
Etapa 3: Una mezcla de 2-bromo-1-(2-oxobutil)-1 H-imidazol-5-carboxilato de metilo (650 mg, 2,36 mmoles) y NH4OAc (727,64 mg, 9,44 mmoles) en 1,4-dioxano (15 mL) se agitó a 90 °C durante 3 días. La mezcla se concentró en vacío. El residuo se purificó por cromatografía en gel de sílice (éter de petróleo/acetato de etilo=10/1, 1/1) para proporcionar 3- bromo-6-etilimidazo[1,5-a]pirazin-8(7H)-ona (500 mg, 88 % de rendimiento). LC-MS: tR = 0,542 min (método 2), m/z = 241,8 [M H]+.
Etapa 4: Una mezcla de 3-bromo-6-etilimidazo[1,5-a]pirazin-8(7H)-ona (500 mg, 2,07 mmoles), 2-(3,6-dihidro-2H-piran-4-il)-4,4,5,5-tetrametil-1,3,2-dioxaborolano (522 mg, 2,48 mmoles), K2CO3 (572 mg, 4,14 mmoles), Pd(dppf)Cl2 (303 mg, 414 pmoles) y H2O (5 mL) en 1,4-dioxano (20 mL) se agitó a 100 °C durante 12 horas. La mezcla se filtró y el filtrado se concentró en vacío. El residuo se purificó por cromatografía en gel de sílice (Diclorometano/Metanol=1/0, 15/1) para proporcionar 3-(3,6-dihidro-2H-piran-4-il)-6-etilimidazo[1,5-a]pirazin-8(7H)-ona (400 mg, 78,78 % de rendimiento). l C-MS: tR = 0,430 min (método 2), m/z = 245,8 [M H]+.
Etapa 5: Una mezcla de 3-(3,6-dihidro-2H-piran-4-il)-6-etilimidazo[1,5-a]pirazin-8(7H)-ona (400 mg, 1,63 mmoles) y Pd/C (seco, Pd al 10 %, 20 mg) en THF (15 mL) se agitó a 15 °C durante 4 horas bajo H2 (15 psi). La mezcla se filtró y el filtrado se concentró en vacío para rendir 6-etil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (300 mg, 74 % de rendimiento). LC-MS: tR = 1,324 min (método 9), m/z = 248,0 [M H]+.
Etapa 6: Una mezcla de 6-etil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (300 mg, 1,21 mmoles), 1-(bromometil)-3-fluorobenceno (297 mg, 1,57 mmoles) y K2CO3 (334 mg, 2,42 mmoles) en DMF (20 mL) se agitó a 60 °C durante 12 horas. La mezcla se filtró y el filtrado se concentró en vacío. El residuo se purificó por cromatografía en gel de sílice (Éter de petróleo/Acetato de etilo=3/1, 0/1) para rendir 6-etil-7-(3-fluorobencil)-3-(tetrahidro-2H-piran-4- il)imidazo[1,5-a]pirazin-8(7H)-ona (150 mg, 34 % de rendimiento).
1H RMN (CDCl3400 MHz): 5 7,94 (s, 1H), 7,32 - 7,27 (m, 1H), 6,98 - 6,94 (m, 2H), 6,86 (d, J = 9,6 Hz, 1H), 6,72 (s, 1H), 5,25 (s, 2H), 4,14 (d, J = 11,2 Hz, 2H), 3,64 - 3,58 (m, 2H), 3,15 - 3,09 (m, 1H), 2,53 - 2,48 (m, 2H), 2,18 - 2,13 (m, 2H), 1,91 (d, J = 13,2 Hz, 2H), 1,25 (t, J = 7,2 Hz, 2H).
LC-MS: tR = 2,475 min (método 3), m/z = 356,1 [M H]+.
Ejemplo 55:

7-(4-metoxibencil)-6-metil-3-(2-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona, estereoisómeros 1,2, 3 y 4:
Etapa 1: A una disolución enfriada (0 °C) de ácido 2-metiltetrahidrofuran-3-carboxílico (110 mg, 845 pmoles) en DCM seco (2 mL) se añadió dicloruro de oxalilo (107 mg, 845 pmoles) gota a gota. Después, se añadió una gota de DMF y la mezcla se agitó a 26 °C durante 1 hora. La disolución de cloruro de 2-metiltetrahidrofuran-3-carbonilo (126 mg) en DCM (2 mL) se usó directamente para la siguiente etapa.
Etapa 2: A una disolución enfriada (0 °C) de cloruro de 2-metiltetrahidrofuran-3-carbonilo (160 mg, 844 pmoles, HCl) en DCM seco (5 mL) se añadió trietilamina (256 mg, 2,53 mmoles) y (3-metoxi-5-metilpirazin-2-il)metanamina (125 mg, 843,70 pmoles) en DCM (2 mL) gota a gota. La mezcla se agitó a 26 °C durante 1 hora. La LCMS mostró que la reacción se había completado. Se añadió agua (5 mL) a la mezcla. La mezcla se extrajo con DCM (30 mL x 2). La capa orgánica combinada se lavó con H2O (20 mL), salmuera (20 mL), se secó sobre Na2SO4, se filtró y se concentró. El producto crudo se purificó por cromatografía flash en gel de sílice (acetato de etilo al 10~50 % en éter
de petróleo) para proporcionar W-((3-metoxi-5-metilpirazin-2-il)metil)-2-metiltetrahidrofuran-3-carboxamida (100 mg, 45 % de rendimiento) como un aceite amarillo claro.
Etapa 3: A una disolución de W-((3-metoxi-5-metilpirazin-2-il)metil)-2-metiltetrahidrofuran-3-carboxamida (150 mg, 565 gmoles) en dioxano seco (5 mL) se añadió POCh (173 mg, 1,13 mmoles). La mezcla se calentó a 80 °C durante 2 horas. La mezcla se enfrió hasta 26 °C y la disolución marrón de 8-metoxi-6-metil-3-(2-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazina (140 mg) en dioxano (5 mL) se usó directamente para la siguiente etapa.
Etapa 4: A una disolución de 8-metoxi-6-metil-3-(2-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazina (140 mg, 566 gmoles) en dioxano (5 mL) se añadió HCl 2 N (2 M, 2 mL). La mezcla se calentó a 80 °C durante 1 hora. La mezcla se enfrió hasta 25 °C, se ajustó a pH=7 con NaHCO3 acuoso saturado y se extrajo con DCM (20 mL x 2). Las fases orgánicas combinadas se lavaron con H2O (20 mL), salmuera (20 mL), se secaron sobre Na2SO4, se filtraron y se concentraron para proporcionar 6-metil-3-(2-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona (131 mg). Etapa 5: A una disolución de 6-metil-3-(2-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona (131 mg, 562 gmoles) en DMF seco (5 mL) se añadió 1-(bromometil)-4-metoxibenceno (169 mg, 842 gmoles) y Cs2CO3 (366 mg, 1,12 mmoles). La mezcla se calentó a 60 °C durante 16 horas. La mezcla se concentró y se añadió agua (10 mL). La mezcla se extrajo con DCM (30 mL x 2). La capa orgánica combinada se lavó con H2O (30 mL x 2), salmuera (30 mL), se secó sobre Na2SO4, se filtró y se concentró. El residuo se purificó por cromatografía flash en gel de sílice (acetato de etilo al 10 %~100 % en éter de petróleo) para proporcionar 7-(4-metoxibencil)-6-metil-3-(2-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona (90 mg, 45 % de rendimiento).
Se purificó 7-(4-metoxibencil)-6-metil-3-(2-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona (100 mg, 283 gmoles) por SFC.
Estereoisómero 1 : (9,4 mg, 9 % de rendimiento).
1H RMN (CDCl3400 MHz): 57,92 (s, 1H), 7,15 (d, J= 8,8 Hz, 2H), 6,84 (d, J= 8,4 Hz, 2H), 6,74 (s, 1H), 5,16 (s, 2H), 4,30 4,26 (m, 1H), 4,12-4,04 (m, 2H), 3,78 (s, 3H), 3,09-3,03 (m, 1H), 2,46-2,34 (m, 2H), 2,20 (s, 3H), 1,32 (d, J= 6,0 Hz, 3H). LC-MS: tR = 2,074 min (método 13), m/z = 354,1 [M H]+. SFC: tR = 5,177 min, % ee > 99 %.
[a]D20 -26 (c = 0,10, DCM).
Estereoisómero 2 : (7,3 mg, 7 % de rendimiento).
1H RMN (CDCl3400 MHz): 57,92 (s, 1H), 7,15 (d, J= 8,8 Hz, 2H), 6,84 (d, J= 8,4 Hz, 2H), 6,73 (s, 1H), 5,16 (s, 2H), 4,30-4,26 (m, 1H), 4,10-4,02 (m, 2H), 3,78 (s, 3H), 3,09-3,03 (m, 1H), 2,44-2,35 (m, 2H), 2,20 (s, 3H), 1,32 (d, J= 6,0 Hz, 3H).
LC-MS: tR = 2,072 min (método 13), m/z = 354,1 [M H]+. SFC: tR = 5,458 min, % ee = 99,7 %. [a]D20 24 (c = 0,10, DCM). Estereoisómero 3 : (39,7 mg, 40 % de rendimiento).
1H RMN (CDCl3400 MHz): 57,92 (s, 1H), 7,15 (d, J= 8,4 Hz, 2H), 6,84 (d, J= 8,4 Hz, 2H), 6,78 (s, 1H), 5,22-5,10 (m, 2H), 4,31-4,26 (m, 2H), 3,88-3,82 (m, 1H), 3,77 (s, 3H), 3,70-3,60 (m, 1H), 2,71-2,67 (m, 1H), 2,43-2,40 (m, 1H), 2,19 (s, 3H), 0,87 (d, J= 4,4 Hz, 3H).
LC-MS: tR = 1,983 min (método 13), m/z = 354,1 [M H]+. SFC: tR = 5,932 min, % ee = 98,8 %. [a]D20 48 (c = 0,10, DCM). Estereoisómero 4 : (26,4 mg, 26 % de rendimiento).
1H RMN (CDCl3400 MHz): 57,92 (s, 1H), 7,15 (d, J= 8,8 Hz, 2H), 6,84 (d, J= 8,4 Hz, 2H), 6,78 (s, 1H), 5,22-5,09 (m, 2H), 4,32-4,26 (m, 2H), 3,87-3,85 (m, 1H), 3,77 (s, 3H), 3,68-3,64 (m, 1H), 2,70-2,65 (m, 1H), 2,42-2,37 (m, 1H), 2,19 (s, 3H), 0,87 (d, J= 6,4 Hz, 3H).
LC-MS: tR = 1,983 min (método 13), m/z = 354,1 [M H]+. SFC: tR = 6,570 min, % ee = 99,6 %. [a]D20 -64 (c = 0,10, DCM). Ejemplo 56:
7-(4-metoxibencil)-6-metil-3-propilimidazo[1,5-a]pirazin-8(7H)-ona:
A una disolución de 3-bromo-7-(4-metoxibencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (500 mg, 1,44 mmoles, 1 eq) en THF (10 mL) se añadió Ni(dppp)Cl2 (327,83 mg, 604,80 pmoles, 0,42 eq) a 0 °C durante 10 min, después a -78 °C durante 10 min. Se añadió gota a gota bromuro de propilmagnesio (1 M, 3 mL, 2,1 eq) a 0 °C. La mezcla resultante se agitó a 0 °C durante 1,5 horas. La reacción se paró por disolución acuosa saturada de NH4Cl (5 mL). El residuo se purificó por cromatografía en columna de sílice (Gradiente: 0~50, EtOAc en PE con trietilamina al 1 %) para proporcionar 220 mg de producto crudo. El producto crudo se purificó por TLC preparativa (PE:EtOAc=1:1 con trietilamina al 1 %). El residuo se lavó con hexano (3 mL) y se filtró y la torta del filtro se secó en vacío para proporcionar 7-(4-metoxibencil)-6-metil-3-propilimidazo[1,5-a]pirazin-8(7H)-ona (82 mg, 18 % de rendimiento).
1HRMN (CDCla, 400 MHz): 57,90 (s, 1H), 7,16 (d, J = 8,8, 2H), 6,85 (d, J = 8,8, 2H), 6,68 (s, 1H), 5,17 (s, 2H), 3,78 (s, 3H), 2,82 (t, J = 7,2, 2H), 2,19 (s, 3H), 1,90-1,81 (m, 2H), 1,03 (t, J = 7,2, 3H).
LC-MS: tR = 1,927 min (método 13), m/z = 312,1 [M H]+.
Ejemplo 57:

7-((6-metoxipiridin-3-il)metil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:
En un vial se añadió 6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (200mg, 0,86 mmoles), 5-(clorometil)-2-metoxipiridina (162 mg, 1,03 mmoles), carbonato de cesio (559 mg, 1,72 mmoles) y yoduro de sodio (154 mg, 1,03 mmoles) en DMF (9,44 g, 10 ml, 129 mmoles). La reacción se agitó toda la noche a 70 °C. A la reacción se añadió acetato de etilo, y se filtró y se concentró. La reacción se purificó por cromatografía en gel de sílice para obtener 7-((6-metoxipiridin-3-il)metil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (127 mg, 0,358 mmoles) con 42 % de rendimiento.
1H RMN (600 MHz, CDCla) 5 8,06 (dd, J = 2,5, 0,8 Hz, 1H), 7,91 (d, J = 0,6 Hz, 1H), 7,55 (dd, J = 8,6, 2,5 Hz, 1H), 6,74 (t, J = 1,1 Hz, 1H), 6,71 (dd, J = 8,6, 0,7 Hz, 1H), 5,15 (s, 2H), 4,12 (m, 2H), 3,91 (s, 3H), 3,58 (td, J = 11,7, 2,2 Hz, 2H), 3,07 (tt, J = 11,4, 3,9 Hz, 1H), 2,25 (d, J = 1,2 Hz, 3H), 2,12 (dtd, J = 13,7, 11,6, 4,3 Hz, 2H), 1,87 (ddd, J = 13,5, 4,2, 2,1 Hz, 2H).
LC-MS: tR = 0,38 min (método 6), m/z = 355,2 [M H]+.
Ejemplo 58:

6,7-dimetil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:
A una disolución de 6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (100 mg, 428,69 pmoles, 1 eq) en DMSO (2 mL) se añadió Cs2CO3 (139,68 mg, 428,69 pmoles, 1 eg) y yoduro de metilo (121,70 mg, 858 pmoles, 53,38 pL, 2 eq). La mezcla se agitó a 50 °C durante 12 horas. La mezcla de reacción se diluyó con H2O (25mL) y se extrajo con DCM (50 mL x 2). Las capas orgánicas combinadas se lavaron con salmuera (15 mL), se secaron sobre Na2SO4, se filtraron y se concentraron bajo presión reducida. El residuo se purificó por TLC preparativa (acetato de etilo). Se obtuvo 6,7-dimetil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (73 mg, 68 % de rendimiento, 98 % de pureza).
1H RMN (CDCI3400 MHz) 5: 7,87 (s, 1H), 6,76 (s, 1H), 4,13 (d, J = 11,2 Hz, 2H), 3,59 (t, J = 11,2 Hz, 2H), 3,46 (s, 3H), 3,11-3,05 (m, 1H), 2,28 (s, 3H), 2,16-2,08 (m, 2H), 1,88 (d, J = 14,0 Hz, 2H).
LC-MS: tR = 1,300 min (método 13), m/z = 248,1 [M H]+.
Ejemplo 59:
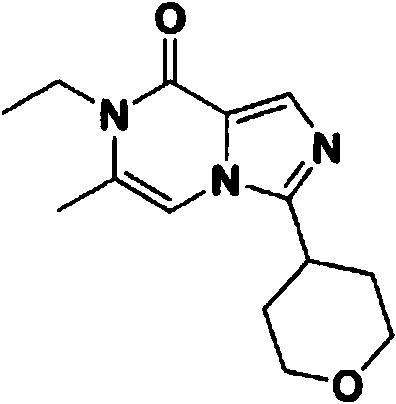
7-etil-6-met¡l-3-(tetrah¡dro-2H-p¡ran-4-¡l)¡midazo[1,5-a]pirazin-8(7H)-ona:
A una disolución de 6-metil-3-(tetrah¡dro-2H-p¡ran-4-¡l)¡m¡dazo[1,5-a]p¡raz¡n-8(7H)-ona (100 mg, 429 jmoles, 1 eq) en DMF anhidro (2 mL) se añadió K2CO3 (119 mg, 858 jmoles, 2 eq) y yodoetano (134 mg, 858 jmoles, 69 |jL, 2 eq). La mezcla se agitó a 50 °C durante 12 horas. La mezcla de reacción se concentró bajo presión reducida. El residuo se diluyo con H2O (20 mL) y se extrajo con EA (40 mL x 2). Las capas orgánicas combinadas se lavaron con salmuera (15 mL), se secaron sobre Na2SO4, se filtraron y se concentraron bajo presión reducida para rendir un residuo. Se obtuvo 7-etil-6-met¡l-3-(tetrah¡dro-2H-p¡ran-4-¡l)¡midazo[1,5-a]pirazin-8(7H)-ona (40 mg, 34 % de rendimiento, 96 % de pureza).
1H RMN (CDCl3400 MHz): 57,85 (s, 1H), 6,73 (s, 1H), 4,12 (d, J = 10,8 Hz, 2H), 4,03-3,99 (m, 2H), 3,58 (t, J = 10,8 Hz, 2H), 3,10-3,04 (m, 1H), 2,30 (s, 3H), 2,16-2,07 (m, 2H), 1,87 (d, J = 13,2 Hz, 2H), 1,29 (t, J = 6,8 Hz, 3H).
LC-MS: tR = 1,490 min (método 11), m/z = 262,1 [M H]+.
Ejemplo 60:
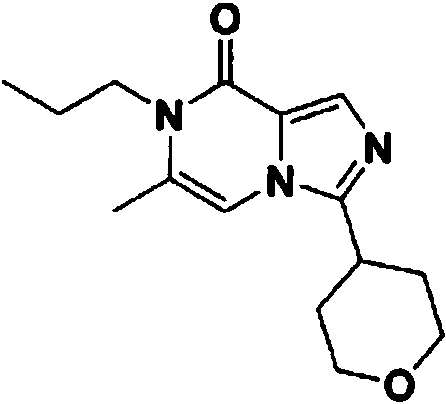
6-metil-7-prop¡l-3-(tetrah¡dro-2H-p¡ran-4-¡l)¡midazo[1,5-a]pirazin-8(7H)-ona:
A una disolución de 6-metil-3-(tetrah¡dro-2H-p¡ran-4-¡l)¡m¡dazo[1,5-a]p¡raz¡n-8(7H)-ona (100 mg, 429 jmoles, 1 eq) en DMF anhidro (2 mL) se añadió K2CO3 (119 mg, 858 jmoles, 2 eq) y 1-bromopropano (105 mg, 858 jmoles, 78,11 j L, 2 eq). La mezcla se agitó a 50 °C durante 12 horas. La mezcla de reacción se concentró bajo presión reducida para eliminar el DMF. El residuo se diluyó con H2O (20 mL) y se extrajo con EA (40 mL x 2). Las capas orgánicas combinadas se lavaron con salmuera (15 mL), se secaron sobre Na2SO4, se filtraron y se concentraron bajo presión reducida. El residuo se purificó por TLC preparativa (acetato de etilo). Se obtuvo 6-metil-7-propil-3-(tetrahidro-2H-piran-4-il)¡m¡dazo[1,5-a]pirazin-8(7H)-ona (45 mg, 37 % de rendimiento, 98 % de pureza).
1H RMN (CDCl3400 MHz): 5 7,85 (s, 1H), 6,72 (s, 1H), 4,12 (d, J = 10,4 Hz, 2H), 3,88 (t, J = 8,0 Hz ,2H), 3,62-3,56 (m, 2H), 3,10-3,07 (m, 1H), 2,28 (s, 3H), 2,14-2,09 (m, 2H), 1,87 (d, J = 13,2 Hz, 2H), 1,73-1,67 (m, 2H), 1,00 (t, J = 7,2 Hz, 3H).
LC-MS: tR = 1,666 min (método 11), m/z = 276,1 [M H]+.
Ejemplo 61:

7-isopropil-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:
A una disolución de 6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (350 mg, 1,50 mmoles, 1 eq) en DMF anhidro (4 mL) se añadió Cs2CO3 (978 mg, 3 mmoles, 2 eq) y 2-yodopropano (510 mg, 3 mmoles, 300 pL, 2 eq). La mezcla se agitó a 50 °C durante 12 horas. La mezcla de reacción se concentró bajo presión reducida para eliminar el DMF. El residuo se diluyó con H2O (15mL) y se extrajo con EtOAc (40mL x 2). Las capas orgánicas combinadas se lavaron con salmuera (10 mL), se secaron sobre Na2SO4, se filtraron y se concentraron bajo presión reducida. El residuo se purificó por TLC preparativa (acetato de etilo). Se obtuvo 7- isopropil-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (68 mg, 16 % de rendimiento, 96 % de pureza).
1H RMN (CDCls 400 MHz): 57,81 (s, 1H), 6,67 (s, 1H), 4,40 (m, 1H), 4,12 (d, J = 11,6 Hz, 2H), 3,58 (t, J = 11,3 Hz , 2H), 3,09-3,03 (m, 1H), 2,27 (s, 3H), 2,14-2,06 (m, 2H), 1,86 (d, J = 13,2 Hz, 2H), 1,62-1,61 (m, 6H).
LC-MS: tR = 1,576 min (método 13), m/z = 276,1 [M H]+.
Ejemplo 62:

7-isopentil-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:
A una disolución de 6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (100 mg, 428,69 pmoles, 1 eq) en DMF anhidro (2 mL) se añadió K2CO3 (118,50 mg, 858 pmoles, 2 eq) y 1-bromo-3-metilbutano (129,50 mg, 858 pmoles, 108 pL, 2 eq). La mezcla se agitó a 50 °C durante 12 horas. La mezcla de reacción se concentró bajo presión reducida para eliminar el DMF. El residuo se diluyó con H2O (20 mL) y se extrajo con EtOAc (40 mL x 2). Las capas orgánicas combinadas se lavaron con salmuera (15 mL), se secaron sobre Na2SO4, se filtraron y se concentraron bajo presión reducida. El residuo se purificó por TLC preparativa (acetato de etilo). Se obtuvo 7-isopentil-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (65 mg, 50 % de rendimiento).
1H RMN (CDCla 400 MHz): 5 7,84 (s, 1H), 6,73 (s, 1H), 4,12 (d, J = 10,4 Hz, 2H), 3,93 (t, J = 8,0 Hz ,2H), 3,61-3,55 (m, 2H), 3,08-3,04 (m, 1H), 2,28 (s, 3H), 2,10-2,09 (m, 2H), 1,87 (d, J = 14,0 Hz, 2H), 1,74-1,69 (m, 1H), 1,56-1,52 (m, 2H), 1,00-0,98 (m, 6H).
LC-MS: tR = 1,968 min (método 13), m/z = 304,2 [M H]+.
Ejemplo 63:
7-(ciclopentilmetil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:
A una mezcla de 6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (150 mg, 0,64 mmoles) y (bromometil)ciclopentano (157 mg, 0,96 mmoles) en DMSO (2 mL) se añadió Cs2CO3 (419 mg, 1,29 mmoles). La mezcla se agitó a 60 °C durante 12 horas. La mezcla se diluyó con agua (10 mL) y se extrajo con DCM (5 mL x 3). La capa orgánica combinada se lavó con agua (5 mL x 2) y se secó sobre Na2SO4. Las capas orgánicas se evaporaron en vacío. El residuo se purificó por TLC preparativa (acetato de etilo) para proporcionar 7-(ciclopentilmetil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (95 mg, 46 % de rendimiento). 1h Rm N (CDCl3, 400 MHz): 5 7,85 (s, 1H), 6,72 (s, 1H), 4,14- 4,11 (m, 2H), 3,91 (d, J = 7,6 Hz, 2H), 3,61-3,56 (m, 2H), 3,12-3,05 (m, 1H), 2,29 (s, 3H), 2,27-2,25 (m, 1H), 2,14-2,10 (m, 2H), 1,90-1,86 (m, 2H), 1,71-1,54 (m, 6H), 1,34-1,32 (m, 2H).
LC-MS: tR = 1,98 min (método 13), m/z = 316,2 [M H]+.
Ejemplo 64:
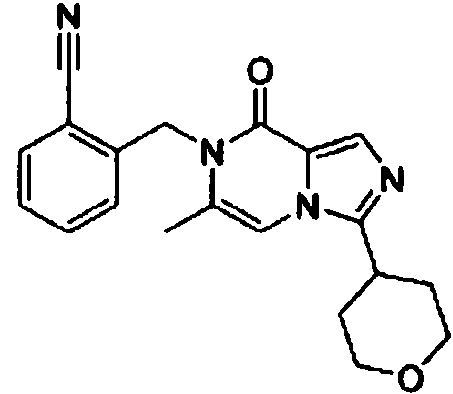
2-((6-metil-8-oxo-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-7(8H)-il)metil)benzonitrilo:
Una mezcla de 6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (100 mg, 429 pmoles), 2-(bromometil)benzonitrilo (126 mg, 643 pmoles) y Cs2CO3 (279 mg, 857 pmoles) en DMF (3,0 mL) se agitó a 70 °C durante 6 horas. La mezcla se filtró y el filtrado se concentró en vacío. El residuo se purificó por cromatografía en gel de sílice (Éter de petróleo/Acetato de etilo=10/1, 0/1) para rendir 2-((6-metil-8-oxo-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-7(8H)-il)metil)benzonitrilo (92 mg, 59 % de rendimiento).
1H RMN (CDCla 400 MHz): 5 7,96 (s, 1H), 7,72 (d, J = 7,6 Hz, 1H), 7,55 (t, J = 7,2 Hz, 1H), 7,40 (t, J = 7,2 Hz, 1H), 7,19 (d, J = 8,0 Hz, 1H), 6,80 (s, 1H), 5,45 (s, 2H), 4,14 (d, J = 12,0 Hz, 2H), 3,63 - 3,57 (m, 2H), 3,14 - 3,08 (m, 1H), 2,20 - 2,09 (m, 5H), 1,90 (d, J = 13,2 Hz, 2H).
LC-MS: tR = 2,202 min (método 3), m/z = 349,1 [M H]+.
Ejemplo 65:
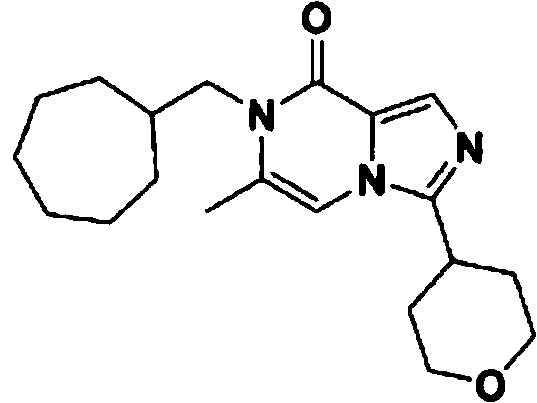
7-(cicloheptilmetil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:
Etapa 1: A una disolución de ácido cicloheptanocarboxílico (500 mg, 3,52 mmoles) en THF (40 mL) se añadió LiAlH4 (401 mg, 10,6 mmoles) en partes a 0 °C. La mezcla se agitó a 65 °C durante 3 horas. La reacción se paró con H2O (0,4 mL) y NaOH al 10 % (0,4 mL, ac). A la mezcla se añadió Na2SO4. La mezcla se filtró. El filtrado se concentró. El residuo se purificó por cromatografía flash en gel de sílice para proporcionar cicloheptilmetanol (383 mg, 85 % de rendimiento).
1H RMN (CDCla 400 MHz): 53,43 (d, J = 6,4 Hz, 2H), 1,80 - 1,62 (m, 5H), 1,56 - 1,40 (m, 4H), 1,31 (br, s, 1H), 1,84 (s, 3H), 1,10 - 1,22 (m, 2H).
Etapa 2: A una disolución de cicloheptilmetanol (313 mg, 2,44 mmoles) y trietilamina (494 mg, 4,88 mmoles) en DCM (5 mL) se añadió MsCl (490 mg, 4,28 mmoles) a 0 °C y se agitó a 20 °C durante 40 min. La disolución se lavó con NaHCO3 (acuoso saturado 5mL x 4), agua (5 mL x 2), salmuera (3 mL) y entonces se secó sobre Na2SO4, se filtró y
se concentró para proporcionar metanosulfonato de cicloheptilmetilo (381 mg) que se usó en la siguiente etapa directamente sin más purificación.
Etapa 3: A una disolución de 6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (150 mg, 643 pmoles) y metanosulfonato de cicloheptilmetilo (159 mg, 772 pmoles) en DMF (3 mL) se añadió Cs2CO3 (419 mg, 1,29 mmoles). La mezcla se agitó a 60 °C durante 6 horas. La mezcla se diluyó con DCM (20 mL) y se lavó con agua (5 mL x 2), salmuera (10 mL), se secó sobre Na2SO4, se filtró y se concentró. El residuo se purificó por cromatografía flash en gel de sílice y entonces se purificó por LC-MS preparativa para proporcionar 7-(cicloheptilmetil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (25,15 mg, 11 % de rendimiento).
1H RMN (CDCla 400 MHz): 57,84 (s, 1H), 6,71 (s, 1H), 4,12 (d, J = 5,2 Hz, 2H), 3,77 (d, J = 3,6 Hz, 2H), 3,65 - 3,52 (m, 2H), 3,15 - 3,02 (m, 1H), 2,26 (s, 3H), 2,17 - 2,07 (m, 2H), 2,07 - 2,98 (m, 1H), 1,87 (d, J = 6,6 Hz, 2H), 1,63 - 1,75 (m, 4H), 1,55 - 1 ,62 (m, 2H), 1,54 - 1,45 (m, 2H), 1,44 - 1,33 (m, 2H), 1,30 - 1,18 (m, 2H).
LC-MS: tR = 2,227 min (método 13), m/z = 344,2 [M H]+.
Ejemplo 69:

Etapa 1: A una disolución de hidrocloruro de (3-metoxi-5-metilpirazin-2-il)metanamina (150 mg, 0,79 mmoles, 1 eq) y ácido 3-metiltetrahidro-2H-piran-4-carboxílico (125 mg, 870 pmoles, 1,1 eq) en DCM (5 mL) se añadió HATU (451 mg, 1,19 mmoles, 1,5 eq) y DIPEA (204 mg, 1,58 mmoles, 276 pL, 2 eq). La mezcla se agitó a 25 °C durante 18 horas. La mezcla se concentró. El producto crudo se purificó por cromatografía flash con éter de petróleo: acetato de etilo = 3:1 ~ 2:1. Se obtuvo W-((3-metoxi-5-metilpirazin-2-il)metil)-3-metiltetrahidro-2H-piran-4-carboxamida (220 mg, 780 pmoles, 99 % de rendimiento).
Etapa 2: A una disolución de W-((3-metoxi-5-metilpirazin-2-il)metil)-3-metiltetrahidro-2H- piran-4-carboxamida (220 mg, 788 pmoles, 1 eq) en dioxano (8 mL) se añadió POCl3 (242 mg, 1,58 mmoles, 146 pL, 2 eq). La mezcla se agitó a 80 °C durante 2 horas. La mezcla se concentró. Se usó 6-metil-3-(3-metiltetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8- ol (223 mg, sal hidrocloruro) en la siguiente etapa sin más purificación.
Etapa 3: A una disolución de 6-metil-3-(3-metiltetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin- 8-ol (170 mg, 0,6 mmoles) en DMF (5 mL) se añadió Cs2CO3 (586 mg, 1,80 mmoles, 3 eq) y 1-(clorometil)-4-metoxibenceno (113 mg, 719 pmoles, 98 pL, 1,20 eq). La mezcla se agitó a 60 °C durante 18 horas. La mezcla de reacción se filtró, y el filtrado se concentró. La mezcla cruda se purificó por LC-MS preparativa, y después por TLC preparativa con acetato de etilo como eluyente. Se obtuvo 7-(4-metoxibencil)-6-metil-3-(3-metiltetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (76 mg, 207 pmoles, 35 % de rendimiento).
1H RMN, una mezcla de diastereoisómeros (CDCl3400 MHz): 57,96 - 7,94 (m, 1H), 7,18 (d, J = 8,0 Hz, 2H), 6,86 (d, J = 8,0 Hz, 2H), 6,76 (s, 0,73H), 6,71 (s, 0,28H), 5,22 - 5,12 (m, 2H), 4,17 - 4,01 (m, 2H), 3,79 (s, 3H), 3,55 - 3,49 (m, 1H), 3,19 - 3,14 (m, 1H), 2,66 - 2,64 (m, 1H), 2,36 - 2,33 (m, 1H), 2,21 (s, 3H), 2,13 - 2,10 (m, 1H), 1,78 - 1,61 (m, 1H), 0,87 (d, J = 7,2 Hz, 1H), 0,71 (d, J = 6,8 Hz, 2H).
LC-MS: tR = 2,370 min (método 11), m/z =368,1 [M H]+.
Ejemplo 70:
7-(4-metoxibencil)-6-metil-3-((1 R,2R,4S)-2-metil-7-oxabiciclo[2.2.1]heptan-2-il)imidazo[1,5-a]pirazin-8(7H)-ona racémica:
Etapa 1: A una disolución de hidrocloruro de (3-metoxi-5-metilpirazin-2-il)metanamina (150 mg, 791 pmoles, 1 eq) en DCM seco (5 mL) se añadió trietilamina (240 mg, 2,37 mmoles, 329 pL, 3 eq), ácido (1 R,2s,4S)-2-metil-7-oxabiciclo[2.2.1]heptano-2-carboxílico racémico (124 mg, 791 pmoles, 1 eq) y HATU (361 mg, 949 pmoles, 1,20 eq). La mezcla se agitó a 15 °C durante 16 horas. Se añadió H2O (5 mL) y la mezcla se extrajo con DCM (20 mL x 2). La capa orgánica combinada se lavó con H2O (20 mL), salmuera (20 mL), se secó sobre Na2SO4, se filtró y se concentró. El residuo se purificó por cromatografía flash en gel de sílice (acetato de etilo al 0 %~50 % en éter de petróleo) para proporcionar (1 R,2S,4S)-W-((3-metoxi-5-metilpirazin-2-il)metil)-2-metil-7-oxabiciclo[2.2.1]heptano-2-carboxamida racémica (200 mg, 87 % de rendimiento).
Etapa 2: A una disolución de (1 R,2S,4S)-W-((3-metoxi-5-metilpirazin-2-il)metil)-2-metil-7-oxabiciclo[2.2.1]heptano-2-carboxamida racémica (150 mg, 515 pmoles, 1 eq) en dioxano seco (5 mL) se añadió POCh (158 mg, 1,03 mmoles, 96 pL, 2 eq). La mezcla se calentó a 80 °C durante 2 horas. La mezcla se enfrió hasta 15 °C y se vertió en agua (5 mL). La mezcla se ajustó a pH 8 con NaHCO3 acuoso saturado y se extrajo con DCM (20 mL x 2). Los orgánicos combinados se lavaron con H2O (20 mL), salmuera (20 mL), se secaron sobre Na2SO4, se filtraron y se concentraron para proporcionar 6-metil-3-((1 R,2R,4S)-2-metil-7-oxabiciclo[2.2.1]heptan-2-il)imidazo[1,5-a]pirazin-8-ol racémico (120 mg).
Etapa 3: A una disolución de 6-metil-3-((1 R,2R,4S)-2-metil-7-oxabiciclo[2.2.1]heptan-2-il)imidazo[1,5-a]pirazin-8-ol racémico (100 mg, 386 pmoles, 1 eq) en DMF seco (5 mL) se añadió 1-(clorometil)-4-metoxi-benceno (72 mg, 463 pmoles, 63 pL, 1,20 eq) y Cs2CO3 (251 mg, 771 pmoles, 2 eq). La mezcla se calentó a 60 °C durante 2 horas. La mezcla se concentró. Se añadió DCM (20 mL) y H2O (10 mL). La mezcla se extrajo con DCM (20 mL). La capa orgánica se lavó con H2O (20 mL), se secó sobre Na2SO4, se filtró y se concentró. El residuo se purificó por LC-MS preparativa para proporcionar 7-(4-metoxibencil)-6-metil-3-((1 R,2R,4S)-2-metil-7-oxabiciclo[2.2.1]heptan-2-il)imidazo[1,5-a]pirazin-8(7H)-ona racémica (40 mg, 27 % de rendimiento).
1H RMN (CDCl3400 MHz): 57,87 (s, 1H), 7,17 (d, J = 8,4 Hz, 2H), 6,84 (d, J = 8,8 Hz, 2H), 6,69 (s, 1H), 5,23-5,07 (m, 2H), 4,67 (d, J = 4,8 Hz, 2H), 3,78 (s, 3H), 2,71 (d, J = 12,0 Hz, 1H), 2,20 (s, 3H), 1,90-1,86 (m, 1H), 1,70-1,66 (m, 2H), 1,57 (s, 3H), 1,39-1,37 (m, 1H), 1,25-1,22 (m, 1H).
LC-MS: tR = 2,228 min (método 13), m/z = 380,2 [M H]+.
Ejemplo 71:

(S)-7-(4-metoxibencil)-6-metil-3-(1-feniletil)imidazo[1,5-a]pirazin-8(7H)-ona:
Etapa 1: A una disolución de hidrocloruro de (3-metoxi-5-metilpirazin-2-il)metanamina (300 mg, 1,58 mmoles) y ácido (S)-2-fenilpropanoico (261 mg, 1,74 mmoles, 237 pL, 1,10 eg) en DCM (10 mL) se añadió trietilamina (400 mg, 3,95 mmoles, 548 pL, 2,50 eg) y HATU (901 mg, 2,37 mmoles, 1,50 eq). La mezcla se agitó a 15 °C durante 12 horas. Se añadió agua (50 mL) a la disolución. La mezcla se extrajo con DCM (50 mL x 2). Las capas orgánicas combinadas se lavaron con salmuera (50 mL), se secaron sobre Na2SO4, se filtraron y se concentraron bajo presión reducida. El residuo se purificó por cromatografía flash en gel de sílice (Eluyente de Acetato de etilo al 0~20 %/éter de petróleo). Se obtuvo (S)-W-((3-metoxi-5-metilpirazin-2-il)metil)-2-fenilpropanamida (450 mg, 1,56 mmoles, 99 % de rendimiento).
1H RMN (CDCl3400 MHz): 57,81 (s, 1H), 7,24-7,37 (m, 5H), 6,74 (s, 1H), 4,37-4,52 (m, 2H), 3,92 (s, 3H), 3,67 (q, J = 7,1 Hz, 1H), 2,40 (s, 3H), 1,56 (d, J = 7,2 Hz, 3H).
Etapa 2: A una disolución de (S)-W-((3-metoxi-5-metilpirazin-2-il)metil)-2-fenilpropanamida (450 mg, 1,58 mmoles, 1 eq) en dioxano (10 mL) se añadió POCl3 (485 mg, 3,16 mmoles, 294 pL, 2 eg). La mezcla se agitó a 90 °C durante 12 horas. La mezcla de reacción se concentró bajo presión reducida para eliminar el dioxano. El residuo se paró por la adición de H2O (50 mL) a 0 °C, se basificó por la adición de NaHCO3 acuoso saturado (10 mL) y entonces se extrajo con EtOAc (50 mL x 2). Las capas orgánicas combinadas se lavaron con salmuera (50 mL), se secaron sobre Na2SO4, se filtraron y se concentraron bajo presión reducida. El residuo se purificó por cromatografía flash en gel de sílice (Eluyente de Acetato de etilo al 0~100 %/éter de petróleo). (S)-6-metil-3-(1 -feniletil)imidazo[1,5-a]pirazin-8-ol (128 mg, 505 pmoles, 32 % de rendimiento).
Etapa 3: A una disolución de (S)-6-metil-3-(1-feniletil)imidazo[1,5-a]pirazin-8-ol (128 mg, 505 pmoles, 1 eq) y 1-(clorometil)-4-metoxibenceno (95 mg, 0,61 mmoles, 83 pL, 1,20 eq) en DMF (10 mL) se añadió Cs2CO3 (329,29 mg, 1,01 mmoles, 2 eq). La mezcla se agitó a 60 °C durante 3 horas. La mezcla de reacción se concentró bajo presión reducida. El residuo se purificó por LC-MS preparativa, se purificó adicionalmente por SFC. Se obtuvo (S)-7-(4-metoxibencil)-6-metil-3-(1-feniletil)imidazo[1,5-a]pirazin-8(7H)-ona (76,40 mg, 203 pmoles, 40 % de rendimiento).
1H RMN (CDCla 400 MHz): 57,98 (s, 1H), 7,17-7,31 (m, 5H), 7,12 (d, J = 8,4 Hz, 2H), 6,82 (d, J = 8,4 Hz, 2H), 6,46 (s, 1H), 5,10 (dd, J = 15,6 Hz, 2H), 4,26 (q, J = 7,2 Hz, 1H), 3,76 (s, 3H), 2,05 (s, 3H), 1,81 (d, J = 7,6 Hz, 3H).
LC-MS: tR = 2,218 min (método 17), m/z = 374,2 [M H]+. SFC: tR = 1,641 min, % ee > 99 %, [a]D20 = -51,4 (c=0,11, MeOH).
Ejemplo 72:

(R)-7-(4-metoxibencil)-6-metil-3-(1-feniletil)imidazo[1,5-a]pirazin-8(7H)-ona:
Etapa 1: A una disolución de hidrocloruro de (3-metoxi-5-metilpirazin-2-il)metanamina (200 mg, 1,05 mmoles, 1 eq) y ácido (R)-2-fenilpropanoico (190 mg, 1,27 mmoles, 173 pL, 1,20 eq) en DCM (10 mL) se añadió trietilamina (267 mg, 2,64 mmoles, 365 pL, 2,50 eq) y HATU (602 mg, 1,58 mmoles, 1,50 eq). La mezcla se agitó a 15 °C durante 12 horas. Se añadió agua (50 mL) a la disolución. La mezcla se extrajo con DCM (50 mL x 2). Las capas orgánicas combinadas se lavaron con salmuera (50 mL), se secaron sobre Na2SO4, se filtraron y se concentraron bajo presión reducida. El residuo se purificó por cromatografía flash en gel de sílice (Eluyente de Acetato de etilo al 0~20 %/éter de petróleo). Se obtuvo (R)-W-((3-metoxi-5-metilpirazin-2-il)metil)-2-fenilpropanamida (300 mg, 1,02 mmoles, 97 % de rendimiento).
1H RMN (CDCla 400 MHz): 57,81 (s, 1H), 7,24-7,37 (m, 5H), 6,75 (s, 1H), 4,37-4,52 (m, 2H), 3,91 (s, 3H), 3,67 (q, J = 7,2 Hz, 1H), 2,40 (s, 3H), 1,56 (d, J = 7,2 Hz, 3H).
Etapa 2: A una disolución de (R)-W-((3-metoxi-5-metilpirazin-2-il)metil)-2-fenilpropanamida (450 mg, 1,58 mmoles, 1 eq) en dioxano (10 mL) se añadió POCta (727 mg, 4,74 mmoles, 440 pL, 3 eq). La mezcla se agitó a 90 °C durante 12 horas. La mezcla de reacción se concentró bajo presión reducida para eliminar el dioxano. El residuo se paró por la adición de H2O (50 mL) a 0 °C, se basificó por la adición de NaHCO3 acuoso saturado (10 mL) y entonces se extrajo con EtOAc (50 mL x 2). Las capas orgánicas combinadas se lavaron con salmuera (50 mL), se secaron sobre Na2SO4, se filtraron y se concentraron bajo presión reducida. El residuo se purificó por cromatografía flash en gel de sílice (Eluyente de Acetato de etilo al 0~100 %/éter de petróleo). Se obtuvo (R)-6-metil-3-(1-feniletil)imidazo[1,5-a]pirazin-8-ol (150 mg, 592 pmoles, 37 % de rendimiento).
Etapa 3: A una disolución de (R)-6-metil-3-(1-feniletil)imidazo[1,5-a]pirazin-8-ol (100 mg, 395 pmoles, 1 eq) y 1-(clorometil)-4-metoxibenceno (74 mg, 474 pmoles, 65 pL, 1,20 eq) en DMF (5 mL) se añadió Cs2CO3 (257 mg, 790 pmoles, 2 eq). La mezcla se agitó a 60 °C durante 3 horas. La mezcla de reacción se concentró bajo presión reducida. El residuo se purificó por LC-MS preparativa. Se obtuvo (R)-7-(4-metoxibencil)-6-metil-3-(1-feniletil)imidazo[1,5-a]pirazin-8(7H)-ona (140 mg, 95 % de rendimiento).
1H RMN (CDCls 400 MHz): 57,98 (s, 1H), 7,17-7,32 (m, 5H), 7,12 (d, J = 8,4 Hz, 2H), 6,82 (d, J = 8,4 Hz, 2H), 6,46 (s, 1H), 5,10 (dd, J = 16,0 Hz, 2H), 4,26 (q, J = 7,2 Hz, 1H), 3,76 (s, 3H), 2,05 (s, 3H), 1,81 (d, J = 6,8 Hz, 3H).
LC-MS: tR = 2,184 min (método 18), m/z = 374,1 [M H]+. SFC: tR = 1,324 min, % ee = 97,8 %, [a]D20 = 50,7 (c=0,11, MeOH).
Ejemplo 73:
3-(1,4-dimetilpiperidin-4-il)-7-(4-metoxibencil)-6-metilimidazo[1,5-a]pirazin- 8(7H)-ona:
Etapa 1: A una disolución de hidrocloruro de (3-metoxi-5-metilpirazin-2-il)metanamina (150 mg, 791 pmoles) en DCM (5 mL) se añadió hidrocloruro del ácido 1,4-dimetilpiperidina-4-carboxílico (169 mg, 870 pmoles, 1,10 eq, HCl), trietilamina (240 mg, 2,37 mmoles, 329 pL, 3 eq) y h At U (361 mg, 949 pmoles, 1,20 eq). La mezcla se agitó a 15 °C durante 16 horas. La mezcla se concentró. El residuo se purificó por TLC preparativa (DCM/MeOH=10/1) para proporcionar /V-((3-metoxi-5-metilpirazin-2-il)metil)-1,4-dimetilpiperidina-4-carboxamida (150 mg, 65 % de rendimiento).
Etapa 2: A una disolución de W-((3-metoxi-5-metilpirazin-2-il)metil)-1,4-dimetilpiperidina-4-carboxamida (100 mg, 342 pmoles, 1 eq) en dioxano seco (5 mL) se añadió POCI3 (105 mg, 684 pmoles, 64 pL, 2 eq). La mezcla se calentó a 80 °C durante 4 horas. La mezcla se enfrió hasta 15 °C y se vertió en agua (5 mL). La mezcla se ajustó a pH 8 con NaHCO3 acuoso saturado y se concentró. Se añadió MeOH al 10 % en DCM (20 mL) al residuo y se filtró, el filtrado se concentró para proporcionar 3-(1,4-dimetilpiperidin-4-il)-6-metilimidazo[1,5-a]pirazin-8-ol (90 mg).
Etapa 3: A una disolución de 3-(1,4-dimetilpiperidin-4-il)-6-metilimidazo[1,5-a]pirazin-8-ol (60 mg, 230 pmoles, 1 eq) en DMF (2 mL) se añadió 1-(clorometil)-4-metoxibenceno (54 mg, 346 pmoles, 47 pL, 1,50 eq) y Cs2CO3 (150 mg, 461 pmoles, 2 eq). La mezcla se calentó a 60 °C durante 2 horas. La mezcla se concentró. Se añadieron DCM (20 mL) y H2O (10 mL). La mezcla se extrajo con DCM (20 mL). La capa orgánica se lavó con H2O (20 mL), se secó sobre Na2SO4, se filtró y se concentró. El residuo se purificó por LC-MS preparativa para proporcionar 3-(1,4-dimetilpiperidin-4-il)-7-(4-metoxibencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (20 mg, 23 % de rendimiento).
1H RMN (CDCl3400 MHz): 57,92 (s, 1H), 7,17 (d, J = 8,8 Hz, 2H), 6,94 (s, 1H), 6,84 (d, J = 8,8 Hz, 2H), 5,15 (s, 2H), 3,78 (s, 3H), 2,58-2,56 (m, 2H), 2,49-2,45 (m, 2H), 2,38-2,35 (m, 2H), 2,25 (s, 3H), 2,19 (s, 3H), 1,86-1,80 (m, 2H), 1,42 (s, 3H).
LC-MS: tR = 1,747 min (método 13), m /z = 381,2 [M H]+.
Ejemplo 74:

3-(6-cloro-2,3-dihidro-1 H-inden-1-il)-7-(4-metoxibencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona:
Etapa 1: A una disolución de hidrocloruro de (3-metoxi-5-metilpirazin-2-il)metanamina (200 mg, 1,05 mmoles) y ácido 6-cloro-2,3-dihidro-1H-inden-1-carboxílico (206 mg, 1,05 mmoles, 1 eq) en DCM (5 mL) se añadió HATU (479 mg, 1,26 mmoles, 1,20 eq) y DIPEA (407 mg, 3,15 mmoles, 550 pL, 3 eq). La mezcla se agitó a 18 °C durante 16 horas. La mezcla se lavó con H2O (20 mL) y se extrajo con DCM (20 mL x 3). El orgánico combinado se secó sobre Na2SO4 y se concentró en vacío. Se obtuvo 6-cloro-W-((3-metoxi-5-metilpirazin-2-il)metil)-2,3-dihidro-1H-inden-1-carboxamida (312 mg, 864 pmoles, 82 % de rendimiento).
Etapa 2: A una disolución de 6-cloro-W-((3-metoxi-5-metilpirazin-2-il)metil)-2,3-dihidro-1H-inden-1-carboxamida (0,262 g, 1eq) en dioxano (10 mL) se añadió pOc I3 (363 mg, 2,37 mmoles, 220 pL, 3 eq). La mezcla se agitó a 80 °C durante 3 horas. La mezcla se paró con H2O (20 mL) y se ajustó a pH > 7 con NaHCO3 acuoso saturado. La mezcla se extrajo con DCM (25 mL x 3). El orgánico combinado se secó sobre Na2SO4 y se concentró en vacío. Se obtuvo 3-(6-cloro-2,3-dihidro-1H-inden-1-il)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (236 mg) y se usó directamente para la siguiente etapa.
Etapa 3: A una disolución de 3-(6-cloro-2,3-dihidro-1H-inden-1-il)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (236 mg, 787 pmoles, 1 eq) y 1-(clorometil)-4-metoxibenceno (148 mg, 945 pmoles, 1,20 eq) en DMF (12 mL) se añadió Cs2CO3 (513 mg, 1,57 mmoles, 2 eq). La mezcla se agitó a 60 °C durante 2,5 horas. La mezcla se concentró en vacío. El residuo se paró con H2O (15 mL) y se extrajo con DCM (20 mL x 3). Las fases orgánicas combinadas se secaron sobe Na2SO4 y se concentraron en vacío. El residuo se purificó por HPLC preparativa. Se obtuvo 3-(6-cloro-2,3-dihidro-1 H-inden-1-il)-7-(4-metoxibencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (99 mg, 237 pmoles, 30 % de rendimiento).
1H RMN (CDCI3400 MHz): 57,93 (s, 1H), 7,30-7,18 (m, 4H), 6,98 (s, 1H), 6,88 (d, J =8,8 HZ, 2H), 6,64 (s, 1H), 5,18 (s, 2H), 4,71 (t, J =8,4 HZ 1H), 3,79 (s, 3H), 3,16-3,13 (m, 1H), 3,05-2,96 (m, 1H), 2,64-2,61 (m, 1H), 2,52-2,49 (m, 1H), 2,17 (s, 3H).
LC-MS: tR = 2,358 min (método 17), m /z = 420,1 [M H]+.
E je m p lo 75:

7-(4-metoxibencil)-6-metil-3-(3-metil-5-oxopirrolidin-3-il)imidazo[1,5-a]pirazin-8(7H)-ona:
Etapa 1: A una disolución de 3-metil-5-oxopirrolidina-3-carboxilato de etilo (200 mg, 1,17 mmoles, 1 eq) en THF (4 mL) y H2O (2 mL) se añadió L iO H ^ O (147,06 mg, 3,50 mmoles, 3 eq). La mezcla se agitó a 20 °C durante 16 horas. La mezcla se acidificó a pH=2 con HCl 1 M y se extrajo con acetato de etilo (20 mL x 3). Las capas orgánicas combinadas se lavaron con H2O (20 mL), salmuera (20 mL), se secaron sobre Na2SO4, se filtraron y se concentraron para proporcionar ácido 3-metil-5-oxopirrolidina-3-carboxílico (100 mg, 60 % de rendimiento).
Etapa 2: A una disolución enfriada (0 °C) de ácido 3-metil-5-oxopirrolidina-3-carboxílico (100 mg, 0,7 mmoles, 1 eq) en DCM (2 mL) se añadió dicloruro de oxalilo (98 mg, 768 pmoles, 67 pL, 1,10 eq) y se añadió una gota de DMF seco. La mezcla se agitó a 20 °C durante 1 hora. La disolución incolora de cloruro de 3-metil-5-oxopirrolidina-3-carbonilo (112,89 mg) en DCM (2 mL) se usó directamente para la siguiente etapa.
Etapa 3: A una disolución enfriada (0 °C) de (3-metoxi-5-metilpirazin-2-il)metanamina (120 mg, 633 pmoles, 1 eq, HCl) en DCM seco (3 mL) se añadió trietilamina (192 mg, 1,90 mmoles, 263 pL, 3 eq) y una disolución de cloruro de 3-metil-5-oxopirrolidina-3-carbonilo ( 112 mg, 696 pmoles, 1,10 eq) en DCM seco (2 mL) gota a gota. La mezcla se agitó a 20 °C durante 1 hora. Se añadió H2O (5 mL) y la mezcla se extrajo con DCM (20 mL x 2). La capa orgánica combinada se lavó con H2O (20 mL), salmuera (20 mL), se secó sobre Na2SO4, se filtró y se concentró. El residuo se purificó por TLC preparativa (DCM/MeOH=10/1) para proporcionar W-((3-metoxi-5-metilpirazin-2-il)metil)-3-metil-5-oxopirrolidina-3-carboxamida (75 mg, 42 % de rendimiento).
Etapa 4: Una disolución de W-((3-metoxi-5-metilpirazin-2-il)metil)-3-metil-5-oxopirrolidina-3-carboxamida (100 mg, 359 pmoles, 1 eq) en reactivo de Eaton (disolución de pentóxido de fósforo al 7,7 % en peso en ácido metanosulfónico) (2 mL) se calentó a 60 °C durante 16 horas. La mezcla se enfrió hasta 15 °C y se vertió en hielo (5 g). La mezcla se ajustó a pH=8 con NH37 M/MeOH y se concentró. Se añadió MeOH al 10 % en DCM (20 mL) al residuo y se filtró, el filtrado se concentró para proporcionar 4-(8-hidroxi-6-metilimidazo[1,5-a]pirazin-3-il)-4-metilpirrolidin-2-ona (100 mg).
Etapa 5: A una disolución de 4-(8-hidroxi-6-metilimidazo[1,5-a]pirazin-3-il)-4-metilpirrolidin-2-ona (100 mg, 406 pmoles, 1 eq) en DMF (5 mL) se añadió 1-(clorometil)-4-metoxibenceno (76 mg, 487 pmoles, 66 pL, 1,20 eq) y Cs2CO3 (265 mg, 812 pmoles, 2 eq). La mezcla se calentó a 60 °C durante 2h. La mezcla se concentró. Se añadió DCM (20 mL) y H2O (10 mL). La mezcla se extrajo con DCM (20 mL). Las capas orgánicas se lavaron con H2O (20 mL), se secaron sobre Na2SO4, se filtraron y se concentraron. La mezcla se purificó por HPLC preparativa para proporcionar 7-(4-metoxibencil)-6-metil-3-(3-metil-5-oxopirrolidin-3-il)imidazo[1,5-a]pirazin-8(7H)-ona (40 mg, 27 % de rendimiento).
1H RMN (CDCl3400 MHz): 57,87 (s, 1H), 7,15 (d, J = 8,4 Hz, 2H), 6,84 (d, J = 8,4 Hz, 2H), 6,65 (s, 1H), 5,93(brs, 1H), 5,16 (s, 2H), 4,25 (d, J = 9,6 Hz, 1H), 3,77 (s, 3H), 3,53 (d, J = 10,4 Hz, 1H), 3,00 (d, J = 16,8 Hz, 1H), 2,57 (d, J = 16,4 Hz, 1H), 2,20 (s, 3H), 1,63 (s, 3H).
LC-MS: tR = 1,764 min (método 11), m/z = 367,1 [M H]+.
Ejemplo 76:

3-(1-metoxi-2-metilpropan-2-il)-7-(4-metoxibencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona:
Etapa 1: A una disolución de ácido 3-metoxi-2,2-dimetilpropanoico (115 mg, 870 pmoles, 1 eq) en DCM (5 mL) se añadió dicloruro de oxalilo (121 mg, 957 pmoles, 84 pL, 1,10 eq) a 0 °C, seguido de una gota de DMF. La mezcla se agitó a 20 °C durante 1 hora. La mezcla se usó directamente para la siguiente etapa.
Etapa 2: A una disolución de cloruro de 3-metoxi-2,2-dimetilpropanoilo (150 mg, 791 pmoles, 1 eq, HCl) y trietilamina (120 mg, 1,19 mmoles, 164 pL, 1,50 eq) en DCM (10mL) se añadió (3- metoxi-5-metilpirazin-2-il)metanamina (131 mg, 870 pmoles, 1,10 eq) en DCM (5mL). La mezcla se agitó a 20 °C durante 1 hora. La mezcla se paró con H2O (20 mL) y se extrajo con DCM (15 mL x 3). Las capas orgánicas combinadas se secaron sobre Na2SO4 y se concentraron en vacío. Se obtuvo 3-metoxi-W-((3-metoxi-5-metilpirazin-2-il)metil)-2,2-dimetilpropanamida (142 mg, 522 pmoles, 66 % de rendimiento).
Etapa 3: A una disolución de 3-metoxi-W-((3-metoxi-5-metilpirazin-2-il)metil)-2,2-dimetilpropanamida (102 mg, 382 pmoles, 1 eq) en dioxano (5 mL) se añadió POCl3 (117 mg, 763 pmoles, 71 pL, 2 eq). La mezcla se agitó a 80 °C durante 2 horas. La mezcla se paró con agua (20 mL) y se extrajo con DCM (20 mL x 3). Las capas orgánicas combinadas se secaron con Na2SO4 y se concentraron en vacío. Se obtuvo 3-(1-metoxi-2-metilpropan-2-il)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (45 mg, 191 pmoles, 50 % de rendimiento).
Etapa 4: A una disolución de 3-(1-metoxi-2-metilpropan-2-il)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (62 mg, 264 pmoles, 1 eq) y 1-(clorometil)-4-metoxibenceno (49,52 mg, 316 pmoles, 43,06 pL, 1,20 eq) en DMF (5 mL) se añadió Cs2CO3 (171,72 mg, 527 pmoles, 2 eq). La mezcla se agitó a 60 °C durante 3 horas. La mezcla se lavó con H2O (20 mL) y se extrajo con DCM (20 mL x 3). Las capas orgánicas combinadas se secaron sobre Na2SO4 y se concentraron en vacío. El residuo se purificó por HPLC preparativa. Se obtuvo 3-(1-metoxi- 2-metilpropan-2-il)-7-(4-metoxibencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (25,15 mg, 71 pmoles, 27 % de rendimiento).
1H RMN (CDCl3400 MHz): 57,91 (s, 1H), 7,20-7,17 (m, 3H), 6,86 (d, J =8,4 Hz, 2H), 5,15 (s, 2H), 3,79 (s, 3H), 3,59 (s, 2H), 3,35 (s, 3H), 2,18 (s, 3H), 1,52 (s, 6H).
LC-MS: tR = 2,050 min (método 13), m /z = 356,1 [M H]+.
Ejemplo 77:

3-isopropil-7-(4-metoxibencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona:
Etapa 1: A una disolución fría (0 °C) de hidrocloruro de (3-metoxi-5-metilpirazin-2-il)metanamina (150 mg, 791 pmoles), Et3N (176 mg, 1,74 mmoles, 241 pL, 2,20 eq) en DCM anhidro (5 mL) se añadió cloruro de isobutirilo (93 mg, 870 pmoles, 91 pL, 1,10 eq). La disolución se agitó a 0 °C durante 0,5h. La mezcla se diluyó con agua (20 mL), se extrajo con DCM (20 mL x 2). La capa orgánica se lavó con salmuera (20 ml), se secó sobre Na2SO4 y se concentró en vacío. Se obtuvo W-((3-metoxi-5-metilpirazin-2-il)metil)isobutiramida (160 mg, 717 pmoles, 91 % de rendimiento).
Etapa 2: A una disolución de W-((3-metoxi-5-metilpirazin-2-il)metil)isobutiramida (160 mg, 717 pmoles, 1 eq) en dioxano (5 mL) se añadió POCl3 (220 mg, 1,43 mmoles, 133 pL, 2 eq). La mezcla se agitó a 90 °C 2 horas. La mezcla se concentró en vacío. Se obtuvo 3-isopropil-8-metoxi-6-metilimidazo[1,5-a]pirazina (130 mg, 633 pmoles, 88 % de rendimiento).
Etapa 3: Una disolución de 3-isopropil-8-metoxi-6-metilimidazo[1,5-a]pirazina (130 mg, 633 pmoles, 1 eq) en HCl(ac) 2 M (4 mL) y dioxano (8 mL) se agitó a 90 °C durante 2 horas. La mezcla se concentró en vacío. El residuo se purificó por cromatografía en gel de sílice (DCM:MeOH=10:1). Se obtuvo 3-isopropil-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (120 mg, 628 pmoles, 99 % de rendimiento).
Etapa 4: A una disolución de 3-isopropil-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (120 mg, 628 pmoles, 1 eq) en DMF (8 mL) se añadió 1-(clorometil)-4-metoxi-benceno (118 mg, 753 pmoles, 103 pL, 1,20 eq) y Cs2CO3 (307 mg, 941 pmoles, 1,50 eq). La mezcla se agitó a 60 °C durante 16 horas. La mezcla se filtró. El filtrado se purificó por HPLC preparativa (base). Se obtuvo 3-isopropil-7-(4-metoxibencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (90 mg, 287 pmoles, 46 % de rendimiento).1
1H RMN (CDCl3400 MHz): 5 7,90 (s, 1H), 7,16 (d, J=8,4 Hz, 1H), 6,85 (d, J=8,4 Hz, 1H), 6,71 (s, 1H), 5,16 (s, 2H), 3,78 (s, 2H), 3,18-3,11 (m, 1H), 2,19 (s, 3H), 1,41-1,79 (d, J=7,2 Hz, 6H).
LC -M S : tR = 1,92 m in (m é tod o 13), m/z = 312,1 [M H]+.
E je m p lo 78:
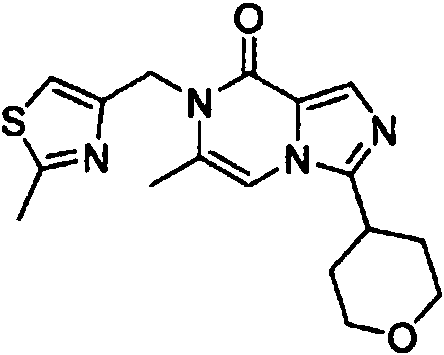
6-metil-7-((2-metiltiazol-4-il)metil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:
En un vial se añadió 6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (30mg, 0,13 mmoles), carbonato de cesio (84 mg, 0,26 mmoles), 4-(clorometil)-2-metiltiazol (23 mg, 0,15 mmoles) y yoduro de sodio (23 mg, 0,15 mmoles) en DMF (2 mL). La reacción se calentó hasta 70 °C, y se agitó toda la noche. La mezcla se filtró y se evaporó y posteriormente se cromatografió en gel de sílice para obtener el producto crudo. La purificación final en LC-MS preparativa rindió 6-metil-7-((2-metiltiazol-4-il)metil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (3,9 mg, 0,013 mmoles) con 10 % de rendimiento.
1H RMN (600 MHz, DMSO-d6) 58,17 (s, 1H), 7,63 (m, 1H), 7,30 (m, 1H), 5,17 (s, 2H), 3,99 (t, J = 3,3 Hz, 1H), 3,97 (t, J = 3,1 Hz, 1H), 3,49 (m, 3H), 2,61 (s, 3H), 2,36 (d, J = 1,2 Hz, 3H), 1,83 (m, 4H).
LC-MS: tR = 0,39 min (método 5), m/z = 344,9 [M H]+.
Ejemplo 79:

6-metil-3-(tetrahidro-2H-piran-4-il)-7-(tiofen-3-ilmetil)imidazo[1,5-a]pirazin-8(7H)-ona:
En un vial se añadió 6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (30mg, 0,129 mmoles), carbonato de cesio (84 mg, 0,26 mmoles), 3-(clorometil)tiofeno (20 mg, 0,15 mmoles) y yoduro de sodio (23 mg, 0,15 mmoles) en DMF (2 mL). La reacción se calentó hasta 70 °C, y se agitó toda la noche. La mezcla se filtró y se evaporó y posteriormente se cromatografió en gel de sílice para obtener el producto crudo. La purificación final en LC-MS preparativa rindió 6-metil-3-(tetrahidro-2H-piran-4-il)-7-(tiofen-3-ilmetil)imidazo[1,5-a]pirazin-8(7H)-ona (10 mg, 0,0324 mmoles) con 25 % de rendimiento.
1H RMN (600 MHz, DMSO-ds) 5 8,21 (s, 1H), 7,66 (m, 1H), 7,53 (dd, J = 5,0, 2,9 Hz, 1H), 7,35 (dq, J = 2,2, 1,0 Hz, 1H), 7,05 (dd, J = 5,0, 1,3 Hz, 1H), 5,17 (s, 2H), 3,98 (dt, J = 11,4, 3,4 Hz, 2H), 3,49 (m, 3H), 2,26 (d, J = 1,2 Hz, 3H), 1,84 (m, 4H).
LC-MS: tR = 0,45 min (método 5), m/z = 329,9 [M H]+.
Ejemplo 80:

6-metil-3-(tetrahidro-2H-piran-4-il)-7-(tiazol-4-ilmetil)imidazo[1,5-a]pirazin-8(7H)-ona:
En un vial se añadió 6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (30mg, 0,129 mmoles), carbonato de cesio (147 mg, 0,450 mmoles), hidrocloruro de 4-(clorometil)tiazol (26 mg, 0,15 mmoles) y yoduro de
sodio (23 mg, 0,15 mmoles) en DMF (2 mL). La reacción se calentó hasta 70 °C, y se agitó toda la noche. La mezcla se filtró y se evaporó y posteriormente se cromatografió en gel de sílice para obtener el producto crudo. La purificación final en LC-MS preparativa rindió 6-metil-3-(tetrahidro-2H-piran-4-il)-7-(tiazol-4-ilmetil)imidazo[1,5-a]pirazin-8(7H)-ona (7 mg, 0,0153 mmoles) con 12 % de rendimiento.
1H RMN (600 MHz, DMSO-ds) 59,07 (d, J = 1,9 Hz, 1H), 8,12 (s, 1H), 7,63 (m, 1H), 7,57 (dd, J = 1,9, 0,9 Hz, 1H), 5,27 (s, 2H), 3,97 (dt, J = 11,3, 3,3 Hz, 2H), 3,48 (m, 3H), 2,36 (d, J = 1,2 Hz, 3H), 1,84 (m, 4H).
LC-MS: tR = 0,34 min (método 5), m/z = 331,0 [M H]+.
Ejemplo 81:

7-((3,5-dimetilisoxazol-4-il)metil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:
En un vial se añadió 6-metil-3-(tetrahidro-2H-piran-4-il)irnidazo[1,5-a]pirazin-8(7H)-ona (25mg, 0,107 mmoles), carbonato de cesio (70 mg, 0,21 mmoles),4-(clorometil)-3,5-dimetilisoxazol (19 mg, 0,129 mmoles) y yoduro de sodio (19 mg, 0,129 mmoles) en DMF (1,6 mL). La reacción se calentó hasta 70 °C, y se agitó toda la noche. La mezcla se filtró y se evaporó y posteriormente se cromatografió en gel de sílice para obtener el producto crudo. La purificación final en LC-MS preparativa rindió 7-((3,5-dimetilisoxazol-4-il)metil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (11 mg, 0,024 mmoles) con 23 % de rendimiento.
1H RMN (600 MHz, DMSO-ds) 58,11 (s, 1H), 7,63 (m, 1H), 5,00 (s, 2H), 3,97 (dt, J = 11,3, 3,3 Hz, 2H), 3,46 (m, 3H), 2,27 (s, 3H), 2,24 (d, J = 1,2 Hz, 3H), 2,08 (s, 3H), 1,82 (dd, J = 7,8, 3,5 Hz, 4H).
LC-MS: tR = 0,38 min (método 5), m/z = 343,0 [M H]+.
Ejemplo 82:

6-metil-7-((5-metilisoxazol-3-il)metil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:
En un vial se añadió 6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (25mg, 0,107 mmoles), carbonato de cesio (70 mg, 0,21 mmoles), 3-(clorometil)-5-metilisoxazol (34 mg, 0,13 mmoles, al 50 %) y yoduro de sodio (19 mg, 0,13 mmoles) en DMF (1,6 mL). La reacción se calentó hasta 70 °C, y se agitó toda la noche. La mezcla se filtró y se evaporó y posteriormente se cromatografió en gel de sílice para obtener el producto crudo. La purificación final en LC-MS preparativa rindió 6-metil-7-((5-metilisoxazol-3-il)metil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (9 mg, 0,0194 mmoles) con 18 % de rendimiento.
1H RMN (600 MHz, DMSO-d6) 5 8,05 (s, 1H), 7,60 (m, 1H), 6,19 (q, J = 0,8 Hz, 1H), 5,18 (s, 2H), 3,97 (dt, J = 11,3, 3,4 Hz, 2H), 3,45 (m, 3H), 2,37 (d, J = 0,9 Hz, 3H), 2,27 (d, J = 1,2 Hz, 3H), 1,86 - 1,79 (m, 4H).
LC -M S : tR = 0 ,38 m in (m é tod o 5), m/z = 328 ,9 [M H]+.
Ejemplo 83:

6-metil-7-((3-metilisoxazol-5-il)metil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:
En un vial se añadió 6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (25mg, 0,107 mmoles), carbonato de cesio (70 mg, 0,21 mmoles), 5-(clorometil)-3-metilisoxazol (17 mg, 0,13 mmoles) y yoduro de sodio (19 mg, 0,13 mmoles) en DMF (1,6 mL). La reacción se calentó hasta 70 °C, y se agitó toda la noche. La mezcla se filtró y se evaporó y posteriormente se cromatografió en gel de sílice para obtener el producto crudo. La purificación final en LC-MS preparativa rindió 6-metil-7-((3-metilisoxazol-5-il)metil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (14 mg, 0,0313 mmoles) con 29 % de rendimiento.
1H RMN (600 MHz, DMSO-d6) 58,20 (s, 1H), 7,70 (m, 1H), 6,31 (s, 1H), 5,29 (m, 2H), 3,98 (dt, J = 11,4, 3,5 Hz, 2H), 3,49 (m, 3H), 2,32 (d, J = 1,2 Hz, 3H), 2,19 (s, 3H), 1,85 (m, 4H).
LC-MS: tR = 0,37 min (método 5), m/z = 328,9 [M H]+.
Ejemplo 84:

3-(2,6-dimetiltetrahidro-2H-piran-4-il)-7-(4-metoxibencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona:
Etapa 1: A una disolución de hidrocloruro de (3-metoxi-5-metilpirazin-2-il)metanamina (200 mg, 1,05 mmoles, 1 eq) y ácido 2,6-dimetiltetrahidro-2H-piran-4-carboxílico (167 mg, 1,05 mmoles, 1 eq) en DCM (10 mL) se añadió HATU (481 mg, 1,27 mmoles, 1,20 eq) y DIPEA (409 mg, 3,16 mmoles, 552 pL, 3 eq). La mezcla se agitó a 18 °C durante 16 horas. La mezcla se lavó con H2O (20 mL) y se extrajo con DCM (20 mL x 3). Las capas orgánicas combinadas se secaron sobre Na2SO4 y se concentraron en vacío. Se obtuvo W-((3-metoxi-5-metilpirazin-2-il)metil)-2,6-dimetiltetrahidro-2H-piran-4-carboxamida (300 mg, 0,95 mmoles, 90 % de rendimiento).
Etapa 2: A una disolución de W-((3-metoxi-5-metilpirazin-2-il)metil)-2,6-dimetiltetrahidro-2H-piran-4-carboxamida (280 mg, 954 pmoles, 1 eq) en dioxano (10 mL) se añadió POCl3 (293 mg, 1,91 mmoles, 177 pL, 2 eq). La mezcla se agitó a 80 °C durante 2 horas. La mezcla se lavó con H2O (20 mL) y se extrajo con DCM (20 mL x 3). Las capas orgánicas combinadas se secaron sobre Na2SO4 y se concentraron en vacío. Se obtuvo 3-(2,6-dimetiltetrahidro-2H-piran-4-il)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (249 mg).
Etapa 3: A una disolución de 3-(2,6-dimetiltetrahidro-2H-piran-4-il)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (200 mg, 765 pmoles, 1 eq) y 1 -(clorometil)-4-metoxibenceno (156 mg, 995 pmoles, 135 pL, 1,30 eq) en DMF (10 mL) se añadió Cs2COa (500 mg, 1,53 mmoles, 2 eq). La mezcla se agitó a 60 °C durante 4 horas. La mezcla se concentró en vacío. La mezcla se lavó con H2O (20 mL) y se extrajo con DCM (15 mL x 3). Las capas orgánicas combinadas se lavaron con H2O (40 mL x 3), salmuera, se secaron sobre Na2SO4 y se concentraron en vacío. El residuo se purificó por LC-MS preparativa para proporcionar 3-(2,6-dimetiltetrahidro-2H-piran-4-il)-7-(4-metoxibencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (48 mg, 126 pmoles, 16 % de rendimiento).
1H RMN (CDCl3400 MHz): 57,91 (s, 1H), 7,16 (d, J =8,4 HZ, 2H), 6,85 (d, J =8,4 HZ, 2H), 6,73 (s, 2H), 5,17 (s, 2H), 3,79 (s, 3H), 3,67-3,63 (m, 2H), 3,14-3,08 (m, 1H), 2,21 (s, 3H), 1,90-1,87 (m, 3H), 1,75-1,62 (m, 2H), 1,28 (d, J =6,0 HZ, 6H).
LC-MS: tR = 2,133 min (método 17), m/z = 382,1 [M H]+.
E je m p lo 85:

7-(ciclohexilmetil)-6-metil-3-propilimidazo[1,5-a]pirazin-8(7H)-ona:
Etapa 1: A una disolución de 3-bromo-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (1 g, 4,39 mmoles, 1 eq) y Cs2CO3 (2,86 g, 8,78 mmoles, 2 eq) en DMF (20 mL) se añadió (bromometil)ciclohexano (1,55 g, 8,78 mmoles, 1,22 mL, 2 eq). La mezcla se agitó a 60 °C durante 18 horas. La mezcla de reacción se filtró y el filtrado se concentró. La mezcla cruda se purificó por cromatografía flash con éter de petróleo:acetato de etilo = 5:1 ~ 3:1. Se obtuvo 3-bromo-7-(ciclohexilmetil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (1 g, 3,02 mmoles, 69 % de rendimiento).
1H RMN (CDCla, 400 MHz): 57,86 (s, 1H), 6,80 (s, 1H), 3,79 (d, J = 7,2 Hz, 2H), 2,29 (s, 3H), 1,75 -1,66 (m, 6H), 1,22 - 1,04 (m, 5H).
LC-MS: tR = 0,791 min (método 15), m /z = 325,9 [M H]+.
Etapa 2: A una disolución de 3-bromo-7-(ciclohexilmetil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (200 mg, 617 pmoles, 1 eq) y (E)-4,4,5,5-tetrametil-2-(prop-1-en-1-il)-1,3,2-dioxaborolano (155 mg, 925 pmoles, 1,50 eq) en dioxano (4 mL) y H2O (1 mL) se añadió Pd(dppf)Cl2 (90 mg, 123 pmoles, 0,20 eq) y Cs2CO3 (402 mg, 1,23 mmoles, 2 eq) bajo una atmósfera de N2. La mezcla se agitó a 90 °C durante 2 horas en condiciones de microondas. Se añadió agua (50 mL) y la mezcla se extrajo con EtOAc (50 mL x 3), las capas orgánicas combinadas se lavaron con salmuera (20 mL), se secaron sobre Na2SO4 y se concentraron. La mezcla cruda se purificó por cromatografía flash con éter de petróleo:acetato de etilo= 1:1. Se obtuvo (E)-7-(ciclohexilmetil)-6-metil-3-(prop-1-en-1-il)imidazo[1,5-a]pirazin-8(7H)-ona (150 mg, 504,59 pmoles, 82 % de rendimiento).
Etapa 3: A una disolución de (E)-7-(ciclohexilmetil)-6-metil-3-(prop-1-en-1-il)imidazo[1,5-a]pirazin-8(7H)-ona (150 mg, 526 pmoles, 1 eq) en EtOAc (30 mL) se añadió Pd-C (al 10 %, 40 mg, húmedo) bajo N2. La suspensión se desgasificó en vacío y se purgó con H2 varias veces. La mezcla se agitó bajo H2 (15 psi) a 25 °C durante 18 horas. La mezcla se filtró y el residuo se lavó con EtOAc (20 mL x 2). Las capas orgánicas combinadas se concentraron. El residuo se purificó por LC-MS preparativa para proporcionar 7-(ciclohexilmetil)-6-metil-3-propilimidazo[1,5-a]pirazin-8(7H)-ona (95.5 mg, 321 pmoles, 61 % de rendimiento).
1H RMN (400 MHz): 57,83 (s, 1H), 6,66 (s, 1H), 3,78 (d, J = 7,2 Hz, 2H), 2,81 (t, J = 7,6 Hz, 2H), 2,26 (s, 3H), 1,86 -1,84 (m, 2H), 1,76 - 1,67 (m, 6H), 1,19 - 1,17 (m, 3H), 1,05 - 1,01 (m, 5H).
LC-MS: tR = 1,68 min (método 17), m /z = 288,3 [M H]+.
Ejemplo 86:

3-(2-hidroxipropan-2-il)-7-(4-metoxibencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona:
Etapa 1: Se añadió NaH (2,64 g, 66 mmoles, al 60 % en aceite mineral, 2,20 eq) a ácido 2-hidroxi-2-metilpropanoico (3,12 g, 30 mmoles, 1 eq) en DMF (30 mL) a 0 °C. La mezcla se agitó a 20 °C durante 30 mins. Se añadió (bromometil)benceno (10,26 g, 60 mmoles, 7,13 mL, 2 eq) a la mezcla de reacción a 20 °C y se agitó a 20 °C durante 16 horas. La mezcla se paró con H2O (30 mL) y se ajustó a pH=7 con HCl (1 M, ac). La mezcla se extrajo con acetato de etilo (30 mL x 3). Las capas orgánicas combinadas se lavaron con H2O (20 mL) y salmuera. El residuo se secó sobre Na2SO4 y se concentró en vacío. La 1H RMN mostró que el compuesto era el producto deseado. Se obtuvo 2-(benciloxi)-2-metilpropanoato de bencilo (3,98 g, 14 mmoles, 47 % de rendimiento).
1H RMN (CDCla 400 MHz): 57,40-7,32 (m, 10H), 5,24 (s, 2H), 4,48 (s, 2H), 1,58 (s, 6H).
Etapa 2: A una disolución de 2-(benciloxi)-2-metilpropanoato de bencilo (2 g, 7,03 mmoles, 1 eq) en H2O (20 mL), THF (20 mL) y MeOH (20 mL) se añadió NaOH (1,12 g, 27,98 mmoles, 3,98 eq). La mezcla se agitó a 80 °C durante 1 hora. La mezcla se ajustó a pH=2 con HCl ac. (1 M) y se extrajo con DCM (10 mL x 3). Las capas orgánicas combinadas se concentraron en vacío. El residuo se lavó con NaOH ac. (1 M, 5 mL) y se extrajo con DCM (15 mL x 3). La disolución acuosa se ajustó a pH=2 con HCl ac. (1 M, ac) y se extrajo con DCM (10 mL x 3). Las capas orgánicas combinadas se lavaron con H2O (15 mL x 2) y salmuera. La mezcla se secó sobre Na2SO4 y se concentró en vacío. Se obtuvo ácido 2-(benciloxi)-2-metilpropanoico (1,36 g, 7 mmoles, 100 % de rendimiento).
Etapa 3: A una disolución de ácido 2-(benciloxi)-2-metilpropanoico (500 mg, 2,64 mmoles, 1 eq, HCl) en DCM (10 mL) se añadió DIPEA (1,02 g, 7,92 mmoles, 1,38 mL, 3 eq). A la mezcla se añadió hidrocloruro de (3-metoxi-5-metilpirazin-2-il)metanamina (513 mg, 2,64 mmoles, 1 eq) y HATu (1,20 g, 3,17 mmoles, 1,20 eq). La mezcla se agitó a 18 °C durante 16 horas. La mezcla se lavó con H2O (20 ml.) y se extrajo con DCM (20 mL x 3). Las capas orgánicas combinadas se secaron sobre Na2SO4 y se concentraron en vacío. Se obtuvo 2-(benciloxi)-W-((3-metoxi-5-metilpirazin-2-il)metil)-2-metilpropanamida (617 mg, 1,75 mmoles, 66 % de rendimiento).
Etapa 4: A una disolución de 2-(benciloxi)-W-((3-metoxi-5-metilpirazin-2-il)metil)-2-metilpropanamida (1,40 g, 4,25 mmoles, 1 eq) en dioxano (20 mL) se añadió POCh (1,30 g, 8,50 mmoles, 790 pL, 2 eq). La mezcla se agitó a 80 °C durante 2 horas. La mezcla se paró con H2O (15 mL) y se ajustó a pH >7 con NaHCO3 acuoso saturado. La mezcla se extrajo con DCM (20 mL x 3). Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 y se concentraron en vacío. Se obtuvo 3-(2-(benciloxi)propan-2-il)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (375 mg, 1,26 mmoles, 30 % de rendimiento).
Etapa 5: A una disolución de 3-(2-(benciloxi)propan-2-il)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (264 mg, 888 pmoles, 1 eq) en DMF (10 mL) se añadió Cs2CO3 (579 mg, 1,78 mmoles, 2 eq) y 1-(clorometil)-4-metoxibenceno (180,76 mg, 1,15 mmoles, 1.578 pL, 1,30 eq). La mezcla se agitó a 60 °C durante 4 horas. La mezcla se concentró en vacío. La mezcla se lavó con H2O (25 mL) y se extrajo con DCM (20mL). Las capas orgánicas combinadas se secaron sobre Na2SO4 y se concentraron en vacío. Se obtuvo 3-(2-(benciloxi)propan-2-il)-7-(4-metoxibencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (445 mg, 998 pmoles, 64 % de rendimiento).
Etapa 6 : A una disolución de 3-(2-(benciloxi)propan-2-il)-7-(4-metoxibencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (345 mg, 826 pmoles, 1 eq) en MeOH (60 mL) se añadió Pd/C (al 10 %, húmedo) (40 mg). La mezcla se agitó a presión de H2 (30 psi). La mezcla se agitó a 18 °C durante 8 horas. La mezcla se filtró. La disolución filtrada se concentró en vacío. El residuo se purificó por TLC (éter de petróleo:acetato de etilo=1:1). Se obtuvo 3-(2-hidroxipropan-2-il)-7-(4-metoxibencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (98 mg, 292 pmoles, 35 % de rendimiento).
1H RMN (CDCl3400 MHz): 5 7,86 (s, 1H), 7,47 (s, 1H), 7,17 (d, J =12 Hz, 2H), 6,85 (d, J =8,4 Hz, 2H), 5,17 (s, 2H), 3,79 (s, 3H), 2,216-2,192 (m, 4H), 1,76 (s, 3H).
LC-MS: tR = 1,906 min (método 13), m/z = 328,2 [M H]+.
Ejemplo 87:

3-(2-fluoropropan-2-il)-7-(4-metoxibencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona:
A una disolución de 3-(2-hidroxipropan-2-il)-7-(4-metoxibencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (75 mg, 229,09 pmoles, 1 eg) en DCM (10 ml) se añadió DAST (40,6 mg, 252 pmoles, 33 pL, 1,10 eg) a -78 °C. La mezcla se agitó a 18 °C durante 2 horas. La mezcla se paró con H2O (10mL) y se extrajo con DCM (15 mL x 3). Las capas orgánicas combinadas se secaron sobre Na2SO4 y se concentraron en vacío. El residuo se purificó con TLC (éter de petróleo:acetato de etilo=1:1). Se obtuvo 3-(2-fluoropropan-2-il)-7-(4-metoxibencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (68 mg, 206 pmoles, 64 % de rendimiento).1
1H RMN (CDCl3400 MHz): 57,89 (s, 1H), 7,19-7,16 (m, 3H), 6,86 (d, J =8,4 Hz, 2H), 5,18 (s, 2H), 3,79 (s, 3H), 2,20 (s, 3H), 1,91 (s, 3H), 1,86 (s, 3H).
LC -M S : tR = 1 ,906 m in (m é tod o 13), m/z = 328 ,2 [M H]+.
E je m p lo 88:

7-(4-metoxibencil)-6-metil-3-(7-oxoazepan-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:
Etapa 1: A una disolución de hidrocloruro de (3-metoxi-5-metilpirazin-2-il)metanamina (500 mg, 2,64 mmoles, 1 eg) y ácido 7-oxoazepan-4-carboxílico (456 mg, 2,90 mmoles, 1,10 eg) en DCM (45 mL) se añadió HATU (1,20 g, 3,17 mmoles, 1,20 eg) y DIPEA (1,02 g, 7,92 mmoles, 1,38 mL, 3 eg). La mezcla se agitó a 18 °C durante 16 horas. La mezcla se paró con H2O (30 mL) y se extrajo con DCM (25 mL x 3). Las capas orgánicas combinadas se secaron sobre Na2SO4 y se concentraron en vacío. Se obtuvo W-((3-metoxi-5-metilpirazin-2-il)metil)-7-oxoazepan-4-carboxamida (493 mg, 1,58 mmoles, 60 % de rendimiento).
Etapa 2: Se añadió /V-((3-metoxi-5-metilpirazin-2-il)metil)-7-oxoazepan-4-carboxamida (463 mg, 1,58 mmoles, 1 eq) a reactivo de Eaton (disolución de pentóxido de fósforo al 7,7 % en peso en ácido metanosulfónico) (3,04 g, 12,77 mmoles, 2 mL, 8,08 eq). La mezcla se agitó a 60 °C durante 7 horas. La mezcla se añadió a hielo (30 g). La mezcla se ajustó a pH>7 con NH3 (MeOH). La mezcla se concentró en vacío. El residuo se lavó con DCM:MeOH =10:1. La mezcla se filtró. La disolución filtrada se concentró en vacío. Se obtuvo 6-metil-3-(7-oxoazepan-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (45 mg, 165,83 pmoles, 11 % de rendimiento).
1H RMN (MeOD 400 MHz): 57,77 (s, 1H), 7,23 (s, 2H), 3,49-3,39 (m, 3H), 2,83-2,79 (m, 2H), 2,55-2,51 (m, 1H), 2,19 (s, 3H), 2,11 -2,09 (m, 2H), 1,93-1,86 (m, 2H).
Etapa 3: A una disolución de 6-metil-3-(7-oxoazepan-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (45 mg, 173 pmoles, 1 eq) y 1-(clorometil)-4-metoxibenceno (32 mg, 207 pmoles, 28 pL, 1,20 eq) en DMF (3 mL) se añadió Cs2CO3 (113 mg, 346 pmoles, 2 eq). La mezcla se agitó a 60 °C durante 2 horas. La mezcla se lavó con H2O (10 mL) y se extrajo con DCM (15 mL x 3). Las capas orgánicas combinadas se lavaron con agua (30 mL x 3), se secaron sobre Na2SO4 y se concentraron en vacío. El residuo se purificó por TLC (DCM:MeOH=10:1). Se obtuvo 7-(4-metoxibencil)-6-metil-3-(7-oxoazepan-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (12 mg, 32 pmoles, 18 % de rendimiento).
1H RMN (CDCl3400 MHz): 57,89 (s, 1H), 7,15 (d, J =8,8 HZ, 2H), 6,84 (d, J =8,4 HZ, 2H), 6,70 (s, 1H), 6,36 (brs, 1H), 5,16 (s, 2H), 5,16 (s, 2H), 3,77 (s, 3H),3,62-3,49 (m, 1H), 3,42- 3,30 (m, 1H), 3,20-3,10 (m, 1H), 2,77-2,72 (m, 1H), 2,61-2,55 (m, 1H), 2,21 (s, 3H), 2,10- 2,01 (m, 4H).
LC-MS: tR = 1,748 min (método 13), m /z = 381,2 [M H]+.
Ejemplo 89:

7-(4-metoxibencil)-6-metil-3-(5-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona:
Etapa 1: A una disolución de (3-metoxi-5-metilpirazin-2-il)metanamina (200 mg, 1,05 mmoles, 1 eq, HCl) y ácido 5-metiltetrahidrofuran-3-carboxílico (137 mg, 1,05 mmoles, 1 eq) en DCM (10 mL) se añadió HATU (481 mg, 1,27 mmoles, 1,20 eq) y DIPEA (409 mg, 3,16 mmoles, 553 pL, 3 eq). La mezcla se agitó a 18 °C durante 16 horas. La mezcla se lavó con H2O (20 mL) y se extrajo con DCM (20 mL x 3). Las capas orgánicas combinadas se secaron sobre Na2SO4 y se concentraron en vacío. Se obtuvo W-((3-metoxi-5-metilpirazin-2-il)metil)-5-metiltetrahidrofuran-3-carboxamida (211 mg, 795 pmoles, 76 % de rendimiento).
Etapa 2: A una disolución de W-((3-metoxi-5-metilpirazin-2-il)metil)-5-metiltetrahidrofuran-3-carboxamida (191 mg, 720 pmoles, 1 eq) en dioxano (5 mL) se añadió POCh (221 mg, 1,44 mmoles, 134 pL, 2 eq). La mezcla se agitó a 80 °C durante 3 horas. La mezcla se lavó con H2O (20 mL) y se extrajo con DCM (20 mL x 3). Las capas orgánicas combinadas se secaron sobre Na2SO4 y se concentraron en vacío. Se obtuvo 6-metil-3-(5-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona (167 mg).
Etapa 3: A una disolución de 6-metil-3-(5-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona (138 mg, 592 pmoles, 1 eq) y 1 -(clorometil)-4-metoxibenceno (120 mg, 769 pmoles, 1,30 eq) en DMF (10 mL) se añadió Cs2CO3 (386 mg, 1,18 mmoles, 2 eq). La mezcla se agitó a 60 °C durante 4 horas. La mezcla se concentró en vacío. La mezcla se lavó con H2O (20 mL) y se extrajo con DCM (15 mL x 3). Las capas orgánicas combinadas se lavaron con H2O (40 mL x 3), salmuera, se secaron sobre Na2SO4 y se concentraron en vacío. El residuo se purificó por HPLC preparativa (base). Se obtuvo 7-(4-metoxibencil)-6-metil-3-(5-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona (10 mg, 28 pmoles, 5 % de rendimiento).
1H RMN (CDCla 400 MHz): 57,90 (s, 1H), 7,16 (d, J =8,4 HZ, 2H), 6,87-6,82 (m, 3H), 5,17 (s, 2H), 4,19-4,14 (m, 3H), 3,79-3,71 (m, 4H), 2,54-2,47 (m, 1H), 2,20 (s, 3H), 2,04-1,96 (m, 1H), 1,40 (s, J =6,0 HZ, 3H).
LC-MS: tR = 2,036 min (método 13), m/z = 354,2 [M H]+.
Ejemplo 90:

7-(4-metoxibencil)-6-metil-3-(1-(4-metiltiazol-2-il)etil)imidazo[1,5-a]pirazin-8(7H)-ona:
Etapa 1: A una disolución de hidrocloruro de (3-metoxi-5-metilpirazin-2-il)metanamina (150 mg, 791 pmoles, 1 eg) en DMF seco (5 mL) se añadió trietilamina (240 mg, 2,37 mmoles, 329 pL, 3 eg), 2-(4-metiltiazol-2-il)propanoato de sodio (153 mg, 791 pmoles, 1 eg) y HATU (361 mg, 949 pmoles, 1,20 eg). La mezcla se agitó a 15 °C durante 16 horas. La mezcla se concentró. Se añadió H2O (5 mL) y la mezcla se extrajo con DCM (20 mL x 2). La capa orgánica combinada se lavó con H2O (20 mL), salmuera (20 mL), se secó sobre Na2SO4, se filtró y se concentró. El residuo se purificó por cromatografía flash en gel de sílice (acetato de etilo al 0 %~50 % en éter de petróleo) para proporcionar W-((3-metox¡-5-metilpirazin-2-il)metil)-2-(4-metiltiazol-2-il)propanamida (210 mg, 87 % de rendimiento).
Etapa 2: A una disolución de W-((3-metoxi-5-metilpirazin-2-il)metil)-2-(4-metiltiazol-2-il)propanamida (200 mg, 653 pmoles, 1 eg) en dioxano seco (5 mL) se añadió POCh (200 mg, 1,31 mmoles, 121 pL, 2 eg). La mezcla se calentó a 80 °C durante 4 horas. La mezcla se enfrió hasta 15 °C y se vertió en agua (5 mL). La mezcla se ajustó a pH 8 con NaHCO3 acuoso saturado y se extrajo con DCM (20 mL x 2). Los orgánicos combinados se lavaron con H2O (20 mL), salmuera (20 mL), se secaron sobre Na2SO4, se filtraron y se concentraron para proporcionar 6-metil-3-(1-(4-metiltiazol-2-il)etil)imidazo[1,5-a]pirazin-8-ol (160 mg).
Etapa 3: A una disolución de 6-metil-3-(1-(4-metiltiazol-2-il)etil)imidazo[1,5-a]pirazin-8-ol (180 mg, 656 pmoles, 1 eg) en DMF (5 mL) se añadió 1-(clorometil)-4-metoxi-benceno (123 mg, 787 pmoles, 107 pL, 1,20 eg) y Cs2CO3 (428 mg, 1,31 mmoles, 2 eg). La mezcla se calentó a 60 °C durante 2h. La mezcla se concentró. Se añadió DCM (20 mL) y H2O (10 mL). La mezcla se extrajo con DCM (20 mL). La capa orgánica se lavó con H2O (20 mL), se secó sobre Na2SO4, se filtró y se concentró. El residuo se purificó por LC-MS preparativa para proporcionar 7-(4-metoxibencil)-6-metil-3-(1-(4-metiltiazol-2-il)etil)imidazo[1,5-a]pirazin-8(7H)-ona (30 mg, 12 % de rendimiento).
1H RMN (CDCla 400 MHz): 57,95 (s, 1H), 7,13 (d, J = 8,4 Hz, 2H), 6,91 (s, 1H), 6,82 (d, J = 8,4 Hz, 2H), 6,79 (s, 1H), 5,19-5,06 (m, 2H), 4,76 (q, J = 7,2 Hz, 1H), 3,76 (s, 3H), 2,41 (s, 3H), 2,14 (s, 3H), 1,92 (d, J = 7,2 Hz, 1H).
LC-MS: tR = 2,532 min (método 11), m/z = 395,1 [M H]+.
E je m p lo 91:
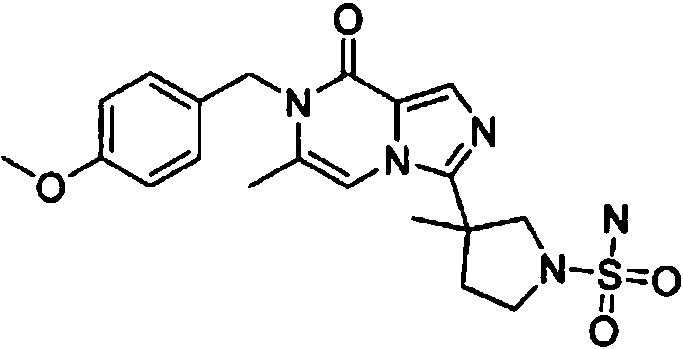
3-(7-(4-metoxibencil)-6-metil-8-oxo-7,8-dihidroimidazo[1,5-a]pirazin-3-il)-3-metilpirrolidina-1-sulfonamida:
Etapa 1: A una disolución de (3-metoxi-5-metilpirazin-2-il)metanamina (200 mg, 1,05 mmoles, 1 eq, HCl) y ácido 1-((benciloxi)carbonil)-3-metilpirrolidina-3-carboxílico (304 mg, 1,16 mmoles, 1,10 eq) en DCM (10 mL) se añadió HATU (479 mg, 1,26 mmoles, 1,20 eq) y DIPEA (407 mg, 3,15 mmoles, 550 pL, 3 eq). La mezcla se agitó a 18 °C durante 16 horas. La mezcla se paró con H2O (30 mL) y se extrajo con DCM (25 mL x 3). Las capas orgánicas combinadas se secaron sobre Na2SO4 y se concentraron en vacío. Se obtuvo 3-(((3-metoxi-5-metilpirazin-2-il)metil)carbamoil)-3-metilpirrolidina-1 -carboxilato de bencilo (420 mg, 1,01 mmoles, 96 % de rendimiento).
Etapa 2: A una disolución de 3-(((3-metoxi-5-metilpirazin-2-il)metil)carbamoil)-3-metilpirrolidina-1-carboxilato de bencilo (390 mg, 979 pmoles, 1 eq) en dioxano (15 mL) se añadió POCh (300 mg, 1,96 mmoles, 182 pL, 2 eq). La mezcla se agitó a 80 °C durante 3 horas. La disolución se paró con H2O (20 mL) y se extrajo con DCM (20 mL x 3). Las capas orgánicas combinadas se secaron sobre Na2SO4 y se concentraron en vacío. Se obtuvo 3-metil-3-(6-metil-8-oxo-7,8-dihidroimidazo[1,5-a]pirazin-3-il)pirrolidina-1-carboxilato de bencilo (170 mg, 423 pmoles, 43 % de rendimiento).
Etapa 3: A una disolución de 3-metil-3-(6-metil-8-oxo-7,8-dihidroimidazo[1,5-a]pirazin-3-il)pirrolidina-1-carboxilato de bencilo (290 mg, 791 pmoles, 1 eq) y dicarbonato de di-ferc-butilo (207 mg, 950 pmoles, 218 pL, 1,20 eq) en MeOH (200 mL) se añadió Pd/C (al 10 %, húmedo) (140 mg). La suspensión se desgasificó en vacío y se purgó con H2 varias veces. La mezcla se agitó bajo H2 (30 psi) a 25 °C durante 5 horas. La mezcla se filtró. La disolución filtrada se concentró en vacío. Se obtuvo 3-metil-3-(6-metil-8-oxo-7,8-dihidroimidazo[1,5-a]pirazin-3-il)pirrolidina-1 -carboxilato de ferc-butilo (204 mg, 614 pmoles, 78 % de rendimiento).
Etapa 4: A una disolución de 3-metil-3-(6-metil-8-oxo-7,8-dihidroimidazo[1,5-a]pirazin-3-il)pirrolidina-1-carboxilato de ferc-butilo (274 mg, 824 pmoles, 1 eq) y 1-(clorometil)-4-metoxibenceno (155 mg, 989 pmoles, 135 pL, 1,20 eq) en DMF (15 mL) se añadió Cs2CO3 (537 mg, 1,65 mmoles, 2 eq). La mezcla se agitó a 80 °C durante 14 horas. La mezcla se concentró en vacío. La mezcla se lavó con H2O (15 mL) y se extrajo con DCM (15 mL x 3). Las capas orgánicas combinadas se lavaron con H2O (30 mL x 2), se secaron sobre Na2SO4 y se concentraron en vacío, se obtuvo 3-(7-(4-metoxibencil)-6-metil-8-oxo-7,8-dihidroimidazo[1,5-a]pirazin-3-il)-3-metilpirrolidina-1 -carboxilato de ferc-butilo (291 mg, 537 pmoles, 65 % de rendimiento).
Etapa 5: A una disolución de 3-(7-(4-metoxibencil)-6-metil-8-oxo-7,8-dihidroimidazo[1,5-a]pirazin-3-il)-3-metilpirrolidina-1-carboxilato de ferc-butilo (288 mg, 636 pmoles, 1 eq) en acetato de etilo (4 mL) se añadió HCl/EtOAc (4 M, 4 mL, 25 eq). La mezcla se agitó a 18 °C durante 4 horas. La mezcla se concentró en vacío. Se obtuvo 7-(4-metoxibencil)-6-metil-3-(3-metilpirrolidin-3-il)imidazo[1,5-a]pirazin-8(7H)-ona (247 mg, cruda, HCl). El producto se usó directamente para la siguiente etapa.
Etapa 6 : A una disolución de 7-(4-metoxibencil)-6-metil-3-(3-metilpirrolidin-3-il)imidazo[1,5-a]pirazin-8(7H)-ona (250 mg, 709 pmoles, 1 eq) y diamida sulfúrica (82 mg, 851 pmoles, 51 pL, 1,20 eg) en dioxano (20 mL) se añadió DIPEA (183 mg, 1,42 mmoles, 248 pL, 2 eq). La mezcla se agitó a 100 °C durante 24 horas. La mezcla se paró con H2O (20 mL) y se extrajo con DCM (20 mL x 3). Las capas orgánicas combinadas se secaron sobre Na2SO4 y se concentraron en vacío. El residuo se purificó por HPLC preparativa (base). Se obtuvo 3-(7-(4- metoxibencil)-6-metil-8-oxo-7,8-dihidroimidazo[1,5-a]pirazin-3-il)-3-metilpirrolidina-1-sulfonamida (14 mg, 32 pmoles, 5 % de rendimiento).
1H RMN (CDCla 400 MHz): 57,91 (s, 1H), 7,16 (d, J =8,4 HZ, 2H), 7,85 (s, 1H), 7,18 (d, J =8,0 HZ, 2H), 6,88-6,82 (m, 3H), 5,17 (s, 2H), 4,79 (m, 1H), 4,30 (d, J =10,4 HZ, 1H), 3,79 (s, 3H), 3,52-3,47 (m, 2H), 3,26-3,20 (m, 1H), 2,73-2,64 (m, 1H), 2,23-2,17 (m, 5H), 1,65- 1,59 (m, 4H).
LC -M S : tR = 2 ,055 m in (m é tod o 13), m/z = 432 ,2 [M H]+.
E je m p lo 92:

6-(ciclopentilmetil)-7-(4-metoxibencil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:
Se disolvió 6-(bromometil)-7-(4-metoxibencil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (250 mg, 0,439 mmoles) en THF (20 ml), a -78 °C. Se añadió bromuro de ciclopentilmagnesio (0,9 ml, 1,8 mmoles, 2 molar en éter) y la reacción se dejó calentar hasta temperatura ambiente toda la noche. Se añadió bromuro de ciclopentilmagnesio (0,9 ml, 1,8 mmoles, 2 molar en éter). Después de 1 hora, la reacción se calentó hasta temperatura ambiente. Después de dos horas, la mezcla de reacción se paró con NH4Cl sat., se extrajo con AcOEt y los orgánicos se lavaron con salmuera. Las capas orgánicas combinadas se secaron con Na2SO4, se filtraron y se concentraron. La mezcla se purificó mediante LC-MS preparativa y se aisló 6-(ciclopentilmetil)-7-(4-metoxibencil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (26 mg, 0,049 mmoles) con 11 % de rendimiento, como la sal de TFA.
1H RMN (500 MHz, Cloroformo-d) 5 13,26 (bs, 1H), 8,14 (s, 1H), 7,12 (d, J = 8,2 Hz, 2H), 6,88 (m, 3H), 5,21 (s, 2H), 4,16 (d, J = 11,7 Hz, 2H), 3,80 (s, 3H), 3,60 (t, J = 11,8 Hz, 2H), 3,46 (t, J = 12,4 Hz, 1H), 2,74 (s, 1H), 2,57 (d, J = 7,0 Hz, 2H), 2,25 - 2,07 (m, 2H), 1,90 (m, 4H), 1,65 (m, 4H), 1,23 (dt, J = 13,2, 7,1Hz, 2H).
LC-MS: tR = 0,69 min (método 5), m/z = 422,0 [M H]+.
Ejemplo 93:

7-(4-metoxibencil)-6-metil-3-morfolinoimidazo[1,5-a]pirazin-8(7H)-ona:
A una mezcla de 3-bromo-7-(4-metoxibencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (300 mg, 0,86 mmoles) y morfolina (150 mg, 1,72 mmoles) en DMSO (5 mL) se añadieron CsF (261 mg, 1,72 mmoles) y K2CO3 (238 mg, 1,72 mmoles). La mezcla se agitó a 100 °C durante 24 horas. La mezcla se diluyó con agua (20 mL) y se extrajo con DCM (10 mL x 3). Las capas orgánicas combinadas se lavaron con agua (10 mL x 2 ); se secaron sobre Na2SO4 y se evaporaron en vacío. El residuo se purificó por TLC preparativa (acetato de etilo) para proporcionar 7-(4-metoxibencil)-6-metil-3-morfolinoimidazo[1,5-a]pirazin-8(7H)-ona (18 mg, 6 % de rendimiento).
1H RMN (CDCla, 400 MHz): 57,76 (s, 1H), 7,16 (d, J = 8,8 Hz, 2H), 6,85 (d, J = 8,8 Hz, 2H), 6,63 (s, 1H), 5,15 (s, 2H), 3,89 (t, J = 4,8 Hz, 4H), 3,79 (s, 3H), 3,21 (t, J = 4,8 Hz, 4H), 2,18 (s, 3H).
LC-MS: tR = 2,03 min (método 13), m/z = 355,1 [M H]+.
Ejemplo 94:
7-(4-metoxibencil)-6-metil-3-((tetrahidrofuran-3-il)amino)imidazo[1,5-a]pirazin-8(7H)-ona:
Se mezclaron 3-bromo-7-(4-metoxibencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (60 mg, 0,172 mmoles) y tetrahidrofuran-3-amina (0,04 ml, 0,465 mmoles) en NMP (2 mL) y DIPEA (0,23 ml, 1,317 mmoles).
La reacción se calentó durante 4 horas a 250 °C en el horno microondas.
La reacción se purificó en gel de sílice, mediante LC-MS preparativa, y TLC preparativa (EtOH al 10 % en acetato de etilo) para proporcionar 7-(4-metoxibencil)-6-metil-3-((tetrahidrofuran-3-il)amino)imidazo[1,5-a]pirazin-8(7H)-ona (2 mg, 0,005 mmoles) con 3 % de rendimiento.
1H RMN (600 MHz, Dimetilsulfóxido-d6) 58,50 (m, NH), 7,68 (s, 1H), 7,31 (s, 1H) 7,22 (d, J = 7,1 Hz, 2H), 6,88 (d, J = 7,1 Hz, 2H), 4,87 (m, 1H), 4,35 (d, J = 14 Hz, 2H), 4,11 (m, 1H), 4,02 (m, 1H), 3,92 (m, 1H), 3,86 (m, 1H), 3,71 (s, 3H), 2,60 (m, 1H), 2,5 (m, 1H) 2,31 (s, 3H).
LC-MS: tR = 0,54 min (método 19), m/z = 355,2 [M H]+.
Ejemplo 95:

(R)-7-(4-metoxibencil)-6-metil-3-(3-metilmorfolino)imidazo[1,5-a]pirazin-8(7H)-ona:
Se mezclaron 3-bromo-7-(4-metoxibencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (80 mg, 0,23 mmoles) y (R)-3-metilmorfolina (34,7 mg, 0,039 ml, 0,343 mmoles) en NMP (2,0 mL) y se añadió DIPEA (0,2 ml, 1,1 mmoles).
La reacción se calentó durante 6,5 horas a 250 °C en el horno microondas.
La reacción se purificó directamente por LC-MS preparativa para proporcionar (R)-7-(4-metoxibencil)-6-metil-3-(3-metilmorfolino)imidazo[1,5-a]pirazin-8(7H)-ona (10 mg, 0,024 mmoles) con 10 % de rendimiento.
1H RMN (500 MHz, Cloroformo-d) 58,00 (s, 1H), 7,18 (d, J = 7,1 Hz, 2H), 6,90 (d, J = 7,1 Hz, 2H), 6,79 (s, 1H), 5,23 (d, J = 14,0 Hz, 1H), 5,13 (d, J = 15,7 Hz, 1H), 3,94 (m, 1H), 3,85 (m, 1H), 3,81 (s, 3H), 3,60 (m, 1H), 3,48 (dd, J = 11,7, 7,9 Hz, 1H), 3,32 (t, J = 10,3 Hz, 1H), 3,22 (d, J = 12,2 Hz, 1H), 2,28 (d, J = 1,6 Hz, 3H), 1,29 (td, J = 7,1, 1,6 Hz, 1H), 1,00 (dd, J = 6,4, 1,5 Hz, 3H).
LC-MS: tR = 0,65 min (método 7), m/z = 369,1 [M H]+.
Ejemplo 96:

(S)-7-(4-metoxibencil)-6-metil-3-(3-metilmorfolino)imidazo[1,5-a]pirazin-8(7H)-ona:
Se mezclaron 3-bromo-7-(4-metoxibencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (80 mg, 0,230 mmoles) y (S)-3-metilmorfolina (0,039 ml, 0,343 mmoles) en NMP (2 mL) y DIPeA (148 mg, 0,2 ml, 1,1 mmoles).
La reacción se calentó durante 6,5 horas a 250 °C, en el horno microondas. La reacción se purificó directamente por LC-MS preparativa para proporcionar (S)-7-(4-metoxibencil)-6-metil-3-(3-metilmorfolino)imidazo[1,5-a]pirazin-8(7H)-ona (10 mg, 0,028 mmoles) con 12 % de rendimiento.
1H RMN (500 MHz, Cloroformo-d) 58,00 (s, 1H), 7,18 (d, J = 7,1 Hz, 2H), 6,90 (d, J = 7,1 Hz,, 2H), 6,79 (s, 1H), 5,23 (d, J = 14,0 Hz, 1H), 5,13 (d, J = 15,7 Hz, 1H), 3,94 (m, 1H), 3,85 (m, 1H), 3,81 (s, 3H), 3,60 (m, 1H), 3,48 (dd, J =
11,7, 7,9 Hz, 1H), 3,32 (t, J = 10,3 Hz, 1H), 3,22 (d, J = 12,2 Hz, 1H), 2,28 (d, J = 1,6 Hz, 3H), 1,29 (td, J = 7,1, 1,6 Hz, 1H), 1,00 (dd, J = 6,4, 1,5 Hz, 3H).
LC-MS: tR = 0,65 min (método 7), m/z = 369,1 [M H]+.
Ejemplo 97:

7-(4-metoxibencil)-6-metil-3-(1,4-oxazepan-4-il)imidazo[1,5-a]pirazin-8(7H)-ona:
Se mezclaron 3-bromo-7-(4-metoxibencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (149 mg, 0,428 mmoles) e hidrocloruro de 1,4-oxazepano (100 mg, 0,727 mmoles) en NMP (2,2 mL) y DIPEA (296 mg, 0,4 mL, 2,29 mmoles). La reacción se calentó durante 3 horas a 250 °C.
La reacción se purificó directamente mediante LC-MS preparativa para rendir 7-(4-metoxibencil)-6-metil-3-(1,4-oxazepan-4-il)imidazo[1,5-a]pirazin-8(7H)-ona (27 mg, 0,073 mmoles) con 17 % de rendimiento.
1H RMN (500 MHz, Cloroformo-d) 57,84 (s, 1H), 7,14 (m, 2H), 6,87 (m, 2H), 6,64 (s, 1H), 5,13 (s, 2H), 3,93 (m, 2H), 3,88 (m, 2H), 3,80 (d, J = 1,7 Hz, 3H), 3,73 (m, 4H), 2,22 (s, 3H), 2,09 (m, 2H).
LC-MS: tR = 0,56 min (método 7), m/z = 369,1 [M H]+.
Ejemplo 98:

3-(2,2-dimetilmorfolino)-7-(4-metoxibencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona:
A una mezcla de 3-bromo-7-(4-metoxibencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (500 mg, 1,44 mmoles) y 2,2-dimetilmorfolina (331 mg, 2,88 mmoles) en d Ms O (5 mL) se añadieron CsF (328 mg, 2,88 mmoles) y K2CO3 (299 mg, 2,88 mmoles). La mezcla se agitó a 100 °C durante 24 horas. La mezcla se diluyó con agua (20 mL) y se extrajo con DCM (10 mL x 3). La capa orgánica combinada se lavó con agua (10 mL x 2); se secó sobre Na2SO4 y se evaporó en vacío. El residuo se purificó por TLC preparativa (acetato de etilo) para proporcionar 3-(2,2-dimetilmorfolino)-7-(4-metoxibencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (15 mg, 3 % de rendimiento).
1H RMN (CDCla, 400 MHz): 57,75 (s, 1H), 7,15 (d, J = 8,8 Hz, 2H), 6,85 (d, J = 8,8 Hz, 2H), 6,60 (s, 1H), 5,15 (s, 2H), 3,93 (t, J = 4,8 Hz, 2H), 3,79 (s, 3H), 3,16 (t, J = 4,8 Hz, 2H), 2,98 (s, 2H), 2,18 (s, 3H), 1,37 (s, 6H).
LC-MS: tR = 2,26 min (método 13), m/z = 383,1 [M H]+.
Ejemplo 99:
7-(3-fluorobencil)-3-(hexahidro-4H-furo[3,2-b]pirrol-4-il)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona, estereoisómero 1 y 2:
Etapa 1: A una disolución de 3-bromo-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (3 g, 13,16 mmoles) y 1 -(bromometil)-3-fluorobenceno (2,99 g, 15,79 mmoles) en DMF (50 mL) se añadió K2CO3 (3,64 g, 26,3 mmoles). La mezcla se agitó a 60 °C durante 12 horas. La mezcla se diluyó con agua (100 mL) y se extrajo con EtOAc (50 mL x 3). La capa orgánica combinada se lavó con agua (50 mL x 2); se secó sobre Na2SO4 y se evaporó en vacío. El residuo se lavó con EtOAc (10 mL) y se filtró. La torta del filtro se secó en vacío para proporcionar 3-bromo-7-(3-fluorobencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (2 g, 45 % de rendimiento).
Etapa 2: A una mezcla de 3-bromo-7-(3-fluorobencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (1,2 g, 3,57 mmoles) y hexahidro-2H-furo[3,2-b]pirrol (0,5 sal del ácido oxálico) (678 mg, 4,28 mmoles) en DMSO (20 mL) se añadieron CsF (542 mg, 3,57 mmoles) y K2CO3 (1,23 g, 8,92 mmoles). La mezcla se agitó a 120 °C durante 72 horas. La mezcla se diluyó con agua (100 mL) y se extrajo con DCM (50 mL x 3). La capa orgánica combinada se lavó con agua (50 mL x 2); se secó sobre Na2SO4 y se evaporó en vacío. El residuo se purificó por HPLC preparativa para proporcionar el compuesto 3 (200 mg, 15 % de rendimiento).
Etapa 3: Se purificó 7-(3-fluorobencil)-3-(hexahidro-4H-furo[3,2-b]pirrol-4-il)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona (200 mg, 0,54 mmoles) por SFC para proporcionar el estereoisómero 1 (48 mg).
1H RMN (CDCla, 400 MHz): 5 7,74 (s, 1H), 7,33 - 7,29 (m, 1H), 7,01 - 6,89 (m, 3H), 6,73 (s, 1H), 5,19 (s, 2H), 4,72 -4.66 (m, 2H), 3,92 - 3,86 (m, 2H), 3,75 - 3,73 (m, 1H), 3,50 - 3,47 (m, 1H), 2,16 - 2,09 (m, 6H), 1,91 - 1,86 (m, 1H).
LC-MS: tR = 1,90 min (método 12), m/z = 369,1 [M H]+. SFC-MS: tR = 4,44 min, % ee > 99 %. [a]D20 133,00 (c = 0,10, DCM).
estereoisómero 2 (32 mg).
1H RMN (CDCla, 400 MHz): 5 7,74 (s, 1H), 7,32 - 7,28 (m, 1H), 7,01 - 6,89 (m, 3H), 6,73 (s, 1H), 5,19 (s, 2H), 4,74 -4.67 (m, 2H), 3,92 - 3,87 (m, 2H), 3,75 - 3,72 (m, 1H), 3,50 - 3,47 (m, 1H), 2,16 - 2,09 (m, 6H), 1,91 - 1,86 (m, 1H).
LC-MS: tR = 1,89 min (método 12), m/z = 369,1 [M H]+. SFC-MS: tR = 5,71 min, % ee > 99 %. [a]D20 -82,00 (c = 0,10, DCM).
Ejemplo 100:

7-(3-fluorobencil)-6-metil-3-(tetrahidro-2H-piran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona, estereoisómero 1 y 2:
Etapa 1: A una disolución de (3-metoxi-5-metilpirazin-2-il)metanamina (150 mg, 979,2 pmoles), ácido tetrahidro-2H-piran-3-carboxílico (127.4 mg, 979.2 pmoles) en DCM (10 mL) se añadió HATU (670,2 mg, 1,8 mmoles) y trietilamina (198,2 mg, 1,9 mmoles). La mezcla se agitó a 25 °C durante 16 horas. La mezcla se diluyó con agua (15 mL), se extrajo con DCM (3 x 30 mL). La capa orgánica combinada se lavó con salmuera (30 mL), se secó sobre Na2SO4, se filtró y se concentró. El residuo se purificó por TLC preparativa (EA/MeOH=20/1) para proporcionar W-((3-metox¡-5-metilpirazin-2-il)metil)tetrahidro-2H-piran-3-carboxamida (130 mg, 50 % de rendimiento).
Etapa 2: A una disolución de W-((3-metoxi-5-metilpirazin-2-il)metil)tetrahidro-2H-piran-3-carboxamida (130 mg, 490 pmoles) en dioxano (3 mL) se añadió POCl3 (1,28 g, 490 pmoles). La mezcla se agitó a 80 °C durante 3h. La mezcla se enfrió hasta 25 °C y se concentró. El residuo se neutralizó con NaHCO3 ac. saturado, se extrajo con acetato de etilo (2 x 20 mL). La capa orgánica combinada se lavó con salmuera (20 mL), se secó sobre Na2SO4, se filtró y se concentró para proporcionar 8-metoxi-6-metil-3-(tetrahidro-2H-piran-3-il)imidazo[1,5-a]pirazina cruda (120 mg, 99 % de rendimiento). El crudo se usó directamente para la siguiente etapa.
Etapa 3: A una disolución de 8-metoxi-6-metil-3-(tetrahidro-2H-piran-3-il)imidazo[1,5-a]pirazina (120 mg, 485,3 pmoles) en dioxano (3 mL) se añadió HCl (2 M, 3 mL). La mezcla se agitó a 80 °C durante 3h. La mezcla se enfrió hasta 25 °C y se concentró, se neutralizó con NaHCO3 ac. saturado, se extrajo con DCM (3 x 30 mL). La capa orgánica combinada se lavó con salmuera (20 mL), se secó sobre Na2SO4, se filtró y se concentró para proporcionar 6-metil-3-(tetrahidro-2H-piran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona cruda (110 mg, 97 % de rendimiento). El producto crudo se usó directamente para la siguiente etapa.
Etapa 4: A una disolución de 6-metil-3-(tetrahidro-2H-piran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona (100,0 mg, 428,7 pmoles) y 1-(bromometil)-3-fluorobenceno (121,6 mg, 643,0 pmoles) en DMF (5 mL) se añadió K2CO3 (118,5 mg, 857,4 pmoles). La mezcla se agitó a 80 °C durante 2h. La mezcla se enfrió hasta 25 °C y se diluyó con agua (20 mL), se extrajo con acetato de etilo (3 x 30 mL). La capa orgánica combinada se lavó con salmuera (30 mL), se secó sobre Na2SO4, se filtró y se concentró. El residuo se purificó por TLC preparativa (PE/EA=1/1) para proporcionar 7-(3-fluorobencil)-6-metil-3-(tetrahidro-2H-piran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona (80 mg, 54 % de rendimiento).
Se purificó 7-(3-fluorobencil)-6-metil-3-(tetrahidro-2H-piran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona (80 mg, 234,3 pmoles) por SFC.
Se obtuvo 7-(3-fluorobencil)-6-metil-3-(tetrahidro-2H-piran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona, estereoisómero 1 (26 mg, 33 % de rendimiento).
1H RMN (CDCla 400 MHz): 57,92 (s, 1H), 7,33-7,27 (m, 1H), 7,00-6,89 (m, 3H), 6,81 (s, 1H), 5,23 (s, 2H), 4,11-4,03 (m, 2H), 3,69 (t, J = 11,2 Hz, 1H), 3,57-3,53 (m, 1H), 3,17-3,14 (m, 1H), 2,18-2,11 (m, 5H), 1,85-1,79 (m, 2H).
LC-MS: tR = 2,06 min (método 3), m/z = 342,1 [M H]+. SFC: tR = 5,286 min, % ee > 99 %. aD20 = -3,0 (c = 0,10, CHCh).
Se obtuvo 7-(3-fluorobencil)-6-metil-3-(tetrahidro-2H-piran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona, estereoisómero 2 (28 mg, rendimiento: 35 %).
1H RMN (CDCla 400 MHz): 57,92 (s, 1H), 7,31-7,27 (m, 1H), 7,00-6,89 (m, 3H), 6,81 (s, 1H), 5,22 (s, 2H), 4,11-4,03 (m, 2H), 3,68 (t, J = 11,2 Hz, 1H), 3,57-3,53 (m, 1H), 3,17-3,14 (m, 1H), 2,18-2,11 (m, 5H), 1,84-1,78 (m, 2H).
LC-MS: tR = 2,06 min (método 3), m/z = 342,2 [M H]+. SFC: tR = 6,404 min, % ee > 99 %. aD20 = 3,0 (c = 0,10, CHCls).
Ensayo in vitro
Ensayo de la inhibición de PDE1
Los ensayos de PDE1A, PDE1B y PDE1C se realizaron como sigue: los ensayos se realizaron en muestras de 60 pL que contenían una cantidad fija de la enzima PDE1 (suficiente para convertir el 20-25 % del sustrato nucleótido cíclico), un tampón (HEPES 50 mM pH 7,6; MgCl210 mM; Tween20 al 0,02 %), 0,1 mg/ml de BSA, AMPc marcado con tritio 15 nM y cantidades variadas de inhibidores. Las reacciones se iniciaron por la adición del sustrato nucleótido cíclico, y se dejó que las reacciones continuaran durante 1 h a temperatura ambiente antes de terminarlas mediante mezclado con 20 pL (0,2 mg) de lechos SPA con silicato de itrio (PerkinElmer). Se dejó que los lechos sedimentaran durante 1 h en oscuridad antes de contar las placas en un contador Wallac 1450 Microbeta. Las señales medidas se convirtieron en actividad respecto a un control no inhibido (100 %) y los valores de CI50 se calcularon usando XIFit (modelo 205, IDBS).
Ensayo de inhibición de PDE9
Un ensayo de PDE9 puede realizarse, por ejemplo, como sigue: El ensayo se realiza en muestras de 60 pL que contienen una cantidad fija de la enzima PDE relevante (suficiente para convertir el 20-25 % del sustrato nucleótido cíclico), un tampón (HEp Es 50 mM pH 7,6; MgCl210 mM; Tween20 al 0,02 %), 0,1 mg/ml de BSA, 225 pCi de sustrato nucleótido cíclico marcado con 3H, AMPc marcado con tritio a una concentración final de 5 nM y cantidades variadas de inhibidores. Las reacciones se inician por la adición del sustrato nucleótido cíclico, y se deja que las reacciones continúen durante una hr a temperatura ambiente antes de terminarlas mediante mezclado con 15 pL 8 mg/mL de lechos SPA con silicato de itrio (Amersham). Se deja que los lechos sedimenten durante una hr en oscuridad antes de contar las placas en un contador Wallac 1450 Microbeta. La señal medida puede convertirse en actividad respecto a un control no inhibido (100 %) y los valores de CI50 pueden calcularse usando la extensión Xlfit de EXCEL.
En el contexto de la presente invención, el ensayo se realizó en 60 pL de tampón de ensayo (HEPES 50 mM pH 7,6; MgCl210mM; Tween20 al 0,02 %) que contenía suficiente PDE9 como para convertir el 20-25 % de 3H-AMPc 10 nM y cantidades variadas de inhibidores. Después de una incubación de 1 hr, las reacciones se terminaron por la adición de 15 pL 8 mg/mL de lechos SPA con silicato de itrio (Amersham). Se dejó que los lechos sedimentaran durante una hr en oscuridad antes de contar las placas en un contador Wallac 1450 Microbeta.
Claims (10)
1. Un compuesto de Fórmula (I)

en donde
n es 0 o 1;
q es 0 o 1;
R1 se selecciona del grupo que consiste en bencilo, indanilo, indolina y heteroarilos de 5 miembros; todos los cuales pueden estar sustituidos con un sustituyente seleccionado del grupo que consiste en halógeno y alquilo C1-C3; o R1 se selecciona del grupo que consiste en anillos monocíclicos saturados que contienen 4-6 átomos de carbono y 1 2 átomos de nitrógeno; todos los cuales pueden estar sustituidos una o más veces con uno o más sustituyentes seleccionados del grupo que consiste en metilo, flúor y sulfonamida; o
R1 se selecciona del grupo que consiste en lactamas que contienen 4-6 átomos de carbono; todos los cuales pueden estar sustituidos una o más veces con uno o más sustituyentes seleccionados del grupo que consiste en metilo y flúor; o R1 se selecciona del grupo que consiste en éteres bicíclicos tales como, 7-oxabiciclo[2.2.1]heptano; todos los cuales pueden estar sustituidos una o más veces con uno o más sustituyentes seleccionados del grupo que consiste en metilo y flúor; o
R1 se selecciona del grupo que consiste en alquilo C1-C8 lineal o ramificado, cicloalquilo C3-C8 monocíclico saturado, oxetanilo, tetrahidrofuranilo y tetrahidropiranilo; todos los cuales pueden estar sustituidos una o más veces con uno o más sustituyentes seleccionados del grupo que consiste en metilo, flúor, hidroxi, ciano o metoxi; o
R1 es un alquilo C1-C3 lineal o ramificado, que está sustituido con un sustituyente seleccionado de fenilo y heteroarilo de 5 miembros, en donde dicho heteroarilo de 5 miembros puede estar sustituido con uno o más alquilos C1-C3; o R1 se selecciona del grupo que consiste en morfolina, tetrahidrofuran-3-amina, hexahidro-2H-furo[3,2-b]pirrol y homomorfolina; todos los cuales pueden estar sustituidos con uno o más sustituyentes seleccionados del grupo que consiste en alquilo C1-C3;
R2 se selecciona del grupo que consiste en hidrógeno, alquilo C1-C8 lineal o ramificado, fenilo, cicloalquilo C3-C8 monocíclico saturado, oxetanilo, benzo[d][1,3]dioxolilo, tetrahidrofuranilo y tetrahidropiranilo; o
R2 es fenilo o piridilo sustituido con uno o más sustituyentes seleccionados del grupo que consiste en hidroxilo, amino, ciano, halógeno, alquilo C1-C3, alcoxi C1-C3, cicloalcoxi C3-C5, cicloalquil C3-C5-metoxi, fluoroalcoxi C1-C3, y -NC(O)CH3; o
R2 es un heteroarilo de 5 miembros que puede estar sustituido con uno o más alquilo C1-C3;
R3 se selecciona del grupo que consiste en hidrógeno, halógeno, alquilo C1-C5, cicloalquilo C3-C5 y fenilo; o R3 se selecciona del grupo que consiste en fenilo sustituido una o más veces con alquilo C1-C3; metilo sustituido una, dos o tres veces con flúor; etilo sustituido una, dos o tres veces con flúor;
R4 es hidrógeno;
y tautómeros y sales de adición farmacéuticamente aceptables del mismo;
con la condición de que R2 y R3 no pueden ser hidrógeno al mismo tiempo; y
2) un segundo compuesto, compuesto que es útil en el tratamiento de un trastorno psiquiátrico; en donde dicho segundo compuesto se selecciona de la lista que consiste en clozapina, risperidona, paliperidona, olanzapina, quetiapina, amisulprido, ziprasidona, aripiprazol, brexpiprazol, asenapina, haloperidol, iloperidona, lurasidona, clorpromazina, blonanserina, perfenazina, levomepromazina, sulpirido, flufenazina, zuclopentixol, flupentixol y cariprazina; en donde
1) y 2) son para uso combinado en el tratamiento de un trastorno psiquiátrico y/o cognitivo.
2. Los compuestos para uso según la reivindicación 1, en donde
R1 se selecciona del grupo que consiste en alquilo C1-C8 lineal o ramificado, cicloalquilo C3-C8 monocíclico saturado, oxetanilo, tetrahidrofuranilo, y tetrahidropiranilo;
R2 se selecciona del grupo que consiste en, alquilo C1-C8 lineal o ramificado, fenilo, y cicloalquilo C3-C8 monocíclico saturado; o
R2 se selecciona del grupo que consiste en fenilo sustituido con un sustituyente seleccionado del grupo que consiste en halógeno, alquilo C1-C3 y metoxi;
R3 se selecciona del grupo que consiste en hidrógeno, alquilo C1-C3 y halógeno;
R4 es hidrógeno.
3. Los compuestos para uso según la reivindicación 1, en donde dicho compuesto de fórmula (I) se selecciona del grupo que consiste en:
7-(3-Fluorobencil)-3-propilimidazo[1,5-a]pirazin-8(7H)-ona;
6-Bencil-7-(3-fluorobencil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
6- Bencil-7-(ciclohexilmetil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7- (Ciclohexilmetil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(3-Fluorobencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
3-Ciclopropil-7-(3-fluorobencil)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(Ciclopentilmetil)-3-ciclopropilimidazo[1,5-a]pirazin-8(7H)-ona;
7-(Ciclohexilmetil)-3-ciclopropilimidazo[1,5-a]pirazin-8(7H)-ona;
7-(3-Fluorobencil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(Ciclopentilmetil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(Ciclohexilmetil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(Cicloheptilmetil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(Cicloheptilmetil)-3-ciclopropilimidazo[1,5-a]pirazin-8(7H)-ona;
7-(4-Clorobencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
6- Bromo-7-(3-fluorobencil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7- Bencil-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(2-Fluorobencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(3-Clorobencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(2-Clorobencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(3-Metoxibencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
6-Metil-7-(2-metilbencil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
6- Metil-7-(4-metilbencil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7- (4-Metoxibencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(4-Fluorobencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
6- Metil-7-(3-metilbencil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7- (3-fluorobencil)-6-metil-3-(4-metiltetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
4-(7-(3-fluorobencil)-6-metil-8-oxo-7,8-dihidroimidazo[1,5-a]pirazin-3-il)tetrahidro-2H-piran-4-carbonitrilo; 7-(3-fluorobencil)-3-(4-metoxitetrahidro-2H-piran-4-il)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona;
7-(3-fluorobencil)-3-(4-fluorotetrahidro-2H-piran-4-il)-6-metilimidazo[1,5-a]pirazin-8(7H) ona;
(R) -7-(3-fluorobencil)-6-metil-3-(tetrahidro-2H-piran-2-il)imidazo[1,5-a]pirazin-8(7H)-ona;
(S) -7-(3-fluorobencil)-6-metil-3-(tetrahidro-2H-piran-2-il)imidazo[1,5-a]pirazin-8(7H)-ona;
(R) -7-(3-fluorobencil)-6-metil-3-(tetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona;
(S) -7-(3-fluorobencil)-6-metil-3-(tetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona;
(R) -7-(3-fluorobencil)-6-metil-3-(3-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H) ona;
(S) -7-(3-fluorobencil)-6-metil-3-(3-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H) ona;
7-(3-fluorobencil)-6-metil-3-(1-metilciclopropil)imidazo[1,5-a]pirazin-8(7H)-ona;
3-(2,2-difluorociclopropil)-7-(3-fluorobencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona;
7-(3-fluorobencil)-6-metil-3-((1 R,2S)-2-metilciclopropil)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(3-fluorobencil)-6-metil-3-((1 R,2R)-2-metilciclopropil)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(3-fluorobencil)-6-metil-3-((1 S,2S)-2-metilciclopropil)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(3-fluorobencil)-6-metil-3-((1 S,2R)-2-metilciclopropil)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(3-fluorobencil)-6-metil-3-((2S,3R)-2-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(3-fluorobencil)-6-metil-3-((2S,3S)-2-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(3-fluorobencil)-6-metil-3-((2R,3R)-2-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(3-fluorobencil)-6-metil-3-((2R,3S)-2-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(3-fluorobencil)-3-((1 R,2S)-2-fluorociclopropil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona;
7-(3-fluorobencil)-3-((1 R,2R)-2-fluorociclopropil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona;
7-(3-fluorobencil)-3-((1 S,2S)-2-fluorociclopropil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona;
7-(3-fluorobencil)-3-((1 S,2R)-2-fluorociclopropil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona;
7-(4-ciclopropoxibencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(4-(difluorometoxi)bencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
6- metil-3-(tetrahidro-2H-piran-4-il)-7-(4-(trifluorometoxi)bencil)imidazo[1,5-a]pirazin-8(7H)-ona;
7- (4-(ciclopropilmetoxi)bencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona; 7-bencil-6-etil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
6- etil-7-(4-metoxibencil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
3- ((6-metil-8-oxo-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-7(8H)-il)metil)benzonitrilo;
4- ((6-metil-8-oxo-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-7(8H)-il)metil)benzonitrilo;
W-(4-((6-metil-8-oxo-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-7(8H)-il)metil)fenil)acetamida; 7- (4-cloro-3-metoxibencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(2-etilbencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(benzo[d][1,3]dioxol-5-ilmetil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(3-cloro-4-metoxibencil)-6-metil-3-(tetrahidro-2H-piran-4-yi)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(4-aminobencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(4-hidroxibencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
6- etil-7-(3-fluorobencil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7- (4-metoxibencil)-6-metil-3-((2S,3R)-2-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(4-metoxibencil)-6-metil-3-((2S,3S)-2-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(4-metoxibencil)-6-metil-3-((2R,3R)-2-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(4-metoxibencil)-6-metil-3-((2R,3S)-2-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(4-metoxibencil)-6-metil-3-propilimidazo[1,5-a]pirazin-8(7H)-ona;
7-((6-metoxipiridin-3-il)metil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
6,7-dimetil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-etil-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
6- metil-7-propil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7- isopropil-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-isopentil-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(ciclopentilmetil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
2- ((6-metil-8-oxo-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-7(8H)-il)metil)benzonitrilo;
7-(cicloheptilmetil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(4-metoxibencil)-6-metil-3-(3-metiltetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(4-metoxibencil)-6-metil-3-((1 R,2R,4S)-2-metil-7-oxabiciclo[2.2.1]heptan-2-il)imidazo[1,5-a]pirazin-8(7H)-ona; (S)-7-(4-metoxibencil)-6-metil-3-(1-feniletil)imidazo[1,5-a]pirazin-8(7H)-ona;
(R)-7-(4-metoxibencil)-6-metil-3-(1-feniletil)imidazo[1,5-a]pirazin-8(7H)-ona;
3- (1,4-dimetilpiperidin-4-il)-7-(4-metoxibencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona;
3-(6-cloro-2,3-dihidro-1 H-inden-1-il)-7-(4-metoxibencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona;
7-(4-metoxibencil)-6-metil-3-(3-metil-5-oxopirrolidin-3-il)imidazo[1,5-a]pirazin-8(7H)-ona;
3-(1-metoxi-2-metilpropan-2-il)-7-(4-metoxibencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona;
3-isopropil-7-(4-metoxibencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona;
6-metil-7-((2-metiltiazol-4-il)metil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
6-metil-3-(tetrahidro-2H-piran-4-il)-7-(tiofen-3-ilmetil)imidazo[1,5-a]pirazin-8(7H)-ona;
6- metil-3-(tetrahidro-2H-piran-4-il)-7-(tiazol-4-ilmetil)imidazo[1,5-a]pirazin-8(7H)-ona;
7- ((3,5-dimetilisoxazol-4-il)metil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
6-metil-7-((5-metilisoxazol-3-il)metil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
6- metil-7-((3-metilisoxazol-5-il)metil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
3-(2,6-dimetiltetrahidro-2H-piran-4-il)-7-(4-metoxibencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona;
7- (ciclohexilmetil)-6-metil-3-propilimidazo[1,5-a]pirazin-8(7H)-ona;
3-(2-hidroxipropan-2-il)-7-(4-metoxibencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona;
3-(2-fluoropropan-2-il)-7-(4-metoxibencil)-6-methytimidazo[1,5-a]pirazin-8(7H)-ona;
7-(4-metoxibencil)-6-metil-3-(7-oxoazepan-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(4-metoxibencil)-6-metil-3-(5-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(4-metoxibencil)-6-metil-3-(1-(4-metiltiazol-2-il)etil)imidazo[1,5-a]pirazin-8(7H)-ona;
3-(7-(4-metoxibencil)-6-metil-8-oxo-7,8-dihidroimidazo[1,5-a]pirazin-3-il)-3-metilpirrolidina-1-sulfonamida;
6- (ciclopentilmetil)-7-(4-metoxibencil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
3-(morfolino)-7-(4-metoxibencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona;
7- (4-metoxibencil)-6-metil-3-((tetrahidrofuran-3-il)amino)imidazo[1,5-a]pirazin-8(7H)-ona;
(R) -7-(4-metoxibencil)-6-metil-3-(3-metilmorfolino)imidazo[1,5-a]pirazin-8(7H)-ona;
(S)-7-(4-metoxibencil)-6-metil-3-(3-metilmorfolino)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(4-metoxibencil)-6-metil-3-(1,4-oxazepan-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
3-(2,2-dimetilmorfolino)-7-(4-metoxibencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona;
7-(3-fluorobencil)-3-((3aS,6aS)-hexahidro-4H-furo[3,2-b]pirrol-4-il)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona;
7-(3-fluorobencil)-3-((3aR,6aR)-hexahidro-4H-furo[3,2-b]pirrol-4-il)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona;
(R)-7-(3-fluorobencil)-6-metil-3-(tetrahidro-2H-piran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona;
(S) -7-(3-fluorobencil)-6-metil-3-(tetrahidro-2H-piran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona;
o una sal farmacéuticamente aceptable de cualquiera de estos compuestos.
4. Los compuestos para uso según cualquiera de las reivindicaciones 1-3, en donde dicho trastorno psiquiátrico y/o cognitivo se selecciona del grupo que consiste en Trastorno de déficit de atención con hiperactividad (TDAH), depresión, ansiedad, narcolepsia, esquizofrenia, alteración cognitiva y alteración cognitiva asociada con esquizofrenia (CIAS).
5. Una composición farmacéutica que comprende:
1) un compuesto de Fórmula (I)

en donde
n es 0 o 1;
q es 0 o 1;
R1 se selecciona del grupo que consiste en bencilo, indanilo, indolina y heteroarilos de 5 miembros; todos los cuales pueden estar sustituidos con un sustituyente seleccionado del grupo que consiste en halógeno y alquilo C1-C3; o R1 se selecciona del grupo que consiste en anillos monocíclicos saturados que contienen 4-6 átomos de carbono y 1 2 átomos de nitrógeno; todos los cuales pueden estar sustituidos una o más veces con uno o más sustituyentes seleccionados del grupo que consiste en metilo, flúor y sulfonamida; o
R1 se selecciona del grupo que consiste en lactamas que contienen 4-6 átomos de carbono; todos los cuales pueden estar sustituidos una o más veces con uno o más sustituyentes seleccionados del grupo que consiste en metilo y flúor; o
R1 se selecciona del grupo que consiste en éteres bicíclicos tales como, 7-oxabiciclo[2.2.1]heptano; todos los cuales pueden estar sustituidos una o más veces con uno o más sustituyentes seleccionados del grupo que consiste en metilo y flúor; o
R1 se selecciona del grupo que consiste en alquilo C1-C8 lineal o ramificado, cicloalquilo C3-C8 monocíclico saturado, oxetanilo, tetrahidrofuranilo y tetrahidropiranilo; todos los cuales pueden estar sustituidos una o más veces con uno o más sustituyentes seleccionados del grupo que consiste en metilo, flúor, hidroxi, ciano o metoxi; o
R1 es un alquilo C1-C3 lineal o ramificado, que está sustituido con un sustituyente seleccionado de fenilo y heteroarilo de 5 miembros, en donde dicho heteroarilo de 5 miembros puede estar sustituido con uno o más alquilos C1-C3; o R1 se selecciona del grupo que consiste en morfolina, tetrahidrofuran-3-amina, hexahidro-2H-furo[3,2-b]pirrol y homomorfolina; todos los cuales pueden estar sustituidos con uno o más sustituyentes seleccionados del grupo que consiste en alquilo C1-C3;
R2 se selecciona del grupo que consiste en hidrógeno, alquilo C1-C8 lineal o ramificado, fenilo, cicloalquilo C3-C8 monocíclico saturado, oxetanilo, benzo[d][1,3]dioxolilo, tetrahidrofuranilo y tetrahidropiranilo; o
R2 es fenilo o piridilo sustituido con uno o más sustituyentes seleccionados del grupo que consiste en hidroxilo, amino, ciano, halógeno, alquilo C1-C3, alcoxi C1-C3, cicloalcoxi C3-C5, cicloalquil C3-C5-metoxi, fluoroalcoxi C1-C3, y -NC(O)CH3; o
R2 es un heteroarilo de 5 miembros que puede estar sustituido con uno o más alquilo C1-C3;
R3 se selecciona del grupo que consiste en hidrógeno, halógeno, alquilo C1-C5, cicloalquilo C3-C5 y fenilo; o R3 se selecciona del grupo que consiste en fenilo sustituido una o más veces con alquilo C1-C3; metilo sustituido una, dos o tres veces con flúor; etilo sustituido una, dos o tres veces con flúor;
R4 es hidrógeno;
y tautómeros y sales de adición farmacéuticamente aceptables del mismo;
con la condición de que R2 y R3 no pueden ser hidrógeno al mismo tiempo; y
2) un segundo compuesto, compuesto que es útil en el tratamiento de un trastorno psiquiátrico en donde dicho segundo compuesto se selecciona de la lista que consiste en clozapina, risperidona, paliperidona, olanzapina, quetiapina, amisulprido, ziprasidona, aripiprazol, brexpiprazol, asenapina, haloperidol, iloperidona, lurasidona, clorpromazina, blonanserina, perfenazina, levomepromazina, sulpirido, flufenazina, zuclopentixol, flupentixol y cariprazina; y uno o más vehículos, diluyentes y excipientes farmacéuticamente aceptables.
6. La composición farmacéutica según la reivindicación 5, en donde
R1 se selecciona del grupo que consiste en alquilo C1-C8 lineal o ramificado, cicloalquilo C3-C8 monocíclico saturado, oxetanilo, tetrahidrofuranilo, y tetrahidropiranilo;
R2 se selecciona del grupo que consiste en, alquilo C1-C8 lineal o ramificado, fenilo, y cicloalquilo C3-C8 monocíclico saturado; o
R2 se selecciona del grupo que consiste en fenilo sustituido con un sustituyente seleccionado del grupo que consiste en halógeno, alquilo C1-C3 y metoxi;
R3 se selecciona del grupo que consiste en hidrógeno, alquilo C1-C3 y halógeno;
R4 es hidrógeno.
7. La composición farmacéutica según la reivindicación 5, en donde el compuesto de fórmula (I) se selecciona del grupo que consiste en:
7-(3-Fluorobencil)-3-propilimidazo[1,5-a]pirazin-8(7H)-ona;
6-Bencil-7-(3-fluorobencil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
6- Bencil-7-(ciclohexilmetil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7- (Ciclohexilmetil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(3-Fluorobencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
3-Ciclopropil-7-(3-fluorobencil)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(Ciclopentilmetil)-3-ciclopropilimidazo[1,5-a]pirazin-8(7H)-ona;
7-(Ciclohexilmetil)-3-ciclopropilimidazo[1,5-a]pirazin-8(7H)-ona;
7-(3-Fluorobencil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(Ciclopentilmetil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(Ciclohexilmetil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(Cicloheptilmetil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(Cicloheptilmetil)-3-ciclopropilimidazo[1,5-a]pirazin-8(7H)-ona;
7-(4-Clorobencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
6- Bromo-7-(3-fluorobencil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7- Bencil-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(2-Fluorobencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(3-Clorobencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(2-Clorobencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(3-Metoxibencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
6-Metil-7-(2-metilbencil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
6- Metil-7-(4-metilbencil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7- (4-Metoxibencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(4-Fluorobencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
6- Metil-7-(3-metilbencil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7- (3-fluorobencil)-6-metil-3-(4-metiltetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
4-(7-(3-fluorobencil)-6-metil-8-oxo-7,8-dihidroimidazo[1,5-a]pirazin-3-il)tetrahidro-2H-piran-4-carbonitrilo; 7-(3-fluorobencil)-3-(4-metoxitetrahidro-2H-piran-4-il)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona;
7-(3-fluorobencil)-3-(4-fluorotetrahidro-2H-piran-4-il)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona;
(R) -7-(3-fluorobencil)-6-metil-3-(tetrahidro-2H-piran-2-il)imidazo[1,5-a]pirazin-8(7H)-ona;
(S) -7-(3-fluorobencil)-6-metil-3-(tetrahidro-2H-piran-2-il)imidazo[1,5-a]pirazin-8(7H)-ona;
(R) -7-(3-fluorobencil)-6-metil-3-(tetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona;
(S) -7-(3-fluorobencil)-6-metil-3-(tetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona;
(R) -7-(3-fluorobencil)-6-metil-3-(3-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona;
(S) -7-(3-fluorobencil)-6-metil-3-(3-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(3-fluorobencil)-6-metil-3-(1-metilciclopropil)imidazo[1,5-a]pirazin-8(7H)-ona;
3-(2,2-difluorociclopropil)-7-(3-fluorobencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona;
7-(3-fluorobencil)-6-metil-3-((1 R,2S)-2-metilciclopropil)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(3-fluorobencil)-6-metil-3-((1 R,2R)-2-metilciclopropil)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(3-fluorobencil)-6-metil-3-((1S,2S)-2-metilciclopropil)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(3-fluorobencil)-6-metil-3-((1 S,2R)-2-metilciclopropil)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(3-fluorobencil)-6-metil-3-((2S,3R)-2-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(3-fluorobencil)-6-metil-3-((2S,3S)-2-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(3-fiuorobencil)-6-metil-3-((2R,3R)-2-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona; 7-(3-fluorobencil)-6-metil-3-((2R,3S)-2-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona; 7-(3-fluorobencil)-3-((1 R,2S)-2-fluorociclopropil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona;
7-(3-fluorobencil)-3-((1 R,2R)-2-fluorociclopropil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona;
7-(3-fluorobencil)-3-((1 S,2S)-2-fluorociclopropil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona;
7-(3-fluorobencil)-3-((1 S,2R)-2-fluorociclopropil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona;
7-(4-ciclopropoxibencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(4-(difluorometoxi)bencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona; 6- metil-3-(tetrahidro-2H-piran-4-il)-7-(4-(trifluorometoxi)bencil)imidazo[1,5-a]pirazin-8(7H)-ona; 7- (4-(ciclopropilmetoxi)bencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona; 7-bencil-6-etil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
6- etil-7-(4-metoxibencil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
3- ((6-metil-8-oxo-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-7(8H)-il)metil)benzonitrilo; 4- ((6-metil-8-oxo-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-7(8H)-il)metil)benzonitrilo; W-(4-((6-metil-8-oxo-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-7(8H)-il)metil)fenil)acetamida; 7- (4-cloro-3-metoxibencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona; 7-(2-etilbencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(benzo[d][1,3]dioxol-5-ilmetil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona; 7-(3-cloro-4-metoxibencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona; 7-(4-aminobencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(4-hidroxibencil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
6- etil-7-(3-fluorobencil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7- (4-metoxibencil)-6-metil-3-((2S,3R)-2-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona; 7-(4-metoxibencil)-6-metil-3-((2S,3S)-2-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona; 7-(4-metoxibencil)-6-metil-3-((2R,3R)-2-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona; 7-(4-metoxibencil)-6-metil-3-((2R,3S)-2-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona; 7-(4-metoxibencil)-6-metil-3-propilimidazo[1,5-a]pirazin-8(7H)-ona;
7-((6-metoxipiridin-3-il)metil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona; 6,7-dimetil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-etil-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
6- metil-7-propil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7- isopropil-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-isopentil-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(ciclopentilmetil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
2-((6-metil-8-oxo-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-7(8H)-il)metil)benzonitrilo; 7-(cicloheptilmetil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(4-metoxibencil)-6-metil-3-(3-metiltetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(4-metoxibencil)-6-metil-3-((1 R,2R,4S)-2-metil-7-oxabiciclo[2.2.1]heptan-2-il)imidazo[1,5-a]pirazin-8(7H)-ona; (S)-7-(4-metoxibencil)-6-metil-3-(1-feniletil)imidazo[1,5-a]pirazin-8(7H)-ona;
(R)-7-(4-metoxibencil)-6-metil-3-(1-feniletil)imidazo[1,5-a]pirazin-8(7H)-ona;
3-(1,4-dimetilpiperidin-4-il)-7-(4-metoxibencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona;
3-(6-cloro-2,3-dihidro-1 H-inden-1-il)-7-(4-metoxibencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona;
7-(4-metoxibencil)-6-metil-3-(3-metil-5-oxopirrolidin-3-il)imidazo[1,5-a]pirazin-8(7H)-ona;
3-(1-metoxi-2-metilpropan-2-il)-7-(4-metoxibencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona;
3-isopropil-7-(4-metoxibencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona;
6-metil-7-((2-metiltiazol-4-il)metil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
6-metil-3-(tetrahidro-2H-piran-4-il)-7-(tiofen-3-ilmetil)imidazo[1,5-a]pirazin-8(7H)-ona;
6- metil-3-(tetrahidro-2H-piran-4-il)-7-(tiazol-4-ilmetil)imidazo[1,5-a]pirazin-8(7H)-ona;
7- ((3,5-dimetilisoxazol-4-il)metil)-6-metil-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
6-metil-7-((5-metilisoxazol-3-il)metil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
6- metil-7-((3-metilisoxazol-5-il)metil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
3-(2,6-dimetiltetrahidro-2H-piran-4-il)-7-(4-metoxibencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona;
7- (ciclohexilmetil)-6-metil-3-propilimidazo[1,5-a]pirazin-8(7H)-ona;
3-(2-hidroxipropan-2-il)-7-(4-metoxibencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona;
3-(2-fluoropropan-2-il)-7-(4-metoxibencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona;
7-(4-metoxibencil)-6-metil-3-(7-oxoazepan-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(4-metoxibencil)-6-metil-3-(5-metiltetrahidrofuran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(4-metoxibencil)-6-metil-3-(1-(4-metiltiazol-2-il)etil)imidazo[1,5-a]pirazin-8(7H)-ona;
3-(7-(4-metoxibencil)-6-metil-8-oxo-7,8-dihidroimidazo[1,5-a]pirazin-3-il)-3-metilpirrolidina-1-sulfonamida;
6- (ciclopentilmetil)-7-(4-metoxibencil)-3-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
3-(morfolino)-7-(4-metoxibencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona;
7- (4-metoxibencil)-6-metil-3-((tetrahidrofuran-3-il)amino)imidazo[1,5-a]pirazin-8(7H)-ona;
(R) -7-(4-metoxibencil)-6-metil-3-(3-metilmorfolino)imidazo[1,5-a]pirazin-8(7H)-ona;
(S) -7-(4-metoxibencil)-6-metil-3-(3-metilmorfolino)imidazo[1,5-a]pirazin-8(7H)-ona;
7-(4-metoxibencil)-6-metil-3-(1,4-oxazepan-4-il)imidazo[1,5-a]pirazin-8(7H)-ona;
3-(2,2-dimetilmorfolino)-7-(4-metoxibencil)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona;
7-(3-fluorobencil)-3-((3aS,6aS)-hexahidro-4H-furo[3,2-b]pirrol-4-il)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona;
7-(3-fluorobencil)-3-((3aR,6aR)-hexahidro-4H-furo[3,2-b]pirrol-4-il)-6-metilimidazo[1,5-a]pirazin-8(7H)-ona;
(R) -7-(3-fluorobencil)-6-metil-3-(tetrahidro-2H-piran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona;
(S) -7-(3-fluorobencil)-6-metil-3-(tetrahidro-2H-piran-3-il)imidazo[1,5-a]pirazin-8(7H)-ona;
o una sal farmacéuticamente aceptable de cualquiera de estos compuestos.
8. La composición farmacéutica según cualquiera de las reivindicaciones 5-7, que comprende el compuesto de fórmula (I) y cualquiera de los compuestos listados en el ítem 2) en la reivindicación 5, en cantidades terapéuticamente efectivas.
9. La composición farmacéutica según cualquiera de las reivindicaciones 5-8 para uso en el tratamiento de un trastorno psiquiátrico y/o cognitivo.
10. La composición farmacéutica para uso según la reivindicación 9, en donde dicho trastorno psiquiátrico y/o cognitivo se selecciona del grupo que consiste en Trastorno de déficit de atención con hiperactividad (TDAH), depresión, ansiedad, narcolepsia, esquizofrenia, alteración cognitiva y alteración cognitiva asociada con esquizofrenia (CIAS).
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DKPA201600659 | 2016-10-28 | ||
| PCT/EP2017/077497 WO2018078038A1 (en) | 2016-10-28 | 2017-10-26 | Combination treatments comprising imidazopyrazinones for the treatment of psychiatric and/or cognitive disorders |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2816005T3 true ES2816005T3 (es) | 2021-03-31 |
Family
ID=60186299
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES17791080T Active ES2816005T3 (es) | 2016-10-28 | 2017-10-26 | Tratamientos de combinación que comprenden imidazopirazinonas para el tratamiento de trastornos psiquiátricos y/o cognitivos |
Country Status (30)
| Country | Link |
|---|---|
| US (1) | US10905688B2 (es) |
| EP (1) | EP3532064B1 (es) |
| JP (1) | JP2019536762A (es) |
| KR (1) | KR20190075068A (es) |
| CN (1) | CN109803658A (es) |
| AR (1) | AR109991A1 (es) |
| AU (1) | AU2017350473A1 (es) |
| BR (1) | BR112018013247A2 (es) |
| CA (1) | CA3041607A1 (es) |
| CL (1) | CL2019001091A1 (es) |
| CO (1) | CO2019002909A2 (es) |
| CY (1) | CY1123396T1 (es) |
| DK (1) | DK3532064T3 (es) |
| ES (1) | ES2816005T3 (es) |
| HR (1) | HRP20201326T1 (es) |
| HU (1) | HUE050403T2 (es) |
| IL (1) | IL265635A (es) |
| LT (1) | LT3532064T (es) |
| MA (1) | MA46611B1 (es) |
| ME (1) | ME03821B (es) |
| MX (1) | MX2019004763A (es) |
| PL (1) | PL3532064T3 (es) |
| PT (1) | PT3532064T (es) |
| RS (1) | RS60716B1 (es) |
| RU (1) | RU2019109039A (es) |
| SG (1) | SG11201903770UA (es) |
| SI (1) | SI3532064T1 (es) |
| SM (1) | SMT202000440T1 (es) |
| WO (1) | WO2018078038A1 (es) |
| ZA (1) | ZA201902091B (es) |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TW201629064A (zh) | 2014-10-10 | 2016-08-16 | H 朗德貝克公司 | 作爲pde1抑制劑之三唑並吡酮 |
| JO3627B1 (ar) | 2015-04-30 | 2020-08-27 | H Lundbeck As | إيميدازو بيرازينونات على هيئة مثبطات pde1 |
| TWI729109B (zh) | 2016-04-12 | 2021-06-01 | 丹麥商H 朗德貝克公司 | 作爲PDE1抑制劑的1,5-二氫-4H-吡唑并[3,4-d]嘧啶-4-酮和1,5-二氫-4H-吡唑并[4,3-c]吡啶-4-酮 |
| WO2018073251A1 (en) | 2016-10-18 | 2018-04-26 | H. Lundbeck A/S | Imidazopyrazinones, pyrazolopyrimidinones and pyrazolopyridinones as pde1 inhibitors |
| WO2018078042A1 (en) | 2016-10-28 | 2018-05-03 | H. Lundbeck A/S | Combination treatments comprising administration of imidazopyrazinones |
| AU2017350473A1 (en) | 2016-10-28 | 2019-04-18 | H. Lundbeck A/S | Combination treatments comprising imidazopyrazinones for the treatment of psychiatric and/or cognitive disorders |
| AR114237A1 (es) | 2018-02-01 | 2020-08-05 | Japan Tobacco Inc | Compuesto de amida heterocíclica que contiene nitrógeno y su uso farmacéutico |
Family Cites Families (52)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB973361A (en) | 1960-05-11 | 1964-10-28 | Ciba Ltd | Pyrazolo-pyrimidines and process for their manufacture |
| DE19709877A1 (de) | 1997-03-11 | 1998-09-17 | Bayer Ag | 1,5-Dihydro-pyrazolo[3,4-d]-pyrimidinon-derivate |
| MY144532A (en) | 2001-08-20 | 2011-09-30 | Lundbeck & Co As H | Novel method for down-regulation of amyloid |
| DE10238724A1 (de) | 2002-08-23 | 2004-03-04 | Bayer Ag | Alkyl-substituierte Pyrazolpyrimidine |
| DE10238723A1 (de) | 2002-08-23 | 2004-03-11 | Bayer Ag | Phenyl-substituierte Pyrazolyprimidine |
| DE10238722A1 (de) | 2002-08-23 | 2004-03-11 | Bayer Ag | Selektive Phosphodiesterase 9A-Inhibitoren als Arzneimittel zur Verbesserung kognitiver Prozesse |
| WO2004099211A1 (de) | 2003-05-09 | 2004-11-18 | Bayer Healthcare Ag | 6-cyclylmethyl- und 6-alkylmethyl-substituierte pyrazolopyrimidine |
| JP2008524331A (ja) | 2004-12-21 | 2008-07-10 | 武田薬品工業株式会社 | ジペプチジルペプチダーゼ阻害剤 |
| US20080194592A1 (en) | 2005-08-23 | 2008-08-14 | Intra-Cellular Therapies, Inc. | Organic Compounds |
| US8012936B2 (en) | 2006-03-29 | 2011-09-06 | New York University | Tau fragments for immunotherapy |
| WO2008070095A1 (en) | 2006-12-05 | 2008-06-12 | Intra-Cellular Therapies, Inc. | Novel uses |
| RS52166B (sr) | 2007-05-11 | 2012-08-31 | Pfizer Inc. | Amino-heterociklična jedinjenja |
| EP2227472A1 (en) | 2007-11-30 | 2010-09-15 | Elbion Gmbh | Aryl and heteroaryl fused imidazo (1,5-a) pyrazines as inhibitors of phosphodiesterase 10 |
| UA105362C2 (en) | 2008-04-02 | 2014-05-12 | Бьорингер Ингельхайм Интернациональ Гмбх | 1-heterocyclyl-1, 5-dihydro-pyrazolo [3, 4-d] pyrimidin-4-one derivatives and their use as pde9a modulators |
| KR20110063447A (ko) | 2008-09-08 | 2011-06-10 | 베링거 인겔하임 인터내셔날 게엠베하 | 피라졸로피리미딘 및 cns 장애의 치료를 위한 이들의 용도 |
| EP2367430B1 (en) | 2008-12-06 | 2014-08-13 | Intra-Cellular Therapies, Inc. | Organic compounds |
| TWI404721B (zh) | 2009-01-26 | 2013-08-11 | Pfizer | 胺基-雜環化合物 |
| CN106390107B (zh) | 2009-06-10 | 2019-12-31 | 纽约大学 | 病理tau蛋白的免疫靶向 |
| WO2011153136A1 (en) | 2010-05-31 | 2011-12-08 | Intra-Cellular Therapies, Inc. | Organic compounds |
| PL3042917T3 (pl) | 2010-08-12 | 2018-07-31 | Eli Lilly And Company | Przeciwciała przeciwko peptydowi beta amyloidu N3pGlu i ich zastosowanie |
| EP2619208B1 (en) | 2010-09-20 | 2016-11-09 | Ironwood Pharmaceuticals, Inc. | Imidazotriazinone compounds |
| EP2619204A4 (en) | 2010-09-21 | 2014-08-27 | Merck Sharp & Dohme | TRIAZOLOPYRAZINONE AS P2X7 RECEPTOR ANTAGONISTS |
| JO3089B1 (ar) | 2010-11-19 | 2017-03-15 | H Lundbeck As | مشتقات ايميدازول كمثبطات لانزيمات pde10a |
| TWI636784B (zh) * | 2011-04-05 | 2018-10-01 | 大塚製藥股份有限公司 | 含7-〔4-(4-苯並〔b〕噻吩-4-基-哌-1-基)丁氧基〕-1H-喹啉-2-酮之醫藥組成物與套組,以及彼之用途 |
| US9499610B2 (en) | 2011-04-08 | 2016-11-22 | H. Lundbeck A/S | Antibodies specific to pyroglutamated Aβ |
| JP6051210B2 (ja) | 2011-06-10 | 2016-12-27 | イントラ−セルラー・セラピーズ・インコーポレイテッドIntra−Cellular Therapies, Inc. | 有機化合物 |
| BR112015007731B1 (pt) | 2011-10-10 | 2022-05-31 | H. Lundbeck A/S | Composto, composição farmacêutica que o compreende e uso do mesmo |
| BR112014018199A8 (pt) | 2012-01-26 | 2021-03-02 | H Lundbeck As | inibidores da pde9 com cadeia principal imidazo triazinona, composição farmacêutica que os compreende, uso dos referidos inibidores e processo para sua preparação |
| CA2877146C (en) | 2012-06-18 | 2020-10-20 | Dart Neuroscience (Cayman) Ltd | Substituted thiophene- and furan-fused azolopyrimidine-5-(6h)-one compounds |
| KR102240326B1 (ko) | 2013-03-15 | 2021-04-13 | 인트라-셀룰라 써래피스, 인코퍼레이티드. | 유기 화합물 |
| JP5783297B2 (ja) | 2013-08-06 | 2015-09-24 | 株式会社デンソー | 力学量センサ |
| KR20160115936A (ko) | 2014-02-19 | 2016-10-06 | 하. 룬드벡 아크티에셀스카브 | 알츠하이머병의 치료를 위한 bace1 저해제로서의 2-아미노-3,5,5-트리플루오로-3,4,5,6-테트라하이드로피리딘 |
| JP5797815B1 (ja) | 2014-06-27 | 2015-10-21 | 細田建設株式会社 | 後施工アンカー及びその施工方法 |
| WO2016042775A1 (en) | 2014-09-18 | 2016-03-24 | Sunovion Pharmaceuticals Inc. | Tricyclic derivative |
| US20160083391A1 (en) | 2014-09-18 | 2016-03-24 | Sunovion Pharmaceuticals Inc. | Tricyclic piperazine derivative |
| US20160083400A1 (en) | 2014-09-18 | 2016-03-24 | Sunovion Pharmaceuticals Inc. | Tricyclic guanidine derivative |
| TW201629064A (zh) | 2014-10-10 | 2016-08-16 | H 朗德貝克公司 | 作爲pde1抑制劑之三唑並吡酮 |
| MA40941A (fr) | 2014-11-10 | 2017-09-19 | H Lundbeck As | 2-amino-5,5-difluoro-6-(fluorométhyl)-6-phényl-3,4,5,6-tétrahydropyridines comme inhibiteurs de bace1 |
| JO3458B1 (ar) | 2014-11-10 | 2020-07-05 | H Lundbeck As | 2- أمينو-6- (دايفلوروميثيل) – 5، 5- ديفلورو-6-فينيل-3،4، 5، 6-تيتراهيدروبيريدين كمثبطات bace1 |
| CR20170187A (es) | 2014-11-10 | 2018-02-01 | H Lundbeck As | 2-Amino-3,5-difluoro-6-metil-6-fenil-3,4,5,6-tetrahidropiridinas en calidad de inhibidores de BACE1 para el tratamiento de la enfermedad de Alzheimer |
| TW201639851A (zh) | 2015-03-16 | 2016-11-16 | 大日本住友製藥股份有限公司 | 雙環咪唑衍生物 |
| TW201643167A (zh) * | 2015-04-22 | 2016-12-16 | H 朗德貝克公司 | 作爲pde1抑制劑之咪唑並三酮 |
| JO3627B1 (ar) | 2015-04-30 | 2020-08-27 | H Lundbeck As | إيميدازو بيرازينونات على هيئة مثبطات pde1 |
| JO3711B1 (ar) | 2015-07-13 | 2021-01-31 | H Lundbeck As | أجسام مضادة محددة لبروتين تاو وطرق استعمالها |
| EP3334720A1 (en) | 2015-08-12 | 2018-06-20 | H. Lundbeck A/S | 2-amino-3-fluoro-3-(fluoromethyl)-6-methyl-6-phenyl-3,4,5,6-tetrahydropyridins as bace1 inhibitors |
| CN105254635A (zh) * | 2015-10-30 | 2016-01-20 | 中国药科大学 | 一类咪唑并吡嗪类化合物及其药物组合物和用途 |
| TWI609870B (zh) | 2016-02-12 | 2018-01-01 | 美國禮來大藥廠 | Pde1抑制劑 |
| TWI729109B (zh) | 2016-04-12 | 2021-06-01 | 丹麥商H 朗德貝克公司 | 作爲PDE1抑制劑的1,5-二氫-4H-吡唑并[3,4-d]嘧啶-4-酮和1,5-二氫-4H-吡唑并[4,3-c]吡啶-4-酮 |
| JP2018076285A (ja) | 2016-09-14 | 2018-05-17 | 大日本住友製薬株式会社 | 二環性イミダゾロ誘導体を含む医薬 |
| WO2018073251A1 (en) | 2016-10-18 | 2018-04-26 | H. Lundbeck A/S | Imidazopyrazinones, pyrazolopyrimidinones and pyrazolopyridinones as pde1 inhibitors |
| AU2017350473A1 (en) | 2016-10-28 | 2019-04-18 | H. Lundbeck A/S | Combination treatments comprising imidazopyrazinones for the treatment of psychiatric and/or cognitive disorders |
| WO2018078042A1 (en) | 2016-10-28 | 2018-05-03 | H. Lundbeck A/S | Combination treatments comprising administration of imidazopyrazinones |
-
2017
- 2017-10-26 AU AU2017350473A patent/AU2017350473A1/en not_active Abandoned
- 2017-10-26 PL PL17791080T patent/PL3532064T3/pl unknown
- 2017-10-26 SI SI201730388T patent/SI3532064T1/sl unknown
- 2017-10-26 EP EP17791080.9A patent/EP3532064B1/en active Active
- 2017-10-26 DK DK17791080.9T patent/DK3532064T3/da active
- 2017-10-26 ME MEP-2020-170A patent/ME03821B/me unknown
- 2017-10-26 MX MX2019004763A patent/MX2019004763A/es unknown
- 2017-10-26 RS RS20200975A patent/RS60716B1/sr unknown
- 2017-10-26 MA MA46611A patent/MA46611B1/fr unknown
- 2017-10-26 JP JP2019522523A patent/JP2019536762A/ja active Pending
- 2017-10-26 SG SG11201903770UA patent/SG11201903770UA/en unknown
- 2017-10-26 CA CA3041607A patent/CA3041607A1/en not_active Abandoned
- 2017-10-26 ES ES17791080T patent/ES2816005T3/es active Active
- 2017-10-26 HU HUE17791080A patent/HUE050403T2/hu unknown
- 2017-10-26 HR HRP20201326TT patent/HRP20201326T1/hr unknown
- 2017-10-26 PT PT177910809T patent/PT3532064T/pt unknown
- 2017-10-26 KR KR1020197011509A patent/KR20190075068A/ko not_active Ceased
- 2017-10-26 RU RU2019109039A patent/RU2019109039A/ru not_active Application Discontinuation
- 2017-10-26 WO PCT/EP2017/077497 patent/WO2018078038A1/en not_active Ceased
- 2017-10-26 US US16/345,136 patent/US10905688B2/en not_active Expired - Fee Related
- 2017-10-26 BR BR112018013247A patent/BR112018013247A2/pt not_active Application Discontinuation
- 2017-10-26 CN CN201780063182.9A patent/CN109803658A/zh active Pending
- 2017-10-26 SM SM20200440T patent/SMT202000440T1/it unknown
- 2017-10-26 LT LTEP17791080.9T patent/LT3532064T/lt unknown
- 2017-10-27 AR ARP170102997A patent/AR109991A1/es unknown
-
2019
- 2019-03-26 IL IL265635A patent/IL265635A/en unknown
- 2019-03-27 CO CONC2019/0002909A patent/CO2019002909A2/es unknown
- 2019-04-03 ZA ZA2019/02091A patent/ZA201902091B/en unknown
- 2019-04-22 CL CL2019001091A patent/CL2019001091A1/es unknown
-
2020
- 2020-08-24 CY CY20201100788T patent/CY1123396T1/el unknown
Also Published As
| Publication number | Publication date |
|---|---|
| HRP20201326T1 (hr) | 2020-11-27 |
| SG11201903770UA (en) | 2019-05-30 |
| MA46611A (fr) | 2019-09-04 |
| US10905688B2 (en) | 2021-02-02 |
| RU2019109039A (ru) | 2020-11-30 |
| ZA201902091B (en) | 2020-10-28 |
| WO2018078038A1 (en) | 2018-05-03 |
| CN109803658A (zh) | 2019-05-24 |
| EP3532064B1 (en) | 2020-07-29 |
| BR112018013247A2 (pt) | 2018-12-04 |
| IL265635A (en) | 2019-05-30 |
| AR109991A1 (es) | 2019-02-13 |
| JP2019536762A (ja) | 2019-12-19 |
| LT3532064T (lt) | 2020-09-25 |
| KR20190075068A (ko) | 2019-06-28 |
| RS60716B1 (sr) | 2020-09-30 |
| CA3041607A1 (en) | 2018-05-03 |
| SMT202000440T1 (it) | 2020-09-10 |
| CO2019002909A2 (es) | 2019-06-19 |
| PT3532064T (pt) | 2020-09-03 |
| CL2019001091A1 (es) | 2019-07-05 |
| PL3532064T3 (pl) | 2020-11-16 |
| DK3532064T3 (da) | 2020-08-24 |
| US20190282571A1 (en) | 2019-09-19 |
| RU2019109039A3 (es) | 2021-01-29 |
| AU2017350473A1 (en) | 2019-04-18 |
| MA46611B1 (fr) | 2020-08-31 |
| SI3532064T1 (sl) | 2020-10-30 |
| HUE050403T2 (hu) | 2020-12-28 |
| CY1123396T1 (el) | 2021-12-31 |
| MX2019004763A (es) | 2019-07-01 |
| EP3532064A1 (en) | 2019-09-04 |
| ME03821B (me) | 2021-04-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2816005T3 (es) | Tratamientos de combinación que comprenden imidazopirazinonas para el tratamiento de trastornos psiquiátricos y/o cognitivos | |
| US11472810B2 (en) | Imidazopyrazinones as PDE1 inhibitors | |
| US10912773B2 (en) | Combinations comprising substituted imidazo[1,5-a]pyrazinones as PDE1 inhibitors | |
| ES2814289T3 (es) | 1,5-Dihidro-4H-pirazolo[3,4-d]pirimidin-4-onas y 1,5-dihidro-4H-pirazolo[4,3-c]piridin-4-onas como inhibidores de la PDE1 | |
| HK40005946B (en) | Combination treatments comprising imidazopyrazinones for the treatment of psychiatric and/or cognitive disorders | |
| HK40005946A (en) | Combination treatments comprising imidazopyrazinones for the treatment of psychiatric and/or cognitive disorders | |
| OA18771A (en) | Imidazopyrazinones as PDE1 Inhibitors |