ES2806206T3 - Derivado de 2-hidrógeno pirazol sustituido que sirve como fármaco anticanceroso - Google Patents
Derivado de 2-hidrógeno pirazol sustituido que sirve como fármaco anticanceroso Download PDFInfo
- Publication number
- ES2806206T3 ES2806206T3 ES16761108T ES16761108T ES2806206T3 ES 2806206 T3 ES2806206 T3 ES 2806206T3 ES 16761108 T ES16761108 T ES 16761108T ES 16761108 T ES16761108 T ES 16761108T ES 2806206 T3 ES2806206 T3 ES 2806206T3
- Authority
- ES
- Spain
- Prior art keywords
- mmol
- group
- equivalents
- reaction
- give
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 CC1=C*2=C(*)*(*)*=C2C(**)=C1 Chemical compound CC1=C*2=C(*)*(*)*=C2C(**)=C1 0.000 description 3
- DPMWUYXSSIHTBR-UHFFFAOYSA-N CC(C)c1c(CN(CC2)c3nc(Nc(cc4C)ncc4N(CC4)CCN4C(OC(C)(C)C)=O)ncc3F)c2n[n]1C Chemical compound CC(C)c1c(CN(CC2)c3nc(Nc(cc4C)ncc4N(CC4)CCN4C(OC(C)(C)C)=O)ncc3F)c2n[n]1C DPMWUYXSSIHTBR-UHFFFAOYSA-N 0.000 description 1
- MAVWLWPCBLIDCH-UHFFFAOYSA-N CC(C)c1c(CN(CC2)c3nc(Nc4ncc(C5CCNCC5)cc4)ncc3F)c2n[n]1C Chemical compound CC(C)c1c(CN(CC2)c3nc(Nc4ncc(C5CCNCC5)cc4)ncc3F)c2n[n]1C MAVWLWPCBLIDCH-UHFFFAOYSA-N 0.000 description 1
- NPZWUIOVWKTBRT-UHFFFAOYSA-N CC(C)c1c(cc(C)cc2F)c2n[n]1C Chemical compound CC(C)c1c(cc(C)cc2F)c2n[n]1C NPZWUIOVWKTBRT-UHFFFAOYSA-N 0.000 description 1
- OGBIXFWCPVKSKC-UHFFFAOYSA-N CC(C)c1c(cc(cc2)-c3cc(Nc(nc4)ccc4N(CC4)CCN4C(OC(C)(C)C)=O)ncc3F)c2n[n]1C Chemical compound CC(C)c1c(cc(cc2)-c3cc(Nc(nc4)ccc4N(CC4)CCN4C(OC(C)(C)C)=O)ncc3F)c2n[n]1C OGBIXFWCPVKSKC-UHFFFAOYSA-N 0.000 description 1
- HAYDXAQWAZWJLP-UHFFFAOYSA-N CC(C)c1c(cc(cc2)-c3nc(Nc(cc4)nnc4N4CCNCCC4)ncc3F)c2n[n]1C Chemical compound CC(C)c1c(cc(cc2)-c3nc(Nc(cc4)nnc4N4CCNCCC4)ncc3F)c2n[n]1C HAYDXAQWAZWJLP-UHFFFAOYSA-N 0.000 description 1
- CPVWPXJKCUNTDK-UHFFFAOYSA-N CC(C)c1c(cc(cc2)-c3nc(Nc(nc4)cc(C)c4N(CC4)CCN4C(OC(C)(C)C)=O)ncc3F)c2n[n]1C Chemical compound CC(C)c1c(cc(cc2)-c3nc(Nc(nc4)cc(C)c4N(CC4)CCN4C(OC(C)(C)C)=O)ncc3F)c2n[n]1C CPVWPXJKCUNTDK-UHFFFAOYSA-N 0.000 description 1
- CRRKUTPFXCLRFN-UHFFFAOYSA-N CC(C)c1c(cc(cc2)-c3nc(Nc(nc4)ccc4N4CC(CO)NCC4)ncc3F)c2n[n]1C Chemical compound CC(C)c1c(cc(cc2)-c3nc(Nc(nc4)ccc4N4CC(CO)NCC4)ncc3F)c2n[n]1C CRRKUTPFXCLRFN-UHFFFAOYSA-N 0.000 description 1
- JKHFYUXEZRRJDO-DUMVDREWSA-N CC(C)c1c(cc(cc2)C(/C(/F)=C\N)=N)c2n[n]1C Chemical compound CC(C)c1c(cc(cc2)C(/C(/F)=C\N)=N)c2n[n]1C JKHFYUXEZRRJDO-DUMVDREWSA-N 0.000 description 1
- QZQICIPMRPPSRL-UHFFFAOYSA-N CC(C)c1c(cc(cc2F)-c3nc(Nc(nc4)ccc4N4CCNCC4)ncc3F)c2n[n]1C Chemical compound CC(C)c1c(cc(cc2F)-c3nc(Nc(nc4)ccc4N4CCNCC4)ncc3F)c2n[n]1C QZQICIPMRPPSRL-UHFFFAOYSA-N 0.000 description 1
- VZYIGGHUAZXVGH-UHFFFAOYSA-N CC(c1c(cc(cc2F)-c3nc(Cl)ncc3F)c2n[n]1C)=C Chemical compound CC(c1c(cc(cc2F)-c3nc(Cl)ncc3F)c2n[n]1C)=C VZYIGGHUAZXVGH-UHFFFAOYSA-N 0.000 description 1
- URTHLXXARWPJCU-UHFFFAOYSA-N C[n]1nc2c(F)cc(Br)cc2c1 Chemical compound C[n]1nc2c(F)cc(Br)cc2c1 URTHLXXARWPJCU-UHFFFAOYSA-N 0.000 description 1
- OZAONQSRAXEGMW-UHFFFAOYSA-N Cc(cc(nc1)[N+]([O-])=O)c1Br Chemical compound Cc(cc(nc1)[N+]([O-])=O)c1Br OZAONQSRAXEGMW-UHFFFAOYSA-N 0.000 description 1
- HJVDYBUDQLXLKH-UHFFFAOYSA-N Ic(cn1)cnc1I Chemical compound Ic(cn1)cnc1I HJVDYBUDQLXLKH-UHFFFAOYSA-N 0.000 description 1
- MMKAQOLNUBDHRF-UHFFFAOYSA-N Ic(cn1)ncc1I Chemical compound Ic(cn1)ncc1I MMKAQOLNUBDHRF-UHFFFAOYSA-N 0.000 description 1
- GMIRDNVYYLFXHK-UHFFFAOYSA-N Nc(nc1)ccc1N1CCN(CCO)CC1 Chemical compound Nc(nc1)ccc1N1CCN(CCO)CC1 GMIRDNVYYLFXHK-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/415—1,2-Diazoles
- A61K31/416—1,2-Diazoles condensed with carbocyclic ring systems, e.g. indazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/437—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having nitrogen as a ring hetero atom, e.g. indolizine, beta-carboline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4985—Pyrazines or piperazines ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5383—1,4-Oxazines, e.g. morpholine ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/08—Bridged systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/10—Spiro-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D498/04—Ortho-condensed systems
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Epidemiology (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Oncology (AREA)
- Hematology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo **(Ver fórmula)** en donde**(Ver fórmula)**se selecciona entre el grupo que consiste en**(Ver fórmula)**y **(Ver fórmula)** R1 se selecciona entre el grupo que consiste en isopropilo, 2-propenilo y alilo; R2 se selecciona entre el grupo que consiste en metilo y fenilo; R3 se selecciona entre el grupo que consiste en H y halógeno; R4, R5 y R6 se selecciona cada uno independientemente entre el grupo que consiste en H, halógeno, OH, NH2, CN, NO2 y =O o se selecciona entre el grupo que consiste en alquilo C1-8, alquilamino C1-8, N,N-di(alquil)amino C1-8, alcoxil C1-8-alquilo C1-8-, hidroxialquilo C1-8, alquenilo C2-8, alquinilo C2-8, cicloalquilo C3-7 y un grupo heterocicloalquilo de 3 a 7 miembros, cada uno de los cuales está opcionalmente sustituido con 1, 2 o 3 Rs; opcionalmente, dos cualesquiera de R4, R5 y R6 juntos pueden formar un anillo de 3 a 7 miembros; R7 se selecciona entre el grupo que consiste en H, halógeno, -OR8, -SR8, -N(R8)(R9) y cicloalquilo C3-7; cada uno de X1, X2, X3 y X4 se selecciona independientemente entre un grupo que consiste en N y C(R10); X7 se selecciona entre el grupo que consiste en carbonilo y C(R11)(R12); W es un enlace sencillo; T se selecciona entre el grupo que consiste en N y C(R10); Q se selecciona entre el grupo que consiste en N y C(R10); cada uno de m y n se selecciona independientemente entre el grupo que consiste en 0, 1 y 2; cada uno de R8 y R9 se selecciona independientemente entre el grupo que consiste en H, alquilo C1-8 y cicloalquilo C3-7; R se selecciona entre el grupo que consiste en F, Cl, Br, I, NH2, CN, OH, CF3, CHF2, CH2F, NHCH3 y N(CH3)2; opcionalmente, R8 y R9 están unidos al mismo átomo y forman un anillo de 3 a 7 miembros con 1-4 heteroátomos; el término "hetero" o "heteroátomo" representa O, S, S(=O), S(=O)2 o N; R10 se selecciona entre el grupo que consiste en H, halógeno, OH, NH2, CN, alquilo C1-6, alcoxilo C1-6, cicloalquilo C3- 5, CN, -OR8, -SR8, -N(R8)(R9), -C(=O)R8, -C(=O)OR8, -C(=O)N(R8)(R9), -S(=O)R8, -S(=O)2R8, -S(=O)N(R8)(R9) y - S(=O)2N(R8)(R9); cada uno de R11 y R12 se selecciona independientemente entre el grupo que consiste en H, OH, halógeno, alquilo C1- 8 y cicloalquilo C3-7; opcionalmente, R4 y R10 están unidos al mismo átomo y forman un anillo de 3 a 7 miembros y, opcionalmente, la unidad estructural**(Ver fórmula)**se puede sustituir por**(Ver fórmula)**
Description
DESCRIPCIÓN
Derivado de 2-hidrógeno pirazol sustituido que sirve como fármaco anticanceroso
Campo técnico
La presente invención se refiere a un derivado de 2H-pirazol sustituido que sirve como inhibidor de CDK4/6 selectivo. De manera específica, la presente invención se refiere a un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo que sirve como inhibidor de CDK4/6 selectivo.

Antecedentes de la invención
La regulación del ciclo celular está influenciada principalmente por una familia de serina/treonina quinasas, dichas serina/treonina quinasas también se conocen como quinasas dependientes de ciclina (cyclin-dependent kinases, CDK). Promueven la progresión del ciclo celular, la transcripción de la información genética y la división y proliferación normales de las células mediante la unión a las ciclinas correspondientes que regulan las subunidades. CDK4/6 es el factor regulador crítico del ciclo celular y es capaz de desencadenar la transición del ciclo celular desde la fase de crecimiento (fase G1) a la fase de replicación del ADN (fase S1). Durante la proliferación celular, El complejo formado por la ciclina D y CDK4/6 puede fosforilar la proteína del retinoblastoma (Rb). Tras la fosforilación de la proteína supresora de tumores Rb, puede liberarse el factor de transcripción E2F que se ha unido estrechamente a la Rb no fosforilada. La activación de E2F transcribe más, que promueve el ciclo celular para pasar el punto de restricción (punto R) y pasar de la fase G1 a la fase S, conduciendo al ciclo de proliferación celular. De esta manera, inhibir que CDK4/6 forme el complejo Ciclina D-CDK4/6 puede evitar la progresión del ciclo celular de la fase G1 a la fase S y, por lo tanto, realizar el fin de inhibir la proliferación tumoral. En el cáncer de mama (BC, del inglés "breast cancer") positivo al receptor de estrógeno (ER+), la hiperactividad de CDK4/6 es bastante frecuente, mientras que CDK4/6 es una diana crítica posterior de la señalización del receptor de estrógenos (estrogen receptor, ER). Los datos preclínicos sugieren que la inhibición dual de CDK4/6 y la señalización de ER produce un efecto sinérgico y es capaz de inhibir el crecimiento de células de cáncer de mama (breast cancer, BC) positivas al receptor de estrógeno (ER+) en la fase G1.
CDK4/6 como diana ha sido un área de desarrollo con una feroz competencia. En 2010, Pietzsch resumió el desarrollo en este campo (Mini-Rev. Med. Chem. 2010, 10, 527-539). En 2014, Malorni también resumió los últimos logros de los inhibidores de CDK4/6 en la investigación preclínica y clínica sobre el cáncer de mama (Curr. Opin. Oncol. 2014, 26, 568-575). La extensa investigación sobre CDK4/6 promovió el desarrollo de una serie de inhibidores de CDK con diferente selectividad y también condujo al descubrimiento de algunos inhibidores de CDK4/6 con efectividad y alta selectividad, Palbociclib (PD0332991) es uno de estos inhibidores de CDK4/6 con efectividad y alta selectividad, ha entrado en las pruebas clínicas en humanos y se ha aplicado al tratamiento de mujeres con cáncer de mama avanzado o metastásico positivo al receptor de estrógenos (ER+) y negativo al receptor 2 del factor de crecimiento epidérmico humano (HER2-). Sobre la base de los datos a medio plazo de PALOMA-1, Pfizer presentó una Solicitud de Nuevo Medicamento (NDA) de palbociclib a la United States Food and Drug Administration (FDA) en agosto de 2014. En febrero de 2015, la FDA aprobó la solicitud de lanzamiento de palbociclib. Abemaciclib (LY2835219) y FEE-011, otros dos inhibidores de CDK4/6, han comenzado a reclutar pacientes para sus pruebas clínicas de fase 3. Estos compuestos heterocíclicos de molécula pequeña son aplicables para el tratamiento de una diversidad de otros tipos de cáncer además del cáncer de mama. Estas patentes incluyen el documento WO2012018540, el documento WO2012129344, el documento WO2011101409, el documento WO2011130232, el documento W02010075074, el documento WO2009126584, el documento W02008032157, el documento W02008/003766 y el documento W02003062236.

Los presentes inventores esperan desarrollar una nueva generación de inhibidores de CDK4/6 con alta selectividad, seguridad más alta y efectividad más alta para satisfacer mejor las demandas del mercado y lograr una mejor efectividad terapéutica del tumor. La presente invención proporciona un inhibidor selectivo de CDK4/6 con una estructura novedosa y se encuentra que los compuestos con la estructura muestran un excelente efecto anticancerígeno.
Sumario
El objetivo de la presente invención es proporcionar un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo,


Ri se selecciona entre un grupo que consiste  en isopropilo, 2-propenilo y alilo;
en isopropilo, 2-propenilo y alilo;
R2 se selecciona entre un grupo que consiste en metilo y fenilo;
R3 se selecciona entre un grupo que consiste en H y halógeno;
cada uno de R4, R5 y R6 se selecciona independientemente entre un grupo que consiste en H, halógeno, OH, NH2, CN, NO2 y =O o se selecciona entre un grupo que consiste en alquilo C1-8, alquilamino C1-8, W,A/-di(alquil)amino C1-8, alcoxil C1_8-alquilo C1-8-, hidroxialquilo C1-8, alquenilo C2-8, alquinilo C2-8, cicloalquilo C3-7 y un grupo heterocicloalquilo de 3 a 7 miembros, cada uno de los cuales está opcionalmente sustituido con 1, 2 o 3 Rs;
opcionalmente, dos cualesquiera de R4, R5 y R6 juntos pueden formar un anillo de 3 a 7 miembros;
R7 se selecciona entre un grupo que consiste en H, halógeno, -OR8, -SR8, -N(R8)(Rg) y cicloalquilo C3-7;
cada uno de X1, X2, X3 y X4 se selecciona independientemente entre un grupo que consiste en N y C(R10);
X7 se selecciona entre un grupo que consiste en carbonilo y C(R ii)(R i2);
W es un enlace sencillo;
T se selecciona entre un grupo que consiste en N y C(Rio);
Q se selecciona entre un grupo que consiste en N y C(Rio);
cada uno de m y n se selecciona independientemente entre un grupo que consiste en 0, 1 y 2;
cada uno de R8 y Rg se selecciona independientemente entre un grupo que consiste en H, alquilo C1-8 y cicloalquilo C3-7;
R se selecciona entre un grupo que consiste en F, Cl, Br, I, NH2, CN, OH, CF3, CHF2, CH2F, NHCH3 y N(CH3)2; opcionalmente, R8 y Rg están unidos al mismo átomo y forman un anillo de 3 a 7 miembros con 1-4 heteroátomos; el término "hetero" o "heteroátomo" representa O, S, S(=O), S(=O)2 o N;
R10 se selecciona entre un grupo que consiste en H, halógeno, O h , NH2, CN, alquilo C1-6, alcoxilo C1-6, cicloalquilo C3-5, CN, -OR8, -SR8, -N(R8)(Rg), -C(=O)R8, -C(=O)OR8, -C(=O)N(Rs)(Rg), -S(=O)R8, -S(=O)2R8, -S(=O)N(R8)(Rg) y -S(=O)2N(R8)(Rg);
cada uno de R11 y R12 se selecciona independientemente entre un grupo que consiste en H, OH, halógeno, alquilo C1-8 y cicloalquilo C3-7;
opcionalmente, R4 y R10 están unidos al mismo carbono y forman un anillo de 3 a 7 miembros y, opcionalmente,


la unidad estructural se puede sustituir por la unidad estructural •
En algunas realizaciones de la presente invención, se proporciona un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo



en donde se selecciona entre el grupo que consiste en y

Ri se selecciona entre el grupo que consiste en isopropilo, 2-propenilo y alilo;
R2 se selecciona entre el grupo que consiste en metilo y fenilo;
R3 se selecciona entre un grupo que consiste en H y halógeno;
cada uno de R4, R5 y R6 se selecciona independientemente entre el grupo que consiste en H, halógeno, OH, NH2, CN, NO2 y =O o se selecciona entre el grupo que consiste en alquilo C1-8, alquilamino C1-8, W,W-di(alquil)amino C1-8, alcoxil C1-8-alquilo C1-8-, hidroxialquilo C1-8, alquenilo C2-8, alquinilo C2-8, cicloalquilo C3-7 y un grupo heterocicloalquilo de 3 a 7 miembros, cada uno de los cuales está opcionalmente sustituido con 1,2 o 3 Rs;
opcionalmente, dos cualesquiera de R4, R5 y R6 juntos pueden formar un anillo de 3 a 7 miembros;
R7 se selecciona entre el grupo que consiste en H, halógeno, -OR8, -SR8, -N(R8)(Rg) y cicloalquilo C3-7;
cada uno de Xi, X2, X3 y X4 se selecciona independientemente entre un grupo que consiste en N y C(Riü);
X7 se selecciona entre el grupo que consiste en carbonilo y C(R11)(R12);
W es un enlace sencillo;
T se selecciona entre el grupo que consiste en N y C(R10),;
Q se selecciona entre el grupo que consiste en N y C(Ri0);
cada uno de m y n se selecciona independientemente entre el grupo que consiste en 0, 1 y 2 ;
cada uno de R8 y Rg se selecciona independientemente entre el grupo que consiste en H, alquilo C1-8 y cicloalquilo C3-7;
R se selecciona entre el grupo que consiste en F, Cl, Br, I, NH2, CN, OH, CF3, CHF2, CH2F, NHCH3 y N (CH)2; opcionalmente, R8 y Rg están unidos al mismo átomo y forman un anillo de 3 a 7 miembros con 1-4 heteroátomos; el término "hetera" o "heteroátomo" representa O, S, S(=O), S(=O)2 o N;
R10 se selecciona entre el grupo que consiste en H, halógeno, OH, NH2, CN, alquilo C1-6, alcoxilo C1-6, cicloalquilo C3-5, CN, -OR8, -SR8, -N(R8)(Rg), -C(=O)R8, -C(=O)OR8,
-C(=O)N(R8)(Rg), -S(=O)R8, -S(=O)2R8, -S(=O)N(R8)(Rg) y -S(=O)2N(R8)(Rg);
cada uno de R11 y R12 se selecciona independientemente entre el grupo que consiste en H, OH, halógeno, alquilo C1-8 y cicloalquilo C3-7;
opcionalmente, R4 y R10 están unidos al mismo átomo y forman un anillo de 3 a 7 miembros.
En algunas realizaciones de la presente invención, el R3 mencionado anteriormente es F.
En algunas realizaciones de la presente invención, cada uno de los R4, R5 y R6 mencionados anteriormente se

selecciona independientemente entre un grupo que consiste en H, halógeno, OH, NH2,

En algunas realizaciones de la presente invención, el R7 mencionado anteriormente se selecciona entre un grupo que consiste en H, F y Cl.

En algunas realizaciones de la presente invención, en donde la unidad estructural se selecciona entre un
grupo que consiste en 
En algunas realizaciones de la presente invención, el R10 mencionado anteriormente se selecciona entre un grupo

que consiste en H, OH, NH2, F, Cl, CN, y Me.
En algunas realizaciones de la presente invención, el X4 mencionado anteriormente se selecciona entre un grupo que consiste en N y CH.
En algunas realizaciones de la presente invención, la unidad estructural mencionada anteriormente




En algunas realizaciones de la presente invención, la unidad estructural mencionada anteriormente
se selecciona entre un grupo que consiste e

El compuesto de la presente invención se selecciona entre un grupo que consiste en


Definiciones y explicaciones
En el presente documento los siguientes términos y frases pretenden tener los significados siguientes a menos que se indique otra cosa. Si un término o frase específico no está particularmente definido, no se debe considerar que esté sin definir o poco claro, sino más bien debe entenderse con su significado ordinario. Cuando aparece un nombre comercial en el presente documento, este pretende referirse al producto correspondiente o al componente activo del mismo.
C1-8 representa el número de átomos de carbono contenido en un grupo hidrocarburo. Por ejemplo, C1 representa que solo contiene un átomo de carbono, C2 representa que contiene dos átomos de carbono, etc.
En el compuesto de fórmula (I), el término "alquilo C W representa un grupo hidrocarburo, lineal o ramificado, que tiene 1-8 átomos de carbono, incluyendo, por ejemplo, metilo, etilo, n-propilo, isopropilo, n-butilo, sec-butilo, isobutilo, t-butilo, n-pentilo, 2,2-dimetilpropilo, n-hexilo, n-heptilo, n-octilo y similares.
El término "alquenilo C2-8" representa un hidrocarburo, lineal o ramificado, de 2-8 átomos de carbono y un doble enlace, incluyendo etenilo, 1-propenilo, 2-propenilo, 1 -butenilo, 2-butenilo y similares.
El término "alquinilo C2-8" representa un grupo hidrocarburo, lineal o ramificado, que tiene 2-8 átomos de carbono y un triple enlace, incluyendo etinilo, propinilo, 1 -butinilo, 2-butinilo y similares.
El término "cicloalquilo C3-7" representa un grupo hidrocarburo monocíclico o dicíclico que tiene 3-7 átomos de carbono, incluyendo ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, cicloheptilo y similares.
En el presente documento la expresión "farmacéuticamente aceptable" significa, para compuestos, materiales, composiciones y/o formas de dosificación, que es adecuado para usar en contacto con tejidos humanos y animales dentro de un intervalo de juicio médico confiable, sin excesiva toxicidad, irritación, anafilaxia, otros problemas o complicaciones y en proporción con una relación beneficio/riesgo razonable.
La expresión "sal farmacéuticamente aceptable" se refiere a las sales de los compuestos de la presente invención que se preparan usando los compuestos de la presente invención que tienen sustituyentes específicos y ácidos o álcalis relativamente no tóxicos. Cuando un compuesto de la presente invención contiene grupos funcionales relativamente ácidos, su sal de adición de álcali se puede obtener poniendo en contacto una cantidad adecuada de álcali con la forma neutra del compuesto en una solución pura o un disolvente inerte adecuado. Las sales de adición de álcali farmacéuticamente aceptables incluyen sales de sodio, potasio, calcio, amonio, amina orgánica, magnesio o similares. Cuando un compuesto de la presente invención contiene grupos funcionales relativamente alcalinos, su sal de adición de ácido se puede obtener poniendo en contacto una cantidad adecuada de ácido con la forma neutra del compuesto en una solución pura o un disolvente inerte. Los ejemplos de sales de adición de ácido farmacéuticamente aceptables incluyen las sales de ácidos inorgánicos y las sales de ácidos orgánicos. Dichos ácidos inorgánicos incluyen, por ejemplo, ácido clorhídrico, ácido bromhídrico, ácido nítrico, ácido carbónico, ion bicarbonato, ácido fosfórico, ion monohidrogenofosfato, ion dihidrogenofosfato, ácido sulfúrico, ion hidrosulfato, ácido yohídrico, ácido fosforoso y similares. Dichos ácidos orgánicos incluyen, por ejemplo, ácido acético, ácido propiónico, ácido isobutírico, ácido maleico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido láctico, ácido mandélico, ácido Itálico, ácido benceno sulfónico, ácido p-tolueno sulfónico, ácido cítrico, ácido tartárico, ácido metano sulfónico y similares. Las sales de adición de ácido farmacéuticamente aceptables incluyen también las sales de aminoácidos (por ejemplo, arginina, etc.) y las sales de ácidos orgánicos tales como ácido glucurónico (véase Berge et al., "Pharmaceutical Salts", Journal of Pharmaceutical Science 66: 1 19 (1977)). Algunos compuestos determinados de la presente invención tienen grupos funcionales alcalinos o ácidos, por lo tanto, pueden convertirse en sales de adición de álcali o ácido.
Preferentemente, la forma neutra de un compuesto se regenera poniendo en contacto una sal con un álcali o ácido de un modo convencional y separando después el compuesto precursor. La forma precursora de un compuesto difiere de sus distintas formas de sal en algunas propiedades físicas, por ejemplo, las distintas solubilidades en disolventes polares.
"Una sal farmacéuticamente aceptable" en el presente documento pertenece a los derivados de los compuestos de la presente invención, en donde dicho compuesto precursor se modifica haciéndolo reaccionar con un ácido o un álcali para formar una sal. Los ejemplos de sales farmacéuticamente aceptables incluyen, pero sin limitación, las sales de ácidos inorgánicos u orgánicos de un álcali tal como una amina y las sales de metal alcalino u orgánicas de un ácido tal como un ácido carboxílico. Las sales farmacéuticamente aceptables incluyen las sales no tóxicas o las sales de amonio cuaternario convencionales de los compuestos parentales, tales como las sales formadas por ácidos inorgánicos u orgánicos no tóxicos. Las sales no tóxicas convencionales incluyen, pero sin limitación, aquellas sales obtenidas a partir de ácidos inorgánicos u orgánicos. Dichos ácidos inorgánicos u orgánicos se seleccionan entre un grupo que consiste en ácido 2-acetoxibenzoico, ácido 2-hidroxietanosulfónico, ácido acético, ácido ascórbico, ácido benceno sulfónico, ácido benzoico, bicarbonato, ácido carbónico, ácido cítrico, ácido edético,
ácido etano disulfónico, ácido etano sulfónico, ácido fumárico, glucoheptosa, ácido glucónico, ácido glutámico, ácido glicólico, ácido bromhídrico, ácido clorhídrico, ácido yohídrico, hidroxi naftaleno, ácido isetiónico, ácido láctico, lactosa, ácido dodecilsulfónico, ácido maleico, ácido málico, ácido mandélico, ácido metano sulfónico, ácido nítrico, ácido oxálico, ácido pamoico, ácido pantoténico, ácido fenilacético, ácido fosfórico, ácido poligalactaldehído, ácido propiónico, ácido salicílico, ácido esteárico, folinato, ácido succínico, ácido sulfámico, ácido paminobencenosulfónico, ácido sulfúrico, tanino, ácido tartárico y ácido p-toluenosulfónico.
Las sales farmacéuticamente aceptables de la presente invención se pueden sintetizar usando los compuestos precursores que contienen grupos ácidos o alcalinos mediante un proceso químico convencional. En general, el método de preparación de dichas sales es como sigue: hacer reaccionar los compuestos precursores en su forma de ácido o álcali libre con una cantidad estequiométrica de un álcali o ácido adecuado en agua, un disolvente orgánico o una mezcla de los mismos. En general, se prefiere un medio no acuoso tal como éter, acetato de etilo, etanol, isopropanol, acetonitrilo o similar.
Además de en la forma de sal, un compuesto proporcionado por la presente invención también puede estar presente en forma de un profármaco. Los profármacos de los compuestos descritos en el presente documento pueden experimentar fácilmente cambios químicos en condiciones fisiológicas, convirtiéndose de este modo en los compuestos de la presente invención. Además, los profármacos se pueden convertir en los compuestos de la presente invención mediante métodos químicos o bioquímicos en un entorno in vivo.
Algunos compuestos de la presente invención pueden existir en formas no solvatadas o en formas solvatadas incluyendo los hidratos. En general, las formas solvatadas son equivalentes a las formas no solvatadas, estando ambas dentro del alcance de la presente invención.
Algunos compuestos de la presente invención pueden comprender átomos de carbono asimétricos (centros ópticos) o dobles enlaces. Los racematos, diastereómeros, isómeros geométricos e isómeros individuales están dentro del alcance de la presente invención.
En el presente documento, los compuestos racémicos, ambiescalémicos y escalémicos o enantioméricamente puros se representan gráficamente usando el método de Maehr., J. Chem. Ed., 1985, 62: 114-120. Los enlaces con forma de cuña y los enlaces punteados se usan para indicar la configuración absoluta de un centro estereogénico a menos que se indique otra cosa. Cuando los compuestos descritos en el presente documento contienen dobles enlaces etilénicos u otros centros geométricamente asimétricos, estos incluyen los isómeros geométricos E y Z a menos que se indique otra cosa. De manera similar, todas las formas tautoméricas están dentro del alcance de la presente invención.
Los compuestos de la presente invención pueden existir en formas geométricas o estereoisoméricas particulares. Los compuestos de la presente invención en cualquier forma concebible, incluyendo isómeros trans y cis, enantiómeros (-) y (+), enantiómeros (R) y (S), diastereómeros, enantiómeros (D) y enantiómeros (L), sus racematos y otras mezclas tales como una mezcla rica en enantiómeros o diastereómeros, están dentro del alcance de la presente invención. Pueden existir otros átomos de carbono asimétricos en sustituyentes tales como grupos alquilo. Todos estos isómeros y una mezcla de los mismos está dentro del alcance de la presente invención.
Los enantiómeros (R) y (S) y los enantiómeros (D) y (L) ópticamente activos can se pueden preparar por síntesis quiral, usando reactivos quiral o por otras técnicas convencionales. Se puede preparar un enantiómero de un compuesto de la presente invención por síntesis asimétrica o la derivación con un auxiliar quiral, en donde la mezcla diastereomérica se separa y el grupo auxiliar se escinde para proporcionar el enantiómero puro deseado. Como alternativa, cuando existe un grupo alcalino (por ejemplo, un grupo amina) o un grupo ácido (por ejemplo, un grupo carboxilo) en la molécula, las sales del diaestereoisómeros se pueden formar con ácidos o álcalis ópticamente activos adecuados. Después, los diastereómeros se resuelven usando un método conocido en este campo y se recuperan los enantiómeros puros. Además, la separación de los enantiómeros y diastereómeros normalmente se realiza usando un método cromatográfico. Dicho método cromatográfico emplea fase estacionaria quiral y opcionalmente se combina con derivatización química (por ejemplo, derivatizar un carbamato de una amina).
Los compuestos de la presente invención pueden contener proporciones no naturales de isótopos en uno o más de los átomos que constituyen los compuestos. Por ejemplo, se pueden usar isótopos radiactivos tales como tritio (3H), yodo-125 (125I) o carbono-14 (14C) para etiquetar los compuestos. Todas las variaciones isotópicas de los compuestos de la presente invención, sean o no radiactivas, entran dentro del alcance de la presente invención.
La expresión "vehículo farmacéuticamente aceptable" se refiere a cualquier formulación o medio transportador que es capaz de administrar una cantidad eficaz de un principio activo de la presente invención, que no interfiere con la bioactividad del principio activo y no tiene efectos secundarios para los hospedadores o pacientes. Los vehículos representativos incluyen agua, aceite, vegetales y minerales, base de crema, base de loción, base de pomada, etc. Estos materiales de base incluyen agentes de suspensión, viscosificadores, potenciadores transdérmicos y similares. Sus preparaciones son aquellas conocidas por un experto en la técnica de la cosmética o los remedios tópicos. Véase Remington: The Science and Practice of Pharmacy, 21a ed., Lippincott, Williams & Wilkins (2005)
para más detalles sobre vehículos.
El término "excipiente" se refiere a los vehículos, diluyentes y/o medios necesarios para formulas composiciones farmacéuticas eficaces.
Para fármacos o principios activos farmacológicamente, la expresión "una cantidad eficaz" o "una cantidad terapéuticamente eficaz" se refiere a una cantidad adecuada del fármaco que no es tóxica pero es capaz de conseguir el efecto esperado. Para la forma de dosificación oral en la presente invención, "una cantidad eficaz" de un principio activo en la composición se refiere a la cantidad del principio activo que se necesita para conseguir el efecto esperado cuando se usa en combinación con otro agente activo en la composición. La determinación de la cantidad eficaz varía de una persona a otra y depende de la edad y de la condición general de los sujetos. También depende del principio activo específico. Los expertos en la técnica pueden determinar la cantidad eficaz adecuada para cada individuo mediante experimentación rutinaria.
La expresión "componente activo", "agente terapéutico", "material activo" o "principio activo" se refiere a una entidad química que es capaz de tratar de manera eficaz los trastornos, enfermedades o afecciones a los que se dirige.
El término "sustituido" se refiere a la sustitución de uno o más átomos de hidrógeno (incluyendo las variantes de hidrógeno y deuterio) en un átomo específico por sustituyentes, con la condición de que la valencia del átomo específico se mantenga normal y que el compuesto sustituido sea estable. Cuando el sustituyente es un grupo acetona (es decir, =O), esto significa que dos átomos de hidrógeno están sustituidos. La sustitución de un grupo acetona nunca sucederá en un grupo arilo. La expresión "opcionalmente sustituido" significa que la sustitución puede tener lugar o no. Salvo que se especifique otra cosa, los sustituyentes pueden ser de cualquier tipo y número alcanzable.
Cuando cualquier variable (por ejemplo, R) aparece en la composición o la estructura de un compuesto más de una vez, la variable varía en su definición de una situación a otra. Por lo tanto, por ejemplo, si un grupo está sustituido con de 0 a 2 Rs, entonces dicho grupo puede estar opcionalmente sustituido con como máximo dos Rs y R tiene opciones independientes en cada situación. Además, la combinación de los sustituyentes y/o sus variantes solo se permite cuando dicha combinación da como resultado compuestos estables.
Cuando los enlaces de un sustituyente pueden estar reticulados a dos átomos de un anillo, dicho sustituyente se puede unir a cualquier átomo del anillo. Cuando no se especifica que ningún átomo del sustituyente listado esté unido al compuesto pero no se especifica, dicho sustituyente se puede unir por cualquiera de sus átomos. La combinación de los sustituyentes y/o sus variantes solo se permite cuando dicha combinación da como resultado
compuestos estables. Por ejemplo, la unidad estructural indica que la sustitución puede ocurrir en cualquier sitio del ciclohexilo o ciclohexadieno.
indica que la sustitución puede ocurrir en cualquier sitio del ciclohexilo o ciclohexadieno.
Un sustituyente de un grupo alquilo o heteroalquilo generalmente se denomina "un sustituyente alquilo", que se puede seleccionar entre, aunque no de forma limitativa, uno o más de los grupos siguientes: -R', -OR', =O, =NR', =N-OR', -NR'R", -SR', halógeno, -SiR'R"R'", OC(O)R', -C(O)R', -CO2R', -CONR'R", -OC(O)NR'R", -NR"C(O)R', NR'C(O)NR"R"', -NR"C(O)2R', -NR -C(NR'R"R'")=NR"", NR""C(NR'R")=NR'", -S(O)R', -S(O)2R', -S(O)2NR'R", NR"SO2R', -CN, -NO2, -N3, -CH(Ph)2 y fluoro-alquilo (C1-C4). El número de sustituyentes varía desde 0 hasta 2m'+1, en donde m' es el número total de átomos de carbono en dicho grupo atómico. Se prefiere que cada uno de R', R", R'", R"" y R...sea hidrógeno, un grupo heteroalquilo sustituido o sin sustituir, un grupo arilo sustituido o sin sustituir (por ejemplo, un arilo sustituido con de 1 a 3 átomos de halógeno), un grupo alquilo sustituido o sin sustituir, un grupo alcoxilo, un grupo tioalcoxi o un grupo aralquilo. Cuando un compuesto de la presente invención contiene uno o más R, por ejemplo, cada R se selecciona independientemente, como en el caso con uno o más R', R", R'", R"" y R.... Cuando R' y R" están unidos al mismo átomo de nitrógeno, estos pueden estar combinados con el átomo de nitrógeno y formar un anillo de 5, 6 o 7 miembros. Por ejemplo, -NR'R" pretende incluir, pero sin limitación, 1-pirrolidinilo y 4-morfolinilo. Basándose en el análisis anterior sobre los sustituyentes, los expertos en la materia pueden entender que el término "alquilo" pretende incluir un grupo cuyos átomos de carbono están unidos a grupos distintos de átomos de hidrógeno, tales como un grupo haloalquilo (por ejemplo, -CF3, -CH2CF3) y un grupo acilo (por ejemplo, -C(O)CH3, -C(O)CF3, -C(O)CH2OCH3, etc.).
De manera similar a dichos sustituyentes de un grupo atómico alquilo, un sustituyente de un grupo arilo o heteroarilo se denomina normalmente "un sustituyente arilo", que se selecciona entre, por ejemplo, R', -OR', -NR'R", -SR', halógeno, -SiR'R"R"', -OC(O)R', -C(O)R', -CO2R', -CONR'R", -OC(O)NR'R", -NR"C(O)R', -NR'C(O)NR"R'", -NR"C(O)2R', -NR""'-C(NR'R"R'")=NR'"', NR'"'C(NR'R")=NR'", -S(O)R', -S(O)2R', -S(O)2NR'R", -NR"SO2R', -CN, -NO2, -N3, -CH(Ph)2, fluoro-alcoxilo (C1-C4) y fluoro-alquilo (C1-C4). El número de sustituyentes varía desde 0 hasta el número total de valencias abiertas en el anillo arilo, en donde se prefiere que cada uno de R', R", R'", R"" y R...se seleccione entre hidrógeno, un grupo alquilo sustituido o sin sustituir, un grupo heteroalquilo sustituido o sin sustituir, un grupo arilo sustituido o sin sustituir y un grupo heteroarilo sustituido o sin sustituir. Cuando un compuesto de la
presente invención contiene uno o más R, cada R se selecciona independientemente, como en el caso con uno o más R', R", R'", R"" o R....
Dos sustituyentes en átomos adyacentes de un grupo arilo o un anillo heteroarilo pueden estar opcionalmente sustituidos por un sustituyente cuya fórmula general es -T-C(O)-(CRR')q-U-, en donde T y U se seleccionan independientemente entre -NR-, -O-, CRR'- o un enlace sencillo y q es un número entero entre 0 y 3. Como alternativa, dos sustituyentes en átomos adyacentes de un grupo arilo o un anillo heteroarilo pueden estar opcionalmente sustituidos por un sustituyente cuya fórmula general es -A(CH2)rB-, en donde A y B se seleccionan independientemente entre -CRR'-, -O-, -NR-, -S-, -S(O)-, S(O)2-, -S(O)2NR'- o un enlace sencillo y r es un número entero entre 1 y 4. Opcionalmente, un enlace sencillo en el nuevo anillo así formado se puede sustituir por un doble enlace. Como alternativa, dos sustituyentes en átomos adyacentes de un grupo arilo o un anillo heteroarilo pueden estar sustituidos por un sustituyente cuya fórmula general es -A(CH2)rB-, en donde s y d se seleccionan independientemente entre un número entero entre 0 y 3, respectivamente X es -O-, -NR', -S-, -S(O)-, S(O)2- o S(O)2NR'-. Los sustituyentes R, R', R" y R'" se prefiere que se seleccionen independientemente entre hidrógeno y un grupo alquilo (C1-C6) sustituido o sin sustituir, respectivamente.
El término "halo" o "halógeno", por sí solo o como parte de otro sustituyente, representa flúor, cloro, bromo o yodo a menos que se indique otra cosa. Además, el término "haloalquilo" pretende incluir monohaloalquilo y polihaloalquilo. Por ejemplo, el término "haloalquilo (C1-C4)" pretende incluir, pero sin limitación, trifluorometilo, 2,2,2-trifluoroetilo, 4-clorobutilo, 3-bromopropilo y similares.
Los ejemplos de grupos haloalquilo incluyen, pero sin limitación, trifluorometilo, triclorometilo, pentafluoroetilo y pentacloroetilo. Un "alcoxilo" representa los grupos alquilo mencionados anteriormente que tienen un número específico de átomos de carbono y están unidos mediante un puente de oxígeno. El grupo alcoxilo C1-6 incluye alcoxilo C1, alcoxilo C2, alcoxilo C3, alcoxilo C4, alcoxilo C5 y alcoxilo C6. Los ejemplos de alcoxilo incluyen, pero sin limitación, metoxilo, etoxilo, n-propoxilo, isopropoxilo, n-butoxilo, sec-butoxilo, ferc-butoxilo, n-pentiloxilo y spentiloxilo. Un "cicloalquilo" incluye un grupo cíclico saturado tal como ciclopropilo, ciclobutilo o ciclopentilo. Cicloalquilo C3-7 incluye cicloalquilo C3, cicloalquilo C4, cicloalquilo C5, cicloalquilo C6 y cicloalquilo C7. Un "alquenilo" incluye un grupo hidrocarburo de cadena lineal o ramificada, en donde uno o más dobles enlaces carbono-carbono (tal como etenilo y propenilo) existe en cualquier lugar estable de la cadena.
El término "halo" o "halógeno" se refiere a flúor, cloro, bromo y yodo.
El término "hetero" representa heteroátomos o grupos heteroatómicos (es decir, grupos atómicos que contienen uno o varios heteroátomos), incluyendo átomos distintos de átomo de carbono (C) y átomo de hidrógeno (H), por ejemplo, incluyendo oxígeno (O), nitrógeno (N), azufre (S), silicio (Si), germanio (Ge), aluminio (Al), boro (B), -O-, -S-, =O, =S, -C(=O)O-, -C(=O)-, -C(=S)-, -S(=O), -S(=O)2- y -C(=O)N(H)- opcionalmente sustituido, -N(H)-, -C(=NH)-, -S(=O)2N(H)- o -S(=O)N(H)-.
El término "anillo" se refiere a cicloalquilo, heterocicloalquilo, cicloalquenilo, heterocicloalquenilo, cicloalquinilo, heterocicloalquinilo, arilo o heteroarilo sustituido o sin sustituir. Los denominados anillos incluyen un anillo individual, un anillo enlazado, un anillo en espiral, un anillo combinado o un anillo puenteado. El número de átomos de un anillo normalmente se define como el número de miembros del anillo. Por ejemplo, un "anillo de 5 a 7 miembros" se refiere al hecho de que de 5 a 7 átomos forman un anillo. Salvo que se especifique otra cosa, el anillo opcionalmente contiene de 1 a 3 heteroátomos. Por lo tanto, un "anillo de 5 a 7 miembros" incluye, por ejemplo, fenilpiridina y piperidinilo. Por otro lado, la expresión "anillo heterocicloalquilo de 5 a 7 miembros" incluye piridilo y piperidinilo y no incluye fenilo. El término "anillo" también incluye el sistema de anillos que contiene al menos un anillo, en donde cada "anillo" independientemente está de acuerdo con la definición anterior.
Salvo que se especifique otra cosa, el término "heterociclo" o "heterocíclico" significa un anillo monocíclico, bicíclico o tricíclico estable, que contiene uno o más heteroátomos o grupos heteroatómicos. Puede estar saturado, parcialmente insaturado o insaturado (aromático) y contener átomos de carbono y 1, 2, 3 o 4 heteroátomos cíclicos seleccionados independientemente entre N, O y S, en donde cualquiera de los anillos heterocíclicos anteriores puede estar condensado con un anillo de benceno para formar un anillo bicíclico. Los heteroátomos de nitrógeno y azufre opcionalmente pueden estar oxidados (es decir, NO y S(O)p). El átomo de nitrógeno puede estar sustituido o sin sustituir (es decir, N o NR, en donde R es H u otro sustituyente que se ha definido en el presente documento). El heterociclo puede estar unido a los grupos laterales de cualquier heteroátomo o átomo de carbono para formar una estructura estable. Si el compuesto resultante es estable, el heterociclo descrito en el presente documento puede estar sustituido en la posición de carbono o la posición de nitrógeno. El o los átomos de nitrógeno en el o los heterociclos están opcionalmente cuaternizados. Una realización preferida es aquella en la que el número total de átomos S y O en el heterociclo excede de 1, estos átomos no son adyacentes entre sí. Otra realización preferida es aquella en la que el número total de átomos S y O en el heterociclo no excede de 1. Tal como se usa en el presente documento, el término "grupo heterocíclico aromático" o "heteroarilo" significa un anillo monocíclico o bicíclico de 5, 6 o 7 miembros estable o un anillo aromático heterocíclico, bicíclico de 7, 8, 9 o 10 miembros, que comprende átomos de carbono y 1, 2, 3 o 4 heteroátomos cíclicos seleccionados independientemente entre N, O y S. El átomo de nitrógeno puede estar sustituido o sin sustituir (es decir, N o NR, en donde R es H u otro sustituyente que se ha
definido en el presente documento). Los heteroátomos de nitrógeno y azufre opcionalmente pueden estar oxidados (es decir, NO y S(O)p). Merece la pena señalar que el número total de átomos de S y O en el anillo heterocíclico aromático no excede de 1. El anillo puenteado también está incluido en la definición del heterociclo. Cuando uno o más átomos (es decir, C, O, N o S) conectan con dos átomos de carbono o nitrógeno no adyacentes para formar un anillo puenteado, los anillos puenteados preferidos incluyen, pero sin limitación, un átomo de carbono, dos átomos de carbono, un átomo de nitrógeno, dos átomos de nitrógeno y un grupo carbono-nitrógeno. Merece la pena señalar que un puente siempre convierte un anillo individual en un anillo tricíclico. En el anillo puenteado, los sustituyentes en el anillo también pueden estar presentes en el puente.
Los ejemplos de compuestos heterocíclicos incluyen, pero sin limitación, acridinilo, azocinilo, benzoimidazolilo, benzofuranilo, benzotiofuranilo, benzotiofenilo, benzoxazolilo, benzoxazolinilo, benzotiazolilo, benzotriazolilo, benzotetrazolilo, benzoisoxazolilo, benzoisotiazolilo, benzoimidazolinilo, carbazolilo, 4aH-carbazolilo, carbolinilo, cromanilo, cromeno, cinnolinildecahidroquinolilo, 2H,6H-1,5,2-ditiazinilo, dihidrofuro[2,3-6]tetrahidrofuranilo, furanilo, furazanilo, imidazolidinilo, imidazolinilo, imidazolilo, 1 H-indolilo, indolenilo, dihidroindolilo, indenilo, indolilo, 3H-indolilo, isatinoílo, isobenzofuranilo, pirano, isoindolilo, isoindolilo, iso dihidroindolilo, isoindolilo, indolilo, isoquinolilo, isotiazolilo, isoxazolilo, metilenodioxifenilo, morfolinilo, naftiridinilo, octahidroisoquinolilo, oxadiazolilo, 1,2,3-oxadiazolilo, 1,2,4-oxadiazolilo, 1,2,5-oxadiazolilo, 1,3,4-oxadiazolilo, oxazolidinilo, oxazolilo, isoxazolilo, oxindolilo, pirimidinilo, fenantridilo, fenantrolinilo, fenazina, fenotiazina, benzoxantinilo, fenoxazinilo, ftalazinilo, piperazinilo, piperidinilo, piperidonilo, 4-piperidonilo, piperonilo, pteridinilo, purinilo, piranilo, pirazinilo, pirazolidinilo, pirazolinilo, pirazolilo, piridazinilo, piridoxazol, piridimidazol, piridotiazol, piridilo, pirimidinilo, pirrolidinilo, pirrolinilo, 2H-pirrolilo, pirrolilo, pirazolilo, quinazolinilo, quinolinilo, 4H-quinolizinilo, quinoxalinilo, quinuclidinilo, tetrahidrofurilo, tetrahidroisoquinolilo, tetrahidroquinolilo, tetrazolilo, 6H-1,2,5-tiadiazinilo, 1,2,3-tiadiazolilo, 1,2,4-tiadiazolilo, 1,2,5-tiadiazolilo, 1,3,4-tiadiazolilo, tiantrenilo, tiazolilo, isotiazoliltienilo, tienilo, tieniloxazolilo, tienotiazolilo, tienoimidazolilo, tienilo, triazinilo, 1,2,3-triazolilo, 1,2,4-triazolilo, 1,2,5-triazolilo, 1,3,4-triazolilo y xanteno. También están incluidos los compuestos de anillo condensado y en espiral.
Salvo que se especifique otra cosa, la expresión "grupo hidrocarburo"o su término específico (por ejemplo, alquilo, alquenilo, alquinilo, fenilo, etc.), por sí solo o como parte de otro sustituyente, representa un grupo atómico hidrocarburo, de cadena lineal, ramificado o cíclico, o una combinación de los mismos, que puede estar completamente saturado, monoinsaturado o poliinsaturado, puede estar monosustituido, disustituido o polisustituido y puede ser monovalente (por ejemplo, metilo), divalente (por ejemplo, metileno) o polivalente (por ejemplo, metenilo). Puede incluir grupos atómicos divalentes o polivalentes y tiene un número especificado de átomos de carbono (por ejemplo, C1-C10 representa de 1 a 10 átomos de carbono). Un "grupo hidrocarburo" incluye, pero sin limitación, grupos hidrocarburo alifáticos y aromáticos. Dichos grupos hidrocarburo alifáticos incluyen grupos hidrocarburo alifáticos y cíclicos, incluyendo de manera específica, aunque no de forma limitativa, alquilo, alquenilo y alquinilo. Dichos grupos hidrocarburo aromáticos incluyen, pero sin limitación, grupos hidrocarburo aromáticos de 6 a 12 miembros tales como fenilo, naftilo y similares. En algunas realizaciones, el término "alquilo" significa un grupo atómico de cadena lineal o ramificada o una combinación de las mismas, que puede estar completamente saturado, monoinsaturado o poliinsaturado y que puede incluir grupos atómicos divalentes y multivalentes. Los ejemplos de grupos atómicos hidrocarburo saturados incluyen, pero sin limitación, metilo, etilo, n-propilo, isopropilo, n-butilo, tbutilo, iso-butilo, sec-butilo, ciclohexilo, (ciclohexil)metilo, ciclopropilmetilo y los homólogos o isómeros de grupos atómicos tales como n-pentilo, n-hexilo, n-heptilo, n-octilo y similares. Un alquilo insaturado tiene uno o más dobles o triples enlaces, cuyos ejemplos incluyen, pero sin limitación, vinilo, 2-propenilo, butenilo, crotonilo, 2-isopentenilo, 2-(butadienilo), 2,4-pentadienilo, 3-(1,4-pentadienilo), etinilo, 1 y 3-propinilo, 3-butinilo y homólogos e isómeros mayores.
Salvo que se especifique otra cosa, la expresión "grupo heterohidrocarburo" o su término específico (por ejemplo, heteroalquilo, heteroalquenilo, heteroalquinilo, heteroarilo, etc.), solo o en combinación con otro término, indica un grupo atómico hidrocarburo, de cadena lineal, ramificada o cíclico, estable o una combinación de las mismas, que tiene cierto número de átomos de carbono y al menos un heteroátomo. En algunas realizaciones, el término "heteroalquilo", solo o en combinación con otro término, indica un grupo atómico hidrocarburo, de cadena lineal o ramificada o una combinación de las mismas, que tiene cierto número de átomos de carbono y al menos un heteroátomo. En una realización habitual, los heteroátomos se seleccionan entre B, O, N y S, donde los átomos de nitrógeno y de azufre opcionalmente están oxidados y los heteroátomos de nitrógeno opcionalmente está cuaternizados. Los heteroátomos B, O, N y S pueden estar ubicados en cualquier posición interna (incluyendo la posición del resto de la molécula donde el grupo hidrocarburo estaba unido) del grupo heterohidrocarburo. Los ejemplos incluyen, pero sin limitación, -CH2-CH2-O-CH3, -CH2-CH2-NH-CH3, -CH2-CH2-N(CH3)-CH3, -CH2-S-CH2-CH3, -CH2-CH2, -S(O)-CH3, -CH2-CH2-S(O)2-CH3, -CH=CH-O-CH3, -CH2-CH=N-OCH3 y -CH=CH-N(CH3)-CH3. Como máximo, dos hetereoátomos pueden ser continuos, por ejemplo, -CH2-NH-OCH3.
Los términos "alcoxi", "alquilamino" y "alquiltio" (o tioalcoxi) son idiomáticos y se refieren a aquellos grupos alquilo que están unidos al resto de la molécula mediante un átomo de oxígeno, un grupo amino o un átomo de azufre, respectivamente.
Salvo que se especifique otra cosa, las expresiones "grupo hidrocarburo cíclico", "grupo hidrocarburo heterociclo" o su término específico (tal como arilo, heteroarilo, cicloalquilo, heterocicloalquilo, cicloalquenilo, heterocicloalquenilo,
cicloalquinilo, heterocicloalquinilo, etc.), por sí solos o en combinación con otros términos, representan respectivamente un "grupo hidrocarburo" o un "grupo heterohidrocarburo" cíclico. Además, para grupos heterohidrocarburo o grupos heterociclocarburo (tal como heteroalquilo, heterocicloalquilo), los heteroátomos pueden ocupar la posición del resto de la molécula donde el heterociclo se une. Los ejemplos de cicloalquilo incluyen, pero sin limitación, ciclopentilo, ciclohexilo, 1-ciclohexenilo, 3-ciclohexenilo, cicloheptilo y similares. Los ejemplos no limitantes de grupos heterocíclicos incluyen 1-(1,2,5,6-tetrahidropiridilo), 1 -piperidinilo, 2-piperidinilo, 3-piperidinilo, 4-morfolinilo, 3-morfolinilo, tetrahidrofuran-2-ilo, tetrahidrofuranindol-3-ilo, tetrahidrotiofen-2-ilo, tetrahidrotiofen-3-ilo, 1-piperazinilo y 2-piperazinilo.
Salvo que se especifique otra cosa, el término "arilo" se refiere a un sustituyente hidrocarburo aromático, poliinsaturado, que puede estar monosustituido, disustituido o polisustituido, puede ser monovalente, divalente o polivalente y puede ser monocíclico o policíclico (por ejemplo, de 1 a 3 anillos, al menos uno de cuales es aromático). Y los anillos están condensados juntos o unidos covalentemente. El término "heteroarilo" se refiere a un grupo arilo (o anillo) que contiene de uno a cuatro heteroátomos. En una realización ilustrativa, los heteroátomos se seleccionan entre B, N, O y S, en donde los átomos de nitrógeno y azufre opcionalmente están oxidados y el átomo de nitrógeno opcionalmente está cuaternizado. El grupo heteroarilo puede estar unido al resto de la molécula mediante un heteroátomo. Los ejemplos no limitantes de grupos arilo o heteroarilo incluyen fenilo, 1-naftilo, 2-naftilo, 4-bifenilo, 1 -pirrolilo, 2-pirrolilo, 3-pirrolilo, 3-pirazolilo, 2-imidazolilo, 4-imidazolilo, pirazinilo, 2-oxazolilo, 4-oxazolilo, 2-fenil-4-oxazolilo, 5-oxazolilo, 3-isoxazolilo, 4-isoxazolilo, 5-isoxazolilo, 2-tiazolilo, 4-tiazolilo, 5-tiazolilo, 2-furilo, 3-furilo, 2-tienilo, 3-tienilo, 2-piridilo, 3-piridilo, 4-piridilo, 2-pirimidinilo, 4-pirimidinilo, 5-benzotiazolilo, purinilo, 2-benzoimidazolilo, 5-indolilo, 1 -isoquinolinilo, 5-isoquinolinilo, 2-quinoxalinilo, 5-quinoxalinilo, 3-quinolinilo y 6-quinolinilo. Los sustituyentes de uno cualquiera de los sistemas de anillo arilo y heteroarilo anteriores se seleccionan entre los sustituyentes aceptables descritos a continuación.
Por motivos de simplicidad, los grupos arilo, cuando se usan en combinación con otros términos (por ejemplo, ariloxilo, ariltiol, aralquilo), incluyen los arillos arilo y heteroarilo tal como se define anteriormente. Por lo tanto, el término "aralquilo" pretende incluir aquellos grupos atómicos (por ejemplo, bencilo, fenetilo, piridilmetilo, etc.) en los que un grupo arilo está unido a un grupo alquilo, incluyendo aquellos en los que un átomo de carbono (tal como metileno) se ha reemplazado por un átomo de oxígeno, tal como fenoximetilo, 2-piridiloximetilo, 3-(1-naftiloxi)propilo y similares.
El término "grupo saliente" se refiere a un grupo funcional o átomo que puede estar sustituido por otro grupo funcional o átomo mediante una reacción de sustitución (por ejemplo, reacción de sustitución nucleófila). Por ejemplo, los grupos salientes representativos incluyen trifluorometanosulfonato; cloro, bromo, yodo; grupos sulfonato tales como metanosulfonato, tosilato, p-bromobencenosulfonato, p-toluenosulfonato, etc.; grupos aciloxi tales como acetoxi, trifluoroacetoxi y similares.
El término "grupo protector" incluye, pero sin limitación, "grupo protector de amino", "grupo protector de hidroxi" o "grupo protector de mercapto". El término "grupo protector de amino" se refiere a los grupos protectores adecuados para prevenir las reacciones secundarias del nitrógeno amino. Los grupos protectores de amino representativos incluyen, pero sin limitación, formilo; acilo tal como alcanoílo (tal como acetilo, tricloroacetilo o trifluoroacetilo); alcoxicarbonilo tal como t-butoxicarbonilo (Boc); arilmetiloxicarbonilo tal como benciloxicarbonilo (Cbz) y 9-fluorenilmetoxicarbonilo (Fmoc); arilmetilo tal como bencilo (Bn), tritilo (Tr), 1,1-bis(4'-metoxifenil)metilo; sililo tal como trimetilsililo (TMS) y t-butildimetilsililo (TBS) y similares. La expresión "grupo protector de hidroxi" se refiere a los grupos protectores adecuados para prevenir las reacciones secundarias del hidroxilo. Los grupos protectores de hidroxi incluyen, pero sin limitación, alquilo tal como metilo, etilo y t-butilo; acilo tal como alcanoílo (por ejemplo, acetilo); arilmetilo tal como bencilo (Bn), p-metoxibencilo (PMB), 9-fluorenilmetilo (Fm) y difenilmetilo (benzhidrilo, DPM); sililo tal como trimetilsililo (TMS) y t-butildimetilsililo ( T b S ) y similar.
Los compuestos de la presente invención se pueden preparar por una diversidad de métodos de síntesis bien conocidos por los expertos en la técnica, que incluyen las realizaciones específicas enumeradas a continuación, las realizaciones formadas combinando las realizaciones enumeradas con otros métodos de síntesis química y los equivalentes alternativos bien conocidos por los expertos en la técnica. Las realizaciones preferidas incluyen, pero sin limitación, los ejemplos de la presente invención.
Los disolventes usados en la presente invención están disponibles en el mercado. La reacción normalmente se realiza en un disolvente anhidro en atmósfera inerte de nitrógeno. Los datos de resonancia magnética nuclear de protones se registraron en un espectrómetro Bruker Avance III 400 (400 MHz) con los desplazamientos químicos representados en ppm con respecto al campo inferior del tetrametilsilano. El espectro de masas se midió en el Agilent 1200 Series plus 6110 (& 1956A). La LC/MS o la MS Shimadzu contiene un DAD:SPD-M20A (LC) y un detector Shimadzu Micromass 2020. El espectrómetro de masas está equipado con una fuente de iones para electropulverización (electrospray ion source, ESI) operando en modo positivo o negativo.
La cromatografía líquida de alta resolución (high performance liquid chromatography, HPLC) se realizó en un sistema Shimadzu LC20AB equipado con un automuestreador Shimadzu SIL-20A y un detector japonés Shimadzu DAD:SPD-M20A, se adoptó una columna Xtimate C18 (filtro de 3 pm, 2,1x300 mm). Un método 0-60AB de 6 min. fue
como sigue: durante más de 4,2 minutos, una elución con gradiente lineal comenzó con A al 100% (A es TFA al 0,0675 % en agua) y terminó con B al 60 % (B es TFA al 0,0625 % en MeCN), después la elución procedió con B al
60 % durante 1 minuto. Después la columna se equilibró durante 0,8 minutos para conseguir 100:0. El tiempo total de ejecución fue de 6 minutos. Un método 10-80AB de 6 min. fue como sigue: durante más de 4,2 minutos, una elución en gradiente lineal comenzó con A al 90 % (A es TFA al 0,0675 % en agua) y terminó con B al 80 % (B es
TFA al 0,0625 % en acetonitrilo), después la elución procedió con B al 80 % durante 1 minuto. Después la columna se equilibró durante 0,8 minutos para conseguir 90:10. El tiempo total de ejecución fue de 6 minutos. La temperatura de la columna era de 50 °C y el caudal era de 0,8 ml/min. La longitud de onda por barrido del detector de matriz de diodos fue de 200-400 nm.
La cromatografía de capa fina (thin layer chromatography,TLC) se realizó en un Silica Gel GF254 del grupo Sanpont.
Las manchas normalmente se detectaron mediante irradiación UV. En algunos casos, las manchas se examinaron también por otros métodos. En estos casos, yodo (obtenido añadiendo aproximadamente 1 g de yodo a 10 g de gel de sílice y mezclando vigorosamente), vanilina (obtenida disolviendo aproximadamente 1 g de vanilina en 100 ml de H2SO4 al 10%), ninhidrina (disponible en Aldrich) o un revelador especial (obtenido mezclando vigorosamente (NH4)6Mo7O24^4H2O, 5 g de (NH4)2Ce(IV)(NO3)6, 450 ml de H2O y 50 ml de H2SO4 concentrado) se usaron para desarrollar la placa de capa fina de modo que los compuestos se pudieran examinar. La cromatografía en columna ultrarrápida se realizó en una columna de gel de sílice Silicicle de 40-63 pm (malla de 230-400) usando un método similar a la técnica divulgada en Still, W. C.; Kahn, M.; y Mitra, M. Journal of Organic Chemistry, 1978, 43, 2923 2925. Los disolventes usados habitualmente en cromatografía en columna ultrarrápida o cromatografía de capa fina incluyen una mezcla de diclorometano/metanol, una mezcla de acetato de etilo/metanol y una mezcla de hexano/acetato de etilo.
El análisis cromatográfico preparativo se realizó usando un detector Gilson UV/VIS-156 en un sistema Gilson-281
Prep LC 322. La columna adoptada fue Agella Venusil ASB Prep C18, 5 pm, 150 x 21,2 mm; Phenomenex Gemini
C18 , 5 pm, 150 x 30 mm; Boston Symmetrix C18, 5 pm, 150 x 30 mm o Phenomenex Synergi C18, 4 pm, 150 x
30 mm. A un caudal de aproximadamente 25 ml/min, los compuestos se eluyeron con un bajo gradiente de acetonitrilo/agua que contenía HCl al 0,05 %, HCOOH al 0,25 % o NH3^ H2O al 0,5 % en agua, el tiempo total de ejecución fue de 8-15 minutos.
Los inhibidores selectivos de CDK4/6 informados en la presente solicitud se pueden usar en el tratamiento de una serie de cánceres incluyendo cáncer de mama, cáncer de pulmón no microcítico, cáncer de esófago, cáncer de recto y leucemia mieloide aguda. Los inhibidores selectivos de CDK4/6 se pueden usar como un único agente o en combinación con otros agentes quimioterapéuticos.
La presente solicitud usa las abreviaturas siguientes: MW representa microondas; t.a. representa temperatura ambiente; ac. representa solución acuosa; DCM representa cloruro de metileno; THF representa tetrahidrofurano;
DMF representa N,N-dimetilformamida; DMSO representa dimetilsulfóxido; EtOAc representa acetato de etilo; EtOH representa etanol; MeOH representa metanol; BOC representa terc-butoxicarbonilo, que es un grupo protector de amina; Boc2O representa dicarbonato de di-terc-butilo; HOAc representa ácido acético; TEA representa trifluoroetilamina; DlPEA representa diisopropiletilamina; TEA o Et3N representa trietilamina; BnNH2 representa bencilamina; PMBNH2 representa p-metoxibencilamina; MnO2 representa dióxido de manganeso; HATU representa hexafluorofosfato de 0-(7-azabenzotriazol-1-il)-N,N,N'N-tetrametiluronio; POCl3 representa oxicloruro de fósforo;
NaH representa hidruro de sodio; LiAlH4 representa hidruro de litio y aluminio; Pd2(dba)3 representa tris(dibencilidenoacetona)dipaladio; Pd(dppf)Cl2 representa dicloruro de [1,1'-bis(difenilfosfino)ferroceno]paladio;
Pd(OAc)2 representa acetato de paladio; Pd(PPh3)4 representa (trifenilfosfin)-paladio; PPh3 representa trifenilfosfina;
Xantphos representa 4,5-bis(difenilfosfin)-9,9-dimetilxanteno; Xphos representa 2-diciclohexilfosfin-2',4',6'-triisopropilbifenilo; BINAP representa (±)-2,2'-bis(difenilfosfin)-1,1'-binaftilo; Xphos-PD-G2 representa cloro(2-diciclohexilfosfin-2',4',6'-triisopropil-1,1'-bifenil)[2-(2'-amino-1,1'-bifenil)]paladio (II); NIS representa N-yodosuccinimida; NBS representa N-bromosuccinimida; NCS representa N-clorosuccinimida; t-BuOK representa terc-butóxido de potasio; t-BuONa representa terc-butóxido de sodio; CS2CO3 representa carbonato de cesio; K2CO3 representa carbonato de potasio; NaBH(OAc)3 representa triacetoxiborohidruro sódico; NaBH3CN representa cianoborohidruro sódico; NaHCO3 representa bicarbonato sódico; Na2SO4 representa sulfato sódico; KOAc representa acetato potásico; Xantphos representa 4,5-bis(difenilfosfin)-9,9-dimetilxanteno.
Los compuestos de la presente invención se pueden preparar por una diversidad de métodos de síntesis bien conocidos por los expertos en la técnica, que incluyen las realizaciones específicas enumeradas a continuación, las realizaciones formadas combinando las realizaciones enumeradas con otros métodos de síntesis química y los equivalentes alternativos bien conocidos por los expertos en la técnica. Las realizaciones preferidas incluyen, pero sin limitación, los ejemplos de la presente invención.
Los compuestos de la presente invención se pueden preparar por una diversidad de etapas de síntesis, donde R1,
R2, R3, R4, R5, R6, R7, X1, X2, X3, X4, X5, X6, X7, m, n, Q, T y W son los mismos que se h

Preparación del compuesto de fórmula I mediante el esquema de reacción 1

Cuando Y=R4, el compuesto representado por la fórmula I se prepara haciendo reaccionar el derivado de 2H-pirazol (A) con la 2-arilamina (B) en la reacción anterior mostrada en el esquema de reacción 1 para dar el compuesto de fórmula I. La reacción requiere un catalizador adecuado (por ejemplo, Pd2(dba)3), un ligando adecuado (por ejemplo, Xantphos), un álcali adecuado (por ejemplo, Cs2CÜ3) y un disolvente adecuado (por ejemplo, 1,4-dioxano). De acuerdo con el esquema de reacción 1, es más probable que la reacción se produzca a temperatura elevadas. Cuando Q-Y es N-Boc, el compuesto representado por la fórmula I aún se puede preparar haciendo reaccionar el derivado de 2H-pirazol (A) con la 2-arilamina (B) en la reacción inferior mostrada en el esquema de reacción 1. Sin embargo, el grupo Boc necesita ser eliminado en un ácido fuerte (por ejemplo, TFA) para dar la amina (C) y finalmente la amina (C) experimenta una reacción de alquilación en condiciones de aminación reductora o de sustitución nucleófila (tal como NaBH3CN o haloalcano) para dar el compuesto de fórmula I.
Preparación del derivado de 2H-pirazol (A) mediante el esquema de reacción 2

Donde

En la reacción mostrada en el esquema de reacción 2, el 5-bromo-2H-indazol (F) se puede preparar haciendo reaccionar el 5-bromo-2H-indazol (D) con el haloalcano R2X (E). La reacción requiere un álcali adecuado (por ejemplo, NaH o MeONa) y un disolvente adecuado (por ejemplo, THF). El 5-bromo-2H-indazol (G) se puede preparar por halogenación del 5-bromo-2H-indazol (F), la reacción requiere un agente de halogenación adecuado (por ejemplo, Br2, NBS o NIS), un disolvente adecuado (por ejemplo, DMF o MeCN). El 5-bromo-2H-indazol (I) se puede preparar mediante acoplamiento catalizado con paladio del borato (H), la reacción requiere un catalizador adecuado (por ejemplo, Pd(dppf)Ch), un álcali adecuado (por ejemplo, K2CO3), un disolvente adecuado (por ejemplo, dioxano y agua). El 5-bromo-2H-indazol (K) se puede preparar mediante acoplamiento catalizado con paladio del bis(pinacolato)diboro (J), la reacción requiere un catalizador adecuado (por ejemplo, Pd(dppf)Ch), un álcali adecuado (por ejemplo, KOAc) y un disolvente adecuado (por ejemplo, dioxano). El derivado de 2H-pirazol (A) se puede preparar mediante acoplamiento catalizado con paladio del compuesto (F), la reacción requiere un catalizador adecuado (por ejemplo, Pd(dppf)Ch), un álcali adecuado (por ejemplo, K2CO3) y un disolvente adecuado (por ejemplo, dioxano). De acuerdo con el esquema de reacción 2, es más probable que la reacción se produzca a temperatura elevadas. El derivado de 2H-pirazol (A') se puede preparar mediante hidrogenación del compuesto (A) en condiciones de catálisis con rodio, la reacción requiere un catalizador adecuado (por ejemplo, Rh(PPh3)3Cl) y un disolvente adecuado (por ejemplo, tetrahidrofurano) R1' es el grupo alquilo después de la reducción de R1.
Preparación del derivado de 2H-pirazol (A) mediante el esquema de reacción 3

Donde es

En la reacción mostrada en el esquema de reacción 3, el compuesto (O) se puede preparar haciendo reaccionar el compuesto (J) con el cloruro de acilo (N). La reacción requiere un álcali adecuado (por ejemplo, LiHMDS) y un disolvente adecuado (por ejemplo, THF). El compuesto (O) puede experimentar ciclación para producir el 2H-pirazol (Q), la reacción requiere una alquil hidrazina (P) adecuada y un disolvente adecuado (por ejemplo, EtOH). El 2H-pirazol (R) se puede preparar por hidrogenación catalítica, que requiere un catalizador adecuado (por ejemplo, Pd/C) y un disolvente adecuado (por ejemplo, MeOH). El derivado de 2-H-pirazol (A) se puede preparar usando el compuesto (L), la reacción requiere un álcali adecuado (por ejemplo, Et3N) y un disolvente adecuado (por ejemplo, THF). De acuerdo con el esquema de reacción 3, es más probable que la reacción se produzca a temperatura elevadas.
Preparación de la 2-arilamina (B) mediante el esquema de reacción 4 (en el que W es S u O y T es C)

En la reacción mostrada en el esquema de reacción 4, el bromuro de piridina (U) se puede preparar haciendo reaccionar la 2-bromo-5-hidroxi-piridina (S) con un tiol o un alcohol (T) disponible en el mercado mediante una reacción de Mitsunobu. El bromuro de piridina (U) se puede convertir en la 2-arilamina (B) en condiciones catalizadas por paladio, lo que requiere un catalizador adecuado (por ejemplo, Pd2(dba)3), un álcali adecuado (por ejemplo, LiHMDS) y un disolvente adecuado (por ejemplo, tolueno). De acuerdo con el esquema de reacción 4, es más probable que la reacción se produzca a temperatura elevadas.
Preparación de la 2-arilamina (B) mediante el esquema de reacción 5 (en el que W es un enlace sencillo directamente enlazado)
En la reacción mostrada en el esquema de reacción 5, cuando W es un enlace sencillo directamente enlazado, la 2-arilamina (B) se puede preparar mediante los dos procesos siguientes: (1) el 4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-5,6-dihidropiridin-1(2H)-carboxilato de íerc-butilo (X)
y la nitropiridina (W) experimentan primero una reacción de acoplamiento en condiciones de catálisis de paladio, seguido de la reducción del grupo nitro y del doble enlace y (2) el átomo de bromo de la nitropiridina (W) se sustituye con la amina (V) disponible en el mercado, seguido de la reducción del grupo nitro.
Realizaciones
Los siguientes ejemplos se proporcionan para describir la presente invención con más detalle, pero el alcance de la presente invención no se limita a los mismos.
Enfoque A
A continuación se muestra un método general para la preparación de Intermedio A, el intermedio B y el intermedio C.

Intermedio A Intermedio C
Etapa 1:
5-bromo-2 H -indazol

Se añadió 5-bromo-2-fluorobenzaldehído (18,00 g, 88,67 mmol, 1,00 equivalente) lentamente a una solución acuosa al 85 % de hidrato de hidrazina (103,00 g, 2,06 mol, 23,20 equivalentes) y durante la adición precipitó lentamente un sólido de color blanco. La mezcla se agitó a 110 °C durante 16 horas. La LC/MS mostró que la mayoría era el compuesto objetivo. La mezcla se enfrió a 16 °C y se filtró. La torta de filtro se lavó con agua (100 ml) para dar un producto en bruto. El producto en bruto se purificó por cromatografía en columna (éter de petróleo:acetato de etilo = 3:2) para dar el compuesto del título (6,50 g, 32,99 mmol, 37,21 % de rendimiento) en forma de un sólido de color blanco. LC/MS (ESI) m/z: 197,1 (M+1).
Etapa 2:
5-bromo -2-metil-2H-indazol

A una solución de 5-bromo-2H-indazol (20,00 g, 101,51 mmol, 1,00 equivalente) metóxido sódico (5,48 g, 5,48 mmol, 5,48 equivalentes) en metanol (150,00 ml) se le añadió yoduro de metilo (57,00 g, 401,58 mmol, 3,96 equivalentes) gota a gota a 30 °C en atmósfera de nitrógeno y se controló que la duración del goteo fuera de 1 hora. Después, la mezcla se calentó a 85 °C y se agitó durante 5 horas. La LC/Ms mostró que el material de partida se consumió casi por completo y se detectó el compuesto deseado. La mezcla se enfrió a 16 °C y se concentró para dar un producto en bruto. El producto en bruto se diluyó con una solución acuosa al 3 % de NaHCO3 (30 ml) y se sometió a extracción usando acetato de etilo (80 ml x 2). La fase orgánica se concentró a presión reducida. El residuo se purificó por cromatografía en columna (éter de petróleo:acetato de etilo = 30:1 a 1:1) para dar el compuesto del título (8,40 g, 39,80 mmol, 39,21 % de rendimiento) en forma de un sólido de color gris. RMN 1H (400 m Hz , DMSO-cfó) 8 8,33 (s, 1H), 7,95 (s, 1H), 7,58 (d, J= 8,0 Hz, 1H), 7,30 (dd, J= 1,8 Hz, 8 Hz, 1H), 4,18 (s, 1H). LC/MS (ESI) m/z: 210,8 (M+1).
Etapa 3:
5-bromo-3-yodo-2-metil-2H-¡ndazol
I

A una solución de 5-bromo-2-metil-2H-indazol (8,40 g, 39,80 mmol, 1,00 equivalente) en diclorometano (90 ml) se le añadió piridina (4,72 g, 59,70 mmol, 1,5 equivalentes) y bis(trifluoroacetoxi)yodobenceno (20,54 g, 47,76 mmol, 1,20 equivalentes) a 30 °C. La mezcla se agitó durante 0,5 horas y después se le añadió yodo (12,12 g, 47,76 mmol, 1,20 equivalentes) y se volvió a agitar durante 23,5 horas. La LC/MS mostró que la reacción se había completado. La mezcla se filtró para dar el compuesto del título (8,20 g, producto en bruto) en forma de un sólido de color amarillo. LC/MS (ESI) m/z: 336,9 (M+1).
Etapa 4:
5-bromo-2-metil-3-¡sopropen¡l-2H-¡ndazol

A una solución de 5-bromo-3-yodo-2-metil-2H-indazol (7,68 g, 22,79 mmol, 1,00 equivalente) y borato de isopropenilo (4,21 g, 25,07 mmol, 1,11 equivalentes) en dioxano (90,00 ml) se le añadió una solución acuosa saturada (30 ml) de K2CO3 (9,45 g, 68,38 mmol, 3,00 equivalentes) y Pd(dppf)Cl2'CHCl2 (1,86 g, 2,28 mmol, 0,10 equivalentes.). La mezcla se agitó a 100 °C durante 3 horas. La TLC mostró que el material de partida reaccionó casi por completo. La mezcla se enfrió a 30 °C y se filtró, el filtrado se sometió a extracción usando acetato de etilo (100 ml x 3), se lavó con agua (50 ml x 3), se lavó con salmuera saturada (20 ml x 3), se secó sobre Na2SO4 anhidro, se filtró y se concentró. El residuo se purificó por cromatografía en columna (éter de petróleo:acetato de etilo = 1:1) para dar el compuesto del título (5,36 g, 21,34 mmol, 93,66 % de rendimiento) en forma de un aceite de color amarillo.
Etapa 5:
2-metil-3-¡sopropen¡l-5-borato-2H-¡ndazol

En atmósfera de nitrógeno, a una solución de 5-bromo-2-metil-3-isopropenil-2H-indazol (2,80 g, 11,15 mmol, 1,00 equivalente) y bis(pinacolato)diboro (3,40 g, 13,38 mmmol, 1,20 equivalentes) en dioxano (56,00 ml) se le añadió k Oac (3,28 g, 33,45 mmol, 3,00 equivalentes) y Pd(dppf)ChCHCl2 (1,82 g, 2,23 mmol, 0,20 equivalentes). La mezcla se agitó a 100 °C durante 5 horas. La LC/MS mostró que la reacción se había completado y detectó la MS del producto objetivo. La mezcla se enfrió a 16 °C. La mezcla se diluyó con acetato de etilo (20 ml) y se filtró para dar el filtrado. El filtrado se purificó por cromatografía en columna (éter de petróleo:acetato de etilo = 1:1) para dar el producto del título (3,30 g, 9,96 mmol, 89,33 % de rendimiento, 90 % de pureza) en forma de un sólido de color púrpura. LC/MS (ESI) m/z: 299,1 (M+1).
Etapa 6:
5-(2-cloro-5
Intermedio A
En atmósfera de nitrógeno, a una solución de 2,4-dicloro-5-fluoro-pirimidina (147,83 mg, 885,34 pmol, 1,20 equivalentes) y 2-metil-3-isopropenil-5-borato-2H-indazol (220,00 mg, 737,78 pmol, 1,00 equivalente) en dioxano (4 ml) se le añadió K2CO3 (305,91 mg, 2,21 mmol, 3,00 equivalentes) y Pd(dppf)Cl2'CH2Cl2 (120,50 mg, 147,56 pmol, 0,20 equivalentes). La mezcla se agitó a 100 °C durante 3,5 horas. La TLC mostró que la mayoría del material de partida reaccionó completamente y la LC/MS mostró que la mayoría era el producto objetivo. La mezcla se enfrió a 30 °C y se filtró. La torta de filtro se lavó con acetato de etilo (5 ml) y el filtrado se concentró. El residuo se purificó por cromatografía en columna (éter de petróleo:acetato de etilo = 1:0 a 6:1 ) para dar el producto del título (Intermedio A) (210,0 mg, 693,69 pmol, 94,02 % de rendimiento) en forma de un sólido de color amarillo brillante. LC/MS (ESI) m/z: 303,0 (M+1).
Etapa 7:
5-fluoro-4-(2-metil-3-(prop-1-en-2-¡l)-2H-¡ndazol-5-¡l) pirimidin-2-il-carbamato de ferc-butilo

A una solución de 5-(2-cloro-5-fluoropirimidin-4-il)-2-metil-3-(prop-1-en-2-il)-2H-indazol (Intermedio A) (1,00 g, 3,30 mmol, 1,00 equivalente) en dioxano (15,00 ml) se le añadió carbamato de ferc-butilo (966,49 mg, 8,25 mmol, 2,50 equivalentes), carbonato potásico (1,37 g, 9,90 mmol, 3,00 equivalentes), acetato de paladio (74,09 mg, 330,00 pmol, 0,10 equivalentes) y Xantphos (381,89 mg, 660,00 pmol, 0,20 equivalentes). El recipiente se purgó tres veces con nitrógeno y se agitó a 100 °C durante 18 horas. La LC/MS mostró que la reacción se había completado y se detectó el producto. La solución se enfrió a 20 °C y se filtró. El filtrado se concentró a presión reducida para dar el compuesto del título (3,00 g, producto en bruto) en forma de un sólido de color amarillo claro. El producto en bruto se usó en la etapa siguiente sin purificación. LC/MS (ESI) m/z: 384,1 (M+1).
Etapa 8:
5-fluoro-4-(2-metil-3-(prop-1-en-2-il)-2H-indazol-5-il)pirimidin-2-amina
H2N 
A una solución de 5-fluoro-4-(2-metil-3-(prop-1-en-2-il)-2H-indazol-5-il)pirimidin-2-il-carbamato de ferc-butilo (3,00 g, 3,31 mmol, 1,00 equivalente) en diclorometano (30,00 ml) se le añadió ácido trifluoroacético (10,00 ml) gota a gota. La solución se agitó a 15 °C durante 1 hora. La TLC (éter de petróleo:acetato de etilo =1:1) mostró que el material de partida había reaccionado por completo. La solución se secó usando un secador rotatorio al vacío, a presión reducida, a 30 °C. El producto en bruto se diluyó con diclorometano (100 ml) y se lavó con bicarbonato sódico saturado (100 ml) y salmuera saturada (100 ml). La fase orgánica se secó sobre sulfato sódico anhidro y se concentró. El producto en bruto se purificó por columna sobre gel de sílice (éter de petróleo:acetato de etilo = 5:1 a 3:1) para dar el compuesto del título (770,00 mg, 2,72 mmol, 82,11 % de rendimiento) en forma de un sólido de color pardo.
Etapa 9:
5-fluoro-4-(3-¡soprop l-2-met l-2H- ndazol-5-il)p¡r¡m¡d¡n-2-am¡na

Intermedio B
A una solución de 5-fluoro-4-(2-metil-3-(prop-1-en-2-il)-2H-indazol-5-il) pirimidin-2-amina (770,00 mg, 2,72 mmol, 1,00 equivalente) en metanol (20,00 ml) se le añadió paladio sobre carbono (250,00 mg). La solución se calentó a 50 °C y se agitó durante 24 horas en atmósfera de hidrógeno 103,42 kPa (15 psi). La LC/MS mostró que el material de partida había reaccionado por completo y se detectó el producto. La solución de reacción se enfrió a 20 °C y se filtró. El filtrado se concentró para dar el compuesto del título (Intermedio B) (600,00 mg, 2,10 mmol, 77,26 % de rendimiento) en forma de un sólido de color amarillo. RMN 1H (400 MHz, CDCh) 88,55 (s, 1H), 8,20 (d, J = 3,9 Hz, 1H), 7,96 (d, J= 9,2 Hz, 1H), 7,69 (d, J= 9,7 Hz, 1H), 5,06 (s a, 2H), 4,17 (s, 3H), 3,54-3,45 (m, 1H), 1,57 (d,J= 7,2 Hz, 6H).
Etapa 10:
5-(2-cloro-5
Intermedio C
A una solución de 5-(2-cloro-5-fluoropirimidin-4-il)-2-metil-3-isopropenil-2H-indazol (Intermedio A) (36,50 g, 120,57 mmol, 1,00 equivalente) en tetrahidrofurano (182,5 ml) se le añadió Rh(PPh3)3Cl (11,16 g, 12,06 mmol, 0,10 equivalente). El sistema de reacción se purgó con hidrógeno varias veces. La mezcla se agitó a 50 °C en atmósfera de hidrógeno de 344,74 kPa (50 psi) durante 24 horas. La LC/MS mostró que la reacción se había completado. La mezcla se enfrió a 25 °C y se concentró a presión reducida. Al residuo se le añadió metanol (100 ml) y la mezcla se batió y se agitó durante 16 horas y después se filtró. La torta de filtro se lavó con metanol (15 ml x 3) y se secó a presión reducida para dar el compuesto del título (Intermedio C) (28,00 g, 91,88 mmol, 76,20 % de rendimiento) en forma de un sólido de color amarillento. RMN 1H (400 MHz, CDCh) 8,69 (s, 1H), 8,49 (d, J= 3,5 Hz, 1H), 8,06 (d, J= 9,3 Hz, 1H), 7,73 (d, J = 9,3 Hz, 1H), 4,19 (s, 3H), 3,58-3,49 (m, 1H), 1,59 (d, J= 7,2 Hz, 6H). LC/MS (ESI) m/z: 305,2 (M+1).
Ejemplo 1
A/-(5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-il)-6-(4-metilpiperazin-1 -il)piridazin-3-amina

Etapa 1:

Una mezcla de 6-cloro-3-aminopiridazina (3,00 g, 23,16 mmol, 1,00 equivalente) y 1-metilpiperazina (8,10 g, 80,87 mmol, 3,49 equivalentes) se añadió a un tubo de microondas y el tubo se cerró herméticamente. La mezcla se calentó a 170 °C y se agitó durante 1,5 horas con microondas. La LC/MS mostró que la reacción se había completado. La mezcla se enfrió a 20 °C y se concentró a presión reducida. El residuo se purificó por HPLC preparativa (alcalina) para dar el compuesto del título (3,53 g, 18,27 mmol, 78,87 % de rendimiento) en forma de un sólido de color blanco. RMN 1H (400 MHz, DMSO-de) 87,11 (d, J= 9,6 Hz, 1H), 6,73 (d, J= 9,6 Hz, 1H), 5,63 (s, 2H), 3,30-3,24 (m, 4H), 2,42-2,36 (m, 4H), 2,19 (s, 3H). LC/MS (ESI) m/z: 194,1 (M+1).
Etapa 2:
W-(5-fluoro-4-(2-metil-3-isopropenil-2H-indazol-5-il)pirimidin-2-il)-6-(4-metilpiperazin-1-il)piridazin-3-amina

En atmósfera de nitrógeno, a una solución de 5-(2-doro-5-fluoropirimidin-4-il)-2-metil-3-isopropenil-2H-indazol (Intermedio A) (350,00 mg, 1,16 mmol, 1,00 equivalente) y 6-(4-metilpiperazin-1-il)piridazin-3-amina (228,65 mg, 1,18 mmol, 1,02 equivalentes) en dioxano (8 ml) se le añadió CS2CO3 (753,39 mg, 2,31 |jmol, 2,00 equivalentes), Xantphos (267,59 mg, 462,46 jmol, 0,40 equivalente) y Pd2(dba)3 (211,74 mg, 231,21 jmol, 0,20 equivalentes). La mezcla se agitó a 100-110 °C durante 16 horas. La TLC y la LC/MS mostraron que el material de partida había reaccionado por completo. La mezcla se enfrió a 20 °C, se diluyó con acetato de etilo (20 ml) y se filtró. El filtrado se concentró para dar un producto en bruto. El producto en bruto se añadió a metanol (10 ml) y se dejó reposar a 25 °C hasta que apareció un precipitado de color amarillo y se filtró. La torta de filtro se lavó con una pequeña cantidad de metanol y se secó para dar el compuesto del título (120,00 mg, 258,53 jmol, 22,29 % de rendimiento, 99% de pureza) en forma de un sólido de color amarillo. LC/MS (ESI) m/z: 460,3 (M+1).
Etapa 3:
A/-(5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-il)-6-(4-metilpiperazin-1 -il)piridazin-3-amina

A una solución de Pd/C (50 mg) en metanol (10 ml) se le añadió W-(5-fluoro-4-(2-metil-3-isopropenil-2H-indazol-5-il)pirimidin-2-il)-6-(4-metilpiperazin-1-il)piridazin-3-amina (120,00 mg, 261,14 jmol, 1,00 equivalente). Se introdujo hidrógeno gaseoso en el sistema de reacción y se mantuvo a una presión de 103,42 kPa (15 psi). La mezcla se agitó a 40-50 °C durante 8 horas. La LC/MS mostró que la reacción se había completado. Después de la filtración, el filtrado se concentró. El residuo se purificó por HpLC preparativa (ácido clorhídrico) para dar el compuesto del título (57,00 mg, 123,50 jmol, 47,29 % de rendimiento). RMN 1H (400 MHz, metanol-d4) 88,72 (s, 1H), 8,46 (d, J= 4,0 Hz, 1H), 8,41 (d, J= 9,6 Hz, 1H), 8,06 (d, J= 9,2 Hz, 1H), 7,65 (d, J= 9,2 Hz, 1H), 7,40 (d, J= 9,2 Hz, 1H), 4,20 (s, 3H), 3,68-3,61 (m, 5H), 2,63 (t, J= 4,8 Hz, 4H), 2,39 (s, 3H), 1,59 (d, J= 7,2 Hz, 6H). LC/MS (ESI) m/z: 462,2 (M+1).
Ejemplo 2
5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)-A/-(5-(piperazin-1 -il)pirazin-2-M)pirimidin-2-amina

Etapa 1:

A una solución de 2,5-dibromopirazina (10,00 g, 42,04 mmol, 1,00 equivalente) en W-metilpirrolidona (100 ml) se le añadió piperazin-1-carboxilato de ferc-butilo (7,83 g, 42,04 mmol, 1,00 equivalente) y K2CO3 (8,72 g, 63,06 mmol, 1,50 equivalentes). La mezcla se calentó a 100 °C y se agitó durante 18 horas. La TLC (éter de petróleo:acetato de etilo = 10:1) mostró que la reacción se había completado. La mezcla se enfrió a 20 °C, se diluyó con agua (200 ml), se sometió a extracción usando acetato de etilo (200 ml x 2), se secó sobre Na2SO4 anhidro, se filtró y se concentró. El residuo se purificó por cromatografía en columna (éter de petróleo:acetato de etilo = 20:1 a 5:1) para dar el compuesto del título (11,00 g, 2,05 mmol, 76,24 % de rendimiento) en forma de un sólido de color amarillo. RMN 1H (400 MHz, CDCl3) 88,15 (d, J= 1,38 Hz, 1H) 7,87 (d, J= 1,38 Hz, 1H) 3,56 (s, 8H) 1,49 (s, 9H).
Etapa 2:
4-(5-aminopirazin-2-il)piperazin-1-carboxilato de ferc-butilo

En atmósfera de nitrógeno, a una solución de 4-(5-bromopirazin-2-il)piperazin-1-carboxilato de ferc-butilo (10,00 g, 29,14 mmol, 1,00 equivalente) y tetrafluoroborato de tri-ferc-butilfosfina (2,54 g, 8,74 mmol, 0,30 equivalente) en tolueno (100 ml) se le añadió LHMDS (1 M, 60,00 ml, 2,06 equivalentes) y Pd2(dba)3 (2,60 g, 2,84 mmol, 0,10 equivalente). La mezcla se agitó a 65 °C durante 16 horas. La LC/MS mostró que la reacción se había completado. La mezcla se enfrió a 20 °C, se inactivó con agua (50 ml) y se sometió a extracción usando acetato de etilo (100 ml x 3). Las fases orgánicas se combinaron y se concentraron. El residuo se disolvió en acetato de etilo (100 ml) y se purificó por HPLC preparativa (alcalina) para dar el compuesto del título (5,00 g, 17,90 mmol, 61,43 % de rendimiento) en forma de un sólido de color naranja. LC/MS (ESI) m/z: 280,1 (M+1).
Etapa 3:
4-(5-((5-fluoro-4-(2-metil-3-(isopropenil)-2H-indazol-5-il)pirimidin-2-il)amino)pirazin-2-il)piperazin-1-carboxilato de ferc-butilo

En atmósfera de nitrógeno, a una solución de 5-(2-cloro-5-fluoropirimidin-4-il)-2-metil-3-isopropenil-2H-indazol (200,00 mg, 660,65 jmol, 1,00 equivalente) y 4-(5-aminopirazin-2-il)piperazin-1-carboxilato de ferc-butilo (246,06 mg, 792,78 |jmol, 1,20 equivalentes) en dioxano (10 ml) se le añadió CS2CO3 (430,51 mg, 1,32 mmol, 2,00 equivalentes), Xantphos (152,91 mg, 264,26 jmol, 0,40 equivalente) y Pd2(dba)3 (120,99 mg, 132,13 jmol, 0,20 equivalentes). La mezcla se agitó a 110-120 °C durante 16 horas. La LC/MS mostró que la reacción se había completado. La mezcla se enfrió a 25 °C y se filtró. La torta de filtro se lavó con acetato de etilo y el filtrado se concentró. El residuo se purificó por TLC preparativa (diclorometano:metanol = 20:1) para dar el compuesto del título (150,00 mg, 247,43 jmol, 37,45 % de rendimiento, 90 % de pureza) en forma de un sólido de color verde. LC/MS (ESI) m/z: 546,2 (M+1).
Etapa 4:
4-(5-((5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-il)amino)pirazin-2-il)piperazin-1-carboxilato de ferc-butilo

A una solución de 4-(5 ((5-fluoro-4-(2-metil-3-(isopropenil)-2H-indazol-5-il)pirimidin-2-il)amino)pirazin-2-il)piperazin-1-carboxilato de ferc-butilo (90,00 mg, 164,95 jmol, 1,00 equivalente) en tetrahidrofurano (10 ml) se le añadió Pd/C (100 mg) y una cantidad catalítica de ácido acético. Se introdujo hidrógeno gaseoso en el sistema de reacción y se mantuvo a una presión de 103,42 kPa (15 psi). La mezcla se agitó a 50-60 °C durante 16 horas. La LC/MS mostró que la reacción se había completado. La mezcla se enfrió a 20 °C y se filtró. El filtrado se concentró para dar el compuesto del título (90,00 mg, producto en bruto), el producto en bruto se usó directamente en la etapa siguiente. LC/MS (ESI) m/z: 548,3 (M+1).
Etapa 5:
5-fluoro-4-(3-¡soprop¡l-2-met¡l-2H-¡ndazol-5-¡l)-A/-(5-(p¡peraz¡n-1-¡l)p¡raz¡n-2-¡l)p¡r¡m¡d¡n-2-am¡na

A una solución de 4-(5-((5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-il)amino)pirazin-2-il)piperazin-1-carboxilato de ferc-butilo (90,00 mg, 164,34 |jmol, 1,00 equivalente) en metanol (5 ml) se le añadió ácido clorhídricometanol (4 M, 1 ml, 20 mmol, 1,00 equivalente). La mezcla se agitó a 30-40 °C durante 3 horas. TLC (diclorometano:metanol = 30:1) mostró consumición completa del material de partida. La LC/MS mostró que el producto representaba el 49 % y el subproducto representaba el 44 %. La mezcla se enfrió a 25 °C y se concentró. El residuo se purificó por HPLC preparativa (ácido clorhídrico) para dar el compuesto del título (6,00 mg, 13,41 jmol, 8,16 % de rendimiento). RMN 1H (400 MHz, metanol-d4) 88,96 (s, 1H), 8,68 (d, J= 4,4 Hz, 1H), 8,31 (d, J= 9,2 Hz, 1H), 8,12 (s, 1H), 7,79 (d, J= 8,8 Hz, 1H), 4,29 (s, 3H), 3,91 (t, J= 4,8 Hz, 4H), 3,74-3,69 (m, 1H), 3,43 (t, J = 5,2 Hz, 4H), 1,65 (d, J= 7,2 Hz, 6H). LC/MS (ESI) m/z: 448,1 (M+1).
Ejemplo 3
5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)-A/-(5-(piperazin-1-il)piridin-2-il)pirimidin-2-amina

Etapa 1:
4-(6-nitropiridin-3-il)piperazin-1 -carboxilato de ferc-butilo

A una solución de 5-bromo-2-nitropiridina (20,00 g, 98,53 mmol, 1,00 equivalente) en dimetilsulfóxido (52 ml) se le añadió piperazin-1-carboxilato de ferc-butilo (24,00 g, 128,86 mmol, 1,31 equivalentes) y trietilamina (20,00 g, 197,65 mmol, 2,01 equivalentes). La solución se calentó a 60 °C y se agitó durante 18 horas. La TLC (éter de petróleo:acetato de etilo = 3:1) mostró que la reacción se había completado. La solución se diluyó con agua (200 ml), se agitó durante 30 minutos y después se filtró. La torta de filtro se lavó con agua y se secó al vacío para dar un producto en bruto. El producto en bruto se purificó por columna sobre gel de sílice (éter de petróleo:acetato de etilo = 50:1 a 20:1) para dar el compuesto del título (27,00 g, 87,57 mmol, 88,87 % de rendimiento) en forma de un sólido de color amarillo. RMN 1H (400 MHz, CDCla) 88,18 (d, J= 9,03 Hz, 1H), 8,13 (d, J= 2,89 Hz, 1H), 7,21 (dd, J= 9,10, 2,95 Hz, 1H), 3,69-3,59 (m, 4H), 3,51-3,40 (m, 4H), 1,49 (s, 9H).
Etapa 2:
4-(6-aminopiridin-3-il)piperazin-1-carboxilato de ferc-butilo

En atmósfera de nitrógeno, a una solución de 4-(6-nitropiridin-3-il)piperazin-1-carboxilato de ferc-butilo (28,00 g, 90,81 mmol, 1,00 equivalente) en metanol (600 ml) se le añadió paladio sobre carbono (6 %, 1,7 g). La suspensión se evacuó y se llenó con hidrógeno varias veces. La solución se agitó a 50 °C en atmósfera de hidrógeno 344,74 kPa (50 psi) durante 18 horas. La TLC (diclorometano:metanol= 10:1) mostró que el material de partida reaccionó por completo. La suspensión se filtró y el filtrado se secó usando un secador rotatorio al vacío para dar el compuesto del título (24,13 g, 86,69 mmol, 95,46 % de rendimiento) en forma de un sólido de color púrpura. RMN 1H (400 MHz, CDCla) 87,78 (d, J= 2,64 Hz, 1H) 7,18 (dd, J= 8,78, 2,89 Hz, 1H) 6,50 (d, J = 8,78 Hz, 1H) 4,21 (s a, 2H) 3,60-3,54 (m, 4H) 3,00-2,92 (m, 4H) 1,48 (s, 9H).
Etapa 3:
4-(6-((5-fluoro-4-(2-metil-3-(prop-1-en-2-il)-2H-indazol-5-il)pirimidin-2-il)amino)piridin-3-il)piperazin-1-carboxilato de ferc-butilo

A una solución de 5-(2-doro-5-fluoropirimidin-4-il)-2-metil-3-(prop-1-en-2-il)-2H-indazol (Intermedio A) (200,00 mg, 660,65 |jmol, 1,00 equivalente) en dioxano (10,00 ml) se le añadió 4-(6-aminopiridin-3-il)piperazin-1-carboxilato de ferc-butilo (220,67 mg, 792,79 jmol, 1,20 equivalentes), Pd2(dba)3 (60,50 mg, 66,07 jmol, 0,10 equivalentes), Xantphos (76,45 mg, 132,13 jmol, 0,20 equivalentes) y carbonato de cesio (430,51 mg, 1,32 mmol, 2,00 equivalentes). La solución se calentó a 110 °C en atmósfera de nitrógeno y se agitó durante 16 horas. La LC/MS mostró que la reacción se había completado. La solución se enfrió a 25 °C, se filtró y se concentró para dar un producto en bruto. El producto en bruto se purificó por TLC preparativa (acetato de etilo:éter de petróleo = 1:2) para dar el compuesto del título (320,00 mg, 587,57 jmol, 88,94 % de rendimiento) en forma de un sólido de color amarillo claro. LC/MS (ESI) m/z: 545,3 (M+1).
Etapa 4:
4-(6-((5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-il)amino)piridin-3-il)piperazin-1-carboxilato de fercbutilo

En atmósfera de nitrógeno, a una solución de 4-(6-((5-fluoro-4-(2-metil-3-(prop-1-en-2-il)-2H-indazol-5-il)pirimidin-2-il)amino)piridin-3-il)piperazin-1 -carboxilato de ferc-butilo en metanol (20,00 ml) se le añadió paladio sobre carbono (200,00 mg) y ácido acético (2,10 g, 34,97 mmol, 59,52 equivalentes). La suspensión se evacuó y se llenó con hidrógeno varias veces. La solución se agitó a 50 °C en atmósfera de hidrógeno 220,63 kPa (32 psi) durante 96 horas. La LC/MS mostró que la reacción se había completado. La suspensión se enfrió a 25 °C, se filtró y se concentró para dar el compuesto del título (500,00 mg, producto en bruto) en forma de un sólido de color blanquecino. LC/MS (ESI) m/z: 547,1 (M+1).
Etapa 5:
5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)-W-(5-(piperazin-1-il)piridin-2-il)pirimidin-2-amina

A 25 °C, a una solución de 4-(6-((5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5 -il)pirimidin-2-il)amino)piridin-3-il)piperazin-1-carboxilato de ferc-butilo (500,00 mg, 914,68 jmol, 1,00 equivalente) en diclorometano (5,00 ml) se le añadió ácido trifluoroacético (2,09 g, 18,29 mmol, 20,00 equivalentes) en una porción. La solución se agitó durante 0,5 h. La LC/MS mostró que la reacción se había completado. La solución se concentró a presión reducida para dar un producto en bruto. El producto en bruto se purificó por HPLC preparativa (ácido clorhídrico) para dar el compuesto del título (99,69 mg, 223,26 jmol, 24,41 % de rendimiento). RMN 1H (400 MHz, metanol-d4) 88,98 (s, 1H), 8,85 (d, J= 3,39 Hz, 1H), 8,53 (d, J= 9,03 Hz, 1H), 8,32 (dd, J = 9,72, 2,20 Hz, 1H), 8,01-7,96 (m, 1H), 7,93 (d, J= 9,03 Hz, 1H), 7,60 (d, J = 9,66 Hz, 1H), 4,39 (s, 3H), 3,79 (dt, J= 13,90, 6,92 Hz, 1H), 3,65-3,56 (m, 4H), 3,49 (d, J = 5,02 Hz, 4H), 1,69 (d, J= 7,03 Hz, 6H). LC/MS (ESI) m/z: 447,1 (M+1).
Ejemplo 4
W-(5-(4-etilpiperazin-1-il)piridin-2-il)-5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-amina
A una solución de 5-fluoro-4-(3-¡soprop¡l-2-met¡l-2H-¡ndazol-5-¡l)-N-(5-p¡peraz¡n-1-¡l-2-p¡r¡d¡l)p¡r¡m¡d¡n-2-am¡na (210,00 mg, 470,30 jmol, 1,00 equ¡valente) en metanol (5,00 ml) se le añad¡ó acetaldehído (77,69 mg, 705,46 jmol, 1,50 equivalentes), NaBHsCN (59,11 mg, 940,61 jmol, 2,00 equ¡valentes) y ác¡do acét¡co (14,12 mg, 235,15 jmol, 0,50 equivalentes). La soluc¡ón se ag¡tó a 20 °C durante 2 horas. La lC/MS mostró que la reacc¡ón se había completado. La soluc¡ón se f¡ltró y el f¡ltrado se concentró a pres¡ón reduc¡da para dar un producto en bruto. El producto en bruto se pur¡f¡có por HPLC preparativa (ác¡do clorhídr¡co) para dar el compuesto del título (66,50 mg, 128,83 |jmol, 27,39 % de rend¡m¡ento, 99 % de pureza, clorh¡drato). RMN 1H (400 MHz, metanol-d4) 68,97 (s, 1H), 8,84 (d, J= 3,6 Hz, 1H), 8,52 (d, J= 9,2 Hz, 1H), 8,32 (dd, J= 9,7, 2,8 Hz, 1H), 7,98 (d, J= 2,8 Hz, 1H), 7,92 (d, J= 9,0 Hz, 1H), 7,59 (d, J= 9,5 Hz, 1H), 4,38 (s, 3H), 4,02-3,93 (m, 2H), 3,82-3,74 (m, 3H), 3,38-3,34 (m, 4H), 3,32-3,28 (m, 2H), 1,69 (d, J= 7,0 Hz, 6H), 1,46 (t, J= 7,3 Hz, 3H). LC/MS (ESI) m/z: 475,2 (M+1).
Ejemplo 5
N-(6-( 1,4-d¡azepan-1-¡l)p¡r¡daz¡n-3-¡l)-5-fluoro-4-(3-¡soprop¡l-2-met¡l-2H-¡ndazol-5-¡l)p¡r¡m¡d¡n-2-am¡na

Etapa 1:
4-(6-clorop¡r¡daz¡n-3-¡l)-1,4-d¡azepan-1-carbox¡lato de terc-but¡lo

A una soluc¡ón de 3,6-d¡clorop¡r¡daz¡na (2,00 g, 13,42 mmol, 1,05 equivalentes), 1,4-d¡azepan-1-carbox¡lato de terc-but¡lo (2,56 g, 12,78 mmol, 1,00 equivalente) en d¡met¡lsulfóx¡do (15,00 ml) se le añad¡ó tr¡et¡lam¡na (3,88 g, 38,34 mmol, 3,00 equivalentes) en una porc¡ón. La soluc¡ón se calentó a 80 °C y se ag¡tó durante 7 horas. La LC/MS mostró que la reacc¡ón se completó y la MS detectó el producto. La soluc¡ón se enfr¡ó a 25 °C y se concentró para dar un producto en bruto. El producto en bruto se pur¡f¡có por cromatografía en columna (éter de petróleo:acetato de et¡lo = 30:1 a 20:1) para dar el compuesto del título (1,20 g, producto en bruto). El producto en bruto era un sól¡do de color amar¡llo. RMN 1H (400 MHz, CDCl3) 6 7,18 (d, J = 9,4 Hz, 1H), 6,79 (d, J = 9,4 Hz, 1H), 3,87-3,57 (m, 6H), 3,39-3,23 (m, 2H), 1,99-1,93 (m, 2H), 1,40 (s, 9H). LC/MS (ESI) m/z: 313,1 (M+1).
Etapa 2:
4-(6-(5-fluoro-4-(3-¡soprop¡l-2-met¡l-2H-¡ndazol-5-¡l)p¡r¡m¡d¡n-2-¡l-am¡no)p¡r¡daz¡n-3-¡l)-1,4-d¡azepan-1-carbox¡lato de terc-but¡lo

A una soluc¡ón de 5-fluoro-4-(3-¡soprop¡l-2-met¡l-2H-¡ndazol-5-¡l)p¡r¡m¡d¡n-2-am¡na (Intermedio B) (200,00 mg, 700,97 jmol, 1,00 equivalente) en d¡oxano (3,00 ml) se le añad¡ó 4-(6-clorop¡r¡daz¡n-3-¡l)-1,4-d¡azepan-1-carbox¡lato de terc-but¡lo, carbonato de ces¡o (575,00 mg, 1,76 mmol, 2,52 equivalentes), Pd2(dba)3 (65,00 mg, 70,98 jmol, 0,10 equivalentes) y Xantphos (85,00 mg, 146,90 jmol, 0,21 equivalentes). El a¡re del rec¡p¡ente se reemplazó con n¡trógeno tres veces. La mezcla se calentó a 100 °C y se ag¡tó durante 18 horas. La LC/Ms mostró que el mater¡al de part¡da reacc¡onó completamente y se detectó el producto. La soluc¡ón se enfr¡ó a 20 °C y se concentró para dar un producto en bruto. El producto en bruto se pur¡f¡có por TLC preparativa (acetato de et¡lo) para dar el compuesto del título (120,00 mg, 213,66 jmol, 30,48 % de rend¡m¡ento) en forma de un sól¡do de color amar¡llo claro. LC/MS (ESI) m/z: 562,2 (M+1).
Etapa 3:
N-(6-( 1,4-d¡azepan-1-¡l)p¡r¡daz¡n-3-¡l)-5-fluoro-4-(3-¡soprop¡l-2-met¡l-2H-¡ndazol-5-¡l)p¡r¡m¡d¡n-2-am¡na

A una solución de 4-(6-(5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-il-amino)piridazin-3-il)-1,4-diazepan-1-carboxilato de ferc-butilo (120,00 mg, 213,66 |jmol, 1,00 equivalente) en diclorometano (2,00 ml) se le añadió ácido trifluoroacético (1,00 ml) gota a gota. La solución se agitó a 25 °C durante 1 hora. La LC/MS mostró que el material de partida reaccionó completamente y se detectó el producto. La solución se concentró a presión reducida para dar un producto en bruto. El producto en bruto se purificó por HPLC preparativa (ácido clorhídrico) para dar el compuesto del título (45,38 mg, 98,32 jmol, 46,02 % de rendimiento). RMN 1H (400 MHz, Metanol-d4) 88,96 (s, 1H) 8,85 (d, J= 3,4 Hz, 1H) 8,48 (d, J= 9,2 Hz, 1H) 8,08 (d, J= 10,0 Hz, 1H) 7,90 (d, J= 9,2 Hz, 2H) 4,36 (s, 3H) 4,11 (t, J= 5,0 Hz, 2H) 3,89 (t, J= 5,9 Hz, 2H) 3,76 (quint., J = 7,0 Hz, 1H) 3,52 (t, J= 5,1 Hz, 2H) 3,48-3,36 (m, 2H) 2,28 (s a, 2H) 1,68 (d, J= 7,0 Hz, 6H).
Ejemplo 6
/V-(6-(3-(dimetilamino)pirrolidin-1 -il)piridazin-3-il)-5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-amina

Etapa 1:
1-(6-cloropiridazin-3-il)-W,W-dimetilpirrolidin-3-amina

Una solución de 3,6-dicloropiridazina (1,20 g, 8,05 mmol, 1,00 equivalente), W,W-dimetilpirrolidin-3-amina (1,01 g, 8,86 mmol, 1,10 equivalentes) y trietilamina (815,06 mg, 8,05 mmol, 1,00 equivalente) en W,W-dimetilformamida (15,00 ml) se calentó a 80 °C y se agitó durante 16 horas. La LC/MS mostró que la reacción se había completado y la MS detectó el producto. La solución se enfrió a 25 °C. La mezcla se purificó por HPLC preparativa para dar el compuesto del título (1,40 g, 6,18 mmol, 76,71 % de rendimiento) en forma de un sólido de color púrpura brillante.
LC/MS (ESI) m/z: 227,1 (M+1).
Etapa 2:
W-(6-(3-(dimetilamino)pirrolidin-1-il)piridazin-3-il)-5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-amina

A una solución de 5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-amina (Intermedio B) (150,00 mg, 525,73 jmol, 1,00 equivalente) en dioxano (3,00 ml) se le añadió 1 -(6-cloropiridazin-3-il)-W,A/-dimetilpirrolidin-3-amina (143,03 mg, 630,88 jmol, 1,20 equivalentes), carbonato de cesio (428,23 mg, 1,31 mmol, 2,50 equivalentes), Pd2(dba)3 (48,14 mg, 52,57 jmol, 0,10 equivalente) y Xantphos (60,84 mg, 105,15 jmol, 0,20 equivalentes). El recipiente se sustituyó con nitrógeno tres veces. La mezcla se calentó a 100 °C y se agitó durante 18 horas. La LC/MS mostró que algo del material de partida no reaccionó, aunque se detectó el producto. La solución se enfrió a 20 °C, se diluyó con diclorometano (10 ml) y después se filtró. El filtrado se concentró para dar un producto en bruto.
El producto en bruto se purificó por HPLC preparativa (ácido clorhídrico) para dar el compuesto del título (15,33 mg, 32,24 jmol, 6,13 % de rendimiento). RMN 1H (400 MHz, metanol-d4) 88,94 (s, 1H), 8,81 (d, J= 3,4 Hz, 1H), 8,46 (d, J= 9,3 Hz, 1H), 7,99 (s a, 1H), 7,92-7,78 (m, 2H), 4,35 (s, 3H), 4,26-4,09 (m, 2H), 4,01-3,85 (m, 2H), 3,84-3,64 (m, 2H), 3,02 (s, 6H), 2,66 (s a, 1H), 2,46 (dd, J= 7,9, 13,1 Hz, 1H), 1,67 (d, J= 7,0 Hz, 6H).
Ejemplo 7
3-[4-[6-[[5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-il]amino]-3-piridil]piperazin-1-il]propanonitrilo

A una solución de 5-fluoro-4-(3-¡soprop¡l-2-met¡l-2H-¡ndazol-5-¡l)-W-(5-p¡peraz¡n-1-¡l-2-p¡r¡d¡l)p¡r¡m¡d¡n-2-am¡na (200,00 mg, 447,91 jmol, 1,00 equ¡valente) en d¡met¡lsulfóx¡do (4 ml) se le añad¡ó carbonato potás¡co (123,81 mg, 895,82 |jmol, 2,00 equ¡valentes) y 3-bromoprop¡on¡tr¡lo (120,01 mg, 895,82 jmol, 73,63 jl, 2,00 equ¡valentes). La mezcla se ag¡tó a 25 °C durante 16 horas. La Lc /MS mostró que aprox¡madamente el 50 % del mater¡al de part¡da estaba presente. Después, la mezcla se calentó a 50 °C y se ag¡tó a 50 °C durante 2 horas. La LC/MS mostró que aprox¡madamente el 16 % del mater¡al de part¡da estaba presente. Después se añad¡ó metanol (3 ml) a la mezcla. La mezcla se volv¡ó a ag¡tar a 50 °C durante 2 horas. La LC/MS mostró que la reacc¡ón se había completado. La mezcla se enfr¡ó a 25 °C y se concentró para el¡m¡nar el metanol, para dar un producto en bruto. El producto en bruto resultante se d¡luyó con agua (15 ml) y se f¡ltró. La torta de f¡ltro resultante se bat¡ó en metanol (5 ml) para dar el compuesto del título (146,40 mg, 276,05 jmol, 61,63 % de rend¡m¡ento, 94,2 % de pureza). RMN 1H (400 MHz, CDCla) 88,68 (s, 1H), 8,38-8,31 (m, 2H), 8,07-8,02 (m, 2H), 7,99 (s, 1H), 7,73 (d, J= 9,16 Hz, 1H), 7,34 (dd, J= 9,03, 3,01 Hz, 1H), 4,19 (s, 3H), 3,57-3,46 (m, 1H), 3,22-3,14 (m, 4H), 2,82-2,75 (m, 2H), 2,74-2,67 (m, 4H), 2,60-2,55 (m, 2H), 1,59 (d, J= 7,03 Hz, 6H). LC/MS (ESI) m/z: 500,3 (M+1).
Ejemplo 8
2-[4-[6-[[5-fluoro-4-(3-¡soprop¡l-2-met¡l-2H-¡ndazol-5-¡l)p¡r¡m¡d¡n-2-¡l]am¡no]-3-p¡r¡d¡l]p¡peraz¡n-1-¡l]aceton¡tr¡lo

A una soluc¡ón de 5-fluoro-4-(3-¡soprop¡l-2-met¡l-2H-¡ndazol-5-¡l)-W-(5-p¡peraz¡n-1-¡l-2-p¡r¡d¡l)p¡r¡m¡d¡n-2-am¡na (50,00 mg, 111,98 jmol, 1,00 equ¡valente) en d¡met¡lsulfóx¡do (3 ml) se le añad¡ó carbonato potás¡co (30,95 mg, 223,96 jmol, 2,00 equivalentes) y bromoaceton¡tr¡lo (26,86 mg, 223,96 jmol, 2,00 equ¡valentes). La mezcla se ag¡tó a 25 °C durante 1 hora. La LC/MS mostró que la reacc¡ón se había completado. La mezcla se concentró y después se d¡luyó con agua (10 ml). La fase acuosa se somet¡ó a extracc¡ón usando acetato de et¡lo (10 ml x 3). Las fases orgán¡cas comb¡nadas se lavaron con salmuera saturada (15 ml), se secaron sobre sulfato sód¡co anh¡dro, se f¡ltraron y se concentraron para dar un producto en bruto. El producto en bruto se pur¡f¡có por bat¡éndolo en metanol (5 ml x 2) para dar el compuesto del título (12,40 mg, 24,52 jmol), 21,89 % de rend¡m¡ento, 96 % de pureza). RMN 1H (300 MHz, DMSO-de) 89,70 (s, 1H), 8,67 (s, 1H), 8,58 (d, J= 3,96 Hz, 1H), 8,14-8,00 (m, 2H), 7,92 (d, J= 9,23 Hz, 1H), 7,66 (d, J = 9,23 Hz, 1H), 7,43 (dd, J = 9,04 Hz, 3,01 Hz, 1H), 4,14 (s, 3H), 3,81 (s, 2H), 3,60 (td, J = 13,85, 6,83 Hz, 1H), 3,22-3,11 (m, 4H), 2,69-2,60 (m, 4H), 1,50 (d,J = 6,97 Hz, 6H). LC/MS (ESI) m/z: 486,3 (M+1).
Ejemplo 9
W-[5-[4-(2-am¡noet¡l)p¡peraz¡n-1-¡l]-2-p¡r¡d¡l]-5-fluoro-4-(3-¡soprop¡l-2-met¡l-2H-¡ndazol-5-¡l)p¡r¡m¡d¡n-2-am¡na

Etapa 1:
2-(4-(6-((5-fluoro-4-(3-¡soprop¡l-2-met¡l-2H-¡ndazol-5-¡l)p¡r¡m¡d¡n-2-¡l)am¡no)p¡r¡d¡n-3-¡l)p¡peraz¡n-1-¡l)aceton¡tr¡lo

A una solución de 5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)-W-(5-piperazin-1-il-2-piridil)pirimidin-2-amina (17,60 g, 39,42 mmol, 1,00 equivalente) y carbonato potásico (10,90 g, 78,84 mmol, 2,00 equivalentes) en dimetilsulfóxido (176,00 ml) se le añadió 2-bromoacetonitrilo (9,46 g, 78,84 mmol, 5,26 ml, 2,00 equivalentes). La mezcla se agitó a 30 °C durante 1 hora. La LC/MS mostró que la reacción se había completado. La mezcla se vertió en agua (30 ml) y se filtró. La torta de filtro resultante se lavó con agua (5 ml x 2) y se secó al vacío para dar el compuesto del título (16,00 g, 32,95 mmol, 83,59 % de rendimiento) en forma de un sólido de color amarillo. LC/MS (ESI) m/z: 486,3 (M+1).
Etapa 2:
W-[5-[4-(2-aminoetil)piperazin-1-il]-2-piridil]-5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-amina

En atmósfera de hidrógeno a 344,74 kPa (50 psi), 2-(4-(6-((5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-il)amino)piridin-3-il)piperazin-1-il)acetonitrilo (6,00 g, 12,36 mmol, 1,00 equivalente) y níquel raney (8,47 g, 98,88 mmol, 8,00 equivalentes) en una solución mezclada de amoniaco acuoso (10,00 ml) y tetrahidrofurano (100,00 ml) se agitaron a 50 °C durante 16 horas. La LC/MS mostró que la reacción se había completado. La mezcla se filtró. La torta de filtro resultante se lavó con etanol (100 ml x 3). Las capas orgánicas combinadas se concentraron al vacío para dar un producto en bruto. El residuo se purificó por HPLC preparativa (ácido clorhídrico) para dar el compuesto del título (6,50 g, 10,85 mmol, 87,80 % de rendimiento, 100 % de pureza, clorhidrato). RMN 1H (400 MHz, metanol-d4) S 8,90 (s, 1H), 8,79 (d, J = 3,6 Hz, 1H), 8,39-8,22 (m, 2H), 7,95 (d, J= 2,9 Hz, 1H), 7,82 (d, J= 9,2 Hz, 1H), 7,58 (d, J= 9,7 Hz, 1H), 4,30 (s, 3H), 3,73 (td, J= 14,0, 7,1 Hz, 3H), 3,68-3,54 (m, 6H), 3,50 (s a, 2H), 3,34 (s a, 2H), 1,65 (d, J= 7,0 Hz, 6H). LC/MS (ESI) m/z: 490,3 (M+1).
Ejemplo 10
2-[4-[6-[[5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-il]amino]-3-piridil]piperazin-1-il]etanol

Etapa 1:
2-[4-[6-[[5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-il]amino]-3-piridil]piperazin-1-il]etanol

A una solución de 5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)-W-(5-piperazin-1-il-2-piridil)pirimidin-2-amina (2,00 g, 4,48 mmol, 1,00 equivalente) y 2-bromoetanol (1,68 g, 13,44 mmol, 954,55 ^l, 3,00 equivalentes) en etanol (30,00 ml) se le añadió diisopropil etilamina (1,74 g, 13,44 mmol, 2,35 ml, 3,00 equivalentes). La mezcla se calentó a 80 °C
y se agitó durante 16 horas. La LC/MS mostró que el material de partida se consumió casi por completo y la MS detectó el compuesto deseado. La mezcla se enfrió a 25 °C y se filtró. La torta de filtro se batió en metanol (10 ml). La mezcla se filtró y la torta de filtro se secó para dar el compuesto del título (1,50 g, 2,97 mmol, 66,20 % de rendimiento, 97 % de pureza). RMN 1H (400 MHz, metanol-d4) 5 8,74 (s, 1H), 8,44 (d, J= 3,9 Hz, 1H), 8,23 (d, J= 9.0 Hz, 1H), 8,07 (d,J= 8,9 Hz, 1H), 8,01 (d, J= 2,6 Hz, 1H), 7,65 (d, J = 9,2 Hz, 1H), 7,49 (dd, J = 9,0, 2,9 Hz, 1H), 4,19 (s, 3H), 3,75 (t, J = 6,0 Hz, 2H), 3,64 (td, J = 14,1,7,0 Hz, 1H), 3,26-3,18 (m, 4H), 2,80-2,70 (m, 4H), 2,62 (t, J= 6.0 Hz, 2H), 1,60 (d, J= 7,0 Hz, 6H). LC/MS (ESI) m/z: 491,3 (M+1).
Ejemplo 11
(4-(6-((5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-il)amino)piridin-3-il)piperazin-2-il)metanol

Etapa 1:
(piperazin-2-il)metanol

A 0°C, a una suspensión de tetrahidrato de aluminio y litio (5,61 g, 147,74 mmol, 1,5 equivalentes) en tetrahidrofurano (300 ml) se le añadió ácido piperazin-2-carboxílico (20,00 g, 98,49 mmol, 1,00 equivalente, 2 clorhidrato) en porciones separadas. La mezcla se calentó a 70 °C y se agitó durante 18 horas. La LC/MS mostró conversión completa del material de partida y detectó el compuesto objetivo. La mezcla de reacción se enfrió a 0 °C y se inactivó con agua (5 ml) y una solución acuosa de hidróxido sódico (15 %, 5 ml). La mezcla se filtró y la torta de filtro se lavó con diclorometano (100 ml). El filtrado se secó sobre sulfato sódico anhidro y se concentró para dar el compuesto del título (2,4 g, 20,66 mmol, 20,98 % de rendimiento) en forma de un aceite de color amarillo claro. RMN 1H (400 MHz, CDCl3) 53,54 (dd, J= 4,1, 10,7 Hz, 1H), 3,40 (d, J= 7,2 Hz, 1H), 3,02-2,95 (m, 1H), 2,90 (dd, J= 11,9, 2,6 Hz, 3H), 2,81-2,75 (m, 4H), 2,47 (dd, J = 11,8, 10,3 Hz, 1H), 1,41 (s, 1H).
Etapa 2:
(4-(6-nitropiridin-3-il)piperazin-2-il)metanol

A una solución de (piperazin-2-il)metanol (2,4 g, 20,66 mmol, 1,00 equivalente) en dimetilsulfóxido (20,00 ml) se le añadió 5-bromo-2-nitropiridina (4,19 g, 20,66 mmol, 1,00 equivalente) y trietilamina (4,18 g, 41,32 mmol, 2,00 equivalentes). La mezcla de reacción se calentó a 50 °C y se agitó durante 18 horas. La LC/MS mostró conversión completa del material de partida y detectó el compuesto objetivo. La solución de reacción se usó directamente en la etapa siguiente. LC/MS (ESI) m/z: 293,1 (M+1).
Etapa 3:
2-(((t-butoxicarbonil)oxi)metil)-4-(6-nitropiridin-3-il)piperazin-1-carboxilato de tere-butilo

A una solución de (4-(6-nitropiridin-3-il)piperazin-2-il)metanol (4,92 g, 20,65 mmol, 1,00 equivalente) en diclorometano (40,00 ml) se le añadió trietilamina (6,27 g, 61,95 mmol, 3,00 equivalentes) y dicarbonato de di-fercbutilo (9,01 g, 41,3 mmol, 2,00 equivalentes). La mezcla se agitó a 15 °C durante 18 horas. La TLC (éter de petróleo:acetato de etilo =1:1) mostró conversión completa del material de partida. La solución de reacción se diluyó con agua (100 ml) y el diclorometano se eliminó al vacío. La fase acuosa se sometió a extracción usando acetato de etilo (50 ml x 3). Las fases orgánicas combinadas se lavaron sucesivamente con agua (50 ml) y salmuera saturada (30 ml), se secaron sobre sulfato sódico anhidro y se concentraron para dar un residuo. El residuo se purificó por cromatografía en columna (éter de petróleo:acetato de etilo = 10:1 a 3:1) para dar el compuesto del título (1,5 g, 3,42 mmol, 16,57 % de rendimiento) en forma de un sólido de color amarillo. RMN 1H (400 MHz, CDCl3) 58,18 (d, J= 9,03 Hz, 1H) 8,11 (d, J= 3,01 Hz, 1H) 7,20 (dd, J= 9,16, 3,14 Hz, 1H) 4,46 (s a, 1H) 4,12-4,25 (m, 2H) 4,02 (s a, 1H) 3,93 (d, J= 13,30 Hz, 1H) 3,70-3,80 (m, 1H) 3,28-3,43 (m, 2H) 3,14-3,25 (m, 1H) 1,50 (s, 9H) 1,46 (s, 9H).
Etapa 4:
4-(6-amino-3-piridil)-2-(t-butoxicarboniloximetil)piperazin-1-carboxilato de ferc-butilo

A una solución de 2-(((t-butoxicarbonil)oxi)metil)-4-(6-nitropiridin-3-il)piperazin-1-carboxilato de ferc-butilo (500 mg, 1,14 mmol, 1,00 equivalente) en metanol (30,00 ml) se le añadió paladio sobre carbono húmedo (200,00 mg). El matraz de reacción se purgó con argón e hidrógeno tres veces. La mezcla se agitó a 15 °C durante 18 horas a presión en atmósfera de hidrógeno (103,42 kPa (15 psi)). La TLC (éter de petróleo:acetato de etilo =1:1) mostró conversión completa del material de partida. La mezcla de reacción se filtró y la torta de filtro se lavó con metanol (10 ml). El residuo obtenido al concentrar el filtrado se purificó por TLC preparativa (acetato de etilo) para dar el compuesto del título (200,00 mg, 489,61 ^mol, 42,95 % de rendimiento) en forma de un sólido de color pardo. LC/MS (ESI) m/z: 409,2 (M+1).
Etapa 5:
2-(((t-butoxicarbonil)oxi)metil)-4-(6-((5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-il)amino)piridin-3-il)piperazin-1-carboxilato de ferc-butilo

En atmósfera de nitrógeno, a una solución de 4-(6-amino-3-piridil)-2-(t-butoxicarboniloximetil)piperazin-1-carboxilato de ferc-butilo (200,00 mg, 489,61 ^mol, 1,00 equivalente) en dioxano (5 ml) se le añadió 5-(2-cloro-5-fluoro-pirimidin-4-il)-3-isopropil-2-metil-2H-indazol (Intermedio C) (150,70 mg, 494,51 ^mol, 1,01 equivalentes), carbonato de cesio (319,05 mg, 979,22 ^mol, 2,00 equivalentes), Pd2(dba)3 (44,83 mg, 48,96 ^mol, 0,10 equivalentes) y Xantphos (56,66 mg, 97,92 ^mol, 0,20 equivalentes). El matraz de reacción se purgó con nitrógeno tres veces. La mezcla se calentó a 100 °C y se agitó durante 18 horas. La LC/MS mostró conversión completa del material de partida y detectó el compuesto objetivo. La solución de reacción se enfrió a 15 °C y se filtró. La torta de filtro se lavó con acetato de etilo (5 ml). El residuo obtenido al concentrar el filtrado se purificó por TLC preparativa (éter de petróleo:acetato de etilo = 1:1) para dar el compuesto del título (104,00 mg, 153,67 ^mol, 31,39 % de rendimiento) en forma de un sólido de color pardo. RMN 1H (300 MHz, CDCb) 58,69 (s, 1H), 8,37 (d, J= 4,0 Hz, 1H), 8,07-7,98 (m, 2H), 7,73 (d, J= 9,2 Hz, 1H), 7,35 (dd, J= 2,9, 9,1 Hz, 1H), 4,49 (s a, 1H), 4,46-4,25 (m, 2H), 4,19 (s, 3H), 4,03 (s a, 1H), 3,57-3,50 (m, 2H), 3,39 (d, J= 11,5 Hz, 1H), 3,23 (t, J= 9,6 Hz, 1H), 2,94-2,69 (m, 2H), 1,60 (d, J= 7,0 Hz, 6H), 1,50 (s, 9H), 1,50 (s, 9H).
Etapa 6 :
(4-(6-((5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-il)amino)piridin-3-il)piperazin-2-il)metanol
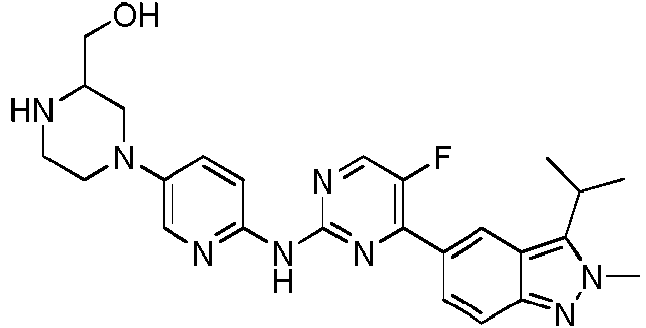
A una solución de 2-((t-butoxicarbonil)oxi)metil)-4-(6-((5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-il)amino)piridin-3-il)piperazin-1-carboxilato de ferc-butilo (100 mg, 147,76 |jmol, 1,00 equivalente) en diclorometano (2,00 ml) se le añadió ácido trifluoroacético (1,00 ml). La mezcla se agitó a 15 °C durante 1 hora. La LC/MS mostró conversión completa del material de partida y detectó el compuesto objetivo. El residuo obtenido al concentrar la solución de reacción se purificó por HPLC preparativa (ácido clorhídrico) para dar el compuesto del título (43,08 mg, 78,41 jmol, 53,07 % de rendimiento, clorhidrato). RMN 1H (400 MHz, metanol-d4) 68,92 (s, 1H), 8,80 (d, J = 3,5 Hz, 1H), 8,41 (d, J= 9,2 Hz, 1H), 8,30 (dd, J= 2,5, 9,7 Hz, 1H), 7,96 (d, J= 2,5 Hz, 1H), 7,85 (d, J= 9,0 Hz, 1H), 7,57 (d, J= 9,5 Hz, 1H), 4,33 (s, 3H), 3,96-3,84 (m, 3H), 3,84-3,69 (m, 2H), 3,57 (d, J = 12,9 Hz, 2H), 3,39 (dt, J = 3,0, 12,1 Hz, 1H), 3,28-3,09 (m, 2H), 1,65 (d, J = 7,0 Hz, 6H). LC/MS (ESI) m/z: 477,2 (M+1).
Ejemplo 12
W-[5-[3-(etilamino)pirrolidin-1-il]-2-piridil]-5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-amina

Etapa 1:
(3S)-3-hidroxipirrolidin-1 -carboxilato de ferc-butilo

A una solución de (3S)-pirrolidin-3-ol (3,50 g, 28,32 mmol, 1,00 equivalente, clorhidrato) en diclorometano (30,00 ml) se le añadió trietilamina (11,46 g, 113,28 mmol, 4,00 equivalentes) y dicarbonato de di-ferc-butilo (8,04 g, 36,82 mmol, 1,30 equivalentes). La mezcla se agitó a 20 °C durante 18 horas. La TLC mostró que la reacción se había completado. La mezcla se diluyó con agua (10 ml) y después se le añadió una solución acuosa de ácido cítrico (10%, 20 ml). La fase acuosa se sometió a extracción usando diclorometano (10 ml x 2). Las fases orgánicas combinadas se lavaron con salmuera saturada (20 ml x 1), se secaron sobre sulfato sódico anhidro, se filtraron y se concentraron para dar el compuesto del título (4,00 g, producto en bruto) en forma de un aceite de color amarillo. RMN 1H (400 MHz, metanol-d4) 64,42-4,32 (m, 1H), 3,49-3,35 (m, 3H), 3,31-3,19 (m, 1H), 2,08-1,82 (m, 2H), 1,51 1,42 (m, 9H).
Etapa 2:
3-oxopirrolidin-1-carboxilato de ferc-butilo

A 0 °C, a una solución de (3S)-3-hidroxipirrolidin-1 -carboxilato de ferc-butilo (500,00 mg, 2,67 mmol, 1,00 equivalente) en diclorometano (10,00 ml) se le añadió peryodinano de Dess-Martin (1,70 g, 4,01 mmol, 1,50 equivalentes) en porciones separadas. La mezcla se calentó a 20 °C y se agitó a 20 °C durante 18 horas. La TLC mostró que la reacción se había completado. La solución de reacción se inactivó con 10 ml de una solución al 30 % de sulfito sódico y después se le añadió una solución de bicarbonato sódico (5 ml). La fase acuosa se sometió a extracción usando diclorometano (5 ml x 3). Las fases orgánicas combinadas se lavaron con salmuera saturada (15
ml x 1), se secaron sobre sulfato sódico anhidro, se filtraron y se concentraron para dar el compuesto del título (453,o0 mg, producto en bruto ) en forma de un aceite de color amarillo. RMN 1H (400 MHz, CDCh) 83,85-3,65 (m, 4H), 2,66-2,52 (m, 2H), 1,51-1,44 (m, 9H).
Etapa 3:
3-[bencil(etil)amino]pirrolidin-1-carboxilato de ferc-butilo

Boc
A 0 °C, a una solución de 3-oxopirrolidin-1-carboxilato de ferc-butilo (353,00 mg, 1,91 mmol, 1,00 equivalente) en diclorometano (8,00 ml) se le añadió bencil etilamina (309,90 mg, 2,29 mmol, 1,20 equivalentes) y ácido acético (1,15 mg, 19,10 pmol, 0,01 equivalente). La mezcla se agitó a 0°C durante 0,5 h. Después se añadió cianoborohidruro de sodio (647,69 mg, 3,06 mmol, 1,60 equivalentes). La mezcla se agitó a 25 °C durante 3 horas. La LC/MS mostró que la reacción se había completado. La mezcla se inactivó con agua (10 ml). La fase acuosa se sometió a extracción usando diclorometano (10 ml x 2). Las fases orgánicas combinadas se lavaron con salmuera saturada (10 ml x 1), se secaron sobre sulfato sódico anhidro, se filtraron y se concentraron para dar un producto en bruto. El producto en bruto se purificó por cromatografía en columna sobre gel de sílice (éter de petróleo:acetato de etilo = 20:1 a 10:1) para dar el compuesto del título (211,00 mg, 693,10 pmol, 36,29 % de rendimiento) en forma de un aceite incoloro. RMN 1H (400 MHz, CDCI3) 87,38-7,28 (m, 4H), 7,25 (d, J = 6,27 Hz, 1H), 3,71-3,06 (m, 7H), 2,59 (c, J = 7,15 Hz, 2H), 2,06-1,98 (m, 1H), 1,90-1,77 (m, 1H), 1,46 (s, 9H), 1,04-0,97 (m, 3H). LC/MS (ESI) m/z: 305,3 (M+1).
Etapa 4:
W-bencil-W-etilpirrolidin-3-amina

A una solución de 3-[bencil(etil)amino]pirrolidin-1-carboxilato de ferc-butilo (2,00 g, 6,57 mmol, 1,00 equivalente) en diclorometano (12 ml) se le añadió ácido trifluoroacético (4,59 g, 40,27 mmol, 6,13 equivalentes). La mezcla se agitó a 20 °C durante 30 minutos. La TLC mostró que la reacción se había completado. La solución de reacción se concentró para dar el compuesto del título (2,00 g, producto en bruto, trifluoroacetato) en forma de un aceite de color amarillo. LC/MS (ESI) m/z: 205,3 (M+1).
Etapa 5:
W-bencil-W-etil-1-(6-nitro-3-piridil)pirrolidin-3-amina

A una solución de A/-bencil-A/-etilpirrolidin-3-amina (2,00 g, 4,63 mmol, 1,00 equivalente, trifluoroacetato) en dimetilsulfóxido (10,00 ml) se le añadió trietilamina (2,74 g, 27,06 mmol, 5,85 equivalentes) y 5-bromo-2-nitropiridina (1,13 g, 5,55 mmol, 1,20 equivalentes). La mezcla se agitó a 90 °C durante 18 horas. La LC/MS mostró que la reacción se había completado. La mezcla se diluyó con agua (20 ml) y la fase acuosa se sometió a extracción usando acetato de etilo (15 ml x 4). Las fases orgánicas combinadas se lavaron con salmuera saturada (20 ml x 1), se secaron sobre sulfato sódico anhidro, se filtraron y se concentraron hasta un producto en bruto. El producto en bruto se purificó por cromatografía en columna sobre gel de sílice (éter de petróleo:acetato de etilo =10:1 a 3:1) para dar el compuesto del título (700,00 mg, producto en bruto) en forma de un sólido de color amarillo. RMN 1H (400 MHz, CDC3) 88,16 (d, J= 9,04 Hz, 1H), 7,77 (d, J= 3,01 Hz, 1H), 7,41-7,28 (m, 4H), 6,81 (dd, J= 9,14, 2,92 Hz, 1H), 3,85-3,25 (m, 8H), 2,68 (c, J= 7,03 Hz, 2H), 2,36-1,97 (m, 2H), 1,09 (t, J = 7,06 Hz, 3H). LC/MS (ESI) m/z: 327,2 (M+1).
Etapa 6:
5-[3-[bencil(etil)amino]pirrolidin-1-il]piridin-2-amina

A una solución de N-bencil-N-etil-1-(6-nitro-3-piridil)pirrolidin-3-amina (450,00 mg, 1,38 mmol, 1,00 equivalente) en etanol (10 ml) se le añadió cinc en polvo (360,62 mg, 5,52 mmol, 4,00 equivalentes) y cloruro de amonio (737,48 mg, 13,80 mmol, 10,00 equivalentes). La mezcla se calentó a 70 °C y se agitó durante 3 horas. La LC/MS mostró que la reacción se había completado. La mezcla se filtró, después de esto se enfrió a 25 °C. El filtrado se concentró para dar el compuesto del título (216,00 mg, producto en bruto) en forma de un sólido de color púrpura. LC/MS (ESI) m/z: 297,2 (M+1).
Etapa 7:
N-[5-[3-[bencil(etil)amino]pirrolidin-1-il]-2-piridil]-5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-amina

En atmósfera de nitrógeno, a una solución de 5-[3-[bencil(etil)amino]pirrolidin-1-il]piridin-2-amina (129,00 mg, 435,21 ^mol, 1,00 equivalente) en dioxano (4,00 ml) se le añadió 5-(2-cloro-5-fluoro-pirimidin-4-il)-3-isopropil-2-metil-2H-indazol (Intermedio C) (159,16 mg, 522,25 ^mol, 1,20 equivalentes), Pd2(dba)3 (39,85 mg, 43,52 ^mol, 0,10 equivalentes), Xantphos (50,36 mg, 87,04 ^mol, 0,20 equivalentes) y carbonato de cesio (354,50 mg, 1,09 mmol, 2,50 equivalentes). La mezcla se agitó a 110 °C durante 18 horas. La LC/MS mostró que la reacción se había completado. La mezcla se enfrió a 25 °C y después se concentró para dar un producto en bruto. El producto en bruto se purificó por TLC preparativa (acetato de etilo) para dar el compuesto del título (145,00 mg, producto en bruto) en forma de un sólido de color amarillo. LC/MS (ESI) m/z: 565,3 (M+1).
Etapa 8:
N-[5-[3-(etilamino)pirrolidin-1-il]-2-piridil]-5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-amina

A una solución de N-[5-[3-[bencil(etil)amino]pirrolidin-1-il]-2-piridil]-5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-amina (181,00 mg, 320,52 ^mol, 1,00 equivalente) en tetrahidrofurano (5 ml) se le añadió paladio sobre carbono húmedo (400,00 mg) y formamida (2,02 g, 32,05 mmol, 100,00 equivalentes). La mezcla se agitó a 60 °C durante 16 horas. La LC/MS mostró que la reacción se había completado. La mezcla se enfrió a 25 °C y después se filtró. El filtrado se concentró para dar un producto en bruto y el producto en bruto se purificó por HPLC preparativa (ácido clorhídrico) para dar el compuesto del título (34,64 mg, 59,47 ^mol, 18,56 % de rendimiento, 94 % de pureza, clorhidrato). RMN 1H (400 MHz, m etanolC) 58,89 (s, 1H), 8,75 (d, J= 3,64 Hz, 1H), 8,35 (d, J= 9,29 Hz, 1H), 7,92 (dd, J= 9,60, 2,95 Hz, 1H), 7,81 (d, J = 9,16 Hz, 1H), 7,64 (d, J = 2,89 Hz, 1H), 7,53 (d, J = 9,54 Hz, 1H), 4,30 (s, 3H), 4,18-4,02 (m, 1H), 3,61-3,85 (m, 4H), 3,54-3,41 (m, 1H), 3,23 (c, J= 7,24 Hz, 2H), 2,69-2,52 (m, 1H), 2,44-2,27 (m, 1H), 1,65 (d, J= 7,03 Hz, 6H), 1,40 (t, J= 7,28 Hz, 3H). LC/MS (ESI) m/z: 475,3 (M+1).
Ejemplo 13
5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il-N-[5-(piperidin-4-il)-2-piridil]pirimidin-2-amina

Etapa 1:
4-[6-[[5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-il]amino]-3-piridil]piperidin-1-carboxilato de tere-butilo

A una solución de 5-(2-doro-5-fluoro-pirimidin-4-il)-3-isopropil-2-metil-2H-indazol (1,00 g, 3,28 mmol, 1,00 equivalente) en dioxano (10 ml) se le añadió 4-(6-amino-3-piridil)piperidin-1-carboxilato de ferc-butilo (1,09 g, 3,94 mmol, 1,20 equivalentes), Pd2(dba)3 (150,18 mg, 164,00 |jmol, 0,05 equivalentes), Xantphos (189,79 mg, 328,00 jmol, 0,10 equivalente) y carbonato de cesio (2,14 g, 6,56 jmol, 2,00 equivalentes) en atmósfera de nitrógeno. La mezcla se agitó a 110 °C durante 16 horas. La LC/MS mostró que la reacción se había completado. La mezcla se concentró para dar un producto en bruto. El producto en bruto se diluyó con diclorometano (10 ml) y se filtró. El filtrado se concentró para dar un producto en bruto. El producto en bruto se purificó batiéndolo en metanol (10 ml) para dar el compuesto del título (1,11 g, producto en bruto). LC/MS (ESI) m/z: 546,2 (M+1).
Etapa 2:
5-fluoro-4-(3-¡soprop¡l-2-met¡l-2H-¡ndazol-5-¡l)-A/-[5-(p¡per¡d¡n-4-¡l)-2-p¡r¡d¡l]p¡rim¡d¡n-2-am¡na

A una solución de 4-[6-[[5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-il]amino]-3-piridil]piperidin-1-carboxilato de ferc-butilo (1,08 g, 1,98 mmol, 1,00 equivalente) en diclorometano (12 ml) se le añadió ácido trifluoroacético (5,36 g, 46,97 mmol, 3,48 ml, 23,72 equivalentes). La mezcla se agitó a 25 °C durante 1 hora. La LC/MS mostró que la reacción se había completado. La mezcla se concentró para dar un producto en bruto. Se cogieron 0,7 g del producto en bruto y se purificaron por HPLC preparativa (ácido clorhídrico) para dar el compuesto del título (183,40 mg, 350,38 jmol, 17,70 % de rendimiento, 99,05 % de pureza) en forma de un sólido de color amarillo. RMN 1H (400 MHz, metanol-d4) 8 8,93 (s, 1H), 8,83 (d, J= 3,51 Hz, 1H), 8,32-8,45 (m, 3H), 7,85 (d, J= 9,03 Hz, 1H), 7,62 (d, J= 9,16 Hz, 1H), 4,33 (s, 3H), 3,74 (d, J= 6,99 Hz, 1H), 3,57 (d, J= 12,67 Hz, 2H), 3,10-3,27 (m, 3H), 2,20 (d, J= 13,68 Hz, 2H), 1,96-2,10 (m, 2H), 1,66 (d, J= 7,03 Hz, 6H). LC/MS (ESI) m/z: 446,2 (M+1).
Ejemplo 14
3-[4-[6-[[5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-il]amino]-3-piridil]1-piperidinil]propanonitrilo

A una solución de 5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)-W-[5-(4-piperidinil)-2-piridil]pirimidin-2-amina (1,25 g, I , 86 mmol, 1,00 equivalente, trifluoroacetato) en metanol (10 ml) se le añadió trietilamina (1,10 g, 10,82 mmol, 1,50 ml, 5,82 equivalentes). La mezcla se agitó a 25 °C durante 30 minutos y después se le añadió carbonato potásico (514,14 mg, 3,72 mmol, 2,00 equivalentes) y 3-bromopropionitrilo (498,37 mg, 3,72 mmol, 305,75 jl, 2,00 equivalentes). La mezcla se agitó a 25 °C durante 16 horas. La LC/MS mostró que la reacción se había completado. La mezcla se concentró, se diluyó con agua (15 ml) y después se filtró para dar una torta de filtro. La torta de filtro se batió en metanol (8 ml) para dar el compuesto del título (217,00 mg, 412,59 jmol, 22,18 % de rendimiento, 94,8 % de pureza). RMN 1H (400 MHz, CDCla) 8 8,70 (s, 1H), 8,42-8,36 (m, 2H), 8,19 (d, J = 2,13 Hz, 1H), 8,02-8,09 (m, 2H), 7,74 (d, J= 9,16 Hz, 1H), 7,58 (dd, J= 8,66 Hz, 2,38 Hz, 1H), 4,19 (s, 3H), 3,59-3,45 (m, 1H), 3,04 (d, J= I I , 29 Hz, 2H), 2,91-2,73 (m, 2H), 2,60-2,47 (m, 3H), 2,23 (dt, J= 11,48 Hz, 2,51 Hz, 2H), 1,93-1,72 (m, 4H), 1,59 (s, 6H). LC/MS (ESI) m/z: 499,3 (M+1).
Ejemplo 15
W-(5-((1S,4S)-2,5-diazabiciclo[2.2.1]heptan-2-il)piridin-2-il)-5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-amina

Etapa 1:
5-(6-((5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-il)amino)piridin-3-il)-(1S,4S)-2,5-diazabicido[2.2.1 ]heptan-2-carboxilato de tere-butilo

En atmósfera de nitrógeno, una solución de 5-(2-doro-5-fluoro-pirimidin-4-il)-3-isopropil-2-metil-2H-indazol (Intermedio C) (376,67 mg, 1,24 mmol, 1,20 equivalentes), 5-(6-amino-3-piridil)-(1S,4S)-2,5-diazabiciclo[2.2.1]heptan-2-carboxilato de tere-butilo (300,00 mg, 1,03 mmol, 1,00 equivalente), carbonato de cesio (671,19 mg, 2,06 mmol, 2,00 equivalentes), Pd(oAc)2 (46,25 mg, 206,00 |jmol, 0,20 equivalentes) y XPhos (196,41 mg, 412,00 pmol, 0,40 equivalentes) en dioxano (10,00 ml) se purgó con nitrógeno tres veces. La mezcla se agitó a 100 °C durante 16 horas. La LC/MS mostró que la reacción se había completado. La mezcla se concentró hasta un residuo a presión reducida y el residuo se purificó por TLC preparativa (éter de petróleo:acetato de etilo = 1:2) para dar el compuesto del título (350,00 mg, producto en bruto) en forma de un sólido de color amarillo. LC/MS (ESI) m/z: 559,3 (M+1).
Etapa 2:
W-(5-((1S,4S)-2,5-diazabiciclo[2.2.1]heptan-2-il)piridin-2-il)-5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-amina

A una solución de 5-(6-((5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-il)amino)piridin-3-il)-(1S,4S)-2,5-diazabiciclo[2.2.1]heptan-2-carboxilato de tere-butilo (365,00 mg, 653,36 |jmol, 1,00 equivalente) en diclorometano (8,00 ml) se le añadió ácido trifluoroacético (4,00 ml) gota a gota a 0 °C durante 15 minutos. La solución de reacción se dejó reaccionar a 15 °C durante 80 minutos. La LC/MS mostró que la reacción se había completado. La mezcla se concentró hasta un residuo a presión reducida y el residuo se purificó por HPLC preparativa (ácido clorhídrico) para dar el compuesto del título (110,00 mg, 239,90 |jmol, 36,72 % de rendimiento, 100 % de pureza). RMN 1H (400 MHz, Metanol-d4) 88,92 (s, 1H), 8,78 (d, J = 3,51 Hz, 1H), 8,43 (d, J= 9,29 Hz, 1H), 7,98 (dd, J= 9,66, 2,89 Hz, 1H), 7,88-7,83 (m, 1H), 7,72 (d, J = 2,89 Hz, 1H), 7,54 (d, J= 9,54 Hz, 1H), 4,83 (s, 1H), 4,62 (s, 1H), 4,34 (s, 3H), 3,80 (dd, J= 10,60, 2,32 Hz, 1H), 3,77-3,70 (m, 1H), 3,55 (d, J= 10,54 Hz, 1H), 3,50-3,41 (m, 2H), 2,36 (d, J= 11,29 Hz, 1H), 2,17 (d,J= 11,42 Hz, 1H), 1,66 (d, J= 7,03 Hz, 6H). LC/MS (ESI) m/z: 459,3 (M+1).
Ejemplo 16
W-('5-('1,7-diazaespiro[4.4]nonan-7-il)piridin-2-il)-5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-amina

Etapa 1:
Metil éster de (S)-1-benzoilprolina

A una solución de clorhidrato de éster metílico de L-prolina (20,00 g, 120,76 mmol, 1,00 equivalente) en diclorometano (300,00 ml) se le añadió trietilamina (36,66 g, 362,28 mmol, 3,00 equivalentes) y cloruro de benzoílo (16,98 g, 120,76 mmol, 1,00 equivalente). La mezcla de reacción se agitó a 15 °C durante 18 horas. La LC/MS mostró conversión completa del material de partida y detectó el compuesto objetivo. El residuo obtenido al concentrar la solución de reacción se diluyó con acetato de etilo (200 ml) y se lavó sucesivamente con agua (100 ml), una solución acuosa de ácido cítrico (2,5 %, 100 ml) y bicarbonato sódico saturado (100 ml). La fase orgánica se secó sobre sulfato sódico anhidro y se concentró para dar el compuesto del título (24,00 g, 102,89 mmol, 85,20 % de rendimiento) en forma de un sólido de color blanco. RMN 1H (400 MHz, CDCh) 67,63-7,52 (m, 2H), 7,45-7,38 (m, 3H), 4,68 (dd, J = 5,2, 8,2 Hz, 1H), 3,82-3,75 (m, 3H), 3,66 (td, J = 7,0, 10,2 Hz, 1H), 3,57-3,53 (m, 1H), 2,40-2,17 (m, 1H), 2,07-1,98 (m, 2H), 1,95-1,82 (m, 1H).
Etapa 2:
1-benzoil-2-(nitrilometil)pirrolidin-2-carboxilato de metilo

A -78 °C, a una solución de (S)1-benzoilpirrolidin-2-carboxilato de metilo (10,00 g, 42,87 mmol, 1,00 equivalente) en tetrahidrofurano (150,00 ml) se le añadió una solución de diisopropilamida de litio en tetrahidrofurano (2 moles por litro, 25,72 ml, 1,20 equivalentes) y la mezcla se agitó durante 1 hora. A esta mezcla se le añadió después una solución de 2-bromoacetonitrilo (6,17 g, 51,44 mmol, 1,20 equivalentes) en tetrahidrofurano (30,00 ml) gota a gota. La mezcla se agitó a 15 °C durante 18 horas. La LC/MS mostró conversión completa del material de partida y detectó el compuesto objetivo. La mezcla de reacción se inactivó con una solución saturada acuosa de cloruro de amonio (100 ml) y se sometió a la fase de separación, la fase acuosa se sometió a extracción usando acetato de etilo (100 ml). Las fases orgánicas se combinaron, se secaron sobre sulfato sódico anhidro y se concentraron para dar el compuesto del título (12,00 g, producto en bruto) en forma de un aceite de color pardo. El producto en bruto se usó directamente en la etapa siguiente. RMN 1H (400 MHz, CDCls) 67,60-7,54 (m, 2H), 7,50-7,40 (m, 3H), 3,81 (s, 3H), 3,80-3,77 (m, 1H), 3,77-3,73 (m, 1H), 3,71-3,64 (m, 1H), 3,15 (d, J = 17,2 Hz, 1H), 2,44-2,27 (m, 2H), 2,17-2,05 (m, 2H).
Etapa 3:
1-benzoil-1,7-diazaespiro[4,4]nonan-6-ona

A una solución de 1-benzoil-2-(nitrilometil)pirrolidin-2-carboxilato de metilo (5,00 g, 18,36 mmol, 1,00 equivalente) y amoniaco acuoso (5,00 ml) en metanol (100,00 ml) se le añadió níquel raney (5,00 g). El matraz de reacción se purgó con argón e hidrógeno tres veces. La mezcla se calentó a 70 °C en atmósfera de hidrógeno a una presión de 379,21 kPa (55 psi) y se agitó durante 18 horas. La LC/MS mostró conversión completa del material de partida y detectó el compuesto objetivo. La mezcla de reacción se enfrió a 25 °C y se filtró. El residuo obtenido al concentrar el filtrado se purificó por columna sobre gel de sílice (éter de petróleo:acetato de etilo = 3:1 a diclorometano:metanol = 30:1) para dar el compuesto del título (2,25 g, 50,17 % de rendimiento) en forma de un sólido de color pardo claro. RMN 1H (400 MHz, CDCla) 67,62-7,50 (m, 2H), 7,45-7,35 (m, 3H), 5,84 (s a, 1H), 3,71-3,53 (m, 3H), 3,42-3,34 (m, 1H), 2,95 (ddd, J = 12,8, 9,7, 6,8 Hz, 1H), 2,34 (td, J = 6,7, 12,5 Hz, 1H), 2,20-1,96 (m, 3H), 1,94-1,82 (m, 1H).
Etapa 4:
1-bencil-1,7-diazaespiro[4,4]nonano

A 0 °C, a una suspensión de hidruro de litio y aluminio (7,13 g, 187,99 mmol, 5,60 equivalentes) en tetrahidrofurano (300,00 ml) se le añadió 1-benzoil-1,7-diazaespiro[4,4]nonan-6-ona (8,2 g, 33,57 mmol, 1,00 equivalente) en porciones separadas. Después de que se completara la adición, la mezcla se calentó a 70 °C y se agitó durante 2 horas. La LC/MS mostró conversión completa del material de partida y detectó el compuesto objetivo. La mezcla de reacción se enfrió a 15 °C y se inactivó con agua (7 ml), después se añadió una solución acuosa de hidróxido sódico (15 %, 7 ml) y agua (21 ml). La mezcla se filtró. El filtrado se secó sobre sulfato sódico anhidro y el residuo obtenido por concentración se purificó por columna sobre gel de sílice (diclorometano:metanol = 30:1 a 10:1)para dar el compuesto del título (3,67 g, 16,97 mmol, 50,54 % de rendimiento) en forma de un aceite de color amarillo claro. RMN 1H (400 MHz, CDCls) 67,36-7,28 (m, 4H), 7,26-7,20 (m, 1H), 3,69 (d, J = 13,2 Hz, 1H), 3,59 (d, J = 13,2 Hz, 1H), 3,13-3,04 (m, 2H), 2,99 (td, J = 10,8, 7,7 Hz, 1H), 2,77 (d, J = 11,2 Hz, 1H), 2,70-2,62 (m, 1H), 2,59-2,51 (m, 1H), 2,01 (td, J = 12,9, 8,1 Hz, 2H), 1,92-1,85 (m, 2H), 1,80-1,71 (m, 2H), 1,59 (ddd, J = 12,6, 7,6, 4,6 Hz, 1H).
Etapa 5:
1-bencil-7-(6-nitropiridin-3-il)-1,7-diazaespiro[4,4]nonano

A una solución de 1-bencil-1,7-diazaespiro[4,4]nonano (1,80 g, 8,32 mmol, 1,00 equivalente) y 5-bromo-2-nitropiridina (1,81 g, 8,90 mmol, 1,07 equivalentes) en W,A/-dimetilformamida (20,00 ml) se le añadió trietilamina (1,80 g, 17,80 mmol, 2,14 equivalentes). La mezcla se calentó a 100 °C y se agitó durante 18 horas. La LC/MS mostró conversión completa del material de partida y detectó el compuesto objetivo. La mezcla de reacción se enfrió a 15 °C, se diluyó con acetato de etilo (40 ml) y después se lavó dos veces con agua (50 ml x 2). La fase acuosa se sometió a extracción usando acetato de etilo (20 ml). Las fases orgánicas combinadas se secaron sobre sulfato sódico anhidro y el residuo obtenido por concentración se purificó por cromatografía sobre gel de sílice (éter de petróleo:acetato de etilo = 10:1 a 1:1) para dar el compuesto del título (1,65 g, 4,88 mmol, 58,60 % de rendimiento) en forma de un sólido de color amarillo. RMN 1H (400 MHz, CDCI3) 68,18 (d, J = 9,0 Hz, 1H), 7,83 (d, J= 2,9 Hz, 1H), 7,37-7,29 (m, 5H), 7,27-7,22 (m, 1H), 6,86 (dd, J= 9,1, 2,9 Hz, 1H), 3,78-3,70 (m, 1H), 3,70-3,61 (m, 2H), 3,57 3,45 (m, 2H), 3,27 (d, J = 10,3 Hz, 1H), 2,83-2,74 (m, 1H), 2,69 (td, J= 7,4, 9,3 Hz, 1H), 2,35 (td, J= 12,5, 9,0 Hz, 1H), 1,98-1,82 (m, 5H).
Etapa 6:
5-(1-bencil-1,7-diazaespiro[4,4]nonan-7-il)piridin-2-amina

A una solución de 1-bencil-7-(6-nitro-3-piridil)-1,7-diazaespiro[4,4]nonano (2,60 g, 7,68 mmol, 1,00 equivalente) en etanol (40,00 ml) se le añadió cinc en polvo (2,01 g, 30,73 mmol, 4,00 equivalentes) y cloruro de amonio (4,11 g, 76,83 mmol, 10,00 equivalentes). La mezcla se calentó a 80 °C y se agitó durante 3 horas. La LC/MS mostró conversión completa del material de partida y detectó el compuesto objetivo. La mezcla de reacción se enfrió a 15 °C, se diluyó con etanol (30 ml) y después se filtró. La torta de filtro se lavó con etanol (30 ml). El producto en bruto obtenido al concentrar el filtrado se purificó por HPLC preparativa (alcalina) para dar el compuesto del título (560,00 mg, 1,72 mmol, 22,34 % de rendimiento, 94,5 % de pureza) en forma de un aceite de color rojo. RMN 1H (400 MHz, CDCla) 67,52 (d, J= 2,8 Hz, 1H), 7,36-7,28 (m, 4H), 7,21-7,14 (m, 1H), 6,86 (dd, J = 8,8, 3,0 Hz, 1H), 6,51 (d, J= 8,7 Hz, 1H), 3,98 (s a, 2H), 3,74 (d, J= 13,3 Hz, 1H), 3,63 (d, J= 13,2 Hz, 1H), 3,47-3,38 (m, 2H), 3,24 (c, J= 8,6 Hz, 1H), 3,04 (d, J= 9,4 Hz, 1H), 2,77-2,59 (m, 2H), 2,37 (s, 1H), 2,27 (td, J= 12,5, 8,6 Hz, 1H), 2,02-1,75 (m, 6H).
Etapa 7:
W-(5-(1-benc¡l-1,7-d¡azaesp¡ro[4,4]nonan-7-¡l)p¡r¡d¡n-2-¡l)-5-fluoro-4-(3-¡soprop¡l-2-met¡l-2H-¡ndazol-5-¡l)p¡r¡m¡d¡n-2-amina

En atmósfera de n¡trógeno, a una soluc¡ón de 5-(1-benc¡l-1,7-d¡azaesp¡ro[4,4]nonan-7-¡l)p¡r¡d¡n-2-am¡na (370 mg, 1,20 mmol, 1,00 equivalente) y 5-(2-cloro-5-fluoro-p¡r¡m¡d¡n-4-¡l)-3-¡soprop¡l-2-met¡l-2H-¡ndazol (438,84 mg, 1,44 mmol, 1,20 equivalentes) en tetrah¡drofurano (20,00 ml) se le añad¡ó ferc-butóx¡do de potas¡o (403,84 mg, 3,60 mmol, 3,00 equ¡valentes) y cloro[2-(d¡c¡clohex¡lfosf¡n)-2',4',6'-tr¡¡soprop¡l-1,1'-b¡fen¡l][2-(2-am¡noet¡l)fen¡l]palad¡o (II) (88,63 mg, 120,00 mmol, 0,10 equivalente). La mezcla se calentó a 80 °C y se ag¡tó durante 18 horas. La LC/MS mostró que quedó un 9,9 % del mater¡al de part¡da y se detectó un 33,6 % del producto objetivo. La mezcla de reacc¡ón se enfr¡ó a 15 °C, se d¡luyó con agua (20 ml) y acetato de et¡lo (20 ml) y se f¡ltró. Después de someter el f¡ltrado a la fase de separac¡ón, la fase acuosa se somet¡ó a extracc¡ón dos veces usando acetato de et¡lo (20 ml x 2). Las fases orgán¡cas comb¡nadas se lavaron con salmuera saturada (10 ml) y después se concentraron. El res¡duo resultante se pur¡f¡có por TLC preparat¡va (acetato de et¡lo:metanol = 10:1) para dar el compuesto del título (220,00 mg, 381,47 pmol, 31,79 % de rend¡m¡ento) en forma de un sól¡do de color pardo. RMN 1H (400 MHz, CDCI3) 8 8,69 (s, 1H), 8,34 (d, J = 4,0 Hz, 1H), 8,22 (s a, 1H), 8,06 (d, J = 9,3 Hz, 1H), 7,89 (s a, 1H), 7,78-7,69 (m, 2H), 7,40-7,29 (m, 4H), 7,27-7,22 (m, 1H), 7,00 (dd, J= 2,9, 9,0 Hz, 1H), 4,17 (s, 3H), 3,74 (s a, 1H), 3,70-3,59 (m, 1H), 3,56-3,49 (m, 3H), 3,33 (c, J= 8,4 Hz, 1H), 3,13 (d, J= 9,2 Hz, 1H), 2,75 (s a, 1H), 2,69 (s a, 1H), 2,31 (s a, 1H), 1,95 1,84 (m, 5H), 1,60 (d, J= 7,0 Hz, 6H).
Etapa 8:
W-('5-('1,7-d¡azaesp¡ro[4.4]nonan-7-¡l)p¡r¡d¡n-2-¡l)-5-fluoro-4-(3-¡soprop¡l-2-met¡l-2H-¡ndazol-5-¡l)p¡r¡m¡d¡n-2-am¡na

A 80 °C, A/-(5-(1-benc¡l-1,7-d¡azaesp¡ro[4,4]nonan-7-¡l)-2-p¡r¡d¡l)-5-fluoro-4-(3-¡soprop¡l-2-met¡l-2H-¡ndazol-5-¡l)p¡r¡m¡d¡n-2-am¡na (220,00 mg, 381,47 pmol, 1,00 equivalente), form¡ato de amon¡o (481,12 mg, 7,63 mmol, 20,00 equivalentes) y Pd/C húmedo (220,00 mg) se ag¡taron en una mezcla de tetrah¡drofurano (22,00 ml) y metanol (22,00 ml) durante 16 horas. La LC/MS mostró que la reacc¡ón se había completado. La mezcla se f¡ltró y el f¡ltrado se concentró al vacío para dar un producto en bruto. El producto en bruto se pur¡f¡có por HPLC preparativa (ác¡do clorhídr¡co) para dar el compuesto del título (30,00 mg, 80,56 pmol, -21,12 % de rend¡m¡ento, 98 % de pureza). RMN 1H (400 MHz, Metanol-d4) 88,92 (s, 1H), 8,78 (d, J = 3,5 Hz, 1H), 8,41 (d, J= 9,2 Hz, 1H), 8,00-7,81 (m, 2H), 7,66 (d, J= 2,5 Hz, 1H), 7,55 (d, J= 9,5 Hz, 1H), 4,34 (s, 3H), 3,93 (d, J = 10,9 Hz, 1H), 3,80-3,67 (m, 2H), 3,65-3,47 (m, 4H), 2,64-2,54 (m, 1H), 2,53-2,43 (m, 1H), 2,35-2,18 (m, 4H), 1,67 (d, J = 7,0 Hz, 6H). LC/MS (ESI) m/z: 487,4 (M+1).
Ejemplo 17
5-fluoro-W-(5-(hexah¡drop¡rrolo[3,4-b][1,4]oxaz¡n-6(2H)-¡l)p¡r¡d¡n-2-¡l)-4-(3-¡soprop¡l-2-met¡l-2H-¡ndazol-5-¡l)p¡r¡m¡d¡n-2-am¡na

Etapa 1:
3-bromo-4-(2-h¡drox¡etox¡)p¡rrol¡d¡n-1 -carbox¡lato de ferc-but¡lo

A una solución de 2,5-dihidropirroM-carboxilato de tere-butilo (8,00 g, 47,28 mmol, 1,00 equivalente) en etilenglicol (30,00 ml) se le añadió N-bromosuccinimida (9,26 g, 52,01 mmol, 1,10 equivalentes) en porciones separadas. La mezcla se agitó a 15 °C durante 16 horas. La LC/MS y la TLC mostraron que la reacción se había completado. Se añadió agua (100 ml) a la mezcla. La mezcla resultante se agitó a 15 °C durante 0,5 h y se sometió a extracción usando acetato de etilo (100 ml x 3). Las capas orgánicas combinadas se secaron sobre sulfato sódico y se concentraron al vacío para dar el compuesto del título (14,00 g, producto en bruto) en forma de un aceite de color blanco. RMN 1H (400 MHz, CDCh) 54,31 (s a, 1H), 4,17 (d, J= 5,5 Hz, 1H), 3,98-3,92 (m, 1H), 3,87-3,82 (m, 1H), 3,77-3,72 (m, 2H), 3,71-3,58 (m, 2H), 3,57-3,39 (m, 1H), 2,06-2,01 (m, 1H), 1,49 (d, J= 6,7 Hz, 9H). LC/MS (ESI) m/z: 264,0 (M+1-56).
Etapa 2:
3-bromo-4-(2-(tosiloxi)etoxi)pirrolidin-1-carboxilato de tere-butilo

A 0 °C, a una solución de 3-bromo-4-(2-hidroxietoxi)pirrolidin-1-carboxilato de tere-butilo (14,00 g, 45,14 mmol, 1,00 equivalente), trietilamina (6,85 g, 67,71 mmol, 1,50 equivalentes) y 4-dimetilaminopiridina (551,48 mg, 4,51 mmol, 0,10 equivalente) en tolueno (100,00 ml) se le añadió cloruro de p-toluenosulfonilo (11,19 g, 58,68 mmol, 1,30 equivalentes). La mezcla se agitó a 10 °C durante 16 horas. La LC/m S y la TLC mostraron que la reacción se había completado. La mezcla se diluyó con agua (100 ml) y la capa acuosa se sometió a extracción usando acetato de etilo (100 ml). Las capas orgánicas combinadas se lavaron con una solución saturada acuosa de cloruro sódico (30 ml x 2), se secaron sobre sulfato sódico y se concentraron al vacío para dar un producto en bruto, que se purificaron después por cromatografía en columna (éter de petróleo:acetato de etilo = 4:1) para dar el compuesto del título (11,10 g, 23,90 mmol, 52,95 % de rendimiento) en forma de un aceite de color blanco. RMN 1H (400 MHz, CDCh) 5 7,81 (d, J = 8,3 Hz, 2H), 7,38 (d, J = 8,0 Hz, 2H), 4,19-4,11 (m, 3H), 4,10-4,02 (m, 1H), 3,90-3,66 (m, 5H), 3,45-3,29 (m, 1H), 2,48 (s, 3H), 1,49 (s, 9H). LC/MS (ESI) m/z: 408,0 (M+1-56).
Etapa 3:
4-bencilhexahidropirrolo[3,4-b][1,4]oxazin-6(2H)-carboxilato de tere-butilo

A 140 °C, 3-bromo-4-(2-(tosiloxi)etoxi)pirrolidin-1-carboxilato de tere-butilo (11,10 g, 23,90 mmol, 1,00 equivalente) y bencilamina (7,68 g, 71,70 mmol, 3,00 equivalentes) en xileno (150,00 ml) se agitaron durante 16 horas. La TLC y la LC/MS mostraron que la reacción se había completado. La mezcla se concentró al vacío para dar un residuo. El residuo se diluyó con acetato de etilo (100 ml) y agua (100 ml). La capa acuosa se sometió a extracción usando acetato de etilo (100 ml x 2). Las capas orgánicas combinadas se secaron sobre sulfato sódico y se concentraron al vacío para dar un producto en bruto, que se purificó después por cromatografía en columna (PE:EA = 20:1 a 4:1) para dar el compuesto del título (5,80 g, 14,21 mmol, 59,45 % de rendimiento, 78 % de pureza) en forma de un aceite de color amarillo. RMN 1H (400 MHz, CDCh) 57,81 (d, J = 8,3 Hz, 2H), 7,38 (d, J= 8,0 Hz, 2H), 4,19-4,11 (m, 3H), 4,10-4,02 (m, 1H), 3,90-3,66 (m, 5H), 3,45-3,29 (m, 1H), 2,48 (s, 3H), 1,49 (s, 9H). LC/MS (ESI) m/z: 319,1 (M+1).
Etapa 4:
4-bencilhexahidropirrolo[3,4-b][1,4]oxazina

A 0°C, a una solución de 4-bencilhexahidropirrolo[3,4-b][1,4]oxazin-6(2H)-carboxilato de ferc-butilo (2,90 g, 9,11 mmol, 1,00 equivalente) en acetato de etilo (30,00 ml) se le añadió ácido clorhídrico/acetato de etilo (30,00 ml). La mezcla se agitó a 10 °C durante 16 horas. La LC/MS mostró que la reacción se había completado. La mezcla se concentró al vacío para dar el compuesto del título (2,60 g, 8,93 mmol, 98,00 % de rendimiento, clorhidrato) en forma de un sólido de color blanco. LC/MS (ESI) m/z: 219,3 (M+1).
Etapa 5:
4-bencil-6-(6-nitropiridin-3-il)octahidropirrolo[3,4-b][1,4]oxazina

A 80 °C, una mezcla de 4-bencilhexahidropirrolo[3,4-b][1,4]oxazina (2,60 g, 8,93 mmol, 1,00 equivalente, 2HCl), 5-bromo-2-nitro-piridina (1,81 g, 8,93 mmol, 1,00 equivalente) y trietilamina (3,61 g, 35,72 mmol, 4,00 equivalentes) en W,W-dimetilformamida (30,00 ml) se agitó durante 16 horas. La LC/MS mostró que la reacción se había completado. La mezcla se diluyó con acetato de etilo (40 ml) y agua (120 ml) y se sometió a extracción usando acetato de etilo (40 ml x 3). Las capas orgánicas combinadas se secaron sobre sulfato sódico y se concentraron al vacío para dar un producto en bruto, que se purificó después por cromatografía en columna (éter de petróleo:acetato de etilo 3:1 a 1:1) para dar el compuesto del título (2,00 g, 5,88 mmol, 65,80 % de rendimiento) en forma de un sólido de color amarillo. LC/MS (ESI) m/z: 341,1 (M+1).
Etapa 6:
5-(4-bencilhexahidropirrolo[3,4-b][1,4]oxazin-6(2H)-il)piridin-2-amina

A 80 °C, a mezcla de 4-bencil-6-(6-nitropiridin-3-il)octahidropirrolo[3,4-b][1,4]oxazina (2,00 g, 5,88 mmol, 1,00 equivalente), polvo de hierro (1,64 g, 29,40 mmol, 5,00 equivalentes) y cloruro de amonio (3,15 g, 58,80 mmol, 10,00 equivalentes) en etanol (50,00 ml) y agua (10,00 ml) se agitó durante 16 horas. La LC/MS mostró que la reacción se había completado. La mezcla se filtró y el filtrado se concentró al vacío para dar un producto en bruto, que se purificó después por HPLC preparativa (ácido trifluoroacético) para dar el compuesto del título (1,00 g, 3,13 mmol, 53,15 % de rendimiento, 97 % de pureza) en forma de un aceite de color pardo. LC/MS (ESI) m/z: 311,2 (M+1).
Etapa 7:
W-(5-(4-bencilhexahidropirrolo[3,4-b][1,4]oxazin-6(2H)-il)piridin-2-il)-5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-amina

En atmósfera de nitrógeno, una solución de 5-(4-bencilhexahidropirrolo[3,4-b][1,4]oxazin-6(2H)-il)piridin-2-amina (300,00 mg, 966,53 pmol, 1,00 equivalente), 5-(2-cloro-5-fluoro-pirimidin-4-il)-3-isopropil-2-metilindazol (Intermedio C) (294,55 mg, 966,53 pmol, 1,00 equivalente), cloro[2-(diciclohexilfosfin)-2'-4'-6'-triisopropil-1,1'-bifenil][2-(2-aminoetil)fenil]paladio (II) (71,40 mg, 96,65 pmol, 0,10 equivalentes) y ferc-butóxido de potasio (325,36 mg, 2,90 mmol, 3,00 equivalentes) en tetrahidrofurano (30,00 ml) se agitó a 80 °C durante 16 horas.
La LC/MS mostró que la reacción se había completado. A la mezcla se le añadió sucesivamente agua (30 ml) y acetato de etilo (30 ml). La mezcla resultante se agitó a 20 °C durante 15 minutos, se filtró y se sometió a la fase de separación. La capa acuosa se sometió a extracción usando acetato de etilo (20 ml x 2). Las capas orgánicas combinadas se secaron sobre sulfato sódico anhidro y se concentraron al vacío para dar un producto en bruto, que se purificó después mediante placa de TLC preparativa (diclorometano:metanol = 10:1) para dar el compuesto del
título (190,00 mg, 265,95 |jmol, 27,52 % de rendimiento, 81 % de pureza) en forma de un sólido de color amarillo. LC/MS (ESI) m/z: 579,4 (M+1).
Etapa 8:
5-fluoro-W-(5-(hexahidropirrolo[3,-4-b][1,4]oxazin-6(2H)-il)piridin-2-il)-4-(3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-amina

Una solución mezclada de W-(5-(4-bencilhexahidropirrolo[3,4-b][1,4]oxazin-6(2H)-il)piridin-2-il)-5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-amina (160,00 mg, 276,49 jmol, 1,00 equivalente), formiato de amonio (348,38 mg, 5,53 mmol, 20,00 equivalentes) y Pd/C húmedo (15,00 mg) en tetrahidrofurano (16,00 ml) y metanol (16,00 ml) se agitó a 80 °C durante 16 horas. La LC/MS mostró que se produjo el producto deseado. La mezcla se filtró y el filtrado se concentró al vacío para dar un producto en bruto, que se produjo después por HPLC preparativa (ácido clorhídrico) para dar el compuesto del título (20,00 mg, 40,94 jmol, 14,81 % de rendimiento, 100 % de pureza). RMN 1H (400 MHz, Metanol-d4) 68,89 (s, 1H), 8,75 (d, J= 3,6 Hz, 1H), 8,33 (d, J= 9,0 Hz, 1H), 7,90 (d, J= 1.1 Hz, 1H), 7,81 (d, J= 9,0 Hz, 1H), 7,62 (s, 1H), 7,54 (d,J= 9,4 Hz, 1H), 4,59 (s a, 1H), 4,30 (s, 3H), 4,19 (s a, 1H), 4,11 (d, J= 11,9 Hz, 1H), 3,97-3,79 (m, 3H), 3,79-3,67 (m, 2H), 3,58 (d, J= 11,0 Hz, 1H), 3,51 (s a, 1H), 3,28 (d, J= 13.1 Hz, 1H), 1,65 (d, J= 6,9 Hz, 6H). LC/MS (ESI) m/z: 489,4 (M+1).
Ejemplo 18
1-(6-((5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-il)amino)piridin-3-il)piperazin-2-ona

Etapa 1:
W-(5-bromo-2-piridil)-W-ferc-butoxicarbonilcarbamato de ferc-butilo

A 0 °C, a una solución de 5-bromopiridin-2-amina (2,00 g, 11,56 mmol, 1,00 equivalente), W,A/-diisopropiletilamina (4,48 g, 34,68 mmol, 6,05 ml, 3,00 equivalentes) y dimetilaminopiridina (282,46 mg, 2,31 mmol, 0,20 equivalente) en diclorometano (30,00 ml) se le añadió carbonato de di-ferc-butilo (7,57 g, 34,68 mmol, 7,97 ml, 3,00 equivalentes). La mezcla de reacción se agitó a 30 °C durante 16 horas. La LC/MS y la TLC mostraron que la reacción se había completado. La mezcla se concentró al vacío para dar un producto en bruto. El producto en bruto se purificó por cromatografía en columna (acetato de etilo:éter de petróleo = 50:1) para dar el compuesto del título (2,00 g, 5,36 mmol, 46,35 % de rendimiento) en forma de un sólido de color blanco. RMN 1H (400 Mhz , CDCh) 68,55 (d, J = 2,3 Hz, 1H), 7,86 (dd, J= 2,5, 8,4 Hz, 1H), 7,20 (d, J= 8,5 Hz, 1H), 1,47 (s, 18H). LC/MS (ESI) m/z: 373,1 (M+1).
Etapa 2:
3-oxopiperazin-1-carboxilato de bencilo

A 0 °C, a una solución mezcla de carbonato sódico (24,77 g, 233,73 mmol, 3,00 equivalentes) y piperazin-2-ona (7,80 g, 77,91 mmol, 1,00 equivalente) en acetato de etilo (70,00 ml) y agua (70,00 ml) se le añadió cloroformiato de
bencilo (16,79 g, 93,49 mmol, 13,99 ml, 95 % de pureza, 1,20 equivalentes). La mezcla de reacción se agitó a 30 °C durante 16 horas. La TLC mostró que la reacción se había completado. La mezcla se sometió a extracción usando acetato de etilo (80 ml x 3). Las capas orgánicas combinadas se lavaron con una solución saturada acuosa de cloruro sódico (80 ml x 3), se secaron sobre sulfato sódico y se concentraron al vacío para dar un producto en bruto. El producto en bruto se batió en (éter de petróleo:acetato de etilo = 20:1, 80 ml). La mezcla resultante se agitó a 30 °C durante 15 minutos y se filtró. El sólido se secó al vacío para dar el compuesto del título (15,50 g, 66,17 mmol, 84,93 % de rendimiento) en forma de un sólido de color blanco.
Etapa 3:
4-[6-[bis(terc-butoxicarbonil)amino]-3-piridil]-3-oxo-piperazin-1 -carboxilato de bencilo

En atmósfera de nitrógeno, una mezcla de 3-oxopiperazin-1-carboxilato (4,00 g, 17,08 mmol, 1,00 equivalente), N-(5-bromo-2-piridil)-N-terc-butoxicarbonilcarbamato de terc-butilo (6,37 g, 17,08 mmol, 1,00 equivalente), N,N-dimetiletan-1,2-diamina (602,09 mg, 6,83 mmol, 734,26 ^l, 0,40 equivalente), carbonato potásico (7,08 g, 51,24 mmol, 3,00 equivalentes) y Cul (650,42 mg, 3,42 mmol, 0,20 equivalentes) en dioxano (80,00 ml) se agitó a 100 °C durante 16 horas. La LC/MS mostró que la reacción se había completado. La mezcla se filtró y el filtrado se concentró al vacío para dar un producto en bruto, que después se purificó por TLC preparativa (éter de petróleo:acetato de etilo = 1:1) para dar una mezcla del compuesto del título y producto mono-Boc (3,20 g, producto en bruto). LC/MS (ESI) m/z: 527,2 (M+1).
Etapa 4:
4-(6-aminopiridin-3-il)-3-oxo-piperazin-1 -carboxilato de bencilo

A una solución de 4-[6-[bis(terc-butoxicarbonil)amino]-3-piridil]-3-oxo-piperazin-1-carboxilato de bencilo (3,20 g, 6.08 mmol, 1,00 equivalente) en diclorometano (20,00 ml) se le añadió ácido trifluoroacético (10,00 ml) a 0 °C. La mezcla de reacción se agitó a 30 °C durante 2 horas. La LC/MS mostró que la reacción se había completado. La mezcla de reacción se concentró al vacío para dar un producto en bruto, que se diluyó con diclorometano. Se añadió carbonato potásico (10 g) a la mezcla. La mezcla resultante se agitó a 30 °C durante 15 minutos y se filtró. El filtrado se concentró al vacío para dar el compuesto del título (2,00 g, producto en bruto) en forma de un aceite de color pardo. RMN 1H (400 MHz, Metanol-d4) 5 7,87 (d, J= 2,5 Hz, 1H), 7,42-7,36 (m, 4H), 7,25-7,13 (m, 2H), 6,63 (d, J= 8.8 Hz, 1H), 5,21 (s, 2H), 4,28 (s a, 2H), 3,88 (s a, 2H), 3,73 (t, J= 5,3 Hz, 2H). LC/MS (ESI) m/z: 327,1 (M+1).
Etapa 5:
4-(6-((5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-il)amino)piridin-3-il)-3-oxopiperazin-1-carboxilato de bencilo

En atmósfera de nitrógeno, una mezcla de 5-(2-cloro-5-fluoro-pirimidin-4-il)-3-isopropil-2-metilindazol (Intermedio C) (1,60 g, 5,25 mmol, 1,00 equivalente), 4-(6-aminopiridin-3-il)-3-oxo-piperazin-1-carboxilato de bencilo (2,06 g, 6,30 mmol, 1,20 equivalentes), carbonato de cesio (5,13 g, 15,75 mmol, 3,00 equivalentes), XPhos (500,58 mg, 1,05 mmol, 0,20 equivalentes) y Pd(OAc)2 (117,87 mg, 525,00 ^mol, 0,10 equivalentes) en dioxano (50,00 ml) se agitó a 110 °C durante 16 horas. La LC/MS mostró que la reacción se había completado. La mezcla se filtró y el filtrado se concentró al vacío para dar un producto en bruto. El producto en bruto se purificó por cromatografía en columna (diclorometano:metanol = 20:1) para dar el compuesto del título (2,40 g, 3,59 mmol, 68,42 % de rendimiento, 89 % de pureza) en forma de un sólido de color amarillo. RMN 1H (400 MHz, CDCh) 58,65 (s, 1H), 8,70 (s, 1H), 8,57 (d, J= 9,0 Hz, 1H), 8,45 (d, J= 3,8 Hz, 1H), 8,33 (d, J = 2,5 Hz, 1H), 8,05 (d, J= 9,2 Hz, 1H), 7,75 (d, J=
9,2 Hz, 1H), 7,67 (dd, J = 8,9, 2,6 Hz, 1H), 7,47-7,31 (m, 5H), 5,22 (s, 2H), 4,37 (s, 2H), 4,20 (s, 3H), 3,93-3,86 (m, 2H), 3,82-3,72 (m, 2H), 3,58-3,46 (m, 1H), 1,61 (d, J= 7,0 Hz, 6H). LC/MS (ESI) m/z: 595,2 (M+1).
Etapa 6:
1-(6-((5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-il)amino)piridin-3-il)piperazin-2-ona

En atmósfera de hidrógeno, una mezcla de 4-(6-((5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-il)amino)piridin-3-il)-3-oxopiperazin-1-carboxilato de bencilo (2,40 g, 3,59 mmol, 1,00 equivalente) y Pd/C húmedo (400,00 mg, 10 % de pureza) en metanol (20,00 ml) y tetrahidrofurano (20,00 ml) se agitó a 30 °C durante 16 horas. La LC/MS mostró que la reacción se había completado. La mezcla se filtró y el filtrado se concentró al vacío para dar un producto en bruto, que se diluyó con diclorometano (20 ml). A la mezcla se le añadió ácido clorhídrico/acetato de etilo (5 ml, 4 M) gota a gota. La mezcla resultante se concentró después al vacío para dar un producto en bruto, que después se purificó por HPLC preparativa (ácido clorhídrico) para dar el compuesto del título (1,25 g, 2,32 mmol, 64,62 % de rendimiento, 99 % de pureza, clorhidrato). RMN 1H (400 MHz, metanol-c^) 8 8,92 (s, 1H), 8,83 (d, J= 3,6 Hz, 1H), 8,63 (d, J= 2,1 Hz, 1H), 8,45 (dd, J= 2,5, 9,4 Hz, 1H), 8,36 (d, J= 9,3 Hz, 1H), 7,86-7,78 (m, 1H), 7,67 (d, J= 9,5 Hz, 1H), 4,31 (s, 3H), 4,20-4,10 (m, 4H), 3,83-3,68 (m, 3H), 1,66 (d, J= 7,0 Hz, 6H). LC/MS (ESI) m/z: 461,2 (M+1).
Ejemplo 19
W-[5-(3,8-diazabiciclo[3.2.1]octan-3-il)-2-piridil]-5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-amina

Etapa 1:
3-(6-nitro-3-piridil)-3,8-diazabiciclo[3.2.1]octan-8-carboxilato de ferc-butilo

A una solución de 3,8-diazabiciclo[3.2.1]octan-8-carboxilato de ferc-butilo (1,00 g, 4,71 mmol, 1,00 equivalente) en dimetilsulfóxido (20,00 ml) se le añadió trietilamina (1,43 g, 14,13 mmol, 3,00 equivalentes) y 5-bromo-2-nitro-piridina (1,15 g, 5,65 mmol, 1,20 equivalentes). La mezcla se agitó a 100 °C durante 16 horas. La LC/MS mostró que la reacción se había completado. Se añadieron 40 ml de agua a la mezcla hasta que la mezcla se enfrió a 20 °C. La fase acuosa se sometió a extracción usando diclorometano (50 ml x 2), se lavó con salmuera saturada (50 ml x 1), se secó sobre sulfato sódico anhidro, se filtró y se concentró. El producto en bruto se purificó batiéndolo en metanol (2 ml x 3) para dar el compuesto del título (639,00 mg, producto en bruto) en forma de un sólido de color amarillo. RMN 1H (400 MHz, CDCla) 88,18 (d, J= 9,2 Hz, 1H), 8,09 (d, J= 3,0 Hz, 1H), 7,15 (dd, J= 9,2 Hz, 3,0 Hz, 1H), 4,47 (s a, 2H), 3,58 (d, J = 10,0 Hz, 2H), 3,27 (s a, 2H), 2,12-1,99 (m, 2H), 1,86-1,76 (m, 2H), 1,49 (s, 9H). LC/MS (ESI) m/z: 335,2 (M+1).
Etapa 2:
3-(6-amino-3-piridil)-3,8-diazabiciclo[3.2.1]octan-8-carboxilato de ferc-butilo

A una solución de 3-(6-nitro-3-piridil)-3,8-diazabiciclo[3.2.1]octan-8-carboxilato de ferc-butilo (339,00 mg, 1,01 mmol, 1,00 equivalente) en metanol (11,00 ml) se le añadió paladio sobre carbono (50,00 mg) en atmósfera de hidrógeno
(103,42 kPa (15 psi)). La mezcla de reacción se agitó a 20 °C durante 18 horas. La LC/MS mostró que la reacción se había completado. La mezcla se filtró y se concentró para dar el compuesto del título (287,00 mg, producto en bruto) en forma de un sólido de color púrpura. LC/MS (ESI) m/z: 305,2 (M+1).
Etapa 3:
3-[6-[[5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-il]amino]-3-piridil]-3,8-diazabiciclo[3.2.1]octan-8-carboxilato de tere-butilo

A una solución de 3-(6-amino-3-piridil)-3,8-diazabiciclo[3.2.1]octan-8-carboxilato de tere-butilo (140,00 mg, 459,94 ^mol, 1,00 equivalente) en dioxano se le añadió 5-(2-cloro-5-fluoro-pirimidin-4-il)-3-isopropil-2-metil-2H-indazol (Intermedio C) (249,50 mg, 818,69 ^mol, 1,78 equivalentes), Xantphos (79,84 mg, 137,98 ^mol, 0,30 equivalentes), carbonato de cesio (446,58 mg, 1,37 mmol, 2,98 equivalentes) y Pd2(dba)3 (63,18 mg, 68,99 ^mol, 0,15 equivalentes) en atmósfera de nitrógeno. La mezcla se agitó a 110 °C durante 18 horas. La LC/MS mostró que el material de partida reaccionó completamente y no se detectó producto. La TLC mostró que el material de partida reacción completamente y se produjeron dos puntos nuevos. La mezcla de reacción se filtró y la torta de filtro se disolvió en 10 ml de agua. La fase acuosa se sometió a extracción usando diclorometano (10 ml x 2). Las fases orgánicas combinadas se lavaron con salmuera saturada (10 ml), se secaron sobre sulfato sódico anhidro, se filtraron y se concentraron para dar el compuesto del título (25,00 mg, producto en bruto) en forma de un sólido de color amarillo. LC/MS (ESI) m/z: 573,3 (M+1).
Etapa 4:
W-[5-(3,8-diazabiciclo[3.2.1]octan-3-il)-2-piridil]-5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-amina

A una solución de 3-[6-[[5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-il]amino]-3-piridil]-3,8-diazabiciclo[3.2.1]octan-8-carboxilato de tere-butilo (25,00 mg, 43,66 ^mol, 1,00 equivalente) en diclorometano (2 ml) se le añadió ácido trifluoroacético (765,00 mg, 6,71 mmol, 153,67 equivalentes). La mezcla se agitó a 20 °C durante 0,5 horas. La LC/MS mostró que la reacción se había completado. La mezcla de reacción se concentró para dar un producto en bruto. El producto en bruto se purificó por HPLC preparativa (ácido clorhídrico) para dar el compuesto del título (11,80 mg, 22,82 ^mol, 52,27 % de rendimiento, 91,39 % de pureza). RMN 1H (400 MHz, Metanol-d4) 5 8,89 (s, 1H), 8,77 (d, J= 3,5 Hz, 1H), 8,35 (d, J= 9,2 Hz, 1H), 8,25 (dd, J= 9,5, 2,1 Hz, 1H), 7,87 (d, J = 1,9 Hz, 1H), 7,81 (d, J = 9,0 Hz, 1H), 7,55 (d, J = 9,5 Hz, 1H), 4,30 (s, 5H), 3,84-3,67 (m, 3H), 3,36-3,32 (m, 2H), 2,20 (s, 4H), 1,64 (d, J= 6,9 Hz, 6H). LC/MS (ESI) m/z: 473,3 (M+1).
Ejemplo 20
5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)-A/-(4-metil-5-piperazin-1-il-2-piridil)pirimidin-2-amina

Etapa 1:
4-[6-[[5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-il]amino]-4-metil-3-piridil]piperazin-1-carboxilato de tere-butilo

A una solución de 4-(6-ammo-4-metil-3-pmdil)piperazm-1-carboxilato de rere-butilo (200,00 mg, 684,04 ^mol, 1,00 equivalente) en dioxano (5,00 ml) se le añadió 5-(2-cloro-5-fluoro-pirimidin-4-il)-3-isopropil-2-metil-2H-indazol (Intermedio C) (250,15 mg, 820,85 ^mol, 1,20 equivalentes), Pd2(dba)3 (62,64 mg, 68,40 ^mol, 0,10 equivalentes), Xantphos (79,16 mg, 136,81 ^mol, 0,20 equivalentes) y carbonato de cesio (445,75 mg, 1,37 mmol, 2,00 equivalentes) en atmósfera de nitrógeno. La mezcla se agitó a 110 °C durante 16 horas. La LC/MS mostró que la reacción se había completado. La mezcla se concentró, el producto en bruto se purificó por TLC preparativa (acetato de etilo:éter de petróleo = 2:1) para dar el compuesto del título (150,00 mg, producto en bruto) en forma de un sólido de color amarillo. LC/MS (ESI) m/z: 561,4 (M+1).
Etapa 2:
5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)-W-(4-metil-5-piperazin-1-il-2-piridil)pirimidin-2-amina

A una solución de 4-[6-[[5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-il]amino]-4-metil-3-piridil]piperazin-1-carboxilato de tere-butilo (150,00 mg, 267,54 ^mol, 1,00 equivalente) en diclorometano (4 ml) se le añadió ácido trifluoroacético (1,53 g, 13,42 mmol, 50,16 equivalentes) a 20 °C. La mezcla de reacción se agitó durante 0,5 horas. La LC/MS mostró que la reacción se había completado. La mezcla se concentró para dar un producto en bruto, que se purificó después por HPLC preparativa (ácido clorhídrico) para dar el compuesto del título (111,60 mg, 224,54 ^mol, 83,93 % de rendimiento, 100 % de pureza, clorhidrato). RMN 1H (400 MHz, metanol-d4) 58,93 (s, 1H), 8,83 (d, J= 3,51 Hz, 1H), 8,32-8,45 (m, 3H), 7,85 (d, J= 9,03 Hz, 1H), 7,62 (d, J = 9,16 Hz, 1H), 4,33 (s, 3H), 3,74 (d, J= 6,99 Hz, 1H), 3,57 (d, J= 12,67 Hz, 2H), 3,10-3,27 (m, 3H), 2,20 (d, J= 13,68 Hz, 2H), 1,96-2,10 (m, 2H), 1,66 (d, J= 7,03 Hz, 6H). LC/MS (ESI) m/z: 446,2 (M+1).
Enfoque B
Preparación de 5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)-W-(5-(piperazin-1-il)piridin-2-il)piridin-2-amina

5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)-W-(5-(piperazin-1-il)piridin-2-il)piridin-2-amina
Etapa 1:
5-(2-doro-5-fluoropiridin-4-il)-2-metil-3-isopropenil-2H-indazol

En atmósfera de nitrógeno, a una solución mezclada de 2-metil-3-isopropenil-5-borato-2H-indazol (500,00 mg, 1,68 mmol, 1,00 equivalente) en dioxano (10 ml) y agua (2 ml) se le añadió 2-doro-5-fluoro-4-yodo-piridina (518,98 mg, 2,02 mmol, 1,20 equivalentes), K2CO3 (696,58 mg, 5,04 mmol, 3,00 equivalentes) y Pd(dppf)Cl2 (122,93 mg, 168,00 jmol, 0,10 equivalentes). La mezcla se agitó a 110 °C durante 3 horas. La LC/MS mostró que la reacción se había completado. La mezcla se enfrió a 20 °C y después se le añadió agua (10 ml). Se realizó la extracción con acetato de etilo (15 ml x 3). Las fases orgánicas se combinaron y se lavaron con salmuera (15 ml x 2), se secaron sobre sulfato sódico anhidro y se filtraron. El residuo obtenido al concentrar el filtrado se purificó por columna sobre gel de sílice (éter de petróleo:acetato de etilo = 30:1 a 5:1) para dar el producto del título (500 mg, 1,66 mmol, 98,63 % de rendimiento) en forma de un aceite de color amarillo. RMN 1H (400 MHz, CDCh) 88,32 (d, J= 2,1 Hz, 1H), 7,89 (s, 1H), 7,77 (dd, J= 9,0, 0,8 Hz, 1H), 7,52-7,47 (m, 2H), 5,69 (t, J = 1,5 Hz, 1H), 5,35 (s, 1H), 4,22 (s, 3H), 2,30 (d, J= 1,0 Hz, 3H). LC/MS (ESI) m/z: 302,0 (M+1).
Etapa 2:
5-(2-cloro-5-fluoropiridin-4-il)-3-isopropil-2-metil-2H-indazol

A una solución mezclada de 5-(2-cloro-5-fluoropiridin-4-il)-2-metil-3-isopropenil-2H-indazol (500 mg, 1,66 mmol, 1,00 equivalente) en metanol (5 ml) y tetrahidrofurano (5 ml) se le añadió Rh(PPh3)3Cl (153,59 mg, 166,00 jmol, 0,1 equivalentes). Se introdujo hidrógeno gaseoso en el sistema y la presión se mantuvo a 344,74 kPa (50 psi). La mezcla se agitó a 50 °C durante 16 horas. La LC/MS mostró que la reacción se había completado. La mezcla se enfrió a 20 °C y se concentró para dar un residuo. El residuo se purificó por cromatografía en columna sobre gel de sílice (éter de petróleo:acetato de etilo = 20:1 a 5:1) para dar el compuesto del título (460,00 mg, 1,51 mmol, 91,23 % de rendimiento) en forma de un aceite de color blanco. RMN 1H (400 MHz, CDCI3) 88,33 (d, J = 2,3 Hz, 1H), 8,05 (s, 1H), 7,75 (dd, J = 9,0, 0,8 Hz, 1H), 7,50 (d, J= 5,6 Hz, 1H), 7,45 (td, J = 9,0, 1,7 Hz, 1H), 4,20 (s, 3H), 3,55-3,48 (m, 1H), 1,58 (d, J= 7,0 Hz, 6H). LC/MS (ESI) m/z: 304,0 (M+1).
Etapa 3:
4-(5-((5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-il)piridin-2-il)amino)piridin-2-il)piperazin-1-carboxilato de ferc-butilo

En atmósfera de nitrógeno, a una solución de 5-(2-cloro-5-fluoropiridin-4-il)-3-isopropil-2-metil-2H-indazol (200,00 mg, 658,41 jmol, 1,00 equivalente) y 4-(6-amino-3-piridin)piperazin-1-carboxilato de ferc-butilo (274,90 mg, 987,62 |jmol, 1,50 equivalentes ) en dioxano (8 ml) se le añadió CS2CO3 (643,57 mg, 1,98 mmol, 3,00 equivalentes), Xantphos (76,19 mg, 131,68 jmol, 0,20 equivalentes) y Pd2(dba)3 (60,29 mg, 65,84 jmol, 0,10 equivalentes). La mezcla se agitó a 110 °C durante 16 horas. La LC/MS mostró que la reacción se había completado. La mezcla se enfrió a 20 °C y se filtró. La torta de filtro se lavó con acetato de etilo y el filtrado se concentró para dar el compuesto del título (350,00 mg, producto en bruto) en forma de un aceite de color negro, que se usó directamente en la etapa siguiente sin purificación. LC/MS (ESI) m/z: 546,4 (M+1).
Etapa 4:
5-fluoro-4-(3-¡soprop¡l-2-met¡l-2H-¡ndazol-5-¡l)-A/-(5-(p¡peraz¡n-1-¡l)p¡r¡d¡n-2-il)p¡r¡d¡n-2-am¡na

A una solución de 4-(5-((5-fluoro-4-(3-isopropil-2-metil-2H-indazol-5-M)piridin-2-il)amino)piridin-2-il) piperazin-1-carboxilato de /ere-butilo (350,00 mg, 641,44 ^mol, 1,00 equivalente) en diclorometano (5 ml) se le añadió ácido trifluoroacético (2,0 ml). La mezcla se agitó a 20 °C durante 0,5 h. La LC/MS mostró que la reacción se había completado. La mezcla se concentró a presión reducida para eliminar el diclorometano y el ácido trifluoroacético para dar un residuo. El residuo se purificó por HPLC preparativa (ácido clorhídrico) para dar el compuesto del título (250,00 mg, 558,27 pmol, 87,03 % de rendimiento, 99,49 % de pureza). RMN 1H (400 MHz, Metanol-d4) 58,47 (d, J= 2,6 Hz, 1H), 8,39 (s, 1H), 8,21 (dd, J= 9,7, 2,9 Hz, 1H), 7,89 (d, J = 2,8 Hz, 1H), 7,87-7,80 (m, 2H), 7,44 (d, J= 9,5 Hz, 1H), 7,40 (d, J= 5,5 Hz, 1H), 4,31 (s, 3H), 3,76-3,69 (m, 1H), 3,56-3,51 (m, 4H), 3,50-3,45 (m, 4H), 1,64 (d, J= 7,0 Hz, 6H). LC/MS (ESI) m/z: 304,0 (M+1).
Enfoque C
A continuación se muestra un método general para la preparación del intermedio D.

Etapa 1:
5-bromo -2,3-difluorobenzaldehído

Se disolvió 2,3-difluorobenzaldehído (3,00 g, 21,22 mmol, 1,00 equivalente) en ácido sulfúrico (18,4 mol/l, 10,20 ml, 8,89 equivalentes) y se calentó a 60 °C durante 40 minutos. En este tiempo, se añadió 1-bromopirrolidin-2,5-diona (4,51 g, 25,33 mmol, 1,20 equivalentes) en tres porciones durante 20 minutos. La mezcla se calentó durante 3 horas en atmósfera de nitrógeno. La TLC y la HPLC mostraron que la reacción se había completado. La mezcla de reacción se vertió en agua enfriada con hielo, se sometió a extracción dos veces usando éter de petróleo (30 ml x 2, se lavó con agua (30 ml x 2) y salmuera saturada (30 ml x 2) y después se concentra a presión reducida. El residuo concentrado se purificó por cromatografía en columna (éter de petróleo) para dar el compuesto del título (2,10 g, 9,50 mmol, 45,00 % de rendimiento) en forma de un líquido amarillo. RMN 1H (400 MHz, CDCl3) 5 10,29 (s, 1H), 7,77 (s a, 1H), 7,65-7,54 (m, 1H).
Etapa 2:
(trans)-5-bromo-2,3-difluorobenzaldehído-oxo-met¡lox¡ma
Una mezcla de 5-bromo-2,3-difluoro-benzaldehído (1,50 g, 6,79 mmol, 1,00 equivalente), O-metilhidroxilamina (680,52 mg, 8,15 mmol, 1,20 equivalentes) y carbonato potásico (1,13 g, 8,15 mmol, 1,2 equivalentes) en dimetil éter (20,00 ml) se calentó a 40 °C y se agitó durante 14 horas. La TLC mostró que aproximadamente el 1% del material de partida no se consumió. La mezcla de reacción se filtró y el filtrado se concentró a presión reducida para dar el compuesto del título (1,00 g, producto en bruto), que se usó directamente en la etapa siguiente sin más purificación. RMN 1H (400 MHz, CDCls) 58,22 (s, 1H), 7,77-7,75 (m, 1H), 7,35-7,27 (m, 1H), 4,04 (s, 3H).
Etapa 3:
5-bromo-7-fluoro-2H-indazol

Una solución de (trans)-5-bromo-2,3-difluorobenzaldehído-oxo-metiloxima (4,90 g, 19,60 mmol, 1,00 equivalente) en tetrahidrofurano (35,00 ml) e hidrato de hidrazina (20,60 g, 411,51 mmol, 21,00 equivalentes, 85% de fuerza) se calentó a 90 °C y se agitó durante 72 h. La LC/MS mostró que el 20 % del material de partida no se consumió. El disolvente orgánico se concentró y la mezcla resultante se diluyó con acetato de etilo (20 ml), se lavó con agua (20 ml x 2) y después se concentró a presión reducida para dar un residuo. El residuo se purificó por cromatografía en columna (éter de petróleo:acetato de etilo = 60:1 a 40:1) para dar el compuesto del título (3,25 g, 12,09 mmol, 61,69 % de rendimiento, 80 % de pureza) en forma de un sólido de color blanco. LC/MS (ESI) m/z: 215,0 (M+1).
Etapa 4:
5-bromo-7-fluoro-2-metil-2H-indazol

En atmósfera de nitrógeno, a una solución de 5-bromo-7-fluoro-2H-indazol (1,10 g, 5,12 mmol, 1,00 equivalente) y metóxido sódico (552,71 mg, 10,23 mmol, 2,00 equivalentes) en metanol (20,00 ml) se le añadió yoduro de metilo (1,09 g, 7,67 mmol, 1,50 equivalentes) gota a gota a 30 °C durante 30 minutos. La mezcla se calentó a 85 °C durante 30 minutos y se agitó durante 5 horas. La LC/MS mostró que aproximadamente el 3 % del material de partida no se consumió. La mezcla de reacción se enfrió a 30 °C, se concentró a presión reducida, se diluyó con una solución acuosa al 3 % de bicarbonato sódico y después se sometió a extracción usando acetato de etilo (20 ml x 2). La fase orgánica se concentró a presión reducida para dar un residuo y el residuo se purificó por cromatografía en columna (éter de petróleo:acetato de etilo = 20:1 a 5:1) para dar el compuesto del título (360,00 mg, 1,57 mmol, 30,66 % de rendimiento) en forma de un sólido de color amarillo. LC/MS (ESl) m/z: 229,0 (M+1).
Etapa 5:
5-bromo-7-fluoro-3-yodo-2-metil-2H-indazol

A 30 °C, a una solución de 5-bromo-7-fluoro-2-metil-2H-indazol (500,00 mg, 2,18 mmol, 1,00 equivalente) en diclorometano (5,00 ml) se le añadió piridina (259,00 ml, 3,27 mmol, 1,50 equivalentes) y bis(trifluoroacetoxi)yodobenceno (1,13 g, 2,62 mmol, 1,20 equivalentes) y la mezcla se agitó durante 30 minutos. Se añadió yodo (664,86 mg, 2,62 mmol, 1,20 equivalentes) a 30 °C y se agitó durante 23,5 horas. La LC/MS mostró conversión completa del material de partida. La solución de reacción se filtró para dar una torta de filtro. La torta de filtro se lavó con un disolvente (éter de petróleo:diclorometano = 2:1, 10 ml) para dar el compuesto del título (300,00 mg, 828,31 ^mol, 38,00 % de rendimiento, 98 % de pureza) en forma de un sólido de color blanco. LC/MS (ESl) m/z: 354,8 (M+1).
Etapa 6:
6-bromo-4-fluoro-2-metil-3-(propen-2-il)-2H-indazol

En atmósfera de nitrógeno, a una solución de 5-bromo-7-fluoro-3-yodo-2-metil-2H-indazol (320,00 mg, 901,56 ^mol, 1,00 equivalente) y 2-isopropenil-4,4,5,5-tetrametil-1,3,2-dioxaborolano (181,80 mg, 1,08 mmol, 1,20 equivalentes) en tetrahidrofurano (8,00 ml) y agua (5,00 ^l-) se le añadió carbonato potásico (373,81 mg, 2,70 mmol, 3,00 equivalentes) y Pd(dppf)Cl2 (131,93 mg, 180,31 ^mol, 0,20 equivalentes). La mezcla de reacción se agitó a 50-60 °C durante 16 horas. La LC/MS mostró el 55 % del producto objetivo. La TLC mostró conversión completa del material de partida. La mezcla se filtró y el filtrado se concentró a sequedad. El residuo concentrado se purificó por cromatografía en columna (éter de petróleo:acetato de etilo = 10:1) para dar el compuesto del título (210,00 mg, 780,35 ^mol, 86,56 % de rendimiento) en forma de un líquido de color pardo claro. LC/MS (ESI) m/z: 268,9 (M+1). Etapa 7:
4-fluoro-2-metil-6-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-3-(propen-2-il)-2H-indazol

En atmósfera de nitrógeno, a una solución de 5-bromo-7-fluoro-3-isopropenil-2-metil-2H-indazol (302,00 mg, 1,12 mmol, 1,00 equivalente) y bis(pinacolato)diboro (341,30 mg, 1,34 mmol, 1,20 equivalentes) en tetrahidrofurano (10,00 ml) se le añadió 3 gotas de agua, Pd(dppf}Cl2 (163,90 mg, 224,00 ^mol, 0,20 equivalente) y acetato potásico (329,75 mg, 3,36 mmol, 3,00 equivalentes). La mezcla se agitó a 80-90 °C durante 14 horas. TLC (éter de petróleo:acetato de etilo = 5:1) mostró que se había completado la reacción de la mayoría del material de partida. La mezcla se enfrió a 25 °C y se filtró. El filtrado se concentró a sequedad. El residuo concentrado se purificó por cromatografía en columna (éter de petróleo:acetato de etilo = 8:1) para dar el compuesto del título (306,00 mg, 967,80 ^mol, 86,41 % de rendimiento) en forma de un sólido de color blanco. LC/MS (ESI) m/z: 317,2 (M+1).
Etapa 8:
5-(2-cloro-5-fluoropirimidin-4-il)-7-fluoro-2-metil-3-(propen-2-il)-2H-indazol

En atmósfera de nitrógeno, a una solución de 2,4-dicloro-5-fluoro-pirimidina (177,44 mg, 1,06 mmol, 1,20 equivalentes) y 7-fluoro-3-isopropenil-2-metil-5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)indazol (280,00 mg, 885,57 ^mol, 1,00 equivalente) en tetrahidrofurano se le añadió carbonato potásico (367,18 mg, 2,66 mmol, 3,00 equivalentes), Pd(dppf)Cl2 (129,59 mg, 177,11 ^mol, 0,20 equivalentes) y 3 gotas de agua. La mezcla se agitó a 80 90 °C durante 16 horas. TLC (éter de petróleo:acetato de etilo = 5:1) mostró conversión de la mayoría del material de partida. La mezcla se enfrió a 25 °C y se filtró. La torta de filtro se lavó con acetato de etilo (3 ml x 2) y el filtrado se concentró a sequedad. El residuo obtenido por concentración se purificó por cromatografía en columna (éter de petróleo:acetato de etilo = 7:1) para dar el compuesto del título (Intermedio D) (260,00 mg, 802,57 ^mol, 90,63 % de rendimiento, 99 % de pureza). LC/MS (ESI) m/z: 320,9 (M+1).
Ejemplo 22
5-fluoro-4-(7-fluoro-3-isopropil-2-metil-2H-indazol-5-il)-W-(5-(piperazin-1-il)piridin-2-il)pirimidin-2-amina

Etapa 1:
4-(6-((5-fluoro-4-(7-fluoro-2-metil-3-(isopropen-2-il)-2H-indazol-5-il)pirimidin-2-il)amino)piridin-3-il)piperazin-1-carboxilato de ferc-butilo

En atmósfera de nitrógeno, a una solución de 5-(2-cloro-5-fluoropirimidin-4-il)-7-fluoro-2-metil-3-(isopropen-2-il)-2H-indazol (Intermedio D) (100 mg, 311,80 mmol, 1,00 equivalente) en dioxano (3 ml) se le añadió 4-(6-amino-3-piridil)piperazin-1-carboxilato de ferc-butilo (99,81 mg, 358,57 mmol, 1,15 equivalentes), carbonato de cesio (203,18 mg, 623,6 mmol, 2,00 equivalentes), Pd2(dba)3 (57,1 mg, 62,36 jmol, 0,2 equivalentes) y Xantphos (72,17 mg, 124,72 jmol, 0,40 equivalentes). La mezcla se purgó con nitrógeno tres veces y se calentó a 100 °C y se agitó durante 18 horas. La LC/MS mostró conversión completa del material de partida y detectó el compuesto objetivo. La solución de reacción se enfrió a 25 °C, se diluyó con diclorometano (5 ml) y se filtró. El residuo obtenido al concentrar el filtrado se purificó por TLC preparativa (acetato de etilo) para dar el compuesto del título (100,00 mg, 177,74 |jmol, 57,01 % de rendimiento) en forma de un sólido de color amarillo. RMN 1H (400 MHz, CDCh) 88,38 (d, J = 3,9 Hz, 1H), 8,30 (s, 1H), 8,03 (d, J= 2,8 Hz, 1H), 7,80 (d, J= 12,7 Hz, 1H), 7,38 (dd, J= 9,0, 2,9 Hz, 1H), 5,75 5,69 (m, 1H), 5,37 (s, 1H), 4,23 (s, 3H), 3,67-3,56 (m, 4H), 3,17-3,04 (m, 4H), 2,31 (s, 3H), 1,50 (s, 9H).
Etapa 2:
4-(6-((5-fluoro-4-(7-fluoro-3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-il)amino)piridin-3-il)piperazin-1-carboxilato de ferc-butilo

A una solución mezclada de 4-(6-((5-fluoro-4-(7-fluoro-2-metil-3-(isopropen-2-il)-2H-indazol-5-il)pirimidin-2-il)amino)piridin-3-il)piperazin-1 -carboxilato de ferc-butilo (100,00 mg, 177,74 jmol, 1,00 equivalente) en metanol (10,00 ml) y ácido acético (500 j l) se le añadió cloruro de tris(trifenilfosfina)rodio (49,33 mg, 53,32 jmol, 0,3 equivalentes). El matraz de reacción se purgó con argón e hidrógeno tres veces. En atmósfera de hidrógeno a presión (244,74 kPa (50 psi)), la mezcla se calentó a 50 °C y se agitó durante 18 horas. La LC/MS mostró conversión completa del material de partida y detectó el compuesto objetivo. La solución de reacción se enfrió a 25 °C y se filtró. El residuo obtenido concentrar el filtrado se purificó por TLC preparativa (acetato de etilo) para dar el compuesto del título (15 mg, producto en bruto) en forma de un sólido de color amarillo. LC/MS (ESI) m/z: 565,3 (M+1).
Etapa 3:
5-fluoro-4-(7-fluoro-3-isopropil-2-metil-2H-indazol-5-il)-W-(5-(piperazin-1-il)piridin-2-il)pirimidin-2-amina

A una solución de 4-(6-((5-fluoro-4-(7-fluoro-3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-il)amino)piridin-3-il)piperazin-1-carboxilato de ferc-butilo (15 mg, 26,57 jmol, 1,00 equivalente) en diclorometano (1 ml) se le añadió ácido trifluoroacético (500 jl). La mezcla se agitó a 25 °C durante 1 hora. La LC/MS mostró conversión completa del material de partida y detectó el compuesto objetivo. El residuo obtenido al concentrar la solución de reacción se purificó por HPLC preparativa (ácido clorhídrico) para dar el compuesto del título (3,88 mg, 8,27 jmol), 31,12 % de rendimiento, 99 % de pureza). RMN 1H (400 MHz, Metanol-d4) 88,73 (d, J = 3,9 Hz, 1H), 8,63 (s, 1H), 8,27 (dd, J= 2,6, 9,7 Hz, 1H), 7,91 (d, J= 2,4 Hz, 1H), 7,84 (d, J= 13,2 Hz, 1H), 7,55 (d, J= 9,7 Hz, 1H), 4,22 (s, 3H), 3,71-3,61 (m, 1H), 3,60-3,51 (m, 4H), 3,49-3,41 (m, 4H), 1,59 (d, J= 7,0 Hz, 6H) LC/MS (ESI)m/z: 465,2 (M+1).
Ejemplo 23
2-[4-[6-[[5-fluoro-4-(7-fluoro-3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-il]amino]-3-piridinil]piperazin-1-il]etanol

Etapa 1:
2-[4-[6-[[5-fluoro-4-(7-fluoro-3-isopropil-2-metil-2H-indazol-5-il)pirimidin-2-il]amino]-3-piridinil]piperazin-1-il]etanol

A una solución de 5-fluorc>-4-(7-fluoro-3-isoprc>pil-2-metil-2H-indazol-5-il)-W-(5-piperazin-1-il-2-piridil)pirimidin-2-amina (80,00 mg, 172,22 ^mol, 1,00 equivalente) y 2-bromoetanol (64,56 mg, 516,66 ^mol, 3,00 equivalentes) en acetonitrilo (5,00 ml) se le añadió diisopropiletilamina (66,77 mg, 516,66 ^mol, 3,00 equivalentes). La mezcla se calentó a 70 °C y se agitó durante 16 horas. La LC/MS mostró consumición casi completa del material de partida y la MS detectó el compuesto deseado. La mezcla de reacción se concentró a presión reducida para dar un residuo. El residuo se diluyó con acetato de etilo (10 ml) y se lavó sucesivamente con agua (5 x 3 ml) y salmuera (3 X 5 ml). La fase orgánica se secó sobre sulfato sódico y se filtró. El filtrado se concentró para dar un residuo. Este residuo se purificó por HPLC preparativa (condiciones alcalinas) para dar el compuesto del título (20,31 mg, 39,14 ^mol, 22,72 % de rendimiento, 98 % de pureza). RMN 1H (400 MHz, metanol-cf4) 58,59 (s, 1H), 8,48 (d, J= 3,5 Hz, 1H), 8,18 (d, J= 8,8 Hz, 1H), 8,01 (s a, 1H), 7,80 (d, J= 12,5 Hz, 1H), 7,73-7,63 (m, 1H), 4,22 (s, 3H), 3,76 (t, J= 5,8 Hz, 2H), 3,71 3,60 (m, 1H), 3,24 (s a, 5H), 2,76 (s a, 4H), 2,64 (t, J= 5,5 Hz, 2H), 1,60 (d, J= 7,0 Hz, 6H). LC/MS (ESI) m/z: 509,3 (M+1).
Enfoque D
A continuación se muestra un método general para la preparación del intermedio E.

Etapa 1:
3-isobutiril-4-oxopiperidin-1-carboxilato de bencilo

A 0 °C, a una solución de 4-oxopiperidin-1-carboxilato de bencilo (128,07 g, 549,04 mmol, 1,95 equivalentes) en tolueno (600,00 ml) se le añadió lentamente LiHMDS (550 ml, 550,00 mmol, 1,95 equivalentes) gota a gota. La mezcla se agitó a 0 °C durante 1 hora. Después, se añadió cloruro de 2-metilpropionilo (30,00 g, 281,56 mmol, 1,00 equivalente) lentamente gota a gota a la mezcla de reacción a 0 °C. Después de que se completara la adición, la mezcla se calentó a 25 °C y se agitó durante 18 horas. La TLC (éter de petróleo:acetato de etilo = 5:1) mostró que se había completado la reacción del material de partida. La reacción se interrumpió añadiendo una solución acuosa al 10 % de ácido acético (100 ml) a la mezcla de reacción. La mezcla resultante se sometió a la fase de separación. La capa orgánica se lavó sucesivamente con agua (500 ml) y salmuera saturada (500 ml). La capa orgánica resultante se secó sobre sulfato sódico anhidro y se filtró. El filtrado se concentró para dar un residuo, que se purificó después por cromatografía en columna sobre gel de sílice (éter de petróleo:acetato de etilo = 50:1 a 20:1) para dar el compuesto del título (57,00 g, producto en bruto) en forma de un aceite de color amarillo claro. RMN 1H (400 MHz, CDCla) 816,10 (s, 1H), 7,38-7,36 (m, 5H), 5,18 (s, 2H), 4,33 (s, 2H), 3,69-3,66 (m, 2H), 2,68-2,57 (m, 1H), 2,48 (s a, 2H), 1,14 (d, J= 6,4 Hz, 6H).
Etapa 2:
6,7-dihidro-3-isopropil-2-metil-2H-pirazolo[4,3-c]piridin-5(4H)-carboxilato de bencilo

A 25 °C, a una solución de 3-isobutiril-4-oxopiperidin-1-carboxilato de bencilo (57,00 g, 187,90 mmol, 1,00 equivalente) en etanol (600,00 ml) se le añadió una solución acuosa al 40% de metilhidrazina (110,00 g, 954,53 mmol, 5,08 equivalentes). La mezcla se agitó durante 1 hora. La LC/MS mostró que se había completado la reacción del material de partida. La solución de reacción se concentró para dar un producto en bruto. El producto en bruto se purificó por HPLC preparativa (ácido trifluoroacético) para dar una solución integral. El acetonitrilo de la solución integral se eliminó al concentrar la solución. El pH de la fase acuosa se ajustó a 8 con bicarbonato sódico y después la fase acuosa se sometió a extracción dos veces usando diclorometano (500 ml x 2). Las capas orgánicas combinadas se secaron sobre sulfato sódico y se filtraron. El filtrado se concentró para dar el compuesto del título (6,50 g, 20,74 mmol, 11,4 % de rendimiento) en forma de un aceite incoloro. RMN 1H (400 MHz, CDCla) 87,43-7,29 (m, 5H), 5,18 (s, 2H), 4,58 (s, 2H), 3,82-3,71 (m, 5H), 3,03 (quint., J= 7,0 Hz, 1H), 2,82-2,67 (m, 2H), 1,28 (d, J= 6,4 Hz, 6H).
Etapa 3:
3-isopropil-2-metil-4,5,6,7-tetrahidro-2H-pirazolo[4,3-c]piridina

A 25 °C, a una solución de 6,7-dihidro-3-isopropil-2-metil-2H-pirazolo[4,3-c]piridin-5(4H)-carboxilato de bencilo (6,50 g, 20,74 mmol, 1,00 equivalente) en metanol (100,00 ml) se le añadió Pd/C (1,00 g) y una solución de ácido clorhídrico (1,00 ml) con una concentración de 12 mol/l. La mezcla se purgó sucesivamente con nitrógeno e hidrógeno tres veces. Se introdujo hidrógeno en la solución de reacción y la mezcla se agitó a una presión de 244,74 kPa (50 psi) durante 2 horas. La TLC (éter de petróleo: acetato de etilo =1:1) mostró que se había completado la reacción del material de partida. La mezcla de reacción se filtró y el filtrado se concentró para dar el compuesto del título (5,25 g, producto en bruto) en forma de un sólido de color blanquecino. RMN 1H (400 MHz, CDCh) 89,93 (s a, 2H), 4,31 (s a, 2H), 3,76 (s, 3H), 3,09-2,98 (m, 3H), 2,10 (s a, 2H), 1,26 (d, J= 7,0 Hz, 6H).
Etapa 4:
5-(2-cloro-5-fluoropirimidin-4-il)-3-isopropil-2-metil-4,5,6,7-tetrahidro-2H-pirazolo[4,3-c]piridina

Intermedio E
A 25 °C, a una solución de 3-isopropil-2-metil-4,5,6,7-tetrahidro-2H-pirazolo[4,3-c]piridina (5,25 g, 20,75 mmol, 1,00 equivalente) en tetrahidrofurano (60,00 ml) se le añadió 2,4-didoro-5-fluoropirimidina (3,50 g, 20,96 mmol, 1,01 equivalentes ) y trietilamina (9,51 g, 94,01 mmol, 4,53 equivalentes). La mezcla de reacción se agitó durante 18 horas. La TLC (éter de petróleo:acetato de etilo = 3:1) mostró que se había completado la reacción del material de partida. La solución de reacción se diluyó con agua (200 ml) y se sometió a extracción usando acetato de etilo (200 ml x 2). Las capas orgánicas combinadas se secaron sobre sulfato sódico anhidro, se filtraron y se concentraron para dar un producto en bruto. El producto en bruto se purificó por cromatografía en columna (éter de petróleo:acetato de etilo = 5:1 a 2:1) para dar el compuesto del título (Intermedio E) (5,60 g, 18,08 mmol, 87,12 % de rendimiento) en forma de un sólido de color blanquecino. RMN 1H (300 MHz, CDCh) 8 7,94 (d, J= 6,0 Hz, 1H), 4,84 (s, 2H), 4,03 (t, J= 5,8 Hz, 2H), 3,79 (s, 3H), 3,07 (spt, J= 7,1 Hz, 1H), 2,88 (t, J= 5,7 Hz, 2H), 1,32 (d, J= 7,2 Hz, 6H).
Ejemplo 24
5-fluoro-4-(6,7-d¡h¡dro-3-¡soprop¡l-2-met¡l-2H-p¡razolo[4,3-c]p¡r¡d¡n-5(4H)-¡l)-A/-(5-(1-met¡lp¡perid¡n-4-¡l)p¡r¡d¡n-2-il)piridin-2-amina

Etapa 1:
6-nitro-5',6'-dihidro-[3,4'-bipiridil]-1'(2'H)carboxilato de tere-butilo

En atmósfera de nitrógeno, a una solución mezclada de 5-bromo-2-nitropiridina (10,00 g, 49,26 mmol, 1,02 equivalentes) en dioxano (120 ml) y agua (10 ml) se le añadió 4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-3,6-dihidro-2H-piridin-1 -carboxilato de tere-butilo (15,00 g, 48,51 mmol, 1,00 equivalente), carbonato potásico (5,9 g, 72,77 mmol, 1,50 equivalentes) y Pd(dppf)Ch (1,77 g, 2,43 mmol, 0,05 equivalente). La mezcla de reacción se calentó a 80 °C y se agitó durante 18 horas. La TLC (éter de petróleo:acetato de etilo = 3:1) mostró que se había completado la reacción del material de partida. La solución de reacción se filtró, se secó sobre sulfato sódico anhidro, se filtró y se concentró para dar un producto en bruto. El producto en bruto se purificó por cromatografía en columna (éter de petróleo:acetato de etilo = 3:1) para dar el compuesto del título (18,70 g, producto en bruto) en forma de un sólido de color amarillo claro. RMN 1H (400 MHz, CDCla) 88,64 (d, J= 2,0 Hz, 1H) 8,24 (d, J= 8,4 Hz, 1H) 7,95 (dd, J= 8,5, 2,3 Hz, 1H) 6,33 (s a, 1H) 4,16 (d, J= 2,6 Hz, 2H) 3,69 (t, J= 5,6 Hz, 2H) 2,57 (s a, 2H) 1,50 (s, 9H).
Etapa 2:
4-(6-aminopiridin-3-il)piperidin-1 -carboxilato de tere-butilo

A una solución de 4-(6-nitro-3-piridil)-3,6-dihidro-2H-piridin-1 -carboxilato de tere-butilo (18,70 g, 48,51 mmol, 1,00 equivalente) en metanol (500 ml) se le añadió Pd/C (2,00 g). El sistema de reacción se purgó sucesivamente con nitrógeno e hidrógeno tres veces. El hidrógeno se introdujo en la solución de reacción para mantener el sistema de reacción a una presión de 244,74 kPa (50 psi). La mezcla de reacción se calentó a 50 °C y se agitó durante 18 horas. La LC/MS mostró que se había completado la reacción del material de partida. La mezcla se filtró y el filtrado se concentró para dar el compuesto del título (10,40 g, 37,50 mmol, 77,31 % de rendimiento) en forma de un sólido de color blanquecino. RMN 1H (400 MHz, CDCla) 87,92 (d, J= 2,4 Hz, 1H) 7,31-7,27 (m, 1H) 6,48 (d, J = 8,5 Hz, 1H) 4,35 (s a, 2H) 4,23 (s a, 2H) 2,78 (t, J= 12,1 Hz, 2H) 2,60-2,48 (m, 1H) 1,78 (s a, 2H) 1,61-1,52 (m, 2H) 1,48 (s, 9H). Etapa 3:
5-(1-metilpiperidin-4-il)piridin-2-amina

A 0 °C, a una solución de hidruro de litio y aluminio (2,05 g, 54,09 mmol, 3,00 equivalentes) en tetrahidrofurano (100 ml) se le añadió lentamente una solución de 4-(6-amino-3-piridinil)piperidin-1-carboxilato de ferc-butilo (5,00 g, 3,18 mmol, 1,00 equivalente) en tetrahidrofurano (50 ml) gota a gota. Después de que se completara la adición, la mezcla se calentó a 70 °C y se agitó durante 2 horas. La LC/MS mostró que se había completado la reacción del material de partida. A 0 °C, la reacción se interrumpió añadiendo una solución acuosa al 20 % de hidróxido potásico (4 ml) a la mezcla de reacción para producir una gran cantidad de sólido. La mezcla de reacción se filtró y el filtrado se concentró para dar el compuesto del título (4,00 g, producto en bruto) en forma de un sólido de color blanco. RMN 1H (400 MHz, CDCla) 87,93 (d, J= 2,3 Hz, 1H) 7,31 (dd, J= 8,4, 2,4 Hz, 1H) 6,46 (d, J= 8,4 Hz, 1H) 4,33 (s a, 2H) 2,95 (d, J= 11,7 Hz, 2H) 2,41-2,32 (m, 1H) 2,31 (s, 3H) 2,04-1,98 (m, 2H) 1,80-1,67 (m, 4H).
Etapa 4:
5-fluoro-4-(3-¡soprop¡l-2-met¡l-6,7-d¡h¡dro-2H-p¡razolo[4,3-c]p¡r¡d¡n-5(4H)-¡l)-A/-(5-(1-met¡lp¡perid¡n-4-¡l)p¡r¡d¡n-2-il)pirimidin-2-amina

En atmósfera de nitrógeno, a una solución de 5-(2-cloro-5-fluoropirimidin-4-il)-3-isopropil-2-metil-4,5,6,7-tetrahidro-2H-pirazolo[4,3-c]piridina (Intermedio E) (2,00 g, 6,46 mmol, 1,00 equivalente) en tolueno (20,00 ml) se le añadió 5-(1-metil-4-piperidinil)piridin-2-amina (1,36 g, 7,10 mmol, 1,10 equivalentes), carbonato de cesio (4,21 g, 12,91 mmol, 2,00 equivalentes), Pd2(dba)3 (400,00 mg, 436,81 pmol, 0,07 equivalentes) y Xantphos (520,00 mg, 898,69 pmol, 0,14 equivalentes). La mezcla se calentó a 110 °C y se agitó durante 18 horas. La LC/MS mostró que se había completado la reacción del material de partida. La mezcla de reacción se enfrió a 20 °C y se concentró para dar un residuo. El residuo se purificó por cromatografía en columna (acetato de etilo a diclorometano:metanol = 10:1) para dar un producto en bruto. El producto en bruto se purificó por HPLC preparativa (alcalina) para dar el compuesto del título (2,48 g, 4,61 mmol, 71,42 % de rendimiento). RMN 1H (400 MHz, M etano l^) 88,27 (d,J= 1,9 Hz, 1H), 8,13 (d, J = 7,2 Hz, 1H), 8,01 (d, J = 8,5 Hz, 1H), 7,45 (d, J= 8,8 Hz, 1H), 5,01 (s, 2H), 4,16 (t, J= 5,8 Hz, 2H), 3,77 (s, 3H), 3,65 (d, J= 12,3 Hz, 2H), 3,26-3,12 (m, 3H), 3,09-2,98 (m, 1H), 2,93 (s, 3H), 2,86 (t, J= 5,8 Hz, 2H), 2,22-1,98 (m, 4H), 1,34 (d, J= 7,0 Hz, 6H).
Ejemplo 25
W-(5-fluoro-4-(3-isopropil-2-metil-6,7-dihidro-2H-pirazolo[4,3-c]piridin-5(4H)-il)pirimidin-2-il)-6-(4-metilpiperazin-1-il)piridazin-3-amina

En atmósfera de nitrógeno, a una solución de 5-(2-cloro-5-fluoropirimidin-4-il)-3-isopropil-2-metil-4,5,6,7-tetrahidro-2H-pirazolo[4,3-c]piridina (Intermedio E) (200,00 mg, 645,64 pmol, 1,00 equivalente) y 6-(4-metilpiperazin-1-il)piridazin-3-amina (124,77 mg, 645,64 pmol, 1,00 equivalente) en dioxano (5,00 ml) se le añadió carbonato de cesio (420,72 mg, 1,29 mmol, 2,00 equivalentes), Pd2(dba)3 (59,12 mg, 64,56 pmol, 0,10 equivalentes) y Xantphos (74,72 mg, 129,13 pmol, 0,20 equivalentes). Después, la mezcla de reacción se calentó a 110 °C y se agitó durante 16 horas. La LC/MS mostró que se había completado la reacción del material de partida. La mezcla de reacción se enfrió a 25 °C, se filtró y se concentró a presión reducida para dar un producto en bruto. El producto en bruto se purificó por HPLC preparativa (alcalina) para dar el compuesto del título (132,29 mg, 283,54 pmol, 43,92 % de rendimiento). RMN 1H (400 MHz, Metanol-d4) 8 8,23 (d, J= 9,91 Hz, 1H), 7,93 (d, J= 6,90 Hz, 1H), 7,34 (d,J= 9,91 Hz, 1H), 4,85 (s, 2H), 4,02 (t, J= 5,84 Hz, 2H), 3,77 (s, 3H), 3,63-3,57 (m, 4H), 3,17 (c, J= 7,00 Hz, 1H), 2,81 (t, J= 5,77 Hz, 2H), 2,65-2,59 (m, 4H), 2,38 (s, 3H), 1,32 (d, J= 7,03 Hz, 6H). LC/MS (ESI) m/z: 467,2 (M+1).
Ejemplo 26
5-fluoro-4-(6,7-dihidro-3-isopropil-2-metil-2H-pirazolo[4,3-c]piridin-5(4H)-il)-A/-(5-(4-metil-1,4-diazepan-1-il)piridin-2
il)pirimidin-2-amina

Etapa 1:
1-metil-4-(6-nitropiridin-3-il)-1,4-diazepano

A una solución de 5-bromo-2-nitro-piridina (2,00 g, 9,85 mmol, 1,00 equivalente) y 1-metil-1,4-diazepano (1,69 g, 14,78 mmol, 1,50 equivalentes) en dimetilsulfóxido (20,00 ml) se le añadió carbonato potásico (2,72 g, 19,70 mmol, 2,00 equivalentes). La mezcla se calentó a 80 °C y se agitó durante 16 horas. La LC/MS mostró que se había completado la reacción del material de partida. La mezcla de reacción se enfrió a 25 °C y se añadió agua (50 ml). La mezcla se sometió después a extracción usando acetato de etilo (100 ml x 3). Las fases orgánicas combinadas se lavaron con salmuera saturada (100 ml x 3), se secaron sobre sulfato sódico anhidro, se filtraron y se concentraron para dar un producto en bruto. El producto en bruto se purificó por cromatografía en columna (éter de petróleo:acetato de etilo = 30:1 a 1:3) para dar el compuesto del título (2,00 g, 8,46 mmol, 85,94 % de rendimiento) en forma de un sólido de color blanco. LC/MS (ESI) m/z: 237,1 (M+1).
Etapa 2:
5-(4-metil-1,4-diazepan-1-il)piridin-2-amina

A 25 °C, a una solución de 1-metil-4-(6-nitropiridin-3-il)-1,4-diazepano (2,00 g, 8,46 mmol, 1,00 equivalente) en metanol (20,00 ml) se le añadió Pd/C (10 %, 500 mg). El sistema de reacción se purgó sucesivamente con nitrógeno e hidrógeno tres veces. El hidrógeno se introdujo en la solución de reacción para mantener el sistema de reacción a una presión de 244,74 kPa (15 psi). La solución de reacción se agitó durante 2 horas. La TLC mostró que se había completado la reacción del material de partida. La mezcla de reacción se filtró y se concentró a presión reducida para dar el compuesto del título (1,70 g, 8,24 mmol, 97,41 % de rendimiento) en forma de un sólido de color blanco. LC/MS (ESI) m/z: 207,1 (M+1).
Etapa 3:
5-fluoro-4-(6,7-d¡h¡dro-3-¡soprop¡l-2-met¡l-2H-p¡razolo[4,3-6]p¡r¡d¡n-5(4H)-¡l)-A/-(5-(4-met¡l-1,4-d¡azepan-1-¡l)p¡r¡d¡n-2-il)pirimidin-2-amina

En atmósfera de nitrógeno, a una solución de 5-(2-cloro-5-fluoropirimidin-4-il)-3-isopropil-2-metil-4,5,6,7-tetrahidro-2H-pirazolo[4,3-c]piridina (Intermedio E) (200,00 mg, 645,64 jmol, 1,00 equivalente) y 5-(4-metil-1,4-diazepan-1-il)piridin-2-amina (159,83 mg, 774,77 |jmol, 1,20 equivalentes) en dioxano (5,00 ml) se le añadió carbonato de cesio (420,72 mg, 1,29 mmol, 2,00 equivalentes), Pd2(dba)3 (59,12 mg, 64,56 jmol, 0,10 equivalentes) y Xantphos (74,72 mg, 129,13 jmol, 0,20 equivalentes). Después, la mezcla de reacción se calentó a 110 °C y se agitó durante 16 horas. La LC/MS mostró que se había completado la reacción del material de partida. La mezcla de reacción se enfrió a 25 °C, se filtró y se concentró a presión reducida para dar un producto en bruto. El producto en bruto se purificó por HPLC preparativa (alcalina) para dar el compuesto del título (93,10 mg, 194,12 jmol, 30,07% de rendimiento). RMN 1H (400 MHz, metanol-d4) 5 7,91-7,84 (m, 2H), 7,77 (d, J= 3,01 Hz, 1H), 7,22 (dd, J= 9,16, 3,14 Hz, 1H), 4,85 (s, 2H), 4,01 (t, J= 5,83 Hz, 2H), 3,77 (s, 3H), 3,62-3,57 (m, 2H), 3,52 (t, J= 6,27 Hz, 2H), 3,17 (c, J= 7,03 Hz, 1H), 2,83-2,75 (m, 4H), 2,67-2,61 (m, 2H), 2,40 (s, 3H), 2,06 (dt, J= 11,51, 5,98 Hz, 2H), 1,36-1,29 (m, 6H). LC/MS (ESI) m/z: 480,2 (M+1).
Ejemplo 27
W-(5-(4-(d¡met¡lam¡no)p¡per¡d¡n-1-¡l)p¡r¡d¡n-2-¡l)-5-fluoro-4-(6,7-d¡h¡dro-3-¡soprop¡l-2-met¡l-2H-p¡razolo[4,3-c]p¡r¡d¡n-5(4H)-il)pirimidin-2-amina

Etapa 1:
W,W-d¡met¡l-1-(6-n¡trop¡r¡d¡n-3-¡l)p¡per¡d¡n-4-am¡na

En atmósfera de n¡trógeno, a una soluc¡ón de W,W-d¡met¡lp¡per¡d¡n-4-am¡na (100,00 mg, 779,97 |jmol, 1,00 equ¡valente) y 5-bromo-2-n¡trop¡r¡d¡na (158,33 mg, 779,97 |jmol, 1,00 equ¡valente) en d¡oxano (5 ml) se le añad¡ó BINAP (48,57 mg, 78,00 pmol, 0,10 equ¡valentes), carbonato de ces¡o (508,26 mg, 1,56 mmol, 2,00 equivalentes) y Pd(OAc)2 (17,51 mg, 78,00 pmol, 0,10 equivalentes). Después, la mezcla de reacc¡ón se calentó a 90 °C y se ag¡tó durante 16 horas. La TLC mostró que se había completado la reacc¡ón del mater¡al de part¡da. La mezcla de reacc¡ón se f¡ltró y se concentró para dar un producto en bruto. El producto en bruto se pur¡f¡có por TLC preparativa (d¡clorometano:metanol = 10:1) para dar el compuesto del título (125,00 mg, 499,40 pmol, 64,03 % de rend¡m¡ento) en forma de un sól¡do de color amar¡llo. RMN 1H (400 MHz, CDCla) 8 8,20-8,14 (m, 2H), 7,21 (dd, J= 9,2, 3,2 Hz, 1H), 3,99 (d, J= 13,2 Hz, 2H), 3,12-3,02 (m, 2H), 2,47-2,39 (m, 1H), 2,33 (s, 6H), 2,01 (d, J = 12,4 Hz, 2H), 1,70-1,64 (m, 2H). LC/MS (ESI) m/z: 251,1 (M+1).
Etapa 2:
5-(4-(d¡met¡lam¡no)p¡per¡d¡n-1-¡l)p¡r¡d¡n-2-am¡na

A una soluc¡ón de W,A/-d¡met¡l-1-(6-n¡trop¡r¡d¡n-3-¡l)p¡per¡d¡n-4-am¡na (525,00 mg, 2,10 mmol, 1,00 equivalente) en metanol (10 ml) se le añad¡ó Pd-C (10 %, 100 mg). El s¡stema de reacc¡ón se purgó sucesivamente con n¡trógeno e h¡drógeno tres veces. El h¡drógeno se ¡ntrodujo en la soluc¡ón de reacc¡ón para mantener el s¡stema de reacc¡ón a una pres¡ón de 244,74 kPa (15 ps¡). La soluc¡ón de reacc¡ón se ag¡tó durante 4 horas. La TLC mostró que se había completado la reacc¡ón del mater¡al de part¡da. La mezcla de reacc¡ón se f¡ltró, y el f¡ltrado se concentró para dar el compuesto del título (450,00 mg, 2,04 mmol, 97,27 % de rend¡m¡ento) en forma de un sól¡do de color amar¡llo. RMN 1H (400 MHz, metanol-d4) 87,65-7,61 (m, 1H), 7,34 (dd, J= 9,2, 2,4 Hz, 1H), 6,59 (dd, J= 8,8, 0,8 Hz, 1H), 3,52-3,44 (m, 2H), 2,65 (dt, J= 12,4, 2,4 Hz, 2H), 2,42-2,38 (m, 6H), 2,05-1,98 (m, 2H), 1,67 (dc, J= 12,4, 4,4 Hz, 2H). LC/MS (ESI) m/z: 221,0 (M+1).
Etapa 3:
W-(5-(4-(d¡met¡lam¡no)p¡per¡d¡n-1-¡l)p¡r¡d¡n-2-¡l)-5-fluoro-4-(6,7-d¡h¡dro-3-¡soprop¡l-2-met¡l-2H-p¡razolo[4,3-c]p¡r¡d¡n-5(4H)-¡l)p¡r¡m¡d¡n-2-am¡na

En atmósfera de n¡trógeno, a una soluc¡ón de 5-(2-cloro-5-fluorop¡r¡m¡d¡n-4-¡l)-3-¡soprop¡l-2-met¡l-4,5,6,7-tetrah¡dro-2H-p¡razolo[4,3-c]p¡r¡d¡na (Intermedio E) (150,00 mg, 484,23 pmol, 1,00 equ¡valente) y 5-[4-(d¡met¡lam¡no)-1-p¡per¡d¡n¡l]p¡r¡d¡n-2-am¡na (128,02 mg, 581,08 pmol, 1,20 equivalentes) en d¡oxano (5,00 ml) se le añad¡ó Pd2(dba)3 (44,34 mg, 48,42 pmol, 0,10 equ¡valentes), carbonato de ces¡o (315,54 mg, 968,46 pmol, 2,00 equivalentes) y Xantphos (56,04 mg, 96,85 pmol, 0,20 equ¡valentes). Después, la mezcla de reacc¡ón se calentó a 110 °C y se ag¡tó durante 16 horas. La LC/MS mostró que se había completado la reacc¡ón del mater¡al de part¡da. La mezcla de
reacción se enfrió a 25 °C, se filtró y se concentró para dar un producto en bruto. El producto en bruto se purificó por HPLC preparativa (ácido cítrico) para dar la mezcla del título (81,64 mg, 165,39 |jmo¡, 34,16 % de rendimiento). RMN 1H (400 MHz, metanol-d4) 88,01-7,86 (m, 3H), 7,51 (dd, J= 9,16, 2,76 Hz, 1H), 4,85 (s, 2H), 4,06-3,96 (m, 2H), 3,84 3,72 (m, 5H), 3,38-3,33 (m, 1H), 3,17 (dt, J= 14,02, 6,98 Hz, 1H), 2,90 (s, 6H), 2,85-2,77 (m, 4H), 2,22 (d, J = 12,05 Hz, 2H), 1,90 (cd, J = 12,03, 3,70 Hz, 2H), 1,32 (d, J = 7,03 Hz, 6H). LC/MS (ESI) m/z: 494,2 (M+1).
Ejemplo 28
W5-(2-(dimetilamino)etil)-W2-(5-fluoro-4-(6,7-dihidro-3-isopropil-2-metil-2H-pirazolo[4,3-c]piridin-5(4H)-il)pirimidin-2-il)-W5-metilpiridin-2,5-diamina

Etapa 1:
W-(2-(dimetilamino)etil)-A/-metil-6-nitropiridin-3-amina

A una solución de 5-bromo-2-nitro-piridina (2,00 g, 9,85 mmol, 1,00 equivalente) y W,W,W-trimetiletan-1,2-diamina (1,51 g, 14,78 mmol, 1,50 equivalentes) en dimetilformamida (20,00 ml) se le añadió carbonato potásico (2,72 g, 19,70 mmol, 2,00 equivalentes) en una porción. La mezcla de reacción se calentó a 90 °C y se agitó durante 16 horas. La LC/MS mostró que la reacción se había completado. Después de enfriar la solución de reacción a 25 °C, se le añadió agua (50 ml). La mezcla de reacción se sometió a extracción usando acetato de etilo (100 ml x 3). Las fases orgánicas se combinaron y se lavaron con salmuera saturada (100 ml x 3), después se secaron sobre sulfato sódico anhidro, se filtraron y se concentraron para dar un producto en bruto. El producto en bruto se purificó por cromatografía en columna (éter de petróleo:acetato de etilo = 30:1 a 1:3) para dar el compuesto del título (1,50 g, 6,69 mmol, 67,91 % de rendimiento) en forma de un sólido de color blanco. RMN 1H (400 MHz, CDCl3): 8, 8,19-8,15 (m, 1H), 7,99 (d, J= 3,14 Hz, 1H), 7,02 (dd, J = 9,29, 3,14 Hz, 1H), 3,63-3,57 (m, 2H), 3,16 (s, 3H), 2,57-2,51 (m, 2H), 2,31 (s, 6H). LC/MS (ESI) m/z: 225,1 (M+1).
Etapa 2:
W3-(2-(dimetilamino)etil)-W3-metilpiridin-3,6-diamina

En atmósfera de nitrógeno, a una solución de W,W,W-trimetil-N'-(6-nitro-3-piridil)etan-1,2-diamina (1,50 g, 6,69 mmol, 1,00 equivalente) en metanol (15,00 ml) se le añadió Pd/C (300 mg). La suspensión se purgó con H2 varias veces y después se agitó a 25 °C en atmósfera de H2 (101,325 kPa (una atmósfera)) durante 2 horas. La TLC mostró que la reacción se había completado. La solución de reacción se filtró y se concentró para dar el compuesto del título (1,20 g, 6,18 mmol, 92,33 % de rendimiento) en forma de un sólido de color blanco que no requirió más purificación. LC/MS (ESI) m/z: 195,1 (M+1).
Etapa 3:
W5-(2-(dimetilamino)etil)-W2-(5-fluoro-4-(6,7-dihidro-3-isopropil-2-metil-2H-pirazolo[4,3-c]piridin-5(4H)-il)pirimidin-2-il)-W5-metilpiridin-2,5-diamina

A un tubo de microondas se le añadió una solución de 5-(2-cloro-5-fluoropirimidin-4-il)-3-isopropil-2-metil-4,5,6,7-tetrahidro-2H-pirazolo[4,3-c]piridina (Intermedio E) (150,00 mg, 484,23 |jmol, 1,00 equivalente), N3-(2-(dimetilamino)etil)-N3-metilpiridin-3,6-diamina (112,89 mg, 581,08 jmol, 1,20 equivalentes), carbonato de cesio (315,54 mg, 968,46 jmol, 2,00 equivalentes), Pd2(dba)3 (88,68 mg, 96,85 jmol, 0,20 equivalentes) y Xantphos (56,04 mg, 96,85 jmol, 0,20 equivalentes) en dioxano (5,00 ml). La solución de reacción se purgó con nitrógeno varias veces, se calentó a 130 °C y se agitó durante 2,5 horas. La LC/MS mostró reacción incompleta del reactivo y el producto representaba aproximadamente el 14%. La solución de reacción se enfrió a 25 °C, se filtró y se concentró para dar un producto en bruto. El producto en bruto se purificó por HPLC preparativa (ácido clorhídrico) para dar el compuesto del título (91,63 mg, 7 jmol, 47 % de rendimiento). RMN 1H (400 MHz, Metanol-c^): 8, 8,25 (d, J= 7,03 Hz, 1H), 8,03 (dd, J= 9,54, 3,01 Hz, 1H), 7,95 (d, J = 2,89 Hz, 1H), 7,46 (d, J= 9,54 Hz, 1H), 5,08 (s, 2H), 4,25 (t, J= 5,58 Hz, 2H), 4,06 (s, 3H), 3,88 (t, J= 7,22 Hz, 2H), 3,45 (t, J = 7,22 Hz, 2H), 3,40-3,35 (m, 1H), 3,15-3,07 (m, 5H), 3,0-22,97 (m, 6H), 1,43 (d, J= 7,03 Hz, 6H). LC/MS (ESI) m/z: 468,3 (M+1).
Ejemplo 29
4-(6-((5-fluoro-4-(6,7-dihidro-3-isopropil-2-metil-2H-pirazolo[4,3-c]piridin-5(4H)-il) pirimidin-2-il)amino)piridin-3-il)piperidin-4-ol

Etapa 1:
4-(6-aminopiridin-3-il)-4-hidroxipiperidin-carboxilato de ferc-butilo

A -70 °C, a una solución de 5-bromopiridin-2-amina (1,00 g, 5,78 mmol, 1,00 equivalente) en tetrahidrofurano (13,00 ml) se le añadió lentamente n-butillitio (2,5 M, 7,07 ml, 3,06 equivalentes) gota a gota. Después de agitar la mezcla a -70 °C durante 1,5 horas, la solución de reacción se calentó a 25 °C y después se agitó durante 2 horas. La LC/MS mostró que aún quedaba aproximadamente el 19 % del reactivo. La LC/Ms mostró varios puntos nuevos y que el producto representaba aproximadamente el 8 %. La solución de reacción se inactivó con agua y una solución saturada de cloruro de amonio a -70 °C y después se calentó a 25 °C. La mezcla se sometió a extracción usando acetato de etilo (50 ml x 3) y las fases orgánicas combinadas se lavaron con salmuera saturada (20 ml x 3), se secaron sobre sulfato sódico anhidro, se filtraron y se concentraron para dar un producto en bruto. El producto en bruto se purificó por HPLC preparativa (alcalina) para dar el compuesto del título (300,00 mg, 1,02 mmol, 17,69 % de rendimiento) en forma de un aceite de color amarillo. RMN 1H (300 MHz, DMSO-d6): 8, 7,98 (d, J= 2,07 Hz, 1H), 7,43 (dd, J= 8,67, 2,45 Hz, 1H), 6,38 (d, J= 8,67 Hz, 1H), 5,75 (s, 2H), 4,89 (s, 1H), 3,79 (d, J= 9,80 Hz, 2H), 3,11 (s a, 2H), 1,77-1,63 (m, 2H), 1,62-1,52 (m, 2H), 1,41 (s, 9H). LC/MS (ESI) m/z: 294,0 (M+1).
Etapa 2:
4-(6-(5-fluoro-4-(6,7-dihidro-3-isopropil-2-metil-2H-pirazolo[4,3-c]piridin-5(4H)-il)pirimidin-2-il-aminopiridin-3-il)-4-hidroxipiperidin-carboxilato de ferc-butilo

En atmósfera de nitrógeno, a una solución de 4-(6-amino-3-piridil)-4-hidroxi-piperidin-1-carboxilato de ferc-butilo (130,00 mg, 443,14 |jmol, 1,00 equivalente) en dioxano (5,00 ml) se le añadió 5-(2-cloro-5-fluoropirimidin-4-il)-3-isopropil-2-metil-4,5,6,7-tetrahidro-2H-pirazolo[4,3-c]piridina (Intermedio E) (164,73 mg, 531,77 jmol, 1,20 equivalentes), Pd2(dba)3 (40,58 mg, 44,31 jmol, 0,10 equivalentes), xantphos (51,28 mg, 88,62 jmol, 0,20 equivalentes) y carbonato de cesio (288,77 mg, 886,28 jmol, 2,00 equivalentes). La mezcla de reacción se agitó a 110 °C durante 16 horas. La LC/MS mostró que la reacción se había completado y se detectó el producto. Cuando la solución de reacción se enfrió a 25 °C, se sometió a filtración por succión. El filtrado se concentró para dar un producto en bruto. El producto en bruto se purificó por TLC preparativa (diclorometano:metanol =10:1) para dar el compuesto del título (220,00 mg, producto en bruto) en forma de un sólido de color amarillo. LC/MS (ESI) m/z: 567,2 (M+1).
Etapa 3:
4-(6-(5-fluoro-4-(6,7-dihidro-3-isopropil-2-metil-2H-pirazolo[4,3-c]piridin-5(4H)-il)pirimidin-2-il-aminopiridin-3-il)piperidin-4-ol

A 0 °C, a una solución de 4-(6-(5-fluoro-4-(6,7-dihidro-3-isopropil-2-metil-2H-pirazolo[4,3-c]piridin-5(4H)-il)pirimidin-2-il-aminopiridin-3-il)-4-hidroxipiperidin-carboxilato de ferc-butilo (220,00 mg, 388,23 jmol, 1,00 equivalente) en diclorometano (4,00 ml) se le añadió ácido trifluoroacético (2,00 ml). La mezcla de reacción se calentó a 25 °C y se agitó durante 1 hora. La LC/MS mostró que la reacción se había completado y se detectó el producto. La solución de reacción se concentró para dar un producto en bruto. El producto en bruto se purificó por HPLC preparativa (ácido clorhídrico) para dar el compuesto del título (115,01 mg, 246,51 jmol, 63,50 % de rendimiento). RMN 1H (300 MHz, Metanol-d4) 88,53 (d, J= 1,9 Hz, 1H), 8,35 (dd, J= 9,0, 2,3 Hz, 1H), 8,29 (d, J= 6,6 Hz, 1H), 7,50 (d, J= 9,0 Hz, 1H), 4,26 (t, J= 5 Hz, 2H), 4,07 (s, 3H), 3,60-3,32 (m, 7H), 3,16-3,06 (m, 2H), 2,47-2,30 (m, 2H), 2,02 (d, J = 13,8 Hz, 2H), 1,49-1,40 (m, 6H). LC/MS (ESI) m/z: 467,2 (M+1).
Ejemplo 30
W-(5-(1,4-diazepan-1-il)piridin-2-il)-5-fluoro-4-(6,7-dihidro-3-isopropil-2-metil-2H-pirazolo[4,3-c]piridin-5(4H)-il)pirimidin-2-amina

Etapa 1:
4-(6-nitropiridin-3-il)-1,4-diazepan-1 -carboxilato de ferc-butilo

A una solución de 5-bromo-2-nitro-piridina (2,00 g, 9,85 mmol, 1,00 equivalente) y 1,4-diazepan-1-carboxilato de ferc-butilo (2,37 g, 11,82 mmol, 1,20 equivalentes) en dimetilsulfóxido (20,00 ml) se le añadió carbonato potásico (2,72 g, 19,70 mmol, 2,00 equivalentes) en una porción. La mezcla de reacción se calentó a 80 °C y se agitó durante 16 horas. La LC/MS mostró que la reacción se había completado y se detectó el producto. La solución de reacción se enfrió a 25 °C y después se añadió agua (50 ml) a la mezcla de reacción. La mezcla resultante se sometió a extracción usando acetato de etilo (100 ml x 3). Las fases orgánicas combinadas se lavaron con salmuera saturada
(100 ml x 3), se secaron sobre sulfato sódico anhidro, se filtraron y se concentraron para dar un producto en bruto. El producto en bruto se purificó por cromatografía en columna (éter de petróleo:acetato de etilo = 30:1 a 1:3) para dar el compuesto del título (2,00 g, 6,20 mmol, 62,99 % de rendimiento) en forma de un sólido de color blanco. LC/MS (ESI) m/z: 323,1 (M+1).
Etapa 2:
4-(6-aminopiridin-3-il)-1,4-diazepan-1-carboxilato de ferc-butilo

En atmósfera de nitrógeno, a una solución de 4-(6-nitropiridin-3-il)-1,4-diazepan-1-carboxilato de ferc-butilo (1,80 g, 5,58 mmol, 1,00 equivalente) en metanol (20,00 ml) se le añadió Pd/C (300 mg). La suspensión se purgó con H2 varias veces y después se agitó a 25 °C en atmósfera de H2 (101,325 kPa (una atmósfera)) durante 2 horas. La TLC mostró que la reacción se había completado y se produjo un punto nuevo. La reacción se filtró y el filtrado se concentró para dar un producto en bruto (1,70 g, producto en bruto) en forma de un sólido de color blanco. El producto en bruto no se purificó y se usó directamente en la etapa siguiente. LC/MS (ESI) m/z: 293,2 (M+1).
Etapa 3:
4-(6-(5-fluoro-4-(6,7-dihidro-3-isopropil-2-metil-2H-pirazolo[4,3-c]piridin-5(4H)-il) pirimidin-2-il-amino)piridin-3-il)-1,4-diazepan-carboxilato de ferc-butilo

En atmósfera de nitrógeno, a una solución de 5-(2-cloro-5-fluoropirimidin-4-il)-3-isopropil-2-metil-4,5,6,7-tetrahidro-2H-pirazolo[4,3-c]piridina (Intermedio E) (200,00 mg, 645,64 |jmol, 1,00 equivalente) en dioxano (5,00 ml) se le añadió 4-(6-aminopiridin-3-il)-1,4-diazepan-1-carboxilato de ferc-butilo (226,53 mg, 774,77 pmol, 1,20 equivalentes), Pd2(dba)3 (59,12 mg, 164,56 pmol, 0,01 equivalente), carbonato de cesio (420,72 mg, 1,29 mmol, 2,00 equivalentes) y Xantphos (74,72 mg, 129,13 pmol, 0,20 equivalentes). La mezcla de reacción se agitó a 110 °C durante 16 horas. La LC/MS mostró que la reacción se había completado y se detectó el producto deseado. La solución de reacción se enfrió a 25 °C, se sometió a filtración por succión. El filtrado se concentró para dar un producto en bruto. El producto en bruto se purificó por TLC preparativa (acetato de etilo) para dar el compuesto del título (250,00 mg, producto en bruto) en forma de un sólido de color blanco. LC/MS (ESI) m/z: 566,2 (M+1).
Etapa 4:
W-(5-(1,4-diazepan-1-il)piridin-2-il)-5-fluoro-4-(6,7-dihidro-3-isopropil-2-metil-2H-pirazolo[4,3-c]piridin-5(4H)-il)pirimidin-2-amina

En atmósfera de nitrógeno, a una solución de 4-(6-(5-fluoro-4-(6,7-dihidro-3-isopropil-2-metil-2H-pirazolo[4,3-c]piridin-5(4H)-il)pirimidin-2-il-amino)piridin-3-il)-1,4-diazepan-1 -carboxilato de ferc-butilo (250,00 mg, 441,95 pmol, 1,00 equivalente) en diclorometano (5,00 ml) se le añadió ácido trifluoroacético (3 ml) a 25 °C. La mezcla de reacción se agitó a 25 °C durante media hora. La LC/MS mostró que la reacción se había completado y se detectó el producto deseado. La solución de reacción se concentró para dar un producto en bruto. El producto en bruto se purificó por HPLC preparativa (ácido clorhídrico) para dar el compuesto del título (127,94 mg, 274,80 pmol, 62,18% de rendimiento). RMN 1H (400 MHz, Metanol-d4) 88,23 (d, J= 6,78 Hz, 1H), 7,94 (dd, J = 9,47, 2,70 Hz, 1H), 7,86 (d, J = 2.64 Hz, 1H), 7,41 (d, J = 9,41 Hz, 1H), 5,04-5,14 (m, 2H), 4,24 (t, J= 5,21 Hz, 2H), 4,06 (s, 3H), 3,83-3,94 (m, 2H), 3.65 (t, J = 6,02 Hz, 2H), 3,45-3,53 (m, 2H), 3,34-3,42 (m, 3H), 3,06-3,13 (m, 2H), 2,24-2,33 (m, 2H), 1,44 (d, J= 7,03 Hz, 6H). LC/MS (ESI) m/z: 466,2 (M+1).
Ejemplo 31
W-(5-fluoro-4-(6,7-d¡h¡dro-3-¡soprop¡l-2-met¡l-2H-p¡razolo[4,3-c]p¡r¡d¡n-5(4H)-¡l) pirimidin-2-il)-5-(piperazin-1-il)pirazin-2-amina

Etapa 1:
4-(5-am¡nop¡raz¡n-2-¡l)p¡peraz¡n-1-carbox¡lato de ferc-but¡lo

En atmósfera de n¡trógeno, a una soluc¡ón de 4-(5-bromop¡raz¡n-2-¡l)p¡peraz¡n-1-carbox¡lato de ferc-but¡lo (10,00 g, 29,14 mmol, 1,00 equ¡valente) y tetrafluoroborato de tr¡-ferc-but¡lfosfon¡o (2,54 g, 8,74 mmol, 0,30 equ¡valentes) en tolueno (100,00 ml) se le añad¡ó L¡HMDS (1 M, 60,00 ml, 2,06 equivalentes) y Pd2(dba)3 (2,60 g, 2,84 mmol, 0,10 equivalente). La soluc¡ón de reacc¡ón se ag¡tó a 65 °C durante 16 horas. La LC/MS mostró que la reacc¡ón se había completado y se detectó el producto deseado. La mezcla de reacc¡ón se enfr¡ó a 25 °C y la reacc¡ón se ¡nterrump¡ó añad¡endo agua (50 ml) a la soluc¡ón de reacc¡ón. La mezcla resultante se somet¡ó a extracc¡ón usando acetato de et¡lo (100 ml x 3). Las fases orgán¡cas comb¡nadas se concentraron a sequedad para dar un producto en bruto. El producto en bruto se pur¡f¡có por HPLC preparativa (alcal¡na) para dar el compuesto del título (5,00 g, 17,90 mmol, 61,43 % de rend¡m¡ento) en forma de un sól¡do de color naranja. LC/MS (ESI) m/z: 280,1 (M+1).
Etapa 2:
4-(5-(5-fluoro-4-(6,7-d¡h¡dro-3-¡soprop¡l-2-met¡l-2H-p¡razolo[4,3-c]p¡r¡d¡n-5(4H)-¡l)p¡r¡m¡d¡n-2-¡l-am¡no)p¡raz¡n-2-¡l)p¡peraz¡n-1-carbox¡lato de ferc-but¡lo

En un tubo de m¡croondas, a una soluc¡ón de 5-(2-cloro-5-fluorop¡r¡m¡d¡n-4-¡l)-3-¡soprop¡l-2-met¡l-4,5,6,7-tetrah¡dro-2H-p¡razolo[4,3-c]p¡r¡d¡na (Intermedio E) (500,00 mg, 1,61 mmol, 1,00 equivalente) y 4-(5-am¡nop¡raz¡n-2-¡l)p¡peraz¡n-1-carbox¡lato de ferc-but¡lo (495,97 mg, 1,78 mmol, 1,10 equ¡valentes) en d¡oxano (16,00 ml) se le añad¡ó Pd2(dba)3 (147,81 mg, 161,41 pmol, 0,10 equivalentes), BINAP (201,01 mg, 322,82 pmol, 0,20 equ¡valentes), tbutóx¡do de sod¡o (232,67 mg, 2,42 mmol, 1,50 equivalentes) y agua (400,00 pl). El n¡trógeno se ¡ntrodujo en el s¡stema de reacc¡ón durante 2 m¡nutos y después el tubo se cerró hermét¡camente, se calentó a 140 °C y se ag¡tó durante 4 horas. La LC/MS mostró que la reacc¡ón se había completado y se detectó el producto deseado. La soluc¡ón de reacc¡ón se f¡ltró a través de d¡atom¡ta (se lavó con acetato de et¡lo) y el f¡ltrado se lavó con agua (20 ml). La fase orgán¡ca se concentró a sequedad para dar un producto en bruto. El producto en bruto se pur¡f¡có por TLC preparat¡va (éter de petróleo:acetato de et¡lo = 2:1) para dar el compuesto del título (800,00 mg, producto en bruto) en forma de un sól¡do de color amar¡llo. LC/MS (ESI) m/z: 553,3 (M+1).
Etapa 3:
W-(5-fluoro-4-(6,7-d¡h¡dro-3-¡soprop¡l-2-met¡l-2H-p¡razolo[4,3-c]p¡r¡d¡n-5(4H)-¡l) p¡r¡m¡d¡n-2-¡l)-5-(p¡peraz¡n-1-¡l)p¡raz¡n-2-am¡na
A una solución de 4-(5-((5-fluoro-4-(3-isopropil-2-metil-6,7-dihidro-2H-pirazolo[4,3-c]piridin-5(4H)-il)pirimidin-2-il)-amino)pirazin-2-il)piperazin-1-carboxilato de ferc-butilo (800,00 mg, 1,45 mmol, 1,00 equivalente) en diclorometano (15,00 ml) se le añadió ácido trifluoroacético (23,03 g, 201,95 mmol, 139,28 equivalentes). La mezcla de reacción se agitó a 30 °C durante 7 horas. La LC/MS mostró que la reacción se había completado y se detectó el producto deseado. La solución de reacción se concentró a sequedad para dar un producto en bruto. El producto en bruto se purificó por HPLC preparativa (ácido clorhídrico) para dar el compuesto del título (213,05 mg, 435,69 pmol, 30,05 % de rendimiento). RMN 1H (400 MHz, metanol -d4): 88,26 (s, 1H), 8,16-8,24 (m, 2H), 5,20 (s, 2H), 4,36 (s a, 2H), 4,07 (s, 3H), 3,84-3,98 (m, 4H), 3,35-3,44 (m, 5H), 3,15 (s a., 2H), 1,33-1,53 (m, 6H). LC/MS (ESI) m/z: 453,2 (M+1).
Ejemplo 32
W-(5-fluoro-4-(6,7-dihidro-3-isopropil-2-metil-2H-pirazolo[4,3-c]piridin-5(4H)-il) pirimidin-2-il)-5-(4-metilpiperazin-1-il)pirazin-2-amina

A 30 °C, a una solución de 5-fluoro-4-(3-isopropil-2-metil-6,7-dihidro-2H-pirazolo[4,3-c]piridin-5(4H)-il)-W-(5-(piperazin-1 -il)pirazin-2-il)pirimidin-2-amina (250,00 mg, 552,45 pmol, 1,00 equivalente) en metanol (25,00 ml) se le añadió NaBHaCN (69,43 mg, 1,10 mmol, 2,00 equivalentes), HCHO (24,89 mg, 828,68 pmol, 1,50 equivalentes) y ácido trifluoroacético (62,99 mg, 552,45 pmol, 1,00 equivalente). La mezcla de reacción se agitó a 30 °C durante 4 horas. La LC/MS mostró que la reacción se había completado. La mezcla de reacción se concentró y se secó. El producto en bruto obtenido se purificó primero por HPLc preparativa (ácido clorhídrico) y después se purificó por cromatografía en columna sobre gel de sílice (éter de petróleo:acetato de etilo =1 :1 ) para dar el compuesto del título (35,03 mg, 75,08 pmol, 13,59 % de rendimiento). RMN 1H (400 MHz, metanol-d4) 88,88 (s, 1H), 7,97 (d, J= 1,13 Hz, 1H), 7,93 (d, J= 6,90 Hz, 1H), 4,04 (t, J = 5,84 Hz, 2H), 3,75-3,79 (m, 3H), 3,57-3,67 (m, 4H), 3,12-3,22 (m, 1H), 2,77-2,85 (m, 6H), 2,52 (s, 3H), 2,03 (s, 1H), 1,33 (d, J = 7,03 Hz, 6H), 1,28-1,32 (m, 1H). LC/MS (ESI) m/z: 467,2 (M+1).
Ejemplo 33
5-fluoro-4-(6,7-dihidro-3-isopropil-2-metil-2H-pirazolo[4,3-c]piridin-5(4H)-il)-A/-(4-metil-5-(piperazin-1-il)piridin-2-il)pirimidin-2-amina
Etapa 1:

5-bromo-4-metil-2-nitropiridina

Se añadió H2O2 (58,06 g, 1,71 mol, 21,28 equivalentes) lentamente gota a gota a H2SO4 (184,00 g, 1,88 mol, 23,39 equivalentes) que se enfrió a 0 °C y la temperatura se mantuvo por debajo de -20 °C durante la adición gota a gota. Una mezcla de 5-bromo-4-metilpiridin-2-amina (15,00 g, 80,20 mmol, 1,00 equivalente) en H2SO4 (100,00 ml) se le añadió después. La mezcla de reacción se agitó primero durante 45 minutos en un baño de hielo y después se calentó a 30 °C. Después de tres horas, el color de la solución de reacción cambió gradualmente del verde hierba al amarillo brillante. Se añadió hielo (500 ml) a la mezcla de reacción. El sólido precipitado se filtró, se lavó con agua y después se secó al vacío para dar el compuesto del título (12,00 g, 55,29 mmol, 68,95 % de rendimiento) en forma de un sólido de color naranja brillante sin más purificación. Lc /MS (ESI) m/z: 216,9 (M+1), 218,9 (M+3).
Etapa 2:
4-(4-metil-6-nitropiridin-3-il)piperazin-1-carboxilato de ferc-butilo

En atmósfera de nitrógeno, a una solución mezclada de 5-bromo-4-metil-2-nitropiridina (500,00 mg, 2,30 mmol, 1,00 equivalente), piperazin-1-carboxilato de ferc-butilo (514,05 mg, 2,76 mmol, 1,20 equivalentes), BINAP (286,43 mg, 460,00 |jmol, 0,20 equivalentes) y CS2CO3 (1,05 g, 3,22 mmol, 1,40 equivalentes) en dioxano (20,00 ml) se le añadió Pd2(dba)3 (210,62 mg, 230,00 jmol, 0,10 equivalentes). La mezcla de reacción se agitó a 110 °C durante 16 horas en atmósfera de nitrógeno. La LC/MS mostró que la reacción se había completado. La mezcla de reacción se enfrió a 20 °C y después se filtró. El filtrado se concentró, se secó y después se purificó mediante placa de TLC (éter de petróleo:acetato de etilo = 4:1) para dar el compuesto del título (420,00 mg, 1,30 mmol, 56,65 % de rendimiento) en forma de un sólido de color amarillo. LC/MS (ESI) m/z: 323,0 (M+1).
Etapa 3:
4-(6-amino-4-metilpiridin-3-il)piperazin-1-carboxilato de ferc-butilo

En atmósfera de nitrógeno, a una solución de 4-(4-metil-6-nitropiridin-3-il)piperazin-1-carboxilato de ferc-butilo (350,00 mg, 651,45 jmol, 1,00 equivalente) en metanol (15,00 ml) se le añadió Pd al 10%-C (200 mg). La suspensión se evacuó y se cargó con hidrógeno, seguido de agitación a 30 °C en atmósfera de H2 (244,74 kPa (15 psi)) durante 1,5 horas. La LC/MS mostró que la reacción se había completado. La mezcla se filtró, se concentró y se secó. El producto en bruto obtenido se purificó por cromatografía en columna sobre gel de sílice (éter de petróleo:acetato de etilo =1:1) para dar el compuesto del título (125,00 mg, 427,53 jmol, 65,63 % de rendimiento) en forma de un sólido de color amarillo. LC/MS (ESI) m/z: 293,2 (M+1).
Etapa 4:
4-(6-(5-fluoro-4-(6,7-dihidro-3-isopropil-2-metil-2H-pirazolo[4,3-c]piridin-5(4H)-il)pirimidin-2-il-amino)-4-metilpiridin-3-il)piperazin-1-carboxilato de ferc-butilo

La mezcla de 4-(6-amino-4-metilpiridin-3-il)piperazin-1-carboxilato de ferc-butilo (124,59 mg, 426,12 jmol, 1,10 equivalentes), 5-(2-cloro-5-fluoropirimidin-4-il)-3-isopropil-2-metil-4,5,6,7-tetrahidro-2H-pirazolo[4,3-c]piridina (Intermedio E) (120,00 mg, 387,38 jmol, 1,00 equivalente), Pd2(dba)3 (35,47 mg, 38,74 jmol, 0,10 equivalentes), Xantphos (44,83 mg, 77,48 jmol, 0,20 equivalentes) y CS2CO3 (252,43 mg, 774,76 jmol, 2,00 equivalentes) en dioxano (20,00 ml) se cargó con N2. Después, la mezcla de reacción se agitó a 110 °C durante 16 horas. La LC/MS mostró que la reacción se había completado. La mezcla de reacción se filtró, se concentró y se secó. El producto en bruto obtenido se diluyó con acetato de etilo (50 ml) y se lavó con agua (20 ml). La fase orgánica se secó usando un secador rotatorio al vacío y se purificó por placa de TLC preparativa (éter de petróleo:acetato de etilo = 1:1) para dar el compuesto del título (150,00 mg, 265,17 jmol, 68,45 % de rendimiento) en forma de un sólido de color amarillo. LC/MS (ESI) m/z: 566,3 (M+1).
Etapa 5:
5-fluoro-4-(6,7-d¡h¡dro-3-¡soprop¡l-2-met¡l-2H-p¡razolo[4,3-c]p¡r¡d¡n-5(4H)-¡l)-A/-(4-metil-5-(p¡peraz¡n-1-¡l)p¡r¡d¡n-2-il)pirimidin-2-amina
A una solución de 4-(6-(5-fluoro-4-(6,7-dihidro-3-isopropil-2-metil-2H-pirazolo[4,3-c]piridin-5(4H)-M)pirimidin-2-M-amino)-4-metilpiridin-3-il)piperazin-1-carboxilato de ¿ere-butilo (150,00 mg, 265,17 pmol, 1,00 equivalente) en diclorometano (7,50 ml) se le añadió ácido trifluoroacético (11,52 g, 101,00 mmol, 380,87 equivalentes). La mezcla de reacción se agitó a 30 °C durante 2 horas. La LC/MS mostró que la reacción se había completado. La mezcla se concentró y se secó. El producto en bruto obtenido se purificó por HPLC preparativa (ácido fórmico) para dar el compuesto del título (121,00 mg, 236,52 |jmol, 89,19 % de rendimiento) en forma de un aceite de color amarillo. RMN 1H (400 MHz, Metanol-^) 58,15 (d, J= 7,2 Hz, 1H), 8,05 (s, 1H), 7,24 (s, 1H), 5,00 (s, 2H), 4,16 (t, J= 5,8 Hz, 2H), 3,81 (s, 3H), 3,51-3,39 (m, 4H), 3,31-3,24 (m, 4H), 3,24-3,16 (m, 1H), 2,89 (t, J= 5,8 Hz, 2H), 2,55-2,45 (m, 3H), 1,36 (d, J= 7,2 Hz, 6H). LC/MS (ESI) m/z: 466,2 (M+1).
Enfoque E
Síntesis de 5-fluoro-4-(6,7-dihidro-3-isopropil-2,7,7-trimetil-2H-pirazolo[4,3-c]piridin-5(4H)-il)-W-(5-(1-metilpiperidin-4-il)piridin-2-il)pirimidin-2-amina:

Ejemplo 34
5-fluoro-4-(6,7-dihidro-3-isopropil-2,7,7-trimetil-2H-pirazolo[4,3-c]piridin-5(4H)-il)-A/-(5 -(1-metilpiperidin-4-il)piridin-2-il)pirimidin-2-amina

Etapa 1:
3,3-dimetil-4-oxopiperidin-1-carboxilato de bencilo

A 0 °C, a una solución de 4-oxopiperidin-1-carboxilato de bencilo (2,00 g, 8,57 mmol, 1,00 equivalente) en tetrahidrofurano (25 ml) se le añadió hidruro sódico (719,88 mg, 18,00 mmol, 2,10 equivalentes) y yoduro potásico (3,80 g, 26,74 mmol, 3,12 equivalentes) en una porción. La mezcla se agitó a 30 °C durante 16 horas. La LC/MS mostró que la reacción se había completado. A la mezcla de reacción se le añadió una solución de cloruro de amonio (50 ml) y después se sometió a extracción usando acetato de etilo (50 ml x 2). La fase orgánica se concentró y se secó. El residuo obtenido se purificó por placa de TLC preparativa (éter de petróleo:acetato de etilo = 6:1) para dar el compuesto del título (870,00 mg, 3,33 mmol, 38,86 % de rendimiento) en forma de un aceite transparente. RMN 1H (400 MHz, Metanol-^) 5 7,38-7,33 (m, 5H), 5,19 (s, 2H), 3,81-3,77 (t, J= 6,4 Hz, 2H), 3,50-3,46 (m, 2H), 2,50 (s, 2H), 1,11-1,04 (m, 6H). LC/MS (ESI) m/z: 284,1 (M+1).
Etapa 2:
5-(isobutiril)-3,3-dimetil-4-oxopiperidin-1-carboxilato de bencilo

A 0 °C, a una solución de 3,3-dimetil4-oxopiperidin-1-carboxilato de bencilo (6,00 g, 22,96 mmol, 1,00 equivalente) en tolueno (150,00 ml) se le añadió LDA (2 M, 22,96 ml, 2,00 equivalentes) en una porción. Después de agitar la mezcla de reacción a esta temperatura durante 30 minutos, se añadió cloruro de isobutirilo (3,67 g, 34,44 mmol, 1,50 equivalentes) y después la mezcla resultante se calentó lentamente a 30 °C y se agitó durante 16 horas. La TLC (éter de petróleo:acetato de etilo = 6: 1) mostró que la reacción se había completado. La mezcla de reacción se inactivó con una solución de cloruro de amonio (80 ml) y después se sometió a extracción usando acetato de etilo (100 ml x 2). Las fases orgánicas se combinaron, se concentraron y se secaron. El producto en bruto obtenido se purificó por cromatografía en columna sobre gel de sílice (éter de petróleo:acetato de etilo = 50:1) para dar el compuesto del título (5,60 g, 16,90 mmol, 73,60 % de rendimiento) en forma de un aceite de color amarillo.
Etapa 3:
6,7-dihidro-3-isopropil-2,7,7-trimetil-2H-pirazolo[4,3-c]piridin-5(4H)-carboxilato de bencilo

A una solución de 5-(isobutiril)-3,3-dimetil-4-oxopiperidin-1-carboxilato de bencilo (3,90 g, 11,77 mmol, 1,00 equivalente) y una solución acuosa al 40 % de metilhidrazina (13,99 g, 121,47 mmol, 10,32 equivalentes) en etanol (35,00 ml) se le añadió ácido acético (706,66 mg, 11,77 mmol, 1,00 equivalente) en una porción. La mezcla de reacción se agitó a 32 °C durante 16 horas. La LC/MS mostró que la reacción se había completado. La mezcla de reacción se concentró y se secó. El producto en bruto obtenido se purificó por placa de TLC preparativa (éter de petróleo:acetato de etilo = 5:1) para dar el compuesto del título (600,00 mg, 1,76 mmol, 14,93 % de rendimiento) en forma de un aceite transparente. LC/MS (ESI) m/z: 342,2 (M+1).
Etapa 4:
3-isopropil-2,7,7-trimetil-4,5,6,7-tetrahidro -2H-pirazolo[4,3-c]piridina

En atmósfera de nitrógeno, a una solución de 6,7-dihidro-3-isopropil-2,7,7-trimetil-2H-pirazolo[4,3-c]piridin-5(4H)-carboxilato de bencilo (600,00 mg, 1,76 mmol, 1,00 equivalente) en metanol (20,00 ml) se le añadió Pd al 10%/C (0,4 g). La suspensión se evacuó, se cargó con hidrógeno y después se agitó a 30 °C en atmósfera de H2 (244,74 kPa (15 psi)) durante 2 horas. La LC/MS mostró que la reacción se había completado. La mezcla se filtró, se concentró y se secó para dar el compuesto del título (323,00 mg, 1,56 mmol, 88,53 % de rendimiento) en forma de un aceite de color amarillo. LC/MS (ESI) m/z: 208,2 (M+1).
Etapa 5:
5-(2-cloro-5-fluoropirimidin-4-il)-4,5,6,7-tetrahidro-3-isopropil-2,7,7-trimetil-2H-pirazolo[4,3-c]piridina

A 30 °C, a una solución de 3-isopropil-2,7,7-trimetil-4,5,6,7-tetrahidro-2H-pirazolo[4,3-c]piridina (323,00 mg, 1,56 mmol, 1,00 equivalente) y 2,4-dicloro-5-fluoropirimidina (338,62 mg, 2,03 mmol, 1,30 equivalentes) en tetrahidrofurano (25,00 ml) se le añadió trietilamina (1,58 g, 15,58 mmol, 10,00 equivalentes). La mezcla de reacción se agitó a 30 °C durante 12 horas. La LC/MS mostró que la reacción se había completado. La mezcla de reacción se diluyó con salmuera saturada (20 ml) y después se sometió a extracción usando acetato de etilo (50 ml x 2). Las fases orgánicas se combinaron y después se secaron. El residuo obtenido se purificó por placa de TLC preparativa
(éter de petróleo:acetato de etilo = 4:1) para dar el compuesto del título (380,00 mg, 1,12 mmol, 72,11 % de rendimiento) en forma de un aceite de color amarillo. LC/MS (ESI) m/z: 338,1 (M+1).
Etapa 6:
5-fluoro-4-(6,7-dihidro-3-isopropil-2,7,7-trimetil-2H-pirazolo[4,3-c]piridin-5(4H)-il)-W-(5 -(1-metilpiperidin-4-M)piridin-2-il)pirimidin-2-amina

En atmósfera de nitrógeno, a una mezcla de 5-(2-cloro-5-fluoropirimidin-4-il)-4,5,6,7-tetrahidro-3-isopropil-2,7,7-trimetil-2H-pirazolo[4,3-c]piridina (380,00 mg, 1,12 mmol, 1,00 equivalente), 5-(1-metilpiperidin-4-il)piridin-2-amina (257,07 mg, 1,34 mmol, 1,20 equivalentes) y CS2CO3 (729,84 mg, 2,24 mmol, 2,00 equivalentes) en dioxano (25,00 ml) se le añadió Pd2(dba)3 (102,56 mg, 112,00 mmol, 0,10 equivalentes) y Xantphos (129,61 mg, 224,00 ^mol, 0,20 equivalentes) en una porción. La mezcla de reacción se cargó con N2 y después se agitó a 110 °C durante 16 horas. La LC/MS mostró que la reacción se había completado. La mezcla de reacción se diluyó con agua (10 ml) y después se sometió a extracción usando acetato de etilo (50 ml x 2). Las fases orgánicas se combinaron y después se secaron. El residuo obtenido se purificó por HPLC preparativa (ácido clorhídrico), seguido de purificación por SFC (AD-3S_5_40_3 ML, Columna: Chiralpak AD-3100 x 4,6 mm ID 3 um, Fase móvil: etanol al 40 % (DEA al 0,05 %) en CO2, Caudal: 3 ml/min, Longitud de onda: 220 nm) para dar el compuesto del título (60,00 mg, 121,80 ^mol, 10,87 % de rendimiento). RMN 1H (400 MHz, metanol-ck) 58,36 (s, 1H), 8,30 (d, J= 6,8 Hz, 1H), 8,27-8,19 (m, 1H), 7,56-7,48 (m, 1H), 5,09 (s, 2H), 4,12 (s, 3H), 4,05 (s, 2H), 3,73-3,60 (m, 2H), 3,44-3,35 (m, 1H), 3,31-3,25 (m, 1H), 3,21-3,09 (m, 1H), 2,96 (s, 3H), 2,35 (s, 1H), 2,26-2,08 (m, 4H), 1,51-1,44 (m, 12H). LC/MS (ESI) m/z: 493,3 (M+1).
Enfoque F
Síntesis de W-(5-(l,4-diazepan-1-il)piridin-2-il)-5-fluoro-4-(6,7-dihidro-3-isopropil-2-fenil-2H-pirazolo[4,3-c]piridin-5(4H)-il)pirimidin-2-amina

Ejemplo 35
W-(5-(1,4-diazepan-1-il)piridin-2-il)-5-fluoro-4-(6,7-dihidro-3-isopropil-2-fenil-2H-pirazolo[4,3-c]piridin-5(4H)-il)pirimidin-2-amina

Etapa 1:
6,7-dihidro-3-isopropil-2-fenil-2H-pirazolo[4,3-c]piridin-5(4H)-carboxilato de bencilo
A una solución de 3-(2-metilpropionil)-4-oxo-piperidin-1-carboxilato de bencilo (500,00 mg, 1,65 mmol, 1,00 equivalente) en etanol (5,00 ml) se le añadió fenilhidrazina (178,24 mg, 1,65 mmol, 1,00 equivalente). La mezcla de reacción se calentó a 80 °C y se agitó a esta temperatura durante 4 horas. La TLC (éter de petróleo:acetato de etilo = 2:1) y la LC/MS mostraron que la reacción se había completado. La mezcla de reacción se enfrió a 25 °C y después se concentró a presión reducida para eliminar el disolvente. El residuo resultante se purificó por cromatografía en columna sobre gel de sílice (éter de petróleo:acetato de etilo = 5:1) para dar el compuesto del título (330,00 mg, 878,92 ^mol, 53,27 % de rendimiento) en forma de un aceite de color rojo. RMN 1H (400 MHz, CDCl3) S 7,50-7,36 (m, 10H), 5,23 (s, 2H), 4,70 (s, 2H), 3,83 (s a, 2H), 3,07 (c, J= 7,0 Hz, 1H), 2,86 (s a, 2H), 1,28-1,20 (m, 6H). LC/MS (ESI) m/z: 376,1 (M+1).
Etapa 2:
4,5,6,7-tetrahidro-3-isopropil-2-fenil-2H-pirazolo[4,3-c]piridina

A una solución de 6,7-dihidro-3-isopropil-2-fenil-2H-pirazolo[4,3-c]piridin-5(4H)-carboxilato de bencilo (330,0 mg, 878,92 ^mol, 1,00 equivalente) en metanol (5,00 ml) se le añadió paladio al 10 % sobre carbono (50 mg). La mezcla de reacción se agitó a 25 °C durante 2 horas en atmósfera de H2 (244,74 kPa (15 psi)). La TLC (diclorometano:metanol = 10:1) mostró que la reacción se había completado. La mezcla de reacción se filtró y el filtrado se concentró para dar el compuesto del título (200,00 mg, producto en bruto) en forma de un aceite de color amarillo.
Etapa 3:
5-(2-cloro-5-fluoropirimidin-4-il)-4,5,6,7-tetrahidro-3-isopropil-2-fenil-2H-pirazolo[4,3-c]piridina

A una solución de 4,5,6,7-tetrahidro-3-isopropil-2-fenil-2H-pirazolo[4,3-c]piridina (200,00 mg, 828,74 ^mol, 1,00 equivalente) en tetrahidrofurano (5,00 ml) se le añadió trietilamina (167,72 mg, 1,66 mmol, 2,00 equivalentes) y 2,4-dicloro-5-fluoro-pirimidina (166,05 mg, 994,49 ^mol, 1,20 equivalentes). La mezcla de reacción se agitó a 25 °C durante 2 horas. La LC/MS mostró que la reacción se había completado. La mezcla de reacción se concentró y se secó. El residuo obtenido se purificó por placa de TLC preparativa (éter de petróleo:acetato de etilo = 2:1) para dar el compuesto del título (190,00 mg, 510,97 ^mol, 61,66 % de rendimiento) en forma de un sólido de color blanco. RMN 1H (400 MHz, CDCh) S 7,99 (d, J= 6,0 Hz, 1H), 7,52-7,43 (m, 3H), 7,40-7,36 (m, 2H), 4,97 (s, 2H), 4,12 (t, J= 5,9 Hz, 2H), 3,10 (c, J= 7,1 Hz, 1H), 2,99 (t, J= 5,9 Hz, 2H), 1,30 (d, J= 7,0 Hz, 6H). LC/MS (ESI) m/z: 372,0 (M+1).
Etapa: 4
4-(6-(5-fluoro-4-(6,7-dihidro-3-isopropil-2-fenil-2H-pirazolo[4,3-c]piridin-5(4H)-il)pirimidin-2-aminopiridin-3-il)-1,4-diazepan-1-carboxilato de ferc-butilo

En atmósfera de nitrógeno, a una solución mezclada de 5-(2-cloro-5-fluoropirimidin-4-il)-4,5,6,7-tetrahidro-3-isopropil-2-fenil-2H-pirazolo[4,3-c]piridina (190,00 mg, 510,97 ^mol, 1,00 equivalente) y 4-(6-amino-3-piridil)-1,4-diazepan-1-carboxilato de ferc-butilo (224,10 mg, 766,46 ^mol, 1,50 equivalentes) en dioxano (5,00 ml) se le añadió CS2CO3 (499,45 mg, 1,53 mmol, 3,00 equivalentes), Xantphos (59,13 mg, 102,19 ^mol, 0,20 equivalentes) y Pd2(dba)3 (46,79 mg, 51,10 ^mol, 0,10 equivalentes). La mezcla de reacción se calentó a 120 °C y se agitó a esta temperatura durante 16 horas. La LC/MS mostró que la reacción se había completado. La mezcla de reacción se filtró y el filtrado se concentró. El residuo obtenido se purificó por placa de TLC preparativa (acetato de etilo puro) para dar el compuesto del título (100,00 mg, producto en bruto) en forma de un sólido de color amarillo. LC/MS (ESI) m/z: 628,3 (M+1).
Etapa 5:
W-(5-(1,4-diazepan-1-il)piridin-2-il)-5-fluoro-4-(6,7-dihidro-3-isopropil-2-fenil-2H-pirazolo[4,3-c]piridin-5(4H)-il)pirimidin
2-amina

A una solución de 4-(6-(5-fluoro-4-(6,7-dihidro-3-isopropil-2-fenil-2H-pirazolo[4,3-c]piridin-5(4H)-il)pirimidin-2-aminopiridin-3-il)-1,4-diazepan-1-carboxilato de ferc-butilo (100,00 mg, 159,30 |jmol, 1,00 equivalente) en diclorometano (3,00 ml) se le añadió ácido trifluoroacético (1 ml). La mezcla de reacción se agitó a 25 °C durante 0,5 h. La LC/MS mostró que la reacción se había completado. La mezcla de reacción se concentró a presión reducida para eliminar el disolvente. El residuo resultante se purificó por HPLC preparativa (ácido clorhídrico) para dar el compuesto del título (50,00 mg, 94,76 jmol, 59,49 % de rendimiento). RMN 1H (400 MHz, Metanol-d4) 58,24 (d,J= 6,8 Hz, 1H), 7,92 (dd, J= 9,4, 2,8 Hz, 1H), 7,86 (d, J= 2,6 Hz, 1H), 7,73-7,68 (m, 3H), 7,65-7,57 (m, 2H), 7,39 (d, J= 9,5 Hz, 1H), 5,17 (s, 2H), 4,31 (t, J= 5,0 Hz, 2H), 3,92-3,84 (m, 2H), 3,65 (t, J= 6,0 Hz, 2H), 3,52-3,45 (m, 2H), 3,41-3,36 (m, 2H), 3,14 (t, J= 5,1 Hz, 2H), 3,06 (td, J= 14,0, 6,9 Hz, 1H), 2,33-2,24 (m, 2H), 1,37 (d, J= 7,0 Hz, 6H). LC/MS (ESI) m/z: 528,2 (M+1).
Experimentos farmacológicos
Los compuestos de la presente invención son inhibidores selectivos de CDK4/6, por ejemplo, sus actividades contra CDK2 fueron inferiores a las de CDK4/6 (comparando sus valores de CI50). Los siguientes resultados experimentales confirmaron que los compuestos enumerados en la presente invención eran inhibidores específicos de CDK4/6 y podrían usarse como posibles fármacos anticancerosos. La CI50 usada en el presente documento se refiere a la concentración correspondiente de un agente cuando el agente produce el 50 % de la inhibición máxima. Los compuestos de la presente invención también tienen actividades biológicas impredecibles. Por ejemplo, el compuesto del Ejemplo 13 no era un sustrato de un transportador de flujo de salida, mientras que se demostró que palbociclib y LY2835219 son sustratos de transportadores de flujo de salida. Además, la exposición del compuesto del Ejemplo 13 en el tejido cerebral fue mucho mayor que la de palbociclib o LY2835219 al mismo tiempo. Puede especularse que el compuesto del Ejemplo 13, en comparación con palbociclib y LY2835219, es más prometedor en el tratamiento de casos de cáncer de mama con metástasis cerebral, que representan aproximadamente el 15 % de los casos de cáncer de mama. Las actividades biológicas mostradas aquí son solo las actividades biológicas representativas de algunos ejemplos individuales.
Experimentos bioquímicos
Materiales experimentales:
Los materiales experimentales incluyeron CDK2/ciclina A, CDK4/ciclina D1, CDK6/ciclina D1 (Life Technologies); sustratos polipeptídicos marcados con ULight, es decir, ULight-4E-BPl y ULight-MBP (PerkinElmer); el anticuerpo marcado con europio contra la proteína básica anti-mielina y anticuerpo de conejo marcado con europio (PerkinElmer); y el Lector de placas Envision® Multilabel (PerkinElmer) usado para la detección de señales.
Método experimental:
Los compuestos a ensayar se diluyeron tres veces para estar a diez concentraciones gradualmente variadas. Las concentraciones finales variaron de 5 jM a 0,25 nM.
• El sistema de reacción enzimática de CDK2/ciclina A
El método convencional Lance Ultra se llevó a cabo en un sistema de reacción enzimática de 10 j l que contenía proteína CDK2/ciclina A 0,5 nM, polipéptido ULight-MBP 100 nM y ATP 25 jM . Las diluciones de gradiente de los compuestos se disolvieron en tampón enzimático, respectivamente. Los componentes del tampón incluían una solución de ácido etanosulfónico de hidroxietilpiperazina 50 mM (pH 7,5), ácido etilendiaminotetraacético 1 mM, cloruro de magnesio 10 mM, Brij-35 al 0,01 % y ditiotreitol 2mM. Después de que comenzó la reacción, la placa OptiPlate de 384 pocillos se selló con la película de sellado térmico superior TopSeal-A y se incubó a temperatura ambiente durante 60 minutos.
• El sistema de reacción enzimática de CDK4/ciclina D1
El método convencional Lance Ultra se llevó a cabo en un sistema de reacción enzimática de 10 j l que contenía proteína CDK4/ciclina D1 0,3 nM, polipéptido ULight-4E-BPl 50 nM y ATP 350 jM . Las diluciones de los compuestos se disolvieron en tampón enzimático, respectivamente. Los componentes del tampón incluían una solución de ácido etanosulfónico de hidroxietilpiperazina 50 mM (pH 7,5), ácido etilendiaminotetraacético 1 mM, cloruro de magnesio 10 mM, Brij-35 al 0,01 % y ditiotreitol 2 mM. Después de que comenzó la reacción, la placa OptiPlate de 384 pocillos se selló con la película de sellado térmico superior TopSeal-A y se incubó a temperatura ambiente durante 180 minutos.
• El sistema de reacción enzimática de CDK6/ciclina D1
El método convencional Lance Ultra se llevó a cabo en un sistema de reacción enzimática de 10 |jl que contenía proteína CDK6/ciclina D1 0,8 nM, polipéptido ULight-4E-BPl 50 nM y ATP 250 jM . Las diluciones de los compuestos se disolvieron en tampón enzimático, respectivamente. Los componentes del tampón incluían una solución de ácido etanosulfónico de hidroxietilpiperazina 50 mM (pH 7,5), ácido etilendiaminotetraacético 1 mM, cloruro de magnesio 10 mM, Brij-35 al 0,01 % y ditiotreitol 2 mM. Después de que comenzó la reacción, la placa OptiPlate de 384 pocillos se selló con la película de sellado térmico superior TopSeal-A y se incubó a temperatura ambiente durante 180 minutos.
El tampón de parada de la reacción enzimática se preparó disolviendo EDTA en una dilución de una vez del tampón de ensayo. La reacción se terminó a temperatura ambiente durante 5 minutos. Para cada uno de los sistemas de reacción de CDK2/ciclina A, CDK4/ciclina D1 y CDK6/ciclina D1 se añadieron 5 j l de mezcla de ensayo (provista con el anticuerpo marcado con europio contra la proteína básica anti-mielina y el anticuerpo de conejo marcado con europio, respectivamente). Se incubaron a temperatura ambiente durante 60 minutos. El instrumento EnVision detectó las señales de reacción de acuerdo con el principio de transferencia de energía de resonancia de fluorescencia resuelta en el tiempo.
Análisis de los datos:
Los datos originales se convirtieron a la tasa de inhibición utilizando la ecuación (Relación-Máx)/(Máx-Mín)* 100%. Los valores de CI50 se obtuvieron mediante ajuste de curvas usando cuatro parámetros (obtenidos del modo 205 en XLFIT5, iDBS). La Tabla 1 proporciona las actividades inhibitorias de los compuestos de la invención contra las quinasas CDK2, CDK4 y Cd K6.
Experimentos celulares
Materiales experimentales:
Medio RPMI 1640, suero bovino fetal, penicilina/estreptomicina se compraron de Promega (Madison, WI). La línea celular MCF-7 y la línea celular MDA-MB-436 se compraron de la European Collection of Authenticated Cell Cultures (ECACC). Se usó el lector de placas EnVision® Multilabel (PerkinElmer).
Método experimental:
Las células MCF-7 se inocularon en una placa negra de 384 pocillos añadiendo 45 j l de la suspensión celular (que contiene 200 células MCF-7) a cada uno de los pocillos. La placa se incubó durante la noche en una incubadora de dióxido de carbono.
Los compuestos a probar se diluyeron tres veces usando Bravo hasta la décima concentración, es decir, diluido de 10 jM a 0,508 nM. Se realizó una prueba de pocillos por duplicado. El medio se añadió a la placa de en medio en una cantidad de 49 j l por pocillo. Las diluciones en gradiente de los compuestos se transfirieron en una cantidad de 1 j l por pocillo a la placa intermedia en las posiciones correspondientes y la mezcla se mezcló y se transfirió a la placa celular en una cantidad de 5 j l por pocillo. La placa celular se incubó en una incubadora de dióxido de carbono durante 6 días. El reactivo Promega CellTiter-Glo se añadió a la placa celular en una cantidad de 25 j l por pocillo. La placa celular se incubó a temperatura ambiente durante 10 minutos para estabilizar la señal de luminiscencia. Se usó el lector de placas EnVision® Multilabel (PerkinElmer) para la detección de la señal.
Análisis de los datos:
Los datos originales se convirtieron a la tasa de inhibición utilizando la ecuación (Relación-Máx)/(Máx-Mín)* 100%. Los valores de CI50 se obtuvieron mediante ajuste de curvas usando cuatro parámetros (obtenidos del modo 205 en XLFIT5, iDBS). La Tabla 1 proporciona las actividades inhibitorias de los compuestos de la invención contra la proliferación de células MCF-7 y las células MDA-MB-436.
Conclusión experimental:
Los compuestos de la presente invención tenían actividades inhibitorias significativas contra CDK4 y CDK6 y tenían una alta selectividad contra CDK2. Por otro lado, los compuestos de la presente invención tenían actividades inhibitorias significativas contra la proliferación de células de cáncer de mama MCF-7 positivas para el receptor de estrógenos y actividades inhibitorias escasas contra la proliferación de células MDA-MB-436 negativas para el receptor de estrógenos. Los compuestos de los Ejemplos 3, 9, 10 y 15 tuvieron actividades inhibitorias más altas contra CDK4 y CDK6 que los compuestos de referencia Palbociclib y LY2835219. Los compuestos de los Ejemplos 3, 7, 9, 10 y 15 tuvieron actividades inhibitorias más altas contra la proliferación de células MCF-7 que los compuestos de referencia Palbociclib y LY2835219. El compuesto del Ejemplo 7 tuvo una selectividad inhibitorias en la inhibición de la proliferación de células de cáncer de mama positivas para el receptor de estrógenos que el compuesto de referencia LY2835219 (MCF-7/MDA-MB-436).
Tabla 1

Experimento de evaluación de permeabilidad bidireccional de células Caco-2
Fin experimental:
Las células Caco-2, células de cáncer de colon humano, son un modelo in vitro ampliamente usado para estudiar la absorción del intestino delgado. El modelo celular Caco-2 en monocapa se ha usado ampliamente para evaluar los procesos de transporte pasivo y activo en el proceso de absorción del intestino delgado. GF120918A es un fuerte inhibidor de los transportadores de flujo de salida, estos transportadores de flujo de salida contienen glicoproteína P (P-gp), proteína de resistencia al cáncer de mama (breast cáncer resistance protein, BCRP) y similares. Este experimento se usó para determinar la permeabilidad bidireccional del compuesto del Ejemplo 7 y los compuestos de referencia Palbociclib y LY2835219 a través del modelo de células Caco-2 y para evaluar el transporte de eflujo de los compuestos de prueba mediante la adición de GF120918A.
Procedimientos experimentales:
Las condiciones experimentales convencionales fueron como sigue:
- Concentración de prueba: 2 pM (DMSO < 1 %);
- Número de replicación: n = 2;
- Dirección: transporte bidireccional, incluyendo dos direcciones, es decir, A ^ B y B ^A ;
- Tiempo de incubación: punto de tiempo único, 2 horas;
- Tampón de transporte: HBSS, pH 7,4;
- Condiciones de incubación: 37 °C, CO2 al 5 %.
Después de la incubación, las soluciones de muestra tomadas de los pocillos de dosificación y los pocillos receptores se mezclaron inmediatamente con una solución de acetonitrilo frío que contenía el patrón interno. Las concentraciones de los compuestos de prueba en todas las muestras (incluyendo las soluciones de dosificación iniciales, los sobrenadantes de los pocillos de dosificación y las soluciones receptoras) se determinaron mediante un método CL/EM/EM. Se calcularon los coeficientes de permeabilidad aparente, las relaciones de flujo de salida y otros parámetros.
Conclusión experimental:
La Tabla 2 enumera los coeficientes de permeabilidad del compuesto del Ejemplo 7 y los compuestos de referencia Palbociclib y LY2835219 en células de monocapa Caco-2. En comparación con los compuestos de referencia Palbociclib y LY2835219, el compuesto del Ejemplo 7 tuvo mayor permeabilidad y su absorción y transporte in vivo eran menos probables a verse afectados por los transportadores de flujo de salida. La permeabilidad más alta podría permitir que el compuesto del Ejemplo 7 se distribuya más en los tejidos del cuerpo (tales como la mama y el pulmón), con lo que se logra una mejor efectividad antitumoral in vivo. La permeabilidad más alta también permitió que el compuesto del Ejemplo 7 tuviera la posibilidad de penetrar la barrera hematoencefálica, logrando de esta manera el fin de tratar metástasis cerebrales de cáncer de mama o cáncer de pulmón.
Tabla 2


El estudio de la permeabilidad cerebral
Fin experimental:
Se usaron ratas SD como los animales de prueba. Las concentraciones de fármaco en cerebro y plasma en diferentes momentos se determinaron por CL/EM/EM después de la administración oral de los compuestos del Ejemplo 7 y los compuestos de referencia Palbociclib y LY2835219 para evaluar sus características de distribución en el tejido cerebral.
Procedimientos experimentales:
Se compraron doce ratas SD macho adultas sanas de 7 a 9 semanas de edad de Shanghai Slack Laboratory Animal Co., Ltd. Se pesó una cantidad apropiada del compuesto de prueba para preparar una suspensión de metilcelulosa al 0,5 % a 1 mg/ml. Las ratas recibieron una dosis oral única de 10 mg/kg. 0,5 h, 2 h, 8 h y 24 horas después de la administración, se mataron 3 ratas, respectivamente, y se recogió toda su sangre y los tejidos cerebrales. El plasma se preparó centrifugando la sangre completa (3000 g, 15 minutos, 4 °C). Las muestras de homogeneizado cerebral se prepararon mezclando los tejidos cerebrales con una mezcla de metanol y un tampón fosfato (metanol: tampón fosfato = 1 : 2) como homogeneizado en una relación de 5 : 1. La concentración de fármaco de cada muestra se determinó por CL/EM/EM y se calculó el AUC para evaluar la exposición.
Conclusión experimental:
La Tabla 3 muestra los datos de la exposición del compuesto del Ejemplo 7 y los compuestos de referencia Palbociclib y LY2835219 en el tejido cerebral cuando la dosis oral es de 10 mg/kg. Los resultados mostraron que el compuesto del Ejemplo 7 tuvo una exposición más alta en el tejido cerebral de rata que los compuestos de referencia Palbociclib y LY2835219 en los dos puntos de tiempo de 0,5 horas y 2 horas, indicando que el compuesto del Ejemplo 7 fue ventajoso para desarrollarse como un medicamento para el tratamiento de metástasis cerebrales a partir de cáncer. Mientras tanto, la exposición del compuesto del Ejemplo 7 de la presente invención en el cerebro disminuyó significativamente después de 8 horas, que cayó entre la exposición de los compuestos de referencia Palbociclib y LY2835219, indicando que el compuesto del Ejemplo 7 de la presente invención no se acumularía en el cerebro durante un largo período de tiempo, y tuvo una mejor seguridad.
Tabla 3

Estudio farmacodinámico in vivo
El estudio farmacodinámico in vivo se llevó a cabo en ratones desnudos BALB/c con xenoinjertos derivados de líneas de células tumorales (CDX) de origen humano, en los cuales se implantó subcutáneamente la línea celular de cáncer de mama humano MCF-7.
Procedimientos experimentales:
Los ratones desnudos BALB/c de 6 a 8 semanas de edad hebras que pesaban aproximadamente 18-22 g se mantuvieron en jaulas ventiladas separadas (10 ratones por jaula) en un entorno especial libre de patógenos. Todas las jaulas, las esterillas y el agua se desinfectaron antes de su uso. Todos los animales tenían acceso libre a una dieta de laboratorio comercial certificada convencional. Se adquirió un total de 100 ratones de Shanghai BK Laboratory Animal Co., LTD para estudio. Se implantó por vía subcutánea un comprimido de estrógeno (0,36 g, liberación sostenida en 60 días) en el abdomen siguiente izquierdo de cada ratón. Después de 3 días, las células tumorales (10x106 en 0,2 ml de tampón fosfato) se implantaron por vía subcutánea a la derecha cerca del abdomen de cada ratón para el crecimiento tumoral. La administración comenzó cuando el volumen tumoral promedio alcanzó aproximadamente 150-200 mm3. Los compuestos de prueba se administraron por vía oral diariamente, las dosis se mostraron como en la Tabla 5. Los volúmenes tumorales se midieron con un calibre bidimensional cada tres días, los volúmenes se expresaron en milímetros cúbicos y se calcularon mediante la fórmula V = 0,5axb2, en donde a y b eran el diámetro mayor y el diámetro menor del tumor, respectivamente. La efectividad antitumoral se determinó dividiendo un aumento promedio de los volúmenes tumorales de los animales tratados con los compuestos entre un aumento de los volúmenes tumorales de los animales no tratados.
Conclusión experimental:
Los compuestos del Ejemplo 3 y el Ejemplo 7 de la presente invención exhibieron actividades antitumorales significativas en el modelo de xenoinjerto derivado de línea celular (CDX) basado en humanos del cáncer de mama MCF-7. Como se muestra en la Tabla 4, 21 días después del inicio del experimento, los volúmenes tumorales del grupo de animales no tratados aumentaron rápidamente desde los 187 mm3 iniciales hasta 1443 mm3, mientras que los volúmenes tumorales del grupo animal al que se administró el compuesto del Ejemplo 3 aumentaron lentamente desde los 187 mm3 iniciales hasta 432 mm3 y la velocidad de crecimiento tumoral del grupo al que se administró el compuesto del Ejemplo 3 fue significativamente más lenta que la de los grupos a los que se administraron los compuestos de referencia Palbociclib y LY2835219. Considerando el hecho de que la dosis administrada del compuesto del Ejemplo 3 (25 mg/kg) era solo la mitad del compuesto de referencia LY2835219 (50 mg/kg) o casi la mitad del compuesto de referencia Palbociclib (45 mg/kg) (dosis altas de Palbociclib demostraron ser intolerables para los animales), los presentes inventores consideran que el compuesto del Ejemplo 3 fue significativamente superior a los dos compuestos de referencia en la actividad antitumoral. Mientras tanto, 21 días después de la administración, los volúmenes tumorales del grupo animal al que se administró el compuesto del Ejemplo 7 aumentaron lentamente desde los 187 mm3 iniciales hasta 350 mm3, y la tasa de crecimiento fue significativamente más lenta que la del grupo al que se administró la misma dosis del compuesto de referencia LY2835219, indicando que el compuesto del Ejemplo 7 tuvo una actividad antitumoral más significativa que el compuesto de referencia LY2835219 de la misma dosis.
Tabla 4

Claims (12)
1. Un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo


R1 se selecciona entre el grupo que consiste en isopropilo, 2-propenilo y alilo;
R2 se selecciona entre el grupo que consiste en metilo y fenilo;
R3 se selecciona entre el grupo que consiste en H y halógeno;
R4, R5 y R6 se selecciona cada uno independientemente entre el grupo que consiste en H, halógeno, OH, NH2, CN, NO2 y =O o se selecciona entre el grupo que consiste en alquilo C1-8, alquilamino C1-8, W,W-di(alquil)amino C1-8, alcoxil C1-8-alquilo C1-8-, hidroxialquilo C1-8, alquenilo C2-8, alquinilo C2-8, cicloalquilo C3-7 y un grupo heterocicloalquilo de 3 a 7 miembros, cada uno de los cuales está opcionalmente sustituido con 1, 2 o 3 Rs;
opcionalmente, dos cualesquiera de R4, R5 y R6 juntos pueden formar un anillo de 3 a 7 miembros;
R7 se selecciona entre el grupo que consiste en H, halógeno, -OR8, -SR8, -N(R8)(Rg) y cicloalquilo C3-7;
cada uno de X1, X2, X3 y X4 se selecciona independientemente entre un grupo que consiste en N y C(R1o); X7 se selecciona entre el grupo que consiste en carbonilo y C(Rn)(R12);
W es un enlace sencillo;
T se selecciona entre el grupo que consiste en N y C(R1o);
Q se selecciona entre el grupo que consiste en N y C(R1o);
cada uno de m y n se selecciona independientemente entre el grupo que consiste en 0, 1 y 2;
cada uno de R8 y R9 se selecciona independientemente entre el grupo que consiste en H, alquilo C1-8 y cicloalquilo
C3-7;
R se selecciona entre el grupo que consiste en F, Cl, Br, I, NH2, CN, OH, CF3, CHF2, CH2F, NHCH3 y N(CH3)2; opcionalmente, R8 y R9 están unidos al mismo átomo y forman un anillo de 3 a 7 miembros con 1-4 heteroátomos;
el término "hetero" o "heteroátomo" representa O, S, S(=O), S(=O)2 o N;
R10 se selecciona entre el grupo que consiste en H, halógeno, OH, NH2, CN, alquilo C1-6, alcoxilo C1-6, cicloalquilo C3-5, CN, -OR8, -SR8, -N(R8)(R9), -C(=O)R8, -C(=O)OR8, -C(=O)N(R8)(R9), -S(=O)R8, -S(=O)2R8, S(=O)2N(R8)(R9);
cada uno de R11 y R12 se selecciona independientemente entre el grupo que consiste en H, OH, halógeno, alquilo C1-8 y cicloalquilo C3-7;
opcionalmente, R4 y R10 están unidos al mismo átomo y forman un anillo de 3 a 7 miembros y, opcionalmente,


la unidad estructural se puede sustituir por '
2. El compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo de acuerdo con la reivindicación 1
en donde 

Ri se selecciona entre el grupo que consiste en isopropilo, 2-propenilo y alilo;
R2 se selecciona entre el grupo que consiste en metilo y fenilo;
R3 se selecciona entre el grupo que consiste en H y halógeno;
R4, R5 y R6 se selecciona cada uno independientemente entre el grupo que consiste en H, halógeno, OH, NH2, CN, NO2 y =O o se selecciona entre el grupo que consiste en alquilo C1-8, alquilamino C1-8,
W,W-di(alquil)amino C1.8, alcoxil C1-8-alquilo C1.8-, hidroxialquilo C1.8, alquenilo C2-8, alquinilo C2-8, cicloalquilo C3-7 y un grupo heterocicloalquilo de 3 a 7 miembros, cada uno de los cuales está opcionalmente sustituido con 1, 2 o 3
Rs;
opcionalmente, dos cualesquiera de R4, R5 y R6 juntos pueden formar un anillo de 3 a 7 miembros;
R7 se selecciona entre el grupo que consiste en H, halógeno, -OR8, -SR8, -N(R8)(Rg) y cicloalquilo C3-7;
cada uno de Xi, X2, X3 y X4 se selecciona independientemente entre un grupo que consiste en N y C(Riü);
X7 se selecciona entre el grupo que consiste en carbonilo y C(R ii)(R i2);
W es un enlace sencillo;
T se selecciona entre el grupo que consiste en N y C(Riü);
Q se selecciona entre el grupo que consiste en N y C(Riü);
cada uno de m y n se selecciona independientemente entre el grupo que consiste en 0, 1 y 2;
cada uno de R8 y R9 se selecciona independientemente entre el grupo que consiste en H, alquilo C1-8 y cicloalquilo
C3-7;
R se selecciona entre el grupo que consiste en F, Cl, Br, I, NH2, CN, OH, CF3, CHF2, CH2F, NHCH3 y N(CH3)2; opcionalmente, R8 y R9 están unidos al mismo átomo y forman un anillo de 3 a 7 miembros con 1-4 heteroátomos;
el término "hetero" o "heteroátomo" representa O, S, S(=O), S(=O)2 o N;
R10 se selecciona entre el grupo que consiste en H, halógeno, OH, NH2, CN, alquilo C1-6, alcoxilo C1-6, cicloalquilo C3-5, CN, -OR8, -SR8, -N(R8)(R9), -C(=O)R8, -C(=O)OR8, -C(=O)N(R8)(R9), -S(=O)R8, -S(=O)2R8, S(=O)2N(R8)(R9);
cada uno de R11 y R12 se selecciona independientemente entre el grupo que consiste en H, OH, halógeno, alquilo C1-8 y cicloalquilo C3-7;
opcionalmente, R4 y R10 están unidos al mismo átomo y forman un anillo de 3 a 7 miembros.
3. El compuesto o la sal farmacéuticamente aceptable del mismo de acuerdo con la reivindicación 1,


donde la unidad estructural está sustituido por
4. El compuesto de acuerdo con la reivindicación 1, onde R3es F.
5. El compuesto o la sal farmacéuticamente aceptable del mismo de acuerdo con la reivindicación 1, donde R4, R5 y R6 se selecciona cada uno independientemente entre el grupo que consiste en H, halógeno, OH, NH2,

6. El compuesto o la sal farmacéuticamente aceptable del mismo de acuerdo con la reivindicación 1, donde R7 se selecciona entre el grupo que consiste en H, F y Cl.
7. El compuesto o la sal farmacéuticamente aceptable del mismo de acuerdo con la reivindicación 1,

8. El compuesto o la sal farmacéuticamente aceptable del mismo de acuerdo con la reivindicación 1, donde Río se

selecciona entre un grupo que consiste en H, OH, NH2, F, Cl, CN, y Me.
9. El compuesto o la sal farmacéuticamente aceptable del mismo de acuerdo con la reivindicación 1, donde X4 se selecciona entre el grupo que consiste en N y CH.
10. El compuesto o la sal farmacéuticamente aceptable del mismo de acuerdo con la reivindicación 1,

donde la unidad estructural se selecciona entre el grupo que consiste en

11. El compuesto o la sal farmacéuticamente aceptable del mismo

donde la unidad estructural se selecciona entre el 

12. Un compuesto de acuerdo con la reivindicación 1, donde el compuesto se selecciona entre el grupo que consiste en


y una sal farmacéuticamente aceptable del mismo.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201510107436 | 2015-03-11 | ||
| CN201610019047 | 2016-01-12 | ||
| PCT/CN2016/076041 WO2016141881A1 (zh) | 2015-03-11 | 2016-03-10 | 作为抗癌药物的取代的2-氢-吡唑衍生物 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2806206T3 true ES2806206T3 (es) | 2021-02-16 |
Family
ID=56878940
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16761108T Active ES2806206T3 (es) | 2015-03-11 | 2016-03-10 | Derivado de 2-hidrógeno pirazol sustituido que sirve como fármaco anticanceroso |
Country Status (13)
| Country | Link |
|---|---|
| US (1) | US9969719B2 (es) |
| EP (1) | EP3269715B1 (es) |
| JP (1) | JP6726677B2 (es) |
| KR (1) | KR102615733B1 (es) |
| CN (2) | CN111333627A (es) |
| AU (1) | AU2016228660B2 (es) |
| BR (1) | BR112017019286B1 (es) |
| CA (1) | CA2978363A1 (es) |
| ES (1) | ES2806206T3 (es) |
| HK (1) | HK1244803A1 (es) |
| RU (1) | RU2722363C2 (es) |
| TW (1) | TWI688559B (es) |
| WO (1) | WO2016141881A1 (es) |
Families Citing this family (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017210489A1 (en) | 2016-06-01 | 2017-12-07 | M3 Biotechnology, Inc. | Compounds |
| CN109311848B (zh) | 2016-06-07 | 2022-02-01 | 北京加科思新药研发有限公司 | 可用作shp2抑制剂的新型杂环衍生物 |
| WO2018045993A1 (zh) * | 2016-09-09 | 2018-03-15 | 正大天晴药业集团股份有限公司 | 一种取代的2-氢-吡唑衍生物的晶型、盐型及其制备方法 |
| CN106588884B (zh) * | 2016-11-10 | 2019-04-09 | 浙江大学 | 2-多取代芳环-嘧啶类衍生物及制备和医药用途 |
| US10988466B2 (en) | 2017-03-23 | 2021-04-27 | Jacobio Pharmaceuticals Co., Ltd. | Heterocyclic derivatives useful as SHP2 inhibitors |
| CN107382974B (zh) * | 2017-06-08 | 2020-06-05 | 扬州市三药制药有限公司 | 一种嘧啶胺类化合物作为周期蛋白依赖性激酶4/6抑制剂的应用 |
| US11351170B2 (en) * | 2017-08-15 | 2022-06-07 | Beijing Xuanyi Pharmasciences Co., Ltd. | Substituted pyrimidines as CDK4/6 inhibitors |
| CN109503573A (zh) * | 2017-09-14 | 2019-03-22 | 昆明圣加南生物科技有限公司 | 2-取代苯胺基嘧啶衍生物及其用途 |
| KR20200057705A (ko) | 2017-10-27 | 2020-05-26 | 프레세니어스 카비 온콜로지 리미티드 | 리보시클립 및 그의 염의 제조를 위한 개선된 방법 |
| KR20200115583A (ko) * | 2018-01-29 | 2020-10-07 | 베타 파마, 인크. | Cdk4 및 cdk6 억제제로서의 2h-인다졸 유도체 및 그의 치료 용도 |
| EP3752491A4 (en) | 2018-02-15 | 2021-12-01 | Nuvation Bio Inc. | HETEROCYCLIC COMPOUNDS USED AS KINASE INHIBITORS |
| CN113507930A (zh) * | 2019-01-29 | 2021-10-15 | 贝达医药公司 | 2h-吲唑衍生物作为脑癌和脑转移的治疗剂 |
| TW202102487A (zh) * | 2019-03-20 | 2021-01-16 | 美商貝達醫藥公司 | N-(5-((4-乙基哌𠯤-1-基)甲基)吡啶-2-基)-5-氟-4-(3-異丙基-2-甲基-2h-吲唑-5-基)嘧啶-2-胺及其鹽的結晶與非晶型以及其製備方法與醫療用途 |
| AU2020269469A1 (en) * | 2019-05-05 | 2021-12-09 | Regor Pharmaceuticals, Inc. | CDK inhibitors |
| CN115768763A (zh) | 2020-06-22 | 2023-03-07 | 正大天晴药业集团股份有限公司 | 一种cdk4/6抑制剂的制备方法 |
| CN114539225B (zh) * | 2020-11-11 | 2024-02-20 | 上海拓界生物医药科技有限公司 | 2-氨基-嘧啶类化合物 |
| WO2022113003A1 (en) | 2020-11-27 | 2022-06-02 | Rhizen Pharmaceuticals Ag | Cdk inhibitors |
| CN114605390A (zh) * | 2020-12-04 | 2022-06-10 | 上海凌达生物医药有限公司 | 具有cdk激酶抑制活性的化合物、其药物组合物和用途 |
| WO2022149057A1 (en) | 2021-01-05 | 2022-07-14 | Rhizen Pharmaceuticals Ag | Cdk inhibitors |
| AU2022231254A1 (en) | 2021-03-03 | 2023-09-28 | Chia Tai Tianqing Pharmaceutical Group Co., Ltd. | Combined pharmaceutical composition containing cdk4/6 inhibitor and use thereof |
| CA3232226A1 (en) | 2021-09-27 | 2023-03-30 | Fan FENG | Combined pharmaceutical composition of cdk4/6 inhibitor and aromatase inhibitor |
| WO2023237055A1 (zh) * | 2022-06-09 | 2023-12-14 | 正大天晴药业集团股份有限公司 | 一种cdk4/6抑制剂治疗去分化脂肪肉瘤的用途 |
| WO2024032751A1 (zh) * | 2022-08-12 | 2024-02-15 | 正大天晴药业集团股份有限公司 | 一种取代的2-氢-吡唑衍生物的晶型及其制备方法 |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2654670A1 (en) * | 2006-07-06 | 2008-01-10 | Boehringer Ingelheim International Gmbh | New compounds |
| KR20090052385A (ko) * | 2006-09-08 | 2009-05-25 | 화이자 프로덕츠 인크. | 2-(피리딘-2-일아미노)-피리도[2,3-d]피리미딘-7-온의합성 |
| US9259399B2 (en) * | 2007-11-07 | 2016-02-16 | Cornell University | Targeting CDK4 and CDK6 in cancer therapy |
| KR101700454B1 (ko) * | 2008-02-15 | 2017-01-26 | 리겔 파마슈티칼스, 인크. | 피리미딘-2-아민 화합물 및 jak 키나제 억제제로서의 그의 용도 |
| JO2885B1 (en) * | 2008-12-22 | 2015-03-15 | ايلي ليلي اند كومباني | Protein kinase inhibitors |
| UY33227A (es) * | 2010-02-19 | 2011-09-30 | Novartis Ag | Compuestos de pirrolopirimidina como inhibidores de la cdk4/6 |
| RU2589696C2 (ru) | 2010-04-13 | 2016-07-10 | Новартис Аг | КОМБИНАЦИЯ, ВКЛЮЧАЮЩАЯ ИНГИБИТОР ЦИКЛИНЗАВИСИМОЙ КИНАЗЫ 4 ИЛИ ЦИКЛИНЗАВИСИМОЙ КИНАЗЫ 6 (CDK4/6) И ИНГИБИТОР mTOR, ДЛЯ ЛЕЧЕНИЯ РАКА |
| PT2937349T (pt) | 2011-03-23 | 2017-03-24 | Amgen Inc | Inibidores duais tricíclicos fusionados de cdk 4/6 e flt3 |
-
2016
- 2016-03-10 JP JP2017547403A patent/JP6726677B2/ja active Active
- 2016-03-10 EP EP16761108.6A patent/EP3269715B1/en active Active
- 2016-03-10 WO PCT/CN2016/076041 patent/WO2016141881A1/zh active Application Filing
- 2016-03-10 BR BR112017019286-1A patent/BR112017019286B1/pt active IP Right Grant
- 2016-03-10 CA CA2978363A patent/CA2978363A1/en active Pending
- 2016-03-10 US US15/557,210 patent/US9969719B2/en active Active
- 2016-03-10 AU AU2016228660A patent/AU2016228660B2/en active Active
- 2016-03-10 CN CN202010144732.3A patent/CN111333627A/zh active Pending
- 2016-03-10 KR KR1020177028357A patent/KR102615733B1/ko active IP Right Grant
- 2016-03-10 RU RU2017131559A patent/RU2722363C2/ru active
- 2016-03-10 CN CN201680014066.3A patent/CN107428731B/zh active Active
- 2016-03-10 ES ES16761108T patent/ES2806206T3/es active Active
- 2016-03-11 TW TW105107614A patent/TWI688559B/zh active
-
2018
- 2018-03-27 HK HK18104212.4A patent/HK1244803A1/zh unknown
Also Published As
| Publication number | Publication date |
|---|---|
| EP3269715A1 (en) | 2018-01-17 |
| CA2978363A1 (en) | 2016-09-15 |
| BR112017019286A2 (pt) | 2018-05-02 |
| AU2016228660B2 (en) | 2020-05-07 |
| CN111333627A (zh) | 2020-06-26 |
| US20180072707A1 (en) | 2018-03-15 |
| RU2017131559A3 (es) | 2019-04-11 |
| BR112017019286B1 (pt) | 2023-02-14 |
| RU2722363C2 (ru) | 2020-05-29 |
| JP6726677B2 (ja) | 2020-07-22 |
| TW201639831A (zh) | 2016-11-16 |
| HK1244803A1 (zh) | 2018-08-17 |
| CN107428731B (zh) | 2020-05-05 |
| WO2016141881A1 (zh) | 2016-09-15 |
| JP2018507883A (ja) | 2018-03-22 |
| KR20180005160A (ko) | 2018-01-15 |
| RU2017131559A (ru) | 2019-04-11 |
| EP3269715B1 (en) | 2020-05-13 |
| AU2016228660A1 (en) | 2017-10-19 |
| CN107428731A (zh) | 2017-12-01 |
| KR102615733B1 (ko) | 2023-12-18 |
| US9969719B2 (en) | 2018-05-15 |
| EP3269715A4 (en) | 2018-08-08 |
| TWI688559B (zh) | 2020-03-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2806206T3 (es) | Derivado de 2-hidrógeno pirazol sustituido que sirve como fármaco anticanceroso | |
| AU2021245168B2 (en) | Amine-substituted aryl or heteroaryl compounds as ehmt1 and ehmt2 inhibitors | |
| ES2954278T3 (es) | Derivados de piperidina como inhibidores de la quinasa dependiente de ciclina 7 (CDK7) | |
| ES2935746T3 (es) | Inhibidores de desmetilasa-1 específica de lisina | |
| AU2009302360B2 (en) | Imidazopyridazinecarbonitriles useful as kinase inhibitors | |
| AU2011253057B2 (en) | Nitrogen heterocyclic compounds useful as PDE10 inhibitors | |
| RU2658912C1 (ru) | АНАЛОГ ПИРИДИНО[1,2-А]ПИРИМИДОНА, ИСПОЛЬЗУЕМЫЙ В КАЧЕСТВЕ ИНГИБИТОРА mTOR/PI3K | |
| WO2010003133A2 (en) | Cdk modulators | |
| JP6680774B2 (ja) | PI3K阻害剤としてのピリド[1,2−a]ピリミドン類似体 | |
| JP2021533179A (ja) | ピラジン化合物およびその使用 | |
| AU2018388439B2 (en) | Carbamate and urea compounds as multikinase inhibitors | |
| EP2935272A1 (en) | Pyrazole substituted imidazopyrazines as casein kinase 1 d/e inhibitors | |
| JP2022530371A (ja) | ピリミド5員複素環化合物及び変異型idh2阻害薬としてのその使用 | |
| CN116323622A (zh) | 双环杂芳基类衍生物及其制备方法和用途 | |
| KR20160086930A (ko) | 피롤로피롤론 유도체 및 bet 억제제로서의 그의 용도 | |
| WO2023046128A1 (en) | A cyclin-dependent kinase inhibitor | |
| WO2024073502A1 (en) | Heterocyclic compounds as e3 ligase inhibitors | |
| WO2023250083A1 (en) | Small molecule inhibitors of dyrk/clk and uses thereof | |
| JP2024509864A (ja) | サイクリン依存性キナーゼ7(cdk7)阻害剤としての三環式ピリジン |