ES2786552T3 - Compuestos de sulfondiimina macrocíclicos nuevos - Google Patents
Compuestos de sulfondiimina macrocíclicos nuevos Download PDFInfo
- Publication number
- ES2786552T3 ES2786552T3 ES16770510T ES16770510T ES2786552T3 ES 2786552 T3 ES2786552 T3 ES 2786552T3 ES 16770510 T ES16770510 T ES 16770510T ES 16770510 T ES16770510 T ES 16770510T ES 2786552 T3 ES2786552 T3 ES 2786552T3
- Authority
- ES
- Spain
- Prior art keywords
- group
- compounds
- formula
- hydrogen atom
- compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- LXFQSRIDYRFTJW-UHFFFAOYSA-M Cc(cc1C)cc(C)c1S([O-])(=O)=O Chemical compound Cc(cc1C)cc(C)c1S([O-])(=O)=O LXFQSRIDYRFTJW-UHFFFAOYSA-M 0.000 description 3
- 0 C[C@@](c(c(*)c1F)ccc1F)N Chemical compound C[C@@](c(c(*)c1F)ccc1F)N 0.000 description 2
- AFCGCODEEUFDAF-UHFFFAOYSA-N CCc(cc(cc1)F)c1N Chemical compound CCc(cc(cc1)F)c1N AFCGCODEEUFDAF-UHFFFAOYSA-N 0.000 description 1
- TVTIFXKLHZKPJD-UHFFFAOYSA-N CN=S(C)(Cc1cc(OCCCOc2cc(F)ccc2-c(cc(N2)nc3)c3F)nc2c1)=N Chemical compound CN=S(C)(Cc1cc(OCCCOc2cc(F)ccc2-c(cc(N2)nc3)c3F)nc2c1)=N TVTIFXKLHZKPJD-UHFFFAOYSA-N 0.000 description 1
- WIGXPGCDKCKVFB-UHFFFAOYSA-N CSCc1cc(Nc(nc2)nc(-c(cccc3F)c3OCCCCO3)c2F)cc3c1 Chemical compound CSCc1cc(Nc(nc2)nc(-c(cccc3F)c3OCCCCO3)c2F)cc3c1 WIGXPGCDKCKVFB-UHFFFAOYSA-N 0.000 description 1
- JESPDIXGDCFSOT-UHFFFAOYSA-N CSCc1cc(OCCCO)nc(Cl)c1 Chemical compound CSCc1cc(OCCCO)nc(Cl)c1 JESPDIXGDCFSOT-UHFFFAOYSA-N 0.000 description 1
- PTCIXNBWMRAOMX-UHFFFAOYSA-N CSCc1cc([N+]([O-])=O)cc(OCCCCO)c1 Chemical compound CSCc1cc([N+]([O-])=O)cc(OCCCCO)c1 PTCIXNBWMRAOMX-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/12—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains three hetero rings
- C07D498/18—Bridged systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/4375—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a six-membered ring having nitrogen as a ring heteroatom, e.g. quinolizines, naphthyridines, berberine, vincamine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/444—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring heteroatom, e.g. amrinone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D498/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D498/08—Bridged systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/12—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains three hetero rings
- C07D498/14—Ortho-condensed systems
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Un compuesto de fórmula general (I), **(Ver fórmula)** en la que L representa un grupo alquileno C2-C5, en el que dicho grupo está opcionalmente sustituido con (i) un sustituyente seleccionado de cicloalquil C3-C4- e hidroximetil-, y/o (ii) uno o dos sustituyentes adicionales, iguales o diferentes, seleccionados de alquil C1-C2-, X, Y representan CH o N con la condición de que uno de X e Y represente CH y uno de X e Y represente N; R1 representa un grupo seleccionado de alquil C1-C4-y cicloalquil C3-C5-, en el que dicho grupo está opcionalmente sustituido con uino o dos o tres sustituyentes, iguales o diferentes, seleccionados del grupo que consiste en hidroxi, ciano, halógeno, alquil C1-C2-, alcoxi C1-C2-, -NH2, -C(=O)OH; R2 representa un grupo seleccionado de un átomo de hidrógeo, un átomo de flúor, un átomo de cloro, metil-, metoxi- , trifluorometil-; R3 representa un grupo seleccionado de un átomo de hidrógeno, un átomo de flúor, un átomo de cloro, ciano, metil- , metoxi-, trifluorometil-, trifluorometoxi-; R4 representa un átomo de hidrógeno o un átomo de flúor; R5 representa un grupo seleccionado de un átomo de hidrógeno, ciano, -C(=O)R8, -C(=O)OR8, -S(=O)2R8, - C(=O)NR6R7, alquil C1-C4-, cicloalquil C3-C5-, en el que dicho grupo alquil C1-C4- o cicloalquil C3-C5- está opcionalmente sustituido con un sustituyente seleccionado del grupo que consiste en flúor, hidroxi, ciano, alcoxi C1-C3-, -NH2, alquilamino-, dialquilamino-; R6, R7 representa, de modo independiente entre sí, un grupo seleccionado de un átomo de hidrógeno, alquil C1- C4- y cicloalquil C3-C5-, en el que dicho grupo C1-C4- o cicloalquil C3-C5- está opcionalmente sustituido con uno o dos sustituyentes, iguales o diferentes, seleccionados del grupo que consiste en hidroxi, alquil C1-C2-, alcoxi C1-C2-, -NH2, alquilamino-, dialquilamino-; R8 representa un grupo seleccionado de alquil C1-C6-, fluoro-alquil C1-C3-, cicloalquil C3-C5- y fenil-, en el que dicho grupo está opcionalmente sustituido con un sustituyente seleccionado del grupo que consiste en hidroxi, alquil C1-C2-, alcoxi C1-C2-, -NH2, o un enantiómero, diastereómero, sal, solvato o sal de solvato del mismo.
Description
DESCRIPCIÓN
Compuestos de sulfondiimina macrocíclicos nuevos
La presente invención se refiere a compuestos de sulfondiimina macrocíclicos de fórmula general (I), como se describen y definen en el presente documento, y procedimientos para prepararlos, su uso para el tratamiento y/o profilaxis de trastornos, particularmente de los trastornos hiperproliferativos y/o enfermedades infecciosas inducidas por virus y/o las enfermedades cardiovasculares. Además, la invención se refiere a compuestos intermedios útiles en la preparación de dichos compuestos de fórmula general (I).
Las proteínas de la familia de las quinasas dependientes de ciclinas (CDK) son reguladores fundamentales del ciclo celular (CDK del ciclo celular), participan en la regulación de la transcripción de los genes (CDK de la transcripción) y cumplen otras funciones. Para ejercer sus efectos, las CDK deben activarse, lo cual puede ocurrir a través de la asociación con una subunidad reguladora, que suele ser una ciclina. Las CDK del ciclo celular, que incluyen el complejo de la CDK1 y la ciclina B, el complejo de la CDK2 y la ciclina A, el complejo de la CDK2 y la ciclina E, el complejo de la CDK4 y la ciclina D y el complejo de la CDK6 y la ciclina D, son activadas consecutivamente para participar en el ciclo celular. Las CDK de la transcripción, que incluyen el complejo de la CDK9 y la ciclina T y el complejo de la CDK7 y la ciclina H, regulan la actividad de la ARN polimerasa II cuando se fosforila el dominio en su extremo carboxilo (CTD). El factor de transcripción positivo b (P-TEFb) es un heterodímero de la CDK9 y una de cuatro ciclinas posibles, la ciclina T1, la ciclina K, la ciclina T2a o la ciclina T2b.
Mientras que la CDK9 (que tiene la identificación del banco de genes del NCBI 1025) solamente participa en la regulación de la transcripción, la CDK7 también participa en la regulación del ciclo celular, donde cumple la función de una quinasa que activa otras CDK (CAK).
La transcripción de los genes por la ARN polimerasa II comienza cuando se ensambla el complejo previo al inicio en la región promotora, después de lo cual los residuos de serina 5 y 7 del CTD son fosforilados por la CDK7/ciclina H. En la mayoría de los genes, la ARN polimerasa II deja de transcribir el ARNm una vez que se ha desplazado 20-40 nucleótidos a lo largo del ADN que hace las veces de molde. Esta pausa que realiza la ARN polimerasa II cerca del promotor es mediada por factores negativos para la continuación de la transcripción, y se reconoce que es un mecanismo de control importante para regular la expresión de los genes que son inducidos rápidamente en respuesta a diversos estímulos (Cho y col., Cell Cycle 2010, 9, 1697). El P-TEFb cumple una función crucial en la resolución de la pausa que se induce cerca del promotor, ya que media en la transición hasta un estado de continuación de la producción, para lo cual se fosforila el residuo de serina 2 del CTD y se fosforilan y se inactivan los factores negativos para la continuación de la transcripción.
La actividad del propio P-TEFb está regulada por diversos mecanismos. Aproximadamente la mitad del P-TEFb en las células toma la forma de un complejo inactivo con el ARN nuclear pequeño 7SK (que se abrevia ARNnp 7SK), la proteína relacionada con La 7 (LARP7/PIP7S) y las proteínas que pueden ser inducidas por la hexametilen bisacetamida 1 y 2 (HEXIM1/2, He y col., Mol. Cell 29, 588, 2008). La porción restante del P-TEFb toma la forma de un complejo activo que contiene una proteína que comprende un dominio con bromo Brd4 (Yang y col., Mol. Cell 19, 535, 2005). A través de una interacción con las histonas acetiladas, la Brd4 recluta el P-TEFb hacia las áreas de la cromatina en las que ha de ocurrir la transcripción de los genes. Mediante una interacción alternativa con los reguladores positivos y negativos, el P-TEFb se mantiene en un equilibrio funcional. El P-TEFb unido al complejo con el ARNnp 7SK constituye una reserva desde la cual puede liberarse el P-TEFb activo sobre la base de la demanda de la transcripción en las células y de la proliferación de las células (Zhou y Yik, Microbiol. Mol. Biol. Rev. 70, 646, 2006). Además, la actividad del P-TEFb es regulada por diversas modificaciones que ocurren después de la traducción, que pueden incluir la fosforilación, la desfosforilación, la ubiquitinación y la acetilación (puede hallarse una revisión en Cho y col., Cell Cycle 9, 1697, 2010).
Una actividad desregulada de la quinasa CDK9 puede provocar una falta de regulación en el heterodímero del P-TEFb, lo que puede observarse en el contexto de diversos trastornos patológicos en los seres humanos, tales como las enfermedades hiperproliferativas (por ejemplo, el cáncer), las enfermedades infecciosas inducidas por virus o las enfermedades cardiovasculares.
Se considera que el cáncer es un trastorno hiperproliferativo que es mediado por un desequilibrio entre la proliferación y la muerte de las células (la apoptosis). Pueden hallarse niveles elevados de las proteínas antiapoptóticas de la familia Bcl-2 en diversos tumores humanos. Estas proteínas dan como resultado una supervivencia prolongada de las células tumorales, lo que implica una resistencia a la terapia. Se demostró que, mediante la inhibición de la actividad de quinasa del P-TEFb, era posible reducir la actividad de transcripción de la ARN polimerasa II, lo que daba como resultado una declinación en la cantidad de proteínas antiapoptóticas, especialmente de Mcl-1 y XIAP, que presentan una vida media breve, con lo que podía inducirse la apoptosis de las células tumorales. Otras proteínas diversas que están asociadas al fenotipo de los tumores transformados (tales como Myc, NF-kB, los transcriptos que responden a los genes o las quinasas mitóticas) son proteínas que tienen vidas medias breves o que están codificadas por transcriptos con vidas medias breves, que son sensibles a las reducciones en la actividad de la ARN polimerasa II que pueden provocarse a través de la inhibición del P-TEFb (puede hallarse una revisión en Wang y Fischer, Trends Pharmacol. Sci. 29, 302, 2008).
Diversos virus aprovechan la maquinaria de transcripción de la célula huésped para transcribir su propio genoma. Por
ejemplo, la ARN polimerasa II del VIH-1 es reclutada hacia la región promotora en la LTR del virus. La proteína activadora de la transcripción del virus (Tat) se une a los transcriptos nacientes del virus, lo que da como resultado la anulación de la pausa en la actividad de la ARN polimerasa II que se induce cerca del promotor. En este procedimiento, se recluta el P-TEFb, que es el responsable de promover la continuación de la transcripción. Además, la proteína Tat provoca un incremento en la fracción del P-TEFb activo, ya que merced a su acción se reemplazan las proteínas que inhiben el P-TEFb, HEXIM1 y 2, en el complejo del ARNnp 7SK. A través de los estudios recientes, se ha demostrado que la inhibición de la actividad de quinasa del P-TEFb es suficiente para bloquear la replicación del VIH-1, y que, para ello, pueden emplearse concentraciones de los inhibidores de quinasas que no resultan tóxicas para las células huésped (puede hallarse una revisión en Wang y Fischer, Trends Pharmacol. Sci. 29, 302, 2008). De manera similar, se ha descrito el reclutamiento del P-TEFb mediado por proteínas de otros virus, tales como el virus de Epstein-Barr que está asociado a las células B cancerosas, donde la proteína antigénica nuclear EBNA2 interactúa con el P-TEFb (Bark-Jones y col., Oncogene 25, 1775, 2006), y el virus linfotrópico de las células T humanas del tipo 1 (HTLV-1), donde el activador de la transcripción Tax es el responsable del reclutamiento del P-TEFb (Zhou y col., J. Virol. 80, 4781, 2006).
La hipertrofia cardíaca, que es la respuesta adaptativa del corazón a la sobrecarga mecánica y a la presión (lo que puede ocurrir en presencia de estrés hemodinámico, por ejemplo, de hipertensión o de un infarto de miocardio), puede dar como resultado una insuficiencia cardíaca y la muerte a largo plazo. Se ha demostrado que la hipertrofia cardíaca está asociada a una actividad de transcripción incrementada y a una fosforilación más elevada del CTD de la ARN polimerasa II en las células del músculo cardiaco. Se descubrió que el P-TEFb es activado cuando se disocia del complejo inactivo con el ARNnp 7SK y HEXIM1 y 2. Sobre la base de estos descubrimientos, puede inferirse que la inhibición farmacológica de la actividad de quinasa del P-TEFb podría servir como un abordaje terapéutico para tratar la hipertrofia cardíaca (puede hallarse una revisión en Dey y col., Cell Cycle 6, 1856, 2007).
En resumen, sobre la base de la abundante evidencia disponible, puede concluirse que la inhibición selectiva de la actividad de la quinasa CDK9 en el heterodímero del P-TEFb (que comprende una quinasa CDK9 y una de las siguientes ciclinas: la ciclina T1, la ciclina K, la ciclina T2a o la ciclina T2b) representa un abordaje innovador para tratar enfermedades como el cáncer, las enfermedades provocadas por virus y/o las enfermedades del corazón. La CDK9 pertenece a una familia que comprende al menos 13 quinasas relacionadas estrechamente, de las cuales las que pertenecen al subgrupo de las CDK del ciclo celular cumplen diversas funciones en la regulación de la proliferación de las células. Por lo tanto, resulta lógico esperar que la inhibición de las CDK del ciclo celular (por ejemplo, la CDK1/ciclina B, la CDK2/ciclina A, la CDK2/ciclina E, la CDK4/ciclina D o la CDK6/ciclina D) y de la c Dk 9 afecte a los tejidos que presentan una proliferación normal, tales como la mucosa intestinal, los órganos linfáticos y hematopoyéticos y los órganos reproductivos. Para maximizar el margen terapéutico de los inhibidores de la quinasa CDK9, se necesitan moléculas con mayor duración de acción y/o alta potencia y eficacia y/o selectividad elevada por la CDK9.
Los inhibidores de las CDK en general, así como los inhibidores de la CDK9 se describen en diversas publicaciones, diferentes:
En el documento WO 200812970 y en el documento WO 200812971, se describen aminopirimidinas 2,4-disustituidas que pueden usarse como inhibidores de las CDK en general. También se menciona que algunos de estos compuestos pueden actuar como inhibidores selectivos de la CDK9 (WO 200812970) y como inhibidores de la CDK5 (WO 200812971), pero no se proporcionan valores específicos para la CI50 sobre la CDK9 (WO 200812970) o sobre la CDK5 (WO 200812971). Estos compuestos no contienen un átomo de flúor en la posición 5 del núcleo de piridina.
En el documento WO 2008129080, se describen aminopirimidinas 4,6-disustituidas y se demuestra que estos compuestos presentan efectos de inhibición sobre la actividad de diversas proteína quinasas, tales como la CDK1, la CDK2, la CDK4, la CDK5, la CDK6 y la CDK9, con una preferencia por la inhibición de la CDK9 (ejemplo 80).
El documento WO 2005026129 desvela aminopirimidinas 4,6-disustituidas y demuestra que estos compuestos presentan un efecto inhibitorio sobre la actividad proteína quinasa de diferentes proteína quinasas, en particular CDK2, CDK4 y CDK9.
En el documento WO 2009118567, se desvelan derivados de las pirimidinas y de las [1,3,5]triazinas como inhibidores de las proteína quinasas, particularmente de la CDK2, de la CDK7 y de la c DK9.
El documento WO 2011116951 desvela derivados de triazina sustituidos como inhibidores selectivos de CDK9. El documento WO 2012117048 desvela derivados de triazina sustituidos como inhibidores selectivos de CDK9. El documento WO 2012117059 desvela derivados de piridina disustituidos como inhibidores selectivos de CDK9. El documento WO 2012143399 desvela 4-aril-N-fenil-1,3,5-triazin-2-aminas sustituidas como inhibidores selectivos de CDK9.
En el documento EP1218360 B1, que corresponde a los documentos US2004116388A1, US7074789B2 y WO 2001025220A1, se describen derivados de triazinas que pueden usarse como inhibidores de quinasas, pero no se desvelan inhibidores potentes o selectivos de la CDK9.
El documento WO 2008079933, desvela derivados de aminopiridinas y de aminopirimidinas que pueden usarse como inhibidores de la CDK1, de la CDK2, de la CDK3, de la CDK4, de la CDK5, de la CDK6, de la CDK7, de la CDK8 o de
la CDK9.
El documento WO 2011012661, describe derivados de aminopiridinas que pueden usarse como inhibidores de las CDK.
El documento WO 2011026917 desvela carboxamidas obtenidas a partir de 4-fenilpiridin-2-aminas sustituidas como inhibidores de CDK9.
El documento WO 2012066065 desvela fenil-heteroarilaminas como inhibidores de CDK9. Se prefiere una selectividad por CDK9 respecto a otras isoformas de CDK, sin embargo, la divulgación de los datos de inhibición de CDK está confinada a c Dk 9. No se desvelan sistemas de anillos bicíclicos unidos a la posición C4 del núcleo pirimidina. Dentro del grupo unido al C4 del núcleo pirimidina, los fenilos alcoxi pueden referirse como abarcados, pero no hay sugerencia de un patrón de sustitución específico caracterizado por un átomo de flúor unido al C5 del anillo pirimidina, y una anilina en el C2 de la pirimidina, exhibiendo un grupo sulfonil-metileno sustituido en posición meta. Los compuestos mostrados en los ejemplos típicamente exhiben un grupo cicloalquilo sustituido como R1, pero no fenilo.
El documento WO 2012066070 desvela compuestos de 3-(aminoaril)-piridina como inhibidores de CDK9. El núcleo de biarilo obligatoriamente consiste en dos anillos heteroaromáticos.
El documento WO 2012101062 desvela compuestos bi-heteroarilo sustituidos que exhiben un núcleo 2-aminopiridina como inhibidores de CDK9. El núcleo biarilo obligatoriamente consiste en dos anillos heteroaromáticos.
El documento WO 2012101063 desvela carboxamidas derivadas de 4-(heteroaril)-piridin-2-aminas sustituidas como inhibidores de CDK9.
El documento WO 2012101064 desvela compuestos biaril-N-acilpirimidina como inhibidores de CDK9.
El documento WO 2012101065 desvela compuestos biarilpirimidina como inhibidores de CDK9. El núcleo biarilo obligatoriamente consiste en dos anillos heteroaromáticos.
El documento WO 2012101066 desvela compuestos biarilpirimidina como inhibidores de CDK9. La sustitución R1 del grupo amino unido al núcleo heteroaromático está confinada a grupos no aromáticos, pero no cubre fenilos sustituidos. Adicionalmente, el núcleo biarilo obligatoriamente consiste en dos anillos heteroaromáticos.
El documento WO 2011077171, desvela derivados 4,6-disustituidos de las aminopirimidinas como inhibidores de la CDK9.
El documento WO 2014031937, desvela derivados 4,6-disustituidos de las aminopirimidina como inhibidores de la CDK9.
El documento WO 2013037896 desvela 5-fluoropirimidinas disustituidas como inhibidores selectivos de CDK9.
El documento WO 2013037894 desvela derivados de 5-fluoropirimidina disustituidos que contienen un grupo sulfoximina como inhibidores selectivos de CDK9.
En Wang y col. (Chemistry and Biology 2010, 17, 1111-1121), se desvelan 2-anilino-4-(tiazol-5-il)pirimidinas que pueden usarse como inhibidores de las CDK de la transcripción, que presentan actividad anticancerosa en diversos modelos basados en animales.
El documento WO 2014060376, desvela derivados sustituidos de las 4-(orto)-fluorofenil-5-fluoropirimidin-2-il aminas que contienen un grupo sulfona como inhibidores selectivos de la CDK9.
El documento WO 2014060375, desvela derivados sustituidos de las 5-fluoro-N-(piridin-2-il)piridin-2-aminas que contienen un grupo sulfona como inhibidores selectivos de la CDK9.
El documento WO 2014060493, desvela derivados sustituidos de las N-(piridin-2-il)pirimidin-4-aminas que contienen un grupo sulfona como inhibidores selectivos de la CDK9.
El documento WO 2014076028, desvela derivados sustituidos de las 4-(orto)-fluorofenil-5-fluoropirimidin-2-il aminas que contienen un grupo sulfoximina como inhibidores selectivos de la CDK9.
El documento WO 2014076091, desvela derivados sustituidos de las 5-fluoro-N-(piridin-2-il)-piridin-2-aminas que contienen un grupo sulfoximina como inhibidores selectivos de la CDK9.
El documento WO 2014076111 desvela derivados sustituidos de las N-(piridin-2-il)-pirimidin-4-aminas que contienen un grupo sulfoximina como inhibidores selectivos de la CDK9.
El documento WO 2015001021 desvela derivados de las 5-fluoro-N-(piridin-2-il)-piridin-2-aminas que contienen un grupo sulfoximina como inhibidores selectivos de la CDK9.
El documento WO 2015136028 desvela derivados de 5-fluoro-N-(piridin-2-il)piridin-2-amina que contienen un grupo sulfona como inhibidores selectivos de CDK9.
El documento WO 2004009562, desvela triazinas sustituidas que pueden usarse como inhibidores de quinasas. Se proporcionan datos sobre determinados compuestos que inhiben la CDK1 y la CDK4, pero no se provee información acerca de la CDK9.
El documento WO 2004072063 describe heteroarilpirroles sustituidos (pirimidinas y triazinas) que pueden usarse como inhibidores de proteína quinasas como la ERK2, la GSK3, la PKA o la CDK2.
El documento WO 2010009155, desvela derivados de triazinas y de pirimidinas que pueden usarse como inhibidores de la histona desacetilasa y/o de las quinasas dependientes de ciclinas (CDK). Se proporcionan datos sobre determinados compuestos que pueden inhibir la CDK2.
El documento WO 2003037346 (que corresponde a los documentos US7618968B2, US7291616B2, US2008064700A1 y US2003153570A1) se refiere a ariltriazinas y sus usos, que abarcan la inhibición de la actividad de la beta aciltransferasa de ácido lisofosfatídico (LPAAT-beta) y/o de la proliferación de determinadas células tumorales.
El documento WO 2005037800 desvela anilino-pirimidinas sustituidas con sulfoximina como inhibidores de VEGFR y quinasas CDK, en particular VEGFR2, CDK1 y CDK2, que no tienen anillo aromático unido directamente al anillo pirimidina y que tienen el grupo sulfoximina directamente unido al grupo anilina. No se describen datos de CDK9.
El documento WO 2008025556, describe carbamilsulfoximidas que presentan un núcleo de pirimidina y que son útiles como inhibidores de quinasas. No se provee información acerca de la CDK9.
El documento WO 2002066481, describe derivados de pirimidinas que pueden usarse como inhibidores de la quinasas dependientes de ciclinas, pero no se menciona la CDK9 y no se provee información acerca de ella.
El documento WO 2008109943, describe compuestos que son fenilaminopiri(mi)dinas y su uso como inhibidores de quinasa, particularmente como inhibidores de la quinasa JAK2. Los ejemplos específicos se centran en compuestos que comprenden núcleos de pirimidinas.
En documento WO 2009032861, describe pirimidinilaminas sustituidas que pueden usarse como inhibidores de la quinasa JNK. Los ejemplos específicos se centran en compuestos que comprenden núcleos de pirimidinas.
El documento WO 2011046970 describe compuestos que son amino-pirimidinas que pueden usarse como inhibidores de TBK1 y/o de IKK épsilon. Los ejemplos específicos se centran en compuestos que comprenden núcleos de pirimidinas.
El documento WO 2012142329 se refiere a compuestos amino-pirimidina como inhibidores de TBK1 y/o IKK épsilon.
El documento WO 2012139499 desvela anilino-pirimidinas sustituidas con urea como inhibidores de diferentes proteína quinasas.
El documento WO 2014106762, desvela derivados de las 4-pirimidinilamino-bencensulfonamidas como inhibidores de las quinasa similares a polo 1.
Se han descrito compuestos macrocíclicos como sustancias con actividad terapéutica, particularmente como inhibidores de una variedad de proteína quinasas, como es el caso de las quinasas que dependen de las ciclinas. Sin embargo, en los documentos que se detallan más adelante no describen compuestos específicos que puedan resultar útiles como inhibidores de la CDK9.
El documento WO 2007147574, desvela sulfonamido-macrociclos como inhibidores de Tie2 que presentan selectividad sobre la CDK2 y sobre la quinasa Aurora C, que pueden resultar útiles en el tratamiento de aquellas enfermedades que están acompañadas por un crecimiento desregulado de los vasos, entre otros propósitos.
El documento WO 2007147575, desvela otros sulfonamido-macrociclos como inhibidores de Tie2 que presentan selectividad sobre la CDK2 y sobre la Plk1, que pueden resultar útiles en el tratamiento de aquellas enfermedades que están acompañadas por un crecimiento desregulado de los vasos, entre otros propósitos.
El documento WO 2006066957/EP 1674470, desvela otros sulfonamido-macrociclos como inhibidores de Tie2 que presentan una citotoxicidad baja, que pueden resultar útiles en el tratamiento de aquellas enfermedades que están acompañadas por un crecimiento desregulado de los vasos, entre otros propósitos.
El documento WO 2006066956/EP 1674469, desvela otros sulfonamido-macrociclos como inhibidores de Tie2 que presentan una citotoxicidad baja, que pueden resultar útiles en el tratamiento de aquellas enfermedades que están acompañadas por un crecimiento desregulado de los vasos, entre otros propósitos.
El documento WO 2004026881/DE 10239042, desvela derivados macrocíclicos de pirimidinas como inhibidores de las quinasas que dependen de las ciclinas, particularmente de la CDK1 y de la CDK2, y también como inhibidores del VEGF-R, que pueden resultar útiles en el tratamiento del cáncer, entre otros propósitos. Los compuestos de la presente
invención difieren de los que se describen en WO 2004026881 porque comprenden una porción biaromática obligatoria en el sistema del anillo macrocíclico. Por otra parte, ninguno de los compuestos que se describen a modo de ejemplo en WO 2004026881 comprende un grupo sulfondiimina.
El documento WO 2007079982/EP 1803723, desvela bencenoaciclononafanos macrocíclicos como inhibidores de una variedad de proteína quinasas, como es el caso de las quinasas Aurora A o C, la CDK1, la CDK2 o c-Kit, que pueden resultar útiles en el tratamiento del cáncer, entre otros propósitos. Los compuestos de la presente invención difieren de los que se describen en WO 2007079982 porque comprenden una porción biaromática obligatoria en el sistema del anillo macrocíclico. Por otro lado, los compuestos de la presente invención no comprenden un grupo sulfondiimina.
El documento WO 2006106895/EP 1710246, desvela compuestos macrocíclicos del tipo de las sulfoximinas como inhibidores de Tie2 que presentan una citotoxicidad baja, que pueden resultar útiles en el tratamiento de aquellas enfermedades que están acompañadas por un crecimiento desregulado de los vasos, entre otros propósitos.
El documento WO 2012009309, desvela compuestos macrocíclicos condensados a anillos de benceno y de piridina que son útiles para disminuir la producción de péptidos amiloides beta.
El documento WO 2009132202, desvela compuestos macrocíclicos como inhibidores de JAK 1, 2 y 3, TYK2 y ALK, que pueden resultar útiles en el tratamiento de aquellas enfermedades que están asociadas a ALK o a JAK, que abarcan las enfermedades inflamatorias, las enfermedades autoinmunes, así como el cáncer.
En ChemMedChem, 2007, 2 (1), 63-77, se describen aminopirimidinas macrocíclicas como agentes para inhibir una variedad de blancos del tipo de las CDK y el VEGF-R con una actividad antiproliferativa potente. Los compuestos de la presente invención difieren de los que se describen en la publicación que se ha citado porque comprenden una porción biaromática obligatoria en el sistema del anillo macrocíclico. Por otra parte, ninguno de los compuestos que se describen en ChemMedChem, 2007, 2 (1), 63-77, comprende un grupo sulfondiimina.
Las sulfondiiminas son compuestos de azufre de alta valencia descritos por primera vez por Coliano y Braude en 1964 (J. A. Cogliano, G. L. Braude, J. Org. Chem. 1964, 29, 1397) y desde su descubrimiento, sólo han tenido un mínimo interés en la comunidad científica (M. Candy, R. A. Bohmann, C. Bolm, Adv. Synth. Catal. 2012, 354, 2928). Así, sólo hay muy pocos ejemplos para el uso del grupo sulfondiimina en los procedimientos de química médica (ver, por ejemplo, a) DE2520230, Ludwig Heumann & Co. GmbH; b) W. L. Mock, J.-T. Tsay, J. Am. Chem. Soc. 1989, 111, 4467).
A pesar de que se conocen diversos inhibidores de las CDK, subsiste la necesidad de inhibidores selectivos de la c DK9, en especial inhibidores de CDK9 que son selectivos a altas concentraciones de ATP, que resulten útiles en el tratamiento de enfermedades como las enfermedades hiperproliferativas, las enfermedades virales y/o las enfermedades en el corazón, que presenten una o más ventajas con relación a los compuestos que se conocen a partir de los antecedentes técnicos, tales como:
• una actividad y/o una eficacia superiores, que, por ejemplo, posibiliten una disminución en las dosis
• un perfil de efectos secundarios mejorado, que, por ejemplo, podrá manifestarse como una menor cantidad de efectos secundarios no deseados, una disminución en la intensidad de los efectos secundarios o una (cito)toxicidad reducida
• una acción con una duración mayor, por ejemplo, como resultado de una mejora a nivel de la farmacocinética y/o de un incremento en la duración de la residencia en el blanco
Un objeto particular de la invención es proporcionar inhibidores selectivos de la quinasa CDK9 que muestran actividad antiproliferativa mejorada en líneas de células tumorales como HeLa, HeLa-MaTu-ADR, NCI-H460, DU145, Caco-2, B16F10, A2780 o Mo LM-13, en comparación con los compuestos conocidos de la técnica anterior.
Otro objeto de la invención es proporcionar inhibidores selectivos de la quinasa CDK9 que muestran una potencia superior en el contexto de la inhibición de la actividad de la CDK9 (lo cual podría manifestarse como una CI50 con relación a la CDK9 o a la ciclina T1 con un valor menor), en comparación con los compuestos conocidos de la técnica anterior.
Otro objeto particular de la invención consiste en proveer inhibidores selectivos de la quinasa CDK9 que muestran una potencia superior en el contexto de la inhibición de la actividad de la CDK9 en presencia de una concentración elevada de ATP, en comparación con los compuestos conocidos de la técnica anterior.
Otro objeto particular de la invención es proporcionar inhibidores selectivos de la quinasa CDK9 que muestran por una residencia en el blanco con una duración mayor, en comparación con los compuestos conocidos de la técnica anterior.
Otro objeto particular de la invención es proporcionar inhibidores selectivos de la quinasa CDK9 que muestran por una acción con una duración mayor, por ejemplo, como resultado de una mejora a nivel de la farmacocinética y/o de un incremento en la duración de la residencia en el blanco.
Además, es un objeto de la presente invención proporcionar inhibidores selectivos de la quinasa CDK9 que, en
comparación con los compuestos conocidos del arte previo, muestran una mayor actividad antiproliferativa en líneas celulares tumorales, tales como HeLa, HeLa-MaTu-ADR, NCI-H460, DU145, Caco-2, B16F10, A2780 o MOLM-13, y/o que muestran una mayor potencia para inhibir la actividad de CDK9 (demostrado por un valor de CI50 inferior para CDK9/ciclina T1), en especial una mayor potencia para inhibir la actividad de CDK9 a altas concentraciones de ATP, y/o que muestran un mayor tiempo de residencia diana en comparación con los compuestos conocidos del arte previo. Los inhibidores de la quinasa CDK9 de acuerdo con la invención deben tener simultáneamente una selectividad por CDK9/ciclina T1 respecto de CDK2/ciclina E, en especial a altas concentraciones de ATP.
Los inhibidores selectivos de quinasa CDK9 de acuerdo con la invención deben tener una aceptable permeabilidad de CaCo-2 y/o una relación de eflujo aceptable de CaCo-2, y/o deben mostrar una aceptable solubilidad acuosa.
La presente divulgación se refiere a compuestos de fórmula general (I)

en la que
L representa un grupo alquileno C2-C5,
en el que dicho grupo está opcionalmente sustituido con
(i) un sustituyente seleccionado de cicloalquil C3-C4- e hidroximetil-, y/o
(ii) uno o dos sustituyentes adicionales, iguales o diferentes, seleccionados de alquil C1-C2-,
X, Y representa CH o N con la condición de que uno de X e Y representen CH y uno de X e Y representen N; R1 representa un grupo seleccionado de alquil C1-C4- y cicloalquil C3-C5-,
en el que dicho grupo está opcionalmente sustituido con uno o dos o tres sustituyentes, idénticos o diferentes, seleccionados del grupo que consiste en hidroxi, ciano, halógeno, alquil C1-C2-, alcoxi C1-C2-, -NH2, -C(=O)O; R2 representa un grupo seleccionado de un átomo de hidrógeno, un átomo de flúor, un átomo de cloro, ciano, metil-, metoxi-, trifluorometil-;
R3 represents a group selected from a hidrogen atom, a fluorine atom, a chlorine atom, cyano, metil-, metoxi-, trifluorometil-, trifluorometoxi-;
R4 representa un átomo de hidrógeno o un átomo de flúor;
R5 representa un grupo seleccionado de un átomo de hidrógeno, ciano, -C(=O)R8, -C(=O)OR8, -S(=O)2R8, -C(=O)NR6R7, alquil C1-C4-, cicloalquil C3-C5-,
en el que dicho grupo alquil C1-C4- o cicloalquil C3-C5- está opcionalmente sustituido con un sustituyente seleccionado del grupo que consiste en flúor, hidroxi, ciano, alcoxi C1-C3-, -NH2 , alquilamino-, dialquilamino-; R6, R7 representan, de modo independiente entre sí, un grupo selecciondo de un átomo de hidrógeno, alquil C1-C4-y cicloalquil C3-C5-,
en el que dicho grupo alquil C1-C4- o cicloalquil C3-C5- está opcionalmente sustituido con uno o dos sustituyentes, iguales o diferentes, seleccionados del grupo que consiste en hidroxi, alquil C1-C2-, alcoxi C1C2-, -NH2, alquilamino-, dialquilamino-;
R8 representa un grupo seleccionado de alquil C1-C6-, fluoro-alquil C1-C3-, cicloalquil C1-C5- y fenil-,
en el que dicho grupo está opcionalmente sustituido con un sustituyente seleccionado del grupo que consiste en halógeno, hidroxi, alquilo C1-C2, alcoxi C1-C2-, -NH2 ,
o un enantiómero, diastereómero, sal, solvato o sal de solvato del mismo. Los compuestos de acuerdo con la invención son los compuestos de la fórmula (I) y las sales, solvatos y solvatos de las sales de los mismos, los compuestos de la fórmula mencionada a continuación en el presente documento que están comprendidos por la fórmula (I) y las sales, solvatos y solvatos de las sales de los mismos y los compuestos que están comprendidos por la fórmula (I) y se mencionan a continuación en el presente documento como formas de realización de ejemplo y las sales, solvatos y solvatos de las sales de los mismos, donde los compuestos que están comprendidos por la fórmula (I) y se mencionan a continuación en el presente documento ya no sean sales, solvatos y solvatos de las sales.
Los compuestos de acuerdo con la invención pueden existir, según su estructura, en formas estereoisoméricas (enantiómeros, diastereómeros). Por ello, la invención se refiere a los enantiómeros o diastereómeros y sus respectivas mezclas. Los constituyentes estereoisoméricamente puros se pueden aislar de una manera conocida de tales mezclas de enantiómeros y/o diastereómeros.
Si los compuestos de acuerdo con la invención pueden estar en formas tautoméricas, la presente invención comprende todas las formas tautoméricas.
Además, los compuestos de la presente invención pueden existir en forma libre, por ejemplo, como una base libre o como un ácido libre o como un zwitterión o puede existir en la forma de una sal. Dicha sal puede ser cualquier sal, ya sea una sal por adición orgánica o inorgánica, en particular cualquier sal por adición orgánica o inorgánica fisiológicamente aceptable, habitualmente usada en farmacia.
Las sales que se prefieren para los fines de la presente invención son sales fisiológicamente aceptables de los compuestos de acuerdo con la invención. Sin embargo, también están comprendidas las sales que no son apropiados para aplicaciones farmacéuticas per se, pero que se pueden usar, por ejemplo, para el aislamiento o la purificación de los compuestos de acuerdo con la invención.
La expresión "sal fisiológicamente aceptable" se refiere a una sal por adición de ácidos inorgánicos u orgánicos relativamente no tóxica de un compuesto de la presente invención, por ejemplo, ver S. M. Berge, y col. "Pharmaceutical Salts", J. Pharm. Sci. 1977, 66, 1-19.
Las sales fisiológicamente aceptables de los compuestos de acuerdo con la invención comprenden sales por adición de ácidos de ácidos minerales, ácidos carboxílicos y ácidos sulfónicos, por ejemplo, sales de ácido clorhídrico, ácido bromhídrico, ácido yodhídrico, ácido sulfúrico, ácido bisulfúrico, ácido sulfámico, ácido fosfórico, ácido nítrico o con un ácido orgánico, tales como ácido fórmico, acético, acetoacético, pirúvico, trifluoroacético, propiónico, butírico, hexanoico, heptanoico, undecanoico, láurico, benzoico, salicílico, ácido 2-(4-hidroxibenzoil)-benzoico, canfórico, cinnámico, ciclopentanpropiónico, diglucónico, 3-hidroxi-2-naftoico, nicotínico, pamoico, pectínico, persulfúrico, 3-fenilpropiónico, piválico, 2-hidroxietansulfónico, itacónico, trifluorometansulfónico, dodecilsulfúrico, etansulfónico, bencensulfónico, para-toluensulfónico, metansulfónico, 2-naftalensulfónico, naftalendisulfónico, canforsulfónico, cítrico, tartárico, esteárico, láctico, oxálico, malónico, succínico, málico, adípico, algínico, maleico, fumárico, D-glucónico, mandélico, ascórbico, glucoheptanoico, glicerofosfórico, aspártico, sulfosalicílico o tiociánico, por ejemplo.
Las sales fisiológicamente aceptables de los compuestos de acuerdo con la invención también comprenden sales de bases convencionales, a modo de ejemplos preferidos, sales de metales alcalinos (por ejemplo, sales de sodio y potasio), sales de metales alcalino-térreos (por ejemplo, sales de calcio y magnesio) y sales de amonio obtenidas a partir de amoníaco o aminas orgánicas con entre 1 y 16 átomos de C, tales como, a modo de ejemplos preferidos, etilamina, dietilamina, trietilamina, etildiisopropilamina, monoetanolamina, dietanolamina, trietanolamina, diciclohexilamina, dimetilaminoetanol, procaína, dibencilamina, N-metilmorfolina, arginina, lisina, etilendiamina, N-metilpiperidina, N-metilglucamina, dimetilglucamina, etilglucamina, 1,6-hexadiamina, glucosamina, sarcosina, serinol, tris(hidroximetil)aminometano, aminopropanodiol, base de Sovak, y 1-amino-2,3,4-butanotriol. Además, los compuestos de acuerdo con la invención pueden formar sales con un ion de amonio cuaternario que se puede obtener por ejemplo, mediante cuaternización de un grupo básico que contiene nitrógeno con agentes, tales como haluros de alquilo inferiores, tales como, cloruros, bromuros y yoduros de metil-, etil-, propilo, y butilo; sulfatos de dialquilo como sulfatos de dimetil-, dietil-, dibutilo y diamilo, haluros de cadena larga como, por ejemplo, cloruros, bromuros y yoduros de decilo, laurilo, miristilo y estearilo, haluros de aralquilo como bromuros de bencilo y fenetil-, y otros. Los ejemplos de iones de amonio cuaternario adecuados son tetrametilamonio, tetraetilamonio, tetra(n-propil)amonio, tetra(nbutil)amonio o N-bencil-N,N,N-trimetilamonio.
La presente invención incluye todas las sales posibles de los compuestos de la presente invención como sales aisladas, o como cualquier mezcla de dichas sales, en cualquier proporción.
Solvatos es el termino usado para los propósitos de la invención para aquellas formas de los compuestos de acuerdo con la invención que forman un complejo con las moléculas del disolvente por coordinación en estado sólido o líquido. Los hidratos son una forma especial de solvatos en los que la coordinación se lleva a cabo con agua. Se prefieren los
hidratos como solvatos dentro del ámbito de la presente invención.
La invención también incluye todas las variaciones isotópicas adecuadas de un compuesto de la invención. Una variación isotópica de un compuesto de la invención se define como una en la cual al menos un átomo se reemplaza por un átomo con el mismo número atómico pero una masa atómica diferente de la masa atómica usualmente o predominantemente encontrada en la naturaleza. Los ejemplos de isótopos que pueden incorporarse en un compuesto de la invención incluyen isótopos de hidrógeno, carbono, nitrógeno, oxígeno, fósforo, azufre, flúor, cloro, bromo y yodo, como, por ejemplo, 2H (deuterio), 3H (tritio), 13C, 14C, 15N, 17O, 18O, 32P, 33P, 33S, 34S, 35S, 129I y 131I, respectivamente. Ciertas variaciones isotópicas de un compuesto de la invención, por ejemplo, aquellas en las que se incorpora uno o más isótopos radiactivos como, por ejemplo, 3H o 14C, son útiles en estudios de distribución tisular de fármacos y/o sustratos. Se prefieren particularmente los isótopos tritiados y de carbono 14, por ejemplo, 14C, por su facilidad de preparación y detectabilidad. Además, la sustitución con isótopos como, por ejemplo, deuterio puede ofrecer ciertas ventajas terapéuticas resultantes de su gran estabilidad metabólica, por ejemplo, alta vida media in vivo o requisitos de dosificación reducidos y por lo tanto pueden ser preferidos en algunas circunstancias. Las variantes isotópicas de un compuesto de la invención pueden prepararse generalmente por procedimientos convencionales conocidos por los especialistas en la materia como, por ejemplo, por medio de los procedimientos ilustrativos o por medio de preparaciones descritas en los ejemplos más adelante, usando las variantes isotópicas apropiadas de los reactivos adecuados.
Además, la presente divulgación también comprende profármacos de los compuestos de acuerdo con la invención. El término "profármacos" comprende compuestos que pueden ser biológicamente activos o inactivos en sí mismos, pero se convierten (por ejemplo, por metabolismo o hidrólisis) a compuestos de acuerdo con la invención durante su estancia en el organismo.
Adicionalmente, la presente invención incluye todas las formas cristalinas posibles, o polimorfos, de los compuestos de la presente invención, como polimorfos simples, o como una mezcla de más de un polimorfo, en cualquier proporción.
En consecuencia, la presente invención incluye todas las sales posibles, polimorfos, metabolitos, hidratos, solvatos, profármacos (por ejemplo: ésteres) de los mismos, y formas diastereoisoméricas de los compuestos de la presente invención como sales simples, polimorfos, metabolitos, hidratos, solvatos, profármaco (por ejemplo: ésteres) de los mismos, o formas diastereoisoméricas, o como mezcla de más de una sal, polimorfo, metabolito, hidrato, solvato, profármaco (por ejemplo: ésteres) del mismo, o forma diastereoisomérica en cualquier proporción.
Para los fines de la presente invención, los sustituyentes tienen los siguientes significados, salvo que se especifique lo contrario:
La expresión "átomo de halógeno", "halógeno" o "halo" representa átomos de flúor, cloro, bromo y yodo, particularmente átomos de bromo, cloro o flúor, preferentemente átomos de cloro o flúor, con mayor preferencia, átomos de flúor.
El término "alquil-" representa un grupo alquilo lineal o ramificado con la cantidad de átomos de carbono indicada específicamente, por ejemplo, en C1-C10, uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve o diez átomos de carbono, por ejemplo, metil-, etil-, n-propil-, isopropil-, n-butil-, isobutil-, sec-butil-, ferc-butil-, pentil-, isopentil-, hexil-, heptil-, octil-, nonil-, decil-, 2-metilbutil-, 1 -metilbutil-, 1 -etilpropil-, 1,2-dimetilpropil-, neo-pentil-, 1,1 -dimetilpropil-, 4-metilpentil-, 3-metilpentil-, 2-metilpentil-, 1 -metilpentil-, 2-etilbutil-, 1 -etilbutil-, 3,3-dimetilbutil-, 2,2-dimetilbutil-, 1,1-dimetilbutil-, 2,3-dimetilbutil-, 1,3-dimetilbutil-, o 1,2-dimetilbutilo. Si la cantidad de átomos de carbono no se indicara específicamente, el término "alquil-" representa un grupo alquil- lineal o ramificado que, como regla general, tiene entre
1 y 9, particularmente entre 1 y 6, preferentemente entre 1 y 4 átomos de carbono. Particularmente, el grupo alquilo tiene 1, 2, 3, 4, 5 o 6 átomos de carbono ("alquil C1-C6-"), por ejemplo, metil-, etil-, n-propil-, isopropil-, n-butil-, fercbutil-, pentil-, isopentil-, hexil-, 2-metilbutil-, 1 -metilbutil-, 1 -etilpropil-, 1,2-dimetilpropil-, neo-pentil-, 1,1 -dimetilpropil-,
4-metilpentil-, 3-metilpentil-, 2-metilpentil-, 1 -metilpentil-, 2-etilbutil-, 1 -etilbutil-, 3,3-dimetilbutil-, 2,2-dimetilbutil-, 1,1-dimetilbutil-, 2,3-dimetilbutil-, 1,3-dimetilbutil-, o 1,2-dimetilbutilo. Preferentemente, el grupo alquilo tiene 1, 2 o 3 átomos de carbono ("alquil C1-C3-"), metil-, etil-, n-propil- o isopropil-.
La expresión "alquileno C2-C8" se ha de entender, con preferencia, como un grupo hidrocarburo linear, bivalente y saturado que tiene 2 a 6, en particular 2, 3, 4 o 5 átomos de carbono, como en "alquileno C2-C5", más en particular, 2,
3 o 4 átomos de carbono, como en "alquileno C2-C4", por ejemplo, etileno, n-propileno, n-butileno, n-pentileno o n-hexileno, con preferencia, n-propileno o n-butileno.
La expresión "alquenil C2-C6-" preferentemente ha de ser interpretada como una referencia a un grupo hidrocarburo monovalente, lineal o ramificado, que contiene un enlace doble y que comprende 2, 3, 4, 5 o 6 átomos de carbono ("alquenil C2-C6-"). En particular, dicho grupo alquenil- es un grupo alquenil C2-C3-, alquenil C3-C6- o alquenil C3-C4-.
Dicho grupo alquenil- es, por ejemplo, un grupo vinil-, alil-, (E)-2-metilvinil-, (Z)-2-metilvinil- o isopropenil-.
La expresión "alquinil C2-C6-" preferentemente ha de ser interpretada como una referencia a un grupo hidrocarburo monovalente, lineal o ramificado, que contiene un enlace triple y que comprende 2, 3, 4, 5 o 6 átomos de carbono. En particular, dicho grupo alquinil- es un grupo alquinil C2-C3-, alquinil C3-C6- o alquinil C3-C4-. Dicho un grupo alquinil C2
C3- es, por ejemplo, un grupo etinil-, prop-1-inil- o prop-2-inil-.
La expresión "cicloalquil C3-C7-" preferentemente ha de ser interpretada como una referencia a un anillo hidrocarburo monocíclico y monovalente, saturado o parcialmente insaturado, que comprende 3, 4, 5, 6 o 7 átomos de carbono. Dicho grupo cicloalquil C3-C7- es, por ejemplo, un anillo hidrocarburo monocíclico, tal como un grupo ciclopropil-, ciclobutil-, ciclopentil-, ciclohexil- o cicloheptil-. Los anillos de cicloalquilo como los que se han descrito no son aromáticos, pero opcionalmente pueden comprender uno o más enlaces dobles, en cuyo caso se los conoce como grupos cicloalquenil-, tales como los grupos ciclopropenil-, ciclobutenil-, ciclopentenil-, ciclohexenil- o cicloheptenil-, donde el enlace entre el anillo y el resto de la molécula puede estar basado en cualquiera de los átomos de carbono que se encuentran en el anillo, independientemente de que se encuentren saturados o no. En particular, dicho grupo cicloalquil- es un grupo cicloalquil C4-C6-, cicloalquil C5-C6- o ciclohexil-.
La expresión "cicloalquil C3-C5-" preferentemente ha de ser interpretada como una referencia a un anillo hidrocarburo monocíclico, monovalente y saturado que comprende 3, 4 o 5 átomos de carbono. En particular, dicho grupo cicloalquil C3-C5- es un anillo hidrocarburo monocíclico, tal como un grupo ciclopropil-, ciclobutil- o ciclopentil-. Preferentemente, dicho grupo cicloalquil C3-C5- es un grupo ciclopropil-.
La expresión "cicloalquil C3-C4-" preferentemente ha de ser interpretada como una referencia a un anillo hidrocarburo monocíclico, monovalente y saturado que comprende 3 o 4 átomos de carbono. En particular, dicho grupo cicloalquil C3-C4- es un anillo hidrocarburo monocíclico, tal como un grupo ciclopropil- o ciclobutil-.
El término "heterociclil-" significa preferentemente un anillo hidrocarburo saturado o parcialmente insaturado monovalente monocíclico o bicíclico que contiene 3, 4, 5, 6, 7, 8 o 9 átomos de carbono y que además contiene 1,2 o 3 grupos que contienen heteroátomos seleccionados entre oxígeno, azufre, nitrógeno. Particularmente, el término "heterociclil-" significa preferentemente un "anillo heterocíclico de entre 4 y 10 miembros".
La expresión "anillo heterocíclico de entre 4 y 10 miembros" significa preferentemente un anillo hidrocarburo saturado o parcialmente insaturado monovalente monocíclico o bicíclico que contiene 3, 4, 5, 6, 7, 8 o 9 átomos de carbono, y que además contiene 1,2 o 3 grupos que contienen heteroátomos seleccionados entre oxígeno, azufre, nitrógeno. Un heterociclil C3-C9- debe entenderse como que se refiere a un heterociclilo que contiene al menos 3, 4, 5, 6, 7, 8 o 9 átomos de carbono y adicionalmente al menos un heteroátomo como átomos del anillo. En consecuencia, en caso de un heteroátomo el anillo es de entre 4 y 10 miembros, en caso de dos heteroátomos el anillo es de entre 5 y 11 miembros y en caso de tres heteroátomos el anillo es de entre 6 y 12 miembros.
Dicho anillo heterocíclico es, por ejemplo, un anillo heterocíclico monocíclico como, por ejemplo, un grupo oxetanil-, azetidinil-, tetrahidrofuranil-, pirrolidinil-, 1,3-dioxolanil-, imidazolidinil-, pirazolidinil-, oxazolidinil-, isoxazolidinil-, 1,4-dioxanil-, pirrolinil-, tetrahidropiranil-, piperidinil-, morfolinil-, 1,3-ditianil-, tiomorfolinil-, piperazinil- o quinuclidinil-. Opcionalmente, dicho anillo heterocíclico puede contener uno o más enlaces dobles, por ejemplo, un grupo 4H-piranil-, 2H-piranil-, 2,5-dihidro-1H-pirrolil-, 1,3-dioxolil-, 4H-1,3,4-tiadiazinil-, 2,5-dihidrofuranil-, 2,3-dihidrofuranil-, 2,5-dihidrotienil-, 2,3-dihidrotienil-, 4,5-dihidrooxazolil-, 4,5-dihidroisoxazolil- o 4H-1,4-tiazinil-, o puede estar benzocondensado.
Particularmente un heterociclil C3-C7- debe entenderse como que se refiere a un heterociclilo que contiene al menos 3, 4, 5, 6 o 7 átomos de carbono y adicionalmente al menos un heteroátomo como átomos del anillo. En consecuencia, en caso de un heteroátomo el anillo es de entre 4 y 8 miembros, en caso de dos heteroátomos el anillo es de entre 5 y 9 miembros y en caso de tres heteroátomos el anillo es de entre 6 y 10 miembros.
Particularmente, un heterociclil C3-C6- debe entenderse como que se refiere a un heterociclilo que contiene al menos 3, 4, 5 o 6 átomos de carbono y adicionalmente al menos un heteroátomo como átomos del anillo. En consecuencia, en caso de un heteroátomo el anillo es de entre 4 y 7 miembros, en caso de dos heteroátomos el anillo es de entre 5 y 8 miembros y en caso de tres heteroátomos el anillo es de entre 6 y 9 miembros.
Particularmente, el término "heterociclil-" debe comprenderse como un anillo heterocíclico que contiene 3, 4 o 5 átomos de carbono, y 1, 2 o 3 de los grupos que contienen heteroátomos que se han mencionado (un "anillo heterocíclico de entre 4 y 8 miembros"), más particularmente dicho anillo puede contener 4 o 5 átomos de carbono, y 1, 2 o 3 de los grupos que contienen heteroátomos que se han mencionado (un "anillo heterocíclico de entre 5 y 8 miembros"), más particularmente dicho heterocíclico anillo es un "anillo heterocíclico de 6 miembros", que debe comprenderse que contiene 4 átomos de carbono y 2 de los grupos que contienen heteroátomos que se han mencionado o 5 átomos de carbono y uno de los grupos que contienen heteroátomos que se han mencionado, preferentemente 4 átomos de carbono y 2 de los grupos que contienen heteroátomos que se han mencionado.
La expresión "alcoxi C1-C6-" significa preferentemente un grupo hidrocarburo lineal o ramificado saturado monovalente de la fórmula -O-alquil-, en el cual el término "alquil-" es como se ha definido anteriormente, por ejemplo, un grupo metoxi-, etoxi, n-propoxi, iso-propoxi, n-butoxi, iso-butoxi, terc-butoxi, sec-butoxi, pentiloxi, iso-pentiloxi, n-hexiloxi o un isómero de los mismos. Particularmente, "alcoxi C1-C6-" es un grupo "alcoxi C1-C4-", un "alcoxi C1-C3-", metoxi-, etoxi, o propoxi, preferentemente un grupo metoxi-, etoxi o propoxi. Más preferible es un grupo "alcoxi C1-C2-", particularmente un grupo metoxi o etoxi.
La expresión "fluoroalcoxi C1-C3-" significa preferentemente un grupo alcoxi C1-C3- lineal o ramificado saturado monovalente, como se ha definido anteriormente, en la cual uno o más de los átomos de hidrógeno se reemplaza, en forma idéntica o diferente, por uno o más átomos de flúor. Dicho grupo fluoroalcoxi C1-C3- es, por ejemplo, un grupo 1,1-difluorometoxi-, 1,1,1-trifluorometoxi-, 2-fluoroetoxi-, 3-fluoropropoxi-, 2,2,2-trifluoroetoxi-o 3,3,3-trifluoropropoxi-, particularmente un grupo "fluoroalcoxi C1-C2-".
El término "alquilamino-" significa preferentemente un grupo alquilamino con un grupo alquilo lineal o ramificado como se ha definido anteriormente. Alquilamino (C1-C3)-, por ejemplo, significa un grupo monoalquilamino con 1, 2 o 3 átomos de carbono, alquilamino (C1-C6)- con 1,2, 3, 4, 5 o 6 átomos de carbono. El término "alquilamino-" comprende, por ejemplo, metilamino-, etilamino-, n-propilamino-, isopropilamino-, ferc-butilamino-, n-pentilamino- o n-hexilamino-.
El término "dialquilamino-" significa preferentemente un grupo alquilamino con dos grupos alquilo lineales o ramificados como se ha definido anteriormente, que son independientes entre sí. Dialquilamino (C1-C3)-, por ejemplo, representa un grupo dialquilamino con dos grupos alquilo donde cada uno tiene entre 1 y 3 átomos de carbono por grupo alquilo. El término "dialquilamino-" comprende por ejemplo: N,N-dimetilamino-, N,N-dietilamino-, N-etil-N-metilamino-, N-metil-N-n-propilamino-, N-isopropil-N-n-propilamino-, N-ferc-butil-N-metilamino-, N-etil-N-n-pentilamino- y N-n-hexil-N-metilamino-.
La expresión "halo-alquil C1-C3-", que es sinónimo de "haloalquil C1-C3-", preferentemente ha de ser interpretada como una referencia a un grupo hidrocarburo saturado y monovalente, lineal o ramificado, en el cual el término "alquil C1-C3-" es como se ha definido anteriormente y en la cual uno o más átomos de hidrógeno han sido sustituidos por átomos halógenos idénticos o diferentes, lo que implica que las sustituciones por átomos halógenos se llevan a cabo de manera independiente. Con preferencia, un grupo halo-alquil C1-C3- es un grupo fluoro-alquil C1-C3- o un grupo fluoroalquil C1-C2-, tal como los grupos -CF3, -CHF2, -CH2F, -CF2CF3 o -CH2CF3, y más preferentemente es un grupo -CF3.
La expresión "hidroxi-alquil C1-C3-" preferentemente ha de ser interpretada como una referencia a un grupo hidrocarburo saturado y monovalente, lineal o ramificado, donde la expresión "alquil C1-C3-" es como se ha definido anteriormente y en el cual uno o más átomos de hidrógeno han sido sustituidos por grupos hidroxilo, donde preferentemente no se ha sustituido más de un átomo de hidrógeno en cada átomo de carbono por un grupo hidroxilo. En particular, un grupo hidroxi-alquil C1-C3- es, por ejemplo, un grupo -CH2OH, CH2-CH2OH, -C(H)OH-CH2OH o -CH2-CH2-CH2OH.
La expresión "fenil-alquil C1-C3-" preferentemente ha de ser interpretada como una referencia a un grupo fenilo en el cual se ha sustituido uno de los átomos de hidrógeno por un grupo alquil C1-C3-, que es como se ha definido anteriormente, cuyo propósito es unir el grupo fenil-alquil C1-C3- al resto de la molécula. En particular, un grupo "fenilalquil C1-C3-" es un grupo fenil-alquil C1-C2-, y preferentemente es un grupo bencil-.
El término "heteroaril-" significa preferentemente un sistema de anillos monovalente con 5, 6, 7, 8, 9, 10, 11, 12, 13 o 14 átomos en el anillo (un grupo "heteroaril- de entre 5 y 14 miembros"), particularmente 5 (un "heteroaril- de 5 miembros") o 6 (un "heteroaril- de 6 miembros") o 9 (un "heteroaril- de 9 miembros") o 10 átomos en el anillo (un "heteroaril- de 10 miembros"), y que contiene al menos un heteroátomo que puede ser idéntico o diferente a los demás, donde dicho heteroátomo es por ejemplo, oxígeno, nitrógeno o azufre, y puede ser monocíclico, bicíclico o tricíclico, y además en cada caso puede ser benzocondensado. Particularmente, heteroaril- se selecciona entre tienil-, furanil-, pirrolil-, oxazolil-, tiazolil-, imidazolil-, pirazolil-, isoxazolil-, isotiazolil-, oxadiazolil-, triazolil-, tiadiazolil-, tetrazolilo etc., y benzoderivados de los mismos, como, por ejemplo, benzofuranil-, benzotienil-, benzoxazolil-, bencisoxazolil-, bencimidazolil-, benzotriazolil-, indazolil-, indolil-, isoindolil-, etc.; o piridil-, piridazinil-, pirimidinil-, pirazinil-, triazinil-, etc., y benzoderivados de los mismos, como, por ejemplo, quinolinil-, quinazolinil-, isoquinolinil-, etc.; o azocinil-, indolizinil-, purinil-, etc., y benzoderivados de los mismos; o cinolinil-, ftalazinil-, quinazolinil-, quinoxalinil-, naftiridinil-, pteridinil-, carbazolil-, acridinil-, fenazinil-, fenotiazinil-, fenoxazinil-, xantenilo u oxepinil-, etc. Preferentemente, heteroaril- se selecciona entre heteroaril- monocíclico, heteroaril- de 5 miembros o heteroaril- de 6 miembros.
La expresión "heteroaril- de 5 miembros" significa preferentemente un sistema de anillos aromático monovalente con 5 átomos en el anillo y que contiene al menos un heteroátomo que puede ser idéntico o diferente a los demás, donde dicho heteroátomo es, por ejemplo, oxígeno, nitrógeno o azufre. Particularmente, "heteroaril- de 5 miembros" se selecciona entre tienil-, furanil-, pirrolil-, oxazolil-, tiazolil-, imidazolil-, pirazolil-, isoxazolil-, isotiazolil-, oxadiazolil-, triazolil-, tiadiazolil-, tetrazolil-.
La expresión "heteroaril- de 6 miembros" significa preferentemente un sistema de anillos aromático monovalente con 6 átomos en el anillo y que contiene al menos un heteroátomo que puede ser idéntico o diferente a los demás, donde dicho heteroátomo es, por ejemplo, oxígeno, nitrógeno o azufre. Particularmente, "heteroaril- de 6 miembros" se selecciona entre piridil-, piridazinil-, pirimidinil-, pirazinil-, triazinil-.
La expresión "heteroaril-alquil C1-C3-" significa preferentemente un grupo heteroarilo, heteroaril- de 5 miembros o heteroarilo de 6 miembros, cada uno como se ha definido anteriormente, en el cual uno de los átomos de hidrógeno se reemplaza por un grupo alquil C1-C3-, como se ha definido, que une el grupo heteroaril-alquil C1-C3- al resto de la molécula. Particularmente, "heteroaril-alquil C1-C3-" es un grupo heteroaril-alquil C1-C2-, piridinil-alquil C1-C3-, piridinilmetil-, piridiniletil-, piridinilpropil-, pirimidinil-alquil C1-C3-, pirimidinilmetil-, pirimidiniletil- o pirimidinilpropil-,
preferentemente un grupo piridinilmetil- o piridiniletil- o pirimidiniletil- o pirimidinilpropil-.
Como se usa en el presente documento, la expresión "grupo saliente" se refiere a un átomo o un grupo de átomos que es desplazado en una reacción química como especies estables llevando consigo los electrones del enlace.
Preferentemente, un grupo saliente se selecciona del grupo que comprende: halo, en particular cloro, bromo o yodo, metansulfoniloxi-, p-toluensulfoniloxi-, trifluorometansulfoniloxi-, nonafluorobutansulfoniloxi-, (4-bromobencen)sulfoniloxi-, (4-nitro-bencen)sulfoniloxi-, (2-nitro-benceno)-sulfoniloxi-, (4-isopropil-bencen)sulfoniloxi-, (2,4,6-tri-isopropil-benceno)-sulfoniloxi-, (2,4,6-trimetil-bencen)sulfoniloxi-, (4-ferc-butil-bencen)sulfoniloxi-, bencensulfoniloxi- y (4-metoxi--bencen)sulfoniloxi-.
Como se usa en el presente documento, la expresión "alquilbenceno C1-C3" hace referencia a un hidrocarburo parcialmente aromático que consiste en un anillo de benceno que está sustituido con uno o dos grupos alquil C1-C3-, como se han definido anteriormente. En particular, "alquilbenceno C1-C3" es tolueno, etilbenceno, cumeno, npropilbenceno, orto-xileno, meta-xileno o para-xileno. Preferentemente "alquilbenceno C1-C3" es tolueno.
Como se usa en el presente documento, la expresión "disolvente a base de una carboxamida" hace referencia a una carboxamida alifática inferior que responde a la fórmula alquil C1-C2-C(=O)-N(alquil C1-C2-)2 o a una carboxamida inferior alifática y cíclica que responde a la fórmula

en la cual G representa -CH2-, -CH2-CH2- o -CH2-CH2-CH2-. En particular, un "disolvente a base de una carboxamida" es la N,N-dimetilformamida, la N,N-dimetilacetamida o la N-metilpirrolidin-2-ona. Con preferencia, un "disolvente a base de una carboxamida" es la W,W-dimetilformamida o W-metil-pirrolidin-2-ona.
La expresión "C1-C10", como se usa en este texto, por ejemplo, en el contexto de la definición de "alquil C1-C1-0" significa preferentemente un grupo alquilo con una cantidad finita de átomos de carbono de entre 1 y 10, por ejemplo,
1, 2, 3, 4, 5, 6, 7, 8, 9 o 10 átomos de carbono. Además, debe comprenderse que dicha expresión "C1-C10" debe interpretarse como cualquier subintervalo comprendido en el mismo, por ejemplo, C1-C10, C1-C9, C1-C8 , C1-C7, C1-C6
C1-C5 , C1-C4, C1-C3, C1-C2, C2-C10, C2-C9, C2-C8 , C2-C7, C2-C6 , C2-C5 , C2-C4, C2-C3, C3-C10, C3-C9 , C3-C C6, C3-C5 , C3-C4 , C4-C10, C4-C9, C4-C8, C4-C7, C4-C6 , C4-C5 , C5-C10, C5-C9, C5-C8, C5-C7, C5-C6, C6-C10, C6-C9, C6-C8,
C6-C7 , C7-C10, C7-C9, C7-C8, C8-C10, C8-C9 , C9-C10.
De manera similar, como se usa en el presente documento, la expresión "C1-C6", por ejemplo, en el contexto de la definición de "alquil C1-C6-", "alcoxi C1-C6" significa preferentemente un grupo alquilo con una cantidad finita de átomos de carbono de entre 1 y 6, por ejemplo, 1, 2, 3, 4, 5 o 6 átomos de carbono. Además, debe comprenderse que dicho término "C1-C6" debe interpretarse como cualquier subintervalo comprendido en el mismo, por ejemplo, C1-C6 C1-C5 ,
C1-C4 , C1-C3, C1-C2, C2-C6 , C2-C5, C2-C4, C2-C3 , C3-C6 , C3-C5, C3-C4 , C4-C6, C4-C5, C5-C6.
De manera similar, como se usa en el presente documento, la expresión "C1-C4", por ejemplo, en el contexto de la definición de un "alquil C1-C4-" o de un "alcoxilo C1-C4", ha de ser interpretada como una referencia a un grupo alquilo o alcoxilo que comprende una cantidad finita de átomos de carbono que varía entre 1 y 4, es decir, 1, 2, 3 o 4 átomos de carbono. Más aun, ha de comprenderse que la expresión "C1-C4" puede ser una referencia a cualquiera de los intervalos intermedios, por ejemplo, C1-C4, C1-C3, C1-C2 , C2-C4 , C2-C3 o C3-C4.
De manera similar, como se usa en el presente documento, la expresión "C1-C3", por ejemplo, en el contexto de la definición de "alquil C1-C3-", "alcoxi C1-C3- " o "fluoroalcoxi C1-C3-" significa preferentemente un grupo alquilo con una cantidad finita de átomos de carbono de entre 1 y 3, por ejemplo, 1, 2 o 3 átomos de carbono. Además, debe comprenderse que dicha expresión "C1-C3" debe interpretarse como cualquier subintervalo comprendido en el mismo, por ejemplo, C1-C3 , C1-C2 , C2-C3.
Además, como se usa en el presente documento, la expresión "C3-C6", como se usa a lo largo de este texto, por ejemplo, en el contexto de la definición de "cicloalquil C3-C6-", debe entenderse como que se refiere a un grupo cicloalquilo que tiene un número finito de átomos de carbono de entre 3 y 6, es decir 3, 4, 5 o 6 átomos de carbono.
Se debe entender además que dicha expresión "C3-C6" se debe interpretar como cualquier subintervalo comprendido dentro del mismo, por ejemplo, C3-C6 , C3-C5 , C3-C4, C4-C6 , C4-C5 , C5-C6. Además, la expresión "C3-C7", como se usa a lo largo de este texto, por ejemplo, en el contexto de la definición de "cicloalquil C3-C7-", significa preferentemente un grupo cicloalquilo con una cantidad finita de átomos de carbono de entre 3 y 7, por ejemplo, 3, 4, 5, 6 o 7 átomos de carbono, particularmente 3, 4, 5 o 6 átomos de carbono. Además, debe comprenderse que dicha expresión "C3-C7" debe interpretarse como cualquier subintervalo comprendido en el mismo, por ejemplo, C3-C7 , C3-C6 , C3-C5 , C3-C4,
C4-C7, C4-C6, C4-C5, C5-C7, C5-C6, C6-C7.
Un símbolo en una unión denota el punto de unión en la molécula.
en una unión denota el punto de unión en la molécula.
Como se usa en el presente documento, la expresión "una o más veces", por ejemplo, en la definición de los sustituyentes de los compuestos de las fórmulas generales de la presente invención, significa una, dos, tres, cuatro o cinco veces, particularmente una, dos, tres o cuatro veces, más particularmente una, dos o tres veces, aún más particularmente una o dos veces.
Cuando en el presente documento se usa la forma plural de los compuestos, sales, hidratos, solvatos y similares, esto también incluye un único compuesto, sal, isómero, hidrato, solvato o similar.
La presente invención se refiere a compuestos de fórmula general (I), en la que
L representa un grupo alquileno C2-C5,
en el que dicho grupo está opcionalmente sustituido con
(i) un sustituyente seleccionado de cicloalquil C3-C4- e hidroximetil-, y/o
(ii) uno o dos sustituyentes adicionales, iguales o diferentes, seleccionados de alquil C1-C2-,
X, Y representan CH o N con la condición de que uno de X e Y represente CH y uno de X e Y represente N; R1 representa un grupo seleccionado de alquil C1-C4- y cicloalquil C3-C5-,
en el que dicho grupo está opcionalmente sustituido con uno o dos o tres sustituyentes, iguales o diferentes, seleccionados del grupo que consiste en hidroxi, ciano, halógeno, alquil C1-C2-, alcoxi C1-C2-, -NH2 , -C(=O)OH;
R2 representa un grupo seleccionado de un átomo de hidrógeno, un átomo de flúor, un átomo de cloro, ciano, metil-, metoxi-, trifluorometilo;
R3 representa un grupo seleccionado de un átomo de hidrógeno, un átomo de flúor, un átomo de cloro, ciano, metil-, metoxi-, trifluorometil-, trifluorometoxi-;
R4 representa un átomo de hidrógeno o un átomo de flúor;
R5 representa un grupo seleccionado de un átomo de hidrógeno, ciano, -C(=O)R8, -C(=O)OR8, -S(=O)2R8, -C(=O)NR6R7, alquil C1-C4-, cicloalquil C3-C5-,
en el que dicho grupo alquil C1-C4- o cicloalquil C3-C5- está opcionalmente sustituido con un sustituyente seleccionado del grupo que consiste en flúor, hidroxi, ciano, alcoxi C1-C3, -NH2 , alquilamino-, dialquilamino; R6, R7 representan, de modo independiente entre sí, un grupo seleccionado de un átomo de hidrógeno, alquil C1-C4- y cicloalquil C3-C5-,
en el que dicho grupo alquil C1-C4- o cicloalquil C3-C5- está opcionalmente sustituido con uno o dos sustituyentes, iguales o diferentes, seleccionados del grupo que consiste en hidroxi, alquil C1-C2-, alcoxi C1-C2-, -NH2, alquilamino-, dialquilamino;
R8 representa un grupo seleccionado de alquil C1-C6-, fluoro-alquil C1-C3-, cicloalquil C3-C5- y fenil-,
en el que dicho grupo está opcionalmente sustituido con un sustituyente seleccionado del grupo que consiste en halógeno, hidroxi, alquil C1-C2-, alcoxi C1-C2-, -NH2,
o sus enantiómeros, diastereómeros, sales, solvatos o sales de solvatos.
En otra forma de realización preferida, la presente invención se refiere a compuestos de fórmula general (I), en la que L representa un grupo alquileno C2-C5,
en el que dicho grupo está opcionalmente sustituido con
(i) un sustituyente seleccionado de cicloalquil C3-C4- e hidroximetil-, y/o
(ii) uno o dos sustituyentes adicionales, iguales o diferentes, seleccionados de alquil C1-C2-,
X, Y representan CH o N con la condición de que uno de X e Y represente CH y uno de X e Y represente N; R1 representa un grupo seleccionado de alquil C1-C4- y cicloalquil C3-C5-,
en el que dicho grupo está opcionalmente sustituido con uno o dos o tres sustituyentes, iguales o diferentes, seleccionados del grupo que consiste en hidroxi, ciano, halógeno, alquil C1-C2-, alcoxi C1-C2-, -NH2 , -C(=O)OH;
R2 representa un grupo seleccionado de un átomo de hidrógeno, un átomo de flúor, un átomo de cloro, ciano, metil-, metoxi-, trifluorometil-;
R3 representa un grupo seleccionado de un átomo de hidrógeno, un átomo de flúor, un átomo de cloro, ciano,
metil-, metoxi-, trifluorometil-, trifluorometoxi-;
R4 representa un átomo de hidrógeno o un átomo de flúor;
R5 representa un grupo seleccionado de un átomo de hidrógeno, ciano, -C(=O)R8, -C(=O)OR8, -S(=O)2R8, -C(=O)NR6R7, alquil C1-C4-,
en el que dicho grupo alquil C1-C4- está opcionalmente sustituido con un sustituyente seleccionado del grupo que consiste en flúor, hidroxi, ciano, alcoxi C1-C3-, -NH2 , alquilamino-, dialquilamino;
R6, R7 representan, de modo independiente entre sí, un grupo seleccionado de un átomo de hidrógeno, alquil C1-C4- y cicloalquil C3-C5-,
en el que dicho grupo alquil C1-C4- o cicloalquil C3-C5- está opcionalmente sustituido con uno o dos sustituyentes, iguales o diferentes, seleccionados del grupo que consiste en hidroxi, alquil C1-C2-, alcoxi C1-C2-, -NH2, alquilamino-, dialquilamino-;
R8 representa un grupo seleccionado de alquil C1-C6-, fluoro-alquil C1-C3-, cicloalquil C3-C5- y fenil-,
en el que dicho grupo está opcionalmente sustituido con un sustituyente seleccionado del grupo que consiste en halógeno, hidroxi, alquil C1-C2-, alcoxi C1-C2-, -NH2,
o sus enantiómeros, diastereómeros, sales, solvatos o sales de solvatos.
En otra forma de realización preferida, la presente invención se refiere a compuestos de fórmula general (I), en la que L representa un grupo alquileno C2-C4;
X, Y representan CH o N con la condición de que uno de X e Y represente CH y uno de X e Y represente N; R1 representa un grupo alquil C1-C4-,
en el que dicho grupo está opcionalmente sustituido con uno o dos sustituyentes, iguales o diferentes, seleccionados del grupo que consiste en hidroxi, alcoxi C1-C2-, -NH2, -C(=O)OH;
R2 representa un átomo de hidrógeno o un grupo ciano;
R3 representa un grupo seleccionado de un átomo de hidrógeno, un átomo de flúor y un grupo metoxi-;
R4 representa un grupo seleccionado de un átomo de hidrógeno y un átomo de flúor;
R5 representa un grupo seleccionado de un átomo de hidrógeno, ciano, alquil C1-C4-, cicloalquil C3-C5-, en el que dicho grupo alquil C1-C4- está opcionalmente sustituido con un grupo hidroxi;
o sus enantiómeros, diastereómeros, sales, solvatos o sales de solvatos.
En otra forma de realización preferida, la presente invención se refiere a compuestos de fórmula general (I), en la que L representa un grupo alquileno C2-C4;
X, Y representan CH o N con la condición de que uno de X e Y represente CH y uno de X e Y represente N; R1 representa un grupo alquil C1-C4-,
en el que dicho grupo está opcionalmente sustituido con uno o dos sustituyentes, iguales o diferentes, seleccionados del grupo que consiste en hidroxi, alcoxi C1-C2-, -NH2, -C(=O)OH;
R2 representa un átomo de hidrógeno;
R3 representa un grupo seleccionado de un átomo de hidrógeno, un átomo de flúor y un grupo metoxi-;
R4 representa un átomo de hidrógeno;
R5 representa un grupo seleccionado de un átomo de hidrógeno, ciano, alquil C1-C4-,
en el que dicho grupo alquil C1-C4- está opcionalmente sustituido con un grupo hidroxi;
o sus enantiómeros, diastereómeros, sales, solvatos o sales de solvatos.
En otra forma de realización preferida, la presente invención se refiere a compuestos de fórmula general (I), en la que L representa un grupo alquileno C2-C4;
X representa N;
Y representa CH;
R1 representa un grupo seleccionado de alquil C1-C4-,
en el que dicho grupo está opcionalmente sustituido con uno o dos sustituyentes, iguales o diferentes, seleccionados del grupo que consiste en hidroxi, alcoxi C1-C2, -NH2 , -C(=O)OH;
R2 representa un átomo de hidrógeno o un grupo ciano;
R3 representa un grupo seleccionado de un átomo de hidrógeno, un átomo de flúor y un grupo metoxi-;
R4 representa un átomo de hidrógeno;
R5 representa un grupo seleccionado de un átomo de hidrógeno, ciano, alquil C1-C4-,
en el que dicho grupo alquil C1-C4- está opcionalmente sustituido con un grupo hidroxi;
o sus enantiómeros, diastereómeros, sales, solvatos o sales de solvatos.
En otra forma de realización preferida, la presente invención se refiere a compuestos de fórmula general (I), en la que L representa un grupo alquileno C2-C4;
X representa N;
Y representa CH;
R1 representa un grupo seleccionado de alquil C1-C4-,
en el que dicho grupo está opcionalmente sustituido con uno o dos sustituyentes, iguales o diferentes, seleccionados del grupo que consiste en hidroxi, alcoxi C1-C2, -NH2 , -C(=O)OH;
R2 representa un átomo de hidrógeno;
R3 representa un grupo seleccionado de un átomo de hidrógeno, un átomo de flúor y un grupo metoxi-;
R4 representa un átomo de hidrógeno;
R5 representa un grupo seleccionado de un átomo de hidrógeno, ciano, alquil C1-C4-,
en el que dicho grupo alquil C1-C4- está opcionalmente sustituido con un grupo hidroxi;
o sus enantiómeros, diastereómeros, sales, solvatos o sales de solvatos.
En otra forma de realización preferida, la presente invención se refiere a compuestos de fórmula general (I), en la que L representa un grupo alquileno C2-C4;
X representa CH;
Y representa N;
R1 representa un grupo seleccionado de alquil C1-C4-,
en el que dicho grupo está opcionalmente sustituido con uno o dos sustituyentes, iguales o diferentes, seleccionados del grupo que consiste en hidroxi, alcoxi C1-C2, -NH2 , -C(=O)OH;
R2 representa un átomo de hidrógeno;
R3 representa un grupo seleccionado de un átomo de hidrógeno, un átomo de flúor y un grupo metoxi-;
R4 representa un grupo seleccionado de un átomo de hidrógeno y un átomo de flúor;
R5 representa un grupo seleccionado de un átomo de hidrógeno, ciano, alquil C1-C4-, cicloalquil C3-C5-, en el que dicho grupo alquil C1-C4- está opcionalmente sustituido con un grupo hidroxi;
o sus enantiómeros, diastereómeros, sales, solvatos o sales de solvatos.
En otra forma de realización preferida, la presente invención se refiere a compuestos de fórmula general (I), en la que L representa un grupo alquileno C2-C4;
X representa CH;
Y representa N;
R1 representa un grupo seleccionado de alquil C1-C4-,
en el que dicho grupo está opcionalmente sustituido con uno o dos sustituyentes, iguales o diferentes,
seleccionados del grupo que consiste en hidroxi, alcoxi C1-C2, -NH2 , -C(=O)OH;
R2 representa un átomo de hidrógeno;
R3 representa un grupo seleccionado de un átomo de hidrógeno, un átomo de flúor y un grupo metoxi-;
R4 representa un átomo de hidrógeno;
R5 representa un grupo seleccionado de un átomo de hidrógeno, ciano, alquil C1-C4-,
en el que dicho grupo alquil C1-C4- está opcionalmente sustituido con un grupo hidroxi;
o sus enantiómeros, diastereómeros, sales, solvatos o sales de solvatos.
En una forma de realización de particular preferencia, la presente invención se refiere a compuestos de fórmula general (I), en la que
L representa un grupo alquileno C3-C4;
X, Y representan CH o N con la condición de que uno de X e Y represente CH y uno de X e Y represente N; R1 representa un grupo metil-;
R2 representa un átomo de hidrógeno o un grupo ciano;
R3 representa un grupo seleccionado de un átomo de hidrógeno, un átomo de flúor;
R4 representa un grupo seleccionado de un átomo de hidrógeno, un átomo de flúor;
R5 representa un grupo seleccionado de un átomo de hidrógeno, ciano, alquil C1-C4-, ciclopropil-,
en el que dicho grupo alquil C1-C4- está opcionalmente sustituido con un grupo hidroxi;
o sus enantiómeros, diastereómeros, sales, solvatos o sales de solvatos.
En otra forma de realización de particular preferencia, la presente invención se refiere a compuestos de fórmula general (I), en la que
L representa un grupo alquileno C3-C4;
X, Y representan CH o N con la condición de que uno de X e Y represente CH y uno de X e Y represente N; R1 representa un grupo metil-;
R2 representa un átomo de hidrógeno;
R3 representa un grupo seleccionado de un átomo de hidrógeno, un átomo de flúor;
R4 representa un átomo de hidrógeno;
R5 representa un grupo seleccionado de un átomo de hidrógeno, ciano, alquil C1-C4-,
en el que dicho grupo alquil C1-C4- está opcionalmente sustituido con un grupo hidroxi;
o sus enantiómeros, diastereómeros, sales, solvatos o sales de solvatos.
En otra forma de realización de particular preferencia, la presente invención se refiere a compuestos de fórmula general (I), en la que
L representa un grupo alquileno C3-C4;
X representa N;
Y representa CH;
R1 representa un grupo metil-;
R2 representa un átomo de hidrógeno o un grupo ciano;
R3 representa un grupo seleccionado de un átomo de hidrógeno, un átomo de flúor;
R4 representa un átomo de hidrógeno;
R5 representa un grupo seleccionado de un átomo de hidrógeno, ciano, alquil C1-C4-,
en el que dicho grupo alquil C1-C4- está opcionalmente sustituido con un grupo hidroxi:
o sus enantiómeros, diastereómeros, sales, solvatos o sales de solvatos.
En otra forma de realización de particular preferencia, la presente invención se refiere a compuestos de fórmula general (I), en la que
L representa un grupo alquileno C3-C4;
X representa N;
Y representa CH;
R1 representa un grupo metil-;
R2 representa un átomo de hidrógeno;
R3 representa un grupo seleccionado de un átomo de hidrógeno, un átomo de flúor;
R4 representa un átomo de hidrógeno;
R5 representa un grupo seleccionado de un átomo de hidrógeno, ciano, alquil C1-C4-,
en el que dicho grupo alquil C1-C4- está opcionalmente sustituido con un grupo hidroxi:
o sus enantiómeros, diastereómeros, sales, solvatos o sales de solvatos.
En otra forma de realización de particular preferencia, la presente invención se refiere a compuestos de fórmula general (I), en la que
L representa un grupo alquileno C3-C4;
X representa CH;
Y representa N;
R1 representa un grupo metil-;
R2 representa un átomo de hidrógeno;
R3 representa un átomo de hidrógeno o un átomo de flúor;
R4 representa un grupo seleccionado de un átomo de hidrógeno, un átomo de flúor;
R5 representa un grupo seleccionado de un átomo de hidrógeno, ciano, alquil C1-C4-, ciclopropilo,
en la que dicho grupo alquil C1-C4- está opcionalmente sustituido con un grupo hidroxi;
o sus enantiómeros, diastereómeros, sales, solvatos o sales de solvatos.
En otra forma de realización de particular preferencia, la presente invención se refiere a compuestos de fórmula general (I), en la que
L representa un grupo alquileno C3-C4;
X representa CH;
Y representa N;
R1 representa un grupo metil-;
R2 representa un átomo de hidrógeno;
R3 representa un grupo seleccionado de un átomo de hidrógeno, un átomo de flúor;
R4 representa un átomo de hidrógeno;
R5 representa un grupo seleccionado de un átomo de hidrógeno, ciano, alquil C1-C4-,
en el que dicho grupo alquil C1-C4- está opcionalmente sustituido con un grupo hidroxi:
o sus enantiómeros, diastereómeros, sales, solvatos o sales de solvatos.
En otra forma de realización de particular preferencia, la presente invención se refiere a compuestos de fórmula general (I), en la que
L representa un grupo alquileno C3-C4;
X, Y representan CH o N con la condición de que uno de X e Y represente CH y uno de X e Y represente N; R1 representa un grupo metilo;
R2 representa un átomo de hidrógeno o un grupo ciano;
R3 representa un átomo de flúor;
R4 representa un átomo de hidrógeno o un átomo de flúor;
R5 representa un grupo seleccionado de un átomo de hidrógeno, ciano, alquil C1-C3-, ciclopropilo,
en el que dicho grupo alquil C1-C3- está opcionalmente sustituido con un grupo hidroxi;
o sus enantiómeros, diastereómeros, sales, solvatos o sales de solvatos.
En otra forma de realización de particular preferencia, la presente invención se refiere a compuestos de fórmula general (I), en la que
L representa un grupo alquileno C3-C4;
X, Y representan CH o N con la condición de que uno de X e Y represente CH y uno de X e Y represente N; R1 representa un grupo metil-;
R2 representa un átomo de hidrógeno;
R3 representa un átomo de flúor;
R4 representa un átomo de hidrógeno;
R5 representa un grupo seleccionado de un átomo de hidrógeno, ciano, alquil C1-C3-,
en el que dicho grupo alquil C1-C3- está opcionalmente sustituido con un grupo hidroxi;
o sus enantiómeros, diastereómeros, sales, solvatos o sales de solvatos.
En otra forma de realización de particular preferencia, la presente invención se refiere a compuestos de fórmula general (I), en la que
L representa un grupo alquileno C3-C4;
X representa N;
Y representa CH;
R1 representa un grupo metilo;
R2 representa un átomo de hidrógeno y un grupo ciano;
R3 representa un átomo de flúor;
R4 representa un átomo de hidrógeno;
R5 representa un grupo seleccionado de un átomo de hidrógeno, ciano, alquil C1-C3-,
en el que dicho grupo alquil C1-C3- está opcionalmente sustituido con un grupo hidroxi;
o sus enantiómeros, diastereómeros, sales, solvatos o sales de solvatos.
En otra forma de realización de particular preferencia, la presente invención se refiere a compuestos de fórmula general (I), en la que
L representa un grupo alquileno C3-C4;
X representa N;
Y representa CH;
R1 representa un grupo metil-;
R2 representa un átomo de hidrógeno;
R3 representa un átomo de flúor;
R4 representa un átomo de hidrógeno;
R5 representa un grupo seleccionado de un átomo de hidrógeno, ciano, alquil C1-C3-,
en el que dicho grupo alquil C1-C3- está opcionalmente sustituido con un grupo hidroxi;
o sus enantiómeros, diastereómeros, sales, solvatos o sales de solvatos.
En otra forma de realización de particular preferencia, la presente invención se refiere a compuestos de fórmula general (I), en la que
L representa un grupo alquileno C3-C4;
X representa CH;
Y representa N;
R1 representa un grupo metilo;
R2 representa un átomo de hidrógeno;
R3 representa un átomo de flúor;
R4 representa un átomo de hidrógeno o un átomo de flúor;
R5 representa un grupo seleccionado de un átomo de hidrógeno, ciano, alquil C1-C3-, ciclopropilo,
en el que dicho grupo alquil C1-C3- está opcionalmente sustituido con un grupo hidroxi;
o sus enantiómeros, diastereómeros, sales, solvatos o sales de solvatos.
En otra forma de realización de particular preferencia, la presente invención se refiere a compuestos de fórmula general (I), en la que
L representa un grupo alquileno C3-C4;
X representa CH;
Y representa N;
R1 representa un grupo metilo;
R2 representa un átomo de hidrógeno;
R3 representa un átomo de flúor;
R4 representa un átomo de hidrógeno;
R5 representa un grupo seleccionado de un átomo de hidrógeno, ciano, alquil C1-C3-,
en el que dicho grupo alquil C1-C3- está opcionalmente sustituido con un grupo hidroxi;
o sus enantiómeros, diastereómeros, sales, solvatos o sales de solvatos.
En otra forma de realización de particular preferencia, la presente invención se refiere a compuestos de fórmula general (I), en la que
L representa un grupo -CH2CH2CH2- o un grupo -CH2CH2CH2CH2-;
X, Y representan CH o N con la condición de que uno de X e Y represente CH y uno de X e Y represente N; R1 representa un grupo metilo;
R2 representa un átomo de hidrógeno o un grupo ciano;
R3 representa un átomo de flúor;
R4 representa un átomo de hidrógeno o un átomo de flúor;
R5 representa un grupo seleccionado de un átomo de hidrógeno, ciano, metil-, 3-hidroxipropil- y ciclopropil-; o sus enantiómeros, diastereómeros, sales, solvatos o sales de solvatos.
L representa un grupo -CH2CH2CH2- o un grupo -CH2CH2CH2CH2-;
X, Y representan CH o N con la condición de que uno de X e Y represente CH y uno de X e Y represente N; R1 representa un grupo metilo;
R2 representa un átomo de hidrógeno;
R3 representa un átomo de flúor, en el que R3 está unido en posición para con el anillo directamente ligado al anillo fenilo al que está unido R3 que es un anillo piridina si Y representa CH y un anillo pirimidina si Y representa N;
R4 representa un átomo de hidrógeno;
R5 representa un grupo seleccionado de un átomo de hidrógeno, ciano, metilo y 3-hidroxipropilo;
o sus enantiómeros, diastereómeros, sales, solvatos o sales de solvatos.
En otra forma de realización de particular preferencia, la presente invención se refiere a compuestos de fórmula general (I), en la que
L representa un grupo -CH2CH2CH2-;
X representa N;
Y representa CH;
R1 representa un grupo metilo;
R2 representa un átomo de hidrógeno o un grupo ciano;
R3 representa un átomo de flúor, en el que R3 está unido en posición para con el anillo directamente ligado al anillo fenilo al que está unido R3 que es un anillo piridina si Y representa CH y un anillo pirimidina si Y representa N;
R4 representa un átomo de hidrógeno;
R5 representa un grupo seleccionado de un átomo de hidrógeno, ciano, metil- y 3-hidroxipropil-;
o sus enantiómeros, diastereómeros, sales, solvatos o sales de solvatos.
En otra forma de realización de particular preferencia, la presente invención se refiere a compuestos de fórmula general (I), en la que
L representa un grupo -CH2CH2CH2-;
X representa N;
Y representa CH;
R1 representa un grupo metil-;
R2 representa un átomo de hidrógeno;
R3 representa un átomo de flúor, en el que R3 está unido en posición para con el anillo directamente ligado al anillo fenilo al que está unido R3 que es un anillo piridina si Y representa CH y un anillo pirimidina si Y representa N;
R4 representa un átomo de hidrógeno;
R5 representa un grupo seleccionado de un átomo de hidrógeno, ciano, metil- y 3-hidroxipropil-;
o sus enantiómeros, diastereómeros, sales, solvatos o sales de solvatos.
En otra forma de realización de particular preferencia, la presente invención se refiere a compuestos de fórmula general (I), en la que
L representa un grupo -CH2CH2CH2CH2-;
X representa CH;
Y representa N;
R1 representa un grupo metil-;
R2 representa un átomo de hidrógeno;
R3 representa un átomo de flúor;
R4 representa un átomo de hidrógeno o un átomo de flúor;
R5 representa un grupo seleccionado de un átomo de hidrógeno, metil- y ciclopropil-;
o sus enantiómeros, diastereómeros, sales, solvatos o sales de solvatos.
En otra forma de realización de particular preferencia, la presente invención se refiere a compuestos de fórmula general (I), en la que
L representa un grupo -CH2CH2CH2CH2-;
X representa CH;
Y representa N;
R1 representa un grupo metil-;
R2 representa un átomo de hidrógeno;
R3 representa un átomo de flúor, en el que R3 está unido en posición para con el anillo directamente ligado al anillo fenilo al que está unido R3 que es un anillo piridina si Y representa CH y un anillo pirimidina si Y representa N;
R4 representa un átomo de hidrógeno;
R5 representa un grupo seleccionado de un átomo de hidrógeno, metil- y ciclopropil-;
o sus enantiómeros, diastereómeros, sales, solvatos o sales de solvatos.
En otra forma de realización de particular preferencia, la presente invención se refiere a compuestos de fórmula general (I), en la que
L representa un grupo -CH2CH2CH2CH2-;
X representa CH;
Y representa N;
R1 representa un grupo metil-;
R2 representa un átomo de hidrógeno;
R3 representa un átomo de flúor, en el que R3 está unido en posición para con el anillo directamente ligado al anillo fenilo al que está unido R3 que es un anillo piridina si Y representa CH y un anillo pirimidina si Y representa N;
R4 representa un átomo de hidrógeno;
R5 representa un átomo de hidrógeno;
o sus enantiómeros, diastereómeros, sales, solvatos o sales de solvatos.
En una forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que L representa un grupo alquileno C2-C5 ,
en el que dicho grupo está opcionalmente sustituido con
(i) un sustituyente seleccionado de cicloalquil C3-C4- e hidroximetil-, y/o
(ii) uno o dos sustituyentes adicionales, iguales o diferentes, seleccionados de alquil C1-C2-.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que L representa un grupo alquileno C2-C4 , en el que dicho grupo está opcionalmente sustituido con uno o dos grupos metil-.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que L representa un grupo alquileno C2-C4.
En una forma de realización de particular preferencia, la invención se refiere a compuestos de fórmula (I), en la que L representa un grupo alquileno C3-C4.
En otra forma de realización de particular preferencia, la invención se refiere a compuestos de fórmula (I), en la que L representa un grupo -CH2CH2CH2- o -CH2CH2CH2CH2-.
En otra forma de realización de particular preferencia, la invención se refiere a compuestos de fórmula (I), en la que L representa un grupo -CH2CH2CH2-.
En otra forma de realización de particular preferencia, la invención se refiere a compuestos de fórmula (I), en la que L representa un grupo -CH2CH2CH2CH2-.
En otra forma de realización, la invención se refiere a compuestos de fórmula (I), en la que X representa N y en la que Y representa CH.
En otra forma de realización, la invención se refiere a compuestos de fórmula (I), en la que X representa CH y en la
que Y representa N.
En otra forma de realización, la invención se refiere a compuestos de fórmula (I), en la que R1 representa un grupo seleccionado de alquil C1-C6- y cicloalquil C3-C5-,
en el que dicho grupo está opcionalmente sustituido con uno o dos o tres sustituyentes, iguales o diferentes, seleccionados del grupo que consiste en hidroxi, ciano, halógeno, alquil C1-C2-, alcoxi C1-C2-, -NH2, -C(=O)OH.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R1 representa un grupo alquil C1-C4-,
en el que dicho grupo está opcionalmente sustituido con uno o dos o tres sustituyentes, iguales o diferentes, seleccionados del grupo que consiste en hidroxi, ciano, un átomo de flúor, alcoxi C1-C2-, -NH2, -C(=O)OH.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R1 representa un grupo alquil C1-C4-,
en la que dicho grupo está opcionalmente sustituido con uno o dos sustituyentes, iguales o diferentes, seleccionados del grupo que consiste en hidroxi, alcoxi C1-C2-, -NH2, -C(=O)OH.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R1 representa un grupo alquil C1-C4-.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R1 representa un grupo alquil C1-C3-.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R1 representa un grupo alquil C1-C2-.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R1 representa un grupo etil-.
En una forma de realización de particular preferencia, la invención se refiere a compuestos de fórmula (I), en la que R1 representa un grupo metil-.
En una forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R1 representa un grupo alquil C1-C4- y R2 representa un átomo de hidrógeno o un átomo de flúor.
En una forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R1 representa un grupo alquil C1-C4- y R2 representa un átomo de hidrógeno o un grupo ciano.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R1 representa un grupo alquil C1-C4- y R2 representa un átomo de hidrógeno.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R1 representa un grupo metilo y R2 representa un átomo de hidrógeno o un grupo ciano.
En una forma de realización de particular preferencia, la invención se refiere a compuestos de fórmula (I), en la que R1 representa un grupo metilo y R2 representa un átomo de hidrógeno.
La invención se refiere a compuestos de fórmula (I), en la que R2 representa un grupo seleccionado de un átomo de hidrógeno, un átomo de flúor, un átomo de cloro, ciano, metil-, metoxi-, trifluorometilo-.
En una forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R2 representa un átomo de hidrógeno o un grupo ciano.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R2 representa un átomo de hidrógeno o un átomo de flúor.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R2 representa un grupo ciano.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R2 representa un átomo de flúor.
En una forma de realización de particular preferencia, la invención se refiere a compuestos de fórmula (I), en la que R2 representa un átomo de hidrógeno.
En otra forma de realización de particular preferencia, la invención se refiere a compuestos de fórmula (I), en la que R2 representa un átomo de hidrógeno, R3 representa un átomo de flúor y R4 representa un átomo de hidrógeno. En otra forma de realización de particular preferencia, la invención se refiere a compuestos de fórmula (I), en la que R1 representa un grupo metil-, R2 representa un átomo de hidrógeno, R3 representa un átomo de flúor y R4 representa un átomo de hidrógeno.
En otra forma de realización de particular preferencia, la invención se refiere a compuestos de fórmula (I), en la que R1 representa un grupo metil-, R2 representa un átomo de hidrógeno y R3 representa un átomo de flúor.
La invención se refiere a compuestos de fórmula (I), en la que R3 representa un grupo seleccionado de un átomo de hidrógeno, un átomo de flúor, un átomo de cloro, ciano, metil-, metoxi-, trifluorometil-, trifluorometoxi-.
En una forma de realización, la invención se refiere a compuestos de fórmula (I), en la que R3 representa, un átomo de hidrógeno, un átomo de flúor o un grupo metoxi-.
En otra forma de realización, la invención se refiere a compuestos de fórmula (I), en la que R3 y R4 representan, de
modo independiente entre sí, un átomo de hidrógeno o un átomo de flúor.
La invención se refiere a compuestos de fórmula (I), en la que R3 representa un grupo seleccionado de un átomo de hidrógeno, un átomo de flúor, un átomo de cloro, ciano, metil-, metoxi-, trifluorometil-, trifluorometoxi- y en la que R4 representa un átomo de hidrógeno.
En una forma de realización, la invención se refiere a compuestos de fórmula (I), en la que R3 representa un átomo de hidrógeno, un átomo de flúor o un grupo metoxi- y en la que R4 representa un átomo de hidrógeno o un átomo de flúor. En otra forma de realización, la invención se refiere a compuestos de fórmula (I), en la que R3 representa un átomo de hidrógeno, un átomo de flúor o un grupo metoxi- y en la que R4 representa un átomo de hidrógeno.
En otra forma de realización, la invención se refiere a compuestos de fórmula (I), en la que R3 representa un átomo de hidrógeno o un átomo de flúor y en la que R4 representa un átomo de hidrógeno.
En otra forma de realización, la invención se refiere a compuestos de fórmula (I), en la que R3 representa un grupo metoxi- y en la que R4 representa un átomo de hidrógeno.
En una forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R3 representa un átomo de flúor y en la que R4 representa un átomo de hidrógeno.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R3 representa un átomo de flúor y en la que R4 representa un átomo de flúor.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R3 representa un átomo de flúor y en la que R4 representa un átomo de flúor.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que

representa un grupo seleccionado de

en las que * es el punto de unión con el anillo piridina (si Y representa CH) o el anillo pirimidina (si Y representa N) al que el anillo fenilo mostrado se une y # es el punto de unión con el resto -O-L-O-.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que

representa un grupo seleccionado de

en las que * es el punto de unión con el anillo piridina (si Y representa CH) o el anillo pirimidina (si Y representa N) al que el anillo fenilo mostrado se une y # es el punto de unión con el resto -O-L-O-.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que

representa un grupo seleccionado de

en las que * es el punto de unión con el anillo piridina (si Y representa CH) o el anillo pirimidina (si Y representa N) al que el anillo fenilo mostrado se une y # es el punto de unión con el resto -O-L-O-.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que

representa un grupo

en la que * es el punto de unión con el anillo piridina (si Y representa CH) o el anillo pirimidina (si Y representa N) al que el anillo fenilo mostrado se une y # es el punto de unión con el resto -O-L-O-.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que

representa un grupo seleccionado de

en la que * es el punto de unión con el anillo piridina (si Y representa CH) o el anillo pirimidina (si Y representa N) al que el anillo fenilo mostrado se une y # es el punto de unión con el resto -O-L-O-.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que

representa un grupo seleccionado de

en la que * es el punto de unión con el anillo piridina (si Y representa CH) o el anillo pirimidina (si Y representa N) al que el anillo fenilo mostrado se une y # es el punto de unión con el resto -O-L-O-.
En otra forma de realización, la invención se refiere a compuestos de fórmula (I), en la que R3 representa un grupo seleccionado de un átomo de hidrógeno, un átomo de flúor, un átomo de cloro, ciano, metil-, metoxi-, trifluorometil-, trifluorometoxi- y en la que R4 representa un átomo de hidrógeno,
en la que R3 está unido en posición para con el anillo directamente ligado al anillo fenilo al que está unido R3 que es un anillo piridina si Y representa CH y un anillo pirimidina si Y representa N.
En una forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R3 representa un átomo de hidrógeno o un átomo de flúor y en la que R4 representa un átomo de hidrógeno,
en la que R3 está unido en posición para con el anillo directamente ligado al anillo fenilo al que está unido R3 que es un anillo piridina si Y representa CH y un anillo pirimidina si Y representa N.
En una forma de realización de particular preferencia, la invención se refiere a compuestos de fórmula (I), en la que R3 representa un átomo de flúor y en la que R4 representa un átomo de hidrógeno,
en la que R3 está unido en posición para con el anillo directamente ligado al anillo fenilo al que está unido R3 que es un anillo piridina si Y representa CH y un anillo pirimidina si Y representa N.
En otra forma de realización, la invención se refiere a compuestos de fórmula (I), en la que R3 representa un grupo seleccionado de un átomo de hidrógeno, un átomo de flúor, un átomo de cloro, ciano, metil-, metoxi-, trifluorometil-, trifluorometoxi-.
En una forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R3 representa un
átomo de hidrógeno o un átomo de flúor.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R3 representa un átomo de hidrógeno.
En una forma de realización de particular preferencia, la invención se refiere a compuestos de fórmula (I), en la que R3 representa un átomo de flúor.
En otra forma de realización de particular preferencia, la invención se refiere a compuestos de fórmula (I), en la que R3 representa un átomo de flúor,
en la que R3 está unido en posición para con el anillo piridina (si Y representa CH) o pirimidina (si Y representa N) directamente unido con el anillo fenilo al que está unido R3
En una forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R4 representa un grupo seleccionado de un átomo de hidrógeno, un átomo de flúor, un átomo de cloro, ciano, metil-, metoxi-, trifluorometil-, trifluorometoxi-.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R4 representa un átomo de hidrógeno o un átomo de flúor.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R4 representa un átomo de flúor.
En una forma de realización de particular preferencia, la invención se refiere a compuestos de fórmula (I), en la que R4 representa un átomo de hidrógeno.
La invención se refiere a compuestos de fórmula (I), en la que R5 representa un grupo seleccionado de un átomo de hidrógeno, ciano, -C(=O)R8, -C(=O)OR8, -S(=O)2R8, -C(=O)NR6R7, alquil C1-C4-, cicloalquil C3-C5-,
en el que dicho grupo alquil C1-C4- o cicloalquil C3-C5- está opcionalmente sustituido con un sustituyente seleccionado del grupo que consiste en flúor, hidroxi, ciano, alcoxi C1-C3-, -NH2 , alquilamino-, dialquilamino-.
En una forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R5 representa un grupo seleccionado de un átomo de hidrógeno, ciano, -C(=O)R8, -C(=O)OR8, -S(=O)2R8, -C(=O)NR6R7, alquil C1-C4-, en la que dicho grupo alquil C1-C4- está opcionalmente sustituido con un sustituyente seleccionado del grupo que consiste en flúor, hidroxi, ciano, alcoxi C1-C3-, -NH2 , alquilamino-, dialquilamino-.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R5 representa un grupo seleccionado de un átomo de hidrógeno, ciano, -C(=O)R8, -C(=O)OR8, -S(=O)2R8, -C(=O)NR6R7, alquil C1-C4-, cicloalquil C3-C5-, en el que dicho grupo alquil C1-C4- está opcionalmente sustituido con un grupo hidroxi.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R5 representa un grupo seleccionado de un átomo de hidrógeno, ciano, -C(=O)R8, -C(=O)OR8, -S(=O)2R8, -C(=O)Nr 6R7, alquil C1-C4-, en el que dicho grupo alquil C1-C4- está opcionalmente sustituido con un grupo hidroxi.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R5 representa un grupo seleccionado de un átomo de hidrógeno, ciano, -C(=O)OR8, -C(=O)NR6R7, alquil C1-C4-, cicloalquil C3-C5-, en el que dicho grupo alquil C1-C4- está opcionalmente sustituido con un grupo hidroxi.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R5 representa un grupo seleccionado de un átomo de hidrógeno, ciano, -C(=O)OR8, -C(=O)NR6R7, alquil C1-C4-,
en el que dicho grupo alquil C1-C4- está opcionalmente sustituido con un grupo hidroxi.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R5 representa un grupo seleccionado de un átomo de hidrógeno, ciano, alquil C1-C4-, cicloalquil C3-C5-,
en el que dicho grupo alquil C1-C4- está opcionalmente sustituido con un grupo hidroxi.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R5 representa un grupo seleccionado de un átomo de hidrógeno, ciano, alquil C1-C4-,
en el que dicho grupo alquil C1-C4- está opcionalmente sustituido con un grupo hidroxi.
En una forma de realización de particular preferencia, la invención se refiere a compuestos de fórmula (I), en la que R5 representa un grupo seleccionado de un átomo de hidrógeno, ciano, alquil C1-C3-, ciclopropil-,
en el que dicho grupo alquil C1-C3- está opcionalmente sustituido con un grupo hidroxi.
En una forma de realización de particular preferencia, la invención se refiere a compuestos de fórmula (I), en la que R5 representa un grupo seleccionado de un átomo de hidrógeno, ciano, alquil C1-C3-,
en el que dicho grupo alquil C1-C3- está opcionalmente sustituido con un grupo hidroxi.
En otra forma de realización de particular preferencia, la invención se refiere a compuestos de fórmula (I), en la que R5 representa un grupo seleccionado de un átomo de hidrógeno, ciano, metil-, 3-hidroxipropil- y ciclopropil-.
En otra forma de realización de particular preferencia, la invención se refiere a compuestos de fórmula (I), en la que R5 representa un grupo seleccionado de un átomo de hidrógeno, ciano, metil- y 3-hidroxipropil-.
En otra forma de realización de particular preferencia, la invención se refiere a compuestos de fórmula (I), en la que R5 representa un átomo de hidrógeno.
En otra forma de realización de particular preferencia, la invención se refiere a compuestos de fórmula (I), en la que R5 representa un grupo ciano.
En otra forma de realización de particular preferencia, la invención se refiere a compuestos de fórmula (I), en la que R5 representa un grupo metil-.
En otra forma de realización de particular preferencia, la invención se refiere a compuestos de fórmula (I), en la que R5 representa un grupo 3-hidroxipropil-,
En otra forma de realización de particular preferencia, la invención se refiere a compuestos de fórmula (I), en la que R5 representa un grupo ciclopropil-,
La invención se refiere a compuestos de fórmula (I), en la que R6 y R7 representan, de modo independiente entre sí, un grupo seleccionado de un átomo de hidrógeno, alquil C1-C4- y cicloalquil C3-C5-,
en el que dicho grupo alquil C1-C4- o cicloalquil C3-C5- está opcionalmente sustituido con uno o dos sustituyentes, iguales o diferentes, seleccionados del grupo que consiste en hidroxi, alquil C1-C2-, alcoxi C1-C2-, -NH2, alquilamino-, dialquilamino-.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R6 representa un grupo seleccionado de un átomo de hidrógeno, alquil C1-C4- y cicloalquil C3-C5-,
en el que dicho grupo alquil C1-C4- o cicloalquil C3-C5- está opcionalmente sustituido con uno o dos sustituyentes, iguales o diferentes, seleccionados del grupo que consiste en hidroxi, alquil C1-C2-, alcoxi C1-C2-, -NH2, alquilamino-, dialquilamino-, y en la que R7 representa un átomo de hidrógeno o un grupo alquil C1-C3-.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R6 representa un grupo seleccionado de un átomo de hidrógeno, alquil C1-C4- y cicloalquil C3-C5-,
en el que dicho grupo alquil C1-C4- o cicloalquil C3-C5- está opcionalmente sustituido con uno o dos sustituyentes, iguales o diferentes, seleccionados del grupo que consiste en hidroxi, alquil C1-C2-, alcoxi C1-C2-, -NH2, alquilamino-, dialquilamino-, y en la que R7 representa un átomo de hidrógeno o un grupo alquil C1-C3-.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R6 y R7 representan, de modo independiente entre sí, un grupo seleccionado de un átomo de hidrógeno y alquil C1-C4-, en el que dicho grupo alquil C1-C4- está opcionalmente sustituido con un sustituyente seleccionado del grupo que consiste en hidroxi, alcoxi C1-C2-, -NH2, alquilamino-, dialquilamino-.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R6 representa un grupo seleccionado de un átomo de hidrógeno y alquil C1-C4-,
en el que dicho grupo alquil C1-C4- está opcionalmente sustituido con un sustituyente seleccionado del grupo que consiste en hidroxi, alcoxi C1-C2 , -NH2 , alquilamino-, dialquilamino-,
y en la que R7 representa un átomo de hidrógeno o un grupo alquil C1-C3-.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R6 y R7 representan, de modo independiente entre sí, un grupo seleccionado de un átomo de hidrógeno, alquil C1-C4- y cicloalquil C3-C5-.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R6 y R7 representan, de modo independiente entre sí, un grupo seleccionado de un átomo de hidrógeno, un grupo metil- y un grupo etil-.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R6 representa un grupo seleccionado de un átomo de hidrógeno, un grupo metil- y un grupo etil- y en la que R7 representa un átomo de hidrógeno.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R6 representa un grupo seleccionado de un átomo de hidrógeno, un grupo metilo-y un grupo etil-.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R7 representa un átomo de hidrógeno.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R6 representa un grupo metilo o un grupo etilo y en la que R7 representa un átomo de hidrógeno.
En otra forma de realización, la invención se refiere a compuestos de fórmula (I), en la que R6 representa un grupo metil- o un grupo etil-.
La invención se refiere a compuestos de fórmula (I), en la que R8 representa un grupo seleccionado de alquil C1-C6-, fluoro-alquil C1-C3-, cicloalquil C3-C5- y fenil-,
en el que dicho grupo está opcionalmente sustituido con un sustituyente seleccionado del grupo que consiste en halógeno, hidroxi, alquil C1-C2-, alcoxi C1-C2-, -NH2.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R8 representa un grupo seleccionado de alquil C1-C3-, fluoro-alquil C1-C2- y fenil-,
en el que dicho grupo está opcionalmente sustituido con un sustituyente seleccionado del grupo que consiste en flúor, hidroxi, metil-, metoxi-.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R8 representa un grupo seleccionado de alquil C1-C3-, fluoro-alquil C1-C2- y fenil-.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R8 representa un
grupo seleccionado de alquil C1-C4-, fluoro-alquil C1-C3-.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R8 representa un grupo alquil C1-C4-.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R8 representa un grupo metil- o un grupo etil-.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R8 representa un grupo metil-.
En otra forma de realización preferida, la invención se refiere a compuestos de fórmula (I), en la que R8 representa un grupo etil-.
Se ha de entender que la presente invención se refiere a cualquier subcombinación dentro de cualquier forma de realización de la presente invención de compuestos de fórmula (I), anterior.
Más particularmente aún, la presente invención cubre compuestos de fórmula (I) que se desvelan en la sección de ejemplos de este texto, posteriormente.
Se prefieren muy especialmente las combinaciones de dos o más de las formas de realización preferidas antes mencionadas.
En particular, un objeto preferido de la presente invención es un compuesto seleccionado de:
- 15,19-difluoro-8-[(S-metilsulfonodiimidoil)metil]-3,4-dihidro-2H,11H-10,6-(azeno)-12,16-(meteno)-1,5,11,13-benzodioxadiazaciclooctadecino;
- (rac)-3-(2-{[15,19-difluoro-3,4-dihidro-2H,11H-10,6-(azeno)-12,16-(meteno)-1,5,11,13-benzodioxadiazaciclooctadecin-8-il]metil}-2-metil-2A6-diazatia-1,2-dien-1-il)propan-1-ol;
- (rac)-[{[15,19-difluoro-3,4-dihidro-2H,1lH-10,6-(azeno)-12,16-(meteno)-1,5,11,13-benzodioxadiazaciclooctadecin-8-il]metil}(imino)metil-A6-sulfaniliden]cianamida;
- (rac)-8-[(N,S-dimetilsulfonodiimidoil)metil]-15,19-difluoro-3,4-dihidro-2H,11H-10,6-(azeno)-12,16-(meteno)-1.5.11.13- benzodioxadiazaciclooctadecino y
- 16,20-difluoro-9-[(S-metilsulfonodiimidoil)metil]-2,3,4,5-tetrahidro-12H-13,17-(azeno)-11,7-(meteno)-1,6,12,14-benzodioxadiazaciclononadecino;
- 16,20,21-trifluoro-9-[(S-metilsulfonodiimidoil)metil]-2,3,4,5-tetrahidro-12H-13,17-(azeno)-11,7-(meteno)-1,6,12,14-benzodioxadiazaciclononadecino;
- 16,21-difluoro-9-[(S-metilsulfonodiimidoil)metil]-2,3,4,5-tetrahidro-12H-13,17-(azeno)-11,7-(meteno)-1,6,12,14-benzodioxadiazaciclononadecino;
- 15,19-difluoro-8-[(S-metilsulfonodiimidoil)metil]-3,4-dihidro-2H,11H-10,6-(azeno)-12,16-(meteno)-1,5,11,13-benzodioxadiazaciclooctadecino-7-carbonitrilo;
- (rac)-9-[(N-ciclopropil-S-metilsulfonodiimidoil)metil]-16,20-difluoro-2,3,4,5-tetrahidro-12H-13,17-(azeno)-11,7-(meteno)-1,6,12,14-benzodioxadiazaciclononadecino;
- (rac)-9-[(N,S-dimetilsulfonodiimidoil)metil]-16,20-difluoro-2,3,4,5-tetrahidro-12H-13,17-(azeno)-11,7-(meteno)-1.6.12.14- benzodioxadiazaciclononadecino;
y los enantiómeros, diastereómeros, sales, solvatos o sales de solvatos de los mismos.
Las definiciones de grupos y radicales anteriores que se detallaron en términos generales o en intervalos preferidos también se aplican a los productos finales de la fórmula (I) y, de manera análoga, a los materiales de partida o intermedios requeridos en cada caso para la preparación.
La presente invención también se refiere a un procedimiento para la preparación de los compuestos de fórmula (10), en la que R1, R2, R3, R4, R5 y L son como se definen para los compuestos de fórmula (I) de acuerdo con la invención, en la que los compuestos de procedimiento de la fórmula (9)

en la que R1, R2, R3, R4 y L son como se definen para el compuesto de fórmula (I) de acuerdo con la invención, se oxidan por tratamiento con un agente seleccionado de diacetato de yodobenceno y W-clorosuccinimida, seguido por adición de una amina seleccionada de una amina primaria de la fórmula R5-NH2, en la que R5 es como se define para los compuestos de fórmula (I) de acuerdo con la invención y hexametildisilaceno, para dar los compuestos de fórmula (10),

y en la que los compuestos resultantes se convierten opcionalmente, de ser apropiado, con los correspondientes (i) disolventes y/o (ii) bases o ácidos a los sus solvatos, sales y/o solvatos de las sales de los mismos.
La presente invención también se refiere a un procedimiento para la preparación de los compuestos de fórmula (23), en la que R1, R2, R3, R4, R5 y L son como se definen para los compuestos de fórmula (I) de acuerdo con la invención, en cuyo procedimiento los compuestos de procedimiento de la fórmula (22)

en la que R1, R2, R3, R4 y L son como se definen para el compuesto de fórmula (I) de acuerdo con la invención, se oxidan por tratamiento con un agente seleccionado de diacetato de yodobenceno y W-clorosuccinimida, seguido por adición de una amina seleccionada de una amina primaria de la fórmula R5-NH2, en la que R5 es como se define para los compuestos de fórmula (I) de acuerdo con la invención y hexametildisilaceno, para dar los compuestos de fórmula (23),

y en cuyo procedimiento los compuestos resultantes se convierten opcionalmente, de ser apropiado, con los correspondientes (i) disolventes y/o (ii) bases o ácidos a los solvatos, sales y/o solvatos de las sales de los mismos.
La invención también se refiere a compuestos de fórmula (9), en la que R1, R2, R3, R4 y L son como se definen para el compuesto de fórmula (I) de acuerdo con la invención,

o los enantiómeros, diastereómeros o solvatos de los mismos.
La invención también se refiere al uso de los compuestos de fórmula (9), en la que R1, R2, R3, R4 y L son como se definen para el compuesto de fórmula (I) de acuerdo con la invención,

o los enantiómeros, diastereómeros o solvatos de los mismos, para la preparación de compuestos de fórmula (I). La invención también se refiere a compuestos de fórmula (22), en la que R1, R2, R3, R4 y L son como se definen para el compuesto de fórmula (I) de acuerdo con la invención,

o los enantiómeros, diastereómeros o solvatos de los mismos.
La invención también se refiere al uso de los compuestos de fórmula (22), en la que R1, R2, R3, R4 y L son como se definen para el compuesto de fórmula (I) de acuerdo con la invención,

o los enantiómeros, diastereómeros o solvatos de los mismos, para la preparación de compuestos de fórmula (I). Los compuestos de acuerdo con la invención presentan un espectro de acción farmacológica y farmacocinética valioso, que no podría haber sido predicho.
Por lo tanto, son adecuados para su uso como medicamentos para el tratamiento y/o la profilaxis de trastornos en seres humanos y animales.
La actividad farmacéutica de los compuestos de acuerdo con la invención se puede explicar por medio de su acción como inhibidores selectivos de CDK9, y, de modo más significativo, como inhibidores selectivos de CDK9 a altas
concentraciones de ATP.
Por tanto, los compuestos de acuerdo con la fórmula general (I) y sus enantiómeros, diastereómeros, sales, solvatos y sales de los solvatos son útiles como inhibidores selectivos de la CDK9.
Además, los compuestos de acuerdo con la invención muestran una potencia particularmente elevada en el contexto de la inhibición selectiva de la actividad de la CDK9 (lo cual puede demostrarse a través del valor reducido de su CI50 sobre el complejo de la CDK9 y la ciclina T1, que puede determinarse con un ensayo apropiado), en particular a altas concentraciones de ATP.
En el contexto de la presente invención, el valor de la CI50 con relación a la CDK9 puede determinarse con los procedimientos que se describen en la sección de procedimientos a continuación.
En comparación con los muchos inhibidores de la CDK9 que se han descrito en los antecedentes técnicos, los compuestos de la presente invención, que responden a la fórmula general (I), presentan una potencia sorprendentemente alta en el contexto de la inhibición de la actividad de la CDK9 en especial a altas concentraciones de ATP, lo cual puede manifestarse como una CI50 con relación a la CDK9 o a la ciclina T1 con un valor menor en un análisis con quinasas en presencia de una concentración elevada de ATP. Por lo tanto, es menos probable que los compuestos de este tipo compitan por el sitio al que puede unirse el ATP en las quinasas del tipo de la CDK9 o la ciclina T1 en presencia de una concentración elevada de ATP en las células (R. Copeland y col., Nature Reviews Drug Discovery, 2006, 5, 730-739). Debido a esta propiedad, los compuestos de la presente invención son particularmente útiles para inhibir la CDK9 o la ciclina T1 en las células durante un período de tiempo más prolongado que los inhibidores de la quinasa clásicos que compiten con el ATP. De esta manera, puede incrementarse la eficacia sobre las células tumorales cuando la concentración del inhibidor en el suero se reduce como resultado de la eliminación farmacocinética después de la administración en un paciente o en un animal.
En comparación con los inhibidores de la CDK9 que se conocen de la técnica anterior, los compuestos de la presente invención pueden residir en el blanco durante un período de tiempo sorprendentemente prolongado. Se ha sugerido que la residencia en el blanco es un parámetro apropiado para predecir la eficacia de un fármaco, y en este contexto, es necesario tener en cuenta el hecho de que los resultados que se obtienen en el equilibrio en los análisis in vitro no reflejan de manera apropiada las situaciones in vivo, donde las concentraciones del fármaco pueden fluctuar debido a los procedimientos de adsorción, de distribución y de eliminación y donde la concentración de la proteína que hace las veces de blanco puede estar regulada de manera dinámica (Tummino, P. J., y Copeland, R. A., Residence time of receptor-ligand complexes and its effect on biological function, Biochemistry, 2008. 47 (20): p. 5481-5492; Copeland, R. A., Pompliano, D. L., y Meek, T. D., Drug-target residence time and its implications for lead optimization, Nature Reviews Drug Discovery, 2006. 5 (9): p. 730-739).
Como consecuencia de lo anterior, el parámetro que suele emplearse para analizar la unión en el equilibrio, la Kd, o el que suele usarse para analizar la función, la CI50, puede no reflejar de manera precisa los requisitos para la eficacia in vivo. Si se asume que una molécula de un fármaco solamente puede actuar en el transcurso del período de tiempo durante el cual permanece unida al blanco, la "vida media" (el período de residencia) del complejo compuesto por el fármaco y el blanco puede servir como un parámetro para predecir de una manera más confiable la eficacia que presentará el fármaco en un sistema in vivo que no se encuentre en el equilibrio. Las implicaciones de lo anterior sobre la eficacia in vivo se mencionan y se discuten en diversas publicaciones (Lu, H., y Tonge, P. J., Drug-target residence time: critical information for lead optimization. Curr. Opin. Chem. Biol., 2010, 14 (4): p. 467-74; Vauquelin, G., y Charlton, S. J., Long-lasting target binding and rebinding as mechanisms to prolong in vivo drug action. Br. J. Pharmacol., 2010, 161 (3): p. 488-508).
Un ejemplo del impacto que puede tener la duración de la residencia en el blanco puede analizarse con el fármaco tiotropio, que suele usarse en el tratamiento de la EPOC. El tiotropio puede unirse con una afinidad comparable a los subtipos M1, M2 y M3 de los receptores muscarínicos, pero presenta selectividad desde el punto de vista cinético, ya que solamente presenta un período de residencia prolongado cuando se une a los receptores M3. El período de residencia de este fármaco en el blanco es suficientemente prolongado para que, después de eliminarla de la tráquea humana in vitro, persista una inhibición de la actividad colinérgica, con una vida media de 9 horas. Esto se traduce en una protección contra los broncoespasmos in vivo durante más de 6 horas (Price, D., Sharma, A., y Cerasoli, F., Biochemical properties, pharmacokinetics and pharmacological response of tiotropium in chronic obstructive pulmonary disease patients, 2009; Dowling, M. (2006), Br. J. Pharmacol., 148, 927-937).
Otro ejemplo puede analizarse con el lapatinib (Tykerb). En función de la determinación del grado de fosforilación de los residuos de tirosina en los receptores, se ha determinado que el período de residencia prolongado que caracteriza al lapatinib en el contexto de la reacción con el dominio intracelular purificado de la enzima que hace las veces de blanco está correlacionado con la inhibición prolongada de la señal que se observa en las células tumorales. Posteriormente, se concluyó que, a través de una unión con una cinética lenta, podría obtenerse una mayor inhibición de la señal en los tumores, lo que podría dar como resultado una probabilidad mayor de afectar la velocidad de crecimiento de los tumores o la eficacia de la administración concurrente de otros agentes quimioterapéuticos (Wood y col. (2004), Cancer Res, 64: 6652-6659; Lackey (2006), Current Topics in Medicinal Chemistry, 2006, vol. 6, n.° 5).
En el contexto de la presente invención, el valor de CI50 respecto a CDK9 a concentraciones altas de ATP puede determinarse por los procedimientos descritos en la sección de procedimientos a continuación. Preferentemente, se determina de acuerdo al procedimiento 1b ("ensayo de quinasa CDK9/CycT1 en alto ATP") como se describe en la sección de materiales y procedimientos a continuación.
Si se desea, el valor de CI50 con respecto a CDK9 a baja concentración de ATP se puede determinar, por ejemplo, por medio de los procedimientos descritos en la sección de procedimientos a continuación, de acuerdo con el Procedimiento 1a. ("Ensayo de quinasa CDK9/CycT1") descrito en la sección de materiales y procedimiento a continuación.
En el contexto de la presente invención, el período de residencia en el blanco que caracteriza a los inhibidores selectivos de la CDK9 de acuerdo con la presente invención puede determinarse con los procedimientos que se describen en la sección de procedimientos más adelante. Con preferencia, se lo determina de acuerdo con el procedimiento 8 ("resonancia de plasmón superficial con el PTEFb"), según se describe en la sección de materiales y procedimientos a continuación.
Además, los compuestos de la presente invención de acuerdo con la fórmula (I), sorprendentemente presentan una actividad antiproliferativa excepcionalmente alta en líneas celulares tumorales como HeLa, HeLa-MaTu-ADR, NCI-H460, DU145, Caco-2, B16F10, A2780 o MOLM-13, en comparación con los inhibidores de la CDK9 que se conocen de la técnica anterior.
En el contexto de la presente invención, la actividad antiproliferativa en líneas de células tumorales como HeLa, HeLa-MaTu-ADR, NCI-H460, DU145, Caco-2, B16F10, A2780 o MOLM-13 preferentemente se determina de acuerdo con el procedimiento 3 ("análisis de la proliferación"), según se describe en la sección de materiales y procedimientos a continuación.
En el contexto de la presente invención, la estabilidad metabólica en los hepatocitos de las ratas preferentemente se determina de acuerdo con el procedimiento 6 ("investigación de la estabilidad metabólica en hepatocitos de rata in vitro"), según se describe en la sección de materiales y procedimientos a continuación.
En el contexto de la presente invención, la vida media en las ratas después de una administración in vivo preferentemente se determina de acuerdo con el procedimiento 7 ("farmacocinética en ratas in vivo"), según se describe en la sección de materiales y procedimientos a continuación.
Además, los compuestos de la presente invención de acuerdo con formula (I) se caracterizan por una permeabilidad aceptable de Caco-2 (Papp A-B) a través de las monocapas de células Caco-2.
Además, los compuestos de la presente invención, que responden a la fórmula (I), se caracterizan por una proporción del flujo de salida aceptable (la proporción del flujo de salida se calcula como Papp B-A/Papp A-B) desde el compartimiento basal hasta el compartimiento apical en monocapas de células Caco-2, en comparación con los compuestos que se conocen de la técnica anterior.
En el contexto de la presente invención, los valores de permeabilidad aparente de Caco-2 del compartimento basal al apical (Papp A-B) o la relación de salida (definida como la relación ((Papp B-A)/(Papp A-B)) preferentemente se determinan de acuerdo al procedimiento 5. ("ensayo de permeación de Caco-2") descrito en la sección de materiales y procedimientos a continuación.
En la presente invención también se describe el uso de los compuestos de acuerdo con la invención, que presentan la fórmula general (I), en el tratamiento y/o la profilaxis de diversos trastornos, preferentemente de aquellos trastornos que están relacionados con la actividad de la CDK9 o que están mediados por dicha actividad, particularmente de los trastornos hiperproliferativos, de las enfermedades infecciosas inducidas por virus y/o de las enfermedades cardiovasculares, y más preferentemente de los trastornos hiperproliferativos. Los compuestos de la presente invención pueden usarse para inhibir selectivamente la actividad o la expresión de la CDK9.
Por lo tanto, se espera que los compuestos de fórmula (I) sean agentes terapéuticos valiosos. De esta manera, en otra forma de realización, en la presente divulgación se proporciona un procedimiento para tratar aquellos trastornos que están relacionados con la actividad de la CDK9 o que están mediados por dicha actividad en un paciente que lo necesita, que comprende administrarle al paciente una cantidad eficaz de un compuesto de fórmula (I) como los que se definieron anteriormente. En determinadas formas de realización, los trastornos que están relacionados con la actividad de la CDK9 son trastornos hiperproliferativos, enfermedades infecciosas inducidas por virus y/o enfermedades cardiovasculares, más preferentemente son trastornos hiperproliferativos, y más particularmente son cánceres.
El término "tratar" o "tratamiento" como se indica a lo largo de este documento se usa convencionalmente, por ejemplo, el manejo o cuidado de un sujeto con el propósito de combatir, aliviar, reducir, aliviar, mejorar la condición de una enfermedad o trastorno, como un carcinoma.
El término "sujeto" o "paciente" incluye los organismos que pueden padecer un trastorno relacionado con la proliferación o un trastorno asociado a una muerte celular programada (una apoptosis) reducida o insuficiente, y también abarcan aquellos organismos que podrían beneficiarse con la administración de un compuesto de acuerdo
con la invención. Estos organismos pueden ser seres humanos o animales no humanos. En particular, los sujetos humanos pueden padecer un trastorno relacionado con la proliferación de las células o un estado asociado como los que se describen en el presente documento, o bien pueden hallarse en riesgo de contraerlo. El término "animales no humanos" abarca los vertebrados, por ejemplo, los animales mamíferos, tales como los primates no humanos, las ovejas, las vacas, los perros, los gatos y los roedores, por ejemplo, los ratones, así como los animales no mamíferos, tales como las aves, particularmente los pollos, los anfibios, los reptiles, etc.
La expresión "trastornos relacionados o mediados por CDK9" incluirá enfermedades asociadas o que impliquen actividad de CDK9, por ejemplo, la hiperactividad de CDK9 y afecciones que acompañan a estas enfermedades. Los ejemplos de "trastornos relacionados o mediados por CDK9" incluyen trastornos que resultan del aumento de la actividad de CDK9 debido a mutaciones en genes que regulan la actividad de CDK9 como LARP7, HEXIM1/2 o 7sk snRNA, o trastornos que resultan de un aumento de la actividad de CDK9 debido a la activación de CDK9/cyclinT/ARN polimerasa II por proteínas virales como HIV-TAT o HTLVTAX o trastornos resultantes del aumento de la actividad de CDK9 debido a la activación de las vías de señalización mitogénicas. La expresión "hiperactividad de CDK9" se refiere al aumento de la actividad enzimática de CDK9 en comparación con las células normales no enfermas, o se refiere al aumento de la actividad de CDK9 que conduce a la proliferación celular no deseada, o a la muerte celular reducida o insuficiente programada (apoptosis), o mutaciones que conduce a la activación constitutiva de CDK9.
La expresión "trastorno hiperproliferativo" abarca aquellos trastornos en los que hay una proliferación indeseable o descontrolada de las células, lo que incluye aquellos trastornos en los que hay una muerte programada de las células (una apoptosis) reducida o insuficiente. Los compuestos de la presente invención pueden usarse para prevenir, inhibir, bloquear, reducir, disminuir o controlar (entre otras acciones) la división de las células y/o para producir la apoptosis. Un procedimiento posible puede comprender administrarle al sujeto que lo necesita, que puede ser un mamífero, tal como un ser humano, una cantidad de un compuesto de acuerdo con la presente invención, o de una sal, un hidrato o un solvato farmacéuticamente aceptable de éste, que sea eficaz para tratar o prevenir el trastorno en cuestión.
En el contexto de esta invención, los trastornos hiperproliferativos incluyen, pero sin limitaciones, la psoriasis, los queloides y otras hiperplasias que afectan a la piel, la endometriosis, los trastornos en el esqueleto, los trastornos relacionados con la angiogénesis o con la proliferación de los vasos sanguíneos, la hipertensión pulmonar, los trastornos fibróticos, los trastornos asociados a la proliferación de las células mesangiales, los pólipos en el colon, la enfermedad poliquística de los riñones, la hiperplasia benigna de la próstata (BPH) y los tumores sólidos, tales como el cáncer de mama, el cáncer del tracto respiratorio, el cáncer de cerebro, el cáncer de los órganos reproductivos, el cáncer del tracto digestivo, el cáncer del tracto urinario, el cáncer de los ojos, el cáncer de hígado, el cáncer de piel, el cáncer de cabeza y cuello, el cáncer de tiroides, el cáncer de paratiroides y sus metástasis distantes. Estos trastornos también abarcan los linfomas, los sarcomas y las leucemias.
Los ejemplos de cánceres de mama incluyen, pero sin limitaciones, los carcinomas invasivos de los conductos, los carcinomas invasivos de los lóbulos, los carcinomas in situ de los conductos, los carcinomas in situ de los lóbulos y los carcinomas en las mamas de los caninos o de los felinos. Los ejemplos de cánceres del tracto respiratorio incluyen, pero sin limitaciones, los carcinomas de las células pulmonares pequeñas o no pequeñas, así como los adenomas bronquiales, los blastomas pleuropulmonares y los mesoteliomas. Los ejemplos de cánceres de cerebro incluyen, pero sin limitaciones, los gliomas en el tallo cerebral o en el hipotálamo, los astrocitomas cerebelares o cerebrales, los glioblastomas, los meduloblastomas, los ependimomas y los tumores en el neuroectodermo o en la glándula pineal. Los tumores en los órganos reproductivos masculinos incluyen, pero sin limitaciones, el cáncer de próstata y el cáncer de testículo. Los tumores en los órganos reproductivos femeninos incluyen, pero sin limitaciones, el cáncer de endometrio, el cáncer de cuello uterino, el cáncer de ovario, el cáncer de vagina, el cáncer de vulva y los sarcomas en el útero.
Los tumores en el tracto digestivo incluyen, pero sin limitaciones, el cáncer de ano, el cáncer de colon, el cáncer colorrectal, el cáncer de esófago, el cáncer de vesícula biliar, el cáncer de estómago, el cáncer de páncreas, el cáncer de recto, el cáncer del intestino delgado, el cáncer de las glándulas salivales, los adenocarcinomas en las glándulas anales y los tumores en los mastocitos.
Los tumores en el tracto urinario incluyen, pero sin limitaciones, el cáncer de vejiga, el cáncer de pene, el cáncer de riñón, el cáncer de la pelvis renal, el cáncer de uréter, el cáncer de uretra y los cánceres papilares hereditarios y esporádicos en los riñones.
Los cánceres en los ojos incluyen, pero sin limitaciones, los melanomas intraoculares y los retinoblastomas.
Los ejemplos de cánceres de hígado incluyen, pero sin limitaciones, los carcinomas hepatocelulares (los carcinomas en las células del hígado, que pueden presentar o no variantes fibrolamelares), los colangiocarcinomas (que son car cinomas en los conductos biliares del hígado) y los colangiocarcinomas hepatocelulares mixtos.
Los cánceres de piel incluyen, pero sin limitaciones, los carcinomas de las células escamosas, los sarcomas de Kaposi, los melanomas malignos, los cánceres de piel que afectan a las células de Merkel, los cánceres de piel diferentes de los melanomas y los tumores en los mastocitos.
Los cánceres de cabeza y cuello incluyen, pero sin limitaciones, el cáncer de laringe, el cáncer de hipofaringe, el cáncer de nasofaringe, el cáncer de orofaringe, el cáncer en los labios y en la cavidad oral, los cánceres que afectan a las células escamosas y los melanomas orales. Los linfomas incluyen, pero sin limitaciones, los linfomas relacionados con el SIDA, los linfomas no Hodgkin, los linfomas cutáneos que afectan a las células T, los linfomas de Burkitt, la enfermedad de Hodgkin y los linfomas en el sistema nervioso central.
Los sarcomas incluyen, pero sin limitaciones, los sarcomas en los tejidos blandos, los osteosarcomas, los histiocitomas
fibrosos malignos, los linfosarcomas, los rabdomiosarcomas, las histiocitosis malignas, los fibrosarcomas, los hemangiosarcomas, los hemangiopericitomas y los leiomiosarcomas.
Las leucemias incluyen, pero sin limitaciones, la leucemia mieloide aguda, la leucemia linfoblástica aguda, la leucemia linfocítica crónica, la leucemia mielógena crónica y la leucemia que afecta a las células pilosas.
Con los compuestos y los procedimientos de la presente invención, es posible tratar determinados trastornos fibróticos relacionados con la proliferación, es decir, determinados trastornos que se caracterizan por la formación de matrices extracelulares anormales, que incluyen la fibrosis pulmonar, la aterosclerosis, la reestenosis, la cirrosis hepática y los trastornos asociados a la proliferación de las células mesangiales, que abarcan enfermedades renales como la glomerulonefritis, la nefropatía diabética, la nefrosclerosis maligna, los síndromes asociados a la microangiopatía trombótica, el rechazo de trasplantes y las glomerulopatías.
Otras afecciones en los seres humanos o en otros mamíferos que pueden tratarse con los compuestos de la presente invención incluyen el desarrollo de tumores, la retinopatía, lo que abarca la retinopatía diabética, la oclusión isquémica de las venas de la retina, la retinopatía de la premadurez y la degeneración macular relacionada con la edad, la artritis reumática, la psoriasis y los trastornos bulbosos asociados a la formación de ampollas subepidérmicas, lo que abarca el penfigoide bulboso, el eritema multiforme y la dermatitis herpetiforme.
Los compuestos de la presente invención también pueden usarse para prevenir o tratar enfermedades en las vías aéreas o en los pulmones, enfermedades en el tracto gastrointestinal o enfermedades en la vejiga o en los conductos biliares.
Los trastornos mencionados anteriormente han sido bien caracterizados en los seres humanos, pero también ocurren con una etiología similar en otros animales, que incluyen otros mamíferos, y es posible tratarlos con las composiciones farmacéuticas de la presente invención.
En otro aspecto de la presente divulgación, los compuestos de acuerdo con la invención se usan en un procedimiento para prevenir y/o tratar enfermedades infecciosas, particularmente enfermedades infecciosas inducidas por virus. Las enfermedades infecciosas inducidas por virus, que pueden ser enfermedades oportunistas, pueden ser provocadas por retrovirus, por hepadnavirus, por virus del herpes, por flavivirus y/o por adenovirus. En otra forma de realización preferida de este procedimiento, los retrovirus se seleccionan entre los lentivirus y los oncorretrovirus, donde los lentivirus pueden seleccionarse del grupo que comprende el VIH-1, el VIH-2, el FIV, el BIV, los SIV, el SHIV, el CAEV, el VMV y el EIAV, y preferentemente se seleccionan entre el VIH-1 y el VIH-2, y donde los oncorretrovirus pueden seleccionarse del grupo que consiste en el HTLV-I, el HTLV-II y el BLV. En otra forma de realización preferida de este procedimiento, los hepadnavirus se seleccionan entre el HBV, el GSHV y el WHV, y preferentemente son e1HBV, los virus del herpes se seleccionan del grupo que consiste en el HSV I, el HSV II, el EBV, el VZV, e1HCMV y e1HHV 8, preferentemente el HCMV y los flavivirus se seleccionan entre el HCV, el virus del Nilo occidental y el virus de la fiebre amarilla.
Los compuestos de acuerdo con la fórmula general (I) también pueden ser útiles para prevenir y/o tratar enfermedades cardiovasculares como la hipertrofia cardíaca, la enfermedad cardíaca congénita que afecta a los adultos, los aneurismas, la angina estable, la angina inestable, la angina de pecho, el edema angioneurótico, la estenosis en la válvula aórtica, los aneurismas aórticos, las arritmias, la displasia arritmogénica en el ventrículo derecho, la arteriosclerosis, las deformaciones arteriovenosas, la fibrilación atrial, el síndrome de Behcet, la bradicardia, las oclusiones cardíacas, la cardiomialgia, la cardiomiopatía congestiva, la cardiomiopatía hipertrófica, la cardiomiopatía restrictiva, la estenosis en la carótida, las hemorragias en el cerebro, el síndrome de Churg-Strauss, la diabetes, la anomalía de Ebstein, el complejo de Eisenmenger, las embolias debidas al colesterol, la endocarditis de origen bacteriano, la displasia fibromuscular, los defectos congénitos en el corazón, las enfermedades en el corazón, la insuficiencia cardíaca congestiva, las enfermedades en las válvulas del corazón, los ataques cardiacos, los hematomas epidurales, los hematomas subdurales, la enfermedad de Hippel-Lindau, la hiperemia, la hipertensión, la hipertensión pulmonar, los desarrollos hipertróficos, la hipertrofia del ventrículo izquierdo, el síndrome hipoplásico en el ventrículo izquierdo del corazón, la hipotensión, la claudicación intermitente, la enfermedad isquémica del corazón, el síndrome de Klippel-Trenaunay-Weber, el síndrome medular lateral, el prolapso de la válvula mitral con un intervalo QT largo, la enfermedad de Moyamoya, el síndrome de los nodos linfáticos mucocutáneos, el infarto de miocardio, las isquemias en el miocardio, la miocarditis, la pericarditis, las enfermedades en los vasos periféricos, la flebitis, la poliarteritis nodosa, la atresia pulmonar, la enfermedad de Raynaud, el síndrome de Sneddon, la estenosis, el síndrome de la vena cava superior, el síndrome X, la taquicardia, la arteritis de Takayasu, la telangiectasia hemorrágica hereditaria, la telangiectasis, la arteritis temporal, la tetralogía de Fallot, la tromboangiitis obliterante, la trombosis, la tromboembolia, la atresia en la válvula tricúspide, las várices en las venas, las enfermedades en los vasos, la vasculitis, el vasoespasmo, la fibrilación ventricular, el síndrome de Williams, las úlceras en las piernas, las trombosis en las venas profundas o el síndrome de Wolff-Parkinson-White.
Se prefieren la hipertrofia cardíaca, la enfermedad cardíaca congénita que afecta a los adultos, los aneurismas, la angina, la angina de pecho, las arritmias, las enfermedades cardiovasculares, las cardiomiopatías, la insuficiencia cardíaca congestiva, el infarto de miocardio, la hipertensión pulmonar, los desarrollos hipertróficos, la reestenosis, la estenosis, la trombosis o la arteriosclerosis.
Un objeto adicional de la presente divulgación es el uso de los compuestos de fórmula general (I) de acuerdo con la
invención como un medicamento.
Un objeto de la presente divulgación es el uso de los compuestos de fórmula general (I) de acuerdo con la invención para el tratamiento y/o la profilaxis de trastornos, en particular de los trastornos mencionados anteriormente.
Un objeto adicional de la presente divulgación es el uso de los compuestos de fórmula general (I) de acuerdo con la invención para el tratamiento y/o la profilaxis de trastornos hiperproliferativos, enfermedades infecciosas viralmente inducidas y/o de enfermedades cardiovasculares.
Un objeto preferido de la presente divulgación es el uso de los compuestos de fórmula general (I) de acuerdo con la invención en el tratamiento y/o la profilaxis de los carcinomas en los pulmones, especialmente de los carcinomas asociados a las células no pequeñas de los pulmones, de los carcinomas en la próstata, especialmente de los carcinomas en la próstata humana que no dependen de hormonas, de los carcinomas cervicales, que abarcan los carcinomas cervicales humanos multirresistentes, de los carcinomas colorrectales, de los melanomas, de los carcinomas en el ovario o de las leucemias, especialmente de las leucemias mieloides agudas.
Un objeto adicional de la presente invención está relacionado con la posibilidad de usar los compuestos de fórmula general (I) de acuerdo con la invención para usar como un medicamento.
Un objeto adicional de la presente invención son los compuestos de fórmula general (I) de acuerdo con la invención para el uso de tratamiento y/o profilaxis de los trastornos mencionados anteriormente.
Un objeto adicional de la presente invención son los compuestos de fórmula general (I) de acuerdo con la invención para el uso de tratamiento y/o profilaxis de trastornos hiperproliferativos, enfermedades infecciosas inducidas por virus y/o enfermedades cardiovasculares.
Un objeto preferido de la presente invención son los compuestos de fórmula general (I) de acuerdo con la invención para el uso de tratamiento y/o profilaxis de carcinomas de pulmón, especialmente carcinomas de pulmón de células no pequeñas, carcinomas de próstata, especialmente humanos independientes de hormonas carcinomas de próstata, carcinomas cervicales, incluidos carcinomas cervicales humanos resistentes a múltiples fármacos, carcinomas colorrectales, melanomas, carcinomas ováricos o leucemias, especialmente leucemias mieloides agudas
Un objeto adicional de la presente invención está relacionado con la posibilidad de usar los compuestos de fórmula general (I) de acuerdo con la invención en un procedimiento para el tratamiento y/o la profilaxis de los trastornos mencionados anteriormente.
Un objeto adicional de la presente invención son los compuestos de fórmula general (I) de acuerdo con la invención para el uso en un procedimiento para el tratamiento y/o la profilaxis de trastornos hiperproliferativos, enfermedades infecciosas viralmente inducidas y/o de enfermedades cardiovasculares.
Un objeto adicional preferido de la presente invención está relacionado con la posibilidad de usar los compuestos de fórmula general (I) de acuerdo con la invención en un procedimiento para tratar y/o prevenir los carcinomas en los pulmones, especialmente de los carcinomas asociados a las células no pequeñas de los pulmones, de los carcinomas en la próstata, especialmente de los carcinomas en la próstata humana que no dependen de hormonas, de los carcinomas cervicales, que abarcan los carcinomas cervicales humanos multirresistentes, de los carcinomas colorrectales, de los melanomas, de los carcinomas en el ovario o de las leucemias, especialmente de las leucemias mieloides agudas.
Un objeto adicional de la presente invención es el uso de los compuestos de fórmula general (I) de acuerdo con la invención en la fabricación de un medicamento para el tratamiento y/o profilaxis de trastornos, en particular los trastornos mencionados anteriormente.
Un objeto de la presente invención es el uso de los compuestos de fórmula general (I) de acuerdo con la invención en la producción de un medicamento para el tratamiento y/o la profilaxis de trastornos hiperproliferativos, enfermedades infecciosas viralmente inducidas y/o de enfermedades cardiovasculares.
Un objeto preferido de la presente invención son los compuestos de fórmula general (I) de acuerdo con la invención para el uso en un procedimiento para el tratamiento y/o la profilaxis de carcinomas de pulmón, especialmente carcinomas de pulmón a células no pequeñas, carcinomas de próstata, especialmente carcinomas de próstata humano independiente de hormonas, carcinomas cervicales, que incluye carcinomas cervicales humanos resistentes a multifármacos, carcinomas colorrectales, melanomas, carcinomas de ovario o leucemias, especialmente leucemia mieloide aguda.
Un objeto de la presente divulgación es un procedimiento para el tratamiento y/o la profilaxis de trastornos, en particular los trastornos mencionados anteriormente, usando una cantidad efectiva de los compuestos de fórmula general (I) de acuerdo con la invención.
Un objeto de la presente divulgación es un procedimiento para el tratamiento y/o la profilaxis de trastornos hiperproliferativos, enfermedades infecciosas viralmente inducidas y/o de enfermedades cardiovasculares, usando
una cantidad efectiva de los compuestos de fórmula general (I) de acuerdo con la invención.
Un objeto preferido de la presente divulgación es un procedimiento para el tratamiento y/o la profilaxis de carcinomas de pulmón, especialmente carcinomas de pulmón a células no pequeñas, carcinomas de próstata, especialmente carcinomas de próstata humano independiente de hormonas, carcinomas cervicales, que incluye carcinomas cervicales humanos resistentes a multifármacos, carcinomas colorrectales, melanomas, carcinomas de ovario o leucemias, especialmente leucemia mieloide aguda mediante el uso de una cantidad eficaz de los compuestos de fórmula general (I) de acuerdo con la invención.
Otro aspecto de la presente invención se refiere a combinaciones farmacéuticas que comprenden un compuesto de acuerdo con la invención, que presenta la fórmula general (I), junto con al menos uno o más ingredientes activos adicionales.
Como se usa en el presente documento, la expresión "combinación farmacéutica" hace referencia a una combinación de al menos un compuesto de acuerdo con la invención, que presenta la fórmula general (I) y que cumple la función de un ingrediente activo, con al menos otro ingrediente activo, con ingredientes adicionales, vehículos, diluyentes y/o disolventes o sin ellos.
Otro aspecto de la presente invención se refiere a composiciones farmacéuticas que comprenden un compuesto de acuerdo con la invención, que presenta la fórmula general (I), en combinación con un coadyuvante inerte, no tóxico y farmacéuticamente aceptable.
Como se usa en el presente documento, la expresión "composición farmacéutica" hace referencia a una formulación galénica que comprende al menos un agente con actividad farmacéutica combinado con al menos un ingrediente adicional, un vehículo, un diluyente y/o un disolvente.
Otro aspecto de la presente invención se refiere al uso de las combinaciones farmacéuticas y/o las composiciones farmacéuticas de acuerdo con la invención en el tratamiento y/o la profilaxis de diversos trastornos, particularmente de los trastornos mencionados anteriormente.
Otro aspecto de la presente invención está relacionado con el uso de las combinaciones farmacéuticas y/o las composiciones farmacéuticas de acuerdo con la invención en el tratamiento y/o la profilaxis de los carcinomas en los pulmones, especialmente de los carcinomas asociados a las células no pequeñas de los pulmones, de los carcinomas en la próstata, especialmente de los carcinomas en la próstata humana que no dependen de hormonas, de los carcinomas cervicales, que abarcan los carcinomas cervicales humanos multirresistentes, de los carcinomas colorrectales, de los melanomas, de los carcinomas en el ovario o de las leucemias, especialmente de las leucemias mieloides agudas.
Otro aspecto de la presente invención está relacionado con la posibilidad de usar las combinaciones farmacéuticas y/o las composiciones farmacéuticas de acuerdo con la invención para el uso del tratamiento y/o la profilaxis de una variedad de trastornos, particularmente de los trastornos mencionados anteriormente.
Otro aspecto de la presente invención está relacionado con la posibilidad de usar las combinaciones farmacéuticas y/o las composiciones farmacéuticas de acuerdo con la invención para el uso del tratamiento y/o la profilaxis de los carcinomas en los pulmones, especialmente de los carcinomas asociados a las células no pequeñas de los pulmones, de los carcinomas en la próstata, especialmente de los carcinomas en la próstata humana que no dependen de hormonas, de los carcinomas cervicales, que abarcan los carcinomas cervicales humanos multirresistentes, de los carcinomas colorrectales, de los melanomas, de los carcinomas en el ovario o de las leucemias, especialmente de las leucemias mieloides agudas.
Los compuestos de fórmula (I) pueden administrarse en forma de agentes farmacéuticos individuales o en combinación con uno o más agentes terapéuticos adicionales, de manera tal que la combinación no provoque efectos colaterales inaceptables. Esta combinación farmacéutica puede comprender una sola forma de dosificación con un compuesto de fórmula (I) y uno o más agentes terapéuticos adicionales. Como alternativa, el compuesto de fórmula (I) y cada agente terapéutico adicional pueden encontrarse en su propia forma de dosificación. Por ejemplo, el compuesto de fórmula (I) y el agente terapéutico adicional pueden administrársele al paciente en una sola forma de dosificación oral, que puede ser una tableta o una cápsula. Como alternativa, cada agente puede administrarse en su propia forma de dosificación.
Cuando se empleen formas de dosificación separadas, el compuesto de fórmula (I) y los uno o más agentes terapéuticos adicionales podrán administrarse de una manera esencialmente simultánea (es decir, al mismo tiempo) o en momentos separados (por ejemplo, de manera consecutiva).
En particular, los compuestos de la presente invención pueden usarse en combinaciones únicas o separadas con otros agentes antitumorales, que pueden ser agentes alquilantes, antimetabolitos, agentes antitumorales de origen vegetal, agentes que pueden usarse en terapias hormonales, inhibidores de la topoisomerasa, derivados de la camptotecina, inhibidores de quinasas, fármacos dirigidas, anticuerpos, interferones, modificadores de las respuestas biológicas, compuestos antiangiogénicos u otros fármacos antitumorales. Con relación a lo anterior, a continuación, se provee
una lista no limitativa de diversos ejemplos de agentes secundarios que pueden usarse en combinación con los compuestos de la presente invención:
• Los agentes alquilantes incluyen, pero sin limitación, los N-óxidos de mostazas de nitrógeno, la ciclofosfamida, la ifosfamida, el tiotepa, la ranimustina, la nimustina, la temozolomida, la altretamina, la apaziquona, la brostalicina, la bendamustina, la carmustina, la estramustina, la fotemustina, la glufosfamida, la mafosfamida, la bendamustina y el mitolactol; los compuestos alquilantes coordinados con platino incluyen, pero sin limitación, cisplatino, carboplatino, eptaplatino, lobaplatino, nedaplatino, oxaliplatino y satraplatino;
• Los antimetabolitos incluyen, pero sin limitación, el metotrexato, los ribósidos de 6-mercaptopurinas, la mercaptopurina, el 5-fluorouracilo, que puede usarse solo o en combinación con leucovorina, el tegafur, la doxifluridina, el carmofur, la citarabina, el octofosfato de citarabina, la enocitabina, la gemcitabina, la fludarabina, la 5-azacitidina, la capecitabina, la cladribina, la clofarabina, la decitabina, la eflornitina, la etinilcitidina, el arabinósido de citosina, la hidroxiurea, el melfalán, la nelarabina, el nolatrexed, la ocfosfita, el premetrexed de disodio, la pentostatina, el pelitrexol, el raltitrexed, la triapina, el trimetrexato, la vidarabina, la vincristina y la vinorelbina;
• Los agentes que pueden usarse en las terapias hormonales incluyen, pero sin limitación, el exemestano, el lupron, el anastrozol, el doxercalciferol, el fadrozol, el formestano, los inhibidores de la 11-beta hidroxiesteroide deshidrogenasa 1, los inhibidores de la 17-alfa hidroxilasa/17,20 liasa, tales como el acetato de abiraterona, los inhibidores de la 5-alfa reductasa, tales como el finasteride y el epristeride, los antiestrógenos, tales como el citrato de tamoxifeno y el fulvestrant, el trelstar, el toremifeno, el raloxifeno, el lasofoxifeno, el letrozol, los antiandrógenos, tales como la bicalutamida, la flutamida, la mifepristona, la nilutamida y el casodex, las antiprogesteronas y las combinaciones de éstos;
• Las sustancias antitumorales de origen vegetal incluyen, por ejemplo, los inhibidores de la mitosis, por ejemplo, las epotilonas, tales como la sagopilona, la ixabepilona o la epotilona B, la vinblastina, la vinflunina, el docetaxel y el paclitaxel;
• Los agentes citotóxicos inhibidores de la topoisomerasa incluyen, pero sin limitaciones, la aclarubicina, la doxorrubicina, el amonafide, el belotecano, la camptotecina, la l0-hidroxicamptotecina, la 9-aminocamptotecina, el diflomotecano, el irinotecano, el topotecano, la edotecarina, la epimbicina, el etopósido, el exatecano, el gimatecano, el lurtotecano, la mitoxantrona, la pirambicina, la pixantrona, el rubitecano, el sobuzoxano, el taflupósido y las combinaciones de éstos;
• Los inmunológicos incluyen interferones como interferón alfa, interferón alfa-2a, interferón alfa-2b, interferón beta, interferón gamma-la e interferón gamma-nl, y otros agentes potenciadores inmunes como L19-IL2 y otros derivados de IL2, filgrastim, lentinan, sizofilan, TheraCys, ubenimex, aldesleukin, alemtuzumab, BAM-002, dacarbazine, daclizumab, denileukin, gemtuzumab, ozogamicin, ibritumomab, imiquimod, lenograstim, lentinan, vacuna contra el melanoma (Corixa), molgramostim, sargramostim, tasonermina, tecleukina, timilalasina, tositumomab, vimlizina, epratuzumab, mitumomab, oregovomab, pemtumomab y Provenge; vacuna contra el melanoma de Merial;
• Los modificadores de las respuestas biológicas abarcan aquellos agentes con los cuales pueden modificarse los mecanismos de defensa de los organismos vivos y aquellos agentes con los cuales pueden modificarse parámetros biológicos como la supervivencia, el crecimiento o la diferenciación de las células que constituyen los tejidos, en cuyo caso pueden conferirles actividad antitumoral. Estos agentes incluyen, por ejemplo, la krestina, el lentinano, el sizofirano, el picibanil, ProMune y el ubenimex;
• Los compuestos antiangiogénicos incluyen, pero sin limitaciones, la acitretina, el aflibercept, la angiostatina, la aplidina, el asentar, el axitinib, la recentina, el bevacizumab, el brivanib, el alaninat, el cilengtide, la combretastatina, DAST, la endostatina, el fenretinide, la halofuginona, el pazopanib, el ranibizumab, el rebimastat, el removab, el revlimid, el sorafenib, el vatalanib, la escualamina, el sunitinib, el telatinib, la talidomida, la ukraína y la vitaxina;
• Los anticuerpos incluyen, pero sin limitaciones, el trastuzumab, el cetuximab, el bevacizumab, el rituximab, el ticilimumab, el ipilimumab, el lumiliximab, el catumaxomab, el atacicept, el oregovomab y el alemtuzumab;
• Los inhibidores del VEGF tales como, por ejemplo, el sorafenib, DAST, el bevacizumab, el sunitinib, la recentina, el axitinib, el aflibercept, el telatinib, el brivanib, el alaninato, el vatalanib, el pazopanib, el ranibizumab y Palladia.
• Los inhibidores del EGFR (HERI) tales como, por ejemplo, el cetuximab, el panitumumab, el vectibix, el gefitinib, el erlotinib y Zactima;
• Los inhibidores de HER2 tales como, por ejemplo, el lapatinib, el tratuzumab y el pertuzumab;
• Los inhibidores de mTOR tales como, por ejemplo, el temsirolimus, el sirolimus, la rapamicina y el everolimus; • Inhibidores de c-Met;
• inhibidores de PI3K y AKT;
• inhibidores de CDK como roscovitina y flavopiridol;
• Inhibidores de los puntos de verificación del ensamblaje del huso y agentes antimitóticos dirigidos, como los inhibidores de PLK, los inhibidores de Aurora (por ejemplo, Hesperadin), los inhibidores de la quinasa de punto de control y los inhibidores de KSP;
• inhibidores de HDAC tales como, por ejemplo, panobinostat, vorinostat, MS275, belinostat y LBH589;
• inhibidores HSP90 y HSP70;
• Inhibidores de proteasoma como bortezomib y carfilzomib;
• Inhibidores de serina/treonina quinasa, incluidos inhibidores de MEK (como, por ejemplo, RDEA 119) e inhibidores de Raf como sorafenib;
• Inhibidores de la farnesil transferasa como, por ejemplo, tipifarnib;
• Inhibidores de la tirosina quinasa que incluyen, p. Ej., Dasatinib, nilotibib, DAST, bosutinib, sorafenib, bevacizumab, sunitinib, AZD2171, axitinib, aflibercept, telatinib, imatinib mesilato, brivanib alaninato, pazopanib, ranibizumab, ceta, cebra, ceta, veib, ceta erlotinib, lapatinib, tratuzumab, pertuzumab e inhibidores de c-Kit; Palladia, masitinib • Agonistas del receptor de vitamina D;
• inhibidores de la proteína Bcl-2, como obatoclax, oblimersen sódico y gosipol;
• Grupo de antagonistas del receptor de diferenciación 20 tales como, por ejemplo, rituximab;
• inhibidores de ribonucleótido reductasa tales como, por ejemplo, gemcitabina;
• Agonistas del receptor 1 del ligando inductor de la apoctosis de necrosis tumoral tales como, por ejemplo, mapatumumab;
• Antagonistas del receptor de 5-hidroxitriptamina tales como, por ejemplo, rEV598, xaliprode, clorhidrato de palonosetrón, granisetrón, Zindol y AB-1001;
• Inhibidores de la integridad, incluidos los inhibidores de la integrina alfa5-beta1 como, por ejemplo, E7820, JSM 6425, volociximab y endostatina;
• Antagonistas de los receptores de andrógenos que incluyen, por ejemplo, decanoato de nandrolona, fluoximesterona, Android, Prost-aid, andromustina, bicalutamida, flutamida, apo-ciproterona, apo-flutamida, acetato de clormadinona, Androcur, Tabi, acetato de ciproterona y nilutamida;
• Inhibidores de aromatasa como, por ejemplo, anastrozol, letrozol, testolactona, exemestano, amino-glutetimida y formestano;
• Inhibidores de la metaloproteinasa de la matriz;
• Otros agentes anticancerosos incluyen, por ejemplo, la alitretinoina, el ampligén, el atrasentán, el bexaroteno, el bortezomib, el bosentán, el calcitriol, el exisulind, la fotemustina, el ácido ibandrónico, la miltefosina, la mitoxantrona, la I-asparraginasa, la procarbazina, la dacarbazina, la hidroxicarbamida, la pegaspargasa, la pentostatina, el tazaroteno, el velcade, el nitrato de galio, la canfosfamida, la darinaparsina y la tretinoína.
Los compuestos de la presente invención también pueden cemplearse en combinación con una terapia con radiación y/o con diversas intervenciones quirúrgicas.
Generalmente, el uso de agentes citotóxicos y/o citostáticos en combinación con un compuesto o composición de la presente invención servirá para:
(1) producir una mejor eficacia para reducir el crecimiento de un tumor o incluso eliminar el tumor en comparación con la administración de cualquier agente solo,
(2) proporcionar la administración de cantidades menores de los agentes quimioterapéuticos administrados, (3) proporcionar un tratamiento quimioterapéutico que sea bien tolerado en el paciente con menos complicaciones farmacológicas perjudiciales que las observadas con quimioterapias de agente único y ciertas otras terapias combinadas,
(4) proporcionar el tratamiento de un espectro más amplio de diferentes tipos de cáncer en mamíferos, especialmente seres humanos,
(5) proporcionar una mayor tasa de respuesta entre los pacientes tratados,
(6) proporcionar un mayor tiempo de supervivencia entre los pacientes tratados en comparación con los tratamientos de quimioterapia convencionales,
(7) proporcionar un tiempo más largo para la progresión del tumor, y/o
(8) producir resultados de eficacia y tolerabilidad al menos tan buenos como los de los agentes usados solos, en comparación con los casos conocidos en los que otras combinaciones de agentes cancerígenos producen efectos antagónicos.
Además, los compuestos de fórmula (I) pueden usarse, por sí solos o en composiciones, en la investigación, en el diagnóstico, como herramientas de referencia en diversos análisis o con fines semejantes bien conocidos en la técnica.
Los compuestos de acuerdo con la invención pueden presentar una acción sistémica y/o local. En este contexto, es posible administrarlos por una vía apropiada, por ejemplo, por vía oral, parenteral, pulmonar, nasal, sublingual, lingual, bucal, rectal, dérmica, transdérmica, ótica o a través de la conjuntiva, o bien en forma de implantes o stents.
Para estas rutas de administración, es posible administrar los compuestos de acuerdo con la invención en formas de aplicación adecuadas.
Son formas de administración adecuadas para la administración oral que funcionan como se describe en la técnica anterior y suministran los compuestos de acuerdo con la invención rápidamente y/o en forma modificada, que comprenden los compuestos de acuerdo con la invención en forma cristalina y/o amorfa y/o disuelta, tales como, por ejemplo, comprimidos (recubiertos o no recubiertos, por ejemplo comprimidos provistos de recubrimientos entéricos o recubrimientos cuya disolución se retrasa o que son insolubles y que controlan la liberación del compuesto de acuerdo con la invención), comprimidos que se descomponen rápidamente en el cavidad oral, o películas/obleas, películas/liofilizados, cápsulas (por ejemplo, cápsulas de gelatina dura o blanda), comprimidos recubiertos de azúcar, gránulos, pellets, polvos, emulsiones, suspensiones, aerosoles o soluciones.
La administración parenteral puede llevarse a cabo evitando una etapa de absorción (por ejemplo, intravenosa, intraarterial, intracardial, intraespinal o intralumbal) o con inclusión de absorción (por ejemplo, intramuscular,
subcutánea, intracutánea, percutánea o intraperitoneal). Las formas de administración adecuadas para la administración parenteral son, entre otras, preparaciones para inyección e infusión en forma de soluciones, suspensiones, emulsiones, liofilizados o polvos estériles.
Ejemplos adecuados para las otras vías de administración son formas farmacéuticas para inhalación (entre otros inhaladores de polvo, nebulizadores), gotas/soluciones/aerosoles nasales; tabletas para administrar lingual, sublingual o bucal, películas/obleas o cápsulas, supositorios, preparaciones para los ojos o los oídos, cápsulas vaginales, suspensiones acuosas (lociones, mezclas de agitación), suspensiones lipofílicas, ungüentos, cremas, sistemas terapéuticos transdérmicos (como yesos, por ejemplo), leche, pastas, espumas, polvos para espolvoreo, implantes o stents.
Los compuestos según la invención pueden convertirse en las formas de administración indicadas. Esto puede tener lugar de una manera conocida per se mezclando con adyuvantes inertes, no tóxicos, farmacéuticamente adecuados. Estos adyuvantes incluyen, entre otros, vehículos (por ejemplo, celulosa microcristalina, lactosa, manitol), disolventes (por ejemplo, polietilenglicoles líquidos), emulsionantes y dispersantes o agentes humectantes (por ejemplo, dodecilsulfato de sodio, oleato de polioxisorbitan), aglutinantes (por ejemplo, polivinilpirrolidona ), polímeros sintéticos y naturales (por ejemplo, albúmina), estabilizadores (por ejemplo, antioxidantes, como por ejemplo, ácido ascórbico), colorantes (por ejemplo, pigmentos inorgánicos, como por ejemplo, óxidos de hierro) y saborizantes y/o agentes para enmascarar olores.
La presente invención además proporciona medicamentos que comprenden al menos un compuesto de acuerdo con la invención, generalmente junto con uno o más adyuvantes farmacéuticamente adecuados, inertes, no tóxicos, y su uso para los fines mencionados anteriormente.
Cuando los compuestos de la presente invención se administran como productos farmacéuticos, a seres humanos o animales, se pueden administrar per se o como una composición farmacéutica que contiene, por ejemplo, del 0,1 % al 99,5 % (más preferentemente del 0,5 % al 90 %) de principio activo en combinación con uno o más adyuvantes inertes, no tóxicos, farmacéuticamente adecuados.
Independientemente de la ruta de administración seleccionada, los compuestos de acuerdo con la invención de fórmula general (I) y/o la composición farmacéutica de la presente invención se formulan en formas de dosificación farmacéuticamente aceptables por métodos convencionales conocidos por los expertos en la materia.
Los niveles de dosificación reales y el curso temporal de la administración de los principios activos en las composiciones farmacéuticas de la invención pueden variarse para obtener una cantidad del ingrediente activo que sea eficaz para lograr la respuesta terapéutica deseada para un paciente particular sin ser tóxico para el paciente.
Materiales y procedimientos
Los datos porcentuales en las siguientes pruebas y los ejemplos son porcentajes en peso, a menos que se indique lo contrario; partes son partes en peso. Las proporciones de disolventes, las proporciones de dilución y los datos de concentración de las soluciones líquido/líquido se basan en cada caso en el volumen.
Los compuestos fueron analizados en ensayos biológicos y/o fisicoquímicos específicos, una o más veces. Cuando se prueban más de una vez, los datos se informan como valores promedio o como valores medios, en los que
• El valor promedio, que también se conoce como la media aritmética, es la suma de los valores obtenidos, dividida por la cantidad de análisis realizados.
• El valor de la mediana es el valor del medio en el grupo de los valores ordenados de manera ascendente o descendente. Si la cantidad de valores en el conjunto de datos es impar, la mediana es el valor del medio. Si la cantidad de valores en el conjunto de datos es par, la mediana es la media aritmética de los dos valores del medio.
Los compuestos de los ejemplos se sintetizaron una o más veces. Cuando se sintetizaron más de una vez, los datos de los ensayos biológicos o fisicoquímicos son los valores promedio o los valores de la mediana, que se calcularon sobre la base de los datos que se obtuvieron cuando se analizó cada lote de compuestos.
Las propiedades farmacológicas, farmacocinéticas y fisicoquímicas de los compuestos pueden determinarse in vitro de acuerdo con los siguientes ensayos y procedimientos.
Cabe destacar que, en los ensayos de la CDK9 que se describen a continuación, la capacidad de resolución está limitada por la concentración de las enzimas, y que el límite inferior para la CI50 es de aproximadamente 1-2 nM en el análisis de la CDK9 en presencia de una concentración alta de ATP y de 2-4 nM en el análisis de las CDK en presencia de una concentración baja de ATP. En el caso de los compuestos que presentan una CI50 que se encuentra dentro del intervalo preferido, la afinidad verdadera por la CDK9 y, por lo tanto, la selectividad por la CDK9 sobre la CDK2, pueden ser incluso más alta, es decir, el valor calculado para la selectividad de estos compuestos, que se detalla en las columnas 4 y 7 de la tabla 2 más adelante, es un valor mínimo, y la selectividad real puede ser mayor.
1a. Ensayo quinasa CDK9/ciclina T1
La actividad de inhibición de la CDK9/ciclina T1 de los compuestos de la presente invención se determinó usando el ensayo basado en la TR-FRET que se describe a continuación.
Se adquirió una CDK9 y una ciclina T1 humanas completas y marcadas con His, que habían sido expresadas en células de insectos y purificadas por medio de una cromatografía de afinidad con Ni-NTA, en Invitrogen (cat. n.° PV4131). Como sustrato para la reacción con la quinasa, se usó el péptido biotinilado biotina-Ttds-YISPLKSPYKISEG (con el extremo C en forma de amida), que puede adquirirse, por ejemplo, en la compañía JERINI Peptide Technologies (Berlín, Alemania).En el ensayo, con una pipeta se colocaron 5o nl de una solución concentrada 100 veces del compuesto de prueba en DMSO en una placa de microtitulación de 384 cavidades con volúmenes reducidos (de Greiner Bio-One, Frickenhausen, Alemania), donde también se agregaron 2 pl de una solución del complejo de la CDK9 y la ciclina T1 en un amortiguador acuoso para el ensayo (que comprendió Tris/HCl 50 mM, pH 8,0, MgCh 10 mM, ditiotreitol 1,0 mM, ortovanadato de sodio 0,1 mM y 0,01 % (volumen en volumen) de Nonidet-P40, de Sigma). La mezcla se incubó a 22 °C durante 15 minutos para permitir que ocurriera la unión preliminar entre los compuestos de prueba y la enzima, antes del inicio de la reacción con la quinasa. Después, la reacción con la quinasa se inició agregando 3 pl de una solución de trifosfato de adenosina (ATP 16,7 pM, lo que, en el volumen del ensayo, 5 pl, representó una concentración de 10 pM) y el sustrato (1,25 pM, lo que, en el volumen del ensayo, 5 pl, representó una concentración de 0,75 pM) en el amortiguador del ensayo. La mezcla resultante se incubó a una temperatura de 22 °C durante un período de 25 minutos. La concentración del complejo de la CDK9 y la ciclina T1 se ajustó sobre la base de la actividad del lote de enzimas, y fue apropiada para mantener un intervalo lineal en el ensayo. Las concentraciones típicas fueron de aproximadamente 1 pg/ml. La reacción se detuvo agregando 5 pl de una solución de los reactivos de terminación para la TR-FRET (estreptavidina-XL665 0,2 pM, de Cisbio Bioassays, Codolet, Francia, un anticuerpo anti-RB(pSer807/pSer811) 1 nM, de BD Pharmingen, n.° 558389, y un anticuerpo anti-IgG de ratón marcado con LANCE EU-W1024 1,2 nM, de Perkin-Elmer, producto n.° AD0077) en una solución acuosa de EDTA (con EDTA 100 mM y 0,2 % (p/v) de albúmina de suero bovino en HEPES 100 mM, pH 7,5).
La mezcla resultante se incubó a 22 °C durante 1 hora para permitir que se formara un complejo con el péptido biotinilado fosforilado y los reactivos de terminación. Después, la cantidad del sustrato fosforilado se determinó midiendo la transferencia de energía de resonancia desde el quelado con Eu hasta la estreptavidina-XL. Se midieron las emisiones de fluorescencia a 620 nm y a 665 nm después de aplicar una excitación a 350 nm en un lector de TR-FRET, por ejemplo, en un lector Rubystar (de BMG Labtechnologies, Offenburg, Alemania) o en un lector Viewlux (de Perkin-Elmer). Se tomó la proporción entre las emisiones a 665 nm y a 622 nm como la medida de la cantidad del sustrato fosforilado. Los datos fueron sometidos a una normalización sobre la base de la reacción que comprendió las enzimas, pero no los inhibidores, que representó una inhibición de 0 %, y sobre la base de la reacción que comprendió todos los otros componentes, pero no la enzima, que representó una inhibición de 100 %. Usualmente, los compuestos de prueba fueron analizados en la misma placa de microtitulación, en 11 concentraciones diferentes en el intervalo de entre 20 pM y 0,1 nM (20 pM, 5,9 pM, 1,7 pM, 0,51 pM, 0,15 pM, 44 nM, 13 nM, 3,8 nM, 1,1 nM, 0,33 nM y 0,1 nM; la serie de diluciones fue preparada por separado, antes del ensayo, con soluciones concentradas 100 veces en DMSO, a través de diluciones en serie de 1 en 3,4), con cada concentración por duplicado. Los valores de la CI50 se determinaron a través de un ajuste de 4 parámetros usando software local.
1b. Ensayo quinasa CDK9/CycT1 en alto ATP
Se cuantificó la actividad inhibitoria de CDK9/CycT1 de los compuestos de la presente invención en una concentración alta de ATP luego de la preincubación de la enzima y compuestos de prueba empleando el ensayo de TR-FRET para CDK9/CycT1 como se describe en los siguientes párrafos.
Se compraron CDK9 y CycT1 humanas de longitud completa recombinantes marcadas con His, expresadas en células de insecto y purificadas por cromatografía de afinidad a Ni-NTA, en Invitrogen (Cat. No PV4131). Como sustrato para la reacción de la quinasa se usó el péptido biotinilado biotin-Ttds-YISPLKSPYKISEG (extremo C terminal forma amida) el cual se adquirió, por ejemplo, de la compañía JERINI peptide technologies (Berlín, Alemania). Para el ensayo se cargaron con pipeta 50 nl de una solución concentrada 100 veces del compuesto de prueba en DMSO en una placa para microtitulación de 384 cavidades de volumen reducido negras (Greiner Bio-One, Frickenhausen, Alemania), se agregaron 2 pl de una solución de CDK9/CycT1 en solución amortiguadora de ensayo acuosa [Tris/HCl 50 mM pH 8,0, MgCl2 10 mM, ditiotreitol 1,0 mM, orto-vanadato de sodio 1,0 mM, Nonidet-P40 0,01 % (volumen en volumen) (Sigma)] y la mezcla se incubó durante 15 minutos a 22°C para permitir la unión preliminar de los compuestos de prueba a la enzima antes del inicio de la reacción de la quinasa. Luego se inició la reacción de la quinasa por el agregado de 3 pl de una solución de adenosina-tri-fosfato (ATP, 3,3 mM, lo que, en el volumen del ensayo, 5 pl, representó una concentración de 2 mM) y sustrato (1,67 pM, lo que en el volumen del ensayo, 5 pl, representó una concentración de 1 pM) en la solución amortiguadora de ensayo y se incubó la mezcla resultante durante un tiempo de reacción de 25 minutos a 22°C. La concentración de CDK9/CycT1 se ajustó dependiendo de la actividad del lote de enzima y se eligió apropiada para tener el ensayo en el intervalo lineal, las concentraciones típicas estuvieron en el intervalo de 0,5 pg/ml. La reacción se detuvo por el agregado de 5 pl de una solución de reactivos de detección de TR-FRET (estreptavidina-XL665 0,2 pM [Cisbio Bioassays, Codolet, Francia] y anticuerpo anti-RB(pSer807/pSer811) 1 nM de BD Pharmingen [# 558389] y anticuerpo anti-IgG de ratón marcado con LANCE EU-W1024 1,2 nM [Perkin-Elmer, no. de producto AD0077]) en una solución acuosa de EDTA (EDTA 100 mM, albúmina sérica bovina 0,2% (peso en volumen) en 100 mM h Ep ES pH 7,5).
La mezcla resultante se incubó 1 hora a 22°C para permitir la formación de complejo entre el péptido biotinilado fosforilado y los reactivos de detección. Posteriormente la cantidad de sustrato fosforilado se evaluó por medida de la transferencia de energía de resonancia del Quelado con Eu a la estreptavidina-XL. Por lo tanto, las emisiones de
fluorescencia a 620 nm y 665 nm luego de la excitación a 350 nm se midieron en un lector de HTRF, por ejemplo, un Rubystar (BMG Labtechnologies, Offenburg, Alemania) o un Viewlux (Perkin-Elmer). La relación de las emisiones a 665 nm y a 622 nm se tomó como la medida para la cantidad de sustrato fosforilado. Los datos se normalizaron (reacción enzimática sin inhibidor = 0 % de inhibición, todos los otros componentes del ensayo, pero sin enzima = 100 % de inhibición). Usualmente los compuestos de prueba se ensayaron en la misma placa para microtitulación en 11 concentraciones diferentes en el intervalo entre 20 j M y 0,1 nM (20 j M, 5,9 j M, 1,7 j M, 0,51 j M, 0,15 j M, 44 nM, 13 nM, 3,8 nM, 1,1 nM, 0,33 nM y 0,1 nM, series de diluciones preparadas separadamente antes del ensayo en el nivel de soluciones concentradas 100 veces en DMSO por diluciones seriales de 1:3,4) en valores duplicados para cada concentración y se calcularon los valores de CI50 por ajuste de 4 parámetros usando un programa de computación propio.
2a. Ensayo quinasa CDK2/ciclina E
La actividad de inhibición de la CDK2/ciclina E de los compuestos de la presente invención se determinó usando el ensayo basado en la TR-FRET que se describe en los siguientes párrafos:
Se adquirieron proteínas de fusión recombinantes que comprendían la GST y la CDK2 humana y la GST y la ciclina E humana, que habían sido expresadas en células de insectos (Sf9) y purificadas por medio de una cromatografía de afinidad con glutatión y sefarosa, en ProQinase GmbH (Friburgo, Alemania). Como sustrato para la reacción con la quinasa, se usó el péptido biotinilado biotina-Ttds-YISPLKSPYKISEG (con el extremo C en forma de amida), que puede adquirirse, por ejemplo, en la compañía JERINI Peptide Technologies (Berlín, Alemania).
En el ensayo, con una pipeta se colocaron 50 nl de una solución concentrada 100 veces del compuesto de prueba en DMSO en una placa de microtitulación de 384 cavidades con volúmenes reducidos (de Greiner Bio-One, Frickenhausen, Alemania), donde también se agregaron 2 jil de una solución del complejo de la CDK2 y la ciclina E en un amortiguador acuoso para el ensayo (que comprendió Tris/HCl 50 mM, pH 8,0, MgCh 10 mM, ditiotreitol 1,0 mM, ortovanadato de sodio 0,1 mM y 0,01 % (volumen en volumen) de Nonidet-P40, de Sigma). La mezcla se incubó a 22 °C durante 15 minutos para permitir que ocurriera la unión preliminar entre los compuestos de prueba y la enzima, antes del inicio de la reacción con la quinasa. Después, la reacción con la quinasa se inició agregando 3 jil de una solución de trifosfato de adenosina (ATP 16,7 j M, lo que, en el volumen del ensayo, 5 jl, representó una concentración de 10 j M) y el sustrato (1,25 j M, lo que, en el volumen del ensayo, 5 jl, representó una concentración de 0,75 j M) en el amortiguador del ensayo. La mezcla resultante se incubó a una temperatura de 22 °C durante un período de 25 minutos. La concentración del complejo de la CDK2 y la ciclina E se ajustó sobre la base de la actividad del lote de enzimas, y fue apropiada para mantener un intervalo lineal en el ensayo. Las concentraciones típicas fueron de aproximadamente 130 ng/ml. La reacción se detuvo agregando 5 j l de una solución de los reactivos de terminación para la TR-FRET (estreptavidina-XL665 0,2 j M, de Cisbio Bioassays, Codolet, Francia, un anticuerpo anti-RB(pSer807/pSer811) 1 nM, de BD Pharmingen, n.° 558389, y un anticuerpo anti-IgG de ratón marcado con LANCE EU-W1024 1,2 nM, de Perkin-Elmer, producto n.° AD0077) en una solución acuosa de EDTA (con EDTA 100 mM y 0,2 % (p/v) de albúmina de suero bovino en HEPES 100 mM, pH 7,5). La mezcla resultante se incubó a 22 °C durante 1 hora para permitir que se formara un complejo con el péptido biotinilado fosforilado y los reactivos de terminación. Después, la cantidad del sustrato fosforilado se determinó midiendo la transferencia de energía de resonancia desde el quelado con Eu hasta la estreptavidina-XL. Se midieron las emisiones de fluorescencia a 620 nm y a 665 nm después de aplicar una excitación a 350 nm en un lector de TR-FRET, por ejemplo, en un lector Rubystar (de BMG Labtechnologies, Offenburg, Alemania) o en un lector Viewlux (de Perkin-Elmer). Se tomó la proporción entre las emisiones a 665 nm y a 622 nm como la medida de la cantidad del sustrato fosforilado. Los datos fueron sometidos a una normalización sobre la base de la reacción que comprendió las enzimas, pero no los inhibidores, que representó una inhibición de 0 %, y sobre la base de la reacción que comprendió todos los otros componentes, pero no la enzima, que representó una inhibición de 100 %. Usualmente, los compuestos de prueba fueron analizados en la misma placa de microtitulación, en 11 concentraciones diferentes en el intervalo de entre 20 j M y 0,1 nM (20 j M, 5,9 j M, 1,7 j M, 0,51 j M, 0,15 j M, 44 nM, 13 nM, 3,8 nM, 1,1 nM, 0,33 nM y 0,1 nM; la serie de diluciones fue preparada por separado, antes del ensayo, con soluciones concentradas 100 veces en DMSO, a través de diluciones en serie de 1 en 3,4), con cada concentración por duplicado. Los valores de la CI50 se determinaron a través de un ajuste de 4 parámetros usando software local.
2b. Ensayo quinasa CDK2/CycE en alto ATP:
Se cuantificó la actividad inhibitoria de CDK2/CycE de compuestos de la presente invención en adenosina-tri-fosfato (ATP) 2 mM empleando el ensayo de TR-FRET para CDK2/CycE (TR-FRET = transferencia de energía de fluorescencia con resolución temporal) como se describe en los siguientes párrafos.
Se compraron proteínas de fusión recombinantes de GST y CDK2 humana y de GST y CycE humana, expresadas en células de insecto (Sf9) y purificadas por cromatografía de afinidad con glutatión-sefarosa, de ProQinase GmbH (Freiburg, Alemania). Como sustrato para la reacción de la quinasa se usó el péptido biotinilado biotin-Ttds-YISPLKSPYKISEG (extremo C terminal en forma de amida) que pudo adquirirse, por ejemplo, de la compañía JERINI peptide technologies (Berlín, Alemania).
Para el ensayo, con una pipeta se colocaron 50 nl de una solución concentrada 100 veces del compuesto de prueba en DMSO en una placa de microtitulación de 384 cavidades con volúmenes reducidos (de Greiner Bio-One, Frickenhausen, Alemania), donde también se agregaron 2 j l de una solución de CDK2/CycE en un amortiguador acuoso para el ensayo (que comprendió Tris/HCl 50 mM, pH 8,0, MgCh 10 mM, ditiotreitol 1,0 mM, ortovanadato de
sodio 0,1 mM y 0,01 % (volumen en volumen) de Nonidet-P40, de Sigma) y la mezcla se incubó a 22 °C durante 15 minutos para permitir que ocurriera la unión preliminar entre los compuestos de prueba y la enzima, antes del inicio de la reacción con la quinasa. Después, la reacción con la quinasa se inició agregando 3 j l de una solución ATP (3,33 mM, lo que, en el volumen del ensayo, 5 jl, representó una concentración de 2 jM ) y el sustrato (1,25 jM , lo que, en el volumen del ensayo, 5 jl, representó una concentración de 0,75 jM ) en el amortiguador del ensayo. La mezcla resultante se incubó a una temperatura de 22 °C durante un período de 25 minutos. La concentración del complejo de CDK2/CycE se ajustó en base de la actividad del lote de enzimas, y fue apropiada para mantener un intervalo lineal en el ensayo. Las concentraciones típicas estuvieron en el intervalo de aproximadamente 15 ng/ml. La reacción se detuvo agregando 5 j l de una solución de los reactivos de terminación para la TR-FRET [estreptavidina-XL665 0,2 jM , de Cisbio Bioassays, Codolet, Francia], un anticuerpo anti-RB(pSer807/pSer811) 1 nM, de BD Pharmingen, n.° 558389, y un anticuerpo anti IgG de ratón marcado con LANCE EU-W1024 1,2 nM (Perkin-Elmer, producto n.° AD0077, como alternativa se puede usar el anticuerpo anti-IgG de ratón marcado con Terbio-criptato de Cisbio Bioassays]) en una solución acuosa de EDTA (EDTA 100 mM y 0,2 % (peso en volumen) de albúmina de suero bovino en HEPES 100 mM, pH 7,5).
La mezcla resultante se incubó a 22 °C durante 1 hora para permitir que se formara un complejo con el péptido biotinilado fosforilado y los reactivos de detección. Después, la cantidad del sustrato fosforilado se determinó midiendo la transferencia de energía de resonancia desde el quelado con Eu a la estreptavidina-XL. Se midieron las emisiones de fluorescencia a 620 nm y a 665 nm después de aplicar una excitación a 350 nm en un lector de TR-FRET, por ejemplo, en un lector Rubystar (de BMG Labtechnologies, Offenburg, Alemania) o en un lector Viewlux (de Perkin-Elmer). Se tomó la proporción entre las emisiones a 665 nm y a 622 nm como la medida de la cantidad del sustrato fosforilado. Los datos se normalizaron (reacción enzimática sin inhibidor = 0 % de inhibición, todos los otros componentes del ensayo, pero sin enzima = 100% de inhibición). Usualmente, los compuestos de prueba fueron analizados en la misma placa de microtitulación, en 11 concentraciones diferentes en el intervalo de entre 20 jM y 0,1 nM (20 jM , 5,9 jM , 1,7 jM , 0,51 jM , 0,15 jM , 44 nM, 13 nM, 3,8 nM, 1,1 nM, 0,33 nM y 0,1 nM; la serie de diluciones fue preparada por separado, antes del ensayo, con soluciones concentradas 100 veces en DMSO, a través de diluciones en serie de 1 en 3,4), con cada concentración por duplicado. Los valores de la CI50 se determinaron a través de un ajuste de 4 parámetros usando un programa de computación propio.
3. Ensayo de proliferación:
Las células tumorales cultivadas (HeLa, células tumorales cervicales humanas, ATCC CCL-2; NCI-H460, células de carcinoma de pulmón de células no pequeñas humanas, ATCC HTB-177; DU 145, células de carcinoma de próstata humano independiente de hormona, ATCC HTB-81; HeLa-MaTu-ADR, células de carcinoma cervical humano resistentes a múltiples fármacos, EPO-GmbH Berlín; Caco-2, células de carcinoma colorrectal humano, ATCC HTB-37; B16F10, células de melanoma de ratón, ATCC CRL-6475) se sembraron a una densidad de 3.500 células/pocillo (DU145), 3.000 células/pocillo (HeLa-MaTu-ADR), 1.500 células/pocillo (NCI-H460), 3.000 células/pocillo (HeLa), 2.000 células/pocillo (Caco-2), o 1000 células/pocillo (B16F10) en una placa de multititulación de 96 pocillos en 150 pl de su respectivo medio de crecimiento suplementado con suero de carnero fetal 10 %. Después de 24 horas, las células de una placa (placa de punto cero) se tiñeron con violeta cristal (ver más adelante), se añadieron las sustancias de ensayo en diversas concentraciones ((0 pM, así como en el intervalo de 0,0001 - 10 pM) empleando el dispensador HP. Las células se incubaron durante 4 días en presencia de sustancias de ensayo. La proliferación celular se determinó mediante la tinción de las células con cristal violeta: las células se fijaron mediante la adición de 20 pl/punto de medición de una solución de aldehído glutárico 11 % durante 15 minutos a temperatura ambiente. Después de tres ciclos de lavado de las células fijadas con agua, las placas se secaron a temperatura ambiente. Las células se tiñeron mediante la adición de 100 pl/punto de medición de una solución de cristal violeta 0,1 % DU 145, Caco-2, HeLa (pH 4,5) B16F10, NCI-H460, HeLa-MaTu-ADR. Después de tres ciclos de lavado de las células teñidas con agua, las placas se secaron a temperatura ambiente. El colorante se disolvió mediante la adición de 100 pl por punto de medición de una solución de ácido acético 10%. La extinción se determinó mediante fotometría a una longitud de onda de 595/550/620 nm en función de la intensidad de la coloración, usualmente a 595 nm. El cambio del número de células, en porcentaje, se calculó por normalización de los valores medidos con los valores de extinción de la placa de punto cero (= 0 %) y la extinción de las células no tratadas (0 pM) (= 100 %). Los valores de CI50 (concentración inhibidora al 50 % del efecto máximo) se determinaron por medio de un ajuste de 4 parámetros.
Las células de leucemia mieloide aguda humanas MOLM-13 (DSMZ ACC 554) y A2780, células de carcinoma de ovario humano (ECACC # 93112519) se sembraron a una densidad de 5.000 células/pocillo (13-MOLM), o 3.000 células/pocillo (A2780) en una placa multititulación de 96 pocillos en 150 pl de medio de crecimiento suplementado de suero de carnero fetal 10 %. Después de 24 horas, la viabilidad celular de una placa (placa de punto cero) se determinó con el ensayo de viabilidad luminiscente Cell Titer-Glo (Promega), mientras que se añadió el compuesto de ensayo a los pocillos de las otras placas que emplean dispensador HP (concentraciones finales en el intervalo de 0,0001 -10 pM y los controles de DMSO). La viabilidad celular se evaluó después de la exposición de 72 horas con el ensayo de viabilidad luminiscente Cell Titer-Glo ter Glo-luminiscente (Promega). Los valores de CI50 (concentración inhibidora a 50 % del efecto máximo) se determinaron por medio de un ajuste de 4 parámetros en los datos de medición que se normalizaron con las células tratadas con vehículo (DMSO) (= 100%) y las lecturas de medición se tomaron inmediatamente antes de la exposición compuesto (= 0 %).
4. Ensayo de la solubilidad en el equilibrio en matraces agitados
4a) Determinación con un desempeño elevado de la solubilidad acuosa de un fármaco (100 mmolar en DMSO)
Este análisis con un desempeño elevado para determinar la solubilidad acuosa de los fármacos se basa en:
El procedimiento de Thomas Onofrey y Greg Kazan para evaluar la solubilidad acuosa con un desempeño elevado en placas con 96 cavidades en función del rendimiento y la correlación, http://www.millipore.com/publications.nsf/a73664f9f981af8c852569b9005b4eee/e565516fb76e743585256da30052db 77/$FILE/AN1731EN00.pdf
El ensayo se ejecutó en un formato que estuvo basado en una placa de 96 cavidades. Cada cavidad fue llenada con un único compuesto.
Todos los pasos de tratamiento en pipetas fueron realizados por una plataforma automatizada.
Se concentraron 100 pl de una solución 10mmolar de cada uno de los fármacos en DMSO por medio de una centrifugación al vacío y se llevó a cabo una disolución en 10 pl de DMSO. Se agregaron 990 pl de un amortiguador a base de fosfato con un pH de 6,5. El contenido de DMSO se incrementó hasta 1 %. La placa de titulación con múltiples cavidades se colocó en un agitador y se mezcló durante 24 horas a temperatura ambiente. Se transfirieron 150 pl de la suspensión a una placa de filtración. Una vez completa la filtración, la cual se llevó a cabo en un recolector al vacío, el filtrado fue sometido a diluciones de 1:400 o de 1:8000. Para la calibración, se empleó una segunda placa de microtitulación donde se colocaron 2 0 pl de una solución de cada uno de los fármacos en una concentración de 10 mM en DMSO. Se prepararon dos concentraciones (0,005 pM y 0,0025 mM) por medio de una dilución en una mezcla 1:1 de DMSO y agua y se las empleó para la calibración. La cuantificación del contenido de las placas que contenían el filtrado y de las placas que se usaron para la calibración se llevó a cabo por medio de un procedimiento de HPLC-MS/MS.
Productos químicos:
Preparación de tampón fosfato 0,1 M pH 6,5:
Se disolvieron 61,86 g de NaCl y 39,54 mg de KH2PO4 en agua y se llevó el volumen hasta 1 l. Se sometió la mezcla a una dilución de 1:10 con agua y se ajustó el pH en 6,5 mediante el uso de NaOH.
Materiales:
Placa Millipore MultiScreenHTS-HV de 0, 45 pm.
Las condiciones cromatografías fueron las siguientes:
Columna HPLC: Ascentis Express C182,7 mm 4,6 x 30 mm
Volumen de inyección: 1 pl
Flujo: 1,5ml/min
Fase móvil: gradiente ácido
A: Agua/HCOOH al 0,05 %
B: Acetonitrilo/HCOOH al 0,05 %
0 min ^ A al 95 % B al 5 %
0,75 min ^ A al 5 % B al 95 %
2.75 min ^ A al 5 % B al 95 %
2.76 min ^ A al 95 % B al 5 %
3 min ^ A al 95 % B al 5 %
Las áreas en las que se inyectaron las muestras que se deseaba analizar y las que se emplearon para la calibración se determinaron mediante el uso de programas informáticos apropiados para análisis de espectrometría de masa (Discovery Quant 2.1.3 y Analyst 1.6.1, de AB Sciex). El cálculo del valor de la solubilidad (en mg/l) se realizó con un macro para Excel que había sido desarrollado en el laboratorio.
4b) Determinación termodinámica de la solubilidad en el agua a partir de un polvo
Se determinó la solubilidad termodinámica de compuestos en agua por un procedimiento en matraz con agitación en equilibrio (véase, por ejemplo: E.H. Kerns, L. Di: Drug-like Properties: Concepts, Structure Design and Methods, 276 286, Burlington, MA, Academic Press, 2008). Se preparó una solución saturada del fármaco y la solución se mezcló durante 24 horas para asegurar que se alcanzara el equilibrio. La solución se centrifugó para eliminar la fracción insoluble y se determinó la concentración del compuesto en solución usando una curva de calibración estándar. Para preparar la muestra, se pesaron 2 mg de compuesto sólido en un vial de vidrio de 4 ml. Se agregó 1 ml de amortiguador fosfato pH 6,5. La suspensión se agitó durante 24 horas a temperatura ambiente. Luego se centrifugó la solución. Para preparar la muestra para la calibración estándar, se disolvieron 2 mg de la muestra sólida en 30 ml de acetonitrilo. Luego del sonicado se diluyó la solución con agua hasta 50 ml. Se cuantificaron la muestra y los estándares por HPLC con detección en UV. Para cada muestra se hicieron dos volúmenes de inyección (5 y 50 pl) por triplicado. Se hicieron tres volúmenes de inyección (5 pl, 10 pl y 20 pl) para el estándar.
Condiciones cromatográficas:
Columna de HPLC: Xterra MS C182,5 pm 4,6 x 30 mm
Volumen de inyección: Muestra: 3x5 pl y 3x50 pl
Estándar: 5 pl, 10 pl, 20 pl
Flujo: 1,5ml/min
Fase móvil: gradiente ácido:
A: Agua/TFA al 0,01 %
B: Acetonitrilo/TFA al 0,01 %
0 min A al 95 % B al 5 %
0 - 3 minutos ^ A al 35 % B al 65 %, gradiente lineal
3 - 5 minutos ^ A al 35 % B al 65 %, isocrático
5 - 6 minutos ^ A al 95 % B al 5 %, isocrático
Detector UV: longitud de onda cercana al máximo de absorción (entre 200 y 400 nm) Las áreas de la muestra e inyecciones estándar, así como el cálculo del valor de solubilidad (en mg/l) se determinaron con el uso de un programa de computación para HPLC (Waters Empower 2 FR).
4c) Solubilidad termodinámica en solución amortiguadora de citrato a pH 4
Se determinó la solubilidad termodinámica mediante un procedimiento de matraz agitado en equilibrio [Literatura: Edward H. Kerns and Li Di (2008) Solubility Methods en: Drug-like Properties: Concepts, Estructura Design and Methods, p276-286. Burlington, MA: Academic Press].
Se preparó una solución saturada del fármaco y se mezcló la solución durante 24 h para asegurarse que se ha alcanzado el equilibrio. La solución se centrifugó para eliminar la fracción insoluble y se determinó la concentración del compuesto en solución usando una curva estándar de calibración.
Para preparar la muestra, se pesó 1,5 mg de compuesto sólido en un vial de vidrio de 4 ml. Se agregó 1 ml de solución amortiguadora de citrato a pH 4. Se puso la suspensión en un agitador y se mezcló durante 24 horas a temperatura ambiente. Se centrifugó la solución después de eso. Para preparar la muestra para la calibración estándar, se disolvieron 0,6 mg de muestra sólida en 19 ml de acetonitrilo/agua 1:1. Después de sonicar, se llevó la solución a 20 ml con acetonitrilo/agua 1:1.
Se cuantificaron la muestra y los estándares por HPLC con detección UV. Para cada muestra se hicieron dos volúmenes de inyección (5 y 50 pl) por triplicado. Se hicieron tres volúmenes de inyección (5 pl, 10 pl y 20 pl) para el estándar.
Productos químicos:
Solución amortiguadora de citrato a pH 4 (MERCK Art. 109435; 1 L consistente en 11,768 g de ácido cítrico, 4,480 g de hidróxido de sodio, 1,604 g de cloruro de hidrógeno)
Las condiciones cromatográficas fueron las siguientes:
Columna de HPLC: Xterra MS C182,5 pm 4,6 x 30 mm
Volumen de inyección: Muestra: 3x5 pl y 3x50 pl
Estándar: 5 pl, 10 pl, 20 pl
Flujo: 1,5ml/min
Fase móvil: gradiente ácido:
A: Agua/TFA al 0,01 %
B: Acetonitrilo/TFA al 0,01 %
0 min: A al 95 % B al 5 %
0 - 3 min: A al 35 %  %, gradiente lineal 3 - 5 min: A al 35 % %, isocrático
%, gradiente lineal 3 - 5 min: A al 35 % %, isocrático
5 - 6 min: A al 95 % B al 5 %, isocrático
Detector UV: longitud de onda cerca del máximo de absorción (entre 200 y 400 nm)
Las áreas de la muestra e inyecciones estándar, así como también el cálculo del valor de solubilidad (en mg/l) se determinaron mediante el uso del programa de la HPLC (Waters Empower 2 FR).
Las áreas de la muestra e inyecciones estándar, así como también el cálculo del valor de solubilidad (en mg/l) se determinaron usando el programa de la HPLC (Waters Empower 2 FR).
5. Ensayo de permeación de Caco-2:
Se sembraron células Caco-2 (adquiridas de DSMZ Braunschweig, Alemania) a una densidad de 4,5 x 104 células por cavidad sobre placas de 24 cavidades, de 0,4 pm de tamaño de poro, y se cultivaron durante 15 días en medio DMEM suplementado con 10 % de suero fetal bovino, 1 % de GlutaMAX (100x, GIBCO), 100 U/ml de penicilina, 100 pg/ml de estreptomicina (GIBCO) y 1 % de aminoácidos no esenciales (100 x). Las células se mantuvieron a 37°C en una atmósfera humidificada con 5 % de CO2. El medio se cambió cada 2-3 días. Antes de conducir el ensayo de permeación, se reemplazó el medio de cultivo por una solución amortiguadora de transporte de hepes-carbonato sin FCS (pH 7,2). Para evaluar la integridad de la monocapa se midió la resistencia eléctrica transepitelial (TEER). Los compuestos de prueba se predisolvieron en DMSO y se agregaron al compartimento apical o basolateral a una concentración final de 2 pM en solución amortiguadora de transporte. Se tomaron muestras antes y después de 2 hs de incubación a 37 °C de ambos compartimentos. El análisis del contenido de compuesto se efectuó luego de una
precipitación con metanol mediante análisis por LC/MS/MS. La permeabilidad (Papp) se calculó en las direcciones apical a basolateral (A ^ B) y basolateral a apical (B ^ A). La permeabilidad aparente se calculó usando la siguiente ecuación:
Papp = (Vr/Po)(1/S)(P2/t)
donde Vr es el volumen del medio en la recámara receptora, Po es el área determinada para los picos o la altura que alcanzó el fármaco que se deseaba analizar en la recámara donante en el tiempo 0, S la superficie de la monocapa, P2 es el área de los picos, determinada después de incubar los fármacos durante 2 horas en la recámara aceptora, y t es la duración de la incubación. La proporción del flujo de salida entre el compartimiento basolateral (B) y el compartimiento apical (A) se calculó dividiendo la Papp B-A por la Papp A-B. Además, se calculó la recuperación compuesto.
6. Análisis de la estabilidad metabólica en hepatocitos de rata in vitro
Los hepatocitos de ratas Wistar Han se aislaron de acuerdo con un procedimiento de perfusión con 2 pasos. Una vez completa la perfusión, se retiró cuidadosamente el hígado de las ratas, se abrió la cápsula del hígado y se agitaron suavemente los hepatocitos en una placa de Petri con un medio E de Williams (WME) enfriado con hielo (que había sido adquirido en Sigma Aldrich Life Science, St. Louis, MO). La suspensión de células que se obtuvo se filtró a través de un tamiz estéril, se colocó en tubos Falcon de 50 ml y se centrifugó a 50 x G, a temperatura ambiente durante 3 minutos. El sedimento celular se suspendió nuevamente en 30 ml del WME y se sometió a dos centrifugaciones a 100 x G a través de un gradiente de Percoll®. Los hepatocitos se lavaron nuevamente con un medio E de Williams y se suspendieron nuevamente en un medio que contenía 5 % de suero fetal bovino (FCS, que había sido adquirido en Invitrogen, Auckland, NZ). La viabilidad de las células se determinó sobre la base de una exclusión con azul de tripano. Para analizar la estabilidad metabólica de las células hepáticas, se las distribuyó en viales de vidrio que contenían un WME con 5 % de FCS, en una densidad de 1,0 x 106 células vitales por ml. El compuesto que se deseaba analizar fue agregado en una concentración final de 1 mM. En el transcurso de la incubación, se agitaron de manera continua las suspensiones de hepatocitos, se tomaron alícuotas una vez transcurridos 2, 8, 16, 30, 45 y 90 minutos y se les agregaron de inmediato volúmenes idénticos de acetonitrilo frío. Las muestras se congelaron a -20 °C durante la noche, luego se las centrifugó a 3000 rpm durante 15 minutos y se analizó el sobrenadante mediante el uso de un sistema de HPLC 1200, de Agilent, con una detección basada en un procedimiento de LCMS/MS.
La vida media de los compuestos que se deseaba analizar fue determinada sobre la base de la representación de la concentración en función del tiempo. A partir de la vida media, se calculó la eliminación intrínseca. Junto con los parámetros útiles para analizar el flujo de la sangre en el hígado, se determinó la cantidad de células hepáticas in vivo e in vitro y se calculó la biodisponibilidad oral máxima (Fmax) con los siguientes parámetros: un flujo de sangre en el hígado (en las ratas) de 4,2 l/h/kg, un peso específico del hígado de 32 g por kg de peso corporal de las ratas, una cantidad de células hepáticas in vivo de 1,1 x 108 por g del hígado y una la cantidad de células hepáticas in vitro de 0,5 x 106 por ml.
7. Análisis farmacocinético en ratas in vivo
Para los experimentos farmacocinéticos in vivo, los compuestos que se deseaba analizar se administraron en ratas Wistar macho por vía intravenosa, en una dosis de entre 0,3 y 1 mg/kg, en forma de soluciones con plasma de rata o con solubilizantes como el PEG 400, en cantidades que pudieran ser bien toleradas.
Para análisis farmacocinético después de una administración intravenosa, se administraron los compuestos que se deseaba analizar por vía intravenosa en forma de bolos y se tomaron muestras de la sangre 2 minutos, 8 minutos, 15 minutos, 30 minutos, 45 minutos, 1 hora, 2 horas, 4 horas, 6 horas, 8 horas y 24 horas después de la administración. En función de la vida media esperada, se tomaron muestras adicionales en momentos posteriores (por ejemplo, después de 48 o 72 horas). La sangre se recolectó en tubos con litio y heparina (Monovetten®, de Sarstedt) y se centrifugó durante a 3000 rpm durante 15 minutos. Se tomó una alícuota de 100 pl del sobrenadante (el plasma), se la hizo precipitar mediante la adición de 400 pl de acetonitrilo enfriado con hielo y se la congeló a -20 °C durante la noche. Posteriormente, se descongelaron las muestras y se las centrifugó a 3000 rpm, a 4°C durante 20 minutos. Se tomaron alícuotas de los sobrenadantes para someterlas a un análisis de HPLC en un sistema Agilent 1200, con una detección basada en un procedimiento de LCMS/MS. Los parámetros farmacocinéticos fueron calculados con un análisis no compartimentalizado, mediante el uso de un programa informático apropiado.
A continuación se detallan los parámetros farmacocinéticos que se obtuvieron a partir de los perfiles de la concentración en función del tiempo después de una administración intravenosa: CLplasma: eliminación total de los compuestos que se deseaba analizar del plasma (en l/kg/hora); CLsangre: eliminación total de los compuestos que se deseaba analizar de la sangre: CLplasma * Cp/Cb (en l/kg/hora), donde Cp/Cb es la proporción entre la concentración en el plasma y la concentración en la sangre, AUCnorm: área bajo la curva de la concentración en función del tiempo, entre t = 0 horas y el infinito (extrapolada), dividida por la dosis administrada (en kg * hora/l); t-i^: vida media terminal (en horas).
8. Resonancia de plasmón superficial con el PTEFb
Definiciones
La expresión "resonancia de plasmón superficial", como se usa en el presente documento, hace referencia a un fenómeno óptico que posibilita el análisis de las asociaciones reversibles entre las moléculas biológicas en una matriz biosensora en tiempo real, para lo cual, por ejemplo, puede usarse un sistema Biacore® (de GE Healthcare Biosciences, Uppsala, Suecia). En el sistema Biacore®, se utisan las propiedades ópticas de resonancia de plasmón superficial (SPR) para detectar las alteraciones en el índice de refracción de un amortiguador, que varía a medida que las moléculas en la solución interactúan con el blanco inmovilizado en la superficie. En resumen, primero se unen las proteínas a través de enlaces covalentes a una matriz de dextrano en una concentración conocida, y luego se inyecta un ligando para las proteínas a través de la matriz de dextrano. La luz infrarroja cercana, dirigida hacia el lado opuesto de la superficie del chip sensor, se refleja y también induce una onda evanescente en una película de oro, lo cual a su vez da como resultado una disminución en la intensidad de la luz que se refleja en un ángulo particular, que se conoce como el ángulo de resonancia. Si se altera el índice de refracción en la superficie del chip sensor (por ejemplo, como resultado de la unión entre un compuesto y una proteína fijada), se produce un cambio en el ángulo de resonancia. Este cambio en el ángulo puede ser medido. Este cambio se representa en función del tiempo a lo largo del eje y de un sensorgrama, que representa la asociación y la disociación de cualquier reacción biológica.
El término "Kd", como se emplea en el presente documento, hace referencia a la constante de disociación en el equilibrio de un complejo formado por un compuesto en particular y la proteína que hace las veces de blanco.
El término "Koff", como se emplea en el presente documento, hace referencia a la velocidad de disociación, es decir, la constante relacionada con la velocidad a la que puede disociarse un complejo formado por un compuesto en particular y la proteína que hace las veces de blanco.
La expresión "período de residencia en el blanco", como se emplea en el presente documento, hace referencia a la inversa de la constante relacionada con la velocidad de disociación (1/Koff) de un complejo formado por un compuesto en particular y la proteína que hace las veces de blanco.
Para descripciones adicionales, ver:
Jonsson, U., y col., 1993, Ann. Biol. Clin., 51(1): 19-26
Johnsson, B., y col., Anal. Biochem., 1991, 198(2): 268-77
Day, Y., y col., Protein Science, 2002, 11, 1017-1025
Myskza, D. G., Anal, Biochem., 2004, 329, 316-323
Tummino y Copeland, Biochemistry, 2008, 47(20): 5481-5492
Actividad biológica
La actividad biológica de los compuestos de acuerdo con la invención (por ejemplo, aplicada a la inhibición del PETFb) pueden determinarse con el análisis de SPR que se ha descrito.
El nivel de actividad que presenta un compuesto determinado en un análisis de SPR puede definirse en términos del valor de la Kd, y los compuestos preferidos de la presente invención son aquellos que se caracterizan por una Kd con un valor inferior a 1 micromolar, más preferentemente inferior a 0,1 micromolar.
Además, el período de residencia de un compuesto de la presente invención en su blanco puede definirse en términos del período de residencia en el blanco (TRT), y los compuestos preferidos de la presente invención son aquellos que se caracterizan por un TRT con un valor superior a 10 minutos, más preferentemente superior a 1 hora.
La capacidad de los compuestos de acuerdo con la invención de unirse al PTEFb humano puede determinarse por medio de un procedimiento de resonancia de plasmón superficial (SPR). El valor de la Kd y de la Koff puede determinarse usando un instrumento Biacore® T200 (de GE Healthcare, Uppsala, Suecia).
En un análisis de SPR, se inmoviliza un PTEFb recombinante humano (una quinasa de proteínas compuesta por la CDK9 y la ciclina T1 humanas, que puede adquirirse en ProQinase, Friburgo, Alemania) sobre la base de una unión de aminas convencional (Johnsson, B., y col., Anal. Biochem., 1 de noviembre de 1991, 198 (2): 268-77). En resumen, se activan chips biosensores con dextrano carboximetilado (CM7, GE Healthcare) con clorhidrato de N-etil-N'-(3-dimetilaminopropil)-carbodiimida (EDC) y N-hidroxisuccinimida (NHS) de acuerdo con las instrucciones del proveedor. El PTEFb humano se diluye en HBS-EP+ 1x (de GE Healthcare) hasta alcanzar una concentración de 30 pg/ml y se inyecta en la superficie del chip activado. Posteriormente, se inyecta una mezcla 1:1 de una solución 1 M de clorhidrato de etanolamina (GE Healthcare) y HBS-EP 1x para bloquear los grupos que no han reaccionado, lo que da como resultado aproximadamente 4000 unidades de respuesta (RU) de la proteína inmovilizada. Se genera una superficie de referencia por medio de un tratamiento con NHS-EDC y clorhidrato de etanolamina. Los compuestos se disuelven en dimetilsulfóxido al 100% (DMSO, de Sigma-Aldrich, Alemania) hasta alcanzar una concentración de 10 mM, y posteriormente se lo diluye en un amortiguador apropiado para llevar a cabo el procedimiento (que es un amortiguador a base de HBS-EP+ 1x con un pH de 7,4, que se genera a partir de un amortiguador compuesto por HBS-EP+ 10X (de GE Healthcare) al que se le agrega He Pe S 0,1 M, Nacl 1,5 M, EDTA 30 mM, 0,5 % v/v del agente tensioactivo P20 y 1 % v/v de DMSO). Para llevar a cabo las mediciones cinéticas, se inyecta una serie de diluciones al cuarto del compuesto (entre 0,39 nM y 100 nM) sobre la proteína inmovilizada. La unión cinética se analiza a 25°C, con un flujo con una velocidad de 50 pl/minuto en el amortiguador que se ha descrito. Cada una de las concentraciones del compuesto que se desea analizar se inyecta durante un período de 60 segundos, a lo que sigue un período de
disociación de 1800 segundos. Como referencia doble para los sensogramas, se emplea una superficie de referencia e inyecciones en blanco.
Los sensogramas con una referencia doble se ajustan a un mecanismo de acción 1:1 simple y reversible de Langmuir mediante el uso del programa informático de evaluación Biacore® T200 2.0 (de GE Healthcare). Cuando no se ha producido una disociación completa de un compuesto al final de la fase de disociación, se ajusta el parámetro Rmax (la respuesta en la saturación) como la variable local. En los casos restantes, se ajusta la Rmax como la variable global.
Síntesis de los compuestos
Las síntesis de los compuestos macrocíclicos de la fórmula (I) de acuerdo con la presente invención se llevan a cabo, con preferencia, según la secuencia de síntesis general como se muestra en los esquemas 1a, 1b, 1c, 1d, 2, 3a, 3b, 3c, 4 y 5.
Además de dichas rutas descritas a continuación, también se pueden usar otras vías para sintetizar los compuestos blanco, de acuerdo con el conocimiento general común de un experto en la materia de las síntesis orgánicas. El orden de las transformaciones ejemplificado en los siguientes esquemas no pretende ser, por ello, limitativo y las etapas de síntesis apropiadas de los diversos esquemas se pueden combinar para formar secuencias de síntesis adicionales. Además, la modificación de cualquiera de los sustituyentes R1, R2, R3, R4 y/o R5 se puede lograr antes y/o después de las transformaciones ejemplificadas. Estas modificaciones pueden ser tales como la introducción de grupos protectores, escisión de grupos protectores, reducción u oxidación de grupos funcionales, halogenación, metalación, reacciones de acoplamiento catalizadas con metal, sustitución u otras reacciones conocidas por los expertos en el arte. Estas transformaciones incluyen aquellas que introducen una funcionalidad que permite una ulterior interconversión de sustituyentes. Los grupos protectores apropiados y su introducción y escisión son bien conocidos por los expertos en el arte (ver, por ejemplo, T.W. Greene y P.G.M. Wuts in Protective Groups in Organic Synthesis, 4a edición, Wiley 2006). Los ejemplos específicos se describen en los párrafos posteriores. Además, es posible que dos o más etapas sucesivas se puedan llevar a cabo sin realizar una elaboración entre dichas etapas, por ejemplo, una reacción "en un solo recipiente", como bien conocerá un experto en la materia.
La geometría del resto de sulfondiimina vuelve quirales a algunos de los compuestos de fórmula general (I). La separación de sulfondiiminas racémicas en sus enantiómeros se puede lograr mediante procedimientos conocidos por el experto en la materia, con preferencia, por medio de HPLC preparativa en fase estacionaria quiral.
La síntesis de los derivados de piridina de fórmula (10), que constituye un subgrupo de fórmula general (I) de acuerdo con la presente invención, se llevan a cabo, con preferencia, según las secuencias de síntesis generales como se muestra en los esquemas 1a, 1b, 1c y 1d.
Esquema 1a

Esquema 1c
Los Esquemas 1a, 1b y 1c, en los que L, R1, R2, R3, R4y R5son como se definen para el compuesto de fórmula general (I) de acuerdo con la presente invención, destacan la preparación de compuestos macrocíclicos a base de piridina de la fórmula (10), a partir de 2-cloro-5-fluoro-4-yodopiridina (1; n.° CAS 884494-49-9). Dicho material de partida (1) se hace reaccionar con un derivado de ácido borónico de la fórmula (2), en la que R3 y R4 son como se definen para el compuesto de fórmula general (I), para dar un compuesto de fórmula (3). El derivado de ácido borónico (2) puede ser un ácido borónico (R = -H) o un éster de ácido borónico, por ejemplo, su éster isopropílico (R = -CH(CH3)2), con preferencia, un éster derivado de pinacol en el que el intermedio de ácido borónico forma un 2-aril-4,4,5,5-tetrametil-1,3,2-dioxaborolano (R-R = -C(CH3)2-C(CH3)2-).
Dicha reacción de acoplamiento es catalizada por catalizadores de paladio, por ejemplo, por catalizadores de Pd(0) tales como tefragu/s(trifenilfosfina)paladio (0) [Pd(PPh3)4], tris(dibencilidenacetona)di-paladio (0) [Pd2(dba)3] o por catalizadores de Pd(II) tales como diclorobis(trifenilfosfina)-paladio (II) [Pd(PPh3)2C y, acetato de paladio (II) y
trifenilfosfina o por dicloruro de [1,1'-bis(difenilfosfino)ferroceno]paladio.
La reacción preferentemente se lleva a cabo en una mezcla de un disolvente como el 1,2-dimetoxietano, el dioxano, la DMF, el THF o el isopropanol y agua, en presencia de una base como el carbonato de potasio, el bicarbonato de sodio o el fosfato de potasio.
(puede hallarse una revisión en D. G. Hall, Boronic Acids, 2005, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, ISBN 3-527-30991-8, y en las referencias que se citan en esta publicación).
La reacción se pone en práctica a una temperatura que varía entre la temperatura ambiente (es decir, aproximadamente 20 °C) y el punto de ebullición del disolvente empleado. También es posible llevar a cabo la reacción a una temperatura superior al punto de ebullición, mediante el uso de tubos a presión y un horno de microondas. La reacción preferentemente se completa después de un período de entre 1 y 36 horas.
En el segundo paso, se convierte un compuesto de fórmula (3) en un compuesto de fórmula (4). Esta reacción puede basarse en una reacción de unión transversal C-N catalizada con paladio (pueden hallarse descripciones de las reacciones de unión transversales C-N, por ejemplo, en (a) L. Jiang y S. L., Buchwald, Metal-Catalyzed Cross-Coupling Reactions, 2a edición, editado por A. de Meijere y F. Diederich, Wiley-VCH: Weinheim, Alemania, 2004). Se prefiere el uso descrito en el presente documento de bis(trimetilsilil)amida de litio, tris(dibencilidenacetona) de di-paladio(0) y 2-(diciclohexilfosfino)-2',4',6'-triisopropil-bifenilo en THF. Las reacciones preferentemente se ponen en práctica en una atmósfera de argón, en un baño de aceite a 40-80 °C durante 3-24 horas.
En el tercer paso, se convierte un compuesto de fórmula (4) en un compuesto de fórmula (5), por medio de la escisión del éter metílico presente en los compuestos de fórmula (4).
Se prefiere el uso descrito en el presente documento de tribromuro de boro en DCM. Las reacciones preferentemente se llevan a cabo durante 1-24 horas de 0 °C a temperatura ambiente.
En el cuarto paso, se acopla un compuesto de fórmula (5) a un compuesto de fórmula (6), en la que R1, R2 y L son como se los ha definido para el compuesto de fórmula general (I), para dar un compuesto de fórmula (7) en presencia de una fosfina terciaria como trifenilfosfina y un dialquildiazodicarboxilato (conocida como reacción de Mitsunobu, ver, por ejemplo, K. C. K, Swamy y col., Chem. Rev., 2009, 109, 2551). Se prefiere el uso descrito en el presente documento de azodicarboxilato de diisopropilo y trifenilfosfina en THF. Las reacciones preferentemente se llevan a cabo durante 1- 24 horas de 0 °C a temperatura ambiente.
Los compuestos de fórmula (6) pueden prepararse como se describe en el esquema 2 más adelante.
En el quinto paso, un compuesto de fórmula (7) se convierte en un macrociclo de la fórmula (8). Esta reacción de ciclación se puede llevar a cabo mediante una reacción de acoplamiento cruzado C-N catalizada con paladio intramolecular (para una revisión de reacciones de acoplamiento cruzado C-N, ver, por ejemplo: a) L. Jiang, S.L. Buchwald en 'Metal-Catalyzed Cross-Coupling Reactions', 2a ed.: A. de Meijere, F. Diederich, Eds.: Wiley-VCH: Weinheim, Alemania, 2004).
Se prefiere el uso descrito en el presente documento de aducto de cloro(2-diciclohexilfosfino-2',4',6'-tri-iso-propil-1,1 '-bifenil)[2-(2-aminoetil)fenil]paladio (II) y metil-ferc-butiléter, 2-(diciclohexilfosfino)-2',4',6'-triisopropilbifenilo como catalizador y ligando, un carbonato alcalino o un fosfato alcalino, con preferencia, fosfato de potasio, como una base, en una mezcla de un alquil C-i-C3-benceno y un disolvente a base de carboxamida, con preferencia, una mezcla de tolueno y NMP, como un disolvente. Las reacciones se ejecutan, con preferencia, en una atmósfera de argón durante 2- 24 horas a 100-130 °C en un horno de microondas o en un baño de aceite.
Como se muestra en el Esquema 1d, los compuestos macrocíclicos de la fórmula (8a), que constituye un subcompartimiento de la fórmula (8) porque R2 de la fórmula (8) representa un átomo de hidrógeno, se pueden usar ventajosamente para la introducción de grupos R2 diferentes de un átomo de hidrógeno, por ejemplo, haciendo reaccionar un compuesto de fórmula (8a), en la que R1, R3, R4 y L son como se definen para el compuesto de fórmula general (I) de acuerdo con la presente invención, con N-yodosuccinimida, en un disolvente a base de carboxamida tales como DMF, para dar intermedios yodados de la fórmula (8b), que, a su vez, se pueden convertir en los compuestos de fórmula (8c), en la que R2 es como se define para el compuesto de fórmula general (I) pero diferente de un átomo de hidrógeno, por procedimientos conocidos por el experto en la materia, ejemplificados, pero sin limitación, a la conversión de un compuesto de fórmula (8b) en el correspondiente carbonitrilo (R2 = CN) por reacción con cianuro de cobre (I) en DMSO, a una temperatura de entre 100 °C y 160 °C.

Esquema 1d
En el sexto paso y como se muestra en el Esquema 1c, anteriorente, un sulfuro de la fórmula (8) se convierte en un compuesto de fórmula (9), por tratamiento con O-(mesitilensulfonil)hidroxilamina (MSH), en un disolvente inerte, tales como un hidrocarburo alifático clorado de la fórmula cloro-alquil C1-C2-H, con mayor preferencia, diclorometano, a una temperatura de entre -20 °C y 80 °C, con preferencia, de entre -10 °C y 60 °C, con mayor preferencia, de entre 0 °C y 40 °C (ver, por ejemplo: C. Bolm y col., Angew. Chem. 2012, 124, 4516).
En el paso final, un compuesto de fórmula (9) se convierte en un compuesto de fórmula (I) en una secuencia en un recipiente por oxidación con A/-clorosuccinimida (NCS), en una carboxamida como un disolvente, con preferencia, W,W-dimetilformamida (DMF), W,W-dimetilacetamida o W-metilpirrolidin-2-ona (NMP) o una de sus mezclas, con mayor preferencia, W,W-dimetilformamida (DMF), en presencia de un carbonato alcalino, con preferencia, carbonato de sodio como una base, seguido por adición de una amina primaria de la fórmula R5-NH2, en la que R5 es como se define para el compuesto de fórmula general (I) o hexametildisilazano en caso de que R5 en el producto de reacción represente un átomo de hidrógeno, a una temperatura de entre -20 °C y 50 °C, con preferencia, de entre -10 °C y 40 °C, con mayor preferencia, de entre 0 °C y 30 °C (ver, por ejemplo: C. Bolm y col., Angew. Chem. 2012, 124, 4516).
De modo alternativo, se puede usar diacetato de yodobenceno en lugar de NCS. Con preferencia, la reacción se ejecuta en un hidrocarburo alifático clorado de la fórmula cloro-alquil C1-C2-H, con mayor preferencia, diclorometano, como un disolvente, si se usa diacetato de yodobenceno en lugar de NCS.
Los compuestos de fórmula (6), en la que R1, R2y L son como se los ha definido para el compuesto de fórmula general (I) de acuerdo con la presente invención, pueden prepararse según se indica el esquema 2, por ejemplo, a partir de un derivado de ácido 2,6-dicloroisonicotínico de la fórmula (11), en la que R2 es como se lo ha definido para el compuesto de fórmula general (I), el cual es reducido por medio de un procedimiento apropiado para obtener el piridinmetanol de la fórmula (12) correspondiente. En este paso, se prefiere el uso de un complejo 1:1 formado por sulfanodiildimetano y borano en tetrahidrofurano.
Los derivados del ácido isonicotínico de fórmula (11) y sus ésteres han de resultar conocidos para aquellos versados en la técnica y suelen estar disponibles en el ámbito comercial.
En un segundo paso, el piridinmetanol de la fórmula (12) se hace reaccionar de manera tal de obtener un compuesto de fórmula (13), en la que LG representa un grupo saliente, tal como el cloro, el bromo, el yodo o un grupo alquil C1-C4--S(=O)2O, trifluorometansulfoniloxilo, bencensulfoniloxilo o paratoluensulfoniloxilo. Las conversiones de este tipo han de resultar conocidas para aquellos versados en la técnica; en este paso, se prefiere el uso de cloruro de metansulfonilo en presencia de trietilamina como base, en diclorometano como disolvente, de manera tal de obtener un compuesto de fórmula (13) en la que LG representa un metansulfoniloxio.
En un tercer paso, un compuesto de fórmula (13) se hace reaccionar con un tiol de la fórmula R1-SH (o una de sus sales), en la que R1 es como se define para el compuesto de fórmula general (I), opcionalmente en presencia de una base como hidróxido de sodio, para dar un derivado de tioéter de la fórmula (14). Los tioles de la fórmula R1SH y sus sales son bien conocidos por el experto en la materia y son comercialmente asequibles en considerable variedad.
En un cuarto paso, se hace reaccionar un derivado de tioéter de la fórmula (14) con un anión formado in situ a partir de un diol de la fórmula HO-L-OH, en la que L es como se ha definido para el compuesto de fórmula general (I), y un metal alcalino, preferentemente sodio, en tetrahidrofurano como disolvente, para dar los compuestos intermedios de fórmula (6), los cuales pueden someterse a un procesamiento adicional, según se describe en los esquemas 1b y 1c.

Esquema 2
La síntesis de los derivados de pirimidina de la fórmula (23), que constituyen otro subconjunto de fórmula general (I) de acuerdo con la presente invención, preferentemente se lleva a cabo de acuerdo con los procedimientos generales que se detallan en los esquemas 3a, 3b y 3c.
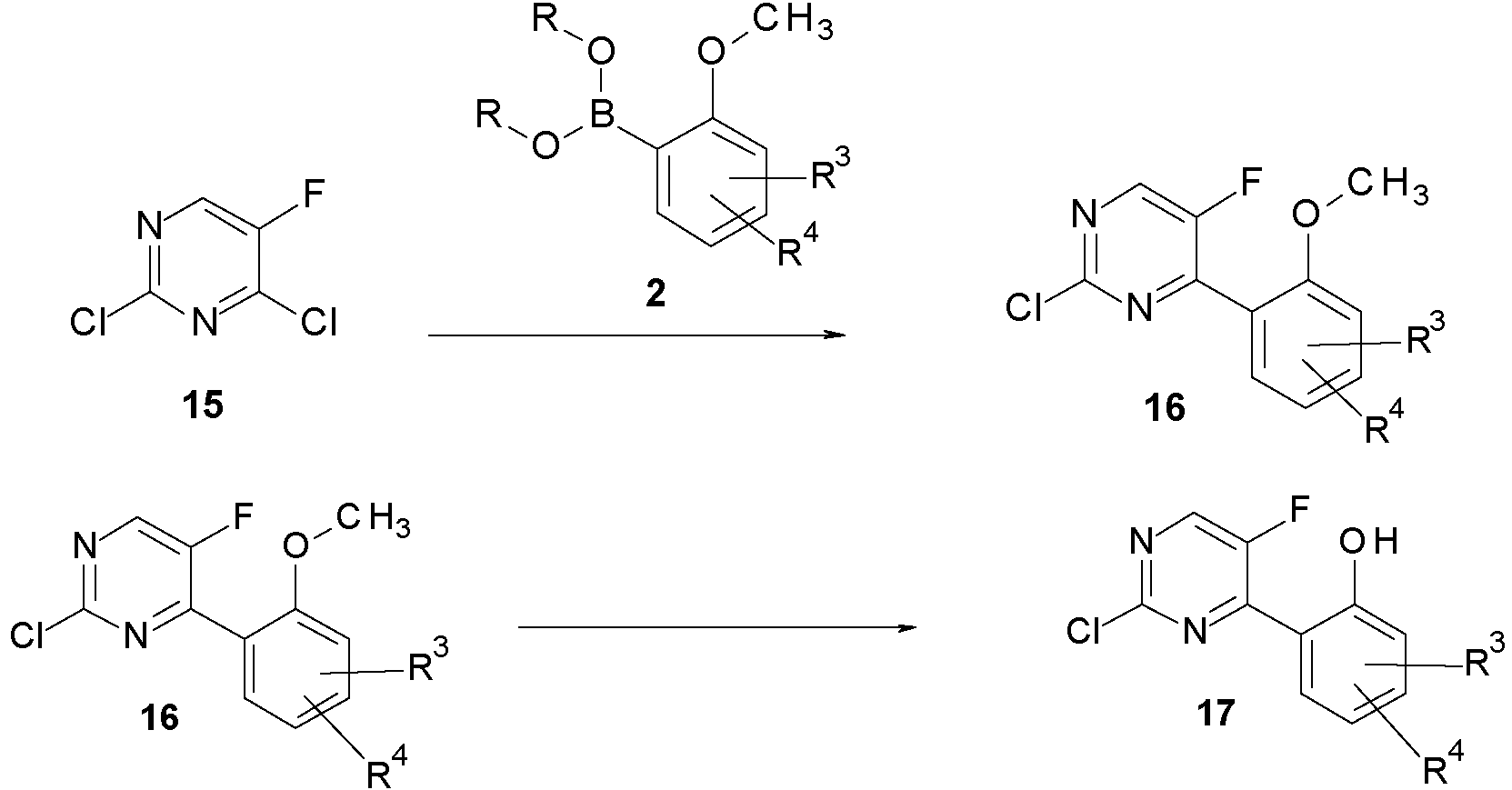

Esquema 3b

Esquema 3c
En los esquemas 3a, 3b y 3c, en los que L, R1, R2, R3, R4 y R5 son como se han definido para el compuesto de fórmula general (I) de acuerdo con la presente invención, se describe la preparación de los compuestos del tipo de las pirimidinas de fórmula general (l) a partir de 2,4-dicloro-5-fluoropirimidinas (n.° CAS 2927-71-1, 15). El material de partida (15) se hace reaccionar con un derivado de ácido borónico de la fórmula (2), de manera tal de obtener un compuesto de fórmula (16). El derivado de ácido borónico (2) puede tomar la forma de un ácido borónico (R = -H) o de un éster de ácido borónico, por ejemplo, de un éster isopropílico (R es -CH(CH3)2), preferentemente de un éster derivado de pinacol donde el intermedio en forma de ácido borónico forma un 2-aril-4,4,5,5-tetrametil-1,3,2-dioxaborolano (R-R = -C(CH3)2-C(CH3)2). Los ácidos borónicos y sus ésteres están disponibles en el ámbito comercial y han de resultar conocidos para aquellos versados en la técnica; véase, por ejemplo, D. G. Hall, Boronic Acids, 2005, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, ISBN 3-527-30991-8, y las referencias que se citan en esta publicación.
La reacción de acoplamiento está catalizada con catalizadores a base de paladio, por ejemplo, catalizadores de Pd(0), tal como tefragu/s(trifenilfosfina) de paladio(0) (Pd (PPh^W o la tris(dibencilidenacetona) de di-paladio(O) (Pd2(dba)3), o por catalizadores de Pd(II), tales como diclorobis(trifenilfosfina) de paladio(II) (Pd(PPh3)2Ch), el acetato de paladio(II) y trifenilfosfina o el dicloruro de [1,1'-bis(difenilfosfino)ferroceno]paladio (Pd(dppf)Ch).
La reacción preferentemente se lleva a cabo en una mezcla de un disolvente como el 1,2-dimetoxietano, el dioxano, la DMF, el DME, el THF o el isopropanol y agua, en presencia de una base como el carbonato de potasio acuoso, el bicarbonato de sodio acuoso o el fosfato de potasio.
La reacción se pone en práctica a una temperatura que varía entre la temperatura ambiente (20 °C) y el punto de ebullición del disolvente. También es posible llevar a cabo la reacción a una temperatura superior al punto de ebullición, mediante el uso de tubos a presión y un horno de microondas (puede hallarse una revisión en D. G. Hall, Boronic Acids, 2005, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, ISBN 3-527-30991-8, y en las referencias que se citan en esta publicación).
La reacción preferentemente se completa después de un período de entre 1 y 36 horas.
En el segundo paso, se convierte un compuesto de fórmula (16) en un compuesto de fórmula (17).
En este paso, se prefiere el uso de tribromuro de boro en DCM. Las reacciones preferentemente se llevan a cabo durante 1-24 horas, a una temperatura que varía entre 0 °C y la temperatura ambiente.
En el tercer paso, un compuesto de fórmula (17) se acopla con un compuesto de fórmula (18) para dar un compuesto de fórmula (19), en presencia de una fosfina terciaria, tales como trifenilfosfina y un diazodicarboxilato de dialquilo (conocida como reacción de Mitsunobu, ver, por ejemplo: K.C.K. Swamy y col., Chem. Rev. 2009, 109, 2551).
Se prefiere el uso descrito en el presente documento de azodicarboxilato de diisopropilo y trifenilfosfina en tetrahidrofurano o diclorometano. Las reacciones se ejecutan, con preferencia, durante 1-24 horas de 0 °C a temperatura ambiente.
Los compuestos de fórmula (19) se pueden reducir para dar anilinas de la fórmula (20). La reducción se puede preparar de manera análoga a procedimientos conocidos (ver, por ejemplo: (a) Sammond y col.; Bioorg. Med. Chem. Lett. 2005, 15, 3519; (b) R.C. Larock, Comprehensive Organic Transformations, VCH, Nueva York, 1989, 411-415). Se prefiere en el presente documento, la hidrogenación descrita en metanol y THF usando platino y vanadio sobre carbón activado como un catalizador.
Un compuesto de fórmula (20) se puede convertir en un macrociclo de la fórmula (21). Esta reacción de ciclación se puede llevar a cabo por una reacción de acoplamiento cruzado C-N catalizado con paladio (para una revisión de reacciones de acoplamiento cruzado C-N, ver, por ejemplo: a) L. Jiang, S.L. Buchwald en 'Metal-Catalyzed Cross
Coupling Reactions', 2a ed.: A. de Meijere, F. Diederich, Eds.: Wiley-VCH: Weinheim, Alemania, 2004). Se prefiere el uso descrito en el presente documento de aducto de cloro(2-diciclohexilfosfino-2',4',6'-tri-iso-propil-1,1'-bifenil)[2-(2-aminoetil)fenil]paladio (II) y metil-ferc-butiléter, 2-(diciclohexilfosfino)-2',4',6'-triisopropilbifenilo como catalizador y ligando, un carbonato alcalino o un fosfato alcalino, con preferencia, fosfato de potasio, como una base, en una mezcla de un alquilbenceno C1-C3 y un disolvente a base de carboxamida, con preferencia, una mezcla de tolueno y NMP, como un disolvente. Las reacciones se ejecutan, con preferencia, en una atmósfera de argón durante 2-24 horas a 100-130 °C en un horno de microondas o en un baño de aceite.
Un sulfuro de la fórmula (21) se puede convertir en un compuesto de fórmula (22), por tratamiento con O-(mesitilensulfonil)hidroxilamina (MSH), en un disolvente inerte, tales como un hidrocarburo alifático clorado de la fórmula cloro-alquil C1-C2-H, con mayor preferencia, diclorometano, a una temperatura de entre -20 °C y 80 °C, con preferencia, entre -10 °C y 60 °C, con mayor preferencia, entre 0 °C y 40 °C (ver, por ejemplo: C. Bolm y col., Angew. Chem. 2012, 124, 4516).
En el paso final, un compuesto de fórmula (22) se puede convertir en un compuesto de fórmula (23) en una secuencia en un recipiente por oxidación con W-clorosuccinimida (NCS), en una carboxamida como un disolvente, con preferencia, W,W-dimetilformamida (DMF), W,W-dimetilacetamida o W-metilpirrolidin-2-ona o una de sus mezclas, con mayor preferencia, W,W-dimetilformamida (DMF), en presencia de un carbonato alcalino, con preferencia, carbonato de sodio como una base, seguido por adición de una amina primaria de la fórmula R5-NH2, en la que R5 es como se define para el compuesto de fórmula general (I) o hexametildisilazano en caso de que R5 en el producto de reacción represente un átomo de hidrógeno, a una temperatura entre -20 °C y 50 °C, con preferencia, entre -10 °C y 40 °C, con mayor preferencia, entre 0 °C y 30 °C (ver, por ejemplo: C. Bolm y col., Angew. Chem. 2012, 124, 4516).
De modo alternativo, se puede usar diacetato de yodobenceno en lugar de NCS. Con preferencia, la reacción se ejecuta en un hidrocarburo alifático clorado de la fórmula cloro-alquil C1-C2-H, con mayor preferencia, diclorometano, como un disolvente, si se usa diacetato de yodobenceno en lugar de NCS.
Los compuestos de fórmula (18), en la que R1, R2 y L son como se ha definido para el compuesto de fórmula general (I) de acuerdo con la presente invención, pueden prepararse según se indica en el esquema 4, por ejemplo, a partir de un derivado del alcohol bencílico de la fórmula (24), en la que R2 es como se ha definido para el compuesto de fórmula general (I), se hace reaccionar para dar un compuesto de fórmula (25), en la que LG representa un grupo saliente, tal como el cloro, el bromo, el yodo o un alquil C1-C4--S(=O)2O, trifluorometansulfoniloxilo-, bencensulfoniloxilo- o para-toluensulfoniloxilo-. Tales conversiones son bien conocidas para la persona experta en la materia; se prefiere el uso descrito en el presente documento de cloruro de tionilo en N,N-dimetilformamida (DMF) como disolvente, para dar un compuesto de fórmula (25) en la que LG representa cloro.
Los derivados del alcohol bencílico de la fórmula (24) o los ácidos carboxílicos correspondientes y sus ésteres son conocidos para la persona experta en la materia y están disponibles en el mercado en determinados casos.
En un segundo paso, un compuesto de fórmula (25) se hace reaccionar con un tiol de la fórmula R1-SH (o una de sus sales), en la que R1 es como se define para el compuesto de fórmula general (I), opcionalmente en presencia de una base, tal como hidróxido de sodio, para dar un derivado de tioéter de la fórmula (26). Los tioles de la fórmula R1SH y sus sales son bien conocidos por el experto en la materia y están disponibles en el mercado en una variedad considerable.
En un tercer paso, un derivado de tioéter de la fórmula (26) se hace reaccionar con un éster carboxílico de la fórmula (27), en la que L' representa un grupo alquileno C1-C5 que caracteriza un átomo de carbono menos en comparación con el correspondiente grupo L en la fórmula (28), siendo L como se define para el compuesto de fórmula general (I), RE representa un grupo alquil C1-C4- y en la que LG representa un grupo saliente, tal como cloro, bromo, yodo, alquil C1-C4-S(=O)2O-, trifluorometansulfoniloxi-, bencensulfoniloxi- o para-toluensulfoniloxi, en presencia de una base, tal como un carbonato alcalino, con preferencia, carbonato de potasio, en W,W-dimetilformamida (DMF) como un disolvente, para dar un compuesto de fórmula (28).
En un cuarto paso, un éster de la fórmula (28) se puede reducir usando un agente de reducción tales como hidruro de litio y aluminio o hidruro de di-iso-butilaluminio (DIBAL), en un éter, con preferencia, tetrahidrofurano, como un disolvente, para dar compuesto de fórmula (18) que se puede seguir procesando como se muestra en los Esquemas 3a, 3b y 3c.
De modo alternativo, un derivado de tioéter de la fórmula (26) se puede convertir directamente en un compuesto de fórmula (18), si se hace reaccionar con un compuesto de fórmula HO-L-LG, en la que L es como se define para el compuesto de fórmula general (I) de acuerdo con la presente invención y en la que Lg representa un grupo saliente, tal como cloro, bromo, yodo, alquil C1-C4-S(=O)2O-, trifluorometansulfoniloxi-, bencensulfoniloxi- o paratoluensulfoniloxi-, en lugar de un compuesto de fórmula (27), en presencia de una base, tales como un carbonato alcalino, con preferencia, carbonato de potasio, en W,W-dimetilformamida (DMF) como un disolvente.

Esquema 4
Abreviaturas usadas en la descripción de la química y en los ejemplos que siguen son:
a. (amplio, señal de RMN 1H); CDCI3 (cloroformo deuterado); cHex (ciclohexano); DCE (dicloroetano); d (doblete, señal de RMN 1H); DCM (diclorometano); DIBAL (hidruro de di-iso-butilaluminio); DIPEA (di-/so-propiletilamina); DMAP (4-N,N-dimetilaminopiridina), DME (1,2-dimetoxietano), DMF (N,N-dimetilformamida); DMSO (dimetilsulfóxido); Es (electropulverización); EtOAc (acetato de etilo); EtOH (etanol); h (hora(s)); RMN 1H (espectroscopia por resonancia magnética nuclear protónica); HPLC (cromatografía líquida de alto rendimiento), iPrOH (/so-propanol); m (multiplete, señal de RMN 1H); mCPBA (ácido mefa-cloroperoxibenzoico), MeCN (acetonitrilo), MeOH (metanol); min (minuto(s)); MS (espectrometría de masa); MSH (O-(mesitilensulfonil) hidroxilamina); MTBE (metil ferc-butil éter); NCS (N-clorosuccinimida); NMP (N-Metilpirrolidin-2-ona); RMN (resonancia magnética nuclear); Pd(dppf)Ch (complejo de [1,1'-bis(difenilfosfino)ferroceno]dicloropaladio (II) con diclorometano); q (cuarteto, señal de RMN 1H); quin (quinteto, señal de RMN 1H); rac (racémico); TA (temperatura ambiente); s (singulete, señal de RMN 1H); sat. ac. (saturada acuosa); SiO2 (gel de sílice); t (triplete, señal de RMN 1H); TFA (ácido trifluoroacético); TFAA (anhidruro trifluoroacético), THF (tetrahidrofurano); UPLC-MS (cromatografía líquida de rendimiento ultraalto combinada con espectrometría de masa, usada para controlar la reacción); UV (ultravioleta); % p (porcentaje en peso).
Espectros de RMN 1H
Las señales de RMN 1H se especifican con su multiplicidad/multiplicidades combinadas como resulta obvio del espectro; no se consideran posibles efectos de orden superior. Los desplazamientos químicos de las señales (8) se especifican como ppm (partes por millón).
Nombres quím icos:
Los nombres químicos se generaron usando el software ACD/Name de ACD/Labs. En algunos casos, los nombres generalmente aceptados de reactivos asequibles en comercios se usaron en lugar de los nombres generados por ACD/Name.
Estequiometría de las sales
En el presente texto, particularmente en la sección experimental, para la síntesis de los productos intermedios y de los ejemplos de la presente invención, cuando se menciona un compuesto como una forma de sal con la base o ácido correspondiente, la composición estequiométrica exacta de dicha forma de sal, como se obtiene mediante el proceso de preparación y/o purificación respectivo, es, en la mayoría de los casos, desconocida.
A menos que se indique lo contrario, los modificadores aplicados a los nombres químicos o a las fórmulas estructurales, tales como "clorhidrato", "trifluoroacetato", "sal de sodio", "x HCl", "x CF3COOH" o "x Na+", por ejemplo, no han de ser interpretados como una especificación estequiométrica, sino únicamente como una referencia a una
forma de sal.
Esto se aplica de manera análoga a los casos en los que se han obtenido intermedios de síntesis o compuestos de ejemplo o sales de los mismos, mediante los procesos de preparación y/o purificación descritos, como solvatos, tales como hidratos con (si se define) una composición estequiométrica desconocida.
HPLC preparativa
Autopurificador: condiciones ácidas

Autopurificador: condiciones básicas

Ejemplo 1:
15,19-difluoro-8-[(S-metNsulfonodMmidoN)metM]-3,4-dihidro-2H,11H-10,6-(azeno)-12,16-(meteno)-1,5,11,13-benzodioxadiazaciclooctadecino

Preparación del intermedio 1.1:
(2,6-Dicloropiridin-4-il)metanol

A una solución agitada de ácido 2,6-dicloroisonicotínico (10,0 g, 52,1 mmol) en THF (300 ml) a 0 °C se le añadió una solución de sulfandiildimetano - borano (1:1) (16,0 g, 210,5 mmol) en THF. La mezcla se dejó reaccionar a temperatura ambiente durante la noche. A continuación, se añadió cuidadosamente MeOH (22 ml) a la mezcla agitada mientras se enfriaba con un baño de hielo. La mezcla de reacción se diluyó con acetato de etilo (300 ml), se lavó con una solución acuosa de hidróxido de sodio (1 N, 100 ml) y una solución acuosa saturada de cloruro de sodio. La capa orgánica se concentró y el residuo se purificó por cromatografía en columna en gel de sílice (hexano/acetato de etilo = 7:1 a 3:1) para dar el compuesto del título deseado (8,3 g; 46,6 mmol).
RMN 1H (300 MHz, CDCla, 300K) 8 = 7,25 (2H); 4,72 (2H); 2,24 (1H).
Preparación del intermedio 1.2:
metanosulfonato de (2,6-dicloropiridin-4-il)metilo

Se disolvió (2,6-dicloropiridin-4-il)metanol (1,0 g; 5,62 mmol) en DCM (20 ml) y se añadió trietilamina (1,0 g; 9,88 mmol). La mezcla resultante se enfrió hasta 0 °C y se añadió cloruro de metansulfonilo (0,9 g, 7,89 mmol). La mezcla se agitó a temperatura ambiente durante 1 hora. Por adición de una solución acuosa de cloruro de hidrógeno (1 N), el valor de pH de la mezcla se ajustó a 3, antes de extraerlo tres veces con acetato de etilo. Las capas orgánicas combinadas se concentraron para dar el compuesto del título en bruto (1,4 g) que se usó sin purificación adicional.
Preparación del intermedio 1.3:
2,6-Dicloro-4-[(metilsulfanil)metil]piridina

El metanosulfonato de (2,6-didoropiridin-4-il)metilo (1,40 g; 5,47 mmol) se disolvió en THF (20 ml) y se añadió una mezcla de tiometóxido de sodio e hidróxido de sodio (p 1/1, 0,70 g, 5 mmol, proporcionado por Shanghai DEMO Medical Tech Co., Ltd). La mezcla resultante se agitó durante la noche a temperatura ambiente. La mezcla de reacción se diluyó con agua (10 ml) y se extrajo tres veces con acetato de etilo. Las capas orgánicas combinadas se concentraron y el residuo se purificó por cromatografía en columna en gel de sílice (hexano/acetato de etilo = 6:1 a 3:1) para dar el compuesto del título deseado (0,54 g; 2,60 mmol).
RMN 1H (300 MHz, CDCh, 300K) 8 = 7,18 (2H), 3,55 (2H), 1,98 (3H).
Preparación del intermedio 1.4:
-({6-Cloro-4-[(metilsulfanil)metil]piridin-2-il}oxi)propan-1-ol

A una solución de 1,3-propanodiol (660 mg; 8,68 mmol) en THF (10 ml) se le añadió sodio (33 mg; 1,43 mmol) y la mezcla de reacción se calentó a reflujo durante 3 horas. Después de enfriar, se añadió 2,6-dicloro-4-[(metilsulfanil)metil]piridina (300 mg, 1,44 mmol) y la mezcla de reacción se calentó a reflujo durante 16 horas. Después de enfriar, la mezcla se diluyó con agua (10 ml) y se extrajo tres veces con acetato de etilo. Las capas orgánicas combinadas se concentraron y el residuo se purificó por cromatografía ultrarrápida en columna en gel de sílice (hexano/acetato de etilo = 5:1 a 2:1) para dar el compuesto del título deseado (180 mg; 0,72 mmol).
RMN 1H (400 MHz, CDCla, 300K) 8 = 6,86 (1H), 6,56 (1H), 4,42 (2H), 3,71 (2H), 3,50 (2H), 3,27 (1H), 1,96 (5H).
Preparación del intermedio 1.5:
-Cloro-5-fluoro-4-(4-fluoro-2-metoxifenil)piridina

Un lote con 2-cloro-5-fluoro-4-yodopiridina (1000 mg; 3,88 mmol; APAC Pharmaceutical, LLC), ácido (4-fluoro-2-metoxifenil)borónico (660 mg; 3,88 mmol; Aldrich Chemical Company Inc.) y tefragu/s(trifenilfosfin)paladio (0) (449 mg; 0,38 mmol) en 1,2-dimetoxietano (10,0 ml) y una solución acuosa 2 M de carbonato de potasio (5,8 ml) se desgasificó usando argón. El lote se agitó en una atmósfera de argón durante 4 horas a 100 °C. Después de enfriar, el lote se diluyó con acetato de etilo y THF y se lavó con una solución acuosa saturada de cloruro de sodio. La capa orgánica se filtró usando un filtro Whatman y se concentró. El residuo se purificó por cromatografía en columna (hexano a hexano/acetato de etilo 50 %) para dar el compuesto del título deseado (947 mg; 3,70 mmol).
RMN 1H (400 MHz, CDCla, 300K) 8 = 8,27 (m, 1H), 7,33 (m, 1H), 7,24 (m, 1H), 6,75 (m, 2H), 3,83 (s, 3H).
Preparación del intermedio 1.6:
5-Fluoro-4-(4-fluoro-2-metoxifenil)piridin-2-amina

Una solución de bis(trimetilsilil)amida de litio en THF (1 M; 20,5 ml; 20,53 mmol; Aldrich Chemical Company Inc.) se añadió a una mezcla de 2-cloro-5-fluoro-4-(4-fluoro-2-metoxifenil)piridina (2,50 g; 9,78 mmol; ver el Intermedio 1.5), tris(dibencilidenacetona)dipaladio (0) (0,18 g; 0,20 mmol; Aldrich Chemical Company Inc.) y 2-(diciclohexilfosfino)-',4',6-triisopropilbifenilo (0,19 g; 0,39 mmol; Aldrich Chemical Company Inc.) en THF (16,3 ml) en una atmósfera de argón a temperatura ambiente. La mezcla se agitó a 60 °C durante 6 horas. La mezcla se enfrió hasta -40 °C y se añadió agua (10 ml). La mezcla se calentó lentamente hasta temperatura ambiente en agitación, se añadió cloruro de sodio sólido y la mezcla se extrajo dos veces con acetato de etilo. Las capas orgánicas combinadas se filtraron usando un filtro Whatman y se concentraron. El residuo se purificó por cromatografía en columna en gel de sílice (hexano a hexano/acetato de etilo al 60 %) para dar el compuesto del título deseado (2,04 g; 8,64 mmol).
RMN 1H (400 MHz, CDCla, 300K) 8 = 7,95 (1H), 7,20 (1H), 6,72 (2H), 6,46 (1H), 4,33 (2H), 3,61 (3H).
Preparación del intermedio 1.7:
2-(2-Amino-5-fluoropiridin-4-il)-5-fluorofenol

Una solución de tribromuro de boro en DCM (1 M; 47,1 ml; 47,1 mmol; Aldrich Chemical Company Inc.) se añadió gota a gota a una solución agitada de 5-fluoro-4-(4-fluoro-2-metoxifenil)piridin-2-amina (2,00 g; 8,47 mmol) en DCM (205 ml) a 0 °C. La mezcla se calentó lentamente hasta temperatura ambiente mientras se agitaba durante la noche. La mezcla se diluyó cuidadosamente con una solución acuosa de bicarbonato de sodio en agitación a 0 °C y se agitó a temperatura ambiente durante 1 hora. Una solución saturada de cloruro de sodio se añadió y la mezcla se extrajo con acetato de etilo. Las capas orgánicas combinadas se filtraron usando un filtro Whatman y se concentraron para dar el compuesto del título en bruto (1,92 g) que se usó sin purificación adicional.
RMN 1H (400 MHz, DMSO-d6, 300K) 8 = 10,21 (1H), 7,84 (1H), 7,19 (1H), 6,71 (2H), 6,39 (1H), 5,80 (2H).
Preparación del intermedio 1.8:
4-{2-[3-({6-Cloro-4-[(metilsulfanil)metil]piridin-2-il}oxi)propoxi]-4-fluorofenil}-5-fluoropiridin-2-amina

Una solución de azodicarboxilato de diisopropilo (1,70 ml; 8,64 mmol) en THF (6,8 ml) se añadió gota a gota a una mezcla de 3-({6-cloro-4-[(metilsulfanil)metil]piridin-2-il}oxi)propan-1-ol (1,96 g; 7,89 mmol, ver el Intermedio 1,4), 2-(2-amino-5-fluoropiridin-4-il)-5-fluorofenol (1,92 g; 8,64 mmol) y trifenilfosfina (2,27 g; 8,64 mmol) en THF (34,0 ml) y el lote se agitó a temperatura ambiente durante 5 horas. Se añadieron más trifenilfosfina (1,04 g; 3,94 mmol) y azodicarboxilato de diisopropilo (0,78 ml; 3,95 mmol) y la mezcla se agitó a temperatura ambiente durante la noche. Se añadió más azodicarboxilato de diisopropilo (0,78 ml; 3,95 mmol) y la mezcla se agitó a temperatura ambiente durante 3 horas. Finalmente, se añadieron más trifenilfosfina (2,07 g; 7,89 mmol) y azodicarboxilato de diisopropilo (1,55 ml; 7,89 mmol) y la mezcla se agitó a temperatura ambiente durante 3 horas antes de concentrarse. El residuo
se purificó por cromatografía en columna en gel de sílice (hexano a hexano/acetato de etilo 75 %) para dar el compuesto del título deseado (2,37 g; 5,24 mmol).
RMN 1H (400 MHz, CDCla, 300K) 8 = 7,98 (1H), 7,25 (1H), 6,92 (1H), 6,76 (2H), 6,59 (1H), 6,51 (1H), 4,41 (4H), 4,16 (2H), 3,56 (2H), 2,21 (2H), 2,04 (3H).
Preparación del intermedio 1.9:
15,19-Difluoro-8-[(metilsulfanil)metil]-3,4-dihidro-2H,11H-10,6-(azeno)-12,16-(meteno)-1,5,11,13-benzodioxadiazaciclooctadecino

Una mezcla de 4-{2-[3-({6-doro-4-[(metilsulfanil)metil]piridin-2-il}oxi)propoxi]-4-fluorofenil}-5-fluoropiridin-2-amina (300 mg; 0,66 mmol), aducto de cloro(2-diciclohexilfosfino-2',4',6'-tri-iso-propil-1,1'-bifenil)[2-(2-aminoetil)fenil]paladio (ll) y metil-ferc-butiléter (55 mg; 0,07 mmol; ABCR GmbH & CO. KG) y 2-(diciclohexilfosfino)-2',4',6'-triisopropilbifenilo (32 mg; 0,07 mmol; Aldrich Chemical Company Inc.) y fosfato de potasio (705 mg; 3,32 mmol) en tolueno (50 ml) y NMP (6 ml) se agitó en una atmósfera de argón a 110 °C en un recipiente cerrado durante 150 minutos. Después de enfriar, el lote se diluyó con DCM y acetato de etilo y se lavó con solución acuosa de cloruro de sodio. La capa orgánica se filtró usando un filtro Whatman y se concentró. El residuo se purificó por cromatografía en columna en gel de sílice (hexano a hexano/acetato de etilo 50 %) para dar el producto deseado (192 mg; 0,46 mmol).
RMN 1H (400 MHz, CDCla, 300K) 8 = 8,81 (1H), 8,18 (1H), 7,63 (1H), 7,11 (1H), 6,79 (1H), 6,72 (1H), 6,23 (2H), 4,63 (2H), 4,07 (2H), 3,55 (2H), 2,29 (2H), 2,06 (3H).
Preparación del intermedio 1.10:
2,4,6-trimetilbencensulfonato de (rac)-[{[15,19-difluoro-3,4-dihidro-2H,11H-10,6-(azeno)-12,16-(meteno)-1,5,11,13-benzodioxadiazaciclooctadecin-8-il]metil}(metil)-A4-sulfaniliden]amonio

A o-(mesitilensulfonil)acetohidroxamato de etilo (69 mg; 0,24 mmol; Aldrich Chemical Company Inc.) en dioxano (0,25 ml) se añadió ácido perclórico (70 %; 0,25 ml) gota a gota a 0 °C. Después de la agitación vigorosa adicional durante 10 minutos a 0 °C, se añadió algo de agua fría y el producto MSH (O-(mesitilensulfonil) hidroxilamina) se
extrajo tres veces con DCM. Las capas orgánicas combinadas se lavaron con salmuera y se secaron sobre sulfato de sodio. Esta solución de MSH en d Cm se añadió lentamente a una solución de 15,19-difluoro-8-[(metilsulfanil)metil]-3,4-dihidro-2H,11H-10,6-(azeno)-12,16-(meteno)-1,5,11,13-benzodioxadiazacidooctadecino (100 mg; 0,24 mmol) en DCM (0,25 ml) a 0 °C. La mezcla de reacción se agitó a TA durante 16 horas. El análisis de UPLC-MS indicó una conversión de aproximadamente el 50 %. Se preparó MSH adicional en DCM de acuerdo con el procedimiento descrito usando o-(mesitilensulfonil)acetohidroxamato de etilo (35 mg; 0,24 mmol) y se añadió a la mezcla de reacción a 0 °C. La mezcla de reacción se agitó a temperatura ambiente durante la noche. La mezcla se enfrió hasta 0 °C y la suspensión se filtró por succión. El sólido se lavó con DCM y se secó al vacío para dar el compuesto del título deseado (117 mg; 0,19 mmol).
RMN 1H (400 MHz, DMSO-d6 ) 8 = 2,10 (2H), 2,17 (3H), 3,07 (3H), 4,09 - 4,16 (2H), 4,29 (1H), 4,44 - 4,58 (3H), 6,02 (2H), 6,25 (1H), 6,58 (1H), 6,74 (2H), 6,92 (1H), 7,10 (1H), 7,50 - 7,62 (1H), 8,36 (1H), 8,69 (1H), 9,96 (1H).
Ejemplo 1 - Preparación del producto final: En un matraz secado al horno, en una atmósfera de argón, se disolvió 2,4,6-trimetilbencensulfonato de (rac)-[{[15,19-difluoro-3,4-dihidro-2H,11H-10,6-(azeno)-12,16-(meteno)-1,5,11,13-benzodioxadiazaciclooctadecin-8 -il]metil}(metil)-A4-sulfaniliden]amonio (125 mg; 0,20 mmol) en DMF (0,5 ml) y se enfrió hasta 0 °C. Se añadió carbonato de sodio (25 mg; 0,24 mmol) seguido por N-clorosuccinimida (32 mg, 0,24 mmol) y la mezcla de reacción se agitó durante 15 min a 0 °C. Hexametildisilazano (96 mg; 0,60 mmol) se añadió y la mezcla de reacción se agitó a temperatura ambiente durante 4 h. La mezcla se diluyó con acetato de etilo y THF, se lavó con solución acuosa de cloruro de sodio, se filtró usando un filtro Whatman y se concentró. El residuo se purificó por HPLC preparativa (Autopurificador: condiciones básicas) para dar el compuesto del título deseado (3,6 mg; 0 , 0 1 mmol).
RMN 1H (400 MHz, DMSO-d6 , 300K) 8 = 2,10 (2H), 2,42 -2,48 (2H), 2,87 (3H), 4,09 -4,16 (2H), 4,19 (2H), 4,45 -4,56 (2H), 6,27 (1H), 6,59 (1H), 6,90 (1H), 7,09 (1H), 7,58 (1H), 8,32 (1H), 8,70 (1H), 9,70 (1H).
Ejemplo 1 - preparación alternativa del producto final:
En un matraz secado al horno, en una atmósfera de argón, el hexametildisilazano (26 mg; 0,16 mmol) se añadió a una suspensión de 2,4,6-trimetilbencensulfonato de (rac)-[{[15,19-difluoro-3,4-dihidro-2H,11H-10,6-(azeno)-12,16-(meteno)-1,5,11,13-benzodioxadiazaciclooctadecin-8-il]metil}(metil)-A4-sulfaniliden]amonio (50 mg; 0,08 mmol) en DCM (0,45 ml) a 0 °C. Se añadió carbonato de sodio (9 mg; 0,09 mmol) y la mezcla de reacción se agitó durante 15 min a 0 °C. El diacetato de yodobenceno (28 mg; 0,09 mmol) se añadió y la mezcla de reacción se agitó a 0 °C durante 4 h antes de agitar la mezcla a temperatura ambiente durante la noche. La mezcla se diluyó con DCM, se lavó con solución acuosa de cloruro de sodio, se filtró usando un filtro Whatman y se concentró. El residuo se purificó por HPLC preparativa para dar el compuesto del título deseado (10 mg; 0,02 mmol).
HPLC preparativa:
Instrumento: Waters Autopurificationsystem; columna: YMC Triart 5 p 100x30 mm;
Eluyente A: H2O 0 , 2 % en vol de NH3 acuoso (32 %), eluyente B: MeCN;
Gradiente: 0,00-0,50 min 19 % de B (25->70 ml/min), 0,51-5,50 min 38-58 % de B (70 ml/min),
Barrido de DAD: 210-400 nm
Ejemplo 2:
(rac)-3-(2-{[15,19-difluoro-3,4-dihidro-2H,11H-10,6-(azeno)-12,16-(meteno)-1,5,11,13-benzodioxadiazaciclooctadecin-8-il]metil}-2-metil-2A6-diazatia-1,2-dien-1-il)propan-1-ol

En un matraz secado al horno, en una atmósfera de argón, se añadió 3-aminopropan-1-ol (23 mg; 0,32 mmol) a una suspensión de 2,4,6-trimetilbencensulfonato de (rac)-[{[15,19-difluoro-3,4-dihidro-2H,11H-10,6-(azeno)-12,16-(meteno)-1,5,11,13-benzodioxadiazacidooctadecin-8-il]metil}(metil)-A4-sulfaniliden]amonio (100 mg; 0,16 mmol; ver el Intermedio 1,10) en DCM (0,90 ml) a 0 °C. Se añadió carbonato de sodio (18 mg; 0,17 mmol) y la mezcla de reacción se agitó durante 15 min a 0 °C. Se añadió diacetato de yodobenceno (56 mg; 0,17 mmol) y la mezcla de reacción se agitó a 0 °C durante 4 h. La mezcla se diluyó con DCM, se lavó con solución acuosa de cloruro de sodio, se filtró usando un filtro Whatman y se concentró. El residuo se purificó por HPLC preparativa para dar el compuesto del título deseado (6 mg; 0,01 mmol).
HPLC preparativa:
Instrumento: Waters Autopurificationsystem; columna: Waters XBrigde C185 p 100x30 mm;
Eluyente A: H2O 0,1 % en vol de ácido fórmico (99 %), eluyente B: MeCN;
Gradiente: 0,00-0,50 min 26 % de B (25-> 70 ml/min), 0,51-5,50 min 26-46 % de B (70 ml/min),
Barrido de DAD: 210-400 nm
RMN 1H (400 MHz, DMSO-d6, 300K) 8 = 1,55 (2H), 2,09 (2H), 2,79 (3H), 2,84 - 3,06 (2H), 3,45 (2H), 4,12 (2H), 4,16 -4,31 (2H), 4,36 -4,59 (2H), 6,27 (1H), 6,57 (1H), 6,90 (1H), 7,05-7,12 (1H), 7,58 (1H), 8,32 (1H), 8,70 (1H), 9,71 (1H).
Ejemplo 3:
(rac)-[{[15,19-difluoro-3,4-dihidro-2H,11H-10,6-(azeno)-12,16-(meteno)-1,5,11,13-benzodioxadiazacidooctadecin-8-il]metil}(im ino)metil-A6-sulfaniliden]cianamida

En un matraz secado al horno, en una atmósfera de argón, se añadió cianoazanuro de sodio (20 mg; 0,32 mmol) a una suspensión de 2,4,6-trimetilbencensulfonato de (rac)-[{[15,19-difluoro-3,4-dihidro-2H,11H-10,6-(azeno)-12,16-(meteno)-1,5,11,13-benzodioxadiazaciclooctadecin-8-il]metil}(metil)-A4-sulfaniliden]amonio (100 mg; 0,16 mmol; ver el Intermedio 1,10) en DCM (0,90 ml) a 0 °C. Se añadió carbonato de sodio (18 mg; 0,17 mmol) y la mezcla de reacción se agitó durante 15 min a 0 °C. Se añadió diacetato de yodobenceno (56 mg; 0,17 mmol) y la mezcla de reacción se agitó a 0 °C durante 4 h. La mezcla se diluyó con DCM, se lavó con solución acuosa de cloruro de sodio, se filtró usando un filtro Whatman y se concentró. El residuo se purificó por HPLC preparativa para dar el compuesto del título deseado (7 mg; 0,01 mmol).
HPLC preparativa:
Instrumento: Waters Autopurificationsystem; columna: Waters XBrigde C18 5 p 100x30 mm;
Eluyente A: H2O 0,1 % en vol de ácido fórmico (99 %), eluyente B: MeCN;
Gradiente: 0,00-0,50 min 37 % de B (25-> 70 ml/min), 0,51-5,50 min 37-59 % de B (70 ml/min),
Barrido de DAD: 210-400 nm
RMN 1H (400 MHz, DMSO-d6, 300K) 8 = 2,07 (2H), 3,23 (3H), 4,08 - 4,16 (2H), 4,46 - 4,55 (3H), 4,65 (2H), 6,31 (1H), 6,63 (1H), 6,90 (1H), 7,09 (1H), 7,58 (1H), 8,33 (1H), 8,68 (1H), 9,83 (1H).
Ejemplo 4:
(rac)-8-[(N,S-dimetilsulfonodiimidoil)metil]-15,19-difluoro-3,4-dihidro-2H,11H-10,6-(azeno)-12,16-(meteno)-1,5,11,13-benzodioxadiazaciclooctadecino

En un matraz secado al horno, en una atmósfera de argón, una solución 2 M de metilamina (0,09 ml; 0,18 mmol) en THF se añadió a una suspensión de 2,4,6-trimetilbencensulfonato de (racH{[15,19-difluoro-3,4-dihidro-2H,11H-10,6-(azeno)-12,16-(meteno)-1,5,11,13-benzodioxadiazacidooctadecin-8-il]metil}(metil)-A4-sulfaniliden]amonio (55 mg; 0,09 mmol; ver el Intermedio 1,10) en DCM (0,50 ml) a 0 °C. Se añadió carbonato de sodio (10 mg; 0,10 mmol) y la mezcla de reacción se agitó durante 15 min a 0 °C. Se añadió diacetato de yodobenceno (31 mg; 0,10 mmol) y la mezcla de reacción se agitó a 0 °C durante 4 h. La mezcla se diluyó con DCM, se lavó con solución acuosa de cloruro de sodio, se filtró usando un filtro Whatman y se concentró. El residuo se purificó por HPLC preparativa para dar el compuesto del título deseado ( 2 mg; 0 , 01 mmol).
HPLC preparativa:
Instrumento: Waters Autopurificationsystem; columna: Waters XBrigde C18 5 p 100x30 mm;
Eluyente A: H2O 0,2 % en vol de NH3 acuoso (32 %), eluyente B: MeCN;
Gradiente: 0,00-0,50 min 36 % de B (25->70 ml/min), 0,51-5,50 min 36-56 % de B (70 ml/min),
Barrido de DAD: 210-400 nm
RMN 1H (400 MHz, DMSO-d6 , 300K) 8 = 2,09 (2H), 2,57 - 2,62 (3H), 2,77 (3H), 4,12 (2H), 4,22 (2H), 4,50 (2H), 6,26 (1H), 6,57 (1H), 6,90 (1H), 7,08 (1H), 7,58 (1H), 8,32 (1H), 8,70 (1H), 9,71 (1H).
Ejemplo 5:
16,20-difluoro-9-[(S-metNsulfonodMmidoN)metM]-2,3,4,5-tetrahidro-12H-13,17-(azeno)-11,7-(meteno)-1,6,12,14-benzodioxadiazaciclononadecino

Preparación del intermedio 5.1:
3-(Clorometil)-5-nitrofenol

Se añadió gota a gota cloruro de tionilo (84,0 g; 712 mmol) a una solución agitada de 3-(hidroximetil)-5-nitrofenol (60,0 g; 355 mmol; n.° CAS 180628-74-4 adquirido de Struchem) en DMF (1200 ml) a 0 °C. La mezcla se agitó a 10 °C durante 3 horas. La mezcla se concentró, se diluyó con agua y se extrajo tres veces con acetato de etilo. Las capas orgánicas combinadas se lavaron dos veces con agua y se concentraron para obtener el compuesto del título en bruto (60,0 g, 320 mmol) que se usó sin purificación adicional.
Preparación del intermedio 5.2:
3-[(Metilsulfanil)metil]-5-nitrofenol

A una solución de 3-(clorometil)-5-nitrofenol en bruto (60,0 g; 320 mmol) en acetona (600 ml) a temperatura ambiente se le añadió una solución acuosa de tiometóxido de sodio (21 %, 180 ml). La mezcla se agitó a temperatura ambiente durante 3 horas antes de añadir solución acuosa adicional de tiometóxido de sodio (21 %, 180 ml) y la mezcla se agitó a temperatura ambiente durante la noche. Finalmente, se añadió la solución acuosa adicional de tiometóxido de sodio (21 %, 90 ml) y la mezcla se agitó a temperatura ambiente durante 6 horas. El lote se diluyó con acetato de etilo y una solución acuosa de cloruro de sodio y se extrajo tres veces con acetato de etilo. Las capas orgánicas combinadas se concentraron y el residuo se purificó por cromatografía en columna en gel de sílice (pentano/acetato de etilo 4:1) para obtener el compuesto del título deseado (60,0 g, 302 mmol).
RMN 1H (300 MHz, CDCla, 300K) 8 = 7,71 (1H), 7,57(1H), 7,15 (1H), 3,66 (2H), 1,99 (3H).
Preparación del intermedio 5.3:
4-{3-[(metilsulfanil)metil]-5-nitrofenoxi}butanoato de etilo

Se añadió gota a gota 4-bromobutanoato de etilo (15,8 g; 81 mmol) a una mezcla agitada de 3-[(metilsulfanil)metil]-5-nitrofenol (15,0 g; 75 mmol) y carbonato de potasio (12,5 g; 90 mmol) en DMF (150 ml) a 0 °C. La mezcla se agitó a temperatura ambiente durante la noche. La mezcla se diluyó con agua y se extrajo tres veces con acetato de etilo. Las capas orgánicas combinadas se lavaron dos veces con agua y se concentraron para obtener el compuesto del título en bruto (17,6 g) que se usó sin purificación adicional.
RMN 1H (300 MHz, DMSO-d6, 300K) 8 = 7,74 (1H), 7,53 (1H), 7,30 (1H), 4,03 (3H), 3,75 (2H), 3,50 (1H), 2,42 (3H), 1,99 (1H), 1,92 (3H), 1,14 (3H).
Preparación del intermedio 5.4:
4-{3-[(Metilsulfanil)metil]-5-nitrofenoxi}butan-1-ol

Se añadió gota a gota una solución de DIBAL en hexano (1 N; 176 ml) a una solución agitada de 4-{3-[(metilsulfanil)metil]-5-nitrofenoxi}butanoato de etilo en bruto (17,6 g) en THF seca (400 ml) a -25 °C La mezcla se agitó a 0 °C durante 150 minutos. Se añadió gota a gota agua (200 ml), la mezcla se acidificó con una solución acuosa de cloruro de hidrógeno (1 N) a pH 4-5 y se extrajo tres veces con acetato de etilo. Las capas orgánicas combinadas se concentraron y el residuo se purificó por cromatografía en columna en gel de sílice (pentano/acetato de etilo = 4:1 a 2:1) para obtener el compuesto del título deseado (14,0 g, 51,7 mmol).
RMN 1H (300 MHz, DMSO-d6, 300K) 8 = 7,71 (1H), 7,50 (1H), 7,28 (1H), 4,43 (1H), 4,03 (2H), 3,73 (2H), 3,43 (2H), 1,92 (3H), 1,74 (2H), 1,54 (2H).
Preparación del intermedio 5.5:
2-Cloro-5-fluoro-4-(4-fluoro-2-metoxifenil)pirimidina

Un lote con 2,4-dicloro-5-fluoropirimidina (200 mg; 1,20 mmol; Aldrich Chemical Company Inc.), ácido (4-fluoro-2-metoxifenil)borónico (224 mg; 1,31 mmol; Aldrich Chemical Company Inc.) y tefragu/s(trifenilfosfin)paladio (0) (138 mg; 0,12 mmol) en 1,2-dimetoxietano (3,6 ml) y una solución acuosa 2 M de carbonato de potasio (1,8 ml) se desgasificó usando argón. El lote se agitó en una atmósfera de argón durante 16 horas a 90 °C. Después de enfriar el lote se diluyó con acetato de etilo y se lavó con solución acuosa saturada de cloruro de sodio. La capa orgánica se filtró usando un filtro Whatman y se concentró. El residuo se purificó por cromatografía en columna (hexano/acetato de etilo 1:1) para dar el compuesto del título deseado (106 mg; 0,41 mmol).
RMN 1H (400 MHz, CDCla, 300K) 8 = 8,47 (1H), 7,51 (1H), 6,82 (1H), 6,73 (1H), 3,85 (3H).
Preparación del intermedio 5.6:
2-(2-Cloro-5-fluoropirimidin-4-il)-5-fluorofenol

Se añadió gota a gota una solución de tribromuro de boro en DCM (1 M; 43,3 ml; 47,1 mmol; Aldrich Chemical Company Inc.) a una solución agitada de 2-cloro-5-fluoro-4-(4-fluoro-2-metoxifenil)pirimidina (2,00 g; 7,79 mmol) en DCM (189 ml) a 0 °C. La mezcla se calentó lentamente hasta temperatura ambiente mientras se agitaba durante la noche. La mezcla se diluyó cuidadosamente con una solución acuosa de bicarbonato de sodio en agitación a 0 °C y se agitó a temperatura ambiente durante 1 hora. Se añadió cloruro de sodio sólido y la mezcla se filtró usando un filtro Whatman. La capa orgánica se concentró para dar el compuesto del título en bruto (1,85 g) que se usó sin purificación adicional.
RMN 1H (400 MHz, DMSO-d6, 300K) 8 = 10,80 (1H), 8,90 (1H), 7,50 (1H), 6,83 (1H), 6,78 (1H).
Preparación del intermedio 5.7:
2-C loro-5-fluoro-4-[4-fluoro-2-(4-{3-[(metilsulfanil)metil]-5-nitrofenoxi}butoxi)fenil]pirim idina

Se añadió gota a gota una solución de azodicarboxilato de diisopropilo (0,41 ml; 2,06 mmol) en THF (1,6 ml) a una mezcla de 4-{3-[(metilsulfanil)metil]-5-nitrofenoxi}butan-1-ol (511 mg; 1,88 mmol; ver el Intermedio 5,4), 2-(2-cloro-5-fluoropirimidin-4-il)-5-fluorofenol (500 mg; 2,06 mmol) y trifenilfosfina (541 mg; 2,06 mmol) en THF (8,1 ml) y el lote se agitó a temperatura ambiente durante la noche. La mezcla se concentró y el residuo se purificó por cromatografía en columna en gel de sílice (hexano a hexano/acetato de etilo 50 %) para dar el compuesto del título deseado (579 mg; 1,11 mmol).
RMN 1H (400 MHz, DMSO-d6, 300K) 8 = 8,87 (1H), 7,77 (1H), 7,54 (2H), 7,31 (1H), 7,16 (1H), 6,97 (1H), 4,14 (2H), 4,08 (2H), 3,78 (2H), 1,95 (3H), 1,79 (4H).
Preparación del intermedio 5.8:
3-{4-[2-(2-cloro-5-fluoropirim idin-4-il)-5-fluorofenoxi]butoxi}-5-[(metilsulfanil)metil]anilina

Se añadieron platino al 1 % y vanadio al 2 %, sobre carbón activado (polvo humectado al 50-70 %, 208 mg) a una solución de 2-cloro-5-fluoro-4-[4-fluoro-2-(4-{3-[(metilsulfanil)metil]-5-nitrofenoxi}butoxi)fenil]pirimidina (1060 mg; 2,14 mmol) en metanol (30 ml) y THF (10 ml) y la mezcla se agitó durante 4 horas a temperatura ambiente en una atmósfera de hidrógeno. La mezcla se filtró y el filtrado se concentró para dar el compuesto del título en bruto (851 mg) que se usó sin purificación adicional.
RMN 1H (400 MHz, DMSO-d6, 300K) 8 = 1,65-1,79 (4H), 1,92 (3H), 3,44 (2H), 3,82 (2H), 4,10 (2H), 5,02 (2H) 5,97 (2H), 6,07 (1H), 6,95 (1H), 7,15 (1H), 7,52 (1H), 8,88 (1H).
Preparación del intermedio 5.9:
16,20-difluoro-9-[(metilsulfanil)metil]-2,3,4,5-tetrahidro-12H-13,17-(azeno)-11,7-(meteno)-1,6,12,14-benzodioxadiazaciclononadecino
Una mezcla de 3-{4-[2-(2-doro-5-fluoropirimidin-4-il)-5-fluorofenoxi]butoxi}-5-[(metilsulfanil)metil]anilina en bruto (760 mg), aducto de doro(2-dicidohexilfosfino-2',4',6'-tri-iso-propil-1,1'-bifenil)[2-(2-aminoetil)fenil]paladio (II) y metilferc-butiléter (135 mg; 0 , 1 6 mmol; ABCR GmbH & CO. KG) y 2-(diciclohexilfosfino)-2',4',6-triisopropilbifenilo (78 mg; 0,16 mmol; Aldrich Chemical Company Inc.) y fosfato de potasio (1731 mg; 8,16 mmol) en tolueno (125 ml) y NMP (15 ml) se agitó en una atmósfera de argón a 110 °C durante 3 horas. Después de enfriar, se añadieron más aducto de cloro(2-diciclohexilfosfino-2',4',6'-tri-iso-propil-1,1'-bifenil)[2-(2-aminoetil)fenil]paladio (II) y metil-ferc-butiléter (135 mg; 0,16 mmol) y 2-(diciclohexilfosfino)-2',4',6'-triisopropilbifenilo (78 mg; 0 , 1 6 mmol) y la mezcla se agitó durante 6 horas a 110 °C. Después de enfriar, más aducto de cloro(2-diciclohexilfosfino-2',4',6-trH so-propil-1,1-bifenil)[2-(2-aminoetil)fenil]paladio (II) y metil-ferc-butiléter ( 6 8 mg; 0,08 mmol) y 2-(diciclohexilfosfino)-2',4',6'-triisopropilbifenilo (39 mg; 0,08 mmol) se añadió y la mezcla se agitó durante 3 horas a 110 °C. Después de enfriar, el lote se diluyó con acetato de etilo y se lavó con solución acuosa de cloruro de sodio. La capa orgánica se filtró usando un filtro Whatman y se concentró. El residuo se purificó por cromatografía en columna en gel de sílice (hexano a hexano/acetato de etilo 50 %) para dar el compuesto del título deseado (207 mg; 0,48 mmol).
RMN 1H (400 MHz, DMSO-d6 ): 8 = 1,78-1,91 (4H), 1,96 (3H), 3,55 (2H), 4,05 - 4,16 (2H), 4,26 (2H), 6,36 (1H), 6,59 (1H), 6,87 (1H), 7,10 -7,18 (1H), 7,39 (1H), 7,86 (1H), 8,65 (1H), 9,70 (1H).
Preparación del intermedio 5.10:
2,4,6-trimetilbencensulfonato de (rac)-[{[16,20-difluoro-2,3,4,5-tetrahidro-12H-13,17-(azeno)-11,7-(meteno)-1,6,12,14-benzodioxadiazaciclononadecin-9-il]metil}(metil)-A4-sulfaniliden]amonio

A o-(mesitilensulfonil)acetohidroxamato de etilo (33 mg; 0,12 mmol; Aldrich Chemical Company Inc.) en dioxano (0,12 ml) se le añadió gota a gota ácido perclórico (70 %; 0,12 ml) a 0 °C. Después de la agitación vigorosa adicional durante 10 minutos a 0 °C, se añadió algo de agua fría y el producto MSH (O-(mesitilensulfonil)hidroxilamina) se extrajo tres veces con DCM. Las capas orgánicas combinadas se lavaron con salmuera y se secaron sobre sulfato de sodio. Esta solución de MSH en d Cm se añadió lentamente a una solución de 16,20-difluoro-9-[(metilsulfanil)metil]-2,3,4,5-tetrahidro-12H-13,17-(azeno)-11,7-(meteno)-1,6,12,14-benzodioxadiazaciclononadecino (50 mg; 0,12 mmol) en DCM (0,12 ml) a 0 °C. La mezcla de reacción se agitó a TA durante 22 horas. El análisis de UPLC-MS indicó aproximadamente el 60 % de la conversión. Se preparó MSH adicional en DCM de acuerdo con el procedimiento descrito usando o-(mesitilensulfonil)acetohidroxamato de etilo (17 mg; 0,06 mmol) y se añadió a la mezcla de reacción a 0 °C. La mezcla de reacción se agitó a temperatura ambiente durante la noche. La mezcla se enfrió hasta 0 °C durante 3 horas y la suspensión se filtró por succión. El sólido se lavó con DCM y se secó al vacío para dar el compuesto del título deseado (60 mg; 0,09 mmol).
RMN 1H (400 MHz, DMSO-d6 ) 8 = 1,86 (4H), 2,16 (3H), 3,01 (3H), 4,14 (2H), 4,28 (3H), 4,49 (1H), 5,93 (2H), 6,50 (1H), 6 , 6 8 (1H), 6,74 (2H), 6,89 (1H), 7,16 (1H), 7,39 (1H), 8,02 (1H), 8,69 (1H), 9,94 (1H).
Ejemplo 5 - Preparación del producto final:
En un matraz secado al horno, en una atmósfera de argón, el hexametildisilazano (29 mg; 0,18 mmol) se añadió a una suspensión de 2,4,6-trimetilbencensulfonato de (rac)-[{[16,20-difluoro-2,3,4,5-tetrahidro-12H-13,17-(azeno)-11,7-(meteno)-1,6,12,14-benzodioxadiazaciclononadecin-9-il]metil}(metil)-A4-sulfaniliden]amonio (58 mg; 0,09 mmol) en DCM (0,50 ml) a 0 °C. Se añadió carbonato de sodio (10 mg; 0,10 mmol) y la mezcla de reacción se agitó durante 15 min a 0 °C. Se añadió diacetato de yodobenceno (32 mg; 0,10 mmol) y la mezcla de reacción se agitó a 0 °C durante 4 h antes de agitar la mezcla a temperatura ambiente durante la noche. La mezcla se diluyó con DCM, se lavó con solución acuosa de cloruro de sodio, se filtró usando un filtro Whatman y se concentró. El residuo se purificó por HPLC preparativa para dar el compuesto del título deseado (21 mg; 0,04 mmol).
HPLC preparativa:
Instrumento: Waters Autopurificationsystem; columna: Waters XBrigde C18 5 p 100x30 mm;
Eluyente A: H2O 0,2 % en vol de NH3 acuoso (32 %), eluyente B: MeCN;
Gradiente: 0,00-0,50 min 28 % de B (25->70 ml/min), 0,51-5,50 min 56-76 % de B (70 ml/min),
Barrido de DAD: 210-400 nm
RMN 1H (400 MHz, DMSO-d6, 300K) 8 = 1,86 (4H), 2,78 (3H), 4,13 (s, 4H), 4,27 (2H), 6,49 -6,51 (1H), 6,67 (1H), 6,87 (1H), 7,14 (1H), 7,38 (1H), 7,93 (1H), 8,65 (1H), 9,75 (1H).
Ejemplo 6:
16,20,21-trifluoro-9-[(S-metilsulfonodiimidoil)metil]-2,3,4,5-tetrahidro-12H-13,17-(azeno)-11,7-(meteno)-1,6,12,14-benzodioxadiazaciclononadecino

Preparación del intermedio 6.1:
2-cloro-4-(3,4-difluoro-2-metoxifenil)-5-fluoropirim idina

Un lote con 2,4-dicloro-5-fluoropirimidina (4,04 g; 24,2 mmol; Aldrich Chemical Company Inc.), ácido (3,4-fluoro-2-metoxifenil)borónico (5,00 g; 26,6 mmol; AOBChem USA) y complejo de [1,1-bis(difenilfosfino)ferroceno]dicloropaladio (II) con diclorometano (1,96 g; 2,4 mmol) en 1,2-dimetoxietano (65 ml) y una solución acuosa 2 M de carbonato de potasio (36 ml) se desgasificó usando argón. El lote se agitó en una atmósfera de argón durante 3 horas a 90 °C. Después de enfriar el lote se diluyó con acetato de etilo y se lavó con solución acuosa saturada de cloruro de sodio. La capa orgánica se filtró usando un filtro Whatman y se concentró. El residuo se purificó por cromatografía en columna (d Cm a DCM/EtOH al 50 %) para dar el compuesto del título deseado (5,1 g; 18,4 mmol).
RMN 1H (400 MHz, DMSO-d6): 8 [ppm]= 3,95 (d, 3H), 7,34 -7,43 (m, 2H), 9,01 (d, 1H).
Preparación del intermedio 6.2:
6-(2-cloro-5-fluoropirim idin-4-il)-2,3-difluorofenol

Se añadió gota a gota una solución de tribromuro de boro en DCM (1M; 5,1 ml; 5,1 mmol; Aldrich Chemical Company Inc.) a una solución agitada de 2-cloro-4-(3,4-difluoro-2-metoxifenil)-5-fluoropirimidina (250 mg; 0,9 mmol) en DCM (26 ml) a 0 °C. La mezcla se calentó lentamente hasta temperatura ambiente mientras se agitaba durante la noche. La mezcla se diluyó cuidadosamente con una solución acuosa de bicarbonato de sodio en agitación a 0 °C y se agitó a temperatura ambiente durante 1 hora. Una solución acuosa saturada de cloruro de sodio se añadió y la mezcla se diluyó con acetato de etilo. La mezcla se filtró usando un filtro Whatman y se concentró para dar el compuesto del título en bruto (196 mg) que se usó sin purificación adicional.
RMN 1H (400 MHz, DMSO-d6): 8 [ppm]= 7,02 -7,10 (m, 1H), 7,27 -7,41 (m, 1H), 8,96 (d, 1H), 11,09 (s a, 1H).
Preparación del intermedio 6.3:
2-cloro-4-[3,4-difluoro-2-(4-{3-[(metilsulfanil)metil]-5-nitrofenoxi}butoxi)fenil]-5-fluoropirim idina

Se añadió gota a gota una solución de azodicarboxilato de diisopropilo (0,83 ml; 4,20 mmol) en DCM (3,0 ml) a una mezcla de 4-{3-[(metilsulfanil)metil]-5-nitrofenoxi}butan-1-ol (1,14 g; 4,20 mmol; ver el Intermedio 5,4), 6-(2-cloro-5-fluoropirimidin-4-il)-2,3-difluorofenol (1,00 g; 3,84 mmol) y trifenilfosfina (1,10 g; 4,20 mmol) en DCM (8,0 ml) a 0 °C y el lote se agitó a temperatura ambiente durante la noche. Se añadieron trifenilfosfina (1,00 g; 3,84 mmol) y una solución de azodicarboxilato de diisopropilo (0,76 ml; 3,84 mmol) en DCM (3,0 ml) a temperatura ambiente y la mezcla se agitó durante 16 horas más. La mezcla se diluyó con agua y se extrajo tres veces con acetato de etilo. La fase orgánica combinada se filtró usando un filtro Whatman y se concentró. El residuo se purificó por cromatografía en columna en gel de sílice (hexano a hexano/acetato de etilo 50 %) para dar el compuesto del título deseado (1,62 g; 3,15 mmol). RMN 1H (400 MHz, DMSO-d6): 8 [ppm] = 1,62 - 1,81 (m, 4H), 1,96 (s, 3H), 3,79 (s, 2H), 4,02 (t, 2H), 4,13 - 4,23 (m, 2H), 7,30 - 7,43 (m, 3H), 7,54 (t, 1H), 7,78 (t, 1H), 8,99 (d, 1H).
Preparación del intermedio 6.4:
3-{4-[6-(2-cloro-5-fluoropirim idin-4-il)-2,3-difluorofenoxi]butoxi}-5-[(metilsulfanil)metil]anilina

Se añadieron platino al 1 % y vanadio al 2 %, sobre carbón activado (polvo humectado al 50-70 %, 200 mg) a una solución de 2-cloro-4-[3,4-difluoro-2-(4-{3-[(metilsulfanil)metil]-5-nitrofenoxi}butoxi)fenil]-5-fluoropirimidina (815 mg; 1,59 mmol) en metanol (30 ml) y la mezcla se agitó durante 1 hora a temperatura ambiente en una atmósfera de hidrógeno. Se añadió más platino al 1 % y vanadio al 2 %, sobre carbón activado (polvo humectado al 50-70 %, 200 mg) y la mezcla se agitó durante 1 hora a temperatura ambiente en una atmósfera de hidrógeno. La mezcla se filtró y el filtrado se concentró para dar el compuesto del título en bruto (793 mg) que se usó sin purificación adicional. RMN 1H (400 MHz, DMSO-d6): 8 [ppm] = 1,57 - 1,77 (m, 4H), 1,94 (s, 3H), 3,46 (s, 2H), 3,78 (t, 2H), 4,17 (t, 2H), 5,04 (s, 2H), 5,95 - 6,00 (m, 2H), 6,09 (t, 1H), 7,34 - 7,44 (m, 2H), 9,00 (d, 1H).
Preparación del intermedio 6.5:
16,20,21-trifluoro-9-[(metilsulfanil)metil]-2,3,4,5-tetrahidro-12H-13,17-(azeno)-11,7-(meteno)-1,6,12,14-benzodioxadiazaciclononadecino

Una mezcla de 3-{4-[6-(2-cloro-5-fluoropirimidin-4-il)-2,3-difluorofenoxi]butoxi}-5-[(metilsulfanil)metil]anilina en bruto (500 mg), aducto de cloro(2-diciclohexilfosfino-2',4',6'-tri-iso-propil-1,1'-bifenil)[2-(2-aminoetil)fenil]paladio (II) y metilferc-butiléter (85 mg; 0,10 mmol; ABCR GmbH & CO. KG) y 2-(diciclohexilfosfino)-2',4',6'-triisopropilbifenilo (49 mg;
0,10 mmol; Aldrich Chemical Company Inc.) y fosfato de potasio (1097 mg; 5,17 mmol) en tolueno (77 ml) y NMP (9 ml) se agitó en una atmósfera de argón a 110 °C durante 4 horas. Después de enfriar, el lote se diluyó con solución acuosa de cloruro de sodio y se extrajo con acetato de etilo/THF (1:1; 2x). La fase orgánica combinada se filtró usando un filtro Whatman y se concentró. El residuo se purificó por cromatografía en columna en gel de sílice (hexano a hexano/acetato de etilo 50 %) para dar el compuesto del título deseado (96 mg; 0,21 mmol).
RMN 1H (400 MHz, DMSO-d6 ): 8 = 1,76 - 1,92 (m, 4H), 1,96 (s, 3H), 3,55 (s, 2H), 4,18 - 4,32 (m, 4H), 6,36 (t, 1H), 6,62 (s, 1H), 7,20 - 7,35 (m, 2H), 8,01 (t, 1H), 8,70 (d, 1H), 9,78 (s, 1H).
Preparación del intermedio 6.6:
(racHmetil{[16,20,21-trifluoro-2,3,4,5-tetrahidro-12H-13,17-(azeno)-11,7-(meteno)-1,6,12,14-benzodioxadiazaciclononadecin-9-il]metil}-A4-sulfaniliden)amonio 2,4,6-trimetilbencensulfonato

A o-(mesitilensulfonil)acetohidroxamato de etilo (32 mg; 0,11 mmol; Aldrich Chemical Company Inc.) en dioxano (0,11 ml) se le añadió gota a gota ácido perclórico (70 %; 0,11 ml) a 0 °C. Después de la agitación vigorosa adicional durante 10 minutos a 0 °C, se añadió algo de agua fría y el producto MSH (O-(mesitilensulfonil)hidroxilamina) se extrajo tres veces con DCM. Las capas orgánicas combinadas se lavaron con salmuera y se secaron sobre sulfato de sodio. Esta solución de MSH en DCM se añadió lentamente a una solución de 16,20,21-trifluoro-9-[(metilsulfanil)metil]-2.3.4.5- tetrahidro-12H-13,17-(azeno)-11,7-(meteno)-1,6,12,14-benzodioxadiazaciclononadecino (50 mg; 0,11 mmol) en DCM (0,12 ml) a 0 °C. La mezcla de reacción se agitó a TA durante 20 horas. La mezcla se mantuvo a 0 °C durante 16 horas. Se añadió dietliéter (1 ml) y la mezcla se mantuvo durante la noche a 0 °C antes de filtrar la suspensión resultante por succión. El sólido se lavó con dietiléter y se secó al vacío para dar el compuesto del título deseado (40 mg; 0,06 mmol).
RMN 1H (400 MHz, DMSO-d6 ) 8 = 1,79 - 1,92 (m, 4H), 2,16 (s, 3H), 3,01 (s, 3H), 4,23 - 4,33 (m, 5H), 4,49 (d, 1H), 5,90 (s a, 2H), 6,51 (s, 1H), 6,69 - 6,74 (m, 3H), 7,22 - 7,29 (m, 1H), 7,31 - 7,39 (m, 1H), 8,17 (s, 1H), 8,74 (d, 1H), 10,02 (s, 1H).
Ejemplo 6 - Preparación del producto final:
En un matraz secado al horno, en una atmósfera de argón, hexametildisilazano (19 mg; 0,12 mmol) se añadió a una suspensión de 2,4,6-trimetilbencensulfonato de 2,4,6-trimetilbencensulfonato de (rac)-(metil{[16,20,21-trifluoro-2.3.4.5- tetrahidro-12H-13,17-(azeno)-11,7-(meteno)-1,6,12,14-benzodioxadiazaciclononadecin-9-il]metil}-A4-sulfaniliden)amonio (40 mg; 0,06 mmol) en DCM (0,40 ml) a 0 °C. Se añadió carbonato de sodio (7 mg; 0,07 mmol) y la mezcla de reacción se agitó durante 15 min a 0 °C. Se añadió diacetato de yodobenceno (21 mg; 0,05 mmol) y la mezcla de reacción se agitó a 0 °C durante 4 h antes de agitar la mezcla a temperatura ambiente durante la noche. La mezcla se diluyó con DCM, se lavó con solución acuosa de cloruro de sodio, se filtró usando un filtro Whatman y se concentró. El residuo se purificó por HPLC preparativa para dar el compuesto del título deseado ( 8 mg; 0,02 mmol).
HPLC preparativa:
Instrumento: Waters Autopurificationsystem; columna: Waters XBrigde C18 5 p 100x30 mm;
Eluyente A: H2O 0,2 % en vol de NH3 acuoso (32 %), eluyente B: MeCN;
Gradiente: 0,00-0,50 min 28 % de B (25->70 ml/min), 0,51-5,50 min 56-76 % de B (70 ml/min),
Barrido de DAD: 210-400 nm
RMN 1H (400 MHz, DMSO-d6 , 300K) 8 = 1,73 - 1,98 (m, 4H), 2,25 -2,37 (m, 2H), 2,79 (s, 3H), 4,14 (s, 2H), 4,21 -4,31 (m, 4H), 6,49 - 6,52 (m, 1H), 6,70 (s, 1H), 7,20 - 7,35 (m, 2H), 8,08 (t, 1H), 8,70 (d, 1H), 9,82 (s, 1H).
Ejemplo 7:
16,21-difluoro-9-[(S-metNsulfonodMmidoN)metM]-2,3,4,5-tetrahidro-12H-13,17-(azeno)-11,7-(meteno)-1,6,12,14-benzodioxadiazaciclononadecino

Preparación del intermedio 7.1:
2-cloro-5-fluoro-4-(3-fluoro-2-metoxifenil)pirim idina

Un lote con 2,4-dicloro-5-fluoropirimidina (4,96 g; 29,7 mmol; Aldrich Chemical Company Inc.), ácido (3-fluoro-2-metoxifenil)borónico (5,56 g; 32,7 mmol; ABCR GmbH & CO. KG) y complejo de [1,1-bis(difenilfosfino)ferroceno]didoropaladio (II) con diclorometano (2,43 g; 2,9 mmol) en 1,2-dimetoxietano (80 ml) y una solución acuosa 2 M de carbonato de potasio (45 ml) se desgasificó usando argón. El lote se agitó en una atmósfera de argón durante 3 horas a 90 °C. Después de enfriar el lote se diluyó con acetato de etilo y se lavó con solución acuosa saturada de cloruro de sodio. La capa orgánica se filtró usando un filtro Whatman y se concentró. El residuo se purificó por cromatografía en columna (d Cm a DCM/EtOH al 50 %) para dar el compuesto del título deseado (6,7 g; 26,0 mmol).
RMN 1H (400 MHz, DMSO-d6): 8 [ppm]= 3,87 (d, 3H), 7,26 -7,37 (m, 2H), 7,55 (ddd, 1H), 9,01 (d, 1H).
Preparación del intermedio 7.2:
2-(2-cloro-5-fluoropirim idin-4-il)-6-fluorofenol

Se añadió gota a gota una solución de tribromuro de boro en DCM (1M; 65,0 ml; 65,0 mmol; Aldrich Chemical Company Inc.) a una solución agitada de 2-cloro-5-fluoro-4-(3-fluoro-2-metoxifenil)pirimidina (3,0 g; 11,69 mmol) en DCM (312 ml) a 0 °C. La mezcla se calentó lentamente hasta temperatura ambiente mientras se agitaba durante la noche. La mezcla se diluyó cuidadosamente con una solución acuosa de bicarbonato de sodio en agitación a 0 °C y se agitó a temperatura ambiente durante 1 hora antes de extraerlo tres veces con DCM. La fase orgánica combinada se filtró usando un filtro Whatman y se concentró para dar el compuesto del título en bruto (2,8 g) que se usó sin purificación adicional.
RMN 1H (400 MHz, DMSO-d6): 8 [ppm]= 6,99 (td, 1H), 7,26 (dt, 1H), 7,41 (ddd, 1H), 8,96 (d, 1H), 10,45 (s, 1H).
Preparación del intermedio 7.3:
2-cloro-5-fluoro-4-[3-fluoro-2-(4-{3-[(metilsulfanil)metil]-5-nitrofenoxi}butoxi)fenil]pirim idina

Se añadió gota a gota una solución de azodicarboxilato de diisopropilo (0,89 ml; 4,51 mmol) en DCM (3,0 ml) a una mezcla de 4-{3-[(metilsulfanil)metil]-5-nitrofenoxi}butan-1-ol (1,23 g; 4,51 mmol; ver el Intermedio 5,4), 2-(2-cloro-5-fluoropirimidin-4-il)-6-fluorofenol (1,00 g; 4,12 mmol) y trifenilfosfina (1,18 g; 4,51 mmol) en DCM (8,0 ml) a 0 °C y el lote se agitó a temperatura ambiente durante la noche. Otra porción de trifenilfosfina (1,08 g; 4,12 mmol) y una solución de azodicarboxilato de diisopropilo (0,81 ml; 4,12 mmol) en DCM (3,0 ml) se añadieron a temperatura ambiente y la mezcla se agitó durante otras 16 horas. La mezcla se concentró y el residuo se purificó por cromatografía en columna en gel de sílice (hexano a hexano/acetato de etilo 50 %) para dar el compuesto del título deseado (2,00 g), que aún contenía algunas impurezas.
Preparación del intermedio 7.4:
3-{4-[2-(2-cloro-5-fluoropirim idin-4-il)-6-fluorofenoxi]butoxi}-5-[(metilsulfanil)metil]anilina

Se añadió platino al 1 % y vanadio al 2 %, sobre carbón activado (polvo humectado al 50-70 %, 200 mg) a una solución de 2-cloro-5-fluoro-4-[3-fluoro-2-(4-{3-[(metilsulfanil)metil]-5-nitrofenoxi}butoxi)fenil]pirimidina (1,04 g) en metanol (30 ml) y la mezcla se agitó durante 80 minutos a temperatura ambiente en una atmósfera de hidrógeno. Se añadió más platino al 1 % y vanadio al 2 %, sobre carbón activado (polvo humectado al 50-70 %, 200 mg) y la mezcla se agitó durante 2 horas a temperatura ambiente en una atmósfera de hidrógeno. La mezcla se filtró y el filtrado se concentró para dar el compuesto del título en bruto (951 mg) que se usó sin purificación adicional.
RMN 1H (400 MHz, DMSO-d6): 8 [ppm] = 1,55 - 1,80 (m, 4H), 1,94 (s, 3H), 3,41 -3,51 (m, 2H), 3,77 (t, 2H), 4,00 -4,13 (m, 2H), 5,04 (s a, 2H), 5,95 - 6,00 (m, 2H), 6,09 (t, 1H), 7,26 - 7,37 (m, 2H), 7,54 (ddd, 1H), 9,00 (d, 1H).
Preparación del intermedio 7.5:
16,21-difluoro-9-[(metilsulfanil)metil]-2,3,4,5-tetrahidro-12H-13,17-(azeno)-11,7-(meteno)-1,6,12,14-benzodioxadiazaciclononadecino

Una mezcla de 3-{4-[2-(2-cloro-5-fluoropirimidin-4-il)-6-fluorofenoxi]butoxi}-5-[(metilsulfanil)metil]anilina en bruto (510 mg), aducto de cloro(2-diciclohexilfosfino-2',4',6'-tri-iso-propil-1,1'-bifenil)[2-(2-aminoetil)fenil]paladio (II) y metilferc-butiléter (91 mg; 0,11 mmol; ABCR GmbH & CO. KG) y 2-(diciclohexilfosfino)-2',4',6'-triisopropilbifenilo (52 mg; 0,11 mmol; Aldrich Chemical Company Inc.) y fosfato de potasio (1162 mg; 5,47 mmol) en tolueno (81 ml) y NMP (10 ml) se agitó en una atmósfera de argón a 110 °C durante la noche. Después de enfriar, el lote se diluyó con solución acuosa de cloruro de sodio y se extrajo dos veces con acetato de etilo/THF (1:1). La fase orgánica combinada se filtró usando un filtro Whatman y se concentró. El residuo se purificó por cromatografía en columna en gel de sílice (hexano a hexano/acetato de etilo 50 %) para dar el compuesto del título deseado (171 mg; 0,40 mmol).
RMN 1H (400 MHz, DMSO-d6): 8 = 1,79 (s a, 2H), 1,86 (br d, 2H), 1,96 (s, 3H), 3,55 (s, 2H), 4,18 (s a, 2H), 4,21 -4,28 (m, 2H), 6,36 (s, 1H), 6,62 (s, 1H), 7,17 (dt, 1H), 7,26 -7,32 (m, 1H), 7,45 (ddd, 1H), 8,03 (s, 1H), 8,70 (d, 1H), 9,76 (s, 1H).
Preparación del intermedio 7.6:
2,4,6-trimetilbencensulfonato de (rac)-[{[16,21-difluoro-2,3,4,5-tetrahidro-12H-13,17-(azeno)-11,7-(meteno)-1,6,12,14-benzodioxadiazaciclononadecin-9-il]metil}(metil)-A4-sulfaniliden]amonio

A o-(mesitilensulfonil)acetohidroxamato de etilo (80 mg; 0,28 mmol; Aldrich Chemical Company Inc.) en dioxano (0,28 ml) se le añadió gota a gota ácido perclórico (70 %; 0,28 ml) a 0 °C. Después de la agitación vigorosa adicional durante 10 minutos a 0 °C, se añadió algo de agua fría y el producto MSH (O-(mesitilensulfonil)hidroxilamina) se extrajo tres veces con DCM. Las capas orgánicas combinadas se lavaron con salmuera y se secaron sobre sulfato de sodio. Esta solución de MSH en d Cm se añadió lentamente a una solución de 16,21-difluoro-9-[(metilsulfanil)metil]-2,3,4,5-tetrahidro-12H-13,17-(azeno)-11,7-(meteno)-1,6,12,14-benzodioxadiazaciclononadecino (120 mg; 0,28 mmol) en DCM (0,28 ml) a 0 °C. La mezcla de reacción se agitó a temperatura ambiente durante la noche. La mezcla se mantuvo a 0 °C durante 16 horas. Se añadió dietiléter (1 ml) y la mezcla se mantuvo durante la noche a 0 °C, antes de filtrar la suspensión resultante por succión. El sólido se lavó con dietiléter y se secó al vacío para dar el compuesto del título deseado (64 mg; 0,10 mmol).
RMN 1H (400 MHz, DMSO-d6) 8 = 1,76 - 1,91 (m, 4H), 2,17 (s, 3H), 3,01 (s, 3H), 4,15 - 4,33 (m, 5H), 4,49 (d, 1H), 5,94 (s, 2H), 6,50 (s, 1H), 6,69 - 6,75 (m, 3H), 7,13 - 7,23 (m, 1H), 7,25 - 7,36 (m, 1H), 7,42 - 7,51 (m, 1H), 8,21 (s, 1H), 8,74 (d, 1H), 10,01 (s, 1H).
Ejemplo 7 - Preparación del producto final:
En un matraz secado al horno, en una atmósfera de argón, se añadió hexametildisilazano (32 mg; 0,20 mmol) a una suspensión de 2,4,6-trimetilbencensulfonato de (rac)-[{[16,21-difluoro-2,3,4,5-tetrahidro-12H-13,17-(azeno)-11,7-(meteno)-1,6,12,14-benzodioxadiazaciclononadecin-9-il]metil}(metil)-A4-sulfaniliden]amonio (64 mg; 0,10 mmol) en DCM (0,60 ml) a 0 °C. Se añadió carbonato de sodio (12 mg; 0,11 mmol) y la mezcla de reacción se agitó durante 15min a 0 °C. Se añadió diacetato de yodobenceno (35 mg; 0,11 mmol) y la mezcla de reacción se agitó a 0 °C durante 4 h antes de agitar la mezcla a temperatura ambiente durante la noche. La mezcla se diluyó con DCM, se lavó con solución acuosa de cloruro de sodio, se filtró usando un filtro Whatman y se concentró. El residuo se purificó por HPLC preparativa para dar el compuesto del título deseado (6 mg; 0,01 mmol).
HPLC preparativa:
Instrumento: Waters Autopurificationsystem; columna: Waters XBrigde C18 5 p 100x30 mm;
Eluyente A: H2O 0,2 % en vol de NH3 acuoso (32 %), eluyente B: MeCN;
Gradiente: 0,00-0,50 min 28 % de B (25->70 ml/min), 0,51-5,50 min 56-76 % de B (70 ml/min),
Barrido de DAD: 210-400 nm
RMN 1H (400 MHz, DMSO-d6, 300K) 8 = 1,76 - 1,93 (m, 4H), 2,26 - 2,33 (m, 2H), 2,79 (s, 3H), 4,11 - 4,29 (m, 6H), 6,50 (s, 1H), 6,70 (s, 1H), 7,16 (td, 1H), 7,25 - 7,30 (m, 1H), 7,45 (ddd, 1H), 8,09 - 8,13 (m, 1H), 8,70 (d, 1H), 9,81 (s, 1H).
Ejemplo 8:
15,19-difluoro-8-[(S-metNsulfonodMmidoN)metM]-3,4-dihidro-2H,11H-10,6-(azeno)-12,16-(meteno)-1,5,11,13-benzodioxadiazaciclooctadecino-7-carbonitrilo

Preparación del intermedio 8.1:
15,19-difluoro-8-[(metilsulfanil)metil]-3,4-dihidro-2H,11H-10,6-(azeno)-12,16-(meteno)-1,5,11,13-benzodioxadiazaciclooctadecino-7-carbonitrilo

Se añadió N-yodosuccinimida (94 mg; 0,42 mmol) a una solución de 15,19-difluoro-8-[(metilsulfanil)metil]-3,4-dihidro-H,11H-10,6-(azeno)-12,16-(meteno)-1,5,11,13-benzodioxadiazacidooctadecino (145 mg; 0,35 mmol; ver el Intermedio 1,9) en DMF (1,0 ml) a temperatura ambiente. La mezcla de reacción se agitó durante 2 horas, antes de diluir con DCM y lavar con agua. La fase orgánica se concentró para dar el producto en bruto 15,19-difluoro-7-yodo-8-[(metilsulfanil)metil]-3,4-dihidro-2H,11H-10,6-(azeno)-12,16-(meteno)-1,5,11,13-benzodioxadiazaciclooctadecino. El producto en bruto se volvió a disolver en DMSO (2,0 ml), se añadió cianuro de cobre (I) (37 mg; 0,42 mmol) y la mezcla de reacción se agitó a 140 °C durante 1 hora. Después de enfriar, la mezcla de reacción se purificó por HPLC preparativa para dar el compuesto del título deseado (70 mg; 0,15 mmol).
HPLC preparativa:
Instrumento: Waters Autopurificationsystem; columna: Waters XBrigde C18 5 p 100x30 mm;
Eluyente A: H2O 0,1 % en vol de ácido fórmico (99 %), eluyente B: MeCN;
Gradiente: 0,00-0,50 min 26 % de B (25-> 70 ml/min), 0,51-5,50 min 26-46 % de B (70 ml/min),
Barrido de DAD: 210-400 nm
RMN 1H (400 MHz, DMSO-d6, 300K) 8 = 2,06 - 2,18 (m, 5H), 3,68 (s, 2H), 4,09 - 4,16 (m, 2H), 4,58 - 4,68 (m, 2H), 6,66 (s, 1H), 6,91 (td, 1H), 7,10 (dd, 1H), 7,59 (ddd, 1H), 8,40 (d, 1H), 8,63 (d, 1H), 10,37 (s, 1H).
Preparación del intermedio 8.2:
2,4,6-trimetilbencensulfonato de (rac)-[{[7-ciano-15,19-difluoro-3,4-dihidro-2H,11H-10,6-(azeno)-12,16-(meteno)-1,5,11,13-benzodioxadiazaciclooctadecin-8-il]metil}(metil)-A4-sulfaniliden]amonio

A o-(mesitilensulfonil)acetohidroxamato de etilo (32 mg; 0,11 mmol; Aldrich Chemical Company Inc.) en dioxano (0,12 ml) se le añadió gota a gota ácido perclórico (70 %; 0,12 ml) a 0 °C. Después de la agitación vigorosa adicional durante 10 minutos a 0 °C, se añadió algo de agua fría y el producto MSH (O-(mesitilensulfonil)hidroxilamina) se extrajo tres veces con DCM. Las capas orgánicas combinadas se lavaron con salmuera y se secaron sobre sulfato de sodio. Esta solución de MSH en DCM se añadió lentamente a una suspensión de 15,19-difluoro-8-[(metilsulfanil)metil]-3,4-dihidro-2H,11H-10,6-(azeno)-12,16-(meteno)-1,5,11,13-benzodioxadiazaciclooctadecino-7-carbonitrilo (50 mg; 0,11 mmol) en DCM (0,11 ml) a 0 °C. La mezcla de reacción se agitó a temperatura ambiente durante la noche. La mezcla se mantuvo a 0 °C durante 16 horas. La mezcla se mantuvo durante la noche a 0 °C, antes de filtrar la suspensión resultante por succión. El sólido se lavó con DCM y se secó al vacío para dar el compuesto del título deseado (66 mg; 0,10 mmol).
RMN 1H (400 MHz, DMSO-d6) 8 = 2,11 -2,21 (m, 5H), 3,17 (s, 3H), 4,11 -4,17 (m, 2H), 4,49 (d, 1H), 4,62 -4,71 (m, 3H), 6,14 (s, 2H), 6,74 (d, 3H), 6,93 (td, 1H), 7,07 - 7,15 (m, 1H), 7,61 (ddd, 1H), 8,46 (d, 1H), 8,61 (d, 1H), 10,70 (s, 1H).
Ejemplo 8 - Preparación del producto final:
En un matraz secado al horno, en una atmósfera de argón, se añadió hexametildisilazano (31 mg; 0,19 mmol) a una suspensión de 2,4,6-trimetilbencensulfonato de (rac)-[{[7-ciano-15,19-difluoro-3,4-dihidro-2H,11H-10,6-(azeno)-12,16-(meteno)-1,5,11,13-benzodioxadiazaciclooctadecin-8-il]metil}(metil)-A4-sulfaniliden]amonio (63 mg; 0,10 mmol) en DCM (0,50 ml) a 0 °C. Se añadió carbonato de sodio (11 mg; 0,11 mmol) y la mezcla de reacción se agitó durante 15min a 0 °C. Se añadió diacetato de yodobenceno (34 mg; 0,11 mmol) y la mezcla de reacción se agitó a 0 °C durante 4 h antes de agitar la mezcla a temperatura ambiente durante la noche. La mezcla se diluyó con DCM, se lavó con solución acuosa de cloruro de sodio, se filtró usando un filtro Whatman y se concentró. El residuo se purificó por HPLC preparativa para dar el compuesto del título deseado (3 mg; 0,01 mmol).
HPLC preparativa:
Instrumento: Waters Autopurificationsystem; columna: Waters XBrigde C18 5 p 100x30 mm;
Eluyente A: H2O 0,2 % en vol de NH3 acuoso (32 %), eluyente B: MeCN;
Gradiente: 0,00-0,50 min 28 % de B (25->70 ml/min), 0,51-5,50 min 56-76 % de B (70 ml/min),
Barrido de DAD: 210-400 nm
RMN 1H (400 MHz, DMSO-d6, 300K) 8 = 2,12 (brd, 2H), 2,96 (s, 3H), 4,05 -4,18 (m, 2H), 4,36 (s, 2H), 4,56 -4,67 (m, 2H), 6,78 (s, 1H), 6,85 -6,94 (m, 1H), 7,08 (br d, 1H), 7,11 (br d, 1H), 7,59 (ddd, 1H), 8,40 (d, 1H), 8,60 (d, 1H), 10,42 (s, 1H).
Ejemplo 9:
(rac)-9-[(N-ciclopropil-S-metilsulfonodNmidoil)metil]-16,20-difluoro-2,3,4,5-tetrahidro-12H-13,17-(azeno)-11,7-(meteno)-1,6,12,14-benzodioxadiazaciclononadecino

En un matraz secado al horno, en una atmósfera de argón, se añadieron carbonato de sodio (17 mg; 0,16 mmol) y N-clorosuccinimida (21 mg; 0,16 mmol) a 2,4,6-trimetilbencensulfonato de (rac)-[{[16,20-difluoro-2,3,4,5-tetrahidro-12H-13,17-(azeno)-11,7-(meteno)-1,6,12,14-benzodioxadiazacidononadecin-9-il]metil}(metil)-A4-sulfaniliden]amonio (84 mg; 0,13 mmol; ver el Intermedio 5,10) en DMF (1,2 ml) a 0 °C y la mezcla se agitó durante 15 min a 0 °C. Se añadió ciclopropanamina (22 mg; 0,39 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante la noche. La mezcla se diluyó con solución acuosa de cloruro de sodio y se extrajo tres veces con DCM. La capa orgánica combinada se filtró usando un filtro Whatman y se concentró. El residuo se purificó por HPLC preparativa para dar el compuesto del título deseado ( 6 mg; 0 , 01 mmol).
HPLC preparativa:
Instrumento: Waters Autopurificationsystem; columna: Waters XBrigde C18 5 p 100x30 mm;
Eluyente A: H2O 0,2 % en vol de NH3 acuoso (32 %), eluyente B: MeCN;
Gradiente: 0,00-0,50 min 28 % de B (25->70 ml/min), 0,51-5,50 min 56-76 % de B (70 ml/min),
Barrido de DAD: 210-400 nm
RMN 1H (400 MHz, DMSO-d6 , 300K) 8 = 0,22 - 0,44 (m, 4H), 1,86 (s a, 4H), 2,09 (s, 1H), 2,42 - 2,48 (m, 1H), 2,72 (s, 3H), 4,09 -4,24 (m, 4H), 4,27 (s a, 2H), 6,51 (s, 1H), 6 , 6 8 (s, 1H), 6,87 (td, 1H), 7,15 (dd, 1H), 7,35 -7,42 (m, 1H), 7,94 (s, 1H), 8 , 6 6 (d, 1H), 9,77 (s, 1H).
Ejemplo 10:
(rac)-9-[(N,S-dimetilsulfonodiimidoil)metil]-16,20-difluoro-2,3,4,5-tetrahidro-12H-13,17-(azeno)-11,7-(meteno)-1,6,12,14-benzodioxadiazaciclononadecino

En un matraz secado al horno, en una atmósfera de argón, se añadió una solución de metilamina en THF (2 M, 0,16 ml; 0,31 mmol) a una suspensión de 2,4,6-trimetilbencensulfonato de (racH{[16,20-difluoro-2,3,4,5-tetrahidro-12H-13,17-(azeno)-11,7-(meteno)-1,6,12,14-benzodioxadiazacidononadecin-9-il]metil}(metil)-A4-sulfaniliden]amonio (ver el Intermedio 5,10; 100 mg; 0,16 mmol) en DCM (0,90 ml) a 0 °C. Se añadió carbonato de sodio (18 mg; 0,17 mmol) y la mezcla de reacción se agitó durante 15 min a 0 °C. Se añadió diacetato de yodobenceno (55 mg; 0,17 mmol) y la mezcla de reacción se agitó a 0 °C durante 4 h antes de agitar la mezcla a temperatura ambiente durante la noche. La mezcla se diluyó con DCM, se lavó con solución acuosa de cloruro de sodio, se filtró usando un filtro Whatman y se concentró. El residuo se purificó por HPLC preparativa (Autopurificador: condiciones ácidas) para dar el compuesto del título deseado (9 mg; 0,02 mmol).
RMN 1H (500 MHz, DMSO-d6): desplazamiento [ppm] = 1,84 - 1,92 (m, 4H), 2,55 (s, 3H), 2,69 (s, 3H), 4,10 -4,15 (m, 2H), 4,17 (s, 2H), 4,26 - 4,29 (m, 2H), 6,48 (s, 1H), 6,66 (s, 1H), 6,87 (td, 1H), 7,15 (dd, 1H), 7,38 (ddd, 1H), 7,94 (s, 1H), 8,65 (d, 1H), 9,76 (s, 1H).
La siguiente Tabla 1 proporciona un resumen de los compuestos descritos en la sección de ejemplos:
Tabla 1

(continuación)

(continuación)


Resultados:
Tabla 2: Inhibición para CDK9 y CDK2 de compuestos de acuerdo con la presente invención
Los valores de CI50 (concentración inhibitoria a 50 % del efecto máximo) se indican en nM, "n.t" significa que los compuestos no han sido evaluados en el ensayo respectivo.
©: Ejemplo Número
©: ensayo de quinasa CDK9: CDK9/CycT1 como se describe bajo el Procedimiento 1a. de materiales y procedimientos
®: ensayo de quinasa CDK2: CDK2/CycE como se describe bajo el Procedimiento 2. de materiales y procedimientos
©: Selectividad de CDK9 sobre CDK2: CI50 (CDK2)/CI50 (CDK9) de acuerdo con los Procedimientos 1a. y 2a. de materiales y procedimientos
©: ensayo de quinasa alto ATP CDK9: CDK9/CycT1 como se describe bajo el Procedimiento 1b. de materiales y procedimientos
©: ensayo de quinasa alto ATP CDK2: CDK2/CycE como se describe bajo el Procedimiento 2b. de materiales y procedimientos
©: Selectividad alto ATP CDK9 sobre alto ATP CDK2: CI50 (alta ATP CDK2)/CI50 (alta ATP CDK9) de acuerdo con los Procedimientos 1b. y 2b. de materiales y procedimientos
Debe notarse que en los ensayos de CDK9, como se describe anteriormente en los Procedimientos 1a. y 1b. de materiales y procedimientos, el poder de resolución se encuentra limitado por las concentraciones de enzima, el límite inferior para CI50 es de aproximadamente 1-2 nM en el ensayo CDK9 alto ATP y 2-4 nM en el ensayo CDK bajo ATP. Para los compuestos que exhiben valores de CI50 en este intervalo, la verdadera afinidad a CDK9 y por lo tanto la selectividad de CDK9 sobre CDK2 pueden ser aún mayores, por ejemplo, para estos compuestos los factores de selectividad calculados en las columnas 4 y 7 de la Tabla 2, posterior, son valores mínimos, también podrían ser mayores.
Tabla 2

(continuación)

(continuación)



Tablas 3a y 3b: Inhibición de la proliferación de células HeLa, HeLa-MaTu-ADR, NCI-H460, DU145, Caco-2, B16F10, A2780 y MOLM-13 por parte de compuestos de acuerdo con la presente invención, determinada como se describe bajo el Procedimiento 3 de materiales y procedimientos. Todos los valores de CI50 (concentración inhibitoria a 50 % del efecto máximo) se indican en nM, "n.t." significa que los compuestos no han sido evaluados en el ensayo respectivo.
© Número de Ejemplo
© Inhibición de proliferación celular HeLa
® Inhibición de proliferación celular HeLa-MaTu-ADR
© Inhibición de proliferación celular NCI-H460
© Inhibición de proliferación celular DU145
© Inhibición de proliferación celular Caco-2
© Inhibición de proliferación celular B16F10
Inhibición de proliferación celular A2780
® Inhibición de proliferación celular MOLM-13
Tabla 3a: Indicaciones re resentadas or las líneas celulares

Tabla 3b: Inhibición de la proliferación

(continuación)

(continuación)



Tabla 4: Permeación de Caco-2 de compuestos de acuerdo con la presente invención, determinada como se describe bajo el Procedimiento 5 de materiales y procedimientos.
©: Número de Ejemplo
©: Concentración de compuesto de ensayo indicado en pM.
®: Papp A-B (Mari) indicado en [nm/s]
©: Papp B-A (Mari) indicado en [nm/s]
©: Flujo de salida (Papp B-A/Papp A-B)
Tabla 4



Tabla 5: Estabilidad en hepatocitos de rata y t i /2 en ratas después de la administración según se determinó mediante el Procedimiento 6 y el Procedimiento 7 como se describe en materiales y procedimientos.
©: Número de Ejemplo
©: La biodisponibilidad oral máxima calculada (Fmax) basada en los datos de estabilidad en hepatocitos de rata. ® t1/2 : vida media terminal (en h) a partir de un estudio in vivo después de la administración de bolos i.v. a ratas.
Tabla 5

(continuación)

Tabla 6a: Constantes de disociación en el equilibrio Kd[1/s], constantes de velocidad de disociación koff[1/s], y tiempos de residencia en el blanco [min] según se determinó mediante el Procedimiento 8 a 25 °C como se describe en materiales y procedimientos. Se indican ligeras variaciones de parámetros experimentales por las letras (A-G): Parámetros A: Descritos en materiales y procedimientos sección 8.
Parámetros B: tasa de flujo: 100 pl/min, tiempo de inyección: 70 s, tiempo de disociación: 1200 s, diluciones seriales de compuesto (3,13 nM hasta 100 nM)
Parámetros C: tasa de flujo: 50 pl/min, tiempo de inyección: 60 s, tiempo de disociación: 1200 s, diluciones seriales de compuesto (0,82 nM hasta 200 nM)
Parámetros D: tasa de flujo: 100 pl/min, tiempo de inyección: 80 s, tiempo de disociación: 1200 s, diluciones seriales de compuesto (3,13 nM hasta 100 nM)
Parámetros E: tasa de flujo: 100 pl/min, tiempo de inyección: 70 s, tiempo de disociación: 1100 s, diluciones seriales de compuesto (0,78 nM hasta 25 nM) y medidas a 37 °C
Parámetros F: tasa de flujo: 100 pl/min, tiempo de inyección: 70 s, tiempo de disociación: 1100 s, diluciones seriales de compuesto (1,56 nM hasta 50 nM)
Parámetros G: tasa de flujo: 100 pl/min, tiempo de inyección: 70 s, tiempo de disociación: 1100 s, diluciones seriales de compuesto (3,13 nM hasta 100 nM)
©: Número de Ejemplo
©: Constante de disociación en el equilibrio Kd [1/s]
®: Constante de velocidad de disociación koff [1/s]
©: Tiempos de residencia en el blanco [min]
©: Parámetros experimentales como se especificó anteriormente [A-G]
Tabla 6a:

(continuación)

(continuación)


Se espera que el prolongado tiempo de residencia de los inhibidores de CDK9 macrocíclicos de acuerdo con la invención resulte en un efecto inhibitorio sostenido contra la señalización de CDK9, contribuyendo a un ataque sostenido del blanco y aumentando la eficacia antitumoral.
Tabla 6 b: Constantes de disociación en equilibrio Kd [1/s], constantes de tasa de disociación koff [1/s] y tiempos de residencia blanco [min] como se determina por el procedimiento 8 a 37°C como se describe en materiales y procedimientos. Ligeras variaciones de los parámetros experimentales se indican por las letras (A-G):
Parámetros A: Descritos en materiales y procedimientos sección 8.
Parámetros B: tasa de flujo: 100 pl/min, tiempo de inyección: 70 s, tiempo de disociación: 1200 s, diluciones seriales de compuesto (3,13 nM hasta l00 nM)
Parámetros C: tasa de flujo: 50 pl/min, tiempo de inyección: 60 s, tiempo de disociación: 1200 s, diluciones seriales de compuesto (0,82 nM hasta 200 nM)
Parámetros D: tasa de flujo: 100 pl/min, tiempo de inyección: 80 s, tiempo de disociación: 1200 s, diluciones seriales de compuesto (3,13 nM hasta 100 nM)
Parámetros E: tasa de flujo: 100 pl/min, tiempo de inyección: 70 s, tiempo de disociación: 1100 s, diluciones seriales de compuesto (0,78 nM hasta 25 nM) y medidas a 37 °C
Parámetros F: tasa de flujo: 100 pl/min, tiempo de inyección: 70 s, tiempo de disociación: 1100 s, diluciones seriales de compuesto (1,56 nM hasta 50 nM)
Parámetros G: tasa de flujo: 100 pl/min, tiempo de inyección: 70 s, tiempo de disociación: 1100 s, diluciones seriales de compuesto (3,13 nM hasta 100 nM)
©: Número de Ejemplo
©: Constante de disociación en equilibrio Kd [1/s]
®: Constante de tasa de disociación koff [1/s]
©: Tiempo de residencia blanco [min]
©: Parámetros experimentales como se especificó anteriormente [A-G]
Tabla 6 b:

(continuación)

(continuación)

Se espera que el tiempo de residencia prolongado de inhibidores macrocíclicos de CDK9 de acuerdo con la invención resultarán en un efecto inhibidor sostenido sobre la señalización de CDK9, contribuyendo por último con el acoplamiento blanco sostenido y la eficacia antitumoral.
Tabla 7: Solubilidad termodinámica de compuestos de acuerdo con la presente invención en agua a pH 6,5 como se determina por los procedimientos de matraces de agitación en equilibrio descritos en el procedimiento 4a. y 4b. de los materiales y procedimientos; "n.t." significa que los compuestos no fueron ensayados en el ensayo respectivo.
©: Número de ejemplo
©: Solubilidad acuosa pH 6,5 [mg/l], termodinámica de la solución de DMSO como se describe bajo el procedimiento 4a. de materiales y procedimientos
®: Solubilidad acuosa pH 6,5 [mg/l], termodinámica de polvo como se describe bajo el procedimiento 4b. de materiales y procedimientos
Tabla 7

Ĩcontinuación)

Claims (23)
1. Un compuesto de fórmula general (I),

en la que
L representa un grupo alquileno C2-C5,
en el que dicho grupo está opcionalmente sustituido con
(i) un sustituyente seleccionado de cicloalquil C3-C4- e hidroximetil-, y/o
(ii) uno o dos sustituyentes adicionales, iguales o diferentes, seleccionados de alquil Ci-C2-,
X, Y representan CH o N con la condición de que uno de X e Y represente CH y uno de X e Y represente N; R1 representa un grupo seleccionado de alquil C1-C4-y cicloalquil C3-C5-, en el que dicho grupo está opcionalmente sustituido con uino o dos o tres sustituyentes, iguales o diferentes, seleccionados del grupo que consiste en hidroxi, ciano, halógeno, alquil C1-C2-, alcoxi C1-C2-, -NH2 , -C(=O)OH; R2 representa un grupo seleccionado de un átomo de hidrógeo, un átomo de flúor, un átomo de cloro, metil-, metoxi-, trifluorometil-;
R3 representa un grupo seleccionado de un átomo de hidrógeno, un átomo de flúor, un átomo de cloro, ciano, metil-, metoxi-, trifluorometil-, trifluorometoxi-;
R4 representa un átomo de hidrógeno o un átomo de flúor;
R5 representa un grupo seleccionado de un átomo de hidrógeno, ciano, -C(=O)R8, -C(=O)OR8, -S(=O)2R8, -C(=o )n R6R7 , alquil C1-C4-, cicloalquil C3-C5-,
en el que dicho grupo alquil C1-C4- o cicloalquil C3-C5- está opcionalmente sustituido con un sustituyente seleccionado del grupo que consiste en flúor, hidroxi, ciano, alcoxi C1-C3-, -NH2, alquilamino-, dialquilamino-; R6, R7 representa, de modo independiente entre sí, un grupo seleccionado de un átomo de hidrógeno, alquil C1-C4- y cicloalquil C3-C5-,
en el que dicho grupo C1-C4- o cicloalquil C3-C5- está opcionalmente sustituido con uno o dos sustituyentes, iguales o diferentes, seleccionados del grupo que consiste en hidroxi, alquil C1-C2-, alcoxi C1-C2-, -NH2, alquilamino-, dialquilamino-;
R8 representa un grupo seleccionado de alquil C1-C6-, fluoro-alquil C1-C3-, cicloalquil C3-C5- y fenil-,
en el que dicho grupo está opcionalmente sustituido con un sustituyente seleccionado del grupo que consiste en hidroxi, alquil C1-C2-, alcoxi C1-C2-, -NH2,
o un enantiómero, diastereómero, sal, solvato o sal de solvato del mismo.
2. El compuesto de fórmula general (I) de acuerdo con la reivindicación 1, en la que
L representa un grupo alquileno C2-C4 ;
X, Y representan c H o N con la condición de que uno de X e Y representen CH y uno de X e Y representen N; R1 representa a alquil C1-C4-, en el que dicho grupo está opcionalmente sustituido con uno o dos sustituyentes, iguales o diferentes, seleccionados del grupo que consiste en hidroxi, alcoxi C1-C2-, -NH2 , -C(=O)OH;
R2 representa un átomo de hidrógeno o un grupo ciano;
R3 representa un grupo seleccionado de un átomo de hidrógeno, un átomo de flúor y un grupo metoxi-;
R4 representa un grupo seleccionado de un átomo de hidrógeno y un átomo de flúor;
R5 representa un grupo seleccionado de un átomo de hidrógeno, ciano, alquil C1-C4-, cicloalquil C3-C5-; en el que dicho grupo alquil C1-C4- está opcionalmente sustituido con un grupo hidroxi;
o un enantiómero, diastereómero, sal, solvato o sal de solvato del mismo.
3. El compuesto de fórmula general (I) de acuerdo con una cualquiera de las reivindicaciones 1 o 2, en la que
L representa un grupo alquileno C3-C4 ;
X, Y representa CH o N con la condición de que uno de X e Y representen CH y uno de X e Y representen N; R1 representa un grupo metil-;
R2 representa un átomo de hidrógeno o un grupo ciano;
R3 representa un átomo de flúor;
R4 representa un grupo seleccionado de un átomo de hidrógeno, un átomo de flúor;
R5 representa un grupo seleccionado de un átomo de hidrógeno, ciano, alquil C1-C3-, ciclopropil- en el que dicho grupo alquil C1-C3- está opcionalmente sustituido con un grupo hidroxi;
o un enantiómero, diastereómero, sal, solvato o sal de solvato del mismo.
4. El compuesto de fórmula general (I) de acuerdo con una cualquiera de las reivindicaciones 1 a 3, en la que
R2 representa un átomo de hidrógeno o un grupo ciano;
o un enantiómero, diastereómero, sal, solvato o sal de solvato del mismo.
5. El compuesto de fórmula general (I) de acuerdo con una cualquiera de las reivindicaciones 1 a 4, en la que
R3 representa un átomo de flúor;
R4 representa un átomo de hidrógeno;
o un enantiómero, diastereómero, sal, solvato o sal de solvato del mismo.
6. El compuesto de fórmula general (I) de acuerdo con la reivindicación 1, en la que
L representa un grupo -CH2CH2CH2- o un -CH2CH2CH2CH2-;
X, Y representan CH o N con la condición de que uno de X e Y representen CH y uno de X e Y representen N; R1 representa un grupo metil-;
R2 representa un átomo de hidrógeno o un grupo ciano;
R3 representa un átomo de flúor;
R4 representa un átomo de hidrógeno o un átomo de flúor;
R5 representa un grupo seleccionado de un átomo de hidrógeno, ciano, metil-, 3-hidroxipropil- y ciclopropil-;
o un enantiómero, diastereómero, sal, solvato o sal de solvato del mismo.
7. El compuesto de acuerdo con la reivindicación 1, el cual se selecciona de:
- 15,19-difluoro-8-[(S-metilsulfonodiimidoil)metil]-3,4-dihidro-2H,11H-10,6-(azeno)-12,16-(meteno)-1,5,11,13-benzodioxadiazaciclooctadecina;
- (rac)-3-(2-{[15,19-difluoro-3,4-dihidro-2H,11H-10,6-(azeno)-12,16-(meteno)-1,5,11,13-benzodioxadiazaciclooctadecin-8-il]metil}-2-metil-2A6-diazatia-1,2-dien-1-il)propan-1-ol;
- (racH{[15,19-difluoro-3,4-dihidro-2H,11H-10,6-(azeno)-12,16-(meteno)-1,5,11,13-benzodioxadiazaciclooctadecin-8-il]metil}(imino)metil-A6-sulfanilidene]cianamida;
- (rac)-8-[(N,S-dimetilsulfonodiimidoil)metil]-15,19-difluoro-3,4-dihidro-2H,11H-10,6-(azeno)-12,16-(meteno)-1.5.11.13- benzodioxadiazaciclooctadecina, and
- -16,20-difluoro-9-[(S-metilsulfonodiimidoil)metil]-2,3,4,5-tetrahidro-12H-13,17-(azeno)-11,7-(meteno)-1,6,12,14-benzodioxadiazaciclononadecina;
- 16,20,21-trifluoro-9-[(S-metilsulfonodiimidoil)metil]-2,3,4,5-tetrahidro-12H-13,17-(azeno)-11,7-(meteno)-1.6.12.14- benzodioxadiazaciclononadecina;
- 16,21-difluoro-9-[(S-metilsulfonodiimidoil)metil]-2,3,4,5-tetrahidro-12H-13,17-(azeno)-11,7-(meteno)-1,6,12,14-benzodioxadiazaciclononadecina;
- 15,19-difluoro-8-[(S-metilsulfonodiimidoil)metil]-3,4-dihidro-2H,11H-10,6-(azeno)-12,16-(meteno)-1,5,11,13-benzodioxadiazaciclooctadecina-7-carbonitrilo;
- (rac)-9-[(N-ciclopropil-S-metilsulfonodiimidoil)metil]-16,20-difluoro-2,3,4,5-tetrahidro-12H-13,17-(azeno)-11,7-(meteno)-1,6,12,14-benzodioxadiazaciclononadecina;
- (rac)-9-[(N,S-dimetilsulfonodiimidoil)metil]-16,20-difluoro-2,3,4,5-tetrahidro-12H-13,17-(azeno)-11,7-(meteno)-1.6.12.14- benzodioxadiazaciclononadecina;
y un enantiómero, diastereómero, sal, solvato o sal de solvato de los mismos.
8. Un compuesto de fórmula general (I) de acuerdo con una cualquiera de las reivindicaciones 1 a 7 para el uso como un medicamento.
9. Un compuesto de fórmula general (I) de acuerdo con una cualquiera de las reivindicaciones 1 a 7 para el uso del
tratamiento y/o la profilaxis de trastornos hiperproliferativos, enfermedades infecciosas inducidas por virus y/o de enfermedades cardiovasculares.
10. Un compuesto de fórmula general (I) de acuerdo con una cualquiera de las reivindicaciones 1 a 7 para el uso del tratamiento y/o la profilaxis de carcinomas de pulmón, carcinomas de próstata, carcinomas cervicales, carcinomas colorrectales, melanomas o carcinomas de ovario.
11. Uso de un compuesto de fórmula general (I) de acuerdo con una cualquiera de las reivindicaciones 1 a 7 en la fabricación de un medicamento para el tratamiento y/o la profilaxis de trastornos hiperproliferativos, enfermedades infecciosas inducidas por virus y/o enfermedades cardiovasculares.
12. Uso de un compuesto de fórmula general (I) de acuerdo con una cualquiera de las reivindicaciones 1 a 7 en la fabricación de un medicamento para el tratamiento y/o la profilaxis de carcinomas de pulmón, carcinomas de próstata, carcinomas cervicales, carcinomas colorrectales, melanomas, carcinomas de ovario o leucemias.
13. Uso de un compuesto de fórmula general (I) de acuerdo con una cualquiera de las reivindicaciones 1 a 7 en la fabricación de un medicamento para el tratamiento y/o la profilaxis de carcinomas de pulmón de células no pequeñas, carcinomas de próstata humanos independientes de hormonas, carcinomas cervicales humanos resistentes a múltiples fármacos o leucemias mieloides agudas humanas.
14. Una combinación farmacéutica que comprende un compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 7 en combinación con al menos uno o más de los principios activos adicionales.
15. La combinación farmacéutica de acuerdo con la reivindicación 14 para uso del tratamiento y/o la profilaxis de trastornos hiperproliferativos, enfermedades infecciosas inducidas por virus y/o de enfermedades cardiovasculares.
16. La combinación farmacéutica de acuerdo con la reivindicación 14 para uso del tratamiento y/o la profilaxis de carcinomas de pulmón, carcinomas de próstata, carcinomas cervicales, carcinomas colorrectales, melanomas, carcinomas de ovario o leucemias.
17. Una composición farmacéutica que comprende un compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 7 en combinación con un adyuvante farmacéuticamente adecuado, no tóxico, inerte.
18. La composición farmacéutica de acuerdo con la reivindicación 17 para uso del tratamiento y/o la profilaxis de trastornos hiperproliferativos, enfermedades infecciosas inducidas por virus y/o de enfermedades cardiovasculares.
19. La composición farmacéutica de acuerdo con la reivindicación 17 para uso del tratamiento y/o la profilaxis de trastornos hiperproliferativos, enfermedades infecciosas inducidas por virus y/o de enfermedades cardiovasculares.
20. Un compuesto de fórmula general (9)

en la que R1, R2, R3, R4 y L son como se definen de acuerdo con una cualquiera de las reivindicaciones 1 a 6 para los compuestos de fórmula general (I),
o un enantiómero, diastereómero o solvato del mismo.
21. Un compuesto de fórmula general (22)

en la que R1, R2, R3, R4y A son como se definen de acuerdo con una cualquiera de las reivindicaciones 1 a 6 para los compuestos de fórmula general (I),
o un enantiómero, diastereómero o solvato del mismo.
22. Un procedimiento para la preparación de un compuesto de fórmula (10), en cuyo procedimiento un compuesto de la fórmula (9), en la que R1, R2, R3, R4 y L son como se definen para el compuesto de fórmula (I) de acuerdo con una cualquiera de las reivindicaciones 1 a 6 ,

se oxida por tratamiento con un agente seleccionado de diacetato de yodobenceno y N-cloro succinimida, seguido por la adición de una amina seleccionada de una amina primaria de fórmula R5-NH2, en la que R5 es como se define para los compuestos de fórmula (I) de acuerdo con la invención, y hexametildisilazeno,

para dar compuestos de la fórmula (1 0 ),
y en cuyo procedimiento el compuesto resultante se convierte opcionalmente, si es apropiado, con los (i) disolventes
y/o (ii) bases o ácidos correspondientes a los solvatos, sales y/o solvatos de los mismos.
23. Un procedimiento para la preparación de un compuesto de fórmula (23), en cuyo procedimiento un compuesto de la fórmula (22), en la que R1, R2, R3, R4y L son como se definen para el compuesto de fórmula (I) de acuerdo con una cualquiera de las reivindicaciones 1 a 6 ,

se oxida por tratamiento con un agente seleccionado de diacetato de yodobenceno y N-cloro succinimida, seguido por la adición de una amina seleccionada de una amina primaria de fórmula R5-NH2, en la que R5 es como se define para los compuestos de fórmula (I) de acuerdo con la invención, y hexametildisilazeno,

para dar compuestos de la fórmula (23),
y en cuyo procedimiento el compuesto resultante se convierte opcionalmente, si es apropiado, con los (i) disolventes y/o (ii) bases o ácidos correspondientes a los solvatos, sales y/o solvatos de los mismos.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2015091056 | 2015-09-29 | ||
| PCT/EP2016/072795 WO2017055196A1 (en) | 2015-09-29 | 2016-09-26 | Novel macrocyclic sulfondiimine compounds |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2786552T3 true ES2786552T3 (es) | 2020-10-13 |
Family
ID=56990449
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16770510T Active ES2786552T3 (es) | 2015-09-29 | 2016-09-26 | Compuestos de sulfondiimina macrocíclicos nuevos |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US10717749B2 (es) |
| EP (1) | EP3356373B1 (es) |
| JP (1) | JP6847099B2 (es) |
| CN (1) | CN108290903B (es) |
| AR (1) | AR106185A1 (es) |
| CA (1) | CA2999931A1 (es) |
| ES (1) | ES2786552T3 (es) |
| TW (1) | TW201718599A (es) |
| UY (1) | UY36923A (es) |
| WO (1) | WO2017055196A1 (es) |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2944251C (en) * | 2014-04-01 | 2022-10-18 | Bayer Pharma Aktiengesellschaft | Disubstituted 5-fluoro pyrimidine derivatives containing a sulfondiimine group |
| JP6888000B2 (ja) * | 2015-10-08 | 2021-06-16 | バイエル ファーマ アクチエンゲゼルシャフト | 新規な修飾された大環状化合物 |
| WO2018177889A1 (en) | 2017-03-28 | 2018-10-04 | Bayer Aktiengesellschaft | Novel ptefb inhibiting macrocyclic compounds |
| ES2900199T3 (es) | 2017-03-28 | 2022-03-16 | Bayer Ag | Novedosos compuestos macrocíclicos inhibidores de PTEFB |
| CN112321604A (zh) * | 2019-08-05 | 2021-02-05 | 华东理工大学 | 大环类jak2抑制剂及其应用 |
| CN113603708B (zh) * | 2021-07-27 | 2023-08-11 | 中国药科大学 | 一种具有大环骨架结构的cdk9抑制剂的制备及其应用 |
Family Cites Families (58)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE2520230A1 (de) | 1975-05-07 | 1976-11-25 | Heumann Ludwig & Co Gmbh | Substituierte sulfodiimide, verfahren zu ihrer herstellung und diese verbindungen enthaltende arzneimittel |
| MXPA02003436A (es) | 1999-10-07 | 2002-08-20 | Amgen Inc | Inhibidores de triazina cinasa. |
| GB0103926D0 (en) | 2001-02-17 | 2001-04-04 | Astrazeneca Ab | Chemical compounds |
| WO2003037346A1 (en) | 2001-10-31 | 2003-05-08 | Cell Therapeutics, Inc. | 6-phenyl-n-phenyl-(1,3,5) -triazine-2,4-diamine derivatives and related compounds with lysophphosphatidic acid acyltransferase beta (lpaat-beta) inhibitory activity for use in the treatment of cancer |
| ES2392426T3 (es) | 2002-07-18 | 2012-12-10 | Janssen Pharmaceutica Nv | Inhibidores de quinasas con triazina sustituida |
| DE10239042A1 (de) | 2002-08-21 | 2004-03-04 | Schering Ag | Makrozyclische Pyrimidine, deren Herstellung und Verwendung als Arzneimittel |
| JP2006518381A (ja) | 2003-02-07 | 2006-08-10 | バーテックス ファーマシューティカルズ インコーポレイテッド | プロテインキナーゼのインヒビターとして有用なヘテロアリール置換ピロール |
| US8084457B2 (en) | 2003-09-15 | 2011-12-27 | Lead Discovery Center Gmbh | Pharmaceutically active 4,6-disubstituted aminopyrimidine derivatives as modulators of protein kinases |
| DE10349423A1 (de) | 2003-10-16 | 2005-06-16 | Schering Ag | Sulfoximinsubstituierte Parimidine als CDK- und/oder VEGF-Inhibitoren, deren Herstellung und Verwendung als Arzneimittel |
| EP1674469A1 (en) | 2004-12-22 | 2006-06-28 | Schering Aktiengesellschaft | Sulfonamido-macrocycles as Tie2 inhibitors |
| EP1674470A1 (en) | 2004-12-22 | 2006-06-28 | Schering Aktiengesellschaft | Sulfonamido-macrocycles as Tie2 inhibitors |
| JP4889335B2 (ja) | 2005-03-30 | 2012-03-07 | 富士フイルム株式会社 | 溶液製膜方法 |
| EP1710246A1 (en) | 2005-04-08 | 2006-10-11 | Schering Aktiengesellschaft | Sulfoximine-pyrimidine Macrocycles and the salts thereof, a process for making them, and their pharmaceutical use against cancer |
| EP1803723A1 (de) | 2006-01-03 | 2007-07-04 | Bayer Schering Pharma Aktiengesellschaft | (2,4,9-triaza-1(2,4)-pyrimidina-3(1,3)-benzenacyclononaphan-3^4-yl)-sulfoximid derivate als selektive inhibitoren der aurora kinase zur behandlung von krebs |
| EP1870416A1 (en) | 2006-06-21 | 2007-12-26 | Bayer Schering Pharma Aktiengesellschaft | Sulphonamido-macrocycles as tie2 inhibitors |
| EP1873159A1 (en) | 2006-06-21 | 2008-01-02 | Bayer Schering Pharma Aktiengesellschaft | Substituted sulphonamido-macrocycles as Tie2 inhibitors and salts thereof, pharmaceutical compositions comprising same, methods of preparing same and uses of same |
| DE102006041382A1 (de) | 2006-08-29 | 2008-03-20 | Bayer Schering Pharma Ag | Carbamoyl-Sulfoximide als Proteinkinaseinhibitoren |
| US20100048597A1 (en) | 2006-12-22 | 2010-02-25 | Novartis Ag | Organic Compounds and Their Uses |
| CN101861313B (zh) | 2007-03-12 | 2014-06-04 | Ym生物科学澳大利亚私人有限公司 | 苯基氨基嘧啶化合物及其用途 |
| JP5379787B2 (ja) | 2007-04-24 | 2013-12-25 | インゲニウム ファーマシューティカルズ ジーエムビーエイチ | プロテインキナーゼの阻害剤 |
| JP5693951B2 (ja) | 2007-04-24 | 2015-04-01 | アストラゼネカ エービー | プロテインキナーゼの阻害剤 |
| WO2008129080A1 (en) | 2007-04-24 | 2008-10-30 | Ingenium Pharmaceuticals Gmbh | 4, 6-disubstituted aminopyrimidine derivatives as inhibitors of protein kinases |
| PT2200436E (pt) | 2007-09-04 | 2015-04-29 | Scripps Research Inst | Pirimidinilaminas substituídas como inibidoras da proteína quinase |
| GB0805477D0 (en) | 2008-03-26 | 2008-04-30 | Univ Nottingham | Pyrimidines triazines and their use as pharmaceutical agents |
| EP2274288A2 (en) | 2008-04-24 | 2011-01-19 | Incyte Corporation | Macrocyclic compounds and their use as kinase inhibitors |
| EP2303881A2 (en) | 2008-07-14 | 2011-04-06 | Gilead Sciences, Inc. | Fused heterocyclyc inhibitors of histone deacetylase and/or cyclin-dependent kinases |
| US8415381B2 (en) | 2009-07-30 | 2013-04-09 | Novartis Ag | Heteroaryl compounds and their uses |
| KR20120049940A (ko) | 2009-09-04 | 2012-05-17 | 노파르티스 아게 | 키나제 억제제로서의 헤테로아릴 화합물 |
| CA2777762A1 (en) | 2009-10-12 | 2011-04-21 | Myrexis, Inc. | Amino - pyrimidine compounds as inhibitors of tbk1 and/or ikk epsilon |
| HU0900798D0 (en) | 2009-12-21 | 2010-03-01 | Vichem Chemie Kutato Kft | 4-phenylamino-pyrimidine derivatives having protein kinase inhibitor activity |
| JP5921525B2 (ja) | 2010-03-22 | 2016-05-24 | リード ディスカバリー センター ゲゼルシャフト ミット ベシュレンクテル ハフツング | 医薬的に活性のある二置換のトリアジン誘導体 |
| TW201206946A (en) | 2010-07-15 | 2012-02-16 | Bristol Myers Squibb Co | Compounds for the reduction of beta-amyloid production |
| JP2013542967A (ja) | 2010-11-17 | 2013-11-28 | ノバルティス アーゲー | 3−(アミノアリール)−ピリジン化合物 |
| WO2012066065A1 (en) | 2010-11-17 | 2012-05-24 | Novartis Ag | Phenyl-heteroaryl amine compounds and their uses |
| US20130303507A1 (en) | 2011-01-28 | 2013-11-14 | Novartis Ag | Substituted hetero-biaryl compounds and their uses |
| WO2012101063A1 (en) | 2011-01-28 | 2012-08-02 | Novartis Ag | N-acyl pyridine biaryl compounds and their uses |
| WO2012101065A2 (en) | 2011-01-28 | 2012-08-02 | Novartis Ag | Pyrimidine biaryl amine compounds and their uses |
| WO2012101066A1 (en) | 2011-01-28 | 2012-08-02 | Novartis Ag | Pyridine biaryl amine compounds and their uses |
| WO2012101064A1 (en) | 2011-01-28 | 2012-08-02 | Novartis Ag | N-acyl pyrimidine biaryl compounds as protein kinase inhibitors |
| WO2012117048A1 (en) | 2011-03-02 | 2012-09-07 | Lead Discovery Center Gmbh | Pharmaceutically active disubstituted triazine derivatives |
| EP2681193B1 (en) | 2011-03-02 | 2016-01-06 | Lead Discovery Center GmbH | Pharmaceutically active disubstituted pyridine derivatives |
| CN103732067A (zh) | 2011-04-12 | 2014-04-16 | 美国阿尔茨海默病研究所公司 | 化合物,组合物及它们的治疗用途 |
| CN102731413A (zh) | 2011-04-15 | 2012-10-17 | 上海医药工业研究院 | 一种脲类化合物、其制备方法、其中间体及其应用 |
| AU2012244745A1 (en) | 2011-04-19 | 2013-09-19 | Bayer Intellectual Property Gmbh | Substituted 4-Aryl-N-phenyl-1,3,5-triazin-2-amines |
| JP5976814B2 (ja) * | 2011-09-16 | 2016-08-24 | バイエル・インテレクチュアル・プロパティ・ゲゼルシャフト・ミット・ベシュレンクテル・ハフツングBayer Intellectual Property GmbH | スルホキシイミン基を含有する二置換5−フルオロピリミジン誘導体 |
| US9108926B2 (en) | 2011-09-16 | 2015-08-18 | Bayer Intellectual Property Gmbh | Disubstituted 5-fluoro-pyrimidines |
| CA2882733C (en) | 2012-08-23 | 2021-07-27 | Virostatics Srl | Novel 4,6-disubstituted aminopyrimidine derivatives having both aromatic and halogenic substituents |
| CN105102434A (zh) | 2012-10-18 | 2015-11-25 | 拜耳药业股份公司 | 含砜基团的4-(邻)-氟苯基-5-氟嘧啶-2-基胺 |
| JP6277196B2 (ja) | 2012-10-18 | 2018-02-07 | バイエル ファーマ アクチエンゲゼルシャフト | スルホン基を含んでいるn−(ピリジン−2−イル)ピリミジン−4−アミン誘導体 |
| WO2014060375A2 (en) | 2012-10-18 | 2014-04-24 | Bayer Pharma Aktiengesellschaft | 5-fluoro-n-(pyridin-2-yl)pyridin-2-amine derivatives containing a sulfone group |
| ME02880B (me) | 2012-11-15 | 2018-04-20 | Bayer Pharma AG | Derivati 5-fluoro- n- (piridin -2-il}piridi n- 2 - amina koji sadrze sulfoksiminsku grupu |
| JP6300818B2 (ja) | 2012-11-15 | 2018-03-28 | バイエル ファーマ アクチエンゲゼルシャフト | スルホキシイミン基を含有する4−(オルト)−フルオロフェニル−5−フルオロピリミジン−2−イルアミン |
| TW201418243A (zh) | 2012-11-15 | 2014-05-16 | Bayer Pharma AG | 含有磺醯亞胺基團之n-(吡啶-2-基)嘧啶-4-胺衍生物 |
| WO2014106762A1 (en) | 2013-01-07 | 2014-07-10 | Vichem Chemie Kutató Kft. | 4-pyrimidinylamino-benzenesulfonamide derivatives and their use for the inhibition of polo-like kinase 1 (plk1) for the treatment of cancer and their use for the treatment of bacterial infections |
| JP6371385B2 (ja) | 2013-07-04 | 2018-08-08 | バイエル ファーマ アクチエンゲゼルシャフト | スルホキシイミン置換5−フルオロ−n−(ピリジン−2−イル)ピリジン−2−アミン誘導体およびそのcdk9キナーゼ阻害薬としての使用 |
| CA2942119A1 (en) | 2014-03-13 | 2015-09-17 | Bayer Pharma Aktiengesellschaft | 5-fluoro-n-(pyridin-2-yl)pyridin-2-amine derivatives containing a sulfone group |
| CA2944251C (en) * | 2014-04-01 | 2022-10-18 | Bayer Pharma Aktiengesellschaft | Disubstituted 5-fluoro pyrimidine derivatives containing a sulfondiimine group |
| EP3129387B1 (en) | 2014-04-11 | 2019-06-26 | Bayer Pharma Aktiengesellschaft | Novel macrocyclic compounds |
-
2016
- 2016-09-26 EP EP16770510.2A patent/EP3356373B1/en active Active
- 2016-09-26 CA CA2999931A patent/CA2999931A1/en active Pending
- 2016-09-26 WO PCT/EP2016/072795 patent/WO2017055196A1/en active Application Filing
- 2016-09-26 JP JP2018516057A patent/JP6847099B2/ja active Active
- 2016-09-26 CN CN201680066997.8A patent/CN108290903B/zh active Active
- 2016-09-26 ES ES16770510T patent/ES2786552T3/es active Active
- 2016-09-26 US US15/764,283 patent/US10717749B2/en active Active
- 2016-09-29 TW TW105131191A patent/TW201718599A/zh unknown
- 2016-09-29 UY UY0001036923A patent/UY36923A/es not_active Application Discontinuation
- 2016-09-29 AR ARP160102970A patent/AR106185A1/es unknown
Also Published As
| Publication number | Publication date |
|---|---|
| WO2017055196A1 (en) | 2017-04-06 |
| CN108290903B (zh) | 2021-09-03 |
| JP6847099B2 (ja) | 2021-03-24 |
| UY36923A (es) | 2017-04-28 |
| EP3356373B1 (en) | 2020-02-19 |
| AR106185A1 (es) | 2017-12-20 |
| CN108290903A (zh) | 2018-07-17 |
| TW201718599A (zh) | 2017-06-01 |
| CA2999931A1 (en) | 2017-04-06 |
| US20180273548A1 (en) | 2018-09-27 |
| JP2018529708A (ja) | 2018-10-11 |
| EP3356373A1 (en) | 2018-08-08 |
| US10717749B2 (en) | 2020-07-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2612978T3 (es) | Derivados de 5-fluoro-n-(piridin-2-il)piridin-2-amina que contienen un grupo sulfoximina | |
| US9856242B2 (en) | 5-fluoro-N-(pyridin-2-yl)pyridin-2-amine derivatives containing a sulfone group | |
| ES2786552T3 (es) | Compuestos de sulfondiimina macrocíclicos nuevos | |
| EP3129387B1 (en) | Novel macrocyclic compounds | |
| ES2819869T3 (es) | Nuevos compuestos macrocíclicos modificados | |
| CA2888371A1 (en) | 5-fluoro-n-(pyridin-2-yl)pyridin-2-amine derivatives containing a sulfone group | |
| ES2900199T3 (es) | Novedosos compuestos macrocíclicos inhibidores de PTEFB | |
| CA2891361A1 (en) | N-(pyridin-2-yl)pyrimidin-4-amine derivatives containing a sulfoximine group | |
| CA2888383A1 (en) | N-(pyridin-2-yl)pyrimidin-4-amine derivatives containing a sulfone group | |
| EP3126338B1 (en) | Disubstituted 5-fluoro pyrimidine derivatives containing a sulfondiimine group |