ES2747423T3 - Compuesto de quinoxalina - Google Patents
Compuesto de quinoxalina Download PDFInfo
- Publication number
- ES2747423T3 ES2747423T3 ES11823576T ES11823576T ES2747423T3 ES 2747423 T3 ES2747423 T3 ES 2747423T3 ES 11823576 T ES11823576 T ES 11823576T ES 11823576 T ES11823576 T ES 11823576T ES 2747423 T3 ES2747423 T3 ES 2747423T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- dihydro
- tetrahydrofuran
- pyrrolo
- methyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4985—Pyrazines or piperazines ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/02—Drugs for disorders of the urinary system of urine or of the urinary tract, e.g. urine acidifiers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/10—Drugs for disorders of the urinary system of the bladder
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Urology & Nephrology (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
Abstract
Compuesto de fórmula (I) o sal del mismo: en la que**Fórmula** A es CH, R1 es 2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-ilo sustituido por fluoro, bromo, metilo, etilo, trifluorometilo o metoxilo en la posición 5, o 5-metoxi-2,3-dihidro-1H-pirrolo[2,3-c]piridin-1-ilo, R2 es metilo o metoximetilo, y R3 es tetrahidrofuran-3-ilo.
Description
DESCRIPCIÓN
Compuesto de quinoxalina
Campo técnico
La presente invención se refiere a un compuesto de quinoxalina que es útil como principio activo para una composición farmacéutica, en particular, una composición farmacéutica para tratar la disfunción del almacenamiento, la disfunción de la evacuación y las enfermedades vesicales/uretrales, y similares.
Antecedentes de la técnica
Los papeles importantes de la función de evacuación son el almacenamiento y la evacuación de la orina, que se regulan mediante una acción coordinada de la vejiga y la uretra. Es decir, durante el almacenamiento de la orina, se relaja el músculo liso de la vejiga y se contrae el esfínter de la uretra, mediante lo cual se mantiene un estado en el que la resistencia uretral es alta, y también se mantiene la continencia urinaria. Por otro lado, durante la evacuación, se contrae el músculo liso de la vejiga, mientras que se relaja el músculo liso de la uretra, y también se inhibe la contracción del esfínter uretral externo. Los ejemplos de disfunción de la evacuación incluyen una disfunción del almacenamiento tal como vejiga hiperactiva y similares en la que la orina no puede retenerse durante el almacenamiento de la orina y una disfunción de la evacuación en la que la orina no puede drenarse suficientemente debido a un aumento en la resistencia uretral y a una disminución en la fuerza contráctil de la vejiga. Estas dos disfunciones pueden expresarse simultáneamente.
En el tratamiento de una disfunción del almacenamiento tal como vejiga hiperactiva y similares, se han usado de manera frecuente agentes anticolinérgicos. Sin embargo, estos agentes no pueden proporcionar un efecto terapéutico suficiente, y además, aparecen efectos secundarios basados en la acción anticolinérgica (sequedad de boca, síntomas gastrointestinales, síntomas oculares, arritmias, o similares), y por consiguiente, a menudo puede interrumpirse la administración de los agentes. Además, los agentes anticolinérgicos reducen la fuerza contráctil de la vejiga, y por tanto, están contraindicados para la polaquiuria/incontinencia urinaria que acompaña a la obstrucción uretral tal como hiperplasia prostática benigna y similares.
La disfunción de la evacuación se produce por un aumento en la resistencia uretral durante la evacuación o una disminución en la fuerza contráctil de la vejiga. Como una enfermedad que produce un aumento en la resistencia uretral, la disfunción de la evacuación que acompaña a la hiperplasia prostática benigna es bien conocida, que se caracteriza por la obstrucción uretral debido a la hipertrofia nodular de los tejidos de la próstata. Un antagonista del receptor a1 ya se ha usado con el fin de tratar la disfunción de la evacuación que acompaña a la hiperplasia prostática benigna (véase, por ejemplo, el documento no de patente 1). Otras causas del aumento en la resistencia uretral incluyen obstrucciones funcionales tales como insuficiencia de la relajación de la uretra durante la evacuación o disinergia detrusor-esfínter uretral externo y similares debido a trastornos neurológicos tales como diabetes, envejecimiento, daño de la médula ósea, cirugía pélvica, y similares. Con pacientes con estas enfermedades, existen muchos casos en los que el antagonista del receptor a1 no es eficaz. Por otro lado, una disminución en la fuerza contráctil de la vejiga durante la evacuación, denominada vejiga hipoactiva, vejiga acontráctil, vejiga neurógena, o similares, también produce una disfunción de la evacuación. Los factores conocidos para disminuir la fuerza contráctil de la vejiga incluyen envejecimiento, enfermedades neurológicas tales como diabetes, enfermedad de Parkinson, esclerosis múltiple y similares, daño de la médula ósea, y trastornos neurológicos debidos a cirugía pélvica. Los ejemplos de un agente para tratar una disminución en la fuerza contráctil de la vejiga durante la evacuación incluyen cloruro de betanecol que es un agonista del receptor muscarínico y bromuro de distigmina que es un inhibidor de la colinesterasa. Ambos fármacos tienen efectos secundarios, y por tanto, su carácter satisfactorio es bajo (véase, por ejemplo, los documentos no de patente 2 y 3). En la disfunción de la evacuación producida por un aumento en la resistencia uretral o una disminución en la fuerza contráctil de la vejiga tal como se describió anteriormente, se observa orina residual después de la evacuación. La orina residual aumentada puede producir una disminución en la capacidad efectiva de la vejiga, y por tanto, producir síntomas de la vejiga hiperactiva tales como polaquiuria y similares, o síntomas graves tales como hidronefrosis en algunos casos, y a este respecto, existe una demanda para un agente terapéutico que sea más eficaz que un agente terapéutico actual.
Se conoce que un sistema de relajación debido a óxido nítrico (NO) está presente en el músculo liso, y se produce NO en las terminaciones nerviosas o se activa de manera local guanilato ciclasa soluble presente en las células del músculo liso. La guanilato ciclasa activada aumenta el monofosfato de guanosina cíclico (GMPc) en las células. Por otro lado, el GMPc se degrada para dar 5'-GMP mediante fosfodiesterasa (PDE) que es una enzima que degrada el GMPc. Un aumento en la concentración de GMPc intracelular se considera que contribuye significativamente a la relajación del músculo liso. Por tanto, la disminución del sistema NO-GMPc provoca el fallo de la relajación del músculo liso. Por ejemplo, en pacientes que muestran obstrucción uretral en hiperplasia prostática benigna o en ancianos tal como se describió anteriormente, se notifica que la producción de NO se disminuye significativamente (documentos no de patente 4 y 5).
Como un subtipo de PDE que degrada específicamente GMPc, se conocen PDE5, PDE6 y PDE9, y entre estas, la
PDE9 tiene una mayor afinidad por el sustrato que la PDE5 y la PDE6 (documento no de patente 6). Además, desde el punto de vista de la distribución de expresión en diversos tejidos, se observa que la PDE9 en su expresión más alta en la próstata humana (documento no de patente 7) desempeña un papel importante en la relajación del músculo liso en el músculo liso de la uretra inferior, y un inhibidor de PDE9 potencia la relajación de la uretra a través del GMPc en el tejido. Por tanto, se considera que el inhibidor de PDE9 presenta un efecto contra la disfunción de la evacuación debido a un aumento en la resistencia uretral. Debido a que el inhibidor de PDE9 disminuye la resistencia uretral, puede esperarse un efecto contra la disfunción de la evacuación en el que se disminuyan las fuerzas contráctiles de la vejiga. Además, la disminución en la orina residual debido a la mejora de la disfunción de la evacuación conducirá a una mejora de los síntomas de la vejiga hiperactiva tales como polaquiuria y similares o la evasión de trastornos renales. Por tanto, se considera que el inhibidor de PDE9 es útil como agente para prevenir y/o tratar la disfunción del almacenamiento, la disfunción de la evacuación y las enfermedades vesicales/uretrales.
Por ejemplo, como un compuesto que tiene una(s) acción/acciones inhibidora(s) de la PDE5 y/o la PDE9, en los documentos de patente 1 y 2, se divulgan compuestos representado por las siguientes fórmulas (A) y (B), respectivamente, pero no hay ninguna divulgación específica de los compuestos de la presente invención. Además, en los documentos de patente 3 y 4, se divulga un derivado de tienopirimidina y un derivado de quinazolina como compuestos que tienen una(s) acción/acciones inhibidora(s) de la PDE5 y/o la PDE9, respectivamente. Además, en los documentos de patente 5 y 6, se divulga un derivado de pirazolopiridina que tiene una acción inhibidora de la PDE9.
Además, en los documentos de patente 7 a 12, se divulgan compuestos representados por las siguientes fórmulas (C) a (H), pero no hay una divulgación específica de los compuestos de la presente invención. Además, no hay ninguna descripción de que el compuesto tenga una acción inhibidora de la PDE9.
[Fórm. quím. 1]

[Fórm. quím. 3]
Ċ 

(Para los símbolos en las fórmulas, véase cada una de las correspondientes publicaciones de patente).
Técnica relacionada
Documento de patente
Documento de patente 1: documento de patente de publicación internacional WO 2008/072779 Documento de patente 2: documento de patente de publicación internacional WO 2008/072778 Documento de patente 3: documento de patente de publicación internacional WO 2006/135080 Documento de patente 4: documento de patente de publicación internacional WO 2008/018306 Documento de patente 5: documento de patente de publicación internacional WO 2010/026214 Documento de patente 6: documento de patente de publicación internacional WO 2010/084438 Documento de patente 7: documento de patente de publicación internacional WO 94/21639 Documento de patente 8: documento de patente de publicación internacional WO 99/09845 Documento de patente 9: documento de patente de publicación internacional WO 96/08492 Documento de patente 10: documento de patente de publicación internacional WO 96/08493 Documento de patente 11: documento de patente de publicación internacional WO 93/17025 Documento de patente 12: documento de patente de publicación internacional WO 93112113 Documento no de patente
Documento no de patente 1: Thiyagarajan, M., Pharmacology, 65:págs. 119-128 (2002) Documento no de patente 2: Shah, P.J.R., et al., Br, J. Urol., 55:págs. 229-232 (1983) Documento no de patente 3: Finkbeiner, A.E., J. Urol., 134:págs. 443-449 (1985)
Documento no de patente 4: Bloch, W., et al., Prostate, 33:págs. 1-8 (1997)
Documento no de patente 5: Toprakqi, M., et al., Int. J Clin. Lab. Res., 30:págs. 83-85 (2000)
Documento no de patente 6: Fisher, D.A., et al., J. Biol. Chem., 273:págs. 15559-15564 (1998)
Documento no de patente 7: Rentero, C., et al., Biochem. Biophys. Res. Commun., 301:págs. 686-692 (2003)
Sumario de la invención
Problemas que van a resolverse mediante la invención
Los presentes inventores pretenden proporcionar un compuesto que tenga una acción inhibidora de la PDE9 y sea útil como principio activo para una composición farmacéutica para prevenir y tratar la disfunción del almacenamiento, la disfunción de la evacuación y las enfermedades vesicales/uretrales, y similares.
Medios para resolver los problemas
Los presentes inventores han investigado exhaustivamente un compuesto que tiene una acción inhibidora de la PDE9, y como resultado, han encontrado que un compuesto de fórmula (1) es útil como compuesto que tiene una acción inhibidora de la PDE9, completando de ese modo la presente invención.
Es decir, la presente invención se refiere a un compuesto de fórmula (I) o una sal del mismo, y a una composición farmacéutica que incluye el compuesto de fórmula (I) o una sal del mismo, y un excipiente.
[Fórm. quím. 9]

A es CH,
R1 es 2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-ilo sustituido con fluoro, bromo, metilo, etilo, trifluorometilo o metoxilo en la posición 5, o 5-metoxi-2,3-dihidro-1H-pirrolo[2,3-c]piridin-1-ilo,
R2 es metilo o metoximetilo, y
R3 es tetrahidrofuran-3-ilo].
Además, a menos que se describa específicamente lo contrario, en el caso en el que los símbolos en cualquiera de las fórmulas en la presente memoria descriptiva también se usen en otras fórmulas, los mismos símbolos indican el mismo significado.
Además, la presente invención se refiere a una composición farmacéutica para prevenir o tratar la disfunción del almacenamiento, la disfunción de la evacuación y las enfermedades vesicales/uretrales, y similares, que incluye un compuesto de fórmula (I) o una sal del mismo. Además, la composición farmacéutica incluye un agente para prevenir o tratar la disfunción del almacenamiento, la disfunción de la evacuación y las enfermedades vesicales/uretrales, y similares, que incluye un compuesto de fórmula (I) o una sal del mismo.
La presente invención se refiere además al uso del compuesto de fórmula (I) o una sal del mismo para la fabricación de una composición farmacéutica para prevenir o tratar la disfunción del almacenamiento, la disfunción de la evacuación y las enfermedades vesicales/uretrales, y similares, al uso del compuesto de fórmula (I) o una sal del mismo para prevenir o tratar la disfunción del almacenamiento, la disfunción de la evacuación y las enfermedades vesicales/uretrales, y similares, al compuesto de fórmula (I) o una sal del mismo para prevenir o tratar la disfunción del almacenamiento, la disfunción de la evacuación y las enfermedades vesicales/uretrales, y similares, y a un método para prevenir o tratar la disfunción del almacenamiento, la disfunción de la evacuación y las enfermedades
vesicales/uretrales, y similares, que incluye administrar a un sujeto una cantidad eficaz del compuesto de fórmula (I) o una sal del mismo. Además, el “sujeto” es un humano u otro animal que necesita tal prevención o tratamiento, y en una determinada realización, un humano que necesita tal prevención o tratamiento.
En la presente memoria descriptiva, la “disfunción del almacenamiento” se refiere a un “trastorno de la función de almacenamiento (disfunción del almacenamiento)” con la que no puede retenerse la orina durante el almacenamiento, y la “disfunción de la evacuación” se refiere a un “trastorno de la función de evacuación (disfunción de la evacuación)” con la que no puede descargarse la orina suficientemente durante la evacuación debida a una resistencia uretral aumentada y una contracción de la vejiga disminuida (Neurourol Urodynam, 21: págs. 167-178 (2002)).
Tal como se usan en la presente memoria descriptiva, las “enfermedades vesicales/uretrales” incluyen “disfunción de las vías urinarias inferiores”, y “síntomas de las vías urinarias inferiores (LUTS)” (Neurourol Urodynam, 21: págs.
167-178 (2002)), que son síntomas derivados de la disfunción de las vías urinarias inferiores. Por consiguiente, las “enfermedades vesicales/uretrales” tal como se usan en el presente documento incluyen la “disfunción del almacenamiento” y la “disfunción de la evacuación”.
En la presente invención, los ejemplos de las enfermedades vesicales/uretrales incluyen, en una determinada realización, vejiga hipoactiva, vejiga hipotónica, vejiga acontráctil, hipoactividad del detrusor, vejiga neurógena, insuficiencia de la relajación de la uretra o disinergia detrusor-esfínter uretral externo, vejiga hiperactiva, y síntomas de las vías urinarias inferiores de las mismas, e hiperplasia prostática benigna, cistitis intersticial, prostatitis crónica, cálculo de uretra, y síntomas de las vías urinarias inferiores que las acompañan, y similares.
En otra realización, los ejemplos de las enfermedades vesicales/uretrales incluyen vejiga hipoactiva, vejiga hipotónica, vejiga acontráctil, hipoactividad del detrusor, vejiga neurógena, insuficiencia de la relajación de la uretra o disinergia detrusor-esfínter uretral externo, y síntomas de las vías urinarias inferiores de las mismas, e hiperplasia prostática benigna, cistitis intersticial, prostatitis crónica, cálculo de uretra, y síntomas de las vías urinarias inferiores que las acompañan, y similares.
En una realización adicional, los ejemplos de las enfermedades vesicales/uretrales incluyen vejiga hipoactiva, vejiga hipotónica, vejiga acontráctil, hipoactividad del detrusor, vejiga neurógena, insuficiencia de la relajación de la uretra o disinergia detrusor-esfínter uretral externo, y síntomas de las vías urinarias inferiores de las mismas, hiperplasia prostática benigna y síntomas de las vías urinarias inferiores que las acompañan, y similares.
En todavía una realización adicional, los ejemplos de las enfermedades vesicales/uretrales incluyen vejiga hipoactiva, vejiga hipotónica, vejiga acontráctil, hipoactividad del detrusor, insuficiencia de la relajación de la uretra o disinergia detrusor-esfínter uretral externo, síntomas de las vías urinarias inferiores de las mismas, hiperplasia prostática benigna y síntomas de las vías urinarias inferiores que las acompañan, y similares.
En la presente invención, los ejemplos específicos de la disfunción del almacenamiento incluyen vejiga hiperactiva, y síntomas de la vejiga hiperactiva tales como necesidad imperiosa de orinar, polaquiuria, incontinencia imperiosa, nicturia, y similares.
En la presente invención, los ejemplos de la disfunción de la evacuación incluyen disfunción de la evacuación debido a un aumento en la resistencia uretral y disfunción de la evacuación debido a una disminución en la fuerza contráctil de la vejiga. En una determinada realización, los ejemplos específicos de la misma incluyen disfunción de la evacuación en la vejiga hipoactiva, disfunción de la evacuación en la vejiga hipotónica, disfunción de la evacuación en la vejiga acontráctil, disfunción de la evacuación en la vejiga neurógena, disfunción de la evacuación en la hipoactividad del detrusor, disfunción de la evacuación en la insuficiencia de la relajación de la uretra o disinergia detrusor-esfínter uretral externo, disfunción de la evacuación que acompaña a la hiperplasia prostática benigna, disfunción de la evacuación que acompaña a la prostatitis crónica, disfunción de la evacuación que acompaña al cálculo de uretra, disfunción de la evacuación que acompaña a la cistitis intersticial, disfunción de la evacuación que acompaña a la hipoactividad del detrusor, y similares.
En una realización adicional, los ejemplos de la disfunción de la evacuación incluyen disfunción de la evacuación en la vejiga hipoactiva, disfunción de la evacuación en la vejiga hipotónica, disfunción de la evacuación en la vejiga acontráctil, disfunción de la evacuación en la hipoactividad del detrusor, disfunción de la evacuación en la insuficiencia de la relajación de la uretra o disinergia detrusor-esfínter uretral externo, disfunción de la evacuación que acompaña a la hiperplasia prostática benigna, y similares.
Efecto de la invención
El compuesto de fórmula (I) o una sal del mismo tiene una acción inhibidora de la PDE9, y puede usarse como agente para prevenir y/o tratar enfermedades relacionadas con la degradación de GMPc mediante PDE9, por ejemplo, disfunción del almacenamiento, disfunción de la evacuación y enfermedades vesicales/uretrales, en otra realización, las enfermedades tales como vejiga hipoactiva, vejiga hipotónica, vejiga acontráctil, hipoactividad del
detrusor, vejiga neurógena, insuficiencia de la relajación de la uretra o disinergia detrusor-esfínter uretral externo, vejiga hiperactiva, y síntomas de las vías urinarias inferiores de las mismas, e hiperplasia prostética benigna, cistitis intersticial, prostatitis crónica, cálculo de uretra, y síntomas de las vías urinarias inferiores que las acompañan, y similares, y en una realización adicional, las enfermedades tales como vejiga hipoactiva, vejiga hipotónica, vejiga acontréctil, vejiga neurógena, hipoactividad del detrusor, vejiga hiperactiva, polaquiuria, nicturia, incontinencia, hiperplasia prostética benigna, síntomas de las vías urinarias inferiores, disfunción de la evacuación que acompaña a la insuficiencia de la relajación de la uretra o disinergia detrusor-esfínter uretral externo, cistitis intersticial, prostatitis crónica, cálculo de uretra, y similares.
Realizaciones para llevar a cabo la invención
(6) El compuesto o una sal del mismo, en el que
A es CH,
R1 es 2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-ilo sustituido con fluoro, bromo, metilo, etilo, trifluorometilo o metoxilo en la posición 5, o 5-metoxi-2,3-dihidro-1H-pirrolo[2,3-c]piridin-1-ilo,
R2 es metilo o metoximetilo, y
R3 es tetrahidrofuran-3-ilo.
(7) El compuesto o una sal del mismo tal como se expone en la reivindicación 1, en el que
A es CH,
R1 es 2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-ilo sustituido con fluoro, bromo, metilo, etilo, trifluorometilo o metoxilo en la posición 5,
R2 es metilo o metoximetilo, y
R3 es tetrahidrofuran-3-ilo.
(8) El compuesto o una sal del mismo tal como se expone en la reivindicación 1, en el que
A es CH,
R1 es 5-metoxi-2,3-dihidro-1H-pirrolo[2,3-c]piridin-1-ilo,
R2 es metilo o metoximetilo, y
R3 es tetrahidrofuran-3-ilo.
(14) El compuesto o una sal del mismo, en el que
A es CH,
R1 es 5-metoxi-2,3-dihidro-1H-pirrolo[2,3-c]piridin-1-ilo,
R2 es metilo, y
R3 es tetrahidro-2H-piran-3-ilo.
Los ejemplos de los compuestos específicos incluidos en el compuesto de fórmula (I) o una sal del mismo incluyen los siguientes compuestos:
7- [(5-metoxi-2,3-dihidro-1H-pirrolo[2,3-c]piridin-1-il)carbonil]-8-(metoximetil)-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
8- (metoximetil)-7-[(5-metil-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
8-metil-7-[(5-metil-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
7-[(5-metoxi-2,3-dihidro-1H-pirrolo[2,3-c]piridin-1-il)carbonil]-8-metil-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin4(5H)-ona,
7-[(5-etil-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-8-metil-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
7-[(5-bromo-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-8-metil-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
7-[(5,6-difluoro-2,3-dihidro-1H-indol-1-il)carbonil]-8-metil-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona, 7-[(5-fluoro-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-8-metil-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
7- [(5-metoxi-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-8-metil-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
8- metil-1-(tetrahidrofuran-3-il)-7-{[(5-(trifluorometil)-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il]carbonil)imidazo[1,5-a]quinoxalin-4(5H)-ona,
7-[(5-metoxi-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-8-(metoximetil)-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
7-[(5-bromo-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-8-(metoximetil)-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
7-[(5-etil-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-8-(metoximetil)-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
y sales de los mismos, e isómeros ópticos aislados de los mismos.
Los ejemplos de los compuestos específicos incluidos en la realización (6) descrita anteriormente incluyen los siguientes compuestos:
7- [(5-metoxi-2,3-dihidro-1H-pirrolo[2,3-c]piridin-1-il)carbonil]-8-(metoximetil)-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
8- (metoximetil)-7-[(5-metil-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
8-metil-7-[(5-metil-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
7-[(5-metoxi-2,3-dihidro-1H-pirrolo[2,3-c]piridin-1-il)carbonil]-8-metil-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
7-[(5-etil-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-8-metil-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
7-[(5-bromo-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-8-metil-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
7-[(5-fluoro-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-8-metil-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
7-[(5-etil-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-8-(metoximetil)-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
7- [(5-metoxi-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-8-metil-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
8- metil-1-(tetrahidrofuran-3-il)-7-{[5-(trifluorometil)-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il]carbonil}imidazo[1,5-a]quinoxalin-4(5H)-ona,
7-[(5-metoxi-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-8-(metoximetil)-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
7-[(5-bromo-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-8-(metoximetil)-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
y sales de los mismos, e isómeros ópticos aislados de los mismos.
En otra realización, los ejemplos de los compuestos específicos incluidos en la realización (6) descrita anteriormente incluyen los siguientes compuestos:
(+)-7-[(5-metoxi-2,3-dihidro-1H-pirrolo[2,3-c]piridin-1-il)carbonil]-8-(metoximetil)-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
(-)-7-[(5-metoxi-2,3-dihidro-1H-pirrolo[2,3-c]piridin-1-il)carbonil]-8-(metoximetil)-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona
(+)-8-(metoximetil)-7-[(5-metil-2,3-dihidro-(1H-pirrolo[2,3-b]piridin-1-il)carbonil]-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
(-)-8-(metoximetil)-7-[(5-metil-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
(+)-7-[(5-metoxi-2,3-dihidro-1H-pirrolo[2,3-c]piridin-1-il)carbonil]-8-metil-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
(-)-7-[(5-metoxi-2,3-dihidro-1H-pirrolo[2,3-c]piridin-1-il)carbonil]-8-metil-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
(+)-7-[(5-bromo-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-8-metil-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
(-)-7-[(5-bromo-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-8-metil-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
(+)-7-[(5-bromo-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-8-(metoximetil)-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
(-)-7-[(5-bromo-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-8-(metoximetil)-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
(+)-7-[(5-etil-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-8-(metoximetil)-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
(-)-7-[(5-etil-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-8-(metoximetil)-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
o sales de los mismos.
En una realización adicional, los ejemplos de los compuestos específicos incluidos en la realización (6) descrita anteriormente incluyen los siguientes compuestos:
(+)-7-[(5-metoxi-2,3-dihidro-1H-pirrolo[2,3-c]piridin-il)carbonil]-8-metil-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
(-)-7-[(5-metoxi-2,3-dihidro-1H-pirrolo[2,3-c]piridin-1-il)carbonil]-8-metil-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
o sales de los mismos.
En todavía una realización adicional, los ejemplos de los compuestos específicos incluidos en la realización (6) descrita anteriormente incluyen los siguientes compuestos:
(+)-7-[(5-metoxi-2,3-dihidro-1H-pirrolo[2,3-c]piridin-1-il)carbonil]-8-(metoximetil)-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
(-)-7-[(5-metoxi-2,3-dihidro-1H-pirrolo[2,3-c]piridin-1-il)carbonil]-8-(metoximetil)-1-(tetrahidrofuran-3-il)imidazol[1,5-a]quinoxalin-4(5H)-ona,
(+)-8-(metoximetil)-7-[(5-metil-2,3-dihidro-pirrolo[2,3-b]piridin-1-il)carbonil]-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
(-)-8-(metoximetil)-7-[(5-metil-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
o sales de los mismos.
En todavía una realización adicional, los ejemplos de los compuestos específicos incluidos en la realización (6) descrita anteriormente incluyen los siguientes compuestos:
(+)-7-[(5-bromo-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-8-metil-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
(-)-7-[(5-bromo-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-8-metil-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
(+)-7-[(5-bromo-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-8-(metoximetil)-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
(-)-7-[(5-bromo-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-8-(metoximetil)-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
o sales de los mismos.
En todavía una realización adicional, los ejemplos de los compuestos específicos incluidos en la realización (6) descrita anteriormente incluyen los siguientes compuestos:
(+)-7-[(5-etil-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-8-(metoximetil)-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
(-)-7-[(5-etil-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-8-(metoximetil)-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
o sales de los mismos.
Los ejemplos de los compuestos específicos incluidos en la realización (7) descrita anteriormente incluyen los siguientes compuestos:
8-(metoximetil)-7-[(5-metil-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
8-metil-7-[(5-metil-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
7-[(5-etil-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-8-metil-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
7-[(5-bromo-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-8-metil-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
7-[(5-fluoro-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-8-metil-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
7-[(5-etil-2,3-dihidro-1H-pirrolo[2,3-b]pirin-1-il)carbonil]-8-(metoximetil)-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
7- [(5-metoxi-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-8-metil-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
8- metil-1-(tetrahidrofuran-3-il)-7-{[5-(trifluorometil)-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il]carbonil}imidazo[1,5-a]quinoxalin-4(5H)-ona,
7-[(5-metoxi-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-8-(metoximetil)-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
7-[(5-bromo-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-8-(metoximetil)-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
y sales de los mismos, e isómeros ópticos aislados de los mismos.
En otra realización, los ejemplos de los compuestos específicos incluidos en la realización (7) descrita anteriormente incluyen los siguientes compuestos:
(+)-8-(metoximetil)-7-[(5-metil-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
(-)-8-(metoximetil)-7-[(5-metil-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-1-(tetrahidrofuran-3-il)imidazo[1,5-a]qumoxalin-4(5H)-ona,
(+)-7-[(5-bromo-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-8-metil-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
(-)-7-[(5-bromo-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-8-metil-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
(+)-7-[(5-bromo-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-8-(metoximetil)-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
(-)-7-[(5-bromo-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-8-(metoximetil)-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
(+)-7-[(5-etil-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-8-(metoximetil)-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
(-)-7-[(5-etil-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-8-(metoximetil)-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
o sales de los mismos.
Los ejemplos de los compuestos específicos incluidos en la realización (8) descrita anteriormente incluyen los siguientes compuestos:
7-[(5-metoxi-2,3-dihidro-1H-pirrolo[2,3-c]piridin-1-il)carbonil]-8-(metoximetil)-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
7-[(5-metoxi-2,3-dihidro-1H-pirrolo[2,3-c]piridin-1-il)carbonil]-8-metil-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
y sales de los mismos, e isómeros ópticos aislados de los mismos.
En otra realización, los ejemplos de los compuestos específicos incluidos en la realización (8) descrita anteriormente incluyen los siguientes compuestos:
(+)-7-[(5-metoxi-2,3-dihidro-1H-pirrolo[2,3-c]piridin-1-il)carbonil]-8-(metoximetil)-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
(-)-7-[(5-metoxi-2,3-dihidro-1H-pirrolo[2,3-c]piridin-1-il)carbonil]-8-(metoximetil)-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
(+)-7-[(5-metoxi-2,3-dihidro-1H-pirrolo[2,3-c]piridin-1-il)carbonil]-8-metil-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
(-)-7-[(5-metoxi-2,3-dihidro-1H-pirrolo[2,3-c]piridin-1-il)carbonil]-8-metil-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,
o sales de los mismos.
El compuesto de fórmula (I) puede existir en forma de tautómeros o isómeros geométricos dependiendo de la clase de sustituyentes. En la presente memoria descriptiva, el compuesto de fórmula (I) debe describirse en una sola forma de isómero, aunque la presente invención incluye los otros isómeros, las formas aisladas de los isómeros, o una mezcla de los mismos.
Además, el compuesto de fórmula (I) puede tener átomos de carbono asimétricos o quiralidad axial en algunos casos, y de manera correspondiente, puede existir en forma de isómeros ópticos. La presente invención incluye tanto una forma aislada de los isómeros ópticos del compuesto de fórmula (I) o una mezcla de los mismos.
Además, la presente invención también incluye un profármaco farmacéuticamente aceptable del compuesto representado por la fórmula (I). El profármaco farmacéuticamente aceptable es un compuesto que tiene un grupo que puede convertirse en un grupo amino, un grupo hidroxilo, un grupo carboxilo, o similar a través de solvólisis o en condiciones fisiológicas. Los ejemplos del grupo que forma el profármaco incluyen los grupos descritos en Prog. Med., 5, 2157-2161 (1985) y Pharmaceutical Research and Development, Drug Design, Hirokawa Publishing Company (1990), vol. 7, 163-198.
Además, la sal del compuesto de fórmula (I) es una sal farmacéuticamente aceptable del compuesto de fórmula (I) y puede formar una sal de adición de ácido o una sal con una base dependiendo de la clase de sustituyentes. Los ejemplos específicos de la misma incluyen sales de adición de ácido con ácidos inorgánicos tales como ácido clorhídrico, ácido bromhídrico, ácido yodhídrico, ácido sulfúrico, ácido nítrico, ácido fosfórico, y similares, y con ácidos orgánicos tales como ácido fórmico, ácido acético, ácido propiónico, ácido oxálico, ácido malónico, ácido succínico, ácido fumárico, ácido maleico, ácido láctico, ácido málico, ácido mandélico, ácido tartárico, ácido dibenzoiltartárico, ácido ditoliltartárico, ácido cítrico, ácido metanosulfónico, ácido etanosulfónico, ácido bencenosulfónico, ácido p-toluenosulfónico, ácido aspártico, ácido glutámico, y similares, y sales con bases inorgánicas tales como sodio, potasio, magnesio, calcio, aluminio, y similares, o bases orgánicas tales como metilamina, etilamina, etanolamina, lisina, arginina, trometamina, ornitina, y similares, sales con diversos aminoácidos o derivados de aminoácidos tales como acetil-leucina y similares, sales de amonio, etc.
Además, la presente invención también incluye diversos hidratos o solvatos, y sustancias cristalinas polimorfas del compuesto de fórmula (I) y una sal del mismo. Además, la presente invención también incluye compuestos marcados con diversos isótopos radiactivos o no radiactivos.
(Métodos de preparación)
El compuesto de fórmula (I) y una sal del mismo pueden prepararse usando las características basadas en la estructura básica o el tipo de sustituyente y aplicando diversos métodos de síntesis conocidos. Durante la preparación, el reemplazo del grupo funcional relevante con un grupo protector adecuado (un grupo que puede convertirse fácilmente en el grupo funcional relevante) en la fase desde un material de partida hasta un producto intermedio puede ser eficaz dependiendo del tipo del grupo funcional en la tecnología de producción en algunos casos. El grupo protector para tal grupo funcional puede incluir, por ejemplo, los grupos protectores descritos en “Greene's Protective Groups in Organic Synthesis (4a ed., 2006)” escrito por P. G. M. Wuts y T. W. Greene, y uno de estos debe seleccionarse y usarse solo según sea necesario dependiendo de las condiciones de reacción. En esta clase de método, puede obtenerse un compuesto deseado introduciendo el grupo protector, llevando a cabo una reacción y eliminando el grupo protector cuando sea necesario.
Además, el profármaco del compuesto de fórmula (I) puede producirse introduciendo un grupo específico o llevando a cabo la reacción usando el compuesto obtenido de la fórmula (I) en la fase desde un material de partida hasta un producto intermedio, igual como en el caso del grupo protector mencionado anteriormente. La reacción puede llevarse a cabo usando métodos conocidos por los expertos en la técnica, tales como esterificación, amidación, deshidratación habituales, y similares.
A continuación en el presente documento, se describirán los métodos de preparación representativos para el compuesto de fórmula (I). Cada uno de los procedimientos de producción también puede llevarse a cabo con referencia a las referencias adjuntas en la presente descripción.
(Procedimiento de producción 1)
[Fórm. quím. 10]
(en la que A, R1, R2 y R3 representan el mismo significado tal como se definió anteriormente. R representa un alquilo inferior. Lo mismo debe aplicarse a continuación en el presente documento.)
El compuesto (I-1) de la presente invención puede obtenerse mediante la reacción de un compuesto (1a) con un compuesto (1b).
En esta reacción, se usan un ácido carboxílico (1a) y una amina (1b) en cantidades equivalentes, o con cualquiera de los mismos en una cantidad en exceso, y se agita una mezcla de los mismos en cualquier condición de temperatura desde enfriamiento hasta calentamiento, preferiblemente a de -20°C a 120°C, habitualmente durante de 0,1 horas a 5 días, en un disolvente que es inerte a la reacción, en presencia de un agente de condensación. El disolvente tal como se usa en el presente documento no está particularmente limitado, pero los ejemplos del mismo incluyen hidrocarburos aromáticos tales como benceno, tolueno, xileno, y similares, hidrocarburos halogenados tales como diclorometano, 1,2-dicloroetano, cloroformo, y similares, éteres tales como dietil éter, tetrahidrofurano, 1,4-dioxano, dimetoxietano, y similares, N,N-dimetilformamida, N-metilpirrolidona, dimetilsulfóxido, acetato de etilo, acetonitrilo, o agua y una mezcla de los mismos. Los ejemplos del agente de condensación incluyen 1-(3-dimetilaminopropil)-3-etilcarbodiimida, 1,3-diciclohexilcarbodiimida, hexafluorofosfato de O-(7-azabenzotriazol-1-il)-N,N,N',N'-tetrametiluronio (HATU), tetrafluoroborato de O-(benzotriazol-1-il)-N,N,N',N'-tetrametiluronio (TBTU), hexafluorofosfato de bromo(tripirrolidin-1-il)fosfonio (PyBrop), 1,1'-carbonildiimidazol (CDI), difenilfosforilazida y cloruro de fosforilo, pero no se limitan a estos, y también puede usarse un agente de condensación soportado sobre una resina de poliestireno, por ejemplo, PS-carbodiimida (Biotage AB, Suecia). Puede ser preferible para la reacción en algunos casos usar un aditivo (por ejemplo, 1-hidroxibenzotriazol). Puede ser ventajoso en algunos casos para el progreso suave de la reacción llevar a cabo la reacción en presencia de una base orgánica tal como trietilamina, N,N-diisopropiletilamina, N-metilmorfolina, y similares, o una base inorgánica tal como carbonato de potasio, carbonato de sodio, hidróxido de potasio, y similares. Además, el uso de un reactor de microondas (Biotage AB) puede permitir el progreso suave de la reacción en algunos casos. Dependiendo del caso, también puede usarse una resina de poliestireno con soporte de isocianato , por ejemplo, PS-isocianato (Biotage AB, Suecia) y similares para retirar una cantidad en exceso de amina después de la finalización de la reacción, y además, también puede usarse una resina de poliestireno con soporte de sal de amonio cuaternario , por ejemplo, MP-carbonato (Biotage AB, Suecia) y similares para retirar una cantidad en exceso de los aditivos después de la finalización de la reacción. Además, también puede usarse un método en el que un ácido carboxílico (1a) se convierte en su derivado reactivo y luego se hace reaccionar con una amina (1b). Los ejemplos del derivado reactivo del ácido carboxílico incluyen haluros de ácido que pueden obtenerse mediante la reacción de un agente de halogenación tal como cloruro de fosforilo, cloruro de tionilo, y similares, anhídridos de ácido mixtos que pueden obtenerse mediante la reacción de cloroformiato de isobutilo o similares, ésteres activos obtenidos mediante la condensación con 1-hidroxibenzotriazol o similares, etc. La reacción del derivado reactivo y el compuesto (1b) puede llevarse a cabo en cualquier condición de temperatura desde enfriamiento hasta calentamiento, preferiblemente a de -20°C a 60°C, en un disolvente que es inerte a la reacción, tal como hidrocarburos halogenados, hidrocarburos aromáticos, éteres, y similares.
Además, también puede usarse un método en el que a un reactivo de amida de aluminio obtenido haciendo reaccionar un éster (1c) con trimetilaluminio y amina (1b) se le deja experimentar una reacción.
Para estas etapas, puede hacerse referencia a los métodos descritos en “Organic Functional Group Preparations”, escrito por S. R. Sandler y W. Karo, 2a edición, vol. 1, Academic Press Inc., 1991, y “Courses in Experimental Chemistry (5a edición)”, editado por The Chemical Society of Japan, vol. 16 (2005) (Maruzen).
Además, algunos de los compuestos representados por la fórmula (I) también pueden producirse a partir del compuesto según la presente invención producido tal como se describió anteriormente combinando de manera apropiada procedimientos habitualmente usados por los expertos en la técnica, tales como alquilación, acilación, sustitución, oxidación, reducción, hidrólisis, desprotección, halogenación conocidos, y similares (véase, por ejemplo, “Courses in Experimental Chemistry” (5a edición), editado por The Chemical Society of Japan, (2005) (Maruzen)). Además, un procedimiento que puede usarse habitualmente por los expertos en la técnica también puede usarse para productos intermedios para la preparación.
(Síntesis del material de partida 1)
[Fórm. quím. 11]

(en la que Li significa un grupo saliente y R’ significa un alquilo inferior. Lo mismo debe aplicarse a continuación en el presente documento).
La etapa representada por la etapa 1-1 es una reacción para obtener un compuesto (2b) mediante una reacción de hidrogenación de un compuesto (2a). En esta reacción, se agita el compuesto (2a) en presencia de un catalizador de metal, habitualmente durante de 1 hora a 5 días, en un disolvente inerte a la reacción, bajo una atmósfera de hidrógeno. Esta reacción se lleva a cabo habitualmente en un intervalo de presión normal a presión aumentada, y en cualquier condición de temperatura desde enfriamiento hasta calentamiento, preferiblemente a presión normal y a temperatura ambiente. Los ejemplos del disolvente usado en el presente documento no están particularmente limitados, pero incluyen alcoholes tales como metanol, etanol, 2-propanol, y similares, éteres tales como dietil éter, tetrahidrofurano, 1,4-dioxano, dimetoxietano, y similares, agua, ácido acético, acetato de etilo, N,N-dimetilformamida, dimetilsulfóxido, y una mezcla de los mismos. Como el catalizador de metal, se usan preferiblemente catalizadores de paladio tales como paladio sobre carbono, negro de paladio, hidróxido de paladio, y similares, catalizadores de platino tales como una placa de platino, óxido de platino, y similares, catalizadores de níquel tales como níquel reducido, níquel Raney, y similares, catalizadores de rodio tales como tetrakistrifenilfosfina clororodio y similares, etc. En lugar de gas hidrógeno, puede usarse ácido fórmico, formiato de amonio o monohidrato de hidrazina en cantidades equivalentes, o con cualquiera de los mismos en una cantidad en exceso como una fuente de hidrógeno, en relación con el compuesto (2a). Esta etapa incluye además una reacción para obtener un compuesto (2b) mediante una reacción de reducción de un compuesto (2a). En esta reacción, se agita el compuesto (2a) en
presencia de un catalizador de hierro tal como hierro reducido, carbón activado/cloruro de hierro (III), y similares, habitualmente durante de 0,5 horas a 5 días, en un disolvente inerte a la reacción. Esta reacción se lleva a cabo habitualmente en cualquier condición de temperatura desde enfriamiento hasta calentamiento, preferiblemente a de 0°C a 100°C. Para esta etapa, puede hacerse referencia a los métodos descritos en “Reductions in Organic Chemistry, 2a ed. (ACS Monograph: 188)” escrito por M. Hudlicky, ACS, 1996, “Courses in Experimental Chemistry” (4a edición), editado por The Chemical Society of Japan, vol. 26 (1992) (Maruzen), y similares.
La etapa representada por la etapa 1-13 es una reacción para obtener un compuesto (2d) mediante la reacción del compuesto (2c) y ferc-butilamina. En esta reacción, se usan el compuesto (2c) y ferc-butilamina en cantidades equivalentes, o con cualquiera de los mismos en una cantidad en exceso, y se agita una mezcla de los mismos en cualquier condición de temperatura desde enfriamiento hasta calentamiento a reflujo, preferiblemente a de 0°C a 80°C, habitualmente durante de 0,1 horas a 5 días, en un disolvente que es inerte a la reacción o sin disolvente. El disolvente tal como se usa en el presente documento no está particularmente limitado, pero los ejemplos del mismo incluyen hidrocarburos aromáticos tales como benceno, tolueno, xileno, y similares, éteres tales como dietil éter, tetrahidrofurano, 1,4-dioxano, dimetoxietano, y similares, hidrocarburos halogenados tales como diclorometano, 1,2-dicloroetano, cloroformo, y similares, N,N-dimetilformamida, N,N-dimetilacetamida, dimetilsulfóxido, acetato de etilo, acetonitrilo, N-metilpirrolidona, N-etilpirrolidona, y una mezcla de los mismos. Puede ser ventajoso en algunos casos para el progreso suave de la reacción llevar a cabo la reacción en presencia de una base orgánica tal como trietilamina, N,N-diisopropiletilamina, piridina, N-metilmorfolina, y similares, o una base inorgánica tal como hidróxido de sodio, carbonato de potasio, carbonato de sodio, hidróxido de potasio, y similares. Para esta etapa, puede hacerse referencia a los métodos descritos en “Organic Functional Group Preparations”, escrito por S. R. Sandler y W. Karo, 2a edición, vol. 1, Academic Press Inc., 1991, “Courses in Experimental Chemistry (5a edición)”, editado por The Chemical Society of Japan, vol. 14 (2005) (Maruzen), y similares.
La etapa representada por la etapa 1-14 es una reacción para obtener un compuesto (2e) mediante una reacción de desalquilación del compuesto (2d). En el presente documento, la reacción de desalquilación puede llevarse a cabo con referencia al método descrito en “Greene's Protective Groups in Organic Synthesis (4a edición, 2006)”.
La etapa representada por la etapa 1-15 es una reacción para obtener un compuesto (2b) mediante una reacción de hidrogenación del compuesto (2e), para la que puede incorporarse el método usado en la etapa 1-1. Además, esta etapa también incluye una reacción para obtener el compuesto (2b) mediante una reacción de reducción del compuesto (2e), para la que puede incorporarse el método usado en la etapa 1-1.
La etapa representada por la etapa 1-2 es una reacción para obtener un compuesto (2f) mediante una reacción de ciclación del compuesto (2b) con oxalato de dietilo ((COOEt)2) o ácido oxálico. Para esta etapa, puede hacerse referencia a los métodos descritos en J. Med. Chem., 38 (19): págs. 3720-3740 (1995), la memoria descriptiva de la publicación de solicitud de patente estadounidense n.° 2004/192698, y similares.
La etapa representada por la etapa 1-3 es una reacción para obtener un compuesto (2g) mediante la cloración del compuesto (2f). En esta reacción, se usa un agente de cloración en cantidades equivalentes, o con cualquiera de los mismos en una cantidad en exceso, en relación al compuesto (2f), y se agita una mezcla de los mismos en cualquier condición de temperatura desde enfriamiento hasta calentamiento a reflujo, preferiblemente desde temperatura ambiente hasta calentamiento a reflujo, habitualmente durante de 0,1 horas a 5 días, en un disolvente que es inerte a la reacción o sin disolvente. El disolvente tal como se usa en el presente documento no está particularmente limitado, pero los ejemplos del mismo incluyen hidrocarburos aromáticos tales como benceno, tolueno, xileno, y similares, éteres tales como tetrahidrofurano, 1,4-dioxano, dimetoxietano, y similares, hidrocarburos halogenados tales como 1,2-dicloroetano, cloroformo, y similares, acetonitrilo, y una mezcla de los mismos. Puede ser ventajoso en algunos casos para el progreso suave de la reacción llevar a cabo la reacción en presencia de N,N-dimetilformamida o N,N-dietilanilina, y similares. Los ejemplos del agente de cloración incluyen cloruro de fosforilo, pentacloruro de fósforo y cloruro de tionilo.
La etapa representada por la etapa 1-4 es una reacción para obtener un compuesto (2h) mediante la reacción del compuesto (2g) con hidrazina (NH2NH2) monohidratada, para la que puede incorporarse el método usado en la etapa 1-13.
Cada una de las etapas representadas por la etapa 1-5 y la etapa 1-10 es una reacción para obtener un compuesto (2k) o un compuesto (2o) mediante una reacción de ciclación, o una reacción de ciclación e hidrólisis del compuesto (2h) y el compuesto (2j). En esta reacción, el compuesto (2j) se usa en una cantidad equivalente o una cantidad en exceso, en relación al compuesto (2h), y se agita una mezcla de los mismos en cualquier condición de temperatura desde enfriamiento hasta calentamiento a reflujo, preferiblemente a de temperatura ambiente a calentamiento a reflujo, habitualmente durante de 0,1 horas a 5 días, en un disolvente que es inerte a la reacción o sin disolvente. Para esta etapa, puede hacerse referencia al método descrito en J. Med. Chem., 33 (8): págs. 2240-2254 (1990).
La etapa representada por la etapa 1-6 es una reacción para obtener un compuesto (2m) mediante la reacción del compuesto (2h) con el compuesto (2l), para la que puede incorporarse el método usado en el (Procedimiento de producción 1).
La etapa representada por la etapa 1-7 es una reacción para obtener un compuesto (2k) mediante la reacción del compuesto (2m) con cloruro de tionilo, para la que puede incorporarse el método usado en la (etapa 1-3).
Cada una de las etapas representadas por la etapa 1-8 y la etapa 1-11 es una reacción para obtener un compuesto (2n) mediante una reacción de hidrólisis del compuesto (2k) o el compuesto (2o). En el presente documento, la reacción de hidrólisis puede llevarse a cabo con referencia al método descrito en “Greene’s Protective Groups in Organic Synthesis (4a edición, 2006)”.
Cada una de las etapas representadas por la etapa 1-9 y la etapa 1-12 es una reacción para obtener un compuesto (2k) o un compuesto (2o) mediante la reacción del compuesto (2h) con el compuesto (2i). En esta reacción, se usan el compuesto (2h) y el compuesto (2i) en cantidades equivalentes, o con cualquiera de los mismos en una cantidad en exceso, y se agita una mezcla de los mismos en cualquier condición de temperatura desde temperatura ambiente hasta calentamiento a reflujo, habitualmente durante de 1 a 5 días, en un disolvente que es inerte a la reacción o sin disolvente, en presencia de un oxidante. El disolvente tal como se usa en el presente documento no está particularmente limitado, pero los ejemplos del mismo incluyen hidrocarburos aromáticos tales como benceno, tolueno, xileno, y similares, hidrocarburos halogenados tales como diclorometano, 1,2-dicloroetano, cloroformo, y similares, éteres tales como dietil éter, tetrahidrofurano, 1,4-dioxano, dimetoxietano, y similares, N,N-dimetilformamida, dimetilsulfóxido, ácido acético, etilenglicol, acetato de etilo, acetonitrilo, agua, ácido clorhídrico, y una mezcla de los mismos. Los ejemplos del oxidante incluyen acetato de cobre, cloruro de cobre, bromo, cloranilo, 2,3-dicloro-5,6-diciano-p-benzoquinona, y similares. En esta reacción, cada una de formación de imina mediante deshidratación, ciclación usando un oxidante e hidrólisis puede llevarse a cabo por etapas. Para esta etapa, puede hacerse referencia al método descrito en Indian J. Chem., 38B: págs. 45-51, 1371-1373 (1999).
(Síntesis del material de partida 2)
[Fórm. quím. 12]

La etapa 2-1 es una etapa para obtener un compuesto (3b) mediante una reacción de nitración del compuesto (3a), y los ejemplos del agente de nitración incluyen ácido nítrico, una mezcla de ácido nítrico y ácido sulfúrico (ácido mixto), una mezcla de un nitrato de metal tal como nitrato de litio, nitrato de sodio, nitrato de potasio, y similares y ácido sulfúrico, sales de nitronio tales como tetrafluoroborato de nitronio y similares, nitrato de acetilo, etc. Para esta etapa, puede hacerse referencia al método descrito en “Courses in Experimental Chemistry (5a edición)”, editado por The Chemical Society of Japan, vol. 14 (2005) (Maruzen), y similares.
La etapa 2-2 es una etapa para obtener un compuesto (3d) mediante la reacción del compuesto (3b) y el compuesto (3c), para la que puede incorporarse el método usado en la etapa 1-4 de (Síntesis del material de partida 1). En el
presente documento, los ejemplos del grupo saliente incluyen halógeno, un grupo metanosulfoniloxilo, un grupo ptoluenosulfoniloxilo, y similares.
La etapa 2-3 es una etapa para obtener un compuesto (3c) mediante una reacción de ciclación del compuesto (2i), que puede llevarse a cabo en presencia de glioxal, o dicloroacetaldehído y amoniaco acuoso. Para esta etapa, puede hacerse referencia al método descrito en Angew. Chem. Int. Ed. Engl., 22 (7): págs. 560-561 (1983).
La etapa 2-4 es una etapa para obtener un compuesto (3e) mediante una reacción de hidrogenación del compuesto (3d), para la que puede incorporarse el método usado en la etapa 1-1 de (Síntesis del material de partida 1).
La etapa 2-5 es una etapa para obtener un compuesto (3f) mediante una reacción de ciclación del compuesto (3e) con 1,1'-carbonildiimidazol (CDI) o trifosgeno. Para esta etapa, puede hacerse referencia al método descrito en J. Med. Chem., 34 (9): págs. 2671-2677 (1991).
La etapa 2-6 es una etapa para obtener un compuesto (3g) mediante la reacción de hidrólisis del compuesto (3f), para la que puede incorporarse el método usado en la etapa 1-11 de (Síntesis del material de partida 1).
Los compuestos de fórmula (I) pueden aislarse y purificarse como sus compuestos, sales, hidratos, solvatos o sustancias cristalinas polimorfas libres de los mismos. Las sales del compuesto de fórmula (1) también pueden prepararse llevando a cabo el tratamiento de una reacción de formación de sal convencional.
El aislamiento y la purificación se llevan a cabo empleando operaciones químicas habituales tales como extracción, cristalización fraccionada, diversos tipos de cromatografía fraccionada, y similares.
Puede prepararse diversos isómeros seleccionando un compuesto de partida apropiado o separarse usando la diferencia en las propiedades fisicoquímicas entre los isómeros. Por ejemplo, los isómeros ópticos pueden obtenerse mediante un método general para diseñar la resolución óptica de productos racémicos (por ejemplo, cristalización fraccionada para inducir sales de diastereómeros con bases o ácidos ópticamente activos, cromatografía usando una columna quiral o similar, y otros), y además, los isómeros también pueden prepararse a partir de un material de partida ópticamente activo apropiado.
La actividad farmacológica del compuesto de fórmula (I) se confirmó mediante las pruebas mostradas a continuación.
Ejemplo de prueba 1: actividad de inhibición de la PDE
(1) Adquisición de PDE9
La PDE9 usada en el presente experimento se expresó/purificó mediante el método tal como en, por ejemplo, Guipponi et al., y Fisher et. al. (Fisher, D.A., et al., J. Biol. Chem., 273: págs. 15559-15564 (1998), Guipponi, M., et al., Hum. Genet., 103: págs. 386-392 (1998)).
(2) Evaluación de la actividad de inhibición de la PDE
La actividad de inhibición de la PDE se midió mediante el siguiente método. Es decir, a una disolución de tampón que contenía ácido tris(hidroximetil)aminometanoclorhídrico (40 mM, pH 8,0), cloruro de magnesio (5 mM) y 2-mercaptoetanol (4 mM) se le añadieron GMPc (1 |iM) y 3H-GMPc (0,33 |iCi/ml) para dar una disolución de tampón del sustrato. Se añadieron a la misma una disolución de sustancia de prueba y una disolución enzimática que se habían ajustado a una concentración óptima para realizar una reacción a 30°C. Se paró la reacción enzimática mediante la adición de perlas de ensayo de proximidad de centelleo (SPA) (Perkin Elmer, EE.UU.) que contenían 3-isobutil-1-metilxantina (IBMX) 5 mM. Para determinar la actividad enzimática, la cantidad de 5'-GMP, que es un producto de degradación de la reacción unido a las perlas de SPA, se midió con un lector de microplacas TopCount (Hewlett Packard, EE.UU.).
La tasa inhibidora se calculó tomando la radiactividad del control que contenía sustancia de prueba como (A), tomando la radiactividad del blanco que no contenía enzima como (B), y tomando la radiactividad de la sustancia de prueba como (C), y usando la siguiente ecuación.
Tasa inhibidora = 100 - {(C)-(B)/(A)-(B)}*100 (%)
Además, el valor de CI50 se calculó como la concentración de compuesto que inhibe los resultados obtenidos por el 50% mediante un método de regresión logística.
(3) Otra evaluación de la actividad de inhibición de la PDE
Para la PDE1, se adquirió una enzima recombinante (BPS Bioscience Inc., EE.UU.). La PDE2 se expresó/purificó
mediante un método de Yang et al. (Yang, Q., et al., Biochem. Biophys. Res. Commun., 205: págs. 1850-1858 (1994)), y la PDE4 se expresó/purificó mediante un método de Nemoz et al. (Nemoz, G., et al., FEBS Lett., 384: págs. 97-102 (1996)). Las PDE3, PDE5 y PDE6 se aislaron a partir de miocardio de conejo, próstata de conejo y retina de rata. Es decir, se seleccionaron los tejidos deseados a partir de cada uno de los animales, y se cortaron en rodajas en una disolución de tampón que contenía bis(2-hidroxietil)iminotris(hidroximetil)aminometano (20 mM), ditioeritritol (5 mM), ácido glicol éter diamina tetraacético (2 mM) y acetato de sodio (50 mM). Luego, se trituraron las células usando un homogeneizador Poritoron. Cada uno de los homogenatos de tejido se ultracentrifugó (100.000 g, 4°C, 60 minutos), y luego, se añadió el sobrenadante a una columna Q Sepharose. Mediante el gradiente de concentración de una disolución de tampón que contenía acetato de sodio de 0,05 a 1,2 M , cloruro de sodio (140 mM), cloruro de potasio (5 mM), glucosa (5 mM) y ácido 2-[4-(2-hidroxietil)-1-piperazinil]etanosulfónico (10 mM), se realizó la elución mediante intercambio iónico para obtener una fracción como una fuente de enzimas. Para cada una de las fracciones de eluato, se identificaron los subtipos de PDE mediante las propiedades enzimáticas y la susceptibilidad del inhibidor selectivo.
Para determinar la actividad enzimática de la PDE, se midió la degradabilidad para AMPc o GMPc mediante la cantidad de 5'-AMP o 5'-GMP, que es un producto de degradación de la reacción unido a las perlas de SPA, mediante el método tal como en la PDE9 anterior.
Para el compuesto de fórmula (I), se confirmó la acción de actividad de inhibición de la PDE mediante el método de prueba anterior. Por ejemplo, los siguientes compuestos de ejemplo presentaron las siguientes acciones de actividad de inhibición de la PDE (valores de CI50: nM).
Ejemplo 1 (3,5), ejemplo 2 (51), ejemplo 3 (22), ejemplo 4(a) (62), ejemplo 4(b) (11), ejemplo 5 (62), ejemplo 6 (64), ejemplo 7 (2,1), ejemplo 8 (2,8), ejemplo 9 (1,3), ejemplo 10 (4,5), ejemplo 11(31), ejemplo 12 (3,6), ejemplo 13 (14), ejemplo 14 (14), ejemplo 15 (2,8), ejemplo 16 (3,3), ejemplo 17 (8,5), ejemplo 18 (5,4), ejemplo 21 (22), ejemplo 22 (56), ejemplo 23 (4,9), ejemplo 24 (17), ejemplo 25(a) (62), ejemplo 25(b) (14), ejemplo 26(a) (152), ejemplo 26(b) (45), ejemplo 27 (84), ejemplo 27(a) (137), ejemplo 27(b) (26), ejemplo 28 (52), ejemplo 28(a) (139), ejemplo 28(b) (47), ejemplo 29 (15), ejemplo 29(a) (13), ejemplo 29(b) (2,2), ejemplo 30 (11), ejemplo 30(a) (38), ejemplo 30(b) (5,8), ejemplo 31 (1,4), ejemplo 32 (2,6), ejemplo 33 (0,81), ejemplo 34 (7,5), ejemplo 35 (19), ejemplo 36 (53), ejemplo 37 (53), ejemplo 38 (28), ejemplo 39(a) (52), ejemplo 39(b) (14), ejemplo 40(a) (77), ejemplo 40(b) (16), ejemplo 41 (18), ejemplo 42 (13), ejemplo 43(a) (25), ejemplo 43(b) (2,8), ejemplo 44 (6,1), ejemplo 45(a) (26), ejemplo 45(b) (8,9), ejemplo 46 (13), ejemplo 47 (2,1), ejemplo 48 (0,8). Los ejemplos 1-3, 5-24, 31-39, 41, 42 y 44 48 son ejemplos de referencia.
Además, se confirmó que los compuestos de fórmula (I), en particular los lotes de los compuestos de ejemplo, tienen una actividad de inhibición de la PDE selectiva. La actividad de inhibición de la PDE selectiva se refiere a una actividad de inhibición más potente que la actividad de inhibición, particularmente en PDE1, PDE3 y PDE6, y es, por ejemplo, un caso en el que el valor de CI50 (nM) es 1/10 o menos, en comparación con cualquiera de PDE1, PDE3 y PDE6, preferiblemente un caso en el que el valor de CI50 (nM) es 1/50 o menos, en comparación con 1, 2 o todas de PDE1, PDE3 y PDE6, y más preferiblemente un caso en el que el valor de CI50 (nM) es 1/100 o menos, en comparación con 1,2 o todas de PDE1, PDE3 y PDE6.
Ejemplo de prueba 2: evaluación de la actividad de inhibición de la PDE en células
Se introdujo de manera transitoria un gen CRE-luc en el que se unió un gen de luciferasa (luc) al gen de la PDE9 y al gen de elemento de respuesta (CRE) del AMP cíclico en la célula HEK293 para preparar una célula de coexpresión de PDE9 y CRE-luc. Al día siguiente, se añadieron IBMX 0,5 mM y una disolución de sustancia de prueba a las células y se cultivaron a 37°C durante 6 horas, y luego se retiró el sobrenadante del cultivo. Se añadió a la misma disolución de tampón de fosfato que contenía Triton X-100 al 0,2% para triturar las células. La actividad de inhibición de la PDE en la célula se evaluó añadiendo un líquido de sustrato de luciferina a la disolución celular obtenida triturando las células y midiendo la actividad de luciferasa en un lector de placas de fluorescencia/iluminación.
Como resultado, se confirmó que existen algunos compuestos que presentan actividad eficaz entre los compuestos de la presente invención.
Ejemplo de prueba 3: acción en modelo de rata de medición simultánea para respuestas de contracción de la vejiga/relajación de la uretra
La medición simultánea de las respuestas de contracción de la vejiga/relajación de la uretra usando una rata se llevó a cabo con una modificación parcial de un método en Wibberley et al., (Wibberley, A., et al., Br. J. Pharmacol., 136: págs. 399-414 (2002)). Es decir, una rata Sprague-Dawley (SD) hembra (Charles River Laboratories Japan, Inc.) se anestesió con uretano, y se expuso la vejiga mediante una incisión de línea media en la región abdominal inferior. Se insertó una cánula de doble vía (una cánula que tenía una estructura doble por PE190 y PE50) desde el vértice vesical en la vejiga, y se fijaron el vértice vesical y la cánula mediante puntos de sutura en un punto en el que la punta alcanzó la uretra proximal. Mientras se infundía solución salina fisiológica en la uretra a través de la cánula
externa, se midió la presión interna de la uretra mediante un transductor de presión a través de la cánula interna con una disolución salina infundida en la uretra a través de la cánula externa. Por otro lado, se insertó una cánula sencilla (PE50) en la vejiga desde el vértice vesical y se colocó en la misma. Se midió la presión interna de la vejiga a través de esta cánula. Después de que había pasado un periodo de estabilización posoperatorio, se infundió una solución salina fisiológica en la vejiga a través de la cánula del vértice vesical para provocar una reacción de contracción de la vejiga, y por tanto provocar una respuesta de relajación de la uretra que acompaña al reflejo de contracción de la vejiga. La sustancia de prueba se administró por vía intravenosa o por vía intraduodenal.
Como resultado, se confirmó que existen algunos compuestos que presentan actividad eficaz entre los compuestos de fórmula (I). Para algunos de los compuestos de fórmula (I), se muestra la razón con tiempo de relajación de la uretra aumentado durante la evacuación a 1 mg/kg (aumento en relación con el grupo de administración del disolvente (frente al vehículo) (%)). Ejemplo 4(b) (167), ejemplo 25(b) (131), ejemplo 27(b) (181), ejemplo 29(b) (149), ejemplo 30(b) (147), ejemplo de referencia 31 (149), ejemplo 43(b) (138), ejemplo de referencia 45(b) (141).
Ejemplo de prueba 4: acción en modelo de rata de disfunción de la evacuación inducida por fármacos
Se anestesió una rata SD macho (Japan SLC, Inc.) para colocar una cánula en la vejiga y en la vena yugular y se despertó más tarde en una jaula Ballman. Después de un periodo de estabilización posoperatorio, se infundió solución salina fisiológica en la vejiga para producir la evacuación. La infusión de la solución salina fisiológica se paró inmediatamente después de la evacuación, y se midió la cantidad de la orina evacuada usando una balanza de platillos colocada bajo la jaula Ballman. Después de la finalización de la evacuación, se recogió la orina residual por gravedad a través de una cánula colocada en la vejiga, y se midió el peso. Además, la presión interna de la vejiga se midió mediante un transductor de presión a través de la cánula de la vejiga. La disfunción de la evacuación se produjo mediante administración intravenosa de uno o una combinación de un agente anticolinérgico, un agonista del receptora! y un inhibidor de la producción de NO, y se observó la dinámica de evacuación después de la administración del fármaco. La sustancia de prueba se administró por vía intravenosa, por vía oral o por vía gástrica.
Como resultado, se confirmó que existen algunos compuestos que presentan actividad eficaz entre los compuestos de fórmula (I).
Como resultado de la prueba anterior, se confirmó que algunos de los compuestos de fórmula (1) tienen una acción inhibidora de la PDE9 y también se confirmó que algunos de los compuestos de fórmula (1) tienen una acción de relajación de la uretra durante la evacuación en los modelos animales. Por consiguiente, el compuesto de fórmula (I) puede usarse para prevenir o tratar enfermedades relacionadas con la degradación de GMPc mediante PDE9, por ejemplo, enfermedades tales como disfunción del almacenamiento, disfunción de la evacuación, enfermedades vesicales/uretrales, en otra realización, vejiga hipoactiva, vejiga hipotónica, vejiga acontráctil, hipoactividad del detrusor, vejiga neurógena, insuficiencia de la relajación de la uretra o disinergia detrusor-esfínter uretral externo, vejiga hiperactiva y síntomas de las vías urinarias inferiores de las mismas, e hiperplasia prostática benigna, cistitis intersticial, prostatitis crónica, cálculo de uretra y síntomas de las vías urinarias inferiores que las acompañan, y similares, y en una realización adicional, vejiga hipoactiva, vejiga hipotónica, vejiga acontráctil, vejiga neurógena, hipoactividad del detrusor, vejiga hiperactiva, polaquiuria, nicturia, incontinencia, hiperplasia prostática benigna, síntomas de las vías urinarias inferiores, disfunción de la evacuación que acompaña a la insuficiencia de la relajación de la uretra o disinergia detrusor-esfínter uretral externo, cistitis intersticial, prostatitis crónica, cálculo de uretra, y similares.
Además, los compuestos de fórmula (I) tienen una actividad de inhibición de la PDE selectiva, y como resultado, pueden evitarse los efectos secundarios derivados de la acción de otros subtipos de PDE, mediante lo cual los compuestos pueden ser excelentes agentes terapéuticos que tienen mayor seguridad. Por ejemplo, puede evitarse el riesgo cardiovascular derivado de la acción inhibidora de la PDE3 o el riesgo de ceguera derivado de la acción inhibidora de la PDE6 (A. M. Laties Drug Safety 2009; 32, 1-18/J. B. Shipley et al., Am. J. Med. Sci., 1996; 311, 286-291/T. M. Vinogradova et al., Circ. Res., 2008; 102, 761-769).
Una composición farmacéutica que contiene una o dos o más clases del compuesto de fórmula (I) o una sal del mismo como principio activo puede prepararse usando excipientes que se usan habitualmente en la técnica, es decir, excipientes para preparación farmacéutica, portadores para preparación farmacéutica, y similares según los métodos habitualmente usados.
La administración puede efectuarse mediante o bien administración oral a través de comprimidos, píldoras, cápsulas, gránulos, polvos, disoluciones, y similares, o bien inyecciones de administración parenteral, tales como inyecciones intraarticulares, intravenosas o intramusculares, y similares, supositorios, disoluciones oftálmicas, pomadas oftálmicas, preparaciones líquidas transdérmicas, pomadas, parches transdérmicos, preparaciones líquidas transmucosas, parches transmucosos, inhaladores, y similares.
La composición sólida para su uso en la administración oral según la presente invención se usa en forma de comprimidos, polvos, gránulos, o similares. En una composición sólida de este tipo, uno o más principio(s) activo(s) se mezclan con al menos un excipiente inactivo. En un método convencional, la composición puede contener
aditivos inactivos, tales como un lubricante, un agente disgregante, un estabilizador o un agente de ayuda de la solubilización. Si es necesario, pueden recubrirse los comprimidos o las píldoras con azúcar o una película de una sustancia de recubrimiento gástrica o entérica.
La composición líquida para la administración oral contiene emulsiones, disoluciones, suspensiones, jarabes, elixires farmacéuticamente aceptables, o similares, y también contiene diluyentes inertes generalmente usados, por ejemplo, agua o etanol purificado. Además del diluyente inerte, la composición líquida también puede contener agentes auxiliares, tales como un agente de ayuda de la solubilización, un agente de humectación y un agente de suspensión, edulcorantes, aromas, compuestos aromáticos y antisépticos.
Las inyecciones para la administración parenteral incluyen preparaciones de disoluciones, suspensiones y emulsiones acuosas o no acuosas estériles. El disolvente acuoso incluye, por ejemplo, agua destilada para inyección y solución salina fisiológica. Los ejemplos del disolvente no acuoso incluyen alcoholes tales como etanol. Una composición de este tipo puede contener además un agente de tonicidad, un antiséptico, un agente de humectación, un agente emulsionante, un agente dispersante, un agente estabilizante o un agente solubilizante. Estos se esterilizan, por ejemplo, mediante filtración a través de un filtro de retención de bacterias, la mezcla de un bactericida o irradiación. Además, éstos también pueden usarse preparando una composición sólida estéril, y disolviéndolo o suspendiéndolo en agua estéril o un disolvente estéril para inyección antes de su uso.
El agente para uso externo incluye pomadas, emplastos, cremas, jaleas, parches, pulverizadores, lociones, gotas oftálmicas, pomadas oftálmicas, y similares. Los agentes contienen bases de pomadas, bases de lociones, preparaciones, suspensiones, emulsiones líquidas acuosas o no acuosas generalmente usadas, y similares.
Como los agentes transmucosos tales como un inhalador, un agente transnasal, y similares, se usan aquellos en forma de un estado sólido, líquido o semisólido, y pueden prepararse según un método conocido convencionalmente. Por ejemplo, puede añadirse de manera apropiada a los mismos un excipiente conocido, y también un agente de ajuste del pH, un antiséptico, un tensioactivo, un lubricante, un agente estabilizante, un agente espesante, o similar. Para su administración, puede usarse un dispositivo apropiado para inhalación o soplado. Por ejemplo, puede administrarse un compuesto solo o como polvo de una mezcla formulada, o como una disolución o suspensión en combinación con un portador farmacéuticamente aceptable, usando un dispositivo o atomizador conocido convencionalmente, tal como un dispositivo de inhalación de administración medida, y similares. Un inhalador de polvo seco o similar puede ser para uso de administración única o múltiple, y puede usarse un polvo seco o una cápsula que contiene polvo. Alternativamente, puede estar en forma tal como un pulverizador de aerosol presurizado que usa un agente de expulsión apropiado, por ejemplo, un gas adecuado tal como clorofluoroalcano, dióxido de carbono, y similares, u otras formas.
En administración oral, la dosis diaria es generalmente desde aproximadamente 0,001 hasta 100 mg/kg, preferiblemente desde 0,1 hasta 30 mg/kg, y más preferiblemente desde 0,1 hasta 10 mg/kg, por peso corporal, administrada en una porción o en de 2 a 4 porciones divididas. En el caso de administración intravenosa, la dosis diaria se administra de manera adecuada desde aproximadamente 0,0001 hasta 10 mg/kg por peso corporal, una vez al día o dos o más veces al día. Además, se administra un agente transmucoso a una dosis de desde aproximadamente 0,001 hasta 100 mg/kg por peso corporal, una vez al día o dos o más veces al día. La dosis se decide de manera apropiada en respuesta al caso individual tomando en consideración los síntomas, la edad y el género, y similares.
Aunque varía dependiendo de las vías de administración, las formas de dosificación, los sitios de administración o los tipos de excipientes y aditivos, la composición farmacéutica de la presente invención contiene del 0,01 al 100% en peso, y en una determinada realización, del 0,01 al 50% en peso de una o más clases del compuesto de fórmula (I) o una sal del mismo, que es un principio activo.
El compuesto de fórmula (I) puede usarse en combinación con diversos agentes para tratar o prevenir enfermedades para las que se considera que es eficaz el compuesto de fórmula (I). La preparación combinada puede administrarse simultáneamente, o de manera separada y continua, o en un intervalo de tiempo deseado. Las preparaciones que van a administrarse conjuntamente pueden ser una combinación, o pueden prepararse de manera individual.
Ejemplos
A continuación en el presente documento, se describirán los métodos de preparación para el compuesto de fórmula (I) en más detalle con referencia a los ejemplos. Además, la presente invención no se limita a los compuestos descritos en los ejemplos tal como se describen a continuación. Además, se describirán los procedimientos de producción para los compuestos de partida en los ejemplos de preparación. Además, el compuesto de fórmula (I) puede prepararse mediante cualquier combinación de los métodos de preparación o los métodos que son evidentes para un experto en la técnica.
Además, para las reacciones de formación de sales que son evidentes para un experto en la técnica, existen casos en los que la adición u omisión de o de los métodos de preparación de los ejemplos.
Las siguientes abreviaturas pueden usarse en algunos casos en los ejemplos, ejemplos de preparación y tablas a continuación. tere-: terciario, Pr.: n.° de ejemplo de preparación, Ej.: n.° de ejemplo, n.°: n.° de compuesto, Estructura: fórmula estructural, Sin.: método de preparación (el numeral muestra que el compuesto del ejemplo de preparó de la misma manera que un compuesto que tiene su número como en el n.° de ejemplo), Datos: datos fisicoquímicos, ESI+: valores m/z en espectroscopia de masas (ionización ESI, que representa (M+H)+ a menos que se especifique lo contrario), ESI-: valores m/z en espectroscopia de masas (ionización ESI, que representa (M-H)- a menos que se especifique lo contrario), EI+: valores m/z en espectroscopia de masas (ionización EI, que representa (M)+ a menos que se especifique lo contrario), FAB+: valores m/z en espectroscopia de masas (ionización FAB, que representa (M+H)+ a menos que se especifique lo contrario), FAB-: valores m/z en espectroscopia de masas (ionización FAB, que representa (M-H)" a menos que se especifique lo contrario), APCI+: valores m/z en espectroscopia de masas (ionización APCI, que representa (M+H)+ a menos que se especifique lo contrario), APCI/ESI+: valores m/z en espectroscopia de masas (ionización APCI y ESI realizadas simultáneamente, que representa (M+H)+ a menos que se especifique lo contrario), APCI/ESI-: valores m/z en espectroscopia de masas (ionización APCI y ESI realizadas simultáneamente, que representa (M-H)- a menos que se especifique lo contrario), pf.: punto de fusión (°C), desc.: descomposición, RMN: 8 (ppm) de pico en 1H-RMN, s: singlete (espectro), d: doblete (espectro), t: triplete (espectro), q: cuadruplete (espectro) y a: linea ancha (espectro) (ejemplo: s a). Además, HCl en la fórmula estructural representa clorhidrato (el prefijo numeral a HCl denota una razón molar), TR representa un tiempo de retención, y el eluyente representa un disolvente para HPLC preparativa. Un caso en el que existe una descripción de “quiral” en la fórmula estructural indica que el compuesto del ejemplo es una forma ópticamente activa, pero existen algunos casos en los que no se determina la estereoquimica. Un caso en el que no existe descripción de “quiral” en la fórmula estructural indica que el compuesto del ejemplo es una mezcla de isómeros geométricos o un racemato. Por consiguiente, un caso en el que existe una descripción de estereoquimica pero no existe descripción de “quiral” indica una mezcla racémica de diastereómeros que tienen configuraciones relativas, y un caso en el que ni existe una descripción de estereoquimica ni una descripción de “quiral” indica una mezcla de isómeros geométricos, o una mezcla de isómeros ópticos. Además, [M] de la concentración representa [mol/l].
Ejemplo de preparación 1
A 649 mg de 4-cloro-8-metil-1-(tetrahidro-2H-piran-4-il)[1,2,4]triazolo[4,3-a]quinoxalin-7-carboxilato de metilo, se le añadieron 12 ml de ácido clorhidrico 6 M, seguido por calentamiento a reflujo durante 20 horas. Después de enfriarse con hielo, se filtró el sólido precipitado, y se lavó con agua y dietil éter. Se secó el sólido obtenido a presión reducida para obtener 307 mg de ácido 8-metil-4-oxo-1-(tetrahidro-2H-piran-4-il)-4,5-dihidro[1,2,4]triazolo[4,3-a]quinoxalin-7-carboxilico como un sólido.
Ejemplo de preparación 2
A un liquido mixto de 4,54 g de 8-(metoximetil)-4-oxo-1-(tetrahidrofuran-3-il)-4,5-dihidroimidazo[1,5-a]quinoxalin-7-carboxilato de metilo y 45,0 ml de metanol, se le añadieron 17,0 ml de una disolución acuosa de hidróxido de sodio 3 M, seguido por agitación a 70°C durante 5 horas. Se le añadieron ácido clorhidrico concentrado y ácido clorhidrico 1 M para ajustar el pH hasta aproximadamente 3, y se recogió el sólido precipitado mediante filtración y se secó a presión reducida para obtener 4,31 g de ácido 8-(metoximetil)-4-oxo-1-(tetrahidrofuran-3-il)-4,5-dihidroimidazo[1,5-a]quinoxalin-7-carboxilico como un sólido blanco.
Ejemplo de preparación 3
A una mezcla de 10 g de 3-cloro-2-hidrazino-7-metilquinoxalin-6-carboxilato de metilo, 5,9 g de ácido tetrahidro-2H-piran-4-carboxilico, 16 ml de N,N-diisopropiletilamina y 100 ml de diclorometano, se le añadieron 21 g de hexafluorofosfato de bromo(tripirrolidin-1-il)fosfonio, seguido por agitación a temperatura ambiente durante 9 horas. A la mezcla de reacción se le añadieron salmuera saturada y cloroformo. Se recogió el sólido mediante filtración y se lavó con acetato de etilo para obtener 9,1 g de 3-cloro-7-metil-2-[2-(tetrahidro-2H-piran-4-ilcarbonil)hidrazino]quinoxalin-6-carboxilato de metilo como un sólido rosa. Además, al filtrado obtenido anteriormente se le sometió a una operación de separación de liquidos mediante la adición de cloroformo, y luego se lavó la fase orgánica obtenida con salmuera saturada y se secó sobre sulfato de magnesio anhidro. Se evaporó el disolvente a presión reducida y se lavó el residuo obtenido con acetato de etilo para obtener 3,58 g de 3-cloro-7-metil-2-[2-(tetrahidro-2H-piran-4-ilcarbonil)hidrazino]quinoxalin-6-carboxilato de metilo como un sólido incoloro. Ejemplo de preparación 4
A una mezcla de 12,7 g de 3-cloro-7-metil-2-[2-(tetrahidro-2H-piran-4-ilcarbonil)hidrazino]quinoxalin-6-carboxilato de metilo y 250 ml de acetonitrilo, se le añadieron 5 ml de cloruro de tionilo, seguido por agitación a 70°C durante la noche. Después de enfriarse hasta temperatura ambiente, se evaporó el disolvente a presión reducida y se lavó el sólido obtenido con dietil éter para obtener 10,4 g de 4-cloro-8-metil-1-(tetrahidro-2H-piran-4-il)[1,2,4]triazolo[4,3-a]quinoxalin-7-carboxilato de metilo como un sólido de color amarillo pálido.
Ejemplo de preparación 5
Se agitó de manera vigorosa una mezcla de 5,3 g de 2-metil-4,5-dinitrobenzoato de metilo, 1,06 g de paladio sobre carbono al 10% (el 50% húmedo), 53 ml de etanol y 53 ml de 1,4-dioxano a temperatura ambiente bajo una atmósfera de hidrógeno. Se filtró el material insoluble y se lavó con metanol. Se combinaron el filtrado y el líquido de lavado, se concentraron y luego se secaron a presión reducida para obtener 3,98 g de 4,5-diamino-2-metilbenzoato de metilo.
Ejemplo de preparación 6
Se agitó una mezcla de 3,98 g de 4,5-diamino-2-metilbenzoato de metilo y 40 ml de oxalato de dietilo a 145°C durante 3 horas. Después de enfriarse hasta temperatura ambiente, se recogió el sólido mediante filtración, se lavó con dietil éter, y se secó a presión reducida para obtener 4,75 g de 7-metil-2,3-dioxo-1,2,3,4-tetrahidroquinoxalin-6-carboxilato de metilo.
Ejemplo de preparación 7
Se calentó a reflujo una mezcla de 4,75 g de 7-metil-2,3-dioxo-1,2,3,4-tetrahidroquinoxalin-6-carboxilato de metilo y 38 ml de cloruro de fosforilo durante 35 horas. Se enfrió la mezcla de reacción hasta temperatura ambiente, luego se concentró, y se sometió a destilación azeotrópica con tolueno. Al residuo obtenido se le añadió dietil éter, seguido por agitación a temperatura ambiente durante 0,5 horas. Se recogió el precipitado mediante filtración y se lavó con dietil éter para obtener 3,23 g de polvo. Se concentró el filtrado y se trató el residuo con dietil éter de la misma manera para obtener 2,22 g de un sólido. Se combinaron 3,23 g del polvo y 2,22 g del sólido y se lavaron con dietil éter para obtener 3,57 g de 2,3-dicloro-7-metilquinoxalin-6-carboxilato de metilo.
Ejemplo de preparación 8
A una suspensión de 3,57 g de 2,3-dicloro-7-metilquinoxalin-6-carboxilato de metilo en 214 ml de metanol, se le añadieron 7,0 ml de piridina y 2,34 ml de monohidrato de hidrazina a 0°C, y luego se agitó la mezcla a temperatura ambiente durante la noche. Se concentró la mezcla hasta que la cantidad total se convirtió en aproximadamente 100 ml, y se le añadió agua, seguido por agitación a temperatura ambiente durante 1 hora. Se recogió el precipitado mediante filtración, se lavó con agua y etanol, y se secó a presión reducida para obtener 2,94 g de 3-cloro-2-hidrazino-7-metilquinoxalin-6-carboxilato de metilo.
Ejemplo de preparación 9
A una mezcla de 1,75 g de 4-fluoro-2-(metoximetil)benzoato de metilo y 8,7 ml de ácido sulfúrico concentrado, se le añadieron 964 mg de nitrato de potasio a lo largo de 5 minutos bajo enfriamiento a -10°C. Después de agitar a la misma temperatura durante 4 horas y media, se añadió la mezcla de reacción a 300 ml de agua helada, seguido por agitación. Se recogió el sólido mediante filtración, se lavó con agua, y se secó a presión reducida para obtener 1,87 g de 4-fluoro-2-(metoximetil)-5-nitrobenzoato de metilo.
Ejemplo de preparación 10
A una mezcla de 6,0 g de 4-fluoro-2-(metoximetil)-5-nitrobenzoato de metilo, 3,5 g de 2-(tetrahidrofuran-3-il)-1H-imidazol y 60 ml de acetonitrilo, se le añadieron 6,9 ml de trietilamina, seguido por agitación a 70°C durante 5,5 horas. Se concentró la mezcla de reacción, y se le añadieron agua y acetato de etilo, seguido por extracción con acetato de etilo. Se lavó la fase orgánica con salmuera saturada y luego se secó sobre sulfato de magnesio anhidro, y se evaporó el disolvente a presión reducida. Se purificó el residuo obtenido mediante cromatografía en columna de gel de sílice (cloroformo/metanol) para obtener 6,65 g de 2-(metoximetil)-5-nitro-4-[2-(tetrahidrofuran-3-il)-1H-imidazol-1-il]benzoato de metilo.
Ejemplo de preparación 11
Se agitó una mezcla de 6,00 g de 5-amino-2-(metoximetil)-4-[2-(tetrahidrofuran-3-il)-1H-imidazol-1-il]benzoato de metilo, 5,87 g de 1,1'-carbonildiimidazol y 30 ml de 1,2-diclorobenceno a 120°C durante 3 horas. Después de enfriarse con aire, se recogió el precipitado mediante filtración. Se lavó el sólido obtenido con metanol y se secó con aire para obtener 4,54 g de 8-(metoximetil)-4-oxo-1-(tetrahidrofuran-3-il)-4,5-dihidroimidazo[1,5-a]quinoxalin-7-carboxilato de metilo.
Ejemplo de preparación 12
A una disolución de 2,5 ml de tetrahidrofuran-3-carboxialdehído (disolución acuosa al 50%) en 40 ml de etanol, se le añadieron 2,6 ml de una disolución acuosa de glioxal al 40% y 14 ml de amoniaco acuoso al 28% en este orden bajo enfriamiento con hielo, seguido por agitación durante 3,5 horas mientras se calentaba gradualmente hasta temperatura ambiente. Se evaporó el etanol a presión reducida, y se añadió cloruro de sodio a una disolución acuosa del residuo hasta saturarlo, seguido por extracción con cloroformo. Se secó la fase orgánica sobre sulfato de
magnesio anhidro y se evaporó el disolvente a presión reducida. Se calentó el residuo obtenido y se disolvió en acetato de etilo, y se dejó enfriar. Se recogió el sólido precipitado mediante filtración para obtener 1,58 g de 2-(tetrahidrofuran-3-il)-1H-imidazol como un sólido blanco.
Ejemplo de preparación 13
A un líquido mixto de 6,65 g de 2-(metoximetil)-5-nitro-4-[2-(tetrahidrofuran-3-il)-1H-imidazol-1-il]benzoato de metilo, 980 mg de cloruro de amonio, 33,0 ml de tetrahidrofurano, 66,0 ml de metanol y 33,0 ml de agua, se le añadieron 4,8 g de hierro reducido, seguido por agitación a 70°C durante 4,5 horas. Se filtró el material insoluble a través de celita. Se concentró el filtrado, y se le añadieron agua y acetato de etilo, seguido por extracción con acetato de etilo. Se lavó la fase orgánica con salmuera saturada y luego se secó sobre sulfato de magnesio anhidro, y se evaporó el disolvente a presión reducida. Se purificó el residuo obtenido mediante cromatografía en columna de gel de sílice (cloroformo/metanol) para obtener 5,30 g de 5-amino-2-(metoximetil)-4-[2-(tetrahidrofuran-3-il)-1H-imidazol-1-il]benzoato de metilo como un sólido marrón.
Ejemplo de preparación 14
A una mezcla de 3 g de 4-fluoro-2-metoxi-5-nitrobenzoato de metilo, 1,99 g de 2-(tetrahidro-2H-piran-4-il)-1H-imidazol y 15 ml de N-etilpirrolidona, se le añadieron 3,6 g de carbonato de potasio, seguido por agitación a 100°C durante 3 horas. A la mezcla de reacción se le añadieron agua y acetato de etilo, seguido por extracción con acetato de etilo. Se lavó la fase orgánica con salmuera saturada y luego se secó sobre sulfato de magnesio anhidro, y se evaporó el disolvente a presión reducida. Se purificó el residuo obtenido mediante cromatografía en columna de gel de sílice (cloroformo/metanol) para obtener 4,12 g de 4-[2-(tetrahidro-2H-piran-4-il)-1H-imidazol-il]-2-metoxi-5-nitrobenzoato de metilo.
Ejemplo de preparación 15
A una disolución de 3,45 g de 1-bromo-4-fluoro-2-(metoximetil)benceno en 20 ml de dimetilsulfóxido, se le añadieron de manera secuencial 353 mg de acetato de paladio, 650 mg de 1,3-bis(difenilfosfino)propano, 4,4 ml de trietilamina y 10 ml de metanol, y se reemplazó la atmósfera en el recipiente de reacción con monóxido de carbono, seguido por agitación a 70°C durante 6 horas. Se vertió la mezcla de reacción en un líquido mixto de agua y acetato de etilo. Se separó la fase acuosa, y se lavó la fase orgánica con salmuera saturada y luego se secó sobre sulfato de magnesio anhidro. Se evaporó el disolvente a presión reducida. Se purificó el residuo obtenido mediante cromatografía en columna de gel de sílice (n-hexano/acetato de etilo) para obtener 1,75 g de 4-fluoro-2-(metoximetil)benzoato de metilo.
Ejemplo de preparación 16
A una disolución de 3 g de 4-fluoro-2-hidroxibenzoato de metilo en 60 ml de tetrahidrofurano, se le añadieron 1,98 g de tetrahidrofuran-3-ilmetanol y 4,28 g de tributilfosfina, seguido por enfriamiento con hielo. A una mezcla se le añadieron de manera gradual 6,67 g de 1,1'-(azodicarbonil)dipiperazina, seguido por agitación durante 20 minutos. Se calentó la mezcla hasta temperatura ambiente, se agitó durante 16 horas, y luego se vertió en un líquido mixto de agua y acetato de etilo. Se separó la fase acuosa, se lavó la fase orgánica con salmuera saturada y luego se secó sobre sulfato de magnesio anhidro, y se evaporó el disolvente a presión reducida. Se purificó el residuo obtenido mediante cromatografía en columna de gel de sílice (n-hexano/acetato de etilo) para obtener 3,14 g de 4-fluoro-2-(tetrahidrofuran-3-ilmetoxi)benzoato de metilo.
Ejemplo de preparación 17
Bajo una atmósfera de nitrógeno, a una mezcla de 8,6 g de cis-2-{[terc-butil(dimetil)silil]oxi}ciclopentanocarboxilato de etilo y 50 ml de dietil éter, se le añadieron gota a gota 32 ml de hidruro de diisobutilaluminio 1,04 M en n-hexano a -78°C, seguido por agitación a la misma temperatura durante 2 horas. Al líquido de reacción se le añadió metanol hasta descomponer el hidruro de diisobutilaluminio en exceso, seguido por calentamiento hasta temperatura ambiente. Al líquido de reacción se le añadió una disolución acuosa de (+)-tartrato de potasio y sodio, seguido por extracción con dietil éter, se lavó la fase orgánica con salmuera saturada y se secó sobre sulfato de magnesio anhidro, y luego se evaporó el disolvente. Se purificó el residuo obtenido mediante cromatografía en columna de gel de sílice (n-hexano/acetato de etilo) para obtener 7,20 g de cis-2-{[tercbutil(dimetil)silil]oxi}ciclopentanocarboxialdehído como una sustancia oleosa incolora.
Ejemplo de preparación 18
A 500 mg de 5-metoxi-2,3-dihidro-1H-pirrolo[2,3-c]piridin-1-carboxilato de terc-butilo, se le añadieron 1,5 ml de ácido trifluoroacético, seguido por agitación a temperatura ambiente durante 0,5 horas. Se vertió el líquido de reacción en agua helada, se neutralizó con una disolución acuosa de hidróxido de sodio 1 M, y luego se sometió a separación de líquidos mediante la adición de cloroformo. Se lavó la fase orgánica con agua y salmuera saturada, y se secó sobre sulfato de magnesio anhidro, y luego se evaporó el disolvente a presión reducida para obtener 262 mg de 5-metoxi2,3-dihidro-1H-pirrolo[2,3-c]piridina como un sólido de color amarillo pálido.
Ejemplo de preparación 19
A una disolución de 200 mg de 4-(metoximetil)-1,3-dihidro-2H-isoindol-2-carboxilato de ferc-butilo en 2 ml de acetato de etilo, se le añadieron 2,3 ml de una disolución de cloruro de hidrógeno-acetato de etilo 4 M bajo enfriamiento con hielo, seguido por agitación a temperatura ambiente durante la noche. Bajo enfriamiento con hielo, al líquido de reacción se le añadió una disolución acuosa de hidróxido de sodio 1 M para ajustar el pH hasta aproximadamente 7. Después de llevar a cabo la operación de separación de líquidos, se lavó la fase orgánica con salmuera saturada y se secó sobre sulfato de magnesio anhidro. Se evaporó el disolvente a presión reducida para obtener 100 mg de 4-(metoximetil)isoindolina como una sustancia oleosa marrón.
Ejemplo de preparación 20
Bajo una atmósfera de nitrógeno, a una disolución de 700 mg de 5-cloro-2-hidroximetil)-2,3-dihidro-1H-indol-1-carboxilato de ferc-butilo en 14 ml de tetrahidrofurano, se le añadieron en porciones 197 mg de hidruro de sodio al 60% bajo enfriamiento con hielo, seguido por calentamiento hasta temperatura ambiente y agitación durante 4 horas. Después de enfriarse con hielo de nuevo, se le añadieron gota a gota 462 |il de yoduro de metilo, seguido por calentamiento hasta temperatura ambiente y agitación durante 2 horas. Se le añadió agua, seguido por extracción con cloroformo, y luego se lavó la fase orgánica obtenida con una disolución acuosa saturada de hidrogenocarbonato de sodio y salmuera saturada. Después de secar sobre sulfato de magnesio anhidro, se eliminó el desecante, se evaporó el disolvente a presión reducida, y se purificó el residuo obtenido mediante cromatografía en columna de gel de sílice (cloroformo/metanol) para obtener 230 mg de 5-cloro-2-(metoximetil)-2,3-dihidro-1H-indol-1-carboxilato de ferc-butilo como una sustancia oleosa marrón.
Ejemplo de preparación 21
A un líquido mixto de 1,17 g de ácido 1-(ferc-butoxicarbonil)-5-cloro-2,3-dihidro-1H-indol-2-carboxílico en 23 ml de tetrahidrofurano, se le añadieron gota a gota 15,7 ml de una disolución 1 M de un complejo de boranotetrahidrofurano en tetrahidrofurano bajo enfriamiento con hielo, seguido por calentamiento hasta temperatura ambiente y agitación durante la noche. Después de enfriarse con hielo de nuevo, se le añadieron gota a gota 15 ml de ácido clorhídrico 1 M, seguido por extracción con cloroformo. Se lavó la fase orgánica obtenida con agua, una disolución acuosa saturada de hidrogenocarbonato de sodio y salmuera saturada, y se secó sobre sulfato de magnesio anhidro. Se eliminó el desecante y se evaporó el disolvente a presión reducida para obtener 1,18 g de 5-cloro-2-(hidroximetil)-2,3-dihidro-1H-indol-1-carboxilato de ferc-butilo como un sólido de color amarillo pálido.
Ejemplo de preparación 22
A un líquido mixto de 1 g de monoclorhidrato de ácido 5-cloro-2,3-dihidro-1H-indol-2-carboxílico y 1,03 g de dicarbonato de di-ferc-butilo en 10 ml de 1,4-dioxano, se le añadieron 9,4 ml de una disolución acuosa de hidróxido de sodio 1 M, seguido por agitación a temperatura ambiente durante la noche. Se evaporó el disolvente a presión reducida y luego se le añadió ácido clorhídrico 1 M para ajustar el pH hasta aproximadamente 3. Luego, se le añadió cloroformo para llevar a cabo una operación de separación de líquidos. Se lavó la fase orgánica obtenida con salmuera saturada y luego se secó sobre sulfato de magnesio anhidro, y se evaporó el disolvente a presión reducida. Se purificó el residuo obtenido mediante cromatografía en columna de gel de sílice (cloroformo/metanol) para obtener 1,18 g de ácido 1-(ferc-butoxicarbonil)-5-cloro-2,3-dihidro-1H-indol-2-carboxílico como una sustancia oleosa marrón.
Ejemplo de preparación 23
Se agitó una mezcla de 238 mg de 4-formil-1,3-dihidro-2H-isoindol-2-carboxilato de ferc-butilo, 612 mg de triacetoxiborohidruro de sodio, 963 |il de una disolución de dimetilamina-tetrahidrofurano 2 M y 12 |il de ácido acético en 14 ml de dicloroetano a 60°C durante la noche. Después de enfriarse hasta temperatura ambiente, al líquido de reacción se le añadió una disolución acuosa saturada de hidrogenocarbonato de sodio, y se le añadió cloroformo para llevar a cabo una operación de separación de líquidos. Se lavó la fase orgánica obtenida con salmuera saturada y luego se secó sobre sulfato de magnesio anhidro, y se evaporó el disolvente a presión reducida. Se purificó el residuo obtenido mediante cromatografía en columna de gel de sílice (n-hexano/acetato de etilo) para obtener 123 mg de 4-[(dimetilamino)metil]-1,3-dihidro-2H-isoindol-2-carboxilato de ferc-butilo como una sustancia oleosa naranja.
Ejemplo de preparación 24
A una disolución de 350 mg de 4-formil-1,3-dihidro-2H-isoindol-2-carboxilato de ferc-butilo en 11 ml de metanol, se le añadieron 134 mg de borohidruro de sodio bajo enfriamiento con hielo, seguido por agitación a temperatura ambiente durante la noche. Al líquido de reacción se le añadieron agua y acetato de etilo para llevar a cabo una operación de separación de líquidos. Se lavó la fase orgánica obtenida con salmuera saturada y luego se secó sobre
sulfato de magnesio anhidro. Se evaporó el disolvente a presión reducida para obtener 213 mg de 4-(hidroximetil)-1.3- dihidro-2H-isoindol-2-carboxilato de ferc-butilo como una sustancia oleosa amarilla.
Ejemplo de preparación 25
Bajo una atmósfera de argón, a una mezcla de 1100 mg de 5-bromo-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-carboxilato de ferc-butilo, 1062 mg de un complejo de 2,4,6-trivinilboroxina/piridina, 1020 mg de carbonato de potasio, 3 ml de 1.4- dioxano/agua (4/1), se le añadieron 215 mg de tetrakis(trifenilfosfina)paladio. Se agitó el líquido de reacción a 80°C durante la noche. Al líquido de reacción se le añadieron agua y acetato de etilo para llevar a cabo una operación de separación de líquidos. Se lavó la fase orgánica con salmuera saturada y se secó sobre sulfato de magnesio anhidro, y se evaporó el disolvente a presión reducida. Se purificó el residuo mediante cromatografía en columna de gel de sílice (n-hexano/acetato de etilo) para obtener 783 mg de 5-vinil-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1 -carboxilato de ferc-butilo como una sustancia oleosa amarilla.
Ejemplo de preparación 26
A una mezcla de 4,10 g de 2,4-difluoro-5-nitrobenzoato de metilo y 40 ml de acetonitrilo, se le añadieron 4,76 ml de trietilamina, y se le añadieron de manera gradual 2,60 g de 2-(tetrahidro-2H-piran-4-il)-1H-imidazol bajo enfriamiento con hielo, seguido por agitación a temperatura ambiente durante 4 horas. A la mezcla de reacción se le añadieron agua y acetato de etilo, seguido por extracción con acetato de etilo, se lavó la fase orgánica con salmuera saturada y luego se secó sobre sulfato de magnesio anhidro, y se evaporó el disolvente a presión reducida. Se purificó el residuo obtenido mediante cromatografía en columna de gel de sílice (cloroformo/metanol) para obtener 3,84 g de 2-fluoro-5-nitro-4-[2-(tetrahidro-2H-piran-4-il)-1H-imidazol-1-il]benzoato de metilo como un sólido amarillo.
Ejemplo de preparación 27
A un líquido mixto de 10,45 g de 4-fluoro-5-nitro-2-vinilbenzoato de metilo y 104,5 ml de piridina, se le añadieron 14,69 ml de ferc-butilamina, seguido por agitación a temperatura ambiente durante la noche. Se evaporó el disolvente a presión reducida, y al residuo se le añadieron agua y acetato de etilo, seguido por extracción con acetato de etilo. Se lavó la fase orgánica con salmuera saturada y se secó sobre sulfato de magnesio anhidro, y se evaporó el disolvente a presión reducida para obtener 14 g de 4-(ferc-butilamino)-5-nitro-2-vinilbenzoato de metilo como una sustancia oleosa marrón.
Ejemplo de preparación 28
A un líquido mixto de 3,74 g de 4-(ferc-butilamino)-5-nitro-2-vinilbenzoato de metilo y 37,4 ml de metanol, se le añadieron 21,7 ml de ácido clorhídrico 6 M, seguido por calentamiento a reflujo durante 1 hora. Se enfrió el líquido de reacción hasta temperatura ambiente y se recogió el sólido precipitado mediante filtración. Se secó el sólido obtenido a 40°C durante 2 horas a presión reducida para obtener 2,56 g de monoclorhidrato de 4-amino-5-nitro-2-vinilbenzoato de metilo como un sólido amarillo.
De la misma manera que en el método del ejemplo de preparación 2, se prepararon los compuestos de los ejemplos de preparación 2-1 a 2-15, de la misma manera que en el método del ejemplo de preparación 3, se prepararon los compuestos de los ejemplos de preparación 3-1 a 3-7, de la misma manera que en el método del ejemplo de preparación 4, se prepararon los compuestos de los ejemplos de preparación 4-1 a 4-7, de la misma manera que en el método del ejemplo de preparación 5, se prepararon los compuestos de los ejemplos de preparación 5-1 a 5-9, de la misma manera que en el método del ejemplo de preparación 6, se prepararon los compuestos de los ejemplos de preparación 6-1 a 6-2, de la misma manera que en el método del ejemplo de preparación 7, se prepararon los compuestos de los ejemplos de preparación 7-1 a 7-2, de la misma manera que en el método del ejemplo de preparación 8, se prepararon los compuestos de los ejemplos de preparación 8-1 a 8-2, de la misma manera que en el método del ejemplo de preparación 9, se prepararon los compuestos de los ejemplos de preparación 9-1 a 9-2, de la misma manera que en el método del ejemplo de preparación 10, se prepararon los compuestos de los ejemplos de preparación 10-1 a 10-6, de la misma manera que en el método del ejemplo de preparación 11, se prepararon los compuestos de los ejemplos de preparación 11-1 a 11-8, de la misma manera que en el método del ejemplo de preparación 12, se prepararon los compuestos de los ejemplos de preparación 12-1 a 12-2, de la misma manera que en el método del ejemplo de preparación 13, se prepararon los compuestos de los ejemplos de preparación 13-1 a 13-2, de la misma manera que en el método del ejemplo de preparación 18, se preparó el compuesto del ejemplo de preparación 18-1, de la misma manera que en el método del ejemplo de preparación 19, se prepararon los compuestos de los ejemplos de preparación 19-1 a 19-3, de la misma manera que en el método del ejemplo de preparación 20, se preparó el compuesto del ejemplo de preparación 20-1, de la misma manera que en el método del ejemplo de preparación 21, se preparó el compuesto del ejemplo de preparación 21-1, de la misma manera que en el método del ejemplo de preparación 25, se prepararon los compuestos de los ejemplos de preparación 25-1 a 25-2, de la misma manera que en el método del ejemplo de preparación 27, se preparó el compuesto del ejemplo de preparación 27-1, de la misma manera que en el método del ejemplo de preparación 28, se preparó el compuesto del ejemplo de preparación 28-1, de la misma manera que en el método del ejemplo 6 tal como se describe más adelante, se preparó el compuesto del ejemplo de preparación 29, de la misma manera que en el método del
ejemplo 3 tal como se describe más adelante, se preparó el compuesto del ejemplo de preparación 30, cada uno de los cuales se preparó usando el correspondiente material de partida. Las estructuras y los datos fisicoquímicos de los compuestos de los ejemplos de preparación se muestran en las tablas 1 a 19 a continuación.
Ejemplo 1 (referencia)
A una mezcla de 150 mg de ácido 4-oxo-8-(tetrahidrofuran-3-ilmetoxi)-1-(tetrahidro-2H-piran-4-il)-4,5-dihidroimidazo[1,5-a]quinoxalin-7-carboxílico y 3 ml de N,N-dimetilformamida, se le añadieron de manera secuencial 122 mg de 5-bromo-2,3-dihidro-1H-pirrolo[2,3-b]piridina, 0,25 ml de N,N-diisopropiletilamina y 220 mg de HATU, seguido por agitación a temperatura ambiente durante 72 horas. Se vertió la mezcla en una disolución acuosa saturada enfriada de hidrogenocarbonato de sodio, seguido por agitación durante 30 minutos y extracción con acetato de etilo. Se lavó la fase orgánica con salmuera saturada y se secó sobre sulfato de magnesio anhidro. Se evaporó el disolvente a presión reducida, y se purificó el residuo mediante cromatografía en columna de gel de sílice (cloroformo/metanol=100/0-92/8) para obtener 81 mg de 7-[(5-bromo-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-8-(tetrahidrofuran-3-ilmetoxi)-1-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]quinoxalin-4(5H)-ona como un sólido blanco.
Ejemplo 2 (referencia)
A una mezcla de 106 mg de ácido 8-fluoro-4-oxo-1-(tetrahidro-2H-piran-4-il)-4,5-dihidroimidazo[1,5-a]quinoxalin-7-carboxílico, 53 mg de 5-metoxi-2,3-dihidro-1H-pirrolo[2,3-c]piridina, 0,22 ml de N,N-diisopropiletilamina y 2 ml de N,N-dimetilformamida, se le añadieron 146 mg de hexafluorofosfato de O-(benzotriazol-1-il)-N,N,N',N'-tetrametiluronio, seguido por agitación a temperatura ambiente durante la noche. Al líquido de reacción se le añadió una disolución acuosa saturada de hidrogenocarbonato de sodio, seguido por extracción con cloroformo. Se lavó la fase orgánica con salmuera saturada y se secó sobre sulfato de magnesio anhidro, y luego se evaporó el disolvente a presión reducida. Se purificó el residuo obtenido mediante HPLC preparativa (metanol/disolución acuosa de ácido fórmico al 0,1%) para obtener 5,9 mg de 8-fluoro-7-[(5-metoxi-2,3-dihidro-1H-pirrolo[2,3-c]piridin-1-il)carbonil]-1-(tetrahidro-2H-piran-4-il)imidazo[1,5-a]quinoxalin-4(5H)-ona como un sólido de color marrón pálido.
Ejemplo 3 (referencia)
A una mezcla de 200 mg de ácido 8-metil-4-oxo-1-(tetrahidro-2H-piran-3-il)-4,5-dihidroimidazo[1,5-a]quinoxalin-7-carboxílico, 100 mg de 5-metoxi-2,3-dihidro-1H-pirrolo[2,3-c]piridina, 0,42 ml de N,N-diisopropiletilamina y 5 ml de N,N-dimetilformamida, se le añadieron 235 mg de TBTu , seguido por agitación a temperatura ambiente durante la noche. Al líquido de reacción se le añadieron agua y una disolución acuosa saturada de hidrogenocarbonato de sodio, seguido por extracción con cloroformo. Se lavó la fase orgánica con salmuera saturada y se secó sobre sulfato de magnesio anhidro, y luego se evaporó el disolvente a presión reducida. Se purificó el residuo obtenido mediante cromatografía en columna de gel de sílice (cloroformo/metanol = 100/0-95/5). Se lavó el sólido obtenido con metanol para obtener 148 mg de 7-[(5-metoxi-2,3-dihidro-1H-pirrolo[2,3-c]piridin-1-il)carbonil]-8-metil-1-(tetrahidro-2H-piran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona.
Ejemplo 4
Se purificaron 373 mg de 7-[(5-metoxi-2,3-dihidro-1H-pirrolo[2,3-c]piridin-1-il)carbonil]-8-(metoximetil)-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona mediante HPLC preparativa (velocidad de flujo: 8 ml/min, acetonitrilo/agua=30/70) usando una columna OJ-RH de CHIRALCEL (marca registrada), 5 |im, 20*150 mm (Daicel Chemical Industries, Ltd ). Como un compuesto con un tiempo de retención corto (tiempo de retención: 9,32 min), se obtuvieron 125 mg de (-)-7-[(5-metoxi-2,3-dihidro-1H-pirrolo[2,3-c]piridin-1-il)carbonil]-8-(metoximetil)-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona, y como un compuesto con un tiempo de retención largo (tiempo de retención: 13,31 min), se obtuvieron 132 mg de (+)-7-[(5-metoxi-2,3-dihidro-1H-pirrolo[2,3-c]piridin-1-il)carbonil]-8-(metoximetil)-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona. A una mezcla de 125 mg de (-)-7-[(5-metoxi-2,3-dihidro-1H-pirrolo[2,3-c]piridin-1-il)carbonil]-8-(metoximetil)-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona, que era un compuesto con un tiempo de retención corto, 5 ml de metanol y 7,4 ml de tetrahidrofurano, se le añadieron 230 |il de una disolución de cloruro de hidrógeno-acetato de etilo 4 M, seguido por agitación a temperatura ambiente durante 30 minutos. Se evaporó el disolvente a presión reducida, al residuo obtenido se le añadieron 2 ml de dietil éter para convertirlo en polvo, y se agitó la suspensión durante 30 minutos. Se recogió el sólido mediante filtración y se secó a presión reducida para obtener 128 mg de diclorhidrato de (-)-7-[(5-metoxi-2,3-dihidro-1H-pirrolo[2,3-c]piridin-1-il)carbonil]-8-(metoximetil)-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona, que era un compuesto con un tiempo de retención corto, como un sólido blanco. A una mezcla de 132 mg de (+)-7-[(5-metoxi-2,3-dihidro-1H-pirrolo[2,3-c]piridin-1-il)carbonil]-8-(metoximetil)-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona, que era un compuesto con un tiempo de retención largo, 5 ml de metanol y 7,4 ml de tetrahidrofurano, se le añadieron 243 |il de una disolución de cloruro de hidrógenoacetato de etilo 4 M, seguido por agitación a temperatura ambiente durante 30 minutos. Se evaporó el disolvente a presión reducida, al residuo obtenido se le añadieron 2 ml de dietil éter para convertirlo en polvo, y se agitó la suspensión durante 30 minutos. Se recogió el sólido mediante filtración y se secó a presión reducida para obtener 125 mg de diclorhidrato de (+)-7-[(5-metoxi-2,3-dihidro-1H-pirrolo[2,3-c]piridin-1-il)carbonil]-8-(metoximetil)-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona, que era un compuesto con un tiempo de retención largo,
como un sólido blanco.
Además, en las tablas, los compuestos con tiempos de retención en columna cortos se denotan como (a) y los compuestos con tiempos de retención en columna largos se denotan como (b).
Además, el ejemplo 25(a), el ejemplo 27(a), el ejemplo 29(a), el ejemplo 30(a), el ejemplo 39(a), el ejemplo 40(a), el ejemplo 43(a) y el ejemplo 45(a) mostraron rotaciones específicas negativas, y el ejemplo 25(b), el ejemplo 27(b), el ejemplo 29(b), el ejemplo 30(b), el ejemplo 39(b), el ejemplo 40(b), el ejemplo 43(b) y el ejemplo 45(b) mostraron rotaciones específicas positivas.
Ejemplo 5 (referencia)
A una mezcla de 369 mg de 7-[(5-bromo-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-1-(cis-2-{[fe/'cbutil(dimetil)silil]oxi}ciclopentil)-8-metilimidazo[1,5-a]quinoxalin-4(5H)-ona y 20,0 ml de metanol, se le añadieron 0,15 ml de ácido clorhídrico concentrado, seguido por agitación a temperatura ambiente durante 3 días. Se evaporó el disolvente a presión reducida, luego se disolvió el residuo en agua, y se le añadió una disolución acuosa saturada de hidrogenocarbonato de sodio para ajustar el pH hasta aproximadamente 8. Se le añadió cloroformo para llevar a cabo la extracción, se secó la fase orgánica sobre sulfato de magnesio anhidro, y luego se evaporó el disolvente a presión reducida. Se purificó el residuo obtenido mediante cromatografía en columna de gel de sílice (cloroformo/metanol=100/0-95/5) y se trituró con metanol para obtener 221 mg de 7-[(5-bromo-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-1-(cis-2-hidroxiciclopentil)-8-metilimidazo[1,5-a]quinoxalin-4(5H)-ona como un sólido incoloro.
Ejemplo 6 (referencia)
Se añadieron 96 mg de ácido 1-(ciclopropilmetil)-8-metil-4-oxo-4,5-dihidro[1,2,4]triazolo[4,3-a]quinoxalin-7-carboxílico, 52 mg de 5-etil-2,3-dihidro-1H-pirrolo[2,3-b]piridina, 336 | l de N,N-diisopropiletilamina y 145 mg de TBTU a 1,6 ml de N-metilpirrolidona, y se hicieron reaccionar a 120°C durante 1 hora usando un reactor de microondas. Al líquido de reacción se le añadió una disolución acuosa saturada de hidrogenocarbonato de sodio, seguido por extracción con acetato de etilo. Se lavó la fase orgánica con salmuera saturada y se secó sobre sulfato de magnesio anhidro, se eliminó el desecante, se evaporó el disolvente a presión reducida, y se purificó el residuo obtenido mediante cromatografía en columna de gel de sílice (cloroformo/metanol=100/0-97/3) para obtener 120 mg de un sólido blanco. A una mezcla del sólido blanco obtenido en 2 ml de metanol se le añadieron 277 | l de una disolución de cloruro de hidrógeno-acetato de etilo 4 M, seguido por agitación a temperatura ambiente durante la noche. Se recogió el sólido precipitado mediante filtración y se secó a presión reducida para obtener 95 mg de diclorhidrato de 1-(ciclopropilmetil)-7-[(5-etil-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-8-metil[1,2,4]triazolo[4,3-a]quinoxalin-4(5H)-ona como un sólido blanco.
De la misma manera que en los métodos de los ejemplos 1 a 6, se prepararon los compuestos de los ejemplos 7 a 48 mostrados en las tablas a continuación. Para los compuestos de ejemplo, las estructuras y los métodos de preparación se muestran en las tablas 20 a 32, y los datos fisicoquímicos se muestran en las tablas 33 a 40. Además, las estructuras de otros compuestos de los compuestos de fórmula (I) se muestran en las tablas 41 a 45. Estos pueden prepararse fácilmente mediante cualquiera de los métodos de preparación anteriores, los métodos descritos en los ejemplos, los métodos evidentes para los expertos en la técnica o los métodos modificados de los mismos.
[Tabla 1]


[Tabla 2]
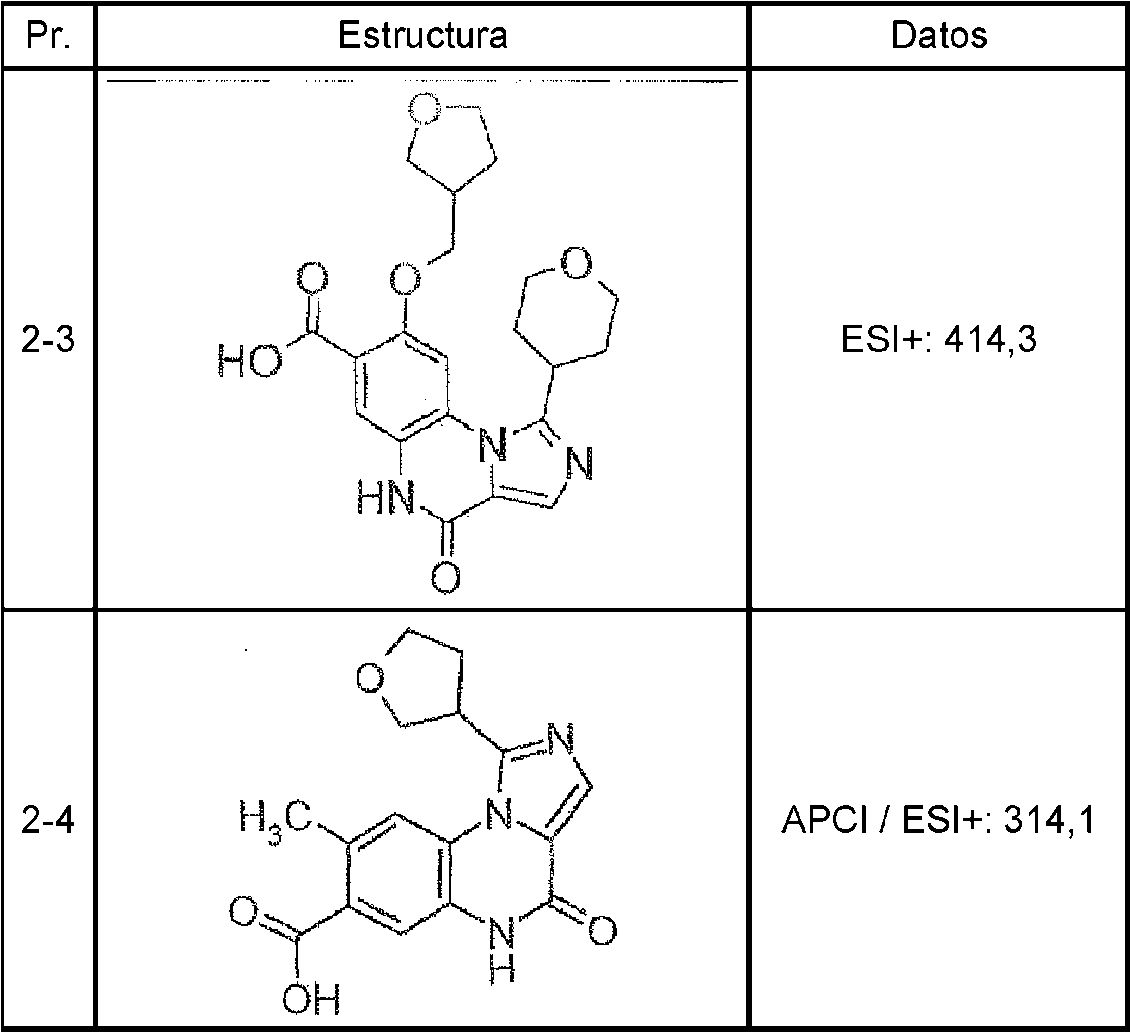

[Tabla 3]


[Tabla 4]


[Tabla 5]


[Tabla 6]


[Tabla 7]

[Tabla 8]
Pr.  Estructura
Estructura  Datos
Datos 

[Tabla 9]
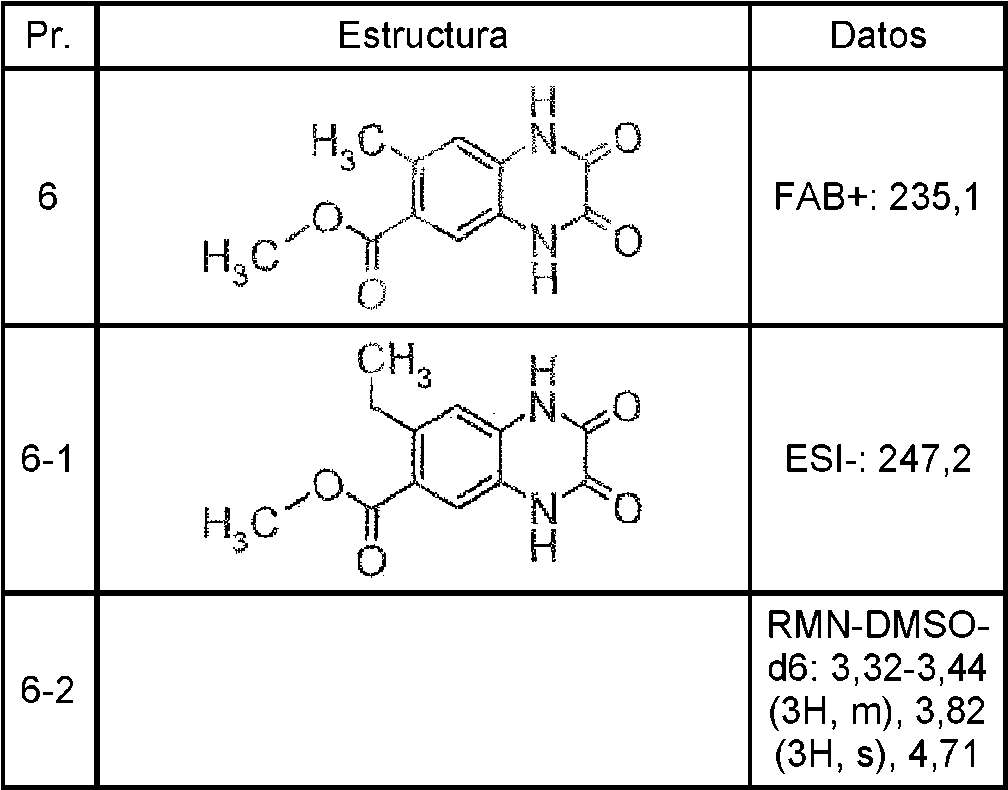

[Tabla 10]


[Tabla 11]


[Tabla 12]
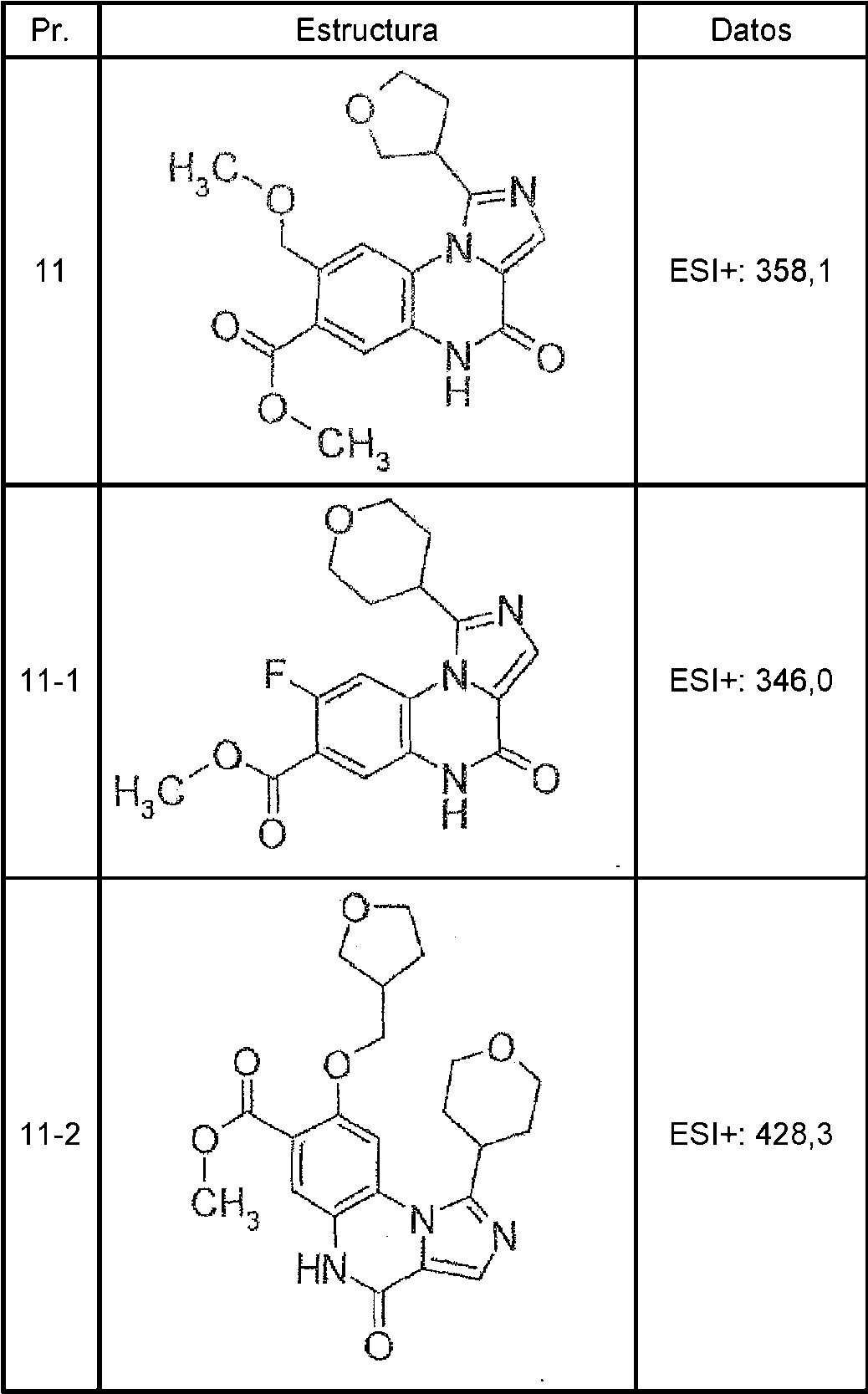

[Tabla 13]


[Tabla 14]

[Tabla 15]

[Tabla 16]


[Tabla 17]


[Tabla 18]


[Tabla 19]

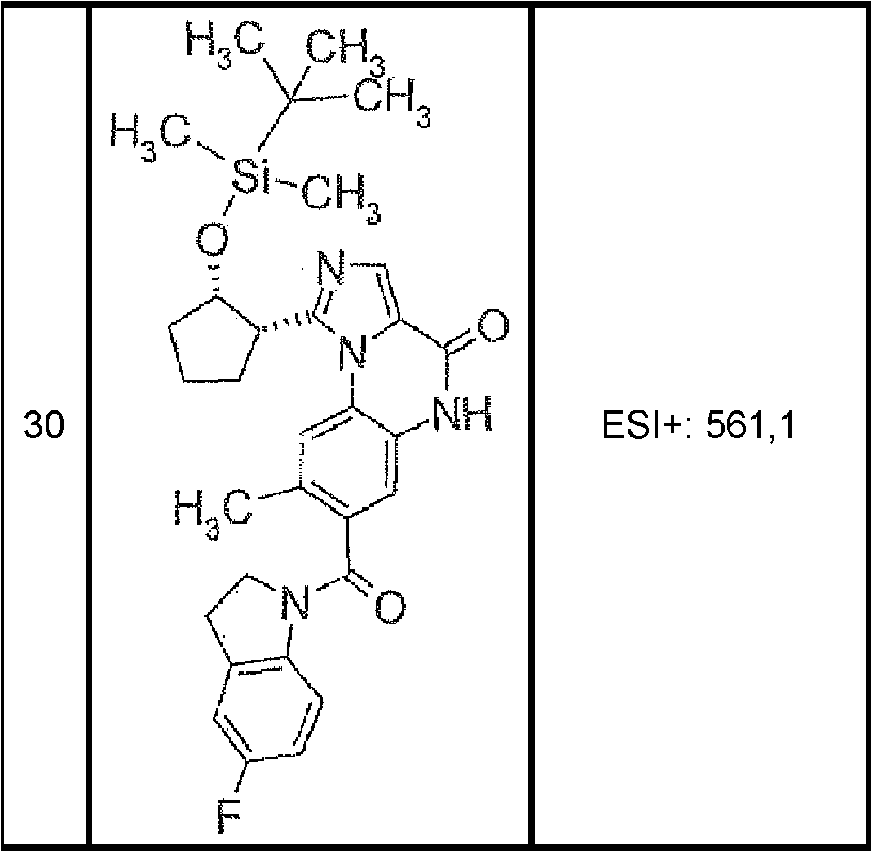
[Tabla 20]
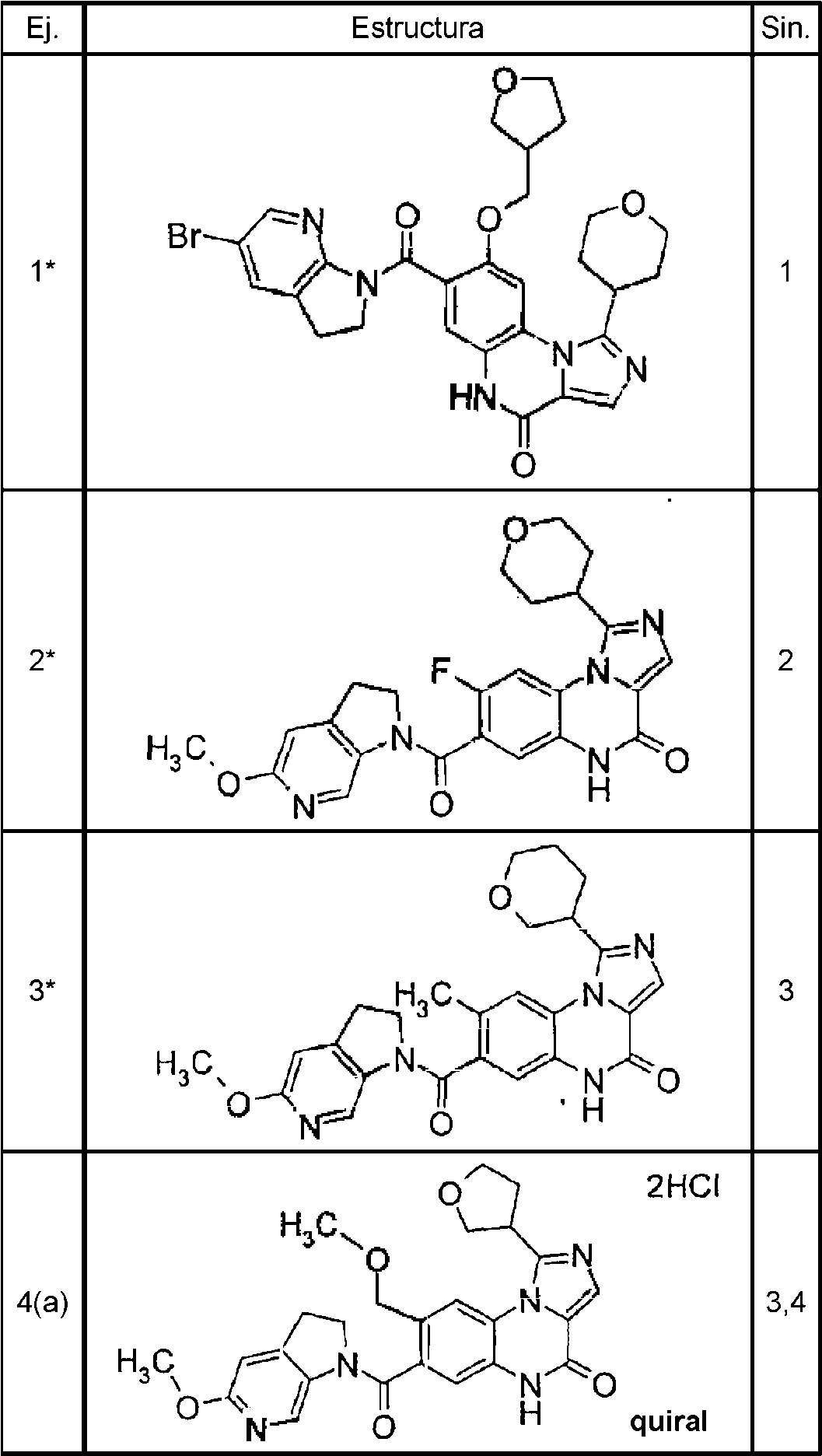

[Tabla 21]

[Tabla 22]

[Tabla 23]
Ej.  Estructura
Estructura  Sin.
Sin.

[Tabla 24]


[Tabla 25]


[Tabla 26]


[Tabla 27]


[Tabla 28]

[Tabla 29]
Ej. Estructura Sin.
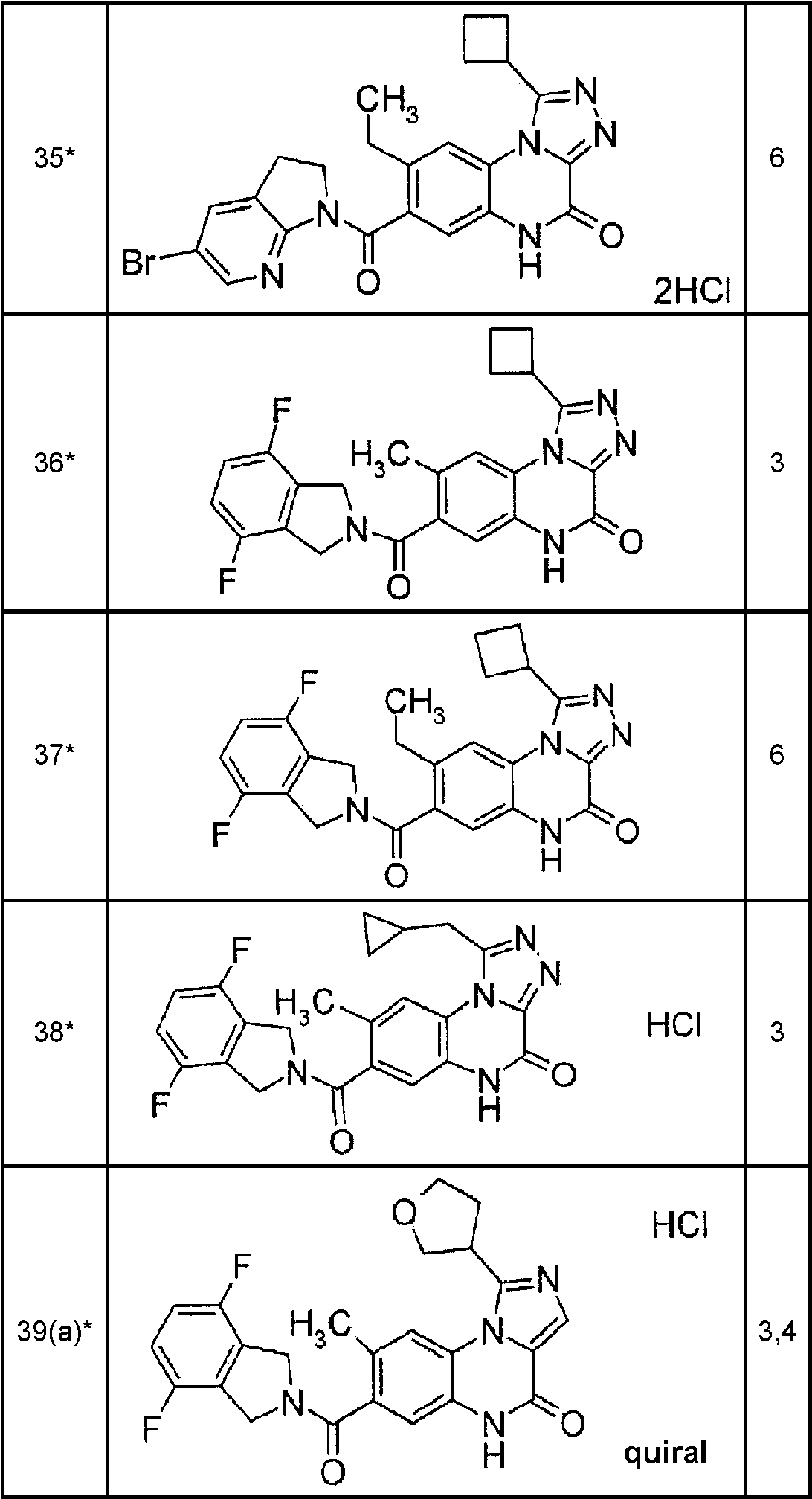
[Tabla 30]
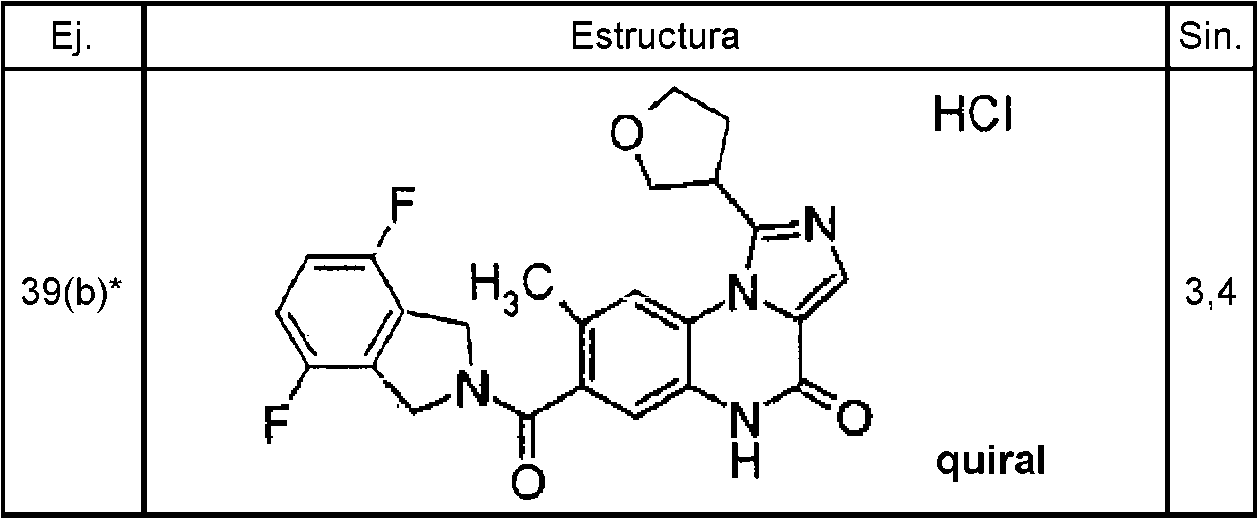

[Tabla 31]


[Tabla 32]


[Tabla 33]


[Tabla 34]


[Tabla 35]


[Tabla 36]


[Tabla 37]


[Tabla 38]



[Tabla 39]


[Tabla 40]

* Referencia
[Tabla 43]
n.° Estructura

Aplicabilidad industrial
El compuesto de fórmula (I) o una sal del mismo tiene una acción inhibidora de la PDE9, y puede usarse como agente para prevenir y/o tratar enfermedades relacionadas con la degradación de GMPc por la PDE9, por ejemplo, vejiga hipoactiva, vejiga hipotónica, vejiga acontráctil, vejiga neurógena, hipoactividad del detrusor, vejiga hiperactiva, polaquiuria, nicturia, incontinencia, hiperplasia prostática benigna, síntomas de las vías urinarias inferiores, disfunción de la evacuación que acompaña a la insuficiencia de la relajación de la uretra o disinergia detrusor-esfínter uretral externo, cistitis intersticial, prostatitis crónica o cálculo de uretra.
Claims (1)
- REIVINDICACIONESi. Compuesto de fórmula (I) o sal del mismo:[Fórm. quím. 13]
 A es CH,R1 es 2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-ilo sustituido por fluoro, bromo, metilo, etilo, trifluorometilo o metoxilo en la posición 5, o 5-metoxi-2,3-dihidro-1H-pirrolo[2,3-c]piridin-1-ilo,R2 es metilo o metoximetilo, yR3 es tetrahidrofuran-3-ilo.2. Compuesto o sal del mismo según la reivindicación 1, que es(+)-7-[(5-metoxi-2,3-dihidro-1H-pirrolo[2,3-c]piridin-1-il)carbonil]-8-metil-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,(+)-7-[(5-metoxi-2,3-dihidro-1H-pirrolo[2,3-c]piridin-1-il)carbonil]-8-(metoximetil)-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,(+)-8-(metoximetil)-7-[(5-metil-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,(+)-7-[(5-bromo-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-8-(metoximetil)-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-onao una sal de los mismos.3. (+)-7-[(5,6-Difluoro-2,3-dihidro-1H-indol-1-il)carbonil]-8-metil-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona o sal del mismo.4. Composición farmacéutica que comprende el compuesto o una sal del mismo según una cualquiera de las reivindicaciones 1 a 3, y un excipiente farmacéuticamente aceptable.5. Compuesto o sal del mismo según una cualquiera de las reivindicaciones 1 a 3, para su uso en un método para prevenir o tratar una vejiga hipoactiva.6. Compuesto o sal del mismo según una cualquiera de las reivindicaciones 1 a 3, para su uso en un método para prevenir o tratar una disfunción de la evacuación en la vejiga hipoactiva.7. Compuesto o sal del mismo según una cualquiera de las reivindicaciones 1 a 3, para su uso en un método para prevenir o tratar una hiperplasia prostética benigna.8. Compuesto o sal del mismo según una cualquiera de las reivindicaciones 1 a 3, para su uso en un método para prevenir o tratar una disfunción de la evacuación que acompaña a la hiperplasia prostética benigna.
A es CH,R1 es 2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-ilo sustituido por fluoro, bromo, metilo, etilo, trifluorometilo o metoxilo en la posición 5, o 5-metoxi-2,3-dihidro-1H-pirrolo[2,3-c]piridin-1-ilo,R2 es metilo o metoximetilo, yR3 es tetrahidrofuran-3-ilo.2. Compuesto o sal del mismo según la reivindicación 1, que es(+)-7-[(5-metoxi-2,3-dihidro-1H-pirrolo[2,3-c]piridin-1-il)carbonil]-8-metil-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,(+)-7-[(5-metoxi-2,3-dihidro-1H-pirrolo[2,3-c]piridin-1-il)carbonil]-8-(metoximetil)-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,(+)-8-(metoximetil)-7-[(5-metil-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona,(+)-7-[(5-bromo-2,3-dihidro-1H-pirrolo[2,3-b]piridin-1-il)carbonil]-8-(metoximetil)-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-onao una sal de los mismos.3. (+)-7-[(5,6-Difluoro-2,3-dihidro-1H-indol-1-il)carbonil]-8-metil-1-(tetrahidrofuran-3-il)imidazo[1,5-a]quinoxalin-4(5H)-ona o sal del mismo.4. Composición farmacéutica que comprende el compuesto o una sal del mismo según una cualquiera de las reivindicaciones 1 a 3, y un excipiente farmacéuticamente aceptable.5. Compuesto o sal del mismo según una cualquiera de las reivindicaciones 1 a 3, para su uso en un método para prevenir o tratar una vejiga hipoactiva.6. Compuesto o sal del mismo según una cualquiera de las reivindicaciones 1 a 3, para su uso en un método para prevenir o tratar una disfunción de la evacuación en la vejiga hipoactiva.7. Compuesto o sal del mismo según una cualquiera de las reivindicaciones 1 a 3, para su uso en un método para prevenir o tratar una hiperplasia prostética benigna.8. Compuesto o sal del mismo según una cualquiera de las reivindicaciones 1 a 3, para su uso en un método para prevenir o tratar una disfunción de la evacuación que acompaña a la hiperplasia prostética benigna.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010200402 | 2010-09-07 | ||
| PCT/JP2011/070287 WO2012033101A1 (ja) | 2010-09-07 | 2011-09-06 | キノキサリン化合物 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2747423T3 true ES2747423T3 (es) | 2020-03-10 |
Family
ID=45810699
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES11823576T Active ES2747423T3 (es) | 2010-09-07 | 2011-09-06 | Compuesto de quinoxalina |
Country Status (16)
| Country | Link |
|---|---|
| US (1) | US8901126B2 (es) |
| EP (1) | EP2615096B1 (es) |
| JP (1) | JP5822080B2 (es) |
| KR (1) | KR20130097178A (es) |
| CN (1) | CN103097393B (es) |
| AU (1) | AU2011299936A1 (es) |
| BR (1) | BR112013005439A2 (es) |
| CA (1) | CA2810659A1 (es) |
| EA (1) | EA201390354A1 (es) |
| ES (1) | ES2747423T3 (es) |
| MX (1) | MX2013002579A (es) |
| PL (1) | PL2615096T3 (es) |
| PT (1) | PT2615096T (es) |
| TW (1) | TW201300390A (es) |
| WO (1) | WO2012033101A1 (es) |
| ZA (1) | ZA201301673B (es) |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102786525B (zh) | 2012-08-08 | 2014-12-17 | 中山大学 | N-取代吡唑并[3,4-d]嘧啶酮类化合物、其制备方法及其应用 |
| WO2017046606A1 (en) * | 2015-09-18 | 2017-03-23 | Redx Pharma Plc | Antibacterial compounds |
| TWI609870B (zh) | 2016-02-12 | 2018-01-01 | 美國禮來大藥廠 | Pde1抑制劑 |
| JOP20170164A1 (ar) | 2016-08-25 | 2019-01-30 | Lilly Co Eli | مشتق ترايازولو بيرازينون مفيد كمثبط لـ pde1 بشري |
| AR112457A1 (es) | 2017-08-02 | 2019-10-30 | Lilly Co Eli | Derivados de [1,2,4]triazolo[4,3-a]pirazin-6(5h)-ona |
| AR112346A1 (es) * | 2017-08-10 | 2019-10-16 | Lilly Co Eli | Derivados de [1,2,4]triazolo |
Family Cites Families (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5055465A (en) * | 1989-05-31 | 1991-10-08 | Berlex Laboratories, Inc. | Imidazoquinoxalinones, their aza analogs and process for their preparation |
| DK162491D0 (da) * | 1991-09-20 | 1991-09-20 | Novo Nordisk As | Heterocycliske forbindelser, deres fremstilling og farmaceutiske praeparater indeholdende forbindelserne |
| AU4035893A (en) | 1991-12-17 | 1993-07-19 | Upjohn Company, The | 3-substituted imidazo (1,5-a) and imidazo (1,5-a)-triazolo (1,5-c) quinoxalines and quinazolines with cns activity |
| JPH07503970A (ja) | 1992-02-19 | 1995-04-27 | ジ・アップジョン・カンパニー | CNS活性を有する3−置換イミダゾ(1,5−a)−キノキサリン類およびキナゾリン類 |
| DK31093D0 (es) * | 1993-03-19 | 1993-03-19 | Novo Nordisk As | |
| RU2149162C1 (ru) | 1994-09-16 | 2000-05-20 | Ново Нордиск А/С | Производные [1,2,4] триазоло [4,3-а]хиноксалинона или их фармацевтически приемлемые соли, способ их получения, фармацевтическая композиция и способ ингибирования рецепторов амра |
| CA2200056A1 (en) | 1994-09-16 | 1996-03-21 | Novo Nordisk A/S | [1,2,4]triazolo[4,3-a]quinoxalinone derivatives, their preparation and use |
| US6235740B1 (en) | 1997-08-25 | 2001-05-22 | Bristol-Myers Squibb Co. | Imidazoquinoxaline protein tyrosine kinase inhibitors |
| HN2002000317A (es) | 2001-11-02 | 2003-05-21 | Pfizer | Inhibidores de pde9 para tratamiento de trastornos cardiovasculares |
| MXPA05010293A (es) | 2003-03-27 | 2005-11-17 | Pfizer Prod Inc | 4-amino[1,2,4]triazolo[4,3-a]quinoxalinas sustituidas. |
| AU2006258461B2 (en) | 2005-06-14 | 2011-08-25 | Aska Pharmaceutical Co., Ltd. | Thienopyrimidine derivative |
| CN101501007B (zh) | 2006-08-08 | 2014-01-29 | Aska制药株式会社 | 喹唑啉衍生物 |
| ES2536906T3 (es) | 2006-12-13 | 2015-05-29 | Aska Pharmaceutical Co., Ltd. | Derivado de quinoxalina |
| CN101557826A (zh) | 2006-12-13 | 2009-10-14 | Aska制药株式会社 | 泌尿系统疾病的治疗药 |
| EP2334684B1 (en) | 2008-09-08 | 2017-08-02 | Boehringer Ingelheim International GmbH | Pyrazolopyrimidines and their use for the treatment of cns disorders |
| TWI404721B (zh) | 2009-01-26 | 2013-08-11 | Pfizer | 胺基-雜環化合物 |
| KR20110132559A (ko) * | 2009-03-05 | 2011-12-08 | 아스텔라스세이야쿠 가부시키가이샤 | 퀴녹살린 화합물 |
-
2011
- 2011-09-06 BR BR112013005439A patent/BR112013005439A2/pt not_active IP Right Cessation
- 2011-09-06 PL PL11823576T patent/PL2615096T3/pl unknown
- 2011-09-06 AU AU2011299936A patent/AU2011299936A1/en not_active Abandoned
- 2011-09-06 TW TW100132077A patent/TW201300390A/zh unknown
- 2011-09-06 MX MX2013002579A patent/MX2013002579A/es active IP Right Grant
- 2011-09-06 KR KR1020137005781A patent/KR20130097178A/ko not_active Application Discontinuation
- 2011-09-06 CN CN201180043069.7A patent/CN103097393B/zh active Active
- 2011-09-06 EA EA201390354A patent/EA201390354A1/ru unknown
- 2011-09-06 PT PT118235761T patent/PT2615096T/pt unknown
- 2011-09-06 CA CA2810659A patent/CA2810659A1/en not_active Abandoned
- 2011-09-06 EP EP11823576.1A patent/EP2615096B1/en active Active
- 2011-09-06 ES ES11823576T patent/ES2747423T3/es active Active
- 2011-09-06 WO PCT/JP2011/070287 patent/WO2012033101A1/ja active Application Filing
- 2011-09-06 JP JP2012532987A patent/JP5822080B2/ja active Active
-
2013
- 2013-03-05 ZA ZA2013/01673A patent/ZA201301673B/en unknown
- 2013-03-07 US US13/788,770 patent/US8901126B2/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| PT2615096T (pt) | 2019-10-15 |
| TW201300390A (zh) | 2013-01-01 |
| CN103097393B (zh) | 2015-09-16 |
| EP2615096B1 (en) | 2019-08-28 |
| JPWO2012033101A1 (ja) | 2014-01-20 |
| BR112013005439A2 (pt) | 2017-09-19 |
| WO2012033101A1 (ja) | 2012-03-15 |
| ZA201301673B (en) | 2014-05-28 |
| US20130296329A1 (en) | 2013-11-07 |
| PL2615096T3 (pl) | 2019-12-31 |
| EA201390354A1 (ru) | 2013-08-30 |
| CN103097393A (zh) | 2013-05-08 |
| KR20130097178A (ko) | 2013-09-02 |
| EP2615096A4 (en) | 2014-03-05 |
| CA2810659A1 (en) | 2012-03-15 |
| AU2011299936A1 (en) | 2013-03-28 |
| EP2615096A1 (en) | 2013-07-17 |
| JP5822080B2 (ja) | 2015-11-24 |
| MX2013002579A (es) | 2013-04-29 |
| US8901126B2 (en) | 2014-12-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5609861B2 (ja) | キノキサリン化合物 | |
| ES2797252T3 (es) | Dihidronaftiridinas y compuestos relacionados útiles como inhibidores de quinasas para el tratamiento de enfermedades proliferativas | |
| CA2806324C (en) | Phthalazinone ketone derivative, preparation method thereof, and pharmaceutical use thereof | |
| ES2747423T3 (es) | Compuesto de quinoxalina | |
| DK2615089T3 (en) | Pyrazoloquinolinforbindelser | |
| US10085983B2 (en) | Azabicyclo derivatives, process for preparation thereof and medical use thereof | |
| BRPI0622030A2 (pt) | Derivados de purina 7-substituída, para imunossupressão | |
| CA2880739A1 (en) | Piperazinotriazole compound, preparation method thereof, and use thereof in drug preparation | |
| JP6307088B2 (ja) | キナゾリノンおよびイソキノリノン誘導体 | |
| CN107922350A (zh) | 新的苯并咪唑化合物及其医药用途 | |
| CN117295734A (zh) | 甲硫氨酸腺苷转移酶抑制剂、其制备方法及应用 | |
| CN116157400A (zh) | 杂环类衍生物及其制备方法和用途 | |
| WO2015135461A1 (zh) | 取代的二氢苯并呋喃-哌啶-甲酮衍生物、其制备及用途 |