ES2726664T3 - Compuestos de quinina e isómeros ópticos, procedimiento de preparación y uso médico de los mismos - Google Patents
Compuestos de quinina e isómeros ópticos, procedimiento de preparación y uso médico de los mismos Download PDFInfo
- Publication number
- ES2726664T3 ES2726664T3 ES14825941T ES14825941T ES2726664T3 ES 2726664 T3 ES2726664 T3 ES 2726664T3 ES 14825941 T ES14825941 T ES 14825941T ES 14825941 T ES14825941 T ES 14825941T ES 2726664 T3 ES2726664 T3 ES 2726664T3
- Authority
- ES
- Spain
- Prior art keywords
- acid
- hydroxyl
- azabicyclo
- ethoxy
- octane
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 C*C(COC1C(CC2)CCN2C1)(O)I Chemical compound C*C(COC1C(CC2)CCN2C1)(O)I 0.000 description 8
- JJFOBACUIRKUPN-UHFFFAOYSA-N BrCCOc1ccccc1 Chemical compound BrCCOc1ccccc1 JJFOBACUIRKUPN-UHFFFAOYSA-N 0.000 description 1
- GIWBRBVGLOZHKF-UHFFFAOYSA-N C1OC1(C1CCCC1)C1=CCCO1 Chemical compound C1OC1(C1CCCC1)C1=CCCO1 GIWBRBVGLOZHKF-UHFFFAOYSA-N 0.000 description 1
- HTYBMJGZGGMGOF-UHFFFAOYSA-N CCN(CCC1C)CC1O Chemical compound CCN(CCC1C)CC1O HTYBMJGZGGMGOF-UHFFFAOYSA-N 0.000 description 1
- LHDMBYWDIBGTAR-UHFFFAOYSA-N IOc1ccccc1 Chemical compound IOc1ccccc1 LHDMBYWDIBGTAR-UHFFFAOYSA-N 0.000 description 1
- HOZDQHPELMDYEM-RGBJRUIASA-N N[C@@](CO[C@@H]1C(CC2)CCN2C1)(C1CCCC1)C1OCCC1 Chemical compound N[C@@](CO[C@@H]1C(CC2)CCN2C1)(C1CCCC1)C1OCCC1 HOZDQHPELMDYEM-RGBJRUIASA-N 0.000 description 1
- IVLICPVPXWEGCA-ZETCQYMHSA-N O[C@@H]1C(CC2)CCN2C1 Chemical compound O[C@@H]1C(CC2)CCN2C1 IVLICPVPXWEGCA-ZETCQYMHSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D453/00—Heterocyclic compounds containing quinuclidine or iso-quinuclidine ring systems, e.g. quinine alkaloids
- C07D453/02—Heterocyclic compounds containing quinuclidine or iso-quinuclidine ring systems, e.g. quinine alkaloids containing not further condensed quinuclidine ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/13—Amines
- A61K31/135—Amines having aromatic rings, e.g. ketamine, nortriptyline
- A61K31/137—Arylalkylamines, e.g. amphetamine, epinephrine, salbutamol, ephedrine or methadone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/439—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom the ring forming part of a bridged ring system, e.g. quinuclidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0043—Nose
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/007—Pulmonary tract; Aromatherapy
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/007—Pulmonary tract; Aromatherapy
- A61K9/0073—Sprays or powders for inhalation; Aerolised or nebulised preparations generated by other means than thermal energy
- A61K9/008—Sprays or powders for inhalation; Aerolised or nebulised preparations generated by other means than thermal energy comprising drug dissolved or suspended in liquid propellant for inhalation via a pressurized metered dose inhaler [MDI]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4808—Preparations in capsules, e.g. of gelatin, of chocolate characterised by the form of the capsule or the structure of the filling; Capsules containing small tablets; Capsules with outer layer for immediate drug release
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/02—Nasal agents, e.g. decongestants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/14—Antitussive agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/02—Drugs for disorders of the urinary system of urine or of the urinary tract, e.g. urine acidifiers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/10—Drugs for disorders of the urinary system of the bladder
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Epidemiology (AREA)
- Pulmonology (AREA)
- Otolaryngology (AREA)
- Urology & Nephrology (AREA)
- Emergency Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
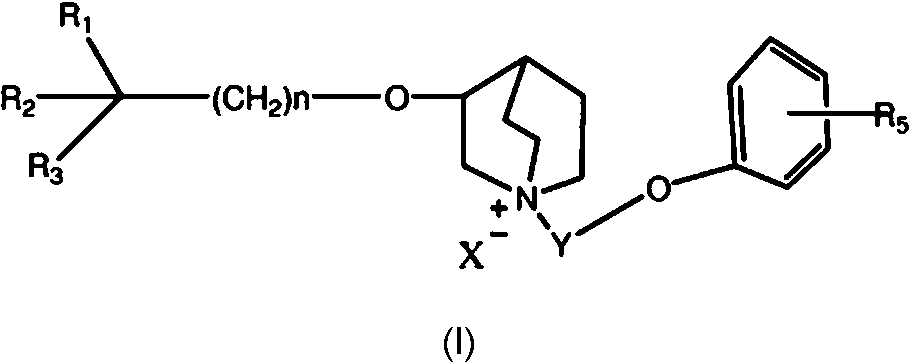
Compuesto de fórmula I:**Fórmula** o una sal, solvato o isómero óptico de la misma farmacéuticamente aceptables, en donde en la fórmula I: n se selecciona entre 1-7, R1 es un hidrocarbilo C3-C7, que puede estar no sustituido u opcionalmente sustituido por halógeno, alcoxi, alcoxihidrocarbilo, heterociclilo o arilo, R2 es un arilo o un heteroarilo que contiene uno o más heteroátomos, que puede estar no sustituido u opcionalmente sustituido, R3 es un hidroxilo, halógeno, alcoxi o aciloxi, en donde el alcoxi o aciloxi puede estar no sustituido u opcionalmente sustituido por halógeno, hidroxilo, alcoxi, hidrocarbilo, alcoxihidrocarbilo, heterociclilo o arilo; R4 y R5 pueden estar presentes o ausentes, y se seleccionan de forma independiente de un grupo que consiste en halógeno, hidroxilo, alcoxi, hidrocarbilo, alcoxihidrocarbilo, heterociclilo o arilo, cuando están presentes; Y es un alquileno C1-C7, lineal o ramificado, o -(CH2-O-CH2)m-, que puede estar opcionalmente sustituido, en donde m es igual a 1-3, X- es un radical ácido o hidroxilo.
Description
DESCRIPCIÓN
Compuestos de quinina e isómeros ópticos, procedimiento de preparación y uso médico de los mismos Campo de la técnica
La presente invención se refiere a compuestos de quinina, isómeros ópticos y procedimientos de fabricación de los mismos y composiciones que comprenden estos compuestos con fines medicinales y, en particular, a nuevos antagonistas del receptor M que tienen un efecto selectivo sobre subtipos del receptor M, que tienen un fuerte efecto sobre los subtipos receptores M3 y M1, pero no tienen ningún efecto significativo sobre el subtipo receptor M2.
Antecedentes de la técnica relacionada
En algunos documentos de patente se encuentra que un compuesto que comprende una estructura de quinina se utiliza para el efecto anti-colinérgico. Por ejemplo, un compuesto divulgado en la patente de invención china CN200810112248.1 y en la patente de invención china CN200910223255.3 (número de publicación CN102070631 A) tiene una estructura como la siguiente,
en donde: R es metilo, etilo, propilo, isopropilo, o ciclopropilo; y X representa un átomo de halógeno.

La patente francesa FR2012964 divulgó una estructura como la siguiente, en donde: R es un átomo de H, hidroxilo o alquilo con 1-4 átomos de carbono; R1 es fenilo o tienilo; y R2 es ciclohexilo, ciclopentilo o tienilo.

La patente de EE.UU. documento US5654314 divulgó una estructura como la siguiente:

La patente WO con documento WO01/04118 divulgó una estructura como la siguiente:

Los compuestos anteriores tienen desventajas significativas, tales como eficacia de corta duración, acción lenta, o efectos tóxicos y secundarios significativos, o similares, en el tratamiento de rinitis, rinitis después de un resfriado, traquitis crónica, hipersensibilidad de las vías respiratorias, asma, enfermedades pulmonares obstructivas crónicas, tos, incontinencia urinaria, micción frecuente, síndrome de vejiga inestable, espasmos vesicales, inflamación de la vejiga y enfermedades gastrointestinales como el síndrome del intestino irritable, colitis espástica, así como úlceras duodenales y gástricas.
Los compuestos de la presente invención superan las desventajas de los compuestos anteriores, y en particular, se caracterizan por una mayor eficacia, acción rápida y menores efectos tóxicos y secundarios en el tratamiento de la traquitis crónica, las hipersensibilidades de las vías respiratorias, asma y enfermedades pulmonares obstructivas crónicas en comparación con los compuestos de la técnica anterior. Debido a su buena estabilidad, los compuestos de la presente invención son adecuados para la fabricación de un inhalante que es administrado una vez al día para tratar enfermedades pulmonares obstructivas crónicas y, en particular, adecuado para fabricar un aerosol de
inhalación de dosis medida de tipo solución que es administrado una vez al día. La presente invención se refiere a la síntesis de dicho compuesto, comprendiendo la fabricación de una composición farmacéutica que comprende dicho compuesto y los usos farmacéuticos del mismo.
El compuesto de la presente invención puede utilizarse también para tratar las enfermedades respiratorias anteriores como rinitis, rinitis después de un resfriado, traquitis crónica, hipersensibilidad de las vías respiratorias, asma, enfermedades pulmonares obstructivas crónicas, y similares, en combinación con agonistas del receptor p2, hormona esteroidea, fármacos antialérgicos, fármacos antiinflamatorios, fármacos antiinfecciosos, antagonistas de la fosfolipasa i.v. y similares.
Contenido de la invención
Un nuevo compuesto antagonista selectivo de subtipos del receptor M de la presente invención puede representarse por la estructura de la fórmula (I):

en donde en la fórmula (I):
n se selecciona entre 1-7, preferiblemente 1-3, lo más preferiblemente 1.
R1 es un hidrocarbilo C3-C7 , que puede estar no sustituido u opcionalmente sustituido por halógenos, alcoxi, alcoxihidrocarbilo, heterociclilo o arilo; preferiblemente un cicloalquilo no sustituido, y lo más preferiblemente un ciclopentilo o ciclohexilo.
R2 es un arilo o un heteroarilo que contiene uno o más heteroátomos (el heteroátomo puede ser N, O o S), por ejemplo, R2 es fenilo, naftilo o bifenilo, que puede estar no sustituido u opcionalmente sustituido por uno o más de entre halógeno, hidroxilo, fenilo, -OR6 , -SR6 , -NR6R7 , -NHCOR6 , -CONR6R7 , -CN, -NO2 , -COOR6 , -CF3 o hidrocarbilo C1-C4 , lineal o ramificado; se prefieren fenilo, piridilo, furilo y tienilo no sustituidos; R6 y R7 pueden ser un átomo de hidrógeno, un hidrocarbilo C1-C4 lineal o ramificado, o pueden formar un ciclohidrocarbilo entre sí.
R3 es un hidroxilo, halógeno, alcoxi o aciloxi, en donde el alcoxi o aciloxi puede estar no sustituido u opcionalmente sustituido por halógeno, hidroxilo, alcoxi, hidrocarbilo, alcoxihidrocarbilo, ciclohidrocarbilo, heterociclilo o arilo; preferiblemente un hidroxilo o metoxilo y lo más preferiblemente un hidroxilo.
R4 y el R5 pueden estar presentes o ausentes, y respectivamente, cuando están presentes, pueden ser sustituyentes como halógenos, hidroxilo, hidrocarbiloxi, hidrocarbilo, hidrocarbiloxihidrocarbilo, heterociclilo y arilo.
Y es un alquileno C1-C7 , lineal o ramificado, o -(CH2-O-CH2V (donde m es igual a 1-3), que puede sustituirse opcionalmente, preferiblemente, sustituirse por halógeno, hidroxilo, alcoxi, alcoxialquilo, hidrocarbilo insaturado, ciclohidrocarbilo, o heterociclilo; preferiblemente un metileno, etileno, propileno o -(CH2-O-CH2)-; y, lo más preferiblemente, un etileno o propileno.
X- es un radical ácido o un hidróxido, preferiblemente un radical ácido farmacéuticamente aceptable, cuyos ejemplos incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato, sulfito, hidrosulfito o fosfito; y una sal derivada de un ácido orgánico relativamente no tóxico como, pero no limitado a, ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
El compuesto representado por la fórmula (I) puede comprender uno o más centros quirales, en los que un único isómero óptico o una mezcla de varios isómeros ópticos entran dentro del alcance reivindicado por la presente invención.
Los siguientes compuestos pueden ilustrar específicamente el contenido de la presente invención pero no limitan el alcance de la presente invención.
1. Bromuro de (2S,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano
2. Bromuro de (2S,3S)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano
3. Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano
4. Bromuro de (2R,3S)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano
5. Bromuro de (2S,3R),(2R,3S)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano 6. Bromuro de (2R,3R),(2S,3S)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano 7. Bromuro de (2S,3R)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano
8. Bromuro de (2S,3S)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano
9. Bromuro de (2R,3R)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano
10. Bromuro de (2R,3S)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano
11. Bromuro de (2R,3S),(2S,3R)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano 12. Bromuro de (2R,3R),(2S,3S)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano 13. Bromuro de (2S,3R)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano
14. Bromuro de (2S,3S)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano
15. Bromuro de (2R,3S)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano
16. Bromuro de (2R,3R)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano
17. Bromuro de (2R,3S),(2S,3R)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano 18. Bromuro de (2R,3R),(2S,3S)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano 19. Bromuro de (2S,3R)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano 20. Bromuro de (2R,3S)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano 21. Bromuro de (2S,3S)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano 22. Bromuro de (2R,3R)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano 23. Bromuro de (2R,3S),(2S,3R)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano 24. Bromuro de (2R,3R),(2S,3S)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano 25. Bromuro de (2S,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(2-fenoxietil)-1-azabiciclo[2,2,2]octano
26. Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(2-fenoxietil)-1-azabiciclo[2,2,2]octano
27. Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-fenoximetil-1-azabiciclo[2,2,2]octano
28. Cloruro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-fenoximetoximetil-1 -azabiciclo[2,2,2]octano 29. Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-naftil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano 30. Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-(o-clorofenil))etoxil]-1-(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano 31. Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-(3-piridil))etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano 32. Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-(2-furil))etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano 33. Bromuro de (2R,3R)-3-[(2-ciclopentil-2-metoxil-2-(3-piridil))etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano
La presente invención proporciona un esquema de proceso para sintetizar el compuesto de fórmula estructural (I) de la siguiente manera:
Etapa 1: (1-R1-1-R2)-oxirano se preparó según los procedimientos descritos en la bibliografía (1-2) (1. Guangling Wen, Peijin Wu, Improvement on the synthesis method of 3-(2-phenyl-2-cyclopentylethoxyl) quinuclidine hydrochloride, Bullet of the Academy of Millitary Medical Sciences, 1988: 470 de vuelta a 402; 2. Peijin Wu, Liuhong Yun, Synthesis of anticholinergic drug of 2-(1-naftil)-2-cyclopentyl-2-hydroxylethoxyl cyclohydrocarbyl amine compounds, Chinese Journal of Medicinal Chemistry 1999.6, 9(2): pág. 102-105), y arilhidrocarbilcetona (algunos tipos de arilhidrocarbilcetona se generaron por reacción de cianuro de arilo con un reactivo de Grignard preparado por reacción de bromuro de hidrocarbilo con magnesio en THF, véase la fórmula (1) a continuación) se hizo reaccionar con presencia de sulfato de dimetilo, sulfuro de dimetilo e hidruro de sodio para generar 1-aril-1-hidrocarbiloxirano, es decir, el intermedio 1 (véase la fórmula (2) a continuación).
(1)
R?^CN Ri— Br

(2)

Etapa 2: Preparación de 3-[(2-R1-2-R2-2-hidroxil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali
El intermedio 2 pudo obtenerse por reacción del intermedio 1 con quinuclidinol (o quinuclidinol sustituido con R4) bajo la condición de NaH.

A los derivados de 3-quinuclidinol comercialmente disponibles se añadió DMSO, seguido por la adición de hidruro de sodio, y la mezcla se hizo reaccionar a 20-60 °C, y después se enfrió a temperatura ambiente, y a ello se añadió una solución de (1-R1-1-R2)-oxirano (auto-preparado) en DMSO, y una vez finalizada la adición, la mezcla resultante se calentó a 20-70 °C en baño de aceite para reaccionar durante 0,5-12 horas. Bajo la condición de baño de hielo, se añadió agua helada a una temperatura interna de 30 °C o menor. La mezcla de reacción se extrajo con éter isopropílico, y la capa de éter se combinó y se lavó con una solución acuosa saturada de NaCl. La capa orgánica se secó sobre sulfato de sodio anhidro durante la noche, el agente desecante se retiró por filtración, y el disolvente se retiró por evaporación rotativa, para obtener el intermedio 2 como una materia aceitosa de color rojo.
Etapa 3: Purificación de 3-[(2-R1-2-R2-2-hidroxil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali por cromatografía de columna y el tratamiento de purificación relacionado
Una muestra del intermedio 2 anterior se separó en una columna de gel de sílice, utilizando diclorometano o triclorometano y metanol, amoniacados, como fase móvil y una placa de TLC para controlar la pureza de la muestra. El intermedio 2 sería una mezcla que contiene diferentes estructuras ópticas dependiendo de la estructura de isómeros ópticos de quinuclidinol, y bajo el sistema de elución anterior, si el quinuclidinol estaba en configuración S, el intermedio 2 comprendería dos tipos de configuraciones (2R,3S) y (2S,3S), y podía ser purificado en dos tipos libre de álcali, (2R,3S) y (2S,3S), dependiendo de la secuencia de elución; si quinuclidinol estaba en configuración R, el intermedio 2 comprendería dos tipos de configuraciones (2R,3R) y (2S,3R), y podía ser purificado en dos tipos libre de álcali, (2S,3r ) y (2R,3R), dependiendo de la secuencia de elución; si quinuclidinol era un racemato, el intermedio 2 comprendería cuatro tipos de configuraciones (2R,3S), (2S,3S), (2R,3R) y (2S,3R), y podía ser purificada en dos tipos libre de álcali, (2R,3S),(2S,3R) y (2S,3S),(2R,3R), dependiendo de la secuencia de elución. El producto separado libre de álcali se refirió como intermedio 3.
Etapa 4: Preparación de 3-Z-Y-oxilbenceno (intermedio 4)

En un matraz de tres bocas se añadió fenol, seguido de la adición de hidróxido de sodio y una solución de Z-Y-Z (donde Z era un átomo de halógeno) en etanol absoluto, y después la mezcla resultante se hizo reaccionar bajo calentamiento y reflujo en baño de aceite, precipitándose un sólido blanco, hasta reacción esencialmente completa de fenol controlada por TLC (condiciones de la TLC: éter de petróleo/acetato de etilo = 5,0 ml/1,0 ml). Una vez finalizada la reacción, el sólido se retiró por filtración, y el disolvente se retiró del filtrado a 50 °C o menor bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa que contenía un sólido blanco, al que se añadió éter de petróleo y después se dejó reposar durante la noche. El sólido precipitado se retiró por filtración, y el disolvente se retiró del filtrado a 25-40 °C bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo. La materia aceitosa se destiló bajo presión reducida, para recoger una materia aceitosa incolora, transparente, es decir, el intermedio 4.
Etapa 5: Preparación del compuesto de la fórmula (I)
El intermedio 2 o el intermedio 3 se añadieron en un matraz, con forma de berenjena, seguido de la adición de cloroformo, para obtener una solución transparente de color amarillo, a la que se añadieron intermedio 4 y acetonitrilo, y la mezcla resultante se agitó a continuación a temperatura ambiente para reaccionar durante 10-90 h
bajo la protección de nitrógeno, siendo controlado el final de la reacción mediante TLC (condiciones de TLC: cloroformo/metanol/amoníaco en agua = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción por evaporación rotativa a 25-40 °C bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener un sólido de color blancuzco, es decir, el compuesto del título de fórmula (I).
El compuesto anterior de fórmula (I) se hizo reaccionar con Ag2Ü para reemplazar el halógeno con hidróxido, que podría después transformarse en otro radical ácido al reaccionar con otro ácido. Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
Cualquier composición farmacéutica que comprenda uno o más compuestos de la fórmula (I) descritos anteriormente está dentro del alcance reivindicado por la presente invención, y la ruta de administración puede ser, por ejemplo, administración oral, tópica, intravenosa, intramuscular, endoarterial, intraperitoneal, rectal, vaginal, endonasal o por inhalación. La formulación de la presente invención puede diseñarse para que sea de acción rápida, liberación rápida o de acción prolongada. Además, el compuesto puede ser administrado por una ruta tópica en lugar de una ruta sistémica y según formas de realización representativas, la composición de la presente invención puede formularse para fármacos administrados para mamíferos, preferentemente humanos.
La composición que comprende uno o más compuestos de la presente invención y excipientes apropiados puede administrarse repetidamente, o la composición se puede administrar de forma continua. Los sitios adecuados de administración incluyen, pero no se limitan a, cavidad nasal, pulmón, vasos sanguíneos, músculos, bronquios, e intestinos y estómago. La formulación puede estar en la forma de una forma de administración líquida, polvo liofilizado, sólido o semisólido, tal como solución, suspensión, emulsión, comprimido, píldora, cápsula, productos pulverizados, supositorio, enemas retentivos, aerosol, aerosol en polvo, o similar, preferiblemente una forma de administración unitaria adecuada para simplemente administrar una dosis exacta. Los ejemplos de excipientes apropiados incluyen, pero no se limitan a, agua, solución salina, lactosa, glucosa, sacarosa, sorbitol, manitol, almidón, goma arábiga, fosfato de calcio, alginato, tragacanto, gelatina, silicato de calcio, celulosa microcristalina, polivinilpirrolidona, celulosa, jarabe, metilcelulosa, etilcelulosa, hidroxipropilmetilcelulosa y poli(ácido acrílico). La composición puede comprender además lubricante como talco, estearato de magnesio y aceite mineral; agente humectante; emulgente; agente de suspensión; conservante tal como metil-, etil-y propil-hidroxilbenzoato; regulador del pH como ácidos y bases inorgánicos y orgánicos; agente edulcorante; y corrector.
Para la administración parenteral, la composición puede estar en la forma de una inyección estéril y polvo envasado asépticamente. Preferiblemente, la inyección se formula a pH 4,5-7,5.
La composición farmacéutica de la presente invención puede ser cualquiera de las formas de administración aceptables por vía oral, incluyendo comprimidos, cápsulas, pastillas, emulsión, suspensión, solución, jarabe, elixir, nebulosa, píldora, troche, productos pulverizados, gránulo y preparado de liberación sostenida. Excipientes adecuados para la administración oral incluyen manitol de calidad farmacéutica, lactosa, almidón, estearato de magnesio, sacarina sódica, talco, celulosa, glucosa, gelatina, sacarosa, carbonato de magnesio y similares. En el caso de comprimidos para administración oral, los portadores normalmente usados incluyen lactosa y celulosa microcristalina, y generalmente se añade un lubricante como estearato de magnesio; para las cápsulas, los diluyentes útiles incluyen lactosa y almidón de maíz seco; cuando se requiere suspensión para administración oral, los ingredientes activos se mezclan con emulgente y agente de suspensión, y también se pueden añadir algunos agentes edulcorantes, correctores o colorantes según corresponda.
Por ejemplo, polvo, solución o suspensión para inhalación pulmonar, pulverizador nasal, administración oral, tópica o intravenosa pueden formarse disolviendo o dispersando uno o más compuestos de la presente invención y, opcionalmente, uno o más materiales auxiliares farmacéuticamente aceptables en un vehículo como una solución salina, solución acuosa de glucosa, glicerol, etanol o similar. Se prepara la composición líquida, y la formulación farmacéutica en forma de suspensión líquida o solución puede prepararse utilizando líquido estéril como aceite, agua, etanol y una combinación de los mismos; para inhalación pulmonar, pulverizador nasal, administración oral o intravenosa, se puede añadir un tensioactivo, agente de suspensión o emulgente farmacéuticamente adecuados; una suspensión puede comprender aceite, como aceite de cacahuete, aceite de sésamo, aceite de semilla de algodón, aceite de maíz y aceite de oliva; una formulación de suspensión también puede comprender ésteres de ácidos grasos, como el oleato de etilo, miristato de isopropilo, glicérido graso y glicérido graso acetilado. Una formulación de suspensión puede comprender alcohol, como etanol, isopropanol, hexadecanol, glicerol y propanodiol; éter, como el polietilenglicol; hidrocarburo del petróleo, como aceite mineral y vaselina. También se puede utilizar agua en una formulación de suspensión.
La composición puede estar en forma de píldora, comprimido o cápsula y, por lo tanto, la composición puede comprender uno o más diluyentes, tales como lactosa, sacarosa, fosfato dicálcico y similares; un disgregante, como
almidón o derivados del mismo; un lubricante, como estearato de magnesio y similares; y/o un aglutinante, como almidón, goma arábiga, polivinilpirrolidona, gelatina, celulosa y derivados de los mismos. La píldora, comprimido o cápsula puede ser preparada por cualquier procedimiento conocido por los expertos en la técnica.
Alternativamente, la composición farmacéutica de la presente invención puede estar en forma de supositorio para la administración rectal. Estos supositorios pueden prepararse mezclando un medicamento con un excipiente no irritante adecuado que sea sólido a temperatura ambiente pero líquido a la temperatura rectal y, así, liberar el medicamento en el recto. Estos materiales incluyen manteca de cacao, cera de abejas, polietilenglicol, estearato de glicerilo y/o cocoglicéridos hidrogenados. La composición adecuada para administración rectal puede comprender también una unidad de enema rectal, que comprende uno o más compuestos de la presente invención y un vehículo farmacéuticamente aceptable (p. ej., solución acuosa de etanol al 50 % o solución salina), y tal vehículo es fisiológicamente compatible con el recto y/o el colon. Una unidad de enema rectal comprende una punta de aplicador protegida por una cubierta inerte, que está preferiblemente compuesta de polietileno, lubricada con un lubricante como vaselina blanca y, preferiblemente, protegida por una válvula antirretorno para evitar el reflujo del medicamento distribuido. La unidad de enema rectal tiene además una longitud suficiente, preferiblemente de 2 pulgadas (5,08 cm), para que se inserte en el colon a través del ano.
La composición farmacéutica de la presente invención también puede estar en forma de administración tópica, especialmente cuando los objetivos terapéuticos incluyen regiones u órganos en los que la composición farmacéutica puede administrarse fácilmente mediante la administración tópica, y las enfermedades de estos órganos incluyen enfermedades de pulmón, de la mucosa nasal y de la tráquea. Las formulaciones tópicas adecuadas para estas regiones u órganos se preparan fácilmente. Para la administración tópica, la composición que comprende uno o más compuestos de la presente invención puede estar en forma de pulverizador nasal, inhalación de solución, inhalación de polvo con dosis medida, inhalación de solución con dosis medida, inhalación de suspensión con dosis medidas y similares.
Para la administración por inhalación, la composición en forma de polvo seco o líquido puede ser entregada mediante un pulverizador. Tales composiciones se preparan según las tecnologías conocidas en el campo de la formulación farmacéutica, y la composición en forma de líquido se puede preparar en solución salina con fenilcarbinol u otros conservantes adecuados, absorbentes que realzan la biodisponibilidad, fluorocarbono y/u otro solubilizante o dispersante convencional.
En el aerosol para inhalación de dosis medida de tipo solución que comprende uno o más compuestos de fórmula (I), el disolvente latente incluye uno o más de entre etanol absoluto, glicerol y dioles, o una mezcla de los mismos, en donde los dioles incluyen, pero no se limitan a, etilenglicol, propanodiol, polietilenglicol 200, polietilenglicol 300, polietilenglicol 400, polietilenglicol 600, polietilenglicol 800 y similares. El propelente en el aerosol anterior incluye uno de entre tetrafluoroetano (HFA-134a) y heptafluoropropano (HFA-227ea), o una mezcla de los mismos. El tensioactivo en el aerosol anterior incluye uno o más de entre ácido oleico; oligómero de ácido láctico (OLA); sorbitanos, tales como Span 20, Span 65, Span 80, Span 85; polioxietilensorbitanos, como Tween 20, Tween 80; alcoholes grasos de polioxietileno, como Brij 30, Brij 35, Cremophor; copolímero de polioxietileno y polioxipropileno, como Pluronic F-68; estearato de polietilenglicol, como Solutol HS15; fosfolípidos, como granulestén y lecitina. Se prefieren ácido oleico, lecitina o una mezcla de los mismos. En el aerosol, el contenido de dicho compuesto de fórmula I es 0,005~1 % en peso, preferiblemente 0,02~0,5 %. El contenido del disolvente latente en el aerosol para inhalación es de 5~40 % en peso, preferiblemente 17,5-29,975 %. El contenido del tensioactivo en el aerosol para inhalación es de 0~5 % en peso, preferiblemente 0,005~2 %. El contenido del propelente en el aerosol para inhalación es de 54~90 % en peso, preferiblemente 70~80 %.
En la inhalación en polvo de dosis medida que comprende uno o más compuestos de la fórmula (I), dicho portador inerte comprende un diluyente y un lubricante, en donde dicho diluyente es uno o más de entre glucano, arabinosa, lactosa, manitol, xilitol, sacarosa, fructosa, sorbitol, maltosa, aminoácido y glucosa, o una mezcla de los mismos, y dicho lubricante es estearato de magnesio o benzoato de sodio.
En naristillae o aerosol nasal de dosis medida que comprende uno o más compuestos de fórmula (I), dicho portador inerte es uno o más seleccionados de cloruro de benzalconio, bromuro de benzalconio, fenilcarbinol, ácido benzoico, tricloro-t-butanol, benzoatos de p-hidroxilo, ácido sórbico, fenol, timol y aceite volátil, o una mezcla de los mismos.
Además, la presente invención proporciona usos de dicha composición farmacéutica que pueden utilizarse para la fabricación de medicamentos para prevenir y tratar diversas enfermedades obstructivas agudas o crónicas de las vías respiratorias, como las enfermedades pulmonares obstructivas crónicas, asma bronquial; así como rinitis aguda o crónica y rinitis después de un resfriado en mamífero y humano.
El compuesto de fórmula (I) y otros ingredientes activos (p. ej., dipropionato de beclometasona, clortrimetón, nafazolina o fenoterol) se formulan en una combinación, que se puede utilizar para el tratamiento de varias enfermedades obstructivas, agudas o crónicas, de las vías respiratorias, como las enfermedades pulmonares obstructivas crónicas, el asma bronquial y varias rinitis.
Para obtener efectos terapéuticos de forma rápida y eficaz, y garantizar los efectos no tóxicos y secundarios en uso, se sugiere que la dosificación diaria del compuesto de la presente invención utilizada debería ser 10-1.000 pg y, de forma óptima, 40-500 pg.
Además de las formas de dosificación representativas, los expertos en la técnica saben también que, en la presente invención, se incluyen, generalmente, otros excipientes, portadores y formas de administración farmacéuticamente aceptables . Se debe entender que la administración específica y el régimen terapéutico para cualquier paciente concreto dependen de diversos factores que incluyen la actividad del compuesto concreto utilizado, la edad, peso corporal, estado de salud general, género, estado dietético del paciente, tiempo de administración, y la tasa de excreción, fármacos de combinación, diagnóstico de terapeutas y la gravedad de la enfermedad concreta a tratar. La cantidad de ingredientes activos puede depender además del compuesto específico y otros fármacos terapéuticos (si están presentes) en la composición.
Descripción detallada de las formas de realización
El contenido de la invención se describe, a continuación, en detalle, mediante los ejemplos de los compuestos e isómeros ópticos puros de los mismos. Debe aclararse que la presente invención no se limita a los ejemplos que se indican a continuación. La proporción de componentes en los ejemplos se expresa en peso, a menos que se indique específicamente lo contrario.
Preparación de compuestos de Ejemplo:
Ejemplo 1. Bromuro de (2S,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxi propil)-1-azabiciclo[2,2,2]octano
Etapa 1: Preparación de 1-fenil-1-ciclopentiloxirano
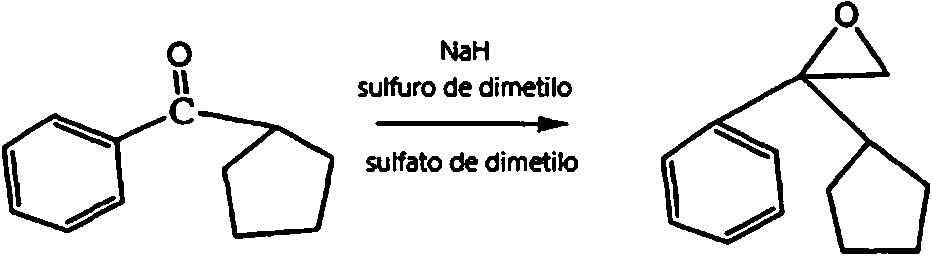
1-fenilo-1-ciclopentiloxirano se obtuvo por reacción de ciclopentilfenilcetona comercialmente disponible como material de partida según la bibliografía(1).
Etapa 2: Preparación de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali a partir de S-3-quinuclidinol

A 18,72 g (147 mmol) de S-3-quinuclidinol disponible comercialmente se añadieron 190 ml de DMSO, seguido de la adición de 7,56 g (190 mmol) de hidruro de sodio, y la mezcla se hizo reaccionar a 20-60 °C durante 0,5-12 h, y después se enfrió a temperatura ambiente, y a ello se añadió una solución de 35,72 g (190 mmol) de 1 -fenil-1 -ciclopentiloxirano (autopreparado) en 45 ml de DMSO, y una vez finalizada la adición, la mezcla resultante se calentó a 20-70 °C en baño de aceite para reaccionar durante 0,5-12 horas. Bajo la condición de baño de hielo, se añadieron 120 ml de agua helada a una temperatura interna de 30 °C o menor. La mezcla de reacción se extrajo con éter isopropílico, 100 ml x 3; y la capa de éter se combinó y se lavó con una solución acuosa saturada de NaCl, 100 ml x 3. La capa orgánica se secó sobre sulfato de sodio anhidro durante la noche, el agente desecante se retiró por filtración, y el disolvente se retiró a presión reducida, para obtener 55,7 g de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali como materia aceitosa de color rojo, con un rendimiento de 97,39 %. El producto obtenido fue el de las configuraciones (2R,3S),(2S,3S) de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali.
Etapa 3: Purificación de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali por cromatografía de columna y el tratamiento de purificación relacionado
La bibliografía(3) (3. Bingdahl B, Resul B y Dahlbom R. Facile preparation of the enantiomers of 3-acetoxiquinuclidine and 3-quinuclidinol. Acta Pharm Suec, 1979; 16: 281-283), y otra bibliografía(4-5) (4. Jianhua Gao, Guangling Wen, Qikai Zhang, Synthesis and separation of optically pure hydroxyl ether compounds. Acta Pharmaceutica Sinica, 1987; 22(9): 708-710; 5. Xiangyu Han. Study on the stereoselective synthesis of chiral M receptor antagonists. Informes de Postdoctoral Research Station en la Academy of Military Medical Sciences, pág. 39-40, 2005, Pekín (Medical Library of Chinese PLA: R914, 20050537)) informaron que la configuración quiral R o S de 3-quinuclidinol como materia prima se hizo reaccionar con racemato de 2-fenil-2-ciclopentiloxirano o enantiómeros R o S del mismo, y se podían obtener cuatro isómeros ópticos por cromatografía de columna. La bibliografía anterior ha determinado sus configuraciones absolutas, y según la nomenclatura, el átomo de carbono quiral en la quinuclidina de la molécula se designa como "3", y el carbono quiral ligado a arilo se designa como "2". La Tabla 1 son datos de rotación específica de cuatro tipos de isómeros ópticos quirales puros de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali.
Tabla 1. Datos de configuración absoluta y rotación específica del compuesto

Condiciones de medición de rotación específica: la temperatura es de 26 °C, el disolvente es metanol, y la concentración de medición es 0,4 %-1 %.
La muestra de la etapa 2 anterior se separó en columna de gel de sílice, utilizando diclorometano o triclorometano y metanol, amoniacados, como fase móvil y una placa de TLC para controlar la pureza de la muestra. Bajo el sistema de elución anterior, una muestra de una mezcla de ambas configuraciones (2R,3S) y (2S,3S) de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali podía ser purificada en dos tipos, (2R,3S) y (2S,3S), libre de álcali dependiendo de la secuencia de elución, obteniéndose de ese modo 23,5 g de (2R,3S)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 86,46 %, y 21,1 g de (2S,3S)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 77,63 %. El valor medido [a]D26 de la rotación específica de (2R,3S) fue de 43,95, y el valor medido de la rotación específica [a]D26 de (2S,3S) fue de -9,33.
La síntesis e identificación de otras bases de isómeros ópticos puros fueron conforme a los procedimientos de síntesis y separación de los anteriores isómeros ópticos puros de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali, y una vez que la base fue cuaternizada, se obtendría el compuesto de título, y el procedimiento de preparación de varios compuestos del título se aclararía, a continuación, en detalle mediante los ejemplos específicos de la síntesis de los compuestos específicos a continuación. Además, cuando se utilizó quinuclidinol racémico como material de partida, la base obtenida fue una mezcla de cuatro tipos de isómeros ópticos antes de la separación por cromatografía de columna, y el compuesto final del título fue también una mezcla de cuatro tipos de isómeros ópticos. Todos estos isómeros ópticos puros y mezclas de isómeros ópticos en diferentes proporciones están dentro del alcance de la presente invención.
Etapa 4: Preparación de 3-bromopropoxibenceno

A un matraz de 150 ml, con tres bocas, se añadieron 9,507 g (101 mmol) de fenol, seguido de la adición de 4,253 g (106 mmol) de hidróxido de sodio y una solución de 52,17 g (258 mmol) de 1,3-dibromopropano en 30 ml de etanol absoluto, y la mezcla resultante se hizo reaccionar después bajo calentamiento y reflujo en baño de aceite, precipitándose un sólido blanco, hasta reacción esencialmente completa de fenol controlada por TLC (condiciones de TLC: éter de petróleo/acetato de etilo = 5,0 ml/1,0 ml). Una vez finalizada la reacción, el sólido se retiró por filtración, y el disolvente se retiró del filtrado a 50 °C o menor bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa que contenía un sólido blanco, al que se añadió éter de petróleo y después se dejó reposar durante la noche. El sólido precipitado se retiró por filtración, y el disolvente se retiró del filtrado a 25-40 °C bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia
aceitosa de color amarillo. La materia aceitosa fue destilada bajo presión reducida, para recoger una fracción a 121 123 °C/8 mmHg, obteniéndose de ese modo 12,786 g de una materia aceitosa incolora, transparente, con un rendimiento de 58,9 % y pureza de 95,60 % detectada por CG.
RMN de 1H (CDCls) (ppm): 57,17-6,77 (m, 5H), 63,96 (t, 2H), 63,32 (t, 2H), 62,21 (m, 2H).
Etapa 5: Bromuro de (2R,3S)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,871 g (9,1 mmol) de la configuración (2R,3S) de la base preparada en la etapa 3 y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,034 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlada por TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción por evaporación rotativa a 25-40 °C bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 3,886 g de (2R,3S)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 80,5 %. El valor medido de la rotación específica [a]ü 26 de (2R,3S) fue de 53,56.
El compuesto preparado en el Ejemplo 1 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes.
Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen, además, una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) (ppm): 57,20-6,65 (m, 10H), 64,15-3,66 (m, 5H), 53,43-3,12 (m, 8H), 52,13 (m, 2H), 2,01 (m, 1H), 51,81-1,40 (m, 13H).
Ejemplo 2. Bromuro de (2S,3S)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1 -(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano
Las etapas 1, 2, 3 y 4 eran las mismas que las etapas 1, 2, 3 y 4 en el Ejemplo 1.
Etapa 5: Bromuro de (2S,3S)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,872 g (9,1 mmol) de la configuración (2R,3S) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,032 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlada por TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción por evaporación rotativa a 25-40 °C bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 3,685 g de bromuro de (2S,3S)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 75,96 %. El valor medido de la rotación específica [a]ü26 de (2S,3S) fue 31,71.
El compuesto preparado en el Ejemplo 2 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes. Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) (ppm): 57,22-6,66 (m, 10H), 64,17-3,67 (m, 5H), 53,45-3,14 (m, 8H), 52,15 (m, 2H), 2,03 (m, 1H), 51,83-1,41 (m, 13H).
Ejemplo 3. Bromuro de (2S,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano
Etapa 1: igual que la etapa 1 del Ejemplo 1
Etapa 2: Preparación de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali a partir de R-3-quinuclidinol

A 18,721 g (147 mmol) de R-3-quinuclidinol disponible comercialmente se añadieron 190 ml de DMSO, seguido de la adición de 7,558 g (190 mmol) de hidruro de sodio, y la mezcla se hizo reaccionar a 20-60 °C durante 0,5-12 h, y después se enfrió a temperatura ambiente, y a ello se añadió una solución de 35,75 g (190 mmol) de 1 -fenil-1-ciclopentiloxirano (autopreparado) en 45 ml de DMSO, y una vez finalizada la adición, la mezcla resultante se calentó a 20-70 °C en baño de aceite para reaccionar durante 0,5-12 horas. Bajo la condición de baño de hielo, se añadieron 120 ml de agua helada a una temperatura interna de 30 °C o menor. La mezcla de reacción se extrajo con éter isopropílico, 100 ml x 3; y la capa de éter se combinó y se lavó con una solución acuosa saturada de NaCl, 100 ml x 3. La capa orgánica se secó sobre sulfato de sodio anhidro durante la noche, el agente desecante se retiró por filtración, y el disolvente se retiró bajo presión reducida, para obtener 54,67 g de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali como materia aceitosa de color rojo, con un rendimiento de 95,6 %. El producto obtenido fue (2R,3R),(2S,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali.
Etapa 3: Purificación de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali por cromatografía de columna y el tratamiento de purificación relacionado
La muestra de la etapa 2 anterior se separó en columna de gel de sílice, utilizando diclorometano o triclorometano y metanol, amoniacados, como fase móvil y una placa de TLC para controlar la pureza de la muestra. Bajo el sistema de elución anterior, una muestra de una mezcla de ambas configuraciones (2R,3R) y (2S,3R) de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali podía ser purificada en dos tipos de (2R,3R) y (2S,3R), libre de álcali dependiendo de la secuencia de elución, obteniéndose de ese modo 21,5 g de la configuración (2R,3R) de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 79,1 % y el valor medido de la rotación específica [a]ü26 de 9,01, y 21,3 g de la configuración (2S,3R) de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 78,37 % y el valor medido de la rotación específica [a]ü26 de -44,20.
Etapa 4: igual que la etapa 4 en el Ejemplo 1
Etapa 5: Bromuro de (2S,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,871 g (9,1 mmol) de la configuración (2S,3R) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,0340 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción por evaporación rotativa a 25-40 °C bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 3,86 g de la configuración (2S,3R) de bromuro de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 79,95 %. El valor medido de la rotación específica [a]ü 26 fue de -58,16.
El compuesto preparado en el Ejemplo 3 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes. Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN 1H (D2O) (ppm): 57,21-6,86 (m, 10H), 54,15-3,66 (m, 5H), 63,43-3,13 (m, 8H), 52,16 (m, 2H), 2,04 (m, 1H), 6 1,79-1,23 (m, 13H).
Ejemplo 4. Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1 -(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano
Etapa 1: igual que la etapa 1 en el Ejemplo 1
Etapas 2 y 3: igual que las etapas 2 y 3 en el Ejemplo 3
Etapa 4: igual que la etapa 4 en el Ejemplo 1
Etapa 5: Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,87 g (9,1 mmol) de la configuración (2R,3R) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,034 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción por evaporación rotativa a 25-40 °C bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 3,685 g de la configuración (2R,3R) de bromuro de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 76,34 %. El valor medido de la rotación específica [a]D26 fue de -31,18.
El compuesto preparado en el Ejemplo 4 se hizo reaccionar con Ag2O para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes.
Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) (ppm): 57,22-6,87 (m, 10H), 54,17-3,65 (m, 5H), 53,45-3,15 (m, 8H), 52,17 (m, 2H), 2,05 (m, 1H), 51,80-1,26 (m, 13H).
Ejemplo 5. Bromuro de (2R,3S),(2S,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1 -(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano
Etapa 1: igual que la etapa 1 en el Ejemplo 1
Etapa 2: Preparación de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali a partir de quinuclidinol racémico

A 18,72 g (147 mmol) de quinuclidinol racémico disponible comercialmente se añadieron 190 ml de DMSO, seguido de la adición de 7,59 g (190 mmol) de hidruro de sodio, y la mezcla se hizo reaccionar a 20-60 °C durante 0,5-12 h, y después se enfrió a temperatura ambiente, y a ello se añadió una solución de 35,68 g (190 mmol) de 1 -fenil-1-ciclopentiloxirano (autopreparado) en 45 ml de DMSO, y una vez finalizada la adición, la mezcla resultante se calentó a 20-70 °C en baño de aceite para reaccionar durante 0,5-12 horas. Bajo la condición de baño de hielo, 120 ml de agua helada se añadieron a una temperatura interna de 30 °C o menor. La mezcla de reacción se extrajo con éter isopropílico, 100 ml x 3; y la capa de éter se combinó y se lavó con una solución acuosa saturada de NaCl, 100 ml x 3. La capa orgánica se secó sobre sulfato de sodio anhidro durante la noche, el agente desecante se retiró por filtración, y el disolvente se retiró bajo presión reducida, para obtener 51,99 g de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali como materia aceitosa de color rojo, con un rendimiento de 90,92 %. El producto obtenido fue (2R,2S),(2R,3R),(2S,3R),(2S,3S)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali.
Etapa 3: Purificación de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali por cromatografía de columna y el tratamiento de purificación relacionado
La muestra de la etapa 2 anterior se separó en columna de gel de sílice, utilizando diclorometano o triclorometano y metanol, amoniacados, como fase móvil y una placa de TLC para controlar la pureza de la muestra. Bajo el sistema de elución anterior, una muestra de una mezcla de cuatro configuraciones de (2R,3S),(2R,3R),(2S,3R),(2S,3S) de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali podía ser purificada en dos tipos
racémicos, (2R,3S),(2S,3R) y (2R,3R),(2S,3S), libres de álcali, dependiendo de la secuencia de elución, en donde la fracción eluida en primer lugar de la columna eran 22,05 g de configuraciones racémicas (2R,3S),(2S,3R) de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali, con un rendimiento de 81,12 %; y la fracción eluida en segundo lugar de la columna eran 21,29 g de configuraciones (2R,3R),(2S,3S) de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali, con un rendimiento de 78,33 %.
Etapa 4: igual que la etapa 4 en el Ejemplo 1
Etapa 5: Configuración (2R,3S),(2S,3R) de bromuro de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,871 g (9,1 mmol) de la configuración (2R,3S),(2S,3R) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,035 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción por evaporación rotativa a 25-40 °C bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 3,46 g de bromuro de (2R,3S),(2S,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 71,66 %.
El compuesto preparado en el Ejemplo 5 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes. Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,19-7,15 (m, 7H), 6,82-6,77 (m, 3H), 3,94-3,45 (m, 5H), 3,24-3,12 (m, 8H), 2,15 (m, 2H), 1,96-1,79 (m, 2H), 1,57-1,25 (m, 12H).
Ejemplo 6. Configuración (2R,3R),(2S,3S) de bromuro de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano
Etapa 1: igual que la etapa 1 en el Ejemplo 1
Etapas 2 y 3: igual que las etapas 2 y 3 en el Ejemplo 5
Etapa 4: igual que la etapa 4 en el Ejemplo 1
Etapa 5: Configuración (2R,3R),(2S,3S) de bromuro de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,87 g (9,1 mmol) de la configuración (2R,3R),(2S,3S) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,034 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción por evaporación rotativa a 25-40 °C bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 3,58 g de la configuración (2R,3R),(2S,3S) de bromuro de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 74,17 %.
Los isómeros ópticos puros de cuatro configuraciones de (2R,3S),(2R,3R),(2S,3R),(2S,3S) de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali podían obtenerse de los Ejemplos 1 y 3, y las mezclas de las configuraciones (2R,3S),(2R,3R),(2S,3R),(2S,3S) de bromuro de 3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano en diferentes proporciones se obtuvieron mezclando estos isómeros en cualquier cantidad y cualquier proporción seguida de cuaternización añadiendo 3-bromopropoxibenceno.
El compuesto preparado en el Ejemplo 6 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podría reaccionar con otros ácidos para transformarlos en las sales correspondientes. Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,18-7,16 (m, 7H), 6,81-6,75 (m, 3H), 3,94-3,45 (m, 5H), 3,25-3,11 (m, 8H), 2,13 (m, 2H), 1,97-1,78 (m, 2H), 1,56-1,24 (m, 12H).
Ejemplo 7. Bromuro de (2S,3R)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1 -(3-fenoxi propil)-1 -azabiciclo[2,2,2]octano
Etapa 1: Preparación de 1-fenil-1-ciclohexiloxirano
Ciclohexilfenilcetona comercialmente disponible como material de partida se hizo reaccionar para obtener 1 -fenil-1 -ciclohexiloxirano según la bibliografía(1).
A un matraz de 3 litros, con tres bocas, se añadieron 1.880 ml de acetonitrilo, seguido de la adición de sulfuro de dimetilo y sulfato de dimetilo, con un leve desprendimiento de calor, y la mezcla resultante se agitó entonces a temperatura ambiente durante 1,5 h y después se dejó reposar durante la noche. A la solución de reacción se añadió NaH (60 %) en porciones adecuadas agitando durante 30-40 min, generándose un gas (préstese atención a tener el matraz externamente conectado a un condensador Allihn y un tubo de secado con cloruro de calcio anhidro), y después se agitó a temperatura ambiente durante 1 hora, seguido por la adición gota a gota de 332,26 g de ciclohexilfenilcetona durante aproximadamente 10 min, y la mezcla resultante se hizo reaccionar posteriormente en baño de aceite a 40-45 °C durante 90 min después de ser agitada a temperatura ambiente durante 20 min, y después se enfrió a temperatura ambiente y se dejó reposar durante la noche. La solución de reacción se transfirió a un matraz con forma de berenjena, el disolvente se retiró por succión usando una bomba de agua (40-42 °C, -0,095 MPa), y el sólido restante se enfrió entonces a 0-5 °C en baño de hielo, seguido de la adición gota a gota de 2.630 ml de agua helada (siendo la temperatura interior controlada a 0-5 °C) durante aproximadamente 1 hora, y la solución de reacción se extrajo con éter isopropílico (650 ml x 3), la capa de éter se combinó y se lavó con agua hasta que es neutra (siendo el pH de los lavados con agua de 7,0), y la capa de éter se secó sobre sulfato de sodio anhidro durante la noche. El agente desecante se retiró por filtración, el disolvente se retiró bajo presión reducida por succión usando una bomba de agua (40-42 °C, -0,095 MPa), y posteriormente se recogieron 291,13 g de fracción a 117-127 °C/3 mmHg bajo presión reducida usando una bomba de aceite.
RMN de 1H (CDCls) 5 (ppm): 7,18-7,10 (m, 5H), 2,91-2,66 (m, 2H), 2,13-1,27 (m, 11H).
Etapa 2: Preparación de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali a partir de S-3-quinuclidinol

A 18,72 g (147 mmol) de S-3-quinuclidinol disponible comercialmente se añadieron 190 ml de DMSO, seguido de la adición de 7,59 g (190 mmol) de hidruro de sodio, y la mezcla se hizo reaccionar a 20-60 °C durante 0,5-12 h, y después se enfrió a temperatura ambiente, y a ello se añadió una solución de 38,38 g (190 mmol) de 1 -fenil-1 -ciclohexiloxirano (autopreparado) en 45 ml de DMSO, y una vez finalizada la adición, la mezcla resultante se calentó a 20-70 °C en baño de aceite para reaccionar durante 0,5-12 horas. Bajo la condición de baño de hielo, se añadieron 120 ml de agua helada a una temperatura interna de 30 °C o menor. La mezcla de reacción se extrajo con éter isopropílico, 100 ml x 3; y la capa de éter se combinó y se lavó con una solución acuosa saturada de NaCl, 100 ml x 3. La capa orgánica se secó sobre sulfato de sodio anhidro durante la noche, el agente desecante se retiró por filtración, y el disolvente se retiró para obtener 47,102 g de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali como una materia aceitosa de color rojo, con un rendimiento de 97,39 %. El producto obtenido fue (2R,3S),(2S,3S)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali.
Etapa 3: Purificación de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali por cromatografía de columna y el tratamiento de purificación relacionado
La muestra de la etapa 2 anterior se separó en columna de gel de sílice, utilizando diclorometano o triclorometano y metanol, amoniacados, como fase móvil y una placa de TLC para controlar la pureza de la muestra. Bajo el sistema de elución anterior, una muestra de una mezcla de ambas configuraciones (2R,3S) y (2S,3S) de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali podía ser purificado en dos tipos (2R,3S) y (2S,3S) libres de álcali, dependiendo de la secuencia de elución, obteniéndose de ese modo 20,282 g de la configuración (2R,3S) de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 86,12 %, y 20,63 g de la configuración (2S,3S) de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 87,6 %.
Etapa 4: igual que la etapa 4 en el Ejemplo 1
Etapa 5: Bromuro de (2R,3S)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,994 g (9,1 mmol) de la configuración (2R,3S) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,034 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción por evaporación rotativa a 25-40 °C bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 4,050 g de bromuro de (2R,3S)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 81,82 %.
El compuesto preparado en el Ejemplo 7 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes. Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,16-7,14 (m, 7H), 6,80-6,77 (m, 3H), 3,93-3,42 (m, 5H), 3,24-3,1 9 (m, 8H), 2,14 (m, 2H), 1,89-1,79 (m, 2H), 1,57-1,27 (m, 14H).
Ejemplo 8. Bromuro de (2S,3S)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1 -(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano
Las etapas 1, 2, 3 y 4 eran iguales que las etapas 1, 2, 3 y 4 en el Ejemplo 7
La etapa 4 era igual que la etapa 4 en el Ejemplo 1
Etapa 5: Bromuro de (2S,3S)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,993 g (9,1 mmol) de la configuración (2S,3S) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,035 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción por evaporación rotativa a 25-40 °C bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 4,115 g de bromuro de (2S,3S)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 83,13 %.
El compuesto preparado en el Ejemplo 8 se hizo reaccionar con Ag2O para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes.
Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,15-7,14 (m, 7H), 6,82-6,76 (m, 3H), 3,94-3,41 (m, 5H), 3,23-3,18 (m, 8H), 2,14 (m, 2H), 1,89-1,79 (m, 2H), 1,58-1,27 (m, 14H).
Ejemplo 9. Bromuro de (2R,3R)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1 -(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano
Etapa 1: igual que la etapa 1 en el Ejemplo 7
Etapa 2: Preparación de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali a partir de R-3-quinuclidinol

A 18,72 g (147 mmol) de R-3-quinuclidinol disponible comercialmente se añadieron 190 ml de DMSO, seguido de la adición de 7,591 g (190 mmol) de hidruro de sodio, y la mezcla se hizo reaccionar a 20-60 °C durante 0,5-12 h, y después se enfrió a temperatura ambiente, y a ello se añadió una solución de 38,38 g (190 mmol) de 1 -fenil-1 -ciclohexiloxirano (autopreparado) en 45 ml de DMSO, y una vez finalizada la adición, la mezcla resultante se calentó a 20-70 °C en baño de aceite para reaccionar durante 0,5-12 horas. Bajo la condición de baño de hielo, se añadieron 120 ml de agua helada a una temperatura interna de 30 °C o menor. La mezcla de reacción se extrajo con éter isopropílico, 100 ml x 3; y la capa de éter se combinó y se lavó con una solución acuosa saturada de NaCl, 100 ml x 3. La capa orgánica se secó sobre sulfato de sodio anhidro durante la noche, el agente desecante se retiró por filtración, y el disolvente se retiró por evaporación rotativa, para obtener 45,89 g de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo [2,2,2]octano libre de álcali como materia aceitosa de color rojo, con un rendimiento de 96,19 %. El producto obtenido fue la configuración (2R,3R),(2S,3R) de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1 -azabiciclo[2,2,2]octano libre de álcali.
Etapa 3: Purificación de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali por cromatografía de columna y el tratamiento de purificación relacionado
La muestra de la etapa 2 anterior se separó en columna de gel de sílice, utilizando diclorometano o triclorometano y metanol, amoniacados, como fase móvil y una placa de TLC para controlar la pureza de la muestra. Bajo el sistema de elución anterior, una muestra de una mezcla de ambas configuraciones (2R,3R) y (2S,3R) de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali podía ser purificada en dos tipos de (2R,3R) y (2S,3R) libre de álcali, dependiendo de la secuencia de elución, obteniéndose de ese modo 19,393 g de (2R,3R)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 84,52 %, y 18,186 g de (2S,3R)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 79,26 %.
Etapa 4: igual que la etapa 4 en el Ejemplo 1
Etapa 5: Configuración (2S,3R) de bromuro de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,993 g (9,1 mmol) de la configuración (2S,3R) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,0340 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción por evaporación rotativa a 25-40 °C bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 4,11 g de bromuro de (2S,3R)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 83,03 %.
El compuesto preparado en el Ejemplo 9 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes. Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,16-7,14 (m, 7H), 6,80-6,77 (m, 3H), 3,93-3,42 (m, 5H), 3,24-3,19 (m, 8H), 2,14 (m, 2H), 1,89-1,79 (m, 2H), 1,57-1,27 (m, 14H).
Ejemplo 10. Bromuro de (2R,3R)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1 -(3-fenoxi propil)-1 -azabiciclo[2,2,2]octano
Etapa 1: igual que la etapa 1 en el Ejemplo 7
Las etapas 2 y 3 eran iguales a las etapas 2 y 3 en el Ejemplo 9
Etapa 4: igual que la etapa 4 en el Ejemplo 1
Etapa 5: Bromuro de (2R,3R)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,995 g (9,1 mmol) de la configuración (2R,3R) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,034 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25-40 °C bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 4,205 g de bromuro de (2R,3R)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 84,95 %.
El compuesto preparado en el Ejemplo 10 se hizo reaccionar con Ag2O para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes. Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico,
como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,16-7,14 (m, 7H), 6,81-6,77 (m, 3H), 3,93-3,42 (m, 5H), 3,24-3,19 (m, 8H), 2,14 (m, 2H), 1,89-1,78 (m, 2H), 1,57-1,27 (m, 14H).
Ejemplo 11. Configuración (2R,3S),(2S,3R) de bromuro de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano
Etapa 1: igual que la etapa 1 en el Ejemplo 7
Etapa 2: Preparación de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali a partir de quinuclidinol racémico

A 18,72 g (147 mmol) de quinuclidinol racémico disponible comercialmente se añadieron 190 ml de DMSO, seguido de la adición de 7,59 g (190 mmol) de hidruro de sodio, y la mezcla se hizo reaccionar a 20-60 °C durante 0,5-12 h, y después se enfrió a temperatura ambiente, y a ello se añadió una solución de 38,42 g (190 mmol) de 1 -fenil-1 -ciclohexiloxirano (autopreparado) en 45 ml de DMSO, y una vez finalizada la adición, la mezcla resultante se calentó a 20-70 °C en baño de aceite para reaccionar durante 0,5-12 horas. Bajo la condición de baño de hielo, se añadieron 120 ml de agua helada a una temperatura interna de 30 °C o menor. La mezcla de reacción se extrajo con éter isopropílico, 100 ml x 3; y la capa de éter se combinó y se lavó con una solución acuosa saturada de NaCl, 100 ml x 3. La capa orgánica se secó sobre sulfato de sodio anhidro durante la noche, el agente desecante se retiró por filtración, y el disolvente se retiró bajo presión reducida, para obtener 43,58 g de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo [2,2,2]octano libre de álcali como materia aceitosa de color rojo, con un rendimiento de 90,12 %. El producto obtenido fue la configuración (2R,3S),(2R,3R),(2S,3R),(2S,3S) de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali.
Etapa 3: Purificación de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali por cromatografía de columna y el tratamiento de purificación relacionado
La muestra de la etapa 2 anterior se separó en columna de gel de sílice, utilizando diclorometano o triclorometano y metanol, amoniacados, como fase móvil y una placa de TLC para controlar la pureza de la muestra. Bajo el sistema de elución anterior, una muestra de una mezcla de cuatro configuraciones de (2R,3S),(2R,3R),(2S,3R),(2S,3S) de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali podía ser purificada en dos tipos (2R,3S),(2S,3R) y (2R,3R),(2S,3S) racémicos, libres de álcali, dependiendo de la secuencia de elución, obteniéndose de ese modo la fracción eluida en primer lugar de 18,06 g de la configuración (2R,3S),(2S,3R) racémica de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 82,88 %, y la fracción eluida en segundo lugar de 17,29 g de la configuración (2R,3R),(2S,3S) de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 79,35 %.
Etapa 4: igual que la etapa 4 en el Ejemplo 1
Etapa 5: Configuración (2R,3S),(2S,3R) de bromuro de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,993 g (9,1 mmol) de la configuración (2R,3S),(2S,3R) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,034 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25 40 °C por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 4,026 g de la configuración (2R,3S),(2S,3R) de bromuro de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 81,36 %.
El compuesto preparado en el Ejemplo 11 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes. Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,19-7,15 (m, 7H), 6,82-6,78 (m, 3H), 3,93-3,42 (m, 5H), 3,24-3,19 (m, 8H), 2,14 (m, 2H), 1,88-1,79 (m, 2H), 1,58-1,26 (m, 14H).
Ejemplo 12. Bromuro de (2R,3R),(2S,3S)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano
Etapa 1: igual que la etapa 1 en el Ejemplo 7
Etapas 2 y 3: igual que las etapas 2 y 3 en el Ejemplo 11
Etapa 4: igual que la etapa 4 en el Ejemplo 1
Etapa 5: Configuración (2R,3R),(2S,3S) de bromuro de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,994 g (9,1 mmol) de la configuración (2R,3R),(2S,3S) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,034 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25 40 °C bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 4,125 g de bromuro de (2R,3R),(2S,3S)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 83,36 %.
Los isómeros ópticos puros de cuatro configuraciones de (2R,3S),(2R,3R),(2S,3R),(2S,3S) de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali podían obtenerse a partir de los Ejemplos 7 y 9, y las mezclas de las configuraciones (2R,3S),(2R,3R),(2S,3R),(2S,3S) de bromuro de 3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano en diferentes proporciones se obtuvieron mezclando estos isómeros en cualquier cantidad y cualquier proporción seguido por cuaternización añadiendo 3-bromopropoxibenceno.
El compuesto preparado en el Ejemplo 12 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes. Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,16-7,14 (m, 7H), 6,80-6,77 (m, 3H), 3,93-3,42 (m, 5H), 3,24-3,19 (m, 8H), 2,14 (m, 2H), 1,89-1,79 (m, 2H), 1,57-1,27 (m, 14H).
Ejemplo 13. Bromuro de (2R,3S)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1 -(3-fenoxi propil)-1 -azabiciclo[2,2,2]octano
Etapa 1: Preparación de 1-fenil-1-ciclobutiloxirano

Se hizo reaccionar ciclobutilfenilcetona comercialmente disponible como material de partida para obtener 1 -fenil-1 -ciclobutiloxirano según la bibliografía(1).
A un matraz de 3 litros, con tres bocas, se añadieron 1.880 ml de acetonitrilo, seguido de la adición de sulfuro de dimetilo y sulfato de dimetilo, con un leve desprendimiento de calor, y la mezcla resultante se agitó entonces a
temperatura ambiente durante 1,5 h y después se dejó reposar durante la noche. Se añadió NaH (60 %) en porciones adecuadas en la solución de reacción con agitación durante 30-40 min, generándose un gas (préstese atención a tener el matraz externamente conectado a un condensador Allihn y un tubo de secado de cloruro de calcio anhidro), y después se agitó a temperatura ambiente durante 1 hora, seguido de la adición gota a gota de 315,6 g de ciclobutilfenilcetona durante aproximadamente 10 min, y la mezcla resultante se hizo reaccionar posteriormente en baño de aceite a 40-45 °C durante 90 min, siendo después agitada a temperatura ambiente durante 20 min, y después se enfrió a temperatura ambiente y se dejó reposar durante la noche. La solución de reacción se transfirió a un matraz con forma de berenjena, el disolvente se retiró por succión usando una bomba de agua (40-42 °C, -0,095 MPa), y el sólido restante se enfrió a 0-5 °C en baño de hielo, seguido de la adición gota a gota de 2.630 ml de agua helada (siendo la temperatura interior controlada a 0-5 °C) durante aproximadamente 1 hora, y la solución de reacción se extrajo con éter isopropílico (650 ml x 3), la capa de éter se combinó y se lavó con agua hasta que es neutra (siendo el pH de los lavados con agua de 7,0), y la capa de éter se secó sobre sulfato de sodio anhidro durante la noche. El agente desecante se retiró por filtración, el disolvente se retiró bajo presión reducida por succión usando una bomba de agua (40-42 °C, -0,095 MPa), y posteriormente se recogieron 282,7 g de fracción a 113-126 °C/3 mmHg bajo presión reducida usando una bomba de aceite, con un rendimiento de 82,37 %.
RMN de 1H (CDCls) 5 (ppm): 7,19 (m, 5H), 2,92-2,65 (m, 2H), 2,61 (m, 1H), 2,03-1,77 (m, 6H).
Etapa 2: Preparación de 3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali a partir de S-3-quinuclidinol

A 18,72 g (147 mmol) de S-3-quinuclidinol disponible comercialmente se añadieron 190 ml de DMSO, seguido de la adición de 7,59 g (190 mmol) de hidruro de sodio, y la mezcla se hizo reaccionar a 20-60 °C durante 0,5-12 h, y después se enfrió a temperatura ambiente, y a ello se añadió una solución de 33,054 g (190 mmol) de 1 -fenil-1-ciclobutiloxirano (autopreparado) en 45 ml de DMSO, y una vez finalizada la adición, la mezcla resultante se calentó a 20-70 °C en baño de aceite para reaccionar durante 0,5-12 horas. Bajo la condición de baño de hielo, se añadieron 120 ml de agua helada a una temperatura interna de 30 °C o menor. La mezcla de reacción se extrajo con éter isopropílico, 100 ml x 3; y la capa de éter se combinó y se lavó con una solución acuosa saturada de NaCl, 100 ml x 3. La capa orgánica se secó sobre sulfato de sodio anhidro durante la noche, el agente desecante se retiró por filtración, y el disolvente se retiró bajo presión reducida para obtener 43,473 g de 3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali como materia aceitosa de color rojo, con un rendimiento de 98,25 %. El producto obtenido fue bromuro de (2R,3S),(2S,3S)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali.
Etapa 3: Purificación de 3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali por cromatografía de columna y el tratamiento de purificación relacionado
La muestra de la etapa 2 anterior se separó en columna de gel de sílice, utilizando diclorometano o triclorometano y metanol, amoniacados, como fase móvil y una placa de TLC para controlar la pureza de la muestra. Bajo el sistema de elución anterior, una muestra de una mezcla de ambas configuraciones (2r ,3S) y (2S,3S) de 3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali podía ser purificada en dos tipos de (2R,3S) y (2S,3S) libre de álcali, dependiendo de la secuencia de elución, obteniéndose de ese modo 17,5548 g de (2R,3S)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 80,42 %, y 17,31 g de bromuro de (2S,3S)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 79,62 %.
Etapa 4: igual que la etapa 4 en el Ejemplo 1
Etapa 5: Bromuro de (2R,3S)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,739 g (9,1 mmol) de la configuración (2R,3S) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,034 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25-40 °C bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 3,828 g de la configuración (2R,3S) de 3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1 -(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 81,52 %.
El compuesto preparado en el Ejemplo 13 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes.
Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,19-7,15 (m, 7H), 6,82-6,77 (m, 3H), 3,94-3,68 (m, 5H), 3,49-3,19 (m, 9H), 2,15 (m, 2H), 2,0-1,79 (m, 11H).
Ejemplo 14. Bromuro de (2S,3S)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano
Las etapas 1, 2 y 3 eran iguales que las etapas 1, 2 y 3 en el Ejemplo 13
La etapa 4 era igual que la etapa 4 en el Ejemplo 1
Etapa 5: Bromuro de (2S,3S)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,739 g (9,1 mmol) de la configuración (2S,3S) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,034 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25-40 °C bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 3,726 g de bromuro de (2S,3S)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 79,34 %.
El compuesto preparado en el Ejemplo 14 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes.
Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado,
sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,19-7,15 (m, 7H), 6,82-6,77 (m, 3H), 3,94-3,68 (m, 5H), 3,49-3,19 (m, 9H), 2,14 (m, 2H), 2,0-1,79 (m, 11H).
Ejemplo 15. Bromuro de (2S,3R)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1 -(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano
La etapa 1 era igual que la etapa 1 en el Ejemplo 13
Etapa 2: Preparación de 3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali a partir de R-3-quinuclidinol

A 18,721 g (147 mmol) de R-3-quinuclidinol disponible comercialmente se añadieron 190 ml de DMSO, seguido de la adición de 7,591 g (190 mmol) de hidruro de sodio, y la mezcla se hizo reaccionar a 20-60 °C durante 0,5-12 h, y después se enfrió a temperatura ambiente, y a ello se añadió una solución de 33,054 g (190 mmol) de 1 -fenil-1-ciclobutiloxirano (autopreparado) en 45 ml de DMSO, y una vez finalizada la adición, la mezcla resultante se calentó a 20-70 °C en baño de aceite para reaccionar durante 0,5-12 horas. Bajo la condición de baño de hielo, se añadieron 120 ml de agua helada a una temperatura interna de 30 °C o menor. La mezcla de reacción se extrajo con éter isopropílico, 100 ml x 3; y la capa de éter se combinó y se lavó con una solución acuosa saturada de NaCl, 100 ml x 3. La capa orgánica se secó sobre sulfato de sodio anhidro durante la noche, el agente desecante se retiró por filtración, y el disolvente se retiró bajo presión reducida para obtener 45,91 g de 3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali como materia aceitosa de color rojo, con un rendimiento de 97,77 %. El producto obtenido fue la configuración (2R,3R),(2S,3R) de 3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali.
Etapa 3: Purificación de 3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali por cromatografía de columna y el tratamiento de purificación relacionado
La muestra de la etapa 2 anterior se separó en columna de gel de sílice, utilizando diclorometano o triclorometano y metanol, amoniacados, como fase móvil y una placa de TLC para controlar la pureza de la muestra. Bajo el sistema de elución anterior, una muestra de una mezcla de ambas configuraciones (2r ,3R) y (2S,3R) de 3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali podía ser purificada en dos tipos de (2R,3R) y (2S,3R) libres de álcali, dependiendo de la secuencia de elución, obteniéndose de ese modo 18,41 g de (2R,3R)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 80,22 %, y 18,27 g de (2S,3R)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 79,59 %.
Etapa 4: igual que la etapa 4 en el Ejemplo 1
Etapa 5: Bromuro de (2S,3R)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,739 g (9,1 mmol) de la configuración (2S,3R) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,0340 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25-40 °C bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 3,790 g de (2S,3R)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 80,72 %.
El compuesto preparado en el Ejemplo 15 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes.
Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,19-7,15 (m, 7H), 6,82-6,77 (m, 3H), 3,94-3,68 (m, 5H), 3,49-3,19 (m, 9H), 2,15 (m, 2H), 2,0-1,79 (m, 11H).
Ejemplo 16. Bromuro de (2R,3R)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1 -(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano
Etapa 1: igual que la etapa 1 en el Ejemplo 13
Las etapas 2 y 3 eran iguales que las etapas 2 y 3 en el Ejemplo 15
Etapa 4: igual que la etapa 4 en el Ejemplo 1
Etapa 5: Bromuro de (2R,3R)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,739 g (9,1 mmol) de la configuración (2R,3R) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,034 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción por evaporación rotativa a 25-40 °C bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 3,805 g de la configuración (2R,3R) de 3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 81,02 %.
El compuesto preparado en el Ejemplo 16 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes. Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,19-7,14 (m, 7H), 6,81-6,77 (m, 3H), 3,94-3,68 (m, 5H), 3,49-3,19 (m, 9H), 2,15 (m, 2H), 2,0-1,77 (m, 11H).
Ejemplo 17. Bromuro de (2R,3S),(2S,3R)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1 -(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano
Etapa 1: igual que la etapa 1 en el Ejemplo 13
Etapa 2: Preparación de 3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali a partir de quinuclidinol racémico

A 18,72 g (147 mmol) de quinuclidinol racémico disponible comercialmente se añadieron 190 ml de DMSÜ, seguido de la adición de 7,59 g (190 mmol) de hidruro de sodio, y la mezcla se hizo reaccionar a 20-60 °C durante 0,5-12 h, y después se enfrió a temperatura ambiente, y a ello se añadió una solución de 33,06 g (190 mmol) de 1 -fenil-1-ciclobutiloxirano (autopreparado) en 45 ml de DMSÜ, y una vez finalizada la adición, la mezcla resultante se calentó a 20-70 °C en baño de aceite para reaccionar durante 0,5-12 horas. Bajo la condición de baño de hielo, se añadieron 120 ml de agua helada a una temperatura interna de 30 °C o menor. La mezcla de reacción se extrajo con éter isopropílico, 100 ml x 3; y la capa de éter se combinó y se lavó con una solución acuosa saturada de NaCl, 100 ml x 3. La capa orgánica se secó sobre sulfato de sodio anhidro durante la noche, el agente desecante se retiró por filtración, y el disolvente se retiró bajo presión reducida, para obtener 42,16 g de 3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo [2,2,2]octano libre de álcali como materia aceitosa de color rojo, con un rendimiento de 95,28 %. El producto obtenido fue (2R,3S),(2R,3R),(2S,3R),(2S,3S)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]-octano libre de álcali.
Etapa 3: Purificación de 3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali por cromatografía de columna y el tratamiento de purificación relacionado
La muestra de la etapa 2 anterior se separó en columna de gel de sílice, utilizando diclorometano o triclorometano y metanol, amoniacados, como fase móvil y una placa de TLC para controlar la pureza de la muestra. Bajo el sistema de elución anterior, una muestra de una mezcla de cuatro configuraciones de (2R,3S),(2R,3R),(2S,3R),(2S,3S) de 3 [(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali podía ser purificada en dos tipos
(2R,3S),(2S,3R) y (2R,3R),(2S,3S) racémicos, libres de álcali, dependiendo de la secuencia de elución, obteniéndose de ese modo la fracción eluida en primer lugar de 17,44 g de configuración racémica (2R,3S),(2S,3R) de 3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 82,72 %, y la fracción eluida en segundo lugar de 16,73 g de la configuración (2R,3R),(2S,3S) de 3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 79,36 %.
Etapa 4: igual que la etapa 4 en el Ejemplo 1
Etapa 5: Bromuro de (2R,3S),(2S,3R)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,739 g (9,1 mmol) de la configuración (2R,3S),(2S,3R) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,0342 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25-40 °C bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 3,916 g de la configuración (2R,3S),(2S,3R) de 3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]-octano como un sólido de color blancuzco, con un rendimiento de 83,38 %.
El compuesto preparado en el Ejemplo 17 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes. Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,19-7,15 (m, 7H), 6,82-6,77 (m, 3H), 3,94-3,68 (m, 5H), 3,49-3,19 (m, 9H), 2,15 (m, 2H), 2,0-1,79 (m, 11H).
Ejemplo 18. Bromuro de (2R,3R),(2S,3S)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1 -(3-fenoxipropil)-1 -azabiciclo[2,2,2]-octano
Las etapas 1, 2 y 3 eran iguales que las etapas 1, 2 y 3 en el Ejemplo 17
Etapa 4: igual que la etapa 4 en el Ejemplo 1
Etapa 5: Bromuro de (2R,3R),(2S,3S)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]-octano
A un matraz de 100 ml, con forma de berenjena, se añadieron 2,739 g (9,1 mmol) de la configuración (2R,3R),(2S,3S) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,034 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de
nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25 40 °C por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 3,918 g de la configuración (2R,3R),(2S,3S) de bromuro de 3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 83,42 %.
Los isómeros ópticos puros de cuatro configuraciones de (2R,3S),(2R,3R),(2S,3R),(2S,3S) de 3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali podían obtenerse a partir de los Ejemplos 13 y 15, y mezclas de configuraciones (2R,3S),(2R,3R),(2S,3R),(2S,3S) de bromuro de 3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano en diferentes proporciones se obtuvieron mezclando estos isómeros en cualquier cantidad y cualquier proporción seguido por cuaternización añadiendo 3-bromopropoxibenceno.
El compuesto preparado en el Ejemplo 18 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes. Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,19-7,15 (m, 7H), 6,82-6,77 (m, 3H), 3,94-3,68 (m, 5H), 3,49-3,19 (m, 9H), 2,15 (m, 2H), 2,0-1,79 (m, 11H).
Ejemplo 19. Bromuro de (2R,3S)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1 -(3-fenoxi propil)-1 -azabiciclo[2,2,2]octano
Etapa 1: Preparación de 1-fenil-1-ciclopropiloxirano

Se hizo reaccionar ciclopropilfenilcetona comercialmente disponible como material de partida para obtener 1 -fenil-1-ciclopropiloxirano según la bibliografía(1).
A un matraz de 3 litros, con tres bocas, se añadieron 1.880 ml de acetonitrilo, seguido de la adición de sulfuro de dimetilo y sulfato de dimetilo, con un leve desprendimiento de calor, y la mezcla resultante se agitó entonces a temperatura ambiente durante 1,5 h y después se dejó reposar durante la noche. Se añadió NaH (60 %) en porciones adecuadas en la solución de reacción con agitación durante 30-40 min, generándose un gas (préstese atención a tener el matraz externamente conectado a un condensador Allihn y un tubo de secado de cloruro de calcio anhidro), y después se agitó a temperatura ambiente durante 1 hora, seguido de la adición gota a gota de 300,5 g de ciclpropilfenilcetona durante aproximadamente 10 min, y la mezcla resultante se hizo reaccionar posteriormente en baño de aceite a 40-45 °C durante 90 min siendo después agitada a temperatura ambiente durante 20 min, y después se enfrió a temperatura ambiente y se dejó reposar durante la noche. La solución de reacción se transfirió a un matraz con forma de berenjena, el disolvente se retiró por succión usando una bomba de agua (40-42 °C, -0,095 MPa), y el sólido restante se enfrió a 0-5 °C en baño de hielo, seguido de la adición gota a gota de 2.630 ml de agua helada (siendo la temperatura interior controlada a 0-5 °C) durante aproximadamente 1 hora, y la solución de reacción se extrajo con éter isopropílico (650 ml * 3), la capa de éter se combinó y se lavó con agua hasta que es neutra (siendo el pH de los lavados con agua de 7,0), y la capa de éter se secó sobre sulfato de sodio anhidro durante la noche. El agente desecante se retiró por filtración, el disolvente se retiró bajo presión reducida por succión usando una bomba de agua (40-42 °C, -0,095 MPa), y posteriormente se recogieron 275,8 g de fracción a 111-124 °C/3 mmHg bajo presión reducida usando una bomba de aceite, con un rendimiento de 83,75 %.
RMN de 1H (CDCls) 5 (ppm): 7,19 (m, 5H), 2,91-2,66 (m, 2H), 0,91 (m, 1H), 0,31-0,07 (m, 4H).
Etapa 2: Preparación de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali a partir de S-3-quinuclidinol

A 18,72 g (147 mmol) de S-3-quinuclidinol disponible comercialmente se añadieron 190 ml de DMSO, seguido de la adición de 7,59 g (190 mmol) de hidruro de sodio, y la mezcla se hizo reaccionar a 20-60 °C durante 0,5-12 h, y después se enfrió a temperatura ambiente, y a ello se añadió una solución de 30,35 g (190 mmol) de 1 -fenil-1-ciclopropiloxirano (autopreparado) en 45 ml de DMSO, y una vez finalizada la adición, la mezcla resultante se calentó a 20-70 °C en baño de aceite para reaccionar durante 0,5-12 horas. Bajo la condición de baño de hielo, se añadieron 120 ml de agua helada a una temperatura interna de 30 °C o menor. La mezcla de reacción se extrajo con éter isopropílico, 100 ml x 3; y la capa de éter se combinó y se lavó con una solución acuosa saturada de NaCl, 100 ml x 3. La capa orgánica se secó sobre sulfato de sodio anhidro durante la noche, el agente desecante se retiró por filtración, y el disolvente se retiró bajo presión reducida para obtener 41,40 g de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali como materia aceitosa de color rojo, con un rendimiento de 98,13 %. El producto obtenido fue configuración (2R,3S),(2S,3S) de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali.
Etapa 3: Purificación de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali por cromatografía de columna y el tratamiento de purificación relacionado
La muestra de la etapa 2 anterior se separó en columna de gel de sílice, utilizando diclorometano o triclorometano y metanol, amoniacados, como fase móvil y una placa de TLC para controlar la pureza de la muestra. Bajo el sistema de elución anterior, una muestra de una mezcla de ambas configuraciones (2R,3S) y (2S,3S) de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali podía ser purificada en dos tipos de (2R,3S) y (2S,3S) libre de álcali, dependiendo de la secuencia de elución, obteniéndose de ese modo 16,44 g de (2R,3S)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 79,43 %, y 16,49 g de bromuro de (2S,3S)-3-[(2-ciclopropil-2-hidroxilo-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 79,68 %.
Etapa 4: igual que la etapa 4 en el Ejemplo 1
Etapa 5: Bromuro de (2R,3S)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,612 g (9,1 mmol) de la configuración (2R,3S) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,034 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25-40 °C por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 3,725 g de bromuro de (2R,3S)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 81,55 %.
El compuesto preparado en el Ejemplo 19 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes.
Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,19-7,15 (m, 7H), 6,82-6,77 (m, 3H), 3,94-3,68 (m, 4H), 3,49-3,19 (m, 9H), 2,15 (m, 2H), 1,82-1,57 (m, 5H), 0,67-0,06 (m, 5H).
Ejemplo 20. Bromuro de (2S,3S)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1 -(3-fenoxi propil)-1 -azabiciclo[2,2,2]octano
Las etapas 1, 2 y 3 eran iguales que las etapas 1, 2 y 3 en el Ejemplo 19
La etapa 4 era igual que la etapa 4 en el Ejemplo 1
Etapa 5: Bromuro de (2S,3S)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano
A un matraz de 100 ml, con forma de berenjena, se añadieron 2,612 g (9,1 mmol) de la configuración (2S,3S) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,034 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25-40 °C por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 3,776 g de la configuración (2S,3S) de bromuro de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 82,66 %.
El compuesto preparado en el Ejemplo 20 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes.
Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,19-7,15 (m, 7H), 6,81-6,77 (m, 3H), 3,94-3,68 (m, 4H), 3,49-3,19 (m, 9H), 2,15 (m, 2H), 1,82-1,57 (m, 5H), 0,66-0,06 (m, 5H).
Ejemplo 21. Bromuro de (2S,3R)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxi propil)-1-azabiciclo[2,2,2]octano
La etapa 1 era igual que la etapa 1 en el Ejemplo 19
Etapa 2: Preparación de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali a partir de R-3-quinuclidinol

A 18,721 g (147 mmol) de R-3-quinuclidinol disponible comercialmente se añadieron 190 ml de DMSO, seguido de la adición de 7,591 g (190 mmol) de hidruro de sodio, y la mezcla se hizo reaccionar a 20-60 °C durante 0,5-12 h, y después se enfrió a temperatura ambiente, y a ello se añadió una solución de 30,36 g (190 mmol) de 1 -fenil-1 -ciclopropiloxirano (autopreparado) en 45 ml de DMSO, y una vez finalizada la adición, la mezcla resultante se calentó a 20-70 °C en baño de aceite para reaccionar durante 0,5-12 horas. Bajo la condición de baño de hielo, se añadieron 120 ml de agua helada a una temperatura interna de 30 °C o menor. La mezcla de reacción se extrajo con éter isopropílico, 100 ml x 3; y la capa de éter se combinó y se lavó con una solución acuosa saturada de NaCl, 100 ml x 3. La capa orgánica se secó sobre sulfato de sodio anhidro durante la noche, el agente desecante se retiró por filtración, y el disolvente se retiró por evaporación rotativa para obtener 40,99 g de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo [2,2,2]octano libre de álcali como materia aceitosa de color rojo, con un rendimiento de 97,15 %. El producto obtenido fue la configuración (2R,3R),(2S,3R) de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali.
Etapa 3: Purificación de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali por cromatografía de columna y el tratamiento de purificación relacionado
La muestra de la etapa 2 anterior se separó en columna de gel de sílice, utilizando diclorometano o triclorometano y metanol, amoniacados, como fase móvil y una placa de TLC para controlar la pureza de la muestra. Bajo el sistema de elución anterior, una muestra de una mezcla de ambas configuraciones (2R,3R) y (2S,3R) de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali podía ser purificada en dos tipos de (2R,3R) y (2S,3R) libres de álcali, dependiendo de la secuencia de elución, obteniéndose de ese modo 16,45 g de (2R,3R)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 80,25 %, y 16,26 g de la configuración (2S,3R) de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 79,34 %.
Etapa 4: igual que la etapa 4 en el Ejemplo 1
Etapa 5: Bromuro de (2S,3R)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,612 g (9,1 mmol) de la configuración (2S,3R) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,0340 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25-40 °C por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 3,599 g de (2S,3R)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 78,78 %.
El compuesto preparado en el Ejemplo 21 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes.
Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,19-7,15 (m, 7H), 6,82-6,77 (m, 3H), 3,94-3,68 (m, 4H), 3,49-3,19 (m, 9H), 2,15 (m, 2H), 1,82-1,57 (m, 5H), 0,67-0,06 (m, 5H).
Ejemplo 22. Bromuro de (2R,3R)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1 -(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano
La etapa 1 era igual que la etapa 1 en el Ejemplo 19
Las etapas 2 y 3 eran iguales que las etapas 2 y 3 en el Ejemplo 21
Etapa 4: igual que la etapa 4 en el Ejemplo 1
Etapa 5: Bromuro de (2R,3R)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,612 g (9,1 mmol) de la configuración (2R,3R) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,034 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25-40 °C por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 3,795 g de bromuro de (2R,3R)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 83,07 %.
El compuesto preparado en el Ejemplo 22 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes.
Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,19-7,15 (m, 7H), 6,82-6,77 (m, 3H), 3,94-3,68 (m, 4H), 3,49-3,19 (m, 9H), 2,15 (m, 2H), 1,82-1,57 (m, 5H), 0,67-0,06 (m, 5H).
Ejemplo 23. Bromuro de (2R,3S),(2S,3R)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1 -(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano
La etapa 1 era igual que la etapa 1 en el Ejemplo 19
Etapa 2: Preparación de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali a partir de quinuclidinol racémico

A 18,72 g (147 mmol) de quinuclidinol racémico disponible comercialmente se añadieron 190 ml de DMSO, seguido de la adición de 7,59 g (190 mmol) de hidruro de sodio, y la mezcla se hizo reaccionar a 20-60 °C durante 0,5-12 h, y después se enfrió a temperatura ambiente, y a ello se añadió una solución de 30,4 g (190 mmol) de 1 -fenil-1 -ciclopropiloxirano (autopreparado) en 45 ml de DMSO, y una vez finalizada la adición, la mezcla resultante se calentó a 20-70 °C en baño de aceite para reaccionar durante 0,5-12 horas. Bajo la condición de baño de hielo, se añadieron 120 ml de agua helada a una temperatura interna de 30 °C o menor. La mezcla de reacción se extrajo con éter isopropílico, 100 ml x 3; y la capa de éter se combinó y se lavó con una solución acuosa saturada de NaCl, 100 ml x 3. La capa orgánica se secó sobre sulfato de sodio anhidro durante la noche, el agente desecante se retiró por filtración, y el disolvente se retiró por evaporación rotativa, para obtener 42,02 g de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali como materia aceitosa de color rojo, con un rendimiento de 99,6 %. El producto obtenido fue la configuración (2R,3S),(2R,3R),(2S,3R),(2S,3S) de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali.
Etapa 3: Purificación de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali por cromatografía de columna y el tratamiento de purificación relacionado
La muestra de la etapa 2 anterior se separó en columna de gel de sílice, utilizando diclorometano o triclorometano y metanol, amoniacados, como fase móvil y una placa de TLC para controlar la pureza de la muestra. Bajo el sistema de elución anterior, una muestra de una mezcla de cuatro configuraciones de (2R,3S),(2R,3R),(2S,3R),(2S,3S) de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali podía ser purificada en dos tipos racémicos, (2R,3S),(2S,3R) y (2R,3R),(2S,3S), libres de álcali, dependiendo de la secuencia de elución, obteniéndose de ese modo la fracción eluida en primer lugar de 17,06 g de la configuración (2R,3S),(2S,3R) racémica de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 81,22 %, y la fracción eluida en segundo lugar de 16,63 g de la configuración (2R,3R),(2S,3S) de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 79,13 %.
Etapa 4: igual que la etapa 4 en el Ejemplo 1
Etapa 5: Configuración (2R,3S),(2S,3R) de bromuro de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,612 g (9,1 mmol) de la configuración (2R,3S),(2S,3R) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,0342 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25-40 °C por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 3,76 g de la configuración (2R,3S),(2S,3R) de bromuro de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 82,31 %.
El compuesto preparado en el Ejemplo 23 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes. Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,19-7,15 (m, 7H), 6,82-6,77 (m, 3H), 3,94-3,68 (m, 4H), 3,49-3,19 (m, 9H), 2,15 (m, 2H), 1,82-1,57 (m, 5H), 0,67-0,06 (m, 5H).
Ejemplo 24. Configuración (2R,3R),(2S,3S) de bromuro de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano
La etapa 1 era igual que la etapa 1 en el Ejemplo 19
Las etapas 2 y 3 eran iguales que las etapas 2 y 3 en el Ejemplo 23
Etapa 4: igual que la etapa 4 en el Ejemplo 1
Etapa 5: Configuración (2R,3R),(2S,3S) de bromuro de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,994 g (9,1 mmol) de la configuración (2R,3R),(2S,3S) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,034 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25 40 °C por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 3,54 g de la configuración (2R,3R),(2S,3S) de bromuro de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 77,49 %.
Los isómeros ópticos puros de cuatro configuraciones de (2R,3S),(2R,3R),(2S,3R),(2S,3S) de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali podían obtenerse a partir de los Ejemplos 19 y 21, y las mezclas de las configuraciones (2R,3S),(2R,3R),(2S,3R),(2S,3S) de bromuro de 3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano en diferentes proporciones se obtuvieron mezclando estos isómeros en cualquier cantidad y cualquier proporción seguida por cuaternización añadiendo 3-bromopropoxibenceno.
El compuesto preparado en el Ejemplo 24 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes. Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,19-7,15 (m, 7H), 6,82-6,77 (m, 3H), 3,94-3,68 (m, 4H), 3,49-3,19 (m, 9H), 2,15 (m, 2H), 1,82-1,57 (m, 5H), 0,67-0,06 (m, 5H).
Ejemplo 25. Bromuro de (2S,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(2-fenoxi Etil)-1-azabiciclo[2,2,2]octano
Etapa 1: igual que la etapa 1 en el Ejemplo 1
Etapas 2 y 3: igual que las etapas 2 y 3 en el Ejemplo 3
Etapa 4: Preparación de 2-bromoetoxilbenceno

A un matraz de 150 ml, con tres bocas, se añadieron 9,5 g (101 mmol) de fenol, seguido de la adición de 4,25 g (106 mmol) de hidróxido de sodio y una solución de 47,11 g (250 mmol) de 1,2-dibromoetano en 30 ml de etanol absoluto, y la mezcla resultante se hizo reaccionar entonces bajo calentamiento y reflujo en baño de aceite, precipitándose un sólido blanco, hasta reacción esencialmente completa de fenol controlada por TLC (condiciones de la TLC: éter de petróleo/acetato de etilo = 5,0 ml/1,0 ml). Una vez finalizada la reacción, el sólido se retiró por filtración, y el disolvente se retiró del filtrado a 50 °C o menor por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa que contenía un sólido blanco, al que se le añadió éter de petróleo y después se dejó reposar durante la noche. El sólido precipitado se retiró por filtración, y el disolvente se retiró del filtrado a 25-40 °C por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo. La materia aceitosa fue destilada bajo presión reducida, para recoger una fracción a 8 mmHg, 118-122 °C, obteniéndose de ese modo 11,31 g de una materia aceitosa incolora, transparente, con un rendimiento de 55,7 % y pureza de 95,6065 % detectada por CG.
RMN de 1H (CDCls) 5 (ppm): 7,15-6,77 (m, 5H), 4,45 (t, 2H), 3,79 (t, 2H).
Etapa 5: Bromuro de (2S,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(2-fenoxietil)-1-azabiciclo[2,2,2]octano

A un matraz de 50 ml, con forma de berenjena, se añadieron 1,436 g (4,55 mmol) de la configuración (2S,3R) de la base y se disolvieron añadiendo 9 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 5,65 g (28,1 mmol) de 2-bromoetoxibenceno y 25 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25-40 °C por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 1,857 g de (2S,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxietil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 79,12 %.
El compuesto preparado en el Ejemplo 25 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía reaccionar con otros ácidos para transformarlos en las sales correspondientes. Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,18-6,76 (m, 10H), 5,86 (t, 2H), 4,39 (t, 2H), 3,98-3,71 (s, 2H), 3,47-3,18 (m, 7H), 1,92 (m, 1H), 1,81-1,66 (m, 5H), 1,59-1,36 (m, 8H).
Ejemplo 26. Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1 -(2-fenoxi Etil)-1 -azabiciclo[2,2,2]octano
Etapa 1: igual que la etapa 1 en el Ejemplo 1
Las etapas 2 y 3 eran iguales que las etapas 2 y 3 en el Ejemplo 3
La etapa 4 era igual que la etapa 4 en el Ejemplo 25
Etapa 5: Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(2-fenoxietil)-1-azabiciclo[2,2,2]octano

A un matraz de 50 ml, con forma de berenjena, se añadieron 1,435 g (4,55 mmol) de la configuración (2R,3R) de la base y se disolvieron añadiendo 9 ml de cloroformo para obtener una solución de color amarillo transparente, a la que se añadieron 5,65 g (28,1 mmol) de 2-bromoetoxibenceno y 25 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25-40 °C por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 1,797 g de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxietil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 76,56 %.
El compuesto preparado en el Ejemplo 26 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes. Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,19-6,78 (m, 10H), 5,88 (t, 2H), 4,38 (t, 2H), 3,99-3,70 (s, 2H), 3,47-3,17 (m, 7H), 1,93 (m, 1H), 1,81-1,67 (m, 5H), 1,60-1,37 (m, 8H).
Ejemplo 27. Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1 -fenoxime thyl-1 -azabiciclo[2,2,2]octano
Etapa 1: igual que la etapa 1 en el Ejemplo 1
Las etapas 2 y 3 eran iguales que las etapas 2 y 3 en el Ejemplo 3
Etapa 4: Preparación del bromometoxilbenceno

A un matraz de 150 ml, con tres bocas, se añadieron 9,5 g (101 mmol) de fenol, seguido de la adición de 4,25 g (106 mmol) de hidróxido de sodio y una solución de 43,51 g (250 mmol) de 1,2-dibromometano en 30 ml de etanol absoluto, y la mezcla resultante se hizo reaccionar después bajo calentamiento y reflujo en baño de aceite, precipitándose un sólido blanco, hasta reacción esencialmente completa de fenol controlada por TLC (condiciones de la TLC: éter de petróleo/acetato de etilo = 5,0 ml/1,0 ml). Una vez finalizada la reacción, el sólido se retiró por filtración, y el disolvente se retiró del filtrado a 50 °C o menor por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa que contenía un sólido blanco, al que se le añadió éter de petróleo y después se dejó reposar durante la noche. El sólido precipitado se retiró por filtración, y el disolvente se retiró del filtrado a 25-40 °C por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo. La materia aceitosa fue destilada bajo presión reducida, para recoger una fracción a 8 mmHg, 116-120 °C, obteniéndose de ese modo 10,75 g de una materia aceitosa incolora, transparente, con un rendimiento de 56,9 % y pureza de 95,6065 % detectada por CG.
RMN de 1H (CDCls) 5 (ppm): 7,16-6,76 (m, 5H), 5,95 (s, 2H)
Etapa 5: Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-fenoximetil-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,871 g (9,1 mmol) de la configuración (2R,3R) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución de color amarillo transparente, a la que se añadieron 10,29 g (55 mmol) de bromometoxilbenceno y 25 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25-40 °C por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 3,751 g de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(fenoximetil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 80,2 %.
El compuesto preparado en el Ejemplo 27 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes. Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 5 (ppm): 7,21-6,76 (m, 10H), 5,89 (s, 2H), 3,97-3,73 (s, 2H), 3,48-3,18 (m, 7H), 1,95 (m, 1H), 1,80 1,58 (m, 5H), 1,61-1,38 (m, 8H).
Ejemplo 28. Cloruro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1 -fenoximetoximetil-1 -azabiciclo[2,2,2]octano
La etapa 1 era igual que la etapa 1 en el Ejemplo 1
Las etapas 2 y 3 eran iguales que las etapas 2 y 3 en el Ejemplo 3
Etapa 4: Preparación del éter de clorometoximetilo y fenilo

A un matraz de 150 ml, con tres bocas, se añadieron 9,5 g (101 mmol) de fenol, seguido de la adición de 4,25 g (106 mmol) de hidróxido de sodio y una solución de 28,75 g (250 mmol) de 1,3-diclorometiléter en 30 ml de etanol absoluto, y la mezcla resultante se hizo reaccionar entonces bajo calentamiento y reflujo en baño de aceite, precipitándose un sólido blanco, hasta reacción esencialmente completa de fenol controlada por TLC (condiciones de la TLC: éter de petróleo/acetato de etilo = 5,0 ml/1,0 ml). Una vez finalizada la reacción, el sólido se retiró por filtración, y el disolvente se retiró del filtrado a 50 °C o menor por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa que contenía un sólido blanco, al cual se le añadió éter de petróleo y después se dejó reposar durante la noche. El sólido precipitado se retiró por filtración, y el disolvente se retiró del filtrado a 25-40 °C por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo. La materia aceitosa fue destilada bajo presión reducida, para recoger una fracción a 8 mmHg, 106-119 °C, obteniéndose de ese modo 8,38 g de una materia aceitosa incolora, transparente, con un rendimiento de 48,1 % y pureza de 97,62 % detectada por CG.
RMN de 1H (CDCls) 5 (ppm): 7,19-6,79 (m, 5H), 6,01 (s, 2H), 5,47 (s, 2H).
Etapa 5: Cloruro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-fenoximetoximetil-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,871 g (9,1 mmol) de la configuración (2R,3R) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 9,49 g (55 mmol) de éter de clorometoximetilo y fenilo y 25 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25 40 °C por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 3,13 g de cloruro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-fenoximetoximetil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 69,2 %.
El compuesto preparado en el Ejemplo 28 se hizo reaccionar con Ag2Ü para retirar el átomo de cloro para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes. Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) 6 (ppm): 7,19-6,77 (m, 10H), 6,02 (s, 2H), 5,32 (s, 2H), 3,98-3,68 (s, 2H), 3,49-3,19 (m, 7H), 1,96 (m, 1H), 1,82-1,57 (m, 5H), 1,60-1,36 (m, 8H).
Ejemplo 29. Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-naftil)etoxil]-1 -(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano
Etapa 1: Preparación de 230 g de 1-naftil-1-ciclopentiloxirano según el procedimiento en la bibliografía(2)
Etapa 2: Preparación de 3-[(2-ciclopentil-2-hidroxil-2-naftil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali a partir de R-3-quinuclidinol

A 18,72 g (147 mmol) de R-3-quinuclidinol disponible comercialmente se añadieron 190 ml de DMSÜ, seguido de la adición de 7,59 g (190 mmol) de hidruro de sodio, y la mezcla se hizo reaccionar a 20-60 °C durante 0,5-12 h, y después se enfrió a temperatura ambiente, y a ello se añadió una solución de 45,15 g (190 mmol) de 1 -naftil-1 -ciclopentiloxirano (autopreparado) en 45 ml de DMSÜ, y una vez finalizada la adición, la mezcla resultante se calentó a 20-70 °C en baño de aceite para reaccionar durante 0,5-12 horas. Bajo la condición de baño de hielo, se añadieron 120 ml de agua helada a una temperatura interna de 30 °C o menor. La mezcla de reacción se extrajo con éter isopropílico, 100 ml x 3; y la capa de éter se combinó y se lavó con una solución acuosa saturada de NaCl, 100 ml x 3. La capa orgánica se secó sobre sulfato de sodio anhidro durante la noche, el agente desecante se retiró por filtración, y el disolvente se retiró bajo presión reducida para obtener 51,71 g de 3-[(2-ciclopentil-2-hidroxil-2naftil)etoxil]-1-azabiciclo [2,2,2]octano libre de álcali como materia aceitosa de color rojo, con un rendimiento de 96,38 %. El producto obtenido fue la configuración (2R,3R),(2S,3R) de 3-[(2-ciclopentil-2-hidroxil-2-naftil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali.
Etapa 3: Purificación de 3-[(2-ciclopentil-2-hidroxil-2-naftil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali por cromatografía de columna y el tratamiento de purificación relacionado
La muestra de la etapa 2 anterior se separó en columna de gel de sílice, utilizando diclorometano o triclorometano y metanol, amoniacados, como fase móvil y una placa de TLC para controlar la pureza de la muestra. Bajo el sistema de elución anterior, una muestra de una mezcla de ambas configuraciones (2R,3R) y (2S,3R) de 3-[(2-ciclopentil-2-hidroxil-2-naftil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali podía ser purificada en dos tipos de (2R,3R) y (2S,3R) libres de álcali, dependiendo de la secuencia de elución, obteniéndose de ese modo 22,36 g de la configuración (2R,3R) de 3-[(2-ciclopentil-2-hidroxil-2-naftil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 86,5 %, y 20,05 g de la configuración (2S,3R) de 3-[(2-ciclopentil-2-hidroxil-2-naftil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 77,55 %.
Etapa 4: igual que la etapa 4 en el Ejemplo 1
Etapa 5: Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-naftil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 3,322 g (9,1 mmol) de la configuración (2R,3R) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,033 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25-40 °C por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 4,238 g de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-naftil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 80,3 %.
El compuesto preparado en el Ejemplo 29 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes.
Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) (ppm): 57,21-6,66 (m, 12H), 54,17-3,65 (m, 5H), 53,44-3,13 (m, 8H), 52,15 (m, 2H), 2,00 (m, 1H), 51,84-1,42 (m, 13H).
Ejemplo 30. Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-o-clorofenil)etoxil]-1 -(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano
Etapa 1: 1-o-clorofenil-1-ciclopentiloxirano

Se hizo reaccionar ciclopentil-o-clorofenilcetona comercialmente disponible como material de partida para obtener 1-o-clorofenil-1-ciclopentiloxirano según la bibliografía(1).
A un matraz de 3 litros, con tres bocas, se añadieron 1.880 ml de acetonitrilo, seguido de la adición de sulfuro de dimetilo y sulfato de dimetilo, con un leve desprendimiento de calor, y la mezcla resultante se agitó entonces a temperatura ambiente durante 1,5 h y después se dejó reposar durante la noche. Se añadió NaH (60 %) en porciones adecuadas a la solución de reacción con agitación durante 30-40 min, generándose un gas (préstese atención a tener el matraz externamente conectado a un condensador Allihn y un tubo de secado de cloruro de calcio anhidro), y se agitó entonces a temperatura ambiente durante 1 hora, seguido de la adición gota a gota de 355 g de ciclopentil-o-clorofenil-cetona durante aproximadamente 10 min, y la mezcla resultante se hizo reaccionar posteriormente en baño de aceite a 40-45 °C durante 90 min siendo después agitada a temperatura ambiente durante 20 min, y después se enfrió a temperatura ambiente y se dejó durante la noche. La solución de reacción se transfirió a un matraz con forma de berenjena, el disolvente se retiró por succión usando una bomba de agua (40 42 °C, -0,095 MPa), y el sólido restante se enfrió a 0-5 °C en baño de hielo, seguido de la adición gota a gota de 2.630 ml de agua helada (siendo la temperatura interior controlada a 0-5 °C) durante aproximadamente 1 hora, y la solución de reacción se extrajo con éter isopropílico (650 ml x 3), la capa de éter se combinó y se lavó con agua hasta que es neutra (siendo el pH de los lavados con agua de 7,0), y la capa de éter se secó sobre sulfato de sodio anhidro durante la noche. El agente desecante se retiró por filtración, el disolvente se retiró bajo presión reducida por succión usando una bomba de agua (40-42 °C, -0,095 MPa) y, posteriormente, se recogieron 315,8 g de fracción a 119-132 °C/3 mmHg bajo presión reducida usando una bomba de aceite, con un rendimiento de 83,36 %.
RMN de 1H (CDCls) 5 (ppm): 7,19-7,07 (m, 4H), 2,92-2,64 (m, 1H), 2,21 (s, 2H), 1,61-1,37 (m, 8H).
Etapa 2: Preparación de 3-[(2-ciclopentil-2-hidroxil-2-o-clorofenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali a partir de R-3-quinuclidinol

A 18,72 g (147 mmol) de R-3-quinuclidinol disponible comercialmente se añadieron 190 ml de DMSO, seguido de la adición de 7,59 g (190 mmol) de hidruro de sodio, y la mezcla se hizo reaccionar a 20-60 °C durante 0,5-12 h, y después se enfrió a temperatura ambiente, y a ello se añadió una solución de 42,317 g (190 mmol) de 1-o-clorofenil-1-ciclopentiloxirano (autopreparado) en 45 ml de DMSO, y una vez finalizada la adición, la mezcla resultante se calentó a 20-70 °C en baño de aceite para reaccionar durante 0,5-12 horas. Bajo la condición de baño de hielo, se añadieron 120 ml de agua helada a una temperatura interna de 30 °C o menor. La mezcla de reacción se extrajo con éter isopropílico, 100 ml x 3; y la capa de éter se combinó y se lavó con una solución acuosa saturada de NaCl, 100 ml x 3. La capa orgánica se secó sobre sulfato de sodio anhidro durante la noche, el agente desecante se retiró por filtración, y el disolvente se retiró bajo presión reducida para obtener 49,48 g de 3-[(2-ciclopentil-2-hidroxil-2-oclorofenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali como materia aceitosa de color rojo, con un rendimiento de 96,31 %. El producto obtenido fue (2R,3R),(2S,3R)-3-[(2-ciclopentil-2-hidroxil-2-o-clorofenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali.
Etapa 3: Purificación de 3-[(2-ciclopentil-2-hidroxil-2-o-clorofenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali por cromatografía de columna y el tratamiento de purificación relacionado
La muestra de la etapa 2 anterior se separó en columna de gel de sílice, utilizando diclorometano o triclorometano y metanol, amoniacados, como fase móvil y una placa de TLC para controlar la pureza de la muestra. Bajo el sistema de elución anterior, una muestra de una mezcla de ambas configuraciones (2R,3R) y (2S,3R) de 3-[(2-ciclopentil-2-hidroxil-2-o-clorofenil)etoxil]-1-azabiciclo[2,2,2]octano libres de álcali podía ser purificada en dos tipos (2R,3R) y (2S, 3R) libres de álcali, dependiendo de la secuencia de elución, obteniendo de este modo 21,03 g de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-o-clorofenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 85,0 %, y 21,15 g de (2S,3R)-3-[(2-ciclopentil-2-hidroxil-2-o-clorofenil)etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 85,485 %.
Etapa 4: igual que la etapa 4 en el Ejemplo 1
Etapa 5: Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-o-clorofenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 3,18 g (9,1 mmol) de la configuración (2R,3R) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,033 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25-40 °C por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 4,25 g de bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-oclorofenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 82,73 %.
El compuesto preparado en el Ejemplo 30 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes.
Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) (ppm): 57,33-6,65 (m, 9H), 54,16-3,63 (m, 5H), 53,45-3,12 (m, 8H), 52,13 (m, 2H), 2,02 (m, 1H), 5 1,83-1,43 (m, 13H).
Ejemplo 31. Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-(3-piridil))etoxil]-1 -(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano
Etapa 1: Preparación de ciclopentil-3-piridil-cetona

En un matraz de 5 litros, con tres bocas, conectado externamente a un condensador Allihn y un tubo de secado de cloruro de calcio anhidro y un termómetro se colocaron 102,06 g de virutas de magnesio, a ello se añadieron y agitaron 1.600 ml de THF y 1,06 g de yodo, y después, a la solución de reacción, se añadieron gota a gota 78,28 g
de bromociclopentano. A aproximadamente 5 minutos después de que la reacción se iniciara por completo, la solución de reacción pasó gradualmente de tener un color marrón rojizo claro a ser incolora, aumentando la temperatura interna automáticamente a 63-65 °C. Después, se añadieron gota a gota 547,93 g de bromociclopentano adicional durante aproximadamente 35 min, aumentando su temperatura de reflujo gradualmente a 75-77 °C. La mezcla de reacción se hizo reaccionar bajo reflujo en baño de aceite durante 2 horas.
A la solución de reacción anterior se añadieron gota a gota 412,74 g de 3-piridilcarbonitrilo diluido en 1.600 ml de THF, y la mezcla resultante se hizo entonces reaccionar bajo reflujo en baño de aceite durante 4 horas. La temperatura interior se redujo a 5-10 °C, se añadieron, gota a gota, 96 ml de agua helada, y después se agitó durante 20 minutos, seguido de la adición de HCl para ajustar el pH = 2,0, y la mezcla resultante se calentó a reflujo durante 3 horas, la capa orgánica fue entonces separada, y una vez enfriada, la fase acuosa se extrajo con éter isopropílico (200 ml x 3). La capa orgánica se combinó y se lavó con Na2CÜ3 1 % (500 x 3) y agua (500 x 3) de forma consecutiva, y la capa orgánica se secó sobre sulfato de sodio anhidro durante la noche. El agente desecante se retiró por filtración, el disolvente se retiró por evaporación (42 °C, -0,095 MPa) y posteriormente se recogieron 332,26 g de materia aceitosa a 122-130 °C/6-7 mmHg por destilación bajo presión reducida usando una bomba de aceite.
RMN de 1H (CDCls) 5 (ppm): 9,31-7,70 (m, 4H), 2,36 (m, 1H), 1,63-1,47 (m, 8H).
Etapa 2: preparación de 1-(3-piridil)-1-ciclopentiloxirano
Se hizo reaccionar ciclopentil-3-piridil-cetona como material de partida para obtener 1 -(3-piridil)-1 -ciclopentiloxirano según la bibliografía(1).
A un matraz de 3 litros, con tres bocas, se añadieron 1.880 ml de acetonitrilo, seguido de la adición de sulfuro de dimetilo y sulfato de dimetilo, con un leve desprendimiento de calor, y la mezcla resultante se agitó entonces a temperatura ambiente durante 1,5 h y después se dejó reposar durante la noche. A la solución de reacción se añadió NaH (60 %) en porciones adecuadas con agitación durante 30-40 min, generándose un gas (préstese atención a tener el matraz externamente conectado a un condensador Allihn y un tubo de secado de cloruro de calcio anhidro), y después se agitó a temperatura ambiente durante 1 hora, seguido de la adición gota a gota de 300 g de ciclopentil-3-piridil-cetona durante aproximadamente 10 min, y la mezcla resultante se hizo reaccionar en baño de aceite a 40-45 °C durante 90 min siendo después agitada a temperatura ambiente durante 20 min, y después se enfrió a temperatura ambiente y se dejó reposar durante la noche. La solución de reacción se transfirió a un matraz con forma de berenjena, el disolvente se retiró por succión usando una bomba de agua (40-42 °C, -0,095 MPa), y el sólido restante se enfrió a 0-5 °C en baño de hielo, seguido de la adición gota a gota de 2.630 ml de agua helada (siendo la temperatura interior controlada a 0-5 °C) durante aproximadamente 1 hora, y la solución de reacción se extrajo con éter isopropílico (650 ml x 3), la capa de éter se combinó y se lavó con agua hasta que es neutra (siendo el pH de los lavados con agua de 7,0), y la capa de éter se secó sobre sulfato de sodio anhidro durante la noche. El agente desecante se retiró por filtración, el disolvente se retiró bajo presión reducida por succión usando una bomba de agua (40-42 °C, -0,095 MPa), y posteriormente se recogieron 285,5 g de fracción a 121-131 °C/3 mmHg bajo presión reducida usando una bomba de aceite.
RMN de 1H (CDCls) 5 (ppm): 8,68-7,50 (m, 4H), 2,91-2,66 (m, 2H), 2,23-1,29 (m, 9H).
Etapa 3: Preparación de 3-[(2-ciclopentil-2-hidroxil-2-(3-piridil))etoxil]-1-azabiciclo[2,2,2]octano libre de álcali a partir de R-3-quinuclidinol

A 18,72 g (147 mmol) de R-3-quinuclidinol disponible comercialmente se añadieron 190 ml de DMSO, seguido de la adición de 7,59 g (190 mmol) de hidruro de sodio, y la mezcla se hizo reaccionar a 20-60 °C durante 0,5-12 h, y después se enfrió a temperatura ambiente, y a ello se añadió una solución de 35,91 g (190 mmol) de 1-(3-piridil)-1-ciclopentiloxirano (autopreparado) en 45 ml de DMSO, y una vez finalizada la adición, la mezcla resultante se calentó a 20-70 °C en baño de aceite para reaccionar durante 0,5-12 horas. Bajo la condición de baño de hielo, se añadieron 120 ml de agua helada a una temperatura interna de 30 °C o menor. La mezcla de reacción se extrajo con éter isopropílico, 100 ml x 3; y la capa de éter se combinó y se lavó con una solución acuosa saturada de NaCl, 100 ml x 3. La capa orgánica se secó sobre sulfato de sodio anhidro durante la noche, el agente desecante se retiró por filtración, y el disolvente se retiró bajo presión reducida para obtener 45,66 g de 3-[(2-ciclopentil-2-hidroxil-2-(3-piridil))etoxil]-1-azabiciclo [2,2,2] octano libre de álcali como materia aceitosa de color rojo, con un rendimiento de 98,3 %. El producto obtenido fue (2R,3R),(2S,3R)-3-[(2-ciclopentil-2-hidroxil-2-(3-piridil))etoxil]-1-azabiciclo[2,2,2]octano libre de álcali.
Etapa 4: Purificación de 3-[(2-ciclopentil-2-hidroxil-2-(3-piridil))etoxil]-1-azabiciclo[2,2,2]octano libre de álcali por cromatografía de columna y el tratamiento de purificación relacionado
La muestra de la etapa 3 anterior se separó en columna de gel de sílice, utilizando diclorometano o triclorometano y metanol, amoniacados, como fase móvil y una placa de TLC para controlar la pureza de la muestra. Bajo el sistema de elución anterior, una muestra de una mezcla de ambas configuraciones (2R,3R) y (2S,3R) de 3-[(2-ciclopentil-2-hidroxil-2-(3-piridil))etoxil]-1-azabiciclo[2,2,2]octano álcali libre podía ser purificada en dos tipos de (2R,3R) y (2S,3R) libre de álcali, dependiendo de la secuencia de elución, obteniéndose de ese modo 19,52 g de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-(3-piridil))etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 85,5 %, y 19,06 g de la configuración (2S,3R) de 3-[(2-ciclopentil-2-hidroxil-2-(3-piridil))etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 83,47 %.
Etapa 5: igual que la etapa 4 en el Ejemplo 1
Etapa 6: Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-(3-piridil))etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 3,18 g (9,1 mmol) de la configuración (2R,3R) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,033 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25-40 °C por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 3,95 g de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-(3-piridil))etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]-octano como un sólido de color blancuzco, con un rendimiento de 81,7 %.
El compuesto preparado en el Ejemplo 31 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes. Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) (ppm): 68,72-6,63 (m, 9H), 64,13-3,61 (m, 5H), 63,44-3,13 (m, 8H), 52,14 (m, 2H), 2,01 (m, 1H), 6 1,82-1,45 (m, 13H).
Ejemplo 32. Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-(2-furil))etoxil]-1 -(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano
Etapa 1: Preparación de ciclopentil-2-furil-cetona

En un matraz de 5 litros, con tres bocas, conectado externamente a un condensador Allihn y a un tubo de secado de cloruro de calcio anhidro y un termómetro, se colocaron 101,5 g de virutas de magnesio, a ello se añadieron y agitaron 1.600 ml de THF y 1,05 g de yodo, y a la solución de reacción se añadieron gota a gota 78,28 g de bromociclopentano. Aproximadamente 5 minutos después de que la reacción se iniciara por completo, la solución de reacción pasó gradualmente de tener un color marrón rojizo claro a ser incolora, aumentando la temperatura interna automáticamente a 62-65 °C. Entonces se añadieron, gota a gota, 548 g de bromociclopentano adicional durante 35 min, aumentando su temperatura de reflujo gradualmente a 75-77 °C. La mezcla de reacción se hizo reaccionar bajo reflujo en baño de aceite durante 2 horas.
A la solución de reacción anterior se añadieron, gota a gota, 404 g de 2-furilcarbonitrilo diluido en 1.600 ml de THF y la mezcla resultante se hizo reaccionar entonces bajo reflujo en baño de aceite durante 4 horas. La temperatura interior se redujo a 5-10 °C, se añadieron, gota a gota, 96 ml de agua helada y después se agitó durante 20 minutos, seguido de la adición de HCl para ajustar el pH = 2,0, y la mezcla resultante se calentó a reflujo durante 3 horas, después se separó la capa orgánica, y una vez enfriada, la fase acuosa se extrajo con éter isopropílico (200 ml x 3). La capa orgánica se combinó y se lavó con Na2CÜ31 % (500 x 3) y agua (500 x 3) de forma consecutiva, y la capa orgánica se secó sobre sulfato de sodio anhidro durante la noche. El agente desecante se retiró por filtración, el disolvente se retiró por evaporación (42 °C, -0,095 MPa), y posteriormente se recogieron 272,6 g de materia aceitosa a 108-119 °C/6-7 mmHg por destilación bajo presión reducida usando una bomba de aceite.
RMN de 1H (CDCls) 5 (ppm): 7,39-6,70 (m, 3H), 2,36 (m, 1H), 1,65-1,45 (m, 8H).
Etapa 2: preparación de 1-(2-furil)-1-ciclopentiloxirano

Se hizo reaccionar ciclopentil-2-furil-cetona como material de partida para obtener 1-(2-furil)-1-ciclopentiloxirano según la bibliografía(1).
A un matraz de 3 litros, con tres bocas, se añadieron 1.780 ml de acetonitrilo seguido de la adición de sulfuro de dimetilo y sulfato de dimetilo, con un leve desprendimiento de calor, y la mezcla resultante se agitó entonces a temperatura ambiente durante 1,5 h y después se dejó reposar durante la noche. A la solución de reacción se añadió NaH (60 %) en porciones adecuadas con agitación durante 30-40 min, generándose un gas (préstese
atención a tener el matraz externamente conectado a un condensador Allihn y un tubo de secado de cloruro de calcio anhidro), y después se agitó a temperatura ambiente durante 1 hora, seguido de la adición gota a gota de 250 g de ciclopentil-2-furil-cetona durante aproximadamente 10 minutos, y la mezcla resultante se hizo reaccionar en baño de aceite a 40-45 °C durante 90 min siendo después agitada a temperatura ambiente durante 20 min, y después se enfrió a temperatura ambiente y se dejó reposar durante la noche. La solución de reacción se transfirió a un matraz con forma de berenjena, el disolvente se retiró por succión usando una bomba de agua (40-42 °C, -0,095 MPa), y el sólido restante se enfrió a 0-5 °C en baño de hielo, seguido de la adición gota a gota de 2.630 ml de agua helada (siendo la temperatura interior controlada a 0-5 °C) durante aproximadamente 1 hora, y la solución de reacción se extrajo con éter isopropílico (650 ml x 3), la capa de éter se combinó y se lavó con agua hasta que es neutra (siendo el pH de los lavados con agua de 7,0), y la capa de éter se secó sobre sulfato de sodio anhidro durante la noche. El agente desecante se retiró por filtración, el disolvente se retiró bajo presión reducida por succión usando una bomba de agua (40-42 °C, -0,095 MPa), y posteriormente se recogieron 231,6 g de fracción a 111-121 °C/3 mmHg bajo presión reducida usando una bomba de aceite.
RMN de 1H (CDCls) 5 (ppm): 7,35-6,50 (m, 3H), 2,93-2,65 (m, 2H), 2,26-1,27 (m, 9H).
Etapa 3: Preparación de 3-[(2-ciclopentil-2-hidroxil-2-(2-furil))etoxil]-1-azabiciclo[2,2,2]octano libre de álcali a partir de R-3-quinuclidinol

A 18,72 g (147 mmol) de R-3-quinuclidinol disponible comercialmente se añadieron 190 ml de DMSO, seguido de la adición de 7,59 g (190 mmol) de hidruro de sodio, y la mezcla se hizo reaccionar a 20-60 °C durante 0,5-12 h, y después se enfrió a temperatura ambiente, y a ello se añadió una solución de 33,88 g (190 mmol) de 1-(2-furil)-1-ciclopentiloxirano (autopreparado) en 45 ml de DMSO, y una vez finalizada la adición, la mezcla resultante se calentó a 20-70 °C en baño de aceite para reaccionar durante 0,5-12 horas. Bajo la condición de baño de hielo, se añadieron 120 ml de agua helada a una temperatura interna de 30 °C o menor. La mezcla de reacción se extrajo con éter isopropílico, 100 ml x 3; y la capa de éter se combinó y se lavó con una solución acuosa saturada de NaCl, 100 ml x 3. La capa orgánica se secó sobre sulfato de sodio anhidro durante la noche, el agente desecante se retiró por filtración, y el disolvente se retiró bajo presión reducida para obtener 42,74 g de 3-[(2-ciclopentil-2-hidroxil-2-(2-furil))etoxil]-1-azabiciclo [2,2,2]octano libre de álcali como materia aceitosa de color rojo, con un rendimiento de 95,32 %. El producto obtenido fue (2R,3R),(2S,3R)-3-[(2-ciclopentil-2-hidroxil-2-(2-furil))etoxil]-1-azabiciclo[2,2,2]octano libre de álcali.
Etapa 4: Purificación de 3-[(2-ciclopentil-2-hidroxil-2-(2-furil))etoxil]-1-azabiciclo[2,2,2]octano libre de álcali por cromatografía de columna y el tratamiento de purificación relacionado
La muestra de la etapa 3 anterior se separó en columna de gel de sílice, utilizando diclorometano o triclorometano y metanol, amoniacados, como fase móvil y una placa de TLC para controlar la pureza de la muestra. Bajo el sistema de elución anterior, una muestra de una mezcla de ambas configuraciones (2R,3R) y (2S,3R) de 3-[(2-ciclopentil-2-hidroxil-2-(2-furil))etoxil]-1-azabiciclo[2,2,2]octano libre de álcali podía ser purificada en dos tipos de (2R,3R) y (2S,3R) libre de álcali, dependiendo de la secuencia de elución, obteniéndose de ese modo 17,84 g de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-(2-furil))etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 83,5 %, y 18,03 g de la configuración (2S,3R) de 3-[(2-ciclopentil-2-hidroxil-2-(2-furil))etoxil]-1-azabiciclo[2,2,2]octano libre de álcali con un rendimiento de 84,37 %.
Etapa 5: igual que la etapa 3 en el Ejemplo 1
Etapa 6: Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-(2-furil))etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,78 g (9,1 mmol) de la configuración (2R,3R) de la base y se disolvieron añadiendo 18 ml de cloroformo para obtener una solución transparente de color amarillo, a la que se añadieron 11,033 g (51,3 mmol) de 3-bromopropoxibenceno y 50 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 h bajo la protección de nitrógeno, siendo el final de la reacción controlado mediante TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25-40 °C por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 3,97 g de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-(2-furil))etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 83,9 %.
El compuesto preparado en el Ejemplo 32 se hizo reaccionar con Ag2Ü para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes.
Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) (ppm): 57,32-6,19 (m, 8H), 54,14-3,60 (m, 5H), 53,42-3,11 (m, 8H), 52,11 (m, 2H), 1,97 (m, 1H), 5 1,83-1,46 (m, 13H).
Ejemplo 33. Bromuro de (2R,3R)-3-[(2-ciclopentil-2-metoxil-2-(3-piridil))etoxil]-1 -(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano
Etapas 1,2, 3 y 4: igual que las etapas 1,2, 3 y 4 en el Ejemplo 31
Etapa 5: igual que la etapa 4 en el Ejemplo 1
Etapa 6: igual que la etapa 6 en Ejemplo 31
Etapa 7: Bromuro de (2R,3R)-3-[(2-ciclopentil-2-metoxil-2-(3-piridil))etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano

A un matraz de 100 ml, con forma de berenjena, se añadieron 2,655 g (4,87 mmol) de bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-(3-piridil))etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano y se disolvieron añadiendo 30 ml de acetonitrilo, y a ello se añadieron 0,5 g de NaH, seguido de la adición gota a gota de una solución de bromometano (0,5 g) en 10 ml de acetonitrilo, y la mezcla resultante se agitó entonces a temperatura ambiente para reaccionar durante 20-90 horas bajo la protección de nitrógeno, siendo el final de la reacción controlada por TLC (condiciones de la TLC: cloroformo/metanol/agua amoniacal = 5,0 ml/1,5 ml/2 d). Una vez finalizada la reacción, el disolvente se retiró de la solución de reacción a 25-40 °C por evaporación rotativa bajo presión reducida mediante una bomba de agua, obteniéndose de ese modo una materia aceitosa de color amarillo, a la que se añadió éter etílico para precipitar una gran cantidad de sólido, y el sólido se recogió mediante filtración por succión para obtener 2,19 g de bromuro de (2R,3R)-3-[(2-ciclopentil-2-metoxil-2-(3-piridil))etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano como un sólido de color blancuzco, con un rendimiento de 80,37 %.
El compuesto preparado en el Ejemplo 33 se hizo reaccionar con Ag2O para retirar el átomo de bromo para obtener hidróxido, que podía hacerse reaccionar con otros ácidos para transformarlos en las sales correspondientes. Ejemplos de una sal de un ácido farmacéuticamente aceptable incluyen una sal derivada de un ácido inorgánico, como hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito o similar; y una sal derivada de un ácido orgánico relativamente no tóxico como ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico o ácido galactónico o similar. Los ejemplos incluyen además una sal de un aminoácido como arginina o similar.
RMN de 1H (D2O) (ppm): 58,77-6,66 (m, 9H), 54,15-3,65 (m, 5H), 53,44-3,13 (m, 11H), 52,16 (m, 2H), 2,03 (m, 1H), 51,85-1,43 (m, 13H).
Ejemplo 1: Intensidad del antagonismo de los compuestos de ejemplo en la respuesta contráctil del músculo liso traqueal de conejillos de Indias inducidos por el carbacol (CCh)
1. Procedimiento:
Preparación del espécimen aislado de músculo liso traqueal de un conejillo de Indias:
Después de que un conejillo de Indias fuera narcotizado con uretano, la tráquea entre la garganta y la carina del conejillo de Indias fue extirpada rápidamente, y colocada en solución Krebs-Henseleit (K-H) (composición (g/l): NaCl 6,92, KCl 0,35, CaCl2 0,28, KH2PO4 0,16, MgSO4'7H2O 0,4568, NaHCO3 2,1, glucosa 2,0) purgada con un gas mezcla de 5 % CO2 y 95 % O2. Después de separar los tejidos conectivos y las grasas alrededor de la tráquea, la tráquea se cortó en trozos de tráquea con una anchura de aproximadamente 3 mm y una longitud de 20 mm con unas tijeras quirúrgicas, con ambos extremos de la pieza de la tráquea ligados con sutura de seda de 4~0, y después se colocaron en un baño termostático conteniendo 5 ml de solución K-H (pH 7,4) a 37 °C, purgada continuamente con el gas mezcla de 5 % CO2 y 95 % de O2. Un transductor de la tensión muscular se conectó al extremo superior para aplicar 1,0 g de tensión de reposo al espécimen, el cambio de tensión se registró, la solución de cultivo se reemplazó cada 20 min, y el ensayo se inició después de estar equilibrado durante 60 min.
Procedimiento de administración: Después de estabilizar la pieza de la tráquea, se añadieron 3 * 10-6 mol/l de CCh en un baño, y después de que la tensión contráctil de la pieza de la tráquea alcanzó un nivel máximo, los compuestos de ejemplo, bromuro de ipratropio y bromuro de tiotropio se añadieron a un baño Magnus mediante un régimen de dosificación acumulativa con una dosificación de 10-9 ~ 10-5 mol, respectivamente, y se observó el estado diastólico de la pieza de la tráquea: Si no se producía respuesta (inferior a la concentración de umbral), continuó añadiéndose una siguiente dosis en secuencia; si se producía respuesta, después de que se alcanzara una meseta diastólica, se añadió posteriormente una siguiente dosis. La operación anterior se repitió hasta que la curva contráctil alcanzaba el valor mínimo. Finalmente, se añadieron 10-6 mol/l de isoprenalina para que se alcanzara la máxima relajación y se registró la curva.
Enfoque estadístico: El resultado se expresó como media ± desviación estándar, y el recuento se hizo mediante el paquete estadístico Sigma stat. Los datos se analizaron mediante análisis de varianza, la comparación entre las medias de una pluralidad de especímenes se realizó mediante la prueba Student-Newman-Keuls (SNK); nivel estadístico significativo a = 0,05 (bilateral). EC50 (95 % de límite de confianza) se calculó mediante el software POMS versión 2.0 de Shanghai Scientific and Technical Publishers.
2. Conclusión:
3 * 10-6 mol/l de CCh podría causar que el músculo liso traqueal del conejillo de Indias generara una respuesta contráctil duradera y estable. En el régimen de dosificación acumulativa, los compuestos de ejemplo y los fármacos de control de bromuro de ipratropio y de bromuro de tiotropio se añadieron en el baño Magnus, y cada uno de los compuestos de ejemplo y cada uno de los fármacos de control podían relajar la contracción del músculo liso traqueal inducido por CCh. Los resultados se mostraron en la tabla 2.
3. Resultados
Tabla 2: Antagonismo in vitro de los compuestos de la presente invención contra la contracción del músculo liso traqueal inducido por una concentración de 3 * 10-6 mol/l de CCh, IC50 (pM)



Los resultados de la Tabla 2 mostraron que cada uno de los compuestos de ejemplo tenía un efecto antagonista significativo en el receptor M, en donde la configuración (2R,3R) del compuesto tenía el mayor efecto, y la intensidad del efecto de una pluralidad de compuestos era comparable a la de los agentes de control positivo de bromuro de ipratropio y bromuro de tiotropio. Los compuestos de la presente invención y bromuro de ipratropio tenían un tiempo de comienzo más corto que el bromuro de tiotropio, y por el contrario, el bromuro de tiotropio actuó más lentamente. Los compuestos de la presente invención y el bromuro de tiotropio tenían una mayor duración de la acción.
Ejemplo 2: Efecto ligante y selectivo de los compuestos de la presente invención en tres subtipos de receptor M
1. Procedimiento: las células mn, m2 y m3 del ovocito de hámster chino (CHO) transfectado se colocaron en un medio de cultivo DMEM (que contenía un 15 % de suero fetal bovino, L-glutamina, 1 % de aminoácido no esencial, 1 % de antibiótico/agente antifúngico), respectivamente, y después se incubaron en una incubadora con 5 % de CO2 a 37 °C. Cuando las células de un matraz de cultivo se cultivaron y proliferaron para formar células en monocapa que se difundieron sobre aproximadamente 90 % del fondo del matraz, se desechó el medio de cultivo, y el matraz de cultivo se lavó dos veces con una solución tampón de PBS (pH 7,4), y después las células fueron raspadas con una solución helada de tampón fosfato (pH 7,7, que contenía 5 mmol/l de MgCh). Las células recogidas fueron homogeneizadas con un homogeneizador de vidrio con Teflón, la solución homogeneizada se centrifugó a baja temperatura, 20.000 r x 20 min, y el precipitado se homogeneizó con una solución tampón de reacción (pH 7,7, que contenía 5 mmol/l de MgCh) para formar una suspensión proteica de membrana. La cantidad de proteína añadida en cada uno de los tubos de reacción era: mu (aproximadamente 0,05 mg), m2 (0,05 mg), m3 (0,1 mg), respectivamente; la concentración de [3H]-QNB era 0,1-2,16 nmol/l; se añadió 1 pmol/l de atropina al tubo de unión no específico; y el volumen total de reacción fue de 300 pl. La solución resultante se hizo reaccionar entonces a 25 °C durante 30 minutos y se enfrió con una solución tampón de reacción helada y, posteriormente, se recogió en una membrana de filtro de fibra de vidrio mediante un colector de células multicabezal. Después de ser secada a 80 °C, la lámina de membrana filtrante se colocó en un vial de centelleo de líquidos, seguido de la adición de 5 ml de agente de centelleo de líquidos, y después se mantuvo en lugar oscuro durante la noche, con cpm medido por un espectrómetro de centelleo de líquidos. Los valores Bmax y Kd se calcularon mediante el software GraphPad Prism. En cada uno de los tubos de ensayo se añadieron ligandos marcados con 3H-QNB que no tiene selectividad sobre el receptor M, en los que las concentraciones eran: mu y m3 (1,042 nmol/l), m2 (1,81 nmol/l), repectivamente; diferentes concentraciones de competidor no marcado, Pirenzepina (PZ, competidor selectivo de mn) o Galamina (GI, competidor selectivo de m2) o 4-DAMP (competidor selectivo de m3) o los compuestos de la presente invención, se añadieron simultáneamente, la concentración final fue de 10"10~10"4 mol/l, con 11 dosis en total; posteriormente se añadió la misma cantidad de espécimen de proteína de membrana; el volumen total de reacción fue de 300 pl, y después se realizó una reacción de unión competitiva. Se proporcionó además un tubo de unión no específico, y la unión no específica se midió por una gran cantidad de atropina (la concentración final fue de 1 pmol/l). La solución resultante se hizo reaccionar a 25 °C durante 30 minutos y se enfrió con una solución tampón de reacción, y después se recogió en una membrana de filtro de fibra de vidrio mediante un colector de células multicabezal. Después de ser secada a 80 °C, la lámina de membrana filtrante se colocó en un vial de centelleo de líquidos, seguido de la adición de 5 ml de agente de centelleo de líquidos, y después se mantuvo en lugar oscuro durante la noche, con cpm medido por un espectrómetro de centelleo de líquidos. Los respectivos valores de Ki de los competidores en tres subtipos de receptor M se calcularon mediante el software GraphPad Prism, y se transformaron en valores pKi, para comparar la selectividad de PZ, GI, 4-DAMP y los compuestos a ensayar en los subtipos de receptor M.
2. Resultados
Tabla 3: Comparación de la selectividad del receptor de tres inhibidores competitivos conocidos y los compuestos de la presente invención en tres subtipos de receptor M (x ± s, n = 6)

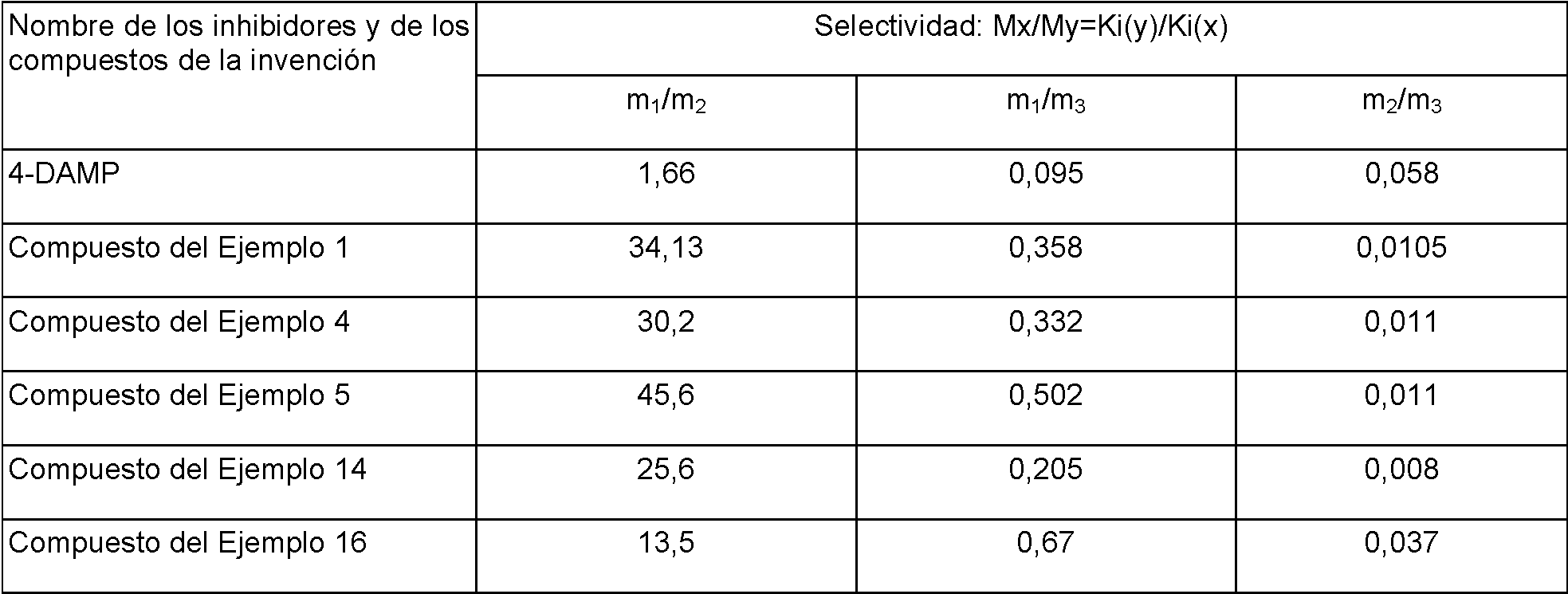
3. Conclusión
La prueba anterior con células de CHO transfectadas mediante ADNc de tres subtipos m1, m2 y m3 de receptor M se utilizó para identificar la selectividad de los compuestos de la presente invención en los subtipos de receptor M. Teniendo en cuenta las características farmacológicas de Pirenzepina, Galamina, y 4-DAMP, los resultados de los ensayos de inhibición competitiva de los tres compuestos con tres tipos de células demostraron que los tres tipos de células podían utilizarse para identificar una selectividad de receptor de un fármaco. Los resultados del ensayo anterior mostraron que los compuestos probados de la presente invención tenían la selectividad más alta en el receptor m3, la segunda selectividad más elevada en el receptor mp, y la selectividad más baja en el receptor m2; en donde tenían los mayores efectos en los receptores m3 y m1. Tenían ventajas significativas en el tratamiento de la rinitis, hipersensibilidad de las vías respiratorias, traquitis crónica senil, EPOC y enfermedades ulcerativas del tubo digestivo, en comparación con la técnica anterior.
Ejemplo 3: Determinación de la intensidad y duración del antagonismo de los compuestos de ejemplo en la respuesta contráctil de los bronquios en conejillos de Indias inducidos por metacolina (Mch)
1. Procedimiento:
Determinación del volumen tidal, caudal de las vías respiratorias y presión transpulmonar:
Se inyectaron intraperitonealmente 1,5 g/kg de uretano para narcotizar al conejillo de Indias. El conejillo de Indias se fijó en posición supina y se trató con intubación traqueal, y la vena yugular externa se separó y se insertó una aguja residente; el conejillo de Indias estaba encerrado en un pletismógrafo corporal, se insertó una aguja sin punta para intubación en la cavidad torácica entre las costillas 4~5 del protórax del conejillo de Indias, y se podía medir la presión intratorácica (con un valor negativo de una columna de agua de un manómetro de agua y una fluctuación con la respiración del conejillo de Indias como marcas). Después de la estabilización, los valores del volumen tidal, el caudal de las vías respiratorias y la presión transpulmonar del conejillo de Indias antes de la administración de Mch se registraron como valores base mediante un sistema MedLab de recogida y procesamiento de señales biológicas. Se inyectaron de forma intravenosa 10 pg de Mch/kg de peso corporal. Los cambios en caudal de las vías respiratorias, volumen tidal y presión transpulmonar del conejillo de Indias se observaron a los 5 minutos. Cálculo de Raw y Cdin: los cambios del aumento en porcentaje del valor Raw y de la disminución en porcentaje de Cdin se calcularon después de la inhalación de Mch.
Las fórmulas de cálculo de Raw y Cdin fueron respectivamente:
Raw después de la inhalación de Mch - Raw antes de la inhalación (valor base)
% de aumento de Raw = ---------------------------------------------------------------------------------------------------------- x 100 % Raw antes de la inhalación (valor base)
Raw antes de la inhalación de Mch (valor base) - Raw después de la inhalación
% de disminución de Cdin = ---------------------------------------------------------------------------------------------------------- x 100 %
Raw antes de la inhalación de Mch (valor base)
Relación dosis-efecto: Para cada uno de los compuestos de ejemplo, 27 conejillos de Indias se dividieron aleatoriamente en 3 grupos: un grupo de control de disolventes con 15 conejillos de Indias, un grupo con 1 pg/kg de los compuestos de ejemplo 4, 10, 14 y 26, un grupo con 3 pg/kg de los compuestos de ejemplo 4, 10, 14 y 26 y un grupo con 10 pg/kg de los compuestos de Ejemplo 4, 10, 14 y 26; después de 30 minutos del goteo en las vías respiratorias de la concentración anterior de fármacos, 10 pg de Mch/kg de peso corporal se inyectaron por vía
intravenosa para la excitación, y se determinó la resistencia de las vías respiratorias (Raw) y la compliancia dinámica pulmonar (Cdin) dentro de 5 min.
Relación tiempo-efecto: Después de que los conejillos de Indias fueron narcotizados, se introdujeron por goteo 10 pg/kg y 5 pg/kg de los compuestos de ejemplo 4, 10, 14, y 26 en las vías respiratorias. Se inyectaron por vía intravenosa 10 pg de Mch/kg de peso corporal para la excitación después de 0,25 h, 0,5 h, 1 h, 1,5 h, 2 h, 4 h, 6 h, 12 h y 24 h después de la dosificación, respectivamente, y se determinaron la resistencia de las vías respiratorias (Raw) y la compliancia dinámica pulmonar (Cdin) a los 5 min.
2. Resultados:
Relación dosis-efecto: las dosis de los compuestos de ejemplo 4, 10, 14, y 26 fueron 1 pg/kg, 3 pg/kg, y 10 pg/kg, respectivamente, y después de 30 min del goteo en las vías respiratorias, se inyectaron 10 pg de Mch/kg de peso corporal por vía intravenosa para la excitación, y se determinó la resistencia de las vías respiratorias (Rw) y la compliancia dinámica pulmonar (Cdin) a los 5 minutos, y los resultados se mostraron en la Tabla siguiente:
Tabla 4. Antagonismo de los compuestos de ejemplo 4, 10, 14 y 26 en la respuesta contráctil de los bronquios en conejillos de Indias inducidos por la relación (Media ± S.E.M) efecto-dosis con Mch

Enfoque estadístico: se usó ANOVA unidireccional, y la comparación entre varios grupos se probó mediante el procedimiento Bonferroni; se realizó la comparación con el grupo de control de disolventes, * P < 0,05, * * P < 0,01, * * * P < 0,001.
Cuando se inyectaron 10 pg de Mch/kg por vía intravenosa, la resistencia de las vías respiratorias de los conejillos de Indias aumentó en un 328 %, la compliancia dinámica pulmonar disminuyó en un 73 %. Una vez 1, 3, y 10 pg/kg de los compuestos en el Ejemplo 4, 10, 14, y 26 fueron sometidos a goteo en las vías respiratorias de los conejillos de Indias, el aumento de la resistencia y la disminución de la complianza dinámica pulmonar fueron inhibidas de forma dependiente de la dosis. Las relaciones de inhibición de los tres grupos de dosificación del compuesto del ejemplo 4 contra el aumento de la resistencia en las vías respiratorias fueron 64,2 % (p<0,01), 86,1 % (p<0,001) y 90,8 % (p<0,001), respectivamente; las relaciones de la inhibición contra la disminución de la complianza dinámica pulmonar fueron 11,2 % (p>0,05), 46,6 % (p<0,001) y 50,0 % (p<0,001), respectivamente. Las relaciones de inhibición de los tres grupos de dosificación del compuesto de ejemplo 10 contra el aumento de la resistencia en las vías respiratorias fueron 63,7 % (p<0,01), 84,5 % (p<0,001) y 91,3 % (p<0,001), respectivamente; las relaciones de
la inhibición contra la disminución de la complianza dinámica pulmonar fueron 10,9 % (p>0,05), 45,9 % (p<0,001) and 49,8 % (p<0,001), respectivamente. Las relaciones de inhibición de los tres grupos de dosificación del compuesto del ejemplo 14 frente al aumento de la resistencia en las vías respiratorias fueron 63,5 % (p<0,01), 85,4 % (p<0,001) y 90,5 % (p<0,001), respectivamente; las relaciones de la inhibición contra la disminución de la complianza dinámica pulmonar fueron 11,0 % (p>0,05), 46,1 % (p<0,001) y 49,5 % (p<0,001), respectivamente. Las relaciones de inhibición de los tres grupos de dosificación del compuesto del ejemplo 26 frente al aumento de la resistencia en las vías respiratorias fueron 64,4 % (p<0,01), 87,2 % (p<0,001) y 92,1 % (p<0,001), respectivamente; las relaciones de la inhibición contra la disminución de la complianza dinámica pulmonar fueron 11,0 % (p>0,05), 47,1 % (p<0,001) y 49,9 % (p<0,001), respectivamente.
Relación tiempo-efecto: Cuando se inyectaron por vía intravenosa 10 pg de Mch/kg, la resistencia de las vías respiratorias de los conejillos de Indias aumentó en un 328 %. Después de 0,25 h del goteo de 10 pg/kg de los compuestos de los ejemplos 4, 10, 14, y 26 en las vías respiratorias, la relación de inhibición contra el aumento de la resistencia de las vías respiratorias fue de 80 % o más; y podía alcanzar el 90 % o más de la relación de inhibición máxima inmediatamente después de 1 h. Con el tiempo, la relación de inhibición contra el aumento de la resistencia de las vías respiratorias fue todavía 85 % o más después de 24 h, y hubo diferencias estadísticas en comparación con el grupo de control de disolventes (p < 0,01-0,001). Para confirmar la exactitud de los resultados, la dosificación de los compuestos de ejemplo 4, 10, 14, y 26 se redujo a 5 pg/kg, cuyos resultados mostraron que cuando 5 pg/kg de los compuestos de ejemplo 4, 10, 14, y 26 se sometieron a goteo en las vías respiratorias, la relación de inhibición contra la resistencia de las vías respiratorias fue de 85 % o más (p < 0,001) después de 12 h, y la relación de inhibición contra la resistencia de las vías respiratorias fue de 65 % o más (p < 0,01) después de 24 h. Después de 12 h del goteo de 10 pg/kg y 5 pg/kg de los compuestos de ejemplo 4, 10, 14, y 26 en las vías respiratorias, no hubo diferencia significativa en la relación de inhibición contra la resistencia de las vías respiratorias entre las dosis de 10 pg/kg y 5 pg/kg. Sin embargo, después de 24 h, hubo diferencias significativas en la relación de inhibición contra la resistencia de las vías respiratorias (p < 0,05) entre 10 pg/kg y 5 pg/kg de los compuestos de ejemplo 4, 10, 14 y 26. Los resultados anteriores indicaron que: los tiempos de acción de 10 pg/kg y 5 pg/kg de los compuestos de ejemplo 4, 10, 14, y 26 administrados por goteo en las vías respiratorias eran más de 24 h, y estos compuestos eran antagonistas de los receptores M superduraderos.
Los compuestos que se muestran en la Tabla 5 se prepararon además según la presente invención, y se determinó su IC50 (mM) de resistencia a la contracción del músculo liso traqueal en conejillos de indias inducidos por una concentración de 3 * 10-6 mol/l de CCh.
Tabla 5. Rotación óptica y la IC50 (mM) de resistencia in vitro para la contracción del músculo liso traqueal de conejillos de indias inducidos por una concentración de 3 * 10-6 mol/l de CCh, de otros compuestos sintetizados según la presente invención




Ejemplo 5: Composiciones y preparación de las mismas
Preparación y uso de naristillae, pulverizador nasal, aerosol, aerosol en polvo, inhalación con atomización y comprimidos
1. Naristillae de prescripción de un solo fármaco
Compuesto del Ejemplo 16 3 mg
Cloruro de benzalconio 3 mg
H2O 10 ml
En condiciones de producción de clase 100.000, el ingrediente activo del compuesto del ejemplo 10 y un bactericida (cloruro de benzalconio) se disolvieron en H2O, y después se rellenaron en un vial de color marrón equipado con un tapón fusiforme que podía ser utilizado para el goteo nasal.
Para este naristillae de prescripción de un solo fármaco, 2,0-20 mg podría disolverse en cada 10 ml de agua, para ser formulado en diferentes concentraciones de naristillae; y la cantidad de bactericida de cloruro de benzalconio podía variar de 1-5 mg.
La dosis de naristillae de la prescripción de un solo fármaco fue de 2-3 gotitas (0,1 ml-0,15 ml)/fosa nasal, aproximadamente 20-300 pg/fosa nasal cada vez.
2. Compuesto naristillae
Compuesto del Ejemplo 10 2,5 mg
Fenoterol 10 mg
Cloruro de benzalconio 5 mg
H2O 10 ml
En condiciones de producción de clase 100.000, los ingredientes activos (compuesto del ejemplo 10 y fenoterol) y un bactericida (cloruro de benzalconio) se disolvieron en H2O, y después se llenaron en un vial de color marrón equipado con un tapón fusiforme que podía ser utilizado para goteo nasal. La cantidad de bactericida de cloruro de
benzalconio podría oscilar entre 1-5 mg. La dosis del compuesto naristillae fue de 2-3 gotitas (0,1 ml-0,15 ml)/fosa nasal, aproximadamente 25 pg/fosa nasal cada vez.
3. Pulverización nasal de la bomba dosificadora
Compuesto del Ejemplo 4 3,33 mg
Cloruro de benzalconio 5.00 mg
HCl 1N añadido hasta pH 4-6
H2O pura 2.0 ml
En condiciones de producción de clase 100.000, los componentes anteriores se disolvieron en H2O pura, y después se llenaron en un frasco equipado con una bomba dosificadora adecuada para pulverización nasal, y el volumen pulverizado de esta bomba cada vez fue de 70-90 pl. La dosis fue 1-2 pulverizaciones/fosa nasal, aproximadamente 21-60 pg/fosa nasal cada vez.
Para obtener una dosis más grande en el tratamiento de pacientes graves, la cantidad de compuesto 4 podría aumentarse.
4. Aerosol de dosis medida que contiene un propulsor
Componente Peso
Compuesto del Ejemplo 4 10 mg
Etanol 2,4 g
Ácido oleico 7,0 mg
HFA-134a 11,75 g
Después de que el compuesto del Ejemplo 4 se disolviera con etanol y ácido oleico, se llenó e1HFA-134a mediante un procedimiento de una sola etapa. Luego, en condiciones de producción de clase 100.000, la mezcla resultante se mezcló con un tensioactivo y propelentes de 1-fluro-3-clorometano, 2-fluro-2-clorometano y 4-fluro-2-cloroetano en proporción, y después se encapsularon en un recipiente a presión cuantitativa mediante una máquina de llenado a presión. Cada frasco contenía 10 g, cada aerosol con 100 mg, y un contenido de 20 pg del compuesto del Ejemplo 4.
5. Aerosol de polvo de tipo cápsula
Un aerosol de polvo de tipo cápsula se componía de los siguientes ingredientes:
Compuesto del Ejemplo 4 0,05 g
Lactosa 98,95 g
Benzoato de sodio 1,0 g
En condiciones de producción de clase 100.000, el compuesto del Ejemplo 4 junto con lactosa y benzoato de sodio se sometieron a micronización, con un diámetro de partícula de 5 pm o menor, y después se mezclaron a fondo y homogéneamente, y finalmente se encapsularon en una cápsula. Cada cápsula contenía 40 mg, y cada inhalación contenía 20 pg del compuesto de la presente invención. En esta composición farmacéutica para el tratamiento de una enfermedad del sistema respiratorio, la lactosa era un diluyente que podría seleccionarse adicionalmente de arabinosa, glucano, manitol, xilitol, sacarosa, fructosa, sorbitol, maltosa, aminoácido o glucosa o similar; el benzoato de sodio era un lubricante, y el estearato de magnesio también podía utilizarse como lubricante.
6. Comprimido
Compuesto del Ejemplo 6 5 g
Lactosa 71,5 g
Celulosa microcristalina 22 g
Estearato de magnesio 1,5 g
El compuesto del Ejemplo 6 se mezcló homogéneamente con lactosa y celulosa microcristalina, y además se mezcló homogéneamente después de añadir estearato de magnesio, y la mezcla resultante se prensó en seco en comprimidos, con un peso por comprimido de 100 mg.
7. Solución de inhalación
Compuesto del Ejemplo 10 100 mg
Solución salina Normal 1.000 ml
El compuesto del Ejemplo 10 se disolvió en 800 ml de solución salina normal, y después se transfirió a un matraz aforado de 1.000 ml, y se complementó con solución salina normal hasta la marca de escala, y después la solución resultante se dividió y se cargó en ampollas, 1 ml/ampolla, y se calentó a 115 °C durante 30 min.
Claims (12)
1. Compuesto de fórmula I:

en donde en la fórmula I:
n se selecciona entre 1-7,
R1 es un hidrocarbilo C3-C7, que puede estar no sustituido u opcionalmente sustituido por halógeno, alcoxi, alcoxihidrocarbilo, heterociclilo o arilo,
R2 es un arilo o un heteroarilo que contiene uno o más heteroátomos, que puede estar no sustituido u opcionalmente sustituido,
R3 es un hidroxilo, halógeno, alcoxi o aciloxi, en donde el alcoxi o aciloxi puede estar no sustituido u opcionalmente sustituido por halógeno, hidroxilo, alcoxi, hidrocarbilo, alcoxihidrocarbilo, heterociclilo o arilo;
R4 y R5 pueden estar presentes o ausentes, y se seleccionan de forma independiente de un grupo que consiste en halógeno, hidroxilo, alcoxi, hidrocarbilo, alcoxihidrocarbilo, heterociclilo o arilo, cuando están presentes;
Y es un alquileno C1-C7, lineal o ramificado, o -(CH2-O-CH2V, que puede estar opcionalmente sustituido, en donde m es igual a 1-3,
X- es un radical ácido o hidroxilo.
2. Compuesto según la reivindicación 1, en donde:
n es 1-3,
R1 es un cicloalquilo no sustituido,
R2 es un fenilo, naftilo o bifenilo, que puede estar no sustituido u opcionalmente sustituido por uno o más halógenos, hidroxilo, fenilo, -OR6, -SR6, -NR6R7, -NHCOR6, -CONR6R7, -CN, -NO2, -COOR6, -CF3 o hidrocarbilo C1-C4, lineal o ramificado, R6 y R7 puede ser un hidrógeno átomo, un hidrocarbilo C1-C4, lineal o ramificado, o puede formar un ciclohidrocarbilo entre sí,
R3 es un hidroxilo o metoxilo
Y es un metileno, etileno, propileno o -(CH2-O-CH2)-.
3. Compuesto según la reivindicación 1 o 2, en donde
n es 1,
R1 es un ciclopentilo o ciclohexilo,
R2 es un fenilo, piridilo, furilo o tienilo, no sustituidos,
Y es un etileno o propileno.
4. Compuesto según una cualquiera de las reivindicaciones anteriores, en donde X-es un radical ácido de un ácido inorgánico farmacéuticamente aceptable o hidróxido, de manera que el compuesto de la fórmula I está en forma de una sal o base de amonio cuaternario farmacéuticamente aceptable, la sal farmacéuticamente aceptable resultante se selecciona de un grupo que consiste en hidrocloruro, bromuro, yoduro, nitrato, carbonato, bicarbonato, fosfato, hidrofosfato, fosfato dihidroxilado, sulfato, disulfato o fosfito.
5. Compuesto según una cualquiera de las reivindicaciones 1-3, en donde X-es un radical ácido de un ácido orgánico farmacéuticamente aceptable, de manera que el compuesto de la fórmula I está en forma de una sal de un ácido orgánico, dicho ácido orgánico es ácido acético, ácido propiónico, ácido isobutírico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido mandélico, ácido ftálico, ácido bencensulfónico, ácido ptoluensulfónico, ácido cítrico, ácido tartárico, ácido metansulfónico, ácido glucurónico, ácido galactónico o aminoácido.
6. Compuesto según la reivindicación 1, que es:
Bromuro de (2S,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano;
Bromuro de (2S,3S)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano;
Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano;
Bromuro de (2R,3S)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano;
Bromuro de (2S,3R),(2R,3S)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano; Bromuro de (2R,3R),(2S,3S)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano; Bromuro de (2S,3R)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1 -(3-fenoxipropil)-1-azabiciclo[2,2,2]octano;
Bromuro de (2S,3S)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano;
Bromuro de (2R,3R)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1 -(3-fenoxipropil)-1-azabiciclo[2,2,2]octano;
Bromuro de (2R,3S)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1 -(3-fenoxipropil)-1-azabiciclo[2,2,2]octano;
Bromuro de (2R,3S),(2S,3R)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano; Bromuro de (2R,3R),(2S,3S)-3-[(2-ciclohexil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano; Bromuro de (2S,3R)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano;
Bromuro de (2S,3S)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano;
Bromuro de (2R,3S)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano;
Bromuro de (2R,3R)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano;
Bromuro de (2R,3S),(2S,3R)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano; Bromuro de (2R,3R),(2S,3S)-3-[(2-ciclobutil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano; Bromuro de (2S,3R)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano;
Bromuro de (2R,3S)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano;
Bromuro de (2S,3S)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano;
Bromuro de (2R,3R)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano;
Bromuro de (2R,3S),(2S,3R)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano; Bromuro de (2R,3R),(2S,3S)-3-[(2-ciclopropil-2-hidroxil-2-fenil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano; Bromuro de (2S,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(2-fenoxietil)-1-azabiciclo[2,2,2]octano;
Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-(2-fenoxietil)-1-azabiciclo[2,2,2]octano;
Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-fenoximetil-1-azabiciclo[2,2,2]octano;
Cloruro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-fenil)etoxil]-1-fenoximetoximetil-1-azabiciclo[2,2,2] cloruro de octano; Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-naftil)etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano;
Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-(o-clorofenil))etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano; Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-(3-piridil))etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano;
Bromuro de (2R,3R)-3-[(2-ciclopentil-2-hidroxil-2-(2-furil))etoxil]-1-(3-fenoxipropil)-1 -azabiciclo[2,2,2]octano; o Bromuro de (2R,3R)-3-[(2-ciclopentil-2-metoxil-2-(3-piridil))etoxil]-1-(3-fenoxipropil)-1-azabiciclo[2,2,2]octano.
7. Una composición farmacéutica que comprende el compuesto según una cualquiera de las reivindicaciones 1-5 y un portador farmacéuticamente aceptable, en donde la composición puede ser cualquier forma de dosificación adecuada, preferiblemente la forma de dosificación para la administración de inhalación o administración nasal; y la composición puede, además, comprender o no comprender, uno o más de los otros fármacos disponibles para la administración combinada.
8. Un procedimiento para la fabricación del compuesto según la reivindicación 1 que comprende las etapas de:
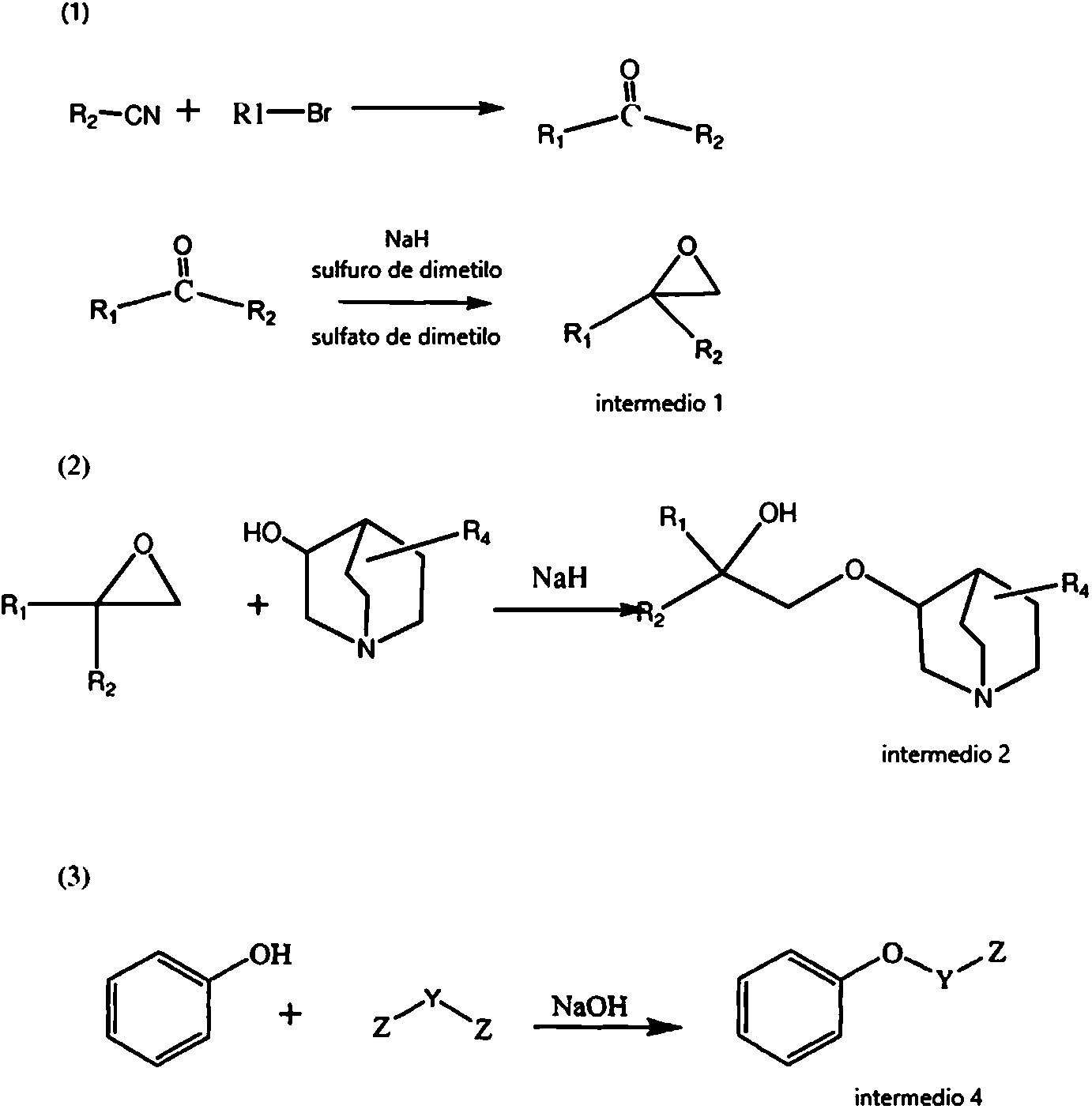
en donde Z es un átomo de halógeno,
(4) hacer reaccionar el intermedio 2 con el intermedio 4, para obtener el compuesto de fórmula (I).
9. El procedimiento según la reivindicación 8, que comprende además la etapa de purificación y separación de los isómeros químicos del intermedio 2 después de terminar la etapa (2).
10. El procedimiento según la reivindicación 8 o 9, en donde después de terminar la etapa (4), el compuesto se hace reaccionar con Ag2O para reemplazar el halógeno con hidróxido, que se puede transformar después en otro radical ácido mediante la reacción con otro ácido.
11. Uso del compuesto según una cualquiera de las reivindicaciones 1-5 para la fabricación de un medicamento capaz de tratar enfermedades asociadas con receptores M.
12. Uso según la reivindicación 11, en donde dichas enfermedades se seleccionan de un grupo que consiste en rinitis, rinitis después de un resfriado, traquitis crónica, hipersensibilidad de las vías respiratorias, asma, enfermedades pulmonares obstructivas crónicas, tos, incontinencia urinaria, micción frecuente, síndrome de vejiga inestable, espasmos vesicales, inflamación de la vejiga y enfermedades gastrointestinales como el síndrome de intestino irritable, colitis espástica, así como úlceras duodenales y gástricas.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201310297901 | 2013-07-13 | ||
| PCT/CN2014/000669 WO2015007073A1 (zh) | 2013-07-13 | 2014-07-11 | 奎宁类化合物、其光学异构体及其制备方法和医药用途 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2726664T3 true ES2726664T3 (es) | 2019-10-08 |
Family
ID=52345746
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES14825941T Active ES2726664T3 (es) | 2013-07-13 | 2014-07-11 | Compuestos de quinina e isómeros ópticos, procedimiento de preparación y uso médico de los mismos |
Country Status (13)
| Country | Link |
|---|---|
| US (1) | US9751875B2 (es) |
| EP (1) | EP3023424B1 (es) |
| JP (1) | JP6218940B2 (es) |
| KR (1) | KR101823451B1 (es) |
| CN (1) | CN105431432B (es) |
| AU (1) | AU2014292722B2 (es) |
| BR (1) | BR112016000629B1 (es) |
| CA (1) | CA2921621C (es) |
| ES (1) | ES2726664T3 (es) |
| RU (1) | RU2641285C2 (es) |
| TR (1) | TR201906532T4 (es) |
| WO (1) | WO2015007073A1 (es) |
| ZA (1) | ZA201600997B (es) |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2976889C (en) * | 2015-03-02 | 2023-06-27 | Ltt Bio-Pharma Co., Ltd. | Quinuclidine derivative |
| CA3047023C (en) | 2016-12-14 | 2022-05-31 | Beijing Showby Pharmaceutical Co., Ltd. | Class of bifunctional compounds with quaternary ammonium salt structure |
| CN109824661B (zh) * | 2019-03-30 | 2020-06-05 | 山东博洛德生物科技有限公司 | 一种盐酸戊乙奎醚中杂质的制备方法 |
| CN109824662A (zh) * | 2019-03-30 | 2019-05-31 | 山东博洛德生物科技有限公司 | 一种奎宁类化合物及其制备方法 |
| CN109776521A (zh) * | 2019-03-30 | 2019-05-21 | 山东博洛德生物科技有限公司 | 一种含有季铵基团的奎宁类化合物及其制备方法 |
| CA3181333A1 (en) * | 2020-04-26 | 2021-11-04 | Beijing Showby Pharmaceutical Co., Ltd. | Crystals, preparation method and application of a muscarinic receptor antagonist |
| WO2023138478A1 (zh) * | 2022-01-19 | 2023-07-27 | 北京硕佰医药科技有限责任公司 | 一种治疗鼻炎的溶液型鼻喷雾剂及制备方法 |
| CN116514800A (zh) * | 2023-05-06 | 2023-08-01 | 湖南一格制药有限公司 | 盐酸戊乙奎醚的制备方法 |
Family Cites Families (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1219606A (en) | 1968-07-15 | 1971-01-20 | Rech S Et D Applic Scient Soge | Quinuclidinol derivatives and preparation thereof |
| DE4108393A1 (de) | 1991-03-15 | 1992-09-17 | Boehringer Ingelheim Kg | Neue ester bi- und tricyclischer aminoalkohole, ihre herstellung und ihre verwendung in arzneimitteln |
| NO2005012I1 (no) * | 1994-12-28 | 2005-06-06 | Debio Rech Pharma Sa | Triptorelin og farmasoytisk akseptable salter derav |
| ES2165768B1 (es) * | 1999-07-14 | 2003-04-01 | Almirall Prodesfarma Sa | Nuevos derivados de quinuclidina y composiciones farmaceuticas que los contienen. |
| HU229306B1 (en) * | 2000-06-27 | 2013-10-28 | L V A T Lab Sa | Carbamates derived from arylalkylamines |
| ATE334128T1 (de) * | 2000-12-28 | 2006-08-15 | Almirall Prodesfarma Ag | Neue chinuclidinderivate und medizinische zusammensetzungen, die diese verbindungen enthalten |
| ES2203327B1 (es) * | 2002-06-21 | 2005-06-16 | Almirall Prodesfarma, S.A. | Nuevos carbamatos de quinuclidina y composiciones farmaceuticas que los contienen. |
| ES2204295B1 (es) * | 2002-07-02 | 2005-08-01 | Almirall Prodesfarma, S.A. | Nuevos derivados de quinuclidina-amida. |
| PE20060259A1 (es) | 2004-04-27 | 2006-03-25 | Glaxo Group Ltd | Compuestos de quinuclidina como antagonistas del receptor de acetilcolina muscarinico |
| JP4637715B2 (ja) * | 2005-10-17 | 2011-02-23 | 日信工業株式会社 | 車両用支持構造体の製造方法 |
| CN1951938A (zh) | 2005-10-21 | 2007-04-25 | 刘丽娅 | 戊乙奎醚季铵盐及其衍生物 |
| US20100113510A1 (en) * | 2006-12-19 | 2010-05-06 | Rhonan Ford | Quinuclidinol derivatives as muscarinic receptor antagonists |
| WO2008096870A1 (ja) * | 2007-02-09 | 2008-08-14 | Astellas Pharma Inc. | アザ架橋環化合物 |
| CN102070631B (zh) * | 2009-11-20 | 2016-09-07 | 中国人民解放军军事医学科学院毒物药物研究所 | 戊乙奎醚光学异构体的季铵盐、药物组合物及其医药用途 |
| CN102850344A (zh) * | 2011-06-28 | 2013-01-02 | 中国人民解放军军事医学科学院毒物药物研究所 | 戊乙奎醚光学异构体衍生物用于抗肿瘤的医药用途 |
-
2014
- 2014-07-11 KR KR1020167003638A patent/KR101823451B1/ko active IP Right Grant
- 2014-07-11 US US14/904,766 patent/US9751875B2/en active Active
- 2014-07-11 RU RU2016104437A patent/RU2641285C2/ru active
- 2014-07-11 WO PCT/CN2014/000669 patent/WO2015007073A1/zh active Application Filing
- 2014-07-11 BR BR112016000629-1A patent/BR112016000629B1/pt active IP Right Grant
- 2014-07-11 CA CA2921621A patent/CA2921621C/en active Active
- 2014-07-11 ES ES14825941T patent/ES2726664T3/es active Active
- 2014-07-11 AU AU2014292722A patent/AU2014292722B2/en active Active
- 2014-07-11 CN CN201480020342.8A patent/CN105431432B/zh active Active
- 2014-07-11 TR TR2019/06532T patent/TR201906532T4/tr unknown
- 2014-07-11 EP EP14825941.9A patent/EP3023424B1/en active Active
- 2014-07-11 JP JP2016524657A patent/JP6218940B2/ja active Active
-
2016
- 2016-02-12 ZA ZA2016/00997A patent/ZA201600997B/en unknown
Also Published As
| Publication number | Publication date |
|---|---|
| AU2014292722B2 (en) | 2017-03-09 |
| EP3023424A4 (en) | 2017-01-18 |
| CA2921621A1 (en) | 2015-01-22 |
| RU2016104437A (ru) | 2017-08-16 |
| JP2016528199A (ja) | 2016-09-15 |
| EP3023424A1 (en) | 2016-05-25 |
| KR20160033146A (ko) | 2016-03-25 |
| CN105431432B (zh) | 2018-01-09 |
| BR112016000629B1 (pt) | 2023-10-31 |
| AU2014292722A1 (en) | 2016-03-03 |
| CN105431432A (zh) | 2016-03-23 |
| BR112016000629A2 (es) | 2017-07-25 |
| RU2641285C2 (ru) | 2018-01-17 |
| ZA201600997B (en) | 2018-05-30 |
| JP6218940B2 (ja) | 2017-10-25 |
| WO2015007073A1 (zh) | 2015-01-22 |
| KR101823451B1 (ko) | 2018-01-30 |
| TR201906532T4 (tr) | 2019-05-21 |
| US9751875B2 (en) | 2017-09-05 |
| US20160244439A1 (en) | 2016-08-25 |
| EP3023424B1 (en) | 2019-02-27 |
| CA2921621C (en) | 2018-08-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2726664T3 (es) | Compuestos de quinina e isómeros ópticos, procedimiento de preparación y uso médico de los mismos | |
| ES2324746T3 (es) | Compuestos de ciclopentano y ciclopenteno sustituidos utiles como inhibidores de neuraminidasa. | |
| JP2013536825A (ja) | ドパミン作動性安定化剤として有用なプリドピジンの重水素化類似体 | |
| RU2624446C2 (ru) | Трициклические соединения, композиции, содержащие указанные соединения, и их применения | |
| ES2802977T3 (es) | Compuesto para la modulación inmunológica, uso del mismo y composición farmacéutica que lo comprende | |
| JP2011527704A (ja) | 経口抗がん製剤 | |
| ES2899367T3 (es) | Forma amorfa de trifenatato de vilanterol y procedimientos para su preparación | |
| EP3877382A1 (en) | Novel compounds for the treatment of respiratory diseases | |
| ES2574277T3 (es) | Derivados de dibenzotiazepina y su uso en el tratamiento de trastornos del SNC | |
| AU2019433734A1 (en) | Treating influenza using substituted polycyclic pyridone derivatives and prodrugs thereof in a subject having influenza and a complication risk factor | |
| US9822117B2 (en) | Pyridoindolobenz[b,d]azepine derivatives and uses thereof | |
| US9409857B2 (en) | Agomelatine sulfuric acid complex, and preparation method and application thereof | |
| EP3984993A1 (en) | Use of aminothiol compounds as cerebral nerve or heart protective agent | |
| WO2024078387A1 (zh) | 取代3-氟苯丙酸酯类化合物及其制备方法、药物组合及用途 | |
| US20240261283A1 (en) | Pharmaceutical preparation for preventing or treating pulmonary fibrosis | |
| US20230100890A1 (en) | Use of combined inhalant cannabinoid therapy in the treatment of migraine | |
| JP2005060311A (ja) | N−(ベンゾイル)アミノ酸誘導体を有効成分とするニューロパシー性疼痛治療剤 | |
| CN115025080A (zh) | 一种抗流感病毒的药物组合物及其用途 |