ES2719145T3 - 1-Piperazino-3-fenil-indanos deuterados para el tratamiento de la esquizofrenia - Google Patents
1-Piperazino-3-fenil-indanos deuterados para el tratamiento de la esquizofrenia Download PDFInfo
- Publication number
- ES2719145T3 ES2719145T3 ES16179882T ES16179882T ES2719145T3 ES 2719145 T3 ES2719145 T3 ES 2719145T3 ES 16179882 T ES16179882 T ES 16179882T ES 16179882 T ES16179882 T ES 16179882T ES 2719145 T3 ES2719145 T3 ES 2719145T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- deuterium
- phenyl
- chloro
- treatment
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 C*(C(C)(C)C*(C1)[C@](C2)c3cc([N-])ccc3[C@@]2c2ccccc2)C1(O)O Chemical compound C*(C(C)(C)C*(C1)[C@](C2)c3cc([N-])ccc3[C@@]2c2ccccc2)C1(O)O 0.000 description 5
- QSYBPOLSZFDUSR-SECBINFHSA-N Cc1ccc([C@@H](CC2=O)c(c(O)c(c(O)c3O)O)c3O)c2c1 Chemical compound Cc1ccc([C@@H](CC2=O)c(c(O)c(c(O)c3O)O)c3O)c2c1 QSYBPOLSZFDUSR-SECBINFHSA-N 0.000 description 2
- ROCVBCBTVOWFTP-PZJWPPBQSA-N CC(C)(CN(CC1)[C@H](C2)c3cc(Cl)ccc3[C@@H]2c2ccccc2)N1C=[IH] Chemical compound CC(C)(CN(CC1)[C@H](C2)c3cc(Cl)ccc3[C@@H]2c2ccccc2)N1C=[IH] ROCVBCBTVOWFTP-PZJWPPBQSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/04—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms
- C07D295/06—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by halogen atoms or nitro radicals
- C07D295/073—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by halogen atoms or nitro radicals with the ring nitrogen atoms and the substituents separated by carbocyclic rings or by carbon chains interrupted by carbocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/451—Non condensed piperidines, e.g. piperocaine having a carbocyclic group directly attached to the heterocyclic ring, e.g. glutethimide, meperidine, loperamide, phencyclidine, piminodine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/454—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. pimozide, domperidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/4545—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring hetero atom, e.g. pipamperone, anabasine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/551—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having two nitrogen atoms, e.g. dilazep
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/554—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having at least one nitrogen and one sulfur as ring hetero atoms, e.g. clothiapine, diltiazem
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B59/00—Introduction of isotopes of elements into organic compounds ; Labelled organic compounds per se
- C07B59/002—Heterocyclic compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/61—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/61—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups
- C07C45/67—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D241/00—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings
- C07D241/02—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings
- C07D241/04—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/05—Isotopically modified compounds, e.g. labelled
Abstract
Un compuesto de fórmula Y:**Fórmula** en donde, R1 - R10 son independientemente hidrógeno o deuterio, en donde R6-R10 son cada uno deuterio, o una sal por adición de ácidos farmacéuticamente aceptable del mismo, para uso como un medicamento.
Description
DESCRIPCIÓN
1-Piperazino-3-fenil-indanos deuterados para el tratamiento de la esquizofrenia
Campo de la invención
La presente invención se refiere a 1-piperazino-3-fenil-indanos deuterados y sales de los mismos con actividad en receptores de dopamina Di y D2, así como a receptores de serotonina 5hT2 en el sistema nervioso central, a medicamentos que comprenden estos compuestos como ingredientes activos, y este tipo de compuestos para usar en el tratamiento de enfermedades en el sistema nervioso central.
Antecedentes de la invención
4-((1R,3S)-6-cloro-3-fenil-indan-1-il)-1,2,2-trimetil-piperazina y sales de la misma, composiciones farmacéuticas que contienen estas sales y el uso médico de las mismas, incluido el tratamiento de la esquizofrenia u otras enfermedades que implican síntomas psicóticos, se describen en el documento W02005/016900. 4-((1R,3S)-6-cloro-3-fenil-indan-1-il)-1,2,2-trimetil-piperazina tiene la fórmula general (X), a la que se alude en lo sucesivo como Compuesto (X)

El documento EP 638073 enumera un grupo de isómeros trans de 3-aril-1-(1-piperazinil)indanos sustituidos en la posición 2 y/o 3 del anillo de piperazina. Los compuestos se describen como que tienen alta afinidad por los receptores de dopamina D1 y D2 y los receptores de serotonina 5 HT2 y se sugiere que son útiles para el tratamiento de varias enfermedades en el sistema nervioso central, incluyendo la esquizofrenia.
El enantiómero de fórmula (X) anterior ha sido descrito por B0 ges0 et al. en J. Med. Chem., 1995, 38, páginas 4380 4392, en forma de la sal fumarato, véase la tabla 5, compuesto (-)-38. Esta publicación concluye que el enantiómero (-) del compuesto 38 es un potente antagonista de D1/D2 que muestra alguna selectividad por D1 in vitro. El compuesto también se describe como un potente antagonista de 5-HT2. También se menciona que el compuesto no induce catalepsia en ratas.
No se conoce la etiología de la esquizofrenia, pero la hipótesis de la dopamina de la esquizofrenia (Carlsson, Am. J. Psychiatry 1978, 135, 164-173), formulada a principios de los años 1960, ha proporcionado un marco teórico para la comprensión de los mecanismos biológicos que subyacen a este trastorno. En su forma más simple, la hipótesis de la dopamina establece que la esquizofrenia está asociada con un estado hiperdopaminérgico, una noción que es apoyada por el hecho de que todos los fármacos antipsicóticos en el mercado hoy en día ejercen algún antagonismo del receptor de dopamina D2 (Seeman Science and Medicine 1995, 2, 28-37). Sin embargo, mientras que en general se acepta que el antagonismo de los receptores de dopamina D2 en las regiones límbicas del cerebro desempeña un papel clave en el tratamiento de los síntomas positivos de la esquizofrenia, el bloqueo de receptores D2 en las regiones del cuerpo estriado del cerebro provoca síntomas extrapiramidales (EPS). Tal como se describe en el documento EP 638073 se ha observado un perfil de inhibición del receptor de dopamina D1/D2 mixto con algunos de los llamados compuestos antipsicóticos "atípicos", en particular con clozapina (8-cloro-11-(4-metilpiperazin-1-il)-5H-dibenzo[b,e][1,4]diazepina), que se utiliza en el tratamiento de pacientes esquizofrénicos.
Además, antagonistas de D1 selectivos han sido relacionados con el tratamiento de trastornos del sueño y el abuso de alcohol (D.N. Eder, Current Opinión in Investigational Drugs, 20023(2): 284-288).
La dopamina también puede jugar un papel importante en la etiología de los trastornos afectivos (P. Willner, Brain Res. Rev. 1983, 6, 211-224, 225-236 y 237-246; B0 ges0 et al, J. Med. Chem., 1985, 28, 1817-1828).
En el documento EP 638 073 se describe cómo los compuestos que tienen afinidad por receptores de 5-HT2, en particular antagonistas del receptor de 5-HT2A, han sido sugeridos para el tratamiento de diferentes enfermedades tales como la esquizofrenia, incluyendo los síntomas negativos en pacientes esquizofrénicos, depresión, ansiedad,
trastornos del sueño, ataques de migraña y parkinsonismo inducido por neurolépticos. También se ha sugerido que el antagonismo del receptor de 5-HT2A reduce la incidencia de efectos secundarios extrapiramidales inducidos por los neurolépticos clásicos (Balsara et al. Psychopharmacology 1979, 62, 67-69).
Una sustitución isotópica de uno o más átomos de hidrógeno (H) por átomos de deuterio (D) en un compuesto puede dar lugar a un efecto isotópico cinético que puede influir en la velocidad de la reacción, p. ej., el metabolismo del compuesto. Este es particularmente el caso cuando la sustitución isotópica es en un enlace químico que se rompe o se forma en una etapa limitante de la velocidad. En tal caso, el cambio se denomina un efecto isotópico primario. Cuando la o las sustituciones isotópicas no están implicadas en uno o más enlaces que se rompen, se puede observar un cambio en la velocidad más pequeño, denominado el efecto isotópico secundario.
Sumario de la invención
La presente invención proporciona compuestos en los que uno o más átomos de átomos de hidrógeno (H) en uno o más de los sitios metabólicos M1, M2 y M3 del Compuesto (X) han sido sustituidos por átomos de deuterio (D). En un aspecto, la invención proporciona un compuesto de fórmula Y:

en donde, R1-R10 son, independientemente, hidrógeno o deuterio, y en donde al menos uno de R1-R10 comprende al menos aproximadamente 50% de deuterio, o una sal por adición de ácidos farmacéuticamente aceptable del mismo. Se describen composiciones farmacéuticas que comprenden un compuesto de fórmula (Y) y uno o más soportes farmacéuticamente aceptables, diluyentes o excipientes.
Se describe la fabricación de un medicamento que comprende un compuesto de fórmula (Y) para el tratamiento de la psicosis, otras enfermedades que implican síntomas psicóticos, trastornos psicóticos o enfermedades que se presentan con síntomas psicóticos.
Se describen métodos para tratar la psicosis, otras enfermedades que implican síntomas psicóticos, trastornos psicóticos o enfermedades con síntomas psicóticos, que comprenden la administración de una cantidad eficaz de un compuesto de fórmula (Y) o una composición farmacéuticamente que comprende un compuesto de fórmula (Y) a un sujeto que lo necesite.
Se describen un compuesto de fórmula

Se describe un procedimiento para la preparación del compuesto

Se describe un procedimiento para la preparación del compuesto tartrato de (1R,3S)-(IV), que comprende el tratamiento de trans-1-(6-cloro-3-fenil(cÍ5)-indan-1-il)-1(cf3), 2, 2-trimetil-piperazina racémica con ácido L-(+)-tartárico. Aún otros objetos y ventajas de la invención resultarán evidentes para los expertos en la técnica a partir de la descripción en esta memoria, que es simplemente ilustrativa y no restrictiva.
Breve descripción de las figuras
FIG. 1 muestra sitios metabólicos principales del Compuesto (X).
FIG. 2 muestra el Compuesto (I) y el Compuesto (XI), cada uno como el enantiómero (1R,3S).
FIG. 3 muestra espectros de RMN del Compuesto (II) y el Compuesto (V). Se muestran regiones seleccionadas de los espectros de 13C-RMN de protón desacoplado y de protón y deuterio desacoplado del Compuesto (II) [Fig. 3A] y del Compuesto (V) [Fig. 3B].
FIG. 4 muestra el espectro de masas del Compuesto (IV).
FIG. 5 muestra la formación del metabolito Compuesto (XI) por el metabolismo de Compuesto (X) y Compuesto (I) (0,1 microM) en hepatocitos criopreservados de perro (n = 2 las barras representan los resultados máximo y mínimo).
FIG. 6 muestra la formación del metabolito Compuesto (XI) por el metabolismo de Compuesto (X) y Compuesto (I) (1 microM) en hepatocitos criopreservados de perro (n = 2 las barras representan los resultados máximo y mínimo). FIG. 7 muestra la formación del metabolito desmetilo por el metabolismo del Compuesto (II), (IV) y (X) (1 micro M) en microsomas de hígado humano (n = 3, las barras representan la desviación estándar).
FIG. 8 muestra la formación del metabolito desmetilo por el metabolismo de los Compuestos (II), (IV) y (X) (10 micro M) en microsomas de hígado humano (n = 3, las barras representan la desviación estándar).
FIG. 9 muestra la formación del metabolito desmetilo por el metabolismo del Compuesto (III) (10 micro M) en microsomas de hígado humano (n = 3, las barras representan la desviación estándar).
FIG. 10 muestra la formación del metabolito desmetilo por el metabolismo del Compuesto (V) (10 microM) en microsomas de hígado humano (n = 3, las barras representan la desviación estándar).
FIG. 11 muestra la formación del metabolito desmetilo por el metabolismo del Compuesto (VI) (10 microM) en microsomas de hígado humano (n = 3, las barras representan la desviación estándar).
FIG. 12 muestra la formación del metabolito desmetilo por el metabolismo del Compuesto (VII) (10 microM) en microsomas de hígado humano (n = 3, las barras representan la desviación estándar).
FIG. 13 muestra la estructura química de los compuestos (I)-(VII), (X)-(XI), y (XIX)-(XXI).
FIG. 14 muestra la formación del metabolito desmetilo por el metabolismo del Compuesto (II) y (X) (10 microM) por CYP2C19 de hígado humano recombinante (n = 3, la desviación estándar).
FIG. 15 muestra la formación del metabolito desmetilo por el metabolismo del Compuesto (IV) y Compuesto (X) (1 microM) por CYP2C19 de hígado humano recombinante (n = 3, las barras representan la desviación estándar). FIG. 16 muestra hiperactividad inducida por PCP en ratones para el compuesto (IV).
FIG. 17 muestra la respuesta cataléptica en ratas para el compuesto (IV).
FIG. 18 muestra difractogramas de rayos X en dos lotes de sal tartrato de hidrógeno del Compuesto (IV).
Descripción detallada de la invención
Los antipsicóticos atípicos han sido objeto de numerosos estudios por parte de la industria farmacéutica, y se han mostrado prometedores en el tratamiento de trastornos mentales tales como la esquizofrenia, el trastorno bipolar, la demencia, el trastorno de ansiedad y el trastorno obsesivo-compulsivo (OCD). Sigue desconociéndose el mecanismo de acción de estos agentes; sin embargo, todos los antipsicóticos funcionan en cierto grado en el sistema de la dopamina. Los antipsicóticos más atípicos exhiben actividad en los receptores de la dopamina de subtipo 1 y 2 (D1 y D2 , respectivamente), y en los receptores de serotonina subtipo 2 (5 -HT2). En algunos casos, la designación "atípicos" se asignó a los antipsicóticos que no inducían efectos secundarios extrapiramidales; sin embargo, se ha demostrado que algunos antipsicóticos atípicos todavía inducen efectos secundarios extrapiramidales, aunque en un grado menor que el observado con los antipsicóticos típicos (Weiden, P.J., "EPS profiles: the atypical antipsychotics are not all the same" J. Psychiatr. Pract. 2007, 13(1): 13-24). Antipsicóticos atípicos aprobados incluyen, por ejemplo, amisulprida (Solian), aripiprazol (Abilify), asenapina (Saphris), blonanserina (Lonasen), clotiapina (Entumine), clozapina (Clozaril), iloperidona (Fanapt), lurasidona (Latuda), mosapramina (Cremin), olanzapina (Zyprexa), paliperidona (Invega), perospirona (Lullan), quetiapina (Seroquel), remoxiprida (Roxiam), risperidona (Risperdal), sertindol (Serdolect), sulpirida (Sulpirid, Eglonyl), ziprasidona (Geodon, Zeldox) y zotepina (Nipolept). Varios otros están actualmente en desarrollo. Debido a que el mecanismo de los antipsicóticos atípicos no se entiende bien, los efectos secundarios asociados con estos fármacos han sido difíciles de diseñar. Por lo tanto, existe la necesidad de terapias antipsicóticas adicionales con potencial para la reducción de efectos secundarios y/o un perfil terapéutico mejorado en relación con las terapias existentes.
En un aspecto, la presente invención proporciona compuestos en los que uno o más átomos de hidrógeno (H) en uno o más de los sitios metabólicos M1, M2 y M3 del Compuesto (X) han sido sustituidos con átomos de deuterio (D). El compuesto (X) y variantes del mismo se describen, por ejemplo, en las Patentes de EE.UU. N°s. 5.807.855; 7.648.991; 7.767.683; 7.772.240; 8.076.342; Publicaciones de Patente de EE.UU. N°s. 2008/0269248; 2010/0069676; 2011/0178094; 2011/0207744; los documentos WO 2005/016900; EP 0638 073; y J. Med. Chem.
1995, 38, 4380-4392.
El efecto isotópico cinético puede influir potencialmente en la tasa de metabolismo en uno o más de los sitios metabólicos M1, M2, y M3 indicados en la Figura 1. Los inventores de la presente invención han identificado tres principales sitios metabólicos de 4-((1R,3S)-6-cloro-3-fenil-indan-1-il)-1,2,2-trimetil-piperazina (Compuesto (X )) designados en esta memoria como M1, M2 y M3 e indicados en la Figura 1.
La deuteración de un compuesto en un sitio sujeto al metabolismo oxidativo puede, en algunos casos, reducir la tasa de metabolismo para un compuesto debido al efecto isotópico primario. Si la etapa de escisión del enlace C-H es limitante de la velocidad, se puede observar un efecto isotópico significativo. Sin embargo, si otras etapas impulsan la tasa de metabolismo de un compuesto, la etapa de escisión del enlace C-H no es limitante de la velocidad y el efecto isotópico puede ser de poca importancia. Adicionalmente, puede observarse un efecto isotópico negativo en los casos en los que se aumenta la velocidad de la reacción tras la sustitución con deuterio. Por lo tanto, la incorporación de deuterio en un lugar sujeto a metabolismo enzimático oxidativo no impacta de forma predecible en la farmacocinética (Véase, por ejemplo, la Patente de EE.UU. N° 7.678.914; Drug Metab. Dispos. 1986, 14, 509; Arch. Toxicol. 1990, 64, 109; Int. Arch. Occup. Environ. Health 1993, 65 (Supl 1.): S139). El impacto de la incorporación de deuterio es impredecible, no funciona para muchos fármacos o clases de fármacos. Se ha observado una disminución del aclaramiento metabólico con algunos compuestos deuterados con respecto a derivados no deuterados; mientras que no se ha visto impactado el metabolismo de otros compuestos. Ejemplos de estudios que indican falta de previsibilidad en relación con la incorporación de deuterio incluyen la Patente de EE.UU. N°. 6.221.335; J. Pharm. Sci. 1975, 64, 367-391; Adv. Drug. Res. 1985, 14, 1-40; J. Med. Chem. 1991, 34, 2871-2876; Can. J. Physiol. Pharmacol. 1999, 79-88; Silverman, R.B., The Organic Chemistry of Drug Design and Drug Action, 2a Ed. (2004), 422; Curr. Opin. Drug Dev. 2006, 9, 101-109; Chemical Res. Tox. 2008, 1672; Harbeson,
S.L. y Tung, R.D. "Deuterium in Drug Discovery and Development", en Ann. Rep. Med. Chem. 2011, 46, 404-418. Incluso la incorporación de deuterio en sitios conocidos del metabolismo tiene un impacto impredecible en el perfil metabólico. La conmutación metabólica puede resultar en que el perfil metabólico de un fármaco particular cambie debido a la incorporación de deuterio, lo que conduce a diferentes proporciones de (o diferentes) metabolitos que las observadas con un análogo no deuterado del mismo fármaco. El nuevo perfil metabólico puede dar lugar a un perfil toxicológico distinto del análogo deuterado. A las posibles complicaciones de la incorporación de deuterio se añade la posibilidad de intercambio de deuterio/hidrógeno en el entorno fisiológico (Adv. Drug. Res. 1985, 14, 1-40; que se incorpora a esta por referencia en su totalidad).
En algunas realizaciones, la sustitución isotópica de uno o más átomos de hidrógeno en el compuesto (X) por átomos de deuterio ha dado lugar a un efecto isotópico cinético que influye en la tasa del metabolismo.
La sustitución isotópica de átomos de hidrógeno en el Compuesto (X) por átomos de deuterio resulta en un menor metabolismo del compuesto deuterado como se demuestra que ocurre en hepatocitos de perro, en donde, por ejemplo, se observó una disminución de aproximadamente 50% en la formación del metabolito desmetilo (Compuesto (XI)) a partir de Compuesto (I) (Figura 2) en comparación con la formación del Compuesto (XI) a partir del metabolismo del Compuesto (X).
La deuteración del fenilo libre, opcionalmente en combinación con la deuteración del grupo 1-metilo (Compuestos (II) y (IV)) reduce, de forma sorprendente, la cantidad del metabolito de desmetilo producido en microsomas de hígado humano en comparación con el compuesto no deuterado (Compuesto (X)). También sorprendentemente, la deuteración del grupo 1-metilo impactó en el metabolismo en hepatocitos de perro pero no humanos, por lo tanto es indicativa de la imprevisibilidad de deuteraciión en las propiedades farmacológicas.
El efecto del metabolismo reducido es una mayor biodisponibilidad del compuesto parental deuterado y una menor formación de metabolitos. Sin estar ligado por la teoría, en base a los resultados descritos en la sección experimental de esta solicitud se espera que se manifieste el mismo efecto después de la dosificación múltiple en seres humanos, lo que permite dosis menores a administrar a los seres humanos, es decir, menos carga para todo el cuerpo, p. ej., el hígado, y una dosificación menos frecuente.
Se sabe que el metabolito de desmetilo (Compuesto (XI)) tiene afinidad por hERG y, por lo tanto, contribuye potencialmente en la prolongación del QTc. Tal como se mencionó anteriormente, la deuteración del fenilo libre, opcionalmente en combinación con la deuteración del grupo 1-metilo (Compuestos (II) y (IV)) reduce de forma sorprendente la cantidad del metabolito de desmetilo producido en microsomas de hígado humano en comparación con el compuesto no deuterado (Compuesto (X)). Por consiguiente, y sin pretender estar ligados por teoría alguna, se anticipa que habrá una menor interacción con el canal de hERG y una menor carga resultante sobre el corazón, cuando la dosificación de las variantes deuteradas del Compuesto (X) [p. ej., compuestos de fórmula (Y)] en comparación con cuando se dosifica Compuesto (X).
La invención se detalla adicionalmente en las realizaciones proporcionadas en esta memoria.
Definiciones
La expresión "compuesto o compuestos de la invención", tal como se utiliza en esta memoria, significa Compuestos (Y), (II), (III), (IV), (V), (VI) y/o (VII), y puede incluir sales, hidratos y/o solvatos de los mismos. Los compuestos de la presente invención se preparan en diferentes formas, tales como sales, hidratos y/o solvatos, y la invención incluye composiciones y métodos que comprenden todas las formas variantes de los compuestos.
La expresión "composición o composiciones de la invención", tal como se utiliza en esta memoria, significa composiciones que comprenden los Compuestos (Y), (II), (III), (IV), (V), (VI) y/o (VII), o sales, hidratos, y solvatos de los mismos. Las composiciones de la invención pueden comprender, además, uno o más componentes químicos tales como, por ejemplo, excipientes, diluyentes, vehículos o soportes.
La expresión "método o métodos de la invención", tal como se utiliza en esta memoria, significa métodos que comprenden el tratamiento con los compuestos y/o composiciones de la invención.
Tal como se utiliza en esta memoria, el término "aproximadamente" se utiliza aquí para dar a entender aproximadamente, grosso modo, alrededor de, o en la región de. Cuando el término "aproximadamente" se utiliza en unión con un intervalo numérico, modifica ese intervalo ampliando los límites por encima y por debajo de los valores numéricos expuestos. En general, el término "aproximadamente" se utiliza aquí para modificar un valor numérico por encima y por debajo del valor indicado por una varianza del 20 por ciento arriba o hacia abajo (mayor o menor). Una "cantidad eficaz", "cantidad suficiente" o "cantidad terapéuticamente eficaz", tal como se utiliza en esta memoria, es una cantidad de un compuesto que es suficiente para obtener resultados beneficiosos o deseados, incluyendo resultados clínicos. Como tal, la cantidad eficaz puede ser suficiente, por ejemplo, para reducir o mejorar la gravedad y/o duración de una aflicción o afección, o uno o más de sus síntomas, evitar el avance de afecciones
relacionadas con una aflicción o afección, prevenir la recurrencia, el desarrollo o la aparición de uno o más síntomas asociados con una aflicción o condición, o mejorar o de otra manera mejorar el o los efectos profilácticos o terapéuticos de otra terapia. Una cantidad efectiva también incluye la cantidad del compuesto que evita o atenúa sustancialmente efectos secundarios indeseables.
Tal como se utiliza en esta memoria, y como es bien entendido en la técnica, "tratamiento" es un enfoque para obtener resultados beneficiosos o deseados, incluyendo resultados clínicos. Resultados clínicos beneficiosos o deseados pueden incluir, pero no se limitan al alivio o la mejora de uno o más síntomas o afecciones, la disminución del grado de la enfermedad, un estado estabilizado (es decir, no empeorado) de la enfermedad, la prevención de la propagación de la enfermedad, el retraso o la ralentización del progreso de la enfermedad, la mejora o paliación del estado de la enfermedad y la remisión (ya sea parcial o total), ya sea detectable o indetectable. “Tratamiento" también puede significar prolongar la supervivencia en comparación con la supervivencia esperada si no se recibe tratamiento.
La expresión "en necesidad del mismo" se refiere a la necesidad de un alivio sintomático o asintomático de una afección tal como, por ejemplo, psicosis o trastorno psicótico. El sujeto en necesidad del mismo puede o puede no ser sometido a tratamiento para afecciones relacionadas con, por ejemplo, psicosis o un trastorno psicótico.
El término "soporte" se refiere a un diluyente, adyuvante, excipiente o vehículo con el que se administra un compuesto. Ejemplos no limitantes de soportes farmacéuticos de este tipo incluyen líquidos tales como agua y aceites, incluyendo los de origen del petróleo, animal, vegetal o sintético tales como aceite de cacahuete, aceite de soja, aceite mineral, aceite de sésamo y similares. Los soportes farmacéuticos también pueden ser solución salina, goma de acacia, gelatina, pasta de almidón, talco, queratina, sílice coloidal, urea, y similares. Además, se pueden utilizar agentes auxiliares, estabilizantes, espesantes, lubricantes y colorantes. Otros ejemplos de soportes farmacéuticos adecuados se describen en Remington: The Science and Practice of Pharmacy, 21a edición (Universidad de Ciencias de Filadelfia, ed, Lippincott Williams & Wilkins 2005).
Los términos "animal", "sujeto" y "paciente", tal como se utilizan en esta memoria, incluyen todos los miembros del reino animal incluyendo, pero no limitados a, mamíferos, animales (p. ej., gatos, perros, caballos, cerdos, etc.) y seres humanos.
La expresión "variante isotópica", tal como se utiliza en esta memoria, significa un compuesto obtenido mediante la sustitución con átomos de deuterio de uno o más hidrógenos en un compuesto parental que no comprende átomos de deuterio.
Se reconoce que los elementos están presentes en abundancias isotópicas naturales en la mayoría de los compuestos sintéticos, y resultan en la incorporación inherente de deuterio. Sin embargo, la abundancia isotópica natural de isótopos de hidrógeno tales como deuterio es inmaterial (aproximadamente 0,015%) con respecto al grado de sustitución isotópica estable de compuestos indicados en esta memoria. Por lo tanto, tal como se utiliza en esta memoria, la designación de un átomo como deuterio en una posición indica que la abundancia de deuterio es significativamente mayor que la abundancia natural de deuterio. Cualquier átomo no designado como un isótopo particular está destinado a representar a cualquier isótopo estable de ese átomo tal como resultará evidente para un experto ordinario en la materia.
Los Compuestos (Y) son variantes isotópicas del Compuesto (X).
En algunas realizaciones, los compuestos (I), (II), (III), (IV), (V), (VI) y (VII) son variantes isotópicas del Compuesto (X).
M1 es un sitio de Compuesto (X) susceptible al metabolismo; M1 consiste en -CH2- en la posición 6 de la piperazina del Compuesto (X).
M2 es un sitio de compuesto (X) susceptible al metabolismo; M2 consiste en el metilo unido a N de la piperazina del Compuesto (X).
M3 es un sitio del Compuesto (X) susceptible al metabolismo; M3 consiste en el grupo fenilo del Compuesto (X). El compuesto parental es el compuesto químico que es la base de sus derivados obtenidos ya sea por sustitución o desintegración, p. ej., desintegración metabólica. En el contexto de la presente invención, el compuesto parental es el Ingrediente Activo Farmacéutico (API).
En algunas realizaciones, cualquier átomo no designado como deuterio está presente en su abundancia isotópica natural. En algunas realizaciones, cualquier átomo de hidrógeno no designado como deuterio está presente a menos de 1% de abundancia isotópica de deuterio.
En un aspecto, la invención proporciona un compuesto de fórmula (Y):

en donde, R 1 - R 10 son independientemente hidrógeno o deuterio, en el que al menos uno de R 1 -R 10 comprende al menos aproximadamente 50% de deuterio, o una sal de adición de ácido farmacéuticamente aceptable del mismo. Se describen composiciones farmacéuticas que comprenden un compuesto de fórmula (Y) y uno o más soportes, diluyentes o excipientes farmacéuticamente aceptables.
Se describe la fabricación de un medicamento que comprende un compuesto de fórmula (Y) para el tratamiento de la psicosis, otras enfermedades que implican síntomas psicóticos, trastornos psicóticos o enfermedades que se presentan con síntomas psicóticos.
En algunas descripciones, el compuesto es racémico. En algunas descripciones, el compuesto está enriquecido en enantiómeros.
En algunas descripciones, el compuesto se selecciona del grupo que consiste en


En algunas descripciones, R1 y R2 comprenden deuterio, R3-R5 comprenden deuterio, o R6-R10 comprenden deuterio. En algunas realizaciones, R1 y R2 comprenden deuterio. En algunas realizaciones, R1 y R2 comprenden deuterio y R3-R5 comprenden hidrógeno.
En algunas realizaciones, R3-R5 comprenden deuterio. En algunas realizaciones, R3-R5 comprenden hidrógeno. R6-R10 comprenden deuterio. En algunas realizaciones, R6-R10 comprenden deuterio y R3-R5 comprenden hidrógeno. En algunas realizaciones, R1-R5 comprenden deuterio.
En algunas realizaciones, R1, R2y R6-R10 comprenden deuterio.
En algunas realizaciones, R3-R10 comprenden deuterio En algunas realizaciones, R1-R10 comprenden deuterio En algunas realizaciones, el compuesto es
En algunas descripciones, el compuesto es
En algunas realizaciones, el compuesto es
En algunas descripciones, el compuesto es
En algunas realizaciones, el compuesto es
En algunas realizaciones, el compuesto es
En algunas realizaciones, al menos aproximadamente 75% del compuesto tiene un átomo de deuterio en cada una de las posiciones designadas como deuterio, y cualquier átomo no designado como deuterio está presente en aproximadamente su abundancia isotópica natural.
En algunas realizaciones, al menos aproximadamente 85% del compuesto tiene un átomo de deuterio en cada una de las posiciones designadas como deuterio, y cualquier átomo no designado como deuterio está presente en aproximadamente su abundancia isotópica natural.
En algunas realizaciones, al menos aproximadamente 90% del compuesto tiene un átomo de deuterio en cada una de las posiciones designadas como deuterio, y cualquier átomo no designado como deuterio está presente en aproximadamente su abundancia isotópica natural.
En algunas realizaciones, el compuesto es una sal seleccionada del grupo que consiste en fumarato, maleato, succinato y tartrato. En algunas realizaciones, el compuesto es una sal fumarato. En algunas realizaciones, el compuesto es una sal de hidrógeno fumarato. En algunas realizaciones, el compuesto es una sal maleato. En algunas realizaciones, el compuesto es una sal hidrógeno maleato. En algunas realizaciones, el compuesto es una sal succinato. En algunas realizaciones, el compuesto es una sal hidrógeno succinato. En algunas realizaciones, el compuesto es una sal tartrato. En algunas realizaciones, el compuesto es la sal hidrógeno tartrato.
En algunas realizaciones, el compuesto es la sal hidrógeno tartrato de (1R,3S)-(IV).
En algunas realizaciones, la psicosis o enfermedad que implica síntomas psicóticos es esquizofrenia, trastorno esquizofreniforme, trastorno esquizoafectivo, trastorno delirante, trastorno psicótico breve, trastorno psicótico compartido, trastorno bipolar, o manía en el trastorno bipolar. En algunas realizaciones, la psicosis o enfermedad que implica síntomas psicóticos es la esquizofrenia.
En algunas realizaciones, los métodos comprenden, además, la administración de uno o más agentes neurolépticos. En algunas realizaciones, los usos comprenden, además, el uso de uno o más agentes neurolépticos.
En algunas realizaciones, el agente neuroléptico se selecciona del grupo que consiste en sertindol, olanzapina, risperidona, quetiapina, aripiprazol, haloperidol, clozapina, ziprasidona y osanetant.
En algunas realizaciones, la administración es oral, sublingual o bucal. En algunas realizaciones, la administración es oral.
En algunas realizaciones, el sujeto es un mamífero. En algunas realizaciones, el sujeto es un roedor, gato, perro, mono, caballo, cerdo, bovino o ser humano. En algunas realizaciones, el sujeto es un roedor, gato, perro, mono, bovino o ser humano. En algunas realizaciones, el sujeto es un ratón, rata, gato, perro, mono o ser humano. En algunas realizaciones, el sujeto es un ratón, rata, perro, mono o ser humano. En algunas realizaciones, el sujeto es un ratón, rata, perro o ser humano. En algunas realizaciones, el sujeto es un ratón, rata o un ser humano. En algunas realizaciones, el sujeto es un perro o un ser humano. En algunas realizaciones, el sujeto es un ser humano. En algunas realizaciones, la designación de una posición como "D" en un compuesto tiene una incorporación de deuterio mínimo mayor que aproximadamente 40% en esa posición. En algunas realizaciones, la designación de una posición como "D" en un compuesto tiene una incorporación de deuterio mínimo mayor que aproximadamente 50% en esa posición. En algunas realizaciones, la designación de una posición como "D" en un compuesto tiene una incorporación de deuterio mínimo mayor que aproximadamente 60% en esa posición. En algunas realizaciones, la designación de una posición como "D" en un compuesto tiene una incorporación de deuterio mínimo mayor que aproximadamente 65% en esa posición. En algunas realizaciones, la designación de una posición como "D" en un compuesto tiene una incorporación de deuterio mínimo mayor que aproximadamente 70% en esa posición. En algunas realizaciones, la designación de una posición como "D" en un compuesto tiene una incorporación de deuterio mínimo mayor que aproximadamente 75% en esa posición. En algunas realizaciones, la designación de una posición como "D" en un compuesto tiene una incorporación de deuterio mínimo mayor que aproximadamente 80% en esa posición. En algunas realizaciones, la designación de una posición como "D" en un compuesto tiene una incorporación de deuterio mínimo mayor que aproximadamente 85% en esa posición. En algunas realizaciones, la designación de una posición como "D" en un compuesto tiene una incorporación de deuterio mínimo mayor que aproximadamente 90% en esa posición. En algunas realizaciones, la designación de una posición como "D" en un compuesto tiene una incorporación de deuterio mínimo mayor que aproximadamente 95% en esa posición. En algunas realizaciones, la designación de una posición como "D" en un compuesto tiene una incorporación de deuterio mínimo mayor que aproximadamente 97% en esa posición. En algunas realizaciones, la designación de una posición como "D" en un compuesto tiene una incorporación de deuterio mínimo mayor que aproximadamente 99% en esa posición.
Sales Farmacéuticamente Aceptables
La presente invención también comprende sales de los compuestos, típicamente sales farmacéuticamente aceptables. Este tipo de sales incluyen sales por adición de ácidos farmacéuticamente aceptables. Sales por adición de ácidos incluyen sales de ácidos inorgánicos, así como de ácidos orgánicos.
Ejemplos representativos de ácidos inorgánicos adecuados incluyen ácido clorhídrico, bromhídrico, yodhídrico, fosfórico, sulfúrico, sulfámico, nítrico y similares. Ejemplos representativos de ácidos orgánicos adecuados incluyen ácido fórmico, acético, tricloroacético, trifluoroacético, propiónico, benzoico, cinámico, cítrico, fumárico, glicólico, itacónico, láctico, metanosulfónico, maleico, málico, malónico, mandélico, oxálico, pícrico, pirúvico, salicílico, succínico , metanosulfónico, etanosulfónico, tartárico, ascórbico, pamoico, bismetilen-salicílico, etanodisulfónico, glucónico, citracónico, aspártico, esteárico, palmítico, EDTA, glicólico, p-aminobenzoico, glutámico, bencenosulfónico, p-toluenosulfónico, ácidos teofilinacéticos, así como las 8-haloteofilinas, por ejemplo 8-bromoteofilina y similares. Otros ejemplos de sales por adición de ácidos inorgánicos u orgánicos farmacéuticamente aceptables incluyen las sales farmacéuticamente aceptables enumeradas en Berge, S.M. et al., J. Pharm. Sci. 1977, 66, 2, y Gould, P.L., Int. J. Pharmaceutics 1986, 33, 201-217; los contenidos de cada una de las cuales se incorpora a la presente por referencia.
Además, los compuestos de esta invención pueden existir en forma no solvatada así como en forma solvatada con disolventes farmacéuticamente aceptables tales como agua, etanol y similares. En general, las formas solvatadas se consideran equiparables a las formas no solvatadas para los fines de esta invención.
Los títulos y subtítulos se utilizan en esta memoria sólo por conveniencia, y no se deben interpretar como limitantes de la invención de modo alguno.
El uso de cualquiera y todos los ejemplos, o del lenguaje a modo de ejemplo (incluyendo "por ejemplo", "p. ej.", y "como tal") en la presente memoria descriptiva pretende meramente iluminar mejor la invención, y no plantea una limitación al alcance de la invención, a menos que se indique lo contrario.
El uso de los términos "un" y "una" y "el" y “la” y referentes similares en el contexto de la descripción de la invención se deben interpretar para cubrir tanto el singular como el plural, a menos que se indique lo contrario en esta memoria o se contradiga claramente por el contexto.
A menos que se indique lo contrario, todos los valores exactos proporcionados en esta memoria son representativos de los valores aproximados correspondientes (p. ej., todos los valores a modo de ejemplo exactos proporcionados con respecto a un factor o medida particular, se puede considerar que también proporcionar una medición aproximada correspondiente, modificada por "aproximadamente", cuando proceda).
La descripción de esta memoria de cualquier aspecto o aspecto de la invención utilizando expresiones tales como "que comprende", "que tiene", "que incluye" o "que contiene" con referencia a un elemento o elementos está destinada a proporcionar apoyo a un aspecto similar o aspecto de la invención que "consiste en", "consiste esencialmente en" o "sustancialmente comprende" ese elemento o elementos particulares, a menos que se indique lo contrario o se contradiga claramente por el contexto.
Síntesis a modo de ejemplo de los compuestos de la invención se pueden conseguir fácilmente por métodos descritos, por ejemplo, en las Patente de EE.UU. N°s 5.807.855; 7,648,991; 7.767.683; 7.772.240;.8.076.342; las Publicaciones de Patente de EE.UU. N°s. 2008/0269248; 2010/0069676; 2011/0178094; 2011/0207744; los documentos WO 2005/016900; EP 0638 073; y J. Med. Chem. 1995, 38, 4380-4392. Tales métodos, y métodos similares pueden llevarse a cabo utilizando reactivos y/o productos intermedios deuterados, y/o introduciendo átomos de deuterio a una estructura química de acuerdo con protocolos conocidos en la técnica.
Métodos de síntesis a modo de ejemplo adicionales incluyen la conversión de indanona A en el compuesto intermedio C a través del tratamiento de 3-bromo-6-cloro-indan-1-ona (A; para referencias en este material, véase: B0 ges0 documento EP 35363 A1 19810909 y Kehler, Juhl, Püschl, documento WO 2008025361) con una base tal como trietilamina en un disolvente tal como tetrahidrofurano a temperatura ambiente (Esquema 1). La separación de la sal hidrobromuro de amina precipitada mediante filtración y concentración del filtrado proporcionará 6-cloro-inden-1-ona (B). Este material se puede hacer reaccionar con ácido fenil-ds-borónico en presencia de aproximadamente 1 equivalente de una base tal como trietilamina y una cantidad catalítica de una mezcla 1:1 de [tetrafluoroborato de Rh(ndb)2]BF4(bis(norbornadieno)rodio(I)) y BINAP (2,2'-bis(difenilfosfino)-1,1'-binaftilo) racémico en un disolvente adecuado (p. ej., mezcla de disolventes aproximadamente 10:1 de 1,4-dioxano y agua) bajo una atmósfera de argón a temperatura elevada (p. ej., aproximadamente 100°C). El tratamiento proporcionará 6-cloro-3-fenil-ds-indan-1-ona (C) racémica.
Esquema 1. Síntesis a modo de ejemplo de compuesto intermedio C

El tratamiento de 6-cloro-3-fenil-ds-indan-1-ona (C) con una base reductora tal como borohidruro de sodio (~ 2 equivalentes) en una mezcla de disolventes ~ 10:1 de tetrahidrofurano y agua a baja temperatura (aproximadamente -15°C) dará lugar a la reducción del grupo carbonilo en el correspondiente alcohol (Esquema 2). El tratamiento proporcionará cis-6-cloro-3-fenil-indan-1-ol (D) racémico. El tratamiento de este material con butirato de vinilo (aproximadamente 5 equivalentes) y Novozym 435® en un disolvente tal como di-iso-propiléter a temperatura ambiente proporcionará (1S,3S)-6-cloro-3-fenil-indan-1-ol (E) después del tratamiento.
Esquema 2. Síntesis a modo de ejemplo de compuesto intermedio E.

Alternativamente, la realización de la secuencia de A a E, utilizando ácido fenilborónico o 4,4,5,5-tetrametil-2-fenil-[1,3,2]dioxaborolano en lugar de 4,4,5,5-tetrametil-2-d5-fenil[1,3,2]dioxaborolano conducirá a (1S,3S)-6-cloro-3-fenilindan-1-ol (E') (Esquema 3).
Esquema 3. Síntesis a modo de ejemplo de compuesto intermedio E'.

Otros métodos de síntesis alternativos para obtener E' se describen en la bibliografía de patentes (Dahl, W 0 hlk Nielsen, Suteu, Robin, Br0 sen, documento WO2006/086984 A1; Bang-Andersen, B0 ges0 , Jensen, Svane, Dahl, Howells, Lyngs0 , Mow documento WO2005/016901 A1). Estos procedimientos se basan en cianuro de bencilo como uno de los sustratos. Utilizando cianuro de bencilod (disponible comercialmente de Aldrich, n° de catálogo 495840) o fenil-ds-acetonitrilo (disponible comercialmente de Aldrich n° de catálogo 495859 o de CDN n° de catálogo D-5340 o de Kanto n° de catálogo 49132-27) el mismo procedimiento puede conducir a E (Esquema 4). Como alternativas a las fuentes comerciales, cianuro de bencilo-dz y fenil-d5-acetonitrilo, se pueden preparar cianuro de sodio y cloruro de bencilo-dz (disponible comercialmente de Aldrich, n° de catálogo 217336) y cloruro de bencilo-2,3,4,5,6-ds (disponible comercialmente de Aldrich, n° de catálogo 485764), respectivamente.
Esquema 4. Síntesis a modo de ejemplo de compuestos intermedios E y E'.

El tratamiento de E con aproximadamente 4 equivalentes de di-iso-propiletMamina y aproximadamente 2 equivalentes de anhídrido metanosulfónico en tetrahidrofurano a aproximadamente - 18°C, seguido de calentamiento lento a aproximadamente -5°C y posterior tratamiento con aproximadamente 4 equivalentes de 2,2-dimetil-piperazina dará lugar a la formación de 1-((1R,3S)-6-cloro-3-fenil-d5-indan-1-il)-3,3-dimetil-piperazina (F) que puede ser purificada después de la reacción (Esquema 5). Alternativamente, el alcohol E se puede convertir en el cloruro correspondiente, predominantemente conservando la configuración en C1 que conduce a (1S,3S)-1-cloro-3-d5-fenilindano (E"; de manera similar, E' se puede convertir en (1S,3S)-1-cloro-3-fenil-indano (E"')). El cloruro E" se puede hacer reaccionar con 2,2-dimetil-piperazina para proporcionar F. La etapa final puede realizarse tal como se describe para la preparación del Compuesto (I) • sal de ácido butanodioico mediante el uso de yodometano para dar el Compuesto (II) o d3-yodometano para dar el Compuesto (IV), respectivamente. Alternativamente, tal como se describe a continuación, el grupo metilo o el grupo d3-metilo se puede instalar por reflujo en HCHO/HCOOH o DCDO/DCOOD, respectivamente.
Esquema 5. Síntesis a modo de ejemplo de compuesto intermedio F y los Compuestos (II) y (IV).

Éster ferc.-butílico del ácido (2-amino-2-metil-propil)-carbámico (G) se puede preparar a partir de 2-metil-propano-1,2-diamina y dicarbonato de di-ferc.-butilo (alternativamente, G se afirma que está disponible comercialmente: catálogo principal n° POI-1362-MB4; catálogo de Rovathin n° NX45401). La reacción de G con un haluro de haloacetilo tal como cualquiera de cloruro de cloroacetilo o bromuro de bromoacetilo dará éster ferc.-butílico del ácido [2-(2-cloro-acetilamino)-2-metil-propil)-carbámico o éster ferc.-butílico del ácido [2-(2-bromo-acetilamino)-2-metil-propil)-carbámico (H), respectivamente
(Esquema 6). Tratamiento de cualquiera de variante de H con ácido seguido por la base conducirá a la formación de 6,6-dimetil-piperazina-2-ona (I). Este material puede ser reducido a 2,2-dimetil-5,5-d2-piperazina (J) mediante tratamiento con deuterio de litio y aluminio.
Esquema 6. Síntesis a modo de ejemplo de compuesto intermedio J.

Alternativamente, J se puede preparar a partir de ácido 2-amino-2-metil-propiónico. La reacción de ácido 2-amino-2-metil-propiónico y dicarbonato de di-ferc.butilo proporcionará ácido 2-ferc.-butoxicarbonilamino-2-metil-propiónico (K) (Esquema 7). La funcionalidad de ácido se puede convertir en la correspondiente amida de Weinreb por reacción con 0,A/-d¡met¡l-h¡droxilam¡na en presencia de un reactivo de acoplamiento adecuado tal como metanaminio de hexafluorofosfato de 2-(1H-7-azabenzotriazol-1-il)-1,1,3,3-tetrametil-uronio (HATU) o 1-etil-3-(3-dimetilaminopropil)-carbodiimida (EDC) para proporcionar éster ferc.-butílico del ácido [1-(metoxi-metil-carbamoil)-1-metiletil]-carbámico (L). La reducción selectiva de la amida de Weinreb conduce a éster ferc.-butílico del ácido (1,1-dimetil-2-oxo-etil)-carbámico (M). La aminación reductora que implica aldehído M y éster metílico del ácido amino-acético se puede utilizar para preparar éster metílico del ácido (2-ferc.-butoxicarbonilamino-2-metil-propilamino)-acético (N). El tratamiento de carbamato-éster N con un ácido adecuado, tal como ácido trifluoroacético, conducirá a la formación de piperazinona I que, tras el tratamiento con deuteruro de litio y aluminio da piperazina J.
Esquema 7. Síntesis a modo de ejemplo alternativa de compuesto intermedio J.

Utilizando J en lugar de 2,2-dimetil-piperazina tal como se describe para la conversión de E en compuestos (II) y (IV), conducirá al Compuesto (VI) y al Compuesto (VII), respectivamente. De manera similar, utilizando E' y J en lugar de 2,2-dimetil-piperazina y E, conducirá al Compuesto (III) y el Compuesto (V).
Se describe un procedimiento para la preparación del compuesto

que comprende tratar el compuesto (XIV) con [(S)-BINAP]Rh(I)BF4.
Se describen un procedimiento de preparación del compuesto tartrato de (1R,3S)-(IV), que comprende el tratamiento de la £rans-1-(6-cloro-3-fenil(d5)-indan-1-il)-1(d3),2,2-trimetil-piperazina racémica con ácido L-(+)-tartárico.
En algunas descripciones, la £rans-1-(6-cloro-3-fenil(d5)-indan-1-il)-1(d3),2,2-trimetil-piperazina racémica se genera a partir de la sal succinato correspondiente de la misma.
En algunas descripciones, succinato de frans-1-(6-cloro-3-fenil(d5)-indan-1-il)-1(d3),2,2-trimetil-piperazina racémico se genera a partir de la sal maleato de la £rans-1-(6-cloro-3-fenil(d5)-indan-1-il)-3,3-dimetil-piperazina racémica. En algunas descripciones, acetofenona-d5 se convierte en un enol-éter. En algunas realizaciones, el enol-éter es un silil-enol-éter. En algunas realizaciones, el enol-éter de acetofenona-d5 se convierte en el boronato de vinilo correspondiente. En algunas realizaciones, el enol-éter de acetofenona-d5 se trata con bis(pinacolato)diboro. En algunas realizaciones, el boronato de vinilo se trata con 2-halo-5-clorobenzaldehído.
En algunas descripciones, los compuestos existen en forma de racematos. En algunas realizaciones, los compuestos existen en más de aproximadamente el 70% de exceso enantiomérico. En algunas realizaciones, los compuestos existen en más de aproximadamente el 75% de exceso enantiomérico. En algunas realizaciones, los compuestos existen en más de aproximadamente el 80% de exceso enantiomérico. En algunas realizaciones, los compuestos existen en más de aproximadamente el 85% de exceso enantiomérico. En algunas realizaciones, los
compuestos existen en más de aproximadamente el 90% de exceso enantiomérico. En algunas realizaciones, los compuestos existen en más de aproximadamente el 92% de exceso enantiomérico. En algunas realizaciones, los compuestos existen en más de aproximadamente el 95% de exceso enantiomérico. En algunas realizaciones, los compuestos existen en más de aproximadamente el 97% de exceso enantiomérico. En algunas realizaciones, los compuestos existen en más de aproximadamente el 99% de exceso enantiomérico.
Composiciones farmacéuticas
Se describen composiciones farmacéuticas que comprenden una cantidad terapéuticamente eficaz de los compuestos de la presente invención y un soporte o diluyente farmacéuticamente aceptable.
Los compuestos de la invención pueden administrarse solos o en combinación con soportes, diluyentes o excipientes farmacéuticamente aceptables, en dosis únicas o múltiples. Las composiciones farmacéuticas de acuerdo con la invención se pueden formular con soportes o diluyentes farmacéuticamente aceptables, así como cualesquiera otros adyuvantes y excipientes conocidos de acuerdo con técnicas convencionales tales como las descritas en Remington: The Science and Practice of Pharmacy, 21a Edición (Universidad de la Ciencias en Filadelfia, ed., Lippincott Williams & Wilkins 2005). Composiciones a modo de ejemplo adicionales de los compuestos de la invención se describen, por ejemplo, en las Patentes de EE.UU. N°s. 5.807.855; 7.648.991; 7.767.683; 7.772.240; 8.076.342; las Publicaciones de Patente de EE.UU. N°s. 2008/0269248; 2010/0069676; 2011/0178094; 2011/0207744; los documentos WO 2005/016900; EP 0638 073; y J. Med. Chem. 1995, 38, 4380 4392.
Las composiciones farmacéuticas se pueden formular específicamente para administración por cualquier vía adecuada tales como las vías oral, nasal, tópica (incluyendo bucal y sublingual), y parenteral (incluyendo subcutánea, intramuscular, intratecal, intravenosa e intradérmica). Se apreciará que la vía dependerá del estado general y de la edad del sujeto a tratar, de la naturaleza de la afección a tratar y del ingrediente activo.
La dosis diaria de los compuestos de la invención, calculada como la base libre, es adecuadamente de aproximadamente 1,0 a aproximadamente 160 mg/día, más adecuadamente de aproximadamente 1 a aproximadamente 100 mg, p. ej., preferiblemente de aproximadamente 2 a aproximadamente 55, tal como de aproximadamente 2 a aproximadamente 15 mg, p. ej., de aproximadamente 3 a aproximadamente 10 mg. En algunas realizaciones, la dosis diaria es de aproximadamente 0,1 mg a aproximadamente 500 mg. En algunas realizaciones, la dosis diaria es de aproximadamente 1 mg a aproximadamente 500 mg. En algunas realizaciones, la dosis diaria es de aproximadamente 1 mg a aproximadamente 400 mg. En algunas realizaciones, la dosis diaria es de aproximadamente 1 mg a aproximadamente 300 mg. En algunas realizaciones, la dosis diaria es de aproximadamente 1 mg a aproximadamente 200 mg. En algunas realizaciones, la dosis diaria es de aproximadamente 1 mg a aproximadamente 160 mg. En algunas realizaciones, la dosis diaria es de aproximadamente 1 mg a aproximadamente 100 mg. En algunas realizaciones, la dosis diaria es de aproximadamente 1 mg a aproximadamente 60 mg. En algunas realizaciones, la dosis diaria es de aproximadamente 2 mg a aproximadamente 30 mg. En algunas realizaciones, la dosis diaria es de aproximadamente 2 mg a aproximadamente 15 mg. En algunas realizaciones, la dosis diaria es de aproximadamente 3 mg a aproximadamente 10 mg. En algunas realizaciones, la dosis diaria es de aproximadamente 60 mg. En algunas realizaciones, la dosis diaria es de aproximadamente 50 mg. En algunas realizaciones, la dosis diaria es de aproximadamente 40 mg. En algunas realizaciones, la dosis diaria es de aproximadamente 30 mg. En algunas realizaciones, la dosis diaria es de aproximadamente 20 mg. En algunas realizaciones, la dosis diaria es de aproximadamente 10 mg. En algunas realizaciones, la dosis diaria es de aproximadamente 5 mg. En algunas realizaciones, la dosis diaria es de aproximadamente 3 mg. En algunas realizaciones, la dosis diaria es de aproximadamente 2 mg. En algunas realizaciones, la dosis diaria es de aproximadamente 1 mg.
Para vías parenterales tales como la intravenosa, intratecal, intramuscular y administración similares, las dosis típicas son del orden de la mitad de la dosis empleada para la administración oral.
Los compuestos de esta invención se utilizan generalmente como la sustancia libre o como una sal farmacéuticamente aceptable del mismo. Ejemplos de ácidos orgánicos e inorgánicos adecuados se describen en esta memoria.
En algunas realizaciones, la composición comprende una ciclodextrina. En algunas realizaciones, la composición comprende una ciclodextrina en agua. En algunas realizaciones, la ciclodextrina es hidroxipropil-p-ciclodextrina. En algunas realizaciones, la composición comprende hidroxipropil-p-ciclodextrina en agua.
Tratamiento de Trastornos
La invención describe el uso médico de compuestos de la presente invención tal como para el tratamiento de una enfermedad en el sistema nervioso central, incluyendo la psicosis, en particular esquizofrenia u otras enfermedades
que implican síntomas psicóticos tales como, p. ej., esquizofrenia, trastorno esquizofreniforme, trastorno esquizoafectivo, trastorno delirante, trastorno psicótico breve, trastorno psicótico compartido, así como otros trastornos psicóticos o enfermedades que se presentan con síntomas psicóticos, p. ej., trastorno bipolar tal como manía en el trastorno bipolar. Los compuestos y/o las composiciones de la invención se pueden utilizar, además, en el tratamiento de trastornos tales como los descritos, por ejemplo, en las Patentes de EE.UU. N°s. 5.807.855; 7.648.991; 7.767.683; 7.772.240; 8.076.342; las Publicaciones de Patente de EE.UU. N°s. 2008/0269248; 2010/0069676; 2011/0178094; 2011/0207744; los documentos WO 2005/016900; EP 0638 073; y J. Med.. Chem.
1995, 38, 4380-4392. La invención también describe el uso médico de compuestos de la presente invención como la terapia de combinación en combinación con otros agentes terapéuticos tales como los descritos, por ejemplo, en las Patentes de EE.UU. N°s. 5.807.855; 7.648.991; 7.767.683; 7.772.240; 8.076.342; las Publicaciones de Patente de EE.UU. N°s. 2008/0269248; 2010/0069676; 2011/0178094; 2011/0207744; los documentos WO 2005/016900; EP 0 638073; y J. Med.. Chem. 1995, 38, 4380-4392.
Se reconocerá que una o más características de cualquier realización descrita en esta memoria se pueden combinar y/o reorganizar dentro del alcance de la invención para producir realizaciones adicionales que están también dentro del alcance de la invención.
La invención se describe adicionalmente mediante los siguientes Ejemplos no limitativos.
EJEMPLOS
A continuación se proporcionan ejemplos para facilitar una comprensión más completa de la invención. Los siguientes ejemplos ilustran los modos a título de ejemplo de hacer y poner en práctica la invención. Sin embargo, el alcance de la invención no se limita a realizaciones específicas descritas en estos Ejemplos, que son para fines de ilustración solamente, ya que métodos alternativos se pueden utilizar para obtener resultados similares.
La purificación de los compuestos mediante cromatografía se refiere a la aplicación de la cromatografía de gel de sílice utilizando cromatografía de resolución instantánea manual o cromatografía de resolución instantánea automatizada, típicamente realizada utilizando gradientes de eluyente desde heptanos a acetato de etilo o mezclas de acetato de etilo, trietilamina y metanol.
Descripción de Métodos LCMS
Los compuestos (I), (II), (III), (IV), (V), (VI) y (VII) se caracterizaron mediante LCMS utilizando los métodos siguientes (Tabla 1):
Tabla 1: Métodos de análisis LCMS
Métodos WXV-AB5, WXV-AB10 y WXV-AB30
Equipo Agilent sistema 1100 LCMS con Detector ELS
[método sistema 1200 LCMS WuXiAB25 Agilent con Detector ELS]
Bomba G1311A
Desgasificador G1379A
Automuestreador pocillo-placa G1367A
Columna Horno G1316A
DAD G1315B
MSD G1946C o G1956A [método WuXiAB256110] ELSD Alltech ELSD 800 [método WuXiAB25 Aligent1200] Columna YMC ODS-AQ [método WuXiAB25 Agilent TC-C18]
Tamaño de partícula 5 micrómetros
Tamaño de poro 12 nm
Métodos WXV-AB5, WXV-AB10 y WXV-AB30

Métodos WXV-AB5, WXV-AB10 y WXV-AB30

Método 131

Métodos WXV-AB5, WXV-AB10 y WXV-AB30

Descripción de los métodos de HPLC Quiral
La pureza enantiomérica se ensayó en un sistema Hewlett Packard serie 1100 equipado con un detector de matrices de diodos y utilizando ChemStation para LC Rev. A.08.03[847]. Los parámetros del método de HPLC se describen en la siguiente tabla (Tabla 2). El compuesto (X) tiene un tiempo de retención de alrededor de 13,6-13,7 min, mientras que su enantiómero, 4-((1S,3R)-6-cloro-3-fenil-indan-1-il)-1,2,2-trimetil-piperazina, eluye a 8,5-8,6 min. Tabla 2: Métodos para el Análisis de HPLC Quiral

Ejemplo de referencia 1 Preparación de 4-((1R,3S)-6-cloro-3-fenil-indan-1-il)-1-metil-d3-2,2-dimetil-piperazina • ácido butanodioico (Compuesto (l) • sal de ácido butanodioico).
Esquema 8. Síntesis de Compuesto (I).

Hidrocloruro de 1-((1R,3S)-6-doro-3-fenil-indan-1-il)-3,3-dimetil-piperazina (11,1 g) se disolvió en una mezcla de tolueno (74 mL) y agua (74 mL). La preparación de hidrocloruro de 1-((1R,3S)-6-cloro-3-fenil-indan-1-il)-3,3-dimetilpiperazina se describe en la bibliografía de patentes (Dahl, W 0 hlk Nielsen, Suteu, Robin, Br0 sen documento WO2006/086984 A1; Bang-Andersen, B0 ges0 , Jensen, Svane, Dahl, Howells, Lyngs0 , Mow documento WO2005/016901 A1; cada una incorporada por referencia en su totalidad). Se añadieron hidróxido de potasio 12,0 M en agua (5,38 mL), bromuro de tetra-W-butilamonio (1,42 g), y cb-yodometano (catálogo Aldrich n° 176036; 2,4 mL) y la mezcla se agitó a temperatura ambiente durante 18 horas (Esquema 8). La mezcla se filtró a través de un filtro de vidrio en un embudo separador. El sólido en el filtro se lavó con tolueno (50 mL) en el embudo de decantación. La capa acuosa se extrajo con tolueno (100 mL) y las capas orgánicas reunidas se lavaron con amoniaco concentrado acuoso (100 mL) y posteriormente con agua (100 mL) antes de que se secaran sobre sulfato de sodio, se filtraran y se concentraran en vacío, proporcionando un aceite ligeramente amarillo. El aceite se enfrió a -78°C bajo vacío que solidificó el aceite. Después de calentar a temperatura ambiente, el aceite se convirtió en un semi-sólido.
Este material se disolvió en acetona (30 mL); en un matraz de ácido butanodioico separado (3,46 g) se suspendió en acetona (30 mL) y se calentó a reflujo (no todo el diácido pasó a disolución). La suspensión de ácido se añadió a la disolución del producto bruto y se añadió acetona adicional (50 mL) al residuo de ácido butanodioico y después se vertió en la disolución. La mezcla se agitó durante la noche. Durante la noche se había producido una precipitación parcial, y la mezcla se concentró en vacío. El residuo se volvió a disolver en acetona (70 mL) y se calentó a reflujo y se dejó enfriar a temperatura ambiente y se agitó durante 2 horas.
La mezcla se filtró proporcionando 4-((1R,3S)-6-cloro-3-fenil-indan-1-il)-1-metil-d3-2,2-dimetil-piperazina • ácido butanodioico (Compuesto (I) • sal del ácido butanodioico; 7,61 g). LC-MS (método 131): RT (UV) 1,57 min; UV/ELS pureza del 100%/100%; masa observada 358,0. Incorporación de tres átomos de deuterio > 99%. El espectro 13C RMN con protón desacoplado mostró un heptete en torno a 36,4 ppm que corresponde al sitio metabólico M2 deuterado; esta señal se derrumbó a un singlete en el espectro 13C RMN con protón y deuterio desacoplado. Todas las demás señales eran singletes en ambos espectros. Pureza óptica > 95% ee.
Ejemplo de referencia 2 Método alternativo de preparación de 4-((1R,3S)-6-cloro-3-fenil-indan-1-il)-1-metil-d3-2,2-dimetil-piperazina • ácido butanodioico (Compuesto (I) • sal del ácido butanodioico)
La base libre de 1-((1R,3S) -6-cloro-3-fenil-indan-1-il)-3,3-dimetil-piperazina se preparó a partir de la sal hidrocloruro correspondiente mediante el reparto de 23,4 g de la sal entre una mezcla de agua (100 mL), hidróxido de potasio acuoso concentrado (40 mL) y tolueno (250 mL). La capa orgánica se lavó con una mezcla de agua (50 mL) e hidróxido de potasio acuoso concentrado (10 mL). Las capas acuosas reunidas se extrajeron con tolueno (75 mL). Las capas orgánicas reunidas se secaron sobre sulfato de sodio, se filtraron y se concentraron en vacío, proporcionando la base libre de 1-((1R,3S)-6-cloro-3-fenil-indan-1-il)-3,3-dimetil-piperazina (21,0 g) en forma de un aceite incoloro. Este material se disolvió en una mezcla de tolueno (150 mL) y agua (150 mL), antes de añadir hidróxido de potasio acuoso 12,0 M (11,3 mL), bromuro de tetra-W-butilamonio (2,98 g), y d3-yodometano (4,9 mL) y la mezcla se agitó a temperatura ambiente durante 18 horas.
El tratamiento y la purificación se realizaron tal como se ha descrito anteriormente y proporcionaron 4-((1R,3S)-6-cloro-3-fenil-indan-1-il)-1-metil-d3-2,2-dimetil-piperazina • ácido butanodioico (Compuesto (I) • sal del ácido butanodioico); 14,34 g; 48,9%).
Ejemplo 3 Preparación de 4-((1R,3S)-6-cloro-3-fenil-d5-indan-1-il)-1,2,2-trimetil-piperazina (Compuesto (II)) y 4-((1R,3S)-6-cloro-3-fenil-d5-indan-1-il)-1-metil-d3-2,2-dimetil-piperazina (Compuesto (IV)).
A una disolución del compuesto A (57 g) en tetrahidrofurano (600 mL) se añadió trietilamina (30 mL) gota a gota durante 30 min. La mezcla de reacción se mantuvo a temperatura ambiente durante 3 horas. El sólido precipitado se filtró y el filtrado se concentró en vacío. El residuo se volvió a precipitar en dietiléter para proporcionar el compuesto B (31 g) en forma de un sólido amarillo. A una disolución del compuesto ácido fenil-ds-borónico (25 g) en 1,4-dioxano/agua (900 mL/90 mL) se añadió [Rh(ndb)2]BF4 (1,3 g), BInAp racémico (2,1 g) y trietilamina (14 mL), y después la mezcla de reacción se mantuvo a temperatura ambiente durante 2 horas bajo N2. A continuación se añadió compuesto indenona (19 g), y la mezcla resultante se calentó a 100°C durante 3 horas. El sólido precipitado se separó por filtración. El filtrado se concentró en vacío. El residuo se purificó por cromatografía para proporcionar indanona C (10 g).
Esquema 9. Síntesis del Compuesto C.

13,4 kg de 3-bromo-6-cloro-indan-1-ona (A; para referencias en este material, véase: B0 ges0 documento EP 35363 A1 19810909 y Kehler, Juhl, Püschl, documento WO 2008025361) se disolvieron en tetrahidrofurano (170,8 L), y la disolución se enfrió a 0-5°C (Esquema 9). Se añadió trietilamina (9,1 L) durante 0,5 h. La mezcla se agitó a 0-5°C durante 5 horas antes de añadir una porción adicional de trietilamina (2,48 L) a lo largo de 0,5 horas, y se continuó la agitación durante 2 horas. La mezcla se filtró, y el filtrado se concentró a 30 L antes de añadir n-heptano (102 L). El volumen se redujo a 60 L. Se añadió más n-heptano (203 L), y la mezcla se agitó durante 1 hora. Se añadió gel de sílice (17,2 kg). La mezcla se filtró, y el sólido residual se lavó con n-heptano (100 L). Los filtrados reunidos se concentraron a 30 L y se agitaron a 0-5°C durante 1 hora. La mezcla se centrifugó y el sólido residual se secó para proporcionar 6-cloro-inden-1-ona (compuesto B; 2,42 kg) suficientemente puro para la siguiente etapa.
2-metil-tetrahidrofurano (85 L) y W,A/-dimetil-acetamida (12,4 L) se añadieron a un reactor, seguido de acetato de potasio (10,9 kg) y bis(pinacolato)diboro (14,8 kg). La mezcla resultante se agitó durante 0,5 horas. Se añadió Pd(dppf)Cl2-DCM (0,91 kg) seguido de bromobenceno-d5 (9,0 kg) y 2-metil-tetrahidrofurano (12,2 L). La mezcla se calentó a 80-85°C durante 3 horas, antes de que la temperatura se redujera a la temperatura ambiente. La mezcla bruta se filtró a través de tierra de diatomeas y gel de sílice. La torta de filtración se lavó con 2-metil-tetrahidrofurano (31 L). Los filtrados reunidos se concentraron a aproximadamente 25 L al tiempo que la temperatura se mantenía por debajo de 35°C. Se añadieron W-heptano (52 L) y NaHCO3 acuosa al 7% (31 L), y la mezcla se agitó durante 0,5 horas. La capa orgánica se agitó con NaHCO3 acuosa al 7% (31 L) durante 0,5 horas. Las capas acuosas reunidas se extrajeron con n-heptano (22 L) a lo largo de 0,5 horas. Los extractos orgánicos reunidos se lavaron con NaCl acuoso al 25% (50 L) a lo largo de 0,5 horas. La capa orgánica se concentró al tiempo que la temperatura se mantenía por debajo de 35°C para proporcionar 4,4,5,5-tetrametil-2-d5-fenil[1,3,2]dioxaborolano (compuesto B'; 10,5 kg) suficientemente puro para la siguiente etapa.
Se añadió a un reactor secuencialmente 1,4-dioxano (85 L), 6-cloro-inden-1-ona (compuesto B; 9,09 kg preparados de una manera similar a la descrita anteriormente), 1,5-ciclooctadieno (0,2 L), tetrafluoroborato de bis(norbornadieno)rodio(I) (0,52 kg), trietilamina (5,5 L), 4,4,5,5-tetrametil-2-d5-fenil-[1,3,2]-dioxaborolano (compuesto B'; 6,5 kg), y 1,4-dioxano (26 L). La mezcla se calentó a 48-53°C y se agitó a esa temperatura durante 5 horas. La
reacción se enfrió bruscamente mediante la adición de HCl acuoso 2 M (13 kg). Después se añadieron n-heptano (110 L), metil-ferc.-butil-éter (32 L) y agua (90 L), y la mezcla resultante se agitó durante 0,3 horas. La capa orgánica se lavó con agua (90 L) a lo largo de 0,3 horas. Las capas acuosas reunidas se extrajeron con una mezcla de metilferc.-butil-éter (30 L) y n-heptano (57 L) a lo largo de 0,3 horas. Las capas orgánicas reunidas se filtraron a través de gel de sílice (13 kg). La torta de filtración se lavó con una mezcla 2:1 de n-heptano y metil-ferc.-butil-éter (19,5 kg). El filtrado se concentró a aproximadamente 25 L. Se añadió n-heptano (45 L), y el volumen se redujo a aproximadamente 25 L. Se añadió n-heptano (45 L), y el volumen se redujo a aproximadamente 35 L. La mezcla se agitó a 0-5°C durante 3 horas. La mezcla se centrifugó, y el sólido residual se secó para dar 6-cloro-3-d5-fenil-indan-1-ona racémica (compuesto C; 8,4 kg) suficientemente puro para la siguiente etapa.
Se añadió tetrahidrofurano (90 L) a un reactor seguido de agua (10 L) y 6-cloro-3-d5-fenil-indan-1-ona (compuesto C; 7,73 kg) (Esquema 10). La mezcla se enfrió a -35 - 30°C. Borohidruro de sodio (1,5 kg) se añadió en porciones manteniendo la temperatura a -35 - -30°C. La mezcla resultante se agitó a -35 - -30°C durante 5 horas antes de dejar que se calentara a temperatura ambiente. Borohidruro de sodio en exceso se enfrió bruscamente mediante la adición de HCl 2 M acuoso (7,6 kg) manteniendo la temperatura por debajo de 45°C. Se añadieron agua (17 L) y metil-ferc.-butil-éter (67 L) y la mezcla se agitó durante 0,3 horas. La capa acuosa se extrajo con metil-ferc.-butil-éter (39 L) a lo largo de 0,3 horas. Las capas orgánicas reunidas se lavaron con salmuera (36 kg) a lo largo de 0,3 horas. La capa orgánica se filtró a través de gel de sílice (6,4 kg). La torta de filtración se lavó con metil-ferc-butil-éter (20 L). Los filtrados reunidos se concentraron a aproximadamente 30 L al tiempo que la temperatura se mantenía por debajo de 45°C. Se añadió n-heptano (55 L) y la mezcla resultante se concentró hasta aproximadamente 30 L al tiempo que la temperatura se mantenía por debajo de 45°C. La mezcla resultante se agitó a 0-5°C durante 2 horas. La mezcla se centrifugó y la torta de filtración se lavó con n-heptano (12 L) antes de centrifugar de nuevo. El sólido residual se secó para proporcionar el D bruto. 4,87 kg de este material se disolvieron en metil-ferc.-butil-éter (20 L) y se secaron sobre Na2SO4(2 kg) a lo largo de 0,25 horas. La mezcla se filtró, y la torta de filtración se lavó con metilferc.-butil-éter (4,4 L). El filtrado reunido se concentró hasta aproximadamente 20 L al tiempo que la temperatura se mantenía por debajo de 45°C. Se añadió n-heptano (32 L) y la mezcla fue de aproximadamente 25 L al tiempo que la temperatura se mantenía por debajo de 45°C. Se añadió n-heptano (16 L) y la mezcla fue de aproximadamente 20 L al tiempo que la temperatura se mantenía por debajo de 45°C. El sólido se separó por filtración y se secó para proporcionar c/s-6-cloro-3-d5-fenil-indan-1-ol racémico (compuesto D; 4,99 kg) suficientemente puro para la siguiente etapa.
Esquema 10. Síntesis y resolución de Compuesto E.

A una disolución de c/s-6-cloro-3-d5-fenil-indan-1-ol racémico (compuesto D; 50 g) en 2-isopropoxipropano (200 mL) se añadió butirato de vinilo (120 mL) y Novozym-435 (15 g). La mezcla se mantuvo a temperatura ambiente durante 2 días. El sólido se separó por filtración. El filtrado se evaporó y se purificó por cromatografía sobre gel de sílice para dar (1S,3S)-6-cloro-3-d5-fenil-indan-1-ol (compuesto E; 13 g) suficientemente puro para la siguiente etapa.
A una disolución de (1S,3S)-6-cloro-3-d5-fenil-indan-1-ol (compuesto E; 7 g) en THF (100 mL) se trató con SOCh(6,6 g) a temperatura ambiente durante la noche. La mezcla se vertió en agua enfriada con hielo y se extrajo con acetato de etilo. La capa orgánica se lavó con salmuera. La capa orgánica se secó sobre Na2SO4 , se filtró y se concentró en vacío para proporcionar el compuesto intermedio de cloruro (7,5 g). 3,5 g de este material se disolvieron en 2 butanona (50 mL) y se hizo reaccionar con 2,2-dimetil-piperazina (1,7 g) en presencia de K2CO3 (2,7 g) a reflujo durante la noche. El sólido se separó por filtración. El filtrado se concentró en vacío y el residuo se purificó mediante HPLC preparativa en un instrumento Shimadzu FRC-10A equipado con una columna Synergi C18 (250 mm * 50 mm, 10 micras) utilizando agua y acetonitrilo (que contiene TFA al 0,1%, v/v) como el eluyente para proporcionar 1-((1R,3S)-6-cloro-3-d5-fenil-indan-1-il)-3,3-dimetil-piperazina (compuesto F; 2,6 g) suficientemente puro para la siguiente etapa.
Una disolución de 1-((1R,3S)-6-cloro-3-d5-fenil-indan-1-il)-3,3-dimetil-piperazina (compuesto F; 2,2 g) en HCHO/HCOOH (3 mL/3 mL) se sometió a reflujo durante la noche. Los componentes volátiles se separaron en vacío. El residuo se repartió entre acetato de etilo y NaOH acuoso al 10%. La capa orgánica se secó sobre Na2SO4, se filtró y se concentró en vacío. El residuo se purificó mediante cromatografía sobre gel de sílice para proporcionar 4-((1R,3S)-6-cloro-3-d5-fenil-indan-1-il)-1,2,2-trimetil-piperazina (Compuesto (II); 1,89 g). LC-m S (método WXV-AB05): rT(UV) 2,43 min; UV/ELS pureza 95,1%/99,6%; masa observada 360,2. La incorporación de cinco átomos de deuterio > 95%. El espectro de 13C-RMN de protón desacoplado mostró tres tripletes en torno a 126,1, 127,2 y 128,2 ppm, que corresponde a los sitios metabólicos deuterados M3; estas señales se derrumbaron a tres singletes en el espectro de 13C-RMN de protón y deuterio desacoplado. Todas las demás señales eran singletes en ambos espectros. Pureza óptica > 95% ee.
Una disolución de 1-((1R,3S)-6-cloro-3-d5-fenil-indan-1-il)-3,3-dimetil-piperazina (compuesto F; 3,0 g) en DCDO/DCOOD (4 mL/4 mL) se sometió a reflujo durante la noche. Los componentes volátiles se separaron en vacío. El residuo se repartió entre acetato de etilo y NaOH acuoso al 10%. La capa orgánica se secó sobre Na2SO4, se filtró y se concentró en vacío. El residuo se purificó mediante cromatografía sobre gel de sílice para proporcionar 4-((1R,3S)-6-cloro-3-d5-fenil-indan-1-il)-1-d3-metil-2,2-dimetil-piperazina (Compuesto (IV); 2,14 g). LC-MS (método w Xv -AB10): RT(UV) 2,06 min; UV/ELS pureza 98%/100%; masa observada 363,3. La incorporación de ocho átomos de deuterio > 94%. El espectro de 13C-RMN de protón desacoplado mostró un heptete en torno a 36,4 ppm, que corresponde al sitio metabólico deuterado M2; esta señal se derrumbó a un singlete en el espectro de 13C-RMN de protón y deuterio-desacoplado. El espectro de 13C-RMN de protón desacoplado mostró, además, tres tripletes en torno a 126,1, 127,2 y 128,2 ppm, que corresponde a los sitios metabólicos deuterados M3; estas señales se derrumbaron a tres singletes en el espectro de 13C-RMN de protón y deuterio desacoplado. Todas las demás señales eran singletes en ambos espectros. Pureza óptica > 95% ee.
Ejemplo de referencia 4: Preparación de 4-((1R,3S)-6-cloro-3-fenil-indan-1-il)-1,2,2-trimetil-piperazina-6,6-d2 (Compuesto (III)), 4-((1R,3S)-6-cloro-3-fenil-indan-1-il)-1-metil-d3-2,2-dimetil-piperazina-6,6-d2 (Compuesto (V)), 4-((1R,3S)-6-cloro-3-fenil-d5-indan-1-il)-1-metil-d3-2,2-dimetil-piperazina-6,6-d2 (Compuesto (VI)) y 4-((1R,3S)-6-cloro-3-fenil-d5-indan-1-il)-1,2,2-trimetil-piperazina-6,6-d2( Compuesto (VII).
Ácido 2-amino-2-metil-propiónico (50,0 g) se suspendió en una mezcla de metanol y trietilamina (9:1, 1,2 L) (Esquema 11). Se añadió NaOH acuoso 1 M (450 mL) con agitación hasta que se disolvió todo el sólido. Se añadió dicarbonato de di-terc.-butilo (Boc2O, 214,0 g) y la mezcla se agitó a temperatura ambiente durante la noche. Los componentes volátiles orgánicos se separaron en vacío. Se añadió EtOAc (500 mL). La capa orgánica se lavó con salmuera y se secó sobre Na2SO4, se filtró y después se concentró para proporcionar ácido 2-terc.-butoxicarbonilamino-2-metil-propiónico (compuesto K; 90 g) en forma de un sólido blanco que se utilizó directamente en la siguiente etapa.
Esquema 11. Síntesis del compuesto intermedio J.

Una mezcla de proporcionar ácido 2-ferc.-butoxicarbonilamino-2-metil-propiónico (compuesto K; 60,0 g) e hidrocloruro de 1-etil-3(3-dimetilaminopropil)carbodiimida (EDCHCl; 86,4 g) en diclorometano (900 mL) se agitó a temperatura ambiente y, a continuación, se añadieron hidrocloruro de N,O-dimetil-hidroxilamina (35,3 g) y trietilamina (150 mL). La mezcla resultante se agitó a temperatura ambiente durante 3 días. Se añadió agua y la mayor parte de los componentes volátiles se separaron en vacío. El residuo se repartió entre DCM y NaHCO3 acuosa. La capa orgánica se lavó con HCl acuoso 3 M, subsiguientemente con salmuera antes de secar sobre Na2SO4, se filtró y se concentró en vacío. El residuo se purificó mediante cromatografía en gel de sílice para dar éster ferc.-butílico del ácido [1-(metoxi-metil-carbamoil)-1-metil-etil]-carbámico (compuesto L; 28,2 g) en forma de un sólido blanco lo suficientemente puro para la siguiente etapa.
Se añadió hidruro de litio y aluminio (7,8 g) a una disolución agitada de éster ferc.-butílico del ácido [1-(metoxi-metilcarbamoil)-1-metil-etil]-carbámico (compuesto L; 42,0 g) en dietiléter seco (1,5 L) a -40°C. A continuación, se agitó a esa temperatura durante aproximadamente 5 min. El exceso de LiAlH4 se enfrió bruscamente con una disolución de hidrógeno-sulfato de potasio en agua. La mezcla resultante se repartió entre EtOAc y HCl acuoso 3M. La capa orgánica se lavó con NaHCO3 acuoso sat., se secó sobre Na2SO4, se filtró y se concentró en vacío para proporcionar éster ferc.-butílico del ácido (1,1-dimetil-2-oxo-etil)-carbámico (compuesto M; 29 g) suficientemente puro para la próxima etapa.
Hidrocloruro de éster metílico del ácido amino-acético (80,6 g) y Et3N (160 mL) se disolvieron en DCM (1000 mL) y se agitaron durante 15 min para liberar la amina de la sal. A continuación, se añadió una disolución de éster ferc.-butílico del ácido (1,1-dimetil-2-oxo-etil)-carbámico (compuesto M; 29,0 g) en DCM (600 mL). La mezcla resultante se agitó durante 0,5 horas a temperatura ambiente antes de añadir NaBH(OAc)3 (102 g) y la mezcla se agitó a temperatura ambiente durante la noche. Se añadió NaHCO3 acuoso sat. La capa acuosa se extrajo con DCM. Las capas orgánicas reunidas se secaron sobre Na2SO4, se filtraron y se concentraron en vacío. El residuo se purificó mediante cromatografía en gel de sílice para dar éster metílico del ácido (2-ferc.-butoxicarbonilamino-2-metilpropilamino)-acético (compuesto N ; 26,5 g) en forma de un sólido blanco que se utilizó directamente en la siguiente etapa.
Una mezcla de ácido (2-ferc.-butoxicarbonilamino-2-metil-propilamino)-acético (compuesto N ; 26,5 g) en DCM (800 mL) se agitó a la temperatura ambiente y TFA (180 mL) se añadió gota a gota . La mezcla se agitó a 30-40°C durante 5 h antes de concentrarla en vacío. El residuo se repartió entre tolueno disuelto y agua. La capa orgánica se secó sobre Na2SO4, se filtró y se concentró en vacío. El sólido residual se disolvió en una mezcla de etanol (400 mL)
y metanol (90 mL). Se añadió K2CO3 (207 g) y la mezcla se sometió a reflujo durante la noche. La mezcla se enfrió a temperatura ambiente. Se añadió DCM (2500 mL), y la mezcla se agitó durante 1 hora a temperatura ambiente. El sólido se separó por filtración, y el filtrado se concentró en vacío para proporcionar 6,6-dimetil-piperazin-2-ona (Compuesto I; 5,85 g) en forma de un sólido blanco suficientemente puro para la siguiente etapa.
Una disolución de 6,6-dimetil-piperazin-2-ona (Compuesto I; 3,6 g) en THF (20 mL) se agitó a 0°C. Se añadió deuteruro de litio y aluminio (LiAlD4; 3,6 g) y después la mezcla se sometió a reflujo durante la noche. La mezcla se enfrió a temperatura ambiente y se añadió Na2SO4. La mezcla se agitó durante 0,5 h antes de separar en vacío la mayor parte de los componentes volátiles. El residuo se suspendió en una disolución saturada de HCl en EtOAc a temperatura ambiente durante 0,5 horas. El sólido se separó por filtración y se secó para proporcionar 2,2-d2-6,6-dimetil-piperazina en forma de la sal bis-hidrocloruro (Compuesto J2HCl; 5,3 g) suficientemente puro para la siguiente etapa.
A una disolución del compuesto E' (5 g) en THF (50 mL) se añadió SOCl2 (4,7 g), y la mezcla resultante se agitó durante una noche a temperatura ambiente (Esquema 12). La mezcla se vertió en agua helada y se extrajo con EtOAc. La capa orgánica se lavó con salmuera, se secó sobre Na2SO4, se filtró y se concentró en vacío para dar el correspondiente cloruro (5,3 g) que se utilizó directamente en la siguiente etapa. 3,3 g de este material se disolvieron en 2-butanona (50 mL) y se hicieron reaccionar con 2,2-d2-6,6-dimetil-piperazina (Compuesto J; 3 g) en presencia de K2CO3 (8,28 g ) a reflujo durante la noche. El sólido se separó por filtración. El filtrado se concentró en vacío. El residuo se purificó mediante HPLC preparativa en un instrumento Shimadzu FRC-10A equipado con una columna Synergy C18 (250 mm * 50 mm, 10 micras) utilizando agua y acetonitrilo (que contiene TFA al 0,1%, v/v) como los eluyentes para proporcionar 1-((1R,3S)-6-cloro-3-fenil-indan-1-il)-3,3-d2-5,5-dimetil-piperazina (Compuesto O; 1,7 g). Esquema 12. Síntesis de Compuesto (III) y Compuesto (V)

Una disolución de 1-((1R,3S)-6-cloro-3-fenil-indan-1-il)-3,3-d2-5,5-dimetil-piperazina (Compuesto O; 0,5 g) en HCHO/HCOOH (1 mL/1 mL) se calentó a reflujo durante la noche. Los componentes volátiles se separaron en vacío. El residuo se repartió entre EtOAc y NaOH acuoso al 10%. La capa orgánica se secó sobre Na2SO4 , se filtró y se concentró en vacío. El residuo se purificó mediante cromatografía sobre gel de sílice para proporcionar 4-((1R,3S)-6-cloro-3-fenil-indan-1-il)-1,2,2-trimetil-piperazina-6,6-d2 (Compuesto (III); 0,33 g). LC-MS (método w Xv -Ab30): RT(UV) 1,42 min; UV/ELS pureza del 100%/100%; masa observada 357,2. La incorporación de dos átomos de deuterio > 97%. El espectro de 13C-RMN de protón desacoplado mostró un quintete en torno a 49,5 ppm, que corresponde al sitio metabólico deuterado M1; esta señal se derrumbó a un singlete en el espectro de 13C-RMN de protón y deuterio desacoplado. El espectro de 13C-RMN de protón desacoplado mostró, además, tres tripletes en torno a 126,1, 127,2 y 128,2 ppm, que corresponden a los sitios metabólicos deuterados M3; estas señales se derrumbaron a tres singletes en el espectro de 13C-RMN de protón y deuterio-desacoplado. Todas las demás señales eran singletes en ambos espectros. Pureza óptica > 95% ee.
Una disolución de 1-((1R,3S)-6-cloro-3-fenil-indan-1-il)-3,3-d2-5,5-dimetil-piperazina (Compuesto O; 0,7 g) en DCDO/DCOOD (1 mL/1 mL) se calentó a reflujo durante la noche. Los componentes volátiles se separaron en vacío. El residuo se repartió entre EtOAc y NaOH acuoso al 10%. La capa orgánica se secó sobre Na2SO4 , se filtró y se concentró en vacío. El residuo se purificó mediante cromatografía sobre gel de sílice para proporcionar 4-((1R,3S)-6-cloro-3-fenil-indan-1-il)-1-metil-d3-2,2-dimetil-piperazina-6,6-d2 (Compuesto (V); 0,49 g). LC-MS (método w Xv -AB25): RT(UV) 2,13 min; UV/ELS pureza del 100%/100%; masa observada 360,2. La incorporación de cinco átomos de deuterio > 95%. El espectro de 13C-RMN de protón desacoplado mostró un heptete en torno a 36,4 ppm, que corresponde al sitio metabólico deuterado M2; esta señal se derrumbó a un singlete en el espectro de 13C-RMN de protón y deuterio desacoplado. El espectro de 13C-RMN de protón desacoplado mostró, además, un quintete en
torno a 49,5 ppm, que corresponden al sitio metabólico deuterado M1; esta señal se derrumbó a un singlete en el espectro de 13C-RMN de protón y deuterio-desacoplado. Todas las demás señales eran singletes en ambos espectros. Pureza óptica > 95% ee.
Una disolución de (1S,3S)-6-cloro-3-d5-fenil-indan-1-ol (compuesto E; 7 g) en THF (100 mL) se trató con SOCh(6,6 g) a temperatura ambiente durante la noche (Esquema 13). La mezcla se vertió en agua enfriada con hielo, y se extrajo con acetato de etilo. La capa orgánica se lavó con salmuera. La capa orgánica se secó sobre Na2SO4, se filtró y se concentró en vacío para dar el compuesto intermedio de cloruro (7,5 g).
Esquema 13. Síntesis de Compuesto (VI) y Compuesto (VII)

1,8 g de este material se disolvieron en 2-butanona (30 mL) y se hicieron reaccionar con 2,2-d2-6,6-dimetilpiperazina (Compuesto J; 1,4 g) en presencia de K2CO3(5,5 g ) a reflujo durante la noche. El sólido se separó por filtración. El filtrado se concentró en vacío. El residuo se purificó mediante HPLC preparativa en un instrumento Shimadzu FRC-10A equipado con una columna Synergy C18 (250 mm * 50 mm, 10 micras) utilizando agua y acetonitrilo (que contiene TF A al 0,1%, v/v) como eluyentes para proporcionar 1-((1R,3S)-6-cloro-3-d5-fenil-indan-1-il)-3,3-d2-5,5-dimetil-piperazina (Compuesto P; 1,7 g).
Una disolución de 1-((1R,3S)-6-cloro-3-d5-fenil-indan-1-il)-3,3-d2-5,5-dimetil-piperazina (Compuesto P; 1 g) en DCDO/DCOOD (1,5 mL/1,5 mL) se sometió a reflujo durante la noche. Los componentes volátiles se separaron en vacío. El residuo se repartió entre EtOAc y NaOH acuoso al 10%. La capa orgánica se secó sobre Na2SO4, se filtró y se concentró en vacío. El residuo se purificó mediante cromatografía sobre gel de sílice para proporcionar 4-((1R,3S)-6-cloro-3-d5-fenil-indan-1-il)-1-d3-metil-2,2-dimetil-piperazina-6,6-d2 (Compuesto (VI); 0,55 g). LC-MS (método WuXiAB25): RT(UV) 2,13 min; UV/ELS pureza 98,2%/100%; masa observada 365,2. La incorporación de diez átomos de deuterio > 91%. El espectro de 13C-RMN de protón desacoplado mostró un heptete en torno a 36,4 ppm, que corresponde al sitio metabólico deuterado M2; esta señal se derrumbó a un singlete en el espectro de 13C-RMN de protón y deuterio desacoplado. El espectro de 13C-RMN de protón desacoplado mostró, además, un quintete en torno a 49,5 ppm, que corresponde al sitio metabólico deuterado M1; esta señal se derrumbó a un singlete en el espectro de 13C-RMN de protón y deuterio-desacoplado. El espectro de 13C-RMN de protón desacoplado mostró, además, tres tripletes en torno a 126,1, 127,2 y 128,2 ppm, que corresponden a los sitios metabólicos deuterados M3; estas señales se derrumbaron a tres singletes en el espectro de 13C-RMN de protón y deuterio-desacoplado. Todas las demás señales eran singletes en ambos espectros. Pureza óptica > 95% ee.
Una disolución de 1-((1R,3S)-6-cloro-3-d5-fenil-indan-1-il)-3,3-d2-5,5-dimetil-piperazina (compuesto P; 0,7 g) en HCHO/HCOOH (1 mL/1 mL) se sometió a reflujo durante la noche. Los componentes volátiles se separaron en vacío. El residuo se repartió entre EtOAc y NaOH acuoso al 10%. La capa orgánica se secó sobre Na2SO4, se filtró y se concentró en vacío. El residuo se purificó mediante cromatografía sobre gel de sílice para proporcionar 4-((1R,3S)-6-cloro-3-d5-fenil-indan-1-il)-1-metil-2,2-dimetil-piperazina-6,6-d2 (Compuesto (VII); 0,47 g). LC-MS (método Wx V-a B30): RT(UV) 1,33 min; UV/ELS pureza 97,4%/100%; masa observada 362,3. La incorporación de siete átomos de deuterio > 93%. El espectro de 13C-RMN de protón desacoplado mostró un quintete en torno a 49,5 ppm, que corresponde al sitio metabólico deuterado M1; esta señal se derrumbó a un quintete en el espectro de 13C-RMN de protón y deuterio desacoplado. El espectro de 13C-RMN de protón desacoplado mostró, además, tres tripletes en torno a 126,1, 127,2 y 128,2 ppm, que corresponde a los sitios metabólicos deuterados M3; estas señales se derrumbaron a tres singletes en el espectro de 13C-RMN de protón y deuterio-desacoplado. Todas las demás señales eran singletes en ambos espectros. Pureza óptica > 95% ee.
Ejemplo 5: Descripción de la determinación de RMN de la o las posiciones que portan deuterio en lugar de hidrógeno
Los espectros de RMN se registraron en un espectrómetro Bruker 600-Avance-III-equipado con una criosonda TCI de 5 mm que funciona a 150,91 MHz para 13C. El disolvente CDCh se utilizó como referencia interna para los experimentos de protones desacoplados, mientras que los espectros de protón y deuterio de sincronización inversa desacoplado se registraron utilizando un cierre sincronizado. La o las diferencias entre los dos espectros para los compuestos de la invención determinan la o las posiciones de los átomos de deuterio. Cuando se combina esta información resumida en la siguiente tabla (Tabla 3) con los datos de espectrometría de masas por electroproyección que determinan el grado de deuteración, las estructuras de los compuestos de la invención se pueden asignar sin ambigüedad.
Tabla 3: Datos de RMN de carbono para compuestos.
M1 ( til

p
Regiones relevantes de los espectros de 13C-RMN de protón-desacoplado (espectro inferior) y 13C-RMN de protón y deuterio-desacoplado (espectro superior) de Compuesto (II) y Compuesto (V) se muestran en la Figura 3 como ejemplos representativos. Regiones seleccionadas de los espectros de 13C-RMN de protón y deuterio-desacoplado de Compuesto (II) [Fig. 3A] y Compuesto (V) [Fig. 3B].
Ejemplo 6: Descripción de la espectrometría de masas por electroproyección para determinar el grado de deuteración
Instrumentación: Los espectros de masas de disoluciones acuosas de carácter ácido de los compuestos se obtuvieron en un espectrómetro de masas cuadrupolar Hewlett Packard modelo 1100 LC-MSD. La cromatografía líquida se realizó en un sistema Agilent HPLC 1100 acoplado al espectrómetro de masas.
Parte Experimental: Disoluciones de las muestras se prepararon disolviendo aprox. 2 mg de sustancia en 2 mL de metanol 18 mL de formiato de amonio 10 mM de pH 3,0. Posteriormente, las disoluciones se diluyeron 100 veces antes del análisis. Con el fin de obtener un pico "limpio", las muestras se sometieron a cromatografía utilizando una columna Waters X-bridge C 18, 3,5 micras (150x2.1mm) y ácido trifluoroacético al 0,1%/acetonitrilo 50/50 como fase móvil. Este procedimiento dio un pico del compuesto de interés que eluye a aprox. 3,6 min, que contiene tanto los compuestos deuterados de la invención como pequeñas cantidades de especies deficientes en deuterio. Los espectros de masas obtenidos a partir de estos picos se utilizaron para evaluar la especiación de las moléculas diana. Se analizaron los resultados en porcentaje de la cantidad total de sustancia, que suman hasta el 100%. La potencia real de los compuestos no se analizó, simplemente el contenido relativo de las especies deficientes en deuterio.
Como ejemplo representativo, el espectro de masas del Compuesto (IV) se muestra en la Figura 4. El patrón isotópico del Compuesto (V) protonado [M H]+ con masa 363.1u (362.1u 1,0 u) y los iones isotópicos 363.1u, 364.1u, 365.1u y 366.1u estaba en la relación de 100:25,3:34,9:7,9; cálculo para C20H22N2ClD8 da la relación de 100:25,2:34,9:8,3. Además de ello, se observaron análogos D7 y análogos D3 en las masas 362.1u y 358.1u, respectivamente. Las señales en 364u, 365u y 366u se deben principalmente a las moléculas protonadas que contienen isótopos 13C y/o 37Cl en lugar de 12C y 35Cl (debido a la distribución natural). Estos datos demuestran que la incorporación de ocho átomos de deuterio era mayor que 94%.
Ejemplo 7: Ensayos de Unión Experimentales
Descripción del ensayo de unión D? humano
El ensayo se realizó como una unión de competencia basado en SPA en un tampón de ensayo Tris 50 mM pH 7,4 que contiene NaCl 120 mM, KCl 5 mM, MgCl2 4 mM, CaCl21,5 mM, EDTA 1 mM.
3H-racloprida 1,5 nM (Perkin Elmer, NET 975) se mezcló con compuesto de ensayo antes de la adición de 20 microg de una preparación de membrana del receptor D2 humano homogeneizado y 0,25 mg de perlas de SPA (WGA RPNQ 0001, Amersham) en un volumen total de 90 microL. Las placas de ensayo se incubaron con agitación durante 60 minutos a temperatura ambiente y posteriormente se contaron en un contador de centelleo (TriLux, Wallac). La unión total, que comprendía aproximadamente el 15% del radioligando añadido, el se definió utilizando tampón de ensayo, mientras que la unión no específica se definió en presencia de haloperidol 10 microM. La unión no específica constituyó aproximadamente el 1 0 % de la unión total.
Los puntos de datos se expresaron en porcentaje de la unión específica de 3H-Racloprida y los valores CI50 (concentración que provoca una inhibición del 50 por ciento de la unión específica de 3H-racloprida) se determinaron mediante análisis de regresión no lineal utilizando una curva de pendiente sigmoidal variable apropiada. La constante de disociación (Ki) se calculó a partir de la ecuación de Cheng Prusoff (Ki= Cl50/(1+ (L/Kd)), en donde la concentración de radioligando libre L se aproxima a la concentración de 3H-racloprida añadida en el ensayo. Se determinó que la Kd de 3H-racloprida era 1,5 nM a partir de dos ensayos de saturación independientes, cada uno realizado con determinaciones por triplicado.
Descripción del ensayo de unión D1 humano
El ensayo se realizó como una unión de competencia basado en SPA en un tampón de ensayo Tris 50 mM pH 7,4 que contiene NaCl 120 mM, KCl 5 mM, MgCl24 mM, CaCl2 1,5 mM, EDTA 1 mM. Aproximadamente 1 nM de 3H-SCH23390 (Perkin Elmer, NET 930) se mezcló con compuesto de ensayo antes de la adición de 2,5 microg de una preparación de membrana del receptor D1 humano homogeneizado y 0,25 mg de perlas de SPA (WGA RPNQ 0001, Amersham) en un volumen total de 60 microL.
Las placas de ensayo se incubaron con agitación durante 60 minutos a temperatura ambiente antes de que las placas se centrifugaran y, posteriormente, se contaron en un contador de centelleo (TriLux, Wallac). La unión total, que comprendía aproximadamente 15% del radioligando añadido, se definió con el tampón de ensayo, mientras que la unión no específica se definió en presencia de haloperidol 10 microM.
Los puntos de datos se expresaron en porcentaje de la unión específica y los valores CI50 (concentración que provoca una inhibición del 50 por ciento de unión específica) se determinaron mediante análisis de regresión no lineal utilizando una curva de pendiente sigmoidal variable apropiada. La constante de disociación (Ki) se calculó a partir de la ecuación de Cheng Prusoff (Ki= CI 50/(1 + (L/Kd)), en donde la concentración de radioligando libre L se aproxima a la concentración de radioligando añadido en el ensayo.
Descripción de la unión de 5-HT2a humano
El experimento se llevó a cabo en Cerep Contract Laboratories (Ref. Cat. n° 471).
El Compuesto (I) también se sometió a ensayo en una instalación in vivo, demostrando efectos centrales del compuesto. Mediante la unión in vivo se evaluó la afinidad in vivo de los compuestos para receptores D2 y se observó la ocupación de 60% del objetivo. La ocupación de los receptores D2 está estrechamente vinculada a efectos antipsicóticos en modelos animales y en pacientes.
Descripción de la unión in vivo a receptores D7 en el cerebro de rata
La unión in vivo se llevó a cabo de acuerdo con Andersen et al (Eur J Pharmacol, (1987) 144:1-6; incorporada a esta memoria por referencia en su totalidad) con unas pocas modificaciones (Kapur S. et al, J Pharm Exp Ther, 2003, 305, 625-631; incorporada a esta memoria por referencia en su totalidad). En síntesis, 6 ratas (Wistar machos, 180 200 g) fueron tratadas con 20 mg/kg de compuesto de ensayo por vía subcutánea 30 minutos antes de recibir 9,4 micro Ci de [3H]-racloprida por vía intravenosa a través de la vena de la cola.
15 minutos después de la inyección del radioligando se sacrificaron los animales por dislocación cervical, el cerebro se retiró rápidamente y el cuerpo estriado y el cerebelo se diseccionaron y se homogeneizaron en 5 mL (cerebelo en 20 mL) de tampón helado (K3PO450 mM, pH 7,4). 1,0 mL del homogeneizado se filtró a través de filtros Whatman GF/C empapados con PEI al 0,1%. Esto se completó en el espacio de los 60 segundos posteriores a la decapitación. Los filtros se lavaron 2 veces con 5 mL de tampón helado y se contaron en un contador de centelleo. Se utilizó un grupo de animales tratados con vehículo para determinar unión total de [3H]-racloprida en el cuerpo estriado y la unión no específica en el cerebelo. El producto homogeneizado se midió en cuanto al contenido de proteínas mediante el ensayo de determinación de proteínas BCA (Smith P.K. et al (1985) Anal. Biochem., 150:6-85; incorporada a esta memoria por referencia en su totalidad).
Ejemplo 8: Investigación del metabolismo de 4-((1R,3S)-6-cloro-3-fenil-indan-1-il)-1,2,2-trimetil-piperazina (Compuesto (X)) y 4-((1R,3S)-6-cloro-3-fenil-indan-1-il)-1-metil-d3-2,2-dimetil-piperazina (Compuesto (I)) Hepatocitos de perro (perro macho Beagle) criopreservados (1 millón de células/mL en suspensión, 50 microL/pocillo) se pre-incubaron durante 15 minutos en una placa de 96 pocillos en un baño de agua a 37°C en DMEM de alto contenido en glucosa tamponado con HEPES 1M. La suspensión celular se añadió con 50 microL de compuestos de ensayo (concentración final 0,1 ó 1 microM de 4-((1R,3S)-6-cloro-3-fenil-indan-1-il)-1,2,2-trimetilpiperazina (Compuesto (X)) o 4-((1R,3S)-6-cloro-3-fenil-indan-1-il)-1-metil-d3-2,2-dimetil-piperazina (Compuesto (I )) y se incubaron adicionalmente durante 0, 15, 45, 75 y 120 minutos. La reacción se detuvo por adición de 100 microL de acetonitrilo a la suspensión celular, y las muestras se retiraron a continuación para el análisis LC-MS del metabolito desmetilo (Compuesto (XI)). Los datos se expresaron como área MS con relación a un patrón interno. Los resultados (Figura 5 y Figura 6) demuestran que la cantidad del metabolito desmetilo (Compuesto (XI)) producido en hepatocitos de perro criopreservados es más baja de la forma deuterada (Compuesto (I)) que del compuesto parental (Compuesto (X)), ambos a una concentración de 0,1 micro M (Figura 5) ya una concentración de 1 micro M (Figura 6).
Ejemplo 9: Ensayo farmacológico de Compuestos
4-((1R,3S)-6-cloro-3-fenil-indan-1-il)-1-d3-metil-2,2-dimetil-piperazina (Compuesto (I)):
4-((1R,3S)-6-cloro-3-fenil-indan-1-il)-1-d3-metil-2,2-dimetil-piperazina (Compuesto (I)) se sometió a ensayo en tres ensayos in vitro para la afinidad a dopamina D1, dopamina D2 y serotonina 5-HT2A.
Los experimentos se llevaron a cabo como en la sección de Ensayos de unión. Los resultados experimentales demostraron las siguientes afinidades para 4-((1R,3S)-6-cloro-3-fenil-indan-1-il)-1-metil-d3-2,2-dimetil-piperazina: D1: Media log Ki = 7,5 nM (pKi 0,88 /- 0,15)
D2 : Media log Ki = 34 nM (pKi 1,54 /- 0,11)
5HT2a: CI50 = 1,14 nM
Estas afinidades de unión indican que el Compuesto (I) tiene actividad biológica probablemente para ejercer un efecto antipsicótico.
Ensayo farmacológico de Compuesto (II) y Compuesto (IV)
Los experimentos se llevaron a cabo tal como se describe en la sección "Ensayos de unión". Los resultados experimentales para los dos compuestos se proporcionan a continuación.
El Compuesto (II) y el Compuesto (IV) se ensayaron en dos ensayos in vitro para la afinidad a dopamina D1 y a dopamina D2.
Compuesto (IV):
D1: Media log Ki = 26,1 nM (pKi 1,42 /-0,03)
D2 : Media log Ki = 26,7 nM (pKi 1,43 /- 0,04)
Compuesto (II):
D1: Media log Ki = 23,2 nM (pKi 1,37 /- 0,03)
D2 : Media log Ki = 26,5 nM (pKi 1,42 /- 0,03)
Estas afinidades de unión indican que Compuestos (II) y (IV) tienen actividad biológica probablemente para ejercer un efecto antipsicótico.
Los Compuestos (II) y (IV) también se ensayaron en una instalación in vivo que demuestra efectos centrales del compuesto. Mediante la unión in vivo, se evaluó la afinidad in vivo de los compuestos para los receptores D2 y se observó la ocupación de 70% (Compuesto (IV)) y 75% (Compuesto (II)) de la diana. La ocupación de los receptores D2 está estrechamente vinculada a efectos antipsicóticos en modelos animales y en pacientes.
Los Compuestos (I) - (VII) y (X) se sometieron a ensayo en un análisis yuxtapuesto en Cerep Contract Laboratories (Refs. de Cat. n° 44, 46 y 471). Los resultados de la unión al receptor se enumeran en la Tabla 4.
Tabla 4. Unión de los compuestos a D1, D2 y 5-HT2a.
unión alternativa de receptor D1 unión alternativa de receptor D2 5-HT2A humano (Ki) humano (Ki) humano(CI50)
(I) 0,10 nM 7,6 nM 0,37 nM; 1,14 nM* (II) 0,20 nM 6,8 nM 1,1 nM
(III) 0,36 nM 7,6 nM 1,1 nM
(IV) 0,05 nM 10 nM 0,25 nM
(V) 0,10 nM 4,8 nM 0,61 nM
(VI) 0,10 nM 3,7 nM 0,24 nM
(VII) 0,14 nM 5,2 nM 0,33 nM
(X) 0,22 nM 7 nM 0,79 nM
* Compuesto (I) se ensayó dos veces en este ensayo.
Ejemplo 10: Investigaciones del Metabolismo en microsomas de hígado humano (HLM) agrupados Microsomas de hígado humano agrupados (50 donantes, de Xenotech) se incubaron con 1 microM o 10 microM de compuesto a 37°C. La mezcla de incubación contenía Tris-HCl50 mM, KCl 154 mM, MgCh 5 mM y un sistema de regeneración de NADPH (NADP+ 1 mM, ácido isocítrico 5 mM, 1 unidad/mL de deshidrogenasa isocítrica, de Sigma-Aldrich). La concentración de proteínas era de 0,2 mg/mL y el volumen final era de 0,5 mL. Después de una pre incubación durante 10 minutos, la reacción se inició mediante la adición de Compuesto. Después de 0, 15, 30, 60, 90, 120 y 180 minutos, las reacciones se terminaron mediante la transferencia de la fracción subcelular a 0,5 mL de reactivo de parada que contenía patrón interno. Las incubaciones se llevaron a cabo por triplicado. Las muestras se centrifugaron a 4000 g (4°C, 15 min) y los sobrenadantes se analizaron mediante HPLC-MS/MS. Los datos se expresaron como área de MS en relación con un patrón interno.
Los resultados se muestran como la media de determinaciones por triplicado ± SD. La Figura 7 y la Figura 8 muestran que la cantidad del metabolito desmetilo producido en microsomas de hígado humano es menor de la forma deuterada (Compuesto (II) y Compuesto (IV)) que del compuesto no deuterado (Compuesto (X)), ambos a una concentración de 1 microM (Figura 7) y a una concentración de 10 microM (Figura 8). Los resultados para el Compuesto (III) se muestran en la Figura 9. Resultados para los compuestos (V) - (VII) se muestran en las Figs. 10 12, respectivamente. Los metabolitos desmetilo de los compuestos (II), (IV) y (X) son compuestos (XX) y (XI), respectivamente (véase la Figura 13).
Investigaciones utilizando CYP2C19 y CYP3A4 de hígado recombinantes humanas
Isoenzimas CYP2C19 y CYP3A4 de hígado recombinantes humanas (de BD biosciences) se incubaron con 1 microM o 10 microM de Compuesto (X), Compuesto (II) o Compuesto (IV) a 37°C. La mezcla de incubación contenía Tris-HCl 50 mM, KCl 154 mM, MgCh 5 mM y un sistema de regeneración de NADPH (NADP+1 mM, ácido isocítrico 5 mM, 1 unidad/mL deshidrogenasa isocítrica, de Sigma-Aldrich). La concentración de proteína era de 0,5 mg/mL y el volumen final era de 0,5 mL. Después de una pre-incubación durante 10 minutos, la reacción se inició añadiendo Compuesto (X), Compuesto (II) y/o Compuesto (IV). Después de 0, 15, 30, 60, 90, 120 y 180 minutos, las reacciones
se terminaron mediante la transferencia de la fracción subcelular a 0,5 mL de reactivo de parada que contiene patrón interno. Las incubaciones se llevaron a cabo por triplicado. Las muestras se centrifugaron a 4000 g (4°C, 15 minutos) y los sobrenadantes se analizaron mediante HPLC-MS/MS. Los datos se expresaron como área MS con relación a un patrón interno.
Los resultados (Figura 14 y Figura 15) demuestran que la cantidad del metabolito desmetilo producida tras la incubación con enzimas CYP2C19 de hígado humanas recombinantes es más baja de las formas deuteradas (Compuesto (II) y Compuesto (IV)) que a partir del compuesto no deuterado (Compuesto (X)), ambos a una concentración de 10 micro M (Figura 14, Compuesto (II)) y a una concentración de 1 micro M (Figura 15, Compuesto (IV)). Se obtuvieron resultados correspondientes para el Compuesto (II) a una concentración de 1 micro M y para el Compuesto (IV) a una concentración de 10 micro M.
Correspondientemente, la cantidad del metabolito desmetilo producido mediante incubación con enzimas CYP3A4 de hígado humanas recombinantes es más baja de las formas deuteradas (Compuesto (II) y (IV)) que a partir del compuesto no deuterado (Compuesto (X)), tanto a una concentración de 1 micro M como de 10 micro M.
Ejemplo 11: Farmacología del Compuesto (IV).
Hiperactividad inducida por PCP
El Compuesto (IV) invierte de forma dependiente de la dosis la hiperactividad inducida por PCP en ratones, indicativo de la eficacia antipsicótica (Figura 16). El Compuesto (IV) tartrato se administró por vía subcutánea (s.c.) 30 minutos antes del ensayo. Hidrocloruro de PCP (2,3 mg/kg) se administró s.c. justo antes del ensayo. La actividad locomotriz se midió durante 60 minutos como número de roturas de haz (recuentos). De ocho a 16 ratones machos se utilizaron en cada uno de los grupos. ## Indica P <0,01 frente a Vehículo-PCP (Análisis de varianza de una vía [ANOVA] seguido del ensayo de Bonferroni post hoc). PCP son los receptores de NMDA de bloqueo y como tal se utiliza para modelar el estado hipo-glutamatérgico relacionados con la esquizofrenia. PCP produce efectos en el comportamiento de los animales que recuerdan a los síntomas positivos, negativos y cognitivos de pacientes con esquizofrenia (Jentsch, J.D. y Roth, R.H. Neuropsychopharmacology 1999; 20: 201-225). La hiperactividad inducida por PCP se utiliza comúnmente como un ensayo para la evaluación de compuestos antipsicóticos (Jackson, D.M. et al., Pharmacol Biochem Behav. 1994; 48:465-471).
Catalepsia
Se piensa que la catalepsia refleja la supresión inducida por fármacos de la capacidad de iniciar una respuesta de comportamiento. La prueba de catalepsia en ratas es una prueba de detección preclínica común y ampliamente utilizada para la responsabilidad EPS de fármacos potencialmente antipsicóticos. Aunque la catalepsia se evalúa generalmente después de la administración aguda de fármacos, la prueba ha demostrado ser un predictor fiable de la propensión de un fármaco antipsicótico utilizado para inducir EPS (es decir, pseudo-parkinsonismo, distonía) en seres humanos (Elliott, P.J. et al, J. Neural Transm. Park. Dis. Dement. Sect. 1990; 2:79-89; incorporada a esta memoria por referencia en su totalidad).
El Compuesto (IV) inducía de forma dependiente de la dosis la catalepsia en ratas sugestivas de responsabilidad EPS. La dosis efectiva mínima que induce catalepsia era de 10 mg/kg (Figura 17). El compuesto (IV) tartrato se administró s.c. 30 minutos antes de la prueba. Ocho ratas Sprague Dawley macho se utilizaron en cada uno de los grupos. # Indica P <0,05 ##, indica P <0,01 frente a vehículo (ANOVA de una vía, seguido por el test de Bonferroni post hoc). Esta dosis es 100 veces mayor que la dosis que indica actividad antipsicótica (Figura 16).
Ejemplo 12: Estudios Farmacocinéticos Humanos.
La farmacocinética de Compuesto (IV) y Compuesto (X) se compararon en un estudio de dosis oral múltiple en hombres jóvenes sanos. Los participantes en el estudio recibieron dosis diarias de 3 mg de Compuesto (IV) y 3 mg de Compuesto (X) durante 18 días y se recogieron muestras de sangre durante 24 horas (un intervalo de dosificación) después de la última dosis para medir la exposición de ambos compuestos y sus metabolitos desmetilados, Compuesto (XX) y Compuesto (XI), respectivamente.
Para todos los participantes en el estudio, el área bajo la curva de concentración de plasma y tiempo para el intervalo de dosificación (AUC 0-24) para el Compuesto (IV) era mayor que para el Compuesto (X), la media de 104 h*ng/mL frente a 98 h*ng/mL. Se observó un desplazamiento consistente en la dirección opuesta para los metabolitos desmetilados con una AUC 0-24 media de 117 h*ng/mL y 120 h*ng/mL para el Compuesto (XX) y el Compuesto (XI), respectivamente.
Ejemplo 13: Síntesis enantioselectiva catalítica de compuestos intermedios de cetona.
Este ejemplo describe la síntesis de (S)-6-cloro-3-fenil(cfe)-indan-1-ona, Compuesto (XV) y (S)-6-cloro-3-fenil-indan-1-ona, Compuesto (XVIII).
(S)-6-cloro-3-fenil(d5)-indan-1-ona, Compuesto (XV), ha demostrado ser un bloque de construcción valioso en la síntesis de variantes deuteradas de Compuesto (X) en el que el grupo fenilo libre está deuterado.
Parte Experimental General
A menos que se establezca lo contrario, todas las reacciones se llevaron a cabo bajo nitrógeno. Las reacciones se controlaron mediante análisis de cromatografía en capa fina (TLC) y LC-MS. Todos los reactivos se adquirieron y se utilizaron sin purificación adicional. Las manchas se visualizaron por exposición a luz ultravioleta (UV) (254 nm), o mediante tinción con una disolución al 5% de ácido fosfomolibdénico (PMA) en etanol o permanganato potásico acuoso de carácter básico (KMnO4) y después calentamiento. La cromatografía en columna se realizó utilizando gel de sílice Merck C60 (40-63 |jm, malla 230-240). Los espectros de RMN se registraron a 500 ó 600 MHz (1H RMN), y se calibraron para el pico de disolvente residual. Las siguientes abreviaturas se utilizan para los datos de RMN: s, singlete; d, doblete; t, triplete; m, multiplete. Las constantes de acoplamiento se redondean a 0,5 Hz más cercano. El exceso enantiomérico se determinó por HPLC quiral.
Método LC-MS:
Columna Acquity UPLC BEH C18 1,7 jm; 2,1 x 50 mm que opera a 60°C con un caudal de 1,2 ml/min de un gradiente binario formado por agua ácido fórmico al 0,1% (A) y acetonitrilo 5% de agua ácido fórmico al 0,1% (B).
Método HPLC quiral:
Columna Phenomenex Lux 5|j Cellulose-2; 250 x 4,6 mm que opera a 30°C con un caudal de 0,6 mL/min de nhexano:isopropanol:dietilamina, 90:10:0,1.
Síntesis de (S)-6-cloro-3-fenil(d5)indan-1-ona (Compuesto (XV)) (Esquema 14)
Esquema 14. Síntesis de Compuesto (XV)

Trifluorometanosulfonato de 1-fenil(ds)vinilo (XII):
A una disolución de acetofenona-d5 (1,56 g, 12,5 mmol) en CH2O 2 (25,0 mL) se añadió anhídrido trifluorometanosulfónico (2,52 mL, 15,0 mmol) a temperatura ambiente. A continuación, se añadió gota a gota N,N-diisopropiletilamina (3,04 mL, 17,5 mmol) al tiempo que la mezcla de reacción se enfriaba en un baño helado. Se
dejó que la mezcla de reacción se calentara a temperatura ambiente, y se agitó durante 1,5 h. Se añadió anhídrido trifluorometanosulfónico (0,63 mL, 3,74 mmol), seguido de A/,W-diisopropiletilamina (1,09 mL, 6,24 mmol). La mezcla de reacción se agitó durante 2 horas a temperatura ambiente. Se añadió tolueno (25 mL) y gel de sílice (5 g). La mezcla se concentró en vacío, y la suspensión resultante se filtró a través de una almohadilla de celite. La torta de filtración se lavó con tolueno (l0 mL), y el filtrado se evaporó a sequedad en vacío para proporcionar el Compuesto (XII) bruto (3,11 g, 82%, pureza (RMN): aprox. 85%) en forma de un aceite oscuro, que se utilizó sin purificación adicional.
1H RMN (600 MHz, CDCls) Sh 5,38 (d, 1H, J = 4,0 Hz), 5,62 (d, 1H, J = 4,0 Hz).
5-cloro-2-(1-fenil(d5)-vinil)benzaldehído (XIV) (Takagi, J.; Takahashi, K.; Ishiyama, T.; Miyaura, N. J. Am. Chem. Soc.
2002, 124, 8001-8006; Simeone, J.P.; Sowa, J.R. Jr. Tetrahedron 2007, 63, 12646-12654).
A una disolución de Compuesto (XII) (3,11 g, 10,3 mmol, pureza (RMN): aprox 85%.) en tolueno se añadió trifenilfosfina (108 mg, 0,685 mmol), bis(pinacolato)diboro (2,61 g, 10,3 mmol), cloruro de bis(trifenilfosfina)paladio(M) (240 mg, 0,342 mmol) y fenolato de potasio (1,92 g, 14,6 mmol). La mezcla de reacción se agitó a 50°C durante 4 horas. Esto produjo el Compuesto (XIII) en la mezcla, que no fue aislado. La mezcla se enfrió a temperatura ambiente y se añadieron etanol (10 mL) y agua (5 mL), seguido de tetrakis(trifenilfosfina)paladio(0) (495 mg, 0,428 mmol), carbonato de potasio (4,73 g, 34,2 mmol) y 2-bromo-5-clorobenzaldehído (1,88 g, 8,56 mmol). La mezcla de reacción se agitó a 80°C durante 16 horas. La mezcla se enfrió a temperatura ambiente, y se repartió entre agua (50 mL) y tolueno (50 mL).
La fase orgánica se separó y se lavó con agua (50 mL) dos veces, y salmuera. La fase orgánica se secó sobre MgSO4 , se filtró y se evaporó a sequedad en vacío. El residuo se sometió a purificación mediante cromatografía en columna, eluyendo con una mezcla 80:1 de n-heptano:EtOAc para proporcionar el Compuesto (XIV) (1,66 g, 74%) en forma de un aceite naranja.
1H RMN (600 MHz, CDCla) Sh 5,28 (d, 1H, J = 0,5 Hz), 6,00 (d, 1H, J = 0,5 Hz), 7,30 (d, 1H, J = 8,0 Hz), 7,56 (dd, 1H; J = 2,5, 8,0 Hz), 7,96 (d, 1H, J = 2,5 Hz); 13C RMN (150 MHz, CDCls) So 118,7, 126,6 (t, J = 24,0 Hz), 127,5, 128,2 (t, J = 24,0 Hz), 128,4 (t, J = 24,0 Hz), 132,5, 133,7, 134,7, 135,7, 140,3, 143,9, 144,8, 190,8; LC-MS (APPI): m/e calc. para C15H7D5CO [M H]+248,1, encontrado 248,1.
(S)-6-cloro-3-fenil(d5)indan-1-ona (XV) (Kundu, K.; McCullagh, J.V.; Morehead, A.T. Jr. J. Am. Chem. Soc. 2005, 127, 16042-16043).
Se burbujeó hidrógeno a través de una disolución de tetrafluoroborato de ((R)-2,2'-bis (difenilfosfino)-1,1'-binaftilo)(norbornadieno)rodio(I) barrida con N2 (37 mg, 0,0404 mmol) en acetona (7,5 mL) durante 10 min a temperatura ambiente, durante lo cual el color de la disolución cambió de naranja a rojo parduzco. El matraz que contiene la disolución fue barrido brevemente a continuación con gas N2. A continuación, una disolución de (XIV) (526 mg, 2,02 mmol, pureza (LC-MS): 95%) en acetona (7,5 mL) se añadió a temperatura ambiente. La mezcla de reacción se agitó durante 24 horas a temperatura ambiente. La mezcla de reacción se mezcló con gel de sílice y se evaporó a sequedad en vacío. El material obtenido se cargó en una columna de gel de sílice y el producto se eluyó con una mezcla 10:1 de n-heptano:EtOAc para obtener el Compuesto (XV) (495 mg, 96%, 96,0% ee) en forma de un sólido.1
1H RMN (500 MHz, CDCls) Sh2,72 (dd, 1H, J = 4,0, 19,5 Hz), 3,27 (dd, 1H, J = 8,0, 19,5 Hz), 4,55 (dd, 1H, J = 4,0, 8,0 Hz), 7,21 (d, 1H; J = 8,0 Hz), 7,52 (dd, 1H, J = 2,0, 8,0 Hz), 7,77 (d, 1H, J = 2,0 Hz); 13 C RMN (125 MHz, CDCls) Sc 44,0, 47,2, 123,2, 126,8 (t, J = 24,0 Hz), 127,3 (t, J = 24,0 Hz), 128,7 (t, J = 24,0 Hz), 134.4, 135.1, 138.2, 142.9, 156,0, 206,4; LC-MS (APPI): m/e calc. para C15H7D5 ClO [M H]+248,1, encontrado 247,6.
Síntesis de (S)-6-cloro-3-fenil-indan-1-ona (XVIII) (Esquema 15)
Esquema 15. Síntesis de Compuesto (XVIII))

(E)-1-(5-cloro-2-hidroxifenil)-3-fenilprop-2-en-1-ona (XVI):
A una disolución helada de hidróxido de sodio (2,34 g, 58,6 mmol) en agua (17,0 mL) se añadió benzaldehído (0.746 g, 7.03 mmol) y luego una disolución de 5-cloro-2-hidroxiacetofenona (1,00 g, 5,86 mmol) en metanol (17,0 mL). Se dejó que la mezcla de reacción se calentara a temperatura ambiente, y se agitó durante 24 horas. La mayor parte del disolvente orgánico se separó por evaporación en vacío. El residuo acuoso se extrajo con EtOAc (3 x 30 mL). Los extractos reunidos se lavaron con agua (50 mL) y salmuera (50 mL), se secaron sobre MgSO4, se filtraron y se evaporaron a sequedad en vacío. El residuo se disolvió en un volumen mínimo de CH2Cl2, y se añadido n-pentano que resultó en la precipitación. La suspensión obtenida se filtró y el precipitado se lavó con un poco de pentano frío, y se secó en vacío para proporcionar el Compuesto (XVI) (695 mg, 46%) en forma de un sólido de color naranja. 1H RMN (500 MHz, CDCh) Sh 6,22 (d, 1H, J = 9,0 Hz), 6,80 (dd, 1H, J = 3,0, 9,0 Hz), 7,33 (t, 1H, J = 7,5 Hz), 7,38 7,42 (m, 4H), 7,60 (d, 2H, J = 7,5 Hz); 8,63 (d, 1H, J = 16,0 Hz); 13C RMN (125 MHz, CDCla) So 110,6, 125,2, 127,8, 128.1, 128,8, 128,9, 129,4, 129,6, 133,0, 136,4, 137,1, 174,5, 188,2.
Éster 4-cloro-2-((E)-(3-fenil-acriloil))-fenílico del ácido trifluorometanosulfónico (XVII):
A una disolución de Compuesto (XVI) (517 mg, 2,00 mmol) en CH2Cl2 (10,0 mL) se añadió A/,W-diisopropiletilamina (697 |jL, 4,00 mmol). Se añadió anhídrido trifluorometanosulfónico (437 j L, 2,60 mmol) gota a gota a 0°C. La mezcla de reacción se agitó durante 45 min a 0°C. Se añadió NH4Cl acuoso sat. (5 mL) y agua (10 mL), y la mezcla se agitó durante 5 minutos. La fase orgánica se separó, y la fase acuosa se extrajo con CH2O 2 (10 mL). Los extractos reunidos se secaron sobre MgSO4, se filtraron y se evaporaron a sequedad en vacío. El residuo se purificó mediante cromatografía en columna eluyendo con 4:1 de n-heptano:EtOAc para dar (XVII) (757 mg, 97%) en forma de un aceite.
1H RMN (500 MHz, CDCh) Sh 7,16 (d, 1H, J = 16,0 Hz), 7,34 (d, 1H, J = 9,0 Hz), 7,40-7,47 (m, 3H), 7,57 (dd, 1H, J = 2,5, 9,0 Hz), 7,60-7,62 (m, 2H), 7,69 (d, 1H, 16,0 Hz), 7,72 (d, 1H, J = 2,5 Hz); 13C RMN (125 MHz, CDCls) So 124,1, 124.2, 129,0, 129,2, 130,7, 131,5, 132,8, 134,1, 134,6, 145,2, 147,8, 188,4.
(S)-6-cloro-3-fenil-indan-1-ona (XVIII) (Minatti, A.; Zheng, X.; Buchwald, S.L. J. Org. Chem. 2007, 72, 9253-9258; incorporada a esta memoria por referencia en su totalidad).
A una disolución del Compuesto (XVII) (195 mg, 0,500 mmol) en DMF (2,0 mL) se añadió una esponja de protones (214 mg, 1,00 mmol), acetato de paladio (6 mg, 0,025 mmol) y (E)-3,5-XylMeoBIPHEP (35 mg, 0,05 mmol) a t.a. La mezcla de reacción se agitó a 85°C durante 45 h. La mezcla se enfrió a t.a., y se diluyó con TBME (15 mL). La mezcla se lavó tres veces con agua (3 x 20 mL), y la fase orgánica se secó sobre MgSO4 , se filtró y se evaporó a sequedad en vacío. El residuo se sometió a cromatografía en columna eluyendo con 10:1 de n-heptano:EtOAc para producir el Compuesto (XVII) (94 mg, 77%, 64,0% ee).
1H RMN (600 MHz, CDCls) Sh 2,71 (dd, 1H, J = 4,0, 19,5 Hz), 3,25 (dd, 1H, J = 8,0, 19,5 Hz), 4,54 (dd, 1H, J = 4,0, 8,0 Hz), 7,10 (d, 2H, J = 7,0 Hz), 7,20 (d, 1H, J = 8,0 Hz), 7,25 (t, 1H, J = 7,5 Hz), 7,31 (t, 2H, J = 7,5 Hz), 7,50 (dd, 1H, J = 2,0, 8,0 Hz), 7,75 (d, 2H, J = 2,0 Hz); 13C RMN (150 MHz, CDCls) So 44,1, 47,2, 123,3, 127,3, 127,6, 128,3, 129,1, 134,4, 135,2, 138,3, 143,1, 156,1, 204,5.
Enriquecimiento en enantiómeros del Compuesto (XVIII) mediante Reprecipitación
El Compuesto (XVII) (940 mg, 3,87 mmol, 96% ee) se disolvió en un volumen mínimo de etanol en ebullición (99% v/v). La disolución resultante se dejó enfriar lentamente a t.a. colocando el matraz de vidrio que contiene la disolución en el aire. Se formó un precipitado que se filtró de la disolución para producir el Compuesto (XVIII) (700 mg, 99,9% ee, 74%). Una segunda cosecha del Compuesto (XVIII) podría obtenerse enfriando el filtrado en el congelador (-8°C) para producir el Compuesto (XVIII) (80 mg, 98,6% ee, 9%).
Los datos analíticos (RMN y LC-MS) para el compuesto (XVIII) eran los mismos que los descritos anteriormente. Ejemplo 14: Producción a gran escala del Compuesto (IV)
Se desarrolló el siguiente procedimiento para la producción a gran escala de la sal tartrato del Compuesto (IV) Esquema 16.: Síntesis de maleato de rac-trans-1-(6-cloro-3-fenil(d5)-indan-1-il)-3,3-dimetil-piperazina

Procedimiento:
1. ) 2,01 kg (16,9 mol) de cloruro de tionilo y 7,2 kg de tetrahidrofurano se mezclan y la reacción se enfría a 10-15°C
2. ) se añade lentamente una disolución de 2,76 kg (11,1 mol) de (XXII) en 7,2 kg de THF y después de la compleción se añaden 5,9 kg de tetrahidrofurano
3. ) la reacción se agita a 15°C durante aproximadamente 90 horas
4. ) 16,7 kg de agua se enfrían a 11°C y se añaden lentamente a la reacción, después se añaden lentamente 7,8 kg de hidróxido de sodio acuoso al 27,7%, seguido de 10 kg de acetato de etilo
5. ) la mezcla se agita durante 20-40 minutos
6. ) las fases se separan y la fase orgánica se reduce a un volumen de aproximadamente 6L
7. ) se añaden 16 kg de metilisobutilcetona y el volumen se reduce a aproximadamente 8 L
8. ) se añaden 1,58 kg (11,4 mol) de carbonato de potasio, 1,69 kg (14,8 mol) de 2,2-dimetilpiperazina y 13,6 kg de metil-isobutil-cetona
9. ) la reacción se agita durante 35 horas a 90-95°C
10. ) después de enfriar a temperatura ambiente, se añaden 11 kg de agua y la mezcla se agita durante 30 -60 minutos
11. ) se separan las fases. se añaden 13,7 kg de agua a la fase orgánica y la mezcla se agita lentamente durante 30 - 60 minutos
12. ) se separan las fases y la fase orgánica se filtra en blanco
13. ) se añaden 5 kg de metil-isobutil-cetona, 7,8 kg de agua y 5,9 kg de cloruro de hidrógeno acuoso al 36% y la mezcla se agita a 50°C durante 30 - 60 minutos
14. ) se separan las fases. se añaden 8 kg de metil-isobutil-cetona a la fase acuosa y la mezcla se enfría a 10-15°C
15. ) se añade lentamente una mezcla de 3,5 kg de metil-isobutil-cetona y 7,8 kg de amoniaco acuoso al 25% a la mezcla y la reacción se agita a 20-25°C durante 60 - 90 minutos
16. ) se separan las fases y la fase orgánica se lava con 10,5 kg de agua
17. ) la fase orgánica se reduce a 8 L
18. ) se añaden 1,19 kg (10,25 mol) de ácido maleico y 9 kg de metil-isobutil-cetona y la reacción se calienta después a 75 - 80°C
19. ) después de enfriar a 10-15°C, el precipitado se separa por filtración y se lava con 10 kg de metilisobutil-cetona
20. ) el sólido se secó en un horno de vacío a 50°C durante aproximadamente 20 horas para dar 3,47 kg (68% de rendimiento) de maleato (XXV).
Datos de RMN para el maleato (XXV):
1H-RMN (DMSO-d6, 600 MHz, ppm): 8,60 (s ancho, 2H, ácido maleico), 7,39 (d, 1H, J = 1,6 Hz), 7,29 (dd, 1H, J = 8,0 Hz, J = 1,8 Hz), 6,98 (d, 1H, J = 8,2 Hz), 6,04 (s, 2H, ácido maleico), 4,56 (dd, 1H, J = 8,1 Hz, J = 4,9 Hz), 4,48 (dd, 1H, J = 8,6 Hz, J = 6,2 Hz), 3,37 (s ancho, 1H), 3,16 (s ancho, 2H), 2,77 (s ancho, 1H), 2,58-2,50 (m, 3H), 2,31 (d, 1H, J = 12,0 Hz), 2,12 (ddd, 1H, J = 13,8 Hz, J = 8,0 Hz, J = 6,0 Hz), 1,33 (s, 3H), 1,31 (s, 3H).
Esquema 17. Síntesis de succinato de rac-trans-1-(6-cloro-3-fenil(ds)-indan-1-il)-1(d3),2,2-trimetil-piperazina

Procedimiento:
1. ) 1,1 kg (2,38 mol) de maleato (XXV), 11 L de metil-terc.-butil-éter, 1.8 L de agua y 1 L de amoníaco acuoso al 25% se agitan durante 1 - 2 horas
2. ) las fases se separan y la fase orgánica se lava dos veces con 2 L de agua
3. ) una disolución de 254 g (3,85 mol) de hidróxido de potasio acuoso al 85% y 1,5 L de agua se añade a la fase orgánica, seguido de la adición de 450 g (3,11 mol) de yoduro de metilo (d3) (CD3 I)
4. ) la reacción se agita a 20-25°C durante 16 - 24 horas
5. ) se añaden 2 L de agua y el subproducto precipitante se separa por filtración
6. ) 0,8 L de agua y 0,2 L de amoniaco acuoso al 25% se añaden al filtrado y la mezcla se agita durante 20 -40 minutos
7. ) las fases se separan y la fase orgánica se lava con 2 L de agua
8. ) las fases se separan y se añaden 38 g (0,48 mol) de cloruro de acetilo a la fase orgánica, que se agita durante 20 - 40 minutos
9. ) se añaden 0,8 L de agua y 0,2 L de amoniaco acuoso al 25% y la mezcla se agita durante 20 - 40 minutos
10. ) las fases se separan y la fase orgánica se lava con 2 L de agua
11. ) la fase orgánica se reduce a sequedad y se añade acetona
12. ) se añaden 225 g (1,91 mol) de ácido succínico y acetona de modo que el volumen de reacción es de aproximadamente 6 - 6,5 L
13. ) la reacción se calienta a reflujo y después se enfría a 5-10°C
14. ) el precipitado se separa por filtración y se lava con 1 L de acetona
15. ) el sólido se seca en un horno de vacío a 50°C durante más de 16 horas para dar 630 g (55% de rendimiento) de succinato (XXVII)
Datos de RMN para succinato (XXVII):
1H-RMN (dmso-d6, 600 MHz, ppm): 7,33 (d, 1H, J = 1,9 Hz), 7,26 (dd, 1H, J = 8,1 Hz, J = 2,0 Hz), 6,95 (d, 1H, J = 8,0 Hz), 4,46 (dd, 1H, J = 8,0 Hz, J = 5,1 Hz), 4,46 (dd, 1H, J = 8,8 Hz, J = 5,8 Hz), 2,65-2,56 (m, 4H), 2,46-2,41 (m, 1H), 2,37 (s, 4H, ácido succínico), 2,31 (s ancho, 1H), 2,13 (d, 1H, J = 10,9 Hz), 2,02 (ddd, 1H, J = 13,7 Hz, J = 7,8 Hz, J = 6,0 Hz), 1,04 (s, 3H), 1,02 (s, 3H).
Esquema 18. Síntesis de L(+)-tartrato de 4-((1R,3S)-6-cloro-3-fenil(ds)-indan-1-il)-1(d3),2,2-trimetil-piperazina

Procedimiento:
1. ) 1,0 kg (2,08 mol) de succinato (XXVII), 8 L de acetato de etilo, 2 L de agua y 1L de amoníaco acuoso al 25% se agitan durante 0,5 -1 horas
2. ) las fases se separan y la fase orgánica se lava con 2 L de agua
3. ) la fase orgánica se reduce a aproximadamente 1,5 L
4. ) se añaden 10 L de acetona y 312 g (2,08 mol) de ácido L(+)-tartárico
5. ) la reacción se calienta a reflujo y después se enfría a 5-10°C
6. ) el precipitado se separa por filtración, se lava con 1,2 L de acetona
7. ) la torta de filtración húmeda se recarga y se añaden 11 L de etanol absoluto
8. ) la reacción se calienta a reflujo y después se enfría a 5-10°C
9. ) el precipitado se separa por filtración y se lava con 1,2 L de etanol absoluto
10. ) el sólido se seca en un horno de vacío a 50°C durante más de 16 horas para dar 395 g (rendimiento 37%) de L(+)-tartrato (IV)
Datos de RMN para L(+)-tartrato (IV):
1H-RMN (DMSO-d6, 600 MHz, ppm): 7,36 (s, 1H), 7,27 (d, 1H, J = 8,2 Hz), 6,96 (d, 1H, J = 8,2 Hz), 4,50 (dd, 1H , J = 8,0 Hz, J = 5,1 Hz), 4,45 (dd, 1H, J = 8,5 Hz, J = 5,8 Hz), 4,07 (s, 2H, tartrato), 2,95 (s ancho, 1H), 2,77 (s ancho, 1H ), 2,61-2,50 (m, 3H), 2,31 (d, 1H, J = 11,7 Hz), 2,04 (ddd, 1H, J = 13,7 Hz, J = 7,8 Hz, J = 6,0 Hz), 1,21 (s, 3H), 1,18 (s, 3H).
Ejemplo 15: Caracterización físico-química de sales del Compuesto (IV)
pKa y log P/D del Compuesto (IV)
pKa se determinó mediante titulación potenciométrica de la base a una fuerza iónica de 0,16 utilizando MeOH como co-disolvente. Tres series de tres titulaciones repetidas en la misma disolución de la muestra se llevaron a cabo de una manera convencional de bajo a alto pH y se creó una curva de diferencia a partir de cada una de estas titulaciones por sustracción en blanco. El valor de pKa aparente en cada una de las relaciones de MeOH:agua se calcula a partir de las curvas de diferencia, y el valor pKa se determina por extrapolación al contenido de MeOH cero. El valor de pKa menor es demasiado bajo para ser determinado por titulación potenciométrica ya que se encontró que los datos sólo eran fiables hasta ~ 3. Se determinó que el pKa alto era de 8,9 0,1.
El pKa bajo se determinó por detección de espectroscopia de absorción con sonda sumergida durante la titulación de la base a una fuerza iónica de 0,16 utilizando MeOH como co-disolvente. El cambio en los espectros de absorción como una función de ionización se utiliza para calcular el valor pKa. Se realizaron dos series de tres titulaciones repetidas en la misma disolución de la muestra de bajo a alto pH, con una matriz de fotodiodos como detección adicional. El valor pKa aparente en cada una de las relaciones MeOH:agua se calcula mediante el análisis de factores de selección en el cambio en los espectros de absorción, y el valor pKa se determina por extrapolación al contenido de MeOH cero.
Resultado: Se determinó que el pKa más bajo era 2,5+0,1.
El perfil de logD se determinó por titulación a 27°C y a una fuerza iónica de aprox. 0.16. Se realizó una serie de tres titulaciones repetidas en la misma muestra en disolución, de bajo a alto pH. La primera titulación se realizó con una pequeña cantidad de n-octanol presente en la disolución, la segunda y la tercera con cantidades crecientes.
Se creó una curva de diferencia a partir de cada una de estas titulaciones por sustracción en blanco y, a partir de estas curvas de diferencia, se calcularon los valores de pKa aparente (p0Ka). A partir del cambio en los valores de pKa aparente (ApKa) con la relación de n-octanol:agua combinada con el valor pKa real, se calculó el valor LogP y se derivó el perfil LogD. Se determinaron los siguientes valores: Log P = 5.4+ 0,4 y Log D7 ,4 = 3.9+0,4.
Punto de fusión determinado mediante DSC
El punto de fusión de la sal (R,R)-hidrógeno-tartrato del Compuesto (IV) se determinó utilizando calorimetría diferencial de barrido (DSC), utilizando un instrumento TA DSC Q1000 calentando la muestra 5°/minuto. La muestra se colocó en una bandeja cubierta con un agujero de alfiler perforado.
La fusión se caracteriza por la aparición y la temperatura pico de la endotermia de fusión, y la entalpía de fusión se calcula a partir del área del pico. Basado en el termograma de DSC se encontró una temperatura de inicio de 187,4°C y un máximo de pico a 189,4°C. La entalpía de fusión era de 96 J/g, correspondiente a 49 kJ/mol, sin embargo, el termograma es indicativo de que la fusión se produce bajo descomposición, lo que significa que la entalpía probablemente contiene energía distinta de la fusión.
Solubilidad
La solubilidad de la sal (R,R)-hidrógeno-tartrato del Compuesto (IV) se midió en disoluciones acuosas y en ciclodextrinas con los siguientes resultados (Tabla 5):
Tabla 5. Solubilidad de la sal (R,R)-hidrógeno-tartrato del Compuesto (IV).
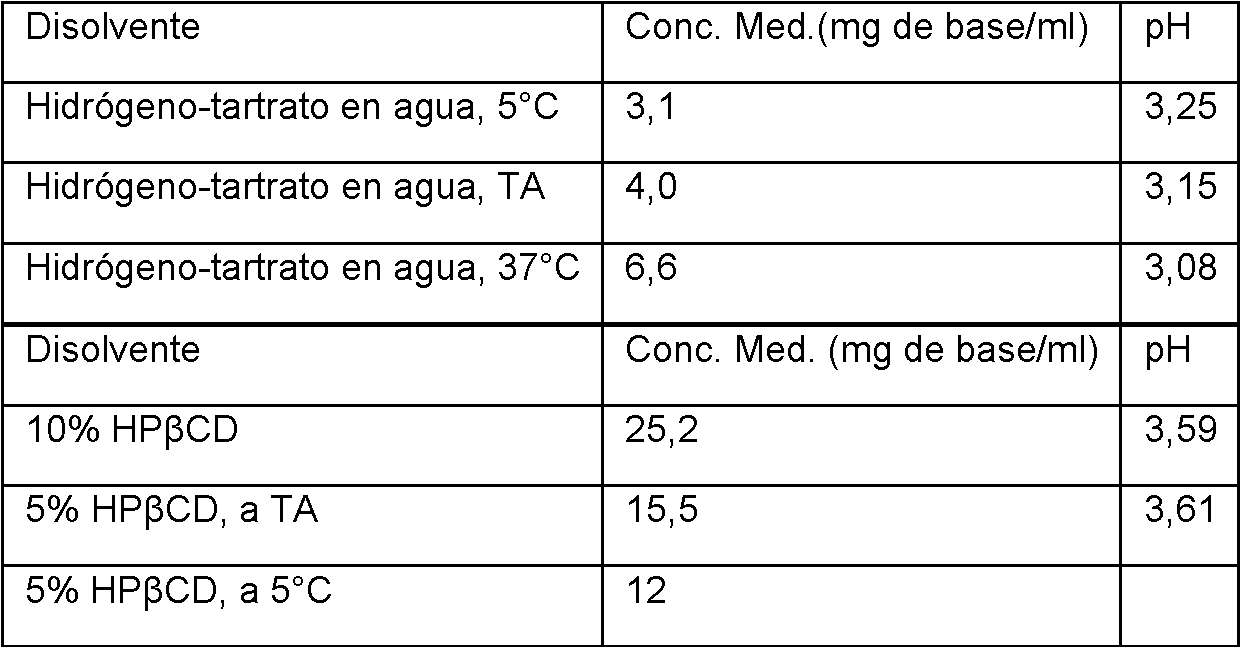
Polimorfismo
Se ha aislado una forma de cristal exenta de disolvente del tartrato. El XRPD de esta forma se muestra en la Figura 18, y designado aquí como "polimorfo A".
Sales de Compuesto (IV)
Se prepararon cuatro sales por precipitación del Compuesto (IV) a partir de EtOH al 99%.
Los datos analíticos se dan en la siguiente tabla (Tabla 6).
Tabla 6. Datos para las sales del Compuesto (IV)

Claims (22)
1. Un compuesto de fórmula Y:

en donde,
R1 - R10 son independientemente hidrógeno o deuterio, en donde R6-R10 son cada uno deuterio, o una sal por adición de ácidos farmacéuticamente aceptable del mismo, para uso como un medicamento.
2. El compuesto de la reivindicación 1 para uso como un medicamento, en donde R3-R5son cada uno hidrógeno.
3. El compuesto de la reivindicación 1 para uso como un medicamento, en donde R3-R5son cada uno deuterio.
4. El compuesto de acuerdo con cualquiera de las reivindicaciones 1 y 2 para uso como un medicamento, en donde el compuesto es

5. El compuesto de acuerdo con cualquiera de las reivindicaciones 1 y 2 para uso como un medicamento, en donde el compuesto es

6. El compuesto de acuerdo con cualquiera de las reivindicaciones 1 y 3 para uso como un medicamento, en donde el compuesto es

7. El compuesto de acuerdo con cualquiera de las reivindicaciones 1 y 3 para uso como un medicamento, en donde el compuesto es
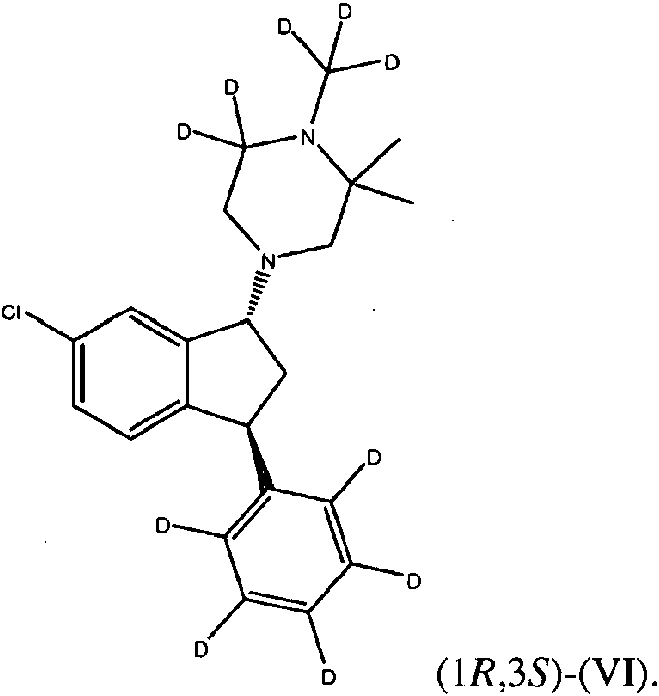
8. El compuesto de acuerdo con cualquiera de las reivindicaciones 1 y 6 para uso como un medicamento, en donde el compuesto es la sal tartrato de hidrógeno de

9. El compuesto de la reivindicación 8 para uso como un medicamento, en donde el compuesto existe en forma polimórfica que tiene un patrón de difracción de XRPD como se indica en la Figura 18.
10. El compuesto de acuerdo con cualquiera de las reivindicaciones 1 a 9 para uso como un medicamento, en donde el compuesto se usa como un medicamento en combinación con uno o más agentes neurolépticos.1
11. El compuesto de acuerdo con cualquiera de las reivindicaciones 1 a 10 para uso como un medicamento, en donde el compuesto se usa como un medicamento en combinación con uno o más agentes neurolépticos, en donde
el uno o más agentes neurolépticos se selecciona del grupo que consiste en sertindol, olanzapina. risperidona, quetiapina, aripiprazol, haloperidol, clozapina, ziprasidona y osanetant.
12. Un compuesto de fórmula Y:

en donde,
R1 - R10son independientemente hidrógeno o deuterio, en donde R6-R10son cada uno deuterio, o una sal por adición de ácidos farmacéuticamente aceptable del mismo, para uso en el tratamiento de la esquizofrenia.
13. El compuesto de la reivindicación 12 para uso en el tratamiento de la esquizofrenia, en donde R3-R5 son cada uno hidrógeno.
14. El compuesto de la reivindicación 12 para uso en el tratamiento de la esquizofrenia, en donde R3-R5 son cada uno deuterio.
15. El compuesto de acuerdo con cualquiera de las reivindicaciones 12 y 13 para uso en el tratamiento de la esquizofrenia, en donde el compuesto es

16. El compuesto de acuerdo con cualquiera de las reivindicaciones 12 y 13 para uso en el tratamiento de la esquizofrenia, en donde el compuesto es

17. El compuesto de acuerdo con cualquiera de las reivindicaciones 12 y 14 para uso en el tratamiento de la esquizofrenia, en donde el compuesto es

18. El compuesto de acuerdo con cualquiera de las reivindicaciones 12 y 14 para uso en el tratamiento de la esquizofrenia, en donde el compuesto es

19. El compuesto de acuerdo con cualquiera de las reivindicaciones 12 y 17 para uso en el tratamiento de la esquizofrenia, en donde el compuesto es la sal tartrato de hidrógeno de

20. El compuesto de la reivindicación 19 para uso en el tratamiento de la esquizofrenia, en donde el compuesto existe en forma polimórfica que tiene un patrón de difracción de XRPD como se indica en la Figura 18.
21. El compuesto de acuerdo con cualquiera de las reivindicaciones 12 a 20 para uso en el tratamiento de la esquizofrenia, en donde el compuesto se usa en el tratamiento de la esquizofrenia en combinación con uno o más agentes neurolépticos.
22. El compuesto de acuerdo con cualquiera de las reivindicaciones 12 a 20 para uso en el tratamiento de la esquizofrenia, en donde el compuesto se usa en el tratamiento de la esquizofrenia en combinación con uno o más agentes neurolépticos, en donde el uno o más agentes neurolépticos se selecciona del grupo que consiste en sertindol, olanzapina, risperidona, quetiapina, aripiprazol, haloperidol, clozapina, ziprasidona y osanetant.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201161498651P | 2011-06-20 | 2011-06-20 | |
| US201161537103P | 2011-09-21 | 2011-09-21 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2719145T3 true ES2719145T3 (es) | 2019-07-08 |
Family
ID=46682855
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16179882T Active ES2719145T3 (es) | 2011-06-20 | 2012-06-19 | 1-Piperazino-3-fenil-indanos deuterados para el tratamiento de la esquizofrenia |
| ES19151618T Active ES2939477T3 (es) | 2011-06-20 | 2012-06-19 | 1-Piperazino-3-fenil-indanos deuterados para el tratamiento de la esquizofrenia |
| ES12748046.5T Active ES2601213T3 (es) | 2011-06-20 | 2012-06-19 | 1-piperazino-3-fenil-indanos deuterados para el tratamiento de la esquizofrenia |
Family Applications After (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES19151618T Active ES2939477T3 (es) | 2011-06-20 | 2012-06-19 | 1-Piperazino-3-fenil-indanos deuterados para el tratamiento de la esquizofrenia |
| ES12748046.5T Active ES2601213T3 (es) | 2011-06-20 | 2012-06-19 | 1-piperazino-3-fenil-indanos deuterados para el tratamiento de la esquizofrenia |
Country Status (40)
| Country | Link |
|---|---|
| US (8) | US8575174B2 (es) |
| EP (4) | EP2720989B1 (es) |
| JP (1) | JP5668177B2 (es) |
| KR (2) | KR101879474B1 (es) |
| CN (1) | CN103649019B (es) |
| AP (1) | AP3310A (es) |
| AR (1) | AR086987A1 (es) |
| AU (1) | AU2012273657B2 (es) |
| BR (1) | BR112013031702B1 (es) |
| CA (1) | CA2837820C (es) |
| CL (1) | CL2013003646A1 (es) |
| CO (1) | CO6821965A2 (es) |
| CR (1) | CR20130654A (es) |
| CY (2) | CY1118158T1 (es) |
| DK (2) | DK3135656T3 (es) |
| DO (1) | DOP2013000305A (es) |
| EA (1) | EA024651B1 (es) |
| EC (1) | ECSP14013155A (es) |
| ES (3) | ES2719145T3 (es) |
| GE (1) | GEP201706655B (es) |
| GT (1) | GT201300304A (es) |
| HK (1) | HK1197228A1 (es) |
| HR (2) | HRP20161348T1 (es) |
| HU (2) | HUE044043T2 (es) |
| IL (1) | IL229640B (es) |
| JO (1) | JO3128B1 (es) |
| LT (2) | LT2720989T (es) |
| MA (1) | MA35268B1 (es) |
| MD (1) | MD4538C1 (es) |
| ME (2) | ME03375B (es) |
| MX (1) | MX339552B (es) |
| MY (1) | MY196998A (es) |
| PE (2) | PE20150928A1 (es) |
| PL (2) | PL2720989T3 (es) |
| PT (2) | PT3135656T (es) |
| RS (2) | RS58546B1 (es) |
| SI (2) | SI3135656T1 (es) |
| SM (1) | SMT201600383B (es) |
| TW (3) | TWI614234B (es) |
| WO (1) | WO2012176066A1 (es) |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2720989B1 (en) * | 2011-06-20 | 2016-08-10 | H. Lundbeck A/S | Deuterated 1-piperazino-3-phenyl indanes for treatment of schizophrenia |
| AR094054A1 (es) * | 2012-12-19 | 2015-07-08 | H Lundbeck As | 6-cloro-3-(fenil-d₅)-inden-1-ona y uso de la misma |
| EP3679018B1 (en) | 2017-09-07 | 2022-03-02 | Otsuka Pharmaceutical Co., Ltd. | Industrial process of mono-alkylating a piperidine nitrogen in piperidine derivatives with deuterated-alkyl |
| WO2020089147A1 (en) | 2018-10-29 | 2020-05-07 | H. Lundbeck A/S | Amorphous compounds of formula (i) and amorphous compounds of formula (i) salts |
| US11535600B2 (en) | 2018-12-03 | 2022-12-27 | H. Lundbeck A/S | Prodrugs of 4-((1R,3S)-6-chloro-3-phenyl-2,3-dihydro-1H-inden-1-yl)-1,2,2-trimethylpiperazine and 4-((1R,3S)-6-chloro-3-(phenyl-d5)-2,3-dihydro-1H-inden-1-yl)-2,2-dimethyl-1-(methyl-d3)piperazine |
| US20220106272A1 (en) * | 2018-12-21 | 2022-04-07 | Terran Biosciences, Inc. | Deuterated Forms And Derivatives Of Volinanserin |
| CA3132953A1 (en) | 2019-03-13 | 2020-09-17 | Otsuka Pharmaceutical Co., Ltd. | Method for introducing deuterated lower alkyl into amine moiety of compound containing secondary amine |
Family Cites Families (30)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| IE50867B1 (en) | 1980-02-29 | 1986-08-06 | Kefalas As | Indane derivatives |
| GB8427125D0 (en) | 1984-10-26 | 1984-12-05 | Lundbeck & Co As H | Organic compounds |
| DK286990D0 (da) | 1990-12-04 | 1990-12-04 | Lundbeck & Co As H | Indanderivater |
| DK55192D0 (da) | 1992-04-28 | 1992-04-28 | Lundbeck & Co As H | 1-piperazino-1,2-dihydroindenderivater |
| US6221335B1 (en) | 1994-03-25 | 2001-04-24 | Isotechnika, Inc. | Method of using deuterated calcium channel blockers |
| KR20040068613A (ko) | 1994-03-25 | 2004-07-31 | 이소테크니카 인코포레이티드 | 중수소화된 화합물 이를 포함하는 고혈압 치료용 조성물 |
| UA81749C2 (uk) | 2001-10-04 | 2008-02-11 | Х. Луннбек А/С | Фенілпіперазинові похідні як інгібітори зворотного захоплення серотоніну |
| AU2004265022A1 (en) | 2003-08-18 | 2005-02-24 | H.Lundbeck A/S | Trans-1(6-chloro-3-phenylindan-1-yl)-3,3-dimethylpiperazine |
| CN101935305B (zh) * | 2003-08-18 | 2013-04-10 | H.隆德贝克有限公司 | 反式-4-((1r,3s)-6-氯-3-苯基茚满-1-基)-1,2,2-三甲基哌嗪及其盐的制备方法及所用中间体 |
| TWI453198B (zh) | 2005-02-16 | 2014-09-21 | Lundbeck & Co As H | 製造反式-1-((1r,3s)-6-氯基-3-苯基茚滿-1-基) -3 , 3 -二甲基六氫吡與其鹽類之方法及製造4-((1r , 3s)-6 -氯基-3-苯基茚滿-1-基 )-1,2,2-三甲基六氫吡與其鹽類之方法 |
| TWI376373B (en) | 2005-02-16 | 2012-11-11 | Lundbeck & Co As H | Crystalline base of a pharmaceutical compound |
| JP2008530039A (ja) | 2005-02-16 | 2008-08-07 | ハー・ルンドベック・アクチエゼルスカベット | トランス−1−((1r,3s)−6−クロロ−3−フェニルインダン−1−イル)−3,3−ジメチルピペラジンの酒石酸塩およびリンゴ酸塩 |
| CN101273024A (zh) | 2005-07-29 | 2008-09-24 | 康瑟特制药公司 | 新颖苯并[d][1,3]-二氧杂环戊烯衍生物 |
| US7863274B2 (en) | 2005-07-29 | 2011-01-04 | Concert Pharmaceuticals Inc. | Deuterium enriched analogues of tadalafil as PDE5 inhibitors |
| EP2998288B1 (en) | 2005-12-01 | 2022-08-24 | ACADIA Pharmaceuticals Inc. | Substituted phenethylamines with serotoninergic and/or norepinephrinergic activity |
| JP4986462B2 (ja) | 2006-01-27 | 2012-07-25 | シャープ株式会社 | 太陽電池ストリングおよびその製造方法、ならびに、その太陽電池ストリングを用いる太陽電池モジュール |
| TW200819426A (en) | 2006-08-31 | 2008-05-01 | Lundbeck & Co As H | Novel indane compounds |
| WO2008128166A1 (en) | 2007-04-13 | 2008-10-23 | Concert Pharmaceuticals Inc. | Deuterated derivatives of 4-(6-fluoro-1, 2-benzisoxazol-3-yl) piperidine compounds |
| KR20100105802A (ko) | 2007-04-19 | 2010-09-29 | 콘서트 파마슈티컬즈, 인크. | 중수소화된 모르폴리닐 화합물 |
| KR101185899B1 (ko) | 2007-06-12 | 2012-09-27 | 콘서트 파마슈티컬즈, 인크. | 아자펩티드 유도체 |
| US20090062303A1 (en) | 2007-08-29 | 2009-03-05 | Protia, Llc | Deuterium-enriched ziprasidone |
| CN102065861B (zh) | 2008-05-07 | 2013-10-16 | H.隆德贝克有限公司 | 反式-4-((1r,3s)-6-氯-3-苯基茚满-1-基)-1,2,2-三甲基哌嗪用于改善认知的用途 |
| EP2344163A1 (en) | 2008-10-03 | 2011-07-20 | H. Lundbeck A/S | Oral formulation |
| WO2010062656A2 (en) | 2008-10-28 | 2010-06-03 | Concert Pharmaceuticals Inc. | Deuterated 2-propylpentanoic acid compounds |
| US8263601B2 (en) | 2009-02-27 | 2012-09-11 | Concert Pharmaceuticals, Inc. | Deuterium substituted xanthine derivatives |
| US8658236B2 (en) * | 2009-08-21 | 2014-02-25 | Deuteria Beverages, Llc | Alcoholic compositions having a lowered risk of acetaldehydemia |
| CN102020522A (zh) * | 2009-09-21 | 2011-04-20 | 陈松源 | 氘代药物的制备方法和应用 |
| US8278460B2 (en) | 2009-10-15 | 2012-10-02 | Concert Pharmaceuticals, Inc. | Substituted benzimidazoles |
| WO2011059080A1 (ja) * | 2009-11-16 | 2011-05-19 | 第一三共株式会社 | 同位体置換されたジアミン誘導体 |
| EP2720989B1 (en) * | 2011-06-20 | 2016-08-10 | H. Lundbeck A/S | Deuterated 1-piperazino-3-phenyl indanes for treatment of schizophrenia |
-
2012
- 2012-06-19 EP EP12748046.5A patent/EP2720989B1/en active Active
- 2012-06-19 EP EP19151618.6A patent/EP3508468B1/en active Active
- 2012-06-19 ES ES16179882T patent/ES2719145T3/es active Active
- 2012-06-19 JO JOP/2012/0161A patent/JO3128B1/ar active
- 2012-06-19 EP EP16179882.2A patent/EP3135656B1/en active Active
- 2012-06-19 ES ES19151618T patent/ES2939477T3/es active Active
- 2012-06-19 BR BR112013031702-7A patent/BR112013031702B1/pt active IP Right Grant
- 2012-06-19 AU AU2012273657A patent/AU2012273657B2/en active Active
- 2012-06-19 CN CN201280029731.8A patent/CN103649019B/zh active Active
- 2012-06-19 RS RS20190429A patent/RS58546B1/sr unknown
- 2012-06-19 PE PE2015000596A patent/PE20150928A1/es active IP Right Grant
- 2012-06-19 EA EA201490045A patent/EA024651B1/ru not_active IP Right Cessation
- 2012-06-19 DK DK16179882.2T patent/DK3135656T3/en active
- 2012-06-19 LT LTEP12748046.5T patent/LT2720989T/lt unknown
- 2012-06-19 ME MEP-2019-93A patent/ME03375B/me unknown
- 2012-06-19 EP EP23150855.7A patent/EP4215512A1/en active Pending
- 2012-06-19 MX MX2013014849A patent/MX339552B/es active IP Right Grant
- 2012-06-19 HU HUE16179882A patent/HUE044043T2/hu unknown
- 2012-06-19 DK DK12748046.5T patent/DK2720989T3/en active
- 2012-06-19 RS RS20160916A patent/RS55304B1/sr unknown
- 2012-06-19 PE PE2013002857A patent/PE20141113A1/es not_active Application Discontinuation
- 2012-06-19 GE GEAP201213350A patent/GEP201706655B/en unknown
- 2012-06-19 WO PCT/IB2012/001386 patent/WO2012176066A1/en active Application Filing
- 2012-06-19 PT PT16179882T patent/PT3135656T/pt unknown
- 2012-06-19 US US13/527,364 patent/US8575174B2/en active Active
- 2012-06-19 CA CA2837820A patent/CA2837820C/en active Active
- 2012-06-19 KR KR1020137033717A patent/KR101879474B1/ko active IP Right Grant
- 2012-06-19 PL PL12748046T patent/PL2720989T3/pl unknown
- 2012-06-19 PT PT127480465T patent/PT2720989T/pt unknown
- 2012-06-19 KR KR1020187018343A patent/KR101939546B1/ko active IP Right Grant
- 2012-06-19 AP AP2013007338A patent/AP3310A/xx active
- 2012-06-19 SI SI201231582T patent/SI3135656T1/sl unknown
- 2012-06-19 ME MEP-2016-226A patent/ME02513B/me unknown
- 2012-06-19 JP JP2014516459A patent/JP5668177B2/ja active Active
- 2012-06-19 MD MDA20140004A patent/MD4538C1/ro active IP Right Grant
- 2012-06-19 PL PL16179882T patent/PL3135656T3/pl unknown
- 2012-06-19 SI SI201230744A patent/SI2720989T1/sl unknown
- 2012-06-19 AR ARP120102179 patent/AR086987A1/es active IP Right Grant
- 2012-06-19 MY MYPI2021002383A patent/MY196998A/en unknown
- 2012-06-19 LT LTEP16179882.2T patent/LT3135656T/lt unknown
- 2012-06-19 HU HUE12748046A patent/HUE030883T2/en unknown
- 2012-06-19 ES ES12748046.5T patent/ES2601213T3/es active Active
- 2012-06-20 TW TW106107563A patent/TWI614234B/zh active
- 2012-06-20 TW TW101122028A patent/TWI627956B/zh active
- 2012-06-20 TW TW107113541A patent/TWI659741B/zh active
-
2013
- 2013-06-24 US US13/924,849 patent/US9012453B2/en active Active
- 2013-11-26 IL IL229640A patent/IL229640B/en active IP Right Grant
- 2013-12-09 GT GT201300304A patent/GT201300304A/es unknown
- 2013-12-13 CR CR20130654A patent/CR20130654A/es unknown
- 2013-12-16 DO DO2013000305A patent/DOP2013000305A/es unknown
- 2013-12-19 CL CL2013003646A patent/CL2013003646A1/es unknown
- 2013-12-19 CO CO13296680A patent/CO6821965A2/es active IP Right Grant
-
2014
- 2014-01-16 MA MA36688A patent/MA35268B1/fr unknown
- 2014-01-17 EC ECSP14013155 patent/ECSP14013155A/es unknown
- 2014-10-22 HK HK14110519A patent/HK1197228A1/zh unknown
-
2015
- 2015-03-13 US US14/656,925 patent/US9216961B2/en active Active
- 2015-11-16 US US14/941,800 patent/US9617231B2/en active Active
-
2016
- 2016-10-17 HR HRP20161348TT patent/HRP20161348T1/hr unknown
- 2016-10-26 CY CY20161101089T patent/CY1118158T1/el unknown
- 2016-10-26 SM SM201600383T patent/SMT201600383B/it unknown
-
2017
- 2017-02-17 US US15/435,826 patent/US10118907B2/en active Active
-
2018
- 2018-09-28 US US16/146,625 patent/US10501427B2/en active Active
-
2019
- 2019-03-27 HR HRP20190593TT patent/HRP20190593T1/hr unknown
- 2019-04-02 CY CY20191100368T patent/CY1121514T1/el unknown
- 2019-11-04 US US16/672,870 patent/US11059798B2/en active Active
-
2021
- 2021-06-07 US US17/340,201 patent/US20220119362A1/en active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2719145T3 (es) | 1-Piperazino-3-fenil-indanos deuterados para el tratamiento de la esquizofrenia | |
| DK201500362A1 (en) | Deuterated 1-piperazino-3-phenyl indanes for treatment of schizophrenia | |
| OA16797A (en) | Deuterated 1-Piperazino-3-Phenyl-Indanes for treatment of Schizophrenia. | |
| NZ618222B2 (en) | Deuterated 1-piperazino-3-phenyl indanes for treatment of schizophrenia |