EP0202471A2 - Wässrige anionische Dispersion - Google Patents
Wässrige anionische Dispersion Download PDFInfo
- Publication number
- EP0202471A2 EP0202471A2 EP86105191A EP86105191A EP0202471A2 EP 0202471 A2 EP0202471 A2 EP 0202471A2 EP 86105191 A EP86105191 A EP 86105191A EP 86105191 A EP86105191 A EP 86105191A EP 0202471 A2 EP0202471 A2 EP 0202471A2
- Authority
- EP
- European Patent Office
- Prior art keywords
- formula
- weight
- dispersion
- product
- compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 0 CC(C(N**)=CC=C1)=C*1N*C=O Chemical compound CC(C(N**)=CC=C1)=C*1N*C=O 0.000 description 1
Classifications
-
- D—TEXTILES; PAPER
- D06—TREATMENT OF TEXTILES OR THE LIKE; LAUNDERING; FLEXIBLE MATERIALS NOT OTHERWISE PROVIDED FOR
- D06M—TREATMENT, NOT PROVIDED FOR ELSEWHERE IN CLASS D06, OF FIBRES, THREADS, YARNS, FABRICS, FEATHERS OR FIBROUS GOODS MADE FROM SUCH MATERIALS
- D06M15/00—Treating fibres, threads, yarns, fabrics, or fibrous goods made from such materials, with macromolecular compounds; Such treatment combined with mechanical treatment
- D06M15/19—Treating fibres, threads, yarns, fabrics, or fibrous goods made from such materials, with macromolecular compounds; Such treatment combined with mechanical treatment with synthetic macromolecular compounds
- D06M15/37—Macromolecular compounds obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds
-
- D—TEXTILES; PAPER
- D06—TREATMENT OF TEXTILES OR THE LIKE; LAUNDERING; FLEXIBLE MATERIALS NOT OTHERWISE PROVIDED FOR
- D06M—TREATMENT, NOT PROVIDED FOR ELSEWHERE IN CLASS D06, OF FIBRES, THREADS, YARNS, FABRICS, FEATHERS OR FIBROUS GOODS MADE FROM SUCH MATERIALS
- D06M13/00—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with non-macromolecular organic compounds; Such treatment combined with mechanical treatment
- D06M13/322—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with non-macromolecular organic compounds; Such treatment combined with mechanical treatment with compounds containing nitrogen
- D06M13/402—Amides imides, sulfamic acids
- D06M13/425—Carbamic or thiocarbamic acids or derivatives thereof, e.g. urethanes
- D06M13/428—Carbamic or thiocarbamic acids or derivatives thereof, e.g. urethanes containing fluorine atoms
Definitions
- the present invention relates to an aqueous anionic dispersion which contains a bis- (2-perfluoroalkyl-ethoxy-carbonylamino) toluene, its preparation and its use for finishing textiles with oleophobic and hydrophobic agents, and the active ingredient contained in the dispersion .
- synthetic fibers can be made dirt-repellent, water-repellent and oil-repellent if they contain a fluorine-containing compound, e.g. 2,4-bis (2-perfluoroalkyl-ethoxycarbonylamino) toluene is applied in such an amount that the film applied contains at least 20% by weight of fluorine.
- a fluorine-containing compound e.g. 2,4-bis (2-perfluoroalkyl-ethoxycarbonylamino) toluene is applied in such an amount that the film applied contains at least 20% by weight of fluorine.
- a commercial product for textile finishing contains such fluorine-containing bisurethanes together with cationic dispersing agents in an aqueous dispersion.
- a disadvantage of using this dispersion is that fibers or textiles treated with it attract dirt particles that are normally anionically charged. For this reason, the cationic dispersions of the fluorine-containing bisurethanes must contain a special antistatic additive if one does not want to be forced to subject the finished goods to additional antistatic treatment later.
- the percentages given above represent percentages by weight.
- the percentages for a, b, c and d are based on the total weight of the aqueous dispersion.
- the perfluoroalkyl radicals or perfluoroalkyl compounds mentioned above and below, according to the above definitions in the w position for Y may also have an H atom.
- the compounds of the formulas II, III and IV can be used in the form of their technical mixtures, which generally contain several compounds of the type mentioned with different index numbers n, p, q and / or r.
- the compounds of formula I are known.
- the active ingredient A which is used according to the invention for the preparation of the aqueous anionic dispersion, is new.
- A is used to produce the product specified under A.
- Suitable tolylene di-isocyanates are, in particular, 2,4-and / or 2,6-tolylene-di-isocyanate, in particular in the form of a commercial product which contains approximately 80% by weight of 2,4-tolylene-di- contains isocyanate and about 20% by weight of 2,6-tolylene di-isocyanate.
- the reaction for the production of product A is generally carried out in such a way that the compound II or the mixture of the compounds II is melted and 2 to 5% by weight, preferably 2 to 3.5% by weight, of N-methyl pyrrolidone is mixed in and then the tolylene di-isocyanate or the mixture of tolylene di-isocyanates is added dropwise with stirring at a temperature which is about 5 to 10 ° C. above the melting point.
- the mixture is then heated to temperatures of approximately 130 ° C. in the course of an hour, the reaction being slightly exothermic from temperatures of approximately 80 ° C.
- the reaction is brought to a conclusion by an approximately three-hour reaction time at approximately 130 ° C.
- the progress of the reaction is continuously checked by IR spectroscopy on the samples for the disappearance of the isocyanate bands. If the reaction occurs within the specified time has not yet been completed, the reaction time must be extended, for example to 6 hours.
- N-methylpyrrolidone presumably forms by-products of still unknown structure, which act as excellent dispersion stabilizers in the inventive dispersion of the product.
- a larger addition of N-methylpyrrolidone in the preparation of product A does not adversely affect the effect of dispersion stabilization, but unnecessarily reduces the yield of the active substance of the formula I.
- the monovalent cation X in the compounds of the formula III generally represents an alkali metal cation, in particular the sodium or potassium cation or the ammonium cation.
- the ammonium cation can optionally also be substituted by organic radicals, for example triethanolammonium.
- emulsifiers of the formula IV are also commercially available, mostly in the form of their technical mixtures.
- r is in particular approximately 6.
- d is preferably a number from 10 to 25.
- components A, B, C and D are dispersed in water, with the supply of a relatively large amount of energy.
- the quantitative ratios for the components are chosen so that the specified composition for the dispersion is achieved after dispersion. It is essential to pre-dissolve product A in at least part of the amount of solvent or solvent mixture to be used, and it is expedient to divide the dispersion into two substeps and first to do a predispersion and then a fine dispersion.
- the predispersion is expediently carried out using high shear forces, for example using a high-speed stirrer, e.g.
- component D water-soluble solvents, such as e.g. Mono- or di-alcohols, lower ketones, polyglycol esters and polyglycol ethers or mixtures of such solvents are used.
- component D contains at least one high-boiling, water-soluble solvent, i.e. a solvent with a boiling point above approx. 150 ° C.
- a solvent mixture used can also include one or more water-insoluble solvents, such as e.g. Contain esters, ethers and / or higher ketones. Low-boiling solvent fractions can optionally be removed again at a later time, e.g. be distilled off.
- water-insoluble solvents such as e.g. Contain esters, ethers and / or higher ketones.
- Low-boiling solvent fractions can optionally be removed again at a later time, e.g. be distilled off.
- Suitable water-soluble, high-boiling solvents are in particular the (C 1 -C 4 ) monoalkyl and dialkyl ether of diethylene glycol and / or dipropylene glycol into consideration.
- a further advantage for the stability of the dispersion is the addition of isopropanol, glycol or glycerin, individually or in a mixture, preferably in an amount of 1 to 5% by weight, based on the final setting.
- Such (meth) acrylic acid ester polymers or copolymers are advantageously added to the dispersions according to the invention in the form of a separately prepared aqueous anionic dispersion. It is also expedient to use the polymer or. Disperse copolymer dispersion using a compound of formula III or a mixture of such compounds.
- the (meth) acrylic acid ester polymers or copolymers normally contain building blocks of esters of acrylic and / or methacrylic acid with C 1 to e 18 alcohols and can be prepared, for example, in a manner known per se.
- Methacrylic ester copolymers are preferred, especially when the monomer mixture used for their preparation contains at least 80% by weight of esters of C 1 to C 4 alcohols.
- Copolymers of methyl and isobutyl methacrylate are particularly preferred, particularly when the methyl ester content predominates in the copolymer.
- a copolymer made from methyl methacrylate and isobutyl ester in a weight ratio of 3: 1 is very particularly preferred.
- the preparation of this copolymer and its dispersion are described in Example 3.
- Other (meth) acrylic ester polymers and copolymers can be prepared and dispersed analogously.
- the aqueous anionic dispersions according to the invention meet all practical requirements and in particular show excellent long-term stability at temperatures from -20 to + 40 ° C. Although they freeze at sub-zero temperatures, the dispersion remains after thawing, in contrast to the previously known dispersions.
- the aqueous anionic dispersions according to the invention have an excellent oleophobic, hydrophobic, dirt-repellent and conductivity-improving effect in textile finishing. They can be used both for textile finishing alone, as well as in combination with other finishing agents, such as textile resins based on glyoxal or their derivatives, plasticizers, PVA and EVA or similar dispersions.
- the aqueous anionic dispersions according to the invention are suitable for finishing textiles made from natural or synthetic fibers, in particular from polyamide, polyester, polyacrylonitrile and wool, or mixtures of these types of fibers.
- the textile material can be in any form, e.g. as thread, fiber, yarn, flake, as fabric, knitted fabric, knitted fabric or fleece, but especially as a carpet.
- the dispersions according to the invention can be applied to the textile material in the form in which they are produced. Normally, however, they will be diluted with water to a solids content of 1 to 10% by weight, preferably 1.5 to 5% by weight, for use.
- the application to the textile material to be treated can be carried out in any suitable manner, for example by spraying, splashing, padding etc.
- the order quantity is chosen so that 0.01 on the textile material up to 1% by weight of fluorine, preferably 0.05 to 0.2% by weight of fluorine. This corresponds approximately to an amount of 0.1 to 10, preferably 0.5 to 2% by weight solids content.
- drying is carried out at temperatures up to approximately 120 ° C., for example at 100 to 120 ° C., and then heat treatment is carried out at temperatures of approximately 130 to 190 ° C., preferably 140 to 180 ° C. , which normally takes about 4 minutes to about 30 seconds.
- the high-boiling organic solvents preferably contained in the dispersion also have an important meaning in fixing the active ingredient of formula I on the fiber in the sense of a kind of carrier effect.
- the progress of the reaction is IR-spectroscopically on removed samples on the disappearance of the isocyanate that controls.
- the response time may be shortened or extended.
- Example 1 is repeated without the addition of N-methyl-pyrrolidone. 1154 g of a pale yellowish melt are obtained which solidify on cooling to a hard, pale yellowish crystal cake.
- the polymerization reaction will then begin immediately, as can be seen from a rise in temperature up to about 57 ° C. and a change in color (bluish fluorescent).
- the monomer solution is added dropwise from the storage vessel and a separately prepared catalyst solution consisting of 0.2 g sodium pyrosulfite (Na 2 S 2 0 5 ) and 10 g water.
- a separately prepared catalyst solution consisting of 0.2 g sodium pyrosulfite (Na 2 S 2 0 5 ) and 10 g water.
- the introduction of nitrogen can be stopped.
- the monomer solution should be metered in after about 1 hour and the catalyst solution a little later.
- the reaction temperature is 55 to 60 ° C with an unchanged bath temperature of 55 ° C.
- the polymer emulsion is heated to 60 to 62 ° C. (bath temperature 65 ° C.) and stirred for 1 h under these conditions, then cooled to room temperature and filtered through a PE sieve bag (105 ⁇ m). 270.4 g of an approximately 40% whitish, opaque dispersion are obtained.
- Example 3 The procedure is as in Example 3, but the 30 g of the isopropanol solution of the emulsifier used there is replaced by 4 g of a commercially available alkane sulfonate used as a wash raw material (e.g. commercial product @Warolat U from Bayer AG).
- a commercially available alkane sulfonate used as a wash raw material (e.g. commercial product @Warolat U from Bayer AG).
- the crude dispersion obtained is then subjected to a final fine treatment, namely by sonication using an ultrasound machine (for example of the Sonifier type from Branson) until 90% of the particles have reached or fallen below a size of 1 ⁇ m. This usually takes 10 to 15 minutes.
- the temperature is first kept at 40 to 45 ° C by water cooling, towards the end by cooling with ice water to 20 lowered to 30 ° C.
- Example 5 is repeated, but product A produced according to example 1 is replaced by 11.8 g of the bisurethane prepared according to example 2.
- the dispersion obtained is not stable in storage, since a clear sediment forms within 24 hours, which further increases over time.
- a solution of 1.25 g of a compound of the formula VII is placed in a 200 ml three-necked flask in the form of a beaker in 48.75 ml of water and 20 g of the approx. 40% anionically dispersed methacrylic ester copolymer according to example 4 submitted.
- This raw dispersion is then subjected to a final fine treatment, namely by sonication using an ultrasound machine (for example of the Sonifier type from Branson) until 90% of the particles have reached or fallen below an average size of 1 ⁇ m. This usually takes 10 to 15 minutes.
- the temperature is initially maintained by water cooling at 40 to 45 0 C, lowered towards the end by cooling with ice water to 20 to 30 ° C.
- the dispersion according to the invention prepared according to Example 5 or 7 is diluted with water to a solids content of 2 to 4% by weight.
- the web to be treated is passed through the liquor thus obtained and squeezed off on a foulard. A repetition of this process promotes the penetration of the substrate and increases the effectiveness of the product according to the invention.
- the textile substrate is dried in a drying unit at temperatures up to 120 ° C. and then fixed in the same or another unit by heat treatment at temperatures from 150 to 180 ° C. for 3 minutes to 3 seconds.
- the dispersion prepared according to Example 5 is diluted with water to a solids content of 3% and sprayed onto a PA tufted carpet.
- the carpet is then dried at 110 ° C and then subjected to a 3 minute heat treatment at 150 ° C.
Landscapes
- Engineering & Computer Science (AREA)
- Textile Engineering (AREA)
- Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Emulsifying, Dispersing, Foam-Producing Or Wetting Agents (AREA)
- Treatments For Attaching Organic Compounds To Fibrous Goods (AREA)
- Colloid Chemistry (AREA)
- Agricultural Chemicals And Associated Chemicals (AREA)
- Polyurethanes Or Polyureas (AREA)
- Materials Applied To Surfaces To Minimize Adherence Of Mist Or Water (AREA)
- Paints Or Removers (AREA)
Abstract
Description
- Die vorliegende Erfindung betrifft eine wäßrige anionische Dispersion, die ein Bis-(2-perfluoralkyl-ethoxy-car- bonylamino)-toluol enthält, ihre Herstellung und ihre Verwendung zur Oleophob- und Hydrophob-ausrüstung von Textilien, sowie den in der Dispersion enthaltenen Wirkstoff.
- Aus der US-P 3 171 861 ist es z.B. bekannt, daß 3-(Per- fluoroctyl)-propanol mit Toluoldiisocyanat zur dem entsprechenden Diurethan umgesetzt werden kann und daß diese Verbindung, aus einer Lösung in einem Aceton/1,1,1-Trichlorethan-Gemisch auf verschiedene Textilien aufgebracht, den behandelten Textilien eine ölabstoßende Eigenschaft verleiht.
- Aus der japanischen Offenlegungsschrift 59 094621 (zitiert nach Textilbericht 10/85) ist es bekannt, daß Synthesefasern schmutzabweisend, wasser- und ölabstoßend ausgerüstet werden können, wenn auf sie vor dem Verstrecken zusammen mit dem Spinnöl eine fluorhaltige Verbindung, z.B. 2,4-Bis(2-perfluoralkyl-ethoxy-carbonyl- amino)-toluol in solcher Menge aufgebracht wird, daß der aufgebrachte Film mindestens 20 Gew.% Fluor enthält.
- Ein Handelsprodukt zur Textilausrüstung enthält derartige fluorhaltige Bisurethane zusammen mit kationischen Dispergierhilfsmitteln in einer wäßrigen Dispersion. Nachteilig bei der Anwendung dieser Dispersion ist jedoch, daß damit behandelte Fasern bzw. Textilien Schmutzpartikel anziehen, die normalerweise anionisch geladen sind. Aus diesem Grund müssen die kationischen Dispersionen der fluorhaltigen Bisurethane einen speziellen Antistatikzusatz enthalten, falls man nicht gezwungen sein will, die ausgerüsteten Waren später einer zusätzlichen Antistatik-Ausrüstung zu unterziehen.
- Aus den genannten Gründen ist es daher wünschenswert, die fluorhaltigen Bisurethane in Form einer anionischen, wäßrigen Dispersion auf die zu behandelnden Textilien aufzubringen. Es war jedoch bisher nicht möglich, fluorhaltige Bisurethane in eine wäßrige anionische Dispersion zu überführen, welche die Forderungen der Praxis, insbesondere der Langzeitstabilität bei Temperaturen von -20°C bis +40°c, erfüllte.
- Es wurde nun überraschenderweise gefunden, daß sich derartige, die Praxisforderungen erfüllende wäßrige anionische Dispersionen von fluorhaltigen Bisurethanen herstellen lassen.
- Die erfindungsgemäße wäßrige anionische Dispersion enthält:
- A) a % eines Produktes, das mindestens ein Bis-(2- fluoralkyl-ethoxy-carbonylamino)-toluol der Formel I


- B) b % mindestens eines Emulgators der Formel III

- C) c % mindestens eines nichtionischen Emulgators der Formel IV

- D) d % eines Lösungsmittels oder Lösungsmittelgemischs, wobei a eine Zahl von 5 bis 25, b eine Zahl von 1 bis 14, c eine Zahl von 1 bis 14, d eine Zahl von 5 bis 30 bedeuten und dabei die Zahlenwerte für b und c so gewählt sind, daß die Summe (b + c) = 2 bis 15 beträgt.
- Die vorstehend angegebenen Prozente stellen Gewichtsprozente dar. Die Prozentangaben für a, b, c und d sind auf das Gesamtgewicht der wäßrigen Dispersion bezogen. Im Rahmen der vorliegenden Erfindung können die vorstehend und nachfolgend erwähnten Perfluoralkyl-reste bzw. Perfluoralkyl-verbindungen nach den vorstehenden Definitionen in w-Stellung für Y auch ein H-Atom besitzen. Die echten Perfluoralkyl-reste bzw. Perfluoralkyl-verbindungen, d.h. diejenigen Verbindungen der Formeln I bis IV, bei denen Y = -F bedeutet, sind jedoch bevorzugt.
- Die Verbindungen der Formel II, III und IV können in Form ihrer technischen Gemische eingesetzt werden, die in der Regel mehrere Verbindungen der genannten Art mit verschiedenen Indexzahlen n, p, q und/oder r enthalten.
- Die Verbindungen der Formel I sind bekannt. Der Wirkstoff A, der erfindungsgemäß zur Herstellung der wäßrigen anionischen Dispersion benutzt wird, ist jedoch neu. Zur Herstellung des unter A angegebenen Produkts wird ein 2-Perfluoralkyl-ethanol der Formel I oder ein Gemisch verschiedener 2-Perfluoralkyl-ethanole der Formel II mit einem Tolylen-di-isocyanat oder einem Gemisch verschiedener Tolylen-di-isocyanate im Mol-Verhältnis (1,8 bis 2) : 1 in Gegenwart von 2 bis 5 Gew.%, vorzugsweise 2 bis 3,5 Gew.%, bezogen auf die Verbindung der Formel II, an N-Methyl-pyrrolidon umgesetzt. Als Ausgangsprodukt sind dabei technische Gemische von 2-Perfluoralkyl-ethanolen der Formel II gut geeignet, die Verbindungen mit n = 5 bis 11 enthalten. Besonders geeignet sind Verbindungen der Formel II mit n = 7 bis 11, und zwar sowohl in Form der Einzelverbindungen als auch in Form ihrer technischen Gemische.
- Als Tolylen-di-isocyanat kommen insbesondere das 2,4-und/oder das 2,6-Tolylen-di-isocyanat in Betracht, insbesondere in Form eines Handelsprodukts, das ca. 80 Gew.% 2,4-Tolylen-di-isocyanat und ca. 20 Gew.% 2,6-Tolylen-di-isocyanat enthält.
- Die Umsetzung zur Herstellung des Produkts A wird in der Regel so durchgeführt, daß die Verbindung II oder das Gemisch der Verbindungen II aufgeschmolzen und zu der Schmelze 2 bis 5 Gew.%, vorzugsweise 2 bis 3,5 Gew.%, N-Methyl-pyrrolidon zugemischt wird und anschließend bei einer ca. 5 bis 10°C über dem Schmelzpunkt liegenden Temperatur das Tolylen-di-isocyanat oder das Gemisch der Tolylen-di-isocyanate unter Rühren zugetropft wird. Anschließend wird im Verlauf einer Stunde auf Temperaturen von ca. 130°C aufgeheizt, wobei die Reaktion ab Temperaturen von ca. 80°C leicht exotherm verläuft. Abschließend wird die Umsetzung durch eine ca. dreistündige Reaktionszeit bei ca. 130°C zum Abschluß gebracht. Das Fortschreiten der Reaktion wird laufend IR-spektroskopisch an entnommenen Proben auf das Verschwinden der Isocyanatbanden kontrolliert. Falls die Reaktion in der angegebenen Zeit noch nicht zum Abschluß gekommen ist, muß die Reaktionszeit, z.B. auf 6 Stunden, verlängert werden.
- Durch den Zusatz von N-Methylpyrrolidon werden vermutlich Nebenprodukte noch unbekannter Struktur gebildet, die bei der erfindungsgemäßen Dispergierung des Produkts als vorzügliche Dispersionsstabilisatoren wirken. Ein größerer Zusatz von N-Methylpyrrolidon bei der Herstellung des Produkts A beeinflußt den Effekt der Dispersionsstabilisierung nicht ungünstig, vermindert aber unnötigerweise die Ausbeute an der Wirksubstanz der Formel I.
- Das einwertige Kation X in den Verbindungen der Formel III stellt in der Regel ein Alkalimetallkation, insbesondere das Natrium- oder Kaliumkation oder das Ammoniumkation dar. Das Ammoniumkation kann gegebenenfalls auch durch organische Reste substituiert sein, beispielsweise Triethanolammonium darstellen. Verbindungen der Formel III sind insbesondere in Form der technischen Gemische mit p = 5 bis 11 oder 7 bis 11 handelsüblich. Vorzugsweise werden die Verbindungen der Formel III mit p = 7 bis 11 in Form der Einzelverbindungen oder in Form der technische Gemische verwendet.
- Auch die Emulgatoren der Formel IV sind im Handel, zumeist in Form ihrer technischen Gemische, erhältlich. Dabei beträgt r insbesondere ca. 6. Es werden Emulgatoren der Formel IV bevorzugt, bei denen q = 5 bis 11, insbesondere 7 bis 11, und r = 4 bis 8 beträgt, und zwar in Form der Einzelverbindungen oder in Form der technischen Gemische.
- Die Zahlen b und c bedeuten vorzugsweise jeweils 1 bis 9, wobei diese Zahlenwerte vorzugsweise so gewählt werden, daß die Summe (b + c) = 2 bis 12 beträgt. d bedeutet vorzugsweise eine Zahl von 10 bis 25.
- Zur Herstellung der erfindungsgemäßen wäßrigen anionischen Dispersionen werden die Komponenten A, B, C und D in Wasser, unter Zufuhr einer verhältnismäßig großen Energiemenge, dispergiert. Die Mengenverhältnisse für die Komponenten werden dabei so gewählt, daß nach der Dispergierung die angegebene Zusammensetzung für die Dispersion erreicht wird. Es ist dabei unerläßlich, das Produkt A zumindest in einem Teil der zur Anwendung kommenden Menge des Lösungsmittels oder Lösungsmittelgemischs vorzulösen, und es ist zweckmäßig, die Dispergierung in zwei Teilschritte aufzuteilen und zuerst eine Vordispergierung und anschließend eine Feindispergierung vorzunehmen. Die Vordispergierung wird zweckmäßigerweise durch Anwendung hoher Scherkräfte, beispielsweise durch die Verwendung eines schnell laufenden Rührers, wie z.B. bei einer Dispergiermaschine vom Typ Ultraturrax, vorgenommen, und die dabei erhaltene Vordispersion wird anschließend z.B. einer Ultraschallbehandlung oder einer Behandlung in einem Hochdruckhomogenisator unterzogen. Nach der Beendigung dieser Behandlung liegt die Teilchengröße in der Dispersion zu über 80 %, vorzugsweise zu über 95 %, bei oder unter 1 gm. Für die Lösungsmittelkomponente D werden wasserlösliche Lösungsmittel, wie z.B. Mono-oder Di-Alkohole, niedere Ketone, Polyglykolester und Polyglykolether oder Gemische derartiger Lösungsmittel verwendet. Vorteilhafterweise enthält die Komponente D mindestens ein hochsiedendes, wasserlösliches Lösungsmittel, d.h. ein Lösungsmittel, dessen Siedepunkt über ca. 150°C liegt. Gegebenenfalls kann ein zur Anwendung kommendes Lösungsmittelgemisch auch ein oder mehrere wasserunlösliche Lösungsmittel, wie z.B. Ester, Ether und/oder höhere Ketone, enthalten. Niedrigsiedende Lösungsmittelanteile können gegebenenfalls zu einem späteren Zeitpunkt wieder entfernt, z.B. abdestilliert werden.
- Als geeignete wasserlösliche, hochsiedende Lösungsmittel kommen insbesondere die (C1-C4)Monoalkyl- und Dialkylether des Diethylenglykols und/oder Dipropylenglykols in Betracht. Günstig für die Stabilität der Dispersion ist ferner ein Zusatz von Isopropanol, Glykol oder Glyzerin, einzeln oder im Gemisch, vorzugsweise in einer Menge von 1 bis 5 Gew.%, bezogen auf die Endeinstellung.
- Besonders günstige Effekte, insbesondere im Hinblick auf die schmutzabweisende Wirkung, werden erhalten, wenn die erfindungsgemäße Dispersion zusätzlich mindestens ein anionisch dispergiertes (Meth)Acrylsäureester-polymerisat oder -copolymerisat in Mengen von e Gew.% enthält, wobei e eine Zahl von 5 bis 25 bedeutet, die zweckmäßigerweise so gewählt ist, daß die Summe (a + e) = 15 bis 30 beträgt. Derartige (Meth)Acrylsäureester-polymerisate bzw. -copolymerisate werden zweckmäßigerweise in Form einer getrennt hergestellten wäßrigen anionischen Dispersion den erfindungsgemäßen Dispersionen zugesetzt. Außerdem ist es zweckmäßig, die Polymerisat-bzw. Copolymerisatdispersion unter Verwendung einer Verbindung der Formel III oder eines Gemisches derartiger Verbindungen zu dispergieren.
- Die (Meth)Acrylsäureester-polymerisate oder -copolymerisate enthalten normalerweise Bausteine von Estern der Acryl- und/oder Methacrylsäure mit C1- bis e18-Alkoholen und können z.B. in an sich bekannter Weise hergestellt werden. Methacrylester-copolymerisate sind bevorzugt, insbesondere dann, wenn das zu ihrer Herstellung benutzte Monomerengemisch mindestens 80 Gew.% Ester von C1- bis C4-Alkoholen enthält. Besonders bevorzugt sind Copolymerisate aus Methacrylsäuremethyl- und -isobutylester, insbesondere dann, wenn in dem Copolymerisat der Methylesteranteil überwiegt. Ganz besonders bevorzugt ist ein Copolymerisat, hergestellt aus Methacrylsäuremethylester und -isobutylester im Gewichtsverhältnis 3 : 1. Die Herstellung dieses Copolymerisats und seine Dispergierung ist in Beispiel 3 beschrieben. Andere (Meth)Acrylester- polymerisate und -copolymerisate können analog hergestellt und dispergiert werden.
- Die erfindungsgemäßen wäßrigen anionischen Dispersionen erfüllen alle Forderungen der Praxis und zeigen insbesondere eine hervorragende Langzeitstabilität bei Temperaturen von -20 bis +40°C. Sie gefrieren zwar bei Minustemperaturen, die Dispersion bleibt aber nach dem Auftauen erhalten, im Gegensatz zu den bisher bekannten Dispersionen. Die erfindungsgemäßen wäßrigen anionischen Dispersionen zeigen bei der Textilausrüstung einen hervorragenden oleophobierenden, hydrophobierenden, schmutzabweisenden und leitfähigkeitsverbessernden Effekt. Sie können sowohl allein für die Textilausrüstung eingesetzt werden, wie auch in Kombination mit anderen Ausrüstungsmitteln, wie Textilharzen auf der Basis Glyoxal oder deren Derivaten, Weichmachern, PVA und EVA oder ähnlichen Dispersionen.
- Die erfindungsgemäßen wäßrigen anionischen Dispersionen sind zur Ausrüstung von Textilien aus natürlichen oder synthetischen Fasern, insbesondere aus Polyamid, Polyester, Polyacrylnitril und Wolle, oder Gemischen dieser Faserarten geeignet. Das Textilmaterial kann in beliebiger Form vorliegen, so z.B. als Faden, Faser, Garn, Flocke, als Gewebe, Gestrick, Gewirk oder Vlies, insbesondere jedoch als Teppich.
- Die erfindungsgemäßen Dispersionen können in der Form auf das Textilmaterial aufgebracht werden, in der sie bei der Herstellung anfallen. Normalerweise wird man sie jedoch zur Anwendung mit Wasser auf einen Feststoffgehalt von 1 bis 10 Gew.%, vorzugsweise von 1,5 bis 5 Gew.%, verdünnen. Die Aufbringung auf das zu behandelnde Textilmaterial kann in jeder geeigneten Weise erfolgen, so z.B. durch Spühen, Pflatschen, Foulardieren ect. Die Auftragsmenge wird so gewählt, daß auf dem Textilmaterial 0,01 bis 1 Gew.% Fluor, vorzugsweise 0,05 bis 0,2 Gew.% Fluor, vorhanden sind. Dies entspricht etwa einer Menge von 0,1 bis 10, vorzugsweise 0,5 bis 2 Gew.% Feststoffgehalt. Nach dem Aufbringen auf das zu behandelnde Textilmaterial erfolgt eine Trocknung bei Temperaturen bis ca. 120°C, z.B. bei 100 bis 120°C, und anschließend wird eine Wärmebehandlung bei Temperaturen von ca. 130 bis 190°C, vorzugsweise 140 bis 180°C, durchgeführt, die normalerweise etwa 4 min bis etwa 30 sec dauert.
- Es hat den Anschein, als ob die in der Dispersion vorzugsweise enthaltenen, hochsiedenden organischen Lösungsmittel auch eine wichtige Bedeutung bei der Fixierung des Wirkstoffs der Formel I auf der Faser im Sinne eines Art Carrier-Effekts besitzen.
- In den nachfolgenden Beispielen bedeuten Prozentangaben, sofern nichts anderes angeben ist, Gewichtsprozente.
- 1080 g (= 2 mol) eines handelsüblichen Gemisches von 2-Perfluoralkyl-ethanolen der Formel II, in der Einzelprodukte mit n = 7 bis 11 enthalten sind und Y = -F bedeutet, werden in einem Reaktionsgefäß über den bei 65°C liegenden Schmelzpunkt aufgeheizt und 30 g N-Methyl-pyrrolidon zugegeben. Bei einer Temperatur von 70 bis 75°C werden dann im Verlauf von 30 min 174.16 g (= 1 mol) eines technischen Gemisches aus 80 % 2,4- und 20 % 2,6-Tolylen-di-isocyanat unter Rühren zugetropft. Anschließend wird im Verlauf von 1 h auf 130°C aufgeheizt, wobei die Reaktion ab 80°C leicht exotherm verläuft, und die Temperatur ca. 3 h bis 130°C gehalten.
- Das Fortschreiten der Reaktion wird an entnommenen Proben IR-spektroskopisch auf das Verschwinden der Isocyanatbanden kontrolliert. Gegebenenfalls wird die Reaktionszeit verkürzt oder verlängert.
- Es werden 1184 g einer gelbbraunen Schmelze erhalten, die beim Erkalten zu einem leicht bräunlichen Kristallkuchen erstarrt.
- Fp: 90 bis 118°C. Durchschnittlicher F-Gehalt: 59 %.
- Das Beispiel 1 wird ohne den Zusatz von N-Methyl-pyrrolidon wiederholt. Es werden 1154 g einer schwach gelblichen Schmelze erhalten, die beim Erkalten zu einem harten, schwach gelblichen Kristallkuchen erstarrt.
- Fp: 116 bis 120°C. Durchschnittlicher F-Gehalt: 60,5 %.
- In einem mit Rührer und Bodenauslauf versehenen 250 ml-Vorratsgefäß werden 75 g Methacrylsäuremethylester und 25 g Methacrylsäure-isobutylester vorgelegt und zu einer homogenen Lösung verrührt, worauf der Rührer abgestellt wird..
- In einem 500ml-Polymerisationskolben, ausgerüstet mit Rührer, Thermometer, Gaseinleitungsrohr, Rückflußkühler, Tropftrichter und Zulaufmöglichkeit aus dem Vorratsgefäß werden 130 g Wasser, 30 g einer 25%igen isopropanolischen Lösung eines handelsüblichen Emulgators der Formel V

- Während dieser gesamten Zutropfphase liegt die Reaktionstemperatur bei 55 bis 60°C bei unveränderter Badtemperatur von 55°C. Nach beendetem Zutropfen wird die Polymeremulsion auf 60 bis 62°C (65°C Badtemperatur) angeheizt und 1 h unter diesen Bedingungen nachgerührt, anschließend auf Raumtemperatur abgekühlt und über einen PE-Siebbeutel (105 µm) filtriert. Es werden 270,4 g einer ca. 40%igen weißlichen, opaken Dispersion erhalten.
- Es wird wie in Beispiel 3 gearbeitet, jedoch werden die 30 g der isopropanolischen Lösung des dort benutzten Emulgators durch 4 g eines handelsüblichen, als Waschrohstoff benutzten Alkansulfonats (z.B. Handelsprodukt @Warolat U der Firma Bayer AG) ersetzt.
- Es werden 244,2 g einer ca. 40%igen weißlichen, opaken Dispersion erhalten.
- In einem 200ml-Schliff-Dreihalskolben in Becherform werden 12,1 g des nach Beispiel 1 hergestellten Produkts A, 5 g Diethylenglykol-dimethylether, 5 g Dipropylenglykolmonomethylether und 3 g Perfluoralkylethyl-polyglykol der Formel VI
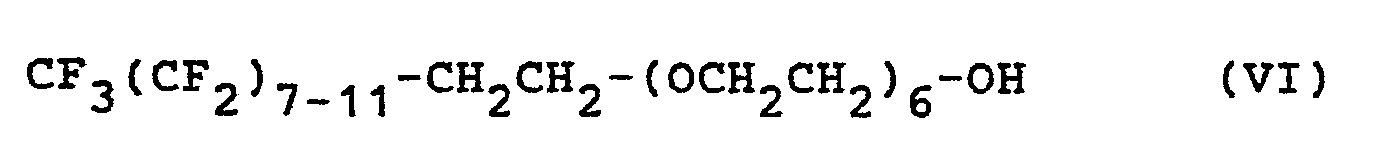

- In die Lösung wird bei 80°C unter der Anwendung von starken Scherkräften einer Dispergiermaschine vom Typ Ultraturrax im Laufe von 2 bis 3 min die Lösung von 1,25 g der Verbindung der Formel VII in 48,75 ml Wasser eingetropft, wobei die Temperatur auf 45 bis 50°C abfällt. Bei dieser Temperatur wird noch 10 bis 15 min weiterdispergiert. Hierbei entsteht bereits eine äußerlich ansprechende Emulsion, die aber in dieser Form noch nicht lagerbeständig ist, sondern sich bald absetzt.
- Die erhaltene Rohdispersion wird dann einer abschließenden Feinbehandlung unterworfen, und zwar durch Beschallung mittels einer Ultraschallmaschine (z.B. vom Typ Sonifier der Firma Branson), bis 90 % der Teilchen eine Größe von 1 µm erreicht oder unterschritten haben. Dies dauert gewöhnlich 10 bis 15 min. Dabei wird die Temperatur zunächst durch Wasserkühlung bei 40 bis 45°C gehalten, gegen Ende durch Kühlung mit Eiswasser auf 20 bis 30°C gesenkt.
- Zu dieser so erhaltenen Feindispersion werden dann 20 g des ca. 40%igen, anionisch dispergierten Methacrylester-copolymerisats nach Beispiel 3 hinzugefügt. Die gesamte Formulierung wird dann nochmals ca 2 min. unter Kühlung bei 20 bis 30°C mit Ultraschall behandelt. Man erhält 100 g einer feinen, milchig opaken Dispersion mit einem Fluorgehalt von 7 % (bezogen auf Wirksubstanz), die auch bei Temperaturen von -20°C und +40°C sehr gut lagerstabil ist.
- Nach einem 24stündigem Abkühlen auf -20°C konnte beim Wiederauftauen keine Veränderung der Dispersion beobachtet werden.
- Das Beispiel 5 wird wiederholt, dabei wird jedoch das gemäß Beispiel 1 hergstellte Produkt A durch 11,8 g des nach Beispiel 2 hergestellten Bisurethans ersetzt.
- Die erhaltene Dispersion ist nicht lagerbeständig, da sich bereits innerhalb von 24 h ein deutlicher Bodensatz bildet, der sich im Laufe der Zeit weiter verstärkt.
- In einem 200 ml-Schliff-Dreihalskolben in Becherform wird eine Lösung von 1,25 g einer Verbindung der Formel VII

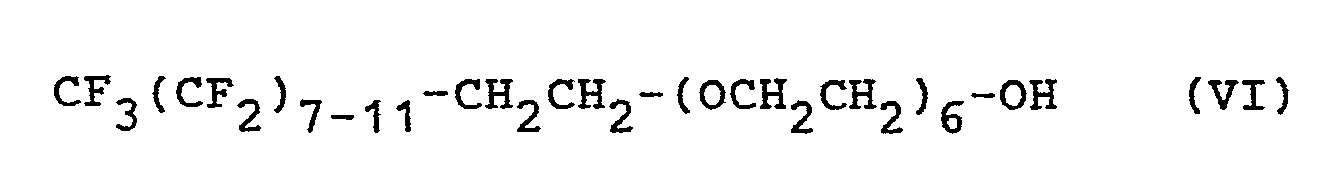
- Hierbei entsteht eine äußerlich ansprechende Emulsion, die aber in dieser Form noch nicht lagerbeständig ist, sondern sich bald absetzt.
- Diese Rohdispersion wird dann einer abschließenden Feinbehandlung unterworfen, und zwar durch Beschallung mittels einer Ultraschallmaschine (z.B. vom Typ Sonifier der Firma Branson), bis 90 % der Teilchen eine mittlere Größe von 1 µm erreicht oder unterschritten haben. Dies dauert gewöhnlich 10 bis 15 min. Dabei wird die Temperatur zunächst durch Wasserkühlung bei 40 bis 450C gehalten, gegen Ende durch Kühlung mit Eiswasser auf 20 bis 30°C gesenkt.
- Man erhält 100 g einer sehr feine, milchig opaken Dispersion mit einem Fluorgehalt von 7 % (bezogen auf Wirksubstanz), die auch bei Temperaturen -20°C und +40 C gut lagerstabil ist.
- Jeweils 100g-Muster der Dispersionen nach den Beispielen 5 und 7 sowie nach dem Vergleichsbeispiel 6 und sowie zum Vergleich 100 g eines kationisch dispergierten Handelsprodukts werden in einem Kühlschrank 24 h auf -20°C abgekühlt. Alle Proben gefrieren dabei. Nach dem Auftauen sind die Muster der Beispiele 5 und 7 noch einwandfrei homogen und genauso wirksam wie vor dem Gefriertest. Die Dispersion des mitgetesteten, kationisch dispergierten Handelsprodukts ist dagegen völlig zerfallen, und auch die Probe des Vergleichsbeispiels 6 zerfällt beim Auftauen.
- Die gemäß Beispiel 5 oder 7 hergestellte erfindungsgemäße Dispersion wird mit Wasser auf einen Feststoffgehalt von 2 bis 4 Gew.% verdünnt. Durch die so erhaltene Flotte wird zu behandelndes, bahnenförmiges Textilgut hindurchgeführt und auf einem Foulard abgequetscht. Eine Wiederholung dieses Prozesses fördert die Durchdringung des Substrats und erhöht die Effektivität des erfindungsgemäßen Produkts.
- Das textile Substrat wird in einem Trockenaggregat bei Temperaturen bis 120°C getrocknet und anschließend im gleichen oder einem anderen Aggregat durch eine Wärmebehandlung bei Temperaturen von 150 bis 180°C 3 min bis 3 sek lang fixiert.
- Die nach Beispiel 5 hergestellte Dispersion wird mit Wasser auf einen Festoffgehalt von 3 % verdünnt und auf einen PA-Tuffted Teppich aufgesprüht. Der Teppich wird dann bei 110°C getrocknet und anschließend einer 3 min langen Wärmebehandlung bei 150°C unterzogen.
- An dem behandelten Teppich werden die Oleophobierung nach der Methode 3M/AATCC 18-1966, die Hydrophobierung/Spray nach der Methode AATCC 22-1952 und die Trockenanschmutzung nach folgender Vorschrift geprüft:
- In einem zylindrischen, mit einem Deckel verschließbaren Gefäß von 20 cm Länge und einem Durchmesser von 10 cm wird die Teppichprobe ausgelegt. Dann werden 200 g Stahlkugeln von 3 mm Durchmesser und 20 g gesiebter Staubsaugerschmutz hinzugegeben, das Gefäß verschlossen und 1 h auf einem Rollgestell gerollt. Dann wird die Probe entnommen, mit einem Staubsauger abgesaugt und beurteilt.
- Bei den Ausprüfungen werden die in der nachfolgenden Tabelle I angegebenen Werte erhalten:
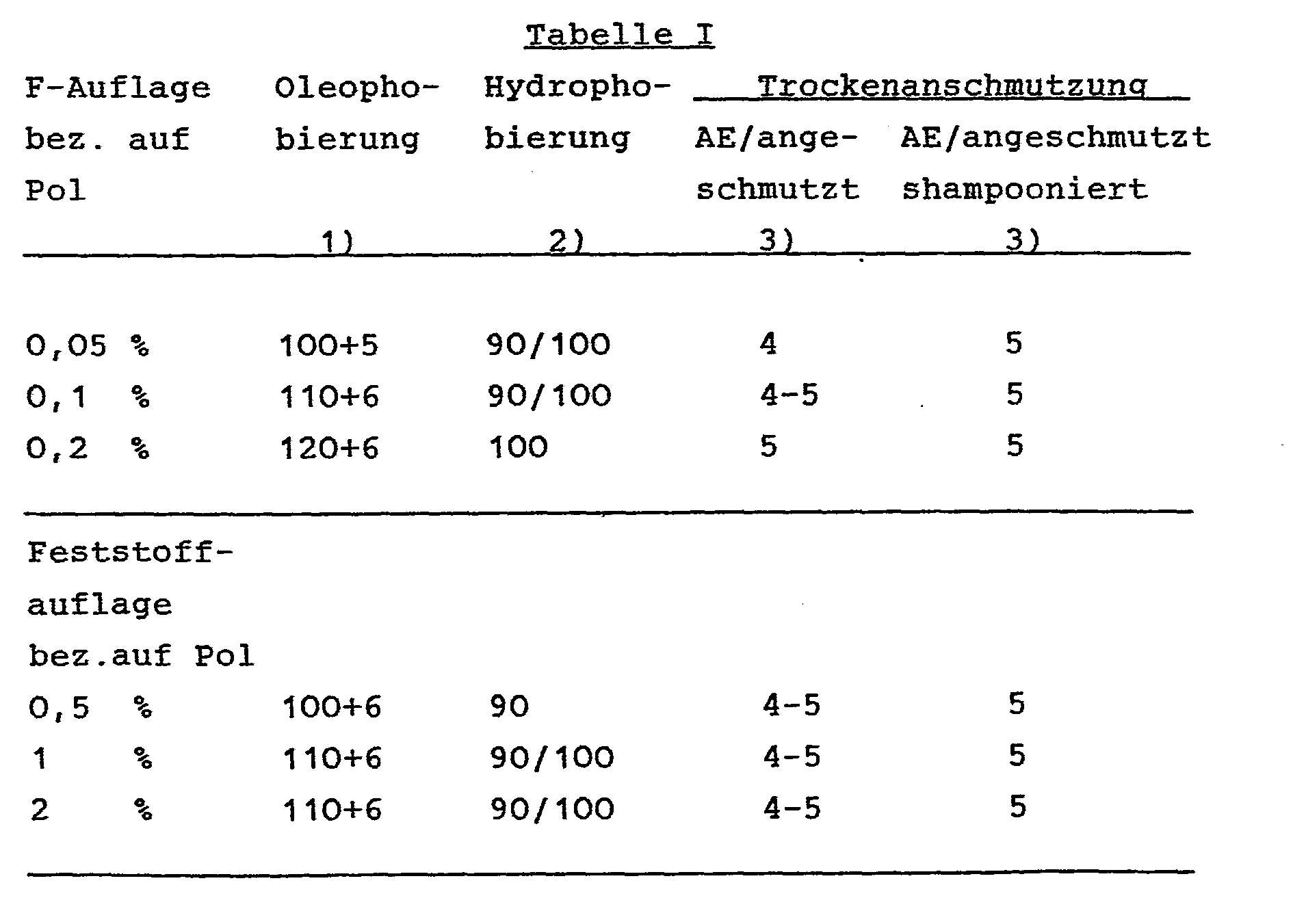
-
- 1. Oleophobierung: 140+8
- 2. Hydrophobierung: 100
- 3. Trockenanschmutzung: 5
- Die mit einem kationisch dispergierten Handelsprodukt erzielten Vergleichsergebnisse sind in Tabelle II angegeben:

Claims (10)





Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AT86105191T ATE65809T1 (de) | 1985-04-20 | 1986-04-15 | Waessrige anionische dispersion. |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE3514373 | 1985-04-20 | ||
| DE19853514373 DE3514373A1 (de) | 1985-04-20 | 1985-04-20 | Waessrige anionische dispersion |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| EP0202471A2 true EP0202471A2 (de) | 1986-11-26 |
| EP0202471A3 EP0202471A3 (en) | 1988-10-12 |
| EP0202471B1 EP0202471B1 (de) | 1991-07-31 |
Family
ID=6268725
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP86105191A Expired - Lifetime EP0202471B1 (de) | 1985-04-20 | 1986-04-15 | Wässrige anionische Dispersion |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US4775488A (de) |
| EP (1) | EP0202471B1 (de) |
| JP (1) | JPH06104192B2 (de) |
| KR (1) | KR860008334A (de) |
| AT (1) | ATE65809T1 (de) |
| AU (1) | AU582229B2 (de) |
| BR (1) | BR8601773A (de) |
| DD (1) | DD248383A5 (de) |
| DE (2) | DE3514373A1 (de) |
| ES (1) | ES8802404A1 (de) |
| NZ (1) | NZ215882A (de) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0213580B1 (de) * | 1985-08-30 | 1990-07-18 | Hoechst Aktiengesellschaft | Perfluoralkyl- und Epichlorhydrin-Gruppen enthaltende Urethane, diese Urethane enthaltende wässrige Dispersionen und ihre Verwendung |
| US5242487A (en) * | 1988-11-11 | 1993-09-07 | Daikin Industries Ltd. | Water- and oil-repellant composition |
| KR20200058622A (ko) | 2018-11-19 | 2020-05-28 | 삼성디스플레이 주식회사 | 다결정 실리콘층의 제조 방법, 표시 장치 및 표시 장치의 제조 방법 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3171861A (en) * | 1957-06-11 | 1965-03-02 | Minnesota Mining & Mfg | Fluorinated aliphatic alcohols |
| DE2115140A1 (de) * | 1971-03-29 | 1972-10-05 |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3993380A (en) * | 1976-01-21 | 1976-11-23 | Lawrence Peska Associates, Inc. | Combined extension cord and grounding device |
| IL59368A (en) * | 1979-02-16 | 1983-03-31 | Fluorocoat Ltd | Fluorocarbon resin compositions and method for coating and coated products thus produced |
| JPS5994621A (ja) * | 1982-11-12 | 1984-05-31 | Unitika Ltd | 防汚性繊維の製造法 |
-
1985
- 1985-04-20 DE DE19853514373 patent/DE3514373A1/de not_active Withdrawn
-
1986
- 1986-04-11 US US06/850,592 patent/US4775488A/en not_active Expired - Fee Related
- 1986-04-15 AT AT86105191T patent/ATE65809T1/de not_active IP Right Cessation
- 1986-04-15 DE DE8686105191T patent/DE3680580D1/de not_active Expired - Fee Related
- 1986-04-15 EP EP86105191A patent/EP0202471B1/de not_active Expired - Lifetime
- 1986-04-18 ES ES554146A patent/ES8802404A1/es not_active Expired
- 1986-04-18 JP JP61088441A patent/JPH06104192B2/ja not_active Expired - Lifetime
- 1986-04-18 AU AU56389/86A patent/AU582229B2/en not_active Ceased
- 1986-04-18 NZ NZ215882A patent/NZ215882A/xx unknown
- 1986-04-18 DD DD86289369A patent/DD248383A5/de unknown
- 1986-04-18 BR BR8601773A patent/BR8601773A/pt unknown
- 1986-04-19 KR KR1019860003042A patent/KR860008334A/ko not_active Application Discontinuation
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3171861A (en) * | 1957-06-11 | 1965-03-02 | Minnesota Mining & Mfg | Fluorinated aliphatic alcohols |
| DE2115140A1 (de) * | 1971-03-29 | 1972-10-05 |
Non-Patent Citations (1)
| Title |
|---|
| PATENT ABSTRACTS OF JAPAN, Unexamined Applications, Sektion C, Band 8, Nr. 205, 19. September 1984 The Patent Office Japanese Government seite 106 C 243 * Kokai-Nr. 59-94 621 (Unitika K.K.) * * |
Also Published As
| Publication number | Publication date |
|---|---|
| KR860008334A (ko) | 1986-11-14 |
| AU5638986A (en) | 1986-10-23 |
| NZ215882A (en) | 1988-09-29 |
| DE3514373A1 (de) | 1986-10-23 |
| ATE65809T1 (de) | 1991-08-15 |
| ES554146A0 (es) | 1988-05-16 |
| ES8802404A1 (es) | 1988-05-16 |
| BR8601773A (pt) | 1986-12-23 |
| JPS61249538A (ja) | 1986-11-06 |
| EP0202471A3 (en) | 1988-10-12 |
| EP0202471B1 (de) | 1991-07-31 |
| JPH06104192B2 (ja) | 1994-12-21 |
| DD248383A5 (de) | 1987-08-05 |
| US4775488A (en) | 1988-10-04 |
| DE3680580D1 (de) | 1991-09-05 |
| AU582229B2 (en) | 1989-03-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE3002369C2 (de) | ||
| EP1485533B1 (de) | Zubereitungen auf basis von wasser und/oder organischen lösemitteln und deren anwendung als appretur auf flächengebilden | |
| DE10325094B4 (de) | Zubereitungen für die öl- und wasserabweisende Ausrüstung von Flächengebilden und deren Anwendung | |
| EP0074335B1 (de) | Lagerstabile Mottenschutzformulierungen | |
| CH657015A5 (de) | Motten- und kaeferschutzmittel. | |
| CH686211A5 (de) | Motten- und Koferschutzmittel. | |
| DE3722375A1 (de) | Fluor und polysiloxan enthaltende urethane, verfahren zu ihrer herstellung und ihre verwendung | |
| EP0304016A1 (de) | Methylolierte und gegebenenfalls veretherte Fluoralkylliganden enthaltende Urethane | |
| EP0213580B1 (de) | Perfluoralkyl- und Epichlorhydrin-Gruppen enthaltende Urethane, diese Urethane enthaltende wässrige Dispersionen und ihre Verwendung | |
| DE3622284A1 (de) | Urethane aus aliphatischen fluoralkoholen, isocyanaten und aromatischen verbindungen, verfahren zu ihrer herstellung und ihre verwendung | |
| EP0105030B1 (de) | Motten- und Käferschutzmittel | |
| EP0491248B1 (de) | Mittel zur Behandlung von Fasermaterialien | |
| DE2225934C3 (de) | Flammschutzmittel fur Textilien, Verfahren zu deren Herstellung und ihre Verwendung | |
| EP0202471B1 (de) | Wässrige anionische Dispersion | |
| DE2926790C2 (de) | ||
| EP0318431B1 (de) | Motten- und Käferschutzmittel | |
| EP0148730B1 (de) | Verfahren zum Schützen von Keratinmaterial vor dem Befall durch keratinfressende Insekten und neue Pyridyloxytrifluormethansulfonanilide | |
| DE2460142A1 (de) | Oel- und wasserabstossend machende zubereitung | |
| EP0023638A1 (de) | Staubfreie Farbstoffe oder Farbstoffzubereitungen | |
| DE2923217A1 (de) | Verfahren zum schuetzen von keratinischen materialien vor dem befall durch keratinfressende insekten | |
| DE3607773C2 (de) | Fluoralkylliganden enthaltende Polyurethane, Verfahren zu ihrer Herstellung und ihre Zubereitungen und ihre Verwendung | |
| DE2658862C3 (de) | Verwendung von Phosphorsäureestern als Faserpräparationsmittel | |
| EP0149423B1 (de) | Verfahren zum Schützen von Keratinmaterial vor dem Befall durch keratinfressende Insekten und neue Phenoxytrifluormethansulfonanilide | |
| DE1619182B2 (de) | Verfahren zum weichmachen von textilien | |
| DE1793357C3 (de) | Verfahren zur Verbesserung der Fleckenbefreiungs- und Fleckenabweisungseigenschaften von Textilien und anderen faserigen Substraten |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| AK | Designated contracting states |
Kind code of ref document: A2 Designated state(s): AT BE CH DE FR GB IT LI NL |
|
| PUAL | Search report despatched |
Free format text: ORIGINAL CODE: 0009013 |
|
| AK | Designated contracting states |
Kind code of ref document: A3 Designated state(s): AT BE CH DE FR GB IT LI NL |
|
| 17P | Request for examination filed |
Effective date: 19890224 |
|
| 17Q | First examination report despatched |
Effective date: 19900907 |
|
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| AK | Designated contracting states |
Kind code of ref document: B1 Designated state(s): AT BE CH DE FR GB IT LI NL |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: IT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRE;WARNING: LAPSES OF ITALIAN PATENTS WITH EFFECTIVE DATE BEFORE 2007 MAY HAVE OCCURRED AT ANY TIME BEFORE 2007. THE CORRECT EFFECTIVE DATE MAY BE DIFFERENT FROM THE ONE RECORDED.SCRIBED TIME-LIMIT Effective date: 19910731 Ref country code: NL Effective date: 19910731 |
|
| REF | Corresponds to: |
Ref document number: 65809 Country of ref document: AT Date of ref document: 19910815 Kind code of ref document: T |
|
| REF | Corresponds to: |
Ref document number: 3680580 Country of ref document: DE Date of ref document: 19910905 |
|
| GBT | Gb: translation of ep patent filed (gb section 77(6)(a)/1977) | ||
| ET | Fr: translation filed | ||
| NLV1 | Nl: lapsed or annulled due to failure to fulfill the requirements of art. 29p and 29m of the patents act | ||
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: AT Effective date: 19920415 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: CH Effective date: 19920430 Ref country code: LI Effective date: 19920430 |
|
| PLBE | No opposition filed within time limit |
Free format text: ORIGINAL CODE: 0009261 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: NO OPPOSITION FILED WITHIN TIME LIMIT |
|
| 26N | No opposition filed | ||
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: PL |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: GB Payment date: 19950301 Year of fee payment: 10 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: FR Payment date: 19950317 Year of fee payment: 10 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: DE Payment date: 19950524 Year of fee payment: 10 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: BE Payment date: 19950530 Year of fee payment: 10 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: GB Effective date: 19960415 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: BE Effective date: 19960430 |
|
| BERE | Be: lapsed |
Owner name: CASSELLA A.G. Effective date: 19960430 |
|
| GBPC | Gb: european patent ceased through non-payment of renewal fee |
Effective date: 19960415 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: FR Effective date: 19961227 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: DE Effective date: 19970101 |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: ST |