EP0016907B1 - Zubereitung zum Schrumpffestmachen von Wolle - Google Patents
Zubereitung zum Schrumpffestmachen von Wolle Download PDFInfo
- Publication number
- EP0016907B1 EP0016907B1 EP80100297A EP80100297A EP0016907B1 EP 0016907 B1 EP0016907 B1 EP 0016907B1 EP 80100297 A EP80100297 A EP 80100297A EP 80100297 A EP80100297 A EP 80100297A EP 0016907 B1 EP0016907 B1 EP 0016907B1
- Authority
- EP
- European Patent Office
- Prior art keywords
- mol
- radicals
- radical
- groups
- atoms
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired
Links
- 238000002360 preparation method Methods 0.000 title claims description 31
- 210000002268 wool Anatomy 0.000 title claims description 27
- -1 mercaptoalkyl radicals Chemical class 0.000 claims description 65
- 229920001296 polysiloxane Polymers 0.000 claims description 30
- 150000003254 radicals Chemical class 0.000 claims description 28
- 125000004432 carbon atom Chemical group C* 0.000 claims description 18
- 125000000129 anionic group Chemical group 0.000 claims description 17
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 12
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 11
- 125000004429 atom Chemical group 0.000 claims description 8
- 239000004215 Carbon black (E152) Substances 0.000 claims description 7
- 239000003995 emulsifying agent Substances 0.000 claims description 7
- 229930195733 hydrocarbon Natural products 0.000 claims description 7
- 150000005840 aryl radicals Chemical class 0.000 claims description 6
- 239000002253 acid Substances 0.000 claims description 5
- DCERHCFNWRGHLK-UHFFFAOYSA-N C[Si](C)C Chemical compound C[Si](C)C DCERHCFNWRGHLK-UHFFFAOYSA-N 0.000 claims description 4
- YZCKVEUIGOORGS-IGMARMGPSA-N Protium Chemical compound [1H] YZCKVEUIGOORGS-IGMARMGPSA-N 0.000 claims description 4
- 125000003545 alkoxy group Chemical group 0.000 claims description 4
- 239000000654 additive Substances 0.000 claims description 3
- 239000001257 hydrogen Substances 0.000 claims description 3
- 229910052739 hydrogen Inorganic materials 0.000 claims description 3
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 3
- 125000004430 oxygen atom Chemical group O* 0.000 claims description 3
- 239000003960 organic solvent Substances 0.000 claims description 2
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 claims 1
- 125000004433 nitrogen atom Chemical group N* 0.000 claims 1
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 15
- 238000003756 stirring Methods 0.000 description 15
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 12
- 239000000839 emulsion Substances 0.000 description 12
- 238000000034 method Methods 0.000 description 12
- 239000000203 mixture Substances 0.000 description 10
- 238000006243 chemical reaction Methods 0.000 description 8
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 7
- 125000000217 alkyl group Chemical group 0.000 description 7
- 239000007795 chemical reaction product Substances 0.000 description 7
- 235000021190 leftovers Nutrition 0.000 description 7
- 238000005406 washing Methods 0.000 description 7
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 6
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 6
- 150000001875 compounds Chemical class 0.000 description 6
- 125000005358 mercaptoalkyl group Chemical group 0.000 description 6
- HMMGMWAXVFQUOA-UHFFFAOYSA-N octamethylcyclotetrasiloxane Chemical compound C[Si]1(C)O[Si](C)(C)O[Si](C)(C)O[Si](C)(C)O1 HMMGMWAXVFQUOA-UHFFFAOYSA-N 0.000 description 6
- UUEWCQRISZBELL-UHFFFAOYSA-N 3-trimethoxysilylpropane-1-thiol Chemical compound CO[Si](OC)(OC)CCCS UUEWCQRISZBELL-UHFFFAOYSA-N 0.000 description 5
- 125000003118 aryl group Chemical group 0.000 description 5
- 239000004205 dimethyl polysiloxane Substances 0.000 description 5
- 239000012071 phase Substances 0.000 description 5
- 229920000435 poly(dimethylsiloxane) Polymers 0.000 description 5
- 239000002904 solvent Substances 0.000 description 5
- QAOWNCQODCNURD-UHFFFAOYSA-N sulfuric acid Substances OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 5
- 238000011282 treatment Methods 0.000 description 5
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 4
- KPUWHANPEXNPJT-UHFFFAOYSA-N disiloxane Chemical class [SiH3]O[SiH3] KPUWHANPEXNPJT-UHFFFAOYSA-N 0.000 description 4
- 239000000835 fiber Substances 0.000 description 4
- 239000000463 material Substances 0.000 description 4
- 150000004756 silanes Chemical class 0.000 description 4
- 229910052710 silicon Inorganic materials 0.000 description 4
- 125000001424 substituent group Chemical group 0.000 description 4
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- 150000002009 diols Chemical class 0.000 description 3
- 230000000694 effects Effects 0.000 description 3
- 239000004744 fabric Substances 0.000 description 3
- 238000002156 mixing Methods 0.000 description 3
- 229910052757 nitrogen Inorganic materials 0.000 description 3
- 229910052760 oxygen Inorganic materials 0.000 description 3
- 239000001301 oxygen Substances 0.000 description 3
- 239000000047 product Substances 0.000 description 3
- DCQBZYNUSLHVJC-UHFFFAOYSA-N 3-triethoxysilylpropane-1-thiol Chemical compound CCO[Si](OCC)(OCC)CCCS DCQBZYNUSLHVJC-UHFFFAOYSA-N 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- 102000011782 Keratins Human genes 0.000 description 2
- 108010076876 Keratins Proteins 0.000 description 2
- BLRPTPMANUNPDV-UHFFFAOYSA-N Silane Chemical compound [SiH4] BLRPTPMANUNPDV-UHFFFAOYSA-N 0.000 description 2
- 125000003277 amino group Chemical group 0.000 description 2
- 125000004103 aminoalkyl group Chemical group 0.000 description 2
- 239000008346 aqueous phase Substances 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- 239000003054 catalyst Substances 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 2
- 150000001282 organosilanes Chemical class 0.000 description 2
- 229920001521 polyalkylene glycol ether Polymers 0.000 description 2
- 238000011085 pressure filtration Methods 0.000 description 2
- 239000000376 reactant Substances 0.000 description 2
- 229910000077 silane Inorganic materials 0.000 description 2
- 239000010703 silicon Substances 0.000 description 2
- 238000001577 simple distillation Methods 0.000 description 2
- ILWRPSCZWQJDMK-UHFFFAOYSA-N triethylazanium;chloride Chemical compound Cl.CCN(CC)CC ILWRPSCZWQJDMK-UHFFFAOYSA-N 0.000 description 2
- GOJUJUVQIVIZAV-UHFFFAOYSA-N 2-amino-4,6-dichloropyrimidine-5-carbaldehyde Chemical group NC1=NC(Cl)=C(C=O)C(Cl)=N1 GOJUJUVQIVIZAV-UHFFFAOYSA-N 0.000 description 1
- WBIQQQGBSDOWNP-UHFFFAOYSA-N 2-dodecylbenzenesulfonic acid Chemical compound CCCCCCCCCCCCC1=CC=CC=C1S(O)(=O)=O WBIQQQGBSDOWNP-UHFFFAOYSA-N 0.000 description 1
- HXLAEGYMDGUSBD-UHFFFAOYSA-N 3-[diethoxy(methyl)silyl]propan-1-amine Chemical compound CCO[Si](C)(OCC)CCCN HXLAEGYMDGUSBD-UHFFFAOYSA-N 0.000 description 1
- MBNRBJNIYVXSQV-UHFFFAOYSA-N 3-[diethoxy(methyl)silyl]propane-1-thiol Chemical compound CCO[Si](C)(OCC)CCCS MBNRBJNIYVXSQV-UHFFFAOYSA-N 0.000 description 1
- KHLRJDNGHBXOSV-UHFFFAOYSA-N 5-trimethoxysilylpentane-1,3-diamine Chemical compound CO[Si](OC)(OC)CCC(N)CCN KHLRJDNGHBXOSV-UHFFFAOYSA-N 0.000 description 1
- KWIUHFFTVRNATP-UHFFFAOYSA-N Betaine Natural products C[N+](C)(C)CC([O-])=O KWIUHFFTVRNATP-UHFFFAOYSA-N 0.000 description 1
- 239000005046 Chlorosilane Substances 0.000 description 1
- RUPBZQFQVRMKDG-UHFFFAOYSA-M Didecyldimethylammonium chloride Chemical compound [Cl-].CCCCCCCCCC[N+](C)(C)CCCCCCCCCC RUPBZQFQVRMKDG-UHFFFAOYSA-M 0.000 description 1
- MYMOFIZGZYHOMD-UHFFFAOYSA-N Dioxygen Chemical compound O=O MYMOFIZGZYHOMD-UHFFFAOYSA-N 0.000 description 1
- SNRUBQQJIBEYMU-UHFFFAOYSA-N Dodecane Natural products CCCCCCCCCCCC SNRUBQQJIBEYMU-UHFFFAOYSA-N 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- 240000009125 Myrtillocactus geometrizans Species 0.000 description 1
- KWIUHFFTVRNATP-UHFFFAOYSA-O N,N,N-trimethylglycinium Chemical compound C[N+](C)(C)CC(O)=O KWIUHFFTVRNATP-UHFFFAOYSA-O 0.000 description 1
- 239000004721 Polyphenylene oxide Substances 0.000 description 1
- 229910020175 SiOH Inorganic materials 0.000 description 1
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 1
- CIUQDSCDWFSTQR-UHFFFAOYSA-N [C]1=CC=CC=C1 Chemical compound [C]1=CC=CC=C1 CIUQDSCDWFSTQR-UHFFFAOYSA-N 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 238000010539 anionic addition polymerization reaction Methods 0.000 description 1
- 239000012736 aqueous medium Substances 0.000 description 1
- 229960003237 betaine Drugs 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 238000010538 cationic polymerization reaction Methods 0.000 description 1
- KOPOQZFJUQMUML-UHFFFAOYSA-N chlorosilane Chemical class Cl[SiH3] KOPOQZFJUQMUML-UHFFFAOYSA-N 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 238000007334 copolymerization reaction Methods 0.000 description 1
- 229960004670 didecyldimethylammonium chloride Drugs 0.000 description 1
- REZZEXDLIUJMMS-UHFFFAOYSA-M dimethyldioctadecylammonium chloride Chemical compound [Cl-].CCCCCCCCCCCCCCCCCC[N+](C)(C)CCCCCCCCCCCCCCCCCC REZZEXDLIUJMMS-UHFFFAOYSA-M 0.000 description 1
- 238000002845 discoloration Methods 0.000 description 1
- 125000003438 dodecyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 229940060296 dodecylbenzenesulfonic acid Drugs 0.000 description 1
- 238000007720 emulsion polymerization reaction Methods 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 239000002657 fibrous material Substances 0.000 description 1
- 239000010419 fine particle Substances 0.000 description 1
- 239000003063 flame retardant Substances 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 125000001183 hydrocarbyl group Chemical group 0.000 description 1
- 125000001841 imino group Chemical group [H]N=* 0.000 description 1
- 238000005470 impregnation Methods 0.000 description 1
- 239000002609 medium Substances 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- 150000003961 organosilicon compounds Chemical class 0.000 description 1
- 125000005375 organosiloxane group Chemical group 0.000 description 1
- 125000006353 oxyethylene group Chemical group 0.000 description 1
- 229920000570 polyether Polymers 0.000 description 1
- 239000002685 polymerization catalyst Substances 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 239000000344 soap Substances 0.000 description 1
- 239000007787 solid Substances 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 125000004434 sulfur atom Chemical group 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 150000003512 tertiary amines Chemical class 0.000 description 1
- 239000004753 textile Substances 0.000 description 1
Classifications
-
- D—TEXTILES; PAPER
- D06—TREATMENT OF TEXTILES OR THE LIKE; LAUNDERING; FLEXIBLE MATERIALS NOT OTHERWISE PROVIDED FOR
- D06M—TREATMENT, NOT PROVIDED FOR ELSEWHERE IN CLASS D06, OF FIBRES, THREADS, YARNS, FABRICS, FEATHERS OR FIBROUS GOODS MADE FROM SUCH MATERIALS
- D06M15/00—Treating fibres, threads, yarns, fabrics, or fibrous goods made from such materials, with macromolecular compounds; Such treatment combined with mechanical treatment
- D06M15/19—Treating fibres, threads, yarns, fabrics, or fibrous goods made from such materials, with macromolecular compounds; Such treatment combined with mechanical treatment with synthetic macromolecular compounds
- D06M15/37—Macromolecular compounds obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds
- D06M15/643—Macromolecular compounds obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds containing silicon in the main chain
- D06M15/647—Macromolecular compounds obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds containing silicon in the main chain containing polyether sequences
Definitions
- the invention relates to a preparation for shrink-proofing wool.
- GB-A-746 307 describes a method for preventing the shrinkage of the wool, the wool fibers being provided with certain organopolysiloxanes. A certain degree of shrink resistance is achieved in this way, but this effect is not washable.
- DE-A-2 242 297, DE-A-2 335 751 and DE-A-2 523 270 disclose processes for shrink-proofing keratin fibers by applying organopolysiloxane, an essential feature being that these compounds contain amino groups.
- the invention is therefore based on the object of finding a preparation for making wool shrink-proof, which contains both in emulsion form and in the form of an organic solution compounds which are suitable for making wool shrink-proof, these properties even after repeated washing of the wool in the customary manner Washing machines should be preserved. It is in particular an object of the invention to find a preparation which can be used optionally in the various stages of wool processing by means of different types of application. The aim is to ensure that the wool can also be treated after the wool has been dyed and that the known treatment methods, such as the pull-out method and the block method, can be used.
- the active ingredient contained in the preparation must not adversely affect the so-called feel of the wool or the knitted fabrics and fabrics made from the wool.
- the organopolysiloxane skeleton which is formed from the units a 1 ) and a 2 ), is a slightly to moderately branched siloxane.
- the units a 1 ) and a 2 ) are preferably statistically distributed in the siloxane molecule.
- these organopolysiloxanes can have 0.1 to 10 mol% of polyoxyalkylene radicals.
- wool-affine groups are bound to the organopolysiloxane skeleton. These wool-affine groups are mercaptoalkyl or mercaptoaryl residues, aminoalkyl residues or anionic groups bonded to Si atoms via C atoms. It is sufficient if, in addition to the polyoxyalkylene radicals, only one of the three types of wool-affinity groups shown, e.g. B. 0.03 to 3 mol% of mercaptoalkyl or mercaptoaryl radicals are included.
- aminoalkyl radicals are present in the molecule in amounts of 0.06 to 6 mol%.
- 0.1 to 10 mol% of anionic groups bonded to Si atoms via carbon atoms are present.
- these three different groupings can also exist side by side, e.g. B. mercaptoalkyl radicals together with aminoalkyl radicals or mercaptoalkyl radicals with anionic groups. You can also use all three different groupings be built into the molecule at the same time.
- R 1 and R 2 groups are methyl groups, of which, however, up to 10 mol% can be replaced by longer-chain alkyl or aryl radicals.
- alkyl or aryl radicals are the ethyl, propyl, dodecyl or phenyl radical.
- the polyoxyalkylene radical preferably corresponds to the formula in which or the trimethylsilyl radical and y has a value from 4 to 20.
- polyoxyalkylene block contains oxypropylene units in addition to oxyethylene units, polyoxyalkylene radicals with at least 40 mol% of polyoxyethylene units are preferred.
- R 1 and R 2 should consist of 0.1 to 10 mol% of polyoxyalkylene radicals, a range of 0.3 to 5 mol% is particularly preferred.
- Examples of mercaptoalkyl or mercaptoaryl residues attached to the polysiloxane skeleton are the mercaptomethyl, 2-mercaptoethyl, 3-mercaptopropyl, 3-mercaptoisobutyl or mercaptophenyl residue.
- the 3-mercaptopropyl radical is particularly preferred.
- mercaptoalkyl or mercaptoaryl residues are bound to the organopolysiloxane molecule, preferably 0.1 to 1 mol%.
- Suitable aminoalkyl radicals which are also preferably used, are the 3- (2-aminoethyl) aminopropyl or the 3-aminopropyl radical.
- suitable aminoalkyl radicals are the 4- (2-aminoethyl) aminobutyl radical or the 3- (2-aminoethyl) aminoisobutyl radical.
- aminoalkyl radicals form 0.06 to 6 mol% of the radicals R 1 and R 2 .
- the preferred range is 0.2 to 2 mol%.
- Anionic groups can also be bound to the organopolysiloxane molecule. These anionic groups are bound to the silicon atom by a divalent hydrocarbon radical.
- the carbon chain R 3 of the divalent hydrocarbon radical can be interrupted by oxygen, nitrogen or sulfur atoms.
- the groups are particularly suitable as anionic groups where m is 0 or 1 and X is an -O- radical or the group -OR 4 .
- R 4 is a hydrogen radical or an alkyl radical with 1 to 4 carbon atoms.
- Examples of such groups are 0.1 to 10 mol% of the groups R T and R 2 are formed by such anionic groups, but preferably 0.3 to 6 mol%.
- the preparations according to the invention can be produced in two different ways.
- the reaction can also be carried out in the form of cocondensation in an anhydrous medium.
- cyclic siloxanes can be used in a known manner (US Pat. No. 2,891,920), optionally from their mixture with organomodified di (tri) alkoxysilanes, such as, for. B. 3-mercaptopropyl-methyl-diethoxysilane, 3-aminopropyl-methyl-diethoxysilane or silanes of the formula the corresponding organomodified ⁇ , ⁇ -polydimethylsiloxane diols are prepared in the presence of anionic or cationic polymerization catalysts.
- organomodified di (tri) alkoxysilanes such as, for. B. 3-mercaptopropyl-methyl-diethoxysilane, 3-aminopropyl-methyl-diethoxysilane or silanes of the formula the corresponding organomodified ⁇ , ⁇ -polydimethylsiloxane diols are prepared in the presence of anionic or cationic polymerization catalysts.
- siloxane diols can then be reacted further with trialkoxysilanes modified or modified with polyalkylene glycol ether or with SH or amino groups and also with silanes, for example of the formula via which an anionic group can be introduced into the organopolysiloxane.
- the synthesis of the organomodified alkoxysilanes is known to the person skilled in the art.
- organopolysiloxanes in the non-aqueous phase, it is expedient to start from a sulfuric acid ester of an organosilicon alcohol of the formula out.
- the reaction product also contains terminally reactive SiOH groups which are capable of cocondensation with the alkoxy groups of the silanes modified with polyalkylene glycol ether, optionally also with mercaptoalkyl.
- cocondensation which is preferably carried out at higher temperature in the presence of tertiary amines, removed the solvents by simple distillation.
- organopolysiloxanes prepared in this way can also be converted into aqueous preparations according to the invention by incorporating water, possibly in the presence of suitable emulsifiers.
- a suitable solvent such as methylene chloride
- the reactants are diluted with methylene chloride to a volume of 500 ml.
- reaction product After a two-hour stirring phase, the reaction product with 64.8 g (0.071 mol) of a silane of the formula and 7.97 g (0.033 mol HS-) of a slightly condensed partial hydrolyzate of 3-mercaptopropyltriethoxysilane with 15% by weight OH, heated to 60 ° C. and stirred for a further 2 hours.
- the partially condensed partial hydrolyzate was obtained by the reaction of 3-mercaptopropyltriethoxysilane with 2% hydrochloric acid in methylene chloride.
- the fine-particle preparation thus prepared contains an organopolysiloxane in which the radicals R 1 and R 2 are 1.29 mol% and 0.322 mol% consist of - (CH 2 ) 3 -SH residues; the remaining residues are methyl residues.
- the potassium hydroxide solution contained in the emulsion is neutralized by adding 12 g of a 10% acetic acid.
- a 95 ° C. hot emulsifier solution corresponding to Example 3 is added dropwise with vigorous stirring with 167 g (0.56 mol) of octamethylcyclotetrasiloxane and 15.95 g (0.03 mol) transferred. After the dropwise addition, stirring is continued for 1 h and 6.05 g (0.027 mol) of 3- (aminoethyl) aminopropyltrimethoxysilane are added. Following a further 30 minute stirring phase, the mixture is cooled to 40 ° C. and the emulsion is neutralized by adding 16 g of 10% acetic acid.
- an emulsifier solution is prepared, which, after heating to 95 ° C. with vigorous stirring, is added via a dropping funnel to 167 g (0.56 mol) of octamethylcyclotetrasiloxane added. After stirring for a further 60 min, 27.98 g (0.03 mol) are added dropwise and stir again for 30 min. The emulsion is then cooled to 40 ° C. and neutralized by adding 12 g of 10% acetic acid.
- the finely divided aqueous preparation contains an organopolysiloxane, the radicals R 'and R 2 of which are methyl radicals, of which, however, 0.66 mol% are replaced.
- the required amount of this mixture is dissolved in toluene and diluted to the desired application concentration.
- the unfinished sample showed a heavily matted surface.
- the handle of the treated samples was also significantly softer after washing than the handle of the untreated material after washing and corresponded to the handle before washing.
Landscapes
- Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Textile Engineering (AREA)
- Treatments For Attaching Organic Compounds To Fibrous Goods (AREA)
- Silicon Polymers (AREA)
Description
- Die Erfindung betrifft eine Zubereitung zum Schrumpffestmachen von Wolle.
- Es ist bekannt, daß Wolle in unbehandeltem Zustand beim Waschen in wäßriger Flotte schrumpft und verfilzt. Um dieser Verschrumpfung und Verfilzung entgegenzuwirken, hat man bereits chemische Behandlungen empfohlen, bei denen die Struktur der Wolle verändert wird oder Ausrüstungen verwendet werden, welche Harz enthalten, das sich auf der Oberfläche der Wollfaser niederschlägt und diese umhüllt. Durch beide Verfahren werden aber Produkte erhalten, deren sogenannter Griff vom Verbraucher als unangenehm empfunden wird.
- Es ist auch bereits empfohlen worden, das Schrumpfen von Wolle beim Waschen durch Behandeln mit Organosiliciumverbindungen zu vermindern. Derartige Verfahren sind in GB-A-594 901, GB-A-613267 und GB-A-629329 beschrieben. Entsprechend diesen Verfahren wird die Wolle mit bestimmten Silanen behandelt.
- In der GB-A-746 307 ist ein Verfahren zur Verhinderung der Schrumpfung der Wolle beschrieben, wobei die Wollfasern mit bestimmten Organopolysiloxanen ausgerüstet werden. Man erreicht hierdurch zwar einen gewissen Grad an Schrumpffestigkeit, jedoch ist dieser Effekt nicht waschfest.
- In einer Reihe weiterer Veröffentlichungen, z. B. DE-A-2 242 297, DE-A-2 335 751 und DE-A-2 523 270 sind Verfahren zum Schrumpffestmachen von Keratinfasern durch Aufbringen von Organopolysiloxan angegeben, wobei ein wesentliches Merkmal ein Gehalt dieser Verbindungen an Aminogruppen ist.
- So ist beispielsweise das Verfahren gemäß DE-A-2 242 297 dadurch gekennzeichnet, daß man als Organopolysiloxane solche mit Einheiten der allgemeinen Formel

- n einen Durchschnittswert von 1,9 bis 2,1 und
- R einen über eine Silicium-Kohlenstoff-Bindung an Silicium gebundenen organischen Rest, wobei 0,25 bis 50% der Substituenten R einwertige Reste mit weniger als 30 Kohlenstoffatome sind, die im Abstand von wenigstens 3 Kohlenstoffatomen von dem Siliciumatom wenigstens eine Iminogruppe und wenigstens eine primäre oder sekundäre Arrinogruppe -NX2 enthalten, worin X ein Wasserstoffatom, eine Alkylgruppe mit 1 bis 30 Kohlenstoffatomen oder eine Arylgruppe ist, und wobei die übrigen Substituenten R einwertige Kohlenwasserstoffreste, halogenierte Kohlenwasserstoffreste, Carboxyalkylreste oder Cyanalkylreste mit 1 bis 30 Kohlenstoffatomen sind, von denen mindestens 70% aus einwertigen Kohlenwasserstoffresten mit 1 bis 18 Kohlenstoffatomen bestehen.
- Aus der DE-A-2 335 751 ist ein Verfahren zur Behandlung der Keratinfase bekannt, welches dadurch gekennzeichnet ist, daß die Organopolysiloxanzusammensetzung das durch Vermischen von A) und B) erhaltene Produkt enthält, wobei
- A) ein Polydiorganosiloxan mit endständigen, an Siliciumatome gebundenen Hydroxygruppen und einem Molekulargewicht von mindestens 750, wobei mindestens 50% der organischen Substituenten des Polydiorganosiloxans Methylgruppen sind und wobei die weiteren Substituenten einwertige Kohlenwasserstoffgruppen mit 2 bis 30 Kohlenstoffatomen darstellen, ist und
- B) ein Organosilan der allgemeinen Fcrmel

- In dieser DE-A-S ist angegeben, daß man die beiden Mischungspartner umsetzen soll, wenn eine Auftragung aus einem wäßrigen Medium angestrebt wird. Es ist jedoch nicht möglich, wie praktische Versuche gezeigt haben, aus derartigen Umsetzungsprodukten stabile wäßrige Emulsionen herzustellen. Es entstehen gelartige Reaktionsprodukte, die nicht in Emulsionsform überführt werden können. Sie sind deshalb zum Schrumpffestmachen von Wolle ungeeignet.
- Verwendet man diese Umsetzungsprodukte in Form organischer Lösungen, zeigt sich, daß der erzielbare Effekt für einen erfolgreichen Einsatz in der Praxis zu gering ist. Außerdem scheidet sich auch aus den Lösungsmittel enthaltenden Flotten durch die Einwirkung von Luftfeuchtigkeit nach einiger Zeit Siloxangel ab, welches die Vorrichtungen zum Behandeln der Wolle verstopft.
- In der Zeitschrift »Journal of the Textile Institute«, 68 (1977), 163 ff., sind Zubereitungen zur Schrumpffestausrüstung von Wolle beschrieben, welche Polysiloxane enthalten, bei denen Polyätherketten an das Molekül gebunden sind. Der beobachtete Effekt ist aber unzureichend. Außerdem sind die für eine Fixierung dieser Verbindungen auf der Wollfaser erforderlichen Temperaturen von 120°C und Einwirkungszeiten von 60 min so groß, daß bereits eine erhebliche Schädigung und Verfärbung der Wolle verursacht werden kann.
- Aus der DE-A-1 769 249 ist weiterhin ein Verfahren zur Behandlung von Fasermaterial, z. B. Wolle, bekannt, bei dem man in Emulsionsform Organosiloxane verwendet, welche Mercaptopropylgruppen enthalten. Mit diesen Verbindungen gelingt es jedoch lediglich, die Anschmutzbarkeit herabzusetzen. Die Verbindungen sind nicht geeignet, Wolle schrumpffest zu machen.
- Mit den Zubereitungen des Standes der Technik war es somit nicht möglich, eine einwandfreie und voll befriedigende Schrumpffestausrüstung von Wolle durchzuführen.
- Der Erfindung liegt somit die Aufgabe zugrunde, eine Zubereitung zum Schrumpffestmachen von Wolle aufzufinden, welche sowohl in Emulsionsform als auch in Form einer organischen Lösung Verbindungen enthält, welche geeignet sind, Wolle schrumpffest zu machen, wobei diese Eigenschaften auch nach mehrmaliger Wäsche der Wolle in üblichen Waschmaschinen erhalten bleiben sollen. Es ist insbesondere eine Aufgabe der Erfindung, eine Zubereitung aufzufinden, welche durch verschiedene Applikationsarten wahlweise in den verschiedenen Stufen der Wollverarbeitung eingesetzt werden kann. Dabei soll angestrebt werden, daß die Behandlung der Wolle auch nach dem Färben der Wolle erfolgen kann und daß die bekannten Behandlungsverfahren, wie das Auszieh- als auch das Klotzverfahren, angewendet werden können. Der in der Zubereitung enthaltene Wirkstoff darf dabei den sogenannten Griff der Wolle bzw. die aus der Wolle hergestellten Gewirke und Gewebe nicht nachteilig beeinflussen.
- Diese Eigenschaften sind bei der erfindungsgemäßen Zubereitung zu finden, die aus
- a) 1 bis 50 Gew.-% Organopolysiloxanen, welche aus
- ai) 90 bis 99,8 Mol-% Einheiten der Formel

- a2) 0,2 bis 10 Mol-% Einheiten der Formel
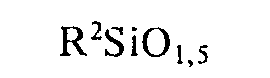
- wobei R1 und R2 einerseits aus
- 0,1 bis 10 Mol-% Polyoxyalkylenresten
- und andererseits aus
- 0,03 bis 3 Mol-% Mercaptoalkyl- oder Mercaptoarylresten und/oder
- 0,06 bis 6 Mol-% Aminoalkylresten und/oder
- 0,1 bis 10 Mol-% über C-Atome an Si-Atome gebundene anionische Gruppen gebildet sind,
- und der verbleibende Anteil an R1- und R2-Gruppen Methylgruppen sind, von denen jedoch bis zu 10 Mol-% durch längerkettige Alkyl-, Aryl- oder Wasserstoffreste ersetzt sein können und bis zu 5 Mol-% der an Si gebundenen Sauerstoffatome durch jeweils zwei niedere Alkoxy- oder Hydroxygruppen ersetzt sein können, und
- ai) 90 bis 99,8 Mol-% Einheiten der Formel
- b) 50 bis 99 Gew.-% Wasser, gegebenenfalls Emulgatoren und/oder organischen Lösungsmitteln und gegebenenfalls üblichen Zusatzstoffen,
- Übliche Zusatzstoffe sind z. B. Flammschutzmittel.
- Das Organopolysiloxangerüst, welches aus den Einheiten a1) und a2) gebildet ist, stellt ein gering bis mäßig verzweigtes Siloxan dar. Die Einheiten a1) und a2) sind dabei im Siloxanmclekül bevorzugt statistisch verteilt.
- Erfindungswesentlich ist nun, daß diese Organopolysiloxane 0,1 bis 10 Mol-% Polyoxyalkylenreste aufweisen können. Außerdem sind an das Organopolysiloxangerüst zusätzlich wollaffine Gruppen gebunden. Diese wollaffinen Gruppen sind Mercaptoalkyl- oder Mercaptoarylreste, Aminoalkylreste oder über C-Atome an Si-Atome gebundene anionische Gruppen. Es genügt, wenn neben den Polyoxyalkylenresten nur eine der drei gezeigten Arten wollaffiner Gruppen, z. B. 0,03 bis 3 Mol-% Mercaptoalkyl- oder Mercaptoarylreste, enthalten sind.
- Es ist deshalb auch ausreichend, wenn neben den Polyoxyalkylenresten Aminoalkylreste in Mengen von 0,06 bis 6 Mol-% im Molekül vorhanden sind. Eine weitere Möglichkeit besteht darin, daß neben den Polyoxyalkylenresten 0,1 bis 10 Mol-% über C-Atome an Si-Atome gebundene anionische Gruppen vorhanden sind. Jedoch können diese drei unterschiedlichen Gruppierungen auch nebeneinander vorliegen, z. B. Mercaptoalkylreste zusammen mit Aminoalkylresten oder Mercaptoalkylreste mit anionischen Gruppen. Es können auch alle drei unterschiedlichen Gruppierungen gleichzeitig im Molekül eingebaut sein.
- Die restlichen Rl- und R2-Gruppen sind Methylgruppen, von denen jedoch bis zu 10 Mol-% durch längerkettige Alkyl- oder Arylreste ersetzt sein können. Beispiele solcher Alkyl- oder Arylreste sind der Äthyl-, Propyl-, Dodecyl- oder Phenylrest.
- Durch die Kombination der an das Polysiloxangerüst gebundenen Polyoxyalkylenreste mit einer oder mehrerer der wollaffinen Gruppen gelingt es, die an die Zubereitung gestellten Anforderungen zu erfüllen.
- Vorzugsweise entsprechen die Polyoxyalkylenreste der Formel
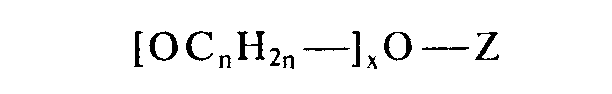
- Z ein Wasserstoffrest, ein einwertiger Alkyl- oder Arylrest, ein Acylrest oder ein Trimethylsilylrest ist,
- n einen Wert von 2 bis 3 und
- x einen Wert von 2 bis 80 hat.
- Vorzugsweise entspricht der Polyoxyalkylenrest der Formel


- Enthält der Polyoxyalkylenblock neben Oxyäthyleneinheiten auch Oxypropyleneinheiten, sind Polyoxyalkylenreste mit mindestens 40 Mol-% Polyoxyäthyleneinheiten bevorzugt.
- Innerhalb des angegebenen Bereiches, daß R1 und R2 zu 0,1 bis 10 Mol-% aus Polyoxyalkylenresten bestehen sollen, ist ein Bereich von 0,3 bis 5 Mol-% besonders bevorzugt.
- Beispiele für an das Polysiloxangerüst gebundene Mercaptoalkyl- oder Mercaptoarylreste sind der Mercaptomethyl-, 2-Mercaptoäthyl-, 3-Mercaptopropyl-, 3-Mercaptoisobutyl- oder Mercaptophenylrest. Besonders bevorzugt ist dabei der 3-Mercaptopropylrest.
- 0,03 bis 3 Mol-% dieser Mercaptoalkyl- oder Mercaptoarylreste sind an das Organopolysiloxanmolekül gebunden, vorzugsweise 0,1 bis 1 Mol-%.
- Beispiele geeigneter Aminoalkylreste, die auch bevorzugt verwendet werden, sind der 3-(2-Aminoäthyl-)aminopropyl- oder der 3-Aminopropylrest.
- Weitere Beispiele für geeignete Aminoalkylreste sind der 4-(2-Aminoäthyl-)aminobutylrest oder der 3-(2-Aminoäthyl-)aminoisobutylrest.
- Diese Aminoalkylreste bilden 0,06 bis 6 Mol-% der Reste R1 und R2. Der bevorzugte Bereich beträgt 0,2 bis 2 Mol-%.
- An das Organopolysiloxanmolekül können ferner anionische Gruppen gebunden sein. Diese anionischen Gruppen sind durch einen zweiwertigen Kohlenwasserstoffrest an das Siliciumatom gebunden. Die Kohlenstoffkette R3 des zweiwertigen Kohlenwasserstoffrestes kann durch Sauerstoff-, Stickstoff- oder Schwefelatome unterbrochen sein. Als anionische Gruppen kommen insbesondere in Frage die Gruppen

- Beispiele derartiger Gruppen sind

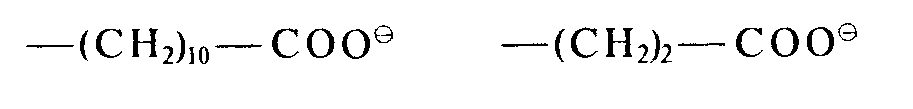

- Die erfindungsgemäßen Zubereitungen können prinzipiell auf zwei verschiedenen Wegen hergestellt werden. Zum einen besteht die Möglichkeit, das Organopolysiloxan durch eine Emulsionscopolymerisation der einzelnen Reaktionspartner in wäßriger Phase herzustellen, zum anderen kann die Umsetzung auch in Form einer Cokondensation in einem wasserfreien Medium erfolgen.
- Wählt man den Weg der Emulsionspolymerisation, können in bekannter Weise (US-A-2 891 920) aus cyclischen Siloxanen, gegebenenfalls aus deren Mischung mit organomodifizierten Di-(Tri)alkoxysilanen, wie z. B. 3-Mercaptopropyl-methyl-diäthoxysilan, 3-Aminopropyl-methyl-diäthoxysilan oder Silanen der Formel


- Bei der Herstellung der Organopolysiloxane in nichtwäßriger Phase geht man zweckmäßigerweise von einem Schwefelsäureester eines siliciumorganischen Alkohols der Formel

- Dieses Ausgangsprodukt, welches auch über eine potentielle anionische Gruppe verfügt, kann in Gegenwart eines Säureakzeptors, wie z. B. eines Amins, mit α,ω-Polydimethylsiloxandiolen, z. B. der Formel

umgesetzt werden. - Das Umsetzungsprodukt enthält neben den anionischen Gruppen noch endständige reaktive SiOH-Gruppen, die zur Cokondensation mit den Alkoxygruppen der mit Polyalkylenglykoläther, gegebenenfalls auch mit Mercaptoalkyl modifizierten Silane befähigt sind. Nach der Cokondensation, welche bevorzugt bei höherer Temperatur in Gegenwart tertiärer Amine durchgeführt wird, entfernt man die Lösungsmittel durch eine einfache Destillation.
- Neben diesem Verfahren, welchem die Umsetzung des beschriebenen Schwefelsäureesters mit Siloxandiolen zugrunde liegt, kann auch folgendermaßen verfahren werden:
- Man mischt α,ω-Polydimethylsiloxandioi mit Di-(Tri)alkoxysilanen, welche die anspruchsgemäßen funktionellen Gruppen enthalten und kondensiert in üblicher Weise z. B. durch Hitzeeinwirkung unter Stickstoffatmosphäre, gegebenenfalls nach Zusatz üblicher Katalysatoren wie Wasser, Organozinnverbindungen und/oder starke Säuren. Ersetzt man einen Teil der organofunktionellen Di-(Tri)alkoxysilane durch Di-(Tri)chlorsilane, erübrigt sich im allgemeinen ein Katalysatorzusatz ebenso wie bei Verwendung des zuerst beschriebenen sauren Schwefelsäureesters.
- Die so hergestellten Organopolysiloxane lassen sich durch Einarbeiten von Wasser, unter Umständen in Gegenwart geeigneter Emulgiermittel, auch in erfindungsgemäße wäßrige Zubereitungen überführen.
- Beispiele von in den erfindungsgemäßen Zubereitungen enthaltenen Organopolysiloxanen sind Verbindungen folgender Struktur




- p ist eine ganze Zahl von 5-1000
- q ist eine ganze Zahl von 1- 20
- r ist eine ganze Zahl von 1- 15
- In 200 ml eines geeigneten Lösungsmittels, wie Methylenchlorid, welche in einem Reaktionsgefäß vorgelegt werden, tropft man unter Rühren aus zwei Tropftrichtern gleichzeitig 29,2 g (0,135 Mol) eines Schwefelsäureesters eines siliciumorganichen Alkohols der Formel

- Nach einer zweistündigen Nachrührphase wird das Reaktionsprodukt mit 64,8 g (0,071 Mol) eines Silans der Formel

- Das teilweise kondensierte partielle Hydrolysat wurde durch die Reaktion von 3-Mercaptopropyltri- äthoxysilan mit 2%iger Salzsäure in Methylenchlorid erhalten.
- Anschließend entfernt man das Lösungsmittel durch eine einfache Destillation. Das Umsetzungsprodukt, welches durch eine Druckfiltration vom Triäthylammoniumchlorid befreit wird, ist ein niedrigviskoses, leicht gelbliches Organopolysiloxan, dessen Reste R1 und R2 Methylreste sind, von denen jedoch
- 1.35 Mol-% durch

- 0,71 Mol% durch

- und 0,32 Mol-% durch

- In einem geeigneten Reaktionsgefäß werden 400 ml Methylenchlorid, 223,3 g (0,3 Mol) α,ω-Polydimethylsiloxandiol (p =9,6) sowie 61,1 g (0,6 Mol) Triäthylamin vorgelegt und unter Rühren über einen Tropftrichter mit 43,3 g (0,2 Mol) eines Schwefelsäureesters eines siliciumorganischen Alkohols der Formel

- Jetzt wird das Lösungsmittel schonend abdestilliert und das Umsetzungsprodukt durch eine Druckfiltration vom Triäthylammoniumchlorid befreit. Aus dem Organopolysiloxan, dessen Reste R1 und R2 zu
- 3,13 Mol-% aus

- zu 1,17 Mol-% aus
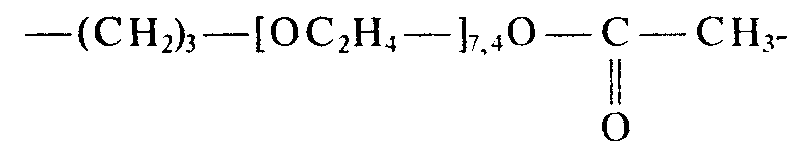
- und zu 0,32 Mol-% aus
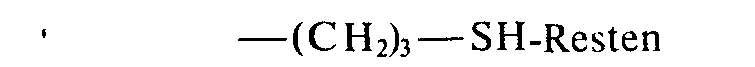
- In einem Reaktionsgefäß werden 480 g Wasser, 3,3 g Didecyldimethylammoniumchlorid, 1,7 g Dioctadecyldimethylammoniumchlorid, 3,5 g eines Betains der Formel
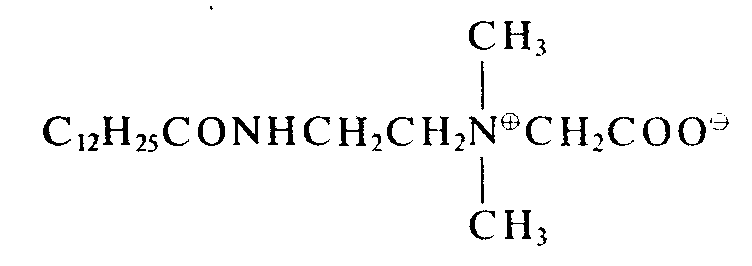
- Über einen Tropftrichter fügt man innerhalb von 45 min 136 g (0,459 Mol) Octamethylcyclotetrasiloxan und 34,4 g (0,0488 Mol)


- In eine dem Beispiel 3 entsprechende 95°C heiße Emulgatorlösung tropft man unter kräftigem Rühren 167 g (0,56 Mol) Octamethylcyclotetrasiloxan. Nach einer einstündigen Nachrührphase fügt man mit 30minütiger Unterbrechung 14,0 g (0,015 Mol)
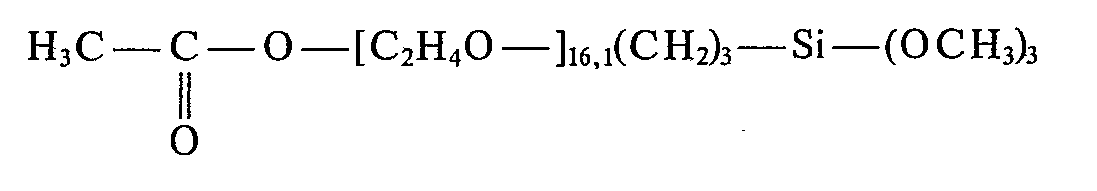
- Nach dem Abkühlen auf 40° C wird die Kalilauge, die in der Emulsion enthalten ist, durch die Zugabe von 12 g einer 10%igen Essigsäure neutralisiert.
- Die feinteilige wäßrige Zubereitung enthält ein Organopolysiloxan, dessen Reste Rl und R2 Methylreste sind, von denen jedoch
- 0,33 Mol-% durch


- Eine dem Beispiel 3 entsprechende 95°C heiße Emulgatorlösung wird unter kräftigem Rühren tropfenweise mit 167 g (0,56 Mol) Octamethylcyclotetrasiloxan und 15,95 g (0,03 Mol)

- Die feinteilige wäßrige Zubereitung enthält ein Organopolysiloxan, bei welchem die Reste R1 und R2 zu
- 0,65 Mol-% aus

- und zu 0,59 Mol-% aus

- Analog zu Beispiel 3 wird eine Emulgatorlösung zubereitet, welcher man nach dem Erhitzen auf 95° C unter kräftigem Rühren über einen Tropftrichter 167 g (0,56 Mol) Octamethylcyclotetrasiloxan hinzugefügt. Nachdem weitere 60 min gerührt wurde, tropft man 27,98 g (0,03 Mol)

- Die feinteilige wäßrige Zubereitung enthält ein Organopolysiloxan, dessen Reste R' und R2 Methylreste sind, von denen jedoch 0,66 Mol-% durch

- In einem Reaktionsgefäß werden 380 g Wasser und 3 g Dodecylbenzolsulfonsäure vorgelegt und unter Rühren auf 95° C erwärmt.
- Zur Herstellung der Organopolysiloxane a), bestehend aus den Einheiten a1) und a2), werden
- 167,00 g (0,56 Mol)
- 5,61 g (0,015 Mol)

- 27,98 g (0,03 Mol)

- 2,95 g (0,015 Mol) 3-Mercaptopropyltrimethoxysilan
- Die feinteilige wäßrige Zubereitung enthält ein Organopolysiloxan, bei welchem die Reste R1 und R2 zu
- 3,33 Mol-% aus

- zu 0,66 Mol-% aus

- und zu 0,33 Mol-% aus

- Es wurden folgende Verbindungen gemischt:

- Von dieser Mischung wird zur Herstellung der Imprägnierlösung die benötigte Menge in Toluol gelöst und auf die gewünschte Anwendungskonzentration verdünnt.
- Gewirktes Feinwollmaterial wird mit den in den Beispielen 1 bis 8 genannten Zubereitungen so behandelt, daß nach einfacher Trocknung des imprägnierten Gewebes bei 90° C eine Festkörperauflage von 1% resultiert. Die Trocknung kann aber bei den erfindungsgemäßen Beispielen 1 bis 5 und 7 auch bei Raumtemperatur erfolgen, da die in diesen Beispielen beschriebenen Zubereitungen Organopolysiloxane enthalten, welche schon bei dieser Temperatur vollständig aushärten. Nach einer 24stündigen Lagerung bei 20°C wurden das ausgerüstete sowie unbehandelte Material in einer Haushaltswaschmaschine bei 40° C unter Zusatz von 5 g/I Perox-Nadelseife und 2 g/I Soda gewaschen und zwischen den Wäschen im Tumbler getrocknet. Nach zwanzig Waschgängen von je 20 min wurde der Flächenfilzschrumpf ermittelt. Die Berechnung des Flächenfilzschrumofes erfolgte nach der folgenden Formel

- % L = prozentualer Längenschrumpf
- % B = prozentualer Breitenschrumpf
- Es wurden folgende Werte bestimmt:

- Im Gegensatz zu den behandelten Materialien zeigte die nicht ausgerüstete Probe eine stark verfilzte Oberfläche. Der Griff der behandelten Proben war außerdem nach der Wäsche wesentlich weicher als der Griff des unbehandelten Materials nach der Wäsche und entsprach dem Griff vor der Wäsche.
besteht.
Octamethylcyclotetrasiloxan,
über einen Tropftrichter der Emulgatorlösung zugefünt. Nach erfolgter Zugabe der verschiedenen Komponenten wird jeweils 60 min lang kräftig gerührt. Danach kühlt man die Emulsion auf 40° C ab und neutralisiert die erhaltene Säure durch den Zusatz von 11 g einer 1molaren Kalilauge.
Claims (7)


Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB7910341 | 1979-03-23 | ||
| GB7910341 | 1979-03-23 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| EP0016907A2 EP0016907A2 (de) | 1980-10-15 |
| EP0016907A3 EP0016907A3 (en) | 1981-03-18 |
| EP0016907B1 true EP0016907B1 (de) | 1982-02-17 |
Family
ID=10504098
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP80100297A Expired EP0016907B1 (de) | 1979-03-23 | 1980-01-22 | Zubereitung zum Schrumpffestmachen von Wolle |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US4283191A (de) |
| EP (1) | EP0016907B1 (de) |
| JP (1) | JPS584114B2 (de) |
| AU (1) | AU529018B2 (de) |
| DE (1) | DE3060191D1 (de) |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE3034380A1 (de) * | 1980-09-12 | 1982-03-25 | Th. Goldschmidt Ag, 4300 Essen | Zubereitung zum schrumpffestmachen von wolle |
| JPS57171768A (en) * | 1981-04-15 | 1982-10-22 | Shinetsu Chem Ind Co | Fiber treating agent |
| DE3417912C1 (de) * | 1984-05-15 | 1985-07-25 | Goldschmidt Ag Th | Betaingruppen enthaltende Siloxane,deren Herstellung und Verwendung in kosmetischen Zubereitungen |
| DE3735086C1 (de) * | 1987-10-16 | 1989-02-02 | Goldschmidt Ag Th | Organopolysiloxane mit Buntesalzgruppen |
| US5269950A (en) * | 1989-06-05 | 1993-12-14 | Sanyo Chemical Industries, Ltd. | Textile treating compositions |
| US5336419A (en) * | 1990-06-06 | 1994-08-09 | The Procter & Gamble Company | Silicone gel for ease of ironing and better looking garments after ironing |
| DE4100703A1 (de) * | 1991-01-11 | 1992-07-16 | Chu Tjoei Ho | Textilausruestungsmittel |
| US5532023A (en) * | 1994-11-10 | 1996-07-02 | The Procter & Gamble Company | Wrinkle reducing composition |
| WO1996015310A2 (en) * | 1994-11-10 | 1996-05-23 | The Procter & Gamble Company | Wrinkle reducing composition |
| US20070190872A1 (en) * | 2006-02-16 | 2007-08-16 | Weber Robert F | Fire retardant silicone textile coating |
| JP6234933B2 (ja) * | 2011-11-04 | 2017-11-22 | ダウ コーニング コーポレーションDow Corning Corporation | 親水性オルガノシラン |
Family Cites Families (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB594901A (en) | 1945-04-11 | 1947-11-21 | Wolsey Ltd | Improvements relating to the treatment of materials consisting wholly or partly of wool |
| GB613267A (en) | 1946-06-14 | 1948-11-24 | Wolsey Ltd | Improvements relating to the shrinkproofing of materials consisting wholly or partlyof wool |
| GB629329A (en) | 1947-05-05 | 1949-09-16 | Wolsey Ltd | Improvements relating to the shrinkproofing of materials consisting wholly or partlyof wool |
| US2728692A (en) | 1953-04-20 | 1955-12-27 | Dow Corning | Method of preventing shrinkage of wool |
| US2891290A (en) * | 1957-07-16 | 1959-06-23 | Gen Bronze Corp | Window scupper arrangement |
| BE618428A (de) * | 1961-06-01 | |||
| NL133796C (de) * | 1965-01-21 | 1900-01-01 | ||
| DE1594953A1 (de) * | 1966-05-28 | 1969-07-03 | Rotta Chem Fab Theodor | Verfahren zur Ausruestung von Fasergut mit Polysiloxanen |
| GB1431321A (en) * | 1973-01-30 | 1976-04-07 | Ici Ltd | Surface treating compositions |
| CH615968D (de) * | 1967-04-26 | 1900-01-01 | ||
| GB1396509A (en) * | 1971-08-28 | 1975-06-04 | Dow Corning Ltd | Treatment of keratinous fibres |
| US3876459A (en) * | 1973-06-29 | 1975-04-08 | Dow Corning | Treatment of fibres |
| GB1502265A (en) * | 1974-05-28 | 1978-03-01 | Dow Corning Ltd | Treatment of fibres |
| AU1823676A (en) * | 1975-10-13 | 1978-04-06 | Commw Scient Ind Res Org | Treatment of wool with polyorganosiloxanes |
| GB1548224A (en) * | 1976-02-12 | 1979-07-04 | Goldschmidt Ag Th | Organosilicon compounds and textile fibre dressings which contain these compounds |
| GB1572397A (en) * | 1976-06-11 | 1980-07-30 | Dow Corning Ltd | Fibre treatment composition and process |
-
1980
- 1980-01-22 EP EP80100297A patent/EP0016907B1/de not_active Expired
- 1980-01-22 DE DE8080100297T patent/DE3060191D1/de not_active Expired
- 1980-03-19 US US06/131,781 patent/US4283191A/en not_active Expired - Lifetime
- 1980-03-21 AU AU56732/80A patent/AU529018B2/en not_active Ceased
- 1980-03-24 JP JP55036316A patent/JPS584114B2/ja not_active Expired
Also Published As
| Publication number | Publication date |
|---|---|
| AU5673280A (en) | 1980-09-25 |
| DE3060191D1 (en) | 1982-03-25 |
| EP0016907A3 (en) | 1981-03-18 |
| JPS584114B2 (ja) | 1983-01-25 |
| JPS55128076A (en) | 1980-10-03 |
| EP0016907A2 (de) | 1980-10-15 |
| US4283191A (en) | 1981-08-11 |
| AU529018B2 (en) | 1983-05-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE3236466C2 (de) | ||
| EP0057937B1 (de) | Verfahren zur Imprägnierung von organischen Fasern | |
| DE3539714A1 (de) | Organosiloxan-oxyalkylen-copolymer, verfahren zu seiner herstellung und dessen verwendung in beschichtungsmitteln | |
| DE69102552T2 (de) | Verfahren zur Behandlung von Fasermaterialien. | |
| DE2728597A1 (de) | Verfahren zur behandlung von cellulose- und synthesefasern | |
| EP0313867B1 (de) | Organopolysiloxane mit Buntesalzgruppen | |
| DE2726108C2 (de) | Verfahren zur Herstellung einer wäßrigen Zubereitung zum Schrumpffestmachen von Wolle | |
| DE69003009T2 (de) | Verfahren zur Behandlung von Fasermaterialien. | |
| DE4211256A1 (de) | Vernetzbare Zusammensetzung auf Aminosiliconbasis | |
| EP0016907B1 (de) | Zubereitung zum Schrumpffestmachen von Wolle | |
| EP1921203B1 (de) | Verfahren zur Behandlung von Füllfasern mit wässrigen Dispersionen von Organopolysiloxanen | |
| DE102007015372A1 (de) | Polysiloxan und Textilhilfsmittel enthaltend ein Polysiloxan | |
| DE3323881C2 (de) | Organopolysiloxane mit Buntesalzgruppen, deren Herstellung und Verwendung zur Oberflächenbehandlung von anorganischen oder organischen Materialien | |
| EP0692567B1 (de) | Aminofunktionelle Organopolysiloxane | |
| EP0738747A1 (de) | Wässrige Dispersionen von Organopolysiloxanen | |
| DE69823711T2 (de) | Verfahren zur Stabilisierung von Siloxanpolymeren | |
| DE2903334C2 (de) | Zubereitung zum Schrumpffestmachen von Wolle | |
| DE1947268A1 (de) | Siloxan-Polyoxyalkylen-Blockcopolymerisate | |
| EP1914261B1 (de) | Neue phosphatierte Organosiloxanylderivate und deren Verwendung zur Behandlung von Leder | |
| DE2844952C2 (de) | Zubereitung zum Schrumpffestmachen von Wolle und Verfahren zur Herstellung der Zubereitung | |
| DE2523270C3 (de) | Verfahren zur Behandlung von Keratinfasern | |
| DE2942786C2 (de) | ||
| DE2365977A1 (de) | Organopolysiloxanzusammensetzung | |
| EP1076078A1 (de) | Silikonhaltige Zusammensetzungen für die Behandlung von Wollematerialien | |
| EP0047922B1 (de) | Zubereitung zum Schrumpffestmachen von Wolle |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| AK | Designated contracting states |
Designated state(s): BE DE FR GB IT NL |
|
| PUAL | Search report despatched |
Free format text: ORIGINAL CODE: 0009013 |
|
| AK | Designated contracting states |
Designated state(s): BE DE FR GB IT NL |
|
| 17P | Request for examination filed |
Effective date: 19810204 |
|
| ITF | It: translation for a ep patent filed | ||
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| AK | Designated contracting states |
Designated state(s): BE DE FR GB IT NL |
|
| REF | Corresponds to: |
Ref document number: 3060191 Country of ref document: DE Date of ref document: 19820325 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: FR Payment date: 19840116 Year of fee payment: 5 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: BE Payment date: 19840331 Year of fee payment: 5 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: DE Payment date: 19850205 Year of fee payment: 6 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: NL Payment date: 19860131 Year of fee payment: 7 |
|
| BERE | Be: lapsed |
Owner name: TH. GOLDSCHMIDT A.G. Effective date: 19870131 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: NL Effective date: 19870801 |
|
| GBPC | Gb: european patent ceased through non-payment of renewal fee | ||
| NLV4 | Nl: lapsed or anulled due to non-payment of the annual fee | ||
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: FR Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 19870930 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: DE Effective date: 19871001 |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: ST |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: GB Effective date: 19881118 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: BE Effective date: 19890131 |
|
| PLBE | No opposition filed within time limit |
Free format text: ORIGINAL CODE: 0009261 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: NO OPPOSITION FILED WITHIN TIME LIMIT |