-
1. Stand der Technik
-
1.1 Technisches Gebiet
-
Allgemein
betrifft die vorliegende Erfindung Expressionsplasmide, die durch
ein Plasmiderhaltungssystem (wie es hierin definiert wird) stabilisiert
werden und ein Protein oder Peptid exprimieren, wie beispielsweise
ein Antigen zur Verwendung als Vektor-Lebendimpfstoff, sowie Verfahren zum
Herstellen und Verwenden der stabilisierten Plasmide. Die Erfindung
optimiert die Stabilisierung (maintenance) von Expressionsplasmiden
auf zwei voneinander unabhängigen
Ebenen, da: (1) die alleinige Abhängigkeit von katalytischen,
balancierten letalen Erhaltungssystemen aufgehoben wird; und (2)
ein Plasmid-Partitionierungssystem eingeschleust wird, das eine
zufällige
Trennung von Expressionsplasmiden verhindert und dadurch die Vererbbarkeit
und Stabilität
erhöht.
-
Das
U.S. Patent Nr. 4,760,022 (Molin
et al.) beschreibt einen Expressionsvektor, der die Elemente parA
und parB umfasst, die beide als Partitionierungsfunktionen gelten,
da sie eine geordnete Aufteilung der Plasmidmoleküle bei der
Zellteilung ermöglichen.
-
Kim
et al. (1996) J. Fermentation and Bioengineering, 82, 495-497 beschreiben
eine Reihe von Plasmiden, die einen par-Locus aus dem Plasmid pSC101
umfassen. Diese Plasmide werden stabil gehalten, wenn die Transkription
von dem PL-Promotor unterdrückt wird,
sie gehen jedoch schnell verloren, wenn die Transkription aktiviert
wird.
-
1.2.1 Bakterielle Vektor-Lebendimpfstoffe.
-
Bakterielle
Vektor-Lebendimpfstoffe übertragen
Antigene auf das Immunsystem eines Wirts, wobei die Antigene aus
dem genetischen Material exprimiert werden, das in einem bakteriellen
Lebendvektor enthalten ist. Das genetische Material ist üblicherweise
ein Replikon, wie beispielsweise ein Plasmid. Die Antigene können eine
breite Vielfalt an Proteinen und/oder Peptiden bakteriellen, viralen,
parasitären
oder anderen Ursprungs einschließen.
-
Unter
den derzeit untersuchten bakteriellen Lebendvektoren befinden sich
attenuierte Darmpathogene (z.B. Salmonella typhi, Shigella, Vibrio
cholerae), Komensale (z.B. Lactobacillus, Streptococcus gordonii)
und zugelassene Impfstoffstämme
(z.B. BCG). S. typhi ist ein zum Impfen von Menschen besonders interessanter Stamm.
-
1.2.2 Attenuierte Salmonella typhi als
Lebendvektorstamm
-
S.
typhi ist ein gut-tolerierter Lebendvektor, der verschiedene, nicht
miteinander verwandte immunogene Antigene auf das humane Immunsystem übertragen
kann. Es wurde gezeigt, dass S. typhi-Lebendvektoren Antikörper und
eine zelluläre
Immunantwort auf ein exprimiertes Antigen hervorrufen. Beispiele
für Antigene,
die erfolgreich von S. typhi übertragen
wurden, schließen
das nicht-toxigene, jedoch stark immunogene C-Fragment des Tetanus-Toxins
und Malaria-Circumsporozoiten-Protein aus Plasmodium falciparum
ein.
-
S.
typhi ist gekennzeichnet durch einen enteralen Infektionsweg, eine
Eigenschaft, die eine orale Verabreichung des Impfstoffs erlaubt.
S. typhi infiziert auch Monozyten und Makrophagen und kann somit
Antigene gegen professionelle APCs richten.
-
Die
Expression eines Antigens durch S. typhi erfordert allgemein den
Einbau eines rekombinanten Plasmids, das für das Antigen codiert. Die
Stabilität
eines Plasmids ist folglich ein Schlüsselfaktor in der Entwicklung
hochqualitativer Impfstoffe aus S. typhi, die die Fähigkeit
besitzen, beständig
fremde Antigene zu exprimieren.
-
Mögliche Kandidaten
für attenuierte
S. typhi-Impfstoffe zur Verwendung in Menschen sollten wenigstens
zwei völlig
voneinander getrennte und genau definierte Mutationen aufweisen,
da die Wahrscheinlichkeit einer Reversion solcher Doppelmutanten
in vivo vernachlässigbar
gering ist. Der attenuierte Impfstoff-Kandidat S. typhi CVD908 besitzt
entsprechende Eigenschaften. CVD908 enthält zwei nicht-revertierende
Deletionsmutationen in den aroC- und aroD-Genen. Diese beiden Gene
codieren für
Enzyme, die für
den Biosyntheseweg entscheidend sind, der die Synthese von Chorismat,
dem Haupt-Precursor, der für
die Synthese der aromatischen Aminosäuren Phenylalanin, Tyrosin
und Tryptophan erforderlich ist, bewirkt. Ebenso ist Chorismat für die Synthese
von p-Aminobenzoesäure
erforderlich; nach Umwandlung desselben in Tetrahydrofolat wird
die p-Aminobenzoesäure
in die Purinnukleotide ATP und GTP umgewandelt.
-
1.2.3 Plasmid-Instabilität
-
Plasmidlose
Bakterienzellen neigen dazu, sich schneller anzuhäufen als
Plasmidtragende Zellen. Ein Grund für die erhöhte Anhäufungsgeschwindigkeit ist,
dass die Transkription und die Translation von Plasmidgenen den
Stoffwechsel belasten, was das Zellwachstum verlangsamt und plasmidlosen
Zellen einen Wettbewerbsvorteil verschafft. Zudem sind fremde Plasmidgene
mitunter für
die Wirtszelle toxisch.
-
Bei
attenuierten bakteriellen Vektor-Lebendimpfstoffen ist eine dauerhafte
Vererbbarkeit von Plasmiden erwünscht,
um dadurch eine erfolgreiche, fortdauernde Antigenproduktion sicherzustellen
sowie bei kommerziellem Betrieb von Bioreaktoren, zu verhindern,
dass der Bioreaktor von plasmidlosen Zellen übernommen wird.
-
Eine
dauerhafte Vererbbarkeit eines Plasmids erfordert generell, dass:
(1) sich das Plasmid einmal in jeder Generation replizieren muss,
(2) die Abweichungen der Kopienzahlen vor einer Zellteilung schnell
korrigiert werden müssen
und (3) die Produkte der Plasmid-Replikation
bei der Zellteilung auf beide Tochterzellen aufgeteilt werden müssen.
-
Obwohl
der Einbau der fremden Gene in die Chromosomen die Stabilität solcher
Sequenzen erhöht, können die
beteiligten genetischen Manipulationen schwierig sein und eine Abnahme
der Kopienzahl der heterologen Gene führt somit oftmals zu einer
Produktion von Mengen an heterologem Antigen, die nicht dazu ausreichen,
eine optimale Immunantwort zu gewährleisten. Der Einbau von heterologen
Genen in Vielkopie-Plasmide, die in einem Lebendvektor-Stamm gehalten
werden, stellt eine in der Natur vorkommende Lösung des Problems der Kopienzahl
dar; eine genetische Manipulation dieser Plasmide für eine kontrollierte
Expression der heterologen Gene ist einfach. Trotzdem können die
resultierenden Plasmide in vivo instabil werden und somit ein Verlust
dieser fremden Gene eintreten.
-
1.2.4 Plasmiderhaltungssysteme
-
In
der Natur werden bakterielle Plasmide oft stabilisiert, auch wenn
sie in der Regel mit sehr geringen Kopienzahlen vorhanden sind.
Eine stabile Vererbbarkeit von natürlich vorkommenden Plasmiden
mit geringer Kopienzahl kann von dem Vorhandensein bestimmter genetischer
Systeme, die aktiv das Auftreten von plasmidfreien Nachkommen verhindern, abhängen. Ein
aktueller Review über
Plasmiderhaltungssysteme ist bei Jensen et al., Molecular Microbiol.,
17:205-210, 1995 zu finden.
-
1.2.5 Antibiotikaresistenz
-
Ein
Mittel zur Erhaltung von Plasmiden ist, ein Antibiotikaresistenzgen
auf dem Plasmid bereitzustellen und die Zellen in einem mit dem
Antibiotikum angereicherten Medium anzuzüchten. Dieses Verfahren bringt jedoch
einige Schwierigkeiten mit sich. Der Antibiotikaresistenz-Ansatz
ist teuer, da die Verwendung teurer Antibiotika erforderlich ist
und, was vielleicht noch wichtiger ist, die Amerikanische Bundesbehörde zur Überwachung
von Nahrungs- und Arzneimitteln (U.S. Food and Drug Administration)
rät derzeit
von der Verwendung von Antibiotika in Verbindung mit der in vivo
Verabreichung von Vektor-Impfstoffen
ab.
-
Bei
großtechnischen
Produktionsansätzen
kann der Einsatz von Antibiotika anderen Einschränkungen unterliegen. Bei kommerziell
verwendeten Bioreaktoren kann das Antibiotikum durch die Mechanismen
der Antibiotikaresistenz abgebaut werden und so das Fortbestehen
einer erheblichen Population an plasmidlosen Zellen in der Kultur
ermöglichen.
Diese plasmidlosen Zellen sind unproduktiv und verringern den Ertrag
des Bioreaktors.
-
In
der Technik besteht daher ein Bedarf nach einem Plasmiderhaltungssystem,
das speziell für
eine Verwendung als Impfstoff aus bakteriellen Lebendvektoren ausgelegt
ist, nicht auf eine Antibiotikaresistenz angewiesen ist und das
vorzugsweise auch bei der Anwendung kommerziell verwendeter Bioreaktoren
von Nutzen ist.
-
1.2.6 Segregationale Plasmiderhaltungsfunktionen
-
Stabile
Plasmide mit geringer Kopienzahl benutzen üblicherweise eine Partitionierungsfunktion,
die aktiv Plasmidkopien zwischen den Tochterzellen aufteilt. Beispielhafte
Partitionierungsfunktionen umfassen, sind aber nicht beschränkt auf,
Systeme von pSC101, den F-Faktor, den P1-Prophagen und IncFII-Arzneimittelresistenzplasmide.
Diese Funktionen werden hierin als „SEG"-Funktionen bezeichnet.
-
1.2.7 Post-segregationale Killing-(PSK-)Funktionen
-
In
der Natur vorkommende PSK-Plasmiderhaltungsfunktionen benutzen üblicherweise
ein aus zwei Komponenten bestehendes Toxin-/Antitoxin-System und
funktionieren generell wie folgt: Das Plasmid codiert sowohl für ein Toxin
als auch für
ein Antitoxin. Die Antitoxine sind weniger stabil als die Toxine,
die dazu neigen, ziemlich stabil zu sein. In einer plasmidlosen
Tochterzelle werden keine Toxine und Antitoxine mehr produziert; die
weniger stabilen Antitoxine werden allerdings schnell abgebaut und
setzen dabei das Toxin frei, wodurch die Zelle getötet wird.
-
Allgemein
sind die Toxine kleine Proteine und die Antitoxine sind entweder
kleine Proteine (Proteinsysteme, wie beispielsweise phd-doc) oder
antisense-RNAs, die an die für
das Toxin codierenden mRNAs binden und so deren Synthese verhindern
(antisense-Systeme, wie beispielsweise hok-sok).
-
Die
später
in Abschnitt 1.2.7.3 diskutierten, balancierten Letalsysteme sind
ein Beispiel für
eine künstliche
PSK-Funktion.
-
1.2.7.1 Protein-Erhaltungssystem: Das
phd-doc-System
-
In
Protein-PSK-Funktionen werden sowohl das Toxin als auch das Antitoxin
aus Operons synthetisiert, in denen das für das Antitoxin codierende
Gen stromaufwärts
von dem für
das Toxin codierenden Gen liegt. Diese Operons autoregulieren die
Transskriptionsniveaus und die Synthese von codierten Proteinen
ist an die Translation gekoppelt. Das Antitoxin wird generell im Überschuss
synthetisiert, um sicherzustellen, dass die Wirkung des Toxins blockiert
ist. Die instabilen Antitoxine werden beständig durch von dem Wirt codierte
Proteasen abgebaut, was, um die Zelle zu schützen, eine kontinuierliche
Synthese des Antitoxins erfordert. Bei einem Verlust des Plasmids
werden keine Antitoxine mehr produziert und die vorhandenen Antitoxine
werden schnell abgebaut, wodurch das Toxin die Wirtszelle töten kann.
-
Das
phd-doc-System ist ein Beispiel für eine Protein-PSK-Funktion.
In der Natur tritt das phd-doc-System in dem temperenten Bakteriophagen
P1, der Escherichia coli lysiert, in Form eines ~ 100 kb großes Plasmid
auf. Der Erhaltungslocus codiert für zwei kleine Proteine: das
toxische, 126 Aminosäuren
lange Doc-Protein bewirkt über
einen unbekannten Mechanismus den Verlust „death an curing" des Plasmids und
das 73 Aminosäuren
lange Phd-Antitoxin verhindert den Tod des Wirts (prevents host
death), indem es vermutlich an Doc bindet und die Wirkung von Doc
blockiert.
-
Phd
und Doc werden von einem einzelnen Transkript codiert, bei dem das
ATG-Startcodon des
stromabwärts
gelegenen doc-Gens mit einer Base des TGA-Stoppcodons des stromaufwärts gelegenen
phd-Gens überlappt.
Die Expression dieser beiden Proteine ist daher an die Translation
gekoppelt, wobei mehr Phd als toxisches Doc-Protein synthetisiert
wird.
-
Daneben
wird die Transkription dieses Operons auf der Transkriptionsebene
von der Bindung eines Phd-Doc-Proteinkomplexes an die Stelle, die,
wenn die Konzentrationen der beiden Proteine einen kritischen Wert
erreichen, den Zugang der RNA-Polymerase an den Promotor des Operons
blockiert, autoreguliert. Obwohl Doc relativ resistent gegen einen
proteolytischen Angriff zu sein scheint, ist Phd für eine Spaltung äußerst anfällig. Der
PSK-Mechanismus
eines von einem Plasmid codierten phd-doc-Locus wird demzufolge
aktiviert, wenn Bakterien spontan das in ihnen befindliche Plasmid
verlieren, wodurch das Phd-Antitoxin
abgebaut und anschließend
das Zelltod verursachende Doc-Toxin aktiviert wird.
-
1.2.7.2 Antisense-Erhaltungssystem: Das
hok-sok-System
-
In
antisense-Stabilisierungssystemen sind die Antitoxine antisense-RNAs,
die die Translation von mRNAs, die für Toxine codieren, hemmen.
Wie die Antitoxinpeptide sind die antisense-RNAs weniger stabil
als die für
Toxine codierende mRNA. Ein Verlust des Plasmids lässt einen
Abbau der vorhandenen Antitoxine und damit die Synthese des Toxins,
das die Wirtszelle tötet,
zu.
-
Ein
Beispiel für
ein antisense-Stabilisierungssystem ist das hok-sok-System, das
von dem parB-Locus des Plasmids R1 codiert wird. Das System umfasst
drei Gene: hok, sok und mok.
-
Hok
ist ein membranassoziiertes Protein, das die Zellmembran irreversibel
schädigt
und dadurch Wirtszellen tötet.
Die Expression von Hok aus hok-mRNA verursacht einen Verlust des
Zellmembranpotentials, ein Aussetzen der Zellatmung, Veränderungen
der Zellmorphologie und den Tod der Zelle.
-
Das
sok-Gen codiert für
eine trans-agierende RNA, die die Translation der hok-mRNA blockiert
und dadurch verhindert, dass Hok Wirtszellen getötet werden. Die sok-RNA ist
weniger stabil als die hok-mRNA und wird von einem relativ schwachen
Promotor exprimiert (Gerdes et al., Annu. Rev. Genet., 31:1-31,
1997). Der Mechanismus, über
den die sok-RNA die Translation von Hok in Plasmid-tragenden Zellen
blockiert, konnte erst durch die Identifikation von mok (modulation
of killing), einem dritten Gen auf dem parB-Locus erkannt werden.
Der offene Leserahmen von mok überlappt
mit hok und ist für
die Expression und Regulation der hok-Translation erforderlich.
-
Die
sok-antisense-RNA bildet mit dem 5'-Ende der mok-hok-Message ein Duplex,
wodurch die Ribosomenbindungsstelle von mok für Ribosomen unzugänglich wird
und eine Spaltung mit RNase III und der Abbau der mRNA begünstigt werden.
Ohne Translation von mok wird hok von einer intakten Message nicht
exprimiert, auch wenn dessen eigene Ribosomenbindungsstelle nicht
direkt von der sok-RNA verdeckt wird.
-
Wenn
eine plasmidfreie Zelle gebildet wird, zerfällt die sok-RNA viel schneller
als die stabile mok-hok-Message. Wenn der durch sok gebotene Schutz
verloren wird, werden Mok und Hok translatiert und die Zelle stirbt.
-
Eine
Einschränkung
des hok-sok-Systems ist, dass eine signifikante Menge an plasmidlosen
Zellen entstehen kann, wenn das hok-sok-System durch Mutationen
im offenen Leserahmen von Hok inaktiviert wird.
-
1.2.7.3 Balancierte Letalsysteme
-
In
einem balancierten Letalsystem (eine PSK-Funktion) wird ein chromosomales
Gen, das für
ein essentielles Strukturprotein oder Enzym codiert, von dem Bakterienchromosom
deletiert oder so mutiert, dass das Gen nicht länger funktionieren kann. Das
entfernte oder beschädigte
Gen wird dann gegen ein Plasmid ausgetauscht, das ein vollständig funktionsfähiges Gen
umfasst. Ein Verlust des Plasmids führt zu einem Mangel an dem
essentiellen Protein und dem Tod der plasmidlosen Zelle.
-
Ein
balanciertes Letalsystem wurde erfolgreich in S. typhimurium verwendet,
das auf der Expression des für
Aspartat-β-semialdehyddehydrogenase
(Asd) codierenden asd-Gens basiert. Asd ist ein entscheidendes Enzym,
das an der Synthese von L-Asparagin-β- Semialdehyd, einem Vorläufer, der
für die
Synthese der Aminosäuren
L-Threonin (und L-Isoleucin),
sowie Diaminopimelinsäure,
einem entscheidenden strukturellen Bestandteil bei der Ausbildung
der Zellwand Gram-negativer Bakterien, essentiell ist, beteiligt
ist. Der Verlust von Plasmiden, die für Asd codieren, wäre für jedes
Bakterium, das nicht in der Lage ist, Asd aus dem Chromosom zu synthetisieren,
tödlich
und würde,
aufgrund des Unvermögens
des Bakteriums, die Peptidoglykanschicht in der Zellwand korrekt
aufbauen zu können,
zur Lyse des Bakteriums führen.
-
Das
asd-System (eine PSK-Funktion) wurde erfolgreich in attenuierten,
auf S. typhimurium basierenden Lebendvektorstämmen zur Immunisierung von
Mäusen
mit einer Vielzahl an prokaryotischen und eukaryotischen Antigenen,
einschließlich
solcher verschiedener Antigene, wie detoxifiziertem Fragment C von
Tetanus-Toxin und dem LT-Enterotoxin,
synthetischen viralen Peptiden von Hepatitis B und Gameten-spezifischen Antigenen,
wie beispielsweise dem Antigen SP 10 von humanem Sperma, durchgeführt.
-
Eine
Immunisierung von Mäusen
mit diesen Lebendvektorstämmen
in die Mucosa hat signifikante Immunreaktionen, an denen Reaktionen
von IgG und sekretorischem IgA beteiligt sind, an den Mucosaoberflächen ausgelöst.
-
Das
asd-System wurde kürzlich
in einem Versuch zur Erhöhung
der Stabilität
von synthetischen viralen Peptiden von Hepatitis B exprimierenden
Plasmiden in attenuierte Salmonella typhii-Impfstoffstämme eingeschleust.
Als die freiwilligen Patienten mit diesen Lebendvektorstämmen immunisiert
wurden, konnte jedoch keine Immunantwort auf das fremde Antigen
nachgewiesen werden.
-
Tatsächlich wurden
nach der Impfung von Menschen mit einem Lebendvektor von attentuiertem
S. typhi bis heute nur sehr wenige Berichte über eine Immunreaktion auf
die auf Plasmiden basierende Expression eines fremden Antigens aus
stabilisierten Plasmiden dokumentiert. In einem Bericht wurde der
Impfstoffstamm Ty21a für
Thymin autotroph gemacht, indem in Gegenwart von Trimethoprim eine
nicht-definierte Mutation in dem thyA-Gen, das für Thymidylatsynthetase codiert,
selektiert wurde.
-
Obwohl
in manchen Fällen
eine übermäßige Attenuierung
des Stammes selbst zu einer Fehlfunktion der Lebendvektorstämme führen kann,
scheint es möglich,
dass die derzeit für Plasmide
verwendeten Killing-Systeme weiteren Einschränkungen unterliegen. In Fällen, wo
die chromosomale Kopie des Gens eher inaktiviert als entfernt wurde,
kann, wenn der verwendete Bakterienstamm für eine Rekombination kompetent ist,
eine Wiederherstellung der chromosomalen Kopie durch homologe Rekombination
mit der aus dem Plasmid stammenden Genkopie erfolgen.
-
Balancierte
Letalsysteme, die auf der Produktion katalytischer Enzyme beruhen,
zeigen eine Reihe erheblicher Mängel.
Da für
eine Komplementierung der chromosomalen Gendeletion nur eine einzelne
Genkopie erforderlich ist, ist es an sich besonders schwierig, mehr
als ein paar Kopien eines Expressionsplasmids zu stabilisieren.
Der plasmidlose Wirtsstamm muss auf Spezialmedien angezüchtet werden,
um den vorhandenen Stoffwechselmangel auszugleichen.
-
Wenn
ein diffusionsfähiger
Wachstumsfaktor wachstumsbedingt ist, können die plasmidlosen Zellen zudem
auch von den Effekten einer „Kreuzfütterung" profitieren.
-
In
der Technik besteht daher der Bedarf nach einem Plasmiderhaltungssystem,
das nicht allein von einem balancierten Letalsystem abhängt und
insbesondere bei bakteriellen Vektor-Lebendimpfstoffen verwendet
werden kann.
-
2. Zusammenfassung
-
Gemäß einem
ersten Aspekt der vorliegenden Erfindung stellen wir einen Expressionsvektor
bereit, der eine Nukleotidsequenz umfasst, die codiert für:
einen
Replikationsursprung, der eine durchschnittliche Kopienzahl zwischen
2 und 75 verleiht, ausgewählt
aus der Gruppe bestehend aus: oriE1 (SEQ ID No. 1), ori101 (SEQ
ID No. 3), ori15A (SEQ ID No. 2) und Derivaten davon und an beiden
Enden von Transkriptionsterminatoren flankiert wird;
wenigstens
eine post-segregationale Killing-Funktion, die ausgewählt ist
aus der Gruppe bestehend aus asd, ssb, phd-doc, kis-kid und hok-sok,
und in der die Transkription flankierender Regionen von Loci, die
die wenigstens eine post-segregationale Funktion umgeben, divergent
ist und das Transkriptionsniveau des Wildtyps nicht in erheblichem
Maße stören wird;
wenigstens
eine Partitionierungsfunktion, die ausgewählt ist aus der Gruppe bestehend
aus dempar-Locus von pSC101 und dem parA von pR1, in der die Transkription
flankierender Regionen von der wenigstens einen Partitionierungsfunktion
aus fortschreitet; und
einen ompC-Promotor mit folgender Sequenz:
AGATCX1X2TAAX3CATCCACAGGAGGATATCTGATG
(SEQ ID No: 36), in der X1 ausgewählt ist
aus der Gruppe bestehend aus G, C und A; X2 ein
aus 1 bis 5 Nukleotiden bestehendes Insert ist; und X3 ausgewählt ist
aus der Gruppe bestehend aus A, T, G und C.
-
Entsprechend
einem zweiten Aspekt der vorliegenden Erfindung wird ein Expressionsvektor
bereitgestellt, der eine Nukleotidsequenz umfasst, die für einen
Expressionsvektor codiert, welcher eine Nukleotidsequenz umfasst,
die codiert für:
eine
Replikationsursprungskassette, die umfasst: eine Nukleotidsequenz,
die für
einen Replikationsursprung codiert, der eine durchschnittliche Kopienzahl
zwischen 2 und 75 verleiht, ausgewählt aus der Gruppe bestehend
aus: oriE1 (SEQ ID No. 1), ori101 (SEQ ID No. 3), ori15A (SEQ ID
No. 2) und Derivaten davon; einer ersten einzigartigen Stelle zur
Spaltung mit einem Restriktionsenzym, das 5' von der für den Replikationsursprung
codierenden Nukleotidsequenz angeordnet ist; und einer zweiten einzigartigen
Stelle zur Spaltung mit einem Restriktionsenzym, das 3' von der für den Replikationsursprung
codierenden Nukleotidsequenz angeordnet ist;
wenigstens eine
post-segregationale Killing-Kassette, die umfasst: eine Nukleotidsequenz,
die für
wenigstens eine post-segregationale Killing-Funktion codiert, die
ausgewählt
ist aus der Gruppe bestehend aus asd, phd-doc, kis-kid und hok-sok;
einer ersten einzigartigen Stelle zur. Spaltung mit einem Restriktionsenzym,
die 5' von der für die wenigstens
eine post-segregationale Killing-Funktion codierenden Nukleotidsequenz
angeordnet ist; und einer zweiten einzigartigen Stelle zur Spaltung
mit einem Restriktionsenzym, die 3' von der für die wenigstens eine post-segregationale
Killing-Funktion
codierenden Nukleotidsequenz angeordnet ist;
wenigstens eine
Partitionierungskassette, die umfasst: (i) eine Nukleotidsequenz,
die für
wenigstens eine Partitionierungsfunktion codiert, die ausgewählt ist
aus der Gruppe bestehend aus dem par-Locus von pSC101 und dem parA
von pR1; einer ersten einzigartigen Stelle zur Spaltung mit einem
Restriktionsenzym, das 5' von der
für die
wenigstens eine Partitionierungsfunktion codierenden Nukleotidsequenz
angeordnet ist; und einer zweiten einzigartigen Stelle zur Spaltung
mit einem Restriktionsenzym, die 3' von der für die wenigstens eine Partitionierungsfunktion
codierenden Nukleotidsequenz angeordnet ist; und
eine Expressionskassette,
die (i) eine Nukleotidsequenz, die für einen ompC-Promotor, der
die folgende Sequenz: AGATCX1X2TAAX3CATCCACAGGAGGATATCTGATG (SEQ ID No. 36)
umfasst, codiert, in der X1 ausgewählt ist
aus der Gruppe bestehend aus G, C und A; X2 ein
aus 1 bis 5 Nukleotiden bestehendes Insert ist; und X3 ausgewählt ist
aus der Gruppe bestehend aus A, T, G und C, (ii) eine erste einzigartige
Stelle zur Spaltung mit einem Restriktionsenzym, die 5' von der für den Promotor
codierenden Nukleotidsequenz angeordnet ist, und (iii) eine zweite
einzigartige Stelle zur Spaltung mit einem Restriktionsenzym, die
3' von der für den Promotor
codierenden Nukleotidsequenz angeordnet ist, umfasst.
-
Gemäß einem
dritten Aspekt der vorliegenden Erfindung stellen wir eine Zelle
bereit, die einen Expressionsvektor gemäß dem ersten oder zweiten Aspekt
der Erfindung umfasst.
-
Als
vierter Aspekt der vorliegenden Erfindung wird ein attenuierter
bakterieller Vektor-Lebendimpfstoff zur Verwendung in einem Verfahren
zum Auslösen
einer Immunantwort in einem Probanden bereitgestellt, wobei der
attenuierte bakterielle Vektor-Lebendimpfstoff
eine Zelle gemäß dem dritten
Aspekt der Erfindung umfasst.
-
Gemäß einem
fünften
Aspekt der vorliegenden Erfindung stellen wir ein Herstellungsverfahren
für einen
attenuierten bakteriellen Vektor-Lebendimpfstoff bereit, das das
Transformieren eines Bakterienstammes mit einem Expressionsvektor
gemäß dem ersten
oder zweiten Aspekt der Erfindung umfasst.
-
In
einem speziellen Aspekt wird ein stabilisiertes Expressionsplasmid
in einem Vektor-Lebendimpfstoff aus Salmonella typhi, wie beispielsweise
CVD908-htrA, verwendet.
-
Die
Stabilisierung von Expressionsplasmiden wird auf zwei voneinander
unabhängigen
Ebenen erreicht, da (1) die alleinige Abhängigkeit von katalytischen,
balancierten Letalstabilisierungssystemen aufgehoben wird; und (2)
ein Plasmid-Partitionierungssystem
eingeschleust wird, das eine zufällige
Segregation von Expressionsplasmiden verhindert und dadurch die
Vererbbarkeit und Stabilität
derselben erhöht.
Das stabilisierte Expressionsplasmid kann rekombinant so hergestellt
werden, dass es ein oder mehrere Antigene, vorzugsweise ein oder
mehrere Shiga-Toxin 2-(Stx 2-) Antigene oder wesentliche Homologe
davon, wie zum Beispiel Pentamere der Untereinheit von Shiga-Toxin oder ein genetisch
detoxifiziertes Stx 2, exprimiert.
-
Das
stabilisierte Expressionsplasmid umfasst vorzugsweise eine oder
mehrere nichtkatalytische Plasmiderhaltungsfunktionen.
-
Wir
beschreiben Expressionsplasmide, die ein Plasmiderhaltungssystem
umfassen, das wenigstens eine PFK-Funktion und wenigstens eine SFG-Funktion
umfasst. Das Plasmiderhaltungssystem kann beispielsweise ein aus
zwei Komponenten bestehendes Plasmiderhaltungssystem umfassen, das
eine PSK-Funktion und eine SEG-Funktion umfasst. Alternativ dazu
kann das Plasmiderhaltungssystem ein aus drei Komponenten bestehendes
Plasmiderhaltungssystem umfassen, das eine PSK-Funktion, eine SFG-Funktion
und eine weitere PSK umfasst. In einer bevorzugten Alternative umfasst
das Plasmiderhaltungssystem hok-sok + par + parA + phd-doc, wobei
jede der angegebenen Funktionen gegen ein wesentliches Homologon
davon ausgetauscht werden kann.
-
Die
Plasmiderhaltungssysteme können
in Vielkopie-Expressionsplasmide, die für eines oder mehrere Proteine
oder Peptide von Interesse codieren, eingeschlossen sein. Solche
Vielkopie-Expressionsplasmide erzeugen einen Gendosiseffekt, der
die Expressionsmenge des Proteins oder Peptids von Interesse verstärkt. Wenn
das Plasmiderhaltungssystem in einem bakteriellen Vektor-Lebendimpfstoff
verwendet werden soll, ist das Protein oder Peptid von Interesse
ein Antigen oder mehrere Antigene.
-
In
einem Aspekt ist das Expressionsplasmid ein Expressionsplasmid für einen
Impfstoff, das ein Plasmiderhaltungssystem und wenigstens ein Antigen,
zum Beispiel wenigstens ein Shiga-Toxin 2-(Stx 2-) Antigen und/oder
ein wesentliches Homologon davon, umfasst. Wenn das Antigen ein
Shiga-Toxin 2-Antigen ist, kann das Shiga-Toxin 2-Antigen zum Beispiel
entweder ein Pentamer der Untereinheit B oder ein genetisch detoxifiziertes
Stx 2 sein.
-
Wir
beschreiben ferner Expressionsplasmide, die ein Plasmiderhaltungssystem
umfassen, das das balancierte Letalsystem ssb einschließt und in
denen der ssb-Locus des bakteriellen Lebendvektors, der unter Verwenden
eines Suizidvektors, der einen temperatursensitiven Replikationsursprung
umfasst, inaktiviert wurde. In einem Aspekt wird der bakterielle
Lebendvektor S. typhi und der Suizidvektor zur Inaktivierung des ssb-Locus
von S- typhi verwendet. In einem Aspekt ist der Suizidvektor ein
Derivat von pSC101, das das hierin beschriebene sacB trägt.
-
Wir
beschreiben ferner ein Plasmiderhaltungssystem, in dem eine PSK-Funktion
enthalten ist, die ein stilles Plasmid-Addiktionssystem einschließt, das
auf antisense-RNA-Kontroilmechanismen
beruht, die Letalproteine erst dann synthetisieren, wenn ein Plasmidverlust
eingetreten ist.
-
Das
Expressionsplasmid kann eine Reihe von Expressionsplasmiden umfassen,
von denen jedes eine vollständige
genetische Kassette, die für
die regulierte Expression eines heterologen Antigens, eines Replikationsursprungs
und eines selektierbaren Markers zur Rückgewinnung des Plasmids codiert,
umfasst.
-
Das
Expressionsplasmid umfasst ein Plasmiderhaltungssystem, das eine
PSK-Funktion einschließt, die
auf dem ssb-Gen basiert. Mutierte Allele, wie zum Beispiel das hierin
beschriebene ssb-1, können
in die Expressionsplasmide eingebaut werden, um Plasmide mit höherer Kopienzahl
durch Überexpression
von SSB1-gleichen Proteinen zu verstärken, um so die erforderlichen
biologisch aktiven Tetramere von SSB zu bilden.
-
Das
Expressionsplasmid umfasst einen Promotor. Der Promotor ist ein
modifizierter ompC-Promotor mit der Sequenz SEQ ID No. 36. Der induzierbare
Promotor kann der hierin beschriebene, mutierte PompC1- oder
der PopmC3-Promotor sein.
-
Das
Expressionsplasmid kann einen Locus für Plasmidvererbbarkeit (oder – partitionierung);
einen Replikationsursprung, der so ausgewählt ist, dass eine Kopienzahl,
die ein gegebenes Antigen effektiv stabilisiert, bereitgestellt
wird; eine PSK-Funktion; und eine Nukleotidsequenz, die für ein Antigen
und einen Promotor, der letztlich die Translation des Antigens kontrolliert
und eine Stärke
besitzt, die zur Verbesserung der Antigenproduktion ohne Abtöten der
Zelle ausgewählt
ist, umfassen.
-
Wir
beschreiben auch ein Verfahren zur Verwendung des Expressionsplasmids,
welches das Transformieren einer Bakterienzelle unter Verwenden
des Expressionsplasmids und das Kultivieren der Bakteriellenzelle
zur Produktion des Proteins oder Peptids (z.B. des Antigens) und/oder
das Verabreichen der transformierten Zelle oder Zellkultur an einen
Probanden umfasst. Wenn die transformierten Bakterienzellen einem Probanden
verabreicht werden, werden sie in einer Menge verabreicht, die für das Auslösen einer
Immunreaktion, die dem Probanden eine Immunität gegen das Protein oder Peptid
verleiht, notwendig ist. Der Proband ist vorzugsweise ein Mensch,
kann jedoch auch ein anderes Lebewesen, wie beispielsweise ein Hund, Pferd-oder
Huhn, sein.
-
Das
Expressionsplasmid kann wenigstens 3 unabhängig voneinander funktionierende
Expressionskassetten umfassen, von denen eine Kassette für ein Protein
oder Peptid von Interesse codiert und die restlichen Kassetten jeweils
für ein
anderes Plasmiderhaltungssystem codieren.
-
Das
Expressionsplasmid kann für
(1) ein Testantigen, das funktionsfähig mit einem Promotor verknüpft ist,
und (2) ein Plasmiderhaltungssystem codieren.
-
Wir
beschreiben ferner eine Kassette zur regulierten Expression eines
Testantigens, die so funktioniert, dass, wenn die Induktion einer
Antigenexpression erhöht
wird, der Stoffwechsel des Bakteriums belastet wird, und dies phänotypisch
gesehen zu einer Instabilität
des Plasmids führt,
d.h. es wird ein selektiver Vorteil für alle diejenigen Bakterien
geschaffen, die das störende
Plasmid spontan verlieren können.
Das Testantigen kann das grüne
fluoreszierende Protein (GFPuv) sein. Die für das Testantigen codierende
Expressionskassette kann auch einen induzierbaren Promotor, beispielsweise
den ompC-Promotor,
umfassen, der so positioniert ist, dass der induzierbare Promotor
letztlich die Translation des Testantigens steuert.
-
Wir
beschreiben ein Verfahren zur Herstellung eines Expressionsplasmids,
das das Synthetisieren eines Expressionsplasmids umfasst, das wenigstens
3 unabhängig
voneinander funktionierende Expressionskassetten, von denen eine
Kassette für
ein Protein oder Peptid von Interesse codiert und die restlichen
Kassetten jeweils für
ein anderes Plasmiderhaltungssystem codieren, umfasst.
-
Ein
Verfahren zum Screenen von Plasmiderhaltungssystemen kann umfassen:
Bereitstellen
einer Expressionskassette, die für
ein Protein oder Peptid von Interesse codiert, und wenigstens zwei
weiterer Expressionskassetten, von denen jede für eine andere Plasmidstabilisierungsfunktion
codiert und diese in einem Lebendvektor eines bakteriellen Wirts
exprimieren kann; Einschleusen der drei Expressionskassetten in
ein einziges Expressionsplasmid; Transformieren eines bakteriellen
Lebendvektors mit dem einzigen Expressionsplasmid; Kultivieren des
transformierten, bakteriellen Lebendvektors und Bestimmen des Anteils
an in die Kultur eingeschleusten, plasmidlosen Zellen.
-
Wir
beschreiben einen attenuierten bakteriellen Vektor-Lebendimpfstoff,
der einen attenuierten bakteriellen Lebendvektor umfasst, der mit
einem stabilisierten Expressionsplasmid, das ein Plasmiderhaltungssystem,
vorzugsweise ein nicht-katalytisches Plasmiderhaltungssystem, umfasst,
transformiert wurde.
-
Wir
beschreiben auch einen attenuierten bakteriellen Vektor-Lebendimpfstoff,
der einen attenuierten bakteriellen Lebendvektor umfasst, der mit
einem Expressionsplasmid, das ein Plasmiderhaltungssystem umfasst,
das wenigstens ein PSK-System und wenigstens ein SEG-System einschließt, transformiert
wurde. Der attenuierte bakterielle Lebendvektor kann zum Beispiel
S. typhi CVD908-htrA sein.
-
Wir
beschreiben ferner ein Verfahren zum Impfen eines Probanden, welches
das Verabreichen einer Menge an bakteriellem Vektor-Lebendimpfstoff,
die zum Auslösen
einer verstärkten
Immunreaktion ausreicht, an den Probanden umfasst. Wir beschreiben
auch ein Verfahren zur Prophylaxe einer Erkrankung durch Impfen
eines Probanden unter Verwenden einer Menge eines solchen bakteriellen
Lebendvektors, die zum Auslösen
einer schützenden
Immunreaktion gegen eines oder mehrere Pathogene einer solchen Erkrankung
ausreicht. Der Proband ist vorzugsweise ein Mensch, kann jedoch
auch ein anderes Lebewesen, wie beispielsweise ein Pferd, eine Kuh
oder ein Schwein sein. Wir beschreiben zum Beispiel ein Verfahren
zur Prophylaxe von hämorrhagisch-urämischem
Syndrom (HUS), das von Shiga-Toxin
2-produzierenden enterohämorrhagischen
Escherichia coli verursacht wird, durch Verabreichen einer Menge
an bakteriellem Lebendvektor, der mit einem stabilisierten Plasmid,
das für
wenigstens ein Shiga-Toxin 2-Antigen codiert, transformiert wurde,
an einen Probanden.
-
Plasmiderhaltungssysteme
können
auf ihre Effizienz gescreent werden, wobei ein Verfahren verwendet
wird, das umfasst: das Bereitstellen von Expressionsplasmiden, die
die hierin beschriebenen Plasmiderhaltungssysteme umfassen und für ein Protein
oder Peptid von Interesse codieren, wobei die Expressionsplasmide
eine Kopienzahl besitzen, die von einer geringen Kopienzahl (z.B.
~ 5 Kopien pro Zelle) zu einer mittleren Kopienzahl (z.B. ~ 15 Kopien
pro Zelle) zu einer hohen Kopienzahl (z.B. ~ 60 Kopien pro Zelle)
variieren kann; Transformieren von bakteriellen Lebendvektoren mit
diesen Expressionsplasmiden; und Überprüfen des Anteils an eingeschleusten
plasmidlosen Zellen und/oder des Anteils an wachsenden, Plasmid-enthaltenden
Zellen. Die modifizierten Replikationsursprünge können Replikationsursprünge aus
den Plasmiden pSC101 (niedrige Kopienzahl), pACYC184 (mittlere Kopienzahl)
und pAT153 (hohe Kopienzahl) sein. Es können unabhängig voneinander funktionierende
Plasmidreplikationskassetten verwendet werden, die das Überprüfen der
Effizienz von einem oder mehreren Plasmiderhaltungssystemen, wenn
die Kopienzahl zunimmt, zulassen.
-
Wir
beschreiben stabilisierte Expressionsplasmide zur Verwendung in
Lebendvektoren aus attenuierten S. typhi, die einen selektierbaren
Marker enthalten, der leicht gegen einen nicht-arzneimittelresistenten
Locus oder ein Gen, das für
einen verträglichen
Arzneimittelresistenz-Marker, wie beispielsweise asp, der für eine Resistenz
gegen die Aminoglykoside Kanamycin und Neomycin codiert, ausgetauscht
werden kann.
-
Die
hierin beschriebenen Plasmiderhaltungssysteme stellen eine verbesserte
Stabilität
von rekombinanten Plasmiden bereit und räumen damit die Probleme der
Instabilität
von Plasmiden aus dem Stand der Technik, zum Beispiel in Bioreaktoren
und bei der Verwendung von Vektor-Lebendimpfstoffen, aus. Die Plasmide
sind speziell auf Impfstoffanwendungen zugeschnitten, sie sind jedoch
auch zur großtechnischen
Herstellung von Proteinen nützlich.
-
Die
größte Verbesserung
bei den Plasmiden gegenüber
dem Stand der Technik ist, dass sie die Probleme überwinden,
die mit einer Übernahme
durch plasmidlose Zellen und der Instabilität von Plasmiden verbunden sind,
und eine weit reichende Verwendung in Gebieten, wie beispielsweise
der kommerziellen Produktion von Proteinen und der Produktion von
attenuierten bakteriellen Vektor-Lebendimpfstoffen finden.
-
Es
bestand lange Zeit der Bedarf, die Probleme mit einer Übernahme
durch plasmidlose Zellen oder einer Stabilität von Plasmiden, die mit der
Verabreichung von Impfstoffen und der Produktion von Proteinen verbunden
sind, zu lösen.
Die vorliegende Erfindung erfüllt
diesen seit langem bestehenden Bedarf.
-
3. Definitionen
-
Der
Begriff „Plasmiderhaltungssystem" („ plasmid
maintenance System", „PMS"), wie er hierin
verwendet wird, bezeichnet eine Nukleotidsequenz, die wenigstens
eine postsegregationale Killing-Funktion („PSK") und wenigstens ein Partitionierungs-
oder Segregationssystem („SEG") und gegebenenfalls
irgendeine weitere Plasmidstabilisierungsfunktion umfasst.
-
Der
Begriff „Plasmiderhaltungssystem", wie er hierin verwendet
wird, bezeichnet jede die Stabilität von Plasmiden verstärkende Funktion,
die mit einem PMS verbunden ist. Der Begriff schließt sowohl
natürlich
vorkommende Nukleotidsequenzen, die für Plasmiderhaltungsfunktionen
codieren, als auch Nukleotidsequenzen, die im Wesentlichen homolog
zu den natürlich
vorkommenden Plasmiderhaltungsfunktionen sind und die die von der
entsprechenden, natürlich
vorkommenden Plasmidstabilisierungsfunktion gezeigte Funktion beibehalten,
ein.
-
Der
Begriff „post-segregationales
Killing-System" („PSK"), wie er hierin
verwendet wird, bezeichnet jede Funktion, die zum Tod irgendeiner
neu geteilten Bakterienzelle führt,
die das Plasmid von Interesse nicht geerbt hat, und er schließt insbesondere
balancierte Letalsysteme, wie zum Beispiel asd oder ssb, aus Proteinen
bestehende Systeme, wie beispielsweise phd-doc, und antisense-Systeme,
wie beispielsweise hok-sok, ein. Der Begriff schließt sowohl
natürlich
vorkommende Nukleotidsequenzen, die für PSKs codieren, als auch Nukleotidsequenzen,
die im Wesentlichen homolog zu den natürlich vorkommenden Nukleotidsequenzen
sind und die die von den entsprechenden, natürlich vorkommenden Nukleotidsequenzen
gezeigte Funktion beibehalten, ein.
-
Der
Begriff „im
Wesentlichen homolog" oder „wesentliches
Homologon", bezogen
auf eine Nukleotidsequenz oder eine Aminosäuresequenz, gibt an, dass die
Nukleinsäuresequenz
im Vergleich zu einer Referenzsequenz (z.B. einer nativen Sequenz)
ausreichende Homologie besitzt, um zuzulassen, dass die Sequenz die
gleichen grundlegenden Funktionen wie entsprechende Referenzsequenz
ausführen
kann; eine im Wesentlichen homologe Sequenz ist üblicherweise zu wenigstens
ungeführ
70 Prozent sequenzidentisch mit der Referenzsequenz, üblicherweise
zu wenigstens ungefähr
85 Prozent sequenzidentisch, vorzugsweise zu wenigstens ungefähr 95 Prozent
sequenzidentisch und ganz besonders bevorzugt zu ungefähr 96, 97,
98 oder 99 Prozent sequenzidentisch mit der Referenzsequenz. In
der Beschreibung, wo auf spezifische Nukleotidsequenzen und/oder
Aminosäuresequenzen
Bezug genommen wird, können
solche Nukleotidsequenzen und/oder Aminosäuren vorzugsweise gegen im
Wesentlichen homologe Sequenzen ausgetauscht werden.
-
Die
Begriffe „Segregationssystem" und/oder „Partitionierungssystem" (hierin beide als „SEG" bezeichnet) sind
hierbei gegeneinander austauschbar und werden dazu verwendet, jede
beliebige, die Stabilität
von Plasmiden verstärkende
Funktion zu beschreiben, die eine Funktion dafür hat, die Häufigkeit
einer erfolgreichen Übertragung
eines Plasmids auf jede neu geteilte Bakterienzelle, im Vergleich
zu der Häufigkeit
der Übertragung
eines entsprechenden Plasmids ohne ein solches SEG-System, zu erhöhen. SEG-Systeme
schließen zum
Beispiel equipartitionierende Systeme, „pair-site"-Partitionierungssysteme und den par-Locus von pSC101
ein. Der Begriff schließt
sowohl natürlich
vorkommende Nukleotidsequenzen, die für SEG-Systeme codieren, als
auch Nukleotidsequenzen, die im Wesentlichen homolog zu den natürlich vorkommenden
Nukleotidsequenzen sind und die die von den entsprechenden natürlich vorkommenden
Nukleotidsequenzen gezeigte Funktion beibehalten, ein.
-
Der
Begriff „detoxifiziert" ziert" wird hierin zur
Beschreibung eines Toxins mit einer oder mehreren Punktmutation(en),
die die Toxizität,
im Vergleich zu einem entsprechenden Toxin ohne solche Punktmutationen,
erheblich senken, verwendet
-
Der
Begriff „im
Hinblick auf eine Immunisierung effektiv", wie er hierin verwendet wird, bezieht
sich auf eine Immunreaktion, die einem Probanden ein immunologisches
Zellgedächtnis
verleiht, mit dem Effekt, dass eine sekundäre Reaktion (gegen das gleiche
oder ein ähnliches
Toxin) durch eine oder mehrere der folgenden Eigenschaften gekennzeichnet
ist: eine kürzere
Lag-Phase im Vergleich zu der Lag-Phase, die sich aus dem entsprechenden
Aussetzen ohne die vorliegende Immunisierung ergibt; eine Produktion
von Antikörpern,
die länger
andauert als die Produktion von Antikörpern bei einem entsprechenden
Aussetzen ohne eine solche Immunisierung; eine Veränderung
im Typ und in der Menge von Antikörpern, im Vergleich zu dem
Typ und der Menge an Antikörpern,
die bei einem solchen Aussetzen ohne die vorliegende Immunisierung
produziert werden; eine Verschiebung in der an der Reaktion beteiligten
Klasse, wobei IgG-Antikörper
in höheren
Konzentrationen auftreten und länger
bestehen bleiben als IgM; eine erhöhte mittlere Affinität (Bindungskonstante)
der Antikörper
zu dem Antigen, im Vergleich zu der mittleren Affinität von Antikörpern gegen
das Antigen bei einem Aussetzen ohne die vorliegende Immunisierung;
und/oder andere in der Wissenschaft zur Charakterisierung einer
sekundären
Immunantwort bekannte Eigenschaften.
-
4. Kurze Beschreibung der Figuren
-
1A-1C: Genkarten von beispielhaften
pGEN-Expressionsplasmiden (pGEN2, pGEN3 und pGEN4) der vorliegenden
Erfindung.
-
2A-2D: Genkarten von beispielhaften,
auf oriE1 basierenden Expressionsplasmiden (pJN72, pJN51, pJN10
und pJN12) der vorliegenden Erfindung.
-
3A-3H: Durchflusszytometriehistogramme
der Fluoreszenz von GFP für
CVD 908-htrA-tragende Expressionsvektoren mit dem post-segregationalen
Killing-System hoksok.
-
4A-4D: Vollständige Nukleotidsequenz von
pGEN2 (SEQ ID No. 1), die die Nukleotide 1-4196 umfasst.
-
5A-B: Partielle Nukleotidsequenz von pGEN3 (SEQ
ID No. 2), die die Nukleotide 1201-2397 umfasst und die Sequenz
von ori15A zeigt.
-
6A-C: Partielle Sequenz der Nukleotidsequenz von
pGEN4 (SEQ ID No. 3), die die Nukleotide 1201-3848 umfasst und die
Sequenz von ori101 zeigt.
-
7A-7E: Genkarten von beispielhaften,
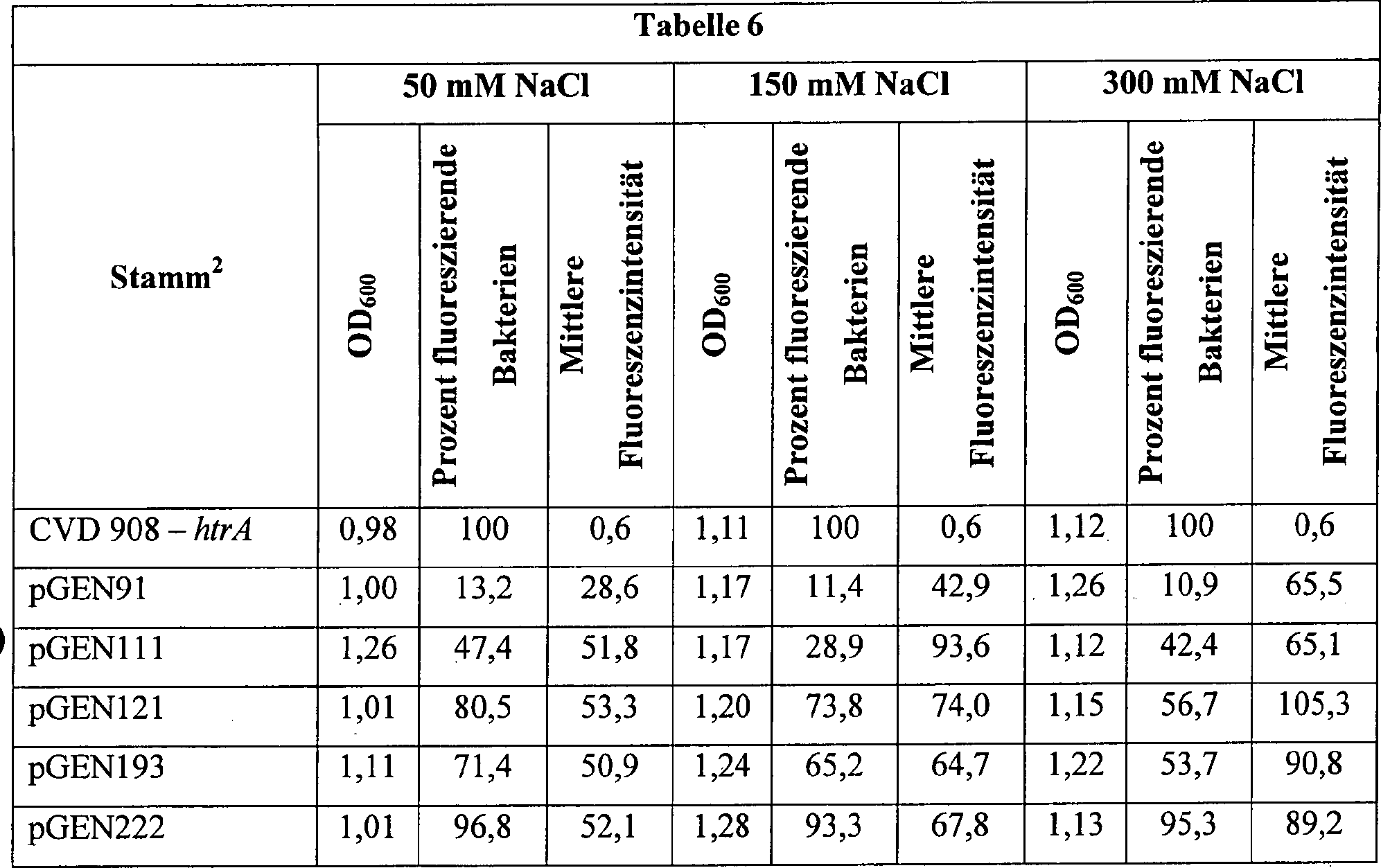
auf ori15A-basierenden pGEN-Expressionsplasmiden (pGEN91,
pGEN111, pGEN121, pGEN193 und pGEN222) der vorliegenden Erfindung.
-
8A-C: Durchflusszytometriehistogramme der Fluoreszenz
von GFP für
die Expressionsplasmide pGEN91, pGEN111, pGEN121, pGEN193 und pGEN222.
-
5. Ausführliche Beschreibung
-
Bakterielle
Vektor-Lebendimpfstoffe verwenden einen bakteriellen Lebendvektor
zur Expression von Genen, die für
schützende
Antigene von bakteriellen, viralen oder parasitären Pathogenen codieren. Die
bakteriellen schützenden
Antigene sind in dem bakteriellen Lebendvektor vorzugsweise nicht
nativ vorhanden, d.h. sie sind heterolog. Der bakterielle Vektor-Lebendimpfstoff
wird einem Wirt verabreicht, wodurch die exprimierten Antigene dem
Immunsystem des Wirts ausgesetzt werden und dadurch eine Immunreaktion
von geeigneter Beschaffenheit auslösen, um so dem Wirt Immunität zu verleihen.
-
Um
eine erhöhte
Immunogenität
zu erreichen, müssen
die Plasmide, die solche schützenden
Antigene exprimieren, stabilisiert werden. Den Erkenntnissen des
Erfinders zufolge wird in keinem derzeit verwendeten auf S. typhi
basierenden Plasmiderhaltungssystem der Vorteil von natürlich vorkommenden
Partitionierungsmechanismen genutzt, die in anderen Stämmen bekanntermaßen die
Stabilität
von Vielkopie-Plasmiden verbessern.
-
Wir
beschreiben ein nichtkatalytisches Plasmiderhaltungssystem zur Stabilisierung
von Expressionsplasmiden, die für
fremde Antigene in einem Vektor-Lebendimpfstoff aus einem S. typhi-Stamm
codieren. Der S. typhi-Stamm kann CVD 908-htrA sein. Wir beschreiben
Verbesserungen und/oder Optimierungen der Stabilisierung von Expressionsplasmiden
durch die Verwendung von Plasmiderhaltungssystemen, die auf zwei voneinander
unabhängigen
Ebenen fungieren: (1) Autheben der alleinigen Abhängigkeit
von katalytischen, balancierten Letalstabilisierungssystemen; und
(2) Einschleusen eines Plasmid-Partitionierungssystems,
das eine zufällige
Segregation der Expressionsplasmide verhindert und dadurch die Vererbbarkeit
und Stabilität derselben
erhöht.
Ein entscheidender Grund für
die Verfolgung dieses speziellen Ansatzes ist, dass dieses Verfahren
zur Verbesserung der Stabilisierung von Plasmiden keine weiteren
Manipulationen des Lebendvektorstammes beinhaltet und daher die
Immunogenität
heterologer Antigene, die mit irgendeinem Lebendvektorstamm exprimiert
werden, verbessern kann.
-
Das
hierin beschriebene Plasmiderhaltungssystem verbessert die Stabilität von Vielkopie-Expressionsplasmiden
in einem bakteriellen Vektor-Lebendimpfstoff, wie beispielsweise
CVD908-htrA.
-
Die
natürlich
vorkommende PSK-Funktion hok-sok aus dem Antibiotikaresistenzfaktor
pR1, oder ein wesentliches Homologon davon, kann in Vielkopie-Expressionsplasmide
eingeschleust werden. Das hok-sok-System ist ein Addiktionssystem
von stillen Plasmiden, das auf antisense-RNA-Kontrollmechanismen beruht,
die nur dann zur Synthese von Letalproteinen führen, wenn der Verlust des
Plasmids eingetreten ist.
-
Wir
beschreiben ferner ein Plasmiderhaltungssystem, das eine auf Komplementierung
basierende PSK-Funktion umfasst, in der das chromosomale Gen ssb,
das für
das essentielle, nicht-katalytische, einzelsträngige Bindungsprotein (SSB),
das für
die DNA-Replikation erforderlich ist, codiert, spezifisch deletiert
und in ein Vielkopie-Expressionsplasmid inseriert ist.
-
Wir
beschreiben auch ein verbessertes Plasmiderhaltungssystem, das ein
Expressionsplasmid umfasst, das für wenigstens einen SEG-Locus
und wenigstens eine PSK-Funktion
codiert.
-
5.1 Suizidvektoren
-
Heterologe
Antigene können
in Lebendvektorstämmen,
wie beispielsweise CVD908-htrA,
aus Genen, die entweder auf Plasmiden liegen oder in das Chromosom
integriert sind, exprimiert werden. Eine Technik zum Integrieren
dieser Gene in das Wirtsgenom beinhaltet die Verwendung von temperatursensitiven „Suizidvektoren", wie beispielsweise
pIB307, der einen temperatursensitiven Replikationsursprung von
pSC101 (ori101ts) enthält. Wir beschreiben einen verbesserten
Suizidvektor zur Verwendung in CVD908 und CVD908-htrA, die von pIB307
stammen, wodurch eine leichtere Konstruktion von Mutagenesekassetten
zur Veränderung
des Lebendvektorchromosoms möglich
ist.
-
Ein
Einbau dieser Suizidvektoren in das Chromosom mittels homologer
Rekombination resultiert aus. der Temperaturinaktivierung des Replikationsproteins
des Plasmids, RepA, einem Protein, das für die Funktion von ori101 essentiell
ist. Eine spontane Auflösung
der resultierenden instabilen, merodiploiden Intermediate wird mittels
Gegenselektion des Verlusts des sacB-Gens, das in dem sich auflösenden Suizidvektor
enthalten ist, nachgewiesen. Das in allen ausgeschnittenen Plasmiden
enthaltene sacB-Gen codiert für
das Enzym Levansucrase, das letal ist, wenn es im Zytoplasma von
Darmbakterien, einschließlich
S. typhi, die in der Gegenwart von Sucrose wachsen, exprimiert wird.
Da sich auflösende
Merodiploide durch Inkubation in Gegenwart von 10% Sucrose selektiert
werden, werden die ausgeschnittenen Plasmide die Wirtsbakterien
solange abtöten,
bis sie spontan verlören
gehen (cure).
-
Dieses
System wurde erfolgreich zum Einbau einer Kanamycinresistenz-Kassette
in den ☐aroC1019-Locus von CVD908 verwendet. Diese Versuche
waren jedoch deshalb erfolgreich, weil das Gen, das in das Chromosom
von S. typhi aktiviert wurde, für
einen selektierbaren Arzneimittelresistenz-Marker codierte. Unter
Verwenden dieser früheren
Vektoren, war ein Austausch der Kanamycinresistenz-Kassette gegen einen
nichtselektierbaren Marker erfolglos, weil, obwohl der eingeführte Marker
als Merodiploid in das Chromosom eingebaut werden könnte, eine
Auflösung
des Merodiploids für
einen Austausch des Arzneimittelresistenzgens niemals nachgewiesen
werden konnte.
-
Wir
beschreiben deshalb ein Verfahren zur Verwendung solcher Suizidvektoren
zum Inaktivieren des ssb-Locus attenuierter Salmonella typhi-Stämme, wie
beispielsweise CVD908-htrA.
-
Suizidvektoren
können
somit eine effiziente Aktivierung von Genen, die Proteine oder Peptide
von Interesse, wie zum Beispiel heterologe Antigene, exprimieren,
in dem Chromosom von S. typhi CVD908-htrA in zwei Stufen ermöglichen.
Der vorliegende Erfinder schleuste zum Beispiel eine sacB-aph-Kassette
in den ☐aroC1019-Locus ein, die dann mit Hilfe von Kanamycin
selektiert wurde. Bei der Bildung von Generationen dieses S. typhi
CVD908-htrA☐aroC1019::sacB-aph-Stamms wird ein nützlicher
Intermediatstamm erzeugt, in dem theoretisch jedes Strukturgen durch
Austausch des Markers effizient in den aroC-Locus inseriert werden kann. Da die
Auflösung
von Merodiploiden in Gegenwart von Sucrose zu einem Verlust des
sacB-Gens führt, wird
das sacB-Gen als Marker für
eine Gegenselektion dafür
verwendet, während
des Passagierens der Merodiploide in Gegenwart von 10% Sucrose einen
Austausch der sacB-aph-Kassette gegen die eingeführte Antigen-Kassette zu selektieren,
um so eine lebensfähige
Nachkommenschaft zu erzeugen. Dieser Intermediatstamm wurde zum
effizienten Einbau der nichttoxischen Mutante LT-K63 des nichthitzebeständigen Enterotoxins
von E. coli verwendet, wobei CVD908☐aroC1019::LT-K63 erzeugt wurde.
-
5.2 Plasmid-basierte Expression von heterologen
Antigenen
-
Obwohl
ein Einbau fremder Gene in das Chromosom den entsprechenden Sequenzen
Stabilität
verleiht, können
die beteiligten genetischen Manipulationen schwierig sein und eine
Abnahme der Kopienzahl des heterologen Gens führt oftmals zu einer Produktion
von Mengen des heterologen Antigens, die nicht ausreichen, um eine
optimale Immunreaktion zu gewährleisten.
-
Die
Stabilität
von Plasmiden ist hingegen ein kompliziertes Phänomen, das von mehreren Faktoren abhängt, die
(1) die Kopienzahl des Plasmids; (2) die genau regulierte Expression
von Genen, die in dem Plasmid enthalten sind; und (3) den selektiven
Druck einschließen,
um eine korrekte Segregation und Vererbbarkeit des Plasmids zu gewährleisten.
-
Um
Stabilität
zu gewährleisten,
müssen
Plasmide in einer regulierten Weise repliziert werden, damit so
verhindert wird, dass ihre Kopienzahl auf letale Mengen ansteigt.
-
Des
Weiteren müssen
Plasmide während
der Teilung eines wachsenden Bakteriums segregiert werden, damit
gewährleistet
ist, dass jede Tochterzelle wenigstens eine Kopie des Plasmids erhält. Die
Segregation kann ein passiver, ein sich zufällig ereignender oder ein aktiver
Prozess sein, der die Synthese von neuartigen Proteinen, die die
Segregation und Vererbung des Plasmids unterstützen, beinhaltet. Eine erfolgreiche Vererbbarkeit
zufällig
segregierender Plasmide ist auf eine ausreichend hohe Kopienzahl
zufällig
verteilter Plasmide in einem sich teilenden Bakterium angewiesen,
so dass die Vererbung von wenigstens einem Plasmid auf jede Tochterzelle
gewährleistet
ist.
-
Die üblicherweise
verwendeten Plasmidklonierungsvektoren, einschließlich pBR322-Derivate mit mittlerer
Kopienzahl und pUC-Plasmide mit hoher Kopienzahl, werden durch zufällige Segregation
vererbt.
-
Eine
aktive Segregation beinhaltet die Synthese von Proteinen, die an
solche Plasmide binden sollen und ferner mit den Membranen von sich
teilenden Bakterien koordinieren, so dass gewährleistet ist, dass jede Tochterzelle
wenigstens eine Plasmidkopie erhält.
Plasmide, die diese Systeme der aktiven Partitionierung verwenden,
sind in der Regel Plasmide mit sehr kleiner Kopienzahl, wie beispielsweise
der Sex-Faktor F von E. coli oder die Antibiotikaresistenz-Faktoren
R, beispielsweise pR1 und pRK2.
-
Wir
beschreiben die Verwendung natürlich
vorkommender SFG-Funktionen zur Erhöhung der Vererbbarkeit von
Vielkopie-Expressionsplasmiden, die ansonsten durch zufällige Segregation
vererbt würden,
um die Stabilität
dieser Plasmide zu erhöhen.
-
Ebenso
verwenden wir natürlich
vorkommende Gensysteme, in denen Tochterzellen, bei denen die Vererbung
eines Expressionsplasmides erfolglos war, getötet und aus der wachsenden
Population entfernt werden, d.h. eine PFK-Funktion. Hierin wird
der Einbau von mehr als einer Klasse an Plasmiderhaltungsfunktionen
als ein Plasmiderhaltungssystem bezeichnet. Beispielsweise wird
mit dem Einbau von sowohl einer SFG-Funktion, wie beispielsweise
einem Partitionslocus, als auch einer PSK-Funktion in ein einziges
Expressionsplasmid ein Plasmiderhaltungssystem erhalten.
-
Es
sollte beachtet werden, dass ein Gen, das Resistenz gegen ein bakterielles
Antibiotikum verleiht, wie beispielsweise das aph-Gen, das für die Resistenz
gegen Kanamycin und Neomycin codiert, genauso als PFK-Funktion zu
betrachten ist, wie das auf asd basierende, balancierte Letalsystem.
-
5.3 Balancierte Letalsysteme
-
Ein
Verfahren, um die Vererbung von Expressionsplasmiden sicherzustellen,
beinhaltet die Konstruktion eines PSK-Systems, oder eines wesentlichen
Homologons davon, als balanciertes Letalsystem bezeichnet, für Plasmide,
die heterologe Antigene exprimieren. In einem auf Plasmiden basierenden,
balancierten Letalsystem exprimieren Plasmide, die im Zytoplasma
des Bakteriums replizieren, ein entscheidendes Protein, das für das Wachstum
und die Replikation des Bakteriums benötigt wird. Ein Verlust solcher
Plasmide nimmt dem Bakterium die Fähigkeit, das entscheidende
Protein zu exprimieren und bewirkt den Tod der Zelle.
-
Das
asd-System wurde vor kurzem in einem Versuch zur Erhöhung der
Stabilität
von Plasmiden, die synthetische Peptide von Hepatitis B-Virus exprimieren,
in attenuierte S. typhi-Impfstoffstämme eingeschleust.
-
Wenn
jedoch die freiwilligen Probanden mit diesen Lebendvektorstämmen immunisiert
wurden, wurde keine Immunreaktion auf das fremde Antigen nachgewiesen.
Tatsächlich
haben bis heute nur sehr wenige Berichte eine Immunreaktion auf
die Plasmidbasierte Expression eines fremdes Antigens aus Plasmiden
(stabilisierten oder anderen) nach der Impfung von Menschen mit
einem attenuierten S. typhi-Lebendvektor belegt.
-
Obwohl
in manchen Fällen
eine übermäßige Attenuierung
des Stammes selbst zu einer Fehlfunktion der Lebendvektorstämme führen kann,
nimmt der Erfinder an, dass die derzeit verwendeten PSK-Funktionen für Plasmide
weiteren Einschränkungen,
insbesondere Einschränkungen
infolge der Segregation und Einschränkungen infolge der katalytischen
Aktivität
unterliegen. Deshalb stellen wir verbesserte Expressionsplasmide
mit verbesserten Segregationsfähigkeiten
bereit, in denen wenigstens ein Partitionierungssystem zusammen
mit wenigstens einem PSK-System eingebaut ist.
-
5.4 Einschränkungen infolge der Segregation
-
Eine
Einschränkung
der Plasmiderhaltungsfunktionen, wie beispielsweise der asd-Funktion (sowie der thyA-Funktion)
ist, dass sie die Vererbbarkeit der in ihnen befindlichen Plasmide,
die mit oder ohne asd-Funktion weiter zufällig segregieren, nicht verstärken. Wenn
die in diesen befindlichen Expressionsplasmide, die asd-Gene tragen,
nicht stabil vererbt werden können,
werden sie deshalb, unabhängig
von dem Bedarf des Bakteriums nach Asd, verloren gehen.
-
Die
stabile Vererbung eines asd-Expressionsplasmids kann durch das Anzüchten von
Plasmid-tragenden Stämmen
in Gegenwart von DAP, das den selektiven Druck als Garantie dafür, dass
alle lebensfähigen Bakterien
das Expressionsplasmid enthalten, wegnimmt, bestimmt werden. Wenn
ein gegebenes Plasmid instabil vererbt wird, wird es in hohen Anteilen
aus den Bakterien verloren gehen und die plasmidlosen Bakterien werden
in Abwesenheit von Wachstumssupplementen lysieren; das Gesamtergebnis
dieses Effekts wird eine Population von Bakterien sein, die viel
langsamer wachst als nicht veränderte
Wildtyp-Stämme.
-
Die
hierin beschriebenen Plasmide besitzen eine verbesserte Plasmidstabilität, da eine
SFG-Funktion, wie beispielsweise ein Partitionslocus, oder ein wesentliches
Homologon einer SFG-Funktion, in das Expressionsplasmid eingeschleust
wurde, um so die Vererbbarkeit der Plasmide bei sich aktiv teilenden
Bakterien zu verbessern. Die Partitionsloci, die in der Natur vorkommen,
befinden sich auf den Virulenzplasmiden von S. typhimurium. Tinge
und Curtiss, Journal of Bacteriology, 172:5266, 1990, berichteten,
dass von den Virulenzplasmiden von S. typhimurium solche Partitionsloci
gut konserviert waren und dass, wenn ein 3,9 kb großes Restriktionsfragment,
das für
diesen Locus codiert, in das Plasmid pACYC 184 mit geringer Kopienzahl
(~ 15 Kopien pro Zelle) eingeschleust wurde, die beobachtete Plasmidstabilität nach 50
Generationen von 34% Plasmid-enthaltende Zellen auf 99% Plasmid-tragende
Zellen zunahm. Die Nukleotidsequenz dieses Locus wurde später von
Cerin und Hackett, Plasmid, 30:30, 1993 (GenBank-Zugangsnummer M97752)
bestimmt.
-
5.5 Einschränkungen infolge der katalytischen
Aktivität
-
Eine
weitere potentielle Einschränkung
einer Plasmidstabilisierungsfunktion, wie beispielsweise der asd-Funktion
(sowie des thyA-Systems) ist dessen Abhängigkeit von einem Enzym mit
katalytischer Aktivität. Angesichts
dessen, dass eine Komplementierung mit nur einer einzigen Kopie
des asd-Gens dazu ausreicht, die Autotrophie aufzuheben, ist nicht
klar, warum alle Kopien eines Vielkopie-Plasmids stabil bleiben
sollten, insbesondere, wenn diese für ein besonders problematisches,
heterologes Antigen codieren, das das Wachstum des Bakteriums hemmt.
-
Obwohl
Expressionsplasmide mit höheren
Kopienzahlen gewünschte
Mengen eines gegebenen heterologen Antigens in vitro exprimieren
können,
können
diese Plasmide aufgrund der Toxizität ferner nicht in vivo mit
der erwarteten Kopienanzahl stabilisiert werden und dürften tatsächlich mit
viel geringeren Kopienzahlen vorliegen, die erwartungsgemäß jede beobachtete
Immunreaktion, die für
das heterologe Antigen spezifisch ist, reduzieren sollten. Die vorliegende
Erfindung stellt daher entsprechend dauerhaft stabilisierte Plasmide
mit geringer und mittlerer Kopienzahl für die Expression heterologer
Antigene bereit.
-
5.6 Die nichtkatalytische ssb-PSK-Funktion
-
Die
potentielle Einschränkung
infolge katalytischer Aktivität,
die mit balancierten Letalsystemen assoziiert ist, wird hierbei
durch die Verwendung von Plasmiden, die das einzelsträngige Bindungsprotein
(SSB) von S. typhi exprimieren, zur Transkomplementierung einer
ansonsten letalen Mutation, die in dem chromosomalen ssb-Gen eingebaut
ist, angegangen. Die Biochemie und die Rolle des SSB-Proteins von
E. coli im Stoffwechsel sind rückblickend
ausführlich
bei Lohman et al., Annual Reviews in Biochemistry, 63:527, 1994
und Chase et al., Annual Reviews in Biochemistry, 55:103, 1986 zusammengefasst.
-
SSB
ist ein nicht-katalytisches, 177 Aminosäuren langes Protein mit einem
relativen Molekulargewicht von 19 kDa, das mit hoher Affinität an einzelsträngige DNA
(ssDNA) bindet und eine wesentliche Rolle als akzessorisches Protein
bei der Replikation, Rekombination und Reparatur von DNA spielt.
Die biologisch relevante Form von SSB, die an der Bindung an ssDNA
beteiligt ist, ist ein Tetramer, das auf zwei Arten an ssDNA bindet,
die eng mit einem Durchschnitt von entweder 35 (SSB35-Bindungsmodus)
oder 65 Basen (SSB65-Bindunsmodus) assoziiert
sind. Die spezifischen Bedingungen, die den bevorzugten Bindungsmodus
steuern, sind kompliziert und hängen
von der Konzentration in der Umgebung vorhandener monovalenter oder
divalenter Salze, dem pH und der Temperatur sowie der Menge an vorliegendem
SSB-Protein ab. Bei gegebenen Bedingungen begünstigen hohe Konzentrationen
an SSB den SSB35-Bindungsmodus und niedrigere
SSB-Konzentrationen
den SSB65-Modus. Es muss jedoch betont werden,
dass die erforderliche Konformation von SSB in beiden Bindungsmodi
ein Tetramer ist.
-
Das
spontane Auftreten von temperatursensitiven Punktmutationen innerhalb
des ssb-Gens wurde nun
auf der biochemischen, der physiologischen und der Nukleotidebene
charakterisiert; eine solche Mutante, ssb-1, enthält die Punktmutation
His 55 bis Tyr und es wurde gefunden, dass sie für eine Assemblierung in Tetramere
bei nicht-toleranten Temperaturen und bei den natürlich vorkommenden
Expressionsmengen instabil ist. Diese mutanten Stämme zeigen
temperatursensitive, letale Defekte bei der Replikation und Rekombination
von DNA.
-
Die
Häufigkeiten
der Segregation von Plasmiden, die ssb tragen, das chromosomale
ssb-Mutationen in E. coli-Bakterien komplementiert, wurden von Porter
et al., Bio/Technology 8:47, 1990, untersucht. Sie beobachteten,
dass in Versuchen, an denen Bioreaktoren beteiligt sind, die Segregationshäufigkeit
in Plasmid-tragenden Stämmen,
die in kontinuierlicher Kultur unter nicht-selektiven Bedingungen
150 Stunden lang angezüchtet
wurden, 1 × 10–7 betrug;
diese Segregationshäufigkeit
hing nicht von der Kopienzahl ab, da sowohl die pACYC184-Plasmide
mit geringerer Kopienzahl als auch die pUC19-Plasmide mit sehr hoher
Kopienzahl bei der gleichen Häufigkeit
stabilisiert wurden. Es muss jedoch beachtet werden, dass die beteiligten
Plasmide zusätzlich
zu dem SSB-Protein nur einen Arzneimittelresistenz-Marker exprimierten.
-
Wir
beschreiben ein verbessertes Plasmiderhaltungssystem, das einen
Partitionslocus, wie beispielsweise den auf pSC101 vorhandenen,
oder ein wesentliches Homologon eines solchen Partitionslocus, einschließt und auch
ein aktives Partitionierungssystem, oder ein wesentliches Homologon
davon, wie beispielsweise das oben für das Virulenzplasmid von S.
typhimurium beschriebene, einschließen kann.
-
Die
vorliegende Erfindung hebt die Abhängigkeit von katalytischen
Enzymen zur Verleihung von Plasmidstabilität auf. Mutierte Allele, die
ssb-1 ähneln,
können
in das Expressionsplasmid eingeschleust werden, um so Plasmide mit
hoher Kopienzahl durch Überexpression
von SSB1-gleichen Proteinen zu verstärken, um die erforderlichen
biologisch aktiven Tetramere von SSB auszubilden. Eine PSK-Funktion,
die ein Addiktionssystem von stillen Plasmiden umfasst, das auf
antisense-RNA-Kontrollmechanismen, die nur dann letale Proteine
synthetisieren, wenn ein Verlust der Plasmide eingetreten ist, basiert,
kann ebenso verwendet werden.
-
5.7 Expressionsplasmide und vollständige Genkassetten
-
Wir
beschreiben außerdem
eine Reihe von Expressionsplasmiden, die hierin als pGEN-Plasmide
bezeichnet werden. pGEN-Plasmide umfassen vollständige Genkassetten, die für die regulierte
Expression eines heterologen Antigens, eines Replikationsursprungs
und eines selektierbaren Markers zur Rückgewinnung des Plasmids codieren.
Diese Reihe von Vektoren wurde speziell dafür konstruiert, zu testen, ob
irgendein Plasmiderhaltungssystem die Stabilität von Plasmiden, zum Beispiel
in einem Ansatz mit einem attenuierten S. typhi-Impfstoff, erhöhen kann.
-
Die
Grundstruktur dieser Vektoren ist in 1 angegeben
und die zusammengesetzte Gensequenz für den Vektor pGEN2 (SEQ ID
No. 1) ist in 4 dargestellt; 5 und 6 zeigen
entsprechend die spezifischen, zusammengesetzten Sequenzen für die Replikationsursprünge in pGEN
3 (SEQ ID No. 2) und pGEN4 (SEQ ID No. 3).
-
Es
ist entscheidend zu beachten, dass die pGEN-Plasmide dazu ausgelegt
sind, 3 unabhängig
voneinander funktionierende Genkassetten zu umfassen. Diese Kassetten
wurden so konstruiert, dass einzelne Komponenten, falls nötig, durch
Austausch optimiert werden können.
Neben den verschiedenen, hierin beschriebenen Plasmiderhaltungssystemen,
können
die Kassetten entsprechend andere viel versprechende Systeme, die
derzeit existieren, oder die in Zukunft zur Verfügung stehen könnten, testen.
Das (die) optimierte(n) Plasmid(e) kann (können) daran angepasst werden,
relevante, schützende,
heterologe Antigene in attenuierten Impfstoffstämmen für eine Imminisierung von Menschen
zu exprimieren.
-
Die
pGEN-Plasmide stellen eine Kassette zur regulierten Expression eines
Testantigens bereit, das so funktionieren kann, dass, wenn die Induktion
der Expression von Antigenen erhöht
ist, der Stoffwechsel des Bakteriums belastet wird, und dies phänotypisch
gesehen zu einer Instabilität
des Plasmids führt,
d.h. es wird ein selektiver Vorteil für alle diejenigen Bakterien
geschaffen, die das störende
Plasmid spontan verlieren können.
Wir beschreiben daher ein hinsichtlich der Bedingungen instabiles
Plasmid, das auf Stabilität
untersucht werden kann, solange Plasmiderhaltungssysteme eingeschlossen
sind.
-
In
einer bevorzugten Ausführungsform
umfasst die Kassette zur regulierten Expression eines Testantigens,
die in den pGEN-Plasmiden enthalten ist, den induzierbaren ompC-Promotor,
oder ein wesentliches Homologon davon, der die Expression eines
nachweisbaren Proteins, wie beispielsweise des codon-optimierten
grünen
fluoreszierenden Proteins (GFPuv, erhältlich bei Clontech), dessen Überexpression
für E.
coli und S. typhi toxisch ist, steuert.
-
Wir
beschreiben auch eine Reihe von Plasmidreplikons mit Kopienzahlen,
die von geringer Kopienzahl (d.h. 1 bis ~ 10, vorzugsweise ~ 5 Kopien
pro Zelle) zu mittleren Kopienzahlen (d.h. ~ 11 bis ~ 25, vorzugsweise ~
15 Kopienzahlen pro Zelle) zu hohen Kopienzahlen (d.h. ~ 26 bis
~ 100, vorzugsweise ~ 60 Kopienzahlen pro Zelle) variieren. Um dies
zu erreichen, wurden Replikationsursprünge der genau charakterisierten
Plasmide pSC101, pACYC184 und pAT153 unter Verwenden von Polymerasekettenreaktions-(PCR-)
Techniken modifiziert, um so unabhängig voneinander funktionierende
Plasmidreplikationskassetten zu erzeugen.
-
Wir
beschreiben auch selektierbare Expressionsplasmide zur Verwendung
in attenuierten S. typhi-Lebendvektoren. Diese Expressionsplasmide
enthalten einen selektierbaren Marker, der letztlich gegen entweder
einen nicht-arzneimittelresistenten Locus, wie beispielsweise ssb,
oder ein Gen, das für
einen verträglichen
Arzneimittelresistenz-Marker,
wie beispielsweise aph, das für
eine Resistenz gegen die Aminoglykoside Kanamycin und Neomycin codiert,
ausgetauscht werden kann.
-
Um
dies zu erreichen, wurden die für
eine Resistenz gegen Carbenicillin und Tetracyclin codierenden Resistenzkassetten,
deren Transkription effizient von einem rrnB-T1T2-Terminator terminiert wird, konstruiert. Eine
ausführliche
Beschreibung der einzelnen Bestandteile, die die Expressions- und
Replikationskassetten umfassen, ist im Folgenden angegeben.
-
Bestimmte
Komponenten des Plasmiderhaltungssystems können systematisch in die Basisexpressionsreplikons
inseriert werden, um so jeden individuellen oder synergistischen
Einfluss von diesen Funktionen auf die Plasmidstabilität mit und
ohne Selektion beurteilen zu können.
Beispielsweise kann eine post-segregationale Killing-Funktion (z.B.
der hok-sok-Locus)
so als EcoRI-XbaI-Kassette inseriert werden, dass die Transkription
flankierender Regionen von umgebenden Loci, wie beispielsweise des
Antigens und der Selektionskassetten, divergent ist und die Transkriptionsebenen
des Wildtyps, die die Letalität
dieses Locus kontrollieren, nicht signifikant stören wird (7B, pGEN111).
-
In ähnlicher
Weise kann der passive Pertitionslocus der Partition par als BamHI-BglII-Fragment zwischen
den Replikationsursprung und Selektionskassetten inseriert werden
(7C, pGEN121). Interessanterweise wurde in der
Arbeit, die zu der vorliegenden Erfindung führte, beobachtet, dass die
Orientierung des par-Locus die Synthese von GFPuv auf festem Medium
verstärkt,
wenn er in der natürlichen,
in ori101 von pSC101 gefundenen Orientierung inseriert wurde; diese
Orientierung wurde bei allen anderen Expressionsplasmiden angepasst.
-
Der
aktive Partitionierungslocus ist vorzugsweise der parA-Locus, der
als XhoI-EcoRI-Kassette
aus dem gleichen pR1-Resistenzplasmid konstruiert wurde, aus dem
hok-sok angepasst worden war. Um die natürlichen Transkriptionsmengen
und die Regulation innerhalb von diesem Locus zu bewahren, wird
die Kassette vorzugsweise so in einem Bereich der Expressionsplasmide
angeordnet, dass die Transkription der flankierenden Regionen von
parA aus fortschreitet (7D und 7E,
pGEN193 und pGEN222).
-
5.8 Bestandteile der Antigenexpressions-
und Replikationskassetten
-
5.8.1 Promotor
-
Fachleute
werden verstehen, dass eine breite Vielfalt an in der Wissenschaft
bekannten Komponenten in die Expressionskassetten eingebaut werden
kann, einschließlich
einer breiten Vielfalt an Transkriptionssignalen, wie beispielsweise
Promotoren und anderen Sequenzen, die die Bindung von RNA-Polymerase
an den Promotor regulieren. Die Funktionsweise von Promotoren ist
im Stand der Technik allgemein bekannt und ist bei Doi, Regulation
of Gene Expression, Modern Microbial. Genetics, Seite 15-39 (1991)
beschrieben. Die folgende Beschreibung verwendet den ompC-Promotor
lediglich als Beispiel und die Erfindung ist nicht darauf beschränkt.
-
Der
Promotor ist vorzugsweise ein durch die Umgebung regulierbarer Promotor,
der von einem biologisch relevanten Signal, wie beispielsweise der
Osmolarität,
kontrolliert wird. In einer bevorzugten Ausführungsform ist der Promotor
der ompC-Promotor. Das ompC-Gen codiert für ein Porinprotein, das als
Trimer in die äußere Membran
einer Bakterienzelle eingeschleust wird. Die Expression und Kontrolle
von ompC ist kompliziert und wurde kürzlich in beachtlicher Ausführlichkeit
von Pratt et al., Molecular Microbiology, 20:911, 1996 und Egger
et al., Genes to Cells, 2:167, 1997 rückblickend zusammengefasst.
-
Die
Synthese des OmpC-Proteins wird auf der Transkriptionsebene zuletzt
durch die Osmolarität
der umliegenden Umgebung kontrolliert, so dass eine Zunahme der
Osmolarität
mit einer erhöhten
Transkription von ompC einhergeht. Die Zunahme der Osmolarität bewirkt
jedoch nicht direkt die erhöhte
Transkription von ompC. Vielmehr erkennt das Bakterium die umliegende
Osmolarität
mit Hilfe eines aus zwei Komponenten bestehenden Signalübertragungssystems,
das von dem ompB-Operon codiert wird. Dieses Operon besteht aus zwei
Genen, die in der Reihenfolge envZ-ompR transkribiert werden. Das
envZ-Gen codiert für
ein 450 Aminosäuren
(AS) langes Protein, das zwei Transmembranregionen enthält, wobei
eine 118 AS lange, osmotisch sensitive Domäne in den periplasmatischen
Raum reicht, und eine 270 AS lange, katalytische Domäne am C-Terminus
in das Zytoplasma reicht; und in die innere Membran des Bakteriums
(vielleicht als Dimer) eingeschleust wird.
-
Die
C-terminale, katalytische Domäne
besitzt sowohl Kinase- als auch Phosphatase-Aktivität, die beide
von der Osmolarität
bestimmt werden, so dass, wenn die Osmolarität zunimmt, die Kinase-Aktivität vorherrscht
und wenn die Osmolarität
abnimmt, die Phosphatase-Aktvität
vorherrscht.
-
Die
Kinase-Aktivität
von EnvZ phosphoryliert den Asparaginsäurerest 55 des 239 AS langen,
zytoplasmatischen Proteins OmpR, wodurch OmpR-P erzeugt wird. Dieses
modifizierte Protein OmpR-P bindet an den ompC-Promotor und aktiviert
die Transkription durch RNA-Polymerase; wenn die Osmolarität zunimmt,
produziert die zunehmende Kinase-Aktivität von EnvZ
somit höhere
Mengen an OmpR-P und das wiederum führt zu einer höheren Transkription
von ompC. OmpR-P bindet an eine Region auf dem ompC-Promotor, die
die Basen –41
(bezogen auf die Stelle des Transkriptionsbeginns mit +1) bis –102 umspannt,
wobei OmpR-P zunächst
an die Basen –78
bis –102
bindet und anschließend
weiter an die bis –41
reichenden Basen bindet, wenn die Konzentration von OmpR-P mit der
Osmolarität
zunimmt. Daneben wurde gezeigt, dass OmpR-P an eine AT-reiche, stromaufwärts gelegene
Region, die bis zu der Base –405
zurückreicht,
bindet, was die Transkription von ompC zusätzlich verstärkt.
-
In
einer bevorzugten Ausführungsform
umspannt das Fragment des ompC-Promotors von E. coli die Nukleotide
+70 bis –389.
Der Promotor kann die Transkription eines Antibiotikaresistenzgens,
wie zum Beispiel des Kanamycinresistenzgens, in attenuierten S.
ryphi-Stämmen
auf eine für
Osmose sensitive Weise steuern. Unsere Versuchen haben zum Beispiel
gezeigt, dass sich die Resistenz gegen Kanamycin von 0 μg/ml auf > 800 μg/ml erhöhte, wenn
die Konzentration von NaCl in dem flüssigen Wachstumsmedium von
0 mM auf 300 mM erhöht
wurde.
-
5.8.2 Replikationsursprung
-
Aufgrund
der verschienen Toxizitätsgrade,
die mit verschiedenen heterologen Antigenen assoziiert sind (d.h.
höhere
Toxizität
bei Antigenen, die von parasitären
Organismen wie Plasmodium falciparum stammen gegenüber einer
nahezu nicht vorhandenen Toxizität
des Fragments C von Tetanus-Toxin), beschreiben wir Vektor-Lebendimpfstoffe,
die solche Antigene vorzugsweise aus entweder Plasmiden mit geringer
Kopienzahl oder aus. Plasmiden mit mittlerer Kopienzahl exprimieren.
Ein Fachmann wird erkennen, dass die Auswahl eines Replikationsursprungs
von dem Toxizitätsgrad
abhängen
wird, d.h. die Kopienzahl sollte kleiner werden, wenn sich die Toxizität des Bakterienstammes
erhöht.
In einer bevorzugten Ausführungsform
sind das (die) verwendeten Plasmiderhaltungssysteme in der Lage,
Replikons mit geringer oder mittlerer Kopienzahl zu stabilisieren.
-
Der
Replikationsursprung verleiht vorzugsweise eine durchschnittliche
Kopienzahl, die zwischen ungefähr
2 und ungefähr
75 liegt. In einer bevorzugten Ausführungsform wird der Replikationsursprung
so ausgewählt,
dass er eine durchschnittliche Kopienzahl verleiht, die zwischen
ungefähr
5 und ungefähr
50 liegt. Besonders bevorzugt liegt sie in einem Bereich zwischen
ungefähr
5 und ungefähr
30. Optimalerweise liegt sie in einem Bereich zwischen ungefähr 15 und
ungefähr
20.
-
Der
Replikationsursprung kann von pSC101, der eine Kopienzahl von ungefähr 5 Kopien
pro Genäquivalent
verleiht, stammen.
-
Der
onE1-Locus bestimmt die Synthese eines 555 Basen großen Iranskripts,
das RNA I genannt wird, und die Synthese eines 110 Basen großen antisense-RNA-Transkripts,
das RNA II genannt wird. Wenn RNA I synthetisiert wird, nimmt die
5'-proximale Region
des Iranskripts eine Stamm-Schleifen-Struktur an, die aus 3 Domänen besteht
und mit einer komplementären
Stamm-Schleifen-Struktur, die von RNA II gebildet wurde, hybridisieren
kann, was zur Bildung einer doppelsträngigen RNA-RNA-Struktur führt, die
einen Abbruch der Plasmidreplikation auslöst.
-
Wenn
die Synthese von RNA I weiter fortschreitet und dabei das 555 Basen
große
Iranskript in voller Länge
erzeugt, zerstört
eine Neuordnung der Sekundärstruktur
des Iranskripts die anfängliche,
aus 3 Domänen
gebildete Stamm-Schleifen-Struktur und es wird eine alternative
Stamm-Schleifen-Konfiguration ausgebildet, die nicht mehr mit RNA
II hybridisiert. Die Ausbildung dieser alternativen Struktur ermöglicht die
Selbst-Hybridisierung des Iranskripts mit einem DNA-Strang des Plasmids,
wodurch ein RNA-DNA-Komplex gebildet wird, der, um die Synthese
des ersten DNA-Strangs des Plasmids und eine Plasmidreplikation
auszulösen,
mit endogener RNAse aufgebrochen wird.
-
Die
Replikation des Plasmids wird somit durch die Synthese von RNA I
kontrolliert, die eine Kaskade von strukturellen Konfigurationen
durchlauft, die eine Initiierung der Replikation bewirken. Das notwendige Fortschreiten
der RNA I-Faltungskaskade (und die daraus resultierende Initiierung
der Replikation) wird durch die Konkurrenz der Domänen mit
RNA II gestört.
Dieser Mechanismus ist in Plasmiden, die entweder oriE1 oder ori15A
enthalten, im Wesentlichen gleich.
-
Der
Grund dafür,
dass diese beiden Typen von Plasmiden nebeneinander in dem gleichen
Bakterium vorliegen, beruht auf Sequenzdivergenz innerhalb der Region
der Hybridisierung zwischen RNA I und RNA II, so dass die RNA II
von ori15A nicht mit RNA I von oriE1 hybridisieren wird; diese Sequenzdivergenz
beeinträchtigt
auch die Stabilität
des RNA I:RNA II-Hybrids, was die Unterschiede in den Kopiezahlen
zwischen den Plasmiden, die die Replikationsursprünge oriE1
oder ori15A tragen, erklärt.
-
Der
strukturelle Organisation der konstruierten Replikationsursprungskassetten
für pSC101
(ori101; ~ 5 Kopien pro Genomäquivalent),
pACYC184 (Derivat von ori15A; ~ 15 Kopien pro Genomäquivalent)
und pAT153 (Derivat von oriE1; ~ 60 Kopien pro Genomäquivalent)
sind hinsichtlich der Struktur und der Funktion analog.
-
5.8.3 Exprimiertes Protein oder Peptid
-
Wenn
die Expressionskassette zum Screenen von Plasmiderhaltungssystemen
verwendet wird, exprimiert sie vorzugsweise ein Protein oder Peptid
ohne Stoffwechselaktivität.
Ein bevorzugtes Protein ist das grün fluoreszierende Protein (GFP)
der biolumineszierenden Qualle Aequoria victoria, ein 238 Aminosäuren langes Protein,
das eine post-translationale Modifikation durchlauft, bei der 3
interne Aminosäuren
(65Ser-Tyr-Gly67) an
einer Zyklisierungs- und Oxidationsreaktion beteiligt sind. Das
resultierende Fluorophor emittiert bei einer Bestrahlung mit langwelligem
ultravioletten Licht bei einer Wellenlänge von 395 nm blau-grünes Licht
bei einer maximalen Wellenlänge
von 509 nm. Daneben ist die Fluoreszenzaktivität über einen weiten pH-Bereich
von 5,5 bis 12 und bis zu einer Temperatur von 70°C bemerkenswert
konstant.
-
Da
GFP keine bekannte katalytische Aktivität besitzt, kann die Höhe der beobachteten
Fluoreszenz direkt auf die Menge der Transkription des in jedem
Bakterium enthaltenen gfp-Gens
zurückgeführt werden. Die
Expression des GFP-Proteins wurde nun in einer Vielzahl von sowohl
prokaryotischen als auch eukaryotischen Zellen quantifiziert und
erfordert keine weiteren Cofaktoren oder Enzyme von A. victoria.
Die Bildung des Fluorophors hängt
offensichtlich entweder von ubiquitären Enzymen ab oder ist ein
autokatalytisches Ereignis.
-
Einzelne
Bakterien, die GFP exprimieren, können mittels Durchflusszytometrie
entweder allein oder in Makrophagen, Epithelzelllinien und infizierten
Geweben von Lebewesen quantifiziert werden. Die Fluoreszenz von
GFP ist absolut unabhängig
von den Resten 2-232 des nichtdenaturierten Proteins. Die Fusion
nicht miteinander verwandter, biologisch aktiver Proteindomänen mit
dem N-Terminus von GFP hat jedoch zu Fusionsproteinen mit der erwarteten,
heterologen, biologischen Aktivität geführt, die auch weiterhin fluoreszieren.
-
Mittels
Sequenzanalyse (Clontech) wurde bestätigt, dass das hierin bevorzugte
gfp-Allel (d.h.
gfpuv) eine GFP-Mutante (GFPuv) exprimiert, das 3 Aminosäuresubstitutionen
(die nicht das Fluorophor betreffen) enthält und die Fluoreszenz gegenüber dem
Wildtyp-GFP um das 18-Fache erhöht.
-
Daneben
wurden 5 selten verwendete Arginincodons für eine effiziente Expression
von GFP in E. coli optimiert. Da ein Vergleich von Expressionsmengen
verschiedener heterologer Proteine in E. coli und CVD908 eine äquivalente
oder höhere
Expression bei CVD908 ergeben hat, wurde erwartet, dass gfpuv effizient
in CVD908-htrA funktionieren würde.
-
Eine
Codierungssequenz wird in einer korrekten Beziehung zu einem Promotor
inseriert, wobei der Promotor und die Codierungssequenz so miteinander
verbunden sind, dass der Promotor die Expression der Codierungssequenz
steuert, so dass das von diesemcodiertes Peptid oder Protein letztlich
produziert wird. Es versteht sich von selbst, dass die Codierungssequenz
auch in einer korrekten Beziehung mit jeder anderen regulatorischen
Sequenz, die vorhanden sein kann, stehen muss.
-
5.8.4 Heterologe Antigene
-
Die
Expressiortsplasmide exprimieren vorzugsweise ein Antigen, das einem
Wirt präsentiert
werden soll, um so eine Immunreaktion auszulösen, was dann zur Immunisierung
und zum Schutz vor einer Erkrankung führt. Während Shiga-Toxine hierin als
Beispiele für
Antigene, die von den hierin offenbarten, als Impfstoff verwendeten
Expressionsplasmiden in nützlicher
Weise exprimiert werden, genannt werden, umfasst die Erfindung in
ihrem breiten Umfang die Expression jedes beliebigen Antigens, das
den bakteriellen Lebendvektor nicht zerstört und eine Immunantwort auslöst, wenn
der bakterielle Lebendvektor, der das (die) Expressionsplasmid(e)
enthält,
einem Wirt, d.h. einem Menschen oder einem Tier, verabreicht wird.
-
Die
hierin als Impfstoffe bereitgestellten Expressionsplasmide werden
dazu verwendet, attenuierte Bakterienstämme, vorzugsweise Stämme zur
Impfung von Menschen, genetisch zu transformieren und ganz besonders
bevorzugt werden sie dazu verwendet, attenuierte S. typhi-Impfstoffstämme, wie
beispielsweise CVD908htrA, zu transformieren, wobei sie vorzugsweise
für entweder
die Untereinheit B von Stx 2 oder ein genetisch detoxifiziertes
Stx 2-Holotoxin codieren.
-
Eine
Untergruppe von STEC, die oft als enterohämorrhagische E. coli (EHEC)
bezeichnet wird, kann ernsthafte klinische Syndrome, die Hämorrhagische
Colitis, Hämolytisch-Urämisches-Syndrom
(HUS) und Thrombotisch-Thrombozytopenische Purpura (TTP) einschließen, in
einem kleinen Teil infizierter Individuen verursachen und daneben
bei den meisten anderen einen nicht-blutigen Durchfall verursachen.
-
Hämorrhagische
Colitis ist durch übermäßigen, blutigen
Durchfall, in der Regel ohne Fieber oder mit leichtem Fieber und
einem relativen Mangel an fäkalen
Leukozyten, wie sich anhand des Durchfallstuhls zeigen lässt, gekennzeichnet.
Diese Eigenschaften unterscheiden eine hämorrhagische Colitis von einer
durch Shigella verursachten Dysenterie, die sich üblicherweise
durch einen geringen, blutigen und schleimigen Stuhlgang, dem hohes
Fieber vorausgegangen ist, und eine große Anzahl an fäkalen Leukozyten,
die unter dem Mikroskop sichtbar sind, kennzeichnet.
-
HUS,
eine potentiell tödliche
Erkrankung, die oft Kleinkinder, aber auch Individuen jeden Alters
befallen kann, ist durch die Dreiergruppe aus mikroangiopathischer
hämolytischer
Anämie,
Thrombozytopenie und Urämie
gekennzeichnet. In Nordamerika ist HUS derzeit die häufigste
Ursache für
akutes Nierenversagen bei Säuglingen
und Kleinkindern. Bei einer Studie von Siegler et al. in Utah von
1970-1994 mit 288 Patienten, die auf HUS nach Diarrhö behandelt
wurden, trat eine ernsthafte Erkrankung (definiert als eine länger als
7 Tage andauernde Anurie, eine länger
als 14 Tage andauernde Oligurie oder extrarenale, strukturelle Schäden, wie zum
Beispiel Herzinfarkt) in 25% aller Fälle auf und war mit Kindern
unter 2 Jahren assoziiert; ungefähr
ein Drittel dieser ernsthaften Fälle
von HUS führten
zum Tod (5%) oder zu ernsthaften Folgeschäden, einschließlich Nierenerkrankung
im Endstadium (5%) oder chronischem Hirnschaden (3-5%) und weniger
ernsthaften chronischen Problemen, die Hypertonie, Proteinurie oder
Azotämie
umfassen.
-
TTP,
die meistens Erwachsene befällt,
ist durch neurologische Komplikationen, wie beispielsweise Herzinfarkt,
neben Thrombozytopenie, hämolytische
Anämie
und Nierenerkrankungen gekennzeichnet.
-
Der
bei weitem häufigste
EHEC-Serotyp ist O157:H7. Dennoch verursachen auch andere EHEC-Serotypen,
einschließlich
O26:H11, O111:H8 und eine Reihe anderer, HUS und hämorrhagische
Colitis. Mit HUS assoziierte EHEC-Stämme vervollkommnen ein oder
mehrere Shiga-Toxine und tragen ein 60 MDa großes Virulenzplasmid. Daneben
beherbergen die meisten auch eine chromosomale Pathogenitätsinsel
(so genannte LEE) mit einer Gruppe von Genen, die für die Fähigkeit
zur Anheftung und Beseitigung codieren. Es wird allgemein angenommen,
dass durch EHEC vervollkommnete Shiga-Toxine eine Schlüsselrolle
in der Pathogenese von hämorrhagischer
Colitis und HUS spielen.
-
Wie
im Folgenden ausführlich
beschrieben wird, besteht die Shiga-Toxin-Familie aus zwei Gruppen von
Toxinen, Stx1 (das im Wesentlichen mit dem von Shigella dysenteriae-Typ 1, dem Shiga-Bacillus,
produzierten Cytotoxin/Neurotoxin/Enterotoxin identisch ist) und
Stx2 (das sich immunologisch von Stx 1 unterscheidet und verschiedene
miteinander verwandte Varianten aufweist). In den USA exprimiert
die überwältigende Mehrheit
von EHEC, die mit Fällen
von HUS assoziiert sind, Stx2, entweder allein oder in Verbindung
mit Stx1.
-
Das
wichtigste Reservoir für
EHEC-Infektionen sind Rinder. Die einzige und maßgeblichste Art der Übertragung
von EHEC auf Menschen erfolgt durch den Verzehr von nicht garem,
kontaminiertem Rindfleisch, am häufigsten
Hackfleisch. Eine Reihe anderer Lebensmittelträger und andere Übertragungsarten
sind dafür weniger
häufig
ursächlich.
EHEC ist vor allem eines der schwierigen bakteriellen Darmpathogene,
die, wie Shigella, durch direkten Kontakt oder durch Kontakt mit
Infektionsträgern übertragen
werden können.
-
Seitens
der meisten Epidemiologen werden große Erwartungen und ein großer Optimismus
darauf gesetzt, dass eine Bestrahlung von Fleisch, das in den USA
verkauft wird, die Übertragung
von EHEC auf Menschen drastisch einschränken wird, da somit die maßgeblichste
Art der Übertragung
eingeschränkt
wird.
-
Dennoch
werden bestimmte Risikogruppen, die anderen Übertragungsarten von EHEC ausgesetzt sind,
nicht von dieser Maßnahme
profitieren. Zum Beispiel erfolgt das Aussetzen der Mitarbeiter
eines Schlachthofs gegen EHEC, was eine berufsbedingte Gefährdung darstellt,
an einem Punkt in der Fleischverarbeitung, der vor dem Einsatz einer
Bestrahlung liegt. Für
solche speziellen Gruppen wie diese, für die das Risiko auch dann
weiterhin besteht, wenn eine Bestrahlung von Fleisch alltäglich wird,
können
anti-EHEC-Impfstoffe
nützlich
sein. Die hierin beschriebenen Plasmide können zur Konstruktion von Impfstoffen gegen
EHEC, die für
die Prophylaxe von Infektionen (in Tierreservaten oder bei Menschen)
und zur Prophylaxe der ernsthaften Komplikationen von EHEC-Infektionen
nützlich
sind, durch Stimulation der Neutralisierung von Shiga-Antitoxin
verwendet werden.
-
Studien
mit attenuierten Vibrio cholerae O1, die die Untereinheit B von
Stx1 exprimieren, haben die Durchführbarkeit des Auslösens von
neutralisierendem Shiga-Antitoxin
durch mucosale Immunisierung mit Lebendvektoren gezeigt. Da jedoch
nahezu alle mit HUS-Fällen
in den USA assoziierte EHEC Stx2, allein oder in Verbindung mit
Stx1, exprimieren, wird bevorzugt, dass ein Impfstoff zur Prophylaxe
von ernsthaften Komplikationen einer EHEC-Infektion durch Auslösen von
das Toxin neutralisierenden Antikörpern anti-Stx2 sowie Stx1
stimulieren sollte. In dem breiten Umfang der vorliegenden Erfindung
ist auch das Bereitstellen eines stabilisierten Plasmidsystems zur
Expression von Stx2-Antigenen, allein oder in Verbindung mit Stx1,
in einem attenuierten S. typhi-Lebendvektor
enthalten.
-
Andere
Antigene, die in gemäß den hierin
beschriebenen Zusammensetzungen und Verfahren, in geeigneter Weise
verabreicht werden können,
schließen
zum Beispiel diejenigen für
Hepatitis B, Haemophilus influenzae Typ B, Hepatitis A, azelluläre Pertussis
(acP), Varicella, Rotavirus, Streptococcus
pneumoniae (Pneumokokken) und Neisseria meningitidis (Meningokokken)
ein. Siehe Ellis et al., Advances in Pharm., 39:393-423, 197.
-
Die
von den Expressionsplasmiden codierten Antigene sind vorzugsweise
Krebsimpfstoffe.
-
Die
von diesen Plasmiden codierten Antigene können so konstruiert werden,
dass sie Immunreaktionen gegen Autoantigene, B-Zellrezeptoren und/oder
T-Zellrezeptoren provozieren, die an Autoimmunerkrankungen oder
immunologischen Erkrankungen beteiligt sind. Wenn beispielsweise
ungünstige
Immunreaktionen gegen Körpergewebe
oder Antigene aus der Umgebung gerichtet werden, können die
erfindungsgemäßen Impfstoffe
gegen die Autoantigene, B-Zellrezeptoren und/oder T-Zellrezeptoren
immunisieren, um so die Reaktionen anzupassen und die Erkrankungen
zu verbessern. Zum Beispiel können
solche Techniken bei der Behandlung von Mysthenia gravis, Lupus
erythematodes, rheumatoider Arthritis, Multipler Sklerose, Allergien und
Asthma wirksam sein.
-
5.8.4.1 Die Shiga-Toxin-Familie
-
1903
berichtete Conradi als erster, dass S. dysenteriae 1 ein starkes
Exotoxin produzierte. Da eine Injektion dieses Toxins zu einer Lähmung der
hinteren Gliedmaßen
eines Kaninchens führte,
wurde es ursprünglich
als Neurotoxin bezeichnet. Später
wurde gezeigt, dass dieses Toxin, das Shiga-Toxin, für bestimmte Zellen
in Gewebekulturen tödlich
war (d.h. es war ein Zytotoxin). Vicari et al. und später Keusch
et al. zeigten, dass es auch als Enterotoxin fungiert.
-
Heute
erkennen Wissenschaftler die Existenz einer Familie von Shiga-Zytotoxinen
an, die die Proteinsynthese hemmen, was den Tod der dafür anfälligen Zellen
bewirkt. Viele Jahre nach der Offenbarung, dass solche Toxine neben
dem ursprünglichen
Shiga-Toxin, das von Shigella dysenteriae Typ 1 produziert wurde, von
bestimmten E. coli-Stämmen
produziert wurden, war die Nomenklatur dieser Familie von Toxinen
verwirrend. Da erste Berichte die Aktivität dieser Toxine auf Vero-Zellen
(eine aus Nierenepithelzellen von afrikanischen grünen Meerkatzen
[African green monkey kidney epithelial cells] stammende Zelllinie)
beschreiben, wurden sie von vielen Forschern Verotoxine genannt.
Andere bezeichneten diese in E. coli exprimierten Toxine als Shiga-gleiche
Toxine.
-
Die
Proteintoxine werden hierin insgesamt als Shiga-Toxine (Stx) bezeichnet
und die Gene, die für
diese Toxine codieren, werden als stx bezeichnet, wobei die tief
gestellten Indices die Gruppe und Variante angeben [d.h. stx1 für
das von E. coli produzierte Shiga-Toxin, das im Wesentlichen mit dem von
Shigella dysenteriae Typ 1 (stx) identisch ist, und stx2,
stx2c, stx2d stx2e für
die sich hinsichtlich der Antigene unterscheidenden Gruppen miteinander
verwandten Toxinen].
-
Die
Struktur, Biochemie und Antigenität von Shiga-Toxinen sind allgemein
bei Melton-Celsa et al., Escherichia coli 0157:H7 and Other Shiga
Toxin producing E. coli Strains, 1998; Takeda, Bacterial and Virulence
Factors in Disease, 1995; Gyles, Canadian J. of Microbiology, 38:734,
1992; und O'Brien
et al., Current Topics in Microbiology and Immunology, 180:165,
1992, beschrieben.
-
Diese
Shiga-Zytotoxine bestehen aus einer einzelnen, katalytischen Untereinheit
A von ungeführ
32 kDa, die nicht-kovalent an eine pentamere Rezeptorbindungsdomäne von ungefähr 7,7 kDa
großen
B-Untereinheiten gebunden ist. Diese Untereinheiten werden von einem
einzelnen Operon mit der Reihenfolge stxA-stxB codiert; die Transkription
der stx- und stx1-Operons wird sowohl in
S. dysenteriae Typ 1 als auch bei E. coli durch Eisen reguliert,
es wurden jedoch bislang keine aus der Umgebung stammenden Kontrollsignale für irgendein
stx2-Operon bestimmt. Keines dieser Toxine
wird auf einem Plasmid codiert; vielmehr werden sie von Phagen (Stx1,
Stx2, Stx2c und Stx2d) oder von Chromosomen (Stx, Stx2e) codiert.
-
Wie
oben angegeben ist, sind alle Mitglieder der Shiga-Toxin-Familie
cytolytische Toxine, die die Proteinsynthese in dafür empfänglichen
Zellen hemmen, indem sie die Bindung der von Elongationsfaktor 1
abhängigen
Aminoacyl-tRNA an Ribosome blockieren. Bei allen Toxinen, die bei
humanen Infektionen identifiziert wurden, folgt eine Penetration
dafür empfänglicher
Zellen durch Endozytose der Bindung des Holotoxins an den notwendigen
Glycolipid-Rezeptor Globotriaosylceramid (Gb3)
auf der Zeltoberfläche,
wodurch das Toxin in den Golgi-Apparat und in das endoplasmatische
Reticulum geleitet und anschließend
in das Zytoplasma freigesetzt wird. Shiga-Toxine sind RNA-N-Glycosidasen,
die ein einzelnes Adenin aus der 28S-RNA der eukaryotischen 60S-ribosomalen
Untereinheit depurinieren, wodurch die 60S-Untereinheit inaktiviert
wird und dies möglicherweise
zum Tod der Zelle führt.
-
Es
gibt sechs prototypische Mitglieder der Shiga-Toxin-Familie: Stx,
Stx1, Stx2, Stx2c, Stx2d und Stx2e, die sich in immunologischer
Hinsicht und in ihrer toxischen Aktivität voneinander unterscheiden.
Es sind hierin signifikante Einzelheiten aufgeführt, die den Hintergrund für das Verständnis der
Bedeutung von Punktmutationen, die nachfolgend erörtert werden
und für
genetisch detoxifizierte Holotoxine erforderlich sind, bereitstellen.
-
Die
Mitglieder der Shiga-Toxin-Familie unterscheiden sich auf 3 grundlegende
Arten voneinander, wie vor kurzem von Melton-Celsa et al., Escherichia
coli O157:H7 and Other Shiga toxin-producing E. coli strains, 1998
zusammengefasst wurde.
- (1) Immunologisch: Die
Shiga-Toxin-Familie besteht aus 2 Serogruppen, Stx/Stx1 und Stx2;
Antiseren, die gegen Stx/Stx1 gerichtet sind, neutralisieren nicht
die Mitglieder der Stx2-Serogruppe, wie mit dem Verozellzytotoxizitätstest bestimmt
wurde.
- (2) Strukturell: Stx und Stx1 sind im Wesentlichen identisch,
unterscheiden sich in einer einzigen Aminosäure an der Position 45 der
reifen Untereinheit A und die Kristallstruktur des Stx-Holotoxins
wurde gelöst. Der
Prototyp Stx2 ist nur 55% homolog zu den Resten der reifen Untereinheit
A von Stx/Stx1 und 57% homolog zu der reifen Untereinheit B, was
erklärt,
warum die gegen Stx/Stx1 gerichteten Antiseren nicht die Mitglieder
der Stx2-Gruppe neutralisieren. In der Stx2-Gruppe ist Stx2e am
weitesten entfernt verwandt und teilt 93% Aminosäurehomologie mit der reifen
Untereinheit A von Stx2 und 84% Homologie mit der reifen Untereinheit
B; Stx2c und Stx2d sind Stx2 sehr ähnlich und teilen 99-100% Homologie
mit den Resten der reifen Untereinheit A und 97% Homologie mit den
Resten der reifen Untereinheit B.
- (3) Zytotoxizität:
Stx2 ist eines der am meisten letalen Shiga-Toxine mit einer in
Mäuse intraperitoneal
injizierten LD50 von 0,5-2 ng. Die LD50 bei Stx1 und Stx2e beträgt 200-400 ng und 1-5 ng
bei Stx2d; Stx2d ist jedoch dahingehend ungewöhnlich, dass dieses Toxin durch
die Darmschleimhaut von Mäusen
aktiviert werden kann, wobei die Toxizität des Toxins zunimmt und dadurch
die LD50 auf 0,5 ng sinkt.
-
5.8.5 Ortsspezifische Mutagenese von Shiga-Toxinen
-
Wir
beschreiben ein genetisch detoxifiziertes Shiga-Toxin. Die Detoxifizierung
wird von einer ortsspezifischen Mutagenese begleitet, die zwei definierte
und genau voneinander abgegrenzte Punktmutationen, die entscheidende
Reste in der katalytischen Stelle der Untereinheit A verändern, einführt. Zwei
weitere definierte und genau voneinander abgegrenzte Punktmutationen
werden in die Untereinheit B eingeführt, um die entscheidenden
Reste in der primären
Bindungsstelle (d.h. SITE I), die innerhalb der von den benachbarten
Untereinheiten B des pentameren Holotoxinrings gebildeten Spalte
liegt, zu verändern.
-
Es
wurden bereits Versuche durchgeführt,
um die Bindungsstelle SITE II mit geringerer Affinität zu verändern. Diese
Bindungsstelle wurde jedoch nur aus Untersuchungen mit Hilfe von
Molekularmodellierung identifiziert und wird weitgehend nicht Untersuchungen
der Mutation gestützt,
die eine Bindung des Gb3-Rezeptors an SITE
I befürworten.
Selbst wenn SITE II eine alternative Bindungsstelle mit geringer
Affinität
ist, die einen Zugang unseres mutanten Holotoxins zu dafür empfänglichen
Zellen zulässt,
kann die Inaktivierung der katalytischen Domäne dennoch den Zelltod verhindern.
-
Auf
der Basis von Aminosäuresequenz-Alignments,
Untersuchungen der Röntgenstrahlkristallographie
und Untersuchungen mit Hilfe von Molekularmodellierung wurden essentielle
Aminosäuren,
die die aktive Stelle in der katalytischen Untereinheit A von Stx
umfassen, sowie diejenigen Reste, die die Bindung von SITE I mit
dem Pentamer der Untereinheit B von Stx/Stx1 umfassen, identifiziert.
Die Schlussfolgerung des Erfinders geht dahin, dass die für die aktive
Stelle essentiellen Aminosäuren
ausgewählt
sind aus der Gruppe bestehend aus Tyr 77, Tyr 114, Glu 167, Arg
170 und Trp 203. Die Reste, die vermutlich für die Bindung des Rezeptors an
die von den benachbarten Untereinheiten B gebildeten Spalte erforderlich
sind, schließen
Lys 13, Asp 16, Asp 17, Asp 18, Thr 21, Glu 28, Phe 30, Gly 60 und
Glu 65 ein. Diese vorhergesagten Orte stimmen mit Untersuchungen
der Funktionen und in-vivo-Versuchen unter Verwenden definierter
Einzel- und Doppelmutationen innerhalb einzelner Domänen des
Holotoxins, die mittels ortsspezifischer Mutagenese eingeschleust
wurden, überein.
Eine Zusammensetzung solcher Mutationen ist in Tabelle 1 angegeben.
Auf der Basis von diesen Daten und kristallographischen Vorhersagen
wird die Bereitstellung von Expressionsplasmiden, die für Shiga-Toxine
codieren, welche zwei spezifische Gruppen von Punktmutationen innerhalb
von sowohl der Untereinheit A als auch der Untereinheit B aufweisen,
zur Erzeugung nicht-toxischer, mutanter Stx2-Holotoxine für eine Verwendung
als Impfstoff, zum Beispiel mittels einer Expression in attenuierten
S. typhi-Lebendvektoren wie beispielsweise CVD908htrA, von der weit
reichenden praktischen Ausführbarkeit
der Erfindung erfasst. Tabelle 1. Untersuchungen der ortsspezifischen
Mutagenese
| Untereinheit | Toxin | Mutation | Abnahme
der Zytotoxi-zität | Abnahme
der Letalität | neutralisierende
Antikörper |
| A | Stx1 | Leu201 → Val + von
Resten 202-213 | keine
Zytotoxizität | - | - |
| Stx1 | Glu167 → Asp | 103 | - | - |
| Stx1 | Arg170 → Leu | 103 | - | - |
| Stx2 | Glul67 → Asp | 103 | - | - |
| Stx2e | Glu167 → Asp | 104 | - | - |
| Stx2e | Arg170 → Lys | 10 | - | - |
| Stx2e | Glu167 → Asp Arg170 → Lys | 104 | | |
| Stx2e | Glu167 → Gin | 106 | 104 | Y |
| B | Stx | Asp 16 → His + Asp17 → His | keine
Zytotoxizität | - | - |
| Stx | Arg33 → Cys | 108 | | - |
| Stx | Gly60 → Asp | 106 | | - |
| Stx1 | Phe30 → Ala | 105 | 10 | Y |
| Stx2 | Ala42 → Thr | 103-104 | Y | Y |
| Stx2 | Gly59 → Asp | 103-104 | Y | Y |
-
5.9 Pharmazeutische Formulierungen
-
Es
wird in Erwägung
gezogen, die hierin beschriebenen bakteriellen Vektor-Lebendimpfstoffe
zur Verwendung bei der Impfung von Individuen, vorzugsweise Menschen,
in pharmazeutischen Formulierungen zu verabreichen. Solche pharmazeutischen
Formulierungen können
pharmazeutisch wirksame Träger
und gegebenenfalls andere therapeutische Bestandteile, wie zum Beispiel
verschiedene, im Stand der Technik bekannte Hilfsstoffe, umfassen.
-
Der
Träger
oder die Träger
müssen
dahingehend pharmazeutisch verträglich
sein, dass sie mit den therapeutischen Bestandteilen kompatibel
sind und den Empfänger
derselben nicht übermäßig schädigen. Der therapeutische
Bestandteil oder die therapeutischen Bestandteile wird (werden)
in einer Menge und Häufigkeit bereitgestellt,
die dazu erforderlich ist, den gewünschten immunologischen Effekt
zu erreichen.
-
Die
Verabreichungsart und die Dosierungsformen werden die therapeutischen
Mengen der Verbindungen, die für
die Anwendung der Impfungen gewünscht
und wirksam sind, beeinflussen. Die bakteriellen Vektor-Lebendmaterialien
werden in einer Menge verabreicht, die eine Immunreaktion dadurch
auslösen
kann, dass sie effektiv darin ist, die Immunreaktion des Patienten
auf das exprimierte, mutante Holotoxin oder auf ein anderes gewünschtes
heterologes Antigen oder andere gewünschte heterologe Antigene
zu erhöhen.
Eine im Hinblick auf die Immunisierung wirksame Menge ist eine Menge,
die ein erhöhtes
Vermögen.
zur Prophylaxe, Verzögerung
oder Senkung der Ernsthaftigkeit bei der Entstehung einer Erkrankung
verleiht, wenn sie mit dem entsprechenden Vermögen ohne eine solche Immunisierung
verglichen wird. Ein Fachmann wird durchaus verstehen, dass die
Menge, die auf den Faktoren, wie beispielsweise dem Gewicht und
dem Gesundheitszustand des Empfängers,
der Art des zu exprimierenden Proteins oder Peptids, der Art des
zu bekämpfenden, infizierenden
Organismus und der Art der Verabreichung und der Art der Zusammensetzungen
basiert, variieren wird.
-
Die
Verabreichungsarten können
die Verwendung irgendeines geeigneten Mittels und/oder Verfahrens zur
Verabreichung der bakteriellen Vektor-Lebendimpfstoffe an eine Körperstelle
des Wirtslebewesens, an dem die bakteriellen Vektor-Lebendimpfstoffe
in immunstimulierender Hinsicht effektiv sind, umfassen.
-
Verabreichungsarten
können,
ohne darauf beschränkt
zu sein, parenterale Verabreichungsverfahren, wie beispielsweise
subcutane (SC) Injektion, intravenöse (IV) Injektion, transdermale,
intramuskuläre
(IM), intradermale (ID), sowie nicht-parenterale, z.B. orale, nasale,
intravaginale, pulmonale, ophtalmische und/oder rektale Verabreichung
einschließen.
-
Der
Dosisanteil und geeignete Dosierungsformen für die bakteriellen Vektor-Lebendimpfstoffzusammensetzungen
können
leicht von Fachleuten ohne übermäßige Versuchsdurchführungen,
mit Hilfe herkömmlicher
Methoden zur Bestimmung der Antikörpertiter und herkömmlicher
Protokolle zur biologischen Wirksamkeit/Biokompatibilität bestimmt
werden. Neben anderen Dingen hängen
der Dosisanteil und geeignete Dosierungsformen von dem speziell
verwendeten Antigen, der gewünschten
therapeutischen Wirkung und der gewünschten Zeitspanne für die Bioaktivität ab.
-
Die
bakteriellen Vektor-Lebendimpfstoffe können einem Wirtslebewesen in
der Regel mit beliebigen anderen geeigneten, pharmazeutischen oder
physiologischen Wirkstoffen, z.B. antigenen und/oder biologisch aktiven
Substanzen verabreicht werden.
-
Die
Formulierungen können
beispielsweise als diskrete Einheiten, wie zum Beispiel Kapseln,
Oblatenkapseln, Tabletten, Pastillen, wobei jedes eine vorher bestimmte
Menge der Vektorübertragungsstruktur
enthält;
oder als Suspension zubereitet werden.
-
6. Beispiele
-
Eine
isogene Reihe von Expressionsplasmiden, die aus einzelnen Kassetten
bestehen, wurde zur Verwendung in bakteriellen Vektor-Lebendimpfstoffen,
wie beispielsweise E. coli und Salmonella, konstruiert. Mit Ausnahme
der ribosomalen Bindungsstellen (RBS), sind die entscheidenden genetischen
Loci, die die Initiierung und Terminierung der Transkription und
Plasmidreplikation kontrollieren oder die für die exprimierten Proteine
codieren, in definierten Restriktionsfragmenten, wie beispielsweise
mit dem repräsentativen
Plasmid-Diagramm
von pGEN2 dargestellt wird, das in 1A zu
sehen ist, enthalten. Zunächst
wird die Basisstruktur dieser Expressionsplasmide herausgestellt
und dann werden die Daten, die die Funktion eines jeden Locus innerhalb
des attenuierten Impfstoffstamms CVD908-htrA angeben, zusammengefasst.
-
6.1 pGEN-Struktur
-
Die
Transkription eines beliebigen heterologen Antigens, das in CVD908-htrA
exprimiert werden soll, wird in erster Linie von einem induzierbaren
Promotor, der auf einer EcoRI – BglII-Kassette
enthalten ist, kontrolliert. Da die Expressionsplasmide zu Beginn
mit pTETnir15 modelliert wurden, trugen frühe Versionen den anaerob aktivierten
Promotor nir15 (Pnir15). Dieser Promotor
wurde jedoch gegen einen strenger regulierten, osmotisch kontrollierten
Promotor PompC ausgetauscht, der leicht
in vivo durch Variieren der Konzentration von NaCl manipuliert werden
kann.
-
Heterologe
Antigene sind auf einer BglII – AvrII-Kassette
enthalten, die von optimierten RBS an dem 5'-proximalen Ende und einem Transkriptionsterminator
trpA am 3'-distalen
Ende dieser Kassette flankiert wird. Der Replikationsursprung für diese
Expressionsplasmide wurde als AvrII – BglII-Kassette konstruiert
und wird vor einer durchlesenden Transkription, die aus flankierenden
Regionen stammt, geschützt.
Diese Kassetten tragen ein extrem effizientes Derivat des Transkriptionsterminators
T1T2 an einem Terminus und den Transkriptionsterminator trpA von
der heterologen Antigenkassette an dem entgegen gesetzten Ende der
Replikationskassette.
-
Die
flankierenden BglII- und SpeI-Orte (siehe 2)
zwischen der Replikationskassette und der Selektionskassette sollen
eine Plasmidstabilisierungsfunktion, wie beispielsweise denpar-Locus
von pSC101, inserieren. Die in den Plasmiden enthaltenen Selektionskassetten
sind innerhalb der SpeI – XbaI-Kassetten
enthalten und können
zum Beispiel zum Codieren der Resistenz gegen Carbenicillin (des
bla-Gens) oder der Resistenz gegen Tetracyclin (des tetA-Gens, siehe 1) verwendet werden.
-
Die
Arzneimittelresistenzkassette kann gegen das ssb-Gen, das für das essentielle,
einzelsträngige Bindungsprotein
von Salmonella typhi CVD908-htrA codiert, ausgetauscht werden.
-
Die
flankierenden XbaI- und EcoRI-Stellen zwischen der Selektionskassette
und PompC sollen weitere Stabilisierungsfunktionen,
einschließlich
eines PSK-Locus, wie beispielsweise hok-sok (siehe 1 und 2), oder eine weitere Partitionsfunktion,
wie beispielsweise den parA-Locus von pR1 (siehe 7)
inserieren.
-
6.2 Modifizierter ompC-Promotor
-
Es
war vorgesehen, dass jeder Promotor, der die Transkription eines
heterologen Gens kontrolliert, für ein
aus der Umgebung stammendes Signal mit biologischer Relevanz verantwortlich
war. Für
die hierin beschriebenen Expressionsplasmide wurde eine ompC-Promotorkassette
(PompC) von E. coli verwendet, die durch zunehmende Osmolarität induziert
wird. Eine Konstruktion dieser Kassette basierte auf der offen gelegten
Sequenz von PompC, die von Norioka et al.
(Norioka et al., 1986) offenbart wurde, und wurde unter Verwenden synthetischer
Primer zur Erzeugung einer 459 bp langen EcoRI-Bg/II-Kassette, in
der die natürliche
RBS entfernt worden war, durchgeführt.
-
Um
zu bestätigen,
dass dieser Promotor in CVD908-htrA osmotisch kontrolliert war,
wurde ein Derivat von pTETnir 15 konstruiert, in dem Pnir15 – toxC gegen
eine Kassette, die aus der von PompC gesteuerten
Expression einer promotorlosen aphA2-Kassette, die Resistenz gegen
Kanamycin verleiht, bestand, ausgetauscht. Dieses Plasmid, als pKompC
bezeichnet, wurde mittels Elektroporation in CVD908-htrA eingeschleust,
und die Empfänger
auf eine Resistenz gegen Kanamycin in LG-Medium gescreent. Die osmotisch regulierte
Expression von aph-2 wurde bestimmt, indem CVD908-htrA(pKompC) in
50 ml supplementiertem Nährmedium
(NB), das von 0 auf 300 μg/ml
ansteigende Konzentrationen an Kanamycin enthielt, angeimpft wurden;
eine parallele Gruppe von Kulturen wurde mit den gleichen Bereichen
von zugegebenem Kanamycin angesetzt, das jedoch auch 10% Sucrose
zur Induktion von PompC enthielt. Die Kulturen
wurden über
Nacht bei 37°C
inkubiert und die OD600 gemessen. Die Ergebnisse
sind in Tabelle 2, Versuch 1 angegeben.
-
Tabelle
2 zeigt die Induktion des Promotors P
ompC,
der die Expression der Resistenz gegen Kanamycin kontrolliert, durch
die Osmolarität
in dem attenuierten S. typhi-Lebendvektor
CVD908-htrA. Tabelle 2
| Versuch
11 | Versuch
22 |
| Konzentration von
Kanamycin (μg/ml) | Geringe
Osmolarität
(OD600) | 10%
Sucrose (OD600) | Konzentration von
Kanamycin /ml (μg/ml) | Geringe
Osmolarität
(OD600) | 300
mM NaCl (OD600) |
| 0 | 0,92 | 0,35 | 0 | 0,95 | 1,04 |
| 50 | 0,13 | 0,35 | 200 | 0,04 | 0,99 |
| 100 | 0,07 | 0,31 | 400 | 0,02 | 0,96 |
| 200 | 0,03 | 0,21 | 600 | 0,01 | 0,92 |
| 300 | 0,02 | 0,19 | 800 | 0,01 | 0,92 |
-
Eine
Kultur von CVD908-htrA(pKompC) wurde in LB-Nährmedium, das mit 0,0001% (w/v)
2,3-Dihydroxybenzoesäure
(DHB) und 50 μg/ml
Kanamycin supplementiert war, angesetzt und 16 Stunden lang bei 37°C inkubiert.
Diese Ausgangskultur wurde dann 1:10 in frischem Medium verdünnt und
2 Stunden lang bei 37°C
inkubiert, um eine Animpfkultur exponentiell wachsender Bakterien
bereitzustellen. 50 μl
dieser Kultur wurden dann in Kulturen von 50 ml Nährmedium
(NB), das wie oben mit DHB supplementiert war, jedoch ansteigende
Konzentrationen von Kanamycin aufwies, angeimpft; eine parallele
Gruppe von Kulturen wurde mit den gleichen Bereichen an dazugegebenem
Kanamycin angesetzt, enthielt jedoch zusätzlich 10% Sucrose, um hoffentlich
PompC zu induzieren. Die Kulturen wurden über Nacht
bei 37°C
inkubiert und die OD600 gemessen.
-
2 Kultur von CVD908-htrA(pKompC) in supplementiertem
NB-Nährmedium
und Kanamycin wurde 16 Stunden lang bei 37°C inkubiert, 1:10 in frischem
Medium verdünnt
und 2 Stunden lang bei 37°C
inkubiert, um eine Animpfkultur exponentiell wachsender Bakterien
bereitzustellen. 100 μl-Aliquots
aus dieser Kultur wurden dann in Kulturen von 50 ml NB-Nährmedium,
das von 200 auf 800 μg/ml
ansteigende Konzentrationen von Kanamycin enthielt, angeimpft; eine
parallele Gruppe von Kulturen, die 300 mM NaCl enthielten, wurde
angesetzt und alle Kulturen wurden 16 Stunden lang bei 37°C inkubiert
und die OD600 gemessen.
-
Trotz
des selektiven Drucks durch die Verwendung von Kanamycin, übte die
Gegenwart von 10% Sucrose einen inhibitorischen Effekt auf das Wachstum
von CVD908-htrA(pKompC)
aus. Die Ergebnisse legen jedoch nahe, dass PompC von
E. coli osmotisch kontrolliert war, wenn er die Expression des aph-2-Gens
in CVD908-htrA(pKompC) steuerte. Um dies zu bestätigen, wurde CVD908-htrA(JKompC)
in 50 ml supplementierter NB-Nährmedium,
das von 200 auf 800 μg/ml
ansteigende Konzentrationen an Kanamycin enthielt, angeimpft; eine
parallele Gruppe, die erneut 300 mM NaCl enthielt, wurde angesetzt,
um PompC zu induzieren. Die Kulturen wurden
16 Stunden lang bei 37°C
inkubiert und die Ergebnisse sind in Tabelle 2, Versuch 2 angegeben.
Es hat sich bestätigt,
dass die durch PompC gesteuerte Expression
des aphA-2-Gens in CVD908-htrA eine Resistenz gegen Kanamycin in
Mengen von bis zu 800 μg/ml
in einer durch Osmolarität
regulierten Weise verleiht.
-
Die
aph-Genkassette wurde dann gegen eine 756 bp große BglII – NheI-Kassette ausgetauscht,
die das für
GFPuv codierende gfpuv-Allel enthielt. Während des visuellen Screenings
der mit ultraviolettem Licht durchleuchteten E. coli-Kolonien wurde
eine sehr hell fluoreszierende Kolonie und eine andere beispielhafte, fluoreszierende
Kolonie zur weiteren Untersuchung ausgesucht und diese entsprechend
Klon 1 und Klon 3 genannt wurden. Nach der Reinigung der beteiligten
Plasmide wurde bestimmt, dass Klon 1 ein Plasmid enthielt, dass
nicht länger
die BglII-Stellen trug, die PompC und gfpuv
voneinander abgrenzen, wohingegen Klon 3 die erwartete BglII-Stelle
trug. Wir untersuchten die Induktion der Expression von GFP, wenn
die Klone 1 und 3 auf Nähragar
in An- oder Abwesenheit von NaCl angezüchtet wurden und bestimmten
durch optische Untersuchung, dass Klon 3 eine nur sehr geringe Fluoreszenz
zeigte, wenn er auf NaCl-haltigem Nähragar angezüchtet wurde,
jedoch hell fluoreszierte, wenn er auf 300 mM NaCl enthaltendem
Nähragar
ausplattiert wurde (Daten sind nicht gezeigt). Klon 1 wies jedoch
eine höhere
Background- Fluoreszenz auf, wenn er nicht induziert wurde, fluoreszierte
jedoch intensiv, wenn er mit 300 mM NaCl induziert wurde. Um die
Mutationen in dem gfpuv-Gen, die die Fluoreszenz beeinträchtigen
könnten,
zu bestimmen, tauschten wir PompC von Klon
1 gegen PompC von Klon 3 aus und bestätigten so
die erwartete Abnahme der Fluoreszenz, wie sie mittels Durchleuchtung
bestimmt wurde (Daten sind nicht gezeigt). Wir schlossen daher,
dass die Unterschiede in der beobachteten Fluoreszenz von zwei genetisch
verschiedenen Versionen des PompC-Promotors,
die wir Pomp1 (höhere Transkriptionsmengen bei
geringerer osmotischer Kontrolle) und PompC3 (mäßige Transkriptionsmengen
bei einer osmotischen Kontrolle, die derjenigen, die bei der oben
beschriebenen PompC – aph-Kassette beobachtet wurde, ähnelte)
nannten, kontrolliert wurden; wir nannten die Plasmide, die diese
Expressionskassetten enthielten, entsprechend pGFPompC1 und pGFPompC3.
-
Um
die Unterschiede in der induzierten und nicht-induzierten Expression
von gfpuv, die von PompC1 und PompC3 kontrolliert
wurden, zu quantifizieren, wurde die Synthese von GFPuv in E. coli
DH5α und
S. typhi CVD 908-htrA mittels Durchflusszytometrie überwacht.
Diese wirkungsvolle Methode besitzt den einzigartigen Vorteil, der
eine schnelle Messung der Expression von GFPuv in großen Anzahlen
von einzelnen Bakterien sowie eine exakte Bestimmung der mittleren
Fluoreszenzintensität
durch die Synthese von GFPuv in jeder der analysiertem Bakterienpopulationen
zulässt.
Um dies zu erreichen, wurden pGFPompC1 und pGFPompC3 mittels Elektroporation
eingeschleust und die Kolonien auf supplementiertem 1X LB-Agar,
der 100 μg/ml
Carbenicillin enthielt, nachdem sie 48 Stunden lang bei 30°C angezüchtet worden
waren, isoliert. Die isolierten Kolonien wurden sodann angezüchtet und
die Kulturen als Master Stocks (Ausgangsstock) schockgefroren. Anschließend wurden
frische Kolonien in entweder supplementiertem Nährmedium oder in supplementiertem
Nährmedium,
das 150 mM NaCl enthielt, angeimpft und 24 Stunden lang bei 37°C mit 250
rpm angezüchtet;
die Unterschiede in der OD600 bei jeder
Kultur betrugen nie mehr als 0,07. Die Induktion der Expression
von gfpuv, die von PompC1 und PompC3 kontrolliert
wurde, wurde mittels Durchflusszytometrie analysiert und die Ergebnisse sind
in Tabelle 3 angegeben.
-
Tabelle
3 zeigt einen Vergleich der Induktion von P
ompC1 und
P
ompC3, die die Expression von GFPuv kontrollieren,
in den Wirtsstämmen
E. coli DH5α und
CVD 908-htrA.
1 Tabelle 3
| Stamm | Geringe Osmolarität (OD600) | Mittlere Fluoreszenz-intensität | 150 mM NaCl (OD600) | Mittlere Fluoreszenzintensität | Induktions-verhältnis2 |
| DH5α | 0,61 | 0,28 | 0,95 | 0,29 | NA3 |
| DH5α (pGFPompC1) | 0,56 | 4,45 | 0,72 | 7,69 | 1,7 |
| DH5α (pGFPompC3) | 0,58 | 1,77 | 0,73 | 4,21 | 2,4 |
| CVD
908-htrA | 0,58 | 0,27 | 0,65 | 0,26 | NA |
| CVD 908-htrA (pGFPompC1) | 0,60 | 5,37 | 0,54 | 23,4 | 4,4 |
| CVD 908-htrA (pGFPompC3) | 0,54 | 2,56 | 0,53 | 17,1 | 6,7 |
- 1Alle Stämme wurden
aus den eingefrorenen Master Stocks aus 2X LB-Agar, der mit DHB
und 50 μg/ml
Carbenicillin supplementiert war, ausgestrichen und 36 Stunden lang
bei 30°C
inkubiert. Die isolierte Kolonien wurden in einem Pool in 300 μl NB-Nährmedium,
das mit DHB und Carbenicillin supplementiert war, gesammelt und
25 μl davon
in 25 ml supplementiertem NB-Nährmedium,
mit und ohne 150 mM NaCl, angeimpft und 24 Stunden lang bei 37°C mit 250
rpm inkubiert. Die Bakterien wurden dann pelletiert, in 1 ml PBS,
pH 7,4, resuspendiert und anschließend für eine Analyse mittels Durchflusszytometrie
1:1000 in PBS verdünnt.
- 2Definiert das Verhältnis der mittleren Fluoreszenzintensität, die nach
der Induktion mit 150 mM NaCl gemessen wurde, geteilt durch die
Grundhöhe
der mittleren Fluoreszenzintensität, die bei geringer Osmolarität gemessen
wurde.
- 3NA = nicht anwendbar.
-
Die
Grundmenge der Expression für
die PompC1 – gfpuv-Kassette ist 2,5-mal
größer als
das der PompC3-Kassette, wenn sie in DH5α exprimiert
wird, und 2,1-mal größer, wenn
sie in CVD 908-htrA exprimiert wird; die Grundhöhe der Fluoreszenz, die bei
der Synthese von GFPuv nachgewiesen wurde, überstieg trotz des Backgrounds
vom Wirt niemals eine Fluoreszenzintensität von 5,37,. Wenn wir das Induktionsverhältnis als
das Verhältnis
der mittleren Fluoreszenzintensität, die nach der Induktion gemessen
wurde, geteilt durch die Grundhöhe
der mittleren Fluoreszenzintensität definieren, wurde beobachtet,
dass PompC1 und PompC3,
wenn sie mit 150 mM NaCl induziert wurden, in DH5α Induktionsverhältnisse
von entsprechend 1,7 und 2,4 zeigten. Das Induktionsverhältnis bei
PompC1, wenn es in CVD 908-htrA gemessen wurde,
betrug überraschenderweise 4,4
und ergab bei diesen Versuchen eine maximale mittlere Fluoreszenzintensität von 23,4.
Obwohl das Induktionsverhältnis
von PompC3 in CVD 908-htrA bei 6,7 lag,
war die mittlere Fluoreszenzintensität von 17,1 geringer als die
bei PompC1 gemessene. Auf der Basis dieser
Daten scheint PompC1 der stärkste und
noch osmotisch kontrollierte der beiden ompC-Promotoren zu sein.
Daher wurde PompC1 für die Synthese des breitest
möglichsten
Bereichs von heterologen Testantigenen zur Untersuchung der Effekte
einer solchen Synthese auf die Plasmidstabilität ausgesucht.
-
Diese
Daten zeigen deutlich, dass, wenn sie die Expression von gfpuv in
dem Lebendvektorstamm CVD 908-htrA steuern, PompC1 und
PompC3 durch die zunehmende Osmolarität induzierbar
sind, obwohl die Grundmenge der Transkription in beiden Fällen noch
erheblich ist. Die unter diesen Bedingungen geringer Osmolarität beobachteten
Ergebnisse belegen zudem unsere Beobachtung unter Verwenden von
festen Medien, bei denen festgestellt wurde, dass PompC1 eine
höhere
Expression von heterologen Antigenen steuert als PompC3. Da
festgestellt wurde, dass PompC3 die gewünschte 3'-terminale BglII-Stelle
besitzt, die bei PompC1 nicht nachgewiesen
werde konnte, bestimmten wir die Nukleotidsequenz von PompC1 für einen
eventuellen Nachweis von (einer) Punktmutation(en), welche die Stärke von
PomC1 erklären könnten. Die einzigen Unterschiede,
die identifiziert werden konnten, waren am 3'-Terminus der Kassette lokalisiert.
Die gewünschte
Sequenz in dieser Region war 5'-...catataacAGATCTtaatcatccacAGGAGGatatctgATG-3' (SEQ ID No. 4)
(von links nach rechts, der obere Fall gibt entsprechend die BglII-Stelle,
die Ribosomenbindungsstelle und das GFPuv-Startcodon an); die aktuelle
Sequenz ergab sich zu 5'-...catataacAGATCGATCTtaaAcatccacAGGAGGAtAtctgATG-3
(SEQ ID No. 5) (inserierte oder geänderte Basen sind im oberen
Fall durch Fettdruck und Unterstreichen angegeben). Diese innerhalb
der Sequenz des ompC1-Promotors nachgewiesenen Änderungen sind offensichtlich
für die
Zunahme der beobachteten Stärke
von PompC1 aufgrund eines unbekannten Mechanismus verantwortlich,
da sich weder die Grundsequenz des ompC-Promotors noch die optimierte Ribosomenbindungsstelle
spontan verändert
haben.
-
6.3 Replikationsursprünge und Selektionskassetten
-
Der
Erfolg der Expression potentiell toxischer oder auf andere Weise
problematischer, heterologer Antigene in CVD908-htrA hängt von
der Kopienzahl des Expressionsplasmids ab. Daneben werden die beobachteten
Immunreaktionen auf ein gegebenes heterologes Antigen von der Kopienzahl
des (der) Gens (e), die für das
Antigen codieren beeinflusst, wobei die von Chromosomen exprimierten
Antigene im Vergleich zu der auf Plasmiden beruhenden Expression
schwächere
Immunreaktionen hervorrufen.
-
Eine
optimierte Immunantwort wird von der auf Vielkopie-Plasmiden beruhenden
Expression des (der) heterologen Antigens (e) von Plasmiden mit
der geeigneten Kopienzahl abhängen.
-
Da
die geeignete Kopienzahl für
ein gegebenes heterologes Gen nicht a priori bekannt sei kann, stellt die
vorliegende Erfindung eine Gruppe. von Expressionsplasmiden bereit,
die die Replikationsursprünge
oriE1 (amplifiziert aus pAT153; Kopienzahl ~ 60), ori15A (amplifiziert
aus pACYC184; Kopienzahl ~ 15) und ori101 (amplifiziert aus pSC101;
Kopienzahl ~ 5) enthalten. Diese vollständigen Replikationskassetten
werden alle von BglII -BamHI-Fragmenten
getragen und sind für
eine Gruppe von 3 Tetracyclinresistenz-tragenden Expressionsplasmiden
in den 1A-1C gezeigt.
-
Die
Expression der durch PompC1 kontrollierten
gfpuv-Expressionskassette, die auf diesen Expressionsplasmiden enthalten
war, wurde mittels Durchflusszytometrie analysiert. Diese Versuche
wurden so konstruiert, dass nachgewiesen werden konnte, ob Unterschiede
in der Höhe
der beobachteten Fluoreszenz mit den erwarteten Kopienzahlen eines
gegebenen Expressionsplasmids in Verbindung gebracht werden konnten. CVD908-htrA-Stämme, die pGEN2,
pGEN3 und pGEN4 trugen, wurden auf reichhaltigem SuperAgar-Medium, das,
wenn geeignet, mit DHB und 20 μg/ml
Tetracyclin supplementiert war, ausgestrichen. Es wurde SuperAgar
verwendet, weil dies ein sehr reichhaltiges Medium ist (3X LB-Agar).
Die Platten wurden bei 30°C
inkubiert, um die Toxizität
der Synthese von GFP zu reduzieren und um die Bakterien üppig auf
den Platten wachsen zu lassen. Die isolierten Kolonien wurden dann
in 45 ml SuperBroth, die, wenn geeignet, mit DHB und 20 μg/ml Tetracyclin
supplementiert war, angeimpft und 16 Stunden lang bei 37°C inkubiert.
Die Bakterien wurde mittels Zentrifugation konzentriert, in 1 ml
sterilem PBS, pH = 7,4, resuspendiert und vor der FACS-Analyse 1:100
in PBS, pH = 7,4, verdünnt.
Die Bakterien wurden mittels Durchflusszytometrie, wie es oben für die beiden
unabhängigen
Versuche zum Wachstum beschrieben ist, analysiert und die Ergebnisse
sind in Tabelle 4 am Ende dieses Abschnitts angegeben.
-
Diese
Daten belegen die Schlussfolgerung, dass eine Überexpression von GFPuv in
CVD908-htrA für die
Bakterien toxisch ist. Wenn die theoretische Kopienzahl für die Plasmide
pGEN4, pGEN3 und pGEN2, die GFPuv unter identischen Wachstumsbedingungen
für den
anfänglichen
Pomp C1-Promotor
exprimieren, nimmt der Prozentsatz der wachsenden Population, die
fluoresziert, ab. Erwartungsgemäß sind die „dim"-Bakterien nicht
lebensfähig
und können
das Expressionsplasmid nicht länger
enthalten, da diese Kulturen in der Gegenwart von 20 μg/ml Tetracyclin
angezüchtet
worden waren. Es wurde jedoch festgestellt, dass CVD908-htrA(pGEN2),
wenn es auf festen Medien ausgestrichen wurde und 24-36 Stunden
lang bei 37°C
angezüchtet wurde,
nur wenig wachst und keine isolierten Kolonien produzieren kann,
wohingegen CVD908-htrA(pGEN3) und CVD908-htrA(pGEN4) ziemlich gut
wachsen und intensiv fluoreszierende, isolierte Kolonien produzieren.
-
GFPuv
wird hierin als ein Beispiel von weiteren problematischen heterologen
Antigenen verwendet, die interessant sind, um in einen bakteriellen
Lebendvektor, wie beispielsweise den auf S. typhi basierenden Lebendvektor,
eingebaut werden zu können;
vorzugsweise kann GFPuv gegen irgendein nicht im Stoffwechsel vorkommendes
Protein- oder Peptidantigen
ausgetauscht werden.
-
Die
obigen Daten zeigen, dass, obwohl die Verwendung von oriE1-Replikons
enthaltenden Expressionsplasmiden mit mittlerer Kopienzahl bei der
Expression einiger Antigene nützlich
sein kann, die Expression von Antigenen mit höherer Toxizität erfolgreicher
aus Plasmiden mit geringerer Kopienzahl exprimiert wird, die Replikationsursprünge verwenden,
die eine mittlere Kopienzahl zwischen 2 und 30 erreichen, wie beispielsweise
die Replikationsursprünge
ori101 oder ori15A.
-
-
6.4 Der antisense-post-segregationale
Killing-Locus von hok-sok
-
Mittels
Polymerasekettenreaktion wurden die PSK-Gene von hok-sok unter Verwenden
des multiplen Antibiotikaresistenz-R-Plasmids pR1 als Templat in
diesen Reaktionen amplifiziert. Alle anfänglichen Versuche, diesen Locus
auf Plasmiden mit entweder hoher oder mittlerer Kopienzahl zu klonieren,
war nicht erfolgreich. Um direkt auf den hok-sok-Locus während des
Subklonierens selektieren zu können,
wurde eine Gruppe von Primern zur Verwendung in sich überlappenden
PCR-Reaktionen konstruiert, so dass das finale Produkt ein Fragment
war, dass eine genetische Fusion des hok-sok-Locus von pR1 und eines
promotorlosen tetA-Gens von pBR322, das für eine Resistenz gegen Tetracyclin
codiert, enthielt. Diese Kassette wurde so konstruiert, dass die
Transkription des hok-Gens in tetA hineinreichen würde; die
beiden Loci in dieser Kassette wurden durch eine XbaI-Restriktionsstelle
für zukünftige Manipulationen
voneinander abgegrenzt.
-
Die
Konstruktion dieser Kassette ermöglichte
nicht nur eine direkte Selektion des hoksok-Locus, sondern auch
die Bestätigung,
dass die PSK-Funktion in S. typhi CVD908-htrA funktionsfähig wäre. Nach
der Elektroporation von Plasmiden, die die Kassette in CVD908-htrA einbringen,
könnten
die Transformanten mit Hilfe von Tetracyclin selektiert werden.
Eine erfolgreiche Rückgewinnung
der isolierte Kolonien belegte die erfolgreiche Synthese der hok – tetA-mRNA
und die erfolgreiche Synthese der antisense-sok-RNA zur Verhinderung
der Synthese von Hok, das die Bakterien töten würde. Die Wiedergewinnung der
hok-soktetA-Kassette war dann einfach und sie war leicht in unsere
Expressionsplasmide einzubauen, um so die Kassette für den selektierbaren
Marker für
die Plasmide pGEN2, pGEN3 und pGEN4, die in den 1A-1C dargestellt sind, zu erzeugen.
-
Dann
wurden Versuche gestartet, um den Effekt der PSK-Funktion von hok-sok
auf die Stabilität
der oriE1 enthaltenden Expressionsplasmide und den für β-Lactamase
codierenden Resistenzmarker bla, der eine Resistenz gegen Carbenicillin
verleiht, zu bestimmen. Die hok-sok-Kassette wurde in das auf pAT153
basierende Expressionsplasmid pTETnir15 inseriert, in dem die heterologe
Antigenkassette Pnir15-toxC gegen unsere PompC1gfpuv-Kassette ausgetauscht
worden war, was die Plasmide pJN72 (ohne hok-sok) und pJN51 (mit
hok-sok) erzeugte. Eine weitere Gruppe von Plasmiden wurde durch
Austausch von Pomp1 gegen den schwächeren Promotor
PompC3 erzeugt, was pJN10 und pJN12 erzeugte;
die Strukturen dieser vier isogenen Plasmide sind in 2 dargestellt. CVD908-htrA-Stämme, die
entweder pJN72, pJN51, pJN10 oder pJN12 trugen, wurden auf reichhaltigem
SuperAgar-Medium,
das mit DHB und 100 μg/ml
Carbenicillin supplementiert war, ausgestrichen und die Platten
wie oben bei den pGEN-Plasmiden bei 30°C inkubiert, um die Toxizität der Synthese
von GFPuv zu reduzieren und die Bakterien üppig auf den Platten wachsen
zu lassen.
-
Die
isolierten Kolonien wurden dann in 45 ml Super-Nährmedium, das mit DHB und 100 μg/ml Carbenicillin
supplementiert war, angeimpft und zur Analyse mittel Durchflusszytometrie
der Fluoreszenz 24 Stunden lang bei 37°C angezüchtet. Ein zweites, davon unabhängiges Experiment
wurde genau so wie das erste durchgeführt, außer dass sie isolierten Klone
in 500 μl
Super-Nährmedium
suspendiert wurden und jeweils 250 μl in 45 ml gepaarten Kulturen
in Super-Nährmedium
angeimpft wurden, wobei 300 mM NaCl zur Induktion der P
ompC-gfpuv-Kassetten
dazugegeben wurden oder nicht; die Kulturen wurden 48 Stunden lang
bei 37°C
inkubiert und erneut mittels Durchflusszytometrie analysiert; die Ergebnisse
der beiden Versuche sind in Tabelle 5 gezeigt. Die Fluoreszenzhistogramme
für nicht-induzierte
und induzierte Expressionsplasmide aus Versuch 2 sind in den
2A-
3H dargestellt.
-
Die
Ergebnisse dieser Durchflusszytometrie lassen sich wie folgt erklären: die
Expression von für
das sekretierte Enzym β-Lactamase
codiert, nimmt der selektive Druck, wenn die GFPuv (oder einem anderen
potentiell schädlichen,
heterologen Antigen) aus einem Vielkopie-Expressionsplasmid, wie
beispielsweise pJN72, stellt eine erhöhte Belastung für den Stoffwechsel
des Lebendvektors CVD 908-htrA(pJN72) dar und verstärkt die
Plasmidstabilität
in Abwesenheit einer Selektion. Da der selektierbare Marker des
Expressionsplasmids Konzentration von Carbenicillin in dem umgebenden
Medium mit der Zeit abnimmt, ab und die Häufigkeit von Plasmidverlusten
nimmt zu; da jedoch Vielkopie-Plasmide
beteiligt sind, gelingt es nur relativ wenigen Bakterien, alle in
ihnen vorhandenen Plasmide zu verlieren, die mittlere Kopienzahl
von pJN72 pro Bakterium nimmt jedoch ab.
-
Eine
Quantifizierung der Produktion von GFPuv mittels Durchflusszytometrie
bei einer nicht-induzierten Population aus gesund wachsendem CVD908-htrA(pJN72)
belegt, dass die Mehrzahl der Bakterien GFPuv exprimieren und nur
wenige, nichtfluoreszierende Zellen nachgewiesen werden (3A). Die ansteigende Produktion von GFPuv durch
die Induktion der PompC1-gfpuv-Kassette
stellt jedoch eine erhöhte
Belastung für
den Stoffwechsel von CVD 908-htrA(pJN72) dar und obwohl sich die
Produktion von GFP verdoppelt, nimmt der Prozentsatz an nicht fluoreszierenden
Bakterien zu, je mehr Plasmide aus der Population verloren gehen
(3B).
-
In
einer ähnlichen
Population aus wachsendem CVD 908-htrA(pJN51) trägt jedes Bakterium Vielkopie-Plasmide,
die für
sowohl GFPuv als auch eine PSK-Funktion codieren. Die Häufigkeit
des Plasmidverlusts bei pJN51 bleibt die gleiche wie bei pJN72,
aber in diesem Fall ist die 1:1-Stöchiometrie, wenn die einzelnen Bakterien
die Kopien des Expressionsplasmids verlieren, zwischen den mRNA-Mengen
von hok und sok gestört
und eine Produktion von Hok führt
zum Tod der Zelle; daher sind die einzigen CVD 908-htrA(pJN51)-Bakterien
die schnell wachsen, solche, die alle ihre Expressionsplasmide behalten.
Entsprechend ist es nicht überraschend,
dass bei einer Quantifizierung der Produktion von GFPuv mittels
Durchflusszytometrie bei einer nicht-induzierten Population gesund
wachsender CVD 908-htrA(pJN51) nun eine Population von fluoreszierenden
Bakterien nachgewiesen wird, die Höhen der Fluoreszenz von GFPuv
zeigen, die mit denjenigen, die bei CVD 908-htrA(pJN72), die unter
induzierenden Bedingungen angezüchtet
wurden, beobachtet wurden, äquivalent
sind (3C gegenüber 3B);
der Prozentsatz an nichtfluoreszierenden Bakterien steigt jedoch bei über der
Hälfte
der Gesamtpopulation der Organismen an.
-
Eine
Erhöhung
der Produktion von GFPuv in dieser Population durch Induktion der
PompC1-gfpuv-Kassette in CVD 908-htrA(pJN51)
stellt wiederum eine Belastung für
den Stoffwechsel des Lebendvektors dar, aber nun übernimmt
der Prozentsatz an nichtfluoreszierenden Bakterien fast vollständig die
wenigen fluoreszierenden Bakterien, da vermutlich viele Plasmide
aus der Population verloren werden und dadurch Bakterien getötet werden
(2D).
-
Es
würde erwartet
werden, dass, wenn ein schwächerer
Promotor zur Kontrolle der Expression von GFPuv verwendet wird,
die Gesamtfluoreszenz der Population abnehmen würde (verglichen mit derjenigen, die
bei einer ähnlichen
Population von Organismen, die mit einem stärkeren, GFPuv exprimierenden
Promotor unter identischen Bedingungen angezüchtet wurden, beobachtet wird),
und der Prozentsatz an nicht-fluoreszierenden Bakterien sollte aufgrund
der Gesamtabnahme der Synthese von GFPuv abnehmen. Wie in den 3E-3H zu sehen ist, verbesserte
die Verwendung der schwächeren
PompC3-gfpuv-Kassette nicht signifikant die Lebensfähigkeit
von induzierten Bakterien, die ein Killing-System trugen, obwohl die gesamte Expression
von GFPuv vermindert war.
-
Daraus
wird geschlossen, dass es, um den Prozentsatz einer Population von
Lebendvektoren, die das heterologe Antigen nach Wahl exprimieren,
zu maximieren, nicht ausreicht, nur eine PSK-Funktion in ein gegebenes
Expressionsplasmid einzuschleusen, unabhängig davon, ob dies ein Arzneimittelresistenzmarker, das
asd-System, ein alternierendes ssb-System oder das hok-sok-Killing-System
ist. Um die Kopienzahl und Expressionsmengen zu optimieren, müssen zusätzlich auch
die Segregationshäufigkeiten
dieser Plasmide verbessert werden, um so sicherzustellen, dass jede
Tochterzelle in einer aktiv wachsenden Population wenigstens ein
Expressionsplasmid erben wird und dass diejenigen, die keines erben,
getötet
und aus der Population entfernt werden. Die Bereitstellung eines
Expressionsplasmids, das eine PSK-Funktion besitzt und ferner eine
optimierte Kopienzahl und/oder optmierte Expressionsmengen aufweist,
gekoppelt mit der Insertion von einer oder mehreren SEG-Funktionen,
ist im Umfang der vorliegenden Erfindung enthalten.
-
6.5 Auf Komplementierung basierendes Killing-System
-
Im
breiten Umfang der vorliegenden Erfindung sind auch die Bereitstellung
eines Expressionsplasmids, das ein auf Komplementierung basierendes
Killing-System, zum Beispiel ein System, das die Deletion des chromosomalen
ssb-Locus von CVD908-htrA durch homologe Rekombination beinhaltet,
und die Transkomplementierung dieser Lücke unter Verwenden von ssb-Funktionen
tragenden Vielkopie-Plasmiden umfasst.
-
Die
Ausführung
solcher Konstruktionen erfordert die Klonierung des relevanten Abschnitts
des S. typhi-Chromosoms, das das ssb-Gen und flankierende Sequenzen
umfasst und in das spezifische Deletionen für chromosomale Mutagenese eingeschleust
werden.
-
Seit
unserer ersten Eingabe wurden wesentliche Fortschritte in der Sequenzierung
des Chromosoms von Salmonella typhi im Sanger Centre in London gemacht.
Das Sanger Centre ist ein Genomforschungszentrum, das 1992 von dem
Wellcome Trust und dem Medical Research Council gegründet wurde,
um unser Wissen über
Genome zu steigern. Neben anderen Projekten sequenziert das Sanger
Centre in Zusammenarbeit mit Gordon Dougan vom Department of Biochemistry,
Imperial College, London das 4,5 Mb große Genom von S. typhi. Sie
sequenzieren den Stamm CT18, einen hoch-pathogenen, gegen viele
Arzneimittel resistenten Stamm, der aus einem Patienten mit Typhus
im Cho Quan Hospital, Ho Chi Minh-Stadt, Vietnam isoliert wurde. Es ist
bekannt, dass dieser Stamm pVN100 (ein 133 kb großes, gegen
viele Arzneimittel resistentes Plasmid) und ein kryptisches 80 kb
Plasmid beherbergt. Das Genom wird in einem Shotgun-Ansatz des vollständigen Genoms
unter Verwenden einer 2 kb großen
pUC-Genbank sequenziert, die firmenintern aus einer aus dem Labor
von Prof. Dougan zur Verfügung
gestellten chromosomalen DNA erzeugt wurde. Jedes Insert wird einmal
von jedem Ende aus sequenziert. Die Shotgun-Phase ist nun beendet
und die Nachbearbeitung hat begonnen. Derzeit liegen 60 Contigs über 1 kb
in der Datenbank vor; insgesamt wurden 5,106 Mb der Sequenz in 87.331
Ablesedurchgängen
assembliert.
-
Basierend
auf den aktualisierten Daten, die am 4. Oktober 1999 bekannt gegeben
wurden, haben wir Contig 343 identifiziert, das den ssb-Locus von
S. typhi und entscheidende flankierende Sequenzen innerhalb einer
205,199 bp großen
Region enthält.
Wir haben die Primer 1 und 4 (unten angegeben) konstruiert, um ein 3535
bp großes
Fragment aus dem Chromosom von S. typhi, in dem der ssb-Locus von
1,5 kb der chromosomalen Sequenz flankiert wird, mit Hilfe von PCR
amplifiziert; die Symmetrie dieser flankierenden Regionen ist für optimale
Häufigkeiten
von Crossing-Over erforderlich, um so die gegen-selektierbare sacB – neo-Kassette einzuschleusen
und gegen ssb auszutauschen. Unter Verwenden der bereits eingereichten
Methodik werden wir die Primer 1 und 2 zu Konstruktion einer 5'-proximalen, 1,5 kb großen EcoRI – XmaI-Kassette,
stromaufwärts
von ssb gelegen, verwenden. Die Primer 3 und 4 werden zur Konstruktion
der 3'-distalen,
1,5 kb großen XmaI – EcoRI-Kassette,
stromabwärts
von ssb gelegen, verwendet; beide 1,5 kb großen Kassetten werden miteinander
ligiert werden, wobei das 3 kb große EcoRI-Fragment, das eine
einzigartige XmaI-Stelle genau in der Mitte der Kassette enthält, ausgebildet
wird. Die sabB-neo-Kassette
kann nun leicht in die XmaI-Stelle inseriert werden, um so die Konstruktion
der Mutagenesekassette, die in pCON (wurde bereits in unserer ersten Eingabe
beschrieben) inseriert werden soll, zu vervollständigen. Die erforderliche,
komplementierende ssb-1-Kassette
wird unter Verwenden der Primer 5 und 6 als NheI-Kassette für einen
Austausch der Arzneimittelresistenzmarker innerhalb der XbaI – SpeI-Kassetten
von pGEN211, pGEN222, pGEN206 oder irgendeiner im Folgenden genannten
Version der hierin ausführlich
beschriebenen Expressionsplasmide, konstruiert werden.
-
- PRIMER 1:
- 5' – gaattcGCGCGCTTCGCGATTCAGTCGCGTTCCTTCACA
GCTGGCGCAGGGGCGATTACTGATGAA – 3' (SEQ ID No. 6)
- PRIMER 2:
- 5' – cccggGAGTCTCCTGAATACGTTTCATAAATAGTGTAAA
ACGCGTGAGTGTACCATTTCCACGTAGC – 3' (SEQ ID No. 7)
- PRIMER 3:
- 5' – cccggGTAAAAAACTCAAAGCGTTATTTGCATTTTCGC
TATAGTTCTCGTCTGCTGAAATGCCTGGTGT – 3' (SEQ ID No. 8)
- PRIMER 4:
- 5' – gaattcCATTTCTATCAATAAATTACTATTAGTTTTGTCT
TCTAACCAAGCCTCTATTTTATGAGTATCCTCTTCAG -3' (SEQ ID No. 9)
- PRIMER 5:
- 5' – gctagcATGGCCAGCAGAGGCGTAAACAAGGTGATTCT
CGTTGGTAATCTGGGCCAGGACCCGGAAGTACGC – 3' (SEQ ID No. 10)
- PRIMER 6:
- 5' – gctagcTCAGAACGGAATGTCGTCGTCAAAATCCATTG
GCGGTTCGTTAGACGGCGCTGGCGCG – 3' (SEQ ID No. 11)
-
6.6 Stabilität von Expressionsplasmiden
in Abwesenheit von Selektion
-
Um
ein nichtkatalytisches Plasmiderhaltungssystem zur Verbesserung
der Stabilität
von Vielkopie-Expressionsplasmiden, die für fremde Antigene in CVD908-htrA
codieren, zu entwickeln, wurden Versuche unternommen, die Plasmidstabilität durch
Quantifizieren der Expression von GFPuv mittels Durchflusszytometrie zu überwachen,
wenn die Stämme
in der Abwesenheit einer Antibiotikaselektion passagiert wurden.
Diese Versuche wurden dazu konstruiert, 3 fundamentale Fragen anzusprechen:
1] Welchen Effekt hat die Induktionsmenge von PompC1 auf
die Stabilität
der Plasmide, die für
ein heterologes Antigen, wie zum Beispiel GFPuv, codieren? 2] Welchen
Effekt hat die Kopienzahl auf die Stabilität der Plasmide, die GFPuv exprimieren?
3] Wie beeinflussen die Stabilisierungsfunktionen von hok-sok, par
und parA die Beibehaltung des Plasmide, sowohl als einzelne Komponenten
als auch aus synergistischer Sicht?
-
Es
wurden erste Versuche zur Durchflusszytometrie durchgeführt, in
denen CVD 908-htrA
Replikons mit entweder dem Replikationsursprung oriE1, ori15A oder
ori101 trugen. Es wurde schnell bestimmt, dass die Replikons, die
die oriE1-Ursprünge
mit höherer
Kopienzahl trugen, sehr instabil waren, selbst wenn die Stämme in Gegenwart
von Antibiotikaselektion angezüchtet
wurden. Die Ergebnisse der Durchflusszytometrie geben an, dass,
wenn die Bakterienpopulationen in Gegenwart von Carbenicillin angezüchtet wurden,
der Prozentsatz dieser Bakterienpopulationen, die nicht länger nachweisbares
GFPuv exprimierten, ungefähr
50% bei pGEN71 (das hok-sok trägt)
und pGEN84 (hok-sok + par) bis 62% bei pGEN211 (hok-sok + par +
parA) betrug. Da Replikons, die einen oriE1-Urspung tragen, deutlich
keine Synthese des heterologen Testantigens GFPuv in der Mehrzahl
der wachsenden Population von Bakterien-Lebendvektor zuließen, wurde
diese Reihe von Expressionsplasmiden nicht weiter untersucht.
-
Danach
wurden CVD 908-htrA, die Expressionsplasmide mit einem ori15A-Ursprung
trugen, untersucht. Die Stämme
wurden in 25 ml-Kulturen aus 1X LB + DHB (keine Antibiotikaselektion),
das entweder 50 mM, 150 mM oder 300 mM NaCl enthielt, angeimpft.
Die Kulturen wurden 24 Stunden lang bei 37°C mit 250 rpm inkubiert, 1:1000
in frischem Medium mit identischer Osmolarität verdünnt und weitere 24 Stunden
lang inkubiert; Proben aus allen Kulturen wurden mittels Durchflusszytometrie
auf die Synthesemengen von GFPuv analysiert. Die Ergebnisse des
ersten Passagierungsdurchgangs in Abwesenheit einer Selektion sind
in Tabelle 6 angegeben und die Histogramme, die diese Daten wiedergeben,
sind in 8 gezeigt.
-
Tabelle
6 zeigt die Stabilität
von ori15A-Replikons, die Plasmiderhaltungssysteme mit zunehmender Komplexizität enthalten
und ohne Selektion und in Gegenwart einer zunehmenden Osmolarität angezüchtet wurden,
in CVD 908-htrA.
1 -
Diese
Daten sind als Histogramme in 8 dargestellt.
-
Alle
Stämme
wurden aus eingefrorenen Master Stocks auf 2X LB-Agar, der mit DHB
und 50 μg/ml
Carbenicillin supplementiert war, ausgestrichen und 36 Stunden lang
bei 37°C
inkubiert. Die isolierten Kolonien wurden in einem Pool in 300 μl 1X LB-Nährmedium,
die mit DHB supplementiert war, gesammelt und jeweils 25 μl davon wurden
in 25 ml 1X LB Nährmedium,
die DHB und entweder 50 mM, 150 mM oder 300 mM NaCl enthielt, angeimpft;
die Kulturen wurden 24 Stunden lang bei 37°C mit 250 rpm inkubiert. Für die in
dieser Tabelle angegebenen Ergebnisse wurden die Bakterien dann
pelletiert, in 1 ml PBS, pH 7,4, resuspendiert und dann zur Analyse
der Durchflusszytometrie 1:1000 in PBS verdünnt.
-
Allgemein
nimmt der Prozentsatz einer Lebendvektorpopulation, die GFPuv exprimiert,
wenn die Osmolarität
zunimmt und die Induktion von PompC1 erhöht wird,
ab; trotzdem nimmt die mittlere Höhe der Fluoreszenzintensität erwartungsgemäß zu. Zum
Beispiel exprimieren in 80,5% einer Population von CVD 908-htrA(pGEN121)
in der Gegenwart von 300 mM NaCl GFPuv mit einer mittleren Fluoreszenzintensität von 53,3. Wenn
die Konzentration von NaCl auf 300 mM NaCl erhöht wird, sinkt der Prozentsatz
der Population, die GFPuv exprimiert, auf 56,7%; trotzdem steigt
die mittlere Fluoreszenzintensität
auf 105,3 an. Es ist jedoch zu beachten, dass bei Stämmen, die pGEN222
mit einem vollständigen
Plasmiderhaltungssystem (d.h. hok-sok + par + parA) tragen, der
Prozentsatz der Population, die das heterologe Antigen produziert,
bei ungefähr
95% bleibt, während
die mittlere Fluoreszenzintensität
von 52,1 (50 mM NaCl) auf 89,2 (300 mM NaCl) ansteigt. Es wurde
festgestellt, dass bei weiterem Passagieren dieser Stämme weitere
24 Stunden lang in Abwesenheit von Antibiotikaselektion, weniger
als 5% der Bakterien weiterhin funktionsfähiges GFPuv exprimierten. Ausstriche
dieser Kulturen auf festem Medium, vor einer Analyse des Durchflusses,
zeigten, dass nichtfluoreszierenden Bakterien nicht lebensfähig, aber
für Antibiotikaselektion
sensitiv blieben. Wenn nichtfluoreszierende Bakterien sortiert und
ausplattiert wurden, wurde bestätigt,
dass sie für
Antibiotika sensitiv waren und nicht fluoreszierten, wenn sie mit
ultraviolettem Licht bestrahlt wurden, was den Verlust der in ihnen
vorhandenen Plasmide anzeigt.
-
Ein
Passagierungsversuch, der CVD 908-htrA, die Expressionsplasmide
mit ori101-Ursprung
trugen, beinhaltete, wies, unabhängig
von der Osmolarität,
keinen signifikanten Verlust der Expression von GFPuv nach dem Passagieren
der Stämme
für 48
Stunden ohne Selektion nach. Die Stämme wurden daher in einem separaten
Versuch 96 Stunden (d.h. 4 × 24
Stunden) lang in Gegenwart von entweder 50, 150 oder 300 mM NaCl
passagiert. Die Populationen wurden nach 3 und 4 Passagierungsdurchgängen mittels
Durchflusszytometrie analysiert und die Ergebnisse sind in Tabelle
7 angegeben.
-
Tabelle
7 zeigt die Stabilität
von ori101-Replikons, die Plasmiderhaltungssysteme mit zunehmender Komplexizität, die ohne
Selektion und in Gegenwart zunehmender Osmolarität angezüchtet wurden, in CVD 908-htrA.
- 1Alle Stämme
wurden aus gefrorenen Master Stocks auf 2X LB-Agar, der mit DHB
und 50 μg/ml
Carbenicillin supplementiert war, ausgestrichen und 36 Stunden lang
bei 30°C
inkubiert. Die isolierten Kolonien wurden in einem Pool in 300 μl 1X LB-Nährmedium,
das mit DHB supplementiert war, gesammelt und 25 μl davon in
25 ml 1X LB-Nährmedium,
das DHB und entweder 50 mM, 150 mM oder 300 mM NaCl enthielt, angeimpft;
die Kulturen wurden 24 Stunden lang bei 37°C mit 250 rpm inkubiert (hier
als Passagierungsdurchgang #1 definiert). Bei dem Passagierungsdurchgang
#2 wurden 25 μl
aus Passagierungsdurchgang #1 in 25 ml (d.h. eine Verdünnung von
1:1000) identischem Medium angeimpft und ohne Selektion für weitere
24 Stunden bei 37°C mit
250 rpm inkubiert. Die Passagierungsdurchgänge #3 und #4 wurden in gleicher
Weise durchgeführt,
jedoch wurden die restlichen Bakterien nach Ansetzen des nächsten Passagierungsdurchgangs
pelletiert, in 1 ml PBS, pH 7,4, resuspendiert und dann zur Analyse
mittels Durchflusszytometrie 1:1000 in PBS verdünnt.
- 2ND nicht durchgeführt
-
Lebendvektoren,
die nicht-stabilisierte ori101-Replikons trugen, verloren möglicherweise
nach 96 Stunden die Fähigkeit,
das heterologe Antigen zu synthetisieren. Beispielsweise exprimierten
nach 96 Stunden Wachstum in Gegenwart von 50 mM NaCl nur 10,9% CVD
908-htrA(PGEN132) GFPuv und fluoreszierten. Wenn die Konzentration
von NaCl in dem Medium auf 150 mM erhöht wurde, wurde die Fluoreszenz
in nur 7,6% der Population nachgewiesen; seltsamerweise ergab der
Prozentsatz bei 300 mM NaCl 51,3% fluoreszierende Bakterien. Beachtenswerter
Weise behielten, unabhängig
von der Osmolarität,
CVD 908-htrA, die entweder pGEN142 (hok-sok) oder pGEN206 (hok-sok
+ parA) trugen, eine Synthese von GFPuv in mehr als 95% der Population
nach 3 Passagierungsdurchgängen
(72 Stunden) bei (siehe Tabelle 7). Der Prozentsatz an fluoreszierenden
CVD 908-htrA(pGEN142)
blieb nach 4 Passagierungsdurchgängen
(96 Stunden) nahezu auf dieser Höhe,
während
er bei CVD 908-htrA(pGEN206) leicht zurückging.
-
Im
Gesamten genommen zeigen diese Daten, dass, wenn die Kopienzahl
verringert ist, die offensichtliche Stabilität der darin vorhandenen Plasmide
und die Fähigkeit
eines Lebendvektors, ein heterologes Antigen, wie beispielsweise
GFPuv, zu synthetisieren, zunehmen; wenn sich die Plasmiderhaltungssysteme
in einem gegebenen Plasmid ansammeln, werden die offensichtliche
Stabilität
und die Synthese des Antigens weiter verstärkt. Wenn die Induktion von
PompC und die damit einhergehende Produktion
des heterologen Antigens erhöht
wird, kann daneben der Prozentsatz einer wachsenden Population,
die in der Lage ist, das Antigen zu synthetisieren, drastisch abnehmen.
-
6.7 Bakterienstämme und Kultivierungsbedingungen
-
Alle
Plasmidkonstruktionen wurden aus dem Escherichia coli-Stamm DH5α oder DH5αF'IQ (Gibco BRL) gewonnen.
Zur Konstruktion der hok-sok-Genkassette wurde Templat-DNA von pR1,
die aus dem E. coli-Stamm J53(pR1) isoliert wurde, eine großzügige Spende
von James B. Kaper, verwendet. Der Lebendvektor S. typhi CVD 908-htrA
ist ein auxotrophes Derivat des Wildtyp-Stamms Ty2 mit Deletionen
in aroC, aroD und htrA (Tacket et al., 1997b). Alle für die Untersuchung
der Plasmidstabilität
verwendeten Stämme
wurden in Medien angezüchtet,
die mit 2,3-Dihydroxybenzoesäure
(DHB) supplementiert waren, wie es oben beschrieben ist (Hone et
al., 1991; Galen et al., 1997). Wenn sie auf festem Medium angezüchtet wurden,
wurden Plasmid-tragende Stämme
von CVD 908-htrA aus gefrorenen (–70°C) Master Stocks auf 2X Luria-Bertani-Agar, der
(pro Liter) 20 g Bacto-Trypton,
10 g Bacto-Hefeextrakt und 3 g NaCl (2X LB-Agar) plus Carbenicillin
mit einer Konzentration von 50 μg/ml
enthielt, ausgestrichen. Die Platten wurden 24-36 Stunden lang bei
30°C inkubiert,
um isolierte Kolonien mit ~ 2 mm Durchmesser zu erhalten; die Stämme wurden
bei 30°C
inkubiert, um die Toxizität
der Expression von GFPuv in CVD 908-htrA zu minimieren.
-
Wenn
sie in flüssigem
Medium angezüchtet
wurden, wurden die Kulturen 16-24 Stunden lang bei 37°C mit 250
rpm inkubiert. Um die osmotische Induktion des ompC-Promotors (PompC) in entweder E. coli DH5α oder CVD
908-htrA zu überprüfen, wurden
die Stämme
in Bacto-Nährbühe (Difco),
das DHB und entweder NaCl oder Sucrose enthält, angezüchtet; die Kulturen waren entweder
mit 50 μg/ml
Carbenicillin oder ansteigenden Konzentrationen von Kanamycin, wenn
die PompC-apha-2-Kassetten untersucht wurden,
supplementiert. Zur Quantifizierung der Synthese von GFPuv mittels
Durchflusszytometrie wurden 6-8 isolierte Kolonien aus Master Stocks,
die, wie oben, auf 2X LB-Agar ausgestrichen wurden, in 25 ml 1X
LB-Nährmedium,
das, wenn gewünscht,
mit 50 μg/ml
Carbenicillin und NaCl in ansteigenden Konzentrationen zur Erhöhung der
Induktion von ompC-Promotoren supplementiert waren, angeimpft. Die
Kulturen wurden vor dem Pelletieren der Bakterien zur Durchflusszytometrie
16-24 Stunden bei 37°C
mit 250 rpm inkubiert.
-
6.8 Molekulargenetische Methoden
-
Zur
Konstruktion der hier angegebenen Plasmide wurden Standardmethoden
verwendet (Sambrook et al., 1989). Soweit nicht anders angegeben
ist, wurde native Taq-DNA-Polymerase
(Gibco BRL) in den Polymerasekettenreaktionen (PCR) verwendet. S.
typhi wurde vor der Ernte aus Millers-LB-Nährmedium (Gibco BRL), das mit
DHB supplementiert war, für
die Elektroporation von rekombinanten Plasmiden vorbereitet; nach dem
Pelletieren der Bakterien wurden die Zellen dreimal mit einem Kulturvolumen
aus sterilem destillierten Wasser gewaschen und auf ein Endvolumen
von 1/100 des ursprünglichen
Kulturvolumens in sterilem destillierten Wasser resuspendiert. Die
Elektroporation der Stämme
wurde mit einer Gene Pulser-Vorrichtung (Bio-Rad), die auf 2,5 kV,
200 Ω und
25 μF eingestellt
war, durchgeführt.
Nach der Elektroporation wurden die Bakterien unter Verwenden von
SOC-Medium repariert
und 45 Minuten lang bei 37°C
mit 250 rpm inkubiert; die Bakterien wurden dann auf 1X LB-Medium,
das DHB plus 50 μg/ml
Carbenicillin enthielt, ausplattiert und 24 Stunden lang bei 30°C inkubiert.
Die isolierten Kolonien wurden dann von dem supplementierten 2X
LB gewischt und 16 Stunden lang bei 30°C inkubiert. Die gefrorenen Master
Stocks wurden durch Ernten der Bakterien in SOC-Medium ohne weitere
Supplementierung und Einfrieren bei –70°C vorbereitet.
-
6.9 Konstruktion der Expressionsvektoren
-
Die
in der folgenden Tabelle 8 aufgelisteten Expressionsvektoren wurden
im Verlauf der neueren Arbeit zubereitet. Tabelle 8
| Plasmid | Größe (kb) | Relevanter
Genotyp | Referenz |
| pTETnir15 | 3,7 | oriE1
toxC bla | Oxer
et al. (1991) |
| pJN1 | 1,9 | oriE1
bla | Diese
Arbeit |
| pJN2 | 3,4 | oriE1
toxC bla | Diese
Arbeit |
| pGFPuv | 3,3 | pUC19ori
gfpuv bla | Clontech |
| pGFPompC | 3,5 | oriE1
gfpuv bla | Diese
Arbeit |
| pNRB1 | 3,5 | oriE1
gfpuv tetA | Diese
Arbeit |
| pGEN2 | 4,2 | oriE1
gfpuv tetA hok-sok | Diese
Arbeit |
| pGEN3 | 4,1 | ori15A
gfpuv tetA hok-sok | Diese
Arbeit |
| pGEN4 | 5,6 | ori
101 gfpuv tetA hok-sok | Diese
Arbeit |
| pJN5 | 3,1 | oriE1
gfpuv bla | Diese
Arbeit |
| pJN6 | 3,7 | oriE1
gfpuv bla hok-sok | Diese
Arbeit |
| pJN7 | 4,1 | oriE1
gfpuv bla hok-sok par | Diese
Arbeit |
| pJN8 | 5,4 | oriE1
gfpuv bla hok-sok parA | Diese
Arbeit |
| pGEN51 | 3,6 | oriE1
gfpuv bla | Diese
Arbeit |
| pGEN71 | 4,2 | oriE1
gfpuv bla hok-sok | Diese
Arbeit |
| pGEN84 | 4,5 | oriE1
gfpuv bla hok-sok par | Diese
Arbeit |
| pGEN183 | 5,9 | oriE1
gfpuv bla hok-sok parA | Diese
Arbeit |
| pGEN211 | 6,2 | oriE1
gfpuv bla hok-sok par parA | Diese
Arbeit |
| pGEN91 | 3,5 | ori15A
gfpuv bla | Diese
Arbeit |
| pGEN111 | 4,1 | ori15A
gfpuv bla hok-sok | Diese
Arbeit |
| pGEN121 | 4,5 | ori15A
gfpuv bla hok-sok par | Diese
Arbeit |
| pGEN193 | 5,8 | ori15A
gfpuv bla hok-sok parA | Diese
Arbeit |
| pGEN222 | 6,2 | ori
15A gfpuv bla hok-sok par parA | Diese
Arbeit |
Tabelle 8-Fortsetzung
| Plasmid | Größe (kb) | Relevanter
Genotyp | Referenz |
| pGEN132 | 4,8 | ori101
gfpuv bla par | Diese
Arbeit |
| pGEN142 | 5,4 | ori101
gfpuv bla par hok-sok | Diese
Arbeit |
| pGEN206 | 7,1 | ori101
gfpuv bla par hok-sok parA | Diese
Arbeit |
-
6.9.1 Konstruktion von pJN1 und pJN2
-
Die
für diese
Untersuchungen konstruierten Expressionsplasmide bestanden aus 3
Basiskassetten, die für
1] die Expression eines heterologen Antigens, 2] eines Replikationsursprung
des Plasmids und 3] Selektions- und Stabilisierungsfunktionen codieren.
Um dies zu erreichen wurde ein Basisreplikon konstruiert, in dem
diese Kassetten durch einzigartige Restriktionsstellen voneinander
abgegrenzt wurden. Die bei der Konstruktion der Plasmidkassetten
verwendeten Primer sind in der folgenden Tabelle 9 angegeben: Tabelle 9
| Primer-Nummer | Sequenz1 | Erzeugte Kassette | GenBank-Zugangs-nummer | Region der Homologie
2 | Region
der Komple-mentarität 3 |
| 1 | 5'-GCAGGAAAGAACATGTGAGCCTAGG
GCCAGCAAAAGGCCAGGAAC-3' (SEQ
ID No. 12) | oriE1 | J01749 | 2463-2507 | |
| 2 | 5'-CATGACCAAAATCCCTTAACTAGTGTT TTAGATCTACTGAGCGTCAGACCCCG-3' (SEQ ID No. 13) | " | " | | 3197-3145 |
| 3 | 5'-CGGGGTCTGACGCTCAGTAGATCTAA
AACACTAGTTAAGGGATTTTGGT CATG-3' (SEQ
ID No. 14) | bla | " | 3145-3197 | |
| 4 | 5'-GCTGTCAAACATGAGAATTCTAGAAG
ACGAAAGGGCCTCGTGATACGCC-3' (SEQ
ID No. 15) | " | " | | 17-1,
4361-4330 |
| 5 | 5'-ACAGCCTGCAGACAGATCTTGACAGC
TGGATCGCACTCTGGTATAATTGGG AA GCCCTGCAAAG-3' (SEQ ID No. 16) | aphA-2 | V00618 | 1-64 | |
Tabelle 9-Fortsetzung
| Primer-Nummer | Sequenz1 | Erzeugte Kassette | GenBank-Zugangsnummer | Region
der Homologie 2 | Region der Komplementarität 3 |
| 6 | 5'-CGAAGCCCAACCTTTCATAGAAGCTA GCGGTGGATCCGAAATCTCGTGAT
GGC AGGTTG-3' (SEQ
ID No. 17) | " | " | | 1044-986 |
| 7 | 5'-AACAAGCGTTATAGGAATTCTGTGGT
AGCA-3' (SEQ ID
No. 18) | PompC | K00541 | 4-33 | |
| 8 | 5'-ACTTTCATGTTATTAAAGATCTGTTAT
ATG-3' (SEQ ID No.
19) | " | " | | 498-469 |
| 9 | 5'-AGATCTTAATCATCCACAGGAGGCTT
TCTGATGAGTAAAGGAGAAGAAC TTTT CACTGG-3' (SEQ ID No. 20) | gfpuv | U62636 | 289-317 | |
| 10 | 5'-GCTAGCTCATTATTTGTAGAGCTCATC
CATGC-3' (SEQ ID
No. 21) | " | " | | 1008-983 |
| 11 | 5'-AGATCTGAATTCTAGATCATGTTTGAC
AGCTTATCATCGATAAGCTTTA ATGCG-3' (SEQ
ID No. 22) | tetA | J01749 | 4-41 | |
| 12 | 5'-AGATCTTATCAGGTCGAGGTGGCCCG
GCTCCATGCACCGCGACGCAACG CG-3' (SEQ
ID No. 23) | " | " | | 1275-1234 |
| 13 | 5'-CGCGAATTCTCGAGACAAACTCCGGG
AGGCAGCGTGATGCGGCAACAA TCACA CGGATTTC-3' (SEQ ID No. 24) | hok-sok-tetA | X05813 | 2-48 | |
| 14 | 5'-ATGAGCGCATTGTTAGATTTCATTTTT
TTTTCCTCCTTATTTTCTAGACAA CATCH GCAAGGAGAAAGG-3' (SEQ ID No. 25) | " | J01749,
X05813 | | 108-86,
580-559 |
| 15 | 5'-CCTTTCTCCTTGCTGATGTTGTCTAGA AAATAAGGAGGAAAAAAAAATGAAATCT AACAATGCGCTCAT-3' (SEQ ID No. 26) | " | X05813,
J01749 | 559-580,
86-108 | |
| 16 | 5'-GCTACATTTGAAGAGATAAATTGCACT GGATCCTAGAAATATTTTATCTGATTAAT
AAGATGATC-3' (SEQ
ID No. 27) | ori15A | X06403 | | 1461-1397 |
| 17 | 5'-CGGAGATTTCCTGGAAGATGCCTAGG
AGATACTTAACAGGGAAGTGAGAG-3' (SEQ
ID No. 28) | " | " | 780-829 | |
Tabelle 9-Fortsetzung
| Primer- Erzeugte Komple- | Sequenz1 | Erzeugte Kassette | GenBank-Zugangsnummer | Region der Homologie
2 | Region
der Komplementarität
3 |
| 18 | 5'-GTCTGCCGGATTGCTTATCCTGGCGG
TCCGGTTGACAGTAAGACGGGTAAGC CTGTTGAT-3' (SEQ ID No. 29) | ori101 | X01654 | 4490-4550 | |
| 19 | 5'-CCTAGGTTTCACCTGTTCTATTAGGTG
TTACATGCTGTTCATCTGTTACATTGTC GATCTG-3' (SEQ ID No. 30) | " | " | | 6464-6408 |
| 20 | 5'-AGGCTTAAGTAGCACCCTCGCAAGAT
CTGGCAAATCGCTGAATATTCCTTTTGTCTCCGAC-3' (SEQ ID No. 31) | par | X01654 | | 4918-4858 |
| 21 | 5'-GAGGGCGCCCCAGCTGGCAATTCTA
GACTCGAGCACTTTTGTTACCCGCCAAA CAAAACCCAAAAACAAC-3' (SEQ ID No. 32) | aphA-2-par A | V00618,
X04268 | 38-16,
1-37 | |
| 22 | 5'-AGAAGAAAAATCGAATTCCAGCATGA AGAGTTTCAGAAAATGACAGAGCGTGA GCAAGTGC-3' (SEQ ID No. 33) | " | X04268 | | 1704-1644 |
| 23 | 5'-CGAAGCCCAACCTTTCATAGAAACTA GTGGTGGAATCGAAATCTCGTGATGGC AGGTTG-3' (SEQ ID No. 3) | " | V00618 | | 1044-986 |
| 24 | 5' -GTTGTTTTTGGGTTTTGTTTGGCGGG
TAACAAAAGTGCTCGAGTCTAGAATTGC CAGCTGGGGCGCCCTC-3' (SEQ ID No. 35) | " | X04268,
V00618 | 37-1,
16-38 | |
- 1Die Restriktionsstellen
sind unterstrichen, auf die im Text Bezug genommen wird; die Ribosomenbindungsstellen
und Startcodons sind kursiv geschrieben.
- 2Bezogen die Sequenz innerhalb des codierenden
Strangs eines gegebenen Gens, zu dem der Primer homolog ist.
- 3Bezogen auf die Sequenz innerhalb des
nicht-codierenden Strangs eines gegebenen Gens, zu dem der Primer
homolog ist.
-
pTETnir15
(siehe Tabelle 8; Oxer et al., 1991) wurde so rekonstruiert, dass
der Replikationsursprung oriE1 und das bla-Gen durch eine einzigartige
Spel-Stelle voneinander abgegrenzt waren. Zu diesem Ende hin wurde
mittels PCR eine oriE1-Kassette unter Verwenden von Vent-Polymerase
mit den Primer 1 und 2 und pVCD315 (Galen et al., 1990) als Templat
synthetisiert. Das resultierende 735 bp große Fragment trägt die konstruierten
SpeI- und BglII-Stellen 5'-proximal
von dem Promotor, die die Transkription von RNA II kontrolliert,
und eine konstruierte AvrII-Stelle 675 Basen von diesen Stellen
entfernt. Eine separate PCR-Reaktion wurde unter Verwenden der Primer
3 und 4 durchgeführt,
um eine 1234 bp große
bla-Kassette zu erzeugen, die eine konstruierte XbaI-Stelle 5'-proximal von der
ursprünglichen
EcoRI-Stelle enthielt. Die Produkte aus diesen beiden PCR-Reaktionen
wurden im Gel gereinigt und in einer überlappenden PCR mit den Primer
1 und 4 zur Erzeugung eines finalen, 1916 bp großen oriEl – bla-Fragments, das zur Erzeugung
von pJNI selbst-ligiert wurde, verwendet. Das Pnir15 –toxC-Fragment
aus pTEPnir15 wurde als EcoRI (partieller Verdau) – AvaI-Fragment,
in dem der AvaI-Terminus geglättet
war, herausgeschnitten und in die multiple Klonierungsregion von
pSL1180 (Brosius, 1989), das mit EcoRI und Stul geschnitten worden
war, inseriert; diese Kassette wurde dann erneut als EcoRI (partieller
Verdau) – AvrII-Fragment
herausgeschnitten und in pJN1, das mit EcoRI-AvrII geschnitten worden war, inseriert,
wodurch pJN2 erzeugt wurde (siehe Tabelle 8).
-
6.9.2 Konstruktion von pGFPompC
-
Um
das Screening eines funktionsfähigen,
osmotisch regulierten PompC-Allels aus Escherichia
coli zu erleichtern, wurde eine aphA-2-Kassette konstruiert, die
für eine
Resistenz gegen die Aminoglykoside Neomycin und Kanamycin codiert
(Shaw et al., 1993). Es wurde eine Polymerasekettenreaktion (PCR)
unter Verwenden der Primer 5 und 6 mit dem Templat pIB279 durchgeführt (Blomfield
et al., 1991), um ein 1044 bp großes Produkt zu erzeugen, aus
dem ein promotorloses, 903 bp großes aphA-2 – BglII – NheI-Fragment für einen Austausch
gegen eine BglII-NheI-toxC-Kassette, die für das Fragment C von Tetanus-Toxin in pTETnir15
codiert, herausgeschnitten wurde. Der anaerob regulierte Pnir15-Promotor wurde gegen ein 459 bp großes EcoRI-BglII-PompC-Allel ausgetauscht, das unter Verwenden
der Primer 7 und 8 mit einer chromosomalen DNA aus E. coli DH5α als Templat
zur Erzeugung von pKompC konstruiert wurde. Nach der Bestätigung der
osmotischen Induktion von PompC durch Untersuchung
der zunehmenden Resistenz gegen Kanamycin mit zunehmender Osmolarität, wurde
dann die aphA-2-Kassette gegen ein gfpuv-Gen ausgetauscht, das für ein prokaryotisches,
Codon-optimiertes GPFuv-Allel (Clontech; Crameri et al., 1996) codierte.
Das gfpuv-Gen wurde mittels PCR unter Verwenden der Primer 9 und
10 mit dem Templat pGFPuv rückgewonnen,
so dass ein 751 bp großes
BglII- NheI-Fragment
erzeugt wurde, das in pKompC inseriert wurde, um dadurch pGFPompC
zu erzeugen. Die Kolonien wurden auf funktionsfähiges GFPuv gescreent und die
am hellsten leuchtenden Kolonien wurden dann auf eine Induktion
der Fluoreszenz mit zunehmenden Konzentrationen von NaCl untersucht.
Eine PompC1-gfpuv-Kassette wurde aus pGFPompC1
als EcoRI – NheI-Fragment
geschnitten und in ein Derivat von pJN2, das mit EcoRI – NheI geschnitten
worden war, inseriert, um pJJ4 zu erzeugen.
-
6.9.3 Konstruktion von pNRB1, pGEN2, pGEN3
und pGEN4
-
Da
die Kopienzahl nicht von einer Transkription, die von Promoter außerhalb
des Replikationsursprungs stammte, beeinflusst werden sollte, musste
sichergestellt werden, dass alle Replikationskassetten an beiden
Enden mit Transkriptionsterminatoren flankiert waren. Da der Ursprung
und die Antigenkassetten von pJN2 durch den Terminator trpA voneinander
abgegrenzt wurden, war es nur nötig,
einen weiteren Terminator zwischen den Ursprung und die bla-Kassetten
zu inserieren.
-
Um
die Konstruktion weiterer Plasmide zu einem späteren Zeitpunkt zu erleichtern,
wurde eine tetA-T1T2-Kassette erzeugt. pYA292 (Galan et al., 1990)
wurde zunächst
mit HindIII und BglII geschnitten und der T1T2-Terminatorfragment
geglättet
und in die SmaI-Stelle
der multiplen Klonierungsregion von pBluescript II KS (Stratagene)
inseriert; wenn die richtige Orientierung identifiziert wurde, wurde
diese Kassette als BamHI – PstI-Fragment
wieder herausgeschnitten und in pIB307 (Blomfield et al., 1991),
das mit BamHI-PstI geschnitten worden war, inseriert, wodurch pJG14
erzeugt wurde. Später
wurde mittels Sequenzanalyse bestimmt, dass die Kassette eine Deletion
von ungefähr
100 bp erlitten hat, bei der der halbe T2-Terminator entfernt wurde.
-
Mit
pBR322 als Templat wurden die Primer 11 und 12 dazu verwendet, ein
1291 bp großes
tetA – BhlII-Fragment
zu synthetisieren. Dieses tetA – BglII-Frgament
wurde dann in die BamHI-Stelle von pJG14 inseriert, so dass die
Transkription des tetA-Gens von dem T1T2-Terminator terminiert wird
und dadurch pJG14tetA erzeugt wird. Schließlich wurde diese tetA – T1T2-Kassette
aus pJG14tetA als EcoRI – PstI-Fragment
herausgeschnitten, wobei die PstI-Stelle durch Glätten entfernt
worden war; das resultierende Fragment wurde in pJJ4 inseriert,
mit SpeI geschnitten, geglättet
und erneut mit EcoRI geschnitten, um die bla-Kassette auszutauschen und pNRB1 zu
erzeugen.
-
Die
nicht-katalytische, post-segregationale Killing-Funktion, die in
die Plasmiderhaltungssysteme der hierin beschriebenen Expressionsplasmide
eingeschleust werden sollte, war der hok-sok-Locus aus dem gegen
viele Arzneimittel resistenten R-Faktor pR1. Anfängliche Versuche zur Rückgewinnung
des hok-sok-Locus nach einer PCR waren erfolglos. Es war daher nötig, eine überlappende
PCR dazu zu verwenden, eine Kassette zu erzeugen, in der hok-sok
transkriptionell mit einem promotorlosen tetA-Gen fusioniert war,
so dass die Transkription, die von dem hok-Promotor stammt, in tetA
weiterreichen würde
und die zu einem Transkript, das sowohl für Hok als auch für eine Resistenz
gegen Tetracyclin codiert, führen
würde.
Die pR1-Plasmid-DNA wurde aus E. coli 153(pR1) gereinigt, in der
pR1 für
eine Resistenz gegen sowohl Carbenicillin als auch Chloramphenicol
codiert. Ein 640 bp großes
hok-sok-Fragment wurde unter Verwenden der Primer 13 und 14 synthetisiert;
ein promotorloses, 1245 bp großes
tetA-Fragment wurde in einer separaten PCR unter Verwenden der Primer
12 und 15 mit pNRB1 als Templat rückgewonnen. Die Produkte aus
diesen beiden PCR-Reaktionen wurden dann in einer überlappenden
PCR mit den Primer 12 und 13 verwendet, um so dass finale, 1816
bp große
hok-sok – tetA-Fragment
zu erhalten. Dieses Fragment wurde als EcoRI – SphI-Fragment in pNRB1, das
mit EcoRI-SphI geschnitten worden war, inseriert, wodurch das tetA-Gen
wiederhergestellt und pGEN1 erzeugt wurde.
-
Dann
wurde eine Gruppe aus 3 isogenen Plasmiden, die sich nur in ihrer
Kopienzahl unterschieden, konstruiert, aus der alle weiteren Expressionsplasmide
abgeleitet werden konnten. Die Replikationsursprungskassette BglI-AvrII
von pGEN1 wurde gegen eine BglII-AvrII – oriE1-Kassette
aus pJN2 ausgetauscht, um pGEN2 zu erzeugen. Eine ori15A-Replikationskassette
wurde mittels PCR unter Verwenden der Primer 16 und 17 mit pACYC184
als Templat synthetisiert, um ein 629 bp großes BamHI – AvrII-Fragment zu erzeugen,
das in pGEN2, das mit BglII-AvrII geschnitten worden war, inseriert
wurde, um pGEN3 zu erzeugen. Schließlich wurde eine ori101-Replikationskassette
mittels PCR unter Verwenden der Primer 18 und 19 mit pSC101 als Templat
synthetisiert, wodurch ein 1949 bp großes BamHI – AvrII-Fragment erzeugt wurde,
das in pGEN2, das mit BglII-AvrII geschnitten worden war, inseriert
wurde, um pGEN4 zu erzeugen.
-
6.9.4 Konstruktion von pJN5, pGEN51, pGEN91
und pGEN132
-
Die
zugrunde liegende Gruppe aus isogenen Expressionsplasmiden, zu denen
die einzelnen Elemente eines Plasmiderhaltungssystems nacheinander
zugegeben wurden, bestand aus pGEN51 (enthält oriE1), pGEN91 (enthält ori15A)
und pGEN132 (enthält
ori101). Das Basis-Replikon, aus dem diese 3 Plasmide konstruiert
wurden, war pJN5, das durch Herausschneiden der PompC – gfpuv-Kassette
als EcoRI – NheI-Fragment aus
pGFPompC zum Austauschen der Pnir15 – toxC-Kassette
von pJN2 assembliert wurde. Die Konstruktion von pGEN51 wurden dann
durch Entfenen der Replikationskassette von pGEN2 als BamHI-Fragment
und Austausch des Replikationsursprungs in pJN5, das mit BglII und
BamHI verdaut worden war, erreicht, wodurch das gfpuv-Gen wiederhergestellt
wurde. Die Konstruktion von pGEN91 und pGEN132 wurde auf identische Weise
durch Herausschneiden von Ursprungskassetten als BamHI-Fragmente
aus entsprechend pGEN3 und pGEN4 erreicht (siehe 7 für die Darstellung
von isogenen Expressionsplasmiden, die auf pGEN91 basieren).
-
6.9.5 Konstruktion von pJN6, pGEN71, pGEN111
und pGEN142
-
Der
hok-sok-Locus wurde dann als XbaI – SalI-Fragment in pJN5, das
mit XbaI und SaiI geschnitten worden war, inseriert, was das gfpuv-Gen
wiederherstellte und pJN6 erzeugte (siehe Tabelle 2). Die Konstruktion
von pGEN71, pGEN111 und pGEN142 wurde dann genauso wie bei pGEN51,
pGEN91 und pGEN32 durch Insertion der Ursprungskassetten als BamHI-Fragmente
aus entsprechend pGEN2, pGEN3 und pGEN4 in pJN6 durchgeführt.
-
6.9.6 Konstruktion von pJN7, pGEN84 und
pGEN121
-
Die
Konstruktion von oriE1- und ori15A-Expressionsplasmiden, die ein
Plasmiderhaltungssystem, das aus sowohl einem post-segregationalen
Killing-System als auch wenigstens einer Partitionsfunktion bestand, wurde
zunächst
unter Verwenden der par-Funktion
aus pSC101 versucht. Ein 377 bp großes BamHI – BglII-Fragment wurde unter
Verwenden der Primer 18 und 20 mit pSC101 als Templat-DNA synthetisiert;
dieses Fragment wurde in pJN6, das mit BglII geschnitten worden
war, inseriert, um pJN7 zu erzeugen. Wie in den obigen Konstruktionen
wurden die Ursprungskassetten von pGEN2 und pGEN3 dann als BamHI-Fragmente
herausgeschnitten und in pJN7, das mit BglII und BamHI verdaut worden
war, inseriert, um pGEN84 nd pGEN121 zu erzeugen.
-
6.9.7 Konstruktion von pJN8, pGEN183,
pGEN206, pGEN211 und pGEN222
-
Die
finalen Expressionsplasmide wurden durch Einschleusen des aktiven
Partitionierungslocus parA aus pR1 konstruiert. Wie bei hok-sok
waren anfängliche
Versuche zur Rückgewinnung
des parA-Locus nach der PCR erfolglos. Es war daher nötig, eine überlappende
PCR zur Erzeugung einer aph – parA-Kassette
zu verwenden, in der aph und parA divergent transkribiert wurden
und durch XbaI- und XhoI-Stellen voneinander abgegrenzt wurden,
um so eine Subklonierung des parA-Locus zu ermöglichen. Ein 1737 bp großes parA-Fragment
wurde unter Verwenden der Primer 21 und 22 mit pR1 als Templat synthetisiert;
ein 1076 bp großes
aphA-2-Fragment wurde in einer separaten PCR unter Verwenden der
Primer 23 und 24 mir pIB279 als Templat rückgewonnen. Die Produkte aus
diesen beiden PCR-Reaktionen wurden dann in einer überlappenden
PCR mit den Primer 22 und 23 verwendet, um das finale, 2743 bp große aphA2 – parA-Fragment
zu erhalten. Dieses Fragment wurde als 2703 bp großes EcoRI – SpeI-Fragment
in pJN6 inseriert. Die parA-Kassette
wurde dann als XhoI-Fragment erneut ausgeschnitten und wieder in
pJN6, das mit ShoI geschnitten worden war, inseriert, was das gfpuv-Gen
wiederherstellte und pJN8 erzeugte.
-
Plasmide,
die ein Plasmiderhaltungssystem trugen, bestanden aus der postsegregationalen
Killing-Funktion hok-sok und parA und wurden durch Herausschneiden
von oriE1 – oder
ori15A – BamHI – Spel-Kassetten
aus entsprechend pGEN51 und pGEN91 und Insertion in pJN8, das mit
BamHI und SpeI geschnitten worden war, zur Erzeugung von entsprechend
pGEN183 und pGEN193 konstruiert. Plasmide, die das vollständige Komplement
von hok-sok-, par- und parA-Stabilisierungsfunktionen enthielten,
wurden durch Insertion von par-enthaltenden Urprungskassetten als
BamHI – SpeI-Kassetten
aus pGEN84, pGEN121 und pGEN132 in pJN8, das mit BamHI und SpeI
geschnitten worden war, konstruiert, um entsprechend pGEN211, pGEN222
und pGEN206 zu erzeugen.
-
6.10 Quantifizierung von GFPuv und Plasmidstabilisierung
-
Die
Quantifizierung von GFPuv und Plasmidstabilisierung wurde durch
Messen der Fluoreszenz von Plasmid-tragenden Lebendvektoren unter
Verwenden eines Epics Elite ESP-Durchflusszytometers/Zellklassifizierungssystems
(Coulter) mit einem Argon-Laser, der die Bakterien bei 488 nm anregte
und Emissionen bei 525 nm erfasste, analysiert. 25 ml 1X LB-Kulturen, die wie
oben beschrieben wurde, angezüchtet
worden waren, wurden pelletiert und die Bakterien wurden in 1 ml
PBS resuspendiert. Anschließend
wurden die Zellen vor der Bestimmung der lebensfähigen Zellzahlen und der Durchflussanalyse
1:1000 in PBS verdünnt.
Eine Lichtstreuung von vorne gegenüber einer Lichtstreuung von
der Seite, die mit logarithmischen Verstärkern gemessen wurde, wurde
dazu verwendet, die Bakterien zu belichten. Ein Minimum von 50.000
Ereignissen wurde aus jeder Probe mit einer Sammelgeschwindigkeit
von ungefähr
3.500 Ereignissen pro Sekunde erfasst. Die mittlere Fluoreszenzintensität bei einer
gegebenen Bakterienpopulation wurde unter Verwenden des Software-Pakets
Epics Elite Software Analysis Package bestimmt. Die Höhe der Autofluoreszenz,
die unter Verwenden von plasmidlosen CVD908-htrA-Stämmen von
S. typhi und DH5α-Stammen
von E. coli bestimmt wurden, wurden dazu verwendet, Marker, die
den Prozentsatz von Bakterien in einer gegebenen Population, die
GFPuv exprimieren, quantifizieren, zu platzieren.
-
6.11 Schlussfolgerungen
-
Das
breit formulierte Forschungsziel, das in den Abschnitten 6.6-6.10
angegeben ist, sollte der Untersuchung der Fähigkeit, ein Plasmiderhaltungssystem
für die
Stabilisierung von Vielkopie-Expressionsplasmiden, die für ein fremdes
Antigen in einem Vektor-Lebendimpstoffstamm
von S. typhi, ohne weitere Modifikation des Chromosoms zu entwickeln,
dienen. Die Stabilisierung der Expressionsplasmide wurde auf zwei
voneinander unabhängigen
Ebenen verbessert. Erstens wurde die Abhängigkeit von balancierten Letalstabilisierungssystemen,
an denen katalytische Enzyme, die von Vielkopie-Plasmiden exprimiert
werden, beteiligt sind, aufgehoben; dies wurde durch Einbau eines
postsegregationalen Killing-Systems, das auf dem Addiktionssystem
des nicht-katalytischen hoksok-Plasmids aus dem Antibiotikaresistenzfaktor
pR1 beruhte, in Expressionsplasmide erreicht. Ebenso wurde wenigstens
eine in der Natur vorkommende Partitionsfunktion in diese Expressionsplasmide
eingeschleust, um eine zufällige
Segregation solcher Plasmide potentiell auszuschließen und
dadurch ihre Vererbbarkeit und Stabilität zu verbessern.
-
Obwohl
diese Expressionsplasmide letztlich immunogene und schützende Antigene
für eine
Verabreichung an das menschliche Immunsystem exprimieren sollen,
wurde GFPuv als Testreporterantigen ausgewählt, da die Quantifizierung
der mittleren Fluoreszenz in einer Population von wachsenden Lebendvektoren als
Maßstab
für die
Stabilität
in dem Lebendvektor enthaltener Plasmide verwendet werden konnte.
Alle Expressionsplasmide trugen eine identische Antigenexpressionskassette,
in der ein PompC1-Allel die Transkription kontrollierte
und eine Translation durch den Einbau einer Consensus- Ribosomenbindungsstelle
optimiert wurde. Da eine katalytische Aktivität nicht mit der Fluoreszenz
von GFPuv in Verbindung steht, könnte
die Höhe der
Fluoreszenzintensität,
das in einzelnen Bakterien mittels Durchflusszytometrie gemessen
wurde, direkt mit der Gendosis und der Kopienzahl in Beziehung gebracht
werden. Daneben ließ die
Verwendung eines osmotisch regulierten ompC-Promotors eine Abschätzung der
Plasmidstabilität
und der Lebensfähigkeit
der Lebendvektoren zu, wenn die zunehmende Osmolarität höhere Mengen
der Synthese von GFPuv und vermutlich eine höhere Belastung für den Stoffwechsel
des Lebendvektors induzierte. Wie in Tabelle 2 zu sehen ist, bestätigten wir,
dass das für
diese Untersuchungen konstruierte PompC1-Allel
für die
zunehmende Osmolarität
verantwortlich war; wenn die Expression eines aph-2-Resistenzgens
gesteuert wurde, wurde eine Resistenz gegen weniger als 50 μg/ml Kanamycin
in der Abwesenheit von osmotischem Druck beobachtet, die Resistenz nahm
jedoch auf gegen mehr als 800μg/ml
in Gegenwart von 300 mM NaCl zu. Es war überraschend, dass, obwohl das
PompC1-Allel aus dem chromosomalen Locus
von E. coli konstruiert wurde, in S. typhi besser zu funktionieren
schien. Die nicht-induzierte Menge der Expression von GFPuv war
bei sowohl DH5α als
auch CVD 908htrA gleich (mittlere Fluoreszenzintensität von entsprechend
4,45 gegenüber
5,37, Tabelle 3). Die Synthese von GFPuv in DH5α wurde jedoch nach der Induktion
auf 70% erhöht,
stieg aber in CVD 908-htrA auf über
300% an (mittlere Fluoreszenzintensität von entsprechend 7,69 gegenüber 23,4).
Dieser Effekt wurde nicht durch das PompC1-Allel
eingeschränkt,
war jedoch genauso beachtlich, wenn PompC3 verwendet
wurde (Tabelle 3). Diese Daten stimmen nicht mit den kürzlich von
Martinez-Flores et al. (1999) gemachten Beobachtungen überein,
die berichten, dass Genfusionen von ompC – lacZ aus E. coli wesentlich
in S. typhi exprimierten und dass diese wesentliche Expressionsmenge
mit den in E. coli induzierten Mengen vergleichbar war. Obwohl wir
einen definierten Locus für
Punktmutationen am 3'-Ende
unseres E. coli-PompC1-Allels identifiziert
haben, das das osmotisch kontrollierte Verhalten desselben in CVD
908-htrA von S. typhi erklären
könnte,
wurden solche Mutationen nicht in PompC3 identifiziert,
das ebenso auf die Osmolarität
in CVD 908-htrA reagiert. Es ist zu beachten, dass die von Martinez-Flores
et al. untersuchten Genfusionen jedoch 1.150 bp der 5' von ompC stromaufwärts gelegenen
Kontrollregion von E. coli umfassten, während die hier konstruierten
PompC-Allele nur 459 bp der zu ompC 5'-proximalen Kontrollregion umfassen.
Ungeachtet dieser Diskrepanz ist es jedoch viel versprechend, dass
die größten Mengen
der regulierten Expression heterologer Gene mit dem Vektor-Lebendimpstoff
aus einem attenuiertem S. typhi-Stamm beobachtet werden.
-
Dann
wurden die Beiträge
verschiedener Plasmiderhaltungssysteme zur Stabilität von Plasmiden
in CVD 908-htrA, die in der Abwesenheit von Antibiotikaselektion
angezüchtet
wurden, untersucht. Keine Kombination von Stabilisierungsfunktionen
konnte die Plasmide, die oriE1-Replikationsursprünge enthielten, stabilisieren;
tatsächlich
waren diese Konstrukte sogar in Gegenwart von Antibiotika schwer
zu vermehren. Diese Beobachtungen ziehen die Gründe für die Verwendung von Plasmiden
mit höherer
Kopienzahl zur Optimierung der Expression von heterologen Antigenen
in dem Zytoplasma von auf S. typhi basierenden Lebendvektoren, eine
Strategie, die von anderen Gruppen, die Salmonella als Lebendvektoren
untersuchten, verfolgt wurde (Covone et al., 1998), in Zweifel.
-
Der
Einbau von Plasmiderhaltungssystemen in Plasmide, die einen ori15A-Replikationsursprung
tragen, war noch viel versprechender. Wenn Lebendvektoren, die solche
Plasmide tragen, ohne Selektion 24 Stunden lang bei 37°C passagiert
wurden, wurden die Effekte verschiedener Kombinationen von Stabilisierungsfunktionen
offensichtlich. In der Abwesenheit von Stabilisierungsfunktionen
wurde das ori15A-Replikon pGEN91, unabhängig von der Höhe der Induktion
von PompC1, aus mehr als 90% der Population
verloren (siehe Tabelle 6 und 8).
Durch den Einbau des post-segregationalen Killing-Locus hok-sok in
pGEN111, verdreifachte sich der Prozentsatz der Bakterien, die GFPuv
exprimiertem, bei allen Induktionsbedingungen, was die Beobachtungen
von anderen, dass der hok-sok-Locus die Stabilität von ori15A-Replikons verbessert
(Gerdes et al., 1985; Gerdes, 1988; Gerdes et al., 1997b), bestätigte. Es
wurde jedoch trotzdem festgestellt, dass, unabhängig von den Induktionsbedingungen,
mehr als 50% der Bakterienpopulation nicht mehr langer fluoreszierten.
Da bestätigt
worden war, dass wenigstens ein Teil dieser nichtfluoreszierenden
Population nicht lebensfähig
war und dieser Arzneimittelresistenz fehlte, bestätigen diese
Daten vorhergehende Berichte (Gerdes et al., 1986; Wu und Wood,
1994; Pecota et al., 1997), die angeben, dass die Gegenwart des
post-segregationalen Killing-Systems
hok-sok an sich nicht dazu ausreicht, um sicherzustellen, dass keine
plasmidlosen, lebensfähigen
Bakterien in einer wachsenden Population auftreten.
-
Ein
möglicher
Mechanismus, der das Umgehen des Einflusses von hok-sok ermöglicht,
beinhaltet spontane Punktmutationen, die dem offenen Leserahmen
des letalen Hok auftreten und Hok bezüglich der Konformation inaktivieren
könnten
und damit das Auftreten eines Plasmidverlusts ohne Letalität zulassen
würden.
Dieser Punkt betont das Erfordernis multipler Mechanismen zur Verbesserung
der Stabilität
von in wachsenden Bakterien enthaltenen Plasmiden; sollte eine Stabilisierungsfunktion
inaktiviert werden, wird die Wahrscheinlichkeit, dass andere davon
unabhängige
Funktionen gleichzeitig inaktiviert werden, vernachlässigbar klein.
Tatsächlich
ist eine solche Redundanz von Stabilisierungsfunktionen in natürlich vorkommenden
Plasmiden mit geringer Kopienzahl weit verbreitet (Nordstrom und
Austin, 1989). Zum Beispiel enthält
der Sex-Faktor F von Escherichia coli eine aktive Partitionierungsfunktion
(sop) und zwei Killing-Systeme (ccd und flm) (Loh et al., 1988;
Golub und Panzer, 1988; Van Melderen et al., 1994, Niki und Hiraga,
1997). In ähnlicher
Weise enthält
des Arzneimittelresistenzplasmid pR1 die aktive Partitionierungsfunktion
parA sowie das post-segregationale Killing-System hok-sok; daneben
trägt es
noch das vor kurzem definierte Killing-System kis-kid (Bravo et
al., 1987; Bravo et al., 1988; Ruiz-Echevarria et al., 1995). In
der hier angegebenen Arbeit zeigen wir, dass die Insertion eines
noch vollständigeren
Stabilisierungssystem, das aus sowohl einem post-segregationalen System
als auch zwei Partitionsfunktionen besteht, in Vielkopie-ori15A-Replikons, unabhängig von
den Induktionsbedingungen für
die Expression des heterologen Antigens, die Stabilität dieser
Expressionsplasmide in Abwesenheit einer Selektion drastisch verbessert.
Nach einem 48-stündigen
Passagierungsdurchgang ohne Selektion wurden die Plasmide, aufgrund
des Umgehens der Letalität
von Hok, jedoch möglicherweise
aus der Bakterienpopulation verloren. Dieses Problem wurde vor kurzem
von Pecota et al. (1997) angegangen, die berichteten, dass der Einbau
von doppelten Killing-Systemen die Plasmidstabilität im Vergleich
zu der Verwendung von hok-sok allein erheblich verbesserte; in diesen
Plasmiden waren keine Partitionsfunktionen vorhanden. Vielleicht
kann der Einbau des kis-kid-Killing-Systems, um dadurch das Komplement
von pR1-Stabilisierungsfunktionen noch vollständiger darzustellen, für eine optimale
Stabilität
von Expressionsplasmiden in Lebensvektoren von S. typhi mit höherer Kopienzahl
erforderlich sein; da kürzlich
phd-doc-PSK-Kassetten
konstruiert wurden, untersuchten wir auch die Kompatibilität dieser
PSK-Funktion mit
unseren Expressionsplasmiden pGEN211, pGEN222 und pGEN206.
-
Ein
Vergleich der Stämme,
die pGEN211 tragen (ein ori15A-Replikon, das hok-sok + par trägt, ~ 15 Kopien
pro chromosomales Äquivalent)
mit dem Plasmid pGEN142 mit viel geringerer Kopienzahl (ein ori101-Replikon,
das hok-sok + par trägt,~
~5 Kopien pro chromosomales Äquivalent)
zeigt, dass unter den Bedingungen einer maximalen Induktion von
PompC1 mit 300 mM NaCl, 57% einer Population
von CVD 908-htrA(pGEN121), die nur 24 Stunden lang ohne Selektion
passagiert wurden), mit einer mittleren Fluoreszenzintensität von 105,3
fluoreszieren; bei einer Population von CVD 908-htrA(pGEN142), die
96 Stunden lang ohne Selektion unter identischen Induktionsbedingungen
passagiert wurden, behalten 94% der Bakterien, die mittels Durchflusszytometrie
analysiert wurden, noch eine mittlere Fluoreszenzintensität von 47,7
bei. Auf der Basis solcher Ergebnisse mit GFPuv als Testantigen
ist die Versuchung groß zu
spekulieren, dass eine optimale Menge für ein heterologes Antigen,
das dem menschlichen Immunsystem durch einen auf attenuiertem S.
typhi basierenden Vektor-Lebendimpfstoff präsentiert wird, durch Senken
der Kopienzahl der darin vorhandenen Expressionsplasmide auf vielleicht
5 Kopien pro chromosomales Äquivalent
erhalten werden kann.
-
Die
Effizienz des Auslösens
einer gegen ein heterologes Antigen gerichteten Immunantwort wird
zum Teil von der Fähigkeit
des Lebendvektors, solche Antigene dem Immunsystem zu präsentieren,
abhängen.
Die Fähigkeit
eines Lebendvektors, Antigene zu präsentieren, wird wiederum von
der Stabilität
der Vielkopie-Expressionsplasmide, die für die heterologen Antigene
codieren, abhängen.
Unsere Ergebnisse zeigen, dass der Einbau eines Plasmiderhaltungssystems
in Vielkopie-Expressionsplasmiden, ohne weitere genetische Manipulation
des Lebendvektors, die Stabilität
solcher Expressionsplasmide verbessert. Die Gegenwart von Vielkopie-Plasmiden
kann jedoch auch die auf den Stoffwechsel bezogene Tauglichkeit
des Lebendvektors beeinflussen. Dies ist deshalb relevant, weil
einige fremde Antigene von Interesse einen schädigenden Effekt auf den Lebendvektor
ausüben.
-
Obwohl
wir uns nicht auf diese Theorie festlegen möchten, stellen wir zusammenfassend
fest, dass der Stoffwechsel von CVD 908-htrA, die ein Vielkopie-Expressionsplasmid
tragen, erheblich belastet wird; wenn die Kopienzahl und/oder die
Menge der Genexpression zunimmt, nimmt die Belastung für den Stoffwechsel
zu. Untersuchungen mit E. coli haben deutlich herausgestellt, dass
Plasmid-tragende Bakterien langsamer wachsen als plasmidlose Bakterien
(Boe et al., 1987; McDermott et al., 1993; Wu und Wood, 1994; Pecota
et al., 1997; Summers, 1998). Es wurde auch gezeigt, dass, wenn
die Kopienzahl zunimmt, die Wachstumsgeschwindigkeit solcher Stämme abnimmt;
in ähnlicher
Weise nimmt die Wachstumsgeschwindigkeit weiter ab, wenn die Induktion
heterologer Gene zunimmt (Wu und Wood, 1994; Pecota et al., 1997).
Natürlich
würde ein spontaner
Verlust des Plasmids jegliche Belastung des Stoffwechsels aufheben
und zulassen, dass plasmidlose Bakterien schnell die Population
von Plasmid-tragenden Bakterien überwachsen.
In eleganten Studien zeigten Wu und Wood (Wu und Wood, 1994), dass
Plasmid-tragende E. coli-Stämme
die Plasmide unter Bedingungen, bei denen die Expression des klonierten
Gens in 100 Stunden gering war, wenn diese in Abwesenheit von Selektion
passagiert wurden, beibehielten; im Gegensatz dazu trat unter maximalen
Induktionsbedingungen ein vollständiger
Plasmidverlust innerhalb von 10 Stunden auf. Interessanter Weise
wurden die Plasmide, wenn der hok-sok-Locus in diese Expressionsplasmide
inseriert wurde, bei nichtinduzierten Bedingungen 300 Stunden lang
und bei induzierenden Bedingungen 30 Stunden lang beibehalten. Eine
solche Verschiebung in der Expression der Antigene innerhalb einer
Population von Bakterien-Lebendvektor würde erwartungsgemäß die Effizienz
der Stimulation irgendeiner für
das fremde Antigen spezifischen Immunantwort reduzieren. Unsere
Analyse führte
uns zu der Schlussfolgerung, dass das Ziel für einen effektiven, multivalenten,
auf S. typhi basierenden Vektor-Lebendimpfstoff ist, die Lebensfähigkeit
mit Hilfe stabilisierter Expressionsvektoren mit geringer Kopienzahl
zu optimieren, die in der Lage sind, hohe Mengen des heterologen
Antigens als Reaktion auf ein aus der Umgebung stammendes Signal,
dem die Impfstofforganismen in vivo wahrscheinlich dann begegnen,
wenn sie eine geeignete ökologische
Nische erreicht haben, zu exprimieren. Wir überprüfen derzeit diese Strategie
unter Verwenden des murinen, intranasalen Modells, um die Immunogenität des Fragments
C von Tetanus-Toxin zu untersuchen, das in CVD 908-htrA von unseren
Expressionsvektoren pGEN211 (oriE1), pGEN222 (ori15A) und pGEN206
(ori101) exprimiert wird, wobei diese alle identische Plasmiderhaltungssysteme
tragen und sich nur in ihrer Kopienzahl unterscheiden. Die hierin
beschriebene Arbeit ermöglicht
die Entwicklung von oralen, auf S. typhi basierenden Vektor-Lebendimpfstoffen
in Einzeldosen, die in der Lage sind, schützende Immunreaktionen gegen
viele, nicht miteinander verwandte, humane Pathogene zu induzieren.
-
7. Literatur
-
- Acheson, D. W. K. 1998. Nomeclature of enterotoxins. Lancet
351:1003.
- Acheson, D. W. K., M. M. Levine, J. B. Kaper und G. T. Keusch.
1996. Protective immunity to Shiga-like toxin I following oral immunization
with Shiga-like toxin I B-subunitproducing Vibrio cholerae CVD 103-HgR.
Infection and Immunity 64:355.
- Austin, S. J. 1988. Plasmid partition. Plasmid 20: 1.
- Austin, S., S. Friedman and D. Ludtke. 1986. Partition functions
of unit-copy plasmids can stabilize the maintenance of plasmid pBR322
at low copy number. J. Bacteriol. 168: 1010-1013.
- Barry, E. M., O. G. Gomez-Duarte, S. Chatfield, R. Rappuoli,
M. Pizza, G. Losonsky, J. E. Galen und M. M. Levine. 1996. Expression
and immunogenicity of pertussis toxin S1 subunit-tetanus toxin fragment
C fusions in Salmonella typhi vaccine strain CVD 908. Infection
and Immunity 64:4172-4181.
- Barth, P. T., H. Richards und N. Datta. 1978. Copy numbers of
coexisting plasmids in Escherichia coli K-12.J. Bacteriol. 135:760-765.
- Bast, D. J., J. L. Brunton, M. A. Karmali und S. E. Richardson.
1997. Toxicity and immunogenicity of a verotoxin 1 mutant with reduced
globotriaosylceramide receptor binding in rabbits. Infection and
Immunity 65:2019.
- Baumler, A. J., J. G. Kusters, I. Stojiljkovic und F. Heffron.
1994. Salmonella typhimurium loci involved in survival within macrophages.
Infection and Immunity 62:1623. Beaucage, S. L., C. A. Miller und
S. N. Cohen. 1991. Gyrase-dependent stabilization of pSC101 plasmid
inheritance by transcriptionally active promoters. EMBOJ 10 :2583- 2588.
- Blattner, F. R., G. Plunkett III, C. A. Bloch, N. T. Perna,
V. Burland, M. Riley, J. Collado-Vides, J. D. Glasner, C. K. Rode,
G. F. Mayhew, J. Gregor, N. W. Davis, H. A. Kirkpatrick, M. A. Goeden,
D. J. Rose, B. Mau und Y. Shao. 1997. The complete genome sequence
of Escherichia coli K-12. Science 277:1453.
- Blomfield, I. C., V. Vaughn, R. F. Rest und B. I. Eisenstein.
1991. Allelic exchange in Escherichia coli using the Bacillus subtilis
sacB gene and a temperature-sensitive pSC101 replicon. Molecular
Microbiology 5:1447-1457. Boe, L. und K. V. Rasmussen. 1996. Suggestions
as to quantitative measurements of plasmid loss. Plasmid 36:153.
- Boe, L., K. Gerdes und S. Molin. 1987. Effects of genes exerting
growth inhibition and plasmid stability on plasmid maintenance.
Journal of Bacteriology 169:4646-4650.
- Bokman, S. H. und W. W. Ward. 1981. Renaturation of Aequorea
green-fluorescent protein. Biochemical and Biophysical Research
Communications 101:1372.
- Bosworth, B. T., J. E. Samuel, H. W. Moon, A. D. O'Brien, V. M. Gordon
und S. C. Whipp. 1996. Vaccination with genetically modified Shiga-like
toxin IIe prevents edema disease in swine. Infection and Immunity
64:55.
- Bouvier, J., C. Richaud, W. Higgins, O. Bogler und P.Stragier.
1992. Cloning, characterization, and expression of the dapE gene
of Escherichia coli. Journal of Bacteriology 174:5265.
- Boyd, B. und C. A. Lingwood. 1989. Verotoxin receptor glycolipid
in human renal tissue. Nephron 51:207.
- Bravo, A., G. de Torrontegui und R. Diaz. 1987. Identification
of components of a new stability system of plasmid R1, ParD, that
is close to the origin of replication of this plasmid. Mol. Gen.
Genet. 210:101-110.
- Bravo, A., S. Ortega, G. de Torrontegui und R. Diaz. 1988. Killing
of Escherichia coli cells modulated by components of the stability
system parD of plasmid R1. Mol. Gen. Genet. 215:146-151.
- Brosius, J. 1989. Superpolylinkers in cloning and expression
vectors. DNA 8: 759- 777.
- Butterton, J. R., E. T. Ryan, D. W. Acheson und S. B.Calderwood.
1997. Coexpression of the B subunit of Shiga toxin 1 and EaeA from
enterohemorrhagic Escherichia coli in Vibrio cholera vaccine strains.
Infection and Immunity 65:2127-2135.
- Cabello, F., K. Timmis und S. N. Cohen. 1976. Replication control
in a composite plasmid constructed by in vitro linkage of two distinct
replicons. Nature 259: 285-290.
- Calderwood, S. B., D. W. K. Acheson, G. T. Keusch, T. J. Barrett,
P. M. Griffan, N. A. Strockbine, B. Swaminathan, J. B. Kaper, M.
M. Levine, B. S. Kaplan, H. Karch, A. D. O'Brien, T. G. Obrig, Y. Takeda, P. I.
Tarr und I. K. Wachsmuth. 1996. Proposed new nomenclature for SLT
(VT) family. ASM News 62:118.
- Calderwood, S. B., F. Auclair, A. Donohue-Rolfe, G. T. Keusch
und J. J. Mekalanos. 1987. Nucleotide sequence of the Shiga-like
toxin genes of Escherichia coli. Proceedings of the National Academy
of Sciences USA 84:4364.
- Carlini, L. E., R. D. Porter, U. Curth und C. Urbanke. 1993.
Viability and preliminary in vivo characterization of site-specific
mutants of Escherichia coli singlestranded DNA-binding protein.
Molecular Microbiology 10:1067.
- Carter, P. B. und F. M. Colins. 1974. Growth of typhoid and
paratyphoid bacilli in intravenously infected mice. Infection and
Immunity 10:816.
- Cerin, H. und J. Hackett. 1989. Molecular cloning and analysis
of the incompatibility and partition functions of the virulence
plasmid of Salmonella typhimurium. Microbial. Pathogenesis 7:85.
- Cerin, H. und J. Hackett. 1993. The par VP region of the Salmonella
typhimurium virulence plasmid pSLT contains four loci required for
incompatibility and partition. Plasmid 30:30.
- Chalfie, M., Y. Tu, G. Euskirchen, W. W. Ward und D. C. Prasher.
1994. Green fluorescent protein as a marker for gene expression.
Science 263:802.
- Chambers, S. P., S. E. Prior, D. A. Barstow und N. P. Minton.
1988. The pMTLnic cloning vectors.1. Improved pUC polylinker regions
to facilitate the use of sonicated DNA for nucleotide sequencing.
Gene 68:139.
- Chang, A. C. Y., und S. N. Cohen. 1978. Construction and characterization
of amplifiable multicopy DNA cloning vehicles derived from the P15A
cryptic miniplasmid. J. Bacteriol 134:1141-1156.
- Chase, J. W. und K. R. Williams. 1986. Single-stranded DNA binding
proteins required for DNA replication. Annual Reviews in Biochemistry
55:103.
- Chase, J. W., J. B. Murphy, R. F. Whittier, E. Lorensen und
J. J. Sninsky. 1983. Amplification of ssb-1 mutant single-stranded
DNA-binding protein in Escherichia coli. Journal of Molecular Biology
163, 164:193.
- Chatfield, S., K. Strahan, D. Pickard, I. G. Charles, C. E.
Hormaeche und G. Dougan. 1992. Evaluation of Salmonella typhimurium
strains harbouring defined mutations in htrA and aroA in the murine
salmonellosis model. Microbial. Pathogenesis 12:145.
- Clark, C., D. Bast, A. M. Sharp, P. M. St. Hilaire, R. Agha,
P. E. Stein, E. J. Toone, R. J. Read und J. L. Brunton. 1996. Phenylalanine
30 plays an important role in receptor binding of verotoxin-1. Molecular
Microbiology 19:891.
- Conradi, H. 1903. Über
lösliche,
durch aseptische Autolyse erhalten Giftstoffe von Ruhr- und Typhusbazillen. Dtsch.
Med. Wochenschr. 29:26.
- Covarrubias, L., L. Cervantes, A. Covarrubias, X.Soberon,l.
Vichido, A. Blanco, Y. M. Kupersztoch-Portnoy und F. Bolivar. 1981.
Construction and characterization of new cloning vehicles. V. Mobilization
and coding properties of pBR322 and several deletion derivatives
including pBR327 and pBR328. Gene 13:25-35.
- Covone, M. G., M. Brocchi, E. Palla, W. D. daSilveira, R. Rappuoli
und C. L. Galeotti. 1998. Levels of expression and immunogenicity
of attenuated Salmonella enterica serovar typhimurium strains expressing
Escherichia coli mutant heat-labile enterotoxin. Infection and Immunity
66:224-231.
- Crameri, A., E. A. Whitehorn, E. Tate und W. P. Stemmer. 1996.
Improved green fluorescent protein by molecular evolution using
DNA shuffling. Nat. Biotechnol. 14:315- 319.
- Dam, M. und K. Gerdes. 1994. Partitioning of plasmid R1: ten
direct repeats flanking the parA promoter constitute acentromere-like
partition site parC, that expresses incompatibility. Journal of
Molecular Biology 236:1289-1298.
- Dopf, J. und T.M. Horiagon. 1996. Deletion mapping of the Aequorea
victoria green fluorescent protein. Gene 173:39.
- Downes, F. P., T. J. Barrett, J. H. Green, C. H.Aloisio, J.
S. Spika, N. A. Strockbine und I. K. Wachsmuth. 1988. Affinity purification
and characterization of Shigalike toxin II and production of toxin-specific
monoclonal antibodies. Infection and Immunity 56:1926.
- Egger, L. A., H. Park und M.Inouye. 1997. Signal transduction
via the histidylaspartyl phosphorelay. Genes to Cells 2:167.
- Endo, Y., K. Tsurugi, T. Yutsudo, Y. Takeda, T. Ogasawara und
K.Igarashi. 1988. Site of action of a Vero toxin (VT2) from Escherichia
coli O157:H7 and of Shiga toxin on eukaryotic ribosomes: RNA N-glycosidase
activity of the toxins. European Journal of Biochemistry 171:45.
- Forrest, B. D., J. T. Labrooy, S. R. Attridge, G. Boehm, L.
Beyer, R. Morona, D. J. C. Shearman und D. Rowley. 1989. Immunogenicity
of a candidate live oral typhoid/cholera hybrid vaccine in humans.
J. Infect Dis. 159:145.
- Fraser, M. E., M. M. Chernaia, Y. V. Kozlov und M. N. G. James.
1994. Crystal structure of the holotoxin from Shigella dysenteriae
at 2.5 A resolution. Nature Structural Biology 1: 59.
- Galen, J. E., K. Nakayama und R.Curtiss III. 1990. Cloning and
characterization of the asd gene of Salmonella typhimurium: use
in stable maintenance of recombinant plasmids in Salmonella vaccine
strains. Gene 94:29-35.
- Galen, J. E. and M. u. Levine. 1995. Improved suicide vectors
for chromosomal mutagenesis in Salmonella typhi. Abstracts of the
Annual Meeting of the American Society of Microbiology H192:(Abstract).
- Galen, J. E. und M. M. Levine. 1996. Further refinements of
suicide vector-mediated chromosomal mutagenesis Salmonella typhi.
Abstracts of the Annual Meeting of the American Society of Microbiology
H260:(Abstract).
- Galen, J. E., O. G. Gomez-Duarte, G. Losonsky, J. L. Halpern,
C. S. Lauderbaugh, S. Kaintuck, M. K. Reymann, and M. M. Levine.
1997. A murine model of intranasal immunization to assess the immunogenicity
of attenuated Salmonella typhi live vector vaccines in stimulating
serum antibody responses to expressed foreign antigens. Vaccine
15:700-708.
- Galen, J. E., E. R. Vimr, L. Lawrisuk und J. B. Kaper. 1990.
Cloning, sequencing, and expression of the gene, nanH, for Vibrio
cholerae neuraminidase. In Advances in research on cholera and related
diarrheas (Edited by Sack R. B. und Zinnake Y. Tokyo: KTK Scientific
Publishers. S. 143-153.
- Gay, P., D. Le Coq, M. Steinmetz, E. Ferrari und J. A. Hoch.
1983. Cloning structural gene sacB, which codes for exoenzyme levansucrase
of Bacillus subtilis: expression of the gen in Escherichia coli.
Journal of Bacteriology 153:1424.
- Gerdes, K. 1988. The parB (hok-sok) locus of plasmid R1 : a
general purpose plasmid stabilization system. Bio/Technology 6:1402-1405.
- Gerdes, K. und S. Molin. 1986. Partitioning of plasmid R1: structural
and functional analysis of the parA locus. Journal of Molecular
Biology 190:269.
- Gerdes, K., A. P. Gultyaev, T. Franch, K. Pedersen und N. D.
Mikkelsen. 1997. Antisense RNA-regulated programmed cell death.
Annual Reviews in Genetics 31:1-31.
- Gerdes, K., J. S. Jacobsen und T. Franch.1997b. Plasmid stabilization
by postsegregational killing. Genet. Eng. (NJ) 19:49-61.
- Gerdes, K., J. E. Larsen und S.Molin. 1985. Stable inheritance
of plasmid R1 requires two different loci. J. Bacteriol 161: 292-298.
- Gerdes, K., P. B. Rasmussen und S.Molin. 1986. Unique type of
plasmid maintenance function: postsegregational killing of plasmid-free
cells. Proc. Natl. Acad. Sci. USA 83:3116-3120.
- Gerichter, C. B. 1960. The dissemination of Salmonella typhi,
S. paratyphi A, and S. paratyphi B through the organs of the white
mouse by oral infection. Journal of Hygiene, Cambridge 58:307.
- Gerichter, C. B. und D. L. Boros. 1962. Dynamics of infection
of the blood stream and internal organs of white mice with Salmonella
typhi by intraperitoneal injection. Journal of Hygiene, Cambridge
60:311.
- Golub, E.I. und H. A. Panzer. 1988. The F factor of Escherichia
coli carries a locus of stable plasmid inheritance stm, similar
to the parB locus of plasmid R1. Mol. Gen. Genet. 214:353-357.
- Gomez-Duarte, O. G., J. E. Galen, S. N. Chatfield, R.Rappuoli,
L. Eidels und M. M. Levine. 1995. Expression of fragment C of tetanus
toxin fused to a carboxyl-terminal fragment of diphtheria toxin
in Salmonella typhi CVD 908 vaccine strain. Vaccine 13:1596.
- Gonzalez, C., D. M. Hone, F. Noriega, C. O. Tacket, J. R. Davis,
G. Losonsky, J. P. Nataro, S. Hoffman, A. Malik, E. Nardin, M. Sztein,
D. G. Heppner, T. R. Fouts, A.Isibasi und M. M. Levine. 1994. Salmonella
typhi vaccine strain CVD 908 expressing the circumsporozoite protein
of Plasmodium falciparum:strain construction and safety and immunogenicity
in humans. Joumal of Infectious Diseases 169:927-931.
- Gordon, V. M., S. C. Whipp, H. W. Moon, A. D. O'Brien und J. E. Samuel.
1992. An enzymatic mutant of Shiga-like toxin II variant is a vaccine
candidate for edema disease of swine. Infection and Immunity 60:485.
- Gottesman, S., W. P. Clark, V. de Crecy-Lagard und M. R. Maurizi.
1993. ClpX, an alternative subunit for the ATP-dependent Clp protease
of Escherichia coli. Journal of Biological Chemistry 268:22618.
- Green, J. M., B. P. Nichols und R. G. Matthews. 1996. Folate
biosynthesis, reduction, and polyglutamylation. In Escherichia coli
and Salmonella: Cellular and molecular biology. 2. Ausg., F. C.
Neidhardt, R. Curtiss III, J. L. Ingraham, E. C. C. Lin, K. B. Low,
B. Magasanik, W. S. Reznikoff, M. Riley, M. Schaechter und H. E.
Umbarger, Hrsg. ASM Press, Washington, D. C. S. 665.
- Griffin, P. M. 1995. Escherichia coli O157:H7 and other enterohemorrhagic
Escherichia coli. In Infections of the gastrointestinal tract. M.
J. Blaser, P. D. Smith, J.I. Ravdin, H. B. Greenberg and R. L. Guerrant,
Hrsg. Raven Press, Ltd, New York, S. 739.
- Gyles, C. L. 1992. Escherichia coli cytotoxins and enterotoxins.
Canadian Journal of Microbiology 38:734.
- Heim, R., D. C. Prasher und R. Y. Tsien. 1994. Wavelength mutations
and posttranscriptional autoxidation of green fluorescent protein.
Proceedings of the National Academy of Sciences USA 91:12501.
- Hiszczynska-Sawicka, E. und J. Kur. 1997. Effect of Escherichia
coli IHF mutations on plasmid p15A copy number. Plasmid 38:174-179.
- Hoiseth, S. K. und B. A. Stocker. 1981. Aromatic-dependent Salmonella
typhimurium are non-virulent and effective as live vaccines. Nature
291:238.
- Hone, D. M., A. M. Harris, S. Chatfield, G. Dougan und M. M.
Levine. 1991. Construction of genetically defined double aro mutants
of Salmonella typhi. Vaccine 9:810- 816.
- Hovde, C. J., S. B. Calderwood, J. J. Mekalanos und R. J. Collier.
1988. Evidence that glutamic acid 167 is an active-site residue
of Shiga-like toxin I. Proceedings of the National Academy of Sciences
USA 85:2568.
- Jackson, M. P., E. A.Wadolkowski, D. L. Weinstein, R. K. Holmes
und A. D. O'Brien.
1990. Functional analysis of the Shiga toxin and Shiga-like toxin
type II variant binding subunits by using site-directed mutagenesis.
Journal of Bacteriology 172:653.
- Jackson, M. P., R. J. Neill, A. D.O'Brien, R. K. Holmes und J. W. Newland.
1987. Nucleotide sequence analysis and comparison of the structural
genes for Shiga-like toxin I and Shiga-like toxin II encoded by
bacteriophages from Escherichia coli. FEMS Microbiology Letters
44:109.
- Jackson, M. P., R. L. Deresiewicz und S. B.Calderwood. 1990.
Mutational analysis of the Shiga toxin and Shiga-like toxin II enzymatic
subunits. Journal of Bacteriology 172: 3346.
- Jarvis, K. G. und J. B. Kaper. 1996. Secretion of extracellular
proteins by enterohemorrhagic Escherichia coli via a putative type
III secretion system. Infection and Immunity 64:4826.
- Jarvis, K. G., J. A. Giron, A. E. Jerse, T. K. McDaniel, M.
S. Donnenberg und J. B. Kaper. 1995. Enteropathogenic Escherichia
coli contains a putative type III secretion system necessary for
the export of proteins involved in attaching and effacing lesion
formation. Proceedings of the National Academy of Sciences USA 92:7996.
- Jensen, R. B. und K. Gerdes. 1995. Programmed cell death in
bacteria:proteic plasmid stabilization systems. Molecular Microbiology
17:205.
- Jensen, R. B. und K. Gerdes. 1997. Partitioning of plasmid R1.
The ParM protein exhibits ATPase activity and interacts with the
centromere-like ParR-parC complex. Journal of Molecular Biology
269:505-513.
- Karem, K. L., S. Chatfield, N. Kuklin und B. T. Rouse. 1995.
Differential induction of carrier antigen-specific immunity by Salmonella
typhimurium live-vaccine strains after single mucosal or intravenous
immunization of BALB/c mice. Infection and Immunity 63: 4557-4563.
- Karmali, M. A. 1989. Infection by verocytotoxin-producing Escherichia
coli. Clinical Microbiological Reviews 2:15.
- Karmali, M. A., M. Petric, C. Lim, P. C. Fleming und B. T. Steele.
1983. Escherichia coli cytotoxin, haemolytic-uraemic syndrome, and
haemorrhagic colitis. Lancet ii:1299.
- Karmali, M. A., M. Petric, C. Lim, P. C. Fleming, G. S. Arbus,
and H. Lior. 1985. The association between idiopathic hemolytic
uremic syndrome and infection by verotoxinproducing Escherichia
coli. Journal of Infectious Diseases 151 :775.
- Karpman, D., H. Connell, M. Svensson, F. Scheutz, P. Alm und
C. Svanborg. 1997. The role of lipopolysaccharide and Shiga-like
toxin in a mouse model of Escherichia coli O157 : H7 infection.
Journal of Infectious Diseases 175:611.
- Keusch, G. T., G. F. Grady, L. J. Mata und J.McIver. 1972. Pathogenesis
of shigella diarrhea. 1. Enterotoxin production by Shigella dysenteriae
1. Journal of Clinical Investigation 51:1212.
- Killeen, K. P., V. Escuyer, J. J. Mekalanos und R. J. Collier.
1992. Reversion of recombinant toxoids: mutations in diphtheria
toxin that partially compensate for active-site deletions. Proceeding
of the National Academy of Sciences USA 89:6207.
- Kim, J. Y., H. A. Kang und D. D. Ryu. 1993. Effects of the par
locus on the growth rate and structural stability of recombinant
cells. Biotechnology Progress 9:548.
- Konowalchuk, J., J. 1. Speirs und S. Stavric. 1977. Vero response
to a cytotoxin of Escherichia coli. Infection and Immunity 18:775.
- Langermann, S., S. Palaszynski, A. Sadziene, C. K. Stover und
S. Koenig. 1994. Systemic and mucosal immunity induced by BCG vector
expressing outer-surface protein A of Borrelia burgdorferi. Nature
372:552-555.
- Lee, S. F., R. J. March, S. A. Halpern, G.Faulkner und L. Gao.
1999. Surface expression of a protective recombinant pertussis toxin
S1 subunit fragment in Streptococcus gordonii. Infect. Immun. 67:1511-1516.
- Lehnherr, H. und M. B. Yarmolinsky. 1995. Addiction protein
Phd of plasmid prophage P1 is a substrate of the ClpXP serine protease
of Escherichia coli. Proceedings of the National Academy of Sciences
USA 92:3274.
- Lehnherr, H., E. Maguin, S. Jafri und M. B. Yarmolinsky. 1993.
Plasmid addiction genes of bacteriophage P1: doc, which causes cell
death on curing of prophage, and phd, which prevents host death
when prophage is retained. Journal of Molecular Biology 233:414.
- Levine, M. M., J. E. Galen, E. M. Barry, F. Noriega, S. Chatfield,
M. Sztein, G. Dougan und C. O. Tacket. 1996. Attenuated Salmonella
as live oral vaccines against typhoid fever and as live vectors.
Journal of Biotechnology 44:193.
- Lindgren, S. W., J. E. Samuel, C. K. Schmitt und A. D. O'Brien. 1994. The
specific activities of Shiga-like toxin type II (SLT-II) andSLT-II-related
toxins of enterohemorrhagic Escherichia coli differ when measured
by Vero cell cytotoxicity but not by mouse lethality. Infection
and Immunity 62:623.
- Lloyd, R. G. und K. B. Low. 1996. Homologous recombination.
In Escherichia coli and Salmonella: Cellular and molecular biology.
2. Ausg., F. C. Neidhardt, R. Curtiss III, J. L. Ingraham, E. C.
C. Lin, K. B. Low, B. Magasanik, W. S. Reznikoff, M. Riley, M. Schaechter
und H. E. Umbarger, Hrsg. ASM Press, Washington, D. C. S. 2236.
- Loh, S. M., D. S. Cram und R. A. Skurray. 1988. Nucleotide sequence
andtranscriptional analysis of a third function (Flm) involved in
F plasmid maintenance. Gene 66:259-268.
- Lohman, T. M. und M. E. Ferrari. 1994. Escherichia coli single-stranded
DNAbinding protein: multiple DNA-binding modes and cooperativities.
Annual Reviews in Biochemistry 63:527.
- Louise, C. B. und T. G. Obrig. 1995. Specific interaction of
Escherichia coli O157: H7-derived Shiga-like toxin II with human
renal endothelial cells. Journal of Infectious Diseases 172:1397.
- Love, C. A., P. E. Lilley und N. E. Dixon. 1996. Stable high-copy-number
bacteriophage lambda promoter vectors for overproduction of proteins
in Escherichia coli. Gene 176:49.
- Lynch, A. S. und E. C. C. Lin. 1996. Responses to molecular
oxygen. In Escherichia coli and Salmonella:Cellular and molecular
biology. 2. Ausg., F. C. Neidhardt, R. Curtiss III, J. L. Ingraham,
E. C. C. Lin, K. B. Low, B. Magasanik, W. S. Reznikoff, M. Riley,
M. Schaechter und H. E. Umbarger, Hrsg. ASM Press, Washington, D. C.
S. 1526.
- Magnuson, R., H. Lehnherr, G. Mukhopadhyay, und M. B. Yarmolinsky.
1996. Autoregulation of the plasmid addiction operon of bacteriophage
P1. Journal of Biological Chemistry 271:18705.
- Makoff, A. J., und A. E. Smallwood. 1988. Heterologous expression
in Escherichia coli: effects of alterations in the sequence 5' to the initiation
codon. Biochem. Soc. Trans. 16: 48-49.
- Mangeney, M., C. A. Lingwood, S. Taga, B. Caillou, T. Tursz
und J. Wiels. 1993. Apoptosis induced in Burkitt's lymphom cells via Gb3/CD77,
a glycolipid antigen. Cancer Research 53:5314.
- Marshall, J., R. Molloy, G. W. J. Moss, J. R. Howe und T. E.
Hughes. 1995. The jellyfish green fluorescent protein: a new tool
for studying ion channel expression and function. Neuron 14:211.
- Martinez-Flores, I., R. Cano, V. H. Bustamante, E. Calva und
J. L. Puente. 1999. The ompB operon partially determines differential
expression of OmpC in Salmonella typhi and Escherichia coli. J.
Bacteriol. 181:556-562.
- Matthews, R. G. 1996. One-carbon metabolism. In Escherichia
coli and Salmonella: Cellular and molecular biology. 2. Ausg., F.
C. Neidhardt, R. Curtiss III, J. L. Ingraham, E. C. C. Lin, K. B.
Low, B. Magasanik, W. S. Reznikoff, M. Riley, M. Schaechter and
H. E. Umbarger, Hrsg. ASM Press, Washington, D. C. S. 600.
- Maurizi, M. R., W. P. Clark, Y. Katayama, S. Rudikoff, J. Pumphrey,
B. Bowers und S. Gottesman. 1990. Sequence and structure of Clp
P, the proteolytic component of the ATP-dependent Clp protease of
Escherichia coli. Journal of Biological Chemistry 265: 12536.
- McClelland, M. und R. Wilson. 1998. Sample sequencing of the
Salmonella typhi genome: comparison to the E. coli K-12 genome.
Infection and Immunity
- McDaniel, T. K., K. G. Jarvis, M. S. Donnenberg und J. B. Kaper.
1995. A genetic locus of enterocyte effacement conserved among diverse
enterobacterial pathogens. Proceedings of the National Academy of
Sciences USA 92:1664.
- McDermott, P. J., P. Gowland und P. C. Gowland. 1993. Adaptation
of Escherichia coli growth rates to the presence of pBR322. Lett.
Appl. Microbiol 17:139-143.
- Meacock, P. A., und S. N. Cohen. 1980. Partitioning of bacterial
plasmids during cell division: a cis-acting locus that accomplishes
stable plasmid inheritance. Cell 20:529-542.
- Medaglini, D., G. Pozzi, T. P. King und V. A. Fischetti. 1995.
Mucosal and systemic immune responses to a recombinant protein expressed
on the surface of the oral commensal bacterium Streptococcus gordonii
after oral colonization. Proc. Natl. Acad. Sci. USA 92:6868-6872.
- Melton-Celsa, A. R. und A. D. O'Brien. 1998. The structure, biology,
and relative toxicity for cells and animals of Shiga toxin family
members. In Escherichia coli O157:H7 and other Shiga toxin producing
E. coli strains. J. B. Kaper und A. D. O'Brien, Hrsg. ASM Press, Washington,
D. C. In Druck.
- Mikkelsen, N. D. und K. Gerdes. 1997. Sok antisense RNA from
plasmid R1 is functionally inactivated by RNaseE and polyadenylated
by poly(A) polymerase I. Molecular Microbiology 26:311.
- Miller, C. A., S. L. Beaucage und S. N. Cohen. 1990. Role of
DNA superhelicity in partitioning of the pSC101 plasmid. Cell 62:
127-133.
- Moxley, R. A. und D. H. Francis. 1998. Overview of Animal Models.
In Escherichia coli O157:H7 and other Shiga toxin producing E. coli
strains. J. B. Kaper und A. D. O'Brien,
Hrsg., ASM Press, Washington, D. C. In Druck.
- Muhldorfer, I., J. Hacker, G. T. Keusch, D. W. Acheson, H. Tschape,
A. V. Kane, A. Ritter, T. Olschlager und A. Donohue-Rolfe. 1996.
Regulation of the Shiga-like toxin II operon in Escherichia coli.
Infection and Immunity 64:495.
- Nakayama, K., S. M. Kelley und R. Curtiss III. 1988. Construction
of an Asd+ expression-cloning vector: stable maintenance
and high level expression of cloned genes in a Salmonella vaccine
strain. Bio/Technology 6:693-697.
- Nakayama, K., S. M. Kelley und R.Curtiss III. 1988. Construction
of an Asd+ expression-cloning vector: stable maintenance
and high level expression of cloned genes in a Salmonella vaccine
strain. Bio/Technology 6:693.
- Nelson, S., S. E. Richardson, C. A. Lingwood, M. Petric, and
M. A.Karmali. 1994. Biological activity of verocytotoxin (VT)2c
and VT1/VT2c chimeras in the rabbit model. In Recent advances in
verocytotoxin producing Escherichia Coli infections. M. A. Karmali
und A. G. Goglio, Hrsg. Elsevier Science, New York, S. 245.
- Niki, H. und S. Hiraga. 1997. Subcellular distribution of actively
partitioning F plasmid during the cell division cycle of E. coli.
Cell 90:951-957.
- Nordstrom, K. und S. J. Austin. 1989. Mechanisms that contribute
to the stable segregation of plasmids. Annual Reviews in Genetics
23:37.
- Noriega, F. R., G. Losonsky, J. Y. Wang, S. B. Formal und M.
M. Levine. 1996. Further characterization of ∆aroA ∆virG Shigella flexneri
2a strain CVD 1203 as a mucosal Shigella vaccine and as a live-vector
vaccine for delivering antigens of enterotoxigenic Escherichia coli.
Infect. Immun. 64:23-27.
- Norioka, S., G. Ramakrishnan, K. Ikenaka und M. Inouye. 1986.
Interaction of a transcriptional activator, OmpR, with reciprocally
osmoregulated genes, ompF and ompC, of Escherichia coli. Journal
of Biological Chemistry 261:17113-17119.
- Nyholm, P., G. Magnusson, Z. Zheng, R. Norel, B. Binnington-Boyd
und C. A. Lingwood. 1996. Two distinct binding sites for globotriaosyl
ceramide on verotoxins: identification by molecular modelling and
confirmation using deoxy analogues and a new glycolipid receptor
for all verotoxins. Chemistry and Biology 3:263.
- Nyholm, P., J. L. Brunton und C. A. Lingwood. 1995. Modelling
of the interaction of verotoxin-1 (VT1) with its glycolipid receptor,
globotriaosylceramide (Gb3). International
Journal of Biological Macromolecules 17:199.
- O'Brien, A.
D. 1982. Innate resistance of mice to Salmonella typhi infection.
Infection and Immunity 38:948.
- O'Brien, A.
D., V. L. Tesh, A.Donohue-Rolfe, M. P. Jackson, S. Olsnes, K. Sandvig,
A. A. Lindberg und G. T. Keusch. 1992. Shiga toxin: biochemistry,
genetics, mode of action, and role in pathogenesis. Current Topics
in Microbiology and Immunology 180:65.
- Olitsky, P. K. und I. J. Kligler. 1920. Toxins and antitoxins
of Bacillus dysenteriae Shiga. Journal of Experimental Medicine
31:19.
- Orosz,A., I. Boros und P. Venetianer. 1991. Analysis of the
complex transcription termination region of the Escherichia coli
rrnB gene. European Journal of Biochemistry 201: 653.
- Oxer, M. D., C. M. Bentley, J. G. Doyle, T. C.Peakman, I. G.
Charles und A. J. Makoff. 1991. High level heterologous expression
in E. coli using the anaerobically-activated nir B promoter. Nucleic
Acids Research 19:2889-2892.
- Pallen, M. J. und B. W. Wren. 1997. The HtrA family of serine
proteases. Molecular Microbiology 26:209.
- Pecota, D. C., C. S. Kim, K. Wu, K. Gerdes und T. K. Wood. 1997.
Combining the hok/sok, parDE, and pnd postsegregational killer loci
to enhance plasmid stability. Applied and Environmental Microbiology 63:1917-1924.
- Perera, L. P., J. E. Samuel, R. K. Holmes und A. D. O'Brien. 1991. Mapping
the minimal contiguous gene segment that encodes functionally active
Shiga-like toxin II. Infection and Immunity 59:829.
- Perera, L. P., J. E. Samuel, R. K. Holmes und A. D. O'Brien. 1991. Identification
of three amino acid residues in the B subunit of Shiga toxin and
Shiga-like toxin type II that are essential for holotoxin activity.
Journal of Bacteriology 173:1151.
- Pittard, A. J. 1996. Biosynthesis of the aromatic amino acids.
In Escherichia coli and Salmonella:Cellular and molecular biology.
2. Ausg., F. C. Neidhardt, R. CurtissIII, J. L. Ingraham, E. C.
C. Lin, K. B. Low, B. Magasanik, W. S. Reznikoff, M. Riley, M. Schaechter
und H. E. Umbarger, Hrsg. ASM Press, Washington, D. C. S. 458.
- Polisky, B. 1986. Replication control of the Co1E1-type plasmids.
In Maximizing gene expression. W. S. Reznikoff und L. Gold, Hrsg.
Butterworths, Boston, S. 143.
- Porter, R. D., S. Black, S. Pannuri, and A. Carlson. 1990. Use
of the Escherichia coli ssb gene to prevent bioreactor takeover
by plasmidless cells. Bio/Technology 8:47.
- Pouwels, P. H., R. J. Leer, M. Shaw, M. J. Heijne den Bak-Glashouwer,
F. D. Tielen, E., Smit, B. Martinez, J. Jore und P. L. Conway. 1998.
Lactic acid bacteria as antigen delivery vehicles for oral immunization
purposes. Int. J. Food Microbiol. 41:155-167.
- Pratt, L. A., W. Hsing, K. E. Gibson und T. J.Silhavy. 1996.
From acids to osmZ: multiple factors influence synthesis of the
OmpF and OmpC porins in Escherichia coli. Molecular Microbiology
20:911.
- Puente, J. L., V. Alvarez-Scherer, G. Gosset und E. Calva. 1989.
Comparative analysis of the Salmonella typhi and Escherichia coli
ompC genes. Gene 83:197.
- Richardson, S. E., T. A. Rotman, V. Jay, C. R. Smith, L. E.
Becker, M. Petric, N. F. Olivieri und M. A.Karmali. 1992. Experimental
verocytotoxemia in rabbits. Infection and Immunity 60:4154.
- Ringquist, S., S.Shinedling, D. Barrick, L. Green, J.Binkley,
G. D. Stormo und L. Gold. 1992. Translation initiation in Escherichia
coli: sequences within the ribosome-binding site. Molecular Microbiology
6:1219.
- Roberts, M., S.Chatfield und G. Dougan. 1994. Salmonella as
carriers of heterologous antigens. In Novel delivery systems for
oral vaccines. D. T. O'Hagan,
Hrsg. CRC Press, Arm Arbor, S. 27-58.
- Ruiz-Echevarria, M. J., G.Gimenez-Gallego, R.Sabariegos-Jareno
und R. Diaz-Orejas.
1995. Kid, a small protein of the parD stability system of plasmid
R1, is an inhibitor of DNA replication acting at the initiation
of DNA synthesis. J. Mol. Biol. 247:568-577.
- Rupp, W. D. 1996. DNA repair mechanisms. In Escherichia coli
and Salmonella: Cellular and molecularbiology. 2. Ausg., F. C. Neidhardt,
R. Curtiss III, J. L. Ingraham, E. C. C. Lin, K. B. Low, B. Magasanik,
W. S. Reznikoff, M. Riley, M. Schaechter und H. E. Umbarger, Hrsg.
ASM Press, Washington, D. C. S. 2277.
- Ryan, E. T., J. R. Butterton, R. N. Smith, P. A. Carroll, T.
I. Crean und S. B. Calderwood. 1997a. Protective immunity against
Clostridium difficile toxin A induced by oral immunization with
a live, attenuated Vibrio cholera vector strain. Infect. Immun.
65: 2941-2949.
- Ryan, E. T., J. R. Butterton, T. Zhang, M. A. Baker, S. L. J.
Stanley und S. B. Calderwood. 1997b. Oral immunization with attenuated
vaccine strains of Vibrio cholerae expressing a dodecapeptide repeat
of the serine-rich Entamoeba histolytica protein fused to the cholera
toxin B subunit induces systemic and mucosal antiamebic and anti-V
cholerae antibody responses in mice. Infect. Immun. 65:3118-3125.
- Sambrook, J., E. F. Fritsch und T. Maniatis. 1989. Molecular
cloning. A Laboratory Manual, 2. Ausgabe. Cold Spring Harbor, New
York: Cold Spring Harbor Laboratory Press.
- Selzer, G., T. Som, T.Itoh und J. Tomizawa. 1983. The origin
of replication of plasmid p15A and comparative studies on the nucleotide
sequences around the origin of related plasmids. Cell 32:119.
- Shaw, K. J., P. N. Rather, R. S. Hare und G. H. Miller. 1993.
Molecular genetics of amino-glycoside resistance genes and familial
relationships of the aminoglycoside-modifying enzymes. Microbiol.
Rev. 57:138-163.
- Siegler, R. L. 1995. The hemolytic uremic syndrome. Pediatric
Nephrology 42:1505. Siegler, R.L., A. T. Pavia, R. D. Christofferson
und M. K. Milligan. 1994. A 20-year
population-based study of postdiarrheal hemolytic uremic syndrome
in Utah. Pediatrics 94:35.
- Sixma, T. K., P. E. Stein, W. G. Hol und R. J. Read. 1993. Comparison
of the B-pentamers
of heat-labile enterotoxin and verotoxin-1: two structures with
remarkable similarity and dissimilarity. Biochemistry 32:191.
- Srinivasan, J., S. A. Tinge, R. Wright, J. C. Herr und R. Curtiss
III. 1995. Oral immunization with attenuated Salmonella expressing
human sperm antigen induces antibodies in serum and the reproductive
tract. Biology of Reproduction 53:462.
- Stein, P. E., A. Boodhoo, G. J. Tyrrell, J. L. Brunton und R.
J. Read. 1992. Crystal structure of the cell-binding B oligomer
of verotoxin-1 from E. coli. Nature 355:748.
- Stoker, N. G., N. F. Fairweather und B. G. Spratt. 1982. Versatile
low-copynumber plasmid vectors for cloning in Escherichia coli.
Gene 18:335-341.
- Streatfield, S. J., M. Sandkvist, T. K. Sixma, M. Bagdasarian,
W. G. Hol und T. R. Hirst. 1992. Intermolecular interactions between
the A and B subunits of heat-labile enterotoxin from Escherichia
coli promote holotoxin assembly and stability in vivo. Proceedings
of the National Academy of Sciences USA 89:12140.
- Strockbine, N. A., L. R. M. Marques, J. W. Newland, H. W. Smith,
R. K. Holmes und A. D. O'Brien.
1986. Two toxin-converting phages from Escherichia coli O157:H7
strain 933 encode antigenically distinct toxins with similar biologic
activities. Infection and Immunity 53:135.
- Strockbine, N. A., M. P. Jackson, L. M. Sung, R. K. Holmes und
A. D. O'Brien. 1988.
Cloning and sequencing of the genes for Shiga toxin from Shigella
dysenteriae Type 1. Journal of Bacteriology 170:1116.
- Strugnell, R. A., D. Maskell, N. F. Fairweather, D. Pickard,
A. Cockayne, C. Penn und G. Dougan. 1990. Stable expression of foreign
antigens from the chromosome of Salmonella typhimurium vaccine strains.
Gene 88:57-63.
- Summers, D. K. The Biology of Plasmids, 65-91,1996.
- Summers, D. K. 1998. Timing, self-control and sense of direction
are the secrets of multicopy plasmid stability. Mol. Microbiol.
29:1137-1145.
- Summers, D. K. und D. J. Sherratt. 1984. Multimerization of
high copy number plasmids causes instability: ColE1 encodes a determinant
essential for plasmid monomerization and stability. Cell 36:1097.
- Tacket, C. O., D. M. Hone, R. Curtiss III, S. M. Kelly, G. Losonsky,
L. Guers, A. M. Harris, R. Edelman und M. M. Levine. 1992. Comparison
of the safety and immunogenicity of ∆aroC∆aroD and ∆cya∆crp
Salmonella typhi strains in adult volunteers. Infection and Immunity
60:536.
- Tacket, C. O., M. Sztein, G. Losonsky, S. S. Wasserman, J. P.
Nataro, R. Edelman, D. Pickard, G. Dougan, S. Chatfield und M. M.
Levine. 1997. Safety of live oral Salmonella typhi vaccine strains
with deletions in htrA and aroC aroD and immune responses in humans.
Infection and Immunity 65:452-456.
- Tacket, C. O., S. M. Kelley, F. Schodel, G. Losonsky, J. P.
Nataro, R. Edelman, M. M. Levine und R. Curtiss III. 1997. Safety
and immunogenicity in humans of an attenuated Salmonella typhi vaccine
vector strain expressing plasmid-encoded hepatitis B antigens stabilized
by the Asd-balanced lethal vector system. Infection and Immunity
65: 3381-3385.
- Takeda, Y. 1995. Shiga and Siga-like (Vero) toxins. In Bacterial
toxins and virulence factors in disease. J. Moss, B. Iglewski, M.
Vaughan und A. Tu, Hrsg. Marcel Dekker, Inc. New York, S. 313.
- Tauxe, R. V. 1998. Public health perspective on immunoprophylactic
strategies for Escherichia coli O157:H7: who or what would we immunize?
In Escherichia coli O157::H7 and other Shiga toxin producing E coli
strains. J. B. Kaper und A. D. O'Brien,
Hrsg. ASM Press, Washington, D. C. In Druck.
- Tesh, V. L., J. A. Burris, J. W. Owens, V. M. Gordon, E. A.
Wadolkowski, A. D. O'Brien
und J. E. Samuel. 1993. Comparison of the relative toxicities of
Shiga-like toxins type I and type II for mice. Infection and Immunity
61 :3392.
- Thisted, T., A. K. Nielsen und K. Gerdes. 1994. Mechanism of
post-segregational killing:translation of Hok, SmB and Pnd mRNAs
of plasmids R1, F and R483 is activated by 3'-end processing. EMBO Journal 13:1950.
- Thisted, T., N. S. Sorensen und K. Gerdes. 1995. Mechanism of
post-segregational killing: secondary structure analysis of the
entire Hok mRNA from plasmid R1 suggests a fold-back structure that
prevents translation and antisense RNA binding. Journal of Molecular
Biology 247:859.
- Thisted, T., N. S. Sorensen, E. G. Wagner und K. Gerdes. 1994.
Mechanism of post-segregational killing: Sok antisense RNA interacts
with Hok mRNA via its 5'-end
singlestranded leader and competes with the 3'-end of Hok mRNA for binding to the
mok translational initiation region. EMBO Journal 13 :1960.
- Tinge, S. A. und R. Curtiss III. 1990. Conservation of Salmonella
typhimurium virulence plasmid maintenance regions among Salmonella
serovars as a basis for plasmid curing. Infection and Immunity 58:3084.
- Tinge, S. A. und R. Curtiss III. 1990. Isolation of the replication
and partitioning regions of the Salmonella typhimurium virulence
plasmid and stabilization of heterologous replicons. Journal of
Bacteriology 172:5266.
- Twigg, A. J., und D. Sherratt. 1980. Trans-complementable copy-number
mutants of plasmid ColE1. Nature 283:216-218.
- Umbarger, H. E. 1978. Amino acid biosynthesis and its regulation.
Annual Reviews in Biochemistry 47:533.
- Valdivia, R. H. und S. Falkow. 1997. Fluorescence-based isolation
of bacterial genes expressed within host cells. Science 277:2007.
- Valdivia, R. H., A. E. Hromockyj, D. Monack, L. Ramakrishnan
und S. Falkow. 1996. Applications for green fluorescent protein
(GFP) in the study of host-pathogen interactions. Gene 173 :47.
- Van Melderen, L., P. Bernard und M. Couturier. 1994. Lon-dependent
proteolysis of CcdA is the key control for activation of CcdB in
plasmid-free segregant bacteria. Mol. Microbiol. 11:1151-1157.
- Vicari, G., A. J. Olitzki und Z. Olitzki. 1960. The action of
the thermolabile toxin of Shigella dysenteriae on cells cultivated
in vitro. British Journal of Experimental Pathology 41:179.
- Wada, K., Y. Wada, F.Ishibashi, T. Gojobori und T.Ikemura. 1992.
Codon usage tabulated from the GenBank genetic sequence data. Nucleic
Acids Research 20:2111.
- Wadolkowski, E. A., L. M. Sung, J. A. Burris, J. E. Samuel und
A. D. O'Brien. 1990.
Acute renal tubular necrosis and death of mice orally infected with
Escherichia coli strains that produce Shiga-like toxin type II.
Infection and Immunity 58:3959.
- Wahle, E. und A. Kornberg. 1988. The partition locus of plasmid
pSC 101 is a specific binding site for DNA gyrase. EMBO J 7:1889-1895.
- Wang, S. und T. Hazelrigg. 1994. Implications for bcd mRNA localization
from spatial distribution of exu protein in Drosophila oogenesis.
Nature 369:400.
- Wang, Y., Z. Zhang, S. Yang und R. Wu. 1992. Cloning of par
region and the effect of par region on the stability of pUC9. Chinese
Journal of Biotechnology 8: 107.
- Williams, K. R., J. B. Murphy und J. W. Chase. 1984. Characterization
of the structural and functional defect in the Escherichia coli
single-stranded DNA binding protein encoded by the ssb-1 mutant
gene. Journal of Biological Chemistry 259:11804.
- Wu, K. und T. K. Wood. 1994. Evaluation of the hok/sok killer
locus for enhanced plasmid stability. Biotechnol. Bioeng. 44:912-921.
- Yamasaki, S., M. Furutani, K. Ito, K.Igarashi, M. Nishibuchi
und Y. Takeda. 1991. Importance of arginine at postion 170 of the
A subunit of Vero toxin 1 produced by enterohemorrhagic Escherichia
coli for toxin activity. Microbial. Pathogenesis 11:1.
- Yanofsky, C., T.Platt, I. P. Crawford, B. P. Nichols, G. E.
Christie, H. Horowitz, M. Van Cleemput und A. M. Wu. 1981. The complete
nucleotide sequence of the tryptophan operon of Escherichia coli.
Nucleic Acids Res. 9:6647-6668.
- Yu, J. und J. B. Kaper. 1992. Cloning and characterization of
the eae gene of enterohaemorrhagic Escherichia coli. Molecular Microbiology
6:411.
- Zalkin, H. und P. Nygaard. 1996. Biosynthesis of purine nucleotides.
In Escherichia coli and Salmonella:Cellular and molecular biology.
2. Ausg., F. C. Neidhardt, J. L. Ingraham, E. C. C. Lin, K. B. Low,
B. Magasanik, W. S. Reznikoff, M. Riley, M. Schaechter und H. E.
Umbarger, Hrsg. ASM Press, Washington, D. C. S. 561.
- Zhang, X., Y. Lou, M. Koopman, T. Doggett, K. S. K. Tung und
R. Curtiss III. 1997. Antibody responses and infertility in mice
following oral immunization with attenuated Salmonella typhimurium
expressing recombinant murine ZP3. Biology of Reproduction 56: 33.
- Zoja, C., D. Corna, C. Farina, G. Sacchi, C. A. Lingwood, M.
P. Doyle, V. V. Padhye, M. Abbate und G. Remuzzi. 1992. Verotoxin
glycolipid receptors determine the localization of microangiopathic
process in rabbits given verotoxin-1. Journal of Laboratory and
Clinical Medicine 120:229.
- Zurita, M., F. Bolivar und X. Soberon. 1984. Construction and
characterization of new cloning vehicles. VII. Construction of plasmid
pBR327par, a completely sequenced, stable derivative of pBR327 containing
the par locus of pSC101. Gene 28:119.
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-