-
Gebiet der
Erfindung
-
Diese
Erfindung betrifft die Verwendung von Inhibitoren der Tyrosinkinase-Familie
und insbesondere auf Inhibitoren der Brutons-Tyrosinkinase (BTK).
-
Hintergrund
der Erfindung
-
Apoptose
ist eine gängige
Art des eukaryontischen Zelltods, der durch eine induzierbare Kaskade
biochemischer Events ausgelöst
wird, die zu einer Aktivierung von Endonukleasen führen, welche
die Kern-DNA in Fragmente mit Oligonukleosom-Länge
spalten. Einige der biochemischen Events, die zum apoptotischen Zelltod
beitragen, wie auch positive und negative Regulatoren der Apoptose
wurden kürzlich
identifiziert (Whyllie, A. et al., (1980), Int. Rev. Cytol. 68,
251-305; Steller, H. (1995), Science 267, 1445-1449; Fraser, A.,
Evan, G. (1996), Cell 85, 781-784; und Korsmeyer, S.J. (1995), Trends
Genet. 11, 101-105). Apoptose spielt in der Entwicklung und der
Aufrechterhaltung eines funktionierenden Immunsystems eine zentrale
Rolle, indem sie die rechtzeitige Selbstzerstörung von autoreaktiven unreifen
und reifen Lymphozyten wie auch von auftretenden neoplastischen
Zielzellen durch cytotoxische T-Zellen sicherstellt.
-
Zusätzlich zu
den vorteilhaften Wirkungen, die mit einer Apoptose verbunden sind,
trägt eine
unpassende Apoptose zur Pathogenese und Arzneimittelresistenz humaner
Leukämie
und Lymphome bei (Cohen, J.J. et al. (1992), Annu. Rev. Immunol.
10, 267-293; Linette, G.P. und Korsmeyer, S.J. (1994), Curr. Opin.
Cell Biol. 6, 509-815 und Thompson, C.B. (1995), Science 367, 1456-1462).
Somit sind Agenzien, die zum Modulieren der Apoptose nützlich sind,
potentiell als therapeutische Agenzien zur Behandlung von Krankheiten
einsetzbar, bei denen eine unpassende Apoptose impliziert ist. Als
Resultat gibt es einen beträchtlichen
Umfang an laufenden Forschungen, die sich mit der Identifizierung
von molekularen Regulatoren der Apoptose beschäftigen, und es besteht ein
fortdauernder Bedarf an neuen Agenzien (z.B. chemische oder biologische)
und neuen therapeutischen Verfahren, die zur Modulierung der Apoptose
einsetzbar sind. Solche Agenzien und Verfahren können zur Behandlung von Krebs
(z.B. Leukämie
und Lymphom) oder Immunkrankheiten bei Säugetieren nützlich sein. Sie können auch
als pharmakologische Werkzeuge zur Verwendung in in vitro- oder
in vivo-Studien einsetzbar sein, um das Verständnis der molekularen Apoptose-Basis
zu verstärken
(z.B. das pro-apoptotische
gegenüber
dem anti-apoptischen Regulatorsignal) sowie das Verständnis für die Pathogenese
von humanen lymphoiden Malignitäten
zu verstärken.
-
WO-A-9117748
offenbart die Verwendung von Isoxazol-4-carboxamid-Derivaten und Hydroxyalkyliden-cyanoacetamid
für die
Behandlung von Krebserkrankungen und rheumatischen Erkrankungen. DE-A-2555685
beschreibt Cyansäureanilid-Derivate und die
Herstellung derselben. EP-A-537742 offenbart Styrol-Derivate, die
Inhibitoraktivität
auf Tyrosinspezifische Proteinkinase und Inhibitoraktivität auf die
Krebszellproliferation haben. Ghosh et al., Clinical Cancer Research,
November 1998, Bd. 4, 2657-68 offenbaren α-Cyano-β-hydroxy-β-methyl-N-[4-(trifluormethoxy)phenyl]propenamid
zur Inhibierung der epidermalen Wachstumsfaktorrezeptor-Tyrosinkinase mit
wirksamer cytotoxischer Aktivität
gegen Brustkrebszellen.
-
BTK
ist ein Apoptose-Regulator mit zweifacher Funktion, der eine strahlungsinduzierte
Apoptose fördert,
aber eine Fasaktivierte Apoptose in B-Zellen inhibiert (Uckum, F.M.
Commentary: Burton's
Tyrosin Kinase (BTK) as a Dual-Function Regulator of Apoptosis;
siehe auch Uckun, F.M. et al., BTK as a Mediator of Radiation-induced
Apoptosis in DT-40 Lymphoma B-Zellen, Science 273: 1096-1100 (1996)).
BTK wirkt in pro-apoptotischer
Weise, wenn B-Zellen reaktiven Sauerstoffzwischenprodukten ausgesetzt
sind, und zwar mindestens teilweise durch "Downregulierung" der anti-apoptotischen Aktivität des STAT3-Stranskriptionsfaktors.
Dagegen assoziiert BTK mit dem Todesrezeptors Fas, verschlechtert
seine Wechselwirkung mit FADD, das für die Rekrutierung und Aktivierung
von FLICE durch Fas während
des apoptotischen Signals essentiell ist, wodurch der Zusammenbau
eines pro-apoptotischen Tod-induzierenden Signalkomplexes (DISC
= death inducing signaling complex) nach Fas-Ligation verhindert
wird.
-
Zusammenfassung
der Erfindung
-
Die
vorliegende Erfindung stellt die Verwendung von Verbindungen der
Formel (I) bereit:
in der:
R
1 für (C
1-3)-Alkyl, (C
3-6)-Cycloalkyl,
Phenyl oder NR
aR
b steht;
R
2 für
Hydroxy, (C
1-6)-Alkoxy, (C
1-6)-Alkanoyloxy,
Amino-(C
2-5)-alkoxy, Hydroxy-(C
2-5)-alkoxy,
Amino-(C
2-5)-alkanoyloxy oder Hydroxy-(C
2-5)-alkanoyloxy
steht;
R
3 für Cyano oder (C
1-3)-Alkanoyl
steht;
R
4 für Hydrogen, (C
1-3)-Alkyl,
Hydroxy-(C
2-5)-alkyl oder Amino-(C
2-5)-alkyl steht;
R
5 für durch
-S(O)
2R
c oder Halogen
und mindestens einen anderen Substituenten substituiertes Phenyl
steht;
R
a und R
b jeweils
unabhängig
voneinander für
Hydrogen oder (C
1-3)-Alkyl stehen oder R
a und R
b zusammen mit
dem Stickstoff, mit welchem sie verbunden sind, für Pyrrolidino,
Piperidino, Morpholino oder Thiomorpholino stehen;
wobei Aryl
oder Heteroaryl für
R
1 und R
5 gegebenenfalls
mit einem oder mehreren (z.B. 1, 2 oder 3) Substituenten, die unabhängig voneinander
ausgewählt
sind unter Halogen, Nitro, Cyano, Hydroxy, Trifluormethyl, Trifluormethoxy,
(C
1-3)-Alkoxy,
(C
1-3)-Alkyl, (C
1-3)-Alkanoyl,
-S(O)
2R
c oder NR
aR
b, wobei R
c für
(C
1-3)-Alkyl oder Aryl steht, substituiert
ist, oder ein pharmazeutisch akzeptables Salz davon, wie es in Anspruch
1 beschrieben ist.
-
Die
Verbindungen der Erfindung sind so entwickelt, daß sie in
ein zusammengesetztes Bindungstaschenmodell der BTK-Domäne passen,
ein Molekülvolumen
haben, das kleiner ist als das Volumen der Bindungstasche (z.B.
weniger als etwa 600 Å3) und vorzugsweise ein Volumen haben, das
etwa 2/3 des Volumens der Tasche, z.B. etwa 400 Å3,
haben. Am vorteilhaftesten sind die Inhibitoren der Erfindung so
entwickelt, daß sie
den Raum der Bindungstasche ausfüllen
und mit den Resten der Tasche für
eine verstärkte
Bindung wechselwirken.
-
Die
Erfindung stellt die Verwendung einer Verbindung der Formel (I)
zur Herstellung eines Medikaments zur Behandlung eines pathologischen
Zustandes oder einer Erkrankung bei einem Säuger, z.B. einem Menschen,
der/die mit BTK-Aktivität
in Verbindung steht, bereit. Diese Zustände oder Erkrankungen sind
unter Störungen
der B-Zell-Lymphozytenproliferation/Autoimmunerkrankungen,
Mastzellenstörungen,
Zuständen, die
mit einer ungeeigneten Blutplättchenaggregation
einhergehen, und Abstoßung
von Xenoplantaten ausgewählt.
-
Kurze Beschreibung
der Figuren
-
1: BTK ist ein Inhibitor für die Fasvermittelte
Apoptose in DT-40-Lymphom-B-Zellen. FACS korrelierter Zwei-Parameter-Display
von Wildtyp (WT)-, BTK-defizienten
(BTK-)-, LYN-defizienten (LYN-)-DT-40-Zellen wie auch von BTK-defizienten
DT-40-Zellen, die mit humanem Wildtyp btk-Gen (BTK; rBTK[WT]) rekonstituiert
waren, gefärbt
mit MC540 und PI 24 Stunden nach Behandlung mit der Kontrolle Maus-IgG
MsIgG (μg/ml)
oder anti-Fas (1 μg/ml).
Die Prozentangaben geben die Zellfraktion in einer frühen Apoptosestufe,
wie sie durch einzelne MC540-Fluoreszenz gemessen wird, und die
Zellfraktion in einer fortgeschrittenen Stufe der Apoptose, wie
sie durch duale MC540/PI-Fluoreszenz gemessen wird, an (Uckun, F.M. et
al. (1996), Science 273, 1096-1100).
-
2A–2C:
Fas-Protein-Expressionslevel in Wildtyp- und BTK-defizienten DT-40-Zellen. 2A zeigt die Expressionslevel von BTK und ACTIN
in Wildtyp-, BTK-difizienten
und mit humanem btk-Gen-rekonstituierten, BTK-defizienten DT-40-Zellen, gemessen durch
Western-Blot-Analyse unter Verwendung geeigneter monoklonaler Antikörper und
des ECL-Chemilumineszenz-Detektionssystem (Amersham Life Sciences, verwendet
nach den Empfehlungen des Herstellers) (Dibirdik, I. et al. (1998),
J. Biol. Chem., 273, 4035-4039; Uckun, F.M. et al. (1997), Blood,
89, 3769-3777). 2B zeigt Membranen nach Immunblotting
mit Anti-BTK- und Anti-ACTIN-Antikörper, die
abgestreift worden waren und mit dem monoklonalen Anti-Fas-Antikörper erneut
geblottet wurden, um die Fas-Protein-Expressionslevel in den einzelnen
Klonen zu vergleichen. 2C zeigt
Fas-Expressionslevel von WT- und BTK-defizienten DT-40-Zellen, die
durch konfokale Mikroskopie untersucht worden waren. Grün: Anti-Fas-Markierung;
Blau: Toto-3-gefärbte
DNA im Kern; Meßbalken
= 10 mm.
-
3A–3D: BTK-inhibiert eine Fas-vermittelte
Apoptose. 3A ist eine Photographie, die
Wildtypzellen (WT) und BTK-defiziente (BTK-)DT-40-Zellen zeigt,
welche für
24 Stunden mit 1 μg/ml
Anti-Fas behandelt worden waren, mit polyklonalem Kanichen-Anti-Tubulin-Antikörper (grüne Fluoreszenz)
und dem DNA-spezifischen Farbstoff Toto-3 (blaue Fluoreszenz) co-gefärbt worden
waren und mit einem konfokalen Laserrastermikroskop untersucht worden
waren, wie es in den Beispielen beschrieben wird. Anders als die WT-Zellen
zeigt die Mehrzahl der BTK-Zellen apoptotische Veränderungen,
einschließlich
einer nukleären Fragmentierung
(a, b, d) und einer Schrumpfung (c). Balken = 10 mm. 3B ist ein DNA-Gel,
das WT- und BTK-DT-40-Zellen zeigt, welche Anti-Fas-Antikörper ausgesetzt
waren, wie es in den Beispielen detailliert beschrieben ist, geerntet
wurden, und DNA aus Triton-X-100-Lysaten wurde auf Fragmentierung
untersucht, wie es beschrieben wurde (Uckun, F.M. et al. (1996),
Science 273, 1096-1100). Die 3C und 3D zeigen BTK-defiziente DT-40-Zellen, die mit
Wildtyp (rWT)-, Kinasedomänenmutanten
(rK-)-, SH2-Domänenmutanten
(rmSH2)- oder PH-Domänenmutanten
(rmPH)-Formen des humanen btk-Gens rekonstituiert worden waren,
und auf Empfindlichkeit gegenüber
einer Anti-Fas-Antikörper-induzierten
Apoptose untersucht wurden, wie es in den Beispielen beschrieben
ist. Kontrollen wurden mit PBS in Kulturmedium für 24 Stunden bei 37°C und 5 %
CO2 vor dem Ernten behandelt.
-
4A–4C:
Anti-apoptotische Eigenschaften von BTK, bestätigt durch BTK-Proteinrekonstitution von
BTK-DT-40-Zellen.
Maltose-Bindungsprotein (MBP) oder MBP-BTK wurde vor Behandlung
mit Anti-Fas-Antikörper
in BTK-DT-40-Zellen elektroporiert, wie es in den Beispielen beschrieben
ist. 4A ist eine Photographie, die
MBP-BTK-elektroporierte, BTK-defiziente DT-40-Zellen und nicht-elektroporierte,
BTK-defiziente DT-40-Zellen,
die mit einem Antikörper
gegen MBP markiert sind, zeigt. Der sekundäre Antikörper war ein FITCkonjugierter
Ziege-Anti-Kaninchen-Antikörper.
Die Zellen wurden unter Verwendung eines Bio-Rad MRC-1024 konfokalen
Laser-Scanning-Mikroskops analysiert; digitale Bilder wurden unter
Verwendung von Adobe Photoshop-Software bearbeitet und unter Verwendung
eines Fuji Pictography-Druckers gedruckt. Es gab keine signifikante
Färbung
gegenüber
dem Hintergrund in nicht-elektroporierten Kontrollzellen (4A.1). Pfeilspitzen zeigen MBP-Antikörper-reaktives
Material im Cytoplasma von Zellen an, die mit dem MBP-BTK-Fusionsprotein
elektroporiert sind (4A.2).
In den MBP-BTK-elektroporierten
Zellen wurden zwei Populationen beobachtet. Einige Zellen hatten
eine sehr helle Markierung an der Peripherie der Zelle, während andere
Zellen eine große
punktierte Fleckenbildung im Inneren des Cytoplasmas hatten. Grün = MBP,
Balken = 10 mm. 4B zeigt einen western-Blot. Lysate wie
auch Überstände von
BTK-defizienten DT-40-Zellen,
die entweder mit MBP oder MBP-BTK elektroporiert waren, wurden einer
Western-Blot-Analyse unter Verwendung von Anti-BTK- und Anti-MBP-Antikörpern unterworfen,
wie es in "Experimentelle
Verfahren" beschrieben
ist. Das 115 kDa-MBP-BTK-Fusionsprotein,
das mit beiden Antikörpern
reaktiv ist, wurde nur in Lysaten (nicht aber in Überständen) von
MBP-BTK-elektroporierten
Zellen detektiert. 4C ist ein Gel. Zellen wurden
24 Stunden nach Behandlung mit Anti-Fas- Antikörper geernetet und DNA aus
Triton-X-100-Lysaten wurde auf Fragmentierung analysiert, wie es
in Uckun, F.M. et al. (1996), Science 273, 1096-1100 und Uckun, F.M.
et al. (1995), Science 267, 886-91 beschrieben ist. Eine Anti-Fas-Behandlung
induzierte eine Apoptose in BTK-defizienten Zellen, nicht aber in
WT-Zellen oder BTK-defizienten Zellen, in die MBP-BTK elektroporiert worden
war. Eine Elektroporation von MBP (negative Kontrolle) hatte keinen
Effekt auf die Apoptose.
-
5A–5D:
BTK-assoziiert mit FAS und beeinflußt FAS-FADD-Wechselwirkungen.
Die Fas-, FLICE-, FADD- und TRADD-Immunkomplexe, die aus Nonidet
P-40-Lysaten von unbehandelten Wildtyp-DT-40-Lymphom-B-Zellen immunpräzipitiert
worden waren, wurden gesammelt, gewaschen, in 2× SDS-Probenpuffer gekocht,
an 12,5 % Polyacrylamidgelen fraktioniert, auf eine Immobilon-PVDF-Membran transferiert
und auf das Vorliegen von BTK-Protein durch Immunblotting untersucht,
wie es in den Beispielen beschrieben ist und in 5A dargestellt ist. Die BTK- und FADD-Immunkomplexe
(wie auch die positiven Kontroll-FAS-Immunkomplexe) immunopräzipitierten
aus unbehandelten, gegen Fas-aktivierten,
BTK-defizienten DT-40-Lymphom-B-Zellen, die mit humanem Wildtyp-btk-Gen
rekonstituiert worden waren, und wurden gesammelt, gewaschen, in
2× SDS-Probenpuffer
gekocht, an 12,5 % Polyacrylamidgel fraktioniert, auf eine Immobilon-PVDF-Membran übertragen
und einem Immunblotting mit einem monoklonalen Anti-Fas-Antikörper unterworfen,
wie es unten beschrieben wird und in 5B dargestellt
ist. FAS-, FLICE-, FADD-, TRADD- und BTK-Immunkomplexe aus Lysaten
BTK-positiver, humaner
NALM-6-UM1-B-Zellen-Vorläuferleukämiezellen
wurden einer Anti-Fas-Westernblot-Analyse unterworfen, wie es in 5C gezeigt ist. FADD wurde aus Nonidet-P-40-Lysaten
von unbehandelten, gegen Fas-aktivierten
(Anti-Fas 1 μg/ml × 1 Stunde),
BTK-defizienten DT-40-Zellen immunpräzipitiert, wie es in den Beispielen
beschrieben ist. Die Immunkomplexe wurden gesammelt, gewaschen,
in 2× SDS-Probenpuffer
gekocht, an 12,5 % Polyacrylamidgelen fraktioniert, auf eine Immobilon-PVDF-Membran transferiert
und mit einem molekularen Anti-Fas-Antikörper einem Immunblotting unterzogen
(5D). In ähnlicher
Weise wurden die FAS-Immunkomplexe aus denselben Zellen durch Immunblotting
auf die Anwesenheit von FADD untersucht.
-
6A–6D:
Apoptotische Reaktionen von humanen Leukämiezellen der B-Linie auf Fas-Ligation in
Beziehung zur BTK-Expression und Funktion. Eine duale Antikörper-Western-Blot-Analyse von
Lysaten ganzer Zellen aus BTK-positiven NALM-6-UM1 (N6-U1)- und
BTK-defizienten RAMOS-1-Zellen gegen BTK und gegen Aktin (5A). Anti-Fas-Western-Blot-Analyse derselben Zelllysate.
[B]-Zellen wurden 24 Stunden nach der Einwirkung von Anti-Fas-Antikörper in
Konzentrationen von 0,1 μg/ml
oder 1,0 μg/ml
geerntet und DNA aus Triton-X-100-Lysaten wurde auf Fragmentierung analysiert,
wie es von Uckun, F.M. et al. (1996), Science 273, 1096-1100 und
Uckun, F.M. et al. (1995), Science 267, 886-91 beschrieben ist.
Eine Anti-Fas-Behandlung induzierte eine Apoptose in BTK-defizienten RAMOS-1-Zellen,
nicht aber in BTK-positiven N6-U1-Zellen. [C.1] und [C.2] Anti-BTK-
und Anti-Fas-Western-Blot-Analyse
von Lysaten ganzer Zellen aus N6-U1-Zellen, die mit dem BTK-Inhibitor
LMA-13 behandelt worden waren (10 μM × 4 Stunden). [C.3] Anti-Fas- und
Anti-BTK-Western-Blot-Analyse von mit Anti-BTK-Antikörper immunpräzipitierten
BTK-Immunkomplexen aus
unbehandelten (= –Inhibitor)
und LMA-13-behandelten
(10 μM × 4 Stunden)
(= +Inhibitor) N6-U1-Zellen. [D] N6-U1-Zellen wurden 24 Stunden
nach Behandlung mit Anti-Fas-Antikörper in
Konzentrationen von 0,1 μg/l oder
1,0 μg/l
geerntet; und DNA aus Triton-X-100-Lysaten wurde auf Fragmentierung
analysiert, wie es von Uckun, F.M. et al. (1996), Science 273, 1096-1100
und Uckun, F.M. et al. (1995), Science 267, 886-91 beschrieben wird.
Eine Anti-Fas-Behandlung
induzierte eine Apoptose in LMA-13-vorbehandelten (10 μM × 4 h) (=
Inhibitor+)N6-U1-Zellen. Dagegen wurde keine Apoptose in N6-U1-Zellen
beobachtet, die nicht mit diesem BTK-Inhibitor vorbehandelt waren
(= Inhibitor-). Ein ECL-Detektionssystem
(Amersham Pharmacia Biotech, Arlington Heights, Illinois, Kat.-Nr.
RPN2106) wurde für
die Western-Blot-Analyse
in [A.1], [A.2], [C.1], [C.2] und [C.3] verwendet.
-
7A–7B:
Kinase-inaktive BTK assoziiert nicht mit Fas. Anti-BTK (A.1)- und
Anti-Fas (A.2)-Western-Blot-Analyse von Ganzzelllysaten aus BTK-defizienten
DT-40-Zellen (7A), rekonstituiert mit humaner
Wildtyp-BTK (BTK-, rBTK[WT]) (Bahn 1) oder Kinasedomäne-mutanter
inaktiver humaner BTK (BTK-, rBTK[K-] (Bahn 2). Die Expressionslevel
von BTK und Fas wurden durch Immunblotting unter Verwendung geeigneter
Antikörper
und des ECL-Chemilumineszenz-Detektionssystems
gemessen. 7B zeigt eine Anti-BTK (B.1)-
und Anti-Fas (B.2)-Western-Blot-Analyse von Fas-Immunkomplexen aus BTK-, rBTK[WT]-Zellen
mit (Bahn 1) oder ohne (Bahn 2) Anti-Fas-Antikörper-Vorbehandlung und aus
BTK-, rBTK[K-]-Zellen mit (Bahn 3) oder ohne (Bahn 4) Anti-Fas-Antikörpervorbehandlung.
Die Immunkomplexe, die aus Nonidet P-40-Ganzzelllysaten immunpräzipitiert
worden waren, wurden gesammelt, gewaschen, in 2 × SDS-Probenpuffer gekocht,
an 12,5 % Polyacrylamidgelen fraktioniert, auf eine Immobilon-PVDF-Membran transferiert
und auf das Vorliegen von BTK- und Fas-Proteinen durch Immunblotting
untersucht, wie es im Abschnitt "Verfahren" beschrieben ist.
-
8A–8F:
Bindung von BTK-Fusionsproteinen an Fas-Protein in Hühner- und humanen Lymphzellen
der B-Linie. 8A zeigt schematische Diagramme
von Vollängen-
und gekürzten
MBP- und GST-Fusionsproteinen, die verschiedenen BTK-Domänen entsprechen.
Die einschließlich
Aminosäure
(AA)-Sequenz ist
für jede
Trunkationsmutante angegeben. BTK 1-659, BTK 408-659, BTK 2-137
wie auch BTK 519-567, die innerhalb der katalytischen Domäne die Y551-Transphorylierungsstelle
enthalten (als Kontrolle verwendet), wurden an MBP fusioniert. BTK
219-377, BTK 281-377 und BTK 219-268 wurden an GST fusioniert. 8B zeigt MBP-BTK- und GST-BTK-Fusionsproteine (7,5 μg/Bahn),
die durch SDS-PAGE unter Verwendung von 12 % Polyacrylamidgelen
analysiert wurden und durch Färben
der Gele mit Coomassie R-250-Blau sichtbar gemacht wurden. 8C ist eine Western-Blot-Analyse von gereinigter
BTK aus Proteinen, die den Kinase (BTK 408-659)-, PH-(BTK 2-137)
und SH2-SH3-Domänen
(BTK 219-377) von BTK entsprechen, wobei Domänen-spezifische Antikörper verwendet
werden, wie es im Abschnitt "Experimentelle
Verfahren" beschrieben wird. 8D–8F zeigen
funktionelle Rollen für
die Kinase- und PH-Domänen
von BTK in BTK-Fas-Wechselwirkungen.
[8D] BTK-defiziente DT-40-Hühner-Lymphom-B-Zellen; [8E] humane NALM-6-pre-B-Leukämiezellen;
[8F]: KL2-humane-EBV-transformierte-Lymphoblastoidzellen.
MBP-BTK- und GST-BTK-Fusionsproteine wurden in "Pull-Down"-Bindungsassay verwendet, um ihre Fähigkeit,
mit Fas in BTK-defizienten DT-40-Zellen wechselzuwirken, zu untersuchen,
wie es in den Beispielen beschrieben wird. Fusionsprotein-Adsorbate
und Kontrollproben C1-C5 (C1(=CON): Zelllysat + Amyloseperlen (kein
zugesetztes Fusionsprotein); C2: Zelllysat + Glutathion-Agarose-Perlen
(kein zugesetztes Fusionsprotein); C3: MBP-BTK 1-659 + Amyloseperlen
(kein Zelllysat); C4: GST-BTK 219-268 + Glutathion-Agaroseperlen
(kein zugesetztes Zelllysat); C5: MBP-BTK 519-567 + Amyloseperlen
+ Zelllysat) wurden durch SDS-PAGE aufgetrennt, mit dem monoklonalen
Anti-Fas-Antikörper einem
Immunblotting unterworfen und mit ECL entwickelt.
-
9A–9B:
Die anti-apoptotische Funktion von BTK. Wildtyp- und BTK-defiziente
(BTK-) DT-40-Lymphom-B-Zellen (9A)
wie auch BTK-DT-40-Zellen, die mit Wildtyp- oder mutanter humaner
BTK rekonstituiert waren (9B),
wurden mit C2-CER, Vincristin (VCR) oder Anti-Fas-Antikörper behandelt,
wie es in den Beispielen beschrieben wird. BTK-defiziente DT-40 (BTK-)-Zellen, die
Wildtyp-BTK, BTK(Arg525®Gln), BTK(Arg28®Cys)
und BTK(Arg307®Ala)
exprimieren, wurden als BTK-, rBTK(WT), BTK-, rBTK(K-), BTK-, rBTK(mPH)
und BTK-, rBTK(mSH2) bezeichnet. Vehikel (0,1 % DMSO in PBS) behandelte
wie auch Arzneimittel-behandelte Zellen wurden für 24 Stunden bei 37°C und 5 %
CO2 in Kulturmedium gehalten, bevor sie geerntet
wurden. DNA aus Triton-X-100-Lysaten wurde wie oben beschrieben
auf Fragmentierung analysiert. Uckun, F.M. et al (1996), Science
22, 1096-1100.
-
10A–10B: Homologiemodell der BTK-Kinase-Domäne. 10A ist eine Banddarstellung des Homologiemodells
der BTK-Kinasedomäne.
Das LFM-A13-Molekül
ist als raumfüllendes
Modell in der katalytischen Stelle von BTK dargestellt. Hergestellt
unter Verwendung von Molscript and Raster 3D-Programmen (Bacon, D.J. und Anderson,
W. F. (1988), J. Molec. Graphics 6, 219-20; Kraulis, P. (1991),
J. Appl. Cryst. 24, 946-50 und Merritt, E.A. und Murphy, M.E.P.
(1994), Acta Cryst. D50, 869-73). 10B ist
eine raumfüllende
Darstellung der Hauptkette der Reste der katalytischen Stelle der
BTK-Kinasedomäne.
Die C-alpha-Kette von BTK ist als blaues Band dargestellt. In Gelb,
Grün, Pink
und Blau sind die Reste an den vier Ecken der rechteckig geformten
Bindungstasche dargestellt. Ein Kugel- und Stabmodell des BTK-Inhibitor
LFM-A13 ist mehrfarbig gezeigt und stellt die günstige Orientierung dieses
Moleküls
in der aktiven Kinasestelle von BTK dar. Hergestellt unter Verwendung
des Insight II-Programms ((1996), Molecular Simulations Inc., San
Diego, CA).
-
11: Bindungsposition des LFM-A13-Moleküls (mehrfarbig)
an der katalytischen Stelle (blaues Band) der Kinasedomäne von BTK.
Die gestrichelten Linien stellen Wasserstoffbindungen zwischen LFM-A13 und
den Kinasedomänenresten
von BTK dar.
-
12: Übereinandergelegte
Docking-Positionen von LFM (purpurfarben), LFM-A12 (rot) und LFM-A13
(mehrfarbig) in der katalytischen Stelle (blaues Band) der Kinasedomäne von BTK.
Anbindungspositionen von LFM und LFM-A12 sind nur zu Vergleichszwecken gezeigt.
-
13: ORTEP-Bild der Kristallstruktur des BTK-Inhibitors LFM-A13.
-
14A–14C: Wirkungen von LFM-A13 auf die Tyrosinkinase-Aktivität von BTK:
Ein hochgereinigtes (> 90
%) Präparat
von BTK, das in einem Baculovirus-Vektor-Expressionssystem produziert worden war,
wurde für
eine Stunde bei Raumtemperatur mit LFM-A13 bei den angegebenen Konzentrationen
behandelt. Die in 14A angegebene enzymatische
Aktivität
von BTK wurde bestimmt, indem die Autophosphorylierung in einem
10-Minuten-Kinaseassay gemessen wurde, wie es in den Beispielen
beschrieben ist. BTK wurde aus B18.2-Zellen (d.h. BTK-DT-40-Zellen,
die mit humaner Wildtyp-BTK-rekonstituiert worden waren) immunpräzipitiert,
mit LFM-A13 oder Vehikel (0,1 % DMSO in PBS) für 1 Stunde behandelt und dann
auf PTK-Aktivität
analysiert, durch Autophosphorylierung wie auch Phosphorylierung
von GST-Iga, das als exogenes Kinasesubstrat verwendet wurde, gemessen.
Die Kinasedaten sind in 14B angegeben.
B18.2-Lymphom-B-Zellen
wurden mit LFM-A13 behandelt, dann lysiert und es wurden BTK-Immunkomplex-Kinase-Assays
und Western-Blots durchgeführt,
wie es in den Beispielen beschrieben ist. Die Daten sind in 14C dargestellt. PIU: Phosphoimager-Einheiten; DSU: densitometrische
Scanningeinheiten; CON: Kontrolle.
-
15: Wirkungen von LFM-A13 auf die Tyrosinkinase-Aktivität von JAK1,
JAK3, HCK und IRK. JAK1 und JAK3, die aus Sf21-Insekteneierstockzellen,
die mit den geeigneten Baculovirus-Expressionsvektoren transfiziert
waren, immunpräzipitiert
waren, HCK, die aus COS-7-Zellen, die mit dem pSV7c-HCK-Plasmid transfiziert
waren, immunpräzipitiert
worden war und IRK, die aus HepG2-Hepatomzellen immunpräzipitiert worden
waren, wurden mit LFM-A13
behandelt, dann in vitro-Kinaseassays unterworfen, wie es in "Experimentelle Verfahren" beschrieben ist.
-
16: Strukturelle Basis für die Selektivität von LFM-A13
für BTK.
In hellem Blau ist eine Spur des BTK-Homologiemodells mit ausgewählten Resten
an den Positionen A, B und C zusammen mit der Verknüpfungsposition
des Leflunomid-Metabolitenanalogon
LFM-A13 (mehrfarbig) dargestellt. Rot ist die Verbindungsposition
von LFM-A13 mit einem EGFR-Modell und die Restedifferenz zwischen
EGFR und BTK in Position B dargestellt. In Gelb ist die Verknüpfungsposition
von LFM-A13 mit der Kristallstruktur von HCK und die Restedifferenz
zwischen HCK und BTK in Position C dargestellt. In Pink ist die
Verknüpfungsposition
von LFM-A13 mit JAK3/JAK1-Modellen und die Restedifferenz zwischen
JAK3/JAK1 und BTK in Position A dargestellt. In Dunkelblau ist die
Verknüpfungsposition
von LFM-A13 mit der Kristallstruktur von IRK und den Restdifferenzen zwischen
IRK und BTK in den Positionen A und B dargestellt.
-
17: Wirkungen von LFM-A13 auf die Ceramid-Empfindlichkeit von
humanen Leukämiezellen.
Drei FACS-korrelierte
Parameter (FSC, Vorwärtsstreuung
= Größe; Fluoreszenz
aus Pl, Propidiumiodid-Fluoreszenz aus MC540-Färbung)-Anzeige
von ALL-1 Ph/t (9; 22)+ humanen ALL-Zellen,
gefärbt
mit MC540 und PI 24 Stunden nach Behandlung mit Vehikel (0,1 % DMSO
in PBS), C2-Ceramid (C2-CER) (10 μM),
LFM-A13 (200 μM)
oder LFM-A13 + C2-CER. Die Prozentangaben bezeichnen die Zellfraktion
in einem frühen
Stadium der Apoptose, gemessen durch Einzel-MC540-Fluoreszenz, und
die Zellfraktion bei fortgeschrittenen Apoptosestadium, gemessen
durch duale MC540/PI-Fluoreszenz.
-
18A–18C: Chemosensibilisierende Wirkungen von LFM-A143.
BTK-defiziente DT-40-Zellen, rekonstituiert mit humanem Wildtyp-BTK-Gen
(d.h. B18.2-Klon) (18A), humane NALM-6-pre-B-ALL-Zellen
(18B) und humane ALL-1-Ph+-ALL-Zellen
(18C) wurden mit LFM-A13 (100 μM), Vincristin (VCR) (10 ng/ml),
C2-Ceramid (C2-CER) (10 μM),
LFM-A13 (100 μM)
+ VCR (10 ng/ml), LFM-A13 (100 μM)
+ C2-CER (10 μM)
für 24
Stunden bei 37°C
behandelt. DNA aus Triton-X-100-Lysaten
wurde so, wie es von Uckun, F.M. et al., (1996) Science 22, 1096-1100,
beschrieben wurde, auf Fragmentierung analysiert.
-
19: Veranschaulicht die Synthese von LFM-13 und
anderen verwandten Verbindungen. LFM, LFM-A1 bis LFM-A12 und LFM-A14
sind lediglich zu Vergleichszwecken enthalten.
-
20A–20C: Erläutern
die Bindungswechselwirkungen von LFM-13 mit der BTK-Bindungstasche.
-
21: Bindungsmerkmale von 5,7-Dihydroxy-8-propanoyl-4-propyl-2H-1-benzopyran-2-on
und LFM-A13. Basierend auf Docking-Studien passen diese Verbindungen
in die ATP-Bindungsstelle von BTK. Beide Verbindungen enthalten
Wasserstoffbindungsgruppen, die mit Arg 525 und Asp 539 wechselwirken.
Bindungsmerkmale von 5,7-Dihydroxy-8-propanoyl-4-propyl-2H-1-benzopyran-2-on
sind lediglich zu Vergleichszwecken dargestellt.
-
Detaillierte
Beschreibung der Erfindung
-
Der
Fas/APO-1 (CD95)-Zelloberflächenrezeptor,
eine Mitglied der Tumornekrosefaktor (TNF)-Rezeptorfamilie, ist
einer der Hauptregulatoren der Apoptose in einer Vielzahl von Zelltypen.
Funktionelle Fas-Abnormalitäten
waren mit pathologischen Zuständen
der Immunsystemhomöostase
verbunden, einschlließlich
lymphoproliferativer Störungen,
Immundefekten und Autoimmunität
(Rieux-Laucat et al. (1995), Science 268, 1347-1349; und Fischer,
G.H. (1995), Cell 81, 935-946). Eine Ligation des Zelloberflächen-Fas-Moleküls induziert
rasch und dramatisch in vielen, aber nicht in allen Fas-positiven
Zelltypen eine Apoptose (Nagata, S. (1997), Cell 88, 355-365). DT-40 ist eine
Hühner-Lymphom-B-Zellinie,
die verwendet wurde, um den molekularen Mechanismus von durch Strahlung
induzierter Apoptose zu klären
(Uckun, F.M. et al. (1996), Science 273, 1096-1100). Trotz ihrer
reichlichen Oberflächenexpression
von Fas sind DT-40-Zellen ähnlich
wie humane B-Zellen-Vorläufer-Leukämiezellen
gegenüber
den cytotoxischen Wirkungen einer Fas-Ligation resistent, was die
Existenz von wirksamen negativen Regulatoren der Fas-vermittelten Apoptose
anzeigt. Bruton-Tyrosin-Kinase (BTK), ein Mitglied der BTK/Tec-Familie
von Protein-Tyrosin-Kinasen (PTKs), ist eine cytoplasmische PTK,
die bei Signaltransduktionswegen involviert ist, welche Wachstum
und Differenzierung von Lymphzellen der B-Zellinie regulieren (Rawlings,
D.J. und Witte, O.N. (1994), Immunol. Rev. 138, 105-119; Kurosaki, T.
(1997), Curr. Opin. Immunol. 9, 309-318; und Uckun, F.M. (1998), Biochemical
Pharmacology et al., 56, 683-691). BTK ist an Signaltransduktionswegen
beteiligt, die durch Bindung einer Vielzahl extrazellulärer Liganden
an ihre Zelloberflächenrezeptoren
initiiert werden: nach Ligation von B-Zell-Antigenrezeptoren (BCR) ist
ein BTK-Aktivierung
durch die gemeinsame Wirkung der PTKs Lys und Syk (Kurosaki, T.
(1997), Curr. Opin. Immunol. 9, 309-318) zur Induzierung einer Phospholipase
C-γ2-vermittelten
Calcium- Mobilisierung
notwendig (Kurosaki, T. (1997), Curr. Opin. Immunol. 9, 309-318).
Mutationen in humanen BTK-Gen sind der Grund für X-verknüpfte Agammaglobulinämie (XLA),
eine männliche
Immundefizienzkrankheit, die durch Fehlen von reifen, Immunglobulin-produzierenden,
peripheren B-Zellen charakterisiert ist (Tsukada, S. et al. (1993),
Cell 72, 279-290;
und Vetrie, D. et al. (1993), Nature 361, 226-233). Bei Mäusen wurden
Mutationen im BTK-Gen als Grund für murine X-verknüpfte Immundefizienz (Xid) identifiziert
(Rawlings, D.J. et al. (1993), Science 261, 358-361).
-
BTK
hat sich als Inhibitor des Fas/APO-1 Tod-induzierenden Signalkomplexes
(DISC) in B-Lymphozyten bzw. Lymphzellen erwiesen (Vassilev, A.,
et al. (1998), J. Biol. chem., 274, 1646-1656). Außerdem wurde derzeit
festgestellt, daß BTK
Ceramid- und Vincristin-induzierte Apoptose verhindert (vorliegende
Studie). Das Schicksal von Leukämie/Lymphom-Zellen kann im Gleichgewicht
zwischen den entgegengesetzten pro-apoptotischen Wirkungen von Kaspasen,
die durch DISC aktiviert werden, und einem "upstream" wirkenden anti-apoptotischen Regulationsmechanismus,
der BTK und/oder ihre Substrat involviert, liegen (Vassilev, A.
et al. (1998), J. Biol. Chem., 274, 1646-1656). Inhibitoren von
BTK verstärken
wahrscheinlich die Arzneimittelempfindlichkeit der Zellen der B-
(z.B. Leukämie/Lymphom)-Zellinie.
Auf diese Weise können
pharmakologische Agenzien mit BTK-Modulatoraktivität als chemosensibilisierende
Modulatoraktivität
als chemosensibilisierende Agenzien zur Behandlung von BTK-exprimierenden Malignitäten oder
Krankheiten, die durch Proliferation und Antikörperproduktion von BTK-exprimierenden
B-Zellen verursacht werden, und als B-Zellen rekonstituierende Mittel
bei humoralen Immundefekten mit verringerter Anzahl oder mit Fehlen
von B-Zellen eingesetzt werden. Außerdem wären BTK-modulierende Agenzien
als immunsupprimierende Agenzien zur Verhinderung einer hyperakuten
Organabstoßung
bei Transplantation, welche durch B-Zellen gesteuert wird, Autoimmunkrankheiten
und Konversion der Immunität
auf Arzneimittel (z.B. Antikörper
oder biologische Präparate) oder
Blutprodukten (z.B. Koagulationsfaktoren, z.B. Faktor VIII) bei
Patienten, die Antikörper
gegen solche Agenzien entwickeln, nützlich.
-
Identifizierung
von Inhibitoren für
BTK
-
Der
wirksame und selektive BTK-Inhibitor LFM-13 und andere BTK-Inhibitoren
wurden unter Verwendung des dreidimensionalen Homologiemodells der
in Beispiel 2 beschriebenen Kinasedomäne identifiziert. Unter Verwendung
dieses Modells und der Größe und der
Kontaktinformation, die in Beispielen 1 und 3 bereitgestellt wird,
wurden zusätzliche
BTK-Inhibitoren entwickelt und untersucht. Unter Verwendung dieses
Modells und dieser Methode können
andere Verbindungen, die günstigerweise
mit der Bindungstasche wechselwirken, identifiziert werden, ebenso
wie Verbindungen, die über
andere verwandte Kinasen selektiv an BTK binden werden. Eine feste
Bindung oder eine gute Einpassung in das Bindungstaschenmodell korreliert
mit wirksamer BTK-Inhibitoraktivität.
-
Die
Fähigkeit
eines Agenzes, die anti-apoptischen Wirkungen von BTK zu inhibieren,
kann unter Verwendung von Assays, die auf dem Fachgebiet bekannt
sind, oder unter Verwendung von Assays, die in den hierin angeführten Beispielen
offenbart sind, gemessen werden. Unter Verwendung der Modellierungsinformation
und der hierin beschriebenen Screens wie auch anderer Informationen,
die auf dem Fachgebiet bekannt sind, kann man Agenzien identifizieren,
die BTK-inhibierende
Eigenschaften besitzen.
-
Inhibitoren
von BTK können
auch solche Inhibitoren umfassen, die durch rekombinante DNA-Verfahren
produziert werden, z.B. Antisense-Moleküle und Transkriptionsinhibitoren.
Anfängliche Untersuchungen über btk-Transkription
haben bewiesen, daß eine
Expression des btk-Gens durch die kombinierte Wirkung von Transkriptionsfaktoren
der Sp1- und PU.1-Familie reguliert wird (S. Muller et al. Oncogene,
1996, 13, 1955-1964;
und A. Himmelmann et al. Blood, 1996, 87, 1036-1044). Transkriptionsregulatorelemente
wurden innerhalb des ersten und des zehnten Introns des btk-Gens
identifiziert und jüngere
Untersuchungen zeigen, daß eine
Regulation der btk-Genexpression
mehrere Transkriptionsfaktoren involviert (J. Rohrer, M.E. Conley, Blood,
1998, 91, 214-221). Neue Agenzien, die die Aktivität dieser
Transkriptionsfaktoren beeinträchtigen,
wären auch
als Modulatoren apoptotischer Signale in Behandlungsprogrammen einsetzbar.
Die Machbarkeit einer Regulierung der btk-Genexpression in humanen
hämatopoetischen
Zellen wurde bereits durch die Fähigkeit
von Retinsäure,
eine brk-Expression in myeloischen Zellen zu erhöhen, und die Fähigkeit
von Phorbolester wie auch von TGF-1, die btk-Expression in B-Zellen zu
verringern, bewiesen (C.I.E. Smith et al., J. Immunol. 1994, 152,
557-565).
-
Somit
kann eines oder mehrerer der rekombinanten DNA-Verfahren verwendet werden, um eine BTK-Expression
zu inhibieren und die hierin diskutierten therapeutischen Wirkungen
zu induzieren. Solche Verfahren umfassen zum Beispiel Antisense-Sequenzen
von Transkriptionsinhibitoren.
-
Beispielsweise
können
BTK-Antisense-Konstrukte direkt gegen eine BTK-Expression gerichtet
sein. Alternativ kann das Antisense-Konstrukt gegen BTK-Regulationssequenzen,
z.B. Antisense-Sequenzen zur Sp1- und PU.1-Familie von Transkriptionsfaktoren,
gerichtet sein. Vorzugsweise zielen die Antisense-Konstrukte auf
Tumorzellen.
-
Verbindungen
-
Verbindungen
der Formel (I) sind spezifische BTK-Inhibitoren, die bevorzugt an
die BTK-Modelltasche, die in den Beispielen beschrieben wird, binden
und die wirksame BTK-Inhibitoraktivität haben.
-
Die
Verbindungen der Formel (I) sind:
in der:
R
1 für (C
1-3)-Alkyl, (C
3-6)-Cycloalkyl,
Phenyl oder NR
aR
b steht;
R
2 für
Hydroxy, (C
1-6)-Alkoxy, (C
1-6)-Alkanoyloxy,
Amino-(C
2-5)-alkoxy, Hydroxy-(C
2-5)-alkoxy,
Amino-(C2-5)-alkanoyloxy
oder Hydroxy-(C
2-5)-alkanoyloxy steht;
R
3 für
Cyano oder (C
1-3)-Alkanoyl steht;
R
4 für
Wasserstoff, (C
1-3)-Alkyl, Hydroxy-(C
2-5)-alkyl oder Amino-(C
2-5)-alkyl
steht;
R
5 für durch -S(O)
2R
c oder Halogen und mindestens einen anderen
Substituenten substituiertes Phenyl steht;
R
a und
R
b jeweils unabhängig voneinander für Wasserstoff
(bzw. Hydrogen) oder (C
1-3)-Alkyl stehen;
oder R
a und R
b zusammen
mit dem Stickstoff, mit welchem sie verbunden sind, für Pyrrolidino,
Piperidino, Morpholino oder Thiomorpholino stehen;
wobei Aryl
oder Heteroaryl als R
1 und R
5 gegebenenfalls
substituiert ist durch einen oder mehrere (z.B. 1, 2 oder 3) Substituenten,
die unabhängig
voneinander ausgewählt
sind unter Halogen, Nitro, Cyano, Hydroxy, Trifluormethyl, Trifluormethoxy,
(C
1-3)-Alkoxy, (C
1-3)-Alkyl,
(C
1-3)-Alkanoyl,
-S(O)
2R
c oder NR
aR
b, wobei R
c für (C
1-3)-Alkyl oder Aryl steht;
oder ein
pharmazeutisch akzeptables Salz davon.
-
Besonders
nützliche
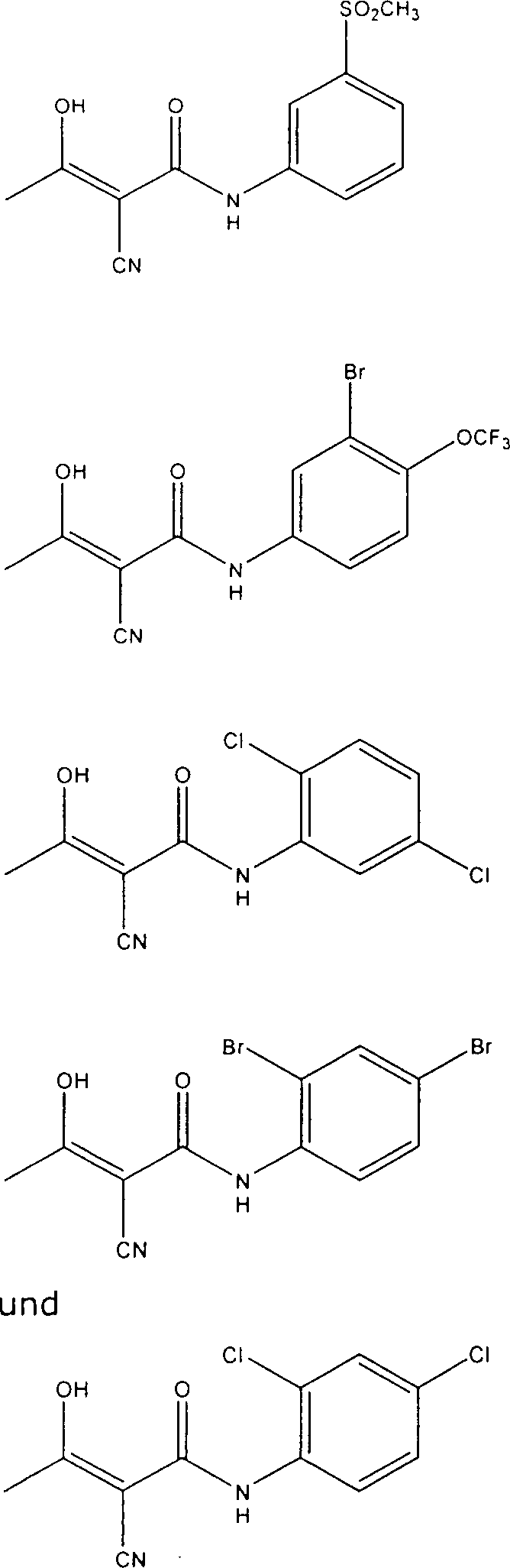
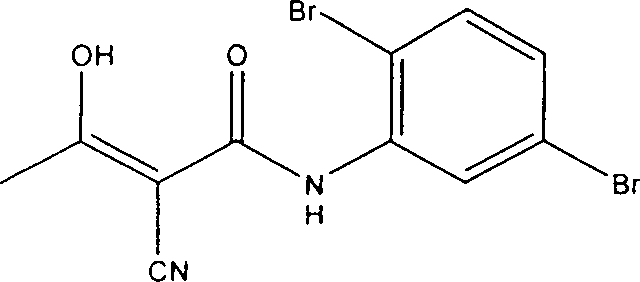
Verbindungen der Formel (I) umfassen:
α-Cyano-β-hydroxy-β-methyl-N-(2,5-dibromphenyl)propenamid;
α-Cyano-β-hydroxy-β-methyl-N-[4-(methylsulfonyl)phenyl]propenamid;
α-Cyano-β-hydroxy-β-methyl-N-[3-(methylsulfonyl)phenyl]propenamid;
α-Cyano-β-hydroxy-β-methyl-N-[3-brom-4-(trifluormethoxy)phenyl]-propenamid;
α-Cyano-β-hydroxy-β-methyl-N-(2,4-dibromphenyl)propenamid;
α-Cyano-β-hydroxy-β-methyl-N-(2,4-dichlorphenyl)propenamid;
α-Cyano-β-hydroxy-β-methyl-N-(2,5-dichlorphenyl)propenamid;
oder
α-Cyano-β-hydroxy-β-methyl-N-(3,4-dichlorphenyl)propenamid;
oder
pharmazeutisch akzeptable Salze davon.
-
Definitionen:
-
Die
folgenden Definitionen werden, soweit nichts anderes beschrieben
wird, hierin verwendet: Halogen ist Fluor, Chlor, Brom oder Iod.
Alkyl, Alkoxy, usw. bezeichnen sowohl geradkettige als auch verzweigte Gruppen;
Bezugnahme auf ein einzelnes Isomer, z.B. "Propyl", umfaßt nur das geradkettige Isomer,
ein verzweigtkettiges Isomer, z.B. "Isopropyl" wird spezifisch bezeichnet. Aryl bezeichnet
eine Phenyl-Gruppe oder eine bicyclische oder tricyclische carbocyclische Gruppe,
die etwa 9 bis 12 Ringatome hat, wobei mindestens ein Ring aromatisch
ist.
-
Dem
Fachmann auf diesem Gebiet wird klar sein, daß Verbindungen der Formel (I),
die ein chirales Zentrum haben, in optisch aktiven und racemischen
Formen existieren können
und isoliert werden können.
Einige Verbindungen können
Polymorphismus aufweisen. Es ist einzusehen, daß die vorliegende Erfindung
die Verwendung einer beliebigen racemischen, optisch aktiven, polymorphen
oder stereoisomeren Form oder Gemischen davon, einer Verbindung
der Formel (I), die die nützlichen
hierin beschriebenen Eigenschaften besitzt, umfaßt, wobei es auf dem Fachgebiet
bekannt ist, wie optisch aktive Formen hergestellt werden (zum Beispiel durch
Trennung der racemischen Form durch Umkristallisationstechniken,
durch Synthese aus optisch aktiven Ausgangsmaterialien, durch chirale
Synthese oder durch chromatographische Trennung unter Verwendung
einer chiralen stationären
Phase) und wie die BTK-inhibierende Aktivität zu bestimmen ist, wobei die
hierin beschriebenen Standardassays verwendet werden oder andere ähnliche
Assays, die auf dem Fachgebiet gut bekannt sind, verwendet werden.
-
Spezifische
und bevorzugte werte, die unten für Substituenten und Bereiche
aufgelistet sind, dienen lediglich der Erläuterung; sie schließen andere
definierte Werte oder andere Werte innerhalb definierter Bereiche
für die
Reste und Substituenten nicht aus.
-
Spezifischerweise
kann (C1-3)-Alkyl Methyl, Ethyl, Propyl
oder Isopropyl sein; (C3-6)-Cycloalkyl kann Cyclopropyl,
Cyclobutyl, Cyclopentyl oder Cyclohexyl sein; (C1-3)-Alkoxy
kann Methoxy, Ethoxy, Propoxy, Isopropropoxy sein; (C1-6)-Alkoxy kann Methoxy,
Ethoxy, Propoxy, Isopropropoxy, Butoxy, Isobutoxy, sek-Butoxy, Pentoxy,
3-Pentoxy oder Hexyloxy sein.
-
Spezifische
und bevorzugte Bedeutungen
-
Eine
spezifische Bedeutung für
R1 ist (C1-3)-Alkyl
oder (C3-6)-Cycloalkyl.
-
Eine
spezifische Bedeutung für
R2 ist Hydroxy.
-
Eine
spezifische Bedeutung für
R3 ist Cyano.
-
Eine
spezifische Bedeutung für
R4 ist Wasserstoff.
-
Eine
spezifische Bedeutung für
R5 ist Phenyl, substituiert mit Halogen
und substituiert mit 1, 2 oder 3 anderen Substituenten, die unabhängig aus
Halogen, Nitro, Cyano, Trifluormethyl, Trifluormethoxy, (C1-3)-Alkoxy, (C1-3)-Alkyl,
(C1-3)-Alkanoyl und NRaRb ausgewählt
sind.
-
Eine
spezifischere Bedeutung für
R1 ist (C1-3)-Alkyl.
-
Eine
spezifischere Bedeutung für
R5 ist Phenyl, substituiert mit Halogen
und substituiert mit 1, 2 oder 3 anderen Substituenten, die unabhängig aus
Halogen, Trifluormethyl, Trifluormethoxy und (C1-3)-Alkoxy
ausgewählt
sind.
-
Eine
spezifischere Bedeutung für
R5 ist Phenyl, das mit 2 oder 3 Halogenen
substituiert ist.
-
Eine
spezifischere Bedeutung für
R5 ist Phenyl, das mit zwei Brom substituiert
ist.
-
Eine
bevorzugt Verbindung der Formel (I) ist α-Cyano-β-hydroxy-β-methyl-N-(2,5-dibromphenyl)propenamid
oder ein pharmazeutisch akzeptables Salz davon.
-
Therapeutische
Verwendung
-
B-Zellen
und B-Zellvorläufer,
die BTK exprimieren, sind in der Pathologie einer Reihe von Krankheiten und
Zuständen
impliziert; diese umfassen B-Zell-Malignitäten (z.B. akute Lymphoblastenleukämie, chronische lymphatische
Leukämie,
Nicht-Hodgkins-Lymphom, EBV-Lymphom und Myelom), andere Krebsarten,
lymphoproliferative B-Zell- Störungen/Autoimmunkrankheiten
(z.B. Lupus, Crohn-Krankheit und chronische oder Transplantat-gegen-Empfänger-Reaktion),
Mastozytose (z.B. Allergien und anaphylaktischer Schock), Zustände, die
mit unpassenden Thrombozytenaggregation in Verbindung stehen und
Abstoßung
von Xenotransplantaten (z.B. Herztransplantate von Schwein zu Menschen).
-
Zusätzlich können die
selektiven BTK-Inhibitoren verwendet werden, um andere Krankheiten
zu identifizieren, in denen BTK eine Rolle spielt, und insbesondere
um eine Genexpression, die durch BTK moduliert ist, zu identifizieren.
Dies kann unter Verwendung von Techniken, die auf dem Fachgebiet
bekannt sind, z.B. unter Verwendung von Gen-Profilierungstechniken, die ähnlich denen
sind, die von A. Sehgal et al., Journal of Surgical Oncology, 1998,
67, 234-241, beschrieben
werden, erfolgen. Eine Inkubation von Zellen in Gegenwart oder Abwesenheit
eines BTK-Inhibitors, gefolgt von einer Profilierung der Gen-Expression
in den Zellen, ist einsetzbar, um eine BTK-regulierte Gen-Expression
zu identifizieren. Materialien, die zur Profilierung der Gen-Expression unter
Verwendung von Atlas-cDNA-Membranen einsetzbar sind, können von
CLONTECH Laboratories, Inc. 1020 East Meadow Circle, Palo Alto,
CA 94303 erhalten werden. cDNA-Mikroarrays können von handelsüblichen
Quellen bestellt werden oder selbst hergestellt werden.
-
Unter
Verwendung solcher Materialien nach den Anweisungen des Herstellers
wurde auch entdeckt, daß BTK
die Expression spezifischer Gene, z.B. MAKPAP-Kinase-Gen und c-myc-Onkogen,
moduliert. Diese Aktivität
legt nahe, daß BTK
bei der Pathologie aller Krebsformen involviert sein kann.
-
BTK
ist ein Glied der Tec-Familie von Tyrosinkinasen. Tec-Kinase wird zum Beispiel
in T-Zellen exprimiert. Die BTK-Inhibitoren
der Erfindung sind auch einsetzbar, um die Aktivität anderer
Glieder der Tec-Kinasefamilie zu inhibieren.
-
BTK-Inhibitoren
(einschließlich
Verbindungen der Formel (I), wie sie hier beschrieben sind) können verwendet
werden, um Störungen
zu behandeln, bei denen die Inhibierung oder Prävention einer Kinase der Tec-Familie,
einschließlich
BTK-Aktivität, indiziert
ist. Es wurde auch festgestellt, daß BTK-Inhibitoren als chemosensibilisierende
Agenzien einsetzbar sind und somit in Kombination mit anderen chemotherapeutischen Arzneimitteln,
insbesondere Arzneimitteln, die eine Apoptose induzieren, einsetzbar
sind. Beispiele für
andere chemotherapeutische Arzneimittel, die in Kombination mit
chemosensibilisierenden BTK-Inhibitoren verwendet werden können, umfassen
Topoisomerase I-Inhibitoren (Camptothesin oder Topotecan), Topoisomerase II-Inhibitoren
(z.B. Daunomycin und Etoposid), Alkylierungsmittel (z.B. Cyclophosphamid,
Melphalan und BCNU), Tubulin-gesteuerte Mittel (z.B. Taxol und Vinblastin)
und biologische Agenzien (z.B. Antikörper wie Anti-CD209-Antikörper, IDEC
8, Immuntoxine und Cytokine).
-
Konjugation
an eine Targeting-Gruppierung
-
Die
Verbindungen der Formel (I) können
zur spezifischen Abgabe an einen zu behandelnden Zelltyp durch Konjugation
des BTK-Inhibitors an eine Targeting-Gruppierung targetiert werden.
Targeting-Gruppierungen, die zur Konjugation mit BTK-Inhibitoren einsetzbar
sind, umfassen Antikörper,
Cytokine und Rezeptorliganden, die für die zu behandelnde Zelle
spezifisch sind.
-
Der
Ausdruck "Konjugat" meint eine Verbindung,
die als Composite zwischen zwei oder mehr Molekülen gebildet wird. Spezifischer
ausgedrückt,
in der vorliegenden Erfindung ist das Chinazolin-Derivat an zellspezifische
Targeting- Gruppierungen,
die eine Konjugatverbindung bilden, zur effizienten und spezifischen
Abgabe des Agenzes an die interessierende Zelle gebunden, z.B, kovalent
gebunden.
-
Der
Ausdruck "Targeting-Gruppierung" meint ein Molekül, das dazu
dient, die erfindungsgemäße Verbindung
für die
gewünschte
Aktivität
an eine spezifische Stelle abzugeben. Targeting-Gruppierungen umfassen
z.B. Moleküle,
die Moleküle
spezifisch eine spezifische Zelloberfläche binden. Solche Targeting-Gruppierungen,
die in der Erfindung einsetzbar sind, umfassen Anti-Zelloberflächen-Antigen-Antikörper. Cytokine,
einschließlich
Interleukine und Faktoren, wie z.B. Granulozyten/Makrophagen-stimulierender
Faktor (GMCSF) sind ebenfalls spezifische Targeting-Gruppierungen,
von denen bekannt ist, daß sie
an spezifische Zellen binden, welche hohe Level ihrer Rezeptoren
exprimieren.
-
Besonders
nützliche
Targeting-Gruppierungen zum Targeting der BTK-Inhibitorverbindungen
der Erfindung auf Zellen zur therapeutischen Aktivität umfassen
solche Liganden, die an Tec-Kinase exprimierenden Zellen vorliegen.
Zum Beispiel können
Antigene, die an B-Zellen und Krebszellen der B-Zellinie, z.B. CD19, vorliegen, mit
Anti-CD19-Antikörpern,
z.B. B43, targetiert werden. Antikörper-Fragmente, einschließlich Einzelkettenfragmente,
können
verwendet werden. Natürliche
Liganden für
die Oberflächenantigene,
z.B. CD19, können
ebenfalls eingesetzt werden. Tec-Kinase exprimierende T-Zellen können z.B.
auf das CD7-Antigen mit Anti-CD7-Antikörpern, beispielsweise TXU,
targetiert werden. Mastzellen können über das
CD48-Antigen mit Anti-CD48-Antikörpern targetiert
werden. Diese und andere Zelloberflächen Antigen-Antikörper sind
z.B. von Pharmingen im Handel erhältlich.
-
Cytokine
sind ebenfalls nützliche
Targeting-Gruppierungen. T-Zellen
können
mit IL2 und IL7 targetiert werden; B-Zellen können mit IL-4 targetiert werden;
Mastzellen können
mit C-KIT, MGH, GMCSF und IL3 targetiert werden. Krebszellen, die
Tec-Kinase exprimieren, können
z.B. mit EGF und IGF targetiert werden.
-
Verbindungen
als Salze
-
In
Fällen,
in denen ein Agens ("Verbindung") ausreichend basisch
oder sauer ist, um stabile, nicht-toxische Säure- oder Basensalze zu bilden, kann eine
Verabreichung der Verbindung als Salz geeignet sein. Beispiele pharmazeutisch
annehmbarer Salze sind Additionssalze mit organischen Säuren, die
z.B. mit Säuren gebildet
werden, welche ein physiologisch akzeptables Anion bilden, z.B.
Tosylat, Methansulfonat, Acetat, Citrat, Malonat, Tartrat, Succinat,
Benzoat, Ascorbat, α-Ketoglutarat
und α-Glycerophosphat.
Geeignete anorganische Salze können
ebenfalls gebildet werden; diese umfassen Hydrochlorid-, Sulfat-,
Nitrat-, Bicarbonat- und Carbonatsalze.
-
Pharmazeutisch
akzeptable Salze können
unter Verwendung von Standardverfahren, die auf dem Fachgebiet gutbekannt
sind, erhalten werden. Beispielsweise indem eine ausreichend basische
Verbindung, z.B. ein Amin, mit einer geeigneten Säure, die
ein physiologisch akzeptables Anion liefert, umgesetzt wird. Alkalimetall
(z.B. Natrium, Kalium oder Lithium)- oder Erdalkalimetall (z.B.
Calcium)-Salze von Carbonsäuren können ebenfalls
hergestellt werden.
-
Prodrug-Derivate
-
Die
Verbindungen der Formel (I) haben daran funktionelle Gruppen gebunden,
um ein Prodrug-Derivat bereitzustellen. Das Prodrug-Derivat erleichtert
eine Verwendung des Arzneimittels im Körper, in dem es z.B. den Eintritt
in Zellen erleichtert. Der Ausdruck "Prodrug-Gruppierung" ist eine Substitutionsgruppe, die die
Verwendung einer erfindungsgemäßen Verbindung
erleichtert, z.B. indem sie den Eintritt des Arzneimittels in Zellen
oder die Verabreichung der Verbindung erleichtert. Die Prodrug-Gruppierung
kann in vivo z.B. durch Spaltung mit Enzymen von der Verbindung
abgespalten werden. Beispiele für
Prodrug-Gruppierungen umfassen Phosphat-Gruppen, Peptidlinker und
Zucker, wobei die Gruppierungen in vivo hydrolysiert werden können.
-
Pharmazeutische
Formulierungen
-
Eine
Verbindung kann als pharmazeutische Zusammensetzungen formuliert
werden und einem Säugerwirt,
z.B. einem menschlichen Patienten, in einer Vielzahl von Formen,
die an die gewählte
Verabreichungsroute, z.B. oral oder parenteral, auf intravenösen, intramuskulären, topischen
oder subkutanen Wegen, angepaßt
sind, verabreicht werden.
-
So
können
Verbindungen systemisch verabreicht werden, z.B. oral in Kombination
mit einem pharmazeutisch akzeptablen Vehikel, beispielsweise einem
inerten Verdünnungsmittel
oder einem assimilierbaren eßbaren
Träger.
Sie können
in Gelatinekapseln mit harter oder weicher Schale eingeschlossen
sein, können
zu Tabletten verpreßt
sein oder können
direkt in das Lebensmittel der Patientennahrung eingearbeitet sein.
Zur oralen therapeutischen Verabreichung kann die Verbindung mit
einem oder mehreren Exzipientien kombiniert werden und in Form von
einnehmbaren Tabletten, bukkalen Tabletten, Lutschbombons, Kapseln,
Elixieren, Suspensionen, Sirupen, Wafern und dgl. verwendet werden.
Solche Zusammensetzungen und Präparate
sollten mindestens 0,1 % aktive Verbindung enthalten. Der Prozentgehalt
der Zusammensetzungen und Präparate
kann verändert
werden und kann zweckdienlicherweise zwischen etwa 2 und 60 % des
Gewichts einer gegebenen Einheitsdosierungsform betragen. Die Menge
der aktiven Verbindung in derartigen therapeutisch nützlichen Zusammensetzungen
ist so, daß ein
wirksamer Dosierungslevel erhalten wird.
-
Die
Tabletten, Lutschtabletten, Pillen, Kapseln und dgl. können auch
folgendes enthalten: Bindemittel, z.B. Tragacanthgummi, Akaziengummi,
Maisstärke
oder Gelatine; Exzipientien, z.B. Dicalciumphosphat; ein Zerfallsmittel,
z.B. Maisstärke,
Kartoffelstärke,
Alginsäure
und dgl.; ein Gleitmittel, z.B. Magnesiumstearat, und ein Süßungsmittel,
wie z.B. Saccharose, Fructose, Lactose oder Aspartam, oder ein Aromamittel,
z.B. Pfefferminz, Wintergrünöl oder Kirscharoma
können
zugesetzt werden. Wenn die Einheitsdosierungsform eine Kapsel ist,
kann sie zusätzlich
zu Materialien des obigen Typs einen flüssigen Träger, z.B. ein Pflanzenöl oder ein Polyethylenglykol,
enthalten. Es können
verschiedene andere Materialien als Beschichtungen oder zur anderweitigen
Modifizierung der physikalischen Form der festen Einheitsdosierungsform
vorliegen. Zum Beispiel können
Tabletten, Pillen oder Kapseln mit Gelatine, Wachs, Schellack, Zucker
oder dgl. überzogen
sein. Ein Sirup oder Elixier kann die aktive Verbindung, Saccharose
oder Fructose als Süßungsmittel,
Methyl- und Propylparabene als Konservierungsstoffe, einen Farbstoff
und ein Aromamittel, z.B. Kirsch- oder Orangenaroma, enthalten.
Jedes Material, das bei der Herstellung einer Einheitsdosierungsform
verwendet wird, sollte pharmazeutisch akzeptabel sein und in den
verwendeten Mengen im wesentlichen nicht toxisch sein. Außerdem kann
die aktive Verbindung in Präparate
und Vorrichtungen mit verzögerter
Freisetzung eingearbeitet sein.
-
Die
Verbindung kann auch intravenös
oder intraperitoneal durch Infusion oder Injektion verabreicht werden.
Lösungen
der Verbindung oder ihres Salzes können in Wasser, gegebenenfalls
mit einem nicht-toxischen oberflächenaktiven
Mittel vermischt, hergestellt werden. Dispersionen können auch
in Glycerin, flüssigen
Polyethylenglykolen, Triacetin und Gemischen davon und in Ölen hergestellt
werden. Unter normalen Lagerungs- und Verwendungsbedingungen enthalten
diese Präparate
ein Konservierungsmittel, um das Wachstum von Mikroorganismen zu
verhindern.
-
Die
pharmazeutischen Dosierungsformen, die zur Injektion oder Infusion
geeignet sind, können
sterile wäßrige Lösungen oder
Dispersionen oder sterile Pulver, die die aktive Verbindung enthalten,
und die für
die außerplanmäßige Herstellung
von sterilen, injizierbaren Lösungen
oder Dispersionen oder Infusionslösungen oder -dispersionen,
gegebenenfalls in Liposome eingekapselt, angepaßt sind, umfassen. In allen
Fällen
sollte die endgültige
Dosierungsform unter den Bedingungen der Herstellung und Lagerung
steril, flüssig
und stabil sein. Der flüssige
Träger
oder das flüssige
Vehikel können
ein Lösungsmittel
oder ein flüssiges
Dispersionsmedium sein, das z.B. Wasser, Ethanol, ein Polyol (z.B.
Glycerin, Propylenglykol, flüssige
Polyethylenglykole und dgl.), Pflanzenöle, nicht-toxische Glycerylester
und geeignete Gemische davon umfaßt. Die geeignete Fluidität kann z.B.
durch Bildung von Liposomen, durch Aufrechterhaltung der erforderlichen
Partikelgröße im Fall von
Dispersionen oder durch die Verwendung von oberflächenaktiven
Mitteln aufrechterhalten werden. Die Verhinderung der Wirkung von
Mikroorganismen kann durch verschiedene antibakterielle und antifungale
Mittel, z.B. Parabene, Chlorbutanol, Phenol, Sorbinsäure, Thimerosal
und dgl., erreicht werden. In vielen Fällen wird es vorteilhaft sein,
daß isotonische
Mittel, z.B. Zucker, Puffer oder Natriumchlorid enthalten sind.
Eine verlängerte
Absorption der injizierbaren Zusammensetzungen kann durch die Verwendung
von Mitteln, die die Absorption verzögern, z.B. Aluminiummonostearat
und Gelatine, erreicht werden.
-
Sterile
injizierbare Lösungen
können
hergestellt werden, indem die aktive Verbindung in der erforderlichen
Menge im geeigneten Lösungsmittel
mit verschiedenen der oben aufgezählten anderen Ingredienzien nach
Bedarf eingearbeitet wird, worauf eine Filtersterilisierung folgt.
Im Fall steriler Pulver zur Herstellung steriler injizierbarer Lösungen sind
die bevorzugten Herstellungsverfahren Vakuumtrocknung und die Gefriertrocknungstechniken,
die zu einem Pulver der aktiven Verbindung plus einem zusätzlichen
gewünschten
Ingrediens, das in den vorher steril filtrierten Lösungen vorlag,
führen.
-
Für eine topische
Verabreichung können
die Verbindungen in reiner Form, d.h. wenn sie Flüssigkeiten sind,
angewendet werden. Allerdings wird es im allgemeinen wünschenswert
sein, sie als Zusammensetzungen oder Formulierungen in Kombination
mit einem dermatologisch akzeptablen Träger, der ein Feststoff oder eine
Flüssigkeit
sein kann, an die Haut zu verabreichen.
-
Verwendbare
feste Träger
umfassen feinverteilte Feststoffe, z.B. Talk, Ton, mikrokristalline
Cellulose, Siliciumdioxid, Aluminiumoxid und dgl. Verwendbare flüssige Träger umfassen
Wasser, Alkohole oder Glykole oder Wasser-Alkohol/Glykol-Mischungen, in denen
die erfindungsgemäßen Verbindungen
in wirksamen Konzentrationen gelöst
oder dispergiert werden können,
gegebenenfalls mit Hilfe nicht-toxischer oberflächenaktiver Mittel. Adjuvantien,
z.B. Duftstoffe und zusätzliche
antimikrobielle Agenzien, können
zugesetzt werden, um die Eigenschaften für eine vorgegebene Verwendung
zu optimieren. Die resultierenden flüssigen Zusammensetzungen können mit
Absorbens-Pads aufgetragen werden, welche verwendet werden, um Verbände und
andere Umschläge
zu imprägnieren,
oder sie können
auf den betroffenen Bereich unter Verwendung von Sprühvorrichtungen
des Pump-Typs oder Aerosol-Sprühvorrichtungen
aufgesprüht
werden.
-
Verdickungsmittel
wie z.B. synthetische Polymere, Fettsäuren, Fettsäuresalze und -ester, Fettalkohole,
modifizierte Cellulosen oder modifizierte Mineralmaterialien können ebenfalls
mit flüssigen
Trägern
eingesetzt werden, um verteilbare Pasten, Gele, Salben, Seifen und
dgl. zur direkten Aufbringung auf die Haut des Verwenders zu bilden.
-
Beispiele
für verwendbare
dermatologische Zusammensetzungen, die verwendet werden können, um die
erfindungsgemäßen Verbindungen
an die Haut abzugeben, sind auf dem Fachgebiet bekannt; siehe z.B. Jacquet
et al. (US-Pat. Nr. 4 608 392), Geria (US-Pat. Nr. 4 992 478), Smith
et al. (US-Pat. Nr. 4 559 157) und Wortzman (US-Pat. Nr. 4 820 508).
-
Nützliche
Dosierungen der erfindungsgemäßen Verbindungen
können
bestimmt werden, indem ihre in vitro-Aktivität und in vivo-Aktivität in Tiermodellen
verglichen wird. Verfahren zur Extrapolation wirksamer Dosierungen
bei Mäusen
und anderen Tieren bis zum Menschen sind auf dem Fachgebiet bekannt;
siehe z.B. US-Patent Nr. 4 938 949.
-
Im
allgemeinen wird die Konzentration der erfindungsgemäßen Verbindung(en)
in einer flüssigen
Zusammensetzung, z.B. einer Lotion, etwa 0,1 bis 25 Gew.%, vorzugsweise
etwa 0,5 bis 10 Gew.% sein. Die Konzentration in einer halbfesten
oder festen Zusammensetzung, z.B. einem Gel oder einem Pulver, wird
etwa 0,1 bis 5 Gew.%, vorzugsweise etwa 0,5 bis 2,5 Gew.% sein.
-
Die
Menge der Verbindung oder eines aktiven Salzes oder Derivats davon,
die zur Verwendung in einer Behandlung erforderlich ist, wird nicht
nur mit dem besonderen Salz, das ausgewählt wurde, sondern auch mit
dem Verabreichungsweg, der Natur des zu behandelnden Zustands und
dem Alter und dem Zustand des Patienten variieren und wird letztlich
in der Entscheidung des behandelnden Arztes oder Klinikers liegen.
-
Im
allgemeinen wird allerdings eine geeignete Dosis im Bereich von
etwa 0,5 bis etwa 100 mg/kg, z.B. von etwa 10 bis etwa 75 mg/kg
Körpergewicht,
pro Tag liegen, z.B. 3 bis etwa 50 mg pro Kilogramm Körpergewicht
des Empfängers
pro Tag, vorzugsweise im Bereich von 6 bis 90 mg/kg pro Tag, am
vorteilhaftesten im Bereich von 15 bis 60 mg/kg/Tag sein.
-
Die
Verbindung wird zweckdienlicherweise in Einheitsdosierungsform,
die z.B. 5 bis 1000 mg, zweckdienlicherweise 10 bis 750 mg, am vorteilhaftesten
50 bis 500 mg aktive Verbindung pro Einheitsdosierungsform enthält, verabreicht.
-
Idealerweise
sollte die aktive Verbindung so verabreicht werden, daß sie Peakplasmakonzentrationen der
aktiven Verbindung von etwa 0,5 bis etwa 75 μM, vorzugsweise etwa 1 bis 50 μM, am vorteilhaftesten
etwa 2 bis etwa 30 μM
erreicht. Dies kann z.B. durch intravenöse Injektion einer 0,05- bis
5 %-Lösung
des aktiven Ingrediens, gegebenenfalls in Salzlösung, oder oral verabreicht
als Bolus, der etwa 1 bis 100 mg des aktiven Ingrediens enthält, erreicht
werden. Wünschenswerte
Blutkonzentrationen können
durch kontinuierliche Infusion unter Zuführung von etwa 0,01 bis 5,0
mg/kg/h oder durch intermittierende Infusionen, die etwa 0,4 bis
15 mg/kg des aktiven Ingrediens (der aktiven Ingredienzien) enthalten,
aufrechterhalten werden.
-
Die
gewünschte
Dosis kann zweckdienlicherweise in einer einzelnen Dosis oder als
aufgeteilte Dosen, die in geeigneten Intervallen verabreicht werden,
z.B. als zwei, drei, vier oder mehr Unterdosen pro Tag, vorliegen.
Die Unterdosis selbst kann weiter aufgeteilt werden, z.B. in eine
Reihe diskreter, lose beabstandeter Verabreichungen; zum Beispiel
als mehrere Inhalationen aus einem Insufflator oder durch Einbringen
einer Vielzahl von Tropfen in das Auge.
-
Wie
hierin offenbart wurde, wurde festgestellt, daß BTK-Inhibitoren als chemosensibilisierender
Agenzien einsetzbar sind und damit zur Erhöhung der Empfindlichkeit einer
Krebszelle für
andere chemotherapeutische Mittel, die eine Apoptose begünstigen,
nützlich
sind. Als solche können
BTK-Inhibitoren
zweckdienlicherweise in Kombination mit anderen chemotherapeutischen
Mitteln verabreicht werden. Zusätzlich
können die
pharmazeutischen Zusammensetzungen, die ein Agens umfassen, welches
BTK inhibiert, außerdem
ein oder mehrere anderen chemotherapeutische Agenzien, welche eine
Apoptose begünstigen,
umfassen.
-
Die
Erfindung wird nun durch die folgenden nichtbeschränkenden
Beispiele erläutert.
-
BEISPIELE
-
BEISPIEL 1: BTK inhibiert
FAS/APO-1 DISC
-
Das
folgende Beispiel liefert einen biochemischen und genetischen Beweis,
daß BTK
in Inhibitor des Fas/APO-1 death inducing signaling complex (DISC)
in Lymphozyten bzw. Lymphzellen der B-Zellinie ist. BTK assoziiert
mit Fas-FADD assoziiert mit Fas über
seine Kinase- und PH-Domänen
und verhindert die FAS-FADD-Wechselwirkung, die für die Rekrutierung
und Aktivierung des Tod-induzierenden proteolytischen Enzyms FLICE
durch Fas während
des apoptotischen Signals essentiell ist. Eine Behandlung von humanen Leukämie-B-Zellen
mit einem wirksamen Inhibitor für
BTK setzt die BTK-Fas-Assoziierung außer Kraft und sensibilisiert
die Zellen gegenüber
einer Fas-vermittelten Apoptose.
-
Zellinien,
Reagenzien und biochemische Assays.
-
Die
Entwicklung von BTK-defizienten DT-40-Lymphom-B-Zellklonen wurde bereits früher von
Uckum, F.M. et al. (1996), Science 273, 1096-1100 beschrieben. Um
das btk-Gen zu unterbrechen, wurden Targetingkonstrukte, die eine
Neomycin-Resistenzgen-Kassette
(d.h. pcBTK-neo) oder eine Histidinol-Resistenzgen-Kassette (d.h. pcBTK-hisD)
enthalten, sequenziell in DT-40-Zellen transfiziert. Die Targetingvektoren, pcBTK-neo
und pcBTK-hisD, wurden konstruiert, indem das 0,75 kb genomischer
BglII-BamHI-Fragment,
das Exons enthält,
welche den humanen BTK-Aminosäureresten
91-124 entsprechen, mit einer neo- oder his-D-Kassette ersetzt wurden. pcBTK-neo
wurde linearisiert und durch Elektroporation in Wildtyp-DT-40-Zellen
eingeführt.
Mittels Southern-Blot-Analyse unter Verwendung einer 3'-flankierenden Sonde (0,5 kb, BglII-Bgl-II-Fragment)
erfolgte ein Screening. Der neo-targetierte Klon wurde erneut mit
pcBTK-hisD transfiziert und mit G418 (2 mg/ml) und Histidinol (1
mg/ml) selektiert. Eine Southern-Blot-Analyse des BTK-defizienten DT-40-Klons
bestätigte
die homologe Rekombination an beiden btk-Loci und eine Hybridisierung
mit einer neo- und hisD-Sonde zeigte an, daß der targetierte Klon eine
einzelne Kopie jedes Konstrukts eingebaut hatte. Das Fehlen einer
BTK-Expression in
BTK-defizienten DT-40-Zellen wurde sowohl durch Immunkomplex-Kinase-Assays
als auch durch Westernblot-Analyse bestätigt (Uckun, F.M. et al. (1996),
Science 273, 1096-1100).
Mutationen in der humanen btk-cDNA wurden durch PCR eingeführt, wobei
Pfu-Polymerase (Stratagene, La Jolla, Californien, #600153) verwendet
wurde, und durch Sequenzierung bestätigt. Wildtyp- und mutante
btk-cDNAs wurden in pApuro-Expressionsvektor subkloniert und in
BTK-defiziente Zelle
elektroporiert. Die PTK-Aktivität
von BTK-Immunkomplexen,
wie sie durch in vitro-Autophosphorylierung gemssen wurde, wurde
durch die Mutation der katalytischen Domäne aufgehoben, durch die Mutation
der PH-Domäne reduziert,
durch die Mutation in der SH2-Domäne allerdings nicht beeinträchtigt.
Gleiche Mengen an BTK-Protein wurden durch Western-Blot-Analyse
in allen BTK-defizienten DT-40-Klonen
detektiert, die mit Wildtyp- oder mutierten humanen btk-Genen transfiziert
waren, allerdings war kein BTK-Protein in nicht-transfizierten BTK-defizienten
DT-40-Zellen nachweisbar (Uckun, F.M., et al. (1996), Science 273,
1096-1100). Die
Entwicklung und Charakterisierung von LYN-defizienten-DT-40-Klonen war bereits
von Uckun, F.M. et al. (1996), Science 273, 1096-1100 beschrieben
worden. Zusätzlich
zu diesen Hühner-Lymphom-B-Zellen
wurden die folgenden humanen B-Lymphozyten-Linien verwendet: NALM-6-UM1,
BTK-positive, humane
B-Zell-Vorläufer (pre-B-akute
Lymphoblastenleukämie)-Zellinie;
RAMOS-1, BTK-defiziente, humane Burkitt's/B-Zell-Leukämielinie und KL2-BTK-positive,
humane EBV-transformierte, normale B-Lymphoblastoid-Zellinien.
-
Antikörper, die
gegen BTK, SYK und LYN gerichtet sind, wurden früher beschrieben (Uckun, F.M.
et al. (1996), Science 273, 1096-1100; Dibirdik, I. et al. (1998),
J. Biol. Chem. 273, 4035-4039 und Uckun, F.M. et al. (1996), J.
Biol. Chem. 271, 6389-6397. Polyklonale Antikörper gegen BTK wurden durch
Immunisierung von Kaninchen mit Glutathion-S-Transferase (GST)-Fusionsproteinen
(Amersham Pharmacia Biotech, Arlington Heights, Illinois), die die
ersten 150 Aminosäuren
von BTK enthalten, erzeugt. Zusätzlich
wurden die folgenden Anti-BTK-Antikörper in
Western-Blots gereinigter Fusionsproteine verwendet: polyklonaler
Ziege-Anti-BTK-Carboxyl-Terminus (Santa Cruz Biotechnology, Inc.,
Santa Cruz, Ca. Kat.# SC1107), polyklonaler Ziege-Anti-BTK-Amino-Terminus
(Santa Cruz Biotechnology, Inc., Santa Cruz, Ca. Kat.# SC1108) und
polyklonales Kaninchenserum gegen die Btk-SH2-SH3-Domänen (aa219-377).
Polyklonaler Anti-MBP-Antikörper
wurden durch Immunisierung von Kaninchen erzeugt. Die polyklonalen
Kaninchen-Anti-Fas-Antikörper
(sc-715, 1:1 mit sc-714 vermischt), die sowohl mit humanen wie auch
mit Hühner-Fas-Proteinen kreuzreagieren,
polyklonaler Ziegen-Anti-FADD (sc-1171)-Antikörper, polyklonaler Ziege-Anti-TRADD
(sc-1163)-Antikörper, polyklonaler
Ziege-Anti-FLICE (sc-6135) wurden von Santa Cruz Biotechnology,
Inc. bezogen und entsprechen den Empfehlungen des Herstellers verwendet.
Der molekulare Anti-Fas-Antikörper
(F22120) wurde von Transduction Laboratories, Inc. (Lexington, KY,
USA) erhalten. Immunpräzipitationen,
Immunkomplex-Proteinkinase-Assays
und Immunblotting unter Verwendung des verstärkten Chemilumineszenz (ECL)-Detektionssystems
(Amersham Pharmacia Biotech, Arlington Heights, Illinois) wurden
wie früher
beschrieben durchgeführt (Uckun,
F.M. et al. (1996), Science 273, 1096-1100; Dibirdik, I. et al.
(1998), J. Biol. Chem., 273, 4035-4039; Uckun, F.M. et al. (1996),
J. Biol. Chem. 271, 6389-6397; Mahajan, S., et al. (1995), Mol.
Cell. Biol. 15, 5304-5311; Uckun, F.M. et al. (1993), J. Biol. Chem.
268, 21172-21184; Uckun, F.M. et al. (1995), Science 267, 886-91
und Uckun, F.M. et al. (1997), Blood, 89, 3769-3777). Der BTK-Inhibitor α-Cyano-β-hydroxy-β-methyl-N-(2,5-dibromphenyl)propenamid
(LMA-13) wurde hergestellt, wie es in Beispiel 2 beschrieben ist.
-
Expression und Reinigung
von MBP-BTK- und GST-BTK-Fusionsproteinen
-
cDNAs,
die für
die Vollängen-BTK
und ihre Kinase- oder PH-Domänen mit
PCR-erzeugten 5'-
und 3'-BamHI-Stellen
codieren, wurden in den E. coli-Expressionsvektor pMAL-C2 mit dem
IPTG-induzierbaren Ptac-Promotor
kloniert, um eine Rasterfusion zwischen diesen codierenden Sequenzen
und dem 3'-Ende
des E. coli-malE-Gens, welches für
das Maltose-Bindungsprotein (MBP) codiert, zu schaffen. cDNAs, die
für die SH2-,
SH3- oder SH2+SH3-Domänen
mit PCR-erzeugten 5'-
und 3'-BamHI-Stellen
codieren, wurden in den E. coli-Expressionsvektor pGEX-2t mit dem
IPTG-induzierbaren Ptac-Promotor kloniert, um eine "im Raster"-Fusion zwischen
diesen codierenden Sequenz und dem 3'-Ende des E. coli-Glutathion-S-Transferase (GST)-Gen
zu schaffen. Die gebildeten rekombinanten Plasmide wurden in den
E. coli-Stamm DH5a transformiert. Einzelne Transformanten wurden
in 5 ml Luria-Burtain (LB)-Medium (1 % Trypton, 1 % NaCl, 0,5 %
Hefeextrakt), das Ampicillin (100 μg/ml) enthielt, durch Übernachtkultur
bei 37°C
vermehrt. Die Expression der Fusionsproteine wurde mit 10 mM IPTG
induziert. Die Zellen wurden durch Zentrifugation mit 4 500 g in
einer Sorvall RC5B-Zentrifuge für
10 Minuten bei 4°C
geerntet, in Saccharose-Lysozym-Puffer (20 mM Tris pH 8,0, 150 mM
NaCl, 10 % Saccharose. 1 mM EDTA, 20 mM Lysozym) lysiert und durch
Beschallung weiter zerstört. Nach
Entfernung der Zellpellets durch Zentrifugation bei 35 000 g für 1 Stunde
bei 4°C,
wurden GST-BTK-Fusionsproteine durch Glutathion-Sepharose-Chromatographie
gereinigt (Uckun, F.M. et al. (1996), J. Biol. Chem. 271, 6389-6397),
während
MBP-BTK-Fusionsproteine
durch Amylose-Affinitätschromatographie
aus den Kulturüberständen gereinigt
wurden (Hsu, D. et al. (1996), Biochemistry 35, 13871-77).
-
Konfokale
Laser-Scanning-Mikroskopie
-
Wildtyp-
und BTK-defiziente DT-40-Zellen, die mit Anti-Fas-Antikörper (1 μg/ml, 24
h bei 37°C)
behandelt worden waren, wurden an mit Poly-l-Lysin überzogene
Deckgläschen
angeheftet und in eiskaltem (–20°C) Methanol
für 15
Minuten fixiert. Nach der Fixierung wurden die Deckgläschen für 15 Minuten
in Phosphat-gepufferter Salzlösung
(PBS) + 0,1 % Triton X-100 gewaschen. Die Zellen wurden nach den
Empfehlungen des Herstellers (Sigma, St. Louis, MO) mit einem polyklonalen
Kaninchen-Anti-Tubulin-Antikörper
gefärbt,
um ihr Cytoplasma sichtbar zu machen. DNA wurde für 10 Minuten
mit Toto-3, einem DNA-spezifischen Farbstoff, (Molecular Probes,
Eugene OR) markiert, um die apoptotischen Veränderungen in den Nuclei sichtbar
zu machen. BTK-MBP-elektroporierte, BTK- defiziente DT-40-Zellen und nicht-elektroporierte,
BTK-defiziente DT-40-Zellen
wurden mit einem Antikörper
gegen Maltose-Bindungsprotein markiert. Der sekundäre Antikörper war
ein Fluorescein-konjugierter Ziege-Anti-Kaninchen-Antikörper. In
einigen Experimenten wurden Zellen durch konfokale Mikroskopie auf
Fas-Expression untersucht. Kurz ausgedrückt, Zellen wurden an mit Poly-L-Lysin überzogen
Deckgläschen
angeheftet und für
40 Minuten in 2 % Paraformaldehyd in Phosphat-gepufferte Salzlösung (PBS)
fixiert. Die Zellen wurden mit PBS + 115 mM Glycin gespült, um den
Formaldehyd abzuschrecken und dann in PBS, die 2 % Rinderserumalbumin
enthielt, (PBS+BSA) blockiert. Ein monoklonaler Antikörper, der
gegen die extrazelluläre
Fas-Domäne gebildet
worden war, (Transduction Labs, Lexington, KY, Katalog #F22120)
wurde in PBS+BSA gegeben und die Deckgläschen wurden für 40 min
bei 37°C
inkubiert, bevor sie wieder in PBS gespült wurden. Ein Fluorescein-markierter
sekundärer
Antikörper
(Ziege-Anti-Maus-FITC H+L, Zymed, South San Francisco, CA, Katalog-Nr.
62-6511), verdünnt
in PBS+BSA, wurde dann zu den Deckgläschen gegeben und diese wurden
erneut für
40 min bei 37°C
inkubiert. Nach einem weiteren Waschen wurde zelluläre DNA durch
Inkubation in 1 mM TOTO-3 (Molecular Probes, Eugene, OR) für 20 min
bei Raumtemperatur markiert. Deckgläschen wurden umgedreht und
auf Objektträgern
in Vectashield (Vector Labs, Burlinghame, CA) montiert, um eine
Lichtbleichung zu verhindern, und mit Nagellack versiegelt. Die
Objektträger
wurden unter Verwendung eines konfokalen Laser-Scanning-Mikroskops
Bio-Rad MRC 1024, montiert an ein Nikon Eclipse E-800 Standmikroskop,
das zur Epifluoreszenz mit Objektiven mit hoher numerischer Blende
ausgestattet war, untersucht (Uckun, F.M. et al. (1998), Clinical
Cancer Research, 4, 901-912). Es wurden optische Schnitte erhalten
und diese wurden unter Verwendung der Lasersharp-Software (Bio-Rad, Hercules,
CA) in Stereo-Mikrophotographien
umgewandelt. Repräsentative
digitale Bilder wurden auf Jaz-Platten gesichert und unter Verwendung der
Photoshop-Software (Adobe Systems, Mountain View CA) bearbeitet. Bilder
wurden mit einem thermischen Transferdrucker von Fuji Pictography
(Fuji Photo, Elmsford, NY) gedruckt. Digitale Daten wurden archiviert
und auf CD-ROM gespeichert.
-
Apoptose-Assays
-
Um
eine Apoptose zu induzieren wurden Zellen mit einem agonistischen
Anti-Fas/APO-1-Antikörper (Biosource
International, Camarillo, Ca., lot. 04/1295) bei Endkonzentrationen
von 0,1 μg/ml,
0,5 μg/ml
oder 1,0 μg/ml
behandelt. Eine MC540-Bindung (als früher Marker der Apoptose) und
die PI-Permeabilität
(als Marker für
das fortgeschrittene Apoptosestadium) wurden 24 Stunden nach Behandlung
mit Anti-Fas-Antikörper, wie
es früher
beschrieben wurden, in DT-40-Zellen gleichzeitig gemessen (Uckun,
F.M. et al. (1996), Science 273, 1096-1100). Ganze Zellen wurden
mit FAC-Star Plus-Strömungscytometer
(Becton Dickinson, San Jose, CA) analysiert. Alle Analysen wurden
unter Verwendung einer 488 nm-Anregung durch einen Argonlaser durchgeführt. MC540- und PI-Emissionen
wurden mit einem 600 nm "Short
Pass"-Kaltlichtspiegel
aufgespalten und ein 575 nm-Bandpaßfilter wurde vor einem Photovervielfacher
angeordnet, um die MC540-Emission zu
messen, und es wurde ein 635 nm-Bandpaßfilter zur PI-Emission verwendet.
-
Um
eine apoptotische DNA-Fragmentierung zu detektieren, wurden DT-40-,
NALM-6-UM1- und RAMOS-1-Zellen 24 Stunden nach Behandlung mit Anti-Fas
geernetet. DNA wurde auf Triton-X-100-Lysaten zur Fragmentierungsanalyse
hergestellt (Uckun, F.M. et al. (1996), Science 273, 1096-1100);
(Uckun, F.M. et al. (1995), Science 267, 886-91 und Uckun, F.M.
et al. (1998) Clinical Cancer Research, 4, 901-912). Zellen wurden
in hypotonischem 10 mmol/l Tris-HCl (pH 7,4), 1 mmol/l EDTA, 0,2
% Triton-X-100-Detergens lysiert und anschießend mit 11 000 g zentrifugiert.
Um eine mit Apoptose assoziierte DNA-Fragmentierung zu detektieren, wurden Überstände an einem
1,2 % Agarosegel einer Elektrophorese unterworfen und die DNA-Fragmente wurden
nach Färbung
mit Ethidiumbromid mit ultraviolettem Licht sichtbar gemacht. In
einigen Experimenten wurden MBP-BTK-Fusionsproteine (100 μg/2,5 × 108 Zellen)
in BTK-defiziente DT-40-Zellen elektroporiert (420V elektrisches
Feld, 125 μF),
wobei ein BioRad-Elektroporator
zum Transfer sowie die Verfahren von Bergland und Starkey (Berland,
D. und Starkey, J. (1991), Cytometry 12, 64-67) mit leichten Modifikationen
4 Stunden vor einer Fas-Ligation und Apoptose-Assays angewendet
wurden.
-
"Pull Down"-Assays mit MBP-BTK- und GST-BTK-Fusionsproteinen
-
GST-BTK-Fusionsproteine
wurden nicht-kovalent an Glutathion-Agarose-Perlen (Sigma Aldrich, Inc., St.
Louis, MO) gebunden und MBP-BTK-Fusionsproteine wurden nicht-kovalent
an Amyloseperlen gebunden, und zwar unter Sättigungsproteinbedingungen,
wie es früher
bereits beschrieben worden war (Uckun, F.M. et al. (1996), J. Biol.
Chem. 271, 6389-6397 und Hsu, D. et al. (1996), Biochemistry 35,
13871-77). Kurz ausgedrückt,
50 μg jedes
Proteins wurden mit 50 μl
der Perlen für
2 Stunden bei 4°C
inkubiert. Die Perlen wurden dreimal mit 1 % Nonidet P-40-Puffer
gewaschen. Nonidet P-40-Lysate von BTK-defizienten DT-40-Zellen,
humanen Leukämie-B-Zellen-NALM-6-UM1-Vorläufern und
humanen EBV-transformierten
lymphoblastoiden KL2-Zellen wurden hergestellt, wie es beschrieben
wurde (Uckun, F.M. et al. (1996), Science 273, 1096-1100 und Uckun,
F.M. et al. (1996), J. Biol. Chem. 271, 6389-6397) und 500 μg des Lysats
wurden mit 50 μl
Fusionsprotein-gekoppelter Perlen für 2 Stunden auf Eis inkubiert.
Dies Fusionsproteinadsorbate wurden mit eiskaltem 1 % Nonidet P-40-Puffer
gewaschen und in reduzierendem SDS-Probenpuffer resuspendiert. Proben wurden
für 5 Minuten
gekocht und dann an SDS-PAGE fraktioniert.
-
Proteine
wurden auf Immobilon-P (Millipore, Bedford, MA)-Membranen transferiert und die Membranen
wurden mit Anti-Fas (Transduction Laboratories, Inc., Lexington,
KY, Kat.# F22120) einem Immunblotting nach früher beschriebenen Verfahren
unterzogen (Uckun, F.M. et al. (1996), Science 273, 1096-1100; Dibirdik, I.
et al. (1998), J. Biol. Chem. 273, 4035-4039; Uckun, F.M. et al
(1996), J. Biol. Chem. 271, 6389-6397 und Uckun, F.M. et al. (1997),
Blood, 89, 3769-3777).
-
In
einer Reihe von Experimenten, die entwickelt wurden, um die potentielle
negative Regulationsrolle von BTK in Fas-vermittlerter Apoptose zu untersuchen,
verglichen wir zuerst die Effekte einer Fas-Ligation an Wildtyp-DT-40-Zellen
mit den Effekten einer Fas-Ligation an einen BTK-defizienten Subklon
von DT-40-Zellen, der durch homologes Rekombinations-"Knockout" entwickelt worden war (Uckun, F.M.
et al. (1996), Science 273, 1096-1100). Zu diesem Zweck verwendeten
wir zuerst einen quantitativen strömungscytometrischen Apoptose-Detektionsassay (Uckun,
F.M. et al. (1996), Science 273, 1096-1100). Die MC540-Bindung und
die Propidiumiodid (PI)-Permeabilität wurde
gleichzeitig vor und nach Behandlung mit dem agonistischen Anti-Fas-Antikörper (1 μg/ml × 24 h)
gemessen. Während
nur 5,0 % der Wildtyp-DT-40-Zellen, die mit dem Anti-Fas-Antikörper behandelt
worden waren, apoptotische Veränderungen
zeigten, machten 96,3 % der BTK-defizienten DT-40-Zellen eine Apoptose
durch, wie es durch MC540-Einezlfluoreszenz
(frühe
Apoptose) oder MC540/PI-Doppelfluoreszenz
(fortgeschrittene Apoptose) nach 24 Stunden bestimmt wurde (1). Bemerkenswert war, daß BTK-defiziente DT-40-Zellen,
die mit einem humanen Wildtyp-btk-Gen rekonstituiert worden waren, einen
sehr schwachen strömungscytometrischen
Beweis für
Apoptose zeigten, was einen formalen Beweis darstellt, daß BTK bei
der Verhinderung des apoptotischen Todessignals, das durch Fas-Ligation
ausgelöst
wird, eine zentrale Rolle spielt. In Übereinstimmung mit den früher veröffentlichten
Informationen bezüglich der
pro-apoptotischen Funktion der Src-Familie-PTK (Waddick, K.G. et
al. (1993), Radiation Research, 136, 313-319; Schlottmann, K.E. et al., Leukocyte
Biology 60, 546-554; und Atkinson, E.A. et al(1996), J. Biol. Chem.
271, 5968-5971)
und der Beeinträchtigung
der Fas-vermittelten Apoptose in B-Zellen aus LYN-defizienten Mäusen (Wang,
J., et al. (1996), J. Exp. Med. 184, 831-838) wurde in einem Anti-Fas-behandelten LYN-defizienten
Subklon von DT-40-Zellen, der als Kontrolle in diesen Experimenten
eingeschlossen war, sehr geringe Apoptose festgestellt (1). Wie in 2A gezeigt
ist, war durch Western Blot-Analyse in den Ganzzellysaten von BTK-defizienten
DT-40-Zellen kein BTK-Protein
nachweisbar, während
BTK-defiziente DT-40-Zellen, die mit einem humanen Wildtyp-btk-Gen
rekonstituiert worden waren, höhere
Level an BTK exprimierten als die Wildtyp-DT-40-Zellen. Allerdings waren die Fas-Protein-Expressionslevel
in diesen drei B-Zellklonen tatsächlich
identisch (2B). Diese Feststellungen wurden
durch konfokale Mikroskopie weiter bestätigt. Wie in C.1 bis C.3 gezeigt
wird, wiesen alle drei Zellinien ähnliche Level einer punktuierten
Fas-Färbung auf.
Dreidimensionale Rekonstruktionen von optischen Reihenschnitten
bestätigten
die Expression von Fas sowohl im Cytoplasma als auch an der Oberflächenmembran
aller drei Zellinien ohne detektierbare Unterschiede bezüglich der
Expressionslevel oder des Expressionsmusters. Somit war die Resistenz
der Wildtyp-DT-40-Zellen oder der BTK-defizienten DT-40-Zellen,
die mit Wildtyp-BTK rekonstituiert worden waren, gegenüber einer
Fas-vermittelten Apoptose nicht in niedrigeren Expressionsleveln
an Fas-Protein begründet
und die Empfindlichkeit von BTK-defizienten DT-40-Zellen gegenüber einer
Fas-Apoptose wurde nicht durch eine erhöhte Fas-Proteinexpression verursacht.
-
Die
vergleichende Untersuchung der morphologischen Merkmale von Wildtyp-
gegenüber
BTK-defizient DT-40-Zellen durch konfokale Laser-Scanning-Mikroskopie
zeigte keinen Apoptosebeweis für
Wildtypzellen nach Behandlung mit dem agonistischen Anti-Fas-Antikörper, wohingegen
BTK-defiziente Zellen eine Schrumpfung und Kernfragmentierung zeigten,
die mit Apoptose übereinstimmt
(3A). Auf Agarosegelen zeigte DNA aus Triton-X-100-Lysaten
von Anti-Fas-behandelten BTK-defizienten DT-40-Zellen ein leiterartiges Fragmentierungsmuster,
das Apoptose entspricht, wohingegen bei Wildtyp-DT-40-Zellen keine
DNA-Fragmentierung beobachtet wurde (3B).
Diese Resultate liefern einen direkten Beweis dafür, daß BTK eine Fas/APO-1-vermittelte
Apoptose inhibieren kann.
-
BTK
hat eine einzigartige N-terminale Region, die Pleckstrin-Homologie (PH)- und
Tec-Homologie (TH)-Domänen,
eine einzelne Src-Homologie-3 (SH3)-Domäne, welche die Autophosphorylierungsstelle
am Tyrosin 223 hat, eine einzelne SH2-Domäne und eine katalytische Kinase-Domäne, welche
die Transphosphorylierungsstelle am Tyrosin 551 enthält, enthält (Rawlings,
D.J., Witte, O.N. (1994) Immunol. Rev. 138, 105-119 und Kurosaki, T. (1997) Curr. Opin.
Immunol. 9, 309-318). Die PH-Domäne
von BTK wechselwirkt mit verschiedenen Isoformen der Proteinkinase
C, heterotrimeren G-Proteinen wie auch dem BAP-135-Protein (Rawlings, D.J.,
Witte, O.N. (1994), Immunol. Rev. 1380 105-119; Kurosaki, T. (1997)
Curr. Opin. Immunol. 9, 309-318; Tsukada, S. (1994), J. Biol. Chem.
269, 10217-10220 und Yang, W., Desiderio, S. (1997), Proc. Natl.
Acad. Sci. USA 94, 604-609). Es hat sich gezeigt, daß SH3-Domänen mit
prolinreichen Sequenzen anderer Proteine wechselwirken, wohingegen
SH2-Domänen
mit Tyrosin-phosphorylierten
Proteinen wechselwirken (Yang, W, Desiderio, S. (1997), Proc. Natl.
Acad. Sci. USA 94, 604-609). Allerdings wurden keine spezifischen
Proteine beschrieben, die mit den BTK-SH2- oder -SH3-Domänen in B-Lymphozyten
wechselwirken. Mutationen in der katalytischen Domäne, der
SH2-Domäne
wie auch der PH-Domäne
der BTK führen erwiesenermaßen zu maturationalen
Blöcken
in frühen
Stadien der B-Zell-Ontogenie in humanem XLA (Pawson, T., Gish, G.D.
(1992), Cell 71, 359-362 und Vihinen, M., et al. (1995) Immunol.
Today 16, 460-465). BTK-defiziente Mäuse, die durch Einführung von
PH-Domänen-
oder katalytischen Domänen-Mutationen in Embryostammzellen
erzeugt worden waren, zeigten eine fehlerhafte B-Zellen-Entwicklung
und -Funktion (Kerner, J.D., et al. (1995), Immunity 3, 301-312).
Demnach sind verschiedene Regionen von BTK für ihre physiologischen Funktionen
wichtig. Um die Beteiligung der verschiedenen BTK-Domänen bei
der negativen Regulierung der Fas-vermittelten Apoptose zu untersuchen,
führten
wird humanes Wildtyp-btk-Gen wie auch humane btk-Gene, die Mutationen
entweder in der katalytischen Domäne (Arg525 zu
Gln), der SH2-Domäne
(Arg307 zu Ala) oder der PH-domäne (Arg28 zu Cys) beherbergten, in die BTK-defizienten
DT-40-Zellen ein (Uckun, F.M. et al. (1996), Science 273, 1096-1100).
Wie in 3C und 3D bewiesen
wurde, erlitten BTK-defiziente DT-40-Zellen, die mit humanem Wildtyp-btk-Gen
(rWT) rekonstituiert worden waren, nach Behandlung mit dem agonistischen
Anti-Fas-Antikörper
keine Apoptose, wohingegen eine Fas-Aktivierung von rekonstituierten
BTK-defizienten DT-40-Zellen, die humane BTK mit Mutationen in der
Kinase (rK-)-, SH2 (rmSH2)- oder PH (rmPH)-Domäne exprimieren, eine Apoptose
induzierte, wie es in nicht-rekonstituierten BTK-defizienten DT-40-Zellen
der Fall ist, was in 3B dargestellt ist. Demnach
sind die Kinase-, SH2- und PH-Domänen von BTK alle wichtig und
offensichtlich für ihre
Funktion als negativer Regulator der Fas-vermittelten Apoptose unverzichtbar.
-
Um
die anti-apoptotische Funktion von BTK weiter zu charakterisieren,
führten
wir ein MBP-Fusionsprotein, das Vollängen-Wildtyp-BTK enthält, 4 Stunden
vor Behandlung mit dem Anti-Fas-Antikörper durch Elektroporation
in BTK-defiziente
Zellen ein. Eine Untersuchung dieser Zellen durch konfokale Laser-Scanning-Mikroskopie
(4A) wie auch durch Western-Blot-Analyse unter
Verwendung von Anti-BTK- und Anti-MBP-Antikörpern (4B)
bestätigte
das Vorliegen des elektroporierten MBP-BTK-Proteins. Wie in 4C gezeigt ist, machte eine Einführung von
Wildtyp-BTK-Protein durch Elektroporation die BTK-defizienten DT-40-Zellen
gegenüber
den apoptotischen Effekten einer Fas-Ligation resistent, was direkte
Protein-Protein-Wechselwirkungen zwischen BTK und Gliedern des Fas-Signaltransducktionswegs
als möglichen
Mechanismus für
die Anti-apoptotische Funktion von BTK nahelegt.
-
Die
pro-apoptotischen "Downstream"-Events, die durch
die Ligation von Fas oder TNF-Rezeptor-1 initiiert werden, werden
derzeit belichtet (Fraser, A., Evan, G. (1996), Cell 85, 781-784; Kischkel, F.C.,
et al. (1995) EMBO J. 14, 5579-5588; Hu, S., Vincenz, et al. (1997),
J. Biol. Chem. 272, 17255-17257; Deveraux, Q.L., et al. (1997),
Nature 388, 300-304; Thome, M., et al. (1997), Nature 386, 517-521;
Boldin, M.P. et al. (1995), J. Biol. Chem. 270, 7795-7798; Los,
M. et al. (1995), Nature 375, 81-83; Boldin, M.P., et al. (1996),
Cell 85, 803-815;
und Chinnaiyan, A.M., et al. (1995), Cell 81, 505-512). Sowohl Fas
als auch TNF-Rezeptor-1 enthält
eine homologe intrazelluläre "Todesdomäne", die im ligandenabhängigen Zusammenbau
eines pro-apoptotischen "death
inducing signaling"-Komplexes
(DISC) eine zentrale Rolle spielt (Kischkel, F.C. et al. (1995), EMBO
J. 14, 5579-5588). Die Todesdomänen
von p55-TNF-Rezeptor-1 und Fas/CD95 dienen als Docking-Stellen,
die eine ligandenabhängige
Rekrutierung von und eine Heteroassoziierung mit einer anderen Todesdomäne, die
mehrwertige Adapterproteine enthält,
vermitteln: Fas-assoziiertes
Protein mit Todesdomäne
(FADD) und mit Rezeptorwechselwirkendes Protein (RIP) im Fall von
CD95; und TNF-Rezeptor-1-assoziiertes
Todesdomänenprotein
(TRADD) und RIP im Fall von TNF-Rezeptor-1 (Fraser, A., Evan, G.
(1996), Cell 85, 781-784; Kischkel, F.C. et al. (1995), EMBO J.
14, 5579-5588; und
Chinnaiyan, A.M. et al. (1995) Cell 81, 505-512).
-
FADD
ist der Konvergenspunkt zwischen den Fas/CD95- und TNF-Rezeptor-1-verknüpften apoptotischen
Signaltransduktionspfaden. Während
Fas/CD95 direkt FADD heranzieht, bindet TNF-Rezeptor-1 TRADD, welches
dann als Adapter-Protein wirkt, um FADD zu rekrutieren. Die Bildung
von CD95-FADD- oder TNF-Rezeptor-1-TRADD-FADD-Komplex nach Ligandenbindung
ist für
die Induktion einer Apoptose wichtig. Die Aneinanderfügung eines
pro-apoptotischen DISC wird durch die Rekrutierung und begleitende
Aktivierung der cytosolischen Caspase FLICE, einem Glied der ICE-Proteasefamilie,
vervollständigt
(Fraser, A., Evan, G. (1996), Cell 85, 781-784; Kischkel, F.C. et
al. (1995), EMBO J. 14, 5579-5588; Hu, S., Vincenz et al. (1997),
J. Biol. Chem. 272, 17255-17257; Deveraux, Q.L. et al. (1997), Nature
388, 300-304; Thome, M. et al. (1997), Nature 386, 517-521; Boldin,
M.P. et al. (1995), J. Biol. Chem. 270, 7795-7798; Los, M. et al.
(1995), Nature 375, 81-83; Boldin, M.P. et al. (1996), Cell 85,
803-815; und Chinnaiyan, A.M., et al. (1995) Cell 81, 505-512). Kürzlich wurde
eine Reihe von Proteinen als Inhibitoren sowohl einer Fas- wie auch
TNF-Rezeptor-1-induzierten
Apoptose identifiziert (Fraser, A., Evan, G. (1996), Cell 85, 781-784;
Kischkel, F.C. et al. (1995), EMBO J. 14, 5579-5588; Hu, S., Vincenz
et al. (1997), J. Biol. Chem. 272, 17255-17257; Deveraux, Q.L. et
al. (1997), Nature 388, 300-304; Thome, M. et al. (1997), Nature
386, 517-521). Diese Proteine wechselwirken direkt mit FADD oder
FLICE, wodurch sie einen DISC-Zusammenbau und dessen Funktion stören. Die
Todesdomäne von
Fas enthält
bemerkenswerterweise ein konserviertes YXXL-Motiv ähnlich den
Sequenzen des Immunrezeptor-Aktivierungsmotiv auf Tyrosin-Basis
(ITAM) als potentielle Bindungsstelle für SH2 enthaltende Proteine; von
Fas wurde kürzlich
gezeigt, daß es
mit Fyn- und Lck-Kinasen als pro-apoptotische Regulatoren, die zur Induktion
einer Fas-vermittelten Apoptose erforderlich sind, assoziiert (Schlottmann,
K.E. et al., Leukocyte Biology 60, 546-554; und Atkinson, E.A. et
al. (1996), J. Biol. Chem. 271, 5968-5971).
-
Wir
haben daher postuliert, daß BTK
mit Fas wechselwirken könnte
und den Zusammenbau eines proapoptotischen DISC nach Fas-Ligation
verhindern könnte.
-
Wir
untersuchten zuerst, ob BTK zu einer physikalischen Assoziierung
mit Fas und anderen DISC-Gliedern fähig ist, indem wir die Fas-,
FLICE-, FADD- und TRADD-Immunkomplexe aus den Nonidet P-40-Lysaten
von unbehandelten DT-40-Zellen auf das Vorliegen von BTK untersuchten.
BTK wurde durch Western-Blot-Analyse
in Fas (aber nicht in den anderen) Immunkomplexen durch Anti-BTK-Immunblotting
detektiert (5A). In ähnlicher weise wurde Fas durch
Anti-Fas-Immunblotting
in BTK-Immunkomplexen aus Wildtyp-DT-40-Zellen wie auch aus BTK-defizienten
DT-40-Zellen, die mit humanem Wildtyp-btk-Gen rekonstituiert worden
waren, detektiert (5B). Die konstitutive Assoziierung
von BTK mit Fas-Protein
wurde auch in der humanen B-Zellvorläufer-Leukämie-Zellinie NALM-6-UM1 gefunden (5C). Zusammengenommen bewiesen diese Resultate,
daß BTK
zur Assoziierung mit Fas-Protein
fähig ist
und daß diese
physikalische Assoziierung kein vorheriges Eingreifen des Fas-Rezeptors
erfordert. Wie in 5D gezeigt ist, ist Fas in BTK-defizienten
DT-40-Zellen mit
FADD assoziiert, wie es durch Detektion von Fas in FADD-Immunkomplexen bewiesen
wird; diese physikalische Wechselwirkung war nach Fas-Ligation deutlich
verstärkt.
In Fas-aktivierten, BTK-defizienten DT-40-Zellen konnten Fas-assoziierte FADD-Moleküle durch
Anti-FADD-Immunblotting detektiert werden (5D).
Im Gegensatz zu BTK-defizienten DT-40-Zellen wurde eine sehr schwache Fas-FADD-Assoziierung
in unbehandelten oder anti-Fas-behandelten, BTK-defizienten DT-40-Zellen,
die mit humaner Wiltyp-BTK rekonstituiert waren, festgestellt (5B). In ähnlicher
Weise konnte eine Fas-Ligation die Fas-FADD-Assoziierung in humanem
NALM-6-UM1-Leukämiezellen
nicht verstärken
(5C). Demnach assoziiert BTK mit Fas und verschlechtert
seine Wechselwirkung mit FADD, einem Protein, welches für die Rekrutierung
und Aktivierung von FLICE durch Fas während des apoptotischen Signals
essentiell ist. Obgleich diese Resultate die Möglichkeit nicht ausschließen, daß BTK das
Schicksal des apoptotischen Signals, das durch Fas-Ligation ausgelöst wird,
durch mehrere Mechanismen, einschließlich Modulation der Funktion
positiver oder negativer Regulatoren der apoptotischen Signaltransduktion,
verändern
kann, liefern sie mindestens eine plausible Erklärung für die anti-apoptotische Funktion von BTK.
-
Um
die physiologische Bedeutung der beobachteten BTK-Fas-Assoziierung in humanen
Leukämie-B-Zell-Vorläufern zu
klären,
verglichen wir die Empfindlichkeiten der BTK-positiven, humanen NALM-6-UM1-pre-B-Leukämie-Zellinie
und der BTK-defizienten
humanen RAMOS-1-B-Zell-Leukämiezellinie gegenüber Fas-vermittelter
Apoptose. Wie in 6A gezeigt ist, exprimieren
diese zwei Zellinien ähnliche
Level an Fas-Protein.
BTK-defiziente RAMOS-1-Zellen machten nach Fas-Ligation mit dem agonistischen Anti-Fas-Antikörper eine
Apoptose durch, allerdings war dies bei BTK-positiven NALM-6-UM1-Zellen nicht
der Fall (6B). Wir untersuchten als nächstes die
Wirkung des Leflunomid-Metabolitenanalogon α-Cyano-β-hydroxy-β-methyl-N-(2,5-dibromphenyl)propenamid
(LMA-13), ein starker
Inhibitor für
BTK auf die BTK-Fas-Assoziierung
und Resistenz gegenüber
Fas-vermittelter Apoptose in NALM-6-UM1-Zellen. Anti-BTK- und Anti-Fas-Western-Blot-Analysen
von Ganzzellysaten von LMA-13-behandelten
NALM-6-UM1-Zellen zeigten keine Verringerung beim BTK (6.C.1)- oder Fas-Protein (6.C.2)-Expressionslevel.
In den BTK-Immunkomplexen war wesentlich weniger Fas-Protein, was
einen direkten Beweis dafür
darstellt, daß eine
BTK-Inhibierung durch LMA-13 die BTK-Fas-Assoziierung außer Kraft setzt (6C.3). Beachtenswerterweise induziert ein Vierstundenbehandlung
mit LMA-13 in NALM-6-UM1-Zellen keine Apoptose, sondern macht diese hochresistenten
humanen Leukämiezellen
gegenüber
Fas- vermitteltert
Apoptose empfindlich (6D). Die bereitgestellten Resultate
zeigen außerdem,
daß BTK
ein physiologisch wichtiger, negativer Regulator der Fas-vermittelten Apoptose
ist.
-
Die
Fähigkeit
des BTK-Inhibitors LFM-A13, die BTK-Fas-Assoziierung außer Kraft zu setzen, lieferte den
Beweis, daß die
Kinaseaktivität
von BTK eine wichtige Rolle für
die Bildung der BTK-Fas-Komplexe liefert. Um weiter zu klären, ob
die Assoziierung von BTK mit Fas von ihrer Kinaseaktivität abhängt oder
nicht, untersuchten wir als nächstes
BTK-Fas-Wechselwirkungen
in BTK-defizienten DT-40-Zellen, die entweder mit humaner Wildtyp-BTK
(BTK-, rBTK[WT]) oder Kinase-inaktiver mutanter (Arg525 zu
Gln), humaner BTK (BTK-, rBTK[K-]) rekonstituiert worden waren.
Wie in 7A dargestellt ist, zeigte
eine Western-Blot-Analyse von Ganzzellysaten dieser zwei Zellinien
mit Anti-BTK- oder Anti-Fas-Antikörpern keine
wesentlichen Differenzen (d.h. in beiden der zwei unabhängigen Experimenten
beobachteten wir eine etwas höhere
BTK und Fas-Expression in BTK, rBTK[K-]-Zellen). Fas-Immunkomplexe aus Lysaten
von BTK-, rBTK[WT]-Zellen,
die BTK-Protein und diese BTK-Fas-Assoziierung enthielten, wurden
durch Behandlung der Zellen mit dem Anti-Fas-Antikörper weiter verstärkt (7B, Bahnen 1 und 2). Dagegen war in Fas-Immunkomplexen
aus BTK-, rBTK[K-]-Zellen ungeachtet einer Behandlung mit dem Anti-Fas-Antikörper kein
BTK-Protein nachweisbar (7B,
Bahnen 3 und 4). Diese Resultate liefern einen bestätigenden
Beweis dafür,
daß die
Assoziierung von BTK mit Fas von der Kinaseaktivität von BTK
abhängt.
-
Als
nächstes
führten
wir Bindungsexperimente mit Vollängen-MBP-BTK- und verkürzten MBP-BTK- und
GST-BTK-Fusionsproteinen,
die verschiedenen BTK-Domänen
entsprechen, (8A-C) durch, um die strukturellen
Anforderungen für
eine BTK-Assoziierung mit Fas zu klären. MBP-BTK 1-659 (Vollängen-BTK) wie
auch MBP-BTK 408-659 ("BTK-Kinasedomäne") und MBP-BTK 2-137
("BTK PH-Domäne") waren zum "pull down" und zum Binden von
Fas aus Lysaten von BTK-defizienten DT-40-Zellen (8D),
humanen NALM-6 pre-B-Leukämiezellen
(8E) und humanen EBV-transformierten B-Lymphoblastenzellen
KL2 (8F) fähig. Allerdings hat Fas nicht
an das Kontroll-MBP-BTK
519-567-Fusionsprotein, das einer verkürzten Kinasedomäne entspricht,
die die Y551-Transphosphorylierungsstelle
enthält
(C5 in 7D) entspricht, GST-BTK 219-377,
das die SH3- plus SH2-Domäne
enthält,
GST-BTK 219-268, das der SH3-Domäne
entspricht oder GST-BTK 281-377, das der SH2-Domäne entspricht, (die zwei letztgenannten
wurden nur mit Lysaten von BTK-defizienten DT-40-Zellen verwendet)
(8D–8F)
gebunden.
-
Obgleich
die Kristallstruktur von Vollängen-BTK
nicht beschrieben worden war, liefern die kürzlich veröffentlichten Strukturen der
PH-Domäne
und des BTK-Motivs (Hyvonen, M., Saraste, M. (1997), EMBO J. 16, 3396-3404)
nützliche
Informationen, die auf die Bindungsfähigkeit von BTK und ihrer PH-Domäne anwendbar sind.
Von FADD wurde berichtet, daß es
mit der cytoplasmischen Fas-Domäne
wechselwirkt, die zum großen Teil
aus einer Todesdomäne
besteht, welche aus sechs antiparallelen α-Helices besteht, die aus den
Resten 230 bis 314 zusammengebaut sind (Huang, B. et al. (1996),
Nature 384, 638-641). Es wurde spekuliert, daß die YXXL-Sequenz der Fas-Todesdomäne ITAMs ähnelt und
durch eine SH2-Domäne
nach Tyrosinphosphorylierung oder durch andere Mechanismen erkannt
wird (Schlottmann, K.E., et al. Leukocyte Biology 60, 546-554; und Atkinson,
E.A., et al. (1996), J. Biol. Chem. 271, 5968-5971). Eine Analyse
der Konformation dieser YXXL-Sequenz zeigt, daß sie in der Mitte einer α-Helix lokalisiert
ist; und wenn keine wesentliche Konformationsänderung dieser α-Helix erfolgen würde, um
den Tyrosin-Rest zugänglicher
zu machen, wäre
sie für eine
Wechselwirkung mit einer PTK zu stark. Somit würde die strukturelle Geometrie
der YXXL- Sequenz
wahrscheinlich verhindern, daß Fas
und BTK einen solchen Bindungsmodus entwickeln, wie der von CD3-ε ITAM/ZAP-70, wie er von Atkinson,
E.A. et al. (1996), J. Biol. Chem. 271, 5968-5971 vorgeschlagen
wurde. Die Unfähigkeit
der BTK-SH2-Domäne zum "pull down" von Fas aus Ganzzellysaten
unterstützt
diese Meinung. Wie assoziiert dann BTK mit Fas? BTK und Fas können über komplementäre elektrostatische
Anziehungen und Wasserstoff-Bindungswechselwirkungen, die die vorher
beschriebenen geladenen Reste an den Oberfläche der α-Helices der Fas-Todesdomäne involvieren
könnten,
assoziieren. Diese Assoziierung könnte durch ein drittes Protein
vermittelt werden, das eine Grenzfläche zwischen Fas und BTK bildet.
die Bedeutung der SH2- und Kinasedomäne von BTK für ihre anti-apoptotische
Funktion legt die Hypothese nahe, daß ein Tyrosin-phosphoryliertes
Substrat von BTK eine solche Grenzfläche bereitstellen kann.
-
Die
Fähigkeit
von BTK, die pro-apoptotischen Effekte einer Fas-Ligation zu inhibieren
weckt die Hypothese, daß eine
Apoptose von sich entwickelnden B-Zellvorläufern während einer normalen humanen
B-Zellontogenese durch Fas und BTK reziprok reguliert werden kann.
Das Fehlen von BTK oder Mutationen in ihren Kinase-, PH- und SH2-Domänen könnte zu
einem unpassenden apoptotischen Zelltod von pre-B-Zellen führen, was
im XLA-Phänotyp
resultiert (Rawlings, D.J., Witte, O.N. (1994), Immunol. Rev. 138,
105-119; Tsukada, S. et al. (1993), Cell 72, 279-290 und Vetrie,
D. (1993) Nature 361, 226-233). Eine unpassende Apoptose kann der
Pathogenese wie auch der Arzneimittelresistenz humaner Leukämie und
Lymphomen zugrundeliegen, was die Apoptosekontrolle zu einem wichtigen
potentiellen Ziel für
eine therapeutische Intervention macht. Das Schicksal von Leukämie/Lymphom-Zellen, die chemotherapeutischen
Mitteln (z.B. Vincristin, Daunorubicin oder Taxol) ausgesetzt sind,
kann auf dem Gleichgewicht zwischen den entgegengesetzten pro-apoptotischen Wirkungen
von durch DISC aktivierten Kaspasen und einem negativen "Upstream"-Regulationsmechanismus, der
BTK und/oder seine Substrate involviert, basieren. Daher können BTK-Inhibitoren
wahrscheinlich die Arzneimittelempfindlichkeit von Leukämie/Lymphom-Zellen
der B-Linie verstärken.
-
Beispiel 2: Synthese spezifischer
Leflunomid-Metabolitenanaloga
-
Chemie
-
Alle
Chemikalien wurden von Aldrich (Milwaukee, WI) bezogen und wurde
ohne weitere Reinigung eingesetzt. Außer, wenn es angegeben ist,
war jeder Reaktionsbehälter
mit einem Kautschukseptum gesichert und wurde die Reaktion unter
Stickstoffatmosphäre
durchgeführt. 1H- und 13C-NMR-Spektren
wurden an einem Varian Mercury 300-Spektrometer (Palo Alto, CA)
bei Umgebungstemperatur in dem spezifizierten Lösungsmittel erhalten. Schmelzpunkte
wurden unter Verwendung einer Fisher-Johns-Schmelzpunktapparatur
bestimmt und sind unkorrigiert. FT-IR-Spektren wurden an einem Nicolet
Protege 460-Spektrometer (Madison, WI) aufgezeichnet. GC/MS-Spektren
wurden mit einem HP 6890-GC-System (Palo Alto, CA), das mit einem massenselektiven
Detektor HP 5973 ausgestattet war, erhalten.
-
MS(EI)-Spektren
wurden an einem HP Series 1100 LL/MSD erhalten.
-
Das
allgemeine Syntheseschema für
die Herstellung von LFM und LFM-A1 bis LFM-A14 ist in 19 dargestellt, wobei LFM, LFM-A1 bis LFM-A12
und LFM-A14 nur zu Vergleichszwecken angegeben sind. Cyanoessigsäure 1 wurde
mit dem gewünschten
Anilin 2 in Gegenwart von Diisopropylcarbodiimid (DIC) unter Bildung
von 3 gekoppelt. Verbindung 3 wurde mit NaH behandelt und dann mit
Acetylchlorid acyliert, wodurch LFM oder LFM-A1 bis LFM-A14 erhalten
wurden.
-
Allgemeine
Syntheseverfahren
-
1,3-Diisopropylcarbodiimid
(1,75 g; 13,9 mmol) wurde zu einer Lösung von Cyanessigsäure 1 (1,70
g; 20,0 mmol) und dem gewünschten
substituierten Anilin 2 (12,6 mmol) in Tetrahydrofuran (25 ml) bei
0°C gegeben.
Das Gemisch wurde für
12 Stunden bei Raumtemperatur gerührt. Das resultierende Harnstoffpräzipitat (Reaktionsnebenprodukt)
wurde durch Filtration entfernt und das Filtrat wurde zwischen Ethylacetat
und 0,5 N HCl verteilt. Die organische Schicht wurde anschließend zweimal
mit Salzlösung
gewaschen, über
wasserfreiem Na2SO4 getrocknet
und durch Rotationsverdampfung konzentriert. Das rohe feste Produkt
wurde aus Ethylalkohol umkristallisiert, wodurch reines 3 erhalten
wurde. Siehe Kuo, E.A. et al. (1996), J. Med. Chem. 39(23), 4608-21
und Sjogren, E.R., et al. (1991), J. Med. Chem. 34, 3295-3301.
-
Natriumhydrid
(0,93 g; 60 % in Mineralöl;
23,2 mmol) wurde langsam zu der Lösung von 3 (12,0 mmol) in Tetrahydrofuran
(40 ml) bei 0°C
gegeben. Nach 30-minütigem
Rühren
bei 0°C
wurde Acetylchlorid (1,04 g; 13,2 mmol) dem Reaktionsgemisch zugesetzt.
Die Reaktion wurde für
eine weitere Stunde bei Raumtemperatur fortgesetzt und durch Zusatz
von Essigsäure
(2 ml) abgeschreckt. Das Gemisch wurde in Eiswasser (100 ml), das
2,5 ml Salzsäure
enthielt, gegossen, um das Rohprodukt auszufällen, welches durch Filtration
gesammelt wurde und mit Wasser gewaschen wurde. Das rohe Endprodukt
wurde durch Umkristallisation erhalten.
-
Physikalische
Daten
-
In
den folgenden Abschnitten werden die physikalischen Daten aller
anderen Verbindungen als LFM-A13 (d.h. LFM, LFM-A1 bis LFM-A12 und
LFM-A14) lediglich zu Vergleichszwecken aufgeführt:
-
α-Cyano-β-hydroxy-β-methyl-N-[4-(trifluormethyl)phenyl]propenamid
(LFM)
-
- Smp.: 230-233°C;
- IR (KBr): 3303, 2218, 1600 und 1555 cm–1;
- 1H-NMR (DMSO-d6) δ: 11,01 (s,
1H, NH), 7,75 (d, J=8,4Hz, 2H, ArH), 7,64 (d, J=8,4Hz, 2H, ArH),
2,22 (s, 3H, CH3);
- GC/MS m/z 270 (M+), 161, 142, 111.
-
α-Cyano-β-hydroxy-β-methyl-N-(4-bromphenyl)propenamid
(LFM-A1)
-
- Smp.: 213-214°C;
- IR (KBr): 3288, 2228, 1615, 1555 cm–1;
- 1H-NMR (DMSO-d6) δ: 10,51 (s, 1H, NH), 7,49 (s,
4H, ArH), 2,25 (s, 3H, CH3);
- MS (EI) m/z 280 (M+).
-
α-Cyano-β-hydroxy-β-methyl-N-(4-chlorphenyl)propenamid
(LFM-A2)
-
- Smp.: 209-211°C;
- IR (KBr): 3298, 2223, 1598 und 1552 cm–1;
- 1H-NMR (DMSO-d6) δ: 10,48 (s,
1H, NH), 7,54 (d, J=8,7Hz, 2H, ArH), 7,45 (s br, 1H, OH), 7,36 (d,
J=8,7Hz, 2H, ArH), 2,25 (s, 3H, CH3);
- MS (CI) m/z 236 (M+), 129, 127.
-
α-Cyano-β-hydroxy-β-methyl-N-(4-fluorphenyl)propenamid
(LFM-A3)
-
- Smp.: 165-166°C;
- IR (KBr): 3298, 2218, 1610 und 1560 cm–1;
- 1H-NMR (DMSO-d6) δ: 10,33 (s,
1H, NH), 7,80 (s br, 1H, OH), 7,53 (m, 2H, ArH), 7,16 (m, 2H, ArH),
2,26 (s, 3H, CH3);
- GC/MS m/z 220 (M+), 111.
-
α-Cyano-β-hydroxy-β-methyl-N-[2-(trifluormethyl)phenyl]propenamid
(LFM-A4)
-
- Smp.: 61-63°C;
- IR (KBr): 3435, 2209, 1619, 1952 und 1548 cm–1;
- 1H-NMR (DMSO-d6) δ: 10,99 (s,
1H, NH), 8,03 (d, J=7,5Hz, 1H, ArH), 7,67 (d, J=7,5Hz, 1H, ArH),
7,60 (dd, J=7,5, 7,5Hz, 1H, ArH), 7,29 (dd, J=7,5, 7,5Hz, 1H, ArH),
5,71 (s br, 1H, OH), 2,20 (s, 3H, CH3);
- GC/MS m/z 270 (M+), 161, 141, 114.
-
α-Cyano-β-hydroxy-β-methyl-N-(2-bromphenyl)propenamid
(LFM-A5)
-
- Smp.: 98-100°C;
- IR (KBr): 3351, 2214, 1609, 1585 und 1536 cm–1;
- 1H-NMR (DMSO-d6) δ: 10,76 (s,
1H, NH), 8,06 (dd, J=8,1, 1,5Hz, 1H, ArH), 7,62 (dd, J=8,1, 1,5Hz,
1H, ArH), 7,03 (m, 1H, ArH), 6,60 (s br, 1H, OH), 2,22 (s, 3H, CH3);
- MS (EI) m/z 280 (M+), 173, 171.
-
α-Cyano-β-hydroxy-β-methyl-N-(2-chlorphenyl)propenamid
(LFM-A6)
-
- Smp.: 93-94°C;
- IR (KBr): 3372, 2208, 1644, 1621 und 1587 cm–1;
- 1H-NMR (DMSO-d6) δ: 10,96 (s,
1H, NH), 8,16 (d, J=8,1Hz, 1H, ArH), 7,46 (dd, J=7,5, 1,5Hz, 1H,
ArH), 7,29 (m, 1H, ArH), 7,08 (m, 1H, ArH), 2,22 (s, 3H, CH3);
- MS (CI) m/z 236 (M+), 129, 127.
-
α-Cyano-β-hydroxy-β-methyl-N-(2-fluorphenyl)propenamid
(LFM-A7)
-
- Smp.: 118-119°C;
- IR (KBr): 3409, 2212, 1613, 1591 und 1532 cm–1;
- 1H-NMR (DMSO-d6) δ: 10,70 (s,
1H, NH), 7,91 (m, 1H, ArH), 7,23 (m, 1H, ArH), 7,13 (m, 2H, ArH),
7,10 (s br, 1H, OH), 2,22 (s, 3H, CH3);
- GC/MS m/z 220 (M+), 111.
-
α-Cyano-β-hydroxy-β-methyl-N-[3-(trifluormethyl)phenyl]propenamid
(LFM-A8)
-
- Smp.: 182-184°C;
- IR (KBr): 3303, 2218, 1619 und 1572 cm–1;
- 1H-NMR (DMSO-d6) δ: 10,79 (s,
1H, NH), 8,05 (s, br, 1H, OH), 8,04 (s, 1H, ArH), 7,75 (d, J=8,1Hz,
1H, ArH), 7,53 (dd, J=8,1, 7,5Hz, 1H, ArH), 7,42 (d, J=7,5Hz, 1H,
ArH), 2,24 (s, 3H, CH3);
- GC/MS m/z 270 (M+), 161.
-
α-Cyano-β-hydroxy-β-methyl-N-(3-bromphenyl)propenamid
(LFM-A9)
-
- Smp.: 184-185°C;
- IR (KBr): 3303, 2228, 1610, 1595 und 1550 cm 1;
- 1H-NMR (DMSO-d6) δ: 10,56 (s,
1H, NH), 7,89 (m, 1H, NH), 7,89 (m, 1H, ArH), 7,47 (m, 1H, ArH),
7,28 (m, 2H, ArH), 6,37 (s br, 1H, OH), 2,26 (s, 3H, CH3);
- MS (EI) m/z 282 (M++H, 81Br),
280 (M++H, 79Br),
173, 171.
-
α-Cyano-β-hydroxy-β-methyl-N-(3-chlorphenyl)propenamid
(LFM-10)
-
- Smp.: 184-187°C;
- IR (KBr): 3293, 2221, 1610, 1595 und 1557 cm–1;
- 1H-NMR (DMSO-d6) Δ: 10,61 (s,
1H, NH), 7,76 (m, 1H, ArH), 7,42 (m, 1H, ArH), 7,33 (dd, J=8,1,
8,4Hz, 1H, ArH), 7,16 (m, 1H, ArH), 6,84 (s br, 1H, OH), 2,25 (s,
3H, CH3);
- MS (CI) m/z 239 (M++H, 37Cl),
237 (M++H 35Cl),
129, 127.
-
α-Cyano-β-hydroxy-β-methyl-N-(3-fluorphenyl)propenamid
(LFM-A11)
-
- Smp.: 136-138°C;
- IR (KBr): 3297, 2221, 1613, 1597 und 1567 cm–1;
- 1H-NMR (DMSO-d6) δ: 10,54 (s,
1H, NH), 7,54 (m, 1H, ArH), 7,33 (m, 2H, ArH), 6,93 (m, 1H, ArH),
2,27 (s, 3H, CH3);
- GC/MS m/z 220 (M+), 111.
-
α-Cyano-β-hydroxy-β-methyl-N-[4-(trifluormethoxy)phenyl]propenamid
(LFM-A12)
-
- Smp.: 182-183°C;
- IR (KBr): 3308, 2213, 1625 und 1580 cm–1;
- 1H-NMR (DMSO-d6) δ: 10,57 (s,
1H, NH), 7,90 (s br, 1H, OH), 7,64 (d, J=8,7Hz, 2H, ArH), 7,32 (d,
J=8,7Hz, 2H, ArH), 2,25 (s, 3H, CH3);
- GC/MS m/z 286 (M+), 177, 108.
-
α-Cyano-β-hydroxy-β-methyl-N-(2,5-dibromphenyl)propenamid
(LFM-A13)
-
- Smp.: 148-150°C;
- IR (KBr): 3353, 2211, 1648 und 1590 cm–1;
- 1H-NMR (DMSO-d6) δ: 11,41 (s,
1H, NH), 8,57 (d, J=2,4Hz, 1H, ArH), 7,55 (d, J=8,7Hz, 1H, ArH),
7,14 (dd, J=8,7, 2,4Hz, 1H, ArH), 7,10 (s br, 1H, OH), 2,17 (s,
3H, CH3);
- MS (EI) m/z 362 (M++4), 360 (M++2), 358 (M+), 253,
251, 249, 150.
-
α-Cyano-β-hydroxy-β-methyl-N-(phenyl)propenamid
(LFM-A14)
-
- Smp.: 134-135°C;
- IR (KBr): 3281, 2214, 1605, 1579 und 1554 cm–1;
- 1H-NMR (DMSO-d6) δ: 10,33 (s,
1H, NH), 7,51 (d, J=7,5Hz, 2H, ArH), 7,40 (s br, 1H, OH), 7,31 (dd,
J=7,5, 7,5Hz, 2H, ArH), 7,11 (m, 1H, ArH), 2,26 (s, 3H, CH3);
- GM/MS m/z 202 (M+), 93.
-
Unter
Verwendung von Verfahren ähnlich
dem oben identifizierten allgemeinen Verfahren wurden auch die folgenden
Verbindungen der Formel (I) hergestellt.
-
α-Cyano-β-hydroxy-β-methyl-N-[4-(methylsulfonyl)phenyl]propenamid,
hergestellt durch Oxidation der entsprechenden 4-Methylthio-Verbindung
mit Peressigsäure
in Essigsäure;
Smp.:
205-206°C;
1H-NMR (DMSO-d6) δ: 11,26 (s,
1), 7,81 (d, 2), 7,76 (d, 2), 3,15 (s, 3), 2,19 (s, 3);
IR
(KBr): 3309, 2225, 1643 und 1586 cm–1;
MS
(EI) m/z 281,0 (M+H+), 172,1.
-
α-Cyano-β-hydroxy-β-methyl-N-[3-(methylsulfonyl)phenyl]propenamid,
hergestellt durch Oxidation der entsprechenden 3-Methylthio-Verbindung
mit Peressigsäure
in Essigsäure;
Smp.:
213-214°C;
1H-NMR (DMSO-d6) δ: 10,98 (s,
1), 8,18 (m, 1), 7,82 (m, 1), 7,60 (m, 2), 3,18 (s, 3), 2,23 (s,
3);
IR (KBr): 3278, 2231, 1607 und 1555 cm–1;
MS
(EI) m/z 281,0 (M+H+), 172,1.
-
α-Cyano-β-hydroxy-β-methyl-N-(3-brom-4-(trifluormethoxy)phenyl]propenamid;
-
- Smp.: 178-179°C;
- 1H-NMR (DMSO-d6) δ: 11,03 (s,
1), 8,12 (d, 1), 7,55 (dd, 1), 7,45 (m, 1), 5,53 (s, 1), 2,20 (s,
3);
- IR (KBr): 3304, 2350, 1620 und 1602 cm–1;
- MS (EI) m/z 365,0 (M+H+), 255,9.
-
α-Cyano-β-hydroxy-β-methyl-N-(2,4-dibromphenyl)propenamid;
-
- Smp.: 181-181°C;
- 1H-NMR (DMSO-d6) δ: 11,03 (s,
1), 8,14 (m, 1), 7,84 (d, 1), 7,51 (dd, 1), 5,65 (s, 1), 2,20 (s,
3);
- IR (KBr): 3360, 2205, 1591 und 1530 cm–1;
- MS (EI) m/z 361,0 (M+H+), 249,9.
-
α-Cyano-β-hydroxy-β-methyl-N-(2,4-dichlorphenyl)propenamid;
-
- Smp.: 143-144°C;
- 1H-NMR (DMSO-d6) δ: 11,23 (s,
1), 8,29 (m, 1), 7,60 (d, 1), 7,35 (dd, 1), 5,17 (s, 1), 2,18 (s,
3);
- IR (KBr): 3371, 2210, 1601 und 1540 cm–1;
- MS (EI) m/z 271,0 (M+H+), 162,0.
-
α-Cyano-β-hydroxy-β-methyl-N-(2,5-dichlorphenyl)propenamid;
-
- Smp.: 143-144°C;
- 1H-NMR (DMSO-d6) δ: 11,70 (s,
1), 8,51 (d, 1), 7,45 (d, 1), 7,05 (dd, 1), 4,97 (s, 1), 2,14 (s,
3);
- IR (KBr): 3370, 2215, 1581 und 1528 cm–1;
- MS (EI) m/z 271,0 (M+H+), 162,0.
-
α-Cyano-β-hydroxy-β-methyl-N-(3,4-dichlorphenyl)propenamid;
-
- Smp.: 216-217°C;
- 1H-NMR (DMSO-d6) δ: 10,74 (s,
1), 7,95 (m, 1), 7,54 (m, 1), 7,47 (m, 1), 5,64 (s, 1), 2,23 (s,
3);
- IR (KBr): 3319, 2225 und 1612 cm–1;
- MS (EI) m/z 271,0 (M+H+), 162,0.
-
Unter
Verwendung eines BTK-Inhibierungs-Assays ähnlich dem in Tabelle 1 beschriebenen,
wurde festgestellt, daß die
obigen Verbindungen als BTK-Inhibitoren wirken und im allgemeinen
IC50-Werte von 20 μm/ml oder weniger haben. Es
wurde auch festgestellt, daß die
Verbindungen Zellen für
eine Apoptose mit Vincristin und Ceramid sensibilisieren, und zwar
unter Verwendung eines Verfahrens ähnlich dem in Beispiel 3 beschriebenen.
-
Beispiel 3:
-
Das
folgende Beispiel liefert Informationen darüber, wie spezifische Inhibitoren
für BTK
entwickelt werden können.
Es wird ein Homologiemodell für
die Kinasedomäne
von BTK offenbart, die für
die Entwicklung und Bestätigung
von BTK-Inhibitoren
nützlich
ist. Die Verwendung des Homologiemodells zur Identifizierung spezifischer
Verbindungen, einschließlich
eines neuen Leflunomid-Metabolitenanalogons, das ein wirksamer und
selektiver BTK-Inhibitor ist, wird ebenfalls offenbart.
-
In
einem Ansatz, wirksame Inhibitoren der anti-apoptotischen Tyrosinkinase
BTK als Mittel gegen Leukämie
mit Apoptosefördernden
und chemosensibilisierenden Eigenschaften zu entwickeln, wurde ein
dreidimensionales Homologiemodell der BTK-Kinasedomäne konstruiert,
wie es nachfolgend vollständiger
beschrieben wird. Modellierungsstudien zeigten eine deutliche rechteckige
Bindungstasche in der Nähe
der Hinge-Region der BTK-Kinasedomäne, wobei Leu460-,
Tyr476-, Arg525-
und Asp539-Reste die Ecken des Rechtecks besetzen.
Die Abmessungen dieses Rechtecks sind etwa 18 Å × 8 Å × 9 Å × 17 Å und die Dicke der Tasche ist
etwa 7 Å.
-
Es
wurden weiterentwickelte Docking-Verfahren für die rationale Planung von
Leflunomid-Metaboliten(LFM)-Analoga mit einer hohen Wahrscheinlichkeit,
günstigerweise
an die katalytische Stelle in der Kinasedomäne von BTK zu binden, verwendet.
Die Verbindung LFM-A13, für
die wir einen Ki-Wert von 1,4 μM errechneten,
inhibierte humane BTK in vitro mit einem IC50-Wert
von 17,2 ± 0,8 μM. Entsprechend
inhibierte LFM-A13 rekombinante BTK, die in einem Baculovirus-Expressionsvektorsystem
exprimiert wurde, mit einem IC50-Wert von
2,5 μM.
Die energetisch günstige
Position von LFM-A13 in der Bindungstasche ist so, daß ihr aromatischer
Ring nahe an Tyr476 ist und ihre Substituentengruppe
zwischen den Resten Arg525 und Asp539 angeordnet ist. Außerdem ist LFM-A13 zu vorteilhaften
Wasserstoff-Bindungswechselwirkungen mit BTK über Asp539-
und Arg525-Reste fähig.
-
Es
wurde auch entdeckt, daß LFM-A13
zusätzlich
zu seiner deutlichen Wirksamkeit in BTK-Kinaseassays ein hochspezifischer
Inhibitor für
BTK ist. Selbst bei Konzentrationen in einer Höhe von 100 μg/ml (–278 μM) beeinträchtigt dieser neue Inhibitor
die enzymatische Aktivität
von anderen Proteintyrosinkinasen, einschließlich JAK1-, JAK3-, HCK-, EGF-Rezeptorkinase
(EGFR) und Insulin-Rezeptorkinase
(IRK) nicht.
-
Entsprechend
der anti-apoptotischen Funktion von BTK verstärkt eine Behandlung von BTK+B-Linien-Leukämiezellen mit LFM-A13 ihre
Empfindlichkeit gegenüber
Ceramid- oder Vincristin-induzierter Apoptose.
-
Kristallstrukturen von
Leflunomid-Metabolit und seinen Analoga
-
Der
Leflunomid-Metabolit (LFM) und zwei seiner Analoga (LFM-A12, LFM-A13) wurden
unter Verwendung verschiedener Lösungsmittel
durch Verdampfungs- oder Flüssigkeits-Flüssigkeits-Diffusion
kristallisiert. LFM und LFM-A12 wurden kristallisiert und anschließend nur
zu Vergleichszwecken analysiert. Röntgendaten von Einkristallen
wurden unter Verwendung eines SMART-CCD-Flächendetektors (Bruker Analytical
X-ray Systems, Madison, WI) mit MoKI-Strahlung (F = 0,7107 Å) gesammelt.
Die Raumgruppe wurde auf der Basis systematischer Absenzen und Intensitätsstatistiken
bestimmt. Eine Lösung
mit direkten Methoden lieferte die meisten der Nicht-Wasserstoffatome
aus der Elektronendichtekarte. Mehrere Vollmatrix-kleinste Quadrate/Differenz-Fourier-Zyklen
wurden durchgeführt,
um die restlichen Nicht-Wasserstoffatome zu lokalisieren. Alle Nicht-Wasserstoffatome
wurden mit anisotropen thermischen Parametern näher bestimmt. Wasserstoffatome wurden
in idealen Positionen angeordnet und als "riding"-Atome mit relativen isotropen Temperaturfaktoren plaziert.
Die Struktur wurde unter Verwendung von Vollmatrixkleinste Quadrate
an F2 verfeinert. Die Kristallstrukturberechnungen wurden unter
Verwendung eines Silicon Graphics INDY R4400-SC-Computers (Silicon Graphics
Inc., Mountain View, CA) oder eines Pentium-Computers unter Verwendung
der SHELXTL V 5.0-Programmfolge (Sheldrick, G., 5.0 Ed., Bruker
Analytical X-ray Systems, Madison, WI) durchgeführt.
-
Konstruktion des Homologiemodells
für die
Kinasedomäne
von BTK
-
Ein
Homologiemodell von BTK wurde konstruiert, indem die Kristallstrukturen
homologer Kinasedomänen
der Proteinkinasen HCK, FGFR, IRK und cAPK verwendet wurden (Sicheri,
F., Moarefi, I. und Kuriyan, J. (1997), Nature 385 (6617), 602-9;
Mohammadi, M., et al. (1997), Science 276 (5314), 955-60; Hubbard,
S.R. (1997), The E. M. B. 0. Journal 16 (18), 5572-5581; und Zheng J.
et al. (1993), Acta Cryst. D49, 362-365). Die Homologiemodellierung
von BTK wurde durchgeführt,
indem zunächst
die Proteinsequenz von BTK (Swiss-Prot # Q06187, Univ. of Geneva,
Genf, Schweiz) von GenBank (National Center for Biotechnology Information,
Bethesda, MD) erhalten wurde. Als nächstes wurde die vernünftigste
Sequenzorientierung zwischen der BTK-Kinase und einer Koordinatenmatrize
bestimmt. Dies geschah, indem zuerst die CI-Koordinaten der Kinasedomänen von
HCK, FGFR, IRK und cAPK unter Verwendung des InsightII-Programms
((1996), Molecular Simulations, Inc., San Diego, CA) übereinandergelegt
wurden, um den besten Gesamtstrukturvergleich bereitzustellen. Alle
vier Sequenzen wurden dann auf der Basis des Übereinanderlegens ihrer Strukturen
(Aminosäuresequenzen
wurden zueinander ausgerichtet, wenn ihre CI-Positionen zueinander
beabstandet waren) ausgerichtet bzw. orientiert. Die Sequenzorientierung
trägt solchen
Merkmalen wie Schleifen in einem Protein, das sich von den anderen
Proteinsequenzen unterscheidet, Rechnung. Die strukturelle Übereinanderlagerung
erfolgte unter Verwendung des Homology module of the Insight II
((1996), Molecular Simulations, Inc., San Diego, CA)-Programms und eines
Silicon Graphics INDIGO2-Computers (Silicon Graphics Inc., Mountain
View, CA). Die Sequenzorientierung wurde manuell auf der Basis der
früher
beschriebenen Betrachtungen eingestellt und produzierte für jede übereinandergelegte
CI-Position ein Sequenzvariationsprofil. Das Sequenzvariationsprofil
diente als Basis für
den nächsten
Arbeitsschritt, der eine Sequenzorientierung aller vier Proteine
mit BTK-Kinase war. Bei diesem Arbeitsgang wurde die Sequenz der
BTK-Kinase in das Programm eingelesen und manuell mit den vier bekannten
Kinaseproteinen auf der Basis des vorstehend beschriebenen Sequenzvariationsprofils
orientiert. Als nächstes
wurde ein Satz von 3D-Koordinaten der BTK-Kinasesequenz zugeordnet,
wobei die 3D-Koordinaten von HCK als Matrize verwendet wurden und
das Homologiemodul innerhalb des Insight II-Programms ((1996), Molecular
Simulations, Inc., San Diego, CA) verwendet wurde. Die Koordinaten
für eine
Schleifenregion, in der eine Sequenzinsertion auftritt (für HCK ohne
die Schleife) wurde aus einer begrenzten Anzahl von Möglichkeiten
ausgewählt,
die automatisch durch das Programm erzeugt wurden und manuell auf
eine idealere Geometrie unter Verwendung des Programms CHAIN (Sack,
J.S. (1988), J. Mol. Graphics 6, 244-245) eingestellt wurden. Schließlich wurde
das konstruierte BTK-Modell einer Energieminimierung unter Verwendung
des Xplor-Programms (Brunger, A.T. (1992), New Haven, CT) unterworfen,
so daß eine
sterische Spannung, die während
des Modellbauprozesses eingeführt wurde,
entspannt werden konnte. Das Modell wurde auf ungünstige sterische
Kontakte durchgemustert und bei Bedarf wurden solche Seitenketten
entweder durch Verwendung einer Rotamer-Bibliothek-Datenbank oder durch
manuelles Drehen der entsprechenden Seitenketten remoduliert. Das
fertige Homologiemodell der BTK-Kinasedomäne hatte nach Energieminimierung
eine RMS-Abweichung von 0,01 Å von
idealen Bindungslängen
und von 2,2° von
idealen Bindungswinkeln. Das Homologiemodell von BTK wurde dann
in Verbindung mit Modellkoordinaten von LFM und seinen Analoga (die
später
mit Kristallstrukturen verglichen wurden) für Modellierungsstudien der
BTK/Inhibitor-Komplexe verwendet.
-
Dockingverfahren unter
Verwendung des Homologiemodells der BTK-Kinasedomäne
-
Eine
Modellierung der BTK/LFM-Analogon-Komplexe erfolgte unter Verwendung
des Dockingmoduls im Programm INSIGHT II und unter Verwendung der
Affinitätsprogrammfolge
für ein
automatisches Docking eines Liganden an den Rezeptor. BTK im Komplex
mit LFM oder LFM-A12 wurde lediglich zu Vergleichszwecken modelliert.
Energie-minimierte Koordinaten wurden für jedes LFM-Molekül erzeugt
und interaktiv in der ATP-Bindungsstelle von BTK auf der Basis der
Quercetin-Position
in HCK/Quercitin-Kristallstruktur verknüpft (Sicheri, F., et al., J.
(1997), Nature 385 (6617), 602-9). Es wurden die Wasserstoffatome
an der Kinasedomäne von
BTK gebildet und vor Beginn des Dockingverfahrens wurden dem Rezeptor
und dem Liganden Potentiale zugeordnet. Das Dockingverfahren im
Insight II-Programm verwendete das CVFF-Kraftfeld und eine Monte Carlo-Suchstrategie,
um nach verknüpften
Strukturen zu suchen und diese zu beurteilen. Während die Koordinaten für die Masse
des Rezeptors fixiert gehalten wurden, wurde eine definierte Region
der Bindungsstelle relaxieren gelassen, wodurch das Protein sich
auf die Bindung verschiedener Inhibitoren einstellen konnte. Innerhalb
eines Abstands von 5 Å vom
Inhibitor wurde ein Bindungssatz definiert, wobei sich Reste innerhalb dieses
Abstands in energetisch günstige
Positionen verschieben und/oder rotieren konnten, um den Liganden anzupassen.
Es wurde eine Anordnung definiert, die aus dem Rezeptor- und Inhibitormolekül besteht
und ein Docking wurde unter Verwendung des festgelegten Dockingmodus
durchgeführt.
Es wurden Berechnungen, die hydrophobe und hydrophile Wechselwirkungen
nähern,
verwendet, um die zehn besten Dockingpositionen für jedes
LFM-Analogon in der katalytischen BTK-Stelle zu bestimmen. Die verschiedenen
Docking-Positionen jedes LFM-Analogon wurden qualitativ beurteilt,
wobei Ludi (Bohm, H.J. (1992), J. Comput. Aided. Mol. Des. (6(6),
593-606 und Bohm,
H.J. (1994), J. Comput. Aided Mol. Des. 8(3), 243-56) in INSIGHT
II verwendet wurde, das eingesetzt wurde, um eine Bindungskonstante
(Ki) für
jede Verbindung zu bestimmen, um ihre relativen Bindungsfähigkeiten
und die vorausgesagte BTK-Inhibierung zu beurteilen. Die Ki-Trends für die LFM-Analoga wurden mit
dem Trend der experimentell bestimmten Tyrosinkinase-Inhibierungs-IC50-Werten für die Verbindungen verglichen,
um die Struktur-Aktivitäts-Beziehungen (SAR)
zu klären,
welche die Wirksamkeit von LFM-Analoga
bestimmen.
-
Rekombinante Baculovirus-Konstruktion
und Protein-Expression
-
Sf21
(IPLB-SF2I-AE)-Zellen (Vassilev, A., et al. (1998), J. Biol. Chem.,
274, 1646-1656), die aus dem Eierstockgewebe von Spodotera frugiperda
stammen, wurden von Invitrogen erhalten und bei 26-28°C in Grace's-Insektenzellmedium,
ergänzt
mit 10 % FBS und 1,0 % Antibiotikum/Antimykotikum, (GIBCO-BRL) gehalten.
Stammzellen wurden mit 0,2 bis 1,6 × 106/ml
in 600 ml Gesamtkulturvolumen in 1 l-Bellco-Zentrifugenkolben bei
60 bis 90 Upm gehalten. Die Zellebensfähigkeit wurde zu 95 bis 100
% aufrechterhalten, was durch Tryptanblau-Farbstoffausschluß bestimmt wurde.
-
Rekombinantes
Baculovirus, das das murine BTK-Gen enthielt, wurde wie beschrieben
konstruiert (Vassilev A., et al. (1998), J. Biol. Chem. 274, 1646-1656).
Kurz ausgedrückt,
das Gen, das für
BTK codiert, wurde aus pBluescript SKII+-Vektor
(Stratagene, La Jolla, CA)-Verdau mit BamHI ausgeschnitten und dieses Fragment
wurde dann in pFastBac1 (Gibco-BRL) ligiert. Der resultierende Vektor,
pFastBacI-BTK, wurde dann verwendet, um das rekombinate Baculovirus
durch ortsspezifische Transposition in E. coli-DH10Bac-Zellen (Gibco-BRL),
die einen Baculovirus-Shuttle-Vektor beherbergen (bacmid), bMON14272,
zu erzeugen. Die resultierende rekombinante bacmid-DNA wurde durch
Transfektion nach dem Liposomen-vermittelten Standardverfahren unter
Verwendung von Cellfectin-Reagens (Gibco-BRL) in Insektenzellen
eingeführt.
Vier Tage später
wurden die Transfektionsüberstände für eine anschließende Plaque-Reinigung
geerntet und wie oben analysiert. Kinase-tote BTK wurde gebildet
wie es beschrieben ist (Vassilev, A. et al. (1998), J. Biol. Chem.,
274, 1646-1656)
und in den Baculovirus-Expressionsvektor kloniert, wie es oben für die Wildtyp-BTK
beschrieben wurde. Baculovirus-Expressionsvektoren
für JAK1-
und JAK3-Kinase wurden konstruiert und in Insektenzellen eingeführt, wie
es vorher beschrieben wurde (Goodman, P.A. et al. (1998), J. Biol.
Chem. 273, 17742-48).
-
Immunpräzipitation
von rekombinanten Proteinen aus Insektenzellen
-
Sf21-Zellen
wurden mit einem Baculovirus-Expressionsvektor für BTK, JAK1 oder JAK3 infiziert,
wie es in der Kurzbeschreibung der Figuren angegeben ist. Zellen
wurden geerntet, lysiert (10 mM Tris pH 6,7, 100 mM NaCl, 1 % Nonidet
P-40, 10 % Glycerin, 50 mM NaF, 100 mM Na3V04, 50 μg/ml Phenylmethylsulfonylfluorid,
10 μg/ml
aprotonin, μg/ml
Leupeptin); und die Kinasen wurden aus den Lysaten immunpräzipitiert,
wie es in der Literatur beschrieben ist (Vassilev, A., et al. (1999),
J. Biol. Chem. 274, 1646-1656). Antikörper, die für Immunpräzipitationen aus Insektenzellen
verwendet wurden, sind folgende: polyklonale Kaninchen-Anti-BTK-Serum-Antikörper (Mahajan,
S. et al. (1995), Mol. Cell. Biol. 15, 5304-11), polyklonale Kaninchen-Anti-JAK1
(HR-785)-Antikörper, Kat.#
sc-277, polyklonales Kaninchen-IgG, affinitätsgereinigt, 0,1 mg/ml, Santa Cruz
Biotechnology und polyklonaler Kaninchen-Anti-JAK3-Antikörper (C-21,
Kat.# sc-513, polyklonales Kaninchen-IgG, affinitätsgereinigt,
0,2 mg/ml, Santa Cruz Biotechnology). Nachdem die immunpräzipitierten
Tyrosinkinasen für
1 Stunde den Testverbindungen ausgesetzt worden waren, wurden Kinase-Assays, wie detailliert
anderswo beschrieben, durchgeführt
(Mahajan, S. et al. (1995), Mol. Cell. Biol. 15, 5304-11 und Uckun, F.M.
et al. (1996), Science 22, 1096-1100). Die Immunpräzipitate
wurden einer Western-Blot-Analyse unterzogen, wie es vorher beschrieben
wurde (Vassilev, A. et al. (1998), J. Biol. Chem. 274, 1646-1656).
-
Zellinien,
Reagenzien und biochemische Assays Die Entwicklung und Charakterisierung
der DT-40-Lymphom-B-Zellinie
wie auch der BTK-defizienten DT-40 und ihrer Derivate, die mit Wildtyp-
oder mutanter humaner BTK rekonstituiert worden waren, wurde bereits
früher
beschrieben (Uckun, F.M. et al. (1996), Science 22, 1096-1100).
Gleiche Mengen an BTK-Protein wurden in allen BTK-defizienten DT-40-Klonen, die mit Wildtyp-
oder mutierten humanen BTK-Genen transfiziert worden waren, nachgewiesen,
allerdings war kein BTK-Protein in den nicht-transfizierten BTK-defizienten
DT-40-Zellen nachweisbar
(Uckun, F.M. et al. (1996), Science 22, 1096-1100). Alle Zellinien,
die von der Hühner-B-Zellinie
DT-40 stammten,
wurden in RPMI 1640-Medium, ergänzt
mit 10 hitzeinaktiviertem fötalem
Rinderserum, 1 % hitzeinaktiviertem Hühnerserum, 2 mM Glutamin, Penicillin
und Streptomycin, gehalten. Zellen wurden bei 37°C in wassergesättigter
Atmosphäre mit
5 % CO2 wachsen gelassen. Die BTK-positiven,
humanen B-Abstammungs-Leukämiezellinien
NALM-6 und ALL-1 wurden in RPMI 1640-Medium, ergänzt mit 10 % hitzeinaktiviertem
fötalem
Rinderserum, gehalten (Uckun, F.M. et al. (1995), Science 267, 886-91).
Die Affennieren-Zellinie COS-7 und die humane Hepatom-Zellinie HepG2
wurden von der ATCC erhalten.
-
Antikörper die
gegen BTK, JAK1, JAK3 und HCK gerichtet sind, wurden früher bereits
beschrieben (Mahajan, S. et al. (1995), Mol. Cell. Biol. 15, 5304-11;
Vassilev, A. et al. (1998), J. Biol. Chem , 274, 1646-1656: Goodman,
P.A. et al. (1998), J. Biol. Chem. 273, 17742-48; und Uckun, F.M.
et al. (1996), Science 22, 1096-1100). Polyklonale Antikörper gegen
BTK wurden durch Immunisierung von Kaninchen mit Glutathion S- Transferase (GST)-Fusionsproteinen
(Pharmacia Biotech Inc.), die die ersten 150 Aminosäuren von
BTK enthalten, gebildet. Der monoklonale Anti-Fas-Antikörper (F22120)
wurde von Transduction Laboratories, Inc. (Lexington, KY) erhalten.
Immunpräzipitationen,
Immunkomplex-Proteinkinase-Assays und Immunblotting unter Verwendung
des ECL-Chemilumineszenz-Detektionssystems
(Amersham Life Sciences) wurden durchgeführt, wie es in der Literatur
beschrieben ist (Mahajan, S. et al. (1995), Mol. Cell. Biol. 15,
5304-11; Vassilev, A. et al. (1998), J. Biol. Chem., 274, 1646-1656:
Goodman, P.A. et al. (1998), J. Biol. Chem. 273, 17742-48; und Uckun,
F.M. et al. (1996), Science 22, 1096-1100). Nach der Elektrophorese
wurden die Kinasegele auf Whatman 3M-Filterpapier getrocknet und einem Phosphoimaging
an einem molekularen Bildgeber (Bio-Rad, Hercules, CA) sowie einer
Autoradiographie auf Film unterzogen. In ähnlicher Weise wurden alle
chemilumineszenten BTK-Western-Blots einem dreidimensionalem densitometrischen
Scanning unterzogen, wobei der Molecular Imager und ein Imaging
Densitometer unter Verwendung der Molecular Analyst/Macintosh-Version 2.1-Software nach den
Beschreibungen des Herstellers (Bio-Rad) verwendet wurden. Für jede Wirkstoffkonzentration
wurde ein BTK-Kinase-Aktivitätsindex
bestimmt, in dem die Verhältnisse
der Kinaseaktivität
in Phosphorimager-Einheiten (PIU) und die Dichte der Proteinbanden
in densitometrischen Scanningeinheiten (DSU) mit denen der Basislinienprobe
verglichen wurden und die folgende Formel verwendet wurde: Aktivitätsindex =
[PIU der Kinasebande/DSU der BTK-Proteinbande]Testprobe: [PIU der Kinasebande/DSU der BTK-Proteinbande]Basislinienkontrollprobe. GST-IGα wurde manchmal
als exogenes Substrat für
BTK-Immunkomplex-Proteinkinase-Assays
verwendet, wie es beschrieben wurde. (Mahajan, S. et al. (1995),
Mol. Cell. Biol. 15, 5304-11). Meerrettichperoxidase-konjugierte
Schaf-Anti-Maus-, Esel-Anti-Kaninchen-sekundärer Antikörper und ECL-Reagenzien
wurden von Amersham (Oakbrook, IL) bezogen. Für Insulin- Rezeptorkinase (IRK)-Assays wurden humane
HepG2-Hepatomzellen,
die zu etwa 80 % Konfluenz gwachsen waren, einmal mit serumfreiem
DMEM gewaschen und für
3 Stunden bei 37°C
in einem CO2-Inkubator ausgehungert. Anschließend wurden
die Zellen mit Insulin (Eli Lilly, Kat.# CP-410; 10 Einheiten/ml/10 × 106 Zellen) für 10 Minuten bei Raumtemperatur
stimuliert. Nach diesem IRK-Aktivierungsschritt
wurden die Zellen einemal mit serumfreiem Medium gewaschen, in NP-40-Puffer
lysiert und IRK wurde aus den Lysaten mit einem Anti-IRb-Antikörper (Santa Cruz,
Kat.# sc-711, polyklonales IgG) immunpräzipitiert. Vor Durchführung der
Immunkomplexkinase-Assays wurden die Perlen mit dem Kinasepuffer
(30 mM Hepes pH 7,4, 30 mM NaCl, 8 mM MgCl2 und
4 mM MnCl2) äquilibriert.
-
Für HCK-Kinase-Assays
verwendeten wir HCK-transfizierte COS-7-Zellen. Die Klonierung und Expression
von HCK in COS-7-Zellen
wurde früher
beschrieben (Saouaf, S.J., et al. (1995), J. Biol. Chem. 270, 27072-8).
Das pSV7c-HCK-Plasmid wurde in 2 × 106 COS-7-Zellen
transfiziert, wobei Lipofectamin (GIBCO/BRL) verwendet wurde; die
Zellen wurden 48 Stunden später
geerntet. Die Zellen wurden in NP-40-Puffer lysiert und HCK wurde
aus den Ganzzelllysaten mit einem Anti-HCK-Antikörper immunpräzipitiert.
-
Apoptose-Assays
-
Um
eine Apoptose zu induzieren, wurden Zellen mit einem agonistischen
Anti-Fas/APO-1-Antikörper (Bender
MedSystems, City/State, Lot. 04/1295) mit 0,1 μg/ml und 0,5 μg/ml Endkonzentrationen,
mit Vincristin (Vincristinsulfat, USP; Pharmacia, NDC 0013-7466-86,
Lot VCB019) mit 10 ng/ml und 100 ng/ml als Endkonzentrationen oder
C2-Ceramid (Biomol, Lot M8107) mit 10 μM, 50 μM und/oder 100 μM als Endkonzentrationen
behandelt. Die MC540-Bindung (als früher Marker für Apoptose)
und die PI-Permeabilität
(als Marker von Apoptose im fortgeschrittenen Stadium) wurden in
DT-40-Zellen gleichzeitig 24 Stunden nach Behandlung mit C2-Ceramid,
Anti-Fas oder Vincristin
gemessen, wie es in der Literatur beschrieben ist (Uckun, F.M. et
al. (1996), Science 22, 1096-1100).
Ganze Zellen wurden unter Verwendung eines FAC-Star Plus-Strömungs-Cytometers
(Becton Dickinson, San Jose, CA) analysiert. Alle Analysen wurden
unter Verwendung einer 488 nm-Anregung auf einem Argonlaser durchgeführt. MC540-
und PI-Emissionen wurden mit einem 600 nm-"Short Pass"-Kaltlichtspiegel
aufgespalten und ein 575 nm-Bandpassfilter wurde vor einer Photomultiplier-Röhre angeordnet,
um die MC540-Emission zu messen, und ein 635 nm-Bandpassfilter wurde
für die PI-Emission
verwendet. Um die Effekte des BTK-Inhibitors auf eine Ceramid-induzierte
Apoptose in BCR-ABLpositive humane ALL-Zellinie ALL-1 zu untersuchen,
wurden Zellen für
4 Stunden bei 37°C
mit 10 μM
C2-Ceramid in Gegenwart oder Abwesenheit des Inhibitors (200 μM LFM-A13)
behandelt. Anschließend
wurden die Zellen gewaschen, mit PI und MC540 gefärbt und
die apoptotischen Fraktionen wurden durch Multiparameter-Strömungscytometrie
bestimmt, wie es in (Uckun, F.M. et al. (1996), Science 22, 1096-1100)
beschrieben ist.
-
Um
eine apoptotische DNA-Fragmentierung zu detektieren, wurden DT-40-Zellen
24 Stunden nach Behandlung mit Anti-Fas, C2-Ceramid oder Vincristin
geerntet. In ähnlicher
Weise wurden B18.2-, NALM-6- und ALL-1-Zellen mit LFM-A13 (100 μM), Vincristin
(VCR) (10 ng/ml), C2-Ceramid (C2-CER) (10 μM), LFM-A13 (100 μM) und VCR
(10 ng/ml), LFM-A13 (100 μM)
+ C2-CER (10 μM) für 24 Stunden
bei 37°C
behandelt. DNA wurde aus Triton-X-100-Lysaten zur Fragmentierungsanalyse
hergestellt (Uckun, F.M. et al. (1996), Science 22, 1096-1100).
Kurz ausgedrückt,
Zellen wurden in hypotonischen 10 mmol/l Tris-HCl (pH 7,4), 1 mmol/1
EDTA, 0,2 % Triton-X-100-Detergens
lysiert und anschließend
mit 11 000 g zentrifugiert. Um eine mit Apoptose assoziierte DNA- Fragmentierung zu
detektieren, wurden Überstände an 1,2
Agarosegel einer Elektrophorese unterworfen und die DNA-Fragmente wurden
nach Färben
mit Ethidiumbromid durch ultraviolettes Licht sichtbar gemacht.
-
BTK ist ein
anti-apoptotisches Enzym
-
Die
anti-apoptotische Aktivität
von BTK wurde beurteilt, indem die Effekte der Apoptose-induzierenden Agenzien
C2-Ceramid, Vincristin
und monoklonaler Anti-Fas-Antikörper
auf Wildtyp-DT-40-Hüher-B-Lymphomzellen
mit denen auf einen BTK-defizienten
Subklon von DT-40-Zellen, die durch homologes Rekombinations-"Knockout"-entwickelt worden
waren, verglichen wurden (Uckun, F.M. et al. (1996), Science 22, 1096-1100).
Es wurde gezeigt, daß Ceramid,
das Produkt von Ceramidsynthase und Sphingomyelinase, als zweiter
Messenger wirkt, der Membran-induzierte apoptotische Signale, einschließlich der
Fas-vermittelten und TNF-Rezeptor-vermittelten Todessignale, zu
stromabwärts
gelegenen Effektoren überträgt (Enari,
M., Hase, A. und Nagana, S. 81995), EMBO J. 14, 5201-5208). Die
Verwendung von Vincristin, einem üblicherweise verwendeten Anti-Leukämie/Lymphom-Arzneimittel,
führt zu
einer zeitabhängigen
Akkumulierung von Ceramid in behandelten Zellen, die zur Apoptose
führt.
An Agarosegelen zeigte DNA aus Triton-X-100-Lysaten von Anti-Fas-behandelten,
BTK-defizienten
DT-40-Zellen ein leiterartiges Fragmentierungsmuster, das mit Apoptose übereinstimmt,
wohingegen in Wildtyp-DT-40-Zellen keine DNA-Fragmentierung beobachtet
wurde (9A, Bahn 7 vs Bahn 14). So
verursachte die Anti-Fas-Antikörper-Behandlung
einen Apoptose in BTK-defizienten DT-40-Zellen, nicht aber in Wildtyp-DT-40-Zellen, was mit der
Tatsache übereinstimmt,
daß BTK
ein Inhibitor des Fas-assoziierten "death inducing signaling"-Komplexes (DISC) ist (Vassilev A., et
al. (1998), J. Biol. Chem. 274, 1646-1656). Die Behandlung von BTK-defizienten
DT-40-Zellen mit
C2-Ceramid (=N-Acetylspingosin; ein synthetisches, zellpermeables
Ceramid-Analogon) oder Vincristin war fähig, die Effekte von Anti-Fas
bei der Induzierung einer oligonukleosomalen DNA-Fragmentierung
bei Agarosegelelektrophorese zu rekapitulieren, wohingegen Wildtyp-DT-40-Zellen
gegenüber
beiden Agenzien resistenz waren, was die Funktion von BTK als negativer
Apoptoseregulator bestätigt
(9A).
-
Um
die Beiteiligung der verschiedenen Domänen von BTK bei ihrer anti-apoptotischen
Funktion zu untersuchen, führten
wird humanes Wildtyp-BTK-Gen wie auch humane BTK-Gene die Mutationen
entweder in der katalytischen Domäne (Arg525 zu
Gln), SH2-Domäne
(Arg307 zu Ala) oder PH-Domäne (Arg28 zu Cys) beherbergten, in die BTK-defizienten
DT-40-Zellen ein (Enari, M. Hase, A. und Nagata, S. (1995), EMBO
J. 14, 5201-5208).
Wie in 9B bewiesen wurde, erleiden
BTK-defiziente DT-40-Zellen,
die mit humanem Wildtyp-BTK-Gen (WT) rekonstituiert waren, nach
Behandlung mit C2-Ceramid (Bahnen 2-4) oder Vincristin (Bahnen 8-10)
keine Apoptose, wohingegen DT-40-Subklone, die humane BTK mit Mutationen
Inder Kinase (K-)- (Bahnen 5-7 und 11-13), SH2 (mSH2)- (Bahnen 15-17
und 21-23) oder PH (mPH)-Domänen
(Bahnen 18-20 und 24-26) dies taten. Demnach sind die Kinase-, SH2-
und PH-Domänen
von BTK für
ihre anti-apoptotische Funktion wichtig.
-
Homologiemodell
der BTK-Kinase-Domäne
-
Die
dreidimensionalen BTK-Koordinaten, die in den Protein/Inhibitor-Modellierungsstudien
verwendet wurden, wurden auf der Basis einer strukturellen Orientierung
mit den bekannten Kristallstrukturen für vier Proteinkinasedomänen (Kinasedomänen von
HCK, FGFR, IRK und cAPK) konstruiert, wie es oben detailliert beschrieben
wurde (Sicheri, F., Moarefi, I. und Kuriyan, J. (1997), Nature 385
(6617), 602-9; Mohammadi, M. et al. (1997), Science 276 (5314),
955-60; Hubbard, S.R. (1997), The E.M.B.O. Journal 16 (18), 5572- 5581; Zheng, J. et
al. (1993), Acta Cryst. D-49, 362-365). Die modellierte BTK-Kinase-Domäne (10A) hat die erwartete Proteinkinasefaltung mit
der katalytischen Stelle in der Mitte, die die Kinase-Domäne in zwei
Lappen aufgeteilt. Sie besteht aus einem kleineren N-terminalen
Lappen, der durch eine flexible Hinge-Region mit dem größeren C-terminalen
Lappen verbunden ist. Der N-terminale Lappen ist reich an β-Strängen, während die C-terminale
Region meist helikal ist. Die katalytische Stelle wird durch zwei β-Faltblätter definiert,
die eine Grenzfläche
an der Spalte zwischen den zwei Lappen bilden. In dieser katalytischen
Region können
kleine Molekülinhibitoren
binden. Unsere Modellierungsstudien zeigten, daß die katalytische Stelle der
BTK-Kinase-Domäne
aus einer getrennten planaren, rechteckigen Bindungstasche nahe
der Hinge-Region besteht. Die rechtwinklige Bindungsregion wird
durch die Reste Leu460, Tyr476,
Arg525 und Asp539 definiert,
die die Ecken des Rechtecks besitzen. Die Abmessungen dieses Rechtecks
sind etwa 18 Å × 8 Å × 9 Å × 17 Å und die
Dicke der Tasche ist etwa 7 Å (10B). Die linke Ecke des Rechtecks kann sichtbar
gemacht werden, da sie nahe an der Hinge-Region bei Leu460 (in 10B gelb gezeigt) beginnt und sich 8 Å nach oben
rechts zu Asp539 (in 10B blau
dargestellt) erstreckt. Dies ist die kürzeste Seite der Bindungstasche
und befindet sich näher zum
inneren Kern des Proteins. Die linke Seite der Tasche, die die längste ist,
erstreckt sich von Leu460 und verläuft 18 Å entlang
der Hinge-Region bis zu Tyr476 (in 10B grün
dargestellt). Die rechte Seite der rechteckigen Tasche erstreckt
sich gegenüber
der Hinge-Region etwa 9 Å von
Asp539 zu Arg525 (in 10B pink dargestellt), was unmittelbar zu den
Bindungsunterstellen für
die Zucker- und Triphosphat-Gruppen von ATP ist. Die Hinge-Region der Bindungsstelle
besteht aus den Resten 472 bis 481. Das Lösungsmittel, das an der vierten
Seite des Rechtecks freigelegt ist, erstreckt sich 17 Å entlang
der schlitzförmigen Öffnung zur
katalytischen Stelle von Tyr476 bis Arg525. Die Bindungstasche ist im Lösungsmittelzugangsbereich
weiter, verengt sich in Richtung der innersten Region der Bindungsstelle
und ist mit einer Dicke von etwa 7 Å insgesamt relativ flach.
Das Volumen der Tasche ist etwa 500 Å3.
-
Während die
meisten Reste der katalytischen Stelle der BTK-Kinase-Domäne im Vergleich zu anderen Tyrosinkinasen
konserviert werden, wurden wenige spezifische Variationen beobachtet.
Die Reste Asn526 und Asp539 (gegenüber der
Hinge-Region) sind
in EGFR, IRK, HCK und BTK konserviert. Der Rest Thr474 in
der Hinge-Region ändert
sich in IRK, JAK1 und JAK3 in Met und der Rest Tyr476 in
der Hinge-Region ändert
sich in EGFR und IRK in Leu. Der Rest Ser538 von
BTK wird in anderen Kinasen nicht konserviert, sondern ändert sich in
JAK1 und IRK zu Gly, in EGFR zu Thr und in FGF-Rezeptor, in JAK3
und HCK in Ala. Eine Region der Bindungsstelle enthält Cys481
in BTK, das hydrophober ist als der entsprechende Rest der PDGF-Rezeptors (Asp),
des FGF-Rezeptors (Asn) und von IRK (Asp). Diese Reste identifizieren
Differenzen, die eine Basis zur Entwicklung selektiver Inhibitoren
der BTK-Kinase-Domäne
liefern.
-
Konzept auf Strukturbasis
und Synthese von LFM-Analoga mit starker BTK-Inhibitoraktivität
-
In
Modellierungsstudien mit dem Ziel, LFM-Analoga mit hoher Wahrscheinlichkeit,
günstigerweise
an die katalytische Stelle der BTK-Kinase-Domäne zu binden, entschieden wir
uns, die bestimmten Ki-Werte zu beurteilen,
die die vorausgesagten Bindungswechselwirkungen zwischen dem Inhibitor
und den Resten in der katalytischen Stelle von BTK quantitativ angeben.
Jedes der kleinen Moleküle
der LFM-Analoga wurde in der katalytischen Stelle der BTK-Kinase-Domäne einsezlen
modelliert, wobei ein weiterentwickeltes Dockingverfahren angewendet
wurde (siehe experimentelles Verfahren, das oben beschrieben wurde).
Die Quercetin-Position in der HCK-Kristallstruktur (Sicheri et al., 1997,
Nature 382:602) wurde als Matrize verwendet, um einen vernünftigen
Ausgangspunkt für
das Dockingverfahren zu erhalten. Die verschiedenen verknüpften Positionen jedes
LFM-Analogon wurden quantitativ beurteilt, wobei ein Punktbewertungsverfahren
verwendet wurde, danach wurden sie mit den IC50-Werten
der Verbindungen in zellfreien BTK-Inhibierungs-Assays verglichen.
Tabelle 1 zeigt die Wechselwirkungspunktbewertungen, die berechneten
Ki-Werte und gemessen IC50-Werte
für LFM
und seine Analoga mit BTK, die Daten für LFM, LFM-A1 bis LFM-A12 und
LFM-A14, die lediglich zu Vergleichszwecken angegeben sind.
-
Die
Inhibitoren in unseren Moedllierungsstudien waren zwischen zwei
Regionen der am stärksten
hydrophoben Reste angeordnet. Die Region oberhalb des gebundenen
Inhibitors bestand aus den Resten Leu408, Val416 und Lys430 und
die Reste unterhalb des gebundenen Inhibitors umfaßten die
BTK-Reste Leu528, Ser538, Gly480 und Cys481. Von
allen beschriebenen Verbindungen, die in unseren Modellierungsstudien
beurteilt wurden (Tabelle 1), sagten wir voraus, daß die Verbindung
LFM-A13 die stärkste
Bindung an BTK liefern würde. Die
Positionen der kritischen Reste in der aktiven Stelle der BTK und
die Docking-Position der Verbindung LFM-A13 ist in 11 gezeigt. Von allen möglichen Orientierungen dieses
Moleküls,
das an die katalytische Stelle gebunden ist, zeigte die in 11 dargestellt die höchste Wechselwirkungsbewertung
mit BTK. Dieser hohe Wechselwirkungsbewertungswert ist für einen
energetisch begünstigten
Bindunsmodus mit entsprechend niedrigem berechneten Ki-Wert
von 1,4 TM hinweisend. Diese Bindungsposition von LFM-A13 ist so, daß der aromatische
Ring des Inhibitors dem Tyr476-Rest gegenüberliegt
und sich die flexible Seitenkette in Richtung der Asp539-
und Arg525-Reste erstreckt. Der aromatische
Ring ist zwischen den hydrophoben Resten Leu408 und
Val416 oben und Gly480 und
Leu528 unten angeordnet. Der Rest Ser538 liegt unter der flexiblen Seitenkette
des Inhibitors und das Ende der Seitenkette liegt zwischen den Resten
Asp539 und Arg525.
Diese Position scheint der des ATP-Analogons sehr ähnlich zu
sein, die in der IRK-Komplex-Kristallstruktur
gefunden wurde (Hubbard, EMBO J, 16 (18): 5572-5581, 1977). Nach
unseren Modellierungsstudien würde
das O3-Atom in der Hydroxyl-Gruppe von LFM-A13 eine Wasserstoffbindung
mit Asp539:O bilden und sein O4-Atom würde eine Wasserstoffbindung
mit Arg525:N bilden.
-
12 stellt die übereinandergelegten
verknüpften
Positionen von LFM-A13 in der katalytischen Stelle von BTK zusammen
mit den Verbindungen LFM und LFM-A12 zum Vergleich dar. Die Moleküle LFM und LFM-A12
sind so verknüpft,
daß sie
entlang der Hinge-Region liegen, was der Quercetin-Position in der HCK-Kristallstruktur
entspricht. Der aromatische Ring dieser Moleküle befindet sich in der Nähe von Tyr476 und das Ende der Seitenkette liegt zwischen
den Resten Asp539 und Thr474.
Die CF3-Gruppe in diesen Molekülen zeigt
zu der Lösungsmittel-Zugangsregion
und ist von Leu408 oben und G1y480 unten
umgeben. Die OH-Gruppe von LFM ist an ein Sauerstoffatom von Asp539 Wasserstoff-gebunden und für LFM-A12 ist dieselbe
Gruppe an ein Sauerstoffatom von Thr474 Wasserstoff-gebunden.
Alle in Tabelle 1 aufgelisteten LFM-Analoga, ausgenommen LFM-A13, liegen
entlang der Hinge-Region wie LFM oder LFM-A12 und ihren Seitenketten
liegen zwischen Asp525 und Thr474.
-
Ein
Vergleich von LFM, LFM-A12 und LFM-A13 bezüglich der aktiven BTK-Stelle
zeigt, daß,
obgleich der aromatische Teil der drei Moleküle grob in derselben Region
ist (was auch für
die anderen inaktiven LFM-Analoga gilt), ist die Seitenkette von
LFM-A13 von denen der anderen weggeneigt und liegt zwischen den Resten
Asp539 und Arg525.
Diese Rotation resultiert wahrscheinlich aus einer günstigen
Orientierung der 2,5-Dibrom-Gruppen von LFM-A13. Diese leicht geneigte Position
und die größeren Br-Gruppen
bieten zwei Vorteile für
die Wechselwirkung von LFM-A13 mit der aktiven Stelle der Reste
von BTK.
-
Der
erste Vorteil ist der, daß LFM-A13
fähig ist,
zwei Wasserstoffbindungen mit den Resten Asp539 und Arg525 der aktiven Stelle zu bilden, wohingegen
die inaktiven LFM-Analoga
nur eine Wasserstoffbindung mit jeweils Thr474 oder
Asp539 von BTK bilden. Der zweite Vorteil
für die
Bindung ist der höhere
Kontaktbereich von LFM-A13 mit den Resten der aktiven Stelle von
BTK im Vergleich zu den anderen 12 inaktiven LFM-Analoga, was zu
einer stärkeren
hydrophoben Wechselwirkung für
LFM-A13 führt.
Diese Merkmal wird durch die entsprechend höhere lipophile Punktbewertung
für LFM-A13
in Tabelle 1 wiedergegeben.
-
Die
Resultate aus den oben diskutierten Modellierungsstudien stützten die
Hypothese, daß LFM-A13 starke
BTK-Inhibitor-Aktivität aufweisen
würden.
Um diese Hypothese zu untersuchen und den Voraussagewert des beschriebenen
BTK-Homologiemodells zu bestätigen,
synthetisierten wird LFM-A13, LFM und 11 andere LFM-Analoga, die
in Tabelle 1 aufgelistet sind. Wiederum werden LFM und alle anderen
Analoga als LFM-A13 lediglich zu Vergleichszwecken synthetisiert
und analysiert. Die Strukturen von LFM, LFM-A12 und LFM-A13 wurden
durch Einkristall-Röntgenbeugung
bestimmt. Es wurde festgestellt, daß alle Strukturen eine planare
Konformation haben und alle Bindungslängen und -winkel im erweiterten
Bereich lagen.
-
13 zeigt eine ORTEP-Darstellung der Verbindung
LFM-A13. Die Kristallstruktur von LFM-A13 zeigte, daß ihre Molekülkonformation
sehr ähnlich
zu den energieminimierten molekularen Koordinaten war, die für Docking-Studien
mit BTK erzeugt und verwendet wurden. Die Konformationsähnlichkeit
mit den Kristallstrukturen zeigt an, daß die zum Docking verwendeten
Molekülmodelle
für Modellierungsstudien
geeignet waren.
-
Spezifische Inhibierung
von BTK durch LFM-A13
-
Zellfreie
Immunkomplex-Kinase-Assays wurden verwendet, um die Wirkungen von
LFM und 12 LFM-Analoga auf die enzymatische Aktivität von humanter
BTK, die aus B18-2-Zellen (Uckun, F.M. et al. (1996), Science 22,
1096-1100) (d.h. BTK-defizienten DT-40-Hühner-Lymphom-B-Zellen, die
mit humanem Wildtyp-BTK-Gen
rekonstituiert worden waren) immunpräzipitiert worden waren. Wie
in Tabelle 1 gezeigt ist, wies nur LFM-A13 deutliche BTK-Inhibitoraktivität mit einem
IC50-Wert von 6,2 ± 0,3 μg/ml (= 17,2 ± 0,8 μM) auf. Keine
der anderen Verbindungen, die nur zum Vergleich enthalten sind,
inhibierte BTK selbst bei hohen Konzentrationen wie 100 μg/ml (d.h.
im Bereich von 349 μM
für LFM-A12
bis 495 μM
für LFM-A14).
-
LFM-A13
war ebenfalls gegen rekombinante BTK, exprimiert in einem Baculovirus-Vektor-Expressionssystem,
mit einem IC50-Wert von 0,9 Tg/ml (–2,5 TM, 14A) wie auch BTK, das aus Zelllysaten der humanen
Zellinie NALM-6-B-ALL (14B)
wirksam. Darüber
hinaus resultierte eine Behandlung von B18.2-Zellen (14C) oder NALM-6-Zellen (Daten nicht gezeigt)
mit LFM-A13 zu einer dosisabhängigen
Inhibierung der zellulären
BTK-Aktivität.
Die Inhibitoraktivität
von LFM-A13 gegen BTK war spezifisch, da sie die enzymatische Aktivität von anderen
Protein-Tyrosinkinasen, einschließlich Janus-Kinasen JAK1 und JAK2, der Kinase HCK
der Src-Familie und der Tyrosinkinase der Rezeptorfamilie IRK, bei
Konzentrationen in einer Höhe von
100 μg/ml
(–278 μM; Tabelle
2, 15) nicht beeinträchtigte.
-
Strukturelle
Basis für
die BTK-Spezifität
von LFM-A13 Biologische Assays haben gezeigt, daß LFM-A13 ein selektiver Inhibitor
von BTK ist, wohingegen es ein schlechter Inhibitor für EGFR,
HCK, JAK1, JAK3 und IRK ist. Um diese Selektivität zu bewerten, konstruierten
wir ein Homologiemodell für
EGFR, JAK1 und JAK3 unter Verwendung der homologen Kristallstrukturkoordinaten
der Proteinkinasen IRK, HCK und cAPK als Matrize. Die Modelle wurden
dann verwendet, um die Bindung kleiner Moleküle, z.B. LFM-A13 in den katalytischen
Stellen dieser Kinasen zu untersuchen und um besser zu verstehen,
wie LFM-A13 BTK inhibieren kann, nicht aber EGFR, IRK, JAK1, JAK3
pder HCK inhibiert. Diese Studien identifizierten drei Faktoren, die
zur Spezifität
von LFM-A13 für
BTK beitragen können.
-
Das
kleine Molekül
LFM-A13 wurde in den Kinase-Domänen
von IRK, HCK, JAK3 und EGFR gebunden. Tabelle 2 zeigt die Wechselwirkungspunktbewertungen,
berechnete Ki-Werte und gemessene IC50-Daten für LFM-A13 mit diesen Kinasen.
Wir pstulieren, daß die
Selektivität
von LFM-A13 für
BTK aus günstigen Wechselwirkungen
mit den Resten der katalytischen Stelle von BTK resultiert, welche
in den anderen untersuchten Kinasen nicht vorliegen. Es gibt einige
Reste in der aktiven BTK-Stelle, die sich von denen anderer PTKs
unterscheiden. Diese Differenzen sind in 16 dargestellt,
die die Hauptkette der katalytisch der BTK-Stelle, die Differenzen
bei den Resten zwischen BTK und anderen Kinasen und die Docking-Positionen von
LFM-A13 in den Kinase-Domänen
von BTK, HCK, JAK3, JAK1, EGFR und IRK darstellt.
-
Es
wird angenommen, daß die
Unterschieden in den Resten, die in Positionen A, B und C in 16 gezeigt sind, zur Spezifität von LFM-A13 für BTK beitragen
können.
Kinasen, die durch LFM-A13 nicht inhibiert werden können, z.B.
IRK (grau) und JAK1/JAK3 (pink) enthalten in Position A einen Methionin- Rest, der in die aktive
Stelle hineinragt und den engen Kontakt von kleinen Molekülen wie
LFM-A13 mit der Hinge-Region
der Bindungsstelle verhindert. Als Resultat kann LFM-A13 einen günstigen
hydrophoben Kontakt mit der Hinge-Region der Kinase-Domänen dieser
Proteine verlieren und bindet nicht fest daran. Allerdings zeigten
Docking-Untersuchungen, daß die
günstige
Position von LFM-A13 in der BTK-Kinase-Domäne so ist, daß die Seitenkette
des kleinen Moleküls
zwischen den Resten Asp539 und Arg525 lokalisiert ist und mit diesen Wasserstoffbindungen
bildet.
-
Außerdem verstärkt ein
hydrophober Rest in Position B von BTK die hydrophobe Wechselwirkung
des LFM-A13-Moleküls
mit dem Rezeptor, eine Wechselwirkung, die in EGFR (rot, 16) und IRK (dunkelblau, 16)
verlorengegangen ist. Während
die Lipo-Punktbewertungen zwischen 457 und 473 für andere Kinasen lagen, war
die Lipo-Punktbewertung für
BTK mit 517 höher
(günstiger).
Schließlich
kann der Arg525-Rest in Position C von BTK
eine Wasserstoffbindung zu LFM-A13 haben. Diese Wechselwirkung geht
in HCK (gelb) verloren, welche ein Ala in der C-Position enthält. Die
günstige
Position von LFM-A13
an der HCK-Kinase-Domäne
(in gelb dargestellt) ist so, daß das kleine Molekül entlang
der Hinge-Region orientiert ist. In dieser Position bildet LFM-A13
keine Wasserstoffbindungen mit HCK, was im Fall für BTK nicht
erfolgt. Die längere
Seitenkette von Arg525 in BTK (Position
C) ist bei der Wasserstoffbindung mit LFM-A13 involviert, wohingegen HCK
ein Ala in dieser Position hat, das nicht fähig ist, dieselbe Wasserstoffbindung
zu bilden.
-
LFM-A13
verstärkt
die Empfindlichkeit akuter Lymphoblastenleukämie (ALL)-Zellen mit B-Abstammung
für eine
Ceramid- und Vincristin-induzierte Apoptose Patienten mit Philadelphia-Chromosom
(Ph+)-ALL haben nach intensiven Multimodalitätsbehandlungsprogrammen ein schlechtes
Ergebnis. Das Versagen einer Behandlung bei diesen Patienten könnte überwunden
werden, wenn die apoptotische Schwelle ihrer Leukämiezellen
gesenkt werden könnten.
Wir versuchten zu bestimmen, ob LFM-A13 durch Inhibierung der anti-apoptotischen
Tyrosinkinase-BTK die Empfindlichkeit der der Ph+ALL-Zellinie ALL-1
für C2-Ceramid ändern könnte. Wie
in 17 gezeigt ist, erhöht eine
Behandlung mit LFM-A13 die Chemosensitivität von ALL-1-Zellen gegenüber Ceramid-induzierter Apoptose
deutlich, wie es durch eine größere prozentuale
Anzahl von Zellen, die mit LFM-A13 plus C2-Ceramid behandelt worden waren, im Vergleich
zu Zellen, die mit C2-Ceramid allein oder LFM-A13 allein behandelt
worden waren, die eine duale PI/MC540 Fluoreszenz (dargestellt in
blauer Farbe) zeigten, was einem fortgeschrittenen Apoptose-Stadium entspricht,
bewiesen wurde. Außerdem zeigte
DNA aus Triton-X-100-Lysaten von B18.2-Hühner-Lymphom-B-Zellen (d.h.
BTK-defizienten DT-40-Zellen, die mit humanem Wildtyp-BTK-Gen rekonstituiert
worden waren; siehe auch 9), human NALM-6-pre-B-ALL-Zellen
und ALL-1-Zellen ein leiterartiges Fragmentierungsmuster, das Apoptose
entsprach, nach Behandlung mit LFM-A13 plus Ceramid oder LFM-A13
plus Vincristin, wobei diese ausgeprägter war als nach Behandlung
mit LFM-A13, Ceramid oder Vincristin allein (18).
Diese Resultate bewiesen, daß LFM-A13
die Empfindlichkeit von Leukämie/Lymphom-B-Zellen
gegenüber
Ceramid-induzierte und Vincristin-induzierter Apoptose verstärkt.
-
Beispiel 4: Modifikationen
an DDE LFM-A13 zur BTK-Inhibierung
-
Das
folgende Beispiel beschreibt, wie das obige Homologiemodell und
Informationen bezüglich
Differenzen zwischen der Bindungsdomäne von BTK und anderen Proteintyrosinkinasen
verwendet wurden, um zusätzliche
Verbindungen der Formel (I), die spezifische BTK-Inhibitoren sind,
zu identifizieren.
-
Strukturelle
und chemische Merkmale von LFM-Analoga, die vorgeschlagen werden,
um die Bindung an die katalytische BTK-Stelle zu unterstützen, werden unten beschrieben
und sind in den 20A bis 20C dargestellt.
Bindungsmodus 1 (20A)–20B zeigt
den wahrscheinlichsten Bindungsmodus der Verbindungen LFM-A13 an
der katalytischen Stelle von BTK. Auf der Basis von Modifikationen
der Verbindung kann auch ein zweiter Bindungsmodus möglich sein,
der in 20C dargestellt ist (Bindungsmodus
2).
-
Tabelle
3 zeigt die Differenzen bei dem Rest an der ATP-Bindungsstelle zwischen den zehn PTKs:
EGFR, Btk, Hck, Jak1, Jak3, IR, FGFR1, PDGFR9, VEGFR2 und Src.
-
-
Ein
Vergleich der Inhibitorbindungstasche von BTK mit der von EGFR,
Hck, Jak1, Jak3, FGFR1, PDGFRβ,
VEGVF2 und Src zeigt, daß SER
538 in der ATP-Bindungsspalte von BTK einzigartig ist (Tabelle 3)
und ein Ziel für
ein spezifisches BTK-Inhibitorenkonzept
darstellt. Liganden, die so konzipiert sind, daß mit diesem Rest wechselwirken,
sollten eine erhöhte
Spezifität
für BTK
haben. 20B zeigt die Abstände (in
Angström) der
NH-, =O- und OH-Gruppe des angekoppelten LFM-13-Liganden aus SER 538. Beim Blick
in die BTK-Bindungsspalte mit dem aromatischen Ring des Liganden
nach außen
gerichtet und der Kette nach Innen gerichtet, erkennt man, daß dieser Rest
unter dem Liganden liegt. Nicht-planare, Einzelkettensubstitutionen,
die mit O oder N enden und die geeigneten Längen (20B)
an den NH-, =O- und OH-Gruppen haben, sollten die Bildung von Wasserstoffbindungen
mit Ser 538 ermöglichen.
Auf der Basis dieser Betrachtung und auf der Modulierung und der
biologischen Daten für
die 13 Leflunomid-Metabolitenanaloga, die oben beschrieben wurden,
können
zusätzliche
Verbindungen der Formel (I) zur besseren Wechselwirkung mit der
Bindungstasche entwickelt werden, von denen erwartet wird, daß sie Inhibitoren
von BTK sind.
-
Beispiel 5: Zweite Generation
an LFM-Analoga
-
Das
folgende Beispiel beschreibt wie neue Verbindungen der Formel (I),
die so konzipiert sind, daß sie auf
SER 538 abzielen, geplant und hergestellt werden.
-
Unter
Verwendung der in den
20A–
20B beschriebenen Konzeptparameter wurden eine
zweite Generation an LFM-Analoga (LFM-A15-20) konzipiert und synthetisiert.
Die Verbindungen haben die folgenden Strukturformeln:
-
Das
folgende Syntheseschema und das in
19 dargestellte
wurden verwendet, um die Verbindungen herzustellen:
-
Syntheseverfahren
A
-
1,3-Diisopropylcarbodiimid
(1,75 g; 13,9 mmol) wurde zu einer Lösung von Cyanessigsäure 1 (1,70
g; 20,0 mmol) und des gewünschten
substituierten Anilin 2 (12,6 mmol) in Tetrahydrofuran (25 ml) bei
0°C gegeben.
Das Gemisch wurde für
12 Stunden bei Raumtemperatur gerührt. Das Harnstoffpräzipitat
(Reaktionsnebenprodukt) wurde durch Filtration entfernt und zwischen
Ethylacetat und 0,5 N HCl verteilt. Die organische Schicht wurde
nacheinander zweimal mit Salzlösung
gewaschen, über
wasserfreiem Na2SO4 getrocknet
und durch Rotationsverdampfung konzentriert. Schließlich wurde
das feste Rohprodukt aus Ethylalkohol umkristallisiert, wobei reine
3 erhalten wurde. Natriumhydrid (0,93 g; 60%ige in Mineralöl; 23,2
mmol) wurde langsam zu der Lösung
von 3 (12,0 mmol) in Tetrahydrofuran (40 ml) bei 0°C gegeben.
Nach Rühren
für 30
Minuten bei 0°C
wurde Acetylchlorid (1,04 g; 13,2 mmol) zu dem Reaktionsgemisch
gegeben. Die Reaktion wurde für
eine weitere Stunde fortgesetzt und wurde dann durch Zusatz von
Essigsäure
(2 ml) abgeschreckt. Das Gemisch wurde in Eiswasser (100 ml), das
2,5 ml Salzsäure
enthielt, gegossen, um das Rohprodukt auszufällen, welches dann durch Filtration
gesammelt wurde und mit Wasser gewaschen wurde. Das reine Produkt
wurde durch Umkristallisation erhalten.
-
Syntheseverfahren
B
-
α-Cyano-β-hydroxy-β-methyl-N-[(methylthio)phenyl]propenamid
(2,48 g, 10,0 mmol) wurde in Essigsäure (150 ml) gelöst und Peressigsäure (8,6
ml einer 32 Gew.%igen Lösung
in Essigsäure)
wurde zugesetzt. Das Gemisch wurde über Nacht bei Raumtemperatur
gerührt
und Wasser (75 ml) wurde zugesetzt. Das Präzipitat wurde filtriert und
mit Wasser gewaschen. Das reine Produkt wurde durch Umkristallisieren
erhalten.
-
Physikalischen
Daten für
die synthetisierten Verbindungen:
-
α-Cyano-β-hydroxy-β-methyl-N-[4-(methylsulfonyl)phenyl]propenamid
-
- Schmelzpunkt: 205-206°C.
- 1H-NMR (DMSO-d6) δ: 11,26 (s,
1H, NH), 7,81 (d, J=9,0Hz, 2H, ArH), 7,76 (d, 9,0Hz, 2H, ArH), 3,15
(s, 3H, SO2CH3).
- IR (KBr): 3309, 2225, 1643 und 1586 cm–1.
- sMS (EI): m/z 281,0 (M+H+), 172,1.
-
α-Cyano-β-hydroxy-β-methyl-N-[3-(methylsulfonyl)phenyl]propenamid
-
- Schmelzpunkt: 213-21406°C.
- 1H-NMR (DMSO-d6) δ: 10,98 (s,
1H, NH), 8,18 (m, 1H, ArH), 7,82 (m, 1H, ArH), 7,60 (m, 2H, ArH),
3,18 (s, 3H, SO2CH3).
- IR (KBr): 3278, 2231, 1607 und 1555 cm–1.
- MS (EI): m/z 281,0 (M+H+), 172,1.
-
α-Cyano-β-hydroxy-β-methyl-N-[3-brom-4-(trifluormethoxy)phenyl]propenamid
-
- Schmelzpunkt: 178-179°C.
- 1H-NMR (DMSO-d6) δ: 11,03 (s,
1H, NH), 8,12 (d, J+2,4Hz, 1H, ArH), 7,55 (dd, J=2,4, 9,0Hz, 1H,
ArH), 7,45 (m, 1H, ArH), 5,53 (s br, 1H, OH), 2,20 (s, 3H, CH3).
- IR (KBr): 3304, 2350, 1620 und 1602 cm–1.
- MS (EI): m/z 365,0 (M+H+), 225,9.
-
α-Cyano-β-hydroxy-β-methyl-N-(2,4-dibromphenyl)propenamid
-
- Schmelzpunkt: 181-182°C.
- 1H-NMR (DMSO-d6) δ: 11,03 (s,
1H, NH), 8,14 (m, 1H, ArH), 7,84 (d, J=2,4Hz, 1H, ArH), 7,51 (dd,
J=2,4, 9,0Hz, 1H, ArH), 5,65 (s br, 1H, OH), 2,20 (s, 3H, CH3).
- IR (KBr): 3360, 2205, 1591 und 1530 cm–1.
- MS (EI): m/z 361,0 (M+H+), 249,9.
-
α-Cyano-β-hydroxy-β-methyl-N-(2,4-Dichlorphenyl)propenamid
-
- Schmelzpunkt: 143-144°C.
- 1H-NMR (DMSO-d6) δ: 11,23 (s,
1H, NH), 8,29 (m, 1H, ArH), 7,6 (d, J=2,4Hz, 1H, ArH), 7,35 (dd,
J=2,4, 9,0Hz, 1H, ArH), 5,17 (s br, 1H, OH), 2,18 (s, 3H, CH3).
- IR (KBr): 3371, 2210, 1601 und 1540 cm–1.
- MS (EI): m/z 271,0 (M+H+), 162,0.
-
α-Cyano-β-hydroxy-β-methyl-N-(2,5-dichlorphenyl)propenamid
-
- Schmelzpunkt: 143-144°C.
- 1H-NMR (DMSO-d6) δ: 11,70 (s,
1H, NH), 8,51 (d, J=3,0Hz, 1H, ArH), 7,45 (d, J=8,7Hz, 1H, ArH),
7,05 (dd, J=3,0, 8,7Hz, 1H, ArH), 4,97 (s br, 1H, OH), 2,14 (s,
3H, CH3).
- IR (KBr): 3402, 2205, 1627 und 1606 cm–1.
- MS (EI): m/z 271 (M+H+), 162,0.
-
Tabelle
4. Wechselwirkungs-Punktbewertungen, geschätzte K
i-Werte und gemessene
IC
50-Daten für LFM-Analoga mit BTK
-
Die
zweite Generation der LFM-Analoga wurde bezüglich der Wechselwirkung mit
dem BTK-Bindungstaschenmodell beurteilt und auch bezüglich der
Inhibitoraktivität
gegenüber
BTK beurteilt, wie es im Beispiel oben beschrieben ist. Die Daten
sind in Tabelle 4 angegeben und beweisen, daß diese neuen Verbindungen
starke Inhibitoren für
BTK sind.
-
Beispiel 7: Neue BTK-Inhibitor-Konzepte
-
21 zeigt die Bindungsmerkmale, die 5,7-Dihydroxy-8-propanoyl-4-propyl-2H-1-benzopyran-2-on (nur
zum Vergleich) und LFM-A13 gemeinsam sind, und zwar auf der Basis
des Dockings der Verbindungen in die ATP-Bindungsstelle von BTK.
Beide Inhibitoren enthalten Wasserstoff-bindende Gruppen (OH, CN),
die so positioniert sind, daß sie
mit Arg 525 und Asp 539 wechselwirken. Verbindungskonzepte, die
die Bindungstasche besser füllen
und günstig
geeigneten Resten in der Tasche wechselwirken, werden entwickelt.
-
Von
diesen Verbindungen wird infolge ihres erhöhten Volumens und der Gruppen,
die dazu bestimmt sind, mit dem Bindungstaschenrest in Wechselwirkung
zu treten, erwartet, daß sie
eine starke BTK-Inhibitoraktivität
haben.
-
-
-
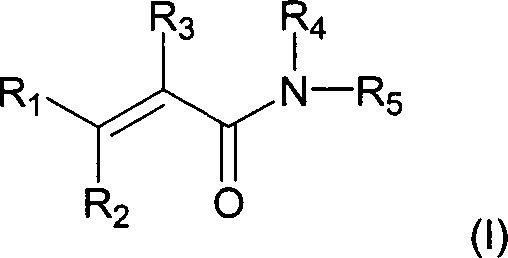
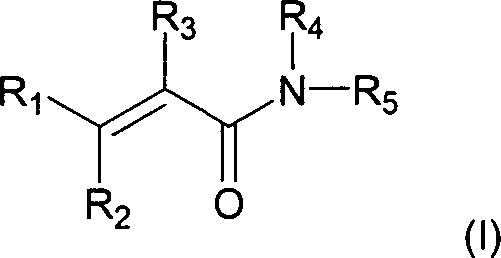
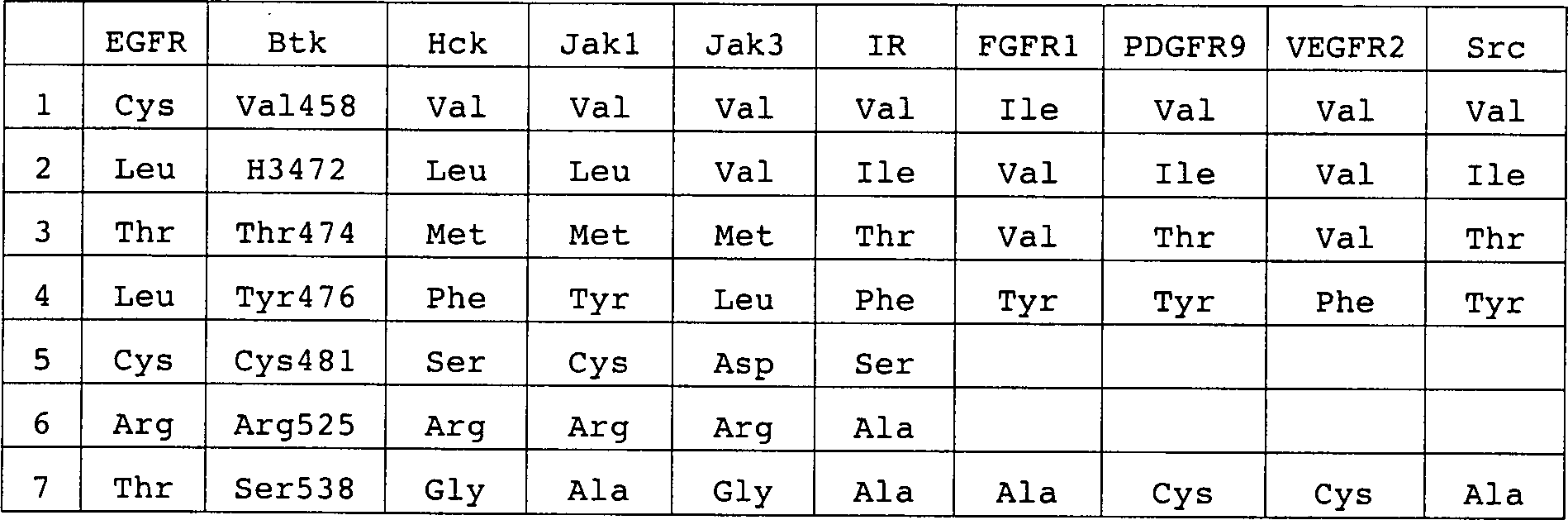




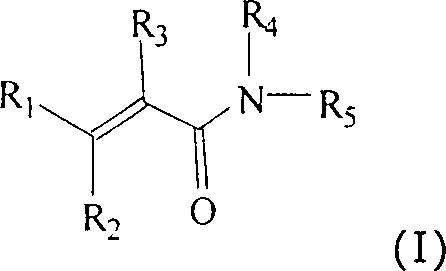
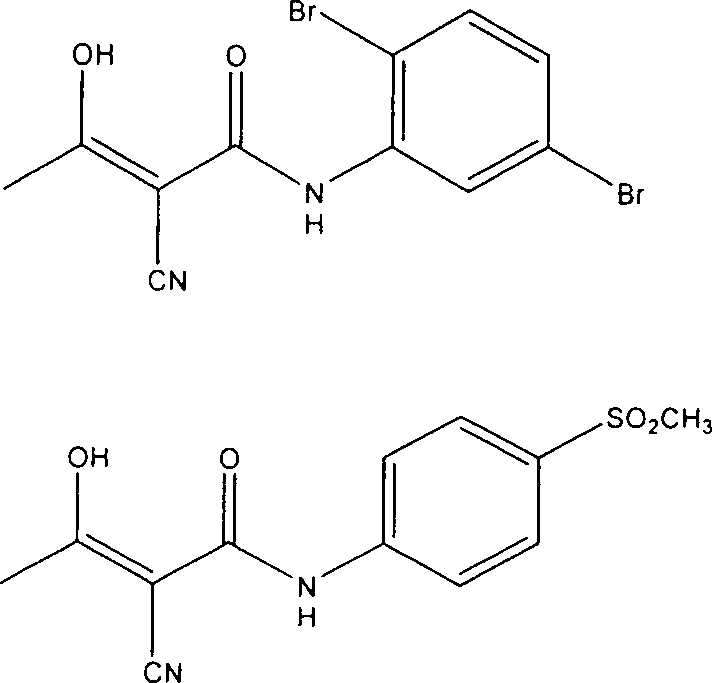 sowie ein pharmazeutisch akzeptables Salz davon umfasst.
sowie ein pharmazeutisch akzeptables Salz davon umfasst.