DE69534718T2 - Substituierte 9-alkyladenine - Google Patents
Substituierte 9-alkyladenine Download PDFInfo
- Publication number
- DE69534718T2 DE69534718T2 DE69534718T DE69534718T DE69534718T2 DE 69534718 T2 DE69534718 T2 DE 69534718T2 DE 69534718 T DE69534718 T DE 69534718T DE 69534718 T DE69534718 T DE 69534718T DE 69534718 T2 DE69534718 T2 DE 69534718T2
- Authority
- DE
- Germany
- Prior art keywords
- pharmaceutically acceptable
- acceptable salt
- endo
- compound according
- compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 150000001875 compounds Chemical class 0.000 claims description 60
- 238000000034 method Methods 0.000 claims description 31
- 150000003839 salts Chemical class 0.000 claims description 27
- 238000002360 preparation method Methods 0.000 claims description 19
- 239000000203 mixture Substances 0.000 claims description 18
- IJGRMHOSHXDMSA-UHFFFAOYSA-N nitrogen Substances N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims description 17
- 125000002868 norbornyl group Chemical group C12(CCC(CC1)C2)* 0.000 claims description 16
- 239000008194 pharmaceutical composition Substances 0.000 claims description 11
- 241000124008 Mammalia Species 0.000 claims description 9
- 229910052757 nitrogen Inorganic materials 0.000 claims description 9
- 229910052739 hydrogen Inorganic materials 0.000 claims description 8
- 230000001225 therapeutic effect Effects 0.000 claims description 8
- 238000002680 cardiopulmonary resuscitation Methods 0.000 claims description 7
- 210000003734 kidney Anatomy 0.000 claims description 7
- 208000009304 Acute Kidney Injury Diseases 0.000 claims description 6
- 108010060263 Adenosine A1 Receptor Proteins 0.000 claims description 6
- 102000030814 Adenosine A1 receptor Human genes 0.000 claims description 6
- 206010022562 Intermittent claudication Diseases 0.000 claims description 6
- 208000033626 Renal failure acute Diseases 0.000 claims description 6
- 230000001154 acute effect Effects 0.000 claims description 6
- 201000011040 acute kidney failure Diseases 0.000 claims description 6
- 125000000217 alkyl group Chemical group 0.000 claims description 6
- 230000001101 cardioplegic effect Effects 0.000 claims description 6
- 208000020832 chronic kidney disease Diseases 0.000 claims description 6
- 208000022831 chronic renal failure syndrome Diseases 0.000 claims description 6
- 230000004217 heart function Effects 0.000 claims description 6
- 239000001257 hydrogen Substances 0.000 claims description 6
- 208000021156 intermittent vascular claudication Diseases 0.000 claims description 6
- 208000004880 Polyuria Diseases 0.000 claims description 5
- 230000004913 activation Effects 0.000 claims description 5
- 230000035619 diuresis Effects 0.000 claims description 5
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 5
- 238000011084 recovery Methods 0.000 claims description 5
- 238000011282 treatment Methods 0.000 claims description 5
- 206010073261 Ovarian theca cell tumour Diseases 0.000 claims description 4
- 208000012998 acute renal failure Diseases 0.000 claims description 4
- 239000003937 drug carrier Substances 0.000 claims description 4
- 230000002401 inhibitory effect Effects 0.000 claims description 4
- 208000001644 thecoma Diseases 0.000 claims description 4
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 3
- 229910052736 halogen Inorganic materials 0.000 claims description 3
- 125000005843 halogen group Chemical group 0.000 claims description 3
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims description 3
- 229910052760 oxygen Inorganic materials 0.000 claims description 3
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 claims description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims description 2
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 claims description 2
- 150000002367 halogens Chemical class 0.000 claims description 2
- 125000005842 heteroatom Chemical group 0.000 claims description 2
- 230000001939 inductive effect Effects 0.000 claims description 2
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims description 2
- 125000002816 methylsulfanyl group Chemical group [H]C([H])([H])S[*] 0.000 claims description 2
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 claims description 2
- 239000001301 oxygen Substances 0.000 claims description 2
- 229910052717 sulfur Inorganic materials 0.000 claims description 2
- 239000011593 sulfur Substances 0.000 claims description 2
- 230000002265 prevention Effects 0.000 claims 2
- 125000004435 hydrogen atom Chemical class [H]* 0.000 claims 1
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 24
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 21
- OIRDTQYFTABQOQ-KQYNXXCUSA-N adenosine Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O OIRDTQYFTABQOQ-KQYNXXCUSA-N 0.000 description 21
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 18
- 239000000243 solution Substances 0.000 description 11
- -1 β-ribofuranosyl rings Chemical group 0.000 description 11
- 239000002126 C01EB10 - Adenosine Substances 0.000 description 10
- 229960005305 adenosine Drugs 0.000 description 10
- 239000000047 product Substances 0.000 description 10
- 108020003175 receptors Proteins 0.000 description 10
- 102000005962 receptors Human genes 0.000 description 10
- 239000004480 active ingredient Substances 0.000 description 8
- 230000008569 process Effects 0.000 description 8
- 210000001519 tissue Anatomy 0.000 description 8
- 238000006243 chemical reaction Methods 0.000 description 7
- 239000000546 pharmaceutical excipient Substances 0.000 description 7
- VFNUTEMVQGLDAG-NKVSQWTQSA-N 2-methoxy-4-[(Z)-(5,6,7,8-tetrahydro-[1]benzothiolo[2,3-d]pyrimidin-4-ylhydrazinylidene)methyl]phenol Chemical compound COC1=CC(\C=N/NC2=NC=NC3=C2C2=C(CCCC2)S3)=CC=C1O VFNUTEMVQGLDAG-NKVSQWTQSA-N 0.000 description 6
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 6
- 0 C(C1)C2CC1*C2 Chemical compound C(C1)C2CC1*C2 0.000 description 6
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 6
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 6
- 239000008298 dragée Substances 0.000 description 6
- 239000000825 pharmaceutical preparation Substances 0.000 description 6
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 6
- 239000000296 purinergic P1 receptor antagonist Substances 0.000 description 6
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 6
- 239000002775 capsule Substances 0.000 description 5
- 231100000673 dose–response relationship Toxicity 0.000 description 5
- 239000003814 drug Substances 0.000 description 5
- 230000000694 effects Effects 0.000 description 5
- 239000011541 reaction mixture Substances 0.000 description 5
- 239000000725 suspension Substances 0.000 description 5
- 238000012360 testing method Methods 0.000 description 5
- UEZNSCCMEMUEMO-UHFFFAOYSA-N 6-chloro-9-methylpurine Chemical compound N1=CN=C2N(C)C=NC2=C1Cl UEZNSCCMEMUEMO-UHFFFAOYSA-N 0.000 description 4
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 4
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 4
- 229920002472 Starch Polymers 0.000 description 4
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 4
- 239000005557 antagonist Substances 0.000 description 4
- ILAHWRKJUDSMFH-UHFFFAOYSA-N boron tribromide Chemical compound BrB(Br)Br ILAHWRKJUDSMFH-UHFFFAOYSA-N 0.000 description 4
- 208000006218 bradycardia Diseases 0.000 description 4
- 229940079593 drug Drugs 0.000 description 4
- 238000001914 filtration Methods 0.000 description 4
- 238000002347 injection Methods 0.000 description 4
- 239000007924 injection Substances 0.000 description 4
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 4
- 239000012528 membrane Substances 0.000 description 4
- 230000002829 reductive effect Effects 0.000 description 4
- GFFGJBXGBJISGV-UHFFFAOYSA-N Adenine Chemical class NC1=NC=NC2=C1N=CN2 GFFGJBXGBJISGV-UHFFFAOYSA-N 0.000 description 3
- 241000700199 Cavia porcellus Species 0.000 description 3
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 3
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 3
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 3
- 239000005977 Ethylene Substances 0.000 description 3
- 108010010803 Gelatin Proteins 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 3
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 3
- JADDQZYHOWSFJD-FLNNQWSLSA-N N-ethyl-5'-carboxamidoadenosine Chemical compound O[C@@H]1[C@H](O)[C@@H](C(=O)NCC)O[C@H]1N1C2=NC=NC(N)=C2N=C1 JADDQZYHOWSFJD-FLNNQWSLSA-N 0.000 description 3
- 239000002202 Polyethylene glycol Substances 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 238000001816 cooling Methods 0.000 description 3
- 238000001035 drying Methods 0.000 description 3
- 239000000706 filtrate Substances 0.000 description 3
- 229920000159 gelatin Polymers 0.000 description 3
- 239000008273 gelatin Substances 0.000 description 3
- 229940014259 gelatin Drugs 0.000 description 3
- 235000019322 gelatine Nutrition 0.000 description 3
- 235000011852 gelatine desserts Nutrition 0.000 description 3
- 238000000338 in vitro Methods 0.000 description 3
- 239000000314 lubricant Substances 0.000 description 3
- 238000002156 mixing Methods 0.000 description 3
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 3
- 230000000144 pharmacologic effect Effects 0.000 description 3
- 229920001223 polyethylene glycol Polymers 0.000 description 3
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 3
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 3
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 3
- NLKNQRATVPKPDG-UHFFFAOYSA-M potassium iodide Chemical compound [K+].[I-] NLKNQRATVPKPDG-UHFFFAOYSA-M 0.000 description 3
- 238000012545 processing Methods 0.000 description 3
- 238000011160 research Methods 0.000 description 3
- 239000012265 solid product Substances 0.000 description 3
- 239000000600 sorbitol Substances 0.000 description 3
- 235000019698 starch Nutrition 0.000 description 3
- 238000003756 stirring Methods 0.000 description 3
- 239000000829 suppository Substances 0.000 description 3
- 239000000454 talc Substances 0.000 description 3
- 229910052623 talc Inorganic materials 0.000 description 3
- 235000012222 talc Nutrition 0.000 description 3
- 150000003626 triacylglycerols Chemical class 0.000 description 3
- 101710169336 5'-deoxyadenosine deaminase Proteins 0.000 description 2
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical compound N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- 102000055025 Adenosine deaminases Human genes 0.000 description 2
- 108050000203 Adenosine receptors Proteins 0.000 description 2
- 102000009346 Adenosine receptors Human genes 0.000 description 2
- 206010049765 Bradyarrhythmia Diseases 0.000 description 2
- 241000700198 Cavia Species 0.000 description 2
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 2
- 206010010071 Coma Diseases 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 2
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 229920002565 Polyethylene Glycol 400 Polymers 0.000 description 2
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 2
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 230000036471 bradycardia Effects 0.000 description 2
- RYYVLZVUVIJVGH-UHFFFAOYSA-N caffeine Chemical compound CN1C(=O)N(C)C(=O)C2=C1N=CN2C RYYVLZVUVIJVGH-UHFFFAOYSA-N 0.000 description 2
- 239000001506 calcium phosphate Substances 0.000 description 2
- 150000001720 carbohydrates Chemical class 0.000 description 2
- 239000001768 carboxy methyl cellulose Substances 0.000 description 2
- 229920002678 cellulose Polymers 0.000 description 2
- 239000001913 cellulose Substances 0.000 description 2
- 238000005119 centrifugation Methods 0.000 description 2
- 239000000460 chlorine Substances 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- 230000000052 comparative effect Effects 0.000 description 2
- 230000001186 cumulative effect Effects 0.000 description 2
- 239000002934 diuretic Substances 0.000 description 2
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 2
- 239000000284 extract Substances 0.000 description 2
- 239000010685 fatty oil Substances 0.000 description 2
- 239000000945 filler Substances 0.000 description 2
- 238000009472 formulation Methods 0.000 description 2
- 230000005714 functional activity Effects 0.000 description 2
- 210000004051 gastric juice Anatomy 0.000 description 2
- 210000002837 heart atrium Anatomy 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 229930195733 hydrocarbon Natural products 0.000 description 2
- 150000002430 hydrocarbons Chemical class 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- 238000011835 investigation Methods 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 230000000670 limiting effect Effects 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 235000019359 magnesium stearate Nutrition 0.000 description 2
- 150000002688 maleic acid derivatives Chemical class 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Chemical compound C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 231100000252 nontoxic Toxicity 0.000 description 2
- 230000003000 nontoxic effect Effects 0.000 description 2
- 239000003921 oil Substances 0.000 description 2
- 235000019198 oils Nutrition 0.000 description 2
- 239000011368 organic material Substances 0.000 description 2
- 239000003791 organic solvent mixture Substances 0.000 description 2
- 239000012188 paraffin wax Substances 0.000 description 2
- 235000011837 pasties Nutrition 0.000 description 2
- SONNWYBIRXJNDC-VIFPVBQESA-N phenylephrine Chemical compound CNC[C@H](O)C1=CC=CC(O)=C1 SONNWYBIRXJNDC-VIFPVBQESA-N 0.000 description 2
- 229960001802 phenylephrine Drugs 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- 239000003379 purinergic P1 receptor agonist Substances 0.000 description 2
- LEHBURLTIWGHEM-UHFFFAOYSA-N pyridinium chlorochromate Chemical compound [O-][Cr](Cl)(=O)=O.C1=CC=[NH+]C=C1 LEHBURLTIWGHEM-UHFFFAOYSA-N 0.000 description 2
- 238000010992 reflux Methods 0.000 description 2
- 230000000284 resting effect Effects 0.000 description 2
- 239000000741 silica gel Substances 0.000 description 2
- 229910002027 silica gel Inorganic materials 0.000 description 2
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 2
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 239000003381 stabilizer Substances 0.000 description 2
- 239000008107 starch Substances 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 239000002511 suppository base Substances 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- 239000003826 tablet Substances 0.000 description 2
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 2
- ZFXYFBGIUFBOJW-UHFFFAOYSA-N theophylline Chemical compound O=C1N(C)C(=O)N(C)C2=C1NC=N2 ZFXYFBGIUFBOJW-UHFFFAOYSA-N 0.000 description 2
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical class [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 2
- 238000001665 trituration Methods 0.000 description 2
- LTBJWRARHRHEGZ-MCDZGGTQSA-N (2r,3r,4s,5r)-2-(6-aminopurin-9-yl)-5-(hydroxymethyl)oxolane-3,4-diol;3,7-dihydropurine-2,6-dione Chemical compound O=C1NC(=O)NC2=C1NC=N2.C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O LTBJWRARHRHEGZ-MCDZGGTQSA-N 0.000 description 1
- LNAZSHAWQACDHT-XIYTZBAFSA-N (2r,3r,4s,5r,6s)-4,5-dimethoxy-2-(methoxymethyl)-3-[(2s,3r,4s,5r,6r)-3,4,5-trimethoxy-6-(methoxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6r)-4,5,6-trimethoxy-2-(methoxymethyl)oxan-3-yl]oxyoxane Chemical compound CO[C@@H]1[C@@H](OC)[C@H](OC)[C@@H](COC)O[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@H]2[C@@H]([C@@H](OC)[C@H](OC)O[C@@H]2COC)OC)O[C@@H]1COC LNAZSHAWQACDHT-XIYTZBAFSA-N 0.000 description 1
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 1
- HZLCGUXUOFWCCN-UHFFFAOYSA-N 2-hydroxynonadecane-1,2,3-tricarboxylic acid Chemical compound CCCCCCCCCCCCCCCCC(C(O)=O)C(O)(C(O)=O)CC(O)=O HZLCGUXUOFWCCN-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- OEPXIQLEKWHAGE-UHFFFAOYSA-N 8-(dicyclopropylmethyl)-1,3-dipropyl-7h-purine-2,6-dione Chemical group N1C=2C(=O)N(CCC)C(=O)N(CCC)C=2N=C1C(C1CC1)C1CC1 OEPXIQLEKWHAGE-UHFFFAOYSA-N 0.000 description 1
- 101150007969 ADORA1 gene Proteins 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- 229930024421 Adenine Natural products 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 206010002383 Angina Pectoris Diseases 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- MOQLCSSZGOILMF-UHFFFAOYSA-N CC(CC(C1)CC2)(C1CC2O)N=C Chemical compound CC(CC(C1)CC2)(C1CC2O)N=C MOQLCSSZGOILMF-UHFFFAOYSA-N 0.000 description 1
- RDTLBOVEOZOBAN-UHFFFAOYSA-N CC(N1C=NC2C(NC(CC3C4)C4CC3O)NC[N]#CC12)=C1CC1 Chemical compound CC(N1C=NC2C(NC(CC3C4)C4CC3O)NC[N]#CC12)=C1CC1 RDTLBOVEOZOBAN-UHFFFAOYSA-N 0.000 description 1
- UIQOBCBLYCNOJQ-UHFFFAOYSA-N Cc(nc[n]c1NC(C23)C=C(C4)C24C3=O)c1/N=C\NC Chemical compound Cc(nc[n]c1NC(C23)C=C(C4)C24C3=O)c1/N=C\NC UIQOBCBLYCNOJQ-UHFFFAOYSA-N 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- LVGKNOAMLMIIKO-UHFFFAOYSA-N Elaidinsaeure-aethylester Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC LVGKNOAMLMIIKO-UHFFFAOYSA-N 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 206010021143 Hypoxia Diseases 0.000 description 1
- LPHGQDQBBGAPDZ-UHFFFAOYSA-N Isocaffeine Natural products CN1C(=O)N(C)C(=O)C2=C1N(C)C=N2 LPHGQDQBBGAPDZ-UHFFFAOYSA-N 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 241000287531 Psittacidae Species 0.000 description 1
- 108010080192 Purinergic Receptors Proteins 0.000 description 1
- 102000000033 Purinergic Receptors Human genes 0.000 description 1
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical class [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- SMEGJBVQLJJKKX-HOTMZDKISA-N [(2R,3S,4S,5R,6R)-5-acetyloxy-3,4,6-trihydroxyoxan-2-yl]methyl acetate Chemical compound CC(=O)OC[C@@H]1[C@H]([C@@H]([C@H]([C@@H](O1)O)OC(=O)C)O)O SMEGJBVQLJJKKX-HOTMZDKISA-N 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- 229940081735 acetylcellulose Drugs 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- YKIOKAURTKXMSB-UHFFFAOYSA-N adams's catalyst Chemical compound O=[Pt]=O YKIOKAURTKXMSB-UHFFFAOYSA-N 0.000 description 1
- 230000006978 adaptation Effects 0.000 description 1
- 229960000643 adenine Drugs 0.000 description 1
- 229940121359 adenosine receptor antagonist Drugs 0.000 description 1
- 150000003838 adenosines Chemical class 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 235000010419 agar Nutrition 0.000 description 1
- 229940040563 agaric acid Drugs 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 150000003973 alkyl amines Chemical class 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 230000008485 antagonism Effects 0.000 description 1
- 210000002376 aorta thoracic Anatomy 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 125000004069 aziridinyl group Chemical group 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 125000005605 benzo group Chemical group 0.000 description 1
- HJHGAEZYELGORM-UHFFFAOYSA-N bicyclo[2.2.1]heptane-2,5-dione;ethene Chemical group C=C.C1C2C(=O)CC1C(=O)C2 HJHGAEZYELGORM-UHFFFAOYSA-N 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 230000000059 bradycardiac effect Effects 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 229960001948 caffeine Drugs 0.000 description 1
- VJEONQKOZGKCAK-UHFFFAOYSA-N caffeine Natural products CN1C(=O)N(C)C(=O)C2=C1C=CN2C VJEONQKOZGKCAK-UHFFFAOYSA-N 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- CJZGTCYPCWQAJB-UHFFFAOYSA-L calcium stearate Chemical compound [Ca+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CJZGTCYPCWQAJB-UHFFFAOYSA-L 0.000 description 1
- 239000008116 calcium stearate Substances 0.000 description 1
- 235000013539 calcium stearate Nutrition 0.000 description 1
- 229910002091 carbon monoxide Inorganic materials 0.000 description 1
- 125000002057 carboxymethyl group Chemical group [H]OC(=O)C([H])([H])[*] 0.000 description 1
- 230000005792 cardiovascular activity Effects 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 229920002301 cellulose acetate Polymers 0.000 description 1
- 239000003610 charcoal Substances 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 238000011281 clinical therapy Methods 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 230000008602 contraction Effects 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 229940099112 cornstarch Drugs 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 238000007405 data analysis Methods 0.000 description 1
- 238000011033 desalting Methods 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- 229910001873 dinitrogen Inorganic materials 0.000 description 1
- MGHPNCMVUAKAIE-UHFFFAOYSA-N diphenylmethanamine Chemical compound C=1C=CC=CC=1C(N)C1=CC=CC=C1 MGHPNCMVUAKAIE-UHFFFAOYSA-N 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- 238000006073 displacement reaction Methods 0.000 description 1
- 238000010494 dissociation reaction Methods 0.000 description 1
- 230000005593 dissociations Effects 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 230000001882 diuretic effect Effects 0.000 description 1
- 229940030606 diuretics Drugs 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- LVGKNOAMLMIIKO-QXMHVHEDSA-N ethyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC LVGKNOAMLMIIKO-QXMHVHEDSA-N 0.000 description 1
- 229940093471 ethyl oleate Drugs 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 238000003818 flash chromatography Methods 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 238000011207 functional examination Methods 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 238000005469 granulation Methods 0.000 description 1
- 230000003179 granulation Effects 0.000 description 1
- 239000008202 granule composition Substances 0.000 description 1
- 238000000227 grinding Methods 0.000 description 1
- 230000026030 halogenation Effects 0.000 description 1
- 238000005658 halogenation reaction Methods 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 150000002431 hydrogen Chemical class 0.000 description 1
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 1
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 1
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 1
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 1
- 229920003132 hydroxypropyl methylcellulose phthalate Polymers 0.000 description 1
- 229940031704 hydroxypropyl methylcellulose phthalate Drugs 0.000 description 1
- 230000007954 hypoxia Effects 0.000 description 1
- 125000002632 imidazolidinyl group Chemical group 0.000 description 1
- 125000002636 imidazolinyl group Chemical group 0.000 description 1
- 230000000297 inotrophic effect Effects 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 239000004922 lacquer Substances 0.000 description 1
- 229940057995 liquid paraffin Drugs 0.000 description 1
- 239000012280 lithium aluminium hydride Substances 0.000 description 1
- 238000012792 lyophilization process Methods 0.000 description 1
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 1
- 235000019341 magnesium sulphate Nutrition 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 150000004702 methyl esters Chemical class 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- LZMRVYPPUVMKOI-UHFFFAOYSA-N n-cyclopentyl-9-methylpurin-6-amine Chemical compound N1=CN=C2N(C)C=NC2=C1NC1CCCC1 LZMRVYPPUVMKOI-UHFFFAOYSA-N 0.000 description 1
- XHFGWHUWQXTGAT-UHFFFAOYSA-N n-methylpropan-2-amine Chemical compound CNC(C)C XHFGWHUWQXTGAT-UHFFFAOYSA-N 0.000 description 1
- 210000001577 neostriatum Anatomy 0.000 description 1
- UMRZSTCPUPJPOJ-KNVOCYPGSA-N norbornane Chemical group C1C[C@H]2CC[C@@H]1C2 UMRZSTCPUPJPOJ-KNVOCYPGSA-N 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 239000012074 organic phase Substances 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- NXJCBFBQEVOTOW-UHFFFAOYSA-L palladium(2+);dihydroxide Chemical compound O[Pd]O NXJCBFBQEVOTOW-UHFFFAOYSA-L 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 239000002831 pharmacologic agent Substances 0.000 description 1
- XNGIFLGASWRNHJ-UHFFFAOYSA-L phthalate(2-) Chemical compound [O-]C(=O)C1=CC=CC=C1C([O-])=O XNGIFLGASWRNHJ-UHFFFAOYSA-L 0.000 description 1
- 230000035790 physiological processes and functions Effects 0.000 description 1
- 230000006461 physiological response Effects 0.000 description 1
- 239000000049 pigment Substances 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 239000004014 plasticizer Substances 0.000 description 1
- 229940068918 polyethylene glycol 400 Drugs 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 229940116317 potato starch Drugs 0.000 description 1
- 230000036515 potency Effects 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 125000003072 pyrazolidinyl group Chemical group 0.000 description 1
- 125000002755 pyrazolinyl group Chemical group 0.000 description 1
- WAJNANMQOPCIPO-UHFFFAOYSA-N pyrazolo[4,3-d]pyrimidin-7-one Chemical class O=C1N=CN=C2C=NN=C12 WAJNANMQOPCIPO-UHFFFAOYSA-N 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- YOWAEZWWQFSEJD-UHFFFAOYSA-N quinoxalin-2-amine Chemical class C1=CC=CC2=NC(N)=CN=C21 YOWAEZWWQFSEJD-UHFFFAOYSA-N 0.000 description 1
- 239000002287 radioligand Substances 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 229940100486 rice starch Drugs 0.000 description 1
- 238000009938 salting Methods 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 210000002027 skeletal muscle Anatomy 0.000 description 1
- 235000010413 sodium alginate Nutrition 0.000 description 1
- 239000000661 sodium alginate Substances 0.000 description 1
- 229940005550 sodium alginate Drugs 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- 239000007901 soft capsule Substances 0.000 description 1
- 239000012439 solid excipient Substances 0.000 description 1
- 239000012453 solvate Substances 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 238000010998 test method Methods 0.000 description 1
- DPKBAXPHAYBPRL-UHFFFAOYSA-M tetrabutylazanium;iodide Chemical compound [I-].CCCC[N+](CCCC)(CCCC)CCCC DPKBAXPHAYBPRL-UHFFFAOYSA-M 0.000 description 1
- 229960000278 theophylline Drugs 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 239000004408 titanium dioxide Substances 0.000 description 1
- 229940078499 tricalcium phosphate Drugs 0.000 description 1
- 235000019731 tricalcium phosphate Nutrition 0.000 description 1
- 229910000391 tricalcium phosphate Inorganic materials 0.000 description 1
- 230000024883 vasodilation Effects 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 229940100445 wheat starch Drugs 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
- C07D473/26—Heterocyclic compounds containing purine ring systems with an oxygen, sulphur, or nitrogen atom directly attached in position 2 or 6, but not in both
- C07D473/32—Nitrogen atom
- C07D473/34—Nitrogen atom attached in position 6, e.g. adenine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/02—Drugs for disorders of the urinary system of urine or of the urinary tract, e.g. urine acidifiers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/02—Drugs for disorders of the nervous system for peripheral neuropathies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/10—Antioedematous agents; Diuretics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmacology & Pharmacy (AREA)
- Engineering & Computer Science (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Endocrinology (AREA)
- Reproductive Health (AREA)
- Urology & Nephrology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
Description
- Gebiet der Erfindung
- Diese Erfindung bezieht sich im Allgemeinen auf neuartige substituierte 9-Alkyladenin-Verbindungen, auf Verfahren zum Herstellen solcher Verbindungen, und Verfahren zum Verabreichen von Zusammensetzungen solcher Verbindungen in Mengen, die effektiv sind, eine gewünschte physiologische Reaktion in einem Säugetier zu induzieren, den Antagonismus des Adenosinrezeptors eingebunden. Insbesondere, bezieht sich die Erfindung auf Verbindungen für die therapeutische Verwendung, speziell als Diuretika, Nierenschutz gegen akutes und chronisches Nierenversagen, sowie als Wirkstoffe um die Erholung aus dem Koma zu fördern, für das Behandeln von oder das Vorbeugen vor Claudicatio intermittens, zum Wiederherstellen der Herzfunktion nach einem kardioplegischen Vorgang, und um das therapeutische Ergebnis zu verbessern, welches von der Defibrillation oder kardiopulmonale Reanimation resultiert, indem eine post-reanimative Bradykardie, Bradyarrhythmie, und Kardioplegie verhindert wird.
- Stand der Technik
- Adenosin (9-β-D-Ribofuranosyl-9H-Purin-6-Amin) wurde in den späten 1920'ern als hypotone oder bradykardische Aktivität aufweisend, charakterisiert. Seit dann, hat beträchtliche Forschung in die molekulare Modifikation von Adenosin zu der allgemeinen Schlussfolgerung geführt, das die kardiovaskuläre Aktivität auf Analoga die intakte Purin- und β-Ribofuranosylringe haben, beschränkt ist.
- Weitere Forschung hat deutlicher gezeigt, wie die Aktivität dieser Adenosinanaloga die purinergen Rezeptoren in peripheren Zellmembranen, insbesondere die A1 und A2 Rezeptoren, beeinflussen.
- Adenosin-Antagonisten haben dabei geholfen, die Rolle des Adenosins in verschiedenen physiologischen Vorgängen zu erklären. Besonders, selektive Antagonisten für den Adenosin A1 Rezeptor waren entscheidend beim Bestimmen der physiologischen Wichtigkeit der A1-Rezeptor Aktivierung. Nicht-selektive Adenosin Antagonisten, wie zum Beispiel Koffein und Theophyllin dienten als Ausgangspunkt für die Strukturaktivitätsforschung mit dem Ziel A1 Rezeptor selektive Antagonisten herzustellen. Ein Beispiel für eine solche Entdeckung ist 8-(Dicyclopropylmethyl)-1,3-Dipropylxanthin (Shimada, J., et al., J. Med. Chem. 34:466-9 (1991)). Alternativ sind verschiedene Nicht-Xanthin-Adenosin-Antagonisten identifiziert worden, einschließlich Triazolo[4,3-a]Chinoxalinamine, Triazolochinazoline, Pyrazolo[4,3-d]pyrimidin-7-one, und Adeninderivate (Williams, M., Med. Res. Rev 9(2):219-43 (1989)). Eine Reihe von Adeninderivaten wurden in dem US Patent Nr. 5.066.655 identifiziert. Die Suche nach wirksamen und selektiven Adenosin A1 Rezeptor selektiven Antagonisten, die als pharmakologische Werkzeuge und als therapeutische Wirkstoffe nützlich sind, geht weiter.
- Zusammenfassung der Erfindung
- Bestimmte neuartige Verbindungen sind nun entdeckt worden, die eine Aktivität als Adenosin-Antagonisten aufweisen. Ein erster Aspekt der vorliegenden Erfindung stellt eine Verbindung mit folgender Struktur bereit:oder ein pharmazeutisch annehmbares Salz davon;
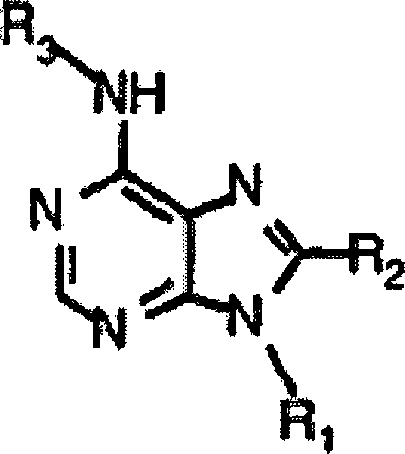
wobei R1 ein C1 bis C4 geradkettiges oder verzweigtes Alkyl ist; R2 ausgewählt ist aus der Gruppe bestehend aus H,-OR1, -SR1, -N(R4)(R5), Aminocarbonyl, Halogen, und -CN, wobei R4 und R5 unabhängig ein C1 bis C6 geradkettiges oder verzweigtes Alkyl, oder R4 und R5 gemeinsam mit dem Stickstoff, an welchen sie gebunden sind, einen 3- bis 7-gliedrigen Heterocycloalkylsubstituenten bilden, welcher gegebenenfalls ein zusätzliches Heteroatom enthält, das ausgewählt ist aus der Gruppe bestehend aus Stickstoff, Sauerstoff und Schwefel; R3ist und R' -OH oder -NH2 ist und R'' H ist, oder ein pharmazeutisch akzeptables Salz davon.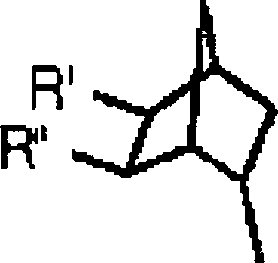
- In der obigen Adenin-bezogenen Verbindung der vorliegenden Erfindung, wird es bevorzugt, dass R1 C1 bis C4 niederes oder verzweigtes Alkyl, am meisten bevorzugt Methyl oder Ethyl ist
- In den bevorzugten Verbindungen, wird es bevorzugt, dass R2 H, -N(R4)(R5), -OR1, -SR1, Halogen oder -CN ist. R2 kann Methylthio oder Methoxy sein.
- In den Verbindungen der vorliegenden Erfindung, kann R' -OH und R'' kann Wasserstoff sein.
- R4 und R5 können unabhängig C1 bis C3 Alkylradikale sein. R4 kann Methyl und R5 kann Isopropyl sein.
- Die Verbindung kann (±)-N6-[endo-2'-(endo-5'-hydroxy)norbornyl]-9-Methyladenin oder (±)-N6-[endo-2'-(endo-5'-hydroxy)norbornyl]-8-isopropylmethylamino-9-Methyladenin sein, oder ein pharmazeutisch akzeptables Salz von jeder dieser Verbindungen.
- Die Verbindungen der vorliegenden Erfindung sind alle therapeutisch wirksame Adenosinrezeptor Antagonisten in Säugetieren. Folglich, sind sie wirksam zum Behandeln von Zuständen, welche auf selektive Andenosin A1 Rezeptorblockierungen reagieren. Dementsprechend, sind die Verbindungen der vorliegenden Erfindungen nützlich als Diuretika, Nierenschutz gegen akutes und chronisches Nierenversagen, als Wirkstoffe um die Erholung aus dem Koma zu fördern (um das Erwachen und um höhere Bewusstseinsniveaus zu induzieren), um die Herzfunktion nach einem kardioplegischen Vorgang wieder herzustellen, für das Behandeln oder das Vorbeugen der Claudicatio intermittens (Angina des Skelettmuskels die durch Hypoxie entsteht), und als Wirkstoffe um das therapeutische Ergebnis, welches von der Defibrillation oder kardiopulmonale Reanimation resultiert, zu verbessern.
- In einem zweiten Aspekt stellt die vorliegende Erfindung eine pharmazeutische Zusammensetzung bereit, die eine Verbindung gemäß dem ersten Aspekt oder ein pharmazeutisch akzeptables Salz davon und einen pharmazeutisch akzeptablen Träger, aufweist.
- In einem dritten Aspekt stellt die vorliegende Erfindung eine Verbindung oder ein pharmazeutisch akzeptables Salz davon gemäß dem ersten Aspekt oder eine pharmazeutische Zusammensetzung gemäß dem zweiten Aspekt bereit, für die Verwendung zum Inhibieren der Adenosin A1 Rezeptoraktivierung in einem Säugetier, das diese benötigt.
- Ein vierter Aspekt stellt eine Verbindung oder eine pharmazeutisch akzeptables Salz davon gemäß dem ersten Aspekt oder eine pharmazeutische Zusammensetzung gemäß dem zweiten Aspekt, für die Verwendung in einem Verfahren um Diurese zu induzieren, um die Nieren gegen akutes oder chronisches Nierenversagen zu schützen, um die Erholung aus dem Koma zu fördern, um das therapeutische Ergebnis zu verbessern, das von der Defibrillation oder kardiopulmonaler Reanimation resultiert, um die Herzfunktion nach einem kardioplegischen Vorgang wieder herzustellen, oder zum Behandeln von oder Vorsorgen vor Claudicatio intermittens.
- In einem fünften Aspekt wird die Verwendung einer Verbindung gemäß dem ersten Aspekt oder eine pharmazeutisch akzeptables Salz davon in der Zubereitung einer pharmazeutischen Zusammensetzung gemäß dem zweiten Aspekt bereitgestellt für die Verwendung zum Inhibieren der Adenosin A1 Rezeptoraktivierung in einem Säugetier, das diese benötigt.
- Ein sechster Aspekt stellt die Verwendung einer Verbindung gemäß dem ersten Aspekt oder ein pharmazeutisch akzeptables Salz davon in der Zubereitung einer pharmazeutischen Zusammensetzung gemäß dem zweiten Aspekt bereit für die Verwendung in einem Verfahren um die Diurese zu induzieren, um die Nieren gegen akutes oder chronisches Nierenversagen zu schützen, um die Erholung aus dem Koma zu fördern, um das therapeutische Ergebnis zu verbessern, die von der Defibrillation oder kardiopulmonaler Reanimation resultiert, um die Herzfunktion nach kardioplegischen Vörgangen wieder herzustellen, oder zum Behandeln von oder Vorsogen vor Claudicatio intermittens.
- Detaillierte Beschreibung der bevorzugten Ausführungsformen
- Illustrative Verbindungen des ersten Aspekts der vorliegenden Erfindung schließen, ohne jedoch darauf beschränkt zu sein, ein: (±)-N6-[endo-2'-(endo-5'-hydroxyl)norbornyl]-9-Methyladenin, (±)-N6-[endo-2'-(endo-5'-hydroxy)norbornyl]-8-isopropylmethylamino-9-Methyladenin, (±)-N6-[endo-2'-(endo-5'-hydroxy)norbornyl]-8-bromo-9-Methyladenin, (±)-N6-[endo-2'-(endo-5'-hydroxy)norbornyl]-8-dimethylamino-9-Methyladenin, und (±)-N6-[endo-2'-(endo-5'-hydroxy)norbornyl]-9-Methyladenin. Die obigen Verbindungen sind nur beispielhaft und nicht dafür bestimmt auf irgendeine Weise einschränkend zu sein.
- Andere Verbindungen, die nicht als Ausführungsformen der vorliegenden Erfindung dargestellt sind, jedoch als Beispiele für das Verständnis der Erfindung nützlich sind, schließen (±)-N6-[endo-2'-norbornyl]-8-isopropylmethylamino-9-Methyladenin, N6-zyclopentyl-8-isopropylmethylamino-9-Methyladenin, N6-cyclopentyl-8-methylthio-9-Methyladenin, (±)-N6-[endo-2'-norbornyl]-8-methylthio-9-Methyladenin, (±)-N6-[endo-2'-norbornyl]-8-cyano-9- Methyladenin, (±)-N6-[endo-2'-norbornyl]-8-methoxy-9-Methyladenin, (±)-N6-[endo-2'-norbornyl]-8-dimethylamino-9-Methyladenin, (±)-N6-[endo-2'-(5'-keto)norbornyl]-8-bromo-9-Methyladenin, (±)-N6-[endo-2'-norbornyl]-8-diethylamino-9Methyladenin, (±)-N6-[endo-2'-norbornyl]-8-piperidinyl-9-Methyladenin, (±)-N6-[endo-2'-norbornyl]-8-amido-9-Methyladenin, (±)-N6-[endo-2'-(5',6'-(6''-hydroxy)benzo)narbornyl]-9-Methyladenin, und N6-cyclopentyl-8-dimethylamino-9-Methyladenin ein.
- Übliche 3 bis 7-gliedrige heterozyklische Substituenten, welche durch R2 dargestellt werden können, schließen Aziridinyl, Piperidinyl, Piperazinyl, Pyrrolidinyl, Imidazolidinyl, Imidazolinyl, Morpholinyl, Pyrazolidinyl, Pyrazolinyl und dergleichen ein.
- Übliche Halogengruppen schließen Fluor, Chlor, Brom, und Iod ein.
- Übliche C1–6 Alkylgruppen schließen Methyl, Ethyl, Propyl, Isopropyl, Butyl, sec-Butyl, tert-Butyl, Pentyl und Hexylgruppen ein.
- Zusammensetzungen innerhalb des Schutzumfanges dieser Erfindung schließen alle Zusammensetzungen ein, wobei die Verbindungen der vorliegenden Erfindung in einer Menge enthalten ist, die wirksam ist, den gewünschten Zweck zu erzielen. Während individuelle Bedürfnisse variieren, ist die Bestimmung des optimalen Bereichs für die wirksame Menge jeder Verbindung innerhalb des Fachkönnens. Üblicherweise, können die Verbindungen einem Säugetier, z.B. einem Menschen oral verabreicht werden, mit einer Dosis von 0,001 bis 100 mg/kg, oder in einer äquivalenten Menge eines pharmazeutisch akzeptablen Salzes davon, pro Tag des Körpergewichtes des Säugetiers, welches für Diurese, Bradykardie, Bradyarrhythmie, und Kardioplegie nach kardiopulmonarer Reanimation behandelt wird oder wenn die Verbindung als Nierenschutz verabreicht wird. Vorzugsweise, ist etwa 0,01 bis etwa 10 mg/kg oral verabreicht um solche Störung zu behandeln oder vorzubeugen. Für eine intramuskuläre Injektion, ist die Dosis im Allgemeinen die Hälfte der oralen Dosis. Zum Beispiel ist eine geeignete intramuskuläre Dosis von etwa 0,0005 bis etwa 50 mg/kg, und am meisten bevorzugt von etwa 0,05 bis etwa 5 mg/kg. Für eine intravenöse Verabreichung, ist die Dosis im Allgemeinen von 0,0001 bis etwa 10 mg/kg, um am meisten bevorzugt von etwa 0,001 bis etwa 1 mg/kg.
- Die orale Einheitsdosis kann von etwa 0,1 bis etwa 7000 mg, vorzugsweise etwa 1 bis etwa 700 mg der Verbindung aufweisen. Die Einheitsdosis kann einmal oder mehrmals täglich als eine oder mehrere Tabletten verabreicht werden, wobei jede von etwa 0,1 bis etwa 500, zweckmäßig von etwa 0,1 bis 100 mg der Verbindung oder ihr Solvat enthält.
- Zusätzlich zum Verabreichen der Verbindung als rohe Chemikalie, können die Verbindung der Erfindung als Teil einer pharmazeutischen Zubereitung verabreicht werden, die geeignete pharmazeutisch akzeptable Träger enthält, welche Excipienten und Hilfsstoffe aufweisen, die das Weiterverarbeiten der Verbindung in eine Zubereitung, welche pharmazeutisch verwendet werden kann, erleichtern. Vorzugsweise, enthalten die Zubereitungen, insbesondere jene Zubereitungen welche oral verabreicht werden können und die für die bevorzugte Art der Verabreichung, wie zum Beispiel Tabletten, Dragees, und Kapseln verwendet werden können, und Zubereitungen die rektal verabreicht werden können, wie zum Beispiel Zäpfchen, sowie geeignete Lösungen für die Verabreichung durch Injektion oder Oral, von etwa 0,01 bis 99 Prozent, vorzugsweise von etwa 0,25 bis 75 Prozent Wirkstoff zusammen mit dem Excipienten.
- Ebenfalls eingeschlossen in den Schutzumfang der vorliegenden Erfindung sind die nichttoxischen pharmazeutisch akzeptablen Salze der Verbindung der vorliegenden Erfindung. Salze werden durch Mischen einer Lösung einer bestimmten Verbindung der vorliegenden Erfindung mit einer Lösung einer pharmazeutisch akzeptablen nichttoxischen Säure, wie zum Beispiel Salzsäure, Essigsäure, Apfelsäure, Phosphorsäure und dergleichen gebildet.
- Die pharmazeutische Zusammensetzung der Erfindung kann jedem Säugetier verabreicht werden, welche die nutzbringenden Wirkungen der Verbindungen der vorliegenden Erfindung erfahren können. Unter diesen sind als Erstes Menschen, obwohl die Erfindung nicht beabsichtigt ist, darauf beschränkt zu sein.
- Die pharmazeutischen Zusammensetzungen der vorliegenden Erfindung kann durch jedes Mittel, welches den beabsichtigten Zweck erzielt, verabreicht werden. Zum Beispiel kann die Verabreichung auf parenteralem, subkutanem, intravenösem, intramuskulärem, intraperitonealem, transdermalem, oder bukkalem Weg erfolgen. Die Verabreicherung kann alternativ oder gleichzeitig auf dem oralen Weg erfolgen. Die verabreichte Dosierung hängt vom Alter, der Gesundheit, und dem Gewicht des Empfängers, der Art von gleichzeitigen Behandlungen, wenn überhaupt, der Häufigkeit der Behandlung, und der Natur der gewünschten Wirkung ab.
- Zusätzlich zu den pharmakologischen Wirkstoffen, können die neue pharmazeutischen Zubereitungen geeignete pharmazeutisch akzeptable Träger, welche Excipienten und Hilfsstoffe aufweisen, die die Weiterverarbeitung der Wirkstoff in pharmazeutisch verwendbare Zubereitungen erleichtert, enthalten. Vorzugsweise, enthalten die Zubereitungen, insbesondere jene Zubereitungen welche oral verabreicht werden können und die für die bevorzugte Art der Verabreichung, wie zum Beispiel Tabletten, Dragees, und Kapseln verwendet werden können, und auch Zubereitungen die rektal verabreicht werden können, wie zum Beispiel Zäpfchen, sowie geeignete Lösungen für die Verabreichung durch Injektion oder Oral, von etwa 0,01 bis 99 Prozent, zusammen mit dem Excipienten.
- Die pharmazeutischen Zubereitungen der vorliegenden Erfindung werden in einer Art und Weise, die selbst bekannt ist, hergestellt, zum Beispiel mittels herkömmlicher Misch-, Granulier-, Dragee-Herstellungs-, Auflösungs- oder Lyophilisierungsverfahren. Folglich können pharmazeutische Zubereitungen für die orale Verwendung durch Kombinieren des Wirkstoffs mit festen Excipienten erreicht werden, gegebenenfalls Mahlen der resultierenden Mischung und Verarbeiten der Granulatmischung, nach dem Hinzufügen geeigneter Hilfsstoffe, wenn gewünscht oder notwendig, um Tabletten oder Dragee-Kerne zu erhalten.
- Geeignete Excipienten sind, insbesondere, Füllstoffe wie zum Beispiel Saccharide, zum Beispiel, Laktose oder Sucrose, Mannitol oder Sorbitol, Cellulose-Zubereitungen und/oder Calciumphosphate, zum Beispiel, Tricalciumphosphat oder Calciumhydrogenphophate, sowie Bindemittel wie zum Beispiel Stärkepaste unter Verwendung von zum Beispiel, Maisstärke, Weizenstärke, Reisstärke, Kartoffelstärke, Gelatin, Targant, Methylcellulose, Hydroxypropylmethylcellulose, Natriumcarboxymethylcellulose, und/oder Polyvinylpyrrolidon. Wenn gewünscht, können Zerfallsmittel wie zum Beispiel die oben genannten Stärken und ebenfalls Carboxymethylstärke, quervernetztes Polyvinylpyrrolidon, Agar, oder Alginsäure oder ein Salz davon, wie zum Beispiel Natriumalginat hinzugefügt werden. Hilfsstoffe sind vor allem das Fließverhalten regulierende Agenzien und Gleitmittel (Lubrikanzien), zum Beispiel Siliciumdioxid, Talkum, Stearinsäure oder Salze davon, wie zum Beispiel Magnesiumstearat oder Calciumstearat, und/oder Polyethylenglycol. Dragee-Kerne werden mit geeigneten Unhüllungen, welche, wenn gewünscht, gegenüber dem Magensaft resistent sind, bereitgestellt. Für diesen Zweck können konzentrierte Saccharidlösungen verwendet werden, welche gegebenenfalls Gummiarabikum, Talkum, Polyvinylpyrrolidon, Polyethylenglykol, und/oder Titandioxid, Lacklösungen und geeignete organische Lösungsmittel oder Lösungsmittelmischungen enthalten. Um Umhüllungen, die gegenüber Magensaft resistent sind, herzustellen werden Lösungen von geeigneten Cellulosezubereitungen, wie zum Beispiel Acetylcellulosephthalat oder Hydroxypropylmethylcellulosephthalat verwendet. Farbstoffe oder Pigmente können den Tabletten oder Dragee-Umhüllungen, zum Beispiel, für die Identifikation oder um Kombinationen von Wirkstoffdosen zu charakterisieren, hinzugefügt werden.
- Andere pharmazeutische Zubereitungen, welche oral verwendet werden können, schließen Steckkapseln, die aus Gelatine hergestellt sind, sowie weiche versiegelte Kapseln, die aus Gelatine und einem Weichmacher, wie zum Beispiel, Glycerol oder Sorbitol hergestellt sind, ein. Die Steckkapseln können die Wirkstoffe in Form von Granulaten enthalten, welche mit Füllstoffen wie zum Beispiel Laktose, Bindemittel wie zum Beispiel Stärken und/oder Gleitmittel wie zum Beispiel Talkum oder Magnesiumstearat und, gegebenenfalls, Stabilisatoren gemischt werden können. In weichen Kapseln, werden die Wirkstoffe bevorzugt in geeigneten Flüssigkeiten, wie zum Beispiel Fettölen oder flüssigem Paraffin aufgelöst oder suspendiert. Zusätzlich können Stabilisatoren verwendet werden.
- Mögliche pharmazeutische Zubereitungen die rektal verwendet werden können schließen zum Beispiel Zäpfchen, welche aus einer Kombination von einem oder mehreren Wirkstoffen) und einer Zäpfchenbasis besteht, ein. Geeignete Zäpfchenbasen sind zum Beispiel natürliche oder synthetische Triglyceride, oder Paraffin-Kohlenwasserstoffe. Zusätzlich ist es möglich, rektale Gelatinekapseln, welche aus einer Kombination von Wirkstoff und einer Basis bestehen, verwendet werden. Mögliche Basismaterialien schließen, zum Beispiel, flüssige Triglyceride, Polyethylenglykol, oder Paraffin-Kohlenwasserstoffe ein.
- Geeignete Formulierungen für die parenterale Verabreichung schließen wässrige Lösungen des Wirkstoffs in wasserlöslicher Form, zum Beispiel, wässerlöslichen Salzen ein. Zusätzlich, können Wirkstoffsuspensionen als entsprechende ölige Injektionssuspensionen verabreicht werden. Geeignete lipophile Lösungsmittel oder Vehikel schließen Fettöle, zum Beispiel, Sesamöl, oder synthetische Fettsäureester, zum Beispiel, Ethyloleat oder Triglyceride oder Polyethylenglycol-400 (die Verbindungen sind in PEG-400 löslich) ein. Wässrige Injektionssuspensionen können Substanzen enthalten, die die Viskosität der Suspension erhöhen, wie zum Beispiel Natrium Carboxyrnethylcellulose, Sorbitol, und/oder Dextran. Gegebenenfalls, kann die Suspension Stabilisatoren enthalten.
- Die folgenden Beispiele sind beispielhaft, und nicht einschränkend, für die Verfahren und Zusammensetzungen der vorliegenden Erfindung. Andere geeignete Modifikationen und Anpassungen von der Vielzahl von Zuständen und Parameter die normalerweise in der klinischen Therapie anzutreffen und für den Fachmann offensichtlich sind, sind innerhalb des Schutzumfanges der Erfindung.
- Beispiele
- Verbindungen der vorliegenden Erfindung, wobei R1 ein niederer Alkyl ist, können wie folgt hergestellt werden (Verfahren A, wobei R1 CH3 ist). Das Verfahren wird in Beispiel Nr. 13 im Detail beschrieben.
- Verfahren A

- Verfahren 1- Detailliertes Beispiel: Herstellung von (±)-N6-(endo-2'-norbornyl)-8-isopropylmethylamino-9-Methyladenin.
- Dies ist ein vergleichendes Beispiel, das nicht als Ausführungsform der Erfindung dargestellt ist, jedoch als Beispiel das für das Verständnis der Erfindung nützlich ist.
- Ein Lösung von 2g (5,42 mmol) (±)-N6-(endo-2'-norbornyl)-8-iodo-9-Methyladenin (hergestellt durch Halogenierung von (±)-N6-(endo-2'-norbornyl)-9-Methyladenin (US Patent Nr. 5.066.655) im Wesentlichen nach Moriarty, R.M., et al, Tet. Lett., 31:5887-90 (1990)) in 5 ml (47 mmol) N-Methylisopropylamin wurde über Nacht auf 135 °C in einer Reaktionsbombe erhitzt. Nach dem Abkühlen, wurde die Reaktionsmischung in Dichlormethan aufgelöst und mit Wasser extrahiert. Die organische Phase wurde getrocknet (MgSO4), filtriert und konzentriert. Der Rückstand wurde auf einer Silicagelsäule (Ethylacetat/Hexan (1:1); gefolgt von Ehtylacetat) chromatographiert um das Produkt zu erhalten, welches in das Maleatsalz umgewandelt wurde, um 347 mg des hellgelben Feststoffs zu erhalten.
-

- Tabelle I
- Die Beispiele in Tabelle I sind nicht als Ausführungsformen der Erfindung dargelegt, jedoch als Beispiele für das Verständnis der Erfindung nützlich.
-

-
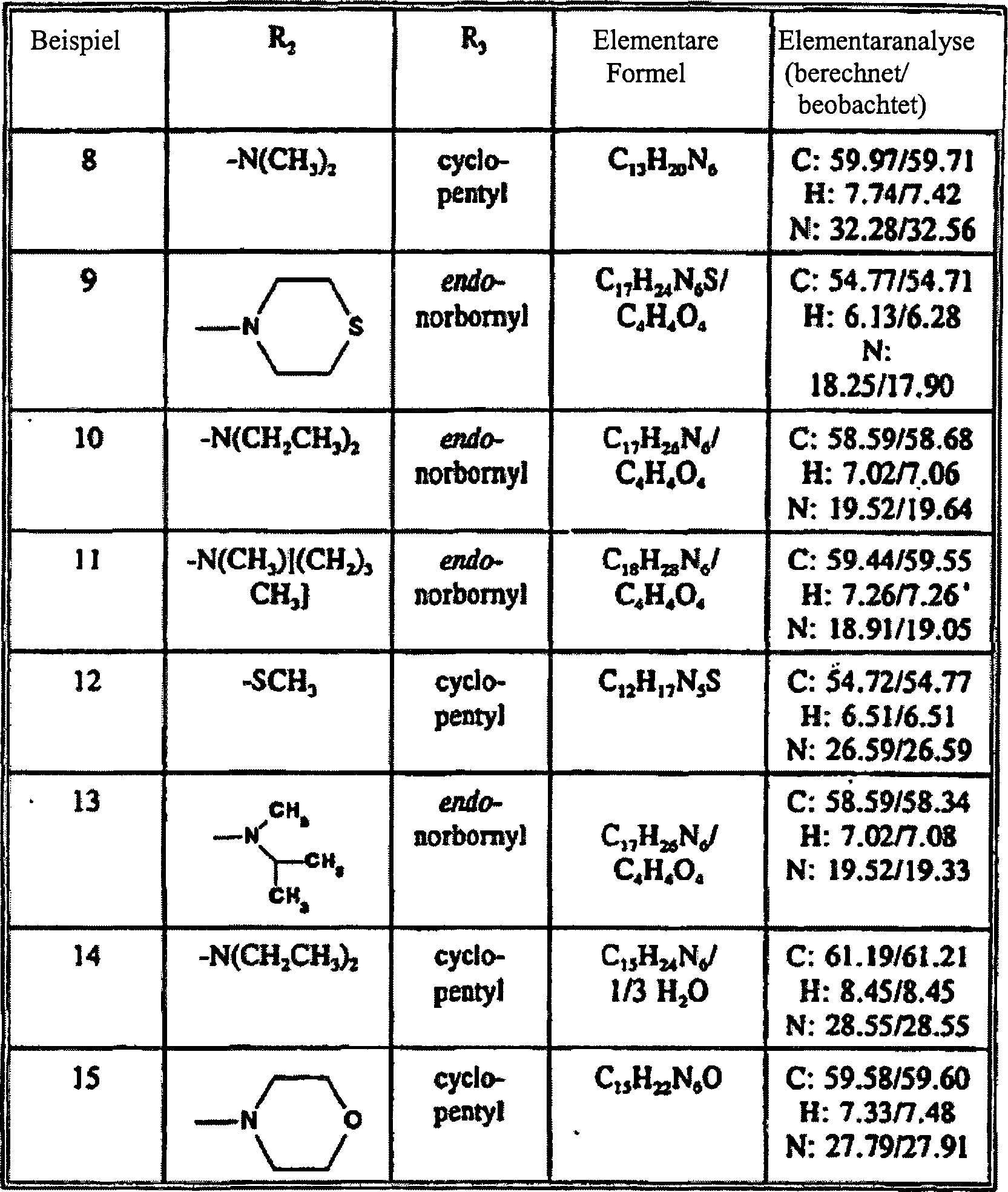
- Bemerkung: Das 8-Cyano-Derivat wurde durch Verdrängung des 8-Halogen-Zwischenprodukts im Verfahren A hergestellt, und das 8-Amino-Derivat wurde durch Oxidation der 8-Cyano-Verbindung mit Schwefelsäure unter Standardbedingungen hergestellt.
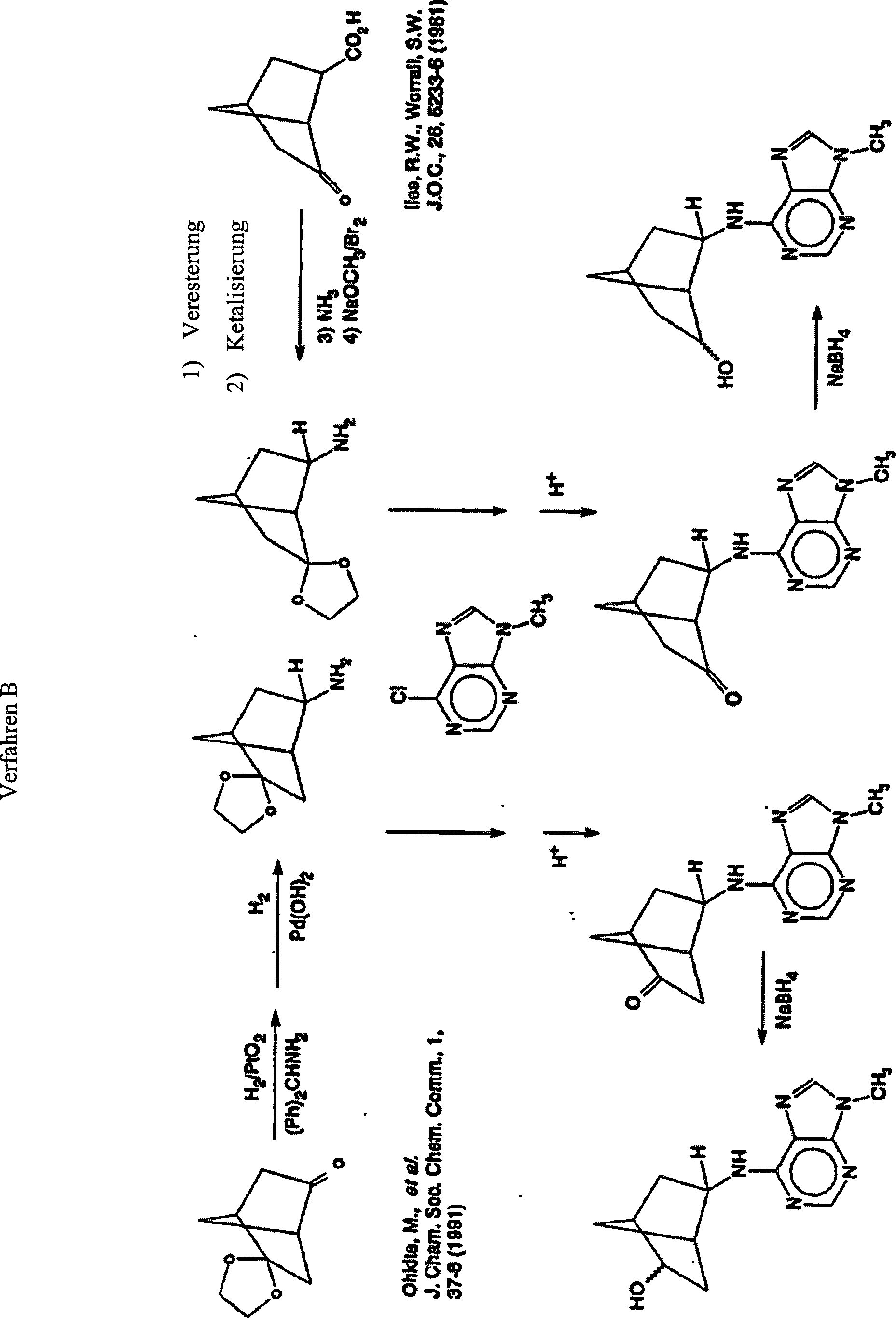
- Verbindungen der vorliegenden Erfindung, wobei R1 durch CH3 dargestellt ist und R3 ist eine substituierte Norbornylgruppe, wie in Tabelle II definiert ist, können wie folgt hergestellt werden (Verfahren B). Das Verfahren ist im Beispiel Nr. 21 im Detail beschrieben.
-
- Um die Derivate in Tabelle II herzustellen, bei welchen R2 nicht H ist, werden die obigen substituierten Norbornanone halogeniert, z.B. mit Chlor, und wie in den letzten zwei Schritten des Verfahrens A dargestellt ist, reagiert.
- Verfahren B – Detailliertes Beispiel: Herstellung von (±)-N6-[endo-2'-(endo-5'-hydroxyl)norbornyl]-9-Methyladenin
-
- A. endo-2-Aminonorbornan-5-on Ethylenketal. Eine Zwei-Liter Parrflasche wurde mit 210 g (1,25 mol) Norbornan-2,5-Dion Monoethylenketal (Ohkita M., et al., J. Chem. Soc. Chem. Comm. 1:37-38 (1991)), 238,3 g (1,25 mol) Aminodiphenylmethan, 108 ml (1,9 mol) Essigsäure, und 1,2 Liter Methanol beladen. 6,3g Platin (IV) oxid wurde unter Stickstoff der Mischung hinzugefügt, und diese wurde auf einem Parrapparat under 50 psi Wasserstoff platziert. Diese Reaktionsmischung wurde bei Raumtemperatur für 2 Stunden geschüttelt, woraufhin die berechnete Menge der Wasserstoffaufnahme erreicht wurde. Die Filtration und Konzentrierung mit einem Rotationsverdampfer ergab 494,4 g (quantitative Ausbeute). Es wurde eine 2 Liter Parrflasche mit diesem Produkt, 1,2 Liter Methanol und 17,5 g Palladiumhydroxid unter Stickstoff beladen. Diese Mischung wurde in einem Parrapparat für 6 Stunden bei 68 °C und 40 psi Wasserstoff hydriert, und dann wurde der Reaktion durch Schütteln bei Raumtemperatur über Nacht, ermöglicht abzukühlen. Der Katalysator wurde durch Filtration entfernt, und das Filtrat wurde konzentriert um einen pastösen Rückstand zu ergeben. Die Zerreibung mit Diethyleter und Trocknung ergab 212,2 g (74%) des endo-2-Aminonorbornan-5-on Ethylenketalacetat.
- B. (±)-N6-[endo-2'-aminonorbornan-5-on]-9-Methyladenin. Eine 5 Liter vierhalsige Rundbodenflasche, die mit einem mechanischen Rührer und einem Kondensator, an welchen ein Stickstoff- Gasbläschenerzeuger angeschlossen ist, ausgestattet ist, wurde mit 201,4 g (0,88 mol) endo-2-aminonorbornan-5-on Ethylenketalacetat, 151,7 g (0,9 mol) 6-Chlor-9-Methylpurin, 364,3 g (3,6 mol) Triethylamin, 3 g Tetrabutylammoniumiodid, und 2 Liter 1-Propanol beladen. Dies wurde unter Rückfluss über Nacht unter Rühren erhitzt, dann auf Raumtemperatur gekühlt. Die Mischung wurde filtriert und unter reduziertem Druck zu einem dicken braunen Sirup konzentriert. Die Zerreibung des Sirups mit Diethylether ergab 207,8 g (78%) eines festen Produktes. Eine 250 ml Rundbodenflasche wurde mit 58,8 g (0,195M) dieses Materials, 195 ml 3 N HCl, und 120 ml Methanol beladen. Diese Mischung wurde in einem Dampfbad für 30 min erhitzt, und dann unter reduziertem Druck konzentriert. Der Rückstand wurde zwischen gesättigtem wässrigem Kaliumcarbonat und Dichlormethan aufgeteilt. Zwei organische Extrakte (2 × 300 ml Methylchlorid) wurden zusammengefasst, mit Magnesiumsulfat getrocknet, und konzentriert um 52,9 g (quantitative Ausbeute) eine blassbraunen viskosen Öls zu erhalten, welches beim Stehen erstarrt.
- C. (±)-N6-[endo-2'-(endo-5'-hydroxyl)norbornyl]-9-Methyladenin Eine 1 Liter Rundbodenflasche wurde mit 48,9 g (0,18M) der obigen Verbindung, aufgelöst in 490 ml Methanol, beladen. Diese Mischung wurde in einem Wasserbad auf ca. 15 °C abgekühlt, woraufhin 3.5 g (0,09M) Natriumborhydrid unter Rühren auf einmal hinzugefügt wurde, und die Reaktionsmischung wurde über Nacht gerührt. Es wurde ein Feststoff (23,6 g) über Nacht gebildet, und wurde durch Filtration entfernt. Das Volumen des Methanfiltrats wurde durch Erwärmung unter einem Stickstoffstrom auf etwa 175 ml reduziert Auf das Abkühlen hin, wurde eine zweite Kristallmenge erhalten (5,2 g). Die kombinierten Feststoffprodukte wurden mit Erwärmen in Methanol aufgelöst, mit Aktivkohle behandelt, und filtriert. Das Filtrat wurde auf ein minimales Volumen reduziert, aus welchem das endgültige Feststoffprodukt heraus zu filtrieren war, welches nach dem Trocknen 23,7 g (50,8 %) Produkt ergab.
-

- Tabelle II
- Beispiele 19, 20, 23, 24, 28 und 29 in Tabelle II sind nicht als Ausführungsformen der Erfindung dargelegt, jedoch als Beispiele für das Verständnis der Erfindung nützlich.
-

-

- Verbindungen der vorliegenden Erfindung, wobei R1 durch CH3 dargestellt ist und R3 ist eine substituierte Norbornangruppe, wie in Tabelle III definiert ist, können wie folgt hergestellt werden (Verfahren C). Das Verfahren ist im Beispiel Nr. 30 im Detail beschrieben (Verfahren C-ii).
- Verfahren C
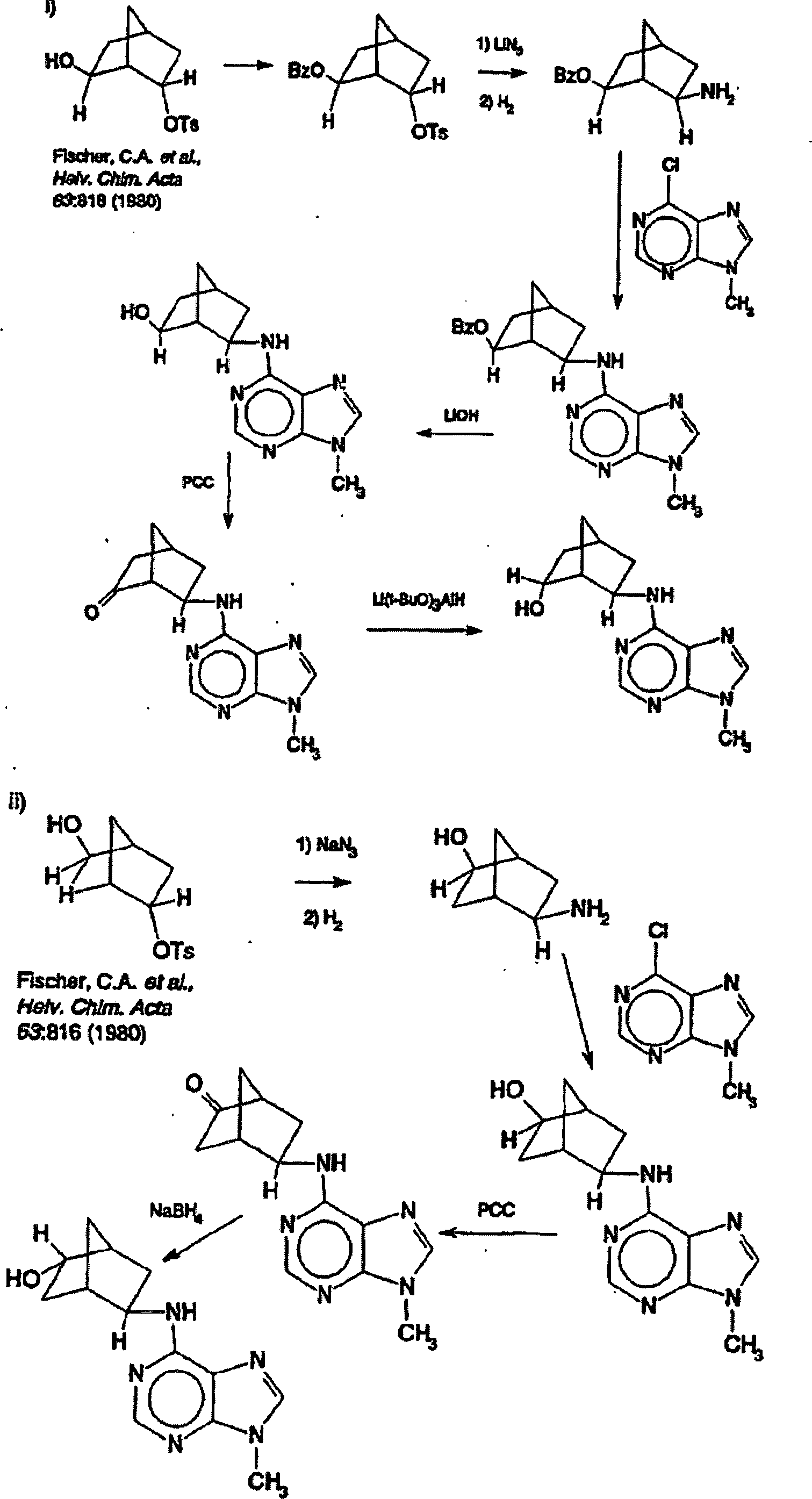
- In dem obigen Verfahren ist Pyridinium Chlorchromat als „PCC" abgekürzt.
- Verfahren C – Detailliertes Beispiel: Herstellung von (±)-N6-[exo-2'-(exo-5'-hydroxy)norbornyl]-9-Methyladenin.
-
- A. exo-2'-amino, exo-5'-Hydroxynorbornan: Eine Mischung aus 9,9 g (0,035 mol) exo-2-Hydroxy-5-endo-Tosyloxynorbornan und 11,4 g (0,175 mol) Natriumazid in 90 ml trockenem Dimethylformamid wurde auf 80 °C mit Rühren unter Stickstoff erhitzt. Nach 28 h, wurde die Reaktionsmischung konzentriert, und der Rückstand zwischen Wasser und Dichlormethan aufgeteilt. Das organische Material wurde abgetrennt und die wässrige Phase wurde nochmals mit Dichlormethan extrahiert, woraufhin die organischen Extrakte zusammengefasst wurden, getocknet (MgSO4), und konzentriert um 5 g Produkt zu erhalten.
- B. (±)-N6-[exo-2'-,(exo-5'-hydroxy)norbornyl]-9-Methyladenin. Eine Lösung des obigen Produkts in 25 ml trockenem Tetrahydrofuran wurde langsam zu 33 ml einer 1 M Lösung von Lithium Aluminiumhydrid in Tetrahydrofuran hinzugefügt. Nach dem Abschluss des Hinzufügens, wurde die Mischung über Nacht gerührt, woraufhin die Mischung mit 1,5 ml Wasser, dann mit 1,5 ml 6 N Natriumhydroxid und 4.5 ml Wasser behandelt wurde. Die resultierende Mischung wurde filtriert, konzentriert und direkt im nächsten Schritt verwendet.
- C. Das Aminprodukt in B) wurde mit 6-Chlor-9-Methylpurin im Wesentlichen wie im Schritt B) des detaillierten Beispiels für Verfahren B reagiert. Die Isolation beinhaltete Flash-Chromatographie, unter anfänglicher Verwendung von Ethylacetat, dann Ethylacetat/Methanol (8:2) als Elutionsmittel. Die Konzentrierung der entsprechenden Fraktionen ergab 100 mg reines Produkt.
-

- Tabelle III
- Beispiele 32, 33, 34, und 35 in Tabelle III sind nicht als Ausführungsformen der Erfindung dargelegt, jedoch als Beispiele für das Verständnis der Erfindung nützlich.
-

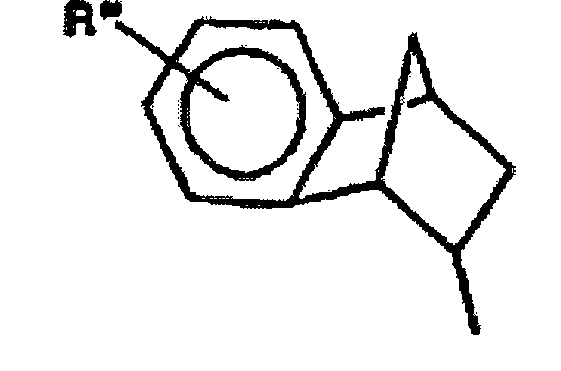
- Verbindungen der gegenwärtigen Erfindung, wobei R3 wie folgt definiert ist:können wie folgt hergestellt werden (Verfahren D). Diese Beispiele sind nicht als Ausführungsformen der Erfindung dargelegt, jedoch als Beispiele für das Verständnis der Erfindung nützlich. Das Verfahren ist im Beispiel Nr. 36 im Detail beschrieben. Verfahren D
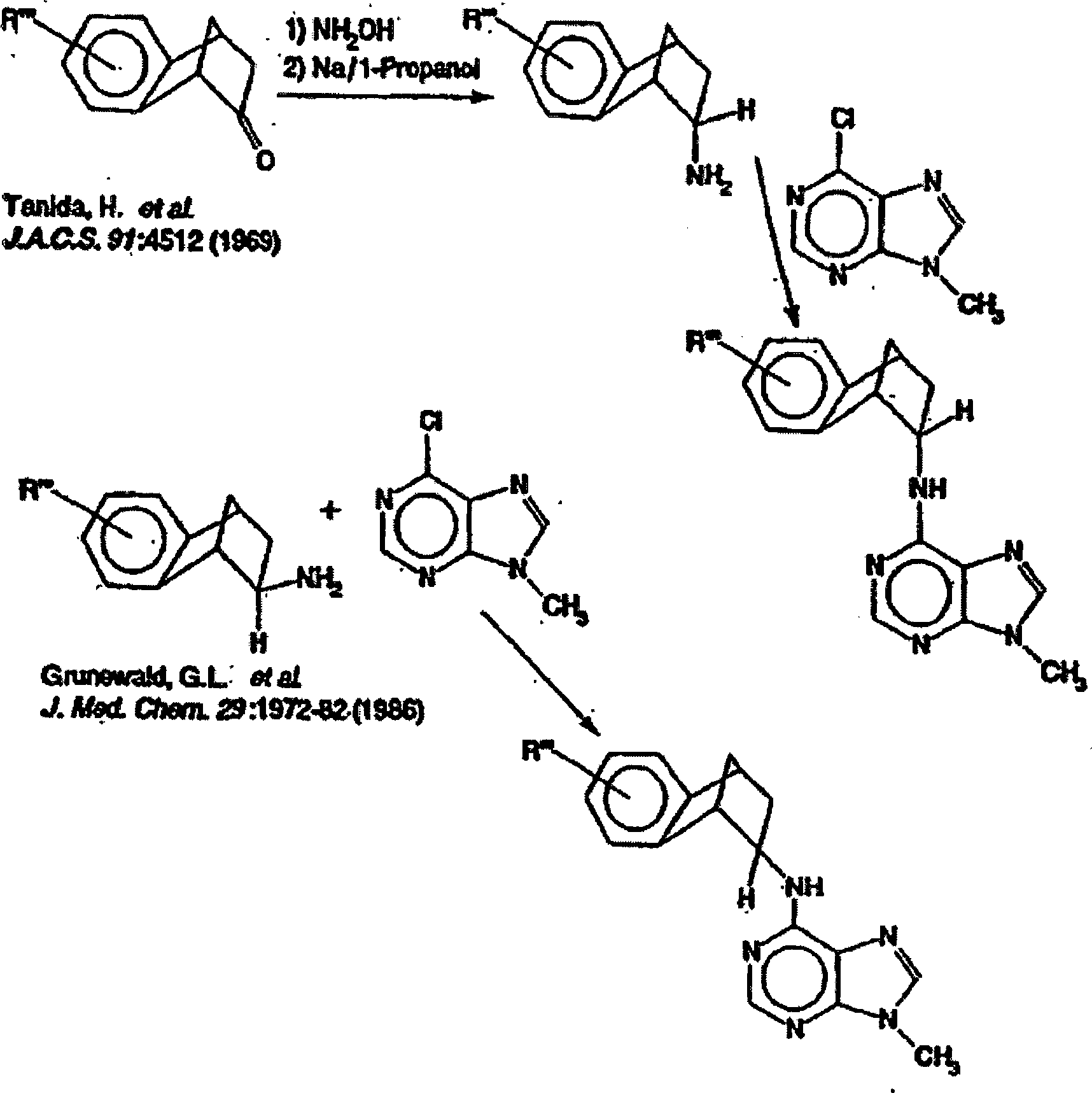
- Verbindungen wobei R''' = OH werden durch Spaltung des Methylesters durch Bortribromid hergestellt.
- Verfahren D – Detailliertes Beispiel: Herstellung von (±)-N6-(exo-2'-amino-6'-hydroxybenzonorbornyl]-9-Methyladenin.
-
- A. (±)-N6-(exo-2'-amino-6'-hydroxybenzonorbornyl]-9-Methyladenin: Die folgenden Reaktanden wurden gemischt und bis zum Rückfluss über Nacht unter Stickstoff erhitzt: 440 mg (2,33 mmol) exo-2-Amino-6-Methoxybenzonorboran, 392 mg (2.33 mmol) 6-Chlor-9-Methylpurin, 476 mg (4.7 mmol) Triethylamin, 48 mg Kaliumiodid und 20 ml 1-Propanol. Die Reaktionsmischung wurde dann zu einem pastiösen Rückstand konzentriert, welcher zwischen Dichlormethan und Wasser aufgeteilt war. Trocknen (MgSO4) und Konzentrierung des organischen Materials ergab ein Öl, welches chromatographiert wurde (Silicagel; Ethylacetat/Methanol Gradient von 100% Ethylacetat bis zu 95:5) um ein Produkt zu ergeben, welches in das Maleatsalz (310 mg) umgewandelt wurde.
- B. (±)-N6-(exo-2'-amino-6'-hydroxybenzonorbornyl]-9-Methyladenin Das vorherige Produkt (88mg) wurde mit 2 ml Dichlormethan und 0,7 ml (2.4 äqu) Bortribromid bei -70 °C unter Stickstoff und Rühren für 30 Minuten gemischt. Die Mischung wurde auf Raumtemperatur gebracht, und für eine Stunde rückflussgekocht. Die Reaktion wurde durch die langsame Zugabe von 1 ml Methanol mit Kühlung gelöscht. Diese wurde dann konzentriert, und nach dem Waschen mit Isopropanol und gesättigtem Natriumcarbonat, wurde die resultierende Mischung auf eine entsalzende Ionenaustauscher XAD-2 Säule geladen. Die Aufbereitung des Eluats ergab ein kristallines Produkt.
-

- Tabelle IV
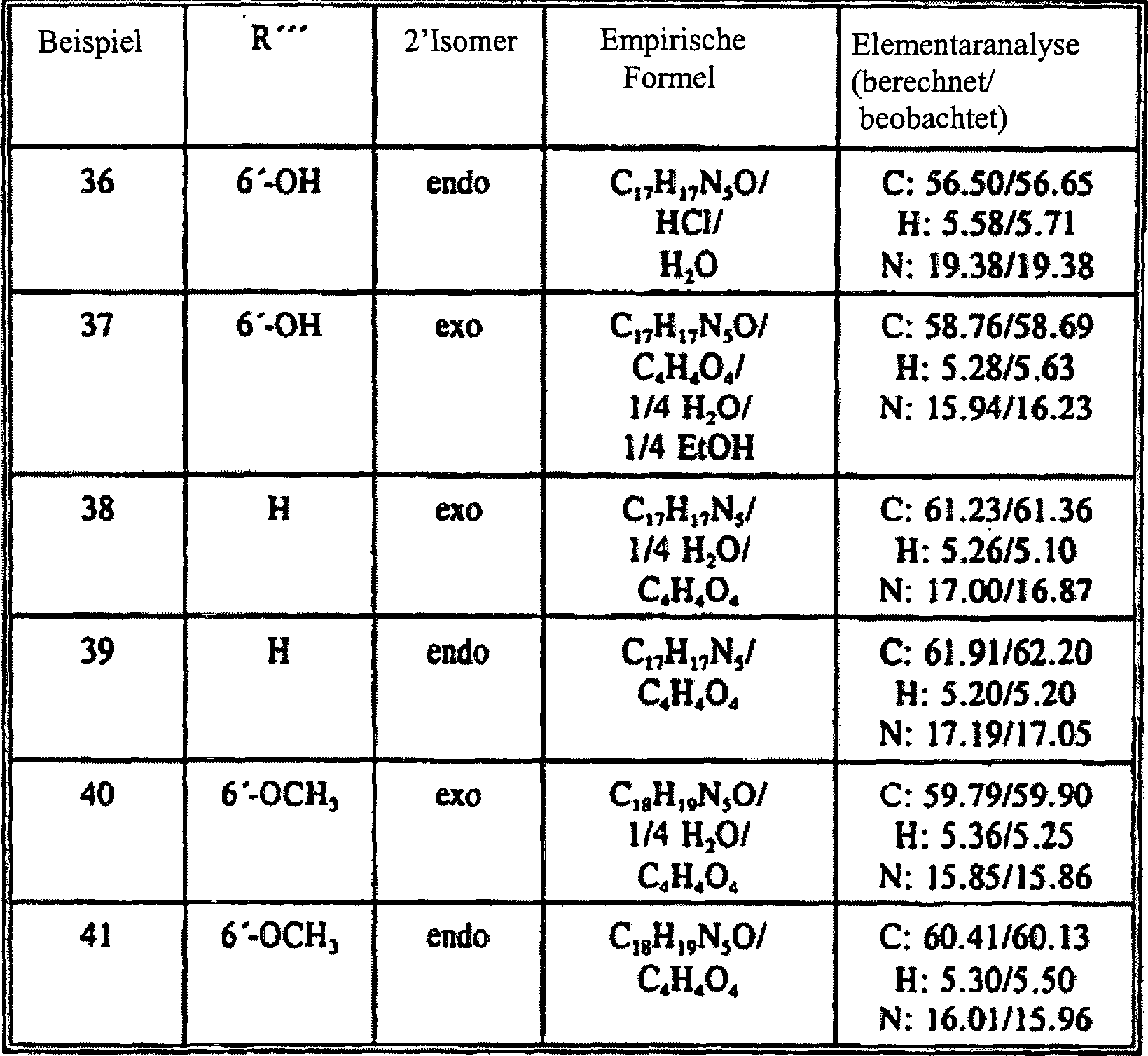
- Verbindungen der vorliegenden Erfindung, wobei R1 nicht Wasserstoff ist, können im Wesentlichen gemäß Verfahren B hergestellt werden, mit der Ausnahme dass anstelle von 9-Methyl-6-Chlorpurin verschiedene 9-Alkyl-6-Chlorpurine verwendet werden können, welche gemäß Robins R, et al, J. Am. Chem. Soc. 79:490-4 (1957) durch Ersetzten von Methylamin mit einem gewünschten Alkylamin hergestellt werden.
-
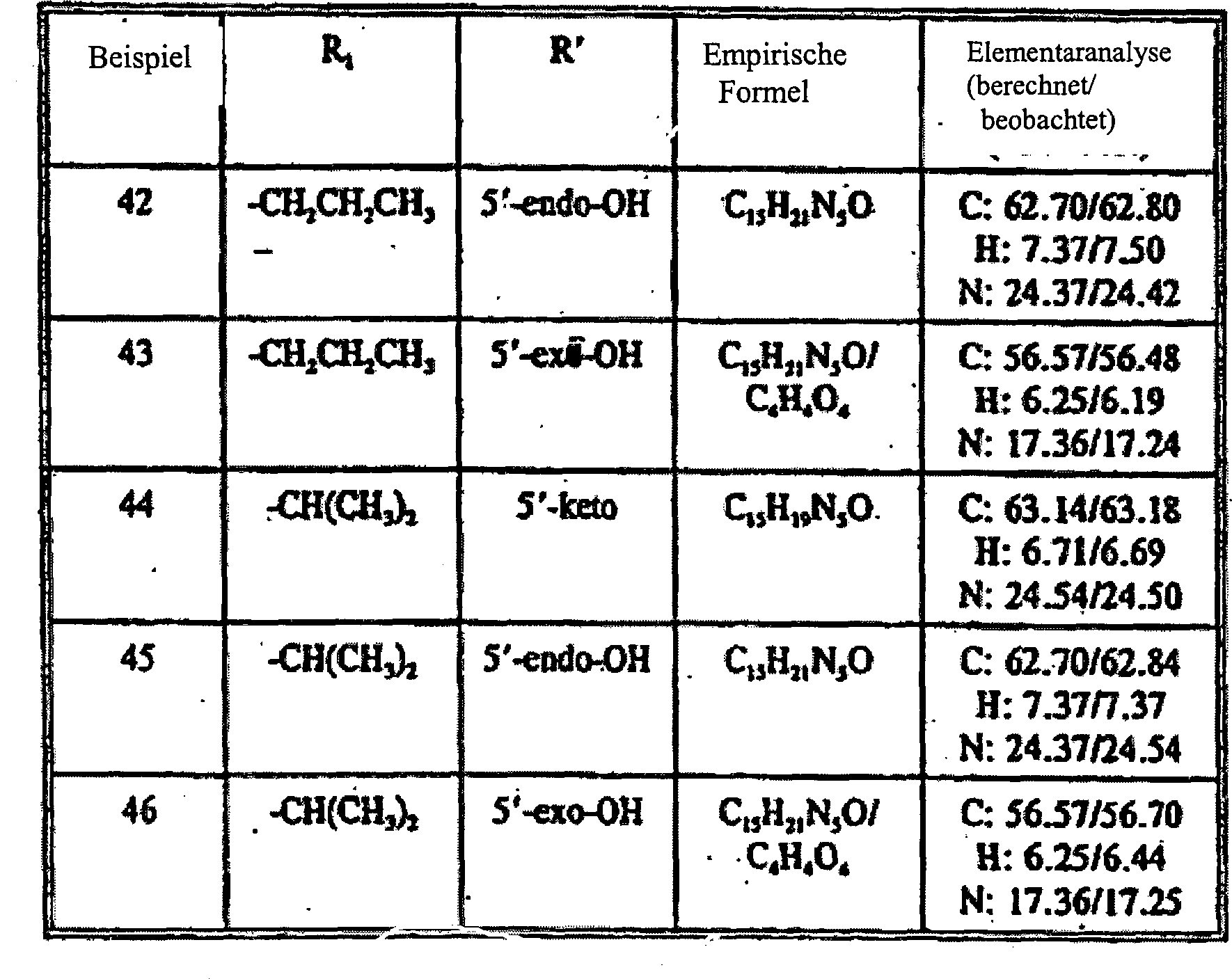
- Tabelle V
- Beispiel 44 der Tabelle V ist nicht als Ausführungsform der Erfindung dargelegt, jedoch als ein Beispiel für das Verständnis der Erfindung nützlich.
-
- Pharmakologische Untersuchungen von beispielhaft erläuterten Verbindungen
- Die pharmakologischen Testverfahren sind nachfolgend beschrieben. Die Verbindungen der vorliegenden Erfindung wurden auf Adenosin-Antagonist-Aktivität untersucht, deren Ergebnisse in den beigelegten Tabellen zusammengefasst sind. Diese Daten wurden erzeugt um ein Maß für die Selektivität der Verbindung (A1/A2) für Adenosin A1 Rezeptor und ihre in vitro funktionelle Wirksamkeit am Adenosin A1 Rezeptor bereitzustellen. Untersuchungsergebnisse für Vergleichsbeispiele des Standes der Technik (siehe Ukena, D., et al., FEBS Lett. 215:203-8 (1987); Thompson, R., et al., J. Med. Chem. 34:2877-82 (1991); Olsson, R., US Patent Nr. 5,066,655) sind für Bezugszwecke eingeschlossen. (Beispiel 47: N6-Cyclohexyl-9-Methyladenin, Beispiel 48: N6-Cyclopentyl-9-Methyladenin, Beispiel 49: N6-(endo 2-norornyl)-9-Methyladenin).
- 1. Rezeptor Affinitätsexperimente (Radioligandbindung)
- Gewebe (Rinderkaudatum, Meerschweinchen-Kortex, Meerschweinchen-Striatum) wurden mit einem Brinkman Polytron homogenisiert und die Membranen wurden durch Zentrifugation bei 40.000 × g bei 4 °C für 10 min gesammelt. Die Membranen wurden gewaschen durch Resuspension und Zentrifugation, resuspendiert und in der Gegenwart von 1 U/ml Adenosindeaminase für 15 bis 30 min bei 37 °C inkubiert. Die Membranen wurden verdünnt, einmal zentrifugiert, und resuspendiert zu einer endgültigen Konzentration von 0,5–2 mg Protein/ml. Adenosindeaminase (0,1 U/ml) wurde der letzten Resuspendierung hinzugefügt. Die Untersuchungen wurden durch die Zugabe der Membranen eingeleitet. Die Verbindungen der vorliegenden Erfindung wurden folgenden experimentellen Bedingungen ausgesetzt: A1 Bestimmungen/inkubiert mit [3H]CHA bei Raumtemperatur für 2 Stunden, A2 Bestimmungen/inkubiert mit [3H]NECA bei Raumtemperatur für 1 Stunde. Die Untersuchungen wurden durch Filtration beendet, und die Filter wurden in einem Szintillationszäliler gezählt. Die Daten wurden mit dem Ligandprogramm von Munson und Rodbard (1980) analysiert, und die Ergebnisse sind in der Tabelle VI unten dargelegt.
- Wie oben angemerkt wurde, sind die Beispiele 1 bis 15, 19, 20, 23, 24, 28, 29, 32 bis 41 und 44 nicht als Ausführungsformen der Erfindung dargelegt, jedoch als ein Beispiel für das Verständnis der Erfindung nützlich
- Tabelle VI Adenosin A1 und A2 Rezeptor Affinität und resultierendes Selektivitätsverhältnis
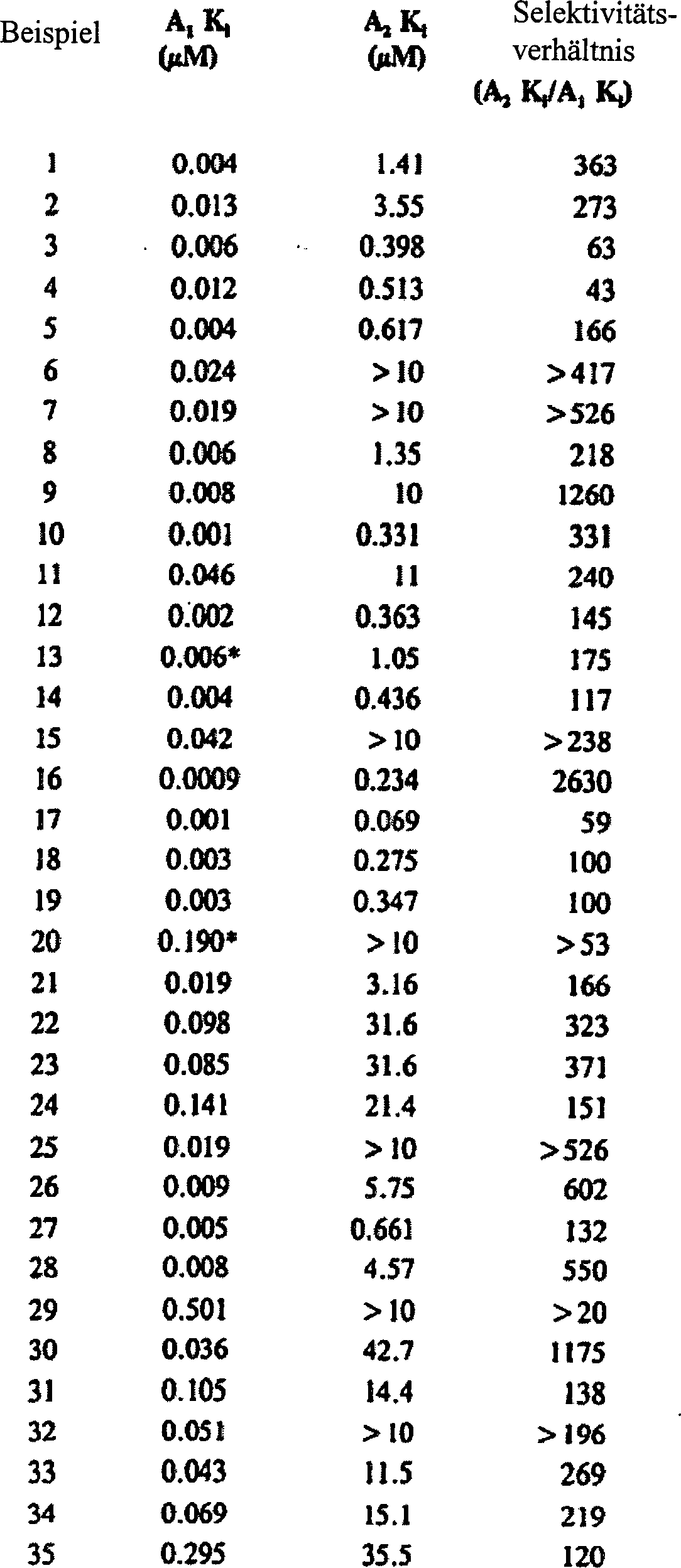
-
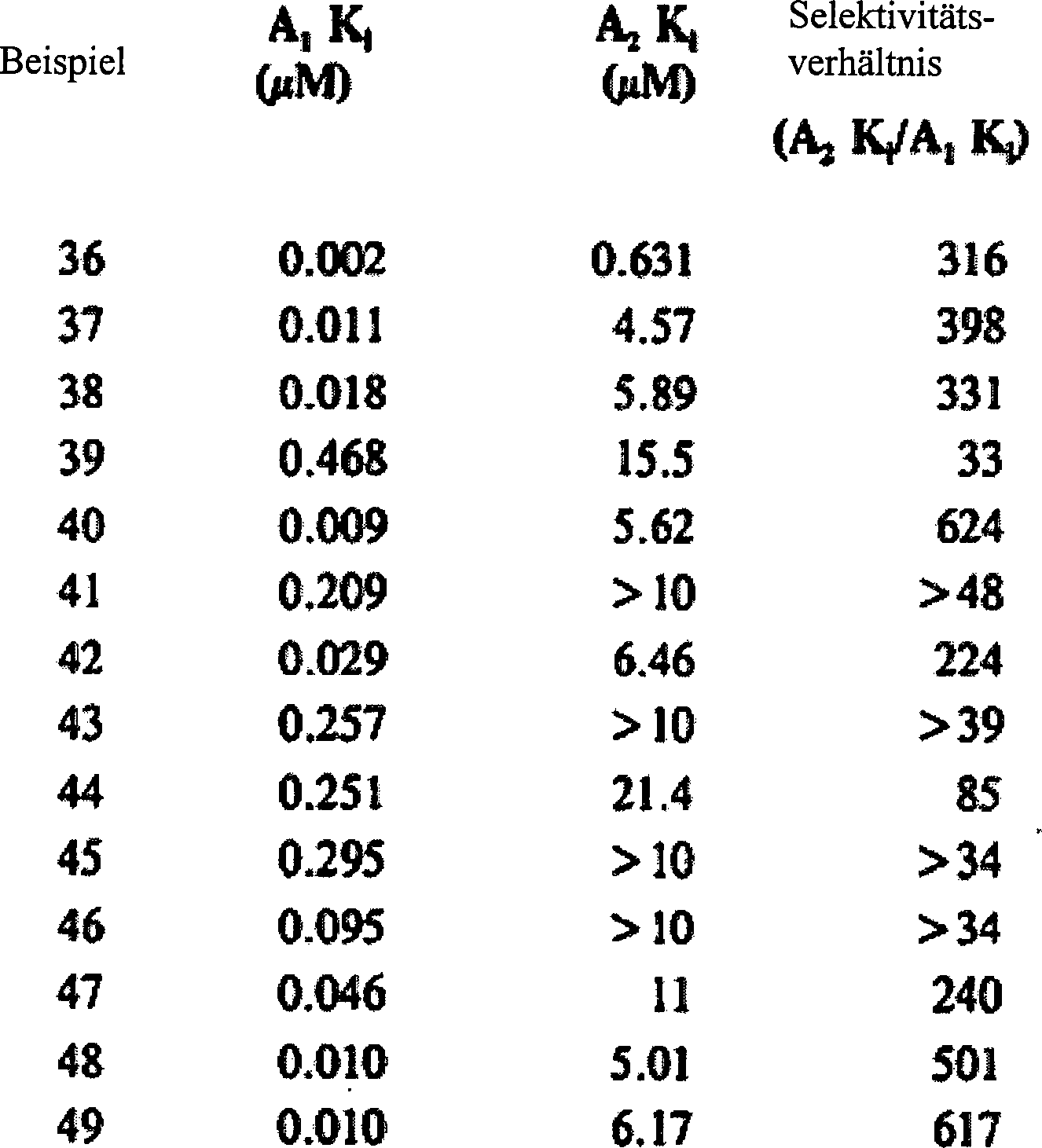
-
- * Diese Werte wurden mittels Meerschweinchengewebe erhalten.
- Diese Daten zeigen die hohen Grade der Adenosin A1 Rezeptoraffinität und Selektivität die mit Verbindungen der vorliegenden Erfindung erreicht wurden.
- 2. Adenosin – A1/A2 In Vitro funktionelle Untersuchung
- (A1 Untersuchung: Negative inotrope Reaktion) – die linken Vorhöfe von Meerschweinchen wurden in Organbäder, die mit Krebs-Henseleit-Lösung gefüllt ist, begast mit 95% O2 und 5% CO2 platziert und bei einer Temperatur von 31 °C gehalten. Eine anfängliche Ruhespannung von 1 g wurde auf jedes Gewebe platziert, welches äquilibrieren gelassen wurde. Die Vorhöfe wurden elektrisch stimuliert um eine Zuckreaktion zu erzeugen. Eine Dosis-Wirkung-Kurve für die Inhibierung der elektrisch-hervorgerufenen Zuckreaktion wurde durch die kumulative Addition des Adenosin Agonisten 5'N-Ethylamidoadenosin erhalten. Nach dem Auswaschen der Gewebe, wurde eine festgelegte Konzentration der Testverbindung hinzugefügt und das Gewebe wurde für 1 Stunde äquilibrieren lassen, vor der kumulativen Zugabe des Adenosin Agonisten 5'-N-Ethylcaroxamidoadenosin (NECA) um eine neue Dosis-Wirkung-Kurve zu erzeugen. Diese Vorgehensweise wurde mit unterschiedlich festgelegten Konzentrationen der Testverbindung wiederholt um eine Familie an Dosis-Wirkung-Kurven zu erzeugen. Schild-Diagramme wurden von den resultierenden Konzentration-Wirkung-Kurven konstruiert, und KB (Antagonist Dissoziationskonstante)-Werte wurden erhalten. Die Ergebnisse (see Tabelle VII) werden als KB-Werte ausgedrückt (Konzentration der Testverbindung so dass eine Hälfte der Gewebe Adenosinrezeptor-Population durch die Testverbindung besetzt ist).
- (A2 Untersuchung: Vasodilation) – Von Meerschweinchen isolierte Brustaorta wurde in Ringsegmente geschnitten und zwischen parallele Drähte für die Messung der isometrischen Kraft in der obigen Lösung bei 37 °C gelegt. Eine anfängliche Ruhespannung von 2 g wurde auf jedes Gewebe platziert, welches dann für 1 Stunde äquilibrieren lassen wurde. Phenylephrin (3 μM) wurde zu jedem Gewebe hinzugefügt um eine kontraktile Reaktion hervorzurufen, und sobald dies erhalten war, wurde die Testverbindung hinzugefügt und für 1 Stunde äquilibrieren lassen. Eine Konzentration-Wirkung-Kurve wurde erzeugt durch die Addition von NECA in 0,5 M Einheitsschrittgrößen. Die Reaktionen wurden als Prozentsatz Inhibierung der Phenylephrin-induzierten Kontraktionen ausgedrückt. Die Datenanalyse wurde in einer analogen Art und Weise wie für die A1-Untersuchung oben durchgeführt, mit den Ergebnissen als KB-Werte ausgedrückt.
- Tabelle VII Funktionelle Wirksamkeiten an Adenosin A1 und A2 Rezeptoren und resultierendes Selektivitätsverhältnis

- Diese Daten zeigen die hohen Grade der Adenosin A1 Rezeptor in vitro funktionelle Aktivität und Selektivität (A1 vs. A2).
- Nachdem die Erfindung vollständig beschrieben wurde, wird der Durchschnittsfachmann verstehen, dass dasselbe innerhalb eines weiten und äquivalenten Bereichs von Zuständen, Formulierungen, und anderen Parameter ohne den Schutzumfang der Erfindung oder jeder Ausführungsform davon zu beeinflussen, durchgeführt werden kann. Alle zitierten Patente und Publikationen werden durch Bezugsnahme in ihrer Gesamtheit hierin eingeschlossen.
Claims (13)
- Verbindung mit der Formeloder ein pharmazeutisch annehmbares Salz davon; worin R1 ein C1 bis C4 geradkettiges oder verzweigtes Alkyl ist; R2 ausgewählt ist aus der Gruppe bestehend aus H, -OR1, -SR1, -N(R4)(R5), Aminocarbonyl, Halogen, und -CN, worin R4 und R5 unabhängig ein C1 bis C6 geradkettiges oder verzweigtes Alkyl, oder R4 und R5 gemeinsam mit dem Stockstoff, an welchen sie gebunden sind, einen 3- bis 7-gliedrigen Heterocycloalkylsubstituenten bilden, welcher gegebenenfalls ein zusätzliches Heteroatom enthält, das ausgewählt ist aus der Gruppe bestehend aus Stickstoff, Sauerstoff und Schwefel; R3
 ist und R' -OH oder -NH2 ist und R'' H ist, oder ein pharmazeutisch annehmbares Salz davon.
ist und R' -OH oder -NH2 ist und R'' H ist, oder ein pharmazeutisch annehmbares Salz davon.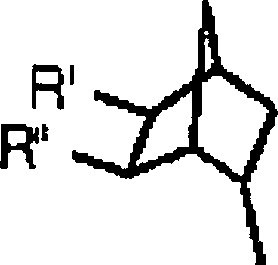
- Verbindung nach Anspruch 1, in welcher R1 Methyl ist, oder ein pharmazeutisch annehmbares Salz davon.
- Verbindung nach Anspruch 1, in welcher R2 H, -N(R4)(R5), -OR1, -SR1, Halogen oder -CN ist, oder ein pharmazeutisch annehmbares Salz davon.
- Verbindung nach Anspruch 3, in welcher R' -OH ist und R'' Wasserstoff ist, oder ein pharmazeutisch annehmbares Salz davon.
- Verbindung nach Anspruch 4, in welcher R2 Methylthio- oder Methoxy- ist, oder ein pharmazeutisch annemhbares Salz davon.
- Verbindung nach Anspruch 4, in welcher R4 und R5 unabhängig C1 bis C3 Alkylradikale sind, oder ein pharmazeutisch annehmbares Salz davon.
- Verbindung nach Anspruch 6, in welcher R4 Methyl ist und R5 Isopropyl ist, oder ein pharmazeutisch annehmbares Salz davon.
- Verbindung welche (±)-N6-[endo-2'-(endo-5'-hydroxyl)norbornyl]-9-Methyladenin oder (±)-N6-[endo-2'-(endo-5'-hydroxy)norbornyl]-8-isopropylmethylamino-9-Methyladenin ist, oder ein pharmazeutisch annehmbares Salz dieser Verbindungen.
- Pharmazeutische Zusammensetzung, die eine Verbindung nach einem der vorstehenden Ansprüche oder ein pharmazeutisch annehmbares Salz derselben und einen pharmazeutisch annehmbaren Träger enthält.
- Verbindung oder ein pharmazeutisch annehmbares Salz derselben nach einem der Ansprüche 1 bis 8 oder eine pharmazeutische Zusammensetzung nach Anspruch 9, für die Verwendung zur Inhibierung der Adenosin A1 Rezeptoraktivierung in einem Säugetier, das diese benötigt.
- Verbindung oder ein ein pharmazeutisch annehmbares Salz derselben nach einem der Ansprüche 1 bis 8 oder eine pharmazeutische Zusammensetzung nach Anspruch 9, für die Verwendung in einem Verfahren zur Auslösung von Diurese, zum Schutz der Nieren gegen akutes oder chronisches Nierenversagen, zur Erleichterung des Erholens aus dem Koma, zur Verbesserung des therapeutischen Ergebnisses nach Defibrillation oder kardiopulmunärer Wiederbelebung, zur Wiederherstellung der Herzfunktion nach kardioplegischen Vorgängen, oder zur Behandlung von oder Vorsorgen vor Claudicatio intermittens.
- Verwendung einer Verbindung nach einem der Ansprüche 1 bis 8 oder eines pharmazeutisch annehmbaren Salzes derselben bei der Herstellung einer pharmazeutischen Zusammensetzung nach Anspruch 9 zur Verwendung bei der Inhibierung der Adenosin A1 Rezeptoraktivierung in einem Säugetier, das diese benötigt.
- Verwendung einer Verbindung nach einem der Ansprüche 1 bis 8 oder eines pharmazeutisch annehmbaren Salzes derselben bei der Herstellung einer pharmazeutischen Zusammensetzung nach Anspruch 9 zur Verwendung in einem Verfahren zur Auslösung von Diurese, zum Schutz der Nieren gegen akutes oder chronisches Nierenversagen, zur Erleichterung des Erholens aus dem Koma, zur Verbesserung des therapeutischen Ergebnisses nach Defibrillation oder kardiopulmunärer Wiederbelebung, zur Wiederherstellung der Herzfunktion nach kardioplegischen Vorgängen, oder zur Behandlung von oder Vorsorgen vor Claudicatio intermittens.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US08/299,992 US5670501A (en) | 1994-09-01 | 1994-09-01 | N-substituted 9-alkyladenines |
| US299992 | 1994-09-01 | ||
| PCT/US1995/010693 WO1996006845A1 (en) | 1994-09-01 | 1995-08-22 | Substituted 9-alkyladenines |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| DE69534718D1 DE69534718D1 (de) | 2006-02-02 |
| DE69534718T2 true DE69534718T2 (de) | 2006-09-07 |
Family
ID=23157204
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| DE69534718T Expired - Fee Related DE69534718T2 (de) | 1994-09-01 | 1995-08-22 | Substituierte 9-alkyladenine |
Country Status (6)
| Country | Link |
|---|---|
| US (2) | US5670501A (de) |
| EP (2) | EP1602657A3 (de) |
| JP (1) | JPH10507744A (de) |
| AU (1) | AU706068B2 (de) |
| DE (1) | DE69534718T2 (de) |
| WO (1) | WO1996006845A1 (de) |
Families Citing this family (32)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| HUP9801177A3 (en) * | 1995-11-14 | 1998-11-30 | Pharmacia & Upjohn Spa | Tetrahydronaphthyl, indanyl and indolyl substituted pyrido[2,3-d]pyrimidine and purine derivatives, process for their preparation and pharmaceutical compositions containing them |
| AU2001280946A1 (en) * | 2000-08-01 | 2002-02-13 | University Of Virginia Patent Foundation | Use of selective adenosine a1 receptor agonists, antagonists and allosteric enhancers to manipulate angiogenesis |
| DE10041478A1 (de) | 2000-08-24 | 2002-03-14 | Sanol Arznei Schwarz Gmbh | Neue pharmazeutische Zusammensetzung |
| DE10041479A1 (de) * | 2000-08-24 | 2002-03-14 | Sanol Arznei Schwarz Gmbh | Neue pharmazeutische Zusammensetzung zur Verabreichung von N-0923 |
| ITRM20010465A1 (it) | 2001-07-31 | 2003-01-31 | Sigma Tau Ind Farmaceuti | Derivati della triazolil-imidazopiridina e delle triazolilpurine utili come ligandi del recettore a2a dell'adenosina e loro uso come medicam |
| EP1465634B1 (de) | 2001-12-12 | 2014-10-22 | The Government of the United States of America, as represented by the Secretary Department of Health and Human Services | Verfahren zur verwendung adenosinrezeptorinhibitoren zur verstärkung der immunantwort und entzündung |
| EP1426049B1 (de) * | 2002-12-02 | 2005-05-18 | Schwarz Pharma Ag | Verabreichung von Rotigotine zur Behandlung der Parkinson'schen Krankheit durch Iontophorese |
| EP1863815A1 (de) * | 2005-03-11 | 2007-12-12 | Aderis Pharmaceuticals, Inc. | Substituierte 9-alkyladenine und deren verwendung |
| US7870903B2 (en) * | 2005-07-13 | 2011-01-18 | Halliburton Energy Services Inc. | Inverse emulsion polymers as lost circulation material |
| KR100869259B1 (ko) | 2007-05-29 | 2008-11-18 | 한국화학연구원 | 광학활성을 갖는 3a-메틸-피롤로[3,4-c]피페리딘의거울상 이성질체의 제조방법 |
| EP2920171B1 (de) | 2012-11-16 | 2018-08-29 | Merck Sharp & Dohme Corp. | Purinhemmer von humaner phosphatidylinosit-3-kinase delta |
| CN104918940B (zh) * | 2012-11-16 | 2017-03-08 | 默沙东公司 | 人磷脂酰肌醇3‑激酶δ的嘌呤抑制剂 |
| JP7227131B2 (ja) | 2016-12-03 | 2023-02-21 | ジュノー セラピューティクス インコーポレイテッド | Car-t細胞の投薬を決定するための方法 |
| WO2018102786A1 (en) | 2016-12-03 | 2018-06-07 | Juno Therapeutics, Inc. | Methods for modulation of car-t cells |
| BR112019011207A2 (pt) | 2016-12-05 | 2019-10-08 | Juno Therapeutics Inc | produção de células modificadas para terapia celular adotiva |
| KR20200054160A (ko) | 2017-06-02 | 2020-05-19 | 주노 쎄러퓨티크스 인코퍼레이티드 | 입양 세포 요법을 사용한 치료를 위한 물품 제조 및 방법 |
| WO2019006427A1 (en) | 2017-06-29 | 2019-01-03 | Juno Therapeutics, Inc. | WALL MODEL FOR ASSESSING TOXICITIES ASSOCIATED WITH IMMUNOTHERAPIES |
| CA3079081A1 (en) | 2017-10-19 | 2019-04-25 | Michael D. Bartberger | Benzimidazole derivatives and their uses |
| KR20200074997A (ko) | 2017-11-01 | 2020-06-25 | 주노 쎄러퓨티크스 인코퍼레이티드 | B-세포 성숙 항원에 특이적인 항체 및 키메릭 항원 수용체 |
| KR102845789B1 (ko) | 2017-11-01 | 2025-08-14 | 주노 쎄러퓨티크스 인코퍼레이티드 | B 세포 성숙 항원에 특이적인 키메라 항원 수용체 및 암호화 폴리뉴클레오타이드 |
| WO2019089858A2 (en) | 2017-11-01 | 2019-05-09 | Juno Therapeutics, Inc. | Methods of assessing or monitoring a response to a cell therapy |
| CN111989106A (zh) | 2017-12-01 | 2020-11-24 | 朱诺治疗学股份有限公司 | 基因工程化细胞的给药和调节方法 |
| WO2019118937A1 (en) | 2017-12-15 | 2019-06-20 | Juno Therapeutics, Inc. | Anti-cct5 binding molecules and methods of use thereof |
| MX2021005024A (es) | 2018-11-01 | 2021-07-21 | Juno Therapeutics Inc | Metodos de tratamiento que utilizan receptores de antigenos quimericos especificos para antigeno de maduracion de celulas b. |
| CA3116413A1 (en) | 2018-11-01 | 2020-05-07 | Juno Therapeutics, Inc. | Chimeric antigen receptors specific for g protein-coupled receptor class c group 5 member d (gprc5d) |
| SG11202105084VA (en) | 2018-11-16 | 2021-06-29 | Juno Therapeutics Inc | Methods of dosing engineered t cells for the treatment of b cell malignancies |
| FI3886875T3 (fi) | 2018-11-30 | 2024-07-23 | Juno Therapeutics Inc | Adoptiivista soluterapiaa käyttäviä hoitomenetelmiä |
| CN113365660A (zh) | 2019-01-29 | 2021-09-07 | 朱诺治疗学股份有限公司 | 对受体酪氨酸激酶样孤儿受体1(ror1)具特异性的抗体及嵌合抗原受体 |
| BR112022020333A2 (pt) | 2020-04-10 | 2022-11-22 | Juno Therapeutics Inc | Métodos e usos relacionados à terapia celular projetada com um receptor de antígeno quimérico que alveja o antígeno de maturação de células b |
| US20250381272A1 (en) | 2022-06-22 | 2025-12-18 | Juno Therapeutics, Inc. | Treatment methods for second line therapy of cd19-targeted car t cells |
| KR20250047766A (ko) | 2022-08-05 | 2025-04-04 | 주노 쎄러퓨티크스 인코퍼레이티드 | Gprc5d 및 bcma에 특이적인 키메라 항원 수용체 |
| CN120712102A (zh) | 2022-12-13 | 2025-09-26 | 朱诺治疗学股份有限公司 | 对baff-r和cd19具特异性的嵌合抗原受体及其方法和用途 |
Family Cites Families (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE2338705A1 (de) * | 1973-07-31 | 1975-02-13 | Boehringer Mannheim Gmbh | Neue n(6)-disubstituierte adenosinderivate und verfahren zur herstellung derselben |
| DE2355536A1 (de) * | 1973-11-07 | 1975-05-22 | Merck Patent Gmbh | Adeninderivate und verfahren zu ihrer herstellung |
| US4364922A (en) * | 1980-10-14 | 1982-12-21 | University Of Virginia Alumni Patents Foundation | Adenosine antagonists in the treatment and diagnosis of A-V node conduction disturbances |
| US4714697A (en) * | 1985-09-09 | 1987-12-22 | Warner Lambert Company | N6 -bicycloadenosines and methods of use |
| US4612315A (en) * | 1984-10-26 | 1986-09-16 | The United States Of America As Represented By The Department Of Health And Human Services | Biologically-active 1,3-dipropyl-8-phenylxanthine derivatives |
| US4751292A (en) * | 1985-07-02 | 1988-06-14 | The Plant Cell Research Institute, Inc. | Adamantyl purines |
| DE3529497A1 (de) * | 1985-08-17 | 1987-02-26 | Boehringer Mannheim Gmbh | N(pfeil hoch)6(pfeil hoch)-disubstituierte purinderivate, verfahren zu deren herstellung sowie diese verbindungen enthaltende arzneimittel |
| US5066655A (en) * | 1987-04-24 | 1991-11-19 | Whitby Research, Inc. | N6-substituted 9-methyladenines: a new class of adenosine receptor antagonists |
| US5565566A (en) * | 1987-04-24 | 1996-10-15 | Discovery Therapeutics, Inc. | N6 -substituted 9-methyladenines: a new class of adenosine receptor antagonists |
| US4980379A (en) * | 1988-06-30 | 1990-12-25 | The University Of Virginia | Use of adenosine antagonists in the treatment of bradyarrhythmias and hemodynamic depression associated with cardiopulmonary resucitation and/or cardiovascular collapse |
| KR910700253A (ko) * | 1989-01-31 | 1991-03-14 | 로버트 제이, 바란 | N_6-치환-9-메틸아데닌: 신규한 부류의 아데노신 수용체 길항질 |
| US5117830A (en) * | 1990-11-08 | 1992-06-02 | Whitby Research, Inc. | Method of determining viability of tissue |
| CA2060138A1 (en) * | 1991-01-29 | 1992-07-30 | Youichi Shiokawa | New use of the adenosine antagonist |
-
1994
- 1994-09-01 US US08/299,992 patent/US5670501A/en not_active Expired - Fee Related
-
1995
- 1995-08-22 JP JP8508833A patent/JPH10507744A/ja not_active Ceased
- 1995-08-22 AU AU33706/95A patent/AU706068B2/en not_active Ceased
- 1995-08-22 WO PCT/US1995/010693 patent/WO1996006845A1/en not_active Ceased
- 1995-08-22 DE DE69534718T patent/DE69534718T2/de not_active Expired - Fee Related
- 1995-08-22 EP EP05012214A patent/EP1602657A3/de not_active Withdrawn
- 1995-08-22 EP EP95930252A patent/EP0778836B1/de not_active Expired - Lifetime
-
1997
- 1997-08-04 US US08/904,553 patent/US5981524A/en not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| AU3370695A (en) | 1996-03-22 |
| US5670501A (en) | 1997-09-23 |
| WO1996006845A1 (en) | 1996-03-07 |
| EP0778836B1 (de) | 2005-12-28 |
| EP1602657A3 (de) | 2006-11-15 |
| DE69534718D1 (de) | 2006-02-02 |
| EP0778836A4 (de) | 1998-02-04 |
| AU706068B2 (en) | 1999-06-10 |
| EP0778836A1 (de) | 1997-06-18 |
| US5981524A (en) | 1999-11-09 |
| EP1602657A2 (de) | 2005-12-07 |
| JPH10507744A (ja) | 1998-07-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE69534718T2 (de) | Substituierte 9-alkyladenine | |
| DE69815378T2 (de) | Adenosin a1-rezeptor agonisten | |
| DE69033915T2 (de) | Xanthinderivate, Verfahren zu ihrer Herstellung und ihre pharmazeutische Anwendung | |
| DE2434911C2 (de) | Phenyläthylamin-Derivate und pharmazeutische Zusammensetzungen | |
| DE3687007T2 (de) | 6-thioxanthinderivate. | |
| DE2056327A1 (de) | Aminopunn Derivate | |
| DE3125471C2 (de) | Piperazinderivate, Verfahren zu ihrer Herstellung und Arzneimittel, die diese Verbindungen enthalten | |
| DE69113284T2 (de) | Chinolinderivat. | |
| DE68910211T2 (de) | Estramustin-ester. | |
| DE3856387T2 (de) | 1,2,3,4,10,14b-HEXAHYDRODIBENZO-c,f]PYRAZINO-1,2-a]AZEPINO-DERIVATE | |
| EP0096279B1 (de) | N-(2-Methoxyethyl)-noroxymorphon, dessen Säureadditionssalze, diese enthaltende Arzneimittel und Verfahren zu deren Herstellung | |
| DE3882571T2 (de) | Sulfonyldecahydro-8H-isochino[2,1-g][1,6]naphthyridine, optische Isomere davon und verwandte Verbindungen. | |
| DE2724478C2 (de) | 5,11-Dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-on-derivate, Verfahren zu ihrer Herstellung und diese Verbindungen enthaltende Arzneimittel | |
| DE3721223C2 (de) | ||
| DE3587138T2 (de) | Polycyclische verbindungen, ihre herstellung und sie enthaltende rezepturen. | |
| EP0665014B1 (de) | 3-Benzoyl-3,7-diazabicyclo(3,3,1)nonan-Verbindungen mit anti-arrhythmischer Wirkung | |
| DE69424816T2 (de) | N-Benzylpiperazinverbindungen, Verfahren zu ihrer Herstellung und sie enthaltende Zusammensetzungen | |
| EP0085893B1 (de) | Pyridobenzodiazepinone, Verfahren zu ihrer Herstellung und diese Verbindungen enthaltende Arzneimittel | |
| DE1620541B2 (de) | Neue in 3-Stellung substituierte 6,8-Dibrom-tetrahydro-chinazoline | |
| DE2410201B2 (de) | 6-substituierte 3-carbaethoxyhydrazinopyridazine beziehungsweise ihre salze sowie solche enthaltende arzneimittel und verfahren zur herstellung derselben | |
| DE69309293T2 (de) | 2-substituierte adenosine mit a-2 rezeptor affinität | |
| EP0647226B1 (de) | 9-AMINO-PYRIDAZINO 4',5' : 3,4]PYRROLO- 2,1-a]ISOCHINOLINE UND DEREN VERWENDUNG FÜR DIE HERSTELLUNG VON PHARMAZEUTISCHEN ZUBEREITUNGEN | |
| AT390062B (de) | Verfahren zur herstellung von neuen trifluormethyl substituierten tetracyclischen chinazolinonen und ihrer saeureadditionssalze | |
| DE602005005308T2 (de) | Verfahren zur herstellung von 3-amino-8-(1-piperazinyl)-2h-1-benzopyran-2-on und seinen salzen und hydraten | |
| DE3017560A1 (de) | 9-amino-6,7-dihydro-4h-pyrido eckige klammer auf 1,2-a eckige klammer zu pyrimidin-4-on-derivate, verfahren zu deren herstellung und diese verbindungen enthaltende pharmazeutische kompositionen |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 8364 | No opposition during term of opposition | ||
| 8339 | Ceased/non-payment of the annual fee |