-
Staatliche Finanzierung
-
Die
hierin beschriebene Erfindung wurde mit Unterstützung der Regierung unter der
Zuschussnummer RO1 HL37942 gemacht, verliehen durch das National
Institute of Health. Die Regierung der Vereinigten Staaten hat bestimmte
Rechte an dieser Erfindung.
-
Querverweis zu verwandten
Anmeldungen
-
Diese
Anmeldung beansprucht die Priorität von der
U.S. vorläufigen
Patentanmeldungsseriennummer 60/371,434 , eingereicht am
10. April 2002, und der
U.S.
vorläufigen
Patentanmeldungsseriennummer 60/387,184 , eingereicht am
7. Juni 2002.
-
Bereich der Erfindung
-
Die
vorliegende Erfindung stellt die Verwendung eines A2A-Adenosinrezeptoragonisten
und ein antipathogenes Mittel für
die Herstellung eines Medikaments zur Behandlung einer Entzündung bereit,
verursacht durch bakterielle, fungale oder virale Infektionen, und
der Entzündung,
die durch die Behandlung dieser Infektionen, zum Beispiel durch
den Tod der bakteriellen oder viralen Zellen, verursacht wird.
-
Hintergrund der Erfindung
-
Bakterielle,
fungale und virale Pathogene können
Infektionen verursachen, die zu einer schweren Erkrankung und sogar
zum Tod führen
können.
Zum Beispiel verursacht die sporenbildenden grampositiven Stäbchen Bacillus
anthracis Anthrax, eine weltweite Erkrankung, die vornehmlich Pflanzenfresser
betrifft. Infektionen des Menschen treten sporadisch durch den Kontakt
mit infizierten Tieren oder kontaminierten tierischen Produkten
auf.
-
Die
Erkrankung ist eine konstante Bedrohung in endemischen Regionen,
weil die Sporen über
Jahre in der Erde fortbestehen können.
Kürzliche
Ereignisse in den Vereinigten Staaten unterstreichen das Potential von
Anthrax als ein bioterroristisches Mittel.
-
Die
Pathogenese von letalen Infektionen ist komplex, sie benötigt die
Keimung des Sporeninokulums, die systemische Invasion, Vermehrung
und Toxinherstellung, die zum Tod führen. Die Symptome der Infektionen
können
die Entwicklung eines fatalen Entzündungs-(septischen)Schocks
einschließen.
Folglich unterliegen die damit verbundenen Therapien einer regen
Erforschung, um die schädlichen
Wirkungen des Entzündungsschocks
zu minimieren.
-
Der
Entzündungsschock
kann direkt durch die Pathogene oder durch den Tod das Pathogen
nach der Behandlung des Patienten mit einem Medikament, dass die
Pathogene abtötet,
verursacht werden. Häufig
begründet
die plötzliche
Entwicklung eines fatalen Entzündungs-(septischen)Schocks
und das Fortschreiten der Erkrankung die hohe Sterblichkeit durch
diese Pathogene, trotz der Verfügbarkeit
eines bakteriologischen oder antiviralen Heilmittels.
-
Der
Entzündungsschock
kann durch bakterielle, Pilz- oder virale Pathogene direkt oder
durch die Behandlung dieser verursacht werden, das heißt der Tod
der Pathogene auf Grund der Behandlung mit antibakteriellen, antifungalen
oder antiviralen Mitteln. Die Agonisten der A2A-Adenosinrezeptoren
hemmen die Entzündung,
die durch sterbende Pathogene verursacht wird. Dementsprechend gibt
es einen Bedarf für
selektive, potente und spezifische A2AAR-Agonisten
für die
Verwendung in einer damit verbundenen Therapie für die Behandlung von entzündlichen
bakteriellen, fungalen und viralen Infektionen.
-
In Übereinstimmung
mit der vorliegenden Erfindung haben selektive, potente und spezifische A2AAR-Agonisten eine Verwendung als ein potenzieller
Zusatz in der Therapie für
die Behandlung in Kombination mit anderen Mitteln, die bakterielle,
fungale und virale Infektionen wie zum Beispiel Anthrax, Tularämie, Escherichia
coli und Pest, abtöten.
-
Es
gibt derzeitig einen Bedarf für
pharmazeutische Mittel, die nützlich
sind die Entzündungsantwort
auf Grund einer Bakterien-, Pilz- oder Vireninvasion zu vermindern
oder die Entzündungsantwort
auf Grund von Toxinen, die von den Bakterien, Pilzen oder Viren,
während
sie leben oder nachdem sie unter Verwendung antibagterieller, antifungaler
oder antiviraler Mittel abgetötet
wurden, freigesetzt werden.
-
Zusammenfassung der Erfindung
-
Die
vorliegende Erfindung stellt die Verwendung eines A2A-Adenosinrezeptoragonisten
bei der Herstellung eines Medikaments zur Behandlung einer Entzündung, verursacht
durch pathogene Organismen, wobei ein antipathogenes Mittel und
der besagte A2A-Adenosinrezeptoragonist
an einen Patienten gleichzeitig oder der Reihe nach verabreicht
werden.
-
Die
vorliegende Erfindung bietet eine Verwendung bei der Herstellung
eines Medikaments für
die Behandlung von biologischen Erkrankungen, was die Verabreichung
einer wirksamen Menge eines geeigneten antibiotischen, antifungalen
oder antiviralen Mittels in Verbindung mit einem A2A-Adenosinrezeptoragonisten umfasst.
Wenn kein antipathogenes Mittel bekannt ist, kann der A2A-Adenosinrezeptoragonist
alleine verwendet werden, um die Entzündung zu verringern, wie es
während
einer Infektion mit antibiotikaresistenten Bakterien vorkommen kann
oder bei bestimmten Viren wie bei jenen, die SARS oder Ebola verursachen.
Optional schließt
das Verfahren die Verabreichgung eines Typ-IV-PDE-Inhibitors ein.
Der A2A-Adenosinrezeptoragonist kann
eine damit verbundene Therapie für
die Behandlungsbedingungen bereitstellen wie für die durch eine Sepsis verursachte
Entzündung,
zum Beispiel ein humanes urämisches
Syndrom, wenn er bei der Behandlung von bioterroristischen Waffen
wie Anthrax, Tularämie,
Escherichia coli und Pest mit Antibiotika verabreicht wird. Die
vorliegende Erfindung stellt auch eine damit verbundene Therapie
für die
Behandlung von letalen bakteriellen, fungale und viralen Infektionen
wie Anthrax, Tularämie,
Escherichia und Pest bereit, umfassend die Verabreichung eines antibakteriellen,
antifungalen oder antiviralen Mittels in Verbindung mit selektiven A2A-Adenosinrezeptoragonisten.
-
Die
vorliegende Erfindung bietet eine Verwendung entweder alleine oder
in Kombination mit einer erkrankungslindernden Medizin bei der Herstellung
eines Medikaments für
die Behandlung von biologischen Erkrankungen, die eine Entzündung hervorrufen.
Diese schließen
Bakterien in Kombination mit Antibiotika ein, einschließlich, aber
nicht darauf beschränkt,
von Bakterien, die Anthrax, Tularämie, Pest, Lyme-Krankheit und Anthrax
verursachen. Es sind auch Viren eingeschlossen, einschließlich jener,
die RSV, schweres akutes respiratorisches Syndrom (SARS), Influenza
und Ebola verursachen, mit oder ohne eine antivirale Therapie. Es sind
auch Hefe- und Pilzinfektionen mit oder ohne Antihefe- oder Antipilzmittel
eingeschlossen.
-
Die
antibakteriellen, antifungalen oder antiviralen Mittel können zusammen
(z.B. gleichzeitig) mit dem A2A-Adenosinrezeptoragonisten
oder entweder gleichzeitig oder als eine Mischung oder sie können der
Reihe nach verabreicht werden. Die Verabreichung des A2A-Adenosinrezeptoragonisten
der Reihe nach kann vor dem Mittel, innerhalb von Minuten oder bis
zu 48 Stunden nach der Verabreichung des Mittels sein. Vorzugsweise
erfolgt die Verabreichung des A2A-Adenosinrezeptoragonisten
innerhalb von 24 Stunden oder besonders vorzugsweise innerhalb von
12 Stunden.
-
Die
Erfindung wird auch für
die Behandlung von Patienten mit einer Sepsis, einer schweren Sepsis und
möglicherweise
für das
systemische inflammatorische Response-Syndrom, zusätzlich zu
einem septischen Schock, nützlich
sein. Die A2AAR-Agonisten setzen vielfache
antientzündliche
Wirkungen früh
in der Entzündungskaskade
ein und folglich könnte
eine kurze Kur mit einem A2AAR-Agonisten
einen großen
Nutzen bei schweren, lebensbedrohlichen Infektionen und inflammatorischen
Fehlfunktionen beim Menschen bewirken, einschließlich Inhalationsanthrax, Tularämie, Escherichia
und Pest.
-
Die
antiinflammatorische Wirkung der A2AAR-Agonisten
wurde in vivo in experimentellen Modellen für Meningitis, Peritonitis und
Arthritis dokumentiert. Das möglicherweise
fatale Syndrom einer bakteriellen Sepsis ist ein zunehmendes Problem
auf Akutstationen. Die Sepsis und der septische Schock, die nun
die elfte der führenden
Todesursachen in den Vereinigten Staaten sind, nehmen an Häufigkeit
zu. Derzeitige Schätzungen weisen
darauf hin, dass in den Vereinigten Staaten jährlich etwa 900000 neue Sepsisfälle (etwa
60 % gramnegativ) mit einer grob geschätzten Sterblichkeitsrate von
35 % auftreten. Darüber
hinaus ist die Sterblichkeitsrate, wie sie in jüngsten klinischen Versuchen
veranschlagt wurde, etwa 25 %, während
etwa 10 % der Patienten an ihrer zu Grunde liegenden Krankheit sterben.
Ein Schock entwickelt sich in etwa 200000 Fällen jährlich mit einer zuordenbaren
Sterblichkeitsrate von 46 % (92000 Tote). Die Sepsis macht geschätzte $ 5-10
Milliarden der jährlichen
Gesundheitsführsorgeausgaben
aus. Es ist nun weitgehend anerkannt, dass unter den sich im Krankenhaus
auf nichtkoronar Intensivstationen befindenden Patienten die Sepsis
die häufigste
Todesursache ist. Das Sepsissyndrom ist ein Volksgesundheitsproblem
von größter Wichtigkeit.
Es wird erwartet, dass die A2AAR-Agonisten
eine Verwendung als einen neuen und einmaligen damit verbundenen
therapeutischen Ansatz haben, um die Morbidität und Mortalität zu verringern.
Es wird angenommen, dass diese Behandlung den Ausgang bei systemischen
Anthrax, Tularämie,
Escherichia und Pest verbessern wird.
-
Die
Agnonisten der A2A-Adenosinrezeptoren der
Erfindung können
die Neutrophilen-, Makrophagen- und T-Zell-Aktivierung hemmen und
dadurch eine Entzündung
verringern, die durch bakterielle und virale Infektionen verursacht
ist. Die Verbindungen, in Verbindung mit Antibiotika oder antiviralen
Mitteln, können
die Mortalität
vorbeugen oder verringern, die durch eine Sepsis oder das hämolytisch-urämische Syndrom
oder durch andere Entzündungsbedingungen
verursacht werden. Die Wirkungen der Adenosin-A2A-Agonisten
werden durch Typ-IV-Phosphodiesteraseinhibitoren wie Rolipram verstärkt.
-
Die
Erfindung stellt auch eine Verbindung mit der Formel I für die Verwendung
in der medizinischen Therapie bereit (z.B. für die Verwendung als ein Zusatz
bei der Behandlung von potentiell letalen bakteriellen Infektionen
wie Anthrax, Tularämie,
Escherichia, Pest oder anderen bakteriellen und/oder viralen Infektionen und
der Behandlung von systemischen Intoxifikationen, verursacht durch
bakterielle und/oder virale Infektionen) sowie die Verwendung einer
Verbindung mit der Formel I für
die Herstellung eines Medikaments zur Verringerung einer Entzündung, verursacht
durch die Bakterien oder Viren oder die Behandlung davon in einem Säugetier
wie einem Menschen. Die Verbindungen der Erfindung sind auch für die Behandlung
von systemischen Intoxifikationen nützlich, worin die bakteriellen
oder viralen Stoffe entweder direkt oder als eine Folge der Behandlung
eine Entzündung
verursachen, z.B. mit einem antibiotischen oder antviralen Mittel.
-
Die
Sepsis ist eine schwere Erkrankung, welche durch eine unbändige Infektion
des Blutstroms mit toxinproduzierenden Bakterien oder Viren verursacht
wird. Die Infektion, welche sich als eine Entzündung offenbaren kann, kann
durch Bakterien- oder Virenpathogene direkt oder durch die Behandlung
von diesen verursacht werden, das heißt, durch den Tod der Pathogene
auf Grund der Behandlung mit antibakteriellen oder antiviralen Mitteln.
Die Sepsis kann auch als die Antwort des Körpers auf eine Infektion betrachtet
werden. Die Infektion kann durch Mikroorganismen oder „Keime" (gewöhnlich Bakterien),
die in den Körper
eindringen, verursacht werden, sie kann auf eine bestimmte Körperregion
begrenzt sein (z.B. einen Zahnabszess) oder kann sich im Blutstrom
ausbreiten (häufig
als „Septikämie" oder „Blutvergiftung" bezeichnet).
-
Die
systemische Intoxifikation oder der Entzündungsschock werden häufig als
septischer Schock; Bakteriämieschock;
Endotoxinschock; Sepsisschock; oder warmer Schock bezeichnet.
-
Der
septische Schock ist ein ernster, abnormaler Zustand, der auftritt,
wenn eine unbändige
Infektion zu einem niedrigen Blutdruck und geringen Blutfluss führt. Die
lebenswichtigen Organe wie das Hirn, Herz, Nieren und Leber können nicht
richtig funktionieren oder können
versagen. Der septische Schock tritt am häufigsten bei den sehr alten
und den sehr jungen Leuten auf. Er tritt auch bei Leuten mit einer
zugrunde liegenden Erkrankung auf. Irgendein bakterieller Organismus
kann einen septischen Schock verursachen. Auch Pilze und Viren können diesen
Zustand verursachen. Toxine, die von den Bakterien, Pilzen oder
Viren freigesetzt werden können
direkt einen Gewebeschaden verursachen und zu einem niedrigen Blutdruck
und/oder schlechter Organfunktion führen. Diese Toxine können auch
eine heftige Entzündungsreaktion
des Körpers
erzeugen, welche zu dem septischen Schock beiträgt.
-
Unter
einem weiteren Gesichtspunkt bietet die vorliegende Erfindung auch
eine Verwendung der beanspruchten Kombination für die Herstellung eines Medikaments
für die
Behandlung eines schweren akuten respiratorischen Syndroms (SARS),
umfassend, dass eine wirksame antiinflammatorische Menge eines Agonisten
des A2A-Adenosinrezeptors und eines antipathogenen
Mittels, optional mit einem PDE-IV-Inhibitor, wie Rolipram, an ein
Säugetier
verabreicht werden, welches diese Therapie benötigt.
-
Beschreibung der Figuren
-
1 stellt
die dosisabhängige
Antwort auf dem A2AAR-Agonisten ATL146e
(ATL) und den dosisabhängigen
Schutz von Mäusen
vor einer E. coli 026:B6 LPS-induzierten Endotoxämie dar. Die Mäuse wurden mit
IP-Injektionen mit
den angezeigten ATL-Dosen eine Stunde vor der LPS (12,5 μg/kg) und
mit 6 Stundenintervallen für
eine Gesamtheit von 8 Dosen/48 Stunden.
-
2 stellt
die dosisabhängige
Antwort von ATL146e (DWH) für
das Überleben
der LPS-behandelten Mäuse
dar. Das Behandlungsschema ist dasselbe wie in 1.
-
3 stellt
dar, dass der A2AAR-Agonist ATL146e (ATL)
die Mäuse
vor einer LPS-induzierten ENsotoxämie nach einer Verzögerung des
Therapiebeginns schützt.
Die Tiere wurden mit LPS und ATL146e (50 μg/kg) wie in 1 behandelt,
mit der Ausnahme, dass die erste Behandlung mit ATL um den angegebenen Zeitraum
verzögert
war.
-
4 stellt
dar, dass der A2AAR-Antagonist ZM241385
(ZM) den Schutz durch ATL146e (ATL) in den LPS-behandelten Mäusen hemmt.
-
5 stellt
dar, dass der A2AAR-Agonist ATL146e einen
geringeren Schutz vor einer E. coli 026:B6 LPS-induzierten Endotoxämie für A2AAR KO-Mäuse bietet,
relativ zu Wildtypmäusen.
-
6 stellt
dar, dass ATL146e (ATL) das Überleben
der Mäuse
erhöhte,
denen lebende E. coli injiziert wurden und die mit einem Antibiotikum
(Ceftriaxon) behandelt wurden, verglichen mit Mäusen, die mit dem Antibiotikum
allein behandelt wurden. Allen Mäusen
wurden 20 Millionen E. coli IP zum Zeitpunkt 0 injiziert. Wo angezeigt
wurden die Mäuse
einmal zum Zeitpunkt 0 mit Ceftriaxon oder mit 50 μg/kg ATL146e
8mal in 6 Stunden Intervallen behandelt.
-
7 stellt
die Verringerung des Nieren IL-6 in Mäusen dar, die für 6 Stunden
LPS/Stx2 unter Verwendung von ATL-146e und ATL-203 ausgesetzt waren.
-
8 stellt
die Verringerung des Chemokins Renal-RANTES in Nierenproben von
Mäusen
dar, die für 6
Stunden LPS/Stx2 unter Verwendung von ATL-146e und ATL-203 ausgesetzt
waren.
-
9 stellt
die Verringerung der Infiltration von Neutrophilen in die Nieren
von C57BL/6-Mäusen
unter Verwendung von ATL-203 dar.
-
Detaillierte Beschreibung
der Erfindung
-
Es
werden, wenn nicht anders beschrieben, die folgenden Definitionen
verwendet. Halo ist Fluor, Chlor, Brom oder Iod. Alkyl, Alkoxy,
Aralkyl, Alkylaryl, ect. bezeichnen sowohl gerade als auch verzweigte
Alkylgruppen; der Verweis aber auf ein individuelles Radikal wie „Propyl" umfasst nur das
gradkettige Radikal, auf ein verzweigtkettiges Isomer wie „Isopropyl" wird spezifisch
Bezug genommen. Aryl schließt
ein Phenylradikal oder ein orthofusioniertes bicyclisches carbocyclisches
Radikal ein, das etwa neun bis 10 Ringatome hat, wobei wenigstens
ein Ringatom aromatisch ist. Heteroaryl umfasst ein Radikal, verbunden über einen
Ringkohlenstoff eines monocyclischen aromatischen Rings, der fünf oder
sechs Ringatome enthält,
bestehend aus einen Kohlenstoff und ein bis vier Heteroatomen, jeweils
ausgewählt
aus der Gruppe bestehend aus einem nichtperoxid Sauerstoff, Schwefel
und N(X), worin X abwesend ist oder H, O, (C1-C4)Alkyl, Phenyl oder Benzyl ist, sowie ein
Radikal eines orthofusionierten bicyclischen Heterocyclus mit etwa
8 bis 10 Ringatomen, abgleitet davon, insbesondere ein Benzderivat
oder eines, das durch die Fusion eines Propylen-, Trimethylen- oder Tetamethylendiradikals
daran erlangt wird.
-
Es
wird von Fachleuten auf dem Gebiet anerkannt werden, dass die Verbindungen
mit den Formeln (I), (II), (III) und (IV) mehr als ein chirales
Zentrum haben und in optisch aktiven und racemischen Formen isoliert
werden können.
Vorzugsweise leitet sich die Ribosehälfte der Verbindungen von D-Ribose
ab, das heißt, die
3',4'-Hydroxylgruppen
sind alpha zu dem Zuckerring und die 2'- und 5'-Gruppen sind beta (3R, 4S, 2R, 5S). Wenn
die zwei Gruppen an der Cyclohexylgruppe in der 1- und 4-Position
sind, sind sie vorzugsweise trans. Einige Verbindungen können einen
Polymorphismus aufweisen. Es versteht sich, dass die vorliegende
Erfindung irgendeine racemische, optisch aktive, polymorphe oder
stereoisomere Form oder Mischungen davon von einer Verbindung der
Erfindung umfasst, welche die hierin beschriebenen nützlichen
Eigenschaften besitzt, wobei es im Stand der Technik gut bekannt
ist, wie man optisch aktive Formen herstellt (zum Beispiel durch
die Auflösung
der racemischen Form mittels Rekristallisationstechniken oder enzymatische
Techniken, durch die Synthese eines optisch aktiven Ausgangsmaterials,
durch chirale Synthese oder durch chromatographische Auftrennung
unter Verwendung einer chiralen stationären Phase) und wie man die
Adenosinagonistenaktivität
unter Verwendung der hierin beschriebenen Tests oder unter Verwendung
anderer ähnlicher
Tests, die im Stand der Technik gut bekannt sind, bestimmt.
-
Die
unten aufgelisteten spezifischen und bevorzugten Werte für Radikale,
Substituenten und Bereiche dienen nur der Illustration; sie schließen nicht
andere definierte Werte oder andere Werte innerhalb der definierten
Bereiche für
die Radikale und Substituenten aus.
-
(C1-C8)Alkyl kann speziell
Methyl, Ethyl, Propyl, Isopropyl, Butyl, Isobutyl, sec-Butyl, Pentyl,
3-Pentyl, Hexyl, Heptyl oder Octyl sein. Wie hierin verwendet umfasst
der Begriff "Cycloalkyl" Bicycloalkyl (Norbornyl, 2.2.2-Bicyclooctyl,
etc.) und Tricycloalkyl (Adamantyl, etc.), optional umfassend 1-2
N, O oder S. Cycloalkyl umfasst auch (Cycloalkyl)alkyl. Folglich
kann (C3-C6)Cycloalkyl
Cyclopropyl, Cyclobutyl, Cyclopentyl, Cyclohexyl und dergleichen
sein. (C1-C8)Alkoxy
kann Methoxy, Ethoxy, Propoxy, Isopropoxy, Butoxy, Isobutoxy, sec-Butoxy,
Pentoxy, 3-Pentoxy oder Hexyloxy sein; (C2-C6)Alkenyl kann Vinyl, Allyl, 1-Propenyl,
2-Propenyl, 1-Butenyl,
2-Butenyl, 3-Butenyl, 1-Pentenyl, 2-Pentenyl, 3-Pentenyl, 4-Pentenyl, 1-Hexenyl, 2-Hexenyl,
3-Hexenyl, 4-Hexenyl oder 5-Hexenyl
sein; (C2-C6)Alkynyl
kann Ethynyl, 1-Propynyl, 2-Propynyl, 1-Butynyl, 2-Butynyl, 3-Butynyl, 1-Pentynyl,
2-Pentynyl, 3-Pentynyl, 4-Pentynyl,
1-Hexynyl, 2-Hexynyl, 3-Hexynyl, 4-Hexynyl oder 5-Hexynyl sein;
(C1-C6)Alkanoyl
kann Acetyl, Propanoyl oder Butanoyl sein; Halo(C1-C6)alkyl kann Iodmethyl, Brommethyl, Chlormethyl,
Fluormethyl, Trifluormethyl, 2-Chlorethyl, 2-Flurethyl, 2,2,2-Trifluorethyl
oder Pentafluorethyl sein; Hydroxy(C1-C6)alkyl kann Hydroxymethyl, 1-Hydroxyethyl, 2-Hydroxyethyl,
1-Hydroxypropyl, 2-Hydroxypropyl, 3-Hydroxypropyl, 1-Hydroxybutyl, 4-Hydroxybutyl,
1-Hydroxypentyl, 5-Hydroxypentyl,
1-Hydroxyhexyl oder 6-Hydroxyhexyl sein; (C1-C6)Alkoxycarbonyl
(CO2R2) kann Methoxycarbonyl,
Ethoxycarbonyl, Propoxycarbonyl, Isopropoxycarbonyl, Butoxycarbonyl,
Pentoxycarbonyl oder Hexyloxycarbonyl sein; (C1-C6)Alkylthio kann Methylthio, Ethylthio, Propylthio,
Isopropylthio, Butylthio, Isobutylthio, Pentylthio oder Hexylthio
sein, (C2-C6)Alkanoyloxy
kann Acetoxy, Propanoyloxy, Butanoyloxy, Isobutanoyloxy, Pentanoyloxy
oder Hexanoyloxy sein; Aryl kann Phenyl, Indenyl oder Naphthyl sein;
und Heteroaryl kann Furyl, Imidazolyl, Triazolyl, Triazinyl, Oxazoyl,
Isoxazoyl, Thiazolyl, Isothiazoyl, Pyraxolyl, Pyrrolyl, Pyrazinyl,
Tetrazolyl, Puridyl (oder seine N-Oxide), Thientyl, Pyrimidinyl
(oder seine N-Oxide), Indolyl, Isoquinolyl (oder seine N-Oxide)
oder Quinolyl (oder seine N-Oxide) sein.
-
Aryl
bezeichnet ein Phenylradikal oder ein orthofusioniertes bicyclisches
carbocyclisches Radikal, das etwa neun bis zehn Ringatome hat, wobei
wenigstens ein Ring aromatisch ist. Heteroaryl bezeichnet ein Radikal
eines monocyclischen aromatischen Rings, der fünf oder sechs Ringatome enthält, bestehend
aus Kohlenstoff und 1, 2, 3 oder 4 Heteroatomen, jeweils ausgewählt aus
der Gruppe bestehend aus nichtperoxid Sauerstoff, Schwefel und N(Y),
worin Y abwesend ist oder H, O, (C1-C8)Alkyl, Phenyl oder Benzyl ist, sowie ein Radikal
eines orthofusionierte bicyclischen Heterocyclus mit etwa 8 bis
10 Ringatomen, abgeleitet davon, insbesondere ein Benzderivat oder
eines, abgeleitet durch die Fusion eines Propylen-, Trimethylen-
oder Tetamethylendiradikals daran.
-
Der
Begriff „Heterocyclus" stellt allgemein
eine nichtaromatische heterocyclische Gruppe dar, die 3 bis etwa
10 Ringatome hat, die gesättigt
oder partiell ungesättigt
sein können
und wenigstens ein Heteroatom enthalten (z.B. 1, 2 oder 3), ausgewählt aus
der Gruppe bestehend aus Sauerstoff, Stickstoff und Schwefel. Genau
schließen „Heterocyclus"-Gruppen monocyclische,
bicyclische oder tricyclische Gruppen ein, die ein oder mehrere
Heteroatome enthalten, ausgewählt
aus der Gruppe bestehend aus Sauerstoff, Stickstoff und Schwefel.
Eine „Heterocyclus"-Gruppe kann auch
ein oder mehrere Oxogruppen (=O), gebunden an ein Ringatom, einschließen. Nicht
einschränkende
Beispiele für
Heterocyclusgruppen schließen
1,3-Dioxolan, 1,4-Dioxan, 1,4-Dithian,
2H-Pyran, 2-Pyrazolin, 4H-Pyran, Chromanyl, Imidazolidinyl, Imidazolinyl,
Indolinyl, Isochromanyl, Isoindolinyl, Morpholin, Piperazinyl, Piperidin,
Piperidyl, Pyrazolidin, Pyrazolidinyl, Pyrazolinyl, Pyrrolidin,
Pyrrolin, Quinuelidin, Thiomorpholin und dergleichen ein.
-
Der
Begriff „Alkylen" bezieht sich auf
eine divalente gerade oder verzweigte Kohlenwasserstoffkette (z.B.
Ethylen -CH2CH2-).
-
Der
Begriff „Aryl(C1-C8)alkylen" schließt zum Beispiel
Benzyl, Phenethyl, 3-Phenylpropyl, Naphthylmethyl ein.
-
Die
Begriffe „systemische
Intoxifikation" oder „Entzündungsschock" beziehen sich auf
die Ansammlung von Toxinen oder eine heftige Entzündungsreaktion
im Körper
auf Grund der Invasion und/oder der Behandlung von Bakterien, Pilzen
oder Viren.
-
Wie
hierin verwendet bezieht sich „antipathogenes
Mittel" auf Verbindungen,
die eine antibakterielle, antifungale oder antivirale Aktivität haben.
-
Wie
hierin verwendet bezieht sich der Begriff "in Verbindung mit" auf die Coverabreichung eines antibakteriellen,
antifungalen oder antiviralen Mittels mit dem A2A-Adenosinrezeptoragonisten.
Die Mittel und A2A-Adenosinrezeptoragonisten können entweder
gleichzeitig oder als eine Mischung oder sie können der Reihe nach verabreicht
werden. Die Verabreichung der A2A-Adenosinrezeptoragonisten
der Reihe nach kann vor dem Mittel, innerhalb von Minuten oder bis
zu etwa 48 Stunden nach der Verabreichung des Mittels sein. Vorzugsweise
erfolgt die Verabreichung der A2A-Adenosinrezeptoragonisten
innerhalb von 24 Stunden und besonders vorzugsweise innerhalb von
12 Stunden.
-
Der
Kohlenstoffatomgehalt der verschiedenen Kohlenwasserstoffreste wird
durch ein Präfix
angezeigt, das die minimale und die maximale Zahl an Kohlenstoffatomen
in dem Rest bezeichnet, das heißt
das Präfix Ci-Cj kennzeichnet
einen Rest mit einschließlich
der ganzen Zahl „i" bis einschließlich der
ganzen Zahl "j" an Kohlenstoffatomen.
Folglich bezieht sich zum Beispiel (C1-C8)Alkyl auf ein Alkyl mit einschließlich ein
bis acht Kohlenstoffatomen.
-
Die
Verbindungen der vorliegenden Erfindung werden allgemein entsprechend
dem IUPAC oder CAS Nomenklatursystem benannt. Es können Abkürzungen
verwendet werden, die Fachleuten auf dem Gebiet gut bekannt sind
(z.B. „Ph" für Phenyl, „Me" für Methyl, „Et" für Ethyl, „h" für Stunde
oder Stunden und „rt" für Raumtemperatur).
-
In
einer Ausführungsform
schließen
die Agonisten der A
2A-Adenosinrezeptoren, die in der Anwendung der
vorliegenden Erfindung nützlich
sind, Verbindungen ein, die die Formel (I) haben:
worin
Z CR
3R
4R
5 oder NR
4R
5 ist;
jeder
R
1 unabhängig
Wasserstoff, Halo, -OR
a, -SR
a,
(C
1-C
8)Alkyl, Cyano,
Nitro, Trifluormethyl, Trifluormethoxy, (C
3-C
8)Cycloalkyl, Heterocyclus, Heterocyclus(C
1-C
8)alkylen-, Aryl,
Aryl(C
1-C
8)alkylen-,
Heteroaryl, Heteroaryl(C
1-C
8)alkylen-,
-CO
2R
a, R
aC(=O)O-, R
aC(=O)-,
-OCO
2R
a, R
bR
cNC(=O)O-, R
aOC(=O)N(R
b)-, R
bR
cN-, R
bR
cNC(=O)-, R
aC(=O)N(R
b)-, R
bR
cNC(=O)N(R
b)-, R
bR
cNC(=S)N(R
b)-, -OPO
3R
a, R
aOC(=S)-, R
aC(=S)-, -SSR
a, R
aS(=O)-, R
aS(=O)
2-, -N=NR
b oder -OPO
2R
a ist;
jeder
R
2 unabhängig
Wasserstoff, Halo, (C
1-C
8)Alkyl,
(C
3-C
8)Cycloalkyl,
Heterocyclus, Heterocyclus(C
1-C
8)alkylen-,
Aryl, Aryl(C
1-C
8)alkylen-,
Heteroaryl oder Heteroaryl(C
1-C
8)alkylen-
ist; oder
R
1 und R
2 und
das Atom, an welches sie gebunden sind, C=O, C=S oder C=NR
d ist;
R
4 und
R
5, zusammen mit den Atomen, an welche sie
gebunden sind, ein gesättigten
oder teilweise ungesättigten,
monocyclischen, bicyclischen oder aromatischen Ring bilden, der
3, 4, 5, 6, 7, 8, 9 oder 10 Ringatome hat, optional umfassend 1,
2, 3 oder 4 Heteroatome, ausgewählt
aus nichtperoxid Oxy (-O-), Thio (-S-), Sulfinyl (-SO-), Sulfonyl
(-S(O)
2-) oder Amin (-NR
b-)
in dem Ring;
worin irgendein Ring, der R
4 und
R
5 umfasst, mit 1 bis 14 R
6-Gruppen
substituiert ist; worin jeder R
6 unabhängig Halo,
OR
a, SR
a, (C
1-C
8)Alkyl, Cyano,
Nitro, Trifluormethyl, Trifluormethoxy, (C
1-C
8)Cycloalkyl, (C
6-C
12)Bicycloalkyl, Heterocyclus oder Heterocyclus(C
1-C
8)alkylen-, Aryl,
Aryl(C
1-C
8)alkylen-,
Heteroaryl, Heteroaryl(C
1-C
8)alkylen-,
-CO
2R
a, R
aC(=O)O-, R
aC(=O)-,
-OCO
2R
a, R
bR
cNC(=O)O-, R
aOC(=O)N(R
b)-, R
bR
cN-, R
bR
cNC(=O)-, R
aC(=O)N(R
b)-, R
bR
cNC(=O)N(R
b)-, R
bR
cNC(=S)N(R
b)-, -OPO
3R
a, R
aOC(=S)-, R
aC(=S)-, -SSR
a, R
aS(=O)-, -NNR
b, -OPO
2R
a ist oder zwei
R
6-Gruppen und das Atom, an welches sie
gebunden sind, C=O, C=S ist oder; zwei R
6-Gruppen,
zusammen mit dem Atom oder den Atomen, an welche sie gebunden sind,
einen carbocyclischen oder heterocyclischen Ring bilden können;
R
3 Wasserstoff, Halo, -OR
a,
SR
a, (C
1-C
8)Alkyl, Cyano, Nitro, Trifluormethyl, Trifluormethoxy,
(C
3-C
8)Cycloalkyl, Heterocyclus,
Heterocyclus(C
1-C
8)alkylen-,
Aryl, Aryl(C
1-C
8)alkylen-,
Heteroaryl, Heteroaryl(C
1-C
8)alkylen-, -CO
2R
a, R
aC(=O)O-,
R
aC(=O)-, -OCO
2R
a, R
bR
cNC(=O)O-,
R
aOC(=O)N(R
b)-,
R
bR
cN-, R
bR
cNC(=O)-, R
aC(=O)N(R
b)-, R
bR
cNC(=O)N(R
b)-, R
bR
cNC(=S)N(R
b)-, -OPO
3R
a, R
aOC(=S)-, R
aC(=S)-, -SSR
a, R
aS(=O)-, R
aS(=O)
2-, -NNR
b, -OPO
2R
a ist; oder, wenn
der Ring, der von CR
4R
5 gebildet
wird, ein Aryl oder ein Heteroaryl oder teilweise ungesättigt ist,
dann kann R
3 fehlen;
jeder R
7 unabhängig
Wasserstoff, (C
1-C
8)Alkyl,
(C
3-C
8)Cycloalkyl,
Aryl oder Aryl(C
1-C
8)alkylen-,
Heteroaryl, Heteroaryl(C
1-C
8)alkylen-
ist;
X -CH
2OR
a,
-CO
2R
a, -OC(O)R
a, -CH
2OC(O)R
a, -C(O)NR
bR
b, -CH
2SR
a, -C(S)OR
a, -OC(S)R
a, -CH
2OC(S)R
a oder -C(S)NR
bR
c oder -CH
2N(R
b)(R
c) ist;
worin
irgendeine der Alkyl-, Cycloalkyl-, Heterocyclus-, Aryl- oder Heteroarylgruppen
von R
1, R
2, R
3, R
6 und R
7 optional an einem Kohlenstoff mit einem
oder mehreren (z.B. 1, 2, 3 oder 4) Substituenten substituiert ist,
ausgewählt
aus der Gruppe, bestehend aus Halo, -OR
a,
-SR
a, (C
1-C
8)Alkyl, Cyano, Nitro, Trifluormethyl, Trifluormethoxy,
(C
3-C
8)Cycloalkyl,
(C
6-C
1 2)Bicycloalkyl,
Heterocyclus oder Heterocyclus(C
1-C
8)alkylen-, Aryl, Aryloxy, Aryl(C
1-C
8)alkylen-, Heteroaryl,
Heteroaryl(C
1-C
8)alkylen-,
-CO
2R
a, R
aC(=O)O-, R
aC(=O)-,
-OCO
2R
a, R
bR
cNC(=O)O-, R
aOC(=O)N(R
b)-, R
bR
cN-, R
bR
cNC(=O)-, R
aC(=O)N(R
b)-, R
bR
cNC(=O)N(R
b)-, R
bR
cNC(=S)N(R
b)-, -OPO
3R
a, R
aOC(=S)-, R
aC(=S)-, -SSR
a, R
aS(=O)
p--, R
bR
cNS(O)
p-,
-N=NR
b und -OPO
2R
a;
worin irgendein (C
1-C
8)Alkyl, (C
3-C
8)Cycloalkyl, (C
6-C
12)Bicycloalkyl, (C
1-C
8)Alkoxy, (C
1-C
8)Alkanoyl, (C
1-C
8)Alkylen oder Heterocyclus optional teilweise
ungesättigt
ist;
jeder R
a, R
b und
R
c unabhängig
Wasserstoff, (C
1-C
8)Alkyl
oder (C
1-C
8)Alkyl
ist, substituiert mit 1-3 (C
1-C
8)Alkoxy,
(C
3-C
8)Cycloalkyl,
(C
1-C
8)Alkylthio,
Aminosäure,
Aryl, Aryl(C
1-C
8)alkylen,
Heteroaryl oder Heteroaryl(C
1-C
8)alkylen;
oder R
b und R
c,
zusammen mit dem Stickstoff, an welchen sie gebunden sind, einen
Pyrrolidino-, Piperidino-, Morpholino- oder Thiomorpholinoring bilden;
und R
d Wasserstoff oder (C
1-C
6)Alkyl ist; m 0 bis 8 und p 0 bis 2 ist;
oder ein pharmazeutisch akzeptables Salz davon.
-
In
einer weiteren Ausführungsform
schließt
die Erfindung die Verwendung von Verbindungen der Formel (I) ein,
vorausgesetzt, dass wenn CR4R5 ein
carbocyclischer Ring ist, wenigstens einer von R1,
R2 oder R3 eine
andere Gruppe als Wasserstoff ist oder wenigsten eine R6-Gruppe
eine andere Gruppe als -CH2OH, -CO2Ra, RaC(=O)O-,
RaC(=O)OCH2- oder
RbRcNC(=O)- ist;
und unter der Voraussetzung, dass m wenigstens 1 ist, wenn Z NR4R5 ist.
-
Die
unten aufgelisteten spezifischen und bevorzugten Werte für Radikale,
Substituenten und Bereiche dienen nur zur Illustration; sie schließen nicht
andere definierte Werte oder andere Werte innerhalb der definierten
Bereiche für
die Radikale und Substituenten aus.
-
Bin
spezifischer Wert für
R1 ist Wasserstoff, -OH, -CH2OH,
-OMe, -OAc, -NH2, -NHMe, -NMe2 oder -NHAc.
-
Ein
weiterer spezifischer Wert für
R1 ist Wasserstoff, -OH, -OMe, -OAc, -NH2, -NHMe, -NMe2 oder -NHAc.
-
Ein
weiterer spezifischer Wert für
R1 ist Wasserstoff, -OH, -OMe oder -NH2.
-
Ein
weiterer spezifischer Wert für
R1 ist Wasserstoff, -OH oder -NH2.
-
Ein
spezifischerer Wert für
R1 ist Wasserstoff oder -OH.
-
Ein
spezifischer Wert für
R1, R2 und das Kohlenstoffatom,
an welches sie gebunden sind, ist Carbonyl (C=O).
-
Ein
spezifischer Wert für
R2 ist Wasserstoff oder (C1-C8)Alkyl, Cyclopropyl, Cyclohexyl oder Benzyl.
-
Ein
weiterer spezifischer Wert für
R2 ist Wasserstoff, Methyl, Ethyl oder Propyl.
-
Ein
weiterer spezifischer Wert für
R2 ist Wasserstoff oder Methyl.
-
Ein
spezifischerer Wert für
R2 ist Wasserstoff.
-
Ein
spezifischer Wert für
R3 ist Wasserstoff, OH, OMe, OAc, NH2, NHMe, NMe2 oder
NHAc.
-
Ein
weiterer spezifischer Wert für
R3 ist Wasserstoff, OH, OMe oder NH2.
-
Ein
weiterer spezifischer Wert für
R3 ist Wasserstoff, OH oder NH2.
-
Ein
spezifischerer Wert für
R3 ist Wasserstoff oder OH.
-
Ein
spezifischer Wert für
den Ring, umfassend R4, R5 und
das Atom, an welches sie gebunden sind, ist Cyclopentan, Cyclohexan,
Piperidin, Dihydro-pyridin, Tetrahydro-pyridin, Pyridin, Piperazin,
Decalin, Tetrahydro-pyrazin,
Dihydro-pyrazin, Pyrazin, Dihydro-pyrimidin, Tetrahydro-pyrimidin,
Hexahydro-pyrimidin, Pyrazin, Imidazol, Dihydro-imidazol, Imidazolidin,
Pyrazol, Dihydro-pyrazol und Pyrazolidin.
-
Ein
spezifischerer Wert für
den Ring, umfassend R4 und R5 und
das Atom, an welches sie gebunden sind, ist Cyclohexan, Piperidin
oder Piperazin.
-
Ein
spezifischer Wert für
R6 ist (C1-C8)Alkyl oder ein substituiertes (C1-C8)Alkyl, -ORa, -CO2Ra, RaC(=O)-, RaC(=O)O-,
RbRcN-, RbRcNC(=O)- oder Aryl.
-
Ein
weiterer spezifischer Wert für
R6 ist (C1-C8)Alkyl, -ORa, -CO2Ra, RaC(=O)-,
RaC(=O)O-, RbRcN-, RbRcNC(=O)-
oder Aryl.
-
Ein
weiterer spezifischer Wert für
R6 ist Methyl, Ethyl, Buthyl, OH, ORa, -CO2Ra,
RaC(=O)-, OC(=O)CH2CH3, -CONRbRc, -NRbRc oder
Phenyl.
-
Ein
weiterer spezifischer Wert für
R6 ist OH, OMe, Methyl, Ethyl, t-Buthyl,
-CO2Ra, -C(=O)NRbRc, -OAc, -NH2, -NHMe, -NMe2,
-NHEt oder -N(Et)2.
-
Ein
weiterer spezifischer Wert für
R6 ist -(CH2)1-2ORa, -(CH2)1-2C(=O)ORa, -(CH2)1-2OC(=O)Ra, -(CH2)1-2C(=O)Ra, -(CH2)1-2OCO2Ra, -(CH2)1-2NHRa,
-(CH2)1-2NRbRc, -(CH2)1-2OC(=O)NHRa oder -(CH2)1-2OC(=O)NRbRc.
-
Ein
weiterer spezifischer Wert für
R6 ist -CH2OH, -CH2OAc, -CH2OCH3, -CH2C(=O)OCH3, -CH2OC(=O)CH3, -CH2C(=O)CH3, -CH2OCO2CH3, -CH2NH(CH3) oder -(CH2)1-2N(CH3)2-
-
Ein
weiterer spezifischer Wert für
R6 ist Methyl, Ethyl, t-Buthyl, Phenyl,
-CO2Ra, -CONRbRc oder RaC(=O)-.
-
Ein
weiterer spezifischer Wert für
R6 ist -CH2OH, -CH2OAc, -C(=O)OCH3,
-C(=O)CH3, OCO2CH3 -OCO2CH3, -CH2NH(CH3) oder -(CH2)1-2N(CH3)2.
-
Ein
spezifischerer Wert für
R6 ist Methyl, Ethyl, CO2Ra -CONRbRc oder RaC(=O).
-
Eine
spezifische Zahl für
die R6-Gruppen, die an dem R4R5-Ring substituiert sind, ist 1 bis 4.
-
Genaue
Werte für
Ra und Rb sind unabhängig Wasserstoff,
(C1-C4)Alkyl, Aryl
oder Aryl(C1-C8)alkylen.
-
Genauere
Werte für
Ra und Rb sind unabhängig Wasserstoff,
Methyl,r Ethyl, Phenyl oder Benzyl.
-
Bin
spezifischer Wert für
Ra ist (C1-C8)Alkyl.
-
Ein
weiterer spezifischer Wert für
Ra ist Methyl, Ethyl, Propyl oder Butyl.
-
Ein
weiterer spezifischer Wert für
Ra ist Methyl, Ethyl, i-Propyl, i-Butyl oder tert-Butyl.
-
Ein
weiterer spezifischer Wert für
Rb und Rc ist ein
Ring.
-
Ein
spezifischer Wert für
R7 ist Wasserstoff, Alkyl, Aryl oder Aryl(C1-C8)alkylen.
-
Ein
weiterer spezifischer Wert für
R7 ist Wasserstoff, Methyl oder Ethyl, Phenyl
oder Benzyl.
-
Ein
spezifischerer Wert für
R7 ist H oder Methyl.
-
Ein
spezifischer Wert für
-N(R7)2 ist Amino,
Methylamino, Dimethylamino, Ethylamino, Pentylamino, Diphenylethylamino,
Pyridylmethylamino, Diethylamino oder Benzylamino.
-
Ein
spezifischer Wert für
-N(R7)2 ist Amino,
Methylamino, Dimethylamino, Ethylamino, Diethylamino, Diphenylethylamino,
Pentylamino oder Benzylamino.
-
Ein
spezifischer Wert für
N(R7)2 ist Amino
oder Methylamino ist.
-
Ein
spezifischer Wert für
X ist -CH2ORa, -CO2Ra, -OC(O)Ra, -CH2OC(O)Ra, -C(O)NRbRc.
-
Ein
spezifischerer Wert für
X ist -CH2ORa oder
-C(O)NRbRc.
-
Ein
weiterer spezifischer Wert für
X ist -CH2OH oder -C(O)NHCH2CH3.
-
Ein
spezifischer Wert für
m ist 0, 1 oder 2.
-
Ein
spezifischerer Wert für
m ist 0 oder 1.
-
Spezifische
Beispiele für
Ringe, die R
4, R
5 und
das Atom, an welches sie gebunden sind, umfassen, schießen ein:
worin
q 0 bis 14 ist und R
d Wasserstoff ist, vorausgesetzt
dass wenn q Null ist R
d dann nicht Wasserstoff
ist.
-
Spezifischere
Beispiele für
Ringe, die R
4, R
5 und
das Atom, an welches sie gebunden sind, umfassen, schießen ein:
-
Spezifische
Werte für
den Ring, der R4, R5 und
das Atom, an welches sie gebunden sind, umfasst, sind 2-Methylcyclohexan,
2,2-Dimethylcyclohexan,
2-Phenylcyclohexan, 2-Ethylcyclohexan, 2,2-Diethylcyclohexan, 2-tert-Butylcyclohexan,
3-Methylcyclohexan, 3,3-Dimethylcyclohexan,
4-Methylcyclohexan, 4-Ethylcyclohexan, 4-Phenylcyclohexan, 4-tert-Butylcyclohexan,
4-Carboxymethylcyclohexan, 4-Carboxyethylcyclohexan, 3,3,5,5-Tetramethylcyclohexan,
2,4-Dimethylcyclopentan,
4-Cyclohexancarbonsäure,
4-Cyclohexancarbonsäureester
oder 4-Methyloxyalkanoyl-cyclohexan.
-
Spezifischere
Werte für
den Ring, der R4, R5 und
das Atom, an welches sie gebunden sind, umfasst, sind 4-Piperidin,
4-Piperidin-1-carbonsäure, 4-Piperidin-1-carbonsäuremethylester,
4-Piperidin-1-carbonsäureethylester,
4-Piperidin-1-carbonsäurepropylester,
4-Piperidin-1-carbonsäure-tert-butylester,
1-Piperidin, 1-Piperidin-4-carbonsäuremethylester,
1-Piperidin-4-carbonsäureethylester,
1-Piperidin-4-carbonsäurepropylester,
1-Piperidin-4-carbonsäure-tert-butylester,
1-Piperidin- 4-carbonsäuremethylester,
3-Piperidin, 3-Piperidin-1-carbonsäure, 3-Piperidin-1-carbonsäuremethylester, 3-Piperidin-1-carbonsäure-tert-butylester,
1,4-Piperazin, 4-Piperazin-1-carbonsäure, 4-Piperazin-1-carbonsäuremethylester,
4-Piperazin-1-carbonsäureethylester,
4-Piperazin-1-carbonsäurepropylester,
4-Piperazin-1-carbonsäure-tert-butylester,
1,3-Piperazin, 3-Piperazin-1-carbonsäure, 3-Piperazin-1-carbonsäuremethylester,
3-Piperazin-1-carbonsäureethylester,
3-Piperazin-1-carbonsäurepropylester,
3-Piperidin-1-carbonsäure-tert-butylester,
1-Piperidin-3-carbonsäuremethylester,
1-Piperidin-3-carbonsäureethylester,
1-Piperidin-3-carbonsäurepropylester
oder 1-Piperidin-3-carbonsäure-tert-butylester.
-
Eine
weitere Gruppe spezifischer Werte für den Ring, der R4 und
R5 umfasst, sind 2-Methylcyclohexan, 2,2-Dimethylcyclohexan,
2-Phenylcyclohexan,
2-Ethylcyclohexan, 2,2-Diethylcyclohexan, 2-tert-Butylcyclohexan, 3-Methylcyclohexan,
3,3-Dimethylcyclohexan, 4-Methylcyclohexan,
4-Ethylcyclohexan, 4-Phenylcyclohexan, 4-tert-Butylcyclohexan, 4-Carboxymethylcyclohexan,
4-Carboxyethylcyclohexan, 3,3,5,5-Tetramethylcyclohexan, 2,4-Dimethylcyclopentan,
4-Piperidin-1-carbonsäuremethylester,
4-Piperidin-1-carbonsäure-tert-butylester,
4-Piperidin, 4-Piperazin-1-carbonsäuremethylester,
4-Piperidin-1-carbonsäure-tert-butylester,
1-Piperidin-4-carbonsäuremethylester,
1-Piperidin-4-carbonsäure-tert-butylester,
tert-Butylester, 1-Piperidin-4-carbonsäuremethylester
oder 1-Piperidin-4-carbonsäure-tert-Butylester,
3-Piperidin-1-carbonsäuremethylester, 3-Piperidin-1-carbonsäure-tert-Butylester, 3-Piperidin,
3-Piperazin-1-carbonsäuremethylester,
3-Piperidin-1-carbonsäure-tert-butylester,
1-Piperidin-3-carbonsäuremethylester,
1-Piperidin-3-carbonsäure-tert-butylester.
-
Spezifische
Verbindungen mit der Formel (I) sind jene, worin jeder R7 H ist, X Ethylaminocarbonyl ist und
R1 Hydroxy ist, R2 Wasserstoff
ist und Z 4-Carboxycyclohexyl ist, worin Ra Wasserstoff
ist, 4; Z 4-Methoxycarbonylcyclohexylmethyl ist, Ra Methyl
ist, 5; R1 und R2 zusammen
Oxo sind, Z eine 4-Carbonylcyclohexylgruppe,
worin Ra Methyl, Methoxy, Ethyl, Ethoxy,
Propyl, Isopropoxy, -Isobutyl, tert-Butyl, Amin, Methylamin oder Dimethylamin
ist, 6.
-
-
Eine
weitere Gruppe spezifischer Verbindungen mit der Formel (I) sind
jene, worin jeder R7 H ist, X Ethylaminocarbonyl
ist, R1 Hydroxy ist, R2 Wasserstoff
ist und Z eine substituierte 4-(Methylenoxycarbonyl)cyclohexyl-Gruppe ist, worin
Ra Methyl, Ethyl, Propyl, tert-Butyl, Methoxy,
Ethoxy, Methylamin oder Dimethylamin ist, 7; oder R1 und
R2 zusammen Oxo sind, Z eine substituierte-(Methylenoxycarbonyl)cyclohexyl-Gruppe
ist, worin Ra Methyl, Ethyl, Propyl, tert-Butyl,
Methoxy, Ethoxy, Methylamin oder Dimethylamin ist, 8.
-
-
Eine
weitere Gruppe spezifischer Verbindungen mit der Formel (I) sind
jene, worin jeder R7 H ist, X Ethylaminocarbonyl
ist, und R1 und R2 jeweils
Wasserstoff sind und Z eine 1-Piperidyl-4-carbonsäure- oder
-estergruppe ist, worin Ra Wasserstoff,
Methyl, Ethyl, Propyl, Isopropyl oder t-Butyl ist, 9; R1 und
R2 zusammen Oxo sind und Z eine 1-Piperidyl-4-carbonsäure- oder
-estergruppe
ist, worin Ra Wasserstoff, Methyl, Ethyl,
Propyl, Isopropyl oder t-Butyl ist, 10; R1 und
R2 jeweils Wasserstoff sind und Z eine 4-(Methylenoxycarbonyl)piperidin-4-yl-Gruppe
ist, worin Ra Methyl, Ethyl, Propyl oder
t-Butyl, Amin, Methylamin, Dimethylamin ist, 11; oder R1 und
R2 zusammen Oxo sind und Z eine 4-(Methylenoxycarbonyl)piperidin-4-yl
ist, worin Ra Methyl, Ethyl, Propyl oder
t-Butyl, Amin, Methylamin, Dimethylamin ist, 12; R1 und
R2 jeweils Wasserstoff sind und Z 4-(Methylenoxycarbonyl)piperidin-4-yl-oxy
ist, worin Ra Wasserstoff, Methyl, Ethyl,
Propyl, Isopropyl, Isobutyl oder t-Butyl ist, 13; oder R1 und R2 zusammen
Oxo sind, Z 4-(Methylenoxycarbonyl)piperidin-4-yl-oxy ist, worin
Ra Wasserstoff, Methyl, Ethyl, Propyl, Isopropyl,
Isobutyl oder t-Butyl ist, 14.
-
-
-
Eine
weitere Gruppe spezifischer Verbindungen mit der Formel (I) sind
jene, worin jeder R7 H ist, X Ethylaminocarbonyl
ist, R1 und R2 jeweils
Wasserstoff sind und Z eine 4-Piperidyl-1-carbonsäure- oder
-estergruppe ist, worin Ra Methyl, Ethyl,
Propyl, Isopropyl, Isobutyl oder t-Butyl ist, 15; R1 Hydroxy
ist, R2 Wasserstoff ist und Z eine 4-Piperidyl-1-carbonsäure- oder -estergruppe
ist, worin Ra Methyl, Ethyl, Propyl, Isopropyl,
Isobutyl oder t-Butyl
ist, 16; oder R1 und R2 zusammen
Oxo sind, Z eine 4-Piperidyl-1-carbonsäure- oder
-estergruppe ist, worin Ra Methyl, Ethyl,
Propyl, Isopropyl, Isobutyl oder t-Butyl ist, 17.
-
-
Eine
weitere Gruppe spezifischer Verbindungen mit der Formel (I) sind
jene, worin jeder R7 H ist, X Ethylaminocarbonyl
ist, R1 und R2 jeweils
Wasserstoff sind, Z eine 4-Piperazin-1-carbonsäure- oder -estergruppe ist,
worin Ra Methyl, Ethyl, Isopropyl, Isobutyl
oder t-Butyl ist, 18; oder R1 und R2 zusammen Oxo sind, Z eine 4-Piperazin-1-carbonsäure- oder
-estergruppe ist, worin Ra Methyl, Ethyl,
Isopropyl, Isobutyl oder t-Butyl ist, 19.
-
-
Zusätzliche
Verbindungen, die in der Anwendung der Erfindung nützlich sind,
sind unten in den Tabellen 1, 2, 3, 4, 5, 6 und 7 dargestellt: Tabelle
1
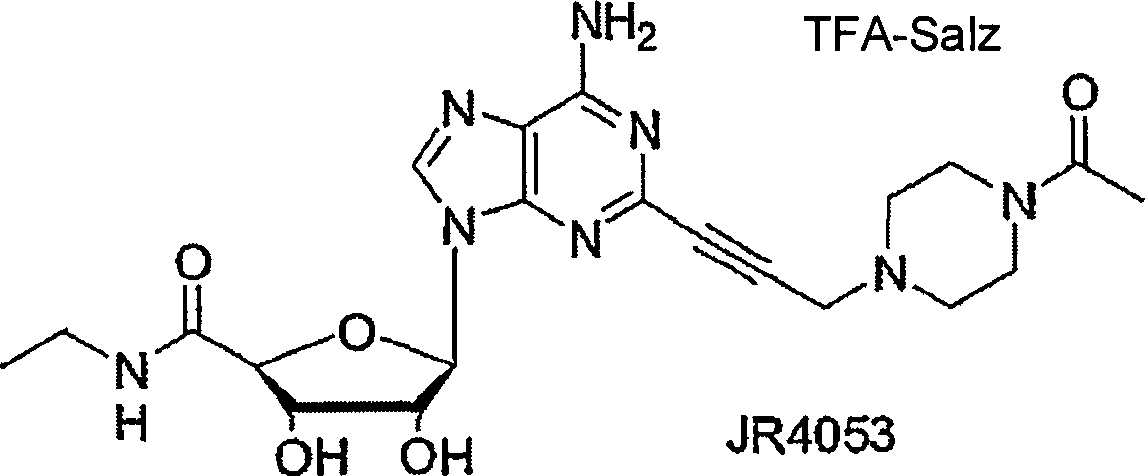
| Verbindung | R | R1 | R2 | R6 |
| ATL2037 | NECA | H | H | CH2OH |
| MP9056 | NECA | OH | H | CH2OH |
| ATL146a | NECA | H | H | CO2H |
| MP9057 | NECA | OH | H | CO2H |
| ATL146e | NECA | H | H | CO2Me |
| MP9058 | NECA | OH | H | CO2Me |
| JR2145 | CH2OH | H | H | CO2Me |
| MP9059 | CH2OH | OH | H | CO2Me |
| ATL193 | NECA | H | H | CH2OAc |
| MP9060 | NECA | OH | H | CH2OAc |
| JR2147 | CH2OH | H | H | CH2OAc |
| MP9061 | CH2OH | OH | H | CH2OAc |
| JR3023 | NECA | H | H | CH2N(CH3)2 |
| MP9062 | NECA | OH | H | CH2N(CH3)2 |
| JR3021 | NECA | H | H | COOCH2CH2NHBoc |
| MP9063 | NECA | OH | H | COOCH2CH2NHBoc |
| JR3033 | NECA | H | H | COOCH2CH2NH2 |
| MP9064 | NECA | OH | H | COOCH2CH2NH2 |
| JR3037 | NECA | H | H | CONHCH2CH3 |
| MP9065 | NECA | OH | H | CONHCH2CH3 |
| JR3055 | NECA | H | H | CONH2 |
| MP9072 | NECA | OH | H | CONH2 |
| JR3065 | NECA | H | H | CONHMe |
| MP9066 | NECA | OH | H | CONHMe |
| JR3067B | NECA | H | H | Me,
cis CO2Me |
| MP9067 | NECA | OH | H | Me,
cis CO2Me |
| JR3067A | NECA | H | H | Me,
trans CO2Me |
| MP9068 | NECA | OH | H | Me,
trans CO2Me |
| JR3087 | NECA | H | H | CH2CH3 |
| MP9069 | NECA | OH | H | CH2CH3 |
| JR3159A | NECA | OH | H | H |
| JR3159B | NECA | OH | H | H |
| JR3119 | NECA | H | H | COCH3 |
| MP9070 | NECA | OH | H | COCH3 |
| JR3121 | NECA | H | H | CHCH3(OH) |
| MP9071 | NECA | OH | H | CHCH3(OH) |
| JR3139 | NECA | OH | C6H11 | H |
NECA = CH
3CH
2N(H)C(O)- Tabelle
2
| Verbindung | R1 | R2 | R6 |
| JR3261 | H | H | H |
| JR3259 | H | H | CO2tBu |
| JR3269 | H | H | CO2Et |
| JR4011 | H | H | CO2iBu |
| JR4009 | H | H | CO2iPr |
| JR4007 | H | H | COMe |
| JR4051 | H | H | COC(CH3)3 |
| JR4047 | H | H | COCH2(CH3)3 |
| MP9047 | H | H | COCH3 |
| MP9048 | H | H | C(O)N(CH3)2 |
| MP9049 | H | H | C(O)N(CH3)Et |
| MP9050 | H | H | C(O)N(CH3)iPr |
| MP9051 | H | H | C(O)N(CH3)iBu |
| MP9052 | H | H | C(O)NH(CH3) |
| MP9053 | H | H | C(O)NH(Et) |
| MP9054 | H | H | C(O)NH(iPr) |
| MP9055 | H | H | C(O)NH(iBu) |
| TX3261 | OH | H | H |
| TX3259 | OH | H | CO2tBu |
| TX3269 | OH | H | CO2Et |
| TX4011 | OH | H | CO2iBu |
| TX4009 | OH | H | CO2iPr |
| TX4007 | OH | H | COMe |
| TX4051 | OH | H | COC(CH3)3 |
| TX4047 | OH | H | COCH2(CH3)3 |
| TX9047 | OH | H | COCH3 |
| TX9048 | OH | H | C(O)N(CH3)2 |
| TX9049 | OH | H | C(O)N(CH3)Et |
| TX9050 | OH | H | C(O)N(CH3)iPr |
| TX9051 | OH | H | C(O)N(CH3)iBu |
| TX9052 | OH | H | C(O)NH(CH3) |
| TX9053 | OH | H | C(O)NH(Et) |
| TX9054 | OH | H | C(O)NH(iPr) |
| TX9055 | OH | H | C(O)NH(iBu) |
Tabelle
3
| Verbindung | n | R3 | R6 |
| JR3135 | 1 | OH | H |
| JR3089 | 2 | OH | H |
| JR3205 | 2 | NH2 | H |
| JR3177A | 2 | OH | 2-CH3 |
| JR3177B | 2 | OH | 2-CH3 |
| JR3181A | 2 | OH | 2-CH3 |
| JR3181B | 2 | OH | 2-CH3 |
| JR3227 | 2 | OH | 2-C(CH3)3 |
| JR9876 | 2 | OH | 2-C6H5 |
| JR3179 | 2 | OH | 3-CH3 |
| JR3221 | 2 | OH
(R) | 3-CH3 (R) |
| ATL
203 | 2 | OH
(S) | 3-CH3 (R) |
| MP9041 | 2 | OH
(R) | 3-CH3 (S) |
| MP9042 | 2 | OH
(S) | 3-CH3 (S) |
| JR3201B | 2 | OH | 3-(CH3)2 |
| MP9043 | 2 | OH
(R) | 3-CH2CH3 (R) |
| MP9044 | 2 | OH
(S) | 3-CH2CH3 (R) |
| MP9045 | 2 | OH
(R) | 3-CH2CH3 (S) |
| MP9046 | 2 | OH
(S) | 3-CH2CH3 (S) |
| JR3163 | 2 | OH | 3-(CH3)2, 5-(CH3)2 |
| JR9875 | 2 | OH | 4-CH3 |
| JR3149 | 2 | OH | 4-C2H5 |
| JR3203 | 2 | OH | 4-C(CH3)3 |
| JR3161 | 2 | OH | 4-C6H5 |
Tabelle
4
| Verbindung | R1 | R2 | R6 |
| JR3231 | H | H | CO2Et |
| JR3218 | H | H | CO2tBu |
| JR3289 | H | H | H |
| JR4025 | H | H | Cyclohexyl |
| JR4053 | H | H | COMe |
| JR4049 | H | H | CO2iBu |
| JR3283 | H | H | 2-Pyrimidinyl |
| MP9029 | H | H | COMe |
| MP9030 | H | H | COC(CH3)3 |
| MP9031 | H | H | COCH2(CH3)3 |
| MP9032 | H | H | COCH3 |
| MP9033 | H | H | C(O)N(CH3)2 |
| MP9034 | H | H | C(O)N(CH3)Et |
| MP9035 | H | H | C(O)N(CH3)iPr |
| MP9036 | H | H | C(O)N(CH3)iBu |
| MP9037 | H | H | C(O)NH(CH3) |
| MP9038 | H | H | C(O)NH(Et) |
| MP9049 | H | H | C(O)NH(iPr) |
| MP9040 | H | H | C(O)NH(iBu) |
Tabelle
5
| Verbindung | R | R1 | R2 | R6 |
| MP9021 | NECA | H | H | CH2OH |
| MP9022 | NECA | H | H | CO2H |
| JR3251 | NECA | H | H | CO2Me |
| JR3279 | NECA | H | H | CO2Et |
| MP9027 | CH2OH | H | H | CO2Me |
| MP9028 | NECA | H | H | CO2MeCH2OAc |
| MP9015 | CH2OH | H | H | CH2OAc |
| MP9016 | NECA | H | H | CH2N(CH3)2 |
| MP9017 | NECA | H | H | COOCH2CH2NHBoc |
| MP9018 | NECA | H | H | COOCH2CH2NH2 |
| MP9019 | NECA | H | H | CONHCH2CH3 |
| MP9020 | NECA | H | H | CONH2 |
| MP9023 | NECA | H | H | CONHMe |
| MP9024 | NECA | H | H | CH2CH3 |
| MP9025 | NECA | H | H | COCH3 |
| MP9026 | NECA | H | H | CHCH3(OH) |
NECA = CH
3CH
2N(H)C(O)- Tabelle
6
| Verbindung | R | R1 | R2 | R6 |
| MP9001 | NECA | H | H | CH2OH |
| MP9002 | NECA | H | H | CO2H |
| JR3253 | NECA | H | H | CO2Me |
| MP9003 | CH2OH | H | H | CO2Me |
| MP9004 | NECA | H | H | CH2OAc |
| MP9005 | CH2OH | H | H | CH2OAc |
| MP9006 | NECA | H | H | CH2N(CH3)2 |
| MP9007 | NECA | H | H | COOCH2CH2NHBoc |
| MP9008 | NECA | H | H | COOCH2CH2NH2 |
| MP9009 | NECA | H | H | CONHCH2CH3 |
| MP9010 | NECA | H | H | CONH2 |
| MP9011 | NECA | H | H | CONHMe |
| MP9012 | NECA | H | H | CH2CH3 |
| MP9013 | NECA | H | H | COCH3 |
| MP9014 | NECA | H | H | CHCH3(OH) |
NECA = CH
3CH
2N(H)C(O)- Tabelle
7
| Verbindung | R | Y | Y' | R6 |
| RJ1111 | NECA | CH | CH | CO2Me |
| RJ1112 | NECA | CH | N | CO2Me |
| RJ1113 | NECA | N | CH | CO2Me |
| RJ1114 | NECA | N | N | CO2Me |
| RJ1115 | NECA | CH | CH | CH2OH |
| RJ1116 | NECA | CH | N | CH2OH |
| RJ1117 | NECA | N | CH | CH2OH |
| RJ1118 | NECA | N | N | CH2OH |
| RJ1119 | NECA | CH | CH | CO2H |
| RJ1120 | NECA | CH | N | CO2H |
| RJ1121 | NECA | N | CH | CO2H |
| RJ1122 | NECA | N | N | CO2H |
| RJ1123 | NECA | CH | CH | CH2OAc |
| RJ1124 | NECA | CH | N | CH2OAc |
| RJ1125 | NECA | N | CH | CH2OAc |
| RJ1126 | NECA | N | N | CH2OAc |
| RJ1127 | NECA | CH | CH | CONH2 |
| RJ1128 | NECA | CH | N | CONH2 |
| RJ1129 | NECA | N | CH | CONH2 |
| RJ1130 | NECA | N | N | CONH2 |
| RJ1131 | NECA | CH | CH | CONHMe |
| RJ1132 | NECA | CH | N | CONHMe |
| RJ1133 | NECA | N | CH | CONHMe |
| RJ1134 | NECA | N | N | CONHMe |
| RJ1135 | NECA | CH | CH | CO2tBu |
| RJ1136 | NECA | CH | N | CO2tBu |
| RJ1137 | NECA | N | CH | CO2tBu |
| RJ1138 | NECA | N | N | CO2tBu |
| RJ1139 | NECA | CH | CH | CO2Et |
| RJ1140 | NECA | CH | N | CO2Et |
| RJ1141 | NECA | N | CH | CO2Et |
| RJ1142 | NECA | N | N | CO2Et |
| RJ1143 | NECA | CH | CH | CO2iBu |
| RJ1144 | NECA | CH | N | CO2iBu |
| RJ1145 | NECA | N | CH | CO2iBu |
| RJ1146 | NECA | N | N | CO2iBu |
| RJ1147 | NECA | CH | CH | CO2iPr |
| RJ1148 | NECA | CH | N | CO2iPr |
| RJ1149 | NECA | N | CH | CO2iPr |
| RJ1150 | NECA | N | N | CO2iPr |
| RJ1151 | NECA | CH | CH | COMe |
| RJ1152 | NECA | CH | N | COMe |
| RJ1153 | NECA | N | CH | COMe |
| RJ1154 | NECA | N | N | COMe |
| RJ1155 | NECA | CH | CH | COC(CH3)3 |
| RJ1156 | NECA | CH | N | COC(CH3)3 |
| RJ1157 | NECA | N | CH | COC(CH3)3 |
| RJ1158 | NECA | N | N | COC(CH3)3 |
| RJ1159 | NECA | CH | CH | COCH2(CH3)3 |
| RJ1160 | NECA | CH | N | COCH2(CH3)3 |
| RJ1161 | NECA | N | CH | COCH2(CH3)3 |
| RJ1162 | NECA | N | N | COCH2(CH3)3 |
| RJ1163 | NECA | CH | CH | C(O)N(CH3)2 |
| RJ1164 | NECA | CH | N | C(O)N(CH3)2 |
| RJ1165 | NECA | N | CH | C(O)N(CH3)2 |
| RJ1166 | NECA | N | N | C(O)N(CH3)2 |
| RJ1167 | NECA | CH | CH | C(O)N(CH3)Et |
| RJ1168 | NECA | CH | N | C(O)N(CH3)Et |
| RJ1169 | NECA | N | CH | C(O)N(CH3)Et |
| RJ1170 | NECA | N | N | C(O)N(CH3)Et |
| RJ1171 | NECA | CH | CH | C(O)N(CH3)iPr |
| RJ1172 | NECA | CH | N | C(O)N(CH3)iPr |
| RJ1173 | NECA | N | CH | C(O)N(CH3)iPr |
| RJ1174 | NECA | N | N | C(O)N(CH3)iPr |
| RJ1175 | NECA | CH | CH | C(O)N(CH3)iBu |
| RJ1176 | NECA | CH | N | C(O)N(CH3)iBu |
| RJ1177 | NECA | N | CH | C(O)N(CH3)iBu |
| RJ1178 | NECA | N | N | C(O)N(CH3)iBu |
| RJ1179 | NECA | CH | CH | C(O)NH(Et) |
| RJ1180 | NECA | CH | N | C(O)NH(Et) |
| RJ1181 | NECA | N | CH | C(O)NH(Et) |
| RJ1182 | NECA | N | N | C(O)NH(Et) |
| RJ1183 | NECA | CH | CH | C(O)NH(iPr) |
| RJ1184 | NECA | CH | N | C(O)NH(iPr) |
| RJ1185 | NECA | N | CH | C(O)NH(iPr) |
| RJ1186 | NECA | N | N | C(O)NR(iPr) |
| RJ1187 | NECA | CH | CH | C(O)NH(iBu) |
| RJ1188 | NECA | CH | N | C(O)NH(iBu) |
| RJ1189 | NECA | N | CH | C(O)NH(iBu) |
| RJ1190 | NECA | N | N | C(O)NH(iBu) |
| RJ1191 | NECA | CH | CH | CH2OCOCH3 |
| RJ1192 | NECA | N | CH | CH2OCOCH3 |
| RJ1193 | NECA | CH | CH | CH2OCOEt |
| RJ1194 | NECA | N | CH | CH2OCOEt |
| RJ1195 | NECA | CH | CH | CH2OCOiPr |
| RJ1196 | NECA | N | CH | CH2OCOiPr |
| RJ1197 | NECA | CH | CH | CH2OCOiBu |
| RJ1198 | NECA | N | CH | CH2OCOiBu |
NECA = CH
3CH
2N(H)C(O)-
-
Beispiele
für antibakterielle
Mittel, die für
die Verwendung in der vorliegenden Erfindung geeignet sind, schließen Acediasulfon,
Acetosulfon, Amikacin, Amoxicillin, Amphotericin B, Ampicillin,
Apramycin, Arbekacin, Aspoxicillin, Aztreonam, Brodimoprim, Butirosin,
Capreomycin, Carumonam, Cefadroxil, Cefatrizin, Cefclidin, Cefdinir,
Cefditoren, Cefepim, Cefetamet, Cefmenoxim, Cefminox, Cefodizim,
Ceforanid, Cefotaxim, Cefotiam, Cefozopran, Cefpirom, Cefprozil,
Cefroxadin, Ceftazidim, Cefteram, Ceftibuten, Ceftriaxon, Cefuzonam,
Cephalexin, Cephaloglycin, Cephalosporin C, Cephradine, Ciprofloxacin,
Clinafloxacin, Colistin, Cyclacillin, Dapson, Diathymosulfon, Dibekacin,
Enviomycin, Epicillin, Fortimicin(s), Gentamicin(s), Gramicidin
S, Isepamicin, Kanamycin(s), Lucensomycin, Lymecyclin, Micronomicin,
Natamycin, Neomycin, Netilmicin, Paromomycin, Pazufloxacin, Penicillin
N, Peplomycin, Perimycin A, Polymyxin, p-Sulfanilylbenzylamin, Ribostamycin,
Ristocetin, Sisomicin, Sparfloxacin, Succisulfon, 2-p-Sulfanilylanilinoethanol,
4,4'-Sulfinyldianilin,
Sulfachrysoidin, Sulfamidochrysoidin, Sulfanilsäure, Sulfoxon, Teicoplanin,
Tetroxoprim, Thiazolsulfon, Tigemonam, Tobramycin, Tosufloxacin,
Trimethoprim, Trovafloxacin, Tuberactinomycin, Vancomycin ein. Bevorzugte
antibiotische Mittel sind Ciprofloxacin und Ceftriaxon.
-
Beispiele
für antifungale
(anti-Hefe) Mittel, die für
die Verwendung in der vorliegenden Erfindung geeignet sind, schließen Amphotericin
B, Azaserin, Candicidin(s), Lucensomycin, Mepartricin, Natamycin,
Nystatin, Tubercidin ein.
-
Beispiele
für antivirale
Mittel die für
die Verwendung in der vorliegenden Erfindung geeignet sind, schließen Abacavir,
Acyclovir, Amantadin, Famciclovir, Focsavir, Ganciclovir, Indinavir,
Lamivudin, Lopinavir, Ritonavir ein.
-
In
einer weiteren Ausführungsform
schließen
die Agonisten der A
2A-Adenosinrezeptoren, die in der Anwendung
der vorliegenden Erfindung nützlich
sind, Verbindungen ein, die die Formel (11) haben:
worin Z CR
3R
4R
5 ist; jeder R
1, R
2 und R
3 Wasserstoff ist; R
4 und
R
5 zusammen mit dem Kohlenstoffatom, an welches
sie gebunden sind, einen Cycloalkylring bilden, der 3, 4, 5, 6,
7, 8, 9 oder 10 Ringatome hat; und
worin der Ring, der R
4 und R
5 umfasst,
mit -(CH
2)
0-6-Y
substituiert ist; worin Y -CH
2OR
a, -CO
2R
a,
-OC(O)R
a, -CH
2OC(O)R
a, -C(O)NR
bR
c, -CH
2SR
a, -C(S)OR
a, -OC(S)R
a, -CH
2OC(S)R
a oder C(S)NR
bR
c oder -CH
2N(R
b)(R
c) ist;
jeder
R
7 unabhängig
Wasserstoff, (C
1-C
8)Alkyl,
(C
3-C
8)Cycloalkyl,
Aryl oder Aryl(C
1-C
8)alkylen
ist;
X -CH
2OR
a,
-CO
2R
a, -OR(O)R
a, -CH
2OC(O)R
a, -C(O)NR
bR
c, -CH
2SR
a, -C(S)OR
a, -OC(S)R
a, -CH
2OC(S)R
a oder C(S)NR
bR
c oder -CH
2N(R
b)(R
c) ist;
jeder
R
a, R
b und R
c unabhängig
Wasserstoff, (C
1-C
8)Alkyl
oder (C
1-C
8)Alkyl
ist, substituiert mit 1-3 (C
1-C
8)Alkoxy,
(C
3-C
8)Cycloalkyl,
(C
1-C
8)Alkylthio,
Aminosäure,
Aryl, Aryl(C
1-C
8)alkylen,
Heteroaryl oder Heteroaryl(C
1-C
8)alkylen;
oder R
b und R
c,
zusammen mit dem Stickstoff, an welchen sie gebunden sind, einen
Pyrrolidino-, Piperidino-, Morpholino- oder Thiomorpholinoring bilden;
und m 0 bis etwa 6 ist; oder ein pharmazeutisch akzeptables Salz
davon.
-
Ein
spezifischer Wert für
-N(R7)2 ist Amido,
Monomethylamino oder Cyclopropylamino.
-
Ein
spezifischer Wert für
Z ist Carboxy- oder -(C1-C4)Alkoxycarbonyl-cyclohexyl(C1-C4)alkyl.
-
Ein
spezifischer Wert für
Ra ist H oder (C1-C4)Alkyl, das heißt Methyl oder Ethyl.
-
Ein
spezifischer Wert für
Rb ist H, Methyl oder Phenyl.
-
Ein
spezifischer Wert für
Rc ist H, Methyl oder Phenyl.
-
Ein
spezifischer Wert für
-(CR1R2)m- ist -CH2- oder
-CH2-CH2-.
-
Ein
spezifischer Wert für
X ist CO2Ra, (C2-C5)Alkanoylmethyl
oder Amido.
-
Ein
spezifischer Wert für
Y ist CO2Ra, (C2-C5)Alkanoylmethyl
oder Amido.
-
Ein
spezifischer Wert für
m ist 1.
-
Spezifische
A
2A-Adenosinrezeptoragonisten, die für die Verwendung
in der vorliegenden Erfindung geeignet sind und die Formel (II)
haben, schließen
jene ein, die in dem
US Patent
Nr: 6,232,297 beschrieben sind. Bevorzugte Verbindungen
mit der Formel (II) sind jene, worin R
7 H
ist, X Ethylaminocarbonyl ist und Z 4-Carboxycyclohexylmethyl (DWH-146a)
ist, Z 4-Methoxycarbonylcyclohexylmethyl (DWH-146e) ist, Z 4-Isopropylcarbonylcyclohexylmethyl
(AB-1) ist, Z 4-Acetoxymethylcyclohexylmethyl (JMR-193) ist oder
Z 4-Pyrrolidin-1- carbonylcyclohexylmethyl
(AB-3) ist. Diese Verbindungen sind unten dargestellt.
-
-
Die
spezifischen A
2A-Adenosinrezeptoragonisten,
die für
die Verwendung in der vorliegenden Erfindung geeignet sind und die
Formel (II) haben, schließen
jene ein, die in dem
U.S. Patent
Nr. 6,232,297 beschrieben sind. Diese Verbindungen, die
die Formel (II) haben, können
entsprechend den hierin beschriebenen Verfahren hergestellt werden.
-
Eine
weitere spezifische Gruppe von Agonisten der A
2A-Adenosinrezeptoren,
die in der Anwendung der vorliegenden Erfindung nützlich sind,
schließt
Verbindungen ein, die die allgemeine Formel (III) haben:
worin Z
2 eine
Gruppe ist, ausgewählt
aus der Gruppe bestehend aus -OR
12, -NR
13R
14, einem -C=C-Z
3 und -NH-N=R
17;
Jeder
Y
2 individuell H, C
1-C
6-Alkyl, C
3-C
7-Cycloalkyl, Phenyl oder Phenyl-C
1-C
3-alkyl ist;
R
12 - a) C1-4-Alkyl
ist;
- b) C1-4-Alkyl ist, substituiert mit
einem oder mehreren C1-4-Alkoxygruppen, Halogenen (Fluor, Chlor
oder Brom), Hydroxygruppen, Aminogruppen, Mono(C1-4-Alkyl)aminogruppen,
Di(C1-4-alkyl)aminogruppen oder C6-10-Arylgruppen, worin die Arylgruppen mit
einem oder mehreren Halogenen (Fluor, Chlor oder Brom), C1-4-Alkylgruppen, Hydroxygruppen, Aminogruppen,
Mono(C1-4-Alkyl)aminogruppen oder Di(C1-4-alkyl)aminogruppen
substituiert sein können;
oder
- c) C6-10-Aryl ist; oder (d) C6-10-Aryl ist, substituiert mit einem oder
mehreren Halogenen (Fluor, Chlor oder Brom), Hydroxygruppen, Aminogruppen,
Mono(C1-4-Alkyl)aminogruppen, Di(C1-4-alkyl)aminogruppen oder C1-4-Alkylgruppen;
einer
von R13 und R14 dieselbe
Bedeutung hat wie R12 und der andere Wasserstoff
ist; und
R17 eine Gruppe ist, die die
Formel (i) hat worin jeder R15 und
R16 unabhängig Wasserstoff, (C3-C7)Cycloalkyl sein
kann oder irgendeine der Bedeutungen von R12 haben
kann, vorausgesetzt, dass R15 und R16 nicht beide Wasserstoff sind;
X2 CH2OH, CH3, CO2R20 oder
C(=O)NR21R22 ist,
worin R20 dieselbe Bedeutung hat wie R13 und worin R21 und R22 dieselben Bedeutungen haben wie R15 und R16 oder R21 und R22 beide
H sind;
Z3 eine der folgenden Bedeutungen
hat: - a) C6-C10-Aryl, optional mit einem bis drei Halogenatomen,
C1-C6-Alkyl, C1-C6-Haloalkyl, C1-C6-Alkoxy, C1-C6-Haloalkoxy, C2-C6-Alkoxycarbonyl,
C2-C6-Alkoxyalkyl,
C1-C6-Alkylthio,
Thio, CHO, Cyanomethyl, Nitro, Cyano, Hydroxy, Carboxy, C2-C6-Acyl, Amino-C1-C3-monoalkyamino, C2-C6-Dialkylamino,
Methylendioxy oder Aminocarbonyl substituiert;
- b) eine Gruppe mit der Formel -(CH2)q-Het, worin q 0 oder eine ganze Zahl von
1 bis 3 ist und Het ein 5- oder 6-gliedriger heterocyclischer aromatischer
oder nichtaromatischer Ring ist, optional benzkondensiert, umfassend
1 bis 3 Heteroatome, ausgewählt
aus der nichtperoxid Sauerstoff, Stickstoff oder Schwefel, verbunden
durch ein Kohlenstoffatom oder ein Stickstoffatom;
- c) C3-C7-Cycloalkyl,
das optional ungesättigt
ist oder C2-C4 Alkenyl
enthält;
- d) worin
R23 Wasserstoff,
Methyl oder Phenyl ist;
R24 Wasserstoff,
C1-C6 lineares oder
verzweigtes Alkyl, C5-C6-Cycloalkyl oder C3-C7-Cycloalkenyl,
Phenyl-C1-C2-alkyl
ist oder R23 und R24 zusammen
genommen einen 5- oder 6-gliedrigen carbocyclischen Ring bilden
oder R25 Wasserstoff ist und R23 und
R24 zusammen genommen eine Oxogruppe oder
ein entsprechendes acetalisches Derivat bilden;
R25 OH,
NH2 Dialkylamino, Halogen, Cyano ist; und
n 0 oder 1 bis 4 ist; oder
- e) C1-C16-Alkyl,
optional 1-2 Doppelbindungen, O, S oder NY2 umfassend;
oder
ein pharmazeutisch akzeptables Salz davon.
-
Spezifische
C6-10-Arylgruppen schließen Phenyl und Naphtyl ein.
-
Vorzugsweise
ist in der Verbindung mit der Formel (I) Z2 eine
Gruppe mit der Formel (iii) -O-(CH2)n-Ar (iii)worin
n eine ganze Zahl von 1-4 ist, vorzugsweise 2, und Ar eine Phenylgruppe,
Tolylgruppe, Naphthylgruppe, Xylylgruppe oder Mesitylgruppe. Besonders
vorzugsweise ist Ar eine para-Tolylgruppe und n = 2.
-
Vorzugsweise
ist in der Verbindung mit der Formel (II) Z2 eine
Gruppe mit der Formel (iv) -NH-N=CHCy (iv)worin Cy
eine C3-7-Cycloalkylgruppe, vorzugsweise
Cyclohexyl oder eine C1-4 Alkylgruppe, vorzugsweise
Isopropyl, ist.
-
Vorzugsweise
ist in der Verbindung mit der Formel (II) Z2 eine
Gruppe mit der Formel (vii) -C=C-Z3 (v)worin
Z3 C3-C16-Alkyl,
Hydroxy C2-C6-Alkyl
oder (Phenyl) (Hydroxymethyl) ist.
-
Spezifische
Beispiele für
solche Verbindungen mit der Formel (I) schließen wie unten gezeigt WRC-0470,
WRC-0474 [SHA 211], WRC-0090 und WRC-0018 ein:
worin
der H an CH
2OH optional durch Ethylaminocarbonyl
ersetzt sein kann. Von diesen spezifischen Beispielen sind WRC-0474
[SHA 211] und WRC-0470
besonders bevorzugt.
-
Solche
Verbindungen können
wie in dem Folgenden beschrieben synthetisiert werden: Olsson et
al. (
U.S. Pat. Nrn. 5,140,015 und
5,278,150 ); Cristalli (
U.S. Pat. Nr. 5,593,975 );
Miyasaka et al. (
U.S. Pat. Nr. 4,956,345 );
Hutchinson, A. J. et al., J. Pharmacol. Exp. Ther., 251, 47 (1989);
Olsson, R. A. et al., J. Med. Chem., 29, 1683 (1986); Bridges, A.
J. et al., J. Med. Chem., 31, 1282 (1988); Hutchinson, A. J. et
al., J. Med. Chem., 33, 1919 (1990); Ukeeda, M. et al., J. Med.
Chem., 34, 1334 (1991); Francis, J. E. et al., J. Med. Chem., 34,
2570 (1991); Yoneyama, F. et al., Eur. J. Pharmacol., 213, 199-204
(1992); Peet, N. P. et al., J. Med. Chem., 35, 3263 (1992); und
Cristalli, G. et al., J. Med. Chem., 35, 2363 (1992).
-
Eine
weitere Ausführungsform
schließt
Verbindungen ein, die die Formel (III) haben, worin Z
2 eine Gruppe
ist, die die Formel (vi) hat:
worin R
34 und
R
35 unabhängig H, C
1-C
6-Alkyl, C
3-C
7-Cycloalkyl, Phenyl, Phenyl-C
1-C
3-alkyl sind oder R
34 und R
35 zusammen genommen mit dem Stickstoffatom
ein 5 oder 6 gliedriger heterocyclischer Ring sind, der 1-2 Heteroatome
enthält,
ausgewählt
aus nichtperoxid Sauerstoff-, Stickstoff-(N(R
13)) oder
Schwefelatomen. Vorzugsweise ist einer von R
34 und
R
35 Wasserstoff und der andere ist Ethyl,
Methyl oder Propyl. Besonders vorzugsweise ist einer von R
34 und R
35 Wasserstoff
und der andere Ethyl oder Methyl.
-
Die
2-(Pyrazol-1-yl)adenosin-Verbindungen der Erfindung, worin Z
2 eine Gruppe ist, die die Formel (vi) hat,
können
durch die Reaktion eines 2-Chlor-
oder 2-Iod-Adenosinderivats mit einer 1H-Pyrazol-4-carboxamid-Verbindung hergestellt
werden, die die Formel (vii) hat:
worin R
34 und
R
35 wie oben beschrieben sind und worin
eine selektiver Schutz/kein Schutz der Amidogruppe je nach Bedarf
verwendet wird. Ein bevorzugtes Pyrazolderivat, welches in der Ausführung dieser
Erfindung nützlich
ist, ist eine Verbindung, die die folgende Formel hat:
-
Die
1H-Pyrazol-4-carboxamide können
ausgehend von einer 1H-Pyrazol-4-carbonsäure hergestellt werden,
die von Aldrich Chemical Co. erhältlich ist. In dem ersten Schritt
wird die Säure
in einen Ester konvertiert, z.B. einen Methyl- oder Ethylester.
Der Ester wird mittels Aminolyse zu dem Amid konvertiert, z.B. mit Methylamin,
um das Methylamid zu bilden. Das Pyrazol-4-carboxamid reagiert in
Anwesenheit einer starken Base mit den 2-Halopurinen, um die 2-(Pyrazol-1-yl)adenosin-Verbindungen
bereitzustellen, die die Formel (III) haben.
-
Eine
weitere spezifische Gruppe von Agonisten der A
2A-Adenosinrezeptoren,
die in der Anwendung der vorliegenden Erfindung nützlich sind,
schließt
Verbindungen ein, die die allgemeine Formel (IV) haben:
worin Z
4 -NR
28R
29 ist;
R
28 Wasserstoff oder (C
1-C
4)Alkyl ist; und R
29 - a) (C1-C4)Alkyl
ist;
- b) (C1-C4)Alkyl
ist, substituiert mit einem oder mehreren (C1-C4)Alkoxy, Halogen, Hydroxy, Amino, Mono((C1-C4)alkyl)amino,
Di((C1-C4)alkyl)amino
oder (C6-C10)Aryl,
worin Aryl optional mit einem oder mehreren Halogen, Hydroxy, Amino,
(C1-C4)Alkyl, R30OCOC-((C1-C4)Alkyl)-, R31R32NC(=O)-, ((C1-C4)Alkyl)-, Mono((C1-C4)alkyl)amino oder Di((C1-C4)alkyl)amino
substituiert ist;
- c) (C6-C10)Aryl
ist; oder
- d) (C6-C10)Aryl
ist, substituiert mit einem oder mehreren Halogen, Hydroxy, Amino,
Mono((C1-C4)alkyl)amino,
Di((C1-C4)alkyl)amino
oder (C1-C4)Alkyl;
worin
jeder Y4 individuell H, (C1-C6)Alkyl, (C3-C7)Cycloalkyl, Phenyl oder Phenyl(C1-C3)alkyl ist; und
X4 -C(=O)NR31R32, -COOR30 oder
-CH2OR30 ist;
worin
jeder R31 und R32 unabhängig; Wasserstoff;
C3-7-Cycloalkyl; (C1-C4)Alkyl; (C1-C4)Alkyl, substituiert mit einem oder mehreren
(C1-C4)Alkoxy, Halogen, Hydroxy, -COOR33, Amino, Mono((C1-C4)alkyl)amino, Di((C1-C4)alkyl)amino oder (C6-C10)Aryl, worin Aryl optional mit einem oder
mehreren Halogen, (C1-C4)Alkyl, Hydroxy,
Amino, Mono((C1-C4)alkyl)amino
oder Di((C1-C4)alkyl)amino
substituiert ist; (C6-C10)Aryl;
oder (C6-C10)Aryl
sind, substituiert mit einem oder mehreren Halogen, Hydroxy, Amino,
Mono((C1-C4)alkyl)amino, Di((C1-C4)alkyl)amino
oder (C1-C4)Alkyl;
R26 und R27 repräsentieren
unabhängig
Wasserstoff, niederes Alkanoyl, niederes Alkoxy-niederes Alkylanoyl, Aroyl,
Carbamoyl oder mono- oder di-niederes Alkylcarbamoyl; und R30 und R33 sind unabhängig Wasserstoff, (C1-C4)Alkyl, (C6-C10)Aryl oder (C6-C10)Aryl((C1-C4)alkyl); oder
ein pharmazeutisches akzeptables Salz davon.
-
In
einer Ausführungsform
der Formel (IV) ist wenigstens einer von R28 und
R29 (C1-C4)Alkyl, substituiert mit einem oder mehreren
(C1-C4)Alkoxy, Halogen,
Hydroxy, Amino, Mono((C1-C4)alkyl)amino,
Di((C1-C4)alkyl)amino
oder (C6-C10)Aryl,
worin Aryl optional mit einem oder mehreren Halogen, Hydroxy, Amino,
(C1-C4)Alkyl, R30OOC-(C1-C4)alkyl, Mono((C1-C4)alkyl)amino oder Di((C1-C4)alkyl)amino substituiert ist.
-
In
einer weiteren Ausführungsform
ist wenigstens einer von R31 und R32 C1-4-Alkyl, substituiert
mit einem oder mehreren (C1-C4)Alkoxy,
Halogen, Hydroxy, Amino, Mono((C1-C4)alkyl)amino, Di((C1-C4)alkyl)amino oder (C6-C10)Aryl, worin Aryl optional mit einem oder
mehreren Halogen, Hydroxy, Amino, (C1-C4)Alkyl, R30OOC-(C1-C4)alkylen-, Mono((C1-C4)alkyl)amino oder Di((C1-C4)alkyl)amino substituiert ist.
-
In
einer weiteren Ausführungsform
ist wenigstens einer von R28 und R29 C6-10-Aryl, substituiert
mit einem oder mehreren Halogen, Hydroxy, Amino, Mono((C1-C4)alkyl)amino,
Di((C1-C4)alkyl)amino
oder (C1-C4)Alkyl.
-
In
einer weiteren Ausführungsform
ist wenigstens einer von R31 und R32 C6-10-Aryl, substituiert
mit einem oder mehreren Halogen, Hydroxy, Amino, Mono((C1-C4)alkyl)amino,
Di((C1-C4)alkyl)amino
oder (C1-C4)Alkyl.
-
In
einer bevorzugten Kombination ist R31 Wasserstoff
und R32 (C1-C4)Alkyl,
Cyclopropyl oder Hydroxy-(C2-C4)alkyl.
Eine bevorzugte R28-Gruppe ist (C1-C4)Alkyl, substitutiert mit (C6-C10)Aryl, das wiederum mit R30 O(O)C-(C1-C4)alkylin- substituiert
ist.
-
Eine
bevorzugte Verbindung, die die Formel (IV) hat, ist:
worin
R
30 Wasserstoff, Methyl, Ethyl, n-Propyl
oder Isopropyl ist. Besonders bevorzugt ist eine Verbindung, worin
die R
30-Gruppe Methyl oder Ethyl ist. Die
am meisten bevorzugte R
30-Gruppe ist Methyl.
-
Zwei
Verbindungen, die insbesondere nützlich
für die
Ausführung
der vorliegenden Erfindung sind, haben die Formel:
worin
R
30 Wasserstoff (Säure, CGS21680) und wo R
30 Methyl (Ester, JR2171) ist.
-
Die
Verbindungen der Erfindung, die die Formel (IV) haben, können wie
in dem
U.S. Patent 4,968,697 oder
dem J. Med. Chem., 33, 1919-1924, (1990) beschrieben synthetisiert
werden.
-
Spezifisch
stellt die Erfindung auch die Verwendung einer Verbindung mit der
Formel (I) oder eines pharmazeutisch akzeptables Salzes davon bereit,
um ein Medikament für
die Behandlung einer systemischen Intoxifikation in Säugetieren
(z.B. Mensch) herzustellen.
-
Spezifisch
stellt die Erfindung auch die Verwendung einer Verbindung mit der
Formel (I) oder eines pharmazeutisch akzeptables Salzes davon bereit,
um ein Medikament für
die Behandlung einer Entzündung, die
durch bakterielle, fungale oder virale Infektionen verursacht wird,
oder der Entzündung
herzustellen, die durch die Behandlung von diesen Infektionen verursacht
wird, z.B. durch den Tod der Bakterien- oder Virenzellen in einem
Säugetier
(z.B. einem Menschen).
-
Das
vorliegende Verfahren schließt
auch die Verabreichung eines Typ-IV-Phosphodiesterase(PDE)-Inhibitors
in Kombination mit Verbindungen ein, die die Formeln (I), (II),
(III) und (IV) haben. Die Kombination von den Verbindungen der Erfindung
mit einem Typ-IV-Phosphodiesteraseinhibitor
stellt synergistische Minderungen der Entzündungsreaktion von Immunzellen
bereit. Bespiele für
Typ-IV-Phosphodiesterase(PDE)-Inhibitoren
schließen
jene ein, die in dem
U.S. Patent
Nr. 4,193,926 und
WO
92-079778 und von Molnar-Kimber, K. L., J. Immunol., 150,
295A (1993), offenbart sind.
-
Geeignete
Typ-IV-Phosphodiesterase(PDE)-Inhibitoren schließen racemische und optisch
aktive 4-(Polyalkoxyphenyl)-2-pyrrolidone mit der allgemeinen Formel
(VI) ein:
(offenbart und beschrieben
in dem
U.S. Patent Nr. 4,193,926 ),
worin R
18 und R
19 unabhängig gleich
oder verschieden sind und Kohlenwasserstoffradikale sind, die bis
zu 18 Kohlenstoffatomen haben, wobei wenigstens eines anders ist
als Methyl, ein heterocyclischer Ring oder ein Alkyl mit 1-5 Kohlenstoffatomen,
welches mit einem oder mehreren Halogenatomen, Hydroxy, Carboxy,
Alkoxy, Alkoxycarbonyl oder einer Aminogruppe oder Amino substituiert
ist.
-
Beispiele
für R18- und R19-Kohlenwasserstoffgruppen
sind gesättigtes
und ungesättigtes,
gradkettiges und verzweigtes Alkyl mit 1-18, vorzugsweise 1-5, Kohlenstoffatomen,
Cycloalkyl und Cycloalkylalkyl, vorzugsweise mit 3-7 Kohlenstoffatomen,
und Aryl und Aralkyl, vorzugsweise mit 6-10 Kohlenstoffatomen, vorzugsweise
monocyclisch.
-
Rolipram
ist einem Beispiel für
einen geeigneten Typ-IV-Phosphodiesterase-
oder PDE-Inhibitor, das in der oben genannten Formel mit eingeschlossen
ist. Rolipram hat die folgende Formel:
-
In
den Fällen,
wo die Verbindungen ausreichend basisch oder sauer sind, um stabile
nichttoxische Säure-
oder Basesalze zu bilden, kann die Verabreichung der Verbindungen
als Salz angemessen sein. Beispiele für pharmazeutisch akzeptable
Salze sind organische Säureadditionssalze,
die mit Säuren
gebildet werden, die ein physiologisch akzeptables Anion bilden,
zum Beispiel Tosylat, Methansulfonat, Acetat, Citrat, Malonate,
Tartarat, Succinat, Benzoat, Ascorbat, α-Ketoglutarat und α-Glycerophosphat.
Es können
auch geeignete organische Salze gebildet werden, einschließlich Hydrochlorid-,
Sulfat-, Nitat-, Bicarbonat- und Carbonatsalze.
-
Pharmazeutisch
akzeptable Salze können
unter Verwendung von Standartverfahren, die im Stand der Technik
gut bekannt sind, erhalten werden, zum Beispiel durch die Reaktion
einer ausreichend basischen Verbindung, wie ein Amin, mit einer
geeigneten Säure,
was ein physiologisch akzeptables Anion hervorbringt. Es können auch
Alkalimetall-(zum Beispiel Natrium, Kalium oder Lithium) oder Erdaikalimetall-(zum
Beispiel Kalzium)-Salz von Carbonsäuren hergestellt werden.
-
Die
Verbindungen der vorliegenden Erfindung können zweckdienlich in einer
pharmazeutischen Zusammensetzung verabreicht werden, die die Verbindung
in Kombination mit einem geeigneten Exzipienten enthält. Solche
pharmazeutischen Zusammensetzungen können durch Verfahren hergestellt
werden und enthalten Exzipienten, die im Stand der Technik gut bekannt
sind. Ein allgemein anerkanntes Kompendium für solche Verfahren und Bestandteile
ist Remington's
Pharmaceutical Sciences von E. W. Martin (Mark Publ. Co., 15. Auflage,
1975). Die Verbindungen und Zusammensetzungen der vorliegenden Erfindung
können
parenteral (zum Beispiel durch eine intravenöse, intrapritoneale oder intramuskuläre Injektion),
topikal, oral oder rektal verabreicht werden.
-
Für eine orale
therapeutische Verabreichung kann die aktive Verbindung mit einem
oder mehreren Exzipienten kombiniert werden und in Form von einnehmbaren
Tabletten, Bukkaltabletten, Troches, Kapseln, Elexieren, Suspensionen,
Sirups, Waffeln und dergleichen verwendet werden. Solche Zusammensetzungen
und Präparate
sollten wenigstens 0,1 % der aktiven Verbindung enthalten. Der Prozentsatz
der Zusammensetzungen und Präparate
kann natürlich
verändert
werden und zweckmäßig zwischen
etwa 2 bis 60 Gew.-% einer gegeben Dosierungseinheitsform sein.
-
Die
Tabletten, Troches, Pillen, Kapseln und dergleichen können auch
das Folgende enthalten: Bindemittel wie Tragantgummi, Acacia, Maisstärke oder
Gelatine; Exzipienten wie Dikalziumphosphat; ein zersetzendes Mittel
wie Maisstärke,
Kartoffelstärke,
Alginsäure
und dergleichen; ein Schmiermittel wie Magnesiumstearat; und ein
Süßungsmittel
wie Saccharose, Fruktose, Lactose oder Aspartam oder es kann ein
Aromastoff wie Pfefferminze, Gaultheriaöl oder Kirschgeschmack zugegeben
werden. Wenn die Dosierungseinheitsform eine Kapsel ist, kann sie
zusätzlich
zu den Materialien des oben genannten Materialtypen, einen flüssigen Träger wie
Pflanzenöl
oder Polyethylenglykol enthalten. Es können verschiedene andere Materialien
als Überzüge oder
um die physikalische Form der festen Dosierungseinheitsform in anderer
Weise zu modifizieren vorhanden sein. Zum Beispiel können Tabletten,
Pillen oder Kapseln mit Gelatine, Wachs, Schellack oder Zucker und
dergleichen überzogen
werden. Ein Sirup oder Elixier kann die aktive Verbindung, Saccharose
oder Fruktose als ein Süßungsmittel,
Methyl- oder Propylparabene als Konservierungsstoffe, einen Farbstoff
und einen Aromastoff wie Kirsch- oder Orangengeschmack enthalten.
Natürlich
sollte irgendein Material, das bei der Herstellung irgendeiner Dosierungseinheitsform
verwendet wird, pharmazeutisch akzeptabel und im Wesentlichen in
den eingesetzten Mengen nichttoxisch sein. Zusätzlich kann die aktive Verbindung
in Präparaten
und Vorrichtungen mit einer verzögerten
Freisetzung enthalten sein.
-
Die
Verbindungen oder Zusammensetzungen können auch intravenös oder intraperitoneal
mittels Infusion oder Injektion verabreicht werden. Die Lösungen der
aktiven Verbindung oder ihrer Salze können in Wasser hergestellt
werden, optional mit einem nichttoxischen oberflächenaktiven Mittel gemischt.
Die Dispersionen können
auch in Glycerol, flüssigen
Polyethylenglykolen, Triacetin und Mischungen davon und in Ölen hergestellt
werden. Unter gewöhnlichen
Aufbewahrungs- und Verwendungsbedingungen enthaltne diese Präparate einen
Konservierungsstoff, um das Wachstum von Mikroorganismen zu verhindern.
-
Pharmazeutische
Dosierungsformen, die für
die Injektion oder Infusion geeignet sind, können sterile wässerige
Lösungen
oder Dispersionen oder sterile Pulver enthalten, die den aktiven
Bestandteil umfassen und welche für die extemporane Herstellung
von sterilen injizierbaren oder infundierbaren Lösungen oder Dispersionen geeignet
sind, optional in Liposomen verpackt. In allen Fällen sollte die entgültige Dosierungsform steril,
flüssig
und unter den Herstellungs- und Aufbewahrungsbedingungen stabil
sein. Der flüssige
Träger
oder das flüssige
Vehikel kann ein Lösungsmittel
oder ein flüssiges
Dispersionsmedium sein, zum Beispiel Wasser, Ethanol, ein Polyol
(zum Beispiel Glycerol, Propylenglycol, flüssige Polyethylenglycole und
dergleichen), Pflanzenöle,
nichttoxische Glycerylestern und geeignete Mischungen davon sein.
Eine angemessene Fluidität
kann zum Beispiel durch die Bildung von Liposomen, durch die Aufrechterhaltung
der benötigten
Partikelgröße im Fall
von Dispersionen oder durch die Verwendung von oberflächenaktiven
Mitteln erhalten werden. Der Schutz vor der Wirkung von Mikroorganismen
kann durch verschiedenen antibakterielle und antifungale Mittel
bewirkt werden, zum Beispiel durch Parabene, Chlorbutanol, Phenol,
Sorbinsäure,
Thimerosal und dergleichen. In vielen Fällen ist es wünschenswert
isotonische Mittel zu umfassen, zum Beispiel Zucker, Puffer oder
Natriumchlorid. Eine anhaltende Absorption der injizierbaren Zusammensetzungen
kann durch die Verwendung von Mitteln in den Zusammensetzungen bewirkt
werden, die die Absorption verzögern,
zum Beispiel Aluminiummonostearat und Gelatine.
-
Sterile
injizierbare Lösungen
können
hergestellt werden, indem die aktive Verbindung in der benötigten Menge
in dem geeigneten Lösungsmittel
mit verschiedenen anderen von den oben aufgelisteten Bestandteilen
je nach Bedarf mit eingeschlossen wird, gefolgt von einer Filtersterilisation.
In dem Fall steriler Pulvern für
die Herstellung von sterilen injizierbaren Lösungen sind die bevorzugten
Verfahren zur Herstellung Vakuumtrocknungs- und Gefriertrocknungstechniken,
welche ein Pulver des aktiven Bestandteils plus irgendeinem zusätzlichen
gewünschten
Bestandteil ergeben, der in den zuvor sterilfiltrierten Lösungen vorhanden
ist.
-
Für topikale
Verabreichungen können
die vorliegenden Verbindungen in der reinen Form angewendet werden,
das heißt
wenn sie Flüssigkeiten
sind. Es wird jedoch im Allgemeinen wünschenswert sein sie auf die Haut
als Zusammensetzungen oder Formulierung in Kombination mit einem
dermatologisch akzeptabeln Träger
zu applizieren, welcher ein Feststoff oder eine Flüssigkeit
sein kann.
-
Brauchbare
feste Träger
schließen
fein verteilte Feststoffe wie Talk, Ton, mikrokristalline Zellulose,
Silica, Aluminiumoxid und dergleichen ein. Brauchbare flüssige Träger schließen Wasser,
Alkohole oder Glykole oder Wasser-Alkohol/Glykol-Mischungen ein,
in welchen die vorliegenden Verbindungen mit wirksamen Leveln gelöst oder
dispergiert werden können,
optional mit der Hilfe von nichttoxischen oberflächenaktiven Stoffen. Es können Adjuvans
wie Düfte
und zusätzliche
antimikrobielle Mittel können
zugegeben werden, um die Eigenschaften für eine gegebene Verwendung
zu verbessern. Die resultierenden flüssigen Zusammensetzungen können mittels
Adsorptionsmitteltupfer, die verwendet werden, um Bandagen oder
andere Verbände
zu imprägnieren,
aufgetragen werden oder auf den betroffenen Bereich unter Verwendung
Pumptyp- oder Aerosolzerstäubern
gesprüht
werden. Verdicker wie synthetische Polymere, Fettsäuren, Fettsäuresalze
und -ester, Fettalkohle, modifizierte Cellulosen oder modifizierte
Mineralmaterialien können
mit flüssigen
Trägern
eingesetzt werden, um streichbare Pasten, Gele, Salben, Seifen und
dergleichen für
die direkte Anwendung auf die Haut des Benutzers zu bilden.
-
Brauchbare
Dosierungen der Verbindungen mit der Formel I können durch den Vergleich ihrer
in vitro-Aktivität
und in vivo-Aktivität
in Tiermodellen bestimmt werden. Die Verfahren für die Extrapolation der wirksamen
Dosierungen in Mäusen
und anderen Tieren auf die Menschen sind im Stand der Technik bekannt;
siehe zum Beispiel
U.S. Pat.
Nr. 4,938,949 .
-
Die
Verbindung wird zweckmäßig in einer
Dosierungseinheitsform verabreicht; zum Beispiel, die etwa 0,05
mg bis etwa 500 mg, zweckmäßig etwa
0,1 mg bis etwa 250 mg, besonders zweckmäßig etwa 1 mg bis etwa 150
mg des aktiven Bestandteils pro Dosierungseinheitsform enthält. Die
gewünschte
Dosis kann zweckmäßig in einer
einzigen Dosis oder als aufgeteilte Dosen in geeigneten Zeitabständen, zum
Beispiel als zwei, drei, vier oder mehr Unterdosen pro Tag, dargeboten
werden. Die Unterdosen selbst können
weiter unterteilt sein, z.B. in eine Zahl eigenständiger,
frei verteilter Verabreichungen.
-
Die
Zusammensetzungen können
zweckmäßig oral,
sublingual, transdermal oder parenteral mit Dosisleveln von etwa
0,01 bis etwa 150 μg/kg,
vorzugsweise von etwa 0,1 bis 50 μg/kg
und besonders vorzugsweise von etwa 0,1 bis etwa 10 μg/kg Säugetierkörpergewicht
verabreicht werden.
-
Für die parenterale
Verabreichung werden die Verbindungen in einer wässerigen Lösung mit einer Konzentration
von etwa 0,1 bis etwa 10 %, besonders vorzugsweise von etwa 0,1
bis etwa 7 % dargeboten. Die Lösung
kann andere Bestandteile wie Emulgatoren, Antioxidationsmittel oder
Puffer enthalten.
-
Die
exakte Kur für
die Verabreichung der Verbindungen und Zusammensetzungen, die hierin
offenbart ist, wird notwendigerweise von den Bedürfnissen des individuellen
Subjekts, das behandelt wird, dem Behandlungstyp und natürlich dem
Ermessen des behandelnden praktischen Arztes abhängen.
-
Die
Herstellung der Verbindungen, die in der Ausführung der vorliegenden Erfindung
nützlich
sind, sind in der
U.S. Patentanmeldungseriennummer
10/236,379 , eingereicht am 1. Oktober 2002, offenbart und können allgemein
wie unten in den Schemata 1A und 1B illustriert hergestellt werden.
Die Ausgangsmaterialien können
durch die in diesen Schemata beschriebenen Verfahren, durch die
Verfahren, die in den unten beschriebenen allgemeinen Verfahren
oder durch die Verfahren, die Fachleuten der organischen Chemie
gut bekannt sind, hergestellt werden. Die Variablen, die in den
Schemata 1A und 1B verwendet werden, sind wie hierin oder wie in
den Ansprüchen
definiert.
-
Die
Herstellung von Alkynylcycloalkanolen ist in dem Schema 1A dargestellt.
Eine Lösung
eines geeigneten Cycloakanons (worin j 0-5 ist) wir in einem Lösungsmittel
wie THF hergestellt. Eine Lösung
einer geeigneten Ethynylmagnesiumhaloid-Verbindung in einem Lösungsmittel
wird zu dem Cycloalkanon zugegeben. Nach der Zugabe der Lösung wird
die Lösung
bei etwa 20°C
für etwa
20 Stunden gerührt.
Die Reaktion wird via TCL überwacht,
bis das Ausgangmaterial aufgebraucht ist. Die Reaktion wird mit Wasser
abgefangen, über einem
Stopfen aus Sand und Silica gefiltert, mit einem Lösungsmittel
wie EtOAc gewaschen und eingedampft, um das Produkt bereitzustellen.
Typischerweise werden zwei Produkte, die Isomere, gebildet, welche
durch die achsiale/äquatoriale
Addition des Alkyns (worin m wie oben definiert ist und die Summe
aus m1 und m2 0 bis etwa 7 ist) zum Keton gebildet werden. Die Verbindungen
werden via Flash-Chromatographie unter Verwendung von EtOAc/Hexanen
gereinigt, um das Produkt bereitzustellen.
-
In Übereinstimung
mit einer Ausführungsform
der vorliegenden Erfindung wir eine Zusammensetzung, die einen Agonisten
des A
2A-AR umfasst, an einem Patienten verabreicht,
um einen septischen Schock und ein systemisches inflammatorisches
Response-Syndrom zu behandeln. Wie hierin verwendet schließt der Begriff „Behandlung" die Prophylaxe der
spezifischen Funktionsstörung
oder des spezifischen Zustands oder die Linderung der Symptome,
die mit einer spezifischen Funktionsstörung oder einem spezifischen
Zustand verbunden sind, und/oder die Vorbeugung oder Beseitigung
der besagten Symptome ein. In einer Ausführungsform wird ein Verfahren
für die
Behandlung des septischen Schocks oder des systemischen inflammatorischen Response-Syndroms
bereitgestellt, worin ein Agonist des A
2A-ARs
an einen Patienten verabreicht wird, um die Entzündung zu verringern und die Überlebensaussichten
eines Patienten, das an einem septischen Schock oder einem systemischen
inflammatorischen Response-Syndrom
leidet, zu verbessern. In einer Ausführungsform wird der A
2A-AR-Agonist
aus der Gruppe ausgewählt,
bestehend aus ATL146e, AB-1, AB-3 und JR-3213. Schema
1A Allgemeiner
Syntheseweg für
Alkynvorläufer
-
Die
Herstellung der 2-Alkynyladenosine ist in dem Schema 1B dargestellt.
Es wird ein flammengetrocknetes Rundbodengefäß unter Stickstoff mit 5-(6-Amino-2-iodpurin-9-yl)-3,4-dihydroxy-tetrahydro-furan-2-carbonsäreethylamid
(NECA-2-Iodadenosin) und einem Lösungsmittel
wie DMF gefüllt.
Das geeignete Alkyn wird gefolgt von Acetonitril und TEA zugegeben.
(Die Lösungsmittel
sind entgast.) Das geeignete Alkyl wird in Actonitril zugegeben,
gefolgt von TEA, 5 Mol-% Pd(PPh
3)
4 und CuI. Die Lösung wird für etwa 24 Stunden bei Raumtemperatur
gerührt
und bis zur Vollständigkeit
mittels HPLC überwacht.
Wenn die Reaktion nach dieser Zeit nicht vollständig ist, werden zusätzlicher
Katalysator, CuI, und TEA zugegeben. Nachdem die Reaktion vollständig ist,
werden die Lösungsmittel
unter Hochvakuum entfernt und der Rest in einer kleinen Menge DMF
aufgenommen. Dieses Produkt wird unter Verwendung einer präparativen
Silica-TLC isoliert. Das Produkt wird mittels RP-HPLC gereinigt. Schema
1B Allgemeines
Kopplungsschema für
die Synthese von 2-Alkynyl-adenosin
-
Die
folgenden Abkürzungen
wurden hierin verwendet:
- 2-Aas
- 2-Alkynyladenosine;
- 125I-ABA
- N6-(4-Amino-3-125iod-benzyl)adenosin
- APCI
- Chemische Ionisierung
bei Atmosphärendruck
- ATL146e
- 4-{3-[6-Amino-9-(5-ethylcarbamoyl-3,4-dihydroxy
tetrahydro-furan-2-yl)-9H-purin-2-yl]-prop-2-ynyl} cyclohexancarbonsäuremethylester;
- CCPA
- 2-Chlor-N6-cyclopentyladenosin;
- CGS21680
- 2-[4-(2-Carboxyethyl)phenethylamino]-5'-N-ethyl carboxamidoadenosin;
- C1-IB-MECA
- N6-3-Iodo-2-chlorbenzyladenosin-5'-N-methyluronamid;
- CPA
- N6-Cyclopentyladenosin
- DMF
- Dimethylformamid
- DMSO
- Dimethylsulfoxid
- DMSO-d6
- Deuteriertes Dimethylsulfoxid
- Et
- OAc Ethylacetat
- eq
- Äquivalent
- GPCR
- G-Proteingekoppelter
Rezeptor;
- hA2AAR
- Rekombinanter humaner
A2A-Adenosinrezeptor;
- IAD
- O 2-Iodadenosin
- 125I-APE
- 2-[2-(4-Amino-3-[125I]iodphenyl)ethylamino]-adenosin;
- NECA
- 5'-N-Ethylcarboxamidoadenosin;
- IB-MECA
- N6-3-Iodbenzyladenosin-5'-N-methyluronamid;
- 2-Iodadenosin
- 5-(6-Amino-2-iod-purin-9-yl)-3,4-dihydroxytetra
hydro-furan-2-carbonsäureethylamid
- HPLC
- Hochleistungsflüssigkeitschromatographie
- HRMS
- Hochauflösende Massenspektroskopie
- 125I-ZM241385
- 125I-4-(2-[7-Amino-2-[2-furyl][1,2,4]triazol[2,3-α] [1,3,5]triazin-5-yl-amino]ethyl)phenol;
- INECA
- 2-Iod-N-ethylcarboxamidoadenosin
- LC/MS
- Flüssigkeitschromatographie/Massenspektrometrie
- m.p.
- Schmelzpunkt
- MHz
- Megahertz
- MRS
- 1220 N-(9-Chlor-2-furan-2-yl-[1,2,4]triazol[1,5-c]
quinazolin-5-yl)-2-phenylacetamid;
- MS
- Massenspektrometrie
- NECA
- N-Ethylcarboxamidoadenosin
- NMR
- Kernresonanzspektroskopie
- RP-HPLC
- Reversphasen-Hochleistungsflüssigkeits
chromatographie
- TRAF
- Tetrabutylammoniumfluorid
- TBS
- tert-Butyldimethylsilyl
- TBDMSCI
- tert-Butyldimethylsilylchlorid
- TEA
- Triethylamin
- TFA
- Trifluoressigsäure
- THF
- Tetrahydrofuran
- TLC
- Dünnschichtchromatographie
- p-TSOH
- para-Toluolsulfonsäure
- XAC
- 8-(4-((2-aminoethyl)aminocarbonyl-methyloxy)
phenyl)-1-3-dipropylxanthin.
-
Synthese von Verbindungen, die bei der
Ausführung
der Erfindung nützlich
sind.
-
Die
Schmelzpunkte wurden mit einem Thomas Hoover Kapillarschmelzpunktapparat
bestimmt und sind nicht korrigiert. Es wurden die Kernresonanzspektren
für das
Proton (
1H NMR) in einem 300 MHz GE-Spektrometer aufgezeichnet.
Die Werte für
die chemische Verschiebung sind in ppm (Parts per Million) relative
zu Tatramethylsilan ausgedrückt.
Für die
Datenauswertung, s = Singulett, d = Duplett, t = Triplett, q = Quartett
und m = Multiplett. Die Massenspektren wurden in einem Finnigan
LcQ Classic gemessen. Es wurden Daten der hochauflösenden Massenspektroskopie
(HRMS) durch das Nebraska Center for Mass Spectronomy bereitgestellt.
Die analytische HPLC wurde in einem Waters 2690 Separation Module
mit einem Waters Symmetry C8(2,1 × 150 mm)-Säule, das bei Raumtemperatur
bedient wurde, durchgerührt.
Die Verbindungen wurden bei 200 μl/min
mit 70:30 Acetonitril:Wasser, das 0,5 % Essigsäure enthielt, mit einem UV-Nachweis
bei 214 nm unter Verwendung eines Waters 486 Tunable Detectors eluiert.
Die Präparative
HPLC wurde mit einer Shimadzu Discovery HPLC mit einer Shim-pack
VP-ODS C18(20 × 100
mm)-Säule,
die bei Raumtemperatur bedient wurde, durchgeführt. Die Verbindungen wurden
mit 30 ml/min mit einem Gradienten 20-80 % Wasser (das 0,1 % TFA
enthielt) zu Methanol über 15
Minuten mit einem UV-Nachweis bei 214 nm unter Verwendung eines
SPD10A VP Tunable Detectors nachgewiesen. Für alle hier dargestellten Endverbindungen
wurde mittels HPLC bestimmt, dass sie zu mehr als 98 % rein sind.
Die Flash-Chromatographie wurde auf einem Silicyle 60A Gel (230-400 Netz) oder unter
Verwendung von wiederverwendbaren Chromatographiesäulen und
des wiederverwendbaren Systems von RT Scientific, Manchester NH,
durchgeführt.
Die analytische Dünnschichtchromatographie
wurde auf Merck Kieselgel 60 F254 Aluminium-Blechplatten gemacht.
Die präparative
Dünnschichtchromatogrpahie
wurde unter Verwendung einer 1000 Mikron Analtech Uniplate mit Silicagel
gemacht. Alle Reaktionen wurden in einer Stickstoffatmosphäre in flammengetrockneter
Glasware gemacht, wenn nicht anders erwähnt. Allgemeines
Verfahren 1: Herstellung von Alkynylcyclohexanolen

-
Zu
einer etwa 10 mmol Lösung
des geeigneten Cyclohexanons in etwa 50 ml THF werden etwa 60 ml (30
mmol) 0,5 M Ethynylmagnesiumbromid in THF zugegeben. Die Lösung wird
bei 20°C
für etwa
20 Stunden gerührt.
Nachdem das Ausgangsmaterial verbraucht ist, überwacht durch TLC, wird die
Reaktion mit etwa 5 ml Wasser abgefangen, über einem Stopfen aus Sand
und Silica gefiltert, mit EtOAc gewaschen und eingedampft, damit
sich ein gelbes Öl
ergibt. Gewöhnlich
enthielt das Öl
mit 20 % EtOAc/Hexanen zwei Flecken auf der TLC, welche mit Vanillin
sichtbar gemacht wurden. Gewöhnlich
sind diese zwei Produkte die unterschiedlichen Isomere, welche durch
die achsiale/äquatoriale
Addition des Alkyns zum Keton gebildet werden. Die Verbindungen
werden via Flash-Chromatographie unter Verwendung von 10 % EtOAc/Hexanen
gereinigt, um klare Öle
oder weiße
Feststoffe mit einer Ausbeute von 50-80 % bereitzustellen. Allgemeines
Verfahren 2: Herstellung von Propargylpiperadinen/Piperazinen
-
Zu
einer Lösung
des geeigneten Piperazins/Piperadins (etwa 10,0 mmol) in etwa 20
ml Acetonitril werden etwa 12,0 mmol Propargylbromid (80 % stabilisiert
in Toluol) und etwa 50,0 mmol wasserfreies Kaliumcarbonat zugegeben.
Die Reaktionsmischung wird gefiltert und bis sie trocken ist eingedampft.
Der Rest wird in 50 ml Dichlormethan/Wasser aufgenommen und die
organischen Schichten entfernt. Die wässerige Schicht wird mit zusätzlichen
3 × 25
ml Dichlormethan gewaschen. Die organische Schicht wird unter Verwendung
von wasserfreiem Natriumsulfat getrocknet, gefiltert und konzentriert,
um das Rohprodukt bereitzustellen, welches unter Verwendung einer
Säulenchromatographie
gereinigt wird. Allgemeines
Verfahren 3: Herstellung von modifizierten Piperadinen/Piperazinen
-
Zu
etwa 100 mg des geeigneten Boc-geschützten Piperazins/Piperadins
werden 2-4 ml reine TFA zugegeben. Die Lösung wird für 6 Stunden gerührt. Die
TFA wird unter reduziertem Druck entfernt, damit sich ein gelbes Öl ergibt.
Das Öl
wird in etwa 10 ml Dichlormethan aufgenommen, zu welchem ein 10-facher Überschuß TEA und
3 Äquivalente
des geeigneten Acylchlorids zugegeben werden. Die gelbe Lösung wird
bei Raumtemperatur für
etwa 12 Stunden gerührt,
nach dieser Zeit werden die Lösungsmittel
entfernt und das Produkt unter Verwendung einer 1,1 × 30 cm
14 g Säule
von Robert Thompson Scientific mit einem 5 %-30 % Gradienten aus
Acetat/Hexanen gereinigt. Allgemeines
Verfahren 4: Herstellung von 2-AAs (2-Alkynyladenosin)
-
Es
wird ein flammengetrocknetes 25 ml Rundbodengefäß unter Stickstoff mit 5-(6-Amino-2-iod-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-carbonsäureethylamid
(2-Iodadenosin) (etwa 40 mg) (X = CH3CH2NHC(O)-) gefüllt und in etwa 2 ml DMF gelöst. Das
geeignete Alkyn (etwa 0,1 ml) wird dann zugegeben, gefolgt von etwa
4 ml Acetonitril und etwa 0,1 ml TEA. Alle drei Lösungsmittel
sind für
mindestens 24 Stunden mit Stickstoff entgast worden. Zu dieser Lösung werden
5 Mol-% Pd(PPh3)4 und
6 Mol-% Kupferiodid zugegeben. Die gelbliche Lösung wird für etwa 24 Stunden bei Raumtemperatur
gerührt
oder bis zur Vollständigkeit
mittels HPLC. Wenn die Reaktion nach dieser Zeit nicht vollständig ist,
werden zusätzlicher
Katalysator, CuI, und TEA zugegeben. Nachdem die Reaktion vollständig ist,
werden die Lösungsmittel
unter Hochvakuum entfernt und der rotschwarze Rest in einer kleinen
Menge DMF aufgenommen. Diese Lösung
wird auf eine präparative
Silica-TLC-Platte (Analtech 1000 Mikron, 20 cm × 20 cm) zugegeben und zuerst
mit 120 ml 40 % Hexanen/CH2Cl2 und
dann nochmals nach der Zugabe von 40 ml MeOH eluiert. Die UV-aktive
Bande (gewöhnlich gelb
in der Farbe) in der Mittel der Platte wird eingesammelt, langsam
mit 4 × 25
ml 20 % MeOH/CH2Cl2 gewaschen
und konzentriert. Dieses Produkt wird mittels RP-HPLC gereinigt.
-
Herstellung
1: [(2R,3R,4R,5R)-3,4-Diacetyloxy-5-(2-amino-6-oxohyropurin-9-yl)oxolan-2-yl]methylacetat
(6.2).
-
Es
wurde eine Suspension aus 113 g (0,4 mol) trockenem Guanosin (6.1),
Essigsäureanhydrid
(240 ml, 2,5 mol), trockenem Pyridin (120 ml) und trockenem DMF
(320 ml) für
3,75 Stunden auf 75°C
erhitzt, ohne das die Temperatur 80°C übersteigt. Die klare Lösung wurde
dann in einen 31 Erlenmeyerkolben transferiert und mit 2-Propanol
gefüllt.
Nach der Abkühlung
der Lösung
auf Raumtemperatur wurde die Kristallisierung initiiert und bei
4°C über Nacht
fortgesetzt. Das weiße
Feststofffiltrat wurde gefiltert, mit 2-Propanol gewaschen und aus
2-Propanol rekristallisiert, um 6.2 (96 %) bereitzustellen: 1H NMR (300 Mhz, CDCl3)
8,20 (s, 1H, H-8), 6,17 (d, J = 5,41 Hz, 1H, H-1) 5,75 (t, J = 5,39
Hz, 1H, H-2), 5,56 (t, J = 5,0, H-3), 4,41 (m, 3 H, H-4,5), 2,14 (s,
3 H, Ac), 2,11 (s, 3 H, Ac), 2,10 (s, 3 H, Ac). 13C
NMR (300 Mhz, CD3OD) 171,0, 170,3, 1702,
157,7, 154,8, 152,4 136,7, 117,7, 85,5, 80,4, 73,0, 71,3, 64,0,
31,3, 21,2, 21,0.
-
Herstellung
2: [(2R,3R,4R,5R)-3,4-diacetyloxy-5-(2-amino-6-chlorpurin-9-yl)oxolan-2-yl]methylacetat
(6.3).
-
Es
wurden in eine 11 Flasche 80 g (0,195 mol) [(2R,3R,4R,5R)-3,4-Diacetyloxy-5-(2-amino-6-oxohyropurin-9-yl)oxolan-2-yl]methylacetat
(6.2), Tetramethylammoniumchlorid (44 g, 0,4 mol), wasserfreies
Acetonitril (400 ml) und N,N-Dimethylanilin (25 ml) gegeben. Die
Flasche wurde in ein Eis-Salz-Bad
gegeben und auf 2°C
gekühlt.
Zu dieser Lösung
wurde tropfenweise POCl3 (107 ml 1,15 mol)
mit einer Geschwindigkeit zugegeben, dass die Temperatur unter 5°C gehalten
wurde (45 Minuten). Die Flasche wurde dann aus dem Eisbad entfernt,
mit einem Kondensator ausgestattet, in ein Ölbad gegeben und für 10 Minuten
refluxiert. Die Lösung änderte sich
zu einer rot/brauen Farbe. Das Lösungsmittel
wurde unter reduziertem Druck entfernt, damit sich ein öliger Rest
ergibt, welcher in ein Becherglas transferiert wurde, das 1000 g
Eis und 400 ml CHCl3 enthielt, und für 1,5 Stunden
gerührt,
um jegliches verbliebene POCl3 zu zersetzen.
Die organische Phase wurde entfernt und die wässerige Phase mit 3 × 50 ml
CHCl3 extrahiert und mit der organischen
Phase vereinigt. Die vereinigten organischen Schichten wurden mit
50 ml Wasser zurückextrahiert,
gefolgt vom Rühren
mit 200 ml gesättigtem
NaHCO3. Die organischen Schichten wurden
ferner mit NaHCO3 extrahiert, bis der wässerige
Extrakt neutral war (2X). Die organische Schicht wurde schließlich mit
Lauge extrahiert und über
MgSO4 für
16 Stunden getrocknet. Zu der Lösung
wurden 800 ml 2-Propanol zugegeben, wonach die Lösung unter reduziertem Druck
konzentriert wurde. Zu dem öligen
Feststoff wurden 200 ml 2-Propanol zugegeben und die Lösung über Nacht
gekühlt.
Das kristalline Produkt wurde gefiltert, gewaschen und über Nacht
getrocknet, um 6.3 (77 %) zu ergeben. 1H
NMR (300 MHz, CD3OD) 8,31 (s, 1H, H-8),
7,00 (s, 2H, NH2) 6,06 (d, J = 5,8 Hz, 1H,
H-1), 5,83 (t, J = 6,16 Hz, 1H, H-2), 5,67 (m, 1H, H-3), 4,29 (m, 3 H,
H-4,5), 2,07 (s, 3 H, Ac), 1,99 (s, 3 H, Ac), 1,98 (s, 3 H, Ac). 13C NMR (300 MHz, CD3OD)
171,0, 170,4, 170,2, 160,8, 154,6, 150,8, 142,2, 124,5, 85,8, 80,6, 72,8,
71,2, 63,9, 21,4, 21,3, 21,1.
-
Herstellung
3: [(2R,3R,4R,5R)-3,4-Diacetyloxy-5-(6-chlor-2-iodpurin-9-yl)oxolan-2-yl]methylacetat
(6.4).
-
Es
wurde Isoamylnitrit (5 ml, 37 mmol) zu einer Mischung aus 5,12 g
(12 mmol) [(2R,3R,4R,5R)-3,4-diacetyloxy-5-(2-amino-6-chlorpurin-9-yl)oxolan-2-yl]methylacetat
(6.3), I2 (3,04 g, 12 mmol), CH2I2 (10 ml, 124 mmol) and CuI (2,4 g, 12,6
mmol) in THF (60 ml) zugegeben. Die Mischung wurde unter Reflux
für 45
Minuten erhitzt und dann auf Raumtemperatur abgekühlt. Zu
dieser Lösung
wurden 100 ml gesättigtes
Na2S2O3 zugegeben.
Dieser Schritt entfernte die rötliche
Farbe. Die wässerige
Schicht wurde 3X mit Chloroform extrahiert, vereinigt und über MgSO4 getrocknet und unter reduziertem Druck
konzentriert. Das Produkt wurde dann über einer Silicagelsäule unter
Verwendung von CHCl3-MeOH (98:2) gereinigt,
um [(2R,3R,4R,5R)-3,4-Diacetyloxy-5-(6-chlor-2-iodpurin-9-yl)oxolan-2-yl]methylacetat (6.4)
(80 % aus EtOH kristallisiert) einzusammeln. 1H NMR
(300 MHz, CDCl3) 8,20 (s, 1H H-8), 6,17
(d, J = 5,41 Hz, 1H, H-1), 5,75 (t, J = 5,39 Hz, 1H, H-2), 5,56
(t, J = 5,40 Hz, 1H, H-3), 4,38 (m, 3 H, H-4,5), 2,14 (s, 1H, Ac),
2,11 (s, 1H, Ac), 2.10 (s, 1H, Ac).
-
Herstellung
4: (4S,2R,3R,5R)-2-(6-Amino-2-iodpurin-9-yl)-5-(hydroxymethyl)oxolan-3,4-diol (6.5).
-
In
eine Flasche, die 6,0 g (11,1 mol) [(2R,3R,4R,5R)-3,4-Diacetyloxy-5-(6-chlor-2-iodpurin-9-yl)oxolan-2-yl]methylacetat
(6.4) enthielt, wurden 100 ml flüssiges
NH3 mit -78°C zugegeben und die Lösung für 6 Stunden
gerührt.
Nach dieser Zeit wurde sie über
Nacht auf Raumtemperatur gebracht mit der gleichzeitigen Verdampfung
des NH3, damit sich ein braunes Öl ergibt.
Das Produkt wurde aus heißem
Isopropanol kristallisiert, um 6.5 (80 %) bereitzustellen, m.p.
143-145°C,
r.f. = 0,6 in 20 % MeOH/CHCl3. 1H
NMR (300 MHz, DMSO-d6) 8,24 (s, 1H), 7,68
(s, 2H), 5,75 (d, J = 6,16, 1H), 5,42 (d, J = 5,40 Hz, 1H), 5,16
(d, J = 4,62 Hz, 1H), 4,99 (t, J = 5,39 Hz, 1H), 4,67 (d, J = 4,81
Hz, 1H), 4,06 (d, J = 3,37 Hz, 1H), 3,89 (m, 1H), 3,54 (m, 2H).
-
Herstellung
5: [(1R,2R,4R,5R)-4-(6-Amino-2-iodpurin-9-yl)-7-7-dimethyl-3,6,8-trioxabicyclo[3.3.0]oct-2-yl]methan-1-ol
(6.6).
-
Zu
einer Lösung
aus 2,0 g (5,8 mmol) (4S,2R,3R,5R)-2-(6-Amino-2-iodpurin-9-yl)-5-(hydroxymethyl)oxolan-3,4-diol
(6.5) in 100 ml Aceton wurden 9,6 g p-Toluolsulfonsäure und
5 ml Dimethoxypropan zugegeben. Die Reaktion wurde bei Raumtemperatur
für 1 Stunde
gerührt.
Es wurden 15 g festes NaHCO3 zu der Lösung zugegeben.
Der Schlamm wurde für
weitere 3 Stunden gerührt.
Der Rest wurde gefiltert und 2X mit EtOAc gewaschen. Das Filtrat
wurde dann unter reduziertem Druck konzentriert. Der Rest wurde
auf einer Silicagelsäule
mit MeOH-CHCl3 (1:99) chromatographiert,
um 6.6 (72 %) als einen Feststoff zu ergeben, m.p. 185-187°C. 1H NMR (300 MHz, DMSO-d6)
8,22 (s, 1H, H-8), 7,69 (s, 2H), NH2), 6,00
(d, J = 2,70 Hz, 1H, H-1), 5,21 (m, 1H, H-2), 5,07 (bs, 1H, OH),
4,88 (m, 1H, H-3), 4,13 (m, 1 H, H-4), 3,47 (m, 2H, H-5), 1,49 und
1,28 (s, 3 H, C(CH3)2).
-
Herstellung
6: (2S,1R,4R,5R)-4-(6-Amino-2-iodpurin-9-yl)-7,7-dimethyl-3,6,8-trioxabicyclo[3.3.0]octan-2-carbonsäure (6.7).
-
Zu
einer gerührten
Lösung
aus 1,6 g (3,7 mmol) [(1R,2R,4R,5R)-4-(6-Amino-2-iodpurin-9-yl)-7,7-dimethyl-3,6,8-trioxabicyclo[3.3.0]oct-2-yl]methan-1-ol (6.6)
in 200 ml H2O wurden 0,60 g KOH und tropfenweise eine
Lösung
aus 1,70 g (10,8 mmol) KMnO4 in 50 ml H2O zugegeben. Die Mischung wurde für 2-4 Tage
bei Raumtemperatur ins Dunkel gegeben. Die Reaktionsmischung wurde
dann auf 5-10°C
gekühlt
und durch eine Lösung
aus 4 ml 30 % H2O2 ind
16 ml Wasser entfärbt,
während
die Temperatur auf unter 10°C
unter Verwendung eines Eis-Salz-Bades gehalten wurde. Die Mischung
wurde durch Celite gefiltert und das Filtrate unter reduziertem
Druck auf etwa 10 ml konzentriert und dann auf pH 4 mit 2 N HCl
angesäuert.
Das resultierende Präzipitat
wurde abgefiltert und mit Ether gewaschen, um nach dem Trocknen
als einen weißen
Feststoff 6.7 (70 %) zu ergeben, m.p. 187-190°C. 1H
NMR (300 MHz, DMSO-d6) 8,11 (s, 1H, H-8),
7,62 (s, 2H, NH2), 7,46 (s, 1H, COOH), 6,22
(s, 1H, H-1), 5,42 (d, J = 5,71 Hz, 1H, H-2), 5,34 (d, J = 6,16 Hz, 1H, H-3),
4,63 (s, 1H, H-4), 1,46 und 1,30 (s, 3 H, C(CH3)2).
-
Herstellung
7: (2S,3S,4R,5R)-5-(6-Amino-2-iodpurin-9-yl)-3,4-dihydroxyoxolan-2-carbonsäure (6.8).
-
Es
wurde eine Lösung
aus 1,72 g (3,85 mmol) (2S,1R,4R,5R)-4-(6-Amino-2-iodpurin-9-yl)-7,7-dimethyl-3,6,8-trioxabicyclo[3.3.0]octan-2-carbonsäure (6.7)
in 80 ml 50 % HCOOH bei 80°C
für 1,5
Stunden gerührt.
Die Reaktionsmischung wurde unter reduziertem Druck eingedampft,
in H2O gelöst und das Lösungsmittel
wieder verdampft. Dieses Verfahren wurde wiederholt, bis der Rest
keinen Geruch nach Ameisensäure mehr
hatte. Die Rekristallisierung aus Wasser lieferte 1,33 g (85 %)
6,8 als einen weißen
Feststoff, m.p. 221-223°C,
abbau. 1H NMR (300 MHz, DMSO-d6)
8,31 (s, 1H, H-8), 7,68 (s, 2H, NH2), 5,90
(d, J = 6,55 Hz, 1H, H-1), 4,42 (m, 1H, H-2), 4,35 (d, J = 2,31 Hz, 1H, H-4),
4,22 (m, 1H, H-3).
-
Herstellung
8: [(2S,3S,4R,5R)-5-(6-Amino-2-iodpurin-9-yl)-3,4-dihydroxyoxolan-2-yl]-N-ethylcarboxamid
(6.9).
-
Zu
einer gekühlten
(5°C) und
gerührten
Lösung
aus 1,29 g (3,17 mmol) (2S,3S,4R,5R)-5-(6-Amino-2-iodpurin-9-yl)-3,4-dihydroxyoxolan-2-carbonsäure (6.8)
in 150 ml absolutem Ethanol wurden tropfenweise 1,15 ml eisgekühltes SOCl2 zugegeben. Die Mischung wurde bei Raumtemperatur über Nacht
gerührt
und dann mit gesättigter
wässeriger
NaHCO3 auf pH 8 gebracht. Die Mischung wurde
gefiltert und das Filtrat dann unter reduziertem Druck konzentriert,
um einen weißen
Feststoff zu ergeben, welcher getrocknet wurde und dann in 20 ml
trockenem Ethylamin bei -20°C
für 3 Stunden
und dann bei Raumtemperatur über
Nacht wieder gelöst
wurde. Die Reaktionsmischung wurde mit absolutam Ethanol verdünnt und
das präzipitierte
Produkt abgefilter und mit trockenem Ether gewaschen, um 530 mg
(72 %) 6.9 als einen reinen Feststoff bereitzustellen, m.p. 232-234°C. 1H NMR (300 MHz, DMSO-d6)
8,34 (s, 1H, H-8), 8,12 (t, 1H, NH), 7,73 (s, 2H, NH2),
5,85, (d, J = 6,93 Hz, 1H, H-1), 4,54 (m, 1H, H-2), 4,25 (d, J =
1,92 Hz, 1H, H-4), 4,13 (m, 1H, H-3), 3,28 (m, 2H, CH2CH3), 1,00 (t, J = 7,2 Hz, 3 H, CH2CH3).
-
Herstellung
9: [4-(tert-Butyl-dimethyl-silanyloxymethyl)-cyclohexyl]-methanol
(83).
-
Zu
einer 100 ml Flasche, die 79 (4,0 g, 27,8 mmol) in DMF (40 ml) enthält) wurden
TBDMSCI (3,56 g, 23,6 mmol) und Imidazol (3,79 g, 55,6 mmol) zugegeben.
Die Reaktion wurde bei 25°C
für 16
Stunden gerührt, nach
welcher Zeit gesättigtes
wässeriges
LiBr (50 ml) zugegeben wurde und die Reaktion mit Ether (2 × 50 ml) extrahiert
wurde. Die Etherschichten wurden vereinigt und wieder mit LiBr (2 × 35 ml)
extrahiert. Die Etherschicht wurdn klar. Die Etherschicht wurde
dann in vacou konzentriert und das Produkt mittel Flash-Chromatographie
auf einer Silicagelsäule
gereinigt, wobei mit 1:2 Ether/Petroleumether eluiert wurde, um
83 (3,80 g, 62 %) als ein homogenes Öl zu erhalten. 1H
NMR (CDCl3) δ 3,46 (d, J = 6,2 Hz, 2H), 3,39
(d, J = 6,2 Hz, 2H), 1,95-1,72 (m, 4H), 1,65 (m, 1H), 1,40 (m, 1H),
1,03-0,89 (m, 4H), 0,88 (s, 9H), 0,04 (s, 6H); 13C
NMR (CDCl3) δ 69,2, 69,1, 41,2, 41,1, 29,5,
26,5, 18,9,-4,8;. APCI m/z (rel Intensität) 259 (MH+,
100).
-
Herstellung
10: Toluol-4-sulfonsäure
4-(tert-butyl-dimethylsilanyloxymethyl)-cyclohexylmethylester (84).
-
Zu
einer 100 ml Flasche, die 83 (3,4 g, 13,2 mmol) in CHCl3 (30
ml) enthielt, wurden Tosylchlorid (3,26 g, 17,1 mmol) und Pyidin
(3,2 ml, 39,6 mmol) zugegeben. Die Reaktion wurde bei 25°C für 14 Stunden
gerührt, nach
welcher Zeit die Reaktion in vacou konzentriert wurde, um einen
nassen weißen
Feststoff zu ergeben. Zu diesem Feststoff wurde Ether (50 ml) zugegeben
und der Feststoff gefiltert und anschließend mit zusätzlichem
Ether (2 × 50
ml) gewaschen. Die Etherschichten wurden vereinigt, in vacou konzentriert,
um ein klaren Öl
zu ergeben, welches mittels Flash-Chromatographie auf einer Silicagelsäure gereinigt
wurde, wobei mit 1:4 Ether/Petroleumether eluiert wurde, um 84 (4,5
g, 83 %) als einen weißen
Feststoff zu ergeben. 1H NMR (CDCl3) δ 7,78
(d, J = 7,7, 2H), 7,33 (d, J = 7,7 Hz, 2H), 3,81 (d, J = 6,2 Hz,
2H), 3,37 (d, J = 6,2, 2H), 2,44 (s, 3H), 1,95-1,72 (m, 4H), 1,65
(m, 1H), 1,40 (m, 1H), 1,03-0,89 (m, 4H), 0.88 (s, 9H), 0,04 (s,
6H); 13C NMR (CDCl3) δ 145,1, 133,7,
130,3, 128,4, 75,8, 68,9, 40,7, 38,0, 29,1, 26,5, 22,1, 18,9-4,9;
APCI m/z (rel Intensität) 413
(MH+, 100).
-
Herstellung
11: (4-Prop-2-ynyl-cyclohexyl)-methanol (86).
-
Eine
dreihalsige 250 ml Flasche, ausgestattet mit einem Gaseinlassrohr
und einem Trockeneiskondensator, wurde auf -78°C gekühlt und mit flüssigem Ammoniak
(40 ml) gefüllt.
Zu der Reaktionsmischung wurde Lithiumdraht (600 mg, 86,4 mmol)
zugegeben, was eine tief blaue Lösung
bildete. Die Lösung
wurde für
1 Stunde gerührt.
Es wurde Acetylen, das durch ein Aktivkohletrocknungsrohr geführt wurde,
zu dem Ammoniak zugegeben, bis alles Lithium reagiert hatte und
die Lösung
farblos wurde, zu welchem Zeitpunkt der Acetylenfluss angehalten
wurde, das Acetylen-Einlassrohr
und der Kondensator entfernt wurden und die Flasche mit einem Thermometer
ausgestattet wurde. Es wurde DMSO (20 ml) zugegeben und das Ammoniak
mit einem Warmwasserbad verdampft, bis die Mischung eine Temperatur
von 30°C
erreichte. Die Lösung
wurde bei dieser Temperatur für
2 Stunden gerührt,
bis die Lösung
aufhörte
Blasen zu bilden. Die Mischung wurde auf 5°C gekühlt und die Verbindung 84 (11,25
g, 27,3 mmol) in DMSO zugegeben. Die Temperatur wurde bei 5°C gehalten.
Die Mischung wurde bei 5°C
für 0,5
Stunden gerührt.
Dann wurde die Lösung
graduell auf Raumtemperatur aufgewärmt und für weitere 18 Stunden gerührt. Die
braunschwarze Reaktionsmischung wurde langsam über Eis (300 g) gegossen und
mit Ether (4 × 100
ml) extrahiert, mit wasserfreiem Natriumsulfat getrocknet und in
vacou konzentriert, um ein gelbes Öl zu ergeben. Das Öl wurde
anschließend
in THF (200 ml) gelöst und änderte sich
nach der Zugabe von TRAF-Hydrat (11,20 g, 35,5 mmol) zu einer bräunlichen
Farbe. Die Lösung
wurde für
24 Stunden unter einer N2-Atmosphäre gerührt. Nach
dem Rühren
wurde die Reaktion mit Wasser (200 ml) abgefangen und mit Ether
(3 × 100
ml) extrahiert. Die Etherextrakte wurden kombiniert und in vacuo
konzentriert. Das Rohprodukt wurde mittels Chromatographie auf einer
Silicagelsäure
gereinigt, wobei mit 1:1 Ether/Petroleumether eluiert wurde, um
86 (3,91 g, 93 %) als ein gelbes Öl zu erhalten. 1H
NMR (CDCl3) δ 3,45 (d, J = 6,2, 2H), 2,10
(d, J = 6,2, 2H), 1,9 (s, 1H), 1,94-1,69 (m, 4H), 1,52-1,34 (m,
2H), 1,16-0,83 (m, 4H); 13C NMR (CDCl3) δ 83,8,
69,5, 69,0, 40,8, 37,7, 32,3, 29,7, 26,5.
-
Herstellung
12: (4-Prop-2-ynylcyclohexyl)methylacetat (87).
-
Zu
einer Lösung
aus 960 mg (6,31 mmol) 86 in 6 ml DMF wurden 0,62 ml (7,57 mmol)
Pyridin und 0,78 ml (8,27 mmol) Essigsäureanhydrid zugegeben. Die
Reaktion wurde über
Nacht bei Raumtemperatur gerührt. Nach
16 Stunden war immer noch Ausgangmaterial verblieben. Die Reaktionsmischung
wurde auf 75°C
für 3 Stunden
erhitzt. Das Lösungsmittel
wurde unter reduzirtem Druck entfernt, damit sich ein gelbes Öl ergibt,
das auf Silcagel mittels Flash-Chromatographie gereinigt wurde,
wobei mit 1:3 Ether/Petroleumether eluiert wurde, um 1,12 g (91
%) 87 als ein Öl
zu erhalten. 1H NMR (CDCl3) δ 3, 87 (d,
J = 6,2 Hz, 2H), 2,06 (d, J = 4,3 Hz, 2H), 2,03 (s, 3H), 1,98-1,93
(m, 1H), 1,92-1,83 (m, 2H), 1,83-1,74 (m, 2H), 1,63-1,36 (m, 2H),
1,12-0,90 (m, 4H); 13C NMR (CDCl3) δ 171,7,
83,7, 69,9, 69,6, 37,4, 37,3, 32,1, 29,7, 26,5, 21,4; APCI m/z (rel
Intensität)
195 (M+, 30), 153 (M+,
70), 135 (M+, 100).
-
Herstellung
13: 4-Prop-2-ynyl-cyclohexancarbonsäure (88).
-
Es
wurde eine Lösung
aus Chromtrioxid (600 mg, 6,0 mmol) in 1,5 M H2SO4 (2,6 ml, 150 mmol) auf 5°C gekühlt und
zu einer Lösung
aus 86 (280 mg, 1,84 mmol) in Aceton (15 ml) zugegeben. Der Mischung wurde
ermöglicht
sich auf Raumtemperatur zu erwärmen
und sie wurde über
Nacht gerührt.
Es wurde Isopropanol (4 ml) zu der grün/schwarzen Lösung zugegeben,
die nach 1 Stunde zu hellblau umgekippt war. Nach der Zugabe von
Wasser (15 ml) wurde die Lösung
mit CHCl3 (6 × 25 ml) extrahiert. Die organischen
Schichten wurde vereinigt und in vacou konzentriert, um einen weißen Feststoff
zu ergeben. Der Feststoff wurde in Ether (50 ml) gelöst und mit
1 M NaOH (2 × 30
ml) extrahiert. Die basischen Extrakte wurden vereinigt, mit w/10
% HCl angesäuert
und mit Ether (3 × 30
ml) reextrahiert. Die Etherschichten wurden kombiniert, mit Natriumsulfat getrocknet
in vacuo konzentriert, um einen weißen Feststoff zu ergeben. Das
Produkt wurde aus Aceton/Wasser rekristallisiert, um 88 (222 mg,
73 %) als weiße
Nadeln zu erhalten. mp 84-85°C; 1H NMR (CDCl3) δ 2,30-2,23
(m,1H), 2,17-2,11 (m, 2 H), 2,07-2,03 (m, 2H), 1,97-1,91 (m, 3H),
1,51-1,39 (m, 3H), 1,13-1,01 (m, 2H); 13C
NMR (CDCl3) δ 182,5, 83,8, 69,6, 40,7, 37,7,
32,3, 29,6, 26,5; APCI m/z (rel Intensität) 165 (M, 100).
-
Herstellung
14: Methyl 4-prop-2-ynylcyclohexancarboxylat (89).
-
Zu
einer Lösung
aus 88 (240 mg, 1,45 mmol) in 7:3 CH2Cl2:MeOH (10 ml) wurde TMS-Diazomethan (2,0
M in Hexanen) (0,9 ml, 1,9 mmol) in 0,2 ml Aliquots zugegeben, bis
die Farbe gelb blieb. Die Reaktion wurde für weitere 0,25 Stunden bei
Raumtemperatur gerührt.
Nach dem Rühren
wurde tropfenweise Eisessig zugegeben, bis die Lösung farblos wurde. Die Reaktion
wurde in vacou zu einem Öl
konzentriert, das mittels Flash-Chromatographie auf einem Silicagel
unter Verwendung von Ether:Petroleumether (1:9) gereinigt wurde,
um 89 (210 mg, 80 %) als ein klares Öl zu erhalten. 1H
NMR (CDCl3) δ 3,60 (s, 3H), 2,25-2,13 (m,
1H), 2,08-1,94 (m, 3H), 1,95-1,90 (m, 2H), 1,49-1,31 (m, 3H), 1,10-0,93
(m, 2H); 13C NMR (CDCl3) δ 176,7, 83,3, 69,8,
51,9, 43,4, 36,7, 31,9, 29,2, 26,3; APCI m/z (rel Intensität) 181 (MH+, 100).
-
Herstellung
15: Trans [4-(1-Propargyl)cyclohexylmethyl]methylcarbonat (90).
-
- Ausbeute: 345 mg, 81 %, 1H NMR (CDCl3) δ 0,98-1,07,
1,40-1,52, 1,57-1,70, 1,78-1,93 (4 × m, 10 H, Cyclohexyl), 1,96
(t, 1H, Acetylen), 2,10 (dd, 2H, -C6H10CH2CCH), 3,78 (s,
3 H, -OCH3), 3,96 (d, -C6H10CH2O-).
-
Herstellung
16: Trans [4-(1-Propargyl)cyclohexylmethyl]-isobutylcarbonat (91).
-
- Ausbeute: 433 mg, 83 %, 1H NMR (CDCl3) δ 0,95
(d, 4 H, -OCH2CH(CH3)2), 0,98-1,09, 1,40-1,51, 1,57-1,70, 1,78-1,93
(4 × m,
10 H, Cyclohexyl), 1,94-2,04
(m, 1H, -OCH2CH(CH3)2), 1,96 (t, 1H, Acetylen), 2,10 (dd, 2H, -C6H10CH2CCH),
3,91, 3,95 (2 × d,
4 H, -OCH2CH(CH3)2, -C6H10CH2O-).
-
Herstellung
17: Trans [4-(1-Propargyl)cyclohexylmethyl]benzylcarbonat(92)
-
- Ausbeute: 340 mg, 69 %. 1H NMR (CDCl3) δ 0,97-1,08,
1,40-1,49, 1,55-1,69, 1,77-1,93 (4 × m, 10 H, Cyclohexyl), 1,96
(t, 1H, Acetylen), 2,10 (dd, 2H, -C6H10CH2CCH), 3,98 (d,
-C6H10CH2O-), 5,15 (s, 2H, -OCH2Ph), 7,33-7,40
(m, 5 H, Ar).
-
Herstellung
18: 4-(Toluol-4-sulfonyloxymethyl)-piperidin-1-carbonsäure-tert-butylester (JR3215).
-
Es
wurde eine Lösung
aus N-Boc-4-piperidinmethanol, 5,0 g (23,2 mmol) in 50 ml Chloroform
hergestellt. Es wurde Toluolsulfonylchlorid, 5,75 g (30,2 mmol),
in 5,6 ml Pyridin (69,6 mmol) zugegeben. Die Lösung wurde unter Stickstoff
für 24
Stunden gerührt.
Die Standardverarbeitung und die chromtographische Aufreinigung
lieferte die Titelverbindung. Ausbeute 6,0 g
-
Herstellung
19: (R)-1-Ethynyl-(R)-3-methyl-cyclohexanol (JR3217A), (S)-1-Ethynyl-(R)-3-methyl-cyclohexanol (JR3217B).
-
Zu
einer Lösung
aus 1,0 g (8,9 mmol) (R)-(+)-3-Methyl-cyclohexanon in 50 ml THF
wurden 54 ml (26,7 mmol) 0,5 M Ethynylmagnesiumbromid in THF zugegeben.
Die Lösung
wurde bei 20°C
für 20
Stunden gerührt. Die
Analyse mittels TLC zeigte an, dass das Ausgangsmaterial verbraucht
worden ist. Die Reaktion wurde mit 5 ml Wasser abgefangen, über einem
Stopfen aus Sand und Silica gefiltert, mit EtOAc gewaschen und eingedampft,
damit sich 1,15 g eines gelben Öls
ergiben, das zwei Flecken (r.f.'s
0,33 (kleiner, JR3217A) und 0,25 (größer, JR3217B), 20 % EtOAc/Hexane)
enthält,
die mit Vanillin sichtbar gemacht wurden. Die Verbindung wurde via
Flash-Chromatographie
unter Verwendung von 10 % EtOAc/Hexanen (225 ml Silica) gereinigt,
um JR3217A und JR3217B zu liefern.
-
Herstellung
20: 1-Prop-2-ynyl-piperidin-2-carbonsäuremethylester(JR3249).
-
Die
Titelverbindung wurde ausgehend von 4,0 g (22,3 mmol) Methylpipecolinathydrochlorid
entsprechend dem allgemeinen Verfahren 2 hergestellt.
-
Herstellung
21: 1-Prop-2-ynyl-piperidin-4-carbonsäuremethylester (JR3245).
-
Zu
einer Lösung
aus Methylisonipecotat 3,5 g (24,4 mmol, 3,30 ml) in 100 ml Dichlormethan
wurden TEA (1,5 eq, 36,6 mmol, 5,1 ml), Propargylbromid (3,0 eq,
73,2 mmol, 6,5 ml) zugegeben, bei Raumtemperatur für 36 Stunden.
Die Reaktion wurde mit 35 ml Wasser abgefangen, um eine klare Lösung bereitzustellen.
Die Lösung
wurde mit Dichlormethyn 2 × 25
ml extrahiert, mit Na2SO4 getrocknet
und das Lösungsmittel
verdampft, um ein gelbes Öl
zu liefern. r.f. (40 % EtOAc/Hexane) 0,26 färbt sich mit Vanillin schwach
weiß,
das Ausgangsmaterial r.f. 0,05 gelb mit Vanillin. Das Produkt erschien
nach der Extraktion rein.
-
Herstellung
22: 1-Prop-2-ynyl-piperidin-4-carbonsäreethylester (JR3271).
-
Die
Titelverbindung wurde ausgehend von 2,0 g (12,7 mmol) Ethylisonipecotat
entsprechend dem allgemeinen Verfahren 2 hergestellt.
-
Herstellung
23: 4-Prop-2-ynyl-piperazin-1-carbonsäure-tert-butylester (JR3275).
-
Zu
einer Lösung
aus 10,0 g (54,8 mmol) tert-Butyl-1-piperazincarboxylat in 60 ml Acetonitil
wurden 5,20 ml (60,4 mmol) Propargylbromide und 37,9 g (274 mmol)
wasserfreies Kaliumcarbonate zugegeben. Zusätzlich wurden nach dem Rühren für 36 Stunden
bei Raumtemperatur 1,5 ml Propargylbromid zugegeben. Der Rest wurde
bis er trocken war eingedampft. Es wurden 50 ml Dichlormethan und
50 ml Wasser zugegeben. Die Reaktionsmischung wurde mit CH2Cl2 4 × 40 ml
extrahiert, über
Magnesiumsulfat getrocknet und eingedampft, um ein braunes Öl bereitzustellen.
Das Öl
wurde in Dichlormethan gelöst
und mit einem RT Scientific System unter Verwendung eines Hexan/Ethylacetatgradienten
gereinigt, um 5,5 g (46 %) gelbes Öl zu ergeben, das nach dem
Stehen schließlich
kristallisierte.
-
Herstellung
24: 4-Cyanomethyl-piperazin-1-carbonsäureethylester (JR3287).
-
Zu
einer Lösung
aus 3 g (19,0 mmol) Ethyl N-Piperazincarboxylat in 25 ml CH3CN wurden 1,57 g (1,32 ml 20,1 mmol) 2-Chloracetonitril
und 15,6 g (95 mmol) K2CO3·1½H2O zugegeben. Die Suspension wurde bei Raumtemperatur
für 16
Stunden gerührt.
Die Reaktion wurde unter Verwendung einer TLC analysiert (35 % Ethylacetat/Hexane,
Produkt r.f. 0,38 vs. sm r.f. von 0,02). Die Analyse zeigte an,
dass die Reaktion vollständig war.
Die goldgelbe Lösung
wurde bis zur Trockenheit eingedampft. Der Rest wurde mit CH2Cl2/H2O
extrahiert, mit MgSO4 getrocknet und konzentriert.
-
Herstellung
25: 5-Prop-2-ynyl-2,5-diaza-bicyclo[2.2.1]heptan-2-carbonsäure-tert-butylester
(JR4013).
-
Die
Titelverbindung wurde ausgehend von 500 mg (2,52 mmol) 2,5-Diaza-bicyclo[2.2.1]heptan-2-carbonsäure-tert-butylester
entsprechend dem allgemeinen Verfahren 2 hergestellt.
-
Herstellung
26: 1-Cyclohexyl-4-prop-2-ynyl-piperazin (JR4019).
-
Die
Titelverbindung wurde ausgehend von 3 g (17,8 mmol) 1-Cyclohexylpiperazin
entsprechend dem allgemeinen Verfahren 2 hergestellt.
-
Herstellung
27: 1-Prop-2-ynyl-piperazin (JR4029).
-
Es
wurden in eine flammengetrocknete 25 ml Rundbodenflasche unter Stickstoff
2,1 g 4-Prop-2-ynyl-piperazin-1-carbonsäure-tert-butylester gegeben.
Zu diesem Feststoff wurden 5 ml 98 % TFA in 1 ml Portionen zugegeben.
Die Lösung
schlug zu weinrot um, warf Blasen und rauchte. Die zusätzlichen TFA-Portionen
wurden zugegeben, wenn die Aktivität nachließ. Nachdem die dritte TFA-Portion
zugegeben worden war, traten nur noch minimal Blasen auf. Die Lösung wurde
unter Stickstoff für
eine weitere Stunde bei Raumtemperatur gerührt und unter reduziertem Druck
eingedampft, um das Produkt als einen dicken roten Sirup zu ergeben.
Vermutete quantitative Ausbeute 1,16 g. Der Rest wurde in 20 ml
Dichlormethan suspendiert und sofort, ohne weitere Reinigung, für die Herstellung
der Verbindungen JR4031, JR4033 und JR4035 verwendet.
-
Herstellung
28: 4-Prop-2-ynyl-piperazin-1-carbonsäuremethylester (JR4031).
-
Die
Titelverbindung wurde ausgehend von 385 g (3,1 mmol) JR4029 und
unter Verwendung von Methylchlorformiat entsprechend dem allgemeinen
Verfahren 3 hergestellt.
-
Herstellung
29: 4-Prop-2-ynyl-piperazin-1-carbonsäureisobutylester (JR4035).
-
Die
Titelverbindung wurde ausgehend von 385 g (3,1 mmol) JR4029 und
unter Verwendung von Isobutylchlorformiat entsprechend dem allgemeinen
Verfahren 3 hergestellt.
-
Herstellung
30: 3,3-Dimethyl-1-(4-prop-2-ynyl-piperidin-1-yl)-butan-1-on (JR4041).
-
Die
Titelverbindung wurde ausgehend von tert-Butylester (JR3257) und
unter Verwendung von tert-Butylacetylchlorid entsprechend dem allgemeinen
Verfahren 3 hergestellt.
-
Herstellung
31: 1-(4-Prop-2-ynyl-piperazin-1-yl)-ethanon (JR4043).
-
Die
Titelverbindung wurde ausgehend von 385 g (3,1 mmol) JR4029 und
unter Verwendung von Acetylchlorid entsprechend dem allgemeinen
Verfahren 3 hergestellt.
-
Herstellung
32: Piperidin-1,4-dicarbonsäure-mono-tert-butylester.
-
Es
wurde eine Lösung
aus Piperidin-4-carbonsäure
(10 g, 77,5 mmol) und Kaliumcarbonat (21,4 g, 155 mmol) in 150 ml
Wasser hergestellt. Es wurde eine Lösung aus Di-tert-butyldicarbonat
(16,9 g, 77,5 mmol) in 40 ml THF tropfenweise mittels eines Trichters
bei 0°C
zugegeben. Der Reaktion wurde ermöglicht sich graduell über 30 Minuten
auf Raumtemperatur anzuwärmen
und für
weitere 4 Stunden gerührt.
Das THF wurde unter reduziertem Druck entfernt und die wässerige
Phase mit 50 ml Ether extrahiert. Die wässerige Phase wurde dann auf
pH 2 mit 10 % HCl eingestellt und mit EtOAc, 4 × 50 ml, extrahiert. Die kombinierten
organischen Schichten wurden über
wasserfreiem Natriumsulfat getrocknet, gefiltert und in vacou konzentriert,
um 17,2 g (97 %) JR3183 als einen weißen Feststoff zu ergeben. Rf
= 0,2 (35 % EtOAc/Hexane, gefärbt
mit Vanillin). 1H NMR (CDCl3) δ 11,83 (s,
1H), 3,98 (d, J = 11,8 Hz, 2H), 2,83 (t, J = 11,8, 2H), 2,46 (m,
1H), 1, 88 (d, J = 12,9 Hz, 2H), 1,2 (m, 2H), 1,42 (s, 9H). 13C NMR (CDCl3) δ 180,0, 154,8,
79,8, 42,9, 40,8, 28,3, 27,7. APCI m/z (rel Intensität) M- 228,2 (100).
-
Herstellung 33:
-
Die
folgenden Zwischenverbindungen werden unter Verwendung des allgemeinen
Verfahrens 1, welches hierin beschrieben ist, und den geeigneten
Ausgangsmaterialien hergestellt. (R)-1-Ethynyl-3-tert-butyl-cyclohexanol
(JR3255A), (S)-1-Ethynyl-3-tert-butyl-cyclohexanol
(JR3255B).
Toloul-4-sulfonsäure-4-prop-2-ynyl-cyclohexylmethylester
(JR3077).
1-Ethyl-4-prop-2-ynyl-cyclohexan
(JR3083).
1-(4-Prop-2-ynyl-cyclohexyl)-ethanon
(JR3115).
1,1-Dicyclohexyl-prop-2-yn-1-ol
(JR3127).
1-Cyclohexyl-prop-2-yn-1-ol
(JR3129).
4-Ethyl-1-ethynyl-cyclohexanol
(JR3143).
1-Ethynyl-3-methyl-cyclohexanol.
1-Ethynyl-3,3,5,5-tetramethyl-cyclohexanol
(JR3151).
1-Ethynyl-4-phenyl-cyclohexanol
(JR3153).
1-Ethynyl-2-methyl-cyclohexanol
(JR3167B)
4-tert-Butyl-1-ethynyl-cyclohexanol
(JR3191).
1-Ethynyl-3,3-dimethyl-cyclohexanol
(JR3193).
Piperidin-1,4-dicarbonsäure-1-tert-butylester-4-methylester
(JR3195).
4-Hydroxymethyl-piperidin-1-carbonsäure-tert-butylester
(JR3199).
4-Prop-2-ynyl-piperazin-1-carbonsäureethylester
(JR3211).
4-Prop-2-ynyl-piperidin-1-carbonsäure-tert-butylester
(JR3257).
4-Prop-2-ynyl-piperidin-1-carbonsäureethylester
(JR3267B).
2-(4-Prop-2-ynyl-piperazin-1-yl)-pyrimidin
(JR3277).
1-(4-Prop-2-ynyl-piperidin-1-yl)-ethanon
(JR4037).
2,2-Dimethyl-1-(4-prop-2-ynyl-piperidin-1-yl)-propan-1-on
(JR4039).
-
Beispiel
1: 4-{3-[6-Amino-9-(5-ethylcarbamoyl-3,4-dihydroxytetrahydrofuran-2-yl)-9H-purin-2-yl]-prop-2-ynyl}cyclohexancarbonsäure (109).
-
Die
Reaktion von 110 mit fünf Äquivalenten
LiOH in THF/Wasser für
6 Stunden ergab 109 (7 mg, 72 %) als einen weißen Feststoff; welcher aus
MeOH/H2O (0,1 % TFA) nach der Reinigung
mittels Reversphasen-HPLC kristallisiert wurde. 1H
NMR (DMSO-d6) δ 8,70 (s, 1H), 8,41 (s, 1H),
7,62 (s, 2H), 5,89 (d, J = 7,25 Hz, 1H), 4,53 (m, 1H), 4,27 (s,
1H), 4,08 (d, J = 3,6 Hz, 1H), 2,29 (d, J = 6,4 Hz, 2H), 2,15-1,99
(m, 1H), 1,92-1,76 (m, 4H), 1,52-1,38 (m, 1H), 1,38-1,19 (m, 2H),
1,02 (t, J = 6,3 Hz 3H); 13C NMR (DMSO-d6) 176,7, 169,2, 155,6, 148,9, 145,2, 141,6,
119,0, 87,7, 85,0, 84,6, 81,6, 73,1, 71,9, 43,2, 35,9, 33,3, 31,2,
28,3, 25,6, 15,0. HRMS (FAB) m/z 474,2196 [(M+H)+ berech.
für C22H29N6O6 474,2182].
-
Beispiel
2: 4-{3-[6-Amino-9-(5-ethylcarbamoyl-3,4-dihydroxytetrahydrofuran-2-yl)-9H-purin-2-yl]-prop-2-ynyl}cyclohexancarbonsäuremethylester
(110).
-
Die
Reaktion von 89 mit 2-Iod-NECA unter den allgemeinen oben beschriebenen
Bedingungen lieferte 110 (74 mg, 60 %) als einen weißen Feststoff. 1H NMR (CD3OD) δ 8,23 (s,
1H), 5,92 (d, J = 7,7 Hz, 1H), 4,69-4,65 (dd, J = 7,7 Hz, 4,6 Hz, 1H), 4,40
(s, 1H), 4,24 (d, J = 4,6 Hz, 1H), 3,59 (s, 3H), 3,49-3,31 (m, 2H), 2,31
(d, J = 6,6 Hz, 2H), 2,10-2,09 (m, 1H), 2,01-1,89 (m, 4H), 1,61-1,32
(m, 5H), 1,13 (t, J = 7,3 Hz, 3H); 13C NMR
(CD3OD) δ 177,1,
171,1, 156,3, 149,3, 146,7, 142,4, 119,7, 89,6, 86,0, 85,5, 81,6,
74,0, 72,2, 51,2, 43,2, 36,8, 34,2, 31,8, 28,9, 26,2, 14,4; HRMS
(FAB) m/z 487,2325 [(M+H)+ berech. für C23H31N6O6 487,2305].
-
Beispiel
3: Essigsäure-4-{3-[6-amino-9-(5-ethylcarbamoyl-3,4-dihydroxytetrahydrofuran-2-yl)-9H-purin-2-yl]-prop-2-ynyl}-cyclohexylmethylester
(111).
-
Die
Reaktion von 87 mit 2-Iod-NECA unter den allgemeinen oben beschriebenen
Bedingungen ergab 111 (78 mg, 62 %) als einen weißen Feststoff. 1H NMR (CD3OD) δ 8,22 (s,
1H), 5,92 (d, J = 8,1 Hz, 1H), 4,70-4,66 (dd, J = 8,1 Hz, 4,6 Hz, 1H), 4,40
(d, J = 1,2 Hz, 1H), 4,25-4,23 (dd, J = 4,6 Hz, 1,2 Hz, 1H), 3,83
(d, J = 6,5, 2H), 3,53-3,31 (m, 2H), 2,29 (d, J = 6,5 Hz, 2H), 1,97
(s, 3H), 1,93-1,89 (m, 2H), 1,79-1,75 (m, 2H), 1,64-1,42 (m, 2H),
1,12 (t, J = 7,3 Hz, 3H), 1,09-0,91 (m, 4H); 13C
NMR (CD3OD) δ 172,0, 171,2, 156,2, 149,3, 146,7,
142,5, 119,7, 89,6, 86,3, 85,5, 81,5, 74,0, 72,2, 69,6, 37,4, 37,2,
34,2, 32,1, 29,4, 26,4, 19,9, 14,5; HRMS (FAB) m/z 501,2469 [(M+H)+ berech. für C24H33N6O6 501,2462]
-
Beispiel
4: 5-{6-Amino-2-[3-(4-hydroxymethyl-cyclohexyl)-prop-1-ynyl]purin-9-yl}-3,4-dihydroxytetrahydrofuran-2-carbonsäureethylamide
(112).
-
Die
Reaktion von 86 (30 mg, 0,2 mmol) mit 2-Iod-NECA (28 mg, 0,07 mmol)
unter den allgemeinen oben beschriebenen Bedingungen ergab 112 (7
mg, 24 %) als einen weißen
Feststoff. 1H NMR (CD3OD) δ 8,22 (s,
1H), 5,92 (d, J = 7,7 Hz, 1H), 4,70-4,66 (dd, J = 7,7 Hz, 4,8 Hz,
1H), 4,40 (d, J = 1,2 Hz, 1H), 4,25-4,23 (dd, J = 4,8 Hz, 1,2 Hz,
1H), 3,51-3,37 (m, 2 H), 3,31 (d, J = 6 Hz, 2H), 2,30 (d, J = 6,8
Hz, 2H), 1,94-1,89 (m, 2H), 1,83- 1,78
(m, 2H), 1,64-1,42 (m, 2H), 1,12 (t, J = 7,3 Hz, 3H), 1,09-0,91
(m, 4 H); 3C NMR (CD3OD) δ 170,3, 155,4,
148,4, 146,0, 141,6, 118,8, 88,7, 85,5, 84,6, 80,6, 73,1, 71,3,
66,8, 39,6, 36,9, 33,3, 31,5, 28,6, 25,6, 13,5; HRMS (FAB) m/z 459,2373
[(M+H)+ berech. für C22H31N6O5 459,2356].
-
Beispiel
5: 5-{6-Amino-2-[3-(4-ethylcarbamoyl-cyclohexyl)-prop-1-ynyl]purin-9-yl}-3,4-dihydroxytetrahydrofuran-2-carbonsäureethylamid
(JR3037).
-
Es
wurden in ein versiegeltes Röhrchen,
das 5 ml frisch destilliertes Ethylamin enthielt, 10 mg (0,02 mmol)
ATL146e zugegeben. Die Flasche wurde versiegelt und für 80 Stunden
bei 60°C
gerührt.
Nach dieser Zeit war die Reaktion mittels HPLC nur zu etwa 50 %
vollständig.
Das Gefäß wurde
auf 0°C
gekühlt,
geöffnet und
das Ethylamin in vacou entfernt, um 4,5 mg (73 %) JR3037 als einen
weißen
Feststoff zu erhalten und 4,0 mg des wiedergewonnen Ausgangmaterials
zu erhalten, nachdem der Rest mittels RP-HPLC gereinigt wurde. 1H
NMR (CD3OD-d4) δ. 13C NMR (CD3OD-d4) δ.
APCI m/z (rel Intensität)
500,8 (MH+, 100), 327,4(3).
-
Beispiel
6: 5-{6-Amino-2-[3-(4-carbamoyl-cyclohexyl)-prop-1-ynyl]purin-9-yl}-3,4-dihydroxytetrahydrofuran-2-carbonsäureethylamid
(JR3055).
-
Es
wurden in ein versiegeltes Röhrchen,
das 10 ml gesättigte
MeOH/NH3-Lösung enthielt, 5 mg (0,01 mmol)
ATL146e zugegeben. Die Flasche wurde versiegelt und für 48 Stunden
bei 25°C
gerührt.
Das Gefäß wurde
auf 0°C
gekühlt,
geöffnet
und das Ammoniak durch blasenbildenden N2 für 1 Stunde
entfernt. Das verbliebende Lösungsmittel
wurde dann in vacou entfernt, um 4,0 mg (83 %) JR3055 als einen
weißen
Feststoff zu erhalten, nachdem der Rest mittels RP-HPLC gereinigt
wurde. 1H NMR (CD3OD-d4) δ 8,41
(s, 1H), 5,98 (d, J = 7,2 Hz, 1H), 4,65 (dd, J = 7,3 Hz, 4,8 Hz,
1H), 4,41 (d, J = 2,0 Hz, 1H), 4,28 (dd, J = 4,6 Hz, 2,0 Hz, 1H), 3,35
(m, 2H), 2,37 (d, J = 6,4 Hz, 2H) 2,10 (m, 1H), 1,90 (m,_H), 1,53
(m,_H_), 1,23 (m,_H), 1,12 (t, J = 7,3 Hz, 3H). 13C
NMR (CD3OD-d4) δ. APCI m/z
(rel Intensität)
472,3 (MH+, 100), 299,4(10).
-
Beispiel
7: 5-{6-Amino-2-[3-(4-methylcarbamoyl-cyclohexyl)-prop-1-ynyl]purin-9-yl}-3,4-dihydroxytetrahydrofuran-2-carbonsäureethylamid
(R3065)
-
Es
wurden in ein versiegeltes Röhrchen,
das 10 ml 2 M Methylamin in Methanol enthielt, 16,5 mg (0,03 mmol)
ATL146e zugegeben. Die Flasche wurde versiegelt und für 120 Stunden
bei 70°C
gerührt.
Das Gefäß wurde
auf 0°C
gekühlt,
geöffnet
und das Lösungsmittel
in vacou entfernt, um 8,0 mg (48 %) JR3065 als einen weißen Feststoff
zu erhalten, nachdem der Rest mittels RP-HPLC gereinigt wurde. 1H NMR (CD3OD-d4) δ. 13C NMR (CD3OD-d4) δ.
APCI m/z (rel Intensität)
486,3 (MH+, 100), 313,4(35).
-
Beispiel
8: 5-[6-Amino-2-(1-hydroxy-cyclopentylethynyl)-purin-9-yl]-3,4- dihydroxytetrahydrofuran-2-carbonsäureethylamid
(JR3135).
-
Die
Titelverbindung wurde unter Verwendung des geeigneten Ausgangsmaterials
und dem hierin beschrieben Verfahren hergestellt. Die Ergebnisse
sind wie folgt: 1H NMR (CD3OD-d4) δ 8,48
(s, 1H), 6,04 (d, J = 6,9 Hz, 1H), 4,72 (dd, J = 6,9 Hz, J = 4,4
Hz, 1H), 4,46 (d, J = 2,3 Hz, 1H), 4,33 (dd, J = 4,6 Hz, J = 1,9
Hz, 1H), 3,42 (m, 2H), 2,04 (m, 4H), 1,83, (m, 4 H), 1,16 (t, J
= 7,3 Hz, 3H). 13C NMR (CD3OD-d4) δ 171,9,
155,3, 150,0, 144,3, 120,6, 95,4, 90,6, 89,5, 86,2, 79,9, 74,9,
74,0, 70,5, 42,9, 35,3, 24,4, 15,3. APCI m/z (rel Intensität) 417,2
(MH+, 100), 399,4(85), 244,3(15), 26,5(25).
HRMS M+ tatsächlich 417,18864, beobachtet 417,18880.
-
Beispiel
9: 5-[6-Amino-2-(3,3-dicyclohexyl-3-hydroxy-prop-1-ynyl)-purin- 9-yl]-3,4-dihydroxytetrahydrofuran-2-carbonsäureethylamid
(JR3139).
-
Die
Titelverbindung wurde unter Verwendung des geeigneten Ausgangsmaterials
und dem hierin beschrieben Verfahren hergestellt. Die Ergebnisse
sind wie folgt: 1H NMR (CD3OD-d4) δ 8,57
(s, 1H), 6,09 (d, J = 6,6 Hz, 1H), 4,77 (dd, J = 6,7, Hz, J = 4,8
Hz, 1H), 4,46 (d, J = 2,3 Hz, 1H), 4,37 (dd, J = 4,6 Hz, J = 2,3
Hz, 1H), 3,42 (m, 2H) 1,80 (m, 13H), 1,28 (m, 9 H), 1,13 (t, J =
7,3 Hz, 3H). 13C NMR (CD3OD-d4) δ.
APCI m/z (rel Intensität)
527,3 (MH+, 60), 509,5(100), 354,4(5), 336,5(5),
279,5(8). HRMS M+ tatsächlich 527,29819, beobachtet
527,29830
-
Beispiel
10: 5-[6-Amino-2-(4-ethyl-1-hydroxycyclohexylethynyl)-purin-9-yl]-3,4-dihydroxytetrahydrofuran-2-carbonsäureethylamid
(JR3149).
-
Die
Titelverbindung wurde unter Verwendung des geeigneten Ausgangsmaterials
und dem hierin beschrieben Verfahren hergestellt. Die Ergebnisse
sind wie folgt: 1H NMR (CD3OD-d4) δ 8,51
(s, 1H), 6,06 (d, J = 7,0 Hz, 1H), 4,75 (dd, J = 6,4 Hz, J = 4,9
Hz, 1H), 4,46 (d, J = 1,9 Hz, 1H), 4,34 (dd, J = 4,9 Hz, J = 2,1
Hz, 1H), 3,42 (m, 2H), 2,12 (d, J = 11,9 Hz, 2 H), 1,80 (d, J =
11, 9 Hz, 2H), 1,58 (t, J = 12,1 Hz, 2H), 1,28 (m, 4H), 1,15 (t,
J = 7,1 Hz, 3H), 0,91 (t, J = 7,1 Hz, 3H). 13C
NMR (CD3OD-d4) δ 171,9, 155,4,
150,0, 144,2, 143,8, 120,6, 94,5, 90,5, 86,1, 81,8, 74,9, 74,1,
70,3, 40,5, 39,8, 35,3, 31,0, 30,2, 15,2, 12,0. APCI m/z (rel Intensität) 459,4
(MH+, 100), 441,4(60), 268,4(10), HRMS M+ tatsächlich
459,23559, beobachtet 459,23550.
-
Beispiel
11: 5-[6-Amino-2-(1-hydroxy-4-phenyl-cyclohexylethynyl)-purin-9-yl]-3,4-dihydroxytetrahydrofuran-2-carbonsäureethylamid
(JR3161).
-
Die
Titelverbindung wurde unter Verwendung des geeigneten Ausgangsmaterials
und dem hierin beschrieben Verfahren hergestellt. Die Ergebnisse
sind wie folgt: 1H NMR (CD3OD-d4) δ 8,45
(s, 1H), 7,26 (m, 4H), 7,14 (m, 1H), 6,05 (d, J = 7,3 Hz, 1H), 4,80
(dd, J = 7,3 Hz, J = 4,8 Hz, 1H), 4,46 (d, J = 1,6 Hz, 1H), 4,34 (dd,
J = 4,7 Hz, J = 1,8 Hz, 1H), 3,44 (m, 2H), 2,58 (m, 1H), 2,23 (d,
J = 11, 7 H, 2H), 1,92 (m, 4H), 1,78, (m, 2H), 1,15 (t, J = 7,2
Hz, 3H). 13C NMR (CD3OD-d4) δ.
APCI m/z (rel Intensität)
507,3 (MH+, 100) 489,4(70), 334,3(5), 316,5(8).
HRMS M+ tatsächlich 507,23559, beobachtet
507,23580.
-
Beispiel
12: 5-[6-Amino-2-(1-hydroxy-3,3,5,5-tetramethylcyclohexylethynyl)purin-9-yl]-3,4-dihydroxytetrahydrofuran-2-carbonsäureethylamid
(JR3163).
-
Die
Titelverbindung wurde unter Verwendung des geeigneten Ausgangsmaterials
und dem hierin beschrieben Verfahren hergestellt. Die Ergebnisse
sind wie folgt: 1H NMR (CD3OD-d4) δ 8,54
(s, 1H), 6,04 (d, J = 6,9 Hz, 1H), 4,74 (dd, J = 6,9 Hz, J = 5,0
Hz, 1H), 4,46 (d, J = 1,9 Hz, 1H), 4,34 (dd, J = 4,7 Hz, J = 1,9
Hz, 1H), 3,44 (m, 2H), 1,74 (s, 4H), 1,13 (m, 17H). APCI m/z (rel
Intensität)
487,3 (MH+, 75), 469,4(100), 296,4(10).
-
Beispiel
13: 5-[6-Amino-2-(1-hydroxy-2-methyl-cyclohexylethynyl)-purin-9-yl]-3,4-dihydroxytetrahydrofuran-2-carbonsäureethylamid
(JR3177A, JR3177B).
-
Die
Reaktion von 1-Ethynyl-2-methyl-cyclohexanol (JR316913) (100 mg,
0,72 mmol) mit 2-Iod-NECA (25 mg, 0,06 mmol) unter den allgemeinen
Kopplungsbedingungen ergab nach der Reinigung mittels eines Silicastopfens
und RP-HPLC als weiße
Feststoffe JR3177A (8,0 mg) und JR3177B (8,2 mg) (Gesamtausbeute 65
%). JR3177A: 1H NMR (CH3OD-d4) δ 8,47
(s, 1H), 6,05 (d, J = 6,9 Hz, 1H), 4,77 (dd, J = 6,9 Hz, J = 4,9
Hz, 1H), 4,45 (d, J = 1,9 Hz, 1H), 4,34 (dd, J = 4,6 Hz, J = 2,1
Hz, 1H), 3,41 (m, 2H), 2,13 (d, J = 12,7 Hz, 2H), 1,65 (M, 5H),
1,32 (m, 2H), 1,14 (t, J = 7,0 Hz, 3H), 1,13 (d, J = 6,6 Hz, 3H). 13C NMR (CD3OD-d4) δ.
APCI m/z (rel Intensität)
445,3 (MH+, 100), 427,4(80), 254,4(14). 1H NMR (CD3OD-d4) δ 8,49
(s, 1H), 6,05 (d, J = 6,9 Hz, 1H), 4,78 (dd, J = 6,4 Hz, J = 4,9
Hz, 1H), 4,45 (d, J = 1,9 Hz, 1H), 4,34 (dd, J = 4,6 Hz, J = 1,6
Hz, 1H), 3,42 (m, 2H), 2,12 (d, J = 12,3 Hz, 2 H), 1,65 (m, 4H),
1,35 (m, 4H), 1,14 (t, J = 7,3 Hz, 3H), 1,12 (d, J = 6,6 Hz, 3H). 13C NMR (CD3OD-d4) δ.
APCI m/z (rel Intensität)
445,7 (MH+, 100), 427,3(35), 254,4(3,5).
-
Beispiel
14: 5-[6-Amino-2-(1-hydroxy-3-methyl-cyclohexylethynyl)-purin-9-yl]-3,4-dihydroxytetrahydrofuran-2-carbonsäureethylamid
(JR3179).
-
Die
Reaktion von 1-Ethynyl-3-methyl-cyclohexanol (JR3149B) (100 mg,
0,72 mmol) mit 2-Iod-NECA (25 mg, 0,06 mmol) unter den allgemeinen
Kopplungsbedingungen ergab nach der Reinigung mittels eines Silicastopfens
und RP-HPLC als einen weißen
Feststoff JR3179 (15) mg, 59 %). 1H NMR
(CD3OD-d4) δ 8,49 (s, 1H),
6,06 (d, J = 6,9 Hz, 1H), 4,75 (dd, J = 6,4 Hz, J = 4,9 Hz, 1H),
4,46 (d, J = 1,9 Hz, 1H), 4,34 (dd, J = 4,9 Hz, J = 2,1 Hz, 1H),
3,42 (m, 2H), 2,09 (d, J = 12,3 Hz, 2H), 1,73 (m, 4H), 1,46 (m,
1H), 1,23 (m, 1H), 1, 16 9 (t, J = 7,1 Hz, 3H), 0,95 (d, J = 6,2
Hz, 3H), 0,89 (m, 1H). 13C NMR (CD3OD-d4) δ. APCI m/z
(rel Intensität) 445,3
(MH+, 100), 427,4(40), 254,4(4).
-
Beispiel
15: 4-{3-[6-Amino-9-(5-ethylcarbamoyl-3,4-dihydroxytetrahydrofuran-2-yl)-9H-purin-2-yl]-prop-2-ynyl}-piperazin-1-carbonsäureethylester
(JR3213).
-
Die
Titelverbindung wurde unter Verwendung des geeigneten Ausgangsmaterials
und der hierin beschrieben Verfahren hergestellt. Die Ergebnisse
sind wie folgt: 1H NMR (CD3OD-d4) δ 8,48
(s, 1H), 6,00 (d, J = 6,9 Hz, 1H), 4,67 (dd, J = 6, 5 Hz, J = 5,0
Hz, 1H), 4,42 (d, J = 1,9 Hz, 1H)), 4,39 (s, 2H), 4,35 (dd, J =
4,7 Hz, J = 1,9 Hz, 1H), 4,13 (q,) 3,42 (m, 2 H). 13C
NMR (CD3OD-d4) δ. APCI m/z
(rel Intensität)
503,4 (MH+, 100), 330,3(6).
-
Beispiel
16: 5-[6-Amino-2-(3-hydroxy-2-oxo-azepan-3-ylethynyl)purin-9-yl]-3,4-dihydroxytetrahydrofuran-2-carbonsäureethylamid
(JR3243A, JR3243B)
-
Es
wurden 35 mg (0,081 mmol) Iod-NECA (62 mg Alkyn, 0,41 mmol), 2 ml
DMF, 4 ml Acetonitril, 0,2 ml TEA, d(PPH3)4, CuI, über
Nacht bei Raumtemperatur gerührt
(11/29/01). Die Reaktion ist ein braun mit einem braunen Präzipitat.
Die TLC (20 % MeOH/CH2Cl2)
zeigt an, dass die Reaktion vollständig ist (r.f. INECA = 0,67,
r.f. Produkt = 0,45). Die Mischung wurde durch Celite gefiltert,
mit 3 × 2
ml DMF gewaschen und unter Vakuum zu einem braunen Öl eingedampft.
(Der Feststoff fällt
nach der Zugabe von MeOH aus, folglich wurde DMF für die Beladung
der Prepplatte verwendet).
-
Die
folgenden Verbindungen können
durch das hierin beschriebene allgemeine Verfahren 4 und die geeigneten
hierin beschriebenen Zwischenverbindungen hergestellt werden.
-
Beispiel
17: N-Ethyl 2-{3-[trans-4-(methoxycarbonyloxamethyl)cyclohexyl]-1-propyn-1-yl}adenosin-5'-uronamid (ATL214):
-
- Ausbeute 3,4 mg, 10 %, 1H NMR (CD3OD) δ 1,18
(t, 3 H, -NHCH2CH3),
1,03-1,20, 1,51-1,70, 1,79-1,85, 1,94-2,01 (4 × m, 10 H, Cyclohexyl), 2,35
(d, 2H, -C6H10CH2CC-), 3,46 (m, 2H, -NHCH2CH3), 3,73 (s, 3 H, -OCH3),
3,94 (d, 2H, -C6H10CH2O-), 4,29 (dd, 1H, 3'-H), 4,45 (d, 1H, 4'-H), 4,72 (dd, 1H, 2'-H), 5,97 (d, 1H, 1'-H), 8,27 (s, 1H, 8-H). APCI m/z 517,4
(M+H+).
-
Beispiel
18: N-Ethyl2-{3-[trans-4-(isobutoxyoxycarbonyloxamethyl)cyclohexyl]-1-propyn-1-yl}adenosin-5'-uronamid (ATL215):
-
- Ausbeute 8,5 mg, 30 %, 1H NMR (CD3OD) δ 0,94
(d, 4 H, -OCH2CH(CH3)2), 1,18 (t, 3 H, -NHCH2CH3), 1,04-1,24, 1,54-1,72, 1,79-2,03 (3 × m, 11H,
Cyclohexyl, -OCH2CH(CH3)2), 2,38 (d, 2H, -C6H10CH2CC-), 3,43 (m,
2H, -NHCH2CH3),
3,89, 3,94 (2 × d,
4 H, -C6H10CH2O-, -OCH2CH(CH3)2), 4,30 (dd, 1H,
3'-H), 4,46 (d,
1H, 4'-H), 4,71
(dd, 1H, 2'-H),
6,00 (d, 1H, 1'-H),
8,37 (br s, 1H, 8-H). APCI m/z 559,5 (M+H+).
-
Beispiel
19: N-Ethyl 2-{3-[trans-4-(benzoxycarbonyloxamethyl)cyclohexyl]-1-propyn-1-yl}adenosin-5'-uronamid (ATL216):
-
- Ausbeute 1,0 mg, 3 %, 1H NMR (CD3OD) δ 1,17
(t, 3 H, -NHCH2CH3),
1,03-1,23, 1,52-1,71, 1,78-1,86, 1,93-2,02 (4 × m, 10 H, Cyclohexyl), 2,35
(d, 2H, -C6H10CH2CC-), 3,45 (m, 2H, -NHCH2CH3), 3,97 (d, 2H, -C6H10CH2O-), 4,29 (dd,
1H, 3'-H), 4,45
(d, 1H, 4'-H), 4,72
(dd, 1H, 2'-H),
5,13 (s, 2H, -OCH2Ph), 5,97 (d, 1H, 1'-H), 7,33-7,37 (m,
5 H, Ar), 8,30 (br s, 1H, 8-H). APCI m/z 593,3 (M+H+).
-
Beispiel
20: 4-{3-[6-Amino-9-(5-ethylcarbamoyl-3,4-dihydroxytetrahydrofuran-2-yl)-9H-purin-2-yl]-prop-2-ynyl}cyclohexancarbonsäure-2-tert-butoxycarbonylamino-ethylester.
-
Beispiel
21: 5-{6-Amino-2-[3-(4-dimethylaminomethyl-cyclohexyl)-prop-1-ynyl]-purin-9-yl}-3,4-dihydroxytetrahydrofuran-2-carbonsäureethylamid
(JR2023).
-
Beispiel
22: 4-{3-[6-Amino-9-(5-ethylcarbamoyl-3,4-dihydroxytetrahydro-furan-2-yl)-9H-purin-2-yl]-prop-2-ynyl}cyclohexancarbonsäure-2-aminoetaylester
(JR3033).
-
Beispiel
23: 4-{3-[6-Amino-9-(5-ethylcarbamoyl-3,4-dihydroxytetrahydrofuran-2-yl)-9H-purin-2-yl]-prop-2-ynyl}-1-methylcyclohexancarbonsäuremethylester
(JR3067A).
-
Beispiel
24: 4-{3-[6-Amino-9-(5-ethylcarbamoyl-3,4-dihydroxytetrahydrofuran-2-yl)-9H-purin-2-yl]-prop-2-ynyl}-1-methylcyclohexancarbonsäuremethylester
(JR3067B).
-
Beispiel
25: 5-{6-Amino-2-[3-(4-ethyl-cyclohexyl)-prop-1-ynyl]-purin-9-yl}-3,4-dihydroxytetrahydrofuran-2-carbonsäureethylamid
(JR3087).
-
Beispiel
26: 5-{2-[3-(4-Acetyl-cyclohexyl)-prop-1-ynyl]-6-aminopurin-9-yl}-3,4-dihydroxytetrahydrofuran-2-carbonsäureethylamid
(JR3119).
-
Beispiel
27: 5-(6-Amino-2-{3-[4-(1-hydroxy-ethyl)-cyclohexyl]-prop-1-ynyl}-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-carbonsäureethylamid.
-
Beispiel
28: 5-[6-Amino-2-(1-hydroxy-2-methyl-cyclohexylethynyl)-purin-9-yl]-3,4-dihydroxytetrahydrofuran-2-carbonsäureethylamid
(JR3181A, JR3181B).
-
Beispiel
29: 5-[6-Amino-2-(1-hydroxy-3,3-dimethylcyclohexyl-ethynyl)-purin-9-yl]-3,4-dihydroxytetrahydrofuran-2-carbonsäureethylamid
(JR3201B).
-
Beispiel
30: 5-[6-Amino-2-(4-tert-butyl-1-hydroxycyclohexylethynyl)-purin-9-yl]-3,4-dihydroxytetrahydrofuran-2-carbonsäureethylamid
(JR3203).
-
Beispiel
31: 5-[6-Amino-2-(1-hydroxy-3-methylcyclohexylethynyl)purin-9-yl]-3,4-dihydroxytetrahydrofuran-2-carbonsäureethylamid
(JR3221).
-
Beispiel
32: 5-[6-Amino-2-(1-hydroxy-3-methylcyclohexylethynyl)purin-9-yl]-3,4-dihydroxytetrahydrofuran-2-carbonsäureethylamid
(JR3223, ATL 203).
-
Beispiel
33: 5-[6-Amino-2-(2-tert-butyl-1-hydroxycyclohexylethynyl)purin-9-yl]-3,4-dihydroxytetrahydrofuran-2-carbonsäureethylamid
(JR3227).
-
Beispiel
34: 1-{3-[6-Amino-9-(5-ethylcarbamoyl-3,4-dihydroxytetrahydrofuran-2-yl)-9H-purin-2-yl]-prop-2-ynyl}-piperidin-4-carbonsäuremethylester
(JR3251).
-
Beispiel
35: 1-{3-[6-Amino-9-(5-ethylcarbamoyl-3,4-dihydroxytetrahydrofuran-2-yl)-9H-purin-2-yl]-prop-2-ynyl}-piperidin-2-carbonsäuremethylester
(JR3253).
-
Beispiel
36: 4-{3-[6-Amino-9-(5-ethylcarbamoyl-3,4-dihydroxytetrahydrofuran-2-yl)-9H-purin-2-yl]-prop-2-ynyl}-piperidin-1-carbonsäure-tert-butylester (JR3259).
-
Beispiel
37: 4-{3-[6-Amino-9-(5-ethylcarbamoyl-3,4-dihydroxytetrahydrofuran-2-yl)-9H-purin-2-yl]-prop-2-ynyl}-piperidin-1-carbonsäureethylester
(JR3269).
-
Beispiel
38: 1-{3-[6-Amino-9-(5-ethylcarbamoyl-3,4-dihydroxytetrahydrofuran-2-yl)-9H-purin-2-yl]-prop-2-ynyl}-piperidin-4-carbonsäureethylester
(JR3279).
-
Beispiel
39: 4-{3-[6-Amino-9-(5-ethylcarbamoyl-3,4-dihydroxytetrahydrofuran-2-yl)-9H-purin-2-yl]-prop-2-ynyl}-piperazin-1-carbonsäure-tert-butylester
(JR3281).
-
Beispiel
40: 5-{6-Amino-2-[3-(4-pyrimidin-2-yl-piperazin-1-yl)-prop-1-ynyl]purin-9-yl}-3,4-dihydroxytetrahydrofuran-2-carbonsäureethylamid
(JR3283).
-
Beispiel
41: 5-[6-Amino-2-(3-piperazin-1-yl-prop-1-ynyl)purin-9-yl]-3,4-dihydroxytetrahydrofuran-2-carbonsäureethylamid
(JR3289).
-
Beispiel
42: 1-{3-[6-Amino-9-(5-ethylcarbamoyl-3,4-dihydroxytetrahydrofuran-2-yl)-9H-purin-2-yl]-prop-2-ynyl}-piperidin-4-carbonsäure (JR3291).
-
Beispiel
43: 4-{3-[6-Amino-9-(5-ethylcarbamoyl-3,4-dihydroxytetrahydrofuran-2-yl)-9H-purin-2-yl]-prop-2-ynyl}-piperidin-1-carbonsäuremethylester
(JR4007).
-
Beispiel
44: 4-{3-[6-Amino-9-(5-ethylcarbamoyl-3,4-dihydroxytetrahydrofuran-2-yl)-9H-purin-2-yl]-prop-2-ynyl}-piperidin-1-carbonsäureisopropylester
(JR4009).
-
Beispiel
45: 4-{3-[6-Amino-9-(5-ethylcarbamoyl-3,4-dihydroxytetrahydrofuran-2-yl)-9H-purin-2-yl]-prop-2-ynyl}-piperidin-1-carbonsäureisobutylester
(JR4011).
-
Beispiel
46: 5-{3-[6-Amino-9-(5-ethylcarbamoyl-3,4-dihydroxytetrahydrofuran-2-yl)-9H-purin-2-yl]prop-2-ynyl}-2,5-diazabicyclo[2.2.1]heptan-2-carbonsäure-tert-butylester
(JR4015).
-
Beispiel
47: 5-(6-Amino-2-{3-[1-(3,3-dimethyl-butyryl)-piperidin-4-yl]prop-1-ynyl}purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-carbonsäureethylamid
(JR4047).
-
Beispiel
48: 5-(6-Amino-2-{3-[1-(2,2-dimethyl-propionyl)-piperidin-4-yl]prop-1-ynyl}-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-carbonsäureethylamid
(JR4051).
-
Beispiel
49: 4-{3-[6-Amino-9-(5-ethylcarbamoyl-3,4-dihydroxytetrahydrofuran-2-yl)-9H-purin-2-yl]-prop-2-ynyl}-piperazin-1-carbonsäureisobutylester
(JR4049).
-
Beispiel
50: 5-{2-[3-(4-Acetyl-piperazin-1-yl)-prop-1-ynyl]-6-amino-purin-9-yl}-3,4-dihydroxytetrahydrofuran-2-carbonsäureethylamid
(JR4053).
-
Beispiel 51:
-
Die
folgenden Verbindungen können
durch die Berücksichtigung
der hierin beschrieben allgemeinen Verfahren und der geeigneten
Zwischenverbindungen hergestellt werden.
-
-
-
-
Beispiel 52: Vorläufige Untersuchungen in vitro
-
Die
Wirkungen der A2A-AR-Agonisten, anfänglich WRC-0470
und in aller jüngster
Zeit ATL146e und ATL193, wurden in phagozytische Zellen in vitro
und in Tiermodellen für
akute Entzündung
untersucht. Die Ergebnisse zeigen an, dass diese Verbindungen potente
Agonisten sind und antiinflammatorische Reaktionen sowohl in vitro
als auch in vivo bereitstellen. Die Wirkung dieser Verbindungen
auf humane PMNL (843 Rezeptoren pro Zelle) wurde charakterisiert
und quantifizierte die A2AARen. Es wurde
belegt, dass die A2AAR-Agonisten die intrazellulären cyclischen
AMP- Konzentrationen
der PMNL erhöhen,
während
sie die TNF-stimulierte Haftung an eine fibronektinbeschichtete
Oberfläche
vermindern. Die A2AAR-Agonisten vermindern die TNF-stimulierte
Superoxidfreisetzung von adhärenten
PMNLen. Die Superoxidfreisetzung wird vollständig durch den A2AAR-Antagonisten
ZM 241385 (ZM) blockiert. Ferner vermindern die A2AAR-Agonisten
die oxidative Aktivität der
PMNLen in einem Gesamtblutassay und verringern die Degranulation
von aktivierten PMNLen, die an einer biologischen Oberfläche haften.
Rolipram hebt synergistisch alle die oben diskutierten Wirkungen
auf die PMNLen hervor. Schließlich
kehrt der Proteinkinase A-Inihibitor H-89 die hemmende Wirkung der
A2AAR-Agonisten
auf den oxidativen Ausbruch der PMNLen vollständig um. Diese Ergebnisse zeigen
an, dass die A2AAR-Agonisten die Entzündung in
vivo durch direkte Wirkungen auf phagozytische Zellen verändern.
-
Es
wurde gezeigt, dass die Aktivierung der A2AARen
auf humanen Monozyten, starkt die TNF-Freisetzung hemmt. Dies zeigt
die antiinflammatorische Wirkung der A2AAR-Agonisten.
ATL146e ist bei der Hemmung der LPS-stimulierten TNF-Produktion
humaner Monozyten wirksamer als CGS21680, eine Wirkung, die durch den
selektiven A2AAR-Antagonisten ZM241385 umgekehrt wird.
Diese Daten zeigen, dass die A2AAR-Agonisten
die A2AAR-vermittelte anitinflammatorische
Wirkung betätigen
und die TNF-Produktion von den Monozyten vermindern.
-
Beispiel 53: Untersuchungen in vivo
-
Dosis-Reaktions-Untersuchungen: murines
Model für
septischen Schock
-
1 stellt
die Sterblichkeit von C57BL/6-Mäusen
in Folge der intraperitonealen (i.p.) Inokulation von E. coli 026:B6
LPS (Difco) dar. Aus diesen Daten wurde einen Dosis von 12,5 mg/kg
LPS für
die murinen Mortalitätsuntersuchungen
ausgewählt.
Diese Untersuchungen erlauben die Herstellung von A2A-KO-Mäusen aus demselben
C57BL/6-Hintergund (siehe unten). Zusätzlich wurde das Model als
ein ausgezeichnetes Model für Multisystemorganversagen
während
einer Endotoxämie
und einem septischen Schock bestätigt.
-
A2A-AR-Agonisten
verringern die Mortalität
in einem murinem Model für
septischen Schock
-
In
diesen Experimenten (n = 15-16 pro Gruppe; LPS 12,5 mg/kg) wurden
Kontrolltiere mit jenen verglichen, die mit ATL146e behandelt wurden.
Der Agonist wurde i.p. in 6-Stunden Abständen für 24 Stunden dosiert, beginnend
gleichzeitig mit der anfänglichen
intraperitonealen LPS-Dosis.
Die anfänglich
verwendete Dosierung von ATL146e war 5 μg/kg; der Schutz vor dem Tod
(p = 0,0002) ist in 2 dargestellt.
-
Mit
der höchsten
Dosierung (von ATL146e) wird ein vollständiger Schutz erzielt. Alle
Tiere, die bis zum 4 Tag überlebten,
erholen sich vollständig.
Mit der höchsten
Dosierung (von ATL146e) ist das Überleben
bei 4 Tagen 100 % gegenüber
75 % bei den Mäusen,
die das Kontrollvehikel erhielten (phosphatgepufferte Saline i.p.).
Die Wirkung eines A2AAR-Agonisten auf die Mortalität in einem
Model für
septischen Schock ist um Größenordnungen
anderen „antiinflammatorischen" Mitteln überlegen,
die in ähnlichen
Modellen für
septischen Schock verwendet werden, einschließlich Corticosteroide, anti-LPS
monoklonale Antikörper,
anti-TNF monoklonale Antikörper,
lösliche
TNF-Rezeptoren und IL-1 Rezeptorantagonisten.
-
ATL146e
verringerte die Mortalität
in einem murinen Model für
einen endotoxininduzierten septischen Schock sogar nach einem verzögerten Therapiebeginn.
In diesen Experimenten (N = 15-16 pro Gruppe: LPS 12,5 mg/kg) wurde
ATL146e mit einer Dosis von 5 μg/kg
i.p. mit sechsstündigen
Intervallen zu verschiedenen Zeiten nach der LPS-Herausforderung
für eine
Gesamtheit von vier Dosen verabreicht. Die Ergebnisse sind in 3 dargestellt.
ATL146e stellte einen Schutz vor dem Tod sogar mit einer Verzögerung von
24 Stunden in Folge der LPS-Herausforderung her. Diese Experimente
sind entscheidend, weil die Patienten mit einem septischen Syndrom
und septischen Schock oft viele Stunden nach dem Beginn der Symptome
beurteilt und behandelt werden. Eine Verzögerung von nur wenigen Stunden
unterband jegliche schützende
Wirkung in diesem Model mit monoklonalen Antikörpern, die gegen LPS und/oder
TNF gerichtet sind. Folglich sind A2A-Agonisten
sogar bei weit fortgeschrittenen, ernsten septischen Syndromen von
Nutzen.
-
Beispiel 54: Schützende Wirkungen
-
Die
schützende
Wirkung von ATL146e auf die Mortalität in dem murinem Model für den endotoxininduzierten
septischen Schock ist spezifisch für den A2A-Rezeptor.
Es wurden zwei experimentelle Strategien eingesetzt, um die Spezifität der schützenden
Wirkung, die mit ATL146e auf die Mortalität und den endotoxininduzierten
Schock durch den A2A-AR-Rezeptor beobachtet
wird zu untersuchen. Es wird ZM 241385 (ZM), ein spezifischer, wirksamer
und hoch selektiver Antagonist des A2A-AR
verwendet. Wie in 4 gezeigt schützt ZM allein
die Mäuse
nicht vor dem Tod in Folge eines endotoxininduziertem septischen
Schocks. Wenn ZM jedoch in äquimolaren
Konzentrationen (3 μg(kg)
mit ATL146e verabreicht wird, wird die Schutzwirksamkeit von ATL146e
beinahe aufgehoben. Folglich wirkt ein spezifischer A2AAR-Antagonist
der Wirkung von ATL146e entgegen und eliminiert beinahe den Überlebensvorteil,
der mit den A2A-Agonisten beobachtet wird.
In diesen Experimenten wurden beide Mittel in Sechsstundenintervallen
für 24
Stunden gegeben, beginnend 12 Stunden nach der LPS-Herausforderung.
-
Homozygote
A2A-KO-Mäuse.
Es wurden A2A-KO-Mäuse aus einem heterozygoten
Zuchtspaar erzeugt. Diesen Mäusen
fehlten die A2A-ARen, was mittels PCR und
durch Lokalisationsuntersuchungen von A2A-ARen in
Wildtyp- und A2A-KO-Mäusenhirnen unter Verwendung
eines selektiven A2A-AR monoklonalen Antikörpers bestätigt wurde
(5). Der mutierte A2A-AR wurde auf einen
C57BL/6J-Hintergund transferiert, wobei eine mikrosatelliten-unterstützte Selektion
verwendet wurde. Diese A2A-KO-Mäuse wurden
verwendet um weiter die Spezifität
der Schutzwirkung von ATL146e in dem murinen Model für septischen
Schock zu untersuchen (1). Die LPS-Dosis war wiederum
12,5 mg/kg und es wurden etwa zehn Tiere in jede Gruppe eingeschlossen.
ATL wurde mit 5 μg/kg
dosiert und in vier Dosen in Sechsstundenintervallen beginnend 12
Stunden in Folge der LPS-Inokulation verabreicht. Es kann in 1 gesehen
werden, dass die Schutzwirkung von ATL146e in A2A-KO-Mäusen vollständig verloren
geht, was stark die Spezifität
des A2A-Agonisten auf dem Level des A2A-AR unterstützt.
-
Beispiel 55: Behandlung von Mäusen mit
E. coli
-
Es
wurden Mäusen
lebende E. coli injiziert und sie wurden mit einem Antibiotikum
(Ceftriaxon) behandelt. Die Kontrollgruppe der Mäuse wurde mit dem Antibiotikum
allein behandelt. Allen Mäusen
wurden 20 Millionen E. coli IP zum Zeitpunkt 0 injiziert. Wie angegeben
wurden die Mäuse
einmal zum Zeitpunkt 0 mit Ceftriaxon oder mit 50 μg/kg ATL146e
8-mal in Sechsstundenintervallen behandelt. Die Ergebnisse sind
in der 6 dargestellt.
-
Beispiel 56: Verminderung der renalen
Produktion von IL-6 und RANTES
-
Es
wurden männlichen
C57BL/6-Mäusen
E. coli LPS (60 ng) und gereinigte E. coli Shigatoxin-2 (Stx2, 12
ng) zur Stunde Null injiziert (i.p.). ATL-146e oder ATL-203 (jeweils
50 μg/kg)
wurden i.p. zur Stunde Null verabreicht. Die Tiere wurden nach 6
Stunden getötet
und die Nieren für
die Verarbeitung und Analyse entfernt. Das IL-6-Protein war im Vergleich
zu der Kochsalzlösungskontrolle
45-fach durch LPS/Stx2 nach 6 Stunden erhöht. Beide ATL-Verbindungen
verminderten von jenen Mäusen,
die LPS/Stx2 ausgesetzt waren, die renalen IL-6-Level stark auf
etwa 16 % (7).
-
Verminderung der renalen Neuetrophilenakkumulation
in Mäusen
-
Die
Mäuse erhielten
2,4 ng gereinigtes Stx2 i.p. zur Stunden Null und wurden entweder
mit oder ohne die ATL-203-Verbindung (i.p.), beginnend zur Stunde
Null und alle 12 Stunden danach, behandelt. Es wurden fixierte und
in Paraffin eingebettete Nierenproben, die in 3 μ dicke Schnitte geschnitten
worden waren, mit einem Neutrophilen-spezifischen Antikörper vor
der Anaylse zur Reaktion gebracht, etc. Die Ergebnisse, dargestellt
in 9, von Nieren, die 48 Stunden postinjektion von
Stx2 erhalten wurden, zeigen, dass Stx2 eine 8,5-fache Zunahme in
den neutrophilenpositiven Glomeruli verursachte. ATL-203 verringerte
diese wirksam auf 40 % von den Stx2-positiven Proben. Es wurde ein ähnliches
Ergebnis erhalten, wenn die 48-Stunden-Proben hinsichtlich der durchschnittlichen
Neurophilenzahl pro leistungsstarkem Feld in dem Nierenkortex ausgewertet
wurden, ausgeschossen der Neutrophilen innerhalb der Glomeruli.
Die Akkumulation der Neutrophilen in den Nieren nach 48 Stunden
ist ein natürliches
Ereignis, dass zu einer weiteren Nierenschädigung führt und dass typischerweise
zu dem Tod solcher Tiere am Tag 4 beiträgt.
-
Diese
Daten zeigen, dass der Adenosin-A2A-Rezeptoragonist
wirksam die Stx2-abhängige
Infiltration der Neutrophilen in die Nieren von C57BL/6-Mäusen vermindert (9).
Dieses Ergebnis übernimmt
eine zusätzliche
Signifikanz, weil die Wirkung von ATL-203 auf Mäuse gerichtet ist, die nur
Stx2 ausgesetzt sind, das heißt
in der Abwesenheit von LPS.

































































 1-Ethyl-4-prop-2-ynyl-cyclohexan (JR3083).
1-Ethyl-4-prop-2-ynyl-cyclohexan (JR3083). 1-(4-Prop-2-ynyl-cyclohexyl)-ethanon (JR3115).
1-(4-Prop-2-ynyl-cyclohexyl)-ethanon (JR3115). 1,1-Dicyclohexyl-prop-2-yn-1-ol (JR3127).
1,1-Dicyclohexyl-prop-2-yn-1-ol (JR3127). 1-Cyclohexyl-prop-2-yn-1-ol (JR3129).
1-Cyclohexyl-prop-2-yn-1-ol (JR3129). 4-Ethyl-1-ethynyl-cyclohexanol (JR3143).
4-Ethyl-1-ethynyl-cyclohexanol (JR3143). 1-Ethynyl-3-methyl-cyclohexanol.
1-Ethynyl-3-methyl-cyclohexanol. 1-Ethynyl-3,3,5,5-tetramethyl-cyclohexanol (JR3151).
1-Ethynyl-3,3,5,5-tetramethyl-cyclohexanol (JR3151). 1-Ethynyl-4-phenyl-cyclohexanol (JR3153).
1-Ethynyl-4-phenyl-cyclohexanol (JR3153). 1-Ethynyl-2-methyl-cyclohexanol (JR3167B)
1-Ethynyl-2-methyl-cyclohexanol (JR3167B) 4-tert-Butyl-1-ethynyl-cyclohexanol (JR3191).
4-tert-Butyl-1-ethynyl-cyclohexanol (JR3191). 1-Ethynyl-3,3-dimethyl-cyclohexanol (JR3193).
1-Ethynyl-3,3-dimethyl-cyclohexanol (JR3193). Piperidin-1,4-dicarbonsäure-1-tert-butylester-4-methylester (JR3195).
Piperidin-1,4-dicarbonsäure-1-tert-butylester-4-methylester (JR3195). 4-Hydroxymethyl-piperidin-1-carbonsäure-tert-butylester (JR3199).
4-Hydroxymethyl-piperidin-1-carbonsäure-tert-butylester (JR3199). 4-Prop-2-ynyl-piperazin-1-carbonsäureethylester (JR3211).
4-Prop-2-ynyl-piperazin-1-carbonsäureethylester (JR3211). 4-Prop-2-ynyl-piperidin-1-carbonsäure-tert-butylester (JR3257).
4-Prop-2-ynyl-piperidin-1-carbonsäure-tert-butylester (JR3257). 4-Prop-2-ynyl-piperidin-1-carbonsäureethylester (JR3267B).
4-Prop-2-ynyl-piperidin-1-carbonsäureethylester (JR3267B). 2-(4-Prop-2-ynyl-piperazin-1-yl)-pyrimidin (JR3277).
2-(4-Prop-2-ynyl-piperazin-1-yl)-pyrimidin (JR3277). 1-(4-Prop-2-ynyl-piperidin-1-yl)-ethanon (JR4037).
1-(4-Prop-2-ynyl-piperidin-1-yl)-ethanon (JR4037). 2,2-Dimethyl-1-(4-prop-2-ynyl-piperidin-1-yl)-propan-1-on (JR4039).
2,2-Dimethyl-1-(4-prop-2-ynyl-piperidin-1-yl)-propan-1-on (JR4039).