-
Zusammenfassung der Erfindung
-
Die
vorliegende Erfindung betrifft eine neue Synthese zur Herstellung
von para-Phenylalkynylbenzaldehyd der allgemeinen Formel (I). Die
Verbindungen der Formel (I) sind nützliche Bausteine, insbesondere
bei der Synthese von Arzneimitteln und elektrisch leitenden Polymeren.
-
Gebiet der Erfindung
-
Die
vorliegende Erfindung betrifft eine neue Synthese zur Herstellung
von para-Phenylalkynylbenzaldehyden der allgemeinen Formel (I):
wobei R aus der Gruppe, die
aus C
1-C
12 Alkyl,
C
1-C
12 Alkylaryl,
C
1-C
12 Alkylheteroaryl,
C
2-C
12 Alkenyl,
C
2-C
12 Alkenylaryl,
C
2-C
12 Alkenylheteroaryl, C
2-C
12 Alkynyl, C
2-C
12 Alkynylaryl, C
2-C
12 Alkynylheteroaryl, C
1-C
12 Alkyl-C
3-C
8 Cycloalkyl, C
3-C
8 Cycloalkyl, C
1-C
12 Alkoxy, Aryl, Heteroaryl, Halogenide besteht,
ausgewählt
wird.
-
Das
Verfahren verwendet handelsübliche
oder leicht erhältliche
Ausgangsverbindungen und umfasst oder besteht aus vier Schritte(n).
-
Hintergrund der Erfindung
-
Der
synthetische Ansatz zur Herstellung von para-Phenylalkynylbenzaldehyden ist gut bekannt.
Mehrere Dokumente zitieren die Verwendung von para-Phenylalkynylbenzaldehyden
als Bausteine in der Synthese von verschiedenen Verbindungen, zum
Beispiel für
die Synthese von elektrisch leitenden Polymeren.
-
Eine
japanische Anmeldung (
JP 07138196 ,
veröffentlicht
am 30. Mai 1995) beschreibt zum Beispiel das folgende spezifische
Verfahren. Das Verfahren schließt
die Verwendung eines Palladiumkatalysators bei zwei getrennten Schritten
ein. Schema
1
-
Eine
weitere Anmeldung, die para-Phenylalkynylbenzaldehyd betrifft, ist
PCT/EP03/00808 (Prioritätsdatum:
29. Januar 2002). Sie schließt
ebenfalls die Verwendung eines Palladiumkatalysators ein und offenbart den
folgenden spezifischen Weg zur Synthese von para-Phenylalkynylbenzaldehyd: Schema
2
-
Die
im Stand der Technik verwendeten Verfahren schließen die
Verwendung von teuren Palladiumkatalysatoren ein. Des Weiteren verursacht
die Verwendung von Palladiumkatalysatoren eine Palladiumverunreinigung
und häufig
die Bildung von unerwünschten
Nebenprodukten. Die vorliegende Erfindung stellt ein neues Verfahren
bereit, das nicht die Verwendung von Palladiumkatalysatoren erfordert.
-
Beschreibung der Erfindung
-
Die
vorliegende Erfindung ermöglicht
es, die oben genannten Probleme durch eine Synthese zu überwinden,
die vier Schritte einschließt
und außerdem
als Ausgangsverbindungen Verbindungen verwendet, die leicht synthetisiert
werden können
oder handelsüblich
sind.
-
Die
folgenden Paragraphen stellen Definitionen der verschiedenen chemischen
Einheiten, die die Verbindungen gemäß der Erfindung bilden, bereit
und sind dazu bestimmt, überall
in der Beschreibung und den Ansprüchen einheitlich verwendet
zu werden, wenn keine ausdrücklich
anders festgelegt Definition eine breitere Definition bereitstellt.
-
"C1-C12 Alkyl" betrifft
Alkylgruppen mit 1 bis 12 Kohlenstoffatomen. Dieser Begriff wird
durch Gruppen, wie zum Beispiel Methyl, Ethyl, n-Propyl, Isopropyl,
n-Butyl, Isobutyl, tert-Butyl, n-Hexyl, Heptyl, Octyl, Nonyl und ähnliche
beispielhaft erläutert.
-
"C1-C12 Alkylaryl" betrifft C1-C12 Alkylgruppen mit einem Arylsubstituenten,
einschließlich
Benzyl, Phenyl und ähnliche.
-
"Aryl" betrifft eine ungesättigte aromatische
carbocyclische Gruppe mit 6 bis 14 Kohlenstoffatomen mit einem einzelnen
Ring (zum Beispiel Phenyl) oder mehreren kondensierten Ringen (zum
Beispiel Naphthyl). Bevorzugt beinhaltet Aryl Phenyl, Naphthyl,
Phenantrenyl und ähnliche.
-
"Heteroaryl" betrifft eine monocyclische
heteroaromatische oder eine bicyclische oder eine tricyclische heteroaromatische
Gruppe mit kondensierten Ringen. Besondere Beispiele von heteroaromatischen
Gruppen beinhalten optional substituiertes Pyridyl, Pyrrolyl, Furyl,
Thienyl, Imidazolyl, Oxazolyl, Isoxazolyl, Thiazolyl, Isothiazolyl,
Pyrazolyl, 1,2,3-Triazolyl, 1,2,4-Triazolyl, 1,2,3-Oxadiazolyl,
1,2,4-Oxadiazolyl,
1,2,5-Oxadiazolyl, 1,3,4-Oxadiazolyl, 1,3,4-Triazinyl, 1,2,3-Triazinyl, Benzofuryl,
[2,3-Dihydro]benzofuryl,
Isobenzofuryl, Benzothienyl, Benzotriazolyl, Isobenzothienyl, Indolyl,
Isoindolyl, 3H-Indolyl,
Benzimidazolyl, Imidazo[1,2-a]pyridyl, Benzothiazolyl, Benzoxazolyl,
Chinolizinyl, Chinazolinyl, Pthalazinyl, Chinoxalinyl, Cinnolinyl,
Napthyridinyl, Pyrido[3,4-b]pyridyl, Pyrido[3,2-b]pyridyl, Pyrido[4,3-b]pyridyl, Chinolyl,
Isochinolyl, Tetrazolyl, 5,6,7,8-Tetrahydrochinolyl,
5,6,7,8-Tetrahydroisochinolyl, Purinyl, Pteridinyl, Carbazolyl,
Xanthenyl oder Benzochinolyl.
-
"Halogen" betrifft Fluor-,
Chlor-, Brom- und Iodatome.
-
"C1-C12 Alkylheteroaryl" betrifft C1-C12 Alkylgruppen mit einem Heteroarylsubstituenten,
einschließlich 2-Furylmethyl,
2-Thienylmethyl,
2-(1H-Indol-3-yl)ethyl und ähnliche.
-
"C2-C12 Alkenyl" betrifft Alkenylgruppen mit bevorzugt
2 bis 12 Kohlenstoffatomen und mit mindestens 1 oder 2 Stellen von
Alkenylungesättigtheit.
Diese Alkenylgruppen beinhalten (-CH=CH2),
n-2-Propenyl (Allyl, -CH2CH=CH2)
und ähnliche.
-
"C2-C12 Alkenylaryl" betrifft C2-C12 Alkenylgruppen mit einem Arylsubstituenten,
einschließlich
2-Phenylvinyl und ähnliche.
-
"C2-C12 Alkenylheteroaryl" betrifft C2-C12 Alkenylgruppen mit einem Heteroarylsubstituenten,
einschließlich
2-(3-Pyridinyl)vinyl
und ähnliche.
-
"C2-C12 Alkynyl" betrifft Alkynylgruppen mit bevorzugt
2 bis 12 Kohlenstoffatomen und mit mindestens 1–2 Stellen an Alkynylungesättigtheit,
bevorzugte Alkynylgruppen beinhalten Ethynyl(-C≡CH), Propargyl(-CH2C≡CH)
und ähnliche.
-
"C2-C12 Alkynylaryl" betrifft C2-C12 Alkynylgruppen mit einem Arylsubstituenten,
einschließlich
Phenylethynyl und ähnliche.
-
"C2-C12 Alkynylheteroaryl" betrifft C2-C12 Alkynylgruppen mit einem Heteroarylsubstituenten,
einschließlich
2-Thienylethynyl
und ähnliche.
-
"C3-C8 Cycloalkyl" betrifft eine gesättigte carbocyclische Gruppe
mit 3 bis 8 Kohlenstoffatomen mit einem einzelnen Ring (zum Beispiel
Cyclohexyl) oder mehreren kondensierten Ringen (zum Beispiel Norbornyl). Bevorzugt
beinhaltet Cycloalkyl Cyclopentyl, Cyclohexyl, Norbornyl und ähnliche.
-
"C1-C12 Cycloalkyl" betrifft C1-C12 Alkylgruppen mit einem Cycloalkylsubstituenten,
einschließlich
Cyclohexylmethyl, Cyclopentylpropyl und ähnliche.
-
"Alkoxy" betrifft die Gruppe
-OR, wobei R "C1-C6 Alkyl", "C2-C6 Alkenyl", "C2-C6 Alkynyl", "C3-C8 Cycloalkyl", "Heterocycloalkyl", "Aryl", "Heteroaryl", "C1-C6 Alkylaryl" oder "C1-C6 Alkylheteroaryl", "C2-C6 Alkenylaryl", "C2-C6 Alkenylheteroaryl", "C2-C6 Alkynylaryl", "C2-C6 Alkynylheteroaryl", "C1-C6 Alkylcycloalkyl", "C1-C6 Alkylheterocycloalkyl".
-
Das
Verfahren gemäß der vorliegenden
Erfindung umfasst und besteht aus den folgenden Schritten 1 bis
4:
Gemäß der Erfindung
kann der Baustein von Formel (I) ausgehend entweder von einer Verbindung
der allgemeinen Formel (II) oder von einer Verbindung der allgemeinen
Formel (III) hergestellt werden, wobei LG eine geeignete Abgangsgruppe
ist. Die Verbindungen (II) und (III) (zum Beispiel Bromid, Chlorid,
Iodid) sind handelsüblich
oder können
gemäß bekannter
Techniken hergestellt werden.
-
Schritt
1: Ein Acylchlorid (III) wird mit einem substituierten Benzol der
Formel (IV) gekoppelt, wobei R aus der Gruppe, die aus C1-C12 Alkyl, C1-C12 Alkylaryl,
C1-C12 Alkylheteroaryl,
C2-C12 Alkenyl,
C2-C12 Alkenylaryl,
C2-C12 Alkenylheteroaryl,
C2-C12 Alkynyl,
C2-C12 Alkynylaryl,
C2-C12 Alkynylheteroaryl,
C3-C8 Cycloalkyl, C1-C12 Alkoxy, Aryl,
Heteroaryl oder einem Halogenid besteht, ausgewählt wird, wobei somit ein Keton
der Formel (V) erhalten wird.
-
LG
ist eine geeignete Abgangsgruppe, wie zum Beispiel ein Halogenid
(Br, Cl, I). Schema
3
-
Bevorzugt
wird die Reaktion in Gegenwart einer Lewis-Säure (zum Beispiel FeCl3, AlCl3) in einem Temperaturbereich
von Raumtemperatur bis 50°C,
typischerweise über
einen Zeitraum von 5 Stunden durchgeführt.
-
Die
Acylchlorid-Ausgangsverbindung (III) in Schema 3 wird typischerweise
durch Umsetzen der Säure (II)
mit einem geeigneten Chlorierungsmittel, zum Beispiel Thionylchlorid,
Oxylylchlorid, PCl
3 oder PCl
5,
erhalten. Schema
4
-
Schritt
2: Dann wird die Verbindung der Formel (V) in eine Verbindung (VI)
unter Verwendung eines geeigneten Halogenierungsmittels, einschließlich Acylchloriden,
zum Beispiel Acetylchlorid, Bromid, umgewandelt. Schema
5
Hal ist Br, Cl.
-
Bevorzugt
wird die Umsetzung mit einem Acylchlorid in einem sauren organischen
Lösungsmittel
(TFA oder Methansulfonsäure),
bevorzugt TFA bei Raumtemperatur, typischerweise über einen
Zeitraum von 40 Stunden durchgeführt.
-
Schritt
3: Die Verbindung (VI) wird dann durch Eliminierung von HCl, bevorzugt
ein einem alkalischen Medium, in eine Verbindung (VII) umgewandelt
(Dehydrohalogenierung). Schema
6
-
Bevorzugt
wird die Umsetzung in einem organischen Lösungsmittel (zum Beispiel einer
Mischung von Dioxan und Methanol), typischerweise in Gegenwart einer
Base (bevorzugte Basen beinhalten NaOH und KOH) bei einer Temperatur
von 80°C,
typischerweise über
einen Zeitraum von 20 Stunden, durchgeführt.
-
Schritt
4: In einem letzten Schritt wird eine Verbindung der Formel (VII)
mit einem Formylierungsagenz (VIII) umgesetzt, um die Verbindung
(I) zu ergeben. In einer Ausführungsform
wird eine Verbindung der Formel (VII), bei der LG ein Halogenid
ist, zuerst in eine aktivierte Spezies, zum Beispiel ein Organometallderivat,
wie zum Beispiel ein Organomagnesium oder Organolithium, unter Verwendung
von Magnesium bzw. Butyllithium umgewandelt. Die aktivierte Spezies,
zum Beispiel das Organometall-Derivat, wird dann in das Aldehyd
der Formel (I) durch Umsetzen mit einem Formylierungsmittel, wie
zum Beispiel DMF, 1-Formylpiperidin, 1-Formylpiperazin, N-Methyl-N-(2-pyridyl)formamid,
N-Methylformanilid,
Weinreb-Formamid (zum Beispiel N-Methoxy-N-methylformamid), umgewandelt. Das Zwei-Stufen-Protokoll
kann in einem Gefäß oder nacheinander durchgeführt werden.
-
In
einer Ausführungsform
wird eine Verbindung (VII) bereitgestellt; Mg in einem organischen
Lösungsmittel,
wie zum Beispiel THF, als auch 1-Formylpiperidin werden hinzugegeben,
um die Ein-Top-Reaktion durchzuführen.
-
In
einer weiteren Ausführungsform
wird eine Verbindung (VII) bereitgestellt; n-Butyllithium in THF
als auch DMF als Formylierungsmittel werden hinzugegeben, um die
Ein-Topf-Reaktion
durchzuführen. Schema
7
-
In
einer spezifischen Ausführungsform
ermögliche
das neue Verfahren die Herstellung von Verbindungen gemäß der Formel
(I), wobei R C1-C6 Alkyl
(zum Beispiel ein Methyl-, Ethyl-, n-Propyl-, Isopropyl-, n-Butyl-, Isobutyl-,
tert-Butyl-, n-Hexylrest)
ist.
-
Der
neue synthetische Ansatz zur Herstellung der Verbindungen der Formel
(I) hat den Vorteil, dass er nicht die Verwendung von Palladium
beinhaltet.
-
Die
vorliegende Erfindung soll mittels der folgenden Beispiele veranschaulicht
werden. Es ist verständlich,
dass dort wo typische oder bevorzugte experimentelle Bedingungen
(d. h. Reaktionstemperaturen, Zeit, Molzahl der Reagenzien, Lösungsmittel
etc.) gegeben sind, ebenfalls andere experimentelle Bedingungen
verwendet werden können,
wenn nicht anders aufgezeigt. Optimale Reaktionsbedingungen können mit
den bestimmten Reaktanten oder verwendeten Lösungsmitteln variieren, aber
diese Bedingungen können
vom Durchschnittsfachmann durch routinemäßige Optimierungsverfahren
bestimmt werden.
-
Beispiel 1: Herstellung von 4-(4-Methoxyphenylethynyl)-benzaldehyd
-
a)
Synthese von 4-(Bromphenyl)acetylchlorid (IIIa)
-
In
einem Kolben, der mit einer HCl-Falle versehen ist, wird SOCl
2 (495 ml; 3 vols) in (4-Bromphenyl)essigsäure (IIA)
(165 g; 767.28 mmol) gegeben. Die Reaktionsmischung wird bei 60°C für 3 h gerührt. Dann
wird sie unter Vakuum aufkonzentriert und mit Toluol (100 ml) co-verdampft.
Das resultierende hellbraune Öl
wurde unter Vakuum für
48 h geschützt
vor Licht unter Verwendung einer Aluminiumfolie getrocknet. Die
Titelverbindung (m = 178.20 g) wurde als Öl in einer Ausbeute von 99.5%
erhalten. b)
Synthese von 2-(4-Bromphenyl)-1-(4-methoxyphenyl)ethanon (Va)
-
In
einen 50 ml Dreihalskolben, der AlCl
3 (4.406
g, 33.05 mmol) unter N
2 enthält, wurde
Anisol (IVa) (4.467 g, 41.31 mmol) in einer Portion bei RT hinzugegeben.
Die Reaktion war exotherm. Zu dieser Suspension wurde (4-Bromphenyl)acetylchlorid
(IIIa) (6.430 g; 27.54 mmol) tropfenweise hinzugegeben, wobei die Temperatur
unterhalb 20°C
gehalten wurde. Dann wurde die resultierende rote Suspension bei
RT für
3 h 30 gerührt.
Die dicke rote Lösung
wurde unter Rühren
in eine Mischung aus Eis und 1N HCl (100 ml) gegossen, dann wurde
der resultierende weiße
Feststoff filtriert und mit Wasser gewaschen. Der Feststoff wurde
mit Pentan (3 × 30
ml) gewaschen und unter Vakuum bei Raumtemperatur getrocknet, um
ein weißes
Pulver (m = 8.51 g) zu ergeben. Die Reinigung wurde durch Kristallisation
aus Aceton (30 ml) durchgeführt,
um die Titelverbindung als ein weißes Pulver (m = 6.113 g) in
73% Ausbeute zu ergeben.
1H-NMR (CDCl
3 = 7.26 ppm): 7.97 (d, J=8.85 Hz, 2H), 7.44
(d, J=8.28 Hz, 2H), 7.13 (d, J=8.28 Hz, 2H), 6.93 (d, J=8.85 Hz,
2H), 4.18 (s, 2H), 3.86 (s, 3H).
Schmelzpunkt: 142°C c)
Schritt 2: Synthese von 4-[(2)-2-(4-Bromphenyl)-1-chlorvinyl]phenylmethylether
(VIa)
-
In
einem 100 ml Kolben wurden TFA (15 ml; 197.30 mmol) und Acetylchlorid
(11.17 ml; 157.81 mmol) in einer Portion zu 2-(4-Bromphenyl)-1-(4-methoxyphenyl)ethanon
(Va) (6.02 g; 19.73 mmol) bei RT hinzugegeben. Die pinkfarbene Reaktionsmischung
wurde bei Raumtemperatur für
20 h heftig gerührt.
Die resultierende braune Suspension wurde auf 0°C abgekühlt, filtriert und mit TFA
(2 × 10
ml) gewaschen. Der cremefarbene Feststoff wurde unter Vakuum bei
30°C getrocknet.
Die Titelverbindung (m = 5.688 g) wurde in 89% Ausbeute erhalten.
Schmelzpunkt: 97°C.
1H-NMR (CDCl
3 = 7.26
ppm): 7.60 (t, J=8.94 Hz, 4H), 7.50 (d, J=8.66 Hz, 2H), 6.92 (d,
J=8.85 Hz, 2H), 6.89 (s, 1H), 3.85 (s, 3H). d)
Schritt 3: Synthese von 4-[(4-Bromphenyl)ethynyl]phenylmethylether
(VIIa)
-
In
einen 100 ml Kolben, der eine Lösung
von 4-[(Z)-2-(4-Bromphenyl)-1-chlorvinyl]phenylmethylether (VIa)
(5.613 g; 17.34 mmol) in 1,4-Dioxan (28 ml; 5 vols) und MeOH (8
ml; 1.4 vols) enthält,
wurde KOH (1.946 g; 34.69 mmol) in einer Portion hinzugegeben. Die
Reaktionsmischung wurde bei 80°C über Nacht
gerührt.
Die Reaktionsmischung wurde in Wasser (200 ml) aufgenommen und die
resultierende Suspension wurde filtriert und mit Wasser gewaschen,
um einen weißen
Feststoff zu ergeben. Trocknen im Vakuum bei 33°C über Nacht ergab die Titelverbindung
(m = 4.786 g) in 96% Ausbeute. Schmelzpunkt: 152°C.
1H-NMR
(CDCl
3 = 7.26 ppm): 7.40 (d, J=2.26 Hz,
2H), 7.37 (d, J=2.26 Hz, 2H), 7.28 (d, J=8.47 Hz, 2H), 6.80 (d,
J=8.85 Hz, 2H), 3.75 (s, 3H). e)
Schritt 4: Synthese von 4-[(4-Methoxyphenyl)ethynyl]benzaldehyd
(Ia)
-
In
einen trocknen 100 ml Dreihalskolben, der Magnesiumspäne (0.447
g; 18.38 mmol) in trockenem THF (8 ml) enthält, wurde unter Rückfluß bei einem
N2-Strom eine kleine Portion 4-[(4-Bromphenyl)ethynyl]phenylmethylether
(VIIa) (0.300 g; 1.044 mmol) in einer Portion hinzugegeben. Der
N2-Strom wurde gestoppt. Die Reaktionsmischung
wurde unter Rückfluß für 5 Minuten
erwärmt,
dann wurden Iodkristalle hinzugegeben, während der Rückfluß aufrechterhalten wurde, um
die Reaktion zu starten. Eine Lösung
der restlichen Menge 4-[(4-Bromphenyl)ethynyl]phenylmethylether
(VIIa) (4.5 g; 15.67 mmol) in trockenem THF (30 ml) wurde tropfenweise
zu der Reaktionsmischung hinzugegeben, während ein sanfter Rückfluß gehalten
wurde. Der Rückfluß wurde
für 15
Minuten aufrechterhalten, dann ließ man die Temperatur unter
Rühren
für 1 h
auf RT abkühlen.
Die Reaktionsmischung wurde auf 3°C
abgekühlt
und eine Lösung
von trockenem 1-Formylpiperidin (2.8 ml; 25.07 mmol) in trockenem
THF (10 ml) wurde tropfenweise hinzugegeben, wobei die Temperatur
bei 5°C
gehalten wurde. Man ließ die
Reaktionsmischung dann auf RT erwärmen und es wurde über Nacht
gerührt.
Die Reaktionsmischung wurde auf 18°C abgekühlt und 3N HCl (30 ml) wurden
hinzugegeben. Wasser (50 ml) wurde hinzugegeben und eine Extraktion
wurde mit MTBE (50 ml × 3)
durchgeführt.
Die organische Phase wurde nacheinander mit Wasser (50 ml × 2), gesättigter
NaHCO3-Lösung
(50 ml × 1)
und Kochsalzlösung
(50 ml × 1)
gewaschen. Sie wurde dann über
MgSO4 getrocknet, filtriert und aufkonzentriert,
um einen gelben Feststoff zu ergeben. Er wurde in Pet-Ether (40
ml) aufgenommen und bei 4°C
stehen gelassen. Nach 16 h wurde die Suspension filtriert und mit
Pet-Ether (2 × 30
ml) gewaschen, um nach dem Trocknen unter Vakuum einen klar-gelben
Feststoff zu ergeben. Die Titelverbindung wurde in 77% Ausbeute
erhalten (m = 3.06 g). Schmelzpunkt: 106°C.
1H-NMR
(CDCl3 = 7.26 ppm): 10.0 (s, 1H), 7.85 (d,
J=8.28 Hz, 2H), 7.64 (d, J=8.28 Hz, 2H), 7.49 (d, J=8.85 Hz, 2H),
6.90 (d, J=8.85 Hz, 2H), 3.84 (s, 3H).
-
Beispiel 2: Herstellung von 4-(4-Hexylphenylethynyl)benzaldehyd
-
a)
Schritt 1: Synthese von 2-(4-Bromphenyl)-1-(4-hexylphenyl)ethanon (Vb)
-
In
einen 2 l Dreihalskolben, der mit einem mechanischen Rührer ausgestattet
ist, der AlCl
3 (85.661 g; 642.42 mmol) unter
N
2 enthält,
wurde 4-Hexylbenzol (IVb) (104.25 g; 642.42 mmol) in einer Portion
bei Raumtemperatur hinzugegeben. Zu dieser resultierenden orangefarbenen
Suspension wurde (4-Bromphenyl)acetylchlorid
(IIIa) (125.000 g; 535.35 mmol) tropfenweise innerhalb von 45 Minuten
ohne Kühlung
hinzugegeben. Das Reaktionsgemisch wurde für 3 h gerührt bis sich die Temperatur
auf Raumtemperatur abgekühlt
hatte, ein Zeitpunkt, bei dem kein weiteres Schäumen beobachtet wurde. Die
tiefbraune Mischung wurde dann bei Raumtemperatur über Nacht
gerührt.
Die schwarze, dicke Lösung
wurde unter Rühren
in eine Mischung aus Eis und 1N HCl (800 ml) gegossen, dann wurde
der resultierende weiß-orangefarbene
Feststoff filtriert und nacheinander mit Wasser, gesättigter
NaHCO
3-Lösung und
schließlich
mit Wasser gewaschen bis der pH des Filtrats 7 war. Der Feststoff
wurde mit Heptan (3 × 200
ml) gewaschen und unter Vakuum bei Raumtemperatur getrocknet, um
die Titelverbindung als ein weißes
Pulver (m = 151.15 g) in 79% Ausbeute zu ergeben. Schmelzpunkt:
108°C.
1H-NMR (CDCl
3 = 7.26
ppm): 7.91 (d, J=8.28 Hz, 2H), 7.44 (d, J=8.28 Hz, 2H), 7.26 (d,
J=8.28 Hz, 2H), 7.13 (d, J=8.47 Hz, 2H), 4.21 (s, 2H), 2.65 (t,
J=7.81 Hz, 2H), 1.62 (quint., J=7.53 Hz, 2H), 1.43-1.22 (br m, 6H), 0.88
(t, J=6.87 Hz, 3H). b)
Schritt 2: Synthese von 1-Brom-4-[(Z)-2-chlor-2-(4-hexylphenyl)vinyl]benzol
(VIb)
-
In
einem 2 l Kolben wurden TFA (464.17 ml; 6065.60 mmol) und Acetylchlorid
(344.71 ml; 4852.42 mmol) in einer Portion zu 2-(4-Bromphenyl)-1-p-tolylethanon
(Vb) (217.940 g; 606.56 mmol) bei Raumtemperatur hinzugegeben. Die
Reaktionsmischung wurde heftig bei Raumtemperatur für 40 h gerührt. Die
resultierende Suspension wurde auf 0°C abgekühlt, filtriert und mit TFA
(100 ml) gewaschen. Der weiße
Feststoff wurde unter Vakuum bei 30°C getrocknet. Die Titelverbindung
(m = 209.79 g) wurde in 93% Ausbeute erhalten. Schmelzpunkt: 52°C.
1H-NMR (CDCl
3 = 7.26
ppm): 7.61 (d, J=3.01 Hz, 2H), 7.58 (d, J=3.01 Hz, 2H), 7.51 (d,
J=8.66 Hz, 2H), 7.21 (d, J=8.47 Hz, 2H), 6.96 (s, 1H), 2.63 (t,
J=7.81 Hz, 2H), 1.70-1.53 (br m, 2H), 1.45-1.20 (br m, 6H), 0.89
(t, J=6.87 Hz, 3H). c)
Schritt 3: Synthese von 1-Brom-4-[(4-hexylphenyl)ethynyl]benzol
(VIIb)
-
In
einen 2 l Kolben, der eine Lösung
von 1-Brom-4-[(Z)-2-chlor-2-(4-hexylphenyl)vinyl]benzol
(VIb) (209.79 g; 555.37 mmol) in 1,4-Dioxan (1000 ml; 4.8 vols)
und MeOH (300 ml; 1.4 vols) enthält,
wurde KOH (62.32 g; 1110.73 mmol) in einer Portion hinzugegeben.
Die Reaktionsmischung wurde über
Nacht bei 80°C gerührt. Das
Volumen wurde unter Vakuum auf 200 ml reduziert und der Rest wurde
in Wasser (2000 ml) aufgenommen. Die resultierende Suspension wurde
filtriert und mit Wasser gewaschen, um einen klar-beigefarbene Feststoff
zu ergeben. Trocknen unter Vakuum bei 33°C über Nacht ergab die Titelverbindung
(m = 173.34 g) in 92% Ausbeute. Schmelzpunkt: 67°C.
1H-NMR
(CDCl
3 = 7.26 ppm): 7.47 (d, J=8.66 Hz,
2H), 7.43 (d, J=8.10 Hz, 2H), 7.37 (d, J=8.28 Hz, 2H), 7.16 (d,
J=8.10 Hz, 2H), 2.61 (t, J=7.81 Hz, 2H), 1.59 (quint., J=7.48 Hz,
2H), 1.42-1.21 (br s, 6H), 0.88 (t, J=6.31 Hz, 3H). d)
Schritt 4: Synthese von 4-(4-Hexylphenylethynyl)benzaldehyd (Ib)
-
In
einen trockenen unter einem N2-Strom stehenden
2 l Dreihalskolben, der Magnesiumspäne (13.579 g; 558.69 mmol)
in trockenem THF (165 ml) unter Rückfluß (Temperatur des Ölbads von
85°C) enthält, wurde eine
aktivierende, kleine Menge von 1-Brom-4-[(4-hexylphenyl)ethynyl]benzol
(VIIb) (10.400 g; 30.474 mmol) in einer Portion gegeben. Der N2-Strom und das Rühren wurden gestoppt. Die Reaktionsmischung
wurde unter Rückfluß für 5 Minuten
erwärmt,
dann wurden mehrere Iodkristalle hinzugegeben, wobei ein heftiger
Rückfluß aufrechterhalten
wurde, um die Reaktion zu starten. Die Reaktionsmischung wurde farblos
nach 5 Minuten und nach einer weiteren Minute wurde die Reaktionsmischung
schwarz-grün.
Eine Lösung
der restlichen Menge von 1-Brom-4[(4-hexylphenyl)ethynyl]benzol (VIIb) (162.94
g; 477.42 mmol) in trockenem THF (360 ml) wurde tropfenweise über 40 Minuten
in die Reaktionsmischung hinzugegeben, während ein leichter Rückfluß gehalten
wurde. Der Rückfluß wurde
für 20
Minuten aufrechterhalten, dann ließ man die Temperatur unter
Rühren innerhalb
von 2 h 30 auf Raumtemperatur abkühlen. Die Reaktionsmischung
wurde auf 3°C
abgekühlt
und eine Lösung
von trockenem 1-Formylpiperidin (84.60 ml; 761.85 mmol) in trockenem
THF (360 ml) wurde tropfenweise über
1 h hinzugegeben, wobei die Temperatur bei 5°C (Maximaltemperatur: 7.3°C) gehalten
wurde. Man ließ die
Reaktionsmischung auf Raumtemperatur erwärmen und es wurde über Nacht
gerührt.
Die Reaktionsmischung wurde auf 18°C abgekühlt und 3N HCl (300 ml) wurden
hinzugegeben bis die Lösung
sauer war (pH = 1). Wasser wurde hinzugegeben (500 ml) und eine
Extraktion wurde mit MTBE (500 ml × 3) wurde durchgeführt. Die
organische Phase wurde nacheinander mit Wasser (500 ml × 2), gesättigter
NaHCO3-Lösung
(500 ml × 1)
und Kochsalzlösung
(500 ml × 1)
gewaschen. Sie wurde dann über
MgSO4 getrocknet, filtriert und aufkonzentriert,
um einen orangefarbenen Feststoff zu ergeben. Er wurde in Pet-Ether (400 ml) aufgenommen und
bei 4°C
stehen gelassen. Nach 16 h wurde die Suspension filtriert und mit
Pet-Ether (2 × 300
ml) gewaschen, um nach dem Trocknen unter Vakuum den ersten Ertrag
m = 105.76 g zu ergeben. Das Filtrat wurde aufkonzentriert und in
Pet-Ether (100 ml) aufgenommen. Der resultierende Feststoff wurde
mit Pet-Ether (2 × 100
ml) gewaschen und getrocknet, um den zweiten Ertrag m = 6.0 g zu
ergeben. Die Titelverbindung wurde in 76% Ausbeute als ein weißer Feststoff
erhalten (m = 111.76 g). Schmelzpunkt: 80°C.
1H-NMR
(DMSO = 2.49 ppm): 10.0 (s, 1H), 7.93 (d, J=8.28 Hz, 2H), 7.74 (d,
J=8.28 Hz, 2H), 7.5 (d, J=8.28 Hz, 2H), 7.26 (d, J=8.28 Hz, 2H),
2.60 (t, J=7.81 Hz, 2H), 1.56 (quint., J=7.44 Hz, 2H), 1.36-1.16
(br s, 6H), 0.84 (t, J=6.78 Hz, 3H).
-
Beispiel 3: Herstellung von 4-(4-Ethylphenylethynyl)-benzaldehyd
-
a)
Schritt 1: Synthese von 2-(4-Bromphenyl)-1-(4-ethylphenyl)ethanon (Vc)
-
In
einen 50 ml Dreihalskolben, der AlCl
3 (7.305
g; 54.79 mmol) unter N
2 enthält, wurde
Ethylbenzol (IVc) (8.40 ml; 68.48 mmol) in einer Portion bei RT
hinzugegeben. Zu dieser Suspension wurde (4-Bromphenyl)acetylchlorid
(IIIa) (10.66 g; 45.65 mmol) tropfenweise hinzugegeben, wobei die
Temperatur unterhalb 40°C
gehalten wurde. Das Protokoll und die Aufarbeitung war dann dem
oben beschriebenen ähnlich.
Die Titelverbindung wurde als ein weißes Pulver (m = 9.923 g) in
68% Ausbeute erhalten. Schmelzpunkt: 146°C.
1H-NMR
(CDCl
3 = 7.26 ppm): 7.92 (d, J=7.91 Hz,
2H), 7.44 (d, J=8.47 Hz, 2H), 7.28 (d, J=8.10 Hz, 2H), 7.13 (d,
J=8.28 Hz, 2H), 4.21 (s, 2H), 2.70 (q, J=7.59 Hz, 2H), 1.25 (t,
J=7.62 Hz, 3H). b)
Schritt 2: Synthese von 1-Brom-4-[(Z)-2-chlor-2-(4-ethylphenyl)vinyl]benzol
(VIc)
-
In
einen 100 ml Kolben wurden TFA (24.7 ml; 322.8 mmol) und Acetylchlorid
(18.34 ml; 258.23 mmol) in einer Portion zu 2-(4-Bromphenyl)-1-(4-ethylphenyl)ethanon
(Vc) (9.787 g; 32.28 mmol) bei Raumtemperatur hinzugegeben. Das
Protokoll und die Aufarbeitung war dann dem oben beschriebenen ähnlich.
Die Titelverbindung (m = 9.60 g) wurde in 92% Ausbeute erhalten.
Schmelzpunkt: 75°C.
1H-NMR (CDCl
3 = 7.26
ppm): 7.60 (d, J=7.53 Hz, 4H), 7.51 (d, J=8.66 Hz, 2H), 7.23 (d,
J=8.28 Hz; 2H), 6.95 (s, 1H), 2.68 (q, J=7.59 Hz, 2H), 1.26 (t,
J=7.53 Hz, 3H). c)
Schritt 4: Synthese von 1-Brom-4-[(4-ethylphenyl)ethynyl]benzol
(VIIc)
-
In
einen 100 ml Kolben, der eine Lösung
von 1-Brom-4-[(Z)-2-chlor-2-(4-ethylphenyl)vinyl]benzol
(VIc) (9.540 g; 29.66 mmol) in 1,4-Dioxan (48 ml; 5 vols) und MeOH
(14 ml; 1.5 vols) enthält,
wurde KOH (3.328 g; 59.32 mmol) in einer Portion hinzugegeben. Das
Protokoll und die Aufarbeitung war dann dem oben beschriebenen ähnlich.
Die Titelverbindung (m = 8.39 g) wurde in 99% Ausbeute erhalten.
Schmelzpunkt: 117°C.
1H-NMR (CDCl
3 = 7.26
ppm): 7.47 (d, J=8.66 Hz, 2H), 7.44 (d, J=8.28 Hz, 2H), 7.37 (d,
J=8.47 Hz, 2H), 7.18 (d, J=8.10 Hz, 2H), 2.66 (q, J=7.59 Hz, 2H),
1.24 (t, J=7.62 Hz, 3H). d)
Schritt 4: Synthese von 4-[(4-Ethylphenyl)ethynyl]-benzaldehyd (Ic)
-
In
einen trockenen unter einem N2-Strom stehenden
100 ml Dreihalskolben, der Magnesiumspäne (0.782 g; 32.17 mmol) in
trockenem THF (10 ml) unter Rückfluß enthält, wurde
eine aktivierende, kleine Menge von 1-Brom-4-[(4-ethylphenyl)ethynyl]benzol (VIIc) (0.500
g; 1.75 mmol) in einer Portion gegeben. Der N2-Strom
und das Rühren
wurden gestoppt. Die Reaktionsmischung wurde unter Rückfluß für 5 Minuten
erwärmt,
dann wurde ein Iodkristall hinzugegeben, wobei ein heftiger Rückfluß aufrechterhalten
wurde, um die Reaktion zu starten. Die Reaktionsmischung wurde farblos
nach 5 Minuten und nach einer weiteren Minute wurde die Reaktionsmischung
schwarz-grün.
Eine Lösung
der restlichen Menge von 1-Brom-4[(4-ethylphenyl)ethynyl]benzol
(VIIc) (7.84 g; 27.49 mmol) in trockenem THF (30 ml) wurde tropfenweise
in die Reaktionsmischung hinzugegeben, während ein leichter Rückfluß gehalten
wurde. Der Rückfluß wurde
für 15
Minuten aufrechterhalten, dann ließ man die Temperatur unter
Rühren
innerhalb von 1 h auf Raumtemperatur abkühlen. Die Reaktionsmischung
wurde auf 3°C
abgekühlt
und eine Lösung
von trockenem 1-Formylpiperidin (4.12 ml; 43.87 mmol) in trockenem
THF (25 ml) wurde tropfenweise hinzugegeben, wobei die Temperatur
bei 5°C gehalten
wurde. Man ließ die Reaktionsmischung
auf Raumtemperatur (RT) erwärmen
und es wurde über Nacht
gerührt.
Das Protokoll und die Aufarbeitung war dann dem oben beschriebenen ähnlich.
Die Titelverbindung (m = 5.77 g) wurde als ein cremefarbener Feststoff
in 84% Ausbeute erhalten. Schmelzpunkt: 89°C.
1H-NMR
(CDCl3 = 7.26 ppm): 10.0 (s, 1H), 7.86 (d,
J=8.28 Hz, 2H), 7.66 (d, J=8.28 Hz, 2H), 7.47 (d, J=8.28 Hz, 2H),
7.21 (d, J=7.91 Hz, 2H), 2.68 (q, J=7.59 Hz, 2H), 1.25 (t, J=7.62
Hz, 3H).
-
Beispiel 4: Herstellung von 4-(4-Chlorphenylethynyl)benzaldehyd
-
a)
Schritt 1: Synthese von 2-(4-Bromphenyl)-1-(4-chlorphenyl)ethanon (Vd)
-
In
einen 100 ml Dreihalskolben, der AlCl
3 (4.797
g; 35.98 mmol) unter N
2 enthält, wurde
Chlorbenzol (IVd) (36.6 ml; 359.76 mmol) in einer Portion bei RT
hinzugegeben. Zu dieser Suspension wurde (4-Bromphenyl)acetylchlorid
(IIIa) (7.0 g; 29.98 mmol) in einer Portion ohne Kühlen hinzugegeben.
Das Protokoll und die Aufarbeitung war dann dem oben beschriebenen ähnlich.
Die Titelverbindung wurde als ein weißes Pulver (m = 7.99 g) in
86% Ausbeute erhalten. Schmelzpunkt: 123°C.
1H-NMR
(CDCl
3 = 7.26 ppm): 7.92 (d, J=8.66 Hz,
2H), 7.46 (d, J=4.89 Hz, 2H), 7.43 (d, J=5.27 Hz, 2H), 7.12 (d,
J=8.47 Hz, 2H), 4.21 (s, 2H). b)
Schritt 2: Synthese von 1-Brom-4-[(Z)-2-chlor-2-(4-chlorphenyl)vinyl]benzol
(VId)
-
In
einem 250 ml Kolben wurden TFA (24.7 ml; 322.8 mmol) und Acetylchlorid
(18.34 ml; 258.23 mmol) in einer Portion zu 2-(4-Bromphenyl)-1-(4-chlorphenyl)ethanon
(Vd) (10.0 g; 32.30 mmol) bei RT hinzugegeben. Das Protokoll und
die Aufarbeitung war dann dem oben beschriebenen ähnlich.
Die Titelverbindung (m = 7.89 g) wurde in 74.5% Ausbeute erhalten.
Schmelzpunkt: 108°C.
1H-NMR (CDCl
3 = 7.26
ppm): 7.62 (d, J=4.70 Hz, 2H), 7.59 (d, J=4.52 Hz, 2H), 7.52 (d,
J=8.66 Hz, 2H), 7.37 (d, J=8.85 Hz, 2H), 6.96 (s, 1H). c)
Schritt 3: Synthese von 1-Brom-4-[(4-chlorphenyl)ethynyl]benzol
(VIId)
-
In
einen 100 ml Kolben, der eine Lösung
von 1-Brom-4-[(Z)-2-chlor-2-(4-chlorphenyl)vinyl]benzol
(VId) (7.89 g; 24.05 mmol) in 1,4-Dioxan (40 ml; 5 vols) und MeOH
(12 ml; 1.5 vols) enthält,
wurde KOH (2.699 g; 48.10 mmol) in einer Portion hinzugegeben. Das
Protokoll und die Aufarbeitung war dann dem oben beschriebenen ähnlich.
Die Titelverbindung (m = 6.598 g) wurde in 94% Ausbeute erhalten.
Schmelzpunkt: 179°C.
1H-NMR (CDCl
3 = 7.26
ppm): 7.48 (d, J=8.47 Hz, 2H), 7.44 (d, J=8.66 Hz, 2H), 7.37 (d,
J=8.47 Hz, 2H), 7.32 (d, J=8.66 Hz, 2H). d)
Schritt 4: Synthese von 4-[(4-Clorphenyl)ethynyl]benzaldehyd (Id)
-
In
einen trockenen unter einem N2-Strom stehenden
100 ml Dreihalskolben, der Magnesiumspäne (0.595 g; 24.50 mmol) in
trockenem THF (10 ml) unter Rückfluß enthält, wurde
eine aktivierende, kleine Menge von 1-Brom-4-[(4-chlorphenyl)ethynyl]benzol (VIId) (0.39
g; 1.33 mmol) in einer Portion gegeben. Der N2-Strom
und das Rühren
wurden gestoppt. Die Reaktionsmischung wurde unter Rückfluß für 5 Minuten
erwärmt,
dann wurde ein Iodkristall hinzugegeben, wobei ein heftiger Rückfluß aufrechterhalten
wurde, um die Reaktion zu starten. Die Reaktionsmischung wurde farblos
nach 5 Minuten und nach einer weiteren Minute wurde die Reaktionsmischung
schwarz-blau. Eine Lösung
der restlichen Menge von 1-Brom-4[(4-chlorphenyl)ethynyl]benzol
(VIId) (6.104 g; 20.93 mmol) in trockenem THF (35 ml) wurde bei
55°C tropfenweise
in die Reaktionsmischung hinzugegeben, während ein leichter Rückfluß gehalten
wurde. Der Rückfluß wurde
für 15 Minuten
aufrechterhalten, dann ließ man
die Temperatur unter Rühren
innerhalb von 1 h auf RT abkühlen.
Die Reaktionsmischung wurde auf 3°C
abgekühlt
und eine Lösung
von trockenem 1-Formylpiperidin (3.71 ml; 33.40 mmol) in trockenem
THF (10 ml) wurde tropfenweise hinzugegeben, wobei die Temperatur
bei 5°C
gehalten wurde. Man ließ die Reaktionsmischung
dann auf RT erwärmen
und es wurde über
Nacht gerührt.
Das Protokoll und die Aufarbeitung war dann dem oben beschriebenen ähnlich.
Die Reinigung wurde durch Flash-Chromatographie (SiO2)
unter Verwendung von (Cyclohexan 9-Ethylacetat 1) durchgeführt. Die
Titelverbindung (m = 1.02 g) wurde als ein weißer Feststoff in 19% Ausbeute
erhalten. Schmelzpunkt: 164°C.
1H-NMR (CDCl3 = 7.26
ppm): 10.0 (s, J=–Hz,
1H), 7.87 (d, J=8.28 Hz, 2H), 7.66 (d, J=8.10 Hz, 2H), 7.48 (d, J=8.47
Hz, 2H), 7.35 (d, J=8.47 Hz, 2H).
-
Beispiel 5: Herstellung von 4-(4-Butylphenylethynyl)benzaldehyd
-
a)
Schritt 1: Synthese von 2-(4-Bromphenyl)-1-(4-butylphenyl)ethanon
(Ve)
-
In
einen 1 l Dreihalskolben, der AlCl
3 (40
g; 0.299 mol) in 1,2-Dichlorethan (600 ml) unter N
2 enthält, wurde
in einer Portion bei –30°C Butylbenzol
(IVe) (49.7 g; 0.299 mol) hinzugegeben. Zu dieser Suspension wurde
(4-Bromphenyl)acetylchlorid
(IIIa) (70 g; 0.299 mol) langsam über einen Zeitraum von 30 min
in einer solchen Geschwindigkeit hinzugegeben, das die Innentemperatur
nicht über –30°C anstieg.
Die Reaktionsmischung wurde bei dieser Temperatur für 45 min
gerührt
und in eine eiskalte Lösung
von 1.5M HCl (1000 ml) gegossen. Das Produkt wurde mit Dichlormethan
(2 × 500
ml) extrahiert, mit 10% Natriumbicarbonatlösung (500 ml), Wasser, Kochsalzlösung gewaschen
und über
Na
2SO
4 getrocknet.
Das Lösungsmittel
wurde unter verringertem Druck verdampft, um die Titelverbindung
als ein weißes
Pulver (m = 90 g) in 90.9% Ausbeute zu erhalten. Schmelzpunkt: 129.4°C–131.1°C.
1H-NMR (CDCl
3 = 7.26
ppm): 1H-NMR 7.92 (d, J=8.16 Hz, 2H), 7.45 (d, J=8.28 Hz, 2H), 7.27
(d, J=8.22 Hz, 2H), 7.14 (d, J=8.19 Hz, 2H), 4.22 (s, 2H), 2.67
(t, J=7.47 Hz, 2H), 1.62 (m, J=7.38 Hz, 2H), 1.37 (m, 2H), 0.93 (t,
J=7.29 Hz, 3H). b)
Schritt 2: Synthese von 1-Brom-4-[(Z)-2-chlor-2-(4-butylphenyl)vinyl]benzol
(VIe)
-
In
einen 2 l Dreihalskolben, der TFA (231 ml; 3.01 mol) und Acetylchlorid
(171.4 ml; 2.41 mol) enthält, wurde
in einer Portion 2-(4-Bromphenyl)-1-(4-butylphenyl)ethanon (Ve)
(100 g; 0.301 mol) bei RT hinzugegeben. Die Reaktionsmischung wurde
bei Raumtemperatur über
Nacht gerührt
und die Aufarbeitung war dem oben beschriebenen ähnlich. Die Titelverbindung
(m = 100 g) wurde in 95% Ausbeute erhalten. Schmelzpunkt: 59–61°C.
1H-NMR (CDCl
3 = 7.26
ppm): 7.6 (m,, 4H), 7.52 (d, J=8.4 Hz, 2H), 7.22 (d, J=8.01 Hz,
2H), 6.97 (s, 1H), 2.65 (t, J=7.59 Hz, 2H), 1.63 (m, J=7.41 Hz,
2H), 1.37 (m, 2H), 0.95 (t, J=7.35 Hz, 3H). c)
Schritt 3: Synthese von 1-Brom-4-[(4-butylphenylethynyl]benzol (VIIe)
-
In
einen 1 l Kolben, der eine Lösung
von 1-Brom-4-[(Z)-2-chlor-2-(4-butylphenyl)vinyl]benzol
(VIe) (100 g; 0.285 mol) in 1,4-Dioxan (500 ml; 5 vols) und MeOH
(200 ml; 2 vols) enthält,
wurde KOH (32 g; 0.571 mol) in einer Portion hinzugegeben. Das Protokoll
und die Aufarbeitung war dann dem oben beschriebenen ähnlich.
Die Titelverbindung (m = 80 g) wurde in 89% Ausbeute erhalten. Schmelzpunkt:
75.6–76.1°C.
1H-NMR (CDCl
3 = 7.26
ppm): 7.4 (m,, 6H), 7.17 (d, J=7.8 Hz, 2H), 2.63 (t, J=7.56 Hz,
2H), 1.63 (m, J=7.38 Hz, 2H), 1.38 (m, 2H), 0.95 (t, J=7.2 Hz, 3H). d)
Schritt 4: Synthese von 4-[(4-Butylphenyl)ethynylbenzaldehyd (Ie)
-
In
einen trockenen unter einem N2-Strom stehenden
2 l Dreihalskolben, der 1-Brom-4-[(4-butylphenylethynyl]benzol (VIIe)
(100 g; 0.319 mol) in trockenem THF (1000 ml) bei –78°C enthält, wurde
n-BuLi (2.5 M in Hexan, 153.25 ml, 0.383 mol) hinzugegeben und die
Reaktionsmischung wurde bei dieser Temperatur für 2 h gerührt. Die Reaktionsmischung
wurde 5 Minuten nach der Zugabe von Butyllithium dunkel-grün. Zu dieser Reaktionsmischung
wurde DMF (29.56 ml, 0.383 mol) hinzugegeben und die resultierende
Mischung würde für zusätzlich 1
h bei –78°C gerührt. Die
Reaktionsmischung wurde nach der Zugabe von DMF schwarz-blau. Die
Reaktionsmischung wurde dann mit 1.5 HCl (750 ml) bei dieser Temperatur
gequencht und das Produkt wurde mit MTBE (3 × 500 ml) extrahiert. Die vereinigte
organische Schicht wurde mit 10% Natriumcarbonat-Lösung (500
ml), Wasser, Kochsalzlösung
gewaschen und getrocknet. Das Lösungsmittel
wurde unter verringertem Druck verdampft, um die Titelverbindung
(m = 65 g) als einen weißen
Feststoff in 77% Ausbeute zu erhalten. Schmelzpunkt: 76–78°C.
1H-NMR (CDCl3 = 7.26
ppm): 10.02 (s, 1H), 7.86 (d, J=7.47 Hz, 2H), 7.66 (d, J=7.98 Hz,
2H), 7.47 (d, J=7.2 Hz, 2H), 7.19 (d, J=7.68 Hz, 2H), 2.64 (t, J=7.5
Hz, 2H), 1.63 (quint., J=7.2 Hz, 2H), 1.36 (m, 2H), 0.94 (t, J=7.2 Hz,
3H).
-
Die
folgenden weiteren Verbindungen können unter Verwendung der oben
dargelegten Protokolle erhalten werden.
Beispiel 6: 4-p-Tolylethynylbenzaldehyd
Beispiel
7: 4-(4-Propylphenylethynyl)benzaldehyd
Beispiel 8: 4-(4-Cyclohexylphenylethynyl)benzaldehyd
Beispiel
9: 4-(4-Propoxyphenylethynyl)benzaldehyd
Beispiel 10: 4-(4-Phenoxyphenylethynyl)benzaldehyd
Beispiel
11: 4-Biphenyl-4-ylethynylbenzaldehyd.










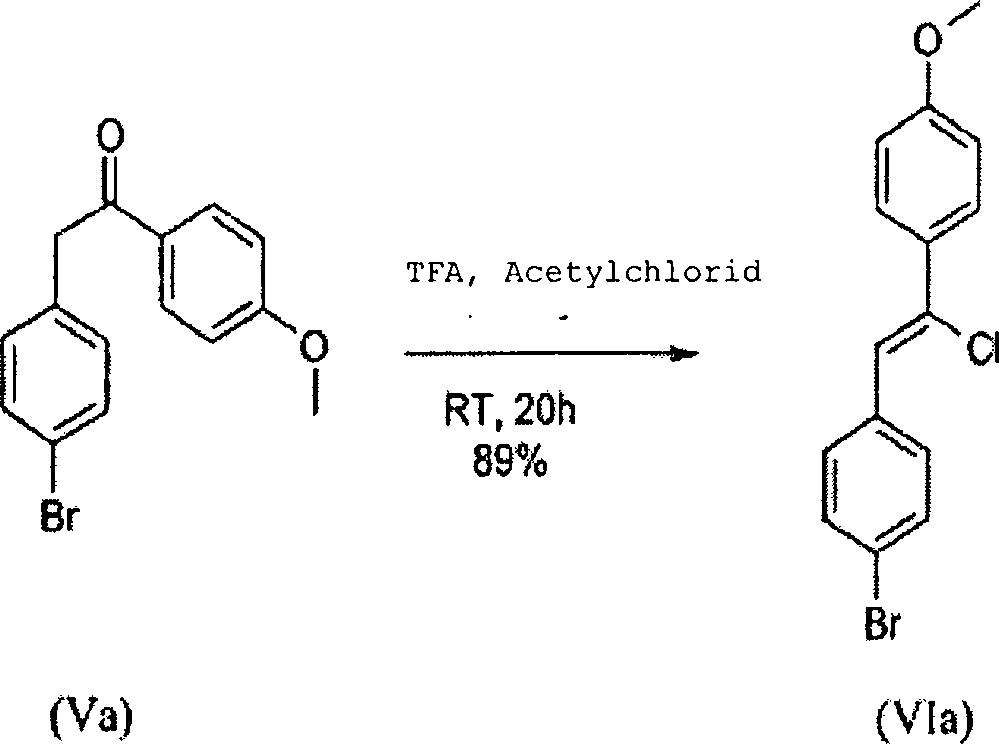


















 wobei LG eine Abgangsgruppe ist; Schritt 2: Umwandlung einer Verbindung der Formel (V) in eine Verbindung der Formel (VI) unter Verwendung eines geeigneten Halogenierungsmittels:
wobei LG eine Abgangsgruppe ist; Schritt 2: Umwandlung einer Verbindung der Formel (V) in eine Verbindung der Formel (VI) unter Verwendung eines geeigneten Halogenierungsmittels: Schritt 3: eine Verbindung (VI) einer Eliminierungsreaktion auszusetzen, um eine Verbindung der Formel (VII) bereitzustellen:
Schritt 3: eine Verbindung (VI) einer Eliminierungsreaktion auszusetzen, um eine Verbindung der Formel (VII) bereitzustellen: Schritt 4: die Verbindung der Formel (VII) oder eine aktivierte Spezies davon einem Formylierungsagenz (VIII) auszusetzen, um die Verbindung (I) zu ergeben:
Schritt 4: die Verbindung der Formel (VII) oder eine aktivierte Spezies davon einem Formylierungsagenz (VIII) auszusetzen, um die Verbindung (I) zu ergeben:
