-
Die vorliegende Erfindung betrifft
makrozyclische Pyrimidinderivate, deren Verfahren zur Herstellung sowie
deren Verwendung als Medikament zur Behandlung verschiedener Erkrankungen.
-
Die CDKs (cyclin-dependent kinase)
ist eine Enzymfamilie, die eine wichtige Rolle bei der Regulation des
Zellzyklus spielt und somit ein besonders interessantes Ziel für die Entwicklung
kleiner inhibitorischer Moleküle
ist. Selektive Inhibitoren der CDKs können zur Behandlung von Krebs
oder anderen Erkrankungen, die Störungen der Zellproliferation
zur Ursache haben, verwendet werden.
-
Pyrimidine und Analoga sind bereits
als Wirkstoffe beschrieben wie beispielsweise die 2-Anilino-Pyrimidine
als Fungizide (
DE 4029650 )
oder substituierte Pyrimidinderivate zur Behandlung von neurologischen oder
neurodegenerativen Erkrankungen (WO 99/19305). Als CDK-Inhibitoren
werden unterschiedlichste Pyrimidinderivate beschrieben, beispielsweise
Bis(anilino)pyrimidinderivate (WO 00/12486), 2-Amino-4-substituierte
Pyrimidine (WO 01/14375), Purine (WO 99/02162), 5-Cyano-Pyrimidine
(WO 02/04429), Anilinopyrimidine (WO 00/12486) und 2-Hydroxy-3-N,N-dimethylaminopropoxy-Pyrimidine
(WO 00/39101).
-
Die Aufgabe der vorliegenden Erfindung
ist es Verbindungen bereitzustellen, die bessere Eigenschaften als
die bereits bekannten Inhibitoren haben. Die hier beschriebenen
Substanzen sind besser wirksam, da sie bereits im nanomolaren Bereich
inhibieren und so von anderen bereits bekannten CDK-Inhibitoren
wie z.B. Olomoucin und Roscovitin zu unterscheiden sind.
-
Es wurde nun gefunden, dass Verbindungen
der allgemeinen Formel I
in der
A für C
3-C
12-Aryl oder C
3-C
18-Heteroaryl
steht,
B für
eine Bindung oder für
gegebenenfalls ein- oder mehrfach, gleich oder verschieden mit Hydroxy,
Halogen, Cyano, Nitro, C
1-C
6-Alkyl,
C
2-C
6-Alkenyl, C
2-C
6-Alkinyl, C
3-C
10-Cycloalkyl, C
1-C
6-Hydroxyalkyl, C
3-C
12-Aryl, C
3-C
18-Heteroaryl,
-(CH
2)
p-C
3-C
12-Aryl, -(CH
2)
p-C
3-C
18-Heteroaryl,
Phenyl-(CH
2)
p-R
10, -(CH
2)
pPO
3(R
10)
2 oder mit der Gruppe -NR
8R
9, -NR
8COR
9, -NR
8CSR
9, -NR
8SOR
9, -NR
8SO
2R
9, -NR
8CONR
9R
10, -NR
8COOR
9, -NR
8C(NH)NR
9R
10, -NR
8CSNR
9R
10, -NR
8SONR
9R
10,
-NR
8SO
2NR
9R
10, -COR
8,-CSR
8,-S(O)R
8, -S(O)
2R
8, -S(O)
2NR
8R
9, -SO
3R
8, -CO
2R
8,
-CONR
8R
9, -CSNR
8R
9, -SR
8 oder
-CR
8(OH)-R
9 substituiertes
C
1-C
12-Alkyl, C
2-C
12-Alkenyl, C
2-C
12-Alkinyl, C
3-C
8-Cycloalkyl,
C
3-C
12-Heterocycloalkyl,
C
3-C
12-Aryl oder
C
3-C
18-Heteroaryl steht,
X
und Y jeweils unabhängig
voneinander für
Sauerstoff, Schwefel oder für
die Gruppe =NR
11, -NR
11O-, -ONR
11-, =CR
6R
7, =C=O, =C=S, =SO, =SO
2,
-C(O)O-, -OC(O)-, -S(O)O-, -OS(O)-, -S(O)
2O-,
-OS(O)
2-, -CONR
8-,
-NR
8CO-, -OCONR
8-,
-NR
8C(O)O-, -CSNR
8-,
-NR
8CS-, -OCSNR
8-,
-NR
8CSO -, -SONR
8-,
-NR
8SO-, -SO
2NR
8-, -NR
8SO
2-, -NR
8CONR
9-, -NR
8CSNR
9-, -NR
8SONR
9-, -NR
8SO
2NR
9-, -NR
8C(O)NR
9- oder -NR
8C(S)NR
9- stehen,
R
1 und R
5 jeweils
unabhängig
voneinander für
Wasserstoff, Hydroxy, Halogen, Nitro, Cyano, C
1-C
6-Alkyl, C
1-C
6-Alkenyl, C
1-C
6-Alkinyl, C
3-C
10- Cycloalkyl,
C
3-C
12-Aryl, C
3-C
18-Heteroaryl
oder für
die Gruppe -(CH
2)
p-C
3-C
12-Aryl, -(CH
2)
p-C
3-C
18-Heteroaryl,
Phenyl-(CH
2)
p-R
10, -(CH
2)
pPO
3(R
10)
2 , -NR
8R
9, -NR
8COR
9, -NR
8CSR
9, -NR
8SOR
9, -NR
8SO
2R
9, -NR
8CONR
9R
10 -NR
8COOR
9, -NR
8C(NH)NR
9R
10, -NR
8CSNR
9R
10, -NR
8SONR
9R
10,
-NR
8SO
2NR
9R
10, -COR
8, -CSR
8, -S(O)R
8, -S(O)
2R
8, -S(O)
2NR
8R
9, -SO
3R
8, -CO
2H, -CO
2R
8, -CONR
8R
9, -CSNR
8R
9, -SR
8 oder
-CR
8(OH)-R
9 stehen,
oder für
ein- oder mehrfach,
gleich oder verschieden mit Hydroxy, C
1-C
6-Alkoxy, Halogen, Phenyl oder mit der Gruppe
-NR
3R
4 substituiertes
C
1-C
10-Alkyl, C
2-C
10-Alkenyl, C
2-C
10-Alkinyl, C
3-C
10-Cycloalkyl, C
3-C
12-Aryl oder C
3-C
18-Heteroaryl stehen und das Phenyl, C
3-C
10-Cycloalkyl,
C
3-C
12-Aryl, C
3-C
18-Heteroaryl, -(CH
2)
p- C
3-C
12-Aryl
und -(CH
2)
p- C
3-C
18-Heteroaryl selbst
gegebenenfalls ein- oder mehrfach, gleich oder verschieden mit Halogen,
Hydroxy, C
1-C
6-Alkyl,
C
1-C
6-Alkoxy, oder
mit der Gruppe -CF
3 oder -OCF
3 substituiert
sein kann, und der Ring des C
3-C
10-Cycloalkyls und das C
1-C
10-Alkyl gegebenenfalls durch ein- oder mehrere
Stickstoff, Sauerstoff und/oder Schwefel-Atome unterbrochen sein
kann und/ oder durch ein oder mehrere =C=O Gruppen im Ring unterbrochen
sein kann und/oder gegebenenfalls ein oder mehrere mögliche Doppelbindungen
im Ring enthalten sein können,
R
2 für
Wasserstoff oder C
1-C
10-Alkyl
steht,
R
3 für Wasserstoff, Halogen, Nitro,
Cyano, C
1-C
10-Alkyl,
Halo-C
1-C
10-Alkyl,
C
2-C
10-Alkenyl,
C
2-C
10-Alkinyl, C
3-C
10-Cycloalkyl,
Hydroxy, C
1-C
6-Alkoxy, C
1-C
6-Alkylthio, Amino,
-NH-(CH
2)
p-C
3-C
10-Cycloalkyl,
C
1-C
6-Hydroxyalkyl, C
1-C
6-Alkoxy-C
1-C
6-Alkyl, C
1-C
6-Alkoxy-C
1-C
6-Alkoxy-C
1-C
6-Alkyl, -NHC
1-C
6-Alkyl, -N(C
1-C
6-Alkyl)
2, -SO(C
1-C
6-Alkyl), -SO
2(C
1-C
6-Alkyl),
C
1-C
6-Alkanoyl,
-CONR
8R
9, -COR
10 , C
1-C
6-AlkylOAc, Carboxy, C
3-C
12-Aryl, C
3-C
18-Heteroaryl, -(CH
2)
p- C
3-C
12-Aryl,
-(CH
2)
p-C
3-C
18-Heteroaryl, Phenyl-(CH
2)
p-R
10, -(CH
2)
pPO
3(R
10)
2 oder für die Gruppe
-NR
8R
9 steht,
oder
für ein-
oder mehrfach, gleich oder verschieden mit Hydroxy, Halogen, C
1-C
6-Alkoxy, C
1-C
6-Alkylthio, Amino,
Cyano, C
1-C
6-Alkyl,
-NH-(CN
2)
p-C
3-C
10-Cycloalkyl,
C
3-C
10-Cycloalkyl,
C
1-C
6-Hydroxyalkyl,
C
2-C
6-Alkenyl, C
2-C
6-Alkinyl, C
1-C
6-Alkoxy-C
1-C
6-Alkyl, C
1-C
6-Alkoxy- C
1-C
6-Alkoxy-C
1-C
6-Alkyl, -NHC
1-C
6-Alkyl, -N(C
1-C
6-Alkyl)
2, -SO(C
1-C
6-Alkyl), -SO
2(C
1-C
6-Alkyl),
C
1-C
6-Alkanoyl,
-CONR
8R
9, -COR
10 , C
1-C
6-AlkylOAc,
Carboxy, C
3-C
12-Aryl,
C
3-C
18-Heteroaryl,
-(CH
2)
p-C
3-C
12-Aryl, -(CH
2)
p-C
3-C
18-Heteroaryl, Phenyl-(CH
2)
p-R
10, -(CH
2)
pPO
3(R
10)
2 oder mit der
Gruppe -NR
8R
9 substituiertes
C
1-C
10-Alkyl, C
2-C
10-Alkenyl, C
2-C
10-Alkinyl, C
3-C
10-Cycloalkyl, C
3-C
12-Aryl oder C
3-C
18-Heteroaryl steht und das Phenyl, C
3-C
10-Cycloalkyl,
C
3-C
12-Aryl, C
3-C
18-Heteroaryl,
-(CH
2)
p- C
3-C
12-Aryl und -(CH
2)
p- C
3-C
18-Heteroaryl selbst gegebenenfalls ein-
oder mehrfach, gleich oder verschieden mit Halogen, Hydroxy, C
1-C
6-Alkyl, C
1-C
6-Alkoxy, oder
mit der Gruppe -CF
3 oder -OCF
3 substituiert
sein kann, und der Ring des C
3-C
10-Cycloalkyls und das C
1-C
10-Alkyl gegebenenfalls durch ein- oder mehrere
Stickstoff, Sauerstoff und/oder Schwefel-Atome unterbrochen sein
kann und/oder durch ein oder mehrere =C=O Gruppen im Ring unterbrochen
sein kann und/oder gegebenenfalls ein oder mehrere mögliche Doppelbindungen
im Ring enthalten sein können,
R
4 für
Wasserstoff, Halogen oder C
1-C
4-Alkyl
steht,
R
6, R
7,
R
8, R
9, R
10 und R
11 jeweils
unabhängig
voneinander für
Wasserstoff oder für
gegebenenfalls ein- oder mehrfach, gleich oder verschieden mit Hydroxy,
Halogen, C
1-C
12-Alkoxy,
C
1-C
6-Alkylthio,
Amino, Cyano, C
1-C
6-Alkyl, -NH-(CH
2)
p-C
3-C
10-Cycloalkyl, C
3-C
10-Cycloalkyl, C
1-C
6-Hydroxyalkyl,
C
2-C
6-Alkenyl, C
2-C
6-Alkinyl, C
1-C
6-Alkoxy-C
1-C
6-Alkyl, C
1-C
6-Alkoxy-C
1-C
6-Alkoxy-C
1-C
6-Alkyl, -NHC
1-C
6-Alkyl, -N(C
1-C
6-Alkyl)
2, -SO(C
1-C
6-Alkyl), -SO
2(C
1-C
6-Alkyl),
C
1-C
6-Alkanoyl,
-CONR
8R
9, -COR
10 , C
1-C
6-AlkylOAc, Carboxy, C
3-C
12-Aryl, C
3-C
8-Heteroaryl, -(CH
2)
p- C
3-C
12-Aryl,
-(CH
2)p- C
3-C
18-Heteroaryl, Phenyl-(CH
2)
p-R
10, -(CH
2)
pPO
3(R
10)
2 oder mit der
Gruppe -NR
8R
9 substituiertes
C
1-C
10-Alkyl, C
2-C
10-Alkenyl, C
2-C
10-Alkinyl, C
3-C
10-Cycloalkyl, C
3-C
12-Aryl oder C
3-C
18-Heteroaryl stehen und das Phenyl, C
3-C
10-Cycloalkyl,
C
3-C
12-Aryl, C
3-C
18-Heteroaryl, -(CH
2)
p C
3-C
12-Aryl
und -(CH
2)
p- C
3-C
18-Heteroaryl selbst
gegebenenfalls ein- oder mehrfach, gleich oder verschieden mit Halogen,
Hydroxy, C
1-C
6-Alkyl,
C
1-C
6-Alkoxy, oder
mit der Gruppe -CF
3 oder -OCF
3 substituiert
sein kann, und der Ring des C
3-C
10-Cycloalkyls und das C
1-C
10-Alkyl gegebenenfalls durch ein- oder mehrere
Stickstoff, Sauerstoff und/oder Schwefel-Atome unterbrochen sein
kann und/oder durch ein oder mehrere =C=O Gruppen im Ring unterbrochen
sein kann und/oder gegebenenfalls ein oder mehrere mögliche Doppelbindungen
im Ring enthalten sein können,
m
für 0 bis
8 steht und
n und p für
0 bis 6 steht, bedeuten, sowie deren Isomeren, Diastereomeren, Enantiomeren
und Salzen, die bekannten Nachteile überwinden.
-
Unter Alkyl ist jeweils ein geradkettiger
oder verzweigter Alkylrest, wie beispielsweise Methyl, Ethyl, Propyl,
Isopropyl, Butyl, Isobutyl, sek. Butyl, tert. Butyl, Pentyl, Isopentyl,
Hexyl, Heptyl, Octyl, Nonyl und Decyl, zu verstehen.
-
Unter Alkoxy ist jeweils ein geradkettiger
oder verzweigter Alkoxyrest, wie beispielsweise Methyloxy, Ethyloxy,
Propyloxy, Isopropyloxy, Butyloxy, Isobutyloxy, sek. Butyloxy, Pentyloxy,
Isopentyloxy, Hexyloxy, Heptyloxy, Octyloxy, Nonyloxy, Decyloxy,
Undecyloxy oder Dodecyloxy zu verstehen.
-
Unter Alkylthio ist jeweils ein geradkettiger
oder verzweigter Alkylthiorest, wie beispielsweise Methylthio, Ethylthio,
Propylthio, Isopropylthio, Butylthio, Isobutylthio, sek. Butylthio,
tert.-Butylthio, Pentylthio, Isopentylthio oder Hexylthio zu verstehen.
-
Unter Cycloalkyl sind monocyclische
Alkylringe wie Cyclopropyl, Cyclobutyl, Cyclopentyl, Cyclohexyl oder
Cycloheptyl, Cyclooctyl, Cyclononyl oder Cyclodecyl, aber auch bicyclische
Ringe oder tricyclische Ringe wie zum Beispiel Adamantanyl zu verstehen.
-
Heterocycloalkyl steht für einen
3–12 Kohlenstoffatome
umfassenden Alkylring, der anstelle des Kohlenstoffes ein oder mehrere,
gleich oder verschiedene Heteroatome, wie z. B. Sauerstoff, Schwefel
oder Stickstoff enthält.
-
Als Heterocycloalkyle seien z. B.
genannt: Oxiranyl, Oxethanyl, Aziridinyl, Azetidinyl, Tetrahydrofuranyl,
Pyrrolidinyl, Dioxolanyl, Imidazolidinyl, Pyrazolidinyl, Dioxanyl, Piperidinyl,
Morpholinyl, Dithianyl, Thiomorpholinyl, Piperazinyl, Trithianyl,
Chinuclidinyl etc.
-
Unter den Ringsystemen, bei denen
gegebenenfalls ein- oder mehrere mögliche Doppelbindungen im Ring
enthalten sein können,
sind zum Beispiel Cycloalkenyle wie Cyclopropenyl, Cyclobutenyl,
Cyclopentenyl, Cyclohexenyl, Cycloheptenyl zu verstehen, wobei die
Anknüpfung
sowohl an der Doppelbindung wie auch an den Einfachbindungen erfolgen
kann.
-
Unter Halogen ist jeweils Fluor,
Chlor, Brom oder Jod zu verstehen.
-
Die Alkenyl- und Alkinyl-Substituenten
sind jeweils geradkettig oder verzweigt, wobei beispielsweise folgenden
Reste gemeint sind: Vinyl, Propen-1-yl, Propen-2-yl, But-1-en-1-yl, But-1-en-2-yl,
But-2-en-1-yl, But-2-en-2-yl, 2-Methyl-prop-2-en-1-yl, 2-Methylprop-1-en-1-yl,
But-1-en-3-yl, Ethinyl, Prop-1-in-1-yl, But-1-in-1-yl, But-2-in-1-yl,
But-3-en-1-yl, Allyl.
-
Der Arylrest hat jeweils 6 – 12 Kohlenstoffatome
wie beispielsweise Naphthyl, Biphenyl und insbesondere Phenyl.
-
Der Heteroarylrest umfaßt jeweils
3 – 18
Ringatome und kann anstelle des Kohlenstoffs ein- oder mehrere,
gleiche oder verschiedene Heteroatome, wie Sauerstoff, Stickstoff
oder Schwefel im Ring enthalten, und kann mono-, bi- oder tricyclisch
sein, und kann zusätzlich
jeweils benzokondensiert sein.
-
Beispielsweise seien genannt:
Thienyl,
Furanyl, Pyrrolyl, Oxazolyl, Thiazolyl, Imidazolyl, Pyrazolyl, Isoxazolyl,
Isothiazolyl, Oxadiazolyl, Triazolyl, Thiadiazolyl, etc. und Benzoderivate
davon, wie z. B. Benzofuranyl, Benzothienyl, Benzoxazolyl, Benzimidazolyl,
Indazolyl, Indolyl, Isoindolyl, etc.; oder Pyridyl, Pyridazinyl,
Pyrimidinyl, Pyrazinyl, Triazinyl, etc. und Benzoderivate davon,
wie z. B. Chinolyl, Isochinolyl, etc.; oder Azocinyl, Indolizinyl,
Purinyl, etc. und Benzoderivate davon; oder Cinnolinyl, Phthalazinyl,
Chinazolinyl, Chinoxalinyl, Naphthyridinyl, Pteridinyl, Carbazolyl, Acridinyl,
Phenazinyl, Phenothiazinyl, Phenoxazinyl, Xanthenyl, Oxepinyl, etc.
-
Ist eine saure Funktion enthalten,
sind als Salze die physiologisch verträglichen Salze organischer und anorganischer
Basen geeignet, wie beispielsweise die gut löslichen Alkali- und Erdalkalisalze
sowie N-Methyl-glukamin, Dimethyl-glukamin, Ethyl-glukamin, Lysin,
1,6-Hexadiamin, Ethanolamin, Glukosamin, Sarkosin, Serinol, Tris-hydroxy-methyl-amino-methan,
Aminopropandiol, Sovak-Base, 1-Amino-2,3,4-butantriol.
-
Ist eine basische Funktion enthalten,
sind die physiologisch verträglichen
Salze organischer und anorganischer Säuren geeignet wie Salzsäure, Schwefelsäure, Phosphorsäure, Zitronensäure, Weinsäure u.a.
-
Besonders wirksam sind solche Verbindungen
der allgemeinen Formel (I), in der
A für C3-C12-Aryl oder C3-C18-Heteroaryl steht,
B für eine Bindung
oder für
gegebenenfalls ein- oder mehrfach, gleich oder verschieden mit Hydroxy,
Halogen, Cyano, Nitro, C1-C6-Alkyl,
C2-C6-Alkenyl, C2-C6-Alkinyl, C3-C10-Cycloalkyl, C3-C12-Aryl, C3-C18-Heteroaryl, -(CH2)p-C3-C12-Aryl, -(CH2)p-C3-C18-Heteroaryl,
Phenyl(CH2)p R10, -(CH2)pPO3(R10)2 oder mit der Gruppe -NR8R9, -NR8COR9, -NR8CSR9, -NR8SOR9, -NR8SO2R9, -NR8CONR9R10, -NR8COOR9, -NR8C(NH)NR9R10, -NR8CSNR9R10, -NR8SONR9R10,
-NR8SO2NR9R10, -COR8, -CSR8, -S(O)R8, -S(O)2R8, -S(O)2NR8R9, -SO3R8, -CO2R8,
-CONR8R9, -CSNR8R9, -SR8 oder
-CR8(OH)-R9 substituiertes
C1-C12-Alkyl, C2-C12-Alkenyl, C2-C12-Alkinyl, C3-C8-Cycloalkyl, C3-C12-Heterocycloalkyl,
C3-C12-Aryl oder
C3-C18-Heteroaryl
steht,
X und Y jeweils unabhängig voneinander für Sauerstoff
oder für
die Gruppe =NR11, -NR8CO-,
-CONR8-, -SO2NR8- oder -NR8SO2- stehen,
R1 und
R5 jeweils unabhängig voneinander für Wasserstoff
oder für
die Gruppe -SO2NR8R9 stehen,
R2 für Wasserstoff
oder C1-C10-Alkyl
steht,
R3 für Wasserstoff, Halogen, Nitro,
Cyano, C1-C10-Alkyl,
Halo-C1-C10-Alkyl,
C2-C10-Alkenyl,
C2-C10-Alkinyl, C3-C10-Cycloalkyl,
Hydroxy, C1-C6-Alkoxy, C1-C6-Alkylthio, Amino,
-NH-(CH2)p-C3-C10-Cycloalkyl,
C1-C6-Hydroxyalkyl, C1-C6-Alkoxy-C1-C6-Alkyl, C1-C6-Alkoxy-C1-C6-Alkoxy-C1-C6-Alkyl, -NHC1-C6-Alkyl, -N(C1-C6-Alkyl)2, -SO(C1-C6-Alkyl), -SO2(C1-C6-Alkyl),
C1-C6-Alkanoyl,
-CONR8R9, -COR10 , C1-C6-AlkylOAc, Carboxy, C3-C12-Aryl, C3-C18-Heteroaryl, -(CH2)p- C3-C12-Aryl,
-(CH2)p- C3-C18-Heteroaryl,
Phenyl-(CH2)p-R10, -(CH2)pPO3(R10)2 oder für
die Gruppe -NR8R9 steht,
oder
für ein-
oder mehrfach, gleich oder verschieden mit Hydroxy, Halogen, C1-C6-Alkoxy, C1-C6-Alkylthio, Amino,
Cyano, C1-C6-Alkyl,
-NH-(CH2)pC3-C10-Cycloalkyl,
C3-C10-Cycloalkyl,
C1-C6-Hydroxyalkyl,
C2-C6-Alkenyl, C2-C6-Alkinyl, C1-C6-Alkoxy-C1-C6-Alkyl, C1-C6-Alkoxy-C1-C6-Alkoxy-C1-C6-Alkyl, -NHC1-C6-Alkyl, -N(C1-C6-Alkyl)2, -SO(C1-C6-Alkyl), -SO2(C1-C6-Alkyl),
C1-C6-Alkanoyl,
-CONR8R9, -COR10 , C1-C6-AlkylOAc,
Carboxy, C3-C12-Aryl,
C3-C18-Heteroaryl,
-(CH2)p- C3-C12-Aryl, -(CH2)p- C3-C18-Heteroaryl, Phenyl-(CH2)p-R10, -(CH2)pPO3(R10)2 oder mit der
Gruppe -NR8R9 substituiertes
C1-C10-Alkyl, C2-C10-Alkenyl, C2-C10-Alkinyl, C3-C10-Cycloalkyl, C3-C12-Aryl oder C3-C18-Heteroaryl steht und das Phenyl, C3-C10-Cycloalkyl,
C3-C12-Aryl, C3-C18-Heteroaryl,
-(CH2)p- C3-C12-Aryl und -(CH2)p- C3-C18-Heteroaryl selbst gegebenenfalls ein-
oder mehrfach, gleich oder verschieden mit Halogen, Hydroxy, C1-C6-Alkyl, C1-C6-Alkoxy, oder
mit der Gruppe -CF3 oder -OCF3 substituiert
sein kann, und der Ring des C3-C10-Cycloalkyls und das C1-C10-Alkyl gegebenenfalls durch ein- oder mehrere
Stickstoff, Sauerstoff und/oder Schwefel-Atome unterbrochen sein
kann und/oder durch ein oder mehrere =C=O Gruppen im Ring unterbrochen
sein kann und/oder gegebenenfalls ein oder mehrere mögliche Doppelbindungen
im Ring enthalten sein können,
R4 für
Wasserstoff, Halogen oder C1-C4-Alkyl
steht,
R6, R7,
R8, R9, R10 und R11 jeweils
unabhängig
voneinander für
Wasserstoff oder für
gegebenenfalls ein- oder mehrfach, gleich oder verschieden mit Hydroxy,
Halogen, C1-C12-Alkoxy,
C1-C6-Alkylthio,
Amino, Cyano, C1-C6-Alkyl, -NH-(CH2)p-C3-C10-Cycloalkyl, C3-C10-Cycloalkyl, C1-C6-Hydroxyalkyl,
C2-C6-Alkenyl, C2-C6-Alkinyl, C1-C6-Alkoxy-C1-C6-Alkyl, C1-C6-Alkoxy-C1-C6-Alkoxy-C1-C6-Alkyl, -NHC1-C6-Alkyl, -N(C1-C6-Alkyl)2, -SO(C1-C6-Alkyl), -SO2(C1-C6-Alkyl),
C1-C6-Alkanoyl,
-CONR8R9, -COR10 , C1-C6-AlkylOAc, Carboxy, C3-C12-Aryl, C3-C8-Heteroaryl, -(CH2)p- C3-C12-Aryl,
-(CH2)p- C3-C18-Heteroaryl,
Phenyl-(CH2)p-R10, -(CH2)pPO3(R10)2 oder mit der Gruppe -NR8R9 substituiertes C1-C10-Alkyl,
C2-C10-Alkenyl,
C2-C10-Alkinyl, C3-C10-Cycloalkyl,
C3-C12-Aryl oder
C3-C18-Heteroaryl
stehen und das Phenyl, C3-C10-Cycloalkyl,
C3-C12-Aryl, C3-C18-Heteroaryl, -(CH2)p- C3-C12-Aryl
und -(CH2)p- C3-C18-Heteroaryl selbst
gegebenenfalls ein- oder mehrfach, gleich oder verschieden mit Halogen,
Hydroxy, C1-C6-Alkyl,
C1-C6-Alkoxy, oder
mit der Gruppe -CF3 oder -OCF3 substituiert
sein kann, und der Ring des C3-C10-Cycloalkyls und das C1-C10-Alkyl gegebenenfalls durch ein- oder mehrere
Stickstoff, Sauerstoff und/oder Schwefel-Atome unterbrochen sein
kann und/oder durch ein oder mehrere =C=O Gruppen im Ring unterbrochen
sein kann und/oder gegebenenfalls ein oder mehrere mögliche Doppelbindungen
im Ring enthalten sein können,
m
für 0 bis
8 steht und
n und p für
0 bis 6 steht, bedeuten, sowie deren Isomeren, Diastereomeren, Enantiomeren
und Salzen, die bekannten Nachteile überwinden.
-
Insbesondere wirksam sind solche
Verbindungen der allgemeinen Formel (I) in der
A für Phenyl
oder Thiophenyl steht,
B für
eine Bindung oder für
gegebenenfalls ein- oder mehrfach, gleich oder verschieden mit Hydroxy,
Halogen, C1-C6-Alkoxy,
C1-C6-Alkylthio,
Amino, Cyano, C1-C6-Alkyl,
C1-C6-Hydroxyalkyl
oder mit der Gruppe -NR8R9 substituiertes
C1-C12-Alkyl steht,
X
und Y jeweils unabhängig
voneinander für
Sauerstoff oder für
die Gruppe =NR11, -NR8CO-,
-CONR8-, -SO2NR8- oder -NR8SO2- stehen,
R1 und
R5 jeweils unabhängig voneinander für Wasserstoff
oder für
die Gruppe -SO2NR8R9 stehen,
R2 für Wasserstoff
oder C1-C10-Alkyl
steht,
R3 für Wasserstoff, Halogen, Nitro,
Cyano, C1-C10-Alkyl
oder für
die Gruppe -CONR8R9 steht,
R4 für
Wasserstoff, Halogen oder C1-C4-Alkyl
steht,
R8, R9,
und R11 jeweils unabhängig voneinander für Wasserstoff
oder C1-C6-Alkyl
stehen,
m für
0 bis 8 steht und
n für
0 bis 6 steht, bedeuten, sowie deren Isomeren, Diastereomeren, Enantiomeren
und Salze.
-
Ausgewählt sind solche Verbindungen
der allgemeinen Formel (1), in der
A für Phenyl oder Thiophenyl steht,
B
für gegebenenfalls
ein- oder mehrfach, gleich oder verschieden mit Hydroxy, C1-C6-Alkyl oder C1-C6-Hydroxyalkyl
substituiertes C1-C12-Alkyl steht,
X
und Y jeweils unabhängig
voneinander für
Sauerstoff oder für
die Gruppe =NR11, -NR8CO-,
-CONR8-, -SO2NR8- oder -NR8SO2- stehen,
R1 und
R5 jeweils unabhängig voneinander für Wasserstoff
oder für
die Gruppe -SO2NR8R9 stehen,
R2 für Wasserstoff
steht,
R3 für Wasserstoff, Halogen, Cyano,
C1-C10-Alkyl oder
für die
Gruppe -CONR8R9 steht,
R4 für
Wasserstoff steht,
R8 und R11 für
Wasserstoff stehen,
R9 für Wasserstoff
oder C1-C6-Alkyl
steht,
m für
0 bis 8 steht und
n für
0 bis 6 steht, bedeuten, sowie deren Isomeren, Diastereomeren, Enantiomeren
und Salze.
-
Soweit die Herstellung der erfindungsgemäßen Verbindungen
der allgemeinen Formel I nicht beschrieben ist, erfolgt diese analog
bekannter Methoden.
-
Die Strukturaufklärung der macrocyclischen Riechstoffe
Muskon und Zibeton durch Ruzicka ((a) Ruzicka, L. Helv. Chim. Acta
1926, 9, 715. (b) Ruzicka, L. Helv. Chim. Acta 1926, 9, 249) im
Jahre 1926 markiert den Beginn der Chemie der Macrocyclen.
-
Im allgemeinen werden mittlere (8 – 11-gliedrige)
und große
(≥ 12-gliedrige)
Ringe als Macrocyclen bezeichnet. Die etablierten Verfahren zur
Synthese von Macrocyclen beruhen zum Teil auf Ringenrveiterungsreaktionen
(Hesse, M. Ring Enlargement in Organic Chemistry, VCH, Weinheim,
1991), seltener auf Ringkontraktionen (Hayashi, T. J. Org. Chem.
1984, 49, 2326).
-
Die am häufigsten angewandte Methode
ist die Cyclisierung von bifunktionellen acyclischen Vorläufern (Reviews
zur Synthese von Macrocyclen: (a) Roxburgh, C.J. Tetrahedron 1995,
51, 9767. (b) Meng, Q. Top. Curr. Chem. 1991, 161, 107. (c) Paterson,
I. Tetrahedron 1985, 41, 3569. (d) Masamune, S. Angew. Chem. 1977,
89, 602. (e) Nicolaou, K.C. Tetrahedron 1977, 33, 683. (f) Ruggli,
P. Liebigs Ann. Chem. 1912, 92).
-
Schnell und mit sehr guten Ausbeuten
kann die Herstellung der erfindungsgemäßen Verbindungen der allgemeinen
Formel I durch den Ringschluß über die
2-Position des Pyrimidins acyclischer Vorläufer der Formeln (IV) oder
(V) erfolgen, indem man
- a) Verbindungen der
allgemeinen Formel IV in der R1,
R2, R3, R4, R5, X, Y, A, B,
m und n die in der allgemeinen Formel I angegebenen Bedeutungen haben
und L für
eine Abgangsgruppe steht, mit einer geeigneten Säure zu Verbindungen der allgemeinen Formel
I cyclisiert, oder
- b) den acyclischen Vorläufer
der allgemeinen Formel (V) in der in der R1, R3, R4,
R5, X, Y, A, B, m und n die in der allgemeinen
Formel I angegebenen Bedeutungen haben und L für eine Abgangsgruppe steht,
in einem geeigneten Lösungsmittel
und einem geeignete Reduktionsmittel bei 0 °C bis Reflux zunächst zum
Amin reduziert und anschließend
das intermediär
gebildete Amin zu den Verbindungen der allgemeinen Formel I cyclisiert.
-
Die Herstellung der erfindungsgemäßen Verbindungen
der allgemeinen Formel I ist ebenfalls Gegenstand der vorliegenden
Erfindung.
-
Geeignete Lösungsmittel sind zum Beispiel
einfachen Ketone wie Aceton, Alkohole wie z.B. Ethanol oder Butanol,
Ester wie zum Beispiel Essigester, aromatische Lösungsmittel wie zum Beispiel
Toluol oder Benzol sowie polar aprotischen Lösungsmittel wie Acetonitril,
DMSO, DMF oder N-Methylpyrrolidine oder Gemischen dieser Lösungsmittel,
auch unter Zusatz von Wasser.
-
Geeignete Reduktionsmittel sind zum
Beispiel Ti(III)Cl oder Sn(II)Cl.
-
Unter Abgangsgruppen in der Bedeutung
von L sind zum Beispiel eine Halogeno- oder Sulphonyloxy-Gruppe, wie Fluor,
Chlor, Brom, Iod, Methansulphonyloxy, Toluol-4-sulphonyloxy, Trifluoromethylsulphonyloxy,
etc. zu verstehen.
-
Für
die Cyclisierung zur Anwendung kommende Säuren sind zum Beispiel geeigneten
Lewis Säuren, wie
anorganische Säuren
wie Hydrogenchlorid, Hydrogenbromid, Schwefelsäure, organische Säuren wie
Essigsäure,
Ameisensäure,
BBr3, Metallsalze wie Ti(III)Cl, Sn(II)Cl,
Ln(III)Otf, etc.
-
Die für die Herstellung der erfindungsgemäßen Verbindungen
der allgemeinen Formel I vorzugsweise verwendeten Zwischenprodukte
der allgemeinen Formeln II und III
in denen R
1,
R
2, R
3, R
4, R
5, R
8,
R
11, A, B und m die in der allgemeinen Formel
I angegebenen Bedeutungen haben und D für -NH
2 oder
-NO
2, q für 1 bis 12, U für die Gruppe
-OH oder
und W für die Gruppe -OH oder -COOH
steht, sind ebenfalls Gegenstand der vorliegenden Erfindung.
-
Die erfindungsgemäßen Verbindungen inhibieren
im wesentlichen Zyklin-abhängige
Kinasen, worauf auch deren Wirkung zum Beispiel gegen Krebs, wie
solide Tumoren und Leukämie,
Autoimmunerkrankungen wie Psoriasis, Alopezie, und Multiple Sklerose,
Chemotherapeutika-induzierte Alopezie und Mukositis, kardiovaskuläre Erkrankungen,
wie Stenosen, Arteriosklerosen und Restenosen, infektiöse Erkrankungen,
wie z. B. durch unizelluläre
Parasiten, wie Trypanosoma, Toxoplasma oder Plasmodium, oder durch
Pilze hervorgerufen, nephrologische Erkrankungen, wie z. B. Glomerulonephritis,
chronische neurodegenerative Erkrankungen, wie Huntington's Erkrankung,
amyotrophe Lateralsklerose, Parkinsonsche Erkrankung, AIDS Dementia und
Alzheimer'sche Erkrankung, akute neurodegenerative Erkrankungen,
wie Ischämien
des Gehirns und Neurotraumata, vitale Infektionen, wie z. B. Cytomegalus-Infektionen,
Herpes, Hepatitis B und C, und HIV Erkrankungen basiert.
-
Der eukaryote Zellteilungszyklus
stellt die Duplikation des Genoms und seine Verteilung auf die Tochterzellen
sicher, indem er eine koordinierte und regulierte Abfolge von Ereignissen
durchläuft.
Der Zellzyklus wird in vier aufeinanderfolgende Phasen eingeteilt:
die G1 Phase repräsentiert
die Zeit vor der DNA-Replikation, in der die Zelle wächst und
für externe
Stimuli empfänglich
ist. In der S Phase repliziert die Zelle ihre DNA, und in der G2
Phase bereitet sie sich auf den Eintritt in die Mitose vor. In der
Mitose (M Phase) wird die replizierte DNA getrennt und die Zellteilung
vollzogen.
-
Die Zyklin-abhängigen Kinasen (CDKs), eine
Familie von Serin/Threonin-Kinasen, deren Mitglieder die Bindung
eines Zyklins (Cyc) als regulatorische Untereinheit zu ihrer Aktivierung
benötigen,
treiben die Zelle durch den Zellzyklus. Unterschiedliche CDK/Cyc
Paare sind in den verschiedenen Phasen des Zellzyklus aktiv. Für die grundlegende
Funktion des Zellzyklus bedeutende CDK/Cyc Paare sind beispielsweise
CDK4(6)/CycD, CDK2/CycE, CDK2/CycA, CDK1/CycA und CDK1/CycB. Einige
Mitglieder der CDK-Enzymfamilie haben eine regulatorische Funktion
indem sie die Aktivität
der vorgenannten Zellzyklus-CDKs beeinflussen, während anderen Mitgliedern der
CDK-Enzymfamlie noch keine bestimmte Funktion zugeordnet werden
konnte. Eine von diesen, CDKS, zeichnet sich dadurch aus, daß sie eine atypische,
von den Zyklinen abweichende, regulatorische Untereinheit besitzt
(p35), und ihre Aktivität
im Gehirn am höchsten
ist.
-
Der Eintritt in den Zellzyklus und
das Durchlaufen des "Restriction Points", der die Unabhängigkeit
einer Zelle von weiteren Wachstumssignalen für den Abschluß der begonnenen
Zellteilung markiert, werden durch die Aktivität der CDK4(6)/CycD und CDK2/CycE
Komplexe kontrolliert. Das wesentliche Substrat dieser CDK-Komplexe
ist das Retinoblastoma-Protein (Rb), das Produkt des Retinoblastoma
Tumorsuppressor Gens. Rb ist ein transkriptionelles Ko-Repressor
Protein. Neben anderen noch weitgehend unverstandenen Mechanismen,
bindet und inaktiviert Rb Transkriptionsfaktoren vom E2F-Typ, und
bildet transkriptionelle Repressorkomplexe mit Histon-Deacetylasen
(HDAC) (Zhang H.S. et al. (2000). Exit from G1 and S phase of the cell
cycle is regulated by repressor complexes containing HDAC-RbhSWI/SNF
and Rb-hSWI/SNF. Cell 101, 79-89). Durch die Phosphorylierung des
Rb durch CDKs werden gebundene E2F Transkriptionsfaktoren freigesetzt
und führen
zu transkriptioneller Aktivierung von Genen, deren Produkte für die DNA
Synthese und die Progression durch die S-Phase benötigt werden.
Zusätzlich
bewirkt die Rb-Phosphorylierung
die Auflösung der
Rb-HDAC Komplexe, wodurch weitere Gene aktiviert werden. Die Phosphorylierung
von Rb durch CDK's ist mit dem Überschreiten
des "Restriction Points" gleichzusetzen. Für die Progression durch die
S-Phase und deren Abschluß ist
die Aktivität
der CDK2/CycE und CDK2/CycA Komplexe notwendig, z. B. wird die Aktivität der Transkriptionsfaktoren
vom E2F-Typ mittels Phosphorylierung durch CDK2/CycA abgeschaltet
sobald die Zellen in die S-Phase
eingetreten sind. Nach vollständiger
Replikation der DNA steuert die CDK1 im Komplex mit CycA oder CycB
den Eintritt und das Durchlaufen der Phasen G2 und M (1).
-
Entsprechend der außerordentlichen
Bedeutung des Zellteilungszyklus ist das Durchlaufen des Zyklus streng
reguliert und kontrolliert. Die Enzyme, die für die Progression durch den
Zyklus notwendig sind, müssen zu
dem richtigen Zeitpunkt aktiviert werden, und auch wieder abgeschaltet
werden sobald die entsprechende Phase durchlaufen ist. Entsprechende
Kontrollpunkte ("Checkpoints") arretieren die Progression durch
den Zellzyklus falls DNA-Schäden
detektiert werden, oder die DNA-Replikation, oder der Aufbau des
Spindelapparates noch nicht beendet ist.
-
Die Aktivität der CDKs wird durch verschiedene
Mechanismen, wie Synthese und Degradation der Zykline, Komplexierung
der CDKs mit den entsprechenden Zyklinen, Phosphorylierung und Dephosphorylierung regulatorischer
Threonin- und Tyrosin-Reste,
und die Bindung natürlicher
inhibitorischer Proteine, direkt kontrolliert. Während die Proteinmenge der
CDKs in einer proliferierenden Zelle relativ konstant ist, oszilliert
die Menge der einzelnen Zykline mit dem Durchlaufen des Zyklus.
So wird zum Beispiel die Expression von CycD während der frühen G1 Phase
durch Wachstumsfaktoren stimuliert, und die Expression von CycE
wird nach Überschreiten
des "Restriction Points" durch die Aktivierung der Transkriptionsfaktoren
vom E2F-Typ induziert. Die Zykline selbst werden durch Ubiquitin-vermittelte
Proteolyse abgebaut. Aktivierende und inaktivierende Phosphorylierungen
regulieren die Aktivität
der CDKs, zum Beispiel phosphorylieren CDK-aktivierende Kinasen
(CAKs) Thr160/161 der CDK1, wohingegen die Familie der Wee1/Myt1
Kinasen CDK1 durch Phosphorylierung von Thr14 und Tyr15 inaktivieren.
Diese inaktivierenden Phosphorylierungen können durch cdc25 Phosphatasen
wieder aufgehoben werden. Sehr bedeutsam ist die Regulation der
Aktivität
der CDK/Cyc-Komplexe durch zwei Familien natürlicher CDK Inhibitorproteine
(CKIs), den Proteinprodukten der p21 Genfamilie (p21, p27, p57)
und der p16 Genfamilie (p15, p16, p18, p19). Mitglieder der p21
Familie binden an Zyklin-Komplexe der CDKs 1,2,4,6, inhibieren aber
nur Komplexe die CDK1 oder CDK2 enthalten. Mitglieder der p16 Familie
sind spezifische Inhibitoren der CDK4- und CDK6-Komplexe.
-
Oberhalb dieser komplexen direkten
Regulation der Aktivität
der CDKs liegt die Ebene der Kontrollpunkt-Regulation. Kontrollpunkte
erlauben der Zelle das geordnete Ablaufen der einzelnen Phasen während des
Zellzykluses zu verfolgen. Die wichtigsten Kontrollpunkte liegen
am Übergang
von G1 nach S und von G2 nach M. Der G1-Kontrollpunkt stellt sicher,
daß die
Zelle keine DNA-Synthese beginnt falls sie nicht entsprechend ernährt ist,
mit anderen Zellen oder dem Substrat korrekt interagiert, und ihre
DNA intakt ist. Der G2/M Kontrollpunkt stellt die vollständige Replikation
der DNA und den Aufbau der mitotischen Spindel sicher, bevor die
Zelle in die Mitose eintritt. Der G1 Kontrollpunkt wird von dem
Genprodukt des p53 Tumorsuppressorgens aktiviert. p53 wird nach
Detektion von Veränderungen
im Metabolismus oder der genomischen Integrität der Zelle aktiviert und kann
entweder einen Stopp der Zellzyklusprogression oder Apoptose auslösen. Dabei
spielt die transkriptionelle Aktivierung der Expression des CDK
Inhibitorproteins p21 durch p53 eine entscheidende Rolle. Ein zweiter
Zweig des G1 Kontrollpunktes umfaßt die Aktivierung der ATM
und Chk1 Kinasen nach DNA-Schädigung
durch UV-Licht oder ionisierende Strahlung und schließlich die
Phosphorylierung und den nachfolgenden proteolytischen Abbau der
cdc25A Phosphatase (Mailand N. et al. (2000). Rapid destruction
of human cdc25A in response to DNA damage. Science 288, 1425-1429). Daraus resultiert
eine Arretierung des Zellzykluses, da die inhibitorische Phosphorylierung
der CDKs nicht entfernt wird. Nach Aktivierung des G2/M Kontrollpunktes
durch Schädigung
der DNA sind beide Mechanismen in ähnlicher Weise daran beteiligt,
die Progression durch den Zellzyklus zu stoppen.
-
Der Verlust der Regulation des Zellzyklusses
und der Verlust der Funktion der Kontrollpunkte sind Charakteristika
von Tumorzellen. Der CDK-Rb-Signalweg ist in über 90% humaner Tumorzellen
von Mutationen betroffen. Diese Mutationen, die schließlich zur
inaktivierenden Phosphorylierung des RB führen, schließen die Überexpression
von D- und E-Zyklinen durch Genamplifikation oder chromosomale Translokationen, inaktivierende
Mutationen oder Deletionen von CDK-Inhibitoren des p16-Typs, sowie
erhöhten
(p27) oder verminderten (CycD) Proteinabbau ein. Die zweite Gruppe
von Genen, die durch Mutationen in Tumorzellen getroffen sind, kodiert
für Komponenten
der Kontrollpunkte. So ist p53, das essentiell für die G1 und G2/M Kontrollpunkte
ist, das am häufigsten
mutierte Gen in humanen Tumoren (ca. 50%). In Tumorzellen, die p53
ohne Mutation exprimieren, wird es häufig aufgrund einer stark erhöhten Proteindegradation
inaktiviert. In ähnlicher Weise
sind die Gene anderer für
die Funktion der Kontrollpunkte notwendiger Proteine von Mutationen
betroffen, zum Beispiel ATM (inaktivierende Mutationen) oder cdc25
Phosphatasen (Überexpression).
-
Überzeugende
experimentelle Daten deuten darauf hin, daß CDK2/Cyc-Komplexe eine entscheidende
Position während
der Zellzyklusprogression einnehmen: (1) Sowohl dominant-negative
Formen der CDK2, wie die transkriptionelle Repression der CDK2 Expression
durch anti-sense Oligonukleotide bewirken einen Stopp der Zellzyklusprogression.
(2) Die Inaktivierung des CycA Gens in Mäusen ist letal. (3) Die Störung der Funktion
des CDK2/CycA Komplexes in Zellen mittels zell permeabler Peptide
führte
zur Tumorzell-selektiven Apoptose (Chen Y.N.P. et al. (1999). Selective
killing of transformed cells by cyclin/cyclin-dependent kinase 2 antagonists.
Proc. Natl. Acad. Sci. USA 96, 4325-4329).
-
Veränderungen der Zellzykluskontrolle
spielen nicht nur bei Krebserkrankungen ein Rolle. Der Zellzyklus
wird durch eine Reihe von Viren, sowohl durch transformierende,
wie durch nicht-transformierende, aktiviert um die Vermehrung der
Viren in der Wirtszelle zu ermöglichen.
Der fälschliche
Eintritt in den Zellzyklus von normalerweise post-mitotischen Zellen
wird mit verschiedenen neurodegenerativen Erkrankungen in Zusammenhang
gebracht.
-
Die Mechanismen der Zellzyklusregulation,
ihrer Veränderungen
in Krankheiten und eine Vielzahl von Ansätzen zur Entwicklung von Inhibitoren
der Zellzyklusprogression und speziell der CDKs wurden bereits in mehreren
Publikationen ausführlich
zusammenfassend beschrieben (Sielecki T.M. et al. (2000). Cyclin-dependent
kinase inhibitors: useful targets in cell cycle regulation. J. Med.
Chem. 43, 1-18; Fry D.W. & Garrett
M.D. (2000). Inhibitors of cyclin-dependent kinases as therapeutic
agents for the treatment of cancer. Curr. Opin. Oncol. Endo. Metab.
Invest. Drugs 2, 40-59; Rosiania G.R. & Chang Y.T. (2000). Targeting hyperproliferative disorders
with cyclin dependent kinase inhibitors. Exp. Opin. Ther. Patents
10, 215-230; Meijer L. et al. (1999). Properties and potential applications
of chemical inhibitors of cyclindependent kinases. Pharmacol. Ther.
82, 279-284; Senderowicz A.M. & Sausville
E.A. (2000). Preclinical and clinical development of cyclin-dependent kinase
modulators. J. Natl. Cancer Inst. 92, 376-387).
-
Zur Verwendung der erfindungsgemäßen Verbindungen
als Arzneimittel werden diese in die Form eines pharmazeutischen
Präparats
gebracht, das neben dem Wirkstoff für die enterale oder parenterale
Applikation geeignete pharmazeutische, organische oder anorganische
inerte Trägermaterialien,
wie zum Beispiel, Wasser, Gelatine, Gummi arabicum, Milchzucker,
Stärke,
Magnesiumstearat, Talk, pflanzliche Öle, Polyalkylenglykole usw.
enthält.
Die pharmazeutischen Präparate
können
in fester Form, zum Beispiel als Tabletten, Dragees, Suppositorien,
Kapseln oder in flüssiger
Form, zum Beispiel als Lösungen,
Suspensionen oder Emulsionen vorliegen. Gegebenenfalls enthalten
sie darüber
hinaus Hilfsstoffe, wie Konservierungs-, Stabilisierungs-, Netzmittel
oder Emulgatoren; Salze zur Veränderung
des osmotischen Drucks oder Puffer.
-
Diese pharmazeutischen Präparate sind
ebenfalls Gegenstand der vorliegenden Erfindung.
-
Für
die parenterale Anwendung sind insbesondere Injektionslösungen oder
Suspensionen, insbesondere wäßrige Lösungen der
aktiven Verbindungen in polyhydroxyethoxyliertem Rizinusöl, geeignet.
-
Als Trägersysteme können auch
grenzflächenaktive
Hilfsstoffe wie Salze der Gallensäuren oder tierische oder pflanzliche
Phospholipide, aber auch Mischungen davon sowie Liposomen oder deren
Bestandteile verwendet werden.
-
Für
die orale Anwendung sind insbesondere Tabletten, Dragees oder Kapseln
mit Talkum und/oder Kohlenwasserstoffträger oder -binder, wie zum Beispiel
Lactose, Mais- oder Kartoffelstärke,
geeignet. Die Anwendung kann auch in flüssiger Form erfolgen, wie zum
Beispiel als Saft, dem gegebenenfalls ein Süßstoff beigefügt ist.
-
Die enteralen, parenteralen und oralen
Applikationen sind ebenfalls Gegenstand der vorliegenden Erfindung.
-
Die Dosierung der Wirkstoffe kann
je nach Verabfolgungsweg, Alter und Gewicht des Patienten, Art und
Schwere der zu behandelnden Erkrankung und ähnlichen Faktoren variieren.
Die tägliche
Dosis beträgt 0,5-1000
mg, vorzugsweise 50-200 mg, wobei die Dosis als einmal zu verabreichende
Einzeldosis oder unterteilt in 2 oder mehreren Tagesdosen gegeben
werden kann.
-
Ebenfalls Gegenstand der vorliegenden
Erfindung ist die Verwendung der Verbindungen der allgemeinen Formel
I, zur Herstellung eines Arzneimittels zur Behandlung von Krebs,
Autoimmunerkrankungen, kardiovaskulären Erkrankungen, Chemotherapeutika-induzierter
Alopezie und Mukositis, infektiösen
Erkrankungen, nephrologischen Erkrankungen, chronischen und akuten
neurodegenerativen Erkrankungen und viralen Infektionen, wobei unter
Krebs solide Tumoren und Leukämie,
unter Autoimmunerkrankungen Psoriasis, Alopezie und Multiple Sklerose,
unter kardiovaskulären
Erkrankungen Stenosen, Arteriosklerosen und Restenosen, unter infektiösen Erkrankungen
durch unizelluläre
Parasiten hervorgerufene Erkrankungen, unter nephrologischen Erkrankungen
Glomerulonephritis, unter chronisch neurodegenerativen Erkrankungen
Huntington's Erkrankung, amyotrophe Lateralsklerose, Parkinsonsche
Erkrankung, AIDS Dementia und Alzheimer'sche Erkrankung, unter akut
neurodegenerativen Erkrankungen Ischämien des Gehirns und Neurotraumata,
und unter viralen Infektionen Cytomegalus-Infektionen, Herpes, Hepatitis
B oder C, und HIV Erkrankungen zu verstehen sind.
-
Ebenfalls Gegenstand der vorliegenden
Erfindung sind Arzneimittel zur Behandlung der oben aufgeführten Erkrankungen,
die mindestens eine Verbindung gemäß der allgemeinen Formel I
enthalten, sowie Arzneimittel mit geeigneten Formulierungs- und Trägerstoffen.
-
Die erfindungsgemäßen Verbindungen der allgemeinen
Formel I sind unter anderem hervorragende Inhibitoren der Zyklin-abhängigen Kinasen,
wie CDK1, CDK2, CDK3, CDK4, CDK5, CDK6, CDK7, CDK8 und CDK9, sowie
der Glycogen-Synthase-Kinase (GSK-3β).
-
Soweit die Herstellung der Ausgangsverbindungen
nicht beschrieben wird, sind diese bekannt oder analog zu bekannten
Verbindungen oder hier beschriebenen Verfahren herstellbar. Es ist
ebenfalls möglich, alle
hier beschriebenen Umsetzungen in Parallel-Reaktoren oder mittels
kombinatorischer Arbeitstechniken durchzuführen. Die Isomerengemische
können
nach üblichen
Methoden wie beispielsweise Kristallisation, Chromatographie oder
Salzbildung in die Enantiomeren bzw. E/Z-Isomeren aufgetrennt werden.
-
Die Herstellung der Salze erfolgt
in üblicher
Weise, indem man eine Lösung
der Verbindung der Formel I mit der äquivalenten Menge oder einem Überschuß einer
Base oder Säure,
die gegebenenfalls in Lösung ist,
versetzt und den Niederschlag abtrennt oder in üblicher Weise die Lösung aufarbeitet.
-
Herstellung der erfindungsgemäßen Verbindungen
-
Die nachfolgenden Beispiele erläutern die
Herstellung der erfindungsgemäßen Verbindungen,
ohne den Umfang der beanspruchten Verbindungen auf diese Beispiele
zu beschränken.
-
Die erfindungsgemäßen Verbindungen der allgemeinen
Formel I lassen sich neben dem bereits oben beschriebenen erfindungsgemäßen Eintopfverfahren
auch gemäß den folgenden
allgemeinen Verfahrensvarianten herstellen:s
-
Verfahrensvariante
1 Herstellung
der 5-Brom-Derivate
-
In den allgemeinen Formeln hat B
die unter der allgemeinen Formel I angegebene Bedeutung.
-
Beispiel
1.0 Herstellung
von 1
5-Brom-4-thia-2,5,11-triaza-1(2,4)-pyrimidina-3(1,3)-benzenacycloundecaphane
4,4-dioxide
-
Eine Lösung von 100 mg (0,22 mmol)
3-Amino-N-[5-(5-brom-2-chlor-pyrimidin-4-ylamino)-pentyl]-benzolsulfonamid in
Acetonitril/Wasser/2-Butanol (8,5 ml/1,5 ml/ 0,5 ml) wird mittels
Spritzenpumpe innerhalb von 2 Stunden zu einer refluxierenden Lösung von
Acetonitril/Wasser/4 molare Lösung
von Chlorwasserstoff in Dioxan (45 ml/5 ml/0,6 ml) gegeben. Nach
weiteren 60 min wird das Acetonitril am Rotationsverdampfer abgezogen
und der Rückstand
mit Wasser (30 ml) versetzt. Man extrahiert mit Essigester (3x).
Die vereinten organischen Phasen werden mit 1M NaHCO3-Lösung, 10%
Zitronensäure,
1M NaHCO3 Lösung gewaschen, getrocknet
(Na2SO4), filtriert
und eingeengt. Man erhält
83 mg (0,20 mmol, entsprechend 90 der Theorie) des Produktes.
1N-NMR (DMSO): 9.65 (s, 1H), 8.78 (s, 1H),
8.03 (s, 1H), 7.43 (m, 2H), 7.30 (m, 2H), 7.22 (t, 1H), 3.42 (m,
2H), 2.75 (m, 2H), 1.65 (m, 2H), 1.42 (m, 4H).
13C-NMR
(DMSO): 158.5s, 158.3s, 156.1d, 140.9s, 139.7s, 130.0d, 122.7d,
118.8d, 117.9d, 93.1s, 66.7t, 41.0t, 27.0t, 26.1t, 22.8t.
MS:
412 (ES).
-
Herstellung
der Zwischenprodukte a)
Herstellung von 3-Amino-N-[5-(5-brom-2-chlor-pyrimidin-4-ylamino)-pentyl]-benzenesulfonamid
-
Eine Lösung von 300 mg (0,63 mmol)
N-[5-(5-Brom-2-chlor-pyrimidin-4-ylamino)pentyl]-3-nitro-benzenesulfonamid
in 6 ml Ethanol wird mit 600 mg Zinn(II)chlorid versetzt und 30
min bei 70°C
gerührt.
Nach dem Abkühlen
wird das Reaktionsgemisch vorsichtig auf Eiswasser gegeben und mit
gesättigter
NaHCO3 Lösung basisch
gestellt. Man extrahiert mit Essigester (3x). Die vereinten organischen
Phasen werden getrocknet (Na2SO4),
filtriert und eingeengt. Der verbleibende Rückstand wird chromatographisch
gereinigt (Essigester/Hexan 4:1). Man erhält 112 mg (0,25 mmol, entsprechend
40% der Theorie) des Produktes.
1H-NMR
(DMSO): 8.20 (s, 1H), 7.70 (br, 1H), 7.31 (br, 1H), 7.15 (t, 1H),
6.94 (m, 1H), 6.85 (m, 1H), 6.71 (m, 1H), 5.52 (s, 2H), 3.30 (m,
2H), 2.71 (m, 2H), 1.45 (m, 4H), 1.21 (m, 2H).
MS: 448 (ES).
-
b)
Herstellung von N-[5-(5-Brom-2-chlor-pyrimidin-4-ylamino)-pentyl]-3-nitro-benzenesulfonamid
-
Eine Lösung von 1,2 g (5,3 mmol) 5-Brom-2,4-dichlor-pyrimidin
in 30 ml Acetonitril wird zu einer Lösung von 1,5 g (5,2 mmol) N-(5-Amino-pentyl)-3-nitro-benzenesulfonamid
in 50 ml Acetonitril gegeben. Das Reaktionsgemisch wird mit 1,0
ml (7,2 mmol) Triethylamin versetzt und 17 Stunden bei Raumtemperatur
gerührt.
Nach der Zugabe von Wasser (50 ml) wird mit Essigester extrahiert
(2x). Die vereinten organischen Phasen werden getrocknet (Na2SO4), filtriert
und eingeengt. Der verbleibende Rückstand wird chromatographisch gereinigt
(Hexan/Essigester 2:1, Flashmaster II). Man erhält 1,5 g (3,1 mmol, entsprechend
60% der Theorie) des Produktes.
1H-NMR
(DMSO): 8.49 (m, 2H), 8.19 (m, 2H), 7.88 (m, 2H), 7.68 (t, 1H),
3.30 (m, 2H), 2.79 (m, 2H), 1.45 (m, 4H), 1.21 (m, 2H).
MS:
478 (ES).
-
c)
Herstellung von N-(5-Amino-pentyl)-3-nitro-benzenesulfonamid
-
5,00 g (12,9 mmol) [5-(3-Nitro-benzenesulfonylamino)-pentyl]-carbamidsäure-tertbutylester
werden mit 15 ml Trifluoressigsäure
versetzt und 90 min bei Raumtemperatur gerührt. Das Reaktionsgemisch wird
eingeengt und der Rückstand
mit gesättigter
NaHCO3 Lösung
basisch gestellt. Anschließend
wird mit Essigester extrahiert (2x). Die vereinten organischen Phasen
werden mit gesättigter
NaCl Lösung
gewaschen, getrocknet (Na2SO4),
filtriert und eingeengt. Man erhält
3,4 g (11,8 mmol, entsprechend 91 % der Theorie) des Produktes.
1N-NMR (DMSO): 8.49 (m, 2H), 8.19 (dd, 1H),
7.90 (t, 1H), 7.60 (br, 3H), 2.73 (m, 4H), 1.35 (m, 6H).
-
d)
Herstellung von [5-(3-Nitro-benzenesulfonylamino)-pentyl]-carbamidsäure-tert-butylester
-
Zu einer Lösung von 3,21 g (14,5 mmol)
3-Nitrobenzolsulfonylchlorid und 3,0 ml (14,4 mmol) N-Boc-1,5-diaminopentan
in 50 ml Aceton und 15 ml Wasser werden 4,2 ml (30,1 mmol) Triethylamin
gegeben. Das Reaktionsgemisch wird eine Stunde bei Raumtemperatur
gerührt.
Anschließend
wird das Aceton am Rotationsverdampfer abgezogen. Nach der Zugabe
von Wasser (20 ml) wird mit Essigester extrahiert (2x).
-
Die vereinten organischen Phasen
werden getrocknet (Na2SO4),
filtriert und eingeengt. Man erhält 5,00
g (12,9 mmol, entsprechend 90% der Theorie) des Produktes als hell
gelbes Öl.
1H-NMR (DMSO): 8.49 (m, 2H), 8.19 (dd, 1H),
7.88 (m, 2H), 6.72 (t, 1H), 2.82 (m, 4H), 1.32 (m, 15H).
-
Beispiel
1.2 Herstellung
von 1
5-Brom-4-thia-2,5,9-triaza-1(2,4)-pyrimidina-3(1,3)-benzenacyclononaphane-4,4-dioxid
-
Methode A
-
Eine Lösung von 200 mg (0,48 mmol)
3-Amino-N-[3-(5-brom-2-chlor-pyrimidin-4-ylamino)-propyl]-benzenesulfonamid in
Acetonitril/Wasser/2-Butanol (9,0 ml/1,0 ml/0,3 ml) wird mittels
Spritzenpumpe innerhalb von 2.5 Stunden zu einer refluxierenden
Lösung
von Acetonitril/Wasser/4 molare Lösung von Chlorwasserstoff in
Dioxan (45 ml/5 ml/0,6 ml) gegeben. Nach weiteren 3 Stunden unter
Rückfluss
wird das Ölbad abgeschaltet
und die Reaktionslösung über Nacht
bei Raumtemperatur gerührt.
Der gebildete Niederschlag wird abfiltriert, mit Wasser gewaschen
und anschließend
im Vakuum getrocknet. Man erhält
112 mg (0,31 mmol) des Produktes. Das Filtrat wird am Rotationsverdampfer
eingeengt. Der gebildete Niederschlag wird mit Wasser gewaschen
und abfiltriert. Nach dem Trocknen erhält man weitere 45 mg (0,12
mmol) des Produktes. Die Gesamtausbeute an Produkt beträgt somit
157 mg (0,41 mmol, entsprechend 85% der Theorie).
-
Methode B
-
Eine Lösung von 450 mg (1,00 mmol)
N-[3-(5-Brom-2-chlor-pyrimidin-4-ylamino)-propyl]-3-nitro-benzenesulfonamid in
9,5 ml Ethanol wird mit 960 mg Zinn(II)chlorid versetzt und 30 min
bei 70°C
gerührt.
Nach dem Abkühlen
wird das Reaktionsgemisch vorsichtig auf Eiswasser gegeben und mit
1N NaOH Lösung
basisch gestellt. Man extrahiert mit Essigester (3x). Die vereinten
organischen Phasen werden getrocknet (Na2SO4), filtriert und eingeengt. Der verbleibende
Rückstand
wird chromatographisch gereinigt (Essigester/Hexan 4:1). Man erhält 72 mg
des Rohproduktes. Man versetzt mit 1N HCl und extrahiert mit Essigester. Aus
der wässrigen
Phase fällt
ein farbloser Feststoff aus. Der Feststoff wird abfiltriert und
getrocknet. Man erhält
20 mg (0,05 mmol, entsprechend 5% der Theorie) des Produktes.
1H-NMR (DMSO): 10.45 (s, 1H), 9.07 (s, 1H),
8.35 (br, 1H), 8.18 (s, 1H), 7.78 (t, 1H), 7.45 (m, 2H), 7.32 (m, 1H),
3.44 (m, 2H), 3.28 (m, 2H), 1.82 (m, 2H).
MS: 384 (ES).
-
Herstellung
der Zwischenprodukte e)
Herstellung von 3-Amino-N-[3-(5-brom-2-chlor-pyrimidin-4-ylamino)-propyl]-benzenesulfonamid
-
Eine Lösung von 1,35 g (2,99 mmol)
N-[3-(5-Brom-2-chlor-pyrimidin-4-ylamino)-propyl]-3-nitro-benzenesulfonamid in
100 ml Tetrahydrofuran wird unter Argon bei Raumtemperatur mit 15
ml einer 15%igen Lösung von
Ti(III)Cl in etwa 10%iger Salzsäure
versetzt. Nach 17 Stunden wird die Reaktionslösung erneut mit 1 ml der Ti(III)Cl-Lösung versetzt
und weitere 3 h gerührt.
Der Ansatz wird mit 1N NaOH Lösung
basisch gestellt und anschließend
filtriert. Der Filterkuchen wird 2x mit jeweils 100 ml Essigester/MeOH
(30 ml/20 ml) nachgewaschen. Das Filtrat wird am Rotationsverdampfer
eingeengt und danach mit Essigester extrahiert (2x). Die vereinten
organischen Phasen werden mit NaCl-Lösung gewaschen, getrocknet
(Na2SO4), filtriert
und eingeengt. Der verbleibende Rückstand wird chromatographisch
gereinigt (Dichlormethan/MeOH 95:5, Flashmaster II). Man erhält 624 mg
(1,48 mmol, entsprechend 49% der Theorie) des Produktes.
1N-NMR (DMSO): 8.21 (s, 1H), 7.63 (t, 1 H),
7.38 (t, 1H), 7.13 (t, 1H), 6.97 (m, 1H), 6.83 (m, 1H), 6.71 (m,
1H), 5.53 (s, 2H), 3.30 (m, 2H), 2.75 (m, 2H), 1.65 (m, 2H).
-
Beispiel
1.3 Herstellung
von rac-1
5-Brom-4-thia-2,5,9-triaza-1(2,4)-pyrimidina-3(1,3)-benzenacyclononaphan-7-ol-4,4-dioxid
-
Eine Lösung von 150 mg (0,34 mmol)
3-Amino-N-[3-(5-brom-2-chlor-pyrimidin-4-ylamino)-2-hydroxy-propyl]-benzolsulfonamid
in Acetonitril/Wasser (9,0 ml/1,0 ml) wird mittels Spritzenpumpe
innerhalb von 2,5 Stunden zu einer refluxierenden Lösung von
Acetonitril/Wasser/4 molare Lösung
von Chlorwasserstoff in Dioxan (45 ml/5 ml/0,6 ml) gegeben. Nach
weiteren 4 Stunden unter Rückfluss
wird das Ölbad
abgeschaltet und die Reaktionslösung über Nacht
bei Raumtemperatur gerührt.
Der gebildete Niederschlag wird abfiltriert, mit MeCN gewaschen
und anschließend
im Vakuum getrocknet. Man erhält
125 mg (0,31 mmol, entsprechend 91 % der Theorie) des Produktes.
1H-NMR (DMSO): 10.65 (br, 1H), 9.03 (s, 1H),
8.41 (br, 1H), 8.22 (s, 1H), 7.93 (m, 1H), 7.46 (m, 2H), 7.34 (m, 1H),
4.14 (m, 1H), 3.94 (dd, 1H), 3.49 (m, 1H), 2.88 (m, 2H).
MS:
402 (ES).
-
Herstellung
der Zwischenprodukte f)
3-Amino-N-[3-(5-brom-2-chlor-pyrimidin-4-ylamino)-2-hydroxy-propyl]-benzenesulfonamid
-
Eine Lösung von 258 mg (0,553 mmol)
N-[3-(5-Brom-2-chlor-pyrimidin-4-ylamino)-2-hydroxy-propyl]-3-nitro-benzenesulfonamid
in 20 ml Tetrahydrofuran wird unter Argon bei Raumtemperatur mit
2,6 ml einer 15%igen Lösung
von Ti(III)Cl in etwa 10%igen Salzsäure versetzt. Nach 2 Stunden
wird die Reaktionslösung erneut
mit 0,2 ml der Ti(III)Cl-Lösung
versetzt und weitere 60 min gerührt.
Der Ansatz wird mit 1 M NaOH Lösung
basisch gestellt und anschließend
filtriert. Der Filterkuchen wird 2x mit jeweils 50 ml Essigester/MeOH (30
ml/20 ml) nachgewaschen. Das Filtrat wird am Rotationsverdampfer
eingeengt und danach mit Essigester extrahiert (2x). Die vereinten
organischen Phasen werden mit NaCl-Lösung gewaschen, getrocknet
(Na2SO4), filtriert
und eingeengt. Der verbleibende Rückstand wird chromatographisch
gereinigt (Dichlormethan/MeOH 95:5, Flashmaster III). Man erhält 155 mg
(0,36 mmol, entsprechend 64% der Theorie) des Produktes.
1N-NMR (DMSO): 8.25 (s, 1H), 7.43 (t, 1H),
7.36 (t, 1H), 7.13 (t, 1H), 6.96 (m, 1H), 6.86 (m, 1 H), 6.71 (m,
1 H), 5.53 (s, 2H), 5.14 (d, 1H), 3.70 (m, 1H), 3.30 (m, 2H), 2.72
(m, 2H).
-
Verfahrensvariante
2 Herstellung
der 5-Carboxamid-Derivate
-
B hat die unter der allgemeinen Formel
I angegebene Bedeutung.
-
Beispiel
2.0 Herstellung
von N-tert-Butyl-4-thia-2,5,9-triaza-1(2,4)-pyrimidina-3(1,3)-benzenacyclononaphan-1
5-carboxamid-4,4-dioxid
-
Eine Lösung von 150 mg (0,32 mmol)
2-Chloro-4-[3-(3-nitro-benzenesulfonylamino)-propylamino]-pyrimidin-5-carboxylsäure-tert-butylamid
in 10 ml THF wird unter Argon bei Raumtemperatur mit 1,6 ml einer 15%igen
Lösung
von Ti(III)Cl in etwa 10%iger Salzsäure versetzt. Nach 17 Stunden
wird die Reaktionslösung erneut
mit 0,3 ml der Ti(III)Cl-Lösung
versetzt und weitere 4 Stunden gerührt. Der Ansatz wird mit 1M
NaOH Lösung
basisch gestellt und anschließend
filtriert. Der Filterkuchen wird 2x mit jeweils 50 ml Essigester/MeOH (30
ml/20 ml) nachgewaschen. Das Filtrat wird am Rotationsverdampfer
eingeengt und danach mit Essigester extrahiert (2x). Die vereinten
organischen Phasen werden getrocknet (Na2SO4), filtriert und eingeengt. Beim Einengen
fällt ein
farbloser Feststoff aus, der abfiltriert und getrocknet wird. Man
erhält
25 mg (0,06 mmol, entsprechen 18% der Theorie) des Produktes.
1N-NMR (DMSO): 9.95 (s, 1H), 9.45 (s, 1H),
8.82 (t, 1H), 8.49 (s, 1H), 7.78 (t, 1H), 7.58 (s, 1H), 7.38 (m,
3H), 3.50 (m, 2H), 3.30 (m, 2H), 1.86 (m, 2H)
MS: 405 (ES)
-
Herstellung
der Zwischenprodukte g)
2-Chlor-4-[3-(3-nitro-benzenesulfonylamino)-propylamino]-pyrimidin-5-carbonsäure-tert-butylamid
-
Eine Lösung von 0,95 g (3,83 mmol)
2,4-Dichlor-pyrimidin-5-carbonsäure-tert-butylamid in 6 ml
THF wird bei Raumtemperatur unter Rühren mit einer Suspension aus
1,00 g (3,86 mmol) N-(3-Amino-propyl)-3-nitro-benzenesulfonamid
in 9 ml THF/ 0,55 ml Triethylamin versetzt. Nach 19 Stunden wird
der gebildete Niederschlag abgesaugt und mit Essigester gewaschen.
Das Filtrat wird einrotiert und der gebildete Rückstand chromatographisch gereinigt
(Hexan/Essigester 2:1, Flashmaster II). Man erhält 0,79 g (1,67 mmol, entsprechend 44%
der Theorie) des Produktes.
1N-NMR
(DMSO): 8.74 (t, 1H), 8.47 (m, 3H), 8.18 (dd, 1H), 8.04 (t, 1H),
7.98 (s, 1H), 7.85 (t, 1H), 3.30 (m, 2H), 2.87 (m, 2H), 1.68 (m,
2H), 1.36 (s, 9H)
-
h)
Herstellung von 2,4-Dichlor-pyrimidin-5-carbonsäure-tert-butylamide
-
Eine Lösung von 24,85 g (117,5 mmol)
2,4-Dichlor-pyrimidin-5-carbonylchlorid in 125 ml THF wird auf –15 °C abgekühlt. Man
versetzt langsam mit einer Lösung
von 13.2 ml (124,5 mmol) tert-Buylamin und 17,4 ml (125,7 mmol)
Triethylamin in 50 ml THF, so dass die Temperatur des Reaktionsgemisches
kleiner –10 °C bleibt.
Es wird weitere 2 Stunden bei –10 °C gerührt, dann
wird das Kühlbad
entfernt und das Reaktionsgemisch unter Rühren auf Raumtemperatur erwärmt. Nach
1 Stunde wird der gebildete Niederschlag abfiltriert und das Filtrat
vollständig
eingeengt. Der erhaltene Rückstand
wird chromatographisch gereinigt (Hexan/Essigester 4:1). Man erhält 14,01
g (56,6 mmol, entsprechend 50% der Theorie) des Produktes.
1N-NMR (DMSO): 8.81 (s, 1H), 8.34 (s, 1H),
1.36 (s, 9H)
-
i)
Herstellung von 2,4-Dichlor-pyrimidin-5-carbonylchlorid
-
Eine Suspension von 21,7 g (139 mmol)
2,4-Dihydroxy-5-carbonsäure-pyrimidin,
96,7 g (463 mmol) Phosphorpentachlorid und 33 ml (348 mmol) Phosphoroxydchlorid
wird 5 Stunden bei 115 °C
gerührt.
Nach dem Abkühlen
wird das Reaktionsgemisch am Rotationsverdampfer eingeengt. Der
gebildete Rückstand
wird durch Vakuumdestillation (Kp0.1mbar :
68 °C) gereinigt.
Man erhält
24,9 g (117 mmol, entsprechend 84 % der Theorie) des Produktes.
1N-NMR (DMSO): 9.11 (s, 1H)
-
Verfahrensvariante
3 Herstellung
der 5-Cyano-Derivate
-
B hat die unter der allgemeinen Formel
I angegebene Bedeutung.
-
Beispiel
3.0 Herstellung
von 1
5-Cyano-4-thia-2,5,9-triaza-1(2,4)-pyrimidina-3(1,3)-benzenacyclononaphan
4,4-dioxid
-
Eine Lösung von 100 mg (0,25 mmol)
N-[3-(2-Chlor-5-cyano-pyrimidin-4-ylamino)-propyl]-3-nitro-benzenesulfonamid in
10 ml THF wird unter Argon bei Raumtemperatur mit 1,2 ml einer 15%igen
Lösung
von Ti(III)Cl in etwa 10%iger Salzsäure versetzt. Nach 3,5 Stunden
wird der Ansatz mit Essigester verdünnt, mit 1M NaOH Lösung basisch
gestellt (pH 13) und anschließend
filtriert. Der Filterkuchen wird mit 50 ml Essigester/MeOH (30 ml/20
ml) und 70 ml Essigester/ MeOH/1N NaOH (40 ml/20 ml/10 ml) nachgewaschen.
Das Filtrat wird am Rotationsverdampfer eingeengt und danach mit
Essigester extrahiert (2x). Die vereinten organischen Phasen werden
getrocknet (Na2SO4),
filtriert und eingeengt. Beim Einengen fällt das Produkt als farbloser
Feststoff aus, der abfiltriert wird. Man erhält 30 mg (0,09 mmol, entsprechend
36% der Theorie) des Produktes.
1N-NMR
(DMSO): 10.29 (s, 1H), 9.29 (s, 1H), 8.39 (s, 1H), 8.15 (br, 1H),
7.78 (br, 1H), 7.38 (m, 3H), 3.30 (m, 4H), 1.85 (m, 2H)
MS:
331 (ES)
-
Herstellung
der Zwischenpodukte j)
Herstellung von N-[3-(2-Chlor-5-cyano-pyrimidin-4-ylamino)-propyl]-3-nitro-benzenesulfonamid
-
125 mg (0,27 mmol) 2-Chlor-4-[3-(3-nitro-benzenesulfonylamino)-propylamino]-pyrimidin-5-carbonsäure-tert-butylamid
werden mit 4 ml Thionylchlorid versetzt und 19 Stunden unter Rückfluss
gerührt.
Das Reaktionsgemisch wird eingeengt. Man versetzt mit Wasser und
Toluol und engt am Rotationsverdampfer bis zur Trockene ein. Man
erhält
110 mg (0,27 mmol, entsprechend 100% der Theorie) des Produktes.
1N-NMR (DMSO): 8.50 (m, 4H), 8.19 (d, 1H),
8.01 (t, 1H), 7.88 (t, 1H), 3.30 (m, 2H), 2.87 (m, 2H), 1.71 (m,
2H)
-
Verfahrensvariante
4 Herstellung
der Thiophen-Derivate
-
B hat die unter der allgemeinen Formel
I angegebene Bedeutung.
-
Beispiel
4.0 Herstellung
von 1
5-Brom-4-thia-2,5,8-triaza-1(2,4)-pyrimidina-3(4,2)-thiophenacyclooctaphan-4,4-dioxid
-
Eine Lösung von 170 mg (0,41 mmol)
4-Amino-thiophen-2-sulfonsäure-[2-(5-brom-2-chlor-pyrimidin-4-ylamino)-ethyl]-amid
in Acetonitril/Wasser (12,m 0 ml/1,5 ml) wird mittels Spritzenpumpe
innerhalb von 2 Stunden zu einer refluxierenden Lösung von
Acetonitril/Wasser/4 molare Lösung
von Chlorwasserstoff in Dioxan (64 ml/7 ml/ 0,8 ml) gegeben. Nach
Beendigung der Zugabe wird das Reaktionsgemisch für weitere
6 Stunden unter Rückfluss
gerührt.
Nach dem Abkühlen
wird das Lösungsmittel
am Rotationsverdampfer abgezogen. Der Rückstand wird mit 2N NaOH versetzt
und mit Essigester extrahiert (2x). Die vereinten organischen Phasen
werden mittels Whatman-Filter filtriert und eingeengt. Der verbleibende
Rückstand
wird aus MeOH/Disopropylether kristallisiert. Man erhält 41 mg
(0,11 mmol, entsprechend 27% der Theorie) des Produktes.
1H-NMR (DMSO): 9.03 (s, 1H), 7.92 (s, 1H),
7.68 (d, 1H), 7.48 (br, 1H), 7.38 (d, 1H), 7.08 (t, 1H), 2.91 (m,
4H)
MS: 376 (ES)
-
Herstellung
der Zwischenprodukte k)
4-Amino-thiophen-2-sulfonsäure-[2-(5-Brom-2-chlor-pyrimidin-4-ylamino)-ethyl]-amid
-
Eine Lösung von 600 mg (1,35 mmol)
eines Gemisches von 4-Nitro-thiophen-2-sulfonsäure [2-(5-brom-2-chlor-pyrimidin-4-ylamino)-ethyl]-amid
und 5-Nitro-thiophen-2-sulfonsäure-[2-(5-brom-2-chlor-pyrimidin-4-ylamino)-ethyl]-amid
(Verhältnis
10/6) in 40 ml THF wird unter Argon bei Raumtemperatur mit 6,4 ml
einer 15%igen Lösung
von Ti(III)Cl in etwa 10%iger Salzsäure versetzt. Nach 46 Stunden
wird die Reaktionslösung
erneut mit 2,0 ml der Ti(III)Cl-Lösung versetzt und weitere 7
Stunden gerührt.
Der Ansatz wird mit 2N NaOH Lösung
basisch gestellt und anschließend
filtriert. Der Filterkuchen wird 2x mit jeweils 50 ml Essigester/MeOH
(30 ml/20 ml) nachgewaschen. Das Filtrat wird am Rotationsverdampfer eingeengt
und danach mit Essigester extrahiert (2x). Die vereinten organischen
Phasen werden über
einem Whatman-Filter filtriert und eingeengt. Der Rückstand
wird chromatographisch (Hexan/Essigester 1 : 4) gereinigt. Man erhält 178 mg
(0,43 mmol, entsprechend 32 % der Theorie) des Produktes.
1N-NMR (DMSO): 8.21 (s, 1H), 7.82 (t, 1H),
7.65 (t, 1H), 7.03 (d, 1H), 6.31 (d, 1H), 5.21 (br, 2H), 3.44 (m,
2H), 3.02 (m, 2H) MS: 412 (ES)
-
l)
Herstellung von 4-Nitro-thiophen-2-sulfonsäure-[2-(5-brom-2-chlor-pyrimidin-4-ylamino)-ethyl]-amide
(A) und 5-Nitro-thiophen-2-sulfonsäure-[2-(5-brom-2-chlor-pyrimidin-4-ylamino)-ethyl]-amid
(B)
-
2,65 g (7,54 mmol) eines Gemisches
von [2-(4-Nitro-thiophen-2-sulfonylamino)ethyl]-carbaminsäure-tert-butylester
und [2-(5-Nitro-thiophen-2-sulfonylamino)-ethyl]carbaminsäure-tert-butylester
im Verhältnis 1/1
werden mit 9 ml TFA versetzt und 2,5 Stunden bei Raumtemperatur
gerührt.
Das Reaktionsgemisch wird am Rotationsverdampfer eingeengt und mit
Wasser und 1N NaOH (pH 13) versetzt. Man extrahiert mit Essigester
(2x). Die vereinten organischen Phasen werden über einen Whatman-Filter filtriert
und eingeengt. Der verbleibende Rückstand wird chromatographisch
(Dichlormethan/MeOH 1:1) aufgereinigt. Das erhaltene Rohprodukt
wird in 3 ml Acetonitril aufgenommen und mit einer Lösung von
1,37 g (3 mmol) 5-Brom-2,4-dichlor-pyrimidine/1 ml Triethylamin
(7 mmol) in 3 ml Acetonitril versetzt. Nach 16 Stunden wird das
Reaktionsgemisch am Rotationsverdampfer eingeengt und der verbleibende
Rückstand
chromatographisch (Hexan/Essigester 2:1, Flashmaster II) gereinigt.
Man erhält
0,87 g (1,97 mmol, entsprechend 26 % der Theorie) eines Gemisches der
Regioisomeren A und B im Verhältnis
10/6.
1H-NMR (DMSO): 8.98 (d, 1H, A),
8.50 (t, 1H, B), 8.32 (t, 2H, A), 8.20 (s, 2H, A+B), 8.05 (d, 1H,
B), 7.98 (d, 1H, A), 7.63 (m, 3H, A+B), 3.47 (m, 4H, A+B), 3.20
(m, 4H, A+B)
MS: 442 (ES)
-
m)
Herstellung von [2-(4-Nitro-thiophen-2-sulfonylamino)-ethyl]-carbaminsäure-tert-butylester
(A) und [2-(5-Nitro-thiophen-2-sulfonylamino)-ethyl]-carbaminsäuretert-butyl-ester
(B)
-
Zu einer Lösung von 2,27 g (10 mmol) eines
Gemisches von 4-Nitro-thiophene-2-sulfonylchlorid und 5-Nitro-thiophen-2-sulfonylchlorid
im Verhältnis
2/1 sowie 1,64 g (10 mmol) (2-Amino-ethyl)-carbaminsäure-tert-butylester
in 40 ml Aceton und 10 ml Wasser werden 2,8 ml (20 mmol) Triethylamin
gegeben. Das Reaktionsgemisch wird 3 Stunden bei Raumtemperatur
gerührt
und anschließend
das Aceton am Rotationsverdampfer abgezogen. Nach der Zugabe von
Wasser (20 ml) wird mit Essigester extrahiert (2x). Die vereinten organischen
Phasen werden über
einen Whatman-Filter filtriert und eingeengt. Man erhält 2,65
g (7,5 mmol, entsprechend 75% der Theorie) eines Gemisches der Verbindungen
A und B im Verhältnis
1/1.
1H-NMR (DMSO): 9.02 (d, 1H, A),
8.85 (t, 1H), 8.15 (m, 2H), 8.02 (d, 1H, A), 7.63 (d, 1H, B), 6.78
(m, 2H), 2.92 (m, 8H), 1.40 (s, 18H)
-
n)
Herstellung von 4-Nitro-thiophen-2-sulfonylchlorid (A) und 5-Nitro-thiophen-2-sulfonylchlorid
(B)
-
Eine Lösung von 25 g (137 mmol) Thiophen-2-sulfonylchlorid
in 20 ml Dichlormethan wird langsam unter Rühren zu 98 ml konz. Salpetersäure getropft.
Das Reaktionsgemisch wird 2 Stunden bei 40 °C gerührt und anschließend auf
Eis gegeben. Man extrahiert mit Dichlormethan (2x). Die vereinten
organischen Phasen werden über
MgSO4 getrocknet, filtriert und eingeengt.
Man erhält
24 g (105 mmol, entsprechend 77 % der Theorie) eines Gemisches der
Produkte A und B im Verhältnis
2/1.
1H-NMR (DMSO): 8.63 (d, 1H, A),
7.93 (d, 1H, B), 7.54 (d, 1H, A), 7.18 (d, 1H, B)
-
Verfahrensvariante
5 Herstellung
der Oxa-phane und Einführung
von Sulfamoylgruppierungen
-
B, R8 und
R9 haben die unter der allgemeinen Formel
I genannten Bedeutungen.
-
Beispiel
5.0 Herstellung
von 1(5)-Brom-N,N'-dimethyl-4-oxa-2,9-diaza-1(2,4)-pyrimidina-3(1,3)-benzenacyclononaphan-3(4),3(6)-disulfonamid
-
10 ml Chlorsulfonsäure werden
unter Kühlung
(4 °C) vorsichtig
mit 75 mg Phosphorpentachlorid versetzt. Man gibt 60 mg (0,18 mmol)
15-Brom-4-oxa-2,9-diaza-1(2,4)-pyrimidina-3(1,3)-benzenacyclononaphan hinzu
und rührt
3 Stunden bei Raumtemperatur. Das Reaktionsgemisch wird vorsichtig
auf Eiswasser gegeben und 1 Stunde gerührt. Der gebildete Feststoff
wird abgesaugt und in 1 ml THF aufgenommen. Man versetzt mit 2 ml
einer Lösung
von Methylamin in Ethanol und rührt
12 Stunden bei Raumtemperatur. Das Reaktionsgemisch wird eingeengt
und der verbleibende Rückstand
chromatographisch (Dichlormethan/Methanol 1 : 1) gereinigt. Man
erhält
8 mg (0,02 mmol, entsprechend 10% der Theorie) des Produktes.
1H-NMR (DMSO): 9.43 (s, 1H), 9.17 (s, 1H),
8.15 (s, 1H), 8.03 (s, 1H), 7.95 (q, 1H), 7.72 (t, 1H), 7.15 (q,
1H), 4.55 (br, 2H), 3.42 (br, 2H), 2.45 (m, 6H), 1.91 (m, 4H)
MS:
521 (ES).
-
Beispiel
5.1 Herstellung
von Brom-4-oxa-2,9-diaza-1(2,4)-pyrimidina-3(1,3)-benzenacyclononaphan
-
Eine Lösung von 295 mg (0,79 mmol)
[4-(3-Amino-phenoxy)-butyl]-(5-brom-2-chlorpyrimidin-4-yl)-amin
in Acetonitril/Wasser (18 ml/2 ml) wird mittels Spritzenpumpe innerhalb
von 4,5 Stunden zu einer refluxierenden Lösung von Acetonitril/Wasser/
4 molare Lösung
von Chlorwasserstoff in Dioxan (105 ml/12 ml/1,4 ml) gegeben. Nach
weiteren 60 min unter Rückfluss
wird das Ölbad
abgeschaltet und die Reaktionslösung über Nacht
bei Raumtemperatur gerührt.
Das Reaktionsgemisch wird mit 2N NaOH basisch gestellt und mit Essigester
extrahiert (3x): Die vereinten organischen Phasen werden mit NaCl – Lösung gewaschen,
getrocknet (MgSO4), filtriert und eingeengt.
Der verbleibende Rückstand
wird mit Methanol digeriert. Man erhält 65 mg (0,19 mmol, entsprechend
24% der Theorie) des Produktes.
1H-NMR
(DMSO): 9.31 (s, 1H), 8.82 (s, 1H), 8.01 (s, 1H), 7.37 (br, 1H),
7.02 (t, 1H), 6.63 (d, 1H), 6.36 (dd, 1H), 4.34 (br, 2H), 3.30 (m,
2H), 1.85 (m, 4H)
MS: 335 (ES).
-
Herstellung
der Zwischenprodukte o)
Herstellung von 4-(3-Amino-phenoxy)-butyl]-(5-brom-2-chlor-pyrimidin-4-yl)-amin
-
Eine Lösung von 401 mg (1,00 mmol)
(5-Brom-2-chlor-pyrimidin-4-yl)-[4-(3-nitrophenoxy)-butyl]-amin in
30 ml THF wird unter Argon bei Raumtemperatur mit 4,5 ml einer 15%igen
Lösung
von Ti(III)Cl in etwa 10%iger Salzsäure versetzt. Nach 3,5 Stunden
wird die Reaktionslösung
erneut mit 0,2 ml der Ti(III)Cl-Lösung versetzt und weitere 12
Stunden gerührt.
Der Ansatz wird mit 2N NaOH Lösung
basisch gestellt und anschließend
filtriert. Der Filterkuchen wird 2x mit jeweils 50 ml Essigester/
MeOH (30 ml/20 ml) nachgewaschen. Das Filtrat wird am Rotationsverdampfer
eingeengt und danach mit Essigester extrahiert (2x). Die vereinten
organischen Phasen werden mit NaCl-Lösung gewaschen, getrocknet
(MgSO4), filtriert und eingeengt. Der verbleibende
Rückstand
wird chromatographisch gereinigt (Hexan/ Essigester 1 : 1, Flashmaster
II). Man erhält
300 mg (0,81 mmol, entsprechend 81 der Theorie) des Produktes.
1H-NMR (CDCl3): 8.10
(s, 1H), 7.03 (t, 1H), 6.36 (m, 3H), 5.66 (br, 1H), 3.97 (m, 2H),
3.60 (m, 2H), 1.86 (m, 2H)
MS: 371 (ES)
-
p)
Herstellung von (5-Brom-2-chlor-pyrimidin-4-yl)-[4-(3-nitro-phenoxy)-butyl]-amin
-
Eine Lösung 2,28 g (10 mmol) 5-Brom-2,4-dichlor-pyrimidin
und 1,4 ml Triethylamin (10 mmol) in 32 ml Acetonitril wird bei
4 °C unter
Rühren
mit einer Lösung
von 2,1 g (10 mmol) 4-(3-Nitro-phenoxy)-butylamin in 5 ml Acetonitril
versetzt. Nach 12 Stunden wird mit Essigester verdünnt und
filtriert. Das Filtrat wird am Rotationsverdampfer eingeengt und
der verbleibende Rückstand
chromatographisch (Hexan/ Essigester 1:1, Flashmaster II) gereinigt.
Man erhält
2,7 g (7 mmol, entsprechend 70 % der Theorie) des Produktes.
1H-NMR (CDCl3): 8.11
(s, 1H), 7.81 (dd, 1H), 7.71 (t, 1H), 7.44 (t, 1H), 7.20 (dd, 1H),
5.62 (br, 1H), 4.11 (m, 2H), 3.62 (m, 2H), 1.92 (m, 4H)
-
q)
Herstellung von 4-(3-Nitro-phenoxy)-butylamin
-
Eine Lösung von 17,0 g (50 mmol) 2-[4-(3-Nitro-phenoxy)-butyl]-isoindol-1,3-dion
in 1000 ml Ethanol wird mit 25 ml Hydrazin versetzt und 2 Stunden
bei 70 °C
gerührt.
Nach dem Abkühlen
wird der gebildete Niederschlag abgesaugt und das Filtrat einrotiert.
Der Rückstand
wird in Dichlormethan aufgenommen. Es wird erneut filtriert und
das Filtrat vollständig
eingeengt. Man erhält
5,8 g (28 mmol, entsprechend 56 der Theorie) des Produktes.
1H-NMR (CDCl3): 7.79
(dd, 1H), 7.71 (t, 1H), 7.39 (t, 1H), 7.20 (dd, 1H), 4.03 (m, 2H),
2.79 (m, 2H), 1.85 (m, 2H), 1.70 (m, 2H)
-
r)
Herstellung von 2-[4-(3-Nitro-phenoxy)-butyl]-isoindol-1,3-dion
-
Zu einer Lösung von 6,96 g (50 mmol) 3-Nitrophenol
in 500 ml DMF werden 9,67 g (70 mmol) Kaliumcarbonat gegeben und
anschließend
10 min bei Raumtemperatur gerührt.
Man versetzt mit 14,1 g (50 mmol) 2-(4-Brom-butyl)-isoindol-1,3-dion
und rührt
4 Stunden bei 60 °C.
Nach dem Erkalten wird mit Wasser versetzt und mit Essigester extrahiert.
Die vereinten organischen Phasen werden getrocknet (MgSO4), filtriert und eingeengt. Man erhält 17,2
g (50 mmol, entsprechend 100% der Theorie) des Produktes.
1H-NMR (CDCl3): 7.85
(m, 3H), 7.70 (m, 3H), 7.40 (t, 1H), 7.18 (dd, 1H), 4.06 (m, 2H),
3.80 (m, 2H), 1.95 (m, 4H)
-
Verfahrensvariante
6 Herstellung
der Sulfonamid-Oxa-Cyclophane
-
B, R8 und
R9 haben die unter der allgemeinen Formel
I angegebenen Bedeutungen.
-
Beispiel
6.0 Herstellung
von 1
5-Brom-4-oxa-2,9-diaza-1(2,4)-pyrimidina-3(1,3)-benzenacyclononaphan-3
4-sulfonamid
-
Eine Lösung von 66 mg (0,15 mmol)
4-Amino-2-[4-(5-brom-2-chlor-pyrimidin-4-ylamino)-butoxy]-benzenesulfonamid in
Acetonitril/Wasser/2-Butanol (8 ml/1 ml/ 1 ml) wird mittels Spritzenpumpe
innerhalb von 3,5 Stunden zu einer refluxierenden Lösung von
Acetonitril/Wasser/4 molare Lösung
von Chlorwasserstoff in Dioxan (45 ml/5 ml/0,6 ml) gegeben. Nach
weiteren 16 Stunden unter Rückfluss
wird das Reaktionsgemisch am Rotationsverdampfer eingeengt. Der
Ansatz wird mit 1 N NaOH basisch gestellt (pH 13) und mit Essigester extrahiert
(2x). Die vereinten organischen Phasen getrocknet (MgSO4),
filtriert und eingeengt. Der verbleibende Rückstand wird mit Hexan und
tert-Butylmethylether digeriert. Man erhält 55 mg (0,13 mmol, entsprechend 87%
der Theorie) des Produktes.
1H-NMR
(DMSO): 9.70 (s, 1H), 9.10 (d, 1H), 8.06 (s, 1H), 7.49 (m, 2H),
6.82 (s, 2H), 6.67 (dd, 1H), 4.45 (br, 2H), 3.40 (m, 2H), 1.85 (m,
4H) MS: 414 (ES)
-
Herstellung
der Zwischenprodukte s)
Herstellung von 4-Amino-2-[4-(5-brom-2-chlor-pyrimidin-4-ylamino)-butoxy]-benzenesulfonamid
-
Eine Lösung von 160 mg (0,33 mmol)
2-[4-(5-Brom-2-chlor-pyrimidin-4-ylamino)butoxy]-4-nitro-benzenesulfonamid
in 10 ml THF wird unter Argon bei Raumtemperatur mit 1,4 ml einer
15%igen Lösung
von Ti(III)Cl in etwa 10%iger Salzsäure versetzt. Nach 4 Stunden
wird die Reaktionslösung
erneut mit 0,2 ml der Ti(III)Cl-Lösung versetzt und weitere 14
Stunden gerührt.
Der Ansatz wird mit 2N NaOH Lösung
basisch gestellt und anschließend
filtriert. Der Filterkuchen wird 2x mit jeweils 50 ml Essigester/MeOH
(30 ml/20 ml) nachgewaschen. Das Filtrat wird am Rotationsverdampfer
eingeengt und danach mit Essigester extrahiert (2x). Die vereinten
organischen Phasen werden mit NaCl-Lösung gewaschen, getrocknet
(MgSO4), filtriert und eingeengt. Der verbleibende
Rückstand
wird chromatographisch gereinigt (Dichlormethan/Methanol 9 : 1;
Flashmaster II). Man erhält
70 mg (0,81 mmol, entsprechend 47 % der Theorie) des Produktes.
1H-NMR (DMSO): 8.22 (s, 1H), 7.73 (t, 1H),
7.36 (d, 1H), 6.44 (s, 2H), 6.27 (d, 1H), 6.13 (dd, 1H), 5.78 (s,
2H), 4.04 (m, 2H), 3.45 (m, 2H), 1.76 (m, 4H)
MS: 450 (ES)
-
t)
Herstellung von 2-[4-(5-Brom-2-chlor-pyrimidin-4-ylamino)-butoxy]-4-nitro-benzenesulfonamid
(A) und 4-[4-(5-Brom-2-chlor-pyrimidin-4-ylamino)-butoxy]-2-nitro-benzenesulfonamid
(B)
-
402 mg (1,01 mmol) (5-Brom-2-chlor-pyrimidin-4-yl)-[4-(3-nitro-phenoxy)-butyl]-amin
werden portionsweise in 4 ml eiskalter Chlorsufonsäure (Kühlung: Eis/Methanol)
eingetragen und anschliessend 3 Stunden bei Raumtemperatur gerührt. Der
Ansatz wird vorsichtig unter Rühren
auf Eiswasser gegeben. Der gebildete Niederschlag wird abgesaugt,
in Aceton aufgenommen und mit konz. Ammoniak versetzt. Man rührt für 2 Stunden bei
Raumtemperatur und engt den Ansatz am Rotationsverdampfer ein. Man
versetzt mit Wasser und extrahiert mit Essigester (2x). Die vereinten
organischen Phasen werden getrocknet (MgSO4),
filtriert und eingeengt. Der verbleibende Rückstand wird chromatographisch
(Hexan/Essigester 1 : 1) gereinigt. Man erhält 190 mg (0,40 mmol, entsprechend
40 % der Theorie) des Produktes A und 110 mg (0,23 mmol, entsprechend
23 % der Theorie) des Produktes B.
-
2-[4-(5-Brom-2-chlor-pyrimidin-4-ylamino)-butoxy]-4-nitro-benzenesulfonamid
(A):
1H-NMR (DMSO): 8.25 (s, 1H), 7.99
(d, 1H), 7.91 (m, 2H), 7.72 (br, 1H), 7.35 (br, 2H), 4.34 (m, 2H),
3.42 (m, 2H), 1.82 (m, 4H)
MS : 480 (ES)
-
4-[4-(5-Brom-2-chlor-pyrimidin-4-ylamino)-butoxy]-2-nitro-benzenesulfonamid
(B):
1H-NMR (DMSO): 8.22 (s, 1H), 7.93
(d, 1H), 7.78 (t, 1H), 7.67 (s, 2H), 7.51 (d, 1H), 7.34 (dd, 1H),
4.16 (m, 2H), 3.45 (m, 2H), 1.73 (m, 4H)
MS : 480 (ES)
-
Verfahrensvariante
7 Ringschluß von bifunktionellen
acyclischen Vorläufern
-
A, B, R2,
R3, R4, X, Y und
n haben die unter der allgemeinen Formel I angegebenen Bedeutungen.
U und V stehen für
Gruppen wie -OH, -CO2N, -CO2R,
-SO2Cl, -SO2F, -SO3H, etc.
-
Der Ringschluss bzw. die Synthese
der Macozyclen kann auch analog bekannter Methoden durchgeführt werden
((a) Roxburgh, C.J. Tetrahedron 1995, 51, 9767. (b) Meng, Q. Top.
Curr. Chem. 1991, 161, 107. (c) Paterson, I. Tetrahedron 1985, 41,
3569. (d) Masamune, S. Angew. Chem. 1977, 89, 602. (e) Nicolaou,
K.C. Tetrahedron 1977, 33, 683. (f) Ruggli, P. Liebigs Ann. Chem.
1912, 92.)
-
Verfahrensvariante
8 Ringschluss
durch Mitsunobu-Reaktion
-
B und R3 haben
die unter der allgemeinen Formel I angegebenen Bedeutungen.
-
Synthese von Macrocylcen unter Verwendung
der Mitsunobu-Reaktion ist allgemein bekannt und kann in (a) Xue,
C.-B. J. Med. Chem. 2001, 44, 2636. (b) Steglich, W. Tet. Lett.
1991, 32, 5781. (c) Mitsunobu, O. Synthesis 1981, 1 nachgelesen
werden.
-
Beispiel
5.1 Herstellung
von 1
5-Brom-4-oxa-2,9-diaza-1(2,4)-pyrimidina-3(1,3)-benzen-acyclononaphan
-
Eine Lösung von 108 mg (0,31 mmol)
3-[5-Brom-4-(4-hydroxy-butylamino)-pyrimidin-2-ylamino]-phenol in THF/N-Methylmorpholin
(9 ml/1 ml) wird unter Rühren
innerhalb von 3 Stunden zu einem Gemisch von 710 mg (2,7 mmol) Triphenylphosphin
und 481 mg (2,8 mmol) DEAD in 100 ml THF bei 40°C gegeben. Nach weiteren 30
min wird das Reaktionsgemisch am Rotationsverdampfer eingeengt.
Nach der Zugabe von Wasser wird mit Essigester extrahiert (2x).
Die vereinten organischen Phasen werden getrocknet (Na2SO4), filtriert und eingeengt. Der gebildete
Rückstand
wird chromatographisch gereinigt (Dichlormethan/ Methanol 9:1) und das
erhaltene Rohprodukt anschließend
mit Diisopropylether digeriert. Man erhält 17 mg (0,05 mmol, entsprechend
17% der Theorie) des Produktes.
1N-NMR
(DMSO): 9.18 (s, 1H), 9.08 (s, 1H), 8.04 (s, 1H), 7.20 (s, 1H),
7.09 (dd, 1H), 6.96 (t, 1H), 6.31 (dd, 1H), 3.30 (m, 4H), 1.90 (m,
4H).
MS: 334 (EI).
-
Herstellung
der Zwischenprodukte u)
3-[5-Brom-4-(4-hydroxy-butylamino)-pyrimidin-2-ylamino]-phenol
-
Ein Reaktionsgemisch von 327 mg (3,0
mmol) 3-Aminophenol und 864 mg (3,1 mmol) 4-(5-Brom-2-chlor-pyrimidin-4-ylamino)-butan-1-ol
in 9 ml Acetonitril wird mit 0,75 ml einer 4M Lösung von Chlorwasserstoff in
Dioxan versetzt und über
Nacht unter Rückfluss
gerührt.
Nach dem Erkalten wird das Reaktionsgemisch filtriert und das Filtrat
vollständig
eingeengt. Das erhaltene Öl
wird aus Essigester/Ethanol umkristallisiert. Der Feststoff wird
abfiltriert und anschließend
in Wasser gelöst.
Durch Zugabe von Triethylamin wird die Lösung basisch gestellt und mit
Essigester extrahiert (2x). Die vereinten organischen Phasen werden getrocknet
(Na2SO4), filtriert
und eingeengt. Man erhält
444 mg (1,2 mmol, entsprechend 40% der Theorie) des Produktes.
1H-NMR (DMSO): 9.18 (s, 1H), 9.06 (s, 1H),
7.97 (s, 1H), 7.19 (m, 2H), 6.99 (m, 2H), 6.32 (m, 1H), 4.45 (t,
1H), 3.40 (m, 4H), 1.60 (m, 2H), 1.47 (m, 2H).
MS: 352 (ES).
-
v)
Herstellung von 4-(5-Brom-2-chlor-pyrimidin-4-ylamino)-butan-1-ol
-
Eine Lösung von 2,28 g (10,0 mmol)
5-Brom-2,4-dichlor-pyrimidin und 1,7 ml (12,0 mmol) Triethylamin in
10 ml Acetonitril wird bei 0°C
mit 1,1 ml (12,0 mmol) 4-Aminobutanol versetzt. Das Reaktionsgemisch
wird durch Entfernen des Eisbades langsam unter Rühren auf
Raumtemperatur erwärmt.
Nach 16 Stunden wird der gebildete Niederschlag abfiltriert. Das
Filtrat wird vollständig
eingeengt und mit Diisopropylether digeriert. Man erhält 2,74
g (9,8 mmol, entsprechend 98% der Theorie) des Produktes.
1H-NMR (DMSO): 8.19 (s, 1H), 7.72 (t, 1H),
4.45 (br, 1H), 3.38 (m, 4H), 1.56 (m, 2H), 1.45 (m, 2H).
MS:
279 (EI).
-
Verfahrensvariante
9 Ringschluss
durch Macrolactamisierung
-
Synthese von Macrolactamen erfolgt
nach gängigen
Verfahren ((a) Xue, C.-B. J. Med. Chem. 2001, 44, 2636. (b) Jackson,
F.W. J. Org. Chem. 2002, 67, 4882).
-
Beispiel
7.0 Herstellung
von 1
5-Brom-2,5,11-triaza-1(2,4)-pyrimidina-3(1,3)-benzenacycloundecaphan-4-on
(A) und 2,5,11-Triaza-1(2,4)-pyrimidina-3(1,3)-benzenacycloundecaphan-4-on (B)
-
Eine Lösung von 300 mg (0,57 mmol)
3-[4-(5-Benzyloxycarbonylamino-pentylamino)-5-brom-pyrimidin-2-ylamino]-benzoesäure in Methanol/Dichlormethan
(40ml/5 ml) wird mit 350 mg Pd/C (10%) versetzt und 150 min in einer
Niederdruck-Apparatur hydriert. Das Reaktionsgemisch wird über Celite
filtriert und vollständig eingeengt.
Der verbleibende Rückstand
wird in DMF/Methanol/Wasser (10,0 ml/1,0 ml/0,2 ml) gelöst und mittels
Spritzenpumpe über
2 Stunden zu einer Lösung
von 410 mg (2,2 mmol) EDC, 330 mg (2,2 mmol) HOBt und 0,25 ml N-Methylmorpholin
in 200 ml DMF gegeben. Nach 72 Stunden wird das Reaktionsgemisch
eingeengt, mit Wasser versetzt und anschließend mit Essigester extrahiert
(2x). Die vereinten organischen Phasen werden mit Wasser gewaschen,
getrocknet (Na2SO4),
filtriert und eingeengt. Der gebildete Rückstand wird chromatographisch
gereinigt (Dichlormethan/ Methanol 1:1). Man erhält 35 mg (0,09 mmol, entsprechend
16% der Theorie) an 15-Brom-2,5,11-triaza-1(2,4)-pyrimidina-3(1,3)-benzenacycloundecaphan-4-on
(A) und 13 mg (0,04 mmol, entsprechend 7% der Theorie) an 2,5,11-Triaza-1(2,4)-pyrimidina-3(1,3)-benzenacycloundecaphan-4-on
(B).
-
15-Brom-2,5,11-triaza-1(2,4)-pyrimidina-3(1,3)-benzenacycloundecaphan-4-on
(A):
1H-NMR (DMSO): 9.45 (s, 1H), 8.81
(s, 1H), 8.12 (t, 1H), 7.98 (s, 1H), 7.20 (m, 4H), 3.30 (m, 4H),
1.78 (m, 2H), 1.60 (m, 2H), 1.30 (m, 2H).
MS: 376 (ES).
-
2,5,11-Triaza-1(2,4)-pyrimidina-3(1,3)-benzenacycloundecaphan-4-on
(B):
1H-NMR (DMSO): 9.21 (s, 1H), 8.97
(s, 1H), 8.10 (t, 1 N), 7.72 (d, 1H), 7.38 (t, 1H), 7.25 (t, 1H),
7.18 (dd, 1H), 7.08 (dd, 1H), 5.84 (d, 1H), 3.30 (m, 2H), 3.17 (m,
2H), 1.75 (m, 2H), 1.53 (m, 2H), 1.30 (m, 2H).MS: 298 (ES).
-
Herstellung
der Zwischenprodukte w)
Herstellung von 3-[4-(5-Benzyloxycarbonylamino-pentylamino)-5-brom-pyrimidin-2-ylamino]-benzoesäure
-
Ein Reaktionsgemisch von 1,20 g (2,8
mmol) [5-(5-Brom-2-chlor-pyrimidin-4-ylamino)-pentyl]-carbaminsäure-benzylester
und 0,37 g (2,7 mmol) 3-Aminobenzoesäure in Acetonitril/Wasser
(8 ml/1,5 ml) wird 20 Stunden unter Rückfluss gerührt. Das Reaktionsgemisch wird
einrotiert und der verbleibende Rückstand chromatographisch (Dichlormethan/Methanol
9:1, Flashmaster II) gereinigt. Man erhält 1,27 g (2,4 mmol, entsprechend
86% der Theorie) des Produktes.
1H-NMR
(DMSO): 10.03 (s, 1H), 8.38 (s, 1H), 8.15 (s, 1H), 7.81 (m, 2H),
7.56 (d, 1H), 7.30 (m, 7H), 4.98 (s, 2H), 3.35 (m, 2H), 2.98 (m,
2H), 1.54 (m, 2H), 1.32 (m, 4H). MS: 528 (CI).
-
x)
Herstellung von [5-(5-Brom-2-chlor-pyrimidin-4-ylamino)-pentyl]-carbaminsäure-benzylester
-
Eine Lösung von 860 mg (3,8 mmol)
5-Brom-2,4-dichlor-pyrimidin und 1,2 ml (8,5 mmol) Triethylamin in
6 ml Acetonitril wird bei 0°C
mit 1,0 g (3,7 mmol) (5-Amino-pentyl)-carbaminsäure-benzylester
versetzt. Das Reaktionsgemisch wird durch Entfernen des Eisbades
langsam unter Rühren
auf Raumtemperatur erwärmt. Nach
60 Stunden wird mit Wasser versetzt und anschließend mit Essigester extrahiert
(2x). Die vereinten organischen Phasen werden getrocknet (Na2SO4), filtriert
und eingeengt. Der gebildete Rückstand
wird chromatographisch gereinigt (Hexan/ Essigester 1:1). Man erhält 1,2 g
(2,8 mmol, entsprechend 77% der Theorie) des Produktes.
1H-NMR (DMSO): 8.21 (s, 1H), 7.72 (t, 1H),
7.35 (m, 5H), 7.23 (t, 1H), 4.99 (s, 2H), 3.30 (m, 2H), 2.97 (m,
2H), 1.47 (m, 4H), 1.27 (m, 2H).
MS: 427 (ES).
-
In analoger Verfahrensweise zu den
oben beschriebenen Verfahrensvarianten werden auch die nachfolgenden
Verbindungen hergestellt:
-
Die nachfolgenden Beispiele beschreiben
die biologische Wirkung der erfindungsgemäßen Verbindungen ohne die Erfindung
auf diese Beispiele zu beschränken.
-
Beispiel 1
-
CDK1/CycB Kinase Assay
-
Rekombinante CDK1- und CycB-GST-Fusionsproteine,
gereinigt aus Bakulovirusinfizierten Insektenzellen (Sf9), wurden
von ProQinase GmbH, Freiburg, gekauft. Das als Kinase-Substrat verwendet
Histon IIIS ist über
die Fa. Sigma käuflich
zu erwerben.
-
CDK1/CycB (200 ng/Meßpunkt)
wurde für
15 min bei 22°C
in Anwesenheit verschiedener Konzentrationen an Testsubstanzen (0 μM, sowie
innerhalb des Bereiches 0,01 – 100 μM) in Assaypuffer
[50 mM Tris/HCl pH8,0, 10 mM MgCl2, 0,1 mM Na ortho-Vanadat, 1,0
mM Dithiothreitol, 0,5 μM
Adenosintrisphosphat (ATP), 10 μg/Meßpunkt Histon
IIIS, 0,2 μCi/Meßpunkt 33P-gamma
ATP, 0,05% NP40, 12,5% Dimethylsulfoxid] inkubiert. Die Reaktion
wurde durch Zugabe von EDTA-Lösung
(250 mM, pH8,0, 14 μl/Meßpunkt)
gestoppt.
-
Von jedem Reaktionsansatz wurden
10 μl auf
P30 Filterstreifen (Fa. Wallac) aufgetragen, und nicht-eingebautes
33P-ATP wurde durch dreimaliges Waschen der Filterstreifen für je 10
min in 0,5%iger Phosphorsäure
entfernt. Nach dem Trocknen der Filterstreifen für 1 Stunde bei 70°C wurden
die Filterstreifen mit Szintillator-Streifen (MeltiLexTM A, Fa. Wallac)
bedeckt und für
1 Stunde bei 90°C
eingebrannt. Die Menge an eingebautem 33P (Substratphosphorylierung)
wurde durch Szintillationsmessung in einem gamma-Strahlungsmeßgerät (Wallac)
bestimmt.
-
Beispiel 2
-
CDK2/CycE Kinase Assay
-
Rekombinante CDK2- und CycE-GST-Fusionsproteine,
gereinigt aus Bakulovirusinfizierten Insektenzellen (Sf9), wurden
von ProQinase GmbH, Freiburg, gekauft. Histon IIIS, das als Kinase-Substrat
verwendet wurde, wurde bei der Fa. Sigma gekauft.
-
CDK2/CycE (50 ng/Meßpunkt)
wurde für
15 min bei 22°C
in Anwesenheit verschiedener Konzentrationen an Testsubstanzen (0 μM, sowie
innerhalb des Bereiches 0,01 – 100 μM) in Assaypuffer
[50 mM Tris/HCl pH8,0, 10 mM MgCl2, 0,1
mM Na ortho-Vanadat, 1,0 mM Dithiothreitol, 0,5 μM Adenosintrisphosphat (ATP), 10 μg/Meßpunkt Histon
IIIS, 0,2 μCi/Meßpunkt 33P-gamma ATP, 0,05% NP40, 12,5% Dimethylsulfoxid]
inkubiert. Die Reaktion wurde durch Zugabe von EDTA-Lösung (250
mM, pH8,0, 14 μl/Meßpunkt)
gestoppt.
-
Von jedem Reaktionsansatz wurden
10 μl auf
P30 Filterstreifen (Fa. Wallac) aufgetragen, und nicht-eingebautes 33P-ATP wurde durch dreimaliges Waschen der
Filterstreifen für
je 10 min in 0,5%iger Phosphorsäure
entfernt. Nach dem Trocknen der Filterstreifen für 1 Stunde bei 70°C wurden
die Filterstreifen mit Szintillator-Streifen (MeltiLexTM A,
Fa. Wallac) bedeckt und für
1 Stunde bei 90°C
eingebrannt. Die Menge an eingebautem 33P
(Substratphosphorylierung) wurde durch Szintillationsmessung in
einem gamma-Strahlungsmeßgerät (Wallac)
bestimmt.
-
Beispiel 3
-
Proliferationsassay
-
Kultivierte humane Tumorzellen (wie
angegeben) wurden in einer Dichte von 5000 Zellen/Meßpunkt in
einer 96-Loch Multititerplatte in 200 μl des entsprechenden Wachstumsmediums
ausplattiert. Nach 24 Stunden wurden die Zellen einer Platte (Nullpunkt-Platte)
mit Kristallviolett gefärbt
(s.u.), während
das Medium der anderen Platten durch frisches Kulturmedium (200 μl), dem die
Testsubstanzen in verschiedenen Konzentrationen (0 μM, sowie
im Bereich 0,01 – 30 μM; die finale
Konzentration des Lösungsmittels
Dimethylsulfoxid betrug 0,5%) zugesetzt waren, ersetzt. Die Zellen
wurden für
4 Tage in Anwesenheit der Testsubstanzen inkubiert. Die Zellproliferation
wurde durch Färbung
der Zellen mit Kristallviolett bestimmt: Die Zellen wurden durch Zugabe
von 20 μl/Meßpunkt einer
11 %igen Glutaraldehyd-Lösung 15
min bei Raumtemperatur fixiert. Nach dreimaligem Waschen der fixierten
Zellen mit Wasser wurden die Platten bei Raumtemperatur getrocknet.
Die Zellen wurden durch Zugabe von 100 μl/Meßpunkt einer 0,1%igen Kristallviolett-Lösung (pH
durch Zugabe von Essigsäure
auf pH3 eingestellt) gefärbt.
Nach dreimaligem Waschen der gefärbten
Zellen mit Wasser wurden die Platten bei Raumtemperatur getrocknet.
Der Farbstoff wurde durch Zugabe von 100 μl/Meßpunkt einer 10%igen Essigsäure-Lösung gelöst. Die
Extinktion wurde photometrisch bei einer Wellenlänge von 595 nm bestimmt. Die
prozentuale Änderung
des Zellwachstums wurde durch Normalisierung der Meßwerte auf
die Extinktionwerte der Nullpunktplatte (=0%) und die Extinktion
der unbehandelten (0 μM)
Zellen (=100%) berechnet.
-
Die Ergebnisse aus den Beispielen
sind in den nachfolgenden Tabellen angegeben.
-
-
Aus den Ergebnissen der Tabelle ist
deutlich ersichtlich, daß sich
die erfindungsgemäßen makrozyklischen
Pyrimidine durch eine hervorragende Inhibition der CDK1/Cyclin B
auszeichnen. Im Durchschnitt wird auch eine gute Selektivität gegenüber CDK2
erreicht. Die Aktivität
gegenüber
CDK1 erklärt
auch die gute zelluläre
Wirkung der Substanzen. Damit sind die makrozyklischen Verbindungen
den bisher bekannten Verbindungen deutlich überlegen.
-



 und W für die Gruppe -OH oder -COOH steht, sind ebenfalls Gegenstand der vorliegenden Erfindung.
und W für die Gruppe -OH oder -COOH steht, sind ebenfalls Gegenstand der vorliegenden Erfindung.
















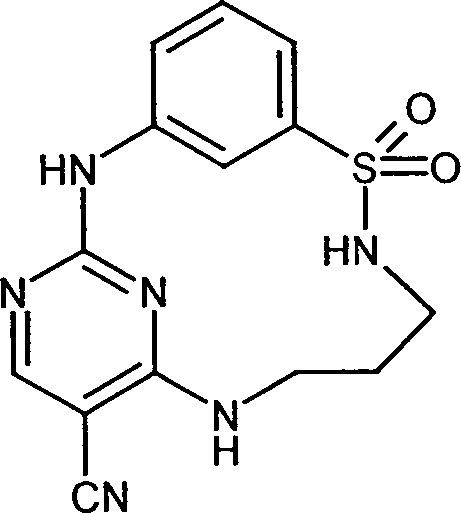














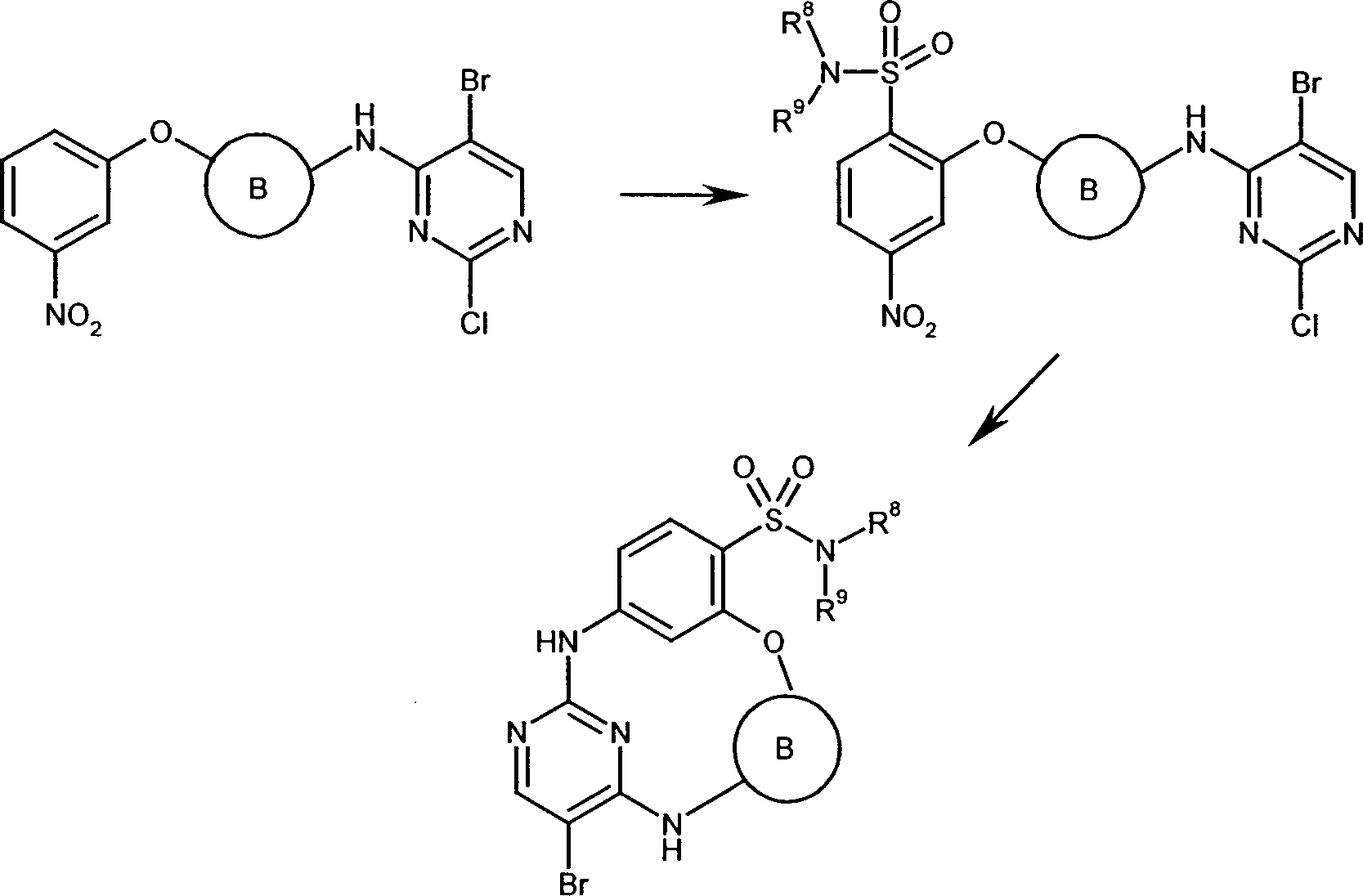

















 in der in der R1, R3, R4, R5, X, Y, A, B, m und n die in der allgemeinen Formel I angegebenen Bedeutungen haben und L für eine Abgangsgruppe steht, in einem geeigneten Lösungsmittel und einem geeignete Reduktionsmittel bei 0 °C bis Reflux zunächst zum Amin reduziert und anschließend das intermediär gebildete Amin zu den Verbindungen der allgemeinen Formel I cyclisiert.
in der in der R1, R3, R4, R5, X, Y, A, B, m und n die in der allgemeinen Formel I angegebenen Bedeutungen haben und L für eine Abgangsgruppe steht, in einem geeigneten Lösungsmittel und einem geeignete Reduktionsmittel bei 0 °C bis Reflux zunächst zum Amin reduziert und anschließend das intermediär gebildete Amin zu den Verbindungen der allgemeinen Formel I cyclisiert.
 und W für die Gruppe -OH oder -COOH steht, als Zwischenprodukte zur Herstellung der erfindungsgemäßen Verbindungen der allgemeinen Formel I.
und W für die Gruppe -OH oder -COOH steht, als Zwischenprodukte zur Herstellung der erfindungsgemäßen Verbindungen der allgemeinen Formel I.