CN113016094A - 正极活性物质、及二次电池 - Google Patents
正极活性物质、及二次电池 Download PDFInfo
- Publication number
- CN113016094A CN113016094A CN201980074923.2A CN201980074923A CN113016094A CN 113016094 A CN113016094 A CN 113016094A CN 201980074923 A CN201980074923 A CN 201980074923A CN 113016094 A CN113016094 A CN 113016094A
- Authority
- CN
- China
- Prior art keywords
- positive electrode
- secondary battery
- active material
- electrode active
- lithium
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images
































































































Classifications
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/13—Electrodes for accumulators with non-aqueous electrolyte, e.g. for lithium-accumulators; Processes of manufacture thereof
- H01M4/131—Electrodes based on mixed oxides or hydroxides, or on mixtures of oxides or hydroxides, e.g. LiCoOx
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M10/00—Secondary cells; Manufacture thereof
- H01M10/05—Accumulators with non-aqueous electrolyte
- H01M10/052—Li-accumulators
- H01M10/0525—Rocking-chair batteries, i.e. batteries with lithium insertion or intercalation in both electrodes; Lithium-ion batteries
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/48—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides
- H01M4/50—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides of manganese
- H01M4/505—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides of manganese of mixed oxides or hydroxides containing manganese for inserting or intercalating light metals, e.g. LiMn2O4 or LiMn2OxFy
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/48—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides
- H01M4/52—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides of nickel, cobalt or iron
- H01M4/525—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides of nickel, cobalt or iron of mixed oxides or hydroxides containing iron, cobalt or nickel for inserting or intercalating light metals, e.g. LiNiO2, LiCoO2 or LiCoOxFy
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M10/00—Secondary cells; Manufacture thereof
- H01M10/05—Accumulators with non-aqueous electrolyte
- H01M10/052—Li-accumulators
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M2004/026—Electrodes composed of, or comprising, active material characterised by the polarity
- H01M2004/028—Positive electrodes
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E60/00—Enabling technologies; Technologies with a potential or indirect contribution to GHG emissions mitigation
- Y02E60/10—Energy storage using batteries
Landscapes
- Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Electrochemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Inorganic Chemistry (AREA)
- Materials Engineering (AREA)
- Manufacturing & Machinery (AREA)
- Battery Electrode And Active Subsutance (AREA)
- Inorganic Compounds Of Heavy Metals (AREA)
Abstract
提供一种大容量且充放电循环特性优良的用于锂离子二次电池的正极活性物质。一种正极活性物质,其包含锂、钴及氧。其中起因于2价的钴离子及4价的钴离子的自旋密度为指定范围内。在正极活性物质中,优选还包含镁。以相对于钴的浓度表示适当的镁的浓度。另外,正极活性物质优选还包含氟。
Description
技术领域
本发明的一个方式涉及一种物品、方法或者制造方法。另外,本发明涉及一种工序(process)、机器(machine)、产品(manufacture)或者组合物(composition of matter)。本发明的一个方式涉及一种半导体装置、显示装置、发光装置、蓄电装置、照明装置或电子设备及其制造方法。尤其涉及一种有关能够用于二次电池的正极活性物质、二次电池及具有二次电池的电子设备。
需要说明的是,在本说明书中,蓄电装置是指具有蓄电功能的所有元件以及装置。例如,锂离子二次电池等蓄电池(也称为二次电池)、锂离子电容器及双电层电容器等都包括在蓄电装置的范畴内。
在本说明书等中,电子设备是指具有蓄电装置的所有装置,具有蓄电装置的电光装置、具有蓄电装置的信息终端装置等都是电子设备。
背景技术
近年来,对锂离子二次电池、锂离子电容器及空气电池等各种蓄电装置的研究开发日益火热。尤其是,随着移动电话、智能手机、平板电脑或笔记本个人计算机等便携式信息终端、便携式音乐播放机、数码相机、医疗设备、新一代清洁能源汽车(混合动力汽车(HEV)、电动汽车(EV)或插电式混合动力汽车(PHEV)等)等的半导体产业的发展,高输出、高能量密度的锂离子二次电池的需求量剧增。另外,作为能够充电的能量供应源,锂离子二次电池成为现代信息化社会的必需品。
作为目前锂离子二次电池被要求的特性,可以举出:更高能量密度化、循环特性的提高、各种工作环境下的安全性及长期可靠性的提高等。
因此,检讨以锂离子二次电池的循环特性的提高及大容量化为目的的正极活性物质的改良(专利文献1及专利文献2)。另外,已经进行了有关正极活性物质的结晶结构的研究(非专利文献1至非专利文献3)。
X射线衍射法(XRD:X-ray Diffraction)是用于分析正极活性物质的晶体结构的方法之一。通过使用非专利文献5所示的无机晶体结构数据库(ICSD:Inorganic CrystalStructure Database),可以分析XRD数据。
[先行技术文献]
[专利文献]
[专利文献1]日本专利申请公开第2002-216760号公报
[专利文献2]日本专利申请公开第2006-261132号公报
[非专利文献]
[非专利文献1]Toyoki Okumura et al,”Correlation of lithium iondistribution and X-ray absorption near-edge structure in O3-and O2-lithiumcobalt oxides from first-principle calculation”,Journal of MaterialsChemistry,2012,22,p.17340-17348
[非专利文献2]Motohashi,T.et al,”Electronic phase diagram of thelayered cobalt oxide system LiXCoO2(0.0≦X≦1.0)”,Physical Review B,80(16);165114
[非专利文献3]Zhaohui Chen et al,“Staging Phase Transitions inLiXCoO2”,Journal of The Electrochemical Society,2002,149(12)A1604-A1609
[非专利文献4]W.E.Counts et al,Journal of the American CeramicSociety,(1953)36[1]12-17.Fig.01471
[非专利文献5]Belsky,A.et al.,“New developments in the InorganicCrystal Structure Database(ICSD):accessibility in support of materialsresearch and design”,Acta Cryst.,(2002)B58 364-369.
发明内容
发明所要解决的技术问题
本发明的一个方式的目的之一是提供一种大容量且充放电循环特性优异的锂离子二次电池用正极活性物质及其制造方法。或者,本发明的一个方式的目的之一是提供一种高生产率的正极活性物质的制造方法。或者,本发明的一个方式的目的之一是提供一种正极活性物质,在将该正极活性物质用于锂离子二次电池中时抑制由充放电循环引起的容量下降。或者,本发明的一个方式的目的之一是提供一种大容量的二次电池。或者,本发明的一个方式的目的之一是提供一种具有良好的充放电特性的二次电池。或者,本发明的一个方式的目的之一是提供一种即使长时间保持高电压充电状态也可以抑制钴等过渡金属的溶解的正极活性物质。或者,本发明的一个方式的目的之一是提供一种安全性或可靠性高的二次电池。
另外,本发明的一个方式的目的之一是提供一种新颖的物质、活性物质粒子、蓄电装置或它们的制造方法。
注意,这些目的的记载不妨碍其他目的的存在。本发明的一个方式并不需要实现所有上述目的。另外,可以从说明书、附图、权利要求书的记载中抽取上述目的以外的目的。
解决技术问题的手段
本发明的一个方式是一种正极活性物质,包含锂、钴及氧,其中起因于2价的钴离子及4价的钴离子的自旋密度为2.0×1017spins/g以上且1.0×1018spins/g以下。
上述正极活性物质优选还包含镁。相对于钴的镁浓度优选为0.1atomic%以上且6.0atomic%以下。
上述正极活性物质优选还包含氟。
在上述正极活性物质中,优选的是,a axis(a轴)的晶格常数为2.8155×10-10m以上且2.8175×10-10m,并且c axis(c轴)的晶格常数为14.045×10-10m以上且14.065×10-10m以下。
本发明的一个方式是包括具有上述正极活性物质的正极及负极的二次电池。
发明效果
根据本发明的一个方式可以提供一种大容量且充放电循环特性优良的用于锂离子二次电池的正极活性物质及其制造方法。另外,根据本发明的一个方式可以提供一种高生产性的正极活性物质的制造方法。另外,根据本发明的一个方式可以提供一种通过被用于锂离子二次电池而抑制充放电循环中的容量减少的正极活性物质。另外,根据本发明的一个方式可以提供一种大容量二次电池。另外,根据本发明的一个方式可以提供一种充放电特性优良的二次电池。另外,根据本发明的一个方式可以提供一种即使长时间保持高电压充电状态也可以抑制钴等过渡金属的溶解的正极活性物质。另外,根据本发明的一个方式可以提供一种安全性或可靠性高的二次电池。根据本发明的一个方式可以提供一种新颖的物质、活性物质粒子、蓄电装置或它们的制造方法。
附图简要说明
图1A和图1B是说明正极活性物质的结构的示意图。
图2A和图2B是说明正极活性物质的结构的示意图。
图3是说明本发明的一个方式的正极活性物质的充电深度与结晶结构的图。
图4是说明现有的正极活性物质的充电深度与结晶结构的图。
图5是根据结晶结构算出的XRD图案。
图6A是说明本发明的一个方式的正极活性物质的结晶结构的图。图6B是说明本发明的一个方式的正极活性物质的磁性的图。
图7A是说明现有的正极活性物质的结晶结构的图。图7B是说明现有的正极活性物质的磁性的图。
图8是说明本发明的一个方式的正极活性物质的制造方法的一个例子的图。
图9是说明本发明的一个方式的正极活性物质的制造方法的一个例子的图。
图10是说明本发明的一个方式的正极活性物质的制造方法的一个例子的图。
图11A和图11B是作为导电助剂使用石墨烯化合物时的活性物质层的截面图。
图12A、图12B、图12C是说明二次电池的充电方法的图。
图13A、图13B、图13C、图13D是说明二次电池的充电方法的图。
图14是说明二次电池的放电方法的图。
图15A、图15B、图15C是说明硬币型二次电池的图。
图16A、图16B、图16C、图16D是说明圆筒型二次电池的图。
图17A和图17B是说明二次电池的例子的图。
图18A1、图18A2、图18B1、图18B2是说明二次电池的例子的图。
图19A和图19B是说明二次电池的例子的图。
图20是说明二次电池的例子的图。
图21A、图21B、图21C是说明层压型二次电池的图。
图22A和图22B是说明层压型二次电池的图。
图23是示出二次电池的外观的图。
图24是示出二次电池的外观的图。
图25A、图25B、图25C是用来说明二次电池的制造方法的图。
图26A、图26B1、图26B2、图26C、图26D是说明可弯曲的二次电池的图。
图27A和图27B是说明可弯曲的二次电池的图。
图28A、图28B、图28C、图28D、图28E、图28F、图28G、图28H是说明电子设备的一个例子的图。
图29A、图29B、图29C是说明电子设备的一个例子的图。
图30是说明电子设备的一个例子的图。
图31A、图31B、图31C是说明车辆的一个例子的图。
图32是示出ESR测定结果的图。
图33是示出ESR测定结果的图。
图34A和图34B是示出自旋密度的图。
图35A和图35B是示出镁的添加量与自旋密度的关系的图。
图36是示出XRD测定结果的图。
图37A和图37B是示出XRD测定结果的图。
图38是示出XRD测定结果的图。
图39A和图39B是示出晶格常数的图。
图40A和图40B是示出循环特性的图。
图41A和图41B是示出循环特性的图。
图42A和图42B是示出连续充电特性的图。
图43A和图43B是示出ESR测定结果的图。
图44A和图44B是示出自旋密度的图。
图45A和图45B是示出镁的添加量与自旋密度的关系的图。
图46A和图46B是示出循环特性的图。
实施发明的方式
下面,参照附图对本发明的实施方式进行详细说明。注意,本发明不局限于以下说明,所属技术领域的普通技术人员可以很容易地理解一个事实就是其方式和详细内容可以被变换为各种形式。此外,本发明不应该被解释为仅限定在下面的实施方式所记载的内容中。
在本说明书等中,以密勒指数表示结晶面和取向。在结晶学上,对数字附上上标横线来表示结晶面和取向。但是,在本说明书等中,由于专利申请中的符号限定,有时对数字前附上-(负数符号)来表示结晶面和取向,代替对数字附上上标横线。另外,以“[]”表示示出结晶内的取向的个别方位,以“<>”表示示出所有等价晶向的集合方位,以“()”表示示出结晶面的个别面,以“{}”表示具有等价对称性的集合面。
在本说明书等中,偏析是指在包含多个元素(例如A、B、C)的固体中某个元素(例如,B)在空间上不均匀地分布的现象。
在本说明书等中,活性物质等的粒子的表层部是指从表面至10nm左右的区域。另外,因裂口或裂缝形成的面也可以称为表面。将比粒子表层部更深的区域称为粒子内部。
在本说明书等中,包含锂及过渡金属的复合氧化物所具有的层状岩盐型结晶结构是指如下结晶结构:具有阳离子和阴离子交替排列的岩盐型离子排列,过渡金属和锂有规律地排列而形成二维平面,其中锂可以二维扩散。另外,也可以包括阳离子或阴离子的空位等缺陷。严格而言,层状岩盐型结晶结构有时为岩盐型结晶的晶格变形的结构。
在本说明书等中,岩盐型结晶结构是指其中阳离子和阴离子交替排列的结构。另外,也可以包括阳离子或阴离子的空位。
在本说明书等中,包含锂和过渡金属的复合氧化物所具有的拟尖晶石型结晶结构是指空间群R-3m,即:虽然不是尖晶石型结晶结构,但是钴、镁等的离子占氧六配位位置,阳离子的配列与尖晶石型具有相似的对称性的结晶结构。另外,有时拟尖晶石型结晶结构存在锂等的轻元素占氧四配位位置的情况,在该情况下离子的配列也具有与尖晶石型相似的对称性。
另外,虽然拟尖晶石型结晶结构在层间无规律地含有锂,但是也可以具有与CdCl2型结晶结构类似的结晶结构。该与CdCl2型类似的结晶结构近似于使镍酸锂充电至充电深度0.94(Li0.06NiO2)的结晶结构,但是纯钴酸锂或含有大量钴的层状岩盐型的正极活性物质通常不具有这样的结晶结构。
层状岩盐型结晶及岩盐型结晶的阴离子分别形成立方最紧密堆积结构(面心立方格子结构)。可以推测拟尖晶石型结晶中的阴离子也具有立方最紧密堆积结构。当这些结晶接触时,存在阴离子所构成的立方最紧密堆积结构的取向一致的结晶面。层状岩盐型结晶及拟尖晶石型结晶的空间群为R-3m,即与岩盐型结晶的空间群Fm-3m(一般的岩盐型结晶的空间群)及Fd-3m(具有最简单的对称性的岩盐型结晶的空间群)不同,因此层状岩盐型结晶及拟尖晶石型结晶与岩盐型结晶的满足上述条件的结晶面的密勒指数不同。在本说明书中,有时在层状岩盐型结晶、拟尖晶石型结晶结构及岩盐型结晶中,阴离子所构成的立方最紧密堆积结构的取向一致是指结晶取向大致一致。
可以根据透射电子显微镜(TEM:Transmission Electron Microscopy)图像、扫描透射电子显微镜(STEM:Scanning Transmission Electron Microscopy)图像、高角度环形暗场-扫描透射电子显微镜(HAADF-STEM:High-angle Annular Dark Field-STEM)图像、环形明场扫描透射电子显微镜(ABF-STEM:Annular bright field-STEM)图像等,判断两个区域的结晶取向大致一致。另外,可以将X射线衍射(XRD)、电子衍射、中子衍射等用作判断依据。在TEM图像等中,阳离子和阴离子的排列被观察为明线和暗线的反复。当在层状岩盐型结晶和岩盐型结晶中,立方最紧密堆积结构的取向对齐时,可以观察到明线和暗线的反复所形成的角度为5度以下,更优选为2.5度以下的样子。注意,在TEM图像等中,有时不能明确地观察到氧、氟等轻元素,在此情况下,可以根据金属元素的排列判断取向的一致。
在本说明书等中,正极活性物质的理论容量是指正极活性物质中的能够嵌入和脱离的锂全部脱离时的电量。例如,LiCoO2的理论容量为274mAh/g、LiNiO2的理论容量为274mAh/g、LiMn2O4的理论容量为148mAh/g。
在本说明书等中,将能嵌入和脱离的锂全部嵌入时的充电深度记作0,将正极活性物质中的能嵌入和脱离的锂全部脱离时的充电深度记作1。
在本说明书等中,充电是指在电池内使锂离子从正极移动到负极而在外部电路中使电子从负极移动至正极。正极活性物质的充电是指锂离子的脱离。另外,将充电深度为0.74以上且0.9以下、更具体地将充电深度为0.8以上且0.83以下的正极活性物质被称为高电压充电的正极活性物质。因此,例如,当LiCoO2被充电至219.2mAh/g就可以说其是被高电压充电的正极活性物质。另外,如下LiCoO2也被为被高电压充电的正极活性物质:在25℃环境下,以充电电压为4.525V以上且4.65V以下(对电极为锂的情况下)进行恒流充电,然后,以电流值变为0.01C或者变为恒流充电时的电流值的1/5至1/100左右的方式进行恒压充电的LiCoO2。
同样地,放电是指在电池内使锂离子从负极移动到正极而在外部电路中使电子从正极移动到负极。正极活性物质的放电是指锂离子的嵌入。另外,将充电深度为0.06以下的正极活性物质或者从已被高电压充电状态将充电容量的90%以上的容量放电的正极活性物质称为已被充分放电的正极活性物质。例如,在LiCoO2中充电容量为219.2mAh/g是指已被高电压充电的状态,从这状态将充电容量的90%的197.3mAh/g以上放电之后的正极活性物质是已被充分放电的正极活性物质。另外,将在LiCoO2中,在25℃环境下直到电池电压成为3V以下(对电极锂时)进行恒流放电之后的正极活性物质也称为已被充分放电的正极活性物质。
在本说明书等中,非平衡相变是指引起物理量非线性变化的现象。例如,在通过容量(Q)与电压(V)的微分(dQ/dV)得到的dQ/dV曲线的峰附近可能出现非平衡相变,而使结晶结构大幅改变。
(实施方式1)
在本实施方式中,对本发明的一个方式的正极活性物质进行说明。
本发明的一个方式是包含锂、钴及氧的正极活性物质。本发明的一个方式的正极活性物质优选还包含镁。由于包含镁而结晶结构稳定,因此能够抑制当反复进行充放电时结晶结构崩塌。此外,本发明的一个方式的正极活性物质在钴酸锂(LiCoO2)中一部分的Li+被Mg2+取代,由此其附近的Co3+被还原而成为Co2+(参照图1A及图2A)。另外,一部分的Co3+被Mg2+取代,由此其附近的Co3+被氧化而成为Co4+(参照图1B及图2A)。因此,本发明的一个方式的正极活性物质包含Co2+及Co4+中的一个或两个。另外,正极活性物质的单位重量的起因于Co2+及Co4+的自旋密度优选为2.0×1017spins/g以上且1.0×1018spins/g以下。通过正极活性物质具有上述自旋密度,尤其在充电状态下结晶结构稳定,这是优选的。注意,图2A是说明不包含镁的正极活性物质的结构的示意图,图1A及图1B是说明本发明的一个方式的包含镁的正极活性物质的结构的示意图。另外,在镁添加量过多的情况下,有时起因于Co2+及Co4 +的自旋密度降低(参照图2B)。另外,通过将本发明的一个方式的正极活性物质用于二次电池,可以实现具有优异的循环特性及速率特性的二次电池。
正极活性物质中的镁浓度优选为相对于钴原子的0.1atomic%以上且6.0atomic%以下,更优选为0.5atomic%以上且5.0atomic%以下,进一步优选为1.0atomic%以上且4.0atomic%以下。注意,上述镁浓度是指正极活性物质的粒子整体的平均值。
例如,通过使用电子自旋共振法(ESR:Electron Spin Resonance)等可以分析正极活性物质中的自旋密度。另外,例如,通过使用感应耦合等离子体质谱分析法(ICP-MS:Inductively Coupled Plasma-Mass Spectrometry)等可以分析正极活性物质的粒子整体的镁浓度的平均值。
本发明的一个方式的正极活性物质优选还包含氟。通过包含氟,可以提高对因电解质的分解而产生的氢氟酸的抗腐蚀性。例如,通过使用X射线光电子能谱(XPS:X-rayPhotoelectron Spectroscopy)等测定正极活性物质的成分。此外,例如,感应耦合等离子体质谱分析法(ICP-MS)等可以分析正极活性物质的粒子整体的氟浓度的平均值。
本发明的一个方式的正极活性物质的a轴的晶格常数优选为2.8155×10-10m以上且2.8175×10-10m且c轴的晶格常数优选为14.045×10-10m以上且14.065×10-10m以下。
[正极活性物质的结构]
参照图3及图4对本发明的一个方式的正极活性物质100及现有的正极活性物质及其不同之处进行说明。在图3及图4中,对作为正极活性物质含有的过渡金属使用钴时的情况进行说明。图3示出本发明的一个方式的正极活性物质100。图4示出现有的正极活性物质。注意,图4中说明的现有的正极活性物质是单纯的钴酸锂(LiCoO2),其中没有对其内部添加锂、钴、氧以外的元素并且没有在正极活性物质的粒子表层部进行涂敷锂、钴、氧以外的元素。
<现有的正极活性物质>
作为现有的正极活性物质之一的钴酸锂LiCoO2,如非专利文献1及非专利文献2等所述,结晶结构根据充电深度改变。图4示出示出典型的钴酸锂的结晶结构。
如图4所示,充电深度是0(放电状态)的钴酸锂包括具有空间群R-3m的结晶结构的区域,在单位晶胞中包括三个CoO2层。由此有时该结晶结构被称为O3型结晶结构。注意,CoO2层是指钴和六个配位氧形成的八面体结构在一个平面上维持棱线共享的状态的结构。
在充电深度是1时,具有空间群P-3m1的结晶结构,并且单位晶胞包括一个CoO2层。由此有时该结晶结构被称为O1型结晶结构。
在充电深度是0.88左右时,钴酸锂具有空间群R-3m的结晶结构,也可以说该结构是如P-3m1(O1)那样的CoO2结构与如R-3m(O3)那样的LiCoO2结构交替地层叠的结构。由此,有时该结晶结构被称为H1-3型结晶结构。实际上,H1-3型结晶结构的每个晶胞中的钴原子的数量为其他结构的2倍。但是,在如图4等本说明书中,为了容易与其他结构进行比较,以单位晶胞的1/2的方式表示H1-3型结晶结构中的c轴。
当反复进行充电深度为0.88左右或更高的高电压的充电和放电时,钴酸锂的结晶结构在H1-3型结晶结构和放电状态的R-3m(O3)的结构之间反复变化(即,非平衡相变)。
但是,上述两种结晶结构的CoO2层的偏离较大。如图4中以虚线及双箭头所示,在H1-3结晶结构中,CoO2层明显偏离于R-3m(O3)。这样动态的结构变化会对结晶结构的稳定性带来不良影响。
并且,H1-3型结晶结构与O3型结晶结构的体积差也较大。在按每相同数量的钴原子进行比较时,H1-3型结晶结构和放电状态的O3型结晶结构的体积差是3.5%以上。
除了上述以外,H1-3型结晶结构所具有的如P-3m1(O1)那样的CoO2层连续的结构不稳定的可能性较高。
由此,当反复进行高电压充放电时,钴酸锂的结晶结构会崩塌。而结晶结构的崩塌会引起循环特性的恶化。这是由于结晶结构崩塌造成锂可稳定存在的位置减少,因此锂的嵌入及脱离变得困难的缘故。
<本发明的一个方式的正极活性物质>
<粒子内部>
本发明的一个方式的正极活性物质100在被充分放电的状态及被高电压充电的状态(充电深度为0.8以上且0.83以下)下的结晶结构的变化及与相同数量的过渡金属原子相比的体积差小。
图3示出正极活性物质100的充放电前后的结晶结构。本发明的一个方式的正极活性物质100是包含锂及钴的复合氧化物。优选的是,除了上述以外还包含镁。另外,优选还包含氟、氯等卤素。
图3所示的充电深度0(放电状态)的结晶结构是与图4相同的R-3m(O3)。另一方面,在被充分充电的充电深度为0.88左右时,本发明的一个方式的正极活性物质100包括与图4不同的结晶结构。在本说明说中将上述空间群R-3m的结晶结构称为拟尖晶石型结晶结构。另外,为了说明钴原子的对称性以及氧原子的对称性,在图3所示的拟尖晶石型结晶结构的图中省略锂的表示,但是实际上在CoO2层间存在有相对于钴的12atomic%左右的锂。另外,在O3型结晶结构和拟尖晶石型结晶结构中,都优选在CoO2层间即在锂位置存在有少量的镁。另外,还在氧位置优选无规律地存在有氟等卤素。
在正极活性物质100中,与现有的LiCoO2相比,高电压充电时大量锂脱离时的结晶结构的变化得到抑制。例如,如图3中虚线所示,在上述结晶结构中几乎没有CoO2层的偏离。
在正极活性物质100中,充电深度是0的O3型结晶结构和充电深度是0.88的拟尖晶石型结晶结构的每个晶胞的体积差是2.5%以下,具体是2.2%以下。
由此,正极活性物质100即使以高电压反复充放电,结晶结构也不容易崩塌。
拟尖晶石型结晶结构的每个晶胞中的钴及氧的坐标分别可以以Co(0,0,0.5)、O(0,0,x)(0.20≤x≤0.25)表示。
在CoO2层间(即,锂位置)无规律地少量存在的镁具有抑制CoO2层的偏离的效果。由此当在CoO2层间存在镁时容易得到拟尖晶石型结晶结构。另外,优选的是,镁分布在整个正极活性物质100的粒子。为了使镁分布在整个粒子,优选在正极活性物质100的制造工序中进行加热处理。
但是,在加热处理的温度过高时,发生阳离子混排(cation mixing)而镁侵入钴位置的可能性增高。当镁存在于钴位置时,有时降低保持R-3m的效果。再者,在加热处理温度过高时,还有产生层状岩盐型的结构不稳定、锂蒸发等不利影响的担忧。
于是,优选在进行用来使镁分布在整个粒子的加热处理之前,对钴酸锂添加氟化合物等卤素化合物。通过添加卤素化合物,使钴酸锂的熔点下降。通过使熔点下降,可以在不容易发生阳离子混排的温度下容易地使镁分布到粒子整体。在此,由于因电解质分解而产生的氢氟酸,有时正极活性物质被腐蚀。本发明的一个方式的正极活性物质100因为包含氟,所以能够提高对因电解液分解而产生的氢氟酸的抗腐蚀性。
注意,在本说明书等中,电解质是指具有导电性的物质。电解质不局限于液体,也可以为凝胶或固体。有时将液体的电解质称为电解液,并且电解液可以通过将溶质溶解于溶剂中而制造。此外,有时将固体的电解质称为固体电解质。
注意,以上对正极活性物质100是包含锂、钴及氧的复合氧化物时的情况进行了说明,但是除了钴之外还可以含有镍。在这种情况下,钴与镍的原子数的和(Co+Ni)中的镍的原子数(Ni)所占的比例Ni/(Co+Ni)优选小于0.1,更优选为0.075以下。
当长时间保持高电压充电状态时,正极活性物质中的过渡金属溶解于电解液中,结晶结构可能会变形。但是,通过以上述比例含有镍,有时可以抑制正极活性物质100中的过渡金属溶解。
通过添加镍充放电电压下降,因此,在相同容量的情况下,可以在较低的电压下进行充放电。由此,可以抑制过渡金属的溶解及电解液的分解。这里,充放电电压例如是指从充电深度0到预定充电深度的范围内的电压。
<粒子表层部>
镁优选在正极活性物质100的粒子整体中分布,但是除此之外,粒子表层部的镁浓度优选高于粒子整体的平均。粒子表层部的镁浓度例如通过X射线光电子能谱(XPS)等可以测定。例如,粒子整体的平均的镁浓度通过感应耦合等离子体质谱分析法(ICP-MS)、辉光放电质谱法(GDMS:Glow Discharge Mass Spectrometry)等可以测定。粒子表面都是结晶缺陷而且由于充电时粒子表面的锂被脱离所以表面的锂浓度比粒子内部的锂浓度低。因此,粒子表面趋于不稳定而结晶结构容易被破坏。在粒子表层部的镁浓度高时,可以更有效地抑制结晶结构的变化。另外,粒子表层部的镁浓度高时,可期待提高对因电解液分解而产生的氢氟酸的抗腐蚀性。
优选的是,正极活性物质100的粒子表层部中的氟等卤素的浓度高于粒子整体的平均。通过在与电解液接触的区域的粒子表层部中存在的卤素,可以有效地提高对氢氟酸的抗腐蚀性。
如此,优选的是:正极活性物质100的粒子表层部的镁及氟的浓度比粒子内部高;具有与粒子内部不同的组成。作为粒子表层部的组成优选采用在常温下稳定的结晶结构。由此,粒子表层部也可以具有与粒子内部不同的结晶结构。例如,正极活性物质100的粒子表层部中的至少一部分可以具有岩盐型结晶结构。注意,在粒子表层部具有与粒子内部不同的结晶结构时,粒子表层部和粒子内部的结晶的取向优选大致一致。
但是,在粒子表层部只有MgO或者只有MgO与CoO(II)固溶的结构时,很难发生锂的嵌入及脱离。由此粒子表层部需要至少包含钴,在放电时还包含锂以具有锂的嵌入及脱离的路径。另外,钴的浓度优选高于镁的浓度。
<晶界>
正极活性物质100所包含的镁或卤素可以无规律且少量地存在于粒子内部,但是更优选的是,其一部分在晶界偏析。
换言之,正极活性物质100的晶界及其附近的镁浓度优选高于粒子内部的其他区域。另外,优选晶界及其附近的卤素浓度高于内部的其他区域。
与粒子表面同样,晶界也是面缺陷。由此,晶界容易不稳定而结晶结构容易开始变化。由此,在晶界及其附近的镁浓度高时,可以更有效地抑制结晶结构的变化。
另外,在晶界及其附近的镁及卤素浓度高时,即使在沿着正极活性物质100的粒子的晶界产生裂缝的情况下,也在因裂缝产生的表面附近镁及卤素浓度变高。因此也可以提高裂缝产生之后的正极活性物质的对氢氟酸的抗腐蚀性。
注意,在本说明书等中,晶界的附近是指从晶界至10nm左右的范围的区域。
<粒径>
在正极活性物质100的粒径过大时有如下问题:锂的扩散变难;在集流体上涂敷时,活性物质层的表面过粗等。另一方面,在正极活性物质100的粒径过小时有如下问题:在集流体上涂敷时不容易担持活性物质层;与电解液的反应过度等。因此,优选平均粒子径(D50)为1μm以上且100μm以下,更优选为2μm以上且40μm以下,进一步优选为5μm以上且30μm以下。
另外,在本说明书等中,平均粒子径(D50)是指以体积为基准时累计50%的粒径。有时将平均粒径(D50)称为中值粒径。
<分析方法>
为了判断某一正极活性物质是否是在以高电压被充电时示出拟尖晶石型结晶结构的本发明的一个方式的正极活性物质100,可以将以高电压被充电的正极通过使用XRD、电子衍射、中子衍射、电子自旋共振法(ESR:Electron Spin Resonance)、核磁共振法(NMR:Nuclear Magnetic Resonance)及磁化测定等分析进行判断。尤其是,XRD具有如下优点,所以是优选的:对正极活性物质所具有的钴等过渡金属的对称性可以以高分辨率进行分析;可以比较结晶性的高度与结晶的取向性;可以分析晶格的周期性畸变及晶粒尺寸;在直接测量通过将二次电池拆开而得到的正极时也可以获得足够的精度等。
如上所述,本发明的一个方式的正极活性物质100的特征是:高电压充电状态与放电状态之间的结晶结构变化少。高电压充电时与放电时之间的变化大的结晶结构占50wt%以上的材料因为不能承受高电压充放电,所以不是优选的。注意,有时只通过添加元素不能实现想要的结晶结构。例如,作为包含镁及氟的钴酸锂的正极活性物质,在以高电压进行充电的状态下,有时具有60wt%以上的拟尖晶石型结晶结构,有时具有50wt%以上的H1-3型结晶结构。另外,在使用规定的电压时拟尖晶石型结晶结构几乎成为100wt%,并且在进一步增高该规定的电压时有时产生H1-3型结晶结构。由此,在判断是否是本发明的一个方式的正极活性物质100时,需要进行XRD等的对结晶结构的分析。
但是,有时高电压充电状态或放电状态的正极活性物质遇空气结晶结构发生变化。例如,有时从拟尖晶石型结晶结构变为H1-3型结晶结构。因此,所有样品优选在氩气氛等惰性气氛中处理。
<充电方法>
作为用来判断某复合氧化物是否是本发明的一个方式的正极活性物质100的高电压充电,例如可以制造作为对电极使用锂的硬币电池(CR2032型、直径为20mm,高度为3.2mm)并对其进行充电。
更具体而言,作为正极可以使用将正极活性物质、导电助剂及粘合剂混合而成的浆料涂敷在铝箔的正极集流体而成的正极。
作为对电极可以使用锂金属。注意,作为对电极使用锂金属以外的材料时的正极的电位与二次电池不同。在没有特别说明时,本说明书等中的电压及电位是正极的电位。
作为电解液所包含的电解质,使用1mol/L的六氟磷酸锂(LiPF6)。作为溶剂,可以使用将体积比为3:7的碳酸乙烯酯(EC)和碳酸二乙酯(DEC)以及2wt%的碳酸亚乙烯酯(VC)混合而成的电解液。
作为隔离体可以使用厚度为25μm的聚丙烯。
正极罐及负极罐可以由不锈钢(SUS)形成。
对在上述条件下制造的硬币电池以4.6V且0.5C进行恒流充电,然后直到电流值成为0.01C为止继续恒压充电。在此将1C设定为137mA/g。将温度设定为25℃。通过如上所述那样充电之后在氩气氛的手套箱内将硬币电池拆开而取出正极,由此可以得到被高电压充电的正极活性物质。在之后进行各种分析时,为了防止与外部成分起反应,优选在氩气氛下进行密封。例如,XRD可以在封入在氩气氛的密封容器的条件下进行。
<XRD>
图5示出从拟尖晶石型结晶结构和H1-3型结晶结构的模型算出的以CuKα1线表示的理想的粉末XRD图案。另外,为了进行比较,一并示出从充电深度为0的LiCoO2(O3)和充电深度为1的CoO2(O1)的结晶结构算出的理想的XRD图案。LiCoO2(O3)及CoO2(O1)的图案通过从ICSD(Inorganic Crystal Structure Database:无机结晶结构数据库)(参照非专利文献5)获得的结晶结构信息使用Materials Studio(BIOVIA)的模块之一的Reflex PowderDiffraction而算出。2θ的范围设定为15°至75°,Step size=0.01,波长λ1=1.540562×10-10m,λ2没有设定,Monochromator设定为single。H1-3型结晶结构的图案参照非专利文献3所记载的结晶结构信息同样地制成。拟尖晶石型结晶结构的图案通过如下方法制出:从本发明的一个方式的正极活性物质的XRD图案推测出结晶结构并利用TOPAS ver.3(Bruker公司制造的结晶结构分析软件)进行拟合,与其他结构同样地制出XRD图案。
如图5所示,在拟尖晶石型结晶结构中,衍射峰在2θ为19.30±0.20°(19.10°以上且19.50°以下)处以及2θ为45.55±0.10°(45.45°以上且45.65°以下)处出现。更详细地说,在2θ为19.30±0.10°(19.20°以上且19.40°以下)处以及2θ为45.55±0.05°(45.50°以上且45.60°以下)处出现尖锐的衍射峰。但是,H1-3型结晶结构及CoO2(P-3m1、O1)在上述位置不出现峰值。由此,可以说在被高电压充电的状态下在2θ为19.28±0.60°处以及2θ为45.55±0.20°处出现峰值是本发明的一个方式的正极活性物质100的特征。
可以说,充电深度为0的结晶结构与高电压充电时的结晶结构的通过XRD观察到的衍射峰值的位置接近。更具体而言,可以说两者的主要衍射峰中的两个以上、优选为三个以上的位置差为2θ=0.7以下、更优选为2θ=0.5以下。
注意,本发明的一个方式的正极活性物质100被高电压充电时具有拟尖晶石型结晶结构,但是不需要所有粒子为拟尖晶石型结晶结构。既可以具有其他结晶结构,一部分也可以为非晶。注意,在对XRD图案进行里特沃尔德分析时,拟尖晶石型结晶结构优选为50wt%以上,更优选为60wt%以上,进一步优选为66wt%以上。在拟尖晶石型结晶结构为50wt%以上,更优选为60wt%以上,进一步优选为66wt%以上时,可以实现循环特性充分优良的正极活性物质。
从测定开始经过100次以上的充放电循环通过里特沃尔德分析的拟尖晶石型结晶结构优选为35wt%以上,更优选为40wt%以上,进一步优选为43wt%以上。
正极活性物质的粒子所具有的拟尖晶石型结晶结构的晶粒尺寸只减小到放电状态的LiCoO2(O3)的1/10左右。由此,即使在与充放电之前的正极相同的XRD测定的条件下也可以在高电压充电之后确认到明显的拟尖晶石型结晶结构的峰。另一方面,即使单纯的LiCoO2中的一部分可具有与拟尖晶石型结晶结构相似的结构,晶粒尺寸也会变小,其峰也会变宽且小。晶粒尺寸可以从XRD峰值的半宽值求出。
优选能够根据XRD图案推定的放电状态的正极活性物质的粒子所具有的层状岩盐型结晶结构中的c轴的晶格常数小。在锂的位置被异质元素(foreign element)取代或者钴进入氧四配位位置(A位置)等时c轴的晶格常数变大。因此,首先制造具有异质元素取代及尖晶石型结晶结构的Co3O4少(即,缺陷少)的层状岩盐型结晶结构的复合氧化物,然后对其混合镁源及氟源使镁插入锂的位置,由此可以制造具有良好的循环特性的正极活性物质。
放电状态的正极活性物质的结晶结构中的a轴的晶格常数优选为2.8155×10-10m以上且2.8175×10-10m,并且c轴的晶格常数优选为14.045×10-10m以上且14.065×10-10m以下。
为了使c轴的晶格常数为上述范围内,优选杂质少,尤其是优选钴、锰、镍以外的过渡金属的添加少。具体而言,优选为3000ppm(wt)以下,更优选为1500ppm(wt)以下。另外,优选锂和钴、锰、镍的阳离子混合少。
从XRD图案可知关于正极活性物质的内部的结构的特征。在平均粒径(D50)为1μm至100μm左右的正极活性物质中,与内部比较起来粒子表层部的体积非常小,所以即使正极活性物质100的粒子表层部具有与粒子内部不同的结晶结构,也有在XRD图案体现不出的可能性。
<ESR>
这里,参照图6及图7说明利用ESR判断拟尖晶石型结晶结构与其他结晶结构的差异的情况。如图3及图6A所示,钴存在于氧六配位的位置。如图6B所示,在氧六配位的钴中,3d轨道分裂成eg轨道及t2g轨道。在氧六配位的钴中,与氧存在的方向而配置的轨道的eg轨道相比,避开氧存在的方向而配置的t2g轨道的能量低,而t2g轨道处于基态。存在于氧六配位的位置的钴的一部分是Co3+,处于基态的Co3+是t2g轨道都被填埋的反磁性Co3+(自旋量子数S=0)。存在于氧六配位的位置的钴的其他一部分也可以为Co2+或Co4+,处于基态的Co2+或Co4+是顺磁性(自旋量子数S=1/2)。上述顺磁性Co2+或Co4+的钴都包括一个(自旋量子数S=1/2)不成对电子,所以不能利用ESR进行判断。
另一方面,有记载说现有的正极活性物质中有一部分在被充电的状态下可以具有粒子表层部不包含锂的尖晶石型结晶结构。此时,具有图7A所示的尖晶石型结晶结构的Co3O4。
在以通式A[B2]O4表示尖晶石时,元素A为氧四配位,元素B为氧六配位。于是,在本说明书等中,有时将氧四配位的位置称为A位置,并且将氧六配位的位置称为B位置。
在尖晶石型结晶结构的Co3O4中,钴除了氧六配位的B位置以外,还存在于氧四配位的A位置。如图7B所示,在氧四配位的钴的3d轨道分裂而成的eg轨道和t2g轨道中,eg轨道的能量低而eg轨道处于基态。由此当氧四配位的Co2+、Co3+以及Co4+处于基态时都包括不成对电子且为顺磁性。由此当利用ESR等分析出是充分包含尖晶石型Co3O4的粒子时,一定会检测出起因于氧四配位的顺磁性Co2+(自旋量子数S=3/2)、Co3+(自旋量子数S=1)或Co4+(自旋量子数S=1/2)的信号。
但是,本发明的一个方式的正极活性物质100的起因于氧四配位的顺磁性钴的信号少到无法确认。因此,本说明书等中的拟尖晶石型结晶结构与尖晶石型结晶结构不同,其不含有能够利用ESR检测出的量的氧四配位的钴。因此,与现有的正极活性物质相比,本发明的一个方式的正极活性物质100有时可以利用ESR等检测出的起因于尖晶石型Co3O4的信号小,或者少到无法确认。由于尖晶石型Co3O4无助于充放电反应,所以尖晶石型Co3O4越少越好。如此,通过ESR分析可以判断正极活性物质100与现有的正极活性物质不同。
本发明的一个方式的正极活性物质包含Co2+及Co4+中的一个或两个。此外,本发明的一个方式的正极活性物质的起因于Co2+及Co4+的自旋密度优选为2.0×1017spins/g以上且1.0×1018spins/g以下,更优选为2.5×1017spins/g以上且9.5×1017spins/g以下,进一步优选为3.0×1017spins/g以上且9.0×1017spins/g以下,更进一步优选为3.5×1017spins/g以上且8.5×1017spins/g以下。例如,正极活性物质的自旋密度通过利用ESR分析可以评价。此外,起因于Co2+及Co4+的ESR信号的g值被观察到2.15附近。上述自旋密度是指通过利用室温的ESR分析而得到的值,为正极活性物质的单位重量的自旋次数。通过ESR分析而得到的自旋次数除以用于ESR分析的样品的重量而可以算出上述自旋密度。
本发明的一个方式的正极活性物质的起因于Co2+及Co4+的自旋密度优选为3.3×10-5spins/Co原子以上且1.6×10-4spins/Co原子以下,更优选为4.1×10-5spins/Co原子以上且1.5×10-4spins/Co原子以下,进一步优选为4.9×10-5spins/Co原子以上且1.5×10- 4spins/Co原子以下,更进一步优选为5.7×10-5spins/Co原子以上且1.4×10-4spins/Co原子以下。上述自旋密度是指通过利用室温的ESR分析而得到的值,为正极活性物质的每个钴原子的自旋次数。通过利用ESR分析而得到的自旋次数除以用于ESR分析的正极活性物质中的钴原子个数而可以算出上述的自旋密度。例如,在为钴酸锂的情况下,将其组成设定为LiCoO2,并且根据其分子量97.87及用于ESR分析的正极活性物质的重量可以算出正极活性物质中的钴原子个数。
具有上述自旋密度的正极活性物质的结晶结构稳定,可以抑制在反复充放电时结晶结构崩塌。另外,通过将本发明的一个方式的正极活性物质用于二次电池,可以实现具有优异的循环特性及速率特性的二次电池。另外,具有上述自旋密度的正极活性物质在充电状态下有时成为拟尖晶石型结晶结构。
<XPS>
X射线光电子能谱(XPS)可以进行从表面到2nm至8nm左右(一般是5nm左右)的深度范围的分析,所以可以定量地分析粒子表层部的约一半区域中的各元素的浓度。另外,通过进行窄扫描分析,可以分析元素的键合状态。XPS的测定精度在很多情况下是±1atomic%左右,虽然根据元素,但是检测下限为1atomic%左右。
在进行正极活性物质100的XPS分析时,钴浓度为1时的镁浓度的相对值优选为0.4以上且1.5以下,更优选为0.45以上且小于1.00。另外,氟等卤素浓度的相对值优选为0.05以上且1.5以下,更优选为0.3以上且1.00以下。
另外,在利用XPS分析正极活性物质100时,优选示出氟与其他元素的键合能量的峰值为682eV以上且小于685eV,更优选为684.3eV左右。该值与氟化锂的键合能量的685eV以及氟化镁的键合能量的686eV都不同。换言之,在正极活性物质100包含氟时,优选为氟化锂及氟化镁以外的键合。
另外,在进行正极活性物质100的XPS分析时,优选示出镁与其他元素的键合能量的峰值为1302eV以上且小于1304eV,更优选为1303eV左右。该值与氟化镁的键合能量的1305eV不同且接近氧化镁的键合能量。换言之,在正极活性物质100包含镁时,优选为氟化镁以外的键合。
<EDX>
例如,通过利用能量分散型X射线分析法(EDX:Energy Dispersive X-raySpectroscopy)可以评价粒子内部、粒子表层部及晶界附近的各种元素的浓度。在EDX测定中,有时将边扫描区域内边进行测定且对区域内进行二维评价的方法称为EDX面分析。另外,有时将从EDX的面分析抽出线状区域的数据,对正极活性物质粒子内的原子浓度分布进行评价的方法称为线分析。
通过EDX面分析(例如元素映射),可以定量分析粒子内部、粒子表层部以及晶界附近的镁及氟的浓度。另外,通过EDX射线分析,可以分析镁及氟的浓度的峰值。
在进行正极活性物质100的EDX分析时,粒子表层部的镁的浓度峰值优选出现在从正极活性物质100的表面到向中心的深度3nm的范围内,更优选出现在到深度1nm的范围,进一步优选出现在到深度0.5nm的范围。
正极活性物质100的氟分布优选与镁分布重叠。因此,在进行EDX分析时,粒子表层部的氟的浓度峰优选出现在从正极活性物质100的表面到向中心的深度3nm的范围,更优选出现在到深度1nm的范围,进一步优选出现在到深度0.5nm的范围。
在进行正极活性物质100的线分析或面分析时,晶界附近的镁和钴的原子个数比(Mg/Co)优选为0.020以上且0.50以下。更优选为0.025以上且0.30以下。进一步优选为0.030以上且0.20以下。
<dQ/dVvsV曲线>
本发明的一个方式的正极活性物质在以高电压充电后,例如,以0.2C以下的低速率进行放电时,在快要结束放电时出现特征性的电压变化。当从放电曲线算出的dQ/dVvsV曲线中至少有一个峰值位于3.5V至3.9V的范围内时,可以清楚地观察到该电压变化。
(实施方式2)
在本实施方式中,本发明的一个方式的正极活性物质的制造方法的一个例子。
[正极活性物质的制造方法1]
参照图8及图9,对本发明的一个方式的正极活性物质100的制造方法的一个例子进行说明。
<步骤S11>
在步骤S11中,作为第一混合物的材料准备卤素源及镁源(图8及图9的步骤S11)。另外,优选还准备锂源。此外,当利用湿式进行接下来的混合及粉碎工序时,准备溶剂。
作为镁源,例如可以使用氟化镁、氧化镁、水氧化镁、碳酸镁等。作为锂源,例如可以使用氟化锂、碳酸锂。
作为卤素源,可以使用包含氟或氯等的材料。可以将包含卤素及镁的材料用作卤素源及镁源。另外,也可以将包含卤素及锂的材料用作卤素源及锂源。作为包含氟的卤素源及锂源,例如可以使用氟化锂等。作为包含氟的卤素源及镁源,例如可以使用氟化镁等。其中,氟化锂的熔点较低为848℃,在后述的退火工序中容易熔化,所以优选地用作卤素源及锂源。作为包含氯的卤素源及锂源,例如,可以使用氯化锂。作为包含氯的卤素源及镁源,例如可以使用氯化镁等。
作为溶剂,可以使用丙酮等酮、乙醇及异丙醇等醇、乙醚、二氧六环、乙腈、N-甲基-2-吡咯烷酮(NMP)等。优选使用不容易与锂发生反应的非质子性溶剂,例如可以优选地使用丙酮。
举出作为卤素源及锂源使用氟化锂(LiF)的例子及作为卤素源及镁源使用氟化镁(MgF2)的例子,具体地进行说明。当氟化锂和氟化镁以LiF:MgF2=65:35(摩尔比)左右混合时,对降低熔点最有效(非专利文献4)。当氟化锂较多时,锂变得过于多而可能导致循环特性恶化。为此,氟化锂LiF和氟化镁MgF2的摩尔比优选为LiF:MgF2=x:1(0≤x≤1.9),更优选为LiF:MgF2=x:1(0.1≤x≤0.5),进一步优选为LiF:MgF2=x:1(x=0.33附近)。另外,在本说明书等中,附近是指大于其值0.9倍且小于1.1倍的值。
<步骤S12>
接着,在步骤S12中,混合步骤S11中准备的材料和溶剂并进行粉碎(图8及图9的步骤S12)。混合可以利用干法或湿法进行,湿法可以将该材料粉碎得更小,所以是优选的。混合例如可以使用球磨机、砂磨机等。当使用球磨机时,例如优选使用锆质球作为介质。优选充分地进行该混合及粉碎工序来使材料微粉化。
<步骤S13、步骤S14>
对在步骤S12中进行了混合、粉碎的材料进行回收(图8及图9的步骤S13)得到第一混合物(图8及图9的步骤S14)。
作为第一混合物的粒子径,例如优选其平均粒子径(D50)为600nm以上且20μm以下,更优选为1μm以上且10μm以下。通过采用被这样微粉化的第一混合物,在后面的工序中与含有锂及过渡金属的复合氧化物混合时,更易于第一混合物均匀地附着于复合氧化物的粒子的表面。当复合氧化物的粒子的表面均匀地附着有第一混合物时,通过加热处理可以使复合氧化物的粒子表层部中含有卤素及镁,所以是优选的。当粒子表层部中存在不含卤素及镁的区域时,在充电状态下不容易形成后述的拟尖晶石型结晶结构。
<步骤S21>
在步骤S21中,作为含有锂及过渡金属的复合氧化物的材料,准备锂源及过渡金属源(图8的步骤S21)。
作为锂源,例如可以使用碳酸锂、氟化锂等。
作为过渡金属可以优选地使用钴。另外,也可以还包含铝及镍中的任一个或两个。
作为过渡金属源,可以使用上述过渡金属的氧化物、氢氧化物等。作为钴源,例如可以使用氧化钴、氢氧化钴等。作为锰源,可以使用氧化锰、氢氧化锰等。作为镍源,可以使用氧化镍、氢氧化镍等。作为铝源,可以使用氧化铝、氢氧化铝等。
<步骤S22>
接着,在步骤S22中混合步骤S21中准备的锂源及过渡金属源(图8的步骤S22)。混合可以利用干法或湿法进行。例如,还可以使用球磨机、砂磨机等进行混合。当利用球磨机时,例如优选使用锆质球作为介质。
<步骤S23>
接着,在步骤S23中,对步骤S22中混合的材料进行加热处理(图8的步骤S23)。为了与后面的加热处理进行区别,有时也将该处理称为焙烧或第一加热处理。加热处理优选以800℃以上且低于1100℃,更优选以900℃以上且1000℃以下,进一步优选以950℃左右进行。加热处理的温度过低时可能导致起始材料分解及熔化不充分。另一方面,加热处理的温度过高时可能导致过渡金属的过度还原,由于锂的蒸发等导致缺陷。
加热处理的时间优选为2小时以上且20小时以下。加热处理的气氛优选为干燥空气等水分少(例如露点为-50℃以下,优选为-100℃以下)。例如,可以以1000℃加热10小时,升温速率可以为200℃/h。此外,干燥气氛的流量优选设定为由于加热处理从材料脱离的气体的分压充分降低的流量。通过将所脱离的气体的分压设定为充分降低的流量,可以促进因材料而发生的反应。
加热处理后,可以将材料冷却至室温。例如,从规定温度到室温的降温时间优选为10小时以上且50小时以下。但是,步骤S23中的冷却不一定必须降至室温。只要能够进行后面的步骤S24、步骤S25及步骤S31至步骤S34的工序,冷却至比室温高的温度也无妨。
<步骤S24、步骤S25>
接着,回收步骤S23中焙烧的材料(图8的步骤S24)得到含有锂及过渡金属的复合氧化物(图8的步骤S25)。具体而言,得到钴酸锂、或包含铝或镍的钴酸锂。
另外,步骤S25中也可以使用预先合成的含有锂及过渡金属的复合氧化物(图9的步骤S25)。此时,可以省略步骤S21至步骤S24。
当使用预先合成的含有锂及过渡金属的复合氧化物时,优选使用杂质少的复合氧化物。在本说明书等中,作为含有锂及过渡金属的复合氧化物以及正极活性物质,将锂、钴、镍、铝及氧看作其主要成分,将上述主要成分以外的元素看作杂质。例如,当利用辉光放电质谱法分析时,总杂质浓度优选为10,000ppm(wt)以下,更优选为5000ppm(wt)以下。
例如,作为预先合成的钴酸锂,可以使用日本化学工业公司(NIPPON CHEMICALINDUSTRIAL CO.,LTD.)制造的钴酸锂粒子(商品名:CELLSEED C-10N)。该钴酸锂的平均粒子径(D50)约为12μm,在利用辉光放电质谱法(GD-MS)的杂质分析中,镁浓度及氟浓度为50ppm(wt)以下、钙浓度、铝浓度及硅浓度为100ppm(wt)以下、镍浓度为150ppm(wt)以下、硫浓度为500ppm(wt)以下、砷浓度为1100ppm(wt)以下、除锂、钴及氧以外的元素的浓度为150ppm(wt)以下。
作为预先合成的钴酸锂,可以使用日本化学工业公司制造的钴酸锂粒子(商品名:CELLSEED C-5H)。该钴酸锂的平均粒子径(D50)约为6.5μm,利用GD-MS进行杂质分析时的除锂、钴及氧以外的元素的浓度与C-10N为相同左右或者更低。
步骤S25中的含有锂及过渡金属的复合氧化物优选具有缺陷及变形少的层状岩盐型结晶结构。为此,优选使用杂质少的复合氧化物。当含有锂及过渡金属的复合氧化物中含有较多杂质时,结晶结构很可能具有大量缺陷或变形。
<步骤S31>
接着,在步骤S31中,混合步骤S14中得到的第一混合物和步骤S25中得到的含有锂及过渡金属的复合氧化物(图8及图9的步骤S31)。含有锂及过渡金属的复合氧化物中的过渡金属TM与第一混合物Mix1中的镁MgMix1的原子个数比优选为TM:MgMix1=1:y(0.0005≤y≤0.03),更优选为TM:MgMix1=1:y(0.001≤y≤0.01),进一步优选为TM:MgMix1=1:0.005左右。
为了不损坏复合氧化物的粒子,步骤S31的混合优选在比步骤S12的混合更温和的条件下进行。例如,优选在比步骤S12的混合的旋转数少或时间短的条件下进行。另外,与湿法相比干法是更为温和的条件。混合例如可以利用球磨机、砂磨机等。当使用球磨机时,例如优选使用锆质球作为介质。
<步骤S32、步骤S33>
接着,回收步骤S31中混合的材料(图8及图9的步骤S32)得到第二混合物(图8及图9的步骤S33)。
注意,虽然本实施方式中对将氟化锂及氟化镁的混合物添加至杂质少的钴酸锂的方法进行说明,但是本发明的一个方式不局限于此,也可以使用将镁源及氟源添加到钴酸锂的起始材料后通过焙烧得到的混合物来代替步骤S33的第二混合物。在这种情况下,不需要分离步骤S11至步骤S14的工序和步骤S21至步骤S25的工序而更为简便,生产率更高。
或者,可以使用预先添加有镁及氟的钴酸锂。使用添加有镁及氟的钴酸锂可以省略到步骤S32为止的工序而更为简便。
再者,可以对预先添加有镁及氟的钴酸锂添加镁源及氟源。
<步骤S34>
接着,在步骤S34中,对步骤S33中得到的第二混合物进行加热处理(图8及图9的步骤S34)。为了与之前的加热处理区别,有时也将该处理称为退火或第二加热处理。
退火优选以适当的温度及时间进行。适当的温度及时间根据步骤S25的含有锂及过渡金属的复合氧化物的粒子的大小及组成等条件不同。在粒子较小的情况下,有时优选在比粒子大时更低的温度或更短时间进行退火。
例如,当步骤S25的粒子的平均粒子径(D50)为12μm左右时,退火温度例如优选为600℃以上且950℃以下。退火时间例如优选为3小时以上,更优选为10小时以上,进一步优选为60小时以上。
当步骤S25的粒子的平均粒子径(D50)为5μm左右时,退火温度例如优选为600℃以上且950℃以下。退火时间例如优选为1小时以上且10小时以下,更优选为2小时左右。
退火后的降温时间例如优选为10小时以上且50小时以下。
可以认为当对第二混合物进行退火时第一混合物中的熔点低的材料(例如,氟化锂,熔点848℃)先熔化而分布在复合氧化物的粒子表层部中。接着,可以推测由于该熔化的材料的存在使其他材料的熔点下降,其他的材料熔化。例如,可以认为氟化镁(熔点1263℃)熔化而分布至复合氧化物的粒子表层部中。
然后,可以认为分布在粒子表层部中的第一混合物含有的元素在含有锂及过渡金属的复合氧化物中形成固溶体。
与复合氧化物的粒子内部相比包含在第一混合物中的元素在粒子表层部及结晶粒界附近扩散得更快。为此,粒子表层部及结晶粒界附近的镁及卤素的浓度高于复合氧化物的粒子内部的镁及卤素的浓度。如后面所述,粒子表层部及结晶粒界附近的镁浓度越高,越可以有效地抑制结晶结构的变化。
<步骤S35>
接着,在步骤S35中,回收步骤S34的退火后的材料得到本发明的一个方式的正极活性物质100。
当以图8及图9那样的方法制造时,可以制造出以高电压进行充电时具有缺陷少的拟尖晶石型结晶结构的正极活性物质100。另外,通过将本发明的一个方式的正极活性物质100用于二次电池,可以实现具有优异的循环特性及速率特性的二次电池。例如,在正极活性物质中拟尖晶石型结晶结构所占的比率为50%以上,由此可以实现具有优异的循环特性及速率特性的二次电池。例如,通过XRD测定中采用里特沃尔德分析,可以确认在正极活性物质中所占的拟尖晶石型结晶结构的比率。
为了制造出高电压充电后具有拟尖晶石型结晶结构的正极活性物质,有效的制造方法是:使正极活性物质包含镁及氟;以适当的温度及时间进行退火。镁源及氟源也可以添加至复合氧化物的起始材料中。但是,当将镁源及氟源添加至复合氧化物的起始材料中时,当镁源及氟源的熔点高于焙烧温度时,镁源及氟源可能不熔化而导致扩散不充分。这会导致层状岩盐型结晶结构有可能有很多缺陷或变形。由此,高电压充电后的拟尖晶石型结晶结构也可能存在缺陷或变形。
因此,优选首先获得具有杂质少、缺陷或变形少的层状岩盐型结晶结构的复合氧化物。然后,优选在后面的工序中混合复合氧化物和镁源及氟源并进行退火以在复合氧化物的粒子表层部中使镁和氟固溶。通过该方法可以制造高电压充电后具有缺陷或变形少的拟尖晶石型结晶结构的正极活性物质。
另外,通过上述工序制造的正极活性物质100也可以被其他材料覆盖。另外,可以进行进一步加热处理。
例如,可以将正极活性物质100和含有磷酸的化合物进行混合。另外,可以在混合后进行加热处理。通过混合含有磷酸的化合物,可以形成即使长时间保持高电压充电状态也可以抑制钴等过渡金属的溶解的正极活性物质100。另外,通过混合后进行加热处理,可以使磷酸更均匀地覆盖。
作为含有磷酸的化合物,例如可以使用磷酸锂、磷酸二氢铵等。作为混合,例如可以利用固相法进行。作为加热,例如可以以800℃以上进行2小时。
[正极活性物质的制造方法2]
参照图10对与上述的[正极活性物质的制造方法1]不同的正极活性物质100的制造方法进行说明。
<步骤S41>
在步骤S41中,准备作为混合物的材料的锂源、过渡金属源、卤素源及镁源(图10的步骤S41)。另外,当利用湿法进行接下来的混合及粉碎工序时,准备溶剂。
关于锂源、过渡金属源、卤素源、镁源及溶剂可以参照上述[正极活性物质的制造方法1]的记载,省略其详细说明。
<步骤S42>
接着,在步骤S42中,混合步骤S41中准备的材料和溶剂并进行粉碎(图10的步骤S42)。关于混合及粉碎,可以参照上述[正极活性物质的制造方法1]的记载,省略其详细说明。
<步骤S43、步骤S44>
对步骤S42中进行了混合、粉碎的材料进行回收(图10的步骤S43)得到混合物(图10的步骤S44)。
<步骤S45>
接着,在步骤S45中,对步骤S44中得到的混合物进行第一加热处理(图10的步骤S45)。第一加热处理优选以800℃以上且小于1100℃,更优选以900℃以上且1000℃以下,进一步优选以950℃左右进行。第一加热处理的温度过低时可能导致起始材料分解及熔化不充分。另一方面,第一加热处理的温度过高时可能导致过渡金属的过度还原,由于锂的蒸发等导致缺陷。
第一加热处理的时间优选为2小时以上且20小时以下。第一加热处理的气氛优选为干燥空气等水分少(例如露点为-50℃以下,优选为-100℃以下)。例如,以1000℃加热10小时,升温速率为200℃/h,干燥气氛的流量优选设定为10L/min。
第一加热处理后,可以将材料冷却至室温。例如,从规定温度到室温的降温时间优选为10小时以上且50小时以下。但是,步骤S45中的冷却不一定必须降至室温。只要能够进行后面的步骤S46及步骤S47的工序,冷却至比室温高的温度也无妨。
<步骤S46>
接着,对步骤S46中焙烧了的混合物进行回收(图10的步骤S46)。
<步骤S47>
接着,在步骤S47中,对步骤S46中得到的第二混合物进行加热处理(图10的步骤S47)。
第二加热处理优选以适当的温度及时间进行。适当的温度及时间根据混合物的粒子的大小及组成等条件不同。在粒子较小的情况下,有时优选在比粒子大时更低的温度或更短时间进行第二加热处理。
例如,当混合物的粒子的平均粒子径(D50)为12μm左右时,第二加热处理的温度例如优选为600℃以上且950℃以下。第二加热处理的时间例如优选为3小时以上,更优选为10小时以上,进一步优选为60小时以上。
当混合物的粒子的平均粒子径(D50)为5μm左右时,第二加热处理的温度例如优选为600℃以上且950℃以下。第二加热处理的时间例如优选为1小时以上且10小时以下,更优选为2小时左右。
第二加热处理后的降温时间例如优选为10小时以上且50小时以下。
<步骤S48>
接着,在步骤S48中,回收步骤S34的退火后的材料而得到本发明的一个方式的正极活性物质100。
以上是本发明的一个方式的正极活性物质100的制造方法。
(实施方式3)
在本实施方式中,对可用于包括上述实施方式所说明的正极活性物质100的二次电池的材料的例子进行说明。在本实施方式中,以正极、负极及电解液被外包装体包围的二次电池为例子进行说明。
[正极]
正极包括正极活性物质层及正极集流体。
<正极活性物质层>
正极活性物质层至少包含正极活性物质。此外,正极活性物质层除了正极活性物质以外,还可以包含活性物质表面的覆膜、导电助剂或粘合剂等其他物质。
作为正极活性物质,可以使用上述实施方式所说明的正极活性物质100。通过使用上述实施方式所说明的正极活性物质100,可以实现高容量且循环特性优异的二次电池。
作为导电助剂,可以使用碳材料、金属材料或导电性陶瓷材料等。此外,作为导电助剂,也可以使用纤维状的材料。在活性物质层总量中导电助剂所占的比率优选为1wt%以上且10wt%以下,更优选为1wt%以上且5wt%以下。
通过利用导电助剂,可以在活性物质层中形成导电网络。通过利用导电助剂,可以维持正极活性物质彼此之间的导电路径。通过对活性物质层添加导电助剂,可以实现具有高电导性的活性物质层。
作为导电助剂,例如可以使用天然石墨、中间相碳微球等人造石墨、碳纤维等。作为碳纤维,例如可以使用中间相沥青类碳纤维、各向同性沥青类碳纤维等碳纤维。作为碳纤维,可以使用碳纳米纤维或碳纳米管等。例如,可以通过气相生长法等制造碳纳米管。作为导电助剂,例如可以使用碳黑(乙炔黑(AB)等)、石墨(黑铅)粒子、石墨烯或富勒烯等碳材料。此外,例如可以使用铜、镍、铝、银、金等的金属粉末或金属纤维、导电性陶瓷材料等。
作为导电助剂也可以使用石墨烯化合物。
石墨烯化合物有时具有高导电性这样的优良的电特性以及高柔软性和高机械强度这样的优良的物理特性。另外,石墨烯化合物具有平面形状。石墨烯化合物可以形成接触电阻低的面接触。石墨烯化合物有时即使薄也具有非常高的导电性,因此可以在活性物质层中以少量高效率地形成导电路径。因此,通过将石墨烯化合物用作导电助剂,可以增大活性物质与导电助剂之间的接触面积,所以是优选的。优选的是,通过利用喷雾干燥装置,可以以覆盖活性物质的表面整体的方式形成被用作覆膜的导电助剂的石墨烯化合物。此外,可以减少电阻,所以是优选的。在此,特别优选的是,作为石墨烯化合物例如使用石墨烯、多层石墨烯或者RGO。在此,RGO例如是指还原氧化石墨烯(graphene oxide:GO)而得到的化合物。
在使用粒径小的活性物质,例如使用粒径为1μm以下的活性物质时,活性物质的比表面积大,所以需要更多的连接活性物质彼此之间的导电路径。因此,导电助剂的量趋于变多,有活性物质的含量相对减少的趋势。当活性物质的含量减少时,二次电池的容量也减少。在这种情况下,作为导电助剂,因为不需要减少活性物质的含量,所以特别优选使用即使是少量也可以高效地形成导电路径的石墨烯化合物。
以下,作为一个例子说明作为导电助剂包含石墨烯化合物的活性物质层200的截面结构例子。
图11A是活性物质层200的纵向截面图。活性物质层200包括粒状正极活性物质100、用作导电助剂的石墨烯化合物201以及粘合剂(未图示)。在此,作为石墨烯化合物201,例如可以使用石墨烯或多层石墨烯。此外,石墨烯化合物201优选具有片状。石墨烯化合物201可以以多个多层石墨烯或(和)多个单层石墨烯部分地重叠的方式形成一个片状。
在活性物质层200的纵向截面中,如图11B所示,片状的石墨烯化合物201大致均匀地分散在活性物质层200的粒子内部。在图11B中,虽然示意性地以粗线表示石墨烯化合物201,但实际上石墨烯化合物201为具有碳分子的单层或多层的厚度的薄膜。由于多个石墨烯化合物201以覆盖多个粒状正极活性物质100的一部分的方式或者以贴在多个粒状正极活性物质100的表面的方式形成,所以石墨烯化合物201与正极活性物质100形成面接触。
在此,通过使多个石墨烯化合物彼此结合,可以形成网状的石墨烯化合物薄片(以下称为石墨烯化合物网或石墨烯网)。当石墨烯网覆盖活性物质时,石墨烯网可以被用作使活性物质彼此结合的粘合剂。因此,可以减少粘合剂的量或不使用粘合剂,由此可以增高电极体积或电极重量中活性物质所占的比例。也就是说,可以提高二次电池的容量。
在此,优选的是,作为石墨烯化合物201使用氧化石墨烯,混合该氧化石墨烯和活性物质来形成将成为活性物质层200的层,然后进行还原。通过在石墨烯化合物201的形成中使用极性溶剂中的分散性极高的氧化石墨烯,石墨烯化合物201可以大致均匀地分散在活性物质层200的粒子内部。使溶剂从包含均匀分散的氧化石墨烯的分散介质中挥发而除去,且将氧化石墨烯还原,因此残留在活性物质层200中的石墨烯化合物201相互部分重叠,以形成面接触的方式分散,由此可以形成三维导电路径。另外,氧化石墨烯的还原例如也可以通过加热处理或者使用还原剂进行。
因此,不同于与活性物质形成点接触的乙炔黑等粒状导电助剂,石墨烯化合物201能够形成接触电阻低的面接触,所以可以以比一般的导电助剂少的石墨烯化合物201提高粒状正极活性物质100与石墨烯化合物201之间的导电性。因此,可以增加活性物质层200中的正极活性物质100所占的比率。由此,可以增加二次电池的放电容量。
另外,通过预先使用喷雾干燥装置,可以以覆盖活性物质的表面整体的方式形成用作覆膜的导电助剂的石墨烯化合物,并且由石墨烯化合物形成活性物质之间的导电路径。
作为粘合剂优选例如使用丁苯橡胶(SBR:styrene-butadiene rubber)、苯乙烯-异戊二烯-苯乙烯橡胶(styrene-isoprene-styrene rubber)、丙烯腈-丁二烯橡胶(acrylonitrile-butadiene rubber)、丁二烯橡胶(butadiene rubber)、乙烯-丙烯-二烯共聚物(ethylene-propylene-diene copolymer)等橡胶材料。作为粘合剂也可以使用氟橡胶。
作为粘合剂例如优选使用水溶性高分子。作为水溶性高分子,例如可以使用多糖类等。作为多糖类,可以使用羧甲基纤维素(CMC)、甲基纤维素、乙基纤维素、羟丙基纤维素、二乙酰纤维素、再生纤维素等纤维素衍生物、淀粉等。更优选并用这些水溶性高分子和上述橡胶材料。
作为粘合剂优选使用聚苯乙烯、聚丙烯酸甲酯、聚甲基丙烯酸甲酯(PMMA)、聚丙烯酸钠、聚乙烯醇(PVA)、聚氧化乙烯(PEO)、聚环氧丙烷、聚酰亚胺、聚氯乙烯、聚四氟乙烯、聚乙烯、聚丙烯、聚异丁烯、聚对苯二甲酸乙二醇酯、尼龙、聚偏二氟乙烯(PVDF)、聚丙烯腈(PAN)、三元乙丙聚合物、聚醋酸乙烯酯、硝酸纤维素等材料。
作为粘合剂,也可以组合使用上述材料中的多种。
例如,也可以组合粘度调节功能特别高的材料与其它材料而使用。例如,虽然橡胶材料等具有高粘结力、高弹性,但是在混合在溶剂中的情况下有时难以进行粘度调节。在这样的情况下,例如,优选与粘度调节功能特别高的材料混合。作为粘度调节功能特别高的材料,例如可以使用水溶性高分子。此外,作为粘度调节功能特别好的水溶性高分子,可以使用上述多糖类,例如可以使用羧甲基纤维素(CMC)、甲基纤维素、乙基纤维素、羟丙基纤维素及二乙酰纤维素、再生纤维素等纤维素衍生物、淀粉。
注意,羧甲基纤维素等纤维素衍生物例如通过转换成羧甲基纤维素的钠盐、铵盐等盐,溶解度得到提高,而容易发挥作为粘度调节剂的效果。由于溶解度增高,而在形成电极的浆料时,可以提高活性物质与其他构成要素的分散性。在本说明书中,用作电极的粘合剂的纤维素及纤维素衍生物包含它们的盐。
氟类树脂具有优异的机械强度、高耐化学品性、高耐热性等的优点。在氟类树脂中,氟类树脂之一的PVDF具有极为优异的特性、机械强度、良好的加工性及高耐热性。
另一方面,在对活性物质层进行涂敷工序时制造的浆料变为碱性时,PVDF有时被凝胶化。或者PVDF有时具有不溶解性。由于粘合剂的凝胶化或不溶解性,有时导致集电体和活性物质层的粘合性的降低。通过使用本发明的一个方式的正极活性物质,有时可以降低浆料的pH而抑制凝胶化或不溶解性,这是优选的。
例如,正极活性物质层的厚度为10μm以上且200μm以下。或者,50μm以上且150μm以下。例如,在正极活性物质含有具有钴的层状岩盐型结晶结构的材料的情况下,正极活性物质层的担载量为1mg/cm2以上且50mg/cm2以下。或者,5mg/cm2以上且30mg/cm2以下。例如,在正极活性物质含有具有钴的层状岩盐型结晶结构的材料的情况下,正极活性物质层的密度为2.2g/cm3以上且4.9g/cm3以下或者为3.8g/cm3以上且4.5g/cm3以下。
<正极集流体>
作为正极集流体,可以使用不锈钢、金、铂、铝、钛等金属及它们的合金等导电性高的材料。此外,用于正极集流体的材料优选不因正极的电位而溶解。此外,还可以使用添加有硅、钛、钕、钪、钼等提高耐热性的元素的铝合金。另外,也可以使用与硅起反应形成硅化物的金属元素形成。作为与硅起反应形成硅化物的金属元素,有锆、钛、铪、钒、铌、钽、铬、钼、钨、钴、镍等。集流体可以适当地具有箔状、板状(片状)、网状、冲孔金属网状、拉制金属网状等形状。集流体的厚度优选为5μm以上且30μm以下。
[负极]
负极包括负极活性物质层及负极集流体。负极活性物质层也可以包含导电助剂及粘合剂。
<负极活性物质>
作为负极活性物质,例如可以使用合金类材料或碳类材料等。
作为负极活性物质,可以使用能够通过与锂的合金化/脱合金化反应进行充放电反应的元素。例如,可以使用包含硅、锡、镓、铝、锗、铅、锑、铋、银、锌、镉和铟等中的至少一个的材料。这种元素的容量比碳大,尤其是硅的理论容量大,为4200mAh/g。因此,优选将硅用于负极活性物质。另外,也可以使用含有这些元素的化合物。例如可以举出SiO、Mg2Si、Mg2Ge、SnO、SnO2、Mg2Sn、SnS2、V2Sn3、FeSn2、CoSn2、Ni3Sn2、Cu6Sn5、Ag3Sn、Ag3Sb、Ni2MnSb、CeSb3、LaSn3、La3Co2Sn7、CoSb3、InSb和SbSn等。有时将能够通过与锂的合金化/脱合金化反应进行充放电反应的元素及包含该元素的化合物等称为合金类材料。
在本说明书等中,SiO例如是指一氧化硅。或者SiO也可以表示为SiOx。在此,x优选表示1附近的值。例如x优选为0.2以上且1.5以下,更优选为0.3以上且1.2以下。
作为碳类材料,可以使用石墨、易石墨化碳(软碳)、难石墨化碳(硬碳)、碳纳米管、石墨烯、碳黑等。
作为石墨,可以举出人造石墨或天然石墨等。作为人造石墨例如可以举出中间相碳微球(MCMB)、焦炭基人造石墨(coke-based artificial graphite)、沥青基人造石墨(pitch-based artificial graphite)等。在此,作为人造石墨可以使用具有球状形状的球状石墨。例如,MCMB有时具有球状形状,所以是优选的。另外,MCMB比较容易减小其表面积,所以有时是优选的。作为天然石墨,例如可以举出鳞片状石墨、球状化天然石墨等。
当锂离子被嵌入在石墨中时(锂-石墨层间化合物的生成时)石墨示出与锂金属相同程度的低电位(0.05V以上且0.3V以下vs.Li/Li+)。由此,锂离子二次电池可以示出高工作电压。石墨还有如下优点:每单位体积的容量较大;体积膨胀比较小;较便宜;与锂金属相比安全性高等,所以是优选的。
作为负极活性物质,可以使用氧化物诸如二氧化钛(TiO2)、锂钛氧化物(Li4Ti5O12)、锂-石墨层间化合物(LiXC6)、五氧化铌(Nb2O5)、氧化钨(WO2)、氧化钼(MoO2)等。
作为负极活性物质,可以使用包含锂和过渡金属的氮化物的具有Li3N型结构的Li3-xMxN(M=Co、Ni、Cu)。例如,Li2.6Co0.4N3示出较大的充放电容量(900mAh/g,1890mAh/cm3),所以是优选的。
当作为负极活性物质使用包含锂和过渡金属的氮化物时,在负极活性物质中含有锂离子,因此可以将该负极活性物质与用作正极活性物质的V2O5、Cr3O8等不包含锂离子的材料组合,所以是优选的。注意,当将含有锂离子的材料用作正极活性物质时,通过预先使包含在正极活性物质中的锂离子脱离,作为负极活性物质,也可以使用包含锂和过渡金属的氮化物。
可以将引起转化反应的材料用于负极活性物质。例如,将氧化钴(CoO)、氧化镍(NiO)、氧化铁(FeO)等不与锂形成合金的过渡金属氧化物用于负极活性物质。作为引起转化反应的材料,还可以举出Fe2O3、CuO、Cu2O、RuO2、Cr2O3等氧化物、CoS0.89、NiS、CuS等硫化物、Zn3N2、Cu3N、Ge3N4等氮化物、NiP2、FeP2、CoP3等磷化物、FeF3、BiF3等氟化物。
作为负极活性物质层可包含的导电助剂及粘合剂,可以使用与正极活性物质层可包含的导电助剂及粘合剂同样的材料。
<负极集流体>
作为负极集流体,可以使用与正极集流体同样的材料。此外,作为负极集流体,优选使用不与锂等载体离子合金化的材料。
[电解液]
电解液包含溶剂及电解质。作为电解液的溶剂,优选使用非质子有机溶剂,例如可以使用碳酸乙烯酯(EC)、碳酸丙烯酯(PC)、碳酸丁烯酯、碳酸氯乙烯酯、碳酸亚乙烯酯、γ-丁内酯、γ-戊内酯、碳酸二甲酯(DMC)、碳酸二乙酯(DEC)、碳酸甲乙酯(EMC)、甲酸甲酯、乙酸甲酯、乙酸乙酯、丙酸甲酯、丙酸乙酯、丙酸丙酯、丁酸甲酯、1,3-二氧六环、1,4-二氧六环、乙二醇二甲醚(DME)、二甲亚砜、二乙醚、甲基二甘醇二甲醚(methyl diglyme)、乙腈、苯腈、四氢呋喃、环丁砜、磺内酯等中的一种,或者可以以任意组合及比率使用上述中的两种以上。
通过作为电解液的溶剂使用一种或多种具有阻燃性及难挥发性的离子液体(室温熔融盐),即使因二次电池的内部短路、过充电等而使内部温度上升也可以防止二次电池的破裂或起火等。离子液体由阳离子和阴离子构成,包含有机阳离子和阴离子。作为用于电解液的有机阳离子,可以举出季铵阳离子、叔锍阳离子及季鏻阳离子等脂肪族鎓阳离子或咪唑鎓阳离子及吡啶鎓阳离子等芳香族阳离子。此外,作为用于电解液的阴离子可以举出一价酰胺类阴离子、一价甲基化物类阴离子、氟磺酸阴离子、全氟烷基磺酸阴离子、四氟硼酸阴离子、全氟烷基硼酸阴离子、六氟磷酸阴离子或全氟烷基磷酸阴离子等。
作为溶解于上述溶剂中的电解质,例如可以使用LiPF6、LiClO4、LiAsF6、LiBF4、LiAlCl4、LiSCN、LiBr、LiI、Li2SO4、Li2B10Cl10、Li2B12Cl12、LiCF3SO3、LiC4F9SO3、LiC(CF3SO2)3、LiC(C2F5SO2)3、LiN(CF3SO2)2、LiN(C4F9SO2)(CF3SO2)、LiN(C2F5SO2)2等锂盐中的一种,或者可以以任意组合及比率使用上述中的两种以上。
作为用于二次电池的电解液,优选使用粒状的尘埃或电解液的构成元素以外的元素(以下,简称为“杂质”)的含量少的高纯度化的电解液。具体而言,杂质在电解液的重量中所占的比率为1%以下,优选为0.1%以下,更优选为0.01%以下。
也可以对电解液添加碳酸亚乙烯酯、丙磺酸内酯(PS)、叔丁基苯(TBB)、氟代碳酸乙烯酯(FEC)、双乙二酸硼酸锂(LiBOB)或丁二腈、己二腈等二腈化合物等添加剂。将添加的材料的浓度可以设定为例如在溶剂整体中占0.1wt%以上且5wt%以下。
可以使用用电解液使聚合物溶胀了的聚合物凝胶电解质。
此外,通过使用聚合物凝胶电解质,针对漏液性的安全性得到提高。而且,可以实现二次装置的薄型化以及轻量化。
作为凝胶化的聚合物,可以使用硅酮凝胶、丙烯酸类胶、丙烯腈类凝胶、聚氧化乙烯类凝胶、聚氧化丙烯类凝胶、氟类聚合物凝胶等。
作为聚合物,例如,可以使用聚氧化乙烯(PEO)等具有聚氧化烷烯结构的聚合物、PVDF及聚丙烯腈等、以及包含这些的共聚物等。例如,可以使用作为PVDF及六氟丙烯(HFP)的共聚物的PVDF-HFP。此外,所形成的聚合物也可以具有多孔形状。
可以使用包含硫化物类或氧化物类等的无机材料的固体电解质、包含PEO(聚氧化乙烯)类等的高分子材料的固体电解质代替电解液。当使用固体电解质时,不需要设置隔离体或间隔物。另外,由于可以使电池整体固态化,所以没有漏液的担忧而显著提高安全性。
[隔离体]
二次电池优选包括隔离体。作为隔离体,例如可以使用如下材料:纸、无纺布、玻璃纤维、陶瓷或包含尼龙(聚酰胺)、维尼纶(聚乙烯醇类纤维)、聚酯、丙烯酸树脂、聚烯烃、聚氨酯的合成纤维等。优选将隔离体加工为袋状,并以包围正极和负极中的任一个的方式配置。
隔离体可以具有多层结构。例如,可以对聚丙烯、聚乙烯等有机材料薄膜涂敷陶瓷类材料、氟类材料、聚酰胺类材料或其混合物。作为陶瓷类材料,例如可以使用氧化铝粒子、氧化硅粒子等。作为氟类材料,例如可以使用PVDF、聚四氟乙烯等。作为聚酰胺类材料,例如可以使用尼龙、芳族聚酰胺(间位芳族聚酰胺、对位芳族聚酰胺)等。
通过涂敷陶瓷类材料可以提高抗氧化性,由此可以抑制高电压充放电时隔离体劣化,从而可以提高二次电池的可靠性。通过涂敷氟类材料易于使隔离体与电极密接,而可以提高输出特性。通过涂敷聚酰胺类材料(尤其是芳族聚酰胺)可以提高耐热性,由此可以提高二次电池的安全性。
例如,可以对聚丙烯薄膜的两面涂敷氧化铝与芳族聚酰胺的混合材料。或者,也可以对聚丙烯薄膜的与正极接触的面涂敷氧化铝与芳族聚酰胺的混合材料而对与负极接触的面涂敷氟类材料。
通过采用多层结构的隔离体即使隔离体的总厚度较小也可以确保二次电池的安全性,因此可以增大二次电池的单位体积的容量。
[外包装体]
作为二次电池所包括的外包装体例如可以使用铝等金属材料及树脂材料等。另外,也可以使用薄膜状的外包装体。作为薄膜,例如可以使用如下三层结构的薄膜:在由聚乙烯、聚丙烯、聚碳酸酯、离聚物、聚酰胺等的材料构成的膜上设置铝、不锈钢、铜、镍等的柔性优良的金属薄膜,在该金属薄膜上还可以设置聚酰胺类树脂、聚酯类树脂等的绝缘性合成树脂膜作为外包装体的外表面。
[充放电方法]
二次电池的充放电例如可以如下述那样进行。
<CC充电>
首先,作为充电方法的一个说明恒流(CC:Constant current)充电。CC充电是指在充电期间的整个期间中使恒定电流流过二次电池,并且在二次电池的电压成为规定电压时停止充电的充电方法。如图12A所示那样,将二次电池假设为内部电阻R与二次电池容量C的等效电路。在此情况下,二次电池电压VB是施加到内部电阻R的电压VR和施加到二次电池容量C的电压VC的总和。
在进行CC充电期间,如图12A所示那样,开关开启,恒定电流I流过二次电池。在此期间,因为电流I恒定,所以施加到内部电阻R的电压VR根据VR=R×I的欧姆定律而恒定。另一方面,施加到二次电池容量C的电压VC随着时间推移而上升。因此,二次电池电压VB随着时间推移而上升。
并且,当二次电池电压VB成为规定电压,例如4.3V时,停止充电。当停止CC充电时,如图12B所示那样,开关关闭,成为电流I=0。因此,施加到内部电阻R的电压VR成为0V。因此,二次电池电压VB下降。
图12C示出进行CC充电期间及停止CC充电之后的二次电池电压VB与充电电流的例子。由图12C可知,在进行CC充电期间上升的二次电池电压VB在停止CC充电之后稍微降低。
<CCCV充电>
接着,对与上述不同的充电方法,即恒流恒压(CCCV:Constant Current ConstantVoltage)充电进行说明。CCCV充电是指首先进行CC充电而充电到规定电压,然后进行CV(恒压)充电而充电到流过的电流变少,具体而言,充电到成为终止电流值的充电方法。
在进行CC充电期间,如图13A所示那样,恒流电源的开关开启,恒压电源的开关关闭,因此恒定的电流I流过二次电池。在此期间,因为电流I恒定,所以施加到内部电阻R的电压VR根据VR=R×I的欧姆定律而恒定。另一方面,施加到二次电池容量C的电压VC随着时间推移而上升。因此,二次电池电压VB随着时间推移而上升。
并且,当二次电池电压VB成为规定电压,例如4.3V时,从CC充电切换为CV充电。在进行CV充电期间,如图13B所示那样,恒流电源的开关开启,恒压电源的开关关闭,因此二次电池电压VB为恒定。另一方面,施加到二次电池容量C的电压VC随着时间推移而上升。因为满足VB=VR+VC,所以施加到内部电阻R的电压VR随着时间推移而变小。随着施加到内部电阻R的电压VR变小,流过二次电池的电流I根据VR=R×I的欧姆定律而变小。
并且,当流过二次电池的电流I成为规定电流,例如相当于0.01C的电流时,停止充电。当停止CCCV充电时,如图13C所示那样,所有开关关闭,成为电流I=0。因此,施加到内部电阻R的电压VR成为0V。但是,因为通过CV充电充分地降低了施加到内部电阻R的电压VR,所以即使内部电阻R的电压不再下降,二次电池电压VB也几乎不下降。
图13D示出进行CCCV充电期间及停止CCCV充电之后的二次电池电压VB与充电电流的例子。由图13D可知,二次电池电压VB即使在停止CCCV充电之后也几乎不下降。
<CC放电>
接着,说明放电方法之一的CC放电。CC放电是指在放电期间的整个期间中从二次电池放出恒定电流,并且在二次电池电压VB成为规定电压,例如2.5V时,停止放电的放电方法。
图14示出进行CC放电期间的二次电池电压VB与放电电流的例子。从图14可知,二次电池电压VB随着放电的进展而下降。
在此,对放电率及充电率进行说明。放电率是指放电时的电流相对于电池容量的比率,并且由单位C表示。在额定容量X[Ah]的电池中,相当于1C的电流是X[A]。在以2X[A]的电流放电的情况下,可以说以2C放电,并且在以0.2X[A]的电流放电的情况下,可以说以0.2C放电。另外,充电率也是同样的,在以2X[A]的电流充电的情况下,可以说以2C充电,并且在以0.2X[A]的电流充电的情况下,可以说以0.2C充电。
(实施方式4)
在本实施方式中,对包括上述实施方式所说明的正极活性物质100的二次电池的形状的例子进行说明。用于本实施方式所说明的二次电池的材料可以参照上述实施方式的记载。
[硬币型二次电池]
首先,说明硬币型二次电池的一个例子。图15A是硬币型(单层扁平型)二次电池的外观图,图15B是其截面图。
在硬币型二次电池300中,兼用作正极端子的正极罐301和兼用作负极端子的负极罐302由使用聚丙烯等形成的垫片303绝缘并密封。正极304由正极集流体305和以与此接触的方式设置的正极活性物质层306形成。负极307由负极集流体308和以与此接触的方式设置的负极活性物质层309形成。
在用于硬币型二次电池300的正极304及负极307分别包括的活性物质层可以只形成在正极和负极中的一个表面。
作为正极罐301及负极罐302,可以使用对电解液具有抗腐蚀性的镍、铝、钛等金属、它们的合金或者它们和其他金属的合金(例如不锈钢等)。另外,为了防止因电解液所引起的腐蚀,正极罐301及负极罐302优选被镍或铝等覆盖。正极罐301与正极304电连接,并且负极罐302与负极307电连接。
通过将这些负极307、正极304及隔离体310浸渗在电解质中,如图15B所示,将正极罐301设置下方按顺序层叠正极304、隔离体310、负极307及负极罐302,并且夹着垫片303压合正极罐301和负极罐302来制造硬币型二次电池300。
通过将上述实施方式所说明的正极活性物质用于正极304,可以实现高容量且循环特性优异的硬币型二次电池300。
在此,参照图15C说明在对二次电池进行充电时电流如何流过。当将使用锂的二次电池看作一个闭路时,锂离子迁移的方向和电流流动的方向相同。注意,在使用锂的二次电池中,由于阳极及阴极、氧化反应及还原反应根据充电或放电调换,所以将反应电位高的电极称为正极,而将反应电位低的电极称为负极。由此,在本说明书中,即使在充电、放电、供应反向脉冲电流以及供应充电电流时也将正极称为“正极”或“+极”,而将负极称为“负极”或“-极”。如果使用与氧化反应及还原反应有关的阳极及阴极的术语,则充电时和放电时的阳极与阴极是相反的,这有可能引起混乱。因此,在本说明书中,不使用阳极及阴极的术语。当使用阳极及阴极的术语时,明确表示是充电时还是放电时,并示出是对应正极(+极)还是负极(-极)。
图15C所示的两个端子与充电器连接,对二次电池300进行充电。随着二次电池300的充电的进展,电极之间的电位差增大。
[圆筒型二次电池]
接着,参照图16对圆筒型二次电池的例子进行说明。图16A示出圆筒型二次电池600的外观图。图16B是示意性地示出圆筒型二次电池600的截面图。如图16B所示,圆筒型二次电池600在顶面具有正极盖(电池盖)601,并在侧面及底面具有电池罐(外装罐)602。上述正极盖与电池罐(外装罐)602通过垫片(绝缘垫片)610绝缘。
在中空圆柱状电池罐602的内侧设置有电池元件,在该电池元件中,带状的正极604和带状的负极606夹着隔离体605被卷绕。虽然未图示,但是电池元件以中心销为中心被卷绕。电池罐602的一端关闭且另一端开着。作为电池罐602可以使用对电解液具有抗腐蚀性的镍、铝、钛等金属、它们的合金或者它们和其他金属的合金(例如不锈钢等)。另外,为了防止电解液所引起的腐蚀,电池罐602优选被镍或铝等覆盖。在电池罐602的内侧,正极、负极及隔离体被卷绕而成的电池元件由对置的一对绝缘板608和绝缘板609夹着。另外,在设置有电池元件的电池罐602的内部中注入有非水电解液(未图示)。作为非水电解液,可以使用与硬币型二次电池相同的电解液。
因为用于圆筒型蓄电池的正极及负极被卷绕,从而活性物质优选形成在集流体的两个表面。正极604与正极端子(正极集电导线)603连接,而负极606与负极端子(负极集电导线)607连接。正极端子603及负极端子607都可以使用铝等金属材料。将正极端子603电阻焊接到安全阀机构612,而将负极端子607电阻焊接到电池罐602底。安全阀机构612与正极盖601通过PTC(Positive Temperature Coefficient:正温度系数)元件611电连接。当电池的内压上升到超过规定的阈值时,安全阀机构612切断正极盖601与正极604的电连接。另外,PTC元件611是在温度上升时其电阻增大的热敏感电阻元件,并通过电阻的增大来限制电流量以防止异常发热。作为PTC元件,可以使用钛酸钡(BaTiO3)类半导体陶瓷等。
另外,如图16C所示那样,也可以将多个二次电池600夹在导电板613和导电板614之间而构成模块615。多个二次电池600可以被并联连接、被串联连接或者被并联连接后再被串联连接。通过构成包括多个二次电池600的模块615,可以提取较大电力。
图16D是模块615的俯视图。为了明确起见,以虚线表示导电板613。如图16D示出,模块615可以包括使多个二次电池600电连接的导线616。可以以与导线616重叠的方式在导线616上设置导电板。另外,也可以在多个二次电池600之间包括温度控制装置617。在二次电池600过热时可以通过温度控制装置617冷却,在二次电池600过冷时可以通过温度控制装置617加热。由此模块615的性能不容易受到外部气温的影响。温度控制装置617所包括的热媒体优选具有绝缘性及不燃性。
通过将上述实施方式所说明的正极活性物质用于正极604,可以实现高容量且循环特性优异的圆筒型二次电池600。
[二次电池的结构例]
参照图17至图21对二次电池的其他结构例子进行说明。
图17A及图17B是电池组的外观图。二次电池913通过电路板900与天线914连接。在二次电池913上贴合有签条910。电路板900被密封材料915固定于签条910。再者,如图17B所示,二次电池913与端子951和端子952连接。
电路板900包括端子911和电路912。端子911与端子951、端子952、天线914及电路912连接。另外,也可以设置多个端子911,将多个端子911分别用作控制信号输入端子、电源端子等。
电路912也可以设置在电路板900的背面。另外,天线914的形状不局限于线圈状,例如也可以为线状、板状。另外,还可以使用平面天线、口径天线、行波天线、EH天线、磁场天线或介质天线等天线。或者,天线914也可以为平板状的导体。该平板状的导体也可以用作电场耦合用导体之一。换言之,也可以将天线914用作电容器所具有的两个导体中之一。由此,不但利用电磁、磁场,而且还可以利用电场交换电力。
电池组在天线914与二次电池913之间包括层916。层916例如具有可遮蔽来自二次电池913的电磁场的功能。作为层916,例如可以使用磁性体。
二次电池的结构不局限于图17所示的结构。
例如,如图18A1及图18A2所示,也可以在图17A及图17B所示的二次电池913的对置的一对表面分别设置天线。图18A1是示出上述一对表面中的一个表面一侧的外观图,图18A2是示出上述一对表面中的另一个表面一侧的外观图。另外,与图17A和图17B所示的二次电池相同的部分可以适当地援用图17A和图17B所示的二次电池的说明。
如图18A1所示,在二次电池913的一对表面中的一个表面上夹着层916设置有天线914,如图18A2所示,在二次电池913的一对表面中的另一个表面上夹着层917设置有天线918。层917例如具有可遮蔽来自二次电池913的电磁场的功能。作为层917,例如可以使用磁性体。
通过采用上述结构,可以增大天线914和天线918双方的尺寸。天线918例如具有与外部设备进行数据通信的功能。作为天线918,例如可以使用具有能应用于天线914的形状的天线。作为利用天线918的二次电池与其他设备之间的通信方法,可以使用NFC(近距离无线通讯)等能够在二次电池与其他设备之间使用的响应方式等。
或者,如图18B1所示,也可以在图17A及图17B所示的二次电池913上设置显示装置920。显示装置920与端子911电连接。另外,也可以在设置有显示装置920的部分不贴合有签条910。此外,与图17A及图17B所示的二次电池相同的部分可以适当地援用图17A及图17B所示的二次电池的说明。
在显示装置920上,例如可以显示示出是否正在进行充电的图像、示出蓄电量的图像等。作为显示装置920,例如可以使用电子纸、液晶显示装置、电致发光(也称为EL)显示装置等。例如,通过使用电子纸可以降低显示装置920的耗电量。
或者,如图18B2所示,也可以在图17A和图17B所示的二次电池913中设置传感器921。传感器921通过端子922与端子911电连接。此外,与图17A和图17B所示的二次电池相同的部分可以适当地援用图17A和图17B所示的二次电池的说明。
传感器921例如可以具有测量如下因素的功能:位移、位置、速度、加速度、角速度、转动数、距离、光、液、磁、温度、化学物质、声音、时间、硬度、电场、电流、电压、电力、辐射线、流量、湿度、斜率、振动、气味或红外线。通过设置传感器921,例如可以检测出示出设置有二次电池的环境的数据(温度等),而将其储存在电路912中的存储器。
再者,参照图19及图20对二次电池913的结构例子进行说明。
图19A所示的二次电池913在框体930的内部包括设置有端子951和端子952的卷绕体950。卷绕体950在框体930的内部浸渗在电解液中。端子952与框体930接触,端子951由于有绝缘材料等而不与框体930接触。注意,为了方便起见,虽然在图19A中分离地图示框体930,但是,实际上卷绕体950被框体930覆盖,端子951及端子952延伸在框体930的外侧。作为框体930,可以使用金属材料(例如铝等)或树脂材料。
另外,如图19B所示,也可以使用多个材料形成图19A所示的框体930。例如,在图19B所示的二次电池913中,框体930a和框体930b是贴合在一起的,在由框体930a及框体930b围绕的区域中设置有卷绕体950。
作为框体930a,可以使用有机树脂等绝缘材料。尤其是,通过将有机树脂等的材料用于形成天线的面,可以抑制由于二次电池913造成的电场屏蔽。另外,如果由于框体930a造成的电场屏蔽小,则也可以在框体930a的内部设置天线914等天线。作为框体930b,例如可以使用金属材料。
再者,图20示出卷绕体950的结构。卷绕体950包括负极931、正极932和隔离体933。卷绕体950是夹着隔离体933使负极931和正极932彼此重叠来形成叠层片,并且将该叠层片卷绕而形成的。另外,也可以进一步层叠多个负极931、正极932和隔离体933的叠层。
负极931通过端子951及端子952中的一方与图17所示的端子911连接。正极932通过端子951及端子952中的另一方与图17所示的端子911连接。
通过将上述实施方式所说明的正极活性物质用于正极932,可以实现高容量且循环特性优异的二次电池913。
[层压型二次电池]
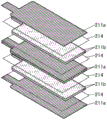
接着,参照图21至图27对层压型二次电池的例子进行说明。在将具有柔性的层压型二次电池安装在至少一部分具有柔性的电子设备时,可以沿着电子设备的变形使二次电池弯曲。
参照图21说明层压型二次电池980。层压型二次电池980包括图21A所示的卷绕体993。卷绕体993包括负极994、正极995以及隔离体996。与图20所说明的卷绕体950同样,卷绕体993是夹着隔离体996使负极994和正极995互相重叠来形成叠层片,并且将该叠层片卷绕而形成的。
另外,由负极994、正极995以及隔离体996构成的叠层的叠层个数可以根据所需的容量和元件体积适当地设计。负极994通过导线电极997和导线电极998中的一个与负极集流体(未图示)连接,正极995通过导线电极997和导线电极998中的另一个与正极集流体(未图示)连接。
如图21B所示,在通过热压合等贴合将成为外包装体的薄膜981和具有凹部的薄膜982而形成的空间中容纳上述卷绕体993,由此可以制造图21C所示的二次电池980。卷绕体993包括导线电极997和导线电极998,并使薄膜981和具有凹部的薄膜982所形成的空间浸渗在电解液中。
薄膜981和具有凹部的薄膜982例如由铝等金属材料或树脂材料构成。当作为薄膜981及具有凹部的薄膜982的材料使用树脂材料时,可以在从外部被施加力量时使薄膜981及具有凹部的薄膜982变形,而可以制造具有柔性的蓄电池。
在图21B和图21C中示出使用两个薄膜的例子,但是也可以将一个薄膜弯折形成空间,并且在该空间中容纳上述卷绕体993。
通过将上述实施方式所说明的正极活性物质用于正极995,可以实现高容量且循环特性优异的二次电池980。
虽然在图21中示出在成为外包装体的薄膜所形成的空间中包括卷绕体的二次电池980的例子,但是也可以采用如图22所示那样在成为外包装体的薄膜所形成的空间中包括长方形的多个正极、隔离体及负极的二次电池。
图22A所示的层压型二次电池500包括:包含正极集流体501及正极活性物质层502的正极503;包含负极集流体504及负极活性物质层505的负极506;隔离体507;电解液508;以及外包装体509。在设置于外包装体509内的正极503与负极506之间设置有隔离体507。此外,在外包装体509内充满了电解液508。作为电解液508,可以使用实施方式2所示的电解液。
在图22A所示的层压型二次电池500中,正极集流体501及负极集流体504还用作与外部电接触的端子。因此,也可以配置为正极集流体501及负极集流体504的一部分露出到外包装体509的外侧。此外,使用导线电极对该导线电极与正极集流体501或负极集流体504进行超声波焊接来使导线电极露出到外包装体509的外侧,而不使正极集流体501及负极集流体504露出到外包装体509的外侧。
在层压型二次电池500中,作为外包装体509,例如可以使用如下三层结构的层压薄膜:在由聚乙烯、聚丙烯、聚碳酸酯、离聚物、聚酰胺等的材料构成的膜上设置铝、不锈钢、铜、镍等的高柔性的金属薄膜,并且在该金属薄膜上作为外包装体的外表面设置聚酰胺类树脂、聚酯类树脂等的绝缘性合成树脂薄膜。
另外,图22B示出层压型二次电池500的截面结构的一个例子。为了简化起见,图22A示出包括两个集流体的例子,但是实际上如图22B所示那样电池包括多个电极层。
图22B中的一个例子包括16个电极层。另外,即使包括16个电极层,二次电池500也具有柔性。图22B示出具有8层的负极集流体504和8层的正极集流体501的总和16层的结构。另外,图22B示出负极的提取部的截面,对8层的负极集流体504进行超声波焊接。当然,电极层的个数不局限于16,可以更多或更少。在电极层的个数多的情况下,可以制造具有更多容量的二次电池。此外,在电极层的个数少的情况下,可以制造实现薄型化且具有优良的柔性的二次电池。
在此,图23及图24示出层压型二次电池500的外观图的一个例子。在图23及图24中包括:正极503;负极506;隔离体507;外包装体509;正极导线电极510;以及负极导线电极511。
图25A示出正极503及负极506的外观图。正极503包括正极集流体501,正极活性物质层502形成在正极集流体501的表面。另外,正极503具有正极集流体501的一部分露出的区域(以下,称为极耳区域(tab region))。负极506具有负极集流体504,负极活性物质层505形成在负极集流体504的表面。此外,负极506具有负极集流体504的一部分露出的区域,即极耳区域。正极及负极所具有的极耳区域的面积或形状不局限于图25A所示的例子。
[层压型二次电池的制造方法]
在此,参照图23对在图25B及图25C中示出其外观的层压型二次电池的制造方法的一个例子进行说明。
首先,层叠负极506、隔离体507和正极503。图25B示出层叠的负极506、隔离体507和正极503。在此,示出使用5组负极和4组正极的例子。接着,使正极503的极耳区域彼此接合,并且使正极导线电极510与最表面的正极的极耳区域接合。作为接合,例如可以利用超声波焊接等。与此同样,使负极506的极耳区域彼此接合,并且使负极导线电极511与最表面的负极的极耳区域接合。
接着,在外包装体509上配置负极506、隔离体507及正极503。
下面,如图25C所示,使外包装体509沿着以虚线表示的部分折叠。然后,使外包装体509的外周部接合。作为接合,例如可以使用热压合等。此时,为了后面注入电解液508,设置不与外包装体509的一部分(或一个边)接合的区域(以下,称为导入口)。
接着,将电解液508(未图示)从设置在外包装体509中的导入口导入到外包装体509的内侧。优选在减压气氛下或惰性气体气氛下导入电解液508。最后,使导入口接合。如此,可以制造层压型二次电池500。
通过将上述实施方式所说明的正极活性物质用于正极503,可以实现高容量且循环特性优异的二次电池500。
[可弯曲的二次电池]
接着,参照图26及图27对可弯曲的二次电池的例子进行说明。
图26A示出可弯曲的二次电池250的俯视示意图。图26B1、图26B2及图26C分别是沿着图26A中的截断线C1-C2、截断线C3-C4、截断线A1-A2的截面示意图。二次电池250包括外包装体251、容纳于外包装体251内部的电极叠层体210。电极叠层体210至少具有层叠有正极211a及负极211b的结构。与正极211a电连接的导线212a以及与负极211b电连接的导线212b延伸在外包装体251的外侧。此外,在由外包装体251围绕的区域中除了正极211a及负极211b以外还密封有电解液(未图示)。
参照图27说明二次电池250所包括的正极211a及负极211b。图27A是说明正极211a、负极211b及隔离体214的叠层顺序的立体图。图27B是除了正极211a及负极211b以外还示出导线212a及导线212b的立体图。
如图27A所示,二次电池250包括多个长方形正极211a、多个长方形负极211b以及多个隔离体214。正极211a及负极211b分别包括突出的极耳部分以及极耳以外的部分。在正极211a的一个面的极耳以外的部分形成有正极活性物质层,在负极211b的一个面的极耳以外的部分形成有负极活性物质层。
以正极211a的没有形成正极活性物质层的面互相接触且负极211b的没有形成负极活性物质层的面互相接触的方式层叠正极211a及负极211b。
另外,正极211a的形成有正极活性物质层的面与负极211b的形成有负极活性物质层的面之间设置有隔离体214。为方便起见,在图27中以虚线表示隔离体214。
如图27B所示,多个正极211a与导线212a在接合部215a中电连接。此外,多个负极211b与导线212b在接合部215b中电连接。
接着,参照图26B1、图26B2、图26C、图26D说明外包装体251。
外包装体251具有薄膜形状,以夹着正极211a及负极211b的方式被对折。外包装体251包括折叠部分261、一对密封部262及密封部263。一对密封部262以夹着正极211a及负极211b的方式设置并也可以称为侧部密封。此外,密封部263包括与导线212a及导线212b重叠的部分并也可以称为顶部密封。
外包装体251优选具有在与正极211a及负极211b重叠的部分交替配置棱线271及谷底线272的波形形状。此外,外包装体251的密封部262及密封部263优选为平坦。
图26B1是在与棱线271重叠的部分截断的截面,图26B2是在与谷底线272重叠的部分截断的截面。图26B1及图26B2都对应于二次电池250以及正极211a和负极211b的宽度方向的截面。
这里,正极211a及负极211b的宽度方向的端部,即正极211a及负极211b的端部与密封部262之间的距离为距离La。当使二次电池250弯曲等变形时,如后面所述,正极211a及负极211b在长度方向上互相错开地变形。此时,在距离La过短时,则有可能外包装体251与正极211a及负极211b强烈地摩擦,而造成外包装体251损坏。尤其是,在外包装体251的金属薄膜露出时,该金属薄膜有可能因电解液发生腐蚀。因此,优选将距离La尽可能地设定为长。另一方面,在距离La过长时,会导致二次电池250的体积增大。
优选的是,层叠的正极211a及负极211b的总厚度越大,正极211a及负极211b与密封部262之间的距离La越长。
更具体而言,在层叠的正极211a、负极211b和未图示的隔离体214的总厚度为厚度t时,距离La为厚度t的0.8倍以上且3.0倍以下,优选为0.9倍以上且2.5倍以下,更优选为1.0倍以上且2.0倍以下。通过使距离La在上述范围内,可以实现小巧且对弯曲具有高可靠性的电池。
当一对密封部262之间的距离为距离Lb时,优选的是距离Lb比正极211a及负极211b的宽度(在此,负极211b的宽度Wb)充分大。由此,在使二次电池250反复弯曲等变形时,由于即使正极211a及负极211b与外包装体251接触,正极211a及负极211b的一部分也可以在宽度方向上错开,所以可以有效地防止正极211a及负极211b与外包装体251摩擦。
例如,一对密封部262之间的距离Lb与负极211b的宽度Wb之差为正极211a及负极211b的厚度t的1.6倍以上且6.0倍以下,优选为1.8倍以上且5.0倍以下,更优选为2.0倍以上且4.0倍以下。
换言之,距离Lb、宽度Wb及厚度t优选满足如下算式1。
[算式1]

这里,a为0.8以上且3.0以下,优选为0.9以上且2.5以下,更优选为1.0以上且2.0以下。
另外,图26C是包括导线212a的截面,对应于二次电池250、正极211a及负极211b的长度方向的截面。如图26C所示,优选在折叠部分261中在正极211a及负极211b的长度方向的端部与外包装体251之间包括空间273。
图26D示出使电池250弯曲时的截面示意图。图26D相当于沿着图26A中的截断线B1-B2的截面。
当二次电池250弯曲时,位于弯曲部外侧的外包装体251的一部分变形为延伸,位于弯曲部内侧的外包装体251的其它一部分变形为收缩。更具体而言,外包装体251的位于弯曲的外侧的部分以波的振幅小且波的周期大的方式变形。另一方面,外包装体251的位于弯曲的内侧的部分以波的振幅大且波的周期小的方式变形。通过上述方式使外包装体251变形,可以缓和因弯曲施加到外包装体251的应力,由此构成外包装体251的材料本身不一定需要具有可伸缩性。其结果是,可以以较小的力量使二次电池250弯曲而不损伤外包装体251。
另外,如图26D所示,当使二次电池250弯曲时,正极211a及负极211b分别相对错开。此时,由于多个层叠的正极211a及负极211b在密封部263一侧的端部由固定构件217固定,因此,它们以离折叠部分261越近其错开量越大的方式错开。由此,可以缓和施加到正极211a及负极211b的应力,并且,正极211a及负极211b本身不一定需要具有可伸缩性。其结果是,可以使二次电池250弯曲而不损伤正极211a及负极211b。
另外,由于在正极211a及负极211b与外包装体251之间包括空间273,所以在弯曲时位于内侧的正极211a及负极211b可以以不与外包装体251接触的方式相对错开。
图26及图27所例示的二次电池250是即使反复弯曲伸展也不容易发生外包装体的破损以及正极211a和负极211b的破损等并且电池特性也不容易劣化的电池。通过将上述实施方式所说明的正极活性物质用于二次电池250所包括的正极211a,可以实现高容量且循环特性优异的电池。
(实施方式5)
在本实施方式中,说明将本发明的一个方式的二次电池安装在电子设备的例子。
首先,图28A至图28G示出将实施方式4的一部分所说明的可弯曲的二次电池安装在电子设备的例子。作为应用可弯曲的二次电池的电子设备,例如可以举出电视装置(也称为电视或电视接收机)、用于计算机等的显示器、数码相机、数码摄像机、数码相框、移动电话机(也称为移动电话、移动电话装置)、便携式游戏机、便携式信息终端、声音再现装置、弹珠机等大型游戏机等。
可以将具有柔性的二次电池沿着在房屋及高楼的内壁或外壁、汽车的内部装修或外部装修的曲面组装。
图28A示出移动电话机的一个例子。移动电话机7400除了组装在框体7401中的显示部7402之外还具备操作按钮7403、外部连接端口7404、扬声器7405、麦克风7406等。另外,移动电话机7400具有二次电池7407。通过将本发明的一个方式的二次电池用作上述二次电池7407,可以提供轻量且使用寿命长的移动电话机。
图28B示出使移动电话机7400弯曲的状态。在因外部的力量使移动电话机7400变形而使其整体弯曲时,设置在其内部的二次电池7407也被弯曲。图28C示出此时被弯曲的二次电池7407的状态。二次电池7407是薄型蓄电池。二次电池7407在弯曲状态下被固定。二次电池7407具有与集流体电连接的导线电极。例如,集电体是铜箔,使其一部分与镓合金化,提高与接触于集流体的活性物质层的密接性,使得二次电池7407在被弯曲的状态下的可靠性得到提高。
图28D示出手镯型显示装置的一个例子。便携式显示装置7100具备框体7101、显示部7102、操作按钮7103及二次电池7104。另外,图28E示出被弯曲的二次电池7104。当将弯曲的二次电池7104戴上使用者的胳膊时,二次电池7104的框体变形,使得二次电池7104的一部分或全部的曲率发生变化。以等价圆半径的值表示曲线的任一点的弯曲程度的值是曲率半径,并且将曲率半径的倒数称为曲率。具体而言,框体或二次电池7104的主表面的一部分或全部在曲率半径为40mm以上且150mm以下的范围变形。只要二次电池7104的主表面中的曲率半径在40mm以上且150mm以下的范围内,就可以保持高可靠性。通过将本发明的一个方式的二次电池用作上述二次电池7104,可以提供轻量且使用寿命长的便携式显示装置。
图28F是手表型便携式信息终端的一个例子。便携式信息终端7200包括框体7201、显示部7202、带子7203、带扣7204、操作按钮7205、输入输出端子7206等。
便携式信息终端7200可以执行移动电话、电子邮件、文章的阅读及编写、音乐播放、网络通信、电脑游戏等各种应用程序。
显示部7202的显示面是弯曲的,能够沿着弯曲的显示面进行显示。另外,显示部7202具备触摸传感器,可以用手指或触屏笔等触摸屏幕来进行操作。例如,通过触摸显示于显示部7202的图标7207,可以启动应用程序。
操作按钮7205除了时刻设定之外,还可以具有电源开关、无线通信的开关、静音模式的设置及取消、省电模式的设置及取消等各种功能。例如,通过利用组装在便携式信息终端7200中的操作系统,可以自由地设定操作按钮7205的功能。
便携式信息终端7200可以执行被通信标准化的近距离无线通信。例如,通过与可无线通信的耳麦通信,可以进行免提通话。
便携式信息终端7200具备输入输出端子7206,可以通过连接器直接向其他信息终端发送数据或从其他信息终端接收数据。另外,也可以通过输入输出端子7206进行充电。另外,充电工作也可以利用无线供电进行,而不利用输入输出端子7206。
便携式信息终端7200的显示部7202包括本发明的一个方式的二次电池。通过使用本发明的一个方式的二次电池,可以提供轻量且使用寿命长的便携式信息终端。例如,可以将弯曲状态的图28E所示的二次电池7104组装在框体7201的内部,或者,将二次电池7104以能够弯曲的状态组装在带子7203的内部。
便携式信息终端7200优选包括传感器。作为传感器例如优选安装指纹传感器、脉搏传感器、体温传感器等人体传感器、触摸传感器、压力传感器、加速度传感器等。
图28G示出袖章型显示装置的一个例子。显示装置7300具备显示部7304以及本发明的一个方式的二次电池。显示装置7300也可以在显示部7304具备触摸传感器,并被用作便携式信息终端。
显示部7304的显示面是弯曲的,能够沿着弯曲的显示面进行显示。另外,显示装置7300可以利用被通信标准化的近距离无线通信等改变显示情况。
显示装置7300具备输入输出端子,可以通过连接器直接向其他信息终端发送数据或从其他信息终端接收数据。另外,也可以通过输入输出端子进行充电。另外,充电工作也可以利用无线供电进行,而不利用输入输出端子。
通过将本发明的一个方式的二次电池用作显示装置7300所包括的二次电池,可以提供轻量且使用寿命长的显示装置。
参照图28H、图29及图30说明将上述实施方式所示的循环特性优异的二次电池安装在电子设备的例子。
通过将本发明的一个方式的二次电池用作日用电子设备的二次电池,可以提供轻量且使用寿命长的产品。例如,作为日用电子设备,可以举出电动牙刷、电动剃须刀、电动美容器等。这些产品中的二次电池被期待为了便于使用者容易握持而具有棒状形状且为小型、轻量、大容量。
图28H是被称为烟液容纳式吸烟装置(电子烟)的装置的立体图。在图28H中,电子烟7500包括:包括加热元件的雾化器(atomizer)7501;对雾化器供电的二次电池7504;包括液体供应容器及传感器等的烟弹(cartridge)7502。为了提高安全性,也可以将防止二次电池7504的过充电及过放电的保护电路电连接到二次电池7504。图28H所示的二次电池7504包括用来与充电器连接的外部端子。在取拿时,二次电池7504位于顶端部,因此优选其总长度较短且重量较轻。由于本发明的一个方式的二次电池为高容量且循环特性优异,所以可以提供在长期间能够长时间使用的小型轻量的电子烟7500。
接着,图29A和图29B示出能够进行对折的平板终端的一个例子。图29A和图29B所示的平板终端9600包括框体9630a、框体9630b、连接框体9630a和框体9630b的可动部9640、包括显示部9631a和显示部9631b的显示部9631、开关9625至开关9627、扣件9629以及操作开关9628。通过将具有柔性的面板用于显示部9631,可以实现显示部更大的平板终端。图29A示出打开平板终端9600的状态,图29B示出合上平板终端9600的状态。
平板终端9600在框体9630a及框体9630b的内部具备蓄电体9635。蓄电体9635穿过可动部9640设置在框体9630a及框体9630b。
在显示部9631中,可以将其整体或一部分用作触摸屏的区域,并且可以通过接触包含在上述区域上所显示的图标的图像、文字、输入框等来输入数据。例如,使框体9630a一侧的显示部9631a的整个面显示键盘并使框体9630b一侧的显示部9631b显示文字、图像等的信息而使用。
使框体9630b一侧的显示部9631a显示键盘并使框体9630a一侧的显示部9631b显示文字、图像等的信息而使用。此外,也可以通过使显示部9631显示触摸屏上的键盘显示切换按钮而使用手指或触屏笔等接触,在显示部9631上显示键盘。
可以同时对框体9630a一侧的显示部9631a的触摸屏区域和框体9630b一侧的显示部9631b的触摸屏区域进行触摸输入。
开关9625至开关9627除了被用于操作平板终端9600的接口以外,还可以被用作可进行各种功能的切换的接口。例如,开关9625至开关9627中的至少一个可以被用作切换平板终端9600的电源的开启/关闭的开关。另外,例如,开关9625至开关9627中的至少一个可以具有:切换竖屏显示和横屏显示等显示的方向的功能;以及切换黑白显示或彩色显示等的功能。另外,例如,开关9625至开关9627中的至少一个可以具有调节显示部9631的亮度的功能。另外,根据通过平板终端9600所内置的光传感器所检测的使用时的外光的光量,可以使显示部9631的亮度最优化。注意,平板终端除了光传感器以外还可以内置陀螺仪和加速度传感器等检测倾斜度的传感器等的其他检测装置。
另外,图29A示出框体9630a一侧的显示部9631a与框体9630b一侧的显示部9631b的显示面积基本相同的例子,但是对显示部9631a及显示部9631b的显示面积没有特别的限定,其中一方的大小可以与另一方的大小不同,显示质量也可以不同。例如,显示部9631a和9631b中的一个可以显示比另一个更高清晰的图像。
图29B是平板终端9600被对折的状态,并且平板终端9600包括框体9630、太阳能电池9633、具备DCDC转换器9636的充放电控制电路9634。作为蓄电体9635使用本发明的一个方式的二次电池。
此外,如上所述,平板终端9600能够对折,因此不使用时可以以彼此重叠的方式折叠框体9630a及框体9630b。通过折叠框体9630a及框体9630b,可以保护显示部9631,而可以提高平板终端9600的耐久性。此外,由于使用本发明的一个方式的二次电池的蓄电体9635为高容量且循环特性优异,所以可以提供在长期间能够长时间使用的平板终端9600。
图29A和图29B所示的平板终端9600还可以具有如下功能:显示各种各样的信息(静态图像、动态图像、文字图像等);将日历、日期或时刻等显示在显示部上;对显示在显示部上的信息进行触摸输入操作或编辑的触摸输入;通过各种各样的软件(程序)控制处理等。
通过利用安装在平板终端9600的表面上的太阳能电池9633,可以将电力供应到触摸屏、显示部或图像信号处理部等。注意,太阳能电池9633可以设置在框体9630的一个表面或两个表面,可以高效地对蓄电体9635进行充电。通过作为蓄电体9635使用锂离子电池,有可以实现小型化等的优点。
另外,参照图29C所示的方框图而对图29B所示的充放电控制电路9634的结构和工作进行说明。图29C示出太阳能电池9633、蓄电体9635、DCDC转换器9636、转换器9637、开关SW1至开关SW3以及显示部9631,蓄电体9635、DCDC转换器9636、转换器9637、开关SW1至开关SW3对应图29B所示的充放电控制电路9634。
首先,说明在利用外光使太阳能电池9633发电时的工作的例子。使用DCDC转换器9636对太阳能电池所产生的电力进行升压或降压以使它成为用来对蓄电体9635进行充电的电压。并且,当利用来自太阳能电池9633的电力使显示部9631工作时使开关SW1导通,并且,利用转换器9637将其升压或降压到显示部9631所需要的电压。另外,可以采用在不进行显示部9631中的显示时使开关SW1断开且使开关SW2导通来对蓄电体9635进行充电的结构。
注意,作为发电单元的一个例子示出太阳能电池9633,但是不局限于此,也可以使用压电元件(piezoelectric element)或热电转换元件(珀耳帖元件(Peltier element))等其他发电单元进行蓄电体9635的充电。例如,也可以使用以无线(非接触)的方式能够收发电力来进行充电的非接触电力传输模块或组合其他充电方法进行充电。
图30示出其他电子设备的例子。在图30中,显示装置8000是使用根据本发明的一个方式的二次电池8004的电子设备的一个例子。具体地说,显示装置8000相当于电视广播接收用显示装置,包括框体8001、显示部8002、扬声器部8003及二次电池8004等。根据本发明的一个方式的二次电池8004设置在框体8001的内部。显示装置8000既可以接收来自商业电源的电力供应,又可以使用蓄积在二次电池8004中的电力。因此,即使当由于停电等不能接收来自商业电源的电力供应时,通过将根据本发明的一个方式的二次电池8004用作不间断电源,也可以利用显示装置8000。
作为显示部8002,可以使用半导体显示装置诸如液晶显示装置、在每个像素中具备有机EL元件等发光元件的发光装置、电泳显示装置、DMD(数字微镜装置:DigitalMicromirror Device)、PDP(等离子体显示面板:Plasma Display Panel)及FED(场致发射显示器:Field Emission Display)等。
另外,除了电视广播接收用的显示装置之外,显示装置还包括所有显示信息用显示装置,例如个人计算机用显示装置或广告显示用显示装置等。
在图30中,安镶型照明装置8100是使用根据本发明的一个方式的二次电池8103的电子设备的一个例子。具体地说,照明装置8100包括框体8101、光源8102及二次电池8103等。虽然在图30中例示出二次电池8103设置在安镶有框体8101及光源8102的天花板8104的内部的情况,但是二次电池8103也可以设置在框体8101的内部。照明装置8100既可以接收来自商业电源的电力供应,又可以使用蓄积在二次电池8103中的电力。因此,即使当由于停电等不能接收来自商业电源的电力供应时,通过将根据本发明的一个方式的二次电池8103用作不间断电源,也可以利用照明装置8100。
另外,虽然在图30中例示出设置在天花板8104的安镶型照明装置8100,但是根据本发明的一个方式的二次电池可以用于设置在天花板8104以外的例如侧壁8105、地板8106或窗户8107等的安镶型照明装置,也可以用于台式照明装置等。
作为光源8102,可以使用利用电力人工性地得到光的人工光源。具体地说,作为上述人工光源的例子,可以举出白炽灯泡、荧光灯等放电灯以及LED或有机EL元件等发光元件。
在图30中,具有室内机8200及室外机8204的空调器是使用根据本发明的一个方式的二次电池8203的电子设备的一个例子。具体地说,室内机8200包括框体8201、送风口8202及二次电池8203等。虽然在图30中例示出二次电池8203设置在室内机8200中的情况,但是二次电池8203也可以设置在室外机8204中。或者,也可以在室内机8200和室外机8204的双方中设置有二次电池8203。空调器可以接收来自商业电源的电力供应,也可以使用蓄积在二次电池8203中的电力。尤其是,当在室内机8200和室外机8204的双方中设置有二次电池8203时,即使当由于停电等不能接收来自商业电源的电力供应时,通过将根据本发明的一个方式的二次电池8203用作不间断电源,也可以利用空调器。
另外,虽然在图30中例示由室内机和室外机构成的分体式空调器,但是也可以将根据本发明的一个方式的二次电池用于在一个框体中具有室内机的功能和室外机的功能的一体式空调器。
在图30中,电冷藏冷冻箱8300是使用根据本发明的一个方式的二次电池8304的电子设备的一个例子。具体地说,电冷藏冷冻箱8300包括框体8301、冷藏室门8302、冷冻室门8303及二次电池8304等。在图30中,二次电池8304设置在框体8301的内部。电冷藏冷冻箱8300可以接收来自商业电源的电力供应,也可以使用蓄积在二次电池8304中的电力。因此,即使当由于停电等不能接收来自商业电源的电力供应时,通过将根据本发明的一个方式的二次电池8304用作不间断电源,也可以利用电冷藏冷冻箱8300。
上述电子设备中,微波炉等高频加热装置、电饭煲等的电子设备在短时间内需要高电力。因此,通过将根据本发明的一个方式的蓄电装置用作用来辅助商业电源不能充分供应的电力的辅助电源,在使用电子设备时可以防止商业电源的总开关跳闸。
在不使用电子设备的时间段,尤其是在商业电源的供应源能够供应的电力总量中的实际使用的电力量的比率(称为电力使用率)低的时间段中,将电力蓄积在二次电池中,由此可以抑制在上述时间段以外的时间段中电力使用率增高。例如,在为电冷藏冷冻箱8300时,在气温低且不进行冷藏室门8302或冷冻室门8303的开关的夜间,将电力蓄积在二次电池8304中。并且,在气温高且进行冷藏室门8302或冷冻室门8303的开关的白天,将二次电池8304用作辅助电源,由此可以抑制白天的电力使用率。
通过采用本发明的一个方式,可以提高二次电池的循环特性并提高可靠性。此外,通过采用本发明的一个方式,可以实现高容量的二次电池而可以提高二次电池的特性,而可以使二次电池本身小型化及轻量化。因此,通过将本发明的一个方式的二次电池安装在本实施方式所说明的电子设备,可以提供使用寿命更长且更轻量的电子设备。本实施方式可以与其他实施方式适当地组合而实施。
(实施方式6)
在本实施方式中,示出将本发明的一个方式的二次电池安装在车辆的例子。
当将二次电池安装在车辆时,可以实现混合动力汽车(HEV)、电动汽车(EV)或插电式混合动力汽车(PHEV)等新一代清洁能源汽车。
在图31中,例示出使用本发明的一个方式的二次电池的车辆。图31A所示的汽车8400是作为用来行驶的动力源使用电发动机的电动汽车。或者,汽车8400是作为用来行驶的动力源能够适当地使用电发动机或引擎的混合动力汽车。通过使用本发明的一个方式的二次电池,可以实现行驶距离长的车辆。另外,汽车8400具备二次电池。作为二次电池,可以将图16C及图16D所示的二次电池模块排列在车内的地板部分而使用。另外,可以将组合多个图19所示的二次电池而成的电池组设置在车内的地板部分。二次电池不但驱动电发动机8406,而且还可以将电力供应到车头灯8401或室内灯(未图示)等发光装置。
二次电池可以将电力供应到汽车8400所具有的速度表、转速计等显示装置。此外,二次电池可以将电力供应到汽车8400所具有的导航系统等半导体装置。
在图31B所示的汽车8500中,可以通过利用插电方式或非接触供电方式等从外部的充电设备接收电力,来对汽车8500所具有的二次电池进行充电。图31B示出从地上设置型的充电装置8021通过电缆8022对安装在汽车8500中的二次电池8024进行充电的情况。当进行充电时,作为充电方法或连接器的规格等,可以根据CHAdeMO(注册商标)或联合充电系统“Combined Charging System”等的规定的方式而适当地进行。作为充电装置8021,也可以使用设置在商业设施的充电站或家庭的电源。例如,通过利用插电技术从外部供应电力,可以对安装在汽车8500中的二次电池8024进行充电。可以通过AC/DC转换器等转换装置将交流电力转换成直流电力来进行充电。
虽然未图示,但是也可以将受电装置安装在车辆中并从地上的送电装置非接触地供应电力来进行充电。当利用非接触供电方式时,通过在公路或外壁中组装送电装置,不但停车中而且行驶中也可以进行充电。此外,也可以利用该非接触供电方式,在车辆之间进行电力的发送及接收。再者,还可以在车辆的外部设置太阳能电池,在停车时或行驶时进行二次电池的充电。可以利用电磁感应方式或磁场共振方式实现这样的非接触供电。
图31C是使用本发明的一个方式的二次电池的两轮车的例子。图31C所示的小型摩托车8600包括二次电池8602、后视镜8601及方向灯8603。二次电池8602可以对方向灯8603供电。
在图31C所示的小型摩托车8600中,可以将二次电池8602收纳在座位下收纳箱8604中。即使座位下收纳箱8604为小型,也可以将二次电池8602收纳在座位下收纳箱8604中。二次电池8602是可拆卸的,因此在充电时将二次电池8602搬到室内,对其进行充电,行驶之前将二次电池8602收纳即可。
通过采用本发明的一个方式,可以提高二次电池的循环特性及容量。由此,可以使二次电池本身小型轻量化。另外,如果可以使二次电池本身小型轻量化,就有助于实现车辆的轻量化,从而可以延长行驶距离。另外,可以将安装在车辆中的二次电池用作车辆之外的电力供应源。此时,例如可以避免在电力需求高峰时使用商业电源。如果可以避免在电力需求高峰时使用商业电源,就有助于节省能量以及二氧化碳排放量的减少。此外,如果循环特性优异,就可以长期间使用二次电池,从而可以降低钴等稀有金属的使用量。
本实施方式可以与其他实施方式适当地组合而实施。
[实施例1]
在本实施例中,作为本发明的一个方式的正极活性物质制造钴酸锂(sample A1至sample A3),进行XRD分析及ESR分析。作为比较例使用市售的钴酸锂(sample A4)。另外,通过使用这些钴酸锂制造二次电池,对高电压充电时的循环特性及连续充电耐性进行评价。在此,使sample A1至sample A3所包含的镁及卤素的添加量彼此不同。
[正极活性物质的制造方法]
<sample A1>
作为起始材料使用包含卤素源、锂源、镁源、以及含有锂及过渡金属的复合氧化物,制造sample A1(参照图9)。
首先,称量起始材料。作为卤素源及镁源使用氟化镁(MgF2)。作为卤素源及锂源使用氟化锂(LiF)。
接着,对含有锂及过渡金属的复合氧化物进行称量。作为含有锂及过渡金属的复合氧化物使用锂钴酸(LiCoO2)的日本化学工业公司制造的CELLSEED C-10N。CELLSEED C-10N是D50为12μm左右的杂质少的钴酸锂。
具体而言,以MgF2为0.47585g(7.6378mmol),LiF为0.06602g(2.54526mmol),CELLSEED C-10N为149.45813g(1527.109mmol)的方式进行称量。这是相对于钴原子数的镁原子数为0.5atomic%的量。另外,这是相对于MgF2的LiF的摩尔比为0.33的量。
注意,在本说明书等中,镁添加量是指起始材料中的相对于钴原子数的镁原子数的比率。
接着,混合起始材料。在混合时使用湿式球磨机。具体而言,利用干法进行混合。利用使用锆质球的球磨机以150rpm进行混合1小时。
接着,对混合物进行加热处理。将混合物放入氧化铝坩埚,在氧气氛的马弗炉中以850℃进行处理60小时。加热处理时对氧化铝坩埚盖上盖。氧的流量设定为10L/min。升温以200℃/hr进行,降温进行了10小时以上。
接着,将被加热的材料冷却至室温并回收而得到sample A1。
<sample A2>
在sample A2中,以MgF2为0.12553g(2.0149mmol),LiF为0.01742g(0.67146mmol),CELLSEED C-10N为9.85705g(100.716mmol)的方式进行称量。这是相对于钴原子数的镁原子数为2.0atomic%的量。另外,这是相对于MgF2的LiF的摩尔比为0.33的量。关于其他工序可以参照sample A1的记载,省略详细说明。
<sample A3>
在sample A3中,以MgF2为0.36613g(5.8767mmol)、LiF为0.05080g(1.9583mmol)、CELLSEED C-10N为9.58307g(97.9163mmol)的方式进行称量。这是相对于钴原子数的镁原子数为6.0atomic%的量。另外,这是相对于MgF2的LiF的摩尔比为0.33的量。关于其他工序可以参照sample A1的记载,省略详细说明。
<sample A4>
将不进行处理的市售的钴酸锂(CELLSEED C-10N)用作sample A4(比较例)。
[ESR分析]
接下来,对sample A1至sample A4进行ESR分析。在ESR分析中,9.15GHz的高频电力(微波的功率)为1mW,将磁场从0mT扫描到800mT,测定温度为300K(27℃左右)、200K(-73℃左右)、113K(-160℃左右)。关于在ESR分析时使用的各样品的重量,sample A1至sampleA4都为0.005g。另外,通过使用Mn2+标记,进行磁场的校正及检测灵敏度。在算出自旋次数时,作为标准样品使用TEMPOL(4-Hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl)。
图32示出sample A1至sample A4的ESR测定结果。在图32中,横轴表示磁场(Magnetic Field),纵轴表示ESR信号的强度(Intensity)。另外,信号强度表示微波的吸收量的一次微分的值。
如图32所示,在每个样品中,在305mT(g=2.15)附近都观察到尖锐的信号。305mT(g=2.15)附近的信号起因于Co2+及Co4+。305mT(g=2.15)附近的信号的线宽度ΔHpp约为4mT。在此,线宽度ΔHpp是指信号的最大值和最小值的磁场之差。另外,在起因于Co3O4的130mT(g=5.1)附近观察不到信号。在sample A1至sample A4中,确认到不存在Co3O4或Co3O4极少。
接着,将磁场从200mT扫描到400mT来进行ESR分析。在ESR分析中,测定温度为300K(27℃左右)、200K(-73℃左右)、113K(-160℃左右)。
图33示出sample A1至sample A4的ESR测定结果。在图33中,横轴表示磁场(Magnetic Field),纵轴表示ESR信号的强度(Intensity)。另外,信号强度表示微波的吸收量的一次微分的值。
如图33所示,在每个样品中,在305mT(g=2.15)附近都观察到尖锐的信号。表1示出根据305mT(g=2.15)附近的信号强度算出的自旋次数。表1还示出根据自旋次数算出的自旋密度1及自旋密度2。表1所示的自旋密度1是自旋次数除以ESR分析中使用的样品的重量(0.005g)的值。另外,表1所示的自旋密度2是以各样品的组成为LiCoO2且根据其分子量97.87算出各样品中的钴原子数,自旋次数除以该钴原子数的值。图34A示出sample A1至sample A4的自旋密度1,图34B示出自旋密度2。在图34A及图34B中,横轴表示样品名称,纵轴分别表示自旋密度1(Spin Density1)及自旋密度2(Spin Density2)。
[表1]


如表1、图34A及图34B所示那样,可以确认到根据居里-外斯定律,本发明的一个方式的sample A1至sample A3的ESR分析的测定温度越低自旋密度越高,并具有顺磁性。另一方面,可以确认到被用作比较样品的sample A4的自旋密度的测定温度依赖性小,并示出与顺磁性不同的举动。
图35A示出以300K(27℃左右)进行ESR分析的自旋密度1和相对于钴的镁添加量的相关。图35B示出自旋密度2和相对于钴的镁添加量的相关。在图35A及图35B中,横轴表示镁添加量,纵轴分别表示自旋密度1及自旋密度2。
如图35A及图35B所示,确认到随着镁添加量的增加,起因于Co2+及Co4+的自旋密度增高。另外,在镁添加量过多的情况下,有时起因于Co2+及Co4+的自旋密度降低。另外,在进行了镁添加的sample A1、A2、A3中,比g=2.1更低的磁场一侧观察到小信号,这表示存在有由于镁的置换而发生的晶体场的各向异性。
如图35A所示那样,在没有添加镁的sample A4中,起因于正极活性物质的单位重量的Co2+及Co4+的自旋密度为1.4×1017spins/g。相比之下,镁添加量为0.5atomic%的sample A1的自旋密度为2.8×1017spins/g,镁添加量为2.0atomic%的sample A2的自旋密度为4.8×1017spins/g,镁添加量为6.0atomic%的sample A3的自旋密度为2.1×1017spins/g,并为2.0×1017spins/g以上且1.0×1018spins/g以下。
如图35B所示那样,在没有添加镁的sample A4中,起因于Co原子个数的Co2+及Co4+的自旋密度为2.3×10-5spins/Co原子。相比之下,镁添加量为0.5atomic%的sample A1的自旋密度为4.6×10-5spins/Co原子,镁添加量为2.0atomic%的sample A2的自旋密度为7.8×10-5spins/Co原子,镁添加量为6.0atomic%的sample A3的自旋密度为3.5×10- 5spins/Co原子,并为3.3×10-5spins/Co原子以上且1.6×10-4spins/Co原子以下。
[XRD分析]
接下来,对sample A1至sample A4进行XRD分析。在XRD分析中,作为X射线源使用波长为0.15418nm的CuKα线。
图36示出sample A1至sample A4的XRD测定结果。在此确认到sample A1至sampleA4中观察的XRD峰大致来源于LiCoO2。
图37A、图37B及图38示出放大图36的图表。在图36、图37A、图37B及图38中,横轴表示回折角度2θ,纵轴表示衍射了的X射线的强度(Intensity)。
如图37A、图37B及图38所示,确认到根据镁添加量,XRD峰的位置不同。注意,在sample A3中,确认到在2θ=37deg附近有不来源于LiCoO2的峰(参照图38)。可以估计到如下可能性,当镁添加量增多时,例如在两个Mg2+分别置换为Li+及Co3+的情况下,LiCoO2整体的化合价不变化,因此不引起磁性自旋。由此,当Mg浓度过高时,有可能不容易引起磁性自旋。
对图36所示的XRD测定结果进行里特沃尔德(Rietveld)分析,求得晶粒尺寸及晶格。在进行里特沃尔德分析时使用Bruker公司的软件(TOPAS),作为空间群使用R-3m的结构。
表2示出利用里特沃尔德分析而得到的sample A1至sample A4的各晶粒尺寸及晶格。另外,图39A及图39B示出晶格常数。在图39A及图39B中,横轴表示镁添加量,纵轴分别表示晶格常数a及晶格常数c。
[表2]

如表2所示,在sample A1至sample A4中,a轴的晶格常数为2.8155×10-10m以上且2.8175×10-10m,c轴的晶格常数为14.045×10-10m以上且14.065×10-10m以下。此外,具有随着镁添加量的增加而晶格常数增大的趋向,然而样品之间的晶格常数的差异小。
接下来,使用sample A1至sample A4制造二次电池,然后对循环特性进行评价。
[二次电池的制造方法]
通过将sample A1至sample A4用作正极材料来制造正极。
[循环特性1]
接下来,对sample A1至sample A4的循环特性进行评价。正极的担持量设定为20mg/cm2,充电的上限电压设定为4.5V。
首先,在25℃下进行2次CCCV充电(速率0.05C,4.5V,终止电流0.005C)及CC放电(0.05C,2.5V)的循环测试。然后,在25℃反复进行CCCV充电(速率0.2C,4.5V,终止电流0.02C)及CC放电(0.2C,2.5V)的充放电,对循环特性进行评价。
图40A及图40B示出sample A1至sample A4的循环特性。在图40A中,横轴表示循环次数(Cycle Number),纵轴表示放电时的容量(Capacity)。在图40B中,横轴表示循环次数(Cycle Number),纵轴表示放电时的容量保持率(Capacity Retention Rate)。放电时的容量保持率是相对于放电时的容量的最大值的各循环中的容量的比率。
如图40A及图40B所示那样,可以确认到与没有添加镁的sample A4相比,添加镁的sample A1至sample A3的循环特性良好。尤其是,可以确认到sample A1及sample A2的容量大且具有优异的循环特性。
[循环特性2]
接下来,正极的担持量设定为20mg/cm2,充电的上限电压设定为4.6V,对循环特性进行评价。
首先,在25℃下进行2次CCCV充电(速率0.05C,4.6V,终止电流0.005C)及CC放电(速率0.05C,2.5V)的循环测试。然后,在25℃反复进行CCCV充电(速率0.2C,4.6V,终止电流0.02C)及CC放电(速率0.2C,2.5V)的充放电,对循环特性进行评价。
图41A及图41B示出sample A1至sample A4的循环特性。
如图41A及图41B所示那样,可以确认到与没有添加镁的sample A4相比,添加镁的sample A1至sample A3的循环特性良好。尤其是,可以确认到sample A1及sample A2的容量大且具有优异的循环特性。
[连续充电耐性]
接着,对使用所制造的各正极活性物质的每个二次电池进行连续充电耐性的评价。首先,在25℃下进行2次CCCV充电(速率0.05C,4.5V或4.6V,终止电流0.005C)及CC放电(速率0.05C,2.5V)的循环测试。
然后,在60℃进行CCCV充电(速率0.05C)。上限电压设定为4.55V或4.65V,进行测定直到二次电池的电压降低到低于通过从上限电压减去0.01V所得的值(当上限电压为4.55V时则为4.54V)为止。当二次电池的电压低于上限电压时,例如有可能发生短路等现象。1C设定为200mA/g。
图42A及图42B示出连续充电耐性的评价结果。图42A示出充电电压设定为4.55V时的连续充电耐性的评价结果。图42B示出充电电压设定为4.65V时的连续充电耐性的评价结果。在图42A及图42B中,横轴表示时间(Time),纵轴表示电流值(Current)。
如图42A及图42B所示那样,根据镁添加量的增加,连续充电耐性增高。另外,可以确认到镁添加量为6.0atomic%的sample A3的连续充电耐性低的趋向。
[实施例2]
在本实施例中,通过与上述实施例1不同的制造方法制造本发明的一个方式的钴酸锂(sample B1),并且进行ESR分析。作为比较例,制造不添加镁的钴酸锂(sample B2)。此外,通过使用这些钴酸锂制造二次电池,对高电压充电时的循环特性进行评价。
[正极活性物质的制造方法]
<sample B1>
通过将卤素源、锂源、镁源及钴源用作起始材料制造sample B1(参照图10)。
首先称量起始材料。作为锂源使用碳酸锂(Li2CO3),作为钴源使用四氧化三钴(Co3O4),作为镁源使用氧化镁(MgO),作为氟源使用氟化锂(LiF)。
具体而言,以Li2CO3为3.1489g(42.62mmol),Co3O4为6.7726g(28.13mmol),MgO为0.0344g(0.85mmol),LiF为0.0442g(1.70mmol)的方式进行称量。这是相对于钴原子数的镁原子数为1.0atomic%的量。注意,作为Li2CO3使用高纯度化学研究所制造(目录号码:LIH06XB)。作为MgO使用高纯度化学研究所制造(目录号码:MGO12PB)。作为LiF使用高纯度化学研究所制造(LIH10XB)。
接着,混合各起始材料。在混合时使用湿式球磨机。具体而言,使用 的球及被用作溶剂的丙酮,以300rpm的旋转数进行粉碎及混合2小时。
的球及被用作溶剂的丙酮,以300rpm的旋转数进行粉碎及混合2小时。
接着,对混合了的材料进行第一加热处理。第一加热处理在马弗炉中以200℃/hr的昇温速率从室温升温至1000℃,以1000℃加热10小时。第一加热处理的气氛设定为干燥空气,其流量设定为10L/min。
接着,将受到第一加热处理的材料冷却至室温。在冷却后,对材料进行研碎处理来减小材料的粒径。在研碎处理时使用53μm的丝网。
接着,对材料进行第二加热处理。第二加热处理在马弗炉中以200℃/hr的升温速率从室温升温至800℃,以800℃加热2小时。第二加热处理的气氛设定为干燥空气,其流量设定为10L/min。
接着,将受到加热处理的材料冷却至室温,然后回收而得到sample B1。
<sample B2>
通过将锂源及钴源用作起始材料制造sample B2。作为比较例的sample B2不使用镁源及卤素源。
首先称量起始材料。作为锂源使用碳酸锂(Li2CO3),作为钴源使用四氧化三钴(Co3O4),作为镁源使用氧化镁(MgO),作为氟源使用氟化锂(LiF)。
具体而言,以Li2CO3为3.1521g(42.66mmol),Co3O4为6.8479g(28.44mmol)的方式进行称量。关于其他工序可以参照sample B1的记载,省略详细说明。
[ESR分析]
接下来,对sample B1及sample B2进行ESR分析。在ESR分析中,9.15GHz的高频电力(微波的功率)为1mW,将磁场从0mT扫描到500mT,测定温度为300K(27℃左右)、200K(-73℃左右)、113K(-160℃左右)。在ESR分析时使用的样品的sample B1的重量为0.0044g,sample B2的重量为0.0045g。
图43A示出sample B1及sample B2的ESR测定结果。另外,图43B示出放大图43A的图。在图43A及图43B中,横轴表示磁场(Magnetic Field),纵轴表示ESR信号的强度(Intensity)。另外,信号强度表示微波的吸收量的一次微分的值。
如图43A所示那样,在没有添加镁的比较例的sample B2中,在130mT(g=5.1)附近观察到较宽的峰。sample B2示出被估计存在有氧四配位的Co的结果。
如图43A及图43B所示,在每个样品中,在305mT(g=2.15)附近都观察到尖锐的信号。305mT(g=2.15)附近的信号起因于Co2+及Co4+。表3示出根据305mT(g=2.15)附近的信号强度算出的自旋次数。表3示出根据自旋次数算出的自旋密度1及自旋密度2。表1所示的自旋密度1是自旋次数除以ESR分析中使用的样品的重量(0.005g)的值。另外,表3所示的自旋密度2是以各样品的组成为LiCoO2且根据其分子量97.87算出各样品中的钴原子数,自旋次数除以该钴原子数的值。图44A示出sample B1及sample B2的自旋密度1,图44B示出自旋密度2。在图44A及图44B中,横轴表示样品名称,纵轴分别表示自旋密度1(Spin Density1)及自旋密度2(Spin Density2)。
[表3]


如表3、图44A及图44B所示那样,可以确认到根据居里-外斯定律,本发明的一个方式的sample B1的ESR分析的测定温度越低自旋密度越高,并具有顺磁性。另一方面,被用作比较样品的sample B2虽然具有顺磁性,然而其自旋密度低。
图45A示出以300K(27℃左右)进行ESR分析的自旋密度1和相对于钴的镁添加量的相关。图45B示出自旋密度2和相对于钴的镁添加量的相关。在图45A及图45B中,横轴表示镁添加量,纵轴分别表示自旋密度1及自旋密度2。
如图45A所示那样,在没有添加镁的sample B2中,起因于正极活性物质的单位重量的Co2+及Co4+的自旋密度为1.1×1017spins/g。相比之下,镁添加量为1.0atomic%的sample B1的自旋密度为2.1×1017spins/g,并为2.0×1017spins/g以上且1.0×1018spins/g以下。
如图45B所示那样,在没有添加镁的sample B2中,起因于Co原子个数的Co2+及Co4+的自旋密度为1.7×10-5spins/Co原子。相比之下,镁添加量为1.0atomic%的sample B1的自旋密度为3.4×10-5spins/Co原子,并为3.3×10-5spins/Co原子以上且1.6×10-4spins/Co原子以下。
[二次电池的制造方法]
通过将sample B1及sample B2用作正极材料来制造正极。
[循环特性]
接下来,对sample B1及sample B2的循环特性进行评价。将正极的担持量设定为7mg/cm2左右,将充电的上限电压设定为4.6V。
首先,在25℃下进行2次CCCV充电(0.05C,4.6V,终止电流0.005C)及CC放电(0.05C,2.5V)的循环测试。然后,在25℃反复进行CCCV充电(0.2C,4.6V,终止电流0.02C)及CC放电(0.2C,2.5V)的充放电,对循环特性进行评价。
图46A及图46B示出sample A1至sample A4的循环特性。在图46A中,横轴表示循环次数(Cycle Number),纵轴表示放电时的容量(Capacity)。在图46B中,横轴表示循环次数(Cycle Number),纵轴表示放电时的容量保持率(Capacity Retention Rate)。放电时的容量保持率是相对于放电时的容量的最大值的各循环中的容量的比率。
如图46A及图46B所示那样,可以确认到与没有添加镁的sample B2相比,添加镁的sample B1循环特性良好。
[符号说明]
100:正极活性物质、200:活性物质层、201:石墨烯化合物、210:电极叠层体、211a:正极、211b:负极、212a:导线、212b:导线、214:隔离体、215a:接合部、215b:接合部、217:固定构件、250:二次电池、251:外包装体、261:折叠部分、262:密封部、263:密封部、271:棱线、272:谷底线、273:空间、300:二次电池、301:正极罐、302:负极罐、303:垫片、304:正极、305:正极集流体、306:正极活性物质层、307:负极、308:负极集流体、309:负极活性物质层、310:隔离体、500:二次电池、501:正极集流体、502:正极活性物质层、503:正极、504:负极集流体、505:负极活性物质层、506:负极、507:隔离体、508:电解液、509:外包装体、510:正极导线电极、511:负极导线电极、600:二次电池、601:正极盖、602:电池罐、603:正极端子、604:正极、605:隔离体、606:负极、607:负极端子、608:绝缘板、609:绝缘板、611:PTC元件、612:安全阀机构、613:导电板、614:导电板、615:模块、616:导线、617:温度控制装置、900:电路板、910:签条、911:端子、912:电路、913:二次电池、914:天线、915:密封材料、916:层、917:层、918:天线、920:显示装置、921:传感器、922:端子、930:框体、930a:框体、930b:框体、931:负极、932:正极、933:隔离体、950:卷绕体、951:端子、952:端子、980:二次电池、981:薄膜、982:薄膜、993:卷绕体、994:负极、995:正极、996:隔离体、997:导线电极、998:导线电极、7100:便携式显示装置、7101:框体、7102:显示部、7103:操作按钮、7104:二次电池、7200:便携式信息终端、7201:框体、7202:显示部、7203:导线、7204:带扣、7205:操作按钮、7206:输入输出端子、7207:图标、7300:显示装置、7304:显示部、7400:移动电话机、7401:框体、7402:显示部、7403:操作按钮、7404:外部连接端口、7405:扬声器、7406:麦克风、7407:二次电池、7500:电子烟、7501:雾化器、7502:烟弹、7504:二次电池、8000:显示装置、8001:框体、8002:显示部、8003:扬声器部、8004:二次电池、8021:充电装置、8022:电缆、8024:二次电池、8100:照明装置、8101:框体、8102:光源、8103:二次电池、8104:天花板、8105:侧壁、8106:地板、8107:窗户、8200:室内机、8201:框体、8202:送风口、8203:二次电池、8204:室外机、8300:电冷藏冷冻箱、8301:框体、8302:冷藏室门、8303:冷冻室门、8304:二次电池、8400:汽车、8401:车头灯、8406:电发动机、8500:汽车、8600:小型摩托车、8601:后视镜、8602:二次电池、8603:方向灯、8604:座位下收纳箱、9600:平板终端、9625:开关、9627:开关、9628:操作开关、9629:扣件、9630:框体、9630a:框体、9630b:框体、9631:显示部、9631a:显示部、9631b:显示部、9633:太阳能电池、9634:充放电控制电路、9635:蓄电体、9636:DCDC转换器、9637:转换器、9640:可动部
Claims (5)
1.一种正极活性物质,包含:
锂、钴及氧,
其中起因于2价的钴离子及4价的钴离子的自旋密度为2.0×1017spins/g以上且1.0×1018spins/g以下。
2.根据权利要求1所述的正极活性物质,还包含镁,
其中相对于所述钴的所述镁的浓度为0.1atomic%以上且6.0atomic%以下。
3.根据权利要求1或2所述的正极活性物质,还包含氟。
4.根据权利要求1至3中任一项所述的正极活性物质,
其中a轴的晶格常数为2.8155×10-10m以上且2.8175×10-10m,
并且c轴的晶格常数为14.045×10-10m以上且14.065×10-10m以下。
5.一种二次电池,包括:
具有权利要求1至4中任一项所述的正极活性物质的正极;以及
负极。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2018-218362 | 2018-11-21 | ||
| JP2018218362 | 2018-11-21 | ||
| PCT/IB2019/059561 WO2020104881A1 (ja) | 2018-11-21 | 2019-11-07 | 正極活物質、および二次電池 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN113016094A true CN113016094A (zh) | 2021-06-22 |
Family
ID=70773570
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201980074923.2A Pending CN113016094A (zh) | 2018-11-21 | 2019-11-07 | 正极活性物质、及二次电池 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US20220006082A1 (zh) |
| JP (1) | JPWO2020104881A1 (zh) |
| KR (1) | KR20210092253A (zh) |
| CN (1) | CN113016094A (zh) |
| TW (1) | TW202036968A (zh) |
| WO (1) | WO2020104881A1 (zh) |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20190140072A (ko) * | 2017-05-03 | 2019-12-18 | 가부시키가이샤 한도오따이 에네루기 켄큐쇼 | 양극 활물질 입자의 제작 방법 및 이차 전지 |
| JPWO2022038451A1 (zh) * | 2020-08-20 | 2022-02-24 | ||
| WO2022090843A1 (ja) * | 2020-10-26 | 2022-05-05 | 株式会社半導体エネルギー研究所 | 二次電池、電子機器および車両 |
| JPWO2022185152A1 (zh) * | 2021-03-05 | 2022-09-09 |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4240242B2 (ja) * | 1998-06-23 | 2009-03-18 | Agcセイミケミカル株式会社 | 非水電解質二次電池用正極活物質及び非水電解質二次電池 |
| JP2000277117A (ja) * | 1999-03-25 | 2000-10-06 | Sony Corp | 非水電解質電池 |
| JP3777988B2 (ja) | 2001-01-23 | 2006-05-24 | 日亜化学工業株式会社 | リチウム二次電池用正極活物質及びその製造方法 |
| JP4736943B2 (ja) | 2006-05-17 | 2011-07-27 | 日亜化学工業株式会社 | リチウム二次電池用正極活物質およびその製造方法 |
| ES2536250T3 (es) * | 2010-10-20 | 2015-05-21 | Council Of Scientific & Industrial Research | Material catódico y batería de iones de litio del mismo |
| US10351440B2 (en) * | 2011-05-31 | 2019-07-16 | Freeport Colbalt Oy | Lithium cobalt oxide material |
| EP2720981B1 (en) * | 2011-06-17 | 2016-11-16 | Umicore | Lithium metal oxide partcles coated with a mixture of the elements of the core material and one or more metal oxides |
| CN115966676A (zh) * | 2016-11-24 | 2023-04-14 | 株式会社半导体能源研究所 | 正极活性物质粒子及正极活性物质粒子的制造方法 |
| US20220190319A1 (en) * | 2019-03-29 | 2022-06-16 | Semiconductor Energy Laboratory Co., Ltd. | Positive electrode active material and secondary battery |
| WO2021130599A1 (ja) * | 2019-12-27 | 2021-07-01 | 株式会社半導体エネルギー研究所 | 正極活物質、二次電池、電子機器 |
-
2019
- 2019-11-07 WO PCT/IB2019/059561 patent/WO2020104881A1/ja active Application Filing
- 2019-11-07 JP JP2020557010A patent/JPWO2020104881A1/ja active Pending
- 2019-11-07 US US17/291,463 patent/US20220006082A1/en active Pending
- 2019-11-07 CN CN201980074923.2A patent/CN113016094A/zh active Pending
- 2019-11-07 KR KR1020217018052A patent/KR20210092253A/ko active Search and Examination
- 2019-11-14 TW TW108141320A patent/TW202036968A/zh unknown
Also Published As
| Publication number | Publication date |
|---|---|
| KR20210092253A (ko) | 2021-07-23 |
| JPWO2020104881A1 (ja) | 2021-10-14 |
| WO2020104881A1 (ja) | 2020-05-28 |
| TW202036968A (zh) | 2020-10-01 |
| US20220006082A1 (en) | 2022-01-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR102529620B1 (ko) | 양극 활물질의 제작 방법 및 이차 전지 | |
| CN115036578B (zh) | 锂离子二次电池的制造方法 | |
| US20220246931A1 (en) | Positive electrode active material and manufacturing method of positive electrode active material | |
| CN113165910A (zh) | 正极活性物质的制造方法 | |
| CN113016094A (zh) | 正极活性物质、及二次电池 | |
| CN112292776A (zh) | 正极活性物质、正极、二次电池以及正极的制造方法 | |
| CN113165908A (zh) | 正极活性物质及二次电池 | |
| WO2020099978A1 (ja) | 正極活物質、二次電池、電子機器及び車両 | |
| CN112753115A (zh) | 锂离子二次电池正极材料、二次电池、电子设备及车辆、以及锂离子二次电池正极材料的制造方法 | |
| CN113646266A (zh) | 正极活性物质以及二次电池 | |
| CN114270566A (zh) | 正极活性物质、正极、二次电池以及其制造方法 | |
| CN112997340A (zh) | 正极活性物质、二次电池、电子设备及车辆 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination |