CN112752570A - 生物可利用口服剂型 - Google Patents
生物可利用口服剂型 Download PDFInfo
- Publication number
- CN112752570A CN112752570A CN201980065062.1A CN201980065062A CN112752570A CN 112752570 A CN112752570 A CN 112752570A CN 201980065062 A CN201980065062 A CN 201980065062A CN 112752570 A CN112752570 A CN 112752570A
- Authority
- CN
- China
- Prior art keywords
- compound
- sdi
- subject
- pharmaceutical composition
- dose
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 239000006186 oral dosage form Substances 0.000 title claims description 22
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 118
- 235000013305 food Nutrition 0.000 claims abstract description 30
- 239000000203 mixture Substances 0.000 claims description 261
- 229940125904 compound 1 Drugs 0.000 claims description 235
- 238000000034 method Methods 0.000 claims description 185
- 229920000642 polymer Polymers 0.000 claims description 94
- 150000001875 compounds Chemical class 0.000 claims description 83
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 claims description 83
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 claims description 83
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 claims description 83
- 239000002904 solvent Substances 0.000 claims description 75
- 230000036470 plasma concentration Effects 0.000 claims description 53
- 239000002775 capsule Substances 0.000 claims description 49
- 229920000036 polyvinylpyrrolidone Polymers 0.000 claims description 47
- 239000001267 polyvinylpyrrolidone Substances 0.000 claims description 47
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 claims description 47
- 239000003814 drug Substances 0.000 claims description 43
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 34
- 239000007962 solid dispersion Substances 0.000 claims description 28
- 239000007921 spray Substances 0.000 claims description 27
- 238000001694 spray drying Methods 0.000 claims description 25
- 238000004519 manufacturing process Methods 0.000 claims description 16
- 238000002360 preparation method Methods 0.000 claims description 14
- 229920001477 hydrophilic polymer Polymers 0.000 claims description 12
- 239000006185 dispersion Substances 0.000 claims description 11
- 208000032839 leukemia Diseases 0.000 claims description 10
- 239000007788 liquid Substances 0.000 claims description 10
- 208000031261 Acute myeloid leukaemia Diseases 0.000 claims description 6
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 claims description 5
- 208000027866 inflammatory disease Diseases 0.000 claims description 5
- 206010000830 Acute leukaemia Diseases 0.000 claims description 4
- 208000024207 chronic leukemia Diseases 0.000 claims description 4
- 229920006316 polyvinylpyrrolidine Polymers 0.000 claims description 3
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 claims description 2
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 claims description 2
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 claims description 2
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 claims description 2
- 201000003793 Myelodysplastic syndrome Diseases 0.000 claims description 2
- 208000006664 Precursor Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 claims description 2
- 208000017733 acquired polycythemia vera Diseases 0.000 claims description 2
- 208000032852 chronic lymphocytic leukemia Diseases 0.000 claims description 2
- 201000009277 hairy cell leukemia Diseases 0.000 claims description 2
- 201000006417 multiple sclerosis Diseases 0.000 claims description 2
- 208000037244 polycythemia vera Diseases 0.000 claims description 2
- 206010039073 rheumatoid arthritis Diseases 0.000 claims description 2
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 claims 3
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 claims 2
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 claims 2
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 claims 2
- 238000004090 dissolution Methods 0.000 abstract description 45
- 238000011068 loading method Methods 0.000 abstract description 40
- 230000001965 increasing effect Effects 0.000 abstract description 38
- 230000001976 improved effect Effects 0.000 abstract description 13
- 238000009472 formulation Methods 0.000 description 173
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 147
- 239000000523 sample Substances 0.000 description 99
- 206010028980 Neoplasm Diseases 0.000 description 97
- 230000001613 neoplastic effect Effects 0.000 description 86
- HSHXDCVZWHOWCS-UHFFFAOYSA-N N'-hexadecylthiophene-2-carbohydrazide Chemical compound CCCCCCCCCCCCCCCCNNC(=O)c1cccs1 HSHXDCVZWHOWCS-UHFFFAOYSA-N 0.000 description 80
- 239000003826 tablet Substances 0.000 description 76
- 239000004094 surface-active agent Substances 0.000 description 69
- 239000003795 chemical substances by application Substances 0.000 description 66
- 239000000463 material Substances 0.000 description 51
- 239000000243 solution Substances 0.000 description 51
- WHNWPMSKXPGLAX-UHFFFAOYSA-N N-Vinyl-2-pyrrolidone Chemical compound C=CN1CCCC1=O WHNWPMSKXPGLAX-UHFFFAOYSA-N 0.000 description 45
- 229920003081 Povidone K 30 Polymers 0.000 description 45
- 239000000543 intermediate Substances 0.000 description 44
- 150000002632 lipids Chemical class 0.000 description 42
- 241001465754 Metazoa Species 0.000 description 41
- 208000035269 cancer or benign tumor Diseases 0.000 description 40
- 238000000113 differential scanning calorimetry Methods 0.000 description 40
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 40
- 238000002560 therapeutic procedure Methods 0.000 description 39
- 229940079593 drug Drugs 0.000 description 35
- RVGRUAULSDPKGF-UHFFFAOYSA-N Poloxamer Chemical compound C1CO1.CC1CO1 RVGRUAULSDPKGF-UHFFFAOYSA-N 0.000 description 33
- 238000011374 additional therapy Methods 0.000 description 33
- 238000000634 powder X-ray diffraction Methods 0.000 description 32
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 31
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 30
- 238000010171 animal model Methods 0.000 description 30
- 239000011159 matrix material Substances 0.000 description 30
- 210000002381 plasma Anatomy 0.000 description 27
- 108010052167 Dihydroorotate Dehydrogenase Proteins 0.000 description 26
- 102100032823 Dihydroorotate dehydrogenase (quinone), mitochondrial Human genes 0.000 description 26
- 239000002245 particle Substances 0.000 description 26
- 208000024891 symptom Diseases 0.000 description 25
- -1 but not limited to Polymers 0.000 description 22
- 230000000694 effects Effects 0.000 description 21
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 20
- 230000006870 function Effects 0.000 description 19
- 229920001992 poloxamer 407 Polymers 0.000 description 19
- 229940044476 poloxamer 407 Drugs 0.000 description 19
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 18
- 230000009246 food effect Effects 0.000 description 17
- 238000002425 crystallisation Methods 0.000 description 16
- 239000008177 pharmaceutical agent Substances 0.000 description 16
- 238000011282 treatment Methods 0.000 description 16
- 241000282472 Canis lupus familiaris Species 0.000 description 15
- 230000008025 crystallization Effects 0.000 description 15
- 238000001727 in vivo Methods 0.000 description 15
- 241000700159 Rattus Species 0.000 description 14
- 230000002411 adverse Effects 0.000 description 14
- 238000000540 analysis of variance Methods 0.000 description 14
- 239000000047 product Substances 0.000 description 14
- 239000007787 solid Substances 0.000 description 14
- 229920003082 Povidone K 90 Polymers 0.000 description 13
- 235000021471 food effect Nutrition 0.000 description 13
- 230000002496 gastric effect Effects 0.000 description 13
- 230000002829 reductive effect Effects 0.000 description 13
- 230000007704 transition Effects 0.000 description 13
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 12
- 239000005557 antagonist Substances 0.000 description 12
- 230000008859 change Effects 0.000 description 12
- 239000002552 dosage form Substances 0.000 description 12
- 239000000843 powder Substances 0.000 description 12
- 238000010521 absorption reaction Methods 0.000 description 11
- 210000004369 blood Anatomy 0.000 description 11
- 239000008280 blood Substances 0.000 description 11
- HBENZIXOGRCSQN-VQWWACLZSA-N (1S,2S,6R,14R,15R,16R)-5-(cyclopropylmethyl)-16-[(2S)-2-hydroxy-3,3-dimethylpentan-2-yl]-15-methoxy-13-oxa-5-azahexacyclo[13.2.2.12,8.01,6.02,14.012,20]icosa-8(20),9,11-trien-11-ol Chemical compound N1([C@@H]2CC=3C4=C(C(=CC=3)O)O[C@H]3[C@@]5(OC)CC[C@@]2([C@@]43CC1)C[C@@H]5[C@](C)(O)C(C)(C)CC)CC1CC1 HBENZIXOGRCSQN-VQWWACLZSA-N 0.000 description 10
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 10
- NKANXQFJJICGDU-QPLCGJKRSA-N Tamoxifen Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 NKANXQFJJICGDU-QPLCGJKRSA-N 0.000 description 10
- 239000007963 capsule composition Substances 0.000 description 10
- 239000013078 crystal Substances 0.000 description 10
- 238000002156 mixing Methods 0.000 description 10
- 239000007916 tablet composition Substances 0.000 description 10
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 9
- 238000002648 combination therapy Methods 0.000 description 9
- 238000010438 heat treatment Methods 0.000 description 9
- GVOISEJVFFIGQE-YCZSINBZSA-N n-[(1r,2s,5r)-5-[methyl(propan-2-yl)amino]-2-[(3s)-2-oxo-3-[[6-(trifluoromethyl)quinazolin-4-yl]amino]pyrrolidin-1-yl]cyclohexyl]acetamide Chemical compound CC(=O)N[C@@H]1C[C@H](N(C)C(C)C)CC[C@@H]1N1C(=O)[C@@H](NC=2C3=CC(=CC=C3N=CN=2)C(F)(F)F)CC1 GVOISEJVFFIGQE-YCZSINBZSA-N 0.000 description 9
- 229920001983 poloxamer Polymers 0.000 description 9
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 9
- 239000000126 substance Substances 0.000 description 9
- 230000001225 therapeutic effect Effects 0.000 description 9
- 238000002411 thermogravimetry Methods 0.000 description 9
- 102000005789 Vascular Endothelial Growth Factors Human genes 0.000 description 8
- 108010019530 Vascular Endothelial Growth Factors Proteins 0.000 description 8
- 239000007864 aqueous solution Substances 0.000 description 8
- 230000015572 biosynthetic process Effects 0.000 description 8
- 238000001938 differential scanning calorimetry curve Methods 0.000 description 8
- 239000003085 diluting agent Substances 0.000 description 8
- 239000007789 gas Substances 0.000 description 8
- 229940125697 hormonal agent Drugs 0.000 description 8
- 238000002844 melting Methods 0.000 description 8
- 230000008018 melting Effects 0.000 description 8
- 230000008569 process Effects 0.000 description 8
- 206010061218 Inflammation Diseases 0.000 description 7
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 7
- 229920002472 Starch Polymers 0.000 description 7
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 7
- 230000000340 anti-metabolite Effects 0.000 description 7
- 229940100197 antimetabolite Drugs 0.000 description 7
- 239000002256 antimetabolite Substances 0.000 description 7
- 239000002246 antineoplastic agent Substances 0.000 description 7
- 239000011248 coating agent Substances 0.000 description 7
- 238000000576 coating method Methods 0.000 description 7
- 239000007884 disintegrant Substances 0.000 description 7
- 229920001903 high density polyethylene Polymers 0.000 description 7
- 239000004700 high-density polyethylene Substances 0.000 description 7
- 230000004054 inflammatory process Effects 0.000 description 7
- 239000003112 inhibitor Substances 0.000 description 7
- 230000002401 inhibitory effect Effects 0.000 description 7
- 238000005259 measurement Methods 0.000 description 7
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 7
- AICOOMRHRUFYCM-ZRRPKQBOSA-N oxazine, 1 Chemical compound C([C@@H]1[C@H](C(C[C@]2(C)[C@@H]([C@H](C)N(C)C)[C@H](O)C[C@]21C)=O)CC1=CC2)C[C@H]1[C@@]1(C)[C@H]2N=C(C(C)C)OC1 AICOOMRHRUFYCM-ZRRPKQBOSA-N 0.000 description 7
- 229960000502 poloxamer Drugs 0.000 description 7
- 229920001993 poloxamer 188 Polymers 0.000 description 7
- 229940044519 poloxamer 188 Drugs 0.000 description 7
- 239000008107 starch Substances 0.000 description 7
- 229940032147 starch Drugs 0.000 description 7
- 235000019698 starch Nutrition 0.000 description 7
- 238000012360 testing method Methods 0.000 description 7
- 206010019233 Headaches Diseases 0.000 description 6
- 108010079943 Pentagastrin Proteins 0.000 description 6
- 238000004458 analytical method Methods 0.000 description 6
- 201000011510 cancer Diseases 0.000 description 6
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 6
- 230000000875 corresponding effect Effects 0.000 description 6
- 229940127089 cytotoxic agent Drugs 0.000 description 6
- 239000012530 fluid Substances 0.000 description 6
- 231100000869 headache Toxicity 0.000 description 6
- 235000009200 high fat diet Nutrition 0.000 description 6
- 238000000338 in vitro Methods 0.000 description 6
- 239000000314 lubricant Substances 0.000 description 6
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 6
- 229960000444 pentagastrin Drugs 0.000 description 6
- ANRIQLNBZQLTFV-DZUOILHNSA-N pentagastrin Chemical compound C([C@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=1[C]2C=CC=CC2=NC=1)NC(=O)CCNC(=O)OC(C)(C)C)CCSC)C(N)=O)C1=CC=CC=C1 ANRIQLNBZQLTFV-DZUOILHNSA-N 0.000 description 6
- 238000001556 precipitation Methods 0.000 description 6
- 238000001953 recrystallisation Methods 0.000 description 6
- 210000002784 stomach Anatomy 0.000 description 6
- 238000001356 surgical procedure Methods 0.000 description 6
- 230000004083 survival effect Effects 0.000 description 6
- 239000000725 suspension Substances 0.000 description 6
- 230000002195 synergetic effect Effects 0.000 description 6
- 239000003981 vehicle Substances 0.000 description 6
- 206010030113 Oedema Diseases 0.000 description 5
- 230000002491 angiogenic effect Effects 0.000 description 5
- 238000004581 coalescence Methods 0.000 description 5
- 239000002274 desiccant Substances 0.000 description 5
- 239000003534 dna topoisomerase inhibitor Substances 0.000 description 5
- 239000003937 drug carrier Substances 0.000 description 5
- 238000011156 evaluation Methods 0.000 description 5
- 239000000835 fiber Substances 0.000 description 5
- 230000009477 glass transition Effects 0.000 description 5
- 239000008187 granular material Substances 0.000 description 5
- 230000002757 inflammatory effect Effects 0.000 description 5
- 230000003993 interaction Effects 0.000 description 5
- 239000008108 microcrystalline cellulose Substances 0.000 description 5
- 229940016286 microcrystalline cellulose Drugs 0.000 description 5
- 229940064639 minipress Drugs 0.000 description 5
- 230000035699 permeability Effects 0.000 description 5
- WFXFYZULCQKPIP-UHFFFAOYSA-N prazosin hydrochloride Chemical compound [H+].[Cl-].N=1C(N)=C2C=C(OC)C(OC)=CC2=NC=1N(CC1)CCN1C(=O)C1=CC=CO1 WFXFYZULCQKPIP-UHFFFAOYSA-N 0.000 description 5
- 230000009467 reduction Effects 0.000 description 5
- 238000004062 sedimentation Methods 0.000 description 5
- 239000003381 stabilizer Substances 0.000 description 5
- 238000003860 storage Methods 0.000 description 5
- 229960001603 tamoxifen Drugs 0.000 description 5
- 229940044693 topoisomerase inhibitor Drugs 0.000 description 5
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- 239000004322 Butylated hydroxytoluene Substances 0.000 description 4
- NLZUEZXRPGMBCV-UHFFFAOYSA-N Butylhydroxytoluene Chemical compound CC1=CC(C(C)(C)C)=C(O)C(C(C)(C)C)=C1 NLZUEZXRPGMBCV-UHFFFAOYSA-N 0.000 description 4
- 208000005443 Circulating Neoplastic Cells Diseases 0.000 description 4
- 102000004127 Cytokines Human genes 0.000 description 4
- 108090000695 Cytokines Proteins 0.000 description 4
- 230000000970 DNA cross-linking effect Effects 0.000 description 4
- 108010010803 Gelatin Proteins 0.000 description 4
- 108010063738 Interleukins Proteins 0.000 description 4
- 102000015696 Interleukins Human genes 0.000 description 4
- YASAKCUCGLMORW-UHFFFAOYSA-N Rosiglitazone Chemical compound C=1C=CC=NC=1N(C)CCOC(C=C1)=CC=C1CC1SC(=O)NC1=O YASAKCUCGLMORW-UHFFFAOYSA-N 0.000 description 4
- 229930006000 Sucrose Natural products 0.000 description 4
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 4
- 239000000654 additive Substances 0.000 description 4
- 230000002776 aggregation Effects 0.000 description 4
- 239000004037 angiogenesis inhibitor Substances 0.000 description 4
- 239000000051 antiandrogen Substances 0.000 description 4
- 239000003963 antioxidant agent Substances 0.000 description 4
- 239000012736 aqueous medium Substances 0.000 description 4
- 239000011230 binding agent Substances 0.000 description 4
- 235000010354 butylated hydroxytoluene Nutrition 0.000 description 4
- 229940095259 butylated hydroxytoluene Drugs 0.000 description 4
- 230000015556 catabolic process Effects 0.000 description 4
- 235000010980 cellulose Nutrition 0.000 description 4
- 229920002678 cellulose Polymers 0.000 description 4
- 210000001175 cerebrospinal fluid Anatomy 0.000 description 4
- 238000002512 chemotherapy Methods 0.000 description 4
- 238000005345 coagulation Methods 0.000 description 4
- 230000015271 coagulation Effects 0.000 description 4
- 238000005056 compaction Methods 0.000 description 4
- 238000006731 degradation reaction Methods 0.000 description 4
- 235000005911 diet Nutrition 0.000 description 4
- 230000037213 diet Effects 0.000 description 4
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 4
- 229960004679 doxorubicin Drugs 0.000 description 4
- 238000001647 drug administration Methods 0.000 description 4
- 238000002651 drug therapy Methods 0.000 description 4
- 239000000945 filler Substances 0.000 description 4
- 210000001035 gastrointestinal tract Anatomy 0.000 description 4
- 229920000159 gelatin Polymers 0.000 description 4
- 239000008273 gelatin Substances 0.000 description 4
- 239000007903 gelatin capsule Substances 0.000 description 4
- 235000019322 gelatine Nutrition 0.000 description 4
- 235000011852 gelatine desserts Nutrition 0.000 description 4
- 229940047122 interleukins Drugs 0.000 description 4
- 229940043355 kinase inhibitor Drugs 0.000 description 4
- 230000007774 longterm Effects 0.000 description 4
- 230000004060 metabolic process Effects 0.000 description 4
- 239000011859 microparticle Substances 0.000 description 4
- 230000025090 microtubule depolymerization Effects 0.000 description 4
- 239000002105 nanoparticle Substances 0.000 description 4
- 230000001575 pathological effect Effects 0.000 description 4
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 4
- 229940068196 placebo Drugs 0.000 description 4
- 239000000902 placebo Substances 0.000 description 4
- 229920001223 polyethylene glycol Polymers 0.000 description 4
- ZAHRKKWIAAJSAO-UHFFFAOYSA-N rapamycin Natural products COCC(O)C(=C/C(C)C(=O)CC(OC(=O)C1CCCCN1C(=O)C(=O)C2(O)OC(CC(OC)C(=CC=CC=CC(C)CC(C)C(=O)C)C)CCC2C)C(C)CC3CCC(O)C(C3)OC)C ZAHRKKWIAAJSAO-UHFFFAOYSA-N 0.000 description 4
- 239000002464 receptor antagonist Substances 0.000 description 4
- 229940044551 receptor antagonist Drugs 0.000 description 4
- 239000013557 residual solvent Substances 0.000 description 4
- 229940095743 selective estrogen receptor modulator Drugs 0.000 description 4
- 239000000333 selective estrogen receptor modulator Substances 0.000 description 4
- 210000002966 serum Anatomy 0.000 description 4
- 229960002930 sirolimus Drugs 0.000 description 4
- QFJCIRLUMZQUOT-HPLJOQBZSA-N sirolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 QFJCIRLUMZQUOT-HPLJOQBZSA-N 0.000 description 4
- 239000005720 sucrose Substances 0.000 description 4
- 238000002562 urinalysis Methods 0.000 description 4
- 239000004066 vascular targeting agent Substances 0.000 description 4
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 3
- MAUCONCHVWBMHK-UHFFFAOYSA-N 3-[(dimethylamino)methyl]-N-[2-[4-[(hydroxyamino)-oxomethyl]phenoxy]ethyl]-2-benzofurancarboxamide Chemical compound O1C2=CC=CC=C2C(CN(C)C)=C1C(=O)NCCOC1=CC=C(C(=O)NO)C=C1 MAUCONCHVWBMHK-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 3
- MLDQJTXFUGDVEO-UHFFFAOYSA-N BAY-43-9006 Chemical compound C1=NC(C(=O)NC)=CC(OC=2C=CC(NC(=O)NC=3C=C(C(Cl)=CC=3)C(F)(F)F)=CC=2)=C1 MLDQJTXFUGDVEO-UHFFFAOYSA-N 0.000 description 3
- 229920002785 Croscarmellose sodium Polymers 0.000 description 3
- 102100021704 Cytochrome P450 2D6 Human genes 0.000 description 3
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 3
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 3
- ZBNZXTGUTAYRHI-UHFFFAOYSA-N Dasatinib Chemical compound C=1C(N2CCN(CCO)CC2)=NC(C)=NC=1NC(S1)=NC=C1C(=O)NC1=C(C)C=CC=C1Cl ZBNZXTGUTAYRHI-UHFFFAOYSA-N 0.000 description 3
- 206010012735 Diarrhoea Diseases 0.000 description 3
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 3
- 229940102550 Estrogen receptor antagonist Drugs 0.000 description 3
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 3
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 3
- 208000032843 Hemorrhage Diseases 0.000 description 3
- 241000282412 Homo Species 0.000 description 3
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 3
- 206010020772 Hypertension Diseases 0.000 description 3
- 102000004889 Interleukin-6 Human genes 0.000 description 3
- 108090001005 Interleukin-6 Proteins 0.000 description 3
- 239000005411 L01XE02 - Gefitinib Substances 0.000 description 3
- 239000002147 L01XE04 - Sunitinib Substances 0.000 description 3
- 239000005511 L01XE05 - Sorafenib Substances 0.000 description 3
- 239000002067 L01XE06 - Dasatinib Substances 0.000 description 3
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 3
- 229920000881 Modified starch Polymers 0.000 description 3
- 206010028813 Nausea Diseases 0.000 description 3
- 239000004698 Polyethylene Substances 0.000 description 3
- 239000002202 Polyethylene glycol Substances 0.000 description 3
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 3
- 208000006011 Stroke Diseases 0.000 description 3
- 238000000692 Student's t-test Methods 0.000 description 3
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 3
- 239000004480 active ingredient Substances 0.000 description 3
- 230000000996 additive effect Effects 0.000 description 3
- 238000004220 aggregation Methods 0.000 description 3
- 101150023881 agl11 gene Proteins 0.000 description 3
- 230000033115 angiogenesis Effects 0.000 description 3
- 239000003945 anionic surfactant Substances 0.000 description 3
- 230000002280 anti-androgenic effect Effects 0.000 description 3
- 239000003886 aromatase inhibitor Substances 0.000 description 3
- 229940046844 aromatase inhibitors Drugs 0.000 description 3
- 238000003556 assay Methods 0.000 description 3
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 3
- 239000013060 biological fluid Substances 0.000 description 3
- 230000000740 bleeding effect Effects 0.000 description 3
- 230000017531 blood circulation Effects 0.000 description 3
- 230000037396 body weight Effects 0.000 description 3
- 229960000590 celecoxib Drugs 0.000 description 3
- RZEKVGVHFLEQIL-UHFFFAOYSA-N celecoxib Chemical compound C1=CC(C)=CC=C1C1=CC(C(F)(F)F)=NN1C1=CC=C(S(N)(=O)=O)C=C1 RZEKVGVHFLEQIL-UHFFFAOYSA-N 0.000 description 3
- 210000004027 cell Anatomy 0.000 description 3
- 239000001913 cellulose Substances 0.000 description 3
- 239000003153 chemical reaction reagent Substances 0.000 description 3
- 230000000052 comparative effect Effects 0.000 description 3
- 238000002591 computed tomography Methods 0.000 description 3
- 235000010947 crosslinked sodium carboxy methyl cellulose Nutrition 0.000 description 3
- 229960002448 dasatinib Drugs 0.000 description 3
- 230000003247 decreasing effect Effects 0.000 description 3
- 238000013535 dynamic contrast enhanced MRI Methods 0.000 description 3
- 230000002708 enhancing effect Effects 0.000 description 3
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 3
- 238000002474 experimental method Methods 0.000 description 3
- 230000037406 food intake Effects 0.000 description 3
- 229960002584 gefitinib Drugs 0.000 description 3
- XGALLCVXEZPNRQ-UHFFFAOYSA-N gefitinib Chemical compound C=12C=C(OCCCN3CCOCC3)C(OC)=CC2=NC=NC=1NC1=CC=C(F)C(Cl)=C1 XGALLCVXEZPNRQ-UHFFFAOYSA-N 0.000 description 3
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 239000004615 ingredient Substances 0.000 description 3
- 102000006495 integrins Human genes 0.000 description 3
- 108010044426 integrins Proteins 0.000 description 3
- 239000008101 lactose Substances 0.000 description 3
- 230000000670 limiting effect Effects 0.000 description 3
- 150000002634 lipophilic molecules Chemical class 0.000 description 3
- 229920001684 low density polyethylene Polymers 0.000 description 3
- 239000004702 low-density polyethylene Substances 0.000 description 3
- 235000019359 magnesium stearate Nutrition 0.000 description 3
- 229960001924 melphalan Drugs 0.000 description 3
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 3
- 230000008693 nausea Effects 0.000 description 3
- 229920006112 polar polymer Polymers 0.000 description 3
- 229920000573 polyethylene Polymers 0.000 description 3
- 239000002244 precipitate Substances 0.000 description 3
- 239000002243 precursor Substances 0.000 description 3
- 229960004618 prednisone Drugs 0.000 description 3
- XOFYZVNMUHMLCC-ZPOLXVRWSA-N prednisone Chemical compound O=C1C=C[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 XOFYZVNMUHMLCC-ZPOLXVRWSA-N 0.000 description 3
- 239000003755 preservative agent Substances 0.000 description 3
- 230000035755 proliferation Effects 0.000 description 3
- 230000005855 radiation Effects 0.000 description 3
- 238000001959 radiotherapy Methods 0.000 description 3
- 230000001105 regulatory effect Effects 0.000 description 3
- 238000011160 research Methods 0.000 description 3
- 230000004044 response Effects 0.000 description 3
- 238000005070 sampling Methods 0.000 description 3
- 239000012047 saturated solution Substances 0.000 description 3
- 238000009097 single-agent therapy Methods 0.000 description 3
- 229910052708 sodium Inorganic materials 0.000 description 3
- 239000011734 sodium Substances 0.000 description 3
- 229940083542 sodium Drugs 0.000 description 3
- 229960003787 sorafenib Drugs 0.000 description 3
- 239000000600 sorbitol Substances 0.000 description 3
- 235000010356 sorbitol Nutrition 0.000 description 3
- 238000012453 sprague-dawley rat model Methods 0.000 description 3
- 238000011301 standard therapy Methods 0.000 description 3
- 150000003431 steroids Chemical class 0.000 description 3
- 238000003756 stirring Methods 0.000 description 3
- 235000000346 sugar Nutrition 0.000 description 3
- 229960001796 sunitinib Drugs 0.000 description 3
- WINHZLLDWRZWRT-ATVHPVEESA-N sunitinib Chemical compound CCN(CC)CCNC(=O)C1=C(C)NC(\C=C/2C3=CC(F)=CC=C3NC\2=O)=C1C WINHZLLDWRZWRT-ATVHPVEESA-N 0.000 description 3
- 239000000454 talc Substances 0.000 description 3
- 229910052623 talc Inorganic materials 0.000 description 3
- 235000012222 talc Nutrition 0.000 description 3
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 3
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 3
- 229940124597 therapeutic agent Drugs 0.000 description 3
- 238000001757 thermogravimetry curve Methods 0.000 description 3
- 210000001519 tissue Anatomy 0.000 description 3
- 231100000419 toxicity Toxicity 0.000 description 3
- 230000001988 toxicity Effects 0.000 description 3
- 210000004881 tumor cell Anatomy 0.000 description 3
- 210000002700 urine Anatomy 0.000 description 3
- 230000003442 weekly effect Effects 0.000 description 3
- DIWRORZWFLOCLC-HNNXBMFYSA-N (3s)-7-chloro-5-(2-chlorophenyl)-3-hydroxy-1,3-dihydro-1,4-benzodiazepin-2-one Chemical compound N([C@H](C(NC1=CC=C(Cl)C=C11)=O)O)=C1C1=CC=CC=C1Cl DIWRORZWFLOCLC-HNNXBMFYSA-N 0.000 description 2
- FPVKHBSQESCIEP-UHFFFAOYSA-N (8S)-3-(2-deoxy-beta-D-erythro-pentofuranosyl)-3,6,7,8-tetrahydroimidazo[4,5-d][1,3]diazepin-8-ol Natural products C1C(O)C(CO)OC1N1C(NC=NCC2O)=C2N=C1 FPVKHBSQESCIEP-UHFFFAOYSA-N 0.000 description 2
- WHTVZRBIWZFKQO-AWEZNQCLSA-N (S)-chloroquine Chemical compound ClC1=CC=C2C(N[C@@H](C)CCCN(CC)CC)=CC=NC2=C1 WHTVZRBIWZFKQO-AWEZNQCLSA-N 0.000 description 2
- ODIGIKRIUKFKHP-UHFFFAOYSA-N (n-propan-2-yloxycarbonylanilino) acetate Chemical compound CC(C)OC(=O)N(OC(C)=O)C1=CC=CC=C1 ODIGIKRIUKFKHP-UHFFFAOYSA-N 0.000 description 2
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 2
- UEJJHQNACJXSKW-UHFFFAOYSA-N 2-(2,6-dioxopiperidin-3-yl)-1H-isoindole-1,3(2H)-dione Chemical compound O=C1C2=CC=CC=C2C(=O)N1C1CCC(=O)NC1=O UEJJHQNACJXSKW-UHFFFAOYSA-N 0.000 description 2
- JVKUCNQGESRUCL-UHFFFAOYSA-N 2-Hydroxyethyl 12-hydroxyoctadecanoate Chemical compound CCCCCCC(O)CCCCCCCCCCC(=O)OCCO JVKUCNQGESRUCL-UHFFFAOYSA-N 0.000 description 2
- AOJJSUZBOXZQNB-VTZDEGQISA-N 4'-epidoxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-VTZDEGQISA-N 0.000 description 2
- CLPFFLWZZBQMAO-UHFFFAOYSA-N 4-(5,6,7,8-tetrahydroimidazo[1,5-a]pyridin-5-yl)benzonitrile Chemical compound C1=CC(C#N)=CC=C1C1N2C=NC=C2CCC1 CLPFFLWZZBQMAO-UHFFFAOYSA-N 0.000 description 2
- XXJWYDDUDKYVKI-UHFFFAOYSA-N 4-[(4-fluoro-2-methyl-1H-indol-5-yl)oxy]-6-methoxy-7-[3-(1-pyrrolidinyl)propoxy]quinazoline Chemical compound COC1=CC2=C(OC=3C(=C4C=C(C)NC4=CC=3)F)N=CN=C2C=C1OCCCN1CCCC1 XXJWYDDUDKYVKI-UHFFFAOYSA-N 0.000 description 2
- IDPUKCWIGUEADI-UHFFFAOYSA-N 5-[bis(2-chloroethyl)amino]uracil Chemical compound ClCCN(CCCl)C1=CNC(=O)NC1=O IDPUKCWIGUEADI-UHFFFAOYSA-N 0.000 description 2
- XAUDJQYHKZQPEU-KVQBGUIXSA-N 5-aza-2'-deoxycytidine Chemical compound O=C1N=C(N)N=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 XAUDJQYHKZQPEU-KVQBGUIXSA-N 0.000 description 2
- WYWHKKSPHMUBEB-UHFFFAOYSA-N 6-Mercaptoguanine Natural products N1C(N)=NC(=S)C2=C1N=CN2 WYWHKKSPHMUBEB-UHFFFAOYSA-N 0.000 description 2
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 2
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical compound N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 2
- MKBLHFILKIKSQM-UHFFFAOYSA-N 9-methyl-3-[(2-methyl-1h-imidazol-3-ium-3-yl)methyl]-2,3-dihydro-1h-carbazol-4-one;chloride Chemical compound Cl.CC1=NC=CN1CC1C(=O)C(C=2C(=CC=CC=2)N2C)=C2CC1 MKBLHFILKIKSQM-UHFFFAOYSA-N 0.000 description 2
- BUROJSBIWGDYCN-GAUTUEMISA-N AP 23573 Chemical compound C1C[C@@H](OP(C)(C)=O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 BUROJSBIWGDYCN-GAUTUEMISA-N 0.000 description 2
- 241000220479 Acacia Species 0.000 description 2
- 108010012934 Albumin-Bound Paclitaxel Proteins 0.000 description 2
- OGSPWJRAVKPPFI-UHFFFAOYSA-N Alendronic Acid Chemical compound NCCCC(O)(P(O)(O)=O)P(O)(O)=O OGSPWJRAVKPPFI-UHFFFAOYSA-N 0.000 description 2
- 108010006654 Bleomycin Proteins 0.000 description 2
- FVLVBPDQNARYJU-XAHDHGMMSA-N C[C@H]1CCC(CC1)NC(=O)N(CCCl)N=O Chemical compound C[C@H]1CCC(CC1)NC(=O)N(CCCl)N=O FVLVBPDQNARYJU-XAHDHGMMSA-N 0.000 description 2
- DLGOEMSEDOSKAD-UHFFFAOYSA-N Carmustine Chemical compound ClCCNC(=O)N(N=O)CCCl DLGOEMSEDOSKAD-UHFFFAOYSA-N 0.000 description 2
- HVXBOLULGPECHP-WAYWQWQTSA-N Combretastatin A4 Chemical compound C1=C(O)C(OC)=CC=C1\C=C/C1=CC(OC)=C(OC)C(OC)=C1 HVXBOLULGPECHP-WAYWQWQTSA-N 0.000 description 2
- 102000004420 Creatine Kinase Human genes 0.000 description 2
- 108010042126 Creatine kinase Proteins 0.000 description 2
- 102000003903 Cyclin-dependent kinases Human genes 0.000 description 2
- 108090000266 Cyclin-dependent kinases Proteins 0.000 description 2
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 2
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 2
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 2
- 108020004414 DNA Proteins 0.000 description 2
- 108010092160 Dactinomycin Proteins 0.000 description 2
- 208000032131 Diabetic Neuropathies Diseases 0.000 description 2
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- 238000002965 ELISA Methods 0.000 description 2
- HTIJFSOGRVMCQR-UHFFFAOYSA-N Epirubicin Natural products COc1cccc2C(=O)c3c(O)c4CC(O)(CC(OC5CC(N)C(=O)C(C)O5)c4c(O)c3C(=O)c12)C(=O)CO HTIJFSOGRVMCQR-UHFFFAOYSA-N 0.000 description 2
- VWUXBMIQPBEWFH-WCCTWKNTSA-N Fulvestrant Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3[C@H](CCCCCCCCCS(=O)CCCC(F)(F)C(F)(F)F)CC2=C1 VWUXBMIQPBEWFH-WCCTWKNTSA-N 0.000 description 2
- 102100039619 Granulocyte colony-stimulating factor Human genes 0.000 description 2
- 206010019280 Heart failures Diseases 0.000 description 2
- 102000003745 Hepatocyte Growth Factor Human genes 0.000 description 2
- 108090000100 Hepatocyte Growth Factor Proteins 0.000 description 2
- 102100021866 Hepatocyte growth factor Human genes 0.000 description 2
- 102000003964 Histone deacetylase Human genes 0.000 description 2
- 108090000353 Histone deacetylase Proteins 0.000 description 2
- 101000896586 Homo sapiens Cytochrome P450 2D6 Proteins 0.000 description 2
- 101000896576 Homo sapiens Putative cytochrome P450 2D7 Proteins 0.000 description 2
- XDXDZDZNSLXDNA-TZNDIEGXSA-N Idarubicin Chemical compound C1[C@H](N)[C@H](O)[C@H](C)O[C@H]1O[C@@H]1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2C[C@@](O)(C(C)=O)C1 XDXDZDZNSLXDNA-TZNDIEGXSA-N 0.000 description 2
- XDXDZDZNSLXDNA-UHFFFAOYSA-N Idarubicin Natural products C1C(N)C(O)C(C)OC1OC1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2CC(O)(C(C)=O)C1 XDXDZDZNSLXDNA-UHFFFAOYSA-N 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- 108090000723 Insulin-Like Growth Factor I Proteins 0.000 description 2
- 102000004218 Insulin-Like Growth Factor I Human genes 0.000 description 2
- 108090001007 Interleukin-8 Proteins 0.000 description 2
- SHGAZHPCJJPHSC-NUEINMDLSA-N Isotretinoin Chemical compound OC(=O)C=C(C)/C=C/C=C(C)C=CC1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-NUEINMDLSA-N 0.000 description 2
- 239000005517 L01XE01 - Imatinib Substances 0.000 description 2
- 239000002136 L01XE07 - Lapatinib Substances 0.000 description 2
- 239000002118 L01XE12 - Vandetanib Substances 0.000 description 2
- 239000002145 L01XE14 - Bosutinib Substances 0.000 description 2
- XNRVGTHNYCNCFF-UHFFFAOYSA-N Lapatinib ditosylate monohydrate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1.CC1=CC=C(S(O)(=O)=O)C=C1.O1C(CNCCS(=O)(=O)C)=CC=C1C1=CC=C(N=CN=C2NC=3C=C(Cl)C(OCC=4C=C(F)C=CC=4)=CC=3)C2=C1 XNRVGTHNYCNCFF-UHFFFAOYSA-N 0.000 description 2
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 2
- GQYIWUVLTXOXAJ-UHFFFAOYSA-N Lomustine Chemical compound ClCCN(N=O)C(=O)NC1CCCCC1 GQYIWUVLTXOXAJ-UHFFFAOYSA-N 0.000 description 2
- 229930195725 Mannitol Natural products 0.000 description 2
- 229930192392 Mitomycin Natural products 0.000 description 2
- PCZOHLXUXFIOCF-UHFFFAOYSA-N Monacolin X Natural products C12C(OC(=O)C(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 PCZOHLXUXFIOCF-UHFFFAOYSA-N 0.000 description 2
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 2
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 2
- 229930012538 Paclitaxel Natural products 0.000 description 2
- 208000034038 Pathologic Neovascularization Diseases 0.000 description 2
- KMSKQZKKOZQFFG-HSUXVGOQSA-N Pirarubicin Chemical compound O([C@H]1[C@@H](N)C[C@@H](O[C@H]1C)O[C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1CCCCO1 KMSKQZKKOZQFFG-HSUXVGOQSA-N 0.000 description 2
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 2
- 239000004743 Polypropylene Substances 0.000 description 2
- RJKFOVLPORLFTN-LEKSSAKUSA-N Progesterone Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H](C(=O)C)[C@@]1(C)CC2 RJKFOVLPORLFTN-LEKSSAKUSA-N 0.000 description 2
- IIDJRNMFWXDHID-UHFFFAOYSA-N Risedronic acid Chemical compound OP(=O)(O)C(P(O)(O)=O)(O)CC1=CC=CN=C1 IIDJRNMFWXDHID-UHFFFAOYSA-N 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- 229920002125 Sokalan® Polymers 0.000 description 2
- 229920001304 Solutol HS 15 Polymers 0.000 description 2
- RAHZWNYVWXNFOC-UHFFFAOYSA-N Sulphur dioxide Chemical compound O=S=O RAHZWNYVWXNFOC-UHFFFAOYSA-N 0.000 description 2
- 229940123445 Tricyclic antidepressant Drugs 0.000 description 2
- 108010053099 Vascular Endothelial Growth Factor Receptor-2 Proteins 0.000 description 2
- 102100033177 Vascular endothelial growth factor receptor 2 Human genes 0.000 description 2
- 229950008805 abexinostat Drugs 0.000 description 2
- 229930183665 actinomycin Natural products 0.000 description 2
- 230000001154 acute effect Effects 0.000 description 2
- 229940062527 alendronate Drugs 0.000 description 2
- SHGAZHPCJJPHSC-YCNIQYBTSA-N all-trans-retinoic acid Chemical compound OC(=O)\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-YCNIQYBTSA-N 0.000 description 2
- 229960003437 aminoglutethimide Drugs 0.000 description 2
- ROBVIMPUHSLWNV-UHFFFAOYSA-N aminoglutethimide Chemical compound C=1C=C(N)C=CC=1C1(CC)CCC(=O)NC1=O ROBVIMPUHSLWNV-UHFFFAOYSA-N 0.000 description 2
- 239000002280 amphoteric surfactant Substances 0.000 description 2
- 229960002550 amrubicin Drugs 0.000 description 2
- VJZITPJGSQKZMX-XDPRQOKASA-N amrubicin Chemical compound O([C@H]1C[C@](CC2=C(O)C=3C(=O)C4=CC=CC=C4C(=O)C=3C(O)=C21)(N)C(=O)C)[C@H]1C[C@H](O)[C@H](O)CO1 VJZITPJGSQKZMX-XDPRQOKASA-N 0.000 description 2
- 229960002932 anastrozole Drugs 0.000 description 2
- YBBLVLTVTVSKRW-UHFFFAOYSA-N anastrozole Chemical compound N#CC(C)(C)C1=CC(C(C)(C#N)C)=CC(CN2N=CN=C2)=C1 YBBLVLTVTVSKRW-UHFFFAOYSA-N 0.000 description 2
- 230000003288 anthiarrhythmic effect Effects 0.000 description 2
- 238000011122 anti-angiogenic therapy Methods 0.000 description 2
- 230000000118 anti-neoplastic effect Effects 0.000 description 2
- 230000000692 anti-sense effect Effects 0.000 description 2
- 239000012296 anti-solvent Substances 0.000 description 2
- 239000003416 antiarrhythmic agent Substances 0.000 description 2
- 239000000935 antidepressant agent Substances 0.000 description 2
- 229940005513 antidepressants Drugs 0.000 description 2
- 239000000427 antigen Substances 0.000 description 2
- 108091007433 antigens Proteins 0.000 description 2
- 102000036639 antigens Human genes 0.000 description 2
- 229940034982 antineoplastic agent Drugs 0.000 description 2
- 239000000164 antipsychotic agent Substances 0.000 description 2
- 229940005529 antipsychotics Drugs 0.000 description 2
- MCGDSOGUHLTADD-UHFFFAOYSA-N arzoxifene Chemical compound C1=CC(OC)=CC=C1C1=C(OC=2C=CC(OCCN3CCCCC3)=CC=2)C2=CC=C(O)C=C2S1 MCGDSOGUHLTADD-UHFFFAOYSA-N 0.000 description 2
- 229950005529 arzoxifene Drugs 0.000 description 2
- 238000000889 atomisation Methods 0.000 description 2
- 229960003005 axitinib Drugs 0.000 description 2
- RITAVMQDGBJQJZ-FMIVXFBMSA-N axitinib Chemical compound CNC(=O)C1=CC=CC=C1SC1=CC=C(C(\C=C\C=2N=CC=CC=2)=NN2)C2=C1 RITAVMQDGBJQJZ-FMIVXFBMSA-N 0.000 description 2
- 231100001125 band 2 compound Toxicity 0.000 description 2
- 230000004888 barrier function Effects 0.000 description 2
- 229960000817 bazedoxifene Drugs 0.000 description 2
- UCJGJABZCDBEDK-UHFFFAOYSA-N bazedoxifene Chemical compound C=1C=C(OCCN2CCCCCC2)C=CC=1CN1C2=CC=C(O)C=C2C(C)=C1C1=CC=C(O)C=C1 UCJGJABZCDBEDK-UHFFFAOYSA-N 0.000 description 2
- 239000011324 bead Substances 0.000 description 2
- 229960002707 bendamustine Drugs 0.000 description 2
- YTKUWDBFDASYHO-UHFFFAOYSA-N bendamustine Chemical compound ClCCN(CCCl)C1=CC=C2N(C)C(CCCC(O)=O)=NC2=C1 YTKUWDBFDASYHO-UHFFFAOYSA-N 0.000 description 2
- 230000009286 beneficial effect Effects 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 102000012740 beta Adrenergic Receptors Human genes 0.000 description 2
- 108010079452 beta Adrenergic Receptors Proteins 0.000 description 2
- 239000000227 bioadhesive Substances 0.000 description 2
- 239000012472 biological sample Substances 0.000 description 2
- 229960001561 bleomycin Drugs 0.000 description 2
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 description 2
- 238000004820 blood count Methods 0.000 description 2
- 229960001467 bortezomib Drugs 0.000 description 2
- GXJABQQUPOEUTA-RDJZCZTQSA-N bortezomib Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)B(O)O)NC(=O)C=1N=CC=NC=1)C1=CC=CC=C1 GXJABQQUPOEUTA-RDJZCZTQSA-N 0.000 description 2
- 229960003736 bosutinib Drugs 0.000 description 2
- UBPYILGKFZZVDX-UHFFFAOYSA-N bosutinib Chemical compound C1=C(Cl)C(OC)=CC(NC=2C3=CC(OC)=C(OCCCN4CCN(C)CC4)C=C3N=CC=2C#N)=C1Cl UBPYILGKFZZVDX-UHFFFAOYSA-N 0.000 description 2
- 239000008364 bulk solution Substances 0.000 description 2
- KVUAALJSMIVURS-ZEDZUCNESA-L calcium folinate Chemical compound [Ca+2].C1NC=2NC(N)=NC(=O)C=2N(C=O)C1CNC1=CC=C(C(=O)N[C@@H](CCC([O-])=O)C([O-])=O)C=C1 KVUAALJSMIVURS-ZEDZUCNESA-L 0.000 description 2
- 239000001506 calcium phosphate Substances 0.000 description 2
- OSGAYBCDTDRGGQ-UHFFFAOYSA-L calcium sulfate Chemical compound [Ca+2].[O-]S([O-])(=O)=O OSGAYBCDTDRGGQ-UHFFFAOYSA-L 0.000 description 2
- 239000001768 carboxy methyl cellulose Substances 0.000 description 2
- 229960005243 carmustine Drugs 0.000 description 2
- 239000003093 cationic surfactant Substances 0.000 description 2
- 229960002412 cediranib Drugs 0.000 description 2
- 229960005395 cetuximab Drugs 0.000 description 2
- 238000006243 chemical reaction Methods 0.000 description 2
- 229960004630 chlorambucil Drugs 0.000 description 2
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 description 2
- 229960003677 chloroquine Drugs 0.000 description 2
- WHTVZRBIWZFKQO-UHFFFAOYSA-N chloroquine Natural products ClC1=CC=C2C(NC(C)CCCN(CC)CC)=CC=NC2=C1 WHTVZRBIWZFKQO-UHFFFAOYSA-N 0.000 description 2
- 230000001684 chronic effect Effects 0.000 description 2
- 229960000928 clofarabine Drugs 0.000 description 2
- WDDPHFBMKLOVOX-AYQXTPAHSA-N clofarabine Chemical compound C1=NC=2C(N)=NC(Cl)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@@H]1F WDDPHFBMKLOVOX-AYQXTPAHSA-N 0.000 description 2
- GKIRPKYJQBWNGO-OCEACIFDSA-N clomifene Chemical compound C1=CC(OCCN(CC)CC)=CC=C1C(\C=1C=CC=CC=1)=C(\Cl)C1=CC=CC=C1 GKIRPKYJQBWNGO-OCEACIFDSA-N 0.000 description 2
- 229960003608 clomifene Drugs 0.000 description 2
- 239000003086 colorant Substances 0.000 description 2
- 229960005537 combretastatin A-4 Drugs 0.000 description 2
- HVXBOLULGPECHP-UHFFFAOYSA-N combretastatin A4 Natural products C1=C(O)C(OC)=CC=C1C=CC1=CC(OC)=C(OC)C(OC)=C1 HVXBOLULGPECHP-UHFFFAOYSA-N 0.000 description 2
- 239000002131 composite material Substances 0.000 description 2
- 238000013329 compounding Methods 0.000 description 2
- 238000007906 compression Methods 0.000 description 2
- 230000006835 compression Effects 0.000 description 2
- 238000013270 controlled release Methods 0.000 description 2
- 230000001276 controlling effect Effects 0.000 description 2
- 238000007796 conventional method Methods 0.000 description 2
- 238000011443 conventional therapy Methods 0.000 description 2
- 229920001577 copolymer Polymers 0.000 description 2
- 230000002596 correlated effect Effects 0.000 description 2
- 229960001681 croscarmellose sodium Drugs 0.000 description 2
- 239000003431 cross linking reagent Substances 0.000 description 2
- 230000001351 cycling effect Effects 0.000 description 2
- 229960004397 cyclophosphamide Drugs 0.000 description 2
- 229960000684 cytarabine Drugs 0.000 description 2
- 229960000975 daunorubicin Drugs 0.000 description 2
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 2
- 229960003603 decitabine Drugs 0.000 description 2
- 238000000354 decomposition reaction Methods 0.000 description 2
- 230000007423 decrease Effects 0.000 description 2
- 239000007857 degradation product Substances 0.000 description 2
- 230000003111 delayed effect Effects 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 229960003957 dexamethasone Drugs 0.000 description 2
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 2
- 238000009792 diffusion process Methods 0.000 description 2
- 201000010099 disease Diseases 0.000 description 2
- 208000035475 disorder Diseases 0.000 description 2
- 239000002270 dispersing agent Substances 0.000 description 2
- 239000012738 dissolution medium Substances 0.000 description 2
- 229960003668 docetaxel Drugs 0.000 description 2
- GVGUFUZHNYFZLC-UHFFFAOYSA-N dodecyl benzenesulfonate;sodium Chemical compound [Na].CCCCCCCCCCCCOS(=O)(=O)C1=CC=CC=C1 GVGUFUZHNYFZLC-UHFFFAOYSA-N 0.000 description 2
- 229940088679 drug related substance Drugs 0.000 description 2
- 239000000975 dye Substances 0.000 description 2
- 229960001484 edetic acid Drugs 0.000 description 2
- 230000002526 effect on cardiovascular system Effects 0.000 description 2
- 239000003995 emulsifying agent Substances 0.000 description 2
- 238000005538 encapsulation Methods 0.000 description 2
- 102000052116 epidermal growth factor receptor activity proteins Human genes 0.000 description 2
- 108700015053 epidermal growth factor receptor activity proteins Proteins 0.000 description 2
- 229960001904 epirubicin Drugs 0.000 description 2
- 229960001842 estramustine Drugs 0.000 description 2
- FRPJXPJMRWBBIH-RBRWEJTLSA-N estramustine Chemical compound ClCCN(CCCl)C(=O)OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 FRPJXPJMRWBBIH-RBRWEJTLSA-N 0.000 description 2
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 2
- 229960005420 etoposide Drugs 0.000 description 2
- 239000000374 eutectic mixture Substances 0.000 description 2
- 229950011548 fadrozole Drugs 0.000 description 2
- 230000002349 favourable effect Effects 0.000 description 2
- 238000011049 filling Methods 0.000 description 2
- 239000010419 fine particle Substances 0.000 description 2
- 239000000796 flavoring agent Substances 0.000 description 2
- 229960000961 floxuridine Drugs 0.000 description 2
- ODKNJVUHOIMIIZ-RRKCRQDMSA-N floxuridine Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(F)=C1 ODKNJVUHOIMIIZ-RRKCRQDMSA-N 0.000 description 2
- 229960000390 fludarabine Drugs 0.000 description 2
- GIUYCYHIANZCFB-FJFJXFQQSA-N fludarabine phosphate Chemical compound C1=NC=2C(N)=NC(F)=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@@H]1O GIUYCYHIANZCFB-FJFJXFQQSA-N 0.000 description 2
- 229960002074 flutamide Drugs 0.000 description 2
- MKXKFYHWDHIYRV-UHFFFAOYSA-N flutamide Chemical compound CC(C)C(=O)NC1=CC=C([N+]([O-])=O)C(C(F)(F)F)=C1 MKXKFYHWDHIYRV-UHFFFAOYSA-N 0.000 description 2
- 235000013355 food flavoring agent Nutrition 0.000 description 2
- 229960004421 formestane Drugs 0.000 description 2
- OSVMTWJCGUFAOD-KZQROQTASA-N formestane Chemical compound O=C1CC[C@]2(C)[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CCC2=C1O OSVMTWJCGUFAOD-KZQROQTASA-N 0.000 description 2
- 238000004108 freeze drying Methods 0.000 description 2
- 229960002258 fulvestrant Drugs 0.000 description 2
- 229960005277 gemcitabine Drugs 0.000 description 2
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- 229960003607 granisetron hydrochloride Drugs 0.000 description 2
- 238000005469 granulation Methods 0.000 description 2
- 230000003179 granulation Effects 0.000 description 2
- 238000000227 grinding Methods 0.000 description 2
- 230000012010 growth Effects 0.000 description 2
- 230000002489 hematologic effect Effects 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- 238000009474 hot melt extrusion Methods 0.000 description 2
- QYZRTBKYBJRGJB-UHFFFAOYSA-N hydron;1-methyl-n-(9-methyl-9-azabicyclo[3.3.1]nonan-3-yl)indazole-3-carboxamide;chloride Chemical compound Cl.C1=CC=C2C(C(=O)NC3CC4CCCC(C3)N4C)=NN(C)C2=C1 QYZRTBKYBJRGJB-UHFFFAOYSA-N 0.000 description 2
- 230000002209 hydrophobic effect Effects 0.000 description 2
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 2
- 229960000908 idarubicin Drugs 0.000 description 2
- 229960001101 ifosfamide Drugs 0.000 description 2
- HOMGKSMUEGBAAB-UHFFFAOYSA-N ifosfamide Chemical compound ClCCNP1(=O)OCCCN1CCCl HOMGKSMUEGBAAB-UHFFFAOYSA-N 0.000 description 2
- 238000003384 imaging method Methods 0.000 description 2
- 229960002411 imatinib Drugs 0.000 description 2
- KTUFNOKKBVMGRW-UHFFFAOYSA-N imatinib Chemical compound C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 KTUFNOKKBVMGRW-UHFFFAOYSA-N 0.000 description 2
- 239000012729 immediate-release (IR) formulation Substances 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- 229960005280 isotretinoin Drugs 0.000 description 2
- 229960004891 lapatinib Drugs 0.000 description 2
- 229950005692 larotaxel Drugs 0.000 description 2
- SEFGUGYLLVNFIJ-QDRLFVHASA-N larotaxel dihydrate Chemical compound O.O.O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@@]23[C@H]1[C@@]1(CO[C@@H]1C[C@@H]2C3)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 SEFGUGYLLVNFIJ-QDRLFVHASA-N 0.000 description 2
- 229960002367 lasofoxifene Drugs 0.000 description 2
- GXESHMAMLJKROZ-IAPPQJPRSA-N lasofoxifene Chemical compound C1([C@@H]2[C@@H](C3=CC=C(C=C3CC2)O)C=2C=CC(OCCN3CCCC3)=CC=2)=CC=CC=C1 GXESHMAMLJKROZ-IAPPQJPRSA-N 0.000 description 2
- 229960004942 lenalidomide Drugs 0.000 description 2
- GOTYRUGSSMKFNF-UHFFFAOYSA-N lenalidomide Chemical compound C1C=2C(N)=CC=CC=2C(=O)N1C1CCC(=O)NC1=O GOTYRUGSSMKFNF-UHFFFAOYSA-N 0.000 description 2
- 229960003881 letrozole Drugs 0.000 description 2
- HPJKCIUCZWXJDR-UHFFFAOYSA-N letrozole Chemical compound C1=CC(C#N)=CC=C1C(N1N=CN=C1)C1=CC=C(C#N)C=C1 HPJKCIUCZWXJDR-UHFFFAOYSA-N 0.000 description 2
- 238000001294 liquid chromatography-tandem mass spectrometry Methods 0.000 description 2
- 229960002247 lomustine Drugs 0.000 description 2
- 229960004391 lorazepam Drugs 0.000 description 2
- PCZOHLXUXFIOCF-BXMDZJJMSA-N lovastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 PCZOHLXUXFIOCF-BXMDZJJMSA-N 0.000 description 2
- 229960004844 lovastatin Drugs 0.000 description 2
- QLJODMDSTUBWDW-UHFFFAOYSA-N lovastatin hydroxy acid Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(C)C=C21 QLJODMDSTUBWDW-UHFFFAOYSA-N 0.000 description 2
- 238000002595 magnetic resonance imaging Methods 0.000 description 2
- 239000000594 mannitol Substances 0.000 description 2
- 235000010355 mannitol Nutrition 0.000 description 2
- 235000012054 meals Nutrition 0.000 description 2
- 229960004961 mechlorethamine Drugs 0.000 description 2
- HAWPXGHAZFHHAD-UHFFFAOYSA-N mechlorethamine Chemical compound ClCCN(C)CCCl HAWPXGHAZFHHAD-UHFFFAOYSA-N 0.000 description 2
- 239000002609 medium Substances 0.000 description 2
- 229920000609 methyl cellulose Polymers 0.000 description 2
- 239000001923 methylcellulose Substances 0.000 description 2
- 235000010981 methylcellulose Nutrition 0.000 description 2
- 238000000386 microscopy Methods 0.000 description 2
- CFCUWKMKBJTWLW-BKHRDMLASA-N mithramycin Chemical compound O([C@@H]1C[C@@H](O[C@H](C)[C@H]1O)OC=1C=C2C=C3C[C@H]([C@@H](C(=O)C3=C(O)C2=C(O)C=1C)O[C@@H]1O[C@H](C)[C@@H](O)[C@H](O[C@@H]2O[C@H](C)[C@H](O)[C@H](O[C@@H]3O[C@H](C)[C@@H](O)[C@@](C)(O)C3)C2)C1)[C@H](OC)C(=O)[C@@H](O)[C@@H](C)O)[C@H]1C[C@@H](O)[C@H](O)[C@@H](C)O1 CFCUWKMKBJTWLW-BKHRDMLASA-N 0.000 description 2
- 229960004857 mitomycin Drugs 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 230000004001 molecular interaction Effects 0.000 description 2
- 239000004570 mortar (masonry) Substances 0.000 description 2
- 230000003232 mucoadhesive effect Effects 0.000 description 2
- YOHYSYJDKVYCJI-UHFFFAOYSA-N n-[3-[[6-[3-(trifluoromethyl)anilino]pyrimidin-4-yl]amino]phenyl]cyclopropanecarboxamide Chemical compound FC(F)(F)C1=CC=CC(NC=2N=CN=C(NC=3C=C(NC(=O)C4CC4)C=CC=3)C=2)=C1 YOHYSYJDKVYCJI-UHFFFAOYSA-N 0.000 description 2
- 229960002653 nilutamide Drugs 0.000 description 2
- XWXYUMMDTVBTOU-UHFFFAOYSA-N nilutamide Chemical compound O=C1C(C)(C)NC(=O)N1C1=CC=C([N+]([O-])=O)C(C(F)(F)F)=C1 XWXYUMMDTVBTOU-UHFFFAOYSA-N 0.000 description 2
- 229960001420 nimustine Drugs 0.000 description 2
- VFEDRRNHLBGPNN-UHFFFAOYSA-N nimustine Chemical compound CC1=NC=C(CNC(=O)N(CCCl)N=O)C(N)=N1 VFEDRRNHLBGPNN-UHFFFAOYSA-N 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- 239000002736 nonionic surfactant Substances 0.000 description 2
- 231100000252 nontoxic Toxicity 0.000 description 2
- 230000003000 nontoxic effect Effects 0.000 description 2
- 238000010606 normalization Methods 0.000 description 2
- 229960000770 ondansetron hydrochloride Drugs 0.000 description 2
- 229940126701 oral medication Drugs 0.000 description 2
- 239000003960 organic solvent Substances 0.000 description 2
- 229960001592 paclitaxel Drugs 0.000 description 2
- WRUUGTRCQOWXEG-UHFFFAOYSA-N pamidronate Chemical compound NCCC(O)(P(O)(O)=O)P(O)(O)=O WRUUGTRCQOWXEG-UHFFFAOYSA-N 0.000 description 2
- 229940046231 pamidronate Drugs 0.000 description 2
- 229960001972 panitumumab Drugs 0.000 description 2
- 230000036961 partial effect Effects 0.000 description 2
- 239000008188 pellet Substances 0.000 description 2
- 229960002340 pentostatin Drugs 0.000 description 2
- FPVKHBSQESCIEP-JQCXWYLXSA-N pentostatin Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(N=CNC[C@H]2O)=C2N=C1 FPVKHBSQESCIEP-JQCXWYLXSA-N 0.000 description 2
- 239000000049 pigment Substances 0.000 description 2
- 229960001221 pirarubicin Drugs 0.000 description 2
- 229960003073 pirfenidone Drugs 0.000 description 2
- ISWRGOKTTBVCFA-UHFFFAOYSA-N pirfenidone Chemical compound C1=C(C)C=CC(=O)N1C1=CC=CC=C1 ISWRGOKTTBVCFA-UHFFFAOYSA-N 0.000 description 2
- 239000004014 plasticizer Substances 0.000 description 2
- 229960003171 plicamycin Drugs 0.000 description 2
- 229920001155 polypropylene Polymers 0.000 description 2
- 229920000136 polysorbate Polymers 0.000 description 2
- 229960000688 pomalidomide Drugs 0.000 description 2
- UVSMNLNDYGZFPF-UHFFFAOYSA-N pomalidomide Chemical compound O=C1C=2C(N)=CC=CC=2C(=O)N1C1CCC(=O)NC1=O UVSMNLNDYGZFPF-UHFFFAOYSA-N 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- 238000009163 protein therapy Methods 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- 108090000623 proteins and genes Proteins 0.000 description 2
- 229960004622 raloxifene Drugs 0.000 description 2
- GZUITABIAKMVPG-UHFFFAOYSA-N raloxifene Chemical compound C1=CC(O)=CC=C1C1=C(C(=O)C=2C=CC(OCCN3CCCCC3)=CC=2)C2=CC=C(O)C=C2S1 GZUITABIAKMVPG-UHFFFAOYSA-N 0.000 description 2
- 229960002633 ramucirumab Drugs 0.000 description 2
- 230000003134 recirculating effect Effects 0.000 description 2
- 229960001302 ridaforolimus Drugs 0.000 description 2
- 229940089617 risedronate Drugs 0.000 description 2
- 238000009490 roller compaction Methods 0.000 description 2
- 229960004586 rosiglitazone Drugs 0.000 description 2
- 229960003440 semustine Drugs 0.000 description 2
- 235000012239 silicon dioxide Nutrition 0.000 description 2
- 238000004513 sizing Methods 0.000 description 2
- 239000002002 slurry Substances 0.000 description 2
- 235000010413 sodium alginate Nutrition 0.000 description 2
- 239000000661 sodium alginate Substances 0.000 description 2
- 229940005550 sodium alginate Drugs 0.000 description 2
- 235000002639 sodium chloride Nutrition 0.000 description 2
- 229940080264 sodium dodecylbenzenesulfonate Drugs 0.000 description 2
- 229920003109 sodium starch glycolate Polymers 0.000 description 2
- 239000008109 sodium starch glycolate Substances 0.000 description 2
- 229940079832 sodium starch glycolate Drugs 0.000 description 2
- WQQPDTLGLVLNOH-UHFFFAOYSA-M sodium;4-hydroxy-4-oxo-3-sulfobutanoate Chemical class [Na+].OC(=O)CC(C([O-])=O)S(O)(=O)=O WQQPDTLGLVLNOH-UHFFFAOYSA-M 0.000 description 2
- 238000000935 solvent evaporation Methods 0.000 description 2
- 239000011877 solvent mixture Substances 0.000 description 2
- 238000005563 spheronization Methods 0.000 description 2
- 230000006641 stabilisation Effects 0.000 description 2
- 238000011105 stabilization Methods 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 230000003637 steroidlike Effects 0.000 description 2
- 239000006228 supernatant Substances 0.000 description 2
- 239000000375 suspending agent Substances 0.000 description 2
- MODVSQKJJIBWPZ-VLLPJHQWSA-N tesetaxel Chemical compound O([C@H]1[C@@H]2[C@]3(OC(C)=O)CO[C@@H]3CC[C@@]2(C)[C@H]2[C@@H](C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C(=CC=CN=4)F)C[C@]1(O)C3(C)C)O[C@H](O2)CN(C)C)C(=O)C1=CC=CC=C1 MODVSQKJJIBWPZ-VLLPJHQWSA-N 0.000 description 2
- 229950009016 tesetaxel Drugs 0.000 description 2
- 229960005353 testolactone Drugs 0.000 description 2
- BPEWUONYVDABNZ-DZBHQSCQSA-N testolactone Chemical compound O=C1C=C[C@]2(C)[C@H]3CC[C@](C)(OC(=O)CC4)[C@@H]4[C@@H]3CCC2=C1 BPEWUONYVDABNZ-DZBHQSCQSA-N 0.000 description 2
- 229960003433 thalidomide Drugs 0.000 description 2
- 229960003087 tioguanine Drugs 0.000 description 2
- MNRILEROXIRVNJ-UHFFFAOYSA-N tioguanine Chemical compound N1C(N)=NC(=S)C2=NC=N[C]21 MNRILEROXIRVNJ-UHFFFAOYSA-N 0.000 description 2
- 229960005026 toremifene Drugs 0.000 description 2
- XFCLJVABOIYOMF-QPLCGJKRSA-N toremifene Chemical compound C1=CC(OCCN(C)C)=CC=C1C(\C=1C=CC=CC=1)=C(\CCCl)C1=CC=CC=C1 XFCLJVABOIYOMF-QPLCGJKRSA-N 0.000 description 2
- 231100000331 toxic Toxicity 0.000 description 2
- 230000002588 toxic effect Effects 0.000 description 2
- 238000012546 transfer Methods 0.000 description 2
- 229960000575 trastuzumab Drugs 0.000 description 2
- 229960001727 tretinoin Drugs 0.000 description 2
- 239000003029 tricyclic antidepressant agent Substances 0.000 description 2
- 229960000875 trofosfamide Drugs 0.000 description 2
- UMKFEPPTGMDVMI-UHFFFAOYSA-N trofosfamide Chemical compound ClCCN(CCCl)P1(=O)OCCCN1CCCl UMKFEPPTGMDVMI-UHFFFAOYSA-N 0.000 description 2
- 229940121358 tyrosine kinase inhibitor Drugs 0.000 description 2
- 239000005483 tyrosine kinase inhibitor Substances 0.000 description 2
- 238000002604 ultrasonography Methods 0.000 description 2
- 229960001055 uracil mustard Drugs 0.000 description 2
- 229960000653 valrubicin Drugs 0.000 description 2
- ZOCKGBMQLCSHFP-KQRAQHLDSA-N valrubicin Chemical compound O([C@H]1C[C@](CC2=C(O)C=3C(=O)C4=CC=CC(OC)=C4C(=O)C=3C(O)=C21)(O)C(=O)COC(=O)CCCC)[C@H]1C[C@H](NC(=O)C(F)(F)F)[C@H](O)[C@H](C)O1 ZOCKGBMQLCSHFP-KQRAQHLDSA-N 0.000 description 2
- 229960000241 vandetanib Drugs 0.000 description 2
- UHTHHESEBZOYNR-UHFFFAOYSA-N vandetanib Chemical compound COC1=CC(C(/N=CN2)=N/C=3C(=CC(Br)=CC=3)F)=C2C=C1OCC1CCN(C)CC1 UHTHHESEBZOYNR-UHFFFAOYSA-N 0.000 description 2
- 230000000007 visual effect Effects 0.000 description 2
- 238000009528 vital sign measurement Methods 0.000 description 2
- 229960001771 vorozole Drugs 0.000 description 2
- XLMPPFTZALNBFS-INIZCTEOSA-N vorozole Chemical compound C1([C@@H](C2=CC=C3N=NN(C3=C2)C)N2N=CN=C2)=CC=C(Cl)C=C1 XLMPPFTZALNBFS-INIZCTEOSA-N 0.000 description 2
- 230000004580 weight loss Effects 0.000 description 2
- 239000000080 wetting agent Substances 0.000 description 2
- 229960000641 zorubicin Drugs 0.000 description 2
- FBTUMDXHSRTGRV-ALTNURHMSA-N zorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(\C)=N\NC(=O)C=1C=CC=CC=1)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 FBTUMDXHSRTGRV-ALTNURHMSA-N 0.000 description 2
- NOOLISFMXDJSKH-UTLUCORTSA-N (+)-Neomenthol Chemical compound CC(C)[C@@H]1CC[C@@H](C)C[C@@H]1O NOOLISFMXDJSKH-UTLUCORTSA-N 0.000 description 1
- JNYAEWCLZODPBN-JGWLITMVSA-N (2r,3r,4s)-2-[(1r)-1,2-dihydroxyethyl]oxolane-3,4-diol Chemical compound OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O JNYAEWCLZODPBN-JGWLITMVSA-N 0.000 description 1
- IDXCXSCCZNCXCL-XMADEQCMSA-N (2s)-2-[[(2s)-2-[[(2s)-1-[(2s)-2-[[(2s)-2-[[2-[[(2s,4r)-1-[(2s)-1-[(2s)-2-amino-5-(diaminomethylideneamino)pentanoyl]pyrrolidine-2-carbonyl]-4-hydroxypyrrolidine-2-carbonyl]amino]acetyl]amino]-3-thiophen-2-ylpropanoyl]amino]-3-hydroxypropanoyl]pyrrolidine Chemical compound C1=CC(OC)=CC=C1C[C@@H](CN[C@@H](CCCN=C(N)N)C(O)=O)NC(=O)[C@H]1N(C(=O)[C@H](CO)NC(=O)[C@H](CC=2SC=CC=2)NC(=O)CNC(=O)[C@H]2N(C[C@H](O)C2)C(=O)[C@H]2N(CCC2)C(=O)[C@@H](N)CCCN=C(N)N)CCC1 IDXCXSCCZNCXCL-XMADEQCMSA-N 0.000 description 1
- OQANPHBRHBJGNZ-FYJGNVAPSA-N (3e)-6-oxo-3-[[4-(pyridin-2-ylsulfamoyl)phenyl]hydrazinylidene]cyclohexa-1,4-diene-1-carboxylic acid Chemical compound C1=CC(=O)C(C(=O)O)=C\C1=N\NC1=CC=C(S(=O)(=O)NC=2N=CC=CC=2)C=C1 OQANPHBRHBJGNZ-FYJGNVAPSA-N 0.000 description 1
- VHZPUDNSVGRVMB-RXDLHWJPSA-N (8s,11r,13s,14s,17s)-11-(4-acetylphenyl)-17-hydroxy-13-methyl-17-(1,1,2,2,2-pentafluoroethyl)-1,2,6,7,8,11,12,14,15,16-decahydrocyclopenta[a]phenanthren-3-one Chemical compound C1=CC(C(=O)C)=CC=C1[C@@H]1C2=C3CCC(=O)C=C3CC[C@H]2[C@H](CC[C@@]2(O)C(F)(F)C(F)(F)F)[C@]2(C)C1 VHZPUDNSVGRVMB-RXDLHWJPSA-N 0.000 description 1
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 1
- NWUYHJFMYQTDRP-UHFFFAOYSA-N 1,2-bis(ethenyl)benzene;1-ethenyl-2-ethylbenzene;styrene Chemical compound C=CC1=CC=CC=C1.CCC1=CC=CC=C1C=C.C=CC1=CC=CC=C1C=C NWUYHJFMYQTDRP-UHFFFAOYSA-N 0.000 description 1
- OKMWKBLSFKFYGZ-UHFFFAOYSA-N 1-behenoylglycerol Chemical compound CCCCCCCCCCCCCCCCCCCCCC(=O)OCC(O)CO OKMWKBLSFKFYGZ-UHFFFAOYSA-N 0.000 description 1
- FDCJDKXCCYFOCV-UHFFFAOYSA-N 1-hexadecoxyhexadecane Chemical compound CCCCCCCCCCCCCCCCOCCCCCCCCCCCCCCCC FDCJDKXCCYFOCV-UHFFFAOYSA-N 0.000 description 1
- SMZOUWXMTYCWNB-UHFFFAOYSA-N 2-(2-methoxy-5-methylphenyl)ethanamine Chemical compound COC1=CC=C(C)C=C1CCN SMZOUWXMTYCWNB-UHFFFAOYSA-N 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-N 2-Propenoic acid Natural products OC(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 1
- POAOYUHQDCAZBD-UHFFFAOYSA-N 2-butoxyethanol Chemical compound CCCCOCCO POAOYUHQDCAZBD-UHFFFAOYSA-N 0.000 description 1
- CTXGTHVAWRBISV-UHFFFAOYSA-N 2-hydroxyethyl dodecanoate Chemical compound CCCCCCCCCCCC(=O)OCCO CTXGTHVAWRBISV-UHFFFAOYSA-N 0.000 description 1
- RFVNOJDQRGSOEL-UHFFFAOYSA-N 2-hydroxyethyl octadecanoate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCCO RFVNOJDQRGSOEL-UHFFFAOYSA-N 0.000 description 1
- VCNPGCHIKPSUSP-UHFFFAOYSA-N 2-hydroxypropyl tetradecanoate Chemical compound CCCCCCCCCCCCCC(=O)OCC(C)O VCNPGCHIKPSUSP-UHFFFAOYSA-N 0.000 description 1
- NDMPLJNOPCLANR-UHFFFAOYSA-N 3,4-dihydroxy-15-(4-hydroxy-18-methoxycarbonyl-5,18-seco-ibogamin-18-yl)-16-methoxy-1-methyl-6,7-didehydro-aspidospermidine-3-carboxylic acid methyl ester Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 NDMPLJNOPCLANR-UHFFFAOYSA-N 0.000 description 1
- TVZGACDUOSZQKY-LBPRGKRZSA-N 4-aminofolic acid Chemical compound C1=NC2=NC(N)=NC(N)=C2N=C1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 TVZGACDUOSZQKY-LBPRGKRZSA-N 0.000 description 1
- LGZKGOGODCLQHG-CYBMUJFWSA-N 5-[(2r)-2-hydroxy-2-(3,4,5-trimethoxyphenyl)ethyl]-2-methoxyphenol Chemical compound C1=C(O)C(OC)=CC=C1C[C@@H](O)C1=CC(OC)=C(OC)C(OC)=C1 LGZKGOGODCLQHG-CYBMUJFWSA-N 0.000 description 1
- RYYCJUAHISIHTL-UHFFFAOYSA-N 5-azaorotic acid Chemical compound OC(=O)C1=NC(=O)NC(=O)N1 RYYCJUAHISIHTL-UHFFFAOYSA-N 0.000 description 1
- ZGMLACURZVGTPK-UHFFFAOYSA-N 5-fluoranyl-1h-pyrimidine-2,4-dione Chemical compound OC1=NC=C(F)C(O)=N1.FC1=CNC(=O)NC1=O ZGMLACURZVGTPK-UHFFFAOYSA-N 0.000 description 1
- VVIAGPKUTFNRDU-UHFFFAOYSA-N 6S-folinic acid Natural products C1NC=2NC(N)=NC(=O)C=2N(C=O)C1CNC1=CC=C(C(=O)NC(CCC(O)=O)C(O)=O)C=C1 VVIAGPKUTFNRDU-UHFFFAOYSA-N 0.000 description 1
- WLCZTRVUXYALDD-IBGZPJMESA-N 7-[[(2s)-2,6-bis(2-methoxyethoxycarbonylamino)hexanoyl]amino]heptoxy-methylphosphinic acid Chemical compound COCCOC(=O)NCCCC[C@H](NC(=O)OCCOC)C(=O)NCCCCCCCOP(C)(O)=O WLCZTRVUXYALDD-IBGZPJMESA-N 0.000 description 1
- 208000030507 AIDS Diseases 0.000 description 1
- 206010000060 Abdominal distension Diseases 0.000 description 1
- 206010067484 Adverse reaction Diseases 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- 102100032187 Androgen receptor Human genes 0.000 description 1
- YUWPMEXLKGOSBF-GACAOOTBSA-N Anecortave acetate Chemical compound O=C1CC[C@]2(C)C3=CC[C@]4(C)[C@](C(=O)COC(=O)C)(O)CC[C@H]4[C@@H]3CCC2=C1 YUWPMEXLKGOSBF-GACAOOTBSA-N 0.000 description 1
- 206010002388 Angina unstable Diseases 0.000 description 1
- 108091023037 Aptamer Proteins 0.000 description 1
- BFYIZQONLCFLEV-DAELLWKTSA-N Aromasine Chemical compound O=C1C=C[C@]2(C)[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CC(=C)C2=C1 BFYIZQONLCFLEV-DAELLWKTSA-N 0.000 description 1
- 206010003178 Arterial thrombosis Diseases 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 206010003591 Ataxia Diseases 0.000 description 1
- 238000012935 Averaging Methods 0.000 description 1
- 208000008035 Back Pain Diseases 0.000 description 1
- 208000035143 Bacterial infection Diseases 0.000 description 1
- 229940122361 Bisphosphonate Drugs 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- WDNDFFXELCXYOB-UHFFFAOYSA-L CCOS(OC(C(C(=O)[O-])S(=O)(=O)O)(C(=O)[O-])OS(=O)(=O)OCC)(=O)=O.[Na+].[Na+] Chemical compound CCOS(OC(C(C(=O)[O-])S(=O)(=O)O)(C(=O)[O-])OS(=O)(=O)OCC)(=O)=O.[Na+].[Na+] WDNDFFXELCXYOB-UHFFFAOYSA-L 0.000 description 1
- 101100223811 Caenorhabditis elegans dsc-1 gene Proteins 0.000 description 1
- 101100123850 Caenorhabditis elegans her-1 gene Proteins 0.000 description 1
- 241000759909 Camptotheca Species 0.000 description 1
- GAGWJHPBXLXJQN-UORFTKCHSA-N Capecitabine Chemical compound C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1[C@H]1[C@H](O)[C@H](O)[C@@H](C)O1 GAGWJHPBXLXJQN-UORFTKCHSA-N 0.000 description 1
- GAGWJHPBXLXJQN-UHFFFAOYSA-N Capecitabine Natural products C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1C1C(O)C(O)C(C)O1 GAGWJHPBXLXJQN-UHFFFAOYSA-N 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- 206010007559 Cardiac failure congestive Diseases 0.000 description 1
- 241000700199 Cavia porcellus Species 0.000 description 1
- ZEOWTGPWHLSLOG-UHFFFAOYSA-N Cc1ccc(cc1-c1ccc2c(n[nH]c2c1)-c1cnn(c1)C1CC1)C(=O)Nc1cccc(c1)C(F)(F)F Chemical compound Cc1ccc(cc1-c1ccc2c(n[nH]c2c1)-c1cnn(c1)C1CC1)C(=O)Nc1cccc(c1)C(F)(F)F ZEOWTGPWHLSLOG-UHFFFAOYSA-N 0.000 description 1
- 229920000623 Cellulose acetate phthalate Polymers 0.000 description 1
- PTHCMJGKKRQCBF-UHFFFAOYSA-N Cellulose, microcrystalline Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC)C(CO)O1 PTHCMJGKKRQCBF-UHFFFAOYSA-N 0.000 description 1
- 206010008190 Cerebrovascular accident Diseases 0.000 description 1
- LZZYPRNAOMGNLH-UHFFFAOYSA-M Cetrimonium bromide Chemical compound [Br-].CCCCCCCCCCCCCCCC[N+](C)(C)C LZZYPRNAOMGNLH-UHFFFAOYSA-M 0.000 description 1
- PTOAARAWEBMLNO-KVQBGUIXSA-N Cladribine Chemical compound C1=NC=2C(N)=NC(Cl)=NC=2N1[C@H]1C[C@H](O)[C@@H](CO)O1 PTOAARAWEBMLNO-KVQBGUIXSA-N 0.000 description 1
- 235000013162 Cocos nucifera Nutrition 0.000 description 1
- 244000060011 Cocos nucifera Species 0.000 description 1
- 102000008186 Collagen Human genes 0.000 description 1
- 108010035532 Collagen Proteins 0.000 description 1
- 206010011224 Cough Diseases 0.000 description 1
- 241000699800 Cricetinae Species 0.000 description 1
- QASFUMOKHFSJGL-LAFRSMQTSA-N Cyclopamine Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H](CC2=C3C)[C@@H]1[C@@H]2CC[C@@]13O[C@@H]2C[C@H](C)CN[C@H]2[C@H]1C QASFUMOKHFSJGL-LAFRSMQTSA-N 0.000 description 1
- 108010001237 Cytochrome P-450 CYP2D6 Proteins 0.000 description 1
- NOOLISFMXDJSKH-UHFFFAOYSA-N DL-menthol Natural products CC(C)C1CCC(C)CC1O NOOLISFMXDJSKH-UHFFFAOYSA-N 0.000 description 1
- 239000012623 DNA damaging agent Substances 0.000 description 1
- 102100021202 Desmocollin-1 Human genes 0.000 description 1
- 235000019739 Dicalciumphosphate Nutrition 0.000 description 1
- 206010013952 Dysphonia Diseases 0.000 description 1
- 229940122558 EGFR antagonist Drugs 0.000 description 1
- 208000005189 Embolism Diseases 0.000 description 1
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 1
- HKVAMNSJSFKALM-GKUWKFKPSA-N Everolimus Chemical compound C1C[C@@H](OCCO)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 HKVAMNSJSFKALM-GKUWKFKPSA-N 0.000 description 1
- 208000010201 Exanthema Diseases 0.000 description 1
- 102100023593 Fibroblast growth factor receptor 1 Human genes 0.000 description 1
- 101710182386 Fibroblast growth factor receptor 1 Proteins 0.000 description 1
- 108010029961 Filgrastim Proteins 0.000 description 1
- MPJKWIXIYCLVCU-UHFFFAOYSA-N Folinic acid Natural products NC1=NC2=C(N(C=O)C(CNc3ccc(cc3)C(=O)NC(CCC(=O)O)CC(=O)O)CN2)C(=O)N1 MPJKWIXIYCLVCU-UHFFFAOYSA-N 0.000 description 1
- 102100021022 Gastrin Human genes 0.000 description 1
- 108010052343 Gastrins Proteins 0.000 description 1
- 244000060234 Gmelina philippensis Species 0.000 description 1
- 239000000579 Gonadotropin-Releasing Hormone Substances 0.000 description 1
- 108010017080 Granulocyte Colony-Stimulating Factor Proteins 0.000 description 1
- 229940127403 HER1 Antagonists Drugs 0.000 description 1
- 208000031886 HIV Infections Diseases 0.000 description 1
- 208000037357 HIV infectious disease Diseases 0.000 description 1
- 229940121710 HMGCoA reductase inhibitor Drugs 0.000 description 1
- 208000010473 Hoarseness Diseases 0.000 description 1
- 101000968043 Homo sapiens Desmocollin-1 Proteins 0.000 description 1
- 101000880960 Homo sapiens Desmocollin-3 Proteins 0.000 description 1
- 101000746367 Homo sapiens Granulocyte colony-stimulating factor Proteins 0.000 description 1
- 101001012157 Homo sapiens Receptor tyrosine-protein kinase erbB-2 Proteins 0.000 description 1
- 101000851007 Homo sapiens Vascular endothelial growth factor receptor 2 Proteins 0.000 description 1
- VSNHCAURESNICA-UHFFFAOYSA-N Hydroxyurea Chemical compound NC(=O)NO VSNHCAURESNICA-UHFFFAOYSA-N 0.000 description 1
- 206010061598 Immunodeficiency Diseases 0.000 description 1
- 102100032817 Integrin alpha-5 Human genes 0.000 description 1
- 108010041014 Integrin alpha5 Proteins 0.000 description 1
- 102000014150 Interferons Human genes 0.000 description 1
- 108010050904 Interferons Proteins 0.000 description 1
- UETNIIAIRMUTSM-UHFFFAOYSA-N Jacareubin Natural products CC1(C)OC2=CC3Oc4c(O)c(O)ccc4C(=O)C3C(=C2C=C1)O UETNIIAIRMUTSM-UHFFFAOYSA-N 0.000 description 1
- 229940121730 Janus kinase 2 inhibitor Drugs 0.000 description 1
- 238000003109 Karl Fischer titration Methods 0.000 description 1
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 1
- 239000005551 L01XE03 - Erlotinib Substances 0.000 description 1
- UIARLYUEJFELEN-LROUJFHJSA-N LSM-1231 Chemical compound C12=C3N4C5=CC=CC=C5C3=C3C(=O)NCC3=C2C2=CC=CC=C2N1[C@]1(C)[C@](CO)(O)C[C@H]4O1 UIARLYUEJFELEN-LROUJFHJSA-N 0.000 description 1
- 208000034800 Leukoencephalopathies Diseases 0.000 description 1
- 108010000817 Leuprolide Proteins 0.000 description 1
- 239000000867 Lipoxygenase Inhibitor Substances 0.000 description 1
- 206010027476 Metastases Diseases 0.000 description 1
- CERQOIWHTDAKMF-UHFFFAOYSA-N Methacrylic acid Chemical compound CC(=C)C(O)=O CERQOIWHTDAKMF-UHFFFAOYSA-N 0.000 description 1
- DUGOZIWVEXMGBE-UHFFFAOYSA-N Methylphenidate Chemical compound C=1C=CC=CC=1C(C(=O)OC)C1CCCCN1 DUGOZIWVEXMGBE-UHFFFAOYSA-N 0.000 description 1
- VFKZTMPDYBFSTM-KVTDHHQDSA-N Mitobronitol Chemical compound BrC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CBr VFKZTMPDYBFSTM-KVTDHHQDSA-N 0.000 description 1
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 1
- PAWIYAYFNXQGAP-UHFFFAOYSA-N N-hydroxy-2-[4-[[(1-methyl-3-indolyl)methylamino]methyl]-1-piperidinyl]-5-pyrimidinecarboxamide Chemical compound C12=CC=CC=C2N(C)C=C1CNCC(CC1)CCN1C1=NC=C(C(=O)NO)C=N1 PAWIYAYFNXQGAP-UHFFFAOYSA-N 0.000 description 1
- 208000003420 Nasal Septal Perforation Diseases 0.000 description 1
- 206010028735 Nasal congestion Diseases 0.000 description 1
- 239000004677 Nylon Substances 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 108091000080 Phosphotransferase Proteins 0.000 description 1
- 102000018967 Platelet-Derived Growth Factor beta Receptor Human genes 0.000 description 1
- 108010051742 Platelet-Derived Growth Factor beta Receptor Proteins 0.000 description 1
- 229940127354 Platelet-derived Growth Factor Receptor Inhibitors Drugs 0.000 description 1
- 241001495452 Podophyllum Species 0.000 description 1
- 229920002845 Poly(methacrylic acid) Polymers 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- HFVNWDWLWUCIHC-GUPDPFMOSA-N Prednimustine Chemical compound O=C([C@@]1(O)CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)[C@@H](O)C[C@@]21C)COC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 HFVNWDWLWUCIHC-GUPDPFMOSA-N 0.000 description 1
- 229940079156 Proteasome inhibitor Drugs 0.000 description 1
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 description 1
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 description 1
- 208000010378 Pulmonary Embolism Diseases 0.000 description 1
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 1
- 108700031422 RMP 7 Proteins 0.000 description 1
- AHHFEZNOXOZZQA-ZEBDFXRSSA-N Ranimustine Chemical compound CO[C@H]1O[C@H](CNC(=O)N(CCCl)N=O)[C@@H](O)[C@H](O)[C@H]1O AHHFEZNOXOZZQA-ZEBDFXRSSA-N 0.000 description 1
- 101100288143 Rattus norvegicus Klkb1 gene Proteins 0.000 description 1
- 229940127361 Receptor Tyrosine Kinase Inhibitors Drugs 0.000 description 1
- 102100030086 Receptor tyrosine-protein kinase erbB-2 Human genes 0.000 description 1
- DWAQJAXMDSEUJJ-UHFFFAOYSA-M Sodium bisulfite Chemical compound [Na+].OS([O-])=O DWAQJAXMDSEUJJ-UHFFFAOYSA-M 0.000 description 1
- 102000005157 Somatostatin Human genes 0.000 description 1
- 108010056088 Somatostatin Proteins 0.000 description 1
- 101000857870 Squalus acanthias Gonadoliberin Proteins 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 208000007271 Substance Withdrawal Syndrome Diseases 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 229940123237 Taxane Drugs 0.000 description 1
- BPEGJWRSRHCHSN-UHFFFAOYSA-N Temozolomide Chemical compound O=C1N(C)N=NC2=C(C(N)=O)N=CN21 BPEGJWRSRHCHSN-UHFFFAOYSA-N 0.000 description 1
- 208000009205 Tinnitus Diseases 0.000 description 1
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 1
- IVTVGDXNLFLDRM-HNNXBMFYSA-N Tomudex Chemical compound C=1C=C2NC(C)=NC(=O)C2=CC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)S1 IVTVGDXNLFLDRM-HNNXBMFYSA-N 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- 102000004887 Transforming Growth Factor beta Human genes 0.000 description 1
- 108090001012 Transforming Growth Factor beta Proteins 0.000 description 1
- 208000032109 Transient ischaemic attack Diseases 0.000 description 1
- 208000007814 Unstable Angina Diseases 0.000 description 1
- 102000009524 Vascular Endothelial Growth Factor A Human genes 0.000 description 1
- 108010073929 Vascular Endothelial Growth Factor A Proteins 0.000 description 1
- 102000016549 Vascular Endothelial Growth Factor Receptor-2 Human genes 0.000 description 1
- 206010047249 Venous thrombosis Diseases 0.000 description 1
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 1
- 229940122803 Vinca alkaloid Drugs 0.000 description 1
- 206010047700 Vomiting Diseases 0.000 description 1
- JMGXJHWTVBGOKG-UHFFFAOYSA-N [4-chloro-3-[5-methyl-3-[4-(2-pyrrolidin-1-ylethoxy)anilino]-1,2,4-benzotriazin-7-yl]phenyl] benzoate Chemical compound N1=C2C(C)=CC(C=3C(=CC=C(OC(=O)C=4C=CC=CC=4)C=3)Cl)=CC2=NN=C1NC(C=C1)=CC=C1OCCN1CCCC1 JMGXJHWTVBGOKG-UHFFFAOYSA-N 0.000 description 1
- 206010000059 abdominal discomfort Diseases 0.000 description 1
- GZOSMCIZMLWJML-VJLLXTKPSA-N abiraterone Chemical compound C([C@H]1[C@H]2[C@@H]([C@]3(CC[C@H](O)CC3=CC2)C)CC[C@@]11C)C=C1C1=CC=CN=C1 GZOSMCIZMLWJML-VJLLXTKPSA-N 0.000 description 1
- 229960000853 abiraterone Drugs 0.000 description 1
- 230000005856 abnormality Effects 0.000 description 1
- 229940028652 abraxane Drugs 0.000 description 1
- 239000002250 absorbent Substances 0.000 description 1
- 230000002745 absorbent Effects 0.000 description 1
- 230000001133 acceleration Effects 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 229920006243 acrylic copolymer Polymers 0.000 description 1
- 239000008186 active pharmaceutical agent Substances 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 230000006838 adverse reaction Effects 0.000 description 1
- 229950003105 afimoxifene Drugs 0.000 description 1
- TXUZVZSFRXZGTL-QPLCGJKRSA-N afimoxifene Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=C(O)C=C1 TXUZVZSFRXZGTL-QPLCGJKRSA-N 0.000 description 1
- 229960002833 aflibercept Drugs 0.000 description 1
- 108010081667 aflibercept Proteins 0.000 description 1
- 238000005054 agglomeration Methods 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 125000000217 alkyl group Chemical group 0.000 description 1
- 150000008051 alkyl sulfates Chemical class 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 229960000473 altretamine Drugs 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- CEGOLXSVJUTHNZ-UHFFFAOYSA-K aluminium tristearate Chemical compound [Al+3].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CEGOLXSVJUTHNZ-UHFFFAOYSA-K 0.000 description 1
- SNAAJJQQZSMGQD-UHFFFAOYSA-N aluminum magnesium Chemical compound [Mg].[Al] SNAAJJQQZSMGQD-UHFFFAOYSA-N 0.000 description 1
- 229940063655 aluminum stearate Drugs 0.000 description 1
- 230000003569 amebicidal effect Effects 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 229920003144 amino alkyl methacrylate copolymer Polymers 0.000 description 1
- 229960003896 aminopterin Drugs 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 229960001694 anagrelide Drugs 0.000 description 1
- OTBXOEAOVRKTNQ-UHFFFAOYSA-N anagrelide Chemical compound N1=C2NC(=O)CN2CC2=C(Cl)C(Cl)=CC=C21 OTBXOEAOVRKTNQ-UHFFFAOYSA-N 0.000 description 1
- 239000002269 analeptic agent Substances 0.000 description 1
- 108010080146 androgen receptors Proteins 0.000 description 1
- 229960001232 anecortave Drugs 0.000 description 1
- 230000006481 angiogenic pathway Effects 0.000 description 1
- 125000000129 anionic group Chemical group 0.000 description 1
- 208000022531 anorexia Diseases 0.000 description 1
- RGHILYZRVFRRNK-UHFFFAOYSA-N anthracene-1,2-dione Chemical class C1=CC=C2C=C(C(C(=O)C=C3)=O)C3=CC2=C1 RGHILYZRVFRRNK-UHFFFAOYSA-N 0.000 description 1
- 229940045799 anthracyclines and related substance Drugs 0.000 description 1
- 230000003474 anti-emetic effect Effects 0.000 description 1
- 230000003510 anti-fibrotic effect Effects 0.000 description 1
- 229940121363 anti-inflammatory agent Drugs 0.000 description 1
- 239000002260 anti-inflammatory agent Substances 0.000 description 1
- 230000000078 anti-malarial effect Effects 0.000 description 1
- 239000003472 antidiabetic agent Substances 0.000 description 1
- 229940125708 antidiabetic agent Drugs 0.000 description 1
- 229940125683 antiemetic agent Drugs 0.000 description 1
- 239000002111 antiemetic agent Substances 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 239000003429 antifungal agent Substances 0.000 description 1
- 239000003430 antimalarial agent Substances 0.000 description 1
- 229940027983 antiseptic and disinfectant quaternary ammonium compound Drugs 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- 210000001367 artery Anatomy 0.000 description 1
- 230000001174 ascending effect Effects 0.000 description 1
- 238000000376 autoradiography Methods 0.000 description 1
- KLNFSAOEKUDMFA-UHFFFAOYSA-N azanide;2-hydroxyacetic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OCC(O)=O KLNFSAOEKUDMFA-UHFFFAOYSA-N 0.000 description 1
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 1
- 208000022362 bacterial infectious disease Diseases 0.000 description 1
- 231100001124 band 1 compound Toxicity 0.000 description 1
- 231100001126 band 3 compound Toxicity 0.000 description 1
- 231100001127 band 4 compound Toxicity 0.000 description 1
- 229960000686 benzalkonium chloride Drugs 0.000 description 1
- UREZNYTWGJKWBI-UHFFFAOYSA-M benzethonium chloride Chemical compound [Cl-].C1=CC(C(C)(C)CC(C)(C)C)=CC=C1OCCOCC[N+](C)(C)CC1=CC=CC=C1 UREZNYTWGJKWBI-UHFFFAOYSA-M 0.000 description 1
- 229960001950 benzethonium chloride Drugs 0.000 description 1
- CADWTSSKOVRVJC-UHFFFAOYSA-N benzyl(dimethyl)azanium;chloride Chemical compound [Cl-].C[NH+](C)CC1=CC=CC=C1 CADWTSSKOVRVJC-UHFFFAOYSA-N 0.000 description 1
- 229960000397 bevacizumab Drugs 0.000 description 1
- 210000000941 bile Anatomy 0.000 description 1
- 239000003833 bile salt Substances 0.000 description 1
- 229940093761 bile salts Drugs 0.000 description 1
- 230000000975 bioactive effect Effects 0.000 description 1
- 239000000090 biomarker Substances 0.000 description 1
- 229960000074 biopharmaceutical Drugs 0.000 description 1
- FSWNJZRQQVVVJT-UHFFFAOYSA-L bis(sulfanylidene)molybdenum;2-hydroxyethyl(trimethyl)azanium;sulfanide Chemical compound [SH-].[SH-].S=[Mo]=S.C[N+](C)(C)CCO.C[N+](C)(C)CCO FSWNJZRQQVVVJT-UHFFFAOYSA-L 0.000 description 1
- 150000004663 bisphosphonates Chemical class 0.000 description 1
- 208000034158 bleeding Diseases 0.000 description 1
- 230000008499 blood brain barrier function Effects 0.000 description 1
- 210000001218 blood-brain barrier Anatomy 0.000 description 1
- 210000000988 bone and bone Anatomy 0.000 description 1
- 210000001185 bone marrow Anatomy 0.000 description 1
- 235000021152 breakfast Nutrition 0.000 description 1
- 239000007853 buffer solution Substances 0.000 description 1
- 229960002092 busulfan Drugs 0.000 description 1
- VTYYLEPIZMXCLO-UHFFFAOYSA-L calcium carbonate Substances [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- 235000008207 calcium folinate Nutrition 0.000 description 1
- 239000011687 calcium folinate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- CJZGTCYPCWQAJB-UHFFFAOYSA-L calcium stearate Chemical compound [Ca+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CJZGTCYPCWQAJB-UHFFFAOYSA-L 0.000 description 1
- 235000013539 calcium stearate Nutrition 0.000 description 1
- 239000008116 calcium stearate Substances 0.000 description 1
- 230000000711 cancerogenic effect Effects 0.000 description 1
- 229960004117 capecitabine Drugs 0.000 description 1
- 229960004562 carboplatin Drugs 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 150000007942 carboxylates Chemical class 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 231100000315 carcinogenic Toxicity 0.000 description 1
- 229940082638 cardiac stimulant phosphodiesterase inhibitors Drugs 0.000 description 1
- 230000007211 cardiovascular event Effects 0.000 description 1
- 125000002091 cationic group Chemical group 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 229940081734 cellulose acetate phthalate Drugs 0.000 description 1
- 208000026106 cerebrovascular disease Diseases 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- AOXOCDRNSPFDPE-UKEONUMOSA-N chembl413654 Chemical compound C([C@H](C(=O)NCC(=O)N[C@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@H](CCSC)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC=1C=CC=CC=1)C(N)=O)NC(=O)[C@@H](C)NC(=O)[C@@H](CCC(O)=O)NC(=O)[C@@H](CCC(O)=O)NC(=O)[C@@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H]1N(CCC1)C(=O)CNC(=O)[C@@H](N)CCC(O)=O)C1=CC=C(O)C=C1 AOXOCDRNSPFDPE-UKEONUMOSA-N 0.000 description 1
- 229950009003 cilengitide Drugs 0.000 description 1
- AMLYAMJWYAIXIA-VWNVYAMZSA-N cilengitide Chemical compound N1C(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](C(C)C)N(C)C(=O)[C@H]1CC1=CC=CC=C1 AMLYAMJWYAIXIA-VWNVYAMZSA-N 0.000 description 1
- 230000004087 circulation Effects 0.000 description 1
- 229960004316 cisplatin Drugs 0.000 description 1
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 1
- 229960002436 cladribine Drugs 0.000 description 1
- 238000005354 coacervation Methods 0.000 description 1
- 229920001436 collagen Polymers 0.000 description 1
- LGZKGOGODCLQHG-UHFFFAOYSA-N combretastatin Natural products C1=C(O)C(OC)=CC=C1CC(O)C1=CC(OC)=C(OC)C(OC)=C1 LGZKGOGODCLQHG-UHFFFAOYSA-N 0.000 description 1
- 238000010954 commercial manufacturing process Methods 0.000 description 1
- 238000010668 complexation reaction Methods 0.000 description 1
- 229940125782 compound 2 Drugs 0.000 description 1
- 230000001010 compromised effect Effects 0.000 description 1
- 239000012050 conventional carrier Substances 0.000 description 1
- 229920006037 cross link polymer Polymers 0.000 description 1
- 239000001767 crosslinked sodium carboxy methyl cellulose Substances 0.000 description 1
- QASFUMOKHFSJGL-UHFFFAOYSA-N cyclopamine Natural products C1C=C2CC(O)CCC2(C)C(CC2=C3C)C1C2CCC13OC2CC(C)CNC2C1C QASFUMOKHFSJGL-UHFFFAOYSA-N 0.000 description 1
- 229960003901 dacarbazine Drugs 0.000 description 1
- 206010061428 decreased appetite Diseases 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- 239000002781 deodorant agent Substances 0.000 description 1
- 239000005547 deoxyribonucleotide Substances 0.000 description 1
- 125000002637 deoxyribonucleotide group Chemical group 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 230000006866 deterioration Effects 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- 201000002342 diabetic polyneuropathy Diseases 0.000 description 1
- NEFBYIFKOOEVPA-UHFFFAOYSA-K dicalcium phosphate Chemical compound [Ca+2].[Ca+2].[O-]P([O-])([O-])=O NEFBYIFKOOEVPA-UHFFFAOYSA-K 0.000 description 1
- 229910000390 dicalcium phosphate Inorganic materials 0.000 description 1
- 229940038472 dicalcium phosphate Drugs 0.000 description 1
- FKGKZBBDJSKCIS-UHFFFAOYSA-N diethyl-[[6-[[4-(hydroxycarbamoyl)phenyl]carbamoyloxymethyl]naphthalen-2-yl]methyl]azanium;chloride;hydrate Chemical compound O.[Cl-].C1=CC2=CC(C[NH+](CC)CC)=CC=C2C=C1COC(=O)NC1=CC=C(C(=O)NO)C=C1 FKGKZBBDJSKCIS-UHFFFAOYSA-N 0.000 description 1
- 238000007865 diluting Methods 0.000 description 1
- 238000007907 direct compression Methods 0.000 description 1
- 208000037765 diseases and disorders Diseases 0.000 description 1
- KSDGSKVLUHKDAL-UHFFFAOYSA-L disodium;3-[2-carboxylatoethyl(dodecyl)amino]propanoate Chemical compound [Na+].[Na+].CCCCCCCCCCCCN(CCC([O-])=O)CCC([O-])=O KSDGSKVLUHKDAL-UHFFFAOYSA-L 0.000 description 1
- 238000011978 dissolution method Methods 0.000 description 1
- 238000007922 dissolution test Methods 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- POULHZVOKOAJMA-UHFFFAOYSA-M dodecanoate Chemical compound CCCCCCCCCCCC([O-])=O POULHZVOKOAJMA-UHFFFAOYSA-M 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 229940115080 doxil Drugs 0.000 description 1
- 239000008298 dragée Substances 0.000 description 1
- 239000000890 drug combination Substances 0.000 description 1
- 238000009509 drug development Methods 0.000 description 1
- 206010013781 dry mouth Diseases 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 238000002565 electrocardiography Methods 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 238000004945 emulsification Methods 0.000 description 1
- 230000003511 endothelial effect Effects 0.000 description 1
- 239000002308 endothelin receptor antagonist Substances 0.000 description 1
- 238000012407 engineering method Methods 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- 208000001780 epistaxis Diseases 0.000 description 1
- 229930013356 epothilone Natural products 0.000 description 1
- HESCAJZNRMSMJG-KKQRBIROSA-N epothilone A Chemical class C/C([C@@H]1C[C@@H]2O[C@@H]2CCC[C@@H]([C@@H]([C@@H](C)C(=O)C(C)(C)[C@@H](O)CC(=O)O1)O)C)=C\C1=CSC(C)=N1 HESCAJZNRMSMJG-KKQRBIROSA-N 0.000 description 1
- 229960001433 erlotinib Drugs 0.000 description 1
- AAKJLRGGTJKAMG-UHFFFAOYSA-N erlotinib Chemical compound C=12C=C(OCCOC)C(OCCOC)=CC2=NC=NC=1NC1=CC=CC(C#C)=C1 AAKJLRGGTJKAMG-UHFFFAOYSA-N 0.000 description 1
- 229960001766 estramustine phosphate sodium Drugs 0.000 description 1
- IIUMCNJTGSMNRO-VVSKJQCTSA-L estramustine sodium phosphate Chemical compound [Na+].[Na+].ClCCN(CCCl)C(=O)OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)OP([O-])([O-])=O)[C@@H]4[C@@H]3CCC2=C1 IIUMCNJTGSMNRO-VVSKJQCTSA-L 0.000 description 1
- MVPICKVDHDWCJQ-UHFFFAOYSA-N ethyl 3-pyrrolidin-1-ylpropanoate Chemical compound CCOC(=O)CCN1CCCC1 MVPICKVDHDWCJQ-UHFFFAOYSA-N 0.000 description 1
- 235000019325 ethyl cellulose Nutrition 0.000 description 1
- 229920001249 ethyl cellulose Polymers 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 229960005167 everolimus Drugs 0.000 description 1
- 201000005884 exanthem Diseases 0.000 description 1
- 229960000255 exemestane Drugs 0.000 description 1
- 229960004177 filgrastim Drugs 0.000 description 1
- 239000013020 final formulation Substances 0.000 description 1
- 238000011010 flushing procedure Methods 0.000 description 1
- 229940014144 folate Drugs 0.000 description 1
- 235000019152 folic acid Nutrition 0.000 description 1
- 239000011724 folic acid Substances 0.000 description 1
- OVBPIULPVIDEAO-LBPRGKRZSA-N folic acid Chemical compound C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-LBPRGKRZSA-N 0.000 description 1
- 235000008191 folinic acid Nutrition 0.000 description 1
- 239000011672 folinic acid Substances 0.000 description 1
- 235000012631 food intake Nutrition 0.000 description 1
- 229960004783 fotemustine Drugs 0.000 description 1
- YAKWPXVTIGTRJH-UHFFFAOYSA-N fotemustine Chemical compound CCOP(=O)(OCC)C(C)NC(=O)N(CCCl)N=O YAKWPXVTIGTRJH-UHFFFAOYSA-N 0.000 description 1
- 239000012634 fragment Substances 0.000 description 1
- 229910021485 fumed silica Inorganic materials 0.000 description 1
- 230000004927 fusion Effects 0.000 description 1
- 230000027119 gastric acid secretion Effects 0.000 description 1
- 238000002682 general surgery Methods 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 239000003862 glucocorticoid Substances 0.000 description 1
- 235000001727 glucose Nutrition 0.000 description 1
- 150000004676 glycans Chemical class 0.000 description 1
- 229940049654 glyceryl behenate Drugs 0.000 description 1
- 229940075507 glyceryl monostearate Drugs 0.000 description 1
- 229940075529 glyceryl stearate Drugs 0.000 description 1
- XLXSAKCOAKORKW-AQJXLSMYSA-N gonadorelin Chemical compound C([C@@H](C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1[C@@H](CCC1)C(=O)NCC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)C1=CC=C(O)C=C1 XLXSAKCOAKORKW-AQJXLSMYSA-N 0.000 description 1
- 229940035638 gonadotropin-releasing hormone Drugs 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- UUVWYPNAQBNQJQ-UHFFFAOYSA-N hexamethylmelamine Chemical compound CN(C)C1=NC(N(C)C)=NC(N(C)C)=N1 UUVWYPNAQBNQJQ-UHFFFAOYSA-N 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 238000001794 hormone therapy Methods 0.000 description 1
- 239000012943 hotmelt Substances 0.000 description 1
- 208000033519 human immunodeficiency virus infectious disease Diseases 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 229920001600 hydrophobic polymer Polymers 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 229960001330 hydroxycarbamide Drugs 0.000 description 1
- 229920003063 hydroxymethyl cellulose Polymers 0.000 description 1
- 229940031574 hydroxymethyl cellulose Drugs 0.000 description 1
- 239000002471 hydroxymethylglutaryl coenzyme A reductase inhibitor Substances 0.000 description 1
- 229920003132 hydroxypropyl methylcellulose phthalate Polymers 0.000 description 1
- 229940031704 hydroxypropyl methylcellulose phthalate Drugs 0.000 description 1
- 229940071676 hydroxypropylcellulose Drugs 0.000 description 1
- 229920000639 hydroxypropylmethylcellulose acetate succinate Polymers 0.000 description 1
- 230000002989 hypothyroidism Effects 0.000 description 1
- 208000003532 hypothyroidism Diseases 0.000 description 1
- 239000002955 immunomodulating agent Substances 0.000 description 1
- 229940121354 immunomodulator Drugs 0.000 description 1
- 238000002650 immunosuppressive therapy Methods 0.000 description 1
- 238000009169 immunotherapy Methods 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 229910052738 indium Inorganic materials 0.000 description 1
- APFVFJFRJDLVQX-UHFFFAOYSA-N indium atom Chemical compound [In] APFVFJFRJDLVQX-UHFFFAOYSA-N 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 230000004968 inflammatory condition Effects 0.000 description 1
- 230000004941 influx Effects 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 239000002348 inosinate dehydrogenase inhibitor Substances 0.000 description 1
- 229940079322 interferon Drugs 0.000 description 1
- 229940047124 interferons Drugs 0.000 description 1
- 201000004332 intermediate coronary syndrome Diseases 0.000 description 1
- 230000000968 intestinal effect Effects 0.000 description 1
- 210000004347 intestinal mucosa Anatomy 0.000 description 1
- 230000003870 intestinal permeability Effects 0.000 description 1
- 210000000936 intestine Anatomy 0.000 description 1
- 238000007917 intracranial administration Methods 0.000 description 1
- 238000007919 intrasynovial administration Methods 0.000 description 1
- 238000007913 intrathecal administration Methods 0.000 description 1
- 230000002601 intratumoral effect Effects 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- 229960004768 irinotecan Drugs 0.000 description 1
- UWKQSNNFCGGAFS-XIFFEERXSA-N irinotecan Chemical compound C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 UWKQSNNFCGGAFS-XIFFEERXSA-N 0.000 description 1
- 229960002014 ixabepilone Drugs 0.000 description 1
- FABUFPQFXZVHFB-CFWQTKTJSA-N ixabepilone Chemical compound C/C([C@@H]1C[C@@H]2O[C@]2(C)CCC[C@@H]([C@@H]([C@H](C)C(=O)C(C)(C)[C@H](O)CC(=O)N1)O)C)=C\C1=CSC(C)=N1 FABUFPQFXZVHFB-CFWQTKTJSA-N 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 229940070765 laurate Drugs 0.000 description 1
- 229940094506 lauryl betaine Drugs 0.000 description 1
- IZWSFJTYBVKZNK-UHFFFAOYSA-N lauryl sulfobetaine Chemical compound CCCCCCCCCCCC[N+](C)(C)CCCS([O-])(=O)=O IZWSFJTYBVKZNK-UHFFFAOYSA-N 0.000 description 1
- 229950001845 lestaurtinib Drugs 0.000 description 1
- 229960001691 leucovorin Drugs 0.000 description 1
- GFIJNRVAKGFPGQ-LIJARHBVSA-N leuprolide Chemical compound CCNC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)CC1=CC=C(O)C=C1 GFIJNRVAKGFPGQ-LIJARHBVSA-N 0.000 description 1
- 229960004338 leuprorelin Drugs 0.000 description 1
- XZEUAXYWNKYKPL-WDYNHAJCSA-N levormeloxifene Chemical compound C1([C@H]2[C@@H](C3=CC=C(C=C3OC2(C)C)OC)C=2C=CC(OCCN3CCCC3)=CC=2)=CC=CC=C1 XZEUAXYWNKYKPL-WDYNHAJCSA-N 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 229950001947 lonaprisan Drugs 0.000 description 1
- 210000003750 lower gastrointestinal tract Anatomy 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 229940124302 mTOR inhibitor Drugs 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 239000003628 mammalian target of rapamycin inhibitor Substances 0.000 description 1
- 230000000873 masking effect Effects 0.000 description 1
- 238000010309 melting process Methods 0.000 description 1
- 229940041616 menthol Drugs 0.000 description 1
- GLVAUDGFNGKCSF-UHFFFAOYSA-N mercaptopurine Chemical compound S=C1NC=NC2=C1NC=N2 GLVAUDGFNGKCSF-UHFFFAOYSA-N 0.000 description 1
- 229960001428 mercaptopurine Drugs 0.000 description 1
- 208000037819 metastatic cancer Diseases 0.000 description 1
- 208000011575 metastatic malignant neoplasm Diseases 0.000 description 1
- 206010061289 metastatic neoplasm Diseases 0.000 description 1
- 229920003145 methacrylic acid copolymer Polymers 0.000 description 1
- 125000005395 methacrylic acid group Chemical group 0.000 description 1
- 229960000485 methotrexate Drugs 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- 229960001344 methylphenidate Drugs 0.000 description 1
- 108091070501 miRNA Proteins 0.000 description 1
- 239000003094 microcapsule Substances 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- 239000008185 minitablet Substances 0.000 description 1
- 229960005485 mitobronitol Drugs 0.000 description 1
- 229960001156 mitoxantrone Drugs 0.000 description 1
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 239000001788 mono and diglycerides of fatty acids Substances 0.000 description 1
- 239000000178 monomer Substances 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- 208000010125 myocardial infarction Diseases 0.000 description 1
- 125000001419 myristoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- WPHXYKUPFJRJDK-AHWVRZQESA-N n-[(3s,5s)-1-(1,3-benzodioxol-5-ylmethyl)-5-(piperazine-1-carbonyl)pyrrolidin-3-yl]-n-[(3-methoxyphenyl)methyl]-3,3-dimethylbutanamide Chemical compound COC1=CC=CC(CN([C@@H]2CN(CC=3C=C4OCOC4=CC=3)[C@@H](C2)C(=O)N2CCNCC2)C(=O)CC(C)(C)C)=C1 WPHXYKUPFJRJDK-AHWVRZQESA-N 0.000 description 1
- DVEKCXOJTLDBFE-UHFFFAOYSA-N n-dodecyl-n,n-dimethylglycinate Chemical compound CCCCCCCCCCCC[N+](C)(C)CC([O-])=O DVEKCXOJTLDBFE-UHFFFAOYSA-N 0.000 description 1
- QRGHOAATPOLDPF-VQFNDLOPSA-N nanatinostat Chemical compound N1=CC(C(=O)NO)=CN=C1N1C[C@@H]([C@@H]2NCC=3N=C4C=CC(F)=CC4=CC=3)[C@@H]2C1 QRGHOAATPOLDPF-VQFNDLOPSA-N 0.000 description 1
- 238000002663 nebulization Methods 0.000 description 1
- 229950007221 nedaplatin Drugs 0.000 description 1
- 230000009826 neoplastic cell growth Effects 0.000 description 1
- 230000007658 neurological function Effects 0.000 description 1
- 229940085033 nolvadex Drugs 0.000 description 1
- 239000000041 non-steroidal anti-inflammatory agent Substances 0.000 description 1
- 229940021182 non-steroidal anti-inflammatory drug Drugs 0.000 description 1
- 239000003956 nonsteroidal anti androgen Substances 0.000 description 1
- 235000021590 normal diet Nutrition 0.000 description 1
- 229920001778 nylon Polymers 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 229920002114 octoxynol-9 Polymers 0.000 description 1
- 239000008184 oral solid dosage form Substances 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 229920000620 organic polymer Polymers 0.000 description 1
- 229960003327 ormeloxifene Drugs 0.000 description 1
- BWKDAMBGCPRVPI-ZQRPHVBESA-N ortataxel Chemical compound O([C@@H]1[C@]23OC(=O)O[C@H]2[C@@H](C(=C([C@@H](OC(C)=O)C(=O)[C@]2(C)[C@@H](O)C[C@H]4OC[C@]4([C@H]21)OC(C)=O)C3(C)C)C)OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)CC(C)C)C(=O)C1=CC=CC=C1 BWKDAMBGCPRVPI-ZQRPHVBESA-N 0.000 description 1
- 229950001094 ortataxel Drugs 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 229950000193 oteracil Drugs 0.000 description 1
- 229960001756 oxaliplatin Drugs 0.000 description 1
- DWAFYCQODLXJNR-BNTLRKBRSA-L oxaliplatin Chemical compound O1C(=O)C(=O)O[Pt]11N[C@@H]2CCCC[C@H]2N1 DWAFYCQODLXJNR-BNTLRKBRSA-L 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 239000003002 pH adjusting agent Substances 0.000 description 1
- 239000006179 pH buffering agent Substances 0.000 description 1
- 238000004806 packaging method and process Methods 0.000 description 1
- 229940124641 pain reliever Drugs 0.000 description 1
- 238000010951 particle size reduction Methods 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 229960003407 pegaptanib Drugs 0.000 description 1
- 229960005079 pemetrexed Drugs 0.000 description 1
- QOFFJEBXNKRSPX-ZDUSSCGKSA-N pemetrexed Chemical compound C1=N[C]2NC(N)=NC(=O)C2=C1CCC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 QOFFJEBXNKRSPX-ZDUSSCGKSA-N 0.000 description 1
- 230000010412 perfusion Effects 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 230000002572 peristaltic effect Effects 0.000 description 1
- 238000011170 pharmaceutical development Methods 0.000 description 1
- 229940124531 pharmaceutical excipient Drugs 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 229940127557 pharmaceutical product Drugs 0.000 description 1
- 238000005191 phase separation Methods 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 239000002571 phosphodiesterase inhibitor Substances 0.000 description 1
- 102000020233 phosphotransferase Human genes 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 230000035479 physiological effects, processes and functions Effects 0.000 description 1
- 229950005566 picoplatin Drugs 0.000 description 1
- IIMIOEBMYPRQGU-UHFFFAOYSA-L picoplatin Chemical compound N.[Cl-].[Cl-].[Pt+2].CC1=CC=CC=N1 IIMIOEBMYPRQGU-UHFFFAOYSA-L 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 229960004403 pixantrone Drugs 0.000 description 1
- PEZPMAYDXJQYRV-UHFFFAOYSA-N pixantrone Chemical compound O=C1C2=CN=CC=C2C(=O)C2=C1C(NCCN)=CC=C2NCCN PEZPMAYDXJQYRV-UHFFFAOYSA-N 0.000 description 1
- YJGVMLPVUAXIQN-XVVDYKMHSA-N podophyllotoxin Chemical compound COC1=C(OC)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@H](O)[C@@H]3[C@@H]2C(OC3)=O)=C1 YJGVMLPVUAXIQN-XVVDYKMHSA-N 0.000 description 1
- 229920000058 polyacrylate Polymers 0.000 description 1
- 229920002523 polyethylene Glycol 1000 Polymers 0.000 description 1
- 229940056099 polyglyceryl-4 oleate Drugs 0.000 description 1
- 229920002959 polymer blend Polymers 0.000 description 1
- 229920001451 polypropylene glycol Polymers 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 229950008882 polysorbate Drugs 0.000 description 1
- 229940068965 polysorbates Drugs 0.000 description 1
- 229940100467 polyvinyl acetate phthalate Drugs 0.000 description 1
- 239000011148 porous material Substances 0.000 description 1
- 238000002600 positron emission tomography Methods 0.000 description 1
- 230000002980 postoperative effect Effects 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- JHDKZFFAIZKUCU-ZRDIBKRKSA-N pracinostat Chemical compound ONC(=O)/C=C/C1=CC=C2N(CCN(CC)CC)C(CCCC)=NC2=C1 JHDKZFFAIZKUCU-ZRDIBKRKSA-N 0.000 description 1
- 229960004694 prednimustine Drugs 0.000 description 1
- 230000002028 premature Effects 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 230000003449 preventive effect Effects 0.000 description 1
- 229960000624 procarbazine Drugs 0.000 description 1
- CPTBDICYNRMXFX-UHFFFAOYSA-N procarbazine Chemical compound CNNCC1=CC=C(C(=O)NC(C)C)C=C1 CPTBDICYNRMXFX-UHFFFAOYSA-N 0.000 description 1
- 229940002612 prodrug Drugs 0.000 description 1
- 239000000651 prodrug Substances 0.000 description 1
- 239000000186 progesterone Substances 0.000 description 1
- 229960003387 progesterone Drugs 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- 210000002307 prostate Anatomy 0.000 description 1
- 239000003207 proteasome inhibitor Substances 0.000 description 1
- 239000003528 protein farnesyltransferase inhibitor Substances 0.000 description 1
- 201000001474 proteinuria Diseases 0.000 description 1
- 230000000541 pulsatile effect Effects 0.000 description 1
- 238000010926 purge Methods 0.000 description 1
- 239000008213 purified water Substances 0.000 description 1
- 150000003856 quaternary ammonium compounds Chemical class 0.000 description 1
- 229950010654 quisinostat Drugs 0.000 description 1
- 229960004432 raltitrexed Drugs 0.000 description 1
- 229960003876 ranibizumab Drugs 0.000 description 1
- 229960002185 ranimustine Drugs 0.000 description 1
- 206010037844 rash Diseases 0.000 description 1
- 238000004064 recycling Methods 0.000 description 1
- 231100000628 reference dose Toxicity 0.000 description 1
- 238000012827 research and development Methods 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 102000027483 retinoid hormone receptors Human genes 0.000 description 1
- 108091008679 retinoid hormone receptors Proteins 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- VHXNKPBCCMUMSW-FQEVSTJZSA-N rubitecan Chemical compound C1=CC([N+]([O-])=O)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 VHXNKPBCCMUMSW-FQEVSTJZSA-N 0.000 description 1
- 229950009213 rubitecan Drugs 0.000 description 1
- 238000005185 salting out Methods 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- 229960005399 satraplatin Drugs 0.000 description 1
- 190014017285 satraplatin Chemical compound 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 238000007789 sealing Methods 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 229940124834 selective serotonin reuptake inhibitor Drugs 0.000 description 1
- 239000012896 selective serotonin reuptake inhibitor Substances 0.000 description 1
- BTIHMVBBUGXLCJ-OAHLLOKOSA-N seliciclib Chemical compound C=12N=CN(C(C)C)C2=NC(N[C@@H](CO)CC)=NC=1NCC1=CC=CC=C1 BTIHMVBBUGXLCJ-OAHLLOKOSA-N 0.000 description 1
- 229950000055 seliciclib Drugs 0.000 description 1
- 229950003647 semaxanib Drugs 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 239000003772 serotonin uptake inhibitor Substances 0.000 description 1
- RMAQACBXLXPBSY-UHFFFAOYSA-N silicic acid Chemical compound O[Si](O)(O)O RMAQACBXLXPBSY-UHFFFAOYSA-N 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 238000005549 size reduction Methods 0.000 description 1
- 239000012748 slip agent Substances 0.000 description 1
- WXMKPNITSTVMEF-UHFFFAOYSA-M sodium benzoate Chemical compound [Na+].[O-]C(=O)C1=CC=CC=C1 WXMKPNITSTVMEF-UHFFFAOYSA-M 0.000 description 1
- 235000010234 sodium benzoate Nutrition 0.000 description 1
- 239000004299 sodium benzoate Substances 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 1
- 235000010267 sodium hydrogen sulphite Nutrition 0.000 description 1
- 229940045902 sodium stearyl fumarate Drugs 0.000 description 1
- 239000007909 solid dosage form Substances 0.000 description 1
- 239000006104 solid solution Substances 0.000 description 1
- 230000007928 solubilization Effects 0.000 description 1
- 238000005063 solubilization Methods 0.000 description 1
- NHXLMOGPVYXJNR-ATOGVRKGSA-N somatostatin Chemical compound C([C@H]1C(=O)N[C@H](C(N[C@@H](CO)C(=O)N[C@@H](CSSC[C@@H](C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CC=2C3=CC=CC=C3NC=2)C(=O)N[C@@H](CCCCN)C(=O)N[C@H](C(=O)N1)[C@@H](C)O)NC(=O)CNC(=O)[C@H](C)N)C(O)=O)=O)[C@H](O)C)C1=CC=CC=C1 NHXLMOGPVYXJNR-ATOGVRKGSA-N 0.000 description 1
- 229960000553 somatostatin Drugs 0.000 description 1
- 229940100515 sorbitan Drugs 0.000 description 1
- 230000002269 spontaneous effect Effects 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 238000010972 statistical evaluation Methods 0.000 description 1
- SFVFIFLLYFPGHH-UHFFFAOYSA-M stearalkonium chloride Chemical compound [Cl-].CCCCCCCCCCCCCCCCCC[N+](C)(C)CC1=CC=CC=C1 SFVFIFLLYFPGHH-UHFFFAOYSA-M 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 125000004079 stearyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000003270 steroid hormone Substances 0.000 description 1
- 229960001052 streptozocin Drugs 0.000 description 1
- ZSJLQEPLLKMAKR-GKHCUFPYSA-N streptozocin Chemical compound O=NN(C)C(=O)N[C@H]1[C@@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O ZSJLQEPLLKMAKR-GKHCUFPYSA-N 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 229960001940 sulfasalazine Drugs 0.000 description 1
- NCEXYHBECQHGNR-UHFFFAOYSA-N sulfasalazine Natural products C1=C(O)C(C(=O)O)=CC(N=NC=2C=CC(=CC=2)S(=O)(=O)NC=2N=CC=CC=2)=C1 NCEXYHBECQHGNR-UHFFFAOYSA-N 0.000 description 1
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 1
- 230000003319 supportive effect Effects 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 208000011580 syndromic disease Diseases 0.000 description 1
- 229920001059 synthetic polymer Polymers 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 229940037128 systemic glucocorticoids Drugs 0.000 description 1
- 239000003760 tallow Substances 0.000 description 1
- 229960001674 tegafur Drugs 0.000 description 1
- WFWLQNSHRPWKFK-ZCFIWIBFSA-N tegafur Chemical compound O=C1NC(=O)C(F)=CN1[C@@H]1OCCC1 WFWLQNSHRPWKFK-ZCFIWIBFSA-N 0.000 description 1
- 229960004964 temozolomide Drugs 0.000 description 1
- NRUKOCRGYNPUPR-QBPJDGROSA-N teniposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@@H](OC[C@H]4O3)C=3SC=CC=3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 NRUKOCRGYNPUPR-QBPJDGROSA-N 0.000 description 1
- 229960001278 teniposide Drugs 0.000 description 1
- ZRKFYGHZFMAOKI-QMGMOQQFSA-N tgfbeta Chemical compound C([C@H](NC(=O)[C@H](C(C)C)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CC(C)C)NC(=O)CNC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CCSC)C(C)C)[C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(O)=O)C1=CC=C(O)C=C1 ZRKFYGHZFMAOKI-QMGMOQQFSA-N 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 231100000886 tinnitus Toxicity 0.000 description 1
- PLHJCIYEEKOWNM-HHHXNRCGSA-N tipifarnib Chemical compound CN1C=NC=C1[C@](N)(C=1C=C2C(C=3C=C(Cl)C=CC=3)=CC(=O)N(C)C2=CC=1)C1=CC=C(Cl)C=C1 PLHJCIYEEKOWNM-HHHXNRCGSA-N 0.000 description 1
- 229950009158 tipifarnib Drugs 0.000 description 1
- OGIDPMRJRNCKJF-UHFFFAOYSA-N titanium oxide Inorganic materials [Ti]=O OGIDPMRJRNCKJF-UHFFFAOYSA-N 0.000 description 1
- 238000004448 titration Methods 0.000 description 1
- 229960003989 tocilizumab Drugs 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 229960000303 topotecan Drugs 0.000 description 1
- UCFGDBYHRUNTLO-QHCPKHFHSA-N topotecan Chemical compound C1=C(O)C(CN(C)C)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 UCFGDBYHRUNTLO-QHCPKHFHSA-N 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 201000010875 transient cerebral ischemia Diseases 0.000 description 1
- 230000001052 transient effect Effects 0.000 description 1
- 230000000472 traumatic effect Effects 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- 229950002860 triplatin tetranitrate Drugs 0.000 description 1
- 190014017283 triplatin tetranitrate Chemical compound 0.000 description 1
- 230000005747 tumor angiogenesis Effects 0.000 description 1
- 230000004614 tumor growth Effects 0.000 description 1
- 229960005486 vaccine Drugs 0.000 description 1
- 238000001291 vacuum drying Methods 0.000 description 1
- 230000002792 vascular Effects 0.000 description 1
- 230000004862 vasculogenesis Effects 0.000 description 1
- 229960003048 vinblastine Drugs 0.000 description 1
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 1
- 229960004528 vincristine Drugs 0.000 description 1
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 1
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 1
- 229960004355 vindesine Drugs 0.000 description 1
- UGGWPQSBPIFKDZ-KOTLKJBCSA-N vindesine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(N)=O)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1N=C1[C]2C=CC=C1 UGGWPQSBPIFKDZ-KOTLKJBCSA-N 0.000 description 1
- GBABOYUKABKIAF-GHYRFKGUSA-N vinorelbine Chemical compound C1N(CC=2C3=CC=CC=C3NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC GBABOYUKABKIAF-GHYRFKGUSA-N 0.000 description 1
- 229960002066 vinorelbine Drugs 0.000 description 1
- 229960004449 vismodegib Drugs 0.000 description 1
- BPQMGSKTAYIVFO-UHFFFAOYSA-N vismodegib Chemical compound ClC1=CC(S(=O)(=O)C)=CC=C1C(=O)NC1=CC=C(Cl)C(C=2N=CC=CC=2)=C1 BPQMGSKTAYIVFO-UHFFFAOYSA-N 0.000 description 1
- 238000011179 visual inspection Methods 0.000 description 1
- 230000008673 vomiting Effects 0.000 description 1
- WAEXFXRVDQXREF-UHFFFAOYSA-N vorinostat Chemical compound ONC(=O)CCCCCCC(=O)NC1=CC=CC=C1 WAEXFXRVDQXREF-UHFFFAOYSA-N 0.000 description 1
- 229960000237 vorinostat Drugs 0.000 description 1
- 239000001993 wax Substances 0.000 description 1
- 238000005550 wet granulation Methods 0.000 description 1
- 238000009736 wetting Methods 0.000 description 1
- 230000029663 wound healing Effects 0.000 description 1
Images


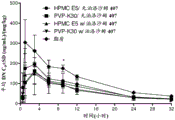


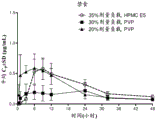




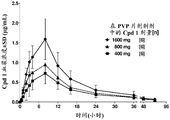

Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1629—Organic macromolecular compounds
- A61K9/1635—Organic macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyvinyl pyrrolidone, poly(meth)acrylates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/437—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having nitrogen as a ring hetero atom, e.g. indolizine, beta-carboline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0053—Mouth and digestive tract, i.e. intraoral and peroral administration
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1629—Organic macromolecular compounds
- A61K9/1652—Polysaccharides, e.g. alginate, cellulose derivatives; Cyclodextrin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/2027—Organic macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyvinyl pyrrolidone, poly(meth)acrylates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/205—Polysaccharides, e.g. alginate, gums; Cyclodextrin
- A61K9/2054—Cellulose; Cellulose derivatives, e.g. hydroxypropyl methylcellulose
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2095—Tabletting processes; Dosage units made by direct compression of powders or specially processed granules, by eliminating solvents, by melt-extrusion, by injection molding, by 3D printing
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4841—Filling excipients; Inactive ingredients
- A61K9/4866—Organic macromolecular compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4883—Capsule finishing, e.g. dyeing, aromatising, polishing
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
Landscapes
- Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Physiology (AREA)
- Nutrition Science (AREA)
- Rheumatology (AREA)
- Pain & Pain Management (AREA)
- Oncology (AREA)
- Hematology (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Immunology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Physical Education & Sports Medicine (AREA)
- Medicinal Preparation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
本发明涉及具有增加的剂量负载和改进的溶解而较少受到食物影响的生物可利用药物组合物。
Description
技术领域
本发明描述可用于药物组合物中的一种形式的亲脂性化合物,以及用所述形式的化合物形成固体分散体(如喷雾干燥的中间体)的方法。本发明还描述所述固体分散体提供生物可利用口服剂型的用途,所述生物可利用口服剂型具有增加的剂量负载和改进的溶解而较少受到食物的影响。
背景技术
经口给予治疗剂的生物可利用性是药剂在人体中吸收且在目标部位(例如细胞等之中或之上)变成可利用的体内标靶(例如用于相互作用或复合作用等)的程度。为了使治疗剂具有生物可利用性,治疗剂通常需要相对于给予的剂量具有一定的水溶性,因此,希望所述药剂与可溶于脂肪(亲脂性)相比更可溶于水(亲水性)。通常,亲脂性药剂在水中溶解不佳。因此,在其他因素之中,药剂的亲脂性程度决定药剂的生物可利用性。
因而,在本领域中仍持续需要且市场仍不断需求可用于特定药剂的易于给药、具有增加的剂量负载和改进的溶解以及生物可利用性的药物组合物。
发明内容
本文涵盖具有本文阐述的式(I)的一种形式的亲脂性化合物,称为(S)-6-氯-1-(4-甲氧基苯基)-1,3,4,9-四氢-2H-吡啶并[3,4-b]吲哚-2-甲酸4-氯苯酯(“化合物1”)。
在一个方面中,化合物1的形式为非晶形式。
在另一方面中,化合物1的形式为结晶形式。
描述所述形式的化合物1在制备固体分散体(如喷雾干燥的中间体)中的用途,所述固体分散体包含非晶形式的化合物1和聚合物,其中所用的聚合物为亲水性聚合物。
在一个方面中,所用的聚合物为聚乙烯吡咯烷酮(PVP)或羟丙基甲基纤维素(HPMC)。
在一个方面中,用于制备喷雾干燥的中间体的化合物1的形式为非晶形式。在另一方面中,用于制备中间体的化合物1的形式为结晶形式。
还涵盖一种用于制备固体分散体(如喷雾干燥的中间体)的方法,所述固体分散体包含非晶形式的化合物1和聚合物。
在一个方面中,所述方法包括使化合物1和聚合物共同溶解于溶剂体系中以形成液体分散体,随后去除溶剂。
在一个方面中,所形成的中间体为固体分散体。
在一个方面中,溶剂通过喷雾干燥去除。在另一方面中,当获得喷雾干燥的中间体时,形成非晶形式的化合物1。
还描述喷雾干燥的中间体在药物组合物中的用途,所述药物组合物包含喷雾干燥的中间体与一种或多种药学上可接受的赋形剂的紧密混杂物以提供生物可利用口服剂型。
在另一方面中,中间体为包含非晶形式的化合物1和亲水聚合物的喷雾干燥的中间体。在另一方面中,亲水聚合物为PVP或HPMC。在另一方面中,PVP为聚乙烯吡咯烷酮K-30(PVP K-30)。在另一方面中,HPMC为HPMC E5。
在一个方面中,剂型为口服固体剂型。在另一方面中,口服剂型为片剂。在另一方面中,口服剂型为胶囊。
还描述生物可利用口服剂型在基于重量的给药方案中的用途,其中所述给药方案维持目标血浆浓度。
还描述生物可利用口服剂型在固定剂量方案中的用途,其中所述方案维持目标血浆浓度。
还描述随食物给予口服剂型以增强生物可利用性。
因此,本申请提供具有增加的剂量负载和改进的溶解性的药物组合物。
附图说明
图1显示在用各种聚合物和赋形剂组合的情况下在各种剂量负载量下,作为时间的函数的喷雾干燥的中间体(SDI)的囊封的干式共混制剂在含有1.5%十二烷基硫酸钠(SDS)的0.1N HCl中的溶解速率;
图2显示在用各种聚合物和赋形剂组合的情况下在各种剂量负载量下,作为时间的函数的SDI的囊封的干式共混制剂在两种不同体积的溶解流体中的含有1.5% SDS的0.1NHCl中的比较性溶解速率;
图3显示临床前体内口服生物可利用性药代动力学动物研究中测试的SDI的囊封的干式共混制剂的作为时间的函数的剂量标准化血浆浓度;
图4显示在临床前体内口服生物可利用性药代动力学动物研究中作为时间的函数的在片剂和胶囊制剂中使用的SDI的剂量标准化血浆浓度;
图5显示在临床前体内药代动力学食物影响动物研究中的喂饲动物中作为时间的函数的在片剂和胶囊制剂中使用的SDI的剂量标准化血浆浓度;
图6显示在临床前体内药代动力学食物影响研究中的禁食动物中作为时间的函数的在片剂和胶囊制剂中使用的SDI的剂量标准化血浆浓度;
图7显示在临床前体内药代动力学食物影响研究中的喂饲动物和禁食动物中作为时间的函数的在片剂和胶囊制剂中使用的SDI的平均血浆浓度;
图8显示在体内药代动力学食物影响临床研究的第1阶段作为时间的函数的脂质胶囊制剂的平均血浆浓度;
图9显示在体内药代动力学食物影响临床研究中的喂饲受试者和禁食受试者中作为时间的函数的脂质胶囊制剂的平均血浆浓度;
图10显示体内药代动力学食物影响临床研究中作为时间的函数的脂质胶囊制剂和PVP片剂制剂的平均血浆浓度;
图11显示体内药代动力学食物影响临床研究中作为时间的函数的在给予剂量水平为400 mg、800 mg和1000 mg的PVP片剂制剂之后获得的平均化合物1 (“Cpd 1”)血浆浓度;
图12显示在体内药代动力学食物影响临床研究中的喂饲受试者和禁食受试者中作为时间的函数的在剂量水平为400 mg和1000 mg的PVP片剂制剂下的平均血浆浓度。
具体实施方式
本文涵盖一种形式的化合物(化合物I),其具有式(I):

在一个方面中,化合物1为非晶形式。
在另一方面中,化合物1的形式为结晶形式。
描述化合物1在制备固体分散体(如喷雾干燥的中间体)中的用途,所述固体分散体包含非晶形式的化合物1和聚合物,其中所用的聚合物为亲水性聚合物。
在一个方面中,所用的聚合物为PVP或HPMC。在另一方面中,PVP为PVP-K30。在另一方面中,HPMC为HPMC E5。
在一个方面中,用于制备中间体的化合物1的形式为非晶形式。在另一方面中,用于制备中间体的化合物1的形式为结晶形式。
还涵盖一种用于制备固体分散体的方法,所述固体分散体包含非晶形式的化合物1和聚合物。
在另一方面中,所述方法包括使化合物1和聚合物共同溶解于溶剂体系中以形成液体分散体,随后去除溶剂。
在另一方面中,所形成的中间体为固体分散体。
在另一方面中,溶剂通过喷雾干燥去除。在另一方面中,当获得喷雾干燥的中间体时,形成非晶形式的化合物1。
在另一方面中,中间体为包含非晶形式的化合物1和亲水聚合物的喷雾干燥的中间体。在另一方面中,亲水聚合物为PVP或HPMC。在另一方面中,PVP为聚乙烯吡咯烷酮K-30(PVP K-30)。在另一方面中,HPMC为HPMC E5。
还描述喷雾干燥的中间体在药物组合物中的用途,所述药物组合物包含喷雾干燥的中间体与一种或多种药学上可接受的赋形剂的紧密混杂物以提供生物可利用口服剂型。
在另一方面中,口服剂型为片剂。
还描述生物可利用口服剂型在基于重量或固定重量的给药方案中的用途,其中所述给药方案维持目标血浆浓度。
定义
如本文所用,术语“一种或多种共晶体”是指在晶体内具有两种或更多种不同分子组分的晶体,通常是指大分子晶体,所述组分包含本文提供的化合物和一种或多种合适的药学上可接受的无毒平衡离子。
如本文所用,术语“化合物1”是指本文所述的式(I)化合物及其药学上可接受的多晶型物或非晶形式。在某些方面中,所述术语是指式(I)的多晶型物。在某些方面中,所述术语是指式(I)的非晶形式。制造化合物1的方法提供于国际申请号WO 2005/089764中。
本文提供的化合物1进一步描述于美国专利7,601,840 (具有相应国际申请公开案号WO2005/089764)、美国专利7,767,689 (具有相应国际申请公开案号WO2006/113703)、国际申请公开案号WO2010/138758;美国专利8,076,352 (具有相应国际申请公开案号WO2008/127715);美国专利8,076,353;美国专利8,367,694;美国公开案号2010/0158858(具有相应国际申请公开案号WO2008/127714)中;其各自以引用的方式整体并入本文中。如本文所用,在将化合物给予具有本文所述病况的受试者的情形下,术语“有效量”是指产生有益或治疗效果的化合物的量。在特定方面中,化合物的“有效量”是指足以达成以下效果中的至少一项、两项、三项、四项或更多项的化合物的量:(i)减轻或改善与本文所述病况相关的一种或多种症状的严重程度;(ii)减少与本文所述病况相关的一种或多种症状的持续期间;(iii)预防肿瘤或与本文所述病况相关的一种或多种症状的复发;(iv)消退本文所述病况和/或与其相关的一种或多种症状;(v)减少受试者住院;(vi)减少住院时间;(vii)增加受试者的存活率;(viii)抑制本文所述病况和/或与其相关的一种或多种症状的进展;(ix)增强或改进另一疗法的治疗效果;(x)在手术前减轻白血病增殖;(xiv)根除、去除或控制白血病增殖;(xv)减小白血病增殖率;(xvi)降低死亡率;(xvii)增加患者的无肿瘤存活率;(xviii)增加无进展存活率;(xix)增加缓解的患者数目;(xx)降低住院率;(xxi)如通过本领域的技术人员可获得的常规方法所测量,在给予标准疗法之后肿瘤尺寸得以维持且不增加或较少增加,所述常规方法是如磁共振成像(MRI)、动态对比增强的MRI (DCE-MRI)、X射线、计算机断层(CT)扫描、正电子发射断层扫描或其他成像模式;(xxii)预防本文所述病况或与其相关的一种或多种症状的发展或发作;(xxiii)增加患者缓解的时间;(xxiv)减少与本文所述病况相关的一种或多种症状的数目;(xxv)增加具有本文所述病况的患者的无症状存活率;(xxv.i)增加具有本文所述病况的患者的无病存活率;(xxvi)降低具有本文所述病况的受试者的血浆、血清或其他生物流体中循环DHODH (二氢乳清酸去氢酶)的浓度;(xxvii)减少具有本文所述病况的受试者的血液中循环肿瘤细胞(CTC);(xxvii.i)减少具有本文所述病况的受试者的血液中与肿瘤细胞相关的循环DNA或RNA;(xxviii)降低具有本文所述病况的受试者的生物样本(例如血浆、血清、尿液、脑脊髓液(CSF))或其他生物流体中DHODH的浓度;(xxviii)预防手术后的肿瘤血管结构;(xxix)改进神经功能,例如听觉、平衡、耳鸣或视觉;(xxx)抑制或减少DHODH的病理性制造;(xxxi)稳定化或减轻受试者中的瘤周发炎或水肿;(xxxii)降低生物样本(例如血浆、血清、大脑脊髓液、尿液或任何其他生物流体)中DHODH或其他血管生成或发炎介质(例如细胞因子或白细胞介素)的浓度;(xxxiii)抑制或减少肿瘤代谢或灌注;(xxxiv)抑制或减少血管生成或血管形成;(xxxv)如通过本领域中熟知的方法(例如通过症状或生活品质调查表)所评估,改进生活品质。在特定方面中,化合物的“有效量”是指以下指定的化合物的量。
如本文所用,术语“老年人”是指65岁或更大年龄的人。
如本文所用,术语“中年人”是指30岁与64岁之间的人。
如本文所用,术语“成年人”是指18岁或更大年龄的人。
如本文所用,术语“孩子”是指1岁至18岁的人。
如本文所用,术语“幼儿”是指1岁至3岁的人。
如本文所用,术语“婴儿”是指新生儿至1岁的人。
如本文所用,术语“亲水性聚合物”是指含有亲水性基团(如羟基)的重复单体的有机聚合物。在本文所述的本发明的情形下,聚合物的长度与相关粘度关联,即具有较高分子量的聚合物趋于更粘稠。对于在制备包含所述形式的化合物和聚合物的中间体中的用途,可用的聚合物的长度受粘度限制。所选聚合物具有低粘度,并且可具有一些表面活性剂性质;即,聚合物具有降低表面张力和与疏水性物质和亲水性物质两者相互作用的能力。在本文的情形下,聚合物具有增强的两亲性特征的能力可通过存在一种或多种表面活性剂增强。因此,在所述化合物和存在任选的赋形剂的情形下,平衡所选聚合物的性质以确保克服化合物粒子的水不溶亲脂性质,同时避免纤维的聚集和形成。
如本文所用,术语“本文所述的病况”是指急性骨髓白血病(AML),包括能够受DHODH抑制影响的急性髓细胞性白血病、急性骨髓性白血病、急性粒细胞白血病和急性非淋巴细胞性白血病。还是指炎性疾病,包括(但不限于)类风湿性关节炎和多发性硬化症。如本文所用,术语“受试者”和“患者”可互换使用,以指针对本文所述病况进行治疗的个体。在一个特定方面中,所述个体是人。
如本文所用,术语“疗法(therapies/therapy)”可指可用于预防、治疗、处理或改善本文所述的病况或病症或其症状(例如本文所述病况;病况或与其相关的症状或病况)的任何一种或多种协议、一种或多种方法、组合物、制剂和/或一种或多种药剂。在某些方面中,术语“疗法(therapies/therapy)”是指可用于治疗、处理、预防或改善本文所述的病况或病症或其症状(例如本文所述的与其相关的症状或病况;病况或本文所述与其相关的症状或病况)的生物疗法、支持疗法和/或其他疗法。在某些方面中,术语“疗法”是指除化合物或其药物组合物外的疗法。在特定方面中,“另外疗法(additional therapy/additionaltherapies)”是指除使用化合物或药物组合物治疗外的疗法。
如本文所用,在制造本文所述的DHODH的情形下,术语“病理”、“病理性”或“病理诱发”是指所述术语涵盖通过肿瘤细胞或肿瘤环境中的其他细胞以致癌性转化诱发方式表达DHODH。在另一方面中,所述术语涵盖DHODH在慢性或创伤性发炎病况中的表达。在另一方面中,响应于环境刺激,所述术语还涵盖错误调节或过度制造DHODH的细胞。适当时,DHODH的表达支持发炎、血管生成和肿瘤生长。通过化合物抑制或减少DHODH的病理性制造可在如本文所述的细胞培养物和/或动物模型、肿瘤组织匀浆、血液样品、尿液样品、CSF等中评估。
如本文所用,术语“约”意指给定值周围的范围,其中所得值与明确叙述的值大体上相同。在一个方面中,“约”意指在给定值或范围的25%内。例如,短语“约70重量%”至少包含52重量%至88重量%的所有值。在另一方面中,术语“约”意指在给定值或范围的10%内。例如,短语“约70重量%”至少包含63重量%至77重量%的所有值。在另一方面中,术语“约”意指在给定值或范围的7%内。例如,短语“约70重量%”至少包含65重量%至75重量%的所有值。
浓度、量、百分比和其他数值在本文中可以范围格式来呈现。应理解,这类范围格式仅是出于便利和简洁目的而使用,且应以灵活方式解释为不仅包括如所述范围的界限所明确叙述的数值,并且还包括所述范围内涵盖的所有个别数值或子范围,如同每个数值和子范围都被明确叙述一样。
1. 制剂
经口给予的治疗剂的生物可利用性根据生物药物分级系统(BCS)分类,所述生物药物分级系统是由美国食品和药物管理局(FDA)提供的将药物物质基于其水溶性和肠渗透性分类的指导。这种系统允许估计溶解、溶解性和渗透性的因素对口服药物吸收的影响。这些因素对口服药物吸收的影响十分重要,因为在美国和欧洲,85%的最畅销药物是经口给予的。根据BCS系统,BCS第I类药物是高度可渗透和可溶解、以通常高于分泌速率的吸收速率充分吸收的那些药剂。BCS第II类药物高度可渗透,但具有低溶解性,其中生物可利用性受水溶性或溶解速率中的任一者或两者限制。在一些情况下,BCS II药剂的体内生物可利用性与体外溶解速率之间可具有相关性。BCS第III类药物高度可溶解,但具有低渗透性。尽管药物可快速溶解,但吸收相反还可受渗透率限制。如果制剂不改变渗透性或胃肠(GI)传输时间,则可适用第I类标准。BCS第IV类药物具有低渗透性和溶解性以及不良生物可利用性,在整个肠粘膜上不充分吸收或具有高度可变的吸收。基于质量平衡或与静脉内参考剂量相比,BCS类别边界在最高剂量浓度在1至7.5的pH范围上可溶于小于250 mL水时将药物定义为高度可溶解,且在确定在人中的吸收程度大于90%的给予剂量时将药物定义为高度可渗透。当在30分钟内使用USP设备I或II有大于85%的标记量的药物物质溶解时,药品被视为快速溶解于小于900 mL体积的缓冲溶液中。
BCS II药剂的不良溶解性和所得不良生物可利用性成为显著药学开发挑战,因为水溶性不佳的药剂趋于在吸收到循环中之前自胃肠道排除。因为BCS II药剂在胃和胃肠道中的溶解不佳,所以取决于食物的存在或不存在,即当药剂经口给予时受试者是喂饲状态还是禁食状态,其还趋于显示其生物可利用性和所得血浆浓度的显著差异(在本文中通常称为“食物影响”)。
例如,在膳食之后给予药剂时,药剂吸收可显著高于给药之前受试者尚未进食时的药剂吸收。不受理论束缚,喂饲或禁食状态的不同吸收药代动力学可归因于亲脂性化合物在脂肪中的较高溶解性或由因食物摄入而分泌的胆汁盐辅助的溶解中的任一种或两种。尽管这种药代动力学作用在不存在食物的情况下可减至最小,但在食物存在下药剂的所得血浆浓度可导致高于预期血浆浓度。对于具有窄治疗性和毒性窗口的药物,不需要这一考量。相反,在不存在食物的情况下,可能达不到所需治疗性血浆浓度。因此,食物影响为BCSII药剂的FDA批准提供显著管理障碍,并且必须在早期药物开发中解决。
理想地,为了使食物影响减至最小、维持恒定的血浆水平且获得所需治疗效果,BCS II药剂的制剂必须增强亲脂性药剂的水溶性且必须使食物影响减至最小。设计成增强BCS II药剂的水溶性的配制方法可涉及组合药学上可接受的有机溶剂或共溶剂、表面活性剂和调节pH条件。尽管存在这类制剂的实例,但其通常具有一些关于胃耐受性的缺点。
此外,取决于制剂平衡药剂的亲脂性与对亲水性需求的能力,传统制剂通常不可在便于经口给予的剂型中容纳足够量(剂量负载)的药剂。例如,受试者可需要消耗多个单位的剂型以获得提供所需治疗效果的亲脂性药剂的血浆浓度。具有这种类型的限制的制剂因此不符合所需给药方案。
增加亲脂性药剂的溶解性的若干技术除了增强制剂之外还包括鉴别和选择更可溶的药剂的多晶型物、水合物或盐。
其他技术包括使用粒度减小(即微米尺寸化或纳米粒子体系)以增加与介质接触的溶解固体的分子表面积,因此加速溶解和潜在的生物可利用性。微米尺寸化和其他粒子工程改造方法可包括精细研磨结晶形式的药剂、自溶液沉淀极精细形式的药剂或通过自溶液喷雾干燥或冷冻干燥药剂形成较小粒子或非晶形式。某些尺寸减小技术会更大程度地减小天然产生或微米化粒度的药剂,产生比原始粒子小高达1000倍的纳米粒子。某些其他技术将药剂涂覆到小粒子上以形成分散体。
还存在基于使用溶剂体系的溶解方法,由此增溶剂将BCS II药剂“拖曳”到溶液中并增加药剂与水性介质的混溶性。本领域的技术人员已知的这些和其他技术可与不同结晶形式的药剂(例如非晶形式)或共熔混合物组合以减少溶解的热力学障碍。
尽管这些溶解技术集中于药剂的结构、结晶形式和粒度,但其在制造生物可利用口服剂型中的有用性取决于与药物制剂中药剂与赋形剂的分子相互 作用相关的多个因素。药剂相关因素对这些溶解技术的有用性的影响需要大量评价,通常显示所述技术单独有时可能不足以达成在最终制剂中提供或改进令人满意的溶解性和剂量负载的所需结果,或可销售性的折衷结果是可获得的所有结果。
例如,这些技术在达成所需溶解性改进结果方面的常见问题在于在形成具有降低的粒度的药剂之后,非常小粒子的物理性质使其趋于聚结在一起且妨碍粉末流动。尽管BCSII药剂的溶解性可因降低的粒度而增加,但松散形式的药剂的便利和实际可用性降低。但最常用于本领域中以减少或防止聚结的许多技术中的一种除了涂覆粒子还会增加另一步骤到配制过程中。
用以在不使用微米尺寸化或纳米粒子的情况下增加BCS II药剂的表面积且因此提供或改进药剂的溶解性的替代方法是制造药剂在合适的高分子量水溶性聚合基质中的固体分散体。固体分散体含有至少两种组分:基质和活性剂在所述基质内的分子分散体。药剂(呈结晶粒子或非晶粒子形式,任选微米化或纳米粒子)均匀分散于聚合物基质内。这类制剂在不可溶药剂与水性介质(例如GI液)之间提供溶解性桥且在暴露于介质时改进药剂的溶解性质。
不受理论束缚,当药剂均匀分散于基质内时,水性溶剂与药剂之间的分子相互作用改进药剂的溶解性。非限制性地,固体分散体可以物理方式分类为共熔混合物、固体溶液、玻璃溶液或悬浮液、在玻璃态或结晶载剂中的非晶沉淀物、复合物、不同体系的复合形式或组合。此外,固体分散体剂型可使用本领域技术人员熟知的各种技术配制,例如通过将药剂和聚合物共同溶解于溶剂中,随后喷雾干燥、喷雾凝结、蒸发、固化或微波处理、共混和直接压制、在升高但非熔融的温度下机械混杂、湿式粒化、挤压-球形化、熔体融合、热熔挤压等配制。
在适当选择一种或多种聚合物的情况下,BCS II药剂与所得制剂两者的溶解性可显著增加。通常使用聚合物(例如但不限于聚乙烯吡咯烷酮(PVP))以与药剂一起形成聚合基质
制备固体分散体的典型方法包括使聚合物和药剂共同溶解于溶剂中。材料可形成悬浮或不饱和混合物或饱和或过饱和基质-溶剂混合物。随后去除溶剂以留下药剂和作为基质的聚合物的复合混合物。非限制性地,溶剂去除方法包括沉淀、冷冻干燥、真空干燥或喷雾干燥。然而,鉴别有效溶解药剂和基质的常见溶剂或溶剂体系需要大量评价。
例如,如果聚合物和药剂的化学要求需要用以共同溶解其的溶剂的量较大,则去除溶剂的过程昂贵且不切实际。尽管可发现合适体积的合适溶剂,但配制者视为合适的那些溶剂可被FDA视为有毒,由此使得其药物用途不切实际。或者,使用表面活性剂和增溶剂减少所用溶剂的量可导致剂型中的药剂负载不足和表面活性剂的浓度高。制剂的这类变化的性质在最好的情况下可能是商业上不可行的,或者是耐受性不佳的,或者在最坏的情况下甚至是有毒的。
尽管固体分散技术已用以改进多种用于药物用途的市售BCS II药剂的溶解性,但对固体分散制剂的需求阻碍有效实施所述技术的能力,所述固体分散制剂在基质形成之后呈溶液形式以及呈固态时在聚合物基质与药剂之间形成物理和化学稳定混合物。
例如,极性聚合基质可增强溶解,但当极性聚合物与亲脂性药剂共同溶解时,材料本身可易于发生相分离。如果极性聚合物也具有吸湿性,则这种趋势可被放大。两种情况的结果是物理稳定性降低。相反,防止药剂在基质内发生相变化的稳定基质需要低分子移动性。提供低分子移动性的聚合物通常具有高分子量,增加了寻找药剂与聚合物两者的常见溶剂的困难。如果使用极性较小的聚合物制造基质以更易于寻找常见溶剂,则可损害溶解速率。此外,十分需要寻找最佳固体分散制剂与可行的市售制造方法的组合。
尽管经过了多年的研究和开发并且尽管其理论上有前景,但固体分散方法的实际应用仍受需要使用试误法以开发用于具体药剂的具体基质所限制,这是因为不能科学地理解在形成聚合基质中药剂与聚合物的相互作用。此外,构建这类制剂的许多要求彼此不相容,包括BCS II药剂的低吸湿性与聚合物具有高吸湿性的组合、需要快速溶解同时维持药剂-聚合物基质的长期物理和化学稳定性、当按比例扩大固体分散体时需要易于工业制造。
例如,国际专利公开案WO2005/084639描述含有具有低口服生物可利用性的第II类药物以及共同溶解于常见溶剂中的疏水性聚合物的制剂,其中溶液形成为小固体粒子且分散于聚合基质中。生物可利用性的增强经由由于稳定的微米尺寸化而增加溶解动力学和在胃肠道中自聚合物快速释放发生。
在另一实施例中,美国专利公开案US2009/0098200描述包含分散且特征化于聚合物基质中的可溶性不佳的生物活性化合物的固体分散体,所述聚合物基质可包含多于一种聚合物。
尽管已提出多种且不同的方法以制备可溶性不佳的药剂的制剂,使用所提出的制剂中的任一种的可行性需要针对各药剂进行大量评价。
因此,在本领域中仍持续需要且市场不断需求易于给药、具有增加的剂量负载和改进的溶解以减少食物影响的可用于特定药剂的药物组合物。
制剂可使用由被视为安全且有效的材料构成的药学上可接受的载剂制备,并且可向受试者给药而不产生不良生物学副作用或不合需要的相互作用。
如本文所用,术语“载剂”是指除活性成分外的药学制剂中所存在的所有组分,并且包括(但不限于)稀释剂、粘合剂、润滑剂、崩解剂、稳定剂、表面活性剂、着色剂或填充剂。
固体分散体(如本文提供的化合物1的喷雾干燥的中间体)可以制剂的常规形式向患者经口或肠胃外给药,所述制剂的常规形式是如胶囊、微胶囊、片剂、颗粒剂、粉剂、糖衣锭、丸剂、栓剂、混悬剂和糖浆。合适的制剂可通过通常采用的方法使用常规有机或无机添加剂制备,所述添加剂是诸如选自以下的赋形剂:填充剂或稀释剂、粘合剂、崩解剂、润滑剂、调味剂、防腐剂、稳定剂、悬浮剂、分散剂、表面活性剂、抗氧化剂或增溶剂。
在一个方面中,本文提供的化合物1的喷雾干燥的中间体使用胶囊剂型组合物经口给予,其中胶囊含有本文提供的化合物1的喷雾干燥的中间体且有或没有另外载剂、赋形剂或媒介物。胶囊可通过混合本文提供的化合物1的喷雾干燥的中间体与合适的载剂或稀释剂且将适当量的化合物1的喷雾干燥的中间体或混合物填充在胶囊中来制备。
在另一方面中,本文提供包含有效量的本文提供的化合物1和药学上可接受的载剂或媒介物的组合物,其中药学上可接受的载剂或媒介物可包含赋形剂、稀释剂或其混合物。组合物可配制成在剂量单位中含有日剂量或日剂量的便利部分。通常,组合物根据已知方法制备为片剂。
在一个方面中,组合物为药物组合物。
本文所述的药物组合物包含喷雾干燥的中间体与一种或多种药学上可接受的赋形剂的紧密混杂物以提供生物可利用口服剂型。
本文所述的喷雾干燥的中间体包含非晶形式的化合物1和聚合物。
在一个方面中,用于制备中间体的化合物1的形式为非晶多晶型物。非晶形式的优势在于某些性质,所述性质使所述形式可修改以用于与另外赋形剂的干燥共混物中。有利的非晶形式性质包括降低的粒度、增加的粒子分布和较佳的流量特征、最终剂型中的分散性和含量均一性。
本文所述的非晶形式可使用本领域的技术人员已知的多种方法制备。制备非晶形式的技术是在本领域中熟知的且描述于本文中。喷雾干燥是选择用以制备包含非晶形式的化合物和聚合物的喷雾干燥的中间体的技术。
在一个方面中,用于制备喷雾干燥的中间体的化合物1的形式为结晶形式。结晶形式的优势在于更高效制造中间体,其中液体分散体包含所述结晶形式,聚合物和任选的赋形剂共同溶解于溶剂体系中以形成液体分散体。
所用的溶剂体系可包含某些比率的一种或多种溶剂,其中所述溶剂比率提供溶解化合物1和聚合物以制备液体分散体的最佳过程。溶剂体系中溶剂的最佳混合物和比率取决于剂量负载与所用聚合物的量和类型、尤其是聚合物的分子量的平衡。
本文所述的方面包括一种或多种选自以下的溶剂:THF (四氢呋喃)、MeOH (甲醇)、EtOH (乙醇)、丙酮、EtOH-95 (95%酒精纯度的乙醇)、无水EtOH (99.99%酒精纯度的乙醇)、DCM (二氯甲烷)、IPA (异丙醇)、DMSO (二甲亚砜)、DMF (二甲基甲酰胺)、水或其混合物。
用于如本文所述用途的溶剂体系的方面可包含DCM与至少一种其他溶剂的混合物。用于如本文所述用途的包含DCM与至少一种其他溶剂的混合物的溶剂体系的一个方面可包含选自以下的溶剂体系:DCM:丙酮、DCM:DMSO、DCM:EtOH-95、DCM:EtOH-无水、DCM:IPA、DCM:MeOH或DCM:THF。某些方面可包含选自DCM:DMSO或DCM:MeOH的溶剂的混合物。
用于如本文所述的溶剂体系混合物中的DCM的量包括约5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%、95%或100%的量。在一个方面中,用于如本文所述的溶剂体系混合物中的DCM的量包括在约5%至约10%、约10%至约15%、约15%至约20%、约20%至约25%、约25%至约30%、约30%至约35%、约35%至约40%、约40%至约45%、约45%至约50%、约50%至约55%、约55%至约60%、约60%至约65%、约65%至约70%、约70%至约75%、约75%至约80%、约80%至约85%、约85%至约90%、约90%至约95%或约95%至约100%的范围内的量。
用于如本文所述的与DCM的溶剂体系混合物中的其他溶剂的量包括约0%、5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%或95%的量。在一个方面中,用于如本文所述的与DCM的溶剂体系混合物中的其他溶剂的量包括在约0%至约5%、约5%至约10%、约10%至约15%、约15%至约20%、约20%至约25%、约25%至约30%、约30%至约35%、约35%至约40%、约40%至约45%、约45%至约50%、约50%至约55%、约55%至约60%、约60%至约65%、约65%至约70%、约70%至约75%、约75%至约80%、约80%至约85%、约85%至约90%或约90%至约95%的范围内的量。
在一个方面中,用于如本文所述的溶剂体系中的DCM的量包括约10%至约100%的范围内的量、约30%至约87%的范围内的量或约50%至约86%的范围内的量、约33%至约87%的范围内的量、约65%至约87%的范围内的量。
在一个方面中,用于如本文所述的与DCM的溶剂体系混合物中的其他溶剂的量包括约0%至约100%的范围内的量、约5%至约13%的范围内的量或约50%至约86%的范围内的量、约33%至约87.5%的范围内的量、约65%至约87.5%的范围内的量。
在一个方面中,用于如本文所述的与DCM的溶剂体系混合物中的DCM和其他溶剂的量的比率(其中所述比率表示为DCM:溶剂)包括DCM:丙酮、DCM:DMSO、DCM:EtOH-95、DCM:EtOH-无水、DCM:IPA、DCM:MeOH或DCM:THF的比率。
在一个方面中,DCM:丙酮的比率可为约50:50。在一个方面中,DCM:DMSO的比率可为约50:50、约65:35、约77:23、约80:20或约95:5。在一个方面中,DCM:EtOH的比率可为约80:20。在一个方面中,DCM:EtOH-95的比率可为约50:50、约80:20、约86:14、87:13或约87.5:12.5。在一个方面中,DCM:IPA的比率可为约50:50。在一个方面中,DCM:MeOH的比率可为约50:50、约80:20、约86:14、87:13或约87.5:12.5。在一个方面中,DCM:THF的比率可为约33:67。
最佳溶剂体系设计中的另一因素包括所用赋形剂、尤其是影响聚合物和相应喷雾干燥的中间体聚合物基质的两亲性特征的赋形剂的量和类型,其中聚合物与化合物1的疏水性和亲水性相互作用最终在胃环境中受影响。
在一个方面中,所用聚合物为亲水性聚合物。
影响药物自亲水性基质释放的因素中的一种包括聚合物的粘度、聚合物与药物的比率、聚合物的混合物、压制压力、片剂的厚度、药物的粒度、基质的pH、片剂中留截的空气、药物的分子尺寸、药物的分子几何形状、药物的溶解性、赋形剂或添加剂的存在和这些物质的并入模式(Patel VF, Patel NM. Statistical Evaluation of Influence ofViscosity and Content of Polymer on Dipyridamole Release From Floating MatrixTablets: A Technical Note. AAPS PharmSciTech. 2007; 8(3): Article 69)。
在一个方面中,所用聚合物选自PVP。
还涵盖一种用于制备固体分散体的方法,所述固体分散体包含非晶形式的化合物1和聚合物。
固体分散体可使用本领域的技术人员已知的基质形成方法中的任一种制成,所述基质形成方法包括(但不限于):溶剂蒸发、溶剂去除、喷雾干燥、相转化封装、自发乳化、凝聚、热熔封装、热熔挤压、喷雾-凝结、造粒和研磨。应理解,固体分散体可使用标准制药技术中的任一种进一步加工成口服剂型,所述标准制药技术包括(但不限于)压片、挤压-球形化和用于多微粒剂型的流体化床涂覆和胶囊填充。
尽管粘着性和预防聚集的主要来源是形成基质的一种或多种聚合物的性质且制备方法为本领域的技术人员已知的,所以需要大量评价以制备包含非晶形式的化合物1和聚合物的固体分散体的聚合基质。
在一个方面中,所述方法包括使化合物1和聚合物共同溶解于溶剂体系中以形成液体分散体,随后去除溶剂。
在一个方面中,所形成的中间体为固体分散体。
在一个方面中,本文中形成固体分散体的方法包括通过合适的方式去除溶剂,所述方式包括喷雾干燥含有溶解的聚合物和分散的化合物1的精细粒子的溶液。另一方法涉及溶解聚合物和溶解或悬浮化合物,并且随后用大体积的用于聚合物和化合物1的反溶剂稀释溶液,其中溶剂大体上可与反溶剂混溶。
在一个方面中,溶液包含共同溶解于互溶剂中,并且随后喷雾干燥以形成微粒的化合物-聚合物混合物。聚合物体系充当用于由于通过维持微米尺寸化化合物粒度来增加表面积而使化合物1更快速溶解的基质。
所得喷雾干燥的中间体(SDI)随后与用于制备用于口服给药的片剂或胶囊剂型的合适药物赋形剂合并。
喷雾干燥溶液中化合物1的剂量负载可在约1%至约90% (w/w)、约1%至约50% w/w、约20%至约70% w/w、约20%至约60% w/w、约30%至约40% w/w或约20%至约30% w/w范围内。
在一个方面中,当获得喷雾干燥的中间体时,形成非晶形式的化合物1。
还描述喷雾干燥的中间体在药物组合物中的用途,所述药物组合物包含中间体与一种或多种药学上可接受的赋形剂的紧密混杂物以提供生物可利用口服剂型。
赋形剂
制剂可包含一种或多种赋形剂。合适的赋形剂包括溶剂、共溶剂、乳化剂、塑化剂、表面活性剂、增稠剂、pH改性剂、润滑剂、抗氧化剂和螯合剂、润湿剂和吸水剂。制剂还可包含一种或多种添加剂,例如染料、有色颜料、珠光剂、除臭剂和气味遮蔽剂。
可选择的其他合适赋形剂为本领域的技术人员已知的且包括(但不限于)填充剂或稀释剂(例如蔗糖、淀粉、甘露糖醇、山梨糖醇、乳糖、葡萄糖、纤维素、滑石、磷酸钙或碳酸钙等)、粘合剂(例如纤维素、羧甲基纤维素、甲基纤维素、羟甲基纤维素、羟丙基甲基纤维素、聚丙基吡咯烷酮、聚乙烯吡咯烷酮、明胶、阿拉伯胶、聚乙二醇或淀粉等)、崩解剂(例如羟基乙酸淀粉钠、交联羧甲基纤维素钠等)、润滑剂(例如硬脂酸镁、轻质无水硅酸、滑石或月桂基硫酸钠等)、调味剂(例如柠檬酸或薄荷醇等)、防腐剂(例如苯甲酸钠、亚硫酸氢钠、对羟基苯甲酸甲酯或对羟基苯甲酸丙酯等)、稳定剂(例如柠檬酸、柠檬酸钠或乙酸等)、悬浮剂(例如甲基纤维素、聚乙烯吡咯烷酮或硬脂酸铝等)、分散剂(例如羟丙基甲基纤维素等)、表面活性剂(例如月桂基硫酸钠、泊洛沙姆(polaxamer)、聚山梨醇酯等)、抗氧化剂(例如乙二胺四乙酸(EDTA)、丁基化羟基甲苯(BHT)等)和增溶剂(例如聚乙二醇、SOLUTOL®、GELUCIRE®等)。药物组合物中本文提供的化合物1的有效量可处于将发挥所需效果的水平。
稀释剂,在本文中也称为“填充剂”,通常需要增加固体剂型的体积,以使得提供实际尺寸用于压制片剂或形成珠粒和颗粒。合适稀释剂包括(但不限于)脱水磷酸二钙、硫酸钙、乳糖、蔗糖、甘露糖醇、山梨糖醇、纤维素、微晶纤维素、高岭土、氯化钠、干淀粉、水解淀粉、预胶凝化淀粉、二氧化硅、氧化钛、硅酸镁铝和粉末糖。
分散剂除其他水之外还包括磷酸盐缓冲盐水(PBS)、生理盐水、葡萄糖、月桂基硫酸钠(SLS)、聚乙烯吡咯烷酮(PVP)、聚乙二醇(PEG)和羟丙基甲基纤维素(HPMC,使用粘合剂为固体剂型赋予粘聚性,且因此确保片剂、珠粒或颗粒在形成剂型之后仍保持完整。合适的粘合剂材料包括(但不限于)淀粉、预胶凝化淀粉、明胶、糖(包括蔗糖、葡萄糖、右旋糖、乳糖和山梨糖醇)、聚乙二醇、蜡、天然和合成胶状物(如阿拉伯胶、黄蓍胶、海藻酸钠)、纤维素(包括羟丙基甲基纤维素(“HPMC”)、微晶纤维素(“MCC”)、羟丙基纤维素、乙基纤维素和维格姆(veegum))和合成聚合物(如丙烯酸和甲基丙烯酸共聚物、甲基丙烯酸共聚物、甲基丙烯酸甲酯共聚物、甲基丙烯酸氨基烷基酯共聚物、聚丙烯酸/聚甲基丙烯酸和聚乙烯吡咯烷酮(PVP))。
使用润滑剂有助于片剂制造。合适润滑剂的实例包括(但不限于)硬脂酸镁、硬脂酸钙、硬脂酸、山嵛酸甘油酯、聚乙二醇、滑石、硬脂酰反丁烯二酸钠、烟雾状二氧化硅和矿物油。
使用崩解剂以有助于剂型崩解或在给予之后“分解”,且通常包括(但不限于)淀粉、羟基乙酸淀粉钠、羧甲基淀粉钠、羧甲基纤维素钠、羟丙基纤维素、预胶凝化淀粉、粘土、纤维素、海藻素、胶状物或交联聚合物,如交联PVP (交联聚维酮,POLYPLASDONE XL)、交联羧甲基纤维素钠。
使用稳定剂以抑制或延缓活性成分分解反应,其包括例如氧化反应。
表面活性剂可为阴离子型、阳离子型、两性或非离子型表面活性剂。合适阴离子型表面活性剂包括(但不限于)含有羧酸根、磺酸根和硫酸根离子的那些表面活性剂。阴离子型表面活性剂的实例包含长链烷基磺酸和烷基芳基磺酸的钠、钾、铵盐,如十二烷基苯磺酸钠;磺基丁二酸二烷基钠,如十二烷基苯磺酸钠;磺基丁二酸二烷基钠,如双-(2-乙基硫氧基)-磺基丁二酸钠;和烷基硫酸盐,如月桂基硫酸钠。
阳离子型表面活性剂包括(但不限于)季铵化合物,如氯化苯甲烃铵、苄索氯铵、溴化十六烷基三甲基铵、氯化硬酯酰基二甲基苯甲基铵、聚氧化乙烯和椰子胺。非离子型表面活性剂的实例包括乙二醇单硬脂酸酯、丙二醇肉豆蔻酸酯、单硬脂酸甘油酯、硬脂酸甘油酯、聚甘油基-4-油酸酯、脱水山梨糖醇酰化物、蔗糖酰化物、PEG-150月桂酸酯、PEG-400单月桂酸酯、聚氧化乙烯单月桂酸酯、聚山梨醇酯、聚氧化乙烯辛基苯基醚、PEG-1000鲸蜡基醚、聚氧化乙烯十三烷基醚、聚丙二醇丁基醚、I泊洛沙姆401 (I Poloxamer 401)、硬脂酰基单异丙醇酰胺和聚氧化乙烯氢化动物脂酰胺。两性表面活性剂的实例包括N-十二烷基-p-丙氨酸钠、N-月桂基-亚氨基二丙酸钠、肉豆蔻酰两性乙酸酯、月桂基甜菜碱和月桂基磺基甜菜碱。
必要时,片剂、珠粒、颗粒或粒子还可含有少量无毒辅助物质,如润湿剂或乳化剂、染料、pH缓冲剂或防腐剂。
制剂可呈片剂、胶囊、迷你片(minitab)、填充片剂、渗透装置、浆料、分散体或悬浮液的形式。在一个优选方面中,制剂为固体口服剂型,如片剂、多微粒组合物或胶囊。
化合物1可以制剂给予,其中包含呈非晶形式的化合物1和亲水聚合物(如PVP)的喷雾干燥的中间体呈与一种或多种药学上可接受的载剂、赋形剂或稀释剂的混杂物形式。药物制剂可使用本领域的技术人员已知的标准程序制造。
立即释放
在一个方面中,组合物包括在立即释放制剂中。化合物1优选呈包含非晶形式的化合物1和聚合物的喷雾干燥的中间体的微粒的形式。微粒通过聚合物而对聚集稳定;因此,可使用标准片剂或胶囊口服剂型中的任一种。微粒可进一步配制成用于经口给予的片剂、浆料或分散体,或置于胶囊(如明胶胶囊)中。
喷雾干燥的中间体的聚合物的基质优选为多孔的,或另外使化合物1便于溶解于胃肠道液中。这种情况使得化合物1快速溶解,而不因未溶解粒子的聚结而减小有效粒子面积。具有生物粘着性的基质通过趋于使粒子保留于胃或肠上段中而进一步增强吸收,同时化合物得以吸收。
控制释放
延时释放/缓释药物组合物可通过将SDI与药学上可接受的离子交换树脂复合和涂覆这类复合物而获得。SDI涂覆有将充当障壁以控制化合物1自其核心复合物向胃肠液中扩散的物质。任选地,SDI涂覆有不可溶于胃的酸环境中但可溶于下段胃肠道的碱性环境中的聚合物膜,以获得在胃内释放小于10%的剂量负载的最终剂型。
合适的控制释放涂层材料的实例包括(但不限于)纤维素聚合物,如邻苯二甲酸乙酸纤维素、羟丙基纤维素、羟丙基甲基纤维素、羟丙基甲基纤维素邻苯二甲酸酯和羟丙基甲基纤维素乙酸酯丁二酸酯;聚乙酸乙烯酯邻苯二甲酸酯、多糖、丙烯酸聚合物和共聚物或甲基丙烯酸树脂。
另外,涂覆材料可含有常规载剂;诸如塑化剂、颜料、着色剂、滑动剂、稳定剂、成孔剂和表面活性剂。
因此,释放速率可通过用糖、肠聚合物或明胶涂覆片剂以改变片剂的溶解来改变。可通过用亲水性聚合物(如HPMC (其是具有增加的聚合物长度和较高粘度的等级)或明胶)涂覆来防止片剂在口腔中过早溶解,从而致使溶解于胃中。
组合物还可设计成以通过增加化合物1与载剂的比率而延长释放时间段,释放拉长至在约90分钟内达到约80% (体外)。据信增加的相对化合物浓度具有增加聚合物基质内有效药物功能域尺寸的作用。增加的药物功能域尺寸致使药物较慢溶解。在聚合物基质含有某些类型的亲水性聚合物的情况下,聚合物将充当粘膜粘着性材料且增加药物在胃肠道中的滞留时间。增加的药物溶解速率与聚合物基质的粘膜粘着性质组合致使(1)药物吸收增加和(2) BCS第II类药物在喂饲和禁食状态所见的差异减少。
本文所述的口服剂型可用以治疗多种疾病和病症。相比于不含生物粘着性聚合物的制剂,这些制剂具有改进的生物可利用性。制剂被设计成有助于使药物扩散到肠组织中。制剂可设计成以缓慢、快速或逐步(脉动)方式释放药物。
因此,本申请提供具有增加的剂量负载和改进的溶解性以不受食物影响的药物组合物。
使用方法
本文还涵盖治疗本文所述的病况的方法。在一个方面中,治疗本文所述的病况的方法涉及向有需要的患者给予作为单一药剂疗法的化合物1。在一个特定方面中,本文提供一种治疗本文所述的病况的方法,其包括向有需要的患者给予有效量的作为单一药剂的化合物1。在另一方面中,本文提供一种治疗本文所述的病况的方法,其包括向有需要的患者给予包含作为单一活性成分的化合物1和药学上可接受的载剂、赋形剂或媒介物的药物组合物。
在另一方面中,治疗本文所述的病况的方法涉及向有需要的患者给予化合物1与另一疗法的组合。这类方法可涉及在给予另外疗法之前、同时或之后给予化合物1。在某些方面中,这类方法具有累加或协同效应。在一个特定方面中,本文提供一种治疗本文所述的病况的方法,其包括向有需要的患者给予有效量的化合物1和有效量的另一疗法。
在特定方面中,任何可抑制DHODH产生的病况均可根据本文提供的方法治疗。在另一特定方面中,根据本文提供的方法治疗的病况为选自由以下组成的组的白血病:急性或慢性白血病,其中急性白血病选自急性淋巴细胞性白血病、急性髓细胞性白血病(如成髓细胞性、原髓细胞性、髓单核细胞性、单核细胞性、红细胞白血病性白血病)或骨髓增生异常综合征;且其中慢性白血病选自慢性髓细胞(myclocytic)(粒细胞(granulocytic))白血病、慢性淋巴细胞性白血病、毛细胞白血病;或真性红细胞增多症;等等。
剂量和给药
根据治疗本文所述的病况的方法,可通过多种途径向有需要的受试者给予产生有益或治疗效果的量的化合物或其药物组合物。化合物1或其药物组合物可根据治疗本文所述的病况的方法经口给予有需要的受试者。经口给予化合物1或其药物组合物可有助于需要这类治疗的受试者遵循获取化合物或药物组合物的方案。因此,在一个特定方面中,化合物1或其药物组合物经口给予有需要的受试者。
本文提供的化合物1的药物组合物或形式可在有或没有食物或水的情况下经口给予。
其他给予途径包括(但不限于)静脉内、皮内、鞘内、肌肉内、皮下、鼻内、吸入、经皮、局部、经粘膜、颅内、肿瘤内、硬膜外和滑膜内给予途径。在一个方面中,化合物1或其药物组合物全身性(例如肠道外)给予有需要的受试者。在另一方面中,化合物1或其药物组合物局部(例如瘤内)给予有需要的受试者。在一个方面中,化合物1或其药物组合物经由允许化合物穿过血脑障壁的途径(例如经口)给予。
根据涉及给予化合物1与一种或多种另外疗法组合的治疗本文所述的病况的方法,化合物和一种或多种另外疗法可通过相同途径或不同给予途径给予。
根据治疗本文所述的病况的方法将有效同时使任何副作用降至最低,向有需要的受试者给予一定给予剂量和频率的化合物1或其药物组合物。化合物1或其药物组合物的确切给予剂量和频率可由从业者根据与需要治疗的受试者相关的因素来确定。可考虑的因素包括疾病病况的严重程度、受试者的一般健康状况、受试者的年龄、体重和性别、膳食、给药的时间和频率、一种或多种药物组合、反应敏感性和对疗法的耐受性/反应。化合物1或其药物组合物的给予剂量和频率可随时间调节以提供足够量的化合物或维持所需效果。
在某些方面中,化合物1或其药物组合物根据治疗本文所述的病况的方法一天一次、一天两次、一天三次或一天四次向受试者给予。在一些方面中,化合物1或其药物组合物根据治疗本文所述的病况的方法每隔一天(即隔天)一次、两次、三次或四次,每两天一次、两次、三次或四次,每三天一次,每四天一次、两次、三次或四次,每5天一次、两次、三次或四次,一周一次、两次、三次或四次,每两周一次、两次、三次或四次,每三周一次、两次、三次或四次,每四周一次、两次、三次或四次,每5周一次、两次、三次或四次,每6周一次、两次、三次或四次,每7周一次、两次、三次或四次,或每8周一次、两次、三次或四次向受试者给予。在特定方面中,化合物1或其药物组合物根据治疗本文所述的病况的方法周期性给予受试者,其中给予所述化合物或药物组合物一段时间,之后停药一段时间(即一段时间不给予化合物或药物组合物)。
在某些方面中,化合物1或其药物组合物根据治疗本文提供的赘瘤的方法以实现以下中的一项或多项的给予剂量和频率向有需要的受试者给予:(i)降低DHODH或其他血管生成或炎症介质的产生和/或浓度或肿瘤血流或代谢的变化,或患有本文所述的病况的受试者或具有预先建立的人肿瘤的动物模型的瘤周发炎或水肿;(ii)降低以下患有赘瘤的受试者或具有预先建立的人肿瘤的动物模型中的一个、两个、三个或更多个或所有的DHODH浓度;(iii)减轻或改善患有赘瘤的受试者的赘瘤和/或与其相关的一种或多种症状的严重程度;(iv)减少患有赘瘤的受试者中与赘瘤相关的症状的数目和/或一种或多种症状的持续期间;(v)防止患有赘瘤的受试者或具有预先建立的人肿瘤的动物模型中与赘瘤相关的一种或多种症状的发作、进展或复发;(vi)减小患有赘瘤的受试者或具有预先建立的人肿瘤的动物模型中的肿瘤的尺寸;(vii)减少受试者或具有预先建立的人肿瘤的动物模型中与恶性赘瘤相关的血管生成;和/或(vii)增强或改进患有赘瘤的受试者或具有预先建立的人肿瘤的动物模型中另一疗法的治疗效果。
在某些方面中,化合物1或其药物组合物根据治疗本文提供的赘生性或非赘生性病况的方法以产生以下中的一项或多项的给予剂量和频率向有需要的受试者给予:(i)减少患有赘生性或非赘生性病况的受试者或具有预先建立的人肿瘤的动物模型的血液中的循环肿瘤细胞(CTC)的数目;(ii)减少与具有病况的受试者的血液中的肿瘤细胞相关的循环DNA或RNA;(iii)患有赘生性或非赘生性病况的患者的存活期是约6个月或更长、约7个月或更长、约8个月或更长、约9个月或更长、或约12个月或更长;(iv)消退患有赘生性病况的受试者或具有预先建立的人肿瘤的动物模型中与赘生性病况相关的肿瘤和/或抑制与赘生性病况相关的肿瘤的进展;(v)降低患有赘瘤的受试者或具有预先建立的人肿瘤的动物模型中赘瘤的生长和/或减小与所述赘瘤相关的肿瘤的肿瘤尺寸(例如体积、横截面积或直径);(vi)在患有赘瘤的受试者或具有预先建立的人肿瘤的动物模型中在给予标准疗法之后维持与赘瘤相关的肿瘤的尺寸和/或肿瘤不增加或其增加小于类似肿瘤的增加,如通过本领域的技术人员可获得的常规方法所测量,如直肠指检、超声波(例如经直肠超声)、CT扫描、PET扫描、DCE-MRI和MRI;(vii)减少患有赘瘤的受试者或具有预先建立的人肿瘤的动物模型中与赘瘤相关的肿瘤形成;(viii)根除、去除或控制患有赘瘤的受试者或具有预先建立的人肿瘤的动物模型中与赘瘤相关的原发性、局部和/或转移性肿瘤;(ix)减少患有赘瘤的受试者或具有预先建立的人肿瘤的动物模型中与恶性赘瘤相关的转移灶的数目或尺寸;(x)减少或抑制肿瘤复发;(xi)减少与肿瘤相关的水肿或发炎;(xii)抑制或减少肿瘤血管形成;(xiii)减少病理血管生成;和/或(x)减少患有恶性赘瘤的受试者或具有预先建立的人肿瘤的动物模型中预先建立的肿瘤或赘瘤的生长和/或减小预先建立的肿瘤的肿瘤尺寸(例如体积、横截面积或直径)。
在某些方面中,化合物1或其药物组合物根据治疗本文提供的非赘生性病况的方法以实现以下中的一项或多项的给予剂量和频率向有需要的受试者给予:(i)降低DHODH或其他血管生成或炎症介质的产生或浓度;(ii)降低以下患有非赘生性病况的受试者或动物模型中的一个、两个、三个或更多个或所有的DHODH的浓度;(iii)减轻或改善患有非赘生性病况的受试者的非赘生性病况和/或与其相关的一种或多种症状的严重程度;(iv)减少患有非赘生性病况的受试者中与非赘生性病况相关的症状的数目和/或一种或多种症状的持续期间;(v)防止患有非赘生性病况的受试者中与非赘生性病况相关的一种或多种症状的发作、进展或复发;(vi)减轻与非赘生性病况相关的发炎;(vii)减少受试者或动物模型中与非赘生性病况相关的病理性血管生成;和/或(vii)增强或改进患有非赘生性病况的受试者或动物模型中另一疗法的治疗效果。
在一个方面中,治疗本文所述的病况的方法涉及给予单位剂量的化合物1或其药物组合物。剂量可以经确定有效的频率给予(例如每天一次、两次或三次,每隔一天,每周一次或两次,两周一次或每月一次)。在某些方面中,治疗本文所述的病况的方法涉及向有需要的受试者给予以下范围内的单位剂量的化合物1或其药物组合物:约0.1毫克(mg)至约30000 mg、约1 mg至约10000 mg、约5 mg至约1000 mg、约10 mg至约500 mg、约100 mg至约500 mg、约150 mg至约500 mg、约150 mg至约8000 mg、约250 mg至约8000 mg、约300 mg至约8000 mg、或约500 mg至约8000 mg或其间的任何范围。在一些方面中,治疗本文所述的病况的方法涉及向有需要的受试者给予以下单位剂量的化合物或其药物组合物:约15 mg、16mg、17 mg、18 mg、19 mg、20 mg、21、mg、22 mg、23 mg、24 mg、25 mg、26 mg、27 mg、28 mg、29mg、30 mg或40 mg。在某些方面中,治疗本文所述的病况的方法涉及向有需要的受试者给予以下单位剂量的化合物1或其药物组合物:约50 mg、60 mg、70 mg、80 mg、90 mg、100 mg、110 mg、120 mg、125 mg、130 mg、140 mg、150 mg、175 mg、200 mg、250 mg、300 mg、350 mg、400 mg、450 mg、500 mg、550 mg、600 mg、650 mg、700 mg、750 mg、800 mg、850 mg、900 mg、1000 mg、1600 mg、3200 mg、4000 mg、4500 mg、5000 mg、5500 mg、6000 mg、7000 mg、7500mg、8000 mg等。
在一些方面中,治疗本文所述的病况的方法涉及向有需要的受试者给予以下单位剂量的化合物1或其药物组合物:至少约0.1 mg、1 mg、5 mg、10 mg、20 mg、30 mg、40 mg、50mg、60 mg、70 mg、80 mg、90 mg、100 mg、110 mg、120 mg、125 mg、130 mg、140 mg、150 mg、175 mg、200 mg、250 mg、300 mg、350 mg、400 mg、450 mg、500 mg、550 mg、600 mg、650 mg、700 mg、750 mg、800 mg、850 mg、900 mg、1000 mg、1600 mg、3200 mg、4000 mg、4500 mg、5000 mg、5500 mg、6000 mg、7000 mg、7500 mg、8000 mg等。在某些方面中,治疗本文所述的病况的方法涉及向有需要的受试者给予以下单位剂量的化合物1或其药物组合物:少于约35 mg、少于约40 mg、少于约45 mg、少于约50 mg、少于约60 mg、少于约70 mg或少于约80mg。
在特定方面中,治疗本文所述的病况的方法涉及向有需要的受试者给予以下单位剂量的化合物1或其药物组合物:约20 mg至约500 mg、约40 mg至约500 mg、约40 mg至约200 mg、约40 mg至约150 mg、约75 mg至约500 mg、约75 mg至约450 mg、约75 mg至约400mg、约75 mg至约350 mg、约75 mg至约300 mg、约75 mg至约250 mg、约75 mg至约200 mg、约100 mg至约200 mg或其间的任何范围。在其他特定方面中,治疗本文所述的病况的方法涉及向有需要的受试者给予以下单位剂量的化合物1或其药物组合物:约20 mg、35 mg、40mg、50 mg、60 mg、75 mg、100 mg、125 mg、150 mg、175 mg、200 mg、225 mg、250 mg或300mg。在一些方面中,治疗本文所述的病况的方法涉及向有需要的受试者给予以下单位剂量的化合物或其药物组合物:约350 mg、400 mg、500 mg、600 mg、700 mg、800 mg、900 mg、1000 mg、1600 mg、3200 mg、4000 mg、4500 mg、5000 mg、5500 mg、6000 mg、7000 mg、7500mg、8000 mg等。在一些方面中,单位剂量的化合物或其药物组合物每天一次、每天两次、每天三次;每隔一天(即隔天)一次、两次或三次;每两天一次、两次或三次;每三天一次、两次或三次;每四天一次、两次或三次;每五天一次、两次或三次;每一周、每两周或每月一次、两次或三次给予受试者,且所述剂量可经口给予。
在某些方面中,治疗本文所述的病况的方法涉及向有需要的受试者经口给予每天约20 mg至约500 mg范围内的单位剂量的化合物1或其药物组合物。在一些方面中,治疗本文所述的病况的方法涉及向有需要的受试者经口给予以下范围内的单位剂量的化合物1或其药物组合物:每天约80 mg至约500 mg、每天约100 mg至约500 mg、每天约80 mg至约400mg、每天约80 mg至约300 mg、每天约80 mg至约200 mg、每天约200 mg至约300 mg、每天约200 mg至约400 mg、每天约200 mg至约500 mg或其间的任何范围。
在一个特定方面中,治疗本文所述的病况的方法涉及每天一次经口给予约200 mg的单位剂量的化合物1或其药物组合物。在另一特定方面中,治疗本文所述的病况的方法涉及向有需要的受试者每天两次经口给予约100 mg的单位剂量的化合物1或其药物组合物。在另一具体面中,治疗本文所述的病况的方法涉及每天四次经口给予约50 mg的单位剂量的化合物1或其药物组合物。在特定方面中,治疗本文所述的病况的方法涉及向有需要的受试者每天两次经口给予约100 mg至约250 mg、约150 mg至约250 mg、约175 mg至约250 mg、约200 mg至约250 mg、或约200 mg至约225 mg的单位剂量的化合物1或其药物组合物。
在一些方面中,治疗本文所述的病况的方法涉及给予表示为毫克/平方米(mg/m2)的剂量的化合物1或其药物组合物。化合物的mg/m2可例如通过使动物的转化因子乘以以毫克/千克(mg/kg)为单位的动物剂量以获得以mg/m2为单位的人剂量等效物的剂量来确定。对于监管性意见,FDA可推荐以下转化因子:小鼠=3,仓鼠=4.1,大鼠=6,天竺鼠=7.7。(基于Freireich等,Cancer Chemother. Rep. 50(4):219-244 (1966))。人的高度和重量可应用博伊德氏体表面积公式(Boyd's Formula of Body Surface Area)用以计算人体表面积。在特定方面中,治疗本文所述的病况的方法涉及向有需要的受试者给予量在约0.1mg/m2至约1000 mg/m2的范围内或其间的任何范围的化合物或其药物组合物。
可用于治疗本文所述的病况的方法中的化合物1或药物组合物的其他非限制性示例性剂量包括每千克受试者或样品重量的毫克量。在某些方面中,治疗本文所述的病况的方法涉及向有需要的受试者给予以下范围内的剂量的化合物1或其药物组合物:约0.001mg/kg至约500 mg/kg、约0.01 mg/kg至约500 mg/kg、约0.1 mg/kg至约500 mg/kg、约1 mg/kg至约500 mg/kg、约10 mg/kg至约500 mg/kg、约100 mg至约500 mg/kg、约150 mg/kg至约500 mg/kg、约250 mg/kg至约500 mg/kg或约300 mg/kg至约500 mg/kg。在一些方面中,治疗本文所述的病况的方法涉及向有需要的受试者给予以下范围内的剂量的化合物1或其药物组合物:约0.001 mg/kg至约100 mg/kg、约0.001 mg/kg至约50 mg/kg、约0.001 mg/kg至约25 mg/kg、约0.001 mg/kg至约10 mg/kg、约0.001 mg/kg至约5 mg/kg;约0.001 mg/kg至约1 mg/kg;或约0.001 mg/kg至约0.01 mg/kg。根据这些方面,剂量可每天一次、两次或三次,每隔一天,或每周一次或两次给予,且剂量可经口给予。
在某些方面中,治疗本文所述的病况的方法涉及向有需要的受试者给予以下范围内的剂量的化合物1或其药物组合物:约0.01 mg/kg至约100 mg/kg、约0.01 mg/kg至约50mg/kg、约0.01 mg/kg至约25 mg/kg、约0.01 mg/kg至约10 mg/kg、约0.01 mg/kg至约5 mg/kg、约0.01 mg至约1 mg/kg或约0.01 mg/kg至约0.1 mg/kg。在一些方面中,治疗本文所述的病况的方法涉及向有需要的受试者给予以下范围内的剂量的化合物1或其药物组合物:约0.1 mg/kg至约100 mg/kg、约0.1 mg/kg至约50 mg/kg、约0.1 mg/kg至约25 mg/kg、约0.1 mg/kg至约10 mg/kg、约0.1 mg/kg至约5 mg/kg、约0.1 mg/kg至约4 mg/kg;约0.1 mg/kg至约3 mg/kg;约0.1 mg/kg至约2 mg/kg;约0.1 mg至约1.5 mg/kg、约0.1 mg至约1.2mg/kg、约0.1 mg至约1 mg/kg或约0.5 mg/kg至约1.5 mg/kg。根据这些方面,剂量可每天一次、两次或三次,每隔一天,或每周一次或两次给予,且剂量可经口给予。
在特定方面中,治疗本文所述的病况的方法涉及向有需要的受试者经口给予以下剂量的化合物1或其药物组合物:每天两次给予约0.1 mg/kg至约5 mg/kg、约0.1 mg/kg至约4 mg/kg、约0.1 mg/kg至约3 mg/kg、约0.1 mg/kg至约2 mg/kg、约0.5 mg/kg至约2 mg/kg或约1 mg/kg至约1.5 mg/kg。在某些方面中,治疗本文所述的病况的方法涉及向有需要的受试者每天两次经口给予以下剂量的化合物1或其药物组合物:约0.1 mg/kg、约0.2 mg/kg、约0.3 mg/kg、约0.4 mg/kg、约0.5 mg/kg、约0.6 mg/kg、约0.7 mg/kg、约0.8 mg/kg、约0.9 mg/kg或约1 mg/kg。在某些特定方面中,治疗本文所述的病况的方法涉及向有需要的受试者每天两次经口给予以下剂量的化合物1或其药物组合物:约1.1 mg/kg、约1.2 mg/kg、约1.3 mg/kg、约1.4 mg/kg、约1.5 mg/kg、约1.6 mg/kg、约1.7 mg/kg、约1.8 mg/kg,1.9 mg/kg或约2 mg/kg。
在特定方面中,治疗本文所述的病况的方法涉及向有需要的受试者给予一定剂量的化合物1或其药物组合物,所述剂量在患有赘生性或非赘生性病况的受试者或动物模型(例如具有预先建立的人肿瘤的动物模型)中实现化合物的目标血浆浓度。在一个特定方面中,治疗本文所述的病况的方法涉及向有需要的受试者给予一定剂量的化合物1或其药物组合物,所述剂量在患有赘生性或非赘生性病况的受试者或动物模型(例如具有预先建立的人肿瘤的动物模型)中实现大约0.001 μg/mL至大约100 mg/mL、大约0.01 μg/mL至大约100 mg/mL、大约0.01 μg/mL至大约10 mg/mL、大约0.1 μg/mL至大约10 mg/mL、大约0.1 μg/mL至大约500 μg/mL、大约0.1 μg/mL至大约200 μg/mL、大约0.1 μg/mL至大约100 μg/mL或大约0.1 μg/mL至大约75 μg/mL范围内的化合物血浆浓度。在特定方面中,治疗本文所述的病况的方法涉及向有需要的受试者给予一定剂量的化合物1或其药物组合物,所述剂量在患有赘生性或非赘生性病况的受试者或动物模型(例如具有预先建立的人肿瘤的动物模型)中实现大约0.1至大约50 μg/mL、大约0.1 μg/mL至大约25 μg/mL、大约0.1 μg/mL至大约20 μg/mL或大约5 μg/mL至大约10 μg/mL范围内的化合物血浆浓度。为获得这类血浆浓度,可视给予途径而定给予剂量在0.001 μg至100,000 mg内变化的化合物1或其药物组合物。在某些方面中,因此可基于向受试者给予初始剂量的化合物或其药物组合物下达到的化合物的血浆浓度调节化合物1的后续剂量。
在特定方面中,治疗本文所述的病况的方法涉及向有需要的受试者给予一定剂量的化合物1或其药物组合物,所述剂量在患有赘生性或非赘生性病况的受试者或动物模型(例如具有预先建立的人肿瘤的动物模型)中实现DHODH的目标血浆浓度。在一个特定方面中,治疗本文所述的病况的方法涉及向有需要的受试者给予一定剂量的化合物1或其药物组合物,所述剂量在患有所述病况的受试者或动物模型(例如具有预先建立的人肿瘤的动物模型)中实现大约0.1 pg/mL至大约100 mg/mL、大约0.1 pg/mL至大约1 mg/mL、大约0.1pg/mL至大约500 μg/mL、大约0.1 pg/mL至大约500 μg/mL、大约0.1 pg/mL至大约100 μg/mL或大约4 pg/mL至大约10 μg/mL范围内的DHODH血浆浓度。为了获得这类血浆浓度,可视给予途径而定给予剂量在0.1 pg至100,000 mg内变化的化合物1或其药物组合物。在某些方面中,因此可基于向受试者给予初始剂量的化合物或其药物组合物下达到的DHODH的血浆浓度调节化合物1或其药物组合物的后续剂量。
在特定方面中,治疗本文所述的病况的方法涉及向有需要的受试者给予一定剂量的化合物1或其药物组合物,所述剂量在患有赘生性或非赘生性病况的受试者或动物模型(如具有预先建立的人肿瘤的动物模型)中实现所需组织/血浆化合物浓度比率,如例如通过本领域中已知的任何成像技术(如整体放射自显影术)所确定。
在一些方面中,治疗本文所述的病况的方法涉及向有需要的受试者给予一次或多次剂量的有效量的化合物1或药物组合物,其中对于各剂量所述有效量可相同或可不相同。在特定方面中,向有需要的受试者给予第一剂量的化合物1或其药物组合物持续第一时间段,且随后向所述受试者给予第二剂量的化合物1持续第二时间段。第一剂量可大于第二剂量,或者第一剂量可小于第二剂量。还可向有需要的受试者给予第三剂量的化合物1持续第三时间段。
在一些方面中,本文所述的剂量是指总给予量;即,如果给予多于一种化合物,则在一些方面中,剂量与总给予量相对应。在一个特定方面中,口服组合物含有约5重量%至约95重量%的化合物。
有需要的受试者根据治疗本文所述的病况的方法给予化合物1或药物组合物的时间长度将为确定有效的时间段。在某些方面中,治疗本文所述的病况的方法涉及给予化合物1或其药物组合物一段时间直至与赘生性或非赘生性病况相关的一种或多种症状的严重程度和/或数目降低。
在一些方面中,治疗本文所述的病况的方法涉及给予化合物1或其药物组合物长达48周。在其他方面中,治疗本文所述的病况的方法涉及给予化合物1或其药物组合物长达4周、8周、12周、16周、20周、24周、26周(0.5年)、52周(1年)、78周(1.5年)、104周(2年)或130周(2.5年)或更长。在某些方面中,治疗本文所述的病况的方法涉及给予化合物1或其药物组合物持续不确定的时间段。在一些方面中,治疗本文所述的病况的方法涉及给予化合物1或其药物组合物持续一段时间,之后在恢复化合物或其药物组合物的给药之前停药一段时间(即其中不给予化合物的时段)。在特定方面中,治疗本文所述的病况的方法涉及周期给予化合物或其药物组合物,例如1周周期、2周周期、3周周期、4周周期、5周周期、6周周期、8周周期、9周周期、10周周期、11周周期或12周周期。在这类周期中,化合物1或其药物组合物可每天一次、两次、三次或四次给予。在特定方面中,治疗本文提供的前列腺病况的方法涉及在4周周期内每天两次给予化合物1或其药物组合物。
在特定方面中,给予化合物1或其药物组合物的时间段可由一个或多个生物标记监测参数决定,所述参数例如为DHODH或其他血管生成或炎症介质(例如细胞因子或白细胞介素,如IL-6或IL-8)的浓度;肿瘤尺寸、血流或代谢;瘤周发炎或水肿。在特定方面中,给予化合物1或其药物组合物的时间段可基于一个或多个监测参数调节,所述参数例如为DHODH或其他血管生成或炎症介质(例如细胞因子或白细胞介素,如IL-6或IL-8)的浓度;肿瘤尺寸、血流或代谢;和/或瘤周发炎或水肿。
在某些方面中,根据治疗本文所述的病况的方法,化合物1或其药物组合物在膳食(例如早餐、午餐或晚餐)之前、同时或之后给予有需要的受试者。在特定方面中,根据治疗本文所述的病况的方法,化合物1或其药物组合物在上午(例如在5 am与12 pm之间)给予有需要的受试者。在某些方面中,根据治疗本文所述的病况的方法,化合物1或其药物组合物在中午(即12 pm)给予有需要的受试者。在特定方面中,根据治疗本文所述的病况的方法,化合物1或其药物组合物在下午(例如在12 pm与5 pm之间)、夜晚(例如在5 pm与上床时间之间)和/或在上床时间之前给予有需要的受试者。
在特定方面中,一定剂量的化合物1或其药物组合物每天一次、每天两次、每天三次;每隔一天(即隔天)一次、两次或三次;每两天一次、两次或三次;每三天一次、两次或三次;每四天一次、两次或三次;每五天一次、两次或三次;每一周、每两周或每月一次、两次或三次给予受试者。
组合疗法
本文提供治疗本文所述的病况的组合疗法,其涉及向有需要的受试者给予化合物1与一种或多种另外疗法的组合。在一个特定方面中,本文提供治疗本文所述的病况的组合疗法,其涉及向有需要的受试者给予有效量的化合物与有效量的另一疗法的组合。
如本文所用,在给予化合物的情形下,术语“组合”是指在给予一种或多种用于治疗本文所述的病况的另外疗法(例如药剂、手术或辐射)之前、同时或之后给予化合物。使用术语“组合”不限制向受试者给予一种或多种化合物和一种或多种另外疗法的次序。在特定方面中,给予化合物与给予一种或多种另外疗法之间的时间间隔可为约1-5分钟、1-30分钟、30分钟至60分钟、1小时、1-2小时、2-6小时、2-12小时、12-24小时、1-2天、2天、3天、4天、5天、6天、7天、1周、2周、3周、4周、5周、6周、7周、8周、9周、10周、15周、20周、26周、52周、11-15周、15-20周、20-30周、30-40周、40-50周、1个月、2个月、3个月、4个月、5个月、6个月、7个月、8个月、9个月、10个月、11个月、12个月、1年、2年或其间的任何时间段。在某些实施方案中,化合物和一种或多种另外疗法相隔小于1天、1周、2周、3周、4周、一个月、2个月、3个月、6个月、1年、2年或5年给予。
在一些方面中,本文提供的组合疗法涉及每天给予化合物1,和一周一次、每2周一次、每3周一次、每4周一次、每月一次、每2个月(例如大约8周)一次、每3个月(例如大约12周)一次或每4个月(例如大约16周)一次给予一种或多种另外疗法。在某些方面中,化合物1和一种或多种另外疗法循环给予受试者。循环疗法涉及化合物的给予持续一段时间,之后给予一种或多种另外疗法持续一段时间且重复此连续给予。在某些方面中,循环疗法还可包括停药的时段,其中不给予化合物或另外疗法一段时间(例如2天、3天、4天、5天、6天、7天、1周、2周、3周、4周、5周、10周、20周、1个月、2个月、3个月、4个月、5个月、6个月、7个月、8个月、9个月、10个月、11个月、12个月、2年或3年)。在一个方面中,给予周期数为1至12个周期、2至10个周期、或2至8个周期。
在一些方面中,治疗本文所述的病况的方法包括给予作为单一药剂的化合物1一段时间,之后给予化合物与另外疗法的组合。在某些方面中,治疗本文所述的病况的方法包括单独给予另外疗法一段时间,之后给予化合物1与另外疗法的组合。
在一些方面中,根据本文提供的方法给予化合物1和一种或多种另外疗法相对单独给予化合物或所述一种或多种另外疗法具有累加效应。在一些方面中,根据本文提供的方法给予化合物和一种或多种另外疗法相对于单独给予化合物或所述一种或多种另外疗法具有协同效应。
如本文所用,术语“协同”是指给予化合物与一种或多种另外疗法(例如药剂)组合的效应,所述组合比任何两种或更多种单一疗法(例如药剂)的累加效应更有效。在一个特定方面中,组合疗法的协同效应允许使用较低剂量(例如次佳剂量)的化合物或另外疗法和/或以较低频率向受试者给予化合物或另外疗法。在某些方面中,利用较低剂量的化合物或另外疗法和/或以较低频率给予化合物或所述另外疗法的能力会降低与向受试者分别给予化合物或所述另外疗法相关的毒性,而不分别降低化合物或所述另外疗法在治疗本文所述的病况中的功效。在一些方面中,协同效应致使化合物和所述另外疗法中的每一种在治疗本文所述的病况中的功效改进。在一些方面中,化合物和一种或多种另外疗法的组合的协同效应避免或减少与使用任何单一疗法相关的不良或不想要的副作用。
化合物1和一种或多种另外疗法的组合可在相同药物组合物中给予受试者。或者,化合物和一种或多种另外疗法可以单独的药物组合物同时给予受试者。化合物1和一种或多种另外疗法可以单独的药物组合物依序给予受试者。化合物1和一种或多种另外疗法还可通过相同或不同的给予途径给予受试者。
本文提供的组合疗法涉及向有需要的受试者给予化合物1与治疗本文所述的病况的常规或已知疗法的组合。本文所述的病况或与其相关的病况的其他疗法旨在控制或减轻一种或多种症状。因此,在一些方面中,本文提供的组合疗法涉及向有需要的受试者给予疼痛舒解剂或旨在缓解或控制与本文所述的病况或与其相关的病况相关的一种或多种症状的其他疗法。
可与化合物1组合使用的抗赘生剂的具体实例包括:激素剂(例如芳香酶抑制剂、选择性雌激素受体调节剂(SERM)、雌激素受体拮抗剂或雄激素拮抗剂)、化学治疗剂(例如微管拆散阻断剂、抗代谢物、拓朴异构酶抑制剂和DNA交联剂或损伤剂)、抗血管生成剂(例如VEGF拮抗剂、受体拮抗剂、整合素拮抗剂、血管靶向剂(VTA)/血管破坏剂(VDA))、辐射疗法和常规手术。
可与化合物1组合使用的激素剂的非限制性实例包括芳香酶抑制剂、SERM和雌激素受体拮抗剂。作为芳香酶抑制剂的激素剂可为类固醇或非类固醇。非类固醇激素剂的非限制性实例包括来曲唑(letrozole)、阿那曲唑(anastrozole)、氨鲁米特(aminoglutethimide)、法屈唑(fadrozole)和伏罗唑(vorozole)。类固醇激素剂的非限制性实例包括阿诺新(aromasin)(依西美坦(exemestane))、福美司坦(formestane)和睾内酯(testolactone)。作为SERM的激素剂的非限制性实例包括他莫昔芬(tamoxifen)(以Nolvadex®为商标/出售)、阿莫昔芬(afimoxifene)、阿佐昔芬(arzoxifene)、巴多昔芬(bazedoxifene)、克罗米芬(clomifene)、芙婷宝(femarelle)、拉索昔芬(lasofoxifene)、奥美昔芬(ormeloxifene)、雷洛昔芬(raloxifene)和托瑞米芬(toremifene)。作为雌激素受体拮抗剂的激素剂的非限制性实例包括氟维司群(fulvestrant)。其他激素剂包括(但不限于)阿比特龙(abiraterone)和洛那立生(lonaprisan)。
可与化合物1组合使用的化学治疗剂的非限制性实例包括微管拆散阻断剂、抗代谢物、拓朴异构酶抑制剂和DNA交联剂或损伤剂。作为微管拆散阻断剂的化学治疗剂包括(但不限于)紫杉烷(例如太平洋紫杉醇(paclitaxel))、多烯紫杉醇(docetaxel)、白蛋白结合型紫杉醇(abraxane)、拉洛他赛(larotaxel)、奥他赛(ortataxel)和替司他赛(tesetaxel));埃博霉素(例如伊沙匹隆(ixabepilone));和长春花生物碱(例如长春瑞宾(vinorelbine)、长春花碱(vinblastine)、长春地辛(vindesine)和长春新碱(vincristine))。
作为抗代谢物的化学治疗剂包括(但不限于)叶酸抗代谢物(例如甲氨喋呤(methotrexate)、氨基喋呤(aminopterin)、培美曲塞(pemetrexed)、雷替曲塞(raltitrexed));嘌呤抗代谢物(例如克拉屈滨(cladribine)、氯法拉滨(clofarabine)、氟达拉滨(fludarabine)、巯基嘌呤(mercaptopurine)、喷司他丁(pentostatin)、硫鸟嘌呤(thioguanine));嘧啶抗代谢物(例如5-氟尿嘧啶(5-fluorouracil)、卡培他滨(capcitabine)、吉西他滨(gemcitabine)、阿糖胞苷(cytarabine)、地西他滨(decitabine)、氟尿苷(floxuridine)、替加氟(tegafur));和脱氧核糖核苷酸抗代谢物(例如羟基脲)。
作为拓朴异构酶抑制剂的化学治疗剂包括(但不限于)第I类(喜树(camptotheca))拓朴异构酶抑制剂(例如拓朴替康(topotecan)、伊立替康(irinotecan)、卢比替康(rubitecan)和巴塞替康(besampleecan));第II类(鬼臼属(podophyllum))拓朴异构酶抑制剂(例如依托泊苷(etoposide)或VP-16和替尼泊苷(teniposide);蒽环类(例如多柔比星(doxorubicin)、表柔比星(epirubicin)、阿霉素(Doxil)、阿柔比星(aclarubicin)、氨柔比星(amrubicin)、柔红霉素(daunorubicin)、伊达比星(idarubicin)、吡柔比星(pirarubicin)、戊柔比星(valrubicin)和佐柔比星(zorubicin));和蒽二酮类(例如米托蒽醌(mitoxantrone)和匹蒽醌(pixantrone))。
作为DNA交联剂(或DNA损伤剂)的化学治疗剂包括(但不限于)烷基化剂(例如环磷酰胺(cyclophosphamide)、氮芥(mechlorethamine)、异环磷酰胺(ifosfamide)、曲磷胺(trofosfamide)、苯丁酸氮芥(chlorambucil)、美法仑(melphalan)、泼尼氮芥(prednimustine)、苯达莫司汀(bendamustine)、乌拉莫司汀(uramustine)、雌莫司汀(estramustine)、卡莫司汀(carmustine)、洛莫司汀(lomustine)、司莫司汀(semustine)、福莫司汀(fotemustine)、尼莫司汀(nimustine)、雷莫司汀(ranimustine)、链尿佐菌素(streptozocin)、白消安(busulfan)、甘露舒凡(mannosulfan)、曲奥舒凡(treosulfan)、卡波醌(carboquone)、N,N'N'-三亚乙基硫代磷酰胺、三亚胺醌(triaziquone)、三亚乙基蜜胺);类烷基化剂(例如卡铂(carboplatin)、顺铂(cisplatin)、奥沙利铂(oxaliplatin)、奈达铂(nedaplatin)、四硝酸三铂(triplatin tetranitrate)、赛特铂(satraplatin)、吡铂(picoplatin));非经典DNA交联剂(例如丙卡巴肼(procarbazine)、达卡巴嗪(dacarbazine)、替莫唑胺(temozolomide))、六甲蜜胺(altretamine)、二溴甘露醇(mitobronitol));和嵌入剂(例如放线菌素(actinomycin)、博莱霉素(bleomycin)、丝裂霉素(mitomycin)和普卡霉素(plicamycin))。
可与化合物1组合使用的抗血管生成剂的非限制性实例包括VEGF拮抗剂、受体拮抗剂、整合素拮抗剂(例如维他欣(vitaxin)、西仑吉肽(cilengitide)和S247)和VTA/VDA(例如磷酸康普瑞汀(fosbretabulin))。VEGF拮抗剂包括(但不限于)抗VEGF抗体(例如贝伐单抗(bevacizumab)和雷珠单抗(ranibizumab))、VEGF捕获剂(例如阿柏西普(aflibercept))、VEGF反义链或siRNA或miRNA和适体(例如派加他尼(pegaptanib))。作为受体拮抗剂的抗血管生成剂包括(但不限于)抗体(例如雷莫芦单抗(ramucirumab))和激酶抑制剂(例如舒尼替尼(sunitinib))、索拉非尼(sorafenib))、西地尼布(cediranib)、帕唑尼布(panzopanib)、凡德他尼(vandetanib)、阿西替尼(axitinib)和AG-013958),如酪氨酸激酶抑制剂。抗血管生成剂的其他非限制性实例包括ATN-224、醋酸阿奈可他(anecortaveacetate)、微管解聚合抑制剂(如康普瑞汀A4前药(combretastatin A4 prodrug))和蛋白质或蛋白质片段(如胶原蛋白18 (内皮生长抑素))。
可与化合物1组合给予受试者的其他疗法的非限制性实例包括:
(1)他汀(statin),如洛伐他汀(lovostatin);
(2) mTOR抑制剂,如西罗莫司(sirolimus) (其还被称为雷帕霉素(Rapamycin))、依维莫司(evorolimus)和地磷莫司(deforolimus);
(3)法尼基转移酶抑制剂,如替吡法尼(tipifarnib);
(4)抗纤维化剂,如吡非尼酮(pirfenidone);
(5)聚乙二醇化干扰素,如PEG-干扰素α-2b;
(6) CNS刺激剂,如哌甲酯(methylphenidate);
(7) HER-2拮抗剂,如抗HER-2抗体(例如曲妥珠单抗(trastuzumab))和激酶抑制剂(例如拉帕替尼(lapatinib));
(8) IGF-1拮抗剂,如抗IGF-1抗体(例如AVE1642和IMC-A11)或IGF-1激酶抑制剂;
(9) EGFR/HER-1拮抗剂,如抗EGFR抗体(例如西妥昔单抗(cetuximab)、帕尼图单抗(panitumamab))或EGFR激酶抑制剂(例如厄沙非尼(ersampleinib)、吉非替尼(gefitinib));
(10) SRC拮抗剂,如博舒替尼(bosutinib)或达沙替尼(dasatinib);
(11)周期蛋白依赖激酶(CDK)抑制剂,如塞利西利(seliciclib);
(12) Janus激酶2抑制剂,如来他替尼(lestaurtinib);
(13)蛋白酶体抑制剂,如硼替佐米(bortezomib);
(14)磷酸二酯酶抑制剂,如阿那格雷(anagrelide);
(15)肌苷单磷酸脱氢酶抑制剂,如噻唑呋林(tiazofurine);
(16)脂肪加氧酶抑制剂,如马索罗酚(masoprocol);
(17)内皮素拮抗剂;
(18)类视黄素受体拮抗剂,如维甲酸(tretinoin)或阿利维甲酸(alitretinoin);
(19)免疫调节剂,如来那度胺(lenalidomide)、泊马度胺(pomalidomide)或沙利度胺(thalidomide);
(20)激酶(例如酪氨酸激酶)抑制剂,如伊马替尼(imatinib)、达沙替尼、厄沙非尼、尼沙非尼(nisampleinib)、吉非替尼、索拉非尼、舒尼替尼、拉帕替尼或TG100801;
(21)非类固醇消炎剂,如塞内昔布(celecoxib)\;
(22)人粒细胞集落刺激因子(G-CSF),如非格司亭(filgrastim);
(23)亚叶酸或亚叶酸钙;
(24)整合素拮抗剂,如整合素α5β1-拮抗剂;
(25)核因子κβ (NF-κβ)拮抗剂,如OT-551,其也是抗氧化剂。
(26)刺猬抑制剂,如CUR61414、环巴胺、GDC-0449和抗刺猬抗体;
(27)组蛋白脱乙酰酶(HDAC)抑制剂,如SAHA (还称为伏立诺他(vorinostat))、PCI-24781、SB939、CHR-3996、CRA-024781、ITF2357、JNJ-26481585或PCI-24781;
(28)类视黄素,如异维A酸(isotretinoin);
(29)肝细胞生长因子/分散因子(HGF/SF)拮抗剂,如HGF/SF单克隆抗体;
(30)合成化学物质,如抗瘤酮(antineoplaston);
(31)抗糖尿病剂,如罗格列酮(rosaiglitazone);
(32)抗疟疾药和杀阿米巴(amebicidal)药,如氯奎(chloroquine);
(33)合成缓激肽,如RMP-7;
(34)血小板源性生长因子受体抑制剂,如SU-101;
(35) Flk-1/KDR/VEGFR2、FGFR1和PDGFR β的受体酪氨酸激酶抑制剂,如SU5416和SU6668;
(36)消炎剂,如柳氮磺胺吡啶(sulfasalazine);
(37) IL-6路径抑制剂,如托珠单抗(tocilizumab);和
(38) TGF-β反义疗法。
可与化合物1组合给予受试者的其他疗法的非限制性实例包括:天然产生的促性腺激素释放激素的合成九肽类似物,如醋酸亮丙立德(leuprolide acetate);非类固醇抗雄激素,如氟他胺(flutamide)或尼鲁胺(nilutamide);非类固醇雄激素受体抑制剂,如类固醇激素,如孕酮(progesterone);抗真菌剂,如糖皮质激素,如泼尼松(prednisone);磷酸雌莫司汀钠;和双膦酸盐,如帕米膦酸盐(pamidronate)、阿仑膦酸盐(alendronate)和利塞膦酸盐(risedronate)。
可与化合物1组合使用的疗法的其他具体实例包括(但不限于)特异性结合至肿瘤特异性抗原或肿瘤相关抗原的抗体,例如抗EGFR/HER-1抗体。
可与化合物1组合使用的疗法的另外具体实例包括(但不限于)与免疫疗法相关的药剂,例如细胞因子、白细胞介素和疫苗。
可与化合物1组合用作疗法的缓解与本文所述的病况相关的副作用的药剂的具体实例包括(但不限于):止吐药,例如盐酸昂丹司琼(Ondansetron hydrochloride)、盐酸格拉司琼(Granisetron hydrochloride)、劳拉西泮(Lorazepam)和地塞米松(Dexamethasone)。
在某些方面中,本文提供的用于治疗本文所述的病况的组合疗法包括给予化合物1与用以治疗和/或处理副作用的一种或多种药剂组合,所述副作用诸如为出血(通常是短暂低级的流鼻血)、动脉和静脉血栓形成、高血压、伤口愈合延迟、无症状蛋白尿、鼻隔膜穿孔、与高血压相关的可逆后脑白质病综合征、轻度头痛、共济失调、头痛、嘶哑、恶心、呕吐、腹泻、皮疹、指甲下出血、骨髓抑制、疲劳、甲状腺功能低下、QT间隔延长或心脏衰竭。
在某些实施方案中,化合物1不与主要通过CYP2D6代谢的药物(如抗抑郁剂(例如三环抗抑郁剂、选择性血清素再吸收抑制剂等)、抗精神病药、β-肾上腺素能受体阻断剂或某些类型的抗心律不齐药)组合用以治疗本文所述的病况。
试剂盒
本文提供药物包装或试剂盒,其包含一个或多个填充有化合物1或其药物组合物的容器。另外,药物包装或试剂盒中还可包括一种或多种可用于治疗病况的其他疗法或其他相关药剂。本文还提供药物包装或试剂盒,其包含一个或多个填充有本文所述的药物组合物的成分中的一种或多种的容器。任选与这类试剂盒相关的可为呈由管理药物或生物产品的制造、使用或销售的政府机构所规定的形式的注意事项,所述注意事项反映所述机构批准制造、使用或销售用于人给药。
患者群体
在一些实施方案中,根据本文提供的方法进行本文所述的病况治疗的受试者为患有或被诊断患有本文所述的病况的人。在其他方面中,根据本文提供的方法进行本文所述的病况治疗的受试者为易感或易患本文所述的病况的人。在一些方面中,根据本文提供的方法进行本文所述的病况治疗的受试者为处于显现本文所述的病况的风险中的人。
在一个方面中,根据本文提供的方法进行本文所述的病况治疗的受试者为婴儿。在另一方面中,根据本文提供的方法进行本文所述的病况治疗的受试者为幼儿。在另一方面中,根据本文提供的方法进行本文所述的病况治疗的受试者为儿童。在另一方面中,根据本文提供的方法进行本文所述的病况治疗的受试者为成人。在另一方面中,根据本文提供的方法进行本文所述的病况治疗的受试者为中年人。在另一方面中,根据本文提供的方法进行本文所述的病况治疗的受试者为老年人。
在某些方面中,根据本文提供的方法进行赘瘤治疗的受试者具有转移至身体的其他区域(如骨骼、肺和肝脏)的恶性赘瘤。在某些方面中,根据本文提供的方法进行赘瘤治疗的受试者具有缓解的赘瘤。在一些方面中,根据本文提供的方法进行赘瘤治疗的受试者具有赘生性病况的复发。在某些方面中,根据本文提供的方法进行治疗的受试者经历一种或多种与赘瘤相关的肿瘤的复发。
某些方面中,根据本文提供的方法进行赘瘤或非赘生性病况治疗的受试者为约1岁至约5岁、约5岁至10岁、约10岁至约18岁、约18岁至约30岁、约25岁至约35岁、约35岁至约45岁、约40岁至约55岁、约50岁至约65岁、约60岁至约75岁、约70岁至约85岁、约80岁至约90岁、约90岁至约95岁、或约95岁至约100岁或其间的任何年龄的人。在一个特定方面中,根据本文提供的方法进行赘瘤或非赘生性病况治疗的受试者为18岁或更年长的人。在一个特定方面中,根据本文提供的方法进行赘瘤或非赘生性病况治疗的受试者为年龄在1岁至18岁之间的儿童。在某一方面中,根据本文提供的方法进行赘瘤或非赘生性病况治疗的受试者为年龄在12岁与18岁之间的人。在某一方面中,受试者为男人。在另一方面中,受试者为女人。在一个方面中,受试者为未怀孕或不进行母乳哺育的女人。在一个方面中,受试者为怀孕,或将/可怀孕,或进行母乳哺育的女性。
在特定方面中,根据本文提供的方法进行赘瘤或非赘生性病况治疗的受试者为处于免疫功能不全状态或免疫抑制状态的人。在某些方面中,根据本文提供的方法进行赘瘤或非赘生性病况治疗的受试者为接受免疫抑制疗法或自其中恢复的人。在某些方面中,根据本文提供的方法进行赘瘤或非赘生性病况治疗的受试者为患有恶性赘瘤(例如转移性癌症)、AIDS或细菌感染或具有患上其的风险的人。在某些方面中,根据本文提供的方法进行赘瘤或非赘生性病况治疗的受试者为经历、将经历或已经历手术、药物疗法(如化学疗法)、激素疗法和/或辐射疗法的人。
在特定方面中,根据本文提供的方法进行赘瘤或非赘生性病况治疗的受试者罹患例如中风或心脏血管病况的可需要VEGF疗法的病况,其中除化合物外可禁止给予抗血管生成疗法。例如,在某些方面中,根据本文提供的方法进行赘瘤或非赘生性病况治疗的受试者遭受中风或罹患心脏血管病况。在一些方面中,根据本文提供的方法进行赘瘤或非赘生性病况治疗的受试者为经历循环问题的人。在某些方面中,根据本文提供的方法进行赘瘤或非赘生性病况治疗的受试者为患有糖尿病性多发性神经病或糖尿病性神经病的人。在一些方面中,根据本文提供的方法进行赘瘤或非赘生性病况治疗的受试者为接受VEGF蛋白质疗法的人。在其他方面中,根据本文提供的方法进行赘瘤或非赘生性病况治疗的受试者不是接受VEGF蛋白质疗法的人。
在一些方面中,根据本文提供的方法进行赘瘤或非赘生性病况治疗的受试者在除化合物外的疗法的任何不良作用或不耐受性显现之前给予化合物或其药物组合物或组合疗法。在一些方面中,根据本文提供的方法进行赘瘤或非赘生性病况治疗的受试者为难治性患者。在某一方面中,难治性患者为难以用标准疗法(例如手术、辐射、抗雄激素疗法和/或药物疗法(如化学疗法))治疗的患者。在某些方面中,当赘瘤或非赘生性病况尚未显著根除和/或一种或多种症状尚未显著缓解时,患有赘瘤或非赘生性病况的患者难以用疗法治疗。可通过测定赘瘤或非赘生性病况的治疗有效性的领域中已知的任何方法,使用在这类情形下本领域可接受的“难治性”的含义,在体内或体外确定患者是否是难治性的。在各个方面中,当一种或多种与赘瘤相关的肿瘤未减小或有所增大时,患有赘瘤的患者是难治性的。在各个方面中,当一种或多种肿瘤转移和/或扩散至另一器官时,患有赘瘤的患者是难治性的。
在一些方面中,根据本文提供的方法进行赘瘤或非赘生性病况治疗的受试者为已证实难以用除用化合物治疗外的疗法治疗但也不再用这些疗法的人。在某些方面中,根据本文提供的方法进行赘瘤或非赘生性病况治疗的受试者为已接受一种或多种常规抗赘生性疗法(如手术、药物疗法(如化学疗法)、抗雄激素疗法或辐射)的人。这些患者之中存在难治性患者、太幼小而不适用常规疗法的患者和尽管用现有疗法治疗但肿瘤仍复发的患者。
在一些方面中,根据本文提供的方法进行赘瘤或非赘生性病况治疗的受试者为易对常规疗法产生不良反应的人。在一些方面中,根据本文提供的方法进行赘瘤或非赘生性病况治疗的受试者是在给予化合物1或其药物组合物之前尚未接受疗法,例如药物疗法(如化学疗法)、手术、抗雄激素疗法或辐射疗法的人。在其他方面中,根据本文提供的方法进行赘瘤或非赘生性病况治疗的受试者是在给予化合物1之前已接受疗法的人。在一些方面中,根据本文提供的方法进行赘瘤或非赘生性病况治疗的受试者是已经历先前疗法的不良副作用或先前疗法由于人不可接受的毒性水平而中断的人。
在一些方面中,根据本文提供的方法进行赘瘤或非赘生性病况治疗的受试者之前尚未暴露于其他抗血管生成疗法(例如抗VEGF单克隆抗体、抗VEGFR单克隆抗体、酪氨酸激酶抑制剂或其他血管生成路径调节剂)。在特定方面中,根据本文提供的方法进行赘瘤或非赘生性病况治疗的受试者不具有不受控制的高血压、大出血、HIV感染或近期急性心脏血管事件。在一些方面中,根据本文提供的方法进行赘瘤或非赘生性病况治疗的受试者具有心肌梗塞、不稳定绞痛、冠状动脉/外周动脉搭桥、充血性心脏衰竭、脑血管意外、短暂缺血发作、动脉血栓栓塞事件或肺栓塞。
在一些方面中,根据本文提供的方法进行赘瘤或非赘生性病况治疗的受试者未接受、尚未接受和/或将不接受主要由CYP2D6代谢的药物。在特定方面中,根据本文提供的方法进行赘瘤或非赘生性病况治疗的受试者在接受化合物或其药物组合物之前1、2、3或4周和在接受化合物或药物组合物之后1、2、3或4周尚未接受且将不接受主要通过CYP2D6代谢的药物。这类药物的实例包括(但不限于)一些抗抑郁剂(例如三环抗抑郁剂和选择性血清素吸收抑制剂)、一些抗精神病药、一些β-肾上腺素能受体阻断剂和某些抗心律不齐药。在特定方面中,根据本文提供的方法进行赘瘤或非赘生性病况治疗的受试者不接受、尚未接受和/或将不接受他莫昔芬。在特定方面中,根据本文提供的方法进行赘瘤或非赘生性病况治疗的受试者在接受化合物或其药物组合物之前1、2、3或4周和在接受化合物或药物组合物之后1、2、3或4周尚未接受且将不接受他莫昔芬。在特定方面中,根据本文提供的方法进行赘瘤或非赘生性病况治疗的受试者例如在接受化合物或其药物组合物之前已接受他莫昔芬持续1、2、3或4周。
具体实施例
将通过参考以下非限制性具体实施例来进一步理解本发明。特定地,实施例表明胶囊和片剂剂型的用途,和喂饲或禁食状态下赋形剂选择对化合物(Cpd)1负载、溶解性和生物可利用性的影响,和包含化合物1和聚合物的SDI在各种组合物和制剂中的稳定性。
实施例1
喷雾干燥的中间体溶解性研究
材料和方法
使用以下赋形剂和其他材料(表2)制备本文所述的制剂。
表2
活性材料和非活性材料

制备喷雾干燥的中间体的方法
对于以所述组合物和制剂使用,结晶形式的化合物1a或化合物1b和任选的赋形剂的溶液使用小型Büchi B-290实验室规模喷雾干燥器通过喷雾干燥共同沉淀。-溶液通过蠕动泵经由喷嘴喷雾以提供包含呈非晶形式的化合物1和任选的共同沉淀的赋形剂的SDI。在室温下使获得的SDI样品保持在真空下持续24小时的时段以去除残余溶剂。
术语“组合物”是指使用本领域的技术人员已知的技术以溶液形式制备的包含如本文所述的SDI和任选的赋形剂的产品。
术语“制剂”是指使用本领域的技术人员已知的干式共混技术制备的包含如本文所述的组合物和另外赋形剂的产品。
射线粉末衍射(XRPD):SDI的结晶或多晶特征通过使用Siemens D-5000 X-射线衍射仪用Co αK辐射(λ=1.7890 Å)经3-40° 2θ的范围以0.017° 2θ s-1的扫描速度进行XRPD来测定。
差示扫描量热法(DSC):DSC测量结果使用DSC1型Mettler Toledo差示扫描量热计在25℃至240℃的温度范围以及10℃ min-1和25℃ min-1的加热速率下在50 mL min-1的氮气吹扫下获得。铟用作校正标准物。使用密封铝盘(40 µL)分析样品。
热解重量分析(TGA):TGA分析通过自动模块化Mettler Toledo TGA/DSC1执行。在以50 mL min-1吹扫的受控氮气氛围中进行所述分析。温度范围为25℃至120℃,加热速率为10℃ min-1。对于TGA,将样品置于氧化铝坩埚(70 µL)中。
水测定:水测定使用795型Metrohm Titrino通过卡尔费歇尔(Karl Fisher)滴定执行。商业制备的试剂效价含有一定比例的咪唑、碘、二氧化硫和乙醚,使得1 mL试剂将与大约2 mg H2O反应。系统配备有合适的干燥剂,且在各系列样品分析之前使用恰好10 μL纯化水执行3次测量来校准溶液。这些测量结果的相对标准偏差限制在小于或等于2.0%。
聚合物选择和化合物负载
SDI批料(表3)使用上文所述的方法在40%至70%的范围内的化合物1负载下制备。对于用PVP K30制得的SDI批料,最高化合物1负载为80%。选择50%至70%的范围内的负载以评价SDI中作为聚合物的PVP K90和HPMC E5。每个SDI批料通过在室温(RT)下搅拌使化合物1和聚合物溶解在DCM:EtOH 80:20溶剂体系(200 mL)中获得。
表3
聚合物选择和负载

如本文所述,SDI样品在加速条件40℃/75%相对湿度下在敞口和聚丙烯(PP)封盖的高密度聚乙烯(HDPE)瓶子中温育以测定非晶形式稳定性。
样品通过以下评价:
1- 零时和稳定站台的后续时间点的非晶状态的XRPD
2- 零时在SDS、泊洛沙姆188、泊洛沙姆407、Gelucire 44/14和Gelucire 50/13的水溶液(5% w/w)中长达24小时的动力学溶解性
3- 零时的DSC和TGA热谱图和稳定站台的后续时间点的DSC
4- 零时0.4 mg/mL的体内代表性浓度的化合物1在水中以及在SDS、泊洛沙姆188、泊洛沙姆407、Gelucire44/14和Gelucire 50/13的水溶液(5% w/w)中的溶解性
喷雾干燥方法和结果
为了获得具有改进的溶解性的稳定SDI并且为了避免化合物1的体内沉淀或再结晶,使用上文所述的喷雾干燥技术以获得包含与根据表3的聚合物复合的沉淀的非晶化合物1的SDI。
为了产生SDI,使用表4中所示的喷雾干燥方法参数使结晶化合物1a或化合物1b与PVP-K30和HPMC复合。每种SDI的组成以及所获得的产率示于表5中。
如表4中所示,在较大浓度的化合物1a或化合物1b下和/或当使用PVP90时,修改方法参数以使产率达到最大。通常,所选聚合物容易地溶解于溶剂体系中,产生非粘性澄清/透明液体,但SDI样品9 (HPMC 50%)除外,在所述样品中,一些极精细粒子仍保持悬浮。
在表4中,入口温度(In)和出口温度(Out)以℃显示,雾化气压(Air)以mm显示,喷雾气体流量(Gas)以L/h显示,抽风机速率(Asp)和泵速率(Pmp)以%显示,体积流量(Vol)以m3/h显示,且进料流量(Feed)以mL/min显示。喷雾气体流量根据制造商的说明书确定,且实际进料流量通过根据泵运行时间使用的溶液的体积计算。
术语“产率tot”是指通过组合自喷雾筒、旋风分离器和收集容器回收的材料计算的总产率的百分比(%)。术语“产率ccv”是指自旋风分离器/收集容器回收的组合量的百分比(%)。
表4
喷雾干燥方法参数

表5
SDI产率

通常,沉淀的SDI样品具有所需粒度,但粒子具有粘性性质。
仅自收集容器中检索到来自样品9和10的SDI样品。
来自样品7和8的SDI样品提供流动性相对不良的精细的粘性粘聚性聚结物。此外,这些样品在喷雾干燥期间形成纤维。50%的PVP K90 (样品7)的浓度致使在干燥腔室中形成完整长丝。在表5中所示范围内的不同方法参数不能避免纤维形成。纤维形成似乎取决于PVP K90浓度。降低至30% PVP K90 (样品h)在收集容器中提供相对低的产率(30.8%)。
SDI分析结果
XRPD
共同沉淀的SDI批料的非晶含量通过XRPD评价。每种SDI样品的XRPD图案是非晶材料典型的。
TGA
TGA测量结果以样品温度和时间的函数显示质量损失的速率,在25℃与120℃之间并没有显著质量损失。在测试产物中没有检测到残余溶剂。残余溶剂的挥发通常与TGA期间样品的最初重量损失相关。
DSC
在10℃/min的加热速率下获得DSC热谱图。显示玻璃转变温度(GTT)(以℃计)和热容量(HC)(以J g-1 k-1计)下在85℃与105℃之间的吸热峰(EndoPeak)拐点的每种SDI示于表6中。通常,结果显示,玻璃转变温度为组合物中所用的聚合物的量和类型的函数。
吸热峰(Endo)(以℃计)可与具有不同熔点的各种晶体改变的发生相关。放热峰(Exo)(以℃计)可通过结晶、固-固转变、分解或化学反应阐述。每个峰的焓(Ey)(以J g-1计)也示于表6中。
对于样品3,未见到可检测的放热峰转变。对于样品5和6,可见通常与材料结晶相关的放热转变。对于样品6,表示熔融过程的吸热转变发生在158℃与224℃之间。放热转变峰的面积与SDI化合物1浓度成正比。
对于样品8、9和10,未见到可检测的放热峰。
对于样品4和5,分别观察到具有207℃和221℃的熔融温度的第二小吸热峰。
对于样品3和6,在25℃/min的加热速率下获得的比较性DSC热谱图通常显示观察到的峰的面积增加。对于样品6,25℃/min热谱图与10℃/min热谱图类似,其中在25℃/min下放热峰温度具有略高的变换。
术语“NO”表示“未观察到”,且术语“ND”表示“未测定”。
表6
DSC结果

SDI和表面活性剂(5% w/w)在水溶液中的溶解性
在室温下,在0小时和24小时的取样时间下,在不同水性介质中测量结晶化合物1b和非晶SDI的溶解性(表7和8)。制备结晶化合物1b和非晶SDI在具有选自SDS、泊洛沙姆188、泊洛沙姆407、Gelucire 44/14和Gelucire 50/13的表面活性剂的饱和溶液中的水性悬浮液(5% w/w)。
SDS Gelucire 50/13溶液不透明,指示并非所有Gelucire均溶解。所述溶液的半透明度并不足以确定添加足够的材料以提供饱和溶液的时间。因而,使溶液静置以使得过量Gelucire沉降,且将上清液回收且用于进行溶解性研究。Gelucire 50/13在溶液中的确切浓度未知。SDI在Gelucire 50/13中的溶液显示在24小时之后大量沉淀。
术语“NR”表示“不可再现”,且术语“SP”表示“显著沉淀”。
表7
溶解性(μg/mL)

表8
溶解性(μg/mL)

验证样品c和e在SDS中的溶解性(表9)。SDS溶液澄清且使SDI过量添加到容器中。超声处理溶液5分钟,振荡2分钟且静置5分钟。经由具有0.45 μm尼龙过滤器的注射器将上清液转移到HPLC小瓶中。即刻注射样品(系统在开始溶解性测试之前预条件化)。
表9
溶解性(μg/mL)

*这个值不可再现。
表8和9中所示的结果指示在表面活性剂存在下在SDI中PVP和HPMC聚合物的选择影响溶解性。
和表面活性剂(5% w/w)在水溶液中的溶解性
根据所需剂量负载,在37℃下使各种量的SDI溶解于具有选自SDS、泊洛沙姆188、泊洛沙姆407、Gelucire 44/14和Gelucire 50/13的表面活性剂的饱和溶液中(如表10中所示)。
例如,使量在75% (0.3 mg/mL)与95% (0.4 mg/mL)之间的SDI溶解于具有泊洛沙姆407或Gelucire 50/13的溶液中。能够使相对较小量的SDI溶解于SDS、泊洛沙姆188和Gelucire 44/14溶液中。在0.4 mg/mL的标称SDI浓度下,SDS、泊洛沙姆407和Gelucire 50/13溶液使SDI保持于溶液中长达2小时。
对于PVP K30样品5和PVP K90样品8,在70%化合物1负载下达到较大溶解性。样品5和8能够在Gelucire 50/13和泊洛沙姆407中维持所述SDI浓度且在SDS中以一定程度维持。
对于HPMC E5样品9和10,在除泊洛沙姆188外的所有溶液中维持约0.2 mg/mL至约0.3 mg/mL的SDI浓度。对于样品9和10,在化合物1负载与溶解性之间没有相关性。
表10
400 µg/mL下的溶解性(µg/mL)

实施例2
所选SDI-表面活性剂组合物的溶解性
具有50-60%的最大化合物1负载的组合物显示根据DSC和热台显微术数据在高温下非晶形式的结晶受限。
短期稳定性研究指示关于化合物1结晶/再结晶使用PVP K30或HPMC E5之间并没有显著差异。两者显示SDI的有利溶解性特征。然而,PVP SDI与HPMC E5 SDI之间的溶解比较显示HPMC E5 SDI的溶解曲线大大改进。
HPMC E5溶解性
在各种有机溶剂中测试HPMC E5、HPMC E5/Cpd 1和HPMC E5/Cpd 1/泊洛沙姆407溶解性(参见表11和12中所示的结果)。组合物在室温下在搅拌下个别地分散于溶剂中。首先溶解HPMC E5和泊洛沙姆407 (在使用时)提供50:50溶液,随后添加化合物1且完全溶解,之后添加DCM以提供比率为87.5:12.5和86:14的溶液。
在表12中,DCM:DMSO (77:23)溶剂体系使用DCM:DMSO (65:35) HPMC E5溶液通过添加另外DCM来制备。
表11
HPMC E5 (mg/mL)溶解性


表12
SDI (化合物1/HPMC E5/泊洛沙姆407)溶解性

喷雾干燥溶液中的固体含量
在不同溶剂体系中评价用于大规模制造的喷雾干燥溶液中可接受的固体的最大量(即,组合起始材料的溶解性)。在室温下在搅拌下将HPMC E5聚合物添加到溶剂体系中,之后添加化合物1b。在30分钟之后和在使样品静置长达72小时的时段之后视觉上评价溶解性。
如表13至16中所示,溶液中可接受的固体的量取决于时间、每种起始材料的量和溶剂体系。
如表13中所示,溶剂体系中所用的SDI使用在以下实施例6中所述的用于SDI样品28的程序以两个步骤制备。
如表14中所示,溶剂体系中所用的SDI使用用于SDI样品28的程序以两个步骤制备。在DCM:MeOH体系中,将化合物1b (3 gm)和HPMC E5 (2 gm)的组合溶解(5% w/v)于100mL体积的体系中。术语“NT”表示“未测试”,术语“VSS”表示“极轻微沉降”且术语“SS”表示“轻微沉降”。
如表16中所示,将化合物1b (6 gm)和HPMC E5 (4 gm)的组合溶解(10% w/v)于DCM:DMSO (50:50) (100 mL)中。术语“VSS”表示“极轻微沉降”,且术语“SS”表示“轻微沉降”。
如表15中所示,将化合物1b (4.5 gm)和HPMC E5 (3 gm)的组合溶解(7.5% w/v)于DCM:DMSO (65:35) (100 mL)中。术语“SS”表示“轻微沉降”。
表13
固体含量(2.5% w/v)

表14
固体含量(5% w/v)

表15
固体含量(7.5% w/v)

表16
固体含量(10.0% w/v)

SDI-表面活性剂喷雾干燥方法和结果
化合物1/聚合物/表面活性剂共同沉淀的SDI组合物使用先前描述的喷雾干燥技术通过固体分散体获得。方法参数设定为表17中所列举的条件。SDI的组成和产率示于表18中。
在表17中,入口温度(In)和出口温度(Out)以℃显示,雾化气压(Air)以mm显示,喷雾气体流量(Gas)以L/h显示,抽风机速率(Asp)和泵速率(Pmp)以%显示,体积流量(Vol)以m3/h显示,且进料流量(Feed)以mL/min显示。喷雾气体流量根据制造商的说明书确定,且实际进料流量通过根据泵运行时间使用的溶液的体积计算。
表17
喷雾干燥方法参数

如表1中所示,使材料溶解于溶剂体系中,产生非粘性完全透明的液体。样品11、12、13、14、15、16、17和20通过使材料溶解于溶剂混合物(200 mL)中来制备。
对于样品18和129,将HPMC E5溶解于50:50 DCM:MeOH (25 mL:25 mL)混合物中,随后添加表面活性剂,接着添加纯净的DCM (150 mL),逐步分散直至完全溶解。
来自样品12和19的SDI样品提供在约78%至约80%的范围内的产率。在35 mm的雾化气压(414标准升/小时)下来自14和16的SDI样品提供在约76%至约77%的范围内的产率。
术语“产率ccv”是指自旋风分离器/收集容器回收的组合量的百分比(%)。
表18
SDI (化合物1/聚合物/表面活性剂)组合物

XRPD
来自样品11、12、13、14、15、16、17和20的共同沉淀的表面活性剂-SDI的非晶结构通过XRPD评价(结果未示出)。每种样品的XRPD图案的特征在于非晶材料。
DSC
如表19中所示,在25℃/min的加热速率下获得DSC热谱图。仅样品11、14、15、16和19在85℃与105℃之间清晰地观察到表示玻璃转变温度(GTT)的吸热拐点。
样品11可见放热转变(Exo)且样品13、14、19和20可见较小程度的放热转变(Exo)。样品12、13、15和17可见吸热转变(Endo),其中每种样品分别在约210℃、207℃、205℃和218℃观察到一个熔融峰。
术语“NO”表示“未观察到”,且术语“ND”表示“未测定”。
表19
DSC结果

SDI-表面活性剂组合物在(5% w/w)水溶液中的溶解性
在37℃下,在2小时和6小时之后确定化合物1/聚合物/表面活性剂SDI组合物在不同水性介质中的溶解性(参见表20至23)。在每个表中,T.C.表示假设所有材料均溶解的溶液的基于样品重量的理论浓度。72小时结果仅为视觉观察结果。
在没有表面活性剂但有泊洛沙姆407或Gelucire 50/13的情况下,含有HPMC E5的SDI组合物在2小时与6小时之间显示较高且较稳定的溶解性,提供387 μg/mL至436 μg/mL之间的浓度,代表理论浓度(TC)的93.5-100%。术语“理论浓度”是指如由材料重量所确定的溶解有所有材料的溶液的浓度。
对于含有PVP K30和泊洛沙姆407或Gelucire 50/13的SDI组合物,溶解浓度在225μg/mL至446 μg/mL之间(代表T.C.的55.8%至99.8%)。
表20
在水(400 μg/mL)中的溶解性(μg/mL)

表21
在含1% SLS的水(400 μg/mL)中的溶解性(μg/mL)

表22
在含5%泊洛沙姆407的水(400 μg/mL)中的溶解性(μg/mL)

表23
在含5% Gelucire 50/13的水(400 μg/mL)中的溶解性(μg/mL)

实施例3
所选干式共混制剂的溶解性
SDI制剂样品4、11、12和17用表面活性剂微晶纤维素(MCC-102)或泊洛沙姆407(Pol 407)和崩解剂A型交联羧甲基纤维素钠(CCS)配制(如表24中所示)。
术语“内相”(IP)是指并入SDI中的一种或多种赋形剂;例如SDI由喷雾干燥组合的化合物1a或化合物1b、聚合物和表面活性剂而形成。含有IP表面活性剂的SDI随后与表24中所示的其他成分混合以提供干式共混制剂。术语“外相”(EP)是指如同一种或多种其他任选赋形剂一样包含在干式共混制剂中的表面活性剂。
每种制剂通过以下来制备:使用研钵和研杵轻柔地干式共混,随后将粉末手动填充到00号(0.91 mL)明胶胶囊中,总计105 mg/盖帽。
将泊洛沙姆407表面活性剂在IP SDI实验5和e中或在用于制剂中的EP SDI样品1和3中的作用与不存在表面活性剂的制剂(实验2和4)相比。
表24
干式共混制剂

实施例4
干式共混制剂的溶解
进行关于干式共混制剂的体外溶解研究以确定喂饲或禁食状态是否对制剂溶解性和溶解速率有影响。
使用USP溶解设备II使含有根据本文提供的实施例制备的制剂的胶囊溶解于禁食状态模拟胃液(FastSSGF)中(图1和2)。
如图1中所示,使来自表24中的实验1至6的干式共混制剂样品21、22、23、24、25和26的胶囊在桨叶速度100转/分钟(RPM)下分别溶解于含0.1N HCl的1.5% SDS水溶液的FastSSGF (1000 mL)中。通过视觉观察,制剂样品21略微浑浊;样品22浑浊;样品23形成略微浑浊的悬浮液;样品24具有极略微浑浊的悬浮液;样品25浑浊且样品26形成不透明悬浮液。
制剂样品23、24和26表明比样品21、22和25高的溶解速率。样品23经一至六个小时具有在83%与92%之间的最高溶解速率。IP表面活性剂的使用显示对样品26溶解没有影响,但当在样品21、22和25中以组合(IP或EP)使用时导致溶解较慢。
如图2中所示,使样品23、24和26在桨叶速度100 RPM下溶解于800 mL FastSSGF中,且将其与溶解于1000 mL FastSSGF中的相同样品相比。
比较在800 mL和1000 mL FastSSGF中的样品23、24和26显示,所有制剂的溶解速率都随溶解介质体积减小而降低。
总体而言,观察到某些制剂的释放曲线差异。
另外结果
在溶解测试之后,观察到样品21、22和25的沉淀。通过XRPD评估样品22和25沉淀物(数据未示出)。对于样品25,XRPD显示化合物1部分再结晶。对于样品22,一种没有泊洛沙姆表面活性剂的制剂,XRPD显示沉淀物仍保持以非晶形式。
将干式共混制剂中所用的结晶化合物1a、结晶化合物1b和表面活性剂及崩解剂中的每一种的XRPD图案与部分再结晶样品25的XRPD图案相比。所述比较指示对于样品25观察到的结晶峰并不与制剂中所用的材料中的任一种的XRPD结晶峰相对应。不受理论束缚,样品25峰可归因于与来自体外溶解介质的IP SDI表面活性剂或SLS的相互作用。
实施例5
SDI制剂的药代动力学研究
使用52%负载的SDI组合物与MCC-102或Pol 407和CCS组合来制备制剂以进行大鼠PK研究。在共混之前将SDI和赋形剂经30目筛网筛分。每一种制剂(1克)通过使用研钵和研杵轻柔地干式共混来制备。制备制剂样品29至33 (参见表25)和样品34至39 (参见表26)用于PK研究。
表25
制剂SDI共混物

表26
制剂SDI共混物

在表27中,52%负载的制剂通过HPLC测定且发现具有在95%与100.6%之间的值。对于样品34和36,由于PVP K30的聚结,添加另外泊洛沙姆和SDI以维持用于1克批量大小的化合物1负载。样品39受的影响比样品34大。使用与先前所述相同的共混方法。
表27
测定

实施例6
SDI稳定性研究
SDI样品27和28由结晶化合物1a使用与用于样品11至20相同的喷雾干燥方法步骤和参数制造以用于长期稳定性研究(参见表28)。
在制备(0小时时间)之后立即通过XRPD和DSC分析SDI样品27和28。根据XRPD,两种SDI样品均具有非晶性质。表29中所示的DSC结果在10℃/min的加热速率下获得。
对于稳定性研究,将每种SDI样品的松散粉末封装在含有干燥剂的双内衬LDPE袋(MiniPax MultiSorb™干燥封包1克50/50 AC/SG)中。随后将LDPE袋置于密闭HDPE瓶子中。
将样品在长期(25℃/60% RH)和加速(40℃/75% RH)稳定条件下温育。
术语“NO”表示“未观察到”,且术语“ND”表示“未测定”。
表28
SDI组合物

表29
DSC结果

加速和长期稳定性
非表面活性剂SDI XRPD
使非表面活性剂SDI样品3至10暴露于敞口HPDE容器中的40℃/75% RH。在一周和三周时间点,XRPD显示样品3至10在0小时时间点在100计数的强度标度下没有可检测的结晶迹象(数据未示出)。在6周时间点,SDI样品3至6 (含有PVP K30)显示结晶迹象。SDI样品7至10 (含有PVP K90和HPMC E5)保持非晶并且在1000计数与100计数两种XRPD强度标度下没有可检测的结晶迹象(数据未示出)。在12周时间点在100计数下对于SDI样品3至6和7至10获得类似结果。
使非表面活性剂SDI样品27和28暴露于密闭袋/HPDE容器中的25℃/60% RH和40℃/75% RH。在四周和八周时间点在两种条件下,在1000计数与100计数两种强度标度下没有观察到SDI样品27和28的XRPD图案变化(数据未示出)。在12周时间点,对于SDI样品28,在25℃/60% RH与40℃/75% RH两者下,都没有观察到XRPD图案的变化。在12周时间点,对于SDI样品27,在25℃/60% RH下,没有观察到XRPD图案变化。在12周时间点,对于SDI样品27,在40℃/75% RH下,XRPD图案指示非晶材料部分再结晶(数据未示出)。
使SDI-表面活性剂组合物样品12至20暴露于敞口HPDE容器中的40℃/75% RH。
在3周40℃/75% RH时间点,对于样品12、23、14、15、18和20,XRPD图案指示非晶材料的显著再结晶(数据未示出)。对于样品17,发生微小结晶。对于样品16和19,XRPD图案显示组合物保持非晶。
在6周40℃/75% RH时间点,对于样品16和19,XRPD图案显示组合物在1000计数与100计数两种强度标度下保持非晶(数据未示出)。
非表面活性剂SDI DSC
表30至32中关于置于敞口HPDE容器中且暴露于40℃/75% RH的SDI样品3至10的样品所示的DSC数据在10℃/min的加热速率下获得。
所有SDI样品显示若干随时间推移的峰,在50%负载下使用PVP K30和HPMC E5的样品具有新颖吸热峰。峰强度(表示焓的总体增加)取决于稳定的时间长度、封装(敞口/密闭容器)和稳定性条件而增加。相对于PVP K30,HPMC E5的峰强度较低,表明非晶形式稳定性改进。
术语“NO”表示“未观察到”,术语“ND”表示“未测定”,术语“NS”表示“样品不可获得”,且术语“D1”表示“高于170-180℃时的降解”。
表30
3周打开盖帽的DSC结果

表31
6周打开盖帽的DSC结果

表32
12周打开盖帽的DSC结果

表33
12周封闭盖帽的DSC结果

非表面活性剂SDI DSC
表34至36中所示的稳定性结果在敞口容器中在40℃/75% RH的条件下获得。内相中包含表面活性剂会影响SDI稳定性,如通过DSC峰的数目和强度所示。对于SDI样品19,在3周时间点许多高于200℃的热事件指示降解增加。对于在25℃/min(表34)或10℃/min (表35和36)的加热速率下获得的DSC热谱图,放热焓和吸热焓显著增加。另外,SDI样品13和15显示低于40℃的温度下的热事件。
术语“NO”表示“未观察到”,术语“ND”表示“未测定”且术语“D2”表示“高于190℃时的降解”。
表34
3周打开盖帽的DSC结果

表35
3周打开盖帽的DSC结果

表36
6周打开盖帽的DSC结果

SDI组合物DSC
封装于密闭HDPE瓶子中的含有干燥剂的双内衬LDPE袋中的指定为s和t的SDI稳定性样品在储存于40℃/75% RH下之后2、4、8和12周的时间点(参见表37至41)和在储存于25℃/60% RH下之后12周时间点(参见表42)下进行测定。
对于SDI样品28,40℃/75% RH下2周时间点的DSC与0周时间点的DSC类似。储存于25℃/60% RH和40℃/75% RH两者下在12周时间点的SDI样品28样品的DSC热谱图与0周时间点的DSC也类似,但进一步储存时储存于40℃/75% RH下的那些样品的焓值略微增加。
对于SDI样品,吸热转变随储存时间增长而增加,吸热峰变换至较低温度。在储存于40℃/75% RH下的样品中,转变略微较高。
SDI样品t通常比SDI样品28更稳定。术语“NO”表示“未观察到”;术语“ND”表示“未测定”。
表37
0周的DSC结果

表38
2周的DSC结果

表39
4周的DSC结果

表40
8周的DSC结果

表41
12周的DSC结果

表42
12周的DSC结果

外观
还在3周和6周时间点检查储存于40℃/75% RH下的SDI打开盖帽样品的外观(参见表43)。观察到SDI的颜色由白色粉末变为灰白色至黄色粉末,SDI-表面活性剂样品13、14、12、20和18伴随聚结。自样品12、15、12、20和21的颜色变化和聚结的程度呈递减次序,样品13具有最大的程度且样品18具有最小的程度。样品13、15和20在0周时间点具有放热峰(参见表19)。通常,在40℃/75% RH下在6周之后在非表面活性剂SDI样品中未观察到视觉颜色变化。
表43
SDI外观

溶解性和水含量
在3周40℃/75% RH稳定性时间点在敞口HPDE容器中,评价样品9和10的溶解性(参见表44)。在2小时、6小时和24小时在400 μg/mL和37℃的溶液中进行评价。与0周稳定性时间点的溶解性结果相比(参见表10),溶解性不受储存条件影响。
在4周40℃/75% RH稳定性时间点在敞口HPDE容器中,通过卡尔费歇尔滴定确定SDI样品的含水量(参见表45)。样品9和10具有较低水分含量。
表44
SDI溶解性(μg/mL)

表45
水含量

测定、水含量和未知产物的值
通常,发现测定值(90-110%)、水含量和未知产物(即降解产物或相关物质)值(总未知物:≤2.0%;单一未知物:≤0.2%)各自在预计范围内。对于SDI-表面活性剂样品,通常发现未知产物的量增加。
更具体地,SDI-表面活性剂样品17、18和19在敞口容器中在3周时间点在40℃/75%RH稳定性条件下的值(表46)、SDI-表面活性剂样品16和19在敞口容器中在6周时间点下的值(表46)、SDI样品27和28在密闭容器中在8周时间点下的值(表47)和非表面活性剂SDI样品4、5、9和10在敞口和密闭容器中在12周时间点的值(表48)各自与0周时间点的值类似且在预计范围内。
如前所示,术语“敞口容器”是指将SDI置于未封盖的高密度聚乙烯(HDPE)瓶子中。此外,如前所示,术语“密闭容器”是指将呈白色粉末状的SDI置于聚乙烯(PE)袋中,在各袋之间置放干燥剂,这些聚乙烯(PE)袋置于封盖的HDPE瓶子中。
如表47中所示,在0周至12周的稳定性时间点在密闭容器中观察到SDI样品27和28在40℃/75% RH稳定性条件下的测定或未知产物值没有相对变化。2周至12周的时期样品27的水含量增加约2.0%且样品28的水含量增加约1.0%。
如表48中所示,在12月时间点在敞口容器和密闭容器中观察到非表面活性剂SDI样品3、5、9和10在40℃/75% RH稳定性条件下未知产物值没有变化。
敞口容器中样品3和5的相对测定和水含量值与密闭容器中的值相比有变化。在12月时间点下在相同条件下,样品3和5的这些值与样品9和10的值相比也有变化。
术语“NT”表示“未测试”;术语“RRT”表示“相对滞留时间”。
表46
测定、水含量和未知物值(%)

表47
测定、水含量和未知物值(%)


表48
测定、水含量和未知物值(%)

结果和讨论
喷雾干燥是制备含有非晶化合物1和PVP K30或HPMC E5聚合物的共同沉淀的SDI的便利技术。含有HPMC E5的SDI显示非常高的产率,但具有PVP K30和PVP K90的那些SDI分别因聚结和纤维形成而产率较低。
所有沉淀的SDI样品的XRPD图案是非晶材料典型的。在TGA中在高达125℃下未遇到显著SDI重量损失。
DSC热谱图显示取决于所用SDI制剂材料的量和类型的在85℃与105℃之间的玻璃转变峰和在158℃与224℃之间的熔融峰。
对于化合物1负载为70%至80%的PVP K30 SDI,可见指示结晶的DSC放热转变。对于化合物1负载为50%和60%的PVP K30 SDI,未见到可检测的放热转变。
对于PVP K90和HPMC E5 SDI,在所研究的化合物1负载下,未见到可检测的放热转变。
对于HPMC E5 SDI,DSC热谱图显示在85℃与105℃之间的玻璃转变峰和在194℃与218℃之间的熔融峰。
在SDI中使用PVP或HPMC聚合物和任选的表面活性剂和所用溶剂体系的类型各自显示对溶解性和化合物1负载具有影响。
在具有表面活性剂(如Gelucire 50/13、泊洛沙姆407 (Lutrol F127)或SLS)的5%水溶液中,体系使SDI维持于溶液中长达2小时。特定地,对于PVP K30和PVP K90 SDI,存在表面活性剂使得在Gelucire 50/13和泊洛沙姆407中维持SDI浓度为约200 µg/m至300 µg/mL,且在SDS中以一定程度维持。对于HPMC E5 SDI,所测试的5%表面活性剂水溶液(除泊洛沙姆188外)能够使SDI浓度维持处于约200 µg/m至300 µg/mL。
看起来为10% w/v、7.5% w/v和5% w/v喷雾干燥溶液提供有利溶解性的溶剂体系分别包括DCM:DMSO (50:50)、DCM:DMSO (65:35)和DCM:MeOH (87.5/12.5)。DCM:MeOH溶剂体系看起来为有或没有表面活性剂的HPMC E5 SDI提供有利溶解曲线。在HPMC E5 SDI所用的表面活性剂之中,泊洛沙姆407表面活性剂看起来提供有利的溶解曲线。
使用所述喷雾干燥技术由聚合物和任选的表面活性剂形成的SDI通常提供大于67%的产率。使用有或没有表面活性剂的HPMC E5 (30-40% w/w)提供78%至80%的产率和387μg/mL至436 μg/mL之间的在表面活性剂水溶液中的溶解性浓度持续2小时与6小时之间的一段时间。对于具有表面活性剂(如泊洛沙姆407或Gelucire 50/13)的PVP K30 SDI,浓度在225 μg/mL至446 μg/mL之间。
化合物1负载为100 mg的HPMC E5 SDI在禁食状态模拟胃液介质中的体外溶解显示80%至90%之间的SDI在第一个小时溶解且保持稳定持续六小时。
对于非表面活性剂SDI,在40℃/75% RH下在3周时间点,XRPD显示没有可检测的化合物1结晶峰。对于表面活性剂SDI,在相同时间点,在所有PVP K30/Gelucire和PVP K30/泊洛沙姆SDI样品(12至15)中和在HPMC E5/泊洛沙姆SDI样品17中可见再结晶峰。对于HPMCE5/Gelucire SDI样品18,观察到微小结晶峰。对于SLS SDI样品16和19,在3周和6周时间点之后化合物1保持非晶。
对于PVP K30和PVP K90 SDI样品4、5、6和8,在化合物1负载>60%的情况下,作为DSC方法的结果,DSC热谱图在40℃/75% RH下在0周时间点显示热诱导的结晶。
对于HPMC E5 SDI样品10 (70%负载),在40℃/75% RH下在3周时间点,与HPMC E5SDI样品9 (50%负载)在0周时间点相比,DSC显示与化合物1熔融对应的微小吸热峰。
对于非表面活性剂SDI样品,在40℃/75% RH下6周之后,DSC显示没有显著变化。对于表面活性剂SDI样品,观察到热事件数目和强度增加。在3周40℃/75% RH稳定性时间点下,含有Gelucire和泊洛沙姆的表面活性剂SDI样品的吸热峰焓增加。对于SLS SDI样品,在40℃/75% RH下3周之后,DSC显示在25℃/min与10℃/min两种加热速率下高于200℃的热事件,指示非晶形式降解,但在6周时间点未见等效物峰,极有可能是归因于残余溶剂蒸发。对于Gelucire SDI样品13和15,DSC显示低于40℃的稳定温度下的热事件,以及观察到的SDI颜色变化。
SDI样品在各种水溶液中的溶解性看起来没有受40℃/75% RH稳定性条件影响,保持几乎恒定。在40℃/75% RH下在4周时间点,与HPMC E5 SDI样品9和10相比,PVP K30 SDI样品3更吸湿(参见表45)。
对于敞口容器中的SDI样品17、18和19,在40℃/75% RH下在0周时间点和在3周时间点,发现测定值、水含量和未知产物值符合规格。
对于敞口容器中的SDI样品16和19,在40℃/75% RH下在0周时间点和在6周时间点,发现测定值、水含量和未知产物值符合规格。
对于密闭容器中的非表面活性剂SDI样品27和28,在40℃/75% RH下在0周时间点和在2周和4周稳定性时间点,发现测定值、水含量和未知产物值符合规格且没有结晶迹象。
在6周时间点,DSC显示放热焓和吸热焓总体增加。PVP K30 SDI样品27在XRPD图案中显示结晶迹象,而PVP K90样品7和HPMC E5 SDI样品28仍保持非晶。在12周时间点获得类似结果。
60%化合物1负载下的SDI样品27 (PVP K30)和样品28 (HPMC E5)在40℃/75% RH(密闭于具有干燥剂的PE袋中)下仍保持物理和化学稳定,在25℃/60% RH与40℃/75% RH两者下持续长达3个月。在40℃/75% RH下在3个月时间点之后,如XRPD所示,SDI样品27的晶体结构具有轻微变化,与1%的HPMC E5 SDI样品28水分吸收相比,总水分增加2%。
实施例7
脂质胶囊制备
使用表49中所示的材料制备含有50 mg化合物1的制剂样品1胶囊(20.00% w/w剂量负载)。术语“TCQ”表示理论胶囊量(mg),即每一胶囊中每种材料的量。术语“TBQ”表示理论批量(kg),即松散产物中每种材料的量。
表49
脂质制剂

为了使得能够在分配之前熔融且消除所有固体块,将各自处于密闭容器中的Gelucire 44/14和Solutol HS-15置于设定为70±5℃的校准的烘箱中持续最少8小时。将Olsa 150L夹套混合/均质化釜预热到70±5℃持续约15分钟的最小时间段,且Gelucire44/14和Solutol HS-15各自经由在真空(在-0.10巴和-0.51巴之间)下与1”底部粉末进入隔膜阀连接的Vardex 1”管道添加到釜中。
使用设定为24±12 RPM的锚式混合器(操作范围:12-36 RPM)和设定为50±17RPM的叶片式混合器(操作范围:22-69 RPM)搅拌混合物约15分钟的时间段以达到70±5℃的溶液温度。解除釜真空,停止混合器且将丁基化羟基甲苯添加到溶液中。在先前相应设置下起始混合器且在70±5℃的温度下搅拌混合物约15分钟的最小时间段或直至丁基化羟基甲苯溶解。获得冲洗体积(3 kg至8 kg)的溶液。
化合物1经由在真空(在-0.10巴和-0.51巴之间)下与1”底部粉末进入隔膜阀连接的Vardex 1”管道缓慢添加到釜中使用冲洗溶液冲洗管道和阀门,且使用设定为1700±1300 RPM的均质机混合器(操作范围:400-3000 RPM)和设定为24±12 RPM的锚式混合器搅拌混合物。使真空增至-0.51巴和-1.02巴之间,且在所指示的相应设置下搅拌混合物最少8小时,同时维持温度为70±5℃。
将再循环泵和热追踪转移软管连接到三通阀。在溶液再循环之前加热转移软管且维持70±5℃的温度15±0.5分钟的最短时间段。解除釜真空,且同时维持0.45±0.25巴的釜压力和70±5℃的温度,将溶液再循环且使用设定为24±12 RPM的锚式混合器和设定为50±17 RPM的叶片式混合器混合持续60±0.5分钟的最大时间段。当使用构造成用于100倍放大级别下的偏光显微术的Olympus BX40显微镜的第一次溶液样品分析在至少3个区域中证实化合物1不存在晶体完全溶解时,停止混合和再循环。在第一次取样存在晶体的情况下,将溶液混合且再循环持续第二个60±0.5分钟的最大时间段。在后续取样存在晶体的情况下,每次将溶液混合且再循环60±0.5分钟的最大时间段,直至至少2次连续采样证实不存在晶体。在取样证实化合物1完全溶解之后,在先前相应设置和压力下混合和再循环的同时,釜温度降至50±5℃的温度。釜温度维持在50±5℃的温度和0.45±0.25巴的压力下,同时使用设定为20±5 RPM的锚式混合器混合和再循环。
准备胶囊液体填料(Shionogi品牌)以填充00号明胶胶囊且维持在50±5℃的温度下约15分钟的最小时间段,之后用釜散装溶液填充料斗。填充胶囊直至散装溶液耗尽,随后冷却约15分钟的最小时间段且适当储存。制备封边溶液且将其置于设定为55±5℃的温度的校准的烘箱中的密封容器中持续最少8小时。将封边溶液置于胶囊密封机(Shionogi品牌)中且维持在45±10℃的温度下。在125±75 RPM的密封辊速度下将胶囊密封,随后适当储存。
实施例8
临床前体内口服生物可利用性药代动力学大鼠研究
评价囊封的SDI制剂样品28、29、30、32和囊封的脂质制剂样品1经口给予大鼠后的暴露。如前述实施例中所示,预计SDI制剂和脂质制剂基于每种制剂中所用的材料在其溶解性质方面不同。在这种研究中确定SDI样品相对于彼此和相对于脂质制剂样品的口服生物可利用性。
如实施例5中所示,SDI样品制剂胶囊含有52.2% w/w的化合物1。如实施例7中所示,脂质制剂胶囊含有20% w/w的化合物1。两种SDI样品29和31各自含有PVP-K30,且两种SDI样品30和32各自含有HPMC E5,如实施例5,表25中所示。将每种SDI样品与表面活性剂Pol 407 (SDI样品31和32)或与MCC-102 (SDI样品29和30)共混。在所有制剂中添加CCS作为崩解剂。
向5组雄性Sprague Dawley Crl:SD大鼠(Charles River, Kingston, New York)(每组中n=4)给予每种制剂的胶囊。为动物随意提供正常食物和水。
在70 kg人中所给予的剂量与约350 mg相关,此剂量为临床相关剂量。给予的给药量(mg)恒定,但因为动物体重不同,所以整个组中剂量(mg/kg)不同。
例如,以1.5 mg/动物给药SDI制剂胶囊(对于平均300 g的动物,以约5 mg/kg计算)。随后在整个组中对个别剂量取平均值。基于个别动物的重量给予脂质制剂胶囊的剂量,以提供5 mg/kg的递送剂量。
在指定时间点(给药后0.5小时、1小时、3小时、6小时、9小时和24小时)获得血浆样品。在各组之中使用SigmaStat 3.0通过方差分析(ANOVA)比较指定时间的血浆浓度和计算的药代动力学参数。使用WinNonLin 5.2 (Pharsight Corporation, Carey Nc)确定每个个别大鼠的非房室药代动力学参数,并且随后在每个给药组中取平均值。
图3显示作为时间的函数的每种测试制剂的剂量标准化血浆浓度(最终第1组、第3组和第4组,n=4,且最终第2组和第5组,n=3)。计算在每个时间点每个动物的血浆浓度相对于剂量的比率,并且随后在整个组中取平均值。
*指示9小时样品结果的p值<0.05 (ANOVA,多重比较相对于脂质媒介物对照)。术语“Cp”表示“血浆浓度”,术语“DN”表示“剂量标准化”且术语“SD”表示“标准偏差”。
结果和讨论
达到最大浓度的时间(Tmax)为给药SDI制剂样品后约3小时和给药脂质制剂后约1小时。在给药脂质制剂的大鼠中Cmax是最高的。
与PVP K30 SDI制剂相比,在HPMC E5 SDI制剂中生物可利用性趋于较高,但没有获得统计学上显著的p值。与给药PVP K30 SDI的组(第1组和第3组)相比,给药HPMC SDI的组(第2组和第4组)显示略高的Cmax和曲线下面积(AUC)。在外相中添加表面活性剂(Pol407)看起来并不改进SDI制剂暴露。
尽管与以SDI制剂给予时相比,在以脂质制剂样品1(例如脂质制剂包含50% w/vGelucire® 44/14、50% w/v Solutol® HS和2.668% w/v 化合物1)给予时化合物1的暴露较高,但与20%剂量负载的脂质制剂相比,52.2%剂量负载的SDI制剂中的任一种的生物等效性表明当SDI以高剂量负载溶解于以下中时,相较于可变溶解性和沉淀而言SDI制剂显著改进较高剂量负载制剂的溶解性和随之而来的生物可利用性:基于脂质的制剂样品2 (100%负载;非晶形式,没有聚合物)、3 (50%负载;PVP K30 SDI)、4 (60%负载;PVP K30 SDI)、5(70%负载;PVP K30 SDI)、6 (80%负载;PVP K30 SDI)、7 (50%负载;PVP K90 SDI)、8 (70%负载;PVP K90 SDI)、9 (50%负载;HPMC E5 SDI)和10 (70%负载;HPMC E5 SDI)(参见表7、8和10和相关论述)。
实施例9
临床前体内口服生物可利用性药代动力学大鼠研究
评价囊封的SDI制剂样品34、35、36、37和39以及囊封的脂质制剂样品38经口给予大鼠后的暴露。
如表50中所示,使用化合物1a结晶形式与所列举的聚合物和材料一起制备非晶形式。SDI制剂样品根据本文实施例中所述的程序进行干式共混。例如,将POL 407与SDI在外相中共混成颗粒。化合物1a的纯度为86.1%,致使与每动物1.5 mg的目标剂量相比,向每个动物给予1.3 mg剂量。术语“N/A”表示“不适用”。
表50
研究制剂

如表51中所示,使用剂量负载为60%的非晶SDI与所列举材料混合来制备脂质制剂。术语“N/A”表示“不适用”。
表51
第5组脂质制剂样品1

向6组雄性Sprague Dawley Crl:SD大鼠(Charles River, Portage, Michigan)(每组中n=4)给予每种制剂的胶囊。为动物随意提供正常食物和水。
例如,以1.5 mg/动物给药SDI制剂胶囊(对于平均250 g的动物,以约6 mg/kg计算)。随后在整个组中对个别剂量取平均值。基于个别动物的重量给予脂质制剂胶囊的剂量,以提供5 mg/kg的递送剂量。
在指定时间点(给药后0.5小时、1小时、3小时、6小时、9小时、24小时、32小时和48小时)获得血浆样品。在各组之中使用SigmaStat 3.0通过方差分析(ANOVA)比较指定时间的血浆浓度和计算的药代动力学参数。使用WinNonLin 5.2 (Pharsight Corporation,Carey Nc)确定每个个别大鼠的非房室药代动力学参数,并且随后在每个给药组中取平均值。
图4显示作为时间的函数的每种测试制剂的剂量标准化血浆浓度。计算在每个时间点每个动物的血浆浓度相对于剂量的比率,并且随后在整个组中取平均值。
术语“Cp”表示“血浆浓度”,术语“DN”表示“剂量标准化”且术语“SD”表示“标准偏差”。
结果和讨论
在给药脂质制剂之后Tmax趋于最早,之后为40%和52%剂量负载的HPMC E5 SDI制剂。然而,当通过ANOVA比较所有六个组的Tmax时,这些组之中的差异并不显著。
当比较六个组之中剂量标准化最大血浆浓度(Cmax)时,与第5组(脂质制剂)相比时,第1组(40%剂量负载的PVP K30 SDI)显著降低;其他组中的Cmax不显著小于给药脂质制剂的动物中所测量的Cmax。
尽管所有SDI制剂组的剂量标准化AUC显著小于第5组脂质制剂,如图5中所示,但当以HPMC SDI制剂(20%、40%或52.2%剂量负载)给予时,与PVP K30 SDI制剂相比,化合物1的AUC血浆浓度通常更高且在统计学上是显著的(ANOVA;所有逐对多重比较程序;Holm-Sidak方法)。
例如,第2组(40%剂量负载的HPMC E5 SDI)显著高于第1组(40%剂量负载的PVPK30 SDI);第4组(20%剂量负载的HPMC E5 SDI)显著高于第1组(40%剂量负载的PVP K30SDI);第6组(52%剂量负载的HPMC E5 SDI)显著高于第1组(40%剂量负载的PVP K30 SDI);且第3组(20%剂量负载的PVP K30 SDI)大于第1组(40%剂量负载的PVP K30 SDI)。
在另一实施例中,第1组和第2组(分别是40%剂量负载的PVP K30 SDI和40%剂量负载的HPMC E5 SDI)在通过学生t检验(Student's t-test)比较时,HPMC E5 SDI制剂的绝对和剂量标准化Cmax值更高。通常,HPMC E5 SDI制剂暴露高于PVP K30 SDI制剂暴露,其中40%剂量负载的HPMC E5 SDI制剂具有最高暴露。
显示六个组之中的半衰期和平均滞留时间没有显著差异。其他比较并未达到显著性。
实施例10
临床前体内口服生物可利用性药代动力学食物影响研究
评价囊封的颗粒SDI制剂样品40、41和42在经口给予禁食正常食物的大鼠或喂饲高脂肪食物的大鼠后的食物影响。
使用化合物1a结晶形式与聚合物一起制备非晶形式。将SDI制剂样品40和41干式共混(都没有表面活性剂)且根据本文实施例中所述的程序粒化且囊封。特定地,根据实施例12的程序制备SDI制剂共混物样品42颗粒(有表面活性剂)。
如表52中所示,向6组雄性Sprague Dawley Crl:SD大鼠(Charles River,Portage, Michigan) (每组中n=4)给予每种制剂的胶囊。为相应组中的动物提供高脂肪食物(34.9%脂肪)、正常食物(4.3%脂肪)。随意提供水。
高脂肪膳食中的脂肪百分比与通常用于人临床高脂肪膳食的那些脂肪百分比类似(50-60%卡路里来自脂肪;参见FDA Guidance for Industry: Food-EffectBioavailability and Fed Bioequivalence Studies, Food and Drug Administration,Rockville, MD)。
关于六个组,第1组、第3组和第5组喂饲正常食物两天,禁食过夜,之后给药,随后允许在给药之后进食四小时。第2组、第4组和第6组喂饲高脂肪食物两天,且允许在给药之前随意进食。
表52
研究制剂

以1.5 mg/动物给药胶囊(对于平均300 g的动物,以约5 mg/kg计算)。随后在整个组中对个别剂量取平均值。基于个别动物的重量给予脂质制剂胶囊的剂量,以提供5 mg/kg的递送剂量。
在指定时间点(给药后0.5小时、1小时、3小时、6小时、9小时、24小时、32小时和48小时)获得血浆样品。在各组之中使用SigmaStat 3.0通过方差分析(ANOVA)比较血浆浓度差异的显著性和计算的药代动力学参数。使用WinNonLin 5.2 (Pharsight Corporation,Carey Nc)确定每个个别大鼠的药代动力学参数,并且随后在每个给药组中取平均值。通过学生t检验使用Excel,比较在喂饲状态与禁食状态之间指定时间的血浆浓度和计算的药代动力学参数。
图5显示在喂饲动物中作为时间的函数的每种测试制剂的剂量标准化血浆浓度。计算在每个时间点每个动物的血浆浓度相对于剂量的比率,并且随后在整个组中取平均值。
图6显示在禁食动物中作为时间的函数的每种测试制剂的剂量标准化血浆浓度。计算在每个时间点每个动物的血浆浓度相对于剂量的比率,并且随后在整个组中取平均值。
术语“Cp”表示“血浆浓度”,且术语“SD”表示“标准偏差”。
结果和讨论
这个研究中所用的SDI制剂的药代动力学参数没有显著不同,如每个动物的AUC所定义。另外,在喂饲组之间暴露没有显著差异。
对于囊封的HPMC E5 SDI制剂,在喂饲组与禁食组之间暴露和食物影响没有显著差异。
比较禁食动物中的暴露,囊封的HPMC E5 SDI制剂的暴露高于30%剂量负载的PVPK30 SDI制剂的暴露。
对于20%剂量负载与30%剂量负载两者的囊封的PVP K30 SDI制剂,喂饲动物中的暴露显著高于禁食动物中的暴露,如通过剂量标准化Cmax和AUC所测量。
比较20%剂量负载和30%剂量负载的PVP K30 SDI制剂的暴露,30%剂量负载制剂的暴露显著低于20%剂量负载制剂的暴露。然而,两种剂量负载的PVP K30 SDI制剂显示显著的食物影响,如Cmax所定义。
实施例11
临床前体内口服生物可利用性药代动力学食物影响研究
如表53中所制备,在经口给予五肽胃泌素治疗的犬之后,将囊封的SDI制剂样品A、样品B、样品C和样品D (200 mg)的食物影响与囊封的脂质制剂样品E (60 mg)相比。样品A、B、C、D和E分别与表53中的第1组至第5组相对应。
如表53中所示,剂量负载表示60% w/w SDI。向每只犬给予的剂量是基于12 kg的平均动物重量(8 kg至16 kg的范围)。给予的实际剂量使用每个个别动物的体重来计算。
表53
研究制剂

如表54中所示,每种脂质制剂胶囊中的每种材料示为每胶囊的量(Amt) (mg)和%w/w。
表54
脂质制剂

将每种制剂的胶囊给予5组未处理的雄性米格犬(Beagle Dog) (StillmeadowInc., Sugar Land, Texas) (每组中n=4)。在给予6 mg/kg五肽胃泌素(肌肉内)之前将喂饲犬与禁食(隔夜)犬预处理40分钟。喂饲犬在胶囊给予之前接受高脂肪罐装饲料30分钟。禁食犬在给药之后喂饲4小时。研究之前使所有犬适应高脂肪食物至少6天。
因为禁食犬的胃的pH在个别犬之中不同且可达到高达pH 8.0 (Kararli, TT.Comparison of the gastrointestinal anatomy, physiology, and biochemistry ofhumans and commonly used laboratory animals. Biopharm. Drug. Disps. 1995, 16:351-380;Akimoto M, Nagahata N, Furuya A, Fukushima K, Higuchi S, Suwa T.Gastric pH profiles of beagle dogs and their use as an alternative to humantesting. Eur J Pharm Biopharm. 2000, 49:99-102),所以用五肽胃泌素预处理的犬进行研究。禁食人胃中的pH为大约2 (Kararli, 1995)。五肽胃泌素是激素胃泌素的类似物且刺激胃酸分泌,使得胃pH更代表着禁食人受试者(Kararli, 1995;Akimoto等,2000)。然而,使用五肽胃泌素使模型标准化,减少归因于个别犬的胃pH差异的任何变化,且使胃pH与人更类似。五肽胃泌素的剂量(6 µg/kg,肌肉内,在口服给药之前40分钟)基于公布的研究来使用(Kararli, 1995;Akimoto等,2000)。
先前公布的研究显示在喂饲犬相对于禁食犬的研究中使用高脂肪膳食可预测人食物影响(Lentz KA, Quitko M, Morgan DG, Grace JE Jr, Gleason C, Marathe PH.Development and validation of a preclinical food effect model. J Pharm Sci.2007, 96:459-472;Homer LM, Clarke CR, Weingarten AJ. Effect of dietary fat onoral bioavailability of tepoxalin in dogs. Aliment Pharmacol Ther. 2005, 28:287-291)。
例如,SDI制剂胶囊以200 mg给药(对于平均10 kg至15 kg的动物,以约6.7 mg/kg至约10 mg/kg计算)。
在指定时间点(给药后30分钟、1小时、2小时、4小时、6小时、8小时、12小时和24小时)获得血浆样品。在各组之中使用SigmaStat 3.0通过方差分析(ANOVA)比较指定时间的血浆浓度和计算的药代动力学参数。使用WinNonLin 5.2 (Pharsight Corporation,Carey Nc)确定每个个别动物的非房室药代动力学参数,并且随后在每个给药组中取平均值。
图7显示作为时间的函数的每种测试制剂的平均血浆浓度。计算在每个时间点每个动物的血浆浓度相对于剂量的比率,并且随后在整个组中取平均值。
术语“Cp”表示“血浆浓度”,且术语“SD”表示“标准偏差”。
结果和讨论
当给予使用PVP K30或HPMC E5的SDI制剂中的任一种和脂质制剂时,在禁食犬中化合物1的暴露较高。使用PVP K30 SDI的喂饲犬显示暴露明显降低,因此表明当使用PVPK30 SDI时食物影响存在。相比之下,在禁食状态或喂饲状态下使用HPMC E5 SDI的化合物1的暴露出人意料地类似,因此表明当使用HPMC E5 SDI时避免了食物影响。
实施例12
临床体内脂质胶囊口服生物可利用性食物影响研究
在健康成人志愿者中在第1阶段、随机分组、安慰剂对照、递增、单剂量、安全性、耐受性、PK和食物影响研究中评价化合物1。化合物1提供于填充脂质的明胶胶囊中。研究的主要目标是确定安全达到药理学活性目标血浆浓度(如由异种移植研究所确定)且适合用于后续多剂量研究的化合物1的剂量范围。
研究中的受试者登记入2个阶段中。在第1阶段中,将40个受试者加入5个具有8个受试者的群组中,各群组接受剂量水平为0.03 mg/kg、0.1 mg/kg、0.3 mg/kg、1 mg/kg和3mg/kg的相继增高的单次剂量的化合物1。在一个群组内,6个受试者(3个雄性和3个雌性)接受化合物1,且2个受试者(1个雄性和1个雌性)接受安慰剂。将另外12个受试者(6个雄性和6个雌性)登记入第2阶段以评价食物对化合物1的PK的影响。
在研究的第1阶段与第2阶段两者期间,在基线和在给予研究药物之后72小时内反复地和另外在最后一次研究治疗之后7天随访时收集关于不良事件、生命征象、血液计数、凝血评估、血液化学测定、尿检和ECG的数据。在第1阶段与第2阶段两者中,在多个时间点收集用于评估血浆化合物1浓度的血液样品。化合物1浓度使用LC-MS/MS分析,对人血浆进行验证。在多个时间点收集用于测量血浆DHODH水平的血液。使用临床验证的ELISA (R&DSystems)分析血浆DHODH浓度。类似地,在多个时间点收集用于测量血浆VEGF-A水平的血液。使用临床验证的ELISA (R&D Systems)分析血浆VEGF-A浓度。
按计划,40个受试者(20个雄性和20个雌性)完成其参与研究的第1阶段,且12个受试者(6个雄性和6个雌性)完成其参与研究的第2阶段。受试者年龄在20岁至55岁(第1阶段)和18岁至52岁(第2阶段)范围内。其体重在51 kg至98 kg (第1阶段)和59 kg至85 kg (第2阶段)范围内。
化合物1通常具有良好耐受性且不存在严重药物相关的不良事件。在第1阶段中给药的40个受试者之中,最频繁的治疗引发的不良事件为头痛(8个受试者发作9次,全部接受化合物1)和恶心(5个受试者发作5次,4个接受化合物1和1个接受安慰剂)。其他类型的不良事件在少于5个受试者(10%)中发生。在第2阶段期间,最频繁的不良事件为头痛(3个受试者发作3次)和背痛(2个受试者发作2次);仅在单一受试者中注意到其他不良事件。所有不良事件的严重程度均为1级,但其中处于禁食状态的接受1 mg/kg化合物1的受试者中在第2阶段中有1例2级腹泻。不良事件的发生、与研究药物的关系和严重程度并非清晰地具有剂量依赖性,但头痛数目可随剂量而略有增加。在研究期间没有发生死亡或严重不良事件。由于安全原因,受试者都没有提前终止研究。
在两个阶段中,基于受试者的体检、生命征象测量或ECG,不存在安全问题。没有观察到血液学、凝血或化学参数方面的临床上显著的变化。类似地,未见到有临床意义的尿检异常。
第1阶段期间化合物1的平均血浆浓度-时间曲线示于图8中。根据受试者的喂饲或禁食状态的化合物1的平均血浆浓度-时间曲线示于图9中。在约30分钟的停滞期之后,化合物1出现在血浆中。在剂量≥0.30 mg/kg下,化合物1浓度持续存在于血浆中72小时,并且在3.0-mg/kg剂量下,在药物给予之后168小时仍显而易见低浓度的化合物1。在受试者在高脂肪、高卡路里膳食之后接受药物时,受试者的平均Cmax增加。在有或没有食物的情况下,可安全达成动物肿瘤模型中所建立的目标血浆浓度。
化合物1在血浆中的PK参数指示平均Tmax在3小时至6小时的范围内。在第1阶段期间,Cmax和AUC的平均值随剂量稳步上升。平均Cmax值的增加通常是以剂量比例的。平均AUC0-24值增加在某种程度上直至1.00-mg/kg剂量水平大于剂量比例,而在从1.00-mg/kg到3.00-mg/kg剂量水平的转变中小于剂量比例。终末半衰期(t1/2β)在28小时至56小时的范围内。
在第2阶段中,就在给予1 mg/kg化合物1之前摄取高脂肪、高卡路里膳食会使平均Cmax增加约40%,但实质上并不改变其他PK参数。
在第1阶段期间,相对于雄性,雌性的Cmax值略高(p=0.043,ANOVA),但AUC0-24值没有显著不同。在第2阶段期间,与雄性相比,雌性的Cmax和AUC0-24值较高(对于两种比较,p<0.01,ANOVA)。这个研究中这些差异的相关性并不清楚,假定在后续第1阶段多剂量研究中没有观察到类似性别相关差异(数据未示出)。
在来自登记入3-mg/kg组(在第1阶段中)中的受试者的血浆样品中评价DHODH。在取样时段过程中,化合物1组中自基线的平均变化与安慰剂组中类似。
实施例13
PVP片剂制备
使用表55中所示的材料制备用于以下中的共混制剂样品42:含有25 mg化合物1(20% w/w剂量负载)的制剂样品42a片剂、含有100 mg化合物1 (20% w/w剂量负载)的制剂样品42b片剂和含有200 mg化合物1 (20% w/w剂量负载)的制剂样品42c片剂。
表55
PVP制剂共混物样品42

称重材料A-K且使用配备有30目筛网的FitzMill按以下次序筛分:A、C、D、E和B。将筛分的材料装载到PK 1ft3 V-共混器中且在25 RPM下混合约5分钟的时间段。所得干燥共混物使用辊压机TFC-Labo在500±100 psi的压实压力、约1.75 RPM (在约1.25 RPM至约2.00 RPM的范围内)的目标辊速度、约21 RPM (在约16 RPM至约25 RPM的范围内)的目标螺杆速度和约0.055英寸(在约0.05英寸至约0.07英寸的范围内)的目标间隙厚度下粒化。收集未压实粉末,随后将其再循环回辊压机料斗中。收集内相条带,随后使用配备有30目筛网的FitzMill减小成颗粒。根据颗粒产量调节材料F、G、H、I、J和K的重量以维持w/w %,随后使用配备有30目筛网的FitzMill按以下次序筛分:G、H、I、J、K和F。将筛分的材料装载到PK1ft3 V-共混器中且在25 RPM下混合约5分钟的时间段。将松散粒化批料添加到PK 1ft3 V-共混器中且在25 RPM下与筛分的材料混合约10分钟的时间段。
对于制剂样品42a 25 mg片剂,准备具有6 mm圆形标准凹形B-工具冲头尺寸的Mini-Press II制片机。压制片剂以获得1250 mg (在约1188 mg至约1313 mg的范围内)的10个片剂的平均目标重量、125 mg (在约112.5 mg至约137.5 mg的范围内)的目标个别片剂重量、4.5 mm (在约3.5 mm至约5.5 mm的范围内)的目标个别厚度和5 kp (在约3 kp至约8 kp的范围内)的目标个别硬度。
对于制剂样品42b 100 mg片剂,准备具有11 mm圆形标准凹形B-工具冲头尺寸的Mini-Press II制片机。压制片剂以获得5000 mg (在约4750 mg至约5250 mg的范围内)的10个片剂的平均目标重量、500 mg (在约450 mg至约550 mg的范围内)的目标个别片剂重量、5.7 mm (在约4.7 mm至约6.7 mm的范围内)的目标个别厚度和7 kp (在约5 kp至约9kp的范围内)的目标个别硬度。
对于制剂样品42c 200 mg片剂,准备具有18.97×9.91 mm长方形标准凹形B-工具冲头尺寸的Mini-Press II制片机。压制片剂以获得10000 mg (在约9500 mg至约10500 mg的范围内)的10个片剂的平均目标重量、1000 mg (在约900 mg至约1100 mg的范围内)的目标个别片剂重量、7.6 mm (在约6.6 mm至约8.6 mm的范围内)的目标个别厚度和13.5 kp(在约11 kp至约16 kp的范围内)的目标个别硬度。
实施例14
临床体内PVP片剂口服生物可利用性食物影响研究
在各种临床试验中,使用脂质制剂的化合物1安全性显示,在各种给药量(25 mg、50 mg、100 mg、125 mg和200 mg)下测试和如上文所述制备的1.5 mg/kg剂量在直至(且包括) 1000 mg的剂量(测试的最高单次剂量)下是可接受的。然而,在脂质制剂胶囊中可达成的化合物1剂量负载限制可以可接受的剂型量长期递送的剂量强度,其中脂质制剂胶囊的每种剂量需要每一剂给予至少六个胶囊。
在评价食物对PVP SDI制剂片剂的生物可利用性的影响的临床BA/BE (生物等效性/生物可利用性)研究中,使实施例13中制备的PVP SDI制剂片剂与实施例7中制备的脂质制剂胶囊相比。
化合物1以单次剂量于脂质制剂胶囊中或以PVP SDI制剂片剂给予。研究的主要目标为确定以2种制剂给予的化合物1的比较性单次剂量PK和安全性。研究还旨在研究食物对PVP SDI制剂片剂的PK和安全性的影响。
研究中的受试者登记入3个阶段。在第1阶段中,24个受试者随机加入3个具有8个受试者的群组中,其接受剂量水平为0.5 mg/kg、1 mg/kg和2 mg/kg的在脂质制剂胶囊和PVP SDI制剂片剂两者中的化合物1。在第2阶段中,24个受试者加入3个具有8个受试者的群组中,每个群组接受剂量水平为400 mg、800 mg和1600 mg的相继增高的剂量的在PVP SDI制剂片剂中的化合物1。另外12个受试者(6个雄性和6个雌性)登记入第3阶段中以评价化合物1在以400 mg和1000 mg在PVP SDI制剂片剂中给予时食物对化合物1的PK的影响。
在研究期间,在基线和在给予研究药物之后72小时内反复地和另外在最后一次研究治疗之后7天随访时收集关于不良事件、生命征象、血液计数、凝血评估、血液化学测定、尿检和ECG的数据。在多个时间点收集用于评估血浆化合物1浓度的血液样品。化合物1浓度使用LC-MS/MS分析,对人血浆进行验证。
按计划,24个受试者(12个雄性和12个雌性)完成其参与研究的第1阶段,24个受试者(12个雄性和12个雌性)完成其参与研究的第2阶段,且12个受试者(6个雄性和6个雌性)完成其参与研究的第3阶段。受试者年龄在22岁至54岁(第1阶段)、19岁至47岁(第2阶段)和20岁至50岁(第3阶段)范围内。
化合物1通常具有良好耐受性。整体安全性与先前化合物1临床研究中的观察结果一致。在第1阶段中,观察到口干、腹部不适、头痛和腹泻散发性发作;在第2阶段中,观察到恶心、厌食症和腹胀散发性发作;在第3阶段中,观察到眼部不适、鼻充血和咳嗽散发性发作。事件主要为轻度的。在研究期间没有发生死亡或严重不良事件。
在所有3个阶段中,基于受试者的体检、生命征象测量或ECG,不存在安全问题。通常,没有观察到血液学、凝血、化学或尿检参数有临床上显著的变化。在第1阶段中,1个在第1周接受2 mg/kg在脂质胶囊制剂中的化合物1的雄性受试者在登记用于第2周研究时段时偶然发现具有3级血清肌酸激酶升高。考虑到可能导致肌酸激酶自肌肉释放的剧烈活动的历程,认为异常值不大可能与药物相关。然而,作为预防措施,将所述受试者排除在进一步参与研究之外。
在脂质胶囊制剂的情况下在第1阶段期间化合物1的平均血浆浓度-时间曲线与先前第1a阶段研究中观察到的血浆-浓度曲线类似。如图10中所示,在整个3种给药剂量水平中,PVP SDI片剂制剂显示平均Cmax和AUC0-24值分别为用脂质胶囊制剂获得的那些的19%和18%。在固体制剂中较高剂量的化合物1的平均血浆浓度-时间曲线示于图11中。在约30分钟的停滞期之后,化合物1出现在血浆中。化合物1浓度持续存在于血浆中72小时且在药物给予之后168小时仍显而易见。在受试者在高脂肪、高卡路里膳食之后接受药物时,受试者的平均Cmax增加,如图12中所示。
化合物1在血浆中的PK参数表明平均Tmax在3小时至7小时的范围内。在第1阶段期间,PVP SDI制剂片剂中化合物1的相对生物可利用性的范围在14%至28%之间,指示在PVPSDI制剂片剂与脂质制剂胶囊之间化合物1的生物等效性的显著差异。在第2阶段期间,当仅给予PVP SDI制剂片剂时,Cmax和AUC的平均值随剂量而上升。然而,平均Cmax值增加不与剂量成比例(对于两种比较,p<0.05,ANOVA)。半衰期在38小时至65小时的范围内。还表明,在第3阶段中就在给予400 mg或1000 mg化合物1之前摄取高脂肪、高卡路里膳食会使平均Cmax和AUC增加约100%。
实施例15
HPMC 50 mg片剂制备
使用表56中所示的材料制备含有50 mg化合物1的制剂样品43片剂(33.33% w/w剂量负载)。
表56
50 mg片剂制剂样品43

称重材料A-G且使用配备有20目筛网的FitzMill在70%的速度下按以下次序筛分:A、B、C、D和E。将筛分的材料装载到PK 1ft3 V-共混器中且在25 RPM下混合约5分钟至约10分钟的时间段。材料F经由30目筛网手动筛分,随后将其添加到PK 1ft3 V-共混器且在25RPM下混合约2分钟的时间段。所得干燥共混物使用辊压机TFC-Labo在1000±100 psi的压实压力、2.5 RPM (在2.00 RPM至3.00 RPM的范围内)的目标辊速度、37.5 RPM (在30.0RPM至45.0 RPM的范围内)的目标螺杆速度和1.0 mm (在0.8 mm至1.3 mm的范围内)的目标条带厚度下压实以形成条带。收集未压实粉末,随后经由30目筛网手动筛分且再循环回辊压机料斗中。收集条带,随后使用配备有20目筛网的FitzMill在70%的速度下减小成颗粒。材料G经由30目筛网手动筛分且装载到具有松散粒化批料的PK 1ft3 V-共混器中。材料在25 RPM下混合约2分钟的时间段。
准备具有9/32英寸圆形标准凹形B-工具冲头尺寸的Mini-Press II制片机。压制片剂以获得1500 mg (在约1425 mg至约1575 mg或约1425 mg至约1575 mg的范围内)的10个片剂的平均目标重量、150 mg (在约135 mg至约165 mg或约135 mg至约165 mg的范围内)的目标个别片剂重量、3.8 mm (在约3.4 mm至约4.2 mm或约3.4 mm至约4.2 mm的范围内)的目标个别厚度和8 kp (在约4 kp至约12 kp或约4 kp至约12 kp的范围内)的目标个别硬度。
实施例16
HPMC 200 mg片剂制备
使用表57中所示的材料制备含有50 mg化合物1的制剂样品44片剂(33.33% w/w剂量负载)。
表57
200 mg片剂制剂样品44

称重材料A-G且使用配备有20目筛网的FitzMill在70%的速度下按以下次序筛分:B、C、D、E和F。将筛分的材料装载到PK 1ft3 V-共混器中且在25 RPM下混合约5分钟至约10分钟的时间段。材料F经由30目筛网手动筛分,将其添加到PK 1ft3 V-共混器且在25 RPM下混合约2分钟的时间段。所得干燥共混物使用辊压机TFC-Labo在1000±100 psi的压实压力、2.5 RPM (在2.00 RPM至3.00 RPM的范围内)的目标辊速度、37.5 RPM (在30.0 RPM至45.0 RPM的范围内)的目标螺杆速度和1.0 mm (在0.8 mm至1.3 mm的范围内)的目标条带厚度下压实以形成条带。收集未压实粉末,随后经由30目筛网手动筛分且再循环回辊压机料斗中。收集条带,随后使用配备有20目筛网的FitzMill在70%的速度下减小成颗粒。材料G经由30目筛网手动筛分且装载到具有松散粒化批料的PK 1ft3 V-共混器中。材料在25 RPM下混合约2分钟的时间段。
准备具有15/32英寸圆形标准凹形B-工具冲头尺寸的Mini-Press II制片机。压制片剂以获得6000 mg (在约5700 mg至约6300 mg或约5820 mg至约6180 mg的范围内)的10个片剂的平均目标重量、600 mg (在约540 mg至约660 mg或约564 mg至约636 mg的范围内)的目标个别片剂重量、5.8 mm (在约5.4 mm至约6.2 mm或约5.5 mm至约6.1 mm的范围内)的目标个别厚度和14 kp (在约9 kp至约19 kp或约10 kp至约18 kp的范围内)的目标个别硬度。
实施例17
10 mg (样品45)、25 mg (样品46)和另外50 mg片剂(样品47)分别由每片50 mg、125 mg或250 mg具有以上实施例13的表55中所述相同的组成的PVP共混物制剂,使用如以上实施例15和16中对于50 mg和200 mg片剂所述类似的辊压程序制造。
5 mg片剂(样品48)通过辊压以下表58中所示的12.5%化合物1 (40%) SDI制剂使用与实施例15中对于50 mg片剂所述类似的程序制造。将100 mg表58中所示的PVP共混物用于每个片剂。
表58
用于5 mg辊压片剂的PVP共混物

另外批料5 mg片剂(样品49)通过直接压实具有以下表59中所示组成的PVP共混物制造。使用30目筛网将制剂的条目A至F筛分在一起且使用V-共混器混合5分钟。筛分最终成分硬脂酸镁,将其添加到混合物中且再共混2分钟。将100 mg的表59中所示的PVP共混物用于每个片剂。
表59
用于5 mg直接压实的片剂的PVP共混物

压制所有片剂。5 mg和25 mg片剂用6 mm直径圆形凹形工具压制,而10 mg和50 mg片剂分别用5 mm和8 mm直径圆形凹形工具压制。
大于300个片剂由以上样品45至49中的每一种制造。随机选择十个片剂且评价外观、重量、厚度、硬度、脆度和崩解时间。发现所述片剂的重量、厚度和硬度均匀,没有观察到物理缺陷。含有50% SDI (样品45-46)的片剂在9分钟至18分钟之间崩解,而含有12.5% SDI的那些片剂在一分钟内崩解。
通过将样品储存在瓶子中15℃、60%相对湿度下或在40℃、75%相对湿度下18个月对以上10 mg和50 mg片剂(样品45和47)进行稳定性研究。在1、3、6、9、12和18个月测试样品。在样品中的任一种中都没有观察到降解产物或其他劣化迹象增加。在整个研究中所有样品中的水含量保持低于4%。溶解曲线也基本上不变。
本发明的范围不受本文所述的特定方面限制。实际上,根据前文描述和附图,除所述的修改之外,本发明的各种修改对本领域的技术人员而言将变得显而易见。这样的修改旨在属于随附权利要求书的范围内。
本文中引用的所有参考文献均以全文引用的方式并入本文中,且用于所有目的,其引用的程度如同特定地且个别地将每个个别出版物或专利或专利申请出于所有目的而以全文引用的方式并入一样。
Claims (17)
1.喷雾干燥的中间体,其包含非晶形式的具有式(I)的化合物:

和聚合物,其中所述聚合物为亲水性聚合物。
2.如权利要求1所述的喷雾干燥的中间体,其中所述聚合物为聚乙烯吡咯烷酮或羟丙基甲基纤维素。
3.如权利要求2所述的喷雾干燥的中间体,其中所述聚乙烯吡咯烷酮为聚乙烯吡咯烷酮K-30。
4.如权利要求2所述的喷雾干燥的中间体,其中所述羟丙基甲基纤维素为羟丙基甲基纤维素E5。
5.如权利要求1所述的喷雾干燥的中间体,其中化合物1以所述喷雾干燥的中间体的重量计为40%。
6.用于制备如权利要求1所述的喷雾干燥的中间体的方法,其包括以下步骤:将所述形式的化合物1和聚合物共同溶解于溶剂体系中以形成液体分散体,随后通过喷雾干燥去除所述溶剂以提供呈固体分散体形式的所述中间体。
7.如权利要求1所述的喷雾干燥的中间体用于制备药物组合物的用途,所述药物组合物包含所述喷雾干燥的中间体与一种或多种药学上可接受的赋形剂的紧密混杂物以提供选自片剂或胶囊的口服剂型。
8.药物组合物,其包含如权利要求1所述的喷雾干燥的中间体与一种或多种药学上可接受的赋形剂的紧密混杂物以提供选自片剂或胶囊的口服剂型。
9.如权利要求8所述的药物组合物,其中化合物1以所述组合物的重量计为20%。
10.如权利要求8所述的药物组合物,其中所述口服剂型为片剂。
11.如权利要求8所述的药物组合物治疗有需要的受试者的选自白血病或炎性疾病的病况的用途,其包括向所述受试者给予有效量的所述药物组合物。
12.如权利要求8所述的药物组合物的用途,其中所述有效量以基于重量或固定剂量的给药方案给予所述受试者,其中所述给药方案维持目标血浆浓度。
13.如权利要求8所述的药物组合物在制造用于治疗有需要的受试者的选自白血病和炎性疾病的病况的药物中的用途,其包括向所述受试者给予有效量的所述药物。
14.治疗有需要的受试者的选自白血病和炎性疾病的病况的方法,其包括向所述受试者给予有效量的如权利要求8所述的药物组合物。
15.如权利要求14所述的方法,其中所述药物组合物与食物一起给予。
16.如权利要求11、13或14中任一项所述的方法或用途,其中所述治疗的病况为选自急性白血病或慢性白血病的白血病,其中所述急性白血病选自急性淋巴细胞性白血病;急性髓细胞性白血病,选自成髓细胞性、原髓细胞性、髓单核细胞性、单核细胞性或红细胞白血病性白血病;或骨髓增生异常综合征;并且其中所述慢性白血病选自慢性髓细胞白血病;慢性粒细胞白血病;慢性淋巴细胞性白血病;或毛细胞白血病;或真性红细胞增多症。
17.如权利要求11、13或14中任一项所述的方法或用途,其中所述治疗的病况为选自类风湿性关节炎和多发性硬化症的炎性疾病。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201862714182P | 2018-08-03 | 2018-08-03 | |
| US62/714182 | 2018-08-03 | ||
| PCT/US2019/044853 WO2020028778A1 (en) | 2018-08-03 | 2019-08-02 | Bioavailable oral dosage forms |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN112752570A true CN112752570A (zh) | 2021-05-04 |
Family
ID=67766269
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201980065062.1A Pending CN112752570A (zh) | 2018-08-03 | 2019-08-02 | 生物可利用口服剂型 |
Country Status (23)
| Country | Link |
|---|---|
| US (1) | US20210205225A1 (zh) |
| EP (1) | EP3829544A1 (zh) |
| JP (1) | JP2021532193A (zh) |
| KR (1) | KR20210041589A (zh) |
| CN (1) | CN112752570A (zh) |
| AR (1) | AR115913A1 (zh) |
| AU (1) | AU2019316036A1 (zh) |
| BR (1) | BR112021001859A2 (zh) |
| CA (1) | CA3107737A1 (zh) |
| CL (1) | CL2021000271A1 (zh) |
| CO (1) | CO2021001979A2 (zh) |
| CR (1) | CR20210093A (zh) |
| EA (1) | EA202190429A1 (zh) |
| EC (1) | ECSP21011795A (zh) |
| IL (1) | IL280471A (zh) |
| MX (1) | MX2021001364A (zh) |
| NI (1) | NI202100006A (zh) |
| PE (1) | PE20211410A1 (zh) |
| PH (1) | PH12021550237A1 (zh) |
| SG (1) | SG11202100984VA (zh) |
| TN (1) | TN2021000021A1 (zh) |
| TW (1) | TW202019414A (zh) |
| WO (1) | WO2020028778A1 (zh) |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA3080983C (en) | 2009-05-27 | 2023-02-28 | Pct Therapeutics, Inc. | Method of inhibiting and reducing a viral infection |
| WO2019028171A1 (en) | 2017-08-01 | 2019-02-07 | Ptc Therapeutics, Inc. | DHODH INHIBITOR FOR USE IN THE TREATMENT OF HEMATOLOGICAL CANCERS |
| CN112028888B (zh) * | 2020-09-11 | 2021-08-20 | 中国药科大学 | 一种化合物及其制备方法和用途 |
| US20220151938A1 (en) * | 2022-01-28 | 2022-05-19 | Ptc Therapeutics, Inc. | Tablet for use in treating huntington's disease and method of making the same |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20090203709A1 (en) * | 2008-02-07 | 2009-08-13 | Abbott Laboratories | Pharmaceutical Dosage Form For Oral Administration Of Tyrosine Kinase Inhibitor |
| US20100158858A1 (en) * | 2007-04-13 | 2010-06-24 | Liangxian Cao | Administration of carboline derivatives useful in the treatment of cancer and other diseases |
| WO2010138706A1 (en) * | 2009-05-27 | 2010-12-02 | Ptc Therapeutics, Inc. | Methods for treating breast cancer |
| US20120202763A1 (en) * | 2009-05-27 | 2012-08-09 | Ptc Therapeutics, Inc | Methods for treating cancer and non-neoplastic conditions |
| WO2017091373A1 (en) * | 2015-11-25 | 2017-06-01 | Patheon Development Services Inc. | Amorphous dispersion granules and oral dosage forms |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2007526341A (ja) | 2004-03-03 | 2007-09-13 | スフェリックス, インコーポレイテッド | 疎水性薬物のためのポリマー薬物送達システム |
| SG184755A1 (en) | 2004-03-15 | 2012-10-30 | Ptc Therapeutics Inc | Carboline derivatives useful in the inhibition of angiogenesis |
| US8076353B2 (en) | 2004-03-15 | 2011-12-13 | Ptc Therapeutics, Inc. | Inhibition of VEGF translation |
| US7767689B2 (en) | 2004-03-15 | 2010-08-03 | Ptc Therapeutics, Inc. | Carboline derivatives useful in the treatment of cancer |
| US8076352B2 (en) | 2004-03-15 | 2011-12-13 | Ptc Therapeutics, Inc. | Administration of carboline derivatives useful in the treatment of cancer and other diseases |
| WO2009040818A1 (en) | 2007-09-25 | 2009-04-02 | Solubest Ltd | Compositions comprising lipophilic active compounds and method for their preparation |
-
2019
- 2019-08-02 EA EA202190429A patent/EA202190429A1/ru unknown
- 2019-08-02 EP EP19759102.7A patent/EP3829544A1/en active Pending
- 2019-08-02 PE PE2021000157A patent/PE20211410A1/es unknown
- 2019-08-02 KR KR1020217006355A patent/KR20210041589A/ko unknown
- 2019-08-02 BR BR112021001859-0A patent/BR112021001859A2/pt unknown
- 2019-08-02 SG SG11202100984VA patent/SG11202100984VA/en unknown
- 2019-08-02 CR CR20210093A patent/CR20210093A/es unknown
- 2019-08-02 CN CN201980065062.1A patent/CN112752570A/zh active Pending
- 2019-08-02 MX MX2021001364A patent/MX2021001364A/es unknown
- 2019-08-02 US US17/264,940 patent/US20210205225A1/en not_active Abandoned
- 2019-08-02 CA CA3107737A patent/CA3107737A1/en active Pending
- 2019-08-02 WO PCT/US2019/044853 patent/WO2020028778A1/en active Application Filing
- 2019-08-02 JP JP2021529250A patent/JP2021532193A/ja active Pending
- 2019-08-02 TN TNP/2021/000021A patent/TN2021000021A1/en unknown
- 2019-08-02 AU AU2019316036A patent/AU2019316036A1/en active Pending
- 2019-08-05 AR ARP190102223A patent/AR115913A1/es unknown
- 2019-08-05 TW TW108127793A patent/TW202019414A/zh unknown
-
2021
- 2021-01-28 IL IL280471A patent/IL280471A/en unknown
- 2021-02-01 PH PH12021550237A patent/PH12021550237A1/en unknown
- 2021-02-02 CL CL2021000271A patent/CL2021000271A1/es unknown
- 2021-02-03 NI NI202100006A patent/NI202100006A/es unknown
- 2021-02-18 CO CONC2021/0001979A patent/CO2021001979A2/es unknown
- 2021-02-19 EC ECSENADI202111795A patent/ECSP21011795A/es unknown
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20100158858A1 (en) * | 2007-04-13 | 2010-06-24 | Liangxian Cao | Administration of carboline derivatives useful in the treatment of cancer and other diseases |
| US20090203709A1 (en) * | 2008-02-07 | 2009-08-13 | Abbott Laboratories | Pharmaceutical Dosage Form For Oral Administration Of Tyrosine Kinase Inhibitor |
| WO2010138706A1 (en) * | 2009-05-27 | 2010-12-02 | Ptc Therapeutics, Inc. | Methods for treating breast cancer |
| US20120202763A1 (en) * | 2009-05-27 | 2012-08-09 | Ptc Therapeutics, Inc | Methods for treating cancer and non-neoplastic conditions |
| WO2017091373A1 (en) * | 2015-11-25 | 2017-06-01 | Patheon Development Services Inc. | Amorphous dispersion granules and oral dosage forms |
Non-Patent Citations (1)
| Title |
|---|
| 李全林: "《新医药开发与研究 下》", 31 December 2008, 中国医药科技出版社 * |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2020028778A1 (en) | 2020-02-06 |
| EA202190429A1 (ru) | 2021-06-15 |
| CR20210093A (es) | 2021-05-28 |
| PH12021550237A1 (en) | 2021-10-11 |
| TW202019414A (zh) | 2020-06-01 |
| AU2019316036A1 (en) | 2021-03-04 |
| EP3829544A1 (en) | 2021-06-09 |
| SG11202100984VA (en) | 2021-02-25 |
| MX2021001364A (es) | 2021-04-13 |
| CO2021001979A2 (es) | 2021-03-08 |
| AR115913A1 (es) | 2021-03-10 |
| US20210205225A1 (en) | 2021-07-08 |
| NI202100006A (es) | 2021-05-21 |
| TN2021000021A1 (en) | 2022-10-03 |
| ECSP21011795A (es) | 2021-03-31 |
| PE20211410A1 (es) | 2021-08-02 |
| IL280471A (en) | 2021-03-25 |
| BR112021001859A2 (pt) | 2021-04-27 |
| KR20210041589A (ko) | 2021-04-15 |
| CA3107737A1 (en) | 2020-02-06 |
| JP2021532193A (ja) | 2021-11-25 |
| CL2021000271A1 (es) | 2021-08-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN112752570A (zh) | 生物可利用口服剂型 | |
| TWI635863B (zh) | 帕博西里之固態劑型 | |
| AU2015250574B2 (en) | Preparation containing tetracyclic compound at high dose | |
| JP6330118B2 (ja) | ベンダムスチンと組み合わせたヒストンデアセチラーゼ阻害剤の製剤とその使用 | |
| JP6133211B2 (ja) | 骨髄線維症を処置するための組成物及び方法 | |
| AU2008262059B2 (en) | Stabilized amorphous forms of Imatinib mesylate | |
| EA028009B1 (ru) | Фармацевтическая композиция с улучшенной биодоступностью | |
| BRPI0913379B1 (pt) | Intermediário compactado cpmpreendendo dimaleato de bibw 2992, na forma de um pó, e seu método de produção, mistura intermediária ou final, e formulações orais sólidas prontas para o uso/ingestão | |
| TW201102067A (en) | Solid dispersions containing an apoptosis-promoting agent | |
| CN109475553A (zh) | 尼洛替尼的药物组合物 | |
| KR20140129164A (ko) | 히스톤 데아세틸라아제 억제제 및 파조파닙의 조합물 및 이의 용도 | |
| ES2886022T3 (es) | Composiciones farmacéuticas sólidas antidiabéticas | |
| EP3354283B1 (en) | Pharmaceutical capsule composition comprising silodosin | |
| US20220064204A1 (en) | Amorphous form of 5-bromopyridin-3-yl 3-deoxy-3-[4-(3,4,5-trifluorophenyl)-1h-1,2,3-triazol-1-yl]-1-thio-alpha-d-galactopyranoside | |
| EP3525792A1 (en) | Powder for oral suspension containing lamotrigine | |
| EA042654B1 (ru) | Биодоступные пероральные дозированные формы | |
| WO2007102038A1 (en) | Ziprasidone formulations | |
| WO2015199115A1 (ja) | 経口投与用医薬組成物 | |
| US20240343735A1 (en) | Hydrochloride Salt of Inupadenant, Pharmaceutical Compositions and Methods of Use Thereof | |
| KR20190007370A (ko) | 바제독시펜 또는 그의 약제학적으로 허용가능한 염, 및 콜레칼시페롤 또는 그의 약제학적으로 허용가능한 염을 포함하는 우수한 안정성 및 용출률을 갖는 복합제제 | |
| KR20230026415A (ko) | 경구 제제 및 이의 용도 | |
| KR20220103952A (ko) | (s)-2-(2,6-디옥소피페리딘-3-일)-4-((2-플루오로-4-((3-모르폴리노아제티딘-1-일)메틸)벤질)아미노)이소인돌린-1,3-디온을 포함하는 제약 조성물 및 그의 사용 방법 | |
| NZ763604B2 (en) | Preparation containing tetracyclic compound at high dose |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| RJ01 | Rejection of invention patent application after publication |
Application publication date: 20210504 |
|
| RJ01 | Rejection of invention patent application after publication |