CN112250552B - 一种全氟己酮的制备方法 - Google Patents
一种全氟己酮的制备方法 Download PDFInfo
- Publication number
- CN112250552B CN112250552B CN202010966807.6A CN202010966807A CN112250552B CN 112250552 B CN112250552 B CN 112250552B CN 202010966807 A CN202010966807 A CN 202010966807A CN 112250552 B CN112250552 B CN 112250552B
- Authority
- CN
- China
- Prior art keywords
- reaction
- perfluoro
- perfluorohexanone
- epoxy
- methylpentane
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- WVSNNWIIMPNRDB-UHFFFAOYSA-N 1,1,1,3,3,4,4,5,5,6,6,6-dodecafluorohexan-2-one Chemical compound FC(F)(F)C(=O)C(F)(F)C(F)(F)C(F)(F)C(F)(F)F WVSNNWIIMPNRDB-UHFFFAOYSA-N 0.000 title claims abstract description 39
- 238000002360 preparation method Methods 0.000 title claims abstract description 11
- 238000006243 chemical reaction Methods 0.000 claims abstract description 74
- 239000003054 catalyst Substances 0.000 claims abstract description 34
- NOESGFSFSJKFIF-UHFFFAOYSA-N 2-fluoro-2-(1,1,2,2,2-pentafluoroethyl)-3,3-bis(trifluoromethyl)oxirane Chemical compound FC(F)(F)C(F)(F)C1(F)OC1(C(F)(F)F)C(F)(F)F NOESGFSFSJKFIF-UHFFFAOYSA-N 0.000 claims abstract description 30
- 238000000034 method Methods 0.000 claims abstract description 22
- 238000006317 isomerization reaction Methods 0.000 claims abstract description 19
- 239000012044 organic layer Substances 0.000 claims abstract description 18
- 239000002904 solvent Substances 0.000 claims abstract description 17
- 239000012295 chemical reaction liquid Substances 0.000 claims abstract description 13
- 239000007800 oxidant agent Substances 0.000 claims abstract description 10
- 230000001590 oxidative effect Effects 0.000 claims abstract description 10
- 238000003756 stirring Methods 0.000 claims description 43
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 claims description 27
- 239000007788 liquid Substances 0.000 claims description 20
- 239000000243 solution Substances 0.000 claims description 19
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 claims description 14
- FAEGGADNHFKDQX-UHFFFAOYSA-N 1,1,1,3,4,4,5,5,5-nonafluoro-2-(trifluoromethyl)pent-2-ene Chemical compound FC(F)(F)C(C(F)(F)F)=C(F)C(F)(F)C(F)(F)F FAEGGADNHFKDQX-UHFFFAOYSA-N 0.000 claims description 11
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 claims description 10
- 239000003377 acid catalyst Substances 0.000 claims description 10
- 239000005708 Sodium hypochlorite Substances 0.000 claims description 9
- SUKJFIGYRHOWBL-UHFFFAOYSA-N sodium hypochlorite Chemical group [Na+].Cl[O-] SUKJFIGYRHOWBL-UHFFFAOYSA-N 0.000 claims description 9
- ZUHZGEOKBKGPSW-UHFFFAOYSA-N tetraglyme Chemical compound COCCOCCOCCOCCOC ZUHZGEOKBKGPSW-UHFFFAOYSA-N 0.000 claims description 9
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 claims description 8
- 239000000460 chlorine Substances 0.000 claims description 8
- 229910052801 chlorine Inorganic materials 0.000 claims description 8
- 239000007864 aqueous solution Substances 0.000 claims description 7
- HQABUPZFAYXKJW-UHFFFAOYSA-N butan-1-amine Chemical compound CCCCN HQABUPZFAYXKJW-UHFFFAOYSA-N 0.000 claims description 7
- SBZXBUIDTXKZTM-UHFFFAOYSA-N diglyme Chemical compound COCCOCCOC SBZXBUIDTXKZTM-UHFFFAOYSA-N 0.000 claims description 6
- MUDUFRFJYXOJGQ-UHFFFAOYSA-N 8-benzyl-3-prop-2-enyl-3,8-diazabicyclo[3.2.1]octane Chemical compound C1N(CC=C)CC2CCC1N2CC1=CC=CC=C1 MUDUFRFJYXOJGQ-UHFFFAOYSA-N 0.000 claims description 5
- 239000002253 acid Substances 0.000 claims description 5
- IOXXVNYDGIXMIP-UHFFFAOYSA-N n-methylprop-2-en-1-amine Chemical compound CNCC=C IOXXVNYDGIXMIP-UHFFFAOYSA-N 0.000 claims description 5
- UHUUYVZLXJHWDV-UHFFFAOYSA-N trimethyl(methylsilyloxy)silane Chemical compound C[SiH2]O[Si](C)(C)C UHUUYVZLXJHWDV-UHFFFAOYSA-N 0.000 claims description 5
- 230000035484 reaction time Effects 0.000 claims description 4
- 239000012299 nitrogen atmosphere Substances 0.000 claims description 2
- 238000005406 washing Methods 0.000 abstract description 6
- 230000008569 process Effects 0.000 abstract description 5
- 230000000694 effects Effects 0.000 abstract description 4
- 230000007613 environmental effect Effects 0.000 abstract description 3
- 238000010992 reflux Methods 0.000 description 19
- 238000010907 mechanical stirring Methods 0.000 description 18
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 6
- 239000005457 ice water Substances 0.000 description 6
- -1 fluoride ions Chemical class 0.000 description 5
- 239000002994 raw material Substances 0.000 description 5
- XJHCXCQVJFPJIK-UHFFFAOYSA-M caesium fluoride Chemical compound [F-].[Cs+] XJHCXCQVJFPJIK-UHFFFAOYSA-M 0.000 description 4
- 230000003197 catalytic effect Effects 0.000 description 4
- 238000004821 distillation Methods 0.000 description 4
- 238000006735 epoxidation reaction Methods 0.000 description 4
- 239000000203 mixture Substances 0.000 description 4
- 238000004321 preservation Methods 0.000 description 4
- PBVZTJDHQVIHFR-UHFFFAOYSA-N 1,1,2,3,3,3-hexafluoroprop-1-ene Chemical compound FC(F)=C(F)C(F)(F)F.FC(F)=C(F)C(F)(F)F PBVZTJDHQVIHFR-UHFFFAOYSA-N 0.000 description 3
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 3
- 230000009471 action Effects 0.000 description 3
- 239000007789 gas Substances 0.000 description 3
- 238000010438 heat treatment Methods 0.000 description 3
- 238000006459 hydrosilylation reaction Methods 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- 229910052757 nitrogen Inorganic materials 0.000 description 3
- YLCLKCNTDGWDMD-UHFFFAOYSA-N 2,2,3,3,3-pentafluoropropanoyl fluoride Chemical compound FC(=O)C(F)(F)C(F)(F)F YLCLKCNTDGWDMD-UHFFFAOYSA-N 0.000 description 2
- 238000009776 industrial production Methods 0.000 description 2
- 238000001819 mass spectrum Methods 0.000 description 2
- NROKBHXJSPEDAR-UHFFFAOYSA-M potassium fluoride Chemical compound [F-].[K+] NROKBHXJSPEDAR-UHFFFAOYSA-M 0.000 description 2
- KGAHKQBZVJJFBW-UHFFFAOYSA-N 1,1,1,2,2,6,6,7,7,7-decafluoro-3,3,5,5-tetrakis(trifluoromethyl)heptan-4-one Chemical compound FC(F)(F)C(F)(F)C(C(F)(F)F)(C(F)(F)F)C(=O)C(C(F)(F)F)(C(F)(F)F)C(F)(F)C(F)(F)F KGAHKQBZVJJFBW-UHFFFAOYSA-N 0.000 description 1
- SAPOZTRFWJZUFT-UHFFFAOYSA-N 1,1,1,2,3,4,5,5,5-nonafluoro-4-(trifluoromethyl)pent-2-ene Chemical compound FC(F)(F)C(F)=C(F)C(F)(C(F)(F)F)C(F)(F)F SAPOZTRFWJZUFT-UHFFFAOYSA-N 0.000 description 1
- XEZNGIUYQVAUSS-UHFFFAOYSA-N 18-crown-6 Chemical compound C1COCCOCCOCCOCCOCCO1 XEZNGIUYQVAUSS-UHFFFAOYSA-N 0.000 description 1
- LLRDUIXNNHUYLY-UHFFFAOYSA-N 2,2,3,4,4,5-hexafluoro-3,5-bis(trifluoromethyl)oxolane Chemical compound FC(F)(F)C1(F)OC(F)(F)C(F)(C(F)(F)F)C1(F)F LLRDUIXNNHUYLY-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- 229920004449 Halon® Polymers 0.000 description 1
- 238000007259 addition reaction Methods 0.000 description 1
- 229910001508 alkali metal halide Inorganic materials 0.000 description 1
- 150000008045 alkali metal halides Chemical class 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 239000002131 composite material Substances 0.000 description 1
- 150000001875 compounds Chemical class 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- 150000002118 epoxides Chemical class 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- 150000004673 fluoride salts Chemical class 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- IYRWEQXVUNLMAY-UHFFFAOYSA-N fluoroketone group Chemical group FC(=O)F IYRWEQXVUNLMAY-UHFFFAOYSA-N 0.000 description 1
- HCDGVLDPFQMKDK-UHFFFAOYSA-N hexafluoropropylene Chemical group FC(F)=C(F)C(F)(F)F HCDGVLDPFQMKDK-UHFFFAOYSA-N 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 238000002329 infrared spectrum Methods 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- RMLFHPWPTXWZNJ-UHFFFAOYSA-N novec 1230 Chemical compound FC(F)(F)C(F)(F)C(=O)C(F)(C(F)(F)F)C(F)(F)F RMLFHPWPTXWZNJ-UHFFFAOYSA-N 0.000 description 1
- 238000007344 nucleophilic reaction Methods 0.000 description 1
- SBOJXQVPLKSXOG-UHFFFAOYSA-N o-amino-hydroxylamine Chemical compound NON SBOJXQVPLKSXOG-UHFFFAOYSA-N 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- 235000003270 potassium fluoride Nutrition 0.000 description 1
- 239000011698 potassium fluoride Substances 0.000 description 1
- 125000006308 propyl amino group Chemical group 0.000 description 1
- 238000006462 rearrangement reaction Methods 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 229910000077 silane Inorganic materials 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 239000013638 trimer Substances 0.000 description 1
- 238000005292 vacuum distillation Methods 0.000 description 1
- 239000012808 vapor phase Substances 0.000 description 1
- 239000002699 waste material Substances 0.000 description 1
Images




Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/56—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds from heterocyclic compounds
- C07C45/57—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds from heterocyclic compounds with oxygen as the only heteroatom
- C07C45/58—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds from heterocyclic compounds with oxygen as the only heteroatom in three-membered rings
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J31/00—Catalysts comprising hydrides, coordination complexes or organic compounds
- B01J31/02—Catalysts comprising hydrides, coordination complexes or organic compounds containing organic compounds or metal hydrides
- B01J31/0272—Catalysts comprising hydrides, coordination complexes or organic compounds containing organic compounds or metal hydrides containing elements other than those covered by B01J31/0201 - B01J31/0255
- B01J31/0274—Catalysts comprising hydrides, coordination complexes or organic compounds containing organic compounds or metal hydrides containing elements other than those covered by B01J31/0201 - B01J31/0255 containing silicon
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J31/00—Catalysts comprising hydrides, coordination complexes or organic compounds
- B01J31/02—Catalysts comprising hydrides, coordination complexes or organic compounds containing organic compounds or metal hydrides
- B01J31/0272—Catalysts comprising hydrides, coordination complexes or organic compounds containing organic compounds or metal hydrides containing elements other than those covered by B01J31/0201 - B01J31/0255
- B01J31/0275—Catalysts comprising hydrides, coordination complexes or organic compounds containing organic compounds or metal hydrides containing elements other than those covered by B01J31/0201 - B01J31/0255 also containing elements or functional groups covered by B01J31/0201 - B01J31/0269
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D301/00—Preparation of oxiranes
- C07D301/02—Synthesis of the oxirane ring
- C07D301/03—Synthesis of the oxirane ring by oxidation of unsaturated compounds, or of mixtures of unsaturated and saturated compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D303/00—Compounds containing three-membered rings having one oxygen atom as the only ring hetero atom
- C07D303/02—Compounds containing oxirane rings
- C07D303/48—Compounds containing oxirane rings with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/10—Compounds having one or more C—Si linkages containing nitrogen having a Si-N linkage
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2231/00—Catalytic reactions performed with catalysts classified in B01J31/00
- B01J2231/50—Redistribution or isomerisation reactions of C-C, C=C or C-C triple bonds
- B01J2231/52—Isomerisation reactions
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Catalysts (AREA)
Abstract
本发明公开了一种全氟己酮的制备方法,包括:(1)将全氟烯烃与氧化剂在第一溶剂中进行反应,反应结束分出有机层,将有机层洗涤、精馏得到全氟‑2,3‑环氧‑2‑甲基戊烷;(2)将步骤(1)得到的全氟‑2,3‑环氧‑2‑甲基戊烷与二氮杂硅烷胺基催化剂在第二溶剂中进行异构化反应,反应结束后将反应液精馏得到全氟己酮产品。本发明具有工艺简单,异构化催化剂反应活性高,绿色环保等优点。
Description
技术领域
本发明涉及含氟酮的制备方法,具体涉及一种全氟己酮的制备方法。
背景技术
20世纪70年代,苏联的科学家合成了全氟己酮作为中间体,但没有大规模的投入生产,直到2001年美国的3M公司将其作为替代哈龙和氟代烷类的灭火剂后(商业名称为Novec l230),其合成及应用研究才日益得到人们的关注。这种新型替代品针对当前的环境政策趋势有较多的优越性能。
全氟己酮的合成路线已报导有许多种,大都以六氟丙烯、环氧六氟丙烷、六氟丙烯二聚体等为原料。
如美国专利US6478979公布了在无水条件并有氟化物离子的存在下,全氟丙酰氟与六氟丙烯进行加成反应生成全氟己酮。少量的六氟丙烯二聚体以及三聚体杂质通过蒸馏的方法除去。该工艺原料全氟丙酰氟价格高,不易得到,难以满足工业化生产要求。
又如中国专利CN102992986A提出以六氟丙烯二聚体混合物、全氟-4-甲基-2-戊烯或全氟2-甲基-2-戊烯为原料,通过将全氟2-甲基-2-戊烯定位氧化成全氟-2 ,3-环氧基-2-甲基戊烷、再将该环氧化物的结构重排得到全氟乙基异丙基酮。它使用的催化剂为有机碱、氨基氧化物、酰胺类化合物、碱金属卤化物或其它能离解出氟的化合物。在实施例,它具体采用次氯酸钠、氟化铯作为催化剂。虽然这种方法可得到全氟己酮,但是其收率还有进一步提高的余地。
又如中国专利CN103787854A提出以全氟2-甲基-2-戊烯为原料,经环氧化反应和催化异构化反应得到全氟己酮,包括制备中间体全氟-2-甲基-2,3-环氧戊烷为原料,随后在载体型催化剂作用下,在100~250℃的反应温度、0~0 .25MPa的压力下,进行连续气相催化异构化反应,得到全氟己酮粗品,再经精馏得到精制的全氟己酮产品。该方法中采用的载体型催化剂为以活性炭或DMSO溶剂为载体,以氟化铯或氟化钾与18-冠-6的络合物为催化剂的活性组分。在实施例中,其气相法合成全氟-2- 甲基-3-戊酮的反应温度具体为180℃左右。显然这种方法的缺点在于反应温度较高,反应流程复杂,不利于工业化生产。
又如中国专利CN103508868A提出在氟化盐和醚类化合物存在下,在10~70℃反应温度下,全氟-2,3-环氧-2-甲基戊烷发生催化重排反应生成全氟己酮。该专利使用复合催化剂,容易失活,反应收率不稳定。
发明内容
本发明针对现有技术的不足之处,提供一种工艺简单,异构化催化剂反应活性高,绿色环保的全氟己酮的制备方法。
为实现上述目的,本发明采用的技术方案为:一种全氟己酮的制备方法, 包括以下步骤:
(1)将全氟烯烃与氧化剂在第一溶剂中进行反应,所述全氟烯烃与氧化剂的质量比为0.05~0.5:1,反应温度为-10~50℃,反应时间为1~10h,反应结束分出有机层,将有机层洗涤、精馏得到全氟-2,3-环氧-2-甲基戊烷;
(2)将步骤(1)得到的全氟-2,3-环氧-2-甲基戊烷与二氮杂硅烷胺基催化剂在第二溶剂中进行异构化反应,所述全氟-2,3-环氧-2-甲基戊烷与异构化催化剂的质量比为5~70:1,反应温度为20~60℃,反应时间为1~8h,反应结束后将反应液精馏得到釜底液和全氟己酮产品。
优选的,所述全氟烯烃为全氟-2-甲基-2-戊烯。
优选的,所述氧化剂为有效氯含量为5-15wt%(wt%,质量百分含量)的次氯酸钠水溶液。
本发明中,全氟烯烃环氧化反应属于亲核反应,在有机溶剂存在的条件下,全氟烯烃与氧化剂作用,在较短的时间内可获得高收率的环氧化产物。优选的,所述第一溶剂为乙腈、二甘醇二甲醚、N,N-二甲基甲酰胺中的至少一种。
本发明中,所述二氮杂硅烷胺基催化剂是按以下方法制备的:
(1)按重量份,将5~15份的氯铂酸加入到200~300份无水异丙醇中,搅拌溶解10~20分钟,制得氯铂酸催化剂溶液,备用;
(2)按重量份,将600~700份四甲基二硅氧烷,300~400份N-甲基烯丙基胺,30~50份3-烯丙基-8-苄基-3,8-二氮杂双环[3.2.1]辛烷、0.08~0.2份步骤(1)得到的氯铂酸催化剂溶液在氮气氛围下进行反应,反应温度为50~70℃,反应30~40分钟后,加入4~10份正丁胺,继续反应2~5小时,结束反应,将反应液真空蒸馏得到二氮杂硅烷胺基催化剂。
优选的,所述真空蒸馏的真空度为0.096~0.1MPa。
优选的,所述第二溶剂为乙腈、二甘醇二甲醚、N,N-二甲基甲酰胺、四乙二醇二甲醚中的至少一种。
优选的,将步骤(2)所述的釜底液不经处理直接回用参与异构化反应。
本发明的全氟己酮的制备方法,将全氟烯烃与氧化剂经环氧化反应,制备全氟-2,3-环氧-2-甲基戊烷;全氟-2,3-环氧-2-甲基戊烷在异构化催化剂的作用下异构化为全氟己酮。本发明工艺简单,异构化催化剂反应活性高,反应液经本领域常规的精馏提纯即可得到纯度在99.9%以上的全氟己酮产品,精馏后的釜底液为溶剂和催化剂的混合物,可重复使用。
与现有技术相比,本发明具有以下有益效果:
1、工艺简单,本发明将全氟烯烃与氧化剂进行反应,制备全氟-2,3-环氧-2-甲基戊烷;全氟-2,3-环氧-2-甲基戊烷在异构化催化剂的作用下异构化为全氟己酮,反应液经本领域常规的精馏提纯即可得到纯度在99.9%以上的全氟己酮产品,反应步骤短,反应条件温和,操作简单,显著简化了工艺;
2、催化剂活性高,二氮杂硅烷胺基催化剂具有二氮杂双环,以及丙基胺官能团,可以有效提高催化活性,使全氟-2,3-环氧-2-甲基戊烷反应完全,降低分离难度,异构化反应收率在97.2%以上,最高可达98.2%;
3、绿色环保,反应液经本领域常规的精馏提纯即可得到纯度在99.9%以上的全氟己酮产品,精馏后的釜底液为溶剂和异构化催化剂的混合物,可重复套用,套用5次后全氟-2,3-环氧-2-甲基戊烷异构化为全氟己酮的收率仍在96.7%以上,显著减少了三废排放。
附图说明
图1为实施例4制备的全氟己酮产品的气相色谱谱图;
图2为实施例4制备的全氟己酮产品的红外光谱图;
图3为实施例4制备的全氟己酮产品质谱图;
图4为实施例4制备的全氟-2,3-环氧-2-甲基戊烷质谱图。
具体实施方式
以下结合具体实施例对本发明做进一步详细描述,但本发明不仅仅局限于以下实施例。
实施例中的中间体及产品含量均采用配有FID检测器的福立9790型气相色谱仪分析,色谱柱为105 m×0.25mm×0.5μm 的RTX-1毛细管柱。
实施例1~3为二氮杂硅烷胺基催化剂的制备实施例
实施例1
(1)向装有回流冷凝管和机械搅拌装置的1L烧瓶中加入200g无水异丙醇,再加入5g氯铂酸,在400rpm转速下搅拌溶解10分钟,制得氯铂酸催化剂溶液,备用;
(2)向装有回流冷凝管和机械搅拌装置的2L烧瓶中加入600g四甲基二硅氧烷,300gN-甲基烯丙基胺,30g3-烯丙基-8-苄基-3,8-二氮杂双环[3.2.1]辛烷,0.08g步骤(1)得到的氯铂酸催化剂溶液,开启搅拌,在200rpm搅拌转速下通入氮气,升温至55℃进行硅氢加成反应,反应30分钟后,将4g正丁胺加入到反应溶液中,继续反应2小时, 反应完毕的物料打入蒸馏釜,在真空度0.096MPa下蒸馏得到480g二氮杂硅烷胺基催化剂。
实施例2
(1)向装有回流冷凝管和机械搅拌装置的1L烧瓶中加入230g无水异丙醇,再加入12g氯铂酸,在300rpm转速下搅拌溶解15分钟,制得氯铂酸催化剂溶液,备用;
(2)向装有回流冷凝管和机械搅拌装置的2L烧瓶中加入620g四甲基二硅氧烷,350gN-甲基烯丙基胺,38g3-烯丙基-8-苄基-3,8-二氮杂双环[3.2.1]辛烷,0.12g步骤(1)得到的氯铂酸催化剂溶液,开启搅拌,在300rpm搅拌转速下通入氮气,升温至58℃进行硅氢加成反应,反应35分钟后,将6g正丁胺加入到反应溶液中,继续反应3小时, 反应完毕的物料打入蒸馏釜,在真空度0.097MPa下蒸馏得到560g二氮杂硅烷胺基催化剂。
实施例3
(1)向装有回流冷凝管和机械搅拌装置的1L烧瓶中加入300g无水异丙醇,再加入15g氯铂酸,在200rpm转速下搅拌溶解20分钟,制得氯铂酸催化剂溶液,备用;
(2)向装有回流冷凝管和机械搅拌装置的2L烧瓶中加入700g四甲基二硅氧烷,400gN-甲基烯丙基胺,50g3-烯丙基-8-苄基-3,8-二氮杂双环[3.2.1]辛烷,0.2g步骤(1)得到的氯铂酸催化剂溶液,开启搅拌,在400rpm搅拌转速下通入氮气,升温至60℃进行硅氢加成反应,反应40分钟后,将10g正丁胺加入到反应溶液中,继续反应5小时, 反应完毕的物料打入蒸馏釜,在真空度0.098MPa下蒸馏得到640g二氮杂硅烷胺基催化剂。
实施例4~9为全氟己酮的制备实施例
实施例4
(1)在装有回流冷凝管和机械搅拌装置的500ml四口烧瓶中,加入100g有效氯含量为15wt%的次氯酸钠水溶液、10g乙腈,开启搅拌,搅拌转速300rpm,控制反应温度-5℃,滴加30g全氟-2-甲基-2-戊烯,滴加时间为1h,滴加完毕继续保温反应6h小时后,停止搅拌结束反应,分出有机层,有机层用等量的冰水洗涤2次,精馏得24.1g全氟-2,3-环氧-2-甲基戊烷,收率76.1%,纯度99.8%;
(2)在装有回流冷凝管和机械搅拌装置的500ml四口烧瓶中,加入200g按步骤(1)的方法得到的全氟-2,3-环氧-2-甲基戊烷、3g实施例3制备的二氮杂硅烷胺基催化剂、20g乙腈,开启搅拌,搅拌转速400rpm,保持反应温度在20℃反应8h,反应结束后,将反应液精馏,得到釜底液和纯度为99.95%的全氟己酮,收率97.2%。
实施例5
(1)在装有回流冷凝管和机械搅拌装置的500ml四口烧瓶中,加入200g有效氯含量为8wt%的次氯酸钠水溶液、20g二甘醇二甲醚,开启搅拌,搅拌转速350rpm,控制反应温度0℃,滴加30g全氟-2-甲基-2-戊烯,滴加时间为0.5h,滴加完毕后继续保温反应2h小时后,停止搅拌结束反应,分出有机层,有机层用等量的冰水洗涤1次,精馏得24.2g全氟-2,3-环氧-2-甲基戊烷,收率76.6%,纯度99.7%;
(2)在装有回流冷凝管和机械搅拌装置的500ml四口烧瓶中,加入200g按步骤(1)的方法得到的全氟-2,3-环氧-2-甲基戊烷、6g实施例3制备的二氮杂硅烷胺基催化剂、50g二甘醇二甲醚,开启搅拌,搅拌转速450rpm,保持反应温度在30℃反应6h,反应结束后,将反应液精馏,得到釜底液和纯度为99.96%的全氟己酮,收率97.5%。
实施例6
(1)在装有回流冷凝管和机械搅拌装置的500ml四口烧瓶中,加入200g有效氯含量为10wt%的次氯酸钠水溶液、30g N,N-二甲基甲酰胺,开启搅拌,搅拌转速200rpm,控制反应温度10℃,滴加30g全氟-2-甲基-2-戊烯,滴加时间为2h,滴加完毕后继续保温反应5h小时,停止搅拌,结束反应,分出有机层,有机层用等量的冰水洗涤2次,精馏得24.6g全氟-2,3-环氧-2-甲基戊烷,收率77.8%,纯度99.9%;
(2)在装有回流冷凝管和机械搅拌装置的500ml四口烧瓶中,加入200g按步骤(1)的方法得到的全氟-2,3-环氧-2-甲基戊烷、9g实施例3制备的二氮杂硅烷胺基催化剂、100g溶剂N,N-二甲基甲酰胺,开启搅拌,搅拌转速400rpm,保持反应温度在40℃反应3h,反应结束后,将反应液精馏,得到釜底液和纯度为99.98%的全氟己酮,收率97.5%。
实施例7
(1)在装有回流冷凝管和机械搅拌装置的500ml四口烧瓶中,加入300g有效氯含量为10wt%的次氯酸钠水溶液、30g乙腈,开启搅拌,搅拌转速250rpm,控制反应温度20℃,滴加30g全氟-2-甲基-2-戊烯,滴加时间为1h,滴加完毕后继续保温反应4h小时,停止搅拌,结束反应,分出有机层,有机层用等量的冰水洗涤2次,精馏得23.9g全氟-2,3-环氧-2-甲基戊烷,收率75.6%,纯度99.6%;
(2)在装有回流冷凝管和机械搅拌装置的500ml四口烧瓶中,加入200g按步骤(1)的方法得到的全氟-2,3-环氧-2-甲基戊烷、12g实施例2制备的二氮杂硅烷胺基催化剂、100g溶剂四乙二醇二甲醚,开启搅拌,搅拌转速250rpm,保持反应温度在40℃反应3h,反应结束后,将反应液精馏,得到釜底液和纯度为99.94%的全氟己酮,收率97.7%。
实施例8
(1)在装有回流冷凝管和机械搅拌装置的500ml四口烧瓶中,加入300g有效氯含量为12wt%的次氯酸钠水溶液、20g乙腈,开启搅拌,搅拌转速400rpm,控制反应温度40℃,滴加30g全氟-2-甲基-2-戊烯,滴加时间为2h,滴加完毕后继续保温反应3h小时后,停止搅拌,结束反应,分出有机层,有机层用等量的冰水洗涤2次,精馏得22.8g全氟-2,3-环氧-2-甲基戊烷,收率72.2%,纯度99.6%;
(2)在装有回流冷凝管和机械搅拌装置的500ml四口烧瓶中,加入200g按步骤(1)的方法得到的全氟-2,3-环氧-2-甲基戊烷、15g实施例1制备的二氮杂硅烷胺基催化剂、200g溶剂四乙二醇二甲醚,开启搅拌,搅拌转速350rpm,保持反应温度在50℃反应4h,反应结束后,将反应液精馏,得到釜底液和纯度为99.96%的全氟己酮,收率97.7%。
实施例9
(1)在装有回流冷凝管和机械搅拌装置的1000ml四口烧瓶中,加入400g有效氯含量为5wt%的次氯酸钠水溶液、40g乙腈。开启搅拌,搅拌转速400rpm,控制反应温度-10℃,滴加30g全氟-2-甲基-2-戊烯,滴加时间为3h,滴加完毕后继续保温反应1h小时后,停止搅拌,结束反应,分出有机层,有机层用等量的冰水洗涤2次,精馏得20.4g全氟-2,3-环氧-2-甲基戊烷,收率64.5%,纯度99.5%。
(2)在装有回流冷凝管和机械搅拌装置的500ml四口烧瓶中,加入200g按步骤(1)的方法得到的全氟-2,3-环氧-2-甲基戊烷、30g实施例2制备的二氮杂硅烷胺基催化剂、200g溶剂四乙二醇二甲醚,开启搅拌,搅拌转速300rpm,保持反应温度在60℃反应1h,反应结束后,将反应液精馏,得到釜底液和纯度为99.98%的全氟己酮,收率98.2%。
实施例10~14为全氟己酮精馏釜底液套用实施例
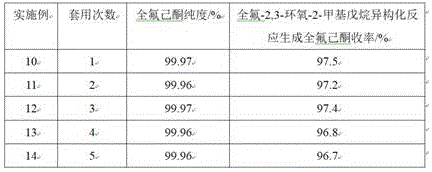
实施例10
在装有回流冷凝管和机械搅拌装置的500ml四口烧瓶中,加入实施例6步骤(2)中精馏后的釜底液、200g的全氟-2,3-环氧-2-甲基戊烷,开启搅拌,搅拌转速400rpm,保持反应温度在40℃反应3h,反应结束后,将反应液精馏,分离出全氟己酮,剩下的釜底液进行套用,试验结果见表1。
实施例11
试验装置和参数同实施例10,不同之处是加入的精馏后的釜底液为实施例10中剩下的釜底液,试验结果见表1。
实施例12
试验装置和参数同实施例10,不同之处是加入的精馏后的釜底液为实施例11中剩下的釜底液,试验结果见表1。
实施例13
试验装置和参数同实施例10,不同之处是加入的精馏后的釜底液为实施例12中剩下的釜底液,试验结果见表1。
实施例14
试验装置和参数同实施例10,不同之处是加入的精馏后的釜底液为实施例13中剩下的釜底液,试验结果见表1。
表1 全氟己酮精馏釜底液套用结果
Claims (2)
1.一种全氟己酮的制备方法, 其特征在于,包括以下步骤:
(1)将全氟烯烃与氧化剂在第一溶剂中进行反应,所述全氟烯烃与氧化剂的质量比为0.05~0.5:1,反应温度为-10~50℃,反应时间为1~10h,反应结束分出有机层,将有机层洗涤、精馏得到全氟-2,3-环氧-2-甲基戊烷,所述全氟烯烃为全氟-2-甲基-2-戊烯,所述氧化剂为有效氯含量为5~15wt%的次氯酸钠水溶液,所述第一溶剂为乙腈、二甘醇二甲醚、N,N-二甲基甲酰胺中的一种;
(2)将步骤(1)得到的全氟-2,3-环氧-2-甲基戊烷与异构化催化剂在第二溶剂中进行异构化反应,所述全氟-2,3-环氧-2-甲基戊烷与异构化催化剂的质量比为5~70:1,反应温度为20~60℃,反应时间为1~8h,反应结束后将反应液精馏得到釜底液和全氟己酮产品,所述第二溶剂为乙腈、二甘醇二甲醚、N,N-二甲基甲酰胺、四乙二醇二甲醚中的一种,所述异构化催化剂是按以下方法制备的:
1)按重量份,将5~15份的氯铂酸加入到200~300份无水异丙醇中,搅拌溶解10~20分钟,制得氯铂酸催化剂溶液,备用;
2)按重量份,将600~700份四甲基二硅氧烷,300~400份N-甲基烯丙基胺,30~50份3-烯丙基-8-苄基-3,8-二氮杂双环[3.2.1]辛烷、0.08~0.2份步骤(1)得到的氯铂酸催化剂溶液在氮气氛围下进行反应,反应温度为50~70℃,反应30~40分钟后,加入4~10份正丁胺,继续反应2~5小时,结束反应,将反应液在真空度为0.096~0.1MPa真空蒸馏得到异构化催化剂。
2.根据权利要求1所述的全氟己酮的制备方法, 其特征在于,将步骤(2)所述的釜底液不经处理直接回用参与异构化反应。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202010966807.6A CN112250552B (zh) | 2020-09-15 | 2020-09-15 | 一种全氟己酮的制备方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202010966807.6A CN112250552B (zh) | 2020-09-15 | 2020-09-15 | 一种全氟己酮的制备方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN112250552A CN112250552A (zh) | 2021-01-22 |
| CN112250552B true CN112250552B (zh) | 2023-02-03 |
Family
ID=74232336
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202010966807.6A Active CN112250552B (zh) | 2020-09-15 | 2020-09-15 | 一种全氟己酮的制备方法 |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN112250552B (zh) |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN114014782B (zh) * | 2021-10-20 | 2023-10-27 | 化学与精细化工广东省实验室 | 胺化-脱水一锅法气相连续制备七氟异丁腈的方法 |
| CN115504871A (zh) * | 2022-10-17 | 2022-12-23 | 内蒙古永和氟化工有限公司 | 一种全氟己酮的制备方法 |
| CN116854574B (zh) * | 2023-09-04 | 2023-12-12 | 哲弗智能系统(上海)有限公司 | 全氟己酮的制备方法 |
| CN116969825B (zh) * | 2023-09-25 | 2023-12-15 | 山东华夏神舟新材料有限公司 | 一种全氟己酮的纯化方法 |
Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3238241A (en) * | 1963-09-20 | 1966-03-01 | Pennsalt Chemicals Corp | Secondary perhaloalkyl chlorosulfates and fluorosulfates |
| JPH06184208A (ja) * | 1992-12-16 | 1994-07-05 | Asahi Glass Co Ltd | 含フッ素重合体の製法 |
| WO2011090992A1 (en) * | 2010-01-25 | 2011-07-28 | 3M Innovative Properties Company | Perfluoroketones as gaseous dielectrics |
| CN103787854A (zh) * | 2014-01-24 | 2014-05-14 | 奥瑞安有限公司 | 一种全氟-2-甲基-3-戊酮的制备工艺 |
| CN104379548A (zh) * | 2012-06-15 | 2015-02-25 | 中化蓝天集团有限公司 | 一种全氟-2-甲基-3-戊酮及中间物的制备方法 |
| CN105439835A (zh) * | 2015-12-02 | 2016-03-30 | 上海三爱富新材料股份有限公司 | 一种全氟己酮的制备方法 |
| CN106542984A (zh) * | 2016-10-13 | 2017-03-29 | 巨化集团技术中心 | 一种全氟‑2‑甲基‑3‑戊酮的制备方法 |
| CN107382692A (zh) * | 2017-09-06 | 2017-11-24 | 北京天康达科技发展有限公司 | 全氟己酮的合成方法与应用 |
| CN107501038A (zh) * | 2017-09-06 | 2017-12-22 | 北京天康达科技发展有限公司 | 一种全氟己酮的制备方法与应用 |
| CN108929212A (zh) * | 2018-08-17 | 2018-12-04 | 浙江利化新材料科技有限公司 | 一种全氟己酮的制备方法 |
| CN111004100A (zh) * | 2019-12-26 | 2020-04-14 | 西安元创化工科技股份有限公司 | 一种合成全氟己酮的方法 |
-
2020
- 2020-09-15 CN CN202010966807.6A patent/CN112250552B/zh active Active
Patent Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3238241A (en) * | 1963-09-20 | 1966-03-01 | Pennsalt Chemicals Corp | Secondary perhaloalkyl chlorosulfates and fluorosulfates |
| JPH06184208A (ja) * | 1992-12-16 | 1994-07-05 | Asahi Glass Co Ltd | 含フッ素重合体の製法 |
| WO2011090992A1 (en) * | 2010-01-25 | 2011-07-28 | 3M Innovative Properties Company | Perfluoroketones as gaseous dielectrics |
| CN104379548A (zh) * | 2012-06-15 | 2015-02-25 | 中化蓝天集团有限公司 | 一种全氟-2-甲基-3-戊酮及中间物的制备方法 |
| CN103787854A (zh) * | 2014-01-24 | 2014-05-14 | 奥瑞安有限公司 | 一种全氟-2-甲基-3-戊酮的制备工艺 |
| CN105439835A (zh) * | 2015-12-02 | 2016-03-30 | 上海三爱富新材料股份有限公司 | 一种全氟己酮的制备方法 |
| CN106542984A (zh) * | 2016-10-13 | 2017-03-29 | 巨化集团技术中心 | 一种全氟‑2‑甲基‑3‑戊酮的制备方法 |
| CN107382692A (zh) * | 2017-09-06 | 2017-11-24 | 北京天康达科技发展有限公司 | 全氟己酮的合成方法与应用 |
| CN107501038A (zh) * | 2017-09-06 | 2017-12-22 | 北京天康达科技发展有限公司 | 一种全氟己酮的制备方法与应用 |
| CN108929212A (zh) * | 2018-08-17 | 2018-12-04 | 浙江利化新材料科技有限公司 | 一种全氟己酮的制备方法 |
| CN111004100A (zh) * | 2019-12-26 | 2020-04-14 | 西安元创化工科技股份有限公司 | 一种合成全氟己酮的方法 |
Non-Patent Citations (2)
| Title |
|---|
| 全氟己酮合成工艺优化研究;屈文良;《中国知网 硕士学位论文》;20140715(第07期);1-44 * |
| 全氟己酮的合成与应用研究进展;丁元胜等;《浙江化工》;20051228(第12期);25-27 * |
Also Published As
| Publication number | Publication date |
|---|---|
| CN112250552A (zh) | 2021-01-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN112250552B (zh) | 一种全氟己酮的制备方法 | |
| CN107501038A (zh) | 一种全氟己酮的制备方法与应用 | |
| CN105330836B (zh) | 一种端环氧基烯丙醇聚氧乙烯醚的合成方法 | |
| CN111116302B (zh) | 一种卤代丁烯的合成方法 | |
| JP5603169B2 (ja) | (e)−3−メチル−2−シクロペンタデセノンの製造方法 | |
| US8039680B2 (en) | Process for producing 4-methyl-2,3,5,6-tetrafluorobenzyl alcohol | |
| CN108383681B (zh) | 一种全氟-2-甲基-2-戊烯的制备方法 | |
| JPWO1999033771A1 (ja) | −ch2−chf−基を有する化合物の製造方法 | |
| CN111039771A (zh) | 一种3,3,3-三氟丙酸的制备方法 | |
| CN106187691B (zh) | 一种从含六氟异丙醇和氢气的混合气中回收六氟异丙醇的方法 | |
| JP6219884B2 (ja) | (z)−3−メチル−2−シクロペンタデセノンの製造方法および(r)−(−)−3−メチルシクロペンタデカノンの製造方法 | |
| CN107011116B (zh) | 一种六氟异丁烯的制备方法 | |
| JPS582210B2 (ja) | 2,3−ジメチル−2,3−ブタンジオ−ルの製法 | |
| US20070191652A1 (en) | Process for production of 1,1,1,2- tetrafluoroethane and/or pentafluorethane and applications of the same | |
| JP2006219419A (ja) | パーフルオロビニルエーテルモノマーの製造法 | |
| JPWO2017022571A1 (ja) | フッ素化炭化水素の製造方法 | |
| JP2002544248A (ja) | 1,1,1,2,3,3,3−ヘプタフルオロプロパンの製造 | |
| KR102913335B1 (ko) | 헥사플루오로-2-부틴 및 1,1,1,4,4,4-헥사플루오로-2-부텐의 제조방법 | |
| JP3613635B2 (ja) | 3,4−カランジオールの製造法 | |
| JP2006008519A (ja) | トリエチレングリコールジビニルエーテルの製造方法 | |
| JP2019034901A (ja) | 組成物の処理方法 | |
| WO2022176837A1 (ja) | シクロヘキセノン化合物の製造方法 | |
| JP3613636B2 (ja) | 3,4−カランジオールの製造法 | |
| JP3673600B2 (ja) | 高純度シクロヘキセンオキサイドの製造法 | |
| JP2017218447A (ja) | 有機化合物の精製方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |