CN107405325B - 用于组合疗法的药物组合物 - Google Patents
用于组合疗法的药物组合物 Download PDFInfo
- Publication number
- CN107405325B CN107405325B CN201680015586.6A CN201680015586A CN107405325B CN 107405325 B CN107405325 B CN 107405325B CN 201680015586 A CN201680015586 A CN 201680015586A CN 107405325 B CN107405325 B CN 107405325B
- Authority
- CN
- China
- Prior art keywords
- ppar
- compound
- liver
- alpha
- agonist
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images
















Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/56—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids
- A61K31/575—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids substituted in position 17 beta by a chain of three or more carbon atoms, e.g. cholane, cholestane, ergosterol, sitosterol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/185—Acids; Anhydrides, halides or salts thereof, e.g. sulfur acids, imidic, hydrazonic or hydroximic acids
- A61K31/19—Carboxylic acids, e.g. valproic acid
- A61K31/192—Carboxylic acids, e.g. valproic acid having aromatic groups, e.g. sulindac, 2-aryl-propionic acids, ethacrynic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/21—Esters, e.g. nitroglycerine, selenocyanates
- A61K31/215—Esters, e.g. nitroglycerine, selenocyanates of carboxylic acids
- A61K31/216—Esters, e.g. nitroglycerine, selenocyanates of carboxylic acids of acids having aromatic rings, e.g. benactizyne, clofibrate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/365—Lactones
- A61K31/366—Lactones having six-membered rings, e.g. delta-lactones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/451—Non condensed piperidines, e.g. piperocaine having a carbocyclic group directly attached to the heterocyclic ring, e.g. glutethimide, meperidine, loperamide, phencyclidine, piminodine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/56—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/12—Antidiarrhoeals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2300/00—Mixtures or combinations of active ingredients, wherein at least one active ingredient is fully defined in groups A61K31/00 - A61K41/00
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Organic Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Emergency Medicine (AREA)
- Urology & Nephrology (AREA)
- Virology (AREA)
- Obesity (AREA)
- Oncology (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Molecular Biology (AREA)
- Communicable Diseases (AREA)
- Gastroenterology & Hepatology (AREA)
- Vascular Medicine (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
本发明涉及一种药物组合物,该药物组合物包含FXR激动剂和至少一种降脂剂(例如,PPAR‑α激动剂、PPAR‑δ激动剂、PPAR‑α和δ双重激动剂、和/或他汀)的组合。还披露了该组合用于治疗或预防FXR介导的疾病或病症的用途,该疾病或病症是如原发性胆汁性肝硬化(PBC)、原发性硬化性胆管炎(PSC)、门静脉高压、胆汁酸腹泻、NAFLD(非酒精性脂肪肝病)、NASH(非酒精诱发性脂肪性肝炎)、以及其他慢性肝病。本发明的组合可用于治疗或预防与脂质和肝酶水平升高相关的病症。本发明还涉及包括该药物组合的包装或试剂盒。
Description
背景技术
血液中循环脂质化合物(如胆固醇和甘油三酯)的浓度升高伴随着许多病症。这些病症包括II型糖尿病、原发性胆汁性肝硬化(PBC)、原发性硬化性胆管炎(PSC)、各种慢性肝炎状态(乙型和丙型肝炎)、NASH(非酒精性脂肪性肝炎)和动脉疾病(包括冠状动脉疾病、脑血管动脉疾病、周围血管疾病、主动脉瘤和颈动脉粥样硬化病症)。过去已经使用各种降脂技术来治疗和预防伴随高血脂状态的血管事件(如心力衰竭、栓塞、心脏病发作和中风)。这样的治疗方法已经包括膳食改变和控制血液中循环的高甘油三酯和胆固醇水平。后者通常在药理学上并且最近用各种“他汀类”进行治疗。包括在用于治疗脂质水平升高的病症的治疗剂中的是各种纤维酸衍生物。包括氯贝特在内的一些较老的纤维酸衍生物在治疗与脂质升高相关的病症中已经占据一个过去的位置,但最近新的贝特类(包括非诺贝特、吉非罗齐、环丙贝特)以及甚至更近的贝特类(包括哌啶、4-羟基哌啶、哌啶-3-烯和哌嗪)已经加入抗脂质疗法的行列。这些较新的分子具有有希望的特性来降低胆固醇和甘油三酯两者。然而,在一些情况下,单独的纤维酸衍生物不足以控制许多患者中存在的严重的高血脂水平。纤维酸衍生物的副作用特征也可以从例如在组合疗法存在下的剂量降低而得到改善。
因此,需要一种改进的疗法用于治疗涉及血液中循环脂质化合物(如胆固醇和甘油三酯)浓度升高的病症。
附图说明
图1A是来自假BDL小鼠的天狼星红(Sirius-red)染色的肝切片的代表性显微照片。
图1B是来自BDL运载体处理的小鼠的天狼星红染色的肝切片的代表性显微照片。
图1C是来自BDL-OCA处理的小鼠的天狼星红染色的肝切片的代表性显微照片。
图1D是BDL-阿托伐他汀处理的小鼠的天狼星红染色的肝切片的代表性显微照片。
图1E是来自BDL-OCA-阿托伐他汀处理的小鼠的天狼星红染色的肝切片的代表性显微照片。
图2是显示用OCA和阿托伐他汀单独和组合处理的BDL小鼠中的天狼星红阳性面积(%)的柱状图。
图3A是显示来自在APOE*3Leiden.CETP小鼠中用OCA、低剂量非诺贝特单独和组合处理的炎症细胞病灶数目的柱状图。
图3B是显示来自在APOE*3Leiden.CETP小鼠中用OCA、高剂量非诺贝特单独和组合处理的炎症细胞病灶数目的柱状图。
图4是显示OCA和阿托伐他汀单独和组合对瘦素-ob/ob小鼠中的纤维化阶段的影响的柱状图。
图5A是显示用OCA和阿托伐他汀单独和组合处理的瘦素-ob/ob小鼠中血浆甘油三酯水平的柱状图。
图5B是显示用OCA和阿托伐他汀单独和组合处理的瘦素-ob/ob小鼠中血浆甘油三酯水平从基线的变化的柱状图。
图6A是显示HFC+OCA相对于HFC维持的APOE*3Leiden.CETP小鼠的经典途径的富集分析的柱状图。
图6B是显示HFC+OCA+低剂量非诺贝特相对于HFC维持的APOE*3Leiden.CETP小鼠的经典途径的富集分析的柱状图。
图7A是显示在APOE*3Leiden.CETP小鼠中由OCA+低剂量非诺贝特的组合与单一疗法调节的新颖的差异表达基因的数量的维恩图。
图7B是显示在APOE*3Leiden.CETP小鼠中由OCA+低剂量非诺贝特的组合相对于单一疗法调节的基因的途径富集的柱状图。
图8是显示OCA、和他汀类的组合对人类中的LDL胆固醇的影响的图。
发明内容
本申请涉及一种药物组合物,该药物组合物包含(i)第一化合物,(ii)至少一种PPAR-α激动剂、PPAR-δ激动剂和/或PPAR-α和δ双重激动剂,以及(iii)任选地一种或多种药学上可接受的载体,其中该第一化合物是FXR激动剂。
本发明还涉及一种药物组合物,该药物组合物包含(i)第一化合物,(ii)至少一种贝特,以及任选地(iii)一种或多种药学上可接受的载体,其中该第一化合物是FXR激动剂。
本发明还涉及一种药物组合物,该药物组合物包含(i)第一化合物,(ii)至少一种降脂剂,以及任选地(iii)一种或多种药学上可接受的载体,其中该第一化合物是FXR激动剂。
本发明还涉及一种药物组合物,该药物组合物包含(i)第一化合物,(ii)至少一种他汀,以及任选地(iii)一种或多种药学上可接受的载体,其中该第一化合物是FXR激动剂。
本发明还涉及一种药物组合物,该药物组合物包含(i)第一化合物,(ii)至少一种PPAR-α激动剂、PPAR-δ激动剂和/或PPAR-α和δ双重激动剂,(iii)至少一种降脂剂,以及任选地(iv)一种或多种药学上可接受的载体,其中该第一化合物是FXR激动剂。
本发明还涉及一种药物组合物,该药物组合物包含(i)第一化合物,(ii)至少一种贝特,(iii)至少一种降脂剂,以及任选地(iv)一种或多种药学上可接受的载体,其中该第一化合物是FXR激动剂。
本发明还涉及一种药物组合物,该药物组合物包含(i)第一化合物,(ii)至少一种PPAR-α激动剂、PPAR-δ激动剂和/或PPAR-α和δ双重激动剂,(iii)至少一种他汀,以及任选地(iv)一种或多种药学上可接受的载体,其中该第一化合物是FXR激动剂。
本发明还涉及一种药物组合物,该药物组合物包含(i)第一化合物,(ii)至少一种贝特,(iii)至少一种他汀,以及任选地(iv)一种或多种药学上可接受的载体,其中该第一化合物是FXR激动剂。
本发明还涉及本发明的药物组合物的治疗用途。
在一个实施例中,该第一化合物是具有化学式A的化合物:

或其药学上可接受的盐或氨基酸缀合物,其中R1、R2、R4、R7和X是如在此所定义的。
本发明还涉及用于治疗或预防FXR介导的疾病或病症或者涉及血液中循环脂质化合物的浓度升高的疾病或病症,用于降低肝酶水平、或者抑制或逆转纤维化的方法,这些方法包括向对其有需要的受试者给予治疗有效量的本发明的药物组合物。
本发明还涉及本发明的药物组合物用于治疗或预防FXR介导的疾病或病症或者涉及血液中循环脂质化合物的浓度升高的疾病或病症的用途,用于降低肝酶水平、或者抑制或逆转纤维化的用途。
本发明还涉及本发明的药物组合物在制造用于治疗或预防FXR介导的疾病或病症或者涉及血液中循环脂质化合物的浓度升高的疾病或病症的药物中的用途,用于降低肝酶水平、或者抑制或逆转纤维化的用途。
本发明的组合物和方法解决在治疗或预防其中涉及血液中循环脂质化合物(如胆固醇和甘油三酯)的浓度升高的疾病或障碍中未满足的需求。
具体实施方式
本申请涉及一种药物组合物,该药物组合物包含第一化合物,至少一种PPAR-α激动剂、PPAR-δ激动剂、和/或PPAR-α和δ或PPAR-α和γ双重激动剂,以及任选地一种或多种药学上可接受的载体,其中该第一化合物是FXR激动剂。
在一个实例中,该药物组合物包含至少一种PPAR-α激动剂。在一个实例中,该药物组合物包含至少一种PPAR-δ激动剂。在一个实例中,该药物组合物包含至少一种PPAR-α和δ双重激动剂。在一个实例中,该药物组合物包含至少一种PPAR-α和γ双重激动剂。在一个实例中,该药物组合物包含至少一种PPAR-α激动剂和至少一种PPAR-δ激动剂。在一个实例中,该药物组合物包含至少一种PPAR-α激动剂以及至少一种PPAR-α和δ双重激动剂。在一个实例中,该药物组合物包含至少一种PPAR-δ激动剂以及至少一种PPAR-α和δ或PPAR-α和γ双重激动剂。在一个实例中,该药物组合物包含至少一种PPAR-α激动剂、至少一种PPAR-δ激动剂、以及至少一种PPAR-α和δ双重激动剂。在一个实例中,该PPAR-α激动剂是贝特,如在此所述的贝特。在一个实例中,该PPAR-δ激动剂是{4-[({4-甲基-2-[4-(三氟甲基)苯基]-1,3-噻唑-5-基}甲基)硫烷基]-2-甲基苯氧基}乙酸(也称为GW501516、GW1516和“Endurabol”)、{2-甲基-4-[5-甲基-2-(4-三氟甲基-苯基)-2H-[1,2,3]三唑-4-基甲基硫烷基]-苯氧基}-乙酸、或[4-[[[2-[3-氟-4-(三氟甲基)苯基]-4-甲基-5-噻唑基]甲基]硫代]-2-甲基苯氧基]-乙酸或其药学上可接受的盐。在一个实例中,该PPAR-α和δ双重激动剂是2-[2,6二甲基-4-[3-[4-(甲基硫代)苯基]-3-氧代-1(E)-丙烯基]苯氧基]-2-甲基丙酸(也称为GFT505)。在一个实例中,该PPAR-α和γ双重激动剂是阿格列扎((2S)-2-甲氧基-3-[4-[2-(5-甲基-2-苯基-4-噁唑基)乙氧基]-7-苯并苯硫基]丙酸)、莫格他唑(N-[(4-甲氧基苯氧基)羰基]-N-{4-[2-(5-甲基-2-苯基-1,3-噁唑-4-基)乙氧基]苄基}甘氨酸)、替格列扎((2S)-2-乙氧基-3-[4-[2-(4-甲基磺酰基氧基苯基)乙氧基]苯基]丙酸)、或沙罗格列扎(saroglitazar)((2S)-2-乙氧基-3-[4-(2-{2-甲基-5-[4-(甲基硫烷基)苯基]-1H-吡咯-1-基}乙氧基)苯基]丙酸)、或其药学上可接受的盐。在一个实例中,该PPAR-α和δ双重激动剂是2-[2,6二甲基-4-[3-[4-(甲基硫代)苯基]-3-氧代-1(E)-丙烯基]苯氧基]-2-甲基丙酸或其药学上可接受的盐。
本申请还涉及一种药物组合物,该药物组合物包含第一化合物、至少一种贝特、和任选地一种或多种药学上可接受的载体,其中该第一化合物是FXR激动剂。该FXR激动剂可以是任何FXR激动剂。该贝特可以是任何贝特。在一个实例中,该贝特选自在此所述的任何贝特。
本申请还涉及一种药物组合物,该药物组合物包含第一化合物、至少一种降脂剂、和任选地一种或多种药学上可接受的载体,其中该第一化合物是FXR激动剂。该FXR激动剂可以是任何FXR激动剂。该降脂剂可以是任何降脂剂。在一个实例中,该降脂剂选自在此所述的任何降脂剂。
本申请还涉及一种药物组合物,该药物组合物包含第一化合物、至少一种他汀、和任选地一种或多种药学上可接受的载体,其中该第一化合物是FXR激动剂。该FXR激动剂可以是任何FXR激动剂。该他汀可以是任何他汀。在一个实例中,该他汀选自在此所述的任何他汀。
本申请还涉及一种药物组合物,该药物组合物包含第一化合物,至少一种PPAR-α激动剂、PPAR-δ激动剂、和/或PPAR-α和δ或PPAR-α和γ双重激动剂,至少一种降脂剂,以及任选地一种或多种药学上可接受的载体,其中该第一化合物是FXR激动剂。本申请还涉及一种药物组合物,该药物组合物包含第一化合物,至少一种PPAR-α激动剂、PPAR-δ激动剂、和/或PPAR-α和δ或PPAR-α和γ双重激动剂,至少一种他汀,以及任选地一种或多种药学上可接受的载体,其中该第一化合物是FXR激动剂。在一个实例中,该PPAR-α激动剂是贝特,如在此所述的贝特。在一个实例中,该PPAR-δ激动剂是{4-[({4-甲基-2-[4-(三氟甲基)苯基]-1,3-噻唑-5-基}甲基)硫烷基]-2-甲基苯氧基}乙酸、{2-甲基-4-[5-甲基-2-(4-三氟甲基-苯基)-2H-[1,2,3]三唑-4-基甲基硫烷基]-苯氧基}-乙酸、或[4-[[[2-[3-氟-4-(三氟甲基)苯基]-4-甲基-5-噻唑基]甲基]硫代]-2-甲基苯氧基]-乙酸、或其药学上可接受的盐。在一个实例中,该PPAR-α和δ双重激动剂是2-[2,6二甲基-4-[3-[4-(甲基硫代)苯基]-3-氧代-1(E)-丙烯基]苯氧基]-2-甲基丙酸。在一个实例中,该PPAR-α和γ双重激动剂是阿格列扎、莫格他唑、替格列扎或沙罗格列扎或其药学上可接受的盐。在一个实例中,该PPAR-α和δ双重激动剂是2-[2,6二甲基-4-[3-[4-(甲基硫代)苯基]-3-氧代-1(E)-丙烯基]苯氧基]-2-甲基丙酸或其药学上可接受的盐。在一个实例中,该降脂剂选自在此所述的任何降脂剂。在一个实例中,该他汀选自在此所述的任何他汀。
在一个实例中,该药物组合物的第一化合物是具有化学式A的化合物:

或其药学上可接受的盐或氨基酸缀合物,其中:
R1是氢或未经取代的C1-C6烷基;
R2是氢或α-羟基;
X是C(O)OH、C(O)NH(CH2)mSO3H、C(O)NH(CH2)nCO2H或OSO3H;
R4是羟基或氢;
R7是羟基或氢;
m是1、2或3;并且
n是1、2或3。
在另外的实例中,该药物组合物的第一化合物选自化学式I和IA:
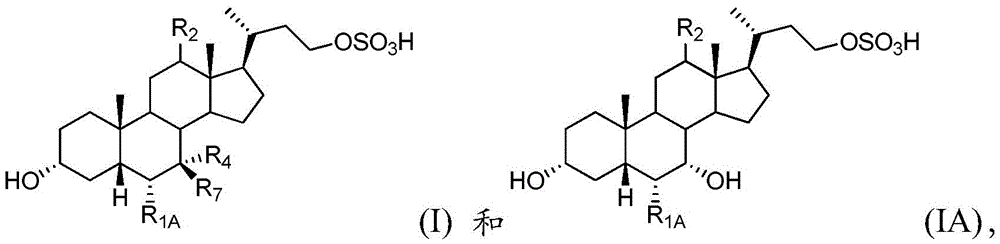
或其药学上可接受的盐或氨基酸缀合物,其中
R1A是氢或未经取代的C1-C6烷基;
R2是氢或α-羟基;
R4是羟基或氢;并且
R7是羟基或氢。
在一个方面,该第一化合物是药学上可接受的盐。在一个实施例中,该第一化合物是具有化学式I或IA的钠盐。在另一个实施例中,该第一化合物是具有化学式I或IA的化合物的铵盐。在另一个实施例中,该第一化合物是具有化学式I或IA的化合物的三乙基铵盐。
在又另一个实例中,该药物组合物的第一化合物选自化学式II和IIA:

或其药学上可接受的盐或氨基酸缀合物,其中:
R1A是氢或未经取代的C1-C6烷基;
R2是氢或α-羟基;
R3是羟基、NH(CH2)mSO3H或NH(CH2)nCO2H;
R4是羟基或氢;
R7是羟基或氢;
m是1、2或3;并且
n是1、2或3。
在一个实例中,该组合物包括具有化学式A、I、IA、II或IIA的第一化合物,其中R2是氢。
在另外的实例中,该组合物包括具有化学式A的第一化合物,其中R1是未经取代的C1-C6烷基。在一个方面,该组合物包括具有化学式A的第一化合物,其中R1是未经取代的C1-C3烷基。在一个方面,该组合物包括具有化学式A的第一化合物,其中R1选自甲基、乙基和丙基。在一个方面,该组合物包括具有化学式A的第一化合物,其中R1是乙基。
在另外的实例中,该组合物包括具有化学式I、IA、II或IIA的第一化合物,其中R1A是未经取代的C1-C6烷基。在一个方面,该组合物包括具有化学式I、IA、II或IIA的第一化合物,其中R1A是未经取代的C1-C3烷基。在一个方面,该组合物包括具有化学式I、IA、II或IIA的第一化合物,其中R1A选自甲基、乙基和丙基。在一个方面,该组合物包括具有化学式I、IA、II或IIA的第一化合物,其中R1A是乙基。
在另外的实例中,该组合物包括具有化学式A的第一化合物,其中X选自C(O)OH、C(O)NH(CH2)mSO3H和C(O)NH(CH2)nCO2H。在一个方面,该组合物包括具有化学式A的第一化合物,其中X选自C(O)OH、C(O)NH(CH2)SO3H、C(O)NH(CH2)CO2H、C(O)NH(CH2)2SO3H、C(O)NH(CH2)2CO2H。在一个方面,该组合物包括具有化学式A的第一化合物,其中X是C(O)OH。在一个方面,该组合物包括具有化学式A的第一化合物,其中X是OSO3H。在一个方面,该组合物包括具有化学式A的第一化合物,其中该第一化合物是药学上可接受的盐。该药学上可接受的盐可以是任何盐。在一个方面,该组合物包括具有化学式A的第一化合物,其中X是OSO3 -Na+。在一个方面,该组合物包括具有化学式A的第一化合物,其中X是OSO3 -NHEt3 +。在一个方面,该氨基酸缀合物是甘氨酸缀合物。在一个方面,该氨基酸缀合物是牛磺酸缀合物。
在又另一个实例中,该组合物包括具有化学式II或IIA的第一化合物,其中R3选自OH、NH(CH2)SO3H、NH(CH2)CO2H、NH(CH2)2SO3H和NH(CH2)2CO2H。在一个方面,该组合物包括具有化学式II或IIA的第一化合物,其中R3是OH。
在另外的实例中,该组合物包括具有化学式A、I或II的第一化合物,其中R4是羟基并且R7是氢。
在另外的实例中,该组合物包括具有化学式A的第一化合物,其中R1选自甲基、乙基和丙基,R4是OH,R7是H,并且R2是H。
在另外的实例中,该组合物包括具有化学式I或II的第一化合物,其中R1A选自甲基、乙基和丙基,R4是OH,R7是H,并且R2是H。
在另外的实例中,该组合物包括具有化学式IA或IIA的第一化合物,其中R1A选自甲基、乙基和丙基,并且R2是H。
在另外的实例中,该组合物包括选自以下各项的第一化合物

或其药学上可接受的盐或氨基酸缀合物。
在又另外的实例中,该组合物包括选自以下各项的作为药学上可接受的盐的第一化合物

具有化学式I、IA、II和IIA的化合物是具有化学式A的化合物的亚组。在此关于具有化学式A的化合物所述的特征同样适用于具有化学式式I、IA、II和IIA的化合物。本申请还描述了这些药物组合物、包装或试剂盒、以及组合的治疗用途。
本发明要解决的问题之一是鉴定用于治疗或预防与血液中循环脂质化合物(如胆固醇和甘油三酯)的浓度升高相关的病症(例如胆汁淤积性肝脏病症,如PBC),以及用于减少血液中循环脂质化合物(例如胆固醇、LDL和甘油三酯),以及用于减少胆红素和/或肝酶(如碱性磷酸酶(ALP、AP或Alk Phos)、丙氨酸氨基转移酶(ALT)、天冬氨酸氨基转移酶(AST)、γ-谷氨酰转肽酶(GGT)、乳酸脱氢酶(LDH)和5'核苷酸酶)的组合疗法。尽管用于与脂质水平和/或肝酶水平升高相关的病症的药物是可获得的,但这些药物经常出于各种原因而不适合用于许多患者。例如,某些药物对于已经发展了对例如熊去氧胆酸有耐药性的患者是无效的。如另一个实例,许多他汀类药物具有诸如肌肉问题、认知丧失、神经病、胰腺和肝功能异常以及性功能障碍等不良影响。当单独给予时,一些药物对于治疗而言可能是不足的。例如,在一些情况下,单独的一种降脂剂不足以控制许多患者中存在的严重的高血脂水平。一些药物由于广泛代谢成无活性或效力更低的代谢物而可能需要高剂量的给予或更频繁的给予。在此所述的组合疗法能够解决上述问题并且可以具有一种或多种以下优点:例如,协同作用,在无药物损失功效的情况下减少每日剂量的次数,在升高的脂质水平抵抗PBC中的治疗的患者中降低脂质(胆固醇和甘油三酯两者),改进的效力、选择性、组织渗透性、半衰期和/或代谢稳定性。
在本发明的组合物、包装或试剂盒、方法、以及用途中,该第一化合物可以是游离酸或者第一化合物可以是药学上可接受的盐或氨基酸缀合物(例如,甘氨酸或牛磺酸缀合物)。在一个方面,该第一化合物是任何FXR激动剂。在一个方面,该第一化合物是具有化学式A的化合物。在一个方面,该第一化合物是具有化学式I或IA的化合物。在一个方面,该第一化合物是具有化学式IA的化合物。在一个方面,该第一化合物是具有化学式II或IIA的化合物。在一个方面,该第一化合物是具有化学式IIA的化合物。在一个方面,该第一化合物是奥贝胆酸(化合物1)。在一个方面,该第一化合物是化合物2。在一个方面,该第一化合物是药学上可接受的盐化合物3。在一个方面,该第一化合物是药学上可接受的盐化合物4。
在本发明的组合物、包装或试剂盒、方法以及用途中,该贝特可以是任何贝特。在一个方面,该贝特选自下组,该组由以下各项组成:非诺贝特、苯扎贝特、苄氯贝特、比尼贝特、环丙贝特、克利贝特、氯贝特、氯贝酸、依托贝特、吉非罗齐、尼可贝特、吡贝特、氯烟贝特、双贝特、氯贝茶碱、托考贝特、普拉贝脲及其药学上可接受的盐和酯,以及2-苯氧基-2-甲基丙酸的衍生物(其中该苯氧基部分被哌啶、4-羟基哌啶、哌啶-3-烯或哌嗪的任选地经取代的残基取代),如欧洲专利申请公开号EP0607536所披露的。在一个方面,该贝特选自下组,该组由以下各项组成:苯扎贝特、环丙贝特、氯贝特、非诺贝特、吉非罗齐、比尼贝特、克利贝特、氯贝酸、尼可贝特、吡贝特、普拉贝脲、氯烟贝特、氯贝茶碱、托考贝特及其药学上可接受的盐和酯,以及2-苯氧基-2-甲基丙酸的衍生物(其中该苯氧基部分被哌啶、4-羟基哌啶、哌啶-3-烯或哌嗪的任选地经取代的残基取代),如欧洲专利申请公开号EP0607536所披露的。后一组物质的实例是2-[3-[1-(4-氟苯甲酰基)哌啶-4-基]苯氧基-2-甲基-丙酸。例如,该贝特是苯扎贝特、非诺贝特、吉非罗齐、环丙贝特、氯贝特、氯贝酸或其药学上可接受的盐或酯。例如,该贝特是选自胆碱、乙醇胺、二乙醇胺、哌嗪、钙和氨丁三醇的非诺贝特或药学上可接受的盐。例如,该贝特是氯贝特或其药学上可接受的盐或酯,如依托贝特或氯贝酸铝。例如,该贝特是苯扎贝特。例如,该贝特是2-苯氧基-2-甲基丙酸的衍生物,如2-[3-[1-(4-氟苯甲酰基)-哌啶-4-基]苯氧基-2-甲基丙酸。
在一个实施例中,该第一化合物是具有化学式A的化合物的游离酸,并且该至少一种贝特选自苯扎贝特、非诺贝特、吉非罗齐、环丙贝特、氯贝特及其药学上可接受的盐或酯。
在一个实施例中,该第一化合物是具有化学式A的化合物的药学上可接受的盐,并且该至少一种贝特选自苯扎贝特、非诺贝特、吉非罗齐、环丙贝特、氯贝特及其药学上可接受的盐或酯。
在一个实施例中,该第一化合物是具有化学式A的化合物的甘氨酸缀合物,并且该至少一种贝特选自苯扎贝特、非诺贝特、吉非罗齐、环丙贝特、氯贝特及其药学上可接受的盐或酯。
在一个实施例中,该第一化合物是具有化学式A的化合物的牛磺酸缀合物,并且该至少一种贝特选自苯扎贝特、非诺贝特、吉非罗齐、环丙贝特、氯贝特及其药学上可接受的盐或酯。
在一个实施例中,该第一化合物是具有化学式A的化合物或药学上可接受的盐或氨基酸缀合物,并且该至少一种贝特是2-[3-[1-(4-氟苯甲酰基)-哌啶-4-基]苯氧基-2-甲基丙酸。
在一个实施例中,该第一化合物是具有化学式A的化合物或药学上可接受的盐或氨基酸缀合物,并且该至少一种PPAR-δ激动剂是{4-[({4-甲基-2-[4-(三氟甲基)苯基]-1,3-噻唑-5-基}甲基)硫烷基]-2-甲基苯氧基}乙酸、{2-甲基-4-[5-甲基-2-(4-三氟甲基-苯基)-2H-[1,2,3]三唑-4-基甲基硫烷基]-苯氧基}-乙酸、或[4-[[[2-[3-氟-4-(三氟甲基)苯基]-4-甲基-5-噻唑基]甲基]硫代]-2-甲基苯氧基]-乙酸、或其药学上可接受的盐。
在一个实施例中,该第一化合物是具有化学式A的化合物或药学上可接受的盐或氨基酸缀合物,并且该至少一种PPAR-α和δ双重激动剂是2-[2,6二甲基-4-[3-[4-(甲基硫代)苯基]-3-氧代-1(E)-丙烯基]苯氧基]-2-甲基丙酸。在一个实施例中,该第一化合物是具有化学式A的化合物或药学上可接受的盐或氨基酸缀合物,并且该至少一种PPAR-α和γ双重激动剂是阿格列扎、莫格他唑、替格列扎、或沙罗格列扎、或其药学上可接受的盐。在一个实例中,该PPAR-α和δ双重激动剂是2-[2,6二甲基-4-[3-[4-(甲基硫代)苯基]-3-氧代-1(E)-丙烯基]苯氧基]-2-甲基丙酸、或其药学上可接受的盐。
在本发明的组合物、包装或试剂盒、方法以及用途中,该他汀可以是任何他汀。在一个方面,该他汀选自下组,该组由以下各项组成:辛伐他汀、氟伐他汀、普伐他汀、利伐他汀、美伐他汀、阿托伐他汀、西立伐他汀、洛伐他汀、匹伐他汀、氟多他汀(fluindostatin)、维洛他汀(velostatin)、达伐他汀、罗素伐他汀、二氢康帕丁和康帕丁。
在一个实施例中,该第一化合物是具有化学式A的化合物的游离酸,并且该至少一种他汀选自辛伐他汀、氟伐他汀、普伐他汀、利伐他汀、美伐他汀、阿托伐他汀、西立伐他汀、洛伐他汀、匹伐他汀、氟多他汀、维洛他汀、达伐他汀、罗素伐他汀、二氢康帕丁和康帕丁。
在一个实施例中,该第一化合物是具有化学式A的化合物的药学上可接受的盐,并且该至少一种他汀选自辛伐他汀、氟伐他汀、普伐他汀、利伐他汀、美伐他汀、阿托伐他汀、西立伐他汀、洛伐他汀、匹伐他汀、氟多他汀、维洛他汀、达伐他汀、罗素伐他汀、二氢康帕丁和康帕丁。
在一个实施例中,该第一化合物是具有化学式A的化合物的甘氨酸缀合物,并且该至少一种他汀选自辛伐他汀、氟伐他汀、普伐他汀、利伐他汀、美伐他汀、阿托伐他汀、西立伐他汀、洛伐他汀、匹伐他汀、氟多他汀、维洛他汀、达伐他汀、罗素伐他汀、二氢康帕丁和康帕丁。
在一个实施例中,该第一化合物是具有化学式A的化合物的牛磺酸缀合物,并且该至少一种他汀选自辛伐他汀、氟伐他汀、普伐他汀、利伐他汀、美伐他汀、阿托伐他汀、西立伐他汀、洛伐他汀、匹伐他汀、氟多他汀、维洛他汀、达伐他汀、罗素伐他汀、二氢康帕丁和康帕丁。
本发明还包含同位素标记的第一化合物或其药学上可接受的盐或氨基酸缀合物,该同位素标记的第一化合物或其药学上可接受的盐或氨基酸缀合物具有与本发明的第一化合物(例如,具有化学式A、I、IA、II或IIA的化合物)的结构相同的结构,但事实是一个或多个原子被具有与最常在自然中发现的原子质量或质量数不同的原子质量或质量数的原子置换。可以掺入该第一化合物或其药学上可接受的盐或氨基酸缀合物中的同位素的实例包括氢、碳、氮、氟的同位素,如3H、11C、14C和18F。
包含上述同位素和/或其他原子的其他同位素的第一化合物或其药学上可接受的盐或氨基酸缀合物在本发明的范围之内。同位素标记的第一化合物或其药学上可接受的盐或氨基酸缀合物,例如掺入放射性同位素如3H和/或14C的第一化合物可用于药物和/或底物组织分布测定中。氚化的(即3H)和碳-14(即14C)同位素因其容易制备和可检测性而被使用。另外,用较重的同位素如氘(即2H)取代可以提供由更大的代谢稳定性引起的某些治疗优点,例如增加的体内半衰期和减少的剂量需求,并且因此可以在一些情况下被使用。同位素标记的第一化合物或其药学上可接受的盐或氨基酸缀合物总体上可以通过执行在本发明的方案和/或实例中披露的程序、通过用容易获得的同位素标记的试剂取代非同位素标记的试剂来制备。在一个实施例中,奥贝胆酸或其药学上可接受的盐或氨基酸缀合物未进行同位素标记。
本发明还提供一种用于治疗或预防一种疾病或病症的方法,该方法包括向对其有需要的受试者给予治疗有效量的本发明的药物组合物。
在一个实施例中,该疾病或病症是FXR介导的疾病或病症。FXR介导的疾病或病症的实例包括但不限于:肝病(包括胆汁淤积性和非胆汁淤积性肝病)(如原发性胆汁性肝硬化(PBC))、原发性硬化性胆管炎(PSC)、胆道闭锁、门静脉高压、胆汁酸腹泻、慢性肝病、非酒精性脂肪肝病(NAFLD)、非酒精性脂肪性肝炎(NASH)、丙型肝炎感染、酒精性肝病、由于进行性纤维化引起的肝损伤、和肝纤维化。FXR介导的疾病的实例还包括高血脂症、高LDL-胆固醇、高HDL-胆固醇、高甘油三酯、和心血管疾病。
NAFLD是表征为肝脏中的脂肪堆积(被称为脂肪浸润)的医学病症。NAFLD是慢性肝病的最常见病因之一,并且涵盖与肝细胞中的脂质沉积相关的一系列病症。它在从脂肪变性(单纯性脂肪肝)至非酒精性脂肪性肝炎(NASH)至晚期纤维化和肝硬化的范围内。该疾病通常是沉默的并且经常是通过偶然升高的肝酶水平而发现的。NAFLD与肥胖症和胰岛素耐受性密切相关并且目前被许多人认为是代谢综合征的肝组分。
非酒精性脂肪性肝炎(NASH)是造成肝中的炎症以及脂肪和纤维(瘢痕)组织积累的一种病症。血液中的肝酶水平可能比在非酒精性脂肪肝(NAFL)情况下观察到的轻微升高更高。虽然类似的病症可以在酗酒的人中发生,但NASH在很少喝酒至不喝酒的那些人中发生。NASH影响2%至5%的美国人,并且最频繁发现于具有一种或多种以下病症的人中:肥胖症、糖尿病、高血脂症、胰岛素耐受性、某些药剂的使用、以及暴露于毒素。NASH是世界范围内慢性肝病的越来越常见的病因,并且与增加的肝相关死亡率和肝细胞癌相关(即使在不存在肝硬化的情况下)。NASH在15%-20%的受影响的个体中进展至肝硬化并且现在是美国的肝移植的主要适应症之一。目前,不存在用于NASH的经批准的疗法。
在一个实施例中,该疾病或病症是高血脂症。在一个实施例中,该疾病或病症是胆汁淤积性肝病。在一个实施例中,该疾病或病症是PBC。在另一个实施例中,该疾病或病症是心血管疾病。在另一个实施例中,该心血管疾病是动脉硬化、血胆固醇过多、或高甘油三酯血症。
本发明还提供了用于治疗或预防NAFLD或NASH的方法。在一个实施例中,本发明提供了用于治疗或预防与高血脂症相关的NAFLD或NASH的方法。在一个实施例中,本发明提供了用于治疗或预防NASH的方法。在一个实施例中,本发明提供了用于治疗或预防与高血脂症相关的NASH的方法。
本发明还提供了用于抑制或逆转纤维化的方法,该方法包括向对其有需要的受试者给予治疗有效量的本发明的药物组合物。在一个实施例中,该受试者不患有胆汁淤积性病症。在另一个实施例中,该受试者患有胆汁淤积性病症。
在一个实施例中,该受试者不患有与选自下组的疾病或病症相关的胆汁淤积性病症,该组由以下各项组成:原发性肝癌和胆管癌(包括肝细胞癌)、结肠直肠癌、转移性癌症、败血症、慢性肠胃外全面营养、囊胞性纤维症、和肉芽肿性肝病。在实施例中,待抑制或逆转的纤维化发生在表达FXR的器官中。
在一个实施例中,胆汁淤积性病症被定义为具有碱性磷酸酶、γ-谷氨酰转肽酶(GGT)、和/或5’核苷酸酶的异常升高的血清水平。在另一个实施例中,胆汁淤积性病症被进一步定义为呈现至少一种临床症状。在一个实施例中,该症状是发痒(瘙痒)。在另一个实施例中,胆汁淤积性病症选自下组,该组由以下各项组成:原发性胆汁性肝硬化(PBC)、原发性硬化性胆管炎(PSC)、药物诱导性胆汁淤积、遗传性胆汁淤积、胆道闭锁、以及妊娠肝内胆汁淤积症。
在一个实施例中,该纤维化选自下组,该组由以下各项组成:肝纤维化、肾纤维化、和肠纤维化。
在一个实施例中,该受试者患有与选自下组的疾病相关的肝纤维化,该组由以下各项组成:乙型肝炎;丙型肝炎;寄生虫肝病;移植后细菌、病毒和真菌感染;酒精性肝病(ALD);非酒精性脂肪肝病(NAFLD);非酒精性脂肪性肝炎(NASH);由甲氨蝶呤、异烟肼、酚丁、甲基多巴、冬眠灵、甲苯磺丁脲、或胺碘酮诱导的肝病;自身免疫性肝炎;结节病;威尔森氏病;血色沉着病;戈谢病;III、IV、VI、IX和X型糖原贮积病α1-抗胰蛋白酶缺乏;脑肝肾综合征;酪氨酸血症;果糖血症;半乳糖血症;与巴德-吉亚利综合征、静脉闭塞性疾病、或门静脉血栓形成有关的血管紊乱;和先天性肝纤维化。
在另一个实施例中,该受试者具有与选自下组的疾病相关的肠纤维化,该组由以下各项组成:克罗恩病、溃疡性结肠炎、放疗后结肠炎、和微观结肠炎。
在另一个实施例中,该患者患有与选自下组的疾病相关的肾纤维化,该组由以下各项组成:糖尿病肾病、高血压性肾硬化、慢性肾小球肾炎、慢性移植肾小球病、慢性间质性肾炎、和多囊肾病。
本发明还提供了用于治疗或预防与升高的脂质水平相关的所有形式的病症。在一个实施例中,该病症是高血脂症,其中它与选自以下各项的病症相关:抗性原发性胆汁性肝硬化;存在相关的肝功能测试升高和高血脂症的原发性胆汁性肝硬化、原发性硬化性胆管炎、非酒精诱发性脂肪性肝炎;以及与乙型肝炎、丙型肝炎或酒精性肝炎相关的慢性肝病。在另一个实施例中,本发明提供了用于治疗或预防高血脂症的方法,其中该高血脂症是具有或不具有遗传成分原发性高血脂症,或者与冠状动脉疾病、脑血管动脉疾病、周围性血管疾病、主动脉瘤、或动脉粥样硬化相关的高血脂症。
在一个方面,针对类似的生化异常、连同由乙型肝炎、丙型肝炎或由酒精性肝炎引起的慢性肝炎,本发明提供了用于治疗或预防原发性硬化性胆管炎的方法。在一个方面,本发明提供了用于治疗或预防与高血脂症相关的其他动脉疾病的方法。在一个方面,本发明提供了用于治疗或预防高甘油三酯血症的方法。
本发明还提供了用于降低如血液中的脂质水平(即脂质的量)的方法,该方法包括向对其有需要的受试者给予治疗有效量的本发明的药物组合物。在一个实施例中,相比于对照受试者(例如,未给予本发明的组合物的受试者),本发明的方法使脂质水平降低至少10%、20%、30%、40%、50%、60%、70%、80%或90%。在一个实施例中,相比于健康受试者(例如,没有疾病或病症(如在此所述的那些)的个体),该受试者具有升高的脂质水平。在一个实施例中,本申请的方法使脂质水平降低到正常水平(例如,类似于没有疾病或病症(如在此所述的那些)的个体中的脂质水平)。
在一个实施例中,该脂质是胆固醇。在一个实施例中,相比于对照受试者(例如,未给予本发明的组合物的受试者),本发明的方法使胆固醇水平降低至少10%、20%、30%、40%、50%、60%、70%、80%或90%。在一个实施例中,相比于健康受试者(例如,没有疾病或病症(如在此所述的那些)的个体),该受试者具有升高的胆固醇水平。在一个实施例中,本发明的方法使胆固醇水平降低到400mg/L、350mg/L、300mg/L、250mg/L、240mg/L、230mg/L、220mg/L、210mg/L、200mg/L、190mg/L、180mg/L、170mg/L、160mg/L、或150mg/L以下。在一个实施例中,本发明的方法使胆固醇水平降低到200mg/L、190mg/L、180mg/L、170mg/L、160mg/L、或150mg/L以下。
在一个实施例中,该胆固醇是LDL。在一个实施例中,相比于对照受试者(例如,未给予本发明的组合物的受试者),本发明的方法使LDL水平降低至少10%、20%、30%、40%、50%、60%、70%、80%或90%。在一个实施例中,相比于健康受试者(例如,没有疾病或病症(如在此所述的那些)的个体),该受试者具有升高的LDL水平。在一个实施例中,本发明的方法使LDL水平降低到300mg/L、200mg/L、190mg/L、180mg/L、170mg/L、160mg/L、150mg/L、140mg/L、130mg/L、120mg/L、110mg/L、100mg/L、90mg/L、80mg/L、70mg/L、60mg/L、或50mg/L以下。在一个实施例中,本发明的方法使LDL水平降低到160mg/L、150mg/L、140mg/L、130mg/L、120mg/L、110mg/L、100mg/L、90mg/L、80mg/L、70mg/L、60mg/L、或50mg/L以下。在一个实施例中,本发明的方法使LDL水平降低到130mg/L、120mg/L、110mg/L、100mg/L、90mg/L、80mg/L、70mg/L、60mg/L、或50mg/L以下。在一个实施例中,本发明的方法使LDL水平降低到100mg/L、90mg/L、80mg/L、70mg/L、60mg/L、或50mg/L以下。在一个实施例中,本发明的方法使LDL水平降低到70mg/L、60mg/L、或50mg/L以下。
在一个实施例中,该脂质是甘油三酯。在一个实施例中,相比于对照受试者(例如,未给予本发明的组合物的受试者),本发明的方法使甘油三酯水平降低至少10%、20%、30%、40%、50%、60%、70%、80%或90%。在一个实施例中,相比于健康受试者(例如,没有疾病或病症(如在此所述的那些)的个体),该受试者具有升高的甘油三酯水平。在一个实施例中,本发明的方法使甘油三酯水平降低到800mg/L、700mg/L、600mg/L、500mg/L、400mg/L、300mg/L、200mg/L、190mg/L、180mg/L、170mg/L、160mg/L、150mg/L、140mg/L、130mg/L、120mg/L、110mg/L、或100mg/L以下。在一个实施例中,本发明的方法使甘油三酯水平降低到200mg/L、190mg/L、180mg/L、170mg/L、160mg/L、150mg/L、140mg/L、130mg/L、120mg/L、110mg/L、或100mg/L以下。在一个实施例中,本发明的方法使甘油三酯水平降低到150mg/L、140mg/L、130mg/L、120mg/L、110mg/L、或100mg/L以下。
本发明还提供了用于减少胆红素和/或一种或多种肝酶的量的方法,该方法包括向对其有需要的受试者给予治疗有效量的本申请的药物组合物。
在一个实施例中,相比于对照受试者(例如,未给予本发明的组合物的受试者),本申请的方法使胆红素的量降低至少10%、20%、30%、40%、50%、60%、70%、80%、或90%。在一个实施例中,相比于健康受试者(例如,没有疾病或病症(如在此所述的那些)的个体),该受试者具有升高的胆红素水平。在一个实施例中,本申请的方法使胆红素的水平降低到一个正常水平(例如,类似于没有疾病或病症(如在此所述的那些)的个体中的胆红素的水平)。在另外的实施例中,本申请的方法使胆红素的水平降低到10mg/L、9mg/L、8mg/L、7mg/L、6mg/L、5mg/L、4mg/L、3mg/L、2mg/L、1.5mg/L、1.2mg/L、或1mg/L以下。在另外的实施例中,本申请的方法使胆红素的水平降低到2mg/L、1.5mg/L、1.2mg/L、或1mg/L以下。
在一个实施例中,肝酶选自下组,该组由以下各项组成:碱性磷酸酶(ALP、AP或AlkPhos)、丙氨酸氨基转移酶(ALT)、天冬氨酸氨基转移酶(AST)、γ-谷氨酰转肽酶(GGT)、乳酸脱氢酶(LDH)、以及5’核苷酸酶。在一个实施例中,相比于对照受试者(例如,未给予本发明的组合物的受试者),本申请的方法使一种或多种肝酶的量减少至少10%、20%、30%、40%、50%、60%、70%、80%、或90%。在一个实施例中,相比于健康受试者(例如,没有疾病或病症(如在此所述的那些)的个体),该受试者具有升高的一种或多种肝酶水平。在一个实施例中,本申请的方法使一种或多种肝酶(例如,ALP、ALT、AST、GGT、LDH、以及5’核苷酸酶)的水平降低到正常水平(例如,类似于没有疾病或病症(如在此所述的那些)的个体中的肝酶的水平)。
在另外的实施例中,本申请的方法使ALP的水平降低到500IU/L(国际单位/升)、400IU/L、300IU/L、200IU/L、180IU/L、160IU/L、或150IU/L以下。在另外的实施例中,本申请的方法使ALP的水平降低到从约40IU/L至约150IU/L。
在另外的实施例中,本申请的方法使ALT的水平降低到200IU/L(国际单位/升)、150IU/L、100IU/L、80IU/L、60IU/L、或50IU/L以下。在另外的实施例中,本申请的方法使ALT的水平降低到从约5IU/L至约50IU/L。
在另外的实施例中,本申请的方法使AST的水平降低到200IU/L(国际单位/升)、150IU/L、100IU/L、80IU/L、60IU/L、50IU/L、或40IU/L以下。在另外的实施例中,本申请的方法使AST的水平降低到从约10IU/L至约50IU/L。
在另外的实施例中,本申请的方法使GGT的水平降低到200IU/L(国际单位/升)、150IU/L、100IU/L、90IU/L、80IU/L、70IU/L、或60IU/L以下。在另外的实施例中,本申请的方法使GGT的水平降低到从约15IU/L至约50IU/L或从约5IU/L至约30IU/L。
在另外的实施例中,本申请的方法使LDH的水平降低到500IU/L(国际单位/升)、400IU/L、300IU/L、200IU/L、180IU/L、160IU/L、150IU/L、140IU/L、或130IU/L以下。在另外的实施例中,本申请的方法使LDH的水平降低到从约120IU/L至约220IU/L。
在另外的实施例中,本申请的方法使5’核苷酸酶的水平降低到50IU/L(国际单位/升)、40IU/L、30IU/L、20IU/L、18IU/L、17IU/L、16IU/L、15IU/L、14IU/L、13IU/L、12IU/L、11IU/L、10IU/L、9IU/L、8IU/L、7IU/L、6IU/L、或5IU/L以下。在另外的实施例中,本申请的方法使5’核苷酸酶的水平降低到从约2IU/L至约15IU/L。
在一个实施例中,本发明的方法包括向对其有需要的受试者给予有效量的为FXR激动剂的第一化合物,与至少一种PPAR-α激动剂、PPAR-δ激动剂、和/或PPAR-α和δ双重激动剂,以及任选地一种或多种药学上可接受的载体的组合。在另外的实施例中,该方法包括向对其有需要的受试者给予有效量的第一化合物与至少一种PPAR-α激动剂、PPAR-δ激动剂、和/或PPAR-α和δ双重激动剂的组合,其中该第一化合物是在此所述的化合物(例如,具有化学式A、I、IA、II或IIA的化合物或化合物1、2、3或4)或其药学上可接受的盐或氨基酸缀合物。
在一个实施例中,本发明的方法包括向对其有需要的受试者给予有效量的为FXR激动剂的第一化合物与至少一种贝特、以及任选地一种或多种药学上可接受的载体的组合。在另外的实施例中,该方法包括向对其有需要的受试者给予有效量的第一化合物与至少一种贝特的组合,其中该第一化合物是在此所述的化合物(例如,具有化学式A、I、IA、II或IIA的化合物或化合物1、2、3或4)或其药学上可接受的盐或氨基酸缀合物。
在一个实施例中,本发明的方法包括向对其有需要的受试者给予有效量的为FXR激动剂的第一化合物与至少一种他汀、以及任选地一种或多种药学上可接受的载体的组合。在另外的实施例中,该方法包括向对其有需要的受试者给予有效量的第一化合物与至少一种他汀的组合,其中该第一化合物是在此所述的化合物(例如,具有化学式A、I、IA、II或IIA的化合物或化合物1、2、3或4)或其药学上可接受的盐或氨基酸缀合物。
在一个实施例中,本发明的方法包括向对其有需要的受试者给予有效量的为FXR激动剂的第一化合物,与至少一种PPAR-α激动剂、PPAR-δ激动剂、和/或PPAR-α和δ双重激动剂,至少一种他汀,以及任选地一种或多种药学上可接受的载体的组合。在另外的实施例中,该方法包括向对其有需要的受试者给予有效量的第一化合物与至少一种PPAR-α激动剂、PPAR-δ激动剂、和/或PPAR-α和δ双重激动剂、至少一种他汀的组合,其中该第一化合物是在此所述的化合物(例如,具有化学式A、I、IA、II或IIA的化合物或化合物1、2、3或4)或其药学上可接受的盐或氨基酸缀合物。
在一个实施例中,本发明的方法包括向对其有需要的受试者给予有效量的为FXR激动剂的第一化合物与至少一种贝特、至少一种他汀、以及任选地一种或多种药学上可接受的载体的组合。在另外的实施例中,该方法包括向对其有需要的受试者给予有效量的第一化合物与至少一种贝特、至少一种他汀的组合,其中该第一化合物是在此所述的化合物(例如,具有化学式A、I、IA、II或IIA的化合物或化合物1、2、3或4)或其药学上可接受的盐或氨基酸缀合物。
在一个实施例中,该受试者是哺乳动物。在一个实施例中,该哺乳动物是人。
在一个实施例中,将该第一化合物和一种或多种PPAR-α激动剂、一种或多种PPAR-δ激动剂、一种或多种PPAR-α和δ或PPAR-α和γ双重激动剂、一种或多种贝特或一种或多种他汀以双向组合进行给予,即除了该第一化合物和一种或多种PPAR-α激动剂、一种或多种PPAR-δ激动剂、一种或多种PPAR-α和δ或PPAR-α和γ双重激动剂、一种或多种贝特或一种或多种他汀以外,没有任何治疗剂。在另外的实施例中,将该第一化合物和一种或多种贝特以双向组合进行给予,即除了该第一化合物和一种或多种贝特以外,没有任何治疗剂。在另一个实施例中,将该第一化合物和一种或多种他汀以双向组合进行给予,即除了该第一化合物和一种或多种他汀以外,没有任何治疗剂。
在另一个实施例中,将该第一化合物和该一种或多种PPAR-α激动剂、一种或多种PPAR-δ激动剂、一种或多种PPAR-α和δ或PPAR-α和γ双重激动剂、或一种或多种贝特以与他汀的三向组合进行给予。在另外的实施例中,将该第一化合物和一种或多种贝特以与他汀的三向组合进行给予。
该第一化合物、连同一种或多种PPAR-α激动剂、一种或多种PPAR-δ激动剂、一种或多种PPAR-α和δ或PPAR-α和γ双重激动剂、或一种或多种贝特和/或一种或多种他汀可以达到意义深远的协同效应,例如在严重的混合型高血脂症状态和对个体疗法有抗性的那些中以及在一种或多种肝酶水平上的协同降低。因此,对于非常难以控制的高血脂症,第一化合物、PPAR-α激动剂、PPAR-δ激动剂、PPAR-α和δ或PPAR-α和γ双重激动剂、或贝特、和/或他汀的组合是有利的。该第一化合物、贝特、和/或他汀的这样一种组合提供在被设计为提高依从性且因此提高有效性的具有药学上可接受的载体的单一药物组合物(如呈单一胶囊形式)中可以是特别有利的。因此,本发明进一步提供了一种药物组合物,该药物组合物包含有效量的第一化合物,有效量的至少一种PPAR-α激动剂、PPAR-δ激动剂、或PPAR-α和δ或PPAR-α和γ双重激动剂,和有效量的至少一种他汀,连同一种或多种药学上可接受的载体、稀释剂、佐剂或赋形剂。在一个实施例中,本发明进一步提供了一种药物组合物,该药物组合物包含有效量的第一化合物,有效量的至少一种贝特,和有效量的至少一种他汀,连同一种或多种药学上可接受的载体、稀释剂、佐剂或赋形剂。
在一个实施例中,将该第一化合物和一种或多种PPAR-α激动剂、一种或多种PPAR-δ激动剂、一种或多种PPAR-α和δ或PPAR-α和γ双重激动剂、或一种或多种贝特同时给予。例如,将该第一化合物和一种或多种PPAR-α激动剂、一种或多种PPAR-δ激动剂、一种或多种PPAR-α和δ或PPAR-α和γ双重激动剂、或一种或多种贝特以与药学上可接受的载体一起的单一药物组合物给予。在另一个实施例中,将该第一化合物和一种或多种PPAR-α激动剂、一种或多种PPAR-δ激动剂、一种或多种PPAR-α和δ或PPAR-α和γ双重激动剂、或一种或多种贝特顺序地给予。例如,将该第一化合物在一种或多种PPAR-α激动剂、一种或多种PPAR-δ激动剂、一种或多种PPAR-α和δ或PPAR-α和γ双重激动剂、或一种或多种贝特之前或之后给予。
在一个实施例中,将该第一化合物和该他汀同时给予。例如,将该第一化合物和该他汀以与药学上可接受的载体一起的单一药物组合物给予。在另一个实施例中,将该第一化合物和该他汀顺序地给予。例如,将该第一化合物在该他汀之前或之后给予。
在一个实施例中,以第一剂量给予该第一化合物持续第一时间段,接着以第二剂量给予该第一化合物持续第二时间段。在一个实施例中,第一化合物或其药学上可接受的盐或氨基酸缀合物以0.1-1500mg、0.2-1200mg、0.3-1000mg、0.4-800mg、0.5-600mg、0.6-500mg、0.7-400mg、0.8-300mg、1-200mg、1-100mg、1-50mg、1-30mg、4-26mg、或5-25mg的每日总量给予持续第一时间段,接着以0.1-1500mg、0.2-1200mg、0.3-1000mg、0.4-800mg、0.5-600mg、0.6-500mg、0.7-400mg、0.8-300mg、1-200mg、1-100mg、1-50mg、1-30mg、4-26mg、或5-25mg的每日总量给予该第一化合物。在一个实施例中,每日一次口服给予该总量。在一个实施例中,第一剂量与第二剂量不同。在另外的实施例中,第一剂量低于第二剂量。在另一个实施例中,第一剂量高于第二剂量。在一个实施例中,第一剂量是约5mg(例如,从4.8mg至5.2mg),并且第二剂量是约10mg(例如,从9.8mg至10.2mg)。在一个实施例中,第一时间段是约6个月。在一个实施例中,第二时间段是约6个月。
在一个实施例中,该药物组合物是口服、胃肠外或局部给予的。在另一个实施例中,该药物组合物是口服给予的。
根据本发明的组合物通常将含有足够的第一化合物或其药学上可接受的盐或氨基酸缀合物、一种或多种PPAR-α激动剂、一种或多种PPAR-δ激动剂、一种或多种PPAR-α和δ或PPAR-α和γ-双重激动剂、或一种或多种贝特、和/或一种或多种他汀,以允许各自的所希望的每日剂量以单一单位剂型(如片剂或胶囊)或以同时或在一天过程中的间隔时间给予的两个或更多个单位剂型给予至对其有需要的受试者。
本发明还提供了一种药物组合物,其中将该第一化合物和一种或多种PPAR-α激动剂、一种或多种PPAR-δ激动剂、一种或多种PPAR-α和δ或PPAR-α和γ双重激动剂、或一种或多种贝特与UDCA组合给予。在一个方面,以三向组合给予UDCA。在另一个方面,向对单独的UDCA具有不足的治疗反应的患者给予第一化合物和一种或多种PPAR-α激动剂、一种或多种PPAR-δ激动剂、一种或多种PPAR-α和δ或PPAR-α和γ双重激动剂、或一种或多种贝特的双向组合替换UDCA用于治疗或预防疾病或病症。
在本发明的方法中,活性物质可以按单次每日剂量或按每日两个、三个、四个或更多个相同或不同的分次剂量给予,并且这些剂量可以同时或在一天过程中的不同时间给予。通常,这些活性物质将同时给予,更通常以单一组合剂型给予。
在一个方面,将该第一化合物、一种或多种PPAR-α激动剂、一种或多种PPAR-δ激动剂、一种或多种PPAR-α和δ或PPAR-α和γ双重激动剂、或一种或多种贝特、和/或一种或多种他汀以与它们在各自单一疗法中给予的剂量基本相同的剂量给予。在一个方面,将该第一化合物以小于(例如,小于90%、小于80%、小于70%、小于60%、小于50%、小于40%、小于30%、小于20%、或小于10%)其单一疗法剂量的剂量给予。在一个方面,将该一种或多种PPAR-α激动剂、一种或多种PPAR-δ激动剂、一种或多种PPAR-α和δ或PPAR-α和γ双重激动剂、或一种或多种贝特以小于(例如,小于90%、小于80%、小于70%、小于60%、小于50%、小于40%、小于30%、小于20%、或小于10%)其单一疗法剂量的剂量给予。在一个方面,将该第一化合物和一种或多种PPAR-α激动剂、一种或多种PPAR-δ激动剂、一种或多种PPAR-α和δ或PPAR-α和γ双重激动剂、或一种或多种贝特以小于(例如,小于90%、小于80%、小于70%、小于60%、小于50%、小于40%、小于30%、小于20%、或小于10%)它们各自单一疗法剂量的剂量给予。在一个方面,将该一种或多种他汀以小于(例如,小于90%、小于80%、小于70%、小于60%、小于50%、小于40%、小于30%、小于20%、或小于10%)其单一疗法剂量的剂量给予。在一个方面,将该第一化合物和一种或多种他汀以小于(例如,小于90%、小于80%、小于70%、小于60%、小于50%、小于40%、小于30%、小于20%、或小于10%)它们各自单一疗法剂量的剂量给予。在一个方面,将该第一化合物、一种或多种PPAR-α激动剂、一种或多种PPAR-δ激动剂、一种或多种PPAR-α和δ或PPAR-α和γ双重激动剂、或一种或多种贝特、和/或一种或多种他汀以小于(例如,小于90%、小于80%、小于70%、小于60%、小于50%、小于40%、小于30%、小于20%、或小于10%)它们各自单一疗法剂量的剂量给予。
本发明的药物组合物可以呈用于口服给予的任何方便的形式,如片剂、胶囊剂、散剂、锭剂、丸剂、糖锭、酏剂、冻干粉、溶液、颗粒剂、混悬剂、乳剂、糖浆剂、或酊剂。还可以制备缓释、改进释放或延迟释放形式,例如呈包衣颗粒剂、多层片剂、胶囊内胶囊剂、胶囊内片剂、或微粒剂的形式。
用于口服给予的固体形式可以含有药学上可接受的粘合剂、甜味剂、崩解剂、稀释剂、调味剂、包衣剂、防腐剂、润滑剂和/或延时剂。适合的粘合剂包括阿拉伯胶、明胶、玉米淀粉、黄蓍胶、海藻酸钠、羧甲基纤维素或聚乙二醇。适合的甜味剂包括蔗糖、乳糖、葡萄糖、阿斯巴甜或糖精。适合的崩解剂包括玉米淀粉、甲基纤维素、聚乙烯吡咯烷酮、黄原胶、膨润土、海藻酸或琼脂。适合的稀释剂包括乳糖、山梨醇、甘露醇、右旋糖、高岭土、纤维素、碳酸钙、硅酸钙或磷酸二钙。适合的调味剂包括薄荷油、冬青油、樱桃、橙子或覆盆子调味剂。适合的包衣剂包括丙烯酸和/或甲基丙烯酸和/或其酯的聚合物或共聚物、蜡、脂肪醇、玉米蛋白、虫胶或谷蛋白。适合的防腐剂包括苯甲酸钠、维生素E、α-生育酚、抗坏血酸、对羟基苯甲酸甲酯、对羟基苯甲酸丙酯或亚硫酸氢钠。适合的润滑剂包括硬脂酸镁、硬脂酸、油酸钠、氯化钠或滑石。适合的延时剂包括单硬脂酸甘油酯或二硬脂酸甘油酯。
除了上述药剂以外,用于口服给予的液体形式还可以含有液体载体。适合的液体载体包括水、油类如橄榄油、花生油、芝麻油、葵花油、红花油、落花生油、椰子油、液体石蜡、乙二醇、丙二醇、聚乙二醇、乙醇、丙醇、异丙醇、甘油、脂肪醇、甘油三酯或其混合物。
用于口服给予的混悬剂可以进一步包括分散剂和/或悬浮剂。适合的悬浮剂包括羧甲基纤维素钠、甲基纤维素、羟丙基甲基纤维素、聚乙烯吡咯烷酮、海藻酸钠或鲸蜡醇。适合的分散剂包括卵磷脂;脂肪酸如硬脂酸的聚氧乙烯酯;聚氧乙烯山梨醇单-或二-油酸酯、-硬脂酸酯或-月桂酸酯;聚氧乙烯脱水山梨醇单-或二-油酸酯、-硬脂酸酯或-月桂酸酯等。
用于口服给予的乳剂可以进一步包括一种或多种乳化剂。适合的乳化剂包括如上文例示的分散剂或天然树胶,如阿拉伯胶或黄蓍胶。
本发明的药物组合物可以通过掺混、研磨、均质化、悬浮、溶解、乳化、分散和/或混合该第一化合物或其药学上可接受的盐或氨基酸缀合物、以及至少一种降脂剂(例如,贝特)、和任选地一种或多种他汀、连同选定的一种或多种赋形剂、一种或多种载体、一种或多种佐剂和/或一种或多种稀释剂来制备。本发明的呈片剂或胶囊剂形式的一种类型的药物组合物可以通过以下方式来制备:(a)制备第一片剂,该第一片剂包含选自该第一化合物或其药学上可接受的盐或氨基酸缀合物的活性物质中的至少一种、以及至少一种降脂剂、连同任何所希望的一种或多种赋形剂、一种或多种载体、一种或多种佐剂和/或一种或多种稀释剂;并且(b)制备第二片剂或胶囊剂,其中该第二片剂或该胶囊剂包含剩余的一种或多种活性物质和该第一片剂。本发明的呈胶囊剂形式的一种类型的药物组合物可以通过以下方式来制备:(a)制备第一胶囊剂,该第一胶囊剂包含选自该第一化合物或其药学上可接受的盐或氨基酸缀合物的活性物质中的至少一种、以及一种或多种降脂剂、连同任何所希望的一种或多种赋形剂、一种或多种载体、一种或多种佐剂和/或一种或多种稀释剂;并且(b)制备第二胶囊剂,其中该第二胶囊剂包含剩余的一种或多种活性物质和该第一胶囊剂。本发明的呈片剂形式的另外的类型的药物组合物可以通过以下方式来制备:(a)制备一种胶囊剂,该胶囊剂包含选自第一化合物或其药学上可接受的盐或氨基酸缀合物的活性物质中的至少一种、以及至少一种降脂剂、连同任何所希望的一种或多种赋形剂、一种或多种载体、一种或多种佐剂和/或一种或多种稀释剂;并且(b)制备一种片剂,其中该片剂包含剩余的一种或多种活性物质和该胶囊剂。
在实施例中,将该一种或多种PPAR-α激动剂、一种或多种PPAR-δ激动剂、一种或多种PPAR-α和δ或PPAR-α和γ双重激动剂、或一种或多种贝特、和/或一种或多种他汀用作立即释放片剂或持续释放片剂。当提供在持续释放片剂中时它是特别有效的。各种降脂剂的持续释放片剂是可商购的。片剂呈持续释放形式的延长作用是优选的。
在另一个实施例中,本发明的药物组合物包含胶囊剂,该胶囊剂含有一种或多种PPAR-α激动剂、一种或多种PPAR-δ激动剂、一种或多种PPAR-α和δ或PPAR-α和γ双重激动剂、或一种或多种贝特、和/或一种或多种他汀,胶囊剂内含有第一化合物或其药学上可接受的盐或氨基酸缀合物。典型地,在该形式中,将该一种或多种PPAR-α激动剂、一种或多种PPAR-δ激动剂、一种或多种PPAR-α和δ或PPAR-α和γ双重激动剂、或一种或多种贝特、和/或一种或多种他汀呈现为立即释放形式。在该事件中,通常每日给予该组合物三次。另一种给予方式是每日两次提供如上所述呈持续释放形式或非持续释放形式的含有该一种或多种PPAR-α激动剂、一种或多种PPAR-δ激动剂、一种或多种PPAR-α和δ或PPAR-α和γ双重激动剂、或一种或多种贝特、和/或一种或多种他汀的组合物,其中所给予的该组合物的每日量包含足够量的活性物质以向患者提供所希望的每日剂量。
在一个实施例中,本发明的药物组合物是包含每日总量为0.1-1500mg、0.2-1200mg、0.3-1000mg、0.4-800mg、0.5-600mg、0.6-500mg、0.7-400mg、0.8-300mg、1-200mg、1-100mg、1-50mg、1-30mg、4-26mg、或5-25mg的第一化合物或其药学上可接受的盐或氨基酸缀合物的剂型。在一个实施例中,每日一次口服给予该总量。
在一个实施例中,本发明的药物组合物是包含每日总量为10-1000mg、20-800mg、50-500mg、80-400mg、或100-300mg、更典型地约200mg的PPAR-α激动剂、PPAR-δ激动剂、PPAR-α和δ或PPAR-α和γ双重激动剂、或贝特、和/或他汀的剂型。在一个实施例中,每日一次口服给予该总量。在一个实施例中,本发明的药物组合物是包含5-1000mg、10-800mg、20-500mg、30-400mg、或40-200mg的量的他汀的剂型。
在实施例中,本发明的组合物是包含于胶囊剂内的包含10-1000mg、20-800mg、50-500mg、80-400mg、或100-300mg、更典型地约200mg的量的PPAR-α激动剂、PPAR-δ激动剂、PPAR-α和δ或PPAR-α和γ双重激动剂、或贝特、和/或他汀的剂型,该胶囊剂包含0.1-1500mg、0.2-1200mg、0.3-1000mg、0.4-800mg、0.5-600mg、0.6-500mg、0.7-400mg、0.8-300mg、1-200mg、1-100mg、1-50mg、1-30mg、4-26mg、或5-25mg的量的第一化合物。在一个实施例中,该PPAR-α激动剂、PPAR-δ激动剂、PPAR-α和δ或PPAR-α和γ双重激动剂、或贝特、和/或他汀处于持续释放形式。
在实施例中,本发明的组合物是包含于胶囊剂内的包含10-1000mg、20-800mg、50-500mg、80-400mg、或100-300mg、更典型地约200mg的量的苯扎贝特的持续释放片剂的剂型,该胶囊剂包含0.1-1500mg、0.2-1200mg、0.3-1000mg、0.4-800mg、0.5-600mg、0.6-500mg、0.7-400mg、0.8-300mg、1-200mg、1-100mg、1-50mg、1-30mg、4-26mg、或5-25mg的量的第一化合物。以这种方式,给予该剂型的患者接受苯扎贝特的持续释放片剂,其随着胶囊剂裂开并释放第一化合物而递送至远端腔。
本发明的药物组合物可以由患者终身使用,延长生存期并延缓肝移植。高血脂和肝酶的减少保证了相关血管疾病的发展的减少。第一化合物和降脂剂(如贝特和/或他汀)都具有非常小的长期副作用特征(除了苯扎贝特以外),因此这种组合可能是伴随高血脂症的原发性胆汁性肝硬化(PBC)以及抗性原发性胆汁性肝硬化(PBC)的治疗选择。由于由本发明提供的简化给药,本发明的组合疗法可能取决于患者的体重和临床反应以增加的剂量使用。
本发明的包含第一化合物或其药学上可接受的盐或氨基酸缀合物、PPAR-α激动剂、PPAR-δ激动剂、PPAR-α和δ或PPAR-α和γ双重激动剂、或贝特、和/或他汀的组合物可以作为单一胶囊剂内的三种活性物质提供。在这样的组合物的一种形式中,他汀可以与内部胶囊中的第一化合物混合,该内部胶囊被包含于外部胶囊内的PPAR-α激动剂、PPAR-δ激动剂、PPAR-α和δ或PPAR-α和γ双重激动剂或贝特包围。胶囊内的位置可以颠倒。也就是说,他汀和第一化合物的混合物可以包含在外部胶囊内,并且PPAR-α激动剂、PPAR-δ激动剂、PPAR-α和δ或PPAR-α和γ双重激动剂或贝特可以包含在内部胶囊内。如果待给予的他汀的量相对较大,则这种安排将是尤其令人满意的。用于给予三种活性物质组合的其他组合是可能的。
在此披露的第一化合物可以通过常规方法(例如,在美国公开号2009/0062526、美国专利号7,138,390和WO 2006/122977中描述的那些),如通过6步骤合成、接着一个纯化步骤来制备,以产生高度纯的化合物1(奥贝胆酸或OCA),如以下方案1中所示。
方案1

以上方法描述于WO 2013/192097中,该专利的内容通过引用以其全文结合在此。该方法是6步骤合成、接着一个纯化步骤。步骤1是7-酮基石胆酸(KLCA)的C-24羧酸的酯化以产生甲酯化合物a。步骤2是从化合物1形成烯醇硅醚以产生化合物c。步骤3是烯醇硅醚化合物c与乙醛的醇醛缩合反应以产生化合物d。步骤4是化合物d的皂化以产生化合物e。步骤5是化合物e的氢化以产生化合物f。步骤6是将化合物f的7-酮基基团选择性还原以产生结晶化合物1。步骤7是结晶化合物转化为非晶形化合物1(奥贝胆酸形式1或OCA形式1)。
可替代地,在此披露的第一化合物可以通过常规方法(例如,美国专利号7,932,244中描述的那些)或经由如方案2所示(和WO 2014/066819披露)的过程来制备。方案2可以用于制备在此披露的化合物2、3或4。

步骤1是具有化学式II的化合物的酯化以获得具有化学式III的化合物。步骤2是从具有化学式III的化合物形成具有化学式IV的化合物的反应。步骤3是在具有化学式IV的化合物的C3位置处的羟基基团的保护以提供具有化学式V的化合物。步骤4是具有化学式V的化合物的氧化裂解以给出具有化学式VI的化合物。步骤5是具有化学式VI的化合物的还原以提供具有化学式VII的化合物。步骤6是具有化学式VII的化合物的磺化以给出具有化学式I-Na的盐。具有化学式I-Na的盐可以转化为其游离碱形式(即,具有化学式I的化合物)或其他盐形式(例如,具有化学式I-(Et)3NH)的盐)。
定义
为了方便起见,在此收集了在说明书、实例和所附权利要求书中使用的某些术语。
如在此所用,术语“贝特”意指可用于在此所述方法中的2-苯氧基-2-甲基丙酸的纤维酸衍生物和药学活性衍生物中的任何一种。贝特的实例包括但不限于,非诺贝特、苯扎贝特、苄氯贝特、比尼贝特、环丙贝特、克利贝特、氯贝特、氯贝酸、依托贝特、吉非罗齐、尼可贝特、吡贝特、氯烟贝特、双贝特、氯贝茶碱、托考贝特、普拉贝脲等。贝特的实例还描述于美国专利号3,781,328、3,948,973、3,869,477、3,716,583、3,262,580、3,723,446、4,058,552、3,674,836、3,369,025、3,984,413、3,971,798、6,384,062、7,119,198和7,259,186;美国公开号20090131395;WO 2008/039829;比利时专利号884722;英国专利号860303;以及欧洲专利申请公开号EP 0607536,将它们中的每一个的全部披露内容通过引用结合在此。
过氧物酶体增殖物激活受体α(PPAR-α),也称为NR1C1(核受体亚家族1,C组,成员1),是核受体蛋白。PPAR-α激动剂结合并激活PPAR-α。PPAR-α激动剂的实例包括但不限于贝特,如在此所述的贝特。
过氧物酶体增殖物激活受体δ(PPAR-δ),也称为NR1C2(核受体亚家族1,C组,成员2),是核受体蛋白。PPAR-δ激动剂结合并激活PPAR-δ。PPAR-δ激动剂的实例包括但不限于,{4-[({4-甲基-2-[4-(三氟甲基)苯基]-1,3-噻唑-5-基}甲基)硫烷基]-2-甲基苯氧基}乙酸(本领域中也称为GW501516、GW1516、和Endurabol)、{2-甲基-4-[5-甲基-2-(4-三氟甲基-苯基)-2H-[1,2,3]三唑-4-基甲基硫烷基]-苯氧基}-乙酸、和[4-[[[2-[3-氟-4-(三氟甲基)苯基]-4-甲基-5-噻唑基]甲基]硫代]-2-甲基苯氧基]-乙酸。
PPAR-α和δ或PPAR-α和γ双重激动剂结合并激活PPAR-α和PPAR-δ两者、或PPAR-α和PPAR-γ两者。PPAR-α和δ双重激动剂的实例包括但不限于,2-[2,6二甲基-4-[3-[4-(甲基硫代)苯基]-3-氧代-1(E)-丙烯基]苯氧基]-2-甲基丙酸(也称为GFT505)。PPAR_α和γ双重激动剂的实例包括但不限于,阿格列扎((2S)-2-甲氧基-3-[4-[2-(5-甲基-2-苯基-4-噁唑基)乙氧基]-7-苯并苯硫基]丙酸,CAS号475479-34-6)、莫格他唑(N-[(4-甲氧基苯氧基)羰基]-N-{4-[2-(5-甲基-2-苯基-1,3-噁唑-4-基)乙氧基]苄基}甘氨酸,CAS号331741-94-7)、替格列扎((2S)-2-乙氧基-3-[4-[2-(4-甲基磺酰基氧基苯基)乙氧基]苯基]丙酸,CAS号251565-85-2)以及沙罗格列扎((2S)-2-乙氧基-3-[4-(2-{2-甲基-5-[4-(甲基硫烷基)苯基]-1H-吡咯-1-基}乙氧基)苯基]丙酸,CAS号495399-09-2)。
如在此所用,术语“FXR激动剂”是指激活FXR的任何化合物。在一个方面,FXR激动剂相对于CDCA实现FXR的至少50%活化,这些测定方法中的适当的阳性对照描述于WO2000/037077中。在另一个方面,FXR激动剂在如WO 2000/037077中所述的闪烁迫近分析法或HTRF测定中实现FXR的100%活化。FXR激动剂的实例包括但不限于以下各项中所述的那些:U.S.7,138,390;7,932,244;20120283234;20120232116;20120053163;20110105475;20100210660;20100184809;20100172870;20100152166;20100069367;20100063018;20100022498;20090270460;20090215748;20090163474;20090093524;20080300235;20080299118;20080182832;20080039435;20070142340;20060069070;20050080064;20040176426;20030130296;20030109467;20030003520;20020132223;以及20020120137。
如在此所用,术语“奥贝胆酸”或“OCA”是指具有以下化学结构的化合物:

奥贝胆酸还被称为奥贝胆酸形式1、INT-747、3α,7α-二羟基-6α-乙基-5β-胆烷-24-酸、6α-乙基-鹅脱氧胆酸、6-乙基-CDCA、6ECDCA、胆烷-24-酸、6-乙基-3,7-二羟基-(3α,5β,6α,7α),并且可以通过在美国公开号2009/0062526A1、美国专利号7,138,390、以及WO2006/122977中描述的方法来制备。奥贝胆酸的CAS登记号是459789-99-2。
如在此所用,术语“结晶奥贝胆酸”是指具有以下化学结构的一种化合物的任何结晶形式:

结晶奥贝胆酸意指该化合物在三个空间维度中结晶成一种特定晶体堆积排列或该化合物具有外表面平面。奥贝胆酸(或其药学上可接受的盐)的结晶形式可以结晶成不同的晶体包装排列,其全部都具有奥贝胆酸的相同元素组成。不同晶体形式通常具有不同的X-射线衍射图案、红外光谱、熔点、密度硬度、晶体形状、光学和电学性质、稳定性、以及溶解度。重结晶溶剂、结晶速率、储存温度、以及其他因素可能造成一种晶体形式占优势。奥贝胆酸的晶体可以通过在不同条件(例如,不同的溶剂、温度等)下结晶来制备。美国专利号9,238,673中描述了OCA的结晶形式的实例。
术语“第一化合物”意指具有化学式A、I、IA、II或IIA的化合物或化合物1、2、3或4、或其药学上可接受的盐或氨基酸缀合物。每当该术语在本发明的上下文中使用时,应当理解的是提及游离碱、同位素标记的化合物、结晶化合物、或其相应药学上可接受的盐或氨基酸缀合物,条件是在这些情况下这样是可能的和/或适当的。
如在此所用,术语“氨基酸缀合物”是指本发明的第一化合物(例如,具有化学式A的化合物)与任何适合的氨基酸的缀合物。例如,具有化学式A的化合物的这样的适合的氨基酸缀合物将具有在胆汁或肠液中完整性增强的附加优点。适合的氨基酸包括但不限于甘氨酸和牛磺酸。因此,本发明涵盖本发明的第一化合物(例如,化合物1)的甘氨酸和牛磺酸缀合物。
术语“他汀”与术语“3-羟基-3-甲基戊二酰基-辅酶A还原酶抑制剂”和“HMG-CoA还原酶抑制剂”同义。这些术语在此可互换使用。正如同义词所示,他汀是3-羟基-3-甲基戊二酰基-辅酶A还原酶的抑制剂,并且因此在降低血浆胆固醇水平方面是有效的,并因此用于治疗或预防心血管疾病。他汀及其药学上可接受的盐在降低哺乳动物、尤其是人类中的低密度脂蛋白胆固醇(LDL-C)水平方面特别有效。结构上,他汀或其衍生物具有共同的4-羟基-6-氧代-2H-吡喃体系,其还可以是处于与HMG-CoA还原酶的活性位点相互作用的二羟酸形式和特别作为多取代的六氢萘体系存在的亲脂性部分,但也可以被多取代的杂芳香族体系代替,如在阿托伐他汀或氟伐他汀中。适合用于在此使用的他汀包括但不限于,辛伐他汀、氟伐他汀、普伐他汀、利伐司他、美伐他汀、阿托伐他汀、西立伐他汀、洛伐他汀、匹伐他汀、氟多他汀、维洛他汀、达伐他汀、罗素伐他汀、二氢康帕丁和康帕丁、或其药学上可接受的盐。
术语“降脂剂”是指能够降低循环中(例如,血液中)的脂质(例如胆固醇、LDL和甘油三酯)浓度的任何试剂。降脂剂包括但不限于(i)胆汁酸螯合剂,如树脂(例如,考来烯胺、考来替泊、考来维仑);(ii)胆固醇吸收型抑制剂,其阻止胆固醇摄取(例如,阻止从小肠进入循环系统),如依泽替米贝(即,(3R,4S)-1-(4-氟苯基)-3-[(3S)-3-(4-氟苯基)-3-羟基丙基]-4-(4-羟基苯基)氮杂环丁-2-酮)和(3R,4S)-1,4-双(4-甲氧基苯基)-3-(3-苯基丙基)-2-氮杂环丁酮);(iii)ω-3脂肪酸乙酯,包括游离脂肪酸衍生物(如 VascepaTM、Epadel、EpanovaTM)或源自海洋的ω-3多不饱和脂肪酸(PUFA);(iv)PCSK9抑制剂;(v)烟酸;(vi)植物甾醇(例如,植物固醇和甾烷醇),如β-谷甾醇、菜油甾醇、豆甾醇、芸苔甾醇、麦角甾醇、β-谷甾烷醇、菜油甾烷醇、豆甾烷醇、环阿屯醇、和羽扇豆醇;(vii)CETP(胆固醇酯转移蛋白)的抑制剂,如安塞曲匹、依塞曲匹(Evacetrapib)、托塞曲匹和达塞曲匹;(viii)角鲨烯合酶抑制剂;(ix)反义寡核苷酸,其影响脂质的合成、降解、吸收和代谢(例如,与编码载脂蛋白B或PCSK9的mRNA结合的反义寡核苷酸)(例如,米泊美生(Mipomersen)(Kynamro));(x)脱辅基蛋白-B抑制剂;(xi)微粒体甘油三酯转运蛋白的抑制剂(例如洛美他派(Lomitapide)(Juxtapid));以及(xii)其他化合物,如考来维纶、阿伐麦布和英普他派。
VascepaTM、Epadel、EpanovaTM)或源自海洋的ω-3多不饱和脂肪酸(PUFA);(iv)PCSK9抑制剂;(v)烟酸;(vi)植物甾醇(例如,植物固醇和甾烷醇),如β-谷甾醇、菜油甾醇、豆甾醇、芸苔甾醇、麦角甾醇、β-谷甾烷醇、菜油甾烷醇、豆甾烷醇、环阿屯醇、和羽扇豆醇;(vii)CETP(胆固醇酯转移蛋白)的抑制剂,如安塞曲匹、依塞曲匹(Evacetrapib)、托塞曲匹和达塞曲匹;(viii)角鲨烯合酶抑制剂;(ix)反义寡核苷酸,其影响脂质的合成、降解、吸收和代谢(例如,与编码载脂蛋白B或PCSK9的mRNA结合的反义寡核苷酸)(例如,米泊美生(Mipomersen)(Kynamro));(x)脱辅基蛋白-B抑制剂;(xi)微粒体甘油三酯转运蛋白的抑制剂(例如洛美他派(Lomitapide)(Juxtapid));以及(xii)其他化合物,如考来维纶、阿伐麦布和英普他派。
“治疗”包括导致病症、疾病、障碍等的改善的任何作用,例如减轻、减少、调节或消除。疾病状态的“治疗(Treating或treatment)”包括:抑制疾病状态,即阻止疾病状态或其临床症状的发展;或减轻疾病状态,即引起疾病状态或其临床症状的暂时或永久消退。
“预防”疾病状态包括使疾病状态的临床症状在可能暴露于或易患该疾病状态、但尚未经历或显示该疾病状态的症状的受试者中不发展。
如在此所用的术语“抑制(inhibiting或inhibition)”是指对疾病或病症的发展或进展的任何可检测的积极效果。这样一种积极作用可以包括疾病或病症的至少一种症状或体征的发作的延迟或预防,该一种或多种症状或一种或多种体征的减轻或逆转,以及该一种或多种症状或一种或多种体征的进一步恶化的减慢或预防。
“疾病状态”意指任何疾病、障碍、病症、症状或适应症。
如在此所用的术语“有效量”或“治疗有效量”是指在适当剂量给予(单独或组合)后产生急性或慢性治疗效果的第一化合物(例如,FXR活化配体)或贝特、或降脂剂、或他汀的量。在一个实施例中,有效量或治疗有效量的第一化合物(例如,FXR活化配体)在与至少一种贝特组合适当剂量给予后产生急性或慢性治疗效果。该作用包括疾病/病症(例如,肝、肾或肠的纤维化)以及达到任何可检测程度的相关并发症的症状、体征和潜在病理的预防、矫正、抑制、或逆转。“有效量”或“治疗有效量”将取决于第一化合物、贝特、降脂剂、他汀、疾病及其严重程度、以及待治疗受试者的年龄、体重等而变化。
治疗有效量的第一化合物可以与一种或多种贝特、以及任选地一种或多种药学上可接受的载体一起配制用于给予至人或非人动物。因此,本发明的药物组合物可以例如经由口服、胃肠外或局部途径给予以提供有效量的第一化合物和一种或多种贝特。在替代性实施例中,本发明的组合物可以用于涂覆或浸渍医疗装置,例如支架。
如在此所用的“药理作用”涵盖了在实现疗法的预期目的的受试者中产生的作用。在一个实施例中,药理作用意指预防、缓解、或减少被治疗的受试者的主要适应症。例如,药理作用将是产生所治疗的受试者中的主要适应症的预防、缓解或减少的一种药理作用。在另一个实施例中,药理作用意指预防、缓解、或减少被治疗的受试者的主要适应症的障碍或症状。例如,药理作用将是产生所治疗的受试者的障碍或症状的预防、缓解或减少的一种药理作用。
应当理解,除非另外说明,由不对称碳原子产生的异构体(例如,所有的对映异构体和非对映异构体)包括在本发明的范围内。可以通过经典分离技术和通过立体化学控制合成获得呈基本上纯的形式的此类异构体。
“药物组合物”是呈适合给予至受试者的形式的、含有治疗剂(如第一化合物)和降脂剂(如,贝特)的配制品。在一个实施例中,该药物组合物是处于散装形式或单位剂型。以易于给予和实现剂量均匀性的剂量单位形式配制组合物可以是有利的。如在此使用的单位剂型是指作为针对待治疗的受试者的单一剂量适合的物理上离散单位;每个单位含有经计算产生与所需药物载体相关的所希望的治疗效果的活性试剂的预定量。本发明的剂量单位形式的规格由如下决定并且直接依赖于:活性试剂的独特特征和待实现的具体治疗效果,和本领域中配制用于治疗个体的这种活性剂的固有局限性。
术语“单位剂型”是指适于作为用于人和其他哺乳动物的单位剂量的物理离散单位,每个单位含有预定量的经计算结合如在此所述的一种适合的药物赋形剂可产生所希望的治疗作用的活性材料。
单位剂型是多种形式中的任一种,包括例如胶囊、IV袋、片剂、气雾剂吸入器上的单个泵、或小瓶。第一化合物或其药学上可接受的盐或氨基酸缀合物在组合物的单位剂量中的量是有效量,并且根据所涉及的特定治疗和/或用于该治疗的一种或多种降脂剂而变化。本领域的技术人员应理解,有时有必要依据患者的年龄和病症对剂量进行常规改变。剂量还将取决于给予途径。考虑了各种途径,包括口服、肺部、直肠、肠胃外、透皮、皮下、静脉内、肌内、腹膜内、吸入、颊部、舌下、胸膜内、鞘内、鼻内等。用于局部或经皮给予本发明的化合物的剂型包括散剂、喷雾剂、软膏、糊剂、乳膏、洗剂、凝胶、溶液、贴剂和吸入剂。在一个实施例中,第一化合物和/或降脂剂在无菌条件下与药学上可接受的载体、以及与所需要的任何防腐剂、缓冲剂或推进剂一起混合。
术语“闪释剂量”是指处于快速分散剂型的配制品。
术语“立即释放”被定义为治疗剂(如第一化合物或降脂剂)在相对较短的时间段(通常至多约60分钟)中从剂型的释放。术语“改良释放”被定义为包括延迟释放、延长释放和脉冲释放。术语“脉冲释放”被定义为药物从剂型的一系列释放。术语“持续释放”或“延长释放”被定义为治疗剂在延长的时间段内从剂型的连续释放。
“受试者”包括哺乳动物,例如,人、伴侣动物(例如,狗、猫、鸟等)、农场动物(例如,牛、绵羊、猪、马、家禽等)、以及实验室动物(例如,大鼠、小鼠、豚鼠、鸟等)。在一个实施例中,该受试者是人。在一个方面,该受试者是雌性。在一个方面,该受试者是雄性。
如在此所用,短语“药学上可接受”是指在正确医学判断的范围内适用于与人类和动物的组织接触而无过多毒性、刺激、过敏反应或其他问题或并发症,与合理益处/风险比相称的那些化合物、材料、组合物、载体和/或剂型。
“药学上可接受的载体或赋形剂”意指总体上安全、无毒,并且在生物学上以及其他方面并非不合需要的适用于制备药物组合物的载体或赋形剂,并且包括可为兽医使用以及人药物使用接受的赋形剂。如在本说明书和权利要求书中使用的“药学上可接受的赋形剂”包括一种和多于一种这样的赋形剂。
虽然有可能在无任何配制品的情况下直接给予第一化合物,但第一化合物可以呈包含药学上可接受的赋形剂的药物配制品的形式给予。这样的配制品可以通过各种途径给予,这些途径包括口服、经颊、直肠、鼻内、经皮、皮下、静脉内、肌内和鼻内。
在一个实施例中,可以经皮给予第一化合物。为了经皮给予,需要经皮递送装置(“贴剂”)。此类经皮贴剂可用于以可控量进行本发明的化合物的连续或间断输注。用于药学试剂递送的经皮贴剂的构造和使用是本领域中熟知的。参见,例如美国专利号5,023,252。此类贴剂可以被制成连续的、脉冲的或者按需递送的药学试剂。
在一个实施例中,本发明的药物组合物适合于经颊和/或舌下或经鼻给予。该实施例提供了以避免胃并发症(如通过胃系统和/或通过肝脏的首过代谢)的方式给予第一化合物。这种给予途径还可以减少吸附时间,从而提供治疗益处的更快起效。
第一化合物可以在宽剂量范围内给予。例如,每日剂量通常在约0.0001至约30mg/kg体重的范围内。在成人的治疗中,可以使用呈单次或分次剂量的约0.1至约15mg/kg/天的范围。在一个实施例中,该配制品包含约0.1mg至约1500mg的第一化合物。在另一个实施例中,该配制品包含约1mg至约100mg的第一化合物。在另一个实施例中,该配制品包含约1mg至约50mg的第一化合物。在另一个实施例中,该配制品包含约1mg至约30mg的第一化合物。在另一个实施例中,该配制品包含约4mg至约26mg的第一化合物。在另一个实施例中,该配制品包含约5mg至约25mg的第一化合物。然而,应当了解,实际给予的第一化合物的量将由医师根据相关情况(包括待治疗的病症、所选择的给予途径、所给予的第一化合物的形式、所给予的一种或多种降脂剂、个体患者的年龄、体重和反应、以及患者症状的严重程度等)来确定。因此,以上剂量范围不旨在以任何方式限制本发明的范围。在一些情况下,低于上述范围下限的剂量水平可能是足够的,而在其他情况下可能使用不引起任何有害副作用的仍然更大的剂量,条件是将这类更大的剂量首先分成数个小剂量以用于全天给予。
“纤维化”是指在组织或器官中涉及过度纤维结缔组织(例如,瘢痕组织)的发展的病症。瘢痕组织的这种产生可以响应于由于疾病、外伤、化学毒性等所致的器官的感染、炎症或损伤而发生。纤维化可以在各种不同的组织和器官(包括肝、肾、肠、肺、心脏等)中发展。
如在此所用,“胆汁淤积性病症”是指来自肝的胆汁排泄受损或被阻断的任何疾病或病症,该疾病或病症可以在肝抑或在胆管中发生。肝内胆汁淤积和肝外淤积是两种类型的胆汁淤积性病症。肝内胆汁淤积(其在肝内部发生)最常见于原发性胆汁性肝硬化、原发性硬化性胆管炎、败血症(全身感染)、急性酒精性肝炎、药物中毒、胃肠外全面营养(静脉内供给)、恶性肿瘤、囊性纤维化、胆道闭锁、以及妊娠中。肝外胆汁淤积(其在肝外部发生)可以由胆管肿瘤、狭窄、囊肿、憩室、胆总管中的结石形成、胰腺炎、胰腺肿瘤或假性囊肿、以及由于附近器官中的团块或肿瘤所致的压缩引起。
胆汁淤积性病症的临床症状和体征包括:发痒(瘙痒)、疲劳、黄疸皮肤或眼睛、不能消化某些食物、恶心、呕吐、白便、小便黄赤、以及右上腹疼痛。患有胆汁淤积性病症的患者可以基于一组标准临床实验室测试进行诊断和临床跟踪,这些测试包括患者血清中的碱性磷酸酶、γ-谷氨酰转肽酶(GGT)、5’核苷酸酶、胆红素、胆汁酸、以及胆固醇水平的测量。通常,如果所有三种诊断标志物(碱性磷酸酶、GGT、和5’核苷酸酶)的血清水平都被认为异常升高,则患者被诊断为患有胆汁淤积性病症。这些标志物的正常血清水平可能取决于测试方案,因实验室和程序而异在一定程度上变化。因此,医师将能够基于具体的实验室和测试程序确定对于这些标志物中的每一种异常升高的血液水平怎么样。例如,患有胆汁淤积性病症的患者通常在血液中具有大于约125IU/L的碱性磷酸酶、大于约65IU/L的GGT、和大于约17NIL的5’核苷酸酶。由于血清标志物的水平的变化性,除了上述症状中的至少一种(如发痒(瘙痒))之外,胆汁淤积性病症可以基于这三种标志物的异常水平进行诊断。
术语“原发性胆汁性肝硬化”通常缩写为PBC,其是以肝的小胆管的缓慢进行性破坏为特点的肝的自身免疫性疾病,其中小叶内导管(赫令管)在疾病早期受到影响。当这些导管被损伤时,胆汁在肝中积累(胆汁淤积)并且随时间推移损伤该组织。这可能会导致疤痕形成、纤维化和肝硬化。原发性胆汁性肝硬化的特征在于小叶间胆管破坏。原发性胆汁性肝硬化的组织病理学发现包括:胆管炎症,其特征在于上皮淋巴细胞和管周上皮样肉芽肿。PBC存在4个阶段。
阶段1-门静脉阶段:正常大小的三联体;门静脉炎症;轻微胆管损伤。通常在此阶段中检测到肉芽肿。
阶段2-门静脉周阶段:扩大的三联体;门静脉周纤维化和/或炎症。典型地,这个阶段的特征在于发现小胆管的增生。
阶段3-中隔阶段:主动和/或被动纤维间隔。
阶段4-胆汁性肝硬化:结节存在;加兰(garland)
术语“原发性硬化性胆管炎”(PSC)是在肝内(肝内部)和肝外(肝外部)水平下引起胆管的炎症和随后阻塞的胆管疾病。炎症阻碍胆汁到肠道的流动,其最终可能导致肝的肝硬化、肝衰竭和肝癌。
术语“非酒精性脂肪性肝炎”(NASH)是由脂肪在肝中的积累引起的肝炎症。在一些人中,脂肪的积累引起肝的炎症。由于炎症,肝不能如其本来那样良好地起作用。NASH可能变得更严重并且引起肝的瘢痕形成,其导致肝硬化。NASH类似于由长期大量饮酒引起的肝病种类。但NASH在不酗酒的人中发生。
术语“器官”是指由细胞和组织组成且执行生物体的一些特定功能的分化的结构(如在心脏、肺、肾、肝等中)。该术语还涵盖执行功能或在活动中协作的身体部分(例如,构成视觉器官的眼睛和相关结构)。术语“器官”进一步涵盖分化的细胞和组织的能够潜在发育成完整结构的任何部分结构(例如,肝的叶或部分)。
在此引用的全部出版物和专利文献通过引用结合在此,如同每一份这样的出版物或者文献都被具体地和分别地指出已通过引用结合在此。出版物和专利文件的引用不欲承认任一个为相关先前技术,并且其也不构成关于其内容或日期的任何承认。现已通过书面说明描述了本发明,本领域的技术人员应认识到,可在各种实施例中实践本发明,且在此提供的描述和实例是出于说明的目的,并不限制所附权利要求书。
在本说明书中,除非上下文另外明确规定,否则单数形式也包括复数。除非另外定义,本文所用的所有技术和科学术语具有与本发明所属领域普通技术人员通常所理解的意义相同的意义。在冲突的情况下,以本说明书为准。除非另外指出,否则在此使用的所有百分比和比率都是按重量计。
实例
实例1:胆管结扎(BDL)模型
进行该实验以评估单独以及组合使用的OCA和阿托伐他汀对由小鼠中胆总管结扎引起的纤维化的影响。
动物、圈养和饮食
从日本SLC获得雄性C57BL/6小鼠(6周龄)。在温度(23℃±2℃)、湿度(45%±10%)、光照(12小时人工光照和黑暗循环;从8:00至20:00光照)、以及换气(换气速率:多于40次/小时)的受控条件下,将动物维持在无特定病原体设备中。在实验室中保持高压(20±4Pa)以防止该设备被污染。将动物圈养在KN-600(Natsume Seisakusho公司,日本)中,每笼最多6只小鼠。将灭菌清洁纸(日本SLC)用于垫料,并且每周更换一次。在手术前3周,任意量提供灭菌的固体高脂肪饮食(HFD)和水。
处理组
组1:假手术
在BDL手术后第0天至第6天,以5mL/kg的体积对假手术小鼠(n=8)每日一次口服给予运载体(0.5%CMC)。
组2:BDL-运载体
在BDL手术后第0天至第6天,以5mL/kg的体积对BDL-手术小鼠(n=12)每日一次口服给予运载体(0.5%CMC)。
组3:BDL-OCA
在BDL手术后第0天至第6天,以5mg/kg的剂量对BDL-手术小鼠(n=12)每日一次口服给予补充有OCA的运载体。
组4:BDL-阿托伐他汀
在BDL手术后第0天至第6天,以10mg/kg的剂量对BDL-手术小鼠(n=12)每日一次口服给予补充有阿托伐他汀的运载体。
组5:OCA-BDL-阿托伐他汀
在BDL手术后第0天至第6天,对BDL-手术小鼠(n=12)每日一次口服给予5mg/kg剂量的补充有OCA的运载体和10mg/kg剂量的补充有阿托伐他汀的运载体。
胆管结扎手术
在第0天进行胆管结扎手术。在戊巴比妥麻醉下通过胆总管结扎在小鼠中建立胆汁淤积,该胆汁淤积随时间导致肝的纤维化。基于手术前一天小鼠的体重,将小鼠分成两个手术组群。剃毛后,打开腹腔,并且将胆总管与5-0手术丝连接两次,并在结扎之间切开胆总管。用缝合线封闭腹膜和皮肤。将小鼠转移到干净的笼子(休息笼)中用于从麻醉中恢复。将假手术小鼠(Sham mice)以与其他组相似的方式进行手术,但胆管未结扎。
动物监测和处死
每日监测生存力、临床体征和行为。处理期间每日记录体重。在处理期间,每笼每周两次测量食物消耗。在第6天,在乙醚麻醉(日本和光化学工业有限公司(Wako PureChemical Industries))下通过直接心脏穿刺通过放血将动物处死。
组织学分析
为了可视化胶原沉积,使用天狼星红(picro-Sirius red)溶液(瓦尔德克公司(Waldeck),德国)染色布安氏(Bouin’s)固定的左侧肝切片。对于纤维化区域的定量分析,使用数码相机(DFC295;莱卡(Leica),德国)以100倍放大率捕获中央静脉周围的天狼星红染色切片的明视野图像,并且使用ImageJ软件(美国国立卫生研究院(National Instituteof Health,USA))测量5个视野/切片的阳性面积。使用Prism软件6(美国图板软件公司(GraphPad Software,Inc.USA))进行统计学分析。
结果
通过天狼星红染色对肝切片进行组织病理学分析(根据常规方法)以估计纤维化面积的百分比。天狼星红染色的肝切片的代表性显微照片显示在图1A-1E中。与BDL-假手术组相比,BDL-运载体组显示天狼星红-阳性面积显著增加。如表1和图2所示,与BDL-运载体组相比,BDL-OCA+阿托伐他汀组显示天狼星红-阳性面积显著减少。
表1

表1中的结果表明,OCA和阿托伐他汀的组合显著降低了纤维化。
实例2:APOE*3Leiden.CETP小鼠中饮食诱导的NASH
进行该实验以评估OCA和非诺贝特单独或组合对APOE*3Leiden.CETP转基因小鼠中饮食诱导的NASH和肝纤维化的发展的影响。进行肝基因表达谱分析和随后的途径分析以确定该组合是否调控未由任一单一疗法治疗调控的新颖基因和/或更强烈地调控也受单一疗法影响的基因。
动物、圈养和饮食
获得雄性APOE*3Leiden.CETP转基因小鼠(9-21周龄),并且每笼圈养2-5只小鼠。给小鼠饲喂含有24%猪油和1%(w/w)胆固醇的高脂肪饮食。在高脂肪饮食中,运行时间为15周。在第16周,禁食4小时后,根据年龄、体重、血浆胆固醇和甘油三酯将小鼠配对。
处理组
组1:HFC参考组起始处理
在第0至14周的运行期间,给小鼠(n=15)饲喂高脂肪饮食。
组2:HFC对照组
从第0至24周给小鼠(n=15)饲喂高脂肪饮食。
组3:HFC+OCA
在第16至24周,每日一次以10mg/kg的剂量给小鼠(n=15)饲喂补充有OCA的高脂肪饮食。
组4:HFC+低剂量非诺贝特
在第16至24周,每日一次以10mg/kg的剂量给小鼠(n=15)饲喂补充有非诺贝特的高脂肪饮食。
组5:HFC+高剂量非诺贝特
在第16至24周,每日一次以40mg/kg的剂量给小鼠(n=15)饲喂补充有非诺贝特的高脂肪饮食。
组6:HFC+OCA+低剂量非诺贝特
在第16至24周,每日一次给小鼠(n=15)饲喂10mg/kg剂量的补充有OCA的和10mg/kg剂量的补充有非诺贝特的高脂肪饮食。
组7:HFC+OCA+高剂量非诺贝特
在第16至24周,每日一次给小鼠(n=15)饲喂10mg/kg剂量的补充有OCA的和40mg/kg剂量的补充有非诺贝特的高脂肪饮食。
组8:食物对照组
从第0至24周给小鼠(n=8)饲喂食物。
研究设计
给小鼠饲喂高脂肪食物(HFC)饮食持续14周。15周的HFC饮食后,禁食4小时后,根据年龄、体重、血浆胆固醇和甘油三酯将HFC小鼠配成7组。从第15周开始,将小鼠用OCA和非诺贝特单独或组合治疗,并在非禁食状态的第25周处死。处死前一周,通过腹膜内(i.p.)注射大量D2O并随后向饮用水中添加4%D2O,将小鼠用D2O标记。通过心脏穿刺获得血浆(EDTA),并储存在-70℃。对肝脏称重并将其分离出4块肝脏:将1块(内侧叶)固定于10%福尔马林中(用于NASH和纤维化组织学),并将3块(左叶)快速冷冻于液N2中,并在-70℃下分别储存。
肝脏炎症评分
炎症是NASH的一个关键特征。根据Liang等人,PlosOne[公共科学图书馆期刊],2014年12月,9(12)的程序对炎症进行分类,并通过定量分析炎症细胞聚集体的数量进行评分。具体地,通过使用100x放大率(视图尺寸为3.1mm2;平均5个不同的视野)计数每个视野的炎症病灶数目来评估炎症水平。
结果:OCA+/-非诺贝特对APOE*3-Leiden.CETP小鼠中炎症的影响的概述
在NASH饮食中,研究了单独OCA 10mg/kg和与非诺贝特(10和40mg/kg)组合对APOE*3-Leiden.CETP小鼠中炎症的影响。在低剂量下药物给予10周后,OCA(10mg/kg)和非诺贝特(10mg/kg)都不减少炎症细胞病灶的数目(图3A和3B以及表2)。相比之下,相对于运载体对照以及每个单一疗法组,该组合显著降低了炎症。相对于运载体对照,较高剂量的非诺贝特(40mg/kg/d)也显著减少了炎症。当与OCA组合时,没有额外的抗炎作用是显而易见的,因为高剂量的非诺贝特对其本身产生接近最大的作用。总之,在OCA+低剂量非诺贝特组合(-63%)下观察到炎症显著减少。此外,在高剂量非诺贝特(-74%)下观察到显著降低,并且与OCA(-79%)的组合观察到相似的降低。参见表2以及图3A和3B。
表2


表2中的结果表明,OCA和高剂量的非诺贝特的组合的疗效是由高剂量的非诺贝特驱动并达到上限。
RNA分离和测序
如先前详细描述地进行核酸提取(Verschuren等人,2014)。简言之,使用玻璃珠和RNAzol(凯博科技公司(Campro Scientific),费嫩达尔(Veenendaal),荷兰)从个体肝样品中提取总RNA。使用片段分析仪(美国先进分析技术公司(Advanced AnalyticalTechnologies,USA))和RNA6000纳米芯片实验室(RNA 6000Nano Lab-on-a-Chip)试剂盒和生物分析器2100(安捷伦科技公司,阿姆斯特尔芬,荷兰)确定RNA浓度和质量。所有样品符合质量要求,并用于RNA测序程序。
使用用于Illumina的NEBNext Ultra定向RNA文库制备试剂盒(Directional RNAlibrary Prep Kit)来处理样品。根据试验方案“用于Illumina的NEBNext Ultra定向RNA文库制备试剂盒”(NEB#E7420S/L)进行样品制备。简言之,使用寡聚-dT磁珠从总RNA中分离mRNA。mRNA片段化后,进行cDNA合成。将其用于测序衔接子的连接和所得产物的PCR扩增。用片段分析仪(美国先进分析技术公司)测量样品制备后的质量和产率。所得产物的尺寸与预期的尺寸分布(300-500bp之间的宽峰)一致。
根据制造商的试验方案使用Illumina NextSeq 2500进行聚集和DNA测序。使用单端读数测序试验方案生成数据,从而获得每个样本约1500万次读数和每个读数75bp。用Illumina数据分析管道RTA v2.4.11进行图像分析、碱基识别和质量检查,以产生原始数据(*.fastq-file)。
使用基于伯罗斯-惠勒(Burrows-Wheeler)变换法的短读数比对器将读数映射到参考序列小家鼠(Mus musculus)GRCm38.p3。使用默认错配率2%(150个碱基读数中3个不匹配)。基于比对文件(*.bam-file)中映射的读数位置,确定了读数多久一次映射在转录物上的频率。将计数保存到计数文件,其作为下游mRNA-seq差异表达分析的输入。将读数计数加载到DESeq软件包中,该软件包是R平台内的统计软件包。专门开发DESeq用于标准化不同样品的RNA-seq数据,并在RNA-seq数据的两种条件之间找到差异表达的基因以估计每个基因的平均值和方差之间的关系(Anders等人,2013)。此外,它允许缩放因子容易地包括在统计测试中。使用显著性为P<0.01的阈值鉴定差异表达的基因,并将基因用作通过独创性途径分析(Ingenuity Pathway Analysis,IPA)套件(2016年访问)进行途径分析的输入。
使用IPA软件(Kramer等人,2014)进行上游调节子分析。该分析基于观察到的差异基因表达来确定转录因子的激活状态。这产生每个转录因子在IPA知识库中的重叠P值和激活z得分。该重叠P值表示转录因子的已知靶基因与在实验中测量的差异表达基因之间重叠的重要性。该激活z得分表示特定转录因子的激活(正z得分)或抑制(负z得分)。激活z得分<-2或>2表明途径或过程的显著抑制或激活。
组学结果
对来自经处理的小鼠的肝mRNA样品进行下一代测序以深入了解潜在的机制和途径。进行了两项分析以深入了解潜在的机制和途径。
首先,标准途径分析的富集分析显示,OCA调节了几种炎症过程(图6A)。该图将每个途径绘图为-log p值的函数(对于参考,p<0.05的转化值为1.3,p<0.0001的转化值为4,p<0.000005的转化值为5.3等)。具有OCA单一疗法的调节途径与T细胞和B细胞信号传导、白细胞外渗信号传导、自然杀伤细胞信号传导等相关。低剂量的非诺贝特对这些途径没有影响。当OCA与低剂量的非诺贝特组合时,一些相同的途径被强有力地调节(在辅助T细胞中的iCOS-iCOSL信号传导、白细胞外渗信号传导、模式识别受体、巨噬细胞中的FC受体介导的吞噬作用和吞噬体形成的情况下)。此外,由该组合调节了由单独的任一试剂未显著调节的其他途径(例如,胆固醇生物合成I和II、脂肪酸b氧化)(图6B)。与组织学数据一样,高剂量的非诺贝特对这些途径具有强大的作用,但用OCA共同给予没有增强。
围绕涉及白细胞外渗信号传导的途径进行更详细的分析。白细胞的外渗对于NASH(和其他疾病)中的病理生理过程是必需的。这些过程包括用于免疫监视的T淋巴细胞的迁移、在急性和慢性炎症反应期间活化的淋巴细胞和粒细胞的募集、以及造血祖细胞的归巢和动员。来自该研究的使小鼠维持高脂肪饮食的作用说明了这些显著上调和下调了基因的过程。表3描述了高脂肪饮食相对于维持标准食物的小鼠对维持高脂肪饮食的小鼠中的白细胞外渗信号传导的影响,以及相对于高脂肪饮食对组合治疗的影响。
表3.





白细胞穿过内皮的迁移和外渗发生在几个不同的步骤中,包括由粘附分子之间的瞬时弱相互作用介导的、白细胞在内皮细胞上的滚动。随后,松散连接的白细胞与内皮如此邻近使得它们被呈现在内皮顶端表面的趋化性细胞因子活化。接下来,活化的白细胞扩散并牢固地附着于内皮形成的对接结构,并最终通过内皮细胞之间的细胞间隙迁移到基础组织。
OCA的给予下调了白细胞内炎症级联过程中涉及的许多基因(WAP、Rac2、RASGRP1、Vav、PKC、PI3K、ERM、ITGAL和PSGL-1)以及内皮细胞内炎症级联过程中涉及的许多基因(VCAM1、PI3K、ERM、NOX、CYBA、PKC、NCF1和NCF2)。这些途径中的基因调控在单独给予低剂量的非诺贝特后不明显。
当OCA与在调节这些途径下无效的剂量的非诺贝特组合时,现在调节多个另外的基因,表明协同效应。在白细胞内,这些另外的基因包括CD43、PSGL-1、CXCR4、ITGAM、ITGB2、Rap1、ITGA4。在内皮细胞内,这些另外的基因包括ICAM1、RhoGAP、VASP、NCF4、ITGAM、ITGB2、ITGA4和ICAM-1。
如上所述,高剂量的非诺贝特对用OCA组合给予没有增强的这个途径有许多影响。所有后续分析集中在低剂量单一疗法和组合(OCA 10mg/kg+/-非诺贝特10mg/kg)。
在第二次分析中,比较了在低剂量组合和每个分别的单一疗法之间差异调控的基因。这与集中于相对于运载体组的比较的第一基因表达分析(如上所述)不同,下面的分析将每个单一疗法与组合进行比较。维恩图(图7A)显示,OCA具有109个独特调节的基因,非诺贝特有92个独特调节的基因和6个共同调节的基因。用OCA的组合调节了517个重叠基因,用非诺贝特的组合调节了75个基因,并且有5个基因是全部共有的。值得注意的是,该组合总共调节了912个独特基因。随后的途径富集突出了组合基因参与的生物学过程(图7B)。
随后对白细胞外渗进行途径分析(例如,如上),但是这次比较是在该组合和每个单一疗法之间。对于白细胞外渗,该组合与每个单一疗法的比较显示,在组合处理的小鼠中有许多独特调节的基因,这与组织学上观察到的增强的抗炎变化一致(表4)。
表4.




鉴于NASH中从炎症到纤维化的进展以及以在组合中显著调节出现的肝纤维化/HSC途径的观察,我们还检测了HSC中的途径。当与单一疗法比较时,很明显,对比单独的非诺贝特,组合中更多的基因被调节,并且对比OCA,组合中更少的基因被调节。换句话说,就这些纤维化途径而言,这两种药剂之间存在明显的相互作用,但该组合的OCA部分可能更强烈地驱使这些效果。
解释和相关性
证明了FXR活化在来自敲除FXR的小鼠的肝脏中预防纤维化和炎症的重要性,该敲除FXR的小鼠展示具有进行性年龄相关性损伤和炎症的炎症基因(Kim 2007)的表达升高(Yang 2007)。与这些报道一致,OCA在HepG2细胞和小鼠原代肝细胞中发挥抗炎特性。用OCA预处理并且然后暴露于促炎症刺激物的HepG2细胞显示TNF-αmRNA水平、环氧合酶-2(COX-2)诱导和TNF-α刺激的诱导型一氧化氮合酶(iNOS)表达降低50%至60%。同样,OCA处理的原代肝细胞显示出对促炎症刺激响应的iNOS和单核细胞趋化蛋白-1(MCP-1)基因表达的钝化诱导(40%至50%)(Wang 2008)。OCA对细胞迁移机制的影响尚未直接研究,但是OCA抑制了由IL-1β诱导的iNOS或COX-2的产生,并消除了药理学诱导的大鼠主动脉平滑肌细胞迁移(Li 2007)。在炎症性肠病的两种动物模型(DSS和三硝基苯磺酸)的肠组织(Gadaleta2011)和1型糖尿病大鼠模型的肾组织(Wang 2010)中,已经证实了OCA对炎症性浸润的类似抑制。因此,由OCA增强的抗炎作用的观察表明,OCA与非诺贝特组合的基因表达变化可以增强许多疾病条件下的炎症和炎症细胞迁移的抑制。
实例3:瘦素缺乏型ob/ob小鼠中饮食诱导的NASH
进行该实验以评估用OCA和阿托伐他汀单独治疗和组合治疗8周对雄性瘦素缺乏型ob/ob-NASH小鼠中纤维化阶段(活组织检查前与活组织检查后)的影响
动物、圈养和饮食
雄性Lepob/Lepob小鼠(5周龄)购自法国让维耶(JanVier)。在驯化和饮食诱导期过程中,将小鼠在12:12光照-黑暗循环下(在从3AM-3PM光照)在受控的温度条件(22℃±1℃;50%±10%相对湿度)下每笼五只分组圈养在定做橱柜中。在整个饮食诱导和研究期间,小鼠随意获取定制的NASH饮食(S8189,Ssniff公司,德国)(40%脂肪、40%糖类(20%果糖)和2%胆固醇)或常规啮齿动物食物(ob/ob-CHOW)(Altromin 1324,Brogaarden,丹麦)和自来水。在介入之前将动物保持饮食持续总共18周,并且在整个研究期间维持饮食。在术后恢复期间和整个研究期间,将动物单独圈养。
处理组
组1:Lepob/Lepob-NASH运载体
从第0至8周,每日一次以5mL/kg的体积向小鼠(n=10)口服给予运载体(0.5%CMC)。
组2:Lepob/Lepob-NASH OCA
在第0至8周,每日一次以30mg/kg剂量向小鼠(n=10)口服给予补充有OCA的运载体。
组3:Lepob/Lepob-NASH
在第0至8周,每日一次以10mg/kg剂量向小鼠(n=11)口服给予补充有阿托伐他汀的运载体。
组4:Lepob/Lepob-NASH OCA+阿托伐他汀
向小鼠(n=9)每日一次口服给予30mg/kg剂量的补充有OCA和10mg/kg剂量的补充有阿托伐他汀的组合的运载体。
分配到研究中、分层随机化和基线监测
饮食诱导15周(研究开始前3周)后,获得了评估纤维化和脂肪变性的肝进展的并且用于肝纤维化阶段评估的肝活组织检查。在第1周,根据肝纤维化阶段、脂肪变性得分和体重进行分层随机分为治疗组。
预活组织检查程序
在手术当天,将小鼠用100%氧气中的异氟烷(2%-3%)麻醉。在中线进行小的剖腹术并且暴露肝的左侧叶。将肝组织的锥形楔(约100mg)从该叶的远端部分切除,称重,并固定在4%多聚甲醛(PFA)中以用于组织学。使用双极电凝(ERBE VIO 100手术电刀)将肝的切割表面立即电凝。将肝返回腹腔并且将腹部缝合且用缝合器闭合皮肤。在手术当天,使小鼠接受温热的盐水(0.5ml)用于再水合。对于术后恢复,手术当天和术后第1天和第2天皮下给予卡洛芬(5mg/ml-0.01ml/10g)和恩诺沙星(5mg/ml-1ml/kg)。
用于评估脂肪变性和纤维化的肝水平的预筛选
用于组织学评定的肝活组织检查切片制备:在4%PFA中过夜储存之后,将肝活组织检查切片过夜浸润在自动Miles Scientific Tissue-TEK VIP-TEK组织处理器中的石蜡中并且随后包埋在石蜡块中。将来自五只不同动物的活检切片包埋在一个块上。然后,对这些块进行修整并且在Microm HM340E超薄切片机(赛默科技公司(Thermo Scientific))上切割两个3μm切片(一个用于天狼星红染色,并且一个用于H&E染色)。两个块被放置在一个载玻片上,从而得到每个载玻片代表10只不同动物的总计10个活检切片。将切片静置干燥过夜。如由Kleiner等人(2005)(参见下文)所概述(参见下文)进行分层且随机化为治疗组的纤维化阶段的评价。
基线和最终血浆生物标志物
在基线和治疗第8周的早上(7-8AM)获得用于测量甘油三酯的非禁食(进食)血浆水平的血液样品。在有意识的状态下从尾静脉(通过剪切)采集血液样品。在血液取样前约18小时给予最近的药物剂量。血液取样后重新饲喂小鼠。
终止和尸体剖检
在第8周在一种非禁食状态下终止动物。最近的药物剂量在终止之前约18小时给予并且动物在终止之前不会接受药物给药。将动物通过CO2/O2诱导并且在麻醉(异氟烷)过程中,打开腹腔并且获得心脏血液用于收集终末血浆。在尸体剖检之后,收集全肝并称重。从左侧叶切除活组织检查切片并且固定于4%PFA中用于组织学和生物化学分析。将中位叶分成片并在液氮中快速冷冻用于生物化学分析(TG)。随后将剩余的肝组织固定于4%PFA中用于以后的任选组织学。
肝组织处理
预研究活组织检查:在研究开始前大约三周,从左侧叶的远端部分切除肝组织的锥形楔(约100mg),称重并立即置于4%PFA中。
终末肝组织:在处理8周之后,收集全肝,称重并从左侧叶切除肝活组织检查切片且立即置于4%PFA(约150-200mg)中。若干片的中叶将在冷冻管(RNAseq)(约100mg)和FastPrep管中快速冷冻,并且用于TG(约100mg)和TC(约50mg)。
用于组织学的固定、包埋和切片:在4%PFA中过夜固定之后,将肝活检切片过夜浸润在自动Miles Scientific Tissue-TEK VIP-TEK组织处理器中的石蜡中并且随后包埋在石蜡块中。将来自五只不同动物的活检切片包埋在一个块中。对这些块进行修整并且在Microm HM340E超薄切片机(赛默科技公司)上切割两个3μm切片/块。将来自两个不同块的一个切片放置在一个物体载玻片上,给出如以上所概述的每个载玻片总共10个活检切片。
纤维化阶段
收集来自左侧叶的肝活组织检查前和活组织检查后组织用于通过使用Kleiner及其同事概述的临床标准来评估纤维化阶段(Design and validation of a histologicalscoring system for nonalcoholic fatty liver disease[非酒精性脂肪肝病的组织学评分系统的设计和验证],Kleiner等人,Hepatology[肝脏病学]41;2005),并重现于下表5中。图4描述了OCA和阿托伐他汀单独和联合对于纤维化评分的影响。OCA和阿托伐他汀的组合显示了降低纤维化得分的趋势,尽管不是以来自运载体的显著的方式(p值=0.09)。
表5

血浆甘油三酯
甘油三酯水平:将100μl血液收集到锂-肝素管中。分离血浆,并且将样品储存在-80摄氏度下直到分析。根据制造商的说明书使用自动分析仪Cobas C-111与商业试剂盒(罗氏诊断公司(Roche Diagnostics),德国)在单次测定中测量甘油三酯水平。如图5A和5B所示,OCA和阿托伐他汀的组合以显著统计学方式降低了甘油三酯水平。
实例4:肝细胞的夹层培养
将进行该实验以评估OCA与PPAR激动剂或他汀组合的作用,以确定它们改变人肝细胞中胶原合成的能力。
试剂和溶液
适合的细胞培养基包括的维莫斯氏(Waymouth's)MB-752/1、汉姆氏(Ham's)F12、RPMI 1640、杜氏改良的伊格尔氏培养基、威廉姆斯氏(Williams')培养基E、莱博维茨氏(Leibovitz's)L15、以及改良的徐氏(Chee's)培养基。IV型胶原酶、I型胶原、珀可、培养基、以及补充剂可以添加至培养基(例如,血清、抗生素、氨基酸、激素如DEX、胰岛素和生长因子),灌注缓冲液和其他溶液是可商购的或由可商购的材料制成。其他类型的胶原(II-IV型)、层粘连蛋白、纤连蛋白、以及硫酸肝素蛋白聚糖可以用于夹层肝细胞培养中。然而,已经表明,I型和IV型胶原优于纤连蛋白和层粘连蛋白。
肝细胞的分离
将使用两步骤原位胶原酶灌注方法来分离肝细胞。简言之,将从雌性路易斯大鼠分离肝细胞。将动物麻醉。首先用灌注缓冲液通过门静脉原位灌注肝。灌注液将在进入肝之前进行平衡。随后会用灌注缓冲液中的胶原酶灌注肝。然后将肝解剖并且转移至冰冷的灌注缓冲液中。肝包膜将被挑开,并且过滤所得细胞悬浮液。将通过离心收集细胞沉淀并且将这些细胞再悬浮。将珀可添加至悬浮液,并且使用一种珀可密度离心技术分离肝细胞。将混合物离心,并且将细胞沉淀用培养基洗涤两次。将通过台盼蓝排除法测定肝细胞活力。可替代地,可以使用冷冻保存的肝细胞来代替新鲜分离的肝细胞。
肝细胞的夹层培养
将分离的肝细胞在涂覆胶原的组织培养板上培养,并且维持在补充有血清、青霉素、链霉素、表皮生长因子、胰岛素、胰高血糖素、以及氢化可的松的培养基中。通过将I型胶原溶液与培养基进行混合来制备胶原胶凝溶液。将组织培养板涂覆该胶凝溶液并且在37℃下孵育以促进凝胶形成。肝细胞将以适当的密度接种,并维持在37℃下。每24小时更换一次培养基。
对于夹层系统,在培养1天之后将一种另外的胶原凝胶溶液分布在这些细胞上。将培养基小心地去除,以便确保胶原凝胶的第二层均匀地散布在整个板上。将这些培养板在37℃下孵育以便允许在更换培养基之前该第二凝胶层的凝胶化和附着。每日更换一次培养基。将培养基样品储存在-20℃下用于进一步分析。
在凝胶胶原的多个层之间培养的肝细胞维持三维立方体形状和类似于在体内所观察到的细胞骨架蛋白的分布。
胆小管网络形成的最优化
为了优化牛磺胆酸盐积聚和胆汁排泄,特定培养基如威廉姆斯氏培养基E和杜氏改良的伊格尔氏培养基可以用于夹层肝细胞培养。
测试样品
旨在用于研究的FXR激动剂是奥贝胆酸,还称为“OCA”和6-乙基鹅去氧胆酸(6-ECDCA)。
旨在用于研究的PPAR-α激动剂包括氯贝特、吉非罗齐、环丙贝特、苯扎贝特和非诺贝特中的一种或多种。
双重PPAR-α/δ激动剂是2-[2,6二甲基-4-[3-[4-(甲基硫代)苯基]-3-氧代-1(E)-丙烯基]苯氧基]-2-甲基丙酸。
旨在用于研究的PPARδ(δ)激动剂是GW501516。
旨在用于研究的他汀(HMG-CoA还原酶抑制剂)包括阿托伐他汀(Lipitor)、罗素伐他汀(Crestor)和辛伐他汀(Zocor)。
实例5:评估单独给予测试样品对脂质谱的影响
将评估5种测试样品,即FXR激动剂、PPAR-α激动剂、PPAR-δ激动剂、双重PPAR-α/δ激动剂(或可替代地,PPAR-α激动剂和PPAR-δ激动剂一起)以及他汀的潜力以确定改变人肝细胞中胆固醇合成和脂质谱的能力。在以3种不同浓度暴露于测试样品72小时后,将在夹层培养的人肝细胞(SCHH)中评估改变。每天将在培养基中新鲜制备给药溶液,并且将每天发生SCHH的给药持续3天。会使用一(1)批Transporter CertifiedTM人肝细胞(N=1)以24孔格式进行该实验。每个测试条件将在三(3)个孔中进行,以提供一式三份的数据(表示为平均值±标准偏差)。溶剂对照处理的板将用作对照并评估基线功能。在测试期结束时,将内标添加到各个孔中,随后添加提取试剂用于全面脂质特征分析。将样品在室温下摇动1小时并离心。将上清液在氮气下蒸发至干燥,重新悬浮并分析。
使用超高效液相色谱(Ultra-Performance Liquid Chromatography,UPLC)和高分辨率MS进行全面脂质特征分析。甲基叔丁基样品提取物将以ESI+和ESI-模式在UPLC-MS(Synapt G2Ion-Mobility QToF)仪器上进行分析以覆盖宽范围的脂质极性和化学成分。最初,UPLC将用于评估大型阵列(5000至8000)脂质(包括多种胆固醇酯)的复合作用。将进行比色分析以测量总胆固醇。基于丰度,大多数将是甘油磷脂,然而,大量的种类将被评估。将对特征进行评估以鉴定对个体脂质的潜在作用。可以使用测定保留时间、准确的质量和片段化处理来对标准品进行特异性脂质的鉴定。根据结果,可以鉴定特定的脂质或脂类用于在实例6和7中的评估。将在两批另外的Transporter CertifiedTM人类肝细胞(N=2)中重复验证性研究。
实例6:评估测试样品的双重组合对脂质谱的影响
将评估FXR激动剂与每个PPAR-α激动剂、PPAR-δ激动剂、PPAR-α和δ双重激动剂(或可替代地,FXR激动剂与PPAR α激动剂和PPAR δ激动剂)和/或他汀的组合改变人肝细胞中胆固醇合成和脂质谱的潜力。评估的具体组合将是:
·FXR激动剂与PPAR-α激动剂
·FXR激动剂与PPAR-δ激动剂
·FXR激动剂与PPAR-α和δ双重激动剂,和/或FXR激动剂与PPAR-α激动剂和PPAR-δ激动剂
·FXR激动剂与他汀
在以3种不同浓度暴露于测试样品72小时后,将在夹层培养的人肝细胞(SCHH)中评估改变。每天将在培养基中新鲜制备给药溶液,并且将每天发生SCHH的给药持续3天。将使用一(1)批Transporter CertifiedTM人肝细胞(N=1)以24孔格式进行该实验。每个测试条件将在三(3)个孔中进行,以提供一式三份的数据(表示为平均值±标准偏差)。将制备样品并分析全面脂质谱,如实例5所详述。将脂质谱和胆固醇合成的变化与实例2中单独给予的效果进行比较。
实例7:评估测试样品的三重组合对脂质谱的影响
将评估FXR激动剂、PPAR-α激动剂、PPAR-δ激动剂、PPAR-α和δ双重激动剂(或PPAR-α激动剂与PPAR-δ激动剂组合)和/或他汀的三重组合改变人肝细胞中的胆固醇合成和脂质谱的潜力。在以3种不同浓度暴露于测试样品72小时后,将在夹层培养的人肝细胞(SCHH)中评估改变。每天将在培养基中新鲜制备给药溶液,并且将每天发生SCHH的给药持续3天。会使用一(1)批Transporter CertifiedTM人肝细胞(N=1)以24孔格式进行该实验。每个测试条件将在三(3)个孔中进行,以提供一式三份的数据(表示为平均值±标准偏差)。将制备样品并分析全面脂质谱,如实例2所详述。将脂质谱和胆固醇合成的变化与实例2中组合给予的效果进行比较。评估的具体组合将是:
·FXR激动剂与PPAR-α激动剂和他汀
·FXR激动剂与PPAR-δ激动剂和他汀
·FXR激动剂与PPAR-α和δ双重激动剂(或者,可替代地,PPAR-α激动剂和PPAR-δ激动剂)和他汀
将制备样品并分析全面脂质谱,如实例4所详述。将脂质谱和胆固醇合成的变化与实例4和5中给予的组合的效果进行比较。
实例8:动物研究
动物
将动物在22℃下在12:12-小时光照-黑暗循环中单独圈养在标准笼子中。将允许雄性C57BL/6小鼠(杰克逊实验室公司(Jackson Laboratories),巴尔港(Bar Harbor),缅因州)随意获得富含脂肪(40%kcal,Primex部分氢化的植物油起酥油)、果糖(按wt计22%)和胆固醇(按wt计2%)的饮食(研究饮食公司(Research Diets),新不伦瑞克(NewBrunswick),新泽西州,目录号D09100301)。无果糖或胆固醇的低脂肪饮食(10%kcal,后文称作LFD)将会用作对照饮食(研究饮食公司,目录号D09100304)。使用这种经验证的LFD建立了一组对照小鼠,其维持“正常”肝表型用于与饲喂实验饮食的动物进行比较。
处理组
对照HFD:
对照LFD:
HFD+FXR激动剂:
HFD+PPARα(即,非诺贝特、吉非罗齐、苯扎贝特、或环丙贝特):
HFD+PPARδ(即,GW501516):
HFD+PPARα+PPARδ:
HFD+双重PPARα/δ(即,GFT505):
HFD+他汀(即,阿托伐他汀、辛伐他汀、罗素伐他汀)
HFD+FXR激动剂+PPARα:
HFD+FXR激动剂+PPARδ:
HFD+FXR激动剂+PPARα+PPARδ:
HFD+FXR激动剂+双重PPARα/δ:
HFD+FXR激动剂+他汀:
组织学和数字成像分析
在终止时,肝的右中叶和/或左侧叶(收获的每个叶的≥50%)将被切除并固定在10%中性缓冲的福尔马林中(在室温下至少7天)。将仔细切除肝组织以选择代表组织边缘和中心两者的相似大小的部分。肝组织将被石蜡包埋、切片(5μm)并封片。苏木精和伊红染色将用于形态学分析,并且马松三色(Masson’s trichrome)和天狼星红染色将用于评估肝纤维化。将由不知道该研究的病理学家进行组织病理学分析。通过使用Kleiner及其同事概述的标准,将对NAFLD和NASH进行评分。对于纤维化的定量评估,将以x20放大率通过使用ScanScope CS整张载玻片扫描系统(阿佩里奥公司(Aperio),比斯塔(Vista),加利福尼亚州)扫描整个天狼星红染色的切片。将提取图像并通过基于立方体的颜色方法使用Image-Pro分析仪软件(MediaCybernetics v.6.2,贝塞斯达,马里兰州)测量来自整个组织的天狼星红染色的胶原轮廓。将评估来自每个动物的三至四个代表性切片的总胶原染色(报告为总面积的%)(除了综合肝纤维化评估实验,其中将评估另外的切片)。所有组织学分析都将以盲法进行。
肝活组织检查
将用100%氧气中的异氟醚(2%-3%)麻醉小鼠。在中线左侧约0.5cm处进行小的剖腹术,并且暴露肝的左侧叶。将一块楔形肝组织(约50mg)从叶的远端部分切除,立即置于小瓶中,并在液氮中快速冷冻。将一块楔形吸收性明胶海绵(明胶海绵(GelFoam),辉瑞公司(Pfizer),纽约)插入肝的切割边缘。一旦达到止血(通常在1分钟内)并且明胶海绵很好地粘附到活组织检查部位,那么将肝返回到腹腔,缝合腹壁并钉合皮肤。在手术时,小鼠将接受单次注射的丁丙诺啡(0.05mg/kg,皮下)以控制术后疼痛。假手术的小鼠将进行相同的流程,除了在肝中没有进行切口。
血浆和血清分析
通过使用Olympus AU400e生物分析仪(奥林巴斯美国诊断公司(Olympus AmericaDiagnostics),中心谷,宾夕法尼亚州)测量血浆葡萄糖、甘油三酯、总胆固醇、丙氨酸转氨酶(ALT、以及天冬氨酸转氨酶(AST)水平。血浆样品将用PBS以1:10稀释,用于测量ALT和AST。将根据制造商的说明书使用可商购的电化学发光试剂盒(细观发现公司(Meso ScaleDiscovery),盖瑟斯堡(Gaithersburg),马里兰州)测量总血浆脂联素和空腹血清胰岛素。
总肝脂质和胶原含量的定量
将使用改编自Folch等人的试验方案从肝中提取总肝脂质。将冷冻的肝组织(约0.3g)在10ml的2:1氯仿-甲醇溶液中均质化。匀浆将使用无脂滤纸过滤,并集中到预先称重的15-ml玻璃小瓶中。将再添加5ml的2:1氯仿-甲醇,然后添加2.5ml的0.9%NaCl。将通过在1,800g、10℃下离心5分钟来分离脂质,丢弃水层,并且用氮气冲洗管子直至脂质颗粒干燥。将含有脂质沉淀的管子重新称重,并计算每克总肝所提取的总脂质。通过胶原(Quickzym公司,莱顿(Leiden),荷兰)的酸水解,通过比色测定羟脯氨酸残基来测量肝中的总胶原含量。
通过蛋白质印迹测定可提取的胶原-1α1蛋白
将从肝的左侧叶收集组织核心(50-100mg),将其在液氮中快速冷冻,并储存在-80℃下直至被处理。组织将在含有蛋白酶抑制剂的裂解缓冲液中均质化。将用一种BCA蛋白质测定试剂盒(皮尔斯公司(Pierce),罗克福德(Rockford),伊利诺伊州)测量澄清的上清液的蛋白质浓度。肝组织裂解物(约50μg)将在还原的4%-12%Nupage凝胶(生命科技公司(Life Technologies),卡尔斯巴德(Carlsbad),加利福尼亚州)上分离并转移到硝化纤维素膜上。在50-kDa和60-kDa标记物之间切割膜,并用5%Blotto阻断。上半部分将用抗胶原-1%1(1:1,000;目录号NBP1-30054;诺维斯生物制剂公司(Novus Biologicals),利特尔顿(Littleton),科罗拉多州))探测,该抗胶原-1%1探测胶原-1%1蛋白质的COOH-末端端肽部分。对于标准化,将用抗甘油醛-3-磷酸脱氢酶(GAPDH,1:7,500;目录号3683;细胞信号传导技术公司(Cell Signaling Technology),丹佛斯(Danvers),马萨诸塞州)探测下半部分。在与辣根过氧化物酶抗兔抗体孵育后,将用增强的化学发光(赛默科技公司(ThermoScientific),罗克福德(Rockford),伊利诺伊州)检测蛋白质表达,并且将用FluorChem系统(细胞生物科学公司(Cell Biosciences),圣克拉拉(Santa Clara),加利福利亚州)进行光密度测定。胶原-1α1的光密度测定将包括140-kDa的成熟蛋白以及对应于于糖基化形式或部分处理的胶原-1α1蛋白的稍大的带。
肝基因表达变化
用6-mm的组织取芯工具或通过活组织检查法收获来自肝左侧叶的组织样品,将其在液氮中快速冷冻,并储存在-80℃下直至被处理。将通过使用TRI试剂(生命科技公司)提取来自肝样品的总RNA(约50-150mg),并且然后用Qiagen RNeasy Plus Mini试剂盒(凯杰公司(Qiagen),巴伦西亚,加利福尼亚)进一步纯化。将在生物分析仪2100(安捷伦科技公司(Agilent Technologies),圣克拉拉,加利福尼亚州)上通过使用安捷伦6000纳米试剂盒来确定RNA完整性。将通过使用高容量cDNA逆转录试剂盒(High Capacity cDNA ReverseTranscription Kit)(生命科技公司)制备cDNA。将通过在ABI Prism 7900HT仪器(应用生物系统公司(Applied Biosystems),福斯特城,加利福尼亚州)上的按需TaqMan基因表达测定和通用预混合物(生命科技公司)来证实基因表达的变化。将通过使用用于标准化的肽基脯氨酰异构酶A(Ppia)和Gapdh的比较阈值循环(CT)方法来计算基因表达的变化。对于基因阵列,通过使用ABI Prism 7900HT快速实时PCR系统(应用生物系统公司),将cDNA样品在小鼠纤维化RT2分析器PCR阵列(PAMM-120C,RT2SYBR Green/ROX qPCR预混合物;SABiosciences公司)上运行。将通过使用DataAssist v3.0软件(应用生物系统公司/生命科技公司)的比较CT方法来计算阵列上基因表达的变化。在包含在小鼠纤维化RT2分析器PCR阵列中的五个管家基因中,根据由DataAssist v3.0软件计算的稳定性评分,次黄嘌呤磷酸核糖基转移酶1(Hprt)和Gapdh具有最稳定的表达。将选择的内源性对照基因的平均值用作标准化因子来计算每个基因的相对表达。为了确认通过使用纤维化阵列获得的结果,将进行TaqMan基因表达测定用于选择由阵列确定的将被上调、将被下调或将不变的基因。
实例9:临床试验
在患有非硬变性、非酒精性脂肪性肝炎的患者中进行多中心、双盲、安慰剂对照、平行组、随机的临床试验,以评估口服给予(每日25mg)奥贝胆酸或安慰剂持续72周的治疗。使用计算机生成的集中管理程序,将患者随机地以1:1分配,通过临床中心和糖尿病状态分层。主要结果测量是中央评分的肝组织学的改善,该中央评分的肝组织学定义为非酒精性脂肪肝病活性得分降低至少2分而从基线至治疗结束时无纤维化恶化。测量24周时丙氨酸氨基转移酶的变化:丙氨酸氨基转移酶的相对变化为-24%,95%CI为-45至-3。
研究设计和参与者
根据以下纳入标准使患者参加该研究:筛选时为18岁或以上,基于在随机分组前90天或以内获得的肝活组织检查的确定性或边界性非酒精性脂肪性肝炎的组织学证据,以及组织学非酒精性脂肪肝病(NAFLD)活性评分为4分或以上,其中在评分的每个组分中评分为1分或以上(脂肪变性评分为0-3分、鼓胀为0-2分、以及小叶炎症为0-3分)。在参加现场进行针对参加目的的活检分级和分期。排除标准包括肝硬化、其他原因的肝病、大量饮酒(女性>20g/天或男性>30g/天)或其他混杂病症(见下文)的存在。
随机分组和掩蔽
将满足合格性标准的患者随机分配(1:1)到口服奥贝胆酸(25mg每日一次)或安慰剂。在标记有代号的相同容器中以相同片剂提供奥贝胆酸和安慰剂。患者、研究员、临床现场工作人员和病理学家对治疗任务为掩蔽的。
流程
随机分组后,患者在第2、4和12周返回进行研究回访,然后每12周返回直至第72周治疗完成,然后以后每24周返回。在这些回访中获得血液样品用于常规生化测试并评估脂质、葡萄糖和胰岛素的空腹浓度。在初始评估和指定的临时时间处测量体重、身高、以及腰围和臀围。所有患者均接受有关健康饮食习惯、体重减轻、运动、以及对高血压、高胆固醇血症和糖尿病的管理的标准建议。
基线和治疗结束的肝活检被集中评估为NAFLD活性评分的对每个组分的一致评分的组,被确定纤维化阶段,并且被分配非酒精性脂肪性肝炎、边界性非酒精性脂肪性肝炎或不是非酒精性脂肪性肝炎的诊断。
纳入和排除标准
满足任何以下排除标准的患者被认为无资格参加:1)筛选前1年内连续3个月以上时段内大量饮酒的现况或病史(大量饮酒定义为平均女性20g/天以上并且男性30g/天以上);2)不能根据现场研究员的判断可靠地量化饮酒;3)在随机分组前一年内,使用历史上与NAFLD相关的药物超过2周;4)先前或计划的减肥手术或方法;5)参加前60天内定义为80.3mmol/mol或更高的HbA1c的不受控制的糖尿病;6)肝活组织检查时肝硬化的存在,7)血小板计数<100×109/L;8)如通过任何以下异常的存在所定义的肝代偿不全的临床证据:血清白蛋白小于32g/L,INR大于1.3,直接胆红素大于22.2μmol/L,或食管静脉曲张、腹水、或肝性脑病的病史;9)其他形式的慢性肝病的证据:如通过乙型肝炎表面抗原(HBsAg)的存在所定义的乙型肝炎、如通过丙型肝炎病毒(HCV)RNA或阳性丙型肝炎抗体(抗HCV)的存在所定义的丙型肝炎、如通过相容的肝组织学所定义的进行性自身免疫性肝病的证据、如通过至少2个标准(主要基于碱性磷酸酶升高的胆汁淤积的生物化学证据、抗线粒体抗体[AMA]的存在、以及非化脓性破坏性胆管炎和小叶间胆管破坏的组织学证据)的存在所定义的原发性胆汁性肝硬化、原发性硬化性胆管炎、如通过低于正常和相容的肝组织学极限的血浆铜蓝蛋白所定义的威尔森氏病(Wilson’s disease)、如通过肝组织学中的诊断特征所定义的α-1-抗胰蛋白酶(A1AT)缺乏(通过α-1抗胰蛋白酶水平低于正常值而确认,由现场研究员自行排除)、如通过肝活组织检查时3+或4+可染色铁的存在所定义的血色素沉着病或铁超负荷的病史、如基于典型暴露和病史所定义的药物诱导性肝病、已知的胆管阻塞、疑似或证实的肝癌、或除了NASH以外的任何其他类型的肝病;10)大于300U/L的血清丙氨酸氨基转移酶(ALT);11)血清肌酸酐为176.8μmol/L或以上;12)不能安全地获得肝活组织检查;13)胆汁转移的病史;14)已知阳性的人类免疫缺陷病毒(HIV)感染;15)预期寿命可能低于5年的活跃的、严重的内科疾病;16)筛选前一年内,包括吸入药物或注射药物的活性物质滥用;17)妊娠、计划妊娠、可能妊娠并且不愿意在试验期间使用有效的生育控制、或哺乳;18)在随机分组前30天内参与IND试验;19)在现场研究员的意见中会阻碍顺从或妨碍完成该研究的任何其他病症;或20)未能给出知情同意书。
统计学分析
使用曼特尔-亨塞尔(Mantel-Haenszel)测试来分析主要结果和二进制次要结果;使用将连续结果从基线至72周的变化与治疗组并与结果的基线值相关联的方差分析(ANCOVA)模型分析连续次要结果。用SAS(SAS协会2011,基础SAS 9.3程序指南)和Stata(StataCorp 2013,Stata统计学软件:发行13)进行统计学分析。
结果
主要结果测量是中央评分的肝组织学的改善,该中央评分的肝组织学的改善被定义为NAFLD活性评分降低至少2分,而从基线至治疗结束时无纤维化恶化。纤维化的恶化被定义为该阶段内的任何数值增加。次要组织学结果包括非酒精性脂肪性肝炎的消退,NAFLD活动评分的变化,以及肝细胞鼓胀、脂肪变性、小叶和门静脉炎症、和纤维化的个体评分变化。纤维化的改善被定义为该阶段内的任何数值减少。出于分析的目的,纤维化阶段1a、1b和1c被认为是第1阶段。其他次要结果包括血清氨基转移酶和γ-谷氨酰转肽酶浓度从基线至72周的变化、评估胰岛素耐受性(HOMA-IR)的空腹体内平衡模型、人体测量(体重、体质指数、腰臀比、腰围)以及健康相关的生活质量评分。
实例10:数据分析
对实例9中获得的数据进行亚分析以评估他汀对低密度脂蛋白胆固醇(LDL-C)水平的影响。这些二级分析的目的是确定OCA与安慰剂在具有更严重NASH的患者亚组中的作用,并评估伴随他汀使用对血清LDL胆固醇的作用。如下以三组评估受试者数据:
·A组(n=64,无他汀)包括奥贝胆酸(OCA)治疗组的受试者,这些受试者在基线时(第0天)不服用他汀以及在整个研究过程中不服用他汀直到并包括第72周。
·B组(n=47,基线他汀)包括OCA治疗组的受试者,这些受试者在基线时服用他汀并且在研究期间持续服用他汀直至并包括第72周。
·C组(n=23,新他汀)包括OCA治疗组的受试者,这些受试者在基线时不服用他汀但在基线之后的一个时间开始他汀治疗直至并包括第72周。对A组、B组和C组中的OCA治疗的受试者进行以下计算。
·下面列出的平均值和中值特征将在基线和第72周进行评估。
o实验室值:LDL-C、高密度脂蛋白胆固醇(HDL-C)、丙氨酸和天冬氨酸氨基转移酶、γ谷氨酰转移酶
o年龄
o性别:男性百分比、女性百分比
o糖尿病百分比
o组织学:脂肪变性、鼓胀、炎症、纤维化
·将评估以上特征在第72周从基线的的平均值和中值百分比变化。结果
在基线时服用他汀的患者的OCA治疗期间,LDL胆固醇升高,但水平不超过安慰剂处理的不服用他汀的患者的水平。在OCA治疗期间,服用他汀将LDL逆转为低于预OCA基线水平。如图8所示,在OCA治疗期间通过开始他汀疗法,OCA相关的LDL增加似乎被逆转。
Claims (5)
1.一种药物组合物,所述药物组合物包含
FXR激动剂和苯扎贝特的双向组合;以及
任选地一种或多种药学上可接受的载体,
其中所述FXR激动剂是具有式(1)的化合物:

其中所述药物组合物不包含除了所述FXR激动剂和苯扎贝特以外的任何治疗剂。
2.权利要求1所述的药物组合物在制造用于治疗或预防FXR介导的疾病或病症的药物中的用途,所述FXR介导的疾病或病症选自原发性胆汁性肝硬化(PBC)、原发性硬化性胆管炎(PSC)、门静脉高压、胆汁酸腹泻、慢性肝病、非酒精性脂肪肝病(NAFLD)、非酒精性脂肪性肝炎(NASH)、丙型肝炎感染、酒精性肝病、由于进行性纤维化引起的肝损伤、肝纤维化、心血管疾病、以及高血脂症。
3.权利要求1所述的药物组合物在制造用于治疗或预防FXR介导的疾病或病症的药物中的用途,所述FXR介导的疾病或病症选自抗性原发性胆汁性肝硬化;存在相关的肝功能测试升高和高血脂症的原发性胆汁性肝硬化;原发性硬化性胆管炎;非酒精诱发性脂肪性肝炎;与乙型肝炎、丙型肝炎或酒精相关的慢性肝病;高血脂症,其中所述高血脂症是具有或不具有遗传成分的原发性高血脂症;以及与冠状动脉疾病、脑血管动脉疾病、周围血管疾病、主动脉瘤、或颈动脉粥样硬化相关的高血脂症。
4.权利要求1所述的药物组合物在制造用于抑制或逆转纤维化的药物中的用途。
5.如权利要求4所述的用途,其中所述纤维化选自肝纤维化、肾纤维化和肠纤维化。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201562113134P | 2015-02-06 | 2015-02-06 | |
| US62/113,134 | 2015-02-06 | ||
| PCT/US2016/016694 WO2016127019A2 (en) | 2015-02-06 | 2016-02-05 | Pharmaceutical compositions for combination therapy |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN107405325A CN107405325A (zh) | 2017-11-28 |
| CN107405325B true CN107405325B (zh) | 2021-11-12 |
Family
ID=56564878
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201680015586.6A Active CN107405325B (zh) | 2015-02-06 | 2016-02-05 | 用于组合疗法的药物组合物 |
Country Status (31)
| Country | Link |
|---|---|
| US (3) | US11311557B2 (zh) |
| EP (2) | EP4035665A1 (zh) |
| JP (2) | JP7306791B2 (zh) |
| KR (1) | KR20170115071A (zh) |
| CN (1) | CN107405325B (zh) |
| AR (1) | AR103624A1 (zh) |
| AU (2) | AU2016215179B2 (zh) |
| BR (1) | BR112017016766B1 (zh) |
| CA (1) | CA2976056C (zh) |
| CL (3) | CL2017001983A1 (zh) |
| CO (1) | CO2017007959A2 (zh) |
| CR (1) | CR20170356A (zh) |
| DK (1) | DK3253382T3 (zh) |
| EA (1) | EA036404B1 (zh) |
| EC (1) | ECSP17057848A (zh) |
| ES (1) | ES2905872T3 (zh) |
| GT (1) | GT201700174A (zh) |
| HK (1) | HK1246191A1 (zh) |
| IL (2) | IL253719B (zh) |
| MA (1) | MA40814A1 (zh) |
| MX (1) | MX2017010131A (zh) |
| NI (1) | NI201700100A (zh) |
| PE (1) | PE20180027A1 (zh) |
| PH (1) | PH12017501384A1 (zh) |
| SG (1) | SG11201706347SA (zh) |
| SV (1) | SV2017005512A (zh) |
| TN (1) | TN2017000344A1 (zh) |
| TW (1) | TW201628625A (zh) |
| UA (1) | UA125744C2 (zh) |
| WO (1) | WO2016127019A2 (zh) |
| ZA (1) | ZA201706007B (zh) |
Families Citing this family (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| PE20180027A1 (es) | 2015-02-06 | 2018-01-09 | Intercept Pharmaceuticals Inc | Composiciones farmaceuticas para terapia combinada |
| AU2016246524B2 (en) | 2015-04-07 | 2021-04-01 | Intercept Pharmaceuticals, Inc. | Pharmaceutical compositions for combination therapy |
| KR20180052756A (ko) * | 2015-09-24 | 2018-05-18 | 인터셉트 파마슈티컬즈, 인크. | 담즙산 유도체 제조를 위한 방법 및 중간체 |
| US12053445B2 (en) | 2016-03-31 | 2024-08-06 | Genfit | Methods of treatment of cholestatic diseases |
| US11185519B2 (en) * | 2016-03-31 | 2021-11-30 | Genfit | Methods of treatment of cholestatic diseases |
| US11331292B2 (en) | 2016-03-31 | 2022-05-17 | Genfit | Methods of treatment of cholestatic diseases |
| WO2018104916A1 (en) | 2016-12-09 | 2018-06-14 | Cadila Healthcare Limited | Treatment for primary biliary cholangitis |
| ES2959842T3 (es) * | 2017-02-21 | 2024-02-28 | Genfit | Combinación de un agonista de PPAR con un agonista de FXR |
| US20190076500A1 (en) * | 2017-09-13 | 2019-03-14 | Novartis Ag | Combinations comprising fxr agonists |
| GB201812382D0 (en) | 2018-07-30 | 2018-09-12 | Nzp Uk Ltd | Compounds |
| WO2020208205A1 (en) * | 2019-04-10 | 2020-10-15 | Genfit | Combination therapy comprising compounds of formula (i) and glp-1 receptor agonists |
| US20220226350A1 (en) * | 2019-05-30 | 2022-07-21 | Intercept Pharmaceuticals, Inc. | Pharmaceutical compositions comprising a fxr agonist and a fibrate for use in the treatment of cholestatic liver disease |
| CN110287493B (zh) * | 2019-06-28 | 2023-04-18 | 中国科学技术信息研究所 | 风险短语识别方法、装置、电子设备及存储介质 |
| EP3999101A1 (en) * | 2019-07-18 | 2022-05-25 | ENYO Pharma | Method for decreasing adverse-effects of interferon |
| WO2021055725A1 (en) * | 2019-09-20 | 2021-03-25 | Reneo Pharmaceuticals, Inc. | Use of a ppar-delta agonist in the treatment of kidney disease |
| US20220387467A1 (en) * | 2019-11-22 | 2022-12-08 | Avolynt | Use of sglt2 inhibitors to treat primary billiary cholangitis |
| CN111690731B (zh) * | 2020-05-22 | 2021-09-28 | 河南大学 | Fxr激动剂在治疗肝性脑病中的应用 |
| CN112885471B (zh) * | 2021-03-12 | 2023-01-24 | 上海中医药大学附属岳阳中西医结合医院 | 基于贝叶斯网络最大熵自学习扩展集对分析的银屑病疗效评价系统 |
| WO2023147141A1 (en) * | 2022-01-28 | 2023-08-03 | Intercept Pharmaceuticals, Inc. | Combination therapy |
| WO2024132001A1 (en) * | 2022-12-21 | 2024-06-27 | Charles University, Faculty Of Pharmacy In Hradec Kralove | Multitarget nuclear receptor ligands based on 2-(4-(quinolin-2-yloxy)phenoxy)propanoic acid and 2-(4-(quinoxalin-2-yloxy)phenoxy)propanoic acid for the treatment of metabolic and liver diseases |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1248916A (zh) * | 1997-01-24 | 2000-03-29 | 加利福尼亚大学董事会 | FXR、PPARα和LXRα激活剂恢复屏障功能、促进表皮分化和抑制增生的用途 |
| WO2000049992A2 (en) * | 1999-02-25 | 2000-08-31 | The Regents Of The University Of California | USE OF FXR, PPARα AND LXRα ACTIVATORS TO TREAT ACNE/ACNEIFORM CONDITIONS |
| US20080096921A1 (en) * | 2006-10-24 | 2008-04-24 | Smithkline Beecham Corporation | Farnesoid x receptor agonists |
| CN101522703A (zh) * | 2006-06-27 | 2009-09-02 | 英特塞普特医药品公司 | 胆酸派生物作为fxr配基预防或治疗fxr-介导的疾病或状况 |
Family Cites Families (61)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB860303A (en) | 1958-06-20 | 1961-02-01 | Ici Ltd | Pharmaceutical compositions comprising ª‡-aryloxy-aliphatic carboxylic acids and/or ª |
| US3262580A (en) | 1964-06-23 | 1966-07-26 | Mcdowell Wellman Eng Co | Slewable gantry crane |
| FR1498459A (zh) | 1965-07-30 | 1968-01-08 | ||
| US3674836A (en) | 1968-05-21 | 1972-07-04 | Parke Davis & Co | 2,2-dimethyl-{11 -aryloxy-alkanoic acids and salts and esters thereof |
| US4058552A (en) | 1969-01-31 | 1977-11-15 | Orchimed Sa | Esters of p-carbonylphenoxy-isobutyric acids |
| FI52570C (fi) | 1969-04-16 | 1977-10-10 | Sumitomo Chemical Co | Menetelmä veren kolesteroli- tai lipoidipitoisuutta alentavien fenoxia lifaattisten karboksyylihappoyhdisteiden ja -esteriyhdisteiden valmist amiseksi. |
| AT296986B (de) | 1969-08-13 | 1972-03-10 | Merz & Co | Verfahren zur Herstellung von neuen α-Halogenphenoxy-isobutyroyl-β-nicotinoyl-glykolen |
| DE2230383C3 (de) | 1971-10-01 | 1981-12-03 | Boehringer Mannheim Gmbh, 6800 Mannheim | Phenoxyalkylcarbonsäurederivate und Verfahren zur Herstellung derselben |
| JPS5118954B2 (zh) | 1972-02-04 | 1976-06-14 | ||
| US3948973A (en) | 1972-08-29 | 1976-04-06 | Sterling Drug Inc. | Halocyclopropyl substituted phenoxyalkanoic acids |
| DE2308826C3 (de) | 1973-02-22 | 1980-03-27 | Ludwig Merckle Kg Chem. Pharm. Fabrik, 7902 Blaubeuren | Phenoxyalkancarbonsäureester von Oxyalkyltheophyllinen, Verfahren zu ihrer Herstellung und Arzneimittel |
| FR2244511B1 (zh) | 1973-07-05 | 1977-07-15 | Roussel Uclaf | |
| ES8101585A1 (es) | 1980-02-15 | 1980-12-16 | Especialidades Farmaco Terape | Procedimiento de obtencion de un nuevo compuesto antiateros-clerotico |
| US5023252A (en) | 1985-12-04 | 1991-06-11 | Conrex Pharmaceutical Corporation | Transdermal and trans-membrane delivery of drugs |
| CA2110095A1 (en) | 1992-12-08 | 1994-06-09 | Teruo Komoto | Arylamide derivatives |
| TWI238064B (en) | 1995-06-20 | 2005-08-21 | Takeda Chemical Industries Ltd | A pharmaceutical composition for prophylaxis and treatment of diabetes |
| HUP0003103A3 (en) * | 1997-08-29 | 2002-03-28 | Pfizer Prod Inc | Combination therapy comprising amlodipine and a statin compound |
| US6054453A (en) | 1997-10-27 | 2000-04-25 | Redd's Research Foundation | Tricyclic compounds and their use in medicine process for their preparation and pharmaceutical compositions containing them |
| US20030109467A1 (en) | 2001-11-15 | 2003-06-12 | Isis Pharmaceuticals Inc. | Antisense modulation of human FXR expression |
| EP1140036A2 (en) | 1998-12-18 | 2001-10-10 | Abbott Laboratories | Novel formulations comprising lipid-regulating agents |
| EP1140079B1 (en) | 1998-12-23 | 2009-06-03 | Glaxo Group Limited | Assays for ligands for nuclear receptors |
| WO2000040965A1 (en) | 1999-01-07 | 2000-07-13 | Tularik, Inc. | Fxr receptor-mediated modulation of cholesterol metabolism |
| US20020132223A1 (en) | 1999-03-26 | 2002-09-19 | City Of Hope | Methods for modulating activity of the FXR nuclear receptor |
| NZ522036A (en) | 2000-04-19 | 2004-04-30 | Borody Thomas J | Compositions and therapies for hyperlipidaemia-associated disorders |
| AU2001288623A1 (en) | 2000-09-05 | 2002-03-22 | Tularik, Inc. | Fxr modulators |
| WO2002024632A2 (en) | 2000-09-18 | 2002-03-28 | Glaxo Group Limited | Substituted aminopropoxyaryl derivatives useful as agonists for lxr |
| JP4021327B2 (ja) | 2001-03-12 | 2007-12-12 | インターセプト ファーマスーティカル インコーポレイテッド | Fxrに対する作用薬としてのステロイド |
| EP1285914B1 (en) | 2001-08-13 | 2007-12-19 | PheneX Pharmaceuticals AG | Nr1h4 nuclear receptor binding compounds |
| US7259186B2 (en) | 2002-12-17 | 2007-08-21 | Abbott Laboratories | Salts of fenofibric acid and pharmaceutical formulations thereof |
| EP1568706A1 (en) | 2004-02-26 | 2005-08-31 | Intercept Pharmaceuticals, Inc. | Novel steroid agonist for FXR |
| GB0405349D0 (en) | 2004-03-10 | 2004-04-21 | Univ Birmingham | Cancer therapy and medicaments therefor |
| HUE024952T2 (en) | 2004-03-12 | 2016-02-29 | Intercept Pharmaceuticals Inc | Treatment of fibrosis using FXR ligands |
| PE20060315A1 (es) * | 2004-05-24 | 2006-05-15 | Irm Llc | Compuestos de tiazol como moduladores de ppar |
| US20060252670A1 (en) * | 2004-10-14 | 2006-11-09 | Intercept Pharmaceuticals Inc. | Method of reducing drug-induced adverse side effects in a patient |
| US20090131395A1 (en) | 2005-05-05 | 2009-05-21 | Microbia, Inc. | Biphenylazetidinone cholesterol absorption inhibitors |
| ITMI20050912A1 (it) | 2005-05-19 | 2006-11-20 | Erregierre Spa | Processo di preparazione di acidi 3-a-ya(b)-diidrossi-6-a(b)-alchil-5b-colanici |
| WO2007095174A2 (en) | 2006-02-14 | 2007-08-23 | Intercept Pharmaceuticals, Inc. | Bile acid derivatives as fxr ligands for the prevention or treatment of fxr-mediated diseases or conditions |
| US20070197606A1 (en) | 2006-02-22 | 2007-08-23 | Burczynski Frank J | Use of ppar agonists as anti-oxidants |
| AU2007230989A1 (en) * | 2006-03-22 | 2007-10-04 | President And Fellows Of Harvard College | Methods and compositions for treating hypercholesterolemia and atherosclerosis |
| EA014720B1 (ru) | 2006-05-24 | 2011-02-28 | Эли Лилли Энд Компани | Соединения и способы модуляции fxr |
| AU2007267606A1 (en) | 2006-05-24 | 2007-12-06 | Eli Lilly And Company | FXR agonists |
| AU2006346853A1 (en) | 2006-08-04 | 2008-02-07 | Aska Pharmaceutical Co., Ltd. | Drug formulation containing fibrate medicament and process for producing the same |
| EP1886685A1 (en) | 2006-08-11 | 2008-02-13 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods, uses and compositions for modulating replication of hcv through the farnesoid x receptor (fxr) activation or inhibition |
| EP1894928A1 (en) | 2006-08-29 | 2008-03-05 | PheneX Pharmaceuticals AG | Heterocyclic fxr binding compounds |
| EP1894924A1 (en) | 2006-08-29 | 2008-03-05 | Phenex Pharmaceuticals AG | Heterocyclic FXR binding compounds |
| WO2008039829A2 (en) | 2006-09-26 | 2008-04-03 | Ironwood Pharmaceuticals, Inc. | Diphenylheterocycle cholesterol absorption inhibitors |
| EP2121621B1 (en) | 2006-12-08 | 2014-05-07 | Exelixis Patent Company LLC | Lxr and fxr modulators |
| US20080300235A1 (en) | 2007-06-01 | 2008-12-04 | Wyeth | FXR Agonists for Reducing LOX-1 Expression |
| US20080299118A1 (en) | 2007-06-01 | 2008-12-04 | Wyeth | FXR Agonists for the Treatment of Malignancies |
| TW200906823A (en) | 2007-07-16 | 2009-02-16 | Lilly Co Eli | Compounds and methods for modulating FXR |
| US8338628B2 (en) | 2007-08-28 | 2012-12-25 | City Of Hope | Method of synthesizing alkylated bile acid derivatives |
| US20090163474A1 (en) | 2007-10-19 | 2009-06-25 | Wyeth | FXR Agonists for the Treatment of Nonalcoholic Fatty Liver and Cholesterol Gallstone Diseases |
| US20090215748A1 (en) | 2007-12-20 | 2009-08-27 | Wyeth | FXR agonists for treating vitamin D associated diseases |
| EP2110374A1 (en) | 2008-04-18 | 2009-10-21 | Merck Sante | Benzofurane, benzothiophene, benzothiazol derivatives as FXR modulators |
| EP2289883A1 (en) | 2009-08-19 | 2011-03-02 | Phenex Pharmaceuticals AG | Novel FXR (NR1H4) binding and activity modulating compounds |
| TR201810393T4 (tr) * | 2009-11-26 | 2018-08-27 | Genfit | Karaciğer rahatıszlıklarının tedavisi için 1,3-difenilprop-2-en-1-on türevlerinin kullanımı. |
| SG11201408501UA (en) | 2012-06-19 | 2015-01-29 | Intercept Pharmaceuticals Inc | Preparation, uses and solid forms of obeticholic acid |
| EP2912013B1 (en) | 2012-10-26 | 2017-10-11 | Intercept Pharmaceuticals, Inc. | Process for preparing bile acid derivatives |
| SMT201800326T1 (it) * | 2013-05-14 | 2018-07-17 | Intercept Pharmaceuticals Inc | Derivati 11-idrossil-6-sostituiti di acidi biliari e relativi coniugati amminoacidici come modulatori del recettore farnesoide x |
| US10077268B2 (en) * | 2014-03-13 | 2018-09-18 | Salk Institute For Biological Studies | FXR agonists and methods for making and using |
| PE20180027A1 (es) | 2015-02-06 | 2018-01-09 | Intercept Pharmaceuticals Inc | Composiciones farmaceuticas para terapia combinada |
-
2016
- 2016-02-05 PE PE2017001322A patent/PE20180027A1/es unknown
- 2016-02-05 ES ES16747312T patent/ES2905872T3/es active Active
- 2016-02-05 CA CA2976056A patent/CA2976056C/en active Active
- 2016-02-05 EP EP21208443.8A patent/EP4035665A1/en active Pending
- 2016-02-05 WO PCT/US2016/016694 patent/WO2016127019A2/en active Application Filing
- 2016-02-05 JP JP2017541334A patent/JP7306791B2/ja active Active
- 2016-02-05 AU AU2016215179A patent/AU2016215179B2/en active Active
- 2016-02-05 SG SG11201706347SA patent/SG11201706347SA/en unknown
- 2016-02-05 KR KR1020177024607A patent/KR20170115071A/ko not_active Application Discontinuation
- 2016-02-05 DK DK16747312.3T patent/DK3253382T3/da active
- 2016-02-05 MA MA40814A patent/MA40814A1/fr unknown
- 2016-02-05 CR CR20170356A patent/CR20170356A/es unknown
- 2016-02-05 TW TW105104019A patent/TW201628625A/zh unknown
- 2016-02-05 BR BR112017016766-2A patent/BR112017016766B1/pt active IP Right Grant
- 2016-02-05 CN CN201680015586.6A patent/CN107405325B/zh active Active
- 2016-02-05 TN TNP/2017/000344A patent/TN2017000344A1/en unknown
- 2016-02-05 EA EA201791770A patent/EA036404B1/ru unknown
- 2016-02-05 EP EP16747312.3A patent/EP3253382B1/en not_active Revoked
- 2016-02-05 AR ARP160100332A patent/AR103624A1/es unknown
- 2016-02-05 US US15/548,537 patent/US11311557B2/en active Active
- 2016-02-05 MX MX2017010131A patent/MX2017010131A/es unknown
- 2016-02-05 UA UAA201708864A patent/UA125744C2/uk unknown
-
2017
- 2017-07-30 IL IL253719A patent/IL253719B/en unknown
- 2017-08-01 PH PH12017501384A patent/PH12017501384A1/en unknown
- 2017-08-03 CL CL2017001983A patent/CL2017001983A1/es unknown
- 2017-08-04 NI NI201700100A patent/NI201700100A/es unknown
- 2017-08-04 GT GT201700174A patent/GT201700174A/es unknown
- 2017-08-04 CO CONC2017/0007959A patent/CO2017007959A2/es unknown
- 2017-08-07 SV SV2017005512A patent/SV2017005512A/es unknown
- 2017-08-31 EC ECIEPI201757848A patent/ECSP17057848A/es unknown
- 2017-09-04 ZA ZA2017/06007A patent/ZA201706007B/en unknown
-
2018
- 2018-04-19 CL CL2018001006A patent/CL2018001006A1/es unknown
- 2018-05-04 HK HK18105766.1A patent/HK1246191A1/zh unknown
- 2018-12-14 CL CL2018003604A patent/CL2018003604A1/es unknown
-
2021
- 2021-02-17 AU AU2021201015A patent/AU2021201015A1/en not_active Abandoned
- 2021-10-11 JP JP2021166629A patent/JP2022000483A/ja not_active Withdrawn
-
2022
- 2022-02-23 IL IL290823A patent/IL290823A/en unknown
- 2022-03-21 US US17/700,317 patent/US20220280533A1/en active Pending
- 2022-03-21 US US17/700,392 patent/US20220280534A1/en active Pending
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1248916A (zh) * | 1997-01-24 | 2000-03-29 | 加利福尼亚大学董事会 | FXR、PPARα和LXRα激活剂恢复屏障功能、促进表皮分化和抑制增生的用途 |
| WO2000049992A2 (en) * | 1999-02-25 | 2000-08-31 | The Regents Of The University Of California | USE OF FXR, PPARα AND LXRα ACTIVATORS TO TREAT ACNE/ACNEIFORM CONDITIONS |
| CN101522703A (zh) * | 2006-06-27 | 2009-09-02 | 英特塞普特医药品公司 | 胆酸派生物作为fxr配基预防或治疗fxr-介导的疾病或状况 |
| US20080096921A1 (en) * | 2006-10-24 | 2008-04-24 | Smithkline Beecham Corporation | Farnesoid x receptor agonists |
Non-Patent Citations (1)
| Title |
|---|
| "法呢醇X受体激动剂研究进展";贾薇;《中国药物化学杂志》;20100116;第20卷(第1期);第64-69页 * |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN107405325B (zh) | 用于组合疗法的药物组合物 | |
| Davidson et al. | Comparative effects of lipid-lowering therapies | |
| US20190111012A1 (en) | Methods of treatment of cholestatic diseases | |
| EA037330B1 (ru) | Лекарственный препарат, полученный путем комбинирования агониста fxr и arb | |
| KR20170132879A (ko) | 조합 요법을 위한 약제학적 조성물 | |
| US11331292B2 (en) | Methods of treatment of cholestatic diseases | |
| Li et al. | Polysorbates as novel lipid-modulating candidates for reducing serum total cholesterol and low-density lipoprotein levels in hyperlipidemic C57BL/6J mice and rats | |
| KR20210079275A (ko) | 고콜레스테롤 관련 병으로 치료 받고 있는 환자에서의 당뇨병의 위험 감소 방법 | |
| US11850223B2 (en) | Methods of treatment of cholestatic diseases | |
| US20210379003A1 (en) | Treatment of diseases associated with biliary system destruction | |
| TW202042826A (zh) | 降脂組成物及其用途 | |
| Shepherd | What is the best way to manage dyslipidemia? | |
| EA044618B1 (ru) | Комбинированный продукт для лечения фиброзного или холестатического заболевания |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| REG | Reference to a national code |
Ref country code: HK Ref legal event code: DE Ref document number: 1246191 Country of ref document: HK |
|
| GR01 | Patent grant | ||
| GR01 | Patent grant | ||
| CP02 | Change in the address of a patent holder |
Address after: new jersey Patentee after: INTERCEPT PHARMACEUTICALS, Inc. Address before: New York, United States Patentee before: INTERCEPT PHARMACEUTICALS, Inc. |
|
| CP02 | Change in the address of a patent holder |