CN103781492A - 用于治疗与masp-2依赖性补体活化相关的状况的方法 - Google Patents
用于治疗与masp-2依赖性补体活化相关的状况的方法 Download PDFInfo
- Publication number
- CN103781492A CN103781492A CN201280028263.2A CN201280028263A CN103781492A CN 103781492 A CN103781492 A CN 103781492A CN 201280028263 A CN201280028263 A CN 201280028263A CN 103781492 A CN103781492 A CN 103781492A
- Authority
- CN
- China
- Prior art keywords
- masp
- inhibitor
- experimenter
- antibody
- compositions
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images






































Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/40—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/39533—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals
- A61K39/3955—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals against proteinaceous materials, e.g. enzymes, hormones, lymphokines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/08—Drugs for disorders of the alimentary tract or the digestive system for nausea, cinetosis or vertigo; Antiemetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/12—Antidiarrhoeals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/02—Drugs for disorders of the urinary system of urine or of the urinary tract, e.g. urine acidifiers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/14—Drugs for dermatological disorders for baldness or alopecia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/16—Emollients or protectives, e.g. against radiation
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/06—Antiglaucoma agents or miotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P39/00—General protective or antinoxious agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P39/00—General protective or antinoxious agents
- A61P39/02—Antidotes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/04—Antihaemorrhagics; Procoagulants; Haemostatic agents; Antifibrinolytic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/06—Antianaemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/54—Medicinal preparations containing antigens or antibodies characterised by the route of administration
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/545—Medicinal preparations containing antigens or antibodies characterised by the dose, timing or administration schedule
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/20—Immunoglobulins specific features characterized by taxonomic origin
- C07K2317/21—Immunoglobulins specific features characterized by taxonomic origin from primates, e.g. man
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/20—Immunoglobulins specific features characterized by taxonomic origin
- C07K2317/24—Immunoglobulins specific features characterized by taxonomic origin containing regions, domains or residues from different species, e.g. chimeric, humanized or veneered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/30—Immunoglobulins specific features characterized by aspects of specificity or valency
- C07K2317/34—Identification of a linear epitope shorter than 20 amino acid residues or of a conformational epitope defined by amino acid residues
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/54—F(ab')2
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/55—Fab or Fab'
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/71—Decreased effector function due to an Fc-modification
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/76—Antagonist effect on antigen, e.g. neutralization or inhibition of binding
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/90—Immunoglobulins specific features characterized by (pharmaco)kinetic aspects or by stability of the immunoglobulin
- C07K2317/92—Affinity (KD), association rate (Ka), dissociation rate (Kd) or EC50 value
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/90—Immunoglobulins specific features characterized by (pharmaco)kinetic aspects or by stability of the immunoglobulin
- C07K2317/94—Stability, e.g. half-life, pH, temperature or enzyme-resistance
Abstract
在一个方面,本发明提供了在有生命受试者中抑制MASP-2依赖性补体活化的影响的方法。本发明包括将有效抑制MASP-2依赖性补体活化的量的MASP-2抑制剂施用于需要其的受试者的步骤。在一些实施方案中,MASP-2抑制剂抑制与MASP-2介导的替代补体途径活化相关的细胞损伤,而使免疫系统的经典(C1q依赖性)途径组分保持完整。在另一个方面,本发明提供了用于抑制凝集素依赖性补体活化的影响的组合物,所述组合物包含治疗有效量的MASP-2抑制剂和药学可接受的载体。
Description
相关申请的交叉引用
本申请主张2011年4月8日提交的临时申请号61/473,698 的权益,其通过引用完整地并入本文。
关于序列表的声明
与本申请相关的序列表以文本格式代替纸印本提供,并且在此通过引用并入本说明书。包含该序列表的文本文件的名称为MP_1_0126_US2_SequenceListingasFiled.txt。该文本文件为110 KB;创建于2012年3月30日;并且正随着本说明书的提交经由EFS-Web提交。
发明背景
补体系统为在人和其他脊椎动物中启动、增强和协调对微生物感染和其他急性损伤的免疫应答提供了早期的作用机制(M.K. Liszewski和J.P. Atkinson, 1993, in Fundamental Immunology, 第三版,W.E. Paul编辑, Raven Press, Ltd., New York)。尽管补体活化提供了重要的针对潜在病原体的第一线防御,但是促进保护性免疫应答的补体活性也可以表现出对宿主的潜在威胁(K.R. Kalli, 等人, Springer Semin. Immunopathol. 15:417-431, 1994; B.P. Morgan, Eur. J. Clinical Investig. 24:219-228, 1994)。例如,C3和C5蛋白水解产物募集并活化嗜中性粒细胞。尽管对宿主防御不可缺少,但是活化的中性粒细胞不加选择地释放破坏性酶,可能造成器官损伤。此外,补体活化可能导致溶胞的补体组分沉积在附近的宿主细胞以及微生物靶上,导致宿主细胞裂解。
补体系统也已经被牵连进许多急性和慢性疾病状况的发病机制,所述疾病包括:心肌梗死、中风、ARDS、再灌注损伤、败血症性休克、热烧伤之后的毛细血管渗漏、心肺分流术后炎症、移植排斥、类风湿性关节炎、多发性硬化、重症肌无力和阿尔茨海默氏病。在几乎所有的这些疾病中,补体都不是病因,而是发病机制所涉及的若干因素之一。尽管如此,补体活化可能是重要的病理机制,并在许多这类疾病状况的临床控制中表现出有效之处。对各种疾病状况中补体介导的组织损伤的重要性的增加的认识增强了对有效补体抑制药物的需求。迄今为止,依库丽单抗(Solaris?),针对C5的抗体,是已被批准用于人使用的唯一补体-靶向药物。然而,C5是位于补体系统“下游”的几个效应分子之一,并且C5的阻断不抑制补体系统的活化。因此,补体激活的起始步骤的抑制剂相对于“下游”补体抑制剂具有显著优势。
目前,普遍认为补体系统可通过三种不同的途径被活化:经典途径、凝集素途径和替代途径。经典途径通常是由宿主抗体与外源颗粒(即抗原)相结合构成的复合物触发的,因此需要预先暴露于该抗原来产生特异性抗体应答。因为经典途径的活化取决于宿主的先前的适应性免疫应答,所以经典途径是获得性免疫系统的一部分。相反,凝集素途径和替代途径两者不依赖于适应性免疫(adaptive immunity),是先天性免疫系统的一部分。
补体系统的活化导致了丝氨酸蛋白酶酶原的序贯活化。经典途径活化的第一步是特异性识别分子C1q与结合了抗原的IgG和IgM分子结合。C1q与C1r和C1s丝氨酸蛋白酶酶原结合成称为C1的复合物。当C1q与免疫复合物结合之后,C1r的Arg-Ile位点进行自我蛋白水解裂解,接着是C1r介导的裂解和C1s的活化,从而获得裂解C4和C2的能力。C4裂解成两个片段,称为C4a和C4b,且类似地,C2裂解成C2a和C2b。C4b片段能够与邻近的羟基或氨基形成共价键,且通过与活化C2的C2a片段进行非共价相互作用而产生C3转化酶(C4b2a)。C3转化酶(C4b2a)通过蛋白水解裂解成C3a和C3b亚组分而活化C3,导致生成C5转化酶(C4b2a3b),所述C5转化酶(C4b2a3b)通过裂解C5而导致形成膜攻击复合物(C5b与C6、C7、C8和C-9组合,也称为“MAC”),所述膜攻击复合物可以破坏细胞膜,导致细胞裂解。C3和C4的活化形式(C3b和C4b)共价沉积在外源靶表面上,其被多种吞噬细胞上的补体受体所识别。
独立地,补体系统通过凝集素途径活化的第一步也是特异性识别分子的结合,接着是所结合的丝氨酸蛋白酶酶原的活化。然而,凝集素途径中的识别分子包含一组碳水化合物结合蛋白(甘露聚糖结合凝集素(MBL)、H-纤维胶凝蛋白(H-ficolin)、M-纤维胶凝蛋白、L-纤维胶凝蛋白和C型凝集素CL-11)(统称为凝集素),而不是通过C1q来结合免疫复合物。参见J. Lu等人,Biochim. Biophys. Acta 1572:387-400, (2002); Holmskov 等人, Annu. Rev. Immunol. 21:547-578 (2003); Teh等人, Immunology 101:225-232 (2000))。还参见J. Luet等人, Biochim Biophys Acta 1572:387-400 (2002); Holmskov等人, Annu Rev Immunol 21:547-578 (2003); Teh等人, Immunology 101:225-232 (2000); Hansen等人, J. Immunol 185(10):6096-6104 (2010)。
Ikeda等人首先证明,与C1q类似,MBL在与酵母甘露聚糖包被的红细胞结合之后能够以C4依赖的方式使补体系统活化(Ikeda等人, J. Biol. Chem. 262:7451-7454, (1987))。MBL是胶原凝集素蛋白家族的成员,是钙依赖性凝集素,与具有定位于吡喃糖环赤道面上的3-羟基和4-羟基的碳水化合物结合。因此MBL的重要配体是D-甘露糖和N-乙酰-D-葡糖胺,而不符合这种空间要求的碳水化合物则对MBL没有可检出的亲和力(Weis等人, Nature 360:127-134, (1992))。MBL和单价糖之间的相互作用是极弱的,解离常数通常在单位数毫摩尔(single-digit millimolar)的范围内。MBL通过亲合力(即通过同时与彼此紧密邻近定位的多个单糖残基相互作用)实现了与聚糖配体的紧密特异性结合(Lee等人, Archiv. Biochem. Biophys. 299:129-136, (1992))。MBL识别通常修饰微生物诸如细菌、酵母、寄生虫和某些病毒的碳水化合物模式。相反,MBL不识别D-半乳糖和唾液酸,即倒数第二位的糖和倒数第一位的糖,它们一般修饰哺乳动物血浆和细胞表面糖蛋白上存在的“成熟”复杂糖缀合物。认为这种结合特异性促进“外源”表面的识别且有助于保护免于“自身活化”。然而,MBL确实以高亲和力结合高甘露糖“前体”聚糖簇,这些簇位于被隔离在哺乳动物细胞内质网和高尔基体内的N-连接的糖蛋白和糖脂上(Maynard等人,J. Biol. Chem. 257:3788-3794,1982)。因此,受损细胞是通过MBL结合的凝集素途径活化的潜在目标。
纤维胶凝蛋白(ficolin)具有类型不同于MBL的凝集素结构域,称为血纤蛋白原样结构域。纤维胶凝蛋白以不依赖Ca++的方式来结合糖残基。在人体中,已经鉴定出纤维胶凝蛋白的三种类型(L-纤维胶凝蛋白、M-纤维胶凝蛋白和H-纤维胶凝蛋白)。L-纤维胶凝蛋白和H-纤维胶凝蛋白这两种血清纤维胶凝蛋白共同对N-乙酰-D-葡糖胺具有特异性;然而,H-纤维胶凝蛋白还结合N-乙酰-D-半乳糖胺。L-纤维胶凝蛋白、H-纤维胶凝蛋白、CL-11和MBL的糖特异性的差异意味着不同的凝集素可能是互补的,尽管有重叠,但是可能靶向不同的糖缀合物。这个观点得到了最新报道的支持,即在凝集素途径的已知凝集素中,只有L-纤维胶凝蛋白与脂磷壁酸特异性结合,脂磷壁酸是一种存在于所有革兰氏阳性菌上的细胞壁糖缀合物(Lynch等人, J. Immunol. 172:1198-1202, (2004))。胶原凝集素(即MBL)和纤维胶凝蛋白在氨基酸序列上没有显著的相似性。然而,两组蛋白质具有类似的结构域组构,与C1q类似,装配成寡聚结构,这样就使得多位点结合的可能性最大化。
MBL的血清浓度在健康人群中是高度可变的,这在遗传上是由MBL基因的启动子和编码区二者的多态性/突变所控制的。作为急性期蛋白,MBL的表达在炎症期间进一步上调。L-纤维胶凝蛋白在血清中存在的浓度与MBL的浓度类似。因此,凝集素途径的L-纤维胶凝蛋白分支在力量上可能与MBL臂相当。MBL和纤维胶凝蛋白也可能作为调理素起作用,这允许吞噬细胞靶向至MBL和纤维胶凝蛋白装饰的表面(参见Jack等人, J Leukoc Biol., 77(3):328-36 (2004), Matsushita和Fujita, Immunobiology, 205(4-5):490-7 (2002), Aoyagi等人, J Immunol, 174(1):418-25(2005)。这种调理素作用需要这些蛋白质与吞噬细胞受体相互作用(Kuhlman等人, J. Exp. Med. 169:1733, (1989); Matsushita等人, J. Biol. Chem. 271:2448-54, (1996)),其身份还未得到确定。
人MBL通过其胶原样结构域与独特的C1r/C1s样丝氨酸蛋白酶(称为MBL相关的丝氨酸蛋白酶(MBL-associated serine proteases,MASP))形成特异性、高亲和力的相互作用。迄今为止已经描述了三种MASP。首先,鉴定出单一的酶“MASP”,其特征是负责启动补体级联(即裂解C2和C4)的酶(Matsushita等人, J Exp Med 176(6):1497-1502 (1992); Ji等人, J. Immunol. 150:571-578, (1993))。随后确定MASP活性,实际上是两种蛋白酶MASP-1和MASP-2的混合物(Thiel等人,Nature 386:506-510, (1997))。然而,有研究表明仅MBL-MASP-2复合物就足以使补体活化(Vorup-Jensen等人, J. Immunol.165:2093-2100, (2000))。此外,只有MASP-2快速裂解C2和C4 (Ambrus等人, J. Immunol. 170:1374-1382, (2003))。因此,MASP-2是负责激活C4和C2以产生C3转化酶C4b2a的蛋白酶。这是不同于经典途径的C1复合物的显著差异,C1复合物中两种特异性丝氨酸蛋白酶(C1r和C1s)协同作用导致了补体系统的活化。此外,已经分离出第三种新的蛋白酶MASP-3 (Dahl, M.R.,等人, Immunity 15:127-35, 2001)。MASP-1和MASP-3是同一基因的可变剪接产物。
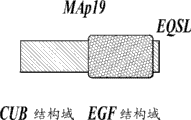
MASP与C1复合物的酶成分C1r和C1s的那些共享相同的结构域组构(Sim等人,Biochem. Soc. Trans. 28:545,2000)。这些结构域包括N末端C1r/C1s/海胆VEGF/骨形成蛋白(CUB)结构域、表皮生长因子样结构域、第二CUB结构域、补体调控蛋白结构域的串联和丝氨酸蛋白酶结构域。与在C1蛋白酶中一样,MASP-2的活化通过丝氨酸蛋白酶结构域附近的Arg-Ile键裂解而产生,这将酶分成二硫键连接的A链和B链,后者由丝氨酸蛋白酶结构域构成。
MBL还可以与MASP-2的可变切片形式(被称作19 kDa的MBL相关蛋白(MAp19)或小MBL相关蛋白(sMAP),其缺乏MASP2的催化活性)结合。(Stover, J. Immunol. 162:3481-90, (1999); Takahashi等人, Int. Immunol. 11:859-863, (1999))。MAp19包括MASP-2的前两个结构域,其后接着是4个独特氨基酸的额外序列。Map19的功能不明确(Degn等人, J Immunol. Methods, 2011)。MASP-1和MASP-2基因分别位于人3号染色体和1号染色体上(Schwaeble等人, Immunobiology 205:455-466, (2002))。
若干证据表明存在不同的MBL-MASP复合物,且血清中MASP的大部分不与MBL复合(Thiel,等人, J. Immunol. 165:878-887, (2000))。H-纤维胶凝蛋白和L-纤维胶凝蛋白都与所有的MASP结合,并且激活凝集素补体途径,同MBL一样(Dahl等人, Immunity 15:127-35, (2001); Matsushita等人, J. Immunol. 168:3502-3506, (2002))。凝集素途径和经典途径都形成共同的C3转化酶(C4b2a),这两条途径在这一步会合。
普遍认为在天然宿主中,凝集素途径在宿主抵抗感染的防御中具有重要作用。MBL参与宿主防御的强有力证据来自于对功能性MBL血清水平降低的患者的分析(Kilpatrick, Biochim. Biophys. Acta1572:401-413, (2002))。这些患者表现出复发性细菌和真菌感染的易感性。这些症状通常在生命早期在易损表观窗期间是明显的,因为从母体获得的抗体效价降低,但处于全部抗体应答发展之前。这种症状经常是由于MBL胶原部分的数个位点突变引起的,其干扰了MBL寡聚体的正确形成。然而,由于MBL可以作为不依赖于补体的调理素起作用,所以还不知道对感染的易感性增加在多大程度上是由于受损的补体活化所致。
与经典途径和凝集素途径相反,没有发现替代途径中完成识别功能的起始因子(initiator),而在其他两种途径中是C1q和凝集素来完成识别功能的。目前普遍接受的是,替代途径自发经历低水平转换活化(turnover activation),其可以在缺乏使自发补体活化保持受检查的正确分子元件的外来表面或其他异常表面(细菌、酵母、病毒感染细胞或者受损组织)上容易地扩增。有四种血浆蛋白直接参与了替代途径的活化:C3、因子B和因子D以及备解素。
尽管大量证据表明在非感染性人类疾病的发病机制中,经典补体途径和替代补体途径两者都存在,但是对凝集素途径作用的评价才刚刚开始。最新研究提供的证据表明,凝集素途径的活化可能是造成缺血/再灌注损伤中补体活化和相关炎症的原因。Collard等人(2000)报告受到氧化应激的培养的内皮细胞结合MBL,且在暴露于人血清之后显示出C3沉积(Collard等人, Am. J. Pathol. 156:1549-1556, (2000))。此外,用阻断性抗MBL单克隆抗体处理人血清抑制了MBL结合和补体活化。将这些发现扩展到心肌缺血-再灌注大鼠模型上,其中比起用对照抗体处理的大鼠,用针对大鼠MBL的阻断性抗体处理的大鼠在冠状动脉闭塞之后显示心肌损伤显著较轻(Jordan等人, Circulation 104:1413-1418, (2001))。尚不清楚氧化应激后MBL与血管内皮结合的分子机制;最近的研究表明,氧化应激后凝集素途径的活化可能是由MBL与血管内皮细胞角蛋白结合而介导的,而不是与糖缀合物结合结合而介导的(Collard等人, Am. J. Pathol. 159:1045-1054, (2001))。其他研究已显示出缺血/再灌注损伤发病机制中的经典途径和替代途径以及凝集素途径在这种疾病中的作用仍然存在争议(Riedermann, N.C.,等人, Am. J. Pathol. 162:363-367, 2003)。
一项新近的研究已经显示,MASP-1(可能还有MASP-3)对于将替代途径活化酶因子D从其酶原形式转换为其酶促活性形式是需要的(参见Takahashi M.等人, J Exp Med 207(1):29-37 (2010))。该过程的生理重要性通过在MASP-1/3-缺陷小鼠的血浆中不存在替代途径功能活性而得到强调。对于替代途径发挥作用,需要由天然C3蛋白水解形成C3b。由于替代途径C3转化酶(C3bBb)含有C3b作为必需亚基,关于通过替代途径的第一个C3b来源的问题代表了令人困扰的问题,且促使了相当多的研究工作。
C3属于含有极少的被称为硫酯键的翻译后修饰的蛋白质家族(还有C4和α-2巨球蛋白)。硫酯基团由形成共价硫脂连接的谷氨酰胺的末端羰基与三个氨基酸距离的半胱氨酸的巯基所形成。该键是不稳定的,并且亲电谷氨酰基-硫酯可以与亲核部分诸如羟基或氨基反应,从而与其他分子形成共价键。当被隔离在完整C3的疏水口袋内部时,硫酯键是相当稳定的。然而,C3被蛋白水解裂解成C3a和C3b,导致C3b上高反应性的硫酯键暴露出来,并且,在包含羟基或氨基的邻近部分的亲核攻击后,C3b变得与目标共价连接。除了有大量文件证明C3b与补体靶共价结合的作用外,还有研究认为C3硫酯具有触发替代途径的关键作用。根据普遍接受的“慢转理论(tick-over theory)”,替代途径是由液相转化酶iC3Bb的形成所启动的,iC3Bb是由C3与水解硫酯(iC3; C3(H2O))和因子B形成的(Lachmann, P.J.,等人, Springer Semin. Immunopathol. 7:143-162, (1984))。C3b样C3由天然C3经蛋白质中内部硫酯的缓慢自发水解而产生(Pangburn, M.K.,等人, J. Exp. Med. 154:856-867, 1981)。通过C3(H2O)Bb转化酶的活性,C3b分子沉积在靶表面,从而启动替代途径。
对替代途径活化的起始因子的了解甚少。认为激活因子包括酵母细胞壁(酵母聚糖)、许多纯的多糖、兔红细胞、某些免疫球蛋白、病毒、真菌、细菌、动物肿瘤细胞、寄生虫和受损细胞。这些激活因子所共有的唯一特征是碳水化合物的存在,但是碳水化合物结构的复杂性和多样性使得难以确定共享的分子决定子,这是公认的。已被广泛接受的是,替代途径活化通过该途径的抑制调节组分诸如因子H、因子I、DAF和CR1和备解素(这是替代途径唯一阳性调节物)之间精细平衡而得到控制(参见Schwaeble W.J. 和Reid K.B., Immunol Today 20(1):17-21 (1999))。
除了上述明显失调的活化机制之外,对于凝集素/经典途径C3转化酶(C4b2a),替代途径也可以提供强大的放大环路(amplification loop),因为所产生的任何C3b都能与因子B一起参与形成额外的替代途径C3转化酶(C3bBb)。替代途径C3转化酶通过备解素的结合而稳定。备解素延长了替代途径C3转化酶的半衰期6-10倍。向替代途径C3转化酶中添加C3b导致替代途径C5转化酶的形成。
认为所有三种途径(即经典、凝集素和替代途径)会合于C5,它被裂解形成具有多种促炎作用的产物。会合后的途径被称为末端补体途径。C5a是最有效的过敏毒素,引起平滑肌和血管紧张度以及血管通透性的改变。它也是嗜中性粒细胞和单核细胞两者的强有力的趋化因子和激活因子。C5a介导的细胞活化能够通过诱导释放多种另外的炎症介质来显著放大炎症反应,另外的炎症介质包括细胞因子、水解酶、花生四烯酸代谢物和活性氧类别。C5裂解导致了C5b-9的形成,它也被称为膜攻击复合物(MAC)。目前强有力的证据表明,亚裂解的MAC沉积除了起裂解成孔复合物的作用外,还可能还在炎症中发挥重要作用。
除了在免疫防御中的基本作用外,补体系统是造成许多临床疾病的组织损伤的原因。因此,迫切需要开发治疗上有效的补体抑制剂以抑制这些副作用。
发明概述
提供本概述,从而以简化形式引入概念的选择,其在以下发明详述中进一步描述。本概述既不欲鉴定请求保护的主题的关键特征,也不欲用于协助确定请求保护的主题的范围。
在一个方面,本发明提供抑制有生命的受试者中MASP-2依赖性补体活化的副作用的方法。该方法包括将有效抑制MASP-2依赖性补体活化的量的MASP-2抑制剂施用于需要其的受试者的步骤。在本发明的另一个方面,MASP-2抑制剂抑制通过凝集素依赖性MASP-2系统的补体活化,但基本上不抑制通过经典或C1q依赖性系统的补体活化,从而C1q依赖性系统仍然具有功能。
在本发明这些方面的一些实施方案中,MASP-2抑制剂是抗MASP-2抗体或其片段。在其他实施方案中,抗MASP-2抗体具有降低的效应子功能。在一些实施方案中,MASP-2抑制剂是MASP-2抑制肽或非肽MASP-2抑制剂。
另一个方面,本发明提供用于抑制MASP-2依赖性补体活化的副作用的组合物,该组合物包含治疗有效量的MASP-2抑制剂和药学可接受的载体。本发明还提供制备用于抑制需要其的有生命的受试者中MASP-2依赖性补体活化的副作用的药物的方法,其包括药物载体中的治疗有效量的MASP-2抑制剂。本发明还提供制备用于抑制MASP-2依赖性补体活化以治疗下文所述病症、疾病和障碍中每一种的药物的方法。
本发明的方法、组合物和药物用于在患有如本文将进一步描述的急性或慢性病理病症或损伤的哺乳动物受试者、包括人中体内抑制MASP-2依赖性补体活化的副作用。
在本发明的另一个方面,提供用于抑制患有阵发性睡眠性血红蛋白尿症(paroxysmal nocturnal hemoglobinuria)的受试者中MASP-2依赖性补体活化的方法,其包括将包含有效抑制MASP-2依赖性补体活化的量的MASP-2抑制剂的组合物施用于所述受试者。
在另一个方面,本发明提供用于抑制患有非因子H依赖性非典型溶血性尿毒综合征(non-Factor H-dependent atypical hemolytic uremic syndrome (aHUS))或处于发生非因子H依赖性非典型溶血性尿毒综合征的风险中的受试者中MASP-2依赖性补体活化的方法,其包括将包含有效抑制MASP-2依赖性补体活化的量的MASP-2抑制剂的组合物施用于所述受试者。
在另一个方面,本发明提供用于降低处于发生非典型溶血性尿毒综合征(aHUS)的风险中的受试者将患有与aHUS相关的临床症状的可能性的方法,其包括:(a) 确定受试者中已知与aHUS相关的基因标志物的存在;(b) 定期监测受试者,以确定选自贫血、血小板减少、肾功能不全和肌酸酐上升的至少一种症状的存在或不存在;和 (c) 在确定贫血、血小板减少、肾功能不全或肌酸酐上升的至少一种的存在之后,将有效抑制MASP-2依赖性补体活化的量的MASP-2抑制剂的组合物施用于所述受试者,其中施用所述组合物的量有效且时间期间足以改善所述一种或多种症状。
在另一个方面,本发明提供抑制患有继发于感染的非典型溶血性尿毒综合征(aHUS)或处于发生继发于感染的非典型溶血性尿毒综合征(aHUS)的风险中的受试者中MASP-2依赖性补体活化的方法,其包括将包含有效抑制MASP-2依赖性补体活化的量的MASP-2抑制剂的组合物施用于所述受试者。
在另一个方面,本发明提供治疗患有非典型溶血性尿毒综合征(aHUS)的受试者的方法,其包括将包含有效抑制MASP-2依赖性补体活化的量的MASP-2抑制剂的组合物施用于所述受试者,其中所述MASP-2抑制剂的施用通过静脉内导管或其他导管递送方法而施用。
在另一个方面,本发明提供用于降低处于发生溶血性尿毒综合征(HUS)的风险中的受试者中发生肾功能受损的可能性的方法,其包括将包含有效抑制MASP-2依赖性补体活化的量的MASP-2抑制剂的组合物施用于所述受试者。
在另一个方面,本发明提供治疗患有溶血性尿毒综合征(HUS)的受试者的方法,其包括将包含有效抑制MASP-2依赖性补体活化的量的MASP-2抑制剂的组合物施用于所述受试者,其中所述MASP-2抑制剂的施用通过静脉内导管或其他导管递送方法而施用于所述受试者。
在另一个方面,本发明提供治疗患有血栓性血小板减少性紫癜(TTP)或表现出与TTP的诊断一致的症状的受试者的方法,其包括将包含有效抑制MASP-2依赖性补体活化的量的MASP-2抑制剂的组合物施用于所述受试者,其中所述MASP-2抑制剂的施用通过静脉内导管或其他导管递送方法而施用于所述受试者。
在另一个方面,本发明提供治疗患有难治性血栓性血小板减少性紫癜(TTP)的受试者的方法,其包括将包含有效抑制MASP-2依赖性补体活化的量的MASP-2抑制剂的组合物施用于所述受试者。
在本发明的另一个方面,提供用于抑制患有冷球蛋白血症的受试者中MASP-2依赖性补体活化的方法,其包括将包含有效抑制MASP-2依赖性补体活化的量的MASP-2抑制剂的组合物施用于所述受试者。
在本发明的另一个方面,提供用于抑制患有冷凝集素病的受试者中MASP-2依赖性补体活化的方法,其包括将包含有效抑制MASP-2依赖性补体活化的量的MASP-2抑制剂的组合物施用于所述受试者。
在本发明的另一个方面,提供用于抑制患有青光眼的受试者中MASP-2依赖性补体活化的方法,其包括将包含有效抑制MASP-2依赖性补体活化的量的MASP-2抑制剂的组合物施用于所述受试者。
在本发明的另一个方面,提供用于抑制处于发生急性放射综合征的风险中或患有急性放射综合征的受试者中MASP-2依赖性补体活化的方法,其包括将包含有效抑制MASP-2依赖性补体活化的量的MASP-2抑制剂的组合物施用于所述受试者。在一些实施方案中,抗MASP-2抑制剂是抗-MASP-2抗体。在一些实施方案中,在辐射暴露(诸如用辐射治疗之前或预期暴露于辐射之前)将MASP-2抑制剂预防性施用于受试者。在一些实施方案中,在暴露于辐射之后24至48小时内施用MASP-2抑制剂。在一些实施方案中,在暴露于辐射之前和/或之后以足以减轻与急性放射综合征相关的一种或多种症状的量施用MASP-2抑制剂。
附图说明
通过参考下面的详细描述连同附图,本发明前述的方面以及许多附带的优点将更容易领会,同样也更好理解,其中:
图1是说明人MASP-2基因组结构的图;
图2A是说明人MASP-2蛋白的结构域结构的示意图;
图2B是说明人MAp19蛋白的结构域结构的示意图;
图3是说明鼠MASP-2敲除策略的图;
图4是说明人MASP-2微基因构建体的图;
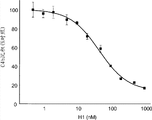
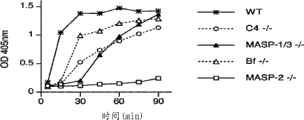
图5A提供了证明MASP-2缺陷导致凝集素途径介导的C4活化丧失的结果,如通过甘露聚糖上C4b沉积缺陷所测定的,如实施例2所述;
图5B提供了证明MASP-2缺陷导致凝集素途径介导的C4活化丧失的结果,如通过酵母聚糖上C4b沉积缺陷所测定的,如实施例2所述;
图5C提供了证明获自MASP-2 +/-、MASP-2 -/-和野生型品系的血清样品的相对C4活化水平的结果,如通过甘露聚糖和酵母聚糖上的C4b沉积所测定的,如实施例2所述;
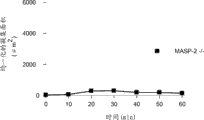
图6提供了证明加入鼠重组MASP-2到MASP-2 -/-血清样品中以蛋白浓度依赖的方式恢复了凝集素途径介导的C4活化的结果,如通过甘露聚糖上的C4b沉积所测定的,如实施例2所述;
图7提供了证明MASP-2 -/-品系中经典途径有功能的结果,如实施例8所述;
图8A提供了证明抗MASP-2 Fab2抗体#11抑制C3转化酶形成的结果,如实施例10所述;
图8B提供了证明抗MASP-2 Fab2抗体#11与天然大鼠MASP-2结合的结果,如实施例10所述;
图8C提供了证明抗MASP-2 Fab2抗体#41抑制C4裂解的结果,如实施例10所述;
图9提供了证明经测试抑制C3转化酶形成的所有抗MASP-2 Fab2抗体还被发现抑制C4裂解的结果,如实施例10所述;
图10是说明从用于抗MASP-2阻断性Fab2抗体表位作图的大鼠MASP-2衍生的重组多肽的示意图,如实施例11所述;
图11提供了证明抗MASP-2 Fab2 #40和#60与大鼠MASP-2多肽结合的结果,如实施例11所述;
图12提供了证明肾缺血/再灌注损伤模型中再灌注后24小时和48小时野生型(+/+)和MASP-2 (-/-)小鼠的血尿素氮清除率的结果,如实施例12所述;
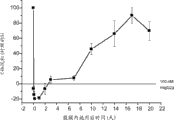
图13A提供了显示从野生型(+/+)和MASP-2 (-/-)小鼠中分离的RPE-脉络膜复合体中的基线VEGF蛋白水平的结果,如实施例13所述;
图13B提供了显示黄斑变性模型中激光诱发的损伤之后第三天野生型(+/+)和MASP-2 (-/-)小鼠的RPE-脉络膜复合体中VEGF蛋白水平的结果,如实施例13所述;
图14提供了显示野生型(+/+)和MASP-2 (-/-)小鼠中激光诱发损伤后第七天平均脉络膜新血管形成(CNV)体积的结果,如实施例13所述;
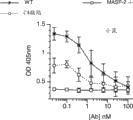
图15A和图15B提供了在正常大鼠血清中施用MASP-2 Fab2抗体之后C4b沉积的抑制(图15A)和凝血酶活化的抑制(图15B)的剂量响应曲线,如实施例14中所述;
图16A和图16B提供了在弥散性血管内凝血的局部Schwartzman反应模型中,与未处理的野生型小鼠和其中补体途径被放血剂眼镜蛇毒因子(cobra venom factor,CVF)和末端途径抑制剂(C5aR拮抗剂)抑制的野生型小鼠中的血小板凝集(图16A)相比,MASP-2(-/-)小鼠中测定的血小板凝集(表示为凝集面积)(图16B),如实施例15中所述;
图17图示说明在WT(+/+)供体肾的WT(+/+)(B6)或MASP-2(-/-)移植受体小鼠中测定的血尿素氮(BUN)水平,如实施例16中所述;
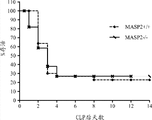
图18将WT(+/+)和MASP-2(-/-)小鼠的百分比存活图示为盲肠结扎和穿刺(CLP)模型中微生物感染后的天数的函数,如实施例17中所述;
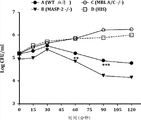
图19图示说明盲肠结扎和穿刺(CLP)模型中微生物感染后在WT(+/+)和MASP-2(-/-)中测定的细菌数,如实施例17中所述;
图20为说明在用绿脓假单胞菌的鼻内施用攻击后6天WT(+/+)、MASP-2(-/-)和C3(-/-)小鼠的百分比存活的Kaplan-Mayer图,如实施例18中所述;
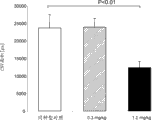
图21图示说明在WT小鼠内小鼠抗MASP-2单克隆抗体的0.3 mg/kg或1.0 mg/kg皮下给药之后,在不同时间点取样的样品中C4b沉积的水平,测定为对照的%,如实施例19中所述;
图22图示说明在WT小鼠内小鼠抗MASP-2单克隆抗体的0.6 mg/kg腹膜内给药之后,在不同时间点取样的样品中C4b沉积的水平,测定为对照的%,如实施例19中所述;
图23图示说明在用0.3 mg/kg或1.0 mg/kg小鼠抗MASP-2单克隆抗体的单次腹膜内注射预处理的WT(+/+)小鼠中激光诱发损伤后第7天的平均脉络膜新生血管形成(CNV)体积,如实施例20中所述;
图24A图示说明在用5×108/100 μl cfu脑膜炎奈瑟氏菌(N. meningitidis)感染后MASP-2(-/-)和WT(+/+)小鼠的百分比存活,如实施例21中所述;
图24B图示采自用5×108 cfu/100 μl脑膜炎奈瑟氏菌感染的MASP-2 KO(-/-)和WT(+/+)小鼠的血样中在不同时间点回收的脑膜炎奈瑟氏菌的log cfu/ml,如实施例21中所述;
图25A图示在用2×108 cfu/100 μl脑膜炎奈瑟氏菌感染后MASP-2 KO(-/-)和WT(+/+)小鼠的百分比存活,如实施例21中所述;
图25B图示说明采自用2×108 cfu/100 μl脑膜炎奈瑟氏菌感染的WT(+/+)小鼠的血样中在不同时间点回收的脑膜炎奈瑟氏菌的log cfu/ml,如实施例21中所述;
图25C图示说明采自用2×108 cfu/100 μl脑膜炎奈瑟氏菌感染的MASP-2(-/-)小鼠的血样中在不同时间点回收的脑膜炎奈瑟氏菌的log cfu / ml,如实施例21中所述。
图26A图示说明证明MASP-2 (-/-)小鼠保留功能性经典途径的C3b沉积测定的结果,如实施例22中所述;
图26B图示说明证明MASP-2 (-/-)小鼠保留功能性经典途径的在酵母多糖包被的板上的C3b沉积测定的结果,如实施例22中所述;
图27A图示说明在C4 (-/-)小鼠(n=6)和匹配的WT同窝小鼠对照(n=7)中冠状动脉的左前降支(LAD)结扎和再灌注之后的心肌缺血/再灌注损伤(MIRI)诱导的组织损失,其显示处于风险中的区域(AAR)和梗死面积(INF),如实施例22中所述;
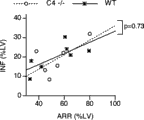
图27B图示说明在如图42A中所述处理的C4 (-/-)和WT小鼠中作为风险中的区域(AAR)的函数的梗死面积(INF),其证明,C4 (-/-)小鼠和WT对照(虚线)一样易受MIRI的影响,如实施例22中所述;
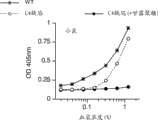
图28A图示说明使用来自WT 小鼠的血清、来自C4 (-/-)小鼠的血清和与甘露聚糖与预孵育的来自C4 (-/-)小鼠的血清的C3b沉积测定的结果,如实施例22中所述;
图28B图示说明对于来自WT的血清和与各种浓度的抗鼠MASP-2 mAb (mAbM11)混合的MASP-2 (-/-)小鼠的血清的C3b沉积测定的结果,如实施例22中所述;
图28C图示说明对来自WT(C4充足)的人血清和C4缺陷血清和与甘露聚糖与预孵育的来自C4缺陷受试者的血清的C3b沉积测定的结果,如实施例22中所述;
图28D图示说明对来自WT(C4充足)的人血清和与抗人MASP-2 mAb (mAbH3)混合的来自C4缺陷受试者的人血清的C3b沉积测定的结果,如实施例22中所述;
图29A图示说明在凝集素活化途径特异性测定条件下或在经典活化途径特异性测定条件下测试的来自各种补体缺陷小鼠品系的血浆中C3转化酶活性的比较分析,如实施例22中所述;
图29B图示说明在凝集素活化途径特异性条件测试的来自各种补体缺陷小鼠品系的血浆中C3转化酶活性的时间分辨的动力学,如实施例22中所述;
图30说明如a'链的存在显示的凝血酶底物FXIa和FXa对人C3的活化的Western印迹分析的结果,如实施例23中所述;
图31显示在从WT、MASP-2 (-/-)、F11(-/-)、F11(-/-)/C4 (-/-) 和C4 (-/-)获得的血清样品上的C3沉积测定的结果,如实施例23中所述;
图32A是显示在对照小鼠和在用抗鼠MASP-2抗体(mAbM11)或抗人MASP-2抗体(mAbH6)处理的小鼠中在暴露于7.0 Gy辐射之后随时间的百分比存活的Kaplain-Meier存活图,如实施例29中所述;
图32B是显示在对照小鼠和在用抗鼠MASP-2抗体(mAbM11)或抗人MASP-2抗体(mAbH6)处理的小鼠中在暴露于6.5 Gy辐射之后随时间的百分比存活的Kaplain-Meier存活图,如实施例29中所述;
图33是图示说明在施用2.6 x 107 cfu的脑膜炎奈瑟氏菌血清群A Z2491的感染剂量之后MASP-2 KO和WT小鼠的百分比存活的Kaplan-Meyer图,其证明,MASP-2缺陷小鼠被保护免于脑膜炎奈瑟氏菌诱导的死亡,如实施例30中所述;
图34是图示说明在施用6 x 106 cfu的脑膜炎奈瑟氏菌血清群B菌株MC58的感染剂量之后MASP-2 KO和WT小鼠的百分比存活的Kaplan-Meyer图,其证明,MASP-2缺陷小鼠被保护免于脑膜炎奈瑟氏菌血清群B菌株MC58诱导的死亡,如实施例30中所述;
图35图示说明在用6x106 cfu的脑膜炎奈瑟氏菌血清组B菌株MC58 i.p.感染后取自MASP-2 KO和WT小鼠的血液样品中在不同时间点回收的脑膜炎奈瑟氏菌血清群B菌株MC58的log cfu/ml(在两组小鼠中不同时间点n=3,结果表示为平均值±SEM),其证明,尽管MASP-2 KO小鼠被与WT小鼠相同剂量的脑膜炎奈瑟氏菌血清群B菌株MC58所感染,但是MASP-2 KO小鼠与WT相比具有增强的菌血症的清除,如实施例30中所述;
图36图示说明在6x106 cfu/100 μl的脑膜炎奈瑟氏菌血清群B菌株MC58的感染后3、6、12和24小时MASP-2和WT小鼠的平均疾病评分,其证明,MASP-2缺陷小鼠显示出对感染的高抗性,在感染后6小时具有低得多的疾病评分,如实施例30中所述;
图37是图示说明在施用4 x 106/100 μl cfu脑膜炎奈瑟氏菌血清群B菌株MC58的感染剂量和随后在感染后3小时施用抑制性抗MASP-2抗体(1 mg/kg)或对照同型抗体之后小鼠的百分比存活的Kaplan-Meyer图,其证明,抗MASP-2抗体对于治疗和改善被脑膜炎奈瑟氏菌感染的受试者的存活是有效的,如实施例31中所述;
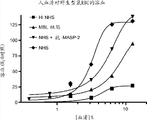
图38图示说明在与(A)正常人血清(NHS)加人抗MASP-2抗体;(B)正常人血清(NHS)加同种型对照抗体;(C) MBL-/-人血清;(D) 正常人血清(NHS)和(E)热失活的正常人血清(NHS)存在的情况下孵育后0、30、60 和90分钟在用6.5x106 cfu/100 μl脑膜炎奈瑟氏菌血清群B菌株MC58 i.p.感染后在20%人血清浓度中在不同时间点回收的脑膜炎奈瑟氏菌血清群B-MC58的活计数的log cfu/ml,其显示,人血清中脑膜炎奈瑟氏菌的补体依赖性杀伤被添加人抗MASP-2抑制性抗体所显著增强,如实施例32中所述;
图39图示说明在小鼠血清样品中在不同时间点回收的脑膜炎奈瑟氏菌血清群B-MC58的活计数的log cfu/ml,其证明,与WT小鼠血清相比,MASP-2 -/-小鼠血清对脑膜炎奈瑟氏菌具有更高水平的杀菌活性,如实施例32中所述;
图40图示说明了一定范围的血清浓度的人血清对甘露聚糖包被的鼠红细胞的溶血(如通过光度法测量的裂解的小鼠红细胞(Cryy/C3-/-)至上清液的血红蛋白释放所测量的)。测试的血清包括热失活(HI)NHS、MBL-/-、NHS +抗-MASP-2抗体和NHS对照,如实施例33中所述;
图41图示说明了一定范围的血清浓度的人血清对未包被的鼠红细胞的溶血(如通过光度法测量的裂解的WT小鼠红细胞至上清液的血红蛋白释放所测量的)。测试的血清包括热失活(HI)NHS、MBL-/-、NHS +抗-MASP-2抗体和NHS对照,其证明,抑制MASP-2抑制未敏化的WT小鼠红细胞的补体介导的裂解,如实施例33中所述;
图42图示说明了一定范围的血清浓度的人血清对未包被的鼠红细胞的溶血(如通过光度法测量的裂解的小鼠红细胞(CD55/59 -/-)至上清液的血红蛋白释放所测量的)。测试的血清包括热失活(HI)NHS、MBL-/-、NHS +抗-MASP-2抗体和NHS对照,如实施例33中所述;
图43图示说明在对照小鼠和在用抗人MASP-2抗体(mAbH6)处理的小鼠中在暴露于8.0 Gy辐射之后随时间的百分比存活,如实施例34中所述;
图44图示说明在MASP-2-/-和WT小鼠中LPS注射后至发生微血管闭塞的时间,显示经60分钟测量的具有血栓形成的小鼠的百分比,这证明,在WT小鼠中在15分钟后检测到血栓形成,其中多达80%的WT小鼠在60分钟时表现出血栓形成;相比之下,MASP-2 -/-中无一在60分钟期间过程中显示任何血栓形成(对数秩:p=0.0005),如实施例35中所述;和
图45图示说明了在STX/LPS诱导的HUS模型中盐水处理的对照小鼠(n=5)和抗MASP-2抗体处理的小鼠(n=5)随时间(小时)的百分比存活,其证明所有对照小鼠到42小时死亡,然而,与之相比,100 %的抗MASP-2抗体处理的小鼠在整个实验的时间进程过程中存活,如实施例36中所述。
序列表描述
SEQ ID NO:1 人MAp19 cDNA
SEQ ID NO:2 人MAp19 蛋白(具有前导序列)
SEQ ID NO:3 人MAp19 蛋白(成熟)
SEQ ID NO:4 人MASP-2 cDNA
SEQ ID NO:5 人MASP-2 蛋白(具有前导序列)
SEQ ID NO:6 人MASP-2 蛋白(成熟)
SEQ ID NO:7 人MASP-2 gDNA (外显子1-6)
抗原:(关于MASP-2成熟蛋白)
SEQ ID NO:8 CUBI序列(氨基酸1-121)
SEQ ID NO:9 CUBEGF序列(氨基酸1-166)
SEQ ID NO:10 CUBEGFCUBII (氨基酸1-293)
SEQ ID NO:11 EGF 区 (氨基酸122-166)
SEQ ID NO:12 丝氨酸蛋白酶结构域 (氨基酸429 – 671)
SEQ ID NO:13 失活的丝氨酸蛋白酶结构域(氨基酸610-625,具有Ser618至Ala的突变 )
SEQ ID NO:14 
SEQ ID NO:15 
SEQ ID NO:16 
SEQ ID NO:17 
SEQ ID NO:18
SEQ ID NO:19  (丝氨酸蛋白酶结合核心)
(丝氨酸蛋白酶结合核心)
详述
肽抑制剂:
SEQ ID NO:20 MBL全长cDNA
SEQ ID NO:21 MBL 全长蛋白
SEQ ID NO:22 OGK-X-GP (共有结合序列)






表达抑制剂:
SEQ ID NO:30 CUBI-EGF结构域的cDNA (SEQ ID NO:4的核苷酸22-680)

SEQ ID NO:4的核苷酸12-45,包括MASP-2翻译起始位点(有义链)

编码含有MASP-2 MBL结合位点区域的SEQ ID NO:4的核苷酸361-396 (有义链)

编码含有CUBII结构域区域的SEQ ID NO:4的核苷酸610-642
克隆引物:


(用于CUBIEGF的3' PCR)

SEQ ID NOS:38-47 是人源化抗体的克隆引物
SEQ ID NO:48 是9个氨基酸的肽键
表达载体:
SEQ ID NO:49是MASP-2小基因插入序列
SEQ ID NO:50是鼠MASP-2 cDNA
SEQ ID NO:51是鼠MASP-2蛋白(w/前导序列)
SEQ ID NO:52是成熟鼠MASP-2蛋白
SEQ ID NO:53是大鼠MASP-2 cDNA
SEQ ID NO:54是大鼠MASP-2蛋白(w/前导序列)
SEQ ID NO:55是成熟大鼠MASP-2蛋白
SEQ ID NO:56-59是用于人MASP-2的定点诱变的寡核苷酸,所述人MASP-2用来产生人MASP-2A
SEQ ID NO:60-63是用于小鼠MASP-2的定点诱变的寡核苷酸,所述小鼠MASP-2用来产生小鼠MASP-2A
SEQ ID NO:64-65是用于大鼠MASP-2的定点诱变的寡核苷酸,所述大鼠MASP-2用来产生大鼠MASP-2A。
发明详述
本发明基于本发明人预料不到的发现,即抑制凝集素介导的MASP-2途径而同时保持经典途径的完整是可能的。本发明也描述了MASP-2作为治疗靶的用途,用于抑制与凝集素介导的补体途径活化有关的细胞损伤而同时保持免疫系统经典(C1q依赖性)途径成分的完整。
I. 定义
除非本文明确规定,否则本文使用的所有术语都具有如本发明领域的普通技术人员所理解的相同含义。为了明确用于本说明书和所附权利要求书中描述本发明的有关术语,提供下列定义。
本文所用术语“MASP-2依赖性补体活化”包括凝集素途径的MASP-2-依赖性活化,其发生在生理条件下(即,在Ca++存在的情况下),导致形成凝集素途径C3转化酶C4b2a,和在C3裂解产物C3b聚集后,形成随后的C5转化酶C4b2a(C3b)n,所述C5转化酶C4b2a(C3b)n已经被确定为主要引起调理作用。
本文所用术语“替代途径”是指例如由酵母聚糖触发的补体活化,所述酵母聚糖来自真菌和酵母细胞壁、革兰氏阴性外膜的脂多糖(LPS)和兔红细胞以及来自多种纯的多糖、兔红细胞、病毒、细菌、动物肿瘤细胞、寄生虫和受损细胞,其在传统上一直认为是由来自补体因子C3自发水解产生C3b而引起的。
本文所用术语“凝集素途径”是指通过血清和非血清碳水化合物结合蛋白(包括甘露聚糖结合凝集素(MBL)、CL-11和纤维胶凝蛋白(H-纤维胶凝蛋白、M-纤维胶凝蛋白或L-纤维胶凝蛋白)的特异性结合而发生的补体活化。
本文所用术语“经典途径”是指由抗体与外源颗粒结合而触发的并且需要结合识别分子C1q的补体活化。
本文所用术语“MASP-2抑制剂”是指与MASP-2结合或直接与MASP-2相互作用并有效抑制MASP-2依赖性补体活化的任何试剂,包括抗MASP-2抗体及其MASP-2结合片段、天然和合成肽、小分子、可溶性MASP-2受体、表达抑制剂和分离的天然抑制剂,还包括在凝集素途径中为结合其他识别分子(例如MBL、H-纤维胶凝蛋白、M-纤维胶凝蛋白或L-纤维胶凝蛋白)而与MASP-2竞争的肽,但是不包括与这些其他的识别分子结合的抗体。用于本发明方法的MASP-2抑制剂可使MASP-2依赖性补体活化降低大于20%,诸如大于50%,诸如大于90%。在一个实施方案中,MASP-2抑制剂使MASP-2依赖性补体活化降低大于90%(即导致仅仅10%或更低的MASP-2补体活化)。
本文所用术语“抗体”包括衍生自产生抗体的任何哺乳动物(例如小鼠、大鼠、兔和包括人在内的灵长类动物),或衍生自杂交瘤、噬菌体选择、重组表达或转基因动物(或产生抗体或抗体片段的其他方法)并与目标多肽诸如例如MASP-2多肽或其部分特异性结合的抗体及其抗体片段。并不意在关于抗体的来源或其制备的方式(例如,通过杂交瘤、噬菌体选择、重组表达、转基因动物、肽合成等)对术语“抗体”进行限定。示例性的抗体包括多克隆抗体、单克隆抗体和重组抗体;全特异性(pan-specific)、多特异性抗体(例如双特异性抗体、三特异性抗体);人源化抗体;鼠抗体;嵌合的、小鼠?人、小鼠?灵长类、灵长类?人单克隆抗体;和抗独特型抗体,且可以是任何完整的抗体或其片段。本文所用术语“抗体”不仅包括完整的多克隆或单克隆抗体,而且包括其片段(诸如dAb、Fab、Fab'、F(ab')2、Fv),单链(ScFv),其合成变体,天然存在的变体,包含抗体部分与具有所需特异性的抗原结合片段的融合蛋白,人源化抗体,嵌合抗体,和包含具有所需特异性的抗原结合位点或片段(表位识别位点)的免疫球蛋白分子的任何其他经修饰后的构型。
“单克隆抗体”是指均质抗体群体,其中所述单克隆抗体是由参与选择性结合表位的氨基酸(天然存在的氨基酸和非天然存在的氨基酸)构成的。单克隆抗体对于目标抗原是高度特异性的。术语“单克隆抗体”不仅包括完整单克隆抗体和全长单克隆抗体,而且包括其片段(诸如Fab、Fab'、F(ab')2、Fv),单链(ScFv),其变体,包含抗原结合部分的融合蛋白,人源化单克隆抗体,嵌合单克隆抗体,和包含具有所需特异性和与表位结合能力的抗原结合片段(表位识别位点)的免疫球蛋白分子的任何其他经修饰的构型。并不意在关于抗体的来源或其制备的方式(例如,通过杂交瘤、噬菌体选择、重组表达、转基因动物等)进行限定。该术语包括“抗体”定义之下的上述完整免疫球蛋白以及片段等。
本文所用术语“抗体片段”是指得自或涉及全长抗体,诸如,例如抗MASP-2抗体的一部分,一般包括抗原结合区或其可变区。抗体片段的说明性实例包括Fab、Fab'、F(ab)2、F(ab')2和Fv片段、scFv片段、双抗体、线性抗体、单链抗体分子和由抗体片段形成的多特异性抗体。
本文所用的“单链Fv”或“scFv”抗体片段包括抗体的VH和VL结构域,其中这些结构域存在于单一多肽链上。Fv多肽一般还包括VH与VL结构域之间的多肽接头,这使得scFv能够形成所需的抗原结合结构。
本文所用的“嵌合抗体”是含有得自非人物种(例如啮齿动物)抗体的可变结构域和互补决定区的重组蛋白,而抗体分子的其余部分来源于人抗体。
本文所用的“人源化抗体”是包含符合源自非人免疫球蛋白的特异性互补决定区的最小序列的嵌合抗体,该特异性互补决定区被植入人抗体构架中。人源化抗体通常是其中只有抗体互补决定区是非人来源的重组蛋白。
本文所用术语“甘露聚糖结合凝集素”(“MBL”)等同于甘露聚糖结合蛋白(“MBP”)。
本文所用的“膜攻击复合物”(“MAC”)是指插入并破坏膜的5种末端补体组分(C5b组合C6、C7、C8和C-9)的复合物(也称为C5b-9)。
本文所用的“受试者”包括所有哺乳动物,包括但不限于人、非人灵长类动物、狗、猫、马、绵羊、山羊、牛、兔、猪和啮齿动物。
本文所用的氨基酸残基的缩写如下:丙氨酸(Ala;A)、天冬酰胺(Asn;N)、天冬氨酸(Asp;D)、精氨酸(Arg;R)、半胱氨酸(Cys;C)、谷氨酸(Glu;E)、谷胺酰胺(Gln;Q)、甘氨酸(Gly;G)、组氨酸(His;H)、异亮氨酸(Ile;I)、亮氨酸(Leu;L)、赖氨酸(Lys;K)、甲硫氨酸(Met;M)、苯丙氨酸(Phe;F)、脯氨酸(Pro;P)、丝氨酸(Ser;S)、苏氨酸(Thr;T)、色氨酸(Trp;W)、酪氨酸(Tyr;Y)和丙氨酸(Val;V)。
从最广意义上看,天然存在的氨基酸可根据各个氨基酸侧链的化学特性来分组。“疏水”氨基酸是指Ile、Leu、Met、Phe、Trp、Tyr、Val、Ala、Cys或Pro。“亲水”氨基酸是指Gly、Asn、Gln、Ser、Thr、Asp、Glu、Lys、Arg或His。氨基酸的这种分组可进一步细分如下。“不带电荷的亲水”氨基酸是指Ser、Thr、Asn或者Gln。“酸性”氨基酸是指Glu或Asp。“碱性”氨基酸是指Lys、Arg或者His。
本文所用术语“保守的氨基酸取代”通过下面每组中氨基酸之间的取代来说明:(1)甘氨酸、丙氨酸、缬氨酸、亮氨酸和异亮氨酸;(2)苯丙氨酸、酪氨酸和色氨酸;(3)丝氨酸和苏氨酸;(4)天冬氨酸和谷氨酸;(5)谷胺酰胺和天冬酰胺;和(6)赖氨酸、精氨酸和组氨酸。
本文所用术语“寡核苷酸”是指核糖核酸(RNA)或脱氧核糖核酸(DNA)或其模拟物的寡聚体或多聚体。该术语还涵盖由天然存在的核苷酸、糖和核苷间(骨架)共价键以及具有非天然存在的修饰的寡核苷酸所组成的那些寡核碱基。
本文所用的“表位”是指由抗体结合的蛋白(例如,人MASP-2蛋白)上的位点。“重叠表位”包括至少一个(例如,二、三、四、五、或六个)共同的氨基酸残基,包括线性和非线性表位。
本文所用术语“多肽”、“肽”和“蛋白”可以互换使用,并且是指氨基酸的任何肽连接链,无论长度或翻译后修饰。本文所述的MASP-2蛋白可以含有或可以是野生型蛋白,或者可以是具有不超过50个(例如,不超过一、两、三、四、五、六、七、八、九、十、12、15、20、25、30、35、40、或50个)保守氨基酸取代的变体。保守取代通常包括下列组内的取代:甘氨酸和丙氨酸;缬氨酸、异亮氨酸和亮氨酸;天冬氨酸和谷氨酸;天冬酰胺、谷氨酰胺、丝氨酸和苏氨酸;赖氨酸、组氨酸和精氨酸;以及苯丙氨酸和酪氨酸。
在一些实施方案中,人MASP-2蛋白的氨基酸序列可以与具有SEQ ID NO:5所示的氨基酸序列的人MASP-2蛋白是或大于70(例如, )%相同。
在一些实施方案中,肽片段可以是至少6(例如,至少







百分比(%)氨基酸序列同一性定义为在比对序列和引入缺口之后,候选序列中,如果需要,达到最大百分比序列同一性的与参考序列中的氨基酸相同的氨基酸的百分比。用于确定百分比序列同一性的目的的比对可以以本领域技术内的各种方式,例如,使用公开获得的计算机软件诸如BLAST、BLAST-2、ALIGN、ALIGN-2 或Megalign (DNASTAR)软件来实现。用于测量比对的适当参数,包括实现待比较序列全长内的最大比对所需的任何算法可以通过已知方法来确定。
II. 发明概述
凝集素(MBP、M-纤维胶凝蛋白、H-纤维胶凝蛋白和L-纤维胶凝蛋白和CL-11)是引发先天性补体系统的特异性识别分子,该系统包括凝集素起始途径和放大末端补体效应分子的凝集素起始的活化的相关末端途径放大环路。C1q是引发获得性补体系统的特异性识别分子,该系统包括经典起始途径和放大末端补体效应分子的C1q起始的活化的相关末端途径放大环路。我们将这两个主要的补体活化系统分别称为凝集素依赖性补体系统和C1q依赖性补体系统。
除了在免疫防御中的基本作用外,补体系统是造成许多临床疾病的组织损伤的原因。因此,迫切需要开发治疗上有效的补体抑制剂以抑制这些副作用。如果认识到其可能抑制凝集素介导的MASP-2途径而使经典途径完整,则就能够理解我们非常需要的是仅仅特异性抑制引起特定病理的补体活化系统而又不完全停止补体的免疫防御能力。例如,在其中补体活化主要由凝集素依赖性补体系统介导的疾病状态中,仅特异性抑制该系统将是有利的。这将保持C1q依赖性补体活化系统的完整性以处理免疫复合物加工以及帮助宿主防御感染。
在特异性抑制凝集素依赖性补体系统的治疗药物的研发中,优选的作为靶标的蛋白成分是MASP-2。在凝集素依赖性补体系统的所有已知的蛋白成分(MBL、H-纤维胶凝蛋白、M-纤维胶凝蛋白、L-纤维胶凝蛋白、MASP-2、C2-C9、因子B、因子D和备解素)中,只有MASP-2是凝集素依赖性补体系统所独有并且是这个系统起作用所必需的。凝集素(MBL、H-纤维胶凝蛋白、M-纤维胶凝蛋白、L-纤维胶凝蛋白和CL-11)也是凝集素依赖性补体系统中独有的成分。然而,这些凝集素成分任一种的丧失并不一定抑制系统活化,这是由于凝集素冗余所致。为了保证抑制凝集素依赖性补体活化系统,抑制所有5种凝集素可能是必要的。此外,由于还已知MBL和纤维胶凝蛋白具有不依赖于补体的调理活性,因此抑制凝集素功能将导致丧失这种有利的抗感染的宿主防御机制。相反,如果MASP-2是抑制靶标的话,这种不依赖于补体的凝集素调理活性会保持完整。MASP-2作为抑制凝集素依赖性补体活化系统的治疗靶的额外好处是,MASP-2的血浆浓度是所有补体蛋白中最低的(约500 ng/ml);因此,为得到完全抑制可能要足够的相对低浓度的高亲和力MASP-2抑制剂(Moller-Kristensen, M.,等人, J. Immunol Methods 282:159-167, 2003)。
III. MASP-2在各种疾病和病症中的作用以及使用MASP-2抑制剂的治疗方法
肾疾病
各种各样的肾疾病的发病机制中涉及补体系统的活化,包括肾小球系膜增生性肾小球肾炎(IgA肾病、贝惹病(Berger's disease)) (Endo, M.,等人, Clin. Nephrology 55:185-191, 2001)、膜性肾小球肾炎(Kerjashki, D., Arch B Cell Pathol. 58:253-71, 1990; Brenchley, P.E.,等人, Kidney Int., 41:933-7, 1992; Salant, D.J.,等人, Kidney Int. 35:976-84, 1989)、膜增生性肾小球肾炎(肾小球系膜毛细血管性肾小球肾炎) (Bartlow, B.G.,等人, Kidney Int. 15:294-300, 1979; Meri, S.,等人, J. Exp. Med. 175:939-50, 1992)、急性感染后肾小球肾炎(链球菌感染后肾小球肾炎)、冷球蛋白血症性肾小球肾炎(Ohsawa, I.,等人, Clin Immunol. 101:59-66, 2001)、狼疮肾炎(Gatenby, P.A., Autoimmunity 11:61-6, 1991)和亨诺赫–舍恩莱因紫癜肾炎(Endo, M.,等人, Am. J. Kidney Dis. 35:401-407, 2000)。对肾疾病中所涉及的补体的认识已有几十年,但是有关它在肾疾病的发作、进展和消退期的确切作用仍存在较多争议。在正常条件下,补体的影响对宿主是有利的,但是补体的不当活化和沉积可能引起组织损伤。
有充分的证据表明,肾小球炎症即肾小球肾炎常常是由于免疫复合物沉积在肾小球或肾小管结构上引发的,继而又引发了补体活化、炎症和组织损伤。Kahn和Sinniah证实,取自各种形式肾小球肾炎患者的活检组织的肾小管基底膜上,C5b-9的沉积增加(Kahn, T.N.,等人, Histopath. 26:351-6, 1995)。在IgA肾病患者的研究中(Alexopoulos, A.,等人, Nephrol. Dial. Transplant10:1166-1172, 1995),C5b-9在肾小管上皮/基底膜结构上的沉积与血浆肌酸酐水平相关。另一项膜性肾病的研究证实了临床结果与尿sC5b-9水平之间的关系(Kon, S.P.,等人, Kidney Int. 48:1953-58, 1995)。sC5b-9水平升高与预后不良呈正相关。Lehto等人测得膜性肾小球肾炎患者尿中CD59以及C5b-9水平升高(Lehto, T.,等人, Kidney Int. 47:1403-11, 1995),CD59是一种抑制质膜中膜攻击复合物的补体调节因子。取自这些相同患者的活检组织样品的组织病理学分析证实,C3和C9蛋白在肾小球中沉积,而C59在这些组织中的表达比正常肾组织的表达减少。这些不同的研究表明,正在发生补体介导的肾小球肾炎导致尿中排泄出补体蛋白,这与组织损伤程度和疾病预后的程度相互关联。
抑制各种肾小球肾炎动物模型的补体活化也证实了该病病因中补体活化的重要性。在膜增生性肾小球肾炎(MPGN)模型中,C6缺陷型大鼠(不能形成C5b-9)中输注抗Thy1抗血清导致肾小球细胞增殖减少90%,血小板和巨噬细胞浸润降低80%,IV型胶原合成减少(肾小球基质扩展的标志)以及蛋白尿比C6+正常大鼠的少50% (Brandt, J.,等人, Kidney Int. 49:335-343, 1996)。这些结果表明在这种大鼠抗胸腺细胞血清模型中,C5b-9是补体导致的组织损伤的主要介质。在另一种肾小球肾炎模型中,输注分级剂量的兔抗大鼠肾小球基底膜,产生了多形核白细胞(PMN)剂量依赖性地流入,这种多形核白细胞流入通过预先用眼镜蛇毒因子处理(消耗补体)而被减弱(Scandrett, A.L.,等人, Am. J. Physiol. 268:F256-F265, 1995)。眼镜蛇毒因子处理的大鼠也显示出组织病理减轻、长期蛋白尿减少以及肌酸酐水平比对照大鼠的降低。Couser等人采用三种GN大鼠模型(抗胸腺细胞血清、Con A抗Con A和被动性海曼肾炎(passive Heymann nephritis)),证实使用重组sCR1蛋白抑制补体这类方法的潜在治疗功效(Couser, W.G.,等人, J. Am. Soc. Nephrol. 5:1888-94, 1995)。比起对照大鼠,用sCR1处理的大鼠则出现PMN、血小板和巨噬细胞流入显著减少、肾小球系膜裂解(mesangiolysis)和蛋白尿减少。通过在NZB/W F1小鼠模型中使用抗C5 MoAb,为补体活化在肾小球肾炎中的重要性提供了进一步的证据。抗C5 MoAb抑制C5的裂解,从而阻断C5a和C5b-9的产生。用抗C5 MoAb连续治疗6个月,导致肾小球肾炎进程显著改善。美国康涅狄格州纽黑文市的Alexion Pharmaceuticals公司正在开发一种防止人补体组分C5裂解成其促炎成分的人源化抗C5 MoAb单克隆抗体(5G1.1),作为肾小球肾炎潜在的治疗方法。
通过对遗传上缺失特定补体组分的患者的研究,为补体在肾脏损伤中的病理学作用提供了直接证据。许多报道证明了肾疾病与补体调节因子H缺陷有关(Ault, B.H., Nephrol.14:1045-1053, 2000; Levy, M.,等人, Kidney Int. 30:949-56, 1986; Pickering, M.C.,等人, Nat. Genet. 31:424-8, 2002)。因子H的缺乏导致因子B和C3的低血浆水平以及C5b-9的消耗。非典型性膜增生性肾小球肾炎(MPGN)和特发性溶血性尿毒综合征(HUS)都与因子H的缺乏有关。因子H缺陷型猪(Jansen, J.H.,等人, Kidney Int. 53:331-49, 1998)和因子H敲除小鼠(Pickering, M.C., 2002)都表现出MPGN样症状,这就证实了因子H在补体调节中的重要性。其他补体组分的缺乏与系统性红斑狼疮(SLE)发生后的继发性肾病有关(Walport, M.J., Davies,等人, Ann. N.Y. Acad. Sci. 815:267-81, 1997)。C1q、C4和C2的缺乏使得非常容易通过与缺乏清除免疫复合物和凋亡物质有关的机制而发生SLE。在这些SLE患者中许多发生了狼疮肾炎,其特征在于免疫复合物沉积在整个肾小球中。
通过鉴定患者体内针对补体组分的自身抗体(其中一些与肾疾病直接相关),为补体活化与肾疾病关联提供了进一步的证据(Trouw, L.A.,等人, Mol. Immunol. 38:199-206, 2001)。这些自身抗体的许多种与肾疾病的关联性如此高,以致于引入了术语肾炎因子(NeF)来表示这种活性。在临床研究中,肾炎因子阳性患者中大约有50%发生MPGN (Spitzer, R.E.,等人, Clin. Immunol. Immunopathol. 64:177-83, 1992)。C3NeF是针对替代途径C3转化酶(C3bBb)的自身抗体,它使这种转化酶稳定,从而促进替代途径活化(Daha, M.R.,等人, J. Immunol. 116:1-7, 1976)。同样地,对经典途径C3转化酶(C4b2a)具有特异性的自身抗体(称为C4NeF)使该转化酶稳定,从而促进经典途径活化(Daha, M.R.等人, J. Immunol. 125:2051-2054, 1980; Halbwachs, L.,等人, J. Clin. Invest. 65:1249-56, 1980)。已表明,抗C1q自身抗体与SLE患者的肾炎有关(Hovath, L.,等人, Clin. Exp. Rheumatol. 19:667-72, 2001; Siegert, C.,等人, J. Rheumatol. 18:230-34, 1991; Siegert, C.,等人, Clin. Exp. Rheumatol. 10:19-23, 1992),还有报道指出这些抗C1q自身抗体的效价升高被用来预测肾炎的突发(Coremans, I.E.,等人, Am. J. Kidney Dis. 26:595-601, 1995)。从SLE患者尸检肾洗脱出的免疫沉积物揭示这些抗C1q自身抗体的累积(Mannick, M.,等人, Arthritis Rheumatol. 40:1504-11, 1997)。所有这些事实都指向这些自身抗体的病理学作用。然而,并不是所有具有抗C1q自身抗体的患者都发生肾病,一些健康个体中也有低效价的抗C1q自身抗体(Siegert, C.E.,等人, Clin. Immunol. Immunopathol. 67:204-9, 1993)。
除了补体活化的替代途径和经典途径以外,凝集素途径在肾疾病中也有重要的病理学作用。通过免疫组织化学技术,从获自诊断为患有几种不同肾病的患者的肾活检材料中,检测出高水平的MBL、MBL相关的丝氨酸蛋白酶及补体活化产物,这些肾病包括亨诺赫–舍恩莱因紫癜肾炎(Endo, M.,等人, Am. J. Kidney Dis. 35:401-407, 2000)、冷球蛋白血症性肾小球肾炎(Ohsawa, I.,等人, Clin. Immunol. 101:59-66, 2001)以及IgA肾病(Endo, M.,等人, Clin. Nephrology 55:185-191, 2001)。因此,尽管已经清楚补体与肾病之间的相关性这一事实几十年,但是关于补体如何确切影响这些肾病的数据仍远远不够完整。
因此,本发明的一个方面涉及通过将包含治疗有效量的MASP-2抑制剂在药物载体中的组合物,施用于患有肾病的受试者以治疗这类疾病,所述疾病包括但不限于肾小球系膜增生性肾小球肾炎、膜性肾小球肾炎、膜增生性肾小球肾炎(肾小球系膜毛细血管性肾小球肾炎)、急性感染后肾小球肾炎(链球菌感染后肾小球肾炎)、冷球蛋白血症性肾小球肾炎、狼疮肾炎、亨诺赫–舍恩莱因紫癜肾炎或者IgA肾病。可以全身性将MASP-2抑制剂施用于受试者,诸如通过动脉内、静脉内、肌内、皮下或其他胃肠外施用,或者对于非肽能药物可通过口服施用。可以在一段长的时间内定期施用MASP-2抑制剂,以治疗或控制慢性病,或者可以在急性创伤或损伤之前、期间或之后的一段时间单次或重复施用。
血液病
脓毒症是由于患者对侵入微生物的过度反应而引起的。补体系统的主要功能是配合对侵入细菌和其他病原体的炎症反应。与这种生理作用相一致,许多研究显示补体活化在脓毒症的发病机制中具有主要作用(Bone, R.C., Annals. Internal. Med. 115:457-469, 1991)。脓毒症临床表现的定义在不断发展中。通常将脓毒症定义为对感染的系统性宿主应答。然而,在许多情况下,有脓毒症症状的患者中没有发现感染的临床证据(如血液培养细菌阳性)。这种矛盾之处最早是在1992年的共识会议上确定术语“全身炎症反应综合征” (systemic inflammatory response syndrome,SIRS)时被注意到的,SIRS不需要确定的细菌感染的存在(Bone, R.C.,等人, Crit. Care Med. 20:724-726, 1992)。目前普遍认为,伴随脓毒症和SIRS的是不能调控炎症反应。出于该简述的目的,我们会考虑脓毒症的临床定义还包括严重脓毒症、败血症性休克和SIRS。
在1980年代末期之前,脓毒症患者的主要感染源是革兰氏阴性菌。已知革兰氏阴性菌细胞壁的主要成分脂多糖(LPS)刺激炎症介质从各种细胞类型中释放出来,并且当注射到动物体内时诱导急性感染症状(Haeney, M.R.,等人, Antimicrobial Chemotherapy 41(Suppl. A):41-6, 1998)。有趣的是,引起感染的微生物的范围似乎从1970年代末期和1980年代主要是革兰氏阴性菌变成了现今的主要是革兰氏阳性菌,其原因目前不清楚(Martin, G.S.,等人, N. Eng. J. Med. 348:1546-54, 2003)。
许多研究表明补体活化在介导炎症和引起休克、特别是败血症性休克和出血性休克特征中的重要性。革兰氏阴性和革兰氏阳性生物一般都促使败血症性休克。LPS是主要经由替代途径的有效的补体活化因子,尽管抗体介导的经典途径活化也有发生(Fearon, D.T.,等人, N. Engl. J. Med. 292:937-400, 1975)。革兰氏阳性细胞壁的主要成分是肽聚糖和脂磷壁酸,两种成分都是替代补体途径的有效活化因子,尽管在特异性抗体存在时,它们也能激活经典补体途径(Joiner, K.A.,等人, Ann. Rev. Immunol. 2:461-2, 1984)。
脓毒症发病机制中最初涉及补体系统是当研究人员注意到过敏毒素C3a和C5a介导各种也可能发生在脓毒症期间的炎症反应时发现的。这些过敏毒素引起血管扩张和微血管通透性增加,这是在败血症性休克中起着重要作用的事件(Schumacher, W.A.,等人, Agents Actions 34:345-349, 1991)。此外,过敏毒素诱导支气管痉挛、组胺从肥大细胞中释放以及血小板聚集。而且,它们对粒细胞发挥了许多作用,诸如趋化、聚集、粘附、溶酶体酶的释放、毒性超氧阴离子的产生和白三烯的形成(Shin, H.S.,等人, Science 162:361-363, 1968; Vogt, W., Complement 3:177-86, 1986)。认为这些生物学作用在脓毒症并发症诸如休克或急性呼吸窘迫综合征(ARDS)的发生中起着作用(Hammerschmidt, D.E.,等人, Lancet 1:947-949, 1980; Slotman, G.T.,等人, Surgery 99:744-50, 1986)。此外,过敏毒素C3a水平的升高与脓毒症的致命结果有关(Hack, C.E.,等人, Am. J. Med. 86:20-26, 1989)。在一些休克动物模型中,某些补体缺陷型品系(如C5缺陷型品系)对LPS的输注效应具有更高的抗性(Hseuh, W.,等人, Immunol. 70:309-14, 1990)。
研究显示,在啮齿动物脓毒症发作期间,用抗体阻断C5a产生极大地提高了存活率(Czermak, B.J.,等人, Nat. Med. 5:788-792, 1999)。当用抗体或小分子抑制剂阻断C5a受体(C5aR)时,得到了类似的结果(Huber-Lang, M.S.,等人, FASEB J. 16:1567-74, 2002; Riedemann, N.C.,等人, J. Clin. Invest. 110:101-8, 2002)。较早期对猴的实验研究表明,C5a的抗体阻断减少了大肠杆菌(E. coli)诱发的败血症性休克和成人呼吸窘迫综合征(Hangen, D.H.,等人, J. Surg. Res. 46:195-9, 1989; Stevens, J.H.,等人, J. Clin. Invest. 77:1812-16, 1986)。与脓毒症不太严重的患者和幸存者相比,C5a在脓毒症病人体内升高,并且与显著降低存活率和多器官衰竭有关(Nakae, H.,等人, Res. Commun. Chem. Pathol.Pharmacol. 84:189-95, 1994; Nakae,等人, Surg. Today 26:225-29, 1996; Bengtson, A.,等人, Arch. Surg. 123:645-649, 1988)。C5a在脓毒症期间发挥其有害作用的机制还有待更详细的研究,但是最近的数据表明,脓毒症期间C5a的产生显著损害血液嗜中性粒细胞的先天免疫功能(Huber-Lang, M.S.,等人, J. Immunol. 169:3223-31, 2002)、它们引起呼吸爆发的能力及其产生细胞因子的能力(Riedemann, N.C.,等人, Immunity 19:193-202, 2003)。此外,脓毒症期间的C5a产生似乎具有促凝血效应(Laudes, I.J.,等人, Am. J. Pathol. 160:1867-75, 2002)。在脓毒症和ARDS动物模型中,补体调控蛋白CI INH也显示出功效(Dickneite, G., Behring Ins. Mitt. 93:299-305, 1993)。
凝集素途径在脓毒症的发病机制中也可能有作用。已显示MBL结合临床上重要的各种微生物,包括革兰氏阴性菌和革兰氏阳性菌,并激活凝集素途径(Neth, O.,等人, Infect. Immun. 68:688, 2000)。脂磷壁酸(LTA)越来越被视作LPS的革兰氏阳性对应物(Gram-positive counterpart)。它是诱导细胞因子从单核吞噬细胞和全血中释放的有效免疫刺激剂(Morath, S.,等人, J. Exp. Med. 195:1635, 2002; Morath, S.,等人, Infect. Immun. 70:938, 2002)。最近证实,L-纤维胶凝蛋白与从许多革兰氏阳性菌(包括金黄色葡萄球菌(Staphylococcus aureus))分离的LTA特异性结合,并且激活凝集素途径(Lynch, N.J.,等人, J. Immunol. 172:1198-02, 2004)。还显示,MBL能结合来自肠球菌(Enterococcus spp)的LTA,其中多聚甘油磷酸链被糖基取代,但是不结合来自其他9种菌种(包括金黄色葡萄球菌)的LTA (Polotsky, V.Y.,等人, Infect. Immun. 64:380, 1996)。
因此,本发明一方面提供用于治疗脓毒症或由脓毒症引起的疾病的方法,该方法通过将包含治疗有效量的MASP-2抑制剂在药物载体中的组合物施用于患有脓毒症或由脓毒症引起的疾病的受试者实施,所述疾病包括但不限于严重脓毒症、败血症性休克、脓毒症引起的急性呼吸窘迫综合征和全身性炎症反应综合征。本发明提供治疗其他血液病的有关方法,该方法通过将包含治疗有效量的MASP-2抑制剂在药物载体中的组合物施用于患此类疾病的受试者实施,所述其他血液病包括出血性休克、溶血性贫血、自身免疫性血栓形成性血小板减少性紫癜(TTP)、溶血性尿毒综合征(HUS)、非典型溶血性尿毒综合征(aHUS)或其他骨髓/血液破坏性疾病。可以将MASP-2抑制剂全身性施用于受试者,例如通过动脉内、静脉内、肌内、吸入(特别是在ARDS的情况下)、皮下或其他胃肠外施用,或者对于非肽能抑制剂可口服施用。MASP-2抑制剂组合物可与一种或多种另外的治疗药联用以对抗脓毒症和/或休克的后遗症。对于晚期脓毒症或休克或者由此引起的疾病状况来说,MASP-2抑制剂组合物可适于以速效剂型施用,例如通过静脉内或动脉内推注含有MASP-2抑制剂组合物的溶液剂来施用。可按照医师规定重复施用,直到疾病消退。
MASP-2在阵发性睡眠性血红蛋白尿症中的作用和使用MASP 2抑制剂的治疗方法
PNH概述
阵发性睡眠性血红蛋白尿症(PNH),有时也称为Marchiafava-Micheli综合征,是一种获得性的潜在威胁生命的血液疾病。PNH可以自身发生(称为“原发性PNH”)或在其他骨髓病症诸如再生障碍性贫血的背景中发生(称为“继发性PNH”)。大多数情况是原发性PNH。PNH的特征在于补体引起的红细胞的破坏(溶血)、低红细胞计数(贫血)、血栓形成和骨髓衰竭。PNH的实验室发现显示与血管内溶血性贫血一致的变化:在作为可能原因的自身反应性RBC-结合抗体不存在的情况下,血红蛋白低、乳酸脱氢酶升高、网织红细胞计数升高(由骨髓释放以取代破坏的细胞的不成熟红细胞)、胆红素升高(血红蛋白的分解产物)。
PNH的标志是由循环RBCs的表面上的末端活化补体组分包括膜攻击复合物的失调活化引起的慢性补体介导的溶血。PNH RBCs经受由于其表面上补体调节物CD55和CD59的不存在而导致的失控的补体活化和溶血(Lindorfer, M.A.,等人, Blood 115(11):2283-91 (2010), Risitano,等人, Mini-Reviews in Medicinal Chemistry, 11:528-535 (2011))。CD55和CD59在正常RBC上大量表达,并且控制补体活化。CD55充当替代途径的负调节物,抑制替代途径C3转化酶(C3bBb)复合物的装配和加快提前形成的转化酶的衰变,从而阻断膜攻击复合物(MAC)的形成。CD59直接通过结合C5b678复合物和防止C9结合和聚合而抑制补体膜攻击复合物。
尽管溶血和贫血是PNH患者的主要临床特征,但是该疾病是进一步包括作为临床发现部分的血栓形成和骨髓衰竭的一种复杂血液病症(Risitano等人, MiniReviews in Med Chem11:528-535 (2011))。在分子水平,PNH由缺乏功能性PIG A基因的造血干细胞的异常克隆扩增所引起。PIG A是编码GPI锚定的A类糖蛋白包括CD55和CD59的稳定表面表达所需的糖基磷脂酰肌醇(GPI)转移酶的X连锁基因。对于目前研究中的原因,由自发体细胞突变导致的具有功能失调的PIG A基因的造血干细胞可以经历克隆扩增到其中其后代构成显著部分的外周血造血干细胞混合物的点。尽管红细胞和突变干细胞克隆的淋巴细胞后代两者都缺乏CD55和CD59,但是只有RBC在它们进入循环后经历裂解。
目前PNH的治疗包括用于贫血的输血、用于血栓形成的抗凝和使用单克隆抗体依库丽单抗(Soliris?),其通过抑制补体系统保护血细胞免于免疫破坏(Hillmen P.等人, N. Engl. J. Med. 350(6):552-559 (2004))。依库丽单抗(Soliris?)是靶向补体组分C5的人源化单克隆抗体,其阻断C5转化酶对其裂解,从而防止C5a的产生和MAC的装配。用依库丽单抗治疗PNH患者已经导致血管内溶血的减少,如通过乳酸脱氢酶(LDH)所测量的,导致约一半患者中的血红蛋白稳定和输血不依赖(Risitano等人, Mini-Reviews in Medicinal Chemistry, 11(6) (2011))。尽管几乎所有经历依库丽单抗治疗的患者达到了正常或几乎正常的LDH水平(由于血管内溶血的控制),但是只有约三分之一患者达到约11克/dL的血红蛋白值,并且约等同比例的依库丽单抗的其余患者继续表现出温和至严重的(即输血依赖性)贫血(Risitano A.M.等人, Blood 113:4094-100 (2009))。如Risitano等人, Mini-Reviews in Medicinal Chemistry 11:528-535 (2011)中所述,表明依库丽单抗的PNH患者含有大量结合至其PNH红细胞的C3片段(而未经治疗的患者则没有)。该发现导致认识到,在依库丽单抗治疗的PNH患者中,由于C5阻断而不再溶血的PNH RBC现在可以聚集丰富量的作为调理素起作用的膜结合的C3片段,导致它们在网状内皮细胞中通过特异性C3受体的捕捉和随后的血管外溶血。因此,在防止血管内溶血和导致的后遗症的同时,依库丽单抗治疗仅仅将这些RBC的倾向从血管内溶血转移到血管外溶血,导致在许多患者中大量残留未治疗的贫血(Risitano A.M.等人, Blood 113:4094-100 (2009))。因此,对于发展C3片段介导的血管外溶血的那些患者需要除了使用依库丽单抗以外的治疗策略,因为他们继续需要红细胞输血。这种C3片段靶向方法已经在实验系统中证明了效用(Lindorfer等人, Blood 115:2283-91, 2010)。
PNH中的补体起始机制
在PNH中阴性补体调节物CD55和CD59的缺陷表达组合依库丽单抗在预防血管内溶血的有效性之间的因果关系,明确地将PNH定义为由补体系统引起的状况。在该范例被广泛接受,但是起始补体活化的事件和补体活化途径的性质仍然没有解决。因为CD55和CD59在对于所有补体起始途径共有的补体级联中阴性调节末端放大步骤,这些分子的缺陷将导致末端补体活化的放大,而无论补体活化是否通过凝集素途径、通过经典途径或通过替代途径的自发转换所起始。因此,在PNH患者中,可以导致RBC表面上的C3b沉积的任何补体活化事件可以触发随后的扩增和病理学溶血(血管内和/或血管外溶血)并且沉淀溶血危象。触发PNH患者中的溶血危象的分子事件的明确机理理解仍然是难懂的。因为在经历溶血危象的PNH患者中没有补体起始事件是公然明显的,所以普遍的观点是,PNH中的补体活化可以由于替代途径的低水平空转活化(tick-over activation)而自发发生,随后其通过由于CD55和CD59的缺乏导致的末端补体活化的不当控制而放大。
然而,重要指出的是,在其自然历史中,PNH通常在某些事件诸如感染或损伤之后发展或恶化(Risitano, Biologics 2:205-222 (2008)),这已经显示触发补体活化。该补体活化应答不依赖于宿主对刺激病原体的先前免疫力,因此可能不涉及经典途径。相反,似乎该补体活化应答由凝集素结合到在微生物剂或受损宿主组织表面上表达的外来或“自我改变的”碳水化合物模式而起始。因此,在PNH中沉淀溶血危象的事件与通过凝集素起始的补体活化紧密相连。这使得非常可能凝集素活化途径提供最终导致PNH患者中的溶血的起始触发。
MASP-2抑制剂通过网状内皮系统阻断调理和PNH RBC的血管外溶血
本部分描述了在PNH的体外模型中MASP-2抑制剂对溶血的抑制作用。该发现支持MASP-2阻断剂(包括但不限于,结合并阻断MASP-2的功能的抗体)治疗患有PNH的各方面的受试者的效用,和还有MASP-2抑制剂在经历用C5-抑制剂诸如依库丽单抗的治疗的PNH患者中改善C3-片段介导的血管外溶血的作用的用途。
如上所详述,PNH患者由于从循环清除RBC的两种不同的机制而变得贫血:通过活化膜攻击复合物(MAC)的血管内溶血,和C3b调理和随后在补体受体结合和网状内皮系统的摄取之后的清除之后的血管外溶血。血管内溶血主要当患者用依库丽单抗治疗时得到防止。由于依库丽单抗阻断补体活化起始事件以及跟着发生的调理作用两者的下游发生的末端裂解效应机制,所以依库丽单抗不会阻断血管外溶血(Risitano A.M.等人, Blood 113:4094-100 (2009))。相反,未治疗的PNH患者中可能已经经历溶血的RBC现在可以在其表面上聚集活化的C3b蛋白,其增强网状内皮系统的摄取并增强它们的血管外溶血。因此,依库丽单抗治疗有效地将RBC倾向从血管内溶血转移到血管外溶血。作为结果,一些依库丽单抗治疗的PNH患者保持贫血。结果就是阻断上游的补体活化和防止PNH RBC的调理的试剂可以特别适合于阻断没有被依库丽单抗阻止的血管外溶血。
此处提供的数据表明,MASP-2依赖性补体活化是凝集素依赖性调理作用的占优势途径。因此,预计MASP-2抑制剂对于PNH中限制调理和抑制血管外溶血是有效的。
使用PNH的体外模型,我们证明,PNH中的补体活化和获得的溶血确实,至少部分地,由MASP-2依赖性补体??活化所起始,并且它不是独立于替代途径的功能。这些研究使用各种品系小鼠的甘露聚糖敏化的RBC,包括来自Crry缺陷小鼠的RBC(小鼠中末端补体途径的重要负调节物),以及来自CD55/CD59缺陷小鼠的RBC(其缺乏PNH患者中不存在的相同补体调节物)。当甘露聚糖敏化的Crry缺陷的RBC暴露于补体足够的人血清时,RBC在3%的血清浓度时有效溶血(图40),而补体缺陷的血清(HI:热灭活的)则不溶血。值得注意的是,在抗MASP-2抗体存在的情况下,补体足够血清具有降低的溶血活性,并且对于有效溶血需要6%血清(图40)。当测试CD55/CD59-缺陷RBC时得到了类似的观察结果(图42)。用抗MASP-2单克隆抗体补充的补体足够的人血清的有效性比未经处理的血清在支持溶血方面差约两倍。此外,与未经处理的血清相比,需要更高浓度的用抗MASP-2单克隆抗体处理的血清来促进未经处理的WT RBC的有效溶血(图40)。总之,这些数据表明,MASP-2依赖性补体活化明显有助于溶血反应。本文提供的数据揭示了以下PNH中贫血的致病机制:由于末端补体组分的失调活化和MAC形成导致的RBC裂解的血管内溶血,和C3b对RBCs的调理作用(其主要由MASP-2依赖性补体活化起始)引起的血管外溶血。因此,预计MASP-2抑制剂显著降低PNH患者中的血管内溶血。
血管外溶血,是导致PNH中贫血的红细胞破坏的不太剧烈、但同样重要的机制,主要是C3b 的调理作用(其主要由MASP-2依赖性补体活化介导)的结果。因此,MASP-2抑制剂在PNH中将优先阻断RBC调理作用和C3b和跟着发生的血管外溶血。预计MASP-2抑制剂的该独特治疗活性为所有PNH患者提供显著的治疗益处,因为目前不存在该致病过程的治疗。
MASP-2抑制剂作为末端补体阻断剂的辅助治疗:
本文提供的数据详细描述了PNH中RBC清除和贫血的两种致病机制:至少部分由MASP-2依赖性补体活化起始并且因此预期被MASP-2抑制剂有效抑制的血管内溶血,和由于MASP-2驱动并且因此被MASP-2抑制剂有效防止的C3b调理作用引起的血管外溶血。
良好证明的是,溶血的血管内和血管外机制两者导致PNH患者中的贫血(Risitano等人, Blood 113:4094-4100 (2009))。因此,在PNH的设置中,预期MASP-2的抑制解决血管内和血管外溶血两者,提供了相比于C5抑制剂依库丽单抗的显著优点。因此,可以预期,抑制血管内溶血并且防止血管外溶血的MASP-2-阻断剂在预防PNH患者中发生贫血的程度是有效的。
还已知C5-阻断剂(诸如依库丽单抗)有效地阻断血管内溶血但不干扰调理作用。这使得抗C5-治疗的PNH患者有大量残留性贫血,这是由于由MASP-2依赖性补体活化介导的血管外溶血仍未经治疗。因此,可以预期,防止血管内溶血的C5-阻断剂(诸如依库丽单抗)与防止血管外溶血的MASP-2抑制剂的组合比任一单独试剂在防止PNH患者中发生贫血中更有效。事实上,预期抗C5和MASP-2抑制剂的组合防止PNH中RBC破坏的所有相关机制,并且因此减少或阻断PNH的所有贫血症状。
还预期阻断导致C5活化和MAC沉积的补体系统的末端放大环路的其他试剂(包括,但并不限于阻断备解素、因子B或因子D或增强因子I、因子H或其他补体抑制因子的抑制活性的试剂)抑制血管内溶血。然而,不预期这些试剂干扰PNH患者中的MASP-2介导的调理作用。这使得此类试剂治疗的PNH患者有大量残留性贫血,这是由于由MASP-2依赖性补体活化介导的血管外溶血仍未经治疗。因此,可以预期,防止血管内溶血的此类试剂与防止血管外溶血的MASP-2抑制剂的组合的治疗比任一单独试剂在防止PNH患者中发生贫血中更有效。事实上,预期此类试剂和MASP-2抑制剂的组合防止PNH中RBC破坏的所有或大部分相关机制,并且因此阻断PNH的所有或大部分贫血症状。
MASP-2的抑制改善被脑膜炎奈瑟氏菌感染的受试者中的存活
如实施例30-32描述和图33-37中显示,MASP-2的抑制不降低被脑膜炎奈瑟氏菌感染后的存活。相反,令人惊讶地发现,在这些研究中,MASP-2抑制显著增强存活(图33和34)以及疾病评分(图36)。施用抗MASP2抗体产生相同结果(图37),在基因敲除小鼠品系中消除了作为可能原因的继发或补偿作用。在MASP-2去除动物中的这些有利结果与从血液中更快速清除奈瑟氏菌相一致(图35)。此外,如本文所述,将奈瑟氏菌与人血清孵育杀死了奈瑟氏菌(图38)。此外,添加阻断MASP-2依赖性凝集素途径补体活化的对于人MASP-2特异的功能性单克隆抗体增强了该杀伤响应,但施用同种型对照单克隆抗体则没有。在奈瑟氏菌对凝集素依赖性补体活化的背景下,MASP-2的阻断在体外增强了生物体的裂解破坏(图38)。因为奈瑟氏菌的裂解是幼稚宿主中的主要保护机制,所以MASP-2的体内阻断增强了奈瑟氏菌清除并且导致杀伤增强。这些结果是令人惊讶的,并且提供了相比于用依库丽单抗治疗的显著优势,这已经显示增加了对于威胁生命和致命脑膜炎球菌感染的易感性(Dmytrijuk A.,等人, The Oncologist 13:993-1000 (2008))。
根据上述,在一个方面,本发明提供了用于治疗阵发性睡眠性血红蛋白尿症(PNH)的方法,其通过将包含药学载体中的治疗有效量的MASP 2抑制剂的组合物施用于患有PNH或由PNH导致的状况(例如贫血、血红蛋白尿和血栓形成)的受试者而实现。将MASP-2抑制剂全身施用于患有PNH或由PNH导致的状况的受试者,诸如通过动脉内、静脉内、肌内、吸入、皮下或其他胃肠外施用,或者对于非肽能抑制剂可口服施用。
MASP-2在血栓性微血管病包括溶血性尿毒综合征(HUS)、非典型溶血性尿毒综合征(AHUS)和血栓性血小板减少性紫癜(TTP)中的作用和使用MASP 2抑制剂的治疗方法。
概述
血栓性微血管病(TMA)是特征在于小血管中的血凝块的病理学(Benz, K.;等人, Curr Opin Nephrol Hypertens 19(3):242–7 (2010))。对基础血管内皮的应力或损伤被认为是主要驱动因素。TMA的临床和实验室发现包括血小板减少、贫血、紫癜和肾功能衰竭。经典的TMA是溶血性尿毒综合征(HUS)和血栓性血小板减少性紫癜(TTP)。TMA的特征性基础病理特征是血小板活化和小动脉和小静脉中微血栓的形成。
通过对遗传上缺失特定补体组分的患者的研究,为补体在大量肾炎(a host of nephritides)中的病理学作用提供了直接证据。许多报道证明了肾脏损伤与补体调节因子H缺乏有关(Ault, B.H., Nephrol.14:1045-1053, 2000; Levy, M.,等人, Kidney Int. 30:949-56, 1986; Pickering, M.C.,等人, Nat. Genet. 31:424-8, 2002)。因子H缺陷导致由于因子B和C3的活化相关消耗导致的因子B和C3的低血浆水平。C5b-9的循环水平也在这些患者的血清中升高,暗示补体活化。膜增生性肾小球肾炎(MPGN)和特发性溶血性尿毒综合征(HUS)与因子H的缺乏或因子H的突变有关。因子H缺陷型猪(Jansen, J.H.,等人, Kidney Int. 53:331-49, 1998)和因子H敲除小鼠(Pickering, M.C., 2002)都表现出MPGN样症状,这就证实了因子H在补体调节中的重要性。其他补体组分的缺乏与系统性红斑狼疮(SLE)发生后的继发性肾病有关(Walport, M.J., Davies,等人, Ann. N.Y. Acad. Sci. 815:267-81, 1997)。C1q、C4和C2的缺乏使得非常容易通过与缺乏清除免疫复合物和凋亡物质有关的机制而发生SLE。在这些SLE患者中许多发生了狼疮肾炎,其特征在于免疫复合物沉积在整个肾小球中。
aHUS
非典型溶血性尿毒综合征(aHUS)是一组被称为“血栓性微血管病”的状况的部分。在非典型形式的HUS(aHUS)中,该疾病与缺陷的补体调节有关,并且可以是偶发性或家族性的。aHUS的家族性病例与编码补体活化或补体调节蛋白,包括补体因子H、因子I、因子B、膜辅因子CD46以及补体因子H-相关蛋白1(CFHR1)和补体因子H相关蛋白3(CFHR3)的基因中的突变相关。(Zipfel, P.F.,等人, PloS Genetics 3(3):e41 (2007))。与aHUS相关的遗传突变的该多样化阵列的统一特征是对细胞或组织表面上增强的补体活化的倾向。因此,本发明的一个方面包括通过施用有效量的MASP-2抑制剂而治疗患有与因子H缺陷相关的aHUS的患者。因此,本发明的另一个方面包括通过施用有效量的MASP-2抑制剂而治疗患有与因子I、因子B、膜辅因子CD46、CFHR1或CFHR3缺陷相关的HUS的患者。
新近已经朝着理解作为由多样组的突变补体因子引起的aHUS中的增强补体活化的基础的分子病理生理学取得了明显进展。该机制对于因子H突变是最好理解的。因子H是包含20个短共有重复(SCR)结构域的丰富血清蛋白,其在溶液中以及宿主细胞表面上都充当补体活化的负调节物。其靶向C3连同因子I和其他辅因子的活化形式,促进其失活,预防进一步补体活化。为了有效地控制在宿主细胞表面上的补体活化,因子H需要与宿主细胞相互作用,其由SCR结构域16-20所介导。迄今描述的与aHUS相关的所有因子H突变都在包括(SCR)结构域16-20的C末端区域中成簇。这些突变因子H蛋白在溶液中对于控制C3活化是完全有功能的,但在细胞表面上不能与宿主细胞表面相互作用,并且因此无法控制C3活化(Exp Med 204(6):1249-56 (2007))。因此,因子H的某些突变与aHUS相关,因为突变因子H蛋白不能与宿主细胞表面相互作用,并且因此不能有效地下调宿主细胞表面包括微血管内皮上的补体活化。作为结果,在具有因子H突变的患者中,一旦初始C3活化已经发生,则随后微血管内皮表面上的补体活化不减弱地前进。补体的这种不受控制的活化最终导致对血管内皮的进行性损伤,随后血小板聚集和微血管内凝血,和由通过部分闭塞微血管的RBC通过的剪切应力引起的溶血。因此,aHUS疾病表现和临床和实验室发现与微血管内皮表面上补体的负调控的缺陷直接相关。
类似于因子H突变,负补体调节物因子I和膜辅因子蛋白(CD46)的丧失功能的突变也与aHUS相关。对于因子B和C3蛋白已观察到相反情况,因为发现aHUS与这些蛋白中的获得功能的突变相关(Pediatr Nephrol 25(12):2431-42 (2010))。因此,许多合并数据涉及aHUS疾病发生中的补体活化。该概念被依库丽单抗(阻断末端补体蛋白C5的单克隆抗体)在aHUS的治疗中的疗效所最令人信服地支持。
尽管补体在aHUS中的效应机制的核心作用已被广泛接受,但是起始补体活化的触发和所涉及的分子途径尚未解决。不是所有携带上述突变的个体都发展aHUS。事实上,家族性研究已经表明,aHUS的外显率只有约50%(Ann Hum Genet 74(1):17-26 (2010))。疾病的自然史表明,aHUS最经常在起始事件诸如感染情节或损伤后发生。众所周知感染性病原体活化补体系统。在预先存在的适应性免疫不存在的情况下,感染性病原体的补体活化可以主要通过凝集素途径而起始。因此,由感染触发的凝集素途径活化可能代表aHUS易感个体中补体活化的随后病理放大的初始触发,这可能最终导致疾病进展。因此,本发明的另一个方面包括通过施用有效量的MASP-2抑制剂而治疗患有继发于感染的aHUS的患者。
对宿主组织的其他形式的损伤将通过凝集素途径活化补体,尤其是对血管内皮的损伤。经受氧化应激的人血管内皮细胞例如通过表达结合凝集素和活化补体的凝集素途径的表面部分而响应(Am J. Pathol 156(6):1549-56 (2000))。在体内,缺血/再灌注后的血管损伤也通过凝集素途径活化补体(Scand J Immunol 61(5):426-34 (2005))。该设置中的凝集素途径活化对于宿主具有病理后果,并且通过阻断MASP-2而抑制凝集素途径防止进一步宿主组织损伤和不良后果(Schwaeble PNAS 2011)。
因此,也已知沉淀aHUS的其他过程活化补体的凝集素途径。因此,有可能凝集素途径可能代表在遗传上易患aHUS的个体中以失调方式不当放大的初始补体活化机制,因此起始aHUS疾病发生。根据推理,预期通过凝集素途径阻断补体活化的试剂、包括抗MASP-2抗体在易患aHUS的个体中防止疾病进展或减少恶化。
为了进一步支持该概念,新近研究已经将肺炎链球菌鉴定为aHUS的儿科病例中的重要病原体。(Nephrology (Carlton), 17:48-52 (2012); Pediatr Infect Dis J. 30(9):736-9 (2011))。该具体病因似乎具有不利的预后,具有显著的死亡率和长期的发病率。值得注意的是,这些病例涉及非肠道感染,导致微血管病变、尿毒症和溶血的显现,而没有已知易患aHUS的补体基因中并发突变的证据。重要注意的是,肺炎链球菌对于活化补体特别有效,并且主要通过凝集素途径做到这一点。因此,在与肺炎球菌感染相关的非肠道HUS的情况下,微血管病变、尿毒症和溶血的显现预期主要由凝集素途径的活化所驱动,并且阻断凝集素途径的试剂,包括抗MASP-2抗体预期在这些患者中防止aHUS的进展或减少疾病严重度。因此,本发明的另一个方面包括通过施用有效量的MASP-2抑制剂而治疗患有与肺炎链球菌感染相关的非肠道aHUS的患者。
根据上述,在一些实施方案中,在处于发生与aHUS相关的肾衰竭的风险中的受试者的设置中,提供了用于降低发生aHUS或发生与aHUS相关的肾衰竭的可能性的方法,其包括持续一定时间期间施用有效改善或预防受试者中的肾衰竭的量的MASP-2抑制剂。在一些实施方案中,该方法进一步包括在与aHUS相关的任何症状发生之前确定受试者是否处于发生aHUS的风险中的步骤。在其他实施方案中,该方法包括在表明aHUS至少一种或多种症状(例如,贫血、血小板减少和/或肾功能不全的存在)的发生和/或在取自受试者的活检组织中血栓性微血管病的存在之后确定受试者是否处于发生aHUS的风险中。确定受试者是否处于发生aHUS的风险中包括确定受试者是否具有发生aHUS的遗传倾向,这可以通过评价遗传信息(例如,来自含有受试者基因型的数据库),或通过基因组测序或基因特异性分析(例如,PCR分析)对受试者进行至少一种基因筛选试验以确定是否存在与aHUS相关的基因标志物(即,确定在编码补体因子H(CFH)、因子I(CFI)、因子B (CFB)、膜辅因子CD46、C3、补体因子H-相关蛋白1(CFHR1)或THBD(编码抗凝蛋白血栓调节蛋白)或补体因子H-相关的蛋白3(CFHR3)或补体因子H-相关的蛋白4(CFHR4)的基因中是否存在与aHUS相关的基因突变),和/或确定受试者是否具有aHUS的家族史来实施。基因筛选是否存在与aHUS相关的基因突变的方法是众所周知的,例如,参见Noris M等人 “Atypical Hemolytic-Uremic Syndrome,” 2007 Nov 16 [Updated 2011 Mar 10]. In: Pagon RA, Bird TD, Dolan CR,等人, editors. GeneReviews?, Seattle (WA): University of Washington, Seattle。
例如,总体而言,具有补体因子H(CFH)的突变的那些中疾病的外显率是48%,并且CD46中的突变的外显率是53%,对于CFI中的突变是50%,对于C3中的突变是56%,并且对于THBD中的突变是64%(Caprioli J.等人, Blood, 108:1267-79 (2006); Noris等人, Clin J Am Soc Nephrol 5:1844-59 (2010))。如上述Caprioli等人(2006)中所述,在补体因子H(CFH)中具有突变的相当多个体从未发生aHUS,并且据推测,这些个体中次优的CFH活性在生理条件下足以保护宿主免于补体活化的影响,然而当暴露于活化补体的试剂产生比正常量更多的C3b时,次优的CFH活性不足以防止C3b沉积在血管内皮细胞上。因此,在一个实施方案中,提供用于抑制患有非因子H依赖性非典型溶血性尿毒综合征或处于发生非因子H依赖性非典型溶血性尿毒综合征的风险中的受试者中MASP-2依赖性补体活化的方法,其包括将包含有效抑制MASP-2依赖性补体活化的量的MASP-2抑制剂的组合物施用于所述受试者。在另一个实施方案中,提供用于抑制处于发生因子H依赖性非典型溶血性尿毒综合征的风险中的受试者中MASP-2依赖性补体活化的方法,其包括定期监测受试者以确定是否存在贫血、血小板减少症或肌酸酐上升,并且在确定贫血、血小板减少症或肌酸酐上升的存在之后用MASP-2抑制剂治疗。在另一个实施方案中,提供用于降低处于发生因子H依赖性aHUS的风险中的受试者将患有与aHUS相关的临床症状的可能性的方法,其包括在已知与触发aHUS临床症状相关的事件(例如,药物暴露(例如,化疗)、感染(例如,细菌感染)、恶性肿瘤、损伤、器官或组织移植或怀孕)之前或期间或之后施用MASP-2抑制剂。
在一个实施方案中,提供用于降低处于发生aHUS的风险中的受试者将患有与aHUS 相关的临床症状的可能性的方法,其包括定期监测受试者以确定是否存在贫血、血小板减少症或肌酸酐上升,并且在确定贫血、血小板减少症或肌酸酐上升的存在之后用MASP-2抑制剂治疗。
在另一个实施方案中,提供用于降低处于发生aHUS的风险中的受试者将患有与aHUS相关的临床症状的可能性的方法,其包括在已知与触发aHUS临床症状相关的事件(例如,药物暴露(例如,化疗)、感染(例如,细菌感染)、恶性肿瘤、损伤、器官或组织移植或怀孕)之前或期间或之后施用MASP-2抑制剂。
在一些实施方案中,MASP-2抑制剂在与触发aHUS临床症状相关的事件之前、期间或之后持续至少一天、两天、三天、四天或更长的时间期间进行施用,并且可以如医生确定进行重复,直到状况已经得到解决或控制。在前aHUS的设置中,MASP-2抑制剂可以全身性,诸如通过动脉内、静脉内、肌内、吸入、鼻腔、皮下或其他肠胃外施用而施用于受试者。
在一些实施方案中,在aHUS的初步诊断的设置中,或在表现出与aHUS的诊断一致的一种或多种症状(例如,贫血、血小板减少和/或肾功能不全的存在)的受试者中,在血浆置换不存在的情况下或与血浆置换组合的情况下用有效量的MASP-2抑制剂(例如,抗MASP-2抗体)治疗受试者作为第一线治疗。作为第一线治疗,MASP-2抑制剂可以全身性,诸如通过动脉内、静脉内、肌内、吸入、鼻腔、皮下或其他肠胃外施用而施用于受试者。在一些实施方案中,为了避免血浆置换的潜在并发症包括出血、感染和暴露于血浆供体固有的病症和/或过敏或另外反对血浆置换的受试者或在其中血浆置换无法获得的设置中,在血浆置换不存在的情况下作为第一线治疗将MASP-2抑制剂施用于受试者。
在一些实施方案中,该方法包括将MASP-2抑制剂通过导管(例如,静脉内)施用于患有aHUS的受试者,持续第一时间期间(例如,至少一天至一周或两周),随后将MASP-2抑制剂皮下施用于受试者,持续第二时间期间(例如,至少两周或更长的慢性期)。在一些实施方案中,第一和/或第二时间期间中的施用发生在血浆置换不存在的情况下。在一些实施方案中,该方法进一步包括在治疗之前,以及任选在治疗期间确定受试者中至少一种补体因子(例如,C3、C5)的水平,其中确定与标准值或健康对照受试者相比至少一种补体因子的降低水平表明需要用MASP-2抑制剂继续治疗。
在一些实施方案中,该方法包括通过静脉内、肌内或优选地皮下将MASP-2抑制剂诸如抗MASP-2抗体施用于患有aHUS或处于发生aHUS的风险中的受试者。治疗可能是长期的,并且每天至每月、但优选每两周进行施用。抗MASP-2抗体可以单独施用,或与C5抑制剂诸如依库丽单抗组合施用。
HUS
像非典型HUS,典型形式的HUS表现出TMA的所有临床和实验室发现。然而,典型HUS经常是儿科疾病并且通常不具有家族性组分或与补体基因的突变直接关联性。典型HUS的病因与某些肠道病原体的感染是紧密相连。患者通常与急性肾衰竭、血红蛋白尿和血小板减少(通常跟随着血性腹泻的发作)存在。该状况由痢疾志贺氏菌(Shigella dissenteria)、沙门氏菌或产生志贺毒素样的大肠杆菌的肠出血性菌株诸如大肠杆菌O157:H7的肠道感染引起。病原体从受污染的食物或水源获得。HUS是一种医疗紧急情况,并且具有5-10%死亡率。显著部分的幸存者发展慢性肾病(Corrigan和Boineau, Pediatr Rev 22 (11):365–9 (2011)),并且可能需要肾移植。
典型HUS中的微血管凝血主要发生,尽管不唯一地,在肾微血管中。潜在的病理生理由志贺毒素(STX)介导。由肠病性微生物排出进入肠腔,STX穿过肠屏障,进入血流,并且通过酰基鞘鞍醇三己糖受体(blobotriaosyl ceramide receptor)CD77结合至血管内皮细胞(Boyd和Lingwood Nephron 51:207 (1989)),CD77优先表达在肾小球内皮上并且介导STX的毒性作用。一旦结合至内皮,STX诱导损伤血管内皮、活化白细胞并且导致vWF依赖性血栓形成的一系列事件(Forsyth等人, Lancet 2: 411–414 (1989); Zoja等人, Kidney Int. 62: 846–856 (2002); Zanchi等人, J. Immunol. 181:1460–1469 (2008); Morigi等人, Blood 98: 1828–1835 (2001); Guessou等人, Infect. Immun., 73: 8306–8316 (2005))。这些微血栓阻塞或闭塞肾脏和其他器官的小动脉和毛细血管。微血栓对小动脉和毛细血管中血流的阻塞增加了当RBC挤压通过狭窄的血管时施加到RBC的剪切力。这可以导致剪切力对RBC的破坏和形成称为裂红细胞的RBC片段。裂红细胞的存在是HUS中的特征性发现。该机制被已知为微血管病性溶血。此外,血流的阻塞导致缺血,起始引起对受影响的器官的额外损害的补体介导的炎症应答。
补体的凝集素途径通过两种主要机制有助于HUS的疾病发生:1) 由内皮损伤引起的凝血级联的MASP-2介导的直接活化,和2) 由微血管血流的初始闭塞导致的缺血诱导的凝集素介导的随后补体活化。
STX损伤微血管内皮细胞,并且已知损伤的内皮细胞活化补体系统。如上所详述,内皮细胞损伤后的补体活化主要由凝集素途径驱动。经受氧化应激的人血管内皮细胞通过表达结合凝集素和活化补体的凝集素途径的表面部分而响应(Collard等人, Am J Pathol. 156(5):1549-56 (2000))。在体内,缺血再灌注后的血管损伤也通过凝集素途径活化补体(Scand J Immunol 61(5):426-34 (2005))。该设置中的凝集素途径活化对于宿主具有病理后果,并且通过MASP-2的阻断而抑制凝集素途径防止进一步宿主组织损伤和不良后果(Schwaeble等人, PNAS (2011))。除了补体活化以外,已经显示MASP-2的凝集素依赖性活化导致凝血酶原的裂解,形成凝血酶并且促进凝血。因此,损伤的内皮细胞对补体的凝集素途径的活化可以直接活化凝血系统。因此,凭借MASP-2介导的凝血酶原活化的补体的凝集素途径可能是将STX的初始内皮损伤与发生在HUS中的凝血和微血管血栓形成连接起来的占优势的分子途径。因此,预期凝集素途径的抑制剂,包括但不限于阻断MASP-2功能的抗体,将防止或减轻患有HUS的患者中的微血管凝血、血栓形成和溶血。事实上,抗MASP-2抗体的施用在典型HUS的模型中深度保护小鼠。如实施例36中描述与图45中显示,暴露于STX和LPS的所有对照小鼠发展严重的HUS,并且在48小时内变得垂死或死亡。另一方面,如图45中进一步显示,用抗MASP-2抗体治疗并且然后暴露于STX和LPS的所有小鼠都存活(Fisher’s精确p<0.01;N=5)。因此,抗MASP-2治疗在该HUS模型中深度保护小鼠。预期MASP-2抑制剂诸如MASP-2抗体的施用在HUS患者的治疗中将是有效的,并且将保护免于由肠病性大肠杆菌或其他产生STX的病原体的感染引起的微血管凝血、血栓形成和溶血。
尽管此处显示由STX引起的HUS,但是预期抗MASP-2治疗对于其他有毒物质引起的内皮损伤导致的HUS样综合征也将是有益的。这包括试剂诸如丝裂霉素、噻氯匹定、顺铂(cycplatin)、奎宁、环孢素、博来霉素以及其他化疗药物和免疫抑制药物。因此,预期抗MASP-2抗体治疗或抑制MASP-2活性的其他方式将有效地防止或限制凝血、血栓形成和RBC破坏和防止HUS和其他TMA相关的疾病(即,aHUS和TTP)中的肾衰竭。
患有HUS的患者经常存在腹泻和呕吐,它们的血小板计数通常降低(血小板减少),并且RBC降低(贫血)。前HUS腹泻阶段通常持续约四天,在此期间,处于发展HUS风险中的受试者通常表现出除了严重腹泻以外的一种或多种下列症状:低于30%的血细胞比容水平,伴有血管内红细胞破坏的涂片证据、血小板减少(血小板计数<150 x 103/mm3)和/或存在肾功能受损(血清肌酸酐浓度大于关于年龄的参考范围的上限)。Oligoanuria(少尿症)(持续>1天的尿排出≤0.5 mL/kg/h)的存在可以用作向发生HUS进展的量度(参见C. Hickey等人, Arch Pediatr Adolesc Med 165(10):884-889 (2011))。通常实施关于被大肠杆菌细菌(大肠杆菌O157:H7)或志贺氏菌或沙门氏菌属物种感染的存在的测试。在对于肠源性大肠杆菌(例如,大肠杆菌0157:H7)的感染测试呈阳性的受试者中,抗生素的使用是禁忌的,因为抗生素的使用可以通过增加的STX产生而增加发生HUS的风险(参见Wong C.等人, N Engl J. Med 342:1930-1936 (2000)。对于对志贺氏菌或沙门氏菌测试呈阳性的受试者,通常施用抗生素以清除感染。HUS的其他公认的第一线治疗包括体积膨胀、透析和血浆置换。
根据上述,在一些实施方案中,在患有与前HUS期相关的一种或多种症状或处于发生HUS的风险中的受试者(即受试者表现出以下的一种或多种:腹泻、低于30%的血细胞比容水平,伴有血管内红细胞破坏的涂片证据、血小板减少(血小板计数<150 x 103/mm3)和/或存在肾功能受损(血清肌酸酐浓度大于关于年龄的参考范围的上限))的设置中,提供了用于降低受试者中发生HUS的风险或降低受试者中肾衰竭的可能性的方法,其包括持续一定时间期间施用有效改善或预防肾功能受损的量的MASP-2抑制剂。在一些实施方案中,MASP-2抑制剂持续至少一天、两天、三天、四天或更多天的时间期间进行施用,并且可以如医生确定进行重复,直到状况已经得到解决或控制。在前HUS的设置中,MASP-2抑制剂可以全身性,诸如通过动脉内、静脉内、肌内、吸入、鼻腔、口服、皮下或其他肠胃外施用而施用于受试者。
杀细菌抗生素、特别是β-内酰胺类治疗大肠杆菌0157:H7感染已经与发生HUS的增加风险相关(Smith等人, Pediatr Infect Dis J 31(1):37-41 (2012)。在一些实施方案中,在患有与前HUS期相关的症状的受试者(其中已知受试者被肠源性大肠杆菌感染,对于该肠源性大肠杆菌使用抗生素是禁忌的(例如,大肠杆菌0157:H7))的设置中,提供了用于降低受试者中发生HUS的风险或降低受试者中肾衰竭的可能性的方法,其包括持续第一时间期间施用有效抑制或预防受试者中的少尿症存在的量的MASP-2抑制剂(例如,至少一天、两天、三天或四天),其中在第一时间期间过程中MASP-2抑制剂的施用在不存在抗生素的情况下进行。在一些实施方案中,该方法进一步包括持续第二时间期间(诸如至少一至两周)将MASP-2抑制剂组合抗生素施用于受试者。
在其他实施方案中,在患有与前HUS期相关的症状的受试者(其中已知受试者被志贺氏菌或沙门氏菌感染)的设置中,提供了用于降低受试者中发生HUS的风险或降低受试者中肾衰竭的可能性的方法,其包括持续一定时间期间施用有效抑制或预防受试者中的少尿症存在的量的MASP-2抑制剂,其中所述MASP-2抑制剂的施用在存在或不存在合适的抗生素的情况下进行。
在一些实施方案中,在HUS的初步诊断的设置中,或在表现出与HUS的诊断一致的一种或多种症状(例如,肾衰竭、或在低纤维蛋白原不存在的情况下微血管病性溶血性贫血或血小板减少)的受试者中,在血浆置换不存在的情况下或与血浆置换组合的情况下用有效量的MASP-2抑制剂(例如,抗MASP-2抗体)治疗受试者作为第一线治疗。作为第一线治疗,MASP-2抑制剂可以全身性,诸如通过动脉内、静脉内、肌内、吸入、鼻腔、皮下或其他肠胃外施用而施用于受试者。在一些实施方案中,为了避免血浆置换的并发症包括出血、感染和暴露于血浆供体固有的病症和/或过敏或在另外反对血浆置换的受试者中或在其中血浆置换无法获得的设置中,在血浆置换不存在的情况下作为第一线治疗将MASP-2抑制剂施用于受试者。
在一些实施方案中,该方法包括将MASP-2抑制剂通过导管(例如,静脉内)施用于患有HUS的受试者,持续第一时间期间(例如,持续至少一天至一周或两周的急性期),随后将MASP-2抑制剂皮下施用于受试者,持续第二时间期间(例如,至少两周或更长的慢性期)。在一些实施方案中,第一和/或第二时间期间中的施用发生在血浆置换不存在的情况下。在一些实施方案中,该方法进一步包括在治疗之前,以及任选在治疗期间确定受试者中至少一种补体因子(例如,C3、C5)的水平,其中确定与标准值或健康对照受试者相比至少一种补体因子的降低水平表明需要治疗,并且其中确定正常水平表明改善。
在一些实施方案中,该方法包括通过皮下或静脉内将MASP-2抑制剂诸如抗MASP-2抗体施用于患有HUS或处于发生HUS的风险中的受试者。治疗优选每天,但可以不频繁如每周或每个月。治疗将继续至少一周且长达3个月。抗MASP-2抗体可以单独施用,或与C5抑制剂诸如依库丽单抗组合施用。
TTP:
血栓性血小板减少性紫癜(TTP)是血液凝固系统的威胁生命的病症,其由活化凝血级联或补体系统的自身免疫或遗传性功能障碍引起(George, JN, N Engl J Med; 354:1927-35 (2006))。这导致在全身小血管中的大量微观凝块或血栓。红细胞经受剪切应力,所述剪切应力损害它们的膜,导致血管内溶血。获得的血流减少和内皮损伤导致器官损伤,包括脑、心脏和肾脏损伤。TTP的临床特征在于血小板减少、微血管病性溶血性贫血、神经系统变化、肾衰竭和发热。在血浆置换之前的时代,在急性发作过程中的致死率为90%。甚至用血浆置换,在六个月时的存活约为80%。
TTP可以由酶ADAMTS-13的遗传性或获得性抑制而产生,所述酶ADAMTS-13是一种负责将von Willebrand因子(vWF)的大多聚体裂解成小单元的金属蛋白酶。ADAMTS-13抑制或缺陷最终导致凝血增加(Tsai, H. J Am Soc Nephrol 14: 1072–1081, (2003))。ADAMTS-13调节vWF的活性;在其不存在的情况下,vWF形成大多聚体,所述大多聚体更可能结合血小板,并且使患者的微血管中易于血小板聚集和血栓形成。
已经在患有TTP的个体中鉴定了ADAMTS13中的许多突变。该疾病也可以由于针对ADAMTS-13的自身抗体而发生。此外,TTP可以在乳腺癌、胃肠道癌或前列腺癌(George JN., Oncology (Williston Park).25:908-14 (2011))、怀孕(第二个三个月或产后),George JN., Curr Opin Hematol 10:339-344 (2003))过程中发生,或者与疾病诸如HIV或自身免疫性疾病如系统性红斑狼疮(Hamasaki K,等人, Clin Rheumatol.22:355-8 (2003))相关。TTP也可以由某些药物治疗,包括肝素、奎宁、免疫介导的成分、癌症化疗剂(博来霉素、顺铂、阿糖胞苷、吉西他滨道诺霉素、丝裂霉素C和他莫昔芬)、环孢素A、口服避孕药、青霉素、利福平和抗血小板药物包括噻氯匹定和氯吡格雷所引起(Azarm, T.等人, J Res Med Sci., 16: 353–357 (2011))。与TTP有关的其他因素或条件是毒素诸如蜂毒、败血症、脾隔离症、移植、血管炎、血管外科手术和感染如肺炎链球菌和巨细胞病毒感染(Moake JL., N Engl J Med., 347:589–600 (2002))。由于短暂功能性ADAMTS-13缺乏引起的TTP可以作为与肺炎链球菌感染有关的内皮细胞损伤的后果而发生(Pediatr Nephrol., 26:631-5 (2011))。
血浆置换是TTP的标准治疗(Rock GA,等人, N Engl J Med 325:393-397 (1991))。血浆置换取代具有遗传缺陷的患者中的ADAMTS-13活性,并且消除具有适应性自身免疫TTP的那些患者中的ADAMTS-13自身抗体(Tsai, H-M, Hematol Oncol Clin North Am., 21(4): 609–v (2007))。通常将额外试剂诸如免疫抑制药物添加至治疗(George, JN, N Engl J Med, 354:1927-35 (2006))。然而,血浆置换对于约20%的患者是不成功的,在超过三分之一的患者中发生复发,并且血浆置换是昂贵的,且技术要求高。此外,许多患者无法耐受血浆置换。因此,仍然存在对TTP的额外和更好的治疗的迫切需要。
因为TTP是血液凝固级联的病症,所以用补体系统的拮抗剂治疗可能有助于稳定和校正该疾病。尽管将替代补体途径的病理活化与aHUS连接起来,但是补体活化在TTP中的作用仍不够清楚。ADAMTS13的功能缺陷对于TTP的易感性是重要的,但是它不足以引起急性发作。环境因素和/或其他遗传变化可能有助于TTP的显现。例如,编码参与凝血级联(vWF、血小板功能、内皮血管表面的组分或补体系统)的调节的蛋白的基因可能牵涉在急性血栓性微血管病的发生中(Galbusera, M.等人, Haematologica, 94: 166–170 (2009))。具体而言,已经显示补体活化发挥关键作用;已经显示来自与ADAMTS-13缺陷相关的血栓性微血管病的血清引起C3和MAC沉积和随后的中性粒细胞活化,这可以被补体失活所消除(Ruiz-Torres MP,等人, Thromb Haemost, 93:443-52 (2005))。此外,新近已经显示,在TTP的急性发作过程中,C4d、C3bBbP和C3a的水平有增加(M. Réti等人, J Thromb Haemost. Feb 28.(2012) doi: 10.1111/j.1538-7836.2012.04674.x. [印刷前电子出版物]),这与经典/凝集素和替代途径的活化是一致的。急性发作中该增加量的补体活化可以起始末端途径活化,并且负责TTP的进一步恶化。
ADAMTS-13和vWF在TTP中的作用负责血小板的活化和聚集以及它们随后在微血管病变中的剪切应力和沉积中的影响。活化的血小板与经典和替代补体途径相互作用,并且触发经典和替代补体途径两者。血小板介导的补体活化增加了炎症介质C3a和C5a(Peerschke E等人, Mol Immunol, 47:2170-5 (2010))。因此血小板可以充当遗传或自身免疫性TTP中经典补体活化的目标。
如上所述,凭借MASP-2介导的凝血酶原活化的补体的凝集素途径是将内皮损伤与发生在HUS中的凝血和微血管血栓形成连接起来的占优势的分子途径。类似地,补体的凝集素途径的活化可以直接驱动TTP中的凝血系统。凝集素途径活化可以响应于TTP中ADAMTS-13缺陷引起的初始内皮损伤而起始。因此,预期凝集素途径的抑制剂,包括但不限于阻断MASP-2功能的抗体,将减轻患有TTP的患者中与微血管凝血、血栓形成和溶血相关的微血管病。
在急诊室中通常存在的患有TTP的患者具有以下中的一种或多种:紫癜、肾衰竭、低血小板、贫血和/或血栓形成,包括中风。TTP的现行护理标准涉及持续两周或更长的期间,通常一周三次,但最多达每天的替换血浆置换的内导管递送(例如,静脉内导管或其他形式的导管)。如果受试者对于ADAMTS13的抑制剂(即,针对ADAMTS13的内源抗体)的存在测试呈阳性,那么血浆置换可以与免疫抑制治疗(例如皮质类固醇、利妥昔单抗或环孢素)组合实施。具有难治性TTP的受试者(TTP患者中约20%)对至少两周的血浆置换治疗不响应。
根据上述,在一些实施方案中,在TTP的初始诊断的设置中,或在表现出与TTP的诊断一致的一种或多种症状(例如,中枢神经系统损害、严重的血小板减少(如果不使用阿司匹林小于或等于5000/μL的血小板计数,如果使用阿司匹林小于或等于20,000/μL的血小板计数)、严重的心脏损害、严重的肺部损害、胃肠梗死或坏疽)的受试者中,提供了在血浆置换不存在的情况下或与血浆置换组合的情况下用有效量的MASP-2抑制剂(例如,抗MASP-2抗体)治疗受试者作为第一线治疗的方法。作为第一线治疗,MASP-2抑制剂可以全身性,诸如通过动脉内、静脉内、肌内、吸入、鼻腔、皮下或其他肠胃外施用而施用于受试者。在一些实施方案中,为了避免血浆置换的潜在并发症诸如出血、感染和暴露于血浆供体固有的病症和/或过敏或在另外反对血浆置换的受试者中或在其中血浆置换无法获得的设置中,在血浆置换不存在的情况下作为第一线治疗将MASP-2抑制剂施用于受试者。在一些实施方案中,将MASP-2抑制剂组合(包括共同施用)免疫抑制剂(例如皮质类固醇、利妥昔单抗或环孢素)和/或组合浓缩的ADAMTS-13施用于患有TTP的受试者。
在一些实施方案中,该方法包括将MASP-2抑制剂通过导管(例如,静脉内)施用于患有TTP的受试者,持续第一时间期间(例如,持续至少一天至一周或两周的急性期),随后将MASP-2抑制剂皮下施用于受试者,持续第二时间期间(例如,至少两周或更长的慢性期)。在一些实施方案中,第一和/或第二时间期间中的施用发生在血浆置换不存在的情况下。在一些实施方案中,该方法用于维持受试者,以防止受试者患有与TTP相关的一种或多种症状。
在另一个实施方案中,提供了通过施用有效降低TTP 的一种或多种症状的量的MASP-2抑制剂而治疗患有难治性TTP的受试者 (即,对至少两周的血浆置换治疗没有响应的受试者)的方法。在一个实施方案中,通过皮下或其他肠胃外施用基于长期经至少两周或更长的时间期间将MASP-2抑制剂(例如,抗MASP-2抗体)施用于患有难治性TTP的受试者。施用如医生决定进行重复,直到状况已经得到解决或控制。
在一些实施方案中,该方法进一步包括在治疗之前,以及任选在治疗期间确定受试者中至少一种补体因子(例如,C3、C5)的水平,其中确定与标准值或健康对照受试者相比至少一种补体因子的降低水平表明需要用MASP-2抑制剂继续治疗。
在一些实施方案中,该方法包括通过皮下或静脉内将MASP-2抑制剂诸如抗MASP-2抗体施用于患有TTP或处于发生TTP的风险中的受试者。治疗优选每天,但可以不频繁如每两周。治疗继续进行,直到受试者的血小板计数持续至少连续2天大于150,000/ml。抗MASP-2抗体可以单独施用,或与C5抑制剂诸如依库丽单抗组合施用。
本发明的另一个方面提供用于治疗冷球蛋白血症的方法,其通过将包含药学载体中的治疗有效量的MASP-2抑制剂的组合物施用于患有冷球蛋白血症或患有由冷球蛋白血症导致的状况的受试者而实现。冷球蛋白血症的特征在于血清中冷球蛋白的存在,其是在低温下经历可逆聚集的单一或混合的免疫球蛋白(通常为IgM抗体)。由冷球蛋白血症导致的状况包括血管炎、肾小球肾炎和全身性炎症。MASP2抑制剂可以全身性,诸如通过动脉内、静脉内、肌内、吸入、皮下或其他肠胃外施用或可能对于非肽能试剂通过口服施用而施用于患有冷球蛋白血症或患有由冷球蛋白血症导致的状况的受试者。
在另一个方面,本发明提供用于治疗冷凝集素病(CAD)的方法,其通过将包含药学载体中的治疗有效量的MASP-2抑制剂的组合物施用于患有CAD或患有由CAD导致的状况的受试者而实现。CAD疾病显现为贫血,并且可以由被称为“继发性CAD”的潜在疾病或病症(诸如感染性疾病、淋巴增生性疾病或结缔组织病症)所引起。这些患者发展针对其红细胞的IgM抗体,其在低温下触发凝集反应。MASP2抑制剂可以全身性,诸如通过动脉内、静脉内、肌内、吸入、皮下或其他肠胃外施用或可能对于非肽能试剂通过口服施用而施用于患有CAD或患有由CAD导致的状况的受试者。
凝血病
已形成补体系统在弥散性血管内凝血("DIC")诸如显著身体外伤继发的DIC中的作用的证据。
前述研究已显示,C4-/-小鼠不受免于肾再灌注损伤的保护。(Zhou, W.,等人, "Predominant role for C5b-9 in renal ischemia/reperfusion injury," J Clin Invest 105:1363-1371 (2000))。为了研究C4-/-小鼠是否仍然能够经由经典或凝集素途径激活补体,在特异性针对经典或凝集素途径激活途径的试验中测定C4-/-血浆中的C3周转。尽管在经由经典途径触发激活时未能观察到C3裂解,但观察到C4缺陷血清中的C3的高效凝集素途径依赖性激活(图30)。能够看出,在MASP-2-/-小鼠体内,甘露聚糖和酵母聚糖上的C3b沉积严重受损,甚至在根据许多先前发表的关于替代途径激活的论文,应该对于所有三条途径而言是允许的实验条件下。当使用由免疫球蛋白复合体包被而不是甘露聚糖或酵母聚糖包被的孔中的相同血清时,在MASP-2+/+小鼠血清和MASP-2-/-血清中看到C3b沉积和因子B裂解,而在C1q耗尽的血清中看不到。这表明,当经由经典活性提供初始的C3b时,在MASP-2-/-血清中促进了替代途径激活。图30C图示以下令人惊讶的发现:C3能够在C4缺陷血浆中以凝集素途径依赖的形式被有效激活。
这个“C4旁路”通过经由用可溶性甘露聚糖或甘露糖预孵育血浆的凝集素途径激活的抑制而废止。
异常、非免疫性的补体系统的激活对人是具有潜在危险性的,并且可能还在血液途径激活中起重要作用,尤其是在其中炎性途径和血液途径均被激活的严重外伤的情况下。在正常的健康者体内,C3转化率是总血浆C3蛋白的<5%。在猛烈的感染(包括败血症和免疫复合物病)中,C3转化率在补体水平常常低于正常水平的情况下以约30%重建自身,这归因于增加的利用和混合物(pool)分布的改变。大于30%的即时C3途径激活通常产生血管舒张和组织体液丧失的明显的临床证据。在30%C3转化率以上,引发机制主要是非免疫性的,并且导致的临床表现对患者有害。健康者体内和受控制的疾病中的补体C5水平似乎远比C3更稳定。C5水平的显著降低和/或转化与患者对非正常多发伤(例如,道路交通事故)和可能形成休克肺综合征的反应相关联。因此,超过30%的血管池的补体C3激活的任何证据、或任何C5参与的任何证据、或两者可被认为可能是患者中有害病理变化的前兆。
C3和C5两者均放出作用于释放血管舒张化合物的肥大细胞和嗜碱细胞的过敏毒素(C3a和C5a)。它们建立趋化性梯度以将多形核细胞(PMN)导向至免疫干扰(一种有益的应答)的中心,但在这里它们不一样,因为C5a对这些吞噬细胞具有特异性凝块(凝集)作用,防止它们远离反应位点的随机移动。在感染的正常对照中,C3激活C5。然而,在多发伤中,C5似乎被广泛激活,系统地生成C5a过敏毒素。此不受控制的活性引起多形白细胞在血管系统内部凝块,并且然后这些凝块被扫入肺的毛细血管,它们由于过氧化物的放出而在其中滞留并且产生局部损伤作用。尽管不愿被理论限制,但该机制在急性呼吸窘迫综合征(ARDS)的发病机理中或许是重要的,不过此见解最近受到了质疑。在体外,C3a过敏毒素能够显示为强有力的血小板凝聚体,但它们的体内参与较少被定义,并且伤口修复中的血小板物质和纤溶酶的释放可仅次要地涉及补体C3。可能的是,C3激活的延时上升为生成DIC所必需。
除以上概述的能够解释外伤与DIC之间的联系的被激活补体组分的细胞和血管效应之外,出现的科学发现鉴定了补体与凝血系统之间的直接分子连接和功能性交叉对话。支持数据已从对C3缺陷小鼠的研究中获得。由于C3是各补体途径的共享成分,所以预测C3缺陷小鼠缺乏全部补体功能。然而,意外地,C3缺陷小鼠能够完美激活末端补体组分(Huber-Lang, M.,等人, "Generation of C5a in the absence of C3: a new complement activation pathway," Nat. Med 12:682-687 (2006))。深入研究揭示,末端补体组分的C3依赖性激活是通过凝血酶(凝血级联的限速酶)介导的。(Huber等人,2006)。初始补体活化之后介导凝血酶激活的分子成分仍然是难以捉摸的。
本发明人阐明了什么被认为是补体与凝血级联之间的交叉对话的分子基础,并且将MASP-2鉴定为连接该两个系统的中心控制点。除众所周知的C2和C4补体蛋白之外,对MASP-2的底物特异性的生化研究已将凝血酶原鉴定为可能的底物。MASP-2在功能相关的位点特异性裂解凝血酶原,生成凝血酶,即凝血级联的限速酶。(Krarup, A.,等人, "Simultaneous Activation of Complement and Coagulation by MBL-Associated Serine Protease 2," PLoS. ONE. 2:e623 (2007))。由MASP-2生成的凝血酶能够促进纤维蛋白在限定的重建的体外系统中的沉积,证明MASP-2裂解的功能相关性。(Krarup 等人,2007)。如以下本文实例中所论述,发明人通过证明凝集素途径激活之后正常啮齿动物血清内的凝血酶激活,进一步印证了这个发现的生理学意义,并且证明了这个过程通过中和MASP-2单克隆抗体而被阻断。
MASP-2可在凝集素途径中代表中心分支点,能够促进补体系统和凝血系统两者的激活。由于凝集素途径激活是对许多类型的外伤性损伤的生理应答,所以本发明人认为,并发的(由补体组分介导的)系统性炎症和(经由凝血途径介导的)弥散性凝血能够通过MASP-2激活两个途径的能力来解释。这些发现清楚地表明MASP-2在DIC产生中的作用和MASP-2抑制在治疗或预防DIC中的治疗益处。MASP-2可提供补体系统与凝血系统之间的分子连接,并且凝集素途径的激活在其发生在外伤的背景下时能够经由MASP-2凝血酶轴直接引发凝血系统的激活,提供外伤和DIC之间的机制联系。根据本发明的一个方面,MASP-2的抑制会抑制凝集素途径激活并且减少过敏毒素C3a和C5a两者的生成。据信,C3激活的延时上升为生成DIC所必需。
所以,本发明的一个方面因而提供用于抑制MASP-2依赖性补体活化以治疗弥散性血管内凝血或其他补体介导的凝血障碍的方法,该方法通过向患有此类病症或处于发生此类病症风险中的受试者施用组合物来实现,所述组合物包含药物载体中的治疗有效量的MASP-2抑制剂(例如,抗MASP-2抗体或其片段、肽抑制剂或小分子抑制剂)。在一些实施方案中,MASP-2抑制剂能够阻断已被激活的MASP-2。将MASP-2抑制组合物适当全身性,诸如通过动脉内、静脉内、肌内、吸入、鼻、皮下或其他肠胃外施用,或对于非肽能剂而言可能通过口服施用而施用于受试者。施用可按照医师决定而重复,直到已消除或控制病症为止。本发明的这个方面的方法可用于治疗DIC,所述DIC继发于败血症、包括神经性创伤(例如,急性脑损伤,参见Kumura, E.,等人, Acta Neurochirurgica85:23-28 (1987)的严重外伤、感染(细菌、病毒、真菌、寄生虫)、癌症、产科并发症、肝病、严重中毒反应(例如,蛇咬伤、昆虫咬伤、输血反应)、休克、热休克、移植排异反应、血管动脉瘤、肝功能衰竭、通过化学治疗或放射治疗的癌症疗法、烧伤、意外辐射暴露及其他病因。参见例如Becker J.U. 和Wira C.R. "Disseminated Intravascular Coagulation" emedicine.medscape.com/9/10/2009。对于外伤或其他急性事件继发的DIC而言,MASP-2抑制组合物可在外伤性损伤之后立即施用,或在外伤诱发损伤或状况(诸如对被认为处于DIC风险中的患者的手术)之前、期间、紧接着或之后一至七天或更长时间内(诸如在24小时至72小时内)施用。在一些实施方案中,MASP-2抑制组合物可以速效剂型适当施用,诸如通过包含MASP-2抑制剂组合物的溶液滴注剂的静脉内或动脉内递送。
在另一个方面,本发明提供治疗患有血栓形成、微循环凝血或继发于微循环凝血的多器官衰竭或处于发生血栓形成、微循环凝血或继发于微循环凝血的多器官衰竭的风险中的受试者的方法。响应于血管损伤(vascular insult)形成生理血栓(血凝块)以防止血液从受损血管渗漏。
凝集素途径可以在与各种病因相关的潜在血管炎症触发的病理血栓形成中发挥作用。例如,血栓可以形成周围动脉粥样硬化斑块,这是凝集素途径的已知起始物。因此,用MASP-2抑制剂治疗可用于在具有潜在动脉粥样硬化的患者中阻断血栓形成。
微循环凝血(毛细血管和小血管中的印迹凝结)发生在设置诸如感染性休克中。凝集素途径在感染性休克中的作用得到确立,如实施例17和图18和19中所述的败血症的MASP-2 (-/-)小鼠模型的受保护表型所证明的。此外,如实施例15和图16A和16B所示,MASP-2 (-/-)小鼠在弥漫性血管内凝血(DIC)的局部Schwartzman反应模型(微血管中的局部凝血模型)中受保护。
围化疗期的施用和恶性肿瘤的治疗
补体系统的活化还可能参与恶性肿瘤的发病机制。最近,使用多克隆抗体或单克隆抗体以及链霉抗生物素-生物素-过氧化物酶技术,对17个乳腺癌样品和6个良性乳腺肿瘤样品中的C5b-9补体复合物新抗原、IgG、C3、C4、S蛋白/玻连蛋白、纤连蛋白和巨噬细胞进行了定位。所有的癌组织样品在每个TNM阶段,都有C5b-9沉积在肿瘤细胞膜上,有细颗粒沉积在细胞残留物上,并且坏死区域中出现弥散性沉积物(Niculescu, F.,等人, Am. J. Pathol. 140:1039-1043, 1992)。
此外,补体活化可能是化学疗法或放射治疗的结果,因此抑制补体活化可能用作恶性肿瘤治疗的辅助治疗以减少医源性炎症。手术之前进行化学治疗和放射治疗时,C5b-9的沉积更致密更久。在所有良性病变样品中都不存在C5b-9沉积物。S蛋白/玻连蛋白呈原纤维状沉积物存在于结缔组织基质中,呈弥散性沉积物存在于肿瘤细胞周围,不如纤连蛋白沉积得致密而持久。IgG、C3和C4沉积物只存在于癌样品中。乳腺癌中C5b-9沉积物的存在表示补体活化及其随后的致病效应(Niculescu, F.,等人, Am. J. Pathol. 140:1039-1043, 1992)。
脉冲式可调谐染料激光(577 nm) (PTDL)治疗诱导了血红蛋白凝固和组织坏死,这主要限于血管中。在经PTDL照射的正常皮肤的研究中,主要发现如下:1) C3片段、C8、C9和MAC沉积在血管壁上;2)这些沉积物不是由于蛋白变性,因为它们仅仅在照射后7分钟就很明显了,与转铁蛋白立即沉积在红细胞凝固部位上不同;3)显示C3沉积物通过替代途径放大了补体活化,这是特异性的反应,因为组织坏死本身并不导致这种放大作用;和4)这些反应是在多形核白细胞局部累积之前发生的。在血管瘤中组织坏死更明显。坏死中心的较大血管瘤血管并不显著地固定补体。相反,位于外周血管中的补体沉积类似于正常皮肤中所观察到的沉积,除了一个例外:在激光治疗后立即在一些血管中检测到C8、C9和MAC,这个发现与没有C5转化酶形成而直接发生MAC装配相一致。这些结果表明,补体在PTDL诱导的血管坏死中被激活,并可能是随后的炎症反应的原因。
肿瘤的光动力学疗法(PDT)引起了强烈的宿主免疫应答,其表现之一是显著的嗜中性粒细胞增多。除了由于PDT诱导的补体活化而释放的补体片段(直接介质)以外,还有至少12种都由于补体活性而引起的间接介质。后者包括细胞因子IL-1β、TNF-α、IL-6、L-10、G-CSF和KC、血栓烷、前列腺素、白三烯、组胺和凝血因子(Cecic, I.,等人, Cancer Lett. 183:43-51, 2002)。
最后,可以展望MASP-2依赖性补体活化的抑制剂与标准治疗方案联用以治疗癌症。例如,用利妥昔单抗(rituximab),即一种嵌合抗CD20单克隆抗体进行治疗可能与中度至严重的首剂副作用相关,特别是在具有大量循环肿瘤细胞的患者中。在最近的首次输注利妥昔单抗的研究中,在5位复发的轻度非霍奇金淋巴瘤(NHL)患者中,测得补体活化产物(C3b/c和C4b/c)和细胞因子(肿瘤坏死因子α (TNF-α)、白介素6 (IL-6)和IL-8)。输注利妥昔单抗诱导快速的补体活化,其先于TNF-α、IL-6和IL-8的释放。尽管研究组小,但是补体活化水平似乎是与输注前的循环B细胞数量有关(r = 0.85;P = 0.07),并与副作用的严重性有关。这些结果表明,补体在利妥昔单抗治疗副作用的发生机制中起着关键作用。因为不能用皮质类固醇防止补体活化,所以研究补体抑制剂在利妥昔单抗首次施用期间的可能作用中具有重大意义(van der Kolk, L.E.,等人, Br. J. Haematol. 115:807-811, 2001)。
在本发明另一个方面,提供用于抑制待接受化学疗法和/或放射疗法治疗的受试者的MASP-2依赖性补体活化的方法,所述疗法包括但不限于癌症状况治疗。该方法包括将包含治疗有效量的MASP-2抑制剂在药物载体中的组合物在围化疗期,即在施用化学疗法和/或放射疗法之前和/或期间和/或之后施用于患者。例如,可以在施用化学疗法或放射疗法之前或者施用化学疗法或放射疗法同时开始本发明的MASP-2抑制剂组合物的施用,并在整个治疗过程中持续,以减轻化学疗法和/或放射疗法在非目标健康组织中的有害影响。此外,可以在化学疗法和/或放射疗法之后施用MASP-2抑制剂组合物。要理解的是,化学疗法和放射疗法方案经常需要重复治疗,因此MASP-2抑制剂组合物的施用也可重复,并与化学疗法和放射疗法相对一致。还认为MASP-2抑制剂可用作化疗药物,单用或与其他化疗药物和/或放射疗法联用,以治疗患有恶性肿瘤的患者。可以适当通过口服(用于非肽能药物)、静脉内、肌内或其他非肠道途径施用。
在另一个实施方案中,MASP-2抑制剂可用于治疗急性放射综合征(也称为放射病或放射中毒)的受试者以降低暴露于电离辐射(意外或其他方式)的有害影响。与急性放射综合征相关的症状包括恶心、呕吐、腹泻、皮肤损伤、脱发、疲劳、发热、痉挛和昏迷。对于急性放射综合征的治疗而言,MASP-2抑制组合物可在放射暴露之后立即施用,或预防地在暴露之前、期间、紧接着或之后一至七天或更长时间内(诸如在24小时至72小时内)施用。在一些实施方案中,该方法可用于在暴露于足以引起急性放射综合征的剂量的电离辐射(即,至少1 Gy、或至少2 Gy、或至少3 Gy、或至少4 Gy、或至少5 Gy、或至少6 Gy、或至少7 Gy或更高的全身剂量的电离辐射)之前或之后治疗受试者。在一些实施方案中,MASP-2抑制组合物可以速效剂型适当施用,诸如通过包含MASP-2抑制剂组合物的溶液滴注剂的静脉内或动脉内递送。
眼科疾病
年龄相关性黄斑变性(AMD)是累及数百万成人的致盲疾病,然而对导致AMD发生的生物化学、细胞和/或分子事件的后遗症了解甚少。AMD导致黄斑的渐进性破坏,这与位于黄斑内及其周围、视网膜后以及视网膜色素上皮(RPE)与脉络膜之间的称为脉络膜小疣(drusen)的胞外沉积物的形成有关。最近的研究揭示,与炎症和免疫介导过程相关的蛋白质在脉络膜小疣相关成分中是普遍的。已经在视网膜、RPE和脉络膜细胞检测到编码许多这类分子的转录物。这些数据还证实,作为有效抗原呈递细胞的树突细胞与脉络膜小疣的发生密切相关,补体活化是脉络膜小疣内部以及沿着RPE–脉络膜界面活跃的关键途径(Hageman, G.S.,等人, Prog. Retin. Eye Res. 20:705-732, 2001)。
若干项独立研究表明,AMD与补体因子H(CFH)基因的遗传多态性之间有强关联性,其中在危险性等位基因纯合的个体中AMD的可能性增加了7.4倍(Klein, R.J.等人, Science 308:362-364, 2005; Haines等人, Science 308:362-364. 2005; Edwards等人, Science 308:263-264, 2005)。CFH基因已经被定位在染色体1q31,这是一个在AMD中涉及6个独立连锁扫描(linkage scan)的区域(参见例如 Schultz, D.W.,等人, Hum. Mol. Genet. 12:3315, 2003)。已知CFH是补体系统的关键调节因子。研究显示,细胞上和循环中的CFH通过抑制将C3激活成为C3a和C3b以及通过失活现有的C3b来调节补体活性。已经在AMD患者的玻璃膜(Brusch's membrane)、毛细血管间支柱(intercapillary pillar)中以及脉络膜小疣内部观察到C5b-9的沉积(Klein等人)。免疫荧光实验表明,在AMD中CFH的多态性可能引起补体沉积在脉络膜毛细血管和脉络膜血管中(Klein等人)。
膜结合补体抑制剂补体受体1也定位在脉络膜小疣中,但是在RPE细胞中用免疫组织化学法无法检测出。相比之下,第二膜结合补体抑制剂膜辅因子蛋白存在于脉络膜小疣结合的RPE细胞中以及脉络膜小疣内部的小的球形亚结构组分中。这些以前未鉴定出的组分对特征性沉积在补体活化部位的补体组分C3的蛋白水解片段也具有强的免疫反应性。有研究认为这些结构代表来自作为补体攻击目标的退行性RPE细胞的残余碎片(Johnson, L.V.,等人, Exp. Eye Res. 73:887-896, 2001)。
这些多补体调节剂以及补体活化产物(C3a、C5a、C3b、C5b-9)的鉴定和定位使研究者断定,慢性补体活化在脉络膜小疣生物发生过程和AMD病因中起着重要作用(Hageman等人, Progress Retinal Eye Res. 20:705-32, 2001)。在脉络膜小疣中鉴定出C3和C5活化产物,并不会让人了解补体是通过经典途径还是通过凝集素途径或替代放大环路活化,正如本发明所了解的一样,因为C3和C5两者是所有三条途径共有的。然而,两项研究使用对经典途径活化必不可少的识别成分C1q特异性的抗体,对脉络膜小疣免疫标记进行了探索(Mullins等人, FASEB J. 14:835-846, 2000; Johnson等人, Exp. Eye Res. 70:441-449, 2000)。两项研究都得出结论,认为在脉络膜小疣中一般观察不到C1q免疫标记。这些有关C1q的阴性结果表明,脉络膜小疣中的补体活化不是通过经典途径产生的。另外,Mullins等人的研究(2000)报道了脉络膜小疣的免疫复合物组分(IgG轻链、IgM)的免疫标记是微弱可变的,这进一步表明经典途径在该疾病过程期间发生的补体活化中起着次要作用。
两篇最近已发表的研究对小鼠(一种人CNV模型)中补体在激光诱导的脉络膜新血管形成(CNV)的发展中的作用进行了评价。Bora及其同事(2005)采用免疫组织学方法发现,在激光处理后,补体活化产物C3b和C5b-9 (MAC)在新血管复合体上的显著沉积(Bora等人, J. Immunol. 174:491-7, 2005)。重要的是,在C3遗传缺陷型小鼠(C3-/-小鼠)中不发生CNV,C3是所有补体活化途径所需的必不可少的成分。在激光诱导的CNV后,对小鼠眼组织中的3种涉及CNV的血管生成因子VEGF、TGF-β2和β-FGF的RNA信息水平进行了评价。值得注意的是,补体耗尽导致这些血管生成因子的RNA水平明显降低。
Nozaki及其同事采用ELISA方法,证实了在激光诱导的CNV进程的早期产生了有效的过敏毒素C3a和C5a (Nozaki等人, Proc. Natl. Acad. Sci. U.S.A. 103:2328-33, 2006)。此外,在野生型小鼠中,将C3和C5的这两种生物活性片段注射到玻璃体腔之后诱导VEGF表达。与这些结果一致,Nozaki及其同事还表明遗传上去除C3a和C5a的受体减少激光损伤后的VEGF表达和CNV形成,抗体介导的C3a或C5a中和或其受体的药理阻断也减少CNV。之前的研究证实,白细胞募集,特别是巨噬细胞募集在激光诱导的CNV中起着关键作用(Sakurai等人, Invest. Opthomol. Vis. Sci. 44:3578-85, 2003; Espinosa-Heidmann,等人, Invest. Opthomol. Vis. Sci. 44:3586-92, 2003)。在Nozaki及其同事的论文(2006)中报道了在激光损伤后,C3aR(-/-)和C5aR(-/-)小鼠的白细胞募集显著减少。
因此,本发明的一方面提供用于抑制MASP-2依赖性补体活化以治疗年龄相关性黄斑变性或其他补体介导的眼科疾病的方法,该方法通过将包含治疗有效量的MASP-2抑制剂在药物载体中的组合物施用于患有这些疾病或其他补体介导的眼科疾病的受试者而实现。可通过诸如凝胶剂、软膏剂或滴剂形式的组合物经冲洗或敷用而经MASP-2抑制剂组合物局部施用于眼部。或者,可全身性,诸如通过动脉内、静脉内、肌内、吸入、经鼻、皮下或其他胃肠外施用,或者对于非肽能抑制剂可口服施用而将MASP-2抑制剂施用于受试者。MASP-2抑制剂组合物可与一种或多种另外治疗药物联用,另外的治疗药物公开于诸如美国专利申请公开号2004-0072809-A1。可按照医师规定重复施用直到疾病消退或得到控制。
在另一个方面,本发明提供用于抑制MASP-2依赖性补体活化以治疗患有青光眼或处于发生青光眼的风险中的受试者的方法。已经显示,不受控制的补体活化有助于青光眼中对视网膜神经节细胞(RGCs)、其突触和轴突的退行性损伤的进展。参见Tezel G.等人, Invest Ophthalmol Vis Sci 51:5071-5082 (2010)。例如,使用不同动物模型的人组织和体内研究的组织病理学研究已经表明,合成了补体组分,包括C1q和C3,并且在青光眼性视网膜中形成末端补体复合物(参见Stasi K.等人, Invest Ophthalmol Vis Sci 47:1024-1029 (2006), Kuehn M.H.等人, Exp Eye Res 83:620-628 (2006))。如Tezel G.等人所述,已经确定,除了经典途径以外,凝集素途径可能参与青光眼性神经变性过程中的补体活化,从而通过并行细胞裂解、炎症和自身免疫促进神经变性损伤的进展。如Tezel G.等人中所述,从有或没有青光眼的供体的眼睛获得的人视网膜样品的蛋白质组分析检测到几种补体组分的表达和差异调节。值得注意的是,与对照相比,在青光眼样品中来自凝集素途径的补体组分(包括MASP-1和MASP-2和C-型凝集素)的表达水平更高,或仅检测到。如Kuehn M.H.等人, Experimental Eye Research 87:89-95 (2008)中进一步描述,补体合成和沉积受视网膜I/R诱导,并且补体级联的破坏延迟RGC变性。在该研究中,当与正常动物相比时,发现携带补体组分C3的靶向破坏的小鼠在短暂性视网膜I/R后表现出延迟的RGC变性。
这些研究的发现表明,在青光眼压力条件下在补体活化和内在调控之间的生理平衡的改变可以对神经退行性损伤的进展具有重要影响,表明补体活化的抑制,诸如通过施用抗MASP-2抗体,可以被用作青光眼患者的治疗剂。
本发明的一个方面因此提供用于抑制MASP-2依赖性补体活化以治疗青光眼的方法,其通过将包含药学载体中的治疗有效量的MASP-2抑制剂的组合物施用于患有青光眼的受试者而实现。可通过例如凝胶剂、软膏剂或滴剂形式的组合物经冲洗或敷用而将MASP-2抑制剂组合物局部施用于眼部。或者,将MASP-2抑制组合物全身性,诸如通过动脉内、静脉内、肌内、吸入、鼻、皮下或其他肠胃外施用,或对于非肽能剂而言可能通过口服施用而施用于受试者。施用可按照医师决定而重复,直到已消除或控制病症为止。
IV. MASP-2抑制剂
在一方面,本发明提供抑制MASP-2依赖性补体活化的副作用的方法。MASP-2抑制剂是以有效抑制有生命的受试者的MASP-2依赖性补体活化的量施用的。在本发明这个方面的实施中,代表性的MASP-2抑制剂包括:抑制MASP-2生物活性的分子(诸如小分子抑制剂、抗MASP-2抗体或者与MASP-2相互作用或干扰蛋白间相互作用的阻断性肽),以及减少MASP-2表达的分子(诸如MASP-2反义核酸分子、MASP-2特异性RNAi分子和MASP-2核酶),从而阻止MASP-2激活凝集素补体途径。MASP-2抑制剂可单独使用作为主要疗法,或者与其他治疗药物联用作为辅助疗法以增强其他药物治疗的治疗益处。
MASP-2依赖性补体活化抑制的特征在于补体系统成分的至少一种以下的变化,所述变化是由于根据本发明的方法施用MASP-2抑制剂所致:MASP-2依赖性补体活化系统产物C4b、C3a、C5a和/或C5b-9 (MAC)形成或产生的抑制(测定法参见例如实施例2)、使用未致敏兔或豚鼠红细胞在溶血分析中评价的补体活化的减少(测定法参见例如实施例33)、C4裂解和C4b沉积的减少(测定法参见例如实施例2),或者C3裂解和C3b沉积的减少(测定法参见例如实施例2)。
根据本发明,使用有效抑制MASP-2依赖性补体活化系统的MASP-2抑制剂。用于实施本发明这个方面的MASP-2抑制剂包括例如抗MASP-2抗体及其片段、MASP-2抑制肽、小分子、MASP-2可溶性受体和表达抑制剂。MASP-2抑制剂可通过阻断MASP-2的生物学功能而抑制MASP-2依赖性补体活化系统。例如,抑制剂可有效阻断MASP-2蛋白间的相互作用,干扰MASP-2二聚化或装配,阻断Ca2+结合,干扰MASP-2丝氨酸蛋白酶活性位点,或者可减少MASP-2蛋白表达。
在一些实施方案中,MASP-2抑制剂选择性抑制MASP-2补体活化,保持了C1q依赖性补体活化系统功能上的完整性。
在一个实施方案中,用于本发明方法的MASP-2抑制剂是特异性的MASP-2抑制剂,其与包含SEQ ID NO:6的多肽特异性结合的亲和力是与补体系统中的其他抗原结合的亲和力的至少10倍高。在另一个实施方案中,MASP-2抑制剂与包含SEQ ID NO:6的多肽特异性结合的结合亲和力是与补体系统中的其他抗原结合的结合亲和力的至少100倍高。MASP-2抑制剂的结合亲和力可使用合适的结合测定法进行测定。
MASP-2多肽具有类似于C1补体系统的蛋白酶MASP-1、MASP-3以及C1r和C1s的分子结构。SEQ ID NO:4所示的cDNA分子编码MASP-2的代表性实例(由SEQ ID NO:5所示的氨基酸序列组成),提供带有前导序列(氨基酸1-15)的人MASP-2多肽,分泌后被裂解,产生成熟形式的人MASP-2 (SEQ ID NO:6)。如图2中所示,人MASP 2基因包括十二个外显子。人MASP-2 cDNA由外显子B、C、D、F、G、H、I、J、K和L编码。可变剪接产生了20 kDa蛋白,称为MBL相关的蛋白19 (“MAp19”,也称“sMAP”) (SEQ ID NO:2),由图2所示外显子B、C、D和E产生的(SEQ ID NO:1)编码。SEQ ID NO:50所示的cDNA分子编码鼠MASP-2 (由SEQ ID NO:51所示的氨基酸序列组成),提供带有前导序列的鼠MASP-2多肽,分泌后被裂解,产生成熟形式的鼠MASP-2 (SEQ ID NO:52)。SEQ ID NO:53所示的cDNA分子编码大鼠MASP-2 (由SEQ ID NO:54所示的氨基酸序列组成),提供带有前导序列的大鼠MASP-2多肽,分泌后被裂解,产生成熟形式的大鼠MASP-2 (SEQ ID NO:55)。
本领域技术人员应理解的是,SEQ ID NO:4、SEQ ID NO:50和SEQ ID NO:53中公开的序列分别代表人、小鼠和大鼠MASP-2的单个等位基因,发生等位基因变异和可变剪接是预料之中的。SEQ ID NO:4、SEQ ID NO:50和SEQ ID NO:53所示核苷酸序列的等位基因变体,包括含有沉默突变的变体以及其中导致氨基酸序列改变的突变的变体,都属本发明的范围。可根据标准方法,通过探测cDNA或基因组文库而从不同个体中克隆MASP-2序列的等位基因变体。
人MASP-2蛋白(SEQ ID NO:6)的结构域见图1和2A,包括N端C1r/C1s/海胆Vegf/骨形成蛋白(CUBI)结构域(SEQ ID NO:6的氨基酸1-121)、表皮生长因子样结构域(氨基酸122-166)、第二CUBI结构域(氨基酸167-293)以及串联的补体调控蛋白结构域和丝氨酸蛋白酶结构域。MASP 2基因的可变剪接产生图1中所示的MAp19。MAp19是含有MASP-2的N端CUB1-EGF区的非酶蛋白,具有得自图1所示外显子E的4个额外残基(EQSL)。
研究表明,一些蛋白质通过蛋白间的相互作用与MASP-2结合或者与MASP-2相互作用。例如,已知MASP-2结合凝集素蛋白MBL、H-纤维胶凝蛋白和L-纤维胶凝蛋白,并与之形成Ca2+依赖性复合物。研究表明,每种MASP-2/凝集素复合物通过蛋白质C4和C2的MASP-2依赖性裂解来激活补体(Ikeda, K.,等人, J. Biol. Chem. 262:7451-7454, 1987; Matsushita, M.,等人, J. Exp. Med.176:1497-2284, 2000; Matsushita, M.,等人, J. Immunol. 168:3502-3506, 2002)。研究表明,MASP-2的CUB1-EGF结构域对于MASP-2与MBL的结合是必不可少的(Thielens, N.M.,等人, J. Immunol. 166:5068, 2001)。研究还表明,CUB1EGFCUBII结构域介导MASP-2的二聚化,这是形成活性MBL复合物所需要的(Wallis, R.,等人, J. Biol. Chem. 275:30962-30969, 2000)。因此,可对结合已知对MASP-2依赖性补体活化重要的MASP-2靶区或者阻碍该区域的MASP-2抑制剂进行鉴定。
抗MASP-2抗体
在本发明这个方面的一些实施方案中,MASP-2抑制剂包括抑制MASP-2依赖性补体活化系统的抗MASP-2抗体。用于本发明这个方面的抗MASP-2抗体包括得自任何产生抗体的哺乳动物的多克隆抗体、单克隆抗体或重组抗体,并且可以是多特异性的、嵌合的、人源化的、抗独特型抗体以及抗体片段。抗体片段包括Fab、Fab'、F(ab)2、F(ab')2、Fv片段、scFv片段和单链抗体,见本文的进一步描述。
文献中记载了几种抗MASP-2抗体,其中的一些列于下表1。可使用本文所述测定法筛选这些前述抗MASP-2抗体抑制MASP-2依赖性补体活化系统的能力。例如,已经鉴定出阻断MASP-2依赖性补体活化的抗大鼠MASP-2 Fab2抗体,详情见本文实施例10和11。一旦鉴定出发挥MASP-2抑制剂作用的抗MASP-2抗体,就可将它用于产生抗独特型抗体,并用于鉴定其他MASP-2结合分子,如见下文进一步描述的。
表1:来自文献的MASP-2特异性抗体

效应子功能降低的抗MASP-2抗体
在本发明这个方面的一些实施方案中,抗MASP-2抗体降低了效应子功能,来减轻可能由经典补体途径活化产生的炎症。研究表明IgG分子引发经典补体途径的能力存在于分子的Fc部分(Duncan, A.R.,等人, Nature332:738-740 1988)。其中通过酶裂解除去分子Fc部分的IgG分子缺少这种效应子功能(参见Harlow, Antibodies: A Laboratory Manual, Cold Spring Harbor Laboratory, New York, 1988)。因此,可通过具有使效应子功能减到最低的基因工程改造的Fc序列,或者成为人IgG2或IgG4同种型,由此去掉分子的Fc部分,从而产生效应子功能降低的抗体。
可对IgG重链的Fc部分进行标准分子生物学操作来产生效应子功能降低的抗体,如本文实施例9所述,也描述于Jolliffe等人, Int'l Rev. Immunol. 10:241-250, 1993, 和Rodrigues等人, J. Immunol. 151:6954-6961, 1998。效应子功能降低的抗体还包括激活补体和/或与Fc受体相互作用的能力降低的人IgG2和IgG4同种型(Ravetch, J.V.,等人, Annu. Rev. Immunol. 9:457-492, 1991; Isaacs, J.D.,等人, J. Immunol.148:3062-3071, 1992; van de Winkel, J.G.,等人, Immunol. Today 14:215-221, 1993)。可通过本领域普通技术人员已知的若干方法之一,来产生由IgG2或IgG4同种型构成的人MASP-2特异性的人源化抗体或完全人抗体,如Vaughan, T.J.,等人, Nature Biotechnical 16:535-539, 1998所述。
抗MASP-2抗体的产生
可使用MASP-2多肽(例如全长MASP-2)或使用带有抗原性MASP-2表位的肽(如MASP-2多肽部分)来产生抗MASP-2抗体。免疫原性肽可以少至五个氨基酸残基。例如,可以使用包括SEQ ID NO:6全部氨基酸序列的MASP-2多肽来诱导用于本发明方法的抗MASP-2多肽。可以将已知参与蛋白间相互作用的特定MASP-2结构域,诸如CUBI和CUBIEGF结构域以及包括丝氨酸蛋白酶活性部位的区域,表达为实施例3中所述的重组多肽并用作抗原。此外,包含MASP-2多肽(SEQ ID NO:6)至少6个氨基酸的部分的肽也可用来诱导MASP-2抗体。下表2中提供了用于诱导MASP-2抗体的MASP-2衍生抗原的其他实例。用于产生抗体的MASP-2肽和多肽可作为天然多肽、或者重组肽或合成肽以及无催化活性的重组多肽(诸如MASP-2A)而被分离出来,如实施例5-7进一步描述的。在本发明这个方面的一些实施方案中,使用实施例8和实施例9中所述转基因小鼠品系获得抗MASP-2抗体,如下文进一步描述。
用于产生抗MASP-2抗体的抗原还包括融合多肽,诸如MASP-2或其部分与免疫球蛋白多肽或者与麦芽糖结合蛋白的融合物。多肽免疫原可以是全长分子或其部分。如果多肽部分是半抗原样的,则最好可将此类部分结合或连接到大分子载体(诸如匙孔血蓝蛋白(KLH)、牛血清白蛋白(BSA)或破伤风类毒素)上用于免疫。
表2:MASP-2衍生的抗原

多克隆抗体
可通过使用本领域普通技术人员众所周知的方法,用MASP-2多肽或其免疫原性部分免疫动物来制备抗MASP-2的多克隆抗体。参见,例如,Green等人, "Production of Polyclonal Antisera," 载于Immunochemical Protocols (Manson主编), 第105页,另详见实施例6。可通过使用佐剂来增加MASP-2多肽的免疫原性,所述佐剂包括无机凝胶(诸如氢氧化铝)或弗氏佐剂(完全或不完全)、表面活性剂(诸如溶血卵磷脂)、普罗尼克多元醇(pluronic polyol)、聚阴离子、油乳液、匙孔 血蓝蛋白和二硝基苯酚。多克隆抗体一般由动物诸如马、牛、狗、鸡、大鼠、小鼠、兔、豚鼠、山羊或绵羊产生。或者,用于本发明的抗MASP-2抗体也可得自近似人类的灵长类。在狒狒中产生诊断用和治疗用抗体的通用技术可参见例如Goldenberg等人的国际专利公开号WO 91/11465和Losman, M.J.,等人, Int. J. Cancer 46:310, 1990。然后采用本领域众所周知的标准方法,从这些被免疫过的动物的血液中产生含有免疫活性抗体的血清。
血蓝蛋白和二硝基苯酚。多克隆抗体一般由动物诸如马、牛、狗、鸡、大鼠、小鼠、兔、豚鼠、山羊或绵羊产生。或者,用于本发明的抗MASP-2抗体也可得自近似人类的灵长类。在狒狒中产生诊断用和治疗用抗体的通用技术可参见例如Goldenberg等人的国际专利公开号WO 91/11465和Losman, M.J.,等人, Int. J. Cancer 46:310, 1990。然后采用本领域众所周知的标准方法,从这些被免疫过的动物的血液中产生含有免疫活性抗体的血清。
单克隆抗体
在一些实施方案中,MASP-2抑制剂是抗MASP-2单克隆抗体。抗MASP-2单克隆抗体是高度特异性的,针对单一MASP-2表位。本文所用的修饰语“单克隆”是指获自基本同质的抗体群的抗体性质,不得理解为需要通过任何特定方法来产生抗体。可采用通过培养物中的连续细胞系以提供抗体分子产生的任何技术来获得单克隆抗体,例如Kohler, G.,等人, Nature 256:495, 1975中所述的杂交瘤方法,或者可以通过重组DNA方法制备单克隆抗体(参见例如Cabilly的美国专利号4,816,567)。也可以采用有关技术从噬菌体抗体文库中分离单克隆抗体(参见Clackson, T.,等人, Nature 352:624-628, 1991, 和Marks, J.D.,等人, J. Mol. Biol. 222:581-597, 1991)。此类抗体可以是任何免疫球蛋白类别,包括IgG、IgM、IgE、IgA、IgD及其任何亚类。
例如,可通过将包含MASP-2多肽或其部分的组合物注射给合适的哺乳动物(例如BALB/c小鼠)而获得单克隆抗体。在预定时间期间之后,从小鼠中取出脾细胞,使之悬浮于细胞培养基中。然后将脾细胞与无限增殖细胞系融合形成杂交瘤。将形成的杂交瘤在细胞培养基中培养,对它们产生抗MASP-2单克隆抗体的能力进行筛选。进一步描述抗MASP-2单克隆抗体产生的实例详见实施例7。(也可参见Current Protocols in Immunology, Vol. 1., John Wiley & Sons, pages 2.5.1-2.6.7, 1991.)。
可通过使用转基因小鼠来获得人单克隆抗体,所述转基因小鼠已被工程改造以在响应抗原攻击时产生特异性人抗体。在这种技术中,将人免疫球蛋白重链和轻链基因座元件引入得自胚胎干细胞系的小鼠品系中,所述胚胎干细胞系含有被定向破坏的内源免疫球蛋白重链和轻链基因座。该转基因小鼠可合成对人抗原(诸如本文所述MASP-2抗原)特异性的人抗体,可使用该小鼠来产生分泌人MASP-2抗体的杂交瘤,其通过采用常规Kohler-Milstein技术,将来自这些动物的B细胞与合适的骨髓瘤细胞系融合(详见实施例7)。具有人免疫球蛋白基因组的转基因小鼠是市售的(例如购自Abgenix, Inc., Fremont, CA, 和Medarex, Inc., Annandale, N.J.)。用于从转基因小鼠中获得人抗体的方法描述于例如Green, L.L.,等人, Nature Genet. 7:13, 1994; Lonberg, N.,等人, Nature 368:856, 1994; 和Taylor, L.D.,等人, Int. Immun. 6:579, 1994。
可通过各种已确立的技术从杂交瘤培养物中分离和纯化单克隆抗体。这些分离技术包括用A蛋白琼脂糖凝胶亲和层析、大小排阻层析和离子交换层析(参见例如Coligan,第2.7.1-2.7.12页和第2.9.1-2.9.3页;Baines等人, "Purification of Immunoglobulin G (IgG),"载于Methods in Molecular Biology, The Humana Press, Inc.,第10卷,第79-104页,1992)。
多克隆抗体、单克隆抗体或得自噬菌体的抗体一旦产生,首先便要测定其对MASP-2结合的特异性。可以利用本领域技术人员已知的各种测定法来检测特异性结合MASP-2的抗体。示例性的测定法包括通过标准方法进行的Western印迹法或免疫沉淀分析法(描述于例如Ausubel等人)、免疫电泳、酶联免疫吸附测定、斑点印迹法、抑制或竞争测定法以及夹心测定法(描述于Harlow和Land, Antibodies: A Laboratory Manual, Cold Spring Harbor Laboratory Press, 1988)。一旦鉴定出特异性结合MASP-2的抗体,便通过以下几种测定法之一,测定抗MASP-2抗体作为MASP-2抑制剂发挥作用的能力:诸如例如凝集素特异性C4裂解测定法(描述于实施例2)、C3b沉积测定法(描述于实施例2)或者C4b沉积测定法(描述于实施例2)。
本领域普通技术人员可以容易地测定抗MASP-2单克隆抗体的亲和力(参见例如Scatchard, A., NY Acad. Sci. 51:660-672, 1949)。在一个实施方案中,用于本发明方法的抗MASP-2单克隆抗体能结合MASP-2,其结合亲和力<100 nM,优选<10 nM,最优选<2 nM。
嵌合/人源化抗体
用于本发明方法的单克隆抗体包括嵌合抗体以及这些抗体的片段,其中重链和/或轻链部分与得自特定物种或者属于特定抗体类别或亚类的抗体的相应序列相同或同源,而链的其余部分与得自另一物种或者属于另一抗体类别或亚类的抗体的相应序列相同或同源(Cabilly的美国专利号4,816,567;和Morrison, S.L.,等人, Proc. Nat'l Acad. Sci.USA 81:6851-6855, 1984)。
用于本发明的一种嵌合抗体的形式是人源化单克隆抗MASP-2抗体。非人(例如鼠)抗体的人源化形式是嵌合抗体,其含有得自非人免疫球蛋白的最小序列。通过将非人(例如小鼠)互补决定区(CDR)从小鼠免疫球蛋白的可变重链和可变轻链转移到人可变区,从而产生人源化单克隆抗体。然后,典型的做法是将人抗体的其余部分代入非人对应部分的构架区。此外,人源化抗体可包括受体抗体或供体抗体中不存在的残基。这些修饰被用来进一步改进抗体性能。一般而言,人源化抗体将包含基本上全部的至少一种、通常两种可变结构域,其中所有或基本上所有的超变环都对应于非人免疫球蛋白的超变环,所有或基本上所有的Fv构架区都是人免疫球蛋白序列的Fv构架区。人源化抗体任选可包含至少一部分免疫球蛋白恒定区(Fc),通常为人免疫球蛋白的恒定区。更多详情可参见Jones, P.T.,等人, Nature 321:522-525, 1986; Reichmann, L.,等人, Nature 332:323-329, 1988; 和Presta, Curr. Op. Struct. Biol. 2:593-596, 1992。
用于本发明的人源化抗体包括至少含有MASP-2结合CDR3区的人单克隆抗体。此外,可以替换Fc部分以便产生IgA或IgM以及人IgG抗体。这些人源化抗体将具有特定临床效用,因为它们特异性地识别人MASP-2,但是却不会引起人体对抗体本身的免疫应答。因此它们更适用于人体的体内施用,尤其是必需重复或长期施用的时候。
由鼠抗MASP-2单克隆抗体获得人源化抗MASP-2抗体的实例见本文实施例6。人源化单克隆抗体的生产技术还记载于例如Jones, P.T.,等人, Nature 321:522, 1986; Carter, P.,等人, Proc. Nat'l. Acad. Sci. USA 89:4285, 1992; Sandhu, J.S., Crit. Rev. Biotech. 12:437, 1992; Singer, I.I.,等人, J. Immun. 150:2844, 1993; Sudhir (主编), Antibody Engineering Protocols, Humana Press, Inc., 1995; Kelley, "Engineering Therapeutic Antibodies,"载于Protein Engineering: Principles and Practice, Cleland等人 (主编), John Wiley & Sons, Inc., 第399-434页, 1996;以及Queen的美国专利号5,693,762,(1997)。此外,还有从特定鼠抗体区合成人源化抗体的商业实体,诸如Protein Design Labs (Mountain View, CA)。
重组抗体
也可使用重组方法制备抗MASP-2抗体。例如,可使用人免疫球蛋白表达文库(可获自例如Stratagene, Corp., La Jolla, CA)产生人抗体片段(VH、VL、Fv、Fd、Fab或F(ab')2)来制备人抗体。然后使用类似于产生嵌合抗体的技术,将这些片段用以构建完整的人抗体。
抗独特型抗体
一旦鉴定出具有所需抑制活性的抗MASP-2抗体,便可采用本领域众所周知的技术将这些抗体用来产生类似部分MASP-2的抗独特型抗体。参见例如Greenspan, N.S.,等人, FASEB J. 7:437, 1993。例如,结合MASP-2并竞争性抑制补体活化所需的MASP-2蛋白的相互作用的抗体可用来产生类似MASP-2蛋白上的MBL结合位点的抗独特型抗体,从而结合并中和MASP-2的结合配体,诸如,例如MBL。
免疫球蛋白片段
用于本发明方法的MASP-2抑制剂不仅包括完整的免疫球蛋白分子,而且还包括众所周知的片段,这些片段包括Fab、Fab'、F(ab)2、F(ab')2和Fv片段、scFv片段、双抗体、线性抗体、单链抗体分子以及由抗体片段形成的多特异性抗体。
本领域众所周知的是,只有小部分的抗体分子即互补位参与抗体与其表位的结合(参见例如Clark, W.R., The Experimental Foundations of Modern Immunology, Wiley & Sons, Inc., NY, 1986)。抗体的pFc'区和Fc区是经典补体途径的效应子,但是不参与抗原结合。其中pFc'区已被酶裂解的抗体,或者所产生的没有pFc'区的抗体被称为F(ab')2片段,它保留了完整抗体的抗原结合部位。分离的F(ab')2片段由于有两个抗原结合部位而被称为二价单克隆片段。类似地,其中Fc区已被酶裂解的抗体,或者所产生的没有Fc区的抗体被称为Fab片段,它保留了完整抗体分子的一个抗原结合部位。
抗体片段可通过蛋白水解而获得,诸如通过常规方法经胃蛋白酶或木瓜蛋白酶消化完整抗体。例如,可通过用胃蛋白酶进行抗体酶裂解来产生抗体片段,从而提供称为F(ab')2的5S片段。该片段可再使用硫醇还原试剂裂解,得到3.5S Fab'单价片段。任选可使用二硫键裂解产生的巯基的封端基团来进行裂解反应。作为替代方法,使用胃蛋白酶的酶裂解直接产生两个单价Fab片段和一个Fc片段。这些方法记载于例如Goldenberg的美国专利号4,331,647;Nisonoff, A.,等人, Arch. Biochem. Biophys. 89:230, 1960; Porter, R.R., Biochem. J. 73:119, 1959; Edelman,等人, 载于Methods in Enzymology 1:422, Academic Press, 1967;以及Coligan的第2.8.1-2.8.10页和第2.10.-2.10.4页。
在一些实施方案中,优选使用缺乏Fc区的抗体片段以避免Fc结合Fcγ受体之后激活经典补体途径。有几种方法可产生避免与Fcγ受体相互作用的MoAb。例如,单克隆抗体的Fc区可通过使用蛋白水解酶部分消化(例如无花果蛋白酶消化),从而用化学法去除,因此产生例如结合抗原的抗体片段,诸如Fab或F(ab)2片段(Mariani, M.,等人, Mol. Immunol. 28:69-71, 1991)。或者,可以在构建本文所述的人源化抗体期间使用不结合Fcγ受体的人γ4 IgG同种型。也可使用本文所述重组技术来工程改造缺少Fc结构域的抗体、单链抗体和结合抗原的结构域。
单链抗体片段
或者,可以制备对MASP-2特异性的单一肽链结合分子,其中重链和轻链Fv区相连接。Fv片段可通过肽接头相连,形成单链抗原结合蛋白(scFv)。通过构建包含编码VH和VL结构域的DNA序列的结构基因来制备这些单链抗原结合蛋白,各结构域之间通过寡核苷酸连接。将结构基因插入表达载体中,随后将其引入宿主细胞(诸如大肠杆菌)中。重组宿主细胞合成了由接头肽桥接两个V结构域的单一多肽链。scFv的制备方法记载于例如Whitlow,等人, "Methods: A Companion to Methods in Enzymology" 2:97, 1991; Bird,等人, Science 242:423, 1988;Ladner的美国专利号4,946,778;Pack, P.,等人, Bio/Technology 11:1271, 1993。
举例来说,可通过将淋巴细胞体外暴露于MASP-2多肽,并在噬菌体或类似载体中选择抗体展示文库(例如通过使用固定化的或标记的MASP-2蛋白或肽),获得MASP-2特异性scFv。可通过对噬菌体或细菌(诸如大肠杆菌)上展示的随机肽文库进行筛选而获得编码具有可能的MASP-2多肽结合结构域的多肽的基因。这些随机肽展示文库可用于筛选与MASP-2相互作用的肽。构建和筛选这些随机肽展示文库的技术是本领域众所周知的(Lardner的美国专利号5,223,409;Lardner的美国专利号4,946,778;Lardner的美国专利号5,403,484;Lardner的美国专利号5,571,698;以及Kay等人, Phage Display of Peptides and Proteins Academic Press, Inc., 1996),且随机肽展示文库和用于筛选这些文库的试剂盒是市售的,例如购自CLONTECH Laboratories, Inc. (Palo Alto, Calif.)、Invitrogen Inc. (San Diego, Calif.)、New England Biolabs, Inc. (Beverly, Mass.)以及Pharmacia LKB Biotechnology Inc. (Piscataway, N.J.)。
用于本发明这个方面的抗MASP-2抗体片段的另一种形式是编码单一互补决定区(CDR)的肽,其结合MASP-2抗原的表位并抑制MASP-2依赖性补体活化。可通过构建编码目标抗体CDR的基因而获得CDR肽(“最小识别单元”)。例如可通过使用聚合酶链式反应从抗体生成细胞的RNA合成可变区,从而制备这些基因(参见例如Larrick等人, Methods: A Companion to Methods in Enzymology 2:106, 1991; Courtenay-Luck, "Genetic Manipulation of Monoclonal Antibodies," 载于Monoclonal Antibodies: Production, Engineering and Clinical Application, Ritter等人 (主编), 第166页, Cambridge University Press, 1995; 以及Ward等人, "Genetic Manipulation and Expression of Antibodies,"载于Monoclonal Antibodies: Principles and Applications, Birch等人 (主编), 第137页, Wiley-Liss, Inc., 1995))。
将本文所述MASP-2抗体施用于需要其的受试者以抑制MASP-2依赖性补体活化。在一些实施方案中,MASP-2抑制剂是效应子功能降低的高亲和力人或人源化单克隆抗MASP-2抗体。
肽抑制剂
在本发明这个方面的一些实施方案中,MASP-2抑制剂包括分离的MASP-2肽抑制剂,其包括抑制MASP-2依赖性补体活化系统的分离的天然肽抑制剂和合成肽抑制剂。本文所用术语“分离的MASP-2肽抑制剂”是指通过结合MASP-2、与MASP-2竞争以结合凝集素途径中的其他识别分子(例如MBL、H-纤维胶凝蛋白、M-纤维胶凝蛋白或L-纤维胶凝蛋白)、和/或直接与MASP-2相互作用以抑制MASP-2依赖性补体活化,从而抑制MASP-2依赖性补体活化的肽,所述肽是基本纯化的,其基本上没有其他与之一起存在于自然界以达到实用和适于其既定用途程度的物质。
已经使用肽抑制剂成功地在体内干扰了蛋白间的相互作用和催化位点。例如,最近,结构上与LFA-1有关的粘着分子的肽抑制剂已获准临床用于凝血病(Ohman, E.M.,等人, European Heart J. 16:50-55, 1995)。研究揭示短的线性肽(<30个氨基酸)防止或干扰整联蛋白依赖性粘附(Murayama, O.,等人, J. Biochem. 120:445-51, 1996)。还使用长度范围为25-200个氨基酸残基的较长肽,成功阻断了整联蛋白依赖性粘附(Zhang, L.,等人, J. Biol. Chem. 271(47):29953-57, 1996)。一般而言,较长的肽抑制剂具有比短肽高的亲和力和/或较慢的解离速度,因此是更有效的抑制剂。研究还表明,环肽抑制剂是有效的整联蛋白体内抑制剂,用于治疗人炎性疾病(Jackson, D.Y.,等人, J. Med. Chem. 40:3359-68, 1997)。产生环肽的一种方法包括其中肽的末端氨基酸是半胱氨酸的肽的合成,从而使得肽能够通过末端氨基酸之间的二硫键以环状形式存在,已经证实用于治疗造血系统肿瘤时,改善了亲和力和体内半衰期(例如Larson的美国专利号6,649,592)。
合成的MASP-2肽抑制剂
通过模拟对于MASP-2功能是重要的靶区的氨基酸序列,来示例性说明用于本发明这个方面方法中的MASP-2抑制肽。用于实施本发明方法的抑制肽的大小范围为约5个氨基酸至约300个氨基酸。表3提供用于实施本发明这个方面的示例性抑制肽的一览表。可通过几种测定法中的一种对候选MASP-2抑制肽作为MASP-2抑制剂起作用的能力进行测定,这些测定法包括例如凝集素特异性的C4裂解测定法(参见实施例2)以及C3b沉积测定法(参见实施例2)。
在一些实施方案中,MASP-2抑制肽得自MASP-2多肽并选自全长的成熟MASP-2蛋白(SEQ ID NO:6),或者得自MASP-2蛋白的特定结构域,诸如例如CUBI结构域(SEQ ID NO:8)、CUBIEGF结构域(SEQ ID N O:9)、EGF结构域(SEQ ID NO:11)以及丝氨酸蛋白酶结构域(SEQ ID NO:12)。如前所述,研究表明CUBEGFCUBII区是二聚化和与MBL结合所需要的(Thielens等人,同上)。特别是在鉴定人体带有Asp105至Gly105纯合突变的研究证实,MASP-2的CUBI结构域中的肽序列TFRSDYN (SEQ ID NO:16)参与结合MBL,所述突变导致从MBL复合物上失去MASP-2 (Stengaard-Pedersen, K.,等人, New England J. Med. 349:554-560, 2003)。
在一些实施方案中,MASP-2抑制肽得自结合MASP-2并参与凝集素补体途径的凝集素蛋白。已经鉴定出参与该途径的几种不同的凝集素,包括甘露聚糖结合凝集素(MBL)、L-纤维胶凝蛋白、M-纤维胶凝蛋白和H-纤维胶凝蛋白(Ikeda, K.,等人, J. Biol. Chem. 262:7451-7454, 1987; Matsushita, M.,等人, J. Exp. Med. 176:1497-2284, 2000; Matsushita, M.,等人, J. Immunol. 168:3502-3506, 2002)。这些凝集素作为同源三聚体亚基的寡聚体而存在于血清中,每个亚基都具有带有碳水化合物识别结构域的N端胶原样纤维。已经证实这些不同的凝集素结合MASP-2,凝集素/MASP-2复合物通过裂解蛋白C4和C2而激活补体。H-纤维胶凝蛋白具有24个氨基酸的氨基端区、带有11个Gly-Xaa-Yaa重复的胶原样结构域、12个氨基酸的颈部结构域和207个氨基酸的血纤蛋白原样结构域(Matsushita, M.,等人, J. Immunol. 168:3502-3506, 2002)。H-纤维胶凝蛋白结合GlcNAc,并使被得自鼠伤寒沙门氏菌(S. typhimurium)、明尼苏达沙门氏菌(S. minnesota)和大肠杆菌的LPS包被的人红细胞凝集。已经证实H-纤维胶凝蛋白结合MASP-2和MAp19并激活凝集素途径,出处同上。L-纤维胶凝蛋白/P35也结合GlcNAc,已被证实与人血清中的MASP-2和MAp19结合,并且证实该复合物激活凝集素途径(Matsushita, M.,等人, J. Immunol. 164:2281, 2000)。因此,用于本发明的MASP-2抑制肽可包括选自MBL蛋白(SEQ ID NO:21)、H-纤维胶凝蛋白(Genbank检索号NM_173452)、M-纤维胶凝蛋白(Genbank检索号O00602)以及L-纤维胶凝蛋白(Genbank检索号NM_015838)的至少5个氨基酸的区域。
更具体地,科学家已经鉴定出MBL上的MASP-2结合部位是在12个Gly-X-Y三联体“
 ”(SEQ ID NO:26)中,该三联体位于MBP胶原样结构域C端部分的铰链和颈之间(Wallis, R.,等人, J. Biol. Chem. 279:14065, 2004)。该MASP-2结合部位区在人H-纤维胶凝蛋白和人L-纤维胶凝蛋白中也是高度保守的。研究指出,所有三种包括氨基酸序列“OGK-X-GP”(SEQ ID NO:22)的凝集素蛋白中存在共有结合部位,该氨基酸序列中字母“O”表示羟脯氨酸,字母“X”表示疏水残基(Wallis等人,2004,同上)。因此,在一些实施方案中,用于本发明这个方面的MASP-2抑制肽长度为至少6个氨基酸并且包含SEQ ID NO:22。已经证实包括氨基酸序列“
”(SEQ ID NO:26)中,该三联体位于MBP胶原样结构域C端部分的铰链和颈之间(Wallis, R.,等人, J. Biol. Chem. 279:14065, 2004)。该MASP-2结合部位区在人H-纤维胶凝蛋白和人L-纤维胶凝蛋白中也是高度保守的。研究指出,所有三种包括氨基酸序列“OGK-X-GP”(SEQ ID NO:22)的凝集素蛋白中存在共有结合部位,该氨基酸序列中字母“O”表示羟脯氨酸,字母“X”表示疏水残基(Wallis等人,2004,同上)。因此,在一些实施方案中,用于本发明这个方面的MASP-2抑制肽长度为至少6个氨基酸并且包含SEQ ID NO:22。已经证实包括氨基酸序列“ ” (SEQ ID NO:24)并得自MBL的肽体外结合MASP-2 (Wallis等人,2004,同上)。为增强与MASP-2的结合,可合成在每端邻接两个GPO三联体的肽(“” SEQ ID NO:25),以增强天然MBL蛋白中所发现的三股螺旋的形成(进一步描述见Wallis, R.,等人, J. Biol. Chem. 279:14065, 2004)。
” (SEQ ID NO:24)并得自MBL的肽体外结合MASP-2 (Wallis等人,2004,同上)。为增强与MASP-2的结合,可合成在每端邻接两个GPO三联体的肽(“” SEQ ID NO:25),以增强天然MBL蛋白中所发现的三股螺旋的形成(进一步描述见Wallis, R.,等人, J. Biol. Chem. 279:14065, 2004)。
MASP-2抑制肽也可得自人H-纤维胶凝蛋白,其包括来自H-纤维胶凝蛋白的共有MASP-2结合区的序列“” (SEQ ID NO:27)。还包括得自人L-纤维胶凝蛋白的肽,其包括来自L-纤维胶凝蛋白的共有MASP-2结合区的序列“
 ” (SEQ ID NO:28)。
” (SEQ ID NO:28)。
MASP-2抑制肽还可得自C4裂解部位,诸如连接到抗凝血酶III C端部分的C4裂解部位“ ” (SEQ ID NO:29) (Glover, G.I.,等人, Mol. Immunol. 25:1261 (1988))。
” (SEQ ID NO:29) (Glover, G.I.,等人, Mol. Immunol. 25:1261 (1988))。
表3:示例性的MASP-2抑制肽
注:字母“O”表示羟脯氨酸。字母“X”为疏水残基。
可对得自C4裂解部位的肽以及抑制MASP-2丝氨酸蛋白酶部位的其他肽进行化学修饰,以使其成为不可逆的蛋白酶抑制剂。例如,合适的修饰可包括但是不必限于C端、Asp或Glu、或者添加到功能侧链上的卤甲基酮(Br、Cl、I、F);在氨基或其他功能侧链上的卤乙酰(或其他α-卤乙酰)基团;在氨基端或羧基端或者功能侧链上的环氧基或含亚胺的基团;或者在氨基端或羧基端或者其他功能侧链上的亚氨酸酯。这些修饰提供了通过肽的共价结合而永久抑制酶的这一优势。这可能导致有效剂量较低和/或施用肽抑制剂需要的频率较低。
除了上述抑制肽以外,用于本发明方法的MASP-2抑制肽包括含有按本文所述方法获得的抗MASP-2 MoAb的结合MASP-2的CDR3区的肽。用于合成肽的CDR区的序列可以通过本领域已知方法来测定。重链可变区是通常长度范围为100-150个氨基酸的肽。轻链可变区是通常长度范围为80-130个氨基酸的肽。在重链和轻链可变区内部的CDR序列包括大约仅3-25个氨基酸序列,本领域普通技术人员很容易对其进行测序。
本领域技术人员应理解的是,上述的MASP-2抑制肽的基本同源的变异也可具有MASP-2抑制活性。示例性的变异包括但不必限于在主题肽的羧基端或氨基端部分有插入、缺失、置换和/或添加的氨基酸的肽及其混合物。因此,我们认为这些具有MASP-2抑制活性的同源肽可用于本发明的方法。所述肽还可包括复制基序和具有保守取代的其他修饰。本文其他部分描述了保守变体,包括一种氨基酸与具有同样电荷、大小或疏水性等性质的另一种氨基酸的交换。
可以对MASP-2抑制肽进行修饰以增加溶解度和/或使正或负电荷最大化,以便使之更类似于完整蛋白的区段。衍生物可以具有或没有本文所公开肽的精确的一级氨基酸结构,只要衍生物在功能上仍保留所需要的MASP-2抑制特性。所述修饰可包括氨基酸取代,即被通常已知的20种氨基酸之一或另一种氨基酸取代、被附加有所需特征(诸如酶降解抗性)的衍生化氨基酸或取代氨基酸取代或者被D氨基酸取代,或者被另外的模拟一种或多种氨基酸或肽的天然构象和功能的分子或化合物(诸如碳水化合物)取代;氨基酸缺失;氨基酸插入,即插入通常已知的20种氨基酸之一或另一种氨基酸、插入附加有所需特征(诸如酶降解抗性)的衍生化氨基酸或取代氨基酸或者插入D氨基酸,或者被另外的模拟一种或多种氨基酸或肽的天然构象和功能的分子或化合物(诸如碳水化合物)取代;或者被另外的模拟母体肽的天然构象、电荷分布和功能的分子或化合物(诸如碳水化合物或核酸单体)取代。肽也可通过乙酰化或酰胺化进行修饰。
可根据肽生物合成、碳水化合物生物合成等已知技术合成衍生的抑制肽。开始时,技术人员可根据合适的计算机程序来确定目标肽的构象。一旦知道本文所公开的肽的构象,然后技术人员便能以合理设计方式来确定能够在一个或多个位点进行什么类别的取代,以便形成的衍生物保留了母体肽的基本构象和电荷分布,但却可以拥有母体肽中不存在的特征或者其特征优于母体肽存在的特征。一旦鉴定出候选衍生分子,就可使用本文所述测定法来测定衍生物,以确定它们是否发挥MASP-2抑制剂的作用。
MASP-2抑制肽的筛选
还可使用分子建模和合理分子设计来产生和筛选模拟MASP-2关键结合区的分子结构并抑制MASP-2的补体活性的肽。用于建模的分子结构包括抗MASP-2单克隆抗体的CDR区,以及已知对MASP-2功能十分重要的目标区域,这些区域包括如前所述的二聚化所需要的区域、涉及结合MBL的区域以及丝氨酸蛋白酶活性部位。用于鉴定结合特定靶的肽的方法是本领域众所周知的。例如,分子印记可用于从头构建大分子结构,诸如结合特定分子的肽。参见例如


举例来说,制备MASP-2结合肽模拟物的一种方法如下。使已知MASP-2结合肽或具有MASP-2抑制作用的抗MASP-2抗体的结合区的功能性单体(模板)聚合。然后去除模板,接着在模板留下的空隙中使第二类单体聚合,得到具有类似于模板的一种或多种所需性质的新分子。除了以这种方式制备肽外,还可制备作为MASP-2抑制剂的其他MASP-2结合分子,诸如多糖、核苷、药物、核蛋白、脂蛋白、碳水化合物、糖蛋白、类固醇、脂质和其他生物活性材料。该方法适用于设计各种各样比其天然对应物更稳定的生物学模拟物,因为它们通常是通过功能单体的自由基聚合而成的,产生了具有生物不能降解骨架的化合物。
肽合成
可采用本领域众所周知的技术制备MASP-2抑制肽,诸如最初由Merrifield提出的固相合成技术(Merrifield,J. Amer. Chem. Soc. 85:2149-2154, 1963)。例如,可根据生产商提供的说明书,使用Applied Biosystems 431A肽合成仪(Foster City, Calif.)实现自动合成。其他技术参见例如Bodanszky, M.,等人, Peptide Synthesis, 第二版, John Wiley & Sons, 1976,以及本领域技术人员已知的其他参考文献。
还可使用本领域技术人员已知的标准遗传工程改造技术来制备肽。例如,可通过酶的方法将编码肽的核酸插入表达载体中,进行DNA表达,在所需要的氨基酸存在下将DNA翻译成肽,从而来制备肽。然后,使用层析技术或电泳技术将肽纯化,或者通过将编码肽的序列与编码载体蛋白的核酸序列同步插入表达载体中的载体蛋白方法来实现,所述载体蛋白可以与肽融合,随后再从肽上切下来。可采用层析技术、电泳技术或免疫学技术(诸如经载体蛋白的抗体而结合到树脂上)分离蛋白-肽融合物。可使用化学方法或酶方法(例如通过水解酶)来裂解肽。
也可按照常规技术,用重组宿主细胞来生产用于本发明方法的MASP-2抑制肽。为了表达MASP-2抑制肽编码序列,必须将编码肽的核酸分子与控制表达载体转录表达的调节序列可操作连接,然后引入宿主细胞中。除了转录调节序列(诸如启动子和增强子)之外,表达载体可包括翻译调节序列和标记基因,标记基因适合于选择携带表达载体的细胞。
可用“基因仪”并用亚磷酰胺法等方法来合成编码MASP-2抑制肽的核酸分子。如果诸如基因或基因片段合成的应用中需要化学合成的双链DNA,则分别制备每条互补链。短基因(60-80个碱基对)的生产在技术上简单易行,可通过合成互补链,然后将它们退火来实现。对于较长基因的生产来说,要由长度为20-100个核苷酸的单链片段按模块形式装配成合成基因(双链)。有关多核苷酸合成的综述参见例如

。
小分子抑制剂
在一些实施方案中,MASP-2抑制剂是小分子抑制剂,包括具有低分子量的天然和合成物质,诸如例如肽、肽模拟物和非肽抑制剂(包括寡核苷酸和有机化合物)。MASP-2的小分子抑制剂可根据抗MASP-2抗体的可变区的分子结构而产生。
小分子抑制剂也可根据MASP-2的晶体结构采用计算机药物设计来进行设计和产生(Kuntz I.D.,等人, Science 257:1078, 1992)。已经证实大鼠MASP-2的晶体结构(Feinberg, H.,等人, EMBO J. 22:2348-2359, 2003)。应用Kuntz等描述的方法,将MASP-2晶体结构坐标输入计算机程序(诸如DOCK),计算机将输出一系列预期结合MASP-2的小分子结构。这些计算机程序的使用为本领域技术人员所众所周知。例如,使用HIV-1蛋白酶抑制剂的晶体结构,通过运用程序DOCK,评价从剑桥晶体学数据库(Cambridge Crystallographic database)中查找到的化合物与酶结合部位的匹配性,来鉴定作为HIV-1蛋白酶抑制剂的独特的非肽配体(Kuntz, I.D.,等人, J. Mol. Biol. 161:269-288, 1982; DesJarlais, R.L.,等人, PNAS 87:6644-6648, 1990)。
应用诸如实施例10所述的MASP-2结合测定法来筛选用计算机方法鉴定为潜在的MASP-2抑制剂的一系列小分子结构。然后在诸如实施例2所述的功能测定法中对被确定为结合MASP-2的小分子进行分析,以确定它们是否抑制MASP-2依赖性补体活化。
MASP-2可溶性受体
认为其他合适的MASP-2抑制剂包括MASP-2可溶性受体,可以采用本领域普通技术人员已知技术进行生产。
MASP-2的表达抑制剂
在本发明这个方面的另一个实施方案中,MASP-2抑制剂是能够抑制MASP-2依赖性补体活化的MASP-2表达抑制剂。在本发明这个方面的实施中,代表性的MASP-2表达抑制剂包括MASP-2反义核酸分子(诸如反义mRNA、反义DNA或反义寡核苷酸)、MASP-2核酶以及MASP-2 RNAi分子。
反义RNA和反义DNA分子通过与MASP-2 mRNA杂交并阻止MASP-2蛋白的翻译而起到直接阻断MASP-2 mRNA翻译的作用。反义核酸分子能以许多不同的方式来构建,只要它能够干扰MASP-2的表达。例如,可通过使MASP-2 cDNA (SEQ ID NO:4)的编码区(或其部分)相对于其正常转录方向进行反转以供其互补序列转录,从而构建反义核酸分子。
反义核酸分子通常与一个或多个靶基因的至少一部分基本相同。然而,核酸并不需要完全相同以抑制表达。较高的同源性一般可用来对较短反义核酸分子的使用予以补偿。最小的百分比同一性通常约65%以上,但是较高的百分比同一性可对内源序列的表达发挥更有效的阻抑作用。通常优选基本为大约80%以上的较大的百分比同一性,尽管通常最优选约95%至完全相同。
反义核酸分子不需要具有与靶基因相同的内含子或外显子模式,靶基因的非编码区段可能在获得靶基因表达的反义抑制方面与编码区段是等效的。至少大约8个左右核苷酸的DNA序列可用作反义核酸分子,尽管优选较长的序列。在本发明中,有用的MASP-2抑制剂的代表性实例是反义MASP-2核酸分子,该分子与由SEQ ID NO:4所示核酸序列组成的MASP-2 cDNA的互补序列有至少90%同一性。SEQ ID NO:4所示核酸序列编码由SEQ ID NO:5所示的氨基酸序列组成的MASP-2蛋白。
反义寡核苷酸靶向结合MASP-2 mRNA是可用于降低MASP-2蛋白合成水平的另一种机制。例如,多聚半乳糖醛酸酶和蝇蕈碱2型乙酰胆碱受体的合成被针对它们相应mRNA序列的反义寡核苷酸所抑制(Cheng的美国专利号5,739,119和Shewmaker的美国专利号5,759,829)。此外,用核蛋白细胞周期蛋白、多药抗药性基因(MDG1)、ICAM-1、E-选择蛋白、STK-1、纹状体GABAA受体和人EGF证实了反义抑制的实例(参见例如Baracchini的美国专利号5,801,154;Baker的美国专利号5,789,573;Considine的美国专利号5,718,709;以及Reubenstein的美国专利号5,610,288)。
文献记载了使得普通技术人员能够确定哪些寡核苷酸可用于本发明的系统,包括使用RNA酶H裂解作为转录物中序列可及性的标志来探测靶mRNA的合适部位。Scherr, M.,等人, Nucleic Acids Res. 26:5079-5085, 1998; Lloyd,等人, Nucleic Acids Res. 29:3665-3673, 2001。将与MASP-2转录物某些区域互补的反义寡核苷酸混合物加到表达MASP-2的细胞提取物(诸如肝细胞)中,进行杂交以产生易受RNA酶H攻击的部位。该方法可以与计算机辅助序列选择相结合,所述计算机辅助的序列选择能够根据序列形成二聚体、发夹结构或其他二级结构的相对能力来预测用于反义组成的最佳序列选择,所述结构可降低或抑制对宿主细胞靶mRNA的特异性结合。可使用OLIGO引物分析软件(Rychlik, I.,1997)和BLASTN 2.0.5算法软件(Altschul, S.F.,等人, Nucl. Acids Res. 25:3389-3402, 1997)来进行这些二级结构分析和靶部位选择考量。针对靶序列的反义化合物的长度优选包括约8个至约50个核苷酸。特别优选包括约9个至约35个左右核苷酸的反义寡核苷酸。本发明人认为,实施本发明基于反义寡核苷酸的方法极优选范围为9个至35个核苷酸(即9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34或35个碱基长度左右的核苷酸)的所有寡核苷酸组成。极优选的MASP-2 mRNA靶区域是位于或邻近AUG翻译起始密码子的区域,以及与mRNA的5’区基本互补的序列,例如MASP 2基因核苷酸序列(SEQ ID NO:4)的-10和+10区域之间的序列。示例性的MASP-2表达抑制剂见表4。
表4:MASP-2的示例性表达抑制剂
| SEQ ID NO:30 (SEQ ID NO:4的核苷酸22-680) | 编码CUBIEGF的MASP-2 cDNA (SEQ ID NO:4) 核酸序列 |
| 包括MASP-2翻译起始位点的SEQ ID NO:4核苷酸12-45 (有义) | |
 |
编码包括MASP-2 MBL 结合位点的区域的SEQ ID NO:4核苷酸361-396(有义) |
 |
编码包括CUBII结构域的区域的SEQ ID NO:4 核苷酸610-642 |
如上所述,本文所用术语“寡核苷酸”是指核糖核酸(RNA)或脱氧核糖核酸(DNA)或其模拟物的寡聚体或多聚体。该术语也包括由天然存在的核苷酸、糖和核苷间(骨架)共价键所组成的那些寡核碱基(oligonucleobase)以及具有非天然存在的修饰的寡核苷酸。这些修饰使得能够引入某些无法通过天然存在的寡核苷酸提供的某些所需性质,诸如毒性降低、抗核酸酶降解的稳定性提高以及细胞摄取增强。在示例性实施方案中,本发明的反义化合物由于磷酸二酯骨架的修饰延长了反义寡核苷酸的寿命而不同于天然DNA,其中磷酸取代基被硫代磷酸酯置换。同样地,寡核苷酸的一端或两端可被一个或多个间插在核酸链内相邻碱基对之间的吖啶衍生物取代。
反义方案的另一替代是使用“RNA干扰”(RNAi)。双链RNA (dsRNA)可在哺乳动物体内引起基因沉默。RNAi的天然功能和共抑制似乎保护基因组不受可移动遗传元件(诸如反转录转座子)以及当其活化时在宿主细胞内产生异常RNA或dsRNA的病毒侵袭(参见例如Jensen, J.,等人, Nat. Genet. 21:209-12, 1999)。可通过合成两条能够形成双链RNA分子的RNA链来制备双链RNA分子,每条链的长度约为19-25 (例如19-23)个核苷酸。例如,用于本发明方法的dsRNA分子可包括相当于表4所列序列及其互补序列的RNA。优选至少一条RNA链具有1-5个核苷酸的3’突出端。合成的RNA链是在形成双链分子的条件下进行组合的。RNA序列可包含SEQ ID NO:4的至少8个核苷酸的部分,总长度为25个核苷酸或以下。特定靶的siRNA序列的设计为本领域普通技术人员所掌握。可获得设计siRNA序列并保证表达有至少70%敲低的商业服务(Qiagen, Valencia, Calif)。
dsRNA可作为药物组合物施用并按已知方法进行,其中核酸被引入所需的靶细胞中。通常使用的基因转移方法包括磷酸钙、DEAE-葡聚糖、电穿孔、显微注射和病毒方法。这些方法在Ausubel等人, Current Protocols in Molecular Biology, John Wiley & Sons, Inc., 1993中教导。
也可利用核酶来降低MASP-2的量和/或生物活性,诸如靶向MASP-2 mRNA的核酶。核酶是催化性RNA分子,能够裂解具有与核酶序列完全或部分同源的序列的核酸分子。可以设计核酶转基因,该核酶转基因编码与靶RNA特异性配对并在特定位置裂解磷酸二酯骨架的RNA核酶,从而使靶RNA功能性失活。在进行这种裂解时,核酶本身并无改变,因此能够再循环并裂解其他分子。在反义RNA中包括核酶序列赋予了反义RNA裂解RNA的活性,从而增加了反义构建体的活性。
用于实施本发明的核酶通常包括杂交区和催化区,杂交区至少约9个核苷酸,与至少部分靶MASP-2 mRNA的核苷酸序列互补,催化区适于裂解靶MASP-2 mRNA (一般参见


核酶可以掺入核酶序列的RNA寡核苷酸的形式直接靶向细胞,或者作为编码所需要的核酶RNA的表达载体而被引入细胞。可与所述用于反义多核苷酸的大致相同的方式使用并应用核酶。
可通过本领域已知的用于DNA和RNA分子合成的任何方法来制备用于本发明方法的反义RNA和DNA、核酶和RNAi分子。这些方法包括本领域众所周知的用于寡脱氧核糖核苷酸和寡核糖核苷酸的化学合成技术,诸如固相亚磷酰胺化学合成。或者,可通过编码反义RNA分子的DNA序列的体外和体内转录来产生RNA分子。这种DNA序列可被掺入各种各样的载体中,载体中插入了合适的RNA聚合酶启动子(诸如T7或SP6聚合酶启动子)。或者,可将取决于所用启动子而组成型或诱导型合成反义RNA的反义cDNA构建体稳定地引入细胞系中。
可引入各种众所周知的DNA分子修饰,从而增加稳定性和半衰期。有用的修饰包括但不限于将核糖核苷酸或脱氧核糖核苷酸侧翼序列加到分子的5’端和/或3’端,或者在寡脱氧核糖核苷酸骨架内使用硫代磷酸酯或2’ O-甲基而不是磷酸二酯键。
V. 药物组合物和递送方法
给药
在另一个方面,本发明提供用于抑制患有本文所述的疾病或状况的受试者中MASP2依赖性补体活化的副作用的组合物,其包括将包含治疗有效量的MASP-2抑制剂和药学可接受的载体的组合物施用于受试者。可以治疗或改善MASP-2依赖性补体活化相关疾病的治疗有效剂量,将MASP-2抑制剂施用于需要其的受试者。治疗有效剂量是指MASP-2抑制剂足以导致改善与疾病或状况相关的症状的量。
可通过标准药学方法,使用实验动物模型(诸如实施例1中所述的表达人MASP-2转基因的鼠MASP-2 -/-小鼠模型),来测定MASP-2抑制剂的毒性和治疗功效。使用这些动物模型,可使用标准方法来确定NOAEL (无观察的副作用水平)和MED (最小有效剂量)。NOAEL/MED效应之间的剂量比是治疗比率,用NOAEL/MED比率表示。最优选的是治疗比率或指数高的MASP-2抑制剂。从细胞培养物分析和动物研究中获得的数据可用来配制用于人体的剂量范围。MASP-2抑制剂的剂量优选在循环浓度的范围之内,包括几乎无毒性或没有毒性的MED。剂量可在这个范围内变化,这取决于所采用的剂型和所使用的施用途径。
对于任何化合物制剂来说,可使用动物模型来评价治疗有效剂量。例如,可在动物模型中配制达得包括MED在内的循环血浆浓度范围的剂量。也可通过例如高效液相层析法来测定血浆中MASP-2抑制剂的定量水平。
除了毒性研究之外,也可根据有生命的受试者中存在的MASP-2蛋白的量以及MASP-2抑制剂的结合亲和力来估计有效剂量。已经证实正常受试人体血清中存在的MASP-2水平在500 ng/ml低水平范围内,可使用MASP-2定量测定法来测定具体受试者的MASP-2水平(描述于Moller-Kristensen M.,等人, J. Immunol. Methods 282:159-167, 2003)。
包含MASP-2抑制剂的组合物的施用剂量一般根据受试者年龄、体重、身高、性别、总体医疗状况和病史而变化。举例来说,可在大约0.010-10.0 mg/kg、优选0.010-1.0 mg/kg、更优选0.010-0.1 mg/kg受试者体重的剂量范围内施用MASP-2抑制剂,诸如抗MASP-2抗体。在某些实施方案中,所述组合物包含抗MASP-2抗体和MASP-2抑制肽的组合。
可根据本领域技术人员众所周知的补体测定法来测定特定受试者的本发明MASP-2抑制剂组合物和方法的治疗功效以及合适的剂量。补体产生多种特定的产物。在最近的十年间,已经研发出灵敏且特异性的测定法,而且大多数的这类活化产物都是市售的,包括小的活化片段C3a、C4a和C5a和大的活化片段iC3b、C4d、Bb和sC5b-9。大多数的这类测定法都利用了与暴露在片段而不是暴露在其所形成的天然蛋白上的新抗原(new antigen/neoantigen)起反应的单克隆抗体,这使得这些测定法非常简单而特异性。大多数都依赖于ELISA技术,尽管有时放射免疫测定法仍用于C3a和C5a。放射免疫测定法测定未经加工的片段及其“desArg”片段,这些片段是存在于循环中的主要形式。未经加工的片段和C5adesArg通过结合细胞表面受体而被迅速清除掉,因而以极低浓度存在,而C3adesArg则不结合细胞,只在血浆中聚积。测定C3a提供了补体活化灵敏的、不依赖途径的标志。可通过测定Bb片段来评价替代途径活化。检测膜攻击途径活化的液相产物sC5b-9,提供了补体被完全激活的证据。因为凝集素途径和经典途径都产生同样的活化产物C4a和C4d,所以测定这两种片段并不提供有关这两条途径中哪一条途径产生了活化产物的任何信息。
MASP-2依赖性补体活化抑制的特征在于补体系统成分的至少一种以下的变化,所述变化是由于根据本发明的方法施用MASP-2抑制剂所致:MASP-2依赖性补体活化系统产物C4b、C3a、C5a和/或C5b-9 (MAC)形成或产生的抑制(测定法参见例如实施例2)、C4裂解和C4b沉积的减少(测定法参见例如实施例10),或者C3裂解和C3b沉积的减少(测定法参见例如实施例10)。
另外的药物
包含MASP-2抑制剂的组合物和方法可任选包括一种或多种另外的治疗药物,它们可增强MASP-2抑制剂的活性,或者以相加或协同的方式提供相关治疗功能。例如,在治疗患有TTP的受试者的背景(其中所述受试者对于ADAM-TS13的抑制剂为阳性)中,可以将一种或多种MASP-2抑制剂与一种或多种免疫试剂组合施用(包括共同施用)。合适的免疫抑制剂包括:皮质类固醇、利妥昔单抗、环孢素等。在治疗患有HUS或aHUS或处于发生HUS或aHUS的风险中的受试者的背景中,可以将一种或多种MASP-2抑制剂与合适的抗生素组合施用(包括共同施用)。在治疗患有aHUS或处于发生aHUS的风险中的受试者的背景中,可以将一种或多种MASP-2抑制剂与其他补体抑制剂诸如依库丽单抗(Soliris)、TT-30、针对因子B的抗体或抑制末端补体组分或替代途径放大的其他试剂组合施用(包括共同施用)。
可对另外药物的包括和选择加以确定以获得所需的治疗结果。在一些实施方案中,MASP-2抑制剂可以与一种或多种抗炎和/或镇痛剂组合施用。合适的消炎和/或镇痛药物包括:血清素受体拮抗剂;血清素受体激动剂;组胺受体拮抗剂;缓激肽受体拮抗剂;激肽释放酶抑制剂;速激肽受体拮抗剂,包括神经激肽1和神经激肽2受体亚型拮抗剂;降钙素基因相关肽(CGRP)受体拮抗剂;白介素受体拮抗剂;花生四烯酸代谢物合成途径中有活性的酶的抑制剂,包括磷脂酶抑制剂,包括PLA2同种型抑制剂和PLCγ同种型抑制剂、环加氧酶(COX)抑制剂(它可以是COX-1、COX-2的抑制剂或非选择性COX-1和COX-2的抑制剂)、脂加氧酶抑制剂;前列腺素类受体拮抗剂,包括类二十烷酸EP-1和EP-4受体亚型拮抗剂以及血栓烷受体亚型拮抗剂;白三烯受体拮抗剂,包括白三烯B4受体亚型拮抗剂和白三烯D4受体亚型拮抗剂;阿片样物质受体激动剂,包括μ-阿片样物质、δ-阿片样物质和κ-阿片样物质受体亚型激动剂;嘌呤受体激动剂和拮抗剂,包括P2X受体拮抗剂和P2Y受体激动剂;腺苷三磷酸(ATP)敏感性钾通道开放剂;MAP激酶抑制剂;烟酸乙酰胆碱抑制剂;以及α肾上腺素能受体激动剂(包括α-1、α-2和非选择性的α-1和α-2激动剂)。
本发明的MASP-2抑制剂也可与一种或多种其他的补体抑制剂联用,诸如C5的抑制剂。迄今为止,依库丽单抗(Solaris?),针对C5的抗体,是已被批准用于人使用的唯一补体-靶向药物。然而已经证明一些药物体内阻断补体。K76COOH和萘莫司他甲磺酸盐是两种在移植动物模型中具有一定疗效的药物(Miyagawa, S.,等人, Transplant Proc. 24:483-484, 1992)。研究还表明低分子量肝素也有效调节补体活性(Edens, R.E.,等人, Complement Today, pp. 96-120, Basel: Karger, 1993)。我们认为这些小分子抑制剂可用作与本发明MASP-2抑制剂联用的药物。
其他天然存在的补体抑制剂可用于与本发明的MASP-2抑制剂联用。补体的生物学抑制剂包括可溶性补体因子1 (sCR1)。这是一种可在人细胞的外膜上找到的天然存在的抑制剂。其他膜抑制剂包括DAF、MCP和CD59。已经对重组形式的体外和体内抗补体活性进行了测定。sCR1表明在异种移植中是有效的,异种移植中补体系统(替代和经典两者)在新移植器官灌注血液几分钟内就引起过度反应排斥综合征(hyperactive rejection syndrome) (Platt, J.L.,等人, Immunol. Today 11:450-6, 1990; Marino, I.R.,等人, Transplant Proc. 1071:6, 1990; Johnstone, P.S.,等人, Transplantation 54:573-6, 1992)。sCR1的使用保护并延长了移植器官的存活时间,表明器官存活机制中的补体途径(Leventhal, J.R.,等人, Transplantation 55:857-66, 1993; Pruitt, S.K.,等人, Transplantation 57:363-70, 1994)。
适于与本发明的组合物联用的其他补体抑制剂还包括例如MoAb,诸如由Alexion Pharmaceuticals, Inc. New Haven Connecticut正在开发的抗C5抗体(例如,依库丽单抗eculizumab),以及抗备解素MoAb。
药物载体和递送媒介物
一般而言,本发明的MASP-2抑制剂组合物当与任何其他所选的治疗药物联用时,适合于包含在药学可接受的载体中。载体是无毒的、生物相容的,其选择不得对MASP-2抑制剂(以及与其联用的任何其他治疗药物)的生物活性产生不利的影响。肽的示例性药学可接受的载体描述于Yamada的美国专利号5,211,657。用于本发明的抗MASP-2抗体和抑制肽可以配制成固体、半固体、凝胶、液体或气体形式制剂,诸如片剂、胶囊剂、粉剂、颗粒剂、软膏剂、溶液剂、栓剂(depositories)、吸入剂和注射剂,供口服、胃肠外或外科手术施用。本发明还包括通过涂敷医疗装置等局部施用组合物。
用于通过注射、输注或冲洗和局部递送的胃肠外递送的合适载体包括蒸馏水、磷酸缓冲生理盐水、标准林格氏液或乳酸盐林格氏液、葡萄糖溶液、Hank氏溶液或丙二醇。此外,无菌不挥发油可用作溶剂或悬浮介质。对于此目的,可采用任何生物相容性油,包括合成的甘油单酯或甘油二酯。此外,脂肪酸(诸如油酸)用于注射剂的制备中。可将载体和药物配制成为液体制剂、混悬剂、可聚合或不可聚合的凝胶剂、糊剂或药膏。
载体也可包括递送媒介物以使药物递送持续(即延长、延缓或调节),或者增强治疗药物的递送、吸收、稳定性或药代动力学特征。这种递送媒介物可包括但不限于以下实例:由蛋白质、脂质体、碳水化合物、合成有机化合物、无机化合物、聚合水凝胶或共聚水凝胶和聚合物胶束组成的微粒、微球、纳米球或纳米粒。合适的水凝胶和胶束递送系统包括WO 2004/009664 A2中公开的PEO:PHB:PEO共聚物和共聚物/环糊精复合物以及美国专利申请公开号2002/0019369 A1中公开的PEO和PEO/环糊精复合物。这些水凝胶可局部注射到期望起效部位,或者皮下或肌内注射以形成缓释贮库。
对于关节内递送,MASP-2抑制剂可被装载于上述可注射的液体或凝胶载体、上述可注射的缓释递送媒介物、或者透明质酸或透明质酸衍生物中。
对于非肽能药物的口服施用,MASP-2抑制剂可装载于惰性填充剂或稀释剂中,诸如蔗糖、玉米淀粉或纤维素中。
对于局部施用,MASP-2抑制剂可装载于软膏剂、洗剂、乳膏剂、凝胶剂、滴剂、栓剂、喷雾剂、液体制剂或粉剂中或者经由透皮贴剂的凝胶或微胶囊递送系统中。
各种经鼻和经肺的递送系统正在研发中,包括气溶胶、定量吸入器、干粉吸入器和雾化吸入器,可分别合适地适于递送气溶胶、吸入剂或雾化递送媒介物中的本发明药物。
对鞘内(IT)或脑室内(ICV)递送,合适的无菌递送系统(例如液体制剂;凝胶剂、混悬剂等)可用来施用本发明的药物。
本发明的组合物还可包括生物相容性赋形剂,诸如分散剂或润湿剂、悬浮剂、稀释剂、缓冲剂、渗透促进剂、乳化剂、粘合剂、增稠剂、矫味剂(用于口服施用)。
抗体和肽的药物载体
更具体地讲,至于抗MASP-2抗体和抑制肽,可以按注射剂量的所述化合物的溶液剂或混悬剂经胃肠外施用示例性剂型,所述化合物包含在生理上可接受的稀释剂与药物载体内,药物载体可以是无菌液体,诸如水、油、盐水、甘油或乙醇。此外,包含抗MASP-2抗体和抑制肽的组合物中可存在辅助物质诸如润湿剂或乳化剂、表面活性剂、pH缓冲物质等。药物组合物的额外组分包括油脂(诸如动物、植物或合成来源的油脂),例如大豆油和矿物油。一般而言,二元醇诸如丙二醇或聚乙二醇是优选的注射溶液剂的液体载体。
还能以长效注射剂或植入制剂的形式施用抗MASP-2抗体和抑制肽,这些制剂可按允许活性剂缓释或脉冲释放的方式来配制。
表达抑制剂的药学可接受的载体
更具体地讲,至于用于本发明方法的表达抑制剂,提供包含上述表达抑制剂和药学可接受的载体或稀释剂的组合物。组合物还可包含胶体分散体系。
包括表达抑制剂的药物组合物可包括但不限于溶液剂、乳剂和含有脂质体的制剂。这些组合物可由各种组分制备,所述组分包括但不限于预制液体、自乳化固体和自乳化半固体。这些组合物的制备通常包括将表达抑制剂与一种或多种以下的成分相混合:缓冲剂、抗氧化剂、低分子量多肽、蛋白质、氨基酸、碳水化合物(包括葡萄糖、蔗糖或糊精)、螯合剂(诸如EDTA)、谷胱甘肽以及其他稳定剂和赋形剂。中性缓冲盐水或者与非特异性血清白蛋白混合的盐水是合适稀释剂的实例。
在一些实施方案中,组合物可被制备和配制成乳剂,乳剂通常是一种液体以液滴形式分散在另一种液体中的非均相系统(参见Idson,载于Pharmaceutical Dosage Forms, 第一卷, Rieger和Banker (主编), Marcek Dekker, Inc., N.Y., 1988)。用于乳剂制剂的天然存在的乳化剂的实例包括阿拉伯树胶、蜂蜡、羊毛脂、卵磷脂和磷脂。
在一个实施方案中,包括核酸的组合物可配制成微乳剂。本文所用的微乳剂是指水、油和两亲物的系统,它是单一的光学各向同性和热力学稳定的液体溶液(参见Rosoff载于Pharmaceutical Dosage Forms, 第一卷)。本发明的方法也可使用脂质体来将反义寡核苷酸转移和递送到所需要的部位。
用于局部施用的表达抑制剂的药物组合物和制剂可包括透皮贴剂、软膏剂、洗剂、乳膏剂、凝胶剂、滴剂、栓剂、喷雾剂、液体制剂和粉剂。可以使用常规药物载体以及水性基质、粉末基质或油性基质和增稠剂等等。
施用方式
可以多种方式施用包含MASP-2抑制剂的药物组合物,这取决于是局部还是全身性施用方式最适于待治疗的疾病。此外,本文上面所述的关于体外再灌注方法,可通过将本发明的组合物引入再循环血液或血浆来施用MASP-2抑制剂。而且,本发明的组合物可通过将组合物涂布或掺入可植入的医疗装置上面或里面而递送。
全身性递送
本文所用术语“全身性递送”和“全身性施用”是指包括但不限于口服和胃肠外途径,包括肌内(IM)、皮下、静脉内(IV)、动脉内、吸入、舌下、口腔、局部、经皮、经鼻、直肠、阴道和其他施用途径,它们将所递送的药物有效地分散到预期治疗作用的一个或多个部位。用于本发明的全身性递送的优选途径包括静脉内、肌内、皮下和吸入。应当理解的是,对于用于本发明具体组合物中所选用的药物,确切的全身性施用途径将部分地考虑药物对与特定施用途径相关的代谢转化途径的敏感性加以确定。例如,肽能药物可能最适于通过口服以外的途径施用。
可通过任何合适的方法将MASP-2抑制抗体和多肽递送到需要其的受试者中。递送MASP-2抗体和多肽的方法包括经口服、肺部、胃肠外(例如肌内、腹膜内、静脉内(IV)或皮下注射)、吸入(诸如经由微细粉制剂)、经皮、经鼻、阴道、直肠或者舌下施用途径而施用,可将其配制成适于各自施用途径的剂型。
举例来说,可以通过将MASP-2抑制抗体和肽应用到能够吸收多肽的身体膜上,例如鼻膜、胃肠膜和直肠膜,而将其引入活体内。通常将多肽和渗透促进剂一起应用到可吸收膜上(参见例如


可以引入与其他分子(诸如脂质)结合的MASP-2抑制抗体和多肽,以保护多肽不被酶降解。例如,聚合物、尤其是聚乙二醇(PEG)的共价结合已被用来保护某些蛋白质不被体内的酶水解,从而延长半衰期(Fuertges, F.,等人, J. Controlled Release 11:139, 1990)。已经报道了许多用于蛋白质递送的聚合物系统

。
最近,开发出血清稳定性和循环半衰期得到改进的脂质体(参见例如Webb的美国专利号5,741,516)。而且,对脂质体和脂质体样制备物作为可能的药物载体的各种方法进行了综述(参见例如Szoka的美国专利号5,567,434;Yagi的美国专利号5,552,157;Nakamori的美国专利号5,565,213;Shinkarenko的美国专利号5,738,868以及Gao的美国专利号5,795,587)。
对于经皮应用,可将MASP-2抑制抗体和多肽与其他合适的成分(诸如载体和/或佐剂)组合。对这些其他成分的性质没有限制,除了对于其期望施用来说必须是药学可接受的,并且不能降低组合物中活性成分的活性。合适媒介物的实例包括含或不含纯化胶原的软膏、乳膏、凝胶或混悬液。MASP-2抑制抗体和多肽也可被浸渍到透皮贴剂、膏药和绷带中,优选液体或半液体形式。
可以在为维持治疗效果所需水平而确定的间隔的周期性基础上,全身性施用本发明的组合物。例如,可按每2-4周或者以更低频率的间隔施用组合物(诸如经皮下注射)。剂量方案将由医师考虑可能影响药物联用的作用的各种因素来确定。这些因素可包括待治疗疾病的进展程度、患者年龄、性别和体重和其他临床因素。各独立药物成分的剂量将随组合物中所包含的MASP-2抑制剂以及任何药物递送媒介物(例如缓释递送媒介物)的存在和性质而变化。此外,可在考虑施用频率和所递送药物的药代动力学特征的变化后对剂量进行调整。
局部递送
本文所用术语“局部”包括药物在预定的局限性作用部位上或其周围的应用,可包括例如局部递送到皮肤或其他受累组织;眼递送;鞘内(IT)、脑室内(ICV)、关节内、腔内、颅内或肺泡内施用、安置或冲洗。可优选能够施用低剂量的局部施用以避免全身性副作用,以及更精确地控制递送时机和局部递送部位的活性剂浓度。不论患者之间在新陈代谢、血流等方面的变化,局部施用都在目标部位获得已知浓度。通过直接递送方式还使剂量控制得到改进。
MASP-2抑制剂的局部递送可在用于治疗疾病或病症的外科手术方法的背景中实现,诸如例如手术诸如在动脉旁路术、动脉粥样硬化斑切除术、激光手术、超声波手术、球囊血管成形术以及支架安置期间。例如,可将MASP-2抑制剂与球囊血管成形手术结合起来施用于受试者。球囊血管成形手术包括将连有已排气球囊的导管插入动脉内。将已排气球囊置于动脉粥样硬化斑块附近,给气囊充气使得斑块向血管壁挤压。结果,球囊表面与血管表面的血管内皮细胞层接触。可以按允许药物在动脉粥样硬化斑块部位释放的方式,将MASP-2抑制剂附着到球囊血管成形术导管上。可按照本领域已知标准方法将药物附着在球囊导管上。例如,可将药物保存在球囊导管的隔室中直到球囊充气,此时药物被释放到局部环境中。或者,可将药物浸渍在球囊表面,从而当球囊充气时,药物就接触到动脉壁的细胞。也可在多孔球囊导管中递送药物,参见例如Flugelman, M.Y.,等人, Circulation 85:1110-1117, 1992。另参见已公开的PCT申请WO 95/23161中对于将治疗蛋白附着到球囊血管成形术导管上的示例性方法。同样地,也可将MASP-2抑制剂包入应用于支架上的凝胶或聚合涂层材料中,或者可将MASP-2抑制剂掺入支架材料中,从而支架在血管安置之后便将MASP-2抑制剂洗脱出来。
用于治疗关节炎和其他肌肉骨骼疾病的MASP-2抑制剂组合物可通过关节内注射来局部递送。这些组合物可适当地包括缓释递送媒介物。作为其中可能需要局部递送的另一个实例,用于治疗泌尿生殖系统疾病的MASP-2抑制剂组合物可适当地滴入膀胱内或者其他泌尿生殖结构中。
医疗装置上的涂层
可将MASP-2抑制剂(诸如抗体和抑制肽)固定在可植入或可连接的医疗装置表面(或内部)。经修饰的表面通常可在植入动物体后与活组织接触。所谓“可植入或可连接的医疗装置”是指在正常操作该装置(例如支架和可植入药物递送装置)时,被植入或连接到动物体组织的任何装置。这些可植入或可连接的医疗装置可由例如以下的材料制成:硝酸纤维素、重氮纤维素、玻璃、聚苯乙烯、聚氯乙烯、聚丙烯、聚乙烯、葡聚糖、琼脂糖凝胶(Sepharose)、琼脂、淀粉、尼龙、不锈钢、钛以及生物可降解的和/或生物相容性聚合物。可通过相连接的蛋白质的生物活性不被破坏的任何技术,来使蛋白质与装置连接,例如通过将蛋白质N端和C端残基的一个或两个连接到装置上。也可在蛋白质内一个或多个内部位点上进行连接。还可以使用多重连接(既在蛋白质内部也在蛋白质两端)。可对可植入或可连接的医疗装置的表面进行修饰以包括用于供蛋白质固定的官能团(例如羧基、酰胺、氨基、醚、羟基、氰基、次氮基、氨磺酰基(sulfanamido)、乙炔基(acetylinic)、环氧基、硅烷、酐、琥珀酰亚胺基、叠氮基)。偶合化学法包括但不限于与MASP-2抗体或抑制肽上可利用的官能团形成酯、醚、酰胺、叠氮化物和氨磺酰衍生物、氰酸酯和其他键。也可通过将亲和标记物序列加到蛋白质上,从而使MASP-2抗体或抑制片段非共价连接,这些标记物诸如GST (D.B. Smith和K.S. Johnson, Gene 67:31, 1988)、多聚组氨酸(E. Hochuli等人, J. Chromatog. 411:77, 1987)或生物素。这些亲和标记物也可用于蛋白质与装置的可逆连接。
还可将蛋白质共价连接到装置体表面上,例如通过医疗装置表面的共价活化。举例来说,可通过以下任一对反应基团将基质细胞蛋白质(matricellular protein)连接到装置体上(配对的一个成员存在于装置体表面,配对的另一个成员则存在于基质细胞蛋白质上):羟基/羧酸,产生酯键;羟基/酸酐,产生酯键;羟基/异氰酸酯,产生氨基甲酸乙酯键。可用射频放电等离子体(RFGD)蚀刻,对不具备有用反应基团的装置体表面进行处理来产生反应基团,以便供基质细胞蛋白质沉积(例如,用氧等离子体处理引入含氧基团;用丙基氨基等离子体处理引入氨基)。
可将包括核酸分子(诸如编码反义核酸、RNAi或DNA的肽抑制剂)的MASP-2抑制剂植入连接有装置体的多孔基质中。可用于制造表面层的代表性多孔基质是由腱或皮肤胶原制备的多孔基质,例如可从各种商业来源获得(例如Sigma和Collagen Corporation)的胶原基质,或者按照参考文献所述方法制备的胶原基质(Jefferies的美国专利号4,394,370和Koezuka的美国专利号4,975,527)。一种胶原材料称为UltraFiberTM,可从Norian Corp. (Mountain View,California)获得。
如果需要,也可采用某些聚合基质,包括丙烯酸酯聚合物和乳酸聚合物,例如参见Urist的美国专利号4,526,909和Urist的美国专利号4,563,489。有用聚合物的具体实例为原酸酯、酸酐、丙烯-共延胡索酸酯的聚合物,或者一种或多种α-羟基羧酸单体的聚合物(例如α-羟基乙酸(乙醇酸)和/或α-羟基丙酸(乳酸))。
治疗方案
在预防性应用中,将药物组合物施用于易患MASP-2依赖性补体活化相关疾病或另外有患MASP-2依赖性补体活化相关疾病风险的受试者,施用量足以消除或降低疾病症状发展的风险。在治疗性应用中,以足以缓解或至少部分减少疾病症状的治疗有效量,将药物组合物施用于疑似患有或已经患有MASP-2依赖性补体活化相关疾病的受试者。在预防兼治疗方案中,可以以若干剂量施用包含MASP-2抑制剂的组合物,直到在受试者中获得充分的治疗结果。可通过组合物的单次施用或有限的连续施用,来应用本发明的MASP-2抑制剂组合物以治疗急性疾病,例如再灌注损伤或其他外伤。或者,可在较长一段时间内按定期间隔施用组合物,用于治疗慢性病,例如关节炎或银屑病。
本发明的方法和组合物可用来抑制通常由诊断性和治疗性内科和外科手术引起的炎症和相关过程。为了抑制这些过程,可以在围手术期应用本发明的MASP-2抑制剂组合物。本文所用“围手术期”是指在手术前和/或手术中和/或手术后施用抑制剂组合物,即手术前,手术前和手术中,手术前和手术后,手术前、手术中和手术后,手术中,手术中和手术后,或者手术后。围手术期应用可通过将组合物局部施用于手术或手术前部位来进行,诸如通过注射或者连续或间歇冲洗手术部位,或者通过全身性施用。用于围手术期局部递送MASP-2抑制剂溶液剂的合适方法参见Demopulos的美国专利号6,420,432和Demopulos的美国专利号6,645,168。用于局部递送包括MASP-2抑制剂的软骨保护组合物的合适方法参见国际PCT专利申请WO 01/07067 A2。用于定向全身性递送包括MASP-2抑制剂的软骨保护组合物的合适方法参见国际PCT专利申请WO 03/063799 A2。
在本发明的一个方面,将药物组合物施用于易患PNH或正处于PNH风险中的受试者,施用量足以消除或降低疾病症状发展的风险。在治疗性应用中,以足以缓解或至少部分减少疾病症状的治疗有效量,将药物组合物施用于疑似患有或已经患有PNH的受试者。
在一个实施方案中,受试者的红细胞在组合物不存在的情况下被C3的片段所调理,并且将包含MASP-2抑制剂的组合物施用于受试者增加了红细胞在受试者中的存活。在一个实施方案中,受试者在组合物不存在的情况下表现出选自以下的一种或多种症状:(i) 低于正常水平的血红蛋白,(ii) 低于正常水平的血小板;(iii)高于正常水平的网织红细胞,和(iv)高于正常水平的胆红素,并且将该组合物施用于受试者改善了至少一种或多种症状,导致(i)增加、正常或接近正常水平的血红蛋白、(ii) 增加、正常或接近正常水平的血小板,(iii)降低、正常或接近正常水平的网织红细胞,和/或(iv) 降低、正常或接近正常水平的胆红素。
在预防兼治疗方案中,可以以若干剂量施用包含MASP-2抑制剂的组合物,直到在受试者中获得充分的治疗结果。在本发明的一个实施方案中,MASP-2抑制剂包含抗MASP-2抗体,其可以合适地以以下剂量施用于成年患者(例如,70 kg的平均成人体重):0.1 mg至10,000 mg、更合适地1.0 mg至5,000 mg、更合适地10.0 mg至2,000 mg、更合适地10.0 mg至1,000 mg和仍更合适地50.0 mg至500 mg。对于儿童患者,剂量可以与患者重量按比例调整。可通过组合物的单次施用或有限的连续施用,来应用本发明的MASP-2抑制剂组合物以治疗PNH。或者,可在延长的时间期间以定期时间间隔(诸如每天、每两周、每周、每隔一周、每月或每两月)施用组合物,用于治疗PNH。
在一些实施方案中,患有PNH的受试者先前已经经历或正在经历用抑制补体蛋白C5的裂解的末端补体抑制剂的治疗。在一些实施方案中,该方法包括将包含MASP-2抑制剂的本发明的组合物施用于受试者,和进一步将抑制补体蛋白C5的裂解的末端补体抑制剂施用于受试者。在一些实施方案中,末端补体抑制剂是人源化抗C5抗体或其抗原结合片段。在一些实施方案中,末端补体抑制剂是依库丽单抗。
在本发明的一个方面,将药物组合物施用于易患aHUS或有患aHUS风险的受试者,施用量足以消除或降低疾病症状发展的风险。在治疗性应用中,以足以缓解或至少部分减少疾病症状的治疗有效量,将药物组合物施用于疑似患有或已经患有aHUS的受试者。在本发明的一个方面,在施用之前,可以检查受试者以确定受试者是否表现出aHUS的一种或多种症状,包括(i)贫血、(ii)血小板减少(iii)肾功能不全和(iv)肌酸酐上升,然后将本发明的组合物以有效量施用足够长的时间期间,以改善这些症状。
在本发明的另一个方面,本发明的MASP-2抑制组合物可用于预防性治疗具有发展aHUS的升高风险的受试者,并且从而降低受试者将发生aHUS的可能性。首先通过对从受试者获得的样品上进行基因筛选测试并且鉴定与aHUS相关的至少一种基因标志物(补体因子H(CFH)、因子I(CFI)、因子B (CFB)、膜辅因子CD46、C3、补体因子H-相关蛋白1(CFHR1)、抗凝蛋白血栓调节蛋白(THBD)、补体因子H-相关蛋白3(CFHR3)或补体因子H-相关蛋白4(CFHR4))的存在而确定已知与aHUS相关的受试者中的基因标志物的存在。然后定期(例如,每个月、每季度、每年两次或每年一次)监测受试者,以确定是否存在aHUS的至少一种症状,诸如贫血、血小板减少、肾功能不全和肌酸酐上升。在确定这些症状中的至少一种的存在之后,将有效抑制MASP-2依赖性补体活化的量的MASP-2抑制剂以有效量施用于受试者足够时间期间,以改善所述一种或多种症状。在本发明的又进一步方面,可以监测由于已被筛选和确定具有与aHUS相关的基因标志物之一而处在发生aHUS的风险中的受试者的与触发aHUS临床症状相关的事件(包括药物暴露、感染(例如细菌感染)、恶性肿瘤、损伤、器官或组织移植和怀孕)的发生。
在本发明的又进一步方面,可以将包含有效抑制MASP-2依赖性补体活化的量的MASP-2抑制剂的组合物施用于患有继发于感染的非典型溶血性尿毒综合征(aHUS)或处于发生继发于感染的非典型溶血性尿毒综合征(aHUS)的风险中的受试者。例如,患有肺炎链球菌感染相关的非肠道aHUS或处于发生与肺炎链球菌感染相关的非肠道aHUS的风险中的患者可以用本发明的组合物进行治疗。
在本发明的又进一步方面,患有aHUS的受试者最初可以用本发明的MASP-2抑制组合物治疗,所述本发明的MASP-2抑制组合物通过导管线(诸如静脉内导管线或皮下导管线)施用第一时间期间,诸如一小时、十二小时、一天、两天或三天。然后受试者可以用通过定期皮下注射(诸如每天、每两周、每周、每隔一周、每月或每两月注射)施用的MASP-2抑制组合物治疗第二时间期间。
在本发明的又进一步方面,为了避免血浆置换的潜在并发症包括出血、感染和暴露于血浆供体固有的病症和/或过敏或在另外反对血浆置换的受试者中或在其中血浆置换无法获得的设置中,可以在血浆置换不存在的情况下将本发明的MASP-2抑制组合物施用于患有aHUS的受试者(即,其aHUS症状在用MASP-2抑制组合物治疗时过去没有用血浆置换治疗并且现在没有用血浆置换治疗的受试者)。
在本发明的又进一步方面,可以将本发明的MASP-2抑制组合物施用于同时用血浆置换治疗患者的患有aHUS的受试者。例如,然后可以在血浆置换之后或与血浆置换交替将MASP-2抑制组合物施用于接受血浆置换治疗的受试者。
在本发明的又进一步方面,可以通过定期(诸如每十二小时或基于每天)确定至少一种补体因子的水平而监测患有aHUS或处于发生aHUS的风险中并且用本发明的MASP-2抑制组合物治疗的受试者,其中确定与标准值或健康受试者相比至少一种补体因子的水平降低表明需要继续用该组合物治疗。
在预防性和治疗方案两者中,可以以几个剂量施用包含MASP-2抑制剂的组合物,直到已经在受试者中实现足够的治疗结果。在本发明的一个实施方案中,MASP-2抑制剂包含抗MASP-2抗体,其可以合适地以以下剂量施用于成年患者(例如,70 kg的平均成人体重):0.1 mg至10,000 mg、更合适地1.0 mg至5,000 mg、更合适地10.0 mg至2,000 mg、更合适地10.0 mg至1,000 mg和仍更合适地50.0 mg至500 mg。对于儿童患者,剂量可以与患者重量按比例调整。可通过组合物的单次施用或有限的连续施用,来应用本发明的MASP-2抑制剂组合物以治疗aHUS。或者,可在较长一段时间内按定期间隔(诸如每天、每两周、每周、每隔一周、每月或每两月)施用组合物,用于治疗aHUS。
在一些实施方案中,患有aHUS的受试者先前已经经历或正在经历用抑制补体蛋白C5的裂解的末端补体抑制剂的治疗。在一些实施方案中,该方法包括将包含MASP-2抑制剂的本发明的组合物施用于受试者,和进一步将抑制补体蛋白C5的裂解的末端补体抑制剂施用于受试者。在一些实施方案中,末端补体抑制剂是人源化抗C5抗体或其抗原结合片段。在一些实施方案中,末端补体抑制剂是依库丽单抗。
在本发明的一个方面,以足以消除或降低发展状况的症状的风险的量将药物组合物施用于易患HUS或以其他方式处于HUS的风险中的受试者。在治疗应用中,将药物组合物以足以减轻或至少部分减少状况的症状的治疗有效量施用于怀疑患有或已经患有HUS的受试者。
在本发明的另一个方面,可以通过以有效抑制MASP-2依赖性补体活化的量将本发明的MASP-2抑制组合物施用于受试者而降低处于发生HUS的风险中的受试者中发生肾功能受损的可能性。例如,处于发展HUS的风险中并且待用本发明的MASP-2抑制组合物治疗的受试者可以表现出与HUS相关的一种或多种症状,包括腹泻、具有血管内红细胞破坏的涂片证据的小于30%的血细胞比容水平、血小板减少和上升的肌酸酐水平。作为进一步实例,处于发展HUS的风险中并且待用本发明的MASP-2抑制组合物治疗的受试者可能感染大肠杆菌、志贺氏菌或沙门氏菌。此类被大肠杆菌、志贺氏菌或沙门氏菌感染的受试者可以用本发明的MASP-2抑制组合物与抗生素治疗同时治疗,或者可以用MASP-2抑制组合物治疗而不同时用抗生素(尤其是对于其中抗生素治疗是禁忌的肠源性大肠杆菌的抗生素)治疗。被已经用抗生素治疗过的肠源性大肠杆菌感染的受试者可能处在发生HUS的高风险中,并且可以用本发明的MASP-2抑制组合物合适地治疗以降低该风险。被肠源性大肠杆菌感染的受试者可以在抗生素不存在的情况下用本发明的MASP-2抑制组合物治疗第一时间期间,并且然后用本发明的MASP-2抑制组合物和抗生素两者治疗第二时间期间。
在本发明的又进一步方面,患有HUS的受试者最初可以用本发明的MASP-2抑制组合物治疗,所述本发明的MASP-2抑制组合物通过导管线(诸如静脉内导管线或皮下导管线)施用第一时间期间,诸如一小时、十二小时、一天、两天或三天。然后受试者可以用通过定期皮下注射(诸如每天、每两周、每周、每隔一周、每月或每两月注射)施用的MASP-2抑制组合物治疗第二时间期间。
在本发明的又进一步方面,为了避免血浆置换的潜在并发症包括出血、感染和暴露于血浆供体固有的病症和/或过敏或在另外反对血浆置换的受试者中或在其中血浆置换无法获得的设置中,可以在血浆置换不存在的情况下将本发明的MASP-2抑制组合物施用于患有HUS的受试者(即,其HUS症状在用MASP-2抑制组合物治疗时过去没有用血浆置换治疗并且现在没有用血浆置换治疗的受试者)。
在本发明的又进一步方面,可以将本发明的MASP-2抑制组合物施用于同时用血浆置换治疗患者的患有HUS的受试者。例如,然后可以在血浆置换之后或与血浆置换交替将MASP-2抑制组合物施用于接受血浆置换治疗的受试者。
在本发明的又进一步方面,可以通过定期(诸如每十二小时或基于每天)确定至少一种补体因子的水平而监测患有HUS或处于发生HUS的风险中并且用本发明的MASP-2抑制组合物治疗的受试者,其中确定与标准值或健康受试者相比至少一种补体因子的水平降低表明需要继续用该组合物治疗。
在预防性和治疗方案两者中,可以以几个剂量施用包含MASP-2抑制剂的组合物,直到已经在受试者中实现足够的治疗结果。在本发明的一个实施方案中,MASP-2抑制剂包含抗MASP-2抗体,其可以合适地以以下剂量施用于成年患者(例如,70 kg的平均成人体重):0.1 mg至10,000 mg、更合适地1.0 mg至5,000 mg、更合适地10.0 mg至2,000 mg、更合适地10.0 mg至1,000 mg和仍更合适地50.0 mg至500 mg。对于儿童患者,剂量可以与患者重量按比例调整。本发明的MASP-2抑制组合物的应用可以通过单一施用该组合物或受限顺序的施用而实施,用于治疗HUS。或者,可在延长的时间期间以定期时间间隔(诸如每天、每两周、每周、每隔一周、每月或每两月)施用组合物,用于治疗HUS。
在一些实施方案中,患有HUS的受试者先前已经经历或正在经历用抑制补体蛋白C5的裂解的末端补体抑制剂的治疗。在一些实施方案中,该方法包括将包含MASP-2抑制剂的本发明的组合物施用于受试者,和进一步将抑制补体蛋白C5的裂解的末端补体抑制剂施用于受试者。在一些实施方案中,末端补体抑制剂是人源化抗C5抗体或其抗原结合片段。在一些实施方案中,末端补体抑制剂是依库丽单抗。
在本发明的一个方面,以足以消除或降低发展状况的症状的风险的量将药物组合物施用于易患TTP或以其他方式处于TTP的风险中的受试者。在治疗应用中,将药物组合物以足以减轻或至少部分减少状况的症状的治疗有效量施用于怀疑患有或已经患有TTP的受试者。
在本发明的另一个方面,表现出TTP的一种或多种症状(包括中枢神经系统损害、血小板减少、严重心脏损害、严重肺损害、胃肠梗死和坏疽)的受试者可以用本发明的MASP-2抑制组合物治疗。在本发明的另一个方面,被确定具有抑制水平的ADAMTS13并且还对于ADAMTS13的抑制剂(即,抗体)的存在检测阳性的受试者可以用本发明的MASP-2抑制组合物治疗。在本发明的又进一步方面,对于ADAMTS13的抑制剂的存在检测阳性的受试者可以用免疫抑制剂(例如皮质类固醇、利妥昔单抗或环孢霉素)与本发明的MASP-2抑制组合物同时治疗。在本发明的又进一步方面,被确定具有降低水平的ADAMTS13并且还对于ADAMTS13的抑制剂的存在检测阳性的受试者可以用ADAMTS13与本发明的MASP-2抑制组合物治疗同时治疗。
在本发明的又进一步方面,患有TTP的受试者最初可以用本发明的MASP-2抑制组合物治疗,所述本发明的MASP-2抑制组合物通过导管线(诸如静脉内导管线或皮下导管线)施用第一时间期间,诸如一小时、十二小时、一天、两天或三天。然后受试者可以用通过定期皮下注射(诸如每天、每两周、每周、每隔一周、每月或每两月注射)施用的MASP-2抑制组合物治疗第二时间期间。
在本发明的又进一步方面,为了避免血浆置换的潜在并发症包括出血、感染和暴露于血浆供体固有的病症和/或过敏或在另外反对血浆置换的受试者中或在其中血浆置换无法获得的设置中,可以在血浆置换不存在的情况下将本发明的MASP-2抑制组合物施用于患有TTP的受试者(即,其TTP症状在用MASP-2抑制组合物治疗时过去没有用血浆置换治疗并且现在没有用血浆置换治疗的受试者)。
在本发明的又进一步方面,可以将本发明的MASP-2抑制组合物施用于同时用血浆置换治疗的患有TTP的受试者。例如,然后可以在血浆置换之后或与血浆置换交替将MASP-2抑制组合物施用于接受血浆置换治疗的受试者。
在本发明的又进一步方面,患有难治性TTP(即对其他治疗诸如血浆置换未有足够响应的TTP症状)的受试者可以在有或没有额外的血浆置换的情况下用本发明的MASP-2抑制组合物治疗。
在本发明的又进一步方面,可以通过定期(诸如每十二小时或基于每天)确定至少一种补体因子的水平而监测患有TTP或处于发生TTP的风险中并且用本发明的MASP-2抑制组合物治疗的受试者,其中确定与标准值或健康受试者相比至少一种补体因子的水平降低表明需要继续用该组合物治疗。
在预防性和治疗方案两者中,可以以几个剂量施用包含MASP-2抑制剂的组合物,直到已经在受试者中实现足够的治疗结果。在本发明的一个实施方案中,MASP-2抑制剂包含抗MASP-2抗体,其可以合适地以以下剂量施用于成年患者(例如,70 kg的平均成人体重):0.1 mg至10,000 mg、更合适地1.0 mg至5,000 mg、更合适地10.0 mg至2,000 mg、更合适地10.0 mg至1,000 mg和仍更合适地50.0 mg至500 mg。对于儿童患者,剂量可以与患者重量按比例调整。本发明的MASP-2抑制组合物的应用可以通过单一施用该组合物或受限顺序的施用而实施,用于治疗TTP。或者,可在延长的时间期间以定期时间间隔(诸如每天、每两周、每周、每隔一周、每月或每两月)施用组合物,用于治疗TTP。
在一些实施方案中,患有TTP的受试者先前已经经历或正在经历用抑制补体蛋白C5的裂解的末端补体抑制剂的治疗。在一些实施方案中,该方法包括将包含MASP-2抑制剂的本发明的组合物施用于受试者,和进一步将抑制补体蛋白C5的裂解的末端补体抑制剂施用于受试者。在一些实施方案中,末端补体抑制剂是人源化抗C5抗体或其抗原结合片段。在一些实施方案中,末端补体抑制剂是依库丽单抗。
在本发明的一个方面,以足以消除或降低发展状况的症状的风险的量将药物组合物施用于易患冷凝集素病或冷球蛋白血症或以其他方式处于冷凝集素病或冷球蛋白血症的风险中的受试者。在治疗应用中,将药物组合物以足以减轻或至少部分减少状况的症状的治疗有效量施用于怀疑患有或已经患有冷凝集素病或冷球蛋白血症的受试者。
在本发明的又进一步方面,可以通过定期(诸如每十二小时或基于每天)确定至少一种补体因子的水平而监测患有冷凝集素病或冷球蛋白血症或处于发生冷凝集素病或冷球蛋白血症的风险中并且用本发明的MASP-2抑制组合物治疗的受试者,其中确定与标准值或健康受试者相比至少一种补体因子的水平降低表明需要继续用该组合物治疗。
在预防性和治疗方案两者中,可以以几个剂量施用包含MASP-2抑制剂的组合物,直到已经在受试者中实现足够的治疗结果。在本发明的一个实施方案中,MASP-2抑制剂包含抗MASP-2抗体,其可以合适地以以下剂量施用于成年患者(例如,70 kg的平均成人体重):0.1 mg至10,000 mg、更合适地1.0 mg至5,000 mg、更合适地10.0 mg至2,000 mg、更合适地10.0 mg至1,000 mg和仍更合适地50.0 mg至500 mg。对于儿童患者,剂量可以与患者重量按比例调整。本发明的MASP-2抑制组合物的应用可以通过单一施用该组合物或受限顺序的施用而实施,用于治疗冷凝集素病或冷球蛋白血症。或者,可在延长的时间期间以定期时间间隔(诸如每天、每两周、每周、每隔一周、每月或每两月)施用组合物,用于治疗冷凝集素病或冷球蛋白血症。
在一些实施方案中,患有冷凝集素病或冷球蛋白血症的受试者先前已经经历或正在经历用抑制补体蛋白C5的裂解的末端补体抑制剂的治疗。在一些实施方案中,该方法包括将包含MASP-2抑制剂的本发明的组合物施用于受试者,和进一步将抑制补体蛋白C5的裂解的末端补体抑制剂施用于受试者。在一些实施方案中,末端补体抑制剂是人源化抗C5抗体或其抗原结合片段。在一些实施方案中,末端补体抑制剂是依库丽单抗。
在本发明的一个方面,以足以消除或降低发展状况的症状的风险的量将药物组合物施用于易患青光眼或以其他方式处于青光眼的风险中的受试者。在治疗应用中,将药物组合物以足以减轻或至少部分减少状况的症状的治疗有效量施用于怀疑患有或已经患有青光眼的受试者。
在本发明的又进一步方面,可以通过定期(诸如每十二小时或基于每天)确定至少一种补体因子的水平而监测患有青光眼或处于发生青光眼的风险中并且用本发明的MASP-2抑制组合物治疗的受试者,其中确定与标准值或健康受试者相比至少一种补体因子的水平降低表明需要继续用该组合物治疗。
在预防性和治疗方案两者中,可以以几个剂量施用包含MASP-2抑制剂的组合物,直到已经在受试者中实现足够的治疗结果。在本发明的一个实施方案中,MASP-2抑制剂包含抗MASP-2抗体,其可以合适地以以下剂量施用于成年患者(例如,70 kg的平均成人体重):0.1 mg至10,000 mg、更合适地1.0 mg至5,000 mg、更合适地10.0 mg至2,000 mg、更合适地10.0 mg至1,000 mg和仍更合适地50.0 mg至500 mg。对于儿童患者,剂量可以与患者重量按比例调整。本发明的MASP-2抑制组合物的应用可以通过单一施用该组合物或受限顺序的施用而实施,用于治疗青光眼。或者,可在延长的时间期间以定期时间间隔(诸如每天、每两周、每周、每隔一周、每月或每两月)施用组合物,用于治疗冷凝集素病。
在一些实施方案中,患有青光眼的受试者先前已经经历或正在经历用抑制补体蛋白C5的裂解的末端补体抑制剂的治疗。在一些实施方案中,该方法包括将包含MASP-2抑制剂的本发明的组合物施用于受试者,和进一步将抑制补体蛋白C5的裂解的末端补体抑制剂施用于受试者。在一些实施方案中,末端补体抑制剂是人源化抗C5抗体或其抗原结合片段。在一些实施方案中,末端补体抑制剂是依库丽单抗。
在本发明的一个方面,以足以消除或降低发展状况的症状的风险的量将药物组合物施用于患有急性放射综合征或处于发生急性放射综合征的风险中的受试者。在治疗应用中,将药物组合物以足以减轻或至少部分减少状况的症状的治疗有效量施用于怀疑患有或已经患有急性放射综合征的受试者。受试者可以在暴露于放射(诸如为了治疗癌性状况而放射暴露)之前或之后用本发明的MASP-2抑制组合物治疗,同时清理在能量生产工厂或实验室用放射性材料工作时或由于核事故、恐怖活动或战争导致的放射暴露的放射污染的部位。在本发明的一个实施方案中,在放射暴露之后24至48小时内施用MASP-2抑制组合物。
在本发明的又进一步方面,可以通过定期(诸如每十二小时或基于每天)确定至少一种补体因子的水平而监测患有急性放射综合征或处于发生急性放射综合征的风险中并且用本发明的MASP-2抑制组合物治疗的受试者,其中确定与标准值或健康受试者相比至少一种补体因子的水平降低表明需要继续用该组合物治疗。
在预防性和治疗方案两者中,可以以几个剂量施用包含MASP-2抑制剂的组合物,直到已经在受试者中实现足够的治疗结果。在本发明的一个实施方案中,MASP-2抑制剂包含抗MASP-2抗体,其可以合适地以以下剂量施用于成年患者(例如,70 kg的平均成人体重):0.1 mg至10,000 mg、更合适地1.0 mg至5,000 mg、更合适地10.0 mg至2,000 mg、更合适地10.0 mg至1,000 mg和仍更合适地50.0 mg至500 mg。对于儿童患者,剂量可以与患者重量按比例调整。本发明的MASP-2抑制组合物的应用可以通过单一施用该组合物或受限顺序的施用而实施,用于治疗青光眼。或者,可在延长的时间期间以定期时间间隔(诸如每天、每两周、每周、每隔一周、每月或每两月)施用组合物,用于治疗急性放射综合征。
在一些实施方案中,患有急性放射综合征的受试者先前已经经历或正在经历用抑制补体蛋白C5的裂解的末端补体抑制剂的治疗。在一些实施方案中,该方法包括将包含MASP-2抑制剂的本发明的组合物施用于受试者,和进一步将抑制补体蛋白C5的裂解的末端补体抑制剂施用于受试者。在一些实施方案中,末端补体抑制剂是人源化抗C5抗体或其抗原结合片段。在一些实施方案中,末端补体抑制剂是依库丽单抗。
VI. 实施例
下面的实施例仅举例说明目前预期的用于实施本发明的最佳方式,但是不应解释为限制本发明。本文所引用的所有文献都通过引用并入本文。
实施例1
本实施例描述了缺乏MASP-2 (MASP-2-/-)但MAp19充足(MAp19+/+)的小鼠品系的产生。
材料与方法:设计靶向载体pKO-NTKV 1901来破坏编码鼠MASP-2 C端的三个外显子,包括编码丝氨酸蛋白酶结构域的外显子,见图3。使用PKO-NTKV 1901转染鼠ES细胞系E14.1a (SV129 Ola)。选出新霉素抗性和胸苷激酶敏感的克隆。筛选出600个ES克隆,在其中鉴定出4个不同克隆,通过Southern印迹证实含有预期选择的靶向事件(targeting event)和重组事件,见图3。通过胚胎转移由这4个阳性克隆产生嵌合体。然后将嵌合体在遗传背景C57/BL6下回交以产生转基因雄性鼠。将转基因雄性鼠与雌性鼠杂交产生F1,50%的后代显示出MASP-2基因被破坏的杂合性。将杂合小鼠互交以产生纯合的MASP-2缺陷型后代,按1:2:1的比例分别产生杂合小鼠和野生型小鼠。
结果和表型:发现所产生的纯合MASP-2-/-缺陷型小鼠是可存活和能育的,通过Southern印迹证实缺乏MASP-2,从而确认了正确的靶向事件,经Northern印迹证实缺乏MASP-2 mRNA,经Western印迹证实缺乏MASP-2蛋白(数据未显示)。使用时间分辨RT-PCR在LightCycler机上进一步证实了存在MAp19 mRNA,缺乏MASP-2 mRNA。MASP-2-/-小鼠确实如预期那样继续表达MAp19、MASP-1和MASP-3 mRNA及蛋白质(数据未显示)。通过LightCycler分析对MASP-2-/-小鼠中备解素、因子B、因子D、C4、C2和C3的mRNA的存在和丰度进行了评价,发现与野生型同窝小鼠对照中的相同(数据未显示)。来自纯合MASP-2-/-小鼠的血浆完全缺乏凝集素途径介导的补体活化,如实施例2进一步描述的。
在纯的C57BL6背景下产生MASP-2-/-品系:将MASP-2-/-小鼠与纯C57BL6品系回交9代后,将MASP-2-/-品系用作实验动物模型。
作为鼠MASP-2-/-、MAp19+/+且表达人MASP-2转基因(鼠MASP-2敲除和人MASP-2敲入)的转基因小鼠品系也生成如下:
材料与方法:构建包括人MASP 2基因启动子区并编码人MASP-2的小基因(SEQ ID NO:49),称为“mini hMASP-2”,见图4,它包括前3个外显子(外显子1至外显子3),接着是代表随后8个外显子的编码序列的cDNA序列,从而编码由其内源启动子驱动的全长MASP-2蛋白。将mini hMASP-2构建体注射到MASP-2-/-的受精卵中,以便通过转基因表达人MASP-2来代替缺陷的鼠MASP 2基因。
实施例2
本实施例证实MASP-2是经由凝集素途径进行补体活化所必需的。
方法与材料:
凝集素途径特异性C4裂解测定法:C4裂解测定法记载于Petersen,等人, J. Immunol. Methods 257:107 (2001),该测定法测定了由来自金黄色葡萄球菌、结合L-纤维胶凝蛋白的脂磷壁酸(LTA)所产生的凝集素途径活化。对Petersen等人, (2001)中所述测定法进行了改进,通过用LPS和甘露聚糖或酵母聚糖包被板后,加入得自MASP-2 -/-小鼠的血清,来测定由MBL引起的凝集素途径活化,如下文所述。还对该测定法进行了改进以消除由经典途径所致的C4裂解的可能性。这通过使用含有1 M NaCl的样品稀释缓冲液来实现,这使得凝集素途径识别组分以高亲和力结合它们的配体,却又阻止了内源C4的活化,从而通过使C1复合物解离而排除了经典途径的参与。简单地说,在改进的测定法中,将血清样品(稀释于高盐(1 M NaCl)缓冲液中)加入到配体包被板上,接着加入缓冲液(具有生理浓度的盐)中的恒量纯C4。含有MASP-2的结合识别复合物裂解C4而引起C4b沉积。
测定方法:
1) 用稀释于包被缓冲液(15 mM Na2CO3,35 mM NaHCO3,pH 9.6)的1 μg/ml甘露聚糖(M7504 Sigma)或任何其他配体(例如诸如下列配体),来包被Nunc Maxisorb微量滴定板(Maxisorb,Nunc, 目录号442404,Fisher Scientific)。
下列试剂用于本测定法中:
a. 甘露聚糖(1 μg/孔甘露聚糖(M7504 Sigma),于100 μl包被缓冲液中);
b. 酵母聚糖(1 μg/孔酵母聚糖(Sigma),于100 μl包被缓冲液中);
c. LTA (1μg/孔,于100 μl包被缓冲液中,或者2 μg/孔,于20 μl甲醇中);
d. 1 μg H-纤维胶凝蛋白特异性Mab 4H5,于包被缓冲液中;
e. 来自浅绿气球菌(Aerococcus viridans)的PSA (2 μg/孔,于100 μl包被缓冲液);
f. 100 μl/孔福尔马林固定的金黄色葡萄球菌DSM20233 (OD550=0.5),于包被缓冲液中。
2) 将板在4℃下孵育过夜。
3) 过夜孵育后,通过将板与0.1% HSA-TBS封闭缓冲液(0.1% (重量/体积) HSA的10 mM Tris-HCl、140 mM NaCl、1.5 mM NaN3溶液(pH 7.4))孵育1-3小时后,然后用TBS/吐温(tween)/Ca2+(含0.05%吐温20和5 mM CaCl2、1 mM MgCl2的TBS (pH 7.4))洗板3次,从而使剩余蛋白结合位点饱和。
4) 将待测试的血清样品稀释于MBL结合缓冲液(1 M NaCl)中,将稀释的样品加到板上,4℃下孵育过夜。只加缓冲液的孔用作阴性对照。
5) 在4℃下孵育过夜之后,将该板用TBS/吐温/Ca2+洗涤3次。然后将人C4 (100 μl/孔,1 μg/ml稀释于BBS (4 mM巴比妥,145 mM NaCl,2 mM CaCl2,1 mM MgCl2,pH 7.4)中)加到板上,在37℃下孵育90分钟。该板用TBS/吐温/Ca2+再洗涤3次。
6) 用碱性磷酸酶缀合的鸡抗人C4c (以1:1000稀释于TBS/吐温/Ca2+中)来检测C4b沉积,将其加到板上,在室温下孵育90分钟。然后该板用TBS/吐温/Ca2+洗涤3次。
7) 通过加入100 μl磷酸对硝基苯酯底物溶液来检测碱性磷酸酶,在室温下孵育20分钟,在微量滴定板读板仪上读取OD405。
结果:图5A-B显示来自MASP-2+/+ (交叉线)、MASP-2+/- (实心圆)和MASP-2-/- (实心三角)的血清稀释液中在甘露聚糖(图5A)和酵母聚糖(图5B)上C4b沉积的量。图5C显示来自MASP-2-/+小鼠(n=5)和MASP-2-/-小鼠(n=4)的酵母聚糖(白色条形柱)或甘露聚糖(阴影条形柱)包被板上相对于野生型小鼠(n=5)的C4转化酶相对活性,所根据的是测定针对野生型血清均一化的C4b沉积的量。误差条代表标准偏差。如图5A-C所示,来自MASP-2-/-小鼠的血浆在甘露聚糖和酵母聚糖包被板上完全缺乏凝集素途径介导的补体活化。这些结果清楚地表明,MASP-2是凝集素途径的效应子成分。
重组MASP-2重构MASP-2-/-小鼠血清中的凝集素途径依赖性C4活化
为了证实MASP-2缺乏是MASP-2-/-小鼠凝集素途径依赖性C4活化丧失的直接原因,在上述C4裂解测定中测试了将重组MASP-2蛋白加到血清样品中的作用。按照以下实施例3中所述方法,制备了有功能活性的鼠MASP-2和无催化活性的鼠MASP-2A (其中丝氨酸蛋白酶结构域活性部位上的丝氨酸残基被丙氨酸残基取代)重组蛋白并进行了纯化。将得自4只MASP-2 -/-小鼠的合并血清与蛋白质浓度递增的重组鼠MASP-2或无活性的重组鼠MASP-2A一起预孵育,按上述方法测定C4转化酶活性。
结果:如图6所示,将有功能活性鼠重组MASP-2蛋白(用空心三角表示)加到获自MASP-2 -/-小鼠的血清中,以蛋白质浓度依赖方式恢复了凝集素途径依赖性C4活化,而无催化活性的鼠MASP-2A蛋白(用星号表示)没有恢复C4活化。将图6所示结果针对用合并的野生型小鼠血清所观察到的C4活化结果均一化(用点线表示)。
实施例3
本实施例描述了重组全长人、大鼠和小鼠MASP-2、MASP-2衍生多肽以及无催化活性的MASP-2突变形式的重组表达和蛋白质生产。
全长人、小鼠和大鼠MASP-2的表达:
将人MASP-2全长cDNA序列(SEQ ID NO:4)同样亚克隆到哺乳动物表达载体pCI-Neo (Promega)中,在CMV增强子/启动子区控制下驱动真核表达(描述于 Kaufman R.J.等人, Nucleic Acids Research19:4485-90, 1991; Kaufman, Methods in Enzymology, 185:537-66 (1991))。将全长小鼠cDNA (SEQ ID NO:50)和大鼠MASP-2 cDNA (SEQ ID NO:53)分别亚克隆到pED表达载体中。然后应用标准磷酸钙转染方法(描述于Maniatis等人,1989),将MASP-2表达载体转染到贴壁的中国仓鼠卵巢细胞系DXB1中。用这些构建体转染的细胞生长得非常缓慢,表明所编码的蛋白酶有细胞毒性。
在另一种方法中,将含有由其内源启动子驱动的人MASP-2 cDNA的小基因构建体(SEQ ID NO:49)瞬时转染到中国仓鼠卵巢细胞(CHO)中。人MASP-2蛋白被分泌到培养基中,如下述进行分离。
全长无催化活性的MASP-2的表达:
基本原理:在识别亚成分MBL或纤维胶凝蛋白(L-纤维胶凝蛋白、H-纤维胶凝蛋白或M-纤维胶凝蛋白)结合它们各自的碳水化合物模式之后,MASP-2通过自催化裂解激活。导致MASP-2活化的自催化裂解经常发生在从血清分离MASP-2的过程中,或者在重组表达后的纯化期间。为获得更稳定的蛋白质制备物用作抗原,通过用丙氨酸残基置换蛋白酶结构域催化三联体中存在的丝氨酸残基,产生无催化活性形式的MASP-2,称为MASP-2A,在大鼠中(SEQ ID NO:55 Ser617变成Ala617);在小鼠中(SEQ ID NO:52 Ser617 变成Ala617);或在人中(SEQ ID NO:3 Ser618变成Ala618)。
为产生无催化活性的人和鼠MASP-2A蛋白,使用表5所示寡核苷酸进行定点诱变。为了将丝氨酸密码子变成丙氨酸密码子,设计了表5中的寡核苷酸使编码有酶活性丝氨酸的人和鼠cDNA区退火,寡核苷酸含有错配。例如,采用PCR寡核苷酸SEQ ID NO:56-59结合人MASP-2 cDNA (SEQ ID NO:4),以便扩增自起始密码子到有酶活性丝氨酸的区域及自丝氨酸到终止密码子的区域,从而产生完整开放可读形式的含有Ser618到Ala618突变的突变型MASP-2A。PCR产物在琼脂糖凝胶电泳和条带制备后被纯化,使用标准加尾方法产生单腺苷重叠。然后将加腺苷尾的MASP-2A克隆到pGEM-T easy载体中,再转化到大肠杆菌中。
通过将SEQ ID NO:64和SEQ ID NO:65两种寡核苷酸激酶化(kinasing),并且通过将这两种寡核苷酸以等摩尔量结合,在100℃下加热2分钟后,缓慢冷却到室温而退火,来产生无催化活性的大鼠MASP-2A蛋白。所得到的退火片段具有Pst1和Xba1相容末端,将该片段插入以替换野生型大鼠MASP-2 cDNA (SEQ ID NO:53)的Pst1-Xba1片段从而产生大鼠MASP-2A。

按下述方法,进一步将人、小鼠和大鼠MASP-2A分别亚克隆到哺乳动物表达载体pED或pCI-Neo并转染到中国仓鼠卵巢细胞系DXB1中。
在另一种方法中,应用Chen等人所述方法构建无催化活性形式的MASP-2 (Chen等人, J. Biol. Chem., 276(28):25894-25902, 2001)。简单来讲,将含有全长人MASP-2 cDNA的质粒(描述于Thiel等人, Nature 386:506, 1997)用Xho1和EcoR1消化,将MASP-2 cDNA (参见本文的SEQ ID NO:4)克隆到pFastBac1杆状病毒转移载体(Life Technologies, NY)相应的限制位点中。然后通过用天然区域氨基酸610-625取代编码肽区域氨基酸610-625 (SEQ ID NO:13)的双链寡核苷酸,将MASP-2丝氨酸蛋白酶活性位点Ser618变成Ala618,从而产生带有无活性蛋白酶结构域的MASP-2全长多肽。含有得自人Masp-2的多肽区的表达质粒的构建。
采用MASP-2信号肽(SEQ ID NO:5的残基1-15)来产生下面的构建体以分泌不同的MASP-2结构域。通过PCR扩增编码MASP-2 (SEQ ID NO:6)残基1-121的区域(相当于N端CUB1结构域)来制备表达人MASP-2 CUBI结构域(SEQ ID NO:8)的构建体。通过PCR扩增编码MASP-2 (SEQ ID NO:6)残基1-166的区域(相当于N端CUB1EGF结构域)来制备表达人MASP-2 CUBIEGF结构域(SEQ ID NO:9)的构建体。通过PCR扩增编码MASP-2 (SEQ ID NO:6)残基1-293的区域(相当于N端CUBIEGFCUBII结构域)来制备表达人MASP-2 CUBIEGFCUBII结构域(SEQ ID NO:10)的构建体。采用VentR聚合酶,pBS-MASP-2作为模板,根据已确立的PCR方法,经PCR扩增上述结构域。有义引物(5'-CGGGATCCATGAGGCTGCTGACCCTC-3' SEQ ID NO:34)的5’引物序列在PCR产物的5’端引入BamHI限制位点(下划线)。下表5中所示的各MASP-2结构域的反义引物被设计用来在各种PCR产物末端的EcoRI位点(下划线)之前引入终止密码子(粗体)。DNA片段一旦被扩增,便用BamHI和EcoRI消化,并克隆到pFastBac1载体相应的位点中。所得构建体用限制酶切作图表征,并通过dsDNA测序来证实。
表5:MASP-2 PCR引物


MASP-2的重组真核表达与无酶活性的小鼠、大鼠和人MASP-2A的蛋白质生产
应用标准磷酸钙转染方法(Maniatis等人,1989),将上述MASP-2和MASP-2A表达构建体转染到DXB1细胞中。在无血清培养基中产生MASP-2A,以确保制备物不被其他血清蛋白污染。每隔一天从汇合细胞中收获培养基(共四次)。三个物种中每一种的重组MASP-2A水平平均约为1.5 mg/升培养基。
MASP-2A蛋白纯化:通过亲和层析法在MBP-A琼脂糖柱上使MASP-2A (上述Ser-Ala突变体)纯化。这种策略能够快速纯化而无需使用外部标记。将MASP-2A (100-200 ml培养基用等体积的加样缓冲液(50 mM Tris-Cl,pH 7.5,含有150 mM NaCl和25 mM CaCl2)稀释)加到MBP-琼脂糖亲和柱(4 ml)上,柱用10 ml加样缓冲液预平衡。另用10 ml加样缓冲液洗涤后,在含有1.25 M NaCl和10 mM EDTA的50 mM Tris-Cl (pH 7.5)的1 ml级分中洗脱蛋白质。用SDS-聚丙烯酰胺凝胶电泳来鉴定含有MASP-2A的级分。必要时,MASP-2A通过离子交换层析在MonoQ柱(HR 5/5)上进一步纯化。蛋白质用含有50 mM NaCl的50 mM Tris-Cl (pH 7.5)透析,加到用同一缓冲液平衡的柱上。洗涤后,另用10 ml的0.05-1 M NaCl梯度洗脱出结合的MASP-2A。
结果:从200 ml培养基中获得产量为0.25-0.5 mg的MASP-2A蛋白。由于糖基化作用,所以经MALDI-MS测得77.5 kDa的分子量比未修饰多肽的计算值(73.5 kDa)大。在每个N-糖基化位点连接有聚糖是所测分子量的原因。MASP-2A作为单一条带在SDS-聚丙烯酰胺凝胶上迁移,证明在生物合成期间没有被蛋白水解加工。通过平衡超速离心法测得的重量平均分子量与糖基化多肽同型二聚体的计算值一致。
重组人MASP-2多肽的生产
生产重组MASP-2和MASP-2A衍生多肽的另一种方法描述于Thielens, N.M.,等人, J. Immunol. 166:5068-5077, 2001。简单来讲,使草地贪夜蛾(Spodoptera frugiperda)昆虫细胞(获自Novagen, Madison, WI的Ready-Plaque Sf9细胞)在Sf900II无血清培养基(Life Technologies)中生长并维持,该培养基补充了50 IU/ml青霉素和50 mg/ml链霉素(Life Technologies)。使粉纹夜蛾(Trichoplusia ni) (High Five)昆虫细胞(由Jadwiga Chroboczek, Institut de Biologie Structurale, Grenoble, France提供)在TC100培养基(Life Technologies)中维持,该培养基含有补充了50 IU/ml青霉素和50 mg/ml链霉素的10% FCS (Dominique Dutscher, Brumath, France)。采用Bac-to-Bac系统(Life Technologies)来产生重组杆状病毒。杆粒(bacmid) DNA采用Qiagen中量制备纯化系统(Qiagen)纯化,并用来按照生产商所述方案,在cellfectin的Sf900 II SFM培养基(Life Technologies)中转染Sf9昆虫细胞。4天后收集重组病毒颗粒,经病毒噬斑测定滴定,按照King和Possee所述方法(King和Possee, 载于The Baculovirus Expression System: A Laboratory Guide, Chapman and Hall Ltd., London,第111-114页,1992)进行扩增。
在Sf900 II SFM培养基中,用含有MASP-2多肽的重组病毒在28℃下感染High Five细胞(1.75 x 107个细胞/175 cm2组织培养瓶) 96小时,感染复数为2。经离心收集上清液,加入二异丙基氟磷酸至终浓度1 mM。
MASP-2多肽被分泌到培养基中。将培养物上清液对50 mM NaCl、1 mM CaCl2、50 mM盐酸三乙醇胺(pH 8.1)透析,以1.5 ml/分钟的速度加到同一缓冲液平衡的Q-琼脂糖凝胶快流速柱(Amersham Pharmacia Biotech) (2.8 x 12 cm)上。在同一缓冲液中,用1.2升的线性梯度(至350 mM NaCl)进行洗脱。通过Western印迹分析法鉴定出含有重组MASP-2多肽的级分,通过加入(NH4)2SO4至60% (重量/体积)进行沉淀,4℃静置过夜。将沉淀重悬浮于145 mM NaCl、1 mM CaCl2、50 mM盐酸三乙醇胺(pH 7.4)中,加到同一缓冲液平衡的TSK G3000 SWG柱(7.5 x 600 mm) (Tosohaas, Montgomeryville, PA)上。然后将已纯化的多肽在Microsep微量浓缩器(截留分子量=10,000) (Filtron, Karlstein, Germany)中通过超滤浓缩至0.3 mg/ml。
实施例4
本实施例描述了抗MASP-2多肽的多克隆抗体的生产方法。
材料与方法:
MASP-2抗原:用以下分离的MASP-2多肽免疫兔,以生产多克隆抗人MASP-2抗血清:分离自血清的人MASP-2 (SEQ ID NO:6);重组人MASP-2 (SEQ ID NO:6),含有无活性的蛋白酶结构域(SEQ ID NO:13)的MASP-2A,见实施例3;表达的重组CUBI (SEQ ID NO:8)、CUBEGFI (SEQ ID NO:9)和CUBEGFCUBII (SEQ ID NO:10),如上文实施例3所述。
多克隆抗体:已用BCG (杆菌卡介苗(bacillus Calmette-Guerin vaccine))免疫的六周龄兔,经注射100 μg MASP-2多肽进行免疫,MASP-2多肽按100 μg/ml溶于无菌盐溶液。每4周进行注射,通过ELISA测定法监测抗体效价,描述于实施例5。收集培养物上清液,用于通过A蛋白亲和层析法进行抗体纯化。
实施例5
本实施例描述了抗大鼠或人MASP-2多肽的鼠单克隆抗体的生产方法。
材料与方法:
将100 μg人或大鼠rMASP-2或rMASP-2A多肽(按实施例3中所述制备)皮下注射到8-12周龄的雄性A/J小鼠(Harlan, Houston, Tex.),所述多肽溶于200 μl磷酸缓冲盐溶液(PBS) (pH 7.4)中的完全弗氏佐剂(Difco Laboratories, Detroit, Mich.)。在两周的时间间隔下,将50 μg溶于不完全弗氏佐剂的人或大鼠rMASP-2或rMASP-2A多肽两次皮下注射到小鼠。在第四周,给小鼠注射溶于PBS的50 μg人或大鼠rMASP-2或rMASP-2A多肽,4天后进行融合。
对于每次融合,从经过免疫的小鼠脾脏制备单细胞悬液,用于与Sp2/0骨髓瘤细胞融合。将5x108个Sp2/0和5x108个脾细胞在含有50%聚乙二醇(分子量1450) (Kodak, Rochester, N.Y.)和5%二甲亚砜(Sigma Chemical Co.,St. Louis, Mo.)的培养基中进行融合。然后将细胞调节到每200 μl的Iscove培养基(Gibco, Grand Island, N.Y.)悬液1.5x105脾细胞的浓度,培养基中补充10%胎牛血清、100单位/ml青霉素、100 μg/ml链霉素、0.1 mM次黄嘌呤、0.4 μM氨基蝶呤和16 μM胸苷。将200微升细胞悬液加到大约二十个96孔微量培养板的各孔中。大约10天后,取出培养物上清液,用于在ELISA测定法中筛选与纯化因子MASP-2的反应性。
ELISA测定法:通过加入50 μl经纯化的50 ng/ml hMASP-2或大鼠rMASP-2 (或rMASP-2A)在室温下过夜,包被Immulon 2 (Dynatech Laboratories, Chantilly, Va.)微量试验板的各孔。用于包被的低浓度MASP-2使得能够选择高亲和力抗体。在轻轻拍打培养板除去包被溶液后,将200 μl的BLOTTO (脱脂奶粉)的PBS溶液加到每个孔中达1小时以封闭非特异性位点。一小时之后,然后各孔用缓冲液PBST (含有0.05%吐温20的PBS)洗涤。从各融合孔中收集50微升的培养物上清液,并与50 μl的BLOTTO混合,然后加到微量试验板的各孔中。孵育1小时之后,各孔用PBST洗涤。然后通过与辣根过氧化物酶(HRP)缀合的山羊抗小鼠IgG (对Fc特异性) (Jackson ImmunoResearch Laboratories, West Grove, Pa.)反应来检测结合的鼠抗体,并以1:2,000稀释于BLOTTO中。将含有0.1% 3,3,5,5-四甲基联苯胺(Sigma, St. Louis, Mo.)和0.0003%过氧化氢(Sigma)的过氧化物酶底物溶液加到各孔中,显色30分钟。每孔加入50 μl 2M H2SO4终止反应。用BioTek ELISA读板仪(BioTek Instruments, Winooski, Vt.)读取反应混合物在450 nm的光密度(OD)。
MASP-2结合测定:
在上述MASP-2 ELISA测定法中测试为阳性的培养物上清液可在结合测定中进行测定,以测定MASP-2抑制剂对MASP-2的结合亲和力。也可使用类似测定法来确定抑制剂是否结合补体系统中的其他抗原。
通过用MASP-2的磷酸缓冲盐溶液(PBS) (pH 7.4) (20 ng/100 μl/孔,Advanced Research Technology, San Diego, CA)在4℃下过夜,来包被聚苯乙烯微量滴定板的各孔(96孔培养基结合板,Corning Costar, Cambridge, MA)。吸出MASP-2溶液后,各孔用含有1%牛血清白蛋白(BSA;Sigma Chemical)的PBS在室温下封闭2小时。无MASP-2包被的孔用作背景对照。将在封闭溶液中不同浓度的杂交瘤上清液或已纯化的抗MASP-2 MoAb等分溶液加到各孔中。在室温下孵育2小时之后,各孔用PBS充分漂洗。通过在封闭溶液中加入过氧化物酶缀合的山羊抗小鼠IgG (Sigma Chemical)在室温下孵育1小时,来检测结合MASP-2的抗MASP-2 MoAb。该板用PBS再次充分漂洗后,加入100 μl 3,3',5,5'-四甲基联苯胺(TMB)底物(Kirkegaard and Perry Laboratories, Gaithersburg, MD)。加入100 μl的1 M磷酸来猝灭TMB反应,将板在微量板读板仪(SPECTRA MAX 250,Molecular Devices, Sunnyvale, CA)中在450 nm下读数。
然后测定来自阳性孔的培养物上清液在功能测定法(诸如实施例2所述C4裂解测定法)中抑制补体活化的能力。然后经有限稀释克隆阳性孔中的细胞。再在上述ELISA测定法中测定MoAb与hMASP-2的反应性。使选出的杂交瘤在转瓶中生长,收集耗尽培养物的上清液,通过A蛋白亲和层析法纯化抗体。
实施例6
本实施例描述了人源化鼠抗MASP-2抗体和抗体片段的产生和生产。
按照实施例5所述方法,在雄性A/J小鼠中产生鼠抗MASP-2单克隆抗体。然后按照下述方法,将鼠恒定区用其人对应物置换来产生IgG与抗体Fab片段的嵌合体,使鼠抗体人源化以降低它的免疫原性,所述嵌合体用于抑制本发明的人受试者体内的MASP-2依赖性补体活化的副作用。
1. 由鼠杂交瘤细胞克隆抗MASP-2可变区基因
按照生产商的方案(Biotech, Houston, Tex.),使用RNAzol由分泌抗MASP-2 MoAb的杂交瘤细胞(按照实施例7中所述方法获得)中分离出总RNA。采用寡聚dT作为引物,由总RNA合成第一cDNA链。使用免疫球蛋白恒定C区衍生的3’引物和得自前导肽或鼠VH或VK基因的第一构架区的简并引物组作为5’引物对来进行PCR。按Chen和Platsucas所述方法(Chen, P.F., Scand. J. Immunol. 35:539-549, 1992)进行锚定PCR。对于克隆VK基因,用Not1-MAK1引物
 来制备双链cDNA。将退火衔接子
来制备双链cDNA。将退火衔接子  和
和 连接到双链cDNA的5’端和3’端。经Notl消化除去3’端衔接子。然后将消化产物用作PCR模板,以AD1寡核苷酸作为5’引物,MAK2作为3’引物。将大约500 bp的DNA片段克隆到pUC19中。选出若干克隆进行序列分析以证实克隆序列包括预期的鼠免疫球蛋白恒定区。Not1-MAK1和MAK2寡核苷酸得自VK区,分别为Cκ基因第一碱基对下游的182 bp和84 bp。选择包括完整VK和前导肽的克隆。
连接到双链cDNA的5’端和3’端。经Notl消化除去3’端衔接子。然后将消化产物用作PCR模板,以AD1寡核苷酸作为5’引物,MAK2作为3’引物。将大约500 bp的DNA片段克隆到pUC19中。选出若干克隆进行序列分析以证实克隆序列包括预期的鼠免疫球蛋白恒定区。Not1-MAK1和MAK2寡核苷酸得自VK区,分别为Cκ基因第一碱基对下游的182 bp和84 bp。选择包括完整VK和前导肽的克隆。
对于克隆VH基因,使用Not1 MAG1引物 来制备双链cDNA。将退火衔接子AD1和AD2连接到双链cDNA的5’端和3’端。经Notl消化除去3’端衔接子。将消化产物用作PCR模板,以AD1寡核苷酸和
来制备双链cDNA。将退火衔接子AD1和AD2连接到双链cDNA的5’端和3’端。经Notl消化除去3’端衔接子。将消化产物用作PCR模板,以AD1寡核苷酸和 作为引物。将长度为500-600 bp的DNA片段克隆到pUC19中。Not1-MAG1和MAG2寡核苷酸得自鼠Cγ.7.1区,分别为鼠Cγ.7.1基因第一碱基对下游的180 bp和93 bp。选择包括完整VH和前导肽的克隆。
作为引物。将长度为500-600 bp的DNA片段克隆到pUC19中。Not1-MAG1和MAG2寡核苷酸得自鼠Cγ.7.1区,分别为鼠Cγ.7.1基因第一碱基对下游的180 bp和93 bp。选择包括完整VH和前导肽的克隆。
2. 嵌合MASP-2 IgG与Fab的表达载体的构建
将上述克隆的VH和VK基因用作PCR反应的模板,以便将Kozak共有序列加到5’端,将剪接供体加到核苷酸序列的3’端。对该序列进行分析确定没有PCR误差后,将VH和VK基因分别插入到含有人C.γ1和C.κ的表达载体盒中,得到pSV2neoVH-huCγ1 和 pSV2neoV-huCγ。使用CsCl梯度纯化的重链和轻链载体的质粒DNA通过电穿孔转染COS细胞。48小时后,通过ELISA测定培养物上清液,证实存在大约200 ng/ml的嵌合IgG。收获细胞,制备总RNA。使用寡聚dT作为引物从总RNA合成第一cDNA链。将该cDNA用作PCR模板以产生Fd和κ DNA片段。对于Fd基因,使用
 作为5’引物以及CH1衍生的3’引物
作为5’引物以及CH1衍生的3’引物 进行PCR。证实DNA序列含有完整的人IgG1的VH和CH1结构域。在用合适的酶消化后,将Fd DNA片段插入表达载体盒pSV2dhfr-TUS的HindIII和BamHI限制位点,得到pSV2dhfrFd。pSV2质粒是市售的,由不同来源的DNA区段组成:pBR322 DNA (细线)含有pBR322 DNA复制起点(pBR ori)以及内酰胺酶氨苄青霉素抗性基因(Amp);SV40 DNA,由较粗影线表示并标记,含有SV40 DNA复制起点(SV40 ori)、早期启动子(dhfr和neo基因的5’端)以及聚腺苷酸化信号(dhfr和neo基因的3’端)。SV40衍生的聚腺苷酸化信号(pA)也位于Fd基因的3’端。
进行PCR。证实DNA序列含有完整的人IgG1的VH和CH1结构域。在用合适的酶消化后,将Fd DNA片段插入表达载体盒pSV2dhfr-TUS的HindIII和BamHI限制位点,得到pSV2dhfrFd。pSV2质粒是市售的,由不同来源的DNA区段组成:pBR322 DNA (细线)含有pBR322 DNA复制起点(pBR ori)以及内酰胺酶氨苄青霉素抗性基因(Amp);SV40 DNA,由较粗影线表示并标记,含有SV40 DNA复制起点(SV40 ori)、早期启动子(dhfr和neo基因的5’端)以及聚腺苷酸化信号(dhfr和neo基因的3’端)。SV40衍生的聚腺苷酸化信号(pA)也位于Fd基因的3’端。
对于κ基因,使用 作为5’引物和CK衍生的3’引物
作为5’引物和CK衍生的3’引物
 进行PCR。验证DNA序列含有完整的VK和人CK区域。在用合适的限制酶消化后,将κ DNA片段插入表达载体盒pSV2neo-TUS的HindIII和BamHI限制位点,得到pSV2neoK。Fd和κ基因的表达均由HCMV衍生的增强子和启动子元件驱动。因为Fd基因不包括涉及到链间二硫键的半胱氨酸氨基酸残基,所以该重组的嵌合Fab就含有非共价连接的重链和轻链。该嵌合Fab被命名为cFab。
进行PCR。验证DNA序列含有完整的VK和人CK区域。在用合适的限制酶消化后,将κ DNA片段插入表达载体盒pSV2neo-TUS的HindIII和BamHI限制位点,得到pSV2neoK。Fd和κ基因的表达均由HCMV衍生的增强子和启动子元件驱动。因为Fd基因不包括涉及到链间二硫键的半胱氨酸氨基酸残基,所以该重组的嵌合Fab就含有非共价连接的重链和轻链。该嵌合Fab被命名为cFab。
为了获得带有重链和轻链间二硫键的重组Fab,可以将上述Fd基因延长以包括来自人IgG1铰链区的额外9个氨基酸(EPKSCDKTH SEQ ID NO:48)的编码序列。编码Fd基因3’端的30个氨基酸的BstEII-BamHI DNA区段可被编码延长的Fd的DNA区段置换,产生pSV2dhfrFd/9aa。
3. 嵌合的抗MASP-2 IgG的表达和纯化
为产生分泌嵌合的抗MASP-2 IgG的细胞系,用经纯化的pSV2neoVH-huC.γ1和pSV2neoV-huC κ的质粒DNA通过电穿孔转染NSO细胞。在0.7 mg/ml G418存在下选出被转染的细胞。将细胞在250 ml转瓶中用含血清的培养基进行培养。
将100 ml旋动培养物的培养物上清液加到10 ml PROSEP-A柱(Bioprocessing, Inc., Princeton, N.J.)上。柱用10个柱床体积的PBS洗涤。结合的抗体用50 mM柠檬酸缓冲液(pH 3.0)洗脱。将等体积的1 M Hepes (pH 8.0)加到含有纯化抗体的级分中,调节pH到7.0。通过Millipore膜超滤(截留分子量:3,000),用PBS进行缓冲液交换来去除残留的盐。通过BCA方法(Pierce)测得经纯化抗体的蛋白质浓度。
4. 嵌合的抗MASP-2 Fab的表达和纯化
为了产生分泌嵌合的抗MASP-2 Fab的细胞系,用经纯化的pSV2dhfrFd (或pSV2dhfrFd/9aa)和pSV2neoκ的质粒DNA通过电穿孔转染CHO细胞。在G418和甲氨蝶呤存在下选出被转染的细胞。在浓度递增的甲氨蝶呤中对所选细胞系进行扩增。经有限稀释对细胞进行单细胞亚克隆。然后将高产单细胞亚克隆细胞系在100 ml转瓶中用无血清培养基进行培养。
采用抗MASP-2 MoAb的小鼠抗独特型MoAb,通过亲和层析法对嵌合的抗MASP-2 Fab进行纯化。可用与匙孔 血蓝蛋白(KLH)缀合的鼠抗MASP-2 MoAb给小鼠免疫,筛选出能够与人MASP-2竞争的特异性MoAb结合,从而来制备抗独特型MASP-2 MoAb。至于纯化,将来自产生cFab或cFab/9aa的旋动培养的CHO细胞的100 ml上清液加到与抗独特型MASP-2 MoAb偶联的亲和柱上。然后该柱用PBS充分洗涤后,结合的Fab用50 mM二乙胺(pH 11.5)洗脱出来。按照上述方法通过缓冲液交换除去残留的盐。通过BCA方法(Pierce)测定经纯化的Fab的蛋白质浓度。
血蓝蛋白(KLH)缀合的鼠抗MASP-2 MoAb给小鼠免疫,筛选出能够与人MASP-2竞争的特异性MoAb结合,从而来制备抗独特型MASP-2 MoAb。至于纯化,将来自产生cFab或cFab/9aa的旋动培养的CHO细胞的100 ml上清液加到与抗独特型MASP-2 MoAb偶联的亲和柱上。然后该柱用PBS充分洗涤后,结合的Fab用50 mM二乙胺(pH 11.5)洗脱出来。按照上述方法通过缓冲液交换除去残留的盐。通过BCA方法(Pierce)测定经纯化的Fab的蛋白质浓度。
可应用实施例2或实施例7中所述的抑制测定法来测定嵌合MASP-2 IgG、cFab和cFAb/9aa抑制MASP-2依赖性补体途径的能力。
实施例7
本实施例描述了用作功能性筛选的体外C4裂解测定法,从而鉴定能够阻断经由L-纤维胶凝蛋白/P35、H-纤维胶凝蛋白、M-纤维胶凝蛋白或甘露聚糖的MASP-2依赖性补体活化的MASP-2抑制剂。
C4裂解测定法:Petersen, S.V.等人描述了C4裂解测定法(Petersen, S.V.,等人, J. Immunol. Methods 257:107, 2001),测定了由结合L-纤维胶凝蛋白的金黄色葡萄球菌的脂磷壁酸(LTA)导致的凝集素途径活化。
试剂:福尔马林固定的金黄色葡萄球菌(DSM20233)如下制备:将细菌在胰胨豆胨血液培养基(tryptic soy blood medium)中于37℃培养过夜,用PBS洗涤三次,然后在室温下于PBS/0.5%福尔马林中固定1小时,再用PBS洗涤三次后,重悬浮于包被缓冲液(15 mM Na2CO3、35 mM NaHCO3,pH 9.6)中。
测定法:Nunc MaxiSorb微量滴定板(Nalgene Nunc International, Rochester, NY)的各孔用以下的成分包被:100 μl福尔马林固定的金黄色葡萄球菌DSM20233 (OD550 = 0.5)的包被缓冲液与1 ug L-纤维胶凝蛋白的包被缓冲液。孵育过夜之后,各孔用0.1%人血清白蛋白(HSA)的TBS溶液(10 mM Tris-HCl,140 mM NaCl,pH 7.4)封闭,然后用含有0.05%吐温20和5 mM CaCl2的TBS溶液(洗涤缓冲液)洗涤。将人血清样品稀释于20 mM Tris-HCl、1 M NaCl、10 mM CaCl2、0.05% Triton X-100、0.1% HSA (pH 7.4)中,这就防止了内源C4的活化,并使C1复合物(由C1q、C1r和C1s组成)解离。将不同浓度的MASP-2抑制剂(包括抗MASP-2 MoAb和抑制肽)加到血清样品中。将稀释的样品加到板中,在4℃下孵育过夜。24小时后,各板用洗涤缓冲液充分洗涤,然后向每个孔中加入0.1 μg溶于100 μl 4 mM巴比妥、145 mM NaCl、2 mM CaCl2、1 mM MgCl2 (pH 7.4)的经纯化的人C4 (按照Dodds, A.W., Methods Enzymol. 223:46, 1993所述方法获得)。37℃1.5小时后,各板再次经洗涤,使用碱性磷酸酶缀合的鸡抗人C4c (获自Immunsystem, Uppsala, Sweden),检测C4b沉积,并使用比色底物磷酸对硝基苯酯进行测定。
关于甘露聚糖的C4测定:修改上述测定法以测定经由MBL的凝集素途径活化,在加入与各种MASP-2抑制剂混合的血清之前,用LSP和甘露聚糖包被测定板。
关于H-纤维胶凝蛋白(Hakata Ag)的C4测定:修改上述测定法以测定经由H-纤维胶凝蛋白的凝集素途径活化,在加入与各种MASP-2抑制剂混合的血清之前,用LSP和H-纤维胶凝蛋白包被测定板。
实施例8
下面的测定法证明了野生型和MASP-2-/-小鼠中存在经典途径活化。
方法:微量滴定板(Maxisorb,Nunc,目录号442404,Fisher Scientific)用0.1%人血清白蛋白的10 mM Tris、140 mM NaCl (pH 7.4)溶液在室温下包被1小时,然后用按1:1000稀释于TBS/吐温/Ca2+中的绵羊抗全血清的抗血清(Scottish Antibody Production Unit, Carluke, Scotland)在4℃下孵育过夜,从而原位产生免疫复合物。从野生型和MASP-2-/-小鼠中获得血清样品,加到包被板中。制备对照样品,其中从野生型和MASP-2-/-血清样品中耗尽C1q。按照供应商的说明书,使用包被了兔抗人C1q IgG (Dako, Glostrup, Denmark)的A蛋白偶联的Dynabead (Dynal Biotech, Oslo, Norway)来制备C1q耗尽的小鼠血清。将各板在37℃下孵育90分钟。用按1:1000稀释于TBS/吐温/Ca++中的多克隆抗人C3c抗体(Dako A 062)来检测结合的C3b。二抗是山羊抗兔IgG。
结果:图7表示用IgG与野生型血清、MASP-2-/-血清、耗尽C1q的野生型和耗尽C1q的MASP-2-/-血清包被板上C3b的相对沉积水平。这些结果证明,经典途径在MASP-2-/-小鼠品系中是完整的。
实施例9
下面的测定法用来通过在经典途径被免疫复合物启动的情况下分析MASP-2抑制剂的作用,从而检测MASP-2抑制剂是否阻断了经典途径。
方法:为了检测MASP-2抑制剂对其中经典途径被免疫复合物启动的补体活化情况的影响,在37℃下,将含有90% NHS的50 μl样品一式三份在10 μg/ml免疫复合物(IC)或PBS存在下孵育,在37℃孵育的还有含有200 nM抗备解素单克隆抗体的平行样品(+/-IC)一式三份。37℃孵育两小时后,将13 mM EDTA加到所有样品中终止进一步的补体活化,立即将样品冷却到5℃。在按照生产商说明书,使用ELISA试剂盒(Quidel, 目录号A015和A009)进行补体活化产物(C3a和sC5b-9)分析之前,将样品保存于-70℃。
实施例10
本实施例描述了阻断MASP-2活性的高亲和力抗MASP-2 Fab2抗体片段的鉴定。
背景与基本原理:MASP-2是具有许多独立功能结构域的复杂蛋白质,包括:MBL和纤维胶凝蛋白的结合部位、丝氨酸蛋白酶催化部位、蛋白水解底物C2的结合部位、蛋白水解底物C4的结合部位、MASP-2酶原自身活化的MASP-2裂解部位和两个Ca++结合部位。鉴定出以高亲和力结合MASP-2的Fab2抗体片段,在功能测定法中测定所鉴定出的Fab2片段,以确定它们是否能够阻断MASP-2功能活性。
为了阻断MASP-2功能活性,抗体或Fab2抗体片段必须结合并阻碍MASP-2功能活性所需要的MASP-2上的结构表位。因此,许多或所有的高亲和力结合抗MASP-2 Fab2不能抑制MASP-2功能活性,除非它们能结合直接参与MASP-2功能活性的MASP-2的结构表位。
测定抑制凝集素途径C3转化酶形成的功能测定法被用来评价抗MASP-2 Fab2的“阻断活性”。已知MASP-2在凝集素途径中的主要生理作用是产生凝集素介导的补体途径的下一个功能成分,即凝集素途径C3转化酶。凝集素途径C3转化酶是蛋白水解裂解C3成为C3a和C3b的关键酶复合物(C4bC2a)。MASP-2不是凝集素途径C3转化酶(C4bC2a)的结构成分;然而,需要MASP-2功能活性以产生组成凝集素途径C3转化酶的两个蛋白质成分(C4b、C2a)。此外,为了使MASP-2产生凝集素途径C3转化酶,似乎需要所有上述MASP-2的独立功能活性。出于这些原因,认为用于评价抗MASP-2 Fab2的“阻断活性”优选的测定法是测定抑制凝集素途径C3转化酶形成的功能测定法。
高亲和力Fab2的产生:应用人可变轻链和重链抗体序列的噬菌体展示文库以及用于鉴定与所选出的目标配体反应的Fab2的自动抗体筛选技术,来产生抗大鼠MASP-2蛋白(SEQ ID NO:55)的高亲和力Fab2。利用已知量的大鼠MASP-2 (~1 mg,纯度>85%)蛋白进行抗体筛选。利用三轮扩增以选出具有最高亲和力的抗体。挑选约250个不同的表达抗体片段的目标(hits)用于ELISA筛选。随后对高亲和力目标进行测序以确定不同抗体的独特性。
将50个独特的抗MASP-2抗体纯化,将250 μg各经纯化的Fab2抗体用于表征MASP-2结合亲和力和补体途径功能测定,详述如下。
用于评价抗MASP-2 Fab2抑制(阻断)活性的测定法
1. 测定抑制凝集素途径C3转化酶形成的测定法:
背景:凝集素途径C3转化酶是蛋白水解裂解C3成为两个有效促炎片段过敏毒素C3a和调理素C3b的酶复合物(C4bC2a)。C3转化酶的形成似乎是在介导炎症方面的凝集素途径中的关键步骤。MASP-2不是凝集素途径C3转化酶(C4bC2a)的结构成分;因此,抗MASP-2抗体(或Fab2)不会直接抑制之前已存在的C3转化酶的活性。然而,为了产生组成凝集素途径C3转化酶的两个蛋白质成分(C4b、C2a),MASP-2丝氨酸蛋白酶活性是必需的。因此,抑制MASP-2功能活性的抗MASP-2 Fab2 (即阻断性抗MASP-2 Fab2)会抑制凝集素途径C3转化酶从头形成。C3含有独特的高反应性硫酯基团作为其结构部分。在该测定法中当C3转化酶裂解C3时,C3b上的硫酯基团可与通过酯键或酰胺键固定在塑料孔底部的大分子上的羟基或氨基形成共价键,从而有利于在ELISA测定法中检测出C3b。
酵母甘露聚糖是凝集素途径的已知激活剂。在测定C3转化酶形成的下列方法中,将用甘露聚糖包被的塑料孔与稀释的大鼠血清在37℃下孵育30分钟以激活凝集素途径。然后洗涤各孔,并采用标准ELISA方法测定固定在孔中的C3b。在该测定法中所产生的C3b的量直接反映了凝集素途径C3转化酶的从头形成。在该测定法中测定选定浓度的抗MASP-2 Fab2抑制C3转化酶形成和随后C3b产生的能力。
方法:
将96孔Costar培养基结合板与稀释于50 mM碳酸盐缓冲液(pH 9.5)的甘露聚糖以1 μg/50 μl/孔在5℃下孵育过夜。过夜孵育后,各孔用200 μl PBS洗涤三次。然后各孔用100 μl/孔的1%牛血清白蛋白的PBS溶液封闭,在室温下轻轻搅动孵育1小时。然后各孔用200 μl PBS洗涤三次。在5℃下,将抗MASP-2 Fab2样品稀释于含Ca++和Mg++的GVB缓冲液(4.0 mM巴比妥,141 mM NaCl,1.0 mM MgCl2,2.0 mM CaCl2,0.1%明胶,pH 7.4)至选定浓度。在5℃下,将0.5%大鼠血清加到上述样品中,将100 μl转移到各孔。将板盖上,在37℃水浴中孵育30分钟以便补体活化。将板从37℃水浴转移装有冰-水混合物的容器中进行终止反应。各孔依次用200 μl PBS-吐温20 (0.05%吐温20的PBS溶液)洗涤5次,用200 μl PBS洗涤2次。加入100 μl/孔按1:10,000稀释的一抗(兔抗人C3c,DAKO A0062),该抗体溶于含有2.0 mg/ml牛血清白蛋白的PBS中,在室温下轻轻搅动孵育1小时。各孔用200 μl PBS洗涤5次。加入100 μl/孔按1:10,000稀释的二抗(过氧化物酶缀合的山羊抗兔IgG,American Qualex A102PU),该抗体溶于含有2.0 mg/ml牛血清白蛋白的PBS中,室温下在振荡器中轻轻搅动孵育1小时。各孔用200 μl PBS洗涤5次。加入100 μl/孔过氧化物酶底物TMB (Kirkegaard & Perry Laboratories),在室温下孵育10分钟。通过加入100 μl/孔1.0 M H3PO4终止过氧化物酶反应,测得OD450。
2. 测定抑制MASP-2依赖性C4裂解的测定法
背景:MASP-2的丝氨酸蛋白酶活性是高度特异性的,仅鉴定出用于MASP-2的两种蛋白质底物:C2和C4。C4裂解产生C4a和C4b。抗MASP-2 Fab2可结合直接参与C4裂解的MASP-2的结构表位(例如C4的MASP-2结合部位;MASP-2丝氨酸蛋白酶催化部位),从而抑制MASP-2的C4裂解功能活性。
酵母甘露聚糖是凝集素途径的已知激活剂。在测定MASP-2的C4裂解活性的下列方法中,将用甘露聚糖包被的塑料孔与稀释的大鼠血清在37℃下孵育30分钟以激活凝集素途径。因为用于该ELISA测定法中的一抗仅识别人C4,所以稀释的大鼠血清还补充了人C4 (1.0 μg/ml)。然后洗涤各孔,采用标准ELISA方法测定固定在孔中的人C4b。在该测定法中,所产生的C4b的量是依赖MASP-2的C4裂解活性的量度。在该测定法中,测定选定浓度的抗MASP-2 Fab2抑制C4裂解的能力。
方法:将96孔Costar培养基结合板与稀释于50 mM碳酸盐缓冲液(pH 9.5)的甘露聚糖按1 μg/50 μl/孔在5℃下孵育过夜。各孔用200 μl PBS洗涤3次。然后各孔用100 μl/孔1%牛血清白蛋白的PBS溶液封闭,在室温下轻轻搅动孵育1小时。各孔用200 μl PBS洗涤3次。在5℃下,将抗MASP-2 Fab2样品稀释于含Ca++和Mg++的GVB缓冲液(4.0 mM巴比妥,141 mM NaCl,1.0 mM MgCl2,2.0 mM CaCl2,0.1%明胶,pH 7.4)至选定浓度。1.0 μg/ml人C4 (Quidel)也包括在这些样品中。在5℃下,将0.5%大鼠血清加到上述样品中,将100 μl转移到各孔。将板盖上,在37℃水浴中孵育30分钟以便补体活化。将板从37℃水浴转移装有冰-水混合物的容器中进行终止反应。各孔用200 μl PBS-吐温20 (0.05%吐温20的PBS溶液)洗涤5次,然后各孔用200 μl PBS洗涤2次。加入100 μl/孔以1:700稀释的生物素缀合的鸡抗人C4c (Immunsystem AB, Uppsala, Sweden)的PBS溶液(含有2.0 mg/ml牛血清白蛋白(BSA)),在室温下轻轻搅动孵育1小时。各孔用200 μl PBS洗涤5次。加入100 μl/孔的0.1 μg/ml过氧化物酶缀合的链霉抗生物素(Pierce Chemical #21126)的PBS溶液(含有2.0 mg/ml BSA),在室温下在振荡器中轻轻搅动孵育1小时。各孔用200 μl PBS洗涤5次。加入100 μl/孔过氧化物酶底物TMB (Kirkegaard & Perry Laboratories),在室温下孵育16分钟。通过加入100 μl/孔1.0 M H3PO4终止过氧化物酶反应,测得OD450。
3. 抗“天然”大鼠MASP-2的抗大鼠MASP-2 Fab2的结合测定法
背景:MASP-2通常作为还包括特异性凝集素分子(甘露糖结合蛋白(MBL)和纤维胶凝蛋白)的MASP-2二聚体复合物存在于血浆中。因此,如果有兴趣研究抗MASP-2 Fab2与生理相关形式的MASP-2结合,则重要的是开发出结合测定法,其中利用的是Fab2与血浆“天然”MASP-2之间,而不是与纯化的重组MASP-2之间的相互作用。在该结合测定法中,先将来自10%大鼠血清的“天然”MASP-2-MBL复合物固定在甘露聚糖包被的孔内。然后采用标准ELISA方法,对各种抗固定化“天然”MASP-2的抗MASP-2 Fab2的结合亲和力进行了研究。
方法:将96孔Costar高结合板与稀释于50 mM碳酸盐缓冲液(pH 9.5)的甘露聚糖按1 μg/50 μl/孔在5℃下孵育过夜。各孔用200 μl PBS洗涤3次。各孔用100 μl/孔的0.5%脱脂奶粉的PBST溶液(PBS与0.05%吐温20)封闭,在室温下轻轻搅动孵育1小时。各孔用200 μl TBS/吐温/Ca++洗涤缓冲液(Tris缓冲盐溶液,0.05%吐温20,含有5.0 mM CaCl2,pH 7.4)洗涤3次。在冰上制备10%大鼠血清的高盐结合缓冲液(20 mM Tris,1.0 M NaCl,10 mM CaCl2,0.05% Triton-X100,0.1% (重量/体积)牛血清白蛋白,pH 7.4)。每孔加入100 μl,在5℃下孵育过夜。各孔用200 μl TBS/吐温/Ca++洗涤缓冲液洗涤3次。然后各孔用200 μl PBS洗涤2次。加入100 μl/孔稀释于含有Ca++和Mg++的GVB缓冲液(4.0 mM巴比妥,141 mM NaCl,1.0 mM MgCl2,2.0 mM CaCl2,0.1%明胶,pH 7.4)的选定浓度的抗MASP-2 Fab2,在室温下轻轻搅动孵育1小时。各孔用200 μl PBS洗涤5次。加入100 μl/孔按1:5000稀释的溶于2.0 mg/ml牛血清白蛋白/PBS的HRP缀合的山羊抗Fab2 (Biogenesis目录号0500-0099),在室温下轻轻搅动孵育1小时。各孔用200 μl PBS洗涤5次。加入100 μl/孔过氧化物酶底物TMB (Kirkegaard & Perry Laboratories),在室温下孵育70分钟。通过加入100 μl/孔的1.0 M H3PO4终止过氧化物酶反应,测得OD450。
结果:
挑选了约250个不同的与抗大鼠MASP-2蛋白进行高亲和力反应的Fab2用于ELISA筛选。对这些高亲和力Fab2进行了测序以确定不同抗体的独特性,对50个独特的抗MASP-2抗体进行纯化用于进一步分析。250 μg各经纯化的Fab2抗体用来表征MASP-2结合亲和力及测定补体途径功能。该分析的结果见下表6。
表6:阻断凝集素途径补体活化的抗MASP-2 FAB2

如上表6所示,50个所测的抗MASP-2 Fab2中,17个Fab2被鉴定为MASP-2阻断性Fab2,其有效抑制C3转化酶形成,IC50 ≤ 10 nM Fab2 (34%阳性选中率)。所鉴定出的17个Fab2中,有8个的IC50范围为纳摩尔以下。此外,表6中所示的所有17个MASP-2阻断性Fab2在凝集素途径C3转化酶测定法中基本上完全抑制了C3转化酶形成。图8A图示说明C3转化酶形成测定法的Fab2抗体#11的结果,在其他所测定的Fab2抗体中具有代表性,其结果见表6。因为甚至当各MASP-2分子被Fab2结合时,“阻断性”Fab2仅可能极微弱地抑制MASP-2功能,这在理论是可能的,因此要慎重考虑。
尽管甘露聚糖是凝集素途径的已知激活剂,但是在大鼠血清存在的抗甘露聚糖抗体还可能激活经典途径,并且通过经典途径C3转化酶产生C3b,这在理论上是可能的。然而,在本实施例所列的17个阻断性抗MASP-2 Fab2中的每一个都有效地抑制了C3b产生(>95%),因此证明了该项测定法对于凝集素途径C3转化酶的特异性。
为了计算各个抗体的表观Kd,对阻断性Fab2的所有17个都进行结合测定。6个阻断性Fab2的抗天然大鼠MASP-2的抗大鼠MASP-2 Fab2的结合测定的结果也见表6。图8B通过图示说明了用Fab2抗体#11的结合测定法的结果。对于其他Fab2也进行了类似的结合测定,其结果见表6。一般而言,对于6个Fab2的每一个结合“天然”MASP-2所获得的表观Kd与在C3转化酶功能测定中Fab2的IC50的对应性颇为适当。有证据表明,在激活其蛋白酶活性之后,MASP-2经历了从“无活性”到“活性”形式的构象变化(Feinberg等人, EMBO J 22:2348-59 (2003); Gal等人, J. Biol. Chem. 280:33435-44 (2005))。在用于C3转化酶形成测定法的正常大鼠血浆中,MASP-2主要以“无活性”酶原构象存在。相比之下,在结合测定法中,MASP-2作为与MBL一起结合到固定化甘露聚糖的复合物的组成部分存在;因此,MASP-2可能为“活性”构象(Petersen等人, J. Immunol Methods 257:107-16, 2001)。因此,对于这两个功能测定法中所测定的17个阻断性Fab2的每一个,可能不必预期IC50与Kd之间确切的对应性,因为在各个测定法中,Fab2可能结合不同构象形式的MASP-2。尽管如此,除了Fab2 #88以外,两种测定法中所测的其他16个Fab2的每一个,IC50与表观Kd之间似乎有相当密切的对应性(参见表6)。
对用于抑制MASP-2介导的C4裂解的若干个阻断性Fab2进行了评价。图8C通过图示说明了C4裂解测定法的结果,表明用Fab2 #41抑制的IC50=0.81 nM (参见表6)。如图9所示,发现所有测试的Fab2都抑制C4裂解,IC50类似于C3转化酶测定法中所获得的IC50 (参见表6)。
尽管甘露聚糖是凝集素途径的已知激活剂,但是大鼠血清中抗甘露聚糖抗体的存在也可能激活经典途径,从而通过C1s介导的C4裂解而产生C4b,这在理论上是可能的。然而,已经鉴定出有效抑制C4b产生(>95%)的若干种抗MASP-2 Fab2,因此证明了该测定法对于MASP-2介导的C4裂解的特异性。C4,如同C3一样,含有独特的高反应性硫酯基团作为其结构部分。在该测定法中当通过MASP-2裂解C4之后,C4b上的硫酯基团可与通过酯键或酰胺键固定在塑料孔底部的大分子上羟基或氨基形成共价键,因此有利于在ELISA测定法中检测出C4b。
这些研究清楚表明,对于大鼠MASP-2蛋白的高亲和力FAB2的产生,其功能性地阻断C4和C3转化酶活性,从而防止了凝集素途径活化。
实施例11
本实施例描述了对若干按照实施例10所述方法产生的阻断性抗大鼠MASP-2 Fab2抗体进行的表位作图。
方法:
如图10所示,使用pED4载体,在CHO细胞中表达下列都具有N端6个His标记的蛋白质:
大鼠MASP-2A,一种全长MASP-2蛋白,通过活性中心上的丝氨酸改变为丙氨酸(S613A)而失活;
大鼠MASP-2K,经改变降低了自身活化的全长MASP-2蛋白(R424K);
CUBI-II,一种仅含有CUBI、EGF样和CUBII结构域的大鼠MASP-2的N端片段;和
CUBI/EGF样,一种仅含有CUBI和EGF样结构域的大鼠MASP-2的N端片段。
按照前述方法将这些蛋白质通过镍亲和层析从培养上清液中纯化出来(Chen等人, J. Biol. Chem. 276:25894-02 (2001))。
采用pTrxFus (Invitrogen),使含有CCPII和大鼠MASP-2丝氨酸蛋白酶结构域的C端多肽(CCPII-SP),在大肠杆菌中表达成为硫氧还蛋白融合蛋白。用Thiobond亲和树脂将蛋白质从细胞裂解物中纯化出来。硫氧还蛋白融合物配偶体由pTrxFus空载体表达作为阴性对照。
将所有重组蛋白透析到TBS缓冲液中,通过测量OD (280 nm),测得其浓度。
斑点印迹法分析:
将连续稀释的上述和图10中所示5个重组MASP-2多肽(以及硫氧还蛋白多肽作为CCPII-丝氨酸蛋白酶多肽的阴性对照)点在硝酸纤维素膜上。蛋白质的点样量在5重步骤中的范围为100 ng-6.4 pg。在稍后的实验中,蛋白质的点样量再次在5重步骤中的范围由50 ng降至16 pg。该膜用5%脱脂奶粉的TBS溶液(封闭缓冲液)封闭,然后与1.0 μg/ml抗MASP-2 Fab2的封闭缓冲液(含有5.0 mM Ca2+)一起孵育。用HRP缀合的抗人Fab (AbD/Serotec;1/10,000稀释)和ECL检测试剂盒(Amersham)检测结合的Fab2。一块膜与作为阳性对照的多克隆兔抗人MASP-2 Ab (参见Stover等人, J Immunol 163:6848-59 (1999))一起孵育。在这种情况下,用HRP缀合的山羊抗兔IgG (Dako;1/2,000稀释),检测出结合的Ab。
MASP-2结合测定法
ELISA板用1.0 μg/孔的重组MASP-2A或CUBI-II多肽的碳酸盐缓冲液(pH 9.0)在4℃下包被过夜。孔用1% BSA的TBS溶液封闭,然后加入连续稀释的抗MASP-2 Fab2的TBS溶液(含有5.0 mM Ca2+)。将所述板在室温下孵育1小时。用TBS/吐温/Ca2+洗涤3次后,加入按1/10,000稀释于TBS/Ca2+的HRP缀合的抗人Fab (AbD/Serotec),将所述板再次在室温下孵育1小时。用TMB过氧化物酶底物试剂盒(Biorad)检测出结合的抗体。
结果:
表明了Fab2与各种MASP-2多肽的反应性的斑点印迹法分析的结果提供在下表7中。表7提供的数值表明,提供大约最大信号强度的一半所需要的蛋白质点样量。如所示,所有多肽(仅硫氧还蛋白融合物配偶体例外)均被阳性对照Ab (多克隆抗人MASP-2血清,在兔中产生)识别。
表7:斑点印迹法中与各重组大鼠MASP-2多肽的反应性

NR = 无反应。阳性对照抗体是在兔中产生的多克隆抗人MASP-2血清。
所有Fab2均与MASP-2A以及MASP-2K反应(数据未显示)。大多数Fab2识别CCPII-SP多肽但不识别N端片段。Fab2 #60和Fab2 #57是两个例外。Fab2 #60识别MASP-2A和CUBI-II片段,但不识别CUBI/EGF样多肽或CCPII-SP多肽,这提示它能结合CUBII的表位或者跨越CUBII和EGF样结构域。Fab2 #57识别MASP-2A但不识别任何所测试的MASP-2片段,这表明这种Fab2识别CCP1的表位。Fab2 #40和#49仅结合完整的MASP-2A。在图11所示的ELISA结合测定法中,Fab2 #60还结合CUBI-II多肽,虽然只具有略微较低的表观亲和力。
这些观察结果表明对于MASP-2蛋白多个区域上的独特阻断性Fab2的鉴定。
实施例12
本实施例描述了鼠肾缺血/再灌注模型中MASP-2-/-小鼠的分析法。
背景/基本原理:体温下,肾缺血-再灌注(I/R)损伤与多种临床病症有关,包括低血容量性休克、肾动脉闭塞和交叉钳夹术。
肾缺血-再灌注(I/R)是急性肾衰竭的重要原因,相关死亡率高达50% (Levy等人, JAMA 275:1489-94, 1996; Thadhani等人, N. Engl. J. Med. 334:1448-60, 1996)。移植后肾衰竭是肾移植之后常见且危险的并发症(Nicholson等人, Kidney Int. 58:2585-91, 2000)。目前还没有肾I/R损伤的有效疗法,血液透析是现有唯一的治疗法。肾I/R损伤的病理生理学十分复杂。最新研究表明,凝集素途径的补体活化可能对肾I/R损伤的发病机制具有重要作用(deVries等人, Am. J. Path. 165:1677-88, 2004)。
方法:
按照实施例1所述方法产生MASP-2(-/-)小鼠,并与C57Bl/6回交至少10代。将Hypnovel (6.64 mg/kg;Roche products Ltd. Welwyn Garden City,UK)注射到称重介于22-25 g之间的6只雄性MASP-2(-/-)和6只野生型(+/+)小鼠腹膜内,随后通过吸入异氟烷(Abbott Laboratories Ltd.,Kent,UK)使之麻醉。选择异氟烷是因为它是温和的吸入麻醉剂,肝毒性最小;浓度准确产生,即使在长时间麻醉后,动物也能迅速恢复。施用Hypnovel是因为它在动物中产生安定麻醉的条件,这意味着只需要施用较少的异氟烷。将保暖垫(warm pad)放置在动物身体下面以便保持恒定体温。接着,沿腹中线切开,用一副牵开器使腹腔保持打开状态。清除掉左右肾的肾静脉和动脉上下的结缔组织,使用微动脉瘤夹钳紧紧夹住肾蒂55分钟。这种一段时间的缺血最初是根据在实验室中进行的先期研究来进行的(Zhou等人, J. Clin. Invest. 105:1363-71 (2000))。另外,在缺血滴定后选择55分钟的标准缺血时间,并且发现55分钟造成仍可逆转的稳定损伤,死亡率低,为5%以下。闭塞后,将0.4 ml热的盐水(37℃)注入腹腔内,然后闭合腹部达缺血时间。取出微动脉瘤夹钳后,观察肾脏直到变色,这是血液回流到肾脏的表示。另将0.4 ml热的盐水注入腹腔内,将切口缝合,之后把动物放回其笼中。取出夹钳后24小时采集尾部血样,48小时时处死小鼠,再次收集血样。
肾损伤的评价:在6只雄性MASP-2(-/-)和6只野生型(WT) (+/+)小鼠再灌注后24小时和48小时,对肾功能进行了评价。通过质谱法测定血液肌酸酐测量值,这提供了可再现的肾功能指标(灵敏度< 1.0 μmol/L)。图12通过图示说明了在再灌注后24小时和48小时,野生型C57Bl/6对照和MASP-2 (-/-)的血尿素氮清除率。如图12所示,与野生型对照小鼠相比,MASP-2(-/-)小鼠在24小时和48小时时呈现出血尿素的量显著降低,这表明在缺血再灌注损伤模型中对肾损伤的保护性功能作用。
总的来说,在手术程序和缺血损伤后24小时和48小时,在WT (+/+)和MASP-2 (-/-)小鼠中均观察到血尿素增加。单独测定了非缺血WT (+/+)手术动物中的血尿素水平为5.8 mmol/L。除了图12所提供的数据外,1只MASP-2 (-/-)动物显示受到几乎完全的保护而未发生缺血损伤,24小时和48小时的值分别为6.8 mmol/L和9.6 mmol/L。这只动物被排除在组别分析之外作为可能的异常值,其中可能存在无缺血损伤。因此,图12所示最终分析包括5只MASP-2(-/-)小鼠和6只WT (+/+)小鼠,在MASP-2 (-/-)小鼠中观察到在24小时和48小时时,血尿素在统计学上有显著降低(Student t-检验p<0.05)。这些观察结果表明抑制MASP-2活性可能将对因缺血损伤所造成的肾损伤产生保护或治疗作用。
实施例13
本实施例描述了MASP-2-/-在鼠黄斑变性模型中的结果。
背景/基本原理:年龄相关性黄斑变性(AMD)是工业化世界55岁以后致盲的主要原因。AMD主要以两种形式出现:新血管性(湿性) AMD和萎缩性(干性) AMD。新血管(湿性)形式占AMD相关的严重视力丧失的90%,即使仅有~20%患有AMD的个体发展成湿性形式。AMD的临床特点包括多发性脉络膜小疣、地区性萎缩(geographic atrophy)和脉络膜新血管形成(CNV)。2004年12月,美国食品和药品管理局(FDA)批准了Macugen (倍加他尼(pegaptanib)),这是一类特异性靶向并阻断血管内皮生长因子(VEGF)的作用新的眼科药物,用于治疗湿性(新血管)形式的AMD (Ng等人, Nat Rev. Drug Discov 5:123-32 (2006))。尽管Macugen对于小部分AMD患者代表着有前景的新的治疗选择,但是对于开发这种复杂疾病的其他治疗方法的迫切需要依然存在。多项独立路线的研究表明了补体活化在AMD发病机制中的重要作用。脉络膜新血管形成(CNV)(是一种最严重的AMD形式)的发病机制可能包括补体途径的活化。
超过25年前,Ryan描述了动物CNV的激光诱导的损伤模型(Ryan, S.J., Tr. Am. Opth. Soc. LXXVII:707-745, 1979)。该模型最初是用猕猴开发的,然而,此后已经应用同一技术在各种研究动物中开发出了类似的CNV模型,包括小鼠(Tobe等人, Am. J. Pathol. 153:1641-46, 1998)。在这一模型中,使用激光凝固来破坏脉胳膜基底层(Bruch's membrane),这种做法将导致形成CNV样膜。激光诱导的模型获取了该人疾病的许多重要特征(有关最新综述可参见Ambati等人, Survey Ophthalmology 48:257-293, 2003)。现已建立了良好的激光诱导的小鼠模型,并且在大量甚至不断增加的多个研究项目中用作实验基础。一般公认的是,激光诱导的模型与人的CNV共有足够的生物相似性,使用该模型的发病机制的临床前研究和药物抑制与人的CNV有关。
方法:
按照实施例1所述方法产生MASP-2-/-小鼠,并与C57Bl/6回交10代。目前的研究对当在激光诱导的CNV的病程中对MASP-2 (-/-)和MASP-2 (+/+)雄性小鼠评价的结果进行了比较,激光诱导的CNV是一种在激光损伤后,通过激光扫描共焦显微镜来衡量组织损伤,并且通过ELISA测定视网膜色素上皮(RPE)/脉络膜的VEGF (一种涉及CNV的有效血管生成因子)水平,焦点集中在激光诱导的CNV体积的新血管AMD的加速模型。
诱导脉络膜新血管形成(CNV):在第0天,由对药物组分配不知情的一个人,对每只动物的双眼实施激光凝固(532 nm,200 mW,100毫秒,75 μm;Oculight GL,Iridex,Mountain View,CA)。使用裂隙灯传递系统和盖玻片作为接触透镜,按标准化方式将激光光斑照在视神经周围。激光损伤形态终点是出现空泡,这是一种被视作与脉胳膜基底层破坏有关的迹象。详细方法和终点数据评价如下。
荧光素血管造影术:在激光凝固后1周,用照相机和成像系统(TRC 50 1A照相机;ImageNet 2.01系统;Topcon,Paramus,NJ)进行荧光素血管造影术。在腹膜内注射0.1 ml 2.5%荧光素钠后,用与眼底照相机镜头接触的20-D镜头抓拍照片。未参与激光凝固或血管造影术的视网膜专家以不知情的方式一次性对荧光素血管造影照片进行评价。
脉络膜新血管形成(CNV)的体积:在激光损伤后一周,摘出双眼,用4%低聚甲醛在4℃下固定30分钟。通过除去前节(anterior segment)获得眼杯,在PBS中洗涤3次,随后通过甲醇系列脱水和再水化。用缓冲液(含有1%牛血清白蛋白和0.5% Triton X-100的PBS)在室温下封闭两次30分钟后,眼杯与稀释于含有0.2% BSA和0.1% Triton X-100的PBS中的0.5% FITC-同工凝集素B4 (Vector laboratories,Burlingame,CA)一起在4℃下孵育过夜,FITC-同工凝集素B4能结合内皮细胞表面的末端β-D-半乳糖残基,并选择性地标记鼠血管系统。用含有0.1% Triton X-100的PBS洗涤2次后,轻轻剥离视网膜感觉神经层(neurosensory retina)并切断视神经。切开4个松驰放射状切口,将剩余的RPE-脉络膜-巩膜复合体平放在antifade培养基(Immu-Mount Vectashield Mounting Medium;Vector Laboratories)中封固,盖上盖玻片。
平放样本(Flatmount)用激光扫描共焦显微镜(TCS SP;Leica,Heidelberg,Germany)观察。通过用蓝色氩波长(488 nm)激发并捕获515 nm和545 nm之间的发射来观测血管。对于所有成像研究都使用40X油浸物镜。从RPE-脉络膜-巩膜复合体表面获取水平光学切片(l μm步进)。可以鉴定出在与病变相连的围绕脉络膜血管网上的最深焦平面,这被认定是病变的基底。在激光定向区域与这一参照平面表面上的任何血管均被断定为CNV。各切片的图像通过数字化予以保存。用显微镜软件(TCS SP;Leica)通过计算机进行的图像分析来测定与CNV相关荧光的面积。在各个水平切片上对整个荧光面积进行汇总,来用作CNV体积的指数。由对处理组分配不知情的操作人员进行成像。
因为各个激光病变发展成CNV的可能性受它所属组别的影响(小鼠、眼和激光光斑),使用具有裂区重复测量设计的线性混合模型对平均病变体积进行比较。主区(whole plot)因子是动物所属的遗传组别,而裂区(split plot)因子则为眼。测得统计上的显著性水平为0.05。用适于多重比较的Bonferroni调整(Bonferroni adjustment)构建平均值的事后比较。
VEGF ELISA。在通过12束激光光斑损伤后的3天,将RPE-脉络膜复合体于裂解缓冲液(20 mM咪唑HCl,10 mM KCl,1 mM MgCl2,10 mM EGTA,1% Triton X-100,10 mM NaF,1 mM钼酸钠和1 mM EDTA与蛋白酶抑制剂)中在冰上用超声处理15分钟。用识别所有切片变体的ELISA试剂盒(R&D Systems, Minneapolis, MN),在450-570 nm (Emax;Molecular Devices,Sunnyvale,CA)下测定上清液中的VEGF蛋白水平,并针对总蛋白均一化。由不参与激光凝固、成像或血管造影术的操作人员以不知情的方式进行重复测量。VEGF数值用至少3次独立实验的平均值+/-SEM表示,采用Mann-Whitney U检验进行比较。在P<0.05时拒绝零假设。
结果:
评价VEGF水平:
图13通过图示说明了在第0天由C57Bl6野生型和MASP-2(-/-)小鼠中分离出来的RPE-脉络膜复合体的VEGF蛋白水平。如图13A所示,对VEGF水平的评价表明,在MASP-2 (-/-)小鼠与C57bl野生型对照小鼠中VEGF的基线水平降低。图13B通过图示说明了激光诱导的损伤后第三天所测VEGF蛋白水平。如图13B所示,在野生型(+/+)小鼠中,在激光诱导的损伤之后的第三天,VEGF水平显著提高,这与已发表的研究(Nozaki等人, Proc. Natl. Acad. Sci. USA 103:2328-33 (2006))一致。然而,预料不到的是,在MASP-2 (-/-)小鼠中观察到VEGF的水平非常低。
评价脉络膜新血管形成(CNV):
除了在激光诱发的黄斑变性之后VEGF水平降低以外,在激光损伤之前和之后测定了CNV面积。图14通过图示说明了在C57bl野生型小鼠和MASP-2(-/-)小鼠中,在激光诱导的损伤后的第7天测得的CNV体积。如图14所示,与野生型对照小鼠相比,MASP-2 (-/-)小鼠的CNV面积在激光诱导的损伤后的第7天呈大约30%的减少。
这些观察结果表明在MASP (-/-)小鼠相比于野生型(+/+)对照中观察到的VEGF和CNV减少,用抑制剂阻断MASP-2可能在黄斑变性的治疗中具有保护或治疗作用。
实施例14
本实施例证明,凝血酶激活能够在生理条件下在凝集素途径激活之后发生,并且证明MASP-2牵涉的程度。在正常大鼠血清中,凝集素途径的激活导致与补体活化(以C4沉积来评估)同时发生的凝血酶激活(以凝血酶沉积来评估)。如在图15A和图15B中可见,这个系统中的凝血酶激活由MASP-2阻断性抗体(Fab2形式)抑制,显示出类似补体活化的曲线(图15A)的抑制浓度-应答曲线(图15B)。这些数据暗示,在外伤中发生时的凝集素途径的激活将在完全依赖于MASP-2的过程中导致补体系统和凝血系统两者的激活。根据推理,MASP-2阻断性抗体在过度系统性凝血例如弥散性血管内凝血的减轻病例中可证明有效,过度系统性凝血是在主要外伤病例中导致死亡率的标志之一。
实施例15
本实施例提供在MASP-2 -/-缺陷小鼠和MASP-2 +/+充足小鼠中使用弥散性血管内凝血("DIC")的局部Schwartzman反应模型以评估凝集素途径在DIC中的作用所产生的结果。
背景/基本原理:
如上文所述,MASP-2的阻断抑制凝集素途径激活并且减少过敏毒素C3a和C5a两者的生成。在体外,C3a过敏毒素能够显示为强有力的血小板凝聚体,但它们的体内牵涉较少被明确定义,并且伤口修复中的血小板物质和纤溶酶的释放可仅次要地牵涉补体C3。在本实施例中,在MASP-2(-/-)和WT(+/+)小鼠中分析了凝集素途径的作用,以便解决C3激活的延时上升是否为引起弥散性血管内凝血所必需。
方法:
如实施例1中所述产生用于本研究的MASP-2(-/-)小鼠和与C57Bl/6回交至少10代。
局部Schwartzman反应模型被用于本实验中。局部Schwartzman反应(LSR)是一种脂多糖(LPS)诱导的应答,该应答具有来自先天性免疫系统的细胞元件和体液元件的充分表征的作用。LSR对补体的依赖是非常确定的(Polak, L.,等人, Nature 223:738-739 (1969); Fong J.S.等人, J Exp Med 134:642-655 (1971))。在LSR模型中,用TNFα(500 ng,阴囊内)预处理小鼠达4小时,然后使小鼠麻醉并且准备用于提睾肌的活体显微镜检查。选择具有良好血流(1-4 mm/s)的毛细血管后微静脉(15-60 μm直径)网络用于观察。用荧光抗体处理动物以选择性标记嗜中性粒细胞或血小板。顺序扫描血管网络,并且数字记录所有血管的图像用于后面的分析。在记录微循环的基本状况之后,小鼠接受了LPS(100 μg)的单次静脉内注射,其单独注射或与下列药剂一同注射。然后每10分钟扫描同一血管网络达1小时。荧光团的特异性累积通过背景荧光的扣除来鉴定,并且通过确定图像的阈值而增强。反应的量级根据记录的图像测定。Schwartzman反应的初步量度是凝集数据。
所述研究比较了暴露于已知的补体途径耗竭剂眼镜蛇毒因子(CVF)或终末途径抑制剂(C5aR拮抗剂)的MASP-2 +/+充足或野生型小鼠。结果(图16A)证明,CVF以及C5aR拮抗剂两者均防止了脉管系统中凝集体的出现。另外,MASP-2 -/-缺陷小鼠(图16B)也证明了局部Schwartzman反应的完全抑制,支持凝集素途径的牵涉。这些结果清楚地证明MASP-2在DIC发生中的作用,并且支持利用MASP-2抑制剂治疗和预防DIC。
实施例16
本实施例描述鼠肾移植模型中的MASP-2 (-/-)小鼠的分析。
背景/基本原理:
利用小鼠模型评估MASP-2在肾移植的功能性后果中的作用。
方法:
利用将单肾同基因移植物移植至单侧肾切除后的受体小鼠中评估肾移植的功能性后果,其中使用六只WT (+/+)移植受体(B6)和六只MASP-2 (-/-)移植受体。为了评估移植肾的功能,在移植后5天从受体除去残存的天然肾,并且通过血尿素氮(BUN)水平的测定在24小时之后评估肾功能。
结果:
图17图示WT (+/+)受体和MASP-2 (-/-)受体中肾移植后6天的肾的血尿素氮(BUN)水平。如图17中所示,在WT(+/+) (B6)移植受体内观察到大大升高的BUN水平(小鼠中的正常BUN水平< 5 mM),指示肾衰竭。相反,MASP-2 (-/-)同基因移植受体小鼠显示显著较低的BUN水平,暗示改善的肾功能。已注意到,这些结果是利用来自WT(+/+)肾供体的移植物而获得的,暗示仅仅移植受体中的功能性凝集素途径的缺少就足以实现治疗益处。
总之,这些结果表明,经由MASP-2抑制的凝集素途径的瞬时抑制在肾移植中提供降低发病率和延迟的移植物功能的方法,并且此方法可能适用于其他移植情况。
实施例17
本实施例证明,MASP-2 (-/-)小鼠在鼠多微生物败血症腹膜炎模型中是抗败血症休克的。
背景/基本原理:
为了评估MASP-2 (-/-)在感染中的潜在作用,评估了盲肠结扎和穿刺(CLP)模型,其为一种多微生物败血症腹膜炎的模型。这个模型被认为最精确地模拟人败血症腹膜炎的过程。盲肠结扎和穿刺(CLP)模型是其中盲肠结扎和由针穿刺、导致细菌连续渗漏进入腹腔、细菌经由淋巴引流到达血液并且然后分布至所有腹器官中、导致多器官衰竭和败血症休克的模型(Eskandari等人, J Immunol 148(9):2724-2730 (1992))。CLP模型模拟患者中观察到的败血症的过程,并且诱发早期高炎性应答,继之以明显的低炎性阶段。在这个阶段期间,动物对细菌攻击高度敏感(Wichterman等人, J. Surg. Res. 29(2):189-201 (1980))。
方法:
在WT (+/+) (n=18)和MASP-2 (-/-) (n=16)小鼠中测定利用盲肠结扎和穿刺(CLP)模型的多微生物感染的死亡率。简述之,将MASP-2缺陷小鼠和它们的野生型同窝小鼠麻醉,并且取出盲肠并且在远端上方30%处结扎。其后,用0.4 mm直径的针穿刺盲肠一次。然后将盲肠放到腹腔中,并且用夹具闭合皮肤。在CLP后14天的时期内监测经受了CLP的小鼠的存活。CLP后16小时在小鼠中收集腹膜灌洗液以测定细菌载量。腹膜灌洗液的连续稀释物在PBS中制备并且在Mueller Hinton板中接种,随后在厌氧条件下37℃孵育达24小时,之后测定细菌载量。
在肺和脾中CLP之后16小时,对细菌感染的TNF-α细胞因子应答也在WT(+/+)和MASP-2(-/-)小鼠中经由定量实时聚合酶链式反应(qRT - PCR)测定。在WT(+/+)和MASP-2(-/-)小鼠中CLP之后16小时,TNF-α的血清水平也通过夹心ELISA定量。
结果:
图18将CLP处理的动物的百分比存活图示为CLP操作之后天数的函数。如图18中所示,与WT (+/+)小鼠相比,MASP-2 (-/-)小鼠中的凝集素途径缺乏不增加利用盲肠结扎和穿刺模型的多微生物感染后的小鼠的死亡率。然而,如图19中所示,与它们的WT (+/+)同窝小鼠相比时,MASP-2 (-/-)小鼠显示了CLP之后腹膜灌洗液中显著更高的细菌载量(细菌数增加大约1000倍)。这些结果表明,MASP-2 (-/-)缺陷小鼠是抗败血症休克的。此模型中的MASP-2缺陷小鼠中减少的细菌清除可能归因于削弱的C3b介导的吞噬作用,因为证明了C3沉积是MASP-2依赖性的。
已测定,与WT (+/+)对照相比,MASP-2 (-/-)小鼠中的对细菌感染的TNF-α细胞因子应答没有升高(未显示数据)。同样已测定,与TNF-α的血清水平保持几乎不变的MASP-2 (-/-)小鼠相反,在CLP后的16小时,WT (+/+)小鼠中存在显著更高的TNF-α的血清浓度。这些结果暗示,对败血症状况的强烈炎性应答在MASP-2 (-/-)小鼠中得到缓和,并且允许动物在更高细菌计数的情况下存活。
总之,这些结果证明就败血症而言凝集素途径补体活化的潜在有害效应和压倒性败血症的患者中增加的死亡率。这些结果进一步证明,MASP-2缺乏调节炎性免疫反应并且降低败血症期间炎性介质的表达水平。因此认为,通过施用针对MASP-2的抑制性单克隆抗体抑制MASP-2 (-/-)将有效减少患有败血症休克的受试者中的炎性应答。
实施例18
本实施例描述鼠鼻内感染性模型中MASP-2 (-/-)小鼠的分析。
背景/基本原理:
绿脓假单胞菌(Pseudomonas aeruginosa)是一种革兰氏阴性机会性人细菌性病原体,该病原体引起大范围的感染,特别是在免疫受损的个体中。它是获得性医院感染的主要来源,特别是医院获得性肺炎。它也在囊性纤维化(CF)患者中造成显著的发病率和死亡率。绿脓假单胞菌肺感染的特征在于强的嗜中性粒细胞募集和导致广泛组织损伤的显著的肺脏炎症(Palanki M.S.等人, J. Med. Chem 51:1546-1559 (2008))。
在本实施例中,进行研究以确定MASP-2 (-/-)小鼠中的凝集素途径的去除是否增加小鼠对细菌感染的易感性。
方法:
用绿脓假单胞菌菌株的鼻内施用攻击22只WT (+/+)小鼠、22只MASP-2 (-/-)小鼠和11只C3 (-/-)小鼠。所述小鼠在感染后受监测超过六天,并且构建Kaplan- Mayer曲线,显示百分比存活。
结果:
图20是感染后六天WT (+/+)、MASP-2 (-/-)或C3 (-/-)小鼠的百分比存活的Kaplan-Mayer曲线。如图20中所示,与WT (+/+)小鼠比较,在MASP-2 (-/-)小鼠中未观察到差异。然而,C3 (-/-)小鼠中经典(C1q)途径的去除导致对细菌感染的严重的易感性。这些结果证明,MASP-2抑制不增加对细菌感染的易感性,表明有可能通过抑制MASP-2来减少外伤患者中不希望的炎性并发症而不减损患者利用经典补体途径对抗感染的能力。
实施例19
本实施例描述如实施例10中所述鉴定的代表性高亲和力抗MASP-2 Fab2抗体的药效动力学分析。
背景/基本原理:
如实施例10中所述,为了鉴定阻断大鼠凝集素途径的高亲和力抗体,利用大鼠MASP-2蛋白淘选噬菌体展示文库。这个文库经设计以提供高免疫多样性并且完全利用人类免疫球蛋白基因序列而构建。如实施例10中所述,通过ELISA筛选鉴定了大约250个独立的噬菌体克隆,所述噬菌体克隆以高亲和力与大鼠MASP-2蛋白结合。这些克隆的测序鉴定了50个独特的MASP-2抗体编码噬菌体。由这些克隆表达Fab2蛋白,纯化和分析Fab2蛋白的MASP-2结合亲和力和凝集素补体途径功能性抑制。
如实施例10的表6中所示,具有功能性阻断活性的17个抗MASP-2 Fab2被鉴定为此分析的结果(对于阻断性抗体而言,34%命中率)。凝集素补体途径通过Fab2的功能性抑制在C4沉积的水平上是明显的,C4沉积是由MASP-2进行的C4裂解的直接量度。重要地,当评估C3转化酶活性时,抑制同样明显,证明凝集素补体途径的功能性阻断。如实施例10中所述鉴定的所述17个MASP-2阻断性Fab2有力地抑制C3转化酶形成,其IC50值等于或小于10 nM。所鉴定的17个Fab2中的八个具有亚纳摩尔范围内的IC50值。此外,所有17个MASP-2阻断性Fab2在凝集素途径C3转化酶试验中给出C3转化酶形成的基本完全的抑制,如图8A-C中所示,并且概括在实施例10的表6中。此外,表6中所示的17个阻断性抗MASP-2 Fab2中的每一个有力地抑制C3b生成(>95%),因而证明本试验针对凝集素途径C3转化酶的特异性。
大鼠IgG2c和小鼠IgG2a全长抗体同种型变体来源于Fab2 #11。本实施例描述这些同种型对于药效动力学参数的体内表征。
方法:
如实施例10中所述,大鼠MASP-2蛋白用于淘选 Fab噬菌体展示文库,Fab2#11是从该文库中鉴定的。大鼠IgG2c和小鼠IgG2a全长抗体同种型变体来源于Fab2 #11。大鼠IgG2c和小鼠IgG2a全长抗体同种型均如下在体内对于药效动力学参数进行表征。
小鼠中的体内研究:
在小鼠中进行药效动力学研究,以研究抗MASP-2抗体给药对体内血浆凝集素途径活性的作用。在此研究中,在凝集素途径试验中,在小鼠抗MASP-2 MoAb(来源于Fab2#11的小鼠IgG2a全长抗体同种型)的0.3 mg/kg或1.0 mg/kg的皮下(sc)和腹膜内(ip)施用之后的不同时间点离体测定C4沉积。
图21图示说明凝集素途径特异性C4b沉积,其在0.3 mg/kg或1.0 mg/kg小鼠抗MASP-2 MoAb皮下给药之后的不同时间点取自小鼠(n=3只小鼠/组)的未稀释的血清样品中离体测定。在抗体给药之前收集的来自小鼠的血清样品充当阴性对照(100%活性),而体外补充了100 nM的同样的阻断性抗MASP-2抗体的血清用作阳性对照(0%活性)。
图21中所示的结果证明在小鼠抗MASP-2 MoAb的1.0 mg/kg剂量皮下施用之后的C4b沉积的快速和完全的抑制。在小鼠抗MASP-2 MoAb的0.3 mg/kg剂量的皮下施用之后,观察到C4b沉积的部分抑制。
小鼠抗MASP-2 MoAb在小鼠中以0.6 mg/kg单次腹膜内施用之后,跟踪凝集素途径恢复的时间过程达三个星期。如图22中所示,凝集素途径活性的急剧下降发生在抗体给药之后,继之以腹膜内施用后持续约7天的完全的凝集素途径抑制。经过第二和第三周,观察到凝集素途径活性的缓慢恢复,小鼠中完全的凝集素途径恢复则是在抗MASP-2 MoAb施用后17天。
这些结果证明,来源于Fab2 #11的小鼠抗MASP-2 MoAb在全身递送时以剂量响应的方式抑制小鼠的凝集素途径。
实施例20
本实施例描述对来源于Fab2 #11的小鼠抗MASP-2 MoAb在年龄相关黄斑变性小鼠模型中效力的分析。
背景/基本原理:
如实施例10中所述,大鼠MASP-2蛋白用于淘选 Fab噬菌体展示文库,Fab2#11从该文库中鉴定为功能活性抗体。大鼠IgG2c和小鼠IgG2a同种型的全长抗体由Fab2 #11产生。如实施例19中所述表征了小鼠IgG2a同种型的全长抗MASP-2抗体的药效动力学参数。在本实施例中,在年龄相关黄斑变性(AMD)的小鼠模型中分析了来源于Fab2 #11的小鼠抗MASP-2全长抗体,该小鼠模型由Bora P.S. 等人,J Immunol 174:491-497 (2005)描述。
方法:
如实施例19中所述的来源于Fab2 #11的小鼠IgG2a全长抗MASP-2抗体同种型在年龄相关黄斑变性(AMD)的小鼠模型中测试,如实施例13中所述并具有以下修改。
小鼠抗MASP-2MoAb的施用
将两种不同剂量(0.3 mg/kg和1.0 mg/kg)的小鼠抗MASP-2 MoAb连同同种型对照 MoAb处理一起在CNV诱发之前16小时腹膜内注射至WT (+/+)小鼠(n=8小鼠/组)。
脉络膜新生血管形成(CNV)的诱发:
利用如实施例13中所述的激光凝固进行脉络膜新生血管形成(CNV)的诱发和CNV体积的测定。
结果:
图23图示说明在用同种型对照 MoAb或小鼠抗MASP-2 MoAb(0.3 mg/kg和1.0 mg/kg)处理的小鼠中激光损伤后7天测定的CNV面积。如图23中所示,在用1.0 mg/kg抗MASP-2 MoAb预处理的小鼠中,激光处理后七天观察到CNV的统计显著的(p<0.01)大约50%减少。如图23中进一步示出,观察到0.3 mg/kg剂量的抗MASP-2 MoAb对减少CNV并不有效。注意到,0.3 mg/kg剂量的抗MASP-2 MoAb显示出在皮下施用之后具有C4b沉积的部分和瞬时抑制,如实施例19中所述和图21中所示。
本实施例中所述的结果证明,利用抑制剂(诸如抗MASP-2 MoAb)的MASP-2的阻断在黄斑变性的治疗中具有预防和/或治疗作用。注意到,这些结果与实施例13中所述的在MASP-2 (-/-)小鼠中进行的研究中观察到的结果一致,其中,与野生型对照小鼠相比,在MASP-2 (-/-)小鼠中观察到激光处理后7天的CNV的30%减少。此外,本实施例中的结果进一步证明,全身递送的抗MASP-2抗体在眼内提供局部的治疗益处,从而强调全身性施用途径治疗AMD患者的潜力。概括而言,这些结果提供支持在AMD的治疗中利用MASP-2 MoAb的证据。
实施例21
本实施例证明,与野生型对照小鼠相比,MASP-2缺陷小鼠受到保护而免于脑膜炎奈瑟氏菌感染后的脑膜炎奈瑟氏菌诱发的死亡率,并且具有增强的菌血症清除。
基本原理:脑膜炎奈瑟氏菌是异养革兰氏阴性双球菌,它在脑膜炎及其他形式的脑膜炎球菌疾病(诸如脑膜炎球菌血症)中的作用是已知的。脑膜炎奈瑟氏菌是儿童期间的致病率和死亡率的主要诱因。严重的并发症包括败血症、华-弗综合征(Waterhouse-Friderichsen syndrome)、肾上腺机能不全和弥散性血管内凝血(DIC)。参见例如Rintala E.等人, Critical Care Medicine28(7):2373-2378 (2000)。在本实施例中,在MASP-2 (-/-)和WT (+/+)小鼠中分析凝集素途径的作用,以便解决MASP-2缺陷小鼠是否会对脑膜炎奈瑟氏菌诱发的死亡率敏感。
方法:
如实施例1中所述产生MASP-2敲除小鼠,并且与C57Bl/6回交至少10代。10周大的MASP-2 KO小鼠(n=10)和野生型C57/B6小鼠(n=10)通过静脉内注射接种5×108 cfu/100 μl、2×108 cfu/100 μl或3×107 cfu/100 μl的溶于400 mg/kg右旋糖酐铁的脑膜炎奈瑟氏菌血清群A Z2491的剂量。监测感染后的小鼠的存活超过72小时时间周期。感染后,以每小时一次的间隔从小鼠采集血样,并且分析所述血样以测定脑膜炎奈瑟氏菌的血清水平(log cfu/ml),以便验证感染并且测定细菌从血清的清除率。
结果:
图24A图示说明在5×108/100 μl cfu感染剂量的脑膜炎奈瑟氏菌施用之后,MASP-2 KO小鼠和WT小鼠的百分比存活。如图24A中所示,在用5×108/100 μl cfu的最高剂量的脑膜炎奈瑟氏菌感染之后,100%的MASP-2 KO小鼠在感染后的72小时期间自始至终存活。相反,仅20%的WT小鼠在感染后的24小时仍然活着。这些结果证明,MASP-2缺陷小鼠受到保护而免于脑膜炎奈瑟氏菌诱发的死亡率。
图24B图示说明取自用5×108 cfu/100 μl脑膜炎奈瑟氏菌感染的MASP-2 KO小鼠和WT小鼠的血样中在不同时间点回收的脑膜炎奈瑟氏菌的log cfu/ml。如图24B中所示,在WT小鼠中,血液中的脑膜炎奈瑟氏菌水平在感染后24小时达到约6.5 log cfu/ml的峰值,并且在感染后48小时下降到零。相反,在MASP-2 KO小鼠中,脑膜炎奈瑟氏菌水平在感染后6小时达到约3.5 log cfu/ml的峰值,并且在感染后36小时下降到零。
图25A图示说明MASP-2 KO小鼠和WT小鼠在由2×108 cfu/100 μl脑膜炎奈瑟氏菌感染之后的百分比存活。如图25A中所示,在用2×108 cfu/100 μl剂量的脑膜炎奈瑟氏菌感染之后,100%的MASP-2 KO小鼠在感染后的72小时期间自始至终存活。相反,仅80%的WT小鼠在感染后24小时仍然活着。与图24A中所示的结果一致,这些结果进一步证明,MASP-2缺陷小鼠受到保护而免于脑膜炎奈瑟氏菌诱发的死亡率。
图25B图示说明取自用2×108 cfu/100 μl脑膜炎奈瑟氏菌感染的WT小鼠的血样中在不同时间点回收的脑膜炎奈瑟氏菌的log cfu/ml。如图25B中所示,用2×108 cfu感染的WT小鼠的血液中的脑膜炎奈瑟氏菌水平在感染后12小时达到约4 log cfu/ml的峰值,并且在感染后24小时下降到零。图25C图示说明取自用2×108 cfu/100 μl脑膜炎奈瑟氏菌感染的MASP-2 KO小鼠的血样中在不同时间点回收的脑膜炎奈瑟氏菌的log cfu/ml。如图25C中所示,用2×108 cfu感染的MASP-2 KO小鼠的血液中的脑膜炎奈瑟氏菌水平在感染后2小时达到约3.5 log cfu/ml的峰水平,并且在感染后3小时下降到零。与图24B中所示的结果一致,这些结果证明,尽管用与WT小鼠相同剂量的脑膜炎奈瑟氏菌感染MASP-2 KO小鼠,但与WT相比,MASP-2 KO小鼠具有增强的菌血症清除。
在用3×107 cfu/100 μl的最低剂量的脑膜炎奈瑟氏菌感染之后,MASP-2 KO 和WT小鼠的百分比存活在72小时时期处为100%(未显示数据)。
讨论
这些结果显示,与WT小鼠相比,MASP-2缺陷小鼠受到保护而免于脑膜炎奈瑟氏菌诱发的死亡率,并且具有增强的菌血症清除。因此,鉴于这些结果预期,MASP-2抑制剂(诸如MASP-2 MoAb)的治疗应用将被期待以有效治疗、预防或减轻脑膜炎奈瑟氏菌感染的影响(即,败血症和DIC)。此外,这些结果表明,MASP-2抑制剂(诸如MASP-2 MoAb)的应用不会将受试者预置于染上脑膜炎奈瑟氏菌感染的增大的风险中。
实施例22
本实施例描述补体C3的新的凝集素途径介导的和MASP-2依赖性C4旁路活化的发现。
基本原理:
利用补体活化的抑制剂来限制心肌缺血/再灌注损伤(MIRI)的主要治疗益处在二十年前在心肌梗死的实验动物模型中令人信服地得到证实:静脉内给予重组sCR1(细胞表面补体受体1型(CR1)的可溶性截短衍生物1(CR1)),并且在MIRI的大鼠体内模型中评估其效果。用sCR1治疗使梗死体积减少超过40%(Weisman, H.F.,等人, Science 249:146-151 (1990))。该重组抑制剂的治疗潜能随后在临床试验中证明,所述临床试验显示在患有MI的患者中施用sCR1阻止了缺血后心脏中的收缩衰竭(Shandelya, S.,等人, Circulation 87:536-546 (1993))。然而,导致缺血组织中补体活化的主要机制还没有最终确定,主要是由于缺乏合适的实验模型,对于导致缺氧细胞的补体活化的分子过程的理解有限,和不同补体活化途径之间的交叉对话(cross-talk)和协同作用。
作为免疫应答的基本组分,补体系统通过抗体依赖性和非依赖性机制提供了针对入侵微生物的保护。其精心安排免疫应答中的许多细胞和体液相互作用,包括趋化性、吞噬作用、细胞粘附和B细胞分化。三种不同的途径引发补体级联:经典途径、替代途径和凝集素途径。经典途径识别亚组分C1q结合到各种目标 - 最显著的是免疫复合物 – 以起始相关丝氨酸蛋白酶C1r和C1s的逐步活化,为适应性免疫系统参与后的病原体和免疫复合物清除提供了主要机制。C1q与免疫复合物的结合将C1r酶原二聚体转换成其活性形式,以裂解并因此活化C1s。C1s在两个裂解步骤将Clq结合转化为补体活化:其首先将C4转化为C4a和C4b,然后裂解C4b结合的C2,以形成C3转化酶C4b2a。该复合物将丰富的血浆组分C3转化为C3a和C3b。C4b2a 复合物紧密邻近处C3b的聚集将对C3的底物特异性转变为对C5的底物特异性,以形成C5转化酶C4b2a(C3b)n。通过经典途径活化生成的C3和C5转化酶复合物与通过凝集素途径活化途径生成的那些是相同的。在替代途径中,组分C3的自发低水平水解导致蛋白片段沉积到细胞表面,触发外来细胞上的补体活化,而宿主组织上的细胞相关的调节蛋白避免活化,因此防止自我损伤。像替代途径,凝集素途径可以在免疫复合物不存在的情况下活化。活化通过多分子凝集素途径活化复合物与病原体相关分子模式(PAMPs),主要是存在于细菌、真菌或病毒性病原体上的碳水化合物结构或存在于凋亡、坏死、恶性或缺氧细胞上的异常糖基化模式的结合而起始(Collard, C.D.,等人, Am. J. Pathol. 156:1549-1556 (2000); Walport, M.J., N. Engl. J. Med. 344:1058-1066 (2001); Schwaeble, W.,等人, Immunobiology 205:455-466 (2002); 和Fujita, T., Nat. Rev. Immunol. 2:346-353 (2002))。
甘露聚糖结合凝集素(MBL)是显示与一组新的丝氨酸蛋白酶(命名为MBL-相关丝氨酸蛋白酶(MASPs))形成复合物的第一碳水化合物识别亚组分,并且根据其发现的顺序编号(即,MASP-1、MASP-2和MASP-3)。在人中,凝集素途径活化复合物可以由具有不同碳水化合物结合特异性的四种替代的碳水化合物识别亚组分(即,MBL 2和纤维胶凝蛋白 (ficolin)家族的三个不同成员,即L-纤维胶凝蛋白、H-纤维胶凝蛋白和M-纤维胶凝蛋白)和MASPs形成。在小鼠和大鼠血浆中,两种形式的MBL(MBL A和MBL C)和纤维胶凝蛋白 -A与MASPs形成凝集素活化途径复合物。我们先前已经在人、小鼠和大鼠中克隆和表征了MASP-2和被称为MAp19或sMAP的19 kDa的额外截短的MASP-2基因产物(Thiel, S.,等人, Nature 386:506-510 (1997);. Stover, C.M.,等人, J. Immunol. 162:3481-3490 (1999); Takahashi, M.,等人, Int. Immunol. 11:859-863 (1999);和Stover, C.M.,等人, J. Immunol. 163:6848-6859 (1999))。MAp19/ sMAP缺乏蛋白酶活性,但可以通过竞争MASPs与碳水化合物识别复合物的结合而调节凝集素途径活化(Iwaki, D.等人, J. Immunol. 177:8626-8632 (2006))。
有证据表明,在三种MASPs中,只有MASP-2对于将凝集素途径识别复合物的结合转化为补体活化是需要的(Thiel, S.,等人 (1997); Vorup-Jensen, T.,等人, J. Immunol. 165:2093-2100 (2000); Thiel, S.,等人, J. Immunol. 165:878-887 (2000); Rossi, V.,等人, J. Biol. Chem. 276:40880-40887 (2001))。该结论被最近描述的MASP-1和MASP-3缺陷小鼠品系的表型所强调。除了凝集素途径介导的补体活化的发生在体外延迟以外,MASP-1/3缺陷小鼠保留了凝集素途径的功能活性。用重组MASP-1重构MASP-1和MASP-3缺陷血清克服了该凝集素途径活化的延迟,暗示MASP-1可以促进MASP-2活化(Takahashi, M.,等人, J. Immunol. 180:6132-6138 (2008))。一项最近的研究已经显示,MASP-1(可能还有MASP-3)对于将替代途径活化酶因子D从其酶原形式转换为其酶促活性形式是需要的(Takahashi, M.,等人, J. Exp. Med. 207:29-37 (2010))。该过程的生理重要性通过在MASP-1/3-缺陷小鼠的血浆中不存在替代途径功能活性而得到强调。
新近生成的具有组合的凝集素途径碳水化合物识别亚组分MBL A和MBL C的靶向缺陷的小鼠品系仍可以通过剩余的鼠凝集素途径识别亚组分纤维胶凝蛋白 A起始凝集素途径活化(Takahashi, K.,等人, Microbes Infect. 4:773-784 (2002))。在MASP-2缺陷小鼠中不存在任何残余凝集素途径功能活性为研究先天体液免疫的该效应臂在健康和疾病中的作用提供了结论性的模型。
C4和MASP-2缺陷小鼠品系的可用性允许我们定义新型凝集素途径特异性、但MASP-2依赖的补体C3的C4-旁路活化途径。该新的凝集素途径介导的C4旁路活化途径对缺血后组织损失的重要贡献通过MIRI 中MASP-2缺陷的显著保护表型而强调,而在相同模型中测试的C4缺陷小鼠则没有显示保护。
在本实施例中,我们描述了补体C3的新的凝集素途径介导的和MASP-2依赖性C4旁路活化。该新的活化途径的生理相关性通过在心肌缺血/再灌注损伤(MIRI)的实验模型中MASP-2缺陷的保护表型而建立,其中C4缺陷动物则没有受保护。
方法:
MASP-2缺陷小鼠没有显示严重的异常情况。MASP-2缺陷小鼠如实施例1中所述生成。杂合(+/-)和纯合(-/-) MASP-2缺陷小鼠两者是健康和可育的,并且显示没有严重的异常情况。它们的平均预期寿命类似于它们的WT同窝小鼠的平均预期寿命(> 18个月)。在疾病的实验模型中研究这些小鼠的表型之前,将我们的MASP-2-/-系回交11代至C57BL/6背景上。MASP-2 mRNA的完全不存在通过聚A+选择的肝RNA制剂的Northern印迹证实,而编码MAp19或sMAP (MASP2基因的截短的替代剪接产物)的1.2kb mRNA是大量表达的。
使用对于MASP-2(B链)的丝氨酸蛋白酶结构域的编码序列或A链的编码序列的剩余部分特异性的引物对的qRT-PCR分析显示,在MASP-2 -/-小鼠中没有检测到编码B链的mRNA,而破坏的A链mRNA转录物的丰度显著增加。同样,编码MAp19/sMAP的mRNA的丰度在MASP-2 +/-和MASP-2 -/-小鼠中增加。对于各基因型的5只动物通过ELISA确定的血浆MASP-2水平,对于WT对照为300ng/ml(范围260-330ng/ml),对于杂合小鼠为360ng/ml(范围330-395ng/ml),并且在MASP-2 -/-小鼠中检测不到。使用qRT-PCR,确立了mRNA表达谱,表明MASP-2-/-小鼠以类似于它们的MASP-2充足同窝小鼠的丰度的丰度表达MBL A、MBL C、纤维胶凝蛋白 A、MASP-1、MASP-3、C1q、C1rA、C1sA、因子B、因子D、C4、和C3的mRNA(数据未显示)。
MASP-2-/- (n=8)和MASP-2+/+ (n=7)同窝小鼠的血浆C3水平使用商售的小鼠C3 ELISA试剂盒(Kamiya, Biomedical, Seattle, WA)来测量。MASP-2缺陷小鼠的C3水平(平均0.84 mg/ml, +/- 0.34)类似于WT对照的C3水平(平均0.92, +/- 0.37)。
结果:
MASP-2对于凝集素途径功能活性是必不可少的。
如实施例2中描述和图5中显示,MASP-2-/-血浆的体外分析显示完全不存在用于活化C4的活化甘露聚糖和酵母聚糖包被的表面上的凝集素途径功能活性。同样,在用N-乙酰葡糖胺包被的表面上都没有在MASP-2-/-血浆中检测到凝集素途径依赖的C4和C3裂解,所述N-乙酰葡糖胺结合MBL A、MBL C和纤维胶凝蛋白 A并通过MBL A、MBL C和纤维胶凝蛋白 A触发活化(数据未显示)。
MASP-2-/-小鼠的血清和血浆的分析清楚地表明,MASP-2对于通过凝集素途径活化补体基本上是需要的。然而,凝集素途径功能活性的总体缺陷使其他补体活化途径完整:MASP-2-/-血浆仍然可以通过经典(图26A )和替代途径(图26B)活化补体。在图26A和26B中,符号“ * ”符号表示来自WT (MASP-2 (+/+))的血清;符号“ ● ”表示来自WT(C1q耗尽)的血清;符号“ □ ”表示来自MASP-2 (-/-)的血清;并且“?”表示来自MASP-2 (-/-) (C1q耗尽)的血清。
图26A图示说明MASP-2-/-小鼠保留功能性经典途径:C3b沉积在用免疫复合物包被的微量滴定板(通过用BSA包被然后添加山羊抗BSA IgG而原位生成)上进行测定。图26B图示说明MASP-2缺陷小鼠保留功能性替代途径:C3b沉积在仅允许替代途径活化(含有Mg2+和EGTA的缓冲液)的条件下在酵母多糖包被的微量滴定板上进行测定。图26A和图26B中显示的结果是重复的平均值,并且是三次独立实验的典型结果。整个过程中对于血浆来源使用相同的符号。这些结果显示,功能性替代途径存在于MASP-2缺陷小鼠,如在设计用于直接触发替代途径、同时失活经典途径和凝集素途径的实验条件下的图26B显示的结果中所证明的。
补体活化的凝集素途径关键性地有助于心肌缺血/再灌注损伤(MIRI)中的炎性组织损失。
为了研究凝集素途径功能活性对MIRI的贡献,我们在MIRI模型中在冠状动脉的左前降支(LAD)的短暂结扎和再灌注之后比较MASP-2-/-小鼠和WT同窝小鼠对照。补体C4的存在与否对MIRI中的缺血组织损失的程度没有影响。我们评估了C4缺陷对实验MIRI之后的梗死面积的影响。如图27A和图27B中显示,在C4-缺陷小鼠和它们的WT同窝小鼠两者中观察到相同的梗死尺寸。图27A图示说明在C4-/-小鼠(n=6)和匹配的WT同窝小鼠对照(n=7)中在LAD结扎和再灌注之后的MIRI诱导的组织损失。图27B图示说明作为AAR的函数的INF,其清楚地证明,C4-/-小鼠和它们的WT对照(虚线)一样易受MIRI的影响。
这些结果证明,C4缺陷小鼠没有被保护免于MIRI。该结果是意想不到的,因为它与广泛接受的观点(即主要C4活化片段C4b是经典途径和凝集素途径C3转化酶C4b2a的重要组分)冲突。因此,我们评估在C4缺陷小鼠和人血浆中是否可以检测到补体C3的残余凝集素途径特异性活化。
凝集素途径可以在C4不存在的情况下通过新的MASP-2依赖性C4旁路活化途径活化补体C3。
受表明C4缺陷豚鼠血清中C4旁路活化途径的存在的历史报道(May, J.E.,和M. Frank, J. Immunol. 111:1671-1677 (1973))的鼓励,我们在排除替代途径的贡献的途径特异性测定条件下分析C4缺陷小鼠是否可以具有残余的经典或凝集素途径功能活性并且监测C3的活化。
C3b沉积使用在禁止替代途径活化的血浆浓度的(1.25%及以下)重新钙化血浆在酵母多糖包被的微量滴定板上进行测定。尽管在用于测试经典途径活化的C4缺陷血浆中没有检测到C3的裂解(数据未显示),但是当通过凝集素途径起始补体活化时,在C4缺陷小鼠血浆中观察到强烈的残余C3裂解活性。凝集素途径依赖性通过在C4缺陷血浆稀释物与可溶性甘露聚糖预孵育之后竞争性抑制C3裂解而证明(参见图28A)。如图28A-D中显示,在C4不存在的情况下观察到C3的MASP-2依赖性活化。图28A图示说明C4+/+ (交叉线)和C4-/-(空心圆)小鼠血浆的C3b沉积。在测定之前将C4-/-血浆与过量(1 μg/ml)流体相甘露聚糖的预孵育完全抑制C3沉积(实心圆)。结果是三次独立实验的典型结果。图28B图示说明以下实验的结果,其中将野生型、MASP-2缺陷(空心方形)和C4-/-小鼠血浆(1%)与各种浓度的抗大鼠MASP-2 mAbM11(横坐标)混合,并且在甘露聚糖包被板上测定C3b沉积。结果是4次测定(每种类型的血浆的2次重复)的平均值(± SD)。图28C图示说明以下实验的结果,其中人血浆为合并的NHS(交叉线)、C4-/-血浆(空心圆)和与1 μg/ml甘露聚糖预孵育的C4-/-血浆(实心圆)。结果是三次独立实验的代表结果。图28D图示说明抗人MASP-2 mAbH3 对C4充足和C4缺陷的人血浆(1%)中的C3b沉积的抑制(三次重复的平均值±SD)。如图28B中显示,在平行测定的MASP-2-/-血浆中没有检测到凝集素途径依赖性C3活化,意味着该C3的C4-旁路活化途径是MASP-2依赖的。
为了进一步证实这些发现,我们通过针对重组人和大鼠MASP-2A(其中通过位点定向诱变用丙氨酸残基替换活性蛋白酶结构域的丝氨酸残基,以防止抗原的自溶降解)的亲和筛选建立了从噬菌体展示抗体文库分离的一系列重组抑制性mAbs。使用作为抗原的重组人和大鼠MASP-2A(Chen, C.B.和Wallis, J. Biol. Chem. 276:25894-25902 (2001)),从组合抗体文库(Knappik, A.,等人, J. Mol. Biol. 296:57-86 (2000))分离针对MASP-2的重组抗体(AbH3和AbM11)。将在小鼠血浆中有效抑制C4和C3的凝集素途径介导的活化的抗大鼠Fab2片段(IC50~1 nM)转化为全长IgG2a抗体。在大鼠中产生多克隆抗鼠MASP-2A抗血清。这些工具允许我们确认MASP-2对该C3的新的凝集素途径特异性的C4-旁路活化途径的依赖性,如下面进一步描述。
如图28B中显示,其选择性结合小鼠和大鼠MASP-2的抑制性单克隆抗体M211以类似的IC50值以浓度依赖的方式通过凝集素途径抑制C4缺陷小鼠中C3的C4旁路活化以及WT小鼠血浆的C3活化。所有测定均在使替代途径活化途径失去功能的高血浆稀释度时实施(具有作为1.25%的最高血浆浓度)。
为了研究人中C3的类似凝集素途径特异性C4-旁路活化的存在,我们分析了两种人C4基因(即,C4A和C4B)遗传缺陷的供体的血浆,导致C4的完全不存在(Yang, Y.,等人, J. Immunol. 173:2803-2814 (2004))。图28C显示,该患者的血浆在高血浆稀释度(使替代活化途径失去功能)下有效地活化C3。通过添加过量浓度的流体相甘露聚糖,在鼠C4-缺陷血浆(图28A )和人C4缺乏血浆(图28C)中证明了甘露聚糖包被板上C3活化的凝集素途径特异性的模式。使用特异性地结合人MASP-2并且去除MASP-2功能活性的单克隆抗体AbH3评估人C4缺陷血浆中该C3活化机制的MASP-2依赖性。如图28D中显示,AbH3在C4充足和C4缺陷人血浆中以相当的效力抑制C3b (和C3dg)的沉积。
为了评估其他补体组分在C3的C4旁路活化中可能的作用,我们在凝集素途径特异性和经典途径特异性测定条件两者下测试MASP-1/3-/-和Bf/C2-/-小鼠的血浆连同MASP-2-/-、C4-/-和C1q-/-血浆(作为对照)。将针对当使用WT血浆时沉积的C3量的C3裂解的相对量进行作图。
图29A图示说明在凝集素活化途径或经典活化途径特异性测定条件下测试的来自各种补体缺陷小鼠品系的血浆中C3转化酶活性的比较分析。平行测试WT小鼠(n=6)、MASP-2-/-小鼠(n=4)、MASP-1/3-/- 小鼠(n=2)、C4-/- 小鼠(n=8)、C4/MASP-1/3-/- 小鼠(n=8)、Bf/C2-/- (n=2) 和C1q-/- 小鼠(n=2)的稀释的血浆样品(1%)。用2.5μg/ml重组大鼠C2 (Bf/C2-/- +C2)重构Bf/C2-/-血浆恢复了C3b沉积。结果是平均值(±SD)。**p<0.01 (相比于WT血浆)。如图29A中显示,在凝集素途径特异性测定条件下测试的C4-/-血浆中看到大量C3沉积,但在经典途径特异性条件下则看不到。再次,在通过凝集素途径活化途径的MASP-2缺陷血浆中没有看到C3沉积,而相同血浆通过经典途径沉积C3。在MASP-1/3-/-血浆中,C3沉积发生在凝集素和经典途径特异性测定条件两者下。使用凝集素途径或经典途径特异性条件,在C4和MASP-1/3组合缺陷的血浆中都没有看到C3沉积。在通过凝集素途径和经典途径的C2/Bf-/-血浆中都没有看到C3沉积。然而用重组C2重构C2/Bf-/-小鼠血浆恢复了凝集素途径和经典途径介导的C3裂解。使用C1q-/-血浆验证测定条件。
图29B图示说明在凝集素活化途径特异性测定条件测试的来自各种补体缺陷小鼠品系的血浆WT、fB-/-、C4-/-、MASP-1/3-/-、和MASP-2-/-血浆中C3转化酶活性的时间分辨的动力学(1%血浆,结果是三次独立实验的代表结果)。如图29B中显示,尽管在MASP-2-/-血浆中没有看到C3裂解,但是fB-/-血浆以类似于WT血浆的动力学裂解C3。在C4-/-以及在MASP-1/3缺陷血浆中看到C3至C3b (和C3dg)的凝集素途径依赖性转化的显著延迟。新近显示MASP-1/3-/-血浆中这种C3活化的延迟是MASP-1、而非MASP-3依赖的(Takahashi, M.,等人, J. Immunol. 180:6132-6138 (2008))。
讨论:
在本实施例中描述的结果强烈表明,MASP-2功能活性对于在C4存在和不存在两者的情况下通过凝集素途径活化C3是必不可少的。此外,C2和MASP-1对于该C3的新的凝集素途径特异性的C4-旁路活化途径起作用是需要的。MASP-2-/-以及C4-/-血浆中凝集素途径功能活性的比较分析揭示了补体C3的先前未认识到的C4非依赖性、但MASP-2依赖性的活化途径的存在,并且显示在C4完全缺乏的情况下C3可以以凝集素途径依赖性模式被活化。尽管该新的MASP-2依赖的C3转化酶的详细分子组成和活化事件的顺序仍有待阐明,但是我们的结果意味着,该C4旁路活化途径还需要补体C2以及MASP-1的存在。具有C4和MASP-1/3组合缺陷的小鼠血浆中凝集素途径介导的C3裂解活性的损失可以通过最近描述的MASP-1通过MASP-2的直接裂解和活化而增强MASP-2依赖的补体活化的作用来解释(Takahashi, M.,等人, J. Immunol. 180:6132-6138 (2008))。同样,MASP-1可以通过其裂解C2的能力来帮助MASP-2功能活性(Moller-Kristensen,等人, Int. Immunol. 19:141-149 (2007))。这两种活性都可以解释MASP-1/3缺陷血浆通过凝集素活化途径裂解C3的降低速率和为什么MASP-1对于通过C4旁路活化途径来维持C3转化是需要的。
C2/fB-/-血浆无法通过凝集素途径来活化C3被显示是C2依赖性的,因为将重组大鼠C2添加至C2/fB-/-血浆恢复了重构的血浆在甘露聚糖包被的板上活化C3的能力。
C4缺陷特异性破坏经典补体活化途径而凝集素途径保留了通过MASP-2依赖的C4旁路活化途径的C3转化酶活性的生理临界水平这一发现要求再次评估凝集素途径在各种疾病模型(包括实验肺炎链球菌感染)(Brown, J. S.,等人, Proc. Natl. Acad. Sci. U. S. A. 99:16969-16974 (2002);实验性变态反应性脑脊髓炎(Boos, L.A.,等人, Glia 49:158-160 (2005);和C3依赖的鼠肝再生模型(Clark, A.,等人, Mol. Immunol. 45:3125-3132 (2008))中的作用。后一组证明,C4缺陷小鼠可以以替代途径非依赖的方式活化C3,因为因子B功能活性的抗体介导的耗尽对替代途径的体内抑制没有影响C4-/-小鼠中的C3裂解依赖的肝再生(Clark, A.,等人 (2008))。该C3的凝集素途径介导的C4旁路活化途径也可以解释在我们的MIRI模型中以及在先前描述的肾同种异体移植物排斥的模型中的C4缺陷的保护表型的缺乏(Lin, T.,等人, Am. J. Pathol. 168:1241-1248 (2006))。相反,我们新近的结果已经独立地证明在肾移植模型中MASP-2-/-小鼠的显著保护表型(Farrar, C.A.,等人, Mol. Immunol. 46:2832 (2009))。
综上所述,本实施例的结果支持以下观点,即C3的MASP-2依赖性C4旁路活化是生理相关的机制,这可能在其中C4的可用性限制C3活化的条件下是重要的。
实施例23
本实施例描述了WT (+/+)、MASP-2 (-/-)、F11 (-/-)、F11/C4 (-/-) 和C4 (-/-)小鼠中凝血酶底物对C3的活化和甘露聚糖上的C3沉积。
基本原理:
如实施例14所述,已确定凝血酶活化可以在生理条件下凝集素途径活化之后发生,并且证明MASP- 2参与的程度。C3在补体系统的活化中起着核心作用。C3活化对于经典和替代补体活化途径两者都是需要的。实施实验,以确定C3是否由凝血酶底物活化。
方法:
凝血酶底物的C3活化
在以下凝血酶底物的活化形式存在的情况下测量C3的活化:人FCXIa、人FVIIa、牛FXa、人FXa、人活化蛋白C和人凝血酶。C3与各种凝血酶底物孵育,然后在10%SDS -聚丙烯酰胺凝胶上在还原条件下分离。使用纤维素膜电泳转移后,将膜与单克隆生物素-偶联的大鼠抗小鼠C3孵育,用链霉亲和素-HRP试剂盒检测,并且用ECL试剂显色。
结果:
C3的活化涉及将完整a-链裂解成截短的a'链和可溶的C3a(在图30中未显示)。图30显示对凝血酶底物对人C3的活化的Western印迹分析的结果,其中未裂解C3α链和活化产物a'链用箭头显示。如图30中显示,C3与人凝血因子Ⅺ和因子Ⅹ的活化形式以及活化的牛凝血因子X的孵育可以在任何补体蛋白酶不存在的情况下体外裂解C3。
甘露聚糖上的C3沉积
在从WT、MASP-2 (-/-)、F11(-/-)、F11(-/-)/C4(-/-) 和C4(-/-)获得的血清样品上实施C3沉积测定。F11是编码凝血因子XI的基因。为了测量C3活化,将微量滴定板用甘露聚糖(1 μg/孔)包被,然后添加TBS/tween/Ca2+中的绵羊抗HSA血清(2 μg/ml)。板用TBS中的0.1% HSA封闭并且如上洗涤。血浆样品稀释在4 mM巴比妥、145 mM NaCl、2 mM CaCl2、1 mM MgCl2、pH 7.4中,添加至板,并且在37℃下孵育1.5小时。洗涤后,结合的C3b使用兔抗人补体C3c (Dako)和随后的碱性磷酸酶缀合的山羊抗兔IgG和pNPP进行检测。
结果:
图31显示在从WT、MASP-2 (-/-)、F11(-/-)、F11(-/-)/C4(-/-) 和C4(-/-)获得的血清样品上的C3沉积测定的结果。如图31中显示,存在功能性凝集素途径,甚至在C4完全不存在的情况下。如图31中进一步显示,该新的凝集素途径依赖的补体活化需要凝血因子XI。
讨论:
在该实验中获得的结果之前,本领域技术人员相信,补体凝集素途径对于活性需要C4。因此,来自C4敲除小鼠(和C4缺陷人)的数据用以下假设进行解释:此类生物体是凝集素途径缺陷的(除了经典途径缺陷以外)。本结果表明,该观点是错误的。因此,表明凝集素途径在基于C4缺陷动物的表型的某些疾病设置中是不重要的过去研究的结论可能是错误的。在本实施例中描述的数据也显示,在全血清的生理背景中,凝集素途径可以活化凝血级联的组分。因此,证明在涉及MASP-2的补体和凝血之间的存在交叉对话。
实施例24
本实施例描述了评价抗MASP-2抗体对来自从阵发性睡眠性血红蛋白尿症(PNH)患者获得的血液样品的红细胞的裂解的影响的方法。
背景/基本原理:
阵发性睡眠性血红蛋白尿症(PNH),也称为Marchiafava-Micheli综合征,是一种获得性的潜在威胁生命的血液疾病,其特征在于补体诱导的血管内溶血性贫血。PNH的标志是慢性血管内溶血,这是补体的替代途径的活化失调的结果。Lindorfer, M.A.,等人, Blood 115(11) (2010)。PNH中的贫血是由于血流中红细胞的破坏。PNH的症状包括红色尿(这是由于尿中出现血红蛋白)和血栓形成。PNH可以自身发生(称为“原发性PNH”)或在其他骨髓病症诸如再生障碍性贫血的背景中发生(称为“继发性PNH”)。PNH的治疗包括用于贫血的输血、用于血栓形成的抗凝和使用单克隆抗体依库丽单抗(Soliris),其通过抑制补体系统保护血细胞免于免疫破坏(Hillmen P.等人, N. Engl. J. Med. 350(6):552-9 (2004))。然而,依库丽单抗治疗的显著部分的PNH患者留下临床显著的免疫介导的溶血性贫血,因为该抗体没有阻断补体的替代途径的活化。
本实施例描述了评价抗MASP-2抗体对来自从PNH患者获得的血液样品(未用Soliris处理)并且与ABO匹配的酸化的正常人血清孵育的红细胞裂解的影响的方法。
方法:
试剂:
来自正常供体和患有PNH(未用Soliris处理)的患者的红细胞通过静脉穿刺获得,并且如Wilcox, L.A.,等人, Blood 78:820-829 (1991)(此处通过引用并入本文)中所述进行制备。具有功能性阻断凝集素途径的活性的抗MASP-2抗体如实施例10中所述生成。
溶血分析:
用于测定抗MASP-2抗体对阻断来自PNH患者的红细胞溶血的能力的影响的方法使用Lindorfer, M.A.,等人, Blood 15(11):2283-91 (2010)和Wilcox, L.A.,等人, Blood 78:820-829 (1991)(两篇参考文献都通过引用并入本文)中所述的方法来实施。如Lindorfer等人中所述,在每次实验之前,将来自PNH患者样品的红细胞离心,吸出血沉棕黄层,并且将细胞在明胶佛罗那缓冲液(GVB)中洗涤。如下测试红细胞对于APC-介导的裂解的敏感性。ABO匹配的正常人血清用含有0.15 mM CaCl2和0.5 mM MgCl2的GVB(GVB+2)稀释,并且酸化至pH 6.4 (酸化NHS,aNHS),并且用来将红细胞重构至50% aNHS中1.6%的血细胞比容。然后将混合物在37℃下孵育,并且在1小时后,通过离心沉淀红细胞。回收上清液的等分试样的光密度在405 nM处测量,并且用于计算百分比裂解。将重构在酸化血清-EDTA中的样品进行类似处理,并且用来定义背景的无补体介导的裂解(通常小于3%)。在蒸馏水中孵育红细胞之后确定完全裂解(100%)。
为了确定抗MASP-2抗体对PNH红细胞溶血的影响,在增量浓度的抗MASP-2抗体存在的情况下将来自PNH患者的红细胞在aNHS中孵育,并且随后定量溶血的存在/量。
鉴于已经显示抗MASP-2抗体阻断替代补体途径的随后活化这一事实,预期抗MASP-2抗体在阻断PNH红细胞的替代途径介导的溶血作用中将是有效的,并且将可用作治疗患有PNH的患者的治疗剂。
实施例25
本实施例描述了评价抗MASP-2阻断性抗体对从患有冷球蛋白血症的患者获得的血液样品中冷球蛋白对补体活化的影响的方法。
背景/基本原理:
冷球蛋白血症的特征在于血清中冷球蛋白的存在。冷球蛋白是在低温下经历可逆聚集的单一或混合的免疫球蛋白(通常为IgM抗体)。聚集导致经典途径补体活化和血管床、特别是外周中的炎症。冷球蛋白血症的临床表现包括血管炎和肾小球肾炎。
冷球蛋白血症可以如下基于冷球蛋白组成进行分类:I型冷球蛋白血症或简单的冷球蛋白血症是单克隆免疫球蛋白、通常是免疫球蛋白M(IgM)的结果;II型和III型冷球蛋白血症(混合型冷球蛋白血症)含有类风湿因子(RFs),这通常是与多克隆IgG的Fc部分的复合物中的IgM。
与冷球蛋白血症相关的状况包括丙型肝炎感染、淋巴增生性病症和其他自身免疫性疾病。含冷球蛋白的免疫复合物导致全身炎症的临床综合征,这可能是由于它们活化补体的能力。尽管IgG免疫复合物通常活化补体的经典途径,但是含IgM的复合物也可以通过凝集素途径活化补体(Zhang, M.,等人, Mol Immunol 44(1-3):103-110 (2007)和Zhang. M.,等人, J. Immunol. 177(7):4727-34 (2006))。
免疫组织化学研究已经进一步证明,冷球蛋白免疫复合物含有凝集素途径的组分,并且来自患有冷球蛋白血症性肾小球肾炎的患者的活检样品显示原位凝集素途径活化的免疫组织化学证据(Ohsawa, I.,等人, Clin Immunol 101(1):59-66 (2001))。这些结果表明,凝集素途径可以有助于冷球蛋白血症中的炎症和不利结果。
方法:
用于测定抗MASP-2抗体对阻断冷球蛋白血症的不利影响的能力的影响的方法使用Ng Y.C.等人, Arthritis and Rheumatism 31(1):99-107 (1988)(此处通过引用并入本文)中所述的流体相C3转化的测定来实施。如在Ng等人中所述,在基本混合型冷球蛋白血症(EMC)中,单克隆类风湿因子(mRF),通常为IgM,与多克隆IgG复合以形成特征性的冷沉淀物免疫复合物(IC)(II型冷球蛋白)。免疫球蛋白和C3已经证明在受影响的组织诸如皮肤、神经和肾的血管壁中。如在Ng等人所述,将125I-标记的mRF添加至血清(正常人血清和从患有冷球蛋白血症的患者获得的血清),在37℃下孵育,并且测定与红细胞的结合。
流体相C3转化在抗MASP-2抗体存在或不存在的情况下并且在以下IC存在或不存在的情况下在血清(正常人血清和从患有冷球蛋白血症的患者获得的血清)中确定:BSA-抗BSA、mRF、mRF加IgG或冷球蛋白。C3和C4至IC的固定使用与F(ab')2抗-C3和F(ab')2抗-C4的共沉淀测定来测定。
鉴于已经显示抗MASP-2抗体阻断凝集素途径的活化这一事实,预期抗MASP-2抗体在阻断与冷球蛋白血症相关的补体介导的不良影响中将是有效的,并且将可用作治疗患有冷球蛋白血症的患者的治疗剂。
实施例26
本实施例描述了评价抗MASP-2抗体对从患有冷凝集素病(显现为贫血)的患者获得的血液样品的影响的方法。
背景/基本原理:
冷凝集素病(CAD)是一类自身免疫性溶血性贫血。冷凝集素抗体(通常为IgM)由低温活化并且结合至红细胞并聚集红细胞。冷凝集素抗体与补体组合,并且攻击红血细胞的表面上的抗原。这导致红细胞的调理作用(溶血),其触发网状内皮系统对其的清除。凝集发生的温度因患者而变化。
CAD显现为贫血。当红细胞破坏的破坏速率超过骨髓产生足够量的携氧细胞的能力时,那么就会发生贫血。CAD可以由被称为“继发性CAD”的潜在疾病或病症(诸如感染性疾病(支原体肺炎、腮腺炎、单核细胞增多),淋巴增生性疾病(淋巴瘤、慢性淋巴细胞性白血病)或结缔组织病症)所引起。原发性CAD患者被认为具有低级别的淋巴增生性骨髓病症。原发性和继发性CAD两者都是获得性状况。
方法:
试剂:
来自正常供体和患有CAD的患者的红细胞通过静脉穿刺获得。具有功能性阻断凝集素途径的活性的抗MASP-2抗体如实施例10中所述生成。
可以如下确定抗MASP-2抗体对于阻断凝集素途径的冷凝集素介导的活化的影响。来自I型阳性患者的血液的红细胞在抗MASP-2抗体存在或不存在的情况下用冷凝集素(即IgM抗体)敏化。然后通过测量C3结合测试红细胞活化凝集素途径的能力。
鉴于已经显示抗MASP-2抗体阻断凝集素途径的活化这一事实,预期抗MASP-2抗体在阻断与冷凝集素病相关的补体介导的不良影响中将是有效的,并且将可用作治疗患有冷凝集素病的患者的治疗剂。
实施例27
本实施例描述了评价抗MASP-2抗体对来自从患有非典型溶血性尿毒综合征(aHUS)的小鼠获得的血液样品中的红细胞的裂解的影响的方法。
背景/基本原理:
非典型溶血性尿毒综合征(aHUS)的特征在于溶血性贫血、血小板减少和在肾和其他器官的微循环中的血小板血栓引起的肾衰竭。aHUS与缺陷的补体调节有关,并且可以是偶发性或家族性的。aHUS与编码补体活化的基因,包括补体因子H、膜辅因子B和因子I以及补体因子H-相关蛋白1(CFHR1)和补体因子H相关蛋白3(CFHR3)中的突变相关。Zipfel, P.F.,等人, PloS Genetics 3(3):e41 (2007)。本实施例描述了评价抗MASP-2抗体对来自从aHUS小鼠获得的血液样品的红细胞的裂解的影响的方法。
方法:
抗MASP-2抗体治疗aHUS的效果可以在该疾病的小鼠模型中确定,在该小鼠模型中,内源小鼠fH基因被替换为编码在aHUS患者中经常发现的fH的突变形式的人同源物。参见Pickering M.C.等人, J. Exp. Med. 204(6):1249-1256 (2007), 在此通过引用并入本文。如Pickering等人中所述,此类小鼠发展出aHUS样病理。为了评价抗MASP-2抗体用于治疗aHUS的效果,将抗MASP-2抗体施用于突变aHUS小鼠,并且比较从抗MASP-2 ab处理和未经处理的对照获得的红细胞的裂解。鉴于已经显示抗MASP-2抗体阻断凝集素途径的活化这一事实,预期抗MASP-2抗体在阻断患有aHUS的哺乳动物受试者中的红细胞的裂解中将是有效的。
实施例28
本实施例描述了评价抗MASP-2抗体治疗青光眼的效果的方法。
基本原理/背景:
已经显示,不受控制的补体活化有助于青光眼中对视网膜神经节细胞(RGCs)、其突触和轴突的退行性损伤的进展。参见Tezel G.等人, Invest Ophthalmol Vis Sci 51:5071-5082 (2010)。例如,使用不同动物模型的人组织和体内研究的组织病理学研究已经表明,合成了补体组分,包括C1q和C3,并且在青光眼性视网膜中形成末端补体复合物(参见Stasi K.等人, Invest Ophthalmol Vis Sci 47:1024-1029 (2006), Kuehn M.H.等人, Exp Eye Res 83:620-628 (2006))。如Kuehn M.H.等人, Experimental Eye Research 87:89-95 (2008)中进一步描述,补体合成和沉积受视网膜I/R诱导,并且补体级联的破坏延迟RGC变性。在该研究中,当与正常动物相比时,发现携带补体组分C3的靶向破坏的小鼠在短暂性视网膜I/R后表现出延迟的RGC变性。
方法:
用于测定抗MASP-2抗体对RGC变性的影响的方法在如Kuehn M.H.等人, Experimental Eye Research 87:89-95 (2008)(此处通过引用并入本文)中所述的视网膜I/R的动物模型中实施。如Kuehn等人中所述,视网膜缺血通过以下来诱导:麻醉动物,然后将连接至含有磷酸盐缓冲盐水的蓄池的30号针头通过角膜插入眼睛的前房。然后升高盐水蓄池,以得到104 mmHg的眼内压,其足以完全防止循环通过视网膜脉管。升高的眼内缺血通过虹膜和视网膜的发白进行证实,并且缺血仅在左眼维持45分钟;右眼充当对照,并且不接受插管。小鼠然后在缺血损伤后1或3周进行安乐死。将抗MASP-2抗体对小鼠局部施用于眼睛或全身施用以评价缺血损伤之前施用的抗MASP抗体的影响。
眼睛的免疫组织化学使用针对C1q和C3的抗体来实施,以检测补体沉积。视神经损伤也可以使用标准电子显微镜方法进行评价。存活的视网膜RGCs的定量使用γ突触核蛋白标记来进行。
结果:
如Kuehn等人中所述,在正常对照小鼠中,短暂视网膜缺血导致视神经的退行性改变和通过免疫组织化学可检测的C1q和C3的视网膜沉积。相反,C3缺陷小鼠表现出轴突变性的显著减少,表现出诱导后1周只有轻微水平的视神经损伤。基于这些结果,可以预期,当该测定在MASP-2敲除小鼠中实施时和当缺血损伤之前将抗MASP-2抗体施用于正常小鼠时可以观察到相似的结果。
实施例29
本实施例证明MASP-2抑制剂诸如抗MASP-2抗体对于治疗放射暴露和/或对于治疗、改善或预防急性放射综合征是有效的。
基本原理:
暴露于高剂量的电离放射通过两种主要机制引起死亡率:对骨髓的毒性和胃肠综合征。骨髓毒性导致所有血液细胞的下降,使生物体倾向于感染和出血引起的死亡。胃肠综合征是更严重的,并且通过由于肠道上皮层的崩溃引起的肠道屏障功能的丧失和肠道内分泌功能的丧失而驱动。这导致可以导致死亡的败血症和相关的全身炎症响应综合征。
补体的凝集素途径是响应于组织损伤和暴露于外表面(例如,细菌)而起始炎症的先天性免疫机制。该途径的阻断导致缺血性肠组织损伤或败血性休克的小鼠模型中更好的结果。据假设,凝集素途径可能触发响应于放射诱导的组织损伤的过度且有害的炎症。因此,凝集素途径的阻断可以减少继发性损伤,并且增强急性放射暴露之后的存活率。
如本实施例中所述实施的本研究的目的是评估通过施用抗鼠MASP-2抗体的凝集素途径阻断对放射损伤的小鼠模型中的存活的影响。
方法与材料:
材料. 在本研究中使用的测试物品是(i)高亲和力的抗鼠MASP-2抗体(mAbM11)和(ii)高亲和力的抗人MASP-2抗体(mAbH6),其阻断在转染的哺乳动物细胞中产生的凝集素补体途径的MASP-2蛋白组分。给药浓度为1 mg/kg抗鼠MASP-2抗体(mAbM11),5mg/kg抗人MASP-2抗体(mAbH6),或无菌盐水。对于每种给药情况,制备足够体积的新鲜给药溶液。
动物. 年轻成年雄性Swiss-Webster小鼠从Harlan Laboratories (Houston, TX)获得。动物饲养具有Alpha-Dri垫的固底笼,并且自由提供认证的PMI 5002 Rodent Diet (Animal Specialties, Inc., Hubbard OR)和水。监测温度,并且以12小时光/12小时暗光周期操作动物饲养室。
放射. 在设施中适应2周之后,使用Therapax X-RAD 320系统以0.78 Gy/min的剂量速率通过在10只的组中的全身暴露以6.5和7.0 Gy辐射小鼠,所述Therapax X-RAD 320系统配备320-kV高稳定性X射线发生器、金属陶瓷X射线管、可变的x射线束准直器和滤片(Precision X-ray Incorporated, East Haven, CT)。基于用相同品系的小鼠进行的先前研究选择剂量水平,其表明LD50/30为6.5至7.0 Gy (数据未显示)。
药物配制和施用。根据方案将适当体积的浓缩原液用冰冷盐水稀释以制备0.2 mg/ml抗鼠MASP-2抗体(mAbM11)或0.5 mg/ml抗人MASP-2抗体(mAbH6)的给药溶液。使用25号针头基于动物体重通过IP注射施用抗MASP-2抗体mAbM11和mAbH6以递送1 mg/kg mAbM11、5mg/kg mAbH6或盐水媒介物。
研究设计. 如表8中所述,将小鼠随机分配到各组。每天测量和记录体重和温度。在辐射后第7天将第7、11和13组中的小鼠处死,并且在深度麻醉下通过心脏穿刺采集血液。在辐射后第30天存活的动物以相同方式处死并且收获血液。血浆根据方案从收集的血液样品制备,并且返回至发起者用于分析。
表 8:研究组

统计分析. 生成Kaplan-Meier存活曲线,并且用于使用对数秩(log-Rank)和Wilcoxon方法比较处理组之间的平均存活时间。报告平均值与标准偏差,或平均值与平均值的标准误差。统计比较使用受控辐射的动物和个体处理组之间的双尾非配对t检验进行。
结果
对于7.0和6.5 Gy暴露组的Kaplan-Meier存活曲线分别提供在图32A和32B中,并且总结在下表9中。总体而言,与媒介物处理的辐射的对照动物相比,在6.5 (20%增加)和7.0 Gy (30%增加)暴露水平两者,用抗鼠MASP-2 ab (mAbM11)预辐射处理增加了经辐射小鼠的存活。在6.5 Gy辐射水平,与媒介物对照辐射动物相比,用抗鼠MASP-2 ab的辐射后处理导致存活的适度增加(15%)。
相比之下,在7.0 Gy暴露水平,与媒介物处理的经辐射对照动物相比,所有处理的动物显示出存活的增加。在接受mAbH6的动物中发生存活的最大变化,与对照动物相比增加45%。此外,在7.0 Gy暴露水平,与媒介物处理的经辐射的对照动物的辐射后第8天相比,mAbH6治疗组的死亡率在辐射后第15天首次发生,比对照动物增加7天。在7.0 Gy暴露水平,与对照动物(20.7 ± 2.0天)相比,接受mAbH6的小鼠的至死亡的平均时间(27.3 ± 1.3天)显著增加。
在整个研究中记录与辐射前一天(第-1天)相比的体重百分比变化。短暂减重发生在所有经辐射的动物中,没有证据证明由于mAbM11或mAbH6治疗导致的与对照相比的差异变化(数据未显示)。在研究结束时,所有存活的动物显示从开始(第-1天)体重的体重增加。
表 9:暴露于辐射的测试动物的存活率
通过在相同辐射暴露水平的受控辐射的动物和处理组之间的双尾非配对t检验,*p = 0.0087。
讨论
急性放射综合征由三种定义的亚综合征组成:造血细胞亚综合征、胃肠道亚综合征和脑血管亚综合征。观察到的综合征取决于放射剂量,其中在具有超过1 Gy的显著部分或全身辐射暴露的人中观察到造血影响。造血综合征的特征在于骨髓功能的严重抑制,导致具有血细胞计数、红细胞和白细胞以及血小板的变化的全血细胞减少,并伴随对免疫系统的损害。随着最低点出现,在外周血中具有少量中性粒细胞和血小板,中性粒细胞减少、发热、败血症的并发症和不可控的出血导致死亡。
在本研究中,发现在以7.0 Gy辐射的Swiss-Webster雄性小鼠中施用mAbH6增加全身X射线辐射的存活能力。值得注意的是,在7.0 Gy暴露水平,与35%的媒介物治疗的对照辐射动物相比,80%接受mAbH6的动物存活至30天。重要的是,在该治疗组中死亡的第一天直到辐射后第15天才出现,比在媒介物治疗的对照辐射动物中观察到的增加7天。好奇的是,在较低的X射线暴露(6.5 Gy)时,与媒介物治疗的对照辐射动物相比,施用mAbH6似乎没有影响存活能力或死亡的延迟。对于暴露水平之间的这种响应差异可能存在多种原因,尽管验证任何假设可能需要额外研究,包括中期样品收集用于微生物培养和血液学参数。一种解释可能仅仅是分配至组的动物数量可能已经排除看到任何细微的治疗相关的差异。例如,在n=20的组大小,在65% (mAbH6,6.5 Gy暴露)和80% (mAbH6,7.0 Gy暴露)之间存活的差异是3只动物。另一方面,在35% (媒介物对照,7.0 Gy暴露)和80% (mAbH6,7.0 Gy暴露)之间的差异是9只动物,并且提供了治疗相关的差异的可靠证据。
这些结果证明,抗MASP-2抗体在治疗处于急性放射综合征的不利影响的风险中或患有急性放射综合征的不利影响的哺乳动物受试者中是有效的。
实施例30
本实施例证明了MASP-2缺陷小鼠在被脑膜炎奈瑟氏菌血清群A或脑膜炎奈瑟氏菌血清群B感染后被保护免于脑膜炎奈瑟氏菌诱导的死亡。
方法:
MASP-2敲除小鼠(MASP-2 KO小鼠)如实施例1中所述生成。用在100 μl体积中的2.6 x 107 CFU脑膜炎奈瑟氏菌血清群A Z2491的剂量通过腹膜内(i.p.)注射接种10周龄MASP-2 KO小鼠(n=10)和野生型(WT) C57/BL6小鼠(n=10)。将感染剂量连同400 mg/kg的终浓度的右旋糖酐铁施用于小鼠。经72小时时间期间监测感染后的小鼠的存活。
在独立的实验中,用在100 μl体积中的6 x 106 CFU脑膜炎奈瑟氏菌血清群B菌株MC58的剂量通过i.p.注射接种10周龄MASP-2 KO小鼠(n=10)和野生型C57/BL6小鼠(n=10)。将感染剂量连同400 mg/kg的最终剂量的右旋糖酐铁施用于小鼠。经72小时时间期间监测感染后的小鼠的存活。还基于下表10中描述的疾病评分参数(其基于Fransen等人 (2010)的方案,进行轻微修改)确定了感染后72小时时间期间过程中WT和MASP-2 KO小鼠的疾病评分。
表 10:在感染小鼠中与临床体征相关的疾病评分
| 体征 | 评分 |
| 正常 | 0 |
| 轻微皱皮 | 1 |
| 皱皮,慢且粘性的眼 | 2 |
| 皱皮,昏睡且闭眼 | 3 |
| 刺激后非常虚弱并且不动 | 4 |
| 死亡 | 5 |
血液样品在感染后以每小时间隔取自小鼠,并且分析以确定脑膜炎奈瑟氏菌的血清水平(log cfu/mL)以验证感染,并且确定细菌从血清中的清除率。
结果:
图33是图示说明在施用2.6 x 107 cfu的脑膜炎奈瑟氏菌血清群A Z2491的感染剂量之后MASP-2 KO和WT小鼠的百分比存活的Kaplan-Meyer图。如图33中显示,100%的MASP-2 KO小鼠在整个感染后72小时期间内存活。相比之下,只有80%的WT小鼠(p=0.012)在感染后24小时仍然活着,并且只有50%的WT小鼠在感染后72小时仍然活着。这些结果证明,MASP-2缺陷小鼠被保护免于脑膜炎奈瑟氏菌血清群A Z2491诱导的死亡。
图34是图示说明在施用6 x 106 cfu的脑膜炎奈瑟氏菌血清群B菌株MC58的感染剂量之后MASP-2 KO和WT小鼠的百分比存活的Kaplan-Meyer图。如图34中显示,90%的MASP-2 KO小鼠在整个感染后72小时期间内存活。相比之下,只有20%的WT小鼠(p=0.0022)在感染后24小时仍然活着。这些结果证明,MASP-2缺陷小鼠被保护免于脑膜炎奈瑟氏菌血清群B菌株MC58诱导的死亡。
图35图示说明在用6x106 cfu的脑膜炎奈瑟氏菌血清群B菌株MC58 i.p.感染后取自MASP-2 KO和WT小鼠的血液样品中在不同时间点回收的脑膜炎奈瑟氏菌血清群B菌株MC58的log cfu/mL(在两组小鼠中不同时间点n=3)。结果表示为平均值±SEM。如图35中显示,在WT小鼠中,血液中的脑膜炎奈瑟氏菌的水平在感染后24小时达到约6.0 log cfu/mL的峰值,到感染后36小时下降至约4.0 log cfu/mL。相比之下,在MASP-2 KO小鼠中,脑膜炎奈瑟氏菌的水平在感染后12小时达到约4.0 log cfu/mL的峰值,并且到感染后36小时下降至约1.0 log cfu/mL(符号“*”表示p<0.05;符号“**”表示p=0.0043)。这些结果证明,尽管MASP-2 KO小鼠被与WT小鼠相同剂量的脑膜炎奈瑟氏菌血清群B菌株MC58所感染,但是MASP-2 KO小鼠与WT相比具有增强的菌血症的清除。
图36图示说明在6x106 cfu的脑膜炎奈瑟氏菌血清群B菌株MC58的感染后3、6、12和24小时MASP-2 KO和WT小鼠的平均疾病评分。如图36中显示,与WT小鼠相比,MASP-2缺陷小鼠显示出对感染的高抗性,在感染后6小时(符号“*”表示p=0.0411)、12小时(符号“**”表示p=0.0049)和24小时(符号“***”表示p=0.0049)具有低得多的疾病评分。图36中的结果表示为平均值±SEM。
总之,本实施例中的结果证明了MASP-2缺陷小鼠在被脑膜炎奈瑟氏菌血清群A或脑膜炎奈瑟氏菌血清群B感染后被保护免于脑膜炎奈瑟氏菌诱导的死亡。
实施例31
本实施例证明了在被脑膜炎奈瑟氏菌感染后施用抗MASP-2抗体增加了被脑膜炎奈瑟氏菌感染的小鼠的存活。
背景/基本原理:
如实施例10中所述,大鼠MASP-2蛋白用于淘选Fab噬菌体展示文库,从所述Fab噬菌体展示文库中将Fab2 #11鉴定为功能活性抗体。从Fab2 #11生成的大鼠IgG2c和小鼠IgG2a同种型的全长抗体。表征小鼠IgG2a同种型的全长抗MASP-2抗体的药效动力学参数(如实施例19中所述)。
在本实施例中,在脑膜炎奈瑟氏菌感染的小鼠模型中分析衍生自Fab2 #11的小鼠抗MASP-2全长抗体。
方法:
如下在脑膜炎奈瑟氏菌感染的小鼠模型中测试如上生成的衍生自Fab2 #11的小鼠IgG2a全长抗MASP-2抗体同种型。
在感染后施用小鼠抗MASP-2单克隆抗体(MoAb)
在用高剂量(4x106 cfu)的脑膜炎奈瑟氏菌血清群B菌株MC58 i.p.注射后3小时,用抑制性小鼠抗MASP-2抗体(1.0 mg/kg) (n=12)或对照同种型抗体(n=10)治疗 9周龄C57/BL6 Charles River小鼠。
结果:
图37是图示说明在施用4x106 cfu的脑膜炎奈瑟氏菌血清群B菌株MC58的感染剂量和随后在感染后3小时施用抑制性抗MASP-2抗体(1.0 mg/kg)或对照同种型抗体之后小鼠的百分比存活的Kaplan-Meyer图。如图37中显示,90%的用抗MASP-2抗体治疗的小鼠在整个感染后72小时期间内存活。相比之下,只有50%的用同种型对照抗体治疗的小鼠在整个感染后72小时期间内存活。如通过两条存活曲线的比较所确定的,符号“*”表示p=0.0301。
这些结果证明,抗MASP-2抗体的施用对于治疗和改善被脑膜炎奈瑟氏菌感染的受试者的存活是有效的。
如本文所证明的,当在感染后3小时内施用时,在被脑膜炎奈瑟氏菌感染的受试者的治疗中使用抗MASP-2抗体是有效的,并且预期在感染后24小时至48小时内是有效的。脑膜炎球菌病(无论是脑膜炎球菌血症或脑膜炎)是医疗紧急状况,并且如果怀疑脑膜炎球菌病(即在脑膜炎奈瑟氏菌阳性鉴定为病原体之前),通常立即启动治疗。
鉴于实施例30中证明的MASP-2 KO小鼠中的结果,可以相信,在被脑膜炎奈瑟氏菌感染之前施用抗MASP-2抗体对于预防或改善感染的严重度也将是有效的。
实施例32
本实施例证明,抗MASP-2抗体的施用对于治疗人血清中的脑膜炎奈瑟氏菌感染是有效的。
基本原理:
具有降低血清水平的功能性MBL的患者表现出对复发性细菌和真菌感染的增加的易感性(Kilpatrick等人, Biochim Biophys Acta 1572:401-413 (2002))。已知脑膜炎奈瑟氏球菌被MBL所识别,并且已经显示,MBL缺陷血清不会裂解奈瑟氏菌。
鉴于实施例30和31中描述的结果,实施一系列实验以确定施用抗MASP-2抗体对于治疗补体缺陷和对照人血清中的脑膜炎奈瑟氏菌感染的功效。在高浓度血清(20%)中实施实验,以保持补体途径。
方法:
1. 在用人抗MASP-2抗体处理的各种补体缺陷的人血清和人血清中的血清杀菌活性
在本实验中使用以下补体缺陷的人血清和对照人血清:
表 11:测试的人血清样品(如图38中显示)
| 样品 | 血清类型 |
| A | 正常人血清(NHS) +人抗MASP-2 Ab |
| B | NHS + 同种型对照Ab |
| C | MBL -/- 人血清 |
| D | NHS |
| E | 热失活的(HI) NHS |
使用作为抗原的重组人MASP-2A,从组合抗体文库(Knappik, A.,等人, J. Mol. Biol. 296:57-86 (2000))分离针对人MASP-2的重组抗体(Chen, C.B.和Wallis, J. Biol. Chem. 276:25894-25902 (2001))。鉴定在人血浆中有效抑制C4和C3的凝集素途径介导的活化的抗人scFv片段(IC50~20 nM),并将其转化为全长人IgG2a抗体。
在37℃下,在有或没有添加抑制人抗MASP-2抗体(3 μg,在100μl总体积中)的情况下,伴随振荡,将脑膜炎奈瑟氏菌血清群B-MC58与表11中显示的不同血清(各自在20%的血清浓度)孵育。在以下时间点:0- 、30- 、60-和90-min间隔取样,铺出,然后确定活计数。热灭活的人血清用作阴性对照。
结果:
图38图示说明在表11中显示的人血清样品中在不同时间点回收的脑膜炎奈瑟氏菌血清群B-MC58的活计数的log cfu/mL。表12提供了图38的Student’s t检验的结果。
表 12:图38的Student’s t检验的结果(时间点60分钟)
| 平均差异(Log) | 显著的? P<0.05? | P值总结 | |
| A vs B | -0.3678 | 是 | ***(0.0002) |
| A vs C | -1.1053 | 是 | ***(p<0.0001) |
| A vs D | -0.2111 | 是 | **(0.0012) |
| C vs D | 1.9 | 是 | ***(p<0.0001) |
如图38和表12中显示,人20%血清中脑膜炎奈瑟氏菌的补体依赖性杀伤被添加人抗MASP-2抑制性抗体所显著增强。
2. 在MASP-2缺陷的20% (v/v)小鼠血清中脑膜炎奈瑟氏菌的补体依赖性杀伤。
在本实验中使用以下补体缺陷的小鼠血清和对照小鼠血清:
表 13:测试的小鼠血清样品(如图39中显示)
| 样品 | 血清类型 |
| A | WT |
| B | MASP-2 -/- |
| C | MBL A/C -/- |
| D | WT热失活的(HIS) |
在37℃下,伴随振荡,将脑膜炎奈瑟氏菌血清群B-MC58与不同补体缺陷的小鼠血清(各自在20%的血清浓度)孵育。在以下时间点:0-、15-、30-、60-、90- 和120-分钟间隔取样,铺出,然后确定活计数。热灭活的人血清用作阴性对照。
结果:
图39图示说明在表13中显示的小鼠血清样品中在不同时间点回收的脑膜炎奈瑟氏菌血清群B-MC58的活计数的log cfu/mL。如图39中显示,与WT小鼠血清相比,MASP-2 -/-小鼠血清对脑膜炎奈瑟氏菌具有更高水平的杀菌活性。符号“**”表示p=0.0058,符号“***”表示p=0.001。表14提供了图39的Student’s t检验的结果。
表 14:图39的Student’s t检验结果。
| 比较 | 时间点 | 平均差异(LOG) | 显著的? (p<0.05)? | P值总结 |
| A vs. B | 60 min. | 0.39 | 是 | ** (0.0058) |
| A vs. B | 90 min. | 0.6741 | 是 | *** (0.001) |
总之,本实施例中的结果证明,与WT血清相比,MASP-2 -/-血清对脑膜炎奈瑟氏菌具有更高水平的杀菌活性。
实施例33
本实施例证明了MASP-2缺陷对来自从阵发性睡眠性血红蛋白尿症(PNH)的小鼠模型获得的血液样品的红细胞的裂解的抑制效果。
背景/基本原理:
阵发性睡眠性血红蛋白尿症(PNH),也称为Marchiafava-Micheli综合征,是一种获得性的潜在威胁生命的血液疾病,其特征在于补体诱导的血管内溶血性贫血。PNH的标志是慢性补体介导的血管内溶血(这是补体的替代途径的活化失调的结果,所述活化失调是由于PNH红细胞上不存在补体调节物CD55和CD59导致的),和随后的血红蛋白尿和贫血。Lindorfer, M.A.,等人, Blood 115(11) (2010), Risitano, A.M, Mini-Reviews in Medicinal Chemistry, 11:528-535 (2011)。PNH中的贫血是由于血流中红细胞的破坏。PNH的症状包括红色尿(这是由于尿中出现血红蛋白)、背痛、疲劳、呼吸急促和血栓形成。PNH可以自身发生(称为“原发性PNH”)或在其他骨髓病症诸如再生障碍性贫血的背景中发生(称为“继发性PNH”)。PNH的治疗包括用于贫血的输血、用于血栓形成的抗凝和使用单克隆抗体依库丽单抗(Soliris?),其通过抑制补体系统保护血细胞免于免疫破坏(Hillmen P.等人, N. Engl. J. Med. 350(6):552-9 (2004))。依库丽单抗(Soliris?)是靶向补体组分C5的人源化单克隆抗体,其阻断C5转化酶对其裂解,从而防止C5a的产生和MAC的装配。用依库丽单抗治疗PNH患者已经导致血管内溶血的减少,如通过乳酸脱氢酶(LDH)所测量的,导致约一半患者中的血红蛋白稳定和输血不依赖(Hillmen P,等人, Mini-Reviews in Medicinal Chemistry, vol 11(6) (2011))。尽管几乎所有经历依库丽单抗治疗的患者达到了正常或几乎正常的LDH水平(由于血管内溶血的控制),但是只有约三分之一患者达到高于11克/dL的血红蛋白值,并且约等同比例的依库丽单抗的其余患者继续表现出温和至严重的(即输血依赖性)贫血(Risitano A.M.等人, Blood 113:4094-100 (2009))。如Risitano等人, Mini-Reviews in Medicinal Chemistry 11:528-535 (2011)中所述,表明依库丽单抗的PNH患者含有结合至显著部分的其PNH红细胞的C3片段(而未经治疗的患者则没有),导致以下结论:膜结合的C3片段作为PNH红细胞上的调理素起作用,通过特异性C3受体和随后的血管外溶血导致它们捕获在网状内皮细胞中。因此,对于发展C3片段介导的血管外溶血的那些患者需要除了使用依库丽单抗以外的治疗策略,因为他们继续需要红细胞输血。
本实施例描述了评价MASP-2缺陷血清和用MASP-2抑制剂处理的血清对来自从PNH的小鼠模型获得的血液样品的红细胞的裂解的影响的方法,并且证明了MASP-2抑制对于治疗患有PNH的受试者的功效,并且还支持MASP-2抑制剂在经历用C5抑制剂诸如依库丽单抗的治疗的PNH患者中改善C3片段介导的血管外溶血的影响的用途。
方法:
PNH动物模型:
从具有Crry和C3缺陷的基因靶向的小鼠(Crry/C3-/-)和CD55/CD59缺陷小鼠获得血液样品。这些小鼠缺少各自的表面补体调节物,并且因此它们的红细胞如同PNH人血细胞一样易于自发补体自我裂解。
为了使这些红细胞甚至更加敏感,在用和不同甘露聚糖包被的情况下使用这些细胞,然后在WT C56/BL6血浆、无MBL血浆、MASP-2 -/-血浆、人NHS、人MBL -/-血浆和用人抗MASP-2处理的NHS中测试溶血。
1. 在MASP-2-缺陷/耗尽血清和对照中Crry/C3和CD55/CD59双缺陷鼠红细胞的溶血测定
第1天. 鼠RBC的制备(±甘露聚糖包被)。
包括的材料:新鲜的小鼠血液、BBS/Mg2+/Ca2+ (4.4 mM巴比妥酸, 1.8 mM巴比妥钠, 145 mM NaCl, pH7.4, 5mM Mg2+, 5mM Ca2+)、氯化铬、CrCl3·6H20 (0.5mg/mL,在BBS/Mg2+/Ca2+中)和甘露聚糖,在BBS /Mg2+/Ca2中100 μg/mL。
在4℃下在冷冻离心机中以2000xg将全血(2mL)离心下来1-2 min。吸去血浆和血沉棕黄层。然后通过将RBC沉淀重悬浮在2ml冰冷BBS/明胶/Mg2+/Ca2+中并且重复离心步骤而将样品洗涤3次。在第三次洗涤后,将沉淀重悬浮在4mL BBS/Mg2+/Ca2+中。旁边设置2 mL等分试样的RBC作为未包被对照。将2 mL CrCl3和2 mL甘露聚糖添加至剩余的2 mL,并且在室温在温和混合下将样品孵育5分钟。通过添加7.5mL BBS/明胶/Mg2+/Ca2+终止反应。将样品如上离心下来,重悬浮在2 mL BBS/明胶/Mg2+/Ca2+中,并且进一步如上洗涤两次,然后保存在4℃。
第2天. 溶血测定
材料包括BBS/明胶/Mg2+/Ca2+(如上),测试血清,96孔圆底板和96孔平底板,和在410-414 nm阅读96孔板的分光光度计。
首先确定RBC的浓度,并且将细胞调整至109/mL,并在该浓度储存。在使用前,将测定缓冲液稀释至108/mL,然后使用100ul/每孔。在410-414 nm测量溶血(允许比541nm更大的灵敏度)。在冰冷BBS/明胶/Mg2+/Ca2+中制备试验血清的稀释物。将100μl各血清稀释物移液到圆底板(参见板布局)。添加100μl适当稀释的RBC制剂(即,108 /mL)(参见板布局),在37℃下孵育约1小时,并且观察裂解。(该板可以在该点拍摄。) 然后将板以最大速度离心下来,持续5分钟。移取100μl流体相,转移到平底板中,并且在410-414 nm记录OD。保留RBC沉淀(这些可以随后用水裂解以获得反结果)。
实验#1:
从CD55/CD59双缺陷小鼠获得新鲜血液,并且如上述方案中详细描述制备Crry/C3双缺陷小鼠的血液和红细胞。将细胞一分为二,将一半细胞包被甘露聚糖,而另一半留下不处理,将最终浓度调节至1x 108/每mL,其中在如上所述实施的溶血测定中使用100 μl。
实验#1的结果: 凝集素途径参与PNH动物模型中的红细胞裂解
在最初实验中,确定未包被的WT小鼠红细胞在任何小鼠血清均未裂解。进一步确定,甘露聚糖包被的Crry-/-小鼠红细胞在WT小鼠血清中缓慢裂解(在37℃超过3小时),但它们并没有在无MBL血清中裂解。(数据未显示)。
确定甘露聚糖包被的Crry-/-小鼠红细胞在人血清但非热失活的NHS中迅速溶解。重要的是,甘露聚糖包被的Crry-/-小鼠红细胞在向下稀释至1/640的NHS中裂解(即,1/40、1/80、1/160、1/320 和1/640稀释物都裂解)。(数据未显示)。在该稀释度中,替代途径不起作用(AP功能活性显著降低低于8%血清浓度)。
来自实验#1的结论
甘露聚糖包被的Crry-/-小鼠红细胞在具有MBL的高度稀释的人血清中非常好地裂解,但在无MBL的高度稀释的人血清中则不是如此。在测试的每种血清浓度中的有效裂解意味着替代途径没有参与该裂解或对于该裂解是不需要的。MBL缺陷小鼠血清和人血清无法裂解甘露聚糖包被的Crry-/-小鼠红细胞表明,经典途径也与观察到的裂解无关。由于凝集素途径识别分子是需要的(即,MBL),所以该裂解由凝集素途径介导。
实验#2:
从Crry/C3和CD55/CD59双缺陷小鼠获得新鲜血液,并且如上所述在以下人血清存在的情况下在溶血测定中分析甘露聚糖包被的Crry-/-小鼠红细胞:无MBL;WT;用人抗MASP-2抗体预处理的NHS;和作为对照的热失活的NHS。
实验#2的结果: MASP-2抑制剂防止PNH动物模型中的红细胞裂解
用甘露聚糖包被的Crry-/-小鼠红细胞,以向下稀释至1/640(即,1/40、1/80、1/160、1/320 和1/640)的稀释物中孵育NHS、人MBL-/-血清、用抗MASP-2 mAb预处理的NHS,和作为对照的热失活的NHS。
将ELISA微量滴定板离心,并且收集在圆孔板的底部上的未裂解的红细胞。收集每孔中的上清液,并且通过在ELISA读数器中阅读OD415 nm而测量从裂解的红细胞释放的血红蛋白的量。
在对照热失活的NHS(阴性对照)中,如预期,没有观察到裂解。MBL-/-人血清在1/8和1/16稀释度裂解甘露聚糖包被的小鼠红细胞。抗MASP-2抗体预处理的NHS在1/8和1/16稀释度裂解甘露聚糖包被的小鼠红细胞,而WT人血清在下降至1/32的稀释度裂解甘露聚糖包被的小鼠红细胞。
图40图示说明了来自热失活(HI)NHS、MBL-/-、抗MASP-2抗体预处理的NHS和NHS对照的血清中在一定范围的血清浓度内人血清对甘露聚糖包被的鼠红细胞的溶血(如通过光度法测量的裂解的鼠红细胞(Cryy/C3-/-)至上清液的血红蛋白释放所测量的)。
从图40中显示的结果,可以证明,抗MASP-2抗体的MASP-2抑制使CH50显著移动,并且抑制免于自体补体活化的保护具有缺陷的敏化红细胞的补体介导的裂解。
实验#3
如上所述在以下人血清存在的情况下在溶血测定中分析未包被的Crry-/-小鼠红细胞中的从Crry/C3和CD55/CD59双缺陷小鼠获得的新鲜血液:MBL -/-;WT血清;用人抗MASP-2抗体预处理的NHS;和作为对照的热失活的NHS。
结果:
图41图示说明了来自热失活(HI)NHS、MBL-/-、抗MASP-2抗体预处理的NHS和NHS对照的血清中在一定范围的血清浓度内人血清对未包被的鼠红细胞的溶血(如通过光度法测量的裂解的WT鼠红细胞至上清液的血红蛋白释放所测量的)。如图41中显示,证明抑制MASP-2抑制未敏化的WT小鼠红细胞的补体介导的裂解。
图42图示说明了来自热失活(HI)NHS、MBL-/-、抗MASP-2抗体预处理的NHS和NHS对照的血清中在一定范围的血清浓度内人血清对未包被的鼠红细胞的溶血(如通过光度法测量的裂解的鼠红细胞(CD55/59 -/-)至上清液的血红蛋白释放所测量的)。
表 12:表示为血清浓度的CH50值
| 血清 | WT | CD55/59 -/- |
| 热失活的NHS | 无裂解 | 无裂解 |
| MBL AO/XX供体(MBL缺陷) | 7.2% | 2.1% |
| NHS + 抗MASP-2抗体 | 5.4% | 1.5% |
| NHS | 3.1% | 0.73% |
注:“CH50”是其中补体介导的溶血达到50%的点。
总之,本实施例中的结果表明,抑制MASP-2抑制免于自体补体活化的保护具有缺陷的敏化红细胞和未敏化红细胞的补体介导的裂解。因此,MASP-2抑制剂可用于治疗患有PNH的受试者,并且还可用于在经历用C5抑制剂诸如依库丽单抗(Soliris?)的治疗的PNH患者中改善(即,抑制、防止或降低其严重度)血管外溶血。
实施例34
本实施例描述了对上述实施例29中的研究的继续研究,提供了验证MASP-2抑制剂诸如MASP-2抗体对于治疗放射暴露和/或对于治疗、改善或预防急性放射综合征是有效的进一步证据。
基本原理:在实施例29中描述的初始研究中,证明与媒介物治疗的辐射的对照动物相比,在6.5 Gy和7.0 Gy暴露水平两者,在小鼠中用抗MASP-2抗体的预辐射治疗增加了经辐射小鼠的存活。在实施例29中进一步证明,在6.5 Gy辐射水平,与媒介物对照辐射动物相比,用抗MASP-2抗体的辐射后治疗导致存活的适度增加。本实施例描述了实施用于证实第一项研究的结果的第二辐射研究。
方法:
研究A的设计:
将Swiss Webster小鼠(n=50)暴露于电离辐射(8.0 Gy)。评估在放射暴露之前18小时和之后2小时施用并且其后每周施用的抗MASP-2抗体治疗(mAbH6 5mg/kg)对死亡率的影响。
研究A的结果:
如图43中显示,确定抗MASP-2抗体mAbH6的施用增加了暴露于8.0 Gy的小鼠中的存活,其中与接受媒介物对照的小鼠相比,经调整的中值存活率从4天增加至6天,并且当与接受媒介物对照的小鼠相比时,死亡率降低12%(对数秩检验,p=0.040)。
研究B的设计:
在以下组中将Swiss Webster小鼠(n=50)暴露于电离辐射(8.0 Gy):(I:媒介物)盐水对照;(II:低) 在辐射之前18小时和辐射之后2小时施用的抗MASP-2抗体mAbH6 (5 mg/kg);(III:高) 在辐射之前18小时和辐射之后2小时施用的mAbH6 (10 mg/kg);和(IV:之后高) 仅在辐射之后2小时施用的mAbH6 (10mg/kg)。
研究B的结果:
与接受媒介物对照的动物相比,辐照之前和之后施用抗MASP-2抗体将平均存活从4天调整至5天。与媒介物对照小鼠相比,抗MASP-2抗体治疗的小鼠中的死亡率降低了6-12%。进一步指出的是,没有观察到显著有害的治疗效果(数据未显示)。
总之,本实施例中显示的结果与实施例29中显示的结果一致,并且进一步证明,抗MASP-2抗体在治疗处于急性放射综合征的不利影响的风险中或患有急性放射综合征的不利影响的哺乳动物受试者中是有效的。
实施例35
本研究研究在LPS(脂多糖)诱导的血栓形成的小鼠模型中MASP-2缺陷的影响。
基本原理:
由产生志贺毒素的大肠杆菌感染引起的溶血性尿毒综合征(HUS)是儿童中急性肾衰竭的首要原因。在本实施例中,在MASP-2-/- (KO)小鼠中实施LPS诱导的血栓形成(微血管凝血)的Schwartzman模型,以确定MASP-2抑制对于抑制或防止血管内血栓的形成是否是有效的。
方法:
在LPS诱导的血栓形成(微血管凝血)的Schwartzman模型中分析MASP-2-/- (n=9)和WT (n=10)小鼠。将沙雷氏菌属LPS施用于小鼠,并且随时间监测血栓形成。实施微血栓和LPS诱导的微血管凝血的发生率的比较。
结果:
值得注意的是,在沙雷氏菌属LPS施用后,所有测试的MASP-2 -/-小鼠(9/9)都没有形成血管内血栓。相比之下,在7/10的并行测试的WT小鼠中检测到微血栓(p=0.0031, Fischer氏精确)。如图44中显示,在MASP-2-/-和WT小鼠中测量LPS注射后至发生微血管闭塞的时间,显示经60分钟测量的具有血栓形成的WT小鼠的百分比,其中早至约15分钟检测到血栓形成。高达80%的WT小鼠在60分钟时表现出血栓形成。相比之下,如图44中显示,MASP-2 -/-中无一在60分钟时具有血栓形成(对数秩:p=0.0005)。
这些结果证明,在HUS模型中,MASP-2抑制可以预防免于血管内血栓的发生。
实施例36
本实施例描述了抗MASP-2抗体在使用纯化的志贺毒素2(STX2)加LPS的腹膜内共注射的HUS小鼠模型中的效果。
背景:
使用纯化的志贺毒素2(STX2)加LPS的腹膜内共注射开发HUS小鼠模型。小鼠肾脏的生化和微阵列分析揭示了STX2加LPS攻击不同于任一单独试剂的作用。这些小鼠的血液和血清分析显示中性粒细胞增多、血小板减少、红细胞溶血和增加的血清肌酸酐和血液尿素氮。此外,小鼠肾脏的组织学分析和电镜证明肾小球纤维蛋白沉积、红细胞充血、微血栓形成和肾小球超微结构改变。确立了该HUS模型在C57BL/6小鼠中诱导定义人疾病的人HUS病理的所有临床症状,包括血小板减少、溶血性贫血和肾衰竭。(J. Immunol 187(1):172-80 (2011))。
方法:
称重为18至20 g的C57BL/6雌性小鼠购自Charles River Laboratories,并且分为2组(每组中5只)。一组小鼠通过腹膜内(i.p.)注射稀释在150 μl盐水的总体积中的重组抗MASP-2抗体mAbM11(100 μg/每只小鼠;对应于5 mg/kg体重的终浓度)而进行预处理。对照组接受没有任何抗体的盐水。i.p注射抗MASP-2抗体mAbM11后六小时,所有小鼠接受在150 μl总体积中的亚致死剂量(3 μg/动物;对应于150 μg/kg体重)的粘质沙雷氏菌的LPS(L6136; Sigma-Aldrich, St. Louis, MO)和一个剂量的4.5 ng/动物(对应于225 ng/kg)的STX2 (两倍于LD50剂量)的组合i.p.注射。盐水注射用于对照。
给药后每6小时监测小鼠的存活。小鼠一旦它们达到HUS病理的昏睡阶段就进行宰杀。36小时之后,将所有小鼠宰杀,并且取出双肾用于免疫组织化学和扫描电子显微镜。在实验结束时通过心脏穿刺采集血液样品。在治疗组和对照组两者中,分离血清并冷冻保存在-80℃,用于测量BUN和血清肌酸酐水平。
免疫组织化学
将三分之一的各小鼠肾脏固定在4%多聚甲醛中24小时,处理,并包埋在石蜡中。切取三微米厚的切片,并置于带电荷的载玻片上,用于后续用H & E染料的染色。
电子显微镜
将肾脏的中间部分切成约1至2 mm3的块,并且在4℃下过夜固定在1×PBS中的2.5%戊二醛中。随后由University of Leicester Electron Microscopy Facility处理固定的组织。
冰冻切片
将另外三分之一的肾脏切成约1至2 mm3的块,并且快速冷冻在液氮中并保存在-80℃下用于冷冻切片和mRNA分析。
结果:
图45图示说明了在STX/LPS诱导的模型中盐水处理的对照小鼠(n=5)和抗MASP-2抗体处理的小鼠(n=5)随时间(小时)的百分比存活。值得注意的是,如图45中显示,所有对照小鼠到42小时死亡。形成鲜明对比的是,100 %的抗MASP-2抗体处理的小鼠在整个实验的时间过程中存活。与图45中显示的结果一致,观察到,具有严重疾病体征的死亡或必须被宰杀的所有未治疗的小鼠具有显著的肾小球损伤,而所有抗MASP-2治疗的小鼠的肾小球看起来正常(数据未显示)。这些结果证明,MASP-2抑制剂,诸如抗MASP-2抗体,可用于治疗患有血栓性微血管病(TMA)诸如溶血性尿毒综合征(HUS)、非典型HUS (aHUS)或血栓性血小板减少性紫癜(TTP)或处于发生血栓性微血管病(TMA)诸如溶血性尿毒综合征(HUS)、非典型HUS (aHUS)或血栓性血小板减少性紫癜(TTP)的风险中的受试者。
虽然描述并说明了优选的实施方案,但是应当理解的是,可以在不偏离本发明的精神和范围的情况下在其中进行各种修改。








































Claims (85)
1.抑制患有阵发性睡眠性血红蛋白尿症(PNH)的受试者中MASP-2依赖性补体活化的方法,其包括将包含有效抑制MASP-2依赖性补体活化的量的MASP-2抑制剂的组合物施用于所述受试者。
2.权利要求1的方法,其中所述MASP-2抑制剂是抗MASP-2抗体或其片段。
3.权利要求2的方法,其中所述MASP-2抑制剂是特异性结合SEQ ID NO:6的部分的抗MASP-2单克隆抗体或其片段。
4.权利要求1的方法,其中所述组合物增加红细胞在所述受试者中的存活。
5.权利要求1的方法,其中所述受试者表现出选自以下的一种或多种症状:(i) 低于正常水平的血红蛋白,(ii) 低于正常水平的血小板;(iii)高于正常水平的网织红细胞,和(iv)高于正常水平的胆红素。
6.权利要求1的方法,其中所述受试者先前已经经历或正在经历用抑制补体蛋白C5的裂解的末端补体抑制剂的治疗。
7.权利要求1的方法,其中所述方法进一步包括将抑制补体蛋白C5的裂解的末端补体抑制剂施用于所述受试者。
8.权利要求7的方法,其中所述末端补体抑制剂是人源化抗C5抗体或其抗原结合片段。
9.权利要求8的方法,其中所述末端补体抑制剂是依库丽单抗。
10.权利要求2的方法,其中所述抗体或其片段选自:重组抗体、具有降低的效应子功能的抗体、嵌合抗体、人源化抗体和人抗体。
11.权利要求1的方法,其中所述组合物通过皮下、肌内、动脉内、静脉内或作为吸入剂施用。
12.权利要求11的方法,其中所述组合物通过皮下施用。
13.抑制患有非因子H依赖性非典型溶血性尿毒综合征(aHUS)或处于发生非因子H依赖性非典型溶血性尿毒综合征(aHUS)的风险中的受试者中MASP-2依赖性补体活化的方法,其包括将包含有效抑制MASP-2依赖性补体活化的量的MASP-2抑制剂的组合物施用于所述受试者。
14.权利要求13的方法,其中在施用所述组合物之前,确定所述受试者表现选自以下的一种或多种症状:(i)贫血、(ii)血小板减少(iii)肾功能不全和(iv)肌酸酐上升,并且将所述组合物以有效量施用足够长的时间期间,以改善所述一种或多种症状。
15.权利要求13的方法,其中所述受试者患有与因子I、因子B或膜辅因子CD46相关的aHUS。
16.权利要求13的方法,其中所述MASP-2抑制剂是抗MASP-2抗体或其片段。
17.权利要求13的方法,其中所述MASP-2抑制剂是特异性结合SEQ ID NO:6的部分的抗MASP-2单克隆抗体或其片段。
18.用于降低处于发生非典型溶血性尿毒综合征(aHUS)的风险中的受试者将患有与aHUS相关的临床症状的可能性的方法,其包括:
(a) 确定所述受试者中已知与aHUS相关的基因标志物的存在;
(b) 定期监测所述受试者,以确定选自贫血、血小板减少、肾功能不全和肌酸酐上升的至少一种症状的存在或不存在;和
(c) 在确定贫血、血小板减少、肾功能不全或肌酸酐上升的至少一种的存在之后,将有效抑制MASP-2依赖性补体活化的量的MASP-2抑制剂的组合物施用于所述受试者,其中将所述组合物以有效量施用足够长的时间期间,以改善所述一种或多种症状。
19.权利要求18的方法,其中所述MASP-2抑制剂是抗MASP-2抗体或其片段。
20.权利要求19的方法,其中所述MASP-2抑制剂是特异性结合SEQ ID NO:6的部分的抗MASP-2单克隆抗体或其片段。
21.权利要求18的方法,其中步骤(a)包括对从所述受试者获得的样品上进行基因筛选测试并且在选自以下的基因中鉴定与aHUS相关的至少一种基因标志物的存在:补体因子H(CFH)、因子I(CFI)、因子B (CFB)、膜辅因子CD46、C3、补体因子H-相关蛋白 (CFHR1)、抗凝蛋白血栓调节蛋白(THBD)、补体因子H-相关蛋白3(CFHR3)和补体因子H-相关蛋白4(CFHR4)。
22.权利要求18的方法,其中所述方法进一步包括监测所述受试者的已知与触发aHUS临床症状相关的事件的发生,并且在所述触发事件之前、期间或之后将所述包含MASP-2抑制剂的组合物施用于所述受试者。
23.权利要求22的方法,其中所述与触发aHUS临床症状相关的事件选自药物暴露、感染、恶性肿瘤、损伤、器官或组织移植和怀孕。
24.权利要求23的方法,其中所述感染是细菌感染。
25.权利要求18的方法,其中所述组合物通过皮下施用。
26.抑制患有继发于感染的非典型溶血性尿毒综合征(aHUS)或处于发生继发于感染的非典型溶血性尿毒综合征(aHUS)的风险中的受试者中MASP-2依赖性补体活化的方法,其包括将包含有效抑制MASP-2依赖性补体活化的量的MASP-2抑制剂的组合物施用于所述受试者。
27.权利要求26的方法,其中所述受试者患有肺炎链球菌感染相关的非肠道aHUS或处于发生与肺炎链球菌感染相关的非肠道aHUS的风险中。
28.权利要求26的方法,其中所述MASP-2抑制剂是抗MASP-2抗体或其片段。
29.权利要求28的方法,其中所述MASP-2抑制剂是特异性结合SEQ ID NO:6的部分的抗MASP-2单克隆抗体或其片段。
30.治疗患有非典型溶血性尿毒综合征(aHUS)的受试者的方法,其包括将包含有效抑制MASP-2依赖性补体活化的量的MASP-2抑制剂的组合物施用于所述受试者,其中所述MASP-2抑制剂的施用通过静脉内导管或其他导管递送方法而施用。
31.权利要求30的方法,其进一步包括用血浆置换治疗所述患者。
32.权利要求30的方法,其中所述包含MASP-2抑制剂的组合物在血浆置换不存在的情况下进行施用。
33.权利要求32的方法,其中通过导管施用所述包含MASP-2抑制剂的组合物持续第一时间期间,进一步包括施用所述包含MASP-2抑制剂的组合物持续第二时间期间,其中所述组合物在所述第二时间期间过程中通过皮下施用。
34.权利要求33的方法,进一步包括定期确定至少一种补体因子的水平,其中确定与标准值或健康受试者相比所述至少一种补体因子的水平降低表明需要继续用所述组合物治疗。
35.权利要求30的方法,其中所述MASP-2抑制剂是抗MASP-2抗体或其片段。
36.权利要求35的方法,其中所述MASP-2抑制剂是特异性结合SEQ ID NO:6的部分的抗MASP-2单克隆抗体或其片段。
37.用于降低处于发生溶血性尿毒综合征(HUS)的风险中的受试者中发生肾功能受损的可能性的方法,其包括将包含有效抑制MASP-2依赖性补体活化的量的MASP-2抑制剂的组合物施用于所述受试者。
38.权利要求37的方法,其中所述处于发生HUS的风险中的受试者表现出选自以下的至少一种或多种症状:腹泻、具有血管内红细胞破坏的涂片证据的小于30%的血细胞比容水平、血小板减少和上升的肌酸酐水平。
39.权利要求37的方法,其中所述受试者被禁忌使用抗生素的肠源性大肠杆菌所感染。
40.权利要求37的方法,其中所述受试者被志贺氏菌或沙门氏菌所感染。
41.权利要求39的方法,其中在抗生素不存在的情况下在第一时间期间过程中将所述包含MASP-2抑制剂的组合物施用于所述受试者。
42.权利要求40的方法,其中在抗生素存在的情况下在第一时间期间过程中将所述包含MASP-2抑制剂的组合物施用于所述受试者。
43.权利要求37、41或43中任一项的方法,其中在所述第一时间期间过程中通过静脉内导管或其他导管递送方法将所述MASP-2抑制剂施用于所述受试者。
44.权利要求41、权利要求42或权利要求43的方法,进一步包括施用所述包含MASP-2抑制剂的组合物持续第二时间期间,其中在所述第二时间期间过程中通过皮下施用所述组合物。
45.权利要求44的方法,进一步包括定期确定至少一种补体因子的水平,其中确定与标准值或健康受试者相比所述至少一种补体因子的水平降低表明需要继续用所述组合物治疗。
46.权利要求37的方法,其中所述MASP-2抑制剂是抗MASP-2抗体或其片段。
47.权利要求46的方法,其中所述MASP-2抑制剂是特异性结合SEQ ID NO:6的部分的抗MASP-2单克隆抗体或其片段。
48.治疗患有溶血性尿毒综合征(HUS)的受试者的方法,其包括将包含有效抑制MASP-2依赖性补体活化的量的MASP-2抑制剂的组合物施用于所述受试者,其中所述MASP-2抑制剂的施用通过静脉内导管或其他导管递送方法而施用于所述受试者。
49.权利要求48的方法,其进一步包括用血浆置换治疗所述受试者。
50.权利要求48的方法,其中所述包含MASP-2抑制剂的组合物在血浆置换不存在的情况下进行施用。
51.权利要求48的方法,其中通过导管施用所述包含MASP-2抑制剂的组合物持续第一时间期间,进一步包括施用所述包含MASP-2抑制剂的组合物持续第二时间期间,其中所述组合物在所述第二时间期间过程中通过皮下施用。
52.权利要求51的方法,进一步包括定期确定至少一种补体因子的水平,其中确定与标准值或健康受试者相比所述至少一种补体因子的水平降低表明需要继续用所述组合物治疗。
53.权利要求48的方法,其中所述MASP-2抑制剂是抗MASP-2抗体或其片段。
54.权利要求53的方法,其中所述MASP-2抑制剂是特异性结合SEQ ID NO:6的部分的抗MASP-2单克隆抗体或其片段。
55.治疗患有血栓性血小板减少性紫癜(TTP)或表现出与TTP的诊断一致的症状的受试者的方法,其包括将包含有效抑制MASP-2依赖性补体活化的量的MASP-2抑制剂的组合物施用于所述受试者,其中所述MASP-2抑制剂的施用通过静脉内导管或其他导管递送方法而施用于所述受试者。
56.权利要求55的方法,其中所述受试者表现出选自以下的一种或多种症状:中枢神经系统损害、血小板减少、严重心脏损害、严重肺损害、胃肠梗死和坏疽。
57.权利要求55的方法,其中所述受试者对于ADAMTS13抑制剂的存在测试呈阳性,并且所述方法进一步包括将免疫抑制剂施用于所述受试者。
58.权利要求55的方法,其中在血浆置换不存在的情况下施用所述包含MASP-2抑制剂的组合物持续第一时间期间。
59.权利要求55的方法,其中所述受试者对于ADAMTS-13抑制剂的存在测试呈阳性,并且所述方法进一步包括施用ADAMTS-13。
60.权利要求55的方法,其进一步包括用血浆置换治疗所述受试者。
61.权利要求55的方法,其中所述包含MASP-2抑制剂的组合物在血浆置换存在的情况下进行施用。
62.权利要求55的方法,其中通过导管施用所述包含MASP-2抑制剂的组合物持续第一时间期间,进一步包括施用所述包含MASP-2抑制剂的组合物持续第二时间期间,其中所述组合物在所述第二时间期间过程中通过皮下施用。
63.权利要求62的方法,进一步包括定期确定至少一种补体因子的水平,其中确定与标准值或健康受试者相比所述至少一种补体因子的水平降低表明需要继续用所述组合物治疗。
64.权利要求55的方法,其中所述MASP-2抑制剂是抗MASP-2抗体或其片段。
65.权利要求54的方法,其中所述MASP-2抑制剂是特异性结合SEQ ID NO:6的部分的抗MASP-2单克隆抗体或其片段。
66.治疗患有难治性血栓性血小板减少性紫癜(TTP)的受试者的方法,其包括将包含有效抑制MASP-2依赖性补体活化的量的MASP-2抑制剂的组合物施用于所述受试者。
67.权利要求66的方法,其中所述组合物通过皮下施用。
68.权利要求66的方法,进一步包括定期确定至少一种补体因子的水平,其中确定与标准值或健康受试者相比所述至少一种补体因子的水平降低表明需要继续用所述组合物治疗。
69.权利要求66的方法,其中所述MASP-2抑制剂是抗MASP-2抗体或其片段。
70.权利要求69的方法,其中所述MASP-2抑制剂是特异性结合SEQ ID NO:6的部分的抗MASP-2单克隆抗体或其片段。
71.抑制患有冷球蛋白血症的受试者中MASP-2依赖性补体活化的方法,其包括将包含有效抑制MASP-2依赖性补体活化的量的MASP-2抑制剂的组合物施用于所述受试者。
72.权利要求71的方法,其中所述MASP-2抑制剂是抗MASP-2抗体或其片段。
73.权利要求72的方法,其中所述MASP-2抑制剂是抗MASP-2单克隆抗体或其片段。
74.抑制患有冷凝集素病的受试者中MASP-2依赖性补体活化的方法,其包括将包含有效抑制MASP-2依赖性补体活化的量的MASP-2抑制剂的组合物施用于所述受试者。
75.权利要求74的方法,其中所述MASP-2抑制剂是抗MASP-2抗体或其片段。
76.权利要求75的方法,其中所述MASP-2抑制剂是抗MASP-2单克隆抗体或其片段。
77.抑制患有青光眼的受试者中MASP-2依赖性补体活化的方法,其包括将包含有效抑制MASP-2依赖性补体活化的量的MASP-2抑制剂的组合物施用于所述受试者。
78.权利要求77的方法,其中所述MASP-2抑制剂是抗MASP-2抗体或其片段。
79.权利要求78的方法,其中所述MASP-2抑制剂是抗MASP-2单克隆抗体或其片段。
80.抑制处于发生急性放射综合征的风险中或正患有急性放射综合征的受试者中MASP-2依赖性补体活化的方法,其包括将包含有效抑制MASP-2依赖性补体活化的量的MASP-2抑制剂的组合物施用于所述受试者。
81.权利要求80的方法,其中所述MASP-2抑制剂是抗MASP-2抗体或其片段。
82.权利要求81的方法,其中所述MASP-2抑制剂是抗MASP-2单克隆抗体或其片段。
83.权利要求81的方法,其中在辐射暴露之前将所述MASP-2抑制剂预防性施用于所述受试者。
84.权利要求81的方法,其中在辐射暴露之后24小时至48小时内将所述MASP-2抑制剂施用于所述受试者。
85.权利要求81的方法,其中在辐射暴露之后将所述MASP-2抑制剂施用于所述受试者。
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201710646474.7A CN107638565B (zh) | 2011-04-08 | 2012-04-06 | 用于治疗与masp-2依赖性补体活化相关的状况的方法 |
| CN201910000825.6A CN110075294A (zh) | 2011-04-08 | 2012-04-06 | 用于治疗与masp-2依赖性补体活化相关的状况的方法 |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201161473698P | 2011-04-08 | 2011-04-08 | |
| US61/473,698 | 2011-04-08 | ||
| PCT/US2012/032650 WO2012139081A2 (en) | 2011-04-08 | 2012-04-06 | Methods for treating conditions associated with masp-2 dependent complement activation |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201910000825.6A Division CN110075294A (zh) | 2011-04-08 | 2012-04-06 | 用于治疗与masp-2依赖性补体活化相关的状况的方法 |
| CN201710646474.7A Division CN107638565B (zh) | 2011-04-08 | 2012-04-06 | 用于治疗与masp-2依赖性补体活化相关的状况的方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN103781492A true CN103781492A (zh) | 2014-05-07 |
Family
ID=46966283
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201280028263.2A Pending CN103781492A (zh) | 2011-04-08 | 2012-04-06 | 用于治疗与masp-2依赖性补体活化相关的状况的方法 |
| CN201910000825.6A Pending CN110075294A (zh) | 2011-04-08 | 2012-04-06 | 用于治疗与masp-2依赖性补体活化相关的状况的方法 |
| CN201710646474.7A Active CN107638565B (zh) | 2011-04-08 | 2012-04-06 | 用于治疗与masp-2依赖性补体活化相关的状况的方法 |
Family Applications After (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201910000825.6A Pending CN110075294A (zh) | 2011-04-08 | 2012-04-06 | 用于治疗与masp-2依赖性补体活化相关的状况的方法 |
| CN201710646474.7A Active CN107638565B (zh) | 2011-04-08 | 2012-04-06 | 用于治疗与masp-2依赖性补体活化相关的状况的方法 |
Country Status (18)
| Country | Link |
|---|---|
| US (5) | US8951522B2 (zh) |
| EP (3) | EP3964233A1 (zh) |
| JP (4) | JP5937197B2 (zh) |
| KR (6) | KR101720562B1 (zh) |
| CN (3) | CN103781492A (zh) |
| AU (1) | AU2012239889B2 (zh) |
| BR (1) | BR112013025917A2 (zh) |
| CA (3) | CA3076975A1 (zh) |
| CL (1) | CL2013002874A1 (zh) |
| DK (2) | DK2694108T3 (zh) |
| ES (2) | ES2894342T3 (zh) |
| HK (1) | HK1250342A1 (zh) |
| IL (3) | IL228758B (zh) |
| MX (2) | MX339002B (zh) |
| NZ (5) | NZ746139A (zh) |
| PL (1) | PL3287142T3 (zh) |
| RU (2) | RU2743409C2 (zh) |
| WO (1) | WO2012139081A2 (zh) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN108699121A (zh) * | 2015-12-23 | 2018-10-23 | 格林诺瓦森生物技术股份有限公司 | 用于抑制补体激活的多肽 |
| CN110506116A (zh) * | 2017-03-09 | 2019-11-26 | 协和麒麟株式会社 | 抑制masp2表达的核酸 |
| CN113674860A (zh) * | 2020-05-15 | 2021-11-19 | 北京大学人民医院 | 一种难治性iTTP风险预测装置、系统及其应用 |
| WO2022228364A1 (zh) * | 2021-04-25 | 2022-11-03 | 江苏恒瑞医药股份有限公司 | 抗masp2抗体、其抗原结合片段及医药用途 |
Families Citing this family (47)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20090010920A1 (en) | 2003-03-03 | 2009-01-08 | Xencor, Inc. | Fc Variants Having Decreased Affinity for FcyRIIb |
| US9644035B2 (en) | 2011-04-08 | 2017-05-09 | Omeros Corporation | Methods for treating conditions associated with MASP-2 dependent complement activation |
| PL3287142T3 (pl) | 2011-04-08 | 2021-12-27 | University Of Leicester | Sposoby leczenia chorób związanych z aktywacją dopełniacza zależną od masp-2 |
| CN106831985A (zh) | 2011-12-21 | 2017-06-13 | 诺华股份有限公司 | 用于抗体靶定p因子的组合物和方法 |
| SI2833907T1 (en) * | 2012-04-06 | 2018-07-31 | Omeros Corporation | COMPOSITION AND METHODS OF INJECTION OF MASP-1 AND / OR MASP-3 FOR TREATMENT OF PAROXYSIMAL NIGHT CHEMOGLOBINURIA |
| BR112014031522A2 (pt) * | 2012-06-18 | 2017-08-01 | Omeros Corp | métodos para inibir a ativação de complemento dependente de masp-3, para inibir a ativação de complemento dependente de masp-2 e para fabricar um medicamento |
| CN104870475B (zh) * | 2012-10-25 | 2019-11-08 | 美国比奥维拉迪维股份有限公司 | 抗补体C1s抗体和其用途 |
| LT2914291T (lt) | 2012-11-02 | 2022-06-10 | Bioverativ Usa Inc. | Antikūnai prieš komplemento c1s ir jų panaudojimo būdai |
| US20140363433A1 (en) | 2013-03-15 | 2014-12-11 | Omeros Corporation | Methods of Generating Bioactive Peptide-bearing Antibodies and Compositions Comprising the Same |
| NZ729747A (en) | 2013-03-15 | 2020-03-27 | Omeros Corp | Methods of generating bioactive peptide-bearing antibodies and compositions comprising the same |
| EP3057993B1 (en) | 2013-10-17 | 2020-08-12 | Omeros Corporation | Methods for treating conditions associated with masp-2 dependent complement activation |
| WO2015070041A1 (en) * | 2013-11-08 | 2015-05-14 | Icahn School Of Medicine At Mount Sinai | Methods for monitoring kidney dysfunction |
| CA2949985C (en) | 2014-06-12 | 2023-10-17 | Ra Pharmaceuticals, Inc. | Modulation of complement activity |
| US9937222B2 (en) | 2015-01-28 | 2018-04-10 | Ra Pharmaceuticals, Inc. | Modulators of complement activity |
| IL254670B2 (en) | 2015-04-06 | 2023-04-01 | Bioverativ Usa Inc | Human anti-c1s antibodies and methods of using them |
| KR102432062B1 (ko) * | 2015-11-09 | 2022-08-12 | 오메로스 코포레이션 | Masp-2 의존성 보체 활성화와 관련된 질병의 치료 방법 |
| KR20180094913A (ko) | 2015-12-16 | 2018-08-24 | 라 파마슈티컬스 인코포레이티드 | 보체 활성의 조절인자 |
| US10736960B2 (en) * | 2016-01-05 | 2020-08-11 | Omeros Corporation | Methods for inhibiting fibrosis in a subject in need thereof |
| IL305200A (en) | 2016-03-31 | 2023-10-01 | Omeros Corp | MASP-2 suppressing agents for use in suppressing angiogenesis |
| JOP20170170B1 (ar) | 2016-08-31 | 2022-09-15 | Omeros Corp | صيغ لجسم مضاد تثبيطية لـ masp-2 بتركيز عالي ولزوجة منخفضة وأطقم، وطرق |
| JOP20190068A1 (ar) * | 2016-10-13 | 2019-04-01 | Omeros Corp | طرق لتقليل البول البروتيني في خاضع بشري يعاني من الاعتلال الكلوي a الناتج عن الجلوبيولين المناعي |
| JP7301741B2 (ja) | 2016-12-07 | 2023-07-03 | ラ ファーマシューティカルズ インコーポレイテッド | 補体活性のモジュレータ |
| JP6442703B2 (ja) * | 2016-12-26 | 2018-12-26 | 日本ビーシージー製造株式会社 | 細菌細胞壁骨格成分を含有する水中油型エマルション製剤 |
| EP3689355A4 (en) * | 2017-07-28 | 2021-09-08 | Lemonex Inc. | PHARMACEUTICAL COMPOSITION FOR THE PREVENTION OR TREATMENT OF LIVER CANCER |
| KR20200095485A (ko) | 2017-12-04 | 2020-08-10 | 라 파마슈티컬스 인코포레이티드 | 보체 활성의 조절인자 |
| GB201800620D0 (en) | 2018-01-15 | 2018-02-28 | Univ Manchester | C3b Binding Polypeptide |
| EP3802489A4 (en) * | 2018-05-29 | 2022-04-13 | Omeros Corporation | MASP -2 INHIBITORS AND METHODS OF USE |
| RU2699040C1 (ru) * | 2018-07-23 | 2019-09-03 | Федеральное государственное бюджетное учреждение науки "Уральский научно-практический центр радиационной медицины Федерального медико-биологического агентства" (ФГБУН УНПЦ РМ ФМБА России) | Способ экстренной профилактики и лечения острой лучевой болезни (варианты) |
| CN111655849A (zh) | 2018-08-21 | 2020-09-11 | 苏州瑞博生物技术有限公司 | 一种核酸、含有该核酸的药物组合物和缀合物及其用途 |
| EP3628735A1 (en) | 2018-09-25 | 2020-04-01 | Centre National De La Recherche Scientifique | Antisense rna targeting pmp22 for the treatment of charcot-marie-tooth 1a disease |
| CN111655297A (zh) | 2018-09-30 | 2020-09-11 | 苏州瑞博生物技术有限公司 | 一种siRNA缀合物及其制备方法和用途 |
| MX2021005758A (es) * | 2018-11-15 | 2021-09-21 | Ionis Pharmaceuticals Inc | Moduladores de la expresión de irf5. |
| CN114729354A (zh) * | 2018-12-25 | 2022-07-08 | 中国医学科学院基础医学研究所 | 炎性相关疾病防治的小rna药物及其组合 |
| WO2020185541A2 (en) | 2019-03-08 | 2020-09-17 | Ra Pharmaceuticals, Inc. | Modulators of complement activity |
| US20220162607A1 (en) * | 2019-03-14 | 2022-05-26 | Rena Therapeutics Inc. | Nucleic acid complex for modulating ihh expression |
| US20230115176A1 (en) | 2019-03-29 | 2023-04-13 | Ra Pharmaceuticals, Inc. | Complement Modulators and Related Methods |
| AU2020261059A1 (en) | 2019-04-24 | 2021-10-14 | Ra Pharmaceuticals, Inc. | Compositions and methods for modulating complement activity |
| JP2022533419A (ja) * | 2019-05-22 | 2022-07-22 | スーチョウ リボ ライフ サイエンス カンパニー、リミテッド | 核酸、薬物組成物及び複合体ならびに調製方法と使用 |
| CN111024947A (zh) * | 2019-11-19 | 2020-04-17 | 江苏美克医学技术有限公司 | 白色念珠菌荧光免疫层析测定试剂盒及其制备方法 |
| WO2021107057A1 (ja) | 2019-11-27 | 2021-06-03 | 京セラ株式会社 | 通信制御方法及びユーザ装置 |
| CN115052862A (zh) | 2019-12-04 | 2022-09-13 | 奥默罗斯公司 | Masp-2抑制剂和使用方法 |
| US20210171461A1 (en) | 2019-12-04 | 2021-06-10 | Omeros Corporation | Masp-2 inhibitors and methods of use |
| JP2023504543A (ja) | 2019-12-04 | 2023-02-03 | オメロス コーポレーション | Masp-2阻害剤および使用方法 |
| IL293588A (en) | 2019-12-04 | 2022-08-01 | Omeros Corp | 2-masp inhibitor compounds, preparations containing them and their uses |
| EP4107165A1 (en) * | 2020-02-19 | 2022-12-28 | Alnylam Pharmaceuticals, Inc. | Mannan binding lectin serine peptidase 2 (masp2) irna compositions and methods of use thereof |
| KR102314642B1 (ko) * | 2020-04-17 | 2021-10-20 | 재단법인 아산사회복지재단 | 파브리 병 진단용 바이오 마커 및 이의 용도 |
| US20240084301A1 (en) * | 2022-07-25 | 2024-03-14 | Amgen Inc. | Rnai constructs and methods for inhibiting fam13a expression |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20050169921A1 (en) * | 2004-02-03 | 2005-08-04 | Leonard Bell | Method of treating hemolytic disease |
| CN101018565A (zh) * | 2004-06-10 | 2007-08-15 | 奥默罗斯公司 | 用于治疗与masp-2依赖的补体活化相关的疾病的方法 |
| CN101460195A (zh) * | 2006-04-03 | 2009-06-17 | 莱斯特大学 | 用于治疗与masp-2依赖性补体活化相关的疾病的方法 |
Family Cites Families (71)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4331647A (en) | 1980-03-03 | 1982-05-25 | Goldenberg Milton David | Tumor localization and therapy with labeled antibody fragments specific to tumor-associated markers |
| US4394370A (en) | 1981-09-21 | 1983-07-19 | Jefferies Steven R | Bone graft material for osseous defects and method of making same |
| US4816567A (en) | 1983-04-08 | 1989-03-28 | Genentech, Inc. | Recombinant immunoglobin preparations |
| US4526909A (en) | 1984-01-09 | 1985-07-02 | Regents Of The University Of California | Polymethylmethacrylate delivery system for bone morphogenetic protein |
| US4563489A (en) | 1984-02-10 | 1986-01-07 | University Of California | Biodegradable organic polymer delivery system for bone morphogenetic protein |
| JPH0662679B2 (ja) | 1985-06-21 | 1994-08-17 | 新田ゼラチン株式会社 | 組織親和性コラ−ゲンとその製法 |
| US5453566A (en) | 1986-03-28 | 1995-09-26 | Calgene, Inc. | Antisense regulation of gene expression in plant/cells |
| US4946778A (en) | 1987-09-21 | 1990-08-07 | Genex Corporation | Single polypeptide chain binding molecules |
| US4987071A (en) | 1986-12-03 | 1991-01-22 | University Patents, Inc. | RNA ribozyme polymerases, dephosphorylases, restriction endoribonucleases and methods |
| EP0640688A1 (en) | 1987-12-15 | 1995-03-01 | Gene Shears Pty. Limited | Ribozymes |
| US5223409A (en) | 1988-09-02 | 1993-06-29 | Protein Engineering Corp. | Directed evolution of novel binding proteins |
| GB8822492D0 (en) | 1988-09-24 | 1988-10-26 | Considine J | Apparatus for removing tumours from hollow organs of body |
| US5211657A (en) | 1988-11-07 | 1993-05-18 | The United States Government As Represented By The Secretary Of The Department Of Health And Human Services | Laminin a chain deduced amino acid sequence, expression vectors and active synthetic peptides |
| US5530101A (en) | 1988-12-28 | 1996-06-25 | Protein Design Labs, Inc. | Humanized immunoglobulins |
| US5549910A (en) | 1989-03-31 | 1996-08-27 | The Regents Of The University Of California | Preparation of liposome and lipid complex compositions |
| US6075181A (en) * | 1990-01-12 | 2000-06-13 | Abgenix, Inc. | Human antibodies derived from immunized xenomice |
| EP0438803B1 (en) | 1990-01-26 | 1997-03-12 | Immunomedics, Inc. | Vaccines against cancer and infectious diseases |
| JP3218637B2 (ja) | 1990-07-26 | 2001-10-15 | 大正製薬株式会社 | 安定なリポソーム水懸濁液 |
| US5789573A (en) | 1990-08-14 | 1998-08-04 | Isis Pharmaceuticals, Inc. | Antisense inhibition of ICAM-1, E-selectin, and CMV IE1/IE2 |
| JP2958076B2 (ja) | 1990-08-27 | 1999-10-06 | 株式会社ビタミン研究所 | 遺伝子導入用多重膜リポソーム及び遺伝子捕捉多重膜リポソーム製剤並びにその製法 |
| DE122004000008I1 (de) | 1991-06-14 | 2005-06-09 | Genentech Inc | Humanisierter Heregulin Antikörper. |
| IL108367A0 (en) | 1993-01-27 | 1994-04-12 | Hektoen Inst For Medical Resea | Antisense polynzcleotide inhibition of human growth factor-sensitive cancer cells |
| US5801154A (en) | 1993-10-18 | 1998-09-01 | Isis Pharmaceuticals, Inc. | Antisense oligonucleotide modulation of multidrug resistance-associated protein |
| US5856121A (en) | 1994-02-24 | 1999-01-05 | Case Western Reserve University | Growth arrest homebox gene |
| JPH07238100A (ja) | 1994-02-25 | 1995-09-12 | Sumitomo Electric Ind Ltd | ヒトのmaspに対するモノクローナル抗体 |
| US5741516A (en) | 1994-06-20 | 1998-04-21 | Inex Pharmaceuticals Corporation | Sphingosomes for enhanced drug delivery |
| JPH10510540A (ja) | 1994-12-12 | 1998-10-13 | オメロス メディカル システムズ,インコーポレーテッド | 灌注用溶液並びに疼痛、炎症及びけいれんの抑制法 |
| US5795587A (en) | 1995-01-23 | 1998-08-18 | University Of Pittsburgh | Stable lipid-comprising drug delivery complexes and methods for their production |
| US5738868A (en) | 1995-07-18 | 1998-04-14 | Lipogenics Ltd. | Liposome compositions and kits therefor |
| US5739119A (en) | 1996-11-15 | 1998-04-14 | Galli; Rachel L. | Antisense oligonucleotides specific for the muscarinic type 2 acetylcholine receptor MRNA |
| US7083786B2 (en) | 1997-04-03 | 2006-08-01 | Jensenius Jens Chr | MASP-2, a complement-fixing enzyme, and uses for it |
| US6297024B1 (en) | 1998-10-15 | 2001-10-02 | Cell Activation, Inc. | Methods for assessing complement activation |
| WO2000035483A1 (en) | 1998-12-15 | 2000-06-22 | The Brigham And Women's Hospital, Inc. | Methods and products for regulating lectin complement pathway associated complement activation |
| US7273925B1 (en) | 1998-12-15 | 2007-09-25 | Brigham And Women's Hospital, Inc. | Methods and products for regulating lectin complement pathway associated complement activation |
| US6235494B1 (en) | 1999-02-08 | 2001-05-22 | The Scripps Research Institute | Substrates for assessing mannan-binding protein-associated serine protease activity and methods using the substrates |
| KR20020012320A (ko) | 1999-07-21 | 2002-02-15 | 그레고리 에이. 데모풀로스 | 동통, 염증 및 연골 퇴화를 억제하기 위한 용액 및 억제방법 |
| US6794132B2 (en) | 1999-10-02 | 2004-09-21 | Biosite, Inc. | Human antibodies |
| CA2391402A1 (en) | 1999-12-02 | 2001-06-07 | Jens Christian Jensenius | Masp-3, a complement-fixing enzyme, and uses for it |
| US6649592B1 (en) | 2000-01-14 | 2003-11-18 | Science & Technology Corporation @ Unm | Peptide inhibitors of LFA-1/ICAM-1 interaction |
| SG98393A1 (en) | 2000-05-19 | 2003-09-19 | Inst Materials Research & Eng | Injectable drug delivery systems with cyclodextrin-polymer based hydrogels |
| AU2001279586A1 (en) | 2000-07-13 | 2002-01-30 | Jens Christian Jensenius | Masp-2, a complement-fixing enzyme, and uses for it |
| WO2003009803A2 (en) | 2001-07-26 | 2003-02-06 | Alexion Pharmaceuticals Inc. | Method of improving cognitive function |
| WO2003063799A2 (en) | 2002-02-01 | 2003-08-07 | Omeros Corporation | Compositions and methods for systemic inhibition of cartilage degradation |
| AU2003223288A1 (en) | 2002-03-18 | 2003-10-08 | Alexion Pharmaceuticals, Inc. | Stratification of patient populations having or suspected of having rheumatoid arthritis |
| ES2601143T3 (es) | 2002-07-19 | 2017-02-14 | Omeros Corporation | Copolímeros tribloque biodegradables, métodos de síntesis de los mismos, e hidrogeles y biomateriales preparados a partir de los mismos |
| AU2003261304A1 (en) | 2002-07-30 | 2004-02-16 | Omeros Corporation | Ophthalmologic irrigation solutions and method |
| ITRM20020511A1 (it) * | 2002-10-09 | 2004-04-10 | Santa Anna Acuto | Uso dell'insulator sns di riccio di mare per la terapia genica di malattie delle cellule eritroidi. |
| AU2003283217A1 (en) | 2002-12-03 | 2004-06-23 | Aarhus Amt | Method for determing predisposition to manifestation of immune system related diseases |
| AU2004216176B2 (en) | 2003-02-21 | 2008-04-03 | Genentech, Inc. | Methods for preventing and treating tissue damage associated with ischemia-reperfusion injury |
| WO2004083250A1 (ja) * | 2003-03-17 | 2004-09-30 | Juridical Foundation The Chemo-Sero-Therapeutic Research Institute | フォンビルブランド因子特異的切断酵素に対する抗体の認識領域からなる構成物 |
| EP2374819B1 (en) | 2003-05-12 | 2017-03-22 | Helion Biotech ApS | Antibodies to MASP-2 |
| GB0412966D0 (en) | 2004-06-10 | 2004-07-14 | Univ Leicester | Genetically modified non-human mammals and cells |
| US8840893B2 (en) | 2004-06-10 | 2014-09-23 | Omeros Corporation | Methods for treating conditions associated with MASP-2 dependent complement activation |
| US20060018896A1 (en) | 2004-06-10 | 2006-01-26 | University Of Leicester | Methods for treating conditions associated with lectin-dependent complement activation |
| ATE544866T1 (de) * | 2005-06-17 | 2012-02-15 | Baxter Int | Adamts13-haltige zusammensetzungen mit thrombolytischer wirkung |
| WO2007086069A1 (en) | 2006-01-27 | 2007-08-02 | Rappaport Family Institute For Research In The Medical Sciences | Methods and kits for determining blood coagulation |
| RU2477137C2 (ru) | 2006-03-08 | 2013-03-10 | АРКЕМИКС Эл Эл Си | Связывающие комплемент аптамеры и средства против с5, пригодные для лечения глазных нарушений |
| CA2742802C (en) * | 2008-11-10 | 2019-11-26 | Alexion Pharmaceuticals, Inc. | Methods and compositions for treating complement-associated disorders |
| US20110002931A1 (en) | 2009-06-23 | 2011-01-06 | Alexion Pharmaceuticals, Inc. | Bispecific antibodies that bind to complement proteins |
| AU2010272483B2 (en) | 2009-07-17 | 2016-07-21 | Omeros Corporation | MASP isoforms as inhibitors of complement activation |
| PT2488203T (pt) | 2009-10-16 | 2017-03-10 | Univ Leicester | Métodos para tratamento de coagulação intravascular disseminada através da inibição da activação do complemento dependente de masp-2 |
| CN102958535A (zh) * | 2009-11-05 | 2013-03-06 | 亚力史剑桥公司 | 阵发性夜间血红蛋白尿、溶血性贫血和涉及血管内和血管外溶血的疾病状态的治疗 |
| MX2012010116A (es) | 2010-03-01 | 2013-02-26 | Alexion Pharma Inc | Metodos y composiciones para el tratamiento de la enfermedad de degos. |
| PL3287142T3 (pl) | 2011-04-08 | 2021-12-27 | University Of Leicester | Sposoby leczenia chorób związanych z aktywacją dopełniacza zależną od masp-2 |
| US20150166676A1 (en) | 2011-04-08 | 2015-06-18 | Omeros Corporation | Methods for Treating Conditions Associated with MASP-2 Dependent Complement Activation |
| US9644035B2 (en) | 2011-04-08 | 2017-05-09 | Omeros Corporation | Methods for treating conditions associated with MASP-2 dependent complement activation |
| CN107011443B (zh) | 2011-05-04 | 2021-04-30 | 奥默罗斯公司 | 用于抑制masp-2依赖的补体活化的组合物 |
| BR112014031522A2 (pt) | 2012-06-18 | 2017-08-01 | Omeros Corp | métodos para inibir a ativação de complemento dependente de masp-3, para inibir a ativação de complemento dependente de masp-2 e para fabricar um medicamento |
| EP3057993B1 (en) | 2013-10-17 | 2020-08-12 | Omeros Corporation | Methods for treating conditions associated with masp-2 dependent complement activation |
| KR102432062B1 (ko) | 2015-11-09 | 2022-08-12 | 오메로스 코포레이션 | Masp-2 의존성 보체 활성화와 관련된 질병의 치료 방법 |
| JPWO2018070521A1 (ja) * | 2016-10-14 | 2019-08-15 | 公立大学法人福島県立医科大学 | 補体の活性化経路を阻害する融合ポリペプチド |
-
2012
- 2012-04-06 PL PL17191208T patent/PL3287142T3/pl unknown
- 2012-04-06 ES ES17191208T patent/ES2894342T3/es active Active
- 2012-04-06 CA CA3076975A patent/CA3076975A1/en active Pending
- 2012-04-06 CN CN201280028263.2A patent/CN103781492A/zh active Pending
- 2012-04-06 EP EP21188935.7A patent/EP3964233A1/en active Pending
- 2012-04-06 MX MX2013011721A patent/MX339002B/es active IP Right Grant
- 2012-04-06 EP EP12767899.3A patent/EP2694108B1/en active Active
- 2012-04-06 KR KR1020137029501A patent/KR101720562B1/ko active IP Right Grant
- 2012-04-06 KR KR1020177007747A patent/KR101870915B1/ko active IP Right Grant
- 2012-04-06 AU AU2012239889A patent/AU2012239889B2/en active Active
- 2012-04-06 DK DK12767899.3T patent/DK2694108T3/en active
- 2012-04-06 MX MX2016006039A patent/MX355648B/es unknown
- 2012-04-06 KR KR1020217004368A patent/KR20210021101A/ko not_active Application Discontinuation
- 2012-04-06 NZ NZ746139A patent/NZ746139A/en unknown
- 2012-04-06 JP JP2014504058A patent/JP5937197B2/ja active Active
- 2012-04-06 KR KR1020227010108A patent/KR20220044616A/ko not_active Application Discontinuation
- 2012-04-06 NZ NZ617298A patent/NZ617298A/en unknown
- 2012-04-06 RU RU2018125514A patent/RU2743409C2/ru active
- 2012-04-06 US US13/441,827 patent/US8951522B2/en active Active
- 2012-04-06 RU RU2013149792A patent/RU2662563C2/ru active
- 2012-04-06 ES ES12767899T patent/ES2683307T3/es active Active
- 2012-04-06 CA CA2832187A patent/CA2832187C/en active Active
- 2012-04-06 BR BR112013025917A patent/BR112013025917A2/pt not_active Application Discontinuation
- 2012-04-06 WO PCT/US2012/032650 patent/WO2012139081A2/en active Application Filing
- 2012-04-06 NZ NZ717517A patent/NZ717517A/en unknown
- 2012-04-06 DK DK17191208.2T patent/DK3287142T3/da active
- 2012-04-06 CN CN201910000825.6A patent/CN110075294A/zh active Pending
- 2012-04-06 CA CA2977009A patent/CA2977009C/en active Active
- 2012-04-06 KR KR1020197037437A patent/KR102217658B1/ko active IP Right Grant
- 2012-04-06 NZ NZ731596A patent/NZ731596A/en unknown
- 2012-04-06 NZ NZ709997A patent/NZ709997A/en unknown
- 2012-04-06 CN CN201710646474.7A patent/CN107638565B/zh active Active
- 2012-04-06 EP EP17191208.2A patent/EP3287142B1/en active Active
- 2012-04-06 KR KR1020187017317A patent/KR20180072851A/ko active Application Filing
-
2013
- 2013-10-06 IL IL228758A patent/IL228758B/en active IP Right Grant
- 2013-10-07 CL CL2013002874A patent/CL2013002874A1/es unknown
-
2014
- 2014-12-23 US US14/581,191 patent/US20150239985A1/en not_active Abandoned
-
2016
- 2016-05-11 JP JP2016095034A patent/JP6239030B2/ja active Active
- 2016-12-07 US US15/371,726 patent/US10202465B2/en active Active
-
2017
- 2017-10-31 JP JP2017209894A patent/JP6584476B2/ja active Active
-
2018
- 2018-07-30 HK HK18109793.0A patent/HK1250342A1/zh unknown
- 2018-12-17 US US16/222,188 patent/US20190292272A1/en not_active Abandoned
-
2019
- 2019-01-09 IL IL264172A patent/IL264172B/en unknown
- 2019-09-03 JP JP2019160040A patent/JP6893539B2/ja active Active
-
2020
- 2020-04-23 IL IL274206A patent/IL274206A/en unknown
-
2021
- 2021-09-09 US US17/470,386 patent/US20220089781A1/en active Pending
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20050169921A1 (en) * | 2004-02-03 | 2005-08-04 | Leonard Bell | Method of treating hemolytic disease |
| CN101018565A (zh) * | 2004-06-10 | 2007-08-15 | 奥默罗斯公司 | 用于治疗与masp-2依赖的补体活化相关的疾病的方法 |
| US20110020337A1 (en) * | 2004-06-10 | 2011-01-27 | Omeros Corporation | Methods for treating conditions associated with masp-2 dependent complement activation |
| CN101460195A (zh) * | 2006-04-03 | 2009-06-17 | 莱斯特大学 | 用于治疗与masp-2依赖性补体活化相关的疾病的方法 |
Non-Patent Citations (1)
| Title |
|---|
| HILL ANITA等: "Recent developments in the understanding and management of paroxysmal nocturnal haemoglobinuria", 《BRITISH JOURNAL OF HAEMATOLOGY》 * |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN108699121A (zh) * | 2015-12-23 | 2018-10-23 | 格林诺瓦森生物技术股份有限公司 | 用于抑制补体激活的多肽 |
| US11591378B2 (en) | 2015-12-23 | 2023-02-28 | eleva GmbH | Polypeptides for inhibiting complement activation |
| CN108699121B (zh) * | 2015-12-23 | 2023-07-04 | 艾丽法有限责任公司 | 用于抑制补体激活的多肽 |
| CN110506116A (zh) * | 2017-03-09 | 2019-11-26 | 协和麒麟株式会社 | 抑制masp2表达的核酸 |
| CN113674860A (zh) * | 2020-05-15 | 2021-11-19 | 北京大学人民医院 | 一种难治性iTTP风险预测装置、系统及其应用 |
| WO2022228364A1 (zh) * | 2021-04-25 | 2022-11-03 | 江苏恒瑞医药股份有限公司 | 抗masp2抗体、其抗原结合片段及医药用途 |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6893539B2 (ja) | Masp−2依存性補体活性化に関連した状態を治療するための方法 | |
| RU2718850C2 (ru) | Способы лечения состояний, ассоциированных с masp-2-зависимой активацией комплемента | |
| CN102781471B (zh) | 通过抑制masp-2依赖性补体活化治疗弥散性血管内凝血的方法 | |
| CN104717975A (zh) | 用于治疗各种疾病和病症的抑制masp-1和/或masp-2和/或masp-3的组合物和方法 | |
| CN108472347B (zh) | 用于治疗与masp-2依赖性补体活化相关的病况的方法 | |
| TWI818919B (zh) | 用於治療和/ 或預防與造血幹細胞移植有關的移植物抗宿主病和/ 或瀰漫性肺泡出血和/ 或靜脈閉塞性病的方法 | |
| AU2015271989B2 (en) | Methods for treating conditions associated with MASP-2 dependent complement activation | |
| AU2013201627B2 (en) | Methods for treating conditions associated with MASP-2 dependent complement activation |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| RJ01 | Rejection of invention patent application after publication | ||
| RJ01 | Rejection of invention patent application after publication |
Application publication date: 20140507 |