CN103179980A - 艾杜糖醛酸-2-硫酸酯酶的cns递送的方法和组合物 - Google Patents
艾杜糖醛酸-2-硫酸酯酶的cns递送的方法和组合物 Download PDFInfo
- Publication number
- CN103179980A CN103179980A CN201180040898XA CN201180040898A CN103179980A CN 103179980 A CN103179980 A CN 103179980A CN 201180040898X A CN201180040898X A CN 201180040898XA CN 201180040898 A CN201180040898 A CN 201180040898A CN 103179980 A CN103179980 A CN 103179980A
- Authority
- CN
- China
- Prior art keywords
- sheath
- preparation
- enzyme
- protein
- brain
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 102100029199 Iduronate 2-sulfatase Human genes 0.000 title claims abstract description 308
- 239000000203 mixture Substances 0.000 title claims abstract description 120
- 238000000034 method Methods 0.000 title claims abstract description 115
- 238000012384 transportation and delivery Methods 0.000 title abstract description 11
- 101710096421 Iduronate 2-sulfatase Proteins 0.000 title abstract description 4
- 238000011282 treatment Methods 0.000 claims abstract description 246
- 108090000623 proteins and genes Proteins 0.000 claims abstract description 128
- 102000004169 proteins and genes Human genes 0.000 claims abstract description 125
- 238000009472 formulation Methods 0.000 claims abstract description 72
- 208000022018 mucopolysaccharidosis type 2 Diseases 0.000 claims abstract description 35
- 201000002273 mucopolysaccharidosis II Diseases 0.000 claims abstract description 32
- 239000004094 surface-active agent Substances 0.000 claims abstract description 17
- 229920000136 polysorbate Polymers 0.000 claims abstract description 12
- 229950008882 polysorbate Drugs 0.000 claims abstract description 12
- 150000003839 salts Chemical class 0.000 claims abstract description 11
- 101000840540 Homo sapiens Iduronate 2-sulfatase Proteins 0.000 claims description 302
- 238000002360 preparation method Methods 0.000 claims description 215
- 210000004556 brain Anatomy 0.000 claims description 208
- 210000001519 tissue Anatomy 0.000 claims description 198
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical group [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 claims description 139
- 235000018102 proteins Nutrition 0.000 claims description 124
- 210000004027 cell Anatomy 0.000 claims description 68
- 210000002569 neuron Anatomy 0.000 claims description 60
- 229910019142 PO4 Inorganic materials 0.000 claims description 57
- 239000010452 phosphate Substances 0.000 claims description 57
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 claims description 55
- 210000003712 lysosome Anatomy 0.000 claims description 53
- 230000001868 lysosomic effect Effects 0.000 claims description 53
- 230000006641 stabilisation Effects 0.000 claims description 52
- 230000000694 effects Effects 0.000 claims description 51
- 238000011105 stabilization Methods 0.000 claims description 51
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 claims description 42
- 210000000278 spinal cord Anatomy 0.000 claims description 42
- 239000011780 sodium chloride Substances 0.000 claims description 41
- 229920001213 Polysorbate 20 Polymers 0.000 claims description 39
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 claims description 39
- 229940068977 polysorbate 20 Drugs 0.000 claims description 38
- 238000001990 intravenous administration Methods 0.000 claims description 36
- 230000002255 enzymatic effect Effects 0.000 claims description 31
- 210000004185 liver Anatomy 0.000 claims description 31
- 208000024891 symptom Diseases 0.000 claims description 31
- 210000004885 white matter Anatomy 0.000 claims description 28
- 230000002490 cerebral effect Effects 0.000 claims description 26
- 239000000872 buffer Substances 0.000 claims description 25
- 239000003795 chemical substances by application Substances 0.000 claims description 25
- 210000003734 kidney Anatomy 0.000 claims description 22
- 210000004705 lumbosacral region Anatomy 0.000 claims description 20
- 210000002216 heart Anatomy 0.000 claims description 18
- 125000003275 alpha amino acid group Chemical group 0.000 claims description 16
- 210000000133 brain stem Anatomy 0.000 claims description 16
- 210000004884 grey matter Anatomy 0.000 claims description 16
- 239000007788 liquid Substances 0.000 claims description 10
- 230000004044 response Effects 0.000 claims description 10
- 229940071643 prefilled syringe Drugs 0.000 claims description 9
- 210000000877 corpus callosum Anatomy 0.000 claims description 8
- 210000004786 perivascular cell Anatomy 0.000 claims description 8
- 210000005250 spinal neuron Anatomy 0.000 claims description 8
- 150000002148 esters Chemical class 0.000 claims description 7
- 238000002650 immunosuppressive therapy Methods 0.000 claims description 7
- 210000004498 neuroglial cell Anatomy 0.000 claims description 7
- 210000000449 purkinje cell Anatomy 0.000 claims description 7
- 239000005388 borosilicate glass Substances 0.000 claims description 6
- 230000002829 reductive effect Effects 0.000 claims description 6
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 claims description 5
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 claims description 5
- 206010003694 Atrophy Diseases 0.000 claims description 4
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 claims description 4
- 239000007983 Tris buffer Substances 0.000 claims description 4
- 206010072731 White matter lesion Diseases 0.000 claims description 4
- 230000037444 atrophy Effects 0.000 claims description 4
- 208000010877 cognitive disease Diseases 0.000 claims description 4
- 239000011347 resin Substances 0.000 claims description 4
- 229920005989 resin Polymers 0.000 claims description 4
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 claims description 3
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 claims description 3
- 239000003708 ampul Substances 0.000 claims description 3
- 239000000088 plastic resin Substances 0.000 claims description 3
- 229920001296 polysiloxane Polymers 0.000 claims description 3
- 229910052710 silicon Inorganic materials 0.000 claims description 3
- 239000010703 silicon Substances 0.000 claims description 3
- 239000004447 silicone coating Substances 0.000 claims description 3
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 claims description 3
- 238000012385 systemic delivery Methods 0.000 claims description 3
- 229920001219 Polysorbate 40 Polymers 0.000 claims description 2
- 229920001214 Polysorbate 60 Polymers 0.000 claims description 2
- 230000001404 mediated effect Effects 0.000 claims description 2
- 239000000244 polyoxyethylene sorbitan monooleate Substances 0.000 claims description 2
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 claims description 2
- 239000000249 polyoxyethylene sorbitan monopalmitate Substances 0.000 claims description 2
- 235000010483 polyoxyethylene sorbitan monopalmitate Nutrition 0.000 claims description 2
- 239000001818 polyoxyethylene sorbitan monostearate Substances 0.000 claims description 2
- 235000010989 polyoxyethylene sorbitan monostearate Nutrition 0.000 claims description 2
- 229940101027 polysorbate 40 Drugs 0.000 claims description 2
- 229940113124 polysorbate 60 Drugs 0.000 claims description 2
- 229920000053 polysorbate 80 Polymers 0.000 claims description 2
- 229940068968 polysorbate 80 Drugs 0.000 claims description 2
- 239000000843 powder Substances 0.000 claims description 2
- 108090000790 Enzymes Proteins 0.000 abstract description 264
- 102000004190 Enzymes Human genes 0.000 abstract description 262
- 208000015439 Lysosomal storage disease Diseases 0.000 abstract description 52
- 230000002132 lysosomal effect Effects 0.000 abstract description 22
- 238000007913 intrathecal administration Methods 0.000 abstract description 16
- 229940088598 enzyme Drugs 0.000 description 261
- 239000003153 chemical reaction reagent Substances 0.000 description 105
- 210000003169 central nervous system Anatomy 0.000 description 90
- 239000011593 sulfur Substances 0.000 description 89
- 229910052717 sulfur Inorganic materials 0.000 description 89
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 88
- 235000010894 Artemisia argyi Nutrition 0.000 description 81
- 244000030166 artemisia Species 0.000 description 81
- 241001465754 Metazoa Species 0.000 description 77
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 74
- 201000010099 disease Diseases 0.000 description 68
- 210000001175 cerebrospinal fluid Anatomy 0.000 description 64
- 238000002347 injection Methods 0.000 description 62
- 239000007924 injection Substances 0.000 description 62
- 241000699670 Mus sp. Species 0.000 description 53
- 239000000243 solution Substances 0.000 description 48
- 238000012360 testing method Methods 0.000 description 48
- 238000004043 dyeing Methods 0.000 description 46
- 239000000523 sample Substances 0.000 description 40
- 238000011160 research Methods 0.000 description 39
- 230000001225 therapeutic effect Effects 0.000 description 35
- 239000003814 drug Substances 0.000 description 31
- 239000000463 material Substances 0.000 description 28
- 230000008929 regeneration Effects 0.000 description 28
- 238000011069 regeneration method Methods 0.000 description 28
- 238000003860 storage Methods 0.000 description 26
- 230000008014 freezing Effects 0.000 description 24
- 238000007710 freezing Methods 0.000 description 24
- 239000003085 diluting agent Substances 0.000 description 22
- 239000008194 pharmaceutical composition Substances 0.000 description 22
- 230000008685 targeting Effects 0.000 description 22
- 239000003981 vehicle Substances 0.000 description 22
- 238000005755 formation reaction Methods 0.000 description 20
- 235000001014 amino acid Nutrition 0.000 description 19
- 238000004458 analytical method Methods 0.000 description 19
- 230000015572 biosynthetic process Effects 0.000 description 19
- 210000001638 cerebellum Anatomy 0.000 description 19
- 239000010410 layer Substances 0.000 description 19
- 210000001103 thalamus Anatomy 0.000 description 19
- 239000000047 product Substances 0.000 description 18
- 238000010186 staining Methods 0.000 description 18
- 150000001413 amino acids Chemical class 0.000 description 17
- 238000004108 freeze drying Methods 0.000 description 17
- 108090000765 processed proteins & peptides Proteins 0.000 description 17
- 229920002683 Glycosaminoglycan Polymers 0.000 description 16
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 16
- 238000003364 immunohistochemistry Methods 0.000 description 16
- 210000002418 meninge Anatomy 0.000 description 16
- 239000002953 phosphate buffered saline Substances 0.000 description 16
- 230000008859 change Effects 0.000 description 15
- 230000008569 process Effects 0.000 description 15
- 239000003381 stabilizer Substances 0.000 description 15
- 238000009826 distribution Methods 0.000 description 14
- 231100000673 dose–response relationship Toxicity 0.000 description 14
- 238000012377 drug delivery Methods 0.000 description 14
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 14
- 230000004071 biological effect Effects 0.000 description 13
- 239000008187 granular material Substances 0.000 description 13
- 238000012744 immunostaining Methods 0.000 description 13
- 238000003998 size exclusion chromatography high performance liquid chromatography Methods 0.000 description 13
- 210000003934 vacuole Anatomy 0.000 description 13
- -1 Sodium octyl glycoside Chemical class 0.000 description 12
- 238000010586 diagram Methods 0.000 description 12
- 210000004248 oligodendroglia Anatomy 0.000 description 12
- 210000000056 organ Anatomy 0.000 description 12
- 230000002093 peripheral effect Effects 0.000 description 12
- 102000004196 processed proteins & peptides Human genes 0.000 description 12
- 238000012545 processing Methods 0.000 description 12
- 210000003710 cerebral cortex Anatomy 0.000 description 11
- 230000000875 corresponding effect Effects 0.000 description 11
- 229940079593 drug Drugs 0.000 description 11
- SGBICHSMBOYOFD-UHFFFAOYSA-N 2,3-dibromo-7-iododibenzo-p-dioxin Chemical compound O1C2=CC=C(I)C=C2OC2=C1C=C(Br)C(Br)=C2 SGBICHSMBOYOFD-UHFFFAOYSA-N 0.000 description 10
- 238000013459 approach Methods 0.000 description 10
- 238000005516 engineering process Methods 0.000 description 10
- 239000000945 filler Substances 0.000 description 10
- 239000012530 fluid Substances 0.000 description 10
- 229920001184 polypeptide Polymers 0.000 description 10
- 239000002243 precursor Substances 0.000 description 10
- 102000005962 receptors Human genes 0.000 description 10
- 108020003175 receptors Proteins 0.000 description 10
- 210000002966 serum Anatomy 0.000 description 10
- 239000001488 sodium phosphate Substances 0.000 description 10
- 229910000162 sodium phosphate Inorganic materials 0.000 description 10
- 238000010257 thawing Methods 0.000 description 10
- 238000002560 therapeutic procedure Methods 0.000 description 10
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 10
- 229930006000 Sucrose Natural products 0.000 description 9
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 9
- 238000009825 accumulation Methods 0.000 description 9
- 230000037396 body weight Effects 0.000 description 9
- 238000009792 diffusion process Methods 0.000 description 9
- 210000002570 interstitial cell Anatomy 0.000 description 9
- 239000005720 sucrose Substances 0.000 description 9
- 210000003462 vein Anatomy 0.000 description 9
- NBSCHQHZLSJFNQ-QTVWNMPRSA-N D-Mannose-6-phosphate Chemical compound OC1O[C@H](COP(O)(O)=O)[C@@H](O)[C@H](O)[C@@H]1O NBSCHQHZLSJFNQ-QTVWNMPRSA-N 0.000 description 8
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 8
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 8
- 230000008499 blood brain barrier function Effects 0.000 description 8
- 210000001218 blood-brain barrier Anatomy 0.000 description 8
- 238000011026 diafiltration Methods 0.000 description 8
- 238000001035 drying Methods 0.000 description 8
- 239000011521 glass Substances 0.000 description 8
- 238000004128 high performance liquid chromatography Methods 0.000 description 8
- 238000002372 labelling Methods 0.000 description 8
- 238000005259 measurement Methods 0.000 description 8
- 230000000926 neurological effect Effects 0.000 description 8
- 230000002159 abnormal effect Effects 0.000 description 7
- 238000000137 annealing Methods 0.000 description 7
- 230000006378 damage Effects 0.000 description 7
- 238000002641 enzyme replacement therapy Methods 0.000 description 7
- 230000000762 glandular Effects 0.000 description 7
- 230000036541 health Effects 0.000 description 7
- 230000001965 increasing effect Effects 0.000 description 7
- 239000000546 pharmaceutical excipient Substances 0.000 description 7
- 230000003442 weekly effect Effects 0.000 description 7
- 241000282472 Canis lupus familiaris Species 0.000 description 6
- 241000282693 Cercopithecidae Species 0.000 description 6
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 6
- 208000033868 Lysosomal disease Diseases 0.000 description 6
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 6
- 238000013461 design Methods 0.000 description 6
- 210000003494 hepatocyte Anatomy 0.000 description 6
- 210000001320 hippocampus Anatomy 0.000 description 6
- 239000007951 isotonicity adjuster Substances 0.000 description 6
- 230000007774 longterm Effects 0.000 description 6
- 208000011045 mucopolysaccharidosis type 3 Diseases 0.000 description 6
- 239000002773 nucleotide Chemical group 0.000 description 6
- 125000003729 nucleotide group Chemical group 0.000 description 6
- 230000007170 pathology Effects 0.000 description 6
- HDTRYLNUVZCQOY-UHFFFAOYSA-N α-D-glucopyranosyl-α-D-glucopyranoside Natural products OC1C(O)C(O)C(CO)OC1OC1C(O)C(O)C(O)C(CO)O1 HDTRYLNUVZCQOY-UHFFFAOYSA-N 0.000 description 5
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 5
- 108090001117 Insulin-Like Growth Factor II Proteins 0.000 description 5
- 102000048143 Insulin-Like Growth Factor II Human genes 0.000 description 5
- HDTRYLNUVZCQOY-WSWWMNSNSA-N Trehalose Natural products O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-WSWWMNSNSA-N 0.000 description 5
- HDTRYLNUVZCQOY-LIZSDCNHSA-N alpha,alpha-trehalose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-LIZSDCNHSA-N 0.000 description 5
- 230000002421 anti-septic effect Effects 0.000 description 5
- 239000000090 biomarker Substances 0.000 description 5
- 210000004369 blood Anatomy 0.000 description 5
- 239000008280 blood Substances 0.000 description 5
- 230000017531 blood circulation Effects 0.000 description 5
- 210000004958 brain cell Anatomy 0.000 description 5
- 159000000007 calcium salts Chemical class 0.000 description 5
- 239000000039 congener Substances 0.000 description 5
- 230000034994 death Effects 0.000 description 5
- 238000001514 detection method Methods 0.000 description 5
- 208000035475 disorder Diseases 0.000 description 5
- 239000012634 fragment Substances 0.000 description 5
- 230000006870 function Effects 0.000 description 5
- 229960003444 immunosuppressant agent Drugs 0.000 description 5
- 239000003018 immunosuppressive agent Substances 0.000 description 5
- 230000006872 improvement Effects 0.000 description 5
- 238000000185 intracerebroventricular administration Methods 0.000 description 5
- 230000004807 localization Effects 0.000 description 5
- 210000001259 mesencephalon Anatomy 0.000 description 5
- 230000004048 modification Effects 0.000 description 5
- 238000012986 modification Methods 0.000 description 5
- 238000010172 mouse model Methods 0.000 description 5
- 229920001542 oligosaccharide Polymers 0.000 description 5
- 150000002482 oligosaccharides Chemical class 0.000 description 5
- 238000012216 screening Methods 0.000 description 5
- 238000012546 transfer Methods 0.000 description 5
- 102100035028 Alpha-L-iduronidase Human genes 0.000 description 4
- WQZGKKKJIJFFOK-QTVWNMPRSA-N D-mannopyranose Chemical compound OC[C@H]1OC(O)[C@@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-QTVWNMPRSA-N 0.000 description 4
- 239000004471 Glycine Substances 0.000 description 4
- 101001019502 Homo sapiens Alpha-L-iduronidase Proteins 0.000 description 4
- 102000038460 IGF Type 2 Receptor Human genes 0.000 description 4
- 108010031792 IGF Type 2 Receptor Proteins 0.000 description 4
- 206010061218 Inflammation Diseases 0.000 description 4
- 208000008955 Mucolipidoses Diseases 0.000 description 4
- 206010056886 Mucopolysaccharidosis I Diseases 0.000 description 4
- 208000028781 Mucopolysaccharidosis type 1 Diseases 0.000 description 4
- 241000699666 Mus <mouse, genus> Species 0.000 description 4
- 230000001133 acceleration Effects 0.000 description 4
- 230000002776 aggregation Effects 0.000 description 4
- 238000004220 aggregation Methods 0.000 description 4
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 4
- 230000008485 antagonism Effects 0.000 description 4
- 210000004204 blood vessel Anatomy 0.000 description 4
- 238000006243 chemical reaction Methods 0.000 description 4
- 230000004087 circulation Effects 0.000 description 4
- 210000001653 corpus striatum Anatomy 0.000 description 4
- 230000008021 deposition Effects 0.000 description 4
- 238000011161 development Methods 0.000 description 4
- 230000018109 developmental process Effects 0.000 description 4
- 238000010790 dilution Methods 0.000 description 4
- 239000012895 dilution Substances 0.000 description 4
- 230000001900 immune effect Effects 0.000 description 4
- 230000001861 immunosuppressant effect Effects 0.000 description 4
- 230000004054 inflammatory process Effects 0.000 description 4
- 238000001802 infusion Methods 0.000 description 4
- 238000007914 intraventricular administration Methods 0.000 description 4
- 238000009593 lumbar puncture Methods 0.000 description 4
- 238000004519 manufacturing process Methods 0.000 description 4
- 230000007246 mechanism Effects 0.000 description 4
- 230000004060 metabolic process Effects 0.000 description 4
- 239000002052 molecular layer Substances 0.000 description 4
- 210000003205 muscle Anatomy 0.000 description 4
- 150000007523 nucleic acids Chemical class 0.000 description 4
- 102000039446 nucleic acids Human genes 0.000 description 4
- 108020004707 nucleic acids Proteins 0.000 description 4
- 210000004940 nucleus Anatomy 0.000 description 4
- 239000002504 physiological saline solution Substances 0.000 description 4
- 238000000746 purification Methods 0.000 description 4
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 4
- 210000000952 spleen Anatomy 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- FWBHETKCLVMNFS-UHFFFAOYSA-N 4',6-Diamino-2-phenylindol Chemical compound C1=CC(C(=N)N)=CC=C1C1=CC2=CC=C(C(N)=N)C=C2N1 FWBHETKCLVMNFS-UHFFFAOYSA-N 0.000 description 3
- 206010067484 Adverse reaction Diseases 0.000 description 3
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 3
- 102100022548 Beta-hexosaminidase subunit alpha Human genes 0.000 description 3
- PMATZTZNYRCHOR-CGLBZJNRSA-N Cyclosporin A Chemical compound CC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C1=O PMATZTZNYRCHOR-CGLBZJNRSA-N 0.000 description 3
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 3
- 238000002965 ELISA Methods 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- 208000001905 GM2 Gangliosidoses Diseases 0.000 description 3
- 201000008905 GM2 gangliosidosis Diseases 0.000 description 3
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Chemical compound OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 3
- WZUVPPKBWHMQCE-UHFFFAOYSA-N Haematoxylin Natural products C12=CC(O)=C(O)C=C2CC2(O)C1C1=CC=C(O)C(O)=C1OC2 WZUVPPKBWHMQCE-UHFFFAOYSA-N 0.000 description 3
- 101001045440 Homo sapiens Beta-hexosaminidase subunit alpha Proteins 0.000 description 3
- 229920001612 Hydroxyethyl starch Polymers 0.000 description 3
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 3
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 3
- 241000124008 Mammalia Species 0.000 description 3
- 229930195725 Mannitol Natural products 0.000 description 3
- 102000018697 Membrane Proteins Human genes 0.000 description 3
- 108010052285 Membrane Proteins Proteins 0.000 description 3
- 239000002202 Polyethylene glycol Substances 0.000 description 3
- 239000004743 Polypropylene Substances 0.000 description 3
- 241000288906 Primates Species 0.000 description 3
- 108010076504 Protein Sorting Signals Proteins 0.000 description 3
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 3
- 230000003213 activating effect Effects 0.000 description 3
- 239000000654 additive Substances 0.000 description 3
- 230000002411 adverse Effects 0.000 description 3
- 230000006838 adverse reaction Effects 0.000 description 3
- 230000001174 ascending effect Effects 0.000 description 3
- 239000001654 beetroot red Substances 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- 210000000988 bone and bone Anatomy 0.000 description 3
- 210000001185 bone marrow Anatomy 0.000 description 3
- 239000007853 buffer solution Substances 0.000 description 3
- 230000003139 buffering effect Effects 0.000 description 3
- 229910052799 carbon Inorganic materials 0.000 description 3
- 150000001768 cations Chemical class 0.000 description 3
- 230000005859 cell recognition Effects 0.000 description 3
- 230000001413 cellular effect Effects 0.000 description 3
- 210000004720 cerebrum Anatomy 0.000 description 3
- 239000012141 concentrate Substances 0.000 description 3
- 239000013078 crystal Substances 0.000 description 3
- 230000002950 deficient Effects 0.000 description 3
- 238000012217 deletion Methods 0.000 description 3
- 230000037430 deletion Effects 0.000 description 3
- 230000001419 dependent effect Effects 0.000 description 3
- 235000005911 diet Nutrition 0.000 description 3
- 230000037213 diet Effects 0.000 description 3
- 239000002552 dosage form Substances 0.000 description 3
- 239000000428 dust Substances 0.000 description 3
- 239000000975 dye Substances 0.000 description 3
- 238000000635 electron micrograph Methods 0.000 description 3
- 238000001914 filtration Methods 0.000 description 3
- 238000003209 gene knockout Methods 0.000 description 3
- 230000008642 heat stress Effects 0.000 description 3
- 230000002440 hepatic effect Effects 0.000 description 3
- 229940027278 hetastarch Drugs 0.000 description 3
- 210000003016 hypothalamus Anatomy 0.000 description 3
- 210000003405 ileum Anatomy 0.000 description 3
- 238000002513 implantation Methods 0.000 description 3
- 238000011534 incubation Methods 0.000 description 3
- 238000007917 intracranial administration Methods 0.000 description 3
- 210000003292 kidney cell Anatomy 0.000 description 3
- 239000008101 lactose Substances 0.000 description 3
- 230000002045 lasting effect Effects 0.000 description 3
- 239000008176 lyophilized powder Substances 0.000 description 3
- 238000003760 magnetic stirring Methods 0.000 description 3
- 210000004962 mammalian cell Anatomy 0.000 description 3
- 239000000594 mannitol Substances 0.000 description 3
- 235000010355 mannitol Nutrition 0.000 description 3
- 210000005036 nerve Anatomy 0.000 description 3
- 230000003204 osmotic effect Effects 0.000 description 3
- 238000012856 packing Methods 0.000 description 3
- 210000003446 pia mater Anatomy 0.000 description 3
- 229920001223 polyethylene glycol Polymers 0.000 description 3
- 229920001155 polypropylene Polymers 0.000 description 3
- 238000011084 recovery Methods 0.000 description 3
- 230000000630 rising effect Effects 0.000 description 3
- QFJCIRLUMZQUOT-HPLJOQBZSA-N sirolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 QFJCIRLUMZQUOT-HPLJOQBZSA-N 0.000 description 3
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 3
- 239000007787 solid Substances 0.000 description 3
- 238000003756 stirring Methods 0.000 description 3
- 238000007920 subcutaneous administration Methods 0.000 description 3
- 238000007910 systemic administration Methods 0.000 description 3
- 231100000027 toxicology Toxicity 0.000 description 3
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 2
- GZCGUPFRVQAUEE-UHFFFAOYSA-N 2,3,4,5,6-pentahydroxyhexanal Chemical compound OCC(O)C(O)C(O)C(O)C=O GZCGUPFRVQAUEE-UHFFFAOYSA-N 0.000 description 2
- 102100027165 Alpha-2-macroglobulin receptor-associated protein Human genes 0.000 description 2
- 101710126837 Alpha-2-macroglobulin receptor-associated protein Proteins 0.000 description 2
- 206010002091 Anaesthesia Diseases 0.000 description 2
- 239000004475 Arginine Substances 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 102100037364 Craniofacial development protein 1 Human genes 0.000 description 2
- 229930105110 Cyclosporin A Natural products 0.000 description 2
- NOQGZXFMHARMLW-UHFFFAOYSA-N Daminozide Chemical group CN(C)NC(=O)CCC(O)=O NOQGZXFMHARMLW-UHFFFAOYSA-N 0.000 description 2
- 229920002307 Dextran Polymers 0.000 description 2
- 241000283070 Equus zebra Species 0.000 description 2
- 101710190709 Eukaryotic translation initiation factor 4 gamma 2 Proteins 0.000 description 2
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 2
- 108090000723 Insulin-Like Growth Factor I Proteins 0.000 description 2
- 102000004218 Insulin-Like Growth Factor I Human genes 0.000 description 2
- LKDRXBCSQODPBY-AMVSKUEXSA-N L-(-)-Sorbose Chemical compound OCC1(O)OC[C@H](O)[C@@H](O)[C@@H]1O LKDRXBCSQODPBY-AMVSKUEXSA-N 0.000 description 2
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 2
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 2
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 2
- 201000011442 Metachromatic leukodystrophy Diseases 0.000 description 2
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 2
- 206010072927 Mucolipidosis type I Diseases 0.000 description 2
- 206010072929 Mucolipidosis type III Diseases 0.000 description 2
- 206010056893 Mucopolysaccharidosis VII Diseases 0.000 description 2
- 102000006386 Myelin Proteins Human genes 0.000 description 2
- 108010083674 Myelin Proteins Proteins 0.000 description 2
- 206010067482 No adverse event Diseases 0.000 description 2
- 239000002033 PVDF binder Substances 0.000 description 2
- ATUOYWHBWRKTHZ-UHFFFAOYSA-N Propane Chemical compound CCC ATUOYWHBWRKTHZ-UHFFFAOYSA-N 0.000 description 2
- 208000025820 Sanfilippo syndrome type B Diseases 0.000 description 2
- 208000017460 Sialidosis type 2 Diseases 0.000 description 2
- 239000004141 Sodium laurylsulphate Substances 0.000 description 2
- 101710132062 Transitional endoplasmic reticulum ATPase Proteins 0.000 description 2
- 230000004913 activation Effects 0.000 description 2
- 230000000996 additive effect Effects 0.000 description 2
- 125000000539 amino acid group Chemical group 0.000 description 2
- 230000003321 amplification Effects 0.000 description 2
- 230000037005 anaesthesia Effects 0.000 description 2
- 238000010171 animal model Methods 0.000 description 2
- 239000000427 antigen Substances 0.000 description 2
- 102000036639 antigens Human genes 0.000 description 2
- 108091007433 antigens Proteins 0.000 description 2
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 2
- 238000002869 basic local alignment search tool Methods 0.000 description 2
- 238000003766 bioinformatics method Methods 0.000 description 2
- 229960002685 biotin Drugs 0.000 description 2
- 239000011616 biotin Substances 0.000 description 2
- 244000144987 brood Species 0.000 description 2
- 238000005251 capillar electrophoresis Methods 0.000 description 2
- 238000005277 cation exchange chromatography Methods 0.000 description 2
- 230000001149 cognitive effect Effects 0.000 description 2
- 238000012790 confirmation Methods 0.000 description 2
- 239000013256 coordination polymer Substances 0.000 description 2
- 210000005257 cortical tissue Anatomy 0.000 description 2
- 238000002425 crystallisation Methods 0.000 description 2
- 210000000805 cytoplasm Anatomy 0.000 description 2
- 230000007423 decrease Effects 0.000 description 2
- 230000007812 deficiency Effects 0.000 description 2
- 210000002451 diencephalon Anatomy 0.000 description 2
- OGGXGZAMXPVRFZ-UHFFFAOYSA-N dimethylarsinic acid Chemical compound C[As](C)(O)=O OGGXGZAMXPVRFZ-UHFFFAOYSA-N 0.000 description 2
- 238000004821 distillation Methods 0.000 description 2
- 239000011363 dried mixture Substances 0.000 description 2
- 230000004064 dysfunction Effects 0.000 description 2
- 210000003725 endotheliocyte Anatomy 0.000 description 2
- 210000000222 eosinocyte Anatomy 0.000 description 2
- 206010015037 epilepsy Diseases 0.000 description 2
- LYCAIKOWRPUZTN-UHFFFAOYSA-N ethylene glycol Natural products OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 2
- 230000003203 everyday effect Effects 0.000 description 2
- 230000029142 excretion Effects 0.000 description 2
- 238000013401 experimental design Methods 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 210000003754 fetus Anatomy 0.000 description 2
- 235000012631 food intake Nutrition 0.000 description 2
- 230000007849 functional defect Effects 0.000 description 2
- 102000037865 fusion proteins Human genes 0.000 description 2
- 108020001507 fusion proteins Proteins 0.000 description 2
- 210000003322 glomus cell Anatomy 0.000 description 2
- 201000008977 glycoproteinosis Diseases 0.000 description 2
- 230000009931 harmful effect Effects 0.000 description 2
- 230000003862 health status Effects 0.000 description 2
- 238000010231 histologic analysis Methods 0.000 description 2
- 229960002396 idursulfase Drugs 0.000 description 2
- 108010072166 idursulfase Proteins 0.000 description 2
- 230000036039 immunity Effects 0.000 description 2
- 238000012151 immunohistochemical method Methods 0.000 description 2
- 238000011532 immunohistochemical staining Methods 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 230000008595 infiltration Effects 0.000 description 2
- 238000001764 infiltration Methods 0.000 description 2
- 239000004615 ingredient Substances 0.000 description 2
- 238000003780 insertion Methods 0.000 description 2
- 230000037431 insertion Effects 0.000 description 2
- 238000010253 intravenous injection Methods 0.000 description 2
- 150000002500 ions Chemical class 0.000 description 2
- 238000011813 knockout mouse model Methods 0.000 description 2
- 208000036546 leukodystrophy Diseases 0.000 description 2
- 230000000670 limiting effect Effects 0.000 description 2
- 125000005645 linoleyl group Chemical group 0.000 description 2
- 210000004072 lung Anatomy 0.000 description 2
- 210000001165 lymph node Anatomy 0.000 description 2
- 229920002521 macromolecule Polymers 0.000 description 2
- 239000000320 mechanical mixture Substances 0.000 description 2
- 208000030159 metabolic disease Diseases 0.000 description 2
- 229930182817 methionine Natural products 0.000 description 2
- 238000001565 modulated differential scanning calorimetry Methods 0.000 description 2
- 239000000178 monomer Substances 0.000 description 2
- 230000002969 morbid Effects 0.000 description 2
- 230000003562 morphometric effect Effects 0.000 description 2
- 238000013425 morphometry Methods 0.000 description 2
- 230000007659 motor function Effects 0.000 description 2
- 208000005340 mucopolysaccharidosis III Diseases 0.000 description 2
- 208000036709 mucopolysaccharidosis type 3B Diseases 0.000 description 2
- 208000025919 mucopolysaccharidosis type 7 Diseases 0.000 description 2
- 208000012227 mucopolysaccharidosis type IIIB Diseases 0.000 description 2
- 238000000569 multi-angle light scattering Methods 0.000 description 2
- 210000005012 myelin Anatomy 0.000 description 2
- 239000013642 negative control Substances 0.000 description 2
- 210000000653 nervous system Anatomy 0.000 description 2
- 238000003199 nucleic acid amplification method Methods 0.000 description 2
- 210000003463 organelle Anatomy 0.000 description 2
- 210000001672 ovary Anatomy 0.000 description 2
- 239000012188 paraffin wax Substances 0.000 description 2
- 230000003836 peripheral circulation Effects 0.000 description 2
- 210000000578 peripheral nerve Anatomy 0.000 description 2
- 230000002085 persistent effect Effects 0.000 description 2
- 150000003016 phosphoric acids Chemical class 0.000 description 2
- 239000004033 plastic Substances 0.000 description 2
- 229920003023 plastic Polymers 0.000 description 2
- 229920001983 poloxamer Polymers 0.000 description 2
- 229920002981 polyvinylidene fluoride Polymers 0.000 description 2
- 239000002244 precipitate Substances 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 230000002265 prevention Effects 0.000 description 2
- 230000004845 protein aggregation Effects 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 230000003252 repetitive effect Effects 0.000 description 2
- 238000001223 reverse osmosis Methods 0.000 description 2
- 238000004007 reversed phase HPLC Methods 0.000 description 2
- 230000035807 sensation Effects 0.000 description 2
- 231100001055 skeletal defect Toxicity 0.000 description 2
- 210000003625 skull Anatomy 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- 239000000758 substrate Substances 0.000 description 2
- 150000005846 sugar alcohols Polymers 0.000 description 2
- 230000001629 suppression Effects 0.000 description 2
- 238000010189 synthetic method Methods 0.000 description 2
- 230000009885 systemic effect Effects 0.000 description 2
- 210000001550 testis Anatomy 0.000 description 2
- 231100000419 toxicity Toxicity 0.000 description 2
- 230000001988 toxicity Effects 0.000 description 2
- YFDSDPIBEUFTMI-UHFFFAOYSA-N tribromoethanol Chemical compound OCC(Br)(Br)Br YFDSDPIBEUFTMI-UHFFFAOYSA-N 0.000 description 2
- 238000011100 viral filtration Methods 0.000 description 2
- 238000005303 weighing Methods 0.000 description 2
- WZUVPPKBWHMQCE-XJKSGUPXSA-N (+)-haematoxylin Chemical compound C12=CC(O)=C(O)C=C2C[C@]2(O)[C@H]1C1=CC=C(O)C(O)=C1OC2 WZUVPPKBWHMQCE-XJKSGUPXSA-N 0.000 description 1
- SNQVCAOGQHOSEN-UHFFFAOYSA-N 2-[methyl(octadecyl)amino]acetic acid Chemical compound CCCCCCCCCCCCCCCCCCN(C)CC(O)=O SNQVCAOGQHOSEN-UHFFFAOYSA-N 0.000 description 1
- HSTOKWSFWGCZMH-UHFFFAOYSA-N 3,3'-diaminobenzidine Chemical compound C1=C(N)C(N)=CC=C1C1=CC=C(N)C(N)=C1 HSTOKWSFWGCZMH-UHFFFAOYSA-N 0.000 description 1
- 101100230376 Acetivibrio thermocellus (strain ATCC 27405 / DSM 1237 / JCM 9322 / NBRC 103400 / NCIMB 10682 / NRRL B-4536 / VPI 7372) celI gene Proteins 0.000 description 1
- 239000012103 Alexa Fluor 488 Substances 0.000 description 1
- 108090001008 Avidin Proteins 0.000 description 1
- 238000011725 BALB/c mouse Methods 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- DHHFDKNIEVKVKS-FMOSSLLZSA-N Betanin Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1OC(C(=C1)O)=CC(C[C@H]2C([O-])=O)=C1[N+]2=C\C=C\1C=C(C(O)=O)N[C@H](C(O)=O)C/1 DHHFDKNIEVKVKS-FMOSSLLZSA-N 0.000 description 1
- DHHFDKNIEVKVKS-MVUYWVKGSA-N Betanin Natural products O=C(O)[C@@H]1NC(C(=O)O)=C/C(=C\C=[N+]/2\[C@@H](C(=O)[O-])Cc3c\2cc(O)c(O[C@H]2[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O2)c3)/C1 DHHFDKNIEVKVKS-MVUYWVKGSA-N 0.000 description 1
- 206010006187 Breast cancer Diseases 0.000 description 1
- 238000011740 C57BL/6 mouse Methods 0.000 description 1
- 241000282465 Canis Species 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 206010008190 Cerebrovascular accident Diseases 0.000 description 1
- 241000282552 Chlorocebus aethiops Species 0.000 description 1
- 208000034030 Chronic neurovisceral acid sphingomyelinase deficiency Diseases 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 241000699802 Cricetulus griseus Species 0.000 description 1
- 108010036949 Cyclosporine Proteins 0.000 description 1
- 206010011777 Cystinosis Diseases 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- 208000027219 Deficiency disease Diseases 0.000 description 1
- 229920000045 Dermatan sulfate Polymers 0.000 description 1
- 206010061818 Disease progression Diseases 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 108010000912 Egg Proteins Proteins 0.000 description 1
- 102000002322 Egg Proteins Human genes 0.000 description 1
- 241000196324 Embryophyta Species 0.000 description 1
- 102000004533 Endonucleases Human genes 0.000 description 1
- 108010042407 Endonucleases Proteins 0.000 description 1
- 102000010911 Enzyme Precursors Human genes 0.000 description 1
- 108010062466 Enzyme Precursors Proteins 0.000 description 1
- 241001411320 Eriogonum inflatum Species 0.000 description 1
- 239000004386 Erythritol Substances 0.000 description 1
- UNXHWFMMPAWVPI-UHFFFAOYSA-N Erythritol Natural products OCC(O)C(O)CO UNXHWFMMPAWVPI-UHFFFAOYSA-N 0.000 description 1
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 1
- 208000035126 Facies Diseases 0.000 description 1
- 208000001948 Farber Lipogranulomatosis Diseases 0.000 description 1
- 208000033149 Farber disease Diseases 0.000 description 1
- 201000008808 Fibrosarcoma Diseases 0.000 description 1
- 201000008892 GM1 Gangliosidosis Diseases 0.000 description 1
- 208000017462 Galactosialidosis Diseases 0.000 description 1
- 208000003098 Ganglion Cysts Diseases 0.000 description 1
- 208000015872 Gaucher disease Diseases 0.000 description 1
- 208000020322 Gaucher disease type I Diseases 0.000 description 1
- 208000020916 Gaucher disease type II Diseases 0.000 description 1
- 208000010055 Globoid Cell Leukodystrophy Diseases 0.000 description 1
- SXRSQZLOMIGNAQ-UHFFFAOYSA-N Glutaraldehyde Chemical compound O=CCCCC=O SXRSQZLOMIGNAQ-UHFFFAOYSA-N 0.000 description 1
- 206010053185 Glycogen storage disease type II Diseases 0.000 description 1
- 208000028782 Hereditary disease Diseases 0.000 description 1
- 101000599951 Homo sapiens Insulin-like growth factor I Proteins 0.000 description 1
- 101001004953 Homo sapiens Lysosomal acid lipase/cholesteryl ester hydrolase Proteins 0.000 description 1
- 108010001336 Horseradish Peroxidase Proteins 0.000 description 1
- 101100273566 Humulus lupulus CCL10 gene Proteins 0.000 description 1
- 206010020651 Hyperkinesia Diseases 0.000 description 1
- 208000000269 Hyperkinesis Diseases 0.000 description 1
- 206010020751 Hypersensitivity Diseases 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 208000028226 Krabbe disease Diseases 0.000 description 1
- 101710192602 Latent membrane protein 1 Proteins 0.000 description 1
- 102100026001 Lysosomal acid lipase/cholesteryl ester hydrolase Human genes 0.000 description 1
- 208000024556 Mendelian disease Diseases 0.000 description 1
- 206010027476 Metastases Diseases 0.000 description 1
- 208000002678 Mucopolysaccharidoses Diseases 0.000 description 1
- 208000025915 Mucopolysaccharidosis type 6 Diseases 0.000 description 1
- 208000000149 Multiple Sulfatase Deficiency Disease Diseases 0.000 description 1
- 208000035032 Multiple sulfatase deficiency Diseases 0.000 description 1
- OVRNDRQMDRJTHS-FMDGEEDCSA-N N-acetyl-beta-D-glucosamine Chemical compound CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O OVRNDRQMDRJTHS-FMDGEEDCSA-N 0.000 description 1
- MBLBDJOUHNCFQT-LXGUWJNJSA-N N-acetylglucosamine Natural products CC(=O)N[C@@H](C=O)[C@@H](O)[C@H](O)[C@H](O)CO MBLBDJOUHNCFQT-LXGUWJNJSA-N 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- 102000008763 Neurofilament Proteins Human genes 0.000 description 1
- 108010088373 Neurofilament Proteins Proteins 0.000 description 1
- 208000002537 Neuronal Ceroid-Lipofuscinoses Diseases 0.000 description 1
- 201000000788 Niemann-Pick disease type C1 Diseases 0.000 description 1
- 201000000785 Niemann-Pick disease type C2 Diseases 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 240000005373 Panax quinquefolius Species 0.000 description 1
- 235000003140 Panax quinquefolius Nutrition 0.000 description 1
- 241000598137 Pedunculus Species 0.000 description 1
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 1
- 206010035226 Plasma cell myeloma Diseases 0.000 description 1
- RVGRUAULSDPKGF-UHFFFAOYSA-N Poloxamer Chemical compound C1CO1.CC1CO1 RVGRUAULSDPKGF-UHFFFAOYSA-N 0.000 description 1
- MUPFEKGTMRGPLJ-RMMQSMQOSA-N Raffinose Natural products O(C[C@H]1[C@@H](O)[C@H](O)[C@@H](O)[C@@H](O[C@@]2(CO)[C@H](O)[C@@H](O)[C@@H](CO)O2)O1)[C@@H]1[C@H](O)[C@@H](O)[C@@H](O)[C@@H](CO)O1 MUPFEKGTMRGPLJ-RMMQSMQOSA-N 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 102000007056 Recombinant Fusion Proteins Human genes 0.000 description 1
- 108010008281 Recombinant Fusion Proteins Proteins 0.000 description 1
- 241000219061 Rheum Species 0.000 description 1
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 1
- 235000013290 Sagittaria latifolia Nutrition 0.000 description 1
- 208000021811 Sandhoff disease Diseases 0.000 description 1
- 208000025816 Sanfilippo syndrome type A Diseases 0.000 description 1
- 201000001828 Sly syndrome Diseases 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- 208000006011 Stroke Diseases 0.000 description 1
- 208000005400 Synovial Cyst Diseases 0.000 description 1
- QJJXYPPXXYFBGM-LFZNUXCKSA-N Tacrolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1\C=C(/C)[C@@H]1[C@H](C)[C@@H](O)CC(=O)[C@H](CC=C)/C=C(C)/C[C@H](C)C[C@H](OC)[C@H]([C@H](C[C@H]2C)OC)O[C@@]2(O)C(=O)C(=O)N2CCCC[C@H]2C(=O)O1 QJJXYPPXXYFBGM-LFZNUXCKSA-N 0.000 description 1
- 208000022292 Tay-Sachs disease Diseases 0.000 description 1
- 239000004809 Teflon Substances 0.000 description 1
- 229920006362 Teflon® Polymers 0.000 description 1
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 description 1
- LFTYTUAZOPRMMI-CFRASDGPSA-N UDP-N-acetyl-alpha-D-glucosamine Chemical compound O1[C@H](CO)[C@@H](O)[C@H](O)[C@@H](NC(=O)C)[C@H]1OP(O)(=O)OP(O)(=O)OC[C@@H]1[C@@H](O)[C@@H](O)[C@H](N2C(NC(=O)C=C2)=O)O1 LFTYTUAZOPRMMI-CFRASDGPSA-N 0.000 description 1
- LFTYTUAZOPRMMI-UHFFFAOYSA-N UNPD164450 Natural products O1C(CO)C(O)C(O)C(NC(=O)C)C1OP(O)(=O)OP(O)(=O)OCC1C(O)C(O)C(N2C(NC(=O)C=C2)=O)O1 LFTYTUAZOPRMMI-UHFFFAOYSA-N 0.000 description 1
- MUPFEKGTMRGPLJ-UHFFFAOYSA-N UNPD196149 Natural products OC1C(O)C(CO)OC1(CO)OC1C(O)C(O)C(O)C(COC2C(C(O)C(O)C(CO)O2)O)O1 MUPFEKGTMRGPLJ-UHFFFAOYSA-N 0.000 description 1
- 244000000188 Vaccinium ovalifolium Species 0.000 description 1
- 206010047700 Vomiting Diseases 0.000 description 1
- 208000027418 Wounds and injury Diseases 0.000 description 1
- 210000001766 X chromosome Anatomy 0.000 description 1
- 238000002441 X-ray diffraction Methods 0.000 description 1
- 206010048215 Xanthomatosis Diseases 0.000 description 1
- TVXBFESIOXBWNM-UHFFFAOYSA-N Xylitol Natural products OCCC(O)C(O)C(O)CCO TVXBFESIOXBWNM-UHFFFAOYSA-N 0.000 description 1
- 230000005856 abnormality Effects 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 238000005903 acid hydrolysis reaction Methods 0.000 description 1
- 239000011149 active material Substances 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- 230000007815 allergy Effects 0.000 description 1
- 201000008333 alpha-mannosidosis Diseases 0.000 description 1
- 210000002821 alveolar epithelial cell Anatomy 0.000 description 1
- 230000000844 anti-bacterial effect Effects 0.000 description 1
- 230000001028 anti-proliverative effect Effects 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 230000003078 antioxidant effect Effects 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 239000013011 aqueous formulation Substances 0.000 description 1
- 210000000576 arachnoid Anatomy 0.000 description 1
- 206010003246 arthritis Diseases 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 208000006673 asthma Diseases 0.000 description 1
- 238000011888 autopsy Methods 0.000 description 1
- 229960002170 azathioprine Drugs 0.000 description 1
- LMEKQMALGUDUQG-UHFFFAOYSA-N azathioprine Chemical compound CN1C=NC([N+]([O-])=O)=C1SC1=NC=NC2=C1NC=N2 LMEKQMALGUDUQG-UHFFFAOYSA-N 0.000 description 1
- 230000003385 bacteriostatic effect Effects 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- 235000012677 beetroot red Nutrition 0.000 description 1
- 230000006399 behavior Effects 0.000 description 1
- 235000019445 benzyl alcohol Nutrition 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- WQZGKKKJIJFFOK-FPRJBGLDSA-N beta-D-galactose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@H]1O WQZGKKKJIJFFOK-FPRJBGLDSA-N 0.000 description 1
- SQVRNKJHWKZAKO-UHFFFAOYSA-N beta-N-Acetyl-D-neuraminic acid Natural products CC(=O)NC1C(O)CC(O)(C(O)=O)OC1C(O)C(O)CO SQVRNKJHWKZAKO-UHFFFAOYSA-N 0.000 description 1
- 201000006486 beta-mannosidosis Diseases 0.000 description 1
- 235000002185 betanin Nutrition 0.000 description 1
- 230000000975 bioactive effect Effects 0.000 description 1
- 229920002988 biodegradable polymer Polymers 0.000 description 1
- 239000004621 biodegradable polymer Substances 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 230000005540 biological transmission Effects 0.000 description 1
- 235000020958 biotin Nutrition 0.000 description 1
- HOQPTLCRWVZIQZ-UHFFFAOYSA-H bis[[2-(5-hydroxy-4,7-dioxo-1,3,2$l^{2}-dioxaplumbepan-5-yl)acetyl]oxy]lead Chemical compound [Pb+2].[Pb+2].[Pb+2].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O.[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O HOQPTLCRWVZIQZ-UHFFFAOYSA-H 0.000 description 1
- 230000037237 body shape Effects 0.000 description 1
- 210000005013 brain tissue Anatomy 0.000 description 1
- 230000005587 bubbling Effects 0.000 description 1
- 239000007978 cacodylate buffer Substances 0.000 description 1
- 229950004243 cacodylic acid Drugs 0.000 description 1
- 238000011088 calibration curve Methods 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 239000003560 cancer drug Substances 0.000 description 1
- 210000000845 cartilage Anatomy 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 238000006555 catalytic reaction Methods 0.000 description 1
- 230000030833 cell death Effects 0.000 description 1
- 210000003198 cerebellar cortex Anatomy 0.000 description 1
- 229930183167 cerebroside Natural products 0.000 description 1
- RIZIAUKTHDLMQX-UHFFFAOYSA-N cerebroside D Natural products CCCCCCCCCCCCCCCCC(O)C(=O)NC(C(O)C=CCCC=C(C)CCCCCCCCC)COC1OC(CO)C(O)C(O)C1O RIZIAUKTHDLMQX-UHFFFAOYSA-N 0.000 description 1
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 1
- 229960001265 ciclosporin Drugs 0.000 description 1
- MYSWGUAQZAJSOK-UHFFFAOYSA-N ciprofloxacin Chemical compound C12=CC(N3CCNCC3)=C(F)C=C2C(=O)C(C(=O)O)=CN1C1CC1 MYSWGUAQZAJSOK-UHFFFAOYSA-N 0.000 description 1
- 238000004140 cleaning Methods 0.000 description 1
- 230000003920 cognitive function Effects 0.000 description 1
- 238000011284 combination treatment Methods 0.000 description 1
- 235000015246 common arrowhead Nutrition 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 239000002131 composite material Substances 0.000 description 1
- 238000004590 computer program Methods 0.000 description 1
- 230000021615 conjugation Effects 0.000 description 1
- 210000001608 connective tissue cell Anatomy 0.000 description 1
- 239000013068 control sample Substances 0.000 description 1
- 230000001276 controlling effect Effects 0.000 description 1
- 229920001577 copolymer Polymers 0.000 description 1
- 230000002596 correlated effect Effects 0.000 description 1
- 239000006184 cosolvent Substances 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- HEBKCHPVOIAQTA-NGQZWQHPSA-N d-xylitol Chemical compound OC[C@H](O)C(O)[C@H](O)CO HEBKCHPVOIAQTA-NGQZWQHPSA-N 0.000 description 1
- 230000002354 daily effect Effects 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 230000000593 degrading effect Effects 0.000 description 1
- 238000002242 deionisation method Methods 0.000 description 1
- AVJBPWGFOQAPRH-FWMKGIEWSA-L dermatan sulfate Chemical compound CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@H](OS([O-])(=O)=O)[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@H](O)[C@H](C([O-])=O)O1 AVJBPWGFOQAPRH-FWMKGIEWSA-L 0.000 description 1
- 229940051593 dermatan sulfate Drugs 0.000 description 1
- 231100000020 developmental retardation Toxicity 0.000 description 1
- 239000012471 diafiltration solution Substances 0.000 description 1
- 238000003745 diagnosis Methods 0.000 description 1
- 238000000502 dialysis Methods 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- 238000007865 diluting Methods 0.000 description 1
- 238000007599 discharging Methods 0.000 description 1
- 208000016097 disease of metabolism Diseases 0.000 description 1
- 230000005750 disease progression Effects 0.000 description 1
- 238000006073 displacement reaction Methods 0.000 description 1
- 238000001647 drug administration Methods 0.000 description 1
- 239000003937 drug carrier Substances 0.000 description 1
- 238000009509 drug development Methods 0.000 description 1
- 210000001951 dura mater Anatomy 0.000 description 1
- 239000003792 electrolyte Substances 0.000 description 1
- 230000005670 electromagnetic radiation Effects 0.000 description 1
- 238000010828 elution Methods 0.000 description 1
- 210000002257 embryonic structure Anatomy 0.000 description 1
- 210000003038 endothelium Anatomy 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 230000009144 enzymatic modification Effects 0.000 description 1
- 210000003059 ependyma Anatomy 0.000 description 1
- 210000000219 ependymocyte Anatomy 0.000 description 1
- 210000000647 epithalamus Anatomy 0.000 description 1
- 235000019414 erythritol Nutrition 0.000 description 1
- UNXHWFMMPAWVPI-ZXZARUISSA-N erythritol Chemical compound OC[C@H](O)[C@H](O)CO UNXHWFMMPAWVPI-ZXZARUISSA-N 0.000 description 1
- 229940009714 erythritol Drugs 0.000 description 1
- 230000005496 eutectics Effects 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 239000006260 foam Substances 0.000 description 1
- 210000001652 frontal lobe Anatomy 0.000 description 1
- 201000008049 fucosidosis Diseases 0.000 description 1
- 230000004927 fusion Effects 0.000 description 1
- 210000000232 gallbladder Anatomy 0.000 description 1
- 238000010353 genetic engineering Methods 0.000 description 1
- 230000009477 glass transition Effects 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 201000004502 glycogen storage disease II Diseases 0.000 description 1
- 229930182470 glycoside Natural products 0.000 description 1
- 238000005469 granulation Methods 0.000 description 1
- 230000003179 granulation Effects 0.000 description 1
- 230000012010 growth Effects 0.000 description 1
- 208000016354 hearing loss disease Diseases 0.000 description 1
- 238000011134 hematopoietic stem cell transplantation Methods 0.000 description 1
- 108010089932 heparan sulfate sulfatase Proteins 0.000 description 1
- 244000144980 herd Species 0.000 description 1
- 238000007489 histopathology method Methods 0.000 description 1
- 102000044162 human IGF1 Human genes 0.000 description 1
- 210000005260 human cell Anatomy 0.000 description 1
- 208000003906 hydrocephalus Diseases 0.000 description 1
- 150000002433 hydrophilic molecules Chemical class 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- 230000003053 immunization Effects 0.000 description 1
- 238000002649 immunization Methods 0.000 description 1
- 230000002055 immunohistochemical effect Effects 0.000 description 1
- 239000003547 immunosorbent Substances 0.000 description 1
- 239000007943 implant Substances 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 230000000968 intestinal effect Effects 0.000 description 1
- 210000000936 intestine Anatomy 0.000 description 1
- 238000004255 ion exchange chromatography Methods 0.000 description 1
- 230000002262 irrigation Effects 0.000 description 1
- 238000003973 irrigation Methods 0.000 description 1
- FZWBNHMXJMCXLU-BLAUPYHCSA-N isomaltotriose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1OC[C@@H]1[C@@H](O)[C@H](O)[C@@H](O)[C@@H](OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C=O)O1 FZWBNHMXJMCXLU-BLAUPYHCSA-N 0.000 description 1
- 210000001865 kupffer cell Anatomy 0.000 description 1
- 239000006193 liquid solution Substances 0.000 description 1
- 210000005229 liver cell Anatomy 0.000 description 1
- 230000033001 locomotion Effects 0.000 description 1
- 238000011866 long-term treatment Methods 0.000 description 1
- 210000001699 lower leg Anatomy 0.000 description 1
- 230000002535 lyotropic effect Effects 0.000 description 1
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 1
- 235000019341 magnesium sulphate Nutrition 0.000 description 1
- 238000002595 magnetic resonance imaging Methods 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 206010025482 malaise Diseases 0.000 description 1
- HEBKCHPVOIAQTA-UHFFFAOYSA-N meso ribitol Natural products OCC(O)C(O)C(O)CO HEBKCHPVOIAQTA-UHFFFAOYSA-N 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 230000037323 metabolic rate Effects 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 239000002905 metal composite material Substances 0.000 description 1
- 230000009401 metastasis Effects 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 238000001471 micro-filtration Methods 0.000 description 1
- 210000000274 microglia Anatomy 0.000 description 1
- 210000003470 mitochondria Anatomy 0.000 description 1
- 150000002772 monosaccharides Chemical class 0.000 description 1
- LPUQAYUQRXPFSQ-DFWYDOINSA-M monosodium L-glutamate Chemical compound [Na+].[O-]C(=O)[C@@H](N)CCC(O)=O LPUQAYUQRXPFSQ-DFWYDOINSA-M 0.000 description 1
- 239000004223 monosodium glutamate Substances 0.000 description 1
- 235000013923 monosodium glutamate Nutrition 0.000 description 1
- 229940126619 mouse monoclonal antibody Drugs 0.000 description 1
- 208000020468 mucolipidosis III alpha/beta Diseases 0.000 description 1
- 206010028093 mucopolysaccharidosis Diseases 0.000 description 1
- 208000000690 mucopolysaccharidosis VI Diseases 0.000 description 1
- 208000036710 mucopolysaccharidosis type 3A Diseases 0.000 description 1
- 208000012226 mucopolysaccharidosis type IIIA Diseases 0.000 description 1
- 210000004400 mucous membrane Anatomy 0.000 description 1
- 201000000050 myeloid neoplasm Diseases 0.000 description 1
- 238000001728 nano-filtration Methods 0.000 description 1
- 210000000478 neocortex Anatomy 0.000 description 1
- 238000004848 nephelometry Methods 0.000 description 1
- 210000004126 nerve fiber Anatomy 0.000 description 1
- 230000007472 neurodevelopment Effects 0.000 description 1
- 210000005044 neurofilament Anatomy 0.000 description 1
- 201000008051 neuronal ceroid lipofuscinosis Diseases 0.000 description 1
- 230000003982 neuronal uptake Effects 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 230000000474 nursing effect Effects 0.000 description 1
- 210000002589 oculomotor nerve Anatomy 0.000 description 1
- 210000002475 olfactory pathway Anatomy 0.000 description 1
- 230000010494 opalescence Effects 0.000 description 1
- 210000001328 optic nerve Anatomy 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 230000008520 organization Effects 0.000 description 1
- 230000010355 oscillation Effects 0.000 description 1
- 210000004681 ovum Anatomy 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 239000006174 pH buffer Substances 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 230000008506 pathogenesis Effects 0.000 description 1
- 230000001717 pathogenic effect Effects 0.000 description 1
- 230000001575 pathological effect Effects 0.000 description 1
- 230000010412 perfusion Effects 0.000 description 1
- 230000035699 permeability Effects 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 238000001050 pharmacotherapy Methods 0.000 description 1
- 239000008363 phosphate buffer Substances 0.000 description 1
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 description 1
- 230000035479 physiological effects, processes and functions Effects 0.000 description 1
- 238000011020 pilot scale process Methods 0.000 description 1
- 239000000902 placebo Substances 0.000 description 1
- 229940068196 placebo Drugs 0.000 description 1
- 229960000502 poloxamer Drugs 0.000 description 1
- 238000003752 polymerase chain reaction Methods 0.000 description 1
- 229920001451 polypropylene glycol Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 239000011148 porous material Substances 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 238000004094 preconcentration Methods 0.000 description 1
- 238000004321 preservation Methods 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 230000003405 preventing effect Effects 0.000 description 1
- 230000003449 preventive effect Effects 0.000 description 1
- 239000001294 propane Substances 0.000 description 1
- 239000012460 protein solution Substances 0.000 description 1
- 230000017854 proteolysis Effects 0.000 description 1
- 230000010349 pulsation Effects 0.000 description 1
- 201000010108 pycnodysostosis Diseases 0.000 description 1
- 238000012372 quality testing Methods 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- MUPFEKGTMRGPLJ-ZQSKZDJDSA-N raffinose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO[C@@H]2[C@@H]([C@@H](O)[C@@H](O)[C@@H](CO)O2)O)O1 MUPFEKGTMRGPLJ-ZQSKZDJDSA-N 0.000 description 1
- ZAHRKKWIAAJSAO-UHFFFAOYSA-N rapamycin Natural products COCC(O)C(=C/C(C)C(=O)CC(OC(=O)C1CCCCN1C(=O)C(=O)C2(O)OC(CC(OC)C(=CC=CC=CC(C)CC(C)C(=O)C)C)CCC2C)C(C)CC3CCC(O)C(C3)OC)C ZAHRKKWIAAJSAO-UHFFFAOYSA-N 0.000 description 1
- 238000013102 re-test Methods 0.000 description 1
- 230000006798 recombination Effects 0.000 description 1
- 238000005215 recombination Methods 0.000 description 1
- 239000012925 reference material Substances 0.000 description 1
- 230000001172 regenerating effect Effects 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 239000012465 retentate Substances 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 210000001202 rhombencephalon Anatomy 0.000 description 1
- 238000005070 sampling Methods 0.000 description 1
- 210000004761 scalp Anatomy 0.000 description 1
- 238000004062 sedimentation Methods 0.000 description 1
- 230000011218 segmentation Effects 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 210000000717 sertoli cell Anatomy 0.000 description 1
- 230000035939 shock Effects 0.000 description 1
- SQVRNKJHWKZAKO-OQPLDHBCSA-N sialic acid Chemical compound CC(=O)N[C@@H]1[C@@H](O)C[C@@](O)(C(O)=O)OC1[C@H](O)[C@H](O)CO SQVRNKJHWKZAKO-OQPLDHBCSA-N 0.000 description 1
- 208000011985 sialidosis Diseases 0.000 description 1
- 238000004088 simulation Methods 0.000 description 1
- 229960002930 sirolimus Drugs 0.000 description 1
- IHQKEDIOMGYHEB-UHFFFAOYSA-M sodium dimethylarsinate Chemical compound [Na+].C[As](C)([O-])=O IHQKEDIOMGYHEB-UHFFFAOYSA-M 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 235000010356 sorbitol Nutrition 0.000 description 1
- 125000006850 spacer group Chemical group 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 238000013112 stability test Methods 0.000 description 1
- 238000007619 statistical method Methods 0.000 description 1
- 230000001954 sterilising effect Effects 0.000 description 1
- 238000004659 sterilization and disinfection Methods 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
- 230000035882 stress Effects 0.000 description 1
- 210000002330 subarachnoid space Anatomy 0.000 description 1
- 239000002344 surface layer Substances 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 238000004114 suspension culture Methods 0.000 description 1
- 230000008961 swelling Effects 0.000 description 1
- 208000011580 syndromic disease Diseases 0.000 description 1
- 229940104261 taurate Drugs 0.000 description 1
- 230000028016 temperature homeostasis Effects 0.000 description 1
- 238000010998 test method Methods 0.000 description 1
- 208000037816 tissue injury Diseases 0.000 description 1
- 239000010936 titanium Substances 0.000 description 1
- 229910052719 titanium Inorganic materials 0.000 description 1
- 230000024664 tolerance induction Effects 0.000 description 1
- 238000012549 training Methods 0.000 description 1
- 230000001131 transforming effect Effects 0.000 description 1
- 230000007723 transport mechanism Effects 0.000 description 1
- 230000017105 transposition Effects 0.000 description 1
- 229950004616 tribromoethanol Drugs 0.000 description 1
- GPRLSGONYQIRFK-MNYXATJNSA-N triton Chemical compound [3H+] GPRLSGONYQIRFK-MNYXATJNSA-N 0.000 description 1
- 210000004926 tubular epithelial cell Anatomy 0.000 description 1
- 210000005239 tubule Anatomy 0.000 description 1
- 238000013060 ultrafiltration and diafiltration Methods 0.000 description 1
- 241000701447 unidentified baculovirus Species 0.000 description 1
- 230000002485 urinary effect Effects 0.000 description 1
- 210000002700 urine Anatomy 0.000 description 1
- 230000002477 vacuolizing effect Effects 0.000 description 1
- 210000004785 virchow-robin space Anatomy 0.000 description 1
- 230000003612 virological effect Effects 0.000 description 1
- 230000000007 visual effect Effects 0.000 description 1
- 230000008673 vomiting Effects 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 239000000811 xylitol Substances 0.000 description 1
- 235000010447 xylitol Nutrition 0.000 description 1
- HEBKCHPVOIAQTA-SCDXWVJYSA-N xylitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)CO HEBKCHPVOIAQTA-SCDXWVJYSA-N 0.000 description 1
- 229960002675 xylitol Drugs 0.000 description 1
- 210000005253 yeast cell Anatomy 0.000 description 1
Images

























































Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0085—Brain, e.g. brain implants; Spinal cord
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/08—Solutions
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/43—Enzymes; Proenzymes; Derivatives thereof
- A61K38/46—Hydrolases (3)
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/43—Enzymes; Proenzymes; Derivatives thereof
- A61K38/46—Hydrolases (3)
- A61K38/465—Hydrolases (3) acting on ester bonds (3.1), e.g. lipases, ribonucleases
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/02—Inorganic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/26—Carbohydrates, e.g. sugar alcohols, amino sugars, nucleic acids, mono-, di- or oligo-saccharides; Derivatives thereof, e.g. polysorbates, sorbitan fatty acid esters or glycyrrhizin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/19—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles lyophilised, i.e. freeze-dried, solutions or dispersions
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y301/00—Hydrolases acting on ester bonds (3.1)
- C12Y301/06—Sulfuric ester hydrolases (3.1.6)
- C12Y301/06013—Iduronate-2-sulfatase (3.1.6.13)
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Epidemiology (AREA)
- Organic Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Biochemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Immunology (AREA)
- Gastroenterology & Hepatology (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Psychology (AREA)
- Inorganic Chemistry (AREA)
- General Engineering & Computer Science (AREA)
- Genetics & Genomics (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Molecular Biology (AREA)
- Hospice & Palliative Care (AREA)
- Psychiatry (AREA)
- Obesity (AREA)
- Hematology (AREA)
- Diabetes (AREA)
- Dermatology (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicinal Preparation (AREA)
Abstract
本发明提供了,除了其它的以外,CNS递送溶酶体酶的组合物和方法,以有效治疗溶酶体贮积症。在一些实施方案中,本发明包括直接CNS鞘内施用的稳定制剂,所述稳定制剂包括艾杜糖醛酸-2-硫酸酯酶(I2S)蛋白质、盐、和聚山梨醇酯表面活性剂,以治疗Hunters综合症。
Description
相关申请的交叉引用
本申请要求美国临时申请序列号61/358,857(2010年06月25日提交);61/360,786(2010年7月1日提交);61/387,862(2010年9月29日提交);61/435,710(2011年1月24日提交);61/442,115(2011年2月11日提交);61/476,210(2011年4月15日提交);以及61/495,268(2011年6月9日提交)的优先权;其中每个的整体通过参考的方式并入本文。
与该申请相关的美国申请:名称为“治疗试剂的CNS递送,”于同日提交;“乙酰肝素N-硫酸酯酶CNS递送的方法和组合物,”于同日提交;“芳基硫酸酯酶A的CNS递送的方法和组合物,”于同日提交;“β-半乳糖脑苷脂酶CNS递送的方法和组合物,”于同日提交;“Sanfilippo综合症类型B的治疗,”于同日提交;其中每个的整体通过参考的方式并入本文。
背景技术
酶替代治疗(ERT)涉及全身施用天然或者重组来源的蛋白质和/或酶至受试者。批准的疗法通常是通过静脉向受试者施用,并且对于治疗潜在的酶缺乏症的躯体症状有效。由于所述静脉施用的蛋白质和/或酶在进入所述中枢神经系统(CNS)的细胞和组织内的有限分布,具有CNS病因学的所述疾病的治疗尤其具有挑战性,因为所述静脉施用的蛋白质和/或酶没有充分穿过血-脑屏障(BBB)。
所述血-脑屏障(BBB)是由内皮细胞构成的体系,其功能是保护所述中枢神经系统(CNS)使免于遭受在所述血流中的有害物质,例如细菌、大分子(例如蛋白质)和其它亲水性分子,通过限制这种物质穿过所述BBB的扩散并进入所述潜在的脑脊髓液(CSF)和CNS。
有几种绕过所述BBB的方式,以增强治疗试剂的脑递送,包括直接的颅内注射、所述BBB的短暂通透性、以及修饰活性剂以改变组织分布。治疗试剂完全绕过所述血管直接注射进入脑组织,但是主要有患并发症(传染、组织损伤、免疫应答)的风险,所述并发症由颅内注射、以及所述活性剂从所述施用位点的轻度扩散所引起。到目前为止,蛋白质直接施用进所述脑物质中不能实现显著的治疗效果,因为扩散障碍和可以被施用的治疗体积有限。对流协助扩散已经通过放置在所述脑实质中的导管研究,使用缓慢的、长期的输液(Bobo,等,Proc.Natl.Acad.Sci.U.S.A91,2076-2080(1994);Nguyen,等J.Neurosurg.98,584-590(2003)),但是没有使用这种方法进行长期治疗的批准疗法。另外,大脑内导管的放置是非常侵入性的,并且作为临床替代不太理想。
鞘内(IT)注射,或者施用蛋白质到所述脑脊髓液(CSF),也已经被尝试,但是没有产生成功的治疗。在这种治疗方法中的主要的挑战是所述活性剂倾向于非常紧密地绑定在脑室室管膜衬里,其阻止了随后的扩散。目前,通过直接施用到所述CSF以治疗脑遗传性疾病,还没有批准的产品。
事实上,很多人都认为在大脑表面扩散的障碍、以及缺乏有效和便捷的递送方式,极大地妨碍了在大脑中任何疾病达到足够的治疗效果。
许多溶酶体贮积症影响神经系统,从而在用传统治疗方法治疗这些疾病中表现出独特的挑战。在受影响的个体的神经元和脑膜中往往在大量聚集糖胺聚糖(GAG),导致各种形式的CNS症状。迄今为止,还没有成功地以任何方式治疗由溶酶体疾病导致的CNS症状。
因此,仍然非常需要有效地将治疗试剂递送到大脑。更具体地讲,非常需要更有效地递送活性剂到中枢神经系统,用于治疗溶酶体贮积症。
发明概述
本发明提供了有效和较少侵入性的直接递送治疗试剂到中枢神经系统(CNS)的方法。部分上,本发明基于意外发现的基础上,即溶酶体贮积症(例如,Hunters综合症)的替代酶(例如艾杜糖醛酸-2-硫酸酯酶(I2S))可以直接以高浓度(例如,大于约3mg/ml,4mg/ml,5mg/ml,10mg/ml或更多)引入到有需要治疗的受试者的脑脊髓液(CSF)中,使得所述酶有效和广泛地在各表面之间扩散并在所述脑的各区域之间贯通,包括深部脑区域。更令人惊讶的是,本发明人已经证明可以使用简单的生理盐水或缓冲液为基础的制剂会达到如此高浓度蛋白的递送,而不诱发实质性的不利影响,如在所述受试者中严重的免疫反应。因此,本发明提供了高效、临床可取的和患者友好型的方法,用于直接CNS递送以治疗具有CNS组分,特别是溶酶体贮积症的各种疾病和紊乱。本发明在CNS靶向领域和酶替代疗法中代表了显著的进步。
如下文详细描述的,本发明人已经成功开发了稳定制剂,以有效鞘内(IT)施用艾杜糖醛酸-2-硫酸酯酶(I2S)蛋白质。可以设想,但是下文描述的多种稳定制剂通常适合于治疗试剂的CNS递送,包括多种其它溶酶体酶。的确,根据本发明的稳定制剂可以通过多种技术和途径,用于CNS递送,包括但不限于,脑实质内的、大脑内的、脑室内的(ICV)、鞘内的(例如,IT-腰椎、IT-小脑延髓池)施用,以及其它直接或间接注射入所述CNS技术和/或CSF的技术和途径。
还可以设想,下文描述的多种稳定制剂通常适合于其它治疗试剂的CNS递送,例如治疗性蛋白质,包括用于溶酶体贮积症的多种替代酶。在一些实施方案中,替代酶可以是合成的、重组的、基因活化的或者天然的酶。
在多种实施方案中,本发明包括直接CNS鞘内施用的稳定制剂,所述稳定制剂包括艾杜糖醛酸-2-硫酸酯酶(I2S)蛋白质、盐和聚山梨醇酯表面活性剂。在一些实施方案中,所述I2S蛋白质存在的浓度范围从大约1-300mg/ml(例如,1-250mg/ml,1-200mg/ml,1-150mg/ml,1-100mg/ml,或者1-50mg/ml)。在一些实施方案中,所述I2S蛋白质存在的浓度为或者至多选自2mg/ml,3mg/ml,4mg/ml,5mg/ml,10mg/ml,15mg/ml,20mg/ml,25mg/ml,30mg/ml,35mg/ml,40mg/ml,45mg/ml,50mg/ml,60mg/ml,70mg/ml,80mg/ml,90mg/ml,100mg/ml,150mg/ml,200mg/ml,250mg/ml,或者300mg/ml。
在多种实施方案中,本发明包括下文描述的任意实施方案的稳定制剂,其中所述I2S蛋白质包括氨基酸序列SEQ ID NO:1。在一些实施方案中,所述I2S蛋白质包括的氨基酸序列与SEQ ID NO:1至少60%、65%、70%、75%、80%、85%、90%、95%、或者98%地一致。在一些实施方案中,所述下文描述的任意实施方案的稳定制剂包括盐。在一些实施方案中,所述盐是NaCl。在一些实施方案中,所述NaCl存在的浓度范围从大约0-300mM(例如,0-250mM,0-200mM,0-150mM,0-100mM,0-75mM,0-50mM,或者0-30mM)。在一些实施方案中,所述NaCl存在的浓度范围从大约137-154mM。在一些实施方案中,所述NaCl存在的浓度大约154mM。
在多种实施方案中,本发明包括下文描述的任意实施方案的稳定制剂,其中所述聚山梨醇酯表面活性剂选自:聚山梨醇酯20、聚山梨醇酯40、聚山梨醇酯60、聚山梨醇酯80及其组合。在一些实施方案中,所述聚山梨醇酯表面活性剂为聚山梨醇酯20。在一些实施方案中,所述聚山梨醇酯20存在的浓度范围大约0-0.02%。在一些实施方案中,所述聚山梨醇酯20以大约0.005%的浓度存在。
在多种实施方案中,本发明包括下文描述的任意实施方案的稳定制剂,其中所述制剂进一步包括缓冲剂。在一些实施方案中,所述缓冲剂选自:磷酸盐、醋酸盐、组氨酸、琥珀酸盐、Tris、及其组合。在一些实施方案中,所述缓冲剂为磷酸盐。在一些实施方案中,所述磷酸盐存在的浓度不大于50mM(例如,不大于45mM、40mM、35mM、30mM、25mM、20mM、15mM、10mM、或者5mM)。在一些实施方案中,所述磷酸盐存在的浓度不大于20mM。在多个方面中本发明包括下文描述的任意实施方案的稳定制剂,其中所述制剂具有pH大约3-8(例如,大约4-7.5、5-8、5-7.5、5-6.5、5-7.0、5.5-8.0、5.5-7.7、5.5-6.5、6-7.5、或者6-7.0)。在一些实施方案中,所述制剂具有pH大约5.5-6.5(例如,5.5,6.0,6.1,6.2,6.3,6.4或者6.5)。在一些实施方案中,所述制剂具有pH大约6.0。
在多种实施方案中,本发明包括本文描述的任意实施方案的稳定制剂,其中所述制剂为液体制剂。在多种实施方案中,本发明包括下文描述的任意实施方案的稳定制剂,其中所述制剂配制成冻干干粉。
在一些实施方案中,本发明包括鞘内施用的稳定制剂,所述稳定制剂包括浓度范围从大约1-300mg/ml的艾杜糖醛酸-2-硫酸酯酶(I2S)蛋白质,在大约154mM浓度的NaCl,在大约0.005%浓度的聚山梨醇酯20,以及大约6.0的pH。在一些实施方案中,所述I2S蛋白质的浓度大约10mg/ml。在一些实施方案中,所述I2S蛋白质的浓度为大约30mg/ml,40mg/ml,50mg/ml,75mg/ml,100mg/ml,150mg/ml,200mg/ml,250mg/ml,或者300mg/ml。
在多个方面中,本发明包括容器,在本文描述的多种实施方案中,所述容器包括单剂型的稳定制剂。在一些实施方案中,所述容器选自:安瓿、小药瓶、瓶子、药筒、贮液囊、(装有冻干粉和稀释剂的)二室注射器给药系统、或预填充的注射器。在一些实施方案中,所述容器为预填充的注射器。在一些实施方案中,所述预填充的注射器选自:具有烘干的硅树脂涂层的硼硅酸盐玻璃注射器,喷涂硅树脂的硼硅酸盐玻璃注射器,或者无硅树脂的塑料树脂注射器。在一些实施方案中,所述稳定制剂存在的体积小于约50mL(例如,小于约45ml,40ml,35ml,30ml,25ml,20ml,15ml,10ml,5ml,4ml,3ml,2.5ml,2.0ml,1.5ml,1.0ml,或者0.5ml)。在一些实施方案中,所述稳定制剂存在的体积小于约3.0mL。
在多个方面中,本发明包括治疗Hunters综合症的方法,所述方法包括根据本文描述的任意实施方案,向需要治疗的受试者鞘内施用制剂的步骤。
在一些实施方案中,本发明包括治疗Hunters综合症的方法,所述方法包括向需要治疗的受试者鞘内施用制剂的步骤,所述制剂包括浓度范围从大约1-300mg/ml的艾杜糖醛酸-2-硫酸酯酶(I2S)蛋白质,在大约154mM浓度的NaCl,在大约0.005%浓度的聚山梨醇酯20,以及大约6的pH。
在一些实施方案中,所述鞘内施用在所述受试者中没有导致实质性副作用(例如,严重的免疫应答)。在一些实施方案中,所述鞘内施用在所述受试者中没有导致实质性适应性T-细胞介导的免疫应答。
在一些实施方案中,所述制剂的所述鞘内施用导致递送所述I2S蛋白质至在所述脑、所述脊髓和/或外周器官中的多个靶组织。在一些实施方案中,所述制剂的所述鞘内施用导致递送所述I2S蛋白质至靶脑组织。在一些实施方案中,所述脑靶组织包括白质和/或在所述灰质中的神经元。在一些实施方案中,所述I2S蛋白质被递送至神经元、胶质细胞、血管周细胞和/或脑膜细胞。在一些实施方案中,所述I2S蛋白质被进一步递送至在所述脊髓中的神经元。
在一些实施方案中,所述制剂的所述鞘内施用进一步导致全身递送所述I2S蛋白质至外周靶组织。在一些实施方案中,所述外周靶组织选自:肝脏、肾脏、脾脏和/或心脏。
在一些实施方案中,所述制剂的所述鞘内施用导致在脑靶组织、脊髓神经元和/或外周靶组织中的细胞溶酶体定位。在一些实施方案中,所述制剂的所述鞘内施用导致GAG储存在所述脑靶组织、脊髓神经元和/或外周靶组织中减少。在一些实施方案中,所述GAG储存与对照相比,以至少10%、20%、30%、40%、50%、60%、70%、80%、90%、1-倍、1.5-倍、或者2-倍地减少(例如,在所述受试者中的所述预处理GAG储存)。在一些实施方案中,所述制剂的所述鞘内施用导致在神经元中液泡化减少(例如,与对照相比,以至少20%,40%、50%、60%、80%、90%、1-倍、1.5-倍、或者2-倍)。在一些实施方案中,所述神经元包括浦肯野细胞。
在一些实施方案中,所述制剂的所述鞘内施用导致在脑靶组织、脊髓神经元和/或外周靶组织中I2S酶活性增加。在一些实施方案中,所述I2S酶活性与对照相比,以至少1-倍、2-倍、3-倍、4-倍、5-倍、6-倍、7-倍、8-倍、9-倍或者10-倍地增加(例如,在所述受试者中的所述预处理内源性酶活性)。在一些实施方案中,所述增加的I2S酶活性为至少大约10nmol/hr/mg,20nmol/hr/mg,40nmol/hr/mg,50nmol/hr/mg,60nmol/hr/mg,70nmol/hr/mg,80nmol/hr/mg,90nmol/hr/mg,100nmol/hr/mg,150nmol/hr/mg,200nmol/hr/mg,250nmol/hr/mg,300nmol/hr/mg,350nmol/hr/mg,400nmol/hr/mg,450nmol/hr/mg,500nmol/hr/mg,550nmol/hr/mg或者600nmol/hr/mg。
在一些实施方案中,所述I2S酶活性在所述腰椎区域中增加。在一些实施方案中,在所述腰椎区域中,所述增加的I2S酶活性为至少大约2000nmol/hr/mg,3000nmol/hr/mg,4000nmol/hr/mg,5000nmol/hr/mg,6000nmol/hr/mg,7000nmol/hr/mg,8000nmol/hr/mg,9000nmol/hr/mg或者10,000nmol/hr/mg。
在一些实施方案中,所述制剂的所述鞘内施用导致所述Hunters综合症的至少一种症状或者特征在强度、严重程度、或者频率方面减少,或者延缓发生。在一些实施方案中,所述Hunters综合症的至少一种症状或者特征为认知障碍;白质病变;在所述脑实质中扩大的血管周围间隙、神经节、胼胝体、和/或脑干;萎缩症;和/或巨脑室。
在一些实施方案中,所述鞘内施用的发生每两个星期一次。在一些实施方案中,所述鞘内施用的发生每月一次。在一些实施方案中,所述鞘内施用的发生每两个月一次。在一些实施方案中,所述施用间隔为每月两次。在一些实施方案中,所述施用间隔为每星期一次。在一些实施方案中,所述施用间隔为每星期两次或者几次。在一些实施方案中,所述施用是持续的,例如通过持续灌注泵。在一些实施方案中,所述鞘内施用被用于连同静脉施用。在一些实施方案中,所述静脉施用频率没有比每隔一周更频繁。在一些实施方案中,所述静脉施用没有比每两个星期一次更频繁。在一些实施方案中,所述静脉施用没有比每月一次更频繁。在一些实施方案中,所述静脉施用没有比每两个月一次更频繁。在某些实施方案中,所述静脉施用比每月一次施用更频繁,例如每周两次、每周一次、每隔一周一次或者每月两次。
在一些实施方案中,静脉和鞘内施用在同一天进行。在一些实施方案中,所述静脉和鞘内施用不在彼此的一定时间内执行,例如不执行:在至少2天以内,在至少3天以内,在至少4天以内,在至少5天以内,在至少6天以内,在至少7天以内,或者在至少一周以内。在一些实施方案中,静脉和鞘内施用按照交替的时间表执行,例如每周一次、每隔一周、每月两次或者每月一次交替施用。在一些实施方案中,鞘内施用替代在施用时间表上的静脉施用,例如在每周一次、每隔一周、每月两次或者每月一次的静脉施用时间表上,在该时间表中的每第三次或第四次或第五次施用可以用鞘内施用代替静脉施用。
在一些实施方案中,静脉和鞘内施用是连续地执行的,例如首先执行静脉施用(例如,每周一次、每隔一周、每月两次或者每月一次给药为期两周、一个月、二个月、三个月、四个月、五个月、六个月或者一年或者更久),然后鞘内施用(例如每周一次、每隔一周、每月两次或者每月一次给药为期多于两周、一个月、二个月、三个月、四个月、五个月、六个月或者一年或者更久)。在一些实施方案中,首先执行鞘内施用(例如,每周一次、每隔一周、每月两次、每月一次、每两个月一次、每隔三个月一次给药为期两周、一个月、二个月、三个月、四个月、五个月、六个月或者一年或者更久),然后静脉施用(例如每周一次、每隔一周、每月两次或者每月一次给药为期多于两周、一个月、二个月、三个月、四个月、五个月、六个月或者一年或者更久)。
在一些实施方案中,所述鞘内施用在缺乏静脉施用时使用。
在一些实施方案中,所述鞘内施用在缺乏并发免疫抑制疗法时使用。
附图简述
所述附图仅仅是解释的目的,而不是为了限制。
图1是示例性的图解,其显示了:在鞘内注射3剂量的I2S后,在所述大脑皮层和小脑皮质(包括覆盖所述脑(箭头头部)表面的一层脑膜细胞)中的神经元(箭头)中检测到IT-递送的I2S。在2剂量注射的脑中,I2S IHC染色较弱(照片未显示)。在媒介物对照动物的所述脑中,对于任何类型的细胞,没有观察到阳性I2S染色,40X。
图2为示例性的图解,其显示了:在鞘内-腰椎I2S注射后,在I2S基因敲除(IKO)小鼠的脑中的病理。H&E染色的脑组织显示了(在所述媒介物对照动物中)大量的细胞储存液泡(箭头)。在2剂量(照片未显示)和3剂量注射小鼠中,在整个脑中,细胞液泡化都减少。在所述3剂量注射小鼠中,发现显著的减少(40X)。
图3为示例性的图解,其显示了LAMP-1的免疫组织化学染色,在2剂量(照片未显示)和3剂量的I2S治疗之后,在所述脑中,溶酶体活性有显著的减少(与媒介物对照小鼠相比)。所述减少的特征为:在所述整个脑所述区域中,LAMP-1阳性细胞的数量较少、以及染色强度变较轻(40X)。
图4为示例性的图解,其显示了通过比较在野生型(WT)、媒介物未处理的、以及I2S(2和3剂量)的小鼠之间的平均LAMP-1阳性区域(在所述大脑皮层(皮层)、尾状核(CP)、丘脑(TH)、白质(WM)和小脑(CBL)中)的形态计量结果,证实了:在被评估脑的所有区域中的所述LAMP-1阳性染色中,有显著的减少。数据以平均值±s.d显示。#=P<0.05;*=P<0.01;**=P<0.001。
图5描述了脑细胞示例性的电子显微照片,其示出了在超微结构水平的病理改善。媒介物处理小鼠神经元具有层状的内含物、斑马体样结构和含有贮存颗粒材料(插入)的液泡,其在I2S注射的小鼠中减少。媒介物处理小鼠的少突胶质细胞示出了大的低电子密度贮存粒(箭头),并且I2S-注射小鼠的少突胶质细胞具有最小的液泡化。比例尺:在神经元中,2μm;在少突胶质细胞中,500nm。
图6描述了示例性免疫组织化学结构,证明:在鞘内注射3剂量的I2S后,在所述肝脏的窦状腺细胞中,检测到I2S。在2剂量注射的肝脏中,2S IHC染色较弱(照片未显示)。在媒介物对照动物的所述肝脏中,没有阳性I2S染色(40X)。
图7描述了来自肝脏的示例性组织。通过H&E染色揭示了严重的细胞液泡化和异常高的溶酶体活性,并且在媒介物对照动物中发现强烈的LAMP-1免疫染色(与WT类型相比)。在用2照片未显示)或者3剂量的I2S治疗进行鞘内治疗后,发现细胞液泡化和LAMP-1免疫染色显著减少。H&E染色揭示了细胞质内液泡化几乎完全消失,伴随接近正常的肝脏细胞结构(H&E,40X;LAMP-1,20X)。
图8A-F图解了示例性的数据,其通过SEC-HPLC对生理盐水和磷酸盐制剂(所有都具有0.01%聚山梨醇酯-20)的聚集进行了比较:1个月,在≤-65℃以及40℃。
图9图解了示例性的数据,其通过SEC-HPLC方法对生理盐水和磷酸盐制剂(所有都具有0.01%聚山梨醇酯-20)的聚集进行了比较:6个月,在≤-65℃以及25℃。
图10A-F图解了示例性的数据,其通过SEC-HPLC方法对生理盐水和磷酸盐制剂(所有都具有0.01%聚山梨醇酯-20)的聚集进行了比较:24个月,在≤-65℃以及2至8℃。
图11图解了示例性的数据,其通过SAX-HPLC方法对生理盐水和磷酸盐制剂(所有都具有0.01%聚山梨醇酯-20)的聚集进行了比较:基线相对于1个月,在40℃。
图12图解了示例性的数据,其通过SAX-HPLC方法对生理盐水和磷酸盐制剂(所有都具有0.01%聚山梨醇酯-20)的电荷进行了比较:基线相对于6个月,在25℃。
图13图解了示例性的数据,其通过SAX-HPLC方法对生理盐水和磷酸盐制剂(所有都具有0.01%聚山梨醇酯-20)的电荷进行了比较:基线相对于24个月,在2至8℃。
图14图解了示例性的数据,其比较了SDS-PAGE,对于生理盐水和磷酸盐制剂(所有都具有0.01%聚山梨醇酯-20)的Coomassie染色,在基线和1个月40℃。
图15A和B图解了示例性的数据,其比较了SDS-PAGE,对于生理盐水和磷酸盐制剂(所有都具有0.01%聚山梨醇酯-20)的Coomassie染色:在6个月、25℃,以及超过16个月,在2-8℃。
图16描述示例性的组织,示出了3mg治疗组动物的大脑。在脑膜细胞中观察到阳性I2S染色(4X)。
图17描述了示例性的组织,示出了30mg治疗组动物的大脑。在神经元和脑膜细胞中观察到阳性I2S染色(4X)。
图18描述了示例性的的组织,示出了100mg治疗组动物的大脑。在神经元和脑膜细胞中观察到阳性I2S染色比在3和30mg处理动物中更强(4X)。
图19描述了示例性的的组织,示出了150mg治疗组动物的大脑。大群体的神经元是I2S阳性,伴随强烈阳性脑膜细胞。
图20描述了示例性的组织,示出了I2S阳性的神经元和胶质细胞、以及脑膜细胞(在所述大脑的层I以内),在30mg治疗组动物中(40X)。
图21描述了示例性的组织,示出了I2S阳性的神经元和胶质细胞、以及血管周细胞(在所述大脑的层III以内),在30mg治疗组动物中(40X)。
图22描述了示例性的组织,示出了I2S阳性的神经元和胶质细胞(在所述大脑的层VI以内,靠近白质),在30mg治疗组动物中(40X)。
图23描述了示例性的组织,示出了强烈阳性的I2S染色,在150mg治疗组动物的神经元中(100X)。
图24描述了示例性的组织,示出了颈部脊髓的I2S免疫染色,在150mg治疗中(4X)。
图25描述了示例性的组织,示出了腰椎脊髓的I2S免疫染色,在150mg治疗组动物中(4X)。
图26描述了示例性的组织,示出了脑膜细胞、胶质细胞和外延的/周边的/神经内膜(结缔组织细胞)的强烈阳性I2S免疫染色在150mg治疗组动物的所述腰椎切片中(40X)中发现。
图27:在150mg治疗组动物的腰椎脊髓中的神经元为强烈的I2S阳性(40X)。
图28描述来自肝脏的示例性结果(从3mg治疗组动物)。只有窦状腺细胞为I2S阳性(40X)。
图29描述来自肝脏的示例性结果(从30mg治疗组动物)。窦状腺细胞和肝细胞为I2S阳性(40X)。
图30描述来自肝脏的示例性结果(从100mg治疗组动物)。在窦状腺细胞和肝细胞中,I2S免疫染色为更加强烈(40X)。
图31描述来自肝脏的示例性结果(从150mg治疗组动物)。在窦状腺细胞和肝细胞中,识别I2S染色为强烈阳性(40X)。
图32描述来自心脏的示例性结果(从3mg治疗组动物)。I2S免疫染色为阴性(40X)。
图33描述来自心脏的示例性结果(从30mg治疗组动物)。间质细胞为I2S阳性(40X)。
图34描述来自心脏的示例性结果(从100mg治疗组动物)。观察到I2S的阳性间质细胞染色(40X)。
图35描述来自心脏的示例性结果(从150mg治疗组动物)。观察到I2S的阳性间质细胞染色(40X)。
图36描述来自肾脏的示例性结果(从3mg治疗组动物)。I2S免疫染色为阴性(40X)。
图37描述来自肾脏的示例性结果(从30mg治疗组动物)。血管小球和间质细胞为I2S阳性。
图38描述来自肾脏的示例性结果(从100mg治疗组动物)。观察到血管小球和间质细胞I2S染色增加(40X)。
图39描述来自肾脏的示例性结果(从150mg治疗组动物)。观察到近端肾小管、血管小球和间质细胞的阳性I2S染色(40X)。
图40图解了免疫组织化学(IHC)研究的结果,其评估了每周施用剂量的艾杜糖醛酸-2-硫酸酯酶(I2S)的食蟹猴CNS组织。正如根据(IHC)所测定的,I2S在整个所述CNS中有广泛的细胞沉积。在所述灰质中I2S被检测到,以剂量依赖型的方式,在所有组的(大脑、小脑、脑干和脊髓的)所述神经元中。在所述更高剂量组的表面灰质中,大量的大脑神经元对I2S染色为阳性,在所述表面皮层中(图40A)。I2S在神经元中检测到(在所述丘脑(图40B)、海马(图40C)、尾状核(图40D)和脊髓(图40E)中)。脑膜和血管周细胞也为I2S染色阳性(图40F)。所述确定的比例尺对应于25μm。
图41通过图表地比较了艾杜糖醛酸-2-硫酸酯酶(I2S)在所述颅池和脊髓池中的清除率,通过标绘I2S在这种池中相对于施用后的时间的量。
图42图解了鞘内施用艾杜糖醛酸-2-硫酸酯酶(I2S)至非人类灵长类,在6个月时间内的剂量依赖的灰质沉积。所述图解的染色强度对应于艾杜糖醛酸-2-硫酸酯酶在所述丘脑中的累积。在目前的图42中,所述细胞核通过DAPI复染,并且似乎为蓝色,所述蛋白质(I2S)似乎为绿色。
图43图解了鞘内施用艾杜糖醛酸-2-硫酸酯酶(I2S)至非人类灵长类,在单一注射后和在多注射后,在6个月时间段内的剂量依赖的累积。所述图解的染色强度对应于I2S蛋白质在所述大脑皮层的累积。
图44表明了艾杜糖醛酸-2-硫酸酯酶(I2S)的细胞定位(在非人类灵长类的整个所述大脑)。图44A图解了脑组织(从所述非人类灵长类大脑提取)的横截面剖视图,而图44B图解了所述范围的特定区域对应于在图44A中识别切片的:白质组织的三个区域(用W1、W2和W3指代),接近脑室的白质(VW)和表面灰质(SG)组织。
图45A-D图解了在6个月的时间段每月注射后,鞘内施用艾杜糖醛酸-2-硫酸酯酶(I2S)至非-人类灵长类的神经元和少突胶质细胞吸收、以及轴突连接。特别地,图45A、图45B、图45C和图45D说明了非-人类灵长类脑组织的细丝染色,在鞘内施用艾杜糖醛酸-2-硫酸酯酶(I2S),并分别对应于所述三个区域的白质(W1、W2和W3)、和表面灰质区域(SG),在图44B中识别。图45A图解了在所述白质(W1)组织中,鞘内施用I2S的少突胶质细胞吸收。图45B和图45C分别图解了在所述W2和W3白质组织中的少突胶质细胞吸收和轴突连接。图45D图解了在所述表面灰质组织中(SG),鞘内施用I2S的神经元吸收。
图46图解了非人类灵长类,在靠近脑室的白质(VW)中,艾杜糖醛酸-2-硫酸酯酶的细胞识别。如在重叠图像中所描述的,所述艾杜糖醛酸-2-硫酸酯酶与髓磷脂不相关联(红色)。在存在的所述图46中,所述核通过DAPI(底部左侧)蛋白质(I2S)复染,在顶部左侧方框出现。
图47图解了健康比格犬组织中的染色,所述比格犬被脑室内地(ICV)或者鞘内地(IT)施用单一注射的艾杜糖醛酸-2-硫酸酯酶(I2S)。如在图47A-47H中描述的,I2S在整个所述灰质(IT和ICV组两者都)广泛地分布,如通过免疫组织化学(IHC)测定的。图47A和47B图解了,在所述大脑皮层中,神经元对于I2S为阳性,在所有6个神经元层中,从所述表面分子层至所述深部内层(IT和ICV组两者都)。图47C和47D图解了,在所述IT和ICV组中的小脑皮层中,在神经元(包括浦肯野细胞)中检测到I2S。相似地,图47E和47F图解了,在IT和ICV组中,在所述海马中都有大量的神经元对于I2S为阳性。最终,图像g和h表明,(在包括IT和ICV组两者中)I2S-阳性神经元在所述丘脑和尾状核中发现。在所述存在的图47中,I2S染色用箭头指示。
图48比较性地图解了艾杜糖醛酸-2-硫酸酯酶基因敲除小鼠(IKO)的胼胝体组织,所述小鼠未处理、或者被鞘内施用I2S(简写V=液泡)。如所描述的,在所述I2S-处理IKO小鼠的胼胝体和穹窿组织中,所述处理的IKO小鼠表现某种溶酶体贮积症的细胞液泡化特征减少。
图49A图解了存在的溶酶体相关膜蛋白1(LAMP1)(一种溶酶体疾病病理学生物标记物)的显著减少,在所述处理IKO小鼠的表面大脑皮层(图49A)中,相对于所述未处理IKO对照小鼠(图49B),在20X和40X的放大倍数。
图50描述了示例性的鞘内药物递送设备(IDDD)。
图51描述了示例性的低调鞘内植入入口系统。
图52描述了示例性的鞘内药物递送设备(IDDD)。
图53描述了示例性的鞘内药物递送设备(IDDD),其允许在家施用以进行CNS酶替代治疗(ERT)。
图54是示例性的图解,其示出了:在神经元(浦肯野细胞)中,在单一大脑内注射艾杜硫酶(idursulfase)的液泡化影响。
图55是示例性的图解,其示出了:在所述脑中的I2S活性(与剂量和区域相关)。
图56是示例性的图解,其示出了艾杜硫酶(idursulfase)(在不同深度的大脑皮层)的免疫组织化学定位。
图57是示例性的图解,其示出了在猴子脊髓中的I2S活性,在鞘内给药艾杜硫酶后。
图58为示例说明,显示了鞘内给药艾杜硫酶后,猴子肝脏、心脏和肾脏的I2S活性。
图59描述了增加的Hunter-IT试验程序的示例性图表。
图60为示例性的图解,显示了在30mg剂量后,在脑组织中的各个部分的I2S浓度测量值。不同图对应于不同的测量时间。
图61为示例性的图解,显示了在施用后随着时间的推移,以各种产品浓度通过不同施用途径的I2S浓度的测量值。
图62描述了在t=5小时,IV、IT-L,或ICV给药后,食蟹猴中124I-标记的艾杜硫酶-IT的PET显像。
图63图解和示例了鞘内药物递送设备IDDD的图表、
图64描绘了IDDD的不同特征,包括受试者体内(图64A)以及展示在平坦表面上的(图64B)。
术语解释
为了使本发明更容易被理解,某些术语在下文首先被定义。下面的术语和其它术语的其它定义在整个说明书提出。
约或者大约:如本文使用的,所述术语“约”或者“大约,”如应用于一个或多个感兴趣的值,其是指与规定的参考值相似的值。在某些实施方案中,所述术语“约”或者“大约”是指某范围的值,其落入所述规定的参考值在任意方向上(大于或小于)的25%、20%、19%、18%、17%、16%、15%、14%、13%、12%、11%、10%、9%、8%、7%、6%、5%、4%、3%、2%、1%、或更小以内,除非另有说明或以其他方式明显从上下文得出(除非其中该数字将超过100%的可能值)。
转佳:如本文使用的,所述术语“转佳”意思是状态的预防、减轻或者缓减,或者受试者状态的改善。转佳包括,但是不要求病情的痊愈或彻底的预防。在一些实施方案中,转佳包括相关蛋白质的水平或其活性的增加,所述相关蛋白质在相关疾病组织中缺乏。
生物活性::如本文使用的,所述短语“生物活性”是指在生物系统中具有活性的任何实际的特性,并且特别地在生物体中。例如,当将试剂施用到生物体中,所述试剂对生物体具有生物效应,这被认为是生物活性。在特别的实施方案中,当蛋白质或多肽是生物活性的,该部分的蛋白或多肽将享有至少所述蛋白或多肽的一种生物活性,这通常被称为“生物活性”部分。
填充剂:如本文使用的,所述术语“填充剂”是指复合物,其为冻干混合物添加质量,并且有助于所述冻干块状物的物理结构(例如促进基本均匀的冻干块状物的生产,使其保持了开放的孔隙结构)。示例性的填充剂包括甘露醇、甘氨酸、氯化钠、羟乙基淀粉、乳糖、蔗糖、海藻糖、聚乙二醇和葡萄聚糖。
阳离子依赖型甘露糖-6-磷酸受体(CI-MPR):如本文使用的,所述术语“阳离子依赖型甘露糖-6-磷酸受体(CI-MPR)”是指细胞受体,所述细胞受体在高尔基体中,在酸水解酶前体上结合甘露糖-6-磷酸(M6P)标签,并被注定运输到所述溶酶体。除了甘露糖-6-磷酸,所述CI-MPR也结合其他蛋白质,包括IGF-II。所述CI-MPR也被称为“M6P/IGF-II受体”、“CI-MPR/IGF-II受体”、“IGF-II受体”或者“IGF2受体”。这些术语和其省略语在本文中互换使用。
并发免疫抑制疗法:如本文使用的,所述术语“并发免疫抑制疗法”包括作为预处理、预调节或者与治疗方法平行的任意免疫抑制疗法。
稀释剂:如本文使用的,所述术语“稀释剂”是指药学上可接受的(例如对施用到人类是安全的和非毒性的)稀释物质,其对于重组制剂的制备是有效的。示例性的稀释剂包括无菌水、注射用制菌水(BWFI)、pH缓冲溶液(例如磷酸盐-缓冲盐水)、无菌生理盐水、林格式溶液或者右旋糖溶液。
剂型:如本文使用的,所述术语“剂型”和“单位剂型”是指待治疗患者的治疗蛋白质的物理上分离的单位。每个单位含有预定量的经计算产生预期治疗效果的活性材料。可以理解,尽管如此,所述组合物的总剂量可以被由主治医生根据合理的医学判断决定。
酶替代治疗(ERT):如本文使用的,所述术语“酶替代治疗(ERT)”是指任意的治疗策略,其可以通过所述丢失的酶以纠正酶缺乏。在一些实施方案中,所述丢失的酶通过鞘内施用提供。在一些实施方案中,所述丢失的酶通过注入到血流中提供。一旦施用,酶被细胞吸收,并运输到所述溶酶体,在所述溶酶体中所述酶发挥清除由于酶缺乏而累积在所述溶酶体中的材料。通常,为了使溶酶体酶替代治疗有效,所述治疗酶被递送到在靶组织合适的细胞中的溶酶体(在所述组织中所述贮积缺陷是明显的)。
改善、增加、或者减少:如本文使用的,所述术语“改善”、“增加”或者“减少”或语法等价表达,包括相对于基线测量的值,例如在开始本文所描述的处理之前在相同个体中的测量,或者在缺乏本文所描述的处理的对照个体(或者多个对照个体)中的测量。“对照个体”是这样的个体:其受到与所述正被治疗的个体相同形式的溶酶体贮积症的折磨,并且其与所述治疗个体年龄相同(以确保在所述正被治疗的个体和所述对照个体之间所述疾病所处的阶段是可比性的)。
个体、受试者、患者:如本文使用的,所述术语“受试者”、“个体”或者“患者”是指人类或者非人类的哺乳动物受试者。所述正被治疗的个体(也称为“患者”或者“受试者”)是患有疾病的个体(胎儿、婴儿、儿童、青少年、成人)。
鞘内施用:如本文使用的,所述术语“鞘内施用”或者“鞘内注射”是指注射进入所述脊椎管(在所述脊髓周围的鞘内空间)。多种技术可以使用,包括但不限于,侧脑室注射(通过钻孔或小脑延髓池或腰椎穿刺或诸如此类)。在一些实施方案中,根据本发明的“鞘内施用”或“鞘内递送”是指IT施用或者递送(通过腰部或腰区),即腰椎IT施用或者递送。如本文使用的,所述术语“腰区”或“腰部”是指在所述第三和第四腰椎(下背部)骨之间的区域,更包含所述脊椎的L2-S1区域。
连接体:如本文使用的,所述术语“连接体”是指,在融合蛋白中,不同于在天然蛋白质中在特定位点出现的氨基酸序列,并且通常设计成柔性的或插入到在两个蛋白质部分之间的结构中(例如α-螺旋)。连接体也被称为间隔区。
冻干保护剂:如本文使用的,所述术语“冻干保护剂”是指这样的分子:其防止或减少蛋白质或其它物质在经过冷冻干燥和后续储存中的化学和/或物理的不稳定性。示例性的冻干保护剂包括糖(例如蔗糖或海藻糖);氨基酸(例如氨基酸(例如谷氨酸一钠或组氨酸);甲胺(例如甜菜碱);感胶离子盐(硫酸镁):多元醇(例如三价或更高的糖醇,例如甘油、赤藻糖醇、甘油、阿拉伯糖醇、木糖醇、山梨糖醇和甘露醇);丙二醇;聚乙二醇;普流罗尼类;及其组合。在一些实施方案中,冻干保护剂是非还原糖,例如海藻糖或蔗糖。
溶酶体酶:如本文使用的,所述术语“溶酶体酶”是指任意的酶,其能够在哺乳动物溶酶体中累积的材料,或者能使一种或多种溶酶体贮积症症状得到拯救或转佳。适合于本发明的溶酶体酶包括野生类型的或者修饰的溶酶体酶,并能使用重组和合成的方法制备,或者从天然来源中纯化。示例性的溶酶体在表1中列出。
溶酶体酶缺乏:如本文使用的,“溶酶体酶缺乏”是指一组遗传性疾病,其由于至少一种酶的缺乏所导致,所述酶是在溶酶体中将大分子(例如酶底物)裂解成肽、氨基酸、单糖、核酸和脂肪酸所需要的。结果,患有溶酶体酶缺乏症的个体在多种组织(例如CNS、肝脏、脾脏、肠、血管壁和其它器官)中具有累积的材料。
溶酶体贮积症:如本文使用的,所述术语“溶酶体贮积症”是指导致一种或多种溶酶体酶缺乏的任意疾病,所述溶酶体酶是代谢天然大分子所需要的。这些疾病通常导致在溶酶体中未降解分子的累积,导致贮藏粒(术语也称为储存囊泡)数量的增加。这些疾病和多种实施例在下文详细地被描述。
多肽:如本文使用的,“多肽”,一般而言,是指由至少两个氨基酸彼此通过肽键连接成的一串。在一些实施方案中,多肽包括至少3-5个氨基酸,其中每个通过至少一个肽键的方式与其余的连接。那些在本领域的普通技术人员会意识到有时多肽包括“非天然”氨基酸或者任选的、其它仍然能合并到多肽链中的其它实体。
替代酶:如本文使用的,所述术语“替代酶”是指任意的酶,其能发挥作用以替代至少部分的所述(在待治疗的疾病中)缺乏或丢失的酶。在一些实施方案中,所述术语“替代酶”是指任意的酶,其能发挥作用以替代至少部分的所述(在待治疗的溶酶体贮积症中)缺乏或丢失的溶酶体酶。在一些实施方案中,替代酶能减少在哺乳动物溶酶体中累积的材料,或者其能拯救或改善一种或多种溶酶体贮积症症状。适合于本发明的替代酶包括野生类型或者修饰的溶酶体酶,并且能使用重组和合成的方法制备或者从天然来源中纯化。替代酶可以是重组的、合成的、基因活化的或者天然的酶。
可溶性:如本文使用的,所述术语“可溶性”是指治疗试剂形成均匀的溶液的能力。在一些实施方案中,所述治疗试剂在所述溶液中的可溶性(其在所述溶液中被施用,并且通过它运输到作用的靶向位点(例如所述脑的所述细胞和组织))是足够的,以允许递送治疗有效量的治疗世界至作用的靶向位点。几种因素可以影响所述治疗试剂的可溶性。例如,可能影响蛋白质可溶性的相关因素包括离子强度、氨基酸序列和存在其它共溶试剂或者盐(例如钙盐)。在一些实施方案中,所述药物组合物被配制,使得所述钙盐从这种组合物中排除。在一些实施方案中,依照本发明的所述试剂是在对应的组合物中可溶性的。可以理解,当等渗溶液通常对于肠胃外施用药物是优选的,使用等渗溶液可能限制一些治疗世界充分的溶解性,特别是某些蛋白质和/或酶。已经表明,轻微高渗溶液(例如高达175mM氯化钠,在5mM磷酸钠中,pH7.0)和含糖溶液(例如高达2%蔗糖,在5mM磷酸钠中,pH7.0)在猴中具有良好的耐受性。例如,最常见的批准的CNS推注制剂组合物是生理盐水(在水中150mM NaCl)。
稳定性:如本文使用的,所述术语“稳定的”是指所述治疗试剂在较长期间保持其治疗效果(例如所有的或者大部分的其预期生物活性和/或理化完整性)的能力。所述治疗试剂的稳定性,以及所述药物组合物保持这种治疗试剂的稳定性的能力,可以在延长的时间周期内进行评估(例如至少1、3、6、12、18、24、30、36月或更久)。通常,本文描述的药物组合物已经被配制,可以使得其能稳定、或者作为选择减慢或阻止与之相配制的一种或多种治疗试剂(例如重组蛋白质)的所述降解。在配制的情况下,稳定的制剂是一种,在这种制剂中所述治疗试剂在储存和在加工过程中(例如冷冻/融化,机械混合和冻干),基本保持其物理的和/或化学完整性和生物活性。对于蛋白质稳定性,其可以通过形成高分子量(HMW)聚集物酶活性损失、肽片段形成和电荷转移性能进行测量。
受试者:如本文使用的,所述术语“受试者”意思是任意的哺乳动物,包括人类。在本发明的某些实施方案中,所述受试者是成年人、青少年或者婴儿。本发明也考虑了所述药物组合物的施用、和/或子宫内治疗方法的性能。
基本的同源性:本文使用的所述短语“基本的同源性”是指,在氨基酸或核酸序列之间的比较。如本领域的普通技术人员的理解,两条序列通常会被认为“基本同源”,如果它们在对应的位点含有同源的残基。同源的残基可以是相同的残基。可选择的,同源的残基也可以是不相同的残基,其具有合适的相似结构和/或功能特性。例如,如本领域的普通技术人员所已知的,某些氨基酸通常被分类为“疏水的”或者“亲水的”氨基酸,和/或具有“极性”或“非极性|”侧链。一个氨基酸取代另一个相同类型的氨基酸,通常被认为是“|同源”取代。
众所周知在本领域技术中,氨基酸或者核酸序列可以使用多种算法比较,包括商业计算机程序,例如核酸序列的BLASTN,氨基酸序列的BLASTP、间隙BLAST(gapped BLAST)和PSI-BLAST。示例性的这种程序在这些文献中进行了描述:Altschul,等,Basic local alignment searchtool,J. Mol.Biol.,215(3):403-410,1990;Altschul,等,Methods inEnzymology;Altschul,等,″Gapped BLAST and PSI-BLAST:a newgeneration of protein database search programs″,Nucleic Acids Res.25:3389-3402,1997;Baxevanis,et al.,Bioinformatics:APractical Guidetothe Analysis of Genes and Proteins,Wiley,1998;and Misener,等,(eds.),Bioinformatics Methods and Protocols(Methods in Molecular Biology,Vol.132),Humana Press,1999.除了识别同源序列,上述提及的所述程序通常提供同源性程度的指示。在一些实施方案中,两个序列被认为基本同源,如果他们相应残基的至少50%、55%、60%、65%、70%、75%、80%、85%、90%、91%、92%、93%、94%、95%、96%、97%、98%、99%或更多在相关延伸的残基之间是同源的。在一些实施方案中,所述相关延伸是完全的序列。在一些实施方案中,所述相关延伸是至少10、15、20、25、30、35、40、45、50、55、60、65、70、75、80、85、90、95、100、125、150、175、200、225、250、275、300、325、350、375、400、425、450、475、500或更多残基。
基本一致性:本文使用的所述短语“基本一致性”是指在氨基酸或者核酸序列之间的比较。如本领域的普通技术人员所理解的,两条序列通常会被认为是“基本一致的”,如果它们在相应的位点含有一致的残基。众所周知在本领域技术中,氨基酸或者核酸序列可以使用多种算法中的任意一个进行比较,包括在商业计算机中的程序,例如核酸序列的BLASTN,以及氨基酸序列的BLASTP、间隙BLAST(gapped BLAST)和PSI-BLAST。示例性的这种程序在下述的文献中进行了描述:Altschul,等,Basic localalignment search tool,J. Mol.Biol.,215(3):403-410,1990;Altschul,等,Methods in Enzymology;Altschul等,Nucleic Acids Res.25:3389-3402,1997;Baxevanis et al.,Bioinformatics:A Practical Guide to the Analysis of Genesand Proteins,Wiley,1998;and Misener,等,(eds.),Bioinformatics Methodsand Protocols(Methods in Molecular Biology,Vol.132),Humana Press,1999.除了识别这些一致的序列,上文所提及的所述程序通常提供了一致性程度的指示。在一些实施方案中,所述序列被认为是基本一致的,如果他们相应残基的至少50%、55%、60%、65%、70%、75%、80%、85%、90%、91%、92%、93%、94%、95%、96%、97%、98%、99%或更多在相关延伸的残基之间是一致的。在一些实施方案中,所述相关延伸是完全的序列。在一些实施方案中,所述相关延伸是至少10、15、20、25、30、35、40、45、50、55、60、65、70、75、80、85、90、95、100、125、150、175、200、225、250、275、300、325、350、375、400、425、450、475、500或更多残基。
合成的CSF:如本文使用的,所述术语“合成的CSF”是指具有与脑脊髓液一致的pH、电解质组成、葡萄糖含量和渗透压的溶液。合成的CSF也被称为人工CSF。在一些实施方案中,合成的CSF是Elliott’s B溶液。
适合于CNS递送:如本文使用的,所述短语“适合于CNS递送”或者“适合于鞘内递送”是指本发明的所述药物组合物,通常是指这些组合物的稳定性、耐受性和可溶性性能,以及这些组合物递送有效量的含于其中的治疗试剂至递送靶向位点(例如所述CSF或者所述脑)的能力。
靶组织:如本文使用的,所述术语“靶组织”是指受到待治疗的所述溶酶体贮积症影响的任意组织,或者在其中所述缺乏的溶酶体酶正常表达。在一些实施方案中,靶组织包括这些组织,在其中具有可检测的或者异常高量的酶底物(例如储存在所述组织的所述细胞溶酶体中),在患有或者易患有溶酶体贮积症的患者中。在一些实施方案中,靶组织包括这些组织:其表现疾病相关病理、症状或特征。在一些实施方案中,靶组织包括这些组织:在其中所述缺乏的溶酶体酶以升高的水平正常表达。如本文使用的,靶组织可以是脑部靶组织、脊髓靶组织和/或周边靶组织。示例性的靶组织在下文详细描述。
治疗部分:如本文使用的,所述术语“治疗部分”是指部分的分子,其为所述分子提供治疗效果。在一些实施方案中,治疗部分是具有治疗活性的多肽。
治疗有效量:如本文使用的,所述术语“治疗有效量”是指治疗性蛋白质(例如替代酶)的量,其给予所述治疗的受试者治疗效果,以合理的效益/风险比率(适合于任意的医疗)。所述治疗效果可以是客观的(例如通过某种测试或者标记物测量)或主观的(例如受试者给出的效果的指示或感觉)。特别地,所述“治疗有效量”是指某种量的治疗蛋白质或者组合物有效治疗、改善或者阻止预期的病或者病情,或者表现可检测的治疗或者预防效果(例如通过改善疾病相关症状、阻止或者延缓所述疾病的发生、和/或减少所述疾病的严重程度或频率)。在给药方案上的治疗有效量通常施用,可以包括多种单位剂量。对任何特定治疗蛋白质,治疗有效量(和/或在有效给药方案内的合适的单位剂量)会有所不同,例如,根据施用途径、以及其它药物试剂。而且,任何特定患者的所述特定的治疗有效量(和/或单位剂量)可以根据多种因素改变,包括被治疗的疾病和所述疾病的严重程度;所采用的特定药物试剂的活性;所采用的特定组合物;所述患者的年龄、体重、一般健康、性别和饮食;施用时间;施用途径;和/或所采用的特定融合蛋白的排泄和代谢;所述治疗持续的时间;以及在医药领域已知的类似因素。
可容忍的:如本文使用的,所述术语“可容忍的”和“耐受性”是指本发明的药物组合物在所述受试者(所述的这些受试者是指被施用组合物的受试者)中不引发有害反应的能力,或者可选择地在所述受试者(所述的这些受试者是指被施用组合物的受试者)中不引发严重的有害反应的能力。在一些实施方案中,本发明所述药物组合物可以被所述受试者(所述受试者是指被施用组合物的受试者)良好地容忍。
治疗:如本文使用的,所述术语“治疗”(也称为“处理”或“治疗”)是指任意施用的治疗性蛋白质(例如溶酶体酶),其部分地或者完全地缓解、改善、减轻、已知或者延缓发生,减少严重程度和/或减少(特定疾病、紊乱和/或病情(例如Hunters综合症、Sanfilippo综合症B)一种或多种症状或特征的发生率。这种治疗可以是这样的受试者(其不表现相关的疾病、紊乱和/或病情的体征),和/或这样的受试者(其仅仅表现相关的疾病、紊乱和/或病情的早期体征)。可选择地或者额外地这种治疗可以是这样的受试者,其表现所述相关的疾病、紊乱和/或病情的一种或多种已建立的体征。
发明详述
本发明提供了,此外,改善的方法和组合物,用于有效直接递送治疗试剂至所述中枢神经系统(CNS)。如上文所讨论的,本发明基于意外的发现:溶酶体贮积症(例如,Hunters综合症)的替代酶(例如I2S蛋白质)可以以高浓度直接引入到有需要治疗的受试者的所述脑脊髓液(CSF)中,而在受试者中不诱发实质性副作用。更加出乎意料的是,本发明人发现:所述替代酶可以在简单的生理盐水或者缓冲液基础制剂中递送,而不使用合成的CSF。甚至更出乎意料的是,根据本发明的鞘内递送在所述受试者中不导致实质性副作用,例如严重的免疫应答。因此,在一些实施方案中,根据本发明的鞘内递送可以在缺乏并发免疫抑制疗法时使用(例如通过预处理或者预调节不诱发免疫耐受性)。
在一些实施方案中,根据本发明的鞘内递送允许在多种脑组织间的有效扩散,导致所述替代酶在在表面、浅部和/或深部的脑区域中多种靶脑组织中的有效递送。在一些实施方案中,根据本发明的鞘内递送产生充分量的替代酶进入所述外周循环。结果,在某些情况下,根据本发明的鞘内递送导致所述替代酶在外周组织中的递送,例如肝脏、心脏、脾脏和肾脏。该发现是预料之外的,并且可以是对具有CNS和外周组件的所述溶酶体贮积症的治疗特别有用,其通常需要常规鞘内施用和静脉施用。这也是想到的:根据本发明的鞘内递送可以允许减少给药和/或iv注射的频率,而不损害周边症状治疗的治疗效果。
本发明提供了多种意外的和有益的特征,其允许替代酶有效和便捷地递送至多种脑靶组织,导致具有CNS指示的溶酶体贮积症的有效治疗。
本发明的多个方面在下面章节进行详细描述。所述章节的使用不是打算限制本发明。每章节可以应用于本发明的任意方面。在这份申请中,使用“或者”意思是“和/或”,除非另有声明。
替代酶
艾杜糖醛酸-2-硫酸酯酶(I2S)蛋白质
在一些实施方案中,本发明提供的发明方法和组合物,用于递送艾杜糖醛酸-2-硫酸酯酶(I2S)蛋白质至所述CNS,以治疗Hunters综合症。合适的I2S蛋白质可以是任意的分子或者分子的部分,其适合于天然发生的艾杜糖醛酸-2-硫酸酯酶(I2S)蛋白质活性或者拯救一种或多种与I2S-缺陷症并发的表现型或症状。在一些实施方案中,适合于本发明的替代酶是这样的多肽:其具有的N-末端和C-末端以及氨基酸序列与成熟人类I2S蛋白质基本相似或者一致。
通常地,所述人类I2S蛋白质以前体形式产生。所述人类I2S的前体形式含有信号肽(所述全长前体的氨基酸残基1-25),前肽(所述全长前体的氨基酸残基26-33),以及链(所述全长前体的残基34-550);所述链可能被进一步加工成42kDa链(所述全长前体的残基34-455)和14kDa链(所述全长前体的残基446-550)。通常地,所述前体形式也称为全长前体或者全长I2S蛋白质,其含有550氨基酸。所述成熟形式(SEQ ID NO:1)的氨基酸序列具有的信号肽被删除,并且典型野生型或天然发生的人类I2S蛋白质的全长前体(SEQ ID NO:2)在表1中示出。
表1.人类艾杜糖醛酸-2-硫酸酯酶
因此,在一些实施方案中,适合于本发明的合适的替代酶为成熟人类I2S蛋白质(SEQ ID NO:1)。在一些实施方案中,合适的替代酶可以是成熟人类I2S蛋白质的同源物或者类似物。例如,成熟人类I2S蛋白质的同源物或者类似物可以是修饰的人类I2S蛋白质,其含有一个或者多个氨基酸替换、删除、和/或插入,与野生型或天然发生的I2S蛋白质(例如,SEQ ID NO:1)相比,同时保留基本的I2S蛋白质活性。因此,在一些实施方案中,适合于本发明的合适的替代酶与成熟人类I2S蛋白质(SEQ IDNO:1)基本同源。在一些实施方案中,适合于本发明的合适的替代酶具有的氨基酸序列与SEQ ID NO:1至少50%、55%、60%、65%、70%、75%、80%、85%、90%、91%、92%、93%、94%、95%、96%、97%、98%、99%或者更多地同源。在一些实施方案中,适合于本发明的合适的替代酶与成熟人类I2S蛋白质(SEQ ID NO:1)基本一致。在一些实施方案中,适合于本发明的合适的替代酶具有的氨基酸序列与SEQ ID NO:1至少50%、55%、60%、65%、70%、75%、80%、85%、90%、91%、92%、93%、94%、95%、96%、97%、98%、99%或者更多地一致。在一些实施方案中,适合于本发明的合适的替代酶含有成熟人类I2S蛋白质的片段或者部分。
可选择地,适合于本发明的合适的替代酶为全长I2S蛋白质。在一些实施方案中,合适的替代酶可以是全长人类I2S蛋白质的同源物或者类似物。例如,全长人类I2S蛋白质的同源物或者类似物可以是修饰的全长人类I2S蛋白质,其含有一个或者多个氨基酸替换、删除、和/或插入,与野生型或天然发生的全长I2S蛋白质(例如,SEQ ID NO:2)相比,同时保留基本的I2S蛋白质活性。因此,在一些实施方案中,适合于本发明的合适的替代酶与全长人类I2S蛋白质(SEQ ID NO:2)基本同源。在一些实施方案中,适合于本发明的合适的替代酶具有的氨基酸序列与SEQ ID NO:2至少50%、55%、60%、65%、70%、75%、80%、85%、90%、91%、92%、93%、94%、95%、96%、97%、98%、99%或者更多地同源。在一些实施方案中,适合于本发明的合适的替代酶与SEQ ID NO:2基本一致。在一些实施方案中,适合于本发明的合适的替代酶具有的氨基酸序列与SEQ IDNO:2至少50%、55%、60%、65%、70%、75%、80%、85%、90%、91%、92%、93%、94%、95%、96%、97%、98%、99%或者更多地一致。在一些实施方案中,适合于本发明的合适的替代酶含有全长人类I2S蛋白质的片段或者部分。如本文所使用的,全长I2S蛋白质通常地含有信号肽序列。
其它溶酶体贮积症和替代酶
可以设想,根据本发明的发明方法和组合物可以用于治疗其它溶酶体贮积症,特别是具有CNS病因和/或症状的溶酶体贮积症,包括但不限于:天冬氨酰葡萄糖胺尿症、胆固醇脂沉积病、沃尔曼病、胱氨酸病、Danon病、法布里病、Farber脂肪肉芽肿病、Farber病,墨角藻糖苷酶缺乏病、半乳糖唾液酸沉积症I/II型、戈谢病类型I/II/III、球细胞脑白质营养不良、克拉伯病、糖原贮积症II、庞贝氏症、GM1-神经节苷脂沉积症类型I/II/III、GM2-神经节苷脂沉积症类型I、泰-萨克斯病、GM2-神经节苷脂沉积症类型II、桑德霍夫病、GM2-神经节苷脂沉积症、α-甘露糖苷过多症类型I/II、β-甘露糖苷过多症、异染性脑白质营养不良、粘脂糖症类型I、唾液酸沉积症类型I/II、唾液酸沉积症类型II/III、I-细胞病、粘脂糖症类型IIIC假胡尔勒氏多种营养不良、黏多糖贮积症类型I、黏多糖贮积症类型II、黏多糖贮积症类型IIIA、Sanfilippo综合症、黏多糖贮积症类型IIIB、黏多糖贮积症类型IIIC、黏多糖贮积症类型IIID、黏多糖贮积症类型IVA、莫基奥综合症、黏多糖贮积症类型IVB、黏多糖贮积症类型VI、黏多糖贮积症类型VII、Sly综合症、黏多糖贮积症类型IX、多种硫酸酯酶缺乏症、神经元蜡样脂褐质沉积症、CLN1Batten病、CLN2Batten病、尼曼匹克病类型A/B、尼曼匹克病类型C1、尼曼匹克病类型C2、致密性成骨不全症、Schindler病类型I/II、戈谢病和唾液酸贮积病。
所述溶酶体贮积症的遗传病因、临床表现和分子生物学的详细评论可以详参于Scriver et al.,eds.,The Metabolic and Molecular Basis of InheritedDisease,7.sup.th Ed.,Vol.II,McGraw Hill,(1995).因此,在上文的疾病中的酶缺陷对本领域的普通技术人员是已知的,其中一些在下文表2中示出。
表2.



根据本发明发明的方法可以用于递送多种其它替代酶。如本文使用的,适合于本发明的替代酶可以包括任何替代酶,其能发挥作用替代在待治疗的溶酶体贮积症中缺乏或者丢失的溶酶体酶的至少部分活性。在一些实施方案中,替代酶能够减少在溶酶体中累积的物质,或者能拯救或改善一种或多种溶酶体贮积症症状。
在一些实施方案中,合适的替代酶可以是已知的与待治疗的所述溶酶体贮积症相关的任何替代酶。在一些实施方案中,合适的替代酶是选自在表2中列出的所述酶中的酶。
在一些实施方案中,适合于本发明的替代酶可以具有野生-类型或者天然发生的序列。在一些实施方案中,适合于本发明的替代酶可以具有修饰的序列,所述修饰的序列与所述野生-类型或者天然发生的序列具有基本的同源性或者一致性(例如与所述野生-类型或者天然发生的序列具有至少50%、55%、60%、65%、70%、75%、80%、85%、90%、95%、98%序列一致性)。
适合于本发明的合适的替代酶可以通过任何可用的方法制备。例如,替代酶可以利用设计表达替代酶编码核酸的宿主细胞系统重组制备。可选择地或者附加地,替代酶可以通过激活内源基因制备。可选择地或者附加地,替代酶可以通过化学合成部分地或者完全地制备。可选择地或者附加地,替代酶可以从天然来源纯化得到。
可以使用任意的表达系统,来重组制备酶。为了给出一些例子,已知的表达系统包括,例如卵、杆状病毒、植物、酵母、或者哺乳动物细胞。
在一些实施方案中,适合于本发明的酶在哺乳动物细胞中制备。可以根据本发明使用的哺乳动物细胞的非限定实施例包括:BALB/c小鼠骨髓瘤系(NSO/l,ECACC No:85110503)、人类成视网膜细胞(PER.C6,CruCell,Leiden,荷兰)、SV40转化的猴肾脏CV1系(COS-7,ATCC CRL1651)、人类胚胎肾脏系(在悬浮培养生长的293或293细胞亚克隆,Graham等,J.Gen Virol.,36:59,1977)、人类纤维肉瘤细胞系(例如HT1080)、幼地鼠肾脏细胞(BHK,ATCC CCL10)、中国仓鼠卵巢细胞+/-DHFR(CHO,Urlaub andChasin,Proc.Natl.Acad.Sci.USA,77:4216,1980)、小鼠睾丸支持细胞(TM4,Mather,Biol.Reprod.,23:243-251,1980)、猴肾脏细胞(CV1ATCC CCL70)、非洲绿猴肾脏细胞(VERO-76,ATCC CRL-1587)、人类宫颈癌细胞(HeLa,ATCC CCL2)、犬肾细胞(MDCK,ATCC CCL34)、布法罗大鼠肝细胞(BRL3A,ATCC CRL1442)、人类肺细胞(W138,ATCC CCL75)、人类肝细胞(HepG2,HB8065)、小鼠乳腺肿瘤(MMT060562,ATCC CCL51)、TRI细胞(Mather et al.,Annals N.Y.Acad.Sci.,383:44-68,1982)、MRC5细胞、FS4细胞、和人类肝癌细胞系(Hep G2)。
在一些实施方案中,根据本发明发明的方法被用来递送从人类细胞制备的替代酶。在一些实施方案中,使用根据本发明发明的方法递送从CHO细胞制备的替代酶。
在一些实施方案中,使用本发明的方法递送的替代酶含有与在脑细胞表面上的受体结合的部分,以促进细胞吸收和/或靶向溶酶体。例如,这种受体可以是阳离子依赖型甘露糖-6-磷酸受体(CI-MPR),其与所述甘露糖-6-磷酸(M6P)残基结合。另外,所述CI-MPR也结合其它蛋白,包括IGF-II。在一些实施方案中,适合于本发明的替代酶在所述蛋白的表面含有M6P残基。在一些实施方案中,适合于本发明的替代酶可以含有双磷酸化的寡糖,其与所述CI-MPR具有更高的结合亲和力。在一些实施方案中,合适的酶含有至多大约平均每酶大约至少20%双磷酸化的寡糖。在其它实施方案中,合适的酶可以含有每酶大约10%、15%、18%、20%、25%、30%、35%、40%、45%、50%、55%、60%双磷酸化的寡糖。而这种双磷酸化的寡糖可以是天然存在在所述酶上的,应该注意,所述酶可以通过某些酶修饰而拥有这种寡糖。例如,合适的替代酶可以被某些酶(其能够催化溶酶体酶上的N-乙酰氨基葡萄糖-L-磷酸盐从UDP-GlcNAc至α-1,2-连接的甘露糖的6′位点)修饰。制备和使用这种酶的方法和组合物被详细描述,例如参见Canfield等(在美国专利号6,537,785中)和美国专利号6,534,300,每个都通过参考的方式并入本文。
在一些实施方案中,在本发明中使用的替代酶可以缀合或融合至能够结合到脑细胞表面上受体的溶酶体靶向部分。合适的溶酶体靶向部分可以是:IGF-I、IGF-II、RAP、p97、和变异型、及其同源物或者片段(例如包括与野生类型成熟人类IGF-I、IGF-II、RAP、p97肽序列具有至少70%、75%、80%、85%、90%或者95%一致性的序列的肽)。
在一些实施方案中,适合于本发明的替代酶未曾被修饰,以增强递送或者转运这种试剂穿过BBB而进入所述CNS。
在一些实施方案中,治疗性蛋白质包括靶向部分(例如溶酶体靶序列)和/或穿膜肽。在一些实施方案中,定向序列和/或穿膜肽是治疗部分的内在部分(例如,通过化学键,通过融合蛋白质)。在一些实施方案中,定向序列含有甘露糖-6-磷酸盐部分。在一些实施方案中,定向序列含有IGF-I部分。在一些实施方案中,定向序列含有IGF-II部分。
制剂
在一些实施方案中,预期的酶在鞘内递送的稳定制剂中进行递送。本发明的某些实施方案是基于,至少部分地,是基于该发现:本文公开的多种制剂促进一种或多种治疗试剂(例如I2S酶)有效递送和分布至所述CNS的靶组织、细胞和/或细胞器官。此外,本文公开的制剂能增溶高浓度的治疗试剂(例如I2S酶),并适合于这种治疗试剂递送至受试者的所述CNS(为了治疗具有CNS组件和/或病因(例如Hunters综合症)的疾病)。本文描述的所述组合物进一步的特征是:当施用到有需要其的受试者的所述CNS(鞘内施用)时,改善其稳定性和改善其耐受性。
在本发明之前,传统的无缓冲等渗盐水和Elliott’s B溶液(其是人工CSF)通常地用于鞘内递送。所述CSF组合物相对于Elliott’s B溶液的比较描述包含在下面的表3中。如表3所示,Elliott’s B溶液的所述浓度密切地与所述CSF相似。Elliott’s B溶液,尽管如此,含有非常低的缓冲液浓度,并且因此可能不能为稳定治疗试剂(例如蛋白质)的需要提供充分的缓冲能力,特别是在一段较长的时间(例如在储存条件下)。而且,Elliott’s B溶液含有某些盐,这些盐可能与所述制剂(所述制剂打算用于递送某些治疗试剂,特别是蛋白质或者酶)不相容。例如,存在于Elliott’s B溶液中的钙盐能够调节蛋白沉淀,并且因此减少了所述制剂的稳定性。
表3

因此,在一些实施方案中,适合于根据本发明的CNS递送的制剂不是合成的或者人工的CSF。
在一些实施方案中,CNS递送的制剂已经配制,使得它们能够稳定化,或者可选择地减缓或者阻止配制在其中的治疗试剂(例如I2S酶)的降解。如本文使用的,所述术语“稳定的”是指所述治疗试剂(例如I2S酶)在一段较长的时间内维持其治疗效果(例如它的预期的生物活性和/或理化完整性的全部或者大部分)的能力。治疗试剂的所述稳定性,和所述药物组合物维持这种治疗试剂稳定性的能力,可以在一段较长的时间内(例如优选至少1、3、6、12、18、24、30、36月或者更多)进行评估。就制剂而言,稳定的制剂是这样的:其中的所述治疗试剂在储存和加工(例如冷冻/解冻、机械混合和冷冻干燥)过程中基本上保持其物理和/或化学完整性和生物活性。对于蛋白质稳定性,它可以通过形成高分子重量(HMW)聚集体,酶活性损失、肽片段的形成和电荷性能的变化进行量度。
治疗试剂的稳定性是特别重要的。所述治疗试剂的稳定性可以进一步评估,相对于所述治疗试剂在较长一段时间内的生物活性或理化完整性。例如,在给定时间点的稳定性可以与较早时间点(例如在配制的第0天)进行比较,或者与未配制的治疗试剂进行比较,并且这种比较结果以百分数表示。优选的,本发明的药物组合物使所述治疗试剂的生物活性或者理化完整性在一端较长的时间(例如在至少大约6-12个月、在室温或者加速储存条件下)内维持至少100%、至少99%、至少98%、至少97%至少95%、至少90%、至少85%、至少80%、至少75%、至少70%、至少65%、至少60%、至少55%或至少50%。
在一些实施方案中,治疗试剂(例如预期的酶)在本发明的制剂中是可溶的。所述术语“可溶性”(如其所提及的本发明的治疗试剂)是指这种治疗试剂形成均匀的溶液的能力。优选地,所述治疗试剂在所述溶液(在其中所述治疗试剂被施用,并且通过所述溶液所述治疗试剂被运送到作用的靶位点)中的可溶性是足够地以允许递送治疗有效量的所述治疗试剂至作用的靶位点。几种因素可以影响所述治疗试剂的可溶性。例如,可能影响蛋白质可溶性的相关因素,包括离子强度、氨基酸序列和存在其它共溶试剂或离子(例如钙盐)。在一些实施方案中,所述药物组合物被配制成使得所述钙盐从所述组合物排除。
合适的制剂,无论是水性的、预冻干的、冻干的或者再生的形式可以含有多种浓度的感兴趣治疗试剂。在一些实施方案中,制剂可以含有感兴趣蛋白质或者治疗试剂的浓度范围为约0.1mg/ml至100mg/ml(例如,约0.1mg/ml至80mg/ml,约0.1mg/ml至60mg/ml,约0.1mg/ml至50mg/ml,约0.1mg/ml至40mg/ml,约0.l mg/ml至30mg/ml,约0.1mg/ml至25mg/ml,约0.1mg/ml至20mg/ml,约0.1mg/ml至60mg/ml,约0.1mg/ml至50mg/ml,约0.1mg/ml至40mg/ml,约0.1mg/ml至30mg/ml,约0.1mg/ml至25mg/ml,约0.1mg/ml至20mg/ml,约0.1mg/ml至15mg/ml,约0.1mg/ml至10mg/ml,约0.1mg/ml至5mg/ml,约1mg/ml至10mg/ml,约1mg/ml至20mg/ml,约1mg/ml至40mg/ml,约5mg/ml至100mg/ml,约5mg/ml至50mg/ml,或者约5mg/ml至25mg/ml)。在一些实施方案中,根据本发明的制剂可以含有治疗试剂的浓度大约1mg/ml,5mg/ml,10mg/ml,15mg/ml,20mg/ml,25mg/ml,30mg/ml,40mg/ml,50mg/ml,60mg/ml,70mg/ml,80mg/ml,90mg/ml,或者100mg/ml。
本发明的所述制剂特征在于它们的耐受性,作为水性的溶液或者再生的冻干的溶液。如本文所使用的,所述术语”可容忍的”和“耐受性”是指本发明的所述药物组合物在被施用这种组合物的所述受试者中不引发不良反应的能力,或者可选择地在被施用这种组合物的所述受试者中不引发严重不良反应的能力。在一些实施方案中,本发明的所述药物组合物被施用这种组合物的受试者良好地容忍。
许多治疗试剂,特别是本发明的蛋白质和酶,要求可控制的pH值和特定的赋形剂,以维持其在本发明所述药物组合物中的可溶性和稳定性。下面的表4认同的蛋白质制剂示例性方面,被认为维持本发明蛋白质治疗试剂的可溶性和稳定性。
表4


缓冲剂
所述制剂的所述pH是能够改变治疗试剂(例如酶或者蛋白质)在水性制剂或者预冻干制剂中的可溶性的额外因素。因此,本发明的所述制剂优选包含一种或多种缓冲剂。在一些实施方案中,所述水性的制剂包含大量的缓冲剂,以充分维持所述组合物最适pH值在约4.0-8.0之间(例如,约4.0、4.5、5.0、5.5、6.0、6.2、6.4、6.5、6.6、6.8、7.0、7.5,或者8.0)。在一些实施方案中,所述制剂的所述pH在约5.0-7.5之间,在约5.5-7.0之间,在约6.0-7.0之间,在约5.5-6.0之间,在约5.5-6.5之间,在约5.0-6.0之间,在约5.0-6.5之间以及在约6.0-7.5之间。合适的缓冲剂包括:例如醋酸盐、柠檬酸盐、组氨酸、磷酸盐、琥珀酸盐、三羟甲基甲胺(“Tris”)和其它有机酸。本发明的所述药物组合物的所述缓冲剂浓度和pH范围是控制或者调解所述制剂的耐受性的因素。在一些实施方案中,缓冲剂存在的浓度范围在约1mM至约150mM之间,或者在约10mM至约50mM之间,或者在约15mM至约50mM之间,或者在约20mM至约50mM之间,或者在约25mM至约50mM之间。在一些实施方案中,合适的缓冲剂存在的浓度范围大约1mM、5mM、10mM、15mM、20mM、25mM、30mM、35mM、40mM、45mM50mM、75mM、100mM、125mM或者150mM。
渗涨度
在一些实施方案中,制剂(无论是水性的、预冻干的、冻干的或者再生的形式)含有保持所述制剂等渗的等渗剂。通常地,“等渗的”意思是所述感兴趣制剂与人类血液具有基本相同的渗透压。等渗的制剂通常具有的渗透压,从大约240mOsm/kg至大约350mOsm/kg。等渗性可以被测量,例如,使用蒸汽压力或者凝固点类型渗透计。示例性的等渗剂包括,但不限于甘氨酸、山梨糖、甘露醇、氯化钠和精氨酸。在一些实施方案中,合适的等渗剂可以存在于水性的和/或预冻干的制剂中,以浓度从大约0.01-5%(例如0.05、0.1、0.15、0.2、0.3、0.4、0.5、0.75、1.0、1.25、1.5、2.0、2.5、3.0、4.0或5.0%),按重量计。在一些实施方案中,冻干的制剂含有等渗剂以维持所述预-冻干制剂或者所述再生的制剂等渗。
而且,通常地对于非肠胃施用的药物需要制备等渗的溶液,使用等渗的溶液可以改变一些治疗试剂的可溶性,并且特别地是一些蛋白质和/或酶。已经被表明,稍微地高渗溶液(例如,至多175mM氯化钠,在5mM磷酸钠中,pH7.0)和含糖溶液(例如,至多2%蔗糖,在5mM磷酸钠中,pH7.0)可以被良好地容忍。最常见的批准CNS推注制剂组合物是生理盐水(约150mM NaCl,在水中)。
稳定剂
在一些实施方案中,制剂可以含有稳定剂或冻干保护剂以保护所述蛋白质。通常地,合适的稳定剂为糖、非还原性糖和/或氨基酸。示例性的糖包括但不限于:葡萄聚糖、乳糖、甘露糖、甘露糖、山梨糖、棉子糖、蔗糖和海藻糖。示例性的氨基酸包括但不限于:精氨酸、甘氨酸和蛋氨酸。附加的稳定剂可以包括氯化钠、羟乙基淀粉和聚乙烯吡咯烷酮。在所述冻干的制剂中的稳定剂的量通常使得所述制剂等渗。无论如何,高渗的再生的制剂也可以是合适的。另外,所述稳定剂的量不能太低,而使所述治疗试剂发生降解/聚集的量不可接受。在所述制剂中的示例性稳定剂浓度范围从约1mM至约400mM(例如,从约30mM至约300mM,以及从约50mM至约100mM),或者可选择地,从0.1%至15%(例如,从1%至10%,从5%至15%,从5%至10%),以重量计。在一些实施方案中,所述稳定剂和所述治疗试剂的质量比例为约1∶1。在一些实施方案中,所述稳定剂和所述治疗试剂的质量比例可以是约0.1∶1,0.2∶1,0.25∶1,0.4∶1,0.5∶1,1∶1,2∶1,2.6∶1,3∶1,4∶1,5∶1,10∶1或者20∶1。在一些实施方案中,适合于冷冻干燥,所述稳定剂也是冻干保护剂。
在一些实施方案中,适合于本发明的液体制剂含有无定形材料。在一些实施方案中,适合于本发明的液体制剂含有基本量的无定形材料(例如,基于蔗糖的制剂)。在一些实施方案中,适合于本发明的液体制剂含有部分晶体/部分无定形材料。
填充剂
在一些实施方案中,适合于冷冻干燥的制剂可以进一步包括一种或多种填充剂。“填充剂”是指复合物,其为冻干混合物添加质量,并且有助于所述冻干块状物的物理结构。例如,填充剂可以改善冻干块状物的所述外观(例如,基本均一的冻干块状物)。合适的填充剂包括但不限于:氯化钠、乳糖、甘露糖、甘氨酸、蔗糖、海藻糖、羟乙基淀粉。示例性的填充剂浓度从约1%至约10%(例如,1.0%、1.5%、2.0%、2.5%、3.0%、3.5%、4.0%、4.5%、5.0%、5.5%、6.0%、6.5%、7.0%、7.5%、8.0%、8.5%、9.0%、9.5%、以及10.0%)。
表面活性剂
在一些实施方案中,理想的是,添加表面活性剂至制剂。示例性的表面活性剂包括非离子表面活性剂,例如聚山梨醇酯(例如聚山梨醇酯20或者80);泊洛沙姆(例如洛沙姆188);Triton;十二烷基硫酸钠(SDS);月桂基硫酸钠;sodium octyl glycoside;十二烷基-,十四烷基-,linoleyl-,或者硬脂基-磺基甜菜碱;十二烷基-,十四烷基-,linoleyl-或者硬脂基-肌氨酸;linoleyl-,十四烷基-,或者鲸蜡基-甜菜碱;lauroamidopropyl-,cocamidopropyl-,linoleamidopropyl-,myristamidopropyl-,palmidopropyl-,或者异硬脂酰丙基-甜菜碱(例如lauroamidopropyl);myristarnidopropyl-,palmidopropyl-,或者异硬脂酰丙基-二甲胺;甲基椰油酰基钠-,或者disodiummethyl ofeyl-taurate;以及MONAQUATTM系列(Mona Industries,Inc.,Paterson,N.J.),聚乙二醇,聚丙二醇,以及乙烯和丙二醇的共聚物(例如Pluronics,PF68等)。通常地,所述添加的表面活性剂的量是使得所述蛋白质聚集减少,并且使颗粒或冒泡最小化。例如,存在于制剂中的表面活性剂可以是从大约0.001-0.5%(例如大约0.005-0.05%,或者0.005-0.01%)。特别的,存在于制剂中的表面活性剂浓度可以是约0.005%、0.01%、0.02%、0.1%、0.2%、0.3%、0.4%或者0.5%等。可选择地,或者另外,所述表面活性剂可以添加至冻干制剂、预冻干制剂和/或再生制剂。
其它药学上可接受的载体、赋形剂或稳定剂,例如在Remington′sPharmaceutical Sciences16th edition,Osol,A.Ed.(1980)中描述的,可以被包含在所述制剂(和/或所述冻干的制剂和/或所述再生的制剂)中,以他们对所述制剂的预期特性没有不良影响为条件。可接受的载体、赋形剂或稳定剂在所采用的剂量和浓度对于接受者是无毒的,并且包括但不限于:附加的缓冲剂;防腐剂;共溶剂;抗氧化剂(包括抗坏血酸和蛋氨酸);螯合剂(例如EDTA);金属复合物(例如,Zn-蛋白质复合物);生物可降解的多聚体(例如多元酯);和/或盐-形成平衡离子(例如钠)。
根据本发明的制剂(无论是水性的、预冻干的、冻干的或者再生的形式)可以基于这些进行评估:产品质量分析、再生时间(如果冻干)、再生质量(如果冻干)、高分子量、湿度、和玻璃转化温度。通常地,蛋白质质量和产品分析包括产品降解率分析,使用的方法包括但不限于,体积排阻HPLC(SE-HPLC)、阳离子交换-HPLC(CEX-HPLC)、X-射线衍射(XRD)、调制式差示扫描量热法(mDSC)、反相HPLC(RP-HPLC)、多角度激光光散射(MALS)、荧光、紫外吸收、浊度测定法、毛细管电泳(CE)、SDS-PAGE、以及其组合。在一些实施方案中,根据本发明的产品的评估可以包括评估外貌的步骤(无论液体或者块状物的外貌)。
通常,制剂(冻干的或者水性的)可以用于在室温下在长时间的储存。储存温度通常的范围可以从0℃至45℃(例如4℃、20℃、25℃、45℃等)。制剂可以在数月至数年的时间段储存。储存时间通常是24月、12月、6月、4.5月、3月、2月或者1月。制剂可以直接在用于施用、消除转移步骤的所述容器中储存。
制剂可以直接在冻干容器中储存(如果冻干),所述冻干容器可以起重组器皿、消除转移步骤的作用。可选择的,冻干产品制剂可以估量成更小的储存增量。储存应该通常避免导致所述蛋白质降解的事件,包括但不限于暴露在阳光、紫外辐射、其它电磁辐射形式、过热或过冷、快速热冲击和机械冲击。
冻干
根据本发明的发明方法可以被用来冻干任何材料,特别是治疗试剂。通常地,预-冻干制剂进一步含有合适选择的赋形剂或者其它成分(例如稳定剂、缓冲剂、填充剂和表面活性剂),以阻止感兴趣的复合物在冷冻干燥和储存期间降解(例如,蛋白质聚集、脱酰胺基、和/或氧化)。对于冷冻干燥的制剂可以包括一种或多种附加的成分,所述附加的成分包括冻干保护剂或者稳定剂、缓冲剂、填充剂、等渗剂和表面活性剂。
在将所述感兴趣的物质和其它附加的成分混合一起后,将所述制剂冻干。冻干通常包括三个主要的阶段:冷冻、初级干燥和次级干燥。冷冻对于将水转变成冰或者将一些无定形制剂组分转变为晶体形式是必要的。初级干燥是在低压和低温下,通过直接升华使冰从所述冷冻产品中除去的加工步骤。次级干燥是利用残余水至所述蒸发面的扩散作用,从所述产品基质除去结合水的加工步骤。在次级干燥期间的产品温度通常高于初级干燥。参见,Tang X.et al.(2004)“Design of freeze-drying processes forpharmaceuticals:Practical advice,”Pharm.Res.,21:191-200;Nail S.L.et al.(2002)“Fundamentals of freeze-drying,”in Development and manufacture ofprotein pharmaceuticals.Nail S.L.editor New York:KluwerAcademic/Plenum Publishers,pp281-353;Wang et al.(2000)“Lyophilizationand development of solid protein pharmaceuticals,”Int.J.Pharm.,203:1-60;Williams N.A.et al.(1984)“The lyophilization of pharmaceuticals;Aliterature review.”J.Parenteral Sci.Technol.,38:48-59.通常,可以将任何的冻干过程与本发明联合使用。
在一些实施方案中,可以在所述产品初始冷冻期间引入退火步骤。所述退火步骤可以减少所述总循环时间。不期望受任何理论的约束,可以设想,所述退火步骤有助于促进赋形剂结晶化、以及大冰晶的形成(由于在过冷期间形成的小结晶体的再-结晶化),这反过来,改善了再生。通常地,退火步骤包括间隔或者冷冻期间的温度振荡。例如,所述冷冻温度可以是-40℃,所述退火步骤的所述温度将增加至,例如,-10℃并维持该温度一段设定的时间。所述退火步骤时间范围可以从0.5小时至8小时(例如,0.5、1.0、1.5、2.0、2.5、3、4、6、和8小时)。所述退火温度可以在所述冷冻温度和0℃之间。
冻干可以在容器中进行,例如管子、袋子、瓶子、盘子、小药瓶(例如,玻璃小药瓶)、注射器或者任意其它合适的容器。所述容器可以是一次性的。冻干也可以是大规模或者小规模进行。在一些情况下,在冻干所述蛋白质制剂的容器中,进行所述蛋白质的再生,以避免转移步骤,是可取的。在这种情况下的所述容器可以例如是:3、4、5、10、20、50或者100cc小药瓶。
为了该目的可以获得许多不同的冷冻干燥器,例如Hull中试规模干燥器(SP Industries,USA),Genesis(SP Industries)实验室冷冻干燥器,或者其它任意能够控制所述给定的冻干加工参数的冷冻干燥器。冷冻-干燥通过所述制剂的冷冻、以及随后在适合初级干燥的温度下使冰从所述冷冻内容物升华来实现。初始冷冻使所述制剂温度低于约-20℃(例如,-50℃,-45℃,-40℃,-35℃,-30℃,-25℃,等),在通常不超过4小时内(例如,不超过约3小时,不超过约2.5小时,不超过约2小时)。在这个条件下,所述产品温度通常地低于所述制剂的共晶点或者崩塌温度。通常地,所述初级干燥的所述搁板温度范围从约-30至25℃(以所述产品在初级干燥期间保持在所述熔点以下为条件),在合适的压力下,范围通常从约20至250mTorr。所述制剂、容纳所述样品的容器(例如玻璃小药瓶)的大小和类型、以及液体体积,将主要决定干燥所需要的时间(其范围可以从几小时至几天)。次级干燥阶段在约0-60℃下执行,主要根据所述容器的类型和大小、以及采用的治疗性蛋白质的类型。再者,液体的体积将主要决定干燥所需要的时间,其范围可以从几小时到几天。
作为一个一般性的命题,冻干将导致冻干制剂其中的所述湿度小于约5%,小于约4%,小于约3%,小于约2%,小于约1%,以及小于约0.5%。
再生
但是,本发明的所述药物组合物在施用至受试者的过程中通常是水性的形式,在一些实施方案中,本发明的所述药物组合物是冻干的。这种组合物在施用至受试者前,必须通过添加一种或多种稀释剂至其中使其再生。在所述预期的阶段,通常在施用至所述患者之前的合适时间点,所述冻干制剂可以用稀释剂再生,使得在所述再生的制剂中的所述蛋白质浓度为理想的。
根据本发明可以使用多种稀释剂。在一些实施方案中,对于再生合适的稀释剂是水。作为稀释剂使用的水可以以多种方式处理,包括:反渗析、蒸馏、消电离作用、过滤(例如,活性炭、微量过滤、纳米过滤)以及这些治疗方法的组合。一般而言,所述水应该是适合于注射的,包括但不限于,无菌水或注射用抑菌水。
附加的示例性稀释剂包括pH缓冲的溶液(例如磷酸盐-缓冲的生理盐水),无菌生理盐水溶液,Elliot’s溶液,Ringer’s溶液或者右旋糖溶液。合适的稀释剂可以优选含有防腐剂。示例性的防腐剂包括芳香醇,例如苯甲基或者苯酚醇。采用的防腐剂的量通过评估不同防腐剂浓度来测定,使其与所述蛋白质和防腐剂功效测试相容。例如,如果所述防腐剂是芳香醇(例如苯甲醇),它能以从约0.1-2.0%,从约0.5-1.5%或者约1.0-1.2%的量存在。
适合本发明的稀释剂包括多种添加剂,包括但不限于,pH缓冲剂,(例如Tris,组氨酸)、盐(例如氯化钠)、以及其它添加剂(例如蔗糖),所述添加剂包括上文描述的这些(例如稳定剂、等渗剂)。
根据本发明,冻干的物质(例如蛋白质)可以被再生至至少25mg/ml的浓度(例如,至少50mg/ml,至少75mg/ml,至少100mg/)以及任何在其间的范围。在一些实施方案中,冻干的物质(例如,蛋白质)可以再生至浓度范围从约1mg/ml至100mg/ml(例如,从约1mg/ml至50mg/ml,从1mg/ml至100mg/ml,从约1mg/ml至约5mg/ml,从约1mg/ml至约10mg/ml,从约1mg/ml至约25mg/ml,从约1mg/ml至约75mg/ml,从约10mg/ml至约30mg/ml,从约10mg/ml至约50mg/ml,从约10mg/ml至约75mg/ml,从约10mg/ml至约100mg/ml,从约25mg/ml至约50mg/ml,从约25mg/ml至约75mg/ml,从约25mg/ml至约100mg/ml,从约50mg/ml至约75mg/ml,从约50mg/ml至约100mg/ml)。在一些实施方案中,在所述再生的制剂中的蛋白质浓度可以高于在所述预-冻干制剂中的浓度。在所述再生制剂中的蛋白质浓度对于打算皮下或者肌肉间递送的再生制剂特别有用。在一些实施方案中,在所述再生制剂中的蛋白质浓度可以是所述预冻干制剂的约2-50倍(例如约2-20,约2-10倍,约2-5倍)。在一些实施方案中,在所述再生制剂中的蛋白质浓度可以是所述预冻干制剂的至少约2倍(例如至少约3、4、5、10、20、40倍)。
根据本发明的再生可以在任意的容器中进行。适合于本发明的示例性容器,包括但不限于:例如管子、小药瓶、注射器(例如,单室的或者二室的)、袋子、瓶子和盘子。合适的容器可以用任意材料制备,例如玻璃、塑料、金属。所述容器可以是一次使用的或者可多次使用的。再生也可以大规模或者小规模进行
在一些情况下,在冻干所述蛋白质制剂的容器中,进行所述蛋白质的再生,以避免转移步骤,是可取的。在这种情况下的所述容器可以例如是:3、4、5、10、20、50或者100cc小药瓶。在一些实施方案中,用于冻干和再生的合适的容器是二室的注射器(例如,(装有冻干粉和稀释剂的)二室注射器给药系统,(Vetter)注射器)。例如,二室的注射器可以含有冻干的物质和所述稀释剂两者,每个在单独的室中,通过阻塞物分开(参见实施例5)。为了再生,可以在所述稀释剂侧将活塞与所述阻塞物连接并且按压让稀释剂移动进入所述产品室,使得所述稀释剂能接触所述冻干的物质,然后再生可以如本文所描述地发生(参见实施例5)。
本发明所述的药物组合物、制剂和相关方法对于递送多种治疗试剂至受试者的所述CNS(例如,鞘内地、脑室内地或者脑池内地)以及治疗所述相关的疾病是有用的。本发明的所述药物组合物对于递送蛋白质和酶(例如,酶替代治疗)至患有溶酶体贮积症的受试者特别有用。所述溶酶体贮积症代表了导致溶酶体功能缺陷的一组相对罕见的遗传性代谢疾病。所述溶酶体疾病的特征在于未消化大分子在所述溶酶体内的累积,其导致这种溶酶体大小和数量的增加,并最终导致细胞功能障碍和临床异常。
CNS递送
可以设想,下文描述的多种稳定制剂通常对于治疗试剂的CNS递送是合适的。根据本发明的稳定制剂可以通过多种技术和途径,用于CNS递送,包括但不限于:脑实质内的、大脑内的、脑室内的(ICV)、鞘内的(例如,IT-腰椎、IT-小脑延髓池)施用,以及其它直接或间接注射入所述CNS和/或CSF的技术和途径。
鞘内递送
在一些实施方案中,替代酶在本文所描述的制剂中被递送至所述CNS。在一些实施方案中,通过施用进入有需要治疗的受试者的所述脑脊髓液(CSF),递送替代酶至所述CNS。在一些实施方案中,使用鞘内施用递送期望的替代酶(例如I2S蛋白质)进入所述CSF。如本文所使用的,鞘内施用(也称为鞘内注射)是指注射进入所述脊髓管(在所述脊髓周围的鞘内空间)。可以使用多种技术,包括但不限于,侧脑室注射(通过钻孔或小脑延髓池或腰椎穿刺或诸如此类)。示例性方法的描述见于Lazorthes et al.Advances in Drug Delivery Systems and Applications in Neurosurgery,143-192and Omaya et al.,Cancer Drug Delivery,1:169-179,其内容通过参考的方式并入本文。
根据本发明,可以在所述脊髓管周围的任意区域注射酶。在一些实施方案中,将酶注射进入所述腰椎区域或者所述小脑延髓池,或者脑室内地注射进入脑室空间。如本文所使用的,所述术语”腰椎部位”或者“腰椎区域”是指在所述第三和第四腰椎(下背部)骨之间的区域,更包含所述脊椎的L2-S1区域。通常地,通过腰椎部位或腰椎区域的鞘内注射也称为“腰椎IT递送”或者“腰椎IT施用”。所述术语“小脑延髓池”是指在所述小脑周围和下方的空间(经由在所述头骨和所述脊椎顶部之间的开口)。通常地,通过小脑延髓池的鞘内注射也称为“小脑延髓池递送”。所述术语“脑室”是指在所述脑中的空腔,其与所述脊髓的中央管是连续的。通常地,经由所述脑室空腔的注射是指脑室内的(intravetricular Cerebral,ICV)递送。
在一些实施方案中,根据本发明的“鞘内施用”或者“鞘内递送”是指腰椎IT施用或者递送,例如,在所述第三和第四腰椎骨(下背部)之间递送,更包含所述脊椎的L2-S1区域。这也是考虑到的,腰部IT施用或者递送区别于在所述小脑延髓池递送,因为根据本发明的腰部IT施用或者递送提供了更好和更有效地递送至远端脊椎管;而此外,小脑延髓池递送通常地不能良好地递送至远端的脊髓管。
鞘内递送的设备
根据本发明鞘内递送可以使用多种设备。在一些实施方案中,鞘内施用的设备含有:流体进入端口(例如可注射的端口);空心体(例如导管),所述空心体具有与所述流体进入端口液体连通的第一流体孔口以及配置插入到脊髓中的第二流体孔口;和固定机构,所述固定机构用于固定在所述脊髓中的空心体的插入。如在图62中示出的非限定性实施例,合适的固定机构含有固定在所述空心体表面的一个或多个球形门柄、以及可调节覆盖所述一个或多个球形门柄的缝合环,所述缝合环用于阻止所述空心体(例如导管)从所述脊髓滑出。在多个实施方案中,所述流体进入端口包括贮液囊。在一些实施方案中,所述流体进入端口包括机械泵(例如灌流泵)。在一些实施方案中,植入的导管连接到贮液囊(例如用于推注递送),或者灌流泵。所述流体进入端口可以是植入的或者外置的。
在一些实施方案中,鞘内施用可以通过腰椎穿刺(缓慢推注)或通过端口-导管递送系统(例如注射或者推注)进行。在一些实施方案中,所述导管插入到所述腰椎之间的薄层,所述前端被拧到预期水平的鞘空间(通常L3-L4)(图63)。
相对于静脉施用,适合鞘内施用的单剂量体积通常地小的。通常地,根据本发明的鞘内递送维持了所述CSF的组合物与所述受试者颅内压力之间的平衡。在一些实施方案中,对于相应的去除了CSF的受试者,鞘内递送没有进行。在一些实施方案中,合适的单剂量体积可以例如小于大约10ml、8ml、6ml、5ml、4ml、3ml、2ml、1.5ml、1ml、或者0.5ml。在一些实施方案中,合适的单剂量体积可以是大约0.5-5ml、0.5-4ml、0.5-3ml、0.5-2ml、0.5-1ml、1-3ml、1-5ml、1.5-3ml、1-4ml、或者0.5-1.5ml。在一些实施方案中,根据本发明的鞘内递送涉及首先去除预期量的CSF的步骤。在一些实施方案中,在IT施用之前,小于大约10ml(例如小于大约9ml、8ml、7ml、6ml、5ml、4ml、3ml、2ml、1ml)的CSF首先被去除。在这些情况下,合适的单剂量体积可以是例如多于大约3ml、4ml、5ml、6ml、7ml、8ml、9ml、10ml、15ml、或者20ml。
可以使用多种其它设备影响治疗组合物的鞘内施用。例如,含有预期酶的制剂的给予可以使用Ommaya贮液囊,所述Ommaya贮液囊通常用于脑膜癌扩散药物的鞘内施用(Lancet2:983-84,1963)。更具体地说,在这种方法中,脑室管通过在所述前角形成的孔中插入,并且连接到安装在所述头皮下的Ommaya贮液囊;并且所述Ommaya贮液囊是皮下穿刺,使其鞘内递送所述特定被替代的酶(所述酶被注射到所述贮液囊中)。治疗组合物或者制剂鞘内施用到个体的其它设备的描述可以参见:美国专利号6,217,552,其通过参考的方式并入本文。可选择地,所述药物可以鞘内施用,例如,可以单一注射,或者持续注射。应该理解,所述剂量治疗可以以单一剂量施用或者多种剂量的形式。
对于注射,本发明的制剂可以以液体溶液配制。另外,所述酶可以以固体形式配制,并且在使用之前重溶或者立即悬浮。冻干形式也包括在内。所述注射,例如以所述酶的推注注射或者持续注射(例如使用灌流泵)的形式。
在本发明的一个实施方案中,所述酶通过侧脑室注射至所述受试者的脑中。所述注射,可以例如通过在所述受试者的头骨中钻孔。在另一实施方案中,所述酶和/或其它药物制剂通过外科插入分流至所述受试者的脑室中。例如,所述注射可以在侧脑室(其更大)进行。在一些实施方案中,也可以注射进入所述第三和第四较小脑室。
在又一个实施方案中,在本发明中使用的所述药物组合物通过注射施用至受试者所述小脑延髓池、或腰椎区域。
在本发明的又一实施方案中,所述药学可接受的制剂提供了持久的递送至受试者(例如:在本发明中使用的所述酶或者其它药物组合物的“缓慢释放”),在所述药学可接受的制剂被施用到所述受试者后,维持至少一周、两周、三周、四周或更长时间段。
如本文使用的,所述术语“持久的递送”是指本发明的药物制剂在施用以后,在体内,在一段时间内持续递送,优选至少几天、一周或数周。所述组合物的持久递送可以被证明,例如所述酶在一段时间内的持久治疗效果(例如所述酶的持久递送可以通过在所述受试者中贮藏粒量的持续减少所证明)。可选择地,所述酶的持久递送可以被证明:通过检测在一定时间内,在体内存在的所述酶。
递送至靶组织
如上文所讨论的,本发明的一种令人惊讶的和重要的特征是:所述治疗试剂,特别的,使用本发明施用的替代酶能有效地、广泛地分散在所述脑表面,并渗透多层脑区域(包括深部脑区域)。另外,本发明的方法和组合有效地递送治疗试剂(例如,I2S酶)至脊髓的多种组织、神经元或细胞,包括腰区,所述腰区难以通过存在的CNS递送方法(例如ICV注射)靶向定位。而且,本发明的发明方法和组合物递送充分的量的治疗试剂(例如,I2S酶)至血流、多种外周器官和组织中。
因此,在一些实施方案中,治疗性蛋白质(例如,I2S酶)被递送至受试者的中枢神经系统。在一些实施方案中,治疗性蛋白质(例如,I2S酶)被递送至脑、脊髓、和/或外周器官的靶组织。如本文使用的,所述术语“靶组织”是指受到待治疗的溶酶体贮积症的影响的任意组织,或者所述缺乏的溶酶体酶正常表达的任意组织。在一些实施方案中,靶组织包括这些组织:在所述组织中具有可检测的或者异常高的量的酶底物,例如储存在所述组织的细胞溶酶体中(在患有或已患有溶酶体贮积症的患者中)。在一些实施方案中,靶组织包括表现具有疾病相关病理、症状或特征的这些组织。在一些实施方案中,靶组织包括这些组织:在所述组织中所述缺乏的溶酶体酶以升高的水平正常表达。如本文使用的,靶组织可以是脑靶组织、脊髓靶组织和/或外周靶组织。示例性的靶组织在下文详细描述。
脑靶组织
一般而言,所述脑可以被分成不同的区域、层和组织。例如,脑膜组织是覆盖所述中枢神经系统(包括所述脑)的膜系统。所述脑膜含有三层,包括硬脑膜、蛛网膜和软脑膜。一般而言,所述脑膜和脑脊髓液的主要功能是保护所述中枢神经系统。在一些实施方案中,根据本发明的治疗性蛋白质被递送至一层或多层所述脑膜。
所述脑具有三个主要的细分区,包括大脑、小脑和脑干。所述大脑半球位于大部分其它脑结构上方,并且被皮层覆盖。在所述大脑下面是脑干,所述脑干看起来像连接在所述大脑上的柄。在所述脑的后部,所述大脑以下和所述脑干后面,是小脑。
间脑(其位于所述脑中线附近,并且在所述中脑上方)含有丘脑、后丘脑、下丘脑、上丘脑、腹侧丘脑和前顶盖。中脑(也称为中脑)含有顶盖、大脑脚盖、脑室中脑水管、大脑脚、红核、和颅神经III核。中脑与视觉、听觉、运动控制、睡眠/唤醒、警觉和温度调节相关。
中枢神经系统组织的区域(包括脑)的特征,可以基于所述组织的深度划分。例如,CNS(例如脑)组织的特征可以描述成表面或者浅部组织、中层深度组织和/或深部组织。
根据本发明,治疗性蛋白质(例如替代酶)可以递送到与在受试者中待治疗的特定疾病相关的任意合适的脑靶组织。在一些实施方案中,根据本发明的治疗性蛋白质(例如替代酶)被递送至表面或浅部的脑靶组织。在一些实施方案中,根据本发明的治疗性蛋白质被递送至中层深度脑靶组织。在一些实施方案中,根据本发明的治疗性蛋白质被递送至深部脑靶组织。在一些实施方案中,根据本发明的治疗性蛋白质被递送至表面或浅部脑靶组织、中层深度脑靶组织、和/或深部脑靶组织的组合。在一些实施方案中,根据本发明的治疗性蛋白质被递送至深部脑组织,其在脑的外部表面以下(或内部)至少4mm、5mm、6mm、7mm、8mm、9mm、10mm或更多。
在一些实施方案中,治疗试剂(例如酶)被递送至大脑的一个或多个表面或者浅部组织。在一些实施方案中,所述靶向的大脑表面或者浅部组织位于距所述大脑表面4mm以内。在一些实施方案中,所述靶向的大脑表面或浅部组织选自:软脑膜组织、大脑皮质带状组织、海马、Virchow Robin空间、VR空间内的血管、海马、在脑下表面上的部分下丘脑、视神经和神经束、嗅球和突起物、以及其组合。
在一些实施方案中,治疗试剂(例如酶)被递送至大脑的一个或多个深部组织。在一些实施方案中,所述靶向的大脑表面或浅部组织位于在所述脑表面以下(或内部)4mm(例如5mm、6mm、7mm、8mm、9mm、或者10mm)。在一些实施方案中,大脑的靶向深部组织包括大脑皮质丝带。在一些实施方案中,大脑的靶向深部组织包括间脑(例如下丘脑、丘脑、腹侧丘脑、底丘脑等)、后脑、豆状核、基底神经节、尾状核、豆状核、杏仁核、苍白球及其组合的一种或多种组织。
在一些实施方案中,治疗试剂(例如酶)被递送至一种或多种的小脑组织。在某些实施方案中,所述靶向的一种多种小脑组织选自:分子层组织、浦肯野(氏)细胞层组织、颗粒细胞层组织、小脑脚、及其组合。在一些实施方案中,治疗试剂(例如酶)被递送至一种或多种小脑组织,包括但不限于浦肯野(氏)细胞层组织、颗粒细胞层组织、小脑深部白质组织(例如相对于颗粒细胞层的深部)、和小脑深部核组织。
在一些实施方案中,治疗试剂(例如酶)被递送至一种或多种脑干组织。在一些实施方案中,所述靶向的一种或多种脑干组织包括脑干白质组织和/或脑干核组织。
在一些实施方案中,治疗试剂(例如酶)被递送至多种脑组织,包括但不限于灰质、白质、脑室周围区域、软膜蛛网膜、脑膜、新皮质、小脑、大脑皮层中的深部组织、分子层、尾状核/豆状核区域、中脑、脑桥或者延髓的深部区域及其组合。
在一些实施方案中,治疗试剂(例如酶)被递送至在脑中的多种细胞,包括但不限于神经元、胶质细胞、血管周细胞和/或脑膜细胞。在一些实施方案中,治疗性蛋白质被递送至深部白质的少突胶质细胞。
脊髓
一般而言,脊髓的区域或者组织可以根据组织深度的特点区分。例如,脊髓组织可以区分为表面或者浅部组织、中层深度组织、和/或深部组织。
在一些实施方案中,治疗试剂(例如酶)被递送至脊髓的一种多种表面或者浅部组织。在一些实施方案中,靶向的脊髓的表面或浅部组织位于距离所述脊髓表面4mm以内。在一些实施方案中,靶向的脊髓的表面或浅部组织含有软脑膜和/或束状的白质。
在一些实施方案中,治疗试剂(例如酶)被递送至所述脊髓的一种或多种组织。在一些实施方案中,脊髓的靶向深部组织位于距所述脊髓表面内部4mm。在一些实施方案中,脊髓的靶向深部组织含有脊髓灰质和/或室管膜细胞。
在一些实施方案中,治疗试剂(例如酶)被递送至脊髓的神经元。
外周靶组织
如本文使用的,外周器官或组织是指那些不是所述中枢神经系统(CNS)组成部分的任意器官或组织。外周靶组织可以包括,但不限于血液系统、肝脏、肾脏、心脏、内皮、骨髓和骨髓来源的细胞、脾脏、肺脏、淋巴结、骨、软骨、卵巢和睾丸。在一些实施方案中,根据本发明的治疗性蛋白质(例如替代酶)被递送至一种或多种外周靶组织。
生物分布和生物药效率
在多种实施方案中,一旦递送至所述靶组织,治疗试剂(例如I2S酶)被定位到细胞内。例如,治疗试剂(例如酶)可以定位至靶细胞(例如神经元,例如浦肯野细胞)的外显子、轴突、溶酶体、线粒体或者液泡。例如,在一些实施方案中,鞘内施用的酶展示易位动态,使得所述酶在所述血管周空间以内移动(例如通过脉动辅助对流机制)。此外,与将所述施用蛋白质或者酶与神经丝结合有关的活性轴突运输机制可能有助于或者促进鞘内施用蛋白质或者酶分布进入所述中枢神经系统的深部组织。
在一些实施方案中,根据本发明递送的治疗试剂(例如I2S酶)可在多种本文描述的靶组织中,实现治疗(或临床)的效果水平(或活性)。如本文使用的,治疗(或临床)的效果水平(或活性)是这样的水平,其能在靶组织中提供足够的治疗效果。所述治疗效果可以是客观的(即通过某种测试或标记物进行可以测量的)或者主观的(即受试者给出的指示或者感觉的效果)。例如,治疗(或临床)的效果水平(或活性)可以是这样的酶水平或者活性:其可以足够地减缓与在所述靶组织中的疾病相关的症状(例如GAG储存)。
在一些实施方案中,根据本发明递送的治疗试剂(例如替代酶)可以实现的酶水平或者活性是:相对于在靶组织中的溶酶体酶的正常水平或者活性的至少5%、10%、20%、30%、40%、50%、60%、70%、80%、90%、95%。在一些实施方案中,根据本发明递送的治疗试剂(例如替代酶)可以实现的酶水平或者活性是:相对于对照(例如没有进行所述治疗时的内源水平或者活性),以至少1-倍、2-倍、3-倍、4-倍、5-倍、6-倍、7-倍、8-倍、9-倍或者10-倍地增加。在一些实施方案中,根据本发明递送的治疗试剂(例如替代酶)可以达到增加的酶水平或者活性是:在靶组织中,至少约10nmol/hr/mg、20nmol/hr/mg、40nmol/hr/mg、50nmol/hr/mg、60nmol/hr/mg、70nmol/hr/mg、80nmol/hr/mg、90nmol/hr/mg、100nmol/hr/mg、150nmol/hr/mg、200nmol/hr/mg、250nmol/hr/mg、300nmol/hr/mg、350nmol/hr/mg、400nmol/hr/mg、450nmol/hr/mg、500nmol/hr/mg、550nmol/hr/mg或者600nmol/hr/mg。
在一些实施方案中,根据本发明发明的方法对于靶向腰区特别有用。在一些实施方案中,根据本发明递送的治疗试剂(例如替代酶)可以达到的增加的酶水平或者活性是:在所述腰区中,至少约500nmol/hr/mg、600nmol/hr/mg、700nmol/hr/mg、800nmol/hr/mg、900nmol/hr/mg、1000nmol/hr/mg、1500nmol/hr/mg、2000nmol/hr/mg、3000nmol/hr/mg、4000nmol/hr/mg、5000nmol/hr/mg、6000nmol/hr/mg、7000nmol/hr/mg、8000nmol/hr/mg、9000nmol/hr/mg或者10,000nmol/hr/mg。
一般而言,根据本发明递送的治疗试剂(例如替代酶)在CSF以及脑、脊髓和外周器官的靶组织中具有充分长的半衰期。在一些实施方案中,根据本发明递送的治疗试剂(例如替代酶)可以具有的半衰期:至少约30分钟、45分钟、60分钟、90分钟、2小时、3小时、4小时、5小时、6小时、7小时、8小时、9小时、10小时、12小时、16小时、18小时、20小时、25小时、30小时、35小时、40小时、多达3天、多达7天、多达14天、多达21天或者多达一月。在一些实施方案中,在一些实施方案中,根据本发明递送的治疗试剂(例如替代酶)可以在CSF或者血流中持有可检测的水平或者活性,在施用的12小时、24小时、30小时、36小时、42小时、48小时、54小时、60小时、66小时、72小时、78小时、84小时、90小时、96小时、102小时、或一周后。可检测的水平或者活性可以使用本领域已知的方法测定。
在某些实施方案中,根据本发明递送的治疗试剂(例如替代酶)达到的浓度是:在施用后(在鞘内施用所述药物组合物至所述受试者后的例如1周、3天、48小时、36小时、24小时、18小时、12小时、8小时、6小时、4小时、3小时、2小时、1小时、30分钟或更少)的所述受试者的所述CNS组织和细胞中,至少30μg/ml。在某些实施方案中,根据本发明递送的治疗试剂(例如替代酶)在施用至受试者(例如在鞘内施用这种组合物至所述受试者1周、3天、48小时、36小时、24小时、18小时、12小时、8小时、6小时、4小时、3小时、2小时、1小时、30分钟或更少时间之后)后,在所述受试者的靶组织或细胞中(例如脑组织或神经元),达到的浓度为:至少20μg/ml、至少15μg/ml、至少10μg/ml、至少7.5g/ml、至少5μg/ml、至少2.5μg/ml、至少1.0μg/ml或者至少0.5μg/ml。
治疗Hunter综合症和其它溶酶体贮积症
所述溶酶体贮积症表示导致溶酶体功能缺陷的一组相对稀少的遗传代谢疾病。所述溶酶体疾病的特征在于在溶酶体内部(参见表1)未消化大分子的累积,包括这些酶底物,这导致这种溶酶体体积和数量的增加,并最终导致细胞机能障碍和临床异常。
本文描述的发明方法能有利地促进一种或多种治疗试剂(一种或多种替代酶)递送至靶细胞器。例如,因为溶酶体贮积症(例如亨特综合症)的特征在于粘多糖(GAG)在受影响细胞的溶酶体中的累积,所述溶酶体表示所述治疗的溶酶体贮积症的预期靶细胞。
本发明的发明方法和组合物对于具有CNS病因或组件的这些疾病的治疗特别有用。具有CNS病因或组件的溶酶体贮积症,包括例如不限于:Sanfilippo综合症类型A、Sanfilippo综合症类型B、亨特综合症、异染性脑白质营养不良和球细胞脑白质营养不良。在本发明之前,传统治疗限于:将其静脉施用至受试者,并且通常仅对治疗潜在的酶缺乏的躯体症状有效。本发明的这些组合物和方法可以有利地直接施用至患有具有CNS病因的疾病的受试者的CNS,从而在所述CNS的受影响细胞和组织(例如脑)中达到治疗浓度,从而克服与传统全身施用这种治疗试剂相关的限制。
在一些实施方案中,本发明的发明方法和组合物对于治疗溶酶体贮积症的(神经病学的和身体的)后遗症或症状有用。例如,本发明的一些实施方案,关于递送(例如鞘内地、脑室内地或者脑池内地)一种或多种治疗试剂至受试者的CNS的组合物和方法,为了治疗所述CNS或者溶酶体贮积症的或者神经病学后遗症和表现,同时也治疗所述溶酶体贮积症的系统的或身体的表现。例如,本发明的一些组合物可以鞘内施用至受试者,从而递送一种或多种治疗试剂至所述受试者的所述CNS,并且治疗所述神经病学后遗症;联合静脉施用一种或多种治疗试剂以递送这种治疗试剂至体循环(例如心脏、肺脏、肝脏、肾脏或者淋巴结的细胞和组织)的细胞和组织(两者都包括),从而治疗所述身体的后遗症。例如,具有或者其它受到溶酶体贮积症(例如亨特综合症)影响的受试者,可以被鞘内施用药物组合物,所述药物组合物包括一种或多种治疗试剂(例如艾杜糖醛酸-2-硫酸酯酶),至少每周、两周,每月、两月或更多时间一次,以治疗所述神经病学的后遗症;同时不同的治疗试剂被静脉施用至所述受试者,更频繁地(例如,每天一次,隔日一次,一周或每周三次),以治疗所述疾病系统的或者身体的表现。
Hunter综合症,或者黏多糖贮积症II(MPS II),是一种由艾杜糖醛酸-2-硫酸酯酶(I2S)缺陷导致的伴X染色体遗传性疾病。I2S定位于溶酶体,并且在粘多糖(GAG)乙酰肝素-以及皮肤素-硫酸盐的代谢中具有重要的作用。在缺乏这种酶的情况下,这些底物累积在细胞内部,最终导致肿胀,然后细胞死亡和组织被破坏。由于酶的广泛表达,在MPS II患者中的多种细胞类型和器官系统受到影响。
这种疾病确定的临床特征是中枢神经系统(CNS)退化,其导致认知障碍(例如,IQ降低)。此外,受到影响个体的MRI扫描显示在所述脑实质中扩大的血管周围间隙、神经节、胼胝体和脑干中的白质病变;萎缩症;和巨脑室(Wang et al.Molecular Genetics and Metabolism,2009)。所述疾病通常在生命第一年表现,并伴有器官巨大症和骨骼异常。一些受影响个体经历了认知功能的进行性丧失,并且大多数受影响个体在他们的第一个或第二个十年死于疾病相关的并发症。
使用本发明组合物和方法可以有效治疗患有或已患有Hunter综合症的个体。所述术语,“处理”或者“治疗”如本文所使用的,是指所述疾病并发的一种或多种症状的转佳,预防或者延缓所述疾病一种或多种症状的发病,和/或减轻所述疾病一种或多种症状的严重程度或者频率。
在一些实施方案中,治疗是指:在Hunter综合症患者中,使神经学损伤的严重程度和/或发生率部分地或者完全地缓解、转佳、减轻、抑制、延缓发生、降低。如本文所使用的,所述术语“神经学损伤”包括:与中枢神经系统(例如脑和脊髓)损伤并发的多种症状。神经学损伤的症状包括,例如,认知障碍;白质病变;在所述脑实质、神经节、胼胝体和/或脑干中扩大的血管周围间隙;萎缩症;和/或巨脑室等等。
在一些实施方案中,治疗是指在多种组织中,溶酶体贮积(例如GAG)的减少。在一些实施方案中,治疗是指在脑靶组织、脊髓神经元、和/或外周靶组织中,溶酶体贮积的减少。在某些实施方案中,溶酶体贮积相对于对照,以大约5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%、95%、100%或更多地减少。在一些实施方案中,溶酶体贮积相对于对照,以至少1-倍、2-倍、3-倍、4-倍、5-倍、6-倍、7-倍、8-倍、9-倍或10-倍地减少。在一些实施方案中,溶酶体储存以溶酶体贮藏粒(例如斑马-条纹形态)的存在进行量度。溶酶体贮藏粒的存在可以通过现有技术的多种方式(例如组织学分析)度量。
在一些实施方案中,治疗是在神经元(例如含有浦肯野细胞的神经元)中液泡化的减少。在某些实施方案中,在神经元中的液泡化相对于对照,以大约5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%、95%、100%或更多地减少。在一些实施方案中,液泡化相对于对照,以至少1-倍、2-倍、3-倍、4-倍、5-倍、6-倍、7-倍、8-倍、9-倍或者10-倍地减少。液泡化的存在和减少可以通过现有技术的多种方式(例如组织学分析)度量。
在一些实施方案中,治疗是指在多种组织中I2S酶活性增加。在一些实施方案中,治疗是指在脑靶组织、脊髓神经元和/或外周靶组织中I2S酶活性增加。在一些实施方案中,I2S酶活性与对照相比,以约5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%、95%、100%、200%、300%、400%、500%、600%、700%、800%、900%、1000%或者更多地增加。在一些实施方案中,I2S酶活性与对照相比,以至少1-倍、2-倍、3-倍、4-倍、5-倍、6-倍、7-倍、8-倍、9-倍或者10-倍地增加。在一些实施方案中,增加的I2S酶活性为至少大约10nmol/hr/mg,20nmol/hr/mg,40nmol/hr/mg,50nmol/hr/mg,60nmol/hr/mg,70nmol/hr/mg,80nmol/hr/mg,90nmol/hr/mg,100nmol/hr/mg,150nmol/hr/mg,200nmol/hr/mg,250nmol/hr/mg,300nmol/hr/mg,350nmol/hr/mg,400nmol/hr/mg,450nmol/hr/mg,500nmol/hr/mg,550nmol/hr/mg,600nmol/hr/mg或者更多。在一些实施方案中,I2S酶活性在所述腰椎区域中或者在所述腰椎区域细胞中增加。在一些实施方案中,在所述腰椎区域中增加的I2S酶活性为至少大约2000nmol/hr/mg,3000nmol/hr/mg,4000nmol/hr/mg,5000nmol/hr/mg,6000nmol/hr/mg,7000nmol/hr/mg,8000nmol/hr/mg,9000nmol/hr/mg,10,000nmol/hr/mg或者更多。在一些实施方案中,I2S酶活性在所述远端脊髓或者远端脊髓细胞中增加。
在一些实施方案中,治疗是指认知能力丧失的进展减少。在某些实施方案中,认知能力丧失的进展相对于对照,以大约5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%、95%、100%或者更多地减少。在一些实施方案中,治疗是指发展迟缓的减少。在某些实施方案中,发育迟缓相对于对照,以大约5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%、95%、100%或者更多地减少。
在一些实施方案中,治疗涉及增加的存活率(例如存活时间)。例如,治疗能导致患者预期寿命的增加。在一些实施方案中,根据本发明的治疗导致患者预期寿命增加,相对于患有类似疾病而没有治疗的一个或多个个体对照的平均预期寿命,以多于大约5%、大约10%、大约15%、大约20%、大约25%、大约30%、大约35%、大约40%、大约45%、大约50%、大约55%、大约60%、大约65%、大约70%、大约75%、大约80%、大约85%、大约90%、大约95%、大约100%、大约105%、大约110%、大约115%、大约120%、大约125%、大约130%、大约135%、大约140%、大约145%、大约150%、大约155%、大约160%、大约165%、大约170%、大约175%、大约180%、大约185%、大约190%、大约195%、大约200%或更多地增加。在一些实施方案中,根据本发明的治疗导致患者预期寿命增加,相对于患有类似疾病而没有治疗的一个或多个个体对照的平均预期寿命,以多于大约6月、大约7月、大约8月、大约9月、大约10月、大约11月、大约12月、大约2月、大约3月、大约4月、大约5月、大约6月、大约7月、大约8月、大约9月、大约10年或者更多地增加。在一些实施方案中,根据本发明的治疗导致患者的长期存活。如本文使用的,所述术语“长期存活”是指存活时间或者预期寿命长于大约40年、45年、50年、55年、60年、或更长时间。
所述术语“改善”、“增加”或“减少”如本文使用的,表示指示相对于对照的值。在一些实施方案中,合适的对照是基线测量,例如在启动本文描述的治疗之前的相同个体内的测量,或者在缺乏本文描述的治疗的对照个体(或多个对照个体)内的测量。“对照个体”是受Hunter综合症折磨的个体,并且其处于与正被治疗的个体相同的年龄和/或者性别(以确保被治疗的所述疾病的阶段和所述对照个体具有可比性)。
正被治疗的所述个体(也称为“患者”或“受试者”)是这样的个体(胎儿、婴儿、儿童、青少年或成年人类):其患有Hunter综合症或者具有发展为Hunter综合症的潜力。所述个体具有残留的内源性I2S表达和/或活性、或者不可测量的活性。例如,患有Hunter综合症的所述个体可以具有I2S表达水平,比正常I2S表达水平,低大约30-50%、低大约25-30%、低大约20-25%、低大约15-20%、低大约10-15%、低大约5-10%、低大约0.1-5%。
在一些实施方案中,所述个体是最近被诊断患有所述疾病的个体。通常地,早期治疗(在诊断后尽可能早地开始治疗)对于使疾病的影响最小化以及使治疗的益处最大化是重要的。
免疫耐受性
通常,鞘内施用根据本发明的治疗试剂(例如替代酶)在所述受试者中不导致严重的不良反应。如本文使用的,严重的不良反应包括但不限于,基本免疫应答、毒性或者死亡。如本文使用的,所述术语“基本免疫应答”是指严重的免疫应答,例如适应性T-细胞免疫应答。
因此,在许多实施方案中,根据本发明发明的方法不涉及并发免疫抑制疗法(例如在与所述方法平行进行的作为预处理/预调节使用的许多免疫抑制剂治疗)。在一些实施方案中,根据本发明发明的方法不包括在所述被治疗的受试者中的免疫耐受诱导。在一些实施方案中,根据本发明发明的方法不涉及使用T-细胞免疫抑制剂进行的预处理或预调节。
在一些实施方案中,治疗试剂的鞘内施用可以发动针对这些试剂的免疫应答。因此,在一些实施方案中,它对于使得接受所述替代酶的所述受试者容忍所述酶替代疗法有用。免疫耐受性可以使用多种现有技术的方法诱导。例如,可以使用的方法:最初30-60天方案的T-细胞免疫抑制剂,例如环孢霉素A(CsA)和抗增殖剂(例如硫唑嘌呤(Aza)),并结合每周鞘内注射低剂量的预期替代酶。
对本领域技术人员已知的任何免疫抑制剂疗法,可以与本发明的组合疗法一起采用。这种免疫抑制剂包括但不限于:环孢霉素、FK506、纳巴霉素、CTLA4-Ig、和抗TNF试剂(例如依那西普)(参见例如Moder,2000,Ann.Allergy Asthma Immunol.84,280-284;Nevins,2000,Curr.Opin.Pediatr.12,146-150;Kurlberg et al.,2000,Scand.J.Immunol.51,224-230;Ideguchi etal.,2000,Neuroscience95,217-226;Potteret al.,1999,Ann.N.Y.Acad.Sci.875,159-174;Slavik et al.,1999,Immunol.Res.19,1-24;Gaziev et al.,1999,Bone Marrow Transplant.25,689-696;Henry,1999,Clin.Transplant.13,209-220;Gummert et al.,1999,J.Am.Soc.Nephrol.10,1366-1380;Qi et al.,2000,Transplantation69,1275-1283)。所述抗IL2受体(.α.-亚单位)抗体赛尼哌(例如Zenapax.TM.),其被表明在移植患者中有效,也能作为免疫抑制剂使用(参见例如Wiseman et al.,1999,Drugs58,1029-1042;Beniaminovitz et al.,2000,N.Engl J.Med.342,613-619;Ponticelli et al.,1999,Drugs R.D.1,55-60;Berard et al.,1999,Pharmacotherapy 19,1127-1137;Eckhoff et al.,2000,Transplantation69,1867-1872;Ekberg et al.,2000,Transpl.Int.13,151-159)。其它免疫抑制剂包括但不限于:抗-CD2(Branco et al.,1999,Transplantation68,1588-1596;Przepiorka et al.,1998,Blood92,4066-4071),抗-CD4(Marinova-Mutafchieva et al.,2000,ArthritisRheum.43,638-644;Fishwild et al.,1999,Clin.Immunol.92,138-152),和抗-CD40配体(Hong et al.,2000,Semin.Nephrol.20,108-125;Chirmule et al.,2000,J.Virol.74,3345-3352;Ito et al.,2000,J.Immunol.164,1230-1235)。
施用
本发明的方法考虑到:单一以及多个治疗有效量的本文所描述的所述治疗试剂(例如替代酶)的施用。治疗试剂(例如替代酶)可以根据所述受试者的病情(例如溶酶体贮积症)的特性、严重程度和范围,以有规律的间隔施用。在一些实施方案中,治疗有效量的本发明治疗试剂(例如替代酶)可以周期性地鞘内施用,以有规律的间隔(例如一年一次、6个月一次、每五个月一次、每三个月一次、两月一次(每两个月一次)、每月一次(每月一次)、两周一次(每两周一次)、每周一次、每天或者持续地)。
在一些实施方案中,鞘内施用可以连同其它施用途径使用(例如静脉内的、皮下地、肌肉间的、肠胃外地、经皮肤地、或者经黏膜地(或者口服地或经鼻地))。在一些实施方案中,这些施用途径(例如静脉施用)执行的频率不多于两周一次、每月一次、每两个月一次、每三个月一次、每四个月一次、每五个月一次、每六个月一次、每年一次施用。在一些实施方案中,所述方法进一步包括静脉施用所述I2S替代酶至所述受试者。在某些实施方案中,所述静脉施用没有比每星期一次施用更加频繁(例如,没有比每两星期一次、每月一次、每两个月一次、每隔三个月一次、每隔四个月一次、每隔五个月一次或者每隔六个月一次更加频繁)。在某些实施方案中,所述静脉施用比每月一次施用更频繁,例如每周两次、每周一次、每隔一周一次或者每月两次。在一些实施方案中,静脉和鞘内施用在同一天进行。在一些实施方案中,所述静脉和鞘内施用不在彼此的一定时间内执行,例如不执行:在至少2天以内,在至少3天以内,在至少4天以内,在至少5天以内,在至少6天以内,在至少7天以内,或者在至少一周以内。在一些实施方案中,静脉和鞘内施用按照交替的时间表执行,例如每周一次、每隔一周、每月两次或者每月一次交替施用。在一些实施方案中,鞘内施用替代在施用时间表上的静脉施用,例如在每周一次、每隔一周、每月两次或者每月一次的静脉施用时间表上,在该时间表中的每第三次或第四次或第五次施用可以用鞘内施用代替静脉施用。在一些实施方案中,静脉施用替代施用时间表上的鞘内施用,例如在每周一次、每隔一周、每月两次或者每月一次鞘内施用的时间表中,在该时间表中的每第三次或第四次或第五次施用可以用静脉施用代替鞘内施用。在一些实施方案中,静脉和鞘内施用是连续地执行的,例如首先执行静脉施用(例如,每周一次、每隔一周、每月两次或者每月一次给药为期两周、一个月、二个月、三个月、四个月、五个月、六个月或者一年或者更久),然后鞘内施用(例如,每周一次、每隔一周、每月两次或者每月一次,给药为期多于两周、一个月、二个月、三个月、四个月、五个月、六个月或者一年或者更久)。在一些实施方案中,首先执行鞘内施用(例如,每周一次、每隔一周、每月两次、每月一次、每两个月一次、每隔三个月一次给药为期两周、一个月、二个月、三个月、四个月、五个月、六个月或者一年或者更久),然后静脉施用(例如每周一次、每隔一周、每月两次或者每月一次,给药为期多于两周、一个月、二个月、三个月、四个月、五个月、六个月或者一年或者更久)。
在一些实施方案中,Hunter综合症与周边症状并发,并且所述邞包括鞘内施用所述替代酶,但是不涉及静脉施用至所述受试者。在某些实施方案中,所述I2S酶的鞘内施用使与所述受试者I2S缺乏症相关的一种或多种周边症状减缓或者减少。
如本文使用的,所述术语“治疗有效量”很大程度上,是基于在所述本发明药物组合物中的治疗试剂的总量决定的。通常的,治疗有效量对于所述受试者足够能实现有意义的好处(例如治疗、调整、治愈、阻止和/或改善所述潜在的疾病或病情)。例如,治疗有效量可以是足够实现预期治疗和/或预防性效果的量,并且这种量足够调节溶酶体受体或者他们的活性,从而治疗这种溶酶体贮积症或者其症状(例如在施用本发明组合物至受试者后,使“斑马小体”或者细胞液泡化的存在或者发生率减少或者消除。通常,治疗试剂(例如溶酶体酶)的量(施用至需要其的受试者)将根据所述受试者的特性变化。这种特性包括:所述受试者的病情、疾病严重程度、健康状况、年龄、性别和体重。一个本领域技术人员将根据这些和其它相关因素,容易地决定合适的剂量。另外,客观的和主观的分析都可以任选地采用,以识别最优剂量范围。
治疗有效量通常施用的剂量方案,包括多个单位剂量。对特定的治疗性蛋白质,治疗有效量(和/或在有效给药方案内的合适的单位剂量)可能会变化,例如根据结合其它药物试剂的施用途径。而且,对于特定患者的特定治疗有效量(和/或单位剂量)可以根据多种因素改变,包括正被治疗的疾病和所述疾病的严重程度、所采用的特定药物试剂的活性;所采用的特定组合物;所述受试者的年龄、体重、健康状况、性别和饮食;所采用的特定融合蛋白的施用时间、施用途径和/或排泄率或代谢率;所述治疗的持续时间;以及在医药领域已知的类似因素。
在一些实施方案中,所述治疗有效剂量的范围:从大约0.005mg/kg脑重量至500mg/kg脑重量,例如,从大约0.005mg/kg脑重量至400mg/kg脑重量,从大约0.005mg/kg脑重量至300mg/kg脑重量,从大约0.005mg/kg脑重量至200mg/kg脑重量,从大约0.005mg/kg脑重量至100mg/kg脑重量,从大约0.005mg/kg脑重量至90mg/kg脑重量,从大约0.005mg/kg脑重量至80mg/kg脑重量,从大约0.005mg/kg脑重量至70mg/kg脑重量,从大约0.005mg/kg脑重量至60mg/kg脑重量,从大约0.005mg/kg脑重量至50mg/kg脑重量,从大约0.005mg/kg脑重量至40mg/kg脑重量,从大约0.005mg/kg脑重量至30mg/kg脑重量,从大约0.005mg/kg脑重量至25mg/kg脑重量,从大约0.005mg/kg脑重量至20mg/kg脑重量,从大约0.005mg/kg脑重量至15mg/kg脑重量,从大约0.005mg/kg脑重量至10mg/kg脑重量。
在一些实施方案中,所述治疗有效剂量是:大于约0.1mg/kg脑重量,大于约0.5mg/kg脑重量,大于约1.0mg/kg脑重量,大于约3mg/kg脑重量,大于约5mg/kg脑重量,大于约10mg/kg脑重量,大于约15mg/kg脑重量,大于约20mg/kg脑重量,大于约30mg/kg脑重量,大于约40mg/kg脑重量,大于约50mg/kg脑重量,大于约60mg/kg脑重量,大于约70mg/kg脑重量,大于约80mg/kg脑重量,大于约90mg/kg脑重量,大于约100mg/kg脑重量,大于约150mg/kg脑重量,大于约200mg/kg脑重量,大于约250mg/kg脑重量,大于约300mg/kg脑重量,大于约350mg/kg脑重量,大于约400mg/kg脑重量,大于约450mg/kg脑重量,大于约500mg/kg脑重量。
在一些实施方案中,所述治疗有效剂量也可以通过mg/kg体重定义。作为本领域技术人员,将理解:所述脑重量和体重是相关的。DekabanAS.“Changes in brain weights during the span of human life:relation of brainweights to body heights and body weights,”Ann Neurol1978;4:345-56。因此,在一些实施方案中,所述剂量可以根据在表5中所示的进行转化。
表5.

在一些实施方案中,所述治疗有效剂量可以通过mg/15cc的CSF定义。作为本领域技术人员,将理解:根据脑重量和体重,将治疗有效量转化成mg/15cc的CSF。例如,在成年人类中的CSF的体积为约150mL(Johanson CE,et al.“Multiplicity of cerebrospinal fluid functions:Newchallenges in health and disease,”cerebrospinal fluid Res.2008May14;5:10).。因此,单剂量注射0.1mg至50mg蛋白质至成年人,在成年人中将会是约0.01mg/15cc的CSF(0.1mg)至5.0mg/15cc的CSF(50mg)剂量。
进一步可以理解,对于任意特定的受试者,随着时间的过去,根据所述个体需要和施用并监督(所述酶替代治疗的施用)的人员的专业判断,应该调整特定剂量方案,并且本文提出的给药范围仅仅是示例性的,并不打算限制本发明要求的范围或实践。
试剂盒
本发明进一步提供了试剂盒或者含有本发明所述制剂的其它制品,并提供了其再生(如果冻干)和/或使用的指导。试剂盒或其它制造业制品可以包括容器、IDDD、导管和其它对于鞘内施用和相关手术有用的制品、设备或装置。合适的容器包括,例如瓶子、小药瓶、注射器(例如预填充的注射器)、安瓿、药筒、贮液囊、或(装有冻干粉和稀释剂的)二室注射器给药系统(lyo-ject)。所述容器可以用多种材料(例如玻璃或者塑料)形成。在一些实施方案中,容器是预填充的注射器。合适的预填充的注射器包括,但不限于,具有烘干的硅树脂涂层的硼硅酸盐玻璃注射器、喷涂硅树脂的硼硅酸盐、或者无硅树脂的塑料树脂注射器。
通常地,所述容器可以容纳制剂,并在其上具有标签,或者与之相关地,所述容器可以指示再生和/或施用的指导。例如,所述标签可以指示:再生所述制剂至上文所描述的蛋白质浓度。所述标签可以进一步指示:所述制剂是有用的,或者打算用于(例如)IT施用。在一些实施方案中,容器可以含有单剂量的稳定制剂,所述制剂含有治疗试剂(例如替代酶)。在多种实施方案中,单剂量的所述稳定的制剂以小于大约15ml、10ml、5.0ml、4.0ml、3.5ml、3.0ml、2.5ml、2.0ml、1.5ml、1.0ml、或者0.5ml的体积存在。可选择地,持有所述制剂的容器可以是多用途的小药瓶,其允许重复施用(例如2-6次施用)所述制剂。试剂盒或者制品可以进一步包括第二容器,所述第二容器包括合适的稀释剂(例如BWFI、生理盐水、缓冲的生理盐水)。在将所述稀释剂和所述制剂混合后,在所述再生的制剂中的所述最终蛋白质浓度通常可以是:至少1mg/ml(例如至少5mg/ml、至少10mg/ml、至少25mg/ml、至少50mg/ml、至少75mg/ml、至少100mg/ml)。试剂盒或者制品可以进一步包括其它(从商业和用户的角度看)理想的材料,其包括其它缓冲剂、稀释剂、过滤器、针头、IDDD、导管、注射器和具有使用指南的包装说明书。
通过参考以下的实施例,本发明将更充分地被理解。但是这些用于解释的实施例不限制本发明的范围。所有文献引用通过参考的方式并入本文。
实施例
实施例1:生物分布
本研究的主要目的是测定是否可以通过鞘内腰椎途径将重组人类的I2S递送到成年MPS II小鼠的大脑。
表6:6组8-12周龄的雄性小鼠按如下治疗:

注射时间表:通过鞘内腰椎的路线,动物接受3次艾杜硫酶(10L)注射:
○组A&D:在1,8,和15天,施用3剂量的I2S
○组B:在1,和8天,施用2剂量的I2S
○组C&E:未治疗的对照(IKO)小鼠
○组F:未治疗的野生型对照小鼠
材料和方法
动物:
每笼4组将小鼠圈养在集群屋中,12-小时的光暗周期。提供鼠类的饮食(LabDiet-5001,St Louis,MO)和水(Lexington,反渗透纯化的MA城市用水),用于实验持续期间的自由采食。按照实验动物护理(美国国家学术出版社,华盛顿特区,1996年)“指南”中描述的爱护和使用准则进行爱护动物。目前从4个载体雌性小鼠杂合IKO突变(获自Dr.JosephMuenzer(北卡罗菜纳大学))建立了IKO繁殖群。饲养载体雌性与C57BL/6背景株(C57BL/6NTac,Taconic,Hudson,NY)的雄性小鼠,生产杂合的雌性和半合子的雄性基因敲除小鼠,以及野生型雄性和雌性同窝小鼠。由PCR分析所有后代基因型的组织DNA。本实验中使用的所有小鼠确定为8和12周龄之间的雄性半合子IKO(-/0)或野生型(WT)的同窝小鼠(+/0)。
艾杜硫酶:
为了更换2L的磷酸盐缓冲生理盐水(PBS),透析22mL I2S[重组人艾杜硫酶]。然后用Vivaspin色谱柱浓缩I2S,而且在悬浮的最终体积为1mL的PBS中,然后通过使用0.2μm的过滤器过滤灭菌。最终浓度为51mg/mL。
鞘内的-腰椎注射:
通过腹膜内注射,使用1.25%2,2,2三溴乙醇(Avertin)(200-300μL/10克体重(250-350mg/kg)),麻醉成年小鼠。除去尾基和肩胛之间的背毛,而且在用异丙醇擦洗之后,用povidine/聚乙烯吡咯酮碘擦洗剃除区域。在腰骶部和背中线的交叉点制作小正中皮肤切口(1-2cm),并确定回肠(单一的回肠)翼部的颅方面。在髂窝内的肌肉(臀中肌)为心形的肌肉,而且“心”顶部两侧接近回肠翼部的位置。32号计量注射针连接到气密10-20μL玻璃Hamilton注射器并插入,直到从底层的骨感觉到阻力。在近似速率为2μL/20秒(10μL/2分钟)下进行注射10μL的测试制品。使用适当的创伤夹关闭皮肤切口,而且在返回到相应的笼中之前,动物允许在恢复室恢复。
组织学程序:
在最后一次注射后的1小时处死动物。
收集脑和肝组织并固定在10%中性缓冲福尔马林中,然后处理并在石蜡中包埋。准备5μm的切片用于苏木素/伊红(H&E)和免疫组织化学(IHC)染色。
苏木精和伊红染色:
用H&E染色脑和肝组织切片。染色结果显示细胞核为紫色而细胞质为粉红色到红色。H&E染色切片用于病理组织学形态评价。
免疫组织化学:
为了I2S生物分布评价,将脱蜡和再水化的脑和肝脏切片与抗重组人的I2S的小鼠单克隆抗体2C4-2B2(Maine Biotechnology Services,Portland,ME)孵育过夜,以检测注射的I2S(或不相关的小鼠IgG作为阴性对照抗体;Vector Laboratories,Burlingame,CA)。在2-8℃下过夜孵育后,加入与辣根过氧化物酶共轭的二次山羊抗小鼠IgG。在37℃下,经过额外的30分钟孵育,加入酪胺-Alexa Fluor488标记溶液(Invitrogen Corp.,Carlsbad,CA)溶液中进行额外的10分钟。使用含有1.5μg/ml4′-6-二脒基-2-苯基吲哚(DAPI)抗褪色安装的介质(VectaShield;Vector Laboratories)封片切片作为核染液,并用多通道Nikon荧光显微镜观察。染色结果显示I2S阳性细胞为绿色,细胞核为蓝色,及背景为黑色。
为了疗效分析,用鼠抗LAMP-1(溶酶体相关的膜蛋白作为溶酶体标记)IgG(Santa Cruz Biotechnology,Santa Cruz,California)染色脑、肝切片作为初级抗体。IgG作为不相关的抗体用作为阴性对照。ABC(亲和素生物素复合物试剂盒来自Vector Labs,Burlingame,California)方法用于扩增目标标记。
简而言之,再水化脱石蜡切片,并用初级抗体孵育。在2-8℃下过夜孵育之后,加入次级生物素化的兔抗鼠IgG(Vector Labs,Burlingame,California),并在37℃下孵育30分钟,然后洗涤样品并用抗生物素蛋白-生物素-过氧化物酶复合物(Vector Laboratories)处理30分钟。为了彩色显影,使用了3,3′-二氨基联苯胺四盐酸盐(DAB)作为发色团。然后用苏木精和封片复染切片。染色结果表明,LAMP-1阳性细胞为棕色而细胞核为蓝色。
拍摄所述代表照片以及用Image-Pro Plus软件(Media Cybernetics,Inc.,Bethesda,MD)分析LAMP-1阳性细胞面积的并使用检定t-检验进行比较性统计分析。
电子显微镜法:
来自3个剂量I2S治疗动物的脑组织被固定在2.5%PFA/2.5%戊二醛(在0.1M二甲胂酸钠缓冲液pH7.4)中,在4度下过夜。然后在二甲砷酸缓冲液(0.1M,pH7.4)中清洗样品,然后固定在四氧化锇中,在酒精和环氧丙烷中脱水,并嵌入在EPON树脂中。在100nm下切成超薄切片,用柠檬酸铅染色,在TECNAITMG2Spirit BioTWIN透射电子显微镜下检查。
结果
在由免疫组化法(IHC)所测定的大脑中,在媒介物对照动物中没有发现I2S。与此相反,在I2S注射的动物中,大脑和小脑的脑膜细胞,神经元对I2S为阳性染色。在施用3剂量的动物(图1)中,染色信号强烈。
与野生型动物相比,在媒介物-治疗的IKO小鼠的脑组织中,在整个大脑中发现细胞空泡化,溶酶体贮积症的组织病理学特点。与未经处理的(图2)小鼠相比,在I2S治疗的IKO小鼠中,从大脑皮层表面,尾状核,丘脑,小脑,到白质的细胞空泡化普遍降低。与野生型动物相比,在媒介物治疗的IKO小鼠中,由溶酶体相关膜蛋白1(LAMP-1)(溶酶体活性和疾病状态的指标)染色,在小胶质细胞,脑膜和血管周围的细胞中,发现异常高的溶酶体活性。I2S鞘内-治疗的小鼠的LAMP-1免疫组化显著减少。此减少的特征在于:LAMP-1阳性细胞数目降低而且染色更轻。在2和3剂量I2S治疗的动物中,从大脑皮质、尾状核、丘脑、小脑到白质((图3),在整个全脑表面发现减少。不同脑区的LAMP-1免疫组化形态计量学分析证实,在评估的大脑的各个区域,LAMP-1阳性(图4)显著减少。
电子显微镜检查媒介物治疗的IKO小鼠的脑细胞发现:扩大后的空泡含有无定形颗粒存储材料和薄片形的内容物和斑马体状结构。在腰椎I2S鞘内注射的小鼠(图5)中,在超微结构水平下,这些典型溶酶体贮积的病理特征减少。
在肝脏中,在媒介物治疗的动物中,没有阳性染色I2S。在I2S鞘内注射的小鼠中,在窦状腺细胞(图6)中清楚地发现注射的大量I2S,这表明注射的I2S与CSF散布在鞘内空间,然后通过蛛网膜颗粒吸收进入循环系统。
与WT小鼠相比,在媒介物治疗的IKO小鼠的肝组织中,由H&E染色表明细胞空泡化严重和异常高的溶酶体活性,以及发现强的LAMP-1免疫组化。在鞘内以I2S治疗后,在肝中发现细胞空泡化和LAMP-1免疫组化显著减少。H&E染色显示的胞浆内空泡形成几乎完全消失,接近正常肝脏细胞结构(图7)。
在IKO小鼠中,通过鞘内腰椎途径,重组人I2S传递到脑,而且在脑中的不同区域,注射的I2S引起广泛的病理组织学改善。
●脑的脑膜细胞和神经元中,注射的I2S是可检测的。
●在光学和电子显微镜水平下,整个脑细胞的空泡化降低。
●在整个脑中,LAMP-1溶酶体标记减少。
●鞘内注射的I2S进入外周循环,而且改善肝脏形态学和组织学标记。
实施例2:毒理学
本实施例说明了在食蟹猴内,通过每月弹丸鞘内腰椎给药艾杜硫酶的临床相关联的体征。为了实现这一目标,将14例雄性食蟹猴随机分配成5个治疗组,如在下面表所示。
表7:实验设计

所有组中的动物,以每月一次的时间间隔在腰椎水平上IT给药3次。从导管系统中用0.3ml的PBS冲洗1ml剂量体积。每个剂量前的1到2天,在小脑延髓池水平,从IT脊椎抽液收集约2ml的CSF。在这个时候,还收集血液样品(2ml)。从组5的预给药动物中,第一次剂量给药后0.5、1、2、4、8,24和48小时后,收集血液(2ml)和CSF(0.1ml)。每天至少记录2次临床体征。第3次给药后约24小时进行尸检,收获并保存选择的组织。
第1天,所有3个组4(150mg)的动物给药后3-12分钟内,表现出倾向于背面的最小倾向,持续5-15分钟,此信号被认为是与测试制品相关联的。体重、食物消费和神经/体检参数没有变化,被认为与测试制品相关联。
分析血清和CSF样品并提供剂量溶液分析。在食蟹猴的不同组织中观察各种内源性艾杜硫酶活性的变化;脑和脊髓比检查的其他外周器官(包括肝脏,心脏和肾脏)的内源性活性较大。艾杜硫酶施用与艾杜硫酶在不同脑区,以及在脑干和脊髓活性的剂量依赖性增加相关联。IT递送不会导致在左、右大脑半球之间可观察到的差异分布。艾杜硫酶活性在以下器官中有明确的剂量依赖性增加:大脑、肝脏,心脏和肾脏。大脑中艾杜硫酶的免疫染色,在染色强度表现出剂量依赖性增加。在3mg组中,在脑膜下方观察到脑膜细胞和神经胶质细胞染色;在3mg治疗组动物中,神经元染色并不明显。艾杜硫酶染色呈阳性,而且在脊髓内具有剂量依赖性,最高的染色强度在腰部,其中发生IT施用艾杜硫酶。艾杜硫酶染色强度在肝、肾和心脏呈剂量依赖性而且在这些器官中与增加的艾杜硫酶活性相一致。
总之,以每月一次的时间间隔,IT施用艾杜硫酶,递送剂量至150mg均无不良影响。因此,没有观察到不利影响水平(NOAEL)被解释为150mg,为本研究中测试的最高剂量。在CNS的艾杜硫酶活性中,艾杜硫酶施用与剂量依赖性增加相关联,并导致在肝、肾、心脏中的全身I2S水平。
在154mM NaCl,0.005%聚山梨酯20,pH值5.3-6.1中,提供测试制品,艾杜硫酶作为剂量溶液。提供的剂量溶液的标称浓度分别为0、3,30或150mg/ml。测试制品保存在-82°至-79℃的冰箱中。磷酸盐缓冲盐水(PBS),pH为7.2,用作为剂量施用和系列脑脊液收集后的冲洗剂。PBS获自Gibco,Invitrogen公司。
测试制品剂量制备
给药第一天的每个时间间隔,从-80℃冷柜取出各浓度的药瓶,在台面上使其解冻至室温。一旦解冻,分别标记为组1,2,和3中的药瓶,称重并通过0.22μm的过滤器取回1ml用于每个预定的动物进行给药。施用完所有的剂量剂量后,再次称重药瓶并放置在冰箱中。
接下来的一天(用于动物003,组4和组5给药的天),从冰箱中取出用于组1和组4的剂量溶液,并在台面上放置至室温。一旦到了室温下,称重用于组1和4的药瓶,标记组4药瓶,并通过过滤器取回用于每只动物预定的1ml,用于给药组1和组4。然后通过在无菌聚丙烯药瓶中注入适量的组4剂量溶液和组1(媒介物)来制备组5剂量溶液。记录来自组1和组4中的添加量。轻轻翻转药瓶混合该溶液而且通过过滤器取回2-1ml剂量用于组5中的动物。给药完成后再次称重组1和组4的药瓶,而且所有的药瓶(组1-5)被放置在冰柜中。
14只动物随机分配到下表中所描述的治疗组。
选择IT施用途径因为这是倾向于对人施用的途径。本研究中(3,30,100,和150mg/ml)被选择的艾杜硫酶剂量,被选择以评估在每月3次连续弹丸IT腰椎注射后,在非人类灵长类动物的中枢神经系统(CNS)中的不同剂量水平的酶的生物分布。
临床观察
临床体征的总发病率是最小的。在组1(对照),组2(3mg),组3(30mg),或组5(100mg)中的动物,没有临床体征,被认为在研究过程中的任何时间上与测试制品相关联。
第1天,所有3个组4(150毫克)的动物(012-014)在给药后3-12分钟内,表现出倾向于背面的最小倾向,持续5-15分钟。本信号被认为是与测试制品相关联的,而且任何的较低剂量组中没有观察到。在紧随第1剂后或在紧随测试制品施用后的期间内,没有其他的临床体征。组4动物中,只观察到其他信号为第35天动物013的单一呕吐事件。
考虑到植入的药物递送设备的固有的变化,单一的每月鞘内弹丸施用测试制品与任何不利的肉眼变化或微观变化不相关联。所有的组,包括对照组,在脑膜中有微观的变化,表明药物递送系统的炎症反应。在动物接受30mg及以上的测试制品剂量时,在脑膜中有炎症反应的倾向,有较明显的嗜酸性粒细胞成分。
由于对照和测试制品处理的动物之间的差异是非常轻微的,在本研究中没有观察到不利影响水平(NOAEL)的最高测试剂量被解释为150mg。
在所有组(包括对照)脑膜中的整体炎症反应比在猴子中进行的此鞘内研究一般遇到的稍加明显。然而,这被认为可能与媒介物的一些特性或尸检前24小时的给药行为相关联。
除了3mg组中的一只动物之外,在所有治疗的动物中,脑艾杜硫酶染色呈阳性,在150mg组(图16、17、18和19)中发现最高的染色强度。在3mg组中,只有脑膜下方的脑膜细胞和少量的神经胶质细胞为阳性;在神经元中没有检测到注射的艾杜硫酶。在高剂量组(30,100和150毫克)中,大量的脑神经元对艾杜硫酶染色呈强阳性,还有脑膜细胞、神经胶质细胞和血管周围细胞。艾杜硫酶免疫组化结果显示:在脑神经元中,注射艾杜硫酶从脑膜表面层I内神经元,到与白质(图20、21和22)相邻的更深层VI广泛分布。在150毫克剂量组(图23)中,也观察到标记染色的神经元。在所有动物中(从30-150mg的剂量组),在神经元的艾杜硫酶染色中,脑的额叶,中部和后部部分之间没有发现显著性差异。
在所有动物的脊髓中,艾杜硫酶染色呈阳性,在所述腰椎区域中(图24和图25)具有最高的染色强度。艾杜硫酶免疫染色也呈剂量依赖性。在150mg组(图26和图27)中,神经元、脑膜细胞、神经胶质细胞、血管周围细胞和外延/周边/神经内膜(结缔细胞)周围的神经纤维,艾杜硫酶染色呈强阳性。
在肝脏中,在所有动物的窦状腺细胞(枯否细胞和内皮细胞)中发现艾杜硫酶的阳性染色。然而,3mg治疗组(图28)的肝细胞中未检测到艾杜硫酶,而在更高剂量的肝细胞中发现阳性艾杜硫酶染色,在150mg治疗组(图29、30和31)中,具最大的染色强度。
在3mg治疗组(图22)的动物中,没有艾杜硫酶的阳性染色。相反,在30,100和150mg组中,间质细胞的艾杜硫酶为阳性染色,在150mg组中观察到标记染色-以阳性细胞数和染色强度(图33、34和35)而言。
肾脏
在从3mg剂量组(图36)的动物中检测到很少或没有检测到注射的艾杜硫酶。然而,在30和100mg组(图37和图38)的肾小球细胞和间质细胞中,发现阳性艾杜硫酶染色。在所述150mg组中,艾杜硫酶免疫组化染色另外揭示近端肾小管上皮细胞艾杜硫酶染色,与显著的肾小球及间质细胞染色(图39)。
讨论
体重、食物消费、体格检查和神经系统检查结果发现:没有测试制品相关的临床体征或影响。在第1天,组4(150mg)的动物,给药后3-12分钟内表现出倾向于背面的最小倾向,持续5到15分钟;此信号被认为是测试制品相关联的。
艾杜硫酶施用与在不同脑区,以及脑干和脊髓中的艾杜硫酶活性的剂量依赖性增加相关联。在脊髓内,最高水平的染色强度是在所述腰椎区域中,其中发生IT施用艾杜硫酶。在肝、肾、心脏中,IT施用艾杜硫酶也导致全身暴露剂量依赖的染色强度。动物接受的测试制品剂量,在30mg和更大时,在脑膜中,具有炎症反应的倾向,具有较明显的嗜酸性粒细胞成分,但是这种差异被认为没有生物显著性。
IT施用艾杜硫酶的剂量至多150mg,以每月一次的间隔递送均无不良影响。因此,在本例中,没有观察到的不良作用水平(NOAEL)被解释为是150mg,为最高测试剂量。在CNS的艾杜硫酶活性中,艾杜硫酶施用与剂量依赖性增加相关联,并导致在在肝、肾、心脏中的系统性水平。
实施例3:IT递送I2S的PK(血清和CSF)
本实施例提供了血清和脑脊液(CSF)分析,通过在食蟹猴中每月弹丸鞘内腰椎注射和每周弹丸静脉注射施用艾杜硫酶的6个月的毒性研究相关联,用于测试制品(TA)浓度。
实验设计
从超过6个月时间的毒理学和安全药理学的角度来看,本研究的目的是评估重复给药鞘内(IT)施用的艾杜硫酶(12s)。该研究的设计见表8。
表8:研究设计

DC-设备对照:在组1中的动物没有给药媒介物或测试制品
测试制品
鉴定:ldursulfase IV给药-(2.0mglmL)
IT给药-艾杜硫酶(0mgImL)
艾杜硫酶(3mg1mL)
艾杜硫酶(30mg/ml)
艾杜硫酶(100mg/ml)
分析方法:
用ELLSA法(酶接免疫吸收剂检测)进行分析,以测定艾杜硫酶浓度。在用稀释因子增加之前,检测限度(LOD)=1.25ng/mL。以1∶50稀释进行样品筛选,因此,检测的灵敏度为62.5ng/mL。进一步稀释超出校准曲线高端的样品,并在一个适当的稀释下重新测试,导致值在曲线的范围内。另外使用的酶活性检测分析选定的样品。在最小的样品稀释液1∶150上,用于此检测的LOD为0.18mU/mL。
组1和组2的动物分别给药以生理盐水或媒介物,在整个IV和IT给药期间内,所有具有的血清艾杜硫酶水平介于138ng/mL和<62.5(或LOD)之间。从组1和组2动物的200个CSF样品测试,62个表现出的水平I2S在检测LOD以上。这些中,7个值较高(>1,000ng/mL)。另外一个CSF样品,从预IT给药3收集的,测试超过I2S的1000ng/mL。
这些测试的样品用于艾杜硫酶活性。在每一种情况下,活性的结果表明,存在I2S以及基于活性水平计算I2S的近似浓度时,结果为通过抗原ELISA法获得的20%的范围内。(见表9),利用酶活性检测,也对其他与随机选择的具有抗原ELISA结果<LOD的CSF样品进行了测试,以排除任何非特异性活性。
表9:CSF样品的调查结果


在本研究中,分析血清和CSF样品用于艾杜硫酶浓度。根据如下进度安排血清样品收集:
IV给药:预给药和给药1至10后2小时,预给药和给药11至23后4小时,以及在尸检时。
IT给药:预给药和给药1至2后2小时,预给药和给药3至6后4小时,以及在尸检时。
根据如下进度安排CSF样品收集:
IV给药:预给药和给药1后2小时,和药3至6给4小时后。
IT给药:预给药和给药1至2后2小时,预给药和给药3至6后4小时,并在尸检。
一般情况下,血清艾杜硫酶的清除速度比CSF艾杜硫酶快。
在所有时间点的测试中,分别组对1和组2动物给药以生理盐水或媒介物时,血清艾杜硫酶水平小于或等于138ng/mL。有些动物有水平低于检测的检测限(LOD)。
组1和组2中较少的CSF样品在检测LOD以上,有7个显著的例外,其导致高(>1000ng/mL)的水平。选自预IT给药3的动物的一个CSF样品,也超过1000ng/mL艾杜硫酶。
对给出了这些趋势结果的样品进行重测,并确认。此外,进行测试这些样品的艾杜硫酶酶活性。这些活性的结果也证实了高的艾杜硫酶水平,在由艾杜硫酶质量检测(表9)获得的20%之内。
通过随机测试低于LOD的艾杜硫酶质量单位的CSF样品证实这些样品队列中活性检测的特异性,并确证在这些样品中艾杜硫酶水平确实为LOD(数据未示出)。
实施例4:制剂
本实验例总结了所述药物开发研究,以建立艾杜硫酶-IT原料药和原料产品的制剂,用于I/II期临床实验。
由于适合于所述CNS递送的所述赋形剂的限制,对于鞘内递送艾杜硫酶制剂开发的努力主要关注于:降低所述磷酸盐和聚山梨醇酯20水平,同时仍然保持与全身递送的所述I2S制剂等价的稳定性。
进行三个关键的筛选胁迫研究,以检测磷酸盐和聚山梨醇酯水平的影响。这些包括冷冻融化、摇动胁迫和热胁迫。所述结果表明,生理盐水制剂对抗所述冷冻融化胁迫,在低蛋白质浓度(2mg/mL)更加稳定。在高蛋白质浓度(100mg/mL),所述冷冻融化胁迫对于含生理盐水和磷酸盐制剂都不产生稳定性问题。所述摇动胁迫研究证实了,0.005%聚山梨醇酯20能保护所述蛋白质抵抗摇动相关的胁迫。所述热稳定性研究表明,所述生理盐水制剂比含磷酸盐制剂更加稳定。另外,所述生理盐水制剂的pH可以维持在6.0,在2-8℃下24小时。所述残留的磷酸盐的量与所述蛋白质、以及增加的蛋白质浓度相关,这种发现可能归因于在所述最终制剂中的pH稳定性。
方法
冷冻/融化胁迫对于在生理盐水和磷酸盐制剂中艾杜硫酶稳定性的影响
为了检测冷冻/融化胁迫对于在不同制剂中艾杜硫酶稳定性的影响,使用微量离心浓缩器Plus(Centricon Plus)将所述病毒SEC池更换/浓缩4次,进入150mM NaCl或者137mM NaCl(具有20mM磷酸钠)(两者都为pH6.0)。所述蛋白质浓度目标定至2mg/ml和100mg/mL。所有溶液通过0.22micron PVDF过滤器过滤。所述溶液等分为1mL,每个1ml装入2mL硼硅酸盐玻璃小药瓶。所述小药瓶放置在冷冻干燥器室的中间搁板上,并且周围围绕安慰剂小药瓶。将所述样品按照程序设定的冷冻/融化循环(在20℃维持1小时,并且以1℃/min冷冻至-50℃)冷冻。然后,以两步法融化,以0.03℃/min的速率,从-50℃至-25℃(在-25℃维持24小时,并允许融化至2-8℃)。在两个或者三个冷冻/融化循环后,通过外观分析和SEC-HPLC分析所述样品。
摇动/剪切胁迫对于在磷酸盐和生理盐水溶液中的影响
在不同的蛋白质浓度,对艾杜硫酶进行摇动研究。测试存在在20mM磷酸盐中的137mM NaCl(pH6.0),以及单独的154mM NaCl(pH6.0)中时,在2mg/mL、8mg/mL、以及90-100mg/mL的蛋白质浓度。为了确定,是否需要聚山梨醇酯,多种量的PS-20在测试条件中被强化。所述溶液等分为每份1.2mL,每份装入2mL玻璃小药瓶,并且然后在轨道摇床上,以250rpm,在室温条件下摇动24小时。在基线和24小时,检测所述外观,并且采集0.1mL等分试样的样品,在低于≤-65℃,在0.5mL聚丙烯管子中冷冻,直到通过SEC-HPLC分析。
首先为了证实聚山梨醇酯20水平的影响,使用3小时卡车运输的材料进行模拟航运研究,然后使用随机测试选项(通过Lansmont(Lansing,MI)进行)在确保等级1下进行1小时空气测试。通过SEC-HPLC,分析所述样品的颗粒外观和可溶性聚集体。
为了检测胁迫搅拌对稳定性的影响,生理盐水制剂(50mg/mL,154mM NaCl,和0.005%PS-20)以1.3mL填充在3mL类型I玻璃小药瓶,并塞有13mm瓶塞,其含有Teflon涂覆的磁力搅拌棒(长度8mm,直径2mm)。所述小药瓶放置在搅拌平板上,其速度设置6(设置6的选址是最大速度,并不导致过多的泡沫)。在0、2、24、48和72小时,测定外观。所述基线和72小时搅拌的样品通过SEC-HPLC方法测试。
热稳定性研究和主要的制剂
比较6种主要制剂的热稳定性。根据两种参数选择这些制剂。第一参数是在CNS递送治疗范围以内的蛋白质浓度。所述第二参数是控制磷酸盐浓度对于稳定性的影响。将所述病毒过滤的SEC池缓冲剂更换,并使用微量离心浓缩器Plus-80浓缩。得到50和100mg/mL蛋白质浓度的目标浓度。所述6种制剂用1%聚山梨醇酯20溶液强化,最终浓度为0.01%PS-20。所述材料通过0.22micron PVDF过滤器过滤,并添加0.5mL至2mL玻璃硼硅酸盐小药瓶中。将这些小药瓶以倒转的姿势放置在胁迫的稳定性(40℃)、加速的稳定性(25℃)、以及实时储藏(2-8℃)上。在每个时间点的样品稳定性通过SEC-HPLC、OD320、SAX-HPLC、SDS-PAGE(Commassie)、pH、和活性进行测试。
在生理盐水制剂中理解所述pH对照
为了理解在所述生理盐水中所述pH是怎样维持,进行下面的研究。
测试在生理盐水制剂中残留的磷酸盐
将所述病毒过滤的SEC池(2mg/mL艾杜硫酶,137mM NaCl,20mM磷酸钠,pH6.0)浓缩,并通过使用Millipore TFF系统和Millipore PelliconBiomax30(50cm2过滤器)渗滤进150mM NaCl中。在7X、10X和15X循环的膜渗滤进入0.9%生理盐水(在TK3制备)之后,与蛋白质相关的磷酸盐的量被测定。另外,也测试了:在10X膜渗滤(含有经过过滤流体的非蛋白质)后的渗透,以及在所述过滤步骤中的所述生理盐水。
测定蛋白质浓度对于pH的影响
为了更好地理解不存在缓冲剂(磷酸盐)时的pH对照,进行了蛋白质效应研究。为了测定所述蛋白质对于pH的贡献,材料在154mM NaCl(生理盐水)中稀释至30mg/mL、10mg/mL、2mg/mL、1mg/mL、0.1mg/mL、0.01mg/mL以及单独的生理盐水。所述材料等分装进2mL聚丙烯管子中,以每管1mL的填充体积。将所述样品在-65℃冷冻1小时,在室温融化30分钟,并且重复所述循环3次。测定所述初始pH,并在3X冷冻/融化循环后进行比较。也在24小时的室温暴露所述样品后,测量所述pH,以测定所述蛋白质浓度对于pH变换可能有的影响。
由于适合于所述CNS递送的赋形剂的限制,开发制剂用于鞘内递送艾杜硫酶的努力主要关注:降低所述磷酸盐和聚山梨醇酯20水平,同时保持其稳定性等价于配制用于全身施用的I2S。进行三个关键的筛选胁迫研究,包括冷冻融化、摇动胁迫和热胁迫。
冷冻/融化对于在生理盐水和磷酸盐制剂中艾杜硫酶的影响
如在表10中所示的,在2mg/mL的低蛋白质浓度下,含有20mM磷酸盐的制剂在冷冻融化胁迫后,形成更多的聚集体。所述生理盐水制剂保持与所述基线相同的聚集体水平。在搞蛋白质浓度(100mg/mL)下,所述冷冻融化胁迫似乎对(表11)中的任一制剂的稳定性没有影响。所述数据表明,单独生理盐水的制剂对抗冷冻融化胁迫具有更好的稳定性。
表10:在低蛋白质浓度的可溶性聚集体

表11:SEC属性,以测定在搞蛋白质浓度下的可溶性聚集体

*调节NaCl的量至137mM,其中所述制剂含有20mM磷酸盐以维持相当的渗涨度
摇动胁迫对于在溶液中艾杜硫酶的影响
在2、8和100mg/mL的三种蛋白中浓度下,进行所述摇动研究。所述数据表明,没有聚山梨醇酯20时,在所有所述蛋白质浓度下发生沉淀,并且在2mg/mL也观察到高水平的可溶性聚集体(表12至表14)。但是,存在低水平的P20(例如0.005%)时,所述沉淀物和可溶性聚集体大部分被阻止。所述数据表明,低水平的聚山梨醇酯对于保护蛋白质对抗摇动胁迫是必需的。
表12-14:在实验室模型中的摇动研究(以250rpm旋转24小时,在室温)
表12:~2mg/ml,在137mM NaCl和20mM磷酸盐中,pH6
| P20浓度 | 外观 | SEC(%单体) |
| 0% | 观察到蛋白质样颗粒 | 95.2% |
| 0.0005% | 观察到蛋白质样颗粒 | 99.4% |
| 0.001% | 观察到蛋白质样颗粒 | 99.4% |
| 0.0025% | 观察到灰尘样颗粒 | 99.7% |
| 0.005% | 观察到灰尘样颗粒 | 99.7% |
| 0.01% | 观察到灰尘样颗粒 | 99.8% |
表13:~8mg/ml,在137mM NaCl和20mM磷酸盐中,pH6


表14:90-100mg/ml,在生理盐水制剂中
*所述对照样品(没有摇动)具有99.8%单体。
为了进一步证实0.005%是否足够对抗摇动的稳定性,进行模拟航运研究,所述航运研究接近于真实的航运条件,其以100mg/mL蛋白质(具有不同水平的聚山梨醇酯20)对生理盐水制剂进行。所述结果证实了0.005%是足够的(表15)。
表15:在模拟航运研究中,聚山梨醇酯20对于在生理盐水中的外观和100mg/mL的可溶性聚集体的影响


使用磁力搅拌棒,研究搅拌对于含50mg/mL艾杜硫酶的生理盐水制剂(具有0.005%聚山梨醇酯20)的影响,其总结在表16中。如所示的,所述蛋白质不易受所述(由磁力搅拌棒搅拌72小时的)胁迫影响。所述结果证实了0.005%足够对抗搅拌胁迫。
表16:聚山梨醇酯20对于53mg/mL艾杜硫酶稳定性(在侵入性搅拌过程中)的影响

主要候选物的热稳定性
检测了6种主要的制剂,在24个月期间的稳定性测试。这些测试结果在这部分进行了描述。
外观
对于所述6种制剂在所述温度和测试时间点,所有所述制剂的外观保持稍微乳光,并且基本上没有颗粒。
OD320
为了检测在浊度上的潜在增加,测定所述OD320值,并在表17中总结。如所示的,在所述冷冻储藏期间,在24个月的储藏后,所有所述制剂的OD320值保持与所述基线相同。在2-8℃条件,所述生理盐水制剂在24个月后,保持与所述基线相同,但是含磷酸盐制剂的OD320值具有增加。在25℃的所述加速条件下,在3-6个月后,所述生理盐水OD320具有稍微增加,但是所述含磷酸盐制剂显示更加显著的增加。这些结果表明,所述生理盐水制剂对于对抗热胁迫更加稳定。
表17:比较生理盐水和磷酸盐制剂的OD320*

*所有含有0.01%聚山梨醇酯20。
SEC-HPLC
通过SEC-HPLC,所有所述制剂的数据总结在表格中。在所述冷冻储藏条件下,在24个月后,没有变化(与基线相比)。
在40℃的胁迫条件下,在两个月后,所有所述制剂可溶性聚集体具有增加的水平。另外,含有磷酸盐的制剂也显示有“12分钟”峰。但是,在1个月之后,在所述含磷酸盐制剂中观察到的所述“12分钟”峰似乎消失。另外,对于所有所述制剂,所述可溶性聚集水平没有进一步增加(与所述2星期时间点比较)(图8和表18)。
在25℃的所述加速条件下,与所述基线相比,对于所有所述制剂,可溶性聚集体的水平增加在6个月后是最小的。但是,所有含磷酸盐的制剂显示有“12分钟”峰(图9和表18)。
在所述2-8℃的长期储藏条件下,在24个月后,对于所有所述制剂的可溶性聚集体增加在24个月储藏之后是最小的。与所有条件一致,所述含磷酸盐的制剂也具有“12分钟”峰,其随着时间稍微增加(图10和表18)。
这些结果表明,在所有储藏条件下,所述生理盐水制剂具有最小的变化(与所述含磷酸盐制剂相比)
表18:通过SEC-HPLC,比较在生理盐水&磷酸盐制剂中的聚集体**All制剂含有0.01%聚山梨醇酯20
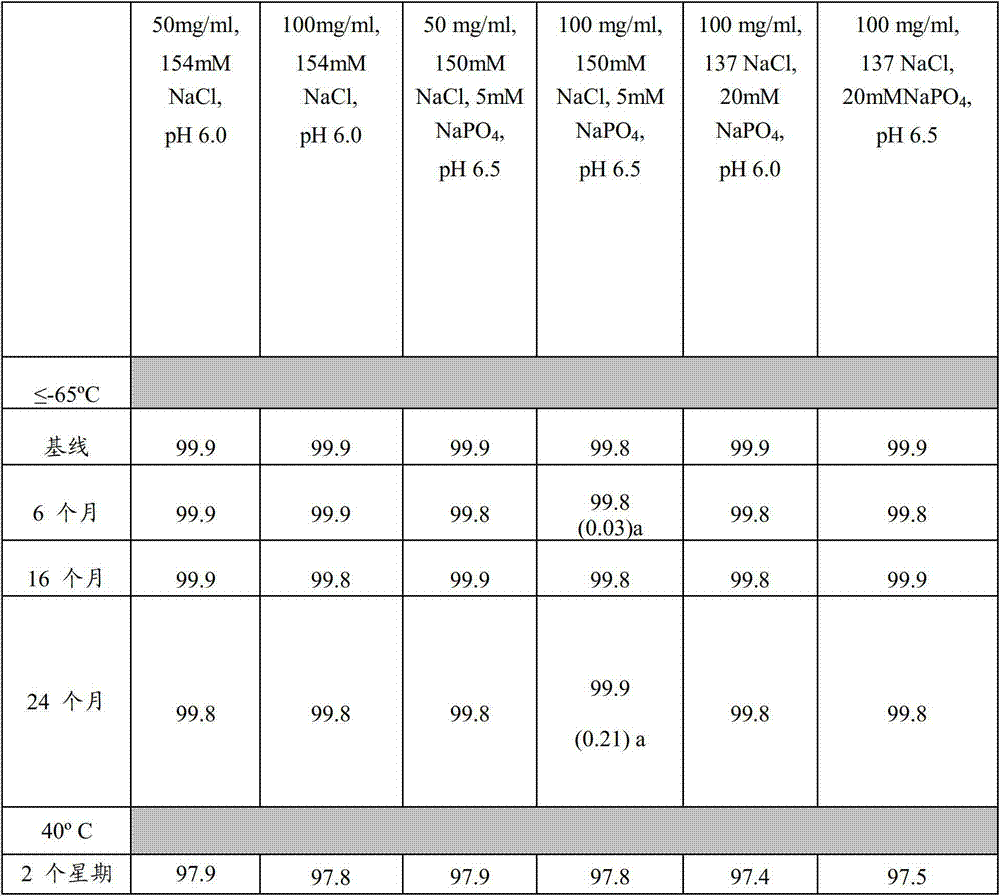

a:表示所述值为高分子种类,其在目前的SEC HPLC方法方法中洗提~约12分钟,通常是指所述“12分钟峰”。所述峰被认为与在所述制剂中存在磷酸盐相关。
SAX-HPLC
SAX-HPLC的所述数据总结在表19中列出。在所述胁迫/加速条件下,所述生理盐水制剂出现稍微多的变化(图11和图12),但是在所述长期储藏条件下,在24个月后,所有所述制剂没有变化(表19和表13)。这表明了,所述生理盐水制剂在2-8C,24个月是稳定的。
表19:通过SAX-HPLC方法,比较生理盐水和磷酸盐制剂(所有都具有0.01%聚山梨醇酯-20)在24个月间的电荷变化。


pH
表20表明了,在2-8℃的24个月,所有所述制剂的pH保持与所述基线相当。对于所述生理盐水制剂,尽管没有缓冲剂,但是所述pH在24个月保持恒定在6.0。
表20:在2-8℃的24个月,比较在生理盐水和磷酸盐制剂(所有都具有0.01%聚山梨醇酯-20)中的pH
酶活性
与参考标准相比,所有所述制剂的特异性活性在2-8℃(24个月后)与所述分析变异相当,其显示了,艾杜硫酶在所述生理盐水制剂中在24个月保持稳定(表21)。
表21:在24个月实时稳定性(2-8℃)之后,通过离子交换色谱法得到的在生理盐水和磷酸盐制剂中的活性结果

*在测试24个月期间的样品,所述参考标准的所述特异性活性为56U/mg。
与所述蛋白质相关的残留磷酸盐的测量
在制备所述生理盐水制剂的所述最终UF/DF步骤,是将所述蛋白质溶液从137mM NaCl,20mM磷酸钠渗滤成150mM NaCl。为了检测所述膜渗滤循环数如何影响在最终产品中残留的磷酸盐浓度,使用原料药(2mg/mL艾杜硫酶,137mM NaCl,20mM磷酸钠,pH6.0)进行实验室规模的研究。首先将所述原料药浓缩成50mg/mL艾杜硫酶,然后渗滤进150mM生理盐水。在7x、10x和15x膜渗滤步骤采集样品,并通过ICP测试磷酸盐含量。所述测试结果在表22中总结。如所示出的,所述生理盐水膜渗滤溶液不含有任何磷酸盐。在7x DF后,所述蛋白质含有约0.22mM磷酸盐,其高于理论计算值。在10x DF后,所述蛋白质滞留物含有约0.16mM磷酸盐,而所述通过的流体为仅仅约0.07mM磷酸盐,其表明所述磷酸盐结合在所述蛋白质上。在15x DF后,所述磷酸盐水平降低到约0.07mM。
来自所述研究的所述结果表明,约0.2mM磷酸盐残留保留在所述原料药中,其可能有助于维持所述生理盐水制剂6.0的pH。
表22:在多膜渗滤步骤后,随所述蛋白质保留的磷酸钠

*测试所述起始生理盐水缓冲剂,并且没有可检测的磷酸盐。
蛋白质浓度对于维持制剂pH的影响
从所述磷酸盐含量分析,显然地,磷酸盐与所述蛋白质结合。因此,可预期的是,高蛋白质可能结合更多的磷酸盐,其可能更好地维持所述pH。为了检验这种假说,在所述生理盐水溶液中的所述蛋白质被浓缩至不同的水平,并且测试了在不同加工条件下的所述溶液pH值。所述结果在表23中总结。
如所示出的,所述溶液的初始pH维持在约6.0,并且不依赖所述蛋白质浓度。但是,在室温暴露24小时或者3个冷冻融化循环后,所述溶液pH含有0.1mg/mL蛋白质或者更少,不能维持溶液约6.0的恒定pH。所述溶液在高于1mg/mL的蛋白质浓度的pH维持在约6.0。这证实了,所述蛋白质浓度是维持所述生理盐水溶液pH的控制因子。
表23:蛋白质浓度对于无缓冲生理盐水制剂pH的影响

*样品储藏在≤-65℃,至少1小时,并且在室温融化0.5小时,并且重复这个循环3次。
来自这项研究的结果表明,在生理盐水制剂(50mg/mL艾杜硫酶,0.005%聚山梨醇酯,150mM NaCl,pH6.0)中的艾杜硫酶,当储藏在2-8C,对于至少24个月是稳定的。该制剂似乎更加稳定,与所述含磷酸盐制剂相比。选择0.005%聚山梨醇酯20,对于保护蛋白质对抗所述摇动胁迫是足够的。另外,所述研究表明,所述生理盐水制剂的pH可以稳定地维持在6.0,在2-8C24个月,部分地由于在所述最终制剂中,所述残留磷酸盐和高蛋白质浓度。
实施例5.生物分布
已成功地证实,鞘内施用为有效的递送I2S到CNS组织的方式,进行了更多的研究以确定是否IT-施用的I2S能分布到脑深部组织以及IT-施用的I2S是否有细胞定位。已经制备重组的人类己醛糖酸盐-2-硫酸酯酶(I2S)制剂并配制在154mM氯化钠,0.005%聚山梨酯20(pH为6.0)媒介物中。
按植入鞘内端口的方式给非人类的灵长类动物按月给予3mg,30mg或100mg的I2S,连续6个月。该研究的设计总结在以下的表24中。
表24

a艾杜硫酶,除非另有规定。DC(设备对照);IT(鞘内);IV(静脉注射);
NS(正常生理盐水);PBS(磷酸盐缓冲盐水,pH值7.2)。
每月重复施用I2S到非人类灵长类动物进行6个月,在最高剂量检测的耐受性良好,不与任何重大不利的毒理学事件相关联。施用第6和最后给药I2S后的24小时,处死受试者非人类灵长类动物,检查此非人类灵长类动物的CNS组织。
如由免疫组化法(IHC)测定的,在整个CNS组织和细胞中,有有广泛的I2S细胞沉积。通过IHC,在所有的脑组织中检测到I2S蛋白,从大脑皮质的脑室白质具有沉积梯度。在剂量依赖式的所有组中的大脑,小脑,脑干和脊髓的神经元的灰质中,可检测到I2S。在高剂量组的灰质表面中,在表面皮层(图40A)内,大量的脑神经元对I2S染色为阳性。在丘脑(图40B)、海马(图40C)、尾状核(图40D)和脊髓(图40E)的神经元中也检测到I2S。脑膜和血管周围细胞也I2S染色(图40F)呈阳性。
如图41和图42中所述,IT-施用的I2S进入CNS组织分布并尤其在受试者非人类灵长类动物的灰质,丘脑和大脑皮质中的沉积是显而易见的。此外,图42和图43示出,IT-施用的I2S受试者非人类灵长类动物CNS组织中的累积为剂量依赖的方式。共定位染色也显示,IT施用的I2S与神经元和少突胶质细胞相关联。IT施用的I2S也在受试者非人类灵长类动物的整个大脑分布和定位,如图44证实。特别地,如图45示出,神经元摄取和轴突关联的I2S(对非人类灵长类动物IT施用I2S)后,如由纤维丝染色表明。而且特别感兴趣的是,目前的研究表明,I2S对神经细胞是有选择性的,而且这些神经细胞协助鞘内-施用I2S在脑深部组织的分布,而且似乎是与轴突结构相关联,表示顺轴突运输I2S。
下表25示出单独动物研究的剂量和各种施用途径的药代动力学数据。
表25
124I标记的I2S施用于试验动物,如下表26所示,以及PET扫描结果见图62、图63中所示。
表26
| 组 | 动物/组 | 途径 | 测试制品 | 剂量 |
| 1 | 1 | ICV | [124I]-艾杜硫酶 | 3mg |
| 2 | 4 | IT-L | [124I]-艾杜硫酶 | 3mg |
| 3 | 4 | IV | [124I]-艾杜硫酶 | 0.1mg/kg |
| 4 | 4 | IV | [124I]-艾杜硫酶 | 1mg/kg |
本研究还显示了IT-施用后,受试者非人类灵长类动物脑室附近,白质脑组织中的IT施用I2S的细胞识别。虽然白质中的I2S染色密度比灰质中的普遍更低,在少突胶质细胞(图46)内可检测到I2S。特别是,图46示出在白质脑组织中I2S的细胞识别,并进一步说明I2S与髓鞘不表现出相关性。
除了展示IT施用的I2S的分布深入到脑组织中,目前的研究也证实了I2S在目标细胞器内的定位,而且重要的是I2S在溶酶体内部的定位,其影响溶酶体贮积症中的细胞器,如Hunter’s综合症。特别地,位于溶酶体内的I2S,而且在轴突也可检测到。图46示出在非人类灵长类动物少突胶质细胞溶酶体内,IT-施用的I2S定位,从而确认IT-施用I2S能够分布到脑深部组织并能够细胞定位。
为了辨别是否递送的I2S保留生物活性,利用特异性活性分析测定I2S在脑中的水平。最后给药后24小时,3mg IT组在大脑中的活性明显不同于设备对照及媒介物对照动物的基础水平。在尸检(给药后24小时)中,30mg和100mg IT给药动物的脑中,酶活性高于基线。
进一步的动物测试,以识别在IT递送I2S至所述脑后的所述生物分布,如在图60中示出,并且样品编号对应于下面表27。
表27:样品定位


实施例6.IT VS.ICV递送
在上述例子中观察到的I2S的分布模式也通过在健康Beagle犬中给予单一的IT或ICV给药来概括。使用计算机生成的数字,雄性Beagle犬随机分为两组(组1(ICV),N=3,组2(IT);N=4)。所有犬具有在腰椎或左侧脑室(用于给药)和在小脑延髓池(采样)内的蛛网膜下腔植入导管。在皮下钛接入端口终止所有的导管。额外的犬用作为不给药的手术对照。
I2S单一的弹丸1ml注射(在20mM磷酸钠中30mg/ml,pH值为6.0;137mM氯化钠;0.02%聚山梨酯-20),施用IT或ICV,然后由0.3ml磷酸缓冲盐水(PBS,pH7.2)冲洗。临床体征进行监测,并且处死发生在给药后24小时。收集脑和脊髓组织样品用于定量I2S分析,如由ELISA所测定的I2S的酶活性和IHC,并在研究组之间进行比较。
I2S广泛分布在IT和ICV组的整个灰质,如由IHC所测定。在大脑皮质的所有6个神经元层中,神经元对I2S呈阳性,从IT和ICV组的表面分子层到深部内层,如由图47所示(A和B)。在IT和ICV组的小脑皮质中,神经元中检测到I2S,包括浦肯野细胞,如由图47(C和D)所示。在IT和ICV组的海马体中,众多数量的神经元对I2S为阳性,如图47(E和F)表明的。在两个组中,也在丘脑和尾状核中发现I2S阳性神经元,如图47(图像G和H)中表明的。
因此,本研究证实了IT-施用酶分配到脑深部细胞和组织的能力,并支持IT-施用的酶的效用,如I2S用于治疗与溶酶体贮积症相关联的CNS的表现,如亨特氏综合症。
实施例7:艾杜糖醛酸盐-2-硫酸酯酶缺陷小鼠M0DEL
进一步研究表明,IT-施用的I2S能够分布到脑深部组织和I2S细胞定位,进行进一步的研究用于测定IT-施用I2S的治疗效果。开发Hunter综合症的基因工程己醛糖酸盐-2-硫酸酯酶基因敲除(IKO)小鼠模型用于研究IT-施用I2S以改变疾病发展的能力。使用目标性的破坏I2S位点,其导致在组织和器官中积累粘多糖(GAG),开发所述I2S基因敲除小鼠模型。IKO小鼠模型显示出许多在人类见到的Hunter综合症的物理特性,包括特征性的粗功能和骨骼缺陷。此外,所述IKO小鼠模型在尿和整个身体组织中演示升高的粘多糖(GAG)水平,以及广泛的细胞空泡化,其为观察的病理组织学。
在本研究中,市售的I2S 为浓缩的,并再悬浮于磷酸盐缓冲盐水(PBS)中。6组雄性IKO小鼠,8-12周龄,用I2S(10μL,26mg/ml)治疗。组A和B(N=3)分别施用3个260μg剂量(1,8,和15天)和260μg剂量(1和8天)的I2S,通过在1天,8,和15鞘内施用260μg剂量,进行组D治疗。组C和E(N=3)为未经治疗的对照,而组F(N=3)为未经治疗的野生型对照。对照小鼠施用的媒介物没有I2S。最后一次注射后1小时处死小鼠,其次为组织制备用于进行免疫组化(IHC)和病理组织学分析。
为浓缩的,并再悬浮于磷酸盐缓冲盐水(PBS)中。6组雄性IKO小鼠,8-12周龄,用I2S(10μL,26mg/ml)治疗。组A和B(N=3)分别施用3个260μg剂量(1,8,和15天)和260μg剂量(1和8天)的I2S,通过在1天,8,和15鞘内施用260μg剂量,进行组D治疗。组C和E(N=3)为未经治疗的对照,而组F(N=3)为未经治疗的野生型对照。对照小鼠施用的媒介物没有I2S。最后一次注射后1小时处死小鼠,其次为组织制备用于进行免疫组化(IHC)和病理组织学分析。
3次注射后,与媒介物治疗过的小鼠相比,在I2S治疗的小鼠中,大脑皮层表面、尾状核,丘脑和小脑的细胞空泡化普遍减少。也在IT处理后的白质中发现细胞空泡化减少。IT-施用后,IKO小鼠脑组织中I2S的分布是明显的。
在IKO小鼠中,3周的IT施用I2S也表明在光和电的微观水平,CNS中的细胞空泡化的显著下降。在IT施用I2S后,相对于未经处理的IKO小鼠,细胞空泡化明显减少,这表明IT-施用I2S能够改变疾病的发展。如图48所示,IT-施用I2S后,IKO小鼠的corpous胼胝体和穹窿中的细胞空泡明显减少。图49说明了显著减少,在所述治疗的IKO小鼠的表面大脑皮层组织中,存在溶酶体相关膜蛋白质(LAMP1)时(一种溶酶体疾病的病理学生物标记物)。
此外,电子显微镜检查表明:存在贮积的减少,包括在灰质中的神经元、和在白质中少突胶质细胞的液泡化。特别是,IKO小鼠IT-施用I2S也表现出栅栏层状体(“斑马小体”)的减少,其特性在于:某些溶酶体贮积症。特别是,图5表示电子显微镜扫描,示出了相对于未治疗的IKO鼠,在施用I2S的IKO小鼠的神经元中,斑马小体的减少。类似地,图5示出了胼胝体中少突胶质细胞的电子显微镜扫描。
此外,IT施用I2S到IKO小鼠,在溶酶体病病理的生物标志物的溶酶体相关的膜蛋白1(LAMP1)免疫染色中,一种溶酶体活性和疾病状态指标,在大脑皮质的表面、尾状核、丘脑,小脑和白质,也表现出显著减少。正如在图49A所示,相对于未处理的IKO对照小鼠大脑皮层组织表面,在治疗的IKO小鼠大脑皮层组织表面,LAMP1免疫染色明显减少,见图49B中所示,反映疾病病理的改进。
图20定量地表明和比较脑组织中μm2区域测量的LAMP1浓度。不同脑区的LAMP-1免疫染色的形态计量学分析证实,在评估的脑的各个领域内,LAMP-1阳性染色显著减少。正如图4所示,在评估的脑组织(皮层,尾状核和豆状核(CP)、丘脑(TH),小脑(CBL)和白质(WM))的每个区域中,相对未治疗的IKO对照小鼠,在治疗的IKO小鼠中,LAMP-阳性区域减少,并接近的野生型小鼠的LAMP-阳性区域。特别值得注意的是:在持续治疗持续时间中,在每个脑组织区域中的分析的LAMP-阳性区域为进一步降低。
在所述脑的所有区域中,异常高溶酶体活性的减少,与剧烈形态改善相关。这些结果证实了:IT-施用I2S能改变溶酶体贮积症的进展,在基因工程的IKO小鼠模型,进一步证实了:IT-施用酶(例如I2S),以治疗与溶酶体贮积症(例如Hunter’s综合症状)并发的CNS表现的能力。
实施例8:Hunter’s疾病患者的治疗
通过例如IT递送的直接CNS施用可以用于有效治疗Hunter’s疾病患者。这个实施例解释了多剂量递增研究,其被设计以评估每隔一周(EOW)高达3剂量水平,持续总共40周,通过鞘内药物递送设备(IDDD)施用rhASA至患有晚婴型Hunter’s疾病患者的安全性。适合于人类治疗的多种示例性的鞘内药物递送设备,在图45-48中描述。
将招募多达20例患者:
队列1:5例患者(最低剂量)
队列2:5例患者(中剂量)
队列3:5例患者(最高剂量)
5例患者被随机分配至没有治疗组。
为本研究选取的患者是根据一下标准的内容:(1)在30个月的年龄之前出现第一症状的外观;(2)在筛选的时候是能走动的(定义为独立站立和用一只手举着向前走10步的能力);(3)在筛选时存在神经学体征。通常地,具有造血干细胞移植历史的患者被排除。
通过IT注射,持续40周,测定在患有晚婴型Hunter’s疾病的儿童中施用I2S的递增剂量的安全性。此外,评估了I2S在粗大运动功能方面的临床活性、以及在血清中单一和重复剂量的药代动力学、以及在脑脊髓液(CSF)中的浓度。
治疗有效量的I2S通过鞘内施用,有规律的间隔,根据所述疾病影响的本性和范围以及发病基础。如本文所使用的,以“间隔”使用,表明,治疗有效量是周期性地(如与一次性剂量区别)。所述间隔可以通过标准临床技术确定。在一些实施方案中,I2S鞘内施用大约每隔一周一次。但是单个个体的所述施用间隔是没有必要固定的,而是可以根据所述个体需求随时间而变化。例如,在身体疾病或者胁迫时,如果抗-I2S抗体存在或者增加,或者如果疾病症状恶化,在所述给药之间的间隔可以减少。
实施例9-Hunter’s疾病患者的治疗
可以使用通过例如IT递送直接CNS施用,以有效治疗Hunter’s疾病患者。这个实施例解释了多剂量递增研究,其被设计以评估:多达3个剂量水平每隔一月,总共持续6个月的I2S施用(通过鞘内药物递送设备(IDDD))至患有晚婴型Hunter’s疾病患者的安全性。适合于人类治疗的多种示例性鞘内药物递送设备在图45-48中描述,以及所述试验示意图在图62中示出。
招募至多16例患者:
队列1:4例患者(最低剂量-10mg)
队列2:4例患者(中剂量-30mg)
队列3:4例患者(最高剂量-100mg)
4例患者被随机分配至没有治疗组或者使用设备。
Hunter’s疾病患者通常表现意识和神经发育损伤,包括早期发育的重要事件(例如,走路、讲话、如厕训练)的延缓、智力缺陷、活动过度、进攻性,听力损伤,癫痫症和脑积水。所有的迹象可以是评判标准的部分。进行此研究的所述患者的选定是基于以下包含的标准:(1)3-18岁的年龄;(2)小于77的智商、或者在过去的3年内15至30IQ点的下降;(3)没有CSF shut或者控制不佳的癫痫症以及(4)没有并发病呈现麻醉和/或手术风险。
在患有晚婴型Hunter’s疾病的儿童中,通过IT注射施用I2S,6个月,测定了递增剂量I2S的安全性。另外,评估了I2S对粗大运动功能的临床活性、血清中单一和重复剂量药代动力学、以及在脑脊髓液(CSF)中的浓度。
本研究的目的是评估I2S递增剂量的安全性和耐受性,以及所述IDDD的安全性、耐受性和长期通畅性。此外,也评估了:在单一和重复IT剂量后的I2S浓度(包括在CSF和血液中),以及I2S对CF生物标记物和泌尿GAG的影响。进一步的评估包括:I2S对临床参数的影响,包括生理学和神经认知评估、神经功能和脑结构容积。此外,也可以评估:治疗对日常生活的影响,以及在生物标记物和症状之间的关系。
Hunter’s疾病患者的治疗,通过IT递送I2S导致硫脑苷酯在多种组织(例如神经系统、心脏、肾脏、肝脏、肾脏、胆囊,、和其它器官)中累积减少。
虽然本文描述的某复合物、组合物和方法已经根据某实施方案进行特定的描述,但是所述实施例仅用于解释本发明的复合物,并且不用于限制必须与其相同。
所述冠词“a”和“an”,如本文在说明书中和权利要求书中使用的,除非清楚地表明与之相反,应该被理解为包括其复数形式。在某组的一个或多个成员之间包括“或者”的权利要求或者描述,被认为:如果一个、一个或多个、或所有组成员存在、被采用或者以其它方式与给定的产品或过程相关,那么就是符合的;除非从上下文中表示相反或者其它方式明显地指出。本发明包括这样的实施方案:其中所述组的只有一个成员存在、或者被采用、或者以其它方式与给定的产品或过程相关。本发明也包括这样的实施方案:其中多个、或者整个组成员存在、或者被采用、或者以其他方式与给定的产品或过程相关。而且,应该理解,本发明包括来自一条或多条所列出的权利要求所有的变型、组合、以及置换,其中一种或多种限制、元素、条款、描述性术语等,其根据所述相同权利基础(或者相关、任意其它权利要求)被引入到另一权利要求,除非其它方式指出或者除非其对于本领域技术人员将明显地出现矛盾或者不一致。当元素以列表形式呈现(例如在Markush基团中或者类似的形式)时,应该理解,所述元素的每个亚组也是包括在内地被公开,并且任何元素可以从所述组删除。应该理解,一般而言,当本发明、或者本发明的某些方面,被指出包括特定的元素、特征等,那么本发明或者本发明的方面的某些实施方案、或者基本含有这些元素、特征等。为了简化这些实施方案,本文没有在每个案例中具体提出这么多的单词。应该理解,本发明的任意实施方案或者方面可以被清楚地从所述权利要求排出,无论所述具体的排除是否在本说明书中叙述。本文所引用的所述公开文本、网址和其它参考材料,为了描述本发明的背景、以及提供关于其实践的其它细节;都通过参考的方式引入本文。
Claims (61)
1.鞘内施用的稳定制剂,包括艾杜糖醛酸-2-硫酸酯酶(I2S)蛋白质、盐、和聚山梨醇酯表面活性剂。
2.如权利要求1所述的稳定制剂,其中所述I2S蛋白质存在的浓度范围从大约1-300mg/ml。
3.如权利要求1或者2所述的稳定制剂,其中所述I2S蛋白质存在的浓度选自:2mg/ml,10mg/ml、30mg/ml、50mg/ml或者100mg/ml。
4.前述权利要求中任意一项所述的稳定制剂,其中所述I2S蛋白质包括氨基酸序列SEQ ID NO:1。
5.前述权利要求中任意一项所述的稳定制剂,其中所述盐是NaCl。
6.权利要求5所述的稳定制剂,其中所述NaCl存在的浓度范围从大约0-300mM。
7.权利要求4所述的稳定制剂,其中所述NaCl存在的浓度范围从大约137-154mM。
8.权利要求7所述的稳定制剂,其中所述NaCl存在的浓度大约154mM。
9.前述权利要求中任意一项所述的稳定制剂,其中所述聚山梨醇酯表面活性剂选自:聚山梨醇酯20、聚山梨醇酯40、聚山梨醇酯60、聚山梨醇酯80及其组合。
10.权利要求9所述的稳定制剂,其中所述聚山梨醇酯表面活性剂选自为聚山梨醇酯20。
11.如权利要求10所述的稳定制剂,其中所述聚山梨醇酯20存在的浓度范围大约0-0.02%。
12.如权利要求11所述的稳定制剂,其中所述聚山梨醇酯20以大约0.005%的浓度存在。
13.前述权利要求中任意一项所述的稳定制剂,其中所述制剂进一步包括缓冲剂。
14.如权利要求13所述的稳定制剂,其中所述缓冲剂选自:磷酸盐、醋酸盐、组氨酸、琥珀酸盐、Tris、及其组合。
15.如权利要求14所述的稳定制剂,其中所述缓冲剂为磷酸盐。
16.如权利要求15所述的稳定制剂,其中所述磷酸盐存在的浓度不大于50mM。
17.如权利要求13所述的稳定制剂,其中所述磷酸盐存在的浓度不大于20mM。
18.前述权利要求中任意一项所述的稳定制剂,其中所述制剂具有pH大约3-8。
19.如权利要求18所述的稳定制剂,其中所述制剂具有pH大约5.5-6.5。
20.如权利要求19所述的稳定制剂,其中所述制剂具有pH大约6.0。
21.前述权利要求中任意一项所述的稳定制剂,其中所述制剂为液体制剂。
22.权利要求1-20任意一项所述稳定制剂,其中所述制剂配制成冻干干粉。
23.鞘内施用的稳定制剂,所述稳定制剂包括:浓度范围从大约1-300mg/ml的艾杜糖醛酸-2-硫酸酯酶(I2S)蛋白质,在大约154mM浓度的NaCl,在大约0.005%浓度的聚山梨醇酯20,以及大约6.0的pH。
24.权利要求23所述的所述稳定制剂,其中所述I2S蛋白质的浓度大约10mg/ml。
25.权利要求23所述的所述稳定制剂,其中所述I2S蛋白质的浓度大约30mg/ml、50mg/ml、100mg/ml,或者300mg/ml。
26.容器,包括单剂型稳定制剂,所述稳定制剂为根据权利要求1-25任意一项所述的稳定制剂。
27.权利要求26所述的容器,其中所述容器选自:安瓿、小药瓶、药筒、贮液囊、二室注射器给药系统或者预填充的注射器。
28.权利要求26-28任意一项所述的容器,其中所述容器为预填充的注射器。
29.权利要求28所述的容器,其中所述预填充的注射器选自:具有烘干的硅树脂涂层的硼硅酸盐玻璃注射器,喷涂硅树脂的硼硅酸盐玻璃注射器,或者无硅树脂的塑料树脂注射器。
30.权利要求27所述的容器,其中所述稳定制剂存在的体积小于约5.0mL。
31.权利要求27所述的容器,其中所述稳定制剂存在的体积小于约3.0mL。
32.治疗Hunters综合症的方法,包括步骤:向需要治疗的受试者鞘内施用根据权利要求1-20任意一项所述的制剂。
33.治疗Hunters综合症的方法,包括步骤:向需要治疗的受试者鞘内施用制剂,所述制剂包括:浓度范围从大约1-300mg/ml的艾杜糖醛酸-2-硫酸酯酶(I2S)蛋白质,在大约154mM浓度的NaCl,在大约0.005%浓度的聚山梨醇酯20,以及大约6的pH。
34.权利要求32或者33所述的方法,其中所述鞘内施用的步骤在所述受试者中没有产生实质性副作用。
35.权利要求34所述的方法,其中所述鞘内施用在所述受试者中没有产生实质性适应性T-细胞介导的免疫应答。
36.权利要求32-35任意一项所述的方法,其中所述制剂的所述鞘内施用导致递送所述I2S蛋白质至靶脑组织。
37.权利要求36所述的方法,其中所述脑靶组织包括白质和/或在灰质中的神经元。
38.权利要求36或者37所述的方法,其中所述I2S蛋白质被递送至神经元、胶质细胞、血管周细胞和/或脑膜细胞。
39.权利要求37-39任意一项所述的方法,其中所述I2S蛋白质被进一步递送至在所述脊髓中的神经元。
40.权利要求37-39任意一项所述的方法,其中所述制剂的所述鞘内施用进一步导致所述I2S蛋白质在外周靶组织中的全身递送。
41.权利要求33所述的方法,其中所述外周靶组织选自:肝脏、肾脏、和/或心脏。
42.权利要求32-41任意一项所述的方法,其中所述制剂的所述鞘内施用导致在脑靶组织、脊髓神经元和/或外周靶组织中的溶酶体定位。
43.权利要求32-42任意一项所述的方法,其中所述制剂的所述鞘内施用导致GAG储存在所述脑靶组织、脊髓神经元和/或外周靶组织中减少。
44.根据权利要求43所述的方法,其中所述GAG储存与对照相比,以少20%、40%、50%、60%、80%、90%、1-倍、1.5-倍,或者2-倍地减少。
45.权利要求32-44任意一项所述的方法,其中所述制剂的所述鞘内施用导致在神经元中液泡化减少。
46.权利要求45所述的方法,其中所述神经元包括浦肯野细胞。
47.权利要求32-35任意一项所述的方法,其中所述制剂的所述鞘内施用导致I2S酶活性在所述脑靶组织、脊髓神经元和/或外周靶组织中增加。
48.权利要求47所述的方法,其中所述I2S酶活性与对照相比,以至少1-倍、2-倍、3-倍、4-倍、5-倍、6-倍、7-倍、8-倍、9-倍或者10-倍增加。
49.权利要求47或者48所述的方法,其中所述增加的I2S酶活性微至少大约10nmol/hr/mg,20nmol/hr/mg,40nmol/hr/mg,50nmol/hr/mg,60nmol/hr/mg,70nmol/hr/mg,80nmol/hr/mg,90nmol/hr/mg,100nmol/hr/mg,150nmol/hr/mg,200nmol/hr/mg,250nmol/hr/mg,300nmol/hr/mg,350nmol/hr/mg,400nmol/hr/mg,450nmol/hr/mg,500nmol/hr/mg,550nmol/hr/mg或者600nmol/hr/mg。
50.权利要求47所述的方法,其中所述I2S酶活性在所述腰椎区域中增加。
51.权利要求50所述的方法,其中在所述腰椎区域中,所述增加的I2S酶活性为至少大约2000nmol/hr/mg,3000nmol/hr/mg,4000nmol/hr/mg,5000nmol/hr/mg,6000nmol/hr/mg,7000nmol/hr/mg,8000nmol/hr/mg,9000nmol/hr/mg或者10,000nmol/hr/mg。
52.权利要求32-51任意一项所述的方法,其中所述制剂的所述鞘内施用导致Hunters综合症的至少一种症状或者特征在强度、严重程度或者频率方面减少,或者延缓发生。
53.权利要求32所述的方法,其中所述Hunters综合症的至少一种症状或者特征为认知障碍;白质病变;在所述脑实质中扩大的血管周围间隙、神经节、胼胝体、和/或脑干;萎缩症;和/或巨脑室。
54.权利要求32-51任意一项所述的方法,其中所述鞘内施用的发生每两个星期一次。
55.权利要求32-51任意一项所述的方法,其中所述鞘内施用的发生每月一次。
56.权利要求32-51任意一项所述的方法,其中所述鞘内施用的发生每两个月一次。
57.权利要求32-56任意一项所述的方法,其中所述鞘内施用被用于连同静脉施用。
58.权利要求57所述的方法,其中所述静脉施用没有比每月一次更频繁。
59.权利要求57所述的方法,其中所述静脉施用没有比每两个月一次更频繁。
60.权利要求32-56任意一项所述的方法,其中所述鞘内施用在缺乏静脉施用时使用。
61.权利要求32-60所述的方法,其中所述鞘内施用在缺乏并发免疫抑制疗法时使用。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201610569581.XA CN106139133B (zh) | 2010-06-25 | 2011-06-25 | 艾杜糖醛酸-2-硫酸酯酶的cns递送的方法和组合物 |
Applications Claiming Priority (15)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US35885710P | 2010-06-25 | 2010-06-25 | |
| US61/358,857 | 2010-06-25 | ||
| US36078610P | 2010-07-01 | 2010-07-01 | |
| US61/360,786 | 2010-07-01 | ||
| US38786210P | 2010-09-29 | 2010-09-29 | |
| US61/387,862 | 2010-09-29 | ||
| US201161435710P | 2011-01-24 | 2011-01-24 | |
| US61/435,710 | 2011-01-24 | ||
| US201161442115P | 2011-02-11 | 2011-02-11 | |
| US61/442,115 | 2011-02-11 | ||
| US201161476210P | 2011-04-15 | 2011-04-15 | |
| US61/476,210 | 2011-04-15 | ||
| US201161495268P | 2011-06-09 | 2011-06-09 | |
| US61/495,268 | 2011-06-09 | ||
| PCT/US2011/041925 WO2011163649A2 (en) | 2010-06-25 | 2011-06-25 | Methods and compositions for cns delivery of iduronate-2-sulfatase |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201610569581.XA Division CN106139133B (zh) | 2010-06-25 | 2011-06-25 | 艾杜糖醛酸-2-硫酸酯酶的cns递送的方法和组合物 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN103179980A true CN103179980A (zh) | 2013-06-26 |
| CN103179980B CN103179980B (zh) | 2016-09-28 |
Family
ID=46888843
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201180040898.XA Active CN103179980B (zh) | 2010-06-25 | 2011-06-25 | 艾杜糖醛酸-2-硫酸酯酶的cns递送的方法和组合物 |
Country Status (24)
| Country | Link |
|---|---|
| US (1) | US8545837B2 (zh) |
| EP (3) | EP3964229A1 (zh) |
| JP (1) | JP6045492B2 (zh) |
| KR (5) | KR20130043165A (zh) |
| CN (1) | CN103179980B (zh) |
| AU (4) | AU2011270669B2 (zh) |
| CA (1) | CA2805448A1 (zh) |
| CY (2) | CY1122307T1 (zh) |
| DK (1) | DK2593131T3 (zh) |
| ES (1) | ES2754234T3 (zh) |
| HR (1) | HRP20191924T1 (zh) |
| IL (3) | IL284599B2 (zh) |
| LT (1) | LT2593131T (zh) |
| ME (1) | ME03649B (zh) |
| MX (2) | MX2013000322A (zh) |
| NZ (2) | NZ605863A (zh) |
| PL (2) | PL2593131T3 (zh) |
| RS (1) | RS59469B1 (zh) |
| RU (1) | RU2660348C2 (zh) |
| SI (1) | SI2593131T1 (zh) |
| TW (1) | TWI611808B (zh) |
| UA (1) | UA115649C2 (zh) |
| WO (1) | WO2011163649A2 (zh) |
| ZA (1) | ZA201300672B (zh) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN108430494A (zh) * | 2015-12-30 | 2018-08-21 | 株式会社绿十字 | 用于治疗亨特综合征的方法和组合物 |
| CN109152847A (zh) * | 2016-01-15 | 2019-01-04 | 桑格摩生物治疗股份有限公司 | 用于治疗神经疾病的方法和组合物 |
| CN110114078A (zh) * | 2016-12-28 | 2019-08-09 | Jcr制药股份有限公司 | 冷冻干燥制剂 |
Families Citing this family (39)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2399586A1 (en) | 2002-01-11 | 2011-12-28 | Jefferies, Dr., Wilfred | Use of P97 as an enzyme delivery system for the delivery of therapeutic lysosomal enzymes |
| EP2182980A4 (en) | 2007-07-27 | 2012-04-18 | Armagen Technologies Inc | METHOD AND COMPOSITIONS FOR INCREASED ALPHA IDURONIDASE ACTIVITY IN THE CNS |
| KR20130043165A (ko) | 2010-06-25 | 2013-04-29 | 샤이어 휴먼 지네틱 테라피즈 인크. | 이듀로네이트-2-설파타제의 cns 전달을 위한 방법들 및 조성물들 |
| PL2585104T3 (pl) | 2010-06-25 | 2020-01-31 | Shire Human Genetic Therapies, Inc. | Sposoby i kompozycje do dostarczania arylosulfatazy a do oun |
| MX354776B (es) * | 2010-06-25 | 2018-03-20 | Shire Human Genetic Therapies | Enzimas de reemplazo lisosomal para usarse en el tratamiento de enfermedades de almacenamiento lisosomal. |
| KR102211960B1 (ko) | 2010-06-25 | 2021-02-05 | 샤이어 휴먼 지네틱 테라피즈 인크. | 치료제들의 cns 전달 |
| BR112012033197A2 (pt) * | 2010-06-25 | 2017-06-06 | Shire Human Genetic Therapies | métodos e composições para liberação de cns de n-sulfatase de heparano. |
| US20160145589A1 (en) | 2011-06-24 | 2016-05-26 | Green Cross Corporation | Composition and formulation comprising recombinant human iduronate-2-sulfatase and preparation method thereof |
| CN103747807B (zh) | 2011-07-05 | 2016-12-07 | 比奥阿赛斯技术有限公司 | P97‑抗体缀合物和使用方法 |
| EP2793922B1 (en) | 2011-12-23 | 2019-10-30 | Shire Human Genetic Therapies, Inc. | Stable formulations for cns delivery of arylsulfatase a |
| ES2728447T3 (es) | 2011-12-23 | 2019-10-24 | Shire Human Genetic Therapies | Tratamiento del déficit cognitivo del síndrome de Hunter para la administración intratecal de iduronato-2-sulfatasa |
| WO2013148277A1 (en) | 2012-03-30 | 2013-10-03 | Shire Human Genetic Therapies, Inc. | Subcutaneous administration of iduronate- 2-sulfatase |
| US20140004097A1 (en) | 2012-06-29 | 2014-01-02 | Shire Human Genetic Therapies, Inc. | Method of producing recombinant iduronate-2-sulfatase |
| US9150841B2 (en) | 2012-06-29 | 2015-10-06 | Shire Human Genetic Therapies, Inc. | Cells for producing recombinant iduronate-2-sulfatase |
| KR101380740B1 (ko) | 2012-06-29 | 2014-04-11 | 쉐어 휴먼 제네텍 세러피스, 인코포레이티드 | 이듀로네이트-2-설파타제의 정제 |
| EP2880156B1 (en) | 2012-07-31 | 2017-08-23 | biOasis Technologies Inc | Dephosphorylated lysosomal storage disease proteins and methods of use thereof |
| EA039246B1 (ru) * | 2012-12-06 | 2021-12-22 | Шир Хьюман Дженетик Терапис, Инк. | Способ лечения когнитивных нарушений, связанных с синдромом хантера |
| AU2014243816B2 (en) | 2013-03-13 | 2019-01-31 | Bioasis Technologies Inc. | Fragments of p97 and uses thereof |
| JP2016523835A (ja) * | 2013-05-15 | 2016-08-12 | リージェンツ オブ ザ ユニバーシティ オブ ミネソタ | 中枢神経系へのアデノ随伴ウイルスを介した遺伝子導入 |
| WO2015117121A1 (en) | 2014-02-03 | 2015-08-06 | Bioasis Technologies, Inc. | P97 fusion proteins |
| DK3107562T3 (da) | 2014-02-19 | 2019-12-16 | Bioasis Technologies Inc | P97-ids-fusionsproteiner |
| JP6847664B2 (ja) | 2014-05-01 | 2021-03-24 | バイオアシス テクノロジーズ インコーポレイテッド | P97−ポリヌクレオチド複合体 |
| MA40954A (fr) | 2014-11-14 | 2017-09-19 | Shire Human Genetic Therapies | Détermination de niveaux de glycosaminoglycane par spectrométrie de masse |
| AU2016256895B2 (en) | 2015-05-07 | 2022-05-26 | Takeda Pharmaceutical Company Limited | Glucocerebrosidase gene therapy for parkinson's disease |
| EP4000631A1 (en) * | 2015-05-15 | 2022-05-25 | REGENXBIO Inc. | Adeno-associated for therapeutic delivery to central nervous system |
| KR20170004814A (ko) * | 2015-07-02 | 2017-01-11 | 주식회사 녹십자 | 헌터증후군 치료제 |
| WO2017003270A1 (ko) * | 2015-07-02 | 2017-01-05 | 주식회사 녹십자 | 헌터증후군 치료제 및 치료방법 |
| AU2017220100B2 (en) * | 2016-02-17 | 2024-04-04 | Takeda Pharmaceutical Company Limited | Methods and compositions for CNS delivery of arylsulfatase A |
| CA3015358A1 (en) * | 2016-02-24 | 2017-08-31 | Biomarin Pharmaceutical Inc. | Targeted therapeutic lysosomal enzyme fusion proteins, associated formulations and uses thereof |
| SG10201912761UA (en) | 2016-04-15 | 2020-02-27 | The Trustees Of The Univ Of Pennsyvania | Gene therapy for treating mucopolysaccharidosis type ii |
| UY37679A (es) * | 2017-04-14 | 2018-11-30 | Regenxbio Inc | Tratamiento de mucopolisacaridosis ii con iduronato-2-sulfatasa (ids) humana recombinante producida por células neurales o gliales humanas |
| SG11202002457RA (en) | 2017-09-22 | 2020-04-29 | Univ Pennsylvania | Gene therapy for treating mucopolysaccharidosis type ii |
| TW201925236A (zh) | 2017-10-02 | 2019-07-01 | 美商戴納立製藥公司 | 包含酶替代療法酶之融合蛋白 |
| JP7060235B2 (ja) * | 2018-05-18 | 2022-04-26 | 国立大学法人広島大学 | 変異型イズロン酸-2-スルファターゼの機能回復薬剤のスクリーニング方法 |
| CN112236164A (zh) * | 2018-05-30 | 2021-01-15 | 株式会社医用基因生物 | 用于通过侧脑室施用治疗亨特综合征的方法和组合物 |
| KR20210024082A (ko) | 2018-06-25 | 2021-03-04 | 제이씨알 파마 가부시키가이샤 | 단백질 함유 수성 액제 |
| KR20200047937A (ko) * | 2018-10-26 | 2020-05-08 | 재단법인 목암생명과학연구소 | Ids를 포함하는 융합 단백질 및 이의 용도 |
| JP7458404B2 (ja) | 2018-12-20 | 2024-03-29 | アーマジェン・インコーポレイテッド | イズロン酸-2-スルファターゼ免疫グロブリン融合タンパク質の精製 |
| BR112023016075A2 (pt) | 2021-02-10 | 2023-11-21 | Regenxbio Inc | Método para tratar hepatoesplenomegalia, método de distribuição de um raav que codifica hids, método para tratar um sujeito humano, método de tratamento de um sujeito humano, método para determinar a eficácia do tratamento, método para identificar um sujeito como tendo mps ii |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20020099025A1 (en) * | 1999-12-30 | 2002-07-25 | Heywood James A. | Treatment of neurological disorders |
| US20050048047A1 (en) * | 2003-08-29 | 2005-03-03 | Kakkis Emil D. | Delivery of therapeutic compounds to the brain and other tissues |
| WO2009073569A2 (en) * | 2007-11-30 | 2009-06-11 | Abbott Laboratories | Protein formulations and methods of making same |
Family Cites Families (32)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6118045A (en) | 1995-08-02 | 2000-09-12 | Pharming B.V. | Lysosomal proteins produced in the milk of transgenic animals |
| US6063378A (en) * | 1997-08-22 | 2000-05-16 | Seikagaku Corporation | Therapeutic agent for herniated intervertebral disc |
| JP4990434B2 (ja) | 1998-12-07 | 2012-08-01 | ジェンザイム・コーポレーション | ポンペ病の処置 |
| US6217552B1 (en) | 1999-03-01 | 2001-04-17 | Coaxia, Inc. | Medical device for selective intrathecal spinal cooling in aortic surgery and spinal trauma |
| US20020052311A1 (en) | 1999-09-03 | 2002-05-02 | Beka Solomon | Methods and compostions for the treatment and/or diagnosis of neurological diseases and disorders |
| US6642038B1 (en) | 1999-09-14 | 2003-11-04 | Genzyme Glycobiology Research Institute, Inc. | GlcNAc phosphotransferase of the lysosomal targeting pathway |
| US7396811B2 (en) | 2001-04-30 | 2008-07-08 | Zystor Therapeutics, Inc. | Subcellular targeting of therapeutic proteins |
| US7629309B2 (en) | 2002-05-29 | 2009-12-08 | Zystor Therapeutics, Inc. | Targeted therapeutic proteins |
| US7560424B2 (en) | 2001-04-30 | 2009-07-14 | Zystor Therapeutics, Inc. | Targeted therapeutic proteins |
| US20040005309A1 (en) | 2002-05-29 | 2004-01-08 | Symbiontics, Inc. | Targeted therapeutic proteins |
| AU2002347910A1 (en) | 2001-10-16 | 2003-04-28 | Symbiontics Inc. | Methods and compositions for targeting proteins across the blood brain barrier |
| US20030072761A1 (en) | 2001-10-16 | 2003-04-17 | Lebowitz Jonathan | Methods and compositions for targeting proteins across the blood brain barrier |
| EP2361631A1 (en) | 2002-04-25 | 2011-08-31 | Shire Human Genetic Therapies, Inc. | Treatment of alpha-galactosidase a deficiency |
| AU2003237314B2 (en) | 2002-05-29 | 2010-03-04 | Biomarin Pharmaceutical Inc. | Targeted therapeutic proteins |
| ATE521701T1 (de) | 2003-01-22 | 2011-09-15 | Univ Duke | Verbesserte konstrukte zur expression lysosomaler polypeptide |
| PT2332567E (pt) * | 2003-01-31 | 2015-09-23 | Sinai School Medicine | Terapia combinada para o tratamento de distúrbios de deficiência proteica |
| WO2005074888A2 (en) | 2004-02-03 | 2005-08-18 | Biodelivery Sciences International, Inc. | Replacement enzyme cochleates |
| US20080014188A1 (en) | 2004-02-06 | 2008-01-17 | Zankel Todd C | Manufacture of Highly Phosphorylated Lysosomal Enzymes and Uses Thereof |
| CA2553955C (en) | 2004-02-10 | 2012-08-28 | Zystor Therapeutics, Inc. | Acid alpha-glucosidase and fragments thereof |
| US20050208090A1 (en) | 2004-03-18 | 2005-09-22 | Medtronic, Inc. | Methods and systems for treatment of neurological diseases of the central nervous system |
| WO2006121199A1 (ja) | 2005-05-11 | 2006-11-16 | Oxygenix Co., Ltd. | 脂質小胞体組成物 |
| AR059089A1 (es) | 2006-01-20 | 2008-03-12 | Genzyme Corp | Administracion intraventricular de una enzima para enfermedades de almacenamiento lisosomal |
| GB0611463D0 (en) | 2006-06-09 | 2006-07-19 | Novartis Ag | Organic compounds |
| WO2008109677A2 (en) | 2007-03-06 | 2008-09-12 | Saint Louis University | Modified enzyme and treatment method |
| US7722865B2 (en) | 2008-01-18 | 2010-05-25 | Biomarin Pharmaceutical Inc. | Manufacture of active highly phosphorylated human lysosomal sulfatase enzymes and uses thereof |
| EP3272773B8 (en) | 2008-05-07 | 2020-11-18 | BioMarin Pharmaceutical Inc. | Lysosomal targeting peptides and uses thereof |
| EP2588132A4 (en) | 2010-06-25 | 2014-10-15 | Shire Human Genetic Therapies | METHOD AND COMPOSITIONS FOR DELIVERING BETA GALACTOCEREBROSIDASE INTO THE CNS |
| BR112012033197A2 (pt) | 2010-06-25 | 2017-06-06 | Shire Human Genetic Therapies | métodos e composições para liberação de cns de n-sulfatase de heparano. |
| KR102211960B1 (ko) | 2010-06-25 | 2021-02-05 | 샤이어 휴먼 지네틱 테라피즈 인크. | 치료제들의 cns 전달 |
| PL2585104T3 (pl) | 2010-06-25 | 2020-01-31 | Shire Human Genetic Therapies, Inc. | Sposoby i kompozycje do dostarczania arylosulfatazy a do oun |
| KR20130043165A (ko) | 2010-06-25 | 2013-04-29 | 샤이어 휴먼 지네틱 테라피즈 인크. | 이듀로네이트-2-설파타제의 cns 전달을 위한 방법들 및 조성물들 |
| JP2012062312A (ja) * | 2010-08-19 | 2012-03-29 | Yoshikatsu Eto | ハンター症候群の治療剤 |
-
2011
- 2011-06-25 KR KR1020137001749A patent/KR20130043165A/ko active Application Filing
- 2011-06-25 AU AU2011270669A patent/AU2011270669B2/en active Active
- 2011-06-25 UA UAA201214667A patent/UA115649C2/uk unknown
- 2011-06-25 PL PL11799036T patent/PL2593131T3/pl unknown
- 2011-06-25 MX MX2013000322A patent/MX2013000322A/es active IP Right Grant
- 2011-06-25 RU RU2012154575A patent/RU2660348C2/ru active
- 2011-06-25 CA CA2805448A patent/CA2805448A1/en not_active Abandoned
- 2011-06-25 PL PL19192679T patent/PL3626258T3/pl unknown
- 2011-06-25 NZ NZ605863A patent/NZ605863A/en unknown
- 2011-06-25 DK DK11799036.6T patent/DK2593131T3/da active
- 2011-06-25 NZ NZ702808A patent/NZ702808A/en unknown
- 2011-06-25 KR KR1020227002252A patent/KR102603430B1/ko active IP Right Grant
- 2011-06-25 EP EP21188176.8A patent/EP3964229A1/en active Pending
- 2011-06-25 EP EP19192679.9A patent/EP3626258B1/en active Active
- 2011-06-25 SI SI201131775T patent/SI2593131T1/sl unknown
- 2011-06-25 KR KR1020197001325A patent/KR102151889B1/ko active Application Filing
- 2011-06-25 CN CN201180040898.XA patent/CN103179980B/zh active Active
- 2011-06-25 LT LT11799036T patent/LT2593131T/lt unknown
- 2011-06-25 US US13/168,966 patent/US8545837B2/en active Active
- 2011-06-25 IL IL284599A patent/IL284599B2/en unknown
- 2011-06-25 ES ES11799036T patent/ES2754234T3/es active Active
- 2011-06-25 RS RSP20191356 patent/RS59469B1/sr unknown
- 2011-06-25 EP EP11799036.6A patent/EP2593131B1/en active Active
- 2011-06-25 ME MEP-2019-300A patent/ME03649B/me unknown
- 2011-06-25 KR KR1020207024827A patent/KR102356192B1/ko active IP Right Grant
- 2011-06-25 KR KR1020237039076A patent/KR20230159646A/ko not_active Application Discontinuation
- 2011-06-25 MX MX2020001395A patent/MX2020001395A/es unknown
- 2011-06-25 WO PCT/US2011/041925 patent/WO2011163649A2/en active Application Filing
- 2011-06-25 JP JP2013516845A patent/JP6045492B2/ja active Active
- 2011-06-27 TW TW100122531A patent/TWI611808B/zh active
-
2012
- 2012-12-18 IL IL223716A patent/IL223716B/en active IP Right Grant
-
2013
- 2013-01-25 ZA ZA2013/00672A patent/ZA201300672B/en unknown
-
2017
- 2017-04-04 AU AU2017202219A patent/AU2017202219B2/en active Active
-
2019
- 2019-05-20 AU AU2019203515A patent/AU2019203515B2/en active Active
- 2019-07-15 IL IL268085A patent/IL268085B/en unknown
- 2019-10-22 HR HRP20191924TT patent/HRP20191924T1/hr unknown
- 2019-11-19 CY CY20191101211T patent/CY1122307T1/el unknown
-
2021
- 2021-04-27 AU AU2021202579A patent/AU2021202579B2/en active Active
- 2021-10-20 CY CY20211100914T patent/CY1124703T1/el unknown
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20020099025A1 (en) * | 1999-12-30 | 2002-07-25 | Heywood James A. | Treatment of neurological disorders |
| US20050048047A1 (en) * | 2003-08-29 | 2005-03-03 | Kakkis Emil D. | Delivery of therapeutic compounds to the brain and other tissues |
| WO2009073569A2 (en) * | 2007-11-30 | 2009-06-11 | Abbott Laboratories | Protein formulations and methods of making same |
Non-Patent Citations (1)
| Title |
|---|
| ZAHRA SHAHROKH 等: "INTRATHECAL DELIVERY OF PROTEIN THERAPEUTICS TO TREAT GENETIC DISEASES INVOLVING THE CNS", 《INJECTABLE DRUG DELIVERY 2010: FORMULATIONS FOCUS》, 31 May 2010 (2010-05-31), pages 16 - 20, XP055074790 * |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN108430494A (zh) * | 2015-12-30 | 2018-08-21 | 株式会社绿十字 | 用于治疗亨特综合征的方法和组合物 |
| CN109152847A (zh) * | 2016-01-15 | 2019-01-04 | 桑格摩生物治疗股份有限公司 | 用于治疗神经疾病的方法和组合物 |
| CN110114078A (zh) * | 2016-12-28 | 2019-08-09 | Jcr制药股份有限公司 | 冷冻干燥制剂 |
| CN110114078B (zh) * | 2016-12-28 | 2024-01-30 | Jcr制药股份有限公司 | 冷冻干燥制剂 |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN103179980B (zh) | 艾杜糖醛酸-2-硫酸酯酶的cns递送的方法和组合物 | |
| US11260112B2 (en) | Methods and compositions for CNS delivery of iduronate-2-sulfatase | |
| CN103282046B (zh) | 芳基硫酸酯酶 a cns 递送的方法和组合物 | |
| CN103260637B (zh) | 乙酰肝素n-硫酸酯酶cns递送的方法和组合物 | |
| CN103096918B (zh) | 治疗试剂的cns递送 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| TR01 | Transfer of patent right |
Effective date of registration: 20230329 Address after: Osaka, Japan Patentee after: TAKEDA PHARMACEUTICAL Co.,Ltd. Address before: Massachusetts Patentee before: SHIRE HUMAN GENETIC THERAPIES, Inc. |
|
| TR01 | Transfer of patent right |