CN102007108A - 趋化因子受体CxCR3的抑制剂 - Google Patents
趋化因子受体CxCR3的抑制剂 Download PDFInfo
- Publication number
- CN102007108A CN102007108A CN2009801138321A CN200980113832A CN102007108A CN 102007108 A CN102007108 A CN 102007108A CN 2009801138321 A CN2009801138321 A CN 2009801138321A CN 200980113832 A CN200980113832 A CN 200980113832A CN 102007108 A CN102007108 A CN 102007108A
- Authority
- CN
- China
- Prior art keywords
- compound according
- replaces
- resin
- dmf
- compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- SRAWVJVPKDOSJQ-UHFFFAOYSA-N CC(Cc(cc1)ccc1Cl)NC(c(cc1)cc(NC(c2ccc[s]2)=O)c1N1CCN(CCN(C)C)CC1)=O Chemical compound CC(Cc(cc1)ccc1Cl)NC(c(cc1)cc(NC(c2ccc[s]2)=O)c1N1CCN(CCN(C)C)CC1)=O SRAWVJVPKDOSJQ-UHFFFAOYSA-N 0.000 description 1
- RLWKNHICPQLFSY-UHFFFAOYSA-N CCN(CC1)CCN1c(ccc(C(NCCc(ccc(Cl)c1)c1Cl)=O)c1)c1NC(c1cc(Cl)ccc1)=O Chemical compound CCN(CC1)CCN1c(ccc(C(NCCc(ccc(Cl)c1)c1Cl)=O)c1)c1NC(c1cc(Cl)ccc1)=O RLWKNHICPQLFSY-UHFFFAOYSA-N 0.000 description 1
- GZMWWZBSMGACGA-UHFFFAOYSA-N CN(C)CCC(N(CC1)CCN1c(ccc(C(NCCc(ccc(Cl)c1)c1Cl)=O)c1)c1NC(c1cccc(C(O)=O)c1)=O)=O Chemical compound CN(C)CCC(N(CC1)CCN1c(ccc(C(NCCc(ccc(Cl)c1)c1Cl)=O)c1)c1NC(c1cccc(C(O)=O)c1)=O)=O GZMWWZBSMGACGA-UHFFFAOYSA-N 0.000 description 1
- AIRYGLYMGRNXLK-UHFFFAOYSA-N CN(C)CCN(CC1)CCN1c(ccc(C(NCCc(cccc1)c1Cl)=O)c1)c1NC(c1cc(Cl)ccc1)=O Chemical compound CN(C)CCN(CC1)CCN1c(ccc(C(NCCc(cccc1)c1Cl)=O)c1)c1NC(c1cc(Cl)ccc1)=O AIRYGLYMGRNXLK-UHFFFAOYSA-N 0.000 description 1
- AHTBAGDMSBHMMO-UHFFFAOYSA-N CN(C)CCN(CC1)CCN1c(ccc(C(NCCc(cccc1)c1F)=O)c1)c1NC(c1cc(Cl)ccc1)=O Chemical compound CN(C)CCN(CC1)CCN1c(ccc(C(NCCc(cccc1)c1F)=O)c1)c1NC(c1cc(Cl)ccc1)=O AHTBAGDMSBHMMO-UHFFFAOYSA-N 0.000 description 1
- FIEKCBNMHDCYHV-UHFFFAOYSA-N CN(C)CCN(CC1)CCN1c(ccc(C(NCCc1cccc(Cl)c1)=O)c1)c1N Chemical compound CN(C)CCN(CC1)CCN1c(ccc(C(NCCc1cccc(Cl)c1)=O)c1)c1N FIEKCBNMHDCYHV-UHFFFAOYSA-N 0.000 description 1
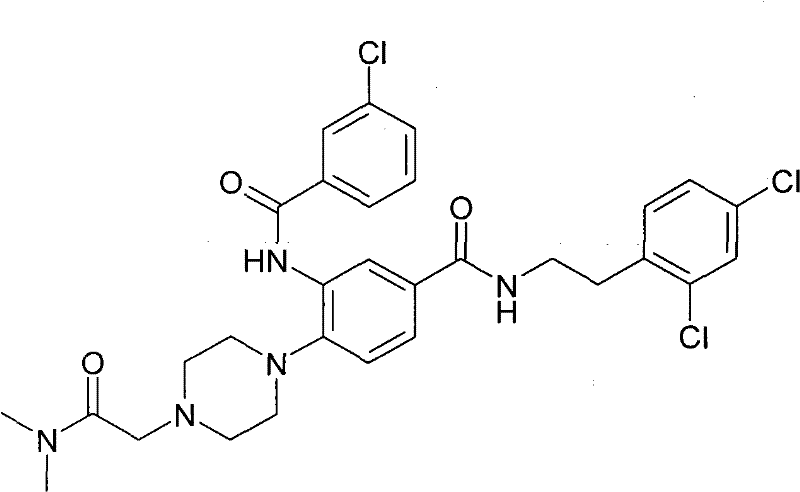
- AQYKMPWZWGIMAZ-LOSJGSFVSA-N CN(C)CCN(CC1)CCN1c(ccc(C(N[C@H](C1)[C@@H]1c1ccccc1)=O)c1)c1NC(c1c[s]cc1)=O Chemical compound CN(C)CCN(CC1)CCN1c(ccc(C(N[C@H](C1)[C@@H]1c1ccccc1)=O)c1)c1NC(c1c[s]cc1)=O AQYKMPWZWGIMAZ-LOSJGSFVSA-N 0.000 description 1
- REZRGVCSIGDUCQ-UHFFFAOYSA-N CN(CC1)CCC1N(CC1)CCN1c(ccc(C(NCCc(ccc(Cl)c1)c1Cl)=O)c1)c1NC(c1cc(Cl)ccc1)=O Chemical compound CN(CC1)CCC1N(CC1)CCN1c(ccc(C(NCCc(ccc(Cl)c1)c1Cl)=O)c1)c1NC(c1cc(Cl)ccc1)=O REZRGVCSIGDUCQ-UHFFFAOYSA-N 0.000 description 1
- KKPGZOBSPZNVBF-UHFFFAOYSA-N COCCN(CC1)CCN1c(ccc(C(NCCc(ccc(Cl)c1)c1Cl)=O)c1)c1NC(c1cccc(Cl)c1)=O Chemical compound COCCN(CC1)CCN1c(ccc(C(NCCc(ccc(Cl)c1)c1Cl)=O)c1)c1NC(c1cccc(Cl)c1)=O KKPGZOBSPZNVBF-UHFFFAOYSA-N 0.000 description 1
- HGILWWCUMUZSIH-UHFFFAOYSA-N O=C(CCN1CC1)N(CC1)CCN1c(ccc(C(NCCc(ccc(Cl)c1)c1Cl)=O)c1)c1NC(c1cc(Cl)ccc1)=O Chemical compound O=C(CCN1CC1)N(CC1)CCN1c(ccc(C(NCCc(ccc(Cl)c1)c1Cl)=O)c1)c1NC(c1cc(Cl)ccc1)=O HGILWWCUMUZSIH-UHFFFAOYSA-N 0.000 description 1
- HMXBPLQWGXADLM-UHFFFAOYSA-N O=C(c(cc1)cc(NC(c2cc(Cl)ccc2)=O)c1N1CCC2(CNCC2)CC1)NCCc(ccc(Cl)c1)c1Cl Chemical compound O=C(c(cc1)cc(NC(c2cc(Cl)ccc2)=O)c1N1CCC2(CNCC2)CC1)NCCc(ccc(Cl)c1)c1Cl HMXBPLQWGXADLM-UHFFFAOYSA-N 0.000 description 1
- VXKVXJNXOWZNQL-UHFFFAOYSA-N O=C(c(cc1)cc(NC(c2cccc(Cl)c2)=O)c1N(CC1)CCC1N1CCCCC1)NCCc(cc1)ccc1Cl Chemical compound O=C(c(cc1)cc(NC(c2cccc(Cl)c2)=O)c1N(CC1)CCC1N1CCCCC1)NCCc(cc1)ccc1Cl VXKVXJNXOWZNQL-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/02—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms containing only hydrogen and carbon atoms in addition to the ring hetero elements
- C07D295/027—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms containing only hydrogen and carbon atoms in addition to the ring hetero elements containing only one hetero ring
- C07D295/033—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms containing only hydrogen and carbon atoms in addition to the ring hetero elements containing only one hetero ring with the ring nitrogen atoms directly attached to carbocyclic rings
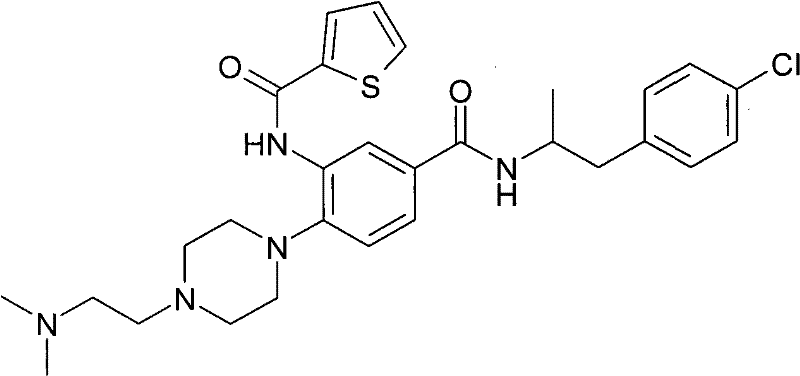
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains three hetero rings
- C07D471/20—Spiro-condensed systems
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Life Sciences & Earth Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Pulmonology (AREA)
- Dermatology (AREA)
- Rheumatology (AREA)
- Heart & Thoracic Surgery (AREA)
- Physical Education & Sports Medicine (AREA)
- Biomedical Technology (AREA)
- Cardiology (AREA)
- Pain & Pain Management (AREA)
- Urology & Nephrology (AREA)
- Neurosurgery (AREA)
- Immunology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Vascular Medicine (AREA)
- Neurology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Furan Compounds (AREA)
- Hydrogenated Pyridines (AREA)
- Pyridine Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Pyrrole Compounds (AREA)
- Heterocyclic Compounds Containing Sulfur Atoms (AREA)
Abstract
本发明涉及一种如本文所定义的3-(酰氨基或磺酰氨基)-4-(4-取代的-吖嗪基)苯甲酰胺或苯磺酰胺化合物。此3-(酰氨基或磺酰氨基)-(4-取代的-吖嗪基)苯甲酰胺或苯磺酰胺化合物可作为趋化因子受体CxCR3的抑制剂使用,并可为需要的患者预防或治疗由CxCR3趋化因子受体介导的疾病或与其相关的病症。
Description
本发明的领域
本发明涉及如本文所定义的3-(酰氨基(amido)或磺酰氨基(sulphamido))-4-(4-取代的-吖嗪基(azinyl))苯甲酰胺化合物。3-(酰氨基或磺酰氨基)-(4-取代的-吖嗪基)苯甲酰胺化合物可作为趋化因子受体CxCR3的抑制剂使用,并可为需要的患者预防或治疗由CxCR3趋化因子受体介导的疾病或与其相关的病症。
本发明的背景
慢性炎症性疾病是一种普遍的医学问题,而且它们的发病率随着衰老而增加。最容易识别的与衰老相关的炎症性疾病是关节炎,这是一种包括明显障碍的综合征概念,如骨关节炎(例如膝盖骨关节炎)和类风湿性关节炎(例如手指关节)。除了关节炎以外,还有以较不明显的发炎症状出现的其他疾病。这些疾病包括慢性阻塞性肺病(COPD)(这是一种主要包括肺气肿、慢性支气管炎和哮喘的综合征名称,是一种反复发作型神经功能障碍)、多发性硬化症(MS)、动脉硬化、牛皮癣(一种脱屑皮肤症状),以及炎症性肠疾病(IBD),如克罗恩氏病(Crohn’s disease)和溃疡性结肠炎,这两者都是分别影响肠道上部和下部的虚弱性症状。而且,溃疡性结肠炎是一种可演变为结肠直肠癌的危险因素,在工业人群中是引起致命疾病和死亡的主要原因。由于在视觉之外,一般人通常不知道这些症状在本质上是属于炎症,但病理学家们早就证实了上述每种疾病的炎症过程。在所有这些疾病中,T淋巴细胞是起作用的重要因素(Westermann,等人,Ann.Intern.Med.2001,135,279)。许多其他疾病涉及炎症现象,但在此不予详述。
自古以来就已经知道炎症的主要特征,罗马医生将其归纳为四种基本迹象,即经身体检查可明显察觉的“四个or”:疼痛(dolor)、发红(rubor)、发热(calor)和肿瘤(tumor)。Dolor是疼痛,在检查患者时这是显而易见的。Rubor是受影响部分的红色外观,这是血管扩张和发炎组织灌注增加而引起的充血。这是身体对代谢刺激的组织的需求所作出的反应。与基础状态相比,细胞和亚细胞成分进出炎症区域的流量急剧增加,而且在能量和营养需要方面需付出显著代价才能痊愈。受影响部位摸起来比周围区域热,这是一种因组织损伤而引起的代谢负担的反映。这种上升的代谢需求和随之引起的供给则导致第三种迹象calor,即发热,因为组织摸起来是热的。最后,炎症的第四种基本迹象是肿瘤或肿胀。炎症过程的净结果是受感染部分物质量的增加。由于血管渗透性增加而使组织肿胀并充有体液,补充的血液提供了一组协调作用的细胞,如白细胞,继之是成纤维细胞,先对付受伤的起因,然后愈合受伤部位。
细胞组分进入炎症组织的运输以及它们在其中的行为是高度受控制和有序的。它并非是一种偶然的过程,对其细节的认识正越来越深。部分炎症组织的运输是促炎性信号分子汇成的一曲真正的交响曲。该组细胞是受某些因素如激素、细胞因子和趋化因子控制的,这些因素提供了治疗干预的机会。特异性白细胞亚群如T细胞的补充是由被称为趋化因子的小蛋白调控的。
趋化因子是参与炎症过程的各种因素中小蛋白激素的一个大家族(Alexander等人,Br.J.Pharmacol.,2007,150(Suppl.1)S25)。趋化因子一词是趋化细胞因子的缩略语,这掩盖了其作用。趋化因子向炎症区域吸引白细胞。趋化因子还有其他作用,如调节与细胞外蛋白的粘附、增殖和其他因子(如干扰素-γ)的分泌。
趋化因子调节不同过程(http://en.wikipedia.org/wiki/Chemokine)。趋化因子的一个主要作用是引导细胞迁移。细胞趋化性涉及细胞从趋化因子浓度低的区域向趋化因子浓度较高的区域移动。某些趋化因子在功能上似乎更是体内稳态的(homeostatic):例如,它们把淋巴细胞引向淋巴结,在那里它们通过在淋巴结内与抗原呈递细胞相互作用而参与免疫监视。某些趋化因子对发育有影响,如促进或抑制新血管的生长-血管生成和血管生成抑制(angiostatic)作用。这种体内稳态趋化因子似乎以日复一日的方式调节细胞的运输。其他的则在对损伤的应答中得到表现,这些炎症趋化因子通常对目标白细胞群具有化学吸引及其他作用。这些趋化因子通常是由各种不同类型细胞的白细胞介素-1或干扰素-γ诱导的。最后,许多趋化因子导致免疫细胞释放酶因子和其他因子。
趋化因子是大小约为8至10kDa的小型蛋白,按照其蛋白序列分层;根据表达在细胞表面的相关受体,不同的细胞群对它们作出响应(Pease和Williams,Br.J.Pharmacol.2006,147,S212)。迄今为止,已知至少有47种趋化因子,它们都有一个由C端α-螺旋覆盖的三个反平行β-折叠片组成的基本“希腊钥匙(Greek key)”蛋白折叠基序。这种蛋白折叠取决于保守的链内二硫键。形成这些桥梁的半胱氨酸是趋化因子命名的基础。在涉及二硫键桥的氨基端附近具有两个连续半胱氨酸的趋化因子被称为CC趋化因子,并具有CCL1至CCl28的系统名称。它们所结合的受体也以类似方式命名,如CCR1、CCR2等,尽管受体的编号与结合趋化因子是不一样的,而且每个受体可与多个趋化因子结合。
在氨基端头两个半胱氨酸之间带一个氨基酸的趋化因子被称为CxC趋化因子。这些激素及其受体的系统名称的形式是CxCL1至CxCL16,且同源受体(cognate receptors)是CxCR1至CxCR16。尽管其他的半胱氨酰基间距似乎也是可能的,下一类已知的趋化因子是Cx3CL1,称为CXXXC趋化分子(fractalkine),它与Cx3CR1受体结合。最后,在涉及二硫键的氨基端区域只有一个半胱氨酸的一类趋化因子至少有一个成员,称为XCL1、淋巴细胞趋化因子,其受体为XCR1。归纳起来,47个已知的趋化因子有18个已知的受体。
重要的是,现在剪接变体已被识别,可进一步细分可能存在的各种趋化因子和受体。对于CxCR3,有两种已知的剪接变体:CxCR3A和CxCR3B。趋化因子受体的长度为340-350个氨基酸,它们都是G蛋白偶联受体(GPCRs)。GPCR是各种治疗剂(包括小分子药物)靶向的一类重要蛋白。长期以来早就感到需要调节炎症,而众所周知GPCR可提供易控制的治疗干预目标。因此,通过使用趋化因子受体的调节剂,有很大希望可实现炎症过程的选择性调节。
CxCR3A受体主要在活化的Th1淋巴细胞上表达,但它也存在于NK(自然杀伤)细胞、巨噬细胞、DC(树突状细胞)和B淋巴细胞上。已知CxCR3A是受三种趋化因子刺激的:CxCL9(干扰素-γ诱导的单核因子,亦称Mig)、CxCL10(干扰素-γ可诱导的10kDa蛋白,亦称IP-10,以及CxCL11(干扰素可诱导的T细胞α-化学吸引因子,亦称I-TAC)。对这些趋化因子的血管生成抑制作用的观察预示了一种不同的受体亚型的可能性,而且确实发现了一种剪接变体。CxCR3A信号传输是由一种对百日咳毒素敏感的G蛋白(Gαi)介导的,它导致了一股钙离子流。经发现,剪接变体CxCR3B在内皮细胞上表达,并介导IP-10、Mig、I-TAC和血小板第4因子的血管生成抑制作用(Lasagni等人,J.Exp.Med.2003,197,1537)。血小板因子4号,待测序的第一种趋化因子,对CxCR3A无作用,且通过CxCR3B的信号传输导致由Gαs介导的cAMP的上升。
长期以来早就知道T淋巴细胞是作为免疫系统的指挥和控制中心起作用。确实,HIV通过选择性破坏T细胞群而导致AIDs。鉴于这一中心调控位置,18个趋化因子受体中有15个在不同的T淋巴细胞亚群中表达,这并不令人惊奇(Pease&Williams Br.J.Pharmacol.2006,147,S212)。
已知其他病原体会破坏趋化因子系统,以逃避免疫监视,抑制免疫反应,并避免消除。趋化因子的作用受到病原体以至少四种不同方式的损害:第一,通过产生作为受体拮抗剂起作用的趋化因子模拟物。第二,通过产生作为不适当激动剂起作用的趋化因子模拟物。第三,通过产生受体模拟物,第四,通过产生约束和中和趋化因子活性的蛋白(Chensue,Clin.Microbiol.Rev.2001,14,821)。CxCR3及其配体属于病原体侵占的因素。因此,大自然找到了利用趋化因子调节免疫系统功能的方式,本发明的主题也利用此方式。
生理系统控制元素的另一个重要性质是正调节输入和负调节输入的存在。经发现,这些整合的平衡点也在趋化因子之中。嗜酸细胞活化趋化因子(Eotaxin)/CCL11和MCP-3/CCL7,在刺激其他趋化因子受体(分别为CCR3和CCR1)的同时,拮抗CxCR3A和另外两个CCRs(CCR2和CCR5)的信号。重要的是,刺激CxCR3A(IP10/CxCL10、I-TAC/CxCL11和Mig/CxCL9)的趋化因子通过在嗜酸性粒细胞、嗜碱性粒细胞和肥大细胞上表达的CCR3来拮抗信号(Alexander等人,Br.J.Pharmacol.,2007,150(Suppl.1)S25)。因此,CxCR3A及其配体在它们是如何受到调节并调控其他趋化因子信号通路的行为方面是独特的。这种受体是在协调炎症的信号分子网络内一个高度相互关联的节点上。确实,CxCR3A和CCR3信号之间达到的平衡确定了炎症反应的方向,使它从Th2(CCR3)种类的抵抗寄生虫的变应性反应极化或向细胞介导的Th1(CxCR3A)型的反应极化。这解释了炎症细胞组分的“极化”(Loetscher,等人,J.Biol.Chem.2001,276,2986)。因此,长期以来早就感到需要发现能够干预这部分免疫反应的治疗剂。
CxCR3A表达的最高水准见于T细胞,它是Th1淋巴细胞的一个标志(Annunziato,等人,Microbes和Infection,1999,1,103;Lasagni等人,J.Exp.Med.2003,197,1537)。T细胞涉及许多疾病,此处按发病率从高至低依次排列:哮喘、格雷夫斯氏病(Grave’s disease)、类风湿性关节炎、特应性皮炎、斯耶格伦氏综合征(Sjogren’s syndrome)、全身性红斑狼疮、多发性硬化症、溃疡性结肠炎、I型糖尿病、克罗恩氏病(Crohn’s disease),肉样瘤病、原发性胆汁性肝硬变化、肾小球性肾炎、重症肌无力、颞动脉炎,以及同种异体器官移植排斥反应(Westermann,等人,Ann.Intern.Med.2001,135,279)。现在将通过几个实验药理学例子来说明CxCR3A调节剂的治疗潜力。
CxCR3及其配体与炎症性肠病相关。IP10的抗体在鼠类结肠炎模型中减少了炎症(Singh,等人,J.Immunol.2003,171,1401)。缺乏IL-10(IL-10-/-)的敲除小鼠自发地罹患类似于克罗恩病的结肠炎。在年龄约为12个星期时,这些小鼠的体重开始减轻,出现慢性腹泻且血清淀粉样蛋白A、IL-6以及其他6个细胞因子的循环浓度上升。用IP10-中和单克隆抗体进行治疗消除了所有这些影响。组织学检查也显示,该抗体显著地降低了淋巴细胞浸润结肠粘膜的程度。
在类风湿关节炎中,与从创伤性关节损伤或骨关节炎采集的样本相比,滑液中CxCR3A激动剂趋化因子浓度升高了100倍(Patel,等人,Clin.Immunol.,2001,98,39)。它们的浓度梯度也与从滑液至血浆由高至低的梯度一致,94%血管周围的T细胞在其表面膜上表达CxCR3A,且在血流中40%在其质膜上带有受体的T细胞中,这种频率被强化。这些结果符合CxCR3A-结合的趋化因子将Th1型T细胞的募集引向发炎关节的理论。因此,对于RA,某些作者认识到对CxCR3A信号进行干预的潜在价值(Houshmand&Zlotnik,Curr.Opinion Chem.Biol.2003,7,457;Proudfoot,Nature ReviewsImmunol.,2002,2,106)。
业已报导了关节炎模型中CxCR3A系统的小分子抑制剂。拮抗CCR5和CxCR3A(两者均选择性在Th1细胞上表达-Hashmand&Zlotnick)的Tak-779抑制小鼠胶原诱导的关节炎(CIA)的病理(Yang,等人,Eur.J.Immunol.2002,32,2124;和Gao,等人,J.Leukoc.Biol.2003,73,273)。胶原诱导的关节炎(CIA)模型是一种公认的小鼠急性关节炎模型,时程为26天。小鼠用胶原免疫,且在注射该增强剂的13天内,几乎所有动物的四肢关节都呈现红肿。这一经典的炎症图像是用TAK-779治疗CIA小鼠而除去的,TAK-779是最初作为阻止HIV感染的药物而开发的一种取代的苯并环庚烯的季胺盐。如组织学评价所确定,TAK-779还阻断了关节的白细胞浸润。虽然TAK-779最初是作为CCR5抑制剂治疗AIDS而开发的,但经竞争性放射配体结合、趋化性和细胞粘附分析测量,发现它也是一种具有类似效力的CxCR3A阻断剂。在Gao等人所研究的细胞类型中表达的其他两个趋化因子对这些参数没有影响。因此,TAK-779是一种针对CCR5和CxCR3A的双重抑制剂。
一种小分子化合物AMG-487,已在转移癌的鼠类模型中经过试验(Walser,等人,Cancer Res.,2006,66,7701)。有趣的是,肿瘤细胞往往异常地表达趋化因子或其受体。研究人员认为,这可能会促进转移性疾病的发展或影响其取向。CxCR3A在人类和小鼠的乳腺癌细胞株中表达,它们在功能上与钙反应关联,并向响应CxCR3A特异性趋化因子的那些细胞赋予趋化活性。在一种转移性乳腺癌的小鼠模型中,AMG-487减少了60%的转移。有趣的是,该化合物对增生现象并没有直接影响,强调了转移取向和增生是可分离的现象。
最后,最近的基因敲除小鼠实验预言了CxCR3抑制剂在治疗动脉硬化中的用途(Veillard,等人,Circulation 2005,112,870)。当给予高脂肪饮食时,缺乏ApoE基因的小鼠在主动脉里迅速出现动脉粥样硬化病变。当这些小鼠的CxCR3的基因被敲除时,胸腹主动脉里脂质沉积的程度从7.9%面积减少至4.5%。
因此,很有理由相信,CxCR3A的调节剂对于不同疾病患者的治疗很可能是有用的。
各种专利申请和已授权的专利披露了趋化因子或CxCR3的抑制剂。
WO 2002/085861、WO 2003/101970、美国专利7,067,662B2、美国专利6,124,319、WO 031070242A1和WO 2007/064553A2分别披露了具有如下结构式的化合物:



作为趋化因子受体抑制剂。
US 20070021611Al、US 20070054919A1、US 20070082913A1、WO2008008453A1以及WO2007/109238A1分别披露了具有如下结构式的化合物:



作为CxCR3抑制剂。所有这些化合物都具有(哌啶-4-基哌嗪-1-基)芳族部分核心结构。
WO 2007/002742A1披露了具有如下结构式的化合物:

该系列的代表性化合物如下所示:
Cole,等人,J.Bioorg.Med.Chem.Lett.16,2006,200-203一文提到,相对于如上所述的系列,需要七元高哌嗪(homopiperazine)环提供活性,因为哌嗪基类似物显示无活性。因此,研究者不同意高哌嗪环向哌嗪环转变的观点。
发明内容
本发明涉及具有式I的3-(酰氨基或磺酰氨基)-4-(4-取代的-吖嗪基)苯甲酰胺或苯磺酰胺化合物

其中
Q和Q1独立地为CO或SO2;
Y是CO或SO2;
X是卤素;
m是1、2或3;
R1是(任选取代的(芳族基或低级环基))低级烷基C1-3;
R2、R4和R6独立地为H或任选取代的低级烷基;
R3是任选取代的芳族基;
A是CH或N,或A和R5一起形成下式的4-7元螺环氮杂环基(spiroazaheterocyclyl)

n和p独立地为0、1、2、3、4或5,只要n和p≥2但≤5;
R5是JGZ、R8R7NQ1低级烷基或任选取代的3-7元氮杂环基;
Z是键、CO或SO2;
G是低级烷基、C3-7环烷基或3-7元杂环基;以及
J是芳族基、低级烷氧基羰基、低级烷基硫基(alkylthio)、低级烷基亚磺酰基(alkylsulfinyl)、低级烷基磺酰基、低级烷氧基、R8R7N或任选(低级烷基或卤素)取代的3-7元杂环基;
R7和R8独立地为H或低级烷基;
R9和R10独立地为H或低级烷基;或
其药学上可接受的盐、溶剂合物、N-氧化物、季铵衍生物或前药,或其任何组合。
本发明还涉及药物组合物,其含有药学上可接受量的式I的化合物,或其药学上可接受的盐、溶剂合物、N-氧化物、季铵衍生物或前药,或其任何组合,以及药学上可接受的添加剂。
本发明还涉及为需要的患者预防或治疗由CxCR3趋化因子受体介导的疾病或相关病症的方法,包括向患者给药药物有效量的式I的化合物,或其药学上可接受的盐、溶剂合物、N-氧化物、季铵衍生物或前药,或其任何组合。
本发明还涉及根据权利要求1所述的一种或多种化合物的用途,以制备用于为需要的患者治疗或预防CxCR3在其中起作用的病症的药物。
本发明还涉及药盒(kit)或药物包(pharmaceutical pack),用于为患者治疗或预防生理状况或疾病状态;该药盒或药物包含有多个单独的容器,其中至少一个所述容器含有一种或多种根据权利要求1所述的化合物(单独或与药学上可接受的载体组合),至少另一个所述容器含有一种或多种能够治疗或预防该生理状况或疾病状态的其他化合物。
发明详述
缩写词表
如上文所用及贯穿本发明的说明,下列缩写词应被理解为具有以下含义,除非另行说明:
DIC=二异丙基碳二亚胺
FMOC=9-芴基甲氧基羰基
TMOF=三甲基原甲酸酯(trimethylorthoformiate)
DIEA=二异丙基乙胺
NaBH3CN=氰基氢硼化钠
DMF:N,N-二甲基甲酰胺
THF:四氢呋喃
DMSO:二甲基亚砜
DCM:二氯甲烷,又可被称为甲叉二氯
NMP=N-甲基吡咯烷酮
MeOH=甲醇
HOAt=1-羟基-7-氮杂苯并三唑
HATU=二甲基氨基-([1,2,3]三唑并[4,5-b]吡啶-3-基氧基)-亚甲基]-二甲基六氟磷酸铵
HOAc=乙酸
AN=乙腈
TFA=三氟乙酸
HPLC=高效液相色谱法
LC/MS=与质谱结合的串联高效液相色谱
NMR=核磁共振光谱法
定义
如上文所用及贯穿本发明的说明,下列术语应被理解为具有以下含义,除非另行说明:
“酸保护基”意为本领域已知的易于除去并可选择性除去的基团,它可保护羧基上酸性的氢,以在合成步骤中抵抗不希望发生的反应,例如,当发生涉及该化合物的其他官能位点的反应时,它能阻断或保护该酸性官能团(functionality)。这样的酸保护基是本领域技术人员众所周知的,并被广泛地用于羧基的保护,如美国专利3,840,556和3,719,66所述。该专利的披露作为参考文献在此引用。欲了解适宜的酸保护基,可参阅T.W.Green和P.G.M.Wuts的“Protective Groups in Organic Chemistry”(有机化学中的保护基),JohnWiley and sons,1991。酸保护基还包括如本文所定义的氢化不稳定的酸保护基。代表性的酸保护基包括酯,如取代的和未取代的C1-8低级烷基,如甲基、乙基、叔丁基、甲氧基甲基、甲基硫基甲基(methylthiomethyl)、2,2,2-三氯乙基等等;四氢吡喃基、取代的和未取代的苯基烷基如苄基,及其取代的衍生物如烷氧基苄基或硝基苄基等等;肉桂基、二烷基氨基烷基,例如二甲基氨基乙基等等;三甲基甲硅烷基,取代的和未取代的酰胺和酰肼,例如N,N-二甲基胺、7-硝基吲哚、肼、N-苯基肼等上的酰胺和酰肼,酰氧基烷基如新戊酰氧基甲基或丙酰氧基甲基等等;芳酰氧基烷基如苯甲酰氧基乙基等等;烷氧基羰基烷基如甲氧基羰基甲基、环己基氧基羰基甲基等等;烷氧基羰基氧基烷基如叔丁氧基羰基氧基甲基等等;烷氧基羰基氨基烷基如叔丁氧基羰基氨基甲基等等;烷基氨基羰基氨基烷基,如甲基氨基羰基氨基甲基等等;酰基氨基烷基如乙酰基氨基甲基等等;杂环基羰基氧基烷基如4-甲基哌嗪基-羰基氧基甲基等等;二烷基氨基羰基烷基如二甲基氨基羰基甲基等等;(5-(低级烷基)-2-氧代-1,3-二氧杂环戊烯-4-基)烷基如(5-叔丁基-2-氧代-1,3-二氧杂环戊烯-4-基)甲基等等;以及(5-苯基-2-氧代-1,3-二氧杂环戊烯-4-基)烷基如(5-苯基-2-氧代-1,3-二氧杂环戊烯-4-基)甲基等等。
“酸不稳定的胺保护基”意为如本文所定义的胺保护基,它可经酸处理而容易地除去,同时对其他试剂维持相对稳定。一种优选的酸不稳定的胺保护基是BOC。
“酰基”意为H-CO-或(脂族基或环基)-CO-基,其中的脂族基如本文所述。优选的酰基含有低级烷基。代表性的酰基包括甲酰基、乙酰基、丙酰基、2-甲基丙酰基、丁酰基、棕榈酰基、丙烯酰基、丙炔酰基(propynoyl)、环己基羰基(cyclohexylcarbonyl),等等。
“链烯酰基”意为链烯基-CO-基,其中的链烯基如本文所定义。
“链烯基”意指含有一个碳碳双键及链上约2至约15个碳原子的直链或支链脂族烃基。优选的链烯基在链上含有2至约12个碳原子;更优选的是在链上含有约2至约4个碳原子。”支链的”意为一个或多个低级烷基如甲基、乙基或丙基与直链烯基链相连。”低级链烯基”意为在链上含有约2至约4个碳原子,它可以是直链或支链。代表性的链烯基包括乙烯基、丙烯基、正丁烯基、异丁烯基、3-甲基丁-2-烯基、正戊烯基、庚烯基、辛烯基、环己基丁烯基、癸烯基,等等。”取代的链烯基”意为如上所定义的链烯基,它被一个或多个相同或不同且如本文所定义的”脂族基取代基”(优选的是1至3个)取代。代表性的链烯基脂族基取代基包括卤素或环烷基。
“链烯氧基”意为链烯基-O-基,其中的链烯基如本文所述。代表性的链烯氧基包括烯丙氧基、3-丁烯氧基,等等。
“烷氧基”意为烷基-O-基,其中的烷基如本文所述。代表性的烷氧基包括甲氧基、乙氧基、正丙氧基、异丙氧基、正丁氧基、庚氧基,等等。
“烷氧基羰基”意为烷基-O-CO-基,其中的烷基如本文所定义。代表性的烷氧基羰基包括甲氧基羰基、乙氧基羰基、叔丁氧基羰基,等等。
“烷基”意为在链上含有约1个至约20个碳原子的脂族烃基,可以是直链或支链。优选的烷基在链上含有1至约12个碳原子。更为优选的是如本文所定义的低级烷基。“支链的”意为一个或多个低级烷基如甲基、乙基或丙基与直链烷基链相连。“低级烷基”意为在链上含有约1个至约4个碳原子,可以是直链或支链。“取代的烷基”意为如上所定义的烷基,它被一个或多个苯基或卤素取代基(优选的是1至3个)取代,包括如本文所定义的全氟取代的烷基。
“烷基亚磺酰基”意为烷基-SO-基,其中的烷基如上所定义。优选的基团是那些其中烷基是低级烷基的基团。
“烷基磺酰基”意为烷基-SO2-基,其中的烷基如上所定义。优选的基团是那些其中烷基是低级烷基的基团。
“烷基磺酰基氨基甲酰基”意为烷基-SO2-NH-C(=O)-基,其中的烷基如本文所述。优选的烷基磺酰基氨基甲酰基是那些其中烷基是低级烷基的基团。
“烷基硫基”意为烷基-S-基,其中的烷基如本文所述。代表性的烷基硫基包括甲基硫基、乙基硫基、异丙基硫基和庚基硫基。
“炔基”意为含有碳碳三键及在链上含有约2至约15个碳原子的的脂族烃基,可以是直链或支链。优选的炔基在链上含有2至约12个碳原子;更优选的是在链上含有约2至约4个碳原子。“支链的”意为一个或多个低级烷基如甲基、乙基或丙基与直链炔基链相连。“低级炔基”意为在链上含有约2至约4个碳原子,可以是直链或支链。炔基可被一个或多个卤素取代。代表性的炔基包括乙炔基、丙炔基、正丁炔基、2-丁炔基、3-甲基丁炔基、正戊炔基、庚炔基、辛炔基、癸炔基,等等。“取代的炔基”意为被一个或多个”脂族基取代基”(优选的是1至3个)取代的如上所定义的炔基,该取代基可为相同或不同且如本文所定义。
“胺保护基”意为本领域已知的易于除去并可选择性除去的基团;它可保护氨基上的氮部分,以在合成步骤中抵抗不希望发生的反应。本领域内众所周知,使用胺保护基可保护基团在合成步骤中抵抗不希望发生的反应,许多这样的保护基是已知的,例如,T.W.Greene和P.G.M.Wuts,Protective groupsin Organic synthesis(有机合成中的保护基),第二版,John Wiley&Sons,NewYork(1991),该文献作为参考文献引用在此。胺保护基还包括“酸不稳定的胺保护基”和”氢化不稳定的胺保护基”。代表性的胺保护基是酰基,包括甲酰基、乙酰基、氯乙酰基、三氯乙酰基、邻硝基苯基乙酰基、邻硝基苯氧基-乙酰基、三氟乙酰基、乙酰乙酰基、4-氯丁酰基、异丁酰基、邻硝基肉桂酰基、吡啶甲酰基(picolinoyl)、酰基异硫氰酸根(acylisothiocyanate)、氨基己酰基、苯甲酰基等等,以及酰氧基,包括甲氧羰基(methoxy-carbonyl)、9-芴基甲氧羰基、2,2,2-三氟乙氧羰基、2-三甲基甲硅烷基乙氧羰基、乙烯基氧基羰基、烯丙氧基羰基、叔丁氧羰基(BOC)、1,1-二甲基-丙炔基氧基羰基、苄氧基羰基(CBZ)、对硝基苄氧基羰基、2,4-二氯-苄氧基羰基,等等。
“酰胺保护基”意为易于除去的基团,据本领域已知它可保护酰胺基上的氮部分,以在合成步骤中抵抗不希望发生的反应,且在它转化为酰胺之后可选择性除去。本领域内众所周知,使用酰胺保护基可保护基团在合成步骤中抵抗不希望发生的反应,许多这样的保护基是已知的,例如,T.W.Greene和P.G.M.Wuts,Protective groups in Organic synthesis(有机合成中的保护基),第二版,John Wiley&Sons,New York(1991),该文献作为参考文献引用在此。酰胺保护基还包括“酸不稳定的酰胺保护基”和”氢化不稳定的酰胺保护基”。代表性的酰胺保护基是邻硝基肉桂酰基、吡啶甲酰基、氨基己酰基、苯甲酰基等等,以及酰氧基,包括甲氧基羰基、9-芴基甲氧基羰基、2,2,2-三氟乙氧基羰基、2-三甲基甲硅烷基乙氧基羰基、乙烯基氧基羰基、烯丙氧基羰基、叔丁氧基羰基(BOC)、1,1-二甲基-丙炔基氧基羰基、苄氧基羰基(CBZ)、对硝基苄氧基羰基、2,4-二氯-苄氧基羰基,等等。
“芳族基”意为如本文所定义的芳基或杂芳基。代表性的芳族基包括苯基、卤素取代的苯基、氮杂杂芳基,等等。
“芳酰基”意为芳基-CO-基,其中的芳基如本文所述。代表性的芳酰基包括苯甲酰基、1-和2-萘甲酰基(naphthoyl),等等。
“芳基”意为芳族单环或多环体系,含有约6至约14个碳原子,优选的是约6至10个碳原子。芳基包括通过芳基部分键合的如本文所定义的稠合环烯基芳基、稠合环烷基芳基、稠合杂环烯基芳基和稠合杂环基芳基。芳基是被一个或多个相同或不同且如本文所定义的“环基取代基”(优选的是1至3个取代基)任选取代的。“取代的芳基”意为如上定义被取代的芳基。代表性的芳基包括苯基、或取代的苯基。
“芳基重氮基”意为芳基-重氮(diazo)-基,其中的芳基和重氮基如本文所定义。
“芳氧基”意为芳基-O-基,其中的芳基如本文所定义。代表性的芳氧基包括苯氧基和2-萘氧基(naphthyloxy)。
“芳氧基羰基”意为芳基-O-CO-基,其中的芳基如本文所定义。代表性的芳氧基羰基包括苯氧羰基和萘氧羰基。
“芳基磺酰基”意为芳基-SO2-基,其中的芳基如本文所定义。
“芳基磺酰基氨基甲酰基”意为芳基-SO2-NH-C(=O)-基,其中的芳基如本文所述。一个代表性的芳基磺酰基氨基甲酰基是苯基磺酰基氨基甲酰基。
“芳基亚磺酰基”意为芳基-SO-基,其中的芳基如本文所定义。
“芳基硫基”意为芳基-S-基,其中的芳基如本文所述。代表性的芳基硫基包括苯基硫基和萘基硫基。
“碱性氮原子”意为能被质子化的含有非键合电子对的sp2或sp3杂化氮原子。代表性的碱性氮原子包括任选取代的亚氨基、任选取代的氨基和任选取代的脒基。
“羧基”意为HO(O)C-(羧酸)基。
“偶联剂”意为与羧基部分上的羟基部分反应从而使它易受亲核攻击的化合物。代表性的偶联剂包括DIC、EDCI、DCC,等等。
“环烯基”意为任选取代的非芳族单环或多环体系,含有约3至约10个碳原子,优选的是约3至约6个碳原子(低级环烯基),并含有至少一个碳碳双键且能与如本文所定义的芳族基任选稠合。“稠合(芳族)环烯基”意为通过其环烯基部分键合的如本文所定义的稠合芳基环烯基和稠合杂芳基环烯基。优选的环体系中环的大小是约含5至约6个环原子;这类优选的环的大小也被称为“低级的”。“取代的环烯基”意为如上所定义的环烯基,它被一个或多个“环基取代基”(优选的是1至3个)取代,该取代基可为相同或不同且如本文所定义。代表性的单环环烯基包括环戊烯基、环己烯基、环庚烯基,等等。一个代表性的多环环烯基是降冰片烯基(norbornylenyl)。
“环烷基”意为非芳族单环或多环体系,含有约3至约10个碳原子,优选的是约3至约6个碳原子(低级环烷基),且能与如本文所定义的芳族基任选稠合。“稠合(芳族)环烷基”意为通过其环烷基部分键合的如本文所定义的稠合芳基环烷基和稠合杂芳基环烷基。“取代的环烷基”意为被一个或多个“环基取代基”(优选的是1至3个)取代的如上所定义的环烷基,该取代基可为相同或不同且如本文所定义。代表性的单环环烷基包括环戊基、环己基、环庚基,等等。代表性的多环环烷基包括1-萘烷基、降冰片基(norbornyl)、金刚烷(adamant)-(1-或2-基),等等。
“环状”或”环基”意为如本文所定义环烷基、环烯基、杂环基或杂环烯基。与术语“环状”连用的术语”低级”与本文中述及环烷基、环烯基、杂环基或杂环烯基时所用的相同。
“环基氧基”意为环基-O-基,其中的环基如本文所述。代表性的环烷氧基包括环戊基氧基、环己基氧基、奎宁环基氧基(quinuclidyloxy)、硫杂己环基氧基(pentamethylenesulfidoxy)、四氢吡喃基氧基、四氢噻吩基氧基(tetrahydrothiophenyloxy)、吡咯烷基氧基、四氢呋喃基氧基,或7-氧杂二环[2.2.1]庚烷基氧基、羟基四氢吡喃基氧基、羟基-7-氧杂二环[2.2.1]庚烷基氧基,等等。
“环基亚磺酰基”意为环基-S(O)-基,其中的环基如本文所述。
“环基磺酰基”意为环基-S(O)2-基,其中的环基如本文所述。
“环基硫基”意为环基-S-基,其中的环基如本文所述。
“重氮基”意为两价的-N=N-基团。
“有效量”意为能有效地产生所希望治疗效果的本发明的化合物/组合物的量。
“稠合芳基环烯基”意为如本文所定义的稠合芳基和环烯基。优选的稠合芳基环烯基,其中的芳基是苯基且该环烯基由约5至约6个环原子组成。稠合芳基环烯基作为变量可通过其环体系中任何有连接能力的原子连接。“取代的稠合芳基环烯基”意为被一个或多个“环基取代基”(优选的是1至3个)取代的如上所定义的稠合芳基环烯基,该取代基可为相同或不同且如本文所定义。代表性的稠合芳基环烯基包括1,2-二氢亚萘基、茚基,等等。
“稠合芳基环烷基”意为如本文所定义的稠合芳基和环烷基。优选的稠合芳基环烷基,其中的芳基是苯基且该环烷基由约5至约6个环原子组成。稠合芳基环烷基作为一个变量可通过其环体系中任何有连接能力的原子连接。“取代的稠合芳基环烷基”意为被一个或多个“环基取代基”(优选的是1至3个)取代的如上所定义的稠合芳基环烷基,该取代基可为相同或不同且如本文所定义。代表性的稠合芳基环烷基包括1,2,3,4-四氢亚萘基,等等。
“稠合芳基杂环烯基”意为如本文所定义的稠合芳基和杂环烯基。优选的稠合芳基杂环烯基,其中的芳基是苯基且该杂环烯基由约5至约6个环原子组成。稠合芳基杂环烯基作为一个变量可通过其环体系中任何有连接能力的原子连接。氮杂、氧杂或硫杂这些名称作为稠合芳基杂环烯基的杂环烯基部分之前的前缀限定了,分别至少有一个氮、氧或硫原子作为环原子存在。“取代的稠合芳基杂环烯基”意为被一个或多个“环基取代基”(优选的是1至3个)取代的如上所定义的稠合芳基杂环烯基,该取代基可为相同或不同且如本文所定义。稠合芳基杂环烯基的氮原子可以是碱性氮原子。稠合芳基杂环烯基的杂环烯基部分的氮或硫原子也可被任选地氧化为对应的N-氧化物、S-氧化物或S,S-二氧化物。代表性的稠合芳基杂环烯基包括3H-二氢吲哚基、IH-2-氧代喹啉基、2H-1-氧代异喹啉基、1,2-二氢喹啉基、3,4-二氢喹啉基、1,2-二氢异喹啉基、3,4-二氢异喹啉基,等等。
“稠合芳基杂环基”意为如本文所定义的稠合芳基和杂环基。优选的稠合芳基杂环基,其中的芳基是苯基且该杂环基由约5至约6个环原子组成。稠合芳基杂环基作为一个变量可通过其环体系中任何有连接能力的原子连接。氮杂、氧杂或硫杂这些名称作为稠合芳基杂环基的杂环基部分之前的前缀限定了,分别至少有一个氮、氧或硫原子作为环原子存在。“取代的稠合芳基杂环基”意为被一个或多个“环基取代基”(优选的是1至3个)取代的如上所定义的稠合芳基杂环基,该取代基可为相同或不同且如本文所定义。稠合芳基杂环基的氮原子可以是碱性氮原子。稠合芳基杂环基的杂环基部分的氮或硫原子还可被任选地氧化为对应的N-氧化物、S-氧化物或S,S-二氧化物。代表性的稠合芳基杂环基环体系包括二氢吲哚基、1,2,3,4-四氢异喹啉、1,2,3,4-四氢喹啉、1H-2,3-二氢异吲哚-2-基、2,3-二氢苯并[f]异吲哚-2-基、1,2,3,4-四氢苯并[g]-异喹啉-2-基,等等。
“稠合杂芳基环烯基”意为如本文所定义的稠合杂芳基和环烯基。优选的稠合杂芳基环烯基,其中的杂芳基是苯基且该环烯基由约5至约6个环原子组成。稠合杂芳基环烯基作为一个变量可通过其环体系中任何有连接能力的原子连接。氮杂、氧杂或硫杂这些名称作为稠合杂芳基环烯基的杂芳基部分之前的前缀限定了,分别至少有一个氮、氧或硫原子作为环原子存在。“取代的稠合杂芳基环烯基”意为被一个或多个“环基取代基”(优选的是1至30个)取代的如上所定义的稠合杂芳基环烯基,该取代基可为相同或不同且如本文所定义。稠合杂芳基环烯基的氮原子可以是碱性氮原子。稠合杂芳基环烯基的杂芳基部分的氮原子还可被任选地氧化为对应的N-氧化物。代表性的稠合杂芳基环烯基包括5,6-二氢喹啉基、5,6-二氢异喹啉基、5,6-二氢喹喔啉基、5,6-二氢喹唑啉基、4,5-二氢-1H-苯并咪唑基、4,5-二氢苯并 唑基,等等。
唑基,等等。
“稠合杂芳基环烷基”意为如本文所定义的稠合杂芳基和环烷基。优选的稠合杂芳基环烷基,其中的杂芳基由约5至约6个环原子组成且该环烷基由约5至约6个环原子组成。稠合杂芳基环烷基作为一个变量可通过其环体系中任何有连接能力的原子连接。氮杂、氧杂或硫杂这些名称作为稠合杂芳基环烷基的杂芳基部分之前的前缀限定了,分别至少有一个氮、氧或硫原子作为环原子存在。“取代的稠合杂芳基环烷基”意为被一个或多个”环基取代基”(优选的是1至3个)取代的如上所定义的稠合杂芳基环烷基,该取代基可为相同或不同且如本文所定义。稠合杂芳基环烷基的氮原子可以是碱性氮原子。稠合杂芳基环烷基的杂芳基部分的氮原子还可被任选地氧化为对应的N-氧化物。代表性的稠合杂芳基环烷基包括5,6,7,8-四氢喹啉基、5,6,7,8-四氢异喹啉基、5,6,7,8-四氢喹喔啉基、5,6,7,8-四氢喹唑啉基、4,5,6,7-四氢-1H-苯并咪唑基、4,5,6,7-四氢苯并 唑基、1H-4-氧杂-1,5-二氮杂萘-2-酮基(1H-4-oxa-1,5-diazanaphthalen-2-only)、1,3-二氢咪唑-[4,5]-吡啶-2-酮基(1,3-dihydroimidizole-[4,5]-pyridin-2-only),等等。
唑基、1H-4-氧杂-1,5-二氮杂萘-2-酮基(1H-4-oxa-1,5-diazanaphthalen-2-only)、1,3-二氢咪唑-[4,5]-吡啶-2-酮基(1,3-dihydroimidizole-[4,5]-pyridin-2-only),等等。
“稠合杂芳基杂环烯基”意为如本文所定义的稠合杂芳基和杂环烯基。优选的稠合杂芳基杂环烯基,其中的杂芳基由约5至约6个环原子组成且该杂环烯基由约5至约6个环原子组成。稠合杂芳基杂环烯基作为一个变量可通过其环体系中任何有连接能力的原子连接。氮杂、氧杂或硫杂这些名称作为稠合杂芳基杂环烯基的杂芳基或杂环烯基部分之前的前缀限定了,分别至少有一个氮、氧或硫原子作为环原子存在。“取代的稠合杂芳基杂环烯基”意为被一个或多个“环基取代基”(优选的是1至3个)取代的如本文所定义的稠合杂芳基杂环烯基,该取代基可为相同或不同且如本文所定义。稠合杂芳基氮杂环烯基的氮原子可以是碱性氮原子。稠合杂芳基杂环基的杂芳基部分的氮或硫原子还可被任选地氧化为对应的N-氧化物。稠合杂芳基杂环基的杂芳基或杂环基部分的氮或硫原子还可被任选地氧化为对应的N-氧化物、S-氧化物或S,S-二氧化物。代表性的稠合杂芳基杂环烯基包括7,8-二氢[1,7]二氮杂萘基、1,2-二氢[2,7]-二氮杂萘基、6,7-二氢-3H-咪唑并[4,5-c]吡啶基、1,2-二氢-1,5-二氮杂萘基、1,2-二氢-1,6-二氮杂萘基、1,2-二氢-1,7-二氮杂萘基、1,2-二氢-1,8-二氮杂萘基、1,2-二氢-2,6-二氮杂萘基,等等。
“稠合杂芳基杂环基”意为如本文所定义的稠合杂芳基和杂环基。优选的稠合杂芳基杂环基,其中的杂芳基由约5至约6个环原子组成且该杂环基由约5至约6个环原子组成。稠合杂芳基杂环基作为一个变量可通过其环体系中任何有连接能力的原子连接。氮杂、氧杂或硫杂这些名称作为稠合杂芳基杂环基的杂芳基或杂环基部分之前的前缀限定了,分别至少有一个氮、氧或硫原子作为环原子存在。“取代的稠合杂芳基杂环基”意为被一个或多个“环基取代基”(优选的是1至3个)取代的如本文所定义的稠合杂芳基杂环基,该取代基可为相同或不同且如本文所定义。稠合杂芳基杂环基的氮原子可以是碱性氮原子。稠合杂芳基杂环基的杂芳基部分的氮或硫原子还可被任选地氧化为对应的N-氧化物。稠合杂芳基杂环基的杂芳基或杂环基部分的氮或硫原子还可被任选地氧化为对应的N-氧化物、S-氧化物或S,S-二氧化物。代表性的稠合杂芳基杂环基包括2,3-二氢-1H吡咯并(pyrrol)[3,4-b]喹啉-2-基、1,2,3,4-四氢苯并[b][1,7]二氮杂萘-2-基、1,2,3,4-四氢苯并[b][1,6]二氮杂萘-2-基、1,2,3,4-四氢-9H-吡啶并[3,4-b]吲哚-2-基、1,2,3,4-四氢-9H-吡啶并[4,3-b]吲哚-2-基、2,3-二氢-1H-吡咯并[3,4-b]吲哚-2-基、1H-2,3,4,5-四氢氮杂 并(tetrahydroazepino)[3,4-b]吲哚-2-基、1H-2,3,4,5-四氢氮杂
并(tetrahydroazepino)[3,4-b]吲哚-2-基、1H-2,3,4,5-四氢氮杂 并[4,3-b]吲哚-3-基、1H-2,3,4,5-四氢氮杂
并[4,3-b]吲哚-3-基、1H-2,3,4,5-四氢氮杂 并[4,5-b]吲哚-2-基、5,6,7,8-四氢[1,7]二氮杂萘基、1,2,3,4-四氢[2,7]二氮杂萘基、2,3-二氢[1,4]二氧杂环己二烯并(dioxino)[2,3-b]吡啶基、2,3-二氢-[1,4]二氧杂环己二烯并[2,3-b]吡啶基、3,4-二氢-2H-1-氧杂[4,6]二氮杂萘基、4,5,6,7-四氢-3H-咪唑并[4,5-c]吡啶基、6,7-二氢[5,8]二氮杂萘基、1,2,3,4-四氢[1,5]-二氮杂萘基、1,2,3,4-四氢[1,6]二氮杂萘基、1,2,3,4-四氢[1,7]二氮杂萘基、1,2,3,4-四氢[1,8]二氮杂萘基、1,2,3,4-四氢[2,6]二氮杂萘基,等等。
并[4,5-b]吲哚-2-基、5,6,7,8-四氢[1,7]二氮杂萘基、1,2,3,4-四氢[2,7]二氮杂萘基、2,3-二氢[1,4]二氧杂环己二烯并(dioxino)[2,3-b]吡啶基、2,3-二氢-[1,4]二氧杂环己二烯并[2,3-b]吡啶基、3,4-二氢-2H-1-氧杂[4,6]二氮杂萘基、4,5,6,7-四氢-3H-咪唑并[4,5-c]吡啶基、6,7-二氢[5,8]二氮杂萘基、1,2,3,4-四氢[1,5]-二氮杂萘基、1,2,3,4-四氢[1,6]二氮杂萘基、1,2,3,4-四氢[1,7]二氮杂萘基、1,2,3,4-四氢[1,8]二氮杂萘基、1,2,3,4-四氢[2,6]二氮杂萘基,等等。
“卤素”意为氟、氯、溴或碘。优选的是氟、氯或溴,更为优选的是氟或氯。
“杂芳酰基”意为杂芳基-CO-基,其中的杂芳基如本文所述。代表性的杂芳酰基包括噻吩甲酰基(thiophenoyl)、烟酰基、吡咯-2-基碳基、1-和2-萘甲酰基、吡啶甲酰基,等等。
“杂芳基”意为芳族单环或多环体系,含有约5至约14个碳原子,优选的是约5至约10个碳原子,其中环体系中一个或多个环原子是碳以外的杂元素,例如氮、氧或硫。优选的是该环体系包括1至3个杂原子。优选的环体系中环的大小是约含5至约6个环原子。杂芳基包括通过其杂芳基部分键合的如本文所定义的稠合杂芳基环烯基、稠合杂芳基环烷基、稠合杂芳基杂环烯基和稠合杂芳基杂环基。“取代的杂芳基”意为被一个或多个”环基取代基”(优选的是1至3个)取代的如上所定义的杂芳基,该取代基可为相同或不同且如本文所定义。氮杂、氧杂或硫杂这些名称作为杂芳基之前的前缀限定了,分别至少有一个氮、氧或硫原子作为环原子存在。杂芳基的氮原子可以是碱性氮原子,还可被任选地氧化为对应的N-氧化物。代表性的杂芳基和取代的杂芳基包括吡嗪基、噻吩基、异噻唑基、 唑基、吡唑基、呋咱基、吡咯基、1,2,4-噻二唑基、哒嗪基、喹喔啉基、酞嗪基(phthalazinyl)、咪唑并[1,2-a]吡啶、咪唑并[2,1-b]噻唑基、苯并呋咱基、氮杂吲哚基、苯并咪唑基、苯并噻吩基、噻吩并吡啶基、噻吩并嘧啶基、吡咯并吡啶基、咪唑并吡啶基、苯并氮杂吲哚基、1,2,4-三嗪基、苯并噻唑基、呋喃基、咪唑基、吲哚基、中氮茚基(indolizinyl)、异
唑基、吡唑基、呋咱基、吡咯基、1,2,4-噻二唑基、哒嗪基、喹喔啉基、酞嗪基(phthalazinyl)、咪唑并[1,2-a]吡啶、咪唑并[2,1-b]噻唑基、苯并呋咱基、氮杂吲哚基、苯并咪唑基、苯并噻吩基、噻吩并吡啶基、噻吩并嘧啶基、吡咯并吡啶基、咪唑并吡啶基、苯并氮杂吲哚基、1,2,4-三嗪基、苯并噻唑基、呋喃基、咪唑基、吲哚基、中氮茚基(indolizinyl)、异 唑基、异喹啉基、异噻唑基、二唑基、吡嗪基、哒嗪基、吡唑基、吡啶基、嘧啶基、吡咯基、喹唑啉基、喹啉基、1,3,4-噻二唑基、噻唑基、噻吩基、三唑基,等等。优选的杂芳基是吡嗪基。
唑基、异喹啉基、异噻唑基、二唑基、吡嗪基、哒嗪基、吡唑基、吡啶基、嘧啶基、吡咯基、喹唑啉基、喹啉基、1,3,4-噻二唑基、噻唑基、噻吩基、三唑基,等等。优选的杂芳基是吡嗪基。
“杂芳基重氮基”意为杂芳基-重氮-基,其中的杂芳基和重氮基如本文所定义。
“杂芳基磺酰基氨基甲酰基”意为杂芳基-SO2-NH-C(=O)-基,其中的杂芳基如本文所述。
“杂环烯基”意为非芳族单环或多环烃环体系,含有约3至约10个碳原子,优选的是约4至约6个碳原子(低级杂环烯基),其中环体系中一个或多个环原子是碳以外的杂元素,例如氮、氧或硫,且其中含有至少一个碳碳双键或碳氮双键。优选的是,该环包括1至3个杂原子。杂环烯基包括通过其杂环烯基部分键合的如本文所定义的稠合芳基杂环烯基和稠合杂芳基杂环烯基。氮杂、氧杂或硫杂这些名称作为杂环烯基之前的前缀限定了,分别至少有一个氮、氧或硫原子作为环原子存在。“取代的杂环烯基”意为被一个或多个“环基取代基”(优选的是1至3个)取代的如上所定义的杂环烯基,该取代基可为相同或不同且如本文所定义。杂环烯基的氮原子可以是碱性氮原子。杂环烯基的氮或硫原子还可被任选地氧化为对应的N-氧化物、S-氧化物或S,S-二氧化物。代表性的单环氮杂环烯基包括1,2,3,4-四氢吡啶(tetrahydrohydropyridine)、1,2-二氢吡啶基、1,4-二氢吡啶基、1,2,3,6-四氢吡啶、1,4,5,6-四氢嘧啶、2-吡咯啉基、3-吡咯啉基、2-咪唑啉基、2-吡唑啉基,等等。代表性的氧杂环烯基包括3,4-二氢-2H-吡喃、二氢呋喃基和氟二氢呋喃基。代表性的多环氧杂环烯基是7-氧杂二环[2.2.1]庚烯基。代表性的单环硫杂杂环烯基环包括二氢噻吩基(dihydrothiophenyl)和二氢硫代吡喃基(dihydrothiopyranyl)。
“杂环基”意为非芳族饱和单环或多环体系,含有约3至约10个碳原子,优选的是约4至约6个碳原子(低级杂环基),其中环体系中一个或多个环原子是碳以外的杂元素,例如氮、氧或硫。优选的是,该环体系含有1至3个杂原子。杂环基包括通过其杂环基部分键合的如本文所定义的稠合芳基杂环基和稠合杂芳基杂环基。氮杂、氧杂或硫杂这些名称作为杂环基之前的前缀限定了,分别至少有一个氮、氧或硫原子作为环原子存在。“取代的杂环基”意为被一个或多个“环基取代基”(优选的是1至3个)取代的如上所定义的杂环基,该取代基可为相同或不同且如本文所定义。杂环基的氮原子可以是碱性氮原子。杂环基的氮或硫原子还可被任选地氧化为对应的N-氧化物、S-氧化物或S,S-二氧化物。代表性的单环杂环基环包括哌啶基、吡咯烷基、哌嗪基、吗啉基、硫代吗啉基、噻唑烷基、1,3-二氧戊环基、1,4-二烷基、四氢呋喃基、四氢噻吩基(tetrahydrothiophenyl)、四氢硫代吡喃基,等等。
“水合物”意为溶剂合物,其中的溶剂分子是H2O。
“氢化不稳定的胺保护基”意为如本文所定义的胺保护基,它容易通过氢化被除去但对其他试剂却保持相对稳定。一种优选的氢化不稳定的胺保护基是Cbz。
“氢化不稳定的酸保护基”意为如本文所定义的酸保护基,它容易通过氢化被除去但对其他试剂却保持相对稳定。一种优选的氢化不稳定的酸保护基是苄基。
“患者”包括人和其他哺乳动物。
本文所述的“药学上可接受的前药”,系指本发明的化合物的这样一些前药:在合理的医学判断范围内,对于那些呈现过度的毒性、刺激和变应性反应等的患者,它们适合用于与身体组织接触,且具有合理的受益/风险比;而且,它们在本发明的化合物的预期用途方面是有效的。术语“前药”是指这样一些化合物,它们在体内迅速地转化,例如通过在血液中水解,从而产生具有上述式的母体化合物。可通过代谢裂解迅速地转化的官能团,在体内形成了一类能与本发明的化合物的羧基反应的基团。它们包括但不限于这样基团,如烷酰基(如乙酰基、丙酰基、丁酰基等)、未取代的和取代的芳酰基(如苯甲酰基和取代的苯甲酰基)、烷氧基羰基(如乙氧基羰基)、三烷基甲硅烷基(如三甲基甲硅烷基和三乙基甲硅烷基)、与二羧酸(如丁二酰基)形成的单酯,等等。由于本发明的化合物的可代谢裂解基团易于在体内裂解,故含有这类基团的化合物可像前药那样起作用。含有可代谢裂解基团的化合物的优点在于,由于该可代谢裂解基团的存在,提高了母体化合物的溶解性和/或吸收速率,故可显示出更好的生物利用度。下列文献提供了详尽的讨论:Design of Prodrugs(前药设计),H.Bundgaard,ed.,Elsevier(1985);Methods inEnzymology(酵素学方法);K.Widder等人,Ed.,Academic Press,42,309-396(1985);A Textbook of Drug Design and Development(药物设计和开发教材),Krogsgaard-Larsen和H.Bandaged,ed.,Chapter 5;“Design and Applications ofProdrugs”(前药设计和应用)113-191(1991);Advanced Drug Delivery Reviews(先进给药方法评述),H.Bundgard,8,1-38,(1992);J.Pharm.Sci.,77.,285(1988);Chem.Pharm.Bull.,N.Nakeya等人,32,692(1984);Pro-drugs asNovel Delivery Systems(作为新颖给药体系的前药),T.Higuchi和V.Stella,14A.C.S.Symposium Series,and Bioreversible Carriers in Drug Design(论文集以及药物设计中生物可逆载体),E.B.Roche,ed.,American PharmaceuticalAssociation and Pergamon Press,1987,其作为参考文献引用在此。
“药学上可接受的盐”是指相对无毒性的本发明的化合物的无机酸和有机酸加成盐以及碱加成盐。这些盐可于该化合物的最终分离和纯化阶段就地制备,尤其是,酸加成盐可通过以游离碱形式存在的纯化后化合物与适当的有机酸或无机酸分别反应、然后分离所形成的盐来制备。代表性的酸加成盐包括氢溴酸盐、盐酸盐、硫酸盐、硫酸氢盐、磷酸盐、硝酸盐、乙酸盐、草酸盐、戊酸盐、油酸盐、棕榈酸盐、硬脂酸盐、月桂酸盐、硼酸盐、苯甲酸盐、乳酸盐、磷酸盐、甲苯磺酸盐、柠檬酸盐、马来酸盐、富马酸盐、琥珀酸盐、酒石酸盐、萘酸盐(naphthylate)、甲磺酸盐、葡庚糖酸盐、乳糖酸盐(lactiobionate)、氨基磺酸盐、丙二酸盐、水杨酸盐、丙酸盐、亚甲基-双-β-羟基萘甲酸盐、龙胆酸盐、羟乙基磺酸盐、二对甲苯酰基酒石酸盐、甲磺酸盐、乙磺酸盐、苯磺酸盐、对甲苯磺酸盐、环己基氨基磺酸盐和月桂基磺酸盐,等等。参阅,例如S.M.Berge,等人,“Pharmaceutical Salts”(药用盐),J. Pharm.Sci.,66,1-19(1977),其作为参考文献引用在此。碱加成盐也可通过以其酸的形式存在的纯化后化合物与适当的有机碱或无机碱分别反应、然后分离所形成的盐来制备。碱加成盐包括药学上可接受的金属和胺盐。适宜的金属盐包括钠、钾、钙、钡、锌、镁以及铝盐。钠和钾盐是优选的。适宜的无机碱加成盐是从金属碱制备的,金属碱包括氢化钠、氢氧化钠、氢氧化钾、氢氧化钙、氢氧化铝、氢氧化锂、氢氧化镁、氢氧化锌等等。适宜的胺的碱加成盐是从某些胺制备的,这些胺具有足够的碱性以形成稳定的盐,优选的包括医药化学中经常使用的那些胺,因为它们具有适合于医学用途的低毒性和可接受性:氨、乙二胺、N-甲基-葡糖胺、赖氨酸、精氨酸、鸟氨酸、胆碱、N,N′-二苄基乙二胺、氯普鲁卡因、二乙醇胺、普鲁卡因、N-苄基苯乙胺、二乙胺、哌嗪、三(羟基甲基)-氨基甲烷、四甲基氢氧化铵、三乙胺、二苄基胺、二苯羟甲胺、脱氢枞胺、N-乙基哌啶、苄基胺、四甲基铵、四乙基铵、甲基胺、二甲基胺、三甲基胺、乙胺、碱性氨基酸,如赖氨酸和精氨酸,以及二环己基胺,等等。
“季胺衍生物”意为用低级烷基烷基化的sp3杂化胺。
“环基取代基”意为与芳族或非芳族环体系连接的取代基,包括芳基、杂芳基、羟基、烷氧基、环基氧基、芳氧基、杂芳氧基、酰基或它的硫代类似物、环基羰基或它的硫代类似物、芳酰基或它的硫代类似物、杂芳酰基或它的硫代类似物、酰氧基、环基羰基氧基、芳酰氧基、杂芳酰氧基、卤素、硝基、氰基、羧基(酸)、-C(O)-NHOH-C(O)-CH2OH、-C(O)-CH2SH-C(O)-NH-CN、磺基(sulfo)、膦酰基(phosphono)、烷基磺酰基氨基甲酰基、四唑基、芳基磺酰基氨基甲酰基、N-甲氧基氨基甲酰基、杂芳基磺酰基氨基甲酰基、3-羟基-3-环丁烯-1,2-二酮、3,5-二氧代-1,2,4- 二唑烷基(oxadiazolidinyl)或羟基杂芳基如3-羟基异
二唑烷基(oxadiazolidinyl)或羟基杂芳基如3-羟基异 唑基、3-羟基-1-甲基吡唑基、烷氧基羰基、环基氧基羰基、芳氧基羰基、杂芳氧基羰基、烷基磺酰基、环基磺酰基、芳基磺酰基、杂芳基磺酰基、烷基亚磺酰基、环基亚磺酰基、芳基亚磺酰基、杂芳基亚磺酰基、烷基硫基、环基硫基、芳基硫基、杂芳基硫基、环基、芳基重氮基、杂芳基重氮基、巯基(thiol)、Y1Y2N-、Y1Y2NC(O)-、Y1Y2NC(O)O-、Y1Y2NC(O)NY3-或Y1Y2NSO2-,其中Y1、Y2和Y3独立地为氢、烷基、芳基或杂芳基,或当取代基是Y1Y2N-时,则Y1和Y2之一可以是如本文所定义的酰基、环基羰基、芳酰基、杂芳酰基、烷氧基羰基、环基氧基羰基、芳氧基羰基或杂芳氧基羰基,而Y1和Y2中的另一个则如前面所定义,或当取代基是Y1Y2NC(O)-、Y1Y2NC(O)O-、Y1Y2NC(O)NY3-或Y1Y2NSO2-时,Y1和Y2也可以与连接Y1和Y2的N原子一起形成4至7元的氮杂环基或氮杂环烯基。当环体系是饱和的或部分饱和的时,“环基取代基”还包括、亚甲基(H2C=)、氧代(O=)和硫代(S=)。酸性的/酰胺环基取代基是羧基(酸)、-C(O)-NHOH、-C(O)-CH2OH、-C(O)-CH2SH、-C(O)-NH-CN、磺基、膦酰基、烷基磺酰基氨基甲酰基、四唑基、芳基磺酰基氨基甲酰基、N-甲氧基氨基甲酰基、杂芳基磺酰基氨基甲酰基、3-羟基-3-环丁烯-1,2-二酮、3,5-二氧代-1,2,4-
唑基、3-羟基-1-甲基吡唑基、烷氧基羰基、环基氧基羰基、芳氧基羰基、杂芳氧基羰基、烷基磺酰基、环基磺酰基、芳基磺酰基、杂芳基磺酰基、烷基亚磺酰基、环基亚磺酰基、芳基亚磺酰基、杂芳基亚磺酰基、烷基硫基、环基硫基、芳基硫基、杂芳基硫基、环基、芳基重氮基、杂芳基重氮基、巯基(thiol)、Y1Y2N-、Y1Y2NC(O)-、Y1Y2NC(O)O-、Y1Y2NC(O)NY3-或Y1Y2NSO2-,其中Y1、Y2和Y3独立地为氢、烷基、芳基或杂芳基,或当取代基是Y1Y2N-时,则Y1和Y2之一可以是如本文所定义的酰基、环基羰基、芳酰基、杂芳酰基、烷氧基羰基、环基氧基羰基、芳氧基羰基或杂芳氧基羰基,而Y1和Y2中的另一个则如前面所定义,或当取代基是Y1Y2NC(O)-、Y1Y2NC(O)O-、Y1Y2NC(O)NY3-或Y1Y2NSO2-时,Y1和Y2也可以与连接Y1和Y2的N原子一起形成4至7元的氮杂环基或氮杂环烯基。当环体系是饱和的或部分饱和的时,“环基取代基”还包括、亚甲基(H2C=)、氧代(O=)和硫代(S=)。酸性的/酰胺环基取代基是羧基(酸)、-C(O)-NHOH、-C(O)-CH2OH、-C(O)-CH2SH、-C(O)-NH-CN、磺基、膦酰基、烷基磺酰基氨基甲酰基、四唑基、芳基磺酰基氨基甲酰基、N-甲氧基氨基甲酰基、杂芳基磺酰基氨基甲酰基、3-羟基-3-环丁烯-1,2-二酮、3,5-二氧代-1,2,4- 二唑烷基或羟基杂芳基如3-羟基异唑基、3-羟基-1-甲基吡唑基和Y1Y2NCO-。非酸性的极性环基取代基是羟基、氧代(O=)、硫代(S=)、酰基或它的硫代类似物、环基羰基或它的硫代类似物、芳酰基或它的硫代类似物、杂芳酰基或它的硫代类似物、烷氧基羰基、环基氧基羰基、芳氧基羰基、杂芳氧基羰基、酰氧基、环基羰基氧基、芳酰氧基、杂芳酰氧基、烷基磺酰基、环基磺酰基、芳基磺酰基、杂芳基磺酰基、烷基亚磺酰基、环基亚磺酰基、芳基亚磺酰基、杂芳基亚磺酰基、巯基、Y1Y2N-、Y1Y2NC(O)-、Y1Y2NC(O)O-、Y1Y2NC(O)NY3-或Y1Y2NSO2。
二唑烷基或羟基杂芳基如3-羟基异唑基、3-羟基-1-甲基吡唑基和Y1Y2NCO-。非酸性的极性环基取代基是羟基、氧代(O=)、硫代(S=)、酰基或它的硫代类似物、环基羰基或它的硫代类似物、芳酰基或它的硫代类似物、杂芳酰基或它的硫代类似物、烷氧基羰基、环基氧基羰基、芳氧基羰基、杂芳氧基羰基、酰氧基、环基羰基氧基、芳酰氧基、杂芳酰氧基、烷基磺酰基、环基磺酰基、芳基磺酰基、杂芳基磺酰基、烷基亚磺酰基、环基亚磺酰基、芳基亚磺酰基、杂芳基亚磺酰基、巯基、Y1Y2N-、Y1Y2NC(O)-、Y1Y2NC(O)O-、Y1Y2NC(O)NY3-或Y1Y2NSO2。
“溶剂合物”意为本发明的化合物与一个或多个溶剂分子的物理性缔合。这种物理性缔合包括氢键合。在某些情况下,例如当结晶固体的晶格内掺入一个或多个溶剂分子时,溶剂合物可以分离。“溶剂合物”包括溶液相以及可分离的溶剂合物。代表性的溶剂合物包括水合物、乙醇合物(ethanolates)、甲醇合物(methanolates),等等。
实施方案
以下是与本文叙述的发明相关的具体实施方案。
本发明的一具体实施方案,其中的R1是任选取代的苯基(C1-3烷基)或任选取代的苯基环丙基。
本发明的另一具体实施方案,其中的R1是任选取代的苯基(C1-3烷基)。
本发明的另一具体实施方案,其中的R1是任选取代的苯基(C2-3烷基)。
本发明的另一具体实施方案,其中的R1是任选取代的苯基(乙基)。
本发明的另一具体实施方案,其中的R1是苯乙基。
本发明的另一具体实施方案,其中的R1是任选取代的苯基环丙基。
本发明的另一具体实施方案,其中的R1中任选取代的苯基是被卤素取代的。
本发明的另一具体实施方案,其中的R1中任选取代的苯基是被氯或氟取代的。
本发明的另一具体实施方案,其中的R1中任选取代的苯基是被氯或氟取代的。
本发明的另一具体实施方案,其中的R1中任选取代的苯基是被氯或氟单取代的。
本发明的另一具体实施方案,其中的R1中任选取代的苯基是被氯邻位单取代的。
本发明的另一具体实施方案,其中的R1中任选取代的苯基是被氯对位单取代的。
本发明的另一具体实施方案,其中的R1中任选取代的苯基是被氯间位单取代的。
本发明的另一具体实施方案,其中的R1中任选取代的苯基是被氟对位单取代的。
本发明的另一具体实施方案,其中的R1中任选取代的苯基是被氯或氟双取代的。
本发明的另一具体实施方案,其中的R1中任选取代的苯基是被氯邻位、对位双取代的。
本发明的另一具体实施方案,其中的R1中任选取代的苯基是被氟邻位、对位双取代的。
本发明的另一具体实施方案,其中的R2是H或甲基。
本发明的另一具体实施方案,其中的R2是H。
本发明的另一具体实施方案,其中的R3是任选取代的苯基、任选取代的噻唑基、吡啶基,或噻吩基。
本发明的另一具体实施方案,其中的R3是任选取代的吲哚满酮基(indolidonyl)。
本发明的另一具体实施方案,其中的R3中任选取代的苯基是被卤素、羧基或烷氧基羰基取代的。
本发明的另一具体实施方案,其中的R3中的任选取代的苯基是被氯或氟单取代的。
本发明的另一具体实施方案,其中的R3中的任选取代的苯基是被氯邻位单取代的。
本发明的另一具体实施方案,其中的R3中的任选取代的苯基是被氯对位单取代的。
本发明的另一具体实施方案,其中的R3中的任选取代的苯基是被氯间位单取代的。
本发明的另一具体实施方案,其中的R3中的任选取代的苯基是被氟双取代的。
本发明的另一具体实施方案,其中的R3中的任选取代的苯基是被氟邻位、对位双取代的。
本发明的另一具体实施方案,其中的R3中的任选取代的苯基是被羧基或烷氧基羰基取代的。
本发明的另一具体实施方案,其中的R3中的任选取代的苯基是被羧基或烷氧基羰基单取代的。
本发明的另一具体实施方案,其中的R3中的任选取代的苯基是被羧基或烷氧基羰基间位单取代的。
本发明的另一具体实施方案,其中的R3中的任选取代的苯基是被羧基或烷氧基羰基对位单取代的
本发明的另一具体实施方案,其中的R3中的任选取代的苯基是2-噻吩基。
本发明的另一具体实施方案,其中的R3中的任选取代的苯基是3-噻吩基。
本发明的另一具体实施方案,其中的R3中的任选取代的苯基是任选取代的噻唑基。
本发明的另一具体实施方案,其中的R3中的任选取代的苯基是甲基取代的噻唑基。
本发明的另一具体实施方案,其中的R4是H。
本发明的另一具体实施方案,其中的R5是JGZ。
本发明的另一具体实施方案,其中的Z是键。
本发明的另一具体实施方案,其中的Z是CO。
本发明的另一具体实施方案,其中的G是低级烷基。
本发明的另一具体实施方案,其中的G是C1-3低级烷基。
本发明的另一具体实施方案,其中的G是C2-3低级烷基。
本发明的另一具体实施方案,其中的J是R8R7N。
本发明的另一具体实施方案,其中的R7和R8是H或低级烷基。
本发明的另一具体实施方案,其中的R7和R8是低级烷基。
本发明的另一具体实施方案,其中的R7和R8是C1-2低级烷基。
本发明的另一具体实施方案,其中的R7和R8是C1低级烷基。
本发明的另一具体实施方案,其中的R7和R8之一是H且R7和R8的另一个是低级烷基。
本发明的另一具体实施方案,其中的R9和R10是H。
本发明的另一具体实施方案,其中的R9和R10是低级烷基。
本发明的另一具体实施方案,其中的R9和R10是甲基。
本发明的另一具体实施方案,其中的J是任选(低级烷基或卤素)取代的3-7元杂环基。
本发明的另一具体实施方案,其中的J是任选(低级烷基或卤素)取代的3-7元氮杂环基。
本发明的另一具体实施方案,其中的J作为任选(低级烷基或卤素)取代的3-7元杂环基是N-甲基哌啶-4-基。
本发明的另一具体实施方案,其中的R5是R8R7NQ1低级烷基。
本发明的另一具体实施方案,其中的Q1是羰基。
本发明的另一具体实施方案,其中的Q1是羰基,且J是R8R7N或任选(低级烷基或卤素)取代的3-7元氮杂环基。
本发明的另一具体实施方案,其中的Y是CO。
本发明的另一具体实施方案,其中的Y是SO2。
本发明的另一具体实施方案,其中的A和R5一起形成具有如下式的4-6元螺环氮杂环基
n和p独立地为0、1、2、3、4或5,只要n和p≥2但≤5。
本发明的另一具体实施方案,其中的A和R5一起形成氮杂环丁烷、吡咯烷或哌啶,其每一个任选被低级烷基N-取代。
本发明的另一优选的实施方案是选自下式的化合物:




以及

或其药学上可接受的盐或前药,或这类化合物的溶剂合物、它的盐或它的前药。
本发明的化合物任选以盐的形式提供。药学上可接受的那些盐是特别感兴趣的,因为它们可在医疗目的中用于前述化合物的给药。非药学上可接受的盐可在制造过程中用于分离和纯化的目的,在一些情况下,可用于分离本发明的化合物的立体异构形式。后者尤其适用于从光学活性胺制备胺盐。
酸加成盐是用其中存在碱性官能团(如亚氨基氮、氨基或单取代的基团或双取代的基团)的本发明的化合物形成的。一种具体的酸加成盐是药学上可接受的酸加成盐,即,在该盐的药用剂量条件下该盐的阴离子对患者无毒性,使得该游离酸内在的有益作用不会因阴离子的副作用而削弱。所选择的盐经过优化选择,与常用医药载体相容,并适合于口服或肠胃外给药。本发明的化合物的酸加成盐可通过应用或改进已知的方法使游离碱与适当的酸进行反应来制备。例如,本发明的化合物的酸加成盐可通过以下制备:或者是将该游离碱溶于水或醇的水溶液或其他含有适当酸的适宜溶剂,并通过蒸发该溶液而分离出该盐;或者是让该游离碱与酸在有机溶剂中反应,在此情况下可直接分离出该盐或可通过浓缩该溶液的方式而获得该盐。一些用于制备这类盐的适宜的酸是盐酸、氢溴酸、磷酸、硫酸、各种有机羧酸和磺酸,如乙酸、柠檬酸、丙酸、琥珀酸、苯甲酸、酒石酸、富马酸、扁桃酸、抗坏血酸、苹果酸、甲磺酸、甲苯磺酸、脂肪酸、己二酸盐(adipate)、海藻酸盐(alginate)、抗坏血酸盐(ascorbate)、天冬氨酸盐(aspartate)、苯磺酸盐(benzenesulfonate)、苯甲酸盐(benzoate)、环戊烷丙酸盐(cyclopentanepropionate)、二葡糖酸盐(digluconate)、十二烷基硫酸盐(dodecylsulfate)、硫酸氢盐(bisulfate)、丁酸盐(butyrate)、乳酸盐(lactate)、月桂酸盐(laurate)、月桂基硫酸盐(lauryl sulfate)、马来酸盐(meleate)、氢碘酸盐(hydroiodide)、2-羟基乙磺酸盐(2-hydroxy-ethanesulfonate)、甘油磷酸盐(glycerophosphate)、苦味酸盐(picrate)、新戊酸盐(pivalate)、双羟萘酸盐(pamoate)、果胶酯酸盐(pectinate)、过硫酸盐(persulfate)、3-苯基丙酸盐(3-phenylpropionate)、硫氰酸盐(thiocyanate)、2-萘磺酸盐(2-naphthalenesulfonate)、十一酸盐(undecanoate)、烟酸盐(nicotinate)、半硫酸盐(hemisulfate)、庚糖酸盐(heptonate)、己酸盐(hexanoate)、樟脑酸盐(camphorate)、樟脑磺酸盐(camphorsulfonate),以及其他。
本发明的化合物的酸加成盐可通过应用或改进已知的方法从盐再生。例如,通过用碱例如碳酸氢钠水溶液或氨水溶液处理,本发明的母体化合物可从它们的酸加成盐再生。
当本发明的化合物含有羧基或具有足够酸性的生物等排物(bioisostere)时,可形成碱加成盐。可用于制备碱加成盐的碱优选包括这样一些碱,当它们与游离酸结合时,将生成药学上可接受的盐,即,在药用剂量条件下该盐的阳离子对患者无毒性,使得该游离碱内在的有益作用不会因阳离子的副作用而削弱。
药学上可接受的盐(包括本发明范围内从碱金属和碱土金属盐衍生的盐),包括从下列各种碱衍生的盐:氢化钠、氢氧化钠、氢氧化钾、氢氧化钙、氢氧化铝、氢氧化锂、氢氧化镁、氢氧化锌、氨、乙二胺、N-甲基-葡糖胺、赖氨酸、精氨酸、鸟氨酸、胆碱、N,N′-二苄基乙二胺、氯普鲁卡因、二乙醇胺、普鲁卡因、N-苄基苯乙胺、二乙胺、哌嗪、三(羟基甲基)-氨基甲烷、四甲基氢氧化铵,等等。
本发明的化合物可通过应用或改进已知的方法从它们的碱加成盐再生。例如,通过用酸例如盐酸处理,本发明的母体化合物可从它们的碱加成盐再生。
在本发明的方法中,本发明的化合物可以溶剂合物(例如水合物)的形式很方便地制备或形成。使用有机溶剂例如二 烷(dioxin)、四氢呋喃或甲醇,通过从水/有机溶剂混合物中重结晶,可很方便地制备本发明的化合物的水合物。
烷(dioxin)、四氢呋喃或甲醇,通过从水/有机溶剂混合物中重结晶,可很方便地制备本发明的化合物的水合物。
式1的化合物可通过应用或改进已知的、迄今为止所使用的或文献中所述的方法制备,或依照本发明的方法制备。
本发明的另一目的是提供一种制备中间体化合物方法,该方法可用于制备式1的化合物。
发明的化合物的制备
本发明的化合物的初始原料和中间体可通过应用或改进已知的方法来制备,例如参比例中所述的那些方法或其显然的等效化学方法。
本发明的化合物可通过应用或改进已知的方法来制备,所谓已知的方法是指迄今为止所使用的或文献中所述的方法,例如R.C.Larock在Comprehensive Organic Transformations(有机转化大全),VCH publishers(1989)中所述的那些方法。
实验部分
一般步骤
用于合成的初始原料是从Aldrich、Acros、Sigma、Fluka、Nova Biochem、Advanced Chemtech、Bachem、Lancaster以及其他化学品供应商获得的。
使用一般的固相合成方法生产本发明的化合物。这类方法在文献中述及,例如Steward和Young,Solid Phase Peptide Synthesis(固相肽合成)(Freeman&Co.,San Francisco,1969),此文作为参考文献引用在此。有时,也使用传统的溶液相合成法。
除非另有说明,化合物是使用FMPE聚苯乙烯HL树脂(01-64-0254或01-64-0399(NovaBiochem,EMD Biosciences,Inc.)合成的。该树脂含有‘Ameba’连接基(linker)。这种连结可以E.Hernandez,等人Tetrahedron Lett.2002,43,4741和D.Weber等人.J.Peptide Sci.2002,8,461所述的步骤引入至任何类型的氨基聚苯乙烯树脂,该文作为参考文献引用在此。
在合成的第一步(关于一般合成示意图,见图1),于环境温度下用胺在DCM/TMOF(1∶1)中的0.5M溶液处理树脂12小时,以产生Schiff碱。用THF洗涤2遍之后,于环境温度下用1份1M NaBH3CN的THF溶液和3份THF∶MeOH∶AcOH(80∶20∶0.5)的混合物处理树脂5小时,实现Schiff碱的还原。洗涤树脂,最后在DMF中溶胀。
图1(合成示意图)

使4-氟-3-硝基苯甲酸与在树脂上生成的仲胺偶联(见示意图1)。偶联(coupling)通常是在DMF/DCM混合物(1∶1)中用DIC/HOAt或HATU/DIEA实现的。此偶联于环境温度(RT)下持续12小时。该树脂依次用一系列溶剂如DMF、THF、AN洗涤数遍,最后在DMF中溶胀。
氟取代反应是于65℃用特定的仲胺在NMP中的0.5M溶液处理树脂48小时实现的。该树脂依次用一系列溶剂如DMF、THF、AN洗涤数遍,最后在DMF中溶胀。
如实施例12,硝基是于环境温度下用SnCl2在DMF中的1M溶液处理树脂12小时而还原的。该树脂依次用一系列溶剂如DMF、THF、AN洗涤数遍,最后在DMF中溶胀。
最终苯胺氮的酰基化是于环境温度下用DIEA作为碱在DMF中通过酸的HATU偶联,反应过夜(12小时)而实现的。
当化合物前体在该树脂上的集结完成后,该树脂依次用一系列溶剂如DMF、THF、AN洗涤数遍,最后在THF中溶胀并在真空中干燥。
裂解是通过于环境温度下用TFA和水的95∶5混合物处理树脂4小时而实现的。然后用相同的混合物萃取树脂3x,将合并的萃取物蒸发至油状残余物。
如果哌嗪亚结构的修饰是必须的,那么适当的反应是在连接的“硝基”构建(construct)的阶段在树脂上实现的(每种化合物如上所述)。
应该理解,本发明的化合物可含有不对称中心。这些不对称中心可以独立地为R构型或S构型。应该理解,本发明包括本发明的化合物的单独的立体异构体及其混合物,包括外消旋混合物。这样的异构体可通过应用或改进已知的方法如色谱技术和重结晶技术,从它们的混合物中分离出来,或者也可从与其中间体的适当异构体分别制备。
出于此处的目的,应该理解,在关于某一特定基团如硫代(thioxo)/巯基或氧基/羟基的叙述的中,在适当的情况下可包括其互变异构形式。
干燥后的化合物经过纯化,此处有两种纯化系统可根据需要选择使用。当提及RP-HPLC Beckman系统时,其意思是在由数据站和Gold Nouveau软件控制的、由Beckman 125P溶剂传送系统(Solvent Deliver System)、Beckamn166可编程检测器组件(Programmable Detector Module),以及YMC ODS-AM20x250mm柱(S-5(5um),YMC,Inc.Wilmington,NC,USA)组成的系统上,在270nm(如果未另行规定)和流速为10ml/min的条件下,使用在水和乙腈(AN)中的0.1%TFA的适当梯度液。当提及Waters质量-触发的-LCMS(mass-triggered-LCMS)纯化时,其意思是在由MassLynx软件数据站控制的、与Waters-Micromass ZQ偶联的Waters 2525梯度溶剂传送系统、以及Waters 2487UV检测器(于220nm(如果未另行规定))上,使用在水和乙腈(AN)中的0.1%TFA的适当梯度液。在流速为32ml/min的条件下使用YMCODS-AM 20x50mm柱(S-5(5um),YMC,Inc.Wilmington,NC,USA)。在收集含有预期合成产物的峰(peak)馏分后,将该化合物溶液冷冻干燥,且将该化合物进行鉴定过程,其包括电喷雾质谱(LC/MS)和/或NMR分析,以确定合成化合物正确无误。
分析的LC/MS是用PE Sciex API 150EX以ES+模式完成的,该仪器配有Sciex MassChrom软件,并设有Gilson 215液体处理器、两个ShimadzuLC-10AD液体模件、Shimadzu SPD-10A检测器以及Keystone Betasil C-18柱(2x30mm,3um,0.7ml/min流速条件下的乙腈/水/0.1%TFA梯度液)。
为了确定结构,对某些化合物测定了NMR谱。在提及NMR时,光谱图是在两种供选择的仪器上获得的,根据需要分别使用。Bruker 300MHz意为Bruker Avance DPX 300MHz仪器,Bruker 600MHz意为Bruker AvanceDPX 600MHz仪器。样本在分别作为溶剂的DMSO-d6(Aldrich)或CDCl3(Aldrich)中测量。
这些初始原料、中间体和产物可通过应用或改进已知的方法来制备,例如实施例中所述的那些方法或其显然的等效化学方法。
实施例1
溶液相前体制备
中间体1

步骤1:于100℃用4-(二甲基氨基-乙基)-哌嗪在DMF中的1M溶液处理3.7g 4-氟-3-硝基苯甲酸120分钟。首先用LCMS检查该反应混合物,然后部分蒸发并从AN结晶。即得纯产物(3g)。
中间体2

步骤2:将322mg(1mmol)前述反应的产物与139mg(1.1mmol)DIC和459mg(=3mmol)HOBt以及171mg(1.1mmol)4-氯-苯乙基胺一起溶于5mlDMF。2小时之后,于环境温度下蒸发2/3溶剂,并通过几次注射用Waters质量-触发的-LCMS纯化系统纯化油状残余物。冷冻干燥适当级分,即得115mg所需的物质。
步骤3:将100mg前述反应的产物溶于8mL甲醇并加入100mg 5%Pd/C。在帕尔设备(Parr apparatus)中抽真空之后,引入35psi的氢。于环境温度下将反应振摇12小时。分析LCMS显示产物和脱卤化副产物。经过5μm多孔玻璃(frit)过滤滤去催化剂并在ROTAVAP上蒸发甲醇。用Waters质量-触发的-LCMS纯化系统,获得两种主要级分,冷冻干燥之后,即得所需产物和脱卤化化合物的副产物。
中间体3
步骤2:将322mg(1mmol)前述反应的产物与139mg(1.1mmol)DIC和459mg(=3mmol)HOBt以及171mg(1.1mmol)3-氯-苯乙基胺一起溶于5mlDMF。2小时之后,于环境温度下蒸发2/3溶剂,并通过几次注射用Waters质量-触发的-LCMS纯化系统纯化油状残余物。冷冻干燥适当级分,即得115mg所需的物质。
步骤3:将100mg前述反应的产物溶于8mL甲醇,并加入100mg 5%Pd/C。在帕尔设备中抽真空之后,引入35psi的氢。于环境温度下将反应振摇2小时。分析LCMS显示产物和脱卤化副产物。通过多孔玻璃过滤滤去催化剂并在ROTAVAP上蒸发甲醇。用Waters质量-触发的-LCMS纯化系统获得主级分,冷冻干燥后即得所需产物。
中间体4

步骤2:将322mg(1mmol)前述反应的产物与139mg(1.1mmol)DIC和459mg(=3mmol)的HOBt以及171mg(1.1mmol)2-氯-苯乙基胺一起溶于5ml DMF。2小时之后,于环境温度下蒸发2/3溶剂,并通过几次注射用Waters质量-触发的-LCMS纯化系统纯化油状残余物。冷冻干燥适当级分,即得115mg所需的物质。
步骤3:将100mg前述反应的产物溶于8mL甲醇,并加入100mg 5%Pd/C。在帕尔设备中抽真空之后,引入35psi的氢。于环境温度下将反应振摇2小时。分析LCMS显示产物和脱卤化副产物。通过多孔玻璃过滤滤去催化剂并在ROTAVAP上蒸发甲醇。用Waters质量-触发的-LCMS纯化系统获得主级分,冷冻干燥之后,即得所需产物。
实施例2
N-{5-[2-(4-氯-苯基)-乙基氨基甲酰基]-2-[4-(2-二甲基氨基-乙基)-哌嗪-1-基]-苯基}-异烟酰胺

从中间体2开始,HATU介导的偶联(其中22mg(0.05mmol)初始原料、0.15mmol(57mg)HATU、0.15mmol(19mg)异烟酸以及0.3mmol(39mg=44μL)DIEA)于环境温度下反应3小时。LCMS检查显示所需产物,其在部分蒸发后经Waters质量-触发的-LCMS纯化系统分离。冷冻干燥,即得所需产物。MW=534.25Da(C29H35ClN6O2单同位素计算值),测量值(M+H)+=535.3Da,带适当Cl-同位素模式,基于UV{220}的纯度为83.7%。
实施例3
N-{5-[2-(3-氯-苯基)-乙基氨基甲酰基]-2-[4-(2-二甲基氨基-乙基)-哌嗪-1-基]-苯基}-异烟酰胺

从中间体3开始,HATU介导的偶联(其中22mg(0.05mmol)初始原料、0.15mmol(57mg)HATU、0.15mmol(19mg)异烟酸以及0.3mmol(39mg=44μL)DIEA)于环境温度下反应3小时。LCMS检查显示所需产物,其在部分蒸发后经Waters质量-触发的-LCMS纯化系统分离。冷冻干燥,即得所需产物。MW=534.25Da(C29H35ClN6O2单同位素计算值),测量值(M+H)+=535.3Da,带适当Cl-同位素模式,UV{220}为基础的纯度为90.9%。
实施例4
N-{5-[2-(2-氯-苯基)-乙基氨基甲酰基]-2-[4-(2-二甲基氨基-乙基)-哌嗪-1-基]-苯基}-异烟酰胺
从中间体4开始,HATU介导的偶联(其中22mg(0.05mmol)初始原料、0.15mmol(57mg)HATU、0.15mmol(19mg)异烟酸以及0.3mmol(39mg=44μL)DIEA)于环境温度下反应3小时。LCMS检查显示所需产物,其在部分蒸发后经Waters质量-触发的-LCMS纯化系统分离。冷冻干燥,即得所需产物。MW=534.25Da(C29H35ClN6O2单同位素计算值),测量值(M+H)+=535.3Da,带适当Cl-同位素模式,UV{220}为基础的纯度为100%。
实施例5
噻吩-2-羧酸{2-[4-(2-二甲基氨基-乙基)-哌嗪-1-基]-5-[2-(2-氯-苯基)-乙基氨基甲酰基]-苯基}-酰胺

从中间体4开始,HATU介导的偶联(其中22mg(0.05mmol)初始原料、0.15mmol(57mg)HATU、0.15mmol(19mg)噻吩-2-羧酸以及0.3mmol(39mg=44μL)DIEA)于环境温度下反应3小时。LCMS检查显示所需产物,其在部分蒸发后经Waters质量-触发的-LCMS纯化系统分离。冷冻干燥,即得所需产物。MW=539.21Da(C28H34ClN5O2S单同位素计算值),测量值(M+H)+=540.2Da,带适当Cl-同位素模式,UV{220}为基础的纯度为97.6%。
实施例7
噻吩-2-羧酸{2-[4-(2-二甲基氨基-乙基)-哌嗪-1-基]-5-[2-(3-氯-苯基)-乙基氨基甲酰基]-苯基}-酰胺

从中间体3开始,HATU介导的偶联(其中22mg(0.05mmol)初始原料、0.15mmol(57mg)HATU、0.15mmol(19mg)噻吩-2-羧酸以及0.3mmol(39mg=44μL)DIEA)于环境温度下反应3小时。LCMS检查显示所需产物,其在部分蒸发后经Waters质量-触发的-LCMS纯化系统分离。冷冻干燥,即得所需产物。MW=539.21Da(C28H34ClN5O2S单同位素计算值),测量值(M+H)+=540.2Da,带适当Cl-同位素模式,UV{220}为基础的纯度为97.1%。
实施例8
噻吩-2-羧酸{2-[4-(2-二甲基氨基-乙基)-哌嗪-1-基]-5-[2-(4-氯-苯基)-乙基氨基甲酰基]-苯基}-酰胺
从中间体2开始,HATU介导的偶联(其中22mg(0.05mmol)初始原料、0.15mmol(57mg)HATU、0.15mmol(19mg)噻吩-2-羧酸以及0.3mmol(39mg=44μL)DIEA)于环境温度下反应3小时。LCMS检查显示所需产物,其在部分蒸发后经Waters质量-触发的-LCMS纯化系统分离。冷冻干燥,即得所需产物。MW=539.21Da(C28H34ClN5O2S单同位素计算值),测量值(M+H)+=540.2Da,带适当Cl-同位素模式,UV{220}为基础的纯度为76.9%。
实施例9
3-(3-氯-苯甲酰基氨基)-N-[2-(2-氯-苯基)-乙基]-4-[4-(2-二甲基氨基-乙基)-哌嗪-1-基]-苯甲酰胺

从中间体4开始,HATU介导的偶联(其中22mg(0.05mmol)初始原料、0.15mmol(57mg)HATU、0.15mmol(24mg)3-氯苯甲酸以及0.3mmol(39mg=44μL)DIEA)于环境温度下反应3小时。LCMS检查显示所需产物,其在部分蒸发后经Waters质量-触发的-LCMS纯化系统分离。冷冻干燥,即得所需产物。MW=567.22Da(C30H35Cl2N5O2单同位素计算值),测量值(M+H)+=568.2Da,带适当Cl-同位素模式,UV{220}为基础的纯度为62.4%。
实施例10
3-(3-氯-苯甲酰基氨基)-N-[2-(3-氯-苯基)-乙基]-4-[4-(2-二甲基氨基-乙基)-哌嗪-1-基]-苯甲酰胺

从中间体3开始,HATU介导的偶联(其中22mg(0.05mmol)初始原料、0.15mmol(57mg)HATU、0.15mmol(24mg)3-氯苯甲酸以及0.3mmol(39mg=44μL)DIEA)于环境温度下反应3小时。LCMS检查显示所需产物,其在部分蒸发后经Waters质量-触发的-LCMS纯化系统分离。冷冻干燥,即得所需产物。MW=567.22Da(C30H35Cl2N5O2单同位素计算值),测量值(M+H)+=568.2Da,带适当Cl-同位素模式,UV{220}为基础的纯度为54.5%。
实施例11
3-(3-氯-苯甲酰基氨基)-N-[2-(4-氯-苯基)-乙基]-4-[4-(2-二甲基氨基-乙基)-哌嗪-1-基]-苯甲酰胺
从中间体2开始,HATU介导的偶联(其中22mg(0.05mmol)初始原料、0.15mmol(57mg)HATU、0.15mmol(24mg)3-氯苯甲酸以及0.3mmol(39mg=44μL)DIEA)于环境温度下反应3小时。LCMS检查显示所需产物,其在部分蒸发后经Waters质量-触发的-LCMS纯化系统分离。冷冻干燥,即得所需产物。MW=567.22Da(C30H35Cl2N5O2单同位素计算值),测量值(M+H)+=568.2Da,带适当Cl-同位素模式,UV{220}为基础的纯度为88.5%。
实施例12
3-(3-氯-苯甲酰基氨基)-N-[2-(2,4-二氯-苯基)-乙基]-4-[4-(2-二甲基氨基-乙基)-哌嗪-1-基]-苯甲酰胺

步骤1:将10mL注射器内的0.2g FMPE聚苯乙烯HL树脂(目录号01-64-0254(NovaBiochem-‘Ameba’S=1.54mmol/g,EMD Biosciences,Inc.))在DCM中溶胀60分钟,然后于环境温度下用1mL 2,4-二氯-苯乙基胺在DCM/TMOF(1∶1)中的0.4M溶液(2mmol)处理该树脂12小时,以产生Schiff碱。用THF快速洗涤2遍之后,加入固态NaBH(OAc)3(5当量)处理树脂然后于环境温度下振摇16小时,实现Schiff碱的还原。洗涤该树脂:MeOH1遍,DMF 2遍,MeOH 2遍,DCM 5遍以及DMF 5遍。
步骤2:将185mg(1mmol)4-氟-3-硝基苯甲酸、1mmol(380mg)HOAt以及1mmol(126mg)DIC在4ml DMF/DCM混合物(1∶1)中的溶液加入该树脂。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤3:氟取代反应是于65℃用3ml 4-[2-(N,N-二甲基氨基)-乙基]-哌嗪在NMP中的0.5M溶液处理树脂48小时实现的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤4:硝基是于环境温度下用SnCl2在DMF中的1M溶液处理树脂12小时而还原的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤5:将156mg(1mmol)3-氯苯甲酸、1mmol(380mg)HATU,以及3mmol(385mg=420μL)DIEA在2mL DMF中的溶液加入该树脂。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 3遍,并在真空中干燥。
为进行裂解,将1.5ml TFA/水95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用混合物AN洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分(General procedures section)”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=601.18Da(C30H34Cl3N5O2单同位素计算值),测量值(M+H)+=602.2Da,带适当Cl-同位素模式,UV{220}为基础的纯度为84.5%。
实施例13
3-(3-氯-苯甲酰基氨基)-N-[2-(2,4-二氯-苯基)-乙基]-4-[4-(3-二甲基氨基-丙基)-哌嗪-1-基]-苯甲酰胺

步骤1:按照实施例12执行
步骤2:按照实施例12执行
步骤3:氟取代反应是于65℃用3ml 4-[2-(N,N-二甲基氨基)-丙基]-哌嗪在NMP中的0.5M溶液处理树脂48小时实现的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤4:如实施例12,硝基是于环境温度下用SnCl2在DMF中的1M溶液处理树脂12小时而还原的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤5:将156mg(1mmol)3-氯苯甲酸、1mmol(380mg)HATU,以及3mmol(385mg=420μL)DIEA在2mL DMF中的溶液加入该树脂。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 3遍,并在真空中干燥。
为进行裂解,将1.5ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用混合物AN洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=615.19Da(C31H36Cl3N5O2单同位素计算值),测量值(M+H)+=616.2Da,带适当Cl-同位素模式,UV{220}为基础的纯度为93.1%。
实施例14
4-[4-(2-氨基-乙基)-哌嗪-1-基]-3-(3-氯-苯甲酰基氨基)-N-[2-(2,4-二氯-苯基)-乙基]-苯甲酰胺

步骤1:按照实施例12执行
步骤2:按照实施例12执行
步骤3:氟取代反应是于65℃用boc-4-(2-氨基-乙基)-哌嗪在NMP中的0.5M溶液处理树脂48小时实现的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤4:如实施例12,硝基是于环境温度下用SnCl2在DMF中的1M溶液处理树脂12小时而还原的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤5:将156mg(1mmol)3-氯苯甲酸、1mmol(380mg)HATU,以及3mmol(385mg=420μL)DIEA在2mL DMF中的溶液加入该树脂。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 3遍,并在真空中干燥。
为进行裂解,将1.5ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用混合物AN洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=573.15Da(C28H30Cl3N5O2单同位素计算值),测量值(M+H)+=574.1Da,带适当Cl-同位素模式,UV{220}为基础的纯度为67.7%。
实施例15
3-(3-氯-苯甲酰基氨基)-N-[2-(2,4-二氯-苯基)-乙基]-4-[4-(2-羟基-乙基)-哌嗪-1-基]-苯甲酰胺

步骤1:按照实施例12执行
步骤2:按照实施例12执行
步骤3:氟取代反应是于65℃用2-(2-哌嗪-1-基)-乙醇在NMP中的0.5M溶液处理树脂48小时实现的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤4:如实施例12,硝基是于环境温度下用SnCl2在DMF中的1M溶液处理树脂12小时而还原的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤5:将156mg(1mmol)3-氯苯甲酸、1mmol(380mg)HATU,以及3mmol(385mg=420μL)DIEA在2mL DMF中的溶液加入该树脂。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 3遍,并在真空中干燥。
为进行裂解,将1.5ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用混合物AN洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=574.13Da(C28H29Cl3N4O3单同位素计算值),测量值(M+H)+=575.1Da,带适当Cl-同位素模式,UV{220}为基础的纯度为92.3%。
实施例16
(4-{2-(3-氯-苯甲酰基氨基)-4-[2-(2,4-二氯-苯基)-乙基氨基甲酰基]-苯基}-哌嗪-1-基)-乙酸乙酯

步骤1:按照实施例12执行
步骤2:按照实施例12执行
步骤3:氟取代反应是于65℃用4-(乙氧基羰基-甲基)-哌嗪在NMP中的0.5M溶液处理树脂48小时实现的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤4:如实施例12,硝基是于环境温度下用SnCl2在DMF中的1M溶液处理树脂12小时而还原的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤5:将156mg(1mmol)3-氯苯甲酸、1mmol(380mg)HATU,以及3mmol(385mg=420μL)DIEA在2mL DMF中的溶液加入该树脂。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 3遍,并在真空中干燥。
为进行裂解,将1.5ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用混合物AN洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=616.14Da(C30H31Cl3N4O4单同位素计算值),测量值(M+H)+=617.1Da,带适当Cl-同位素模式,UV{220}为基础的纯度为87.5%。
实施例17
3-(3-氯-苯甲酰基氨基)-N-[2-(2,4-二氯-苯基)-乙基]-4-(4-乙基-哌嗪-1-基)-苯甲酰胺

步骤1:按照实施例12执行
步骤2:按照实施例12执行
步骤3:氟取代反应是于65℃用4-乙基哌嗪在NMP中的0.5M溶液处理树脂48小时实现的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤4:如实施例12,硝基是于环境温度下用SnCl2在DMF中的1M溶液处理树脂12小时而还原的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤5:将156mg(1mmol)3-氯苯甲酸、1mmol(380mg)HATU,以及3mmol(385mg=420μL)DIEA在2mL DMF中的溶液加入该树脂。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 3遍,并在真空中干燥。
为进行裂解,将1.5ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用混合物AN洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=558.14Da(C28H29Cl3N4O2单同位素计算值),测量值(M+H)+=559.1Da,带适当Cl-同位素模式,UV{220}为基础的纯度为100%。
实施例18
3-(3-氯-苯甲酰基氨基)-N-[2-(2,4-二氯-苯基)-乙基]-4-{4-[2-(2-羟基乙氧基)-乙基]-哌嗪-1-基}-苯甲酰胺

步骤1:按照实施例12执行
步骤2:按照实施例12执行
步骤3:氟取代反应是于65℃用2-(2-哌嗪-1-基-乙氧基)-乙醇在NMP中的0.5M溶液处理树脂48小时实现的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤4:如实施例12,硝基是于环境温度下用SnCl2在DMF中的1M溶液处理树脂12小时而还原的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤5:将156mg(1mmol)3-氯苯甲酸、1mmol(380mg)HATU,以及3mmol(385mg=420μL)DIEA在2mL DMF中的溶液加入该树脂。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 3遍,并在真空中干燥。
为进行裂解,将1.5ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用混合物AN洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=618.16Da(C30H33Cl3N4O4单同位素计算值),测量值(M+H)+=619.2Da,带适当Cl-同位素模式,UV{220}为基础的纯度为97%。
实施例19
3-(3-氯-苯甲酰基氨基)-N-[2-(2,4-二氯-苯基)-乙基]-4-[4-(1-甲基-哌啶-4-基)-哌嗪-1-基]-苯甲酰胺

步骤1:按照实施例12执行
步骤2:按照实施例12执行
步骤3:氟取代反应是于65℃用1-(1-甲基-哌啶-4-基)-哌嗪在NMP中的0.5M溶液处理树脂48小时实现的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤4:如实施例12,硝基是于环境温度下用SnCl2在DMF中的1M溶液处理树脂12小时而还原的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤5:将156mg(1mmol)3-氯苯甲酸、1mmol(380mg)HATU,以及3mmol(385mg=420μL)DIEA在2mL DMF中的溶液加入该树脂。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 3遍,并在真空中干燥。
为进行裂解,将1.5ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用混合物AN洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=627.19Da(C32H36Cl3N5O2单同位素计算值),测量值(M+H)+=628.2Da,带适当Cl-同位素模式,UV{220nm}为基础的纯度为83.2%。
---------------------------------------------------------------------------------------------
预载树脂(preloaded resin)I
此预载树脂用于制备实施例20-21、24、61、63-64以及71中的化合物。
预载树脂I的制备:
步骤1:将250ml塑料瓶中的20g FMPE聚苯乙烯HL树脂(目录号01-64-0254,NovaBiochem-‘Ameba’S=0.92mmol/g,EMD Biosciences,Inc.)用29.1g的2,4-二氯-苯乙基胺和32.6g在DCM/TMOF(1∶1)中的混合物于环境温度下处理16小时,以产生并随后还原Schiff碱。然后洗涤该树脂:MeOH2遍,10%AcOH的DMF溶液3遍,DMF 3遍,DCM 3遍。
步骤2:将前述反应获得的15g树脂,用4.27g的4-氟-3-硝基苯甲酸、8.78g HATU以及12.06ml DIEA在DMF/DCM混合物(1∶1)中处理。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 3遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤3:氟取代反应是于65℃用哌嗪在NMP中的0.5M溶液处理5g树脂48小时实现的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
实施例20
3-(3-氯-苯甲酰基氨基)-N-[2-(2,4-二氯-苯基)-乙基]-4-[4-(1-甲基-哌啶-4-基甲基)-哌嗪-1-基]-苯甲酰胺

200mg预载树脂I(在步骤1和2之后)被转化为如下最终产物:
步骤3:氟取代反应是于80℃用4-(1-甲基-哌啶-4-基甲基)-哌嗪在NMP中的0.5M溶液处理5g树脂16小时实现的。洗涤该树脂:DMF 3遍,DCM2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤4:如实施例12,硝基是于环境温度下用SnCl2在DMF中的1M溶液处理树脂12小时而还原的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤5:将156mg(1mmol)3-氯苯甲酸、1mmol(380mg)HATU,以及3mmol(385mg=420μL)DIEA在3.5mL DMF中的溶液加入该树脂。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 2遍,THF 2遍并在真空中干燥。
为进行裂解,将6ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用TFA/水的混合物洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用RP-HPLC Beckman系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=641.21Da(C33H38Cl3N5O2单同位素计算值),测量值(M+H)+=642.2Da,带适当Cl-同位素模式,UV{220}为基础的纯度为79.7%。
实施例21
4-[1,4′]联哌啶-1′-基-3-(3-氯-苯甲酰基氨基)-N-[2-(4-氯-苯基)-乙基]-苯甲酰胺

200mg预载树脂I(在步骤1和2之后)被转化为如下最终产物:
步骤3:氟取代反应是于80℃用[1,4′]联哌啶([1,4′]Bipiperidinyl)在NMP中的2M溶液处理树脂16小时实现的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤4:如实施例12,硝基是于环境温度下用SnCl2在DMF中的1M溶液处理树脂12小时而还原的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤5:将156mg(1mmol)3-氯苯甲酸、1mmol(380mg)HATU,以及3mmol(385mg=420μL)DIEA在3.5mL DMF中的溶液加入该树脂。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 2遍,THF 2遍并在真空中干燥。
为进行裂解,将6ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用TFA/水的混合物洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用RP-HPLC Beckman系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=578.22Da(C32H36Cl2N4O2单同位素计算值),测量值(M+H)+=579.2Da,带适当Cl-同位素模式,UV{220}为基础的纯度为92.3%。
实施例22
3-(3-氯-苯甲酰基氨基)-N-[2-(2,4-二氯-苯基)-乙基]-4-[4-(2-吗啉-4-基-乙基)-哌嗪-1-基]-苯甲酰胺

步骤1:按照实施例12执行
步骤2:按照实施例12执行
步骤3:氟取代反应是于65℃用4-(2-吗啉-4-基-乙基)-哌嗪在NMP中的0.5M溶液处理树脂48小时实现的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤4:如实施例12,硝基是于环境温度下用SnCl2在DMF中的1M溶液处理树脂12小时而还原的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤5:将156mg(1mmol)3-氯苯甲酸、1mmol(380mg)HATU,以及3mmol(385mg=420μL)DIEA在2mL DMF中的溶液加入该树脂。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 3遍,并在真空中干燥。
为进行裂解,将1.5ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用混合物AN洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=643.19Da(C32H36Cl3N5O3单同位素计算值),测量值(M+H)+=644.2Da,带适当Cl-同位素模式,UV{220}为基础的纯度为62.8%。
实施例23
3-(3-氯-苯甲酰基氨基)-N-[2-(2,4-二氯-苯基)-乙基]-4-(4-二甲基氨基甲酰基甲基-哌嗪-1-基)-苯甲酰胺
步骤1:按照实施例12执行
步骤2:按照实施例12执行
步骤3:氟取代反应是于65℃用哌嗪基乙酸二甲基酰胺(piperazino-aceticacid-dimethylamide)在NMP中的0.5M溶液处理树脂48小时实现的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤4:如实施例12,硝基是于环境温度下用SnCl2在DMF中的1M溶液处理树脂12小时而还原的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤5:将156mg(1mmol)3-氯苯甲酸、1mmol(380mg)HATU,以及3mmol(385mg=420μL)DIEA在2mL DMF中的溶液加入该树脂。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 3遍,并在真空中干燥。
为进行裂解,将1.5ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用混合物AN洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=615.16Da(C30H32Cl3N5O3单同位素计算值),测量值(M+H)+=616.2Da,带适当Cl-同位素模式,UV{220}为基础的纯度为99.1%。
实施例24
3-(3-氯-苯甲酰基氨基)-N-[2-(2,4-二氯-苯基)-乙基]-4-[4-(2-二甲基氨基乙酰基)-哌嗪-1-基]-苯甲酰胺

200mg预载树脂I(在步骤1、2和3之后)被转化为如下最终产物:
步骤4:N,N-二甲基-甘氨酸的HATU偶联是经2mmol N,N-二甲基-甘氨酸、2mmol HATU以及6mmol DIEA在6ml DMF中于环境温度下反应12小时而实现的。洗涤该树脂:DMF 3遍,DCM 3遍,DMF 3遍-最后在DMF中溶胀。
步骤4:如实施例12,硝基是于环境温度下用SnCl2在DMF中的1M溶液处理树脂12小时而还原的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤5:将156mg(1mmol)3-氯苯甲酸、1mmol(380mg)HATU,以及3mmol(385mg=420μL)DIEA在3.5mL DMF中的溶液加入该树脂。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 2遍,THF 2遍并在真空中干燥。
为进行裂解,将6ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用TFA/水的混合物洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用RP-HPLC Beckman系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=615.16Da(C30H32Cl3N5O3单同位素计算值),测量值(M+H)+=616.2Da,带适当Cl-同位素模式,UV {220}为基础的纯度为94.7%。
实施例25
3-(3-氯-苯甲酰基氨基)-N-[2-(2,4-二氯-苯基)-乙基]-4-(4-吡啶-2-基甲基-哌嗪-1-基)-苯甲酰胺
步骤1:按照实施例12执行
步骤2:按照实施例12执行
步骤3:氟取代反应是于65℃用4-(吡啶-2-基-甲基)-哌嗪在NMP中的0.5M溶液处理树脂48小时实现的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤4:如实施例12,硝基是于环境温度下用SnCl2在DMF中的1M溶液处理树脂12小时而还原的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤5:将156mg(1mmol)3-氯苯甲酸、l mmol(380mg)HATU,以及3mmol(385mg=420μL)DIEA在2mL DMF中的溶液加入该树脂。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 3遍,并在真空中干燥。
为进行裂解,将1.5ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用混合物AN洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=621.15Da(C32H30Cl3N5O2单同位素计算值),测量值(M+H)+=622.1Da,带适当Cl-同位素模式,UV{220}为基础的纯度为99.3%。
实施例26
3-(3-氯-苯甲酰基氨基)-N-[2-(2,4-二氯-苯基)-乙基]-4-(4-吡啶-3-基甲基-哌嗪-1-基)-苯甲酰胺

步骤1:按照实施例12执行
步骤2:按照实施例12执行
步骤3:氟取代反应是于65℃用4-(吡啶-3-基-甲基)-哌嗪在NMP中的0.5M溶液处理树脂48小时实现的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤4:如实施例12,硝基是于环境温度下用SnCl2在DMF中的1M溶液处理树脂12小时而还原的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤5:将156mg(1mmol)3-氯苯甲酸、1mmol(380mg)HATU,以及3mmol(385mg=420μL)DIEA在2mL DMF中的溶液加入该树脂。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 3遍,并在真空中干燥。
为进行裂解,将1.5ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用混合物AN洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=621.15Da(C32H30Cl3N5O2单同位素计算值),测量值(M+H)+=622.1Da,带适当Cl-同位素模式,UV{220}为基础的纯度为96.4%。
实施例27
3-(3-氯-苯甲酰基氨基)-N-[2-(2,4-二氯-苯基)-乙基]-4-[4-(四氢呋喃-2-基甲基)-哌嗪-1-基]-苯甲酰胺

步骤1:按照实施例12执行
步骤2:按照实施例12执行
步骤3:氟取代反应是于65℃用1-(四氢呋喃-2-基甲基)-哌嗪在NMP中的0.5M溶液处理树脂48小时实现的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤4:如实施例12,硝基是于环境温度下用SnCl2在DMF中的1M溶液处理树脂12小时而还原的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤5:将156mg(1mmol)3-氯苯甲酸、1mmol(380mg)HATU,以及3mmol(385mg=420μL)DIEA在2mL DMF中的溶液加入该树脂。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 3遍,并在真空中干燥。
为进行裂解,将1.5ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用混合物AN洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=614.16Da(C31H33Cl3N4O3单同位素计算值),测量值(M+H)+=615.2Da,带适当Cl-同位素模式,UV{220}为基础的纯度为96.1%。
实施例28
3-(3-氯-苯甲酰基氨基)-N-[2-(2,4-二氯-苯基)-乙基]-4-[4-(2-二甲基氨基-乙基)-哌啶-1-基]-苯甲酰胺

步骤1:按照实施例12执行
步骤2:按照实施例12执行
步骤3:氟取代反应是于65℃用4-(2-二甲基氨基-乙基)-哌啶在NMP中的0.5M溶液处理树脂48小时实现的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤4:如实施例12,硝基是于环境温度下用SnCl2在DMF中的1M溶液处理树脂12小时而还原的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤5:将156mg(1mmol)3-氯苯甲酸、1mmol(380mg)HATU,以及3mmol(385mg=420μL)DIEA在2mL DMF中的溶液加入该树脂。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 3遍,并在真空中干燥。
为进行裂解,将1.5ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用混合物AN洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=600.18Da(C31H35Cl3N4O2单同位素计算值),测量值(M+H)+=601.2Da,带适当Cl-同位素模式,UV{220}为基础的纯度为62%。
实施例29
3-(3-氯-苯甲酰基氨基)-N-[2-(2,4-二氯-苯基)-乙基]-4-[4-(2-甲氧基-乙基)-哌嗪-1-基]-苯甲酰胺

步骤1:按照实施例12执行
步骤2:按照实施例12执行
步骤3:氟取代反应是于65℃用4-(2-甲氧基-乙基)-哌嗪在NMP中的0.5M溶液处理树脂48小时实现的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤4:如实施例12,硝基是于环境温度下用SnCl2在DMF中的1M溶液处理树脂12小时而还原的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤5:将156mg(1mmol)3-氯苯甲酸、1mmol(380mg)HATU,以及3mmol(385mg=420μL)DIEA在2mL DMF中的溶液加入该树脂。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 3遍,并在真空中干燥。
为进行裂解,将1.5ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用混合物AN洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=588.15Da(C29H31Cl3N4O3单同位素计算值),测量值(M+H)+=589.1Da,带适当Cl-同位素模式,UV{220}为基础的纯度为99.5%。
预载树脂II
以下步骤是为最终制备实施例30-53所述的化合物而制备适当的构建物(construct)-预载树脂。
8支20ml注射器每支均充入1g的FMPE聚苯乙烯HL树脂(目录号01-64-0254,NovaBiochem-‘Ameba’S=1.54mmol/g,EMD Biosciences,Inc.)并任其在DCM中溶胀1小时。采用以下步骤。
步骤1:于环境温度下用12mL对应的胺在DCM/TMOF(1∶1)中的0.4M溶液(4.8mmol)处理每个注射器中的溶胀树脂12小时,以产生Schiff碱。所用的胺:2’-氟-苯乙胺、4’-溴-苯乙胺、4’-氟苯乙基胺、1-(4’-氯苯乙基)-2-氨基丙烷、1,2-二苯基-乙胺、反式-2-苯基-1-氨基-环丙烷、2’,6’-二氯-苯乙胺、2-苯基-丙基胺。用THF快速洗涤2遍之后,加入固态NaBH(OAc)3(5当量)处理树脂然后于环境温度下振摇16小时,实现Schiff碱的还原。然后洗涤该树脂:MeOH 2遍,DMF 2遍,MeOH 2遍,DCM 5遍,以及DMF 5遍。
步骤2:将1110mg(6mmol)4-氟-3-硝基苯甲酸、6mmol(2280mg)HATU以及2ml DIEA在10ml DMF/DCM混合物(1∶1)中的溶液加入该树脂。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 5遍,DCM 5遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤3:氟取代反应是于65℃用12ml 4-[2-(N,N-二甲基氨基]-乙基]-哌嗪在NMP中的0.5M溶液处理树脂48小时实现的。洗涤该树脂:DMF 3遍,DCM 3遍,DMF 3遍,(最后得到在DMF中的溶胀树脂)。
步骤4:如实施例12,硝基是于环境温度下用SnCl2在DMF中的1M溶液处理树脂16小时而还原的。洗涤该树脂:DMF 3遍,DCM 3遍,DMF 3遍,(最后得到在DMF中的溶胀树脂)。
将预载树脂分装入5支注射器,每支使用200mg树脂。
----------------------------------------------------------------------------------------------
实施例30
噻吩-3-羧酸{2-[4-(2-二甲基氨基-乙基)-哌嗪-1-基]-5-[2-(2-氟-苯基)-乙基氨基甲酰基]-苯基}-酰胺

步骤5:将128mg(1mmol)噻吩-3-羧酸、1mmol(380mg)HATU,以及3mmol(385mg=420μL)DIEA在3.5mL DMF中的溶液加入装有含有2’-氟苯乙胺构建物的200mg预载树脂II的注射器。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 2遍,THF 2遍并在真空中干燥。
为进行裂解,将6ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用TFA/水的混合物洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=523.24Da(C28H34FN5O2S单同位素计算值),测量值(M+H)+=524.2Da,UV{220}为基础的纯度为99.1%。
实施例31
噻吩-3-羧酸{5-[2-(4-氯-苯基)-1-甲基-乙基氨基甲酰基]-2-[4-(2-二甲基氨基-乙基)-哌嗪-1-基]-苯基}-酰胺

步骤5:将128mg(1mmol)噻吩-3-羧酸、1mmol(380mg)HATU,以及3mmol(385mg=420μL)DIEA在3.5mL DMF中的溶液加入装有含有1-(4’-氯苯基)-2-氨基丙烷构建物的200mg预载树脂II的注射器。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 2遍,THF 2遍并在真空中干燥。
为进行裂解,将6ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用TFA/水的混合物洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=553.23Da(C29H36ClN5O2S单同位素计算值),测量值(M+H)+=554.2Da,带适当Cl-同位素模式,UV{220}为基础的纯度为99.3%。
实施例32
噻吩-3-羧酸{2-[4-(2-二甲基氨基-乙基)-哌嗪-1-基]-5-[2-(4-氟苯基)-乙基氨基甲酰基]-苯基}-酰胺

步骤5:将128mg(1mmol)噻吩-3-羧酸、1mmol(380mg)HATU,以及3mmol(385mg=420μL)DIEA在3.5mL DMF中的溶液加入装有含有4’-氟苯乙胺构建物的200mg预载树脂II的注射器。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 2遍,THF 2遍并在真空中干燥。
为进行裂解,将6ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用TFA/水的混合物洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=523.24Da(C28H34FN5O2S单同位素计算值),测量值(M+H)+=524.2Da,UV{220}为基础的纯度为95.4%。
实施例33
噻吩-3-羧酸{5-[2-(4-溴-苯基)-乙基氨基甲酰基]-2-[4-(2-二甲基氨基-乙基)-哌嗪-1-基]-苯基}-酰胺

步骤5:将128mg(1mmol)噻吩-3-羧酸、1mmol(380mg)HATU,以及3mmol(385mg=420μL)DIEA在3.5mL DMF中的溶液,加入装有含有4’-溴苯乙胺构建物的200mg预载树脂II的注射器。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 2遍,THF 2遍并在真空中干燥。
为进行裂解,将6ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用TFA/水的混合物洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=583.16Da(C28H34BrN5O2S单同位素计算值),测量值(M+H)+=584.2Da,带适当Br-同位素模式,UV{220}为基础的纯度为98.7%。
实施例34
噻吩-3-羧酸[2-[4-(2-二甲基氨基-乙基)-哌嗪-1-基]-5-(2-苯基-丙基氨基甲酰基)-苯基]-酰胺

步骤5:将128mg(1mmol)噻吩-3-羧酸、1mmol(380mg)HATU,以及3mmol(385mg=420μL)DIEA在3.5mL DMF中的溶液,加入装有含有2-苯基-氨基丙烷构建物的200mg预载树脂II的注射器。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 2遍,THF 2遍并在真空中干燥。
为进行裂解,将6ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用TFA/水的混合物洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=519.27Da(C29H37N5O2S单同位素计算值),测量值(M+H)+=520.3Da,UV{220}为基础的纯度为97.7%。
实施例35
噻吩-3-羧酸{5-[2-(2,6-二氯-苯基)-乙基氨基甲酰基]-2-[4-(2-二甲基氨基-乙基)-哌嗪-1-基]-苯基}-酰胺

步骤5:将128mg(1mmol)噻吩-3-羧酸、1mmol(380mg)HATU,以及3mmol(385mg=420μL)DIEA在3.5mL DMF中的溶液,加入装有含有2’,6’-二氯苯乙胺构建物的200mg预载树脂II的注射器。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 2遍,THF 2遍并在真空中干燥。
为进行裂解,将6ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用TFA/水的混合物洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=573.17Da(C28H33Cl2N5O2S单同位素计算值),测量值(M+H)+=574.2Da,带适当Cl-同位素模式,UV{220}为基础的纯度为99.2%。
实施例36
噻吩-3-羧酸[2-[4-(2-二甲基氨基-乙基)-哌嗪-1-基]-5-(2-苯基-环丙基氨基甲酰基)-苯基]-酰胺
步骤5:将128mg(1mmol)噻吩-3-羧酸、1mmol(380mg)HATU,以及3mmol(385mg=420μL)DIEA在3.5mL DMF中的溶液,加入装有含有反式-2-苯基-1-氨基环丙烷构建物的200mg预载树脂II的注射器。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 2遍,THF2遍并在真空中干燥。
为进行裂解,将6ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用TFA/水的混合物洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=517.25Da(C29H35N5O2S单同位素计算值),测量值(M+H)+=518.3Da,UV{220}为基础的纯度为100%。
实施例37
噻吩-3-羧酸[2-[4-(2-二甲基氨基-乙基)-哌嗪-1-基]-5-(1,2-二苯基-乙基氨基甲酰基)-苯基]-酰胺

步骤5:将128mg(1mmol)噻吩-3-羧酸、1mmol(380mg)HATU,以及3mmol(385mg=420μL)DIEA在3.5mL DMF中的溶液,加入装有含有1,2-二苯基乙胺构建物的200mg预载树脂II的注射器。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 2遍,THF 2遍并在真空中干燥。
为进行裂解,将6ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用TFA/水的混合物洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=581.28Da(C34H39N5O2S单同位素计算值),测量值(M+H)+=582.3Da,UV{220}为基础的纯度为99.2%。
实施例38
噻吩-2-羧酸{5-[2-(4-氯-苯基)-1-甲基-乙基氨基甲酰基]-2-[4-(2-二甲基氨基-乙基)-哌嗪-1-基]-苯基}-酰胺
步骤5:将128mg(1mmol)噻吩-2-羧酸、1mmol(380mg)HATU,以及3mmol(385mg=420μL)DIEA在3.5mL DMF中的溶液,加入装有含有1-(4’-氯苯基)-2-氨基-丙烷构建物的200mg预载树脂II的注射器。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 2遍,THF 2遍并在真空中干燥。
为进行裂解,将6ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用TFA/水的混合物洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=553.23Da(C29H36ClN5O2S单同位素计算值),测量值(M+H)+=554.2Da,带适当Cl-同位素模式,UV{220}为基础的纯度为99.6%。
实施例39
噻吩-2-羧酸{2-[4-(2-二甲基氨基-乙基)-哌嗪-1-基]-5-[2-(4-氟-苯基)-乙基氨基甲酰基]-苯基}-酰胺

步骤5:将128mg(1mmol)噻吩-2-羧酸、1mmol(380mg)HATU,以及3mmol(385mg=420μL)DIEA在3.5mL DMF中的溶液,加入装有含有4’-氟-苯乙胺构建物的200mg预载树脂II的注射器。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 2遍,THF 2遍并在真空中干燥。
为进行裂解,将6ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用TFA/水的混合物洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=523.24Da(C28H34FN5O2S单同位素计算值),测量值(M+H)+=524.2Da,UV{220}为基础的纯度为94.3%。
实施例40
噻吩-2-羧酸{5-[2-(4-溴-苯基)-乙基氨基甲酰基]-2-[4-(2-二甲基氨基-乙基)-哌嗪-1-基]-苯基}-酰胺
步骤5:将128mg(1mmol)噻吩-2-羧酸、1mmol(380mg)HATU,以及3mmol(385mg=420μL)DIEA在3.5mL DMF中的溶液,加入装有含有4’-溴-苯乙胺构建物的200mg预载树脂II的注射器。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 2遍,THF 2遍并在真空中干燥。
为进行裂解,将6ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用TFA/水的混合物洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=583.16Da(C29H36ClN5O2S单同位素计算值),测量值(M+H)+=584.2Da,带适当Br-同位素模式,UV{220}为基础的纯度为99.5%。
实施例41
噻吩-2-羧酸{2-[4-(2-二甲基氨基-乙基)-哌嗪-1-基]-5-[2-(2-氟-苯基)-乙基氨基甲酰基]-苯基}-酰胺

步骤5:将128mg(1mmol)噻吩-2-羧酸、1mmol(380mg)HATU,以及3mmol(385mg=420μL)DIEA在3.5mL DMF中的溶液,加入装有含有2’-氟-苯乙胺构建物的200mg预载树脂II的注射器。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 2遍,THF 2遍并在真空中干燥。
为进行裂解,将6ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用TFA/水的混合物洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=523.24Da(C28H34FN5O2S单同位素计算值),测量值(M+H)+=524.2Da,UV{220}为基础的纯度为100%。
实施例42
噻吩-2-羧酸[2-[4-(2-二甲基氨基-乙基)-哌嗪-1-基]-5-(2-苯基-丙基氨基甲酰基)-苯基]-酰胺

步骤5:将128mg(1mmol)噻吩-2-羧酸、1mmol(380mg)HATU,以及3mmol(385mg=420μL)DIEA在3.5mL DMF中的溶液,加入装有含有2-苯基-氨基-丙烷构建物的200mg预载树脂II的注射器。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 2遍,THF 2遍并在真空中干燥。
为进行裂解,将6ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用TFA/水的混合物洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=519.27Da(C29H37N5O2S单同位素计算值),测量值(M+H)+=520.3Da,UV{220}为基础的纯度为99.3%。
实施例43
噻吩-2-羧酸{5-[2-(2,6-二氯-苯基)-乙基氨基甲酰基]-2-[4-(2-二甲基氨基-乙基)-哌嗪-1-基]-苯基}-酰胺

步骤5:将128mg(1mmol)噻吩-2-羧酸、1mmol(380mg)HATU,以及3mmol(385mg=420μL)DIEA在3.5mL DMF中的溶液,加入装有含有2’,6’-二氯-苯乙胺构建物的200mg预载树脂II的注射器。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 2遍,THF 2遍并在真空中干燥。
为进行裂解,将6ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用TFA/水的混合物洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=573.17Da(C28H33Cl2N5O2S单同位素计算值),测量值(M+H)+=574.2Da,带适当Cl-同位素模式,UV{220}为基础的纯度为98.8%。
实施例44
噻吩-2-羧酸[2-[4-(2-二甲基氨基-乙基)-哌嗪-1-基]-5-(2-苯基-环丙基氨基甲酰基)-苯基]-酰胺

步骤5:将128mg(1mmol)噻吩-2-羧酸、1mmol(380mg)HATU,以及3mmol(385mg=420μL)DIEA在3.5mL DMF中的溶液,加入装有含有反式-2-苯基-1-氨基-环丙烷构建物的200mg预载树脂II的注射器。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 2遍,THF 2遍并在真空中干燥。
为进行裂解,将6ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用TFA/水的混合物洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=517.25Da(C29H35N5O2S单同位素计算值),测量值(M+H)+=518.3Da,UV{220}为基础的纯度为100%。
实施例45
噻吩-2-羧酸[2-[4-(2-二甲基氨基-乙基)-哌嗪-1-基]-5-(1,2-二苯基-乙基氨基甲酰基)-苯基]-酰胺

步骤5:将128mg(1mmol)噻吩-2-羧酸、1mmol(380mg)HATU,以及3mmol(385mg=420μL)DIEA在3.5mL DMF中的溶液,加入装有带有1,2-二苯基-乙胺构建物的200mg预载树脂II的注射器。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 2遍,THF 2遍并在真空中干燥。
为进行裂解,将6ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用TFA/水的混合物洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=581.28Da(C34H39N5O2S单同位素计算值),测量值(M+H)+=582.3Da,UV{220}为基础的纯度为99%。
实施例46
3-(3-氯-苯甲酰基氨基)-N-[2-(4-氯-苯基)-1-甲基-乙基]-4-[4-(2-二甲基氨基-乙基)-哌嗪-1-基]-苯甲酰胺

步骤5:将156mg(1mmol)3-氯苯甲酸、1mmol(380mg)HATU,以及3mmol(385mg=420μL)DIEA在3.5mL DMF中的溶液,加入装有含有1-(4’-氯苯基)-2-氨基-丙烷构建物的200mg预载树脂II的注射器。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 2遍,THF 2遍并在真空中干燥。
为进行裂解,将6ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用TFA/水的混合物洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=581.23Da(C31H37Cl2N5O2单同位素计算值),测量值(M+H)+=582.2Da,带适当Cl-同位素模式,UV{220}为基础的纯度为83.3%。
实施例47
3-(3-氯-苯甲酰基氨基)-4-[4-(2-二甲基氨基-乙基)-哌嗪-1-基]-N-[2-(4-氟-苯基)-乙基]-苯甲酰胺
步骤5:将156mg(1mmol)3-氯苯甲酸、1mmol(380mg)HATU,以及3mmol(385mg=420μL)DIEA在3.5mL DMF中的溶液,加入装有含有4’-氟-苯乙胺构建物的200mg预载树脂II的注射器。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 2遍,THF 2遍并在真空中干燥。
为进行裂解,将6ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用TFA/水的混合物洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=551.25Da(C30H35ClFN5O2单同位素计算值),测量值(M+H)+=552.2Da,带适当Cl-同位素模式,UV{220}为基础的纯度为89.4%。
实施例48
N-[2-(4-溴-苯基)-乙基]-3-(3-氯-苯甲酰基氨基)-4-[4-(2-二甲基氨基-乙基)-哌嗪-1-基]-苯甲酰胺

步骤5:将156mg(1mmol)3-氯苯甲酸、1mmol(380mg)HATU,以及3mmol(385mg=420μL)DIEA在3.5mL DMF中的溶液,加入装有含有4’-溴-苯乙胺构建物的200mg预载树脂II的注射器。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 2遍,THF 2遍并在真空中干燥。
为进行裂解,将6ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用TFA/水的混合物洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=611.17Da(C30H35BrClN5O2单同位素计算值),测量值(M+H)+=612.2Da,带适当Br-Cl-同位素模式,UV{220}为基础的纯度为98.1%。
实施例49
3-(3-氯-苯甲酰基氨基)-4-[4-(2-二甲基氨基-乙基)-哌嗪-1-基]-N-[2-(2-氟-苯基)-乙基]-苯甲酰胺

步骤5:将156mg(1mmol)3-氯苯甲酸、1mmol(380mg)HATU,以及3mmol(385mg=420μL)DIEA在3.5mL DMF中的溶液,加入装有含有2’-氟-苯乙胺构建物的200mg预载树脂II的注射器。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 2遍,THF 2遍并在真空中干燥。
为进行裂解,将6ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用TFA/水的混合物洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=551.25Da(C30H35ClFN5O2单同位素计算值),测量值(M+H)+=552.2Da,带适当Cl-同位素模式,UV{220}为基础的纯度为99.2%。
实施例50
3-(3-氯-苯甲酰基氨基)-4-[4-(2-二甲基氨基-乙基)-哌嗪-1-基]-N-(2-苯基丙基)-苯甲酰胺

步骤5:将156mg(1mmol)3-氯苯甲酸、1mmol(380mg)HATU,以及3mmol(385mg=420μL)DIEA在3.5mL DMF中的溶液,加入装有含有2-苯基-1-氨基-丙烷构建物的200mg预载树脂II的注射器。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 2遍,THF 2遍并在真空中干燥。
为进行裂解,将6ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用TFA/水的混合物洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=547.27Da(C31H38ClN5O2单同位素计算值),测量值(M+H)+=548.3Da,带适当Cl-同位素模式,UV{220}为基础的纯度为96.2
实施例51
3-(3-氯-苯甲酰基氨基)-N-[2-(2,6-二氯-苯基)-乙基]-4-[4-(2-二甲基氨基-乙基)-哌嗪-1-基]-苯甲酰胺

步骤5:将156mg(1mmol)3-氯苯甲酸、1mmol(380mg)HATU,以及3mmol(385mg=420μL)DIEA在3.5mL DMF中的溶液,加入装有含有2’,6’-二氯-苯乙胺构建物的200mg预载树脂II的注射器。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 2遍,THF 2遍并在真空中干燥。
为进行裂解,将6ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用TFA/水的混合物洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=601.18Da(C30H34Cl3N5O2单同位素计算值),测量值(M+H)+=602.2Da,带适当Cl-同位素模式,UV{220}为基础的纯度为90.4%。
实施例52
3-(3-氯-苯甲酰基氨基)-4-[4-(2-二甲基氨基-乙基)-哌嗪-1-基]-N-(2-苯基-环丙基)-苯甲酰胺

步骤5:将156mg(1mmol)3-氯苯甲酸、1mmol(380mg)HATU,以及3mmol(385mg=420μL)DIEA在3.5mL DMF中的溶液,加入装有含有反式-2-苯基-1-氨基-环丙烷构建物的200mg预载树脂II的注射器。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 2遍,THF 2遍并在真空中干燥。
为进行裂解,将6ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用TFA/水的混合物洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=545.26Da(C31H36ClN5O2单同位素计算值),测量值(M+H)+=546.3Da,带适当Cl-同位素模式,UV{220}为基础的纯度为100%。
实施例53
3-(3-氯-苯甲酰基氨基)-4-[4-(2-二甲基氨基-乙基)-哌嗪-1-基]-N-(1,2-二苯基-乙基)-苯甲酰胺

步骤5:将156mg(1mmol)3-氯苯甲酸、1mmol(380mg)HATU,以及3mmol(385mg=420μL)DIEA在3.5mL DMF中的溶液,加入装有含有1,2-二苯基-乙胺构建物的200mg预载树脂II的注射器。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 2遍,THF 2遍并在真空中干燥。
为进行裂解,将6ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用TFA/水的混合物洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=609.29Da(C36H40ClN5O2单同位素计算值),测量值(M+H)+=610.3Da,带适当Cl-同位素模式,UV{220}为基础的纯度为98.9%。
实施例54
3-(3-氯-苯甲酰基氨基)-4-(2,8-二氮杂-螺[4.5]癸-8-基)-N-[2-(2,4-二氯-苯基)-乙基]-苯甲酰胺

将在10mL注射器中的预载2,4-二氯-苯乙基胺和4-氟-3-硝基苯甲酸(见1346JU144)的0.5g FMPE聚苯乙烯HL树脂(目录号01-64-0254,NovaBiochem-‘Ameba’S=1.54mmol/g,EMD Biosciences,Inc.)进行如下的步骤3。
步骤3:氟取代反应是于80℃用Boc-2,8-二氮杂-螺[4.5]癸烷在NMP中的0.5M溶液处理树脂16小时实现的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤4:如实施例12,硝基是于环境温度下用SnCl2在DMF中的1M溶液处理树脂16小时而还原的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤5:将468mg(3mmol)3-氯苯甲酸、3mmol(1140mg)HATU以及9mmol(1155mg=1260μL)DIEA在3.5mL DMF中的溶液加入该树脂。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM2遍,THF 2遍并在真空中干燥。
为进行裂解,将6ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用TFA/水的混合物洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用RP-HPLC Beckman系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=584.15Da(C30H31Cl3N4O2单同位素计算值),测量值(M+H)+=585.2Da,带适当Cl-同位素模式,UV{220}为基础的纯度为95.3%。
实施例55
3-(3-氯-苯甲酰基氨基)-N-[2-(2,4-二氯-苯基)-乙基]-4-(2-甲基-2,8--二氮杂-螺[4.5]癸-8-基)-苯甲酰胺

将40mg实施例54的产物溶于2mL DCM,加入50μL AcOH,再加入50μL 40%甲醛水溶液和200mg氰基-硼氢化物树脂。将反应于环境温度下振摇3小时。LCMS显示所需产物的峰。将该混合物过滤、蒸发并用Waters质量-触发的-LCMS纯化系统纯化。MW=598.17Da(C31H33Cl3N4O2单同位素计算值),测量值(M+H)+=599.2Da,带适当Cl-同位素模式,UV{220}为基础的纯度为98.6%。
实施例56
3-(3-氯-苯甲酰基氨基)-4-(3,9-二氮杂-螺[5.5]十一烷-3-基)-N-[2-(2,4-二氯-苯基)-乙基]-苯甲酰胺

将在10mL注射器中的预载2,4-二氯-苯乙基胺和4-氟-3-硝基苯甲酸(见1346JU144)的0.5g FMPE聚苯乙烯HL树脂(目录号01-64-0254,NovaBiochem-‘Ameba’S=1.54mmol/g,,EMD Biosciences,Inc.)进行如下的步骤3。
步骤3:氟取代反应是于80℃用boc-3,9-二氮杂-螺[5.5]十一烷在NMP中的0.5M溶液处理树脂16小时实现的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤4:如实施例12,硝基是于环境温度下用SnCl2在DMF中的1M溶液处理树脂16小时而还原的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤5:将468mg(3mmol)3-氯苯甲酸、3mmol(1140mg)HATU以及9mmol(1155mg=1260μL)DIEA在3.5mL DMF中的溶液加入该树脂。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM2遍,THF 2遍并在真空中干燥。
为进行裂解,将6ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用TFA/水的混合物洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用RP-HPLC Beckman系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=598.17Da(C31H32Cl3N4O2单同位素计算值),测量值(M+H)+=599.2Da,带适当Cl-同位素模式,UV{220}为基础的纯度为88.9%。
实施例57
3-(3-氯-苯甲酰基氨基)-N-[2-(2,4-二氯-苯基)-乙基]-4-(9-甲基-3,9-二氮杂-螺[5.5]十一烷-3-基)-苯甲酰胺

将35mg实施例56的产物溶于2mL DCM,加入50μL AcOH,再加入50μL 40%甲醛水溶液和200mg氰基-硼氢化物树脂。将反应于环境温度下振摇3小时。LCMS显示所需产物的峰。将混合物过滤、蒸发并用Waters质量-触发的-LCMS纯化系统纯化。MW=612.18Da(C32H35Cl3N4O2单同位素计算值),测量值(M+H)+=613.2Da,带适当Cl-同位素模式,UV{220}为基础的纯度为100%。
实施例58
9-{2-(3-氯-苯甲酰基氨基)-4-[2-(2,4-二氯-苯基)-乙基氨基甲酰基]-苯基}-3,3-二甲基-9-氮杂-3-氮鎓-螺[5.5]十一烷三氟乙酸盐

将35mg实施例57的产物溶于2mL丙酮,加入200μL甲基碘,并将该反应混合物在一密封管内于80℃加热1小时。LCMS显示所需产物的峰。用RP-HPLC Beckman系统并按照“一般步骤部分”描述的步骤蒸发和纯化混合物。MW=627.21Da(C33H38Cl3N4O2单同位素计算值),测量值(M+H)+=628.2Da,带适当Cl-同位素模式,UV{220}为基础的纯度为100%。
实施例59
3-(3-氯-苯甲酰基氨基)-4-(2,7-二氮杂-螺[3.5]壬-7-基)-N-[2-(2,4-二氯-苯基)-乙基]-苯甲酰胺
200mg预载树脂I(在步骤1和2之后)按照以下被转化为最终产物:
步骤3:氟取代反应是于80℃用2,7-二氮杂-螺[3.5]壬烷-2-羧酸叔丁基酯在NMP中的0.4M溶液处理树脂16小时实现的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤4:如实施例12,硝基是于环境温度下用SnCl2在DMF中的1M溶液处理树脂12小时而还原的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤5:将156mg(1mmol)3-氯苯甲酸、1mmol(380mg)HATU,以及3mmol(385mg=420μL)DIEA在3.5mL DMF中的溶液加入该树脂。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 2遍,THF 2遍并在真空中干燥。
为进行裂解,将6ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用TFA/水的混合物洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用RP-FHPLC Beckman系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=570.14Da(C29H29Cl3N4O2单同位素计算值),测量值(M+H)+=571.2Da,带适当Cl-同位素模式,UV{220}为基础的纯度为87%。
实施例60
3-(3-氯-苯甲酰基氨基)-N-[2-(2,4-二氯-苯基)-乙基]-4-(2-甲基-2,7-二氮杂-螺[3.5]壬-7-基)-苯甲酰胺

将8mg实施例59制备的化合物溶于1.5ml DCE/TMOF(2∶1)混合物,加入300ul 40%CH2O水溶液,振摇20分钟,然后加入100mg氰基氢硼化物树脂,然后于环境温度下再振摇4小时。滤去树脂并用1ml MeOH洗涤2遍,蒸发萃取物。在用RP-HPLC Beckman系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=584.15Da(C30H31Cl3N4O2单同位素计算值),测量值(M+H)+=583.2Da,带适当Cl-同位素模式,UV{220}为基础的纯度为87%。
实施例61
4-[4-(3-氨基-丙酰基)-哌嗪-1-基]-3-(3-氯-苯甲酰基氨基)-N-[2-(2,4-二氯-苯基)-乙基]-苯甲酰胺

200mg预载树脂I(在步骤1、2和3之后)按照以下被转化为最终产物:
步骤4:Fmoc-β-丙氨酸的HATU偶联是经2mmol Fmoc-β-丙氨酸、2mmolHATU以及6mmol DIEA在6ml DMF中,于环境温度下反应12小时而实现的。洗涤该树脂:DMF 3遍,DCM 3遍,DMF 3遍-最后在DMF中溶胀。
步骤4:如实施例12,硝基是于环境温度下用SnCl2在DMF中的1M溶液处理树脂12小时而还原的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤5:将156mg(1mmol)3-氯苯甲酸、1mmol(380mg)HATU,以及3mmol(385mg=420μL)DIEA在3.5mL DMF中的溶液加入该树脂。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 2遍,THF 2遍并在真空中干燥。
为进行裂解,将6ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用TFA/水的混合物洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用RP-HPLC Beckman系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=601.14Da(C29H30Cl3N5O3单同位素计算值),测量值(M+H)+=602.1Da,带适当Cl-同位素模式,UV{220}为基础的纯度为91.2%。
实施例62
3-(3-氯-苯甲酰基氨基)-N-[2-(2,4-二氯-苯基)-乙基]-4-[4-(3-二甲基氨基-丙酰基)-哌嗪-1-基]-苯甲酰胺
200mg预载树脂I(在步骤1、2和3之后)按照以下被转化为最终产物:
步骤4:N,N-二甲基-β-丙氨酸的HATU偶联是经2mmol N,N-二甲基-β-丙氨酸、2mmol HATU以及6mmol DIEA在6ml DMF中于环境温度下反应12小时实现的。洗涤该树脂:DMF 3遍,DCM 3遍,DMF 3遍-最后在DMF中溶胀。
步骤4:如实施例12,硝基是于环境温度下用SnCl2在DMF中的1M溶液处理树脂12小时而还原的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤5:将312mg(2mmol)3-氯苯甲酸、2mmol(760mg)HATU,以及6mmol(770mg=840μL)DIEA在7mL DMF中的溶液加入该树脂。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 2遍,FHF 2遍并在真空中干燥。
为进行裂解,将6ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用TFA/水的混合物洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=629.17Da(C31H34Cl3N5O3单同位素计算值),测量值(M+H)+=630.2Da,带适当Cl-同位素模式,UV{220}为基础的纯度为91.4%。
实施例63
3-(3-氯-苯甲酰基氨基)-N-[2-(2,4-二氯-苯基)-乙基]-4-[4-(3-二乙基氨基-丙酰基)-哌嗪-1-基]-苯甲酰胺

与实施例66中的化合物完全一样地在300mg预载树脂上制备,但用N,N-二乙基-β-丙氨酸代替N,N-二甲基-β-丙氨酸。裂解的化合物用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤纯化。MW=657.2Da(C33H38Cl3N5O3单同位素计算值),测量值(M+H)+=658.2Da,带适当Cl-同位素模式,UV{220}为基础的纯度为56.3%。
实施例64
3-(3-氯-苯甲酰基氨基)-N-[2-(2,4-二氯-苯基)-乙基]-4-[4-(3-吡咯烷-1-基-丙酰基)-哌嗪-1-基]-苯甲酰胺

与实施例62中的化合物完全一样地在300mg预载树脂上制备,但用(经迈克尔(Michael)加成)新制备的粗3-吡咯烷基(pyrrolidino)-丙酸代替N,N-二甲基-β-丙氨酸。裂解的化合物用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤纯化。MW=655.19Da(C33H36Cl3N5O3单同位素计算值),测量值(M+H)+=656.2Da,带适当Cl-同位素模式,UV{220}为基础的纯度为96.9%。
实施例65
3-(3-氯-苯甲酰基氨基)-N-[2-(2,4-二氯-苯基)-乙基]-4-{4-[3-(3,3,4,4-四氟-吡咯烷-1-基)-丙酰基]-哌嗪-1-基}-苯甲酰胺
与实施例66中的化合物完全一样地在300mg预载树脂上制备,但用(经迈克尔加成)新制备的粗3-(3’,3’,4’,4’-四氟吡咯烷基)-丙酸代替N,N-二甲基-β-丙氨酸。裂解的化合物用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤纯化。MW=727.15Da(C33H32Cl3F4N5O3单同位素计算值),测量值(M+H)+=728.2Da,带适当Cl-同位素模式,UV{220}为基础的纯度为100%。
实施例66
4-[4-(3-氮杂环丙烷-1-基-丙酰基)-哌嗪-1-基]-3-(3-氯-苯甲酰基氨基)-N-[2-(2,4-二氯-苯基)-乙基]-苯甲酰胺

与实施例66中的化合物完全一样地在300mg预载树脂上制备,但用(经迈克尔加成)新制备的粗3-氮杂环丙烷基(aziridino)-丙酸代替N,N-二甲基β-丙氨酸。裂解的化合物用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤纯化。MW=627.16Da(C31H32Cl3N5O3单同位素计算值),测量值(M+H)+=628.2Da,带适当Cl-同位素模式,UV{220}为基础的纯度为19.1%。
实施例67
3-(3-氯-苯甲酰基氨基)-N-[2-(2,4-二氯-苯基)-乙基]-4-[4-(3-甲基氨基-丙酰基)-哌嗪-1-基]-苯甲酰胺

与实施例62中的化合物完全一样地在300mg预载树脂上制备,但用N-甲基-Fmoc-β-丙氨酸代替N,N-二甲基-β-丙氨酸。裂解的化合物先于环境温度下用3ml哌啶/DMF(1∶1)处理产物30分钟以进行Fmoc脱保护,然后再用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤纯化。MW=615.16Da(C30H32Cl3N5O3单同位素计算值),测量值(M+H)+=616.3Da,带适当Cl-同位素模式。
实施例68
3-(3-氯-苯甲酰基氨基)-4-[4-(2-二甲基氨基-乙基)-哌嗪-1-基]-N-甲基-N-苯乙基-苯甲酰胺
步骤2:将1288mg(4mmol)中间体1与504mg(4mmol)DIC和1836mg(=12mmol)HOBt以及810mg(6mmol)N-甲基-苯乙基胺的4ml DMF溶液一起溶于10mL DMF。2小时之后,于环境温度下蒸发2/3溶剂,用RP-HPLCBeckman系统并按照“一般步骤部分”描述的步骤纯化油状残余物。冷冻干燥适当级分,即得450mg所需的物质。
步骤3:将400mg前述反应的产物溶于40mL甲醇,并加入250mg 10%Pd/C。在帕尔设备中抽真空之后,引入30psi的氢。于环境温度下振摇该反应混合物3小时。分析LCMS显示产物。用二氧化硅垫滤去催化剂,在ROTAVAP上蒸发甲醇。该产物无需纯化即用于下一步反应。
标准的HATU介导的偶联(其中110mg(0.25mmol)初始原料、1mmol(380mg)HATU、1mmol(156mg)3-氯苯甲酸以及3mmol(390mg=440μL)DIEA)于环境温度下反应3小时。LCMS检查显示所需产物,其在部分蒸发后经Waters质量-触发的-LCMS纯化系统分离。冷冻干燥,即得所需产物。MW=547.23Da(C31H38ClN5O2单同位素计算值),测量值(M+H)+=548.3Da,带适当Cl-同位素模式,UV{220}为基础的纯度为98%。
实施例69
4-{2-(3-氯-苯甲酰基氨基)-4-[2-(2,4-二氯-苯基)-乙基氨基甲酰基]-苯基}-哌嗪-1-羧酸乙酰胺

200mg预载树脂I(在步骤1、2和3之后)按照以下被转化为最终产物:
步骤4:将注射器中的树脂于50C用149mg异氰酸乙酯在DMF中处理2小时。洗涤:DMF 3遍,DCM 3遍,DMF3遍-最后在DMF中溶胀。
步骤4:如实施例12,硝基是于环境温度下用SnCl2在DMF中的1M溶液处理树脂12小时而还原的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤5:将312mg(2mmol)3-氯苯甲酸、2mmol(760mg)HATU,以及6mmol(770mg=840μL)DIEA在7mL DMF中的溶液加入该树脂。此偶联反应于环境温度(RT)下持续12小时。洗涤该树脂:DMF 3遍,DCM 2遍,THF 2遍并在真空中干燥。
为进行裂解,将6ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用TFA/水的混合物洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=601.14Da(C29H30Cl3N5O3单同位素计算值),测量值(M+H)+=602.1Da,带适当Cl-同位素模式,UV{220}为基础的纯度为95.4%。
实施例70
N-{5-[2-(2,4-二氯-苯基)-乙基氨基甲酰基]-2-[4-(3-二甲基氨基-丙酰基)-哌嗪-1-基]-苯基}-间氨基甲酰苯甲酸(isophthalamic acid)

200mg预载树脂I(在步骤1、2和3之后)按照以下被转化为最终产物:
步骤4:N,N-二甲基-β-丙氨酸的HATU偶联是经2mmol N,N-二甲基-β-丙氨酸、2mmol HATU以及6mmol DIEA在6ml DMF中于环境温度下反应12小时实现的。洗涤该树脂:DMF 3遍,DCM 3遍,DMF 3遍-最后在DMF中溶胀。
步骤5:如实施例12,硝基是于环境温度下用SnCl2在DMF中的1M溶液处理树脂12小时而还原的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤6:将注射器中的树脂用新制备的间苯二酸的对称酐(sym-anhydride)(通过将2mmol氨基酸与1mmol DIC混合5分钟而制备)在DMF中于环境温度下处理12小时。洗涤该树脂:DMF 3遍,DCM 3遍,THF 3遍-然后在真空中干燥。
为进行裂解,将6ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用TFA/水的混合物洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=639.2Da(C32H35Cl2N5O5单同位素计算值),测量值(M+H)+=640.2Da,带适当Cl-同位素模式,UV{220}为基础的纯度为94.9%。
实施例71
N-{5-[2-(2,4-二氯-苯基)-乙基氨基甲酰基]-2-[4-(3-二甲基氨基-丙酰基)-哌嗪-1-基]-苯基}-对氨基甲酰苯甲酸(terephthalamic acid)
200mg预载树脂I(在步骤1、2和3之后)按照以下被转化为最终产物:
步骤4:N,N-二甲基-β-丙氨酸的HATU偶联是经2mmol N,N-二甲基-β-丙氨酸、2mmol HATU以及6mmol DIEA在6ml DMF中于环境温度下反应12小时实现的。洗涤该树脂:DMF 3遍,DCM 3遍,DMF 3遍-最后在DMF中溶胀。
步骤5:如实施例12,硝基是于环境温度下用SnCl2在DMF中的1M溶液处理树脂12小时而还原的。洗涤该树脂:DMF 3遍,DCM 2遍,DMF 2遍,(最后得到在DMF中的溶胀树脂)。
步骤6:将注射器中的树脂用新制备的对苯二甲酸的对称酐(通过将2mmol氨基酸与1mmol DIC混合5分钟而制备)在DMF中于环境温度下处理12小时。洗涤该树脂:DMF 3遍,DCM 3遍,THF 3遍-然后在真空中干燥。
为进行裂解,将6ml TFA/水的95∶5混合物加入该干树脂,并于室温下振摇4小时。滤去该树脂,用TFA/水的混合物洗涤并将合并的萃取物在真空中蒸发。将粗产物溶于AN/水混合物并冷冻干燥。在用Waters质量-触发的-LCMS纯化系统并按照“一般步骤部分”描述的步骤进行HPLC纯化之后,分离出纯的标题化合物。MW=639.2Da(C32H35Cl2N5O5单同位素计算值),测量值(M+H)+=640.2Da,带适当Cl-同位素模式,UV{220}为基础的纯度为94.2%。
药理学
本文所述的依照本发明的化合物能够用于调节通过CxCR3的信号,因此,还可用于治疗依赖于CxCR3功能的疾病或过程。
相应地,此处所述的发明涉及一种治疗患病患者的方法,包括接触有效量的本发明的结构式的化合物或组合物。
此外,本文叙述的另一项发明涉及治疗患有或遭受炎症的患者的方法,包括给患者施用药学上有效量的式1的化合物。本文提及的治疗应被理解为包括预防性治疗以预防或抑制疾病,以及治疗已确诊的急性或慢性病症或相关的生理状况,以便从本质上治愈该患者,抑制疾病的程度(量)或改善与其相关的生理状况。“有效量”旨在表述本发明的化合物的量,它在合理的生物学判断范围内是有效的,适合用于与人类和其他哺乳动物的细胞接触而不会产生过度的毒性、刺激和变应性反应等,且在治疗中显示出合理的受益/风险比,从而产生所需的治疗效果。
本文讨论的生理状况包括一些可能的有理由进行抗病治疗的临床情况,但并不包括所有情况。本领域内有经验人员很了解那些需要治疗的情况。
本发明的一个特别方面提供了以药物组合物形式给药的本发明的化合物,尽管该化合物也可单独给药。取决于给药方式的性质、剂量和形式,“药物组合物”意为含有式1的化合物以及至少一种选自以下的成分的组合物:药学上可接受的载体、稀释剂、包衣、佐剂、赋形剂或媒介,如防腐剂、填充剂、崩解剂、润湿剂、乳化剂、乳液稳定剂、悬浮剂、等渗剂、甜味剂、调味剂、芳香剂、着色剂、抗细菌剂、抗真菌剂、其他治疗剂、润滑剂、吸附延缓或促进剂、以及分配剂。该组合物可以下列形式给药:片剂、丸剂、颗粒、粉剂、水溶液或悬浮液、注射溶液、酏剂或糖浆。代表性的悬浮剂包括乙氧基化异硬脂醇、聚氧乙烯山梨糖醇和失水山梨糖醇酯、微晶纤维素,偏氢氧化铝(aluminum metahydroxide)、膨润土、琼脂和黄蓍胶,或这些物质的混合物。代表性的预防微生物作用的抗细菌剂和抗真菌剂包括对羟基苯甲酸酯、氯丁醇、苯酚、山梨酸等。代表性的等渗剂包括糖、氯化钠等等。代表性的用于延缓吸收的吸附延缓剂包括单硬脂酸铝和明胶。代表性的用于增加吸收的吸附促进剂包括二甲基亚砜和相关类似物。代表性的载体、稀释剂、溶剂、媒介、增溶剂、乳化剂和乳液稳定剂包括水、氯仿、蔗糖、乙醇、异丙醇、碳酸乙酯、乙酸乙酯、苯甲醇、四氢糠醇、苯甲酸苯甲酯、多元醇、丙二醇、1,3-丁二醇、甘油、聚乙二醇、二甲基甲酰胺、 60,
60, 60,鲸蜡硬脂醇、肉豆蔻醇、单硬脂酸甘油酯和月桂基硫酸钠、失水山梨糖醇脂肪酸酯、植物油(如棉籽油、花生油、玉米胚油、橄榄油、蓖麻油和芝麻油)以及可注射的有机酯如油酸乙酯等,或这些物质的适当混合物。代表性的赋形剂包括乳糖(lactose)、乳糖(milk sugar)、柠檬酸钠、碳酸钙和磷酸二钙。代表性的崩解剂包括淀粉、藻酸以及某些复合硅酸盐类。代表性的润滑剂包括硬脂酸镁、月桂基硫酸钠、滑石,以及高分子量聚乙二醇。
60,鲸蜡硬脂醇、肉豆蔻醇、单硬脂酸甘油酯和月桂基硫酸钠、失水山梨糖醇脂肪酸酯、植物油(如棉籽油、花生油、玉米胚油、橄榄油、蓖麻油和芝麻油)以及可注射的有机酯如油酸乙酯等,或这些物质的适当混合物。代表性的赋形剂包括乳糖(lactose)、乳糖(milk sugar)、柠檬酸钠、碳酸钙和磷酸二钙。代表性的崩解剂包括淀粉、藻酸以及某些复合硅酸盐类。代表性的润滑剂包括硬脂酸镁、月桂基硫酸钠、滑石,以及高分子量聚乙二醇。
其他治疗剂可与本发明的的化合物组合使用,包括其他抗剂(anti agent)。与本发明的化合物组合使用的其他治疗剂可单独、同时或依次给药。除式1的化合物以外药物组合物中其他物质的选择,通常取决于活性化合物的化学性质如溶解性、具体给药方式和用药实践中须遵守的规定。例如,赋形剂如乳糖、柠檬酸钠、碳酸钙、磷酸二钙,崩解剂如淀粉、藻酸以及一些与硬脂酸镁、月桂基硫酸钠及滑石等润滑剂相组合的复合硅酸盐类都可用于制备片剂。
药物组合物可以下列各种形式给药:片剂、丸剂、颗粒、散剂、水性溶液或悬浮液、注射溶液、酏剂或糖浆。
“液体剂型”意为欲给患者施用的活性化合物的剂型是处于液态,例如药学上可接受的乳液、溶液、悬浮液、糖浆和酏剂。除活性化合物的外,液体剂型可含有本领域内常用的惰性稀释剂,例如溶剂、增溶剂和乳化剂。
固体药物组合物也可作为软胶囊和硬胶囊的填充物,使用乳糖(lactose)、乳糖(milk sugar)以及高分子量聚乙二醇等为赋形剂。
当使用水基悬浮液时,它们可含有乳化剂或促进悬浮的试剂。
乳液状药物组合物的油相可以已知的方式由已知的成分组成。虽然该油相可仅由一种乳化剂(另外已知为emulgent)组成,但它最好含有由至少一种乳化剂与脂肪或油,或与脂肪和油两者所组成的混合物。在一项优选的实施方案中,亲水乳化剂与作为稳定剂的亲脂乳化剂一起使用。该乳化剂在有或没有稳定剂的情况下构成乳化蜡,与油和脂肪一起则构成乳化软膏基质,后者形成乳膏制剂的油性分散相。
如果需要,乳膏基质的水相可包括例如至少30%w/w的多元醇,即含有两个或两个以上羟基的醇,如丙二醇、1,3-丁二醇、甘露醇、山梨糖醇、甘油和聚乙二醇(包括PEG 400)及其混合物。局部应用的制剂可理想地含有能促进活性成分吸收或促进活性成分穿透皮肤或其他受影响部位的化合物。
适合于制剂的油类或脂肪的选择是基于能否达到理想的性质。因此,该乳膏最好应是非油脂、不着色及容易洗去的产品,并具有适当的稠度以避免从管或其他容器中渗漏。直链或支链、一元或二元的烷基酯如豆蔻酸二异丙酯、油酸癸酯、棕榈酸异丙酯、硬脂酸丁酯、棕榈酸-2-乙基己酯或被称为Crodamol CAP的支链酯混合物均可使用。取决于所需的性质,这些助剂可单独使用或组合使用。或者,也可使用高熔点脂质如白色软石蜡和/或液体石蜡或其他矿物油。
实际上,本发明的化合物/药物组合物可以一种适合于人和动物的制剂形式,通过局部性或全身性给药方式施用,包括口服、吸入、直肠、鼻腔、口腔、舌下、阴道、结肠、肠胃外(包括皮下、肌内、静脉内、皮内、鞘内和硬膜外)、脑池内,以及腹膜内给药。应该理解,优选的途径可随例如受药者的身体状况而改变。
“药学上可接受的剂型”指本发明的化合物的剂型,包括例如片剂、糖衣丸、散剂、酏剂、糖浆、包括悬浮液在内的液体制剂、喷雾剂、吸入片剂、锭剂、乳液、溶液、颗粒、胶囊和栓剂,以及用于注射的液体制剂,包括脂质体制剂。其技术和制剂通常可在雷氏药学大全(Remington′s PharmaceuticalSciences,Mack Publishing Co.,Easton,PA,最新版)中找到。
“适合于口服给药的制剂”可制成独立的单元如每剂含有预定量活性成分的胶囊、扁囊剂或片剂;或粉末或颗粒;或溶液或水基或非水基液体悬浮液;或水包油液体乳液或油包水液体乳液。活性成分也可制成大丸剂、药糖剂或糊剂。
片剂可以压制或模制的方式制备,还可任选地含有一种或多种辅助成分。压制片剂可通过将活性成分以自由流动形式如粉末或颗粒,任选地与粘合剂、润滑剂、惰性稀释剂、防腐剂、表面活性剂或分散剂混合,再在适当的机器中压制而成。模制片剂可通过用惰性液体稀释剂润湿的粉末状化合物的混合物在适当的机器中模制而成。片剂可任选地包衣或刻痕,也可配制成使所含活性成分得以缓慢地或控制性释放。
用于直肠给药的固体组合物包括按照已知方法配制的栓剂,并含有至少一种本发明的化合物。
如果需要,并为了更有效地分布,该化合物可用微囊密封或附着于缓释或靶向递送体系,例如生物相容的、可生物降解的聚合物基质(如d,l-丙交酯乙交酯共聚物(poly(d,l-lactide co-glycolide))、脂质体和微球体,并通过一种被称为皮下或肌内贮库的技术进行皮下注射或肌内注射,以使该化合物在两周或更长时间内得以持续缓慢地释放。该化合物可以各种方式灭菌,例如,通过除菌过滤器过滤,或通过加入无菌固体组合物形式的灭菌剂,在紧接使用前可将其溶于无菌水或其他无菌注射介质。
“适合于经鼻或吸入途径给药的制剂”意为适合于经鼻或吸入途径向患者给药的制剂形式。此制剂可含有粉末状载体,其粒径为例如1至500微米的范围(包括20和500微米之间的粒径,以5微米为增量,例如30微米、35微米等)。其载体为液体的适当制剂,例如作为鼻腔喷剂或鼻腔滴剂而给药的制剂,包括活性成分的水溶液或油溶液。适合于以气雾剂方式给药的制剂可按照传统方法制备,并可与其他治疗剂一起给药。吸入治疗剂可容易地通过计量吸入器给药。
“适合于口服给药的制剂”意为适合于经口给患者给药的制剂形式。该制剂可制成独立单元如每剂含有预定量活性成分的胶囊、扁囊剂或片剂;或粉末或颗粒;或溶液或水基或非水基液体悬浮液;或水包油液体乳液或油包水液体乳液。活性成分也可制成大丸剂、药糖剂或糊剂。
“适合于肠胃外给药的制剂”意为适合于向患者肠胃外给药的制剂形式。此制剂是无菌的,且包括乳液、悬浮液、水基与非水基注射溶液,可含有悬浮剂和增稠剂以及抗氧化剂、缓冲剂、抑菌剂,以及使该制剂与预期受药者的血液等渗并调节至适当pH值的溶质。
“适合于直肠或阴道给药的制剂”意为适合于经直肠或阴道给患者给药的制剂形式。栓剂是这类制剂的一种具体形式,可将本发明的化合物与适当的无刺激性赋形剂或载体如可可脂、聚乙二醇或某种栓剂用蜡混合的方式来制备。这些赋形剂或载体在常温下是固体但在体温下成为液体,因此可在直肠或阴道腔中融化并释放活性组分。
“适合于全身性给药的制剂”意为适合于给患者全身性给药的制剂形式。此制剂优选是以包括经肌肉、静脉内、腹腔内和皮下注射在内的注射方式给药。为了注射,本发明的化合物配制在液体溶液中,尤其是生理上相容的缓冲液如汉克(Hank)溶液或林格氏(Ringer)溶液中。此外,该化合物可配制成固态形式并在使用之前再重新溶解或悬浮。冻干的形式也包括在内。全身性给药也可采取经粘膜或经皮的方式,或者该化合物也可经口服途径给药。对于经粘膜或经皮方式给药,在制剂中使用适合于欲穿透障碍的渗透剂。这类渗透剂是本领域内周知的,且包括用于经粘膜给药的例如胆盐和梭链孢酸衍生物。此外,还可使用促进渗透的洗净剂。经粘膜给药可通过使用例如鼻腔喷剂或栓剂来实现。对于经口给药方式,该化合物被配制成常规的经口给药形式如胶囊、片剂以及滋补剂(tonics)。
“适合于局部给药的制剂”意为适合于给患者局部给药的制剂形式。此制剂可配制成本领域内众所周知的局部使用的软膏、油膏、粉剂、喷雾剂和吸入剂、凝胶剂(水基或醇基)、乳膏;或者加入至基质中以贴片形式敷用,使得该化合物可经由皮肤障碍控制性释放。当配制成软膏时,活性成分可与石蜡或水混溶性软膏基质一起使用。或者,活性成分可与水包油乳膏基质配制成乳膏。适合于在眼睛里局部给药的制剂包括滴眼剂,其中活性成分溶解或悬浮于适当的载体中,尤其是适合于该活性成分的水性溶剂。适合于在口腔内局部给药的制剂包括调味基质中含有活性成分的锭剂,该调味基质通常是蔗糖和阿拉伯胶或黄蓍胶;还包括惰性基质中含有活性成分的软锭剂(pastille),该惰性基质的例子为明胶和甘油,或蔗糖和阿拉伯胶;还包括在适当液体载体中含有活性成分的漱口剂。
“固体剂型”意为本发明的化合物的剂型是固态形式,例如胶囊、片剂、丸剂、粉末、糖衣丸或颗粒。在这种固体剂型中,本发明的化合物与至少一种常用的惰性赋形剂(或载体)混合,如柠檬酸钠或磷酸二钙或(a)填充剂或增量剂(extender),例如淀粉、乳糖、蔗糖、葡萄糖、甘露醇和硅酸,(b)粘合剂,例如羧甲基纤维素、藻酸盐类、明胶、聚乙烯吡咯烷酮、蔗糖和阿拉伯胶,(c)保湿剂,例如甘油,(d)崩解剂,例如琼脂、碳酸钙、马铃薯淀粉或木薯淀粉、藻酸、某些复合的硅酸盐类和碳酸钠,(e)溶液阻滞剂,例如石蜡,(f)吸收促进剂,例如季铵化合物,(g)润湿剂,例如鲸蜡醇和单硬脂酸甘油酯,(h)吸附剂,例如高岭土和膨润土,(i)润滑剂,例如滑石、硬脂酸钙、硬脂酸镁、固体聚乙二醇、月桂基硫酸钠,(j)遮光剂,(k)缓冲剂,以及在肠道内某一部分以缓释方式释放本发明的化合物的试剂。
本发明的组合物所含活性成分的实际剂量水平可以改变,以便获得活性成分的量,该量使得患者对某种特定的组合物和给药方法产生理想的治疗反应。因此,为任何具体患者选择的剂量水平取决于多种因素,包括所希望的治疗作用、给药途径、所希望的治疗持续时间、疾病的病因和严重性、患者的病情、体重、性别、饮食和年龄、每种活性成分的类型和效能、吸收、代谢和/或排泄的速率及其他因素。
患者每日单次或分次服用的本发明的化合物的日总剂量可以是,例如,每天约0.001至约100mg/kg体重,优选的是0.01至10mg/kg/天。例如,一个成年人每日的吸入剂量通常是约0.001至约100mg/kg体重,优选约0.01至约10mg/kg体重;每日的口服剂量是约0.01至约100mg/kg体重,优选0.1至70mg/kg体重;尤佳的是0.5至10mg/kg体重;每日的静脉内给药剂量是约0.01至约50mg/kg体重,更优选的是0.01至10mg/kg体重。组合物中活性组分的百分比可以改变,但它仍应构成一定的比例,以获得某一适当的剂量。单位剂量医药组合物的含量可以是每日剂量的一部分,由若干单位剂量组成每日剂量。显然,几种单位剂型可以在几乎同时给药。为了获得理想的治疗效果,可以根据需要而尽量频繁地施用某一剂量。某些患者可能会对较高或较低的剂量迅速作出反应,也可能会发现低得多的维持剂量已经足够。对于另一些患者,可能有必要按照每个具体患者的生理要求,进行每日1至4剂的长期治疗。自不待言,对于另一些患者,将有必要规定每日不超过一剂或两剂。
该制剂可用药剂学领域中众所周知的任何方法制备成单位剂量形式。这些方法包括将活性成分与构成一种或多种辅助成分的载体相结合的步骤。通常,这些制剂通过将活性组分与液体载体或磨得很细的固体载体或这两者一起均匀和密切地结合,然后,若有必要,使产品成形而制备。
这些制剂可置于单位剂量或多剂量容器内,例如密封的安瓿剂和带胶塞的瓶,并可在冻干(冷冻干燥)条件下储存,只需在即将使用之前加入无菌液状载体如注射用水。临时调配的注射溶液和悬浮液可从前述的那类无菌粉末、颗粒和片剂制备。
根据文献和下文所述的试验,本发明范围内的化合物展示了显著的药理学活性,该试验结果据信是与在人和其他哺乳动物中的药理学活性相关的。
以上引述的参考文献中所述的化学反应通常以其最广义的应用披露了本发明的化合物制备。有时,对于本文所披露化合物范围内的每种化合物,上述化学反应也可能不适用。发生这种情况的化合物将为本领域熟练技术人员很容易地识别。在所有这类情况下,或者可以通过本领域熟练技术人员周知的常规性修改措施(例如适当保护干扰基团、通过改用替代的常规反应剂、通过常规的修改反应条件等等)而成功地实现这些反应;或者将本文披露的其他反应或其他常规反应可应用于本发明的相应化合物的制备。在所有的制备方法中,所有初始原料均是已知的或可从已知的初始原料制备。
用本发明的化合物和/或组合物治疗患有相应疾病的患者时,所采用的疗法是按照各种因素而选择的,包括年龄、体重、性别、饮食以及患者的病情、感染的严重性、给药途径、药理学方面的考量如所采用具体化合物的活性、功效、药代动力学和毒性,以及是否使用给药系统等因素。本文披露的药物组合的给药通常应持续一段时期直至用药时长可被接受,显示疾病已被控制或根除。接受本文披露的药物组合治疗的患者可常规地通过测量适当临床指标而得到监测,以确定治疗的有效性。这样的衡量标准(metrics)包括例如监视COPD患者肺部功能的FEV1(一秒内用力呼气量)、评估关节炎患者的活动范围,以及定量分析不同炎症状况下热度的热扫描。组织肿胀和外观(肿瘤和发红)的测量也是日常临床实践的有用衡量标准。用这些方法所获资料的连续分析,使得可对治疗期间治疗方案做出修改,从而使组合中每种组分的给药量达到最优,还有助于确定治疗的持续时间。如此,治疗过程中可合理地修改治疗方案/给药计划,使得组合中使用的每种化合物的量达到最低,却又能在一起展现令人满意的效果,并使得组合中这些化合物的给药只持续为成功治愈患者所必需的时间。
本发明包括将其他类型化合物和上述具有CxCR3活性的化合物组合使用以治疗或预防疾病,由于它们的累加或协同作用,其中一种或多种这些化合物以药物有效量存在,而其余的一种或多种化合物则可以低于临床药物有效量或名义(nominally)有效量存在。本文所用的术语“累加作用”描述了两种(或两种以上)药物活性剂的合并作用,它等于每种试剂单独给药时作用的总和。协同作用是指两种(或两种以上)药物活性剂的合并作用大于每种试剂单独给药时作用的总和。当组合给药时可预期多种化合物将有益于患者,这些可能性取决于待治的具体疾病。这些药包括非类固醇抗炎药如乙酰水杨酸及其同类物(cogeners),2-芳基丙酸衍生物如布洛芬。可组合镇痛药如对乙酰氨基酚、阿片剂(opiates)以及它们的类似物。某些疾病的治疗可得益于与类固醇的组合使用,例如泼尼松或其他糖化皮质激素或其同类物。各种蛋白治疗剂也可与本发明组合使用,它们包括针对细胞因子的抗体如 最后,在呼吸道疾病治疗中可结合使用的某些化合物包括β-2肾上腺素能激动剂如沙丁胺醇(albuterol)及其同类物,以及黄嘌呤类如茶碱和相关类似物。对于现述的针对CxCR3的化合物,可以想象其他许许多多当组合给药时可提供累加益处的药物。
最后,在呼吸道疾病治疗中可结合使用的某些化合物包括β-2肾上腺素能激动剂如沙丁胺醇(albuterol)及其同类物,以及黄嘌呤类如茶碱和相关类似物。对于现述的针对CxCR3的化合物,可以想象其他许许多多当组合给药时可提供累加益处的药物。
采用其他药物与本发明的化合物组合的疗法与单一药物疗法相比具有几个主要优点。首先,由于有可能使用比单独使用时较低剂量的各种药物来进行治疗,可以预期与治疗相关的毒性和副作用将会下降。组合疗法的第二个主要优点是,由于两种药物独自起作用,所以可限制治疗应答性的脱敏作用的发展的机会较少。组合疗法的第三个优点是,由于进行有效治疗所需要的治疗剂的量降低,故有可能降低成本。最后,第四个优点是,由于某些患者的临床功效可能取决于以不同方式起作用的适宜化合物的组合,所以能更好地对疾病进行处理。
测得的生物活性
CxCR3A生物鉴定方法
化合物的生物活性是在萤光成像读板器(fluorometric imaging plate reader)(FLIPR;Molecular Devices)中用表达与嵌合G蛋白(Gαi4qi4)偶联的人CxCR3A的工程化CHO细胞测定的。细胞在生物鉴定前一天在添加了10%FBS和0.3mg/ml潮霉素抗生素的F12培养基中铺板,以维持重组体选择。在生物鉴定的当天,洗涤细胞并在汉克平衡盐溶液(Hanks Balanced SaltSolution)(Gibco)中以Fluo-4-AM(Invitrogen)加载染料一小时,该汉克平衡盐溶液用25mM Hepes于pH 7.4缓冲并含有0.36mg/ml 4-(二丙基氨磺酰基)苯甲酸(probenicid)。此缓冲液也用于测试化合物的连续稀释,但不加染料(aka标准缓冲液)。洗去细胞上过量的染料(将它们留在25μl缓冲液中),并在Beckman FX上将每种化合物的稀释液5μl从源板上转移至细胞板上。在FLIPR仪器上,在读取基础读数后,用添加0.1%BSA的标准缓冲液将20μl的46nM IPl0(购自R&D Systems)加入。读取萤光影像共两分钟。取最大FLIPR值/样本报告数据,并用XLfit处理以计算IC50值。在上述FLIPR测试中,本发明范围内的化合物具有低于25微摩尔IC50,优选的是低于1微摩尔IC50,更优选的是低于200纳摩尔IC50的活性。
本发明也可以不背离其精神或基本属性的其他特定形式实施。
Claims (67)
1.式I的3-(酰氨基或磺酰氨基)-4-(4-取代的-吖嗪基)苯甲酰胺或苯磺酰胺化合物

其中
Q和Q1独立地为CO或SO2;
Y是CO或SO2;
X是卤素;
m是1、2或3;
R1是(任选取代的(芳族基或低级环基))低级烷基C1-3;
R2、R4和R6独立地为H或任选取代的低级烷基;
R3是任选取代的芳族基;
A是CH或N,或A和R5一起形成下式的4-7元螺环氮杂环基

n和p独立地为0、1、2、3、4或5,只要n和p≥2但≤5;
R5是JGZ、R8R7NQ1低级烷基或任选取代的3-7元氮杂环基;
Z是键、CO或SO2;
G是低级烷基、C3-7环烷基或3-7元杂环基;以及
J是芳族基、低级烷氧基羰基、低级烷基硫基、低级烷基亚磺酰基、低级烷基磺酰基、低级烷氧基、R8R7N或任选(低级烷基或卤素)取代的3-7元杂环基;
R7和R8独立地为H或低级烷基;
R9和R10独立地为H或低级烷基;或
其药学上可接受的盐、溶剂合物、N-氧化物、季铵衍生物或前药,或其任何组合。
2.根据权利要求1所述的化合物,其中R1是任选取代的苯基(C1-3烷基)或任选取代的苯基环丙基。
3.根据权利要求1所述的化合物,其中R1是任选取代的苯基(C1-3烷基)。
4.根据权利要求1所述的化合物,其中R1是任选取代的苯基(C2-3烷基)。
5.根据权利要求1所述的化合物,其中R1是任选取代的苯基(乙基)。
6.根据权利要求1所述的化合物,其中R1是苯乙基。
7.根据权利要求1所述的化合物,其中R1是任选取代的苯基环丙基。
8.根据权利要求1所述的化合物,其中R1中任选取代的苯基是被卤素取代的。
9.根据权利要求1所述的化合物,其中R1中任选取代的苯基是被氯或氟取代的。
10.根据权利要求1所述的化合物,其中R1中任选取代的苯基是被氯或氟取代的。
11.根据权利要求1所述的化合物,其中R1中任选取代的苯基是被氯或氟单取代的。
12.根据权利要求1所述的化合物,其中R1中任选取代的苯基是被氯邻位单取代的。
13.根据权利要求1所述的化合物,其中R1中任选取代的苯基是被氯对位单取代的。
14.根据权利要求1所述的化合物,其中R1中任选取代的苯基是被氯间位单取代的。
15.根据权利要求1所述的化合物,其中R1中任选取代的苯基是被氟对位单取代的。
16.根据权利要求1所述的化合物,其中R1中任选取代的苯基是被氯或氟双取代的。
17.根据权利要求1所述的化合物,其中R1中任选取代的苯基是被氯邻位、对位双取代的。
18.根据权利要求1所述的化合物,其中R1中任选取代的苯基是被氟邻位、对位双取代的。
19.根据权利要求1所述的化合物,其中R2是H或甲基。
20.根据权利要求1所述的化合物,其中R2是H。
21.根据权利要求1所述的化合物,其中R3是任选取代的苯基、任选取代的噻唑基、吡啶基、吲哚满酮基或噻吩基。
22.根据权利要求1所述的化合物,其中R3中任选取代的苯基是被卤素、羧基或烷氧基羰基取代的。
23.根据权利要求1所述的化合物,其中R3中任选取代的苯基是被氯或氟单取代的。
24.根据权利要求1所述的化合物,其中R3中任选取代的苯基是被氯邻位单取代的。
25.根据权利要求1所述的化合物,其中R3中任选取代的苯基是被氯对位单取代的。
26.根据权利要求1所述的化合物,其中R3中任选取代的苯基是被氯间位单取代的。
27.根据权利要求1所述的化合物,其中R3中任选取代的苯基是被氟双取代的。
28.根据权利要求1所述的化合物,其中R3中任选取代的苯基是被氟邻位、对位双取代的。
29.根据权利要求1所述的化合物,其中R3中任选取代的苯基是被羧基或烷氧基羰基取代的。
30.根据权利要求1所述的化合物,其中R3中任选取代的苯基是被羧基或烷氧基羰基单取代的。
31.根据权利要求1所述的化合物,其中R3中任选取代的苯基是被羧基或烷氧基羰基间位单取代的。
32.根据权利要求1所述的化合物,其中R3中任选取代的苯基是被羧基或烷氧基羰基对位单取代的。
33.根据权利要求1所述的化合物,其中R3中任选取代的苯基是2-噻吩基。
34.根据权利要求1所述的化合物,其中R3中任选取代的苯基是3-噻吩基。
35.根据权利要求1所述的化合物,其中R3中任选取代的苯基是任选取代的噻唑基。
36.根据权利要求1所述的化合物,其中R3中任选取代的苯基是被甲基取代的噻唑基。
37.根据权利要求1所述的化合物,其中R4是H。
38.根据权利要求1所述的化合物,其中R5是JGZ。
39.根据权利要求1所述的化合物,其中Z是键。
40.根据权利要求1所述的化合物,其中Z是CO。
41.根据权利要求1所述的化合物,其中G是低级烷基。
42.根据权利要求1所述的化合物,其中G是C1-3低级烷基。
43.根据权利要求1所述的化合物,其中G是C2-3低级烷基。
44.根据权利要求1所述的化合物,其中J是R8R7N。
45.根据权利要求1所述的化合物,其中R7和R8是H或低级烷基。
46.根据权利要求1所述的化合物,其中R7和R8是低级烷基。
47.根据权利要求1所述的化合物,其中R7和R8是C1-2低级烷基。
48.根据权利要求1所述的化合物,其中R7和R8是C1低级烷基。
49.根据权利要求1所述的化合物,其中R7和R8之一是H,且R7和R8中另一基团是低级烷基。
50.根据权利要求1所述的化合物,其中J是任选(低级烷基或卤素)取代的3-7元杂环基。
51.根据权利要求1所述的化合物,其中J是任选(低级烷基或卤素)取代的3-7元氮杂环基。
52.根据权利要求1所述的化合物,其中J作为任选(低级烷基或卤素)取代的3-7元杂环基是N-甲基哌啶-4-基。
53.根据权利要求1所述的化合物,其中R5是R8R7NQ1低级烷基。
54.根据权利要求1所述的化合物,其中Q1是羰基。
55.根据权利要求1所述的化合物,其中Q1是羰基且J是R8R7N或任选(低级烷基或卤素)取代的3-7元氮杂环基。
56.根据权利要求1所述的化合物,其中A和R5一起形成下式的4-6元螺环氮杂环基

n和p独立地为0、1、2、3、4或5,只要n和p≥2但≤5。
57.根据权利要求1所述的化合物,其中A和R5一起形成氮杂环丁烷、吡咯烷或哌啶,其每一个任选被低级烷基N-取代。
58.根据权利要求1所述的式I的化合物,其中Y是CO。
59.根据权利要求1所述的式I的化合物,其中Y是SO2。
60.根据权利要求1所述的化合物,其中R9和R10是H。
61.根据权利要求1所述的化合物,其中R9和R10是低级烷基。
62.根据权利要求1所述的化合物,其中R9和R10是甲基。
63.根据权利要求1所述的化合物,其选自下式:


以及

或它们药学上可接受的盐、溶剂合物、N-氧化物、季铵衍生物或前药,或其任何组合。
64.药物组合物,其含有药学上可接受量的根据权利要求1所述的式I的化合物,或其药学上可接受的盐、溶剂合物、N-氧化物、季铵衍生物或前药,或其任何组合,以及药学上可接受的添加剂。
65.为需要的患者预防或治疗由CxCR3趋化因子受体介导的疾病或与其相关的病症的方法,包括向患者给药药物有效量的根据权利要求1所述的式I的化合物,或其药学上可接受的盐、溶剂合物、N-氧化物、季铵衍生物或前药,或其任何组合。
66.根据权利要求1所述的式I的一种或多种化合物在制备药物中的用途,该药物用于为需要的患者治疗或预防CxCR3在其中起作用的病症。
67.药盒或药物包,用于治疗或预防患者的生理状况或疾病状态,其中该药盒或药物包包括多个单独的容器,其中至少一个所述容器含有一种或多种根据权利要求1所述的式I的化合物(单独或与药学上可接受的载体组合),至少另一个所述容器含有一种或多种能够治疗或预防该生理状况或疾病状态的其他化合物。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US2973808P | 2008-02-19 | 2008-02-19 | |
| US61/029,738 | 2008-02-19 | ||
| PCT/US2009/034340 WO2009105435A1 (en) | 2008-02-19 | 2009-02-18 | Inhibitors of the chemokine receptor cxcr3 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN102007108A true CN102007108A (zh) | 2011-04-06 |
Family
ID=40527535
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN2009801138321A Pending CN102007108A (zh) | 2008-02-19 | 2009-02-18 | 趋化因子受体CxCR3的抑制剂 |
Country Status (25)
| Country | Link |
|---|---|
| US (1) | US8268828B2 (zh) |
| EP (1) | EP2262784B1 (zh) |
| JP (1) | JP2011512412A (zh) |
| KR (1) | KR20100123835A (zh) |
| CN (1) | CN102007108A (zh) |
| AR (1) | AR070430A1 (zh) |
| AU (1) | AU2009215643A1 (zh) |
| BR (1) | BRPI0908815A2 (zh) |
| CA (1) | CA2715557A1 (zh) |
| CL (1) | CL2009000368A1 (zh) |
| CO (1) | CO6241113A2 (zh) |
| CR (1) | CR11604A (zh) |
| DO (1) | DOP2010000254A (zh) |
| EC (1) | ECSP10010409A (zh) |
| IL (1) | IL207597A0 (zh) |
| MA (1) | MA32191B1 (zh) |
| MX (1) | MX2010008397A (zh) |
| NI (1) | NI201000132A (zh) |
| NZ (1) | NZ587380A (zh) |
| PE (1) | PE20091576A1 (zh) |
| RU (1) | RU2010138577A (zh) |
| SV (1) | SV2010003648A (zh) |
| TW (1) | TW200948791A (zh) |
| WO (1) | WO2009105435A1 (zh) |
| ZA (1) | ZA201005314B (zh) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN107072205A (zh) * | 2014-09-10 | 2017-08-18 | Epizyme股份有限公司 | Smyd抑制剂 |
Families Citing this family (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| PE20091576A1 (es) | 2008-02-19 | 2009-11-05 | Sanofi Aventis | DERIVADOS DE 3-(AMIDO O SULFAMIDO)-4-(4-AZINIL SUSTITUIDO)BENZAMIDA COMO INHIBIDORES DEL RECEPTOR DE QUIMIOQUINAS CxCR3 |
| WO2010126811A1 (en) | 2009-04-27 | 2010-11-04 | Boehringer Ingelheim International Gmbh | Cxcr3 receptor antagonists |
| WO2011084985A1 (en) * | 2010-01-07 | 2011-07-14 | Boehringer Ingelheim International Gmbh | Cxcr3 receptor antagonists |
| KR101251788B1 (ko) | 2010-12-06 | 2013-04-08 | 기아자동차주식회사 | 차량 연비 정보 단말표시 시스템 및 그 방법 |
| US9181226B2 (en) * | 2011-07-18 | 2015-11-10 | Merck Patent Gmbh | Benzamides |
| EP2601950A1 (en) | 2011-12-06 | 2013-06-12 | Sanofi | Cycloalkane carboxylic acid derivatives as CXCR3 receptor antagonists |
| JP6219844B2 (ja) * | 2012-01-10 | 2017-10-25 | メルク パテント ゲゼルシャフト ミット ベシュレンクテル ハフツングMerck Patent Gesellschaft mit beschraenkter Haftung | 卵胞刺激ホルモンの調節因子としてのベンズアミド誘導体 |
| ES2632237T3 (es) | 2012-02-02 | 2017-09-12 | Idorsia Pharmaceuticals Ltd | Compuestos de 4-(benzoimidazol-2-il)-tiazol y derivados aza relacionados |
| EP2666769A1 (en) | 2012-05-23 | 2013-11-27 | Sanofi | Substituted B-amino acid derivatives as CXCR3 receptor antagonist |
| WO2015011099A1 (en) | 2013-07-22 | 2015-01-29 | Actelion Pharmaceuticals Ltd | 1-(piperazin-1-yl)-2-([1,2,4]triazol-1-yl)-ethanone derivatives |
| AR099789A1 (es) | 2014-03-24 | 2016-08-17 | Actelion Pharmaceuticals Ltd | Derivados de 8-(piperazin-1-il)-1,2,3,4-tetrahidro-isoquinolina |
| JP6337218B2 (ja) | 2015-01-15 | 2018-06-06 | イドーシア ファーマシューティカルズ リミテッドIdorsia Pharmaceuticals Ltd | Cxcr3受容体調節剤としてのヒドロキシアルキル−ピペラジン誘導体 |
| AR103399A1 (es) | 2015-01-15 | 2017-05-10 | Actelion Pharmaceuticals Ltd | Derivados de (r)-2-metil-piperazina como moduladores del receptor cxcr3 |
| WO2018183145A1 (en) | 2017-03-26 | 2018-10-04 | Takeda Pharmaceutical Company Limited | Piperidinyl- and piperazinyl-substituted heteroaromatic carboxamides as modulators of gpr6 |
| CN116410159B (zh) * | 2023-06-09 | 2023-08-22 | 济南国鼎医药科技有限公司 | 一种恩曲替尼中间体的制备方法及其应用 |
Family Cites Families (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2052541A5 (zh) | 1969-06-13 | 1971-04-09 | Takeda Chemical Industries Ltd | |
| US3840556A (en) | 1971-05-28 | 1974-10-08 | Lilly Co Eli | Penicillin conversion by halogen electrophiles and anti-bacterials derived thereby |
| US6124319A (en) | 1997-01-21 | 2000-09-26 | Merck & Co., Inc. | 3,3-disubstituted piperidines as modulators of chemokine receptor activity |
| US6469002B1 (en) | 2001-04-19 | 2002-10-22 | Millennium Pharmaceuticals, Inc. | Imidazolidine compounds |
| US6794379B2 (en) | 2001-06-06 | 2004-09-21 | Tularik Inc. | CXCR3 antagonists |
| GB0203994D0 (en) | 2002-02-20 | 2002-04-03 | Celltech R&D Ltd | Chemical compounds |
| US20050272936A1 (en) | 2002-06-03 | 2005-12-08 | Axten Jeffrey M | Imidazolium cxcr3 inhibitors |
| US6734659B1 (en) | 2002-06-13 | 2004-05-11 | Mykrolis Corporation | Electronic interface for use with dual electrode capacitance diaphragm gauges |
| US7417045B2 (en) * | 2005-02-16 | 2008-08-26 | Schering Corporation | Heterocyclic substituted pyridine or phenyl compounds with CXCR3 antagonist activity |
| AU2006214378A1 (en) * | 2005-02-16 | 2006-08-24 | Pharmacopeia, Inc. | Pyridyl and phenyl substituted piperazine-piperidines with CXCR3 antagonist activity |
| EP1856098B1 (en) | 2005-02-16 | 2012-08-01 | Schering Corporation | Pyrazinyl substituted piperazine-piperidines with cxcr3 antagonist activity |
| WO2007002742A1 (en) | 2005-06-28 | 2007-01-04 | Pharmacopeia, Inc. | Substituted [1,4]-diazepanes as cxcr3 antagonists and their use in the treatment of inflammatory disorders |
| CN101341147A (zh) | 2005-10-11 | 2009-01-07 | 先灵公司 | 具有cxcr3拮抗剂活性的取代的杂环化合物 |
| US20090143413A1 (en) | 2005-11-29 | 2009-06-04 | Adams Alan D | Thiazole Derivatives as CXCR3 Receptor Modulators |
| EP1996577B1 (en) | 2006-03-21 | 2016-02-10 | Merck Sharp & Dohme Corp. | Heterocyclic substituted pyridine compounds with cxcr3 antagonist activity |
| AU2007272884A1 (en) | 2006-07-14 | 2008-01-17 | Pharmacopeia, Inc. | Heterocyclic substituted piperazine compounds with CXCR3 antagonist activity |
| PE20091576A1 (es) | 2008-02-19 | 2009-11-05 | Sanofi Aventis | DERIVADOS DE 3-(AMIDO O SULFAMIDO)-4-(4-AZINIL SUSTITUIDO)BENZAMIDA COMO INHIBIDORES DEL RECEPTOR DE QUIMIOQUINAS CxCR3 |
-
2009
- 2009-02-17 PE PE2009000232A patent/PE20091576A1/es not_active Application Discontinuation
- 2009-02-18 JP JP2010547712A patent/JP2011512412A/ja active Pending
- 2009-02-18 CL CL2009000368A patent/CL2009000368A1/es unknown
- 2009-02-18 MX MX2010008397A patent/MX2010008397A/es not_active Application Discontinuation
- 2009-02-18 RU RU2010138577/04A patent/RU2010138577A/ru not_active Application Discontinuation
- 2009-02-18 AR ARP090100561A patent/AR070430A1/es not_active Application Discontinuation
- 2009-02-18 CA CA2715557A patent/CA2715557A1/en not_active Abandoned
- 2009-02-18 WO PCT/US2009/034340 patent/WO2009105435A1/en active Application Filing
- 2009-02-18 NZ NZ587380A patent/NZ587380A/en not_active IP Right Cessation
- 2009-02-18 EP EP09711984.6A patent/EP2262784B1/en active Active
- 2009-02-18 KR KR1020107018467A patent/KR20100123835A/ko not_active Application Discontinuation
- 2009-02-18 AU AU2009215643A patent/AU2009215643A1/en not_active Abandoned
- 2009-02-18 BR BRPI0908815A patent/BRPI0908815A2/pt not_active IP Right Cessation
- 2009-02-18 CN CN2009801138321A patent/CN102007108A/zh active Pending
- 2009-02-19 TW TW098105199A patent/TW200948791A/zh unknown
-
2010
- 2010-07-26 ZA ZA2010/05314A patent/ZA201005314B/en unknown
- 2010-07-28 CR CR11604A patent/CR11604A/es not_active Application Discontinuation
- 2010-07-29 NI NI201000132A patent/NI201000132A/es unknown
- 2010-08-06 US US12/852,076 patent/US8268828B2/en active Active
- 2010-08-12 IL IL207597A patent/IL207597A0/en unknown
- 2010-08-18 EC EC2010010409A patent/ECSP10010409A/es unknown
- 2010-08-18 DO DO2010000254A patent/DOP2010000254A/es unknown
- 2010-08-18 SV SV2010003648A patent/SV2010003648A/es not_active Application Discontinuation
- 2010-08-19 CO CO10102315A patent/CO6241113A2/es not_active Application Discontinuation
- 2010-09-13 MA MA33173A patent/MA32191B1/fr unknown
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN107072205A (zh) * | 2014-09-10 | 2017-08-18 | Epizyme股份有限公司 | Smyd抑制剂 |
Also Published As
| Publication number | Publication date |
|---|---|
| ZA201005314B (en) | 2011-03-30 |
| CA2715557A1 (en) | 2009-08-27 |
| EP2262784B1 (en) | 2014-04-16 |
| AU2009215643A1 (en) | 2009-08-27 |
| PE20091576A1 (es) | 2009-11-05 |
| US8268828B2 (en) | 2012-09-18 |
| CL2009000368A1 (es) | 2009-06-26 |
| IL207597A0 (en) | 2010-12-30 |
| NZ587380A (en) | 2011-09-30 |
| KR20100123835A (ko) | 2010-11-25 |
| WO2009105435A1 (en) | 2009-08-27 |
| JP2011512412A (ja) | 2011-04-21 |
| MA32191B1 (fr) | 2011-04-01 |
| AR070430A1 (es) | 2010-04-07 |
| ECSP10010409A (es) | 2010-09-30 |
| US20100305088A1 (en) | 2010-12-02 |
| BRPI0908815A2 (pt) | 2019-09-24 |
| MX2010008397A (es) | 2010-08-23 |
| RU2010138577A (ru) | 2012-03-27 |
| TW200948791A (en) | 2009-12-01 |
| EP2262784A1 (en) | 2010-12-22 |
| NI201000132A (es) | 2011-03-16 |
| SV2010003648A (es) | 2011-01-31 |
| CR11604A (es) | 2010-10-05 |
| CO6241113A2 (es) | 2011-01-20 |
| DOP2010000254A (es) | 2010-08-31 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN102007108A (zh) | 趋化因子受体CxCR3的抑制剂 | |
| CN101594871B (zh) | 吡咯并嘧啶化合物及其用途 | |
| JP6639497B2 (ja) | ブロモドメインインヒビターおよびその使用 | |
| CN101959516B (zh) | 用以治疗癌症的包含喹喔啉PI3Kα抑制剂的组合治疗 | |
| CN101263130B (zh) | 2-苯胺-4-芳基取代的噻唑衍生物 | |
| CN102056926B (zh) | 吡咯并吡啶基嘧啶-2-基胺衍生物 | |
| EP2609082B1 (de) | Imidazo[4,5-c]chinoline als dna-pk-inhibitoren | |
| CA2875619C (en) | Aminotriazolopyridine for use in the treatment of inflammation, and pharmaceutical compositions thereof | |
| CN102369195A (zh) | 自分泌运动因子抑制剂 | |
| EP2547664A1 (de) | Morpholinylchinazoline | |
| CN103930416A (zh) | 作为激酶抑制剂的苄腈衍生物 | |
| CN102014914A (zh) | 3h-[1,2,3]三唑并[4,5-d]嘧啶化合物、其作为mtor激酶和pi3激酶抑制剂的用途、以及它们的合成 | |
| CN101534824A (zh) | 作为化学活素受体拮抗剂的氨基吡咯烷 | |
| EP2588457B1 (de) | Pyrazolochinolinderivate als dna-pk-inhibitoren | |
| AU2011315498A1 (en) | CXCR4 receptor antagonists | |
| CN101883764A (zh) | 吡唑衍生物及其作为细胞周期蛋白依赖性激酶抑制剂的用途 | |
| US10875863B2 (en) | RIPK2 inhibitors and method of treating cancer with same | |
| EP2794602A1 (de) | 3-cyanaryl-1h-pyrazolo-(2,3-b-)pyridinderivate | |
| CN107001367A (zh) | 用于治疗例如注意力缺陷障碍(add)的作为人多巴胺主动转运体(dat)蛋白抑制剂的2‑[双(4‑氟苯基)甲基]‑2,7‑二氮杂螺[4.5]癸烷‑10‑酮衍生物和有关化合物 | |
| WO2013072015A1 (de) | Morpholinylbenzotriazine zur anwendung in der krebstherapie | |
| CN101827851A (zh) | 用于治疗肿瘤的5-氰基噻吩并吡啶 | |
| CA3176325A1 (en) | Halogenated-heteroaryl and other heterocyclic kinase inhibitors, and uses thereof | |
| CN107530304A (zh) | Olig2活性的抑制 | |
| KR20190025545A (ko) | 특정 단백질 키나제 억제제 | |
| JP2019502680A (ja) | Cxcr4受容体アンタゴニスト |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C02 | Deemed withdrawal of patent application after publication (patent law 2001) | ||
| WD01 | Invention patent application deemed withdrawn after publication |
Application publication date: 20110406 |