CAMPO DA INVENÇÃO
[0001] Em um aspecto, a presente invenção é dirigida a uma dispersão sólida compreendendo: (a) um ingrediente ativo farmacêutico ou um ingrediente ativo nutracêutico com baixa solubilidade; e (b) alginato de sódio ou potássio. Num outro aspecto, a presente invenção é dirigida a uma forma de dosagem de fármaco preparada a partir de uma tal dispersão sólida.
ANTECEDENTES DA INVENÇÃO
[0002] Muitas das novas entidades químicas que estão sendo atualmente desenvolvidas no campo farmacêutico ou no campo nutracêutico são pouco solúveis. Como bem observado por Tiwari et al, Solid Dispersions: An Overview to Modify Bioavailability of Poorly Water Soluble Drugs; International Journal of PharmTech Research, Vol.1, No. 4, pp. 1338-1349 (2009), a administração oral de fármacos é a maneira mais simples e fácil de se administrar fármacos, com formas de dosagem oral sólidas tendo várias vantagens em relação a outros tipos de formas de dosagem oral. Um grande problema associado a muitos fármacos que apresentam baixa solubilidade é que elas são absorvidas apenas no intestino delgado superior e, portanto, possuem apenas uma pequena janela de absorção. Consequentemente, se tais medicamentos não são liberados nesta área gastrointestinal, eles apresentarão baixa biodisponibilidade; uma preocupação que torna seu perfil de lançamento crítico. As dispersões sólidas, que podem ser definidas como misturas moleculares de fármacos pouco solúveis em água em veículos hidrófilos, são uma estratégia que pode ser empregada para aumentar a liberação do fármaco de tais fármacos pouco solúveis.
[0003] Entre os veículos atualmente utilizados para produzir dispersões sólidas estão a polivinilpirrolidona (PVP) e o succinato de acetato de hipromelose (HPMCAS). Embora estes materiais possam ser empregados para produzir dispersões sólidas que exibam perfis de liberação aceitáveis, em certos casos a exposição ao calor, luz e / ou alta umidade pode afetar negativamente o perfil de liberação observado.
[0004] As dispersões sólidas podem ser produzidas por vários processos diferentes, incluindo o método de fusão, o método do solvente e o método do fluido supercrítico (Tiwari et al., citado acima). Poovi et al, Development of Domperidone Solid Dispersion Powders Using Sodium Alginate as Carrier, European Journal of Applied Sciences 5 (2): 36-42 (2013) compara os perfis de liberação de proporções de 1: 1 em peso de fármaco: misturas de alginato de sódio de dispersões produzidas por vários desses métodos.
[0005] Enquanto que Poovi et al. concluem que o alginato de sódio é um veículo adequado em dispersões sólidas para melhorar a solubilidade e dissolução de um fármaco pouco solúvel como a domperidona, tal publicação não proporciona qualquer motivação para aumentar a quantidade de alginato empregada nas composições aqui descritas. Especificamente, Poovi et al indicam (no primeiro parágrafo na página 40) que sua análise de dispersões de alginato e fármaco 1: 1 formadas "indica que o medicamento está uniformemente disperso na formulação em pó". Consequentemente, assumir-se-ia que não haveria nenhum benefício para a adição de alginato adicional a tal formulação. Consequentemente, é completamente inesperado que a taxa de liberação de ingredientes ativos pouco solúveis possa ser aumentada materialmente aumentando-se a quantidade de alginato empregada em tais dispersões sólidas.
SUMARIO DA INVENÇÃO
[0006] Em um aspecto, a presente invenção é dirigida a uma dispersão sólida compreendendo: (a) um ingrediente ativo com baixa solubilidade; e (b) um alginato selecionado do grupo que consiste em alginato de sódio e alginato de potássio;
[0007] em que a proporção em peso do ingrediente ativo: alginato se encontra entre 1: 1,1 e 1:10;
[0008] com a condição de que quando o referido alginato num alginato de sódio com um teor de G inferior a 50% em peso,
[0009] se tal alginato de sódio tiver uma viscosidade inferior a 200 mPa.s numa solução aquosa a 1% a 20 °C utilizando um RV do tipo Brookfield com o veio Brookfield RV 2, a proporção em peso do ingrediente ativo: alginato se encontra entre 1: 2,5 e 1: 5; e
[0010] se tal alginato de sódio tiver uma viscosidade de 200 mPa.s ou mais numa solução aquosa a 1% a 20 °C utilizando um RV do tipo Brookfield com o veio Brookfield RV 2, a proporção em peso do ingrediente ativo: alginato se encontra entre 1: 1,5 e 1: 3.5.
[0011] Ainda num outro aspecto, a presente invenção é dirigida a uma forma de dosagem de fármaco preparada a partir de uma tal dispersão sólida.
DESCRIÇÃO DETALHADA DA INVENÇÃO
[0012] Em um aspecto, a presente invenção é dirigida a uma dispersão sólida compreendendo: (a) um ingrediente ativo com baixa solubilidade; e (b) um alginato selecionado do grupo que consiste em alginato de sódio e alginato de potássio;
[0013] em que a proporção em peso do fármaco: alginato se encontra entre 1: 1., e 1:10;
[0014] com a condição de que quando o referido alginato é um alginato de sódio com um teor de G inferior a 50% em peso,
[0015] se tal alginato de sódio tiver uma viscosidade inferior a 200 mPa.s numa solução aquosa a 1% a 20 °C utilizando um RV do tipo Brookfield com o veio Brookfield RV 2, a proporção em peso do ingrediente ativo: alginato se encontra entre 1: 2,5 e 1: 5; e
[0016] se tal alginato de sódio tiver uma viscosidade de 200 mPa.s ou mais numa solução aquosa a 1% a 20 °C utilizando um RV tipo Brookfield com o veio Brookfield RV 2, a proporção em peso do ingrediente ativo: alginato se encontra entre 1: 1,5 e 1: 3.5.
[0017] Tal como aqui se utiliza, o termo "ingrediente ativo com baixa solubilidade" significa um ingrediente farmacêutico ativo que é classificado como um ativo de Classe II ou Classe IV sob o Sistema de Classificação Biofarmacêutica ou um ingrediente ativo nutracêutico com um perfil de solubilidade semelhante. De acordo com esse Sistema de Classificação, considera-se que uma substância tem baixa solubilidade quando a força de dose mais elevada é solúvel em 250 mL ou mais de água em uma faixa de pH de 1 a 7,5. Numa modalidade, a solubilidade pode ser determinada de acordo com os parâmetros estabelecidos na seguinte matriz a 20 °C:
[0018] Para os propósitos da presente invenção, um ingrediente ativo com baixa solubilidade inclui qualquer ingrediente ativo que cai nas categorias: bem pouco solúvel e praticamente insolúvel como estabelecido na matriz acima, embora se considere que o método de formulação descrito nesta invenção poderia aumentar a solubilidade de um ingrediente ativo que cai nas categorias razoavelmente solúvel e pouco solúvel em uma das categorias mais solúveis descritas acima.
[0019] Como aqui empregados, os termos "ingrediente ativo farmacêutico" incluem fármacos veterinários, bem como aqueles destinados a uso humano. O termo "ingrediente ativo nutracêutico com baixa solubilidade" refere-se a um composto nutracêutico que satisfaz os critérios de solubilidade para um fármaco de baixa solubilidade definido acima.
[0020] O ingrediente ativo de baixa solubilidade pode ser pelo menos um selecionado, por exemplo, a partir de: um fármaco anti-inflamatório não esteróide incluindo acetaminofeno, ácido acetilsalicílico, ibuprofeno, fenbuprofeno, fenoprofeno, flurbiprofeno, indometacina, naproxeno, etodolaco, cetoprofeno, dexibuprofeno, piroxicam ou aceclofenac; um fármaco imunossupressor ou dermatite atópica incluindo ciclosporina, tacrolimus, rapamicina, micofenolato ou pimecrolimus; um bloqueador de canais de cálcio incluindo nifedipina, nimodipina, nitrendipina, nilvadipina, felodipina, amlodipina ou isradipina; um antagonista da angiotensina II incluindo valsartan, eprosartan, irbesartan, candesartan, telmisartan, olmesartan ou losartan; um agente hipolipidêmico inibidor de síntese de colesterol incluindo atorvastatina, lovastatina, simvastatina, fluvastatina, rosuvastatina ou pravastatina; um agente hipolipemiante promotor do metabolismo do colesterol e da secreção, incluindo gemfibrozil, fenofibrato, etofibrato ou bezafibrato; um fármaco antidiabético incluindo pioglitazona, rosiglitazona ou metformina; Um inibidor de lipase incluindo orlistat; Um agente antifúngico que inclui itraconazol, anfotericina B, terbinafina, nistatina, griseofulvina, fluconazol ou cetoconazol; um fármaco hepatoprotector que inclui bifenil dimetil dicarboxilato, silimarina ou ácido ursodesoxicólico; um fármaco gastrointestinal incluindo sofalcone, omeprazole, pantoprazole, famotidina, itoprida ou mesalazina; um agente antiplaquetário incluindo cilostazol ou clopidogrel; um medicamento para osteoporose incluindo raloxifeno; um medicamento antiviral incluindo aciclovir, famciclovir, lamivudina ou oseltamivir; um antibiótico que inclui claritromicina, ciprofloxacina ou cefuroxima; um medicamento antiasmático ou anti- histamínico, incluindo pranlukast, budesonida ou fexofenadina; Um fármaco hormonal incluindo testosterona, prednisolona, estrogênio, cortisona, hidrocortisona ou dexametasona; um fármaco anticancerígeno incluindo paclitaxel, docetaxel, derivados de paclitaxel, doxorrubicina, adriamicina, daunomicina, camptotecina, etoposido, teniposídeo ou busulfon; sais destes; e seus derivados farmacêuticos. Especificamente, pode ser pelo menos um selecionado de naproxeno, tacrolimus, valsartan, simvastatina, fenofibrato, itraconazol, dicarboxilato de bifenil dimetila, silimarina, sofalcona, pantoprazole, cilostazol, seus sais e seus derivados farmacêuticos.
[0021] O alginato utilizado na composição da presente invenção está na forma de um sal de sódio ou potássio. Quando é utilizado o alginato de potássio, a proporção em peso do ingrediente ativo pouco solúvel: alginato pode variar de 1: 1,1 a 1:10 e variará tipicamente entre 1: 1,5 e 1:10. Mais tipicamente, tais dispersões compreendem uma proporção em peso de ingrediente ativo pouco solúvel: alginato entre 1: 2 e 1: 5.
[0022] Quando é utilizado um alginato de sódio com um teor de guluronato ("G") de pelo menos 50 por cento em peso ou mais (com base no peso total de guluronato e manuronato), a proporção em peso de ingrediente ativo fracamente solúvel: alginato pode variar de 1: 1,1 a 1:10, e geralmente variará entre 1: 1,5 e 1:10. Mais tipicamente, tais dispersões compreendem uma proporção em peso de ingrediente ativo pouco solúvel: alginato entre 1: 2 e 1: 5.
[0023] Quando é utilizado um alginato de sódio com um teor de G inferior a 50 por cento em peso, a proporção em peso do ingrediente ativo fracamente solúvel: alginato variará de acordo com a viscosidade do alginato de sódio particular empregada. Especificamente, se tal alginato de sódio tiver uma viscosidade inferior a 200 mPa.s numa solução aquosa a 1% a 20 °C utilizando um RV tipo Brookfield (eg, RVT, RVF, RVTDV) com o veio Brookfield RV 2, a proporção em peso de Ingrediente ativo: alginato pode variar entre 1: 2,5 e 1: 5. Se tal alginato de sódio tiver uma viscosidade de 200 mPa.s ou mais numa solução aquosa a 1% a 20 °C utilizando um RV tipo Brookfield (por exemplo, RVT, RVF, RVTDV) com o veio Brookfield RV 2, a proporção em peso do ingrediente ativo : alginato pode variar entre 1: 1,5 e 1: 3,5.
[0024] As dispersões sólidas da presente invenção podem ser produzidas por qualquer método convencionalmente utilizado para produzir dispersões sólidas. Estes incluem o método de evaporação do solvente, o método de fusão ou fusão, o método do solvente fundido (todos os quais são descritos em Poovi et al, discutidos acima); bem como o método do fluido supercrítico (como descrito em Tiwari et al, discutido acima). A utilização do método de evaporação do solvente é tipicamente empregada.
[0025] A presente descrição proporciona ainda uma forma de dosagem de fármaco produzida a partir da dispersão sólida da presente invenção.
[0026] Essa forma de dosagem de fármaco pode ser um grânulo, um pó, um xarope, um líquido, uma suspensão, um comprimido, uma cápsula, um troche ou uma pastilha para administração oral, ou agente transdérmico, loção, pomada oftálmica, unguento, gesso, cataplasma, creme, pasta, suspensão, líquido, injeção ou supositório para administração parenteral. Essa forma de dosagem de fármaco pode incluir adicionalmente excipientes que são convencionalmente empregados em composições farmacêuticas e podem ser preparados por métodos familiares para um técnico no assunto.
[0027] Os excipientes típicos que podem ser utilizados na forma de dosagem de fármaco da presente invenção incluem enchimentos e lubrificantes. Estes excipientes são empregados em quantidades convencionais que são bem conhecidas para um técnico no assunto.
[0028] As cargas adequadas incluem carbonato de cálcio (Barcroft, Cal-Carb, CalciPure, Destab, MagGran, Millicarb, Pharma-Carb, Precarb, Sturcal, Vivapres Ca), fosfato de cálcio, anidro dibásico (A-TAB, Di-Cafos AN, Emcompress Anhydrous, Fujicalin), fosfato de cálcio, dihidrato dibásico (Cafos, Calipharm, Calstar, Di-Cafos, Emcompress), fosfato de cálcio tribásico (Tri-Cafos, TRI-CAL WG, TRI-TAB), sulfato de cálcio (Destab, Drierite, Branca de Neve, Cal-Tab, Compactrol, USG Terra Alba), celulose em pó (Arbocel, Elcema, Sanacel, Solka-Floc), celulose microcristalina silicificada (ProSolv), acetato de celulose, açúcar compressível (Di-Pac), açúcar de confeiteiro, dextranos (Candex, Emdex), dextrina (Avedex, Caloreen, Crystal Gum, Primogran W), dextrose (Caridex, Dextrofin, Lycadex PF, Roferose, Tab fine D-100), frutose (Advantose, Fructamyl, Fructofin, Krystar), kaolinLion, Sim 90), lactitol (Finlac ACX, Finlac DC, Finlac MCX) 5 lactose (Aero Flo 20, Aero Flo 65, Anhydrox, CapsuLac, Fast-Flo, FlowL Ac, GranuLac, InhaLac, Lactochem, Lactohale, Lactopress, Microfine, Microtose, Pharmatose, Prisma Lac, Respitose, SacheLac, SorboLac, Super-Tab, Tablettose, Wyndale, Zeparox), carbonato de magnésio, óxido de magnésio (MagGran MO), maltodextrina ( C * Dry MD, Glucidex, Glucodry, Lycatab DSH, Maldex, Maltagran, Maltrin, Maltrin QD, Paselli MD 10 PH, Star-Dri), maltose (Advantose 100), manitol (Mannogem, Pearlitol), celulose microcristalina (Avicel PH, Celex, Celphere, Ceolus KG, Emcocel, Ethispheres, Fibrocel, Pharmacel, Tabulose, Vivapur), polidextrose (Litesse), simeticona (Dow Corning Q7-2243 LVA, Cow Corning Q7-2587, Sentry Simethicone), alginato de sódio (Kelcosol, Keltone , Protanal), cloreto de sódio (Alberger), sorbitol (Liponec 70-NC, Liponic 76-NC, Meritol, Neosorb, Sorbifin, Sorbitol Instantâneo, Sorbogem), amido (Aytex P, Fluftex W, Instant Pure- Cote, Melojel, Meritena Paygel 55, Perfectamyl D6PH, Pure-Bind, Pure-Cote, Pure- Dent, Pure-Gel, Pure-Set, Pureza 21, Pureza 826, Tablet White), Amido pré- gelatinizado (Instastarch, Lycatab C, Lycatab PGS, Merigel, National 78-1551, Pharma-Gel, Prejel, Sepistab ST 200, Spress B820, Starch 1500 G, Tablitz, Unipure LD, Unipure WG220), sacarose, trealose e xilitol ( Klinit, Xylifm, Xylitab, Xylisorb, Xilitolo).
[0029] O termo "enchimento" às vezes é usado indistintamente com o termo "diluente". No entanto, o termo "enchimento" é geralmente utilizado para formulações sólidas, enquanto o termo "diluente" é utilizado em formulações líquidas.
[0030] Os lubrificantes adequados incluem estearato de cálcio (HyQual), monoestearato de glicerina (Capmul GMS-50, Cutina GMS, Imwitor 191 e 900, Kessco GMS5 Lipo GMS 410, 450 e 600, Myvaplex 600P, Myvatex, Protachem GMS-450, Rita GMS , Stepan GMS, Tegin, Tegin 503 e 515, Tegin 4100, Tegin M, Unimate GMS), behenato de glicerilo (Compritol 888 ATO), palmitoestearato de glicerilo Precirol ATO 5), óleo de rícino hidrogenado (Castorwax, Castorwax MP 70, Castorwax MP 80, Croduret, Cutina HR, Fancol, Simulsol 1293), óleo vegetal hidrogenado de tipo I (Akofine, Lubritab, Sterotex, Dynasan P60, Softisan 154, Hydrocote, Lipovol HS-K, Sterotex HM), laurilsulfato de magnésio, estearato de magnésio, cadeia média Triglicerídeos (Captex 300, Captex 355, Crodamol GTC / C, Labrafac CC, Miglyol 810, Miglyol 812, Myritol, Neobee M5, Nesatol, Waglinol 3/9280), poloxâmero (Lutrol, Monolan, Pluronic, Supronicm Synperonic), polietilenoglicol ( Carbowax, Carbowax Sentry, Lipo, Lipoxol, Lutrol E, Pluriol E), benzoato de sódio (Antimol), s Cloreto de odio (Alberger), laurilsulfato de sódio (Elfan 240, Texapon K1 2P), fumarato de estearilo de sódio (Pruv, Alubra), ácido esteárico (Crodacid E570, Emersol, Hystrene, Industrene, Kortacid 1895, Pristerene), talco (Altaic, Luzenac, Luzenac Pharma, Magsil Osmanthus, Magsil Star, Superiore), estearato de sacarose (Surfhope SE Pharma D-1803 F) e estearato de zinco (HyQual).
[0031] Em algumas modalidades preferidas, a forma de dosagem de fármaco da presente invenção compreende um revestimento entérico. Os revestimentos entéricos que podem ser utilizados incluem, mas não estão limitados a, aqueles baseados em gelatina, amido e ftalatos de acetato de amilose, copolímeros de estireno-ácido maleico, succinato de acetato de celulose, ftalato de acetato de celulose (CAP), ftalato de acetato de vinila (PVAP), ftalato de hidroxipropilmetilcelulose (Graus HP-50 e HP- 55), etilcelulose, gorduras, estearato de butila e copolímeros de ácido metacrílico- ácido metacrílico com grupos ionizáveis a ácidos (incluindo "ACRYLEZE®" e "EUDRAGIT®").
[0032] Em tais formas de realização entéricas, é útil observar a liberação percentual a 30 minutos (como um critério para o início rápido) e a 90 minutos (para caracterização de liberação completa). A eficácia entérica (ou liberação retardada) normalmente é medida de acordo com a seção de monografia 701, Desintegração, da Convenção Farmacêutica dos Estados Unidos.
[0033] Deve ser entendido que cada componente, composto, substituinte ou parâmetro aqui divulgado deve ser interpretado como sendo divulgado para uso sozinho ou em combinação com um ou mais de cada um dos outros componentes, compostos, substituintes ou parâmetros aqui divulgados.
[0034] Deve também ser entendido que cada quantidade / valor ou intervalo de quantidades / valores para cada componente, composto, substituinte ou parâmetro aqui divulgado deve ser interpretado como também sendo divulgado em combinação com cada quantidade / valor ou intervalo de valores / valores divulgados para qualquer outro componente, composto, substituinte ou parâmetros aqui divulgados e que qualquer combinação de quantidades / valores ou intervalos de quantidades / valores para dois ou mais componentes(s), composto(s), substituinte(s) ou parâmetros aqui divulgados são assim também divulgados em combinação uns com os outros para os propósitos deste relatório descritivo.
[0035] Deve ainda ser entendido que cada limite inferior de cada intervalo aqui divulgado deve ser interpretado como revelado em combinação com cada limite superior de cada intervalo aqui divulgado para o mesmo componente, composto, substituinte ou parâmetro. Assim, a divulgação de dois intervalos deve ser interpretada como uma divulgação de quatro intervalos derivados combinando cada limite inferior de cada intervalo com cada limite superior de cada intervalo. A divulgação de três intervalos deve ser interpretada como uma divulgação de nove intervalos derivados combinando cada limite inferior de cada intervalo com cada limite superior de cada intervalo, etc. Além disso, quantidades / valores específicos de um componente, composto, substituinte ou parâmetro revelado na descrição ou um exemplo deve ser interpretado como uma divulgação de um limite inferior ou superior de um intervalo e, portanto, pode ser combinado com qualquer outro limite inferior ou superior de um intervalo ou quantidade / valor específico para o mesmo componente, composto, substituinte ou parâmetro divulgado em outro lugar no relatório descritivo para formar um intervalo para esse componente, composto, substituinte ou parâmetro.
EXEMPLOS
[0036] Os exemplos a seguir são fornecidos para ilustrar a invenção de acordo com os princípios da presente invenção, mas não devem ser interpretados como limitativos da invenção de qualquer maneira, exceto como indicado nas reivindicações anexas.
Exemplo 1
Preparação dispersões sólidas de Lovastatina: alginato de sódio com alto teor G
[0037] As dispersões sólidas de lovastatina no alginato (Protanal LFR5 / 60, um alginato de sódio com um teor de G de 65 a 75% em peso e uma viscosidade de 3,5 a 7 mPa.s numa solução a 1% e uma viscosidade de 300 a 700 mPa.s numa solução a 10%) foram preparados usando uma técnica de evaporação do solvente nas proporções em peso (lovastatina: alginato) indicadas na Tabela 1 abaixo, como se segue. A quantidade necessária de fármaco foi pesada e dissolvida em 10 mL de etanol a 60 °C seguido por combinação em veículos com amassamento para se preparar uma pasta. A pasta foi, em seguida, seca abaixo de 40 °C ao longo de um período de 12 horas, para remover os solventes. As dispersões sólidas resultantes foram recolhidas e pulverizadas utilizando uma argamassa e pilão e armazenadas num dessecador àem temperatura ambiente.
Estudos de dissolução in vitro
[0038] Estudos in vitro de dissolução da lovastatina: as dispersões sólidas de alginato produzidas acima foram realizados utilizando o método de pás equipadas com um aparelho USP II a 37,5 ± 0,5 °C e uma velocidade das pás de 50 rpm. A quantidade apropriada de dispersão sólida de lovastatina (contendo 60 mg de lovastatina) foi adicionada a um meio de dissolução de 900 mL (tampão de fosfato de pH 6,8 0,05 M com SDS a 0,1% como condição de não imersão). Amostras (6 mL) foram recolhidas em intervalos de tempo predeterminados e filtradas imediatamente através de um filtro de seringa de 0,15 μm, os primeiros 2 mL de filtrado foram descartados e o filtrado restante foi coletado para análise a 237 nm usando um espectrofotômetro UV (UV-2000, instrumento UNIC Corp., Xangai, China). Enquanto isso, adicionou-se um volume igual de meio de dissolução fresco. Todas as experiências foram realizadas em triplicado, e os resultados médios desses estudos estão resumidos na Tabela 1 abaixo. Tabela 1 Lovastatina: Dispersão LFR 5/60
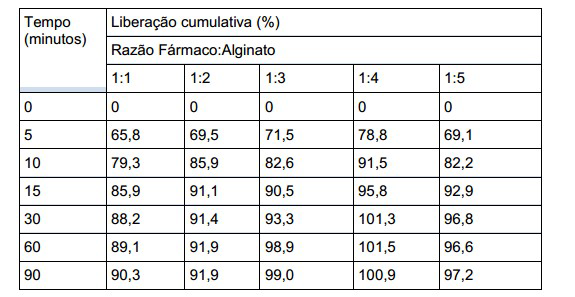
[0039] Os resultados acima demonstram que o aumento das quantidades de alginato de sódio G elevado nas dispersões acima de uma proporção 1: 1 resulta em liberação aumentada do fármaco na dispersão sólida. Exemplo 2 Dispersão sólida de Lovastatina: alginato de potássio
[0040] Utilizando o método descrito no Exemplo 1, dispersões sólidas de lovastatina em alginato de potássio (KF200FTS, um alginato de potássio com um teor de G de 60 a 70% em peso e uma viscosidade de 200 a 400 mPa.s em uma solução a 1%) foram preparadas usando a técnica de evaporação do solvente descrita no Exemplo 1 nas proporções em peso (lovastatina: alginato) indicadas na Tabela 2 abaixo.
[0041] As dispersões sólidas de lovastatina: alginato de potássio preparadas foram testadas in vitro em estudos de dissolução como descritos no Exemplo 1. Os resultados de tais testes estão resumidos na Tabela 2. Tabela 2 Dispersão Lovastatina:KF200FTS
[0042] Os resultados acima demonstram que aumentar as quantidades de alginato de potássio nas dispersões acima de uma proporção de 1:1 resulta em liberação aumentada do fármaco na dispersão sólida. Exemplo 3 Dispersões sólidas de lovastatina: alginato de sódio com baixo teor de viscosidade / baixo G
[0043] Utilizando o método descrito no Exemplo 1, dispersões sólidas de lovastatina em um alginato de sódio com baixo teor de viscosidade / baixo G (Protanal CR8133, alginato de sódio com um teor de G de 37 a 40% em peso e uma viscosidade de 15 a 45 MPa.s numa solução a 1%) foram preparadas usando a técnica de evaporação do solvente descrita no Exemplo 1 nas proporções em peso (lovastatina: alginato) indicadas na Tabela 3 abaixo.
[0044] As dispersões sólidas de lovastatina em um alginato de sódio com baixo teor de viscosidade / baixo G preparadas foram testadas in vitro em estudos de dissolução como descrito no Exemplo 1. Os resultados de tais testes estão resumidos na Tabela 3. Tabela 3 Dispersão Lovastatina:Protanal CR8133
[0045] Os resultados acima demonstram que o aumento das quantidades de um alginato de sódio com baixo teor de viscosidade / baixo G nas dispersões acima de uma proporção de 1: 1 para fármaco: proporções de alginato na faixa de 1: 2,5 a 1: 5 resulta em liberação melhorada do fármaco na dispersão sólida. Exemplo 4 Dispersões sólidas de lovastatina: alginato de sódio com alto teor de viscosidade / baixo G
[0046] Utilizando o método descrito no Exemplo 1, dispersões sólidas de lovastatina em um alginato de sódio com baixo teor de G / alta viscosidade (Protanal CR8223, alginato de sódio com um teor de G de 37 a 40% em peso e uma viscosidade de 300 a 450 MPa.s numa solução a 1%) foram preparadas utilizando a técnica de evaporação do solvente descrita no Exemplo 1 nas proporções em peso (lovastatina: alginato) indicado na Tabela 4 abaixo.
[0047] As dispersões sólidas de lovastatina: alginato de sódio baixo G / alta viscosidade preparadas foram testadas in vitro em estudos de dissolução como descrito no Exemplo 1. Os resultados de tais testes estão resumidos na Tabela 4. Tabela 4
[0048] Os resultados acima demonstram que o aumento das quantidades de um alginato de sódio de baixa viscosidade e baixo G nas dispersões acima de uma proporção de 1: 1 para fármaco: proporções de alginato na faixa de 1: 1,5 a 1: 3,5 resulta em liberação melhorada do fármaco na dispersão sólida. Exemplo 5 Preparação de dispersão sólida itraconazol: alginato
[0049] As dispersões sólidas de itraconazol em alginato LFR5 / 60 foram preparadas utilizando a técnica de evaporação do solvente descrita no Exemplo 1 em proporções em peso (itraconazol: veículo) de 1: 3, 1: 4 e 1: 5. A quantidade necessária de fármaco foi pesada e dissolvida em 6 mL de diclorometano seguido por combinação nos veículos com amassamento para preparar uma pasta. A pasta foi, em seguida, seca abaixo de 40 °C ao longo de um período de 12 horas, para remover os solventes. As dispersões sólidas resultantes foram recolhidas e pulverizadas utilizando uma argamassa e pilão e armazenadas num dessecador em temperatura ambiente. Estudos de dissolução in vitro
[0050] Estudos in vitro de dissolução do itraconazol: as dispersões sólidas de alginato produzidas acima foram realizadas utilizando o método de pás equipadas com um aparelho USP II a 37,5 ± 0,5 °C e uma velocidade das p de 50 rpm. A quantidade apropriada de dispersão sólida (contendo itraconazol 50 mg) foi adicionada a 900 mL de meio de dissolução (tampão de fosfato 0,05 M pH 6,8 com SDS a 0,6% como condição de não lavagem). A concentração de itraconazol foi determinada a 261 nm. Os resultados médios de três repetições estão listados na Tabela 5.
Exemplo 6 Comparação com dispersões de polivinilpirrolidona ("PVP") fármaco bruto Preparação de dispersão de PVP e mistura de fármacos brutos
[0051] Foi preparada uma dispersão sólida de lovastatina em PVP (Kollidon 29/32; BASF) pelo método de evaporação do solvente utilizando um evaporador rotativo RE- 52AA (YARONG Co. Ltd, Xangai, China). A quantidade necessária de lovastatina e PVP para produzir uma mistura 1: 4 foi pesada e dissolvida em álcool num balão de fundo redondo e depois removeu-se o solvente utilizando um evaporador rotativo a 45 °C. A amostra foi ainda seca durante 2 h em um dessecador de vácuo e moída em uma argamassa. O pó foi coletado e armazenado em um dessecador em temperatura ambiente
[0052] A mistura física foi preparada misturando lovastatina e alginato LFR5 / 60 na proporção de 1:4. Estudos de dissolução in vitro
[0053] Um estudo de dissolução in vitro da amostra de PVP foi realizado seguindo o procedimento descrito no Exemplo 1. Os resultados de tais estudos, juntamente com os resultados para dispersões sólidas 1: 4: de lovastatina e alginato preparadas no Exemplo 1 e do fármaco sozinho são resumidos na Tabela 6 abaixo: Tabela 6 Teste 1:4 Lovastatina:Dispersante
Exemplo 7 Teste Comparativo da Dispersão Sólida de Alginato com Succinato de Acetato de Hipromelose (HPMCAS) e Polivinil-Pirrolidona (PVP) Dispersões sob condições de estresse A) Preparação de dispersões sólidas de lovastatina / alginato, lovastatina / PVPS630 e lovastatina / HPMCAS:
[0054] As dispersões sólidas de lovastatina (Hubei Xinyinhe Pharmaceutical) em alginato (Protanal LFR 5/60; FMC Corporation), em succinato de acetato de hipromelose (HPMCAS-MF; Shin-Etsu Chemical Co. Ltd) e polivinil pirolidona (PVPS630; ISP Technologies , Inc.) foram preparadas por um método de evaporação do solvente usando um evaporador rotativo RE-52AA (YARONG Co. Ltd, Xangai, China) em proporções em peso de fármaco / veículos variando de 1: 1 a 1: 5. A quantidade necessária de lovastatina e veículos foi pesada e estes dissolvidos em etanol em um balão de fundo redondo e, em seguida, o solvente foi removido utilizando um evaporador rotativo a 60 °C. As amostras foram adicionalmente secas durante 12 horas em um dessecador de vácuo e moídas em uma argamassa. O pó de dispersão de lovastatina foi recolhido e armazenado num dessecador em temperatura ambiente. B) Determinação de Razões Ótimas de Veículos de Fármacos
[0055] Liberação ótima do fármaco: a taxa de liberação do veículo para as dispersões sólidas de lovastatina/alginato, lovastatina/PVPS630 e lovastatina/HPMCAS produzidas na etapa A foi determinada por um estudo de dissolução in vitro realizado utilizando o método de pás equipadas com um aparelho USP II a 37,5 ± 0,5 °C e uma velocidade das p de 50 rpm. Uma quantidade dessa dispersão contendo 60 mg de lovastatina foi adicionada a um meio de dissolução de 900 mL (tampão de fosfato de pH 6,8 0,05 M com SDS a 0,1% como condição de não imersão, 1X). As amostras (6 mL) foram coletadas em intervalos de tempo predeterminados e filtradas imediatamente através de um filtro de seringa de 0,15 μm, os primeiros 2 mL de filtrado foram descartados e o filtrado restante foi coletado para análise a 237 nm usando um espectrofotômetro UV (Unic2000, Xangai, China). Enquanto isso, adicionou-se um volume igual de meio de dissolução fresco. Todas as experiências foram realizadas em triplicata.
[0056] Os resultados de tais testes indicaram que as razões ótimas de fármaco: peso portador eram: • Lovastatina: Alginato (LFR 5/60) 1: 4 • Lovastatina: HPMCAS 1: 4 • Lovastatina: PVP 1: 3 C) Formação do comprimido:
[0057] Os comprimidos contendo as dispersões sólidas nas proporções ótimas determinadas na etapa B foram formados empregando-se as seguintes composições: • Componente Quantidade (mg) • Dispersão de Lovastatina 100 • Ac-Di-Sol (FMC Corporation) 20 • Crospovidona 20 • Estearato de magnésio 0,8 • Celulose microcristalina 259,2
[0058] Depois de ter sido submetido às condições de armazenamento especificadas nas etapas D, E ou F abaixo, os estudos in vitro de dissolução dos comprimidos produzidos como na etapa C foram realizados utilizando o método de pás equipadas com um aparelho USP II a 37,5 ± 0,5 C e uma velocidade de remo de 50 rpm. Os comprimidos à base de dispersão sólida foram colocados em meio de dissolução de 900 mL (tampão de fosfato pH 6,8 0,05 M com 0,1% de SDS, condição de imersão). Amostras (6 mL) foram recolhidas em intervalos de tempo predeterminados e depois filtradas através de um filtro de seringa de 0,45 μm. Os primeiros 2 mL de filtrado foram descartados e o filtrado restante foi coletado para análise a 237nm usando um espectrofotômetro UV (Unic2000, Xangai, China). Enquanto isso, adicionou-se um volume igual de meio de dissolução fresco. Todas as experiências foram realizadas em triplicata. D) Estabilidade de alta temperatura
[0059] Os comprimidos que contêm a razão ótima de fármaco: veículo determinada na etapa C foram armazenados a 60 °C durante cinco dias e durante dez dias antes de serem submetidos ao teste de dissolução descrito na etapa C. Os resultados de tais testes são apresentados nas Tabelas 7A , 7B e 7C a seguir: Tabela 7A Teste de estabilidade térmica de Lovastatina:LFR 5/60 Alginato (razão em peso 1:4)
Tabela 7B Teste de estabilidade térmica de Lovastatina:HPMCAS (razão em peso 1:4)
Tabela 7C Teste de estabilidade térmica de Lovastatina:PVP (razão em peso 1:3)
[0060] Os dados acima indicam que a dispersão sólida alginato proporcionou melhor estabilidade ao calor do que as formadas usando dispersões HPMCAS ou PVP como um veículo. E) Estabilidade em alta umidade
[0061] Os comprimidos que contêm a razão ótima de fármaco: veículo determinada na etapa C foram armazenados a 92,5% de umidade relativa (HR) durante cinco dias e durante dez dias antes de serem submetidos ao teste de dissolução descrito na etapa C. Os resultados de tais testes são apresentados nas Tabelas 8A, 8B e 8C a seguir: Tabela 8A Teste em alta umidade de Lovastatina:LFR 5/60 Alginato (razão em peso 1:4)
Tabela 8B Teste em alta umidade de Lovastatina:HPMCAS (razão em peso 1:4)
Tabela 8C Teste em alta umidade de Lovastatina:PVP (razão em peso 1:3)
[0062] Os dados acima indicam que a dispersão sólida de alginato proporcionou melhor estabilidade ao calor do que as formadas usando dispersões HPMCAS ou PVP como um veículo. F) Estabilidade à luz reforçada
[0063] Os comprimidos que contêm a razão ótima de fármaco: veículo determinada na etapa C foram armazenados a 4500 + 500 lux durante cinco dias e durante dez dias antes de serem submetidos ao teste de dissolução descrito na etapa C. Os resultados de tais testes são apresentados nas Tabelas 9A, 9B e 9C a seguir: Tabela 9A Teste de estabilidade à luz reforçada de Lovastatina:LFR 5/60 Alginato (razão em peso 1:4)
Tabela 9B Teste de estabilidade à luz reforçada de Lovastatina:HPMCAS (razão em peso 1:4)
Tabela 9C Teste de estabilidade à luz reforçada de Lovastatina:PVP (razão em peso 1:3)
[0064] Os dados acima indicam que a dispersão sólida alginato fornece uma melhor estabilidade á luz do que as formadas usando dispersões de HPMCAS ou PVP como um veículo.


















