WO2020130119A1 - Wntシグナル伝達経路阻害剤 - Google Patents
Wntシグナル伝達経路阻害剤 Download PDFInfo
- Publication number
- WO2020130119A1 WO2020130119A1 PCT/JP2019/049992 JP2019049992W WO2020130119A1 WO 2020130119 A1 WO2020130119 A1 WO 2020130119A1 JP 2019049992 W JP2019049992 W JP 2019049992W WO 2020130119 A1 WO2020130119 A1 WO 2020130119A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- compound
- optionally substituted
- cancer
- substituted
- Prior art date
Links
Images



















Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
- A61K31/4178—1,3-Diazoles not condensed 1,3-diazoles and containing further heterocyclic rings, e.g. pilocarpine, nitrofurantoin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/454—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. pimozide, domperidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/472—Non-condensed isoquinolines, e.g. papaverine
- A61K31/4725—Non-condensed isoquinolines, e.g. papaverine containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- the present invention relates to a novel Wnt signaling pathway inhibitor.
- the present invention also provides a medicament for the prevention or treatment of a disease (for example, cancer, coronary artery disease, acute coronary syndrome, osteoarthritis, etc.) that can alleviate the symptoms by inhibiting the Wnt signaling pathway.
- a disease for example, cancer, coronary artery disease, acute coronary syndrome, osteoarthritis, etc.
- the Wnt/ ⁇ -catenin pathway is one of the Wnt signaling pathways and is a signal pathway conserved in various animal species in evolution, including nematodes, Drosophila, mice and humans.
- ⁇ -catenin which is a major molecule, is a protein of about 92 kDa, which acts as a lining protein of integrin for tissue formation, while translocating ⁇ -catenin into the nucleus leads to transcriptional activation of a target gene. In a state where the secretory ligand Wnt protein does not act, the amount of ⁇ -catenin protein is kept low.
- ⁇ -catenin forms a complex with adenomatous polyposis coli, axin, glycogen synthase kinase 3 ⁇ (GSK-3 ⁇ ) and casein kinase 1 ⁇ , and is phosphorylated by GSK-3 ⁇ .
- GSK-3 ⁇ glycogen synthase kinase 3 ⁇
- ⁇ -TrCP ⁇ -transducin repeat-containing protein
- proteasome phosphorylation of ⁇ -catenin is suppressed via the disheveled (Dsh).
- ⁇ -catenin accumulated in the cytoplasm is translocated into the nucleus and binds to T cell factor/lymphoid enhancer factor (TCF/LEF) family transcription factors.
- TCF/LEF T cell factor/lymphoid enhancer factor
- ⁇ -catenin forms a complex with B-cell chronic leukemia/lymphoma 9 and Pygopus, promotes transcriptional activity of TCF/LEF-related genes such as c-myc, cyclin D1, and c-jun, and promotes cell proliferation. It controls stem cell maintenance, segmentation and organ formation, and plays an important role in life phenomena such as body axis formation in early embryonic maturation, various organ formation, cell proliferation/differentiation, and tissue regeneration.
- Non-Patent Documents 1 and 2 In recent years, it has become clear that the Wnt signal transduction pathway is also involved in the proliferation and maintenance of many cancer stem cells, and has attracted attention as a target for cancer treatment (Non-Patent Documents 1 and 2). In addition to the proliferation/maintenance of cancer stem cells, the Wnt signaling pathway has also been reported to be involved in the onset of various diseases such as various coronary artery diseases, acute coronary syndromes, and osteoarthritis ( Non-Patent Documents 3 and 4).
- Non-Patent Document 5 Non-Patent Document 5
- Non-Patent Documents 7 and 8 include the following formula:
- the object of the present invention is to provide a novel Wnt signaling pathway inhibitor.
- a further object of the present invention is to provide a medicament for the prevention or treatment of diseases, in particular cancer, which can alleviate the symptoms by inhibiting the Wnt signaling pathway.
- R represents an optionally substituted C 6-14 aryl group, an optionally substituted heteroaryl group or an optionally protected hydroxy group
- R 1 represents an optionally substituted C 1-6 alkyl group or an optionally substituted C 6-14 aryl group
- R 2 represents an optionally substituted C 1-6 alkyl group, an optionally substituted C 1-6 alkoxy group or an optionally substituted C 6-14 aryl group
- R 3 and R 4 are each independently a hydrogen atom, a halogen atom, a hydroxy group, an amino group, a nitro group, a cyano group, a carboxy group, an optionally substituted C 1-6 alkyl group or a substituted group.
- a C 1-6 alkoxy group which may also be substituted, or R 3 and R 4 are each optionally substituted, together with the carbon atom to which they are attached, a cyclohexane ring Or it may form a benzene ring;
- Bond b represents a single bond or a double bond;
- X represents CH 2 or carbonyl (C ⁇ O);
- Y represents an oxygen atom or NR 0 (in the formula, R 0 represents a hydrogen atom or a C 1-4 alkoxy group);
- m represents an integer of 1 to 3; and n represents 0 or 1.
- the present invention was found for the first time to find that the compound represented by (hereinafter sometimes referred to as “compound (I)”) or a pharmaceutically acceptable salt thereof exhibits excellent Wnt signaling pathway inhibitory activity. It came to completion.
- R represents an optionally substituted C 6-14 aryl group, an optionally substituted heteroaryl group or an optionally protected hydroxy group
- R 1 represents an optionally substituted C 1-6 alkyl group or an optionally substituted C 6-14 aryl group
- R 2 represents an optionally substituted C 1-6 alkyl group, an optionally substituted C 1-6 alkoxy group or an optionally substituted C 6-14 aryl group
- R 3 and R 4 are each independently a hydrogen atom, a halogen atom, a hydroxy group, an amino group, a nitro group, a cyano group, a carboxy group, an optionally substituted C 1-6 alkyl group or a substituted group.
- a C 1-6 alkoxy group which may also be substituted, or R 3 and R 4 are each optionally substituted, together with the carbon atom to which they are attached, a cyclohexane ring Or it may form a benzene ring;
- Bond b represents a single bond or a double bond;
- X represents CH 2 or carbonyl (C ⁇ O);
- Y represents an oxygen atom or NR 0 (in the formula, R 0 represents a hydrogen atom or a C 1-4 alkoxy group);
- m represents an integer of 1 to 3; and n represents 0 or 1.
- a Wnt signal transduction pathway inhibitor comprising a compound represented by: or a pharmaceutically acceptable salt thereof.
- R is an optionally substituted C 6-10 aryl group, an optionally substituted heteroaryl group or a protected hydroxy group
- R 1 is a C 1-4 alkyl group optionally substituted by 1 or 2 phenyl groups, or an optionally substituted phenyl group
- R 2 is optionally substituted C 1-4 alkyl group, optionally substituted C 1-4 alkoxy group, a phenyl group which may be substituted
- R 3 and R 4 are each independently a hydrogen atom, a hydroxy group or a C 1-6 alkoxy group, or R 3 and R 4 are bonded to each other and are carbon atoms to which they are bonded.
- R is a phenyl group which may be substituted, a naphthyl group which may be substituted, an imidazolyl group which may be substituted, an indolyl group which may be substituted, a trityloxy group or a benzyloxy group.
- R 3 and R 4 are both hydrogen atoms, or R 3 and R 4 are bonded to each other to form a cyclohexane ring together with the carbon atom to which they are bonded.
- the Wnt signal transduction pathway inhibitor according to any one of the above [1] to [4], wherein the bond b is a single bond.
- a medicament comprising the Wnt signal transduction pathway inhibitor according to any one of [1] to [5] above as an active ingredient.
- the medicament according to [6] above which is used for the prevention or treatment of a disease in which symptoms can be alleviated by inhibiting the Wnt signal transduction pathway.
- the medicine according to the above [7], wherein the disease that can alleviate symptoms by inhibiting the Wnt signaling pathway is cancer, coronary artery disease, acute coronary syndrome, or osteoarthritis.
- the drug according to [7] above, wherein the disease in which symptoms can be alleviated by inhibiting the Wnt signal transduction pathway is cancer.
- Cancer is colon cancer, breast cancer, head and neck cancer, lung cancer, gastric cancer, esophageal cancer, malignant melanoma, prostate cancer, hepatocellular carcinoma, ovarian cancer, hepatoblastoma, acute myelogenous leukemia, acute lymphoblastic
- the above-mentioned [9] which is leukemia, chronic myelogenous leukemia, chronic lymphocytic leukemia, multiple myeloma, malignant lymphoma, pancreatic cancer, kidney cancer, bladder cancer, glioblastoma, medulloblastoma, or Wilms tumor.
- R′ is substituted with a phenyl group optionally substituted with a hydroxy group or a C 1-6 alkoxy group, a naphthyl group, an imidazolyl group substituted with a trityl group, an optionally substituted C 6-14 arylsulfonyl group A selected indolyl group, trityloxy group or benzyloxy group;
- R 1 ' is two methyl group which may be substituted with a phenyl group, or a phenyl group;
- R 2' is optionally biphenylyl group which may be substituted by ethynyl group, substituted with a tert- butyl group A phenyl group, a methyl group, or a fluorenylmethoxy group;
- R 3 ′ and R 4 ′ each independently represent a hydrogen atom or a hydroxy group, or R 3 ′ and R 4 ′ are bonded to each other and together with the carbon
- Bond b' represents a single bond or a double bond
- X′ represents CH 2 or carbonyl (C ⁇ O)
- Y′ represents an oxygen atom, NH or N(OCH 3 );
- R 2 'when is unsubstituted biphenylyl group Y'-R 1' represents a methoxy group.
- compound (II) is a pharmaceutically acceptable salt thereof.
- a drug containing a Wnt signal transduction pathway inhibitor comprising the compound (I) according to the present invention as an active ingredient is a disease in which symptoms can be alleviated by inhibiting the Wnt signal transduction pathway, for example, cancer, coronary It is useful for prevention or treatment of arterial disease, acute coronary syndrome, osteoarthritis, and the like, and in particular, various cancers (for example, colon cancer, breast cancer, head and neck cancer, lung cancer, gastric cancer, esophageal cancer, malignant melanoma, prostate cancer).
- various cancers for example, colon cancer, breast cancer, head and neck cancer, lung cancer, gastric cancer, esophageal cancer, malignant melanoma, prostate cancer.
- a novel compound (compound (II)) having excellent Wnt signal transduction pathway inhibitory activity or a pharmaceutically acceptable salt thereof can be provided.
- FIG. 1 shows 6 multiple myeloma (MM) cell lines (NCI-H929, MM1.s, RPMI8226, U266, AMO-1, OPM-2) with different compounds (1) using WST-8 assay. Shows a growth inhibitory effect on.
- FIG. 2 shows changes in mRNA expression by compound (1) in three MM cell lines (NCI-H929, AMO-1, MM1.s).
- FIG. 3 shows a cell cycle analysis for confirming the effect of the compound (1) on the cell cycle of three MM cell lines (NCI-H929, AMO-1, MM1.s) using the FCM method.
- FIG. 1 shows 6 multiple myeloma (MM) cell lines (NCI-H929, MM1.s, RPMI8226, U266, AMO-1, OPM-2) with different compounds (1) using WST-8 assay. Shows a growth inhibitory effect on.
- FIG. 2 shows changes in mRNA expression by compound (1) in three MM cell lines (NCI-
- FIG. 4 shows the induction of apoptosis (cell death) by the compound (1) in the MM cell lines (NCI-H929, AMO-1, MM1.s, U266) using the flow cytometry (FCM) method.
- FIG. 5 shows 6 multiple myeloma (MM) cell lines (MM1.s, RPMI8226, IM-9, OPM-2, AMO-1, NCI-) with different compounds (7) using WST-8 assay. 12 shows the inhibitory effect on proliferation against H929).
- FIG. 6 shows changes in intracellular ⁇ -catenin protein expression in AMO-1 cells due to compound (7).
- FIG. 7 shows the inhibitory effect on proliferation of pancreatic cancer cell line (AsPC-1) by compound (11) using WST-8 assay.
- FIG. 8 shows changes in the expression of intracellular ⁇ -catenin protein, C-MYC, SURVIVIN and GAPDH in pancreatic cancer cell line (AsPC-1) by compound (11).
- FIG. 9 shows the inhibitory effects on proliferation of pancreatic cancer cell line (AsPC-1) by compound (10) and compound (24) using WST-8 assay.
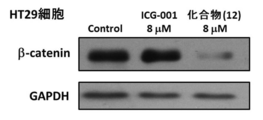
- FIG. 10 shows the growth inhibitory effect of the compound (12) using the WST-8 assay on three types of colon cancer cell lines (HT29, SW480, HCT116/E6).
- FIG. 11 shows changes in mRNA expression by the compound (12) in a colon cancer cell line (HT29).
- FIG. 12 shows changes in intracellular ⁇ -catenin protein and GAPDH expression in a colon cancer cell line (HT29) by the compound (12).
- FIG. 13 shows the anti-resistance to human acute myelogenous leukemia cell line (MV4;11, KG1a, HL-60) and acute lymphoblastic leukemia/lymphoma cell line (RS4; 11, Nalm6, Raji) by compound (19). Tumor effect is shown.
- FIG. 14 shows changes in expression of ⁇ -catenin and Wnt signal downstream molecule in human acute myelogenous leukemia cell line (MV4; 11, HL-60) by compound (19).
- FIG. 15 shows changes in expression of ⁇ -catenin and Wnt signal downstream molecule in human acute myelogenous leukemia cell line (KG1a) by compound (19).
- FIG. 16 shows the expression change of ⁇ -catenin (CTNNB1) mRNA in human acute myelogenous leukemia cell line (MV4; 11) by compound (19).
- FIG. 17 shows induction of apoptosis (cell death) by a compound (19) against a human acute myelogenous leukemia cell line (MV4; 11, KG1a).
- FIG. 18 shows the antitumor effect of compound (19) on acute myeloid leukemia cell lines (KBM5, KBM/STIR).
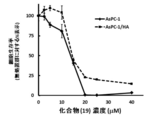
- FIG. 19 shows the antitumor effect of compound (19) on human pancreatic cancer cell line AsPC-1 cells.
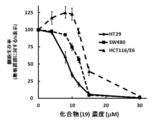
- FIG. 20 shows the antitumor effect against human colon cancer cell lines (SW480, HT29, HCT116/E6) by compound (19) using a luciferase assay.
- FIG. 21 shows the cancer sphere formation in breast cancer cell line (MDA-MB-231) and glioblastoma cell line (U251-MG), and the expression level of ⁇ -catenin (CTNNB1)-mRNA in each cancer stem cell. Show.
- the solid line in FIG. 22 shows the antitumor effect of compound (19) on cancer stem cells (MDA-MB-231, U251-MG).
- halogen atom means a fluorine atom, a chlorine atom, a bromine atom or an iodine atom.
- alkyl (group) means a straight-chain or branched-chain alkyl group having 1 or more carbon atoms, and when there is no particular limitation on the carbon number range, it is preferably C It is a 1-20 alkyl group, and among them, a C 1-6 alkyl group is more preferable, and a C 1-4 alkyl group is particularly preferable.
- C 1-20 alkyl (group) means a linear or branched alkyl group having 1 to 20 carbon atoms, and examples thereof include methyl, ethyl, propyl, isopropyl, butyl and isobutyl.
- C 1-6 alkyl (group) means a linear or branched alkyl group having 1 to 6 carbon atoms, and examples thereof include methyl, ethyl, propyl, isopropyl, butyl, isobutyl. , Sec-butyl, tert-butyl, pentyl, isopentyl, neopentyl, 1-ethylpropyl, hexyl, isohexyl, 1,1-dimethylbutyl, 2,2-dimethylbutyl, 3,3-dimethylbutyl, 2-ethylbutyl, etc. Can be mentioned.
- C 1-4 alkyl (group) means a straight chain or branched chain alkyl group having 1 to 4 carbon atoms, and examples thereof include methyl, ethyl, propyl, isopropyl, butyl and isobutyl. , Sec-butyl, tert-butyl and the like.
- cycloalkyl (group) means a cyclic alkyl group, and is preferably a C 3-8 cycloalkyl group, unless the carbon number range is particularly limited.
- C 3-8 cycloalkyl (group) means a cyclic alkyl group having 3 to 8 carbon atoms, and examples thereof include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl and the like. Are listed. Of these, a C 3-6 cycloalkyl group is preferable.
- the “C 2-6 alkynyl (group)” has one or more carbon-carbon triple bonds and has a carbon number of 2 to 6 which is a linear or branched monovalent carbonization.
- Means a hydrogen group for example, ethynyl, 1-propynyl, 2-propynyl, 1-butynyl, 2-butynyl, 3-butynyl, 1-pentynyl, 2-pentynyl, 3-pentynyl, 4-pentynyl, 1-hexynyl, 2-hexynyl, 3-hexynyl, 4-hexynyl, 5-hexynyl, 4-methyl-2-pentynyl can be mentioned. Among them, ethynyl is preferable.
- alkoxy (group) means a group in which a linear or branched alkyl group is bonded to an oxygen atom, and the carbon number range is not particularly limited, but is preferably C 1-6 alkoxy. A group, and more preferably a C 1-4 alkoxy group.
- C 1-6 alkoxy (group) means a straight chain or branched chain alkoxy group having 1 to 6 carbon atoms, and includes, for example, methoxy, ethoxy, propoxy, isopropoxy, butoxy, Examples thereof include isobutoxy, sec-butoxy, tert-butoxy, pentyloxy, isopentyloxy, neopentyloxy and hexyloxy. Of these, a C 1-4 alkoxy group is preferable.
- alkoxy-carbonyl (group) means a group in which the above alkoxy group is bonded to an oxygen atom and a carbonyl group, and the carbon number range is not particularly limited, but is preferably C 1-6 alkoxy. A carbonyl group.
- alkylsulfanyl (group) means a group in which the above “alkyl” group is bonded to a sulfur atom, and the carbon number range is not particularly limited, but is preferably C 1-6 alkylsulfanyl. is there.
- C 1-6 alkylsulfanyl (group) means a linear or branched alkylsulfanyl group having 1 to 6 carbon atoms, and examples thereof include methylsulfanyl, ethylsulfanyl, propylsulfanyl, Examples thereof include isopropylsulfanyl, butylsulfanyl, isobutylsulfanyl, sec-butylsulfanyl, tert-butylsulfanyl, pentylsulfanyl, isopentylsulfanyl, neopentylsulfanyl, 1-ethylpropylsulfanyl and hexylsulfanyl.
- C 1-6 alkylsulfinyl (group) is a group in which the aforementioned “C 1-6 alkyl” group is bonded to a sulfinyl group, that is, a straight chain or branched chain alkyl having 1 to 6 carbon atoms. Means a sulfinyl group.
- C 1-6 alkylsulfinyl (group) examples include methylsulfinyl, ethylsulfinyl, propylsulfinyl, isopropylsulfinyl, butylsulfinyl, isobutylsulfinyl, sec-butylsulfinyl, tert-butylsulfinyl, pentylsulfinyl, isopentyl. Examples thereof include sulfinyl, neopentylsulfinyl, 1-ethylpropylsulfinyl and hexylsulfinyl.
- C 1-6 alkylsulfonyl (group) is a group in which the above “C 1-6 alkyl” group is bonded to a sulfonyl group, that is, a straight chain or branched chain alkyl group having 1 to 6 carbon atoms. It means a sulfonyl group.
- C 1-6 alkylsulfonyl (group) examples include methylsulfonyl, ethylsulfonyl, propylsulfonyl, isopropylsulfonyl, butylsulfonyl, isobutylsulfonyl, sec-butylsulfonyl, tert-butylsulfonyl, pentylsulfonyl, isopentyl. Examples thereof include sulfonyl, neopentylsulfonyl, 1-ethylpropylsulfonyl, hexylsulfonyl and the like.
- alkylsulfonyloxy (group) means a group in which an alkylsulfonyl group is bonded to an oxygen atom, and the carbon number range is not particularly limited, but is preferably a C 1-6 alkylsulfonyloxy group. Is.
- C 1-6 alkylsulfonyloxy (group) means a group in which a C 1-6 alkylsulfonyl group is bonded to an oxygen atom, and examples thereof include methylsulfonyloxy, ethylsulfonyloxy and propylsulfonyl. Oxy, isopropyl sulfonyloxy, butyl sulfonyloxy, etc. are mentioned.
- aryl (group) means a monocyclic or polycyclic (fused) hydrocarbon group exhibiting aromaticity, and specifically, for example, phenyl, 1-naphthyl, Examples thereof include C 6-14 aryl groups such as 2-naphthyl, biphenylyl, 2-anthryl and fluorenyl, and among them, C 6-10 aryl groups are preferable.
- examples of the “C 6-10 aryl (group)” include phenyl, 1-naphthyl and 2-naphthyl, and phenyl or 1-naphthyl is particularly preferable.
- aralkyl (group) means a group in which an aryl group is substituted on an alkyl group, and the carbon number range is not particularly limited, but is preferably C 7-14 aralkyl.
- C 7-22 aralkyl (group) means a group in which a “C 1-4 alkyl group” is substituted with a “C 6-18 aryl group”, and examples thereof include benzyl and 1-phenyl.
- heteroaryl (group) means a monocyclic, bicyclic or polycyclic aromatic group in which at least one ring atom is a hetero atom and the remaining ring atoms are carbon.
- Monocyclic heteroaryl groups include, but are not limited to, cyclic aromatic groups having 5 or 6 ring atoms, at least one ring atom being a heteroatom and the remaining ring atoms being carbon. Examples include cyclic aromatic groups.
- the nitrogen atom may be optionally quaternized and the sulfur atom may be optionally oxidized.
- heteroaryl group of the present invention examples include furan, imidazole, isothiazole, isoxazole, oxadiazole, oxazole, 1,2,3-oxadiazole, pyrazine, pyrazole, pyridazine, pyridine, pyrimidine, pyrroline, thiazole, 1 ,3,4-thiadiazole, triazole, and tetrazole.
- “Heteroaryl” means one or two heteroaryl rings independently selected from the group consisting of aryl rings, cycloalkyl rings, cycloalkenyl rings, and other monocyclic heteroaryl or heterocycloalkyl rings.
- bicyclic or tricyclic rings fused to one ring.
- bicyclic or tricyclic heteroaryls include benzo[b]furan, benzo[b]thiophene, benzimidazole, imidazo[4,5-c]pyridine, quinazoline, thieno[2,3-c].
- arylsulfanyl (group) means a group in which an “aryl group” is bonded to a sulfur atom, and the carbon number range is not particularly limited, but is preferably C 6-10 arylsulfanyl. ..
- C 6-10 arylsulfanyl (group) means an arylsulfanyl group having 6 to 10 carbon atoms, and examples thereof include phenylsulfanyl, 1-naphthylsulfanyl, 2-naphthylsulfanyl and the like.
- arylsulfonyl (group) means a group in which an aryl group is bonded to a sulfonyl group, and the carbon number range is not particularly limited, but is preferably a C 6-10 arylsulfonyl group.
- C 6-10 arylsulfonyl (group) means a group in which a “C 6-10 aryl group” is bonded to a sulfonyl group, and examples thereof include phenylsulfonyl, 1-naphthylsulfonyl, 2- Naphthyl sulfonyl etc. are mentioned.
- arylsulfonyloxy (group) means a group in which the arylsulfonyl group is bonded to an oxygen atom, and the carbon number range is not particularly limited, but is preferably C 6-10 arylsulfonyloxy. It is a base.
- C 6-10 arylsulfonyloxy (group) means a group in which a C 6-10 arylsulfonyl group is bonded to an oxygen atom, and examples thereof include phenylsulfonyloxy, 1-naphthylsulfonyloxy, 2-naphthylsulfonyloxy and the like can be mentioned.
- acyl (group) means alkanoyl or aroyl, and the carbon number range is not particularly limited, but is preferably a C 1-7 alkanoyl group or C 7-11 aroyl.
- C 1-7 alkanoyl (group) is a linear or branched formyl or alkylcarbonyl having 1 to 7 carbon atoms (that is, C 1-6 alkyl-carbonyl).
- formyl, acetyl, propionyl, butyryl, isobutyryl, pentanoyl, hexanoyl, heptanoyl and the like can be mentioned.
- C 7-11 aroyl (group) is an arylcarbonyl having 7 to 11 carbon atoms (that is, C 6-10 aryl-carbonyl), and examples thereof include benzoyl.
- acyloxy (group) means a group in which the alkanoyl group or aroyl group is bonded to an oxygen atom, and the carbon number range is not particularly limited, but preferably a C 1-7 alkanoyloxy group. Alternatively, it is a C 7-11 aroyloxy group.
- examples of the “C 1-7 alkanoyloxy (group)” include formyloxy, acetoxy, ethylcarbonyloxy, propylcarbonyloxy, isopropylcarbonyloxy, butylcarbonyloxy, isobutylcarbonyloxy, sec-butylcarbonyl.
- Oxy, tert-butylcarbonyloxy (pivaloyloxy), pentylcarbonyloxy, isopentylcarbonyloxy, neopentylcarbonyloxy, hexylcarbonyloxy and the like can be mentioned, with preference given to acetoxy or pivaloyloxy.
- examples of the “C 7-11 aroyloxy (group)” include benzoyloxy, 1-naphthoyloxy, 2-naphthoyloxy and the like.
- the “optionally protected hydroxy group” means a hydroxy group or a hydroxy group protected by a “protecting group”.
- a protecting group of hydroxy group described in Protective Groups in Organic Synthesis, John Wiley and Sons (1980) may be used, and examples thereof include C 1-6 alkyl group and C 7-22.
- the above protecting groups may be further substituted with a halogen atom, a C 1-6 alkyl group, a C 1-6 alkoxy group or a nitro group.
- the “hydroxy group which may be present” is preferably a protected hydroxy group, and particularly preferably a benzyl- or trityl-protected hydroxy group.
- the “protected amino group” means an amino group protected with a “protecting group”.
- a “protecting group” for example, an amino protecting group described in Protective Groups in Organic Synthesis, John Wiley and Sons (1980) can be used, and examples thereof include a C 1-6 alkyl group and a C 7-22.
- the above protecting groups may be further substituted with a halogen atom, a C 1-6 alkyl group, a C 1-6 alkoxy group or a nitro group.
- Specific examples of the amino-protecting group include methyl (monomethyl or dimethyl), benzyl, trityl, acetyl, trifluoroacetyl, pivaloyl, tert-butoxycarbonyl, benzyloxycarbonyl and the like.
- the "optionally substituted” means that it may have one or more substituents
- the "substituent” includes (1) a halogen atom and (2 )Nitro, (3) cyano, (4) C 1-6 alkyl, (5) C 2-6 alkynyl, (6) C 3-8 cycloalkyl, (7) C 1-6 alkoxy, (8) C 6 -10 aryl, (9)C 7-22 aralkyl, (10)C 1-7 alkanoyloxy, (11)C 7-11 aroyloxy, (12)C 1-7 alkanoyloxy-carbonyl, (13)C 7- 11 aroyloxy-carbonyl, (14)C 1-7 alkanoyl, (15)C 7-11 aroyl, (16)C 1-6 alkylsulfanyl, (17)C 6-10 arylsulfanyl, (18)C 1-6 Carbamoyl which may be mono- or di-substituted by an alkyl group, (19)
- halogen atom hydroxy, C 1-6 alkyl, C 1-6 alkoxy, ethynyl, benzyl, fluorenylmethyl, diphenylmethyl, trityl, acetyl, formyl, carbamoyl, triethylsilyl, triisopropylsilyl, tert-butyldimethyl
- silyl dimethylamino, acetylamino, tert-butoxycarbonylamino, benzyloxycarbonylamino and the like.
- each substituent may be the same or different.
- the above substituents may be further substituted with the above substituents.
- the number of substituents is not particularly limited as long as it is a substitutable number, but is preferably 1 to 5 and more preferably 1 to 3. When multiple substituents are present, each substituent may be the same or different.
- the substituent may further be a C 1-6 alkyl group, a C 3-8 cycloalkyl group, a C 6-10 aryl group, a C 7-22 aralkyl group, a halogen atom, a hydroxy group, a protected amino group, a carbamoyl group, It may be substituted with a cyano group, a nitro group, an oxo group or the like.
- the number of substituents is not particularly limited as long as it is a substitutable number, but is preferably 1 to 5 and more preferably 1 to 3. When multiple substituents are present, each substituent may be the same or different.
- the “pharmaceutically acceptable salt” means a salt that can be used as a medicine.
- the compound (I) (or compound (II)) of the present invention has an acidic group or a basic group, it can be converted to a basic salt or an acidic salt by reacting with a base or an acid. Indicates salt.
- Examples of the pharmaceutically acceptable "basic salt" of compound (I) (or compound (II)) of the present invention include, for example, alkali metal salts such as sodium salt, potassium salt and lithium salt; magnesium salt, calcium salt and the like.
- Examples of the pharmaceutically acceptable “acid salt” of the compound (I) (or compound (II)) of the present invention include, for example, hydrofluoric acid salt, hydrochloride, hydrobromide, hydroiodide and the like.
- Inorganic acid salts such as hydrohalides, nitrates, perchlorates, sulfates and phosphates; lower alkane sulfonates such as methane sulfonate, trifluoromethane sulfonate and ethane sulfonate, benzene Allyl sulfonates such as sulfonates and p-toluene sulfonates, acetates, malates, fumarates, succinates, citrates, ascorbates, tartrates, oxalates, maleates
- organic acid salts such as glycine salt, lysine salt, arginine salt, ornithine salt, glutamic acid
- prevention includes prevention of the onset of the disease and delay of the onset of the disease.
- prophylactically effective amount refers to a dose of compound (I) sufficient to achieve such a purpose.
- treatment includes healing of a disease, improvement of a disease state (for example, one or more symptoms), and suppression of progression of (severity of) the disease.
- “Therapeutically effective amount” refers to a dose of compound (I) sufficient to achieve such a purpose.
- the “Wnt signal transduction pathway inhibitor” means an agent for partially or completely inhibiting, suppressing, or neutralizing the biological activity of the Wnt signal transduction pathway.
- a disease capable of alleviating symptoms by inhibiting the Wnt signaling pathway means a disease or a symptom caused by abnormal activation of Wnt signal.
- Specific examples of the “disease in which symptoms can be alleviated by inhibiting the Wnt signaling pathway” are useful, for example, in the prevention or treatment of cancer, coronary artery disease, acute coronary syndrome, osteoarthritis and the like.
- various cancers eg, colon cancer, breast cancer, head and neck cancer, lung cancer, gastric cancer, esophageal cancer, malignant melanoma, prostate cancer, hepatocellular carcinoma, ovarian cancer, hepatoblastoma, acute myelogenous leukemia, acute lymphoblast
- Useful for prevention or treatment of leukemia chronic myelogenous leukemia, chronic lymphocytic leukemia, multiple myeloma, malignant lymphoma, pancreatic cancer, kidney cancer, bladder cancer, glioblastoma, medulloblastoma, Wilms tumor, etc. Is.
- the Wnt signaling pathway inhibitor of the present invention has the following formula (I):
- R represents an optionally substituted C 6-14 aryl group, an optionally substituted heteroaryl group or an optionally protected hydroxy group
- R 1 represents an optionally substituted C 1-6 alkyl group or an optionally substituted C 6-14 aryl group
- R 2 represents an optionally substituted C 1-6 alkyl group, an optionally substituted C 1-6 alkoxy group or an optionally substituted C 6-14 aryl group
- R 3 and R 4 are each independently a hydrogen atom, a halogen atom, a hydroxy group, an amino group, a nitro group, a cyano group, a carboxy group, an optionally substituted C 1-6 alkyl group or a substituted group.
- a C 1-6 alkoxy group which may also be substituted, or R 3 and R 4 are each optionally substituted, together with the carbon atom to which they are attached, a cyclohexane ring Or it may form a benzene ring;
- Bond b represents a single bond or a double bond;
- X represents CH 2 or carbonyl (C ⁇ O);
- Y represents an oxygen atom or NR 0 (in the formula, R 0 represents a hydrogen atom or a C 1-4 alkoxy group);
- m represents an integer of 1 to 3; and n represents 0 or 1.
- R represents an optionally substituted C 6-14 aryl group, an optionally substituted heteroaryl group or an optionally protected hydroxy group.
- R is preferably an optionally substituted C 6-10 aryl group, an optionally substituted heteroaryl group or a protected hydroxy group, and more preferably an optionally substituted phenyl group, An optionally substituted naphthyl group, an optionally substituted imidazolyl group, an optionally substituted indolyl group, a trityloxy group or a benzyloxy group, and particularly preferably an optionally substituted imidazolyl group ( For example, an imidazolyl group substituted with a trityl group).
- R 1 represents an optionally substituted C 1-6 alkyl group or an optionally substituted C 6-14 aryl group.
- R 1 is preferably a C 1-4 alkyl group optionally substituted by 1 or 2 phenyl groups, or an optionally substituted phenyl group, and more preferably 2 phenyl groups.
- substituted C 1-4 alkyl group e.g., diphenylmethyl group
- a C 1-4 alkyl group e.g., methyl group
- a phenyl group particularly preferably, a C 1-4 alkyl group (e.g., methyl Group).
- R 2 represents an optionally substituted C 1-6 alkyl group, an optionally substituted C 1-6 alkoxy group or an optionally substituted C 6-14 aryl group.
- R 2 is preferably an optionally substituted C 1-4 alkyl group, optionally substituted C 1-4 alkoxy group, a phenyl group which may be substituted, more preferably, substituted Optionally substituted C 1-4 alkyl group or optionally substituted phenyl group, particularly preferably substituted with a protected amino group (eg, N-tert-butoxycarbonyl-N-phenyl-amino)
- a C 1-4 alkyl group which may be substituted, or a phenyl group which may be substituted with a halogen atom (eg bromine atom, fluorine atom), a phenyl group or a C 1-4 alkyl group (eg methyl) is there.
- R 3 and R 4 are each independently a hydrogen atom, a halogen atom, a hydroxy group, an amino group, a nitro group, a cyano group, a carboxy group, an optionally substituted C 1-6 alkyl group or a substituted group.
- R 3 and R 4 are preferably each independently a hydrogen atom, a hydroxy group or a C 1-6 alkoxy group, or R 3 and R 4 are bonded to each other and they are bonded. And R 3 and R 4 are bonded to each other to form a cyclohexane ring, more preferably a hydrogen atom together, or a carbon to which they are bonded. Together with the atoms form a cyclohexane ring.
- the bond b represents a single bond or a double bond.
- the bond b is preferably a single bond.
- X represents CH 2 or carbonyl (C ⁇ O).
- X is preferably CH 2 .
- Y represents an oxygen atom or NR 0 (R 0 in the formula represents a hydrogen atom or a C 1-4 alkoxy group).
- Y is preferably an oxygen atom, NH or N(OCH 3 ), and more preferably an oxygen atom or N(OCH 3 ).
- M represents an integer of 1 to 3.
- M is preferably 1.
- N 0 or 1.
- N is preferably 1.
- R is an optionally substituted C 6-10 aryl group, an optionally substituted heteroaryl group or a protected hydroxy group
- R 1 is a C 1-4 alkyl group optionally substituted by 1 or 2 phenyl groups, a C 1-4 alkyl group or an optionally substituted phenyl group
- R 2 is optionally substituted C 1-4 alkyl group, optionally substituted C 1-4 alkoxy group, a phenyl group which may be substituted
- R 3 and R 4 are each independently a hydrogen atom, a hydroxy group or a C 1-6 alkoxy group, or R 3 and R 4 are bonded to each other and are carbon atoms to which they are bonded.
- R is an optionally substituted phenyl group, an optionally substituted naphthyl group, an optionally substituted imidazolyl group, an optionally substituted indolyl group, a trityloxy group or a benzyloxy group
- R 1 is a C 1-4 alkyl group (eg, diphenylmethyl group) substituted with two phenyl groups, a C 1-4 alkyl group (eg, methyl group), or a phenyl group
- R 2 is an optionally substituted C 1-4 alkyl group or an optionally substituted phenyl group
- R 3 and R 4 are both hydrogen atoms, or R 3 and R 4 are bonded to each other and together with the carbon atom to which they are bonded to form a cyclohexane ring
- Bond b is a single bond
- X is CH 2 or carbonyl
- Y is an oxygen atom
- R is an optionally substituted imidazolyl group (eg, an imidazolyl group substituted with a trityl group)
- R 1 is a C 1-4 alkyl group (eg, methyl group)
- R 2 is a C 1-4 alkyl group which may be substituted with a protected amino group (eg, N-tert-butoxycarbonyl-N-phenyl-amino), or a halogen atom (eg, bromine atom, fluorine) Atom), a phenyl group or a C 1-4 alkyl group (eg, methyl), which may be substituted with a phenyl group
- R 3 and R 4 are both hydrogen atoms, or R 3 and R 4 are bonded to each other and together with the carbon atom to which they are bonded to form a cyclohexane ring
- Bond b is a single bond
- X is CH 2
- Y is an oxygen
- preferable compound (I) are the compounds of Examples 1 to 34 (compound (1) to compound (34)) described below, or pharmaceutically acceptable salts thereof.
- More preferred compound (I) is specifically the following compound or a pharmaceutically acceptable salt thereof.
- Particularly preferred compound (I) is specifically the following compound or a pharmaceutically acceptable salt thereof.
- R′ is substituted with a phenyl group optionally substituted with a hydroxy group or a C 1-6 alkoxy group, a naphthyl group, an imidazolyl group substituted with a trityl group, an optionally substituted C 6-14 arylsulfonyl group A selected indolyl group, trityloxy group or benzyloxy group;
- R 1 ' is two methyl group which may be substituted with a phenyl group, or a phenyl group;
- R 2' is optionally biphenylyl group which may be substituted by ethynyl group, substituted with a tert- butyl group A phenyl group, a methyl group, or a fluorenylmethoxy group;
- R 3 ′ and R 4 ′ each independently represent a hydrogen atom or a hydroxy group, or R 3 ′ and R 4 ′ are bonded to each other and together with the carbon
- Bond b' represents a single bond or a double bond
- X′ represents CH 2 or carbonyl (C ⁇ O)
- Y′ represents an oxygen atom, NH or N(OCH 3 )
- R 2 ' When is unsubstituted biphenylyl group, Y'-R 1' represents a methoxy group.
- the compound (compound (II)) represented by or a pharmaceutically acceptable salt thereof is provided.
- R′ is substituted with a phenyl group optionally substituted with a hydroxy group or a C 1-6 alkoxy group, a naphthyl group, an imidazolyl group substituted with a trityl group, an optionally substituted C 6-14 arylsulfonyl group Represents an indolyl group, a trityloxy group or a benzyloxy group.
- R' is preferably a phenyl group optionally substituted with a C 1-4 alkoxy group, a naphthyl group, an imidazolyl group substituted with a trityl group, an indolyl group substituted with an optionally substituted phenylsulfonyl group.
- R 1 ′ represents a methyl group which may be substituted with two phenyl groups, or a phenyl group.
- R 1 ′ is preferably a methyl group.
- R 2 ′ represents a biphenylyl group optionally substituted with an ethynyl group, a phenyl group substituted with a tert-butyl group, a methyl group, or a fluorenylmethoxy group.
- R 2 ′ is preferably a phenyl group substituted with a tert-butyl group or a methyl group, and more preferably a phenyl group substituted with a tert-butyl group.
- R 3 ′ and R 4 ′ each independently represent a hydrogen atom or a hydroxy group, or R 3 ′ and R 4 ′ are bonded to each other and together with the carbon atom to which they are bonded. Therefore, a cyclohexane ring or a benzene ring may be formed.
- R 3 ′ and R 4 ′ are preferably both hydrogen atoms, or R 3 ′ and R 4 ′ are bonded to each other, together with the carbon atom to which they are bonded, cyclohexane Form a ring.
- the bond b' represents a single bond or a double bond.
- the bond b' is preferably a single bond.
- X′ represents CH 2 or carbonyl (C ⁇ O).
- X′ is preferably CH 2 .
- Y′ represents an oxygen atom, NH or N(OCH 3 ).
- Y′ is preferably an oxygen atom or N(OCH 3 ).
- N' represents 0 or 1.
- N' is preferably 1.
- R′ is a phenyl group optionally substituted with a C 1-4 alkoxy group, a naphthyl group, an imidazolyl group substituted with a trityl group, an indolyl group substituted with an optionally substituted phenylsulfonyl group, trityloxy A group or a benzyloxy group
- R 1 ′ is a methyl group substituted with two phenyl groups, or a phenyl group
- R 2 ' is a phenyl group substituted with tert- butyl group
- R 3 ′ and R 4 ′ are both hydrogen atoms, or R 3 ′ and R 4 ′ are bonded to each other to form a cyclohexane ring together with the carbon atom to which they are bonded.
- You may Bond b′ is a single bond
- X′ is CH 2 or carbonyl
- Y is a phenyl group optionally substituted with
- R′ is a phenyl group optionally substituted with a C 1-4 alkoxy group, a naphthyl group, an imidazolyl group substituted with a trityl group, an indolyl group substituted with an optionally substituted phenylsulfonyl group, trityloxy A group or a benzyloxy group, R 1 'is methyl group, R 2 'is a methyl group, biphenylyl group, or tert- butyl-substituted phenyl, R 3 ′ and R 4 ′ are both hydrogen atoms, or R 3 ′ and R 4 ′ are bonded to each other to form a cyclohexane ring together with the carbon atom to which they are bonded.
- You may Bond b′ is a single bond
- X′ is CH 2 or carbonyl
- Still another suitable compound (II) is the following compound.
- R′ is a phenyl group optionally substituted with a C 1-4 alkoxy group, a naphthyl group, an imidazolyl group substituted with a trityl group, an indolyl group substituted with an optionally substituted phenylsulfonyl group, trityloxy A group or a benzyloxy group, R 1 'is methyl group, R 2 'is substituted biphenylyl groups ethynyl group, or a methyl group, R 3 ′ and R 4 ′ are both hydrogen atoms, or R 3 ′ and R 4 ′ are bonded to each other to form a cyclohexane ring together with the carbon atom to which they are bonded.
- You may Bond b′ is a single bond
- X′ is CH 2
- Y′ is N(OCH 3 )
- the compound (II) wherein
- Another suitable compound (II) is the following compound.
- R′ is an imidazolyl group substituted with a trityl group, R 1 'is methyl group, R 2 'is a phenyl group substituted with tert- butyl group, R 3 'and R 4' are both hydrogen atoms, Bond b′ is a single bond, X′ is CH 2 or carbonyl, Y'is an oxygen atom, The compound (II), wherein m′ is 1 and n′ is 0 or 1.
- preferable compound (II) are the compounds (compound (3), compound (12), compound (14) to compound (34)) of Examples 3, 12, 14 to 34 described later, or their pharmaceuticals. It is an acceptable salt.
- More preferred compound (II) is specifically the following compound or a pharmaceutically acceptable salt thereof.
- Compound (I) (for example, compound (1) to compound (15) described in the examples described later) is the same as described in Non-Patent Document 7 (ShimamotoY, et al. Bioorg. Med. Chem., 2015, 23, 876-890) and 8 (Teruya, K, et al. Peptide Science, 2015, 106(4), 391-402), or a method according to a method known per se or a method analogous thereto.
- the compound (I) can also be produced by the methods shown in the following production methods 1 to 4, the examples described later, or methods analogous thereto, and the like.
- Each raw material compound may form a salt as long as it does not inhibit the reaction, and examples of the salt include the same as the salt of compound (I).
- a commercially available compound can be easily obtained and used, or can be produced according to a method known per se or a method analogous thereto.
- the intermediate produced in the following production method may be isolated and purified by a method such as column chromatography, recrystallization and distillation, or may be used in the next step without isolation.
- the reaction scheme is shown below, and each symbol of the compound in the scheme has the same meaning as described above.
- Trt Trityl Fmoc: 9-fluorenylmethyloxycarbonyl homoPro-OH: Homoproline Hyp-OH: hydroxyproline Tic-OH: 1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid HCl H-His(Trt)-OMe: Trityl Histidine Methyl Ester Hydrochloride Ser(Trt): O-Trityl-L-serine Ser(Bn): O-benzyl-L-serine
- Production method 1 is a method in which compound (1a) or (1b) is condensed with tritylhistidine methyl ester hydrochloride (2a), then de-Fmoc-ized, and then condensed with 4-t-butylbenzoic acid (4a). This is a method of obtaining (I).
- the compounds (16) to (19), the compound (23), and the compounds (25) to (33) described in Examples below can be synthesized by this production method.
- compound (1a) or (1b) a commercially available compound can be used, or it can be produced by a method known per se or a method analogous thereto.
- This step is a step for producing compound (3a) or (3b) by condensing compound (1a) or (1b) with trityl histidine methyl ester hydrochloride (2a) in the presence of a condensing agent and a base. .. In this step, the reaction may be carried out in the presence of a condensation additive, if necessary.
- condensing agent used examples include 1-ethyl-3-(3′-dimethylaminopropyl)carbodiimide (WSC), dicyclohexylcarbodiimide (DCC), diisopropylcarbodiimide (DIC), N-ethyl-N′-3-dimethylaminopropyl.
- Carbodiimide and its hydrochloride salt EDC ⁇ HCl
- benzotriazol-1-yloxy-trisdimethylaminophosphonium salt BOP
- BOP benzotriazol-1-yloxy-trisdimethylaminophosphonium salt
- COMPU ⁇ [(1-cyano-2-ethoxy-2-oxoethylidene)amino]oxy ⁇ -4 -Morpholinomethylene ⁇ dimethylammonium hexafluorophosphate
- COMPU hexafluorophosphoric acid
- benzotriazol-1-yloxy)tripyrrolidinophosphonium PyBop
- O-(benzotriazol-1-yl)-N,N, N',N'-tetramethyluronium tetrafluoroborate TBTU
- the condensing agent can be used in an amount of usually 1 to 3 mol, preferably 1 to 1.5 mol, per 1 mol of the compound (1a) or (1b).
- the compound (2a) can be used in an amount of usually 1 to 3 mol, preferably 1 to 1.5 mol, per 1 mol of the compound (1a) or (1b).
- condensation additives include 1-hydroxybenzotriazole (HOBt), 1-hydroxy-1H-1,2,3-triazole-5-carboxylic acid ethyl ester (HOCt), 1-hydroxy-7-azabenzotriazole (HOAt). ) And the like, but it is not necessary when BOP or COMU is used as the condensing agent.
- the amount of the condensation additive used is preferably 0.05 to 1.5 mol per 1 mol of the compound (1a) or (1b).
- the base include organic bases such as triethylamine, pyridine, N,N-diisopropylethylamine and N-methylmorpholine, and inorganic bases such as sodium hydride. Of these, N-methylmorpholine is preferable.
- the amount of base used will usually be 1-10 mol, preferably 5-8 mol, per 1 mol of compound (1a) or (1b).
- reaction solvent is not particularly limited, and examples thereof include aromatic hydrocarbons such as toluene and xylene; amide solvents such as N,N-dimethylformamide and N,N-dimethylacetamide; diethyl ether, tetrahydrofuran, dioxane and the like. Ethers; halogenated hydrocarbons such as chloroform and dichloromethane, and mixtures thereof. Of these, dichloromethane is preferable.
- the reaction temperature is usually -10 to 30°C, preferably 0°C to 20°C, and the reaction time is usually about 1 to 30 hours.
- This step is a step of removing Fmoc (deprotection) of compound (3a) or (3b). This step can be performed in a solvent that does not affect the reaction.
- reaction conditions (reactant, reaction solvent, reaction temperature, reaction time, etc.) in the deprotection reaction are not particularly limited, but for example, ProtectiveGroups in Organic Synthesis, 4th Ed,, Theodora W. Greene, Peter G. M. Wuts , Wiley-Interscience (2007), the examples in this specification, or a method according to these. Specifically, it can be de-Fmoc-ized (de-protected) by treating with an organic base.
- Organic bases include diethylamine, 1,8-diazabicyclo[5.4.0]-7-undecene (DBU), 1,4-diazabicyclo[2.2.2]octane (DABCO), 1,5-diazabicyclo[ 4.3.0]-5-nonene (DBN) and the like are preferable, and diethylamine is preferable.
- the solvent include halogen solvents such as chloroform, dichloromethane and 1,2-dichloroethane; aromatic hydrocarbons such as toluene and xylene; ether solvents such as diethyl ether, CPME, THF and 1,4-dioxane.
- Examples thereof include nitriles such as acetonitrile and the like, or a mixture thereof, and acetonitrile is preferable.
- the amount of the organic base used is usually an excess amount (10 mol or more) with respect to 1 mol of the compound (3a) or (3b).
- the reaction temperature is usually ⁇ 10 to 40° C., preferably 10° C. to 30° C., and the reaction time is usually 0.2 to 2 hours.
- This step is a step of producing the compound (I) by condensing the deprotected compound obtained in step 2 with 4-t-butylbenzoic acid (4a) in the presence of a condensing agent and a base. This step can be performed under the same reaction conditions as in step 1 above.
- the amount of the compound (4a) used can be usually 1 to 3 mol, preferably 1 to 1.5 mol, per 1 mol of the compound (3a) or (3b).
- Production method 2 is a histidine methyl ester hydrochloride after condensation of compound (1a) or (1b) with N-methoxy-N-methylamine hydrochloride (5a), followed by reduction reaction to aldehyde (7a) or (7b).
- Compound (8a) or (8b) is obtained by reductive amination reaction by addition of salt (2a) followed by sodium cyanoborohydride, which is de-Fmocized and then condensed with 4-t-butylbenzoic acid (4a).
- This is a method for obtaining the compound (I).
- Compound (20) to compound (22) and compound (24) described in Examples below can be synthesized by this production method.
- step 4 compound (6a) or (6b) is produced by condensing compound (1a) or (1b) with N-methoxy-N-methylamine hydrochloride (5a) in the presence of a condensing agent and a base. It is a process to do.
- This step can be performed under the same reaction conditions as in step 1 of the above production method 1.
- the amount of the compound (5a) to be used can be generally 1 to 3 mol, preferably 1 to 1.5 mol, per 1 mol of the compound (1a) or (1b).
- the amount of DIBAL-H used can be usually 1 to 3 mol, preferably 1 to 1.5 mol, per 1 mol of compound (6a) or (6b).
- reaction can be carried out in a solvent that does not affect the reaction.
- the reaction solvent is not particularly limited, but examples thereof include halogenated hydrocarbons such as chloroform and dichloromethane. Of these, dichloromethane is preferable.
- the reaction temperature is generally ⁇ 78° C. to 0° C., preferably ⁇ 78° C.
- the reaction time is usually about 0.2 to 2 hours.
- compound (7a) or (7b) is reacted with compound (2a) and then treated with a reducing agent to carry out reductive amination to produce compound (8a) or (8b). is there.
- the amount of the compound (2a) to be used can be generally 1 to 3 mol, preferably 1 to 1.5 mol, based on the compound (7a) or (7b).
- the reducing agent examples include sodium borohydride, sodium cyanoborohydride, sodium triacetoxyborohydride and the like, and preferably sodium cyanoborohydride.
- the reducing agent is used in an amount of usually 1 to 5 equivalents, preferably 1 to 3 equivalents, relative to compound (7a) or (7b).
- This reaction is usually performed in an inert solvent.
- the inert solvent include tetrahydrofuran, diethyl ether, 1,2-dimethoxyethane, 1,4-dioxane, dichloromethane, toluene, benzene, xylene, methanol, ethanol, N,N-dimethylformamide, dimethyl sulfoxide and the like.
- dichloromethane is particularly preferred.
- the reaction temperature is generally ⁇ 20 to 80° C., preferably under ice cooling to 20° C.
- the reaction time is not particularly limited, but is usually 0.1 to 24 hours, preferably 0.5 to 3 hours.
- This step is a step of removing Fmoc (deprotection) of compound (8a) or (8b). This step can be carried out under the same reaction conditions as in step 2 of the above production method 1.
- This step is a step for producing a compound (I) by condensing the deprotected compound obtained in step 7 with 4-t-butylbenzoic acid (4a) in the presence of a condensing agent and a base. This step can be performed under the same reaction conditions as in step 3 of the above production method 1.
- the amount of the compound (4a) used can be generally 1 to 3 mol, preferably 1 to 1.5 mol, per 1 mol of the compound (8a) or (8b).
- This step is a step of producing compound (11a) by condensing compound (9a) and compound (10a) in the presence of a condensing agent and a base. This step can be performed under the same reaction conditions as in step 1 of the above production method 1.
- the amount of the compound (10a) used can be generally 1 to 3 mol, preferably 1 to 2 mol, per 1 mol of the compound (9a).
- This step is a step of removing Fmoc (deprotection) of compound (11a). This step can be carried out under the same reaction conditions as in step 2 of the above production method 1.
- Step 11 This step is a step of producing compound (13a) by condensing the deprotected compound obtained in step 10 with compound (12a) in the presence of a condensing agent and a base. This step can be performed under the same reaction conditions as in step 3 of the above production method 1.
- the amount of the compound (12a) used can be generally 1 to 3 mol, preferably 1 to 1.5 mol, per 1 mol of the compound (11a).
- This step is a step of removing Fmoc (deprotection) of compound (13a). This step can be carried out under the same reaction conditions as in step 2 of the above production method 1.
- step 13 the deprotected compound obtained in step 12 is condensed with compound (14a) in the presence of N,N-dimethyl-4-aminopyridine (DMAP) and a base to produce compound (I). It is a process.
- DMAP N,N-dimethyl-4-aminopyridine
- the amount of the compound (14a) used can be generally 1 to 3 mol, preferably 1 to 1.5 mol, per 1 mol of the compound (13a).
- the amount of DMAP used can be usually 1 to 5 mol, preferably 1 to 3 mol, per 1 mol of compound (13a).
- the base examples include organic bases such as triethylamine, pyridine, N,N-diisopropylethylamine and N-methylmorpholine. Of these, pyridine is preferable.
- the base is usually in excess (10 mol or more) per 1 mol of the compound (13a), and it is particularly preferable to use the base as a solvent.
- reaction solvent is not particularly limited, but examples thereof include aromatic hydrocarbons such as toluene and xylene; amide solvents such as N,N-dimethylformamide and N,N-dimethylacetamide; ethers such as diethyl ether and tetrahydrofuran. And halogenated hydrocarbons such as chloroform and dichloromethane.
- the reaction temperature is usually 0 to 60° C., preferably 10 to 30° C., and the reaction time is usually about 1 to 30 hours.
- Step 14 This step is a step of producing the compound (16a) by condensing the compound (15a) and the compound (2a) in the presence of a condensing agent and a base. This step can be performed under the same reaction conditions as in step 1 of the above production method 1.
- the amount of the compound (2a) used can be generally 1 to 3 mol, preferably 1 to 1.5 mol, per 1 mol of the compound (15a).
- This step is a step of removing Fmoc (deprotection) of compound (16a). This step can be carried out under the same reaction conditions as in step 2 of the above production method 1.
- Step 16 This step is a step of producing compound (I) by condensing the deprotected compound obtained in step 15 with compound (4a) in the presence of a condensing agent and a base. This step can be performed under the same reaction conditions as in step 3 of the above production method 1.
- the amount of the compound (4a) to be used can be usually 1 to 3 mol, preferably 1 to 1.5 mol, per 1 mol of the compound (16a).
- the compound (I) (and the compound (II) included in the compound (I)) produced by the above method is isolated and purified by a usual separation means such as recrystallization, distillation, chromatography and the like. be able to.
- the compound (I) of the present invention contains an optical isomer, a stereoisomer, a positional isomer, and a rotational isomer, these are also included as the compound (I), and a synthesis method known per se and a separation method are known. Each can be obtained as a single product by a method (concentration, solvent extraction, column chromatography, recrystallization, etc.). For example, when the compound (I) has an optical isomer, the optical isomer resolved from the compound is also included in the compound (I).
- compound (I) of the present invention or a pharmaceutically acceptable salt thereof contains optical isomers, stereoisomers, positional isomers and rotamers, all of these isomers and any ratio A mixture of these isomers of is also included as compound (I). Further, each of these isomers can be obtained as a single product by a known synthesis method and separation method (concentration, solvent extraction, column chromatography, recrystallization, etc.).
- the compound (I) of the present invention is a labeled body, that is, one or more atoms constituting the compound (I) of the present invention isotope (for example, 2 H, 3 H, 11 C, 13 C, 14 C, 15 N, 18 O, 18 F, 35 S, etc.) are also included.
- Optical isomers can be produced by a method known per se. Specifically, use an optically active synthetic intermediate, or refer to the final racemate by a conventional method (for example, J. Jacques et al., “Enantiomers, Racemates and Resolution, John Wiley And Sons, Inc.”, etc. Optical isomers can be obtained by performing optical resolution according to (4).
- the compound (I) of the present invention may be in the form of a crystal, and a single crystal form or a mixture of crystal forms is included in the compound (I).
- the crystal can be produced by applying a crystallization method known per se to crystallize.
- the compound (I) of the present invention or a pharmaceutically acceptable salt thereof may also include a solvate thereof.
- a solvate thereof is a compound (I) or a salt thereof in which a molecule of a solvent is coordinated, and a hydrate is also included. It For example, a hydrate, ethanol solvate, dimethyl sulfoxide solvate, etc. of compound (I) or a salt thereof can be mentioned.
- the compound (I) of the present invention exhibits an excellent Wnt signal transduction pathway inhibitory action and a growth inhibitory effect on various tumor cells, as shown in the test examples described later. From the above, for example, it is useful for the prevention or treatment of cancer, coronary artery disease, acute coronary syndrome, osteoarthritis, and the like, and in particular, various cancers (for example, colon cancer, breast cancer, head and neck cancer, lung cancer, gastric cancer, esophageal cancer).
- various cancers for example, colon cancer, breast cancer, head and neck cancer, lung cancer, gastric cancer, esophageal cancer.
- a drug (hereinafter, also referred to as "drug of the present invention") containing a Wnt signal transduction pathway inhibitor comprising the compound (I) of the present invention or a pharmaceutically acceptable salt thereof as an active ingredient.
- the drug may consist of the Wnt signal transduction pathway inhibitor alone, or a drug containing the Wnt signal transduction pathway inhibitor and a pharmaceutically acceptable carrier or the like.
- the preventive or therapeutically effective amount of the medicament of the present invention can be administered to mammals (eg, mouse, rat, hamster, rabbit, cat, dog, cow, sheep, monkey, human etc.).
- the pharmaceutically acceptable carrier examples include excipients (eg, starch, lactose, sugar, calcium carbonate, calcium phosphate, etc.), binders (eg, starch, gum arabic, carboxymethyl cellulose, hydroxypropyl cellulose, crystalline cellulose, etc.).
- excipients eg, starch, lactose, sugar, calcium carbonate, calcium phosphate, etc.
- binders eg, starch, gum arabic, carboxymethyl cellulose, hydroxypropyl cellulose, crystalline cellulose, etc.
- lubricants eg magnesium stearate, talc etc.
- disintegrants eg carboxymethyl cellulose, talc etc.
- solvents eg water for injection, physiological saline, Ringer's solution, alcohol, propylene glycol, polyethylene glycol, Sesame oil, corn oil, olive oil, cottonseed oil, etc.
- solubilizing agents eg polyethylene glycol, propylene glycol, D-mannitol, trehalose, benzyl benzoate, ethanol, trisaminomethane, cholesterol, triethanolamine, sodium carbonate, citric acid Sodium, sodium salicylate, sodium acetate, etc.
- suspending agents eg stearyl triethanolamine, sodium lauryl sulfate, lauryl aminopropionic acid, lecithin, benzalkonium chloride, benzethonium chloride, glyceryl monostearate, etc.
- Hydrophilic polymers such as polyvinyl alcohol, polyvinylpyrrolidone, sodium carboxymethylcellulose, methylcellulose, hydroxymethylcellulose, hydroxyethylcellulose, hydroxypropylcellulose; polysorbates, polyoxyethylene hydrogenated castor oil, etc.), tonicity agent (eg sodium chloride) , Glycerin, D-mannitol, D-sorbitol, glucose, etc.), buffer (eg, phosphate, acetate, carbonate, citrate, etc.), soothing agent (eg, benzyl alcohol, etc.) , Preservatives (eg, paraoxybenzoates, chlorobutanol, benzyl alcohol, phenethyl alcohol, dehydroacetic acid, sorbic acid, etc.), antioxidants (eg, sulfite, ascorbate, etc.), colorants (eg, water-soluble Edible tar dyes (eg, edible red Nos.
- tonicity agent eg sodium chloride
- water-insoluble lake dyes eg, the water-soluble edible tar dyes
- Aluminum salts natural pigments (eg, ⁇ -carotene, chlorophyll, red iron oxide, etc.)
- sweeteners eg, sodium saccharin, dipotassium glycyrrhizinate, aspartame, stevia, etc.
- the medicine of the present invention is prepared by mixing the above-mentioned components, and then mixing the mixture according to a method known per se, for example, for oral administration such as capsules, tablets, fine granules, granules, dry syrup, or injection (for example, subcutaneous Injections, intravenous injections, intramuscular injections, intraperitoneal injections, infusions, etc.), external preparations (eg, transdermal preparations, ointments, lotions, patches), suppositories (eg, rectal) Suppositories, vaginal suppositories), pellets, nasal preparations, pulmonary preparations (inhalation preparations), eye drops and the like can be used for preparations for parenteral administration.
- the pharmaceutical of the present invention is preferably a preparation for parenteral administration such as injection.
- the content of the compound (I) of the present invention or a pharmaceutically acceptable salt thereof in the medicament (formulation) of the present invention varies depending on the form of the preparation, but is usually about 0.01 to 100 relative to the whole preparation.
- the content is in the range of wt%, preferably in the range of about 0.1 to 50 wt%, and more preferably in the range of about 0.5 to 20 wt%.
- the dose of the compound (I) of the present invention or a pharmaceutically acceptable salt thereof varies depending on the administration subject, symptoms, and other factors, but when administered parenterally to an adult cancer patient, it is usually 1
- the dose is about 0.01 to 200 mg/kg body weight, preferably 0.1 to 100 mg/kg body weight, more preferably 1 to 50 mg/kg body weight, and this amount is administered once to three times a day. desirable.
- the compound (I) of the present invention or a pharmaceutically acceptable salt thereof can be used in combination with another drug, for example, an existing anticancer drug (combination drug), as long as the drug effect is not impaired.
- another drug for example, an existing anticancer drug (combination drug)
- the administration timing is not limited, and these may be administered to the administration subject at the same time, or may be administered with a time lag.
- the compound (I) of the present invention and a concomitant drug can be administered in a single preparation in combination.
- the dose of the concomitant drug can be appropriately selected based on the dose clinically used.
- the compounding ratio of the compound of the present invention or a pharmaceutically acceptable salt thereof and the concomitant drug can be appropriately selected according to the administration subject, administration route, target disease, symptom, combination and the like.
- anticancer agents examples include chemotherapeutic agents, hormone therapeutic agents, immunotherapeutic agents, molecular targeting agents, immune checkpoint inhibitors (anti-PD-1 antibody, anti-PD-L1 antibody), and the like.
- chemotherapeutic agent for example, an alkylating agent, an antimetabolite, an anticancer antibiotic, a plant-derived anticancer agent, etc. are used.
- alkylating agent examples include nitrogen mustard, nitrogen mustard hydrochloride-N-oxide, chlorambucil, cyclophosphamide, ifosfamide, thiotepa, carbocon, improsulfan tosylate, busulfan, nimustine hydrochloride, mitbronitol, melamine.
- Phalan dacarbazine, ranimustine, estramustine phosphate phosphate, triethylenemelamine, carmustine, lomustine, streptozocin, pipobroman, etogluside, carboplatin, cisplatin, mivoplatin, nedaplatin, oxaliplatin, altretamine, ambamustine, dibrospidium hydrochloride, fotemuthine, fothetine.
- Predonimustine, pumitepa, ribomustine, temozolomide, threosulfan, trofosfamide, zinostatin stimalamer, adzeresin, systemustin, viserecin, and DDS preparations thereof are used.
- antimetabolite examples include mercaptopurine, 6-mercaptopurine riboside, thioinosine, methotrexate, pemetrexed, enocitabine, cytarabine, cytarabine ocfosphate, ancitabine hydrochloride, 5-FU drugs (eg, fluorouracil, tegafur, UFT, doxyfluridine, carmofur, gallocitabine, emitefur, capecitabine), aminopterin, nerzarabine, leucovorin calcium, tabloid, butosine, folineate calcium, levofolinate calcium, cladribine, emitefur, fludarabine, gemcitabinestatin, hydroxycarbamide, hydroxycarbamide, Idoxyuridine, mitoguazone, thiazofurin, ambamustine, bendamustine, and their DDS preparations are used.
- 5-FU drugs eg, fluorouracil, tegafur
- anticancer antibiotics examples include actinomycin D, actinomycin C, mitomycin C, chromomycin A3, bleomycin hydrochloride, bleomycin sulfate, peplomycin sulfate, daunorubicin hydrochloride, doxorubicin hydrochloride, aclarubicin hydrochloride, pirarubicin hydrochloride, epirubicin hydrochloride.
- Neocarzinostatin, mithramycin, zarcomycin, carcinophylline, mitotan, zorubicin hydrochloride, mitoxantrone hydrochloride, idarubicin hydrochloride, and their DDS preparations are used.
- plant-derived anticancer drug for example, etoposide, etoposide phosphate, vinblastine sulfate, vincristine sulfate, vindesine sulfate, teniposide, paclitaxel, docetaxel, vinorelbine, and DDS preparations thereof are used.
- hormone therapeutic agent examples include phosfestrol, diethylstilbestrol, chlorotrianisene, medroxyprogesterone acetate, megestrol acetate, chlormadinone acetate, cyproterone acetate, danazol, allylestrenol, gestrinone, mepaltricin, Raloxifene, olmeroxifene, levormeroxifene, anti-estrogens (eg, tamoxifen citrate, toremifene citrate), pill preparations, mepithiostane, testrolactone, aminoglutethiimide, LH-RH agonists (eg, goserelin acetate, buserelin acetate) , Leuprorelin), droloxifene, epithiostanol, ethinyl estradiol sulfonate, aromatase inhibitors (eg, fadrozole
- the “immunotherapeutic agent” includes biological response modifiers (eg, picibanil, krestin, schizophyllan, lentinan, ubenimex, interferon, interleukin, macrophage colony stimulating factor, granulocyte colony stimulating factor, erythropoietin, lymphotoxin, BCG vaccine, Corynebacterium parvum, levamisole, polysaccharide K, procodazole, anti-CTLA4 antibody) and the like are used.
- biological response modifiers eg, picibanil, krestin, schizophyllan, lentinan, ubenimex, interferon, interleukin, macrophage colony stimulating factor, granulocyte colony stimulating factor, erythropoietin, lymphotoxin, BCG vaccine, Corynebacterium parvum, levamisole, polysaccharide K, procodazole, anti-CTLA4 antibody
- Examples of the “molecularly targeted drug” include tositumomab, ibritumomab, alemtuzumab, axitinib, bevacizumab, afatinib, bortezomib, bosutinib, carfilzomib, cetuximab, dasatinib, denosumab, edrecolomab, erlotinib, erlotinib, erlotinib, erlotinib, edrecozumab, erlotinib, erlotinib.
- Alectinib, ceritinib, ibrutinib, parvocyclib, regorafenib, pilaralicib and the like are used.
- the “immunity checkpoint inhibitor” for example, nivolumab, pembrolizumab, avelumab, atezolizumab, etc. are used.
- Non-Patent Document 7 Shiamoto Y, et al. Bioorg. Med. Chem., 2015, 23, 876-890
- 8 Teuya, K, et al. Peptide Science, 2015, 106(4). ), 391-402.
- the compound (12) is a non-patent document 7 (Shimamoto Y, et al. Bioorg. Med. Chem., 2015, 23, 876-890) and 8 (Teruya, K, et al. Peptide Science, 2015, 106(4). ), 391-402).
- the compound (14) is a non-patent document 7 (Shimamoto Y, et al. Bioorg. Med. Chem., 2015, 23, 876-890) and 8 (Teruya, K, et al. Peptide Science, 2015, 106(4). ), 391-402).
- the compound (15) is a non-patent document 7 (Shimamoto Y, et al. Bioorg. Med. Chem., 2015, 23, 876-890) and 8 (Teruya, K, et al. Peptide Science, 2015, 106(4). ), 391-402).
- Compound (18) has the following formula in accordance with the method of the above-mentioned production method 1:
- the compound (31) was dissolved in acetonitrile, and diethylamine (Et 2 NH) was added so that the concentration became 20% (V/V). After stirring for 20 minutes, the solvent was distilled off under reduced pressure. The residue was used for the next reaction without purification. The obtained residue was dissolved in dichloromethane, and 4-t-butylbenzoic acid (1.1 equivalent) and NMM (7.0 equivalent) were added. Under ice-cooling stirring, N-[1-(cyano-2-ethoxy-2-oxoethylideneaminooxy)dimethylamino(morpholino)]uronium hexafluorophosphate (COMU) (1.2 equiv.) was added and stirred for 16 hours.
- Et 2 NH diethylamine
- Compound (19) was synthesized by a method similar to that of Production method 1 above using commercially available 1-Fmoc homoproline (Fmoc-homoPro-OH) (12a) (manufactured by Watanabe Chemical Industry Co., Ltd.) as a starting material. ..
- Compound (20) has the following formula in accordance with the method of the above-mentioned production method 2:
- the obtained residue was dissolved in dichloromethane, and diisobutylaluminum hydride (DIBAL-H, 1 M hexane solution) (1.1 equivalent) was added at -78°C. After stirring for 20 minutes, the reaction was stopped with methanol and the temperature was raised to room temperature. The obtained suspension was suction filtered through a silica gel/Celite (registered trademark) pad, the filtrate was concentrated, and the residue was roughly purified by silica gel column chromatography to obtain a crude product of compound (7c). The obtained crude product was dissolved in dichloromethane, the commercially available compound (2a) (1.0 equivalent) was added, and the mixture was stirred for 1 hour.
- DIBAL-H diisobutylaluminum hydride
- Compound (23) was synthesized from a commercially available Fmoc-protected proline (manufactured by Watanabe Chemical Industry Co., Ltd.) according to the method of Production method 1 (specifically, the production method of compound (18) described above).
- Compound (25) has the following formula in accordance with the method of the above-mentioned production method 3:
- the compound (11b) was dissolved in acetonitrile, and diethylamine was added so that the concentration became 20% (V/V). After stirring for 20 minutes, the solvent was distilled off under reduced pressure. The residue was used for the next reaction without purification. The obtained residue was dissolved in dichloromethane, and commercially available Fmoc-protected homoproline (12a) (1.1 equivalent) and NMM (7.0 equivalent) were added. BOP (1.2 equivalents) was added under ice-cooling stirring and the mixture was stirred for 16 hours. The reaction was quenched with saturated aqueous ammonium chloride solution and extracted with ethyl acetate. The extract was washed with saturated saline and dried over anhydrous sodium sulfate.
- Compound (34) has the following formula in accordance with the method of the above-mentioned production method 4:
- Test Example 1 Measurement and evaluation of TCF/ ⁇ -catenin transcription activity
- pGL4.32 is used to perform selective culture of cells after gene transfer using M50 Super 8x TOPFlash.
- luc2P/NF- ⁇ B-RE/Hygro] vector using the restriction enzymes (BamHI 2228 and Sal1 3833), the nucleotide sequence part of the hygromycin resistance gene in the pGL4.32 vector was cloned into p.M50 Super 8x TOPFlash. Was inserted into the cell to prepare a Super TOP Flash plasmid.
- TOP cells were treated with 10% FBS, antibiotics (100 unit/mL penicillin, 100 ⁇ g/mL streptomycin), and low glucose content D-MEM containing hygromycin (containing L-glutamine and phenol red) at 37°C, 5%. It was cultured in an environment of CO 2 . TOP cells used in the luciferase assay have low levels of endogenous ⁇ -catenin. Therefore, in order to activate the Wnt signal and increase the amount of luminescence, an experiment was performed using a medium (Wnt supernatant) rich in Wnt protein derived from Wnt-3A-L cells. Wnt-3A cells are L512 cells into which a Wnt-3A expression vector has been introduced and cloned.
- Wnt-3A cells secrete active Wnt-3A protein and are used for the preparation of Wnt-3A-containing culture supernatant.
- Low glucose content D-MEM L-glutamine and phenol red
- antibiotics 100 unit/mL penicillin, 100 ⁇ g/mL streptomycin
- G418 0.4 mg/mL G418 at 37°C
- 5% CO 2 Cultured and maintained in the environment.
- Wnt-3A cells were seeded at 1.25 x 10 6 cells/dish and cultured in a G418-free medium. After 3 days of confluent culture, the culture supernatant was passed through a syringe filter to collect the supernatant. This Wnt-containing culture supernatant was added to the TOP cell culture system and used as an evaluation system.
- the TOP cells were removed by phosphate buffer (PBS(-)), collected, centrifuged, and the supernatant-free pellet was mixed with Wnt-containing culture supernatant and pipetted into a 96-well plate at 5 x 10 4 cells/ TOP cells were seeded well by well. The next day, the compound was treated and incubated for 24 hours. Then, a cell lysing agent was prepared, 100 ⁇ L of each was added, and the plate was placed on a shaker for 30 minutes to lyse the cells.
- PBS(-) phosphate buffer
- NCI-H929 cells MM1.s cells, RPMI8226 cells, U266 cells, AMO-1 cells and OPM-2 cells (multiple myeloma (MM) cells); AsPC-1 cells and AsPC-1/HA Cells (pancreatic cancer cells); HT29 cells, SW480 cells and HCT116/E6 cells (colon cancer cells); MV4;11 cells, KG1a cells and HL-60 cells (acute myeloid leukemia cells); RS4;11 cells and Nalm6 cells (Acute lymphoblastic leukemia cells); Raji cells (malignant lymphoma cells); KMB5 cells and KMB5/STIR cells (chronic myelogenous leukemia cells); MDA-MB-231 cells (breast cancer cells); U2510MG cells (glioblastoma) Tumor cells))) were seeded in a 96-well plate (Thermo Fisher Scientific, Waltham, MA, USA) at 90 ⁇ L of each suspension at an
- the compound (I) of the present invention was treated at 10 ⁇ L/well each and reacted for 72 hours. After 69 hours, 10 ⁇ L/well of SF (Nacalai Tesque) and Cell Counting Kit-8 (Dojindo Laboratories) were added at each time and reacted for 3 hours, then GloMax Discover System (Promega, Madison, Madison, The absorbance at 450 nm was measured using WI, USA). The ratio of the absorbance at each concentration of compound (I)/the absorbance without compound treatment was calculated, and this was multiplied by 100 to calculate the cell viability.
- Test Example 3 Quantitative RT-PCR (qRT-PCR) method
- RNA expression analysis was analyzed using the qRT-PCR method.
- Various cancer cells were cultured in a 96-well plate (Thermo Fisher Scientific, Waltham, MA, U.S.A.) at appropriate cell concentrations for 3 hours. Then, each compound was dissolved in 1 mL of each culture medium containing no fetal bovine serum and treated. After 1, 4, and 24 hours, total RNA was extracted using the illustra RNAspin Mini RNA Isolation kit (GE Healthcare, Chicago, IL, U.S.A.) and quantified using NanoDrop 2000 (Thermo Fisher Scientific).
- Test Example 4 Western blotting method
- the cells were lysed in the reagent by ice-cooling (20 minutes) and vortexing every 5 minutes for 10 seconds 4 times in total. Then, it was centrifuged (12000 rpm, 20 minutes, 4° C.), and the supernatant was recovered as an extracted protein. The recovered protein was quantified using the Qubit Protein Assay kit (Thermo Fisher Scientific) and Quantus Fluorometer (Promega), and the sample buffer solution (2-mercaptoethanol+) (Wako Pure was used) to adjust the protein concentration to 2 ⁇ g/ ⁇ L. (Pharmaceutical industry) and sterilized ultrapure water were mixed and boiled for 5 minutes to prepare.
- an antibody against each protein is used as a primary antibody, and glycerin is used as an internal control protein.
- Aldehyde-3-phosphate dehydrogenase and ⁇ -actin were selected, and antibodies against them were used as primary antibodies.
- Horseradish peroxidase-conjugated anti-mouse IgG (GE Healthcare) and anti-rabbit IgG (GE Healthcare) were used as secondary antibodies to be bound to the primary antibody.
- ECL Blotting Detection System GE Healthcare
- ECL Prime Western Blotting Detection System GE Healthcare
- Test Example 6 Analysis of apoptosis (cell death)
- Test Example 7 Cancer sphere formation
- the culture was carried out according to the previous report (Takada T, et al. J Physiol Sci, 66:387-396, 2016.), and 3 ml of DMEM/F12 (on the Ultra-Low attachment 6-well plate (Corning International, Corning, NY)). Thermo Fisher Scientific, Waltham, MA) for 7 days. At this time, 1% PC/SM, 2% B-27 [B-27 Serum-Free Supplement (50x): Thermo Fisher Scientific] in the medium, 20 ng/ml epidermal growth factor (EGF). Peprotech, Rocky Hill, NJ), 20 ng/ml of basic fibroblast growth factor (b-FGF; Peprotech) was added. EGF and b-FGF were further added at 20 ng/ml on the third day of culture. The number of cells seeded was 1.0 ⁇ 10 5 cells/ml.
- Test Example 8 CellTiter-Glo (registered trademark) 3D cell viability assay
- the CellTiter-Glo® 3D cell viability assay was used to investigate the cytostatic effect of inhibitors on CSCs, which are floating spheres created by cancer sphere formation.
- the cells were seeded in Ultra-Low attachment 96-well plates (Corning International) at 3.0 ⁇ 10 3 cells/90 ⁇ l per well. Compound (19) was used as an inhibitor, and ICG-001 was used as a control. After culturing for 6 days, the cells in each well were transferred together with the culture solution to a white microplate (96F Nunclon Delta WhiteMicrowell SI; Thermo Fisher Scientific).
- CellTiter-Glo (registered trademark) 3D Reagent (Promega, Madison, WI, USA) was added thereto in an amount of 100 ⁇ l per well, stirred for 5 minutes, and reacted at room temperature for 25 minutes. Then, the luminescence amount was measured using a luminometer (GloMax (registered trademark) Discover System: Promega).
- the compound (I) of the present invention or a pharmaceutically acceptable salt thereof exhibits excellent Wnt signal transduction pathway inhibitory activity. Therefore, according to the present invention, a disease containing Compound (I) or a pharmaceutically acceptable salt thereof as an active ingredient and capable of alleviating symptoms by inhibiting the Wnt signaling pathway, for example, cancer, It is useful for prevention or treatment of coronary artery disease, acute coronary syndrome, osteoarthritis, and the like, and particularly, various cancers (for example, colon cancer, breast cancer, head and neck cancer, lung cancer, gastric cancer, esophageal cancer, malignant melanoma, prostate cancer).
- various cancers for example, colon cancer, breast cancer, head and neck cancer, lung cancer, gastric cancer, esophageal cancer, malignant melanoma, prostate cancer.
- a novel compound (compound (II)) having excellent Wnt signal transduction pathway inhibitory activity or a pharmaceutically acceptable salt thereof can be provided.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Hematology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Immunology (AREA)
- Rheumatology (AREA)
- Physical Education & Sports Medicine (AREA)
- Oncology (AREA)
- Urology & Nephrology (AREA)
- Vascular Medicine (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
本発明は、新規なWntシグナル伝達経路阻害剤に関する。本発明によれば、式(I): [式中の各記号の定義は、明細書に記載の通りである。] で表される化合物、又はその医薬上許容される塩からなるWntシグナル伝達経路阻害剤、及びそれを有効成分として含有する、癌を予防又は治療するための医薬を提供することができる。
Description
本発明は、新規なWntシグナル伝達経路阻害剤に関する。また、本発明は、Wntシグナル伝達経路を阻害することにより症状を緩和させることができる疾患(例えば、癌、冠状動脈疾患、急性冠症候群、変形性関節症等)の予防又は治療のための医薬に関する。
Wnt/β-カテニン経路は、Wntシグナル伝達経路の1つで、線虫、ショウジョウバエ、マウス及びヒトに至るまで、進化上様々な動物種で保存されているシグナル経路で、本経路のシグナル伝達に主要な分子であるβ-カテニンは、約92kDaのタンパク質であり、インテグリンの裏打ちタンパク質として組織形成に働く一方で、β-カテニンが核内移行することで標的遺伝子の転写活性化をもたらす。分泌性のリガンドであるWntタンパク質が作用しない状態では、β-カテニンタンパク量は低く保たれている。β-カテニンは、腺腫様多発結腸ポリープ、アキシン、グリコーゲンシンターゼキナーゼ3β(GSK-3β)、カゼインキナーゼ1αと複合体を作り、GSK-3βによりリン酸化を受け、リン酸化を受けたβ-カテニンは、β-トランスデューシンリピート含有タンパク質(β-TrCP)によりユビキチン化され、プロテアソームで分解される。一方、Wntタンパク質は、膜7回貫通型受容体Fz(Frizzled)に結合しシグナルがON状態になると、Dishevelled(Dsh)を介してβ-カテニンのリン酸化が抑制される。こうして細胞質内に蓄積されたβ-カテニンが核内へ移行し、T細胞因子/リンパ系エンハンサー因子(TCF/LEF)ファミリー転写因子と結合する。さらに、β-カテニンは、B細胞慢性白血球性白血病/リンパ腫9やPygopusと複合体を作り、c-mycやcyclin D1、c-jun等のTCF/LEF関連遺伝子の転写活性を促進し、細胞増殖、幹細胞維持、体節や器官形成を制御し、初期胚成熟における体軸形成、各種器官形成、細胞の増殖・分化、組織再生等の生命現象に重要な役割を担っている。近年、Wntシグナル伝達経路は、多くの癌幹細胞の増殖・維持にも関与することが明らかとなり、癌治療のターゲットとして注目されている(非特許文献1、2)。また、Wntシグナル伝達経路は、癌幹細胞の増殖・維持以外にも、様々な冠状動脈疾患、急性冠症候群、変形性関節症等の様々な疾患の発症等に関与することも報告されている(非特許文献3、4)。
Wntシグナル伝達経路を標的とする阻害剤の開発は、低分子での阻害に適した経路構成因子の数が少数であることが妨げとなっていた。これまでに、Wntシグナル伝達経路を阻害する低分子化合物は、いくつか報告されている(非特許文献5)が、その効果や毒性の面で問題点を有していた。
非特許文献7、8には、下記式:
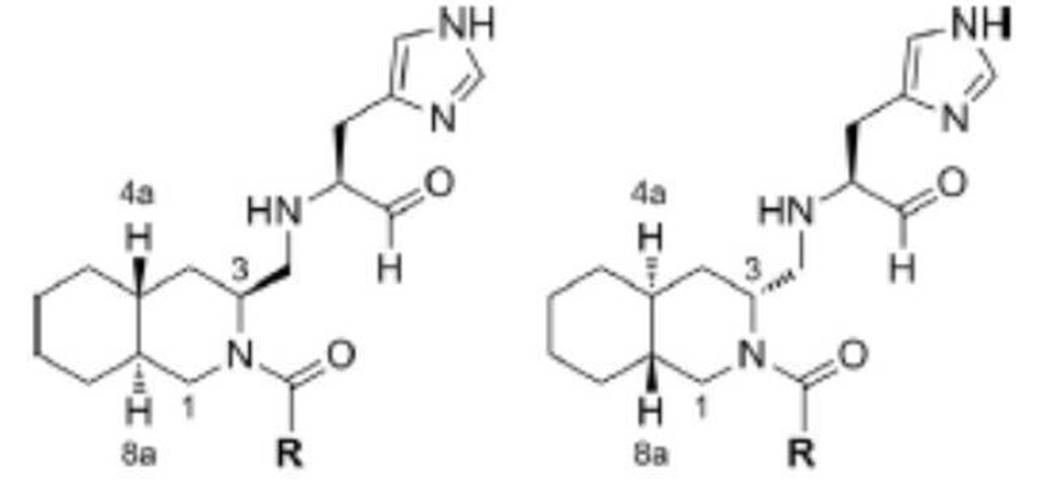
(式中のRの定義は、非特許文献7、8に記載の通りである。)で表されるデカヒドロイソキノリン誘導体がSARS 3CLプロテアーゼ阻害活性を示すことが記載され、また、その合成中間体として、下記式(I)に包含される化合物が記載されている。しかし、非特許文献7、8のいずれにも、当該合成中間体の生物活性については、何ら記載も示唆もされていない。
Anastas JN, et al. Nat Rev Cancer, 2013, 13, 11-26
Yao H, et al. Expert Opin Ther Targets, 2011, 1-15
Hutter R, et al. Circulation, 2013, 128, 2351-2363
Ren J, et al., J. Cell Biochem., 2018, 119, 4514-4527
Voronkov A and Kraussm S, Current Pharmaceutical Design, 2013, 19, 634-664
Shimamoto Y, et al. Bioorg. Med. Chem., 2015, 23, 876-890
Teruya, K, et al. Peptide Science, 2015, 106(4), 391-402
このような背景のもと、より効果的かつ毒性の低い新規Wntシグナル伝達経路阻害剤の開発が切望されている。
本発明の目的は、新規なWntシグナル伝達経路阻害剤を提供することである。本発明の更なる目的は、Wntシグナル伝達経路を阻害することにより症状を緩和させることができる疾患、特に、癌の予防又は治療のための医薬を提供することである。
本発明者らは、かかる状況下、鋭意検討を重ねた結果、下記式(I):
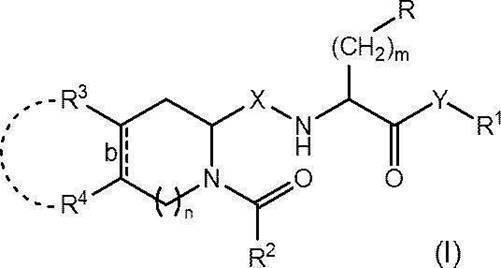
[式中、
Rは、置換されていてもよいC6-14アリール基、置換されていてもよいヘテロアリール基又は保護されていてもよいヒドロキシ基を示し;
R1は、置換されていてもよいC1-6アルキル基、又は置換されていてもよいC6-14アリール基を示し;
R2は、置換されていてもよいC1-6アルキル基、置換されていてもよいC1-6アルコキシ基又は置換されていてもよいC6-14アリール基を示し;
R3及びR4は、それぞれ独立して、水素原子、ハロゲン原子、ヒドロキシ基、アミノ基、ニトロ基、シアノ基、カルボキシ基、置換されていてもよいC1-6アルキル基又は置換されていてもよいC1-6アルコキシ基を示すか、或いは、R3及びR4は、互いに結合して、それらが結合している炭素原子と一緒になって、それぞれ置換されていてもよい、シクロヘキサン環又はベンゼン環を形成してもよく;
結合bは、単結合又は二重結合を示し;
Xは、CH2又はカルボニル(C=O)を示し;
Yは、酸素原子又はNR0(式中のR0は、水素原子又はC1-4アルコキシ基を示す。)を示し;
mは、1~3の整数を示し;並びに
nは、0又は1を示す。]
で表される化合物(以下、「化合物(I)」と称することもある。)、又はその医薬上許容される塩が、優れたWntシグナル伝達経路阻害活性を示すことを初めて見出し、本発明を完成するに至った。
Rは、置換されていてもよいC6-14アリール基、置換されていてもよいヘテロアリール基又は保護されていてもよいヒドロキシ基を示し;
R1は、置換されていてもよいC1-6アルキル基、又は置換されていてもよいC6-14アリール基を示し;
R2は、置換されていてもよいC1-6アルキル基、置換されていてもよいC1-6アルコキシ基又は置換されていてもよいC6-14アリール基を示し;
R3及びR4は、それぞれ独立して、水素原子、ハロゲン原子、ヒドロキシ基、アミノ基、ニトロ基、シアノ基、カルボキシ基、置換されていてもよいC1-6アルキル基又は置換されていてもよいC1-6アルコキシ基を示すか、或いは、R3及びR4は、互いに結合して、それらが結合している炭素原子と一緒になって、それぞれ置換されていてもよい、シクロヘキサン環又はベンゼン環を形成してもよく;
結合bは、単結合又は二重結合を示し;
Xは、CH2又はカルボニル(C=O)を示し;
Yは、酸素原子又はNR0(式中のR0は、水素原子又はC1-4アルコキシ基を示す。)を示し;
mは、1~3の整数を示し;並びに
nは、0又は1を示す。]
で表される化合物(以下、「化合物(I)」と称することもある。)、又はその医薬上許容される塩が、優れたWntシグナル伝達経路阻害活性を示すことを初めて見出し、本発明を完成するに至った。
すなわち、本発明は以下の通りである。
[1]式(I):
[1]式(I):
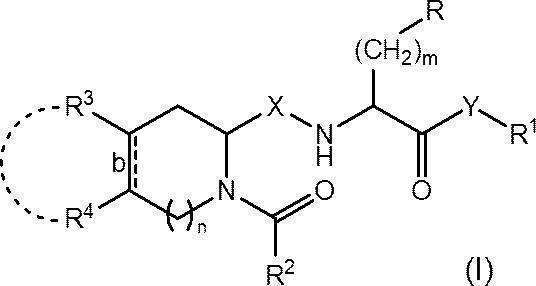
[式中、
Rは、置換されていてもよいC6-14アリール基、置換されていてもよいヘテロアリール基又は保護されていてもよいヒドロキシ基を示し;
R1は、置換されていてもよいC1-6アルキル基、又は置換されていてもよいC6-14アリール基を示し;
R2は、置換されていてもよいC1-6アルキル基、置換されていてもよいC1-6アルコキシ基又は置換されていてもよいC6-14アリール基を示し;
R3及びR4は、それぞれ独立して、水素原子、ハロゲン原子、ヒドロキシ基、アミノ基、ニトロ基、シアノ基、カルボキシ基、置換されていてもよいC1-6アルキル基又は置換されていてもよいC1-6アルコキシ基を示すか、或いは、R3及びR4は、互いに結合して、それらが結合している炭素原子と一緒になって、それぞれ置換されていてもよい、シクロヘキサン環又はベンゼン環を形成してもよく;
結合bは、単結合又は二重結合を示し;
Xは、CH2又はカルボニル(C=O)を示し;
Yは、酸素原子又はNR0(式中のR0は、水素原子又はC1-4アルコキシ基を示す。)を示し;
mは、1~3の整数を示し;並びに
nは、0又は1を示す。]
で表される化合物、又はその医薬上許容される塩からなるWntシグナル伝達経路阻害剤。
[2]Rが、置換されていてもよいC6-10アリール基、置換されていてもよいヘテロアリール基又は保護されたヒドロキシ基であり、
R1が、1又は2個のフェニル基で置換されていてもよいC1-4アルキル基、或いは置換されていてもよいフェニル基であり、
R2が、置換されていてもよいC1-4アルキル基、置換されていてもよいC1-4アルコキシ基、置換されていてもよいフェニル基であり、
R3及びR4が、それぞれ独立して、水素原子、ヒドロキシ基又はC1-6アルコキシ基であるか、或いは、R3及びR4は、互いに結合して、それらが結合している炭素原子と一緒になって、シクロヘキサン環を形成してもよく、
結合bが、単結合であり、
Xが、CH2又はカルボニルであり、
Yが、酸素原子、NH又はN(OCH3)であり、
mが、1であり、且つ
nが、0又は1である、上記[1]に記載のWntシグナル伝達経路阻害剤。
[3]R2が、置換されていてもよいC1-3アルキル基又は置換されていてもよいフェニル基である、上記[1]又は[2]に記載のWntシグナル伝達経路阻害剤。
[4]Rが、置換されていてもよいフェニル基、置換されていてもよいナフチル基、置換されていてもよいイミダゾリル基、置換されていてもよいインドリル基、トリチルオキシ基又はベンジルオキシ基である、上記[1]~[3]のいずれかに記載のWntシグナル伝達経路阻害剤。
[5]R3及びR4が、共に水素原子であるか、または、R3及びR4は、互いに結合して、それらが結合している炭素原子と一緒になって、シクロヘキサン環を形成し、且つ
結合bが、単結合である、上記[1]~[4]のいずれかに記載のWntシグナル伝達経路阻害剤。
[6]上記[1]~[5]のいずれかに記載のWntシグナル伝達経路阻害剤を有効成分として含有する医薬。
[7]Wntシグナル伝達経路を阻害することにより症状を緩和させることができる疾患の予防又は治療のための上記[6]に記載の医薬。
[8]Wntシグナル伝達経路を阻害することにより症状を緩和させることができる疾患が、癌、冠状動脈疾患、急性冠症候群、又は変形性関節症である、上記[7]に記載の医薬。
[9]Wntシグナル伝達経路を阻害することにより症状を緩和させることができる疾患が、癌である、上記[7]に記載の医薬。
[10]癌が、大腸癌、乳癌、頭頸部癌、肺癌、胃癌、食道癌、悪性黒色腫、前立腺癌、肝細胞癌、卵巣癌、肝芽腫、急性骨髄性白血病、急性リンパ芽球性白血病、慢性骨髄性白血病、慢性リンパ性白血病、多発性骨髄腫、悪性リンパ腫、膵臓癌、腎臓癌、膀胱癌、神経膠芽腫、髄芽腫、又はウィルムス腫瘍である、上記[9]に記載の医薬。
[11]式(II):
Rは、置換されていてもよいC6-14アリール基、置換されていてもよいヘテロアリール基又は保護されていてもよいヒドロキシ基を示し;
R1は、置換されていてもよいC1-6アルキル基、又は置換されていてもよいC6-14アリール基を示し;
R2は、置換されていてもよいC1-6アルキル基、置換されていてもよいC1-6アルコキシ基又は置換されていてもよいC6-14アリール基を示し;
R3及びR4は、それぞれ独立して、水素原子、ハロゲン原子、ヒドロキシ基、アミノ基、ニトロ基、シアノ基、カルボキシ基、置換されていてもよいC1-6アルキル基又は置換されていてもよいC1-6アルコキシ基を示すか、或いは、R3及びR4は、互いに結合して、それらが結合している炭素原子と一緒になって、それぞれ置換されていてもよい、シクロヘキサン環又はベンゼン環を形成してもよく;
結合bは、単結合又は二重結合を示し;
Xは、CH2又はカルボニル(C=O)を示し;
Yは、酸素原子又はNR0(式中のR0は、水素原子又はC1-4アルコキシ基を示す。)を示し;
mは、1~3の整数を示し;並びに
nは、0又は1を示す。]
で表される化合物、又はその医薬上許容される塩からなるWntシグナル伝達経路阻害剤。
[2]Rが、置換されていてもよいC6-10アリール基、置換されていてもよいヘテロアリール基又は保護されたヒドロキシ基であり、
R1が、1又は2個のフェニル基で置換されていてもよいC1-4アルキル基、或いは置換されていてもよいフェニル基であり、
R2が、置換されていてもよいC1-4アルキル基、置換されていてもよいC1-4アルコキシ基、置換されていてもよいフェニル基であり、
R3及びR4が、それぞれ独立して、水素原子、ヒドロキシ基又はC1-6アルコキシ基であるか、或いは、R3及びR4は、互いに結合して、それらが結合している炭素原子と一緒になって、シクロヘキサン環を形成してもよく、
結合bが、単結合であり、
Xが、CH2又はカルボニルであり、
Yが、酸素原子、NH又はN(OCH3)であり、
mが、1であり、且つ
nが、0又は1である、上記[1]に記載のWntシグナル伝達経路阻害剤。
[3]R2が、置換されていてもよいC1-3アルキル基又は置換されていてもよいフェニル基である、上記[1]又は[2]に記載のWntシグナル伝達経路阻害剤。
[4]Rが、置換されていてもよいフェニル基、置換されていてもよいナフチル基、置換されていてもよいイミダゾリル基、置換されていてもよいインドリル基、トリチルオキシ基又はベンジルオキシ基である、上記[1]~[3]のいずれかに記載のWntシグナル伝達経路阻害剤。
[5]R3及びR4が、共に水素原子であるか、または、R3及びR4は、互いに結合して、それらが結合している炭素原子と一緒になって、シクロヘキサン環を形成し、且つ
結合bが、単結合である、上記[1]~[4]のいずれかに記載のWntシグナル伝達経路阻害剤。
[6]上記[1]~[5]のいずれかに記載のWntシグナル伝達経路阻害剤を有効成分として含有する医薬。
[7]Wntシグナル伝達経路を阻害することにより症状を緩和させることができる疾患の予防又は治療のための上記[6]に記載の医薬。
[8]Wntシグナル伝達経路を阻害することにより症状を緩和させることができる疾患が、癌、冠状動脈疾患、急性冠症候群、又は変形性関節症である、上記[7]に記載の医薬。
[9]Wntシグナル伝達経路を阻害することにより症状を緩和させることができる疾患が、癌である、上記[7]に記載の医薬。
[10]癌が、大腸癌、乳癌、頭頸部癌、肺癌、胃癌、食道癌、悪性黒色腫、前立腺癌、肝細胞癌、卵巣癌、肝芽腫、急性骨髄性白血病、急性リンパ芽球性白血病、慢性骨髄性白血病、慢性リンパ性白血病、多発性骨髄腫、悪性リンパ腫、膵臓癌、腎臓癌、膀胱癌、神経膠芽腫、髄芽腫、又はウィルムス腫瘍である、上記[9]に記載の医薬。
[11]式(II):
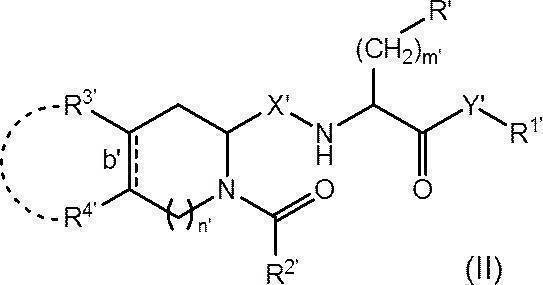
[式中、
R’は、ヒドロキシ基又はC1-6アルコキシ基で置換されていてもよいフェニル基、ナフチル基、トリチル基で置換されたイミダゾリル基、置換されていてもよいC6-14アリールスルホニル基で置換されたインドリル基、トリチルオキシ基又はベンジルオキシ基を示し;
R1’は、2個のフェニル基で置換されていてもよいメチル基、又はフェニル基を示し;R2’は、エチニル基で置換されていてもよいビフェニリル基、tert-ブチル基で置換されたフェニル基、メチル基、又はフルオレニルメトキシ基を示し;
R3’及びR4’は、それぞれ独立して、水素原子又はヒドロキシ基を示すか、或いは、R3’及びR4’は、互いに結合して、それらが結合している炭素原子と一緒になって、シクロヘキサン環又はベンゼン環を形成してもよく;
結合b’は、単結合又は二重結合を示し;
X’は、CH2又はカルボニル(C=O)を示し;
Y’は、酸素原子、NH又はN(OCH3)を示し;
m’は、1を示し;並びに
n’は、0又は1を示す。但し、R2’が、無置換ビフェニリル基である時は、Y’-R1’は、メトキシ基を示す。]
で表される化合物(以下、「化合物(II)」と称することもある。)、又はその医薬上許容される塩。
[12]R3’及びR4’が、共に水素原子であり、且つ結合b’が、単結合である、上記[11]に記載の化合物、又はその医薬上許容される塩。
R’は、ヒドロキシ基又はC1-6アルコキシ基で置換されていてもよいフェニル基、ナフチル基、トリチル基で置換されたイミダゾリル基、置換されていてもよいC6-14アリールスルホニル基で置換されたインドリル基、トリチルオキシ基又はベンジルオキシ基を示し;
R1’は、2個のフェニル基で置換されていてもよいメチル基、又はフェニル基を示し;R2’は、エチニル基で置換されていてもよいビフェニリル基、tert-ブチル基で置換されたフェニル基、メチル基、又はフルオレニルメトキシ基を示し;
R3’及びR4’は、それぞれ独立して、水素原子又はヒドロキシ基を示すか、或いは、R3’及びR4’は、互いに結合して、それらが結合している炭素原子と一緒になって、シクロヘキサン環又はベンゼン環を形成してもよく;
結合b’は、単結合又は二重結合を示し;
X’は、CH2又はカルボニル(C=O)を示し;
Y’は、酸素原子、NH又はN(OCH3)を示し;
m’は、1を示し;並びに
n’は、0又は1を示す。但し、R2’が、無置換ビフェニリル基である時は、Y’-R1’は、メトキシ基を示す。]
で表される化合物(以下、「化合物(II)」と称することもある。)、又はその医薬上許容される塩。
[12]R3’及びR4’が、共に水素原子であり、且つ結合b’が、単結合である、上記[11]に記載の化合物、又はその医薬上許容される塩。
化合物(I)は、優れたWntシグナル伝達経路阻害活性を示す。それ故、本発明に係る化合物(I)からなるWntシグナル伝達経路阻害剤を有効成分として含む医薬は、Wntシグナル伝達経路を阻害することにより症状を緩和させることができる疾患、例えば、癌、冠状動脈疾患、急性冠症候群、変形性関節症等の予防又は治療に有用であり、とりわけ、各種癌(例えば、大腸癌、乳癌、頭頸部癌、肺癌、胃癌、食道癌、悪性黒色腫、前立腺癌、肝細胞癌、卵巣癌、肝芽腫、急性骨髄性白血病、急性リンパ芽球性白血病、慢性骨髄性白血病、慢性リンパ性白血病、多発性骨髄腫、悪性リンパ腫、膵臓癌、腎臓癌、膀胱癌、神経膠芽腫、髄芽腫、ウィルムス腫瘍等)の予防又は治療に特に有用である。また、本発明によれば、優れたWntシグナル伝達経路阻害活性を有する、新規な化合物(化合物(II))、又はその医薬上許容される塩も提供することができる。
以下に本発明の詳細を説明する。
(定義)
本明細書中、「ハロゲン原子」とは、フッ素原子、塩素原子、臭素原子又はヨウ素原子を意味する。
本明細書中、「アルキル(基)」としては、直鎖状または分岐鎖状の炭素原子数1以上のアルキル基を意味し、特に炭素数範囲の限定がない場合には、好ましくは、C1-20アルキル基であり、中でも、C1-6アルキル基がより好ましく、C1-4アルキル基が特に好ましい。
本明細書中、「C1-20アルキル(基)」とは、直鎖又は分岐鎖の炭素原子数1~20のアルキル基を意味し、例えば、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、sec-ブチル、tert-ブチル、ペンチル、イソペンチル、ネオペンチル、1-エチルプロピル、ヘキシル、イソヘキシル、1,1-ジメチルブチル、2,2-ジメチルブチル、3,3-ジメチルブチル、2-エチルブチル、ヘプチル、オクチル、ノニル、デシル、ウンデシル、ドデシル、トリデシル、エイコシル等が挙げられる。
本明細書中、「C1-6アルキル(基)」とは、直鎖又は分岐鎖の炭素原子数1~6のアルキル基を意味し、例えば、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、sec-ブチル、tert-ブチル、ペンチル、イソペンチル、ネオペンチル、1-エチルプロピル、ヘキシル、イソヘキシル、1,1-ジメチルブチル、2,2-ジメチルブチル、3,3-ジメチルブチル、2-エチルブチル等が挙げられる。
本明細書中、「C1-4アルキル(基)」とは、直鎖又は分岐鎖の炭素原子数1~4のアルキル基を意味し、例えば、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、sec-ブチル、tert-ブチル等が挙げられる。
本明細書中、「シクロアルキル(基)」とは、環状アルキル基を意味し、特に炭素数範囲の限定がない場合には、好ましくは、C3-8シクロアルキル基である。
本明細書中、「C3-8シクロアルキル(基)」とは、炭素原子数3~8の環状アルキル基を意味し、例えば、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロオクチル等が挙げられる。中でも、C3-6シクロアルキル基が好ましい。
本明細書中、「C2-6アルキニル(基)」とは、1個以上の炭素-炭素三重結合を有し、炭素数が2~6の直鎖状または分岐鎖状の一価の炭化水素基を意味し、例えば、エチニル、1-プロピニル、2-プロピニル、1-ブチニル、2-ブチニル、3-ブチニル、1-ペンチニル、2-ペンチニル、3-ペンチニル、4-ペンチニル、1-ヘキシニル、2-ヘキシニル、3-ヘキシニル、4-ヘキシニル、5-ヘキシニル、4-メチル-2-ペンチニルが挙げられる。中でも、好ましくは、エチニルである。
本明細書中、「アルコキシ(基)」とは、直鎖または分岐鎖のアルキル基が酸素原子と結合した基を意味し、特に炭素数範囲は限定されないが、好ましくは、C1-6アルコキシ基であり、より好ましくは、C1-4アルコキシ基である。
本明細書中、「C1-6アルコキシ(基)」とは、直鎖又は分岐鎖の炭素原子数1~6のアルコキシ基を意味し、例えば、メトキシ、エトキシ、プロポキシ、イソプロポキシ、ブトキシ、イソブトキシ、sec-ブトキシ、tert-ブトキシ、ペンチルオキシ、イソペンチルオキシ、ネオペンチルオキシ、ヘキシルオキシ等が挙げられる。中でも、C1-4アルコキシ基が好ましい。
本明細書中、「アルコキシ-カルボニル(基)」とは、前記アルコキシ基が酸素原子とカルボニル基に結合した基を意味し、特に炭素数範囲は限定されないが、好ましくは、C1-6アルコキシ-カルボニル基である。
本明細書中、「アルキルスルファニル(基)」とは、硫黄原子に前記「アルキル」基が結合した基を意味し、特に炭素数範囲は限定されないが、好ましくは、C1-6アルキルスルファニルである。
本明細書中、「C1-6アルキルスルファニル(基)」とは、直鎖又は分岐鎖の炭素原子数1~6のアルキルスルファニル基を意味し、例えば、メチルスルファニル、エチルスルファニル、プロピルスルファニル、イソプロピルスルファニル、ブチルスルファニル、イソブチルスルファニル、sec-ブチルスルファニル、tert-ブチルスルファニル、ペンチルスルファニル、イソペンチルスルファニル、ネオペンチルスルファニル、1-エチルプロピルスルファニル、へキシルスルファニル等が挙げられる。
本明細書中、「C1-6アルキルスルフィニル(基)」は、スルフィニル基に前記「C1-6アルキル」基が結合した基、すなわち、炭素数が1~6の直鎖または分岐鎖アルキルスルフィニル基を意味する。該「C1-6アルキルスルフィニル(基)」としては、例えば、メチルスルフィニル、エチルスルフィニル、プロピルスルフィニル、イソプロピルスルフィニル、ブチルスルフィニル、イソブチルスルフィニル、sec-ブチルスルフィニル、tert-ブチルスルフィニル、ペンチルスルフィニル、イソペンチルスルフィニル、ネオペンチルスルフィニル、1-エチルプロピルスルフィニル、へキシルスルフィニル等が挙げられる。
本明細書中、「C1-6アルキルスルホニル(基)」は、スルホニル基に前記「C1-6アルキル」基が結合した基、すなわち、炭素数が1~6の直鎖または分岐鎖アルキルスルホニル基を意味する。該「C1-6アルキルスルホニル(基)」としては、例えば、メチルスルホニル、エチルスルホニル、プロピルスルホニル、イソプロピルスルホニル、ブチルスルホニル、イソブチルスルホニル、sec-ブチルスルホニル、tert-ブチルスルホニル、ペンチルスルホニル、イソペンチルスルホニル、ネオペンチルスルホニル、1-エチルプロピルスルホニル、へキシルスルホニル等が挙げられる。
本明細書中、「アルキルスルホニルオキシ(基)」とは、アルキルスルホニル基が酸素原子に結合した基を意味し、特に炭素数範囲は限定されないが、好ましくは、C1-6アルキルスルホニルオキシ基である。
本明細書中、「C1-6アルキルスルホニルオキシ(基)」とは、C1-6アルキルスルホニル基が酸素原子に結合した基を意味し、例えば、メチルスルホニルオキシ、エチルスルホニルオキシ、プロピルスルホニルオキシ、イソプロピルスルホニルオキシ、ブチルスルホニルオキシ等が挙げられる。
本明細書中、「アリール(基)」とは、芳香族性を示す単環式あるいは多環式(縮合)の炭化水素基を意味し、具体的には、例えば、フェニル、1-ナフチル、2-ナフチル、ビフェニリル、2-アンスリル、フルオレニル等のC6-14アリール基が挙げられ、中でもC6-10アリール基が好ましい。
本明細書中、「C6-10アリール(基)」とは、例えば、フェニル、1-ナフチル、2-ナフチルが挙げられ、フェニル又は1-ナフチルが特に好ましい。
本明細書中、「アラルキル(基)」とは、アルキル基にアリール基が置換した基を意味し、特に炭素数範囲は限定されないが、好ましくは、C7-14アラルキルである。
本明細書中、「C7-22アラルキル(基)」とは、「C1-4アルキル基」に「C6-18アリール基」が置換した基を意味し、例えば、ベンジル、1-フェニルエチル、2-フェニルエチル、(ナフチル-1-イル)メチル、(ナフチル-2-イル)メチル、1-(ナフチル-1-イル)エチル、1-(ナフチル-2-イル)エチル、2-(ナフチル-1-イル)エチル、2-(ナフチル-2-イル)エチル、ジフェニルメチル、フルオレニルメチル、トリチル等が挙げられる。
本明細書中、「ヘテロアリール(基)」とは、少なくとも1つの環原子がヘテロ原子であり、残りの環原子が炭素である、単環式、二環式または多環式芳香族基を意味する。単環式ヘテロアリール基としては、限定されないが、5または6個の環原子を有する環式芳香族基であって、少なくとも1つの環原子がヘテロ原子であり、残りの環原子が炭素である環式芳香族基が挙げられる。窒素原子は任意に四級化されていてもよく、硫黄原子は任意に酸化されていてもよい。本発明のヘテロアリール基としては、フラン、イミダゾール、イソチアゾール、イソオキサゾール、オキサジアゾール、オキサゾール、1,2,3-オキサジアゾール、ピラジン、ピラゾール、ピリダジン、ピリジン、ピリミジン、ピロリン、チアゾール、1,3,4-チアジアゾール、トリアゾール、及びテトラゾールから誘導されるものが挙げられるが、これらに限定されない。「ヘテロアリール」には、ヘテロアリール環が、アリール環、シクロアルキル環、シクロアルケニル環、及び別の単環式ヘテロアリールまたはヘテロシクロアルキル環からなる群から独立して選択される1つまたは2つの環に縮合している、二環式または三環式の環も含まれるが、これらに限定されない。これらの二環式または三環式のヘテロアリールとしては、ベンゾ[b]フラン、ベンゾ[b]チオフェン、ベンズイミダゾール、イミダゾ[4,5-c]ピリジン、キナゾリン、チエノ[2,3-c]ピリジン、チエノ[3,2-b]ピリジン、チエノ[2,3-b]ピリジン、インドリジン、イミダゾ[1,2-a]ピリジン、キノリン、イソキノリン、フタラジン、キノキサリン、ナフチリジン、キノリジン、インドール、イソインドール、インダゾール、インドリン、ベンゾオキサゾール、ベンゾピラゾール、ベンゾチアゾール、イミダゾ[1,5-a]ピリジン、ピラゾロ[1,5-a]ピリジン、イミダゾ[1,2-a]ピリミジン、イミダゾ[1,2-c]ピリミジン、イミダゾ[1,5-a]ピリミジン、イミダゾ[1,5-c]ピリミジン、ピロロ[2,3-b]ピリジン、ピロロ[2,3-c]ピリジン、ピロロ[3,2-c]ピリジン、ピロロ[3,2-b]ピリジン、ピロロ[2,3-d]ピリミジン、ピロロ[3,2-d]ピリミジン、ピロロ[2,3-b]ピラジン、ピラゾロ[1,5-a]ピリジン、ピロロ[1,2-b]ピリダジン、ピロロ[1,2-c]ピリミジン、ピロロ[1,2-a]ピリミジン、ピロロ[1,2-a]ピラジン、トリアゾ[1,5-a]ピリジン、プテリジン、プリン、カルバゾール、アクリジン、フェナジン、フェノチアゼン、フェノキサジン、1,2-ジヒドロピロロ[3,2,1-hi]インドール、ピリド[1,2-a]インドール、及び2(1H)-ピリジノンから誘導されるものが挙げられるが、これらに限定されない。好適なヘテロアリール(基)としては、例えば、インドールから誘導されるもの(インドリル(基))、イミダゾールから誘導されるもの(イミダゾリル(基))等が挙げられる。
本明細書中、「アリールスルファニル(基)」とは、硫黄原子に「アリール基」が結合した基を意味し、特に炭素数範囲は限定されないが、好ましくは、C6-10アリールスルファニルである。
本明細書中、「C6-10アリールスルファニル(基)」とは、炭素原子数6~10のアリールスルファニル基を意味し、例えば、フェニルスルファニル、1-ナフチルスルファニル、2-ナフチルスルファニル等が挙げられる。
本明細書中、「アリールスルホニル(基)」とは、アリール基がスルホニル基に結合した基を意味し、特に炭素数範囲は限定されないが、好ましくは、C6-10アリールスルホニル基である。
本明細書中、「C6-10アリールスルホニル(基)」とは、「C6-10アリール基」がスルホニル基に結合した基を意味し、例えば、フェニルスルホニル、1-ナフチルスルホニル、2-ナフチルスルホニル等が挙げられる。
本明細書中、「アリールスルホニルオキシ(基)」とは、前記アリールスルホニル基が酸素原子に結合した基を意味し、特に炭素数範囲は限定されないが、好ましくは、C6-10アリールスルホニルオキシ基である。
本明細書中、「C6-10アリールスルホニルオキシ(基)」とは、C6-10アリールスルホニル基が酸素原子に結合した基を意味し、例えば、フェニルスルホニルオキシ、1-ナフチルスルホニルオキシ、2-ナフチルスルホニルオキシ等が挙げられる。
本明細書中、「アシル(基)」とは、アルカノイル又はアロイルを意味し、特に炭素数範囲は限定されないが、好ましくは、C1-7アルカノイル基又はC7-11アロイルである。
本明細書中、「C1-7アルカノイル(基)」とは、炭素原子数1~7の直鎖又は分枝鎖状のホルミル又はアルキルカルボニル(すなわち、C1-6アルキル-カルボニル)であり、例えば、ホルミル、アセチル、プロピオニル、ブチリル、イソブチリル、ペンタノイル、ヘキサノイル、ヘプタノイル等が挙げられる。
本明細書中、「C7-11アロイル(基)」とは、炭素原子数7~11のアリールカルボニル(すなわち、C6-10アリール-カルボニル)であり、ベンゾイル等が挙げられる。
本明細書中、「アシルオキシ(基)」とは、前記アルカノイル基又はアロイル基が酸素原子と結合した基を意味し、特に炭素数範囲は限定されないが、好ましくは、C1-7アルカノイルオキシ基又はC7-11アロイルオキシ基である。
本明細書中、「C1-7アルカノイルオキシ(基)」としては、例えば、ホルミルオキシ、アセトキシ、エチルカルボニルオキシ、プロピルカルボニルオキシ、イソプロピルカルボニルオキシ、ブチルカルボニルオキシ、イソブチルカルボニルオキシ、sec-ブチルカルボニルオキシ、tert-ブチルカルボニルオキシ(ピバロイルオキシ)、ペンチルカルボニルオキシ、イソペンチルカルボニルオキシ、ネオペンチルカルボニルオキシ、ヘキシルカルボニルオキシ等が挙げられ、好ましくは、アセトキシ又はピバロイルオキシである。
本明細書中、「C7-11アロイルオキシ(基)」としては、例えば、ベンゾイルオキシ、1-ナフトイルオキシ、2-ナフトイルオキシ等が挙げられる。
本明細書中、「保護されていてもよいヒドロキシ基」とは、ヒドロキシ基、又は「保護基」で保護されたヒドロキシ基を意味する。当該「保護基」としては、例えば、Protective Groups in Organic Synthesis,John Wiley and Sons刊(1980)に記載のヒドロキシ基の保護基を使用し得、例えば、C1-6アルキル基、C7-22アラルキル基、C1-7アルカノイル基、C7-14アラルキルオキシ-カルボニル基、トリC1-6アルキルシリル基(例、トリ-C1-6アルキルシリル基(例、トリメチルシリル、tert-ブチル(ジメチル)シリル)等の保護基が挙げられる。上記の保護基は、ハロゲン原子、C1-6アルキル基、C1-6アルコキシ基又はニトロ基により更に置換されていてもよい。「保護されていてもよいヒドロキシ基」としては、好ましくは、保護されたヒドロキシ基であり、特に好ましくは、ベンジル又はトリチルで保護されたヒドロキシ基である。
本明細書中、「保護されたアミノ基」とは、「保護基」で保護されたアミノ基を意味する。当該「保護基」としては、例えば、Protective Groups in Organic Synthesis,John Wiley and Sons刊(1980)に記載のアミノ基の保護基を使用し得、例えば、C1-6アルキル基、C7-22アラルキル基、C6-10アリール基、C1-7アルカノイル基、C7-14アラルキル-カルボニル基、トリC1-6アルキルシリル基(例、トリ-C1-6アルキルシリル基(例、トリメチルシリル、tert-ブチル(ジメチル)シリル)等の保護基が挙げられる。上記の保護基は、ハロゲン原子、C1-6アルキル基、C1-6アルコキシ基又はニトロ基により更に置換されていてもよい。当該アミノ基の保護基の具体例としては、メチル(モノメチル又はジメチル)、ベンジル、トリチル、アセチル、トリフルオロアセチル、ピバロイル、tert-ブトキシカルボニル、ベンジルオキシカルボニル等が挙げられる。
本明細書中、「置換されていてもよい」とは、1個以上の置換基を有していてもよいことを意味し、該「置換基」としては、(1)ハロゲン原子、(2)ニトロ、(3)シアノ、(4)C1-6アルキル、(5)C2-6アルキニル、(6)C3-8シクロアルキル、(7)C1-6アルコキシ、(8)C6-10アリール、(9)C7-22アラルキル、(10)C1-7アルカノイルオキシ、(11)C7-11アロイルオキシ、(12)C1-7アルカノイルオキシ-カルボニル、(13)C7-11アロイルオキシ-カルボニル、(14)C1-7アルカノイル、(15)C7-11アロイル、(16)C1-6アルキルスルファニル、(17)C6-10アリールスルファニル、(18)C1-6アルキル基でモノ又はジ-置換されていてもよいカルバモイル、(19)C1-6アルキルスルホニルオキシ基、(20)C6-10アリールスルホニルオキシ基、(21)トリC1-6アルキルシリル基、(22)保護されたアミノ基等が挙げられる。中でも、ハロゲン原子、ヒドロキシ、C1-6アルキル、C1-6アルコキシ、エチニル、ベンジル、フルオレニルメチル、ジフェニルメチル、トリチル、アセチル、ホルミル、カルバモイル、トリエチルシリル、トリイソプロピルシリル、tert-ブチルジメチルシリル、ジメチルアミノ、アセチルアミノ、tert-ブトキシカルボニルアミノ、ベンジルオキシカルボニルアミノ等が好ましい。また、複数の置換基が存在する場合、各置換基は、同一でも異なっていてもよい。
上記置換基は、さらに上記置換基で置換されていてもよい。置換基の数は、置換可能な数であれば特に限定されないが、好ましくは1乃至5個、より好ましくは1乃至3個である。複数の置換基が存在する場合、各置換基は、同一でも異なっていてもよい。当該置換基はまたさらにC1-6アルキル基、C3-8シクロアルキル基、C6-10アリール基、C7-22アラルキル基、ハロゲン原子、ヒドロキシ基、保護されたアミノ基、カルバモイル基、シアノ基、ニトロ基、オキソ基等で置換されていてもよい。置換基の数は、置換可能な数であれば特に限定されないが、好ましくは1乃至5個、より好ましくは1乃至3個である。複数の置換基が存在する場合、各置換基は、同一でも異なっていてもよい。
本明細書中、「その医薬上許容される塩」とは、医薬として使用することができる塩を意味する。本発明の化合物(I)(若しくは化合物(II))では、酸性基又は塩基性基を有する場合に、塩基又は酸と反応させることにより、塩基性塩又は酸性塩にすることができるので、その塩を示す。
本発明の化合物(I)(若しくは化合物(II))の医薬上許容される「塩基性塩」としては、例えば、ナトリウム塩、カリウム塩、リチウム塩等のアルカリ金属塩;マグネシウム塩、カルシウム塩等のアルカリ土類金属塩;N-メチルモルホリン塩、トリエチルアミン塩、トリブチルアミン塩、ジイソプロピルエチルアミン塩、ジシクロヘキシルアミン塩、N-メチルピペリジン塩、ピリジン塩、4-ピロリジノピリジン塩、ピコリン塩等の有機塩基塩;グリシン塩、リジン塩、アルギニン塩、オルニチン塩、グルタミン酸塩、アスパラギン酸塩等のアミノ酸塩等が挙げられ、好ましくは、アルカリ金属塩である。
本発明の化合物(I)(若しくは化合物(II))の医薬上許容される「酸性塩」としては、例えば、フッ化水素酸塩、塩酸塩、臭化水素酸塩、ヨウ化水素酸塩等のハロゲン化水素酸塩、硝酸塩、過塩素酸塩、硫酸塩、リン酸塩等の無機酸塩;メタンスルホン酸塩、トリフルオロメタンスルホン酸塩、エタンスルホン酸塩等の低級アルカンスルホン酸塩、ベンゼンスルホン酸塩、p-トルエンスルホン酸塩等のアリ-ルスルホン酸塩、酢酸塩、リンゴ酸塩、フマル酸塩、コハク酸塩、クエン酸塩、アスコルビン酸塩、酒石酸塩、蓚酸塩、マレイン酸塩等の有機酸塩;グリシン塩、リジン塩、アルギニン塩、オルニチン塩、グルタミン酸塩、アスパラギン酸塩のようなアミノ酸塩等が挙げられ、好ましくは、ハロゲン化水素酸塩(特に、塩酸塩)である。
本明細書中、「予防」には、疾患の発症の防止、及び疾患の発症の遅延が含まれる。「予防有効量」とは、かかる目的を達成するに足る化合物(I)の用量をいう。
本明細書中、「治療」には、疾患の治癒、疾患の病態(例えば1つ又は複数の症状)の改善、及び疾患(の重篤度)の進展の抑制が含まれる。「治療有効量」とは、かかる目的を達成するに足る化合物(I)の用量をいう。
本明細書中、「Wntシグナル伝達経路阻害剤」とは、Wntシグナル伝達経路の生物活性を部分的に、又は完全に阻害、抑制、又は中和するための薬剤を意味する。
本明細書中、「Wntシグナル伝達経路を阻害することにより症状を緩和させることができる疾患」とは、Wntシグナルの異常な活性化によって引き起こされる疾患や症状を意味する。「Wntシグナル伝達経路を阻害することにより症状を緩和させることができる疾患」の具体例としては、例えば、癌、冠状動脈疾患、急性冠症候群、変形性関節症等の予防又は治療に有用であり、とりわけ、各種癌(例えば、大腸癌、乳癌、頭頸部癌、肺癌、胃癌、食道癌、悪性黒色腫、前立腺癌、肝細胞癌、卵巣癌、肝芽腫、急性骨髄性白血病、急性リンパ芽球性白血病、慢性骨髄性白血病、慢性リンパ性白血病、多発性骨髄腫、悪性リンパ腫、膵臓癌、腎臓癌、膀胱癌、神経膠芽腫、髄芽腫、ウィルムス腫瘍等)の予防又は治療に有用である。
(本発明の化合物(I))
本発明のWntシグナル伝達経路阻害剤は、下記式(I):
本発明のWntシグナル伝達経路阻害剤は、下記式(I):
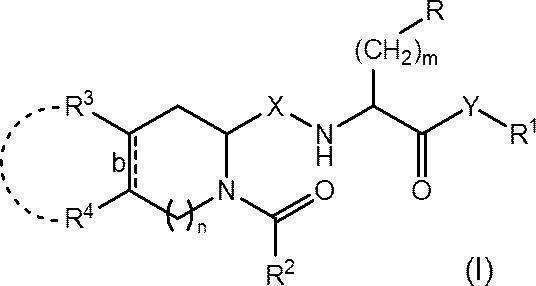
[式中、
Rは、置換されていてもよいC6-14アリール基、置換されていてもよいヘテロアリール基又は保護されていてもよいヒドロキシ基を示し;
R1は、置換されていてもよいC1-6アルキル基、又は置換されていてもよいC6-14アリール基を示し;
R2は、置換されていてもよいC1-6アルキル基、置換されていてもよいC1-6アルコキシ基又は置換されていてもよいC6-14アリール基を示し;
R3及びR4は、それぞれ独立して、水素原子、ハロゲン原子、ヒドロキシ基、アミノ基、ニトロ基、シアノ基、カルボキシ基、置換されていてもよいC1-6アルキル基又は置換されていてもよいC1-6アルコキシ基を示すか、或いは、R3及びR4は、互いに結合して、それらが結合している炭素原子と一緒になって、それぞれ置換されていてもよい、シクロヘキサン環又はベンゼン環を形成してもよく;
結合bは、単結合又は二重結合を示し;
Xは、CH2又はカルボニル(C=O)を示し;
Yは、酸素原子又はNR0(式中のR0は、水素原子又はC1-4アルコキシ基を示す。)を示し;
mは、1~3の整数を示し;並びに
nは、0又は1を示す。]
で表される化合物(化合物(I))、又はその医薬上許容される塩からなることを特徴とする。
Rは、置換されていてもよいC6-14アリール基、置換されていてもよいヘテロアリール基又は保護されていてもよいヒドロキシ基を示し;
R1は、置換されていてもよいC1-6アルキル基、又は置換されていてもよいC6-14アリール基を示し;
R2は、置換されていてもよいC1-6アルキル基、置換されていてもよいC1-6アルコキシ基又は置換されていてもよいC6-14アリール基を示し;
R3及びR4は、それぞれ独立して、水素原子、ハロゲン原子、ヒドロキシ基、アミノ基、ニトロ基、シアノ基、カルボキシ基、置換されていてもよいC1-6アルキル基又は置換されていてもよいC1-6アルコキシ基を示すか、或いは、R3及びR4は、互いに結合して、それらが結合している炭素原子と一緒になって、それぞれ置換されていてもよい、シクロヘキサン環又はベンゼン環を形成してもよく;
結合bは、単結合又は二重結合を示し;
Xは、CH2又はカルボニル(C=O)を示し;
Yは、酸素原子又はNR0(式中のR0は、水素原子又はC1-4アルコキシ基を示す。)を示し;
mは、1~3の整数を示し;並びに
nは、0又は1を示す。]
で表される化合物(化合物(I))、又はその医薬上許容される塩からなることを特徴とする。
以下、化合物(I)の各基について説明する。
Rは、置換されていてもよいC6-14アリール基、置換されていてもよいヘテロアリール基又は保護されていてもよいヒドロキシ基を表す。
Rは、好ましくは、置換されていてもよいC6-10アリール基、置換されていてもよいヘテロアリール基又は保護されたヒドロキシ基であり、より好ましくは、置換されていてもよいフェニル基、置換されていてもよいナフチル基、置換されていてもよいイミダゾリル基、置換されていてもよいインドリル基、トリチルオキシ基又はベンジルオキシ基であり、特に好ましくは、置換されていてもよいイミダゾリル基(例、トリチル基で置換されたイミダゾリル基)である。
R1は、置換されていてもよいC1-6アルキル基、又は置換されていてもよいC6-14アリール基を表す。
R1は、好ましくは、1又は2個のフェニル基で置換されていてもよいC1-4アルキル基、或いは置換されていてもよいフェニル基であり、より好ましくは、2個のフェニル基で置換されたC1-4アルキル基(例、ジフェニルメチル基)、C1-4アルキル基(例、メチル基)、或いはフェニル基であり、特に好ましくは、C1-4アルキル基(例、メチル基)である。
R2は、置換されていてもよいC1-6アルキル基、置換されていてもよいC1-6アルコキシ基又は置換されていてもよいC6-14アリール基を表す。
R2は、好ましくは、置換されていてもよいC1-4アルキル基、置換されていてもよいC1-4アルコキシ基、置換されていてもよいフェニル基であり、より好ましくは、置換されていてもよいC1-4アルキル基、又は置換されていてもよいフェニル基であり、特に好ましくは、保護されたアミノ基(例、N-tert-ブトキシカルボニル-N-フェニル-アミノ)で置換されていてもよいC1-4アルキル基、又は、ハロゲン原子(例、臭素原子、フッ素原子)、フェニル基若しくはC1-4アルキル基(例、メチル)で置換されていてもよいフェニル基である。
R3及びR4は、それぞれ独立して、水素原子、ハロゲン原子、ヒドロキシ基、アミノ基、ニトロ基、シアノ基、カルボキシ基、置換されていてもよいC1-6アルキル基又は置換されていてもよいC1-6アルコキシ基を表すか、或いは、R3及びR4は、互いに結合して、それらが結合している炭素原子と一緒になって、それぞれ置換されていてもよい、シクロヘキサン環又はベンゼン環を形成してもよい。
R3及びR4は、好ましくは、それぞれ独立して、水素原子、ヒドロキシ基又はC1-6アルコキシ基であるか、或いは、R3及びR4は、互いに結合して、それらが結合している炭素原子と一緒になって、シクロヘキサン環を形成してもよく、より好ましくは、共に水素原子であるか、または、R3及びR4は、互いに結合して、それらが結合している炭素原子と一緒になって、シクロヘキサン環を形成する。
結合bは、単結合又は二重結合を表す。
結合bは、好ましくは、単結合である。
Xは、CH2又はカルボニル(C=O)を表す。
Xは、好ましくは、CH2である。
Yは、酸素原子又はNR0(式中のR0は、水素原子又はC1-4アルコキシ基を示す。)を表す。
Yは、好ましくは、酸素原子、NH又はN(OCH3)であり、より好ましくは、酸素原子又はN(OCH3)である。
mは、1~3の整数を表す。
mは、好ましくは、1である。
nは、0又は1を表す。
nは、好ましくは、1である。
化合物(I)としては、以下の化合物が好適である。
[化合物(IA)]
Rが、置換されていてもよいC6-10アリール基、置換されていてもよいヘテロアリール基又は保護されたヒドロキシ基であり、
R1が、1又は2個のフェニル基で置換されていてもよいC1-4アルキル基、C1-4アルキル基又は置換されていてもよいフェニル基であり、
R2が、置換されていてもよいC1-4アルキル基、置換されていてもよいC1-4アルコキシ基、置換されていてもよいフェニル基であり、
R3及びR4が、それぞれ独立して、水素原子、ヒドロキシ基又はC1-6アルコキシ基であるか、或いは、R3及びR4は、互いに結合して、それらが結合している炭素原子と一緒になって、シクロヘキサン環を形成してもよく、
結合bが、単結合であり、
Xが、CH2又はカルボニルであり、
Yが、酸素原子、NH又はN(OCH3)であり、
mが、1であり、且つ
nが、0又は1である、化合物(I)。
[化合物(IA)]
Rが、置換されていてもよいC6-10アリール基、置換されていてもよいヘテロアリール基又は保護されたヒドロキシ基であり、
R1が、1又は2個のフェニル基で置換されていてもよいC1-4アルキル基、C1-4アルキル基又は置換されていてもよいフェニル基であり、
R2が、置換されていてもよいC1-4アルキル基、置換されていてもよいC1-4アルコキシ基、置換されていてもよいフェニル基であり、
R3及びR4が、それぞれ独立して、水素原子、ヒドロキシ基又はC1-6アルコキシ基であるか、或いは、R3及びR4は、互いに結合して、それらが結合している炭素原子と一緒になって、シクロヘキサン環を形成してもよく、
結合bが、単結合であり、
Xが、CH2又はカルボニルであり、
Yが、酸素原子、NH又はN(OCH3)であり、
mが、1であり、且つ
nが、0又は1である、化合物(I)。
より好適な化合物(I)は、以下の化合物である。
[化合物(IB)]
Rが、置換されていてもよいフェニル基、置換されていてもよいナフチル基、置換されていてもよいイミダゾリル基、置換されていてもよいインドリル基、トリチルオキシ基又はベンジルオキシ基であり、
R1が、2個のフェニル基で置換されたC1-4アルキル基(例、ジフェニルメチル基)、C1-4アルキル基(例、メチル基)、或いはフェニル基であり、
R2が、置換されていてもよいC1-4アルキル基、又は置換されていてもよいフェニル基であり、
R3及びR4が、共に水素原子であるか、または、R3及びR4は、互いに結合して、それらが結合している炭素原子と一緒になって、シクロヘキサン環を形成し、
結合bが、単結合であり、
Xが、CH2又はカルボニルであり、
Yが、酸素原子、NH又はN(OCH3)であり、
mが、1であり、且つ
nが、0又は1である、化合物(I)。
[化合物(IB)]
Rが、置換されていてもよいフェニル基、置換されていてもよいナフチル基、置換されていてもよいイミダゾリル基、置換されていてもよいインドリル基、トリチルオキシ基又はベンジルオキシ基であり、
R1が、2個のフェニル基で置換されたC1-4アルキル基(例、ジフェニルメチル基)、C1-4アルキル基(例、メチル基)、或いはフェニル基であり、
R2が、置換されていてもよいC1-4アルキル基、又は置換されていてもよいフェニル基であり、
R3及びR4が、共に水素原子であるか、または、R3及びR4は、互いに結合して、それらが結合している炭素原子と一緒になって、シクロヘキサン環を形成し、
結合bが、単結合であり、
Xが、CH2又はカルボニルであり、
Yが、酸素原子、NH又はN(OCH3)であり、
mが、1であり、且つ
nが、0又は1である、化合物(I)。
更に好適な化合物(I)は、以下の化合物である。
[化合物(IC)]
Rが、置換されていてもよいイミダゾリル基(例、トリチル基で置換されたイミダゾリル基)であり、
R1が、C1-4アルキル基(例、メチル基)であり、
R2が、保護されたアミノ基(例、N-tert-ブトキシカルボニル-N-フェニル-アミノ)で置換されていてもよいC1-4アルキル基、又は、ハロゲン原子(例、臭素原子、フッ素原子)、フェニル基若しくはC1-4アルキル基(例、メチル)で置換されていてもよいフェニル基であり、
R3及びR4が、共に水素原子であるか、または、R3及びR4は、互いに結合して、それらが結合している炭素原子と一緒になって、シクロヘキサン環を形成し、
結合bが、単結合であり、
Xが、CH2であり、
Yが、酸素原子又はN(OCH3)であり、
mが、1であり、且つ
nが、0又は1(好ましくは、1)である、化合物(I)。
[化合物(IC)]
Rが、置換されていてもよいイミダゾリル基(例、トリチル基で置換されたイミダゾリル基)であり、
R1が、C1-4アルキル基(例、メチル基)であり、
R2が、保護されたアミノ基(例、N-tert-ブトキシカルボニル-N-フェニル-アミノ)で置換されていてもよいC1-4アルキル基、又は、ハロゲン原子(例、臭素原子、フッ素原子)、フェニル基若しくはC1-4アルキル基(例、メチル)で置換されていてもよいフェニル基であり、
R3及びR4が、共に水素原子であるか、または、R3及びR4は、互いに結合して、それらが結合している炭素原子と一緒になって、シクロヘキサン環を形成し、
結合bが、単結合であり、
Xが、CH2であり、
Yが、酸素原子又はN(OCH3)であり、
mが、1であり、且つ
nが、0又は1(好ましくは、1)である、化合物(I)。
好適な化合物(I)の具体例は、後述する実施例1~34の化合物(化合物(1)~化合物(34))、又はそれらの医薬上許容される塩である。
より好適な化合物(I)は、具体的には以下の化合物、又はそれらの医薬上許容される塩である。
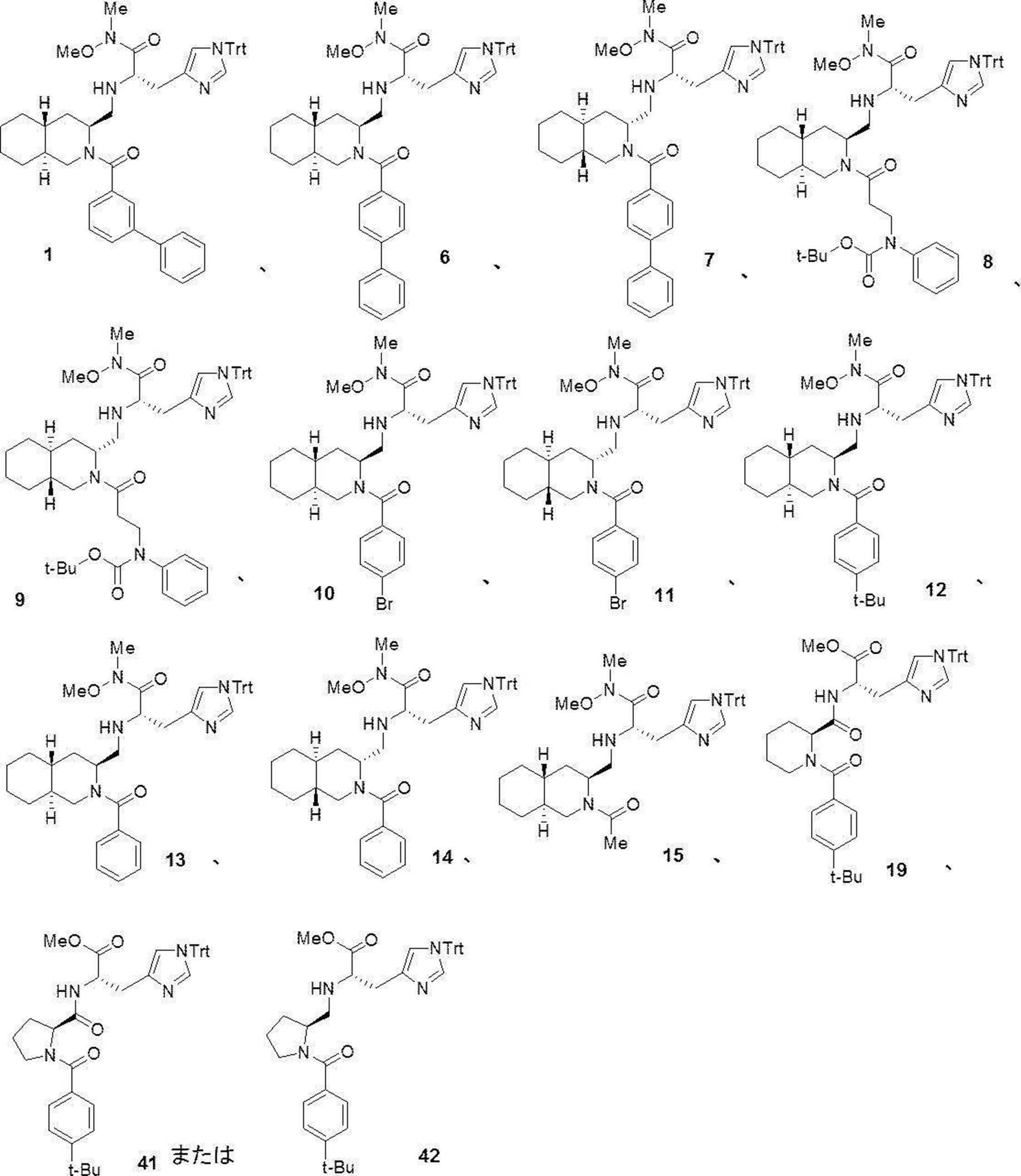
特に好適な化合物(I)は、具体的には以下の化合物、又はそれらの医薬上許容される塩である。

(本発明の化合物(II))
本発明は、前記化合物(I)に包含される新規化合物である、下記式(II):
本発明は、前記化合物(I)に包含される新規化合物である、下記式(II):

[式中、
R’は、ヒドロキシ基又はC1-6アルコキシ基で置換されていてもよいフェニル基、ナフチル基、トリチル基で置換されたイミダゾリル基、置換されていてもよいC6-14アリールスルホニル基で置換されたインドリル基、トリチルオキシ基又はベンジルオキシ基を示し;
R1’は、2個のフェニル基で置換されていてもよいメチル基、又はフェニル基を示し;R2’は、エチニル基で置換されていてもよいビフェニリル基、tert-ブチル基で置換されたフェニル基、メチル基、又はフルオレニルメトキシ基を示し;
R3’及びR4’は、それぞれ独立して、水素原子又はヒドロキシ基を示すか、或いは、R3’及びR4’は、互いに結合して、それらが結合している炭素原子と一緒になって、シクロヘキサン環又はベンゼン環を形成してもよく;
結合b’は、単結合又は二重結合を示し;
X’は、CH2又はカルボニル(C=O)を示し;
Y’は、酸素原子、NH又はN(OCH3)を示し;
m’は、1を示し;並びに
n’は、0又は1を示す。但し、R2’が、無置換ビフェニリル基である時は、Y’-R1’は、メトキシ基を示す。]
で表される化合物(化合物(II))、又はその医薬上許容される塩を提供する。
R’は、ヒドロキシ基又はC1-6アルコキシ基で置換されていてもよいフェニル基、ナフチル基、トリチル基で置換されたイミダゾリル基、置換されていてもよいC6-14アリールスルホニル基で置換されたインドリル基、トリチルオキシ基又はベンジルオキシ基を示し;
R1’は、2個のフェニル基で置換されていてもよいメチル基、又はフェニル基を示し;R2’は、エチニル基で置換されていてもよいビフェニリル基、tert-ブチル基で置換されたフェニル基、メチル基、又はフルオレニルメトキシ基を示し;
R3’及びR4’は、それぞれ独立して、水素原子又はヒドロキシ基を示すか、或いは、R3’及びR4’は、互いに結合して、それらが結合している炭素原子と一緒になって、シクロヘキサン環又はベンゼン環を形成してもよく;
結合b’は、単結合又は二重結合を示し;
X’は、CH2又はカルボニル(C=O)を示し;
Y’は、酸素原子、NH又はN(OCH3)を示し;
m’は、1を示し;並びに
n’は、0又は1を示す。但し、R2’が、無置換ビフェニリル基である時は、Y’-R1’は、メトキシ基を示す。]
で表される化合物(化合物(II))、又はその医薬上許容される塩を提供する。
以下、化合物(II)の各基について説明する。
R’は、ヒドロキシ基又はC1-6アルコキシ基で置換されていてもよいフェニル基、ナフチル基、トリチル基で置換されたイミダゾリル基、置換されていてもよいC6-14アリールスルホニル基で置換されたインドリル基、トリチルオキシ基又はベンジルオキシ基を表す。
R’は、好ましくは、C1-4アルコキシ基で置換されていてもよいフェニル基、ナフチル基、トリチル基で置換されたイミダゾリル基、置換されていてもよいフェニルスルホニル基で置換されたインドリル基、トリチルオキシ基又はベンジルオキシ基であり、より好ましくは、トリチル基で置換されたイミダゾリル基である。
R1’は、2個のフェニル基で置換されていてもよいメチル基、又はフェニル基を表す。
R1’は、好ましくは、メチル基である。
R2’は、エチニル基で置換されていてもよいビフェニリル基、tert-ブチル基で置換されたフェニル基、メチル基、又はフルオレニルメトキシ基を表す。
R2’は、好ましくは、tert-ブチル基で置換されたフェニル基、メチル基であり、より好ましくは、tert-ブチル基で置換されたフェニル基である。
R3’及びR4’は、それぞれ独立して、水素原子又はヒドロキシ基を表すか、或いは、R3’及びR4’は、互いに結合して、それらが結合している炭素原子と一緒になって、シクロヘキサン環又はベンゼン環を形成してもよい。
R3’及びR4’は、好ましくは、共に水素原子であるか、または、R3’及びR4’は、互いに結合して、それらが結合している炭素原子と一緒になって、シクロヘキサン環を形成する。
結合b’は、単結合又は二重結合を表す。
結合b’は、好ましくは、単結合である。
X’は、CH2又はカルボニル(C=O)を表す。
X’は、好ましくは、CH2である。
Y’は、酸素原子、NH又はN(OCH3)を表す。
Y’は、好ましくは、酸素原子又はN(OCH3)である。
m’は、1を表す。
n’は、0又は1を表す。
n’は、好ましくは、1である。
化合物(II)としては、以下の化合物が好適である。
[化合物(IIA)]
R’が、C1-4アルコキシ基で置換されていてもよいフェニル基、ナフチル基、トリチル基で置換されたイミダゾリル基、置換されていてもよいフェニルスルホニル基で置換されたインドリル基、トリチルオキシ基又はベンジルオキシ基であり、
R1’が、2個のフェニル基で置換されたメチル基、又はフェニル基であり、
R2’が、tert-ブチル基で置換されたフェニル基であり、
R3’及びR4’が、共に水素原子であるか、または、R3’及びR4’が、互いに結合して、それらが結合している炭素原子と一緒になって、シクロヘキサン環を形成してもよく、
結合b’が、単結合であり、
X’が、CH2又はカルボニルであり、
Y’が、NHであり、
m’が、1であり、且つ
n’が、0又は1である、化合物(II)。
[化合物(IIA)]
R’が、C1-4アルコキシ基で置換されていてもよいフェニル基、ナフチル基、トリチル基で置換されたイミダゾリル基、置換されていてもよいフェニルスルホニル基で置換されたインドリル基、トリチルオキシ基又はベンジルオキシ基であり、
R1’が、2個のフェニル基で置換されたメチル基、又はフェニル基であり、
R2’が、tert-ブチル基で置換されたフェニル基であり、
R3’及びR4’が、共に水素原子であるか、または、R3’及びR4’が、互いに結合して、それらが結合している炭素原子と一緒になって、シクロヘキサン環を形成してもよく、
結合b’が、単結合であり、
X’が、CH2又はカルボニルであり、
Y’が、NHであり、
m’が、1であり、且つ
n’が、0又は1である、化合物(II)。
別の好適な化合物(II)は、以下の化合物である。
[化合物(IIB)]
R’が、C1-4アルコキシ基で置換されていてもよいフェニル基、ナフチル基、トリチル基で置換されたイミダゾリル基、置換されていてもよいフェニルスルホニル基で置換されたインドリル基、トリチルオキシ基又はベンジルオキシ基であり、
R1’が、メチル基であり、
R2’が、メチル基、ビフェニリル基、又はtert-ブチル基で置換されたフェニル基であり、
R3’及びR4’が、共に水素原子であるか、または、R3’及びR4’が、互いに結合して、それらが結合している炭素原子と一緒になって、シクロヘキサン環を形成してもよく、
結合b’が、単結合であり、
X’が、CH2又はカルボニルであり、
Y’が、酸素原子であり、
m’が、1であり、且つ
n’が、0又は1である、化合物(II)。
[化合物(IIB)]
R’が、C1-4アルコキシ基で置換されていてもよいフェニル基、ナフチル基、トリチル基で置換されたイミダゾリル基、置換されていてもよいフェニルスルホニル基で置換されたインドリル基、トリチルオキシ基又はベンジルオキシ基であり、
R1’が、メチル基であり、
R2’が、メチル基、ビフェニリル基、又はtert-ブチル基で置換されたフェニル基であり、
R3’及びR4’が、共に水素原子であるか、または、R3’及びR4’が、互いに結合して、それらが結合している炭素原子と一緒になって、シクロヘキサン環を形成してもよく、
結合b’が、単結合であり、
X’が、CH2又はカルボニルであり、
Y’が、酸素原子であり、
m’が、1であり、且つ
n’が、0又は1である、化合物(II)。
更に別の好適な化合物(II)は、以下の化合物である。
[化合物(IIC)]
R’が、C1-4アルコキシ基で置換されていてもよいフェニル基、ナフチル基、トリチル基で置換されたイミダゾリル基、置換されていてもよいフェニルスルホニル基で置換されたインドリル基、トリチルオキシ基又はベンジルオキシ基であり、
R1’が、メチル基であり、
R2’が、エチニル基で置換されたビフェニリル基、又はメチル基であり、
R3’及びR4’が、共に水素原子であるか、または、R3’及びR4’が、互いに結合して、それらが結合している炭素原子と一緒になって、シクロヘキサン環を形成してもよく、
結合b’が、単結合であり、
X’が、CH2であり、
Y’が、N(OCH3)であり、
m’が、1であり、且つ
n’が、1である、化合物(II)。
[化合物(IIC)]
R’が、C1-4アルコキシ基で置換されていてもよいフェニル基、ナフチル基、トリチル基で置換されたイミダゾリル基、置換されていてもよいフェニルスルホニル基で置換されたインドリル基、トリチルオキシ基又はベンジルオキシ基であり、
R1’が、メチル基であり、
R2’が、エチニル基で置換されたビフェニリル基、又はメチル基であり、
R3’及びR4’が、共に水素原子であるか、または、R3’及びR4’が、互いに結合して、それらが結合している炭素原子と一緒になって、シクロヘキサン環を形成してもよく、
結合b’が、単結合であり、
X’が、CH2であり、
Y’が、N(OCH3)であり、
m’が、1であり、且つ
n’が、1である、化合物(II)。
別の好適な化合物(II)は、以下の化合物である。
[化合物(IID)]
R’が、トリチル基で置換されたイミダゾリル基であり、
R1’が、メチル基であり、
R2’が、tert-ブチル基で置換されたフェニル基であり、
R3’及びR4’が、共に水素原子であり、
結合b’が、単結合であり、
X’が、CH2又はカルボニルであり、
Y’が、酸素原子であり、
m’が、1であり、且つ
n’が、0又は1である、化合物(II)。
[化合物(IID)]
R’が、トリチル基で置換されたイミダゾリル基であり、
R1’が、メチル基であり、
R2’が、tert-ブチル基で置換されたフェニル基であり、
R3’及びR4’が、共に水素原子であり、
結合b’が、単結合であり、
X’が、CH2又はカルボニルであり、
Y’が、酸素原子であり、
m’が、1であり、且つ
n’が、0又は1である、化合物(II)。
好適な化合物(II)の具体例は、後述する実施例3、12、14~34の化合物(化合物(3)、化合物(12)、化合物(14)~化合物(34))、又はそれらの医薬上許容される塩である。
より好適な化合物(II)は、具体的には、以下の化合物、又はそれらの医薬上許容される塩である。

(化合物(I)の合成)
以下、本発明化合物(化合物(I))の製造法について説明する。
化合物(I)の製造法の例として、代表的な製造法を以下に述べるが、製造法はこれらに限定されない。
以下、本発明化合物(化合物(I))の製造法について説明する。
化合物(I)の製造法の例として、代表的な製造法を以下に述べるが、製造法はこれらに限定されない。
化合物(I)(例えば、後述する実施例に記載の化合物(1)~化合物(15))は、前記した非特許文献7(Shimamoto Y, et al. Bioorg. Med. Chem., 2015, 23, 876-890)及び8(Teruya, K, et al. Peptide Science, 2015, 106(4), 391-402)に記載の自体公知の方法又はそれに準ずる方法に従って、製造することができる。
化合物(I)は、下記の製造法1~4で示される方法、後述する実施例、又はそれらに準じた方法等により製造することも可能である。
各原料化合物は、反応を阻害しないのであれば、塩を形成していてもよく、かかる塩としては、化合物(I)の塩と同様のものが挙げられる。
原料化合物は具体的製法を述べない場合、市販されているものを容易に入手して用いることができるか、又は自体公知の方法、又はそれに準ずる方法に従って製造することもできる。また以下の製造法において生成する中間体はカラムクロマトグラフィー、再結晶、蒸留等の方法で単離精製してもよく、あるいは単離せずに次の工程に用いても良い。
以下にその反応式の略図を示すが、略図中の化合物の各記号は前記と同義を示す。
原料化合物は具体的製法を述べない場合、市販されているものを容易に入手して用いることができるか、又は自体公知の方法、又はそれに準ずる方法に従って製造することもできる。また以下の製造法において生成する中間体はカラムクロマトグラフィー、再結晶、蒸留等の方法で単離精製してもよく、あるいは単離せずに次の工程に用いても良い。
以下にその反応式の略図を示すが、略図中の化合物の各記号は前記と同義を示す。
下記製造法及び実施例においては、以下の略号を使用する。
Trt:トリチル
Fmoc:9-フルオレニルメチルオキシカルボニル
homoPro-OH:ホモプロリン
Hyp-OH:ヒドロキシプロリン
Tic-OH:1,2,3,4-テトラヒドロイソキノリン-3-カルボン酸
HCl H-His(Trt)-OMe:トリチルヒスチジンメチルエステル塩酸塩
Ser(Trt):O-トリチル-L-セリン
Ser(Bn):O-ベンジル-L-セリン
Trt:トリチル
Fmoc:9-フルオレニルメチルオキシカルボニル
homoPro-OH:ホモプロリン
Hyp-OH:ヒドロキシプロリン
Tic-OH:1,2,3,4-テトラヒドロイソキノリン-3-カルボン酸
HCl H-His(Trt)-OMe:トリチルヒスチジンメチルエステル塩酸塩
Ser(Trt):O-トリチル-L-セリン
Ser(Bn):O-ベンジル-L-セリン
[製造法1]
製造法1は、化合物(1a)又は(1b)とトリチルヒスチジンメチルエステル塩酸塩(2a)を縮合させた後、脱Fmoc化後、4-t-ブチル安息香酸(4a)と縮合させることにより化合物(I)を得る方法である。後述する実施例に記載の化合物(16)~(19)、化合物(23)、及び化合物(25)~(33)は、本製造法により合成することができる。
化合物(1a)又は(1b)は、市販のものを用いるか、又は自体公知の方法若しくはこれらに準じた方法で製造することができる。
製造法1は、化合物(1a)又は(1b)とトリチルヒスチジンメチルエステル塩酸塩(2a)を縮合させた後、脱Fmoc化後、4-t-ブチル安息香酸(4a)と縮合させることにより化合物(I)を得る方法である。後述する実施例に記載の化合物(16)~(19)、化合物(23)、及び化合物(25)~(33)は、本製造法により合成することができる。
化合物(1a)又は(1b)は、市販のものを用いるか、又は自体公知の方法若しくはこれらに準じた方法で製造することができる。

(工程1)
本工程は、縮合剤及び塩基存在下で、化合物(1a)又は(1b)とトリチルヒスチジンメチルエステル塩酸塩(2a)を縮合させることにより、化合物(3a)又は(3b)を製造する工程である。本工程は、必要に応じて、縮合添加剤の存在下で反応を行ってもよい。
本工程は、縮合剤及び塩基存在下で、化合物(1a)又は(1b)とトリチルヒスチジンメチルエステル塩酸塩(2a)を縮合させることにより、化合物(3a)又は(3b)を製造する工程である。本工程は、必要に応じて、縮合添加剤の存在下で反応を行ってもよい。
使用する縮合剤としては、1-エチル-3-(3’-ジメチルアミノプロピル)カルボジイミド(WSC)、ジシクロヘキシルカルボジイミド(DCC)、ジイソプロピルカルボジイミド(DIC)、N-エチル-N’-3-ジメチルアミノプロピルカルボジイミド及びその塩酸塩(EDC・HCl)、ベンゾトリアゾール-1-イルオキシ-トリスジメチルアミノホスホニウム塩(BOP)、{{[(1-シアノ-2-エトキシ-2-オキソエチリデン)アミノ]オキシ}-4-モルホリノメチレン}ジメチルアンモニウムヘキサフルオロりん酸塩(COMU)、ヘキサフルオロリン酸(ベンゾトリアゾール-1-イルオキシ)トリピロリジノホスホニウム(PyBop)、O-(ベンゾトリアゾール-1-イル)-N,N,N’,N’-テトラメチルウロニウム テトラフルオロボレート(TBTU)、1-[ビス(ジメチルアミノ)メチレン]-5-クロロ-1H-ベンゾトリアゾリウム3-オキシド ヘキサフルオロホスフェート(HCTU)、O-ベンゾトリアゾール-N,N,N’,N’-テトラメチルウロニウム ヘキサフルオロボレート(HBTU)等が挙げられる。中でも、BOP又はCOMUが好ましい。
縮合剤の使用量は、化合物(1a)又は(1b)1モルに対して、通常1~3モル使用することができ、好ましくは1~1.5モルである。
化合物(2a)の使用量は、化合物(1a)又は(1b)1モルに対して、通常1~3モル使用することができ、好ましくは1~1.5モルである。
縮合添加剤としては、1-ヒドロキシベンゾトリアゾール(HOBt)、1-ヒドロキシ-1H-1,2,3-トリアゾール-5-カルボン酸エチルエステル(HOCt)、1-ヒドロキシ-7-アザベンゾトリアゾール(HOAt)等が挙げられるが、縮合剤として、BOP又はCOMUを使用する場合は不要である。
縮合添加剤の使用量は、化合物(1a)又は(1b)1モルに対して、好ましくは0.05~1.5モルである。
塩基としては、トリエチルアミン、ピリジン、N,N-ジイソプロピルエチルアミン、N-メチルモルホリン等の有機塩基、水素化ナトリウム等の無機塩基が挙げられる。中でも、N-メチルモルホリンが好ましい。
塩基の使用量は、化合物(1a)又は(1b)1モルに対して、通常1~10モル、好ましくは、5~8モルである。
縮合剤の使用量は、化合物(1a)又は(1b)1モルに対して、通常1~3モル使用することができ、好ましくは1~1.5モルである。
化合物(2a)の使用量は、化合物(1a)又は(1b)1モルに対して、通常1~3モル使用することができ、好ましくは1~1.5モルである。
縮合添加剤としては、1-ヒドロキシベンゾトリアゾール(HOBt)、1-ヒドロキシ-1H-1,2,3-トリアゾール-5-カルボン酸エチルエステル(HOCt)、1-ヒドロキシ-7-アザベンゾトリアゾール(HOAt)等が挙げられるが、縮合剤として、BOP又はCOMUを使用する場合は不要である。
縮合添加剤の使用量は、化合物(1a)又は(1b)1モルに対して、好ましくは0.05~1.5モルである。
塩基としては、トリエチルアミン、ピリジン、N,N-ジイソプロピルエチルアミン、N-メチルモルホリン等の有機塩基、水素化ナトリウム等の無機塩基が挙げられる。中でも、N-メチルモルホリンが好ましい。
塩基の使用量は、化合物(1a)又は(1b)1モルに対して、通常1~10モル、好ましくは、5~8モルである。
本反応は、反応に影響を及ぼさない溶媒中で行なうことができる。反応溶媒としては、特に限定されないが、例えば、トルエン、キシレン等の芳香族炭化水素類;N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド系溶媒;ジエチルエーテル、テトラヒドロフラン、ジオキサン等のエーテル類;クロロホルム、ジクロロメタン等のハロゲン化炭化水素類等あるいはそれらの混合物が挙げられる。中でも、ジクロロメタンが好ましい。
反応温度は、通常-10~30℃、好ましくは0℃~20℃であり、反応時間は、通常1~30時間程度である。
(工程2)
本工程は、化合物(3a)又は(3b)を脱Fmoc化(脱保護)する工程である。本工程は、反応に影響を及ぼさない溶媒中で行うことができる。
本工程は、化合物(3a)又は(3b)を脱Fmoc化(脱保護)する工程である。本工程は、反応に影響を及ぼさない溶媒中で行うことができる。
脱保護反応における反応条件(反応剤、反応溶媒、反応温度、反応時間等)は、特に限定されないが、例えば、Protective Groups in Organic Synthesis, 4th Ed., Theodora W. Greene, Peter G. M. Wuts, Wiley-Interscience (2007)に記載の方法、又は本明細書中の実施例、或いはこれらに準ずる方法に従って行うことができる。具体的には、有機塩基で処理することにより脱Fmoc化(脱保護)することができる。
有機塩基としては、ジエチルアミン、1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(DBU)、1,4-ジアザビシクロ[2.2.2]オクタン(DABCO)、1,5-ジアザビシクロ[4.3.0]-5-ノネン(DBN)等が挙げられ、好ましくは、ジエチルアミンである。溶媒としては、例えば、クロロホルム、ジクロロメタン、1,2-ジクロロエタン等のハロゲン系溶媒;トルエン、キシレン等の芳香族炭化水素類;ジエチルエーテル、CPME、THF、1,4-ジオキサン等のエーテル系溶媒;アセトニトリル等のニトリル類等、あるいはそれらの混合物が挙げられ、好ましくは、アセトニトリルである。
有機塩基の使用量は、化合物(3a)又は(3b)1モルに対して、通常、過剰量(10モル以上)である。
有機塩基の使用量は、化合物(3a)又は(3b)1モルに対して、通常、過剰量(10モル以上)である。
反応温度は、通常-10~40℃、好ましくは10℃~30℃であり、反応時間は、通常0.2~2時間程度である。
(工程3)
本工程は、工程2で得られた脱保護体を、縮合剤及び塩基存在下で、4-t-ブチル安息香酸(4a)と縮合させることにより、化合物(I)を製造する工程である。本工程は、前記工程1と同様の反応条件下で行うことができる。
本工程は、工程2で得られた脱保護体を、縮合剤及び塩基存在下で、4-t-ブチル安息香酸(4a)と縮合させることにより、化合物(I)を製造する工程である。本工程は、前記工程1と同様の反応条件下で行うことができる。
化合物(4a)の使用量は、化合物(3a)又は(3b)1モルに対して、通常1~3モル使用することができ、好ましくは1~1.5モルである。
[製造法2]
製造法2は、化合物(1a)又は(1b)とN-メトキシ‐N-メチルアミン塩酸塩(5a)を縮合後、アルデヒド(7a)又は(7b)への還元反応を経て、ヒスチジンメチルエステル塩酸塩(2a)、続くシアノ水素化ホウ素ナトリウムの添加による還元的アミノ化反応により化合物(8a)又は(8b)を得、脱Fmoc化後に4-t-ブチル安息香酸(4a)と縮合させることにより化合物(I)を得る方法である。後述する実施例に記載の化合物(20)~化合物(22)及び化合物(24)は、本製造法により合成することができる。
製造法2は、化合物(1a)又は(1b)とN-メトキシ‐N-メチルアミン塩酸塩(5a)を縮合後、アルデヒド(7a)又は(7b)への還元反応を経て、ヒスチジンメチルエステル塩酸塩(2a)、続くシアノ水素化ホウ素ナトリウムの添加による還元的アミノ化反応により化合物(8a)又は(8b)を得、脱Fmoc化後に4-t-ブチル安息香酸(4a)と縮合させることにより化合物(I)を得る方法である。後述する実施例に記載の化合物(20)~化合物(22)及び化合物(24)は、本製造法により合成することができる。
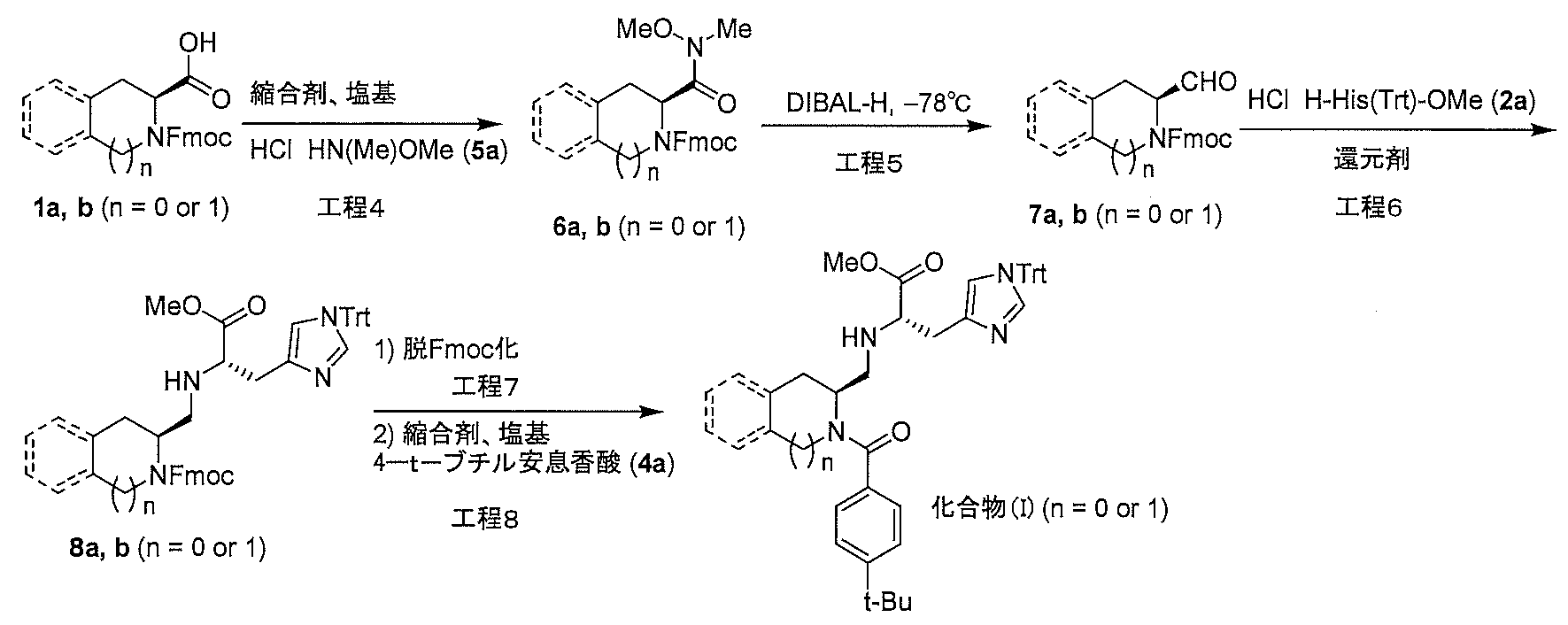
(工程4)
本工程は、縮合剤及び塩基存在下で、化合物(1a)又は(1b)とN-メトキシ‐N-メチルアミン塩酸塩(5a)を縮合させることにより、化合物(6a)又は(6b)を製造する工程である。本工程は、前記製造法1の工程1と同様の反応条件下で行うことができる。
本工程は、縮合剤及び塩基存在下で、化合物(1a)又は(1b)とN-メトキシ‐N-メチルアミン塩酸塩(5a)を縮合させることにより、化合物(6a)又は(6b)を製造する工程である。本工程は、前記製造法1の工程1と同様の反応条件下で行うことができる。
化合物(5a)の使用量は、化合物(1a)又は(1b)1モルに対して、通常1~3モル使用することができ、好ましくは1~1.5モルである。
(工程5)
本工程は、化合物(6a)又は(6b)のアミド基を水素化ジイソブチルアルミニウム(DIBAL-H)で処理することにより、対応するアルデヒド体(7a)又は(7b)に変換する工程である。
本工程は、化合物(6a)又は(6b)のアミド基を水素化ジイソブチルアルミニウム(DIBAL-H)で処理することにより、対応するアルデヒド体(7a)又は(7b)に変換する工程である。
DIBAL-Hの使用量は、化合物(6a)又は(6b)1モルに対して、通常1~3モル使用することができ、好ましくは1~1.5モルである。
本反応は、反応に影響を及ぼさない溶媒中で行なうことができる。反応溶媒としては、特に限定されないが、例えば、クロロホルム、ジクロロメタン等のハロゲン化炭化水素類等が挙げられる。中でも、ジクロロメタンが好ましい。
反応温度は、通常-78℃~0℃であり、好ましくは、-78℃である。
反応時間は、通常0.2~2時間程度である。
反応時間は、通常0.2~2時間程度である。
(工程6)
本工程は、化合物(7a)又は(7b)と化合物(2a)を反応させた後に還元剤で処理することにより、還元的アミノ化を行い、化合物(8a)又は(8b)を製造する工程である。
化合物(2a)の使用量は、化合物(7a)又は(7b)に対して、通常1~3モル使用することができ、好ましくは1~1.5モルである。
本工程は、化合物(7a)又は(7b)と化合物(2a)を反応させた後に還元剤で処理することにより、還元的アミノ化を行い、化合物(8a)又は(8b)を製造する工程である。
化合物(2a)の使用量は、化合物(7a)又は(7b)に対して、通常1~3モル使用することができ、好ましくは1~1.5モルである。
還元剤としては、水素化ホウ素ナトリウム、水素化シアノホウ素ナトリウム、トリアセトキシ水素化ホウ素ナトリウム等が挙げられ、好ましくは、水素化シアノホウ素ナトリウムである。
還元剤の使用量は、化合物(7a)又は(7b)に対して、通常1~5当量、好ましくは1~3当量である。
還元剤の使用量は、化合物(7a)又は(7b)に対して、通常1~5当量、好ましくは1~3当量である。
本反応は、通常、不活性溶媒中で行われる。不活性溶媒としては、例えば、テトラヒドロフラン、ジエチルエーテル、1,2-ジメトキシエタン、1,4-ジオキサン、ジクロロメタン、トルエン、ベンゼン、キシレン、メタノール、エタノール、N,N-ジメチルホルムアミド、ジメチルスルホキシド等が挙げられるが、ジクロロメタンが特に好ましい。
反応温度は、通常、-20~80℃、好ましくは氷冷下~20℃である。
反応時間は、特に限定されないが、通常0.1~24時間、好ましくは0.5~3時間である。
反応時間は、特に限定されないが、通常0.1~24時間、好ましくは0.5~3時間である。
(工程7)
本工程は、化合物(8a)又は(8b)を脱Fmoc化(脱保護)する工程である。本工程は、前記製造法1の工程2と同様の反応条件下で行うことができる。
本工程は、化合物(8a)又は(8b)を脱Fmoc化(脱保護)する工程である。本工程は、前記製造法1の工程2と同様の反応条件下で行うことができる。
(工程8)
本工程は、工程7で得られた脱保護体を、縮合剤及び塩基存在下で、4-t-ブチル安息香酸(4a)と縮合させることにより、化合物(I)を製造する工程である。本工程は、前記製造法1の工程3と同様の反応条件下で行うことができる。
本工程は、工程7で得られた脱保護体を、縮合剤及び塩基存在下で、4-t-ブチル安息香酸(4a)と縮合させることにより、化合物(I)を製造する工程である。本工程は、前記製造法1の工程3と同様の反応条件下で行うことができる。
化合物(4a)の使用量は、化合物(8a)又は(8b)1モルに対して、通常1~3モル使用することができ、好ましくは1~1.5モルである。
[製造法3]
製造法3は、化合物(9a)と化合物(10a)を縮合後、脱Fmoc化を経て、1-Fmoc保護ホモプロリン(Fmoc-homoPro-OH)(12a)と縮合させて化合物 (13a)を得、脱Fmoc化後に4-t-ブチル安息香酸クロリド(14a)と縮合させることにより化合物(I)を得る方法である。後述する実施例に記載の化合物(25)~化合物(33)は、本製造法により合成することができる。
製造法3は、化合物(9a)と化合物(10a)を縮合後、脱Fmoc化を経て、1-Fmoc保護ホモプロリン(Fmoc-homoPro-OH)(12a)と縮合させて化合物 (13a)を得、脱Fmoc化後に4-t-ブチル安息香酸クロリド(14a)と縮合させることにより化合物(I)を得る方法である。後述する実施例に記載の化合物(25)~化合物(33)は、本製造法により合成することができる。

(工程9)
本工程は、縮合剤及び塩基存在下で、化合物(9a)と化合物(10a)を縮合させることにより、化合物(11a)を製造する工程である。本工程は、前記製造法1の工程1と同様の反応条件下で行うことができる。
本工程は、縮合剤及び塩基存在下で、化合物(9a)と化合物(10a)を縮合させることにより、化合物(11a)を製造する工程である。本工程は、前記製造法1の工程1と同様の反応条件下で行うことができる。
化合物(10a)の使用量は、化合物(9a)1モルに対して、通常1~3モル使用することができ、好ましくは1~2モルである。
(工程10)
本工程は、化合物(11a)を脱Fmoc化(脱保護)する工程である。本工程は、前記製造法1の工程2と同様の反応条件下で行うことができる。
本工程は、化合物(11a)を脱Fmoc化(脱保護)する工程である。本工程は、前記製造法1の工程2と同様の反応条件下で行うことができる。
(工程11)
本工程は、工程10で得られた脱保護体を、縮合剤及び塩基存在下で、化合物(12a)と縮合させることにより、化合物(13a)を製造する工程である。本工程は、前記製造法1の工程3と同様の反応条件下で行うことができる。
本工程は、工程10で得られた脱保護体を、縮合剤及び塩基存在下で、化合物(12a)と縮合させることにより、化合物(13a)を製造する工程である。本工程は、前記製造法1の工程3と同様の反応条件下で行うことができる。
化合物(12a)の使用量は、化合物(11a)1モルに対して、通常1~3モル使用することができ、好ましくは1~1.5モルである。
(工程12)
本工程は、化合物(13a)を脱Fmoc化(脱保護)する工程である。本工程は、前記製造法1の工程2と同様の反応条件下で行うことができる。
本工程は、化合物(13a)を脱Fmoc化(脱保護)する工程である。本工程は、前記製造法1の工程2と同様の反応条件下で行うことができる。
(工程13)
本工程は、工程12で得られた脱保護体を、N,N-ジメチル-4-アミノピリジン(DMAP)及び塩基存在下、化合物(14a)と縮合させることにより、化合物(I)を製造する工程である。
本工程は、工程12で得られた脱保護体を、N,N-ジメチル-4-アミノピリジン(DMAP)及び塩基存在下、化合物(14a)と縮合させることにより、化合物(I)を製造する工程である。
化合物(14a)の使用量は、化合物(13a)1モルに対して、通常1~3モル使用することができ、好ましくは1~1.5モルである。
DMAPの使用量は、化合物(13a)1モルに対して、通常1~5モル使用することができ、好ましくは1~3モルである。
塩基としては、トリエチルアミン、ピリジン、N,N-ジイソプロピルエチルアミン、N-メチルモルホリン等の有機塩基等が挙げられる。中でも、ピリジンが好ましい。
塩基は、化合物(13a)1モルに対して、通常、過剰量(10モル以上)であり、塩基を溶媒として使用するのが特に好ましい。
塩基は、化合物(13a)1モルに対して、通常、過剰量(10モル以上)であり、塩基を溶媒として使用するのが特に好ましい。
本反応は、反応に影響を及ぼさない溶媒共存下でも行なうことができる。反応溶媒としては、特に限定されないが、例えば、トルエン、キシレン等の芳香族炭化水素類;N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド系溶媒;ジエチルエーテル、テトラヒドロフラン等のエーテル類;クロロホルム、ジクロロメタン等のハロゲン化炭化水素類等が挙げられる。
反応温度は、通常0~60℃、好ましくは10℃~30℃であり、反応時間は、通常1~30時間程度である。
[製造法4]
製造法4は、1-Fmoc-2-ヒドロキシプロリン(15a)とトリチルヒスチジンメチルエステル塩酸塩(2a)を縮合させて化合物(16a)を得、脱Fmoc化後に4-t-ブチル安息香酸(4a)と縮合させることにより化合物(I)を得る方法である。後述する実施例に記載の化合物(34)は、本製造法により合成することができる。
製造法4は、1-Fmoc-2-ヒドロキシプロリン(15a)とトリチルヒスチジンメチルエステル塩酸塩(2a)を縮合させて化合物(16a)を得、脱Fmoc化後に4-t-ブチル安息香酸(4a)と縮合させることにより化合物(I)を得る方法である。後述する実施例に記載の化合物(34)は、本製造法により合成することができる。

(工程14)
本工程は、縮合剤及び塩基存在下で、化合物(15a)と化合物(2a)を縮合させることにより、化合物(16a)を製造する工程である。本工程は、前記製造法1の工程1と同様の反応条件下で行うことができる。
本工程は、縮合剤及び塩基存在下で、化合物(15a)と化合物(2a)を縮合させることにより、化合物(16a)を製造する工程である。本工程は、前記製造法1の工程1と同様の反応条件下で行うことができる。
化合物(2a)の使用量は、化合物(15a)1モルに対して、通常1~3モル使用することができ、好ましくは1~1.5モルである。
(工程15)
本工程は、化合物(16a)を脱Fmoc化(脱保護)する工程である。本工程は、前記製造法1の工程2と同様の反応条件下で行うことができる。
本工程は、化合物(16a)を脱Fmoc化(脱保護)する工程である。本工程は、前記製造法1の工程2と同様の反応条件下で行うことができる。
(工程16)
本工程は、工程15で得られた脱保護体を、縮合剤及び塩基存在下で、化合物(4a)と縮合させることにより、化合物(I)を製造する工程である。本工程は、前記製造法1の工程3と同様の反応条件下で行うことができる。
本工程は、工程15で得られた脱保護体を、縮合剤及び塩基存在下で、化合物(4a)と縮合させることにより、化合物(I)を製造する工程である。本工程は、前記製造法1の工程3と同様の反応条件下で行うことができる。
化合物(4a)の使用量は、化合物(16a)1モルに対して、通常1~3モル使用することができ、好ましくは1~1.5モルである。
上記のような方法により生成した化合物(I)(及び化合物(I)に包含される化合物(II))は、例えば、再結晶、蒸留、クロマトグラフィー等の通常の分離手段により単離、精製することができる。
本発明の化合物(I)が、光学異性体、立体異性体、位置異性体、回転異性体を含有する場合には、これらも化合物(I)として含有されるとともに、自体公知の合成手法、分離手法(濃縮、溶媒抽出、カラムクロマトグラフィー、再結晶等)によりそれぞれを単品として得ることができる。例えば、化合物(I)に光学異性体が存在する場合には、該化合物から分割された光学異性体も化合物(I)に包含される。
本発明の化合物(I)、又はその医薬上許容される塩が、光学異性体、立体異性体、位置異性体、回転異性体を含有する場合には、これら全ての異性体、及びいずれの比率のこれら異性体混合物をも化合物(I)として包含される。また、これら異性体は、自体公知の合成手法、分離手法(濃縮、溶媒抽出、カラムクロマトグラフィー、再結晶等)によりそれぞれを単品として得ることができる。また、本発明の化合物(I)は、ラベル体、すなわち、本発明の化合物(I)を構成する1又は2以上の原子を同位元素(例えば、2H、3H、11C、13C、14C、15N、18O、18F、35S等)で標識された化合物も包含される。
光学異性体は自体公知の方法により製造することができる。具体的には、光学活性な合成中間体を用いる、又は、最終物のラセミ体を常法(例えば、J.Jacquesらの、「Enantiomers, Racemates and Resolution, John Wiley And Sons,Inc.」等参照)に従って光学分割することにより光学異性体を得ることができる。
本発明の化合物(I)、又はその医薬上許容される塩は、結晶であってもよく、結晶形が単一であっても結晶形混合物であっても化合物(I)に包含される。結晶は、自体公知の結晶化法を適用して、結晶化することによって製造することができる。
本発明の化合物(I)、又はその医薬上許容される塩には、それらの溶媒和物も包含され得る。化合物(I)、又はその医薬上許容される塩には、それらの溶媒和物とは、化合物(I)又はその塩に、溶媒の分子が配位したものであり、水和物も包含される。例えば、化合物(I)またはその塩の水和物、エタノール和物、ジメチルスルホキシド和物等が挙げられる。
本発明の化合物(I)、又はその医薬上許容される塩は、後述する試験例に示されるように、優れたWntシグナル伝達経路阻害作用を示すと共に、各種腫瘍細胞に対する増殖抑制効果を示すことから、例えば、癌、冠状動脈疾患、急性冠症候群、変形性関節症等の予防又は治療に有用であり、とりわけ、各種癌(例えば、大腸癌、乳癌、頭頸部癌、肺癌、胃癌、食道癌、悪性黒色腫、前立腺癌、肝細胞癌、卵巣癌、乳癌、肝芽腫、急性骨髄性白血病、急性リンパ芽球性白血病、慢性骨髄性白血病、慢性リンパ性白血病、多発性骨髄腫、悪性リンパ腫、膵臓癌、腎臓癌、膀胱癌、神経膠芽腫、髄芽腫、ウィルムス腫瘍等)の予防又は治療に特に有用である。
本発明の化合物(I)、又はその医薬上許容される塩からなるWntシグナル伝達経路阻害剤を有効成分として含有してなる医薬(以下、「本発明の医薬」と称することもある。)は、当該Wntシグナル伝達経路阻害剤のみからなる医薬、又は当該Wntシグナル伝達経路阻害剤と医薬上許容される担体等を配合した医薬のいずれでもよい。本発明の医薬は、その予防有効量又は治療有効量を哺乳動物(例えば、マウス、ラット、ハムスター、ウサギ、ネコ、イヌ、ウシ、ヒツジ、サル、ヒト等)に投与することができる。
医薬上許容される担体としては、例えば、賦形剤(例えば、デンプン、乳糖、砂糖、炭酸カルシウム、リン酸カルシウム等)、結合剤(例えば、デンプン、アラビアゴム、カルボキシメチルセルロース、ヒドロキシプロピルセルロース、結晶セルロース等)、滑沢剤(例えば、ステアリン酸マグネシウム、タルク等)、崩壊剤(例えば、カルボキシメチルセルロース、タルク等)、溶剤(例えば、注射用水、生理的食塩水、リンゲル液、アルコール、プロピレングリコール、ポリエチレングリコール、ゴマ油、トウモロコシ油、オリーブ油、綿実油等)、溶解補助剤(例えば、ポリエチレングリコール、プロピレングリコール、D-マンニトール、トレハロース、安息香酸ベンジル、エタノール、トリスアミノメタン、コレステロール、トリエタノールアミン、炭酸ナトリウム、クエン酸ナトリウム、サリチル酸ナトリウム、酢酸ナトリウム等)、懸濁化剤(例えば、ステアリルトリエタノールアミン、ラウリル硫酸ナトリウム、ラウリルアミノプロピオン酸、レシチン、塩化ベンザルコニウム、塩化ベンゼトニウム、モノステアリン酸グリセリン等の界面活性剤;ポリビニルアルコール、ポリビニルピロリドン、カルボキシメチルセルロースナトリウム、メチルセルロース、ヒドロキシメチルセルロース、ヒドロキシエチルセルロース、ヒドロキシプロピルセルロース等の親水性高分子;ポリソルベート類、ポリオキシエチレン硬化ヒマシ油等)、等張化剤(例えば、塩化ナトリウム、グリセリン、D-マンニトール、D-ソルビトール、ブドウ糖等)、緩衝剤(例えば、リン酸塩、酢酸塩、炭酸塩、クエン酸塩等の緩衝液等)、無痛化剤(例えば、ベンジルアルコール等)、防腐剤(例えば、パラオキシ安息香酸エステル類、クロロブタノール、ベンジルアルコール、フェネチルアルコール、デヒドロ酢酸、ソルビン酸等)、抗酸化剤(例えば、亜硫酸塩、アスコルビン酸塩等)、着色剤(例えば、水溶性食用タール色素(例、食用赤色2号及び3号、食用黄色4号及び5号、食用青色1号及び2号等の食用色素)、水不溶性レーキ色素(例、前記水溶性食用タール色素のアルミニウム塩)、天然色素(例、β-カロチン、クロロフィル、ベンガラ)等)、甘味剤(例えば、サッカリンナトリウム、グリチルリチン酸二カリウム、アスパルテーム、ステビア等)等が挙げられる。
本発明の医薬は、上記諸成分を混合した後、混合物を自体公知の手段に従い、例えば、カプセル剤、錠剤、細粒剤、顆粒剤、ドライシロップ等の経口投与用、または注射剤(例えば、皮下注射剤、静脈内注射剤、筋肉内注射剤、腹腔内注射剤、点滴剤等)、外用剤(例、経皮吸収型製剤、軟膏剤、ローション剤、貼付剤)、坐剤(例、直腸坐剤、膣坐剤)、ペレット、経鼻剤、経肺剤(吸入剤)、点眼剤等の非経口投与用の製剤とすることができる。
本発明の医薬としては、中でも、注射剤等の非経口投与用の製剤が好ましい。
本発明の医薬としては、中でも、注射剤等の非経口投与用の製剤が好ましい。
本発明の医薬(製剤)中の本発明の化合物(I)、又はその医薬上許容される塩の含有量は、製剤の形態によって相違するが、通常製剤全体に対して約0.01乃至100重量%の範囲であり、好ましくは約0.1乃至50重量%の範囲であり、さらに好ましくは約0.5乃至20重量%程度の範囲である。
本発明の化合物(I)、又はその医薬上許容される塩の投与量は、投与対象、症状、その他の要因によって異なるが、例えば、成人の癌患者に対して非経口投与する場合、通常1回量として約0.01~200mg/kg体重、好ましくは0.1~100mg/kg体重、さらに好ましくは1~50mg/kg体重であり、この量を1日1回~3回投与するのが望ましい。
本発明の化合物(I)、又はその医薬上許容される塩は、その薬効を損なわない限り、他の薬剤、例えば、既存の抗癌剤(併用薬)と併用することができる。この際、投与時期は限定されず、これらを投与対象に対し、同時に投与してもよいし、時間差をおいて投与してもよい。また、本発明の化合物(I)と併用薬とを組み合わせて含有する単一の製剤として投与することもできる。
併用薬の投与量は、臨床上用いられている用量を基準として適宜選択することができる。また、本発明の化合物またはその医薬上許容される塩等と併用薬の配合比は、投与対象、投与ルート、対象疾患、症状、組み合わせ等に応じて適宜選択することができる。
既存の抗癌剤としては、例えば、化学療法剤、ホルモン療法剤、免疫療法剤、分子標的薬、免疫チェックポイント阻害剤(抗PD-1抗体、抗PD-L1抗体)等が挙げられる。
「化学療法剤」としては、例えば、アルキル化剤、代謝拮抗剤、抗癌性抗生物質、植物由来抗癌剤等が用いられる。
「アルキル化剤」としては、例えば、ナイトロジェンマスタード、塩酸ナイトロジェンマスタード-N-オキシド、クロラムブチル、シクロフォスファミド、イホスファミド、チオテパ、カルボコン、トシル酸インプロスルファン、ブスルファン、塩酸ニムスチン、ミトブロニトール、メルファラン、ダカルバジン、ラニムスチン、リン酸エストラムスチンナトリウム、トリエチレンメラミン、カルムスチン、ロムスチン、ストレプトゾシン、ピポブロマン、エトグルシド、カルボプラチン、シスプラチン、ミボプラチン、ネダプラチン、オキサリプラチン、アルトレタミン、アンバムスチン、塩酸ジブロスピジウム、フォテムスチン、プレドニムスチン、プミテパ、リボムスチン、テモゾロミド、トレオスルファン、トロフォスファミド、ジノスタチンスチマラマー、アドゼレシン、システムスチン、ビゼレシン、及びそれらのDDS製剤等が用いられる。
「化学療法剤」としては、例えば、アルキル化剤、代謝拮抗剤、抗癌性抗生物質、植物由来抗癌剤等が用いられる。
「アルキル化剤」としては、例えば、ナイトロジェンマスタード、塩酸ナイトロジェンマスタード-N-オキシド、クロラムブチル、シクロフォスファミド、イホスファミド、チオテパ、カルボコン、トシル酸インプロスルファン、ブスルファン、塩酸ニムスチン、ミトブロニトール、メルファラン、ダカルバジン、ラニムスチン、リン酸エストラムスチンナトリウム、トリエチレンメラミン、カルムスチン、ロムスチン、ストレプトゾシン、ピポブロマン、エトグルシド、カルボプラチン、シスプラチン、ミボプラチン、ネダプラチン、オキサリプラチン、アルトレタミン、アンバムスチン、塩酸ジブロスピジウム、フォテムスチン、プレドニムスチン、プミテパ、リボムスチン、テモゾロミド、トレオスルファン、トロフォスファミド、ジノスタチンスチマラマー、アドゼレシン、システムスチン、ビゼレシン、及びそれらのDDS製剤等が用いられる。
「代謝拮抗剤」としては、例えば、メルカプトプリン、6-メルカプトプリンリボシド、チオイノシン、メトトレキサート、ペメトレキセド、エノシタビン、シタラビン、シタラビンオクフォスファート、塩酸アンシタビン、5-FU系薬剤(例、フルオロウラシル、テガフール、UFT、ドキシフルリジン、カルモフール、ガロシタビン、エミテフール、カペシタビン)、アミノプテリン、ネルザラビン、ロイコボリンカルシウム、タブロイド、ブトシン、フォリネイトカルシウム、レボフォリネイトカルシウム、クラドリビン、エミテフール、フルダラビン、ゲムシタビン、ヒドロキシカルバミド、ペントスタチン、ピリトレキシム、イドキシウリジン、ミトグアゾン、チアゾフリン、アンバムスチン、ベンダムスチン、及びそれらのDDS製剤等が用いられる。
「抗癌性抗生物質」としては、例えば、アクチノマイシンD、アクチノマイシンC、マイトマイシンC、クロモマイシンA3、塩酸ブレオマイシン、硫酸ブレオマイシン、硫酸ペプロマイシン、塩酸ダウノルビシン、塩酸ドキソルビシン、塩酸アクラルビシン、塩酸ピラルビシン、塩酸エピルビシン、ネオカルチノスタチン、ミスラマイシン、ザルコマイシン、カルチノフィリン、ミトタン、塩酸ゾルビシン、塩酸ミトキサントロン、塩酸イダルビシン、及びそれらのDDS製剤等が用いられる。
「植物由来抗癌剤」としては、例えば、エトポシド、リン酸エトポシド、硫酸ビンブラスチン、硫酸ビンクリスチン、硫酸ビンデシン、テニポシド、パクリタキセル、ドセタクセル、ビノレルビン、及びそれらのDDS製剤等が用いられる。
「ホルモン療法剤」としては、例えば、ホスフェストロール、ジエチルスチルベストロール、クロロトリアニセン、酢酸メドロキシプロゲステロン、酢酸メゲストロール、酢酸クロルマジノン、酢酸シプロテロン、ダナゾール、アリルエストレノール、ゲストリノン、メパルトリシン、ラロキシフェン、オルメロキシフェン、レボルメロキシフェン、抗エストロゲン(例、クエン酸タモキシフェン、クエン酸トレミフェン)、ピル製剤、メピチオスタン、テストロラクトン、アミノグルテチイミド、LH-RHアゴニスト(例、酢酸ゴセレリン、ブセレリン、リュープロレリン)、ドロロキシフェン、エピチオスタノール、スルホン酸エチニルエストラジオール、アロマターゼ阻害薬(例、塩酸ファドロゾール、アナストロゾール、レトロゾール、エキセメスタン、ボロゾール、フォルメスタン)、抗アンドロゲン(例、フルタミド、ビカルタミド、ニルタミド)、5α-レダクターゼ阻害薬(例、フィナステリド、エプリステリド)、副腎皮質ホルモン系薬剤(例、デキサメタゾン、プレドニゾロン、ベタメタゾン、トリアムシノロン)、アンドロゲン合成阻害薬(例、アビラテロン)、レチノイド及びレチノイドの代謝を遅らせる薬剤(例、リアロゾール)等が用いられる。
「免疫療法剤」としては、生物反応修飾物質(例えば、ピシバニール、クレスチン、シゾフィラン、レンチナン、ウベニメクス、インターフェロン、インターロイキン、マクロファージコロニー刺激因子、顆粒球コロニー刺激因子、エリスロポイエチン、リンホトキシン、BCGワクチン、コリネバクテリウムパルブム、レバミゾール、ポリサッカライドK、プロコダゾール、抗CTLA4抗体)等が用いられる。
「分子標的薬」としては、例えば、トシツモマブ、イブリツモマブ、アレムツズマブ、アキシチニブ、ベバシズマブ、アファチニブ、ボルテゾミブ、ボスチニブ、カルフィルゾミブ、セツキシマブ、ダサチニブ、デノスマブ、エドレコロマブ、エルロチニブ、エベロリムス、ビスモデギブ、ゲフィチニブ、ゲムツズマブオゾガマイシン、イマチニブ、イピリムマブ、ラパチニブ、レナリドミド、ニロチニブ、ニモツズマブ、オラパリブ、パニツムマブ、パゾパニブ、ペルツズマブ、リツキシマブ、シルツキシマブ、ソラフェニブ、スニチニブ、タミバロテン、テムシロリムス、サリドマイド、トラスツズマブ、トレチノイン、バンデタニブ、ボリノスタット、カボザンチニブ、トラメチニブ、ダブラフェニブ、アレクチニブ、セリチニブ、イブルチニブ、パルボシクリブ、レゴラフェニブ、ピララリシブ等が用いられる。
「免疫チェックポイント阻害剤」としては、例えば、ニボルマブ、ペムブロリズマブ、アベルマブ、アテゾリズマブ等が用いられる。
実施例
以下に、実施例及び試験例に基づいて本発明をより詳細に説明するが、本発明は、実施例及び試験例により限定されるものではなく、本発明の範囲を逸脱しない範囲で変化させてもよい。また、本発明において使用する試薬や装置、材料は特に言及されない限り、商業的に入手可能である。
以下に、実施例及び試験例に基づいて本発明をより詳細に説明するが、本発明は、実施例及び試験例により限定されるものではなく、本発明の範囲を逸脱しない範囲で変化させてもよい。また、本発明において使用する試薬や装置、材料は特に言及されない限り、商業的に入手可能である。
下記実施例1、2、4~11及び13の化合物(化合物(1)、化合物(2)、化合物(4)~(11)及び化合物(13))は、公知化合物であり、非特許文献7(Shimamoto Y, et al. Bioorg. Med. Chem., 2015, 23, 876-890)及び8(Teruya, K, et al. Peptide Science, 2015, 106(4), 391-402)に記載の方法に従って合成、同定したものを使用した。
実施例1
(S)-2-[({(3S,4aR,8aS)-2-[(1,1'-ビフェニル)-3-カルボニル]デカヒドロイソキノリン-3-イル}メチル)アミノ]-N-メトキシ-N-メチル-3-(1-トリチル-1H-イミダゾール-4-イル)プロパンアミド(化合物(1))
(S)-2-[({(3S,4aR,8aS)-2-[(1,1'-ビフェニル)-3-カルボニル]デカヒドロイソキノリン-3-イル}メチル)アミノ]-N-メトキシ-N-メチル-3-(1-トリチル-1H-イミダゾール-4-イル)プロパンアミド(化合物(1))

実施例2
(S)-2-[({(3S,4aR,8aS)-2-[(1,1'-ビフェニル)-3-カルボニル]デカヒドロイソキノリン-3-イル}メチル)アミノ]-3-(1H-イミダゾール-4-イル)-N-メトキシ-N-メチルプロパンアミド(化合物(2))
(S)-2-[({(3S,4aR,8aS)-2-[(1,1'-ビフェニル)-3-カルボニル]デカヒドロイソキノリン-3-イル}メチル)アミノ]-3-(1H-イミダゾール-4-イル)-N-メトキシ-N-メチルプロパンアミド(化合物(2))
実施例3
(S)-2-({[(3S,4aR,8aS)-2-[(3'-エチニル-1,1'-ビフェニル)-4-カルボニル]デカヒドロイソキノリン-3-イル]メチル}アミノ)-N-メトキシ-N-メチル-3-(1-トリチル-1H-イミダゾール-4-イル)プロパンアミド(化合物(3))
(S)-2-({[(3S,4aR,8aS)-2-[(3'-エチニル-1,1'-ビフェニル)-4-カルボニル]デカヒドロイソキノリン-3-イル]メチル}アミノ)-N-メトキシ-N-メチル-3-(1-トリチル-1H-イミダゾール-4-イル)プロパンアミド(化合物(3))

化合物(3)は、非特許文献7(Shimamoto Y, et al. Bioorg. Med. Chem., 2015, 23, 876-890)及び8(Teruya, K, et al. Peptide Science, 2015, 106(4), 391-402)に記載の方法に準じて合成した。
1H NMR (300 MHz): δ = 7.70 (br d, J = 6.9 Hz, 1.2H), 7.61-7.30 (m, 17.8H), 7.13-7.08 (m, 6H), 6.62 (s, 0.6H), 6.56 (s, 0.4H), 4.93 (br s, 0.6H), 4.43-4.40 (m, 0.4H), 4.16-4.11 (m, 0.6H), 3.91 (m, 1H), 3.77-3.76 (m, 0.4H), 3.68 (s, 1.8H), 3.50 (s, 1.2H), 3.39-3.35 (m, 0.6H), 3.14 (s, 1.8H), 3.08 (s, 1.2H), 2.83-2.81 (m, 2.4H), 2.71-2.69 (m, 1.6H), 2.49-2.42 (m, 0.4H), 2.31-2.29 (m, 0.4H), 2.05-2.01 (m, 1.6H), 1.79-1.54 (m, 3H), 1.39-1.26 (m, 6H), 1.07-0.83 (m, 2H).
1H NMR (300 MHz): δ = 7.70 (br d, J = 6.9 Hz, 1.2H), 7.61-7.30 (m, 17.8H), 7.13-7.08 (m, 6H), 6.62 (s, 0.6H), 6.56 (s, 0.4H), 4.93 (br s, 0.6H), 4.43-4.40 (m, 0.4H), 4.16-4.11 (m, 0.6H), 3.91 (m, 1H), 3.77-3.76 (m, 0.4H), 3.68 (s, 1.8H), 3.50 (s, 1.2H), 3.39-3.35 (m, 0.6H), 3.14 (s, 1.8H), 3.08 (s, 1.2H), 2.83-2.81 (m, 2.4H), 2.71-2.69 (m, 1.6H), 2.49-2.42 (m, 0.4H), 2.31-2.29 (m, 0.4H), 2.05-2.01 (m, 1.6H), 1.79-1.54 (m, 3H), 1.39-1.26 (m, 6H), 1.07-0.83 (m, 2H).
実施例4
(S)-2-({[(3S,4aR,8aS)-2-(4-フルオロベンゾイル)デカヒドロイソキノリン-3-イル]メチル}アミノ) -3-(1H-イミダゾール-4-イル)-N-メトキシ-N-メチルプロパンアミド(化合物(4))
(S)-2-({[(3S,4aR,8aS)-2-(4-フルオロベンゾイル)デカヒドロイソキノリン-3-イル]メチル}アミノ) -3-(1H-イミダゾール-4-イル)-N-メトキシ-N-メチルプロパンアミド(化合物(4))
実施例5
(S)-2-({[(3S,4aR,8aS)-2-ベンゾイルデカヒドロイソキノリン-3-イル]メチル}アミノ)-3-(1H-イミダゾール-4-イル)-N-メトキシ-N-メチルプロパンアミド(化合物(5))
(S)-2-({[(3S,4aR,8aS)-2-ベンゾイルデカヒドロイソキノリン-3-イル]メチル}アミノ)-3-(1H-イミダゾール-4-イル)-N-メトキシ-N-メチルプロパンアミド(化合物(5))

実施例6
(S)-2-({[(3S,4aR,8aS)-2-[(1,1'-ビフェニル)-4-カルボニル]デカヒドロイソキノリン-3-イル]メチル}アミノ)-N-メトキシ-N-メチル-3-(1-トリチル-1H-イミダゾール-4-イル)プロパンアミド(化合物(6))
(S)-2-({[(3S,4aR,8aS)-2-[(1,1'-ビフェニル)-4-カルボニル]デカヒドロイソキノリン-3-イル]メチル}アミノ)-N-メトキシ-N-メチル-3-(1-トリチル-1H-イミダゾール-4-イル)プロパンアミド(化合物(6))
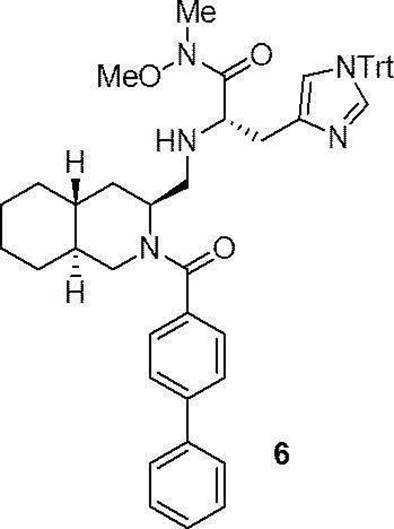
実施例7
(S)-2-[({(3R,4aS,8aR)-2-[(1,1'-ビフェニル)-4-カルボニル]デカヒドロイソキノリン-3-イル}メチル)アミノ]-3-(1H-イミダゾール-4-イル)-N-メトキシ-N-メチルプロパンアミド(化合物(7))
(S)-2-[({(3R,4aS,8aR)-2-[(1,1'-ビフェニル)-4-カルボニル]デカヒドロイソキノリン-3-イル}メチル)アミノ]-3-(1H-イミダゾール-4-イル)-N-メトキシ-N-メチルプロパンアミド(化合物(7))

実施例8
(S)-2-({[(3S,4aR,8aS)-2-[(1,1-ジメチルエトキシ)フェニルアミノエチルカルボニル]デカヒドロイソキノリン-3-イル]メチル}アミノ)-N-メトキシ-N-メチル-3-(1-トリチル-1H-イミダゾール-4-イル)プロパンアミド(化合物(8))
(S)-2-({[(3S,4aR,8aS)-2-[(1,1-ジメチルエトキシ)フェニルアミノエチルカルボニル]デカヒドロイソキノリン-3-イル]メチル}アミノ)-N-メトキシ-N-メチル-3-(1-トリチル-1H-イミダゾール-4-イル)プロパンアミド(化合物(8))
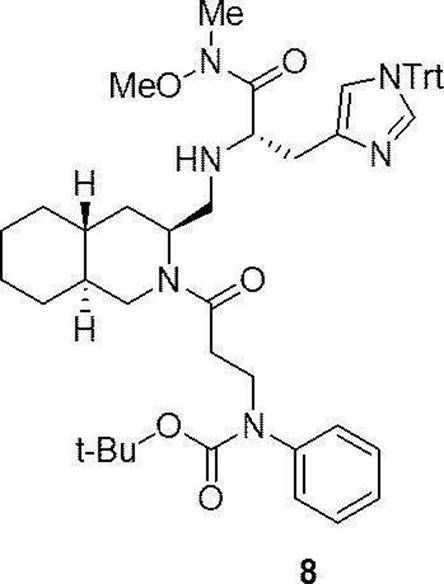
実施例9
(R)-2-({[(3R,4aS,8aR)-2-[(1,1-ジメチルエトキシ)フェニルアミノエチルカルボニル]デカヒドロイソキノリン-3-イル]メチル}アミノ)-N-メトキシ-N-メチル-3-(1-トリチル-1H-イミダゾール-4-イル)プロパンアミド(化合物(9))
(R)-2-({[(3R,4aS,8aR)-2-[(1,1-ジメチルエトキシ)フェニルアミノエチルカルボニル]デカヒドロイソキノリン-3-イル]メチル}アミノ)-N-メトキシ-N-メチル-3-(1-トリチル-1H-イミダゾール-4-イル)プロパンアミド(化合物(9))
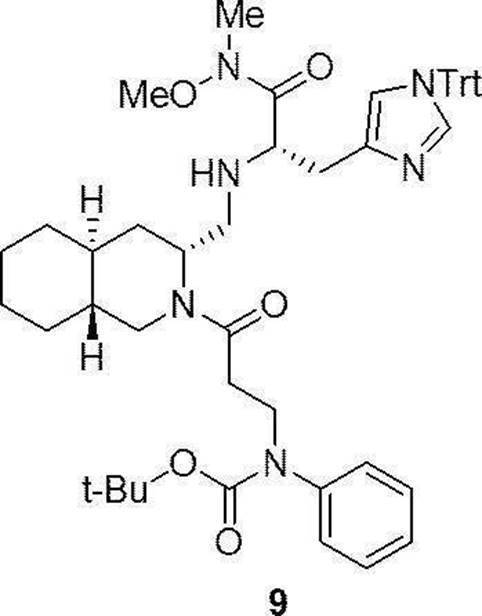
実施例10
(S)-2-({[(3S,4aR,8aS)-2-(4-ブロモベンゾイル)デカヒドロイソキノリン-3-イル]メチル}アミノ)-N-メトキシ-N-メチル-3-(1-トリチル-1H-イミダゾール-4-イル)プロパンアミド(化合物(10))
(S)-2-({[(3S,4aR,8aS)-2-(4-ブロモベンゾイル)デカヒドロイソキノリン-3-イル]メチル}アミノ)-N-メトキシ-N-メチル-3-(1-トリチル-1H-イミダゾール-4-イル)プロパンアミド(化合物(10))

実施例11
(S)-2-({[(3R,4aS,8aR)-2-(4-ブロモベンゾイル)デカヒドロイソキノリン-3-イル]メチル}アミノ)-N-メトキシ-N-メチル-3-(1-トリチル-1H-イミダゾール-4-イル)プロパンアミド(化合物(11))
(S)-2-({[(3R,4aS,8aR)-2-(4-ブロモベンゾイル)デカヒドロイソキノリン-3-イル]メチル}アミノ)-N-メトキシ-N-メチル-3-(1-トリチル-1H-イミダゾール-4-イル)プロパンアミド(化合物(11))
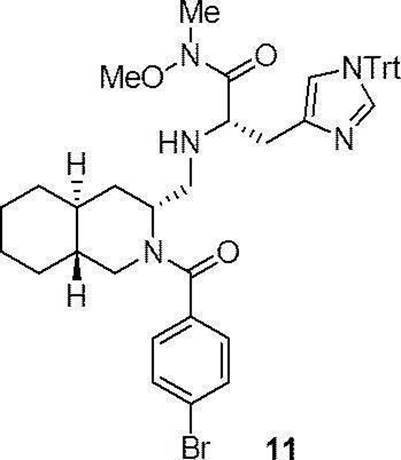
実施例12
{(3S,4aR,8aS)-3-[({(R)-1-[メトキシ(メチル)アミノ]-2-(1-トリチル-1H-イミダゾール-4-イル)エチル}アミノ)メチル]オクタヒドロイソキノリン-2(1H)-yl}(4-tert-ブチルフェニル)メタノン(化合物(12))
{(3S,4aR,8aS)-3-[({(R)-1-[メトキシ(メチル)アミノ]-2-(1-トリチル-1H-イミダゾール-4-イル)エチル}アミノ)メチル]オクタヒドロイソキノリン-2(1H)-yl}(4-tert-ブチルフェニル)メタノン(化合物(12))
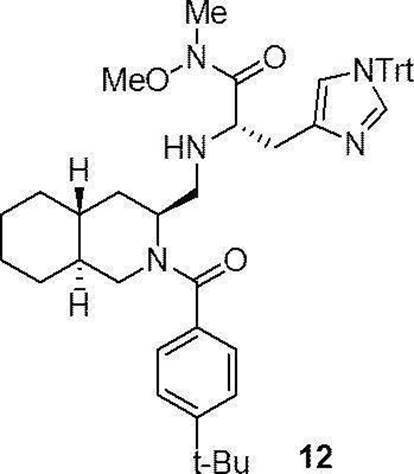
化合物(12)は、非特許文献7(Shimamoto Y, et al. Bioorg. Med. Chem., 2015, 23, 876-890)及び8(Teruya, K, et al. Peptide Science, 2015, 106(4), 391-402)に記載の方法に準じて合成した。
1H NMR (300 MHz): δ = 7.36-7.24 (m, 15H), 7.14-7.11 (m, 6H), 6.61 (s, 0.6H), 6.56 (s, 0.4H), 4.86 (m, 0.6H), 4.39 (dd, J = 12.9, 3.9 Hz, 0.4H), 4.11 (m, 0.6H), 3.94-3.91 (m, 0.8H), 3.71 (s, 1.8H), 3.50 (s, 1.2H), 3.50-3.37 (m, 0.6H), 3.13 (s, 1.8H), 3.08 (s, 1.2H), 2.88-2.81 (m, 3.4H), 2.75-2.68 (m, 1.6H), 1.79-1.52 (m, 4H), 1.31 (s, 5.4H), 1.29 (s, 4.6H), 1.40-1.25 (m, 6H), 1.09-0.80 (m, 2H).
1H NMR (300 MHz): δ = 7.36-7.24 (m, 15H), 7.14-7.11 (m, 6H), 6.61 (s, 0.6H), 6.56 (s, 0.4H), 4.86 (m, 0.6H), 4.39 (dd, J = 12.9, 3.9 Hz, 0.4H), 4.11 (m, 0.6H), 3.94-3.91 (m, 0.8H), 3.71 (s, 1.8H), 3.50 (s, 1.2H), 3.50-3.37 (m, 0.6H), 3.13 (s, 1.8H), 3.08 (s, 1.2H), 2.88-2.81 (m, 3.4H), 2.75-2.68 (m, 1.6H), 1.79-1.52 (m, 4H), 1.31 (s, 5.4H), 1.29 (s, 4.6H), 1.40-1.25 (m, 6H), 1.09-0.80 (m, 2H).
実施例13
{(3S,4aR,8aS)-3-[({(R)-1-[メトキシ(メチル)アミノ]-2-(1-トリチル-1H-イミダゾール-4-イル)エチル}アミノ)メチル]オクタヒドロイソキノリン-2(1H)-yl}(フェニル)メタノン(化合物(13))
{(3S,4aR,8aS)-3-[({(R)-1-[メトキシ(メチル)アミノ]-2-(1-トリチル-1H-イミダゾール-4-イル)エチル}アミノ)メチル]オクタヒドロイソキノリン-2(1H)-yl}(フェニル)メタノン(化合物(13))
実施例14
(S)-2-({[(3R,4aS,8aR)-2-ベンゾイルデカヒドロイソキノリン-3-イル]メチル}アミノ)-N-メトキシ-N-メチル-3-(1-トリチル-1H-イミダゾール-4-イル)プロパンアミド(化合物(14))
(S)-2-({[(3R,4aS,8aR)-2-ベンゾイルデカヒドロイソキノリン-3-イル]メチル}アミノ)-N-メトキシ-N-メチル-3-(1-トリチル-1H-イミダゾール-4-イル)プロパンアミド(化合物(14))

化合物(14)は、非特許文献7(Shimamoto Y, et al. Bioorg. Med. Chem., 2015, 23, 876-890)及び8(Teruya, K, et al. Peptide Science, 2015, 106(4), 391-402)に記載の方法に準じて合成した。
1H NMR (300 MHz): δ = 7.41-7.30 (m, 15H), 7.11 (br s, 7H), 6.57 (br s, 0.5H), 6.54 (s, 0.5H), 4.99 (m, 0.5H), 4.44 (dd, J = 13.8, 2.7 Hz, 0.5H), 4.15 (br s, 0.5H), 3.85-3.69 (m, 0.5H), 3.65 (s, 1.5H), 3.52 (s, 1.5H), 3.39-3.34 (m, 1H), 3.13 (s, 1.5H), 3.10 (s, 1.5H), 3.06-2.88 (m, 1.5H), 2.80-2.63 (m, 1.5H), 2.51-2.32 (m, 2H), 1.6-1.58 (m, 4H), 1.45-1.26 (m, 6H), 1.01-0.81 (m, 2H);
13C NMR (75 MHz): δ= 169.8, 169.6, 169.2, 142.54, 142.48, 138.3, 138.1, 137.3, 129.80, 129.75, 128.0, 119.4, 119.3, 75.6, 61.6, 57.9, 57.5, 54.7, 48.2, 47.7, 47.2, 46.3, 42.5, 42.4, 41.7, 36.5, 36.1, 36.0, 34.2, 33.0, 32.5, 32.0, 30.0, 29.9, 29.7, 26.2, 26.1, 25.8, 22.0, 21.8.
1H NMR (300 MHz): δ = 7.41-7.30 (m, 15H), 7.11 (br s, 7H), 6.57 (br s, 0.5H), 6.54 (s, 0.5H), 4.99 (m, 0.5H), 4.44 (dd, J = 13.8, 2.7 Hz, 0.5H), 4.15 (br s, 0.5H), 3.85-3.69 (m, 0.5H), 3.65 (s, 1.5H), 3.52 (s, 1.5H), 3.39-3.34 (m, 1H), 3.13 (s, 1.5H), 3.10 (s, 1.5H), 3.06-2.88 (m, 1.5H), 2.80-2.63 (m, 1.5H), 2.51-2.32 (m, 2H), 1.6-1.58 (m, 4H), 1.45-1.26 (m, 6H), 1.01-0.81 (m, 2H);
13C NMR (75 MHz): δ= 169.8, 169.6, 169.2, 142.54, 142.48, 138.3, 138.1, 137.3, 129.80, 129.75, 128.0, 119.4, 119.3, 75.6, 61.6, 57.9, 57.5, 54.7, 48.2, 47.7, 47.2, 46.3, 42.5, 42.4, 41.7, 36.5, 36.1, 36.0, 34.2, 33.0, 32.5, 32.0, 30.0, 29.9, 29.7, 26.2, 26.1, 25.8, 22.0, 21.8.
実施例15
(S)-2-({[(3S,4aR,8aS)-2-アセチルデカヒドロイソキノリン-3-イル]メチル}アミノ)-N-メトキシ-N-メチル-3-(1-トリチル-1H-イミダゾール-4-イル)プロパンアミド(化合物(15))
(S)-2-({[(3S,4aR,8aS)-2-アセチルデカヒドロイソキノリン-3-イル]メチル}アミノ)-N-メトキシ-N-メチル-3-(1-トリチル-1H-イミダゾール-4-イル)プロパンアミド(化合物(15))
化合物(15)は、非特許文献7(Shimamoto Y, et al. Bioorg. Med. Chem., 2015, 23, 876-890)及び8(Teruya, K, et al. Peptide Science, 2015, 106(4), 391-402)に記載の方法に準じて合成した。
1H NMR (300 MHz): δ = 7.34-7.31 (m, 10H), 7.14-7.11 (m, 6H), 6.59 (s, 0.5H), 6.58 (s, 0.5H), 4.76-4.75 (m, 0.5H), 4.37 (dd, J = 13.4, 3.5 Hz, 0.5H), 4.00 (br s, 0.5H), 3.89 (m, 0.5H), 3.66 (s, 1.5H), 3.61 (d, J = 3.0 Hz, 0.5H), 3.58 (s, 1.5H), 3.35 (dd, J = 12.6, 2.7 Hz, 0.5H), 3.12 (s, 3H), 2.84-2.49 (m, 3H), 2.21-2.13 (m, 1H), 2.08 (s, 1.5H), 2.00 (s, 1.5H), 1.95 (br s, 1H), 1.72-1.53 (m, 4H), 1.25-1.09 (m, 6H), 1.00-0.82 (m, 2H).
1H NMR (300 MHz): δ = 7.34-7.31 (m, 10H), 7.14-7.11 (m, 6H), 6.59 (s, 0.5H), 6.58 (s, 0.5H), 4.76-4.75 (m, 0.5H), 4.37 (dd, J = 13.4, 3.5 Hz, 0.5H), 4.00 (br s, 0.5H), 3.89 (m, 0.5H), 3.66 (s, 1.5H), 3.61 (d, J = 3.0 Hz, 0.5H), 3.58 (s, 1.5H), 3.35 (dd, J = 12.6, 2.7 Hz, 0.5H), 3.12 (s, 3H), 2.84-2.49 (m, 3H), 2.21-2.13 (m, 1H), 2.08 (s, 1.5H), 2.00 (s, 1.5H), 1.95 (br s, 1H), 1.72-1.53 (m, 4H), 1.25-1.09 (m, 6H), 1.00-0.82 (m, 2H).
実施例16
Fmoc-Tic-His(Trt)-OMe(化合物(16))
Fmoc-Tic-His(Trt)-OMe(化合物(16))
化合物(16)は、前記製造法1の方法に準じた方法により合成した。具体的には、後述する化合物(18)の製造方法に記載の方法により合成した。
1H NMR (300 MHz): δ = 8.50 (br d, J = 7.2 Hz,, 0.55H), 8.11 (br d, J = 7.2 Hz, 0.45H), 7.72 (br d, J = 6.0 Hz, 2H), 7.59-7.51 (m, 2H), 7.35-7.14 (m, 17H), 7.07-7.01 (m, 7H), 6.50 (br s, 0.45H), 6.44 (br s, 0.55H), 5.06-5.00 (m, 1H), 4.86 (br t, J = 16.1 Hz, 1H), 4.74-4.62 (m, 2H), 4.51-4.41 (m, 1H), 4.32-4.21 (m, 2H), 3.49 (br s, 1.35H), 3.43 (s, 1.65H), 3.43-3.35 (m, 1H), 3.17-3.07 (m, 1H), 2.95-2.90 (m, 1H), 2.78-2.62 (m, 1H);
13C NMR (75 MHz): δ= 171.2, 171.0, 170.5, 170.3, 156.0, 155.5, 144.2, 143.8, 143.5, 142.0, 141.1, 140.9, 138.5, 136.3, 133.1, 132.8, 132.6, 129.5, 128.5, 128.1, 127.89, 127.87, 127.5, 127.1, 126.9, 126.5, 126.4, 126.1, 125.8, 125.4, 125.2, 125.0, 119.7, 119.3, 75.1, 68.3, 67.8, 55.4, 54.4, 52.5, 52.3, 51.8, 47.0, 44.6, 31.3, 30.4, 29.3.
1H NMR (300 MHz): δ = 8.50 (br d, J = 7.2 Hz,, 0.55H), 8.11 (br d, J = 7.2 Hz, 0.45H), 7.72 (br d, J = 6.0 Hz, 2H), 7.59-7.51 (m, 2H), 7.35-7.14 (m, 17H), 7.07-7.01 (m, 7H), 6.50 (br s, 0.45H), 6.44 (br s, 0.55H), 5.06-5.00 (m, 1H), 4.86 (br t, J = 16.1 Hz, 1H), 4.74-4.62 (m, 2H), 4.51-4.41 (m, 1H), 4.32-4.21 (m, 2H), 3.49 (br s, 1.35H), 3.43 (s, 1.65H), 3.43-3.35 (m, 1H), 3.17-3.07 (m, 1H), 2.95-2.90 (m, 1H), 2.78-2.62 (m, 1H);
13C NMR (75 MHz): δ= 171.2, 171.0, 170.5, 170.3, 156.0, 155.5, 144.2, 143.8, 143.5, 142.0, 141.1, 140.9, 138.5, 136.3, 133.1, 132.8, 132.6, 129.5, 128.5, 128.1, 127.89, 127.87, 127.5, 127.1, 126.9, 126.5, 126.4, 126.1, 125.8, 125.4, 125.2, 125.0, 119.7, 119.3, 75.1, 68.3, 67.8, 55.4, 54.4, 52.5, 52.3, 51.8, 47.0, 44.6, 31.3, 30.4, 29.3.
実施例17
3-フェニルベンゾイル-Tic-His(Trt)-OMe(化合物(17))
3-フェニルベンゾイル-Tic-His(Trt)-OMe(化合物(17))

化合物(17)は、前記製造法1の方法に準じた方法により合成した。具体的には、後述する化合物(18)の製造方法に準じて合成した。
1H NMR (300 MHz): δ = 8.48 (br d, J = 7.5 Hz, 0.55H), 8.09 (br d, J = 7.5 Hz, 0.45H), 7.76-7.72 (m, 2H), 7.60-7.51 (m, 2H), 7.41-7.30 (m, 3H), 7.28-7.23 (m, 12H), 7.21-7.11 (m, 3H), 7.07-7.01 (m, 7H), 6.50 (br s, 0.45H), 6.43 (br s, 0.55H), 5.05-4.97 (m, 1H), 4.86 (br t, J = 15.6 Hz, 1H), 4.73-4.60 (m, 1H), 4.46-4.41 (m, 1H), 4.31-4.18 (m, 1H), 3.50 (s, 1.35H), 3.43 (s, 1.65H), 3.11 (dt, J = 15.2, 5.9 Hz, 1H), 2.95 (dt, J = 13.9, 4.5 Hz, 1H), 2.76 (dd, J = 13.5, 4.2 Hz, 0.4H), 2.65 (dd, J = 14.7, 4.2 Hz, 0.6H);
13C NMR (75 MHz): δ = 171.3, 171.1, 170.6, 170.4, 156.1, 155.6, 144.25, 143.9, 143.7, 143.6, 142.1, 141.2, 141.0, 138.5, 136.4, 133.2, 132.9, 132.7, 129.6, 128.6, 128.2, 127.98, 127.95, 127.6, 127.2, 127.0, 126.6, 126.2, 125.9, 125.5, 125.2, 125.1, 119.8, 119.4, 75.2, 68.4, 67.9, 55.5, 54.5, 52.6, 52.4, 51.9, 47.1, 44.7, 31.4, 30.5, 29.4.
1H NMR (300 MHz): δ = 8.48 (br d, J = 7.5 Hz, 0.55H), 8.09 (br d, J = 7.5 Hz, 0.45H), 7.76-7.72 (m, 2H), 7.60-7.51 (m, 2H), 7.41-7.30 (m, 3H), 7.28-7.23 (m, 12H), 7.21-7.11 (m, 3H), 7.07-7.01 (m, 7H), 6.50 (br s, 0.45H), 6.43 (br s, 0.55H), 5.05-4.97 (m, 1H), 4.86 (br t, J = 15.6 Hz, 1H), 4.73-4.60 (m, 1H), 4.46-4.41 (m, 1H), 4.31-4.18 (m, 1H), 3.50 (s, 1.35H), 3.43 (s, 1.65H), 3.11 (dt, J = 15.2, 5.9 Hz, 1H), 2.95 (dt, J = 13.9, 4.5 Hz, 1H), 2.76 (dd, J = 13.5, 4.2 Hz, 0.4H), 2.65 (dd, J = 14.7, 4.2 Hz, 0.6H);
13C NMR (75 MHz): δ = 171.3, 171.1, 170.6, 170.4, 156.1, 155.6, 144.25, 143.9, 143.7, 143.6, 142.1, 141.2, 141.0, 138.5, 136.4, 133.2, 132.9, 132.7, 129.6, 128.6, 128.2, 127.98, 127.95, 127.6, 127.2, 127.0, 126.6, 126.2, 125.9, 125.5, 125.2, 125.1, 119.8, 119.4, 75.2, 68.4, 67.9, 55.5, 54.5, 52.6, 52.4, 51.9, 47.1, 44.7, 31.4, 30.5, 29.4.
実施例18
4-tert-ブチルベンゾイル-Tic-His(Trt)-OMe(化合物(18))
4-tert-ブチルベンゾイル-Tic-His(Trt)-OMe(化合物(18))
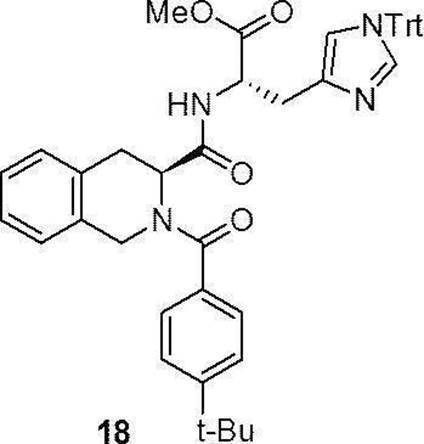
化合物(18)は、前記製造法1の方法に準じて、下記式:
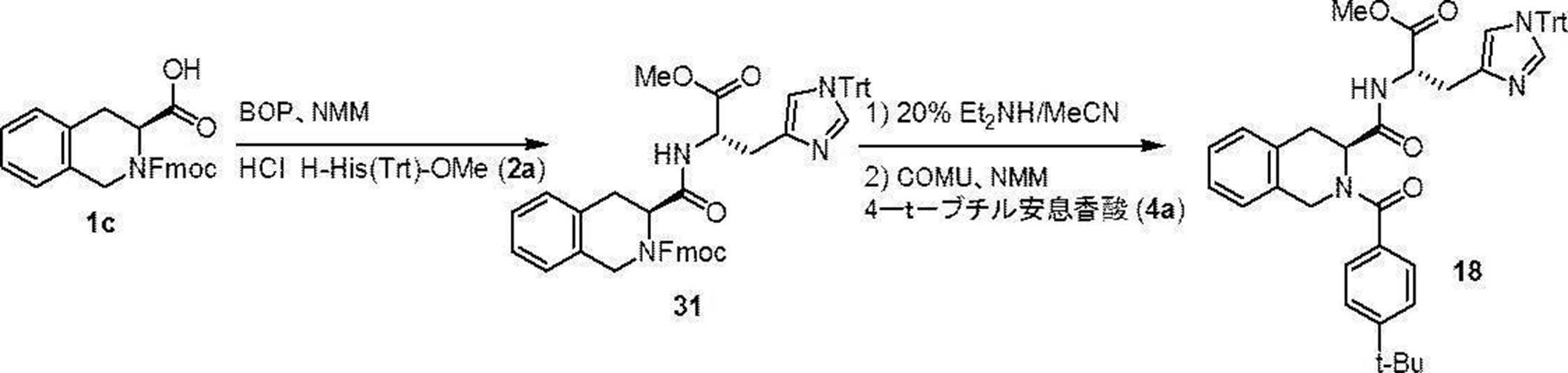
で示される合成スキームに従って合成した。具体的には、市販のFmoc保護アミノ酸(1c)(渡辺化学工業株式会社製)をジクロロメタンに溶解し、市販のトリチルヒスチジンメチルエステル塩酸塩(1.1当量)とN-メチルモルホリン(NMM)(7.0当量)を加えた。氷冷撹拌下、続いて1H-ベンゾトリアゾール-1-イルオキシ-トリス(ジメチルアミノ)ホスホニウムヘキサフルオロホスフェート(BOP)(1.2当量)を加えて16 時間撹拌した。飽和塩化アンモニウム水溶液を用いて反応を停止させ、酢酸エチルで抽出した。有機相を合わせて飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥させた。ろ過後、減圧下溶媒留去を行い、残渣をシリカゲルカラムクロマトグラフィーに付すことにより、化合物(31)を得た。
化合物(31)をアセトニトリルに溶解し、20%(V/V)になるようにジエチルアミン(Et2NH)を加えた。20分撹拌後、減圧下溶媒留去を行った。残渣は精製することなく次の反応に使用した。得られた残渣をジクロロメタンに溶解し4-t-ブチル安息香酸(1.1当量)とNMM(7.0当量)を加えた。氷冷撹拌下、N-[1-(シアノ-2-エトキシ-2-オキソエチリデンアミノオキシ)ジメチルアミノ (モルホリノ)]ウロニウム ヘキサフルオロホスフェート(COMU)(1.2 equiv.)を加えて16時間撹拌した。飽和塩化アンモニウム水溶液を用いて反応を停止させ、酢酸エチルで抽出した。飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥させた。ろ過後、減圧下溶媒留去を行い、残渣をシリカゲルカラムクロマトグラフィーに付すことにより化合物(18)を得た。
1H NMR (300 MHz): δ = 8.96 (br d, J = 7.2 Hz, 0.6H), 8.21 (br d, J = 7.2 Hz, 0.4H), 7.62 (br d, J = 7.5 Hz, 0.8H), 7.52 (br d, J = 8.1 Hz, 1.2H), 7.43-7.37 (m, 2H), 7.31 (br s, 9H), 7.19-7.06 (m, 10.6H), 6.87-6.84 (m, 0.4H), 6.57 (br s, 0.4H), 6.50 (br s, 0.6H), 5.55 (br s, 0.4H), 5.30-5.24 (m, 0.6H), 4.88-4.78 (m, 2H), 4.71-4.41 (m, 0.4H), 4.54-4.18 (m, 0.6H), 3.56 (s, 1.8H), 3.54 (s, 1.2H), 3.43-3.38 (m, 1H), 3.20-3.04 (m, 1H), 2.97-2.92 (m, 1.4H), 2.61-2.56 (m, 0.6H), 1.29 (s, 9H);
13C NMR (75 MHz): δ = 172.2, 171.2, 170.0, 153.0, 144.2, 138.9, 136.2, 133.3, 132.4, 132.2, 129.7, 128.7, 128.1, 127.7, 126.9, 126.7, 126.3, 125.5, 125.2, 119.8, 119.3, 75.3, 58.3, 52.7, 52.1, 43.9, 34.8, 31.9, 31.6, 31.2, 29.7, 29.5, 29.3, 28.9, 24.8, 22.7.
1H NMR (300 MHz): δ = 8.96 (br d, J = 7.2 Hz, 0.6H), 8.21 (br d, J = 7.2 Hz, 0.4H), 7.62 (br d, J = 7.5 Hz, 0.8H), 7.52 (br d, J = 8.1 Hz, 1.2H), 7.43-7.37 (m, 2H), 7.31 (br s, 9H), 7.19-7.06 (m, 10.6H), 6.87-6.84 (m, 0.4H), 6.57 (br s, 0.4H), 6.50 (br s, 0.6H), 5.55 (br s, 0.4H), 5.30-5.24 (m, 0.6H), 4.88-4.78 (m, 2H), 4.71-4.41 (m, 0.4H), 4.54-4.18 (m, 0.6H), 3.56 (s, 1.8H), 3.54 (s, 1.2H), 3.43-3.38 (m, 1H), 3.20-3.04 (m, 1H), 2.97-2.92 (m, 1.4H), 2.61-2.56 (m, 0.6H), 1.29 (s, 9H);
13C NMR (75 MHz): δ = 172.2, 171.2, 170.0, 153.0, 144.2, 138.9, 136.2, 133.3, 132.4, 132.2, 129.7, 128.7, 128.1, 127.7, 126.9, 126.7, 126.3, 125.5, 125.2, 119.8, 119.3, 75.3, 58.3, 52.7, 52.1, 43.9, 34.8, 31.9, 31.6, 31.2, 29.7, 29.5, 29.3, 28.9, 24.8, 22.7.
実施例19
4-tert-ブチルベンゾイル-ホモPro-His(Trt)-OMe(化合物(19))
4-tert-ブチルベンゾイル-ホモPro-His(Trt)-OMe(化合物(19))
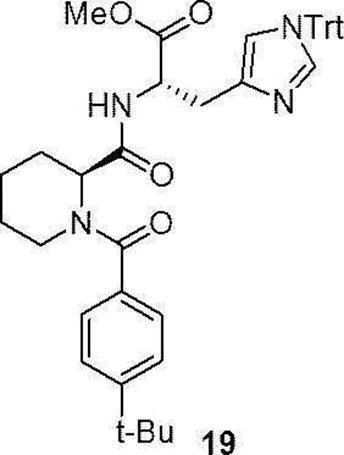
化合物(19)は、前記製造法1の方法に準じた方法により、市販の1-Fmocホモプロリン(Fmoc-homoPro-OH)(12a)(渡辺化学工業株式会社製)を出発原料として用いて合成した。
1H NMR (300 MHz): δ = 8.76 (br d, J = 6.6 Hz, 0.4H), 8.05 (br d, J = 7.5 Hz, 0.6H), 7.51 (br d, J = 8.1 Hz, 1.2H), 7.43 (br d, J = 7.5 Hz, 0.8H), 7.32-7.30 (m, 12H), 7.08 (br s, 6H), 6.572 (br s, 0.4H), 6.570 (br s, 0.6H), 5.47 (br s, 0.6H), 4.85 (m, 1H), 4.76 (br s, 0.4H), 4.53 (br s, 0.4H), 3.77 (br s, 0.4H), 3.73-3.67 (m, 0.4H), 3.63 (s, 1.2H), 3.58 (s, 1.8H), 3.46 (d, J = 4.2 Hz, 0.4H), 3.25-3.22 (m, 1.2H), 3.13 (dd, J = 14.6, 4.7 Hz, 1H), 3.03-2.98 (m, 1H), 2.43 (br d, J = 7.5 Hz, 0.6H), 2.35 (br d, J = 9.0 Hz, 0.6H), 1.97 (br s, 1H), 1.68-1.49 (m,5H), 1.29 (s, 6H), 1.27 (s, 3H);
HRMS (EI) Calcd. For C43H46N4O4 [M]+: 682.3519. Found: 682.3523.
1H NMR (300 MHz): δ = 8.76 (br d, J = 6.6 Hz, 0.4H), 8.05 (br d, J = 7.5 Hz, 0.6H), 7.51 (br d, J = 8.1 Hz, 1.2H), 7.43 (br d, J = 7.5 Hz, 0.8H), 7.32-7.30 (m, 12H), 7.08 (br s, 6H), 6.572 (br s, 0.4H), 6.570 (br s, 0.6H), 5.47 (br s, 0.6H), 4.85 (m, 1H), 4.76 (br s, 0.4H), 4.53 (br s, 0.4H), 3.77 (br s, 0.4H), 3.73-3.67 (m, 0.4H), 3.63 (s, 1.2H), 3.58 (s, 1.8H), 3.46 (d, J = 4.2 Hz, 0.4H), 3.25-3.22 (m, 1.2H), 3.13 (dd, J = 14.6, 4.7 Hz, 1H), 3.03-2.98 (m, 1H), 2.43 (br d, J = 7.5 Hz, 0.6H), 2.35 (br d, J = 9.0 Hz, 0.6H), 1.97 (br s, 1H), 1.68-1.49 (m,5H), 1.29 (s, 6H), 1.27 (s, 3H);
HRMS (EI) Calcd. For C43H46N4O4 [M]+: 682.3519. Found: 682.3523.
実施例20
(2S,2’’’S)-N-(4’-tert-ブチルベンゾイル)-2-メチルアミノ-N’’-[2’’’-(メトキシカルボニル)-1’’’-(1’’’’-トリチルイミダゾール-4’’’’-イル)エタン-2’’’-イル]ピペリジン(化合物(20))
(2S,2’’’S)-N-(4’-tert-ブチルベンゾイル)-2-メチルアミノ-N’’-[2’’’-(メトキシカルボニル)-1’’’-(1’’’’-トリチルイミダゾール-4’’’’-イル)エタン-2’’’-イル]ピペリジン(化合物(20))

化合物(20)は、前記製造法2の方法に準じて、下記式:

で示される合成スキームに従って合成した。具体的には、市販の1-Fmoc保護ホモプロリン(12a)をジクロロメタンに溶解し、市販の化合物(5a)(1.1当量)とNMM(7.0当量)を加えた。氷冷撹拌下、続いてBOP(1.2当量)を加えて16時間撹拌した。飽和塩化アンモニウム水溶液を用いて反応を停止させ、酢酸エチルで抽出した。飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥させた。ろ過後、減圧下溶媒留去を行った。残渣は精製することなく次の反応に使用した。得られた残渣をジクロロメタンに溶解し、-78 ℃でジイソブチルアルミナムヒドリド(DIBAL-H、1 Mヘキサン溶液)(1.1当量)を加えた。20分撹拌後、メタノールで反応を停止して室温まで昇温した。得られた懸濁液をシリカゲル・セライト(登録商標)パッドで吸引濾過し、濾液を濃縮後、残渣をシリカゲルカラムクロマトグラフィーで粗精製することにより化合物(7c)の粗生成物を得た。得られた粗生成物をジクロロメタンに溶解し、市販の化合物(2a)(1.0当量)を加えて1時間撹拌した。水素化シアノホウ素ナトリウム(NaBH3(CN))(2.0当量)を加えてさらに1時間撹拌した。飽和塩化アンモニウム水溶液を用いて反応を停止させ、酢酸エチルで抽出した。飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥させた。ろ過後、減圧下溶媒留去を行い、残渣をシリカゲルカラムクロマトグラフィーに付すことにより、化合物(8c)を得た。
化合物(8c)をアセトニトリルに溶解し、20%(V/V)になるようにジエチルアミンを加えた。20分撹拌後、減圧下溶媒留去を行った。残渣は精製することなく次の反応に使用した。得られた残渣をジクロロメタンに溶解し、4-t-ブチル安息香酸(1.1当量)とNMM(7.0当量)を加えた。氷冷撹拌下、N-[1-(シアノ-2-エトキシ-2-オキソエチリデンアミノオキシ)ジメチルアミノ (モルホリノ)]ウロニウム ヘキサフルオロホスフェート(COMU)(1.2当量)を加えて16時間撹拌した。飽和塩化アンモニウム水溶液を用いて反応を停止させ、酢酸エチルで抽出した。飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥させた。ろ過後、減圧下溶媒留去を行い、残渣をシリカゲルカラムクロマトグラフィーに付すことにより化合物(20)を得た。
1H NMR (300 MHz): δ = 7.79 (br d, J = 1.5 Hz, 0.5H), 7.43-7.28 (m, 13H), 7.14-7.08 (m, 7H), 6.79 (br d, J = 1.8 Hz, 0.5H), 6.55 (br s, 1H), 3.85 (dd, J = 7.7, 5.9 Hz, 1H), 3.67-3.48 (m, 2H), 3.48 (s, 3H), 3.22 (dd, J = 15.8, 5.6 Hz, 0.5H), 2.95 (dd, J = 16.2, 4.7 Hz, 0.5H), 2.89 (br d, J = 7.2 Hz, 2H), 2.80 (br d, J = 6.9 Hz, 2H), 1.78 (br s, 2H), 1.60 (m, 4H), 1.30 (s, 9H);
13C NMR (75 MHz): δ = 174.8, 152.2, 153.9, 142.4, 140.3, 138.4, 138.3, 137.1, 134.7, 134.0, 129.8, 129.6, 129.0, 128.6, 127.99, 127.97, 126.5, 125.3, 122.0, 119.2, 77.6, 75.1, 61.6, 53.1, 52.3, 51.5, 46.6, 34.7, 32.0, 31.9, 31.2, 30.3, 21.0, 19.4;
HRMS (EI) Calcd. For C43H48N4O3 [M]+: 668.3726. Found: 668.3728.
化合物(8c)をアセトニトリルに溶解し、20%(V/V)になるようにジエチルアミンを加えた。20分撹拌後、減圧下溶媒留去を行った。残渣は精製することなく次の反応に使用した。得られた残渣をジクロロメタンに溶解し、4-t-ブチル安息香酸(1.1当量)とNMM(7.0当量)を加えた。氷冷撹拌下、N-[1-(シアノ-2-エトキシ-2-オキソエチリデンアミノオキシ)ジメチルアミノ (モルホリノ)]ウロニウム ヘキサフルオロホスフェート(COMU)(1.2当量)を加えて16時間撹拌した。飽和塩化アンモニウム水溶液を用いて反応を停止させ、酢酸エチルで抽出した。飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥させた。ろ過後、減圧下溶媒留去を行い、残渣をシリカゲルカラムクロマトグラフィーに付すことにより化合物(20)を得た。
1H NMR (300 MHz): δ = 7.79 (br d, J = 1.5 Hz, 0.5H), 7.43-7.28 (m, 13H), 7.14-7.08 (m, 7H), 6.79 (br d, J = 1.8 Hz, 0.5H), 6.55 (br s, 1H), 3.85 (dd, J = 7.7, 5.9 Hz, 1H), 3.67-3.48 (m, 2H), 3.48 (s, 3H), 3.22 (dd, J = 15.8, 5.6 Hz, 0.5H), 2.95 (dd, J = 16.2, 4.7 Hz, 0.5H), 2.89 (br d, J = 7.2 Hz, 2H), 2.80 (br d, J = 6.9 Hz, 2H), 1.78 (br s, 2H), 1.60 (m, 4H), 1.30 (s, 9H);
13C NMR (75 MHz): δ = 174.8, 152.2, 153.9, 142.4, 140.3, 138.4, 138.3, 137.1, 134.7, 134.0, 129.8, 129.6, 129.0, 128.6, 127.99, 127.97, 126.5, 125.3, 122.0, 119.2, 77.6, 75.1, 61.6, 53.1, 52.3, 51.5, 46.6, 34.7, 32.0, 31.9, 31.2, 30.3, 21.0, 19.4;
HRMS (EI) Calcd. For C43H48N4O3 [M]+: 668.3726. Found: 668.3728.
実施例21
4-フェニルベンゾイル-Tic-His(Trt)-OMe(化合物(21))
4-フェニルベンゾイル-Tic-His(Trt)-OMe(化合物(21))
化合物(21)は、前記製造法1の方法(具体的には、前記した化合物(18)の製造方法)に準じて合成した。
1H NMR (300 MHz): δ = 9.04 (br d, J = 7.2 Hz, 0.55H), 8.35 (br d, J = 7.5 Hz, 0.45H), 7.80 (br d, J = 7.5 Hz, 0.9H), 7.68 (br d, J = 7.8 Hz, 1.1H), 7.61-7.35 (m, 8.55H), 7.34-7.28 (m, 8.45H), 7.23-7.07 (m, 9.45H), 6.87 (br d, J = 6.9 Hz, 0.55H), 6.57 (br s, 0.45H), 6.51 (br s, 0.55H), 5.62 (br s, 0.45H), 5.29 (br d, J = 17.4 Hz, 0.55H), 4.95-4.70 (m, 2.45H), 4.56-4.54 (m, 0.55H), 3.57-3.41 (m, 1H), 3.55 (s, 3H), 3.11-2.88 (m, 2.45H), 2.61-2.54 (m, 0.55H);
HRMS (ESI) Calcd. For C49H43N4O4 [M+H]+: 751.3274. Found: 751.3279.
1H NMR (300 MHz): δ = 9.04 (br d, J = 7.2 Hz, 0.55H), 8.35 (br d, J = 7.5 Hz, 0.45H), 7.80 (br d, J = 7.5 Hz, 0.9H), 7.68 (br d, J = 7.8 Hz, 1.1H), 7.61-7.35 (m, 8.55H), 7.34-7.28 (m, 8.45H), 7.23-7.07 (m, 9.45H), 6.87 (br d, J = 6.9 Hz, 0.55H), 6.57 (br s, 0.45H), 6.51 (br s, 0.55H), 5.62 (br s, 0.45H), 5.29 (br d, J = 17.4 Hz, 0.55H), 4.95-4.70 (m, 2.45H), 4.56-4.54 (m, 0.55H), 3.57-3.41 (m, 1H), 3.55 (s, 3H), 3.11-2.88 (m, 2.45H), 2.61-2.54 (m, 0.55H);
HRMS (ESI) Calcd. For C49H43N4O4 [M+H]+: 751.3274. Found: 751.3279.
実施例22
(2S,2’’’S)-N-(4’-tert-ブチルベンゾイル)-2-メチルアミノ-N’’-[2’’’-(メトキシカルボニル)-1’’’-(1’’’’-トリチルイミダゾール-4’’’’-イル)エタン-2’’’-イル]テトラヒドロイソキノリン(化合物(22))
(2S,2’’’S)-N-(4’-tert-ブチルベンゾイル)-2-メチルアミノ-N’’-[2’’’-(メトキシカルボニル)-1’’’-(1’’’’-トリチルイミダゾール-4’’’’-イル)エタン-2’’’-イル]テトラヒドロイソキノリン(化合物(22))

化合物(22)は、前記製造法2の方法(具体的には、前記した化合物(20)の製造方法)に準じて合成した。
1H NMR (300 MHz): δ = 7.77 (br d, J = 1.8 Hz, 1H), 7.43-7.33 (m, 10H), 7.25-7.22 (m, 5H), 7.11-7.04 (m, 8H), 6.84 (br d, J = 1.5 Hz, 1H), 3.74 (d, J = 5.4 Hz, 1H), 3.67 (s, 3H), 3.64 (dd, J = 17.7, 5.7 Hz, 2H), 3.55 (d, J = 12.6 Hz, 1H), 3.15 (dd, J = 15.6, 5.1 Hz, 1H), 3.01 (dd, J = 15.6, 8.1 Hz, 1H), 2.37-2.28 (m, 1H), 1.29 (s, 9H).
1H NMR (300 MHz): δ = 7.77 (br d, J = 1.8 Hz, 1H), 7.43-7.33 (m, 10H), 7.25-7.22 (m, 5H), 7.11-7.04 (m, 8H), 6.84 (br d, J = 1.5 Hz, 1H), 3.74 (d, J = 5.4 Hz, 1H), 3.67 (s, 3H), 3.64 (dd, J = 17.7, 5.7 Hz, 2H), 3.55 (d, J = 12.6 Hz, 1H), 3.15 (dd, J = 15.6, 5.1 Hz, 1H), 3.01 (dd, J = 15.6, 8.1 Hz, 1H), 2.37-2.28 (m, 1H), 1.29 (s, 9H).
実施例23
4-tert-ブチルベンゾイル-Pro-His(Trt)-OMe(化合物(23))
4-tert-ブチルベンゾイル-Pro-His(Trt)-OMe(化合物(23))
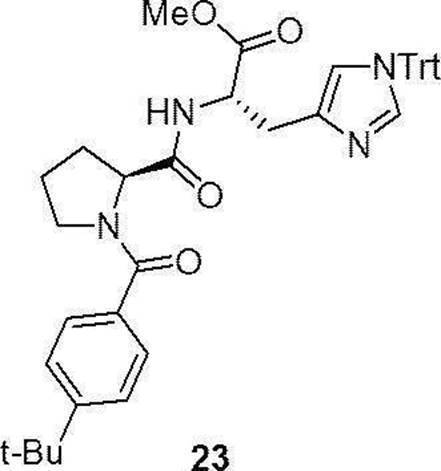
化合物(23)は、前記製造法1の方法(具体的には、前記した化合物(18)の製造方法)に準じて、市販品のFmoc保護プロリン(渡辺化学工業株式会社製)から合成した。
1H NMR (300 MHz): δ = 8.64 (br d, J = 6.9 Hz, 0.33H), 8.00 (br d, J = 7.8 Hz, 0.67H), 7.53 (br d, J = 8.1 Hz, 2H), 7.37-7.23 (m, 12H), 7.07-7.06 (m, 6H), 6.58 (br s, 0.67H), 6.55 (br s, 0.33H), 4.86-4.80 (m, 1H), 4.76 (br t, J = 6.5 Hz, 0.67H), 4.35 (br s, 0.33H), 3.91-3.86 (m, 0.67H), 3.63 (br s, 1H), 3.55 (s, 2H), 3.52 (m, 0.33H), 3.39 (s, 1H), 3.12-2.97 (m, 2H), 2.20-2.18 (m, 1.66H), 1.98-1.96(m, 1.67H), 1.83-1.77 (m, 0.67H), 1.28 (s, 9H);
HRMS (EI) Calcd. For C42H44N4O4 [M]+: 668.3363. Found: 668.3358.
1H NMR (300 MHz): δ = 8.64 (br d, J = 6.9 Hz, 0.33H), 8.00 (br d, J = 7.8 Hz, 0.67H), 7.53 (br d, J = 8.1 Hz, 2H), 7.37-7.23 (m, 12H), 7.07-7.06 (m, 6H), 6.58 (br s, 0.67H), 6.55 (br s, 0.33H), 4.86-4.80 (m, 1H), 4.76 (br t, J = 6.5 Hz, 0.67H), 4.35 (br s, 0.33H), 3.91-3.86 (m, 0.67H), 3.63 (br s, 1H), 3.55 (s, 2H), 3.52 (m, 0.33H), 3.39 (s, 1H), 3.12-2.97 (m, 2H), 2.20-2.18 (m, 1.66H), 1.98-1.96(m, 1.67H), 1.83-1.77 (m, 0.67H), 1.28 (s, 9H);
HRMS (EI) Calcd. For C42H44N4O4 [M]+: 668.3363. Found: 668.3358.
実施例24
(2S,2’’’S)-N-(4’-tert-ブチルベンゾイル)-2-メチルアミノ-N’’-[2’’’-(メトキシカルボニル)-1’’’-(1’’’’-トリチルイミダゾール-4’’’’-イル)エタン-2’’’-イル]ピロリジン(化合物(24))
(2S,2’’’S)-N-(4’-tert-ブチルベンゾイル)-2-メチルアミノ-N’’-[2’’’-(メトキシカルボニル)-1’’’-(1’’’’-トリチルイミダゾール-4’’’’-イル)エタン-2’’’-イル]ピロリジン(化合物(24))

化合物(24)は、前記製造法2の方法(具体的には、前記した化合物(20)の製造方法)に準じて合成した。
1H NMR (300 MHz): δ = 7.45-7.41 (m, 3H), 7.37-7.30 (m, 12H), 7.14-7.11 (m, 6H), 6.59 (br s, 1H), 4.41-4.32 (m, 1H), 3.88-3.74 (m, 1H), 3.67-3.53 (m, 1H), 3.58 (s, 3H), 2.96-2.71 (m, 4H), 2.03-1.61 (m, 5H), 1.33 (s, 2.25H), 1.30 (s, 6.75H);13C NMR (75 MHz): δ = 175.0, 153.0, 142.5, 138.4, 137.4, 134.4, 129.8, 128.0, 127.2, 127.0, 125.2, 125.0, 119.2, 75.1, 61.9, 61.6, 57.3, 51.5, 51.2, 50.7, 50.0, 34.8, 34.7, 32.3, 31.18, 31.16, 29.7, 28.8, 28.6, 25.1, 25.0;
HRMS (EI) Calcd. For C42H46N4O3 [M]+: 654.3570. Found: 654.3575.
1H NMR (300 MHz): δ = 7.45-7.41 (m, 3H), 7.37-7.30 (m, 12H), 7.14-7.11 (m, 6H), 6.59 (br s, 1H), 4.41-4.32 (m, 1H), 3.88-3.74 (m, 1H), 3.67-3.53 (m, 1H), 3.58 (s, 3H), 2.96-2.71 (m, 4H), 2.03-1.61 (m, 5H), 1.33 (s, 2.25H), 1.30 (s, 6.75H);13C NMR (75 MHz): δ = 175.0, 153.0, 142.5, 138.4, 137.4, 134.4, 129.8, 128.0, 127.2, 127.0, 125.2, 125.0, 119.2, 75.1, 61.9, 61.6, 57.3, 51.5, 51.2, 50.7, 50.0, 34.8, 34.7, 32.3, 31.18, 31.16, 29.7, 28.8, 28.6, 25.1, 25.0;
HRMS (EI) Calcd. For C42H46N4O3 [M]+: 654.3570. Found: 654.3575.
実施例25
4-tert-ブチルベンゾイル-ホモPro-Ser(Bn)-アニリド(化合物(25))
4-tert-ブチルベンゾイル-ホモPro-Ser(Bn)-アニリド(化合物(25))

化合物(25)は、前記製造法3の方法に準じて、下記式:

で示される合成スキームに従って合成した。具体的には、化合物(9c)をジクロロメタンに溶解し、アニリン(2.0当量)とNMM(5.0当量)を加えた。氷冷撹拌下、続いてBOP(1.2当量)を加えて16時間撹拌した。飽和塩化アンモニウム水溶液を用いて反応を停止させ、酢酸エチルで抽出した。飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥させた。ろ過後、減圧下溶媒留去を行い、残渣をシリカゲルカラムクロマトグラフィーに付すことにより、化合物(11b)を得た。
化合物(11b)をアセトニトリルに溶解し、20%(V/V)になるようにジエチルアミンを加えた。20分撹拌後、減圧下溶媒留去を行った。残渣は精製することなく次の反応に使用した。得られた残渣をジクロロメタンに溶解し、市販品のFmoc保護ホモプロリン(12a)(1.1当量)とNMM(7.0当量)を加えた。氷冷撹拌下、BOP(1.2当量)を加えて16時間撹拌した。飽和塩化アンモニウム水溶液を用いて反応を停止させ、酢酸エチルで抽出した。飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥させた。ろ過後、減圧下溶媒留去を行い、残渣をシリカゲルカラムクロマトグラフィーに付すことにより、化合物(13b)を得た。
化合物(13b)をアセトニトリルに溶解し、20%(V/V)になるようにジエチルアミンを加えた。20分撹拌後、減圧下溶媒留去を行った。残渣は精製することなく次の反応に使用した。得られた残渣をピリジンに溶解し、4-t-ブチル安息香酸クロリド(14a)(1.5当量)とDMAP(2.0当量)を加えて16時間撹拌した。飽和塩化アンモニウム水溶液を用いて反応を停止させ、酢酸エチルで抽出した。飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥させた。ろ過後、減圧下溶媒留去を行い、残渣をシリカゲルカラムクロマトグラフィーに付すことにより、化合物(25)を得た。
1H NMR (300 MHz): δ = 8.73 (br s, 0.2H), 8.64 (br s, 0.8H), 7.58-7.51 (m, 2H), 7.41-7.24 (m, 11H), 7.12-7.07 (m, 1H), 5.30 (br s, 0.8H), 5.22 (br s, 0.2H), 4.77-4.76 (m, 1H), 4.67-4.51 (m, 2H), 4.06 (m, 1H), 3.77-3.67 (m, 2H), 3.24-3.21 (m, 0.2H), 3.08-3.01 (m, 0.8H), 2.41-2.37 (m, 1H), 1.90 (br s, 0.4H), 1.73 (m, 2.6H), 1.63-1.47 (m, 3H), 1.32 (s, 9H);
13C NMR (75 MHz): δ = 172.9, 171.1, 167.8, 153.6, 137.9, 137.2, 131.9, 128.9, 128.6, 128.5, 127.9, 127.8, 127.1, 125.5, 124.3, 119.9, 73.6, 69.3, 66.6, 53.8, 53.4, 52.9, 47.2, 46.2, 38.4, 34.8, 31.1, 25.3, 20.7;
HRMS (EI) Calcd. For C33H39N3O4 [M]+: 541.2941. Found: 541.2943.
化合物(11b)をアセトニトリルに溶解し、20%(V/V)になるようにジエチルアミンを加えた。20分撹拌後、減圧下溶媒留去を行った。残渣は精製することなく次の反応に使用した。得られた残渣をジクロロメタンに溶解し、市販品のFmoc保護ホモプロリン(12a)(1.1当量)とNMM(7.0当量)を加えた。氷冷撹拌下、BOP(1.2当量)を加えて16時間撹拌した。飽和塩化アンモニウム水溶液を用いて反応を停止させ、酢酸エチルで抽出した。飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥させた。ろ過後、減圧下溶媒留去を行い、残渣をシリカゲルカラムクロマトグラフィーに付すことにより、化合物(13b)を得た。
化合物(13b)をアセトニトリルに溶解し、20%(V/V)になるようにジエチルアミンを加えた。20分撹拌後、減圧下溶媒留去を行った。残渣は精製することなく次の反応に使用した。得られた残渣をピリジンに溶解し、4-t-ブチル安息香酸クロリド(14a)(1.5当量)とDMAP(2.0当量)を加えて16時間撹拌した。飽和塩化アンモニウム水溶液を用いて反応を停止させ、酢酸エチルで抽出した。飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥させた。ろ過後、減圧下溶媒留去を行い、残渣をシリカゲルカラムクロマトグラフィーに付すことにより、化合物(25)を得た。
1H NMR (300 MHz): δ = 8.73 (br s, 0.2H), 8.64 (br s, 0.8H), 7.58-7.51 (m, 2H), 7.41-7.24 (m, 11H), 7.12-7.07 (m, 1H), 5.30 (br s, 0.8H), 5.22 (br s, 0.2H), 4.77-4.76 (m, 1H), 4.67-4.51 (m, 2H), 4.06 (m, 1H), 3.77-3.67 (m, 2H), 3.24-3.21 (m, 0.2H), 3.08-3.01 (m, 0.8H), 2.41-2.37 (m, 1H), 1.90 (br s, 0.4H), 1.73 (m, 2.6H), 1.63-1.47 (m, 3H), 1.32 (s, 9H);
13C NMR (75 MHz): δ = 172.9, 171.1, 167.8, 153.6, 137.9, 137.2, 131.9, 128.9, 128.6, 128.5, 127.9, 127.8, 127.1, 125.5, 124.3, 119.9, 73.6, 69.3, 66.6, 53.8, 53.4, 52.9, 47.2, 46.2, 38.4, 34.8, 31.1, 25.3, 20.7;
HRMS (EI) Calcd. For C33H39N3O4 [M]+: 541.2941. Found: 541.2943.
実施例26
4-tert-ブチルベンゾイル-ホモPro-Tyr(t-Bu)-アニリド(化合物(26))
4-tert-ブチルベンゾイル-ホモPro-Tyr(t-Bu)-アニリド(化合物(26))
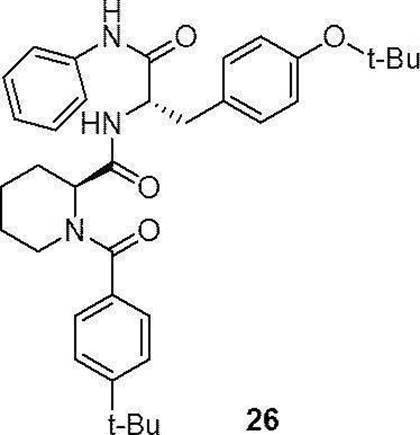
化合物(26)は、前記製造法3の方法(具体的には、前記した化合物(25)の製造方法)に準じて合成した。
1H NMR (300 MHz): δ = 7.47-7.44 (m, 2H), 7.34 (br d, J = 7.8 Hz, 2H), 7.13-7.06 (m, 4H), 6.77 (br d, J = 7.8 Hz, 2H), 6.34-6.28 (m, 1H), 5.67 (br s, 1H), 5.18 (br s, 1H), 4.71-4.69 (m, 1H), 3.47 (br d, J = 12.9 Hz, 1H), 3.21 (dd, J = 14.4, 5.4 Hz, 1H), 3.07 (dd, J = 14.7, 9.3 Hz, 1H), 2.31 (m, 2H), 1.67-1.56 (m, 5H), 1.43 (br s, 3H), 1.35 (s, 9H), 1.21 (s, 9H);
13C NMR (75 MHz): δ = 173.3, 172.9, 171.3, 154.2, 153.9, 131.7, 131.6, 129.6, 127.8, 127.3, 127.2, 125.6, 125.5, 124.2, 78.2, 53.7, 53.4, 50.9, 45.9, 36.0, 34.9, 31.2, 31.1, 30.9, 29.7, 28.7, 25.4, 25.1, 20.6.
1H NMR (300 MHz): δ = 7.47-7.44 (m, 2H), 7.34 (br d, J = 7.8 Hz, 2H), 7.13-7.06 (m, 4H), 6.77 (br d, J = 7.8 Hz, 2H), 6.34-6.28 (m, 1H), 5.67 (br s, 1H), 5.18 (br s, 1H), 4.71-4.69 (m, 1H), 3.47 (br d, J = 12.9 Hz, 1H), 3.21 (dd, J = 14.4, 5.4 Hz, 1H), 3.07 (dd, J = 14.7, 9.3 Hz, 1H), 2.31 (m, 2H), 1.67-1.56 (m, 5H), 1.43 (br s, 3H), 1.35 (s, 9H), 1.21 (s, 9H);
13C NMR (75 MHz): δ = 173.3, 172.9, 171.3, 154.2, 153.9, 131.7, 131.6, 129.6, 127.8, 127.3, 127.2, 125.6, 125.5, 124.2, 78.2, 53.7, 53.4, 50.9, 45.9, 36.0, 34.9, 31.2, 31.1, 30.9, 29.7, 28.7, 25.4, 25.1, 20.6.
実施例27
4-tert-ブチルベンゾイル-ホモPro-Phe-アニリド(化合物(27))
4-tert-ブチルベンゾイル-ホモPro-Phe-アニリド(化合物(27))
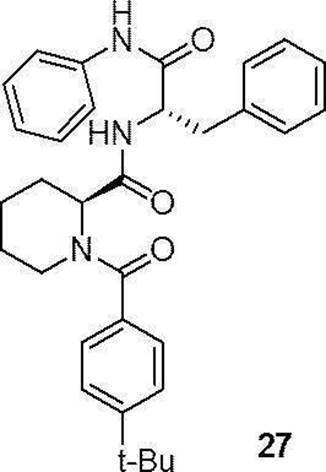
化合物(27)は、前記製造法3の方法(具体的には、前記した化合物(25)の製造方法)に準じて合成した。
1H NMR (300 MHz): δ = 8.65 (br s, 1H), 7.51-7.05 (m, 14H), 5.19 (br s, 1H), 4.90-4.89 (m, 1H), 4.49-4.42 (m, 0.5H), 3.52 (br s, 1H), 3.36-3.31 (m, 0.5H), 3.14-3.06 (m, 1.5H), 2.41-2.27 (m, 1.5H), 1.62-1.45 (m, 6H), 1.35 (s, 9H).
1H NMR (300 MHz): δ = 8.65 (br s, 1H), 7.51-7.05 (m, 14H), 5.19 (br s, 1H), 4.90-4.89 (m, 1H), 4.49-4.42 (m, 0.5H), 3.52 (br s, 1H), 3.36-3.31 (m, 0.5H), 3.14-3.06 (m, 1.5H), 2.41-2.27 (m, 1.5H), 1.62-1.45 (m, 6H), 1.35 (s, 9H).
実施例28
4-tert-ブチルベンゾイル-ホモPro-Trp(Tris)-アニリド(化合物(28))
4-tert-ブチルベンゾイル-ホモPro-Trp(Tris)-アニリド(化合物(28))
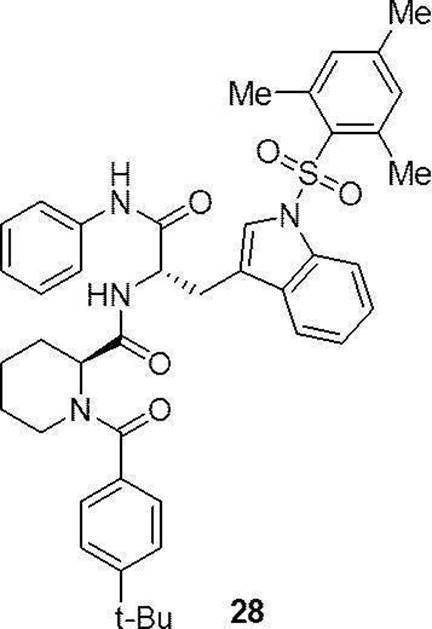
化合物(28)は、前記製造法3の方法(具体的には、前記した化合物(25)の製造方法)に準じて合成した。
1H NMR (300 MHz): δ = 8.35 (br s, 0.7H), 8.00 (dd, J = 6.8, 2.0 Hz, 0.3H), 7.65-7.62 (m, 1H), 7.54-7.05 (m, 12H), 6.92-6.84 (m, 2H), 5.18 (br s, 1H), 5.03-5.00 (m, 1H), 3.58 (br d, J = 14.1 Hz, 1H), 3.35 (d, J = 7.2 Hz, 1H), 2.74-2.66 (m, 1H), 2.42 (s, 6H), 2.34-2.32 (m, 1H), 2.25 (s, 3H), 1.66-1.46 (m, 6H), 1.31 (s, 9H);
13C NMR (75 MHz): δ = 173.1, 171.8, 168.7, 153.6, 144.1, 140.2, 137.5, 134.9, 132.7, 132.4, 131.2, 130.0, 129.8, 128.9, 127.1, 125.5, 124.6, 124.5, 124.3, 122.9, 120.1, 119.5, 115.0, 112.7, 57.8, 54.2, 53.8, 53.6, 46.3, 35.1, 34.8, 31.1, 29.7, 26.8, 25.2, 22.6, 21.0, 20.7;
HRMS (EI) Calcd. For C43H48N4O5S[M]+: 732.3345. Found: 733.3338.
1H NMR (300 MHz): δ = 8.35 (br s, 0.7H), 8.00 (dd, J = 6.8, 2.0 Hz, 0.3H), 7.65-7.62 (m, 1H), 7.54-7.05 (m, 12H), 6.92-6.84 (m, 2H), 5.18 (br s, 1H), 5.03-5.00 (m, 1H), 3.58 (br d, J = 14.1 Hz, 1H), 3.35 (d, J = 7.2 Hz, 1H), 2.74-2.66 (m, 1H), 2.42 (s, 6H), 2.34-2.32 (m, 1H), 2.25 (s, 3H), 1.66-1.46 (m, 6H), 1.31 (s, 9H);
13C NMR (75 MHz): δ = 173.1, 171.8, 168.7, 153.6, 144.1, 140.2, 137.5, 134.9, 132.7, 132.4, 131.2, 130.0, 129.8, 128.9, 127.1, 125.5, 124.6, 124.5, 124.3, 122.9, 120.1, 119.5, 115.0, 112.7, 57.8, 54.2, 53.8, 53.6, 46.3, 35.1, 34.8, 31.1, 29.7, 26.8, 25.2, 22.6, 21.0, 20.7;
HRMS (EI) Calcd. For C43H48N4O5S[M]+: 732.3345. Found: 733.3338.
実施例29
4-tert-ブチルベンゾイル-ホモPro-Tyr(t-Bu)-ベンズヒドリドアミド(化合物(29))
4-tert-ブチルベンゾイル-ホモPro-Tyr(t-Bu)-ベンズヒドリドアミド(化合物(29))
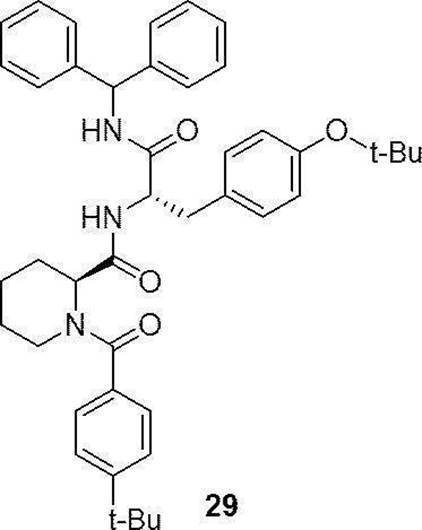
化合物(29)は、前記製造法3の方法(具体的には、前記した化合物(25)の製造方法)に準じて合成した。
1H NMR (300 MHz): δ = 7.43 (br d, J = 7.5 Hz, 2H), 7.32-7.22 (m, 12H), 7.02 (br d, J = 7.8 Hz, 2H), 6.94 (d, J = 8.1 Hz, 2H), 6.72 (br d, J = 7.2 Hz, 2H), 6.24 (d, J = 8.1 Hz, 1H), 5.08 (br s, 1H), 4.78-4.67 (m, 1H), 3.50 (br d, J = 13.2 Hz, 1H), 3.16 (dd, J = 14.3, 5.9 Hz, 1H), 3.04 (dd, J = 14.3, 8.6 Hz, 1H), 2.45-2.42 (m, 1H), 2.23-2.20 (m, 1H), 1.85 (br s, 1H), 1.65-1.59 (m, 2H), 1.46 (br s, 2H), 1.34 (s, 9H), 1.22 (s, 9H);
13C NMR (75 MHz): δ = 172.8, 171.5, 169.9, 154.3, 153.9, 141.2, 131.4, 129.6, 128.6, 127.4, 127.3, 125.4, 124.2, 78.2, 56.9, 54.3, 53.4, 45.9, 36.4, 34.9, 31.6, 28.7, 25.3, 25.2, 20.6.
1H NMR (300 MHz): δ = 7.43 (br d, J = 7.5 Hz, 2H), 7.32-7.22 (m, 12H), 7.02 (br d, J = 7.8 Hz, 2H), 6.94 (d, J = 8.1 Hz, 2H), 6.72 (br d, J = 7.2 Hz, 2H), 6.24 (d, J = 8.1 Hz, 1H), 5.08 (br s, 1H), 4.78-4.67 (m, 1H), 3.50 (br d, J = 13.2 Hz, 1H), 3.16 (dd, J = 14.3, 5.9 Hz, 1H), 3.04 (dd, J = 14.3, 8.6 Hz, 1H), 2.45-2.42 (m, 1H), 2.23-2.20 (m, 1H), 1.85 (br s, 1H), 1.65-1.59 (m, 2H), 1.46 (br s, 2H), 1.34 (s, 9H), 1.22 (s, 9H);
13C NMR (75 MHz): δ = 172.8, 171.5, 169.9, 154.3, 153.9, 141.2, 131.4, 129.6, 128.6, 127.4, 127.3, 125.4, 124.2, 78.2, 56.9, 54.3, 53.4, 45.9, 36.4, 34.9, 31.6, 28.7, 25.3, 25.2, 20.6.
実施例30
4-tert-ブチルベンゾイル-ホモPro-Nal(2)-アニリド(化合物(30))
4-tert-ブチルベンゾイル-ホモPro-Nal(2)-アニリド(化合物(30))
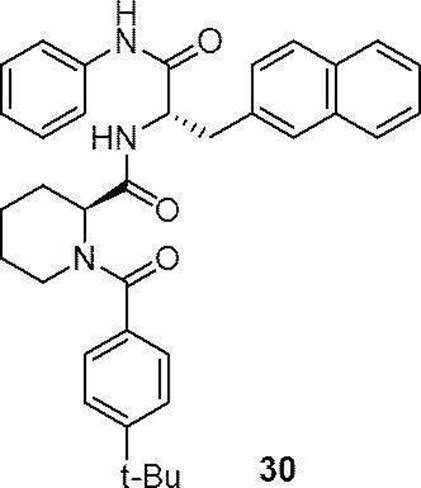
化合物(30)は、前記製造法3の方法(具体的には、前記した化合物(25)の製造方法)に準じて合成した。
1H NMR (300 MHz): δ = 8.49 (br s, 0.8H), 8.02 (d, J = 8.7 Hz, 0.2H), 7.72-7.67 (m, 2H), 7.61 (br s, 1H), 7.53-7.46 (m, 3H), 7.41-7.29 (m, 7H), 7.12-7.07 (m, 4H), 5.15 (br s, 1H), 5.02-5.01 (m, 1H), 3.48 (dd, J = 14.4, 4.8 Hz, 1H), 3.33 (dd, J = 14.4, 9.3 Hz, 1H), 3.22 (br d, J = 13.5 Hz, 1H), 2.28-2.17 (m, 2H), 1.75-1.56 (m, 4H), 1.35-1.19 (m, 1H), 1.34 (s, 9H);
13C NMR (75 MHz): δ = 172.9, 171.9, 169.0, 153.9, 137.6, 134.2, 133.4, 132.4, 131.1, 130.0, 128.9, 128.6, 127.8, 127.6, 127.85, 127.3, 126.9, 126.2, 125.7, 125.4, 125.3, 124.4, 120.1, 54.5, 53.6, 45.8, 37.1, 34.8, 31.2, 25.3, 25.0, 20.5.
1H NMR (300 MHz): δ = 8.49 (br s, 0.8H), 8.02 (d, J = 8.7 Hz, 0.2H), 7.72-7.67 (m, 2H), 7.61 (br s, 1H), 7.53-7.46 (m, 3H), 7.41-7.29 (m, 7H), 7.12-7.07 (m, 4H), 5.15 (br s, 1H), 5.02-5.01 (m, 1H), 3.48 (dd, J = 14.4, 4.8 Hz, 1H), 3.33 (dd, J = 14.4, 9.3 Hz, 1H), 3.22 (br d, J = 13.5 Hz, 1H), 2.28-2.17 (m, 2H), 1.75-1.56 (m, 4H), 1.35-1.19 (m, 1H), 1.34 (s, 9H);
13C NMR (75 MHz): δ = 172.9, 171.9, 169.0, 153.9, 137.6, 134.2, 133.4, 132.4, 131.1, 130.0, 128.9, 128.6, 127.8, 127.6, 127.85, 127.3, 126.9, 126.2, 125.7, 125.4, 125.3, 124.4, 120.1, 54.5, 53.6, 45.8, 37.1, 34.8, 31.2, 25.3, 25.0, 20.5.
実施例31
4-tert-ブチルベンゾイル-ホモPro-Nal(2)-ベンズヒドリルアミド(化合物(31))
4-tert-ブチルベンゾイル-ホモPro-Nal(2)-ベンズヒドリルアミド(化合物(31))
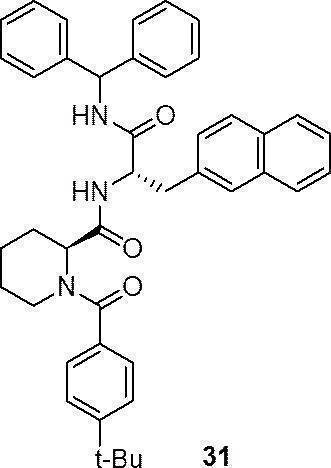
化合物(31)は、前記製造法3の方法(具体的には、前記した化合物(25)の製造方法)に準じて合成した。
1H NMR (300 MHz): δ = 7.72 (br d, J = 7.8 Hz, 1.5H), 7.61-7.30 (m, 6.5H), 7.27-6.93 (m, 15H), 6.24 (br d, J = 8.4 Hz, 1H), 5.08 (br s, 1H), 4.98-4.95 (m, 1H), 3.33 (d, J = 6.6 Hz, 3H), 2.44-2.40 (m, 1H), 2.21-2.16 (m, 1H), 1.82 (br s, 1H), 1.58 (m, 3H), 1.32 (s, 9H) , 1.35-1.26 (m, 1H);
13C NMR (75 MHz): δ = 172.8, 171.3, 169.8, 153.6, 141.3, 141.1, 134.1, 133.4, 132.4, 131.3, 128.6, 128.5, 128.2, 127.9, 127.6, 127.5, 127.31 127.27, 127.1, 127.0, 126.1, 125.7, 125.3, 56.9, 53.9, 53.5, 45.8, 37.5, 34.8, 31.2, 25.3, 25.1, 20.5.
1H NMR (300 MHz): δ = 7.72 (br d, J = 7.8 Hz, 1.5H), 7.61-7.30 (m, 6.5H), 7.27-6.93 (m, 15H), 6.24 (br d, J = 8.4 Hz, 1H), 5.08 (br s, 1H), 4.98-4.95 (m, 1H), 3.33 (d, J = 6.6 Hz, 3H), 2.44-2.40 (m, 1H), 2.21-2.16 (m, 1H), 1.82 (br s, 1H), 1.58 (m, 3H), 1.32 (s, 9H) , 1.35-1.26 (m, 1H);
13C NMR (75 MHz): δ = 172.8, 171.3, 169.8, 153.6, 141.3, 141.1, 134.1, 133.4, 132.4, 131.3, 128.6, 128.5, 128.2, 127.9, 127.6, 127.5, 127.31 127.27, 127.1, 127.0, 126.1, 125.7, 125.3, 56.9, 53.9, 53.5, 45.8, 37.5, 34.8, 31.2, 25.3, 25.1, 20.5.
実施例32
4-tert-ブチルベンゾイル-ホモPro-Ser(Trt)-アニリド(化合物(32))
4-tert-ブチルベンゾイル-ホモPro-Ser(Trt)-アニリド(化合物(32))
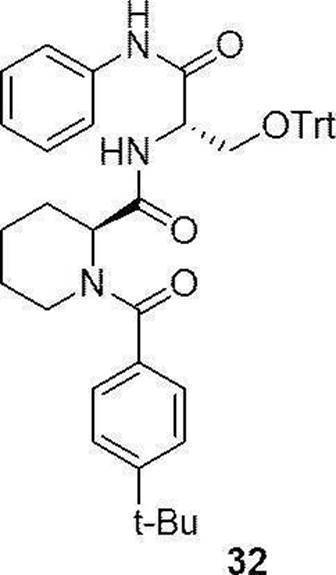
化合物(32)は、前記製造法3の方法(具体的には、前記した化合物(25)の製造方法)に準じて合成した。
1H NMR (300 MHz): δ = 8.61 (br s, 1H), 7.60 (br d, J = 7.5 Hz, 1H), 7.47-7.21 (m, 21H), 7.15-7.05 (m, 2H), 5.22 (br s, 1H), 4.77-4.71 (m, 1H), 3.98-3.96 (m, 1H), 3.75-3.71 (m, 1H), 3.22 (m, 1H), 2.84 (m, 1H), 2.38 (m, 1H), 1.70-1.54 (m, 6H), 1.31 (s, 9H);
13C NMR (75 MHz): δ = 173.1, 170.5, 167.9, 153.9, 149.8, 143.2, 137.8, 135.9, 131.4, 129.0, 128.7, 128.5, 128.1, 127.9, 127.4, 125.5, 124.39, 120.0, 87.2, 63.1, 54.4, 53.8, 46.3, 34.1, 31.1, 25.2, 20.7.
1H NMR (300 MHz): δ = 8.61 (br s, 1H), 7.60 (br d, J = 7.5 Hz, 1H), 7.47-7.21 (m, 21H), 7.15-7.05 (m, 2H), 5.22 (br s, 1H), 4.77-4.71 (m, 1H), 3.98-3.96 (m, 1H), 3.75-3.71 (m, 1H), 3.22 (m, 1H), 2.84 (m, 1H), 2.38 (m, 1H), 1.70-1.54 (m, 6H), 1.31 (s, 9H);
13C NMR (75 MHz): δ = 173.1, 170.5, 167.9, 153.9, 149.8, 143.2, 137.8, 135.9, 131.4, 129.0, 128.7, 128.5, 128.1, 127.9, 127.4, 125.5, 124.39, 120.0, 87.2, 63.1, 54.4, 53.8, 46.3, 34.1, 31.1, 25.2, 20.7.
実施例33
4-tert-ブチルベンゾイル-ホモPro-Ser(Bn)-ベンズヒドリルアミド(化合物(33))
4-tert-ブチルベンゾイル-ホモPro-Ser(Bn)-ベンズヒドリルアミド(化合物(33))
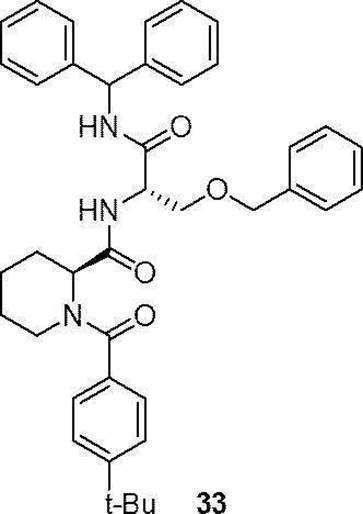
化合物(33)は、前記製造法3の方法(具体的には、前記した化合物(25)の製造方法)に準じて合成した。
1H NMR (300 MHz): δ = 7.35-7.14 (m, 20H), 6.27-6.22 (m, 1H), 5.24 (br s, 1H), 4.70-4.68 (m, 1H), 4.59-4.4.46 (m, 2H), 4.02 (br s, 1H), 3.67-3.62 (m, 2H), 3.04-3.00 (m, 1H), 2.33-2.30 (m, 1H), 1.78-1.49 (m, 6H), 1.29 (s, 9H).
1H NMR (300 MHz): δ = 7.35-7.14 (m, 20H), 6.27-6.22 (m, 1H), 5.24 (br s, 1H), 4.70-4.68 (m, 1H), 4.59-4.4.46 (m, 2H), 4.02 (br s, 1H), 3.67-3.62 (m, 2H), 3.04-3.00 (m, 1H), 2.33-2.30 (m, 1H), 1.78-1.49 (m, 6H), 1.29 (s, 9H).
実施例34
4-tert-ブチルベンゾイル-Hyp-His(Trt)-OMe(化合物(34))
4-tert-ブチルベンゾイル-Hyp-His(Trt)-OMe(化合物(34))
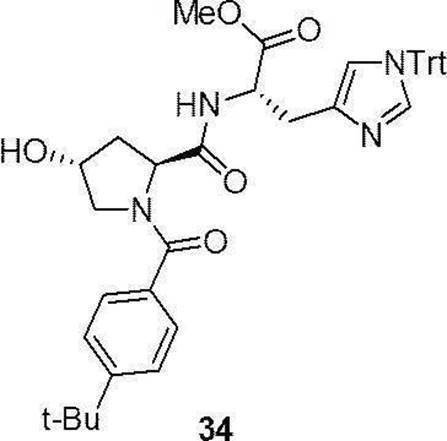
化合物(34)は、前記製造法4の方法に準じて、下記式:
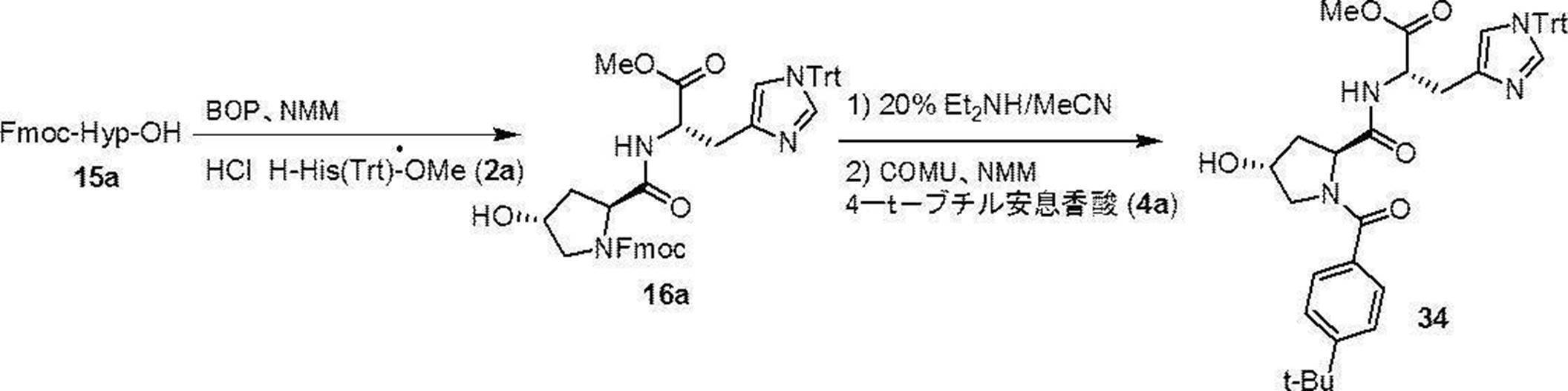
で示される合成スキームに従って合成した。具体的には、市販の1-Fmoc-3-ヒドロキシプロリン (15a)(渡辺化学工業株式会社製)をジクロロメタンに溶解し、市販の化合物(2a)(1.1当量)とNMM(7.0当量)を加えた。氷冷撹拌下、続いてBOP(1.2当量)を加えて16 時間撹拌した。飽和塩化アンモニウム水溶液を用いて反応を停止させ、酢酸エチルで抽出した。飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥させた。ろ過後、減圧下溶媒留去を行い、残渣をシリカゲルカラムクロマトグラフィーに付すことにより、化合物(16a)を得た。
化合物(16a)をアセトニトリルに溶解し、20%(V/V)になるようにジエチルアミンを加えた。20分撹拌後、減圧下溶媒留去を行った。残渣は精製することなく次の反応に使用した。得られた残渣をジクロロメタンに溶解し4-t-ブチル安息香酸(1.1当量)とNMM(7.0当量)を加えた。氷冷撹拌下、COMU(1.2 当量)を加えて16時間撹拌した。飽和塩化アンモニウム水溶液を用いて反応を停止させ、酢酸エチルで抽出した。飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥させた。ろ過後、減圧下溶媒留去を行い、残渣をシリカゲルカラムクロマトグラフィーに付すことにより、化合物(34)を得た。
1H NMR (300 MHz): δ = 8.01 (br s, 1H), 7.48 (d, J = 8.4 Hz, 2H), 7.39 (s, 1H), 7.33-7.28 (m, 11H), 7.09-7.05 (m, 6H), 6.61 (br s, 1H), 4.91-4.79 (m, 4H), 4.44 (br s, 1H), 3.79 (dd, J = 11.7, 3.6 Hz, 1H), 3.58 (s, 3H), 3.05 (br s, 2H), 2.34-2.23 (m, 2H), 1.28 (s, 9H);
13C NMR (75 MHz): δ = 171.4, 153.5, 142.2, 138.6, 136.1, 132.9, 129.7, 128.1, 127.6, 125.0, 119.7, 75.3, 70.5, 58.7, 52.7, 52.1, 34.8, 31.2, 29.7.
化合物(16a)をアセトニトリルに溶解し、20%(V/V)になるようにジエチルアミンを加えた。20分撹拌後、減圧下溶媒留去を行った。残渣は精製することなく次の反応に使用した。得られた残渣をジクロロメタンに溶解し4-t-ブチル安息香酸(1.1当量)とNMM(7.0当量)を加えた。氷冷撹拌下、COMU(1.2 当量)を加えて16時間撹拌した。飽和塩化アンモニウム水溶液を用いて反応を停止させ、酢酸エチルで抽出した。飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥させた。ろ過後、減圧下溶媒留去を行い、残渣をシリカゲルカラムクロマトグラフィーに付すことにより、化合物(34)を得た。
1H NMR (300 MHz): δ = 8.01 (br s, 1H), 7.48 (d, J = 8.4 Hz, 2H), 7.39 (s, 1H), 7.33-7.28 (m, 11H), 7.09-7.05 (m, 6H), 6.61 (br s, 1H), 4.91-4.79 (m, 4H), 4.44 (br s, 1H), 3.79 (dd, J = 11.7, 3.6 Hz, 1H), 3.58 (s, 3H), 3.05 (br s, 2H), 2.34-2.23 (m, 2H), 1.28 (s, 9H);
13C NMR (75 MHz): δ = 171.4, 153.5, 142.2, 138.6, 136.1, 132.9, 129.7, 128.1, 127.6, 125.0, 119.7, 75.3, 70.5, 58.7, 52.7, 52.1, 34.8, 31.2, 29.7.
試験例1:TCF/β-カテニン転写活性の測定と評価
(TOP細胞の作製)
既報(Emami KH, et al. Proc Natl Acad Sci USA, 101:12682-12687, 2004.)を参考にし、M50 Super 8x TOPFlashを用い、遺伝子導入後の細胞の選択培養を行うため、pGL4.32 [luc2P/NF-κB-RE/Hygro] ベクターから、制限酵素 (BamHI 2228とSal1 3833)を用いて、pGL4.32 ベクター内のハイグロマイシン(hygromycin)耐性遺伝子の塩基配列部分をp.M50 Super 8x TOPFlashに挿入し、Super TOPFlash プラスミドを作製した。
次に、Opti-MEMとFuGENE(登録商標) HD トランスフェクション試薬を用いてHEK293細胞にSuper TOPFlash プラスミドをトランスフェクションした後、ハイグロマイシン入りの低グルコース含有D-MEM (L-グルタミン、フェノールレッド含有)で選択培養を行った。Super TOPFlash プラスミド が導入されたTOPバルク細胞を96穴プレートに1 cell/穴になるように播種・培養し、クローン化TOP細胞を樹立した。TOP細胞は、10 % FBS、抗生物質(100 unit/mL ペニシリン、100 μg/mL ストレプトマイシン) 、ハイグロマイシンを含む低グルコース含有D-MEM (L-グルタミン、フェノールレッド含有)にて37 ℃、5 % CO2の環境下で培養した。
ルシフェラーゼアッセイに用いるTOP細胞は、内在性β-cateninレベルが低い。そこでWntシグナルを活性化させ発光量を増加させるために、Wnt-3A-L細胞由来のWntタンパク質を豊富に含むメディウム (Wnt上清) を用いて実験を行った。Wnt-3A細胞はL512細胞にWnt-3A発現ベクターを導入しクローン化した細胞で、Wnt-3A細胞は、活性Wnt-3Aタンパク質を分泌するためWnt-3A含有培養上清の調製に用いられる。10 % FBS、抗生物質 (100 unit/mL ペニシリン、100 μg/mL ストレプトマイシン)、0.4 mg/mL G418を含む低グルコース含有D-MEM (L-グルタミン、フェノールレッド含有) で 37 ℃、5 % CO2の環境下で培養・維持した。Wnt-3A細胞を1.25 x 106 cells/dishで播種し、G418を含まない培地にて培養した。コンフルエントとなる培養3日後、培養上清をシリンジフィルターに通して上清を回収した。このWnt含有培養上清をTOP細胞の培養系に添加し、評価系に用いた。
既報(Emami KH, et al. Proc Natl Acad Sci USA, 101:12682-12687, 2004.)を参考にし、M50 Super 8x TOPFlashを用い、遺伝子導入後の細胞の選択培養を行うため、pGL4.32 [luc2P/NF-κB-RE/Hygro] ベクターから、制限酵素 (BamHI 2228とSal1 3833)を用いて、pGL4.32 ベクター内のハイグロマイシン(hygromycin)耐性遺伝子の塩基配列部分をp.M50 Super 8x TOPFlashに挿入し、Super TOPFlash プラスミドを作製した。
次に、Opti-MEMとFuGENE(登録商標) HD トランスフェクション試薬を用いてHEK293細胞にSuper TOPFlash プラスミドをトランスフェクションした後、ハイグロマイシン入りの低グルコース含有D-MEM (L-グルタミン、フェノールレッド含有)で選択培養を行った。Super TOPFlash プラスミド が導入されたTOPバルク細胞を96穴プレートに1 cell/穴になるように播種・培養し、クローン化TOP細胞を樹立した。TOP細胞は、10 % FBS、抗生物質(100 unit/mL ペニシリン、100 μg/mL ストレプトマイシン) 、ハイグロマイシンを含む低グルコース含有D-MEM (L-グルタミン、フェノールレッド含有)にて37 ℃、5 % CO2の環境下で培養した。
ルシフェラーゼアッセイに用いるTOP細胞は、内在性β-cateninレベルが低い。そこでWntシグナルを活性化させ発光量を増加させるために、Wnt-3A-L細胞由来のWntタンパク質を豊富に含むメディウム (Wnt上清) を用いて実験を行った。Wnt-3A細胞はL512細胞にWnt-3A発現ベクターを導入しクローン化した細胞で、Wnt-3A細胞は、活性Wnt-3Aタンパク質を分泌するためWnt-3A含有培養上清の調製に用いられる。10 % FBS、抗生物質 (100 unit/mL ペニシリン、100 μg/mL ストレプトマイシン)、0.4 mg/mL G418を含む低グルコース含有D-MEM (L-グルタミン、フェノールレッド含有) で 37 ℃、5 % CO2の環境下で培養・維持した。Wnt-3A細胞を1.25 x 106 cells/dishで播種し、G418を含まない培地にて培養した。コンフルエントとなる培養3日後、培養上清をシリンジフィルターに通して上清を回収した。このWnt含有培養上清をTOP細胞の培養系に添加し、評価系に用いた。
(使用化合物)
試験に使用した本発明の化合物(I)は、ジメチルスルホキシドを溶媒として、100 mMに溶解し使用した。
試験に使用した本発明の化合物(I)は、ジメチルスルホキシドを溶媒として、100 mMに溶解し使用した。
(TCF/β-カテニン転写活性の測定)
TOP細胞をリン酸バッファー(PBS(-))ではがして回収し、遠心後、上清を除いたペレットにWnt含有培養上清を加えピペッティングし、96穴プレートに5 x 104 個/wellずつTOP細胞を播種した。さらにその翌日、化合物を処置し、24時間培養した。その後、細胞溶解剤を調製し、100 μLずつ加え、プレートを30分間シェイカー上におき細胞を溶解させた。1 wellずつ細胞溶解液をピペッティングして、100 μLずつ白色ポリスチレン製96穴プレート(Thermo Fisher Scientific, Tokyo, Japan) に移し、調製した発光試薬をマイクロプレートリーダー(GloMax(登録商標)Discover System) を用いて発光試薬のインジェクション及び発光測定を行った。
細胞溶解剤及び発光試薬の成分表は以下の通りである(表1)。試薬は、用時調製して用いた。
なお、評価のポジティブコントロールには、既存のWnt/β-catenin阻害薬(Wntシグナル伝達経路阻害薬)ICG-001を用いた。
TOP細胞をリン酸バッファー(PBS(-))ではがして回収し、遠心後、上清を除いたペレットにWnt含有培養上清を加えピペッティングし、96穴プレートに5 x 104 個/wellずつTOP細胞を播種した。さらにその翌日、化合物を処置し、24時間培養した。その後、細胞溶解剤を調製し、100 μLずつ加え、プレートを30分間シェイカー上におき細胞を溶解させた。1 wellずつ細胞溶解液をピペッティングして、100 μLずつ白色ポリスチレン製96穴プレート(Thermo Fisher Scientific, Tokyo, Japan) に移し、調製した発光試薬をマイクロプレートリーダー(GloMax(登録商標)Discover System) を用いて発光試薬のインジェクション及び発光測定を行った。
細胞溶解剤及び発光試薬の成分表は以下の通りである(表1)。試薬は、用時調製して用いた。
なお、評価のポジティブコントロールには、既存のWnt/β-catenin阻害薬(Wntシグナル伝達経路阻害薬)ICG-001を用いた。

評価結果を以下の通りである(表2)。低い%数値が、Wnt/β-catenin阻害抑制効果が高いことを示す。これらの結果から、本発明の化合物(I)群は、Wnt/β-cateninシグナルを抑制することが確認された。

試験例2:WST-8アッセイ
既報(Imayoshi N, et al. Biochem Biophys Res Commun, 484:262-268, 2017.)に従い、2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt (WST-8) アッセイを用いて細胞生存率の測定を行った。各種癌細胞(NCI-H929細胞、MM1.s細胞、RPMI8226細胞、U266細胞、AMO-1細胞及びOPM-2細胞(多発性骨髄腫(MM)細胞);AsPC-1細胞及びAsPC-1/HA細胞(膵臓癌細胞);HT29細胞、SW480細胞及びHCT116/E6細胞(大腸癌細胞);MV4;11細胞、KG1a細胞及びHL-60細胞(急性骨髄性白血病細胞);RS4;11細胞及びNalm6細胞(急性リンパ芽球性白血病細胞);Raji細胞(悪性リンパ腫細胞);KMB5細胞及びKMB5/STIR細胞(慢性骨髄性白血病細胞);MDA-MB-231細胞(乳癌細胞);U2510MG細胞(神経膠芽腫細胞))を96穴プレート (Thermo Fisher Scientific, Waltham, MA, U.S.A.) にそれぞれの適切な細胞濃度の浮遊液を90μLずつ播種し、3~4時間培養した。その後、本発明の化合物(I)をそれぞれ10μL/wellずつ処置し、72時間反応させた。69時間経過した時点で、生細胞数測定試薬SF (ナカライテスク)、Cell Counting Kit-8 (同仁化学研究所) を10 μL/wellずつ加え、3時間反応させ、GloMax Discover System (Promega, Madison, WI, U.S.A.) を用いて450 nmの吸光度を測定した。化合物(I)の各濃度での吸光度/化合物無処置での吸光度の比を算出し、これに100を乗じて、細胞生存率を算出した。
結果を図1(化合物(1))、図5(化合物(7))、図7(化合物(11))、図9(化合物(10)、化合物(24))、図10(化合物(12))、図13、18、19、20及び22(破線)(化合物(19))に示した。これらの結果から、本発明の化合物(I)群が各種細胞の増殖を濃度依存的に抑制することが確認された。
試験例3:定量的RT-PCR(qRT-PCR)法
既報(Imayoshi N, et al. Biochem Biophys Res Commun, 484:262-268, 2017.)に従いmRNAの発現解析をqRT-PCR法を用いて解析した。各種癌細胞を96穴プレート (Thermo Fisher Scientific, Waltham, MA, U.S.A.) にそれぞれの適切な細胞濃度で3時間培養した。その後、各化合物を、胎仔牛血清を含まない各培養液1 mLに溶解し、処置した。1、4、24時間後に、全RNAをillustra RNAspin Mini RNA Isolation キット (GE Healthcare, Chicago, IL, U.S.A.) を用いて抽出し、NanoDrop 2000 (Thermo Fisher Scientific) を用いて定量した。その後、High Capacity Transcription キット (Applied Biosystems, Foster, CA, U.S.A.) と、Takara PCR Thermal Cycler MP (タカラバイオ) を用いて、逆転写を行い、cDNAを合成した。合成したcDNAを用い、C-MYC、CCND1、CTNNB1 mRNAの発現量をThermal Cycler Dice Real Time System II (タカラバイオ) で解析し、その際の内部標準には18S rRNAを用いた。この時、FastStart Essential DNA Probes Master (Roche Diagnostics, Basel, Switzerland)、Universal Probe Library Probe (Roche)、プライマー (Thermo Fisher Scientific) を使用した(表3)。

結果を図2(化合物(1))、図11(化合物(12))及び図16(化合物(19))に示した。これらの結果によれば、本発明の化合物(I)群は、β-カテニン(CTNNB1) mRNA発現を変化させないが、Wnt/β-カテニンシグナル経路の下流遺伝子であるC-MYCやcyclin D1 (CCND1) mRNA発現を減少させることが確認された。
試験例4:ウェスタンブロッティング(Western blotting)法
RT-PCR法と同様に、各種癌細胞を培養し、各化合物を処置した。既報(Imayoshi N, et al. Biochem Biophys Res Commun, 484:262-268, 2017.)に従い各評価時間、化合物を処理し、全タンパク質を回収した。タンパク質抽出試薬として、protease inhibitor (Sigma-Aldrich)、NaF (和光純薬工業)、酸化バナデート (Sigma-Aldrich) を加えたradio-immunoprecipitation assay buffer (RIPA buffer; 和光純薬工業) を作製した。回収した細胞に抽出試薬を添加した。 なお、試薬の容量は、タンパク質濃度を揃える目的で細胞数に応じて設定した。氷冷 (20分) し、その間に5分ごとに10秒間、計4回ボルテックスをかけ、細胞を試薬中に溶解させた。その後、遠心 (12000 rpm、20分、4℃)し、その上清を抽出タンパク質として回収した。回収したタンパク質はQubit Protein Assay kit (Thermo Fisher Scientific)、Quantus Fluorometer (Promega) を用いて定量を行い、タンパク質濃度が2 μg/μLになるように試料緩衝液(2-メルカプトエタノール+) (和光純薬工業) と滅菌処理をした超純水を混合し、5分間煮沸し調製した。
上記で調製したサンプルを用い、タンパク質の発現をドデシル硫酸ナトリウムポリアクリルアミドゲル電気泳動法を行い解析した。電気泳動 (12.5% ポリアクリルアミドゲル、0.04 A) を行い、ポリビニリデンジフルオリド膜 (Merck Millipore, Burlington, MA, U.S.A.) に転写 (100 V、90分) した。Table 3に示す通りにブロッキング、抗体処置、及び化学発光を行った。化学発光は、X線フィルム (Kodak, Rochester, NY, U.S.A.) を感光させることでバンドとして検出した。ブロッキング剤として、牛血清アルブミン (BSA; 和光純薬工業)、スキムミルク (雪印メグミルク, 東京) を使用した。
なお、発現変化を確認したいタンパク質(β-catenin、c-myc、cyclin D1、caspase-3、cleaved caspase-3)に応じて、それぞれのタンパク質に対する抗体を一次抗体として用い、内部コントロールタンパク質としてグリセルアルデヒド-3-リン酸デヒドロゲナーゼ、β-アクチンを選び、それぞれに対する抗体を一次抗体として用いた。一次抗体に結合させる二次抗体として、horseradish peroxidase抱合型抗mouse IgG (GE Healthcare)、抗rabbit IgG (GE Healthcare)を用いた。発色試薬には、ECL Blotting Detection System (GE Healthcare)、ECL Prime Western Blotting Detection System (GE Healthcare)を使用した。
上記で調製したサンプルを用い、タンパク質の発現をドデシル硫酸ナトリウムポリアクリルアミドゲル電気泳動法を行い解析した。電気泳動 (12.5% ポリアクリルアミドゲル、0.04 A) を行い、ポリビニリデンジフルオリド膜 (Merck Millipore, Burlington, MA, U.S.A.) に転写 (100 V、90分) した。Table 3に示す通りにブロッキング、抗体処置、及び化学発光を行った。化学発光は、X線フィルム (Kodak, Rochester, NY, U.S.A.) を感光させることでバンドとして検出した。ブロッキング剤として、牛血清アルブミン (BSA; 和光純薬工業)、スキムミルク (雪印メグミルク, 東京) を使用した。
なお、発現変化を確認したいタンパク質(β-catenin、c-myc、cyclin D1、caspase-3、cleaved caspase-3)に応じて、それぞれのタンパク質に対する抗体を一次抗体として用い、内部コントロールタンパク質としてグリセルアルデヒド-3-リン酸デヒドロゲナーゼ、β-アクチンを選び、それぞれに対する抗体を一次抗体として用いた。一次抗体に結合させる二次抗体として、horseradish peroxidase抱合型抗mouse IgG (GE Healthcare)、抗rabbit IgG (GE Healthcare)を用いた。発色試薬には、ECL Blotting Detection System (GE Healthcare)、ECL Prime Western Blotting Detection System (GE Healthcare)を使用した。
結果を図6(化合物(7))、図8(化合物(11))、図12(化合物(12))、図14及び図15(化合物(19))に示した。これらの結果によれば、本発明の化合物(I)群は、β-カテニン(CTNNB1)タンパク質、及びWnt/β-カテニンシグナル経路の下流タンパク質であるC-MYC、cyclin D1、SURVIVINタンパク質発現を減少させることが確認された。
試験例5:細胞周期解析
本発明の化合物(I)(化合物(1))の細胞周期への影響を確認するため、flow cytometry (FCM) 法を用いて、既報(Imayoshi N, et al. Biochem Biophys Res Commun, 484:262-268, 2017.)に従い細胞周期解析を行った。上記と同様に各種癌細胞を培養し、化合物(1)で処置した。24、48時間後に1.0×106個の癌細胞を回収し、-20℃に冷却した70%エタノールで固定した。固定した細胞は-20℃で12時間以上保存し、6ヶ月以内にThe BDLSRFortessaセルアナライザー (BD Bioscience, Franklin Lakes, NJ, U.S.A.) を用いて解析を行った。解析の際はエタノールを除き、propidium iodide (PI; フナコシ) 20 μg/mL、RNase 100 μg/mL (Sigma-Aldrich) を含むCa2+, Mg2+-free phosphate buffered saline (PBS(-)) 1 mLを加え、ボルテックスにて混和後、4℃、30分反応させてから行った。結果の解析には、FlowJo (BD Bioscience) を用いた。
結果を図3に記した。図3によれば、化合物(1)はMM細胞を細胞周期のG1期で停止させ、subG1期を増加させることが確認された。
試験例6:アポトーシス(細胞死)解析
本発明の化合物(I)(化合物(1)及び化合物(19))が癌細胞にアポトーシス(細胞死)を誘導しているかを、既報(Imayoshi N, et al. Biochem Biophys Res Commun, 484:262-268, 2017.)に従いFCMを用いてApoptosis assayを行い解析した。上記と同様に癌細胞を培養し、化合物(1)で処置した。Annexin-V/FITC detection kit (BD) を用いて、FCMで解析を行った。データの解析には、FlowJo (BD Bioscience) を用いた。
また、化合物(19)のアポトーシス誘導を、cleaved caspase 3タンパク質の発現によるウェスタンブロッティング法にて解析した。
また、化合物(19)のアポトーシス誘導を、cleaved caspase 3タンパク質の発現によるウェスタンブロッティング法にて解析した。
結果を図4(化合物(1))及び図17(化合物(19))に示した。これらの結果によれば、本発明の化合物(I)群は、癌細胞に対してアポトーシス細胞死を誘導することが確認された。
試験例7:Cancer sphere形成
培養は既報(Takada T, et al. J Physiol Sci, 66:387-396, 2016.)に従い、Ultra-Low attachment 6-well plate (Corning International, Corning, NY)上で3 mlのDMEM/F12(Thermo Fisher Scientific, Waltham, MA)を用いて7日間行った。このとき培地には1%のPC/SM,2%のB-27 [B-27 Serum-Free Supplement (50×): Thermo Fisher Scientific],20 ng/mlの上皮成長因子(epidermal growth factor: EGF; Peprotech, Rocky Hill, NJ)、20 ng/mlの塩基性線維芽細胞成長因子(basic fibroblast growth factor: b-FGF; Peprotech)を添加した。EGFとb-FGFは培養3日目にさらに20 ng/mlずつ添加した。細胞播種数は、1.0 × 105cells/mlで行った。
結果を図21に示した。図21の結果によれば、MDA-MB-231乳癌細胞株及びU251-MB神経膠芽腫細胞株から、それぞれの癌幹細胞を多く含むcancer sphereが形成され、これらの癌幹細胞ではβ-カテニン(CTNNB1)mRNAが高発現していることが確認された。
試験例8:CellTiter-Glo(登録商標)3D 細胞生存率アッセイ
Cancer sphere形成で作製した浮遊状態のsphereであるCSCsに対する阻害剤の細胞増殖抑制効果を調べるため、CellTiter-Glo(登録商標)3D細胞生存率アッセイを用いた。細胞は、Ultra-Low attachment 96-well plates (Corning International)に1 wellあたり3.0 × 103cells/90 μlで播種した。阻害剤として、化合物(19)、コントロールとしてICG-001を用いた。6日間培養を行った後、各wellの細胞を培養液ごと白色マイクロプレート(96F Nunclon Delta WhiteMicrowell SI;Thermo Fisher Scientific)に移し替えた。そこにCellTiter-Glo(登録商標)3D Reagent(Promega, Madison, WI, U.S.A.)を1 wellあたり100 μl加え、5分間撹拌し、室温で25分反応させた。そして、ルミノメーター(GloMax(登録商標)Discover System: Promega)を用いて発光量を測定した。
結果を図22に示した。図22の実線によれば、化合物(19)は、通常の癌細胞に対する細胞増殖抑制効果(破線)よりも、癌幹細胞に対してより効果的に細胞増殖を抑制することが確認された。
本発明の化合物(I)、又はその医薬上許容される塩は、優れたWntシグナル伝達経路阻害活性を示す。それ故、本発明によれば、化合物(I)、又はその医薬上許容される塩を有効成分として含み、Wntシグナル伝達経路を阻害することにより症状を緩和させることができる疾患、例えば、癌、冠状動脈疾患、急性冠症候群、変形性関節症等の予防又は治療に有用であり、とりわけ、各種癌(例えば、大腸癌、乳癌、頭頸部癌、肺癌、胃癌、食道癌、悪性黒色腫、前立腺癌、肝細胞癌、卵巣癌、肝芽腫、急性骨髄性白血病、急性リンパ芽球性白血病、慢性骨髄性白血病、慢性リンパ性白血病、多発性骨髄腫、悪性リンパ腫、膵臓癌、腎臓癌、膀胱癌、神経膠芽腫、髄芽腫、ウィルムス腫瘍等)の予防又は治療に有用な医薬を提供することができる。また、本発明によれば、優れたWntシグナル伝達経路阻害活性を有する、新規な化合物(化合物(II))、又はその医薬上許容される塩も提供することができる。
本出願は、日本国で2018年12月20日に出願された特願2018-237943及び2019年1月9日に出願された特願2019-002015を基礎としており、その内容は本明細書にすべて包含されるものである。
Claims (12)
- 式(I):
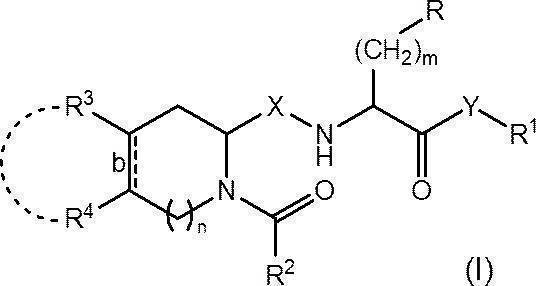
Rは、置換されていてもよいC6-14アリール基、置換されていてもよいヘテロアリール基又は保護されていてもよいヒドロキシ基を示し;
R1は、置換されていてもよいC1-6アルキル基、又は置換されていてもよいC6-14アリール基を示し;
R2は、置換されていてもよいC1-6アルキル基、置換されていてもよいC1-6アルコキシ基又は置換されていてもよいC6-14アリール基を示し;
R3及びR4は、それぞれ独立して、水素原子、ハロゲン原子、ヒドロキシ基、アミノ基、ニトロ基、シアノ基、カルボキシ基、置換されていてもよいC1-6アルキル基又は置換されていてもよいC1-6アルコキシ基を示すか、或いは、R3及びR4は、互いに結合して、それらが結合している炭素原子と一緒になって、それぞれ置換されていてもよい、シクロヘキサン環又はベンゼン環を形成してもよく;
結合bは、単結合又は二重結合を示し;
Xは、CH2又はカルボニル(C=O)を示し;
Yは、酸素原子又はNR0(式中のR0は、水素原子又はC1-4アルコキシ基を示す。)を示し;
mは、1~3の整数を示し;並びに
nは、0又は1を示す。]
で表される化合物、又はその医薬上許容される塩からなるWntシグナル伝達経路阻害剤。 - Rが、置換されていてもよいC6-10アリール基、置換されていてもよいヘテロアリール基又は保護されたヒドロキシ基であり、
R1が、1又は2個のフェニル基で置換されていてもよいC1-4アルキル基、或いは置換されていてもよいフェニル基であり、
R2が、置換されていてもよいC1-4アルキル基、置換されていてもよいC1-4アルコキシ基、置換されていてもよいフェニル基であり、
R3及びR4が、それぞれ独立して、水素原子、ヒドロキシ基又はC1-6アルコキシ基であるか、或いは、R3及びR4は、互いに結合して、それらが結合している炭素原子と一緒になって、シクロヘキサン環を形成してもよく、
結合bが、単結合であり、
Xが、CH2又はカルボニルであり、
Yが、酸素原子、NH又はN(OCH3)であり、
mが、1であり、且つ
nが、0又は1である、請求項1に記載のWntシグナル伝達経路阻害剤。 - R2が、置換されていてもよいC1-4アルキル基又は置換されていてもよいフェニル基である、請求項1又は2に記載のWntシグナル伝達経路阻害剤。
- Rが、置換されていてもよいフェニル基、置換されていてもよいナフチル基、置換されていてもよいイミダゾリル基、置換されていてもよいインドリル基、トリチルオキシ基又はベンジルオキシ基である、請求項1~3のいずれか1項に記載のWntシグナル伝達経路阻害剤。
- R3及びR4が、共に水素原子であるか、または、R3及びR4は、互いに結合して、それらが結合している炭素原子と一緒になって、シクロヘキサン環を形成し、且つ
結合bが、単結合である、請求項1~4のいずれか1項に記載のWntシグナル伝達経路阻害剤。 - 請求項1~5のいずれか1項に記載のWntシグナル伝達経路阻害剤を有効成分として含有する医薬。
- Wntシグナル伝達経路を阻害することにより症状を緩和させることができる疾患の予防又は治療のための請求項6に記載の医薬。
- Wntシグナル伝達経路を阻害することにより症状を緩和させることができる疾患が、癌、冠状動脈疾患、急性冠症候群、又は変形性関節症である、請求項7に記載の医薬。
- Wntシグナル伝達経路を阻害することにより症状を緩和させることができる疾患が、癌である、請求項7に記載の医薬。
- 癌が、大腸癌、乳癌、頭頸部癌、肺癌、胃癌、食道癌、悪性黒色腫、前立腺癌、肝細胞癌、卵巣癌、肝芽腫、急性骨髄性白血病、急性リンパ芽球性白血病、慢性骨髄性白血病、慢性リンパ性白血病、多発性骨髄腫、悪性リンパ腫、膵臓癌、腎臓癌、膀胱癌、神経膠芽腫、髄芽腫、又はウィルムス腫瘍である、請求項9に記載の医薬。
- 式(II):
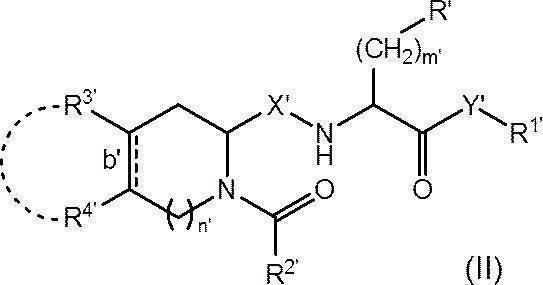
R’は、ヒドロキシ基又はC1-6アルコキシ基で置換されていてもよいフェニル基、ナフチル基、トリチル基で置換されたイミダゾリル基、置換されていてもよいC6-14アリールスルホニル基で置換されたインドリル基、トリチルオキシ基又はベンジルオキシ基を示し;
R1’は、2個のフェニル基で置換されていてもよいメチル基、又はフェニル基を示し;R2’は、エチニル基で置換されていてもよいビフェニリル基、tert-ブチル基で置換されたフェニル基、メチル基、又はフルオレニルメトキシ基を示し;
R3’及びR4’は、それぞれ独立して、水素原子又はヒドロキシ基を示すか、或いは、R3’及びR4’は、互いに結合して、それらが結合している炭素原子と一緒になって、シクロヘキサン環又はベンゼン環を形成してもよく;
結合b’は、単結合又は二重結合を示し;
X’は、CH2又はカルボニル(C=O)を示し;
Y’は、酸素原子、NH又はN(OCH3)を示し;
m’は、1を示し;並びに
n’は、0又は1を示す。但し、R2’が、無置換ビフェニリル基である時は、Y’-R1’は、メトキシ基を示す。]
で表される化合物、又はその医薬上許容される塩。 - R3’及びR4’が、共に水素原子であり、且つ結合b’が、単結合である、請求項11に記載の化合物、又はその医薬上許容される塩。
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2018-237943 | 2018-12-20 | ||
| JP2018237943 | 2018-12-20 | ||
| JP2019-002015 | 2019-01-09 | ||
| JP2019002015 | 2019-01-09 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2020130119A1 true WO2020130119A1 (ja) | 2020-06-25 |
Family
ID=71101376
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2019/049992 WO2020130119A1 (ja) | 2018-12-20 | 2019-12-20 | Wntシグナル伝達経路阻害剤 |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2020130119A1 (ja) |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2007503816A (ja) * | 2003-08-28 | 2007-03-01 | チョンウェ ファーマ コーポレーション | β−カテニン/TCF活性化転写の調節 |
| JP2012505923A (ja) * | 2008-10-17 | 2012-03-08 | エグゼリクシス, インコーポレイテッド | スフィンゴシン−1−リン酸受容体拮抗剤と抗微小管剤との間の相乗効果 |
| US20120190820A1 (en) * | 2011-01-26 | 2012-07-26 | Ming Xian | Facile amide formation via s-nitroso thioacid intermediates |
-
2019
- 2019-12-20 WO PCT/JP2019/049992 patent/WO2020130119A1/ja active Application Filing
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2007503816A (ja) * | 2003-08-28 | 2007-03-01 | チョンウェ ファーマ コーポレーション | β−カテニン/TCF活性化転写の調節 |
| JP2012505923A (ja) * | 2008-10-17 | 2012-03-08 | エグゼリクシス, インコーポレイテッド | スフィンゴシン−1−リン酸受容体拮抗剤と抗微小管剤との間の相乗効果 |
| US20120190820A1 (en) * | 2011-01-26 | 2012-07-26 | Ming Xian | Facile amide formation via s-nitroso thioacid intermediates |
Non-Patent Citations (4)
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2944573T3 (es) | Antagonistas de TLR7/8 y usos de los mismos | |
| JP7307057B2 (ja) | 統合的ストレス経路の調節剤 | |
| JP2019163324A (ja) | Jak及びpi3k阻害剤併用によるb細胞悪性腫瘍の処置 | |
| CA2829188C (en) | Dispiropyrrolidine derivatives | |
| JP2021501785A (ja) | 統合的ストレス経路の調節剤 | |
| CA3025806A1 (en) | Degradation of bromodomain-containing protein 9 (brd9) by conjugation of brd9 inhibitors with e3 ligase ligand and methods of use | |
| JP2021501788A (ja) | 統合的ストレス経路の調節剤 | |
| TW201720816A (zh) | Cxcr2抑制劑 | |
| AU2024200013A1 (en) | Prodrug modulators of the integrated stress pathway | |
| AU2017257211A1 (en) | Polymorphic form of N-{6-(2-Hydroxypropan-2-yl)-2-[2-(methylsulphonyl)ethyl]-2H-indazol-5-yl}-6-(trifluoromethyl)pyridine-2-carboxamide | |
| JP7365332B2 (ja) | Cdk8/19阻害薬としての新規ヘテロ環式化合物 | |
| TW201728579A (zh) | 作為檢測點激酶1 (chk1)抑制劑之3,5-二取代吡唑及其製備及應用 | |
| JP7166028B2 (ja) | 高活性stingタンパク質アゴニスト化合物 | |
| CN112898269A (zh) | 化合物的结晶形式 | |
| WO2019189732A1 (ja) | 光学活性な架橋型環状2級アミン誘導体 | |
| CA2875494A1 (en) | 2h-imidazol-4-amine compounds and their use as bace inhibitors | |
| JP2022516685A (ja) | ホスファターゼ結合化合物およびそれらを使用する方法 | |
| US11986480B2 (en) | Heterocyclic compound | |
| KR20210038590A (ko) | 보론산 유도체 | |
| TW201030011A (en) | Imidazothiazole derivatives bearing the proline ring structure | |
| KR20200040295A (ko) | 보론산 유도체 | |
| CN108137556B (zh) | 1,4-二羰基-哌啶基衍生物 | |
| JP6970684B2 (ja) | クマリン骨格を有するスルホンアミド誘導体 | |
| WO2018028491A1 (zh) | 吲哚胺2,3-双加氧酶抑制剂及其在药学中的用途 | |
| WO2020130119A1 (ja) | Wntシグナル伝達経路阻害剤 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 19899061 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 19899061 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: JP |