WO2020054782A1 - 乳癌細胞存在率の推定方法 - Google Patents
乳癌細胞存在率の推定方法 Download PDFInfo
- Publication number
- WO2020054782A1 WO2020054782A1 PCT/JP2019/035774 JP2019035774W WO2020054782A1 WO 2020054782 A1 WO2020054782 A1 WO 2020054782A1 JP 2019035774 W JP2019035774 W JP 2019035774W WO 2020054782 A1 WO2020054782 A1 WO 2020054782A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- breast cancer
- seq
- nucleotide sequence
- nucleotides
- cytosine residue
- Prior art date
Links
- 206010006187 Breast cancer Diseases 0.000 title claims abstract description 128
- 208000026310 Breast neoplasm Diseases 0.000 title claims abstract description 128
- 238000000034 method Methods 0.000 title claims abstract description 65
- 125000003729 nucleotide group Chemical group 0.000 claims abstract description 122
- 239000002773 nucleotide Substances 0.000 claims abstract description 111
- 230000011987 methylation Effects 0.000 claims abstract description 107
- 238000007069 methylation reaction Methods 0.000 claims abstract description 107
- OPTASPLRGRRNAP-UHFFFAOYSA-N cytosine Chemical group NC=1C=CNC(=O)N=1 OPTASPLRGRRNAP-UHFFFAOYSA-N 0.000 claims abstract description 80
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 claims abstract description 18
- 238000002651 drug therapy Methods 0.000 claims abstract description 17
- 239000000523 sample Substances 0.000 claims description 52
- 239000000439 tumor marker Substances 0.000 claims description 27
- 238000004458 analytical method Methods 0.000 claims description 25
- 108020004711 Nucleic Acid Probes Proteins 0.000 claims description 15
- 239000002853 nucleic acid probe Substances 0.000 claims description 15
- 239000003153 chemical reaction reagent Substances 0.000 claims description 14
- 230000000694 effects Effects 0.000 claims description 14
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 claims description 12
- 108091008146 restriction endonucleases Proteins 0.000 claims description 11
- 238000012175 pyrosequencing Methods 0.000 claims description 8
- 238000003018 immunoassay Methods 0.000 claims description 5
- 230000008018 melting Effects 0.000 claims description 4
- 238000002844 melting Methods 0.000 claims description 4
- 108091029523 CpG island Proteins 0.000 claims description 3
- 241000270295 Serpentes Species 0.000 claims description 3
- 238000001369 bisulfite sequencing Methods 0.000 claims description 3
- 238000004949 mass spectrometry Methods 0.000 claims description 3
- 238000007855 methylation-specific PCR Methods 0.000 claims description 3
- 238000002493 microarray Methods 0.000 claims description 3
- 238000007838 multiplex ligation-dependent probe amplification Methods 0.000 claims description 3
- 210000004027 cell Anatomy 0.000 description 79
- 108091029430 CpG site Proteins 0.000 description 66
- 206010028980 Neoplasm Diseases 0.000 description 50
- 201000011510 cancer Diseases 0.000 description 49
- 101150105781 CCDC181 gene Proteins 0.000 description 25
- 101150029409 CFTR gene Proteins 0.000 description 25
- 101150017499 SIM1 gene Proteins 0.000 description 25
- 239000003550 marker Substances 0.000 description 22
- 210000001519 tissue Anatomy 0.000 description 17
- 108090000623 proteins and genes Proteins 0.000 description 15
- 101001045218 Homo sapiens Peroxisomal multifunctional enzyme type 2 Proteins 0.000 description 12
- 102100022587 Peroxisomal multifunctional enzyme type 2 Human genes 0.000 description 12
- 238000012937 correction Methods 0.000 description 10
- 229940079593 drug Drugs 0.000 description 10
- 239000003814 drug Substances 0.000 description 10
- 101001012157 Homo sapiens Receptor tyrosine-protein kinase erbB-2 Proteins 0.000 description 9
- 102100030086 Receptor tyrosine-protein kinase erbB-2 Human genes 0.000 description 9
- 239000002246 antineoplastic agent Substances 0.000 description 9
- 108020004414 DNA Proteins 0.000 description 8
- 238000011282 treatment Methods 0.000 description 8
- 102100030507 Coiled-coil domain-containing protein 181 Human genes 0.000 description 7
- 108010079245 Cystic Fibrosis Transmembrane Conductance Regulator Proteins 0.000 description 7
- 101000772632 Homo sapiens Coiled-coil domain-containing protein 181 Proteins 0.000 description 7
- 101000703681 Homo sapiens Single-minded homolog 1 Proteins 0.000 description 7
- 102000008371 intracellularly ATP-gated chloride channel activity proteins Human genes 0.000 description 7
- 238000000386 microscopy Methods 0.000 description 7
- 230000007067 DNA methylation Effects 0.000 description 6
- 230000009946 DNA mutation Effects 0.000 description 6
- 102100031980 Single-minded homolog 1 Human genes 0.000 description 6
- ISAKRJDGNUQOIC-UHFFFAOYSA-N Uracil Chemical compound O=C1C=CNC(=O)N1 ISAKRJDGNUQOIC-UHFFFAOYSA-N 0.000 description 6
- 238000002512 chemotherapy Methods 0.000 description 6
- 238000011156 evaluation Methods 0.000 description 6
- 208000017891 HER2 positive breast carcinoma Diseases 0.000 description 5
- 230000004044 response Effects 0.000 description 5
- 238000002560 therapeutic procedure Methods 0.000 description 5
- 239000003534 dna topoisomerase inhibitor Substances 0.000 description 4
- 238000001794 hormone therapy Methods 0.000 description 4
- 239000003112 inhibitor Substances 0.000 description 4
- 230000001575 pathological effect Effects 0.000 description 4
- 238000001050 pharmacotherapy Methods 0.000 description 4
- 238000012163 sequencing technique Methods 0.000 description 4
- 239000007790 solid phase Substances 0.000 description 4
- 229940044693 topoisomerase inhibitor Drugs 0.000 description 4
- 229960000575 trastuzumab Drugs 0.000 description 4
- 238000011144 upstream manufacturing Methods 0.000 description 4
- 229940125497 HER2 kinase inhibitor Drugs 0.000 description 3
- 108091022875 Microtubule Proteins 0.000 description 3
- 102000029749 Microtubule Human genes 0.000 description 3
- 108091028043 Nucleic acid sequence Proteins 0.000 description 3
- 230000002152 alkylating effect Effects 0.000 description 3
- 230000000340 anti-metabolite Effects 0.000 description 3
- 229940100197 antimetabolite Drugs 0.000 description 3
- 239000002256 antimetabolite Substances 0.000 description 3
- 239000003886 aromatase inhibitor Substances 0.000 description 3
- 229940046844 aromatase inhibitors Drugs 0.000 description 3
- 238000005259 measurement Methods 0.000 description 3
- 210000004688 microtubule Anatomy 0.000 description 3
- 238000002360 preparation method Methods 0.000 description 3
- 229940035893 uracil Drugs 0.000 description 3
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 2
- 101000904177 Clupea pallasii Gonadoliberin-1 Proteins 0.000 description 2
- 101800003838 Epidermal growth factor Proteins 0.000 description 2
- 239000000579 Gonadotropin-Releasing Hormone Substances 0.000 description 2
- 101150064023 HSD17B4 gene Proteins 0.000 description 2
- 239000002136 L01XE07 - Lapatinib Substances 0.000 description 2
- PJKKQFAEFWCNAQ-UHFFFAOYSA-N N(4)-methylcytosine Chemical group CNC=1C=CNC(=O)N=1 PJKKQFAEFWCNAQ-UHFFFAOYSA-N 0.000 description 2
- 102100033237 Pro-epidermal growth factor Human genes 0.000 description 2
- RJKFOVLPORLFTN-LEKSSAKUSA-N Progesterone Chemical class C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H](C(=O)C)[C@@]1(C)CC2 RJKFOVLPORLFTN-LEKSSAKUSA-N 0.000 description 2
- 108020004518 RNA Probes Proteins 0.000 description 2
- 239000003391 RNA probe Substances 0.000 description 2
- 101000857870 Squalus acanthias Gonadoliberin Proteins 0.000 description 2
- 239000000556 agonist Substances 0.000 description 2
- 229940046836 anti-estrogen Drugs 0.000 description 2
- 230000001833 anti-estrogenic effect Effects 0.000 description 2
- 238000003556 assay Methods 0.000 description 2
- 238000001574 biopsy Methods 0.000 description 2
- 229940116977 epidermal growth factor Drugs 0.000 description 2
- 239000000328 estrogen antagonist Substances 0.000 description 2
- 230000002496 gastric effect Effects 0.000 description 2
- XLXSAKCOAKORKW-AQJXLSMYSA-N gonadorelin Chemical compound C([C@@H](C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1[C@@H](CCC1)C(=O)NCC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)C1=CC=C(O)C=C1 XLXSAKCOAKORKW-AQJXLSMYSA-N 0.000 description 2
- 229960004891 lapatinib Drugs 0.000 description 2
- BCFGMOOMADDAQU-UHFFFAOYSA-N lapatinib Chemical compound O1C(CNCCS(=O)(=O)C)=CC=C1C1=CC=C(N=CN=C2NC=3C=C(Cl)C(OCC=4C=C(F)C=CC=4)=CC=3)C2=C1 BCFGMOOMADDAQU-UHFFFAOYSA-N 0.000 description 2
- 238000000370 laser capture micro-dissection Methods 0.000 description 2
- 208000026534 luminal B breast carcinoma Diseases 0.000 description 2
- -1 macitinib Chemical compound 0.000 description 2
- 239000012528 membrane Substances 0.000 description 2
- 231100000782 microtubule inhibitor Toxicity 0.000 description 2
- 238000013188 needle biopsy Methods 0.000 description 2
- 230000007170 pathology Effects 0.000 description 2
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 2
- 239000000583 progesterone congener Substances 0.000 description 2
- 230000035945 sensitivity Effects 0.000 description 2
- 210000003765 sex chromosome Anatomy 0.000 description 2
- VBEQCZHXXJYVRD-GACYYNSASA-N uroanthelone Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CS)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CS)C(=O)N[C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C(C)C)[C@@H](C)O)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CCSC)NC(=O)[C@H](CS)NC(=O)[C@@H](NC(=O)CNC(=O)CNC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CS)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CS)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC(N)=O)C(C)C)[C@@H](C)CC)C1=CC=C(O)C=C1 VBEQCZHXXJYVRD-GACYYNSASA-N 0.000 description 2
- CBIAKDAYHRWZCU-UHFFFAOYSA-N 2-bromo-4-[(6,7-dimethoxyquinazolin-4-yl)amino]phenol Chemical compound C=12C=C(OC)C(OC)=CC2=NC=NC=1NC1=CC=C(O)C(Br)=C1 CBIAKDAYHRWZCU-UHFFFAOYSA-N 0.000 description 1
- QQWUGDVOUVUTOY-UHFFFAOYSA-N 5-chloro-N2-[2-methoxy-4-[4-(4-methyl-1-piperazinyl)-1-piperidinyl]phenyl]-N4-(2-propan-2-ylsulfonylphenyl)pyrimidine-2,4-diamine Chemical compound COC1=CC(N2CCC(CC2)N2CCN(C)CC2)=CC=C1NC(N=1)=NC=C(Cl)C=1NC1=CC=CC=C1S(=O)(=O)C(C)C QQWUGDVOUVUTOY-UHFFFAOYSA-N 0.000 description 1
- 229930024421 Adenine Natural products 0.000 description 1
- GFFGJBXGBJISGV-UHFFFAOYSA-N Adenine Chemical group NC1=NC=NC2=C1N=CN2 GFFGJBXGBJISGV-UHFFFAOYSA-N 0.000 description 1
- 230000004544 DNA amplification Effects 0.000 description 1
- MWWSFMDVAYGXBV-RUELKSSGSA-N Doxorubicin hydrochloride Chemical compound Cl.O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 MWWSFMDVAYGXBV-RUELKSSGSA-N 0.000 description 1
- 238000002965 ELISA Methods 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 208000000461 Esophageal Neoplasms Diseases 0.000 description 1
- 102100032191 Guanine nucleotide exchange factor VAV3 Human genes 0.000 description 1
- 101000775742 Homo sapiens Guanine nucleotide exchange factor VAV3 Proteins 0.000 description 1
- 101000942701 Homo sapiens Liprin-alpha-3 Proteins 0.000 description 1
- 101000692455 Homo sapiens Platelet-derived growth factor receptor beta Proteins 0.000 description 1
- 101001121506 Homo sapiens Protein odd-skipped-related 2 Proteins 0.000 description 1
- 101000848744 Homo sapiens Rap guanine nucleotide exchange factor-like 1 Proteins 0.000 description 1
- 101000752241 Homo sapiens Rho guanine nucleotide exchange factor 4 Proteins 0.000 description 1
- 239000005517 L01XE01 - Imatinib Substances 0.000 description 1
- 239000005411 L01XE02 - Gefitinib Substances 0.000 description 1
- 239000005551 L01XE03 - Erlotinib Substances 0.000 description 1
- 239000002147 L01XE04 - Sunitinib Substances 0.000 description 1
- 102100032892 Liprin-alpha-3 Human genes 0.000 description 1
- 238000000585 Mann–Whitney U test Methods 0.000 description 1
- 229940122255 Microtubule inhibitor Drugs 0.000 description 1
- 239000000020 Nitrocellulose Substances 0.000 description 1
- 206010030155 Oesophageal carcinoma Diseases 0.000 description 1
- OYONTEXKYJZFHA-SSHUPFPWSA-N PHA-665752 Chemical compound CC=1C(C(=O)N2[C@H](CCC2)CN2CCCC2)=C(C)NC=1\C=C(C1=C2)/C(=O)NC1=CC=C2S(=O)(=O)CC1=C(Cl)C=CC=C1Cl OYONTEXKYJZFHA-SSHUPFPWSA-N 0.000 description 1
- 229930012538 Paclitaxel Natural products 0.000 description 1
- 102100026547 Platelet-derived growth factor receptor beta Human genes 0.000 description 1
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 1
- 102100025660 Protein odd-skipped-related 2 Human genes 0.000 description 1
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 description 1
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 description 1
- 102100034586 Rap guanine nucleotide exchange factor-like 1 Human genes 0.000 description 1
- 241001495413 Rattus tanezumi Species 0.000 description 1
- 102100021709 Rho guanine nucleotide exchange factor 4 Human genes 0.000 description 1
- 208000005718 Stomach Neoplasms Diseases 0.000 description 1
- 229940123237 Taxane Drugs 0.000 description 1
- 108090001039 Transcription factor AP-2 Proteins 0.000 description 1
- 102100033348 Transcription factor AP-2-beta Human genes 0.000 description 1
- 208000003721 Triple Negative Breast Neoplasms Diseases 0.000 description 1
- 239000007984 Tris EDTA buffer Substances 0.000 description 1
- 239000002250 absorbent Substances 0.000 description 1
- 230000002745 absorbent Effects 0.000 description 1
- 229960000643 adenine Drugs 0.000 description 1
- 229960001686 afatinib Drugs 0.000 description 1
- ULXXDDBFHOBEHA-CWDCEQMOSA-N afatinib Chemical compound N1=CN=C2C=C(O[C@@H]3COCC3)C(NC(=O)/C=C/CN(C)C)=CC2=C1NC1=CC=C(F)C(Cl)=C1 ULXXDDBFHOBEHA-CWDCEQMOSA-N 0.000 description 1
- 230000004520 agglutination Effects 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- 229940045799 anthracyclines and related substance Drugs 0.000 description 1
- 230000000692 anti-sense effect Effects 0.000 description 1
- 239000003972 antineoplastic antibiotic Substances 0.000 description 1
- 229940041181 antineoplastic drug Drugs 0.000 description 1
- 238000003491 array Methods 0.000 description 1
- 238000012575 bio-layer interferometry Methods 0.000 description 1
- 229960002685 biotin Drugs 0.000 description 1
- 235000020958 biotin Nutrition 0.000 description 1
- 239000011616 biotin Substances 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 210000001124 body fluid Anatomy 0.000 description 1
- 239000010839 body fluid Substances 0.000 description 1
- 210000000069 breast epithelial cell Anatomy 0.000 description 1
- 229960005395 cetuximab Drugs 0.000 description 1
- 238000012767 chemiluminescent enzyme immunoassay Methods 0.000 description 1
- 238000010219 correlation analysis Methods 0.000 description 1
- KTEIFNKAUNYNJU-GFCCVEGCSA-N crizotinib Chemical compound O([C@H](C)C=1C(=C(F)C=CC=1Cl)Cl)C(C(=NC=1)N)=CC=1C(=C1)C=NN1C1CCNCC1 KTEIFNKAUNYNJU-GFCCVEGCSA-N 0.000 description 1
- 210000004748 cultured cell Anatomy 0.000 description 1
- 238000010494 dissociation reaction Methods 0.000 description 1
- 230000005593 dissociations Effects 0.000 description 1
- 229940121647 egfr inhibitor Drugs 0.000 description 1
- 238000011984 electrochemiluminescence immunoassay Methods 0.000 description 1
- 229960001433 erlotinib Drugs 0.000 description 1
- AAKJLRGGTJKAMG-UHFFFAOYSA-N erlotinib Chemical compound C=12C=C(OCCOC)C(OCCOC)=CC2=NC=NC=1NC1=CC=CC(C#C)=C1 AAKJLRGGTJKAMG-UHFFFAOYSA-N 0.000 description 1
- 201000004101 esophageal cancer Diseases 0.000 description 1
- 238000004299 exfoliation Methods 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 238000000684 flow cytometry Methods 0.000 description 1
- 238000005194 fractionation Methods 0.000 description 1
- 230000008014 freezing Effects 0.000 description 1
- 238000007710 freezing Methods 0.000 description 1
- 206010017758 gastric cancer Diseases 0.000 description 1
- 229960002584 gefitinib Drugs 0.000 description 1
- XGALLCVXEZPNRQ-UHFFFAOYSA-N gefitinib Chemical compound C=12C=C(OCCCN3CCOCC3)C(OC)=CC2=NC=NC=1NC1=CC=C(F)C(Cl)=C1 XGALLCVXEZPNRQ-UHFFFAOYSA-N 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- UYTPUPDQBNUYGX-UHFFFAOYSA-N guanine Chemical group O=C1NC(N)=NC2=C1N=CN2 UYTPUPDQBNUYGX-UHFFFAOYSA-N 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- KTUFNOKKBVMGRW-UHFFFAOYSA-N imatinib Chemical compound C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 KTUFNOKKBVMGRW-UHFFFAOYSA-N 0.000 description 1
- 229960002411 imatinib Drugs 0.000 description 1
- 230000001900 immune effect Effects 0.000 description 1
- 238000012744 immunostaining Methods 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 229960003881 letrozole Drugs 0.000 description 1
- HPJKCIUCZWXJDR-UHFFFAOYSA-N letrozole Chemical compound C1=CC(C#N)=CC=C1C(N1N=CN=C1)C1=CC=C(C#N)C=C1 HPJKCIUCZWXJDR-UHFFFAOYSA-N 0.000 description 1
- 208000026535 luminal A breast carcinoma Diseases 0.000 description 1
- 238000004020 luminiscence type Methods 0.000 description 1
- 239000006249 magnetic particle Substances 0.000 description 1
- 206010025482 malaise Diseases 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 238000002483 medication Methods 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 238000012164 methylation sequencing Methods 0.000 description 1
- 239000000203 mixture Substances 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 229950003968 motesanib Drugs 0.000 description 1
- RAHBGWKEPAQNFF-UHFFFAOYSA-N motesanib Chemical compound C=1C=C2C(C)(C)CNC2=CC=1NC(=O)C1=CC=CN=C1NCC1=CC=NC=C1 RAHBGWKEPAQNFF-UHFFFAOYSA-N 0.000 description 1
- 229950008835 neratinib Drugs 0.000 description 1
- ZNHPZUKZSNBOSQ-BQYQJAHWSA-N neratinib Chemical compound C=12C=C(NC\C=C\CN(C)C)C(OCC)=CC2=NC=C(C#N)C=1NC(C=C1Cl)=CC=C1OCC1=CC=CC=N1 ZNHPZUKZSNBOSQ-BQYQJAHWSA-N 0.000 description 1
- 229920001220 nitrocellulos Polymers 0.000 description 1
- 108020004707 nucleic acids Proteins 0.000 description 1
- 102000039446 nucleic acids Human genes 0.000 description 1
- 150000007523 nucleic acids Chemical class 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 229940127084 other anti-cancer agent Drugs 0.000 description 1
- 229960001592 paclitaxel Drugs 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 238000010827 pathological analysis Methods 0.000 description 1
- 229960002087 pertuzumab Drugs 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- 108091033319 polynucleotide Proteins 0.000 description 1
- 102000040430 polynucleotide Human genes 0.000 description 1
- 239000002157 polynucleotide Substances 0.000 description 1
- 238000004393 prognosis Methods 0.000 description 1
- 102000027426 receptor tyrosine kinases Human genes 0.000 description 1
- 108091008598 receptor tyrosine kinases Proteins 0.000 description 1
- 238000005057 refrigeration Methods 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 238000007619 statistical method Methods 0.000 description 1
- 201000011549 stomach cancer Diseases 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- WQDSRJBTLILEEK-UHFFFAOYSA-N sulfurous acid Chemical compound OS(O)=O.OS(O)=O WQDSRJBTLILEEK-UHFFFAOYSA-N 0.000 description 1
- 229960001796 sunitinib Drugs 0.000 description 1
- WINHZLLDWRZWRT-ATVHPVEESA-N sunitinib Chemical compound CCN(CC)CCNC(=O)C1=C(C)NC(\C=C/2C3=CC(F)=CC=C3NC\2=O)=C1C WINHZLLDWRZWRT-ATVHPVEESA-N 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- DKPFODGZWDEEBT-QFIAKTPHSA-N taxane Chemical class C([C@]1(C)CCC[C@@H](C)[C@H]1C1)C[C@H]2[C@H](C)CC[C@@H]1C2(C)C DKPFODGZWDEEBT-QFIAKTPHSA-N 0.000 description 1
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 230000004797 therapeutic response Effects 0.000 description 1
- RWQNBRDOKXIBIV-UHFFFAOYSA-N thymine Chemical group CC1=CNC(=O)NC1=O RWQNBRDOKXIBIV-UHFFFAOYSA-N 0.000 description 1
- 229960001612 trastuzumab emtansine Drugs 0.000 description 1
- 208000022679 triple-negative breast carcinoma Diseases 0.000 description 1
- 229940121358 tyrosine kinase inhibitor Drugs 0.000 description 1
- 239000005483 tyrosine kinase inhibitor Substances 0.000 description 1
- 238000001262 western blot Methods 0.000 description 1
Images




Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
- C12Q1/6883—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material
- C12Q1/6886—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material for cancer
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/106—Pharmacogenomics, i.e. genetic variability in individual responses to drugs and drug metabolism
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/112—Disease subtyping, staging or classification
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/154—Methylation markers
Definitions
- the present invention relates to a method for estimating the abundance rate of breast cancer cells.
- Evaluation of the cancer cell fraction in a cancer tissue sample is performed by diagnosing cancer using a cancer marker (eg, determining the risk of developing cancer, determining the presence or absence of cancer, predicting the effect of drug therapy on cancer (for example, It is important to eliminate the decrease in determination accuracy due to the mixture of unnecessary fractions (normal cells) in the literature 1) and the evaluation of prognosis of drug therapy for cancer).
- a cancer marker eg, determining the risk of developing cancer, determining the presence or absence of cancer, predicting the effect of drug therapy on cancer (for example, It is important to eliminate the decrease in determination accuracy due to the mixture of unnecessary fractions (normal cells) in the literature 1) and the evaluation of prognosis of drug therapy for cancer).
- Methods for evaluating the cancer cell fraction include, for example, (1) pathological diagnosis, and (2) a cancer tissue sample (including cancer cells and non-cancerous cells) collected by biopsy using a predetermined method.
- a cancer tissue sample including cancer cells and non-cancerous cells collected by biopsy using a predetermined method.
- the method (1) requires the involvement of a skilled physician, and the method (2) has problems such as complexity.
- a cancer cell fraction by analyzing a cancer tissue sample using a marker ("fraction marker") specific to the cancer cell fraction.
- a marker for example, methylation markers TFAP2B, ARHGEF4, and RAPGEFL1 in esophageal cancer (Non-patent document 2) and methylation markers OSR2, PPFIA3, and VAV3 in gastric cancer (Non-patent document 3) have been reported. I have.
- An object of the present invention is to provide a cancer marker for determining the content of breast cancer cells contained in breast cancer tissue. Another object of the present invention is to improve the accuracy of an index for predicting the effect of drug therapy on breast cancer by determining the content of breast cancer cells.
- the present inventors first evaluated breast cancer cell fractions, so that breast cancer cells that were highly methylated in breast cancer cells but were completely or only slightly methylated in normal cells (non-breast cancer cells) The idea was to search for specific CpG sites, and in this search to select CpG sites that do not have a high incidence of copy number changes between cells and that can target a wide range of patients. . Thus, the present inventors have conducted extensive and comprehensive studies on more than 450,000 methylation candidate sites present on autosomes and sex chromosomes. As a result, such CpG sites were found in the SIM1 gene, CCDC181 gene, and CFTR gene. Specific CpG sites. The present inventors have succeeded in using the methylation rate of these CpG sites as an index of the abundance rate of breast cancer cells, and have completed the present invention.
- a method for estimating the abundance of breast cancer cells including the following (1) and (2): (1) In a sample derived from a human subject, the following: (A) a cytosine residue in the CG portion consisting of nucleotides 1000 and 1001 in the nucleotide sequence of SEQ ID NO: 1; (B) a cytosine residue in the CG portion consisting of nucleotides 1000 and 1001 in the nucleotide sequence of SEQ ID NO: 2; (C) a cytosine residue in the CG portion consisting of nucleotides 1001 and 1002 in the nucleotide sequence of SEQ ID NO: 3; or (d) analyzing the methylation rate of these combinations; and (2) (1) Estimating the abundance of breast cancer cells in a sample derived from a human subject based on the methylation rate analyzed in [1].
- the analysis is performed using one or more means selected from the group consisting of bisulfite, one or more primers, one or more nucleic acid probes, restriction enzymes, anti-methylated cytosine antibodies, and nanopores; The method of [1].
- [3] Analysis includes bisulfite sequencing, bisulfite pyrosequencing, methylation-specific PCR, restriction enzyme landmark genome scanning (RLGS), single nucleotide primer extension (SNuPE) , CpG island microarray method, methylite method, COBRA method, mass spectrometry (mass array) method, use of methylation specific restriction enzyme, high resolution melting analysis (HRM) method, nanopore analysis method, ICON probe method, methylation specific method The method according to [1] or [2], which is performed by the MLPA method or an immunoassay.
- a method for predicting the effect of drug therapy on breast cancer including the following (1) to (4): (1) measuring the value of a breast cancer marker in a sample derived from a human subject; (2) In a sample derived from a human subject, the following: (A) a cytosine residue in the CG portion consisting of nucleotides 1000 and 1001 in the nucleotide sequence of SEQ ID NO: 1; (B) a cytosine residue in the CG portion consisting of nucleotides 1000 and 1001 in the nucleotide sequence of SEQ ID NO: 2; (C) a cytosine residue in the CG portion consisting of nucleotides 1001 and 1002 in the nucleotide sequence of SEQ ID NO: 3; or (d) analyzing the methylation rate of these combinations; (3) calculating the corrected value of the breast cancer marker by correcting the value of the breast cancer marker measured in (1) with the methylation rate analyzed in (2); and (4) correcting the corrected value of (3).
- a method for determining breast cancer including the following (1) and (2): (1) In a sample derived from a human subject, the following: (A) a cytosine residue in the CG portion consisting of nucleotides 1000 and 1001 in the nucleotide sequence of SEQ ID NO: 1; (B) a cytosine residue in the CG portion consisting of nucleotides 1000 and 1001 in the nucleotide sequence of SEQ ID NO: 2; (C) a cytosine residue in the CG portion consisting of nucleotides 1001 and 1002 in the nucleotide sequence of SEQ ID NO: 3; or (d) analyzing the methylation rate of these combinations; and (2) (1) Assessing the likelihood of developing breast cancer based on the methylation rates analyzed in.
- [6] The following: (A) a cytosine residue in the CG portion consisting of nucleotides 1000 and 1001 in the nucleotide sequence of SEQ ID NO: 1; (B) a cytosine residue in the CG portion consisting of nucleotides 1000 and 1001 in the nucleotide sequence of SEQ ID NO: 2; (C) a cytosine residue in the CG portion consisting of nucleotides 1001 and 1002 in the nucleotide sequence of SEQ ID NO: 3; For reagents.
- the estimation method of the present invention is useful, for example, for improving the accuracy of breast cancer determination using breast cancer markers (eg, breast cancer risk determination, breast cancer determination, prediction of the effect of drug therapy, prognostic evaluation).
- the prediction method of the present invention is useful, for example, as an index in judging the necessity of surgery for a breast cancer patient and the selection of drug therapy.
- the determination method of the present invention is useful, for example, for diagnosing breast cancer.
- the reagents and kits of the present invention are useful, for example, for simple implementation of the method of the present invention.
- FIG. 1 is a graph showing the correlation between cancer cell content assessed by microscopy and the methylation levels of SIM1, CCDC181, and CFTR alone.
- FIG. 2 is a graph showing the correlation between the cancer cell content evaluated by microscopy and the methylation level of a combination of two genes selected from SIM1, CCDC181 and CFTR (higher methylation level is selected). is there.
- FIG. 3 is a graph showing the correlation between the cancer cell content evaluated by microscopy and the methylation level of the combination of the three genes SIM1, CCDC181, and CFTR (the highest methylation level was selected).
- FIG. 4 shows the cancer cell content evaluated by microscopic examination of the measured value of the methylation rate of the HSD17B4 gene in a HER2-positive breast cancer tissue sample of pathological complete response (pCR) and non-complete response (Non-pCR).
- 10 is a graph showing values corrected by the methylation rate by a combination of SIM1 and CCDC181.
- the cancer targeted in the present invention is breast cancer.
- Breast cancer can be classified into HER2-positive, luminal A, luminal B, and triple negative subtypes.
- the cancer targeted in the present invention may be any type of breast cancer, but HER2 positive type is preferable in a specific aspect.
- fraction marker refers to a cancer cell fraction specific when a cell population such as a sample derived from a subject is separated into a cancer cell fraction and a non-cancerous cell (normal cell) fraction. Refers to a marker.
- a fraction marker is a gene, it may be referred to as a “fraction marker gene”.
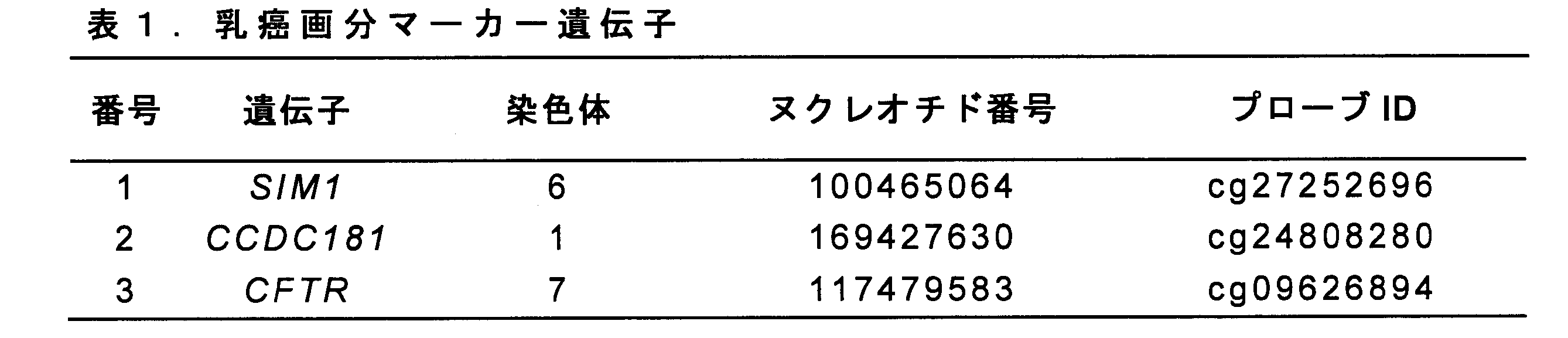
- the information of the CpG site of the SIM1 gene, the CCDC181 gene, and the CFTR gene (breast cancer fraction marker gene), which are mainly intended to be analyzed in the present invention, are as shown in Table 1 below (the genome is located in the human genome assembly). hg38).
- the CpG sites of the SIM1 gene analyzed in the present invention are the 1000th and 1001th nucleotide residues in the nucleotide sequence of SEQ ID NO: 1 (the 4th and 5th nucleotides in the standard nucleotide sequence ggccgact located upstream of exon 1 of the SIM1 gene). Nucleotide portion) (when the 5 ′ ggc portion and the 3 ′ act portion are not mutated).
- the standard nucleotide sequence is intended to be referred to to identify the CpG site corresponding to the CpG site in Table 1 above (as well as the CpG sites of other genes analyzed in the present invention).
- CpG site of SIM1 gene refers to the above CpG site unless otherwise specified.
- the CpG sites of the CCDC181 gene analyzed in the present invention are nucleotides 1000 and 1001 in the nucleotide sequence of SEQ ID NO: 2 (the 4th and 5th nucleotides in the standard nucleotide sequence cagcggca present in intron 2 of the CCDC181 gene). Nucleotide portion) (when the 5′-side cag portion and the 3′-side gca portion are not mutated).
- a person skilled in the art can appropriately determine a CpG site corresponding to the CpG site existing at the above position, in consideration of the genomic DNA mutation, for a human subject having a genomic DNA mutation.
- CpG site of CCDC181 gene when simply referred to as “CpG site of CCDC181 gene” in the present specification, the above CpG site is indicated unless otherwise specified.
- the CpG site of the CFTR gene analyzed in the present invention is the nucleotide residues 1001 and 1002 in the nucleotide sequence of SEQ ID NO: 3 (the 4th and 5th nucleotides in the standard nucleotide sequence ctccgggg located upstream of exon 1 of the CFTR gene). Nucleotide portion) (when the 5 'ctc portion and the 3' ggg portion are not mutated).
- a person skilled in the art can appropriately determine a CpG site corresponding to the CpG site existing at the above position, in consideration of the genomic DNA mutation, for a human subject having a genomic DNA mutation.
- CFTR gene CpG site when simply referred to as “CFTR gene CpG site” in the present specification, the above CpG site is indicated unless otherwise specified.
- the present invention provides a method for estimating the abundance of breast cancer cells.
- the estimation method of the present invention includes the following (1) and (2): (1) In a sample derived from a human subject, the following: (A) a cytosine residue in the CpG site of the SIM1 gene; (B) cytosine residues in the CpG site of the CCDC181 gene; (C) cytosine residues in the CpG site of the CFTR gene; or (d) analyzing the methylation rate of these combinations; and (2) a human test based on the methylation rate analyzed in (1). Estimating the abundance of breast cancer cells in a body-derived sample.
- a tissue sample corresponding to the above-mentioned cancer is preferably used.
- a specific CpG site is analyzed in genomic DNA contained in such a cancer tissue sample.
- Genomic DNA can be appropriately extracted from a cancer tissue sample by any method.
- a sample collected by a biopsy such as a needle biopsy can be used.
- the cancer tissue sample may have been subjected to a preliminary treatment. Examples of such pretreatment include extraction, cell fixation, tissue fixation, tissue exfoliation, cell dissociation treatment, heating, freezing, refrigeration, and liquefaction.
- a cancer tissue sample is usually a sample of a cell population containing cancer cells and normal cells (non-cancerous cells).
- the methylation analyzed in the present invention is methylation at the 5-position of a cytosine residue.
- the analysis of the methylation rate can be performed by any method known in the art.
- the analysis can be performed using one or more analysis means selected from the group consisting of bisulfite, a primer, a nucleic acid probe, a restriction enzyme, an anti-methylated cytosine antibody, and a nanopore.
- bisulfite converts unmethylated cytosine to uracil while not converting methylated cytosine to uracil. Therefore, bisulfite is widely used in combination with other analysis means in the analysis of methylated cytosine due to such properties.
- the primer is a sequencing primer, a gene amplification primer (eg, a PCR primer), or a combination thereof.
- the primer can be appropriately designed so that the target CpG site can be analyzed.
- the primer may be a downstream region of the CpG site of interest (an arbitrary region in the portion consisting of nucleotide residues 1002 to 2001 in the nucleotide sequence of SEQ ID NO: 1 for the SIM1 gene, and a nucleotide of SEQ ID NO: 2 for the CCDC181 gene).
- Anneals to any region in the portion consisting of nucleotides 1002 to 2001 in the sequence, or to the CFTR gene (any region in the portion consisting of nucleotides 1003 to 2001 in the nucleotide sequence of SEQ ID NO: 3)
- any region in the region consisting of nucleotide residues 1 to 999 in the nucleotide sequence of SEQ ID NO: 1 for the SIM1 gene is used.
- the sense and antisense strands can be designed to anneal in any region in the portion) and in the downstream region (any region shown above as a downstream region).
- the nucleic acid probe is a region containing the CpG site of interest (for the SIM1 gene, a portion containing a CG portion consisting of the 1000th and 1001th nucleotide residues in the nucleotide sequence of SEQ ID NO: 1; for the CCDC181 gene, a nucleotide sequence of SEQ ID NO: 2)
- a portion containing a CG portion consisting of the 1000th and 1001th nucleotide residues in the above, a portion containing a CG portion consisting of the 1001th and 1002th nucleotide residues in the nucleotide sequence of SEQ ID NO: 3 for the CFTR gene or It can be designed to hybridize to its upstream region (any region shown above as an upstream region) or downstream region (any region shown above as a downstream region).
- the nucleic acid probe can be used in a free form or a form immobilized on a solid phase.
- the solid phase include particles (eg, magnetic particles); supports such as arrays, membranes (eg, nitrocellulose membranes, filter papers) and columns; and containers such as plates (eg, multiwell plates) and tubes.
- Can be Solid phase materials include, for example, glass, plastic, and metal.
- the nucleic acid probe may also be a nucleic acid probe as detailed in the analysis of methylated cytosine using an anti-methylated cytosine antibody.
- the restriction enzyme is a methylation-specific or non-specific restriction enzyme, and can be used in an appropriate combination with the above-mentioned bisulfite, primer, and nucleic acid probe.
- examples of analysis methods using the above-described analysis means include DNA methylation sequencing, bisulfite sequencing, bisulfite pyrosequencing, methylation-specific PCR, and restriction enzyme land.
- Analysis can also be performed using anti-methylated cytosine antibodies. Analysis of methylated cytosine using an anti-methylated cytosine antibody is well known in the art (eg, WO2015 / 025862; WO2015 / 025863; WO2015 / 025864; WO2016 / 052368; JP-A-2012-230019; DNA @ Research # 13). Chem., 37-42 (2006); Anal. Chem. @ 2012, 84, 7533-7538). Anti-methylated cytosine antibodies may be used in combination with one or more analysis means as described above.
- a method using such analysis means for example, a method using a combination of an anti-methylated cytosine antibody and a heterologous nucleic acid probe (eg, a normal RNA probe, a modified RNA probe) (eg, WO2015 / 025862) A) a method using a combination of an anti-methylated cytosine antibody, and a solid phase probe and a capture probe (eg, WO2015 / 025863); a method using a combination of an anti-methylated cytosine antibody, and an absorbent polynucleotide and a capture probe (eg, WO2015 / 025864); a method using a combination of an anti-methylated cytosine antibody and a modified nucleic acid base-pairing heterologous nucleic acid probe (eg, WO2016 / 052368).
- a heterologous nucleic acid probe eg, a normal RNA probe, a modified RNA probe
- the nucleic acid probe used in combination with the anti-methylated cytosine antibody hybridizes with the DNA chain containing the above-described CpG site analyzed in the present invention, and forms a double-stranded structure portion comprising the DNA chain and the nucleic acid probe.
- it may be designed so that the methylcytosine residue in the CpG site forms an unpaired portion with the nucleic acid probe (in other words, does not bind complementarily) (eg, WO2015 / 025862).
- the nucleic acid probe may be used as a nucleotide residue corresponding to an unpaired portion other than a guanine residue capable of binding complementarily to a methylcytosine residue (eg, a cytosine residue, a thymine residue, an adenine residue). , A uracil residue).
- the nucleic acid probe may be designed so that such an unpaired portion is not formed (eg, WO2016 / 052368).
- the method using an anti-methylated cytosine antibody can be performed by any immunological method known in the art.
- examples of such a method include enzyme immunoassay (EIA) (eg, CLEIA, ELISA), fluorescence immunoassay, chemiluminescence immunoassay, electrochemiluminescence immunoassay, agglutination, Examples include immunostaining, flow cytometry, biolayer interferometry, In situ PLA, chemical amplified luminescence proximity homogeneous assay, line blotting, and western blotting.
- EIA enzyme immunoassay
- fluorescence immunoassay eg, CLEIA, ELISA
- fluorescence immunoassay eg, chemiluminescence immunoassay
- electrochemiluminescence immunoassay agglutination
- Examples include immunostaining, flow cytometry, biolayer interferometry, In situ PLA, chemical amplified luminescence proximity homogeneous as
- the methylation rate analyzed in the present invention is the rate of methylation of cytosine residues in CpG in cancer cells.
- the measurement of the methylation rate is well known in the art (eg, JP-A-2012-090555, JP-A-2014-036672, JP-T-2010-538638, and Table-A-2009 / 136501).
- the methylation rate analyzed in the present invention may be a combination of the methylation rates of cytosine residues in the CpG site of the SIM1 gene, CCDC181 gene, and CFTR gene.
- the combination of the methylation rates as shown in the above (d) is a combination consisting of two or more of these methylation rates (ie, 2 or 3), and specifically, a combination of the SIM1 gene and the CCDC181 gene, The methylation rates of cytosine residues in CpG sites in the combination of the SIM1 gene and the CFTR gene, the combination of the CCDC181 gene and the CFTR gene, and the combination of the SIM1 gene, the CCDC181 gene, and the CFTR gene.
- the methylation rate in the combination for example, any of the highest methylation rate, the lowest methylation rate, and the average methylation rate in the combination may be employed, but the highest methylation rate is preferably employed.
- step (2) the percentage of breast cancer cells in a sample derived from a human subject can be estimated based on the analyzed methylation rate.
- the present invention also provides a method for predicting the effect of drug therapy on breast cancer.
- ⁇ ⁇ Pharmacotherapy for breast cancer includes, for example, treatment with anticancer agents.
- the anticancer agent include a microtubule inhibitor, an anticancer antibiotic, a topoisomerase inhibitor, a platinum preparation, and an alkylating agent.
- the anti-cancer agent may also be a molecular targeted drug.
- molecular targeted drugs examples include HER2 inhibitors, EGFR inhibitors (eg, gefitinib, lapatinib, erlotinib, cetuximab), c-MET inhibitors (eg, PHA-665752, SU11274, XL-880), ALK Inhibitors (eg, WHI-P154, TAE684, PF-2341066), PDGFR inhibitors (eg, imatinib, desatinib, valatinib), c-KIT inhibitors (eg, sunitinib, macitinib, motesanib).
- EGFR inhibitors eg, gefitinib, lapatinib, erlotinib, cetuximab
- c-MET inhibitors eg, PHA-665752, SU11274, XL-880
- ALK Inhibitors eg, WHI-P154, TAE684, PF-2341066
- PDGFR inhibitors
- ⁇ ⁇ Drug therapy for breast cancer can be appropriately selected according to the subtype of breast cancer.
- HER2 positive breast cancer includes, for example, treatment with a HER2 inhibitor.
- HER2 is a receptor tyrosine kinase that belongs to the same family as epidermal growth factor (EGFR).
- EGFR epidermal growth factor
- HER2 has a structure in which an extracellular domain, a transmembrane domain, and an intracellular domain having tyrosine kinase activity are linked by a single chain (eg, Coussens et al., Science, 1985, Vol. 230, No. 4730, pp. 1132). -1139, and Genebank accession number: NP_001005862.1).
- HER2 inhibitor examples include antibodies (eg, trastuzumab, pertuzumab), low molecular weight organic compounds (eg, HER2 tyrosine kinase inhibitors such as lapatinib, neratinib, afatinib, etc.), and drug-binding antibodies (eg, T-DM1).
- antibodies eg, trastuzumab, pertuzumab
- low molecular weight organic compounds eg, HER2 tyrosine kinase inhibitors such as lapatinib, neratinib, afatinib, etc.
- drug-binding antibodies eg, T-DM1.
- Medications for luminal type A breast cancer include, for example, treatment with hormone therapy or chemotherapy.
- Hormone therapy includes, for example, therapy using LH-RH agonist preparations, antiestrogens, progestin, and aromatase inhibitors.
- Chemotherapy includes, for example, therapies using topoisomerase inhibitors, microtubule acting drugs, alkylating drugs, antimetabolites.
- Pharmacotherapy for luminal B-type breast cancer includes, for example, treatment with hormone therapy or chemotherapy.
- Hormone therapy includes, for example, therapy using LH-RH agonist preparations, antiestrogens, progestin, and aromatase inhibitors.
- Chemotherapy includes, for example, therapies using topoisomerase inhibitors, microtubule acting drugs, alkylating drugs, antimetabolites.
- ⁇ ⁇ Pharmacotherapy for triple negative breast cancer includes, for example, treatment with chemotherapy.
- Chemotherapy includes, for example, therapies using topoisomerase inhibitors, microtubule acting drugs, alkylating drugs, antimetabolites.
- a plurality of anticancer drugs can be used in combination.
- the plurality of anticancer agents used in combination include a combination of two or more anticancer agents described above and a combination of one or more anticancer agents described above and another anticancer agent.
- other anticancer agents include microtubule inhibitors such as taxane anticancer agents (eg, paclitaxel), anthracyclines (eg, adriacin), and aromatase inhibitors (eg, letrozole).
- the prediction method of the present invention includes the following (1) to (4): (1) measuring the value of a breast cancer marker in a sample derived from a human subject; (2) In a sample derived from a human subject, the following: (A) a cytosine residue in the CpG site of the SIM1 gene; (B) cytosine residues in the CpG site of the CCDC181 gene; (c) cytosine residues in the CpG site of the CFTR gene; or (d) analyzing the methylation rate of these combinations; (3) calculating the corrected value of the breast cancer marker by correcting the value of the breast cancer marker measured in (1) with the methylation rate analyzed in (2); and (4) correcting the corrected value of (3). Predicting the effect of drug therapy on breast cancer based on corrected values of breast cancer markers.
- the breast cancer marker in step (1) is a marker that serves as an indicator of breast cancer, and examples thereof include a DNA methylation rate (eg, cg15896301 of HSD17B4: Non-Patent Document 3).
- the means for analyzing the CpG site of the fraction marker gene (SIM1 gene, CCDC181 gene, or CFTR gene) in step (2) and the methylation rate are as described above.
- the correction of the value of the breast cancer marker in the step (3) corresponds to a pureer breast cancer cell based on the value of the breast cancer marker measured in a sample (sample containing breast cancer cells and non-breast cancer cells) derived from a human subject. This means calculating the value of the obtained breast cancer marker. Therefore, the correction value of the breast cancer marker can be obtained, for example, by the following equation.
- Correction value of breast cancer marker (value of breast cancer marker measured in sample derived from human subject) / (presence of breast cancer cells in sample derived from human subject)
- the step (4) can be performed by using a correction value of the breast cancer marker instead of the value of the breast cancer marker in a method of predicting the effect of the pharmacotherapy on the breast cancer based on the value of the breast cancer marker.
- a correction value of the breast cancer marker instead of the value of the breast cancer marker in a method of predicting the effect of the pharmacotherapy on the breast cancer based on the value of the breast cancer marker.
- the present invention also provides a method for determining breast cancer.
- Determination of breast cancer refers to evaluating whether a human subject is more or less likely to have breast cancer.
- the determination method of the present invention includes the following (1) and (2): (1) In a sample derived from a human subject, the following: (A) a cytosine residue in the CpG site of the SIM1 gene; (B) cytosine residues in the CpG site of the CCDC181 gene; (C) a cytosine residue in the CpG site of the CFTR gene; or (d) analyzing the methylation rate of these combinations; and (2) a breast cancer tumor based on the methylation rate analyzed in (1). Assess morbidity.
- the human subject-derived sample, the CpG site of the fraction marker gene (SIM1 gene, CCDC181 gene or CFTR gene) in step (1), and the means for analyzing the methylation rate are as described above.
- the human subject may be suffering from breast cancer when the methylation rate is equal to or higher than the reference value, and / or the human subject may have breast cancer when the methylation rate is lower than the reference value. It can be evaluated that the possibility of suffering is low.
- a cutoff value appropriately set so that a breast cancer patient and a healthy person can be distinguished can be used.
- the cut-off value of the methylation rate for example, any percentage value within the range of 10 to 30% can be employed, but any percentage value within the range of 15 to 25% is employed. And preferably 20%.
- the present invention also provides a reagent for estimating the abundance of breast cancer cells.
- the estimation reagent of the present invention includes the following: (A) a cytosine residue in the CpG site of the SIM1 gene; (B) cytosine residues in the CpG site of the CCDC181 gene; (C) a cytosine residue in the CpG site of the CFTR gene; or (d) a means for analyzing the methylation rate of these combinations.
- the means for analyzing the CpG site of the fraction marker gene (SIM1 gene, CCDC181 gene, or CFTR gene) in the estimation reagent of the present invention, and the methylation rate are as described above.
- the estimation reagent of the present invention analyzes the methylation rate of the fraction marker gene in the human subject-derived sample using a methylation rate analysis means, and based on the analyzed methylation rate, the human subject-derived sample It can be used to estimate the abundance of breast cancer cells in.
- the present invention also provides a reagent for determining breast cancer.
- the determination reagent of the present invention includes the following: (A) a cytosine residue in the CpG site of the SIM1 gene; (B) cytosine residues in the CpG site of the CCDC181 gene; (C) a cytosine residue in the CpG site of the CFTR gene; or (d) a means for analyzing the methylation rate of these combinations.
- the means for analyzing the CpG site of the fraction marker gene (SIM1 gene, CCDC181 gene, or CFTR gene) and the methylation rate in the determination reagent of the present invention are as described above.
- the determination reagent of the present invention analyzes the methylation rate of a fraction marker gene in a sample derived from a human subject using a methylation rate analysis means, and based on the analyzed methylation rate, a breast cancer in a human subject. Can be used to determine
- the present invention also provides a kit.
- the kit of the present invention comprises the following (1) and (2): (1) The following: (A) a cytosine residue in the CpG site of the SIM1 gene; (B) cytosine residues in the CpG site of the CCDC181 gene; (C) a cytosine residue in the CpG site of the CFTR gene; or (d) means for analyzing the methylation rate of a combination thereof; and (2) means for measuring a breast cancer marker.
- the means for analyzing the CpG site of the fraction marker gene (SIM1 gene, CCDC181 gene, or CFTR gene) in the reagent for judging breast cancer of the present invention, the methylation rate, and the means for measuring the breast cancer marker are as described above.
- the kit of the present invention can be used for various purposes, and can be used, for example, for predicting the effect of drug therapy on breast cancer.
- Sample 1-1 Biologically-derived sample
- 195 breast cancer tissue samples were obtained from 195 breast cancer patients.
- Breast cancer tissue samples from each patient were collected by needle biopsy from patients diagnosed with breast cancer.
- Samples were fixed using the PAX gene tissue system (Qiagen, Hilden, Germany) and embedded in low melting paraffin.
- Ten 10 ⁇ m thin sections were prepared and DNA was extracted.
- microscopic examination of the samples was performed to determine the cancer cell fraction.
- the therapeutic response to trastuzumab was determined, and the state in which no cancer cells remained in the sample was determined as a complete pathological response (pCR). It was defined as incomplete response (Non-pCR).
- 10 of the 61 samples were separated into cancer and non-cancer cells by laser capture microdissection (LCM).
- HMECs Human breast epithelial cells
- the bisulfite-modified DNA was suspended in 40 ⁇ l of TE buffer, and a 1 ⁇ l aliquot was previously reported (Yoshida T, Yamashita S, Takamura-Enya T, Niwa T, Anato T, Alcoa, and other sickness. pylori-infected gastric mucosae. International journal of cancer. 2011; 128 (1): 33-9).
- Target genomic regions were amplified with biotinylated primers. Biotin-labeled PCR products were annealed to 0.2 ⁇ M pyrosequencing primers and biosequencing was performed using the PSQ 96 pyrosequencing system (QIAGEN, Valencia, CA, USA). Methylation levels were obtained using PSQ assay design software (QIAGEN).
- HSD17B4 methylation level by breast cancer cell fraction 3-1. Measurement of the value of breast cancer marker The methylation level of the HSD17B4 gene was measured for 61 HER2-positive breast cancer samples for which pCR and Non-pCR were determined in 1-1 above. HSD17B4 methylation level was measured by bisulfite pyrosequencing according to 2-1 (see Non-Patent Document 3).
- SEQ ID NO: 1 shows the nucleotide sequence of a 1000 bp genomic region before and after the CpG site for measuring the methylation level of the SIM1 gene (nucleotide residues at positions 1000 and 1001).
- SEQ ID NO: 2 shows the nucleotide sequence of a genomic region of 1000 bp before and after the CpG site (the 1000th and 1001th nucleotide residues) for measuring the methylation level of the CCDC181 gene.
- SEQ ID NO: 3 shows the nucleotide sequence of a 1000 bp genomic region before and after the CpG site for measuring the methylation level of the CFTR gene (nucleotide residues at positions 1001 and 1002).
- SEQ ID NOs: 4 to 6 show the nucleotide sequences of a forward primer, a reverse primer, and a sequencing primer for measuring the methylation level of the CpG site of the SIM1 gene, respectively.
- SEQ ID NOs: 7 to 9 respectively show the nucleotide sequences of a forward primer, a reverse primer, and a sequencing primer for measuring the methylation level of the CpG site of the CCDC181 gene.
- SEQ ID NOs: 10 to 12 show the nucleotide sequences of a forward primer, a reverse primer, and a sequencing primer for measuring the methylation level of the CpG site of the CFTR gene, respectively.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Engineering & Computer Science (AREA)
- Analytical Chemistry (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Genetics & Genomics (AREA)
- Pathology (AREA)
- Immunology (AREA)
- General Health & Medical Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biotechnology (AREA)
- Biophysics (AREA)
- Physics & Mathematics (AREA)
- Hospice & Palliative Care (AREA)
- Microbiology (AREA)
- Oncology (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- General Engineering & Computer Science (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Reproductive Health (AREA)
- Endocrinology (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Investigating Or Analysing Biological Materials (AREA)
Abstract
本発明は、乳癌に対する薬物療法の効果予測をする指標の精度の向上を提供する。具体的には、本発明は、以下(1)および(2)を含む、乳癌細胞存在率の推定方法などを提供する: (1)ヒト被験体由来サンプルにおいて、以下: (a)配列番号1のヌクレオチド配列における1000番目と1001番目のヌクレオチドからなるCG部分中のシトシン残基; (b)配列番号2のヌクレオチド配列における1000番目と1001番目のヌクレオチドからなるCG部分中のシトシン残基; (c)配列番号3のヌクレオチド配列における1001番目と1002番目のヌクレオチドからなるCG部分中のシトシン残基;または (d)これらの組合せ のメチル化率を解析すること;および (2)(1)で解析されたメチル化率に基づいて、ヒト被験体由来サンプル中の乳癌細胞存在率を推定すること。
Description
本発明は、乳癌細胞存在率の推定方法などに関する。
癌組織サンプルにおける癌細胞画分の評価は、癌マーカーを用いた癌の診断(例、癌の発症リスクの判定、癌の罹患の有無判定、癌に対する薬物療法の効果の予測(例えば、非特許文献1)、および癌に対する薬物療法の予後の評価)における不要画分(正常細胞)の混在による判定精度の低下を排除するために重要である。
癌細胞画分を評価する方法としては、例えば、(1)病理診断、および(2)所定の方法を用いて、生検で採取した癌組織サンプル(癌細胞および非癌性細胞を含む)から癌細胞を分離する方法などが存在する。しかし、(1)については、熟練した医師の従事が必要であり、また、(2)の方法については、煩雑であるなどの問題がある。
上記問題を解決するため、癌細胞画分に特異的なマーカー(「画分マーカー」)を用いて癌組織サンプルを解析することにより、癌細胞画分を評価することが提案されている。このような画分マーカーとしては、例えば、食道癌におけるメチル化マーカーTFAP2B、ARHGEF4、およびRAPGEFL1(非特許文献2)、胃癌におけるメチル化マーカーOSR2、PPFIA3、VAV3(非特許文献3)が報告されている。
Fujii S, Yamashita S, Yamaguchi T, Takahashi M, Hozumi Y, Ushijima T et al. Pathological complete response of HER2-positive breast cancer to trastuzumab and chemotherapy can be predicted by HSD17B4 methylation. Oncotarget. 2017;8(12):19039-48. doi:10.18632/oncotarget.15118.
Takahashi T et al., PLoS one, 2013;8(12):e82302
Zong L et al., Gastric cancer, 2016;19(2):361-9
本発明の目的は、乳癌組織に含まれる乳癌細胞含有率を判定する癌マーカーを提供することである。本発明の目的はまた、乳癌細胞含有率を判定することにより、乳癌に対する薬物療法の効果予測をする指標の精度を向上させることである。
本発明者らは、先ず、乳癌細胞画分を評価するため、乳癌細胞では高度にメチル化されているが、正常細胞(非乳癌細胞)では全くまたは低度にしかメチル化されていない乳癌細胞特異的なCpGサイトを探索すること、ならびにこの探索において、細胞間において高発生率のコピー数変化を有さず、かつ広範な患者を対象とすることができるCpGサイトを選択することを着想した。そこで、本発明者らは、常染色体および性染色体に存在する45万を超えるメチル化候補サイトについて包括的に鋭意検討した結果、このようなCpGサイトとして、SIM1遺伝子、CCDC181遺伝子、およびCFTR遺伝子中の特定のCpGサイトを見出した。本発明者らは、これらのCpGサイトのメチル化率を、乳癌細胞存在率の指標として利用することなどに成功し、本発明を完成するに至った。
すなわち、本発明は以下のとおりである。
〔1〕以下(1)および(2)を含む、乳癌細胞存在率の推定方法:
(1)ヒト被験体由来サンプルにおいて、以下:
(a)配列番号1のヌクレオチド配列における1000番目と1001番目のヌクレオチドからなるCG部分中のシトシン残基;
(b)配列番号2のヌクレオチド配列における1000番目と1001番目のヌクレオチドからなるCG部分中のシトシン残基;
(c)配列番号3のヌクレオチド配列における1001番目と1002番目のヌクレオチドからなるCG部分中のシトシン残基;または
(d)これらの組合せ
のメチル化率を解析すること;および
(2)(1)で解析されたメチル化率に基づいて、ヒト被験体由来サンプル中の乳癌細胞存在率を推定すること。
〔2〕解析が、亜硫酸水素塩、1以上のプライマー、1以上の核酸プローブ、制限酵素、抗メチル化シトシン抗体、およびナノポアからなる群より選ばれる1または2以上の手段を用いて行われる、〔1〕の方法。
〔3〕解析が、バイサルファイトシーケンシング法、バイサルファイト・パイロシーケンシング法、メチル化特異的PCR法、制限酵素ランドマーク・ゲノム・スキャニング(RLGS)法、単一ヌクレオチド・プライマー伸長(SNuPE)法、CpGアイランド・マイクロアレイ法、メチライト法、COBRA法、質量分析(マスアレイ)法、メチル化特異的制限酵素の使用、高解像度融解解析(HRM)法、ナノポア解析法、ICONプローブ法、メチル化特異的MLPA法、またはイムノアッセイにより行われる、〔1〕または〔2〕の方法。
〔4〕以下(1)~(4)を含む、乳癌に対する薬物療法の効果の予測方法:
(1)ヒト被験体由来サンプルにおいて、乳癌マーカーの値を測定すること;
(2)ヒト被験体由来サンプルにおいて、以下:
(a)配列番号1のヌクレオチド配列における1000番目と1001番目のヌクレオチドからなるCG部分中のシトシン残基;
(b)配列番号2のヌクレオチド配列における1000番目と1001番目のヌクレオチドからなるCG部分中のシトシン残基;
(c)配列番号3のヌクレオチド配列における1001番目と1002番目のヌクレオチドからなるCG部分中のシトシン残基;または
(d)これらの組合せ
のメチル化率を解析すること;
(3)(1)で測定された乳癌マーカーの値を(2)で解析されたメチル化率で補正して、乳癌マーカーの補正値を計算すること;および
(4)(3)で補正された乳癌マーカーの補正値に基づいて、乳癌に対する薬物療法の効果を予測すること。
〔5〕以下(1)および(2)を含む、乳癌の判定方法:
(1)ヒト被験体由来サンプルにおいて、以下:
(a)配列番号1のヌクレオチド配列における1000番目と1001番目のヌクレオチドからなるCG部分中のシトシン残基;
(b)配列番号2のヌクレオチド配列における1000番目と1001番目のヌクレオチドからなるCG部分中のシトシン残基;
(c)配列番号3のヌクレオチド配列における1001番目と1002番目のヌクレオチドからなるCG部分中のシトシン残基;または
(d)これらの組合せ
のメチル化率を解析すること;および
(2)(1)で解析されたメチル化率に基づいて、乳癌の罹患可能性を評価すること。
〔6〕以下:
(a)配列番号1のヌクレオチド配列における1000番目と1001番目のヌクレオチドからなるCG部分中のシトシン残基;
(b)配列番号2のヌクレオチド配列における1000番目と1001番目のヌクレオチドからなるCG部分中のシトシン残基;
(c)配列番号3のヌクレオチド配列における1001番目と1002番目のヌクレオチドからなるCG部分中のシトシン残基;または
(d)これらの組合せ
のメチル化率の解析手段を含む、乳癌細胞存在率の推定用試薬。
〔7〕以下:
(a)配列番号1のヌクレオチド配列における1000番目と1001番目のヌクレオチドからなるCG部分中のシトシン残基;
(b)配列番号2のヌクレオチド配列における1000番目と1001番目のヌクレオチドからなるCG部分中のシトシン残基;
(c)配列番号3のヌクレオチド配列における1001番目と1002番目のヌクレオチドからなるCG部分中のシトシン残基;または
(d)これらの組合せ
のメチル化率の解析手段を含む、乳癌の判定用試薬。
〔8〕以下(1)および(2)を含むキット:
(1)以下:
(a)配列番号1のヌクレオチド配列における1000番目と1001番目のヌクレオチドからなるCG部分中のシトシン残基;
(b)配列番号2のヌクレオチド配列における1000番目と1001番目のヌクレオチドからなるCG部分中のシトシン残基;
(c)配列番号3のヌクレオチド配列における1001番目と1002番目のヌクレオチドからなるCG部分中のシトシン残基;または
(d)これらの組合せ
のメチル化率解析手段;および
(2)乳癌マーカーの測定手段。
〔1〕以下(1)および(2)を含む、乳癌細胞存在率の推定方法:
(1)ヒト被験体由来サンプルにおいて、以下:
(a)配列番号1のヌクレオチド配列における1000番目と1001番目のヌクレオチドからなるCG部分中のシトシン残基;
(b)配列番号2のヌクレオチド配列における1000番目と1001番目のヌクレオチドからなるCG部分中のシトシン残基;
(c)配列番号3のヌクレオチド配列における1001番目と1002番目のヌクレオチドからなるCG部分中のシトシン残基;または
(d)これらの組合せ
のメチル化率を解析すること;および
(2)(1)で解析されたメチル化率に基づいて、ヒト被験体由来サンプル中の乳癌細胞存在率を推定すること。
〔2〕解析が、亜硫酸水素塩、1以上のプライマー、1以上の核酸プローブ、制限酵素、抗メチル化シトシン抗体、およびナノポアからなる群より選ばれる1または2以上の手段を用いて行われる、〔1〕の方法。
〔3〕解析が、バイサルファイトシーケンシング法、バイサルファイト・パイロシーケンシング法、メチル化特異的PCR法、制限酵素ランドマーク・ゲノム・スキャニング(RLGS)法、単一ヌクレオチド・プライマー伸長(SNuPE)法、CpGアイランド・マイクロアレイ法、メチライト法、COBRA法、質量分析(マスアレイ)法、メチル化特異的制限酵素の使用、高解像度融解解析(HRM)法、ナノポア解析法、ICONプローブ法、メチル化特異的MLPA法、またはイムノアッセイにより行われる、〔1〕または〔2〕の方法。
〔4〕以下(1)~(4)を含む、乳癌に対する薬物療法の効果の予測方法:
(1)ヒト被験体由来サンプルにおいて、乳癌マーカーの値を測定すること;
(2)ヒト被験体由来サンプルにおいて、以下:
(a)配列番号1のヌクレオチド配列における1000番目と1001番目のヌクレオチドからなるCG部分中のシトシン残基;
(b)配列番号2のヌクレオチド配列における1000番目と1001番目のヌクレオチドからなるCG部分中のシトシン残基;
(c)配列番号3のヌクレオチド配列における1001番目と1002番目のヌクレオチドからなるCG部分中のシトシン残基;または
(d)これらの組合せ
のメチル化率を解析すること;
(3)(1)で測定された乳癌マーカーの値を(2)で解析されたメチル化率で補正して、乳癌マーカーの補正値を計算すること;および
(4)(3)で補正された乳癌マーカーの補正値に基づいて、乳癌に対する薬物療法の効果を予測すること。
〔5〕以下(1)および(2)を含む、乳癌の判定方法:
(1)ヒト被験体由来サンプルにおいて、以下:
(a)配列番号1のヌクレオチド配列における1000番目と1001番目のヌクレオチドからなるCG部分中のシトシン残基;
(b)配列番号2のヌクレオチド配列における1000番目と1001番目のヌクレオチドからなるCG部分中のシトシン残基;
(c)配列番号3のヌクレオチド配列における1001番目と1002番目のヌクレオチドからなるCG部分中のシトシン残基;または
(d)これらの組合せ
のメチル化率を解析すること;および
(2)(1)で解析されたメチル化率に基づいて、乳癌の罹患可能性を評価すること。
〔6〕以下:
(a)配列番号1のヌクレオチド配列における1000番目と1001番目のヌクレオチドからなるCG部分中のシトシン残基;
(b)配列番号2のヌクレオチド配列における1000番目と1001番目のヌクレオチドからなるCG部分中のシトシン残基;
(c)配列番号3のヌクレオチド配列における1001番目と1002番目のヌクレオチドからなるCG部分中のシトシン残基;または
(d)これらの組合せ
のメチル化率の解析手段を含む、乳癌細胞存在率の推定用試薬。
〔7〕以下:
(a)配列番号1のヌクレオチド配列における1000番目と1001番目のヌクレオチドからなるCG部分中のシトシン残基;
(b)配列番号2のヌクレオチド配列における1000番目と1001番目のヌクレオチドからなるCG部分中のシトシン残基;
(c)配列番号3のヌクレオチド配列における1001番目と1002番目のヌクレオチドからなるCG部分中のシトシン残基;または
(d)これらの組合せ
のメチル化率の解析手段を含む、乳癌の判定用試薬。
〔8〕以下(1)および(2)を含むキット:
(1)以下:
(a)配列番号1のヌクレオチド配列における1000番目と1001番目のヌクレオチドからなるCG部分中のシトシン残基;
(b)配列番号2のヌクレオチド配列における1000番目と1001番目のヌクレオチドからなるCG部分中のシトシン残基;
(c)配列番号3のヌクレオチド配列における1001番目と1002番目のヌクレオチドからなるCG部分中のシトシン残基;または
(d)これらの組合せ
のメチル化率解析手段;および
(2)乳癌マーカーの測定手段。
本発明の推定方法は、例えば、乳癌マーカーを用いた乳癌の判定(例、乳癌リスク判定、乳癌罹患の判定、薬物療法の効果の予測、予後評価)の精度向上に有用である。
本発明の予測方法は、例えば、乳癌患者に対する外科的手術の要否および薬物療法の選択の判断における指標として有用である。
本発明の判定方法は、例えば、乳癌の診断に有用である。
本発明の試薬およびキットは、例えば、本発明の方法の簡便な実施に有用である。
本発明の予測方法は、例えば、乳癌患者に対する外科的手術の要否および薬物療法の選択の判断における指標として有用である。
本発明の判定方法は、例えば、乳癌の診断に有用である。
本発明の試薬およびキットは、例えば、本発明の方法の簡便な実施に有用である。
(一般的な説明)
本発明において対象とされる癌は、乳癌である。乳癌は、HER2陽性型、ルミナルA型、ルミナルB型、およびトリプルネガティブ型のサブタイプに分類することができる。本発明において対象とされる癌は、いずれのサブタイプの乳癌であってもよいが、特定の局面ではHER2陽性型が好ましい。
本発明において対象とされる癌は、乳癌である。乳癌は、HER2陽性型、ルミナルA型、ルミナルB型、およびトリプルネガティブ型のサブタイプに分類することができる。本発明において対象とされる癌は、いずれのサブタイプの乳癌であってもよいが、特定の局面ではHER2陽性型が好ましい。
本発明において、「乳癌細胞存在率」とは、サンプル中の全細胞の個数(すなわち、乳癌細胞の個数および非乳癌細胞の個数の合計)に対する乳癌細胞の個数の比率をいう。すなわち、乳癌細胞存在率は、下記式で表される。
乳癌細胞存在率=(乳癌細胞の個数)/〔(乳癌細胞の個数)+(非乳癌細胞の個数)〕
乳癌細胞存在率=(乳癌細胞の個数)/〔(乳癌細胞の個数)+(非乳癌細胞の個数)〕
本明細書において、「画分マーカー」とは、被験体由来サンプル等の細胞集団を癌細胞画分と非癌性細胞(正常細胞)画分に分離した場合の癌細胞画分に特異的なマーカーをいう。画分マーカーは、遺伝子である場合に、「画分マーカー遺伝子」と呼ぶこともある。
本発明において主に解析が意図されるSIM1遺伝子、CCDC181遺伝子、およびCFTR遺伝子(乳癌画分マーカー遺伝子)のCpGサイトの情報は、以下の表1に示すとおりである(ゲノムの位置は、ヒトゲノムアセンブリhg38に基づく)。
本発明において解析されるSIM1遺伝子のCpGサイトは、配列番号1のヌクレオチド配列における1000番目と1001番目のヌクレオチド残基(SIM1遺伝子のエクソン1上流に存在する標準ヌクレオチド配列ggccgact中の4および5番目のヌクレオチド)からなるCG部分と定義することができる(5’側のggc部分および3’側のact部分が変異していない場合)。標準ヌクレオチド配列は、上記表1のCpGサイトに対応するCpGサイトを特定するために参照されることを意図するものである(本発明において解析される他の遺伝子のCpGサイトも同様)。当業者であれば、ゲノムDNAの変異が認められるヒト被験体について、ゲノムDNAの変異を考慮して、上記位置に存在するCpGサイトに相当するCpGサイトを適宜決定することができる。以下、本明細書において単に「SIM1遺伝子のCpGサイト」と称する場合は、特に記載がない限り、上記CpGサイトを示す。
本発明において解析されるCCDC181遺伝子のCpGサイトは、配列番号2のヌクレオチド配列における1000番目と1001番目のヌクレオチド残基(CCDC181遺伝子のイントロン2中に存在する標準ヌクレオチド配列cagcggca中の4および5番目のヌクレオチド)からなるCG部分と定義することもできる(5’側のcag部分および3’側のgca部分が変異していない場合)。当業者であれば、ゲノムDNAの変異が認められるヒト被験体について、ゲノムDNAの変異を考慮して、上記位置に存在するCpGサイトに相当するCpGサイトを適宜決定することができる。以下、本明細書において単に「CCDC181遺伝子のCpGサイト」と称する場合は、特に記載がない限り、上記CpGサイトを示す。
本発明において解析されるCFTR遺伝子のCpGサイトは、配列番号3のヌクレオチド配列における1001番目と1002番目のヌクレオチド残基(CFTR遺伝子のエクソン1上流に存在する標準ヌクレオチド配列ctccgggg中の4および5番目のヌクレオチド)からなるCG部分と定義することもできる(5’側のctc部分および3’側のggg部分が変異していない場合)。当業者であれば、ゲノムDNAの変異が認められるヒト被験体について、ゲノムDNAの変異を考慮して、上記位置に存在するCpGサイトに相当するCpGサイトを適宜決定することができる。以下、本明細書において単に「CFTR遺伝子のCpGサイト」と称する場合は、特に記載がない限り、上記CpGサイトを示す。
(乳癌細胞存在率の推定方法)
本発明は、乳癌細胞存在率の推定方法を提供する。
本発明は、乳癌細胞存在率の推定方法を提供する。
本発明の推定方法は、以下(1)および(2)を含む:
(1)ヒト被験体由来サンプルにおいて、以下:
(a)SIM1遺伝子のCpGサイト中のシトシン残基;
(b)CCDC181遺伝子のCpGサイト中のシトシン残基;
(c)CFTR遺伝子のCpGサイト中のシトシン残基;または
(d)これらの組合せ
のメチル化率を解析すること;および
(2)(1)で解析されたメチル化率に基づいて、ヒト被験体由来サンプル中の乳癌細胞存在率を推定すること。
(1)ヒト被験体由来サンプルにおいて、以下:
(a)SIM1遺伝子のCpGサイト中のシトシン残基;
(b)CCDC181遺伝子のCpGサイト中のシトシン残基;
(c)CFTR遺伝子のCpGサイト中のシトシン残基;または
(d)これらの組合せ
のメチル化率を解析すること;および
(2)(1)で解析されたメチル化率に基づいて、ヒト被験体由来サンプル中の乳癌細胞存在率を推定すること。
工程(1)において、サンプルとしては、血液、体液、組織、および細胞からなる群より選択された1または2以上のサンプルを使用することができるが、上述した癌に対応する組織サンプルを好適に使用できる。本発明では、このような癌組織サンプルに含まれるゲノムDNAにおいて、特定のCpGサイトが解析される。ゲノムDNAは、癌組織サンプルから、任意の方法により適宜抽出することができる。癌組織サンプルとしては、例えば、針生検等の生検により採取されたものを用いることができる。癌組織サンプルは、予備処理に付されてもよい。このような予備処理としては、例えば、抽出、細胞固定、組織固定、組織薄片化、細胞解離処理、加熱、凍結、冷蔵、液状化が挙げられる。癌組織サンプルは、通常、癌細胞と正常細胞(非癌性細胞)を含む細胞集団のサンプルである。
本発明において解析されるメチル化は、シトシン残基の5位のメチル化である。
メチル化率の解析は、当該分野において公知である任意の方法により行うことができる。例えば、解析は、亜硫酸水素塩、プライマー、核酸プローブ、制限酵素、抗メチル化シトシン抗体、およびナノポアからなる群より選ばれる1または2以上の解析手段を用いて行うことができる。
亜硫酸水素塩〔バイサルファイト(bisulfite)〕は、非メチル化シトシンをウラシルに変換する一方で、メチル化シトシンをウラシルに変換しない。よって、重亜硫酸水素塩は、このような性質のため、メチル化シトシンの解析では他の解析手段と組み合わせて汎用されている。
プライマーは、シークエンシング用プライマー、若しくは遺伝子増幅用プライマー(例、PCRプライマー)、またはその組み合わせである。プライマーは、目的のCpGサイトを解析できるように適宜設計することができる。例えば、プライマーは、目的のCpGサイトの下流領域(SIM1遺伝子については配列番号1のヌクレオチド配列における1002~2001番目のヌクレオチド残基からなる部分中の任意の領域、CCDC181遺伝子については配列番号2のヌクレオチド配列における1002~2001番目のヌクレオチド残基からなる部分中の任意の領域、CFTR遺伝子については配列番号3のヌクレオチド配列における1003~2001番目のヌクレオチド残基からなる部分中の任意の領域)にアニールするように、または目的のCpGサイトを含む遺伝子領域を増幅するため、目的のCpGサイトの上流領域(SIM1遺伝子については配列番号1のヌクレオチド配列における1~999番目のヌクレオチド残基からなる部分中の任意の領域、CCDC181遺伝子については配列番号2のヌクレオチド配列における1~999番目のヌクレオチド残基からなる部分中の任意の領域、CFTR遺伝子については配列番号1のヌクレオチド配列における1~1000番目のヌクレオチド残基からなる部分中の任意の領域)および下流領域(上記で下流領域として示した任意の領域)においてセンス鎖およびアンチセンス鎖がアニールするように設計することができる。
核酸プローブは、目的のCpGサイトを含む領域(SIM1遺伝子については配列番号1のヌクレオチド配列における1000番目と1001番目のヌクレオチド残基からなるCG部分を含む部分、CCDC181遺伝子については配列番号2のヌクレオチド配列における1000番目と1001番目のヌクレオチド残基からなるCG部分を含む部分、CFTR遺伝子については配列番号3のヌクレオチド配列における1001番目と1002番目のヌクレオチド残基からなるCG部分を含む部分を含む部分)またはその上流領域(上記で上流領域として示した任意の領域)若しくは下流領域(上記で下流領域として示した任意の領域)にハイブリダイズするように設計することができる。核酸プローブは、遊離の形態、または固相に固定された形態において用いることができる。固相としては、例えば、粒子(例、磁性粒子);アレイ、メンブレン(例、ニトロセルロース膜、ろ紙)、カラム等の支持体;およびプレート(例、マルチウェルプレート)、チューブ等の容器が挙げられる。固相の材料としては、例えば、ガラス、プラスチック、および金属が挙げられる。核酸プローブはまた、抗メチル化シトシン抗体を用いるメチル化シトシンの解析において詳述したような核酸プローブであってもよい。
制限酵素は、メチル化特異的または非特異的制限酵素であり、上述したような亜硫酸水素塩、プライマー、核酸プローブと適宜組み合わせて用いることができる。
具体的には、上述したような解析手段を用いる解析方法としては、例えば、DNAメチル化シーケンス法、バイサルファイトシーケンシング法、バイサルファイト・パイロシーケンシング法、メチル化特異的PCR法、制限酵素ランドマーク・ゲノム・スキャニング(RLGS)法、単一ヌクレオチド・プライマー伸長(SNuPE)法、CpGアイランド・マイクロアレイ法、メチライト法、COBRA法、質量分析(マスアレイ)法、メチル化特異的制限酵素の使用、高解像度融解解析(HRM)法、ナノポア解析法、ICONプローブ法、メチル化特異的MLPA法が挙げられる。これらの方法は、当該分野において周知である(例、特開2012-090555号公報、特開2014-036672号公報、特表2010-538638号公報、再表2009/136501号公報)。
解析はまた、抗メチル化シトシン抗体を用いて行うことができる。抗メチル化シトシン抗体を用いるメチル化シトシンの解析は、当該分野において周知である(例、WO2015/025862;WO2015/025863;WO2015/025864;WO2016/052368;特開2012-230019号公報;DNA Research 13,37-42(2006);Anal.Chem. 2012,84,7533-7538)。抗メチル化シトシン抗体は、上述したような1または2以上の解析手段と組み合わされて用いられてもよい。具体的には、このような解析手段を用いる方法としては、例えば、抗メチル化シトシン抗体、および異種核酸プローブ(例、ノーマルRNAプローブ、修飾RNAプローブ)を組み合わせて用いる方法(例、WO2015/025862);抗メチル化シトシン抗体、並びに固相プローブおよび捕捉プローブを組み合わせて用いる方法(例、WO2015/025863);抗メチル化シトシン抗体、並びに吸収剤ポリヌクレオチドおよび捕捉プローブを組み合わせて用いる方法(例、WO2015/025864);抗メチル化シトシン抗体、および修飾核酸塩基対合性異種核酸プローブを組み合わせて用いる方法(例、WO2016/052368)が挙げられる。抗メチル化シトシン抗体と組み合わせて用いられる核酸プローブは、本発明において解析される上記CpGサイトを含むDNA鎖ハイブリダイズして、当該DNA鎖と核酸プローブからなる二本鎖構造部分が形成されたときに、CpGサイト中のメチルシトシン残基が核酸プローブと不対部分を形成するように(換言すれば、相補的に結合しないように)、設計されてもよい(例、WO2015/025862)。したがって、核酸プローブは、不対部分に対応するヌクレオチド残基として、メチルシトシン残基と相補的に結合し得るグアニン残基以外のヌクレオチド残基(例、シトシン残基、チミン残基、アデニン残基、ウラシル残基)を有していてもよい。あるいは、核酸プローブは、このような不対部分が形成されないように設計されてもよい(例、WO2016/052368)。
抗メチル化シトシン抗体を用いる方法は、当該分野において公知である任意の免疫学的方法により行うことができる。具体的には、このような方法としては、例えば、酵素免疫測定法(EIA)(例、CLEIA、ELISA)、蛍光免疫測定法、化学発光免疫測定法、電気化学発光免疫測定法、凝集法、免疫染色、フローサイトメトリー法、バイオレイヤー干渉法、In Situ PLA法、化学増幅型ルミネッセンス・プロキシミティ・ホモジニアス・アッセイ、ラインブロット法、ウエスタンブロット法が挙げられる。
本発明において解析されるメチル化率は、癌細胞における上記CpG中のシトシン残基のメチル化の割合である。メチル化率の測定は、当該分野において周知である(例、特開2012-090555号公報、特開2014-036672号公報、特表2010-538638号公報、再表2009/136501号公報)。
本発明において解析されるメチル化率は、SIM1遺伝子、CCDC181遺伝子、およびCFTR遺伝子のCpGサイト中のシトシン残基のメチル化率の組合せであってもよい。上記(d)で示されるようなメチル化率の組合せは、これらのメチル化率の2以上(すなわち、2または3)からなる組合せであり、具体的には、SIM1遺伝子とCCDC181遺伝子の組合せ、SIM1遺伝子およびCFTR遺伝子の組合せ、CCDC181遺伝子およびCFTR遺伝子の組合せ、ならびに、SIM1遺伝子、CCDC181遺伝子、およびCFTR遺伝子の組合せにおけるCpGサイト中のシトシン残基のメチル化率である。組合せにおけるメチル化率は、例えば、組合せ中の最高メチル化率、最低メチル化率、および平均メチル化率のいずれを採用してもよいが、最高メチル化率を採用するのが好ましい。
工程(2)において、解析されたメチル化率に基づいて、ヒト被験体由来サンプル中の乳癌細胞存在率を推定することができる。
(乳癌に対する薬物療法の効果の予測方法)
本発明はまた、乳癌に対する薬物療法の効果の予測方法を提供する。
本発明はまた、乳癌に対する薬物療法の効果の予測方法を提供する。
乳癌に対する薬物療法としては、例えば、抗癌剤による治療が挙げられる。抗癌剤としては、例えば、微小管阻害剤、抗がん抗生物質、トポイソメラーゼ阻害剤、プラチナ製剤、アルキル化剤が挙げられる。抗癌剤はまた、分子標的薬であってもよい。このような分子標的薬としては、例えば、HER2阻害剤、EGFR阻害剤(例、ゲフィチニブ、ラパチニブ、エルロチニブ、セツキシマブ)、c-MET阻害剤(例、PHA-665752、SU11274、XL-880)、ALK阻害剤(例、WHI-P154、TAE684、PF-2341066)、PDGFR阻害剤(例、イマチニブ、デサチニブ、ヴァラチニブ)、c-KIT阻害剤(例、スニチニブ、マシチニブ、モテサニブ)が挙げられる。
乳癌に対する薬物療法は、乳癌のサブタイプに応じて適宜選択することができる。
HER2陽性型乳癌に対する薬物療法としては、例えば、HER2阻害剤による治療が挙げられる。HER2は、上皮成長因子(EGFR)と同じファミリーに属する受容体型チロシンキナーゼである。HER2は、細胞外ドメイン、膜貫通ドメインおよびチロシンキナーゼ活性を有する細胞内ドメインが一本鎖で連結した構造を有する(例、Coussensら、Science,1985,Vol.230,No.4730,pp.1132-1139、およびGenebankアクセッション番号:NP_001005862.1を参照)。HER2阻害剤としては、例えば、抗体(例、トラスツズマブ、ペルツヅマブ)、低分子有機化合物(例、ラパチニブ、ネラチニブ、アファチニブ等のHER2チロシンキナーゼ阻害剤)、薬物結合抗体(例、T-DM1)が挙げられる。
ルミナルA型乳癌に対する薬物療法としては、例えば、ホルモン療法または化学療法による治療が挙げられる。ホルモン療法としては、例えば、LH-RHアゴニスト製剤、抗エストロゲン薬、黄体ホルモン薬、アロマターゼ阻害薬を用いた療法が挙げられる。化学療法としては、例えば、トポイソメラーゼ阻害薬、微小管作用薬、アルキル化薬、代謝拮抗薬を用いた療法が挙げられる。
ルミナルB型乳癌に対する薬物療法としては、例えば、ホルモン療法または化学療法による治療が挙げられる。ホルモン療法としては、例えば、LH-RHアゴニスト製剤、抗エストロゲン薬、黄体ホルモン薬、アロマターゼ阻害薬を用いた療法が挙げられる。化学療法としては、例えば、トポイソメラーゼ阻害薬、微小管作用薬、アルキル化薬、代謝拮抗薬を用いた療法が挙げられる。
トリプルネガティブ型乳癌に対する薬物療法としては、例えば、化学療法による治療が挙げられる。化学療法としては、例えば、トポイソメラーゼ阻害薬、微小管作用薬、アルキル化薬、代謝拮抗薬を用いた療法が挙げられる。
癌に対する薬物療法においては、複数の抗癌剤を併用することができる。併用される複数の抗癌剤としては、例えば、上述した2以上の抗癌剤の組み合わせ、および上述した1以上の抗癌剤と他の抗癌剤との組み合わせが挙げられる。他の抗癌剤としては、例えば、タキサン系抗癌剤(例、パクリタキセル)等の微小管阻害剤、アントラサイクリン系(例、アドリアシン)、アロマターゼ阻害剤(例、レトロゾール)が挙げられる。
本発明の予測方法は、以下(1)~(4)を含む:
(1)ヒト被験体由来サンプルにおいて、乳癌マーカーの値を測定すること;
(2)ヒト被験体由来サンプルにおいて、以下:
(a)SIM1遺伝子のCpGサイト中のシトシン残基;
(b)CCDC181遺伝子のCpGサイト中のシトシン残基; (c)CFTR遺伝子のCpGサイト中のシトシン残基;または
(d)これらの組合せ
のメチル化率を解析すること;
(3)(1)で測定された乳癌マーカーの値を(2)で解析されたメチル化率で補正して、乳癌マーカーの補正値を計算すること;および
(4)(3)で補正された乳癌マーカーの補正値に基づいて、乳癌に対する薬物療法の効果を予測すること。
(1)ヒト被験体由来サンプルにおいて、乳癌マーカーの値を測定すること;
(2)ヒト被験体由来サンプルにおいて、以下:
(a)SIM1遺伝子のCpGサイト中のシトシン残基;
(b)CCDC181遺伝子のCpGサイト中のシトシン残基; (c)CFTR遺伝子のCpGサイト中のシトシン残基;または
(d)これらの組合せ
のメチル化率を解析すること;
(3)(1)で測定された乳癌マーカーの値を(2)で解析されたメチル化率で補正して、乳癌マーカーの補正値を計算すること;および
(4)(3)で補正された乳癌マーカーの補正値に基づいて、乳癌に対する薬物療法の効果を予測すること。
工程(1)における乳癌マーカーは、乳癌の指標となるマーカーであり、例えば、DNAメチル化率(例、HSD17B4のcg15896301:非特許文献3)が挙げられる。
工程(2)における画分マーカー遺伝子(SIM1遺伝子、CCDC181遺伝子、またはCFTR遺伝子)のCpGサイト、ならびにメチル化率の解析手段は、上述のとおりである。
工程(3)における乳癌マーカーの値の補正とは、ヒト被験体由来サンプル(乳癌細胞および非乳癌細胞を含むサンプル)において測定された乳癌マーカーの値に基づいて、より純粋に乳癌細胞に対応し得る乳癌マーカーの値を算出することをいう。したがって、乳癌マーカーの補正値は、例えば、下記式により求めることができる。
乳癌マーカーの補正値=(ヒト被験体由来サンプルにおいて測定された乳癌マーカーの値)/(ヒト被験体由来サンプル中の乳癌細胞存在率)
乳癌マーカーの補正値=(ヒト被験体由来サンプルにおいて測定された乳癌マーカーの値)/(ヒト被験体由来サンプル中の乳癌細胞存在率)
工程(4)は、乳癌マーカーの値に基づいて乳癌に対する薬物療法の効果を予測する手法において、乳癌マーカーの値の代わりに乳癌マーカーの補正値を用いることにより行うことができる。乳癌マーカーの補正値を用いることにより、薬物療法の効果の高精度な予測が期待される。
(乳癌の判定方法)
本発明はまた、乳癌の判定方法を提供する。
本発明はまた、乳癌の判定方法を提供する。
乳癌の判定とは、ヒト被験体が乳癌に罹患している可能性が高いかまたは低いかを評価することをいう。
本発明の判定方法は、以下(1)および(2)を含む:
(1)ヒト被験体由来サンプルにおいて、以下:
(a)SIM1遺伝子のCpGサイト中のシトシン残基;
(b)CCDC181遺伝子のCpGサイト中のシトシン残基;
(c)CFTR遺伝子のCpGサイト中のシトシン残基;または
(d)これらの組合せ
のメチル化率を解析すること;および
(2)(1)で解析されたメチル化率に基づいて、乳癌の罹患可能性を評価すること。
(1)ヒト被験体由来サンプルにおいて、以下:
(a)SIM1遺伝子のCpGサイト中のシトシン残基;
(b)CCDC181遺伝子のCpGサイト中のシトシン残基;
(c)CFTR遺伝子のCpGサイト中のシトシン残基;または
(d)これらの組合せ
のメチル化率を解析すること;および
(2)(1)で解析されたメチル化率に基づいて、乳癌の罹患可能性を評価すること。
工程(1)におけるヒト被験体由来サンプル、画分マーカー遺伝子(SIM1遺伝子、CCDC181遺伝子、またはCFTR遺伝子)のCpGサイト、ならびにメチル化率の解析手段は、上述のとおりである。
工程(2)において、メチル化率が基準値以上のときにヒト被験体は乳癌に罹患している可能性があり、ならびに/あるいはメチル化率が基準値より少ないときにヒト被験体は乳癌に罹患している可能性が低いと評価することができる。このような基準値としては、乳癌患者および健常人を鑑別し得るように適宜設定されたカットオフ値を利用することができる。メチル化率のカットオフ値としては、例えば、10~30%の範囲内にある任意の百分率の値を採用することができるが、15~25%の範囲内にある任意の百分率の値を採用することが好ましく、20%がより好ましい。
(試薬およびキット)
本発明はまた、乳癌細胞存在率の推定用試薬を提供する。
本発明はまた、乳癌細胞存在率の推定用試薬を提供する。
本発明の推定用試薬は、以下:
(a)SIM1遺伝子のCpGサイト中のシトシン残基;
(b)CCDC181遺伝子のCpGサイト中のシトシン残基;
(c)CFTR遺伝子のCpGサイト中のシトシン残基;または
(d)これらの組合せ
のメチル化率の解析手段を含む。
(a)SIM1遺伝子のCpGサイト中のシトシン残基;
(b)CCDC181遺伝子のCpGサイト中のシトシン残基;
(c)CFTR遺伝子のCpGサイト中のシトシン残基;または
(d)これらの組合せ
のメチル化率の解析手段を含む。
本発明の推定用試薬における画分マーカー遺伝子(SIM1遺伝子、CCDC181遺伝子、またはCFTR遺伝子)のCpGサイト、ならびにメチル化率の解析手段は、上述のとおりである。
本発明の推定用試薬は、メチル化率の解析手段を用いてヒト被験体由来サンプルにおける画分マーカー遺伝子のメチル化率を解析し、解析されたメチル化率に基づいて、ヒト被験体由来サンプル中の乳癌細胞存在率を推定するために用いることができる。
本発明はまた、乳癌の判定用試薬を提供する。
本発明の判定用試薬は、以下:
(a)SIM1遺伝子のCpGサイト中のシトシン残基;
(b)CCDC181遺伝子のCpGサイト中のシトシン残基;
(c)CFTR遺伝子のCpGサイト中のシトシン残基;または
(d)これらの組合せ
のメチル化率の解析手段を含む。
(a)SIM1遺伝子のCpGサイト中のシトシン残基;
(b)CCDC181遺伝子のCpGサイト中のシトシン残基;
(c)CFTR遺伝子のCpGサイト中のシトシン残基;または
(d)これらの組合せ
のメチル化率の解析手段を含む。
本発明の判定用試薬における画分マーカー遺伝子(SIM1遺伝子、CCDC181遺伝子、またはCFTR遺伝子)のCpGサイト、ならびにメチル化率の解析手段は、上述のとおりである。
本発明の判定用試薬は、メチル化率の解析手段を用いてヒト被験体由来サンプルにおける画分マーカー遺伝子のメチル化率を解析し、解析されたメチル化率に基づいて、ヒト被験体の乳癌を判定するために用いることができる。
本発明はまた、キットを提供する。
本発明のキットは、以下(1)および(2)を含む:
(1)以下:
(a)SIM1遺伝子のCpGサイト中のシトシン残基;
(b)CCDC181遺伝子のCpGサイト中のシトシン残基;
(c)CFTR遺伝子のCpGサイト中のシトシン残基;または
(d)これらの組合せ
のメチル化率解析手段;および
(2)乳癌マーカーの測定手段。
(1)以下:
(a)SIM1遺伝子のCpGサイト中のシトシン残基;
(b)CCDC181遺伝子のCpGサイト中のシトシン残基;
(c)CFTR遺伝子のCpGサイト中のシトシン残基;または
(d)これらの組合せ
のメチル化率解析手段;および
(2)乳癌マーカーの測定手段。
本発明の乳癌の判定用試薬における画分マーカー遺伝子(SIM1遺伝子、CCDC181遺伝子、またはCFTR遺伝子)のCpGサイト、メチル化率の解析手段、ならびに乳癌マーカーの測定手段は、上述のとおりである。
本発明のキットは、種々の用途に用いることができるが、例えば、乳癌に対する薬物療法の効果の予測するために用いることができる。
以下、本発明を実施例により詳細に説明するが、本発明は、これらの実施例に限定されるものではない。
1.サンプル
1-1.生体由来サンプル
乳癌患者由来サンプルとして、195検体の乳癌組織サンプルを195人の乳癌患者から取得した。各患者の乳癌組織サンプルは、乳癌と診断された患者から針生検により収集した。サンプルを、PAX遺伝子組織システム(Qiagen,ヒルデン,ドイツ)を用いて固定し、低融点パラフィン中に埋包した。10枚の10μm薄切切片を作成し、DNAを抽出した。同時に、サンプルの顕微鏡検査を実施し、癌細胞画分を決定した。上記の195検体のうち、HER2陽性型乳癌患者由来検体61検体については、トラスツズマブに対する治療応答を決定し、サンプル中に癌細胞が残存しない状態を、病理学的完全奏効(pCR)、それ以外を非完全奏功(Non-pCR)と定義した。さらに、61個のサンプルのうち10個のサンプルを、レーザー捕捉マイクロダイセクション(LCM)により癌細胞および非癌細胞に分離した。
1-1.生体由来サンプル
乳癌患者由来サンプルとして、195検体の乳癌組織サンプルを195人の乳癌患者から取得した。各患者の乳癌組織サンプルは、乳癌と診断された患者から針生検により収集した。サンプルを、PAX遺伝子組織システム(Qiagen,ヒルデン,ドイツ)を用いて固定し、低融点パラフィン中に埋包した。10枚の10μm薄切切片を作成し、DNAを抽出した。同時に、サンプルの顕微鏡検査を実施し、癌細胞画分を決定した。上記の195検体のうち、HER2陽性型乳癌患者由来検体61検体については、トラスツズマブに対する治療応答を決定し、サンプル中に癌細胞が残存しない状態を、病理学的完全奏効(pCR)、それ以外を非完全奏功(Non-pCR)と定義した。さらに、61個のサンプルのうち10個のサンプルを、レーザー捕捉マイクロダイセクション(LCM)により癌細胞および非癌細胞に分離した。
1-2.培養細胞株サンプル
培養乳癌細胞株サンプルとして、全体で20個の乳癌細胞株(BT-474、SK-BR-3、MDA-MB-453、HCC38、MDA-MB-231、T-47D、Hs 578T、MCF7、UACC-3199、ZR-75-1、BT-20、MDA-MB-436、HCC1937、MDA-MB-468、HCC1428、BT-549、AU565、HCC1395、MDA-MB-157、およびHCC1954)を、アメリカン・タイプ・カルチャー・コレクションから購入した(ロックビル,MD)。
培養通常細胞株サンプルとして、ヒト乳房上皮細胞(HMECs)を、Cambrex(イーストラザフォード,NL)から購入した。
培養乳癌細胞株サンプルとして、全体で20個の乳癌細胞株(BT-474、SK-BR-3、MDA-MB-453、HCC38、MDA-MB-231、T-47D、Hs 578T、MCF7、UACC-3199、ZR-75-1、BT-20、MDA-MB-436、HCC1937、MDA-MB-468、HCC1428、BT-549、AU565、HCC1395、MDA-MB-157、およびHCC1954)を、アメリカン・タイプ・カルチャー・コレクションから購入した(ロックビル,MD)。
培養通常細胞株サンプルとして、ヒト乳房上皮細胞(HMECs)を、Cambrex(イーストラザフォード,NL)から購入した。
2.画分マーカーのDNAメチル化レベルの測定
2-1.DNAメチル化レベルの測定方法
ゲノム特異的DNAメチル化レベルは、バイサルファイト・パイロシーケンシングにより解析した。バイサルファイト・パイロシーケンシングは、具体的には、以下の手順により行った。ゲノムDNAをBamHIで消化し、1μgのBamHI消化ゲノムDNAを、既報(Yamashita S, Takahashi S, McDonell N, Watanabe N, Niwa T, Hosoya K et al. Methylation silencing of transforming growth factor-beta receptor type II in rat prostate cancers. Cancer research. 2008;68(7):2112-21)のとおりバイサルファイト修飾に供した。バイサルファイト修飾DNAを40μlのTE緩衝液中に懸濁し、1μlのアリコートを、既報(Yoshida T, Yamashita S, Takamura-Enya T, Niwa T, Ando T, Enomoto S et al. Alu and Satalpha hypomethylation in Helicobacter pylori-infected gastric mucosae. International journal of cancer. 2011;128(1):33-9)のとおりバイサルファイトシーケンシングに供した。標的ゲノム領域を、ビオチニル化プライマーにより増幅した。ビオチン標識PCR産物を、0.2μMパイロシーケンシング・プライマーに対してアニールさせ、バイロシーケンシングを、PSQ 96パイロシーケンシング・システム(QIAGEN,バレンシア,CA,USA)を用いて行った。メチル化レベルは、PSQアッセイ・デザイン・ソフトウェア(QIAGEN)を用いて取得した。
2-1.DNAメチル化レベルの測定方法
ゲノム特異的DNAメチル化レベルは、バイサルファイト・パイロシーケンシングにより解析した。バイサルファイト・パイロシーケンシングは、具体的には、以下の手順により行った。ゲノムDNAをBamHIで消化し、1μgのBamHI消化ゲノムDNAを、既報(Yamashita S, Takahashi S, McDonell N, Watanabe N, Niwa T, Hosoya K et al. Methylation silencing of transforming growth factor-beta receptor type II in rat prostate cancers. Cancer research. 2008;68(7):2112-21)のとおりバイサルファイト修飾に供した。バイサルファイト修飾DNAを40μlのTE緩衝液中に懸濁し、1μlのアリコートを、既報(Yoshida T, Yamashita S, Takamura-Enya T, Niwa T, Ando T, Enomoto S et al. Alu and Satalpha hypomethylation in Helicobacter pylori-infected gastric mucosae. International journal of cancer. 2011;128(1):33-9)のとおりバイサルファイトシーケンシングに供した。標的ゲノム領域を、ビオチニル化プライマーにより増幅した。ビオチン標識PCR産物を、0.2μMパイロシーケンシング・プライマーに対してアニールさせ、バイロシーケンシングを、PSQ 96パイロシーケンシング・システム(QIAGEN,バレンシア,CA,USA)を用いて行った。メチル化レベルは、PSQアッセイ・デザイン・ソフトウェア(QIAGEN)を用いて取得した。
2-2.画分マーカーのメチル化レベルの解析
2-2-1.画分マーカーのスクリーニング
常染色体および性染色体中の477,344のCpGサイトより、1-2で調製した培養通常細胞株(非癌細胞)でメチル化率が低く、培養乳癌細胞株でメチル化率の高いCpGサイトを探索し、3つのCpGサイトを画分マーカーとして見出した。各CpGサイトの詳細を表1に示す。
2-2-1.画分マーカーのスクリーニング
常染色体および性染色体中の477,344のCpGサイトより、1-2で調製した培養通常細胞株(非癌細胞)でメチル化率が低く、培養乳癌細胞株でメチル化率の高いCpGサイトを探索し、3つのCpGサイトを画分マーカーとして見出した。各CpGサイトの詳細を表1に示す。
2-2-2.画分マーカーによる乳癌評価と病理による癌細胞含有率との相関
画分マーカーの乳癌評価の正確性を判定するために、HER2陽性型乳癌サンプル51検体(CFTRについては33検体)について、各画分マーカー(SIM1、CCDC181、およびCFTR)、およびこれらの組み合わせにより評価される癌細胞画分と、顕微鏡検査により評価された癌細胞含有率との間の相関を判定した。画分マーカーを組み合わせる場合は、3つのマーカーの最高メチル化率を採用した。結果を図1~3に示す。画分マーカーにより評価された癌細胞画分および病理による癌細胞含有率の間で有意な相関を確認した(R=0.38~0.77)。したがって、これらの画分マーカーは、乳癌細胞画分を評価することができるマーカーであると考えられた。
画分マーカーの乳癌評価の正確性を判定するために、HER2陽性型乳癌サンプル51検体(CFTRについては33検体)について、各画分マーカー(SIM1、CCDC181、およびCFTR)、およびこれらの組み合わせにより評価される癌細胞画分と、顕微鏡検査により評価された癌細胞含有率との間の相関を判定した。画分マーカーを組み合わせる場合は、3つのマーカーの最高メチル化率を採用した。結果を図1~3に示す。画分マーカーにより評価された癌細胞画分および病理による癌細胞含有率の間で有意な相関を確認した(R=0.38~0.77)。したがって、これらの画分マーカーは、乳癌細胞画分を評価することができるマーカーであると考えられた。
3.乳癌細胞画分によるHSD17B4メチル化レベルの補正
3-1.乳癌マーカーの値の測定
上記1-1でpCRとNon-pCRを判定したHER2陽性型乳癌サンプル61検体について、HSD17B4遺伝子のメチル化レベルを測定した。HSD17B4メチル化レベルは、上記2-1にしたがってバイサルファイト・パイロシーケンシングにより測定した(非特許文献3参照)。
3-1.乳癌マーカーの値の測定
上記1-1でpCRとNon-pCRを判定したHER2陽性型乳癌サンプル61検体について、HSD17B4遺伝子のメチル化レベルを測定した。HSD17B4メチル化レベルは、上記2-1にしたがってバイサルファイト・パイロシーケンシングにより測定した(非特許文献3参照)。
3-2.乳癌マーカーの値の補正
上記3-1で測定したHSD17B4のメチル化レベルを、画分マーカーにより、または顕微鏡検査により評価された乳癌細胞画分により、以下のとおり補正した:[補正HSD17B4メチル化レベル=100×(HSD17B4メチル化レベル)/(癌細胞画分)]。
上記3-1で測定したHSD17B4のメチル化レベルを、画分マーカーにより、または顕微鏡検査により評価された乳癌細胞画分により、以下のとおり補正した:[補正HSD17B4メチル化レベル=100×(HSD17B4メチル化レベル)/(癌細胞画分)]。
3-3.統計解析
相関解析は、ピアソンの積率相関係数を用いて行った。トラスツズマブ応答者(pCR)と非応答者(Non-pCR)との間のHSD17B4の補正メチル化レベルにおける差異を、マン-ホイットニーU検定により評価した。全ての解析は、PASW統計バージョン18.0(SPSS Japan Inc.,東京,日本)を用いて行い、両側p値<0.05を、統計学的に有意とみなした。
相関解析は、ピアソンの積率相関係数を用いて行った。トラスツズマブ応答者(pCR)と非応答者(Non-pCR)との間のHSD17B4の補正メチル化レベルにおける差異を、マン-ホイットニーU検定により評価した。全ての解析は、PASW統計バージョン18.0(SPSS Japan Inc.,東京,日本)を用いて行い、両側p値<0.05を、統計学的に有意とみなした。
3-4.HSD17B4メチル化レベルの補正に対する癌細胞画分マーカーの応用
癌細胞画分を評価することにより癌細胞がHSD17B4メチル化レベルをどの程度補正できるか評価した。メチル化の生データに基づくと、pCRおよびNon-pCR試料間において、HSD17B4メチル化レベルにおいて如何なる有意な差異も認められなかった(p=0.245)(図4)。対照的に、補正後では、メチル化レベルは、Non-pCR試料よりもpCR試料において有意により高かった(顕微鏡検査:p=0.0001;画分マーカー:p=0.0004)。pCRを予測する感度および特異性(表2)に関しては、補正前はそれぞれ13.6%および94.9%であった。DNAメチル化マーカーによる補正後の感度および特異性(59.1%および84.6%)は、顕微鏡検査により補正されたもの(59.1%および87.2%)と同等であった。表2において、HSD17B4メチル化レベルを既報(前述のFujii S et al.)に基づくカットオフ値(50%)を用いて高レベルおよび低レベルに分類した。pCRは、病理学的完全奏効を示す。
癌細胞画分を評価することにより癌細胞がHSD17B4メチル化レベルをどの程度補正できるか評価した。メチル化の生データに基づくと、pCRおよびNon-pCR試料間において、HSD17B4メチル化レベルにおいて如何なる有意な差異も認められなかった(p=0.245)(図4)。対照的に、補正後では、メチル化レベルは、Non-pCR試料よりもpCR試料において有意により高かった(顕微鏡検査:p=0.0001;画分マーカー:p=0.0004)。pCRを予測する感度および特異性(表2)に関しては、補正前はそれぞれ13.6%および94.9%であった。DNAメチル化マーカーによる補正後の感度および特異性(59.1%および84.6%)は、顕微鏡検査により補正されたもの(59.1%および87.2%)と同等であった。表2において、HSD17B4メチル化レベルを既報(前述のFujii S et al.)に基づくカットオフ値(50%)を用いて高レベルおよび低レベルに分類した。pCRは、病理学的完全奏効を示す。

4.各サブタイプの乳癌への適用性の評価
1-1でpCR/Non-pCR判定を行った61検体を除く、134検体の乳癌サンプル(HER2陽性型、ルミナルA型、ルミナルB型、およびトリプルネガティブ型)について、非癌細胞も混入した状態で画分マーカー(SIM1、CCDC181、およびCFTR)メチル化率を測定した。メチル化率20%をカットオフとして、3つのマーカーのいずれかがカットオフ以上のメチル化率を示した検体を「陽性」と判定した。結果を表3に示す。表3中、HER2はHER2陽性型、TNBCはトリプルネガティブ型を表す。
1-1でpCR/Non-pCR判定を行った61検体を除く、134検体の乳癌サンプル(HER2陽性型、ルミナルA型、ルミナルB型、およびトリプルネガティブ型)について、非癌細胞も混入した状態で画分マーカー(SIM1、CCDC181、およびCFTR)メチル化率を測定した。メチル化率20%をカットオフとして、3つのマーカーのいずれかがカットオフ以上のメチル化率を示した検体を「陽性」と判定した。結果を表3に示す。表3中、HER2はHER2陽性型、TNBCはトリプルネガティブ型を表す。
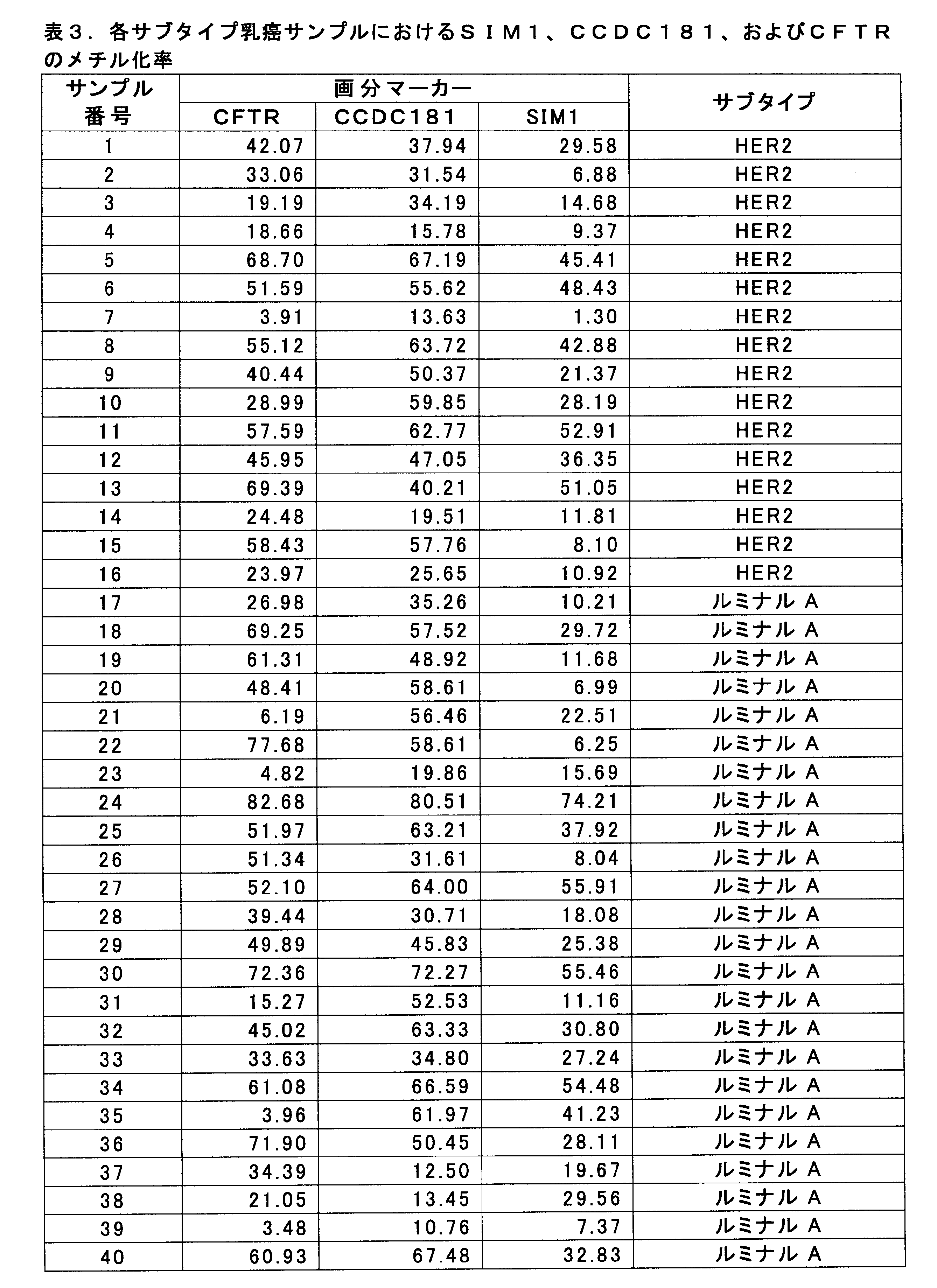
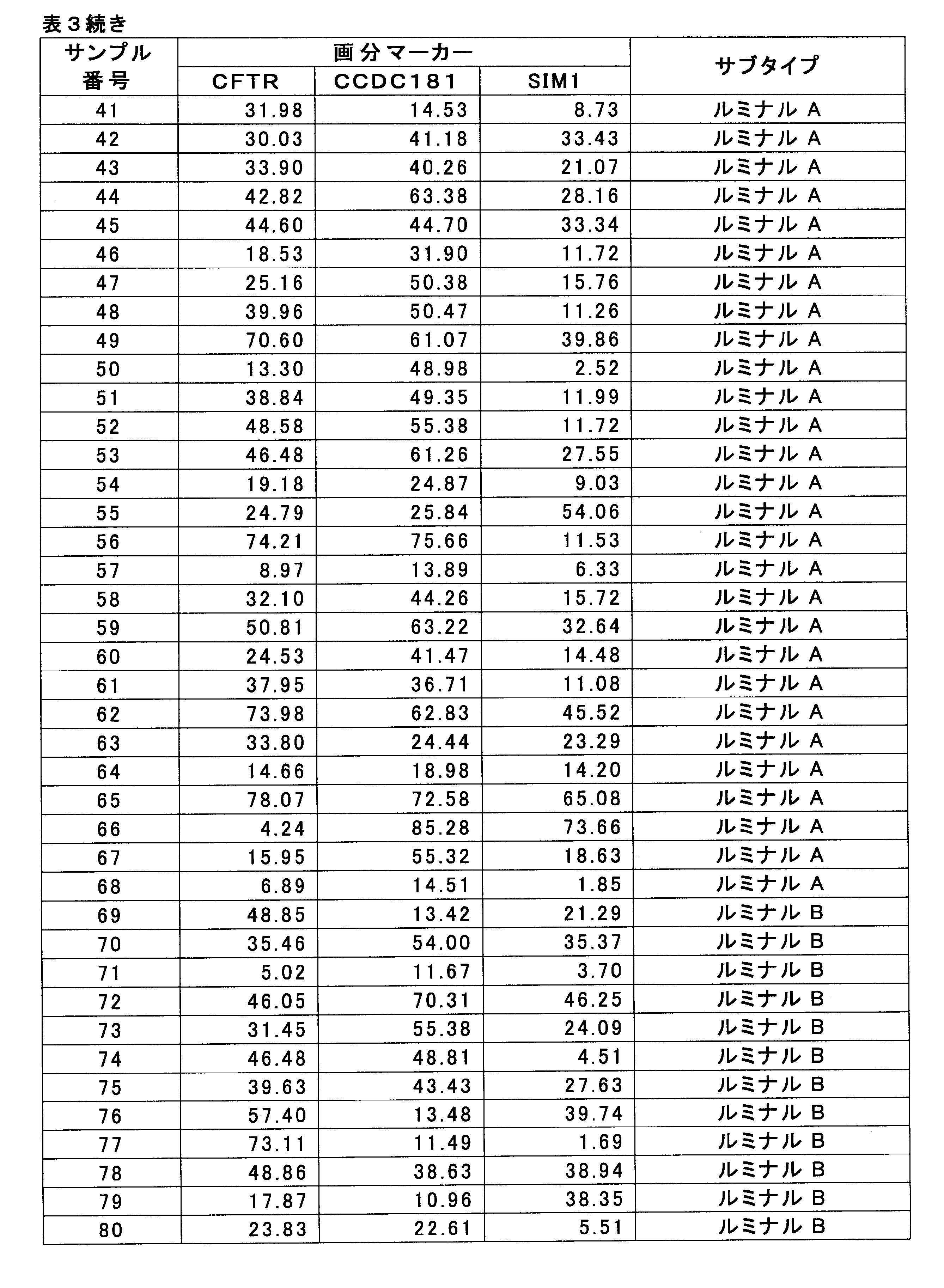

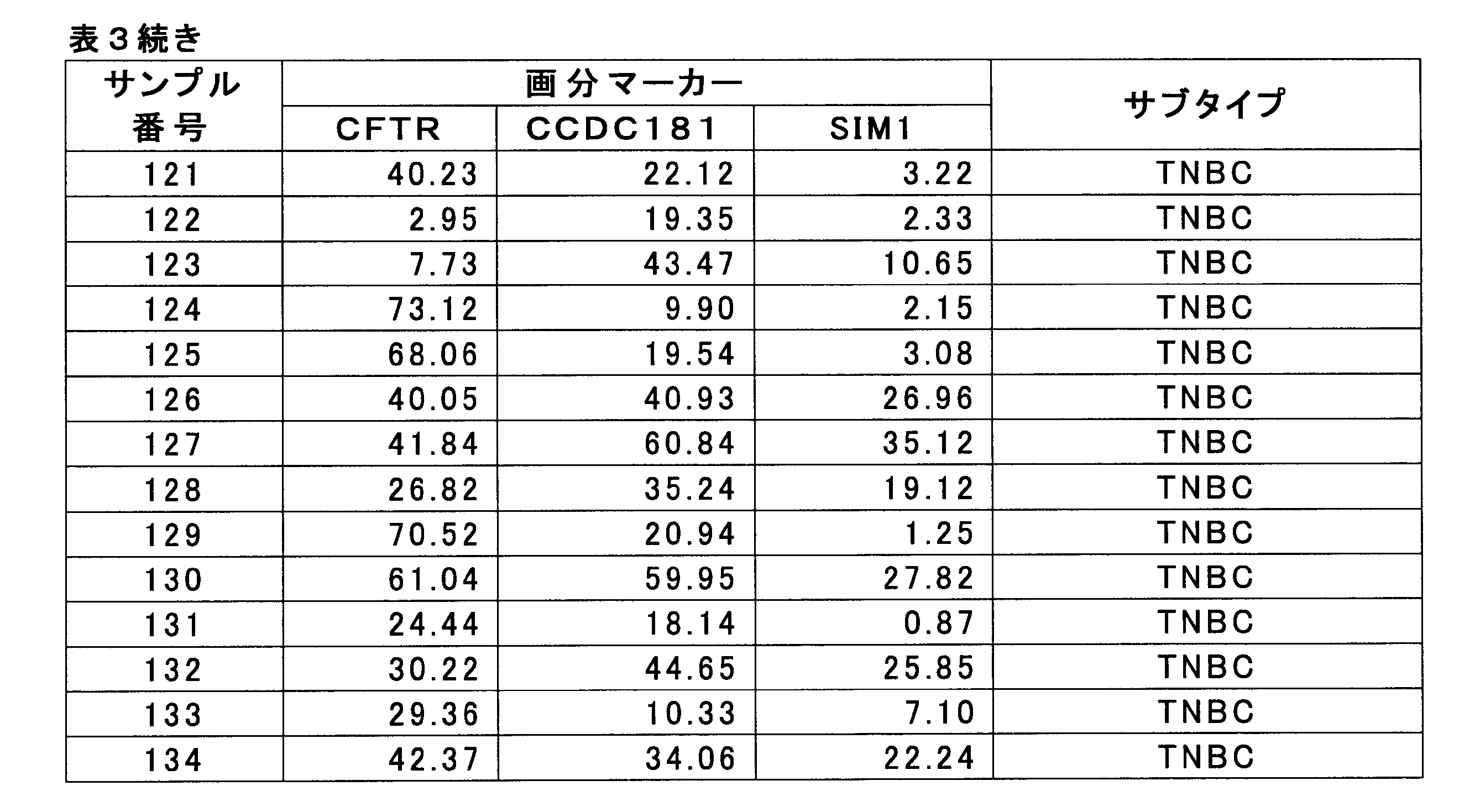
上記の結果において、各乳癌サブタイプにおいて、SIM1、CCDC181、CFTR、およびこれらの組み合わせについてメチル化率(組み合わせの場合は最高メチル化率)が20%を超えるものを陽性と判定した場合、各画分マーカーおよび乳癌判定率(メチル化陽性の乳癌患者の割合)は下記表4に示すとおりとなった。

いずれの画分マーカー、いずれの組み合わせにおいても高い乳癌判定率(メチル化陽性の乳癌患者の割合)を示した。特に2種類以上の画分マーカーを組み合わせることで、高い確率で乳癌を判定し得ることが示唆された。
配列番号1は、SIM1遺伝子のメチル化レベル測定CpGサイト(1000および1001番目のヌクレオチド残基)前後1000bpのゲノム領域の塩基配列を示す。
配列番号2は、CCDC181遺伝子のメチル化レベル測定CpGサイト(1000および1001番目のヌクレオチド残基)前後1000bpのゲノム領域の塩基配列を示す。
配列番号3は、CFTR遺伝子のメチル化レベル測定CpGサイト(1001および1002番目のヌクレオチド残基)前後1000bpのゲノム領域の塩基配列を示す。
配列番号4~6はそれぞれ、SIM1遺伝子のCpGサイトのメチル化レベル測定用フォワードプライマー、リバースプライマー、およびシークエンシングプライマーの塩基配列を示す。
配列番号7~9はそれぞれ、CCDC181遺伝子のCpGサイトのメチル化レベル測定用フォワードプライマー、リバースプライマー、およびシークエンシングプライマーの塩基配列を示す。
配列番号10~12はそれぞれ、CFTR遺伝子のCpGサイトのメチル化レベル測定用フォワードプライマー、リバースプライマー、およびシークエンシングプライマーの塩基配列を示す。
配列番号2は、CCDC181遺伝子のメチル化レベル測定CpGサイト(1000および1001番目のヌクレオチド残基)前後1000bpのゲノム領域の塩基配列を示す。
配列番号3は、CFTR遺伝子のメチル化レベル測定CpGサイト(1001および1002番目のヌクレオチド残基)前後1000bpのゲノム領域の塩基配列を示す。
配列番号4~6はそれぞれ、SIM1遺伝子のCpGサイトのメチル化レベル測定用フォワードプライマー、リバースプライマー、およびシークエンシングプライマーの塩基配列を示す。
配列番号7~9はそれぞれ、CCDC181遺伝子のCpGサイトのメチル化レベル測定用フォワードプライマー、リバースプライマー、およびシークエンシングプライマーの塩基配列を示す。
配列番号10~12はそれぞれ、CFTR遺伝子のCpGサイトのメチル化レベル測定用フォワードプライマー、リバースプライマー、およびシークエンシングプライマーの塩基配列を示す。
Claims (8)
- 以下(1)および(2)を含む、乳癌細胞存在率の推定方法:
(1)ヒト被験体由来サンプルにおいて、以下:
(a)配列番号1のヌクレオチド配列における1000番目と1001番目のヌクレオチドからなるCG部分中のシトシン残基;
(b)配列番号2のヌクレオチド配列における1000番目と1001番目のヌクレオチドからなるCG部分中のシトシン残基;
(c)配列番号3のヌクレオチド配列における1001番目と1002番目のヌクレオチドからなるCG部分中のシトシン残基;または
(d)これらの組合せ
のメチル化率を解析すること;および
(2)(1)で解析されたメチル化率に基づいて、ヒト被験体由来サンプル中の乳癌細胞存在率を推定すること。 - 解析が、亜硫酸水素塩、1以上のプライマー、1以上の核酸プローブ、制限酵素、抗メチル化シトシン抗体、およびナノポアからなる群より選ばれる1または2以上の手段を用いて行われる、請求項1に記載の方法。
- 解析が、バイサルファイトシーケンシング法、バイサルファイト・パイロシーケンシング法、メチル化特異的PCR法、制限酵素ランドマーク・ゲノム・スキャニング(RLGS)法、単一ヌクレオチド・プライマー伸長(SNuPE)法、CpGアイランド・マイクロアレイ法、メチライト法、COBRA法、質量分析(マスアレイ)法、メチル化特異的制限酵素の使用、高解像度融解解析(HRM)法、ナノポア解析法、ICONプローブ法、メチル化特異的MLPA法、またはイムノアッセイにより行われる、請求項1または2に記載の方法。
- 以下(1)~(4)を含む、乳癌に対する薬物療法の効果の予測方法:
(1)ヒト被験体由来サンプルにおいて、乳癌マーカーの値を測定すること;
(2)ヒト被験体由来サンプルにおいて、以下:
(a)配列番号1のヌクレオチド配列における1000番目と1001番目のヌクレオチドからなるCG部分中のシトシン残基;
(b)配列番号2のヌクレオチド配列における1000番目と1001番目のヌクレオチドからなるCG部分中のシトシン残基;
(c)配列番号3のヌクレオチド配列における1001番目と1002番目のヌクレオチドからなるCG部分中のシトシン残基;または
(d)これらの組合せ
のメチル化率を解析すること;
(3)(1)で測定された乳癌マーカーの値を(2)で解析されたメチル化率で補正して、乳癌マーカーの補正値を計算すること;および
(4)(3)で補正された乳癌マーカーの補正値に基づいて、乳癌に対する薬物療法の効果を予測すること。 - 以下(1)および(2)を含む、乳癌の判定方法:
(1)ヒト被験体由来サンプルにおいて、以下:
(a)配列番号1のヌクレオチド配列における1000番目と1001番目のヌクレオチドからなるCG部分中のシトシン残基;
(b)配列番号2のヌクレオチド配列における1000番目と1001番目のヌクレオチドからなるCG部分中のシトシン残基;
(c)配列番号3のヌクレオチド配列における1001番目と1002番目のヌクレオチドからなるCG部分中のシトシン残基;または
(d)これらの組合せ
のメチル化率を解析すること;および
(2)(1)で解析されたメチル化率に基づいて、乳癌の罹患可能性を評価すること。 - 以下:
(a)配列番号1のヌクレオチド配列における1000番目と1001番目のヌクレオチドからなるCG部分中のシトシン残基;
(b)配列番号2のヌクレオチド配列における1000番目と1001番目のヌクレオチドからなるCG部分中のシトシン残基;
(c)配列番号3のヌクレオチド配列における1001番目と1002番目のヌクレオチドからなるCG部分中のシトシン残基;または
(d)これらの組合せ
のメチル化率の解析手段を含む、乳癌細胞存在率の推定用試薬。 - 以下:
(a)配列番号1のヌクレオチド配列における1000番目と1001番目のヌクレオチドからなるCG部分中のシトシン残基;
(b)配列番号2のヌクレオチド配列における1000番目と1001番目のヌクレオチドからなるCG部分中のシトシン残基;
(c)配列番号3のヌクレオチド配列における1001番目と1002番目のヌクレオチドからなるCG部分中のシトシン残基;または
(d)これらの組合せ
のメチル化率の解析手段を含む、乳癌の判定用試薬。 - 以下(1)および(2)を含むキット:
(1)以下:
(a)配列番号1のヌクレオチド配列における1000番目と1001番目のヌクレオチドからなるCG部分中のシトシン残基;
(b)配列番号2のヌクレオチド配列における1000番目と1001番目のヌクレオチドからなるCG部分中のシトシン残基;
(c)配列番号3のヌクレオチド配列における1001番目と1002番目のヌクレオチドからなるCG部分中のシトシン残基;または
(d)これらの組合せ
のメチル化率解析手段;および
(2)乳癌マーカーの測定手段。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201980059667.XA CN112673114A (zh) | 2018-09-13 | 2019-09-11 | 乳腺癌细胞存在率的推定方法 |
| JP2020546062A JP7398712B2 (ja) | 2018-09-13 | 2019-09-11 | 乳癌細胞存在率の推定方法 |
| US17/275,871 US20210285057A1 (en) | 2018-09-13 | 2019-09-11 | Method for estimating breast cancer cell existence ratio |
| EP19860288.0A EP3851543A4 (en) | 2018-09-13 | 2019-09-11 | METHOD FOR ESTIMATING THE ABUNDANCE OF BREAST CANCER CELLS |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2018-171850 | 2018-09-13 | ||
| JP2018171850 | 2018-09-13 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2020054782A1 true WO2020054782A1 (ja) | 2020-03-19 |
Family
ID=69778441
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2019/035774 WO2020054782A1 (ja) | 2018-09-13 | 2019-09-11 | 乳癌細胞存在率の推定方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US20210285057A1 (ja) |
| EP (1) | EP3851543A4 (ja) |
| JP (1) | JP7398712B2 (ja) |
| CN (1) | CN112673114A (ja) |
| WO (1) | WO2020054782A1 (ja) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN114984027A (zh) * | 2022-04-24 | 2022-09-02 | 浙江大学智能创新药物研究院 | 柴胡皂苷a在制备减轻egfr抑制剂毒副作用的药物中的应用 |
| CN117538544A (zh) * | 2023-11-22 | 2024-02-09 | 湛江中心人民医院 | 检测基因Ccdc181表达量的试剂在制备精子异常诊断产品中的应用 |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009136501A1 (ja) | 2008-05-07 | 2009-11-12 | 北海道公立大学法人札幌医科大学 | 癌の検出方法および検出用キット、ならびに癌治療剤 |
| JP2012090555A (ja) | 2010-10-26 | 2012-05-17 | Sapporo Medical Univ | Rasgrf1の定量的メチル化測定を用いた発癌リスク予測 |
| JP2012230019A (ja) | 2011-04-27 | 2012-11-22 | National Institute Of Advanced Industrial & Technology | メチル化核酸検出法 |
| JP2014036672A (ja) | 2007-02-21 | 2014-02-27 | Oslo Universitetssykehus Hf | 新しい癌マーカー |
| WO2015025862A1 (ja) | 2013-08-21 | 2015-02-26 | 富士レビオ株式会社 | 異種核酸プローブを用いた修飾核酸塩基の測定方法およびキット |
| WO2015025864A1 (ja) | 2013-08-21 | 2015-02-26 | 富士レビオ株式会社 | 吸収剤ポリヌクレオチドを用いた修飾核酸塩基の測定方法およびそのキット |
| WO2015025863A1 (ja) | 2013-08-21 | 2015-02-26 | 富士レビオ株式会社 | 固相プローブを用いた修飾核酸塩基の測定方法およびそのキット |
| WO2016052368A1 (ja) | 2014-09-29 | 2016-04-07 | 富士レビオ株式会社 | 修飾核酸塩基を含む標的核酸の測定方法およびキット |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR101142131B1 (ko) | 2009-11-05 | 2012-05-11 | (주)지노믹트리 | 장암 진단을 위한 장암 특이적 메틸화 마커 유전자의 메틸화 검출방법 |
| US20130296328A1 (en) | 2011-01-20 | 2013-11-07 | Université Libre de Bruxelles | Epigenetic portraits of human breast cancers |
| US20150011396A1 (en) * | 2012-07-09 | 2015-01-08 | Benjamin G. Schroeder | Methods for creating directional bisulfite-converted nucleic acid libraries for next generation sequencing |
| EP3224377B1 (en) | 2014-11-25 | 2020-01-01 | AIT Austrian Institute of Technology GmbH | Diagnosis of lung cancer |
| JP6876304B2 (ja) * | 2016-06-10 | 2021-05-26 | 国立研究開発法人国立がん研究センター | 癌に対する薬物療法の効果の予測方法 |
-
2019
- 2019-09-11 EP EP19860288.0A patent/EP3851543A4/en not_active Withdrawn
- 2019-09-11 JP JP2020546062A patent/JP7398712B2/ja active Active
- 2019-09-11 CN CN201980059667.XA patent/CN112673114A/zh active Pending
- 2019-09-11 US US17/275,871 patent/US20210285057A1/en active Pending
- 2019-09-11 WO PCT/JP2019/035774 patent/WO2020054782A1/ja active Application Filing
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2014036672A (ja) | 2007-02-21 | 2014-02-27 | Oslo Universitetssykehus Hf | 新しい癌マーカー |
| WO2009136501A1 (ja) | 2008-05-07 | 2009-11-12 | 北海道公立大学法人札幌医科大学 | 癌の検出方法および検出用キット、ならびに癌治療剤 |
| JP2012090555A (ja) | 2010-10-26 | 2012-05-17 | Sapporo Medical Univ | Rasgrf1の定量的メチル化測定を用いた発癌リスク予測 |
| JP2012230019A (ja) | 2011-04-27 | 2012-11-22 | National Institute Of Advanced Industrial & Technology | メチル化核酸検出法 |
| WO2015025862A1 (ja) | 2013-08-21 | 2015-02-26 | 富士レビオ株式会社 | 異種核酸プローブを用いた修飾核酸塩基の測定方法およびキット |
| WO2015025864A1 (ja) | 2013-08-21 | 2015-02-26 | 富士レビオ株式会社 | 吸収剤ポリヌクレオチドを用いた修飾核酸塩基の測定方法およびそのキット |
| WO2015025863A1 (ja) | 2013-08-21 | 2015-02-26 | 富士レビオ株式会社 | 固相プローブを用いた修飾核酸塩基の測定方法およびそのキット |
| WO2016052368A1 (ja) | 2014-09-29 | 2016-04-07 | 富士レビオ株式会社 | 修飾核酸塩基を含む標的核酸の測定方法およびキット |
Non-Patent Citations (13)
| Title |
|---|
| "Genebank", Database accession no. NP 001005862.1 |
| ANAL. CHEM., vol. 84, 2012, pages 7533 - 7538 |
| COUSSENS ET AL., SCIENCE, vol. 230, no. 4730, 1985, pages 1132 - 1139 |
| DEDEURWAERDER, S.: "DNA methylation profiling reveals a predominant immune component in breast cancers", EMBO MOL. MED., vol. 3, no. 12, December 2011 (2011-12-01), pages 726 - 741, XP055026119, DOI: 10.1002/emmm.201100801 * |
| DING, S. ET AL.: "Methylation profile of the promoter CpG island s of 14 ''drug-resistance'' genes in hepatocellular carcinoma", WORLD J GASTROENTEROLOGY, vol. 10, no. 23, 1 December 2004 (2004-12-01), pages 3433 - 3440, XP055694085 * |
| DNA RESEARCH, vol. 13, 2006, pages 37 - 42 |
| FUJII SYAMASHITA SYAMAGUCHI TTAKAHASHI MHOZUMI YUSHIJIMA T ET AL.: "Pathological complete response of HER2-positive breast cancer to trastuzumab and chemotherapy can be predicted by HSD17B4 methylation", ONCOTARGET, vol. 8, no. 12, 2017, pages 19039 - 48, XP055462043, DOI: 10.18632/oncotarget.15118 |
| MIYAMOTO, KAZUAKI ET AL.: "Identification of 20 genes aberrantly methylated in human breast cancers", INT. J. CANCER, vol. 116, no. 3, September 2005 (2005-09-01), pages 407 - 414, XP002711967, DOI: 10.1002/IJC.21054 * |
| See also references of EP3851543A4 |
| TAKAHASHI T ET AL., PLOS ONE, vol. 8, no. 12, 2013, pages e82302 |
| YAMASHITA STAKAHASHI SMCDONELL NWATANABE NNIWA THOSOYA K ET AL.: "Methylation silencing of transforming growth factor-beta receptor type II in rat prostate cancers", CANCER RESEARCH, vol. 68, no. 7, 2008, pages 2112 - 21, XP055080616, DOI: 10.1158/0008-5472.CAN-07-5282 |
| YOSHIDA TYAMASHITA STAKAMURA-ENYA TNIWA TANDO TENOMOTO S ET AL.: "Alu and Satalpha hypomethylation in Helicobacter pylori-infected gastric mucosae", INTERNATIONAL JOURNAL OF CANCER, vol. 128, no. 1, 2011, pages 33 - 9 |
| ZONG L ET AL., GASTRIC CANCER, vol. 19, no. 2, 2016, pages 361 - 9 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3851543A1 (en) | 2021-07-21 |
| EP3851543A4 (en) | 2023-01-04 |
| JPWO2020054782A1 (ja) | 2021-12-02 |
| CN112673114A (zh) | 2021-04-16 |
| US20210285057A1 (en) | 2021-09-16 |
| JP7398712B2 (ja) | 2023-12-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11143657B2 (en) | Topographic genotyping for determining the diagnosis, malignant potential, and biologic behavior of pancreatic cysts and related conditions | |
| Li et al. | EGFR mutations in lung adenocarcinomas: clinical testing experience and relationship to EGFR gene copy number and immunohistochemical expression | |
| Eberhard et al. | Biomarkers of response to epidermal growth factor receptor inhibitors in non–small-cell lung cancer working group: standardization for use in the clinical trial setting | |
| JP5422120B2 (ja) | 癌患者による上皮成長因子受容体阻害薬に対する臨床転帰の予測方法 | |
| Tomita et al. | Estrogen receptor α gene ESR1 amplification may predict endocrine therapy responsiveness in breast cancer patients | |
| JP2016047044A (ja) | Bcl−2ファミリー阻害剤耐性腫瘍及び癌を有する被験者を同定、分類及びモニターするための方法及び組成物 | |
| CA2765772A1 (en) | Biomarkers and methods for determining efficacy of anti-egfr antibodies in cancer therapy | |
| Roschewski et al. | Circulating tumor DNA in lymphoma: principles and future directions | |
| US20120277110A1 (en) | Parp and adjuvant cisplatin-based chemotherapy in non-small-cell lung cancer | |
| US20120004136A1 (en) | Msh2 and adjuvant cisplatin-based chemotherapy in non-small-cell lung cancer | |
| WO2020054782A1 (ja) | 乳癌細胞存在率の推定方法 | |
| Bergqvist et al. | Quantitative real-time PCR analysis and microarray-based RNA expression of HER2 in relation to outcome | |
| JP5858405B2 (ja) | 肺腺がんの予後予測方法、および、肺腺がんの検出キット | |
| JP2016140262A (ja) | 肝細胞がんの転移性再発リスクの予測方法 | |
| CN109642257B (zh) | 药物疗法对癌的效果的预测方法 | |
| KR102384992B1 (ko) | 대장암 환자의 연령 특이적 바이오마커 및 이의 용도 | |
| JP2012085556A (ja) | 乳がんの診断方法 | |
| JP2012085555A (ja) | 乳がん診断用マーカー | |
| JP2012085554A (ja) | 乳がんのサブタイプの判別方法 | |
| WO2011107664A1 (en) | Method for selecting patients for treatment with an egfr inhibitor | |
| Evans | MOLECULAR DIAGNOSIS | |
| JPWO2015105191A1 (ja) | リンパ節腫脹病変の評価方法 | |
| AU2011265464B8 (en) | Methods for prediction of clinical outcome to epidermal growth factor receptor inhibitors by cancer patients | |
| JPWO2013151026A1 (ja) | Pi3k阻害剤耐性がん細胞用診断マーカー及び診断方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 19860288 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2020546062 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2019860288 Country of ref document: EP Effective date: 20210413 |