WO2020034601A1 - 化合物及其制备和应用 - Google Patents
化合物及其制备和应用 Download PDFInfo
- Publication number
- WO2020034601A1 WO2020034601A1 PCT/CN2019/073473 CN2019073473W WO2020034601A1 WO 2020034601 A1 WO2020034601 A1 WO 2020034601A1 CN 2019073473 W CN2019073473 W CN 2019073473W WO 2020034601 A1 WO2020034601 A1 WO 2020034601A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- group
- compound
- ring
- atom
- Prior art date
Links
- 150000001875 compounds Chemical class 0.000 title claims abstract description 271
- 238000002360 preparation method Methods 0.000 title claims abstract description 24
- 238000006243 chemical reaction Methods 0.000 claims abstract description 285
- 239000002360 explosive Substances 0.000 claims abstract description 59
- 238000000034 method Methods 0.000 claims abstract description 44
- 239000002243 precursor Substances 0.000 claims abstract description 42
- -1 nitroformyl Chemical group 0.000 claims description 190
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 claims description 158
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 claims description 147
- 229910017604 nitric acid Inorganic materials 0.000 claims description 126
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 122
- 239000002904 solvent Substances 0.000 claims description 114
- 125000003367 polycyclic group Chemical group 0.000 claims description 111
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 claims description 93
- 229910052757 nitrogen Inorganic materials 0.000 claims description 91
- 125000001424 substituent group Chemical group 0.000 claims description 91
- 238000006396 nitration reaction Methods 0.000 claims description 85
- 230000035484 reaction time Effects 0.000 claims description 80
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 75
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 claims description 73
- 125000004432 carbon atom Chemical group C* 0.000 claims description 72
- 239000000047 product Substances 0.000 claims description 70
- 239000013067 intermediate product Substances 0.000 claims description 69
- 239000007787 solid Substances 0.000 claims description 69
- 229910052799 carbon Inorganic materials 0.000 claims description 64
- QAEDZJGFFMLHHQ-UHFFFAOYSA-N trifluoroacetic anhydride Chemical compound FC(F)(F)C(=O)OC(=O)C(F)(F)F QAEDZJGFFMLHHQ-UHFFFAOYSA-N 0.000 claims description 62
- 150000001721 carbon Chemical group 0.000 claims description 59
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 claims description 58
- 125000004430 oxygen atom Chemical group O* 0.000 claims description 58
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 claims description 57
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 claims description 57
- 239000002253 acid Substances 0.000 claims description 56
- 239000000460 chlorine Substances 0.000 claims description 53
- KYQCOXFCLRTKLS-UHFFFAOYSA-N Pyrazine Chemical compound C1=CN=CC=N1 KYQCOXFCLRTKLS-UHFFFAOYSA-N 0.000 claims description 52
- YLQBMQCUIZJEEH-UHFFFAOYSA-N Furan Chemical compound C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 51
- 229910052801 chlorine Inorganic materials 0.000 claims description 51
- 239000000203 mixture Substances 0.000 claims description 50
- 239000000243 solution Substances 0.000 claims description 49
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 claims description 48
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical group CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 claims description 47
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 46
- 238000010992 reflux Methods 0.000 claims description 46
- 239000000126 substance Substances 0.000 claims description 46
- 150000001540 azides Chemical class 0.000 claims description 44
- HIFJUMGIHIZEPX-UHFFFAOYSA-N sulfuric acid;sulfur trioxide Chemical compound O=S(=O)=O.OS(O)(=O)=O HIFJUMGIHIZEPX-UHFFFAOYSA-N 0.000 claims description 44
- 239000005457 ice water Substances 0.000 claims description 43
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 43
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 claims description 42
- 235000011114 ammonium hydroxide Nutrition 0.000 claims description 42
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 claims description 41
- 150000003839 salts Chemical class 0.000 claims description 41
- 229910052794 bromium Inorganic materials 0.000 claims description 40
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 claims description 39
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 claims description 38
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 claims description 38
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 claims description 38
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 38
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 claims description 36
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 claims description 36
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 claims description 36
- 229910052739 hydrogen Inorganic materials 0.000 claims description 36
- 230000015572 biosynthetic process Effects 0.000 claims description 35
- 238000003786 synthesis reaction Methods 0.000 claims description 35
- 125000005842 heteroatom Chemical group 0.000 claims description 34
- 239000000843 powder Substances 0.000 claims description 32
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 claims description 31
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 claims description 31
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 claims description 31
- 239000001257 hydrogen Substances 0.000 claims description 31
- 229910052717 sulfur Inorganic materials 0.000 claims description 31
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 claims description 30
- 229910052698 phosphorus Inorganic materials 0.000 claims description 30
- 125000004437 phosphorous atom Chemical group 0.000 claims description 29
- 125000004434 sulfur atom Chemical group 0.000 claims description 29
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 claims description 27
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 claims description 26
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 claims description 26
- PCNDJXKNXGMECE-UHFFFAOYSA-N Phenazine Natural products C1=CC=CC2=NC3=CC=CC=C3N=C21 PCNDJXKNXGMECE-UHFFFAOYSA-N 0.000 claims description 26
- ZWWCURLKEXEFQT-UHFFFAOYSA-N dinitrogen pentaoxide Chemical compound [O-][N+](=O)O[N+]([O-])=O ZWWCURLKEXEFQT-UHFFFAOYSA-N 0.000 claims description 26
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 26
- KZBUYRJDOAKODT-UHFFFAOYSA-N Chlorine Chemical compound ClCl KZBUYRJDOAKODT-UHFFFAOYSA-N 0.000 claims description 25
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 claims description 24
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 claims description 24
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 claims description 24
- NHNBFGGVMKEFGY-UHFFFAOYSA-N nitrate group Chemical group [N+](=O)([O-])[O-] NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 claims description 24
- 229910052731 fluorine Inorganic materials 0.000 claims description 23
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 22
- 125000000623 heterocyclic group Chemical group 0.000 claims description 22
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 claims description 22
- 239000011259 mixed solution Substances 0.000 claims description 22
- AKEJUJNQAAGONA-UHFFFAOYSA-N sulfur trioxide Chemical compound O=S(=O)=O AKEJUJNQAAGONA-UHFFFAOYSA-N 0.000 claims description 22
- JYEUMXHLPRZUAT-UHFFFAOYSA-N 1,2,3-triazine Chemical compound C1=CN=NN=C1 JYEUMXHLPRZUAT-UHFFFAOYSA-N 0.000 claims description 21
- 229910052708 sodium Inorganic materials 0.000 claims description 20
- 239000011734 sodium Substances 0.000 claims description 20
- 150000003852 triazoles Chemical class 0.000 claims description 20
- 125000002029 aromatic hydrocarbon group Chemical group 0.000 claims description 19
- AJXWEJAGUZJGRI-UHFFFAOYSA-N fluorine azide Chemical compound FN=[N+]=[N-] AJXWEJAGUZJGRI-UHFFFAOYSA-N 0.000 claims description 19
- 229910052700 potassium Inorganic materials 0.000 claims description 19
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 claims description 18
- PBMFSQRYOILNGV-UHFFFAOYSA-N pyridazine Chemical compound C1=CC=NN=C1 PBMFSQRYOILNGV-UHFFFAOYSA-N 0.000 claims description 18
- 229910052701 rubidium Inorganic materials 0.000 claims description 18
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 claims description 17
- 229910002651 NO3 Inorganic materials 0.000 claims description 17
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims description 17
- 229910052802 copper Inorganic materials 0.000 claims description 17
- 229910052744 lithium Inorganic materials 0.000 claims description 17
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims description 16
- KRHYYFGTRYWZRS-UHFFFAOYSA-N Fluorane Chemical compound F KRHYYFGTRYWZRS-UHFFFAOYSA-N 0.000 claims description 16
- 125000001183 hydrocarbyl group Chemical group 0.000 claims description 16
- 229910052740 iodine Inorganic materials 0.000 claims description 16
- 150000003536 tetrazoles Chemical class 0.000 claims description 16
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 claims description 15
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 15
- 229910052737 gold Inorganic materials 0.000 claims description 15
- 230000001590 oxidative effect Effects 0.000 claims description 15
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 claims description 14
- 125000000852 azido group Chemical group *N=[N+]=[N-] 0.000 claims description 14
- 229910052793 cadmium Inorganic materials 0.000 claims description 14
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 14
- 229910052742 iron Inorganic materials 0.000 claims description 14
- 125000004971 nitroalkyl group Chemical group 0.000 claims description 14
- 125000005575 polycyclic aromatic hydrocarbon group Chemical group 0.000 claims description 14
- 125000001273 sulfonato group Chemical group [O-]S(*)(=O)=O 0.000 claims description 14
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 claims description 13
- BUGBHKTXTAQXES-UHFFFAOYSA-N Selenium Chemical group [Se] BUGBHKTXTAQXES-UHFFFAOYSA-N 0.000 claims description 13
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical group [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 claims description 13
- 125000000217 alkyl group Chemical group 0.000 claims description 13
- 229910052785 arsenic Inorganic materials 0.000 claims description 13
- RQNWIZPPADIBDY-UHFFFAOYSA-N arsenic atom Chemical group [As] RQNWIZPPADIBDY-UHFFFAOYSA-N 0.000 claims description 13
- 239000011737 fluorine Substances 0.000 claims description 13
- 229910052759 nickel Inorganic materials 0.000 claims description 13
- 229910052711 selenium Inorganic materials 0.000 claims description 13
- 229910052710 silicon Inorganic materials 0.000 claims description 13
- 229910052709 silver Inorganic materials 0.000 claims description 13
- 229910052714 tellurium Inorganic materials 0.000 claims description 13
- PORWMNRCUJJQNO-UHFFFAOYSA-N tellurium atom Chemical group [Te] PORWMNRCUJJQNO-UHFFFAOYSA-N 0.000 claims description 13
- WTDHULULXKLSOZ-UHFFFAOYSA-N Hydroxylamine hydrochloride Chemical compound Cl.ON WTDHULULXKLSOZ-UHFFFAOYSA-N 0.000 claims description 12
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 claims description 12
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 claims description 12
- 229910052760 oxygen Inorganic materials 0.000 claims description 12
- 239000001301 oxygen Substances 0.000 claims description 12
- 229930195734 saturated hydrocarbon Natural products 0.000 claims description 12
- 229930192474 thiophene Natural products 0.000 claims description 12
- 239000008096 xylene Substances 0.000 claims description 12
- POAOYUHQDCAZBD-UHFFFAOYSA-N 2-butoxyethanol Chemical compound CCCCOCCO POAOYUHQDCAZBD-UHFFFAOYSA-N 0.000 claims description 11
- ZNQVEEAIQZEUHB-UHFFFAOYSA-N 2-ethoxyethanol Chemical compound CCOCCO ZNQVEEAIQZEUHB-UHFFFAOYSA-N 0.000 claims description 11
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 claims description 11
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 claims description 11
- GSEJCLTVZPLZKY-UHFFFAOYSA-N Triethanolamine Chemical compound OCCN(CCO)CCO GSEJCLTVZPLZKY-UHFFFAOYSA-N 0.000 claims description 11
- 125000003277 amino group Chemical group 0.000 claims description 11
- YWEUIGNSBFLMFL-UHFFFAOYSA-N diphosphonate Chemical compound O=P(=O)OP(=O)=O YWEUIGNSBFLMFL-UHFFFAOYSA-N 0.000 claims description 11
- 238000000921 elemental analysis Methods 0.000 claims description 11
- 235000019253 formic acid Nutrition 0.000 claims description 11
- 150000002923 oximes Chemical class 0.000 claims description 11
- DLYUQMMRRRQYAE-UHFFFAOYSA-N phosphorus pentoxide Inorganic materials O1P(O2)(=O)OP3(=O)OP1(=O)OP2(=O)O3 DLYUQMMRRRQYAE-UHFFFAOYSA-N 0.000 claims description 11
- 150000004905 tetrazines Chemical class 0.000 claims description 11
- XNWFRZJHXBZDAG-UHFFFAOYSA-N 2-METHOXYETHANOL Chemical compound COCCO XNWFRZJHXBZDAG-UHFFFAOYSA-N 0.000 claims description 10
- FFNKBQRKZRMYCL-UHFFFAOYSA-N 5-amino-1h-pyrazole-4-carbonitrile Chemical compound NC1=NNC=C1C#N FFNKBQRKZRMYCL-UHFFFAOYSA-N 0.000 claims description 10
- IVRMZWNICZWHMI-UHFFFAOYSA-N azide group Chemical group [N-]=[N+]=[N-] IVRMZWNICZWHMI-UHFFFAOYSA-N 0.000 claims description 10
- 229910052792 caesium Inorganic materials 0.000 claims description 10
- KJUGUADJHNHALS-UHFFFAOYSA-N 1H-tetrazole Substances C=1N=NNN=1 KJUGUADJHNHALS-UHFFFAOYSA-N 0.000 claims description 9
- 229930195733 hydrocarbon Natural products 0.000 claims description 9
- AVXURJPOCDRRFD-UHFFFAOYSA-N hydroxylamine group Chemical group NO AVXURJPOCDRRFD-UHFFFAOYSA-N 0.000 claims description 9
- 125000002950 monocyclic group Chemical group 0.000 claims description 9
- 235000006408 oxalic acid Nutrition 0.000 claims description 9
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 9
- 150000003918 triazines Chemical class 0.000 claims description 9
- HLCPWBZNUKCSBN-UHFFFAOYSA-N 2-aminobenzonitrile Chemical compound NC1=CC=CC=C1C#N HLCPWBZNUKCSBN-UHFFFAOYSA-N 0.000 claims description 8
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 claims description 8
- XEPBRDBFOSKYCF-UHFFFAOYSA-N 4-amino-1h-imidazole-5-carbonitrile Chemical compound NC=1N=CNC=1C#N XEPBRDBFOSKYCF-UHFFFAOYSA-N 0.000 claims description 8
- WYLIQDJQLIYWHP-UHFFFAOYSA-N 5-aminopyridazine-4-carbonitrile Chemical compound NC1=CN=NC=C1C#N WYLIQDJQLIYWHP-UHFFFAOYSA-N 0.000 claims description 8
- KLSJWNVTNUYHDU-UHFFFAOYSA-N Amitrole Chemical group NC1=NC=NN1 KLSJWNVTNUYHDU-UHFFFAOYSA-N 0.000 claims description 8
- IOVCWXUNBOPUCH-UHFFFAOYSA-M Nitrite anion Chemical compound [O-]N=O IOVCWXUNBOPUCH-UHFFFAOYSA-M 0.000 claims description 8
- ZCQWOFVYLHDMMC-UHFFFAOYSA-N Oxazole Chemical compound C1=COC=N1 ZCQWOFVYLHDMMC-UHFFFAOYSA-N 0.000 claims description 8
- 125000003172 aldehyde group Chemical group 0.000 claims description 8
- 125000003545 alkoxy group Chemical group 0.000 claims description 8
- 125000003368 amide group Chemical group 0.000 claims description 8
- YADSGOSSYOOKMP-UHFFFAOYSA-N dioxolead Chemical compound O=[Pb]=O YADSGOSSYOOKMP-UHFFFAOYSA-N 0.000 claims description 8
- 125000001033 ether group Chemical group 0.000 claims description 8
- NIHNNTQXNPWCJQ-UHFFFAOYSA-N fluorene Chemical compound C1=CC=C2CC3=CC=CC=C3C2=C1 NIHNNTQXNPWCJQ-UHFFFAOYSA-N 0.000 claims description 8
- 125000005843 halogen group Chemical group 0.000 claims description 8
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 claims description 8
- 229940071870 hydroiodic acid Drugs 0.000 claims description 8
- 150000002443 hydroxylamines Chemical class 0.000 claims description 8
- 229910052745 lead Inorganic materials 0.000 claims description 8
- CUONGYYJJVDODC-UHFFFAOYSA-N malononitrile Chemical compound N#CCC#N CUONGYYJJVDODC-UHFFFAOYSA-N 0.000 claims description 8
- 125000002971 oxazolyl group Chemical group 0.000 claims description 8
- 238000000746 purification Methods 0.000 claims description 8
- QIRZRDVIOUPHLT-UHFFFAOYSA-N 3,3-diamino-2-(2H-tetrazol-5-yl)propanenitrile Chemical compound NC(C(C#N)C1=NN=NN1)N QIRZRDVIOUPHLT-UHFFFAOYSA-N 0.000 claims description 7
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 claims description 7
- 239000004215 Carbon black (E152) Substances 0.000 claims description 7
- 125000003158 alcohol group Chemical group 0.000 claims description 7
- 125000002843 carboxylic acid group Chemical group 0.000 claims description 7
- 230000007613 environmental effect Effects 0.000 claims description 7
- 125000004185 ester group Chemical group 0.000 claims description 7
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 7
- 239000011630 iodine Substances 0.000 claims description 7
- 125000000468 ketone group Chemical group 0.000 claims description 7
- 125000000018 nitroso group Chemical group N(=O)* 0.000 claims description 7
- FYADHXFMURLYQI-UHFFFAOYSA-N 1,2,4-triazine Chemical compound C1=CN=NC=N1 FYADHXFMURLYQI-UHFFFAOYSA-N 0.000 claims description 6
- DWBOSISZPCOPFS-UHFFFAOYSA-N 2-nitroacetonitrile Chemical compound [O-][N+](=O)CC#N DWBOSISZPCOPFS-UHFFFAOYSA-N 0.000 claims description 6
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 claims description 6
- 150000001335 aliphatic alkanes Chemical group 0.000 claims description 6
- 125000001931 aliphatic group Chemical group 0.000 claims description 6
- 229910052782 aluminium Inorganic materials 0.000 claims description 6
- 125000006615 aromatic heterocyclic group Chemical group 0.000 claims description 6
- 150000004945 aromatic hydrocarbons Chemical class 0.000 claims description 6
- 125000002349 hydroxyamino group Chemical group [H]ON([H])[*] 0.000 claims description 6
- 150000004767 nitrides Chemical class 0.000 claims description 6
- 125000000101 thioether group Chemical group 0.000 claims description 6
- 125000003342 alkenyl group Chemical group 0.000 claims description 5
- 229910021529 ammonia Inorganic materials 0.000 claims description 5
- 229910052804 chromium Inorganic materials 0.000 claims description 5
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 5
- 125000001841 imino group Chemical group [H]N=* 0.000 claims description 5
- 229910052748 manganese Inorganic materials 0.000 claims description 5
- 125000002080 perylenyl group Chemical group C1(=CC=C2C=CC=C3C4=CC=CC5=CC=CC(C1=C23)=C45)* 0.000 claims description 5
- CSHWQDPOILHKBI-UHFFFAOYSA-N peryrene Natural products C1=CC(C2=CC=CC=3C2=C2C=CC=3)=C3C2=CC=CC3=C1 CSHWQDPOILHKBI-UHFFFAOYSA-N 0.000 claims description 5
- 229910052725 zinc Inorganic materials 0.000 claims description 5
- 125000001376 1,2,4-triazolyl group Chemical group N1N=C(N=C1)* 0.000 claims description 4
- KFSLWBXXFJQRDL-UHFFFAOYSA-N Peracetic acid Chemical compound CC(=O)OO KFSLWBXXFJQRDL-UHFFFAOYSA-N 0.000 claims description 4
- 150000001350 alkyl halides Chemical group 0.000 claims description 4
- 229910052788 barium Inorganic materials 0.000 claims description 4
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 claims description 4
- 125000001309 chloro group Chemical group Cl* 0.000 claims description 4
- 125000005331 diazinyl group Chemical group N1=NC(=CC=C1)* 0.000 claims description 4
- 150000002244 furazanes Chemical group 0.000 claims description 4
- DHIGSAXSUWQAEI-UHFFFAOYSA-N hydrazine azide Chemical compound NNN=[N+]=[N-] DHIGSAXSUWQAEI-UHFFFAOYSA-N 0.000 claims description 4
- 125000004043 oxo group Chemical group O=* 0.000 claims description 4
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 claims description 4
- 229910052720 vanadium Inorganic materials 0.000 claims description 4
- NSPMIYGKQJPBQR-UHFFFAOYSA-N 4H-1,2,4-triazole Chemical compound C=1N=CNN=1 NSPMIYGKQJPBQR-UHFFFAOYSA-N 0.000 claims description 3
- KJFKKGZKIKNTFN-UHFFFAOYSA-N N(=[N+]=[N-])C=1C2=C(N=[N+](N=1)[O-])NN=C2[N+](=O)[O-] Chemical compound N(=[N+]=[N-])C=1C2=C(N=[N+](N=1)[O-])NN=C2[N+](=O)[O-] KJFKKGZKIKNTFN-UHFFFAOYSA-N 0.000 claims description 3
- 150000001336 alkenes Chemical group 0.000 claims description 3
- 125000000304 alkynyl group Chemical group 0.000 claims description 3
- 238000004458 analytical method Methods 0.000 claims description 3
- 125000000751 azo group Chemical group [*]N=N[*] 0.000 claims description 3
- 229910052791 calcium Inorganic materials 0.000 claims description 3
- 125000000232 haloalkynyl group Chemical group 0.000 claims description 3
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 claims description 3
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims description 3
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 3
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 claims description 3
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 claims description 3
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims description 3
- 229910052712 strontium Inorganic materials 0.000 claims description 3
- MZRUFMBFIKGOAL-UHFFFAOYSA-N 5-nitro-1h-pyrazole Chemical compound [O-][N+](=O)C1=CC=NN1 MZRUFMBFIKGOAL-UHFFFAOYSA-N 0.000 claims description 2
- RHDGNLCLDBVESU-UHFFFAOYSA-N but-3-en-4-olide Chemical compound O=C1CC=CO1 RHDGNLCLDBVESU-UHFFFAOYSA-N 0.000 claims description 2
- 125000002883 imidazolyl group Chemical group 0.000 claims description 2
- 125000003544 oxime group Chemical group 0.000 claims description 2
- 125000003373 pyrazinyl group Chemical group 0.000 claims description 2
- 125000003226 pyrazolyl group Chemical group 0.000 claims description 2
- 159000000000 sodium salts Chemical class 0.000 claims description 2
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 2
- 125000004435 hydrogen atom Chemical class [H]* 0.000 claims 15
- DPOPAJRDYZGTIR-UHFFFAOYSA-N Tetrazine Chemical compound C1=CN=NN=N1 DPOPAJRDYZGTIR-UHFFFAOYSA-N 0.000 claims 3
- 150000002430 hydrocarbons Chemical class 0.000 claims 3
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 claims 1
- KURZCZMGELAPSV-UHFFFAOYSA-N [Br].[I] Chemical compound [Br].[I] KURZCZMGELAPSV-UHFFFAOYSA-N 0.000 claims 1
- 150000008065 acid anhydrides Chemical class 0.000 claims 1
- 238000007306 functionalization reaction Methods 0.000 claims 1
- 239000007789 gas Substances 0.000 claims 1
- 125000000262 haloalkenyl group Chemical group 0.000 claims 1
- WBGPNPZUWVTYAA-UHFFFAOYSA-N methane;dihydrochloride Chemical compound C.Cl.Cl WBGPNPZUWVTYAA-UHFFFAOYSA-N 0.000 claims 1
- 230000001546 nitrifying effect Effects 0.000 claims 1
- 150000004892 pyridazines Chemical class 0.000 claims 1
- 125000004354 sulfur functional group Chemical group 0.000 claims 1
- 230000000977 initiatory effect Effects 0.000 abstract description 20
- 230000035945 sensitivity Effects 0.000 abstract description 8
- 229910052751 metal Inorganic materials 0.000 abstract description 7
- 239000002184 metal Substances 0.000 abstract description 7
- 238000004880 explosion Methods 0.000 abstract description 6
- 230000037452 priming Effects 0.000 abstract 1
- 125000004429 atom Chemical group 0.000 description 38
- 238000001308 synthesis method Methods 0.000 description 25
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 24
- 238000010586 diagram Methods 0.000 description 24
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 23
- 239000011133 lead Substances 0.000 description 23
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 20
- 239000000463 material Substances 0.000 description 19
- 238000005259 measurement Methods 0.000 description 16
- 238000012360 testing method Methods 0.000 description 14
- 238000005474 detonation Methods 0.000 description 13
- 238000001035 drying Methods 0.000 description 13
- 238000001914 filtration Methods 0.000 description 13
- 150000002431 hydrogen Chemical class 0.000 description 13
- 239000010949 copper Substances 0.000 description 12
- 239000012452 mother liquor Substances 0.000 description 11
- 239000010931 gold Substances 0.000 description 10
- XEEYBQQBJWHFJM-UHFFFAOYSA-N iron Substances [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 10
- 239000002585 base Substances 0.000 description 9
- 238000001816 cooling Methods 0.000 description 9
- 239000003814 drug Substances 0.000 description 9
- 238000002329 infrared spectrum Methods 0.000 description 8
- 239000010944 silver (metal) Substances 0.000 description 8
- LPXPTNMVRIOKMN-UHFFFAOYSA-M sodium nitrite Chemical compound [Na+].[O-]N=O LPXPTNMVRIOKMN-UHFFFAOYSA-M 0.000 description 8
- 125000003118 aryl group Chemical group 0.000 description 7
- 239000003795 chemical substances by application Substances 0.000 description 7
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 6
- 230000036961 partial effect Effects 0.000 description 6
- 238000001228 spectrum Methods 0.000 description 6
- 238000003756 stirring Methods 0.000 description 6
- 230000002194 synthesizing effect Effects 0.000 description 6
- 238000005979 thermal decomposition reaction Methods 0.000 description 6
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 5
- GVPFVAHMJGGAJG-UHFFFAOYSA-L cobalt dichloride Chemical compound [Cl-].[Cl-].[Co+2] GVPFVAHMJGGAJG-UHFFFAOYSA-L 0.000 description 5
- 238000010189 synthetic method Methods 0.000 description 5
- LMBFAGIMSUYTBN-MPZNNTNKSA-N teixobactin Chemical compound C([C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H](CCC(N)=O)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H]1C(N[C@@H](C)C(=O)N[C@@H](C[C@@H]2NC(=N)NC2)C(=O)N[C@H](C(=O)O[C@H]1C)[C@@H](C)CC)=O)NC)C1=CC=CC=C1 LMBFAGIMSUYTBN-MPZNNTNKSA-N 0.000 description 5
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 4
- 150000007513 acids Chemical class 0.000 description 4
- 229940079593 drug Drugs 0.000 description 4
- 230000000694 effects Effects 0.000 description 4
- 239000012467 final product Substances 0.000 description 4
- 238000002156 mixing Methods 0.000 description 4
- 239000003960 organic solvent Substances 0.000 description 4
- 239000003208 petroleum Substances 0.000 description 4
- 239000000376 reactant Substances 0.000 description 4
- 235000010288 sodium nitrite Nutrition 0.000 description 4
- 238000005406 washing Methods 0.000 description 4
- YIWGJFPJRAEKMK-UHFFFAOYSA-N 1-(2H-benzotriazol-5-yl)-3-methyl-8-[2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidine-5-carbonyl]-1,3,8-triazaspiro[4.5]decane-2,4-dione Chemical compound CN1C(=O)N(c2ccc3n[nH]nc3c2)C2(CCN(CC2)C(=O)c2cnc(NCc3cccc(OC(F)(F)F)c3)nc2)C1=O YIWGJFPJRAEKMK-UHFFFAOYSA-N 0.000 description 3
- DDFHBQSCUXNBSA-UHFFFAOYSA-N 5-(5-carboxythiophen-2-yl)thiophene-2-carboxylic acid Chemical compound S1C(C(=O)O)=CC=C1C1=CC=C(C(O)=O)S1 DDFHBQSCUXNBSA-UHFFFAOYSA-N 0.000 description 3
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 3
- VCLINDGRTZZTDJ-UHFFFAOYSA-N [Na].[N+](=O)([O-])CC#N Chemical compound [Na].[N+](=O)([O-])CC#N VCLINDGRTZZTDJ-UHFFFAOYSA-N 0.000 description 3
- 235000019270 ammonium chloride Nutrition 0.000 description 3
- UAZDIGCOBKKMPU-UHFFFAOYSA-O azanium;azide Chemical compound [NH4+].[N-]=[N+]=[N-] UAZDIGCOBKKMPU-UHFFFAOYSA-O 0.000 description 3
- 239000011651 chromium Substances 0.000 description 3
- 229940097267 cobaltous chloride Drugs 0.000 description 3
- 238000011161 development Methods 0.000 description 3
- SYMAEXJCESFOLG-UHFFFAOYSA-N furan-3,4-diamine Chemical compound NC1=COC=C1N SYMAEXJCESFOLG-UHFFFAOYSA-N 0.000 description 3
- 238000010438 heat treatment Methods 0.000 description 3
- IXHMHWIBCIYOAZ-UHFFFAOYSA-N styphnic acid Chemical compound OC1=C([N+]([O-])=O)C=C([N+]([O-])=O)C(O)=C1[N+]([O-])=O IXHMHWIBCIYOAZ-UHFFFAOYSA-N 0.000 description 3
- 239000011593 sulfur Substances 0.000 description 3
- PAWQVTBBRAZDMG-UHFFFAOYSA-N 2-(3-bromo-2-fluorophenyl)acetic acid Chemical compound OC(=O)CC1=CC=CC(Br)=C1F PAWQVTBBRAZDMG-UHFFFAOYSA-N 0.000 description 2
- OCKGFTQIICXDQW-ZEQRLZLVSA-N 5-[(1r)-1-hydroxy-2-[4-[(2r)-2-hydroxy-2-(4-methyl-1-oxo-3h-2-benzofuran-5-yl)ethyl]piperazin-1-yl]ethyl]-4-methyl-3h-2-benzofuran-1-one Chemical compound C1=C2C(=O)OCC2=C(C)C([C@@H](O)CN2CCN(CC2)C[C@H](O)C2=CC=C3C(=O)OCC3=C2C)=C1 OCKGFTQIICXDQW-ZEQRLZLVSA-N 0.000 description 2
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 2
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 2
- 125000002252 acyl group Chemical group 0.000 description 2
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 2
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 229910052796 boron Inorganic materials 0.000 description 2
- 238000012937 correction Methods 0.000 description 2
- 239000013078 crystal Substances 0.000 description 2
- 238000000354 decomposition reaction Methods 0.000 description 2
- 230000007547 defect Effects 0.000 description 2
- XHFGWHUWQXTGAT-UHFFFAOYSA-N dimethylamine hydrochloride Natural products CNC(C)C XHFGWHUWQXTGAT-UHFFFAOYSA-N 0.000 description 2
- IQDGSYLLQPDQDV-UHFFFAOYSA-N dimethylazanium;chloride Chemical compound Cl.CNC IQDGSYLLQPDQDV-UHFFFAOYSA-N 0.000 description 2
- 150000002148 esters Chemical class 0.000 description 2
- JKFAIQOWCVVSKC-UHFFFAOYSA-N furazan Chemical group C=1C=NON=1 JKFAIQOWCVVSKC-UHFFFAOYSA-N 0.000 description 2
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 2
- 230000000670 limiting effect Effects 0.000 description 2
- 230000000737 periodic effect Effects 0.000 description 2
- 239000011574 phosphorus Substances 0.000 description 2
- 230000001681 protective effect Effects 0.000 description 2
- 239000011669 selenium Substances 0.000 description 2
- 239000010703 silicon Substances 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 2
- 150000004655 tetrazenes Chemical class 0.000 description 2
- 230000004580 weight loss Effects 0.000 description 2
- 0 *C(C(C([N-]1)=N2)=C(*)[N-][N+]2O)=N[N+]1O Chemical compound *C(C(C([N-]1)=N2)=C(*)[N-][N+]2O)=N[N+]1O 0.000 description 1
- ZFXBERJDEUDDMX-UHFFFAOYSA-N 1,2,3,5-tetrazine Chemical compound C1=NC=NN=N1 ZFXBERJDEUDDMX-UHFFFAOYSA-N 0.000 description 1
- KEQXNNJHMWSZHK-UHFFFAOYSA-L 1,3,2,4$l^{2}-dioxathiaplumbetane 2,2-dioxide Chemical compound [Pb+2].[O-]S([O-])(=O)=O KEQXNNJHMWSZHK-UHFFFAOYSA-L 0.000 description 1
- MUTZAFQFMYGRNL-UHFFFAOYSA-N 2-oxidotriazin-2-ium Chemical compound [O-][N+]1=NC=CC=N1 MUTZAFQFMYGRNL-UHFFFAOYSA-N 0.000 description 1
- XXWYUBYMBOTAGQ-UHFFFAOYSA-N 6-diazo-4,5-dinitrocyclohexa-2,4-dien-1-ol Chemical compound OC1C=CC([N+]([O-])=O)=C([N+]([O-])=O)C1=[N+]=[N-] XXWYUBYMBOTAGQ-UHFFFAOYSA-N 0.000 description 1
- GQOILBFEHZOUGT-UHFFFAOYSA-N 7h-pyrazolo[3,4-d]triazine Chemical compound N1N=NC=C2C=NN=C21 GQOILBFEHZOUGT-UHFFFAOYSA-N 0.000 description 1
- 208000005156 Dehydration Diseases 0.000 description 1
- 102100037709 Desmocollin-3 Human genes 0.000 description 1
- LCGLNKUTAGEVQW-UHFFFAOYSA-N Dimethyl ether Chemical compound COC LCGLNKUTAGEVQW-UHFFFAOYSA-N 0.000 description 1
- 101000968042 Homo sapiens Desmocollin-2 Proteins 0.000 description 1
- 101000880960 Homo sapiens Desmocollin-3 Proteins 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- TZRXHJWUDPFEEY-UHFFFAOYSA-N Pentaerythritol Tetranitrate Chemical compound [O-][N+](=O)OCC(CO[N+]([O-])=O)(CO[N+]([O-])=O)CO[N+]([O-])=O TZRXHJWUDPFEEY-UHFFFAOYSA-N 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- VVRKDUKHRGNPON-UHFFFAOYSA-N [N+](=O)([O-])C1=C(OC2=C1C=CC=C2)[N+](=O)[O-].[K] Chemical compound [N+](=O)([O-])C1=C(OC2=C1C=CC=C2)[N+](=O)[O-].[K] VVRKDUKHRGNPON-UHFFFAOYSA-N 0.000 description 1
- 238000000862 absorption spectrum Methods 0.000 description 1
- 238000001467 acupuncture Methods 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- DSAJWYNOEDNPEQ-UHFFFAOYSA-N barium atom Chemical compound [Ba] DSAJWYNOEDNPEQ-UHFFFAOYSA-N 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 125000002619 bicyclic group Chemical group 0.000 description 1
- 238000009529 body temperature measurement Methods 0.000 description 1
- BDOSMKKIYDKNTQ-UHFFFAOYSA-N cadmium atom Chemical compound [Cd] BDOSMKKIYDKNTQ-UHFFFAOYSA-N 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 230000018044 dehydration Effects 0.000 description 1
- 238000006297 dehydration reaction Methods 0.000 description 1
- 238000001739 density measurement Methods 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 230000005611 electricity Effects 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- BHEPBYXIRTUNPN-UHFFFAOYSA-N hydridophosphorus(.) (triplet) Chemical compound [PH] BHEPBYXIRTUNPN-UHFFFAOYSA-N 0.000 description 1
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- QSHDDOUJBYECFT-UHFFFAOYSA-N mercury Chemical compound [Hg] QSHDDOUJBYECFT-UHFFFAOYSA-N 0.000 description 1
- 229910052753 mercury Inorganic materials 0.000 description 1
- 239000007769 metal material Substances 0.000 description 1
- 150000002739 metals Chemical class 0.000 description 1
- DOTMOQHOJINYBL-UHFFFAOYSA-N molecular nitrogen;molecular oxygen Chemical compound N#N.O=O DOTMOQHOJINYBL-UHFFFAOYSA-N 0.000 description 1
- 230000000802 nitrating effect Effects 0.000 description 1
- 229910052755 nonmetal Inorganic materials 0.000 description 1
- 239000003921 oil Substances 0.000 description 1
- 150000002926 oxygen Chemical class 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 238000003825 pressing Methods 0.000 description 1
- 230000008707 rearrangement Effects 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 230000002829 reductive effect Effects 0.000 description 1
- 230000003014 reinforcing effect Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 239000004065 semiconductor Substances 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 239000012265 solid product Substances 0.000 description 1
- 238000009987 spinning Methods 0.000 description 1
- 230000003068 static effect Effects 0.000 description 1
- 230000001360 synchronised effect Effects 0.000 description 1
- 125000003698 tetramethyl group Chemical group [H]C([H])([H])* 0.000 description 1
- XJPANWOKBWZVHC-UHFFFAOYSA-N tetrazol-2-amine Chemical compound NN1N=CN=N1 XJPANWOKBWZVHC-UHFFFAOYSA-N 0.000 description 1
- 150000003568 thioethers Chemical class 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- ILWRPSCZWQJDMK-UHFFFAOYSA-N triethylazanium;chloride Chemical compound Cl.CCN(CC)CC ILWRPSCZWQJDMK-UHFFFAOYSA-N 0.000 description 1
- 239000002699 waste material Substances 0.000 description 1
- 239000002351 wastewater Substances 0.000 description 1
- 238000005303 weighing Methods 0.000 description 1
Images


























Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C06—EXPLOSIVES; MATCHES
- C06B—EXPLOSIVES OR THERMIC COMPOSITIONS; MANUFACTURE THEREOF; USE OF SINGLE SUBSTANCES AS EXPLOSIVES
- C06B25/00—Compositions containing a nitrated organic compound
- C06B25/34—Compositions containing a nitrated organic compound the compound being a nitrated acyclic, alicyclic or heterocyclic amine
-
- C—CHEMISTRY; METALLURGY
- C06—EXPLOSIVES; MATCHES
- C06C—DETONATING OR PRIMING DEVICES; FUSES; CHEMICAL LIGHTERS; PYROPHORIC COMPOSITIONS
- C06C7/00—Non-electric detonators; Blasting caps; Primers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D253/00—Heterocyclic compounds containing six-membered rings having three nitrogen atoms as the only ring hetero atoms, not provided for by group C07D251/00
- C07D253/08—Heterocyclic compounds containing six-membered rings having three nitrogen atoms as the only ring hetero atoms, not provided for by group C07D251/00 condensed with carbocyclic rings or ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains three hetero rings
- C07D487/14—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D498/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/12—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains three hetero rings
- C07D498/14—Ortho-condensed systems
Definitions
- the preparation method further comprises adding a base and an ester compound after adding the hydroxylamine compound.
- the base is a compound of the general formula M a (OH) b , where M is one or two or more selected from the group consisting of NH 4 , Li, Na, K, Rb, Cs, Ca, Sr, and Ba. kind of mix.
- FIG. 10 is a nuclear magnetic hydrogen spectrum of the compound shown in FIG. 7.
- FIG. 29 is a schematic diagram of a funnel used for angle of repose determination of a compound of the present invention.
- FIG. 30 is a schematic diagram of a tri-scaffold used for determination of a repose angle of a compound of the present invention.
- Figures 12, 14, 16, 18, 20, 22, 24, and 26 are chemical structural diagrams of various embodiments of a compound of the present invention.
- Figures 13, 15, 17, 19, 21, 23, 25, and 27 are chemical structural diagrams of partial structures of various embodiments of a compound of the present invention.
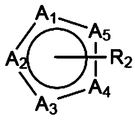
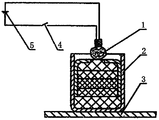
- a 1 to A 11 atoms; I, II, III: compounds; 1: electric ignition head; 2: sample; 3: lead plate; 4: power supply; 5: switch
- this structure is composed of non-metal atoms such as carbon, nitrogen, oxygen, hydrogen, etc., and contains no metal. Therefore, it is possible to produce metal-free compounds for explosives and detonators, which can be prepared. Green and environmentally friendly explosives and explosives.
- These one or more single rings fused with the structure shown in FIG. 1 may be three-membered rings, four-membered rings, five-membered rings, six-membered rings, seven-membered rings, eight-membered rings, or even more membered rings.
- Polycyclic For example, a five-membered ring may be fused with the structure shown in FIG. 1 and share two atoms at positions 5 and 6 in the structure shown in FIG. 1, for example, share two carbon atoms.
- a six-membered ring may be fused with the structure shown in FIG. 1 and share two atoms at positions 5 and 6 in the structure shown in FIG. 1, for example, share two carbon atoms.
- the aromatic ring fused to the structure shown in FIG. 1 may be pyrazine, and the two carbon atoms at positions 2 and 3 are simultaneously the positions 5 and 6 of the structure shown in FIG. 1, respectively. Two carbon atoms.
- the one or more monocyclic rings condensed with the structure shown in FIG. 1 may have no substituents or have one or more substituents.
- substituents may be: (1) a saturated hydrocarbon hydrocarbon group, such as an alkane group; (2) a functionalized saturated hydrocarbon hydrocarbon group, such as a nitroalkane group, a fluorinated dinitroalkane group, a nitroformyl alkyl group, a halogenated alkyl group, and the like; (3) alcohol group; (4) ether group, thioether group; (5) carboxylic acid group, ester group, nitrate group, sulfonate group, sulfonate group, etc .; (6) keto group, amide group, acyl group (7) aldehyde group; (8) aldehyde group; (8) unsaturated aliphatic hydrocarbon group, such as alkenyl group, alkynyl group, halogenated olefin group, nitroalkynyl
- heterocyclic non-aromatic hydrocarbon groups such as aza-bridged cyclic hydrocarbons, oxa-bridged cyclic hydrocarbons, etc.
- halogen atom such as aza-bridged cyclic hydrocarbons, oxa-bridged cyclic hydrocarbons, etc.
- halogen atom such as aza-bridged cyclic hydrocarbons, oxa-bridged cyclic hydrocarbons, etc.
- halogen atom such as aza-bridged cyclic hydrocarbons, oxa-bridged cyclic hydrocarbons, etc.
- halogen atom such as aza-bridged cyclic hydrocarbons, oxa-bridged cyclic hydrocarbons, etc.
- cyano, oxime such as amino, imino, azo, azo oxide, azido, hydrazine
- hydroxyl hydroxylamine, alkoxy Base
- Nitro Nitro, nitroso.
- it may be a
- FIG. 3 is a chemical structure diagram of one embodiment of a compound of the present invention. Please refer to FIG. 3, wherein the structure shown in FIG. 1 is fused with a five-membered ring and has at least one substituent R 3 .
- atoms such as A 1 to A 5 may be carbon atoms, nitrogen atoms, oxygen atoms, phosphorus atoms, or sulfur atoms, and may be the same type or different types.
- the number of substituents on atoms such as A 1 to A 5 may be 0, 1, 2, 3, 4 or 5.
- FIG. 3 is a chemical structure diagram of one embodiment of a compound of the present invention. Please refer to FIG. 3, wherein the structure shown in FIG. 1 is fused with a five-membered ring and has at least one substituent R 3 .
- atoms such as A 1 to A 5 may be carbon atoms, nitrogen atoms, oxygen atoms, phosphorus atoms, or sulfur atoms, and may be the same type or different types.
- the precursor substance to be synthesized here that is, the structure of the general formula shown in FIG. 6 may include two atoms A 10 and A 11 , where A 10 may be connected to at least one cyano group, and A 11 may be connected to at least An amino group.
- these cyano groups and amino groups will participate in the reaction, so that the two atoms A 10 and A 11 become, for example, A 1 and A 2 in the structure shown in FIG. 1, or for example Two of A 1 to A 6 in the structures shown in FIGS. 3 to 5.
- the precursor material includes a polycyclic ring.
- a 10 and A 11 are part of a polycyclic ring.
- the multivalent ring here can be a three-membered ring, a four-membered ring, a five-membered ring, a six-membered ring, a seven-membered ring, an eight-membered ring, or even a multi-membered ring.
- it can be a five-membered ring.
- it could be a six-membered ring.
- it can be a five-membered ring and a six-membered fused ring.
- the precursor material includes an aromatic ring.
- a 10 and A 11 are part of an aroma.
- the precursor material includes a polycyclic ring, but atoms such as A 10 and A 11 are not part of the polycyclic ring.
- These rings may be aromatic heterocycles such as furan, pyrrole, thiophene, imidazole, pyrazole, oxazole, thiazole, pyridine, pyrazine, pyrimidine, pyridazine, furazine, or other triazoles, tetrazoles, triazoles Azines, tetrazines ... and so on.
- aromatic heterocycles such as furan, pyrrole, thiophene, imidazole, pyrazole, oxazole, thiazole, pyridine, pyrazine, pyrimidine, pyridazine, furazine, or other triazoles, tetrazoles, triazoles Azines, tetrazines ... and so on.
- the precursor material is subjected to an off-tetrazolium ring reaction to obtain a first intermediate product.
- the solvent in step two (1) may be N, N-dimethylformamide, dimethyl sulfoxide, tetrahydrofuran, acetonitrile, dichloromethane, chloroform, methanol, ethanol, water, isopropanol, ethylene glycol, Any one of ethylene glycol monomethyl ether, ethylene glycol monoethyl ether, ethylene glycol monobutyl ether, glycerol, acetone, triethanolamine, pyridine, benzene, toluene, xylene, etc., or two or more suitable ones
- the mixture of solvents may be, for example, water, ethanol, dimethyl sulfoxide, N, N-dimethylformamide, or a mixed solution of acetone and methanol in a volume ratio of 4: 1.
- the present invention is not limited thereto, and it should be considered that the present invention can adopt any solvent suitable for the reaction.
- the present invention is not limited thereto, and it should be considered that the present invention can adopt any proportion of solvents suitable for the reaction.
- the salts may, for example, have the general formula M a X b , where: M may be Fe, Co, Ni, Mn, Zn, Cd, Au, Ag, Cu, Cr, Al, V, NH 4 , (CH 3 ) 2 NH 2 , (CH 3 ) 3 NH, (CH 3 CH 2 ) 2 NH 2 , (CH 3 CH 2 ) 3 NH, etc., and X may be F, Cl, Br, I, NO 3 , SO 4 , HSO 4 , CO 3 , HCO 3 , HCOO, CH 3 COO, CF 3 COO, (COO) 2 , CH 2 (COO) 2 and so on.
- the type of the acid to be added may be: nitric acid, sulfuric acid, hydrochloric acid, hydrobromic acid, hydrofluoric acid, hydroiodic acid, formic acid, acetic acid, acetic anhydride, oxalic acid, malonic acid, and trifluoro acid.
- Acetic acid and the like may be used alone or as a mixture of two or more acids.
- it can be hydrochloric acid, formic acid, acetic anhydride, and trifluoroacetic acid; however, the present invention is not limited thereto, and it should be considered that the present invention can use any acid suitable for the reaction.
- the amount of the acid to be added dropwise can be used to adjust the pH of the solution to 1.0 to 4.0, for example, 1.0 to 3.5, for example, 2.0 to 3.0, for example, 1.4, 2.0, 2.4, 2.5 , 3.0, 3.1, 3.3, etc., but the present invention is not limited to this, it should be considered that the amount of acid added dropwise can adjust the solution to any desired pH value.
- the nitration system may be a commercially available fuming nitric acid or 100% nitric acid, or other nitration systems, such as dinitrogen pentoxide, tetrafluoroboron nitrate, 100% nitric acid / trifluoroacetic anhydride, 100 % Nitric acid / concentrated sulfuric acid, fuming nitric acid, 100% nitric acid / acetic anhydride, fuming nitric acid / concentrated sulfuric acid, fuming nitric acid / acetic anhydride, fuming nitric acid / trifluoroacetic anhydride, fuming nitric acid / acetic anhydride, sulfur trioxide / Nitric acid, phosphorus pentoxide / nitric acid, fuming sulfuric acid / fuming nitric acid, fuming sulfuric acid / 100% nitric acid and the like.
- other nitration systems such as dinitrogen pentoxid
- the molar ratio of the amount of the first intermediate product to the tetrafluoroboron nitrate may be 1: 1 to 1:50; the solvent used may not be the same as the nitration system or the first intermediate product,
- the solvent for the second intermediate product reaction may be, for example, acetonitrile.
- the specific method for terminating the reaction in step three (2) may be, for example, pouring the reaction system into ice or ice water, so that the nitration system is diluted and cooled, the reaction is stopped, and the product is precipitated.
- the flow rate of the chlorine gas flow may be between 1 mL and 5 mL per minute, for example, 2 mL and 5 mL per minute, such as 3 mL and 4 mL per minute, or 5 mL and per minute, for example.
- the reaction temperature in step tetramethyl (2) may be 30 ° C to 100 ° C, such as 50 ° C to 90 ° C, such as 60 ° C to 70 ° C, such as 60 ° C, or 70 ° C, for example.
- the reaction time in step 4A (2) is, for example, 1 hour to 10 hours, such as 1 hour to 5 hours, such as 1 hour to 3 hours, such as 1 hour, or 2 hours, for example.
- the reflux reaction time is, for example, 0.5 hours to 24 hours, such as 1 hour to 12 hours, such as 2 hours to 8 hours, such as 2 hours, or 4 hours, for example.
- the reaction time in step tetrapropene (3) may be, for example, 0.5 to 12 hours, for example, may be 0.5 to 4 hours, for example, may be 0.5 hours, 1 hour, 2 hours, or 4 hours.
- the solvent used may be various common organic solvents, as long as it does not react with the second intermediate product or concentrated ammonia water, so that subsequent reactions cannot be performed.
- the solvent used may be methanol.
- step Si Ding (2) the time of the reflux reaction is, for example, 0.5 hour to 24 hours, such as 1 hour to 12 hours, such as 2 hours to 8 hours, such as 2 hours, or 4 hours, for example.
- the reaction time in step tetrabutane (4) may be 0.5 to 12 hours, for example, may be 0.5 to 4 hours, for example, may be 0.5 hours, 1 hour, 2 hours, or 4 hours.
- the present invention is not limited thereto, and it should be considered that the present invention can adopt any reaction time suitable for the reaction.
- the molar ratio of the amount of solids obtained from step tetrabutylene (3) to tetrafluoroboron nitrate may be 1: 1 to 1:50.
- FIG. 7 is a chemical structure diagram of one embodiment of a compound of the present invention.
- the compound shown in FIG. 7 has a specific name: 2-oxo-4-azido-5-nitro-7H-pyrazolo [3,4-d] [1,2,3] triazine, It has the structure shown in FIG. 1, specifically the structure shown in FIG. 3, and more specifically the structure shown in FIG. 8.
- This compound is a white to yellow-green powdery solid, and the pure substance precipitated by recrystallization is white needle-like crystals. Its crystal density is 1.5-1.9 g / cm 3 , and after being subjected to constant temperature dehydration treatment at 70 ° C. to 90 ° C., the measured density at room temperature is 1.6-2.0 g / cm 3 . Good diffusibility, not easy to adhere to glass containers.
- the thermal decomposition temperature is higher than 150 °C, and it is not sensitive to light, water and steam. No obvious color change was observed under direct sunlight for a long time. Stable in nature and does not react with strong acids.
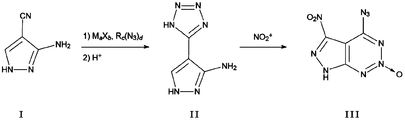
- Example 1 The compound of Example 1 was synthesized by using 3-amino-4-cyanopyrazole as a precursor.
- the specific reaction can be shown in FIG. 9 for example:
- Second step reaction Synthesis of the product 2-oxo-4-azido-5-nitro- from the first intermediate 3-amino-4- [1H-tetrazole] -1H-pyrazole (compound of formula II) 7H-pyrazolo [3,4-d] [1,2,3] triazine (compound of formula III in FIG. 9, ie, compound shown in FIG. 7)
- step (1) The solvent used in step (1), the salts, azides added in step (2), reaction temperature and reaction time, and the acid added in step (3) can be referred to in step 2 of the general synthesis method.
- Second reaction step Synthesis of 2-oxo-4-azido-5-nitro-7H-pyrazolo [3,4-d] [1,2,3] triazine (compound of formula III, Fig. 7 (Compound shown)
- Example 1 The first intermediate product of Example 1, that is, the compound of formula II in FIG. 9 is slowly added to the nitration system. After the addition, the temperature of the reaction system is controlled to maintain the nitration reaction at a certain temperature for a certain time.
- step (6) For the nitration system and reaction temperature added in step (5) and the method for terminating the reaction in step (6), please refer to the reaction conditions described in step 3 of the general synthesis method.
- the nuclear magnetic hydrogen spectrum of the compound of formula III (ie, the compound shown in FIG. 7) prepared in Example 1A-1D is shown in FIG. 10, and the characteristic nuclear magnetic hydrogen spectrum shift is: 1 H NMR (400 MHz, DMSO-d 6 , 25 ° C) ⁇ (ppm): 14.51 (brs, 1H, NH).
- the nuclear magnetic carbon spectrum of the compound of formula III prepared in Example 1A-1D (ie, the compound shown in FIG. 7) is shown in FIG. 11.
- the characteristic nuclear magnetic carbon spectrum shift is: 13 C NMR (100 MHz, DMSO-d 6 , 25 ° C) ⁇ (ppm): 157.95, 157.31, 148.07, 90.03.
- the wavelength of the infrared absorption spectrum of the compound of formula III (ie, the compound shown in FIG. 7) prepared in Examples 1A-1D is: IR (KBr, ⁇ / cm -1 ): 3414 (m), 3231 (m), 2154 ( s), 1600 (s), 1543 (s), 1403 (s), 1384 (s), 1313 (m), 1209 (m), 1131 (m).
- the elemental analysis structure of the compound of formula III (that is, the compound shown in FIG. 7) (molecular formula C 4 HN 9 O 3 ) prepared in Example 1A-1D is (measured value (calculated value)): C: 21.62% (21.53%) , H: 0.47% (0.45%), N: 56.61% (56.50%), O: 21.22% (21.51%).
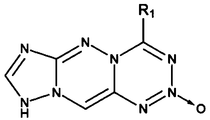
- FIG. 12 is a chemical structure diagram of one embodiment of a compound of the present invention, and its specific name is: 5-oxidized-7-nitro [1,2,5] oxadiazole [3,4-d ] [1,2,3] triazine has the structure shown in FIG. 1, specifically has the structure shown in FIG. 3, and more specifically has the structure shown in FIG.
- the first reaction synthesis of precursors
- the reaction temperature may be -20 ° C to 130 ° C, for example, -20 ° C to 40 ° C, for example, -20 ° C, -10 ° C, 40 ° C.
- the present invention is not limited thereto, and it should be considered that the present invention can adopt any reaction temperature suitable for the reaction.
- the nitrite in step (1) may be a compound of the general formula MNO 2 , where M is one selected from the group consisting of NH 4 , Li, Na, K, Rb, and Cs, or a mixture of two or more.
- M is one selected from the group consisting of NH 4 , Li, Na, K, Rb, and Cs, or a mixture of two or more.
- it may be sodium nitrite.
- the solvent in step (1) may be N, N-dimethylformamide, dimethyl sulfoxide, tetrahydrofuran, acetonitrile, dichloromethane, chloroform, methanol, ethanol, water, isopropanol, ethylene glycol, ethyl acetate Any one of glycol monomethyl ether, ethylene glycol monoethyl ether, ethylene glycol monobutyl ether, glycerol, acetone, triethanolamine, pyridine, benzene, toluene, xylene, etc., or a mixture of two or more thereof.
- the present invention is not limited thereto, and it should be considered that the present invention can adopt any solvent suitable for the reaction.
- the hydroxylamine compound may be a hydroxylamine salt.
- the hydroxylamine salts may be compounds of the general formula (NH 3 OH) a X b , where X is selected from the group consisting of F, Cl, Br, I, NO 3 , SO 4 , HSO 4 , CO 3 , HCO 3 , One of HCOO, CH 3 COO, CF 3 COO, (COO) 2 and CH 2 (COO) 2 , or a mixture of two or more of them.
- the hydroxylamine compound may be a halogenated salt of hydroxylamine; for example, the hydroxylamine compound may be hydroxylamine hydrochloride.
- the reaction time in step (2) (3) may be 2 hours to 24 hours, and may be 2 hours, 3 hours, 4 hours, 5 hours, 10 hours, or 24 hours, for example.
- the present invention is not limited thereto, and it should be considered that the present invention can adopt any reaction time suitable for the reaction.
- the ester compound in step (3) may be a compound of the general formula R 1 COOR 2 , wherein R 1 and R 2 are selected from methyl, ethyl, propyl, isopropyl, primary butyl, sec-butyl, Tert-butyl and one of various pentyl and various hexyl, or a mixture of two or more.
- R 1 and R 2 are selected from methyl, ethyl, propyl, isopropyl, primary butyl, sec-butyl, Tert-butyl and one of various pentyl and various hexyl, or a mixture of two or more.
- R 1 and R 2 are selected from methyl, ethyl, propyl, isopropyl, primary butyl, sec-butyl, Tert-butyl and one of various pentyl and various hexyl, or a mixture of two or more.
- the present invention is not limited to this, and it should be considered that the
- the base in step (3) may be a compound of the general formula M a (OH) b , where M is one selected from NH 4 , Li, Na, K, Rb, Cs, Ca, Sr, Ba, or two A mix of one or more.
- M is one selected from NH 4 , Li, Na, K, Rb, Cs, Ca, Sr, Ba, or two A mix of one or more.
- the present invention is not limited to this, and it should be considered that an appropriate base can be used in the present invention.
- the type of the acid to be added may be: nitric acid, sulfuric acid, hydrochloric acid, hydrobromic acid, hydrofluoric acid, hydroiodic acid, formic acid, acetic acid, acetic anhydride, oxalic acid, malonic acid, trifluoroacetic acid, etc.
- it can be hydrochloric acid, formic acid, acetic anhydride, and trifluoroacetic acid; however, the present invention is not limited thereto, and it should be considered that the present invention can use any acid suitable for the reaction.
- step (12) in this part can refer to step 4c of the general synthesis method The reaction conditions described in the section.
- the obtained solid was dispersed in 10 mL of water together with 3.6 g of ammonium fluoride and 12 g of sodium azide, and then reacted at 75 ° C for 7 hours. After cooling to room temperature, hydrochloric acid was added dropwise thereto until the pH of the solution was 3.1, and then filtered. , Washed with 5 mL of ice water, and dried the solid to obtain 12.2 g of orange powder.
- FIG. 14 is a chemical structure diagram of one embodiment of a compound of the present invention, and its specific name is: 2-oxo-4-chloro-7H-imidazole [4,5-d] [1,2,3 ] Triazine has the structure shown in FIG. 1, specifically has the structure shown in FIG. 3, and more specifically has the structure shown in FIG. 15.
- Example 3 The specific method for synthesizing the compound of Example 3 is specifically using 4-amino-5-cyanoimidazole as a precursor. Details are as follows:
- the first reaction synthesis of the first intermediate substance
- step (1) The solvents used in step (1), salts, azides added in (2), reaction temperature and reaction time, and acids added in step (3) can be referred to in step 2 of the general synthesis method. Reaction conditions.
- Second step reaction synthesis of a second intermediate
- Example 3 The first intermediate product of Example 3 is slowly added to the nitration system, and the temperature of the reaction system is controlled after the addition, and the nitration reaction is maintained at a certain temperature for a certain time.
- step (5) For the nitration system and reaction temperature added in step (4) in this section, and the method for terminating the reaction in step (5), refer to the reaction conditions described in step 3 of the general synthesis method.
- Example 3 The second intermediate product of Example 3 is dispersed in a solvent.
- step (6) in this section the chlorine gas flow rate, reaction temperature, and reaction time used in step (7), refer to the reaction conditions described in step 4A of the general synthesis method.
- the obtained solid was dissolved in 8 mL of dichloromethane, and a stream of chlorine gas (flow rate of 5 mL / min) was passed, reacted at 70 ° C. for 1 hour, and filtered to obtain 0.17 g of a white powdery product with a yield of 30%.
- FIG. 16 is a chemical structure diagram of one embodiment of a compound of the present invention, and its specific name is: 3,5-dioxide-7-amino- [1,2,5] oxadiazole [3, 4-d] [1,2,3] triazine has the structure shown in FIG. 1, specifically has the structure shown in FIG. 3, and more specifically has the structure shown in FIG. 17.
- the first reaction synthesis of precursors
- the reaction temperature may be -20 ° C to 130 ° C, and for example, may be -20 ° C, -10 ° C, or 40 ° C.
- the present invention is not limited thereto, and it should be considered that the present invention can adopt any reaction temperature suitable for the reaction.
- the solvent in step (1) may be N, N-dimethylformamide, dimethyl sulfoxide, tetrahydrofuran, acetonitrile, dichloromethane, chloroform, methanol, ethanol, water, isopropanol, ethylene glycol, ethyl acetate Any one of glycol monomethyl ether, ethylene glycol monoethyl ether, ethylene glycol monobutyl ether, glycerol, acetone, triethanolamine, pyridine, benzene, toluene, xylene, etc., or a mixture of two or more thereof.
- the present invention is not limited thereto, and it should be considered that the present invention can adopt any solvent suitable for the reaction.
- the nitrite in step (1) may be a compound of the general formula MNO 2 , where M is one selected from the group consisting of NH 4 , Li, Na, K, Rb, and Cs, or a mixture of two or more.
- M is one selected from the group consisting of NH 4 , Li, Na, K, Rb, and Cs, or a mixture of two or more.
- it may be sodium nitrite.
- the hydroxylamine compound may be a hydroxylamine salt.
- the hydroxylamine salts may be compounds of the general formula (NH 3 OH) a X b , where X is selected from the group consisting of F, Cl, Br, I, NO 3 , SO 4 , HSO 4 , CO 3 , HCO 3 , One of HCOO, CH 3 COO, CF 3 COO, (COO) 2 and CH 2 (COO) 2 , or a mixture of two or more of them.
- the hydroxylamine compound may be a halogenated salt of hydroxylamine; for example, the hydroxylamine compound may be hydroxylamine hydrochloride.
- the reaction time in steps (2) and (4) may be from 2 hours to 24 hours, and may be, for example, 2 hours, 3 hours, 4 hours, 5 hours, 10 hours, or 24 hours.
- the present invention is not limited thereto, and it should be considered that the present invention can adopt any reaction time suitable for the reaction.
- Example 4 The precursor material of Example 4 is dispersed in a solvent.
- step (6) The solvent used in step (6), the salts, azides added in step (7), reaction temperature and reaction time, and the acid added in step (8) can be referred to in step 2 of the general synthesis method.
- step (9) For the nitration system and reaction temperature added in step (9) and the method for terminating the reaction in step (10), please refer to the reaction conditions described in step 3 of the general synthesis method.
- step (5) For the nitration system and reaction temperature added in step (4) in this section, and the method for terminating the reaction in step (5), refer to the reaction conditions described in step 3 of the general synthesis method.
- FIG. 22 is a chemical structure diagram of one embodiment of a compound of the present invention, and its specific name is: 2,7-oxidized-4,5-diazide- [1,2,3] triazine [ 4,5-d] [1,2,3] triazine has the structure shown in FIG. 1, specifically the structure shown in FIG. 4, and more specifically the structure shown in FIG. 23. .
- Example 7 The specific method for synthesizing the compound of Example 7 is specifically the synthesis using 3,3-diamino-2- (1H-tetrazol-5-yl) propionitrile as the precursor material. Details are as follows:
- step (1) The solvent used in step (1), the salts, azides added in step (2), reaction temperature and reaction time, and the acid added in step (3) can be referred to in step 2 of the general synthesis method.
- FIG. 24 is a chemical structure diagram of one embodiment of a compound of the present invention, and its specific name is: 6-oxidized-8-nitroamino [1,2,5] oxadiazole [3 ', 4' : 5,6] pyrazine [2,3-d] [1,2,3] triazine has the structure shown in FIG. 1, specifically has the structure shown in FIG. 5, and more specifically has The structure shown in FIG. 25.
- the first reaction synthesis of precursors
- the reaction temperature may be -20 ° C to 130 ° C, for example, 40 ° C, 70 ° C, 100 ° C (heating and refluxing), 110 ° C, and 120 ° C.
- the present invention is not limited thereto, and it should be considered that the present invention can adopt any reaction temperature suitable for the reaction.
- the solvent in step (1) or (5) may be N, N-dimethylformamide, dimethylsulfoxide, tetrahydrofuran, acetonitrile, dichloromethane, chloroform, methanol, ethanol, water, isopropanol, ethyl Any one or more of glycol, ethylene glycol monomethyl ether, ethylene glycol monoethyl ether, ethylene glycol monobutyl ether, glycerol, acetone, triethanolamine, pyridine, benzene, toluene, xylene, etc. A mixture of species.
- the present invention is not limited thereto, and it should be considered that the present invention can adopt any solvent suitable for the reaction.
- the present invention is not limited thereto, and it should be considered that the present invention can adopt any proportion of solvents suitable for the reaction.
- the reaction time in step (3) may be 0.5 to 12 hours, for example, may be 0.5 to 4 hours, for example, may be 1 to 4 hours, for example, may be 0.5 hours, 1 hour, 2 hours, or 4 hours.
- the present invention is not limited thereto, and it should be considered that the present invention can adopt any reaction time suitable for the reaction.
- Nitride in step (5) may be a compound of formula R a (N) b wherein R is selected from Li, Na, K, Rb, Cu, Fe, Ag, Au, Pb, Cd, Ni , and One of H, or a mixture of two or more, and select the corresponding a and b.
- the solvent in step (7) may be a solvent insoluble or hardly soluble in ammonia gas flow, or a solvent soluble in concentrated ammonia.
- the solvent may be any one of dichloromethane, chloroform, acetone, benzene, toluene, xylene, etc., or a mixture of two or more kinds;
- the solvent may be any one of methanol, ethanol, water, isopropanol, ethylene glycol, glycerol, acetone, or a mixture of two or more.
- step (7) may use an ammonia gas stream, and the solvent thereof may be dichloromethane.
- the reaction time in step (7) may be 0.5 to 12 hours, for example, 0.5 to 4 hours, for example, 1 to 4 hours, for example, 0.5 hours, 1 hour, 2 hours, or 4 hours.
- the present invention is not limited thereto, and it should be considered that the present invention can adopt any reaction time suitable for the reaction.
- step (9), the salts, azides added in step (10), reaction temperature and reaction time, and the acid added in step (11) can be referred to in step 2 of the general synthesis method.
- Example 8 The first intermediate product of Example 8 is slowly added to the nitration system. After the addition, the temperature of the reaction system is controlled to keep the nitration reaction at a certain temperature for a certain time.
- step (12) For the nitration system and reaction temperature added in step (12) and the method for terminating the reaction in step (13), please refer to the reaction conditions described in step 3 of the general synthesis method.
- step (14) of this section The solvent used in step (14) of this section, the concentration of concentrated ammonia water used in step (15), the dosage ratio and the reaction time, the nitration system and reaction temperature added in step (17), and the way to stop the reaction in step (18)
- the reaction conditions described in Step 4 of General Synthesis please refer to the reaction conditions described in Step 4 of General Synthesis.
- This solid was dissolved in 70 mL of dichlorosulfoxide, 5 mL of N, N-dimethylformamide was added dropwise, the reaction was heated to reflux for 4 hours, poured into 200 g of ice water, and filtered to obtain a solid. Then, it was dissolved in 50 mL of acetone, 3.9 g of sodium nitride was added, and the temperature was raised to reflux for 1 hour, followed by cooling, filtering, and spinning the mother liquor. The obtained solid was dissolved in 60 mL of dichloromethane, and an ammonia gas flow (flow rate of 5 mL / min) was passed. The reaction was refluxed for 4 h, cooled, filtered, and the mother liquor was spin-dried.
- the obtained pink powder was added to 180 mL of acetonitrile with 13.3 g of tetrafluoroboronitrate, and then reacted at 0 ° C for 3 hours.
- the reaction solution was poured into 50 g of ice water, stirred for 0.5 hours, filtered, and 20 mL of ice water was used. After washing, it was washed with 10 mL of petroleum ether. After drying, a pale yellow powdery solid was obtained.
- the obtained solid was dissolved in 90 mL of methanol, 900 mL of concentrated ammonia water was added, and the mixture was reacted at reflux for 2 hours, and cooled and filtered to obtain an orange powdery solid.
- the obtained solid was added to 60 mL of fuming nitric acid, stirred for 2 hours, poured into 200 g of ice, filtered, and washed with water to obtain 7.7 g of a yellow solid.
- FIG. 26 is a chemical structure diagram of one embodiment of a compound of the present invention, and its specific name is: 2-oxo-4-chloro-9H- [1,2,4] triazole [5'-1 ': 3,4] [1,2,4] triazine [6,1-d] [1,2,3,5] tetrazine has the structure shown in FIG. 1, and specifically has the structure shown in FIG. 5. The structure shown has a structure shown in FIG. 27 more specifically.
- the first reaction synthesis of precursors
- the solvent in step (1) may be N, N-dimethylformamide, dimethyl sulfoxide, tetrahydrofuran, acetonitrile, dichloromethane, chloroform, methanol, ethanol, water, isopropanol, ethylene glycol, ethyl acetate Any one of glycol monomethyl ether, ethylene glycol monoethyl ether, ethylene glycol monobutyl ether, glycerol, acetone, triethanolamine, pyridine, benzene, toluene, xylene, etc., or a mixture of two or more thereof. By way of example, it may be acetonitrile.
- the present invention is not limited thereto, and it should be considered that the present invention can adopt any solvent suitable for the reaction.
- the present invention is not limited thereto, and it should be considered that the present invention can adopt any proportion of solvents suitable for the reaction.
- the nitroacetonitrile compound may be a nitroacetonitrile salt.
- the nitroacetonitrile compound may be a compound of the general formula M a (CH (NO 2 ) CN) b , where M is selected from One of Li, Na, K, Rb, Cs, or a mixture of two or more.
- the nitroacetonitrile compound may be a nitroacetonitrile sodium salt.
- the second step reaction synthesis of the first intermediate substance
- Example 9 The precursor material of Example 9 is dispersed in a solvent.
- Example 3 The first intermediate product of Example 3 is slowly added to the nitration system. After the addition, the temperature of the reaction system is controlled to keep the nitration reaction at a certain temperature for a certain time.
- step (7) For the nitration system and reaction temperature added in step (7) and the method for terminating the reaction in step (8), please refer to the reaction conditions described in step 3 of the general synthesis method.
- step (9) in this section the chlorine gas flow rate, reaction temperature, and reaction time used in step (10), refer to the reaction conditions described in step 4A of the general synthesis method.
- the obtained solid was dispersed in 100 mL of water with 3.6 g of ammonium nitrate and 15.1 g of sodium azide, and then reacted at 75 ° C. for 7 hours. After cooling to room temperature, hydrochloric acid was added dropwise thereto until the solution pH was 3.1, and then filtered. The solid was washed with 5 mL of ice water, and the solid was dried to obtain 11.3 g of a brown powder.
- the present invention is not limited to this.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
本发明公开了一种化合物,其结构包括摘要附图中所示的结构。本发明又公开了此化合物的制备方法、制备方法中所用的前体物质,包括此化合物的炸药以及此化合物在炸药领域上的应用,特别是在绿色环保型起爆药上的应用。本发明所公开的化合物,能够提供一种绿色环保型的起爆药,满足军工、火工、民爆工程等众多领域亟需绿色环保型起爆药的问题。而且,本发明所公开的化合物,其制备方法简单,只需简单的反应步骤及条件即可制得;反应过程及使用过程中无污染;化合物结构中不需要金属,绿色可靠;稳定性佳,撞击感度、摩擦感度高;起爆能力高,并具备合适使用的极限起爆药量。
Description
本发明涉及化学领域,更具体涉及一种化合物、其制备方法及其中的前体物质、其应用和具备该化合物的炸药。
起爆药作为火工品和武器中的重要组成部分,对它的研究一直是一个重要的科学课题。但相对于现代火工品和武器的发展而言,它的研究长期以来进展缓慢。一百多年来,叠氮化铅、斯蒂芬酸铅仍然是主要应用于军用火工品的两个起爆药品种。随着武器弹药用火工品安全性的要求越来越高,叠氮化铅、斯蒂芬酸铅在性能上的缺陷也越来越明显。
叠氮化铅机械感度过高,无约束条件下受轻微刺激直接产生爆轰,火焰针刺感度也较差。斯蒂芬酸铅对静电极为敏感,静电放电起爆能量仅10
-4J。这些常常导致火工品及药剂勤务处理和使用中发生重大事故,也阻碍了先进火工品的发展。
从环保角度来看,火工品当前面临没有绿色环保型起爆药可选的困境。据统计,在现有已生产定性的二十余种单质起爆药中,只有二硝基重氮酚(DDNP)、四氮烯两种起爆药不含金属成分,二硝基苯并氧化呋咱钾(KDNBF)含有轻金属钾。其它起爆药均含汞、铅、钡、镉等有毒金属成分。而DDNP由于极限药量大而仅限于在民爆工程雷管中应用,且合成过程中产生大量废水污染环境。四氮烯仅作为机械感度敏化剂使用,用量仅为配方的5%,不能单独使用。二硝基苯并氧化呋咱钾由于起爆能力、感度性能、流散性等多方面的缺陷而极少量地应用于动力源火工品的电点火头。
随着现代战争环境的恶劣和火工品新起爆技术的发展,现役起爆药难以满足先进火工品技术指标要求。特别是精确打击与速射武器弹药引信火工品、耐高过载火工品、激光、半导体桥、冲击片火工品几乎没有适用性的含能材料可选。
因此,在军工、火工、民爆工程等众多领域中,亟需一种绿色环保型的起爆药。
发明内容
为了克服现有技术的不足,本发明所解决的技术问题是:提供一种绿色环保型的起爆药,满足军工、火工、民爆工程等众多领域亟需绿色环保型起爆药的问题。
为解决上述技术问题,本发明所采用的技术方案如下:
一种化合物,其结构包括图1所示的结构。
优选地是,此化合物为稠环化合物,稠环化合物至少包括图1所示的结构及多元环。更优选地,图1所示的结构中,A
1及A
2选自碳原子、氮原子、氧原子、磷原子及硫原子中的一种或多种。其中优选地,图1所示的结构中,A
1及A
2选自碳原子及氮原子中的一种或二种。更优选地,图1所示的结构中,A
1及A
2为碳原子。同样优选地,图1所示的结构中,A
1及A
2其中一者为氮原子,另一者为碳原子。更优选地,图1所示的结构中,A
1为氮原子,A
2为碳原子。
优选地,此化合物为包括二个至三个环的稠环化合物。其中优选地,化合物为包括二个环的稠环化合物。同样优选地,化合物为包括三个环的稠环化合物。优选地,此稠环化合物为图1所示的结构与此多元环的稠环化合物。优选地,此稠环化合物中,图1所示的结构及此多元环共用两个碳原子。
其中优选地,多元环由碳原子所组成。又优选地,多元环为选自三元环、四元环、五元环、六元环、七元环及八元环的其中一种或多种。其中优选地,多元环为选自五元环、六元环及七元环的其中一种或多种。更优选地,多元环为五元环及六元环的其中一种或多种。更优选地,多元环为五元环。同样优选地,多元环为六元环。另一方面优选地,此多元环为多个多元环。其中优选地,多个多元环包括五元环及六元环。更优选地,多个多元环为五元环及六元环。
优选地,多元环为选自茂环、苯环及
 环的其中一种。其中优选地,多元环为苯环。或优选地,此多元环为杂环,杂环的杂原子包括选自氮原子、氧原子、硅原子、磷原子、硫原子、砷原子、硒原子及碲原子的其中一种。优选地,杂原子包括选自氮原子、氧原子、硅原子、磷原子、硫原子、砷原子、硒原子及碲原子的其中二种。更优选地,杂原子为氮原子、氧原子、硅原子、磷原子、硫原子、砷原子、硒原子或碲原子中的其中一种。更优选地,杂原子为氮原子或氧原子。更优选地,杂原子为选自氮原子及氧原子的其中一种。更优选地,杂原子为氮原子。同样优选地,杂原子为氧原子。优选地,杂环包括1个、2个、3个、4个或5个杂原子。其中优选地,杂环包括1个、2个、3个、4个或5个氮原子。同样优选地,杂环包括1个氧原子。其中优选地,此多元环为选自呋喃、吡咯、噻吩、咪唑、吡唑、噁唑、噻唑、吡啶、吡嗪、嘧啶、哒嗪、呋咱、三唑类、四唑类、三嗪类及四嗪类的其中一种。更优选地,多元环为选自咪唑、吡唑、吡啶、吡嗪、嘧啶、哒嗪、呋咱、三唑类、四唑类、三嗪类及四嗪类的其中一种。更优选地,多元环为选自咪唑、吡唑、吡啶、吡嗪、呋咱、三唑类、四唑类、三嗪类及四嗪类的其中一种。更优选地,多元环为选自咪唑、吡唑、吡啶及吡嗪的其中一种。更优选地,多元环为选自咪唑及吡唑的其中一种。更优选 地,多元环为吡唑。或优选地,多元环为咪唑。同样优选地,多元环为选自哒嗪、吡啶及吡嗪的其中一种。更优选地,多元环为哒嗪。或优选地,多元环为吡啶。或优选地,多元环为吡嗪。同样优选地,多元环为三唑类。更优选地,多元环为1,2,4-三唑。同样优选地,多元环为三嗪类。更优选地,多元环为1,2,3-三嗪。或优选地,多元环为1,2,4-三嗪。同样优选地,多元环为选自呋喃、呋咱及噁唑的其中一种。更优选地,多元环为呋喃。或优选地,多元环为呋咱。
环的其中一种。其中优选地,多元环为苯环。或优选地,此多元环为杂环,杂环的杂原子包括选自氮原子、氧原子、硅原子、磷原子、硫原子、砷原子、硒原子及碲原子的其中一种。优选地,杂原子包括选自氮原子、氧原子、硅原子、磷原子、硫原子、砷原子、硒原子及碲原子的其中二种。更优选地,杂原子为氮原子、氧原子、硅原子、磷原子、硫原子、砷原子、硒原子或碲原子中的其中一种。更优选地,杂原子为氮原子或氧原子。更优选地,杂原子为选自氮原子及氧原子的其中一种。更优选地,杂原子为氮原子。同样优选地,杂原子为氧原子。优选地,杂环包括1个、2个、3个、4个或5个杂原子。其中优选地,杂环包括1个、2个、3个、4个或5个氮原子。同样优选地,杂环包括1个氧原子。其中优选地,此多元环为选自呋喃、吡咯、噻吩、咪唑、吡唑、噁唑、噻唑、吡啶、吡嗪、嘧啶、哒嗪、呋咱、三唑类、四唑类、三嗪类及四嗪类的其中一种。更优选地,多元环为选自咪唑、吡唑、吡啶、吡嗪、嘧啶、哒嗪、呋咱、三唑类、四唑类、三嗪类及四嗪类的其中一种。更优选地,多元环为选自咪唑、吡唑、吡啶、吡嗪、呋咱、三唑类、四唑类、三嗪类及四嗪类的其中一种。更优选地,多元环为选自咪唑、吡唑、吡啶及吡嗪的其中一种。更优选地,多元环为选自咪唑及吡唑的其中一种。更优选 地,多元环为吡唑。或优选地,多元环为咪唑。同样优选地,多元环为选自哒嗪、吡啶及吡嗪的其中一种。更优选地,多元环为哒嗪。或优选地,多元环为吡啶。或优选地,多元环为吡嗪。同样优选地,多元环为三唑类。更优选地,多元环为1,2,4-三唑。同样优选地,多元环为三嗪类。更优选地,多元环为1,2,3-三嗪。或优选地,多元环为1,2,4-三嗪。同样优选地,多元环为选自呋喃、呋咱及噁唑的其中一种。更优选地,多元环为呋喃。或优选地,多元环为呋咱。
优选地,此多元环上包括取代基。更优选地,取代基为选自:饱和烃烃基,包括烷烃基;官能团化饱和烃烃基,包括硝基烷烃基、偕二硝基烷烃基、硝仿基烷烃基、卤代烷烃基;醇基;醚基、硫醚基;羧酸基、酯基、硝酸酯基、磺酸基、磺酸酯基;酮基、酰胺基、酰肼基;醛基;不饱和脂肪族烃基,包括烯烃基、炔烃基、卤代烯烃基、硝基炔烃基;不饱和芳香烃烃基,包括单环芳烃基、多环芳烃基、稠环芳烃基和官能团化的单环芳烃基、官能团化的多环芳烃基、官能团化的稠环芳烃基;杂环芳香烃基,包括噻吩、吡啶、呋喃、唑类环、嗪类环、噁唑类环及其对应的单官能团化或多官能团化基团;杂环非芳香烃基,包括氮杂桥环烃、氧杂桥环烃;卤原子;氰基、肟基;氨基、亚氨基、偶氮基、氧化偶氮基、叠氮基、肼基;羟基、羟氨基、烷氧基;硝氨基;及硝基及亚硝基的其中一种。其中优选地,取代基为选自硝基、亚硝基、硝氨基、羟氨基、氰基、肟基、氨基、亚氨基、偶氮基、氧化偶氮基、叠氮基、肼基、氮杂桥环烃、吡啶唑类环、嗪类环、硝基烷烃基、偕二硝基烷烃基、硝仿基烷烃基及硝基炔烃基的其中一种。同样优选地,取代基为选自羟基、羟胺基、烷氧基、醇基、醚基、硫醚基、羧酸基、酯基、硝酸酯基、磺酸基、磺酸酯基、酮基、酰胺基、酰肼基、醛基、硝基炔烃基、硝基烷烃基、偕二硝基烷烃基、硝仿基烷烃基、呋喃、噁唑类环及氧杂桥环烃的其中一种。更优选地,取代基为选自硝基、亚硝基、硝氨基、硝基烷烃基、偕二硝基烷烃基、硝仿基烷烃基及硝基炔烃基的其中一种。更优选地,取代基为选自硝基及亚硝基的其中一种。更优选地,取代基为硝基。同样优选地,取代基为选自氨基、亚氨基、硝氨基、羟氨基及肼基的其中一种。更优选地,取代基为选自氨基及亚氨基的其中一种。更优选地,取代基为氨基。其中优选地,取代基为硝氨基。同样优选地,取代基为选自叠氮基、偶氮基、氧化偶氮基及肼基的其中一种。更优选地,取代基为叠氮基。同样优选地,取代基为卤原子。更优选地,取代基为选自氟、氯、溴及碘的其中一种。更优选地,取代基为氯。另一方面优选地,取代基位于多元环的碳原子上。
优选地,图1所示的结构中的取代基R
1为选自氢、氨基、硝基、硝氨基、叠氮基、氟、氯、溴、碘、C
nH
m(NO
2)
p、图2所示的结构中的其中一种,其中m+p=2n+1。更优选地,图1所示的结构中的取代基R
1为选自氨基、硝基、硝氨基、叠氮基中的其中一种。更优选地,R
1为叠氮基。更优选地,R
1为氨基。更优选地,R
1为硝基。更优选地,R
1为硝氨基。同样优选地,R
1为选自氟、氯、溴、碘中的其中一种。更优选地,R
1为氯。另外优选地,R
1为选自硝基、硝氨基或C
nH
m(NO
2)
p中的其中一种,其中n、p为正整数。更优选地,R
1为选自硝基、硝氨基或C
nH
m(NO
2)
p中的其中一种,其中m、n、p为正整数。更优选地,R
1为选自硝基、硝氨基或C
nH
m(NO
2)
p中的其中一种,其中m、n、p为正整数,且n=1、2、3或4。
另一方面优选地,图2所示的结构中,A
1至A
5选自碳原子、氮原子、氧原子、磷原子及硫原子中的一种或多种。更优选地,图2所示的结构中,A
1至A
5选自碳原子、氮原子及氧原子中的一种、二种或三种。更优选地,图2所示的结构中,A
1至A
5选自碳原子及氮原子中的一种或二种。更优选地,图2所示的结构中,A
1至A
5为碳原子。另一方面优选地,图2所示的结构中,A
1至A
5上的取代基个数为0、1、2、3或4个。并且优选地,R
2为选自氢、氨基、硝基、硝氨基、叠氮基、氟、氯、溴、碘、C
nH
m(NO
2)
p及图2所示的结构中的其中一或多个,其中m+p=2n+1。另外优选地,R
2为选自硝基、硝氨基或C
nH
m(NO
2)
p中的其中一种,其中n、p为正整数。更优选地,R
2为选自硝基、硝氨基或C
nH
m(NO
2)
p中的其中一种,其中m、n、p为正整数。更优选地,R
2为选自硝基、硝氨基或C
nH
m(NO
2)
p中的其中一种,其中m、n、p为正整数,且n=1、2、3或4。
优选地,此稠环化合物至少包括图3所示的结构,其中A
1至A
5选自碳原子、氮原子、氧原子、磷原子及硫原子中的一种或多种。更优选地,图3所示的结构中,A
1至A
5选自碳原子、氮原子及氧原子中的一种、二种或三种。更优选地,图3所示的结构中,A
1至A
5选自碳原子及氮原子中的一种或二种。另优选地,图3所示的结构中,A
1至A
2为碳原子。与此同时优选地,图3所示的结构中,A
3至A
5选自碳原子、氮原子及氧原子中的一种、二种或三种。更优选地,图3所示的结构中,A
3至A
5选自碳原子及氮原子中的一种或二种。其中优选地,图3所示的结构中,A
1至A
5包括0、1、2、3、4或5个氮原子,其余为碳原子。其中优选地,图3所示的结构中,A
3至A
5包括0、1、2或3个氮原子,其他的是碳原子。其中优选地,图3所示的结构中,A
1至A
5组成一个呋咱环。更优选地,图3所示的结构中,A
1至A
2为碳原子,A
3及A
5为氮原子,且A
4为氧原子。更优选地,图3所示的结构中,A
1至A
2为碳原子,A
3及A
5为氮原子,A
4为氧原子,且A
3有一个氧取代基,A
3与氧取代基形成配位键。同样优选地,图3所示的结构中,A
1至A
5组成一个二唑环。其中优选地,图3所示的结构中,A
1至A
5组成一个吡唑环。更优选地,图3所示的结构中,A
1、A
2及A
5为碳原子,且A
3及A
4为氮原子。其中优选地,图3所示的结构中,A
5上有硝基取代基。特别优选地,图3所示的结构中,A
1、A
2及A
5为碳原子,A
3及A
4为氮原子,且图3所示的结构中,A
5上有硝基取代基。同样优选地,图3所示的结构中,A
1至A
5组成一个咪唑环。其中优选地,图3所示的结构中,A
1、A
2及A
4为碳原子,且A
3及A
5为氮原子。另一方面优选地,图3所示的结构中,A
1至A
5上的取代基个数为0、1、2、3或4个。并且优选地,R
3为选自氢、氨基、硝基、硝氨基、叠氮基、氟、氯、溴、碘、氧、C
nH
m(NO
2)
p及图2所示的结构中的其中一或多个,其中m+p=2n+1。另外优选地,R
3为选自硝基、硝氨基或C
nH
m(NO
2)
p中的其中一种,其中n、p为正整数。更优选地,R
3为选自硝基、硝氨基或C
nH
m(NO
2)
p中的其中一种,其中m、n、p为正整数。更优选地,R
3为选自硝基、硝氨基或C
nH
m(NO
2)
p中的其中一种,其中m、n、p为正整数,且n=1、2、3或4。
优选地,此稠环化合物至少包括图4所示的结构,其中A
1至A
6选自碳原子、氮原子、氧原子、磷原子及硫原子中的一种或多种。更优选地,图4所示的结构中,A
1至A
6选自碳原子及氮原子中的一种或二种。另优选地,图4所示的结构中,A
1至A
2为碳原子。与此同时优选地,图4所示的结构中,A
3至A
6选自 碳原子及氮原子中的一种或二种。其中优选地,图4所示的结构中,A
1至A
6包括0、1、2、3、4、5或6个氮原子,其余为碳原子。其中优选地,图4所示的结构中,A
3至A
6包括0、1、2、3或4个氮原子,其余为碳原子。特别优选地,图4所示的结构中,A
1至A
6为碳原子。同样优选地,图4所示的结构中,A
4、A
5为氮原子。更优选地,图4所示的结构中,A
1至A
3、A
6为碳原子,且A
4、A
5为氮原子。同样优选地,图4所示的结构中,A
3至A
5为氮原子。更优选地,图4所示的结构中,A
1至A
2、A
6为碳原子,且A
3至A
5为氮原子。特别优选地,图4所示的结构中,A
1至A
2、A
6为碳原子,A
3至A
5为氮原子,A
4有一个氧取代基,A
4与氧取代基形成配位键,且A
6有一个叠氮取代基。另一方面优选地,图4所示的结构中,A
1至A
6上的取代基个数合计为0、1、2、3、4或5个。并且优选地,R
4为选自氢、氨基、硝基、硝氨基、叠氮基、氟、氯、溴、碘、氧、C
nH
m(NO
2)
p及图2所示的结构中的其中一或多个,其中m+p=2n+1。另外优选地,R
4为选自硝基、硝氨基或C
nH
m(NO
2)
p中的其中一种,其中n、p为正整数。更优选地,R
4为选自硝基、硝氨基或C
nH
m(NO
2)
p中的其中一种,其中m、n、p为正整数。更优选地,R
4为选自硝基、硝氨基或C
nH
m(NO
2)
p中的其中一种,其中m、n、p为正整数,且n=1、2、3或4。
优选地,稠环化合物至少包括图5所示的结构,其中A
1至A
9选自碳原子、氮原子、氧原子、磷原子及硫原子中的一种或多种。更优选地,图5所示的结构中,A
1至A
9选自碳原子、氮原子及氧原子中的一种、二种或三种。更优选地,图5所示的结构中,A
1至A
9选自碳原子及氮原子中的一种或二种。另优选地,图5所示的结构中,A
1至A
2为碳原子。与此同时优选地,图5所示的结构中,A
3至A
9选自碳原子、氮原子及氧原子中的一种、二种或三种。更优选地,图5所示的结构中,A
3至A
9选自碳原子及氮原子中的一种或二种。其中优选地,图5所示的结构中,A
1至A
9包括0、1、2、3、4、5、6、7、8或9个氮原子,其余为碳原子。其中优选地,图5所示的结构中,A
1至A
6包括0、1、2、3、4、5或6个氮原子,其余为碳原子。同样优选地,图5所示的结构中,A
4、A
5、A
7至A
9包括0、1、2、3、4或5个氮原子,其余为碳原子。其中优选地,图5所示的结构中,A
4、A
5、A
7至A
9组成一个呋咱环。同样优选地,图5所示的结构中,A
4、A
5、A
7至A
9组成一个三唑环。更优选地,图5所示的结构中,A
4、A
5、A
7至A
9组成一个1,2,4-三唑环。同样优选地,图5所示的结构中,A
1至A
6组成一个二嗪环。更优选地,图5所示的结构中,A
1至A
6组成一个吡嗪环。同样优选地,图5所示的结构中,A
1至A
6组成一个三嗪环。更优选地,图5所示的结构中,A
1至A
6组成一个1,2,4-三嗪环。更优选地,图5所示的结构中,A
4、A
5、A
7至A
9组成一个呋咱环,且A
1至A
6组成一个二嗪环。特别优选地,图5所示的结构中,A
1、A
2、A
4、A
5为碳原子,A
3、A
6、A
7、A
9为氮原子,且A
8为氧原子。同样优选地,图5所示的结构中,A
4、A
5、A
7至A
9组成一个三唑环,且A
1至A
6组成一个三嗪环。更优选地,图5所示的结构中,A
4、A
5、A
7至A
9组成一个1,2,4-三唑环,且A
1至A
6组成一个1,2,4-三嗪环。特别优选地,图5所示的结构中,A
2、A
3、A
5、A
8为碳原子,且A
1、A
4、A
6、A
7、A
9为氮原子。另一方面优选地,图5所示的结构中,A
1至A
6上的取代基个数合计为0、1、2、3、4或5个。更优选地,图5所示的结构中,A
4、A
5、A
7至A
9上的取代基个数合计为0、1、2、3或4个。并且优选地,R
5为选自氢、氨基、硝基、硝氨基、叠氮基、氟、氯、溴、碘、氧、C
nH
m(NO
2)
p及图2所示的结构中的其中一或多个,其中m+p=2n+1。另外优选地,R
5为选自硝基、硝氨基或C
nH
m(NO
2)
p中的其中一种,其中n、p为正整数。更优选地,R
5为选自硝基、硝氨基或C
nH
m(NO
2)
p中的其中一种,其中m、n、p为正整数。更优选地,R
5为选自硝基、硝氨基或C
nH
m(NO
2)
p中的其中一种,其中m、n、p为正整数,且n=1、2、3或4。
优选地,此稠环化合物包括图1所示的结构及3-硝基吡唑。更优选地,此化合物的结构包括图8所示的结构。更优选地,此化合物的结构包括图7所示的结构。更优选地,此化合物的结构为图7所示的结构。更优选地,图7所示的结构的分析测定结果如下:DSC(160℃,50℃–250℃,5℃·min
-1);
1H NMR(400MHz,DMSO-d
6,25℃)δ(ppm):14.51(brs,1H,NH);
13C NMR(100MHz,DMSO-d
6,25℃)δ(ppm):157.95,157.31,148.07,90.03;IR(KBr,γ/cm
-1):3414(m);3231(m);2154(s);1600(s);1543(s);1403(s);1384(s);1313(m);1209(m);1131(m);元素分析C
4HN
9O
3(%):实测值(计算值)C:21.62(21.53),H:0.47(0.45),N:56.61(56.50),O:21.22(21.51)。
或优选地,此化合物的结构包括图13所示的结构;或优选地,此化合物的结构包括图15所示的结构;或优选地,此化合物的结构包括图17所示的结构;或优选地,此化合物的结构包括图19所示的结构;或优选地,此化合物的结构包括图21所示的结构;或优选地,此化合物的结构包括图23所示的结构;或优选地,此化合物的结构包括图25所示的结构;或优选地,此化合物的结构包括图27所示的结构。其中优选地,上述图所示的结构中的取代基R
1为选自氢、氨基、硝基、硝氨基、叠氮基、氟、氯、溴、碘、C
nH
m(NO
2)
p、图2所示的结构中的其中一种,其中m+p=2n+1。更优选地,上述图所示的结构中的R
1为选自氨基、硝基、硝氨基、叠氮基中的其中一种。更优选地,上述图所示的结构中的R
1为叠氮基。更优选地,上述图所示的结构中的R
1为氨基。更优选地,上述图所示的结构中的R
1为硝基。更优选地,上述图所示的结构中的R
1为硝氨基。同样优选地,上述图所示的结构中的R
1为选自氟、氯、溴、碘中的其中一种。更优选地,上述图所示的结构中的R
1为氯。另外优选地,上述图所示的结构中的R
1为选自硝基、硝氨基或C
nH
m(NO
2)
p中的其中一种,其中n、p为正整数。更优选地,上述图所示的结构中的R
1为选自硝基、硝氨基或C
nH
m(NO
2)
p中的其中一种,其中m、n、p为正整数。更优选地,上述图所示的结构中的R
1为选自硝基、硝氨基或C
nH
m(NO
2)
p中的其中一种,其中m、n、p为正整数,且n=1、2、3或4。另外优选地,优选地,图8所示的结构中的R
3为选自氢、氨基、硝基、硝氨基、叠氮基、氟、氯、溴、碘、氧、C
nH
m(NO
2)
p及所述式(B)结构中的其中一或多个,其中m+p=2n+1。更优选地,图8所示的结构中的所述R
3为硝基。
或优选地,此稠环化合物包括图1所示的结构及2-氧化呋咱。或优选地,此稠环化合物包括二个图1所示的结构;更优选地,此稠环化合物包括二个相同的图1所示的结构;更优选地,此稠环化合物包括二个图1所示的结构,二个图1所示的结构共用A
1及A
2。
或优选地,此化合物的结构包括图12、图14、图16、图18、图20、图22、图24或图26所示的结构;更优选地,此化合物的结构为图12、图14、图16、图18、图20、图22、图24或图26所示的结构。
本发明更公开了此化合物在其制备方法中的一种前体物质。具体来说,此前体物质是包括图6所示的结构的一种化合物。
优选地,图6所示的结构中,A
10及A
11选自碳原子、氮原子、氧原子、磷原子及硫原子中的一种或多种。更优选地,图6所示的结构中,A
10及A
11选自碳原子及氮原子中的一种或二种。更优选地,图6所示的结构中,A
10及A
11为碳原子。同样优选地,图6所示的结构中,A
10及A
11的其中一者为氮原子,另一者为碳原子。
优选地是,此化合物包括多元环。更优选地,多元环为选自三元环、四元环、五元环、六元环、七元环及八元环的其中一种或多种。更优选地,多元环为选自五元环、六元环及七元环的其中一种或多种。更优选地,多元环为五元环及六元环的其中一种或多种。更优选地,多元环为五元环。同样优选地,多元环为六元环。
优选地,多元环由碳原子所组成。更优选地,多元环为选自茂环、苯环及
 环的其中一种。更优选地,多元环为苯环。或优选地,多元环为杂环,杂环的杂原子包括选自氮原子、氧原子、硅原子、磷原子、硫原子、砷原子、硒原子及碲原子的其中一种。更优选地,杂原子包括选自氮原子、氧原子、硅原子、磷原子、硫原子、砷原子、硒原子及碲原子的其中二种。或优选地,杂原子为氮原子、氧原子、硅原子、磷原子、硫原子、砷原子、硒原子或碲原子中的其中一种。更优选地,杂原子为氮原子或氧原子。优选地,杂原子为选自氮原子及氧原子的其中一种。更优选地,杂原子为氮原子。其中优选地,多元环为选自呋喃、吡咯、噻吩、咪唑、吡唑、噁唑、噻唑、吡啶、吡嗪、嘧啶、哒嗪、呋咱、三唑类、四唑类、三嗪类及四嗪类的其中一种。更优选地,多元环为选自咪唑、吡唑、吡啶、吡嗪、嘧啶、哒嗪、呋咱、三唑类、四唑类、三嗪类及四嗪类的其中一种。更优选地,多元环为选自咪唑、吡唑、吡啶、吡嗪、呋咱、三唑类、四唑类、三嗪类及四嗪类的其中一种。更优选地,多元环为选自咪唑、吡唑、吡啶及吡嗪的其中一种。更优选地,多元环为选自咪唑及吡唑的其中一种。更优选地,多元环为吡唑。或优选地,多元环为选自哒嗪、吡啶及吡嗪的其中一种。更优选地,多元环为哒嗪。或优选地,多元环为四唑类。
环的其中一种。更优选地,多元环为苯环。或优选地,多元环为杂环,杂环的杂原子包括选自氮原子、氧原子、硅原子、磷原子、硫原子、砷原子、硒原子及碲原子的其中一种。更优选地,杂原子包括选自氮原子、氧原子、硅原子、磷原子、硫原子、砷原子、硒原子及碲原子的其中二种。或优选地,杂原子为氮原子、氧原子、硅原子、磷原子、硫原子、砷原子、硒原子或碲原子中的其中一种。更优选地,杂原子为氮原子或氧原子。优选地,杂原子为选自氮原子及氧原子的其中一种。更优选地,杂原子为氮原子。其中优选地,多元环为选自呋喃、吡咯、噻吩、咪唑、吡唑、噁唑、噻唑、吡啶、吡嗪、嘧啶、哒嗪、呋咱、三唑类、四唑类、三嗪类及四嗪类的其中一种。更优选地,多元环为选自咪唑、吡唑、吡啶、吡嗪、嘧啶、哒嗪、呋咱、三唑类、四唑类、三嗪类及四嗪类的其中一种。更优选地,多元环为选自咪唑、吡唑、吡啶、吡嗪、呋咱、三唑类、四唑类、三嗪类及四嗪类的其中一种。更优选地,多元环为选自咪唑、吡唑、吡啶及吡嗪的其中一种。更优选地,多元环为选自咪唑及吡唑的其中一种。更优选地,多元环为吡唑。或优选地,多元环为选自哒嗪、吡啶及吡嗪的其中一种。更优选地,多元环为哒嗪。或优选地,多元环为四唑类。
优选地,多元环上包括取代基。更优选地,取代基为选自:饱和烃烃基,包括烷烃基;官能团化饱和烃烃基,包括硝基烷烃基、偕二硝基烷烃基、硝仿基烷烃基、卤代烷烃基;醇基;醚基、硫醚基;羧酸基、酯基、硝酸酯基、磺酸基、磺酸酯基;酮基、酰胺基、酰肼基;醛基;不饱和脂肪族烃基,包括烯烃基、炔烃基、卤代烯烃基、硝基炔烃基;不饱和芳香烃烃基,包括单环芳烃基、多环芳烃基、稠环芳烃基和官能团化的单环芳烃基、官能团化的多环芳烃基、官能团化的稠环芳烃基;杂环芳香烃基,包括噻吩、吡啶、呋喃、唑类环、嗪类环、噁唑类环及其对应的单官能团化或多官能团化基团;杂环非芳香烃基,包括氮杂桥环烃、氧杂桥环烃;卤原子;氰基、肟基;氨基、亚氨基、偶氮基、氧化偶氮基、叠氮基、肼基;羟基、羟氨基、烷氧基;硝氨基;及硝基及亚硝基的其中一种。更优选地,取代基为选自硝基、亚硝基、硝氨基、羟氨基、氰基、肟基、氨基、亚氨基、偶氮基、氧化偶氮基、叠氮基、肼基、氮杂桥环烃、吡啶唑类环、嗪类环、硝基烷烃基、偕二硝基烷烃基、硝仿基烷烃基及硝基炔烃基的其中一种。更优选地,取代基为选自氰基及肟基的其中一种。或优选地,取代基为选自氨基、亚氨基、硝氨基、羟氨基及肼基的其中一种。更优选地,取代基为选自氨基及亚氨基的其中一种。更优选地,取代基为氨基。另一方面优选地,取代基位于多元环的碳原子上。
优选地,化合物包括3-氨基-4-氰基吡唑的结构。同样优选地,化合物包括4-氨基-5-氰基咪唑的结构。同样优选地,化合物包括1-氨基-2-氰基苯的结构。同样优选地,化合物包括4-氨基-5-氰基哒嗪的结构。同样优选地,化合物包括3,3-二氨基-2-(1H-四唑-5-基)丙腈的结构。特别优选地,化合物为3-氨基-4-氰基吡唑。同样优选地,化合物为4-氨基-5-氰基咪唑。同样优选地,化合物为1-氨基-2-氰基苯。同样优选地,化合物为4-氨基-5-氰基哒嗪。同样优选地,化合物为3,3-二氨基-2-(1H-四唑-5-基)丙腈。
本发明更公开了一种化合物的制备方法,包括:将前体物质(即前述包括图6所示的结构的化合物)分散于溶剂中;加入盐类及叠氮化物,在第一反应温度下反应第一反应时间;加入酸;进行纯化,得到中间产物;将中间产物加入硝化体系当中,在第二温度下硝化反应第二反应时间;终止硝化反应;进行纯化。
优选地是,此包括图6所示的结构的化合物为上述优选的前体物质化合物。另一方面优选地,中间产物为3-氨基-4-[1H-四氮唑]-1H-吡唑。优选地是,此化合物制备方法为本发明所公开的,包括图1所示的结构的化合物的制备方法。更优选地是,此化合物制备方法为本发明所公开的,上述优选的包括图1所示的结构的化合物的制备方法。
优选地,溶剂为N,N-二甲基甲酰胺、二甲基亚砜、四氢呋喃、乙腈、二氯甲烷、氯仿、甲醇、乙醇、水、异丙醇、乙二醇、乙二醇单甲醚、乙二醇单乙醚、乙二醇单丁醚、甘油、丙酮、三乙醇胺、吡啶、苯、甲苯、二甲苯等中的其中一种,或二种或多种的混合物。其中优选地,溶剂的用量体积与包括图6所示的结构的化合物质量的比值为2mL/g至2mL/mg。
优选地,盐类为通式M
aX
b的化合物,其中M为Fe、Co、Ni、Mn、Zn、Cd、Au、Ag、Cu、Cr、Al、V、NH
4、(CH
3)
2NH
2、(CH
3)
3NH、(CH
3CH
2)
2NH
2及(CH
3CH
2)
3NH的其中一种,或二种或多种的混合,而X为F、Cl、Br、I、NO
3、SO
4、HSO
4、CO
3、HCO
3、HCOO、CH
3COO、CF
3COO、(COO)
2及CH
2(COO)
2的 其中一种,或二种或多种的混合。其中优选地,盐类与包括图6所示的结构的化合物的摩尔比为1:1至6:1。更优选地,盐类与包括图6所示的结构的化合物的摩尔比为20:9至50:9。另一方面优选地,叠氮化物为通式R
a(N
3)
b的化合物,其中R为选自Li、Na、K、Rb、Cu、Fe、Ag、Au、Pb、Cd、Ni及H的其中一种,或二种或多种的混合。更优选地,叠氮化物与包括图6所示的结构的化合物的摩尔比为1:1至6:1。另一方面优选地,第一温度为介于-20℃至130℃之间的温度。更优选地,第一温度为介于-10℃至130℃之间的温度。另优选地,第一反应时间为介于2小时至24小时。更优选地,第一反应时间为介于2小时至10小时。更优选地,第一反应时间为介于2小时至8小时。
优选地,酸为:硝酸、硫酸、盐酸、氢溴酸、氢氟酸、氢碘酸、甲酸、乙酸、乙酸酐、草酸、丙二酸及三氟乙酸中的任意一者或多者。此外优选地,其中加入酸为加入酸将反应体系调整至第一pH值,第一pH值介于1-4之间。更优选地,第一pH值介于1.0至3.5之间。更优选地,第一pH值介于2.0至3.0之间。
优选地,硝化体系为发烟硝酸、100%硝酸、五氧化二氮、四氟硼硝、100%硝酸/三氟乙酸酐、100%硝酸/浓硫酸、发烟硝酸、100%硝酸/乙酸酐、发烟硝酸/浓硫酸、发烟硝酸/乙酸酐、发烟硝酸/三氟乙酸酐、发烟硝酸/乙酸酐、三氧化硫/硝酸、五氧化二磷/硝酸、发烟硫酸/发烟硝酸或发烟硫酸/100%硝酸。其中优选地,硝化体系为100%硝酸、100%硝酸/乙酸酐、100%硝酸/三氟乙酸酐、硝酸/乙酸酐、100%硝酸/浓硫酸、发烟硝酸、发烟硝酸/浓硫酸、发烟硝酸/乙酸酐、发烟硝酸/三氟乙酸酐、五氧化二氮、三氧化硫/硝酸、五氧化二磷/硝酸、发烟硫酸/发烟硝酸或发烟硫酸/100%硝酸,且中间产物质量与硝化体系体积的比为1g:5mL至1g:50mL。同时优选地,硝化体系为四氟硼硝,且中间产物与硝化体系体积的摩尔比为1:1至1:50。另一方面优选地,第二温度为介于-20℃至130℃之间的温度。更优选地,第二温度为-20℃至110℃之间的温度。更优选地,第二温度为-20℃至100℃之间的温度。更优选地,第二温度为-20℃至室温之间的温度。更优选地,第二温度为室温。同样优选地,第二温度为40℃至80℃之间的温度。更优选地,第二温度为50℃至70℃之间的温度。另外优选地,第二反应时间为0.5至48小时。更优选地,第二反应时间为0.5至12小时。更优选地,第二反应时间为1至4小时。此外优选地,终止硝化反应为将所得产物加入冰水中。
优选地,此制备方法更包括将终止硝化反应后的所得产物分散于溶剂中,通入氯气气流。更优选地,氯气气流不溶或难溶于溶剂。另优选地,通入氯气气流的反应温度为介于30℃至100℃。更优选地,通入氯气气流的反应温度为介于50℃至90℃。更优选地,通入氯气气流的反应温度为介于60℃至70℃。另优选地,通入氯气气流的反应时间为介于1小时至10小时。更优选地,通入氯气气流的反应时间为介于1小时至5小时。更优选地,通入氯气气流的反应时间为介于1小时至3小时。
优选地,此制备方法更包括将终止硝化反应后的所得产物分散于溶剂中,加入浓氨水,并进行回流反应。更优选地,浓氨水溶于溶剂。另优选地,浓氨水的浓度为介于10%至32%。另优选地,终止硝化反应后的所得产物的质量和浓氨水的体积的比介于1g:1mL至1g:100mL。更优选地,终止硝化反应后的所得产物的质量和浓氨水的体积的比介于1g:5mL至1g:20mL。更优选地,终止硝化反应后的所得产物的质量和浓氨水的体积的比介于1g:6mL至1g:12mL。另优选地,回流反应的反应时间为介于0.5小时至24小时。更优选地,回流反应的反应时间为介于1小时至12小时。更优选地,回流反应的反应时间为介于2小时至8小时。
优选地,此制备方法更包括将加入浓氨水并进行回流反应的所得产物加入氧化性溶液。其中优选地,氧化性溶液为选自浓硫酸与30%双氧水的混合溶液、浓硫酸与50%双氧水的混合溶液、浓硫酸与三氟乙酸的混合溶液、浓硫酸与过氧乙酸的混合溶液及浓硫酸与乙酸酐混合溶液中的一种或多种。另优选地,加入氧化性溶液的反应时间为介于0.5至12小时。所述优选地,加入氧化性溶液的反应时间为介于0.5至4小时。
优选地,此制备方法更包括在加入浓氨水并进行回流反应之后,将所得到的产物加入另一个硝化体系当中,在另一个第二温度下硝化反应另一个第二反应时间。
优选地,此制备方法更包括将丙二腈和亚硝酸盐分散于溶剂中。其中优选地,亚硝酸盐为通式MNO
2的化合物,其中M为选自NH
4、Li、Na、K、Rb及Cs的其中一种,或二种或多种的混合。另优选地,此处的溶剂为N,N-二甲基甲酰胺、二甲基亚砜、四氢呋喃、乙腈、二氯甲烷、氯仿、甲醇、乙醇、水、异丙醇、乙二醇、乙二醇单甲醚、乙二醇单乙醚、乙二醇单丁醚、甘油、丙酮、三乙醇胺、吡啶、苯、甲苯、二甲苯等中的任意一种,或二种或多种的混合物。
优选地,此制备方法更包括加入羟胺化合物。其中优选地,羟胺化合物为羟胺盐类。其中优选地,羟胺盐类为通式(NH
3OH)
aX
b的化合物,其中X为选自F、Cl、Br、I、NO
3、SO
4、HSO
4、CO
3、HCO
3、HCOO、CH
3COO、CF
3COO、(COO)
2及CH
2(COO)
2的其中一种,或二种或多种的混合。更优选地,羟胺化合物为羟胺卤化盐类。更优选地,羟胺化合物为盐酸羟胺。另优选地,加入羟胺化合物的反应温度为介于-20℃至130℃之间的温度。更优选地,加入羟胺化合物的反应温度为介于-20℃至40℃之间的温度。更优选地,加入羟胺化合物的反应温度为介于-10℃至40℃之间的温度。另优选地,加入羟胺化合物的反应时间为为介于2-24小时的时间。更优选地,加入羟胺化合物的反应时间为为介于2-10小时的时间。更优选地,加入羟胺化合物的反应时间为为介于3-5小时的时间。
优选地,此制备方法更包括在加入羟胺化合物后,加入碱及酯类化合物。其中优选地,碱为通式M
a(OH)
b的化合物,其中M为选自NH
4、Li、Na、K、Rb、Cs、Ca、Sr、Ba的其中一种,或二种或多种的混合。另一方面优选地,酯类化合物为通式R
1COOR
2的化合物,其中R
1、R
2为选自甲基、乙基、丙基、异丙基、 伯丁基、仲丁基、叔丁基以及各种戊基、各种己基的其中一种,或二种或多种的混合。
优选地,此制备方法更包括在加入碱及酯类化合物后,加入酸及二氧化铅。优选地,此制备方法更包括加入浓盐酸,进行回流反应。优选地,此制备方法更包括将所得产物溶于二氯亚砜并加入N,N-二甲基甲酰胺,进行回流反应。优选地,此制备方法更包括加入氮化物。其中优选地,氮化物为通式R
a(N)
b的化合物,其中R为选自Li、Na、K、Rb、Cu、Fe、Ag、Au、Pb、Cd、Ni及H的其中一种,或二种或多种的混合。优选地,此制备方法更包括加入硝基乙腈化合物。其中优选地,硝基乙腈化合物为硝基乙腈盐类。其中优选地,硝基乙腈化合物为通式M
a(CH(NO
2)CN)
b的化合物,其中M为选自Li、Na、K、Rb、Cs的其中一种,或二种或多种的混合。其中优选地,硝基乙腈化合物为硝基乙腈钠盐。
优选地,此制备方法包括以下步骤:
步骤一:合成3-氨基-4-[1H-四氮唑]-1H-吡唑
在一定温度下,将3-氨基-4-氰基吡唑分散于溶剂中,接着分批加入特定的盐(M
aX
b)、叠氮化物,然后在一定温度下继续反应,再向其中滴加酸,而后过滤,洗涤,将固体干燥,得到白色粉末,即3-氨基-4-[1H-四氮唑]-1H-吡唑。优选地,步骤一中反应温度为-10℃-130℃。优选地,步骤一溶剂是N,N-二甲基甲酰胺、二甲基亚砜、四氢呋喃、乙腈、二氯甲烷、氯仿、甲醇、乙醇、水、异丙醇、乙二醇、乙二醇单甲醚、乙二醇单乙醚、乙二醇单丁醚、甘油、丙酮、三乙醇胺、吡啶、苯、甲苯、二甲苯中的任意一种,溶剂体积用量与3-氨基-4-氰基吡唑投料质量比为2mL/g-20mL/g。优选地,步骤一中盐的物质的量之比为n(M
aX
b):n(3-氨基-4-[1H-四氮唑]-1H-吡唑)=1:1–6:1。盐的组成为:M=Fe、Co、Ni、Mn、Zn、Cd、Au、Ag、Cu、Cr、Al、V、NH
4、(CH
3)
2NH
2、(CH
3)
3NH、(CH
3CH
2)
2NH
2或(CH
3CH
2)
3NH,X=F、Cl、Br、I、NO
3、SO
4、HSO
4、CO
3、HCO
3、HCOO、CH
3COO、CF
3COO、(COO)
2或CH
2(COO)
2。优选地,步骤一中所用叠氮化物物质的量之比为n(R
a(N
3)
b):n(3-氨基-4-[1H-四氮唑]-1H-吡唑)=1:1–6:1。叠氮化物中R是:R=Li、Na、K、Rb、Cu、Fe、Ag、Au、Pb、Cd、Ni或H。优选地,步骤一中酸的种类为:硝酸、硫酸、盐酸、氢溴酸、氢氟酸、氢碘酸、甲酸、乙酸、乙酸酐、草酸、丙二酸或三氟乙酸中的任意一种或几种混合,用量以将溶液pH调整至1-4为准。
步骤二:合成4-叠氮基-5-硝基-7H-吡唑并[3,4-d][1,2,3]三嗪-2-氧化物将3-氨基-4-1H-四氮唑-吡唑缓慢加入硝化体系,加完后控制反应体系温度,保持硝化0.5h-48h,接着将反应体系倾入冰中,依次经过滤洗涤、干燥,得到米白色4-叠氮基-5-硝基-7H-吡唑并[3,4-d][1,2,3]三嗪-2-氧化物粉末。优选地,步骤二中所用硝化体系为市售发烟硝酸或100%硝酸,或其他硝化体系,包括五氧化二氮、四氟硼硝、100%硝酸/三氟乙酸酐、100%硝酸/浓硫酸、发烟硝酸、100%硝酸/乙酸酐、发烟硝酸/浓硫酸、发烟硝酸/乙酸酐、发烟硝酸/三氟乙酸酐、发烟硝酸/乙酸酐、三氧化硫/硝酸、五氧化二磷/硝酸、发烟硫酸/发烟硝酸、发烟硫酸/100%硝酸。当采用的硝化体系为100%硝酸、100%硝酸/乙酸酐、100%硝酸/三氟乙酸酐、硝酸/乙酸酐、100%硝酸/浓硫酸、发烟硝酸、发烟硝酸/浓硫酸、发烟硝酸/乙酸酐、发烟硝酸/三氟乙酸酐、五氧化二氮、三氧化硫/硝酸、五氧化二磷/硝酸、发烟硫酸/发烟硝酸、发烟硫酸/100%硝酸时,3-氨基-4-[1H-四氮唑]-1H-吡唑与硝化体系的质量体积比为1g:(5-50)mL。当采用的硝化体系为四氟硼硝时,3-氨基-4-[1H-四氮唑]-1H-吡唑与四氟硼硝的用量摩尔比为1:(1-50)。优选地,步骤二中反应温度为-20℃至130℃,步骤二中反应时间为0.5小时至48小时。
本发明更公开了如上述的化合物,或以上述的前体物质制备的化合物,或以上述的制备方法所制备的化合物,其在炸药领域的应用,以及包括其的炸药。其中优选地,此处所述的炸药为起爆药。优选地,此处所述的炸药为绿色环保型起爆药。优选地,此处所述的炸药为无金属起爆药。
本发明的化合物、制备方法及应用能够带来以下有益效果:
1、制备方法简单,只需简单的反应步骤及条件即可制得。
2、反应过程及使用过程中无污染。
3、化合物结构中不需要金属,绿色可靠。
4、化合物稳定性佳,撞击感度、摩擦感度高。
5、起爆能力高,并具备合适使用的极限起爆药量。
图1为本发明的一种化合物的部份结构的化学结构图。
图2为本发明的一种化合物的一类实施方式的部份结构的化学结构图。
图3至图5为本发明的一种化合物的一类实施方式的化学结构图。
图6为本发明的一种化合物的合成方法中,前体物质的部份结构的化学结构图。
图7为本发明的一种化合物的一种实施方式的化学结构图。
图8为本发明的一种化合物的一种实施方式的部分结构的化学结构图。
图9为图7所示的化合物的具体合成方法的化学反应式。
[根据细则91更正 20.02.2019]
图10为图7所示的化合物的核磁氢谱。图29为本发明的一种化合物进行安息角测定所用的漏斗的示意图。
图10为图7所示的化合物的核磁氢谱。图29为本发明的一种化合物进行安息角测定所用的漏斗的示意图。
[根据细则91更正 20.02.2019]
图11为图7所示的化合物的核磁碳谱。图30为本发明的一种化合物进行安息角测定所用的三支架的示意图。
图11为图7所示的化合物的核磁碳谱。图30为本发明的一种化合物进行安息角测定所用的三支架的示意图。
图12、14、16、18、20、22、24、26为本发明的一种化合物的多种实施方式的化学结构图。
图13、15、17、19、21、23、25、27为本发明的一种化合物的多种实施方式的部分结构的化学结构图。
图28为本发明的一种化合物进行极限起爆药量测定的装置图。
其中,图1至图30的附图标记说明如下:
A
1至A
11:原子;I、II、III:化合物;1:电点火头;2:试样;3:铅板;4:电源;5:开关
为更进一步阐述本发明为了达成预定发明目的所采取的技术手段及功效,以下结合附图及较佳实施例,对依据本发明的具体实施方式、结构、特征及其功效,详细说明如下:
图1为本发明的一种化合物的部份结构的化学结构图。其中可以看到,此结构中至少包括了一组三个氮相连的结构,中间的氮原子则通过氮-氧的配位键连接一个氧原子。
通过具备以上的图1所示的结构,能够具有强起爆力、稳定性佳等优点。另一方面,本结构仅由碳、氮、氧、氢……等非金属原子构成,不含金属,因此随之可以制造出不含金属的用于作为炸药、起爆药的化合物,从而可以制备绿色、环保的炸药、起爆药。
更进一步,通过图1所示的结构,可以衍生出多种不同的化合物。由于这些化合物均具有图1所示的结构,因此均具有上述的技术效果。
举例来说,在一个实施例当中,在图1所示的结构当中,原子A
1、A
2为碳原子。在这样的实施例中,也就至少包括了一个2-氧化-1,2,3-三嗪的结构。不过,值得注意的是,在图1所示的结构当中,原子A
1、A
2并不限定为碳原子。在一个实施例中,原子A
1、A
2也可以选自于碳原子、氮原子、氧原子、磷原子及硫原子中的一种或多种。举例来说,原子A
1、A
2可以选自于碳原子、氮原子中的一种或多种。在一个实施例当中,原子A
1、A
2可以分别为碳原子及氮原子。在一个实施例当中,原子A
1为碳原子,原子A
2为氮原子。
在另一个方面,举例来说,在图1所示的结构的5,6号位(图1所示杂环的左上、左下两个位置,即原子A
1、A
2所在位置),可以形成其他的键结以形成环,从而形成稠环(并环)化合物,即稠合在一起的多环化合物。例如是,此稠环化合物可以为二环、三环甚至是三环以上的多环化合物。
这些与图1所示的结构稠合的一个或多个单环,其可为三元环、四元环、五元环、六元环、七元环、八元环乃至于更多元的多元环。举例来说,可以是一个五元环与图1所示的结构稠合,共用图1所示的结构中5,6号位的两个原子,例如是共用两个碳原子。或者是,也可以是一个六元环与图1所示的结构稠合,共用图1所示的结构中5,6号位的两个原子,例如是共用两个碳原子。又或者是,可以是一个五元环与六元环的稠环与图1所示的结构稠合,共用图1所示的结构中5,6号位的两个原子,例如是共用两个碳原子。
这些与图1所示的结构稠合的一个或多个单环,可以全部由碳原子组成,举例来说,可以是苯环;但也可以包括一个、二个、三个或多个杂原子,甚至是全部由杂原子组成。这些杂原子可以包括硼原子、氮原子、氧原子、硅原子、磷原子、硫原子、砷原子、硒原子、碲原子……以及可得到合理化学结构的元素周期表中其他的原子。同一个环上所含原子的种类可以是一种(整个环上都由同一种原子构成)、二种、三种或更多种,甚至是整个环上的原子种类都不同。
这些环上除了与图1所示的结构共用的原子外,其他部分的多个原子,其间可以单键或双键,甚至是三键来相互连接;可以是多个共轭单双键,可以使得这些环为芳香环。具体来说,这些环可以是芳香杂环,例如是呋喃、吡咯、噻吩、咪唑、吡唑、噁唑、噻唑、吡啶、吡嗪、嘧啶、哒嗪、呋咱,或其他三唑类、四唑类、三嗪类、四嗪类……等等。举例来说,与图1所示的结构稠合的芳香环可以是吡唑,其4号位、5号位的两个碳原子,分别同时为图1所示的结构的5号位、6号位的两个碳原子。或者是,与图1所示的结构稠合的芳香环可以是呋咱,其2号位、3号位的两个碳原子,分别同时为图1所示的结构的5号位、6号位的两个碳原子。或者是,与图1所示的结构稠合的芳香环可以是哒嗪,其4号位、5号位的两个碳原子,分别同时为图1所示的结构的5号位、6号位的两个碳原子。或者是,与图1所示的结构稠合的芳香环可以是嘧啶,其4号位、5号位的两个碳原子,分别同时为图1所示的结构的5号位、6号位的两个碳原子。或者是,与图1所示的结构稠合的芳香环可以是吡嗪,其2号位、3号位的两个碳原子,分别同时为图1所示的结构的5号位、6号位的两个碳原子。
而这些与图1所示的结构稠合的一个或多个单环,其上可以没有取代基,或是具有一个或多个取代基。这些取代基可以是:(1)饱和烃烃基,如烷烃基等;(2)官能团化饱和烃烃基,如硝基烷烃基、偕二硝基烷烃基、硝仿基烷烃基、卤代烷烃基等;(3)醇基;(4)醚基、硫醚基;(5)羧酸基、酯基、硝酸酯基、磺酸基、磺酸酯基等;(6)酮基、酰胺基、酰肼基等;(7)醛基;(8)不饱和脂肪族烃基,如烯烃基、炔烃基、卤代烯烃基、硝基炔烃基等;(9)不饱和芳香烃烃基,如单环芳烃基、多环芳烃基、稠环芳烃基和官能团化的单环芳烃基、官能团化的多环芳烃基、官能团化的稠环芳烃基;(10)杂环芳香烃基,如噻吩、 吡啶、呋喃、唑类环、嗪类环、噁唑类环等及其对应的单官能团化或多官能团化基团;(11)杂环非芳香烃基,如氮杂桥环烃、氧杂桥环烃等;(12)卤原子;(13)氰基、肟基;(14)氨基、亚氨基、偶氮基、氧化偶氮基、叠氮基、肼基;(15)羟基、羟胺基、烷氧基;(16)硝基、亚硝基。举例来说,可以是硝基,更具体来说可以是在吡唑的5号碳上有一个硝基。或者是,这些与图1所示的结构稠合的一个或多个单环上的原子也可以与取代基形成配位键。举例来说,可以是其上的氮原子与取代基氧原子形成配位键。具体来说,可以是呋咱上的氮原子与取代基氧原子形成配位键。
在图1所示的结构上,图1所示的结构的4号位(图1中杂环的上方位置)上还可以具有一个取代基,在图1上标示为R
1。具体来说,R
1可为氢、氨基、硝基、硝氨基、叠氮基、氟、氯、溴、碘、C
nH
m(NO
2)
p(其中m+p=2n+1),或者是如图2所示结构。在图2所示结构中,A
1至A
5等原子可为碳原子、氮原子、氧原子、磷原子或硫原子,可以是同一种或是不同的多种。在图2所示结构中,A
1至A
5等原子上的取代基个数可为0、1、2、3、4或5个。在图2所示结构中,可包括取代基R
2。取代基R
2可为氢、氨基、硝基、硝氨基、叠氮基、氟、氯、溴、碘、C
nH
m(NO
2)
p,或图2所示结构。
具体来说,如图1所示的通式化合物,具体又可例如为图3、4、5所示的通式化合物。
图3为本发明的一种化合物的其中一类实施方式的化学结构图。请参照图3所示,其中图1所示的结构与一个五元环稠合,并至少具有一个取代基R
3。在图3所示结构中,A
1至A
5等原子可为碳原子、氮原子、氧原子、磷原子或硫原子,可以是同一种或是不同的多种。在图3所示结构中,A
1至A
5等原子上的取代基个数可为0、1、2、3、4或5个。在图3所示结构中,取代基R
3可为氢、氨基、硝基、硝氨基、叠氮基、氟、氯、溴、碘、C
nH
m(NO
2)
p(其中m+p=2n+1),或图2所示结构。取代基R
3可为一个,或相同或不同的多个。
图4为本发明的一种化合物的其中另一类实施方式的化学结构图。请参照图4所示,其中图1所示的结构与一个六元环稠合,并至少具有一个取代基R
4。在图4所示结构中,A
1至A
6等原子可为碳原子、氮原子、氧原子、磷原子、硫原子,可以是同一种或是不同的多种。在图4所示结构中,A
1至A
6等原子上的取代基个数可为0、1、2、3、4、5或6个。在图4所示结构中,取代基R
4可为氢、氨基、硝基、硝氨基、叠氮基、氟、氯、溴、碘、C
nH
m(NO
2)
p(其中m+p=2n+1),或图2所示结构。取代基R
4可为一个,或相同或不同的多个。
图5为本发明的一种化合物的其中又一类实施方式的化学结构图。请参照图5所示,其中图1所示的结构与一个六元环稠合,此六元环又与一个五元环稠合,在此五元环上至少具有一个取代基R
5。在图5所示结构中,A
1至A
9等原子可为碳原子、氮原子、氧原子、磷原子、硫原子,可以是同一种或是不同的多种。在图5所示结构中,A
4至A
5、A
7至A
9等原子上的取代基个数可为0、1、2、3、4或5个。在图5所示结构中,A
1至A
6等原子上的取代基个数可为0、1、2、3、4、5或6个。在图5所示结构中,取代基R
5可为氢、氨基、硝基、硝氨基、叠氮基、氟、氯、溴、碘、C
nH
m(NO
2)
p(其中m+p=2n+1),或图2所示结构。取代基R
5可为一个,或相同或不同的多个。
以上图3至图5所示结构及其所衍生的化合物,其性质各异,不过由于都具有类似的结构,因此也都具有本申请所述的有强起爆力、稳定性佳等等技术效果。
另一方面,如图1所示的结构,其通用合成方式可为如下步骤:
【步骤一】
合成前体物质,即具有图6所示通式的结构。
具体来说,此处所要合成的前体物质,即图6所示通式的结构,可包括两个原子A
10、A
11,其中A
10可至少连接一个氰基,A
11则可至少连接一个氨基。在接下來的关四氮唑环反应当中,这些氰基及氨基会参与反应,使得两个原子A
10、A
11成为例如是图1所示结构中的A
1、A
2,或者是例如是图3至图5所示结构中的A
1至A
6的其中二者。
在一个实施例当中,A
10、A
11等原子可为碳原子、氮原子、氧原子、磷原子、硫原子,可以是同一种或是不同的二种。在一个实施例当中,A
10、A
11等原子为为碳原子。A
10、A
11等原子分别为碳原子及氮原子。
在一个实施例当中,前体物质包括一个多元环。在一个实施例当中,A
10、A
11为多元环的一部分。这里的多元环可为三元环、四元环、五元环、六元环、七元环、八元环乃至于更多元的多元环。举例来说,可以是一个五元环。或者是,也可以是一个六元环。又或者是,可以是一个五元环与六元环的稠环。在一个实施例当中,前体物质包括一个芳香环。在一个实施例当中,A
10、A
11为芳香的一部分。在另一个实施例当中,前体物质包括一个多元环,但A
10、A
11等原子并不是这个多元环的一部分。
这些环可以全部由碳原子组成,举例来说,可以是苯环;但也可以包括一个、二个、三个或多个杂原子,甚至是全部由杂原子组成。这些杂原子可以包括硼原子、氮原子、氧原子、硅原子、磷原子、硫原子、砷原子、硒原子、碲原子……以及可得到合理化学结构的元素周期表中其他的原子。同一个环上所含原子的种类可以是一种(整个环上都由同一种原子构成)、二种、三种或更多种,甚至是整个环上的原子种类都不同。
这些环可以是芳香杂环,例如是呋喃、吡咯、噻吩、咪唑、吡唑、噁唑、噻唑、吡啶、吡嗪、嘧啶、哒嗪、呋咱,或其他三唑类、四唑类、三嗪类、四嗪 类……等等。
而这些环上可以没有取代基,或是具有一个或多个取代基。这些取代基可以是:(1)饱和烃烃基,如烷烃基等;(2)官能团化饱和烃烃基,如硝基烷烃基、偕二硝基烷烃基、硝仿基烷烃基、卤代烷烃基等;(3)醇基;(4)醚基、硫醚基;(5)羧酸基、酯基、硝酸酯基、磺酸基、磺酸酯基等;(6)酮基、酰胺基、酰肼基等;(7)醛基;(8)不饱和脂肪族烃基,如烯烃基、炔烃基、卤代烯烃基、硝基炔烃基等;(9)不饱和芳香烃烃基,如单环芳烃基、多环芳烃基、稠环芳烃基和官能团化的单环芳烃基、官能团化的多环芳烃基、官能团化的稠环芳烃基;(10)杂环芳香烃基,如噻吩、吡啶、呋喃、唑类环、嗪类环、噁唑类环等及其对应的单官能团化或多官能团化基团;(11)杂环非芳香烃基,如氮杂桥环烃、氧杂桥环烃等;(12)卤原子;(13)氰基、肟基;(14)氨基、亚氨基、偶氮基、氧化偶氮基、叠氮基、肼基;(15)羟基、羟胺基、烷氧基;(16)硝基、亚硝基,或者是这些官能基的组合。在一个实施例中,这些取代基可以是A
10、A
11等原子所在的结构。
在步骤一中,可以根据所要合成的产物不同,选择不同的所要合成的前体物质、其合成方式及其合成条件。不过,若是可以直接取得所需要合成的产物的前体物质,也可以直接取得,并跳过此步骤。
【步骤二】
对前体物质进行关四氮唑环反应,得到第一中间产物。
具体来说,可以为:
(1)将前体物质分散于溶剂中。
(2)加入一定量的盐类及一定量的叠氮化物,在一定的反应温度下反应一定时间。
(3)加入酸,纯化并得到第一中间产物。
步骤二(1)中的溶剂可为N,N-二甲基甲酰胺、二甲基亚砜、四氢呋喃、乙腈、二氯甲烷、氯仿、甲醇、乙醇、水、异丙醇、乙二醇、乙二醇单甲醚、乙二醇单乙醚、乙二醇单丁醚、甘油、丙酮、三乙醇胺、吡啶、苯、甲苯、二甲苯等中的任意一种,或合适的两种及多种溶剂的混合物,例如可为水、乙醇、二甲基亚砜、N,N-二甲基甲酰胺,或是丙酮与甲醇体积比4:1的混合溶液。但本发明并不以此为限,应视为本发明可以采用任意适合用于本反应的溶剂。
步骤二(1)中溶剂的用量体积与前体物质的质量比值,例如可为溶剂体积/前体物质质量=2mL/g-2mL/mg;例如可为10mL/g、15mL/g、35mL/g、75mL/g或400mL/g。但本发明并不以此为限,应视为本发明可以采用任意适合用于本反应的溶剂比例。
在步骤二(2)中的反应温度,可以是-20℃至130℃,例如可为70℃至120℃,例如可为70℃、110℃、120℃。但本发明并不以此为限,应视为本发明可以采用任意适合用于本反应的反应温度。
在步骤二(2)中的反应时间,可以为2小时至24小时,例如可为2小时至10小时,例如可为2小时至8小时,例如可以是2小时、3小时、4小时、5小时、7小时、10小时、24小时。但本发明并不以此为限,应视为本发明可以采用任意适合用于本反应的反应时间。
在步骤二(2)中,所加入的盐类,其摩尔比可为盐类:前体物质=1:1至6:1,例如可为20:9至50:9,例如可为1:1、20:9、40:9、50:9,或是其他任意适合的比例。此处盐类可例如具有通式M
aX
b,其中:M可为Fe、Co、Ni、Mn、Zn、Cd、Au、Ag、Cu、Cr、Al、V、NH
4、(CH
3)
2NH
2、(CH
3)
3NH、(CH
3CH
2)
2NH
2、(CH
3CH
2)
3NH等,而X可为F、Cl、Br、I、NO
3、SO
4、HSO
4、CO
3、HCO
3、HCOO、CH
3COO、CF
3COO、(COO)
2、CH
2(COO)
2等。举例来说,M可为NH
4、Al、(CH
3CH
2)
3NH、Co等,X可为F、Cl等,并选择对应的a与b。举例来说,此处盐类可以是氟化铵、氯化铵、硝酸铵、氯化亚钴、二甲胺盐酸盐等。但本发明并不以此为限,应视为本发明可以采用任意适合用于本反应的盐类。
在步骤二(2)中,所加入的叠氮化物,其摩尔比可为所用叠氮化物:前体物质=1:1至6:1。所用叠氮化物可以表示为通式R
a(N
3)
b,其中R可为Li、Na、K、Rb、Cu、Fe、Ag、Au、Pb、Cd、Ni、H、NH
4等,并选择对应的a与b。举例来说,此处叠氮化物可以是叠氮化钠、叠氮化铵等。但本发明并不以此为限,应视为本发明可以采用任意适合用于本反应的叠氮化物。
在步骤二(3)中,所加入的酸的种类可以为:硝酸、硫酸、盐酸、氢溴酸、氢氟酸、氢碘酸、甲酸、乙酸、乙酸酐、草酸、丙二酸、三氟乙酸等,可以使用如上单独一种酸,也可为两种或多种酸混合使用。举例来说,可以为盐酸、甲酸、乙酸酐、三氟乙酸;但本发明并不以此为限,应视为本发明可以采用任意适合用于本反应的酸。
在步骤二(3)中,滴加酸的用量可为以将溶液pH值调整至1.0至4.0,例如可以是1.0至3.5,例如可为2.0至3.0,例如可为1.4、2.0、2.4、2.5、3.0、3.1、3.3等,但本发明并不以此为限,应视为滴加酸的用量可将溶液调整为所需要的任何pH值。
【步骤三】
对步骤二所得的第一中间产物进行重排环反应,并在图1所示结构中对应R
1部分的位置反应产生叠氮基(-N
3),得到产物(所要合成的产物如图1所示结构中对应R
1部分的基团为叠氮基时)或第二中间产物(所要合成的产物如图1所示结构中对应R
1部分的基团不是叠氮基时)。
具体来说,可以为:
(1)将第一中间产物加入硝化体系当中,加完后控制反应体系温度,保持硝化反应在一定温度下进行一定时间。
(2)终止反应,过滤、纯化,得到产物或第二中间产物。
在步骤三(1)中的反应温度,可为-20℃至130℃,例如可为-20℃至110℃,例如可为-20℃至100℃,例如可为-20℃至室温,例如可为-20℃、-10℃、-5℃、0℃、4℃或室温,或也可为例如40℃至80℃,例如是50℃至70℃,例如是50℃,或例如是60℃等。但本发明并不以此为限,应视为本发明可以采用任意适合用于本反应的反应温度。
在步骤三(1)中的反应时间,可为0.5至48小时,例如可为0.5至12小时,例如可为0.5至4小时,例如可为0.5小时、1小时、2小时、3小时、4小时,或也可以是7小时。但本发明并不以此为限,应视为本发明可以采用任意适合用于本反应的反应时间。
在步骤三(1)中,硝化体系可以为市售发烟硝酸或100%硝酸,也可以是其他硝化体系,例如五氧化二氮、四氟硼硝、100%硝酸/三氟乙酸酐、100%硝酸/浓硫酸、发烟硝酸、100%硝酸/乙酸酐、发烟硝酸/浓硫酸、发烟硝酸/乙酸酐、发烟硝酸/三氟乙酸酐、发烟硝酸/乙酸酐、三氧化硫/硝酸、五氧化二磷/硝酸、发烟硫酸/发烟硝酸、发烟硫酸/100%硝酸等。
当采用的硝化体系为100%硝酸、100%硝酸/乙酸酐、100%硝酸/三氟乙酸酐、硝酸/乙酸酐、100%硝酸/浓硫酸、发烟硝酸、发烟硝酸/浓硫酸、发烟硝酸/乙酸酐、发烟硝酸/三氟乙酸酐、五氧化二氮、三氧化硫/硝酸、五氧化二磷/硝酸、发烟硫酸/发烟硝酸、发烟硫酸/100%硝酸等时,第一中间产物与硝化体系的质量体积比可为1g:5mL至1g:50mL;硝化体系的各成分的比例例如可为体积比1:1至3:1,例如可为体积比1:1。举例来说,可以采用体积比为1:1的100%硝酸/三氟乙酸酐,或是体积比为1:1的100%硝酸/浓硫酸作为硝化体系。
当采用的硝化体系为四氟硼硝时,第一中间产物与四氟硼硝的用量摩尔比可为1:1至1:50;所用的溶剂可为不与硝化体系或第一中间产物、第二中间产物反应的溶剂,例如可为乙腈。
在步骤三(2)中终止反应的具体方法,例如可为将反应体系倒入冰或冰水中,使得硝化体系被稀释且冷却,反应停止,产物析出。
【步骤四】
若所要合成的产物,其如图1所示结构中对应R
1部分的基团不是叠氮基,则进行取代基置换反应。以下列举R
1部分的基团为氯、氨基、硝基、硝氨基时的对应反应步骤如下:
[甲、R
1部分为氯(-Cl)时]
(1)将第二中间产物分散于溶剂中。
(2)通入氯气气流,在一定温度下反应一段时间。
(3)过滤、纯化,得到最终产物。
在步骤四甲(1)中,所用的溶剂可为各种常见的有机溶剂,只要不与第二中间产物或氯气发生反应,致使后续反应无法进行即可。举例来说,在在步骤四甲(1)中,所用的溶剂可为二氯甲烷。
在步骤四甲(2)中,氯气气流的流量可以是介于每分钟1mL至5mL之间,例如是每分钟2mL至5mL,例如是每分钟3mL至4mL,或者例如是每分钟5mL。
在步骤四甲(2)中的反应温度可以为30℃至100℃,例如是50℃至90℃,例如是60℃至70℃,例如是60℃,或例如是70℃。
在步骤四甲(2)中的反应时间例如是1小时至10小时,例如是1小时至5小时,例如是1小时至3小时,例如是1小时,或例如是2小时。
[乙、R
1部分为氨基(-NH
2)时]
(1)将第二中间产物分散于溶剂中。
(2)加入一定量的浓氨水,回流反应一段时间,反应完后冷却。
(3)过滤、纯化,得到最终产物。
在步骤四乙(1)中,所用的溶剂可为各种常见的有机溶剂,只要不与第二中间产物或浓氨水发生反应,致使后续反应无法进行即可。举例来说,在在步骤四乙(1)中,所用的溶剂可为甲醇。
在步骤四乙(2)中,浓氨水的浓度例如可以是10%至32%。
在步骤四乙(2)中,第二中间产物的质量和浓氨水的体积的比,例如可以是1g:1mL至1g:100mL,例如可以是1g:5mL至1g:20mL,例如可以是1g:6mL至1g:12mL。
在步骤四乙(2)中,回流反应的时间例如是0.5小时至24小时,例如是1小时至12小时,例如是2小时至8小时,例如是2小时,或例如是4小时。
[丙、R
1部分为硝基(-NO
2)时]
(1)将第二中间产物分散于溶剂中。
(2)加入一定量的浓氨水,回流反应一段时间,冷却过滤得到固体。
(3)将此固体加入氧化性溶液,反应一段时间。
(4)终止反应,纯化,得到最终产物。
在步骤四丙(1)中,所用的溶剂可为各种常见的有机溶剂,只要不与第二中间产物或浓氨水发生反应,致使后续反应无法进行即可。举例来说,在在步骤四丙(1)中,所用的溶剂可为甲醇。
在步骤四丙(2)中,浓氨水的浓度例如可以是10%至32%。
在步骤四丙(2)中,第二中间产物的质量和浓氨水的体积的比,例如可以是1g:1mL至1g:100mL,例如可以是1g:5mL至1g:20mL,例如可以是1g:6mL至1g:12mL。
在步骤四丙(2)中,回流反应的时间例如是0.5小时至24小时,例如是1小时至12小时,例如是2小时至8小时,例如是2小时,或例如是4小时。
在步骤四丙(3)中,氧化性溶液可以是:浓硫酸与30%双氧水的混合溶液、浓硫酸与50%双氧水的混合溶液、浓硫酸与三氟乙酸的混合溶液、浓硫酸与过氧乙酸的混合溶液,或浓硫酸与乙酸酐混合溶液。氧化性溶液中各成分的体积比可以例如是1:1,举例来说,可以是浓硫酸与30%双氧水体积比1:1的混合溶液。
在步骤四丙(3)中的反应时间,例如可为0.5至12小时,例如可为0.5至4小时,例如可为0.5小时、1小时、2小时、4小时。
[丁、R
1部分为硝氨基(-NHNO
2)时]
(1)将第二中间产物分散于溶剂中。
(2)加入一定量的浓氨水,回流反应一段时间。
(3)冷却过滤得固体。
(4)将所得到的固体加入硝化体系中,反应一段时间。
(5)终止反应,过滤、纯化,得到最终产物。
在步骤四丁(1)中,所用的溶剂可为各种常见的有机溶剂,只要不与第二中间产物或浓氨水发生反应,致使后续反应无法进行即可。举例来说,在在步骤四丁(1)中,所用的溶剂可为甲醇。
在步骤四丁(2)中,浓氨水的浓度例如可以是10%至32%。
在步骤四丁(2)中,第二中间产物的质量和浓氨水的体积的比,例如可以是1g:1mL至1g:100mL,例如可以是1g:5mL至1g:20mL,例如可以是1g:6mL至1g:12mL。
在步骤四丁(2)中,回流反应的时间例如是0.5小时至24小时,例如是1小时至12小时,例如是2小时至8小时,例如是2小时,或例如是4小时。
在步骤四丁(4)中的反应温度,可为-20℃至130℃,例如可为-20℃至110℃,例如可为-20℃至100℃,例如可为-20℃至室温,例如可为-20℃、10℃、4℃或室温等。但本发明并不以此为限,应视为本发明可以采用任意适合用于本反应的反应温度。
在步骤四丁(4)中的反应时间,可为0.5至12小时,例如可为0.5至4小时,例如可为0.5小时、1小时、2小时、4小时。但本发明并不以此为限,应视为本发明可以采用任意适合用于本反应的反应时间。
在步骤四丁(4)中,硝化体系可以为市售发烟硝酸或100%硝酸,也可以是其他硝化体系,例如五氧化二氮、四氟硼硝、100%硝酸/三氟乙酸酐、100%硝酸/浓硫酸、发烟硝酸、100%硝酸/乙酸酐、发烟硝酸/浓硫酸、发烟硝酸/乙酸酐、发烟硝酸/三氟乙酸酐、发烟硝酸/乙酸酐、三氧化硫/硝酸、五氧化二磷/硝酸、发烟硫酸/发烟硝酸、发烟硫酸/100%硝酸等。
当采用的硝化体系为100%硝酸、100%硝酸/乙酸酐、100%硝酸/三氟乙酸酐、硝酸/乙酸酐、100%硝酸/浓硫酸、发烟硝酸、发烟硝酸/浓硫酸、发烟硝酸/乙酸酐、发烟硝酸/三氟乙酸酐、五氧化二氮、三氧化硫/硝酸、五氧化二磷/硝酸、发烟硫酸/发烟硝酸、发烟硫酸/100%硝酸等时,步骤四丁(3)所得固 体与硝化体系的质量体积比为1g:5mL至1g:50mL。
当采用的硝化体系为四氟硼硝时,步骤四丁(3)所得固体与四氟硼硝的用量摩尔比可为1:1至1:50。
在步骤四丁(4)中采用的硝化体系,可以是与步骤三相同的硝化体系,或者是不同的硝化体系。
在步骤四丁(5)中终止反应的具体方法,例如可为将反应体系倒入冰或冰水中,使得硝化体系被稀释且冷却,反应停止,产物析出。
根据上述的多种化合物通式及上述的通用合成方法,在此举多个详细实施例,并详细说明如下:
实施例1
图7为本发明的一种化合物的其中一种实施方式的化学结构图。图7所示的化合物,其具体的名称为:2-氧化-4-叠氮基-5-硝基-7H-吡唑并[3,4-d][1,2,3]三嗪,具有如图1所示的结构,具体来说具有如图3所示的结构,更具体来说具有如图8所示的结构。
此化合物为一种白色至黄绿色的粉末状固体,重结晶析出的纯物质则为白色针状晶体。其晶体密度为1.5-1.9g/cm
3,经70℃至90℃恒温脱水处理后,室温下实测密度为1.6-2.0g/cm
3。流散性较好,不易黏附于玻璃类容器。热分解温度高于150℃,对光、水、汽等均不敏感,太阳直射下长时间未观察到明显颜色变化。性质稳定,与强酸不发生反应。75℃下持续加热48小时,失重0.8%,100℃条件下失重增加,分解2%左右,表观颜色变化明显。与铜、铁等在环境条件下不反应,与塑料、纸张接触,不发生反应。
【合成方法】
实施例1的化合物的合成方法,为采用3-氨基-4-氰基吡唑作为前体物质进行合成。具体的反应例如可以是图9所示:
第一步反应:由前体物质3-氨基-4-氰基吡唑(图9中式I化合物)合成第一中间产物3-氨基-4-[1H-四氮唑]-1H-吡唑(图9中式II化合物)
第二步反应:由第一中间产物3-氨基-4-[1H-四氮唑]-1H-吡唑(式II化合物)合成产物2-氧化-4-叠氮基-5-硝基-7H-吡唑并[3,4-d][1,2,3]三嗪(图9中式III化合物,即图7所示的化合物)
具体的合成步骤与反应条件,在此详述如下:
第一步反应:合成第一中间产物3-氨基-4-[1H-四氮唑]-1H-吡唑(图9中式II化合物)
(1)在一定温度下,将实施例1的前体物质3-氨基-4-[1H-四氮唑]-1H-吡唑,即图9中式I化合物分散于溶剂中。
(2)在反应液中加入盐类及叠氮化物,在一定温度下继续反应。
(3)在反应液中加入酸。
(4)过滤,洗涤,将固体干燥,得到实施例1的第一中间产物,即图9中式II化合物。
本部分步骤(1)所用的溶剂、步骤(2)所加入的盐类、叠氮化物,反应温度及反应时间,以及步骤(3)中所加入的酸,可参照通用合成方式步骤二中所述的反应条件。
第二步反应:合成产物2-氧化-4-叠氮基-5-硝基-7H-吡唑并[3,4-d][1,2,3]三嗪(式III化合物,图7所示化合物)
(5)将实施例1的第一中间产物,即图9中式II化合物缓慢加入硝化体系,加完后控制反应体系温度,保持硝化反应在一定温度下进行一定时间。
(6)终止反应,过滤、洗涤、干燥,得到实施例1的产物,即图9中式III化合物(图7所示化合物)。
本部分步骤(5)所加入的硝化体系及反应温度,以及步骤(6)中终止反应的方式,可参照通用合成方式步骤三中所述的反应条件。
关于上述反应的具体范例,请参照以下实施例1A-1D。
[实施例1A]
在室温下,将3-氨基-4-[1H-四氮唑]-1H-吡唑1g(9mmol)分散于10mL水中,接着分批加入氯化铵0.48g(9mmol)、叠氮化钠0.59g(9mmol),然后在70℃下继续反应5小时,冷却至室温,再向其中滴加盐酸,直至溶液pH值为2,而后过滤,以5mL冰水洗涤,将固体干燥,得到白色粉末0.68g,得率50%。
接着取0.3g(2mmol)所得白色粉末,加入到100%硝酸/三氟乙酸酐按照一比一的体积比配置的6mL混合液中,而后在室温下反应1小时,将反应液倒入50g冰水中,搅拌0.5小时后,过滤,用冰水10mL洗涤,干燥之后得到0.31g黄色粉末状固体,得率70%。
[实施例1B]
在冰水浴下,将3-氨基-4-[1H-四氮唑]-1H-吡唑2g(18mmol)分散于70mL乙醇中,接着分批加入氯化铝5.34g(40mmol)、叠氮化钠1.95g(30mmol),然后在120℃下继续反应2小时,冷却至室温,再向其中滴加乙酸而后滴加甲酸,直至溶液pH值为3.3,而后过滤,以20mL冰水洗涤,将固体干燥,得到白色粉末2.20g,得率81%。
接着取0.5g(3.3mmol)所得白色粉末,加入到100%硝酸/三氟乙酸酐按照一比一的体积比配置的20mL混合液中,而后在-10℃下反应4小时,将反应液倒入50g冰水中,搅拌0.5小时后,过滤,用冰水20mL洗涤,干燥之后得到0.28g黄绿色粉末状固体,得率30%。
[实施例1C]
在冰水浴下,将3-氨基-4-[1H-四氮唑]-1H-吡唑2g(18mmol)分散于150mL的二甲基亚砜中,接着分批加入三乙胺盐酸盐11g(80mmol)、叠氮化钠6.5g(100mmol),然后在110℃下继续反应24小时,再向其中滴加乙酸酐,直至溶液pH值为2.5,而后过滤,以20mL冰水洗涤,而后以10mL乙醚洗涤,将固体干燥,得到白色粉末0.81g,得率30%。
接着取0.5g(3.3mmol)所得白色粉末,加入到加有1.1g四氟硼硝的20mL乙腈中,而后在4℃下反应4小时,将反应液倒入50g冰水中,搅拌0.5小时后,过滤,用冰水20mL洗涤,而后以10mL石油醚洗涤,干燥之后得到0.55g米白色粉末状固体,得率75%。
[实施例1D]
在室温下,将3-氨基-4-[1H-四氮唑]-1H-吡唑2g(18mmol)分散于800mL由丙酮和甲醇按照体积比为4:1混合而成的溶剂中,接着分批加入氯化亚钴13g(100mmol)、叠氮化钠5.2g(80mmol),然后在回流条件下继续反应10小时,再向其中滴加三氟乙酸,直至溶液pH值为1.4,而后过滤,以30mL冰水洗涤,而后以30mL乙醚洗涤,将固体干燥,得到白色粉末2.26g,得率83%。
接着取0.5g(3.3mmol)所得白色粉末,加入到加有0.9g五氧化二氮的30mL乙腈中,而后在-20℃下反应5小时,将反应液倒入50g冰水中,搅拌0.5小时后,过滤,用冰水20mL洗涤,干燥之后得到0.58g白色粉末状固体,得率79%。
【产物测定结果】
实施例1A-1D制备得到的式III化合物(即图7所示化合物)的核磁氢谱如图10所示,其特征核磁氢谱的位移为:
1H NMR(400MHz,DMSO-d
6,25℃)δ(ppm):14.51(brs,1H,NH)。
实施例1A-1D制备得到的式III化合物(即图7所示化合物)的核磁碳谱如图11所示,其特征核磁碳谱的位移为:
13C NMR(100MHz,DMSO-d
6,25℃)δ(ppm):157.95,157.31,148.07,90.03。
实施例1A-1D制备得到的式III化合物(即图7所示化合物)的红外线吸收光谱的波长为:IR(KBr,γ/cm
-1):3414(m),3231(m),2154(s),1600(s),1543(s),1403(s),1384(s),1313(m),1209(m),1131(m)。
实施例1A-1D制备得到的式III化合物(即图7所示化合物)的粉末分析测定结果为:DSC(160℃,50℃-250℃,5℃·min
-1)。
实施例1A-1D制备得到的式III化合物(即图7所示化合物)(分子式C
4HN
9O
3)的元素分析结构为(实测值(计算值)):C:21.62%(21.53%),H:0.47%(0.45%),N:56.61%(56.50%),O:21.22%(21.51%)。
实施例2
图12为本发明的一种化合物的其中一种实施方式的化学结构图,其具体的名称为:5-氧化-7-硝基[1,2,5]噁二唑[3,4-d][1,2,3]三嗪,具有如图1所示的结构,具体来说具有如图3所示的结构,更具体来说具有如图13所示的结构。
【合成方法】
实施例2的化合物具体的合成方法,在此详述如下:
第一步反应:合成前体物质
(1)将丙二腈和亚硝酸盐分散于溶剂中。
(2)在反应液中加入羟胺化合物。
(3)加入羟胺化合物完后,加入碱及酯类化合物,继续反应。
(4)将所得化合物加入酸及二氧化铅,加热回流。
(5)过滤、将母液旋干,得到实施例2的前体物质。
步骤(1)至(3)中,反应的温度可以是-20℃至130℃,例如可为-20℃、至40℃,例如可为-20℃、-10℃、40℃。但本发明并不以此为限,应视为本发明可以采用任意适合用于本反应的反应温度。
步骤(1)中亚硝酸盐可为通式MNO
2的化合物,其中M为选自NH
4、Li、Na、K、Rb及Cs的其中一种,或二种或多种的混合。举例来说,可以是亚硝酸钠。
步骤(1)中的溶剂可为N,N-二甲基甲酰胺、二甲基亚砜、四氢呋喃、乙腈、二氯甲烷、氯仿、甲醇、乙醇、水、异丙醇、乙二醇、乙二醇单甲醚、乙二醇单乙醚、乙二醇单丁醚、甘油、丙酮、三乙醇胺、吡啶、苯、甲苯、二甲苯等中的任意一种,或二种或多种的混合物。但本发明并不以此为限,应视为本发明可以采用任意适合用于本反应的溶剂。
步骤(1)中溶剂的用量体积与反应物的质量比,例如可为溶剂体积:丙二腈=2mL/g-2mL/mg;例如可为10mL/g、15mL/g、35mL/g、75mL/g或400mL/g。但本发明并不以此为限,应视为本发明可以采用任意适合用于本反应的溶剂比例。
步骤(2)中,羟胺化合物可为羟胺盐类。举例来说,羟胺盐类可为通式(NH
3OH)
aX
b的化合物,其中X为选自F、Cl、Br、I、NO
3、SO
4、HSO
4、CO
3、HCO
3、HCOO、CH
3COO、CF
3COO、(COO)
2及CH
2(COO)
2的其中一种,或二种或多种的混合。举例来说,所述羟胺化合物可为羟胺卤化盐类;举例来说,所述羟胺化合物可为盐酸羟胺。
步骤(2)(3)中的反应时间可以为2小时至24小时,例如可以是2小时、3小时、4小时、5小时、10小时、24小时。但本发明并不以此为限,应视为本发明可以采用任意适合用于本反应的反应时间。
步骤(3)中的酯类化合物可为通式R
1COOR
2的化合物,其中R
1、R
2为选自甲基、乙基、丙基、异丙基、伯丁基、仲丁基、叔丁基以及各种戊基、各种己基的其中一种,或二种或多种的混合。但本发明并不以此为限,应视为本发明可以采用适当的酯类。
步骤(3)中的碱可为通式M
a(OH)
b的化合物,其中M为选自NH
4、Li、Na、K、Rb、Cs、Ca、Sr、Ba的其中一种,或二种或多种的混合。但本发明并不以此为限,应视为本发明可以采用适当的碱。
步骤(4)中,所加入的酸的种类可以为:硝酸、硫酸、盐酸、氢溴酸、氢氟酸、氢碘酸、甲酸、乙酸、乙酸酐、草酸、丙二酸、三氟乙酸等,可以使用如上单独一种酸,也可为两种或多种酸混合使用。举例来说,可以为盐酸、甲酸、乙酸酐、三氟乙酸;但本发明并不以此为限,应视为本发明可以采用任意适合用于本反应的酸。
步骤(4)中,反应的温度可以是-20℃至130℃,例如可为40℃、70℃、100℃(加热回流)、110℃、120℃。但本发明并不以此为限,应视为本发明可以采用任意适合用于本反应的反应温度。
第二步反应:合成第一中间产物
(6)将实施例2的前体物质分散于溶剂中。
(7)加入一定量的盐类及一定量的叠氮化物,在一定的反应温度下反应一定时间。
(8)加入酸,纯化并得到实施例2的第一中间产物。
本部分步骤(6)所用的溶剂、步骤(7)所加入的盐类、叠氮化物,反应温度及反应时间,以及步骤(8)中所加入的酸,可参照通用合成方式步骤二中所述的反应条件。
第三步反应:合成第二中间产物
(9)将实施例2的第一中间产物缓慢加入硝化体系,加完后控制反应体系温度,保持硝化反应进行。
(10)终止反应,过滤。
(11)纯化、干燥,得到实施例2的第二中间产物。
本部分步骤(9)所加入的硝化体系及反应温度,以及步骤(10)中终止反应的方式,可参照通用合成方式步骤三中所述的反应条件。
第四步反应:合成产物
(12)将实施例2的第二中间产物分散于溶剂中。
(13)加入一定量的浓氨水,回流反应一段时间,冷却过滤得到固体。
(14)将此固体投入氧化性溶液中,反应一段时间。
(15)终止反应,过滤、纯化,得到实施例2的产物,即图11所示的化合物。
本部分步骤(12)中所用的溶剂,步骤(13)所用的浓氨水浓度、用量比例及反应时间,以及步骤(14)中的氧化性溶液选择及反应时间,可参照通用合成方式步骤四丙部分中所述的反应条件。
关于上述反应的具体范例,请参照以下实施例2A。
[实施例2A]
将丙二腈6.66g和亚硝酸钠20.7g分散在100mL水中,冰水浴冷却,而后滴加含27.8g盐酸羟胺的200mL水溶液(半小时加完),滴加完毕后置于-10℃下,加入氢氧化钠(10.1g)的100mL水溶液,加乙酸乙酯100mL,搅拌反应10小时,而后过滤。将所得到固体溶于乙醚200mL,加入乙酸20mL,二氧化铅17.2g,加热回流,过滤,将母液旋干。
将所得到固体与3.6g氟化铵、12g叠氮化钠一起分散于10mL水中,之后在75℃下反应7小时,冷却至室温再向其中滴加盐酸,直至溶液pH值为3.1,而后过滤,以5mL冰水洗涤,将固体干燥,得到橙色粉末12.2g。
接着取0.4g所得橙色粉末,加入到100%硝酸/三氟乙酸酐按照一比一的体积比配置的6mL混合液中,而后在室温下反应1小时,将反应液倒入50g冰水中,搅拌0.5小时后,过滤,用冰水10mL洗涤,干燥之后得到0.51g黄色粉末状固体。
而后将所得到固体溶于5mL甲醇,加入浓氨水4mL,回流反应2小时,冷却过滤得到淡黄色粉末状产物。将此产物投入浓硫酸与30%双氧水以1:1体积比混合的5mL溶剂中,搅拌反应1小时,而后倾入20g冰中,过滤得到橘黄色固体0.44g,得率69%。
【产物测定结果】
元素分析EA:测量值(理论值)C:19.59%(19.58%);N:45.67%(45.66%);O:34.74%(34.77%)
红外光谱IR(KBr,γ/cm
-1):3019(m),2790(s),1598(s),1535(s),1371(s),1341(m),1209(m),871(m),827(m),740(s),688(m),540(w)
实施例3
图14为本发明的一种化合物的其中一种实施方式的化学结构图,其具体的名称为:2-氧化-4-氯-7H-咪唑[4,5-d][1,2,3]三嗪,具有如图1所示的结构,具体来说具有如图3所示的结构,更具体来说具有如图15所示的结构。
【合成方法】
实施例3的化合物具体的合成方法,具体为采用4-氨基-5-氰基咪唑作为前体物质进行合成。在此详述如下:
第一步反应:合成第一中间物质
(1)将4-氨基-5-氰基咪唑分散于溶剂中。
(2)加入一定量的盐类及一定量的叠氮化物,在一定的反应温度下反应一定时间。
(3)加入酸,纯化并得到实施例3的第一中间产物。
本部分步骤(1)所用的溶剂、(2)所加入的盐类、叠氮化物,反应温度及反应时间,以及步骤(3)中所加入的酸,可参照通用合成方式步骤二中所述的反应条件。
第二步反应:合成第二中间产物
(4)将实施例3的第一中间产物缓慢加入硝化体系,加完后控制反应体系温度,保持硝化反应在一定温度下进行一定时间。
(5)终止反应,纯化,得到实施例3的第二中间产物。
本部分步骤(4)所加入的硝化体系及反应温度,以及步骤(5)中终止反应的方式,可参照通用合成方式步骤三中所述的反应条件。
第三步反应:合成产物
(6)将实施例3的第二中间产物分散于溶剂中。
(7)通入氯气气流,在一定温度下反应一段时间。
(8)纯化、干燥,得到实施例3的产物,即图12所示的化合物。
本部分步骤(6)中所用的溶剂,步骤(7)所用的氯气流量、反应温度及反应时间,可参照通用合成方式步骤四甲部分中所述的反应条件。
关于上述反应的具体范例,请参照以下实施例3A。
[实施例3A]
取4-氨基-5-氰基咪唑10.8g,与3.6g氟化铵、12g叠氮化钠一起分散于10mL水中,之后在75℃下反应7小时,冷却至室温再向其中滴加盐酸,直至溶液pH值为3.1,而后过滤,以5mL冰水洗涤,将固体干燥,得到褐色粉末11.3g。
接着取0.5g所得褐色粉末,加入到100%硝酸/三氟乙酸酐按照一比一的体积比配置的20mL混合液中,而后在-10℃下反应4小时,将反应液倒入50g冰水中,搅拌0.5小时后,过滤,用冰水20mL洗涤,干燥之后得到0.28g黄绿色粉末状固体,得率30%。
将所得固体溶于8mL二氯甲烷,通入氯气气流(流量5mL/min),在70℃反应1小时,过滤,得到白色粉末状产物0.17g,收率为30%。
【产物测定结果】
元素分析EA:测量值(理论值)C:27.79%(28.01%);H:1.12%(1.18%);Cl:20.61%(20.67%);N:40.77%(40.83%);O:9.29%(9.33%)
红外光谱IR(KBr,γ/cm
-1):3321(s),3290(m),1577(s),1555(s),1366(s),1332(m),1220(m),996(w),871(m),827(m),765(w),621(m)
实施例4
图16为本发明的一种化合物的其中一种实施方式的化学结构图,其具体的名称为:3,5-二氧化-7-氨基-[1,2,5]噁二唑[3,4-d][1,2,3]三嗪,具有如图1所示的结构,具体来说具有如图3所示的结构,更具体来说具有如图17所示的结构。
【合成方法】
实施例4的化合物具体的合成方法,在此详述如下:
第一步反应:合成前体物质
(1)将丙二腈和亚硝酸盐分散于溶剂中。
(2)在反应液中加入酸。反应结束后,纯化并取得产物。
(3)将所得产物分散于溶剂中。
(4)加入羟胺化合物,回流反应。
(5)过滤、将母液旋干,得到实施例4的前体物质。
步骤(1)至步骤(4)中,反应的温度可以是-20℃-130℃,例如可为-20℃、-10℃、40℃。但本发明并不以此为限,应视为本发明可以采用任意适合用于本反应的反应温度。
步骤(1)中的溶剂可为N,N-二甲基甲酰胺、二甲基亚砜、四氢呋喃、乙腈、二氯甲烷、氯仿、甲醇、乙醇、水、异丙醇、乙二醇、乙二醇单甲醚、乙二醇单乙醚、乙二醇单丁醚、甘油、丙酮、三乙醇胺、吡啶、苯、甲苯、二甲苯等中的任意一种,或二种或多种的混合物。但本发明并不以此为限,应视为本发明可以采用任意适合用于本反应的溶剂。
步骤(1)中溶剂的用量体积与反应物的质量比,例如可为溶剂体积:丙二腈=2mL/g-2mL/mg;例如可为10mL/g、15mL/g、35mL/g、75mL/g或400mL/g。但本发明并不以此为限,应视为本发明可以采用任意适合用于本反应的溶剂比例。
步骤(1)中亚硝酸盐可为通式MNO
2的化合物,其中M为选自NH
4、Li、Na、K、Rb及Cs的其中一种,或二种或多种的混合。举例来说,可以是亚硝酸钠。
步骤(2)中,所加入的酸的种类可以为:硝酸、硫酸、盐酸、氢溴酸、氢氟酸、氢碘酸、甲酸、乙酸、乙酸酐、草酸、丙二酸、三氟乙酸等,可以使用如 上单独一种酸,也可为两种或多种酸混合使用。举例来说,可以为盐酸、甲酸、乙酸酐、三氟乙酸;但本发明并不以此为限,应视为本发明可以采用任意适合用于本反应的酸。
步骤(4)中,羟胺化合物可为羟胺盐类。举例来说,羟胺盐类可为通式(NH
3OH)
aX
b的化合物,其中X为选自F、Cl、Br、I、NO
3、SO
4、HSO
4、CO
3、HCO
3、HCOO、CH
3COO、CF
3COO、(COO)
2及CH
2(COO)
2的其中一种,或二种或多种的混合。举例来说,所述羟胺化合物可为羟胺卤化盐类;举例来说,所述羟胺化合物可为盐酸羟胺。
步骤(2)、(4)中的反应时间可以为2小时至24小时,例如可以是2小时、3小时、4小时、5小时、10小时、24小时。但本发明并不以此为限,应视为本发明可以采用任意适合用于本反应的反应时间。
第二步反应:合成第一中间产物
(6)将实施例4的前体物质分散于溶剂中。
(7)加入一定量的盐类及一定量的叠氮化物,在一定的反应温度下反应一定时间。
(8)加入酸,纯化并得到实施例4的第一中间产物。
本部分步骤(6)所用的溶剂、步骤(7)所加入的盐类、叠氮化物,反应温度及反应时间,以及步骤(8)中所加入的酸,可参照通用合成方式步骤二中所述的反应条件。
第三步反应:合成第二中间产物
(9)将实施例4的第一中间产物缓慢加入硝化体系,加完后控制反应体系温度,保持硝化反应在一定温度下进行一定时间。
(10)终止反应,过滤、纯化,得到实施例4的第二中间产物。
本部分步骤(9)所加入的硝化体系及反应温度,以及步骤(10)中终止反应的方式,可参照通用合成方式步骤三中所述的反应条件。
第四步反应:合成产物
(11)将实施例4的第二中间产物分散于溶剂中。
(12)加入一定量的浓氨水,回流反应一段时间,反应完后冷却。
(13)过滤、纯化,得到实施例4的产物,即图13所示的化合物。
本部分步骤(11)中所用的溶剂以及步骤(12)所用的浓氨水浓度、用量比例及反应时间,可参照通用合成方式步骤四乙部分中所述的反应条件。
关于上述反应的具体范例,请参照以下实施例4A。
[实施例4A]
将丙二腈6.66g和亚硝酸钠20.7g分散在100mL水中,滴加5mL乙酸,室温反应3小时,以200mL乙酸乙酯萃取,之后将乙酸乙酯层旋干。将所得到物质分散于200mL乙醇中,加入盐酸羟胺27.8g,回流反应4小时,过滤,将母液旋干。
在室温下,将式所得到固体分散于300mL由丙酮和甲醇按照体积比为4:1混合而成的溶剂中,接着分批加入氯化亚钴13g、叠氮化钠15.2g,然后在回流条件下继续反应10小时,再向其中滴加三氟乙酸,直至溶液pH值为2.4,而后过滤,以30mL冰水洗涤,而后以30mL乙醚洗涤,将固体干燥,得到白色粉末。
接着取所得白色粉末,加入到加有11.1g四氟硼硝的200mL乙腈中,而后在4℃下反应4小时,将反应液倒入50g冰水中,搅拌0.5小时后,过滤,用冰水20mL洗涤,而后以10mL石油醚洗涤,干燥之后得到12.1g米白色粉末状固体,得率75%。
而后将所得到固体溶于150mL甲醇,加入浓氨水140mL,回流反应2小时,冷却过滤得到最终淡黄色粉末状产物6.97g,收率为40%。
【产物测定结果】
元素分析EA:测量值(理论值)C:21.19%(21.18%);H:1.16%(1.19%);N:49.33%(49.41%);O:28.19%(28.22%)
红外光谱IR(KBr,γ/cm
-1):3434(s),3134(m),3019(m),2790(s),2321(m),1571(s),1512(s),1380(s),1321(m),836(m),721(s),644(m)
实施例5
图18为本发明的一种化合物的其中一种实施方式的化学结构图,其具体的名称为:2-氧化-4-硝氨基苯并[d][1,2,3]三嗪,具有如图1所示的结构,具体来说具有如图4所示的的结构,更具体来说具有如图19所示的结构。
【合成方法】
实施例5的化合物具体的合成方法,具体为采用1-氨基-2-氰基-苯作为前体物质进行合成。在此详述如下:
第一步反应:合成第一中间产物
(1)将1-氨基-2-氰基-苯分散于溶剂中。
(2)加入一定量的盐类及一定量的叠氮化物,在一定的反应温度下反应一定时间。
(3)加入酸,纯化并得到实施例5的第一中间产物。
本部分步骤(1)所用的溶剂、步骤(2)所加入的盐类、叠氮化物,反应温度及反应时间,以及步骤(3)中所加入的酸,可参照通用合成方式步骤二中所述的反应条件。
第二步反应:合成第二中间产物
(4)将实施例5的第一中间产物缓慢加入硝化体系,加完后控制反应体系温度,保持硝化反应在一定温度下进行一定时间。
(5)终止反应,过滤、纯化,得到实施例5的第二中间产物。
本部分步骤(4)所加入的硝化体系及反应温度,以及步骤(5)中终止反应的方式,可参照通用合成方式步骤三中所述的反应条件。
第三步反应:合成产物
(6)将第二中间产物分散于溶剂中。
(7)加入一定量的浓氨水,回流反应一段时间。
(8)冷却过滤得固体。
(9)将所得到的固体加入硝化体系中,搅拌一段时间。
(10)终止反应,过滤、纯化,得到实施例5的产物,即图14所示的化合物。
本部分步骤(6)中所用的溶剂,步骤(7)所用的浓氨水浓度、用量比例及反应时间,步骤(9)所加入的硝化体系及反应温度,以及步骤(10)中终止反应的方式,可参照通用合成方式步骤四丁部分中所述的反应条件。
关于上述反应的具体范例,请参照以下实施例5A。
[实施例5A]
将1-氨基-2-氰基苯11.8g分散于300mL由丙酮和甲醇按照体积比为4:1混合而成的溶剂中,接着分批加入氯化亚钴13g、叠氮化钠15.2g,然后在回流条件下继续反应10小时,再向其中滴加三氟乙酸,直至溶液pH值为3,而后过滤,以30mL冰水洗涤,而后以30mL乙醚洗涤,将固体干燥,得到灰色粉末。
接着取所得灰色粉末,加入到加有13.3g四氟硼硝的180mL乙腈中,而后在-5℃下反应7小时,将反应液倒入50g冰水中,搅拌0.5小时后,过滤,用冰水20mL洗涤,而后以10mL石油醚洗涤,干燥之后得到米黄色粉末状固体。
而后将所得到固体溶于150mL甲醇,加入浓氨水140mL,回流反应2小时,冷却过滤得到淡黄色粉末状固体。将所得到固体加入100mL发烟硝酸中,搅拌2小时,倾入300g冰中,过滤,水洗得到黄色固体11.5g。
【产物测定结果】
元素分析EA:测量值(理论值)C:40.51%(40.59%);H:2.39(2.43);N:33.86%(33.81%);O:23.27%(23.17%)
红外光谱IR(KBr,γ/cm
-1):3435(m),3021(w),2824(s),2708(s),1602(s),1581(s),1535(s),1500(s),1453(m),1369(s),1323(m),908(w),701(m),634(m)
实施例6
图20为本发明的一种化合物的其中一种实施方式的化学结构图,其具体的名称为:2-氧化-4-硝基哒嗪[4,5-d][1,2,3]三嗪,具有如图1所示的结构,具体来 说具有如图4所示的的结构,更具体来说具有如图21所示的结构。
【合成方法】
实施例6的化合物具体的合成方法,具体为采用4-氨基-5氰基哒嗪作为前体物质进行合成。在此详述如下:
第一步反应:合成第一中间产物
(1)将4-氨基-5氰基哒嗪分散于溶剂中。
(2)加入一定量的盐类及一定量的叠氮化物,在一定的反应温度下反应一定时间。
(3)加入酸,纯化并得到实施例6的第一中间产物。
本部分步骤(1)所用的溶剂、步骤(2)所加入的盐类、叠氮化物,反应温度及反应时间,以及步骤(3)中所加入的酸,可参照通用合成方式步骤二中所述的反应条件。
第二步反应:合成第二中间产物
(4)将实施例6的第一中间产物缓慢加入硝化体系,加完后控制反应体系温度,保持硝化反应在一定温度下进行一定时间。
(5)终止反应、过滤、纯化,得到实施例6的第二中间产物。
本部分步骤(4)所加入的硝化体系及反应温度,以及步骤(5)中终止反应的方式,可参照通用合成方式步骤三中所述的反应条件。
第三步反应:合成产物
(6)将实施例6的第二中间产物分散于溶剂中。
(7)加入一定量的浓氨水,回流反应一段时间,冷却过滤得到固体。
(8)将此固体投入氧化性溶液中,反应一段时间。
(9)终止反应,过滤、纯化,得到实施例2的产物,即图11所示的化合物。
本部分步骤(6)中所用的溶剂,步骤(7)所用的浓氨水浓度、用量比例及反应时间,以及步骤(8)中的氧化性溶液选择及反应时间,可参照通用合成方式步骤四丙部分中所述的反应条件。
关于上述反应的具体范例,请参照以下实施例6A。
[实施例6A]
取4-氨基-5-氰基-哒嗪11.1g与5.1g氯化铵、11g叠氮化铵一起分散于50mL水中,之后在85℃下反应7小时,冷却至室温再向其中滴加盐酸,直至溶液pH值为1,而后过滤,以15mL冰水洗涤,将固体干燥,得到褐色粉末12.8g。
接着取0.4g所得褐色粉末,加入到100%硝酸/三氟乙酸酐按照一比一的体积比配置的6mL混合液中,而后在室温下反应1小时,将反应液倒入50g冰水中,搅拌0.5小时后,过滤,用冰水10mL洗涤,干燥之后得到0.55g黄褐色粉末状固体。
而后将所得到的固体溶于5mL甲醇,加入浓氨水4mL,回流反应2小时,冷却过滤得到淡黄色粉末状产物。将此产物投入浓硫酸与30%双氧水以1:1体积比混合的5mL溶剂中,搅拌反应1小时,而后倾入20g冰中,过滤得到黄色固体。
【产物测定结果】
元素分析EA:测量值(理论值)C:30.91%(30.94%);H:1.01%(1.04%);N:43.12%(43.30%);O:24.63%(24.73%)
红外光谱IR(KBr,γ/cm
-1):3091(s),1592(s),1541(s),1366(s),1339(m),881(m),831(m),712(s)
实施例7
图22为本发明的一种化合物的其中一种实施方式的化学结构图,其具体的名称为:2,7-氧化-4,5-二叠氮-[1,2,3]三嗪[4,5-d][1,2,3]三嗪,具有如图1所示的结构,具体来说具有如图4所示的的结构,更具体来说具有如图23所示的结构。
【合成方法】
实施例7的化合物具体的合成方法,具体为采用3,3-二氨基-2-(1H-四唑-5-基)丙腈作为前体物质进行合成。在此详述如下:
第一步反应:合成第一中间产物
(1)将4-氨基-5氰基哒嗪分散于溶剂中。
(2)加入一定量的盐类及一定量的叠氮化物,在一定的反应温度下反应一定时间。
(3)加入酸,纯化并得到实施例7的第一中间产物。
本部分步骤(1)所用的溶剂、步骤(2)所加入的盐类、叠氮化物,反应温度及反应时间,以及步骤(3)中所加入的酸,可参照通用合成方式步骤二中所述的反应条件。
第二步反应:合成产物
(4)将实施例7的第一中间产物缓慢加入硝化体系,加完后控制反应体系温度,保持硝化反应在一定温度下进行一定时间。
(5)终止反应、过滤、纯化,得到实施例7的产物,即图16所示的化合物。
本部分步骤(4)所加入的硝化体系及反应温度,以及步骤(5)中终止反应的方式,可参照通用合成方式步骤三中所述的反应条件。
关于上述反应的具体范例,请参照以下实施例7A。
[实施例7A]
取3,3-二氨基-2-(1H-四唑-5-基)丙腈15.3g,与5.1二甲胺盐酸盐、11g叠氮化铵一起分散于50mLDMF中,之后在85℃下反应3小时,过滤,将母液浓缩至10mL,再向其中加入50mL水,继而滴加盐酸,直至溶液pH值为2,而后过滤,以15mL冰水洗涤,将固体干燥,得到灰白色粉末14.1g。
接着取0.4g所得灰白色粉末,加入到3mL 100%硝酸与3mL浓H2SO4组成的混合溶液中,而后在60℃下反应4小时,将反应液倒入20g冰水中,搅拌0.5小时后,以100mL乙酸乙酯萃取,而后将有几层旋干,干燥之后得到0.50g黄褐色粉末状固体。
【产物测定结果】
元素分析EA:测量值(理论值)C:19.38%(19.38%);H:0.01%(0.00%);N:67.77%(67.74%);O:12.93%(12.90%)
红外光谱IR(KBr,γ/cm
-1):3110(s),2163(s),1855(m),1313(s),899(m),842(m),659(s)
实施例8
图24为本发明的一种化合物的其中一种实施方式的化学结构图,其具体的名称为:6-氧化-8-硝氨基[1,2,5]噁二唑[3',4':5,6]吡嗪[2,3-d][1,2,3]三嗪,具有如图1所示的结构,具体来说具有如图5所示的结构,更具体来说具有如图25所示的结构。
【合成方法】
实施例8的化合物具体的合成方法,在此详述如下:
第一步反应:合成前体物质
(1)将3,4-二氨基呋咱及乙二酸分散于溶剂中。
(2)加入酸,进行反应。
(3)将所得产物溶于二氯亚砜,滴加N,N-二甲基甲酰胺,回流进行反应。
(4)终止反应,过滤,得到固体。
(5)将所得固体分散于溶剂中,加入氮化物,回流进行反应。
(6)冷却、过滤,将母液旋干。
(7)将所得固体分散于溶剂中,加入浓氨水或通入氨气气流,回流进行反应。
(8)冷却、过滤,将母液旋干,得到实施例8的前体物质。
步骤(1)至步骤(3)中,反应的温度可以是-20℃-130℃,例如可为40℃、70℃、100℃(加热回流)、110℃、120℃。但本发明并不以此为限,应视为本发明可以采用任意适合用于本反应的反应温度。
步骤(1)或(5)中的溶剂可为N,N-二甲基甲酰胺、二甲基亚砜、四氢呋喃、乙腈、二氯甲烷、氯仿、甲醇、乙醇、水、异丙醇、乙二醇、乙二醇单甲醚、乙二醇单乙醚、乙二醇单丁醚、甘油、丙酮、三乙醇胺、吡啶、苯、甲苯、二甲苯等中的任意一种,或二种或多种的混合物。但本发明并不以此为限,应视为本发明可以采用任意适合用于本反应的溶剂。
步骤(1)中溶剂的用量体积与反应物的质量比,例如可为溶剂体积:3,4-二氨基呋咱=2mL/g-2mL/mg;例如可为9.9mL/g、10mL/g、15mL/g、35mL/g、75mL/g或400mL/g。但本发明并不以此为限,应视为本发明可以采用任意适合用于本反应的溶剂比例。
步骤(2)中,所加入的酸的种类可以为:浓盐酸、浓氢溴酸、浓氢氟酸、浓氢碘酸等,可以使用如上单独一种酸,也可为两种或多种酸混合使用;但本发明并不以此为限,应视为本发明可以采用任意适合用于本反应的酸。
在步骤(3)中的反应时间,可为0.5至12小时,例如可为0.5至4小时,例如可为1至4小时,例如可为0.5小时、1小时、2小时、4小时。但本发明并不以此为限,应视为本发明可以采用任意适合用于本反应的反应时间。
在步骤(5)中的氮化物,可为通式R
a(N)
b的化合物,其中R为选自Li、Na、K、Rb、Cu、Fe、Ag、Au、Pb、Cd、Ni及H的其中一种,或二种或多种的混合,并选择对应的a与b。
在步骤(5)中的反应时间,可为0.5至12小时,例如可为0.5至4小时,例如可为1至4小时,例如可为0.5小时、1小时、2小时、4小时。但本发明并不以此为限,应视为本发明可以采用任意适合用于本反应的反应时间。
在步骤(7)中的溶剂,可以是氨气气流不溶或难溶的溶剂,或是浓氨水可溶的溶剂。举例来说,当步骤(7)采用氨气气流时,溶剂可为二氯甲烷、氯仿、丙酮、苯、甲苯、二甲苯等中的任意一种,或二种或多种的混合物;当步骤(7)采用浓氨水时,溶剂可为甲醇、乙醇、水、异丙醇、乙二醇、甘油、丙酮等中的任意一种,或二种或多种的混合物。举例来说,步骤(7)可采用氨气气流,其溶剂可为二氯甲烷。
在步骤(7)中的反应时间,可为0.5至12小时,例如可为0.5至4小时,例如可为1至4小时,例如可为0.5小时、1小时、2小时、4小时。但本发明并不以此为限,应视为本发明可以采用任意适合用于本反应的反应时间。
第二步反应:合成第一中间产物
(9)将实施例8的前体物质分散于溶剂中。
(10)加入一定量的盐类及一定量的叠氮化物,在一定的反应温度下反应一定时间。
(11)加入酸,纯化并得到实施例8的第一中间产物。
本部分步骤(9)所用的溶剂、步骤(10)所加入的盐类、叠氮化物,反应温度及反应时间,以及步骤(11)中所加入的酸,可参照通用合成方式步骤二中所述的反应条件。
第三步反应:合成第二中间产物
(12)将实施例8的第一中间产物缓慢加入硝化体系,加完后控制反应体系温度,保持硝化反应在一定温度下进行一定时间。
(13)终止反应,过滤、纯化,得到实施例8的第二中间产物。
本部分步骤(12)所加入的硝化体系及反应温度,以及步骤(13)中终止反应的方式,可参照通用合成方式步骤三中所述的反应条件。
第四步反应:合成产物
(14)将第二中间产物分散于溶剂中。
(15)加入一定量的浓氨水,回流反应一段时间。
(16)冷却过滤得固体。
(17)将所得到的固体加入硝化体系中,搅拌一段时间。
(18)终止反应,过滤、纯化,得到实施例8的产物,即图17所示的化合物。
本部分步骤(14)中所用的溶剂,步骤(15)所用的浓氨水浓度、用量比例及反应时间,步骤(17)所加入的硝化体系及反应温度,以及步骤(18)中终止反应的方式,可参照通用合成方式步骤四丁部分中所述的反应条件。
关于上述反应的具体范例,请参照以下实施例8A。
[实施例8A]
3,4-二氨基呋咱10.1g与15.1g乙二酸分散于100mL水中,而后在冰水浴下加入20mL浓盐酸,升温至回流反应4小时,冷却、过滤,得到固体产物。
将该固体溶于二氯亚砜70mL,滴加5mL N,N-二甲基甲酰胺,升温至回流反应4小时,倒入200g冰水中,过滤得到固体。而后将其溶于50mL丙酮中,加入3.9g氮化钠,升温至回流反应1小时,冷却、过滤,将母液旋干。将所得固体溶于60mL二氯甲烷,通入氨气气流(流量5mL/min),回流反应4h,冷却、过滤,将母液旋干。
将所得固体分散于300mL由丙酮和甲醇按照体积比为4:1混合而成的溶剂中,接着分批加入氯化亚钴13g、叠氮化钠15.2g,然后在回流条件下继续反应10小时,再向其中滴加稀硫酸,直至溶液pH值为3.3,而后过滤,以30mL冰水洗涤,而后以30mL乙醚洗涤,将固体干燥,得到粉色粉末。
接着取所得粉色粉末,加入到加有13.3g四氟硼硝的180mL乙腈中,而后在0℃下反应3小时,将反应液倒入50g冰水中,搅拌0.5小时后,过滤,用冰水20mL洗涤,而后以10mL石油醚洗涤,干燥之后得到淡黄色粉末状固体。
而后将所得到固体溶于90mL甲醇,加入浓氨水900mL,回流反应2小时,冷却过滤得到桔黄色粉末状固体。将所得到固体加入60mL发烟硝酸中,搅拌2小时,倾入200g冰中,过滤,水洗得到黄色固体7.7g。
【产物测定结果】
元素分析EA:测量值(理论值)C:23.89%(23.91%);H:0.35%(0.40%);N:50.23%(50.20%);O:25.44%(25.48%)
红外光谱IR(KBr,γ/cm
-1):3011(s),2182(w),2147(m),1591(s),1522(s),1331(s),1304(m),851(m),671(s)
实施例9
图26为本发明的一种化合物的其中一种实施方式的化学结构图,其具体的名称为:2-氧化-4-氯-9H-[1,2,4]三唑[5'-1':3,4][1,2,4]三嗪[6,1-d][1,2,3,5]四嗪,具有如图1所示的结构,具体来说具有如图5所示的结构,更具体来说具有如图27所示的结构。
【合成方法】
实施例9的化合物具体的合成方法,在此详述如下:
第一步反应:合成前体物质
(1)将3-氨基-1,2,4-三氮唑分散于溶剂中。
(2)加入硝基乙腈钠盐,回流反应。
(3)冷却、过滤,将母液旋干,得到实施例9的前体物质。
步骤(1)中的溶剂可为N,N-二甲基甲酰胺、二甲基亚砜、四氢呋喃、乙腈、二氯甲烷、氯仿、甲醇、乙醇、水、异丙醇、乙二醇、乙二醇单甲醚、乙二醇单乙醚、乙二醇单丁醚、甘油、丙酮、三乙醇胺、吡啶、苯、甲苯、二甲苯等中的任意一种,或二种或多种的混合物。举例来说,可以是乙腈。但本发明并不以此为限,应视为本发明可以采用任意适合用于本反应的溶剂。
步骤(1)中溶剂的用量体积与反应物的质量比,例如可为溶剂体积:3-氨基-1,2,4-三氮唑=2mL/g–2mL/mg;例如可为5.95mL/g、10mL/g、15mL/g、35mL/g、75mL/g或400mL/g。但本发明并不以此为限,应视为本发明可以采用任意适合用于本反应的溶剂比例。
步骤(2)中硝基乙腈化合物可为硝基乙腈盐类,举例来说,所述硝基乙腈化合物可为通式M
a(CH(NO
2)CN)
b的化合物,其中M为选自Li、Na、K、Rb、Cs的其中一种,或二种或多种的混合。具体来说,所述硝基乙腈化合物可为硝基乙腈钠盐。
第二步反应:合成第一中间物质
(4)将实施例9的前体物质分散于溶剂中。
(5)加入一定量的盐类及一定量的叠氮化物,在一定的反应温度下反应一定时间。
(6)加入酸,纯化并得到实施例3的第一中间产物。
本部分步骤(4)所用的溶剂、步骤(5)所加入的盐类、叠氮化物,反应温度及反应时间,以及步骤(6)中所加入的酸,可参照通用合成方式步骤二中 所述的反应条件。
第三步反应:合成第二中间产物
(7)将实施例3的第一中间产物缓慢加入硝化体系,加完后控制反应体系温度,保持硝化反应在一定温度下进行一定时间。
(8)终止反应,纯化,得到实施例3的第二中间产物。
本部分步骤(7)所加入的硝化体系及反应温度,以及步骤(8)中终止反应的方式,可参照通用合成方式步骤三中所述的反应条件。
第三步反应:合成产物
(9)将实施例3的第二中间产物分散于溶剂中。
(10)通入氯气气流,在一定温度下反应一段时间。
(11)纯化、干燥,得到实施例3的产物,即图12所示的化合物。
本部分步骤(9)中所用的溶剂,步骤(10)所用的氯气流量、反应温度及反应时间,可参照通用合成方式步骤四甲部分中所述的反应条件。
关于上述反应的具体范例,请参照以下实施例9A。
[实施例9A]
3-氨基-1,2,4-三氮唑8.4g分散于50mL乙腈中,再加入12.1g硝基乙腈钠盐,升温至回流反应3小时,冷却、过滤,将母液旋干。
将所得到固体与3.6g硝酸铵和15.1g叠氮化钠分散在100mL水中,之后在75℃下反应7小时,冷却至室温再向其中滴加盐酸,直至溶液pH值为3.1,而后过滤,以5mL冰水洗涤,将固体干燥,得到褐色粉末11.3g。
接着取0.5g所得褐色粉末,加入到100%硝酸/三氟乙酸酐按照一比一的体积比配置的20mL混合液中,而后在-10℃下反应4小时,将反应液倒入50g冰水中,搅拌0.5小时后,过滤,用冰水20mL洗涤,干燥之后得到0.28g黄绿色粉末状固体,得率30%。
将所得固体溶于20mL二氯甲烷,通入氯气气流(流量5mL/min),在60℃反应1小时,过滤,得到白色粉末状产物0.17g,收率为30%。
【产物测定结果】
元素分析EA:测量值(理论值)C:26.41%(26.50%);H:1.28%(1.33%);Cl:15.59%(15.65%);N:49.42%(49.45%);O:7.10%(7.06%)
红外光谱IR(KBr,γ/cm
-1):3011(s),2280(w),2096(m),1571(s),1379(s),901(m),735(s),610(w)
起爆药性质测定
【极限起爆药量测定I】
依照GJB 5891.19-2006所规定的标准执行。具体来说,依照以下步骤:
(1)将被测起爆药(例如是实施例1-9的化合物中的待测定者)、三硝基间苯二酚铅(符合WJ 617规范)、黑索今(符合GJB 296A-1995标准)分别放入水浴或油浴烘箱中,在60℃±2℃的温度下烘2小时,取出,放入干燥器中冷却至室温,备用。
(2)用分析天平称取经步骤(1)处理的黑索今0.030g,准确至0.0002g,置于铜管壳(直径5.1mm,高8.2mm)中,以117.6MPa的压力压药。
(3)分别称取经步骤(1)处理的三硝基间苯二酚铅(对于直接能被电点火头点燃的被测药剂,可以不用三硝基间苯二酚铅作点火药)0.030g、黑索今0.020g和待测药量的被测起爆药,准确至0.0002g,依次置于带有绸垫(符合FZ66201标准的规格401平纹绸)的铝加强帽(可与铜管壳紧配合)内,以49.0MPa的压力压药,并做好记录。
(4)称取黑索今0.020g,准确至0.0002g,并依次将此步骤称取的黑索今和经步骤(3)中压好药的铝加强帽装入步骤(2)中的铜管壳内,以49.0MPa的压力压制成试样10发,并编号,记录每一编号的被测起爆药质量,备用。
(5)如图28所示,将装好的试样2置于铅板3(符合WJ 580规范,直径为35mm,厚2mm)上,另一方面连接电点火头1,连接至包括电源4、开关5的直流电源电路中。
(6)打开开关5以接通电源4,起爆试样。检查雷管是否被成功引爆,且铅板是否被击穿。
(7)测量试样在铅板上的炸孔:炸孔直径不小于试样外径时为全爆,小于试样外径时为半爆。若连续试验的数发试样均全爆,则可适当减少被测起爆药的药量再进行试验;若试样有半爆,则可适当增加被测药剂的药量;直至找到数发试样连续试验均为全爆时被测起爆药的最小药量。
(8)依照找出的极限起爆药量,压制试样50发进行试验以进行复核。此时,试样均应全爆。
举例来说,实施例1的极限起爆药量测定结果如表1:
| 起爆药装药量 | 起爆结果 | 全爆或半爆比例 |
| 150mg | 全爆 | 10/10 |
| 140mg | 全爆 | 10/10 |
| 130mg | 全爆 | 10/10 |
| 125mg | 全爆 | 10/10 |
| 120mg | 半爆或不爆 | 6/10 |
表1
由上述表1,可以得到实施例1的极限起爆药量在上述的测试条件下为120mg。本测定中,进行了实施例1-9的极限起爆药量测定,每个实施例选取至少5种起爆药装药量,每一种起爆药量装填10发,结果如后述表2。
【热分解温度测定】
使用梅特勒-托利多公司的TGA/DSC3+型同步热分析仪,在10℃·min
-1升温速率和20mL·min
-1的氮气流量下,在50℃至250℃的温度范围内测得实施例1-9的化合物的热分解温度,结果如后述表2。
【密度测定】
利用Micromeritics Accupyc II 1340比重瓶自动真密度计在25℃下测量实施例1-9的化合物的密度,结果如后述表2。
【安息角测定】
依照以下步骤进行试验准备:
(1)准备如图29所示,金属(例如铜H62)制,表面光滑、角度为30°的漏斗,其出口处内径为4mm,外表面去毛刺、内表面镀铬,余导角为R=1~2;并准备如图30所示,金属材料的三支架,其高度的设计应使漏斗的出口端面到测量面的距离为36mm。
(2)将被测药剂(实施例1-9的化合物的待测试者)按相应规定进行干燥,达到技术要求后,在干燥器中自然冷却30分钟备用。
(3)在防护板后的平面上,放上测量坐标纸(例如是尺寸150mm×150mm)。将漏斗套在支架上后,将其放在测量坐标纸的正中间。
(4)用架盘天平(例如是最大称量为200g,分度值为0.1g)称取经处理的被测药剂3g,准确至0.1g,置于纸质簸箕中备用。
接着依照以下步骤进行试验:
(1)在防护板后,将纸质簸箕中的被测药剂延漏斗壁缓慢倒入漏斗中,使药剂自由落到测量坐标纸上,形成锥体。
(2)移开漏斗及其支架,用高度游标卡尺(例如是测量范围为0mm至150mm,分度值0.02mm)测量圆锥体高度,准确至1mm。
(3)用铅笔在测量坐标纸上标出圓锥体底面互相垂直的两个直径的端点。
(4)将测量坐标纸上的药剂倒回纸质簸箕中,读出测量坐标纸上标出的两个互相垂直的直径长度,准确至1mm取其算术平均值。
(5)用毛刷将漏斗和测量坐标纸上的浮药扫至废药杯中。
如此可通过以下式(1)以圆锥体的高度及其两个直径长度算出安息角:
θ=tan
-1(2h/D) 式(1)
其中,θ为被测药剂的安息角,h为圆锥体高度,D圆锥体底面相互垂直的两个直径的算术平均值。算出的安息角的结果如后述表2。
上述极限起爆药量测定I、热分解温度测定、密度测定、安息角测定中所得各实施例的极限起爆药量、热分解温度、密度及安息角如下表2:
| 极限起爆药量 | 热分解温度 | 密度 | 安息角 | |
| 实施例1 | 125mg | 161℃ | 1.83g/cm 3 | 31.2° |
| 实施例2 | 120mg | 182℃ | 1.83g/cm 3 | 32.2° |
| 实施例3 | 125mg | 190℃ | 1.89g/cm 3 | 32.5° |
| 实施例4 | 130mg | 197℃ | 1.82g/cm 3 | 31.7° |
| 实施例5 | 120mg | 180℃ | 1.87g/cm 3 | 33.3° |
| 实施例6 | 135mg | 184℃ | 1.83g/cm 3 | 32.1° |
| 实施例7 | 100mg | 160℃ | 1.88g/cm 3 | 30.2° |
| 实施例8 | 120mg | 163℃ | 1.81g/cm 3 | 31.8° |
| 实施例9 | 140mg | 188℃ | 1.87g/cm 3 | 34.1° |
表2
一般来说,具有较高的分解温度、较大的密度及较小的安息角为较佳。但本发明并不以此为限。
【极限起爆药量测定II】
依照GJB 5891-2006所规定的标准执行。具体来说,依照以下步骤:
(1)将600mg黑索金分三次以不同的压力压入标准8
#雷管中,每次药量约为200mg、200mg、200mg,使得其装药密度由下而上分别为1.65-1.75g/cm
3、1.6-1.68g/cm
3和1.5-1.6g/cm
3。
(2)以14.28MPa的压力将一定量的实施例1的化合物作为起爆药,压入该雷管的最上层。
(3)将加强帽压入其上,装入电点火头,进行封口。
(4)将装好的雷管置于约5mm厚的铅板上,而后连接电点火装置。
(5)启动点火,观测雷管是否被成功引爆,且铅板是否被击穿。此处的“成功引爆”是指三层黑索金装药均被引爆,且无白色药粉残留,下部铅板被击穿,穿孔直径大于雷管直径。
其中,本测定中,针对实施例1总共选取至少5种起爆药装药量,每一种起爆药量装填10发。因此可以得到实施例1的化合物其极限起爆药量为50mg(52.11MPa,1.75g/cm
3,8
#雷管)
【其他起爆测试】
实施例1的化合物作为起爆药,在15MPa–50MPa压力下,于8
#雷管压装后得到的样品,小于150mg即可稳定起爆500mg的RDX和PETN,并在5mm铅板上形成直径为8–15mm的穿孔。也就是说,其起爆能力约为DDNP的2-3倍,具有强起爆能力。
此外,实施例1的化合物,在装药密度1.85g/cm
3时的计算爆速、爆压与HMX相当,具有强爆轰能力。
上述实施方式仅为本发明的优选实施方式,不能以此来限定本发明保护的范围,本领域的技术人员在本发明的基础上所做的任何非实质性的变化及替换均属于本发明所要求保护的范围。
Claims (8)
- 一种化合物,其特征在于,所述化合物的结构包括式(A)结构,

- 如权利要求1所述的化合物,其特征在于,所述化合物为稠环化合物,所述稠环化合物至少包括所述式(A)结构及多元环;优选地,所述式(A)结构中,A 1及A 2选自碳原子、氮原子、氧原子、磷原子及硫原子中的一种或多种;优选地,所述式(A)结构中,A 1及A 2选自碳原子及氮原子中的一种或二种;优选地,所述式(A)结构中,A 1及A 2为碳原子;优选地,所述式(A)结构中,A 1及A 2其中一者为氮原子,另一者为碳原子;优选地,所述式(A)结构中,A 1为氮原子,A 2为碳原子;优选地,所述化合物为包括二个至三个环的稠环化合物;优选地,所述化合物为包括二个环的稠环化合物;优选地,所述化合物为包括三个环的稠环化合物;优选地,所述稠环化合物为所述式(A)结构与所述多元环的稠环化合物;优选地,所述稠环化合物中,所述式(A)结构及所述多元环共用两个碳原子;优选地,所述多元环为选自三元环、四元环、五元环、六元环、七元环及八元环的其中一种或多种;优选地,所述多元环为选自五元环、六元环及七元环的其中一种或多种;优选地,所述多元环为五元环及六元环的其中一种或多种;优选地,所述多元环为五元环;优选地,所述多元环为六元环;优选地,所述多元环为多个多元环;优选地,所述多个多元环包括五元环及六元环;优选地,所述多个多元环为五元环及六元环;优选地,所述多元环由碳原子所组成;优选地,所述多元环为选自茂环、苯环及环的其中一种;优选地,所述多元环为苯环;优选地,所述多元环为杂环,所述杂环的杂原子包括选自氮原子、氧原子、硅原子、磷原子、硫原子、砷原子、硒原子及碲原子的其中一种;优选地,所述杂原子包括选自氮原子、氧原子、硅原子、磷原子、硫原子、砷原子、硒原子及碲原子的其中二种;优选地,所述杂原子为氮原子、氧原子、硅原子、磷原子、硫原子、砷原子、硒原子或碲原子中的其中一种;优选地,所述杂原子为氮原子或氧原子;优选地,所述杂原子为选自氮原子及氧原子的其中一种;优选地,所述杂原子为氮原子;优选地,所述杂原子为氧原子;优选地,所述杂环包括1个、2个、3个、4个或5个所述杂原子;优选地,所述杂环包括1个、2个、3个、4个或5个氮原子;优选地,所述杂环包括1个氧原子;优选地,所述多元环为选自呋喃、吡咯、噻吩、咪唑、吡唑、噁唑、噻唑、吡啶、吡嗪、嘧啶、哒嗪、呋咱、三唑类、四唑类、三嗪类及四嗪类的其中一种;优选地,所述多元环为选自咪唑、吡唑、吡啶、吡嗪、嘧啶、哒嗪、呋咱、三唑类、四唑类、三嗪类及四嗪类的其中一种;优选地,所述多元环为选自咪唑、吡唑、吡啶、吡嗪、呋咱、三唑类、四唑类、三嗪类及四嗪类的其中一种;优选地,所述多元环为选自咪唑、吡唑、吡啶及吡嗪的其中一种;优选地,所述多元环为选自咪唑及吡唑的其中一种;优选地,所述多元环为吡唑;优选地,所述多元环为咪唑;优选地,所述多元环为选自哒嗪、吡啶及吡嗪的其中一种;优选地,所述多元环为哒嗪;优选地,所述多元环为吡啶;优选地,所述多元环为吡嗪;优选地,所述多元环为三唑类;优选地,所述多元环为1,2,4-三唑;优选地,所述多元环为三嗪类;优选地,所述多元环为1,2,3-三嗪;优选地,所述多元环为1,2,4-三嗪;优选地,所述多元环为选自呋喃、呋咱及噁唑的其中一种;优选地,所述多元环为呋喃;优选地,所述多元环为呋咱;优选地,所述多元环上包括取代基;优选地,所述取代基为选自:饱和烃烃基,包括烷烃基;官能团化饱和烃烃基,包括硝基烷烃基、偕二硝基烷烃基、硝仿基烷烃基、卤代烷烃基;醇基;醚基、硫醚基;羧酸基、酯基、硝酸酯基、磺酸基、磺酸酯基;酮基、酰胺基、酰肼基;醛基;不饱和脂肪族烃基,包括烯烃基、炔烃基、卤代烯烃基、硝基炔烃基;不饱和芳香烃烃基,包括单环芳烃基、多环芳烃基、稠环芳烃基和官能团化的单环芳烃基、官能团化的多环芳烃基、官能团化的稠环芳烃基;杂环芳香烃基,包括噻吩、吡啶、呋喃、唑类环、嗪类环、噁唑类环及其对应的单官能团化或多官能团化基团;杂环非芳香烃基,包括氮杂桥环烃、氧杂桥环烃;卤原子;氰基、肟基;氨基、亚氨基、偶氮基、氧化偶氮基、叠氮基、肼基;羟基、羟氨基、烷氧基;硝氨基;及硝基及亚硝基的其中一种;优选地,所述取代基为选自硝基、亚硝基、硝氨基、羟氨基、氰基、肟基、氨基、亚氨基、偶氮基、氧化偶氮基、叠氮基、肼基、氮杂桥环烃、吡啶唑类环、嗪类环、硝基烷烃基、偕二硝基烷烃基、硝仿基烷烃基及硝基炔烃基的其中一种;优选地,所述取代基为选自羟基、羟胺基、烷氧基、醇基、醚基、硫醚基、羧酸基、酯基、硝酸酯基、磺酸基、磺酸酯基、酮基、酰胺基、酰肼基、醛基、硝基炔烃基、硝基烷烃基、偕二硝基烷烃基、硝仿基烷烃基、呋喃、噁唑类环及氧杂桥环烃的其中一种;优选地,所述取代基为选自硝基、亚硝基、硝氨基、硝基烷烃基、偕二硝基烷烃基、硝仿基烷烃基及硝基炔烃基的其中一种;优选地,所述取代基为选自硝基及亚硝基的其中一种;优选地,所述取代基为硝基;优选地,所述取代基为选自氨基、亚氨基、硝氨基、羟氨基及肼基的其中一种;优选地,所述取代基为选自氨基及亚氨基的其中一种;优选地,所述取代基为氨基;优选地,所述取代基为硝氨基;优选地,所述取代基为选自叠氮基、偶氮基、氧化偶氮基及肼基的其中一种;优选地,所述取代基为叠氮基;优选地,所述取代基为卤原子;优选地,所述取代基为选自氟、氯、溴及碘的其中一种;优选地,所述取代基为氯;优选地,所述取代基位于所述多元环的碳原子上;优选地,所述式(A)结构中的所述取代基R 1为选自氢、氨基、硝基、硝氨基、叠氮基、氟、氯、溴、碘、C nH m(NO 2) p、式(B)结构中的其中一种,其中m+p=2n+1;优选地,所述式(A)结构中的所述取代基R 1为选自氨基、硝基、硝氨基、叠氮基中的其中一种;优选地,所述R 1为叠氮基; 优选地,所述R 1为氨基;优选地,所述R 1为硝基;优选地,所述R 1为硝氨基;优选地,所述R 1为选自氟、氯、溴、碘中的其中一种;优选地,所述R 1为氯;优选地,所述R 1为选自硝基、硝氨基或C nH m(NO 2) p中的其中一种,其中n、p为正整数;优选地,所述R 1为选自硝基、硝氨基或C nH m(NO 2) p中的其中一种,其中m、n、p为正整数;优选地,所述R 1为选自硝基、硝氨基或C nH m(NO 2) p中的其中一种,其中m、n、p为正整数,且n=1、2、3或4;优选地,所述式(B)结构中,所述A 1至A 5选自碳原子、氮原子、氧原子、磷原子及硫原子中的一种或多种;优选地,所述式(B)结构中,所述A 1至A 5选自碳原子、氮原子及氧原子中的一种、二种或三种;优选地,所述式(B)结构中,所述A 1至A 5选自碳原子及氮原子中的一种或二种;优选地,所述式(B)结构中,所述A 1至A 5为碳原子;优选地,所述式(B)结构中,所述A 1至A 5上的取代基个数为0、1、2、3或4个;优选地,所述R 2为选自氢、氨基、硝基、硝氨基、叠氮基、氟、氯、溴、碘、C nH m(NO 2) p及所述式(B)结构中的其中一或多个,其中m+p=2n+1;优选地,所述R 2为选自硝基、硝氨基或C nH m(NO 2) p中的其中一种,其中n、p为正整数;优选地,所述R 2为选自硝基、硝氨基或C nH m(NO 2) p中的其中一种,其中m、n、p为正整数;优选地,所述R 2为选自硝基、硝氨基或C nH m(NO 2) p中的其中一种,其中m、n、p为正整数,且n=1、2、3或4;优选地,所述稠环化合物至少包括式(C)结构,其中A 1至A 5选自碳原子、氮原子、氧原子、磷原子及硫原子中的一种或多种;优选地,所述式(C)结构中,所述A 1至A 5选自碳原子、氮原子及氧原子中的一种、二种或三种;优选地,所述式(C)结构中,所述A 1至A 5选自碳原子及氮原子中的一种或二种;优选地,所述式(C)结构中,所述A 1至A 2为碳原子;优选地,所述式(C)结构中,所述A 3至A 5选自碳原子、氮原子及氧原子中的一种、二种或三种;优选地,所述式(C)结构中,所述A 3至A 5选自碳原子及氮原子中的一种或二种;优选地,所述式(C)结构中,所述A 1至A 5包括0、1、2、3、4或5个氮原子,其余为碳原子;优选地,所述式(C)结构中,所述A 3至A 5包括0、1、2或3个氮原子,其他的是碳原子;优选地,所述式(C)结构中,所述A 1至A 5组成一个呋咱环;优选地,所述式(C)结构中,所述A 1至A 2为碳原子,所述A 3及A 5为氮原子,且所述A 4为氧原子;优选地,所述式(C)结构中,所述A 1至A 2为碳原子,所述A 3及A 5为氮原子,所述A 4为氧原子,且所述A 3有一个氧取代基,所述A 3与所述氧取代基形成配位键;优选地,所述式(C)结构中,所述A 1至A 5组成一个二唑环;优选地,所述式(C)结构中,所述A 1至A 5组成一个吡唑环;优选地,所述式(C)结构中,所述A 1、A 2及A 5为碳原子,且所述A 3及A 4为氮原子;优选地,所述式(C)结构中,所述A 5上有硝基取代基;优选地,所述式(C)结构中,所述A 1、A 2及A 5为碳原子,所述A 3及A 4为氮原子,且所述式(C)结构中,所述A 5上有硝基取代基;优选地,所述式(C)结构中,所述A 1至A 5组成一个咪唑环;优选地,所述式(C)结构中,所述A 1、A 2及A 4为碳原子,且所述A 3及A 5为氮原子;优选地,所述式(C)结构中,A 1至A 5上的取代基个数为0、1、2、3或4个;优选地,所述R 3为选自氢、氨基、硝基、硝氨基、叠氮基、氟、氯、溴、碘、氧、C nH m(NO 2) p及所述式(B)结构中的其中一或多个,其中m+p=2n+1;优选地,所述R 3为选自硝基、硝氨基或C nH m(NO 2) p中的其中一种,其中n、p为正整数;优选地,所述R 3为选自硝基、硝氨基或C nH m(NO 2) p中的其中一种,其中m、n、p为正整数;优选地,所述R 3为选自硝基、硝氨基或C nH m(NO 2) p中的其中一种,其中m、n、p为正整数,且n=1、2、3或4;优选地,所述稠环化合物至少包括式(D)结构,其中A 1至A 6选自碳原子、氮原子、氧原子、磷原子及硫原子中的一种或多种;优选地,所述式(D)结构中,所述A 1至A 6选自碳原子及氮原子中的一种或二种;优选地,所述式(D)结构中,所述A 1至A 2为碳原子;优选地,所述式(D)结构中,所述A 3至A 6选自碳原子及氮原子中的一种或二种;优选地,所述式(D)结构中,所述A 1至A 6包括0、1、2、3、4、5或6个氮原子,其余为碳原子;优选地,所述式(D)结构中,所述A 3至A 6包括0、1、2、3或4个氮原子,其余为碳原子;优选地,所述式(D)结构中,所述A 1至A 6为碳原子;优选地,所述式(D)结构中,所述A 4、A 5为氮原子;优选地,所述式(D)结构中,所述A 1至A 3、A 6为碳原子,且A 4、A 5为氮原子;优选地,所述式(D)结构中,所述A 3至A 5为氮原子;优选地,所述式(D)结构中,所述A 1至A 2、A 6为碳原子,且A 3至A 5为氮原子;优选地,所述式(D)结构中,所述A 1至A 2、A 6为碳原子,A 3至A 5为氮原子,所述A 4有一个氧取代基,所述A 4与所述氧取代基形成配位键,且所述A 6有一个叠氮取代基;优选地,所述式(D)结构中,A 1至A 6上的取代基个数合计为0、1、2、3、4或5个;优选地,所述R 4为选自氢、氨基、硝基、硝氨基、叠氮基、氟、氯、溴、碘、氧、C nH m(NO 2) p及所述式(B)结构中的其中一或多个,其中m+p=2n+1;优选地,所述R 4为选自硝基、硝氨基或C nH m(NO 2) p中的其中一种,其中n、p为正整数;优选地,所述R 4为选自硝基、硝氨基或C nH m(NO 2) p中的其中一种,其中m、n、p为正整数;优选地,所述R 4为选自硝基、硝氨基或C nH m(NO 2) p中的其中一种,其中m、n、p为正整数,且n=1、2、3或4;优选地,所述稠环化合物至少包括式(E)结构,其中A 1至A 9选自碳原子、氮原子、氧原子、磷原子及硫原子中的一种或多种;优选地,所述式(E)结构中,所述A 1至A 9选自碳原子、氮原子及氧原子中的一种、二种或三种;优选地,所述式(E)结构中,所述A 1至A 9选自碳原子及氮原子中的一种或二种;优选地,所述式(E)结构中,所述A 1至A 2为碳原子;优选地,所述式(E)结构中,所述A 3至A 9选自碳原子、氮原子及氧原子中的一种、二种或三种;优选地,所述式(E)结构中,所述A 3至A 9选自碳原子及氮原子中的一种或二种;优选地,所述式(E)结构中,所述A 1至A 9包括0、1、2、3、4、5、6、7、8或9个氮原子,其余为碳原子;优选地,所述式(E)结构中,所述A 1至A 6包括0、1、2、3、4、5或6个氮原子,其余为碳原子;优选地,所述式(E)结构中,所述A 4、A 5、A 7至A 9包括0、1、2、3、4或5个氮原子,其余为碳原子;优选地,所述式(E)结构中,所述A 4、A 5、A 7至A 9组成一个呋咱环;优选地,所述式(E)结构中,所述A 4、A 5、A 7至A 9组成一个三唑环;优选地,所述式(E)结构中,所述A 4、A 5、A 7至A 9组成一个1,2,4-三唑环;优选地,所述式(E)结构中,所述A 1至A 6组成一个二嗪环;优选地,所述式(E)结构中,所述A 1至A 6组成一个吡嗪环;优选地,所述式(E)结构中,所述A 1至A 6组成一个三嗪环;优选地,所述式(E)结构中,所述A 1至A 6组成一个1,2,4-三嗪环;优选地,所述式(E)结构中,所述A 4、A 5、A 7至A 9组成一个呋咱环,且所述A 1至A 6组成一个二嗪环;优选地,所述式(E)结构中,所述A 1、A 2、A 4、A 5为碳原子,所述A 3、A 6、A 7、A 9为氮原子,且所述A 8为氧原子;优选地,所述式(E) 结构中,所述A 4、A 5、A 7至A 9组成一个三唑环,且所述A 1至A 6组成一个三嗪环;优选地,所述式(E)结构中,所述A 4、A 5、A 7至A 9组成一个1,2,4-三唑环,且所述A 1至A 6组成一个1,2,4-三嗪环;优选地,所述式(E)结构中,所述A 2、A 3、A 5、A 8为碳原子,且所述A 1、A 4、A 6、A 7、A 9为氮原子;优选地,所述式(E)结构中,A 1至A 6上的取代基个数合计为0、1、2、3、4或5个;优选地,所述式(E)结构中,A 4、A 5、A 7至A 9上的取代基个数合计为0、1、2、3或4个;优选地,所述R 5为选自氢、氨基、硝基、硝氨基、叠氮基、氟、氯、溴、碘、氧、C nH m(NO 2) p及所述式(B)结构中的其中一或多个,其中m+p=2n+1;优选地,所述R 5为选自硝基、硝氨基或C nH m(NO 2) p中的其中一种,其中n、p为正整数;优选地,所述R 5为选自硝基、硝氨基或C nH m(NO 2) p中的其中一种,其中m、n、p为正整数;优选地,所述R 5为选自硝基、硝氨基或C nH m(NO 2) p中的其中一种,其中m、n、p为正整数,且n=1、2、3或4;优选地,所述稠环化合物包括所述式(A)结构及3-硝基吡唑;优选地,所述化合物的结构为式(F′)结构;优选地,所述式(F′)结构中的所述R 1为选自氢、氨基、硝基、硝氨基、叠氮基、氟、氯、溴、碘、C nH m(NO 2) p、式(B)结构中的其中一种,其中m+p=2n+1;优选地,所述式(F′)结构中的所述R 1为选自氨基、硝基、硝氨基、叠氮基中的其中一种;优选地,所述式(F′)结构中的所述R 1为叠氮基;优选地,所述式(F′)结构中的所述R 3为选自氢、氨基、硝基、硝氨基、叠氮基、氟、氯、溴、碘、氧、C nH m(NO 2) p及所述式(B)结构中的其中一或多个,其中m+p=2n+1;优选地,所述式(F′)结构中的所述R 3为硝基;优选地,所述化合物的结构为式(F)结构;优选地,所述式(F)结构的分析测定结果如下:DSC(160℃,50℃–250℃,5℃·min -1); 1H NMR(400MHz,DMSO-d 6,25℃)δ(ppm):14.51(brs,1H,NH); 13C NMR(100MHz,DMSO-d 6,25℃)δ(ppm):157.95,157.31,148.07,90.03;IR(KBr,γ/cm -1):3414(m);3231(m);2154(s);1600(s);1543(s);1403(s);1384(s);1313(m);1209(m);1131(m);元素分析C 4HN 9O 3(%):实测值(计算值)C:21.62(21.53),H:0.47(0.45),N:56.61(56.50),O:21.22(21.51);优选地,所述化合物的结构包括式(G′)结构;优选地,所述式(G′)结构中的所述R 1为选自氢、氨基、硝基、硝氨基、叠氮基、氟、氯、溴、碘、C nH m(NO 2) p、式(B)结构中的其中一种,其中m+p=2n+1;优选地,所述式(G′)结构中的所述R 1为选自氨基、硝基、硝氨基、叠氮基中的其中一种;优选地,所述式(G′)结构中的所述R 1为选自硝基、硝氨基或C nH m(NO 2) p中的其中一种,其中n、p为正整数;优选地,所述式(G′)结构中的所述R 1为选自硝基、硝氨基或C nH m(NO 2) p中的其中一种,其中m、n、p为正整数;优选地,所述式(G′)结构中的所述R 1为选自硝基、硝氨基或C nH m(NO 2) p中的其中一种,其中m、n、p为正整数,且n=1、2、3或4;优选地,所述式(G′)结构中的所述R 1为硝基;优选地,所述化合物的结构为式(G)结构;优选地,所述化合物的结构包括式(H’)结构;优选地,所述式(H′)结构中的所述R 1为选自氢、氨基、硝基、硝氨基、叠氮基、氟、氯、溴、碘、C nH m(NO 2) p、式(B)结构中的其中一种,其中m+p=2n+1;优选地,所述式(H′)结构中的所述R 1为选自氨基、硝基、硝氨基、叠氮基中的其中一种;优选地,所述式(H′)结构中的所述R 1为叠氮基;优选地,所述式(H′)结构中的所述R 1为氨基;优选地,所述式(H′)结构中的所述R 1为硝基;优选地,所述式(H′)结构中的所述R 1为硝氨基;优选地,所述式(H′)结构中的所述R 1为选自氟、氯、溴、碘中的其中一种;优选地,所述式(H′)结构中的所述R 1为氯;优选地,所述式(H′)结构中的所述R 1为选自硝基、硝氨基或C nH m(NO 2) p中的其中一种,其中n、p为正整数;优选地,所述式(H′)结构中的所述R 1为选自硝基、硝氨基或C nH m(NO 2) p中的其中一种,其中m、n、p为正整数;优选地,所述式(H′)结构中的所述R 1为选自硝基、硝氨基或C nH m(NO 2) p中的其中一种,其中m、n、p为正整数,且n=1、2、3或4;优选地,所述化合物的结构为式(H)结构;优选地,所述稠环化合物包括所述式(A)结构及2-氧化呋咱;优选地,所述化合物的结构包括式(J′)结构;优选地,所述式(J′)结构中的所述R 1为选自氢、氨基、硝基、硝氨基、叠氮基、氟、氯、溴、碘、C nH m(NO 2) p、式(B)结构中的其中一种,其中m+p=2n+1;优选地,所述式(J′)结构中的所述R 1为选自氨基、硝基、硝氨基、叠氮基中的其中一种;优选地,所述式(J′)结构中的所述R 1为氨基;优选地,所述化合物的结构为式(J)结构;优选地,所述化合物的结构包括式(K′)结构;优选地,所述式(K′)结构中的所述R 1为选自氢、氨基、硝基、硝氨基、叠氮基、氟、氯、溴、碘、C nH m(NO 2) p、式(B)结构中的其中一种,其中m+p=2n+1;优选地,所述式(K′)结构中的所述R 1为选自氨基、硝基、硝氨基、叠氮基中的其中一种;优选地,所述式(K′)结构中的所述R 1为硝氨基;优选地,所述化合物的结构为式(K)结构;优选地,所述化合物的结构包括式(L′)结构;优选地,所述式(L′)结构中的所述R 1为选自氢、氨基、硝基、硝氨基、叠氮基、氟、氯、溴、碘、C nH m(NO 2) p、式(B)结构中的其中一种,其中m+p=2n+1;优选地,所述式(L′)结构中的所述R 1为选自氨基、硝基、硝氨基、叠氮基中的其中一种;优选地,所述式(L′)结构中的所述R 1为硝基;优选地,所述式(L′)结构中的所述R 1为硝氨基;优选地,所述化合物的结构为式(L)结构;优选地,所述稠环化合物包括二个所述式(A)结构;优选地,所述稠环化合物包括二个相同的所述式(A)结构;优选地,所述稠环化合物包括二个所述式(A)结构,所述二个所述式(A)结构共用A 1及A 2;优选地,所述化合物的结构包括式(M′)结构;优选地,所述式(M′)结构中的所述R 1为选自氢、氨基、硝基、硝氨基、叠氮基、氟、氯、溴、碘、C nH m(NO 2) p、式(B)结构中的其中一种,其中m+p=2n+1;优选地,所述式(M′)结构中的所述R 1为选自氨基、硝基、硝氨基、叠氮基中的其中一种;优选地,所述式(M′)结构中的所述R 1为叠氮基;优选地,所述式(M′)结构中的所述二个R 1为二个相同的基团;优选地,所述化合物的结构为式(M)结构;优选地,所述化合物的结构包括式(N′)结构;优选地,所述式(N′)结构中的所述R 1为选自氢、氨基、硝基、硝氨基、叠氮基、氟、氯、溴、碘、C nH m(NO 2) p、式(B)结构中的其中一种,其中m+p=2n+1;优选地,所述式(N′)结构中的所述R 1为选自氨基、硝基、硝氨基、叠氮基中的其中一种;优选地,所述式(N′)结构中的所述R 1为硝氨基;优选地,所述式(N′)结构中的所述R 1为选自硝基、硝氨基或C nH m(NO 2) p中的其中一种,其中n、p为正整数;优选地,所述式(N′)结构中的所述R 1为选自硝基、硝氨基或C nH m(NO 2) p中的其中一种,其中m、n、p为正整数;优选地,所述式(N′)结构中的所述R 1为选自硝基、硝氨基或C nH m(NO 2) p中的其中一种,其中m、n、p为正整数,且n=1、2、3或4;优选地,所述化合物的结构为式(N)结构;优选地,所述化合物的结构包括式(P′)结构,优 选地,所述式(P′)结构中的所述R 1为选自氢、氨基、硝基、硝氨基、叠氮基、氟、氯、溴、碘、C nH m(NO 2) p、式(B)结构中的其中一种,其中m+p=2n+1;优选地,所述式(P′)结构中的所述R 1为选自氟、氯、溴、碘中的其中一种;优选地,所述式(P′)结构中的所述R 1为氯;优选地,所述化合物的结构为式(P)结构。

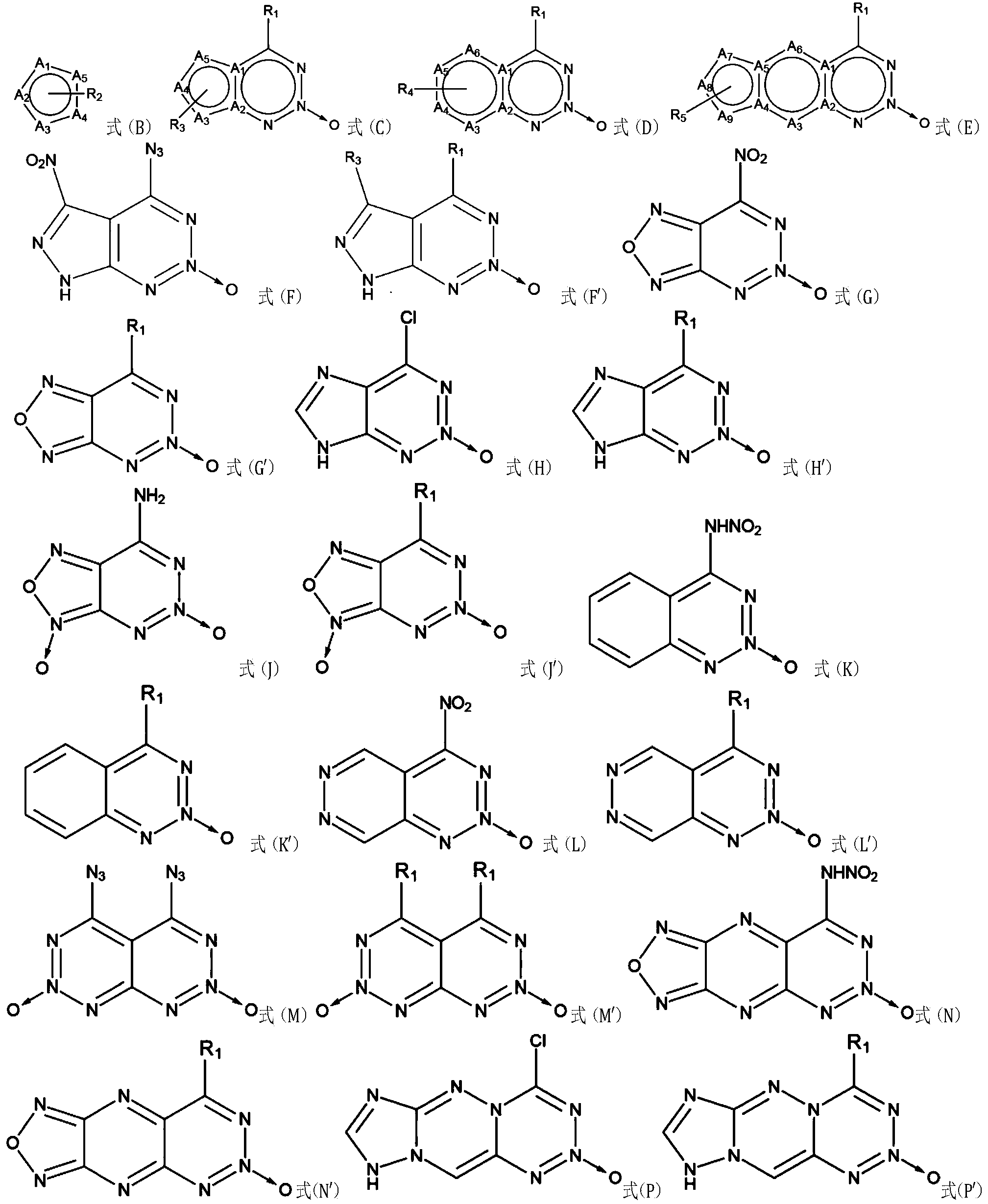
- 一种化合物,其特征在于,所述化合物包括式(Q)结构,

- 如权利要求3所述的化合物,其特征在于,所述化合物包括多元环;优选地,所述式(Q)结构中,A 10及A 11选自碳原子、氮原子、氧原子、磷原子及硫原子中的一种或多种;优选地,所述式(Q)结构中,A 10及A 11选自碳原子及氮原子中的一种或二种;优选地,所述式(Q)结构中,A 10及A 11为碳原子;优选地,所述式(Q)结构中,A 10及A 11的其中一者为氮原子,另一者为碳原子;优选地,所述多元环为选自三元环、四元环、五元环、六元环、七元环及八元环的其中一种或多种;优选地,所述多元环为选自五元环、六元环及七元环的其中一种或多种;优选地,所述多元环为五元环及六元环的其中一种或多种;优选地,所述多元环为五元环;优选地,所述多元环为六元环;优选地,所述多元环由碳原子所组成;优选地,所述多元环为选自茂环、苯环及环的其中一种;优选地,所述多元环为苯环;优选地,所述多元环为杂环,所述杂环的杂原子包括选自氮原子、氧原子、硅原子、磷原子、硫原子、砷原子、硒原子及碲原子的其中一种;优选地,所述杂原子包括选自氮原子、氧原子、硅原子、磷原子、硫原子、砷原子、硒原子及碲原子的其中二种;优选地,所述杂原子为氮原子、氧原子、硅原子、磷原子、硫原子、砷原子、硒原子或碲原子中的其中一种;优选地,所述杂原子为氮原子或氧原子;优选地,所述杂原子为选自氮原子及氧原子的其中一种;优选地,所述杂原子为氮原子;优选地,所述多元环为选自呋喃、吡咯、噻吩、咪唑、吡唑、噁唑、噻唑、吡啶、吡嗪、嘧啶、哒嗪、呋咱、三唑类、四唑类、三嗪类及四嗪类的其中一种;优选地,所述多元环为选自咪唑、吡唑、吡啶、吡嗪、嘧啶、哒嗪、呋咱、三唑类、四唑类、三嗪类及四嗪类的其中一种;优选地,所述多元环为选自咪唑、吡唑、吡啶、吡嗪、呋咱、三唑类、四唑类、三嗪类及四嗪类的其中一种;优选地,所述多元环为选自咪唑、吡唑、吡啶及吡嗪的其中一种;优选地,所述多元环为选自咪唑及吡唑的其中一种;优选地,所述多元环为吡唑;优选地,所述多元环为选自哒嗪、吡啶及吡嗪的其中一种;优选地,所述多元环为哒嗪;优选地,所述多元环为四唑类;优选地,所述多元环上包括取代基;优选地,所述取代基为选自:饱和烃烃基,包括烷烃基;官能团化饱和烃烃基,包括硝基烷烃基、偕二硝基烷烃基、硝仿基烷烃基、卤代烷烃基;醇基;醚基、硫醚基;羧酸基、酯基、硝酸酯基、磺酸基、磺酸酯基;酮基、酰胺基、酰肼基;醛基;不饱和脂肪族烃基,包括烯烃基、炔烃基、卤代烯烃基、硝基炔烃基;不饱和芳香烃烃基,包括单环芳烃基、多环芳烃基、稠环芳烃基和官能团化的单环芳烃基、官能团化的多环芳烃基、官能团化的稠环芳烃基;杂环芳香烃基,包括噻吩、吡啶、呋喃、唑类环、嗪类环、噁唑类环及其对应的单官能团化或多官能团化基团;杂环非芳香烃基,包括氮杂桥环烃、氧杂桥环烃;卤原子;氰基、肟基;氨基、亚氨基、偶氮基、氧化偶氮基、叠氮基、肼基;羟基、羟氨基、烷氧基;硝氨基;及硝基及亚硝基的其中一种;优选地,所述取代基为选自硝基、亚硝基、硝氨基、羟氨基、氰基、肟基、氨基、亚氨基、偶氮基、氧化偶氮基、叠氮基、肼基、氮杂桥环烃、吡啶唑类环、嗪类环、硝基烷烃基、偕二硝基烷烃基、硝仿基烷烃基及硝基炔烃基的其中一种;优选地,所述取代基为选自氰基及肟基的其中一种;优选地,所述取代基为选自氨基、亚氨基、硝氨基、羟氨基及肼基的其中一种;优选地,所述取代基为选自氨基及亚氨基的其中一种;优选地,所述取代基为氨基;优选地,所述取代基位于所述多元环的碳原子上;优选地,所述化合物包括3-氨基-4-氰基吡唑的结构;优选地,所述化合物包括4-氨基-5-氰基咪唑的结构;优选地,所述化合物包括1-氨基-2-氰基苯的结构;优选地,所述化合物包括4-氨基-5-氰基哒嗪的结构;优选地,所述化合物包括3,3-二氨基-2-(1H-四唑-5-基)丙腈的结构;优选地,所述化合物为3-氨基-4-氰基吡唑;优选地,所述化合物为4-氨基-5-氰基咪唑;优选地,所述化合物为1-氨基-2-氰基苯;优选地,所述化合物为4-氨基-5-氰基哒嗪;优选地,所述化合物为3,3-二氨基-2-(1H-四唑-5-基)丙腈。

- 一种化合物的制备方法,其特征在于,包括:将包括式(Q)结构的化合物分散于溶剂中;加入盐类及叠氮化物,在第一反应温度下反应第一反应时间;加入酸;进行纯化,得到中间产物;将所述中间产物加入硝化体系当中,在第二温度下硝化反应第二反应时间;终止所述硝化反应;以及进行纯化。
- 如权利要求5所述的化合物的制备方法,其特征在于,所述包括式(Q)结构的化合物为如权利要求3所述的化合物;优选地,所述包括式(Q)结构的化合物为如权利要求4所述的化合物;优选地,所述包括式(Q)结构的化合物为3-氨基-4-氰基吡唑;优选地,所述包括式(Q)结构的化合物为4-氨基-5-氰基咪唑;优选地,所述包括式(Q)结构的化合物为1-氨基-2-氰基苯;优选地,所述包括式(Q)结构的化合物为4-氨基-5-氰基哒嗪;优选地,所述包括式(Q)结构的化合物为3,3-二氨基-2-(1H-四唑-5-基)丙腈;优选地,所述中间产物为3-氨基-4-[1H-四氮唑]-1H-吡唑;优选地,所述溶剂为N,N-二甲基甲酰胺、二甲基亚砜、四氢呋喃、乙腈、二氯甲烷、氯仿、甲醇、乙醇、水、异丙醇、乙二醇、乙二醇单甲醚、乙二醇单乙醚、乙二醇单丁醚、甘油、丙酮、三乙醇胺、吡啶、苯、甲苯、二甲苯等中的其中一种,或二种或多种的混合物优选地,所述溶剂的用量体积与所述包括式(Q) 结构的化合物质量的比值为2mL/g至2mL/mg;优选地,所述盐类为通式M aX b的化合物,其中M为Fe、Co、Ni、Mn、Zn、Cd、Au、Ag、Cu、Cr、Al、V、NH 4、(CH 3) 2NH 2、(CH 3) 3NH、(CH 3CH 2) 2NH 2及(CH 3CH 2) 3NH的其中一种,或二种或多种的混合,而X为F、Cl、Br、I、NO 3、SO 4、HSO 4、CO 3、HCO 3、HCOO、CH 3COO、CF 3COO、(COO) 2及CH 2(COO) 2的其中一种,或二种或多种的混合;优选地,所述盐类与所述包括式(Q)结构的化合物的摩尔比为1:1至6:1;优选地,所述盐类与所述包括式(Q)结构的化合物的摩尔比为20:9至50:9;优选地,所述叠氮化物为通式R a(N 3) b的化合物,其中R为选自Li、Na、K、Rb、Cu、Fe、Ag、Au、Pb、Cd、Ni及H的其中一种,或二种或多种的混合;优选地,所述叠氮化物与所述包括式(Q)结构的化合物的摩尔比为1:1至6:1;优选地,所述第一温度为介于-20℃至130℃之间的温度;优选地,所述第一温度为介于-10℃至130℃之间的温度;优选地,所述第一反应时间为介于2小时至24小时;优选地,所述第一反应时间为介于2小时至10小时;优选地,所述第一反应时间为介于2小时至8小时;优选地,所述酸为:硝酸、硫酸、盐酸、氢溴酸、氢氟酸、氢碘酸、甲酸、乙酸、乙酸酐、草酸、丙二酸及三氟乙酸中的任意一者或多者;优选地,其中所述加入所述酸为加入所述酸将反应体系调整至第一pH值,所述第一pH值介于1-4之间;优选地,所述第一pH值介于1.0至3.5之间;优选地,所述第一pH值介于2.0至3.0之间;优选地,所述硝化体系为发烟硝酸、100%硝酸、五氧化二氮、四氟硼硝、100%硝酸/三氟乙酸酐、100%硝酸/浓硫酸、发烟硝酸、100%硝酸/乙酸酐、发烟硝酸/浓硫酸、发烟硝酸/乙酸酐、发烟硝酸/三氟乙酸酐、发烟硝酸/乙酸酐、三氧化硫/硝酸、五氧化二磷/硝酸、发烟硫酸/发烟硝酸或发烟硫酸/100%硝酸;优选地,所述硝化体系为100%硝酸、100%硝酸/乙酸酐、100%硝酸/三氟乙酸酐、硝酸/乙酸酐、100%硝酸/浓硫酸、发烟硝酸、发烟硝酸/浓硫酸、发烟硝酸/乙酸酐、发烟硝酸/三氟乙酸酐、五氧化二氮、三氧化硫/硝酸、五氧化二磷/硝酸、发烟硫酸/发烟硝酸或发烟硫酸/100%硝酸,所述中间产物质量与所述硝化体系体积的比为1g:5mL至1g:50mL;优选地,所述硝化体系为四氟硼硝,所述中间产物与所述硝化体系体积的摩尔比为1:1至1:50;优选地,所述第二温度为介于-20℃至130℃之间的温度;优选地,所述第二温度为-20℃至110℃之间的温度;优选地,所述第二温度为-20℃至100℃之间的温度;优选地,所述第二温度为-20℃至室温之间的温度;优选地,所述第二温度为室温;优选地,所述第二温度为40℃至80℃之间的温度;优选地,所述第二温度为50℃至70℃之间的温度;优选地,所述第二反应时间为0.5至48小时;优选地,所述第二反应时间为0.5至12小时;优选地,所述第二反应时间为1至4小时;优选地,所述终止所述硝化反应为将所得产物加入冰水中;优选地,所述方法更包括将所述终止所述硝化反应后的所得产物分散于溶剂中,通入氯气气流;优选地,所述氯气气流不溶或难溶于所述溶剂;优选地,所述通入氯气气流的反应温度为介于30℃至100℃;优选地,所述通入氯气气流的反应温度为介于50℃至90℃;优选地,所述通入氯气气流的反应温度为介于60℃至70℃;优选地,所述通入氯气气流的反应时间为介于1小时至10小时;优选地,所述通入氯气气流的反应时间为介于1小时至5小时;优选地,所述通入氯气气流的反应时间为介于1小时至3小时;优选地,所述方法更包括将所述终止所述硝化反应后的所得产物分散于溶剂中,加入浓氨水,并进行回流反应;优选地,所述浓氨水溶于所述溶剂;优选地,所述浓氨水的浓度为介于10%至32%;优选地,所述终止所述硝化反应后的所得产物的质量和浓氨水的体积的比介于1g:1mL至1g:100mL;优选地,所述终止所述硝化反应后的所得产物的质量和浓氨水的体积的比介于1g:5mL至1g:20mL;优选地,所述终止所述硝化反应后的所得产物的质量和浓氨水的体积的比介于1g:6mL至1g:12mL;优选地,所述回流反应的反应时间为介于0.5小时至24小时;优选地,所述回流反应的反应时间为介于1小时至12小时;优选地,所述回流反应的反应时间为介于2小时至8小时;优选地,所述方法更包括将所述加入所述浓氨水并进行所述回流反应的所得产物加入氧化性溶液;优选地,所述氧化性溶液为选自浓硫酸与30%双氧水的混合溶液、浓硫酸与50%双氧水的混合溶液、浓硫酸与三氟乙酸的混合溶液、浓硫酸与过氧乙酸的混合溶液及浓硫酸与乙酸酐混合溶液中的一种或多种;优选地,所述加入所述氧化性溶液的反应时间为介于0.5至12小时;优选地,所述加入所述氧化性溶液的反应时间为介于0.5至4小时;优选地,所述方法更包括在所述加入所述浓氨水并进行所述回流反应之后,将所得到的产物加入另一个所述硝化体系当中,在另一个所述第二温度下硝化反应另一个所述第二反应时间;优选地,所述方法更包括将丙二腈和亚硝酸盐分散于溶剂中;优选地,所述亚硝酸盐为通式MNO 2的化合物,其中M为选自NH 4、Li、Na、K、Rb及Cs的其中一种,或二种或多种的混合;优选地,所述溶剂为N,N-二甲基甲酰胺、二甲基亚砜、四氢呋喃、乙腈、二氯甲烷、氯仿、甲醇、乙醇、水、异丙醇、乙二醇、乙二醇单甲醚、乙二醇单乙醚、乙二醇单丁醚、甘油、丙酮、三乙醇胺、吡啶、苯、甲苯、二甲苯等中的任意一种,或二种或多种的混合物;优选地,所述方法更包括加入羟胺化合物;优选地,所述羟胺化合物为羟胺盐类;优选地,所述羟胺盐类为通式(NH 3OH) aX b的化合物,其中X为选自F、Cl、Br、I、NO 3、SO 4、HSO 4、CO 3、HCO 3、HCOO、CH 3COO、CF 3COO、(COO) 2及CH 2(COO) 2的其中一种,或二种或多种的混合;优选地,所述羟胺化合物为羟胺卤化盐类;优选地,所述羟胺化合物为盐酸羟胺;优选地,所述加入羟胺化合物的反应温度为介于-20℃至130℃之间的温度;优选地,所述加入羟胺化合物的反应温度为介于-20℃至40℃之间的温度;优选地,所述加入羟胺化合物的反应温度为介于-10℃至40℃之间的温度;优选地,所述加入羟胺化合物的反应时间为为介于2-24小时的时间;优选地,所述加入羟胺化合物的反应时间为为介于2-10小时的时间;优选地,所述加入羟胺化合物的反应时间为为介于3-5小时的时间;优选地,所述方法更包括在加入所述羟胺化合物后,加入碱及酯类化合物;优选地,所述碱为通式M a(OH) b的化合物,其中M为选自NH 4、Li、Na、K、Rb、Cs、Ca、Sr、Ba的其中一种,或二种或多种的混合;优选地,所述酯类化合物为通式R 1COOR 2的化合物,其中R 1、R 2为选自甲基、乙基、丙基、异丙基、伯丁基、仲丁基、叔丁基以及各种戊基、各种己基的其中一种,或二种或多种的混合;优选地,所述方法更包括在加入所述碱及酯类化合物后,加入酸及二氧化铅;优选地,所述方法更包括加入浓盐酸,进行回流反应;优选地,所述方法更包括将所得产物溶于二氯亚砜并加入N,N-二甲基甲酰胺,进行回流反应;优选地,所述方法更包括加入氮化物;优选地,所述氮化物为通式R a(N) b的化合物,其中R为选自Li、Na、K、Rb、Cu、Fe、Ag、Au、Pb、Cd、Ni及H的其中一种,或二 种或多种的混合;优选地,所述方法更包括加入硝基乙腈化合物;优选地,所述硝基乙腈化合物为硝基乙腈盐类;优选地,所述硝基乙腈化合物为通式M a(CH(NO 2)CN) b的化合物,其中M为选自Li、Na、K、Rb、Cs的其中一种,或二种或多种的混合;优选地,所述硝基乙腈化合物为硝基乙腈钠盐;优选地,所述化合物的制备方法为权利要求1所述的化合物的制备方法;优选地,所述化合物的制备方法为权利要求2所述的化合物的制备方法;优选地,所述方法包括以下步骤:步骤一:合成3-氨基-4-[1H-四氮唑]-1H-吡唑:在一定温度下,将3-氨基-4-氰基吡唑分散于溶剂中,接着分批加入特定的盐(M aX b)、叠氮化物,然后在一定温度下继续反应,再向其中滴加酸,而后过滤,洗涤,将固体干燥,得到白色粉末,即3-氨基-4-[1H-四氮唑]-1H-吡唑;步骤二:合成4-叠氮基-5-硝基-7H-吡唑并[3,4-d][1,2,3]三嗪-2-氧化物:将3-氨基-4-1H-四氮唑-吡唑缓慢加入硝化体系,加完后控制反应体系温度,保持硝化0.5h-48h,接着将反应体系倾入冰中,依次经过滤洗涤、干燥,得到米白色4-叠氮基-5-硝基-7H-吡唑并[3,4-d][1,2,3]三嗪-2-氧化物粉末;优选地,所述步骤一中反应温度为-10℃-130℃;优选地,所述步骤一溶剂是N,N-二甲基甲酰胺、二甲基亚砜、四氢呋喃、乙腈、二氯甲烷、氯仿、甲醇、乙醇、水、异丙醇、乙二醇、乙二醇单甲醚、乙二醇单乙醚、乙二醇单丁醚、甘油、丙酮、三乙醇胺、吡啶、苯、甲苯、二甲苯中的任意一种,溶剂体积用量与3-氨基-4-氰基吡唑投料质量比为2mL/g-20mL/g;优选地,所述步骤一中盐的物质的量之比为n(M aX b):n(3-氨基-4-[1H-四氮唑]-1H-吡唑)=1:1–6:1;盐的组成为:M=Fe、Co、Ni、Mn、Zn、Cd、Au、Ag、Cu、Cr、Al、V、NH 4、(CH 3) 2NH 2、(CH 3) 3NH、(CH 3CH 2) 2NH 2或(CH 3CH 2) 3NH,X=F、Cl、Br、I、NO 3、SO 4、HSO 4、CO 3、HCO 3、HCOO、CH 3COO、CF 3COO、(COO) 2或CH 2(COO) 2;优选地,所述步骤一中所用叠氮化物物质的量之比为n(R a(N 3) b):n(3-氨基-4-[1H-四氮唑]-1H-吡唑)=1:1–6:1;叠氮化物中R是:R=Li、Na、K、Rb、Cu、Fe、Ag、Au、Pb、Cd、Ni或H;优选地,所述步骤一中酸的种类为:硝酸、硫酸、盐酸、氢溴酸、氢氟酸、氢碘酸、甲酸、乙酸、乙酸酐、草酸、丙二酸或三氟乙酸中的任意一种或几种混合,用量以将溶液pH调整至1-4为准;优选地,所述步骤二中所用硝化体系为市售发烟硝酸或100%硝酸,或其他硝化体系,包括五氧化二氮、四氟硼硝、100%硝酸/三氟乙酸酐、100%硝酸/浓硫酸、发烟硝酸、100%硝酸/乙酸酐、发烟硝酸/浓硫酸、发烟硝酸/乙酸酐、发烟硝酸/三氟乙酸酐、发烟硝酸/乙酸酐、三氧化硫/硝酸、五氧化二磷/硝酸、发烟硫酸/发烟硝酸、发烟硫酸/100%硝酸;当采用的硝化体系为100%硝酸、100%硝酸/乙酸酐、100%硝酸/三氟乙酸酐、硝酸/乙酸酐、100%硝酸/浓硫酸、发烟硝酸、发烟硝酸/浓硫酸、发烟硝酸/乙酸酐、发烟硝酸/三氟乙酸酐、五氧化二氮、三氧化硫/硝酸、五氧化二磷/硝酸、发烟硫酸/发烟硝酸、发烟硫酸/100%硝酸时,3-氨基-4-[1H-四氮唑]-1H-吡唑与硝化体系的质量体积比为1g:(5-50)mL;当采用的硝化体系为四氟硼硝时,3-氨基-4-[1H-四氮唑]-1H-吡唑与四氟硼硝的用量摩尔比为1:(1-50);优选地,所述步骤二中所述反应温度为-20℃至130℃,步骤二中所述反应时间为0.5小时至48小时。
- 一种如权利要求1-2其中一者所述的化合物,或一种以如权利要求3-4其中一者所述的化合物为前体物质制备的化合物,或一种如权利要求5-6其中一者所述的制备方法所制备的化合物,其在炸药领域的应用;优选地,所述炸药为起爆药;优选地,所述炸药为绿色环保型起爆药;优选地,所述炸药为无金属起爆药。
- 一种炸药,包括如权利要求1-2其中一者所述的化合物,或包括一种如权利要求3-4其中一者所述的化合物为前体物质制备的化合物,或包括一种如权利要求5-6其中一者所述的制备方法所制备的化合物;优选地,所述炸药为起爆药;优选地,所述炸药为绿色环保型起爆药;优选地,所述炸药为无金属起爆药。
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP19734222.3A EP3660019B1 (en) | 2018-08-15 | 2019-01-28 | 7h-pyrazolo[3,4-d]triazine-2-oxides as explosives |
| US16/480,674 US11247997B2 (en) | 2018-08-15 | 2019-01-28 | Compound and preparation and application thereof |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201810929060.XA CN108752349B (zh) | 2018-08-15 | 2018-08-15 | 一种绿色环保型起爆药及其制备方法 |
| CN201810929060.X | 2018-08-15 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2020034601A1 true WO2020034601A1 (zh) | 2020-02-20 |
Family
ID=63970084
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2019/073473 WO2020034601A1 (zh) | 2018-08-15 | 2019-01-28 | 化合物及其制备和应用 |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US11247997B2 (zh) |
| EP (1) | EP3660019B1 (zh) |
| CN (1) | CN108752349B (zh) |
| WO (1) | WO2020034601A1 (zh) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN114539010A (zh) * | 2022-03-09 | 2022-05-27 | 中北大学 | 一种mtnp/dntf/dnan低共熔物的制备方法 |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN108752349B (zh) * | 2018-08-15 | 2020-06-16 | 中国工程物理研究院化工材料研究所 | 一种绿色环保型起爆药及其制备方法 |
| CN111153757B (zh) * | 2020-01-06 | 2021-09-21 | 郑州大学 | 高性能叠氮化铜复合起爆药及其制备方法 |
| CN112079838B (zh) * | 2020-09-27 | 2021-11-16 | 中国工程物理研究院化工材料研究所 | 一种耐热炸药材料及其制备方法 |
| CN115124471B (zh) * | 2021-03-24 | 2024-02-13 | 中国工程物理研究院化工材料研究所 | 一类具有高能和低熔点特性的吡唑环类炸药及其合成方法 |
| CN115141190B (zh) * | 2022-07-26 | 2024-01-12 | 南京理工大学 | 1,2,4-三唑连-1,3,4-恶二唑类含能化合物及其制备方法 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101492424A (zh) * | 2009-03-06 | 2009-07-29 | 北京工业大学 | 一种1h-咪唑-5-羧酸酯类化合物的光化学合成方法 |
| CN101591356A (zh) * | 2009-07-01 | 2009-12-02 | 合肥工业大学 | 一种手性膦酰胺化合物及其合成方法 |
| CN105622615A (zh) * | 2015-12-30 | 2016-06-01 | 上海天慈国际药业有限公司 | 一种合成扎莱普隆的新方法 |
| CN105949219A (zh) * | 2016-05-13 | 2016-09-21 | 西安近代化学研究所 | 4-氨基-1,2,3-三氮唑[4,5-e]呋咱并[3,4-b]吡嗪-6-氧化物 |
| CN108752349A (zh) * | 2018-08-15 | 2018-11-06 | 中国工程物理研究院化工材料研究所 | 一种绿色环保型起爆药及其制备方法 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE3222960A1 (de) * | 1982-06-19 | 1983-12-22 | Bayer Ag, 5090 Leverkusen | Annellierte 1,2,3-triazin-4-on-2-n-oxide und verfahren zu ihrer herstellung |
| CN103373968A (zh) * | 2012-04-23 | 2013-10-30 | 由国峰 | 3-氨基-1,2,4-苯并三嗪-1,4-二氧化物的合成方法 |
| CN103193560A (zh) * | 2013-03-20 | 2013-07-10 | 南京理工大学 | 一种硝胺炸药及其制备方法 |
| CN105111213B (zh) * | 2015-08-19 | 2017-04-12 | 西安近代化学研究所 | 1,4‑二羟基‑3,6‑二硝基吡唑[4,3‑c]并吡唑化合物 |
| CN108299442B (zh) * | 2018-03-15 | 2019-12-10 | 西安近代化学研究所 | 1,4-二硝胺基-3,6-二硝基吡唑[4,3-c]并吡唑脒基脲盐化合物 |
| CN112079838B (zh) * | 2020-09-27 | 2021-11-16 | 中国工程物理研究院化工材料研究所 | 一种耐热炸药材料及其制备方法 |
-
2018
- 2018-08-15 CN CN201810929060.XA patent/CN108752349B/zh active Active
-
2019
- 2019-01-28 WO PCT/CN2019/073473 patent/WO2020034601A1/zh unknown
- 2019-01-28 US US16/480,674 patent/US11247997B2/en active Active
- 2019-01-28 EP EP19734222.3A patent/EP3660019B1/en active Active
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101492424A (zh) * | 2009-03-06 | 2009-07-29 | 北京工业大学 | 一种1h-咪唑-5-羧酸酯类化合物的光化学合成方法 |
| CN101591356A (zh) * | 2009-07-01 | 2009-12-02 | 合肥工业大学 | 一种手性膦酰胺化合物及其合成方法 |
| CN105622615A (zh) * | 2015-12-30 | 2016-06-01 | 上海天慈国际药业有限公司 | 一种合成扎莱普隆的新方法 |
| CN105949219A (zh) * | 2016-05-13 | 2016-09-21 | 西安近代化学研究所 | 4-氨基-1,2,3-三氮唑[4,5-e]呋咱并[3,4-b]吡嗪-6-氧化物 |
| CN108752349A (zh) * | 2018-08-15 | 2018-11-06 | 中国工程物理研究院化工材料研究所 | 一种绿色环保型起爆药及其制备方法 |
Non-Patent Citations (4)
| Title |
|---|
| BOULTON, A.J. ET AL.: "1, 2, 3-Benzotriazine 2-oxides", J. CHEM. SOC. PERKIN TRANS., 31 December 1988 (1988-12-31), pages 1509 - 1512, XP055644420, DOI: 10.1039/P19880001509 * |
| DATABASE Registry 19 June 2015 (2015-06-19), Database accession no. 1784648-12-9 * |
| LECH STEFANIAK: "Nitrogen-14 Nuclear Magnetic Resonance of Azine-N-oxides", SPECTROCHIMICA ACTA, PART A: MOLECULAR AND BIOMOLECULAR SPECTROSCOPY, vol. 32 A, no. 2, 1 January 1976 (1976-01-01), pages 345 - 349, XP055644414, ISSN: 0584-8539, DOI: 10.1016/0584-8539(76)80087-6 * |
| See also references of EP3660019A4 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN114539010A (zh) * | 2022-03-09 | 2022-05-27 | 中北大学 | 一种mtnp/dntf/dnan低共熔物的制备方法 |
| CN114539010B (zh) * | 2022-03-09 | 2022-11-11 | 中北大学 | 一种mtnp/dntf/dnan低共熔物的制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3660019C0 (en) | 2023-12-20 |
| EP3660019A4 (en) | 2021-09-08 |
| US11247997B2 (en) | 2022-02-15 |
| CN108752349A (zh) | 2018-11-06 |
| EP3660019A1 (en) | 2020-06-03 |
| CN108752349B (zh) | 2020-06-16 |
| EP3660019B1 (en) | 2023-12-20 |
| US20210155624A1 (en) | 2021-05-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2020034601A1 (zh) | 化合物及其制备和应用 | |
| Manzoor et al. | Nitro-tetrazole based high performing explosives: Recent overview of synthesis and energetic properties | |
| Tang et al. | Enforced planar FOX-7-like molecules: a strategy for thermally stable and insensitive π-conjugated energetic materials | |
| Lei et al. | A facile strategy for synthesizing promising pyrazole-fused energetic compounds | |
| Tang et al. | Nitrogen-rich tricyclic-based energetic materials | |
| Gao et al. | Recent progress in taming FOX-7 (1, 1-diamino-2, 2-dinitroethene) | |
| Türker | Azo-bridged triazoles: Green energetic materials | |
| Liu et al. | Intermolecular weak hydrogen bonding (Het-HN/O): An effective strategy for the synthesis of monosubstituted 1, 2, 4, 5-Tetrazine-based energetic materials with excellent sensitivity | |
| CN109627235B (zh) | 一种高能钝感含能化合物及其合成方法 | |
| Du et al. | Review on the synthesis and performance for 1, 3, 4‐oxadiazole‐based energetic materials | |
| Lei et al. | The Unique Synthesis of a Green Metal‐Free Primary Explosive: 3, 3′‐azo‐5, 5′‐diazido‐1, 2, 4‐triazole | |
| Zeng et al. | From N atom to C–NO2 group: Achieving a high energy density material with a “NH2–NO2–NH2” block | |
| Fershtat | Recent advances in the synthesis and performance of 1, 2, 4, 5-tetrazine-based energetic materials | |
| Zhang et al. | Heat‐resistant energetic materials deriving from benzopyridotetraazapentalene: Halogen bonding effects on the outcome of crystal structure, thermal stability and sensitivity | |
| Wang et al. | 4, 4′-Bis (trinitromethyl)-3, 3′-azo/azoxy-furazan: High-energy dense oxidizers | |
| Deshmukh et al. | Cyclodextrin nitrate Ester/H2SO4 as a novel nitrating system for efficient synthesis of insensitive high explosive 3-nitro-1, 2, 4-triazol-5-one | |
| EP1119544A1 (en) | Energetic nitramine-linked azoles and hydroxylammonium salts as oxidizers, initiators and gas generators | |
| CZ305403B6 (cs) | Energetické materiály na bázi bismutu | |
| Cao et al. | Synthesis and Characterization of Azido-Functionalized 1, 2, 3-Triazole and Fused 1, 2, 3-Triazole | |
| Bhatia et al. | N-Acetonitrile functionalized 3-nitrotriazole: Precursor to nitrogen rich stable and insensitive energetic materials | |
| Shaferov et al. | Energetic 1, 2, 4-oxadiazoles: synthesis and properties | |
| CN103121977A (zh) | 重(n-二硝基乙基)呋咱类含能离子盐及其制备方法 | |
| Wang et al. | An unexpected method to synthesise 1, 2, 4-oxadiazolone derivatives: a class of insensitive energetic materials | |
| CN115141190B (zh) | 1,2,4-三唑连-1,3,4-恶二唑类含能化合物及其制备方法 | |
| Bhatia et al. | Zwitterionic Energetic Materials: Synthesis, Structural Diversity and Energetic Properties |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2019734222 Country of ref document: EP Effective date: 20190709 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |