WO2019189147A1 - 細胞の有する二本鎖dnaの標的部位を改変する方法 - Google Patents
細胞の有する二本鎖dnaの標的部位を改変する方法 Download PDFInfo
- Publication number
- WO2019189147A1 WO2019189147A1 PCT/JP2019/012807 JP2019012807W WO2019189147A1 WO 2019189147 A1 WO2019189147 A1 WO 2019189147A1 JP 2019012807 W JP2019012807 W JP 2019012807W WO 2019189147 A1 WO2019189147 A1 WO 2019189147A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- dna
- cell
- nucleic acid
- sequence
- double
- Prior art date
Links
Images











Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/85—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
- C12N15/102—Mutagenizing nucleic acids
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/111—General methods applicable to biologically active non-coding nucleic acids
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/113—Non-coding nucleic acids modulating the expression of genes, e.g. antisense oligonucleotides; Antisense DNA or RNA; Triplex- forming oligonucleotides; Catalytic nucleic acids, e.g. ribozymes; Nucleic acids used in co-suppression or gene silencing
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/80—Vectors or expression systems specially adapted for eukaryotic hosts for fungi
- C12N15/81—Vectors or expression systems specially adapted for eukaryotic hosts for fungi for yeasts
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/16—Hydrolases (3) acting on ester bonds (3.1)
- C12N9/22—Ribonucleases RNAses, DNAses
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/24—Hydrolases (3) acting on glycosyl compounds (3.2)
- C12N9/2497—Hydrolases (3) acting on glycosyl compounds (3.2) hydrolysing N- glycosyl compounds (3.2.2)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/78—Hydrolases (3) acting on carbon to nitrogen bonds other than peptide bonds (3.5)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/10—Type of nucleic acid
- C12N2310/20—Type of nucleic acid involving clustered regularly interspaced short palindromic repeats [CRISPRs]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2800/00—Nucleic acids vectors
- C12N2800/80—Vectors containing sites for inducing double-stranded breaks, e.g. meganuclease restriction sites
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y302/00—Hydrolases acting on glycosyl compounds, i.e. glycosylases (3.2)
- C12Y302/02—Hydrolases acting on glycosyl compounds, i.e. glycosylases (3.2) hydrolysing N-glycosyl compounds (3.2.2)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y305/00—Hydrolases acting on carbon-nitrogen bonds, other than peptide bonds (3.5)
- C12Y305/04—Hydrolases acting on carbon-nitrogen bonds, other than peptide bonds (3.5) in cyclic amidines (3.5.4)
- C12Y305/04005—Cytidine deaminase (3.5.4.5)
Definitions
- the present invention enables modification of a target site in a specific region of a double-stranded DNA of a cell using homologous recombination without double-strand breaks of DNA (with no break or single-strand break).
- the present invention relates to a method for modifying double-stranded DNA.
- CRISPR clustered regularly interspaced short palindromic repeats
- Cas CRISPR-associated proteins cleave target DNA in a manner dependent on a single guide RNA (sgRNA) and protospacer adjacent motif (PAM)
- sgRNA single guide RNA
- PAM protospacer adjacent motif
- HDR homologous recombination repair
- Non-patent Documents 1 and 2 Although homologous recombination using Cas9 nickase (nCas9) has been reported (Non-patent Documents 1 and 2), the recombination induction efficiency is often very low compared to Cas9 nuclease (Non-patent Document 3). ). In addition, as far as the present inventors know, no homologous recombination using Cas9 (dCas9) in which both nuclease activities are inactivated has been reported.
- the subject of the present invention is not limited by the type or site of mutation that can be introduced, but can switch the orientation and combination of genes or knock in a gene fragment, or a nucleobase converting enzyme such as deaminase or the like. It is to provide a novel DNA modification technique using DNA glycosylase.
- NER nucleotide excision repair
- the toxicity to the cells is suppressed by bringing the target DNA into contact with the donor DNA containing the complex that combines the nucleic acid sequence recognition module, the nucleobase converting enzyme, and the inserted sequence.
- homologous recombination of DNA is possible, and in a preferred embodiment, surprisingly, close to 100% homologous recombination activity occurs at the target site.
- the present inventor has completed the present invention.
- a method for modifying a target site of double-stranded DNA possessed by a cell comprising a nucleic acid sequence recognition module that specifically binds to a target nucleotide sequence in a selected double-stranded DNA, a nucleobase converting enzyme, A donor DNA containing a complex bound with a DNA glycosylase and an insertion sequence is brought into contact with the double-stranded DNA, and the target site is released without cleaving at least one strand of the double-stranded DNA at the target site. Replacing the inserted sequence or inserting the inserted sequence into the target site.
- the nucleic acid sequence recognition module is selected from the group consisting of a CRISPR-Cas system, a zinc finger motif, a TAL effector, and a PPR motif in which at least one DNA cleavage ability of a Cas effector protein is inactivated, [1] or The method according to [2].
- nucleic acid sequence recognition module is a CRISPR-Cas system in which the DNA cleavage ability of both Cas effector proteins is inactivated.
- nucleobase converting enzyme is deaminase.
- deaminase is cytidine deaminase.
- cytidine deaminase is PmCDA1.
- a nucleobase converting enzyme such as deaminase or the like that can switch the orientation and combination of genes or knock in a gene fragment without being restricted by the type of mutation that can be introduced and the mutation site.
- a novel DNA modification technique using DNA glycosylase is provided. Since the DNA modification technology of the present invention can modify the target site without cleaving double-stranded DNA, unexpected rearrangement and toxicity associated with the cleaving can be kept low, and much more efficient than conventional methods. The target site can be modified well.
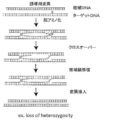
- FIG. 1 shows a schematic diagram of the mechanism of genome modification by complementary strand modification.
- a complex in which a nucleic acid sequence recognition module, a nucleobase converting enzyme or a DNA glycosylase are bound, and a donor DNA for recombination By introducing a complex in which a nucleic acid sequence recognition module, a nucleobase converting enzyme or a DNA glycosylase are bound, and a donor DNA for recombination, a highly efficient knock-in of an inserted sequence is possible.
- FIG. 2 shows that a target site mutation could be introduced using dCas9-CDA or nCas9-CDA and donor DNA.
- FIG. 3 shows a recombination evaluation system using a budding yeast (BY4741 strain) into which a marker switch has been introduced in advance between the promoter regions of Ade1 and Ade1. When the marker switch is inverted by recombination in the homologous region, the function of Ade1 is restored and the color of the colony changes from red to white.
- FIG. 3 shows a recombination evaluation system using a budding yeast (BY4741 strain) into which a marker switch has been introduced in advance between the promoter regions of Ade1 and Ade1. When the marker switch is inverted by recombination in the homologous region, the function of Ade1 is restored and the color of the colony changes from red to white.
- FIG. 3 shows a recombination evaluation system using a budding yeast (BY4741 strain) into which a marker switch has been introduced in advance between the promoter regions of Ade1 and Ade1. When the marker switch is inverted by recombination in the homologous
- vector 1559 nCas9-CDA_UraAde target 8 (target nucleotide sequence: ttggcggataatgcctttag (SEQ ID NO: 11)); vector 1560: nCas9-CDA_UraAde target 9 (target nucleotide sequence: tgcagttgggttaagaatac (SEQ ID NO: 12)) nCas9-CDA_UraAde target 11 (target nucleotide sequence: gctaacatcaaaaggcctct (SEQ ID NO: 13)); vector 1565: dCas9-CDA_UraAde target 3 (target nucleotide sequence: tggcggataatgcctttag (SEQ ID NO: 14)).
- the vectors (1553, 1557, 1559, 1560, 1562, 1565) correspond to vectors obtained by substituting the nucleotide sequences 3890 to 3909 of the sequence of the vector 1059 (SEQ ID NO: 5) with the respective target nucleotide sequences. Further, the last two digits of the vector number correspond to the numbers of the target sites in FIG.
- FIG. 5 shows a schematic diagram of a knock-in or knock-out method using the DNA modification method of the present invention.
- FIG. 6 shows the results of a demonstration experiment of knock-in or knock-out using the method of FIG.
- FIG. 7 shows a schematic diagram of an evaluation system for recombination using animal cells and experimental conditions performed in Example 5.
- FIG. 8 shows the results of a demonstration experiment of the recombination reaction using the recombination evaluation system of FIG.
- the horizontal axis of the graph represents the homologous recombination rate (%).
- FIG. 9 shows a schematic diagram and experimental conditions of a recombination evaluation system using animal cells performed in Example 6.
- FIG. 10 shows the results of a demonstration experiment of the recombination reaction using the recombination evaluation system of FIG.
- the vertical axis of the graph represents the homologous recombination rate (%).
- FIG. 11 shows a schematic diagram of an evaluation system for recombination using animal cells and experimental conditions performed in Example 7.
- FIG. 12 shows the results of a demonstration experiment of the recombination reaction using the recombination evaluation system of FIG.
- the horizontal axis of the graph represents the homologous recombination rate (%).
- the present invention does not cut at least one strand of double-stranded DNA to be modified (for example, chromosomal DNA, mitochondrial DNA, chloroplast DNA; hereinafter collectively referred to as “genomic DNA”).
- genomic DNA for example, chromosomal DNA, mitochondrial DNA, chloroplast DNA; hereinafter collectively referred to as “genomic DNA”.
- the method includes a nucleic acid sequence recognition module that specifically binds to a target nucleotide sequence in the double-stranded DNA, a nucleobase converting enzyme or a DNA glycosylase (hereinafter sometimes abbreviated as “nucleobase converting enzyme etc.”), And a step of bringing a donor DNA containing an inserted sequence into contact with the double-stranded DNA.
- a nucleic acid sequence recognition module that specifically binds to a target nucleotide sequence in the double-stranded DNA
- a nucleobase converting enzyme or a DNA glycosylase hereinafter sometimes abbreviated as “nucleobase converting enzyme etc.”
- “modification” of double-stranded DNA means that a certain nucleotide (eg, dA, dC, dG or dT) or nucleotide sequence on the DNA strand is replaced with another nucleotide or nucleotide sequence, or It means that another nucleotide or nucleotide sequence is inserted between certain nucleotides on the DNA strand.
- the double-stranded DNA to be modified is not particularly limited, but is preferably genomic DNA.
- the “donor DNA” means a DNA containing a foreign insertion sequence, and the donor DNA usually has two regions adjacent to the target site upstream and downstream of the target site (hereinafter “adjacent”). 2 types of sequences (hereinafter also referred to as “homology arms”). When distinguishing each homology arm, “5 ′ homology arm” and “3 ′ homology arm” may be distinguished.
- the “target site” of double-stranded DNA means a region to be replaced with the insertion sequence contained in the donor DNA, or between the nucleotides into which the insertion sequence is to be inserted. Does not include the adjacent sequence.
- sequence homologous to the adjacent region of the target site is preferably not less than 80% (for example, 85%) with respect to the completely identical sequence as long as homologous recombination can occur in the cell as well as the completely identical sequence. % Or more, 90% or more, 95% or more, 96% or more, 97% or more, 98% or more, 99% or more).
- Insertion sequences include drug resistance genes (eg, kanamycin resistance gene, ampicillin resistance gene, puromycin resistance gene, etc.), thymidine kinase gene, diphtheria toxin gene and other selectable marker sequences as necessary, green fluorescent protein (GFP) , Reporter gene sequences such as red fluorescent protein, ⁇ -glucuronidase (GUS), and FLAG.
- GFP green fluorescent protein
- GUS ⁇ -glucuronidase
- FLAG FLAG
- a LoxP sequence, an FRT sequence, or a transposon-specific terminal inverted sequence may be present before and after these genes so that these genes can be excised after cell selection is completed. .
- Preferred transposons include, for example, piggyBac, a transposon derived from lepidopterous insects (Kaji, K. et al., Nature, 458: 771-775 (2009), Woltjen et al., Nature, 458: 766 -770 (2009), WO 2010/012077).
- piggyBac a transposon derived from lepidopterous insects
- Woltjen et al. Nature, 458: 766 -770 (2009), WO 2010/012077.
- co-introducing an expression vector containing the above-mentioned drug resistance gene, and transient (several days) drug selection May be performed.
- Whether the inserted sequence is inserted into the target site or whether it is replaced with the target site is confirmed by decoding the sequence and screening chromosomal DNA extracted from cells by Southern hybridization or PCR. In the case where the drug resistance gene or the like is present in the donor DNA, its expression can be confirmed as an index.
- the donor DNA may be linear (eg, synthetic double-stranded DNA), circular (eg, plasmid DNA), or single-stranded DNA (eg, single-stranded oligodeoxynucleotide). (SsODN)) or double-stranded DNA.
- the donor DNA can be appropriately designed depending on the base length of the inserted sequence, the homologous recombination activity of the host cell, and the like. For example, when the insertion sequence is 100 bases or less, ssODN or synthetic double-stranded DNA is usually used, and when it is longer than that, synthetic double-stranded DNA or plasmid DNA is usually used.
- the length of the donor DNA is not particularly limited, and can be appropriately designed depending on the length of the inserted sequence.
- the length of the inserted sequence is not particularly limited, and is usually in the range of 1 to tens of thousands of bases (for example, in the case of ssODN, 100 bases or less (eg, 70 bases or less, 50 bases or less)) It can be designed appropriately according to the purpose.
- the length of each homology arm is not particularly limited.
- the length is usually 10 to 150 bases
- the donor DNA is a synthetic double-stranded DNA
- a DNA having a length of 5000 bases is used and the donor DNA is a plasmid DNA
- a DNA having a length of 100 bases to 5000 bases, preferably 500 bases to 1000 bases is usually used.
- the “nucleic acid sequence recognition module” means a molecule or molecular complex having the ability to specifically recognize and bind to a specific nucleotide sequence (ie, a target nucleotide sequence) on a DNA strand.
- the nucleobase converting enzyme or the like linked to the module becomes a target site of the double-stranded DNA nucleobase converting enzyme (ie, the target nucleotide sequence and its nucleotide sequence). It is possible to act specifically on neighboring nucleotides).
- a target site can be modified by introducing a complex of a nucleobase converting enzyme, a nucleic acid sequence recognition module, and donor DNA into a cell.
- a complex of a nucleobase converting enzyme, a nucleic acid sequence recognition module, and donor DNA into a cell.
- the mechanism of target site modification by this method is presumed as follows.
- the base present in the target site of the nucleobase converting enzyme is converted into another base, and the converted base is removed by DNA glycosylase, and the base-free site (apurinic / apyrimidic (AP ) Site) is processed by enzymes downstream of the base excision repair (BER) pathway, such as AP endonuclease, DNA polymerase, and DNA ligase.
- BER base excision repair
- nucleobase converting enzyme refers to a target nucleotide without cleaving a DNA strand by catalyzing a reaction for converting a substituent on a purine or pyrimidine ring of a DNA base into another group or atom. Is an enzyme capable of converting to other nucleotides.
- DNA glycosylase means an enzyme that hydrolyzes an N-glycoside bond of DNA.
- DNA glycosylase plays a role of removing bases that are originally damaged in BER from DNA, in the present invention, normal bases in DNA (that is, dC, dT, dA, or dG, or they undergo epigenetic modification). Those that can act on those received) are preferred.
- a mutant DNA glycosylase that originally does not react with a normal base or has low reactivity, but has acquired or improved reactivity with a normal base by mutation, is also included in the DNA glycosylase of the present invention and is preferably used. Can be done.
- the base-free site (apurinic / apyrimidic (AP) site) resulting from the abasic reaction by the enzyme is treated with an enzyme downstream of the BER pathway, such as AP endonuclease, DNA polymerase, or DNA ligase.
- an enzyme downstream of the BER pathway such as AP endonuclease, DNA polymerase, or DNA ligase.
- the strain is not sufficiently reactive to DNA with a double-helix structure without strain” means that the double-helix structure without strain is only at a frequency at which cytotoxicity is suppressed to the extent that it does not affect cell survival. This means that an abasic reaction does not occur in the region where the DNA is formed.
- strain-free double-helical DNA means that a strong double-helical structure is formed (that is, unrelaxed double-helical DNA (or simply referred to as unrelaxed DNA)), Not only in the state of single-stranded DNA in which paired bases are completely dissociated, but also in the state of relaxed double-stranded DNA in which double-helix structure is unfolded although base pairs are formed Things are not included.
- a DNA glycosylase that is sufficiently low in reactivity to DNA with a double-helix structure without strain is relatively low compared to a DNA glycosylase that is sufficiently low in reactivity to DNA with a double-helix structure that is not naturally strained, or the wild type.
- Examples thereof include mutant DNA glycosylase into which a mutation that reduces the reactivity to DNA having a strain-free double helix structure is introduced. Furthermore, it is a DNA glycosylase divided into two fragments, and each fragment binds to one of the two nucleic acid sequence recognition modules divided into two to form two complexes, and both complexes are ligated. When folded, the nucleic acid sequence recognition module is a split enzyme designed to specifically bind to the target nucleotide sequence, which allows the DNA glycosylase to catalyze an abasic reaction. A certain DNA glycosylase is also included in the “DNA glycosylase having a sufficiently low reactivity to DNA having an undistorted double helix structure” in the present invention.
- the “nucleic acid modifying enzyme complex” means a nucleobase having a specific nucleotide sequence recognition ability, comprising a complex in which the nucleic acid sequence recognition module is linked to a nucleobase converting enzyme or a DNA glycosylase. It means a molecular complex having a catalytic function for conversion reaction or abasic reaction.
- the “complex” includes not only a complex composed of a plurality of molecules but also a complex protein having a nucleic acid sequence recognition module, a nucleobase converting enzyme and the like in a single molecule.
- the nucleobase converting enzyme used in the present invention is not particularly limited as long as it can catalyze the above reaction.
- a nucleic acid / nucleotide deaminase superfamily that catalyzes a deamination reaction for converting an amino group into a carbonyl group.
- Deaminase belonging to Preferred examples include cytidine deaminase that can convert cytosine or 5-methylcytosine to uracil or thymine, adenosine deaminase that can convert adenine to hypoxanthine, and guanosine deaminase that can convert guanine to xanthine.
- cytidine deaminase include activation-induced cytidine deaminase (hereinafter also referred to as AID), which is an enzyme that introduces a mutation into an immunoglobulin gene in acquired immunity of vertebrates.
- AID activation-induced cytidine deaminase
- the origin of the nucleobase converting enzyme is not particularly limited.
- PmCDA1 derived from lamprey (Petromyzon marinus cytosine deaminase 1), AID (Activation-induced cytidine derived from mammals (eg, human, pig, cow, horse, monkey, etc.) deaminase; AICDA) can be used.
- the base sequence and amino acid sequence of PmCDA1 cDNA can refer to GenBank accession No. EF094822 and ABO15149
- the base sequence and amino acid sequence of human AID cDNA can refer to GenBank accession No. NM_020661 and NP_065712, respectively.
- PmCDA1 is preferable from the viewpoint of enzyme activity.
- the DNA glycosylase used in the present invention is not particularly limited as long as it can catalyze the reaction of hydrolyzing the N-glycoside bond of DNA to eliminate the base, but means to increase versatility as a genome editing technique And those capable of acting on normal bases (that is, dC, dT, dA or dG, or those subjected to epigenetic modification, such as 5-methylcytosine, etc.) are preferred.
- an enzyme having CDG activity that catalyzes a reaction that desorbs cytosine
- an enzyme that has TDG activity that catalyzes a reaction that desorbs thymine
- a reaction that desorbs 5-methylcytosine examples include enzymes having activity (5-mCDG activity).
- thymine DNA glycosylase oxoguanine glucosylase
- alkyladenine DNA glycosylase eg, yeast 3-methyladenine-DNA glycosylase (MAG1), etc.
- the inventor previously used DNA glycosylase, a DNA glycosylase that is sufficiently low in reactivity to undistorted double helix DNA (unrelaxed DNA) to reduce cytotoxicity and efficiently modify the target sequence. It is reported that it can be done (International Publication No. 2016/072399). Therefore, it is preferable to use a DNA glycosylase having a sufficiently low reactivity to DNA having a double helix structure without strain as the DNA glycosylase.
- UNG uracil-DNA glycosylase
- CDG cytosine-DNA glycosylase
- TDG thymine-DNA glycosylase
- UNG mutant examples include yeast UNG1 N222D / L304A double mutant, N222D / R308E double mutant, N222D / R308C double mutant, Y164A / L304A double mutant, Y164A / R308E two Double mutant, Y164A / R308C double mutant, Y164G / L304A double mutant, Y164G / R308E double mutant, Y164G / R308C double mutant, N222D / Y164A / L304A triple mutant, N222D / Y164A / R308E Triple mutant, N222D / Y164A / R308C triple mutant, N222D / Y164G / L304A triple mutant, N222D / Y164G / R308E triple mutant, N222D / Y164G / R308E triple mutant, N222D / Y164G / R308C triple mutant and the like.
- a mutant in which the same mutation is introduced into the amino acid corresponding to each mutant may be used.
- the UNG mutation of E. coli corresponding to the Y164A or Y164G mutation of yeast UNG1, which is a mutation imparting TDG activity includes Y66A or Y66G
- the human UNG mutation includes Y147A or Y147G.
- N123D is a mutation of E. coli corresponding to the N222D mutation of yeast UNG1, which is a mutation that imparts CDG activity
- N204D is a mutation of human UNG.
- the UNG1 mutation corresponding to the L304A, R308E, or R308C mutation in yeast UNG1, which reduces the reactivity of DNA to an undistorted double helix structure, is L191A, R195E, or R195C, a human UNG mutation.
- Vaccinia virus-derived UDG mutants include N120D mutant (providing CDG activity ability), Y70G mutant (confering TDG activity ability), Y70A mutant (confering TDG activity ability), N120D / Y70G double mutant, N120D / Y70A double mutant and the like.
- the nucleic acid sequence recognition module is a split enzyme designed to specifically bind to the target nucleotide sequence, which allows the DNA glycosylase to catalyze an abasic reaction.
- split enzymes are designed and produced by referring to, for example, International Publication No. 2016/072399, Nat Biotechnol. 33 (2): 139-142 (2015), PNAS 112 (10): 2984-2989 (2015) can do.
- UNG is not particularly limited.
- E. coli Veshney, U. et al. (1988) J. Biol. Chem., 263, 7776-7784
- yeast mammals
- UNG1 or UNG2 derived from pigs, cows, horses, monkeys, etc.
- UDG derived from viruses eg, poxviridae (vaccinia virus etc.), herpesviridae, etc.
- viruses eg, poxviridae (vaccinia virus etc.), herpesviridae, etc.
- the target nucleotide sequence in the double-stranded DNA that is recognized by the nucleic acid sequence recognition module of the nucleic acid modifying enzyme complex of the present invention is not particularly limited as long as the module can specifically bind, and the target nucleotide sequence in the double-stranded DNA is not limited. It can be any sequence.
- the length of the target nucleotide sequence only needs to be sufficient for the nucleic acid sequence recognition module to specifically bind. For example, when a mutation is introduced at a specific site in mammalian genomic DNA, it depends on the genome size. 12 nucleotides or more, preferably 15 nucleotides or more, more preferably 17 nucleotides or more.
- the upper limit of the length is not particularly limited, but is preferably 25 nucleotides or less, more preferably 22 nucleotides or less. As shown in the examples described later, high modification efficiency has been demonstrated in any of the experimental systems in which the target nucleotide sequence exists in a target site, a sequence homologous to the homology arm, and a region partially including a sequence homologous to the homology arm. Therefore, the target nucleotide sequence may be present in the target site, may be present in at least a partial region of the sequence homologous to the homology arm, or may be present in the vicinity region of the sequence homologous to the homology arm. .
- Examples of the nucleic acid sequence recognition module of the nucleic acid modifying enzyme complex of the present invention include a CRISPR-Cas system (hereinafter referred to as “CRISPR-mutated Cas”) in which at least one DNA cleavage ability of a Cas effector protein (also referred to as Cas nuclease) is inactivated.
- CRISPR-mutated Cas a CRISPR-Cas system
- Cas effector protein also referred to as Cas nuclease
- TAL transcription activator-like effector
- PPR penentatricopeptide repeat
- CRISPR-mutated Cas Preferably, CRISPR-mutated Cas, zinc finger motif, TAL effector, PPR motif and the like can be mentioned.
- the Cas effector protein in which at least one DNA cleavage ability is inactivated is also referred to as a Cas effector protein mutant.
- the zinc finger motif is a linkage of 3 to 6 different zinc finger units of the Cys2His2 type (one finger recognizes about 3 bases), and can recognize a target nucleotide sequence of 9 to 18 bases.
- Zinc finger motifs are: Modular assembly method (Nat Biotechnol (2002) 20: 135-141), OPEN method (Mol Cell (2008) 31: 294-301), CoDA method (Nat Methods (2011) 8: 67-69) In addition, it can be prepared by a known method such as E. coli one-hybrid method (Nat Biotechnol (2008) 26: 695-701). Japanese Patent No. 4968498 can be referred to for details of the production of the zinc finger motif.
- the TAL effector has a repeating structure of modules of about 34 amino acids, and the binding stability and base specificity are determined by the 12th and 13th amino acid residues (called RVD) of one module.
- RVD 12th and 13th amino acid residues
- the PPR motif consists of 35 amino acids, and is constructed to recognize a specific nucleotide sequence by a series of PPR motifs that recognize one nucleobase.
- the 1st, 4th, and ii (-2) th amino acids of each motif Only recognize the target base. Since there is no dependence on the motif structure and there is no interference from the motifs on both sides, it is possible to produce a PPR protein specific to the target nucleotide sequence by linking the PPR motifs just like the TAL effector. JP, 2013-128413, A can be referred to for details of preparation of a PPR motif.
- DNA-binding domain of these proteins is well known, so it is easy to design a fragment that contains this domain and does not have the ability to cleave DNA double strands. And can be built.
- nucleic acid sequence recognition modules can be provided as a fusion protein with the above nucleobase converting enzyme or the like, or a protein binding domain such as SH3 domain, PDZ domain, GK domain, GB domain and their binding A partner may be fused with a nucleic acid sequence recognition module, a nucleobase converting enzyme, or the like, respectively, and provided as a protein complex through the interaction between the domain and its binding partner.
- intein can be fused to a nucleic acid sequence recognition module, a nucleobase converting enzyme, etc., and both can be linked by ligation after synthesis of each protein.
- nucleic acid modifying enzyme complex of the present invention comprising a complex (including a fusion protein) in which a nucleic acid sequence recognition module and a nucleobase converting enzyme, etc. are bound to the double-stranded DNA, This is carried out by introducing a nucleic acid encoding the complex into a cell having strand DNA (eg, genomic DNA).
- a nucleic acid encoding a nucleic acid modifying enzyme complex includes a base sequence encoding a nucleic acid sequence recognition module and a base sequence encoding a nucleobase converting enzyme or a DNA glycosylase, and the nucleic acid sequence recognition module is a CRISPR.
- the nucleic acid sequence recognition module and the nucleobase converting enzyme form a complex in the host cell after translating into a protein as a nucleic acid encoding the fusion protein or using a binding domain or intein. It is preferable to prepare them as nucleic acids encoding them in such a form that they can be made.
- the nucleic acid may be DNA or RNA.
- DNA it is preferably double-stranded DNA and is provided in the form of an expression vector placed under the control of a promoter functional in the host cell.
- RNA it is preferably a single-stranded RNA.
- a cell into which a nucleic acid encoding a nucleic acid sequence recognition module and / or a nucleobase converting enzyme or the like is introduced is derived from a microbial cell such as a prokaryote such as Escherichia coli or a lower eukaryote such as yeast.
- a microbial cell such as a prokaryote such as Escherichia coli or a lower eukaryote such as yeast.
- Cells of any species can be included, including vertebrates including mammals such as mammals, higher eukaryotic cells such as insects and plants.
- a DNA encoding a nucleic acid sequence recognition module such as a zinc finger motif, a TAL effector, and a PPR motif can be obtained by any of the methods described above for each module.
- DNA encoding sequence recognition modules such as restriction enzymes, transcription factors, RNA polymerase, etc. covers the region encoding the desired part of the protein (part containing the DNA binding domain) based on the cDNA sequence information.
- oligo DNA primers can be synthesized and cloned using the total RNA or mRNA fraction prepared from cells producing the protein as a template and amplified by RT-PCR.
- DNA encoding nucleobase converting enzyme ie, DNA encoding nucleobase converting enzyme or DNA encoding DNA glycosylase
- an oligo DNA primer is synthesized based on the cDNA sequence information of the enzyme used. Cloning can be performed by amplifying by RT-PCR using the total RNA or mRNA fraction prepared from cells producing the enzyme as a template.
- DNA encoding lamprey PmCDA1 is designed based on the cDNA sequence (accession No. EF094822) registered in the NCBI database, and appropriate primers are designed upstream and downstream of CDS. Can be cloned by RT-PCR.
- DNA encoding human AID is designed based on the cDNA sequence (accession No. AB040431) registered in the NCBI database, and appropriate primers are designed upstream and downstream of CDS.
- human lymph node-derived mRNA Can be cloned by RT-PCR.
- the donor DNA can also be cloned in the same manner as described above based on the sequence information of the target site.
- the cloned DNA can be digested as is or, if desired, with restriction enzymes, or with an appropriate linker and / or nuclear translocation signal (if the desired double-stranded DNA is mitochondrial or chloroplast DNA, each organelle translocation signal ), And then ligated with DNA encoding the nucleic acid sequence recognition module to prepare DNA encoding the fusion protein.
- a DNA encoding a nucleic acid sequence recognition module and a DNA encoding a nucleobase converting enzyme or the like are each fused with a DNA encoding a binding domain or its binding partner, or both DNAs are fused with a DNA encoding a separated intein.
- a complex may be formed after the nucleic acid sequence recognition / conversion module, the nucleobase converting enzyme and the like are translated in the host cell.
- a linker and / or a nuclear translocation signal can be linked to an appropriate position of one or both DNAs as desired.
- the donor DNA may be prepared as a single DNA, or may be provided as a single DNA with a nucleic acid encoding a nucleic acid sequence recognition module and / or a nucleobase converting enzyme.
- DNA encoding nucleic acid sequence recognition module DNA encoding nucleobase converting enzyme, etc., donor DNA, chemically synthesize DNA strands or partially synthesized oligo DNA short strands, PCR method or By connecting using the Gibson Assembly method, it is also possible to construct DNA that encodes the full length.
- the donor DNA is a single-stranded nucleic acid
- plasmid DNA containing the DNA is digested with a restriction enzyme into a single strand, and RNA is synthesized with RNA polymerase. Thereafter, it can be prepared by synthesizing cDNA with reverse transcriptase and differentiating the RNA strand with RNaseH.
- a plasmid containing donor DNA can be digested with a nickase-type restriction enzyme and prepared by separation and purification by electrophoresis.
- the advantage of constructing full-length DNA by chemical synthesis or in combination with PCR method or Gibson Assembly method is that the codon used can be designed over the entire CDS according to the host into which the DNA is introduced.
- an increase in protein expression level can be expected by converting the DNA sequence into a codon frequently used in the host organism.
- Data on the frequency of codon usage in the host to be used is, for example, the genetic code usage frequency database (http://www.kazusa.or.jp/codon/index.html) published on the Kazusa DNA Research Institute website.
- An expression vector containing a DNA encoding a nucleic acid sequence recognition module and / or a nucleobase converting enzyme can be produced, for example, by ligating the DNA downstream of a promoter in an appropriate expression vector.
- Expression vectors include plasmids derived from E.
- coli eg, pBR322, pBR325, pUC12, pUC13
- plasmids derived from Bacillus subtilis eg, pUB110, pTP5, pC194
- yeast-derived plasmids eg, pSH19, pSH15
- insect cell expression Plasmid eg, pFast-Bac
- animal cell expression plasmid eg, pA1-11, pXT1, pRc / CMV, pRc / RSV, pcDNAI / Neo
- bacteriophage such as ⁇ phage
- insect virus vector such as baculovirus ( Examples: BmNPV, AcNPV); animal virus vectors such as retrovirus, vaccinia virus, adenovirus, etc.
- the promoter may be any promoter as long as it is appropriate for the host used for gene expression. In conventional methods involving DSB, the viability of host cells may be significantly reduced due to toxicity, so it is desirable to use an inducible promoter to increase the number of cells by the start of induction. Since sufficient cell growth can be obtained even if the enzyme complex is expressed, a constitutive promoter can also be used without limitation.
- SR ⁇ promoter when the host is an animal cell, SR ⁇ promoter, SV40 promoter, LTR promoter, CMV (cytomegalovirus) promoter, RSV (rous sarcoma virus) promoter, MoMuLV (Moloney murine leukemia virus) LTR, HSV-TK (herpes simplex) Virus thymidine kinase) promoter and the like are used.
- CMV promoter, SR ⁇ promoter and the like are preferable.
- the host is E. coli, trp promoter, lac promoter, recA promoter, .lambda.P L promoter, lpp promoter, T7 promoter and the like are preferable.
- SPO1 promoter When the host is Bacillus, SPO1 promoter, SPO2 promoter, penP promoter and the like are preferable.
- yeast When the host is yeast, the Gal1 / 10 promoter, PHO5 promoter, PGK promoter, GAP promoter, ADH promoter and the like are preferable.
- the host When the host is an insect cell, a polyhedrin promoter, a P10 promoter and the like are preferable.
- CaMV35S promoter When the host is a plant cell, CaMV35S promoter, CaMV19S promoter, NOS promoter and the like are preferable.
- an expression vector containing an enhancer, a splicing signal, a terminator, a poly A addition signal, a drug resistance gene, an auxotrophic complementary gene or other selection marker, an origin of replication, etc. can do.
- RNA encoding the nucleic acid sequence recognition module and / or nucleobase converting enzyme is, for example, known in vitro using a vector encoding the DNA encoding the nucleic acid sequence recognition module and / or nucleobase converting enzyme as a template. It can be prepared by transcription into mRNA in a transcription system.
- a complex of a nucleic acid sequence recognition module and a nucleobase converting enzyme or the like by introducing an expression vector containing a DNA encoding a nucleic acid sequence recognition module and / or a nucleobase converting enzyme into a host cell and culturing the host cell Can be expressed intracellularly.
- a host for example, Escherichia, Bacillus, yeast, insect cells, insects, animal cells and the like are used.
- Examples of the genus Escherichia include, for example, Escherichia coli K12 / DH1 [Proc. Natl. Acad. Sci.
- Bacillus bacteria include Bacillus subtilis MI114 [Gene, 24, 255 (1983)], Bacillus subtilis 207-21 [Journal of Biochemistry, 95, 87 (1984)] and the like.
- yeast examples include Saccharomyces cerevisiae (Saccharomyces cerevisiae) AH22, AH22R - , NA87-11A, DKD-5D, 20B-12, Schizosaccharomyces pombe (Schizosaccharomyces pombe) NCYC1913, NCYC2036, Pichia pastoris (Pichia pastoris) KM71 etc. are used.
- Insect cells include, for example, when the virus is AcNPV, larvae-derived cell lines (Spodoptera frugiperda cells; Sf cells), MG1 cells derived from the midgut of Trichoplusia ni, High Five TM cells derived from eggs of Trichoplusia ni , Cells derived from Mamestra brassicae, cells derived from Estigmena acrea, and the like are used.
- Sf cells include Sf9 cells (ATCC CRL1711), Sf21 cells [above, In Vivo, 13, 213-217 (1977)].
- insects include silkworm larvae, Drosophila and crickets [Nature, 315, 592 (1985)].
- animal cells examples include monkey COS-7 cells, monkey Vero cells, Chinese hamster ovary (CHO) cells, dhfr gene-deficient CHO cells, mouse L cells, mouse AtT-20 cells, mouse myeloma cells, rat GH3 cells, and humans.
- Fetal kidney-derived cells eg: HEK293 cells
- human liver cancer-derived cells eg: HepG2
- cell lines such as human FL cells
- pluripotent stem cells such as human and other mammalian iPS cells and ES cells
- various Primary cultured cells prepared from tissue are used.
- zebrafish embryos, Xenopus oocytes, and the like can also be used.
- Plant cells were prepared from various plants (for example, grains such as rice, wheat and corn, commercial crops such as tomato, cucumber and eggplant, garden plants such as carnation and eustoma, experimental plants such as tobacco and Arabidopsis thaliana). Suspension culture cells, callus, protoplasts, leaf sections, root sections and the like are used.
- the expression vector may be introduced by a known method (eg, lysozyme method, competent method, PEG method, CaCl 2 coprecipitation method, electroporation method, microinjection method, particle gun method, lipofection method, Agrobacterium method, etc.).
- Donor DNA can also be introduced into cells by the same method.
- the expression vector and the donor DNA may be introduced at the same time or at different timings.
- E. coli can be transformed according to the method described in, for example, Proc. Natl. Acad. Sci. USA, 69, 2110 (1972), Gene, 17, 107 (1982).
- Bacillus can be introduced into a vector according to the method described in, for example, Molecular & General Genetics, 168, 111 (1979).
- Yeast can be introduced into a vector according to a method described in, for example, Methods in Enzymology, 194, 182-187 (1991), Proc. Natl. Acad. Sci. USA, 75, 1929 (1978).
- Insect cells and insects can be introduced into a vector according to the method described in, for example, Bio / Technology, 6, 47-55 (1988).
- Animal cells can be introduced into a vector according to the method described in, for example, Cell Engineering Supplement 8 New Cell Engineering Experimental Protocol, 263-267 (1995) (published by Shujunsha), Virology, 52, 456 (1973).
- Culturing of cells into which a vector and donor DNA have been introduced can be performed according to a known method depending on the type of host.
- a liquid medium is preferable as the medium used for the culture.
- a culture medium contains a carbon source, a nitrogen source, an inorganic substance, etc. which are required for the growth of a transformant.
- Examples of the carbon source include glucose, dextrin, soluble starch, and sucrose; examples of the nitrogen source include ammonium salts, nitrates, corn steep liquor, peptone, casein, meat extract, soybean meal, Inorganic or organic substances such as potato extract; examples of inorganic substances include calcium chloride, sodium dihydrogen phosphate, magnesium chloride, and the like.
- yeast extract, vitamins, growth promoting factors and the like may be added to the medium.
- the pH of the medium is preferably about 5 to about 8.
- M9 medium containing glucose and casamino acids As a medium for culturing Escherichia coli, for example, M9 medium containing glucose and casamino acids [Journal of Experiments in Molecular Genetics, 431-433, Cold Spring Harbor Laboratory, New York 1972] is preferable. If necessary, an agent such as 3 ⁇ -indolylacrylic acid may be added to the medium in order to make the promoter work efficiently.
- Cultivation of E. coli is usually performed at about 15 to about 43 ° C. If necessary, aeration or agitation may be performed.
- the culture of Bacillus is usually performed at about 30 to about 40 ° C. If necessary, aeration or agitation may be performed.
- a medium for culturing yeast for example, a Burkholder minimum medium [Proc. Natl. Acad. Sci. USA, 77, 4505 (1980)] or an SD medium containing 0.5% casamino acid [Proc. Natl. Acad. Sci. USA, 81, 5330 (1984)].
- the pH of the medium is preferably about 5 to about 8.
- the culture is usually performed at about 20 ° C to about 35 ° C. Aeration and agitation may be performed as necessary.
- a medium for culturing insect cells or insects for example, a medium obtained by appropriately adding an additive such as 10% bovine serum inactivated to Grace's Insect Medium [Nature, 195, 788 (1962)] is used.
- the pH of the medium is preferably about 6.2 to about 6.4.
- the culture is usually performed at about 27 ° C. You may perform ventilation
- a medium for culturing animal cells for example, a minimum essential medium (MEM) containing about 5 to about 20% fetal bovine serum [Science, 122, 501 (1952)], Dulbecco's modified Eagle medium (DMEM) [ Virology, 8, 396 (1959)], RPMI 1640 medium [The Journal of the American Medical Association, 199, 519 (1967)], 199 medium [Proceeding of the Society for the Biological Medicine, 73, 1 (1950)], etc. Is used.
- the pH of the medium is preferably about 6 to about 8.
- the culture is usually performed at about 30 ° C to about 40 ° C. You may perform ventilation
- a medium for culturing plant cells MS medium, LS medium, B5 medium and the like are used.
- the pH of the medium is preferably about 5 to about 8.
- the culture is usually performed at about 20 ° C to about 30 ° C. You may perform ventilation
- a complex of a nucleic acid sequence recognition module and a nucleobase converting enzyme, that is, a nucleic acid modifying enzyme complex can be expressed in a cell.
- RNA encoding a nucleic acid sequence recognition module and / or nucleobase converting enzyme into a host cell can be performed by a microinjection method, a lipofection method, or the like.
- RNA can be introduced once or repeatedly several times (for example, 2 to 5 times) at an appropriate interval.
- Zinc finger motifs are not efficient in producing zinc fingers that specifically bind to the target nucleotide sequence, and the selection of zinc fingers with high binding specificity is complicated, so many zinc finger motifs that actually function are created. It's not easy. TAL effectors and PPR motifs have a higher degree of freedom in target nucleic acid sequence recognition than zinc finger motifs, but it is necessary to design and construct a huge protein each time depending on the target nucleotide sequence, which is problematic in terms of efficiency. Remains. In contrast, the CRISPR-Cas system recognizes the target double-stranded DNA sequence with a guide RNA complementary to the target nucleotide sequence, so it can synthesize oligo DNA that can specifically hybridize with the target nucleotide sequence.
- CRISPR-Cas system in which only one or both of the Cas effector proteins are inactivated is used as the nucleic acid sequence recognition module.
- the nucleic acid sequence recognition module of the present invention using CRISPR-mutated Cas is a trans-activating necessary for recruitment of CRISPR-RNA (crRNA) containing a sequence complementary to the target nucleotide sequence and, if necessary, the mutant Cas effector protein.
- crRNA CRISPR-RNA
- tracrRNA RNA molecule consisting of a crRNA alone or a chimeric RNA of crRNA and tracrRNA that constitutes a nucleic acid sequence recognition module in combination with a mutant Cas effector protein.
- guide RNA An RNA molecule consisting of a crRNA alone or a chimeric RNA of crRNA and tracrRNA that constitutes a nucleic acid sequence recognition module in combination with a mutant Cas effector protein is collectively referred to as “guide RNA”. The same applies when using a CRISPR / Cas system into which no mutation has been introduced.
- the Cas effector protein used in the present invention is not particularly limited as long as it forms a complex with the guide RNA and can recognize and bind to the target nucleotide sequence in the target gene and the adjacent protospacer adjacent motif (PAM).
- PAM adjacent protospacer adjacent motif
- Is preferably Cas9 (also referred to as Cas9 nuclease) or Cpf1 (also referred to as Cpf1 nuclease).
- Cas9 examples include Cas9 derived from Streptococcus pyogenes (Streptococcus pyogenes) (SpCas9; PAM sequence NGG (N is A, G, T, or C; the same applies hereinafter)), Cas9 derived from Streptococcus thermophilus (StCas9; PAM sequence NNAGAAW), Cas9 derived from Neisseria meningitidis (MmCas9; PAM sequence NNNNGATT), and the like, but are not limited thereto. SpCas9, which is less constrained by PAM, is preferred (substantially 2 bases and can be theoretically targeted almost anywhere on the genome).
- Cpf1 for example, Cpf1 (FnCpf1; PAM sequence NTT) derived from Francisella AMnovicida, Cpf1 (AsCpf1; PAM sequence NTTT) derived from Acidaminococcus sp. Cpf1 (LbCpf1; PAM sequence NTTT) derived from (Lachnospiraceae) bacterium) and the like are included, but not limited thereto.
- the mutant Cas effector protein used in the present invention (sometimes abbreviated as mutant Cas) includes those in which the ability to cleave both strands of the double-stranded DNA of the Cas effector protein and the ability to cleave one strand.
- the 10th Asp residue is converted to an Ala residue and lacks the ability to cleave the opposite strand of the strand that forms a complementary strand with the guide RNA (thus, against the strand that forms the complementary strand with the guide RNA).
- the H840A mutant (which has nickase activity against the opposite strand of the chain), and also its double mutant (dCas9) can be used.
- dCas9 a mutant lacking the ability to cleave both strands, with the 917th Asp residue converted to an Ala residue (D917A) or the 1006th Glu residue converted to an Ala residue (E1006A) can be used.
- Other mutant Cass can be used similarly as long as they lack the ability to cleave at least one strand of the double-stranded DNA.
- DNA encoding a Cas effector protein can be cloned from cells producing the enzyme by methods similar to those described above for DNA encoding inhibitors of base excision repair.
- the mutant Cas is obtained by using a site-directed mutagenesis method known per se to the cloned Cas-encoding DNA, and the amino acid residue at the site important for DNA cleavage activity (for example, in the case of SpCas9, the 10th position).
- Asp residue, 840th His residue, and FnCpf1 such as, but not limited to, the 917th Asp residue and the 1006th Glu residue
- the DNA encoding the Cas effector protein can be synthesized by a method similar to that described above for the DNA encoding the nucleic acid sequence recognition module and the DNA encoding the nucleobase converting enzyme, by chemical synthesis or in combination with the PCR method or Gibson Assembly method. , Can also be constructed as DNA with codon usage suitable for expression in the host cell used.
- the DNA encoding the obtained Cas effector protein and / or nucleic acid-modifying enzyme and / or inhibitor of base excision repair can be inserted downstream of the promoter of the same expression vector as described above depending on the target cell.
- a DNA encoding a guide RNA is a crRNA sequence (for example, as a Cas effector protein) containing a nucleotide sequence complementary to a target nucleotide sequence (also referred to herein as a “targeting sequence”).
- crRNA containing SEQ ID NO: 1; AAUU UCUAC UGUU GUAGA U on the 5 'side of the targeting sequence can be used, and the underlined sequence forms a base pair to form a stem-loop structure) Coding sequence, or crRNA coding sequence and known tracrRNA coding sequence as necessary (for example, as a crcr coding sequence for recruiting Cas9 as a Cas effector protein, gttttagagctagaaatagcaagttaaaataggctagtccgttatcaacttgaaaaagtggcaccgagtcggtgcttttttttttt; SEQ ID No.
- the oligo DNA sequence linked to No. 3 can be designed and chemically synthesized using a DNA / RNA synthesizer.
- the “target strand” means the strand that hybridizes with the crRNA of the target nucleotide sequence, and the strand that becomes a single strand by hybridization of the target strand with the crRNA at the opposite strand is referred to as the “non-target strand”. (Non-targeted strand) ”.
- the target nucleotide sequence is expressed by one strand (for example, when a PAM sequence is described or when the positional relationship between the target nucleotide sequence and PAM is expressed), the sequence is represented by the sequence of the non-target strand.
- the length of the targeting sequence is not particularly limited as long as it can specifically bind to the target nucleotide sequence, and is, for example, 15 to 30 nucleotides, preferably 18 to 25 nucleotides.
- the targeting sequence is designed using a public guide RNA design website (CRISPR Design Tool, CRISPRdirect, etc.), from the CDS sequence of the target gene, such as PAM (for example, SpCas9 When NGG) is listed on the 3 'adjacent 20mer sequence and C within 7 nucleotides is converted to T in the 3' direction from its 5 'end, the amino acid changes in the protein encoded by the target gene. This can be done by selecting such a sequence. In addition, when using a length of a targeting sequence other than 20mer, the sequence can be appropriately selected.
- CRISPR Design Tool CRISPRdirect, etc.
- a candidate sequence having a small number of off-target sites in the target host genome can be used as a targeting sequence. If the guide RNA design software to be used does not have a function to search the off-target site of the host genome, for example, about the 8 to 12 nucleotides on the 3 ′ side of the candidate sequence (seed sequence with high discrimination ability of the target nucleotide sequence), the host genome You can search off-target sites by performing a Blast search on.
- a DNA encoding a guide RNA can also be inserted into the same expression vector as above, but as a promoter, a pol III promoter (eg, SNR6, SNR52, SCR1, RPR1, U3, U6, H1 promoter, etc.) and a terminator (e.g., poly T sequence (T 6 sequence, etc.)) is preferably used.
- a promoter e.g, SNR6, SNR52, SCR1, RPR1, U3, U6, H1 promoter, etc.
- a terminator e.g., poly T sequence (T 6 sequence, etc.)
- the DNA encoding the guide RNA includes a sequence complementary to the target strand of the target nucleotide sequence and a known tracrRNA sequence (when recruiting Cas9) or a direct repeat sequence of crRNA (Cpf1 Can be synthesized chemically using a DNA / RNA synthesizer.
- DNA or RNA encoding mutant Cas and / or nucleobase converting enzyme, guide RNA-tracrRNA or DNA encoding the same can be introduced into a host cell by the same method as described above depending on the host.
- DSBs DNA double-strand breaks
- off-target breaks random chromosome breaks
- the modification of the target site is not performed by DNA cleavage, but by using a conversion reaction (especially deamination reaction) or abasic reaction of a substituent on the DNA base, and a subsequent repair mechanism. Significant reduction can be realized.
- the target site can be modified using a plurality of target nucleotide sequences at different positions.
- two or more nucleic acid sequence recognition modules that specifically bind to different target nucleotide sequences can be used.
- one of each of these nucleic acid sequence recognition modules and a nucleobase converting enzyme form a nucleic acid modifying enzyme complex.
- a common nucleobase converting enzyme or the like can be used.
- a common complex including fusion protein of Cas effector protein and nucleobase converting enzyme is used, and guide RNA (crRNA or crRNA-tracrRNA chimera)
- crRNA guide RNA
- Two or more crRNAs each forming a complementary strand with a different target nucleotide sequence, or each of two or more crRNAs and two or more chimeric RNAs of tracrRNA can be prepared and used.
- nucleic acid sequence recognition module when a zinc finger motif, a TAL effector, or the like is used as the nucleic acid sequence recognition module, for example, a nucleobase converting enzyme or the like can be fused to each nucleic acid sequence recognition module that specifically binds to a different target nucleotide.
- an expression vector containing DNA encoding the nucleic acid modifying enzyme complex is introduced into the host cell as described above. In order to do so, it is desirable to maintain the expression of the nucleic acid modifying enzyme complex at a certain level or more for a certain period or more. From this point of view, it is certain that the expression vector is integrated into the host genome, but since the persistent expression of the nucleic acid modifying enzyme complex increases the risk of off-target cleavage, the modification of the target site has been successfully achieved. After that, it is preferable to remove it promptly. Examples of means for removing DNA integrated into the host genome include a method using a Cre-loxP system and a FLP-FRT system, a method using a transposon, and the like.
- the nucleic acid modification enzyme complex of the present invention is transiently expressed in the host cell for a period of time necessary for the nucleic acid reaction to occur at a desired time and the modification of the target site to be fixed.
- the host genome can be efficiently edited while avoiding the risk of cleavage.
- Those skilled in the art can appropriately determine a suitable expression induction period based on the culture conditions used.
- the expression induction period of the nucleic acid encoding the nucleic acid-modifying enzyme complex of the present invention is extended beyond the above-mentioned "period necessary for fixing the target site modification" in a range that does not cause side effects in the host cell. Also good.
- a nucleic acid encoding the nucleic acid modifying enzyme complex in the mutant CRISPR-Cas system, a guide RNA is used.
- examples include a method of preparing a construct (expression vector) containing a DNA encoding and a DNA encoding a Cas effector protein and a nucleic acid modifying enzyme in a form in which the expression period can be controlled, and introducing the construct into a host.
- Specific examples of the “form capable of controlling the expression period” include a nucleic acid encoding the nucleic acid-modifying enzyme complex of the present invention under the control of an inducible regulatory region.
- the “inducible regulatory region” is not particularly limited, and examples thereof include an operon of a temperature sensitive (ts) mutation repressor and an operator controlled thereby.
- the ts mutation repressor include, but are not limited to, a ts mutant of a cI repressor derived from ⁇ phage.
- ⁇ phage cI repressor ts
- binding to the operator is suppressed at 30 ° C or lower (eg, 28 ° C), but downstream gene expression is suppressed, but at a high temperature of 37 ° C or higher (eg, 42 ° C), the operator Gene expression is induced to dissociate from.
- homologous recombination is performed by culturing host cells into which a nucleic acid encoding a nucleic acid-modifying enzyme complex has been introduced, usually at a temperature of 30 ° C. or lower, and by raising the temperature to 37 ° C. or higher at an appropriate time for a certain period. If the target gene is targeted to a host cell, the period during which expression of the target gene is suppressed can be minimized by quickly returning to 30 ° C or less after the mutation has been introduced into the target gene. However, editing can be done efficiently while suppressing side effects.
- thermosensitive mutant proteins include, but are not limited to, Rep101 ori temperature-sensitive mutants required for pSC101 ori replication.
- Rep101 ori acts on pSC101 ori at 30 ° C or lower (eg, 28 ° C) to allow autonomous replication of the plasmid, but at 37 ° C (eg, 42 ° C) it loses its function, and the plasmid Autonomous replication becomes impossible. Therefore, by using together with the cI repressor (ts) of the above-mentioned ⁇ phage, transient expression of the nucleic acid modifying enzyme complex of the present invention and plasmid removal can be performed simultaneously.
- DNA encoding the nucleic acid-modifying enzyme complex of the present invention is under the control of an inducible promoter (eg, lac promoter (induced by IPTG), cspA promoter (induced by cold shock), araBAD promoter (induced by arabinose), etc.).
- an inducer is added to the medium (or removed from the medium) at an appropriate time to induce the expression of the nucleic acid modifying enzyme complex, followed by culturing for a certain period of time to carry out the nucleic acid modification reaction.
- the transient expression of the nucleic acid modifying enzyme complex can be realized after the mutation is introduced into the target gene.
- the budding yeast Saccharomyces cerevisiae BY4741 strain (requiring leucine and uracil) was used and cultured in a Dropout composition adapted to the auxotrophy of a standard YPDA medium or SD medium.
- the culture was performed at 25 ° C. to 30 ° C. by stationary culture on an agar plate or shaking culture on a liquid medium.
- the lithium acetate method was used, and selection was performed on an SD medium suitable for appropriate auxotrophy.
- the mutation rate was calculated and evaluated using the number of surviving colonies on the SD plate as the total number of cells and the number of surviving colonies on the Canavanine plate as the number of resistant mutants. Mutation sites are amplified by DNA fragments containing the target gene region of each strain by colony PCR, followed by DNA sequencing and alignment analysis based on the sequence of the Saccharomyces Genome Database (http://www.yeastgenome.org/). Identified.
- HEK293T cells Human fetal kidney-derived cells (HEK293T cells) using DME-glutamax medium (Thermo Fisher Scientific) supplemented with 10 ⁇ g / mL puromycin (Life Technologies) and 10% fetal bovine serum (FBS) (Biosera, Nuaille, France) The culture was performed under the conditions of 37 ° C. and 5% CO 2 . 5% trypsin was used for cell recovery.
- HEK293T cells stored in a deep freezer were lysed in a 37 ° C. water bath and seeded on 75 T-flask to 5 ⁇ 10 6 cells.
- Cells were collected after 1-3 days of culture and seeded in each well of a 24-well plate at 0.5 ⁇ 10 5 cells / well.
- the cells in each well in a 60-80% confluent state after 1-3 days of culture were each 500 ng / well of the following plasmids (effector plasmid and reporter plasmid) (1 ⁇ g / well in total), 200 nM donor DNA, Transfected with 1.5 ⁇ l FugeneHD (Promega).
- the donor DNA used in each example is shown in Table 1.
- Cells were collected 72 hours after transfection, and iRFP and EGFP fluorescence was detected using FACS. From the number of detected cells, recombination efficiency (%) was calculated by the following formula.
- DNA was processed and constructed by PCR, restriction enzyme treatment, ligation, Gibson Assembly, or artificial chemical synthesis.
- Plasmids used as a backbone were pRS415 for leucine selection and pRS426 for uracil selection as yeast / E. Coli shuttle vectors.
- the plasmid was amplified with E. coli strain XL-10 gold or DH5 ⁇ and introduced into yeast by the lithium acetate method.
- ⁇ Construction of budding yeast construct Sequences such as homology arms, guide RNAs, and insertion sequences were designed with reference to the yeast genome database (https://www.yeastgenome.org/). Vector construction was performed according to the method described in Nishida K. et al., Science 16: 353 (6305) (2016) doi: 10.1126 / science.aaf8729.
- the 1 ⁇ gRNA vector corresponds to a vector in which the nucleotide sequence from positions 5871 to 5890 of the sequence of SEQ ID NO: 15 is replaced with a complementary sequence of the target nucleotide sequence of L86 or M4.
- the nucleotide sequence from 2638 to 2657 of the sequence of SEQ ID NO: 16 is replaced with a complementary sequence of any of the target nucleotide sequences of L86, L87, L88, L93 and R90, and SEQ ID NO: 16 This corresponds to the nucleotide sequence of Nos. 6293 to 6312 substituted with a complementary sequence of any of the target nucleotide sequences of L87, R89, R90, R91 and R92.
- the target nucleotide is as follows.
- pcDNA3.1 vector backbone and CMV, PmCDA1, Cas9, H1, and sgRNA sequences are derived from a paper by Nishida et al 2016. Each mutation was introduced by PCR. EF1, iRFP and mEGFP fragments were produced by artificial gene synthesis. Fragments were inserted / replaced by Gibson assembly or ligation reaction.
- vector SY4 H1_sgRNA, CMV_mEGFP
- vector SY45 CMV_Cas9-PmCDA1, EF1_iRFP
- vector SY45 CMV_Cas9, EF1_iRFP
- SEQ ID NOs: 42 to 44 respectively.
- Vector SY45 CMV_nCas9 (D10A) -PmCDA1, EF1_iRFP
- the vector SY45 (CMV_nCas9 (H840A) -PmCDA1, EF1_iRFP) corresponds to SEQ ID NO: 43 with the 3260th to 3262th bases replaced with gct.
- the vector SY45 (CMV_dCas9-PmCDA1, EF1_iRFP) corresponds to SEQ ID NO: 43 with the bases 770 to 772 replaced with gct and the bases 3260 to 3262 replaced with gct.
- the vector SY45 (CMV_nCas9 (D10A), EF1_iRFP) corresponds to the one obtained by substituting the 3724th to 3726th bases with gct in SEQ ID NO: 44.
- the vector SY45 (CMV_nCas9 (H840A), EF1_iRFP) corresponds to the one obtained by substituting the 6214th to 6216th bases with gct in SEQ ID NO: 44.
- Vector SY45 (CMV_dCas9, EF1_iRFP) corresponds to SEQ ID NO: 44 with the 3724th to 3726th bases replaced with gct and the 6214th to 6216th bases replaced with gct.
- ⁇ Sequencing of cellular DNA> iRFP positive cells were collected by FACS, and then genomic DNA and introduced plasmid DNA were extracted. Then, the following samples were prepared, and PCR was performed under the following conditions to amplify the target site.
- Sample preparation gDNA 1 ⁇ L 1 ⁇ L of each primer rTaq 10x Buffer 5 ⁇ L 25 mM MgCl 2 3 ⁇ L 2 mM dNTP 5 ⁇ L rTaq (TOYOBO) 0.5 ⁇ L ddH 2 O 33.5 ⁇ L 50 ⁇ L in total PCR conditions: After maintaining at 94 ° C. for 2 minutes, 33 cycles of 94 ° C. for 45 seconds, 55 ° C. for 45 seconds, and 72 ° C.
- SY157 and SY182 were used as primers for amplification.
- the size of the amplification product is 1554 bp.
- each purified product and pGEM-t easy vecter were TA cloned, and Escherichia coli (JM109) was transformed with the vector. Then 24 colonies were picked for each sample (with blue and white selection) and the plasmid DNA was purified by Mini prep (using Fastgene).
- Example 1 Insertion of Insertion Sequence into Target Site Using dCas9-CDA or nCas9-CDA and Donor DNA Saccharomyces cerevisiae BY4741 strain, plasmid vector 1525 (the 6036th base of SEQ ID NO: 4 is g, 6037th) The base is c) or 1526 (the 6036th base of SEQ ID NO: 4 is c, the 6037th base is a), 1059 (SEQ ID NO: 5) or 1149 (3890th to 3909 of the sequence of SEQ ID NO: 5) Double-transformation with TCCAATAACGGAATCCAACT (SEQ ID NO: 6) in the base sequence of No.) and selected with an auxotrophic medium (SD-Leu-Ura).
- plasmid vector 1525 the 6036th base of SEQ ID NO: 4 is g, 6037th
- the base is c) or 1526 (the 6036th base of SEQ ID NO: 4 is c
- the cells were cultured overnight in S-Leu-Ura 2% raffinose medium.
- the solution was diluted 1/32 in S-Leu-Ura 2% raffinose + 0.02% galactose medium and cultured overnight at 30 ° C.
- SD-Ura-Leu and SD-Ura-Leu + Canavanine plates were spotted at a 10-fold dilution. Two days later, Canavanine resistant colonies were sequenced. As a result, insertion of the mutation at the target site was confirmed (FIG. 2).
- Example 2 Construction of an evaluation system for recombination A DNA fragment treated with SmaI / HpaI of plasmid vector 1548 (SEQ ID NO: 7) was transformed into BY4741 strain and selected with SD-Ura medium. Sequence analysis confirmed the incorporation into the Ade1 region.
- Example 3 Demonstration Experiment of Recombination Reaction Using Recombination Evaluation System
- Any of the above plasmid vectors was transformed into a demonstration experimental strain and selected on SD-Leu-Ura medium.
- the cells were cultured overnight in S-Leu-Ura 2% raffinose medium.
- S-Leu 2% raffinose + 0.02% (or 0.2%) diluted 1/32 in galactose medium and cultured overnight at 30 ° C. were defined as 5 generations. For 20 generations, 1/32 dilution was repeated a total of 4 times.
- the SD-Leu plate was spotted at a 10-fold dilution, and the colony number and color were evaluated after 2 days. As a result, colonies that became white after restoration of Ade1 function appeared frequently, and thus the method of the present invention showed induction of homologous recombination at the target site (FIG. 4).
- Example 4 Demonstration experiment of knock-in or knock-out according to the present invention Saccharomyces cerevisiae BY4741 strain was double-transformed with plasmid vector 1251 (SEQ ID NO: 8) and 2x gRNA vector, and auxotrophic medium (SD-Leu-Ura) was used. Selected. The cells were cultured overnight in S-Leu-Ura 2% raffinose medium. The solution was diluted 1/32 in S-Leu-Ura 2% raffinose + 0.2% galactose medium and cultured overnight at 30 ° C.
- Example 5 Demonstration experiment of recombination reaction in animal cells Using single-stranded oligo DNA (70 base length) (Table 1) as donor DNA, it was verified whether or not a recombination reaction occurred in animal cells (HEK293T cells). .
- a schematic diagram of the experiment is shown in FIG.
- vector SY4 H1_sgRNA, CMV_mEGFP
- vector SY45 CMV_Cas9-PmCDA1, EF1_iRFP
- vector SY45 CMV_nCas9 (D10A) -PmCDA1, EF1_iRFP
- vector SY45 CH_ACas9P
- EF1_iRFP vector SY45
- vector SY45 CMV_dCas9-PmCDA1, EF1_iRFP
- vector SY45 CV_Cas9, EF1_iRFP
- vector SY45 CV_nCas9 (D10A), EF1_iRFP
- vector SY45 CV_nCas9 (H840i), EF1_EF4, EF1_iRFP)
- Fw2 and Fw3 are used as donor DNAs, if homologous recombination occurs successfully, an initiation codon is generated in the sequence encoding EGFP, and EGFP expression is observed.
- Fw1 is a donor DNA designed so that an initiation codon does not occur in the sequence encoding EGFP even when homologous recombination occurs, and was used as a negative control.
- Fw3 is a substitution of one base of the homology arm of Fw2 (c ⁇ g), verifying whether homologous recombination reaction occurs even if the homology arm is not completely homologous to the adjacent region of the target site Therefore, it was used to verify whether or not mutations at a plurality of different locations can also be introduced.
- Example 6 Verification of influence on homologous recombination reaction by the number of bases of donor DNA and the type of complementary strand (front (Fw) or reverse (Rv)) Single-stranded oligo DNA (50 base length) as donor DNA ( Table 1) was used to verify whether a recombination reaction occurred in animal cells (HEK293T cells). A schematic diagram of the experiment is shown in FIG.
- Vector SY4 H1_sgRNA, CMV_mEGFP was used as a reporter plasmid, and vector SY45 (CMV_nCas9 (D10A))-PmCDA1, EF1_iRFP) or vector SY45 (CMV_nCas9 (H840A))-PmCDA1 was used as an effector plasmid.
- Example 7 Verification of donor DNA homology arms Using donor DNA having homology arms for different homologous regions (Table 1), changes in the efficiency of homologous recombination reactions were verified by homologous regions.
- a schematic diagram of the experiment is shown in FIG.
- Vector SY4 H1_sgRNA, CMV_mEGFP
- vector SY45 CMV_nCas9 (D10A))-PmCDA1, EF1_iRFP
- vector SY45 CMV_nCas9 (H840A)
- Example 8 Verification of modification in DNA of mammalian cells Modification in DNA was verified using the same gRNA and donor DNA as those used in the experiment using Fw2 in Example 5. The results are shown in Table 2 below. When nCas9 (D10A) -PmCDA1 and nCas9 (H840A) -PmCDA1 are used, the occurrence of indel as a by-product is significantly suppressed, that is, the cytotoxicity is reduced, compared with the case where Cas9 is used. It was proved. Note that the term DNA used in this example includes both genomic DNA and plasmid DNA.
- nCas9-CDA when nCas9-CDA is used, the efficiency of homologous recombination is about the same as or higher than when Cas9 is used, and avoids Indel as a by-product and high cytotoxicity that occurs when Cas9 is used. Therefore, the method using nCas9-CDA can be more beneficial and useful than the conventional method. Furthermore, by using nCas9-CDA, higher efficiency can be achieved than nCas9 used for the purpose of avoiding the above-mentioned problems that occur when Cas9 is used.
- a novel method using a nucleobase converting enzyme such as deaminase or a DNA glycosylase that can switch the direction and combination of genes or knock in a gene fragment without being restricted by the type or site of mutation that can be introduced.
- DNA modification technology is provided. Since the DNA modification technology of the present invention can modify the target site without cleaving double-stranded DNA, unexpected rearrangement and toxicity associated with the cleaving can be kept low, and much more efficient than conventional methods. Since the target site can be modified well, it is extremely useful.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Genetics & Genomics (AREA)
- Engineering & Computer Science (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biomedical Technology (AREA)
- Biotechnology (AREA)
- General Engineering & Computer Science (AREA)
- Molecular Biology (AREA)
- Microbiology (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- Plant Pathology (AREA)
- Biophysics (AREA)
- Physics & Mathematics (AREA)
- Medicinal Chemistry (AREA)
- Crystallography & Structural Chemistry (AREA)
- Mycology (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Enzymes And Modification Thereof (AREA)
Abstract
Description
[1] 細胞の有する二本鎖DNAの標的部位を改変する方法であって、選択された二本鎖DNA中の標的ヌクレオチド配列と特異的に結合する核酸配列認識モジュールと、核酸塩基変換酵素又はDNAグリコシラーゼとが結合した複合体、及び挿入配列を含むドナーDNAを該二本鎖DNAと接触させ、該標的部位において該二本鎖DNAの少なくとも一方の鎖を切断することなく、該標的部位を該挿入配列に置換する、又は該標的部位に該挿入配列を挿入する工程を含む、方法。
[2] 前記ドナーDNAが、標的部位の隣接領域に相同な配列を含む、[1]に記載の方法。
[3] 前記核酸配列認識モジュールが、Casエフェクタータンパク質の少なくとも1つのDNA切断能が失活したCRISPR-Casシステム、ジンクフィンガーモチーフ、TALエフェクター及びPPRモチーフからなる群より選択される、[1]又は[2]に記載の方法。
[4] 前記核酸配列認識モジュールが、Casエフェクタータンパク質の2つのDNA切断能のうちの一方のみのDNA切断能が失活したCRISPR-Casシステムである、[1]~[3]のいずれかに記載の方法。
[5] 前記核酸配列認識モジュールが、Casエフェクタータンパク質の両方のDNA切断能が失活したCRISPR-Casシステムである、[1]~[3]のいずれかに記載の方法。
[6] 前記核酸塩基変換酵素がデアミナーゼである、[1]~[5]のいずれかに記載の方法。
[7] 前記デアミナーゼがシチジンデアミナーゼである、[6]に記載の方法。
[8] 前記シチジンデアミナーゼがPmCDA1である、[7]に記載の方法。
[9] 二本鎖DNAと複合体との接触が、該細胞への、該複合体をコードする核酸の導入により行われる、[1]~[8]のいずれかに記載の方法。
[10] 前記細胞が原核生物細胞又は真核生物細胞である、[1]~[9]のいずれかに記載の方法。
[11] 前記細胞が微生物細胞である、[10]に記載の方法。
[12] 前記細胞が植物細胞、昆虫細胞又は動物細胞である、[10]に記載の方法。
[13] 前記動物細胞が脊椎動物細胞である、[12]に記載の方法。
[14] 前記脊椎動物細胞が哺乳類動物細胞である、[13]に記載の方法。
また、「ひずみのない二重らせん構造のDNAへの反応性が十分に低い」とは、細胞の生存に影響を与えない程度に細胞毒性が抑えられる頻度でしか、ひずみのない二重らせん構造のDNAを形成する領域内での脱塩基反応を起こさないことを意味する。ここで「ひずみのない二重らせん構造のDNA」とは、強固な二重らせん構造を形成した状態にあること(即ち、unrelaxed double-helical DNA(又は単にunrelaxed DNAともいう))を意味し、対形成する塩基同士が完全に解離した一本鎖DNAの状態のみならず、塩基対は形成しているものの二重らせん構造がほどけた緩んだ二本鎖(relaxed double-stranded DNA)の状態のものも含まれない。ひずみのない二重らせん構造のDNAへの反応性が十分に低いDNAグリコシラーゼとしては、生来的にひずみのない二重らせん構造のDNAに対する反応性が十分に低いDNAグリコシラーゼや、野生型に比べてひずみのない二重らせん構造のDNAに対する反応性を低下させる変異が導入された変異DNAグリコシラーゼ等が挙げられる。さらに、2つの断片に分断されたDNAグリコシラーゼであって、それぞれの断片が2つに分断された核酸配列認識モジュールのいずれか一方と結合して2つの複合体を形成し、両複合体がリフォールディングすると該核酸配列認識モジュールは標的ヌクレオチド配列と特異的に結合することができ、該特異的な結合によって該DNAグリコシラーゼが脱塩基反応を触媒することが可能となるようにデザインされたスプリット酵素であるDNAグリコシラーゼも、本発明における「ひずみのない二重らせん構造のDNAへの反応性が十分に低いDNAグリコシラーゼ」に包含される。
従って、核酸配列認識モジュールと、核酸塩基変換酵素等とは、それらの融合タンパク質をコードする核酸として、あるいは、結合ドメインやインテイン等を利用してタンパク質に翻訳後、宿主細胞内で複合体を形成し得るような形態で、それらをそれぞれコードする核酸として調製することが好ましい。ここで核酸は、DNAであってもRNAであってもよい。DNAの場合は、好ましくは二本鎖DNAであり、宿主細胞内で機能的なプロモーターの制御下に配置した発現ベクターの形態で提供される。RNAの場合は、好ましくは一本鎖RNAである。
核酸配列認識モジュールと核酸塩基変換酵素等とが結合した本発明の複合体は、DNA二重鎖切断(DSB)を伴わないため、毒性の低いゲノム編集が可能であり、本発明の方法は幅広い生物材料に適用することができる。従って、核酸配列認識モジュール及び/又は核酸塩基変換酵素等をコードする核酸が導入される細胞は、原核生物である大腸菌などの細菌や下等真核生物である酵母などの微生物の細胞から、ヒト等の哺乳動物を含む脊椎動物、昆虫、植物など高等真核生物の細胞にいたるまで、あらゆる生物種の細胞をも包含し得る。
核酸塩基変換酵素等をコードするDNA(即ち、核酸塩基変換酵素をコードするDNA又はDNAグリコシラーゼをコードするDNA)も、同様に、使用する酵素のcDNA配列情報をもとにオリゴDNAプライマーを合成し、当該酵素を産生する細胞より調製した全RNAもしくはmRNA画分を鋳型として用い、RT-PCR法によって増幅することにより、クローニングすることができる。例えば、ヤツメウナギのPmCDA1をコードするDNAは、NCBIデータベースに登録されているcDNA配列(accession No. EF094822)をもとに、CDSの上流及び下流に対して適当なプライマーを設計し、ヤツメウナギ由来mRNAからRT-PCR法によりクローニングできる。また、ヒトAIDをコードするDNAは、NCBIデータベースに登録されているcDNA配列(accession No. AB040431)をもとに、CDSの上流及び下流に対して適当なプライマーを設計し、例えばヒトリンパ節由来mRNAからRT-PCR法によりクローニングできる。また、ドナーDNAも、標的部位の配列情報等に基づき、上記と同様にクローニングできる。
クローン化されたDNAは、そのまま、または所望により制限酵素で消化するか、適当なリンカー及び/又は核移行シグナル(目的の二本鎖DNAがミトコンドリアや葉緑体DNAの場合は、各オルガネラ移行シグナル)を付加した後に、核酸配列認識モジュールをコードするDNAとライゲーションして、融合タンパク質をコードするDNAを調製することができる。あるいは、核酸配列認識モジュールをコードするDNAと、核酸塩基変換酵素等をコードするDNAに、それぞれ結合ドメインもしくはその結合パートナーをコードするDNAを融合させるか、両DNAに分離インテインをコードするDNAを融合させることにより、核酸配列認識変換モジュールと核酸塩基変換酵素等とが宿主細胞内で翻訳された後に複合体を形成できるようにしてもよい。これらの場合も、所望により一方もしくは両方のDNAの適当な位置に、リンカー及び/又は核移行シグナルを連結することができる。また、ドナーDNAは、単一DNAとして作製してもよいし、核酸配列認識モジュール及び/又は核酸塩基変換酵素等をコードする核酸と単一なDNAとして提供されてもよい。
また、標的ヌクレオチド配列とPAM配列以外の部位を標的部位とする場合は、改変後もこれらの配列が残り、核酸改変酵素等により核酸塩基変換反応又は脱塩基反応が生じる可能性があるため、これらの配列が除かれるようにドナーDNAを設計するか、ホモロジーアーム上の標的ヌクレオチド配列又はPAM配列に、サイレント変異を導入することが好ましい。
発現ベクターとしては、大腸菌由来のプラスミド(例、pBR322,pBR325,pUC12,pUC13);枯草菌由来のプラスミド(例、pUB110,pTP5,pC194);酵母由来プラスミド(例、pSH19,pSH15);昆虫細胞発現プラスミド(例:pFast-Bac);動物細胞発現プラスミド(例:pA1-11、pXT1、pRc/CMV、pRc/RSV、pcDNAI/Neo);λファージなどのバクテリオファージ;バキュロウイルスなどの昆虫ウイルスベクター(例:BmNPV、AcNPV);レトロウイルス、ワクシニアウイルス、アデノウイルスなどの動物ウイルスベクターなどが用いられる。
プロモーターとしては、遺伝子の発現に用いる宿主に対応して適切なプロモーターであればいかなるものでもよい。DSBを伴う従来法では毒性のために宿主細胞の生存率が著しく低下する場合があるので、誘導プロモーターを使用して誘導開始までに細胞数を増やしておくことが望ましいが、本発明の核酸改変酵素複合体を発現させても十分な細胞増殖が得られるので、構成プロモーターも制限なく使用することができる。
例えば、宿主が動物細胞である場合、SRαプロモーター、SV40プロモーター、LTRプロモーター、CMV(サイトメガロウイルス)プロモーター、RSV(ラウス肉腫ウイルス)プロモーター、MoMuLV(モロニーマウス白血病ウイルス)LTR、HSV-TK(単純ヘルペスウイルスチミジンキナーゼ)プロモーターなどが用いられる。なかでも、CMVプロモーター、SRαプロモーターなどが好ましい。
宿主が大腸菌である場合、trpプロモーター、lacプロモーター、recAプロモーター、λPLプロモーター、lppプロモーター、T7プロモーターなどが好ましい。
宿主がバチルス属菌である場合、SPO1プロモーター、SPO2プロモーター、penPプロモーターなどが好ましい。
宿主が酵母である場合、Gal1/10プロモーター、PHO5プロモーター、PGKプロモーター、GAPプロモーター、ADHプロモーターなどが好ましい。
宿主が昆虫細胞である場合、ポリヘドリンプロモーター、P10プロモーターなどが好ましい。
宿主が植物細胞である場合、CaMV35Sプロモーター、CaMV19Sプロモーター、NOSプロモーターなどが好ましい。
宿主としては、例えば、エシェリヒア属菌、バチルス属菌、酵母、昆虫細胞、昆虫、動物細胞などが用いられる。
エシェリヒア属菌としては、例えば、エシェリヒア・コリ(Escherichia coli)K12・DH1〔Proc. Natl. Acad. Sci. USA,60,160 (1968)〕,エシェリヒア・コリJM103〔Nucleic Acids Research,9,309 (1981)〕,エシェリヒア・コリJA221〔Journal of Molecular Biology,120,517 (1978)〕,エシェリヒア・コリHB101〔Journal of Molecular Biology,41,459 (1969)〕,エシェリヒア・コリC600〔Genetics,39,440 (1954)〕などが用いられる。
バチルス属菌としては、例えば、バチルス・サブチルス(Bacillus subtilis)MI114〔Gene,24,255 (1983)〕,バチルス・サブチルス207-21〔Journal of Biochemistry,95, 87 (1984)〕などが用いられる。
酵母としては、例えば、サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)AH22,AH22R-,NA87-11A,DKD-5D,20B-12、シゾサッカロマイセス・ポンベ(Schizosaccharomyces pombe)NCYC1913,NCYC2036,ピキア・パストリス(Pichia pastoris)KM71などが用いられる。
昆虫としては、例えば、カイコの幼虫、ショウジョウバエ、コオロギなどが用いられる〔Nature,315,592 (1985)〕。
大腸菌は、例えば、Proc. Natl. Acad. Sci. USA,69,2110 (1972)やGene,17,107 (1982)などに記載の方法に従って形質転換することができる。
バチルス属菌は、例えば、Molecular & General Genetics,168,111 (1979)などに記載の方法に従ってベクター導入することができる。
酵母は、例えば、Methods in Enzymology,194,182-187 (1991)、Proc. Natl. Acad. Sci. USA,75,1929 (1978)などに記載の方法に従ってベクター導入することができる。
昆虫細胞および昆虫は、例えば、Bio/Technology,6,47-55 (1988)などに記載の方法に従ってベクター導入することができる。
動物細胞は、例えば、細胞工学別冊8 新細胞工学実験プロトコール,263-267 (1995)(秀潤社発行)、Virology,52,456 (1973)に記載の方法に従ってベクター導入することができる。
例えば、大腸菌またはバチルス属菌を培養する場合、培養に使用される培地としては液体培地が好ましい。また、培地は、形質転換体の生育に必要な炭素源、窒素源、無機物などを含有することが好ましい。ここで、炭素源としては、例えば、グルコース、デキストリン、可溶性澱粉、ショ糖などが;窒素源としては、例えば、アンモニウム塩類、硝酸塩類、コーンスチープ・リカー、ペプトン、カゼイン、肉エキス、大豆粕、バレイショ抽出液などの無機または有機物質が;無機物としては、例えば、塩化カルシウム、リン酸二水素ナトリウム、塩化マグネシウムなどがそれぞれ挙げられる。また、培地には、酵母エキス、ビタミン類、生長促進因子などを添加してもよい。培地のpHは、好ましくは約5~約8である。
大腸菌を培養する場合の培地としては、例えば、グルコース、カザミノ酸を含むM9培地〔Journal of Experiments in Molecular Genetics, 431-433, Cold Spring Harbor Laboratory, New York 1972〕が好ましい。必要により、プロモーターを効率よく働かせるために、例えば、3β-インドリルアクリル酸のような薬剤を培地に添加してもよい。大腸菌の培養は、通常約15~約43℃で行なわれる。必要により、通気や撹拌を行ってもよい。
バチルス属菌の培養は、通常約30~約40℃で行なわれる。必要により、通気や撹拌を行ってもよい。
酵母を培養する場合の培地としては、例えば、バークホールダー(Burkholder)最小培地〔Proc. Natl. Acad. Sci. USA,77,4505 (1980)〕や0.5%カザミノ酸を含有するSD培地〔Proc. Natl. Acad. Sci. USA,81,5330 (1984)〕などが挙げられる。培地のpHは、好ましくは約5~約8である。培養は、通常約20℃~約35℃で行なわれる。必要に応じて、通気や撹拌を行ってもよい。
昆虫細胞または昆虫を培養する場合の培地としては、例えばGrace's Insect Medium〔Nature,195,788 (1962)〕に非働化した10%ウシ血清等の添加物を適宜加えたものなどが用いられる。培地のpHは、好ましくは約6.2~約6.4である。培養は、通常約27℃で行なわれる。必要に応じて通気や撹拌を行ってもよい。
動物細胞を培養する場合の培地としては、例えば、約5~約20%の胎児ウシ血清を含む最小必須培地(MEM)〔Science,122,501 (1952)〕,ダルベッコ改変イーグル培地(DMEM)〔Virology,8,396 (1959)〕,RPMI 1640培地〔The Journal of the American Medical Association,199,519 (1967)〕,199培地〔Proceeding of the Society for the Biological Medicine,73,1 (1950)〕などが用いられる。培地のpHは、好ましくは約6~約8である。培養は、通常約30℃~約40℃で行なわれる。必要に応じて通気や撹拌を行ってもよい。
植物細胞を培養する培地としては、MS培地、LS培地、B5培地などが用いられる。培地のpHは好ましくは約5~約8である。培養は、通常約20℃~約30℃で行なわれる。必要に応じて通気や撹拌を行ってもよい。
以上のようにして、核酸配列認識モジュールと核酸塩基変換酵素等との複合体、即ち核酸改変酵素複合体を細胞内で発現させることができる。
これに対し、CRISPR-Casシステムは、標的ヌクレオチド配列に対して相補的なガイドRNAにより目的の二本鎖DNAの配列を認識するので、標的ヌクレオチド配列と特異的にハイブリッド形成し得るオリゴDNAを合成するだけで、任意の配列を標的化することができる。
従って、本発明のより好ましい実施態様においては、核酸配列認識モジュールとして、Casエフェクタータンパク質の1つのみ、又は両方のDNA切断能が失活したCRISPR-Casシステム(CRISPR-変異Cas)が用いられる。
あるいはCasエフェクタータンパク質をコードするDNAは、核酸配列認識モジュールをコードするDNAや核酸塩基変換酵素をコードするDNAについて上記したのと同様の方法により、化学合成又はPCR法もしくはGibson Assembly法との組み合わせで、用いる宿主細胞での発現に適したコドン使用を有するDNAとして構築することもできる。
ここで「標的鎖」とは、標的ヌクレオチド配列のcrRNAとハイブリッド形成する方の鎖を意味し、その反対鎖で標的鎖とcrRNAとのハイブリッド形成により一本鎖状になる鎖を「非標的鎖(non-targeted strand)」と呼ぶこととする。標的ヌクレオチド配列を片方の鎖で表現する場合(例えばPAM配列を表記する場合や、標的ヌクレオチド配列とPAMとの位置関係を表す場合等)、非標的鎖の配列で代表させるものとする。
温度感受性変異を利用する場合、例えば、ベクターの自律複製に必要なタンパク質の温度感受性変異体を、本発明の核酸改変酵素複合体をコードするDNAを含むベクターに搭載することにより、該核酸改変酵素複合体の発現後、速やかに自律複製が出来なくなり、細胞分裂に伴って該ベクターは自然に脱落する。このような温度感受性変異タンパク質としては、pSC101 oriの複製に必要なRep101 oriの温度感受性変異体が挙げられるが、これに限定されない。Rep101 ori (ts)は30℃以下(例、28℃)では、pSC101 oriに作用してプラスミドの自律複製を可能にするが、37℃以上(例、42℃)になると機能を失い、プラスミドは自律複製できなくなる。従って、上記λファージのcIリプレッサー(ts)と併用することで、本発明の核酸改変酵素複合体の一過的発現と、プラスミド除去とを、同時に行うことができる。
出芽酵母Saccharomyces cerevisiae BY4741株(ロイシン及びウラシル要求性)を用い、標準的なYPDA培地ないしSD培地の栄養要求性に合わせたDropout組成で培養した。培養は25℃から30℃の間で、寒天プレートでの静置培養または液体培地での振とう培養を行った。形質転換は酢酸リチウム法を用い、適切な栄養要求性に合わせたSD培地で選抜を行った。ガラクトースによる発現誘導には、適切なSD培地で一晩予備培養した後、炭素源を2%グルコースから2%ラフィノースに代えたSR培地に植え継いで一晩培養、さらに炭素源を0.2%ガラクトースに代えたSGal培地に植え継いで3時間から二晩程度培養して発現誘導を行った。
生存細胞数及びCan1変異率の測定には、細胞懸濁液をSDプレート培地及びSD-Arg+60mg/l Canavanineプレート培地あるいはSD+300mg/l Canavanineプレート培地に適宜希釈して塗布し、3日後に出現するコロニー数を生存細胞数としてカウントした。SDプレートでの生存コロニー数を全細胞数とし、Canavanineプレートでの生存コロニー数を耐性変異株数として、変異率を算出・評価した。変異導入箇所はコロニーPCR法によって各株のターゲット遺伝子領域を含むDNA断片を増幅した後DNAシーケンスを行い、Saccharomyces Genome Database (http://www.yeastgenome.org/)の配列をベースにアラインメント解析を行って同定した。
ヒト胎児腎臓由来細胞(HEK293T細胞)を、10μg/mL ピューロマイシン(Life Technologies)及び10%胎仔ウシ血清(FBS)(Biosera, Nuaille, France)を添加したDME-glutamax培地(Thermo Fisher Scientific)を用いて、37℃、5% CO2条件で培養を行った。細胞の回収には5%トリプシンを用いた。ディープフリーザーで保存したHEK293T細胞を37℃のウォーターバスにて溶解し、5x106 cellsになるように75 T-flaskに播種した。1-3日間培養後に細胞を回収し、0.5x105cells/wellになるように24ウェルプレートの各ウェルに播種した。1-3日培養後に60-80%コンフルエント状態の各ウェルの細胞に対して、各500 ng/wellの下記プラスミド(エフェクタープラスミド及びレポータープラスミド)(計1 μg/well)、200 nMのドナーDNA、1.5 μlのFugeneHD(プロメガ)を用いてトランスフェクションした。各実施例で用いたドナーDNAを表1に示す。トランスフェクション72時間後に細胞を回収し、FACSを用いてiRFP及びEGFPの蛍光を検出した。検出された細胞数から、以下の式により組み換え効率(%)を算出した。

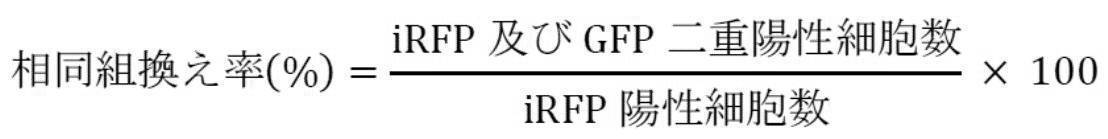
DNAは、PCR法、制限酵素処理、ライゲーション、Gibson Assembly法、人工化学合成のいずれかによって、加工・構築した。プラスミドは酵母・大腸菌シャトルベクターとしてロイシン選抜用のpRS415、及びウラシル選抜用のpRS426をバックボーンとして用いた。プラスミドは大腸菌株XL-10 gold又はDH5αで増幅し、酢酸リチウム法で酵母に導入した。
ホモロジーアーム、ガイドRNA、挿入配列等の配列は、酵母ゲノムのデータベース(https://www.yeastgenome.org/)を参照して設計した。ベクター構築は、Nishida K. et al., Science 16:353(6305) (2016) doi: 10.1126/science.aaf8729に記載する方法に準じて行った。1 x gRNAベクターは、配列番号15の配列の5871番~5890番の塩基配列を、L86又はM4の標的ヌクレオチド配列の相補配列に置換したベクターに相当する。2 x gRNAベクターは、配列番号16の配列の2638番~2657番の塩基配列を、L86、L87、L88、L93及びR90のいずれかの標的ヌクレオチド配列の相補配列に置換し、かつ配列番号16の6293番~6312番の塩基配列を、L87、R89、R90、R91及びR92のいずれかの標的ヌクレオチド配列の相補配列に置換したものに相当する。上記標的ヌクレオチドは、次の通り。
L86:CGAACAGAGTAAACCGAATC(配列番号17)
L87:AGCACTATCAAGGCTAATAA(配列番号18)
L88:GCGAACTTGAAGAATAACCA(配列番号19)
R89:TCACCTAACTCAGACATTAT(配列番号20)
R90:TTGCTGATTCTATTTACAAA(配列番号21)
R91:GCAAACTCTATTCTTGGTGC(配列番号22)
R92:ACCAGAGTATCATCCATGTC(配列番号23)
L93:AATTCGGACACTTTAGGGTT(配列番号24)
M4 :AGATATTATACCTGGACCCC(配列番号25)
pcDNA3.1ベクターバックボーンおよびCMV, PmCDA1, Cas9, H1, sgRNAの各配列はNishida et al 2016の論文より由来する。各変異はPCR法によって導入した。EF1,iRFPおよびmEGFP断片は人工遺伝子合成によって作製した。ギブソンアッセンブリーないしライゲーション反応によって断片を挿入・置換した。
iRFP陽性細胞をFACSにより分取し、次いでGenomic DNA及び導入したプラスミドDNAを抽出した後、以下のサンプルを調製し、以下の条件でPCRを行い目的の箇所を増幅した。
サンプル調製:
gDNA 1μL
プライマー 各1μL
rTaq 10x Buffer 5μL
25mM MgCl2 3μL
2mM dNTP 5μL
rTaq(TOYOBO) 0.5μL
ddH 2 O 33.5μL
計50μL
PCRの条件:94℃に2分間維持した後、94℃で45秒、55℃で45秒、72℃で1分30秒のサイクルを33サイクル行い、最後に72℃に5分間維持した。
増幅用のプライマーとして、以下のSY157及びSY182を使用した。増幅産物のサイズは、1554 bpである。
SY157: TTCTGCTTGTCGGCCATGAT(配列番号47)
SY182: AGGCAAGGCTTGACCGACAATT(配列番号48)
増幅産物を切り出し、Fastgeneを用いて精製した後、各精製産物とpGEM-t easy vecterとをTAクローニングし、該ベクターで大腸菌(JM109)をトランスフォーメーションした。次いで、各サンプルにつき24個のコロニーをピックアップし(青白選抜あり)、プラスミドDNAをMini prep (Fastgene使用)により精製した。
各サンプル 2.5μL
プライマー SY157 (10 pmol/μL) 2.5μL
ddH 2 O 10μL
計15μL
最後に、入手した配列情報をSnapgeneを用いてアライメントした。
出芽酵母BY4741株を、プラスミドベクター1525(配列番号4の6036番目の塩基がg、6037番目の塩基がcである)又は1526(配列番号4の6036番目の塩基がc、6037番目の塩基がaである)と、1059(配列番号5)又は1149(配列番号5の配列の3890番~3909番の塩基配列をTCCAATAACGGAATCCAACT(配列番号6)に置換したベクターに相当する)とで二重形質転換し、栄養要求性培地(SD-Leu-Ura)により選抜した。S-Leu-Ura 2% ラフィノース培地で一晩培養した。S-Leu-Ura 2% ラフィノース + 0.02% ガラクトース培地に1/32希釈し、30℃で一晩培養した。SD-Ura-Leu及びSD-Ura-Leu+Canavanineプレートに10倍希釈でスポッティングした。二日後にCanavanine耐性コロニーをシーケンス解析した。その結果、標的部位への変異の挿入が確認された(図2)。
プラスミドベクター1548(配列番号7)を、SmaI/HpaIで処理したDNA断片をBY4741株に形質転換し、SD-Ura培地で選抜した。シーケンス解析により、Ade1領域への組み込みが確認された。
上記のプラスミドベクターのいずれかを実証実験株に形質転換し、SD-Leu-Ura培地で選抜した。S-Leu-Ura 2% ラフィノース培地で一晩培養した。S-Leu 2% ラフィノース + 0.02% (又は0.2%) ガラクトース培地に1/32希釈し、30℃で一晩培養したものを5 generationsとした。20 generationsには、1/32希釈を計4回繰り返した。SD-Leuプレートに10倍希釈でスポッティングし、2日後にコロニー数と色を評価した。その結果、Ade1機能が回復して白くなったコロニーが高頻度で現れたことから、本発明の方法により、標的部位での相同組換えの誘導が示された(図4)。
出芽酵母BY4741株を、プラスミドベクター1251(配列番号8)及び2x gRNA ベクターで二重形質転換し、栄養要求性培地(SD-Leu-Ura)により選抜した。S-Leu-Ura 2% ラフィノース培地で一晩培養した。S-Leu-Ura 2% ラフィノース + 0.2% ガラクトース培地に1/32希釈し、30℃で一晩培養した。SD-Ura-LeuおよびSD-Ura-Leu (+Canavanine)プレートに10倍希釈でスポッティングし、二日後にCanavanine耐性コロニーをシーケンス解析した。その結果、本発明の方法により高い効率でノックインが実現できた(図6)。
ドナーDNAとして一本鎖オリゴDNA(70塩基長)(表1)を用いて、動物細胞(HEK293T細胞)において組み換え反応が生じるか否かを検証した。実験の概略図を図7に示す。レポータープラスミドとして、ベクターSY4(H1_sgRNA、CMV_mEGFP)を用い、エフェクタープラスミドとして、ベクターSY45(CMV_Cas9-PmCDA1、EF1_iRFP)、ベクターSY45(CMV_nCas9(D10A)-PmCDA1、EF1_iRFP)、ベクターSY45(CMV_nCas9(H840A)-PmCDA1、EF1_iRFP)、ベクターSY45(CMV_dCas9-PmCDA1、EF1_iRFP)、ベクターSY45(CMV_Cas9、EF1_iRFP)、ベクターSY45(CMV_nCas9(D10A)、EF1_iRFP)、ベクターSY45(CMV_nCas9(H840A)、EF1_iRFP)又はベクターSY45(CMV_dCas9、EF1_iRFP)を用いた。ドナーDNAとしてFw2、Fw3を用いた場合、首尾よく相同組換えが生じた場合には、EGFPをコードする配列に開始コドンが生じる結果、EGFPの発現が認められる。Fw1は、相同組換えが生じてもEGFPをコードする配列に開始コドンが生じないように設計されたドナーDNAであり、ネガティブコントロールとして用いた。Fw3は、Fw2のホモロジーアームの1塩基が置換(c→g)されたものであり、ホモロジーアームが標的部位の隣接領域と完全に相同でない場合でも、相同組換え反応が生じるか否かを検証するため、及び複数の異なる箇所の変異も導入可能であるか否かを検証するために用いた。
ドナーDNAとして一本鎖オリゴDNA(50塩基長)(表1)を用いて、動物細胞(HEK293T細胞)において組み換え反応が生じるか否かを検証した。実験の概略図を図9に示す。レポータープラスミドとして、ベクターSY4(H1_sgRNA、CMV_mEGFP)を用い、エフェクタープラスミドとして、ベクターSY45(CMV_nCas9(D10A))-PmCDA1、EF1_iRFP)又はベクターSY45(CMV_nCas9(H840A))-PmCDA1を用いた。
異なる相同領域に対するホモロジーアームを有するドナーDNA(表1)を用いて、相同領域により相同組換え反応の効率の変化を検証した。実験の概略図を図11示す。レポータープラスミドとして、ベクターSY4(H1_sgRNA、CMV_mEGFP)を用い、エフェクタープラスミドとして、ベクターSY45(CMV_nCas9(D10A))-PmCDA1、EF1_iRFP)又はベクターSY45(CMV_nCas9(H840A))-PmCDA1を用いた。
実施例5のFw2を用いた実験と同じgRNA及びドナーDNAを用いて、DNAにおける改変を検証した。結果を以下の表2に示す。nCas9(D10A)-PmCDA1及びnCas9(H840A)-PmCDA1を用いた場合には、Cas9を用いた場合に比べて、副産物であるIndelの発生が顕著に抑制されること、即ち細胞毒性が低減されることが実証された。なお、本実施例で用いるDNAとの用語には、ゲノムDNAとプラスミドDNAの両方が含まれる。

Claims (14)
- 細胞の有する二本鎖DNAの標的部位を改変する方法であって、選択された二本鎖DNA中の標的ヌクレオチド配列と特異的に結合する核酸配列認識モジュールと、核酸塩基変換酵素又はDNAグリコシラーゼとが結合した複合体、及び挿入配列を含むドナーDNAを該二本鎖DNAと接触させ、該標的部位において該二本鎖DNAの少なくとも一方の鎖を切断することなく、該標的部位を該挿入配列に置換する、又は該標的部位に該挿入配列を挿入する工程を含む、方法。
- 前記ドナーDNAが、標的部位の隣接領域に相同な配列を含む、請求項1に記載の方法。
- 前記核酸配列認識モジュールが、Casエフェクタータンパク質の少なくとも1つのDNA切断能が失活したCRISPR-Casシステム、ジンクフィンガーモチーフ、TALエフェクター及びPPRモチーフからなる群より選択される、請求項1又は2に記載の方法。
- 前記核酸配列認識モジュールが、Casエフェクタータンパク質の2つのDNA切断能のうちの一方のみのDNA切断能が失活したCRISPR-Casシステムである、請求項1~3のいずれか1項に記載の方法。
- 前記核酸配列認識モジュールが、Casエフェクタータンパク質の両方のDNA切断能が失活したCRISPR-Casシステムである、請求項1~3のいずれか1項に記載の方法。
- 前記核酸塩基変換酵素がデアミナーゼである、請求項1~5のいずれか1項に記載の方法。
- 前記デアミナーゼがシチジンデアミナーゼである、請求項6に記載の方法。
- 前記シチジンデアミナーゼがPmCDA1である、請求項7に記載の方法。
- 二本鎖DNAと複合体との接触が、該細胞への、該複合体をコードする核酸の導入により行われる、請求項1~8のいずれか1項に記載の方法。
- 前記細胞が原核生物細胞又は真核生物細胞である、請求項1~9のいずれか1項に記載の方法。
- 前記細胞が微生物細胞である、請求項10に記載の方法。
- 前記細胞が植物細胞、昆虫細胞又は動物細胞である、請求項10に記載の方法。
- 前記動物細胞が脊椎動物細胞である、請求項12に記載の方法。
- 前記脊椎動物細胞が哺乳類動物細胞である、請求項13に記載の方法。
Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201980003718.7A CN111148833B (zh) | 2018-03-26 | 2019-03-26 | 改变细胞具有的双链dna的目标部位的方法 |
| CA3095291A CA3095291C (en) | 2018-03-26 | 2019-03-26 | Method for modifying target site in double-stranded dna in cell |
| EP19776369.1A EP3660152A4 (en) | 2018-03-26 | 2019-03-26 | METHOD FOR MODIFYING THE DESTINATION IN DOUBLE-STRANDED DNA IN A CELL |
| US16/643,402 US11041169B2 (en) | 2018-03-26 | 2019-03-26 | Method for modifying target site in double-stranded DNA in cell |
| JP2019568280A JP6727680B2 (ja) | 2018-03-26 | 2019-03-26 | 細胞の有する二本鎖dnaの標的部位を改変する方法 |
| KR1020207029183A KR102287880B1 (ko) | 2018-03-26 | 2019-03-26 | 세포에서 이중 가닥 dna의 표적 부위를 변형시키기 위한 방법 |
| BR112020019301-1A BR112020019301A2 (pt) | 2018-03-26 | 2019-03-26 | Método para modificar um sítio alvo de um dna de fita dupla de uma célula. |
| SG11202009319YA SG11202009319YA (en) | 2018-03-26 | 2019-03-26 | Method for modifying target site in double-stranded dna in cell |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2018059073 | 2018-03-26 | ||
| JP2018-059073 | 2018-03-26 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2019189147A1 true WO2019189147A1 (ja) | 2019-10-03 |
Family
ID=68061818
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2019/012807 WO2019189147A1 (ja) | 2018-03-26 | 2019-03-26 | 細胞の有する二本鎖dnaの標的部位を改変する方法 |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US11041169B2 (ja) |
| EP (1) | EP3660152A4 (ja) |
| JP (3) | JP6727680B2 (ja) |
| KR (1) | KR102287880B1 (ja) |
| CN (1) | CN111148833B (ja) |
| BR (1) | BR112020019301A2 (ja) |
| CA (1) | CA3095291C (ja) |
| SG (1) | SG11202009319YA (ja) |
| WO (1) | WO2019189147A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2022050413A1 (ja) | 2020-09-04 | 2022-03-10 | 国立大学法人神戸大学 | 小型化シチジンデアミナーゼを含む二本鎖dnaの改変用複合体 |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN115725650A (zh) * | 2021-08-26 | 2023-03-03 | 华东师范大学 | 实现a到c和/或a到t碱基突变的碱基编辑系统及其应用 |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010012077A1 (en) | 2008-07-28 | 2010-02-04 | Mount Sinai Hospital | Compositions, methods and kits for reprogramming somatic cells |
| JP4968498B2 (ja) | 2002-01-23 | 2012-07-04 | ユニバーシティ オブ ユタ リサーチ ファウンデーション | ジンクフィンガーヌクレアーゼを用いる、標的化された染色体変異誘発 |
| JP2013513389A (ja) | 2009-12-10 | 2013-04-22 | リージェンツ オブ ザ ユニバーシティ オブ ミネソタ | Talエフェクターに媒介されるdna修飾 |
| JP2013128413A (ja) | 2010-03-11 | 2013-07-04 | Kyushu Univ | Pprモチーフを利用したrna結合性蛋白質の改変方法 |
| WO2015133554A1 (ja) | 2014-03-05 | 2015-09-11 | 国立大学法人神戸大学 | 標的化したdna配列の核酸塩基を特異的に変換するゲノム配列の改変方法及びそれに用いる分子複合体 |
| WO2016072399A1 (ja) | 2014-11-04 | 2016-05-12 | 国立大学法人神戸大学 | 脱塩基反応により標的化したdna配列に特異的に変異を導入する、ゲノム配列の改変方法、並びにそれに用いる分子複合体 |
| JP2018059073A (ja) | 2016-10-04 | 2018-04-12 | 信越化学工業株式会社 | (メタ)アクリル酸トリイソプロピルシリルと(メタ)アクリル酸誘導体の共重合体およびその製造方法 |
| WO2018230731A1 (ja) * | 2017-06-16 | 2018-12-20 | 国立大学法人 長崎大学 | がん遺伝子の転写調節領域 |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DK3401400T3 (da) | 2012-05-25 | 2019-06-03 | Univ California | Fremgangsmåder og sammensætninger til rna-styret mål-dna-modifikation og til rna-styret transskriptionsmodulering |
| WO2015021426A1 (en) | 2013-08-09 | 2015-02-12 | Sage Labs, Inc. | A crispr/cas system-based novel fusion protein and its application in genome editing |
| US10767173B2 (en) | 2015-09-09 | 2020-09-08 | National University Corporation Kobe University | Method for converting genome sequence of gram-positive bacterium by specifically converting nucleic acid base of targeted DNA sequence, and molecular complex used in same |
| DK3348636T3 (da) | 2015-09-09 | 2022-02-28 | Univ Kobe Nat Univ Corp | Fremgangsmåde til modificering af en genomsekvens, der specifikt omdanner nukleobasen af en targetteret DNA-sekvens, og molekylær sekvens anvendt i fremgangsmåden |
| US20180237800A1 (en) * | 2015-09-21 | 2018-08-23 | The Regents Of The University Of California | Compositions and methods for target nucleic acid modification |
| AU2016342380B2 (en) * | 2015-10-23 | 2022-04-07 | President And Fellows Of Harvard College | Nucleobase editors and uses thereof |
| ES2914623T3 (es) * | 2015-11-27 | 2022-06-14 | Univ Kobe Nat Univ Corp | Método para convertir una secuencia genómica de una planta monocotiledónea en la que una base de ácido nucleico en la secuencia de ADN diana se convierte de manera específica y el complejo molecular utilizado en el mismo |
| MX2019014497A (es) | 2017-06-08 | 2020-10-12 | Univ Osaka | Metodo para producir celula eucariota de adn editado, y kit usado en el mismo. |
-
2019
- 2019-03-26 BR BR112020019301-1A patent/BR112020019301A2/pt unknown
- 2019-03-26 EP EP19776369.1A patent/EP3660152A4/en active Pending
- 2019-03-26 WO PCT/JP2019/012807 patent/WO2019189147A1/ja unknown
- 2019-03-26 SG SG11202009319YA patent/SG11202009319YA/en unknown
- 2019-03-26 CN CN201980003718.7A patent/CN111148833B/zh active Active
- 2019-03-26 KR KR1020207029183A patent/KR102287880B1/ko active IP Right Grant
- 2019-03-26 JP JP2019568280A patent/JP6727680B2/ja active Active
- 2019-03-26 CA CA3095291A patent/CA3095291C/en active Active
- 2019-03-26 US US16/643,402 patent/US11041169B2/en active Active
-
2020
- 2020-06-26 JP JP2020110705A patent/JP7250349B2/ja active Active
-
2023
- 2023-03-14 JP JP2023039794A patent/JP2023063448A/ja active Pending
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4968498B2 (ja) | 2002-01-23 | 2012-07-04 | ユニバーシティ オブ ユタ リサーチ ファウンデーション | ジンクフィンガーヌクレアーゼを用いる、標的化された染色体変異誘発 |
| WO2010012077A1 (en) | 2008-07-28 | 2010-02-04 | Mount Sinai Hospital | Compositions, methods and kits for reprogramming somatic cells |
| JP2013513389A (ja) | 2009-12-10 | 2013-04-22 | リージェンツ オブ ザ ユニバーシティ オブ ミネソタ | Talエフェクターに媒介されるdna修飾 |
| JP2013128413A (ja) | 2010-03-11 | 2013-07-04 | Kyushu Univ | Pprモチーフを利用したrna結合性蛋白質の改変方法 |
| WO2015133554A1 (ja) | 2014-03-05 | 2015-09-11 | 国立大学法人神戸大学 | 標的化したdna配列の核酸塩基を特異的に変換するゲノム配列の改変方法及びそれに用いる分子複合体 |
| WO2016072399A1 (ja) | 2014-11-04 | 2016-05-12 | 国立大学法人神戸大学 | 脱塩基反応により標的化したdna配列に特異的に変異を導入する、ゲノム配列の改変方法、並びにそれに用いる分子複合体 |
| JP2018059073A (ja) | 2016-10-04 | 2018-04-12 | 信越化学工業株式会社 | (メタ)アクリル酸トリイソプロピルシリルと(メタ)アクリル酸誘導体の共重合体およびその製造方法 |
| WO2018230731A1 (ja) * | 2017-06-16 | 2018-12-20 | 国立大学法人 長崎大学 | がん遺伝子の転写調節領域 |
Non-Patent Citations (46)
| Title |
|---|
| "Journal of Experiments in Molecular Genetics", vol. 431-433, 1972, COLD SPRING HARBOR LABORATORY |
| BIO/TECHNOLOGY, vol. 6, 1988, pages 47 - 55 |
| CELL ENGINEERING ADDITIONAL, vol. 8 |
| CONG, L. ET AL., SCIENCE, vol. 339, 2013, pages 819 - 823 |
| CONG, L. ET AL.: "Multiplex genome engineering using CRISPR/Cas systems", SCIENCE, vol. 339, 2013, pages 819 - 823, XP055400719, doi:10.1126/science.1231143 * |
| GENE, vol. 17, 1982, pages 107 |
| GENE, vol. 24, 1983, pages 255 |
| GENETICS, vol. 39, 1954, pages 440 |
| HOCKEMEYER D ET AL., NAT BIOTEFCHNOL, vol. 27, 2009, pages 851 - 857 |
| IN VIVO, vol. 13, 1977, pages 213 - 217 |
| JOURNAL OF BIOCHEMISTRY, vol. 95, 1984, pages 87 |
| JOURNAL OF MOLECULAR BIOLOGY, vol. 120, 1978, pages 517 |
| JOURNAL OF MOLECULAR BIOLOGY, vol. 41, 1969, pages 459 |
| KAJI, K. ET AL., NATURE, vol. 458, 2009, pages 766 - 770 |
| KOMOR, A. C. ET AL., NATURE, vol. 61, 2016, pages 5985 - 91 |
| MA, Y. ET AL., NAT. METHODS, vol. 1-9, 2016 |
| MALI, P. ET AL.: "RNA-guided human genome enginieering via Cas9", SCIENCE, vol. 339, 2013, pages 823 - 826, XP055636507 * |
| METHODS IN ENZYMOLOGY, vol. 194, 1991, pages 182 - 187 |
| MOL CELL, vol. 31, 2008, pages 294 - 301 |
| MOLECULAR & GENERAL GENETICS, vol. 168, 1979, pages 111 |
| NAT BIOTECHNOL, vol. 20, 2002, pages 135 - 141 |
| NAT BIOTECHNOL, vol. 26, 2008, pages 695 - 701 |
| NAT BIOTECHNOL, vol. 30, 2012, pages 460 - 465 |
| NAT BIOTECHNOL., vol. 33, no. 2, 2015, pages 139 - 142 |
| NAT METHODS, vol. 8, 2011, pages 67 - 69 |
| NATURE, vol. 195, 1962, pages 788 |
| NATURE, vol. 315, 1985, pages 592 |
| NISHIDA, K. ET AL., SCIENCE, vol. 102, no. 6305, 2016, pages 353 - 563 |
| NUCLEIC ACIDS RES, vol. 39, 2011, pages e82 |
| NUCLEIC ACIDS RESEARCH, vol. 9, 1981, pages 309 |
| OCHIAI H, INT J MOL SCI, vol. 16, 2015, pages 21128 - 21137 |
| OJI A ET AL., SCI REP, vol. 6, 2016, pages 31666 |
| PNAS, vol. 112, no. 10, 2015, pages 2984 - 2989 |
| PROC. NATL. ACAD. SCI. USA, vol. 60, 1968, pages 160 |
| PROC. NATL. ACAD. SCI. USA, vol. 69, 1972, pages 2110 |
| PROC. NATL. ACAD. SCI. USA, vol. 75, 1978, pages 929 |
| PROC. NATL. ACAD. SCI. USA, vol. 77, 1980, pages 4505 |
| PROC. NATL. ACAD. SCI. USA, vol. 81, 1984, pages 5330 |
| PROCEEDING OF THE SOCIETY FOR THE BIOLOGICAL MEDICINE, vol. 73, 1950, pages 1 |
| RAN, F.A. ET AL., NAT PROTOC, vol. 8, 2013, pages 2281 - 2308 |
| SCIENCE, vol. 122, 1952, pages 501 |
| SHUJUNSHA, NEW CELL ENGINEERING EXPERIMENT PROTOCOL, vol. 263-267, 1995 |
| THE JOURNAL OF THE AMERICAN MEDICAL ASSOCIATION, vol. 199, 1967, pages 519 |
| VARSHNEY, U. ET AL., J. BIOL. CHEM., vol. 263, 1988, pages 7776 - 7784 |
| VIROLOGY, vol. 52, 1973, pages 456 |
| VIROLOGY, vol. 8, 1959, pages 396 |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2022050413A1 (ja) | 2020-09-04 | 2022-03-10 | 国立大学法人神戸大学 | 小型化シチジンデアミナーゼを含む二本鎖dnaの改変用複合体 |
Also Published As
| Publication number | Publication date |
|---|---|
| CA3095291C (en) | 2022-10-11 |
| KR102287880B1 (ko) | 2021-08-09 |
| JP6727680B2 (ja) | 2020-07-22 |
| US11041169B2 (en) | 2021-06-22 |
| BR112020019301A2 (pt) | 2021-01-05 |
| CN111148833A (zh) | 2020-05-12 |
| KR20200123255A (ko) | 2020-10-28 |
| CN111148833B (zh) | 2021-04-02 |
| JPWO2019189147A1 (ja) | 2020-04-30 |
| EP3660152A4 (en) | 2020-07-15 |
| EP3660152A1 (en) | 2020-06-03 |
| JP7250349B2 (ja) | 2023-04-03 |
| JP2023063448A (ja) | 2023-05-09 |
| US20200270631A1 (en) | 2020-08-27 |
| SG11202009319YA (en) | 2020-10-29 |
| JP2020191879A (ja) | 2020-12-03 |
| CA3095291A1 (en) | 2019-10-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20210171935A1 (en) | Method for modifying genome sequence to introduce specific mutation to targeted dna sequence by base-removal reaction, and molecular complex used therein | |
| KR102647766B1 (ko) | 클래스 ii, 타입 v crispr 시스템 | |
| JP2022123889A (ja) | 標的化したdna配列の核酸塩基を特異的に変換するゲノム配列の改変方法及びそれに用いる分子複合体 | |
| JP2023063448A (ja) | 細胞の有する二本鎖dnaの標的部位を改変する方法 | |
| JPWO2017183724A1 (ja) | ゲノム配列改変技術における変異導入効率の向上方法、及びそれに用いる分子複合体 | |
| US20240117384A1 (en) | Method for converting nucleic acid sequence of cell specifically converting nucleic acid base of targeted dna using cell endogenous dna modifying enzyme, and molecular complex used therein | |
| CA3192224A1 (en) | Base editing enzymes | |
| WO2021100731A1 (ja) | Cas9ヌクレアーゼを用いて相同組み換えを誘導する方法 | |
| EP4347816A1 (en) | Class ii, type v crispr systems | |
| WO2022050413A1 (ja) | 小型化シチジンデアミナーゼを含む二本鎖dnaの改変用複合体 | |
| CN116867897A (zh) | 碱基编辑酶 | |
| WO2024086845A2 (en) | Engineered casphi2 nucleases | |
| WO2022056301A1 (en) | Base editing enzymes |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 19776369 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2019568280 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2019776369 Country of ref document: EP Effective date: 20200227 |
|
| ENP | Entry into the national phase |
Ref document number: 3095291 Country of ref document: CA |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20207029183 Country of ref document: KR Kind code of ref document: A |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112020019301 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 112020019301 Country of ref document: BR Kind code of ref document: A2 Effective date: 20200924 |