WO2022050413A1 - 小型化シチジンデアミナーゼを含む二本鎖dnaの改変用複合体 - Google Patents
小型化シチジンデアミナーゼを含む二本鎖dnaの改変用複合体 Download PDFInfo
- Publication number
- WO2022050413A1 WO2022050413A1 PCT/JP2021/032689 JP2021032689W WO2022050413A1 WO 2022050413 A1 WO2022050413 A1 WO 2022050413A1 JP 2021032689 W JP2021032689 W JP 2021032689W WO 2022050413 A1 WO2022050413 A1 WO 2022050413A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- amino acid
- acid sequence
- deaminase
- complex
- nucleic acid
- Prior art date
Links
- 108020004414 DNA Proteins 0.000 title claims abstract description 175
- 102100026846 Cytidine deaminase Human genes 0.000 title claims abstract description 97
- 108010031325 Cytidine deaminase Proteins 0.000 title claims abstract description 97
- 102000053602 DNA Human genes 0.000 title claims abstract description 76
- 230000000051 modifying effect Effects 0.000 title claims abstract description 28
- 150000007523 nucleic acids Chemical group 0.000 claims abstract description 134
- 125000003275 alpha amino acid group Chemical group 0.000 claims abstract description 104
- 125000000539 amino acid group Chemical group 0.000 claims abstract description 102
- 108091028043 Nucleic acid sequence Proteins 0.000 claims abstract description 95
- 108090000623 proteins and genes Proteins 0.000 claims abstract description 94
- 230000004048 modification Effects 0.000 claims abstract description 88
- 238000012986 modification Methods 0.000 claims abstract description 88
- 102000004169 proteins and genes Human genes 0.000 claims abstract description 75
- 239000002773 nucleotide Substances 0.000 claims abstract description 72
- 125000003729 nucleotide group Chemical group 0.000 claims abstract description 72
- 150000001413 amino acids Chemical class 0.000 claims abstract description 63
- 210000004027 cell Anatomy 0.000 claims description 104
- 238000000034 method Methods 0.000 claims description 56
- 102000039446 nucleic acids Human genes 0.000 claims description 46
- 108020004707 nucleic acids Proteins 0.000 claims description 46
- 239000013598 vector Substances 0.000 claims description 33
- 230000002209 hydrophobic effect Effects 0.000 claims description 28
- 108091033409 CRISPR Proteins 0.000 claims description 27
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 claims description 20
- 239000011701 zinc Substances 0.000 claims description 20
- 229910052725 zinc Inorganic materials 0.000 claims description 20
- 238000012217 deletion Methods 0.000 claims description 18
- 230000037430 deletion Effects 0.000 claims description 18
- 108010073062 Transcription Activator-Like Effectors Proteins 0.000 claims description 17
- 125000001165 hydrophobic group Chemical group 0.000 claims description 17
- 210000004899 c-terminal region Anatomy 0.000 claims description 16
- 210000004900 c-terminal fragment Anatomy 0.000 claims description 15
- 210000004898 n-terminal fragment Anatomy 0.000 claims description 15
- 238000006467 substitution reaction Methods 0.000 claims description 15
- 230000007018 DNA scission Effects 0.000 claims description 11
- 241000702421 Dependoparvovirus Species 0.000 claims description 6
- 230000009437 off-target effect Effects 0.000 abstract description 23
- 235000001014 amino acid Nutrition 0.000 description 74
- 239000002609 medium Substances 0.000 description 70
- 235000018102 proteins Nutrition 0.000 description 67
- 229940024606 amino acid Drugs 0.000 description 63
- FSBIGDSBMBYOPN-VKHMYHEASA-N L-canavanine Chemical compound OC(=O)[C@@H](N)CCONC(N)=N FSBIGDSBMBYOPN-VKHMYHEASA-N 0.000 description 40
- FSBIGDSBMBYOPN-UHFFFAOYSA-N O-guanidino-DL-homoserine Natural products OC(=O)C(N)CCON=C(N)N FSBIGDSBMBYOPN-UHFFFAOYSA-N 0.000 description 40
- 230000035772 mutation Effects 0.000 description 40
- 108020005004 Guide RNA Proteins 0.000 description 32
- 101710172430 Uracil-DNA glycosylase inhibitor Proteins 0.000 description 31
- 239000013612 plasmid Substances 0.000 description 29
- 230000000694 effects Effects 0.000 description 27
- 238000010586 diagram Methods 0.000 description 26
- 230000014509 gene expression Effects 0.000 description 23
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 20
- 101000910035 Streptococcus pyogenes serotype M1 CRISPR-associated endonuclease Cas9/Csn1 Proteins 0.000 description 20
- 235000014680 Saccharomyces cerevisiae Nutrition 0.000 description 19
- 239000012636 effector Substances 0.000 description 19
- MZZYGYNZAOVRTG-UHFFFAOYSA-N 2-hydroxy-n-(1h-1,2,4-triazol-5-yl)benzamide Chemical compound OC1=CC=CC=C1C(=O)NC1=NC=NN1 MZZYGYNZAOVRTG-UHFFFAOYSA-N 0.000 description 17
- 230000002829 reductive effect Effects 0.000 description 17
- 101000755690 Homo sapiens Single-stranded DNA cytosine deaminase Proteins 0.000 description 16
- 239000013604 expression vector Substances 0.000 description 16
- 241000238631 Hexapoda Species 0.000 description 15
- 101000658622 Homo sapiens Testis-specific Y-encoded-like protein 2 Proteins 0.000 description 15
- 102100034917 Testis-specific Y-encoded-like protein 2 Human genes 0.000 description 15
- 238000012258 culturing Methods 0.000 description 15
- 102000004190 Enzymes Human genes 0.000 description 14
- 108090000790 Enzymes Proteins 0.000 description 14
- 238000002474 experimental method Methods 0.000 description 14
- 238000003780 insertion Methods 0.000 description 14
- 230000037431 insertion Effects 0.000 description 14
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 13
- 241000588724 Escherichia coli Species 0.000 description 13
- 230000027455 binding Effects 0.000 description 13
- 108091079001 CRISPR RNA Proteins 0.000 description 11
- 239000012634 fragment Substances 0.000 description 11
- 230000008685 targeting Effects 0.000 description 11
- 239000013607 AAV vector Substances 0.000 description 10
- 230000004568 DNA-binding Effects 0.000 description 10
- 238000003776 cleavage reaction Methods 0.000 description 10
- 230000007017 scission Effects 0.000 description 10
- 241000196324 Embryophyta Species 0.000 description 9
- 230000000295 complement effect Effects 0.000 description 9
- 108020001507 fusion proteins Proteins 0.000 description 9
- 102000037865 fusion proteins Human genes 0.000 description 9
- 238000006481 deamination reaction Methods 0.000 description 8
- 230000010076 replication Effects 0.000 description 8
- 101000808011 Homo sapiens Vascular endothelial growth factor A Proteins 0.000 description 7
- 102100039037 Vascular endothelial growth factor A Human genes 0.000 description 7
- 241000700605 Viruses Species 0.000 description 7
- 238000003556 assay Methods 0.000 description 7
- 239000002299 complementary DNA Substances 0.000 description 7
- 230000001939 inductive effect Effects 0.000 description 7
- 239000013641 positive control Substances 0.000 description 7
- 241000193830 Bacillus <bacterium> Species 0.000 description 6
- 241001465754 Metazoa Species 0.000 description 6
- 210000004102 animal cell Anatomy 0.000 description 6
- 238000006243 chemical reaction Methods 0.000 description 6
- 230000005782 double-strand break Effects 0.000 description 6
- 108091008146 restriction endonucleases Proteins 0.000 description 6
- 238000012795 verification Methods 0.000 description 6
- 108091026890 Coding region Proteins 0.000 description 5
- 108020004705 Codon Proteins 0.000 description 5
- 241000701022 Cytomegalovirus Species 0.000 description 5
- 108010008532 Deoxyribonuclease I Proteins 0.000 description 5
- 102000007260 Deoxyribonuclease I Human genes 0.000 description 5
- 108010055325 EphB3 Receptor Proteins 0.000 description 5
- 102100021601 Ephrin type-A receptor 8 Human genes 0.000 description 5
- 102100031982 Ephrin type-B receptor 3 Human genes 0.000 description 5
- 102220567867 Fatty acid-binding protein 5_K34A_mutation Human genes 0.000 description 5
- 102220567879 Fatty acid-binding protein 5_R33A_mutation Human genes 0.000 description 5
- 101000898676 Homo sapiens Ephrin type-A receptor 8 Proteins 0.000 description 5
- 238000004458 analytical method Methods 0.000 description 5
- 230000009615 deamination Effects 0.000 description 5
- 238000013461 design Methods 0.000 description 5
- 238000001415 gene therapy Methods 0.000 description 5
- 238000010362 genome editing Methods 0.000 description 5
- 238000001890 transfection Methods 0.000 description 5
- 238000009423 ventilation Methods 0.000 description 5
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- 244000063299 Bacillus subtilis Species 0.000 description 4
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 4
- 102000004163 DNA-directed RNA polymerases Human genes 0.000 description 4
- 108090000626 DNA-directed RNA polymerases Proteins 0.000 description 4
- 206010059866 Drug resistance Diseases 0.000 description 4
- 102100037964 E3 ubiquitin-protein ligase RING2 Human genes 0.000 description 4
- 241000701959 Escherichia virus Lambda Species 0.000 description 4
- 101001095815 Homo sapiens E3 ubiquitin-protein ligase RING2 Proteins 0.000 description 4
- ISAKRJDGNUQOIC-UHFFFAOYSA-N Uracil Chemical compound O=C1C=CNC(=O)N1 ISAKRJDGNUQOIC-UHFFFAOYSA-N 0.000 description 4
- -1 aromatic amino acids Chemical class 0.000 description 4
- 229910052799 carbon Inorganic materials 0.000 description 4
- 208000031752 chronic bilirubin encephalopathy Diseases 0.000 description 4
- 235000019877 cocoa butter equivalent Nutrition 0.000 description 4
- OPTASPLRGRRNAP-UHFFFAOYSA-N cytosine Chemical compound NC=1C=CNC(=O)N=1 OPTASPLRGRRNAP-UHFFFAOYSA-N 0.000 description 4
- 108020004999 messenger RNA Proteins 0.000 description 4
- 238000002715 modification method Methods 0.000 description 4
- 230000036961 partial effect Effects 0.000 description 4
- 238000011160 research Methods 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- 101100139907 Arabidopsis thaliana RAR1 gene Proteins 0.000 description 3
- 239000004475 Arginine Substances 0.000 description 3
- 241000255789 Bombyx mori Species 0.000 description 3
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 3
- 101150008604 CAN1 gene Proteins 0.000 description 3
- 241000282693 Cercopithecidae Species 0.000 description 3
- 108091092236 Chimeric RNA Proteins 0.000 description 3
- 241000206602 Eukaryota Species 0.000 description 3
- 101000860092 Francisella tularensis subsp. novicida (strain U112) CRISPR-associated endonuclease Cas12a Proteins 0.000 description 3
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 3
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 3
- 108010043121 Green Fluorescent Proteins Proteins 0.000 description 3
- 102000004144 Green Fluorescent Proteins Human genes 0.000 description 3
- 241000282412 Homo Species 0.000 description 3
- 241000235789 Hyperoartia Species 0.000 description 3
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 3
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 3
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 3
- 241000124008 Mammalia Species 0.000 description 3
- 101710163270 Nuclease Proteins 0.000 description 3
- 101100028790 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) PBS2 gene Proteins 0.000 description 3
- 108091023040 Transcription factor Proteins 0.000 description 3
- 102000040945 Transcription factor Human genes 0.000 description 3
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 3
- 102000006943 Uracil-DNA Glycosidase Human genes 0.000 description 3
- 108010072685 Uracil-DNA Glycosidase Proteins 0.000 description 3
- 238000013019 agitation Methods 0.000 description 3
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 3
- 235000009697 arginine Nutrition 0.000 description 3
- 125000000637 arginyl group Chemical group N[C@@H](CCCNC(N)=N)C(=O)* 0.000 description 3
- 238000004422 calculation algorithm Methods 0.000 description 3
- 230000003197 catalytic effect Effects 0.000 description 3
- 230000008859 change Effects 0.000 description 3
- 239000000470 constituent Substances 0.000 description 3
- 210000004748 cultured cell Anatomy 0.000 description 3
- 238000012350 deep sequencing Methods 0.000 description 3
- 229940079593 drug Drugs 0.000 description 3
- 239000003814 drug Substances 0.000 description 3
- 238000005516 engineering process Methods 0.000 description 3
- 239000012091 fetal bovine serum Substances 0.000 description 3
- 238000011049 filling Methods 0.000 description 3
- 230000004927 fusion Effects 0.000 description 3
- 229930182830 galactose Natural products 0.000 description 3
- 239000008103 glucose Substances 0.000 description 3
- 125000000291 glutamic acid group Chemical group N[C@@H](CCC(O)=O)C(=O)* 0.000 description 3
- 239000005090 green fluorescent protein Substances 0.000 description 3
- 230000006801 homologous recombination Effects 0.000 description 3
- 238000002744 homologous recombination Methods 0.000 description 3
- 230000006698 induction Effects 0.000 description 3
- 239000003112 inhibitor Substances 0.000 description 3
- 230000017730 intein-mediated protein splicing Effects 0.000 description 3
- 238000002360 preparation method Methods 0.000 description 3
- 238000003757 reverse transcription PCR Methods 0.000 description 3
- 241000894007 species Species 0.000 description 3
- 238000003756 stirring Methods 0.000 description 3
- 239000000758 substrate Substances 0.000 description 3
- 230000001629 suppression Effects 0.000 description 3
- 230000002194 synthesizing effect Effects 0.000 description 3
- 125000000430 tryptophan group Chemical group [H]N([H])C(C(=O)O*)C([H])([H])C1=C([H])N([H])C2=C([H])C([H])=C([H])C([H])=C12 0.000 description 3
- GOJUJUVQIVIZAV-UHFFFAOYSA-N 2-amino-4,6-dichloropyrimidine-5-carbaldehyde Chemical group NC1=NC(Cl)=C(C=O)C(Cl)=N1 GOJUJUVQIVIZAV-UHFFFAOYSA-N 0.000 description 2
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 description 2
- 101000860090 Acidaminococcus sp. (strain BV3L6) CRISPR-associated endonuclease Cas12a Proteins 0.000 description 2
- 108091093088 Amplicon Proteins 0.000 description 2
- 235000014469 Bacillus subtilis Nutrition 0.000 description 2
- 241000409811 Bombyx mori nucleopolyhedrovirus Species 0.000 description 2
- 108700010070 Codon Usage Proteins 0.000 description 2
- 108020001019 DNA Primers Proteins 0.000 description 2
- 230000008836 DNA modification Effects 0.000 description 2
- 239000003155 DNA primer Substances 0.000 description 2
- 241000588722 Escherichia Species 0.000 description 2
- 102000053187 Glucuronidase Human genes 0.000 description 2
- 108010060309 Glucuronidase Proteins 0.000 description 2
- 101001135589 Homo sapiens Tyrosine-protein phosphatase non-receptor type 22 Proteins 0.000 description 2
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 description 2
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 2
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 2
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 2
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 2
- 241000700584 Simplexvirus Species 0.000 description 2
- 102100022433 Single-stranded DNA cytosine deaminase Human genes 0.000 description 2
- 241000193996 Streptococcus pyogenes Species 0.000 description 2
- 101150073336 Sycp1 gene Proteins 0.000 description 2
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 2
- 239000004473 Threonine Substances 0.000 description 2
- 108020004440 Thymidine kinase Proteins 0.000 description 2
- 102100033138 Tyrosine-protein phosphatase non-receptor type 22 Human genes 0.000 description 2
- 241000251539 Vertebrata <Metazoa> Species 0.000 description 2
- 240000008042 Zea mays Species 0.000 description 2
- 235000005824 Zea mays ssp. parviglumis Nutrition 0.000 description 2
- 235000002017 Zea mays subsp mays Nutrition 0.000 description 2
- 108010017070 Zinc Finger Nucleases Proteins 0.000 description 2
- 239000002253 acid Substances 0.000 description 2
- 239000006285 cell suspension Substances 0.000 description 2
- 239000013611 chromosomal DNA Substances 0.000 description 2
- 238000010276 construction Methods 0.000 description 2
- 235000005822 corn Nutrition 0.000 description 2
- 229940104302 cytosine Drugs 0.000 description 2
- 230000001419 dependent effect Effects 0.000 description 2
- 238000011156 evaluation Methods 0.000 description 2
- 239000000284 extract Substances 0.000 description 2
- 235000013922 glutamic acid Nutrition 0.000 description 2
- 239000004220 glutamic acid Substances 0.000 description 2
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 2
- 235000004554 glutamine Nutrition 0.000 description 2
- 125000000404 glutamine group Chemical group N[C@@H](CCC(N)=O)C(=O)* 0.000 description 2
- 230000012010 growth Effects 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 230000003993 interaction Effects 0.000 description 2
- 238000001638 lipofection Methods 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- XIXADJRWDQXREU-UHFFFAOYSA-M lithium acetate Chemical compound [Li+].CC([O-])=O XIXADJRWDQXREU-UHFFFAOYSA-M 0.000 description 2
- 239000003550 marker Substances 0.000 description 2
- 230000001404 mediated effect Effects 0.000 description 2
- 244000005700 microbiome Species 0.000 description 2
- 238000000520 microinjection Methods 0.000 description 2
- 239000000203 mixture Substances 0.000 description 2
- 238000002703 mutagenesis Methods 0.000 description 2
- 231100000350 mutagenesis Toxicity 0.000 description 2
- 210000004897 n-terminal region Anatomy 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- 230000009438 off-target cleavage Effects 0.000 description 2
- RXWNCPJZOCPEPQ-NVWDDTSBSA-N puromycin Chemical compound C1=CC(OC)=CC=C1C[C@H](N)C(=O)N[C@H]1[C@@H](O)[C@H](N2C3=NC=NC(=C3N=C2)N(C)C)O[C@@H]1CO RXWNCPJZOCPEPQ-NVWDDTSBSA-N 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- 235000008521 threonine Nutrition 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- 230000009466 transformation Effects 0.000 description 2
- 230000010474 transient expression Effects 0.000 description 2
- 241001515965 unidentified phage Species 0.000 description 2
- 238000011144 upstream manufacturing Methods 0.000 description 2
- 229940035893 uracil Drugs 0.000 description 2
- 210000005253 yeast cell Anatomy 0.000 description 2
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 description 1
- VKTCMMONRNFKJD-BYPYZUCNSA-N (2r)-3-aminosulfanyl-2-(ethylamino)propanoic acid Chemical compound CCN[C@H](C(O)=O)CSN VKTCMMONRNFKJD-BYPYZUCNSA-N 0.000 description 1
- SMZOUWXMTYCWNB-UHFFFAOYSA-N 2-(2-methoxy-5-methylphenyl)ethanamine Chemical compound COC1=CC=C(C)C=C1CCN SMZOUWXMTYCWNB-UHFFFAOYSA-N 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-N 2-Propenoic acid Natural products OC(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 1
- 108010029988 AICDA (activation-induced cytidine deaminase) Proteins 0.000 description 1
- 241000093740 Acidaminococcus sp. Species 0.000 description 1
- 241001655883 Adeno-associated virus - 1 Species 0.000 description 1
- 241000702423 Adeno-associated virus - 2 Species 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- 241000589158 Agrobacterium Species 0.000 description 1
- 241000252073 Anguilliformes Species 0.000 description 1
- 101100028789 Arabidopsis thaliana PBS1 gene Proteins 0.000 description 1
- 101100365087 Arabidopsis thaliana SCRA gene Proteins 0.000 description 1
- 101100365680 Arabidopsis thaliana SGT1B gene Proteins 0.000 description 1
- DCXYFEDJOCDNAF-UHFFFAOYSA-N Asparagine Natural products OC(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-N 0.000 description 1
- 101100007857 Bacillus subtilis (strain 168) cspB gene Proteins 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 1
- 101100417900 Clostridium acetobutylicum (strain ATCC 824 / DSM 792 / JCM 1419 / LMG 5710 / VKM B-1787) rbr3A gene Proteins 0.000 description 1
- 241000699802 Cricetulus griseus Species 0.000 description 1
- 240000008067 Cucumis sativus Species 0.000 description 1
- 235000010799 Cucumis sativus var sativus Nutrition 0.000 description 1
- 102000000311 Cytosine Deaminase Human genes 0.000 description 1
- 108010080611 Cytosine Deaminase Proteins 0.000 description 1
- 101150074155 DHFR gene Proteins 0.000 description 1
- 230000006463 DNA deamination Effects 0.000 description 1
- 230000033616 DNA repair Effects 0.000 description 1
- 241000252212 Danio rerio Species 0.000 description 1
- 229920001353 Dextrin Polymers 0.000 description 1
- 239000004375 Dextrin Substances 0.000 description 1
- 240000006497 Dianthus caryophyllus Species 0.000 description 1
- 235000009355 Dianthus caryophyllus Nutrition 0.000 description 1
- 108010053187 Diphtheria Toxin Proteins 0.000 description 1
- 241000255581 Drosophila <fruit fly, genus> Species 0.000 description 1
- 102100031780 Endonuclease Human genes 0.000 description 1
- 241000283086 Equidae Species 0.000 description 1
- 241001452028 Escherichia coli DH1 Species 0.000 description 1
- 241001131785 Escherichia coli HB101 Species 0.000 description 1
- 241001646716 Escherichia coli K-12 Species 0.000 description 1
- 108700039887 Essential Genes Proteins 0.000 description 1
- 241000589601 Francisella Species 0.000 description 1
- 241000233866 Fungi Species 0.000 description 1
- 108010001498 Galectin 1 Proteins 0.000 description 1
- 102100021736 Galectin-1 Human genes 0.000 description 1
- 235000011201 Ginkgo Nutrition 0.000 description 1
- 244000194101 Ginkgo biloba Species 0.000 description 1
- 235000008100 Ginkgo biloba Nutrition 0.000 description 1
- 239000012571 GlutaMAX medium Substances 0.000 description 1
- 206010020649 Hyperkeratosis Diseases 0.000 description 1
- 244000283207 Indigofera tinctoria Species 0.000 description 1
- 241000235058 Komagataella pastoris Species 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- 241001112693 Lachnospiraceae Species 0.000 description 1
- 241000904817 Lachnospiraceae bacterium Species 0.000 description 1
- 239000012097 Lipofectamine 2000 Substances 0.000 description 1
- 235000007688 Lycopersicon esculentum Nutrition 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 241000555303 Mamestra brassicae Species 0.000 description 1
- 108020005196 Mitochondrial DNA Proteins 0.000 description 1
- 108010021466 Mutant Proteins Proteins 0.000 description 1
- 102000008300 Mutant Proteins Human genes 0.000 description 1
- 210000004460 N cell Anatomy 0.000 description 1
- 241000588650 Neisseria meningitidis Species 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- 244000061176 Nicotiana tabacum Species 0.000 description 1
- 235000002637 Nicotiana tabacum Nutrition 0.000 description 1
- 108010077850 Nuclear Localization Signals Proteins 0.000 description 1
- 241000238814 Orthoptera Species 0.000 description 1
- 240000007594 Oryza sativa Species 0.000 description 1
- 235000007164 Oryza sativa Nutrition 0.000 description 1
- 101150034686 PDC gene Proteins 0.000 description 1
- 102000000470 PDZ domains Human genes 0.000 description 1
- 108050008994 PDZ domains Proteins 0.000 description 1
- 101150012394 PHO5 gene Proteins 0.000 description 1
- 239000001888 Peptone Substances 0.000 description 1
- 108010080698 Peptones Proteins 0.000 description 1
- 241000251745 Petromyzon marinus Species 0.000 description 1
- 206010035226 Plasma cell myeloma Diseases 0.000 description 1
- 101710182846 Polyhedrin Proteins 0.000 description 1
- 108010092799 RNA-directed DNA polymerase Proteins 0.000 description 1
- 239000012980 RPMI-1640 medium Substances 0.000 description 1
- MUPFEKGTMRGPLJ-RMMQSMQOSA-N Raffinose Natural products O(C[C@H]1[C@@H](O)[C@H](O)[C@@H](O)[C@@H](O[C@@]2(CO)[C@H](O)[C@@H](O)[C@@H](CO)O2)O1)[C@@H]1[C@H](O)[C@@H](O)[C@@H](O)[C@@H](CO)O1 MUPFEKGTMRGPLJ-RMMQSMQOSA-N 0.000 description 1
- 108700008625 Reporter Genes Proteins 0.000 description 1
- 101150105073 SCR1 gene Proteins 0.000 description 1
- 102000000395 SH3 domains Human genes 0.000 description 1
- 108050008861 SH3 domains Proteins 0.000 description 1
- 229910003797 SPO1 Inorganic materials 0.000 description 1
- 229910003798 SPO2 Inorganic materials 0.000 description 1
- 101100134054 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) NTG1 gene Proteins 0.000 description 1
- 101100150136 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) SPO1 gene Proteins 0.000 description 1
- 241000793189 Saccharomyces cerevisiae BY4741 Species 0.000 description 1
- 241000235343 Saccharomycetales Species 0.000 description 1
- 206010039491 Sarcoma Diseases 0.000 description 1
- 241000235347 Schizosaccharomyces pombe Species 0.000 description 1
- 101100478210 Schizosaccharomyces pombe (strain 972 / ATCC 24843) spo2 gene Proteins 0.000 description 1
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 1
- 108020004682 Single-Stranded DNA Proteins 0.000 description 1
- 101710143275 Single-stranded DNA cytosine deaminase Proteins 0.000 description 1
- 240000003768 Solanum lycopersicum Species 0.000 description 1
- 244000061458 Solanum melongena Species 0.000 description 1
- 235000002597 Solanum melongena Nutrition 0.000 description 1
- 244000061456 Solanum tuberosum Species 0.000 description 1
- 235000002595 Solanum tuberosum Nutrition 0.000 description 1
- 238000002105 Southern blotting Methods 0.000 description 1
- 235000019764 Soybean Meal Nutrition 0.000 description 1
- 241000256251 Spodoptera frugiperda Species 0.000 description 1
- 241000191967 Staphylococcus aureus Species 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 241000194020 Streptococcus thermophilus Species 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 241000282887 Suidae Species 0.000 description 1
- 238000010459 TALEN Methods 0.000 description 1
- 102000006601 Thymidine Kinase Human genes 0.000 description 1
- 108010043645 Transcription Activator-Like Effector Nucleases Proteins 0.000 description 1
- 108700019146 Transgenes Proteins 0.000 description 1
- 241000255993 Trichoplusia ni Species 0.000 description 1
- 235000021307 Triticum Nutrition 0.000 description 1
- 244000098338 Triticum aestivum Species 0.000 description 1
- 102000004142 Trypsin Human genes 0.000 description 1
- 108090000631 Trypsin Proteins 0.000 description 1
- 102220528040 Tyrosine-protein phosphatase non-receptor type 22_D10A_mutation Human genes 0.000 description 1
- MUPFEKGTMRGPLJ-UHFFFAOYSA-N UNPD196149 Natural products OC1C(O)C(CO)OC1(CO)OC1C(O)C(O)C(O)C(COC2C(C(O)C(O)C(CO)O2)O)O1 MUPFEKGTMRGPLJ-UHFFFAOYSA-N 0.000 description 1
- 241000700618 Vaccinia virus Species 0.000 description 1
- 241000589634 Xanthomonas Species 0.000 description 1
- 241000269370 Xenopus <genus> Species 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 101150063416 add gene Proteins 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 238000005273 aeration Methods 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- AVKUERGKIZMTKX-NJBDSQKTSA-N ampicillin Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@H]3SC([C@@H](N3C2=O)C(O)=O)(C)C)=CC=CC=C1 AVKUERGKIZMTKX-NJBDSQKTSA-N 0.000 description 1
- 229960000723 ampicillin Drugs 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- PYMYPHUHKUWMLA-WDCZJNDASA-N arabinose Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)C=O PYMYPHUHKUWMLA-WDCZJNDASA-N 0.000 description 1
- PYMYPHUHKUWMLA-UHFFFAOYSA-N arabinose Natural products OCC(O)C(O)C(O)C=O PYMYPHUHKUWMLA-UHFFFAOYSA-N 0.000 description 1
- 235000009582 asparagine Nutrition 0.000 description 1
- 229960001230 asparagine Drugs 0.000 description 1
- 125000000613 asparagine group Chemical group N[C@@H](CC(N)=O)C(=O)* 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- 230000033590 base-excision repair Effects 0.000 description 1
- 238000002869 basic local alignment search tool Methods 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- SRBFZHDQGSBBOR-UHFFFAOYSA-N beta-D-Pyranose-Lyxose Natural products OC1COC(O)C(O)C1O SRBFZHDQGSBBOR-UHFFFAOYSA-N 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- 239000012620 biological material Substances 0.000 description 1
- 238000010170 biological method Methods 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 239000012888 bovine serum Substances 0.000 description 1
- 238000009395 breeding Methods 0.000 description 1
- 230000001488 breeding effect Effects 0.000 description 1
- 239000001110 calcium chloride Substances 0.000 description 1
- 229910001628 calcium chloride Inorganic materials 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 229940041514 candida albicans extract Drugs 0.000 description 1
- 239000005018 casein Substances 0.000 description 1
- BECPQYXYKAMYBN-UHFFFAOYSA-N casein, tech. Chemical compound NCCCCC(C(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(CC(C)C)N=C(O)C(CCC(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(C(C)O)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(COP(O)(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(N)CC1=CC=CC=C1 BECPQYXYKAMYBN-UHFFFAOYSA-N 0.000 description 1
- 235000021240 caseins Nutrition 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 230000032823 cell division Effects 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 230000003833 cell viability Effects 0.000 description 1
- 210000003169 central nervous system Anatomy 0.000 description 1
- 235000013339 cereals Nutrition 0.000 description 1
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 1
- 238000000975 co-precipitation Methods 0.000 description 1
- 238000010835 comparative analysis Methods 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 101150110403 cspA gene Proteins 0.000 description 1
- 101150068339 cspLA gene Proteins 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 230000000593 degrading effect Effects 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 235000019425 dextrin Nutrition 0.000 description 1
- 238000007865 diluting Methods 0.000 description 1
- 235000013601 eggs Nutrition 0.000 description 1
- 238000001962 electrophoresis Methods 0.000 description 1
- 238000004520 electroporation Methods 0.000 description 1
- 210000002257 embryonic structure Anatomy 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 238000006911 enzymatic reaction Methods 0.000 description 1
- 239000013613 expression plasmid Substances 0.000 description 1
- 230000001605 fetal effect Effects 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 239000006451 grace's insect medium Substances 0.000 description 1
- 230000005661 hydrophobic surface Effects 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 239000000411 inducer Substances 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 210000000936 intestine Anatomy 0.000 description 1
- 229960000310 isoleucine Drugs 0.000 description 1
- AGPKZVBTJJNPAG-UHFFFAOYSA-N isoleucine Natural products CCC(C)C(N)C(O)=O AGPKZVBTJJNPAG-UHFFFAOYSA-N 0.000 description 1
- 125000000741 isoleucyl group Chemical group [H]N([H])C(C(C([H])([H])[H])C([H])([H])C([H])([H])[H])C(=O)O* 0.000 description 1
- BPHPUYQFMNQIOC-NXRLNHOXSA-N isopropyl beta-D-thiogalactopyranoside Chemical compound CC(C)S[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O BPHPUYQFMNQIOC-NXRLNHOXSA-N 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 231100001231 less toxic Toxicity 0.000 description 1
- 125000001909 leucine group Chemical group [H]N(*)C(C(*)=O)C([H])([H])C(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 208000032839 leukemia Diseases 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 231100000053 low toxicity Toxicity 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 235000018977 lysine Nutrition 0.000 description 1
- 229910001629 magnesium chloride Inorganic materials 0.000 description 1
- 210000004962 mammalian cell Anatomy 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 235000013372 meat Nutrition 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 239000007758 minimum essential medium Substances 0.000 description 1
- 230000002438 mitochondrial effect Effects 0.000 description 1
- 238000006011 modification reaction Methods 0.000 description 1
- 229910000403 monosodium phosphate Inorganic materials 0.000 description 1
- 235000019799 monosodium phosphate Nutrition 0.000 description 1
- 239000006870 ms-medium Substances 0.000 description 1
- 230000036438 mutation frequency Effects 0.000 description 1
- 201000000050 myeloid neoplasm Diseases 0.000 description 1
- 150000002823 nitrates Chemical class 0.000 description 1
- 230000005937 nuclear translocation Effects 0.000 description 1
- 229940046166 oligodeoxynucleotide Drugs 0.000 description 1
- 238000010397 one-hybrid screening Methods 0.000 description 1
- 210000000287 oocyte Anatomy 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 210000001672 ovary Anatomy 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 230000001717 pathogenic effect Effects 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 101150019841 penP gene Proteins 0.000 description 1
- 235000019319 peptone Nutrition 0.000 description 1
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 1
- 230000003032 phytopathogenic effect Effects 0.000 description 1
- 210000001778 pluripotent stem cell Anatomy 0.000 description 1
- 102000040430 polynucleotide Human genes 0.000 description 1
- 108091033319 polynucleotide Proteins 0.000 description 1
- 239000002157 polynucleotide Substances 0.000 description 1
- 229920001184 polypeptide Polymers 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 239000013615 primer Substances 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 108090000765 processed proteins & peptides Proteins 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- 230000029983 protein stabilization Effects 0.000 description 1
- 238000001243 protein synthesis Methods 0.000 description 1
- 210000001938 protoplast Anatomy 0.000 description 1
- 229950010131 puromycin Drugs 0.000 description 1
- MUPFEKGTMRGPLJ-ZQSKZDJDSA-N raffinose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO[C@@H]2[C@@H]([C@@H](O)[C@@H](O)[C@@H](CO)O2)O)O1 MUPFEKGTMRGPLJ-ZQSKZDJDSA-N 0.000 description 1
- 101150079601 recA gene Proteins 0.000 description 1
- 230000006798 recombination Effects 0.000 description 1
- 238000005215 recombination Methods 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 230000007115 recruitment Effects 0.000 description 1
- 108010054624 red fluorescent protein Proteins 0.000 description 1
- 230000008439 repair process Effects 0.000 description 1
- 210000001525 retina Anatomy 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 235000009566 rice Nutrition 0.000 description 1
- GHSJKUNUIHUPDF-UHFFFAOYSA-N s-(2-aminoethyl)-l-cysteine Chemical compound NCCSCC(N)C(O)=O GHSJKUNUIHUPDF-UHFFFAOYSA-N 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 238000012163 sequencing technique Methods 0.000 description 1
- 235000004400 serine Nutrition 0.000 description 1
- 230000035939 shock Effects 0.000 description 1
- 239000013605 shuttle vector Substances 0.000 description 1
- 230000037432 silent mutation Effects 0.000 description 1
- 210000002027 skeletal muscle Anatomy 0.000 description 1
- 101150068906 snr-6 gene Proteins 0.000 description 1
- AJPJDKMHJJGVTQ-UHFFFAOYSA-M sodium dihydrogen phosphate Chemical compound [Na+].OP(O)([O-])=O AJPJDKMHJJGVTQ-UHFFFAOYSA-M 0.000 description 1
- 239000004455 soybean meal Substances 0.000 description 1
- 230000006641 stabilisation Effects 0.000 description 1
- 238000011105 stabilization Methods 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 230000003068 static effect Effects 0.000 description 1
- 238000005728 strengthening Methods 0.000 description 1
- 229960005322 streptomycin Drugs 0.000 description 1
- 238000012916 structural analysis Methods 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 125000000341 threoninyl group Chemical group [H]OC([H])(C([H])([H])[H])C([H])(N([H])[H])C(*)=O 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 230000002103 transcriptional effect Effects 0.000 description 1
- 230000001052 transient effect Effects 0.000 description 1
- 230000007704 transition Effects 0.000 description 1
- 230000014616 translation Effects 0.000 description 1
- 230000005945 translocation Effects 0.000 description 1
- 239000012588 trypsin Substances 0.000 description 1
- 241000701161 unidentified adenovirus Species 0.000 description 1
- 241000701447 unidentified baculovirus Species 0.000 description 1
- 241001430294 unidentified retrovirus Species 0.000 description 1
- 210000003501 vero cell Anatomy 0.000 description 1
- 108700026220 vif Genes Proteins 0.000 description 1
- 239000013603 viral vector Substances 0.000 description 1
- 230000003612 virological effect Effects 0.000 description 1
- 239000011782 vitamin Substances 0.000 description 1
- 235000013343 vitamin Nutrition 0.000 description 1
- 229940088594 vitamin Drugs 0.000 description 1
- 229930003231 vitamin Natural products 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 1
- 239000012138 yeast extract Substances 0.000 description 1
- 229910052727 yttrium Inorganic materials 0.000 description 1
- VWQVUPCCIRVNHF-UHFFFAOYSA-N yttrium atom Chemical compound [Y] VWQVUPCCIRVNHF-UHFFFAOYSA-N 0.000 description 1
Images


















































Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/78—Hydrolases (3) acting on carbon to nitrogen bonds other than peptide bonds (3.5)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
- C12N15/102—Mutagenizing nucleic acids
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/85—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells
- C12N15/86—Viral vectors
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/16—Hydrolases (3) acting on ester bonds (3.1)
- C12N9/22—Ribonucleases RNAses, DNAses
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y305/00—Hydrolases acting on carbon-nitrogen bonds, other than peptide bonds (3.5)
- C12Y305/04—Hydrolases acting on carbon-nitrogen bonds, other than peptide bonds (3.5) in cyclic amidines (3.5.4)
- C12Y305/04005—Cytidine deaminase (3.5.4.5)
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/10—Type of nucleic acid
- C12N2310/20—Type of nucleic acid involving clustered regularly interspaced short palindromic repeats [CRISPRs]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2750/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA ssDNA viruses
- C12N2750/00011—Details
- C12N2750/14011—Parvoviridae
- C12N2750/14111—Dependovirus, e.g. adenoassociated viruses
- C12N2750/14141—Use of virus, viral particle or viral elements as a vector
- C12N2750/14143—Use of virus, viral particle or viral elements as a vector viral genome or elements thereof as genetic vector
Definitions
- the present disclosure uses a modified complex of the double-stranded DNA and the complex that allows modification of the targeted site of the double-stranded DNA of the cell without double-strand breaks in the DNA. Regarding the method of modifying the double-stranded DNA.
- ZFN zinc finger nuclease
- Patent Document 1 or using TALEN in which a transcriptional activator-like (TAL) effector, which is a DNA binding module of the phytopathogenic fungus Xanthomonas genus, and a DNA endonuclease are linked, within a specific nucleotide sequence or A method of cleaving / modifying a targeted gene at a site adjacent thereto (Patent Document 2) has been reported.
- TAL transcriptional activator-like
- Cas9 nucleases from Streptococcus pyogenes are widely used as powerful genome editing tools in eukaryotes with repair pathways for DNA double-strand breaks (DSBs) (eg, Patent Document 3, 3. Non-Patent Documents 1 and 2).
- Non-Patent Document 3 target base editing mediated by cytidine deaminase, which directly edits nucleotides at the target locus without using a donor DNA containing a homology arm for the target region. Since this technique utilizes DNA deamination instead of nuclease-mediated DNA cleavage, it is less toxic to cells and can introduce mutations pinpointly. Therefore, it is expected to be applied not only as a molecular biological tool for producing genetically modified animals but also in medical treatment such as gene therapy.
- Non-Patent Document 4 an attempt has been made to reduce the molecular weight of cytidine deaminase by deleting a part of the region of cytidine deaminase.
- Non-Patent Document 4 reduces the modification efficiency of the target site as the portion deleted from the wild-type cytidine deaminase increases. ing.
- the complex also contains a uracil-DNA glycosylase inhibitor (UGI), which inhibits the function of uracil DNA glycosylase, which is important for DNA repair, thus enhancing the unwanted off-target effect. It is expected that.
- UMI uracil-DNA glycosylase inhibitor
- the present disclosure provides a complex for modifying double-stranded DNA containing miniaturized cytidine deaminase, which can reduce the size of cytidine deaminase while suppressing a decrease in the modification efficiency of the target site and can also achieve suppression of the off-target effect.
- the present inventors prepared a complex excluding UGI from the complex disclosed in Non-Patent Document 4 above.
- a complex in which the N-terminal region of the complex was further deleted was also prepared, and the modification efficiency of the target site in these complexes was verified.
- CDA1 consisting of the 1-position to 161-position regions (that is, the CDA lacking the C-terminal 32 amino acid region of the wild-type CDA), which was found to have high modification efficiency in Non-Patent Document 4, (Also also referred to as CDA1 ⁇ 161), when UGI is not used, the modification efficiency is less than two-thirds of that of the complex using wild-type CDA1.
- the present disclosure provides: (Item 1) A complex in which a nucleic acid sequence recognition module and deaminase are bound,
- the nucleic acid sequence recognition module specifically binds to a target nucleotide sequence in double-stranded DNA and
- the deaminase is smaller in size than the wild-type deaminase corresponding to the deaminase, and is modified so that the area of the cross section exposed as a result of the modification or the index indicating the area is not more than a predetermined value.
- a complex having the ability to modify the targeted site of the double-stranded DNA.
- the amino acid sequence of (3) is the hydrophilicity of the amino acid residue at the position selected from the group consisting of positions 122, 126 and 139 in the amino acid sequence shown in SEQ ID NO: 1 or the amino acid residue corresponding to the position.
- the amino acid sequence of (3) is 2 to the amino acid residue at position 122 and 139 in the amino acid sequence shown in SEQ ID NO: 1, or the hydrophilic amino acid residue of the amino acid residue corresponding to the position.
- the complex according to any one of the above items, which comprises substitutions at or above.
- the nucleic acid sequence recognition module is selected from the group consisting of the CRISPR-Cas system in which at least one DNA cleavage ability of the Cas protein has been deactivated, a zinc finger motif, a TAL effector, and a PPR motif, according to any one of the above items. The complex described.
- nucleic acid sequence recognition module is a CRISPR-Cas system in which one DNA cleaving ability of Cas protein is deactivated.
- Cas protein is a Cas9 protein.
- Item 15 A complex in which the N-terminal fragment of the nucleic acid sequence recognition module, deaminase, and the C-terminal fragment of the nucleic acid sequence recognition module are bound.
- the nucleic acid sequence recognition module When the N-terminal fragment and the C-terminal fragment of the nucleic acid sequence recognition module are refolded, the nucleic acid sequence recognition module specifically binds to the target nucleotide sequence in the double-stranded DNA and of the double-stranded DNA.
- a complex that has the ability to modify the targeted site. (Item 16)
- the deaminase is described in the above item, which is smaller in size than the wild-type deaminase corresponding to the deaminase, and is modified so that the area of the cross section exposed as a result of the modification or the index indicating the area is not more than a predetermined value.
- the deaminase is modified so that the number of hydrophobic amino acid residues appearing in the cross section exposed as a result of the modification of the deaminase is equal to or less than a predetermined value, and the modification includes deletion, any one of the above items.
- the complex described in the section. (Item 18) The deaminase is modified so that the ratio of hydrophobic residues appearing in the cross section exposed as a result of the modification to the number of modified amino acid residues is not more than a predetermined value, and the modification includes a deletion.
- the complex according to any one of the above items.
- the nucleic acid sequence recognition module is selected from the group consisting of the CRISPR-Cas system in which at least one DNA cleavage ability of the Cas protein has been deactivated, a zinc finger motif, a TAL effector, and a PPR motif, according to any one of the above items.
- the complex described. (Item 26) A nucleic acid encoding the complex according to any one of the above items.
- (Item 27) A vector containing the nucleic acid described in the above item.
- (Item 28) The vector according to the above item, which is an adeno-associated virus vector.
- (Item 29) A method for modifying a targeted site of a double-stranded DNA possessed by a cell, comprising contacting the complex according to any one of the above items with the double-stranded DNA.
- (Item 30) The method according to the above item, wherein the contact between the double-stranded DNA and the complex is carried out by introducing the nucleic acid or the vector according to any one of the above items into the cells.
- the disclosure also provides: [1] A complex in which a nucleic acid sequence recognition module and cytidine deaminase are bound,
- the nucleic acid sequence recognition module specifically binds to a target nucleotide sequence in double-stranded DNA and
- the cytidine deaminase (1) An amino acid sequence consisting of regions of amino acid residues at positions 30 to 150 in the amino acid sequence represented by SEQ ID NO: 1.
- (2) An amino acid sequence consisting of a region corresponding to the region (1), which is an ortholog of a protein consisting of the amino acid sequence shown in SEQ ID NO: 1.
- amino acid sequence of (3) An amino acid sequence in which one or several amino acids are deleted, substituted, inserted and / or added in the amino acid sequence of (1) or (2), or the amino acid of (4) (1) or (2). Amino acid sequence having 90% or more similarity or identity with the sequence, Consists of A complex that modifies the targeted site of the double-stranded DNA.
- the amino acid sequence of (3) is the hydrophilicity of the amino acid residue at the position selected from the group consisting of positions 122, 126 and 139 in the amino acid sequence shown in SEQ ID NO: 1 or the amino acid residue corresponding to the position.
- the complex according to [1] which comprises one or more substitutions with amino acid residues.
- the amino acid sequence of (3) is 2 to the amino acid residue at position 122 and 139 in the amino acid sequence shown in SEQ ID NO: 1, or the hydrophilic amino acid residue of the amino acid residue corresponding to the position.
- the nucleic acid sequence recognition module is selected from the group consisting of the CRISPR-Cas system, the zinc finger motif, the TAL effector and the PPR motif in which at least one DNA cleavage ability of the Cas protein is deactivated, [1] to [3].
- nucleic acid sequence recognition module is a CRISPR-Cas system in which one DNA cleavage ability of Cas protein is deactivated.
- Cas protein is a Cas9 protein.
- It is a complex in which the N-terminal fragment of the nucleic acid sequence recognition module, cytidine deaminase, and the C-terminal fragment of the nucleic acid sequence recognition module are bound.
- the nucleic acid sequence recognition module is characterized in that when the N-terminal fragment and the C-terminal fragment of the nucleic acid sequence recognition module are refolded, the nucleic acid sequence recognition module specifically binds to a target nucleotide sequence in the double-stranded DNA. A complex that modifies the targeted site of double-stranded DNA.
- the cytidine deaminase (1) An amino acid sequence consisting of regions of amino acid residues at positions 30 to 150 in the amino acid sequence represented by SEQ ID NO: 1.
- nucleic acid sequence recognition module is selected from the group consisting of the CRISPR-Cas system, the zinc finger motif, the TAL effector and the PPR motif in which at least one DNA cleavage ability of the Cas protein has been deactivated, in [7] or [8].
- [12] The vector according to [11], which is an adeno-associated virus vector.
- [13] A method for modifying a targeted site of double-stranded DNA possessed by a cell, comprising contacting the complex according to any one of [1] to [9] with the double-stranded DNA. .. [14] The method according to [13], wherein the contact between the double-stranded DNA and the complex is performed by introducing the nucleic acid or vector according to any one of [10] to [12] into the cell.
- a complex for modifying double-stranded DNA which is smaller in size than the conventional one, has high modification efficiency, and suppresses the off-target effect.
- the complex can be used to more safely modify the targeted site of DNA without breaking the double-stranded DNA.
- the nucleic acid encoding the complex is also mounted on an adeno-associated virus vector, which facilitates delivery of the complex to a target site, and thus may be particularly useful in application aspects such as gene therapy.
- FIG. 1 shows the alignment results of the sequences of human AID (denoted as HsAID), wild-type PmCDA1 and one aspect of deaminase of the present disclosure (PmCDA1-36).
- the sequence of HsAID is shown as SEQ ID NO: 3
- the sequence of wild-type PmCDA1 is shown as SEQ ID NO: 1
- the sequence of PmCDA1-36 is shown as SEQ ID NO: 2.
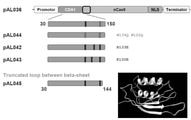
- FIG. 2 is a schematic diagram of the plasmid construct used in Example 1. The numbers in each construct indicate the amino acid positions of the proteins encoded by each sequence.
- FIG. 3 shows the results of Example 1.
- -Canavanine indicates a Canavanine-free medium
- + Canavanine indicates a Canavanine-containing medium.
- FIG. 4 shows the results of Example 1.
- -Canavanine indicates a Canavanine-free medium
- + Canavanine indicates a Canavanine-containing medium.
- FIG. 5 shows a graph of the results of Example 1. The vertical axis shows the mutation introduction rate (%). The rightmost KN1251 is a positive control.
- FIG. 6 shows the result of the three-dimensional structure analysis of CDA1. The figure on the left shows the three-dimensional structure of wild-type CDA1, and the figure on the right shows the three-dimensional structure of CDA1 ⁇ 161.
- FIG. 7 shows the exposed internal amino acid residues of CDA1 ⁇ 161 (white part in the right figure).
- FIG. 8 is a schematic diagram of the plasmid construct used in Example 2.
- FIG. 9 shows the results of Example 2.
- -Canavanine indicates a Canavanine-free medium, and + Canavanine indicates a Canavanine-containing medium.
- FIG. 10 shows the results of Example 2.
- -Canavanine indicates a Canavanine-free medium, and + Canavanine indicates a Canavanine-containing medium.
- FIG. 11 shows a graph of the results of Example 2.
- the vertical axis shows the mutation introduction rate (%).
- the rightmost KN1252 is a positive control.
- FIG. 12 is a schematic diagram of the plasmid construct used in Example 2.
- the numbers in each construct indicate the amino acid positions of the proteins encoded by each sequence.
- FIG. 13 shows a mutation-introduced portion (white portion) in the three-dimensional structure of Example 2.
- FIG. 14 shows the results of Example 2.
- -Canavanine indicates a Canavanine-free medium, and + Canavanine indicates a Canavanine-containing medium.
- FIG. 15 shows the results of Example 2.
- -Canavanine indicates a Canavanine-free medium, and + Canavanine indicates a Canavanine-containing medium.
- FIG. 16 shows a graph of the results of Example 2.
- the vertical axis shows the mutation introduction rate (%).
- the rightmost KN1252 is a positive control.
- FIG. 17 is a schematic diagram of the plasmid construct used in Example 2. The numbers in each construct indicate the amino acid positions of the proteins encoded by each sequence.
- FIG. 17 is a schematic diagram of the plasmid construct used in Example 2. The numbers in each construct indicate the amino acid positions of the proteins encoded by each sequence.
- FIG. 18 shows the results of Example 2.
- -Canavanine indicates a Canavanine-free medium, and + Canavanine indicates a Canavanine-containing medium.
- FIG. 19 shows the results of Example 2.
- -Canavanine indicates a Canavanine-free medium, and + Canavanine indicates a Canavanine-containing medium.
- FIG. 20 shows a graph of the results of Example 2.
- the target sequence of the guide RNA is different between the left figure and the right figure.
- the vertical axis of each figure shows the mutation introduction rate (%).
- the KN1252 on the far right of each figure is a positive control.
- FIG. 21 is a schematic diagram of the plasmid construct used in Example 3. The numbers in each construct indicate the amino acid positions of the proteins encoded by each sequence.
- FIG. 21 is a schematic diagram of the plasmid construct used in Example 3. The numbers in each construct indicate the amino acid positions of the proteins encoded by each sequence.
- FIG. 22 shows the results of Example 3.
- -Canavanine indicates a Canavanine-free medium
- + Canavanine indicates a Canavanine-containing medium.
- FIG. 23 shows the results of Example 3.
- -Canavanine indicates a Canavanine-free medium
- + Canavanine indicates a Canavanine-containing medium.
- FIG. 24 shows a graph of the results of Example 3.
- the target sequence of the guide RNA is different between the left figure and the right figure.
- the vertical axis of each figure shows the mutation introduction rate (%).
- the KN1252 on the far right of each figure is a positive control.
- FIG. 25 shows the schematic of the plasmid construct used in Example 3 and the tertiary structure of one aspect of the deaminase of the present disclosure.
- FIG. 26 shows a schematic diagram of the plasmid construct used in Example 3. The numbers in each construct indicate the amino acid positions of the proteins encoded by each sequence.
- FIG. 27 shows the results of Example 3. -Canavanine indicates a Canavanine-free medium, and + Canavanine indicates a Canavanine-containing medium.
- FIG. 28 shows the results of Example 3. -Canavanine indicates a Canavanine-free medium, and + Canavanine indicates a Canavanine-containing medium.
- FIG. 29 shows a graph of the results of Example 3.
- the target sequence of the guide RNA is different between the left figure and the right figure.
- the vertical axis of each figure shows the mutation introduction rate (%).
- FIG. 30 shows the results of Example 3.
- -Canavanine indicates a Canavanine-free medium
- + Canavanine indicates a Canavanine-containing medium
- FIG. 31 shows the results of Example 3.
- -Canavanine indicates a Canavanine-free medium
- + Canavanine indicates a Canavanine-containing medium
- FIG. 32 shows a graph of the results of Example 3.
- the target sequence of the guide RNA is different between the left figure and the right figure.
- the vertical axis of each figure shows the mutation introduction rate (%).
- the KN1252 on the far right of each figure is a positive control.
- FIG. 33 shows a schematic diagram of the plasmid construct used in Example 4.
- FIG. 34 shows the results of Example 4.
- -Canavanine indicates a Canavanine-free medium
- + Canavanine indicates a Canavanine-containing medium
- FIG. 35 shows the results of Example 4.
- -Canavanine indicates a Canavanine-free medium
- + Canavanine indicates a Canavanine-containing medium.
- FIG. 36 shows the results of Example 5.
- FIG. 37 shows the results of Example 5.
- FIG. 38 shows the results of Example 5.
- FIG. 39 shows the results of Example 5.
- FIG. 40 shows the results of Example 5.
- FIG. 41 shows the results of Example 5.
- FIG. 42 shows a schematic diagram of the plasmid construct used in Example 6.
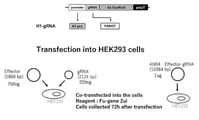
- FIG. 43 is a schematic diagram (upper diagram) of the plasmid encoding the guide RNA and a schematic diagram (lower diagram) of the experimental procedure of Example 6.
- FIG. 44 shows the results of Example 6. The nucleotides after mutation and the mutation efficiency in each nucleotide of the sequence are shown.
- FIG. 45 shows a graph of the results of Example 6. The vertical axis corresponds to the mutation efficiency, and the horizontal axis corresponds to the numerical value in the 24 well column and the arrangement in the Reference column in FIG.
- FIG. 46a is a ribbon model showing the structure of the complex of human AID and dsDNA.
- the non-catalytic double-stranded DNA binding domain is shown in green (N-terminus) and red (C-terminus), and its amino acid sequence is compared with that of PmCDA1 at the bottom.
- FIG. 46a is a prediction diagram of the space-filling structure of PmCDA1 before and after modification. In addition to the direct DNA binding sites (green and red), the segments shown in blue were trimmed to minimize the protein cross section. Mutant amino acids (W122 and W139) are shown in yellow.
- FIG. 46c is a graph showing the on-target editing efficiency of UGI-free Target-AID, AID-2S, and AID-3S in a yeast canavanine resistance assay.
- FIG. 46d is a schematic diagram showing the domain arrangement of the CBE mutant used in this example.
- the structure of BE is common to YE1, YE2, and R33A + K34A except for the point mutation of rAPOBEC1.
- 46e and 46f are graphs showing on-target editing profiles of CBE variants analyzed by deep sequencing at the HEK2, HEK3, RNF2, and VEGFA sites of HEK293T.
- the nucleotide positions having the highest C ⁇ T conversion frequency of each target are shown.
- FIG. 46f shows an average edit window for the four targets.
- the mean score (square bar) and standard deviation (error bars) are shown, and in the case of n ⁇ 9, each biological replication is shown by dots.
- FIG. 46g is a schematic diagram showing the domain structures of SaAID and SaAID-3S. A gRNA expression cassette is attached to each effector plasmid.
- FIG. 46h is a graph showing the on-target editing frequency of SaAID and SaAID-3S in HEK293T in the same manner as in FIG. 46e. To normalize transfection efficiency, cells were screened by expressing iRFP670 from the plasmid backbone.
- FIG. 47a is a graph in which the incidence of on-target mutation (canavanine resistance) and off-target mutation (thialicin resistance) was measured after inducing each construct (AID-2S, -3S, rAPOBEC1) as shown in yeast. Is. Biological repeat values were plotted for the target sites of CAN1-2 (blue dots) and CAN1-3 (orange dots).
- FIG. 47b is a schematic diagram of off-target evaluation of orthogonal R-loops.
- FIG. 47c is a graph showing the results of selection of seven off-target R-loop sites (1 to 7), co-introduction with one of the on-target sites (HEK2, HEK3, RNF2, VEGFA) and analysis by a deep sequencer. ..
- FIG. 47d is a graph showing the on-target edit vs. average off-target edit profile of all CBEs of this embodiment. The y-axis represents the mean on-target edits of the four on-target sites (HEK2, HEK3, RNF2, VEGFA) used in the R-loop assay, and the x-axis represents the mean off-target edits of the seven orthogonal R-loop sites. ing.
- FIG. 48 is a diagram showing the effect when the C-terminal of PmCDA1 is deleted.
- FIG. 48a is a prediction diagram of a series of space-filling structures of PmCDA1 with the C-terminus deleted. Non-catalytic dsDNA binding domains are shown in green (N-terminus) and red (C-terminus), respectively.
- FIG. 48b is a schematic diagram showing the C-terminal truncated Target-AID construct verified in FIG. 48c.
- FIG. 48a is a prediction diagram of a series of space-filling structures of PmCDA1 with the C-terminus deleted. Non-catalytic dsDNA binding domains are shown in green (N-terminus) and red (C-terminus), respectively.
- FIG. 48b is a schematic diagram showing the C-terminal truncated Target-AID construct verified in FIG. 48c.
- FIG. 48c is a graph showing the transition of on-target editing efficiency of the truncated construct in yeast. The incidence of Canavanine-resistant mutants was measured as a CAN1 gene mutant. Trend lines for different datasets (pink and gray dots) were plotted for the target sites of CAN1-1 and CAN1-2.
- FIG. 49 is a diagram showing the effect of deleting the N-terminal and C-terminal of PmCDA1.
- FIG. 49a is a prediction diagram of a series of space-filling structures of PmCDA1 with N-terminal and C-terminal deleted. The non-catalytic dsDNA binding domain is shown in green (N-terminus) and the blue segment indicates adjacent sites that are cleaved to smooth the shape of the protein and minimize cross section.
- FIG. 49b is a schematic diagram showing the Target-AID construct with the N-terminus and C-terminus cleaved as tested in FIG. 49c.
- FIG. 49c is a graph showing the trend of on-target editing efficiency of cleavage constructs in yeast. The incidence of Canavanine-resistant mutants was measured as a CAN1 gene mutant. Trend lines for different datasets (pink and gray dots) were plotted for the target sites of CAN1-1 and CAN1-2.
- FIG. 50 is a diagram showing the effect of amino acid substitution on cleaved PmCDA1 (30-150). Hydrophobic residues exposed after cleavage were replaced with hydrophilic residues. On-target editing efficiency was measured by yeast canavanine assay as shown in FIG.
- FIG. 51 is a diagram showing a domain-embedded Target-AID3S.
- FIG. 51a is a schematic diagram showing a domain-embedded AID-3S (1054-tCDA1EQ-1055) at the position of the RuvC domain of Cas9.
- FIG. 51b shows the on-target editing efficiency evaluated with yeast.
- Biological repeat values were plotted for the target sites of CAN1-2 (blue dots) and CAN1-3 (orange dots).
- FIG. 52 is a diagram showing an on-target editing profile of the SaCas9-AID variant.
- nucleic acid sequence recognition module specifically binds to a target nucleotide sequence in double-stranded DNA.
- the deaminase is smaller in size than the wild-type deaminase corresponding to the deaminase, and is modified so that the area of the cross section exposed as a result of the modification or the index indicating the area is not more than a predetermined value.
- a complex is provided that has the ability to modify the targeted site of the main strand DNA.
- the area of the cross section exposed as a result of modification or the index indicating the area can be set to a predetermined value or less appropriately set depending on the type of deaminase.
- the exponent and the number of hydrophobic amino acid residues can be used.
- the object of the present disclosure is to provide a base editing system that can be mounted on a single AAV vector by downsizing the deaminase.
- the present inventors preferably make it smaller so that the area of the cross section exposed when the amino acid is deleted or modified, or the index indicating the area is equal to or less than a predetermined value. Found that minimizing it leads to structural stabilization.
- the number of hydrophobic amino acids appearing in the cross section exposed when the amino acid is deleted or substituted is set to a predetermined value or less, and is reduced as compared with the case where other modifications are made, for example. , More preferably to a minimum.
- the number of hydrophobic amino acid residues appearing on the cross section exposed on the N-terminal side and the C-terminal side of the wild-type deaminase as a result of modifying the deaminase is less than or equal to a predetermined value, or is the minimum. Can be modified to be.
- the amino acid residues of the modified deaminase are not limited to the N-terminal or C-terminal, and the number of hydrophobic amino acid residues appearing in the exposed cross section as a result of modifying the deaminase is less than or equal to a predetermined value, or Amino acids inside the sequence (not at the terminus) can also be modified as long as they are modified to minimize.
- the deaminase is modified so as to reduce or minimize the ratio of the number of hydrophobic amino acid residues appearing in the cross section exposed as a result of the modification to the number of modified amino acid residues. Can be done.
- the index can be reduced by substituting the hydrophobic residue appearing in the cross section exposed as a result of the modification with the hydrophilic residue.
- at least one of the exposed hydrophobic internal amino acid residues in deaminase can be replaced with a hydrophilic amino acid residue.
- the ratio of hydrophobic residues appearing in the cross section exposed as a result of modification to the number of modified amino acid residues is, for example, about 3% or less, about 4% or less, about 5% or less.
- the number of hydrophobic amino acid residues appearing in the cross section exposed as a result of modifying the deaminase is, for example, about 1 or less, about 2 or less, about 3 or less, about 4 or less, and about 5.
- the deaminase is modified.
- the modification may be such that the number of hydrophobic amino acid residues appearing in the exposed cross section is 100 or more.
- size means the physical or chemical size of a molecule such as a protein, and includes the size such as molecular weight, occupied volume, and mass. Reducing the size includes reducing the molecular weight, volume, mass, etc. of the molecule. Preferably the molecular weight can be a more appropriate indicator.
- minimize means that a value is at least reduced or smaller than before or with other modifications, and is minimal. It does not have to be a value.
- modification includes the deletion or substitution of an amino acid.
- Whether or not a specific amino acid in a protein (such as deaminase) is exposed when modified can be accurately calculated by modeling, for example, I-TASSER (https://zhanggroup.org/I-TASSER/). ) Etc. can be used to predict the structure of proteins.
- the structure of the protein that is the basis for the structure prediction can be obtained from, for example, RCSBPDB (https://www.rcsb.org/), and if it is AID, it is 5W1C (https://www.rcsb). .org / structure / 5W1C) can be used
- the present disclosure discloses a complex for modifying double-stranded DNA, in which a nucleic acid sequence recognition module that specifically binds to a target nucleotide sequence in double-stranded DNA and a miniaturized citidine deaminase are bound.
- miniaturization complex of the present disclosure Sometimes referred to as "miniaturization complex of the present disclosure”).
- miniaturization complex of the present disclosure The following 3.
- a target double-stranded DNA eg, genomic DNA
- complex (split) of the present disclosure a complex in which the N-terminal fragment of the nucleic acid sequence recognition module, citidine deaminase, and the C-terminal fragment of the nucleic acid sequence recognition module are bound (hereinafter, "complex (split) of the present disclosure". Type) "may be referred to.) Is provided.
- the cytidine deaminase constituting the complex (split type) of the present disclosure exhibits high modification efficiency even if it is a wild type, but from the viewpoint of miniaturization and suppression of off-target effect, it is a miniaturized cytidine deaminase. Is preferable.
- the complex (split type) of the present disclosure the N-terminal fragment and the C-terminal fragment of the nucleic acid sequence recognition module are refolded so that the nucleic acid sequence recognition module is specific to the target nucleotide sequence in the double-stranded DNA. Can be combined with.
- the term “complex of the present disclosure” may be used as a term that includes both the “miniaturized complex of the present disclosure” and the “complex of the present disclosure (split type)”.
- the complex (split type) of the present disclosure contains an N-terminal fragment of a nucleic acid sequence recognition module, citidine deaminase, and a C-terminal fragment of a nucleic acid sequence recognition module from the N-terminal to the C-terminal in this order or in reverse order. It may be provided as a fusion protein, in which case at least one of the elements may be linked via a suitable linker (eg, 3xFlag linker, GS linker, etc.), without the linker. It may be combined.
- a suitable linker eg, 3xFlag linker, GS linker, etc.
- the nucleic acid sequence recognition module and citidine deaminase are each divided into two fragments, and one of the fragments is linked to form two partial complexes, which are associated to form a functional nucleic acid sequence recognition.
- Split enzymes designed to reconstitute functional citidine deaminase when the module is reconstituted and bound to the target nucleotide sequence can also be used.
- the nucleic acid sequence recognition module and cytidine deaminase were divided into N-terminal fragments and C-terminal fragments, respectively, and for example, a partial complex in which N-terminal fragments were linked and C-terminal fragments were linked.
- a functional nucleic acid sequence recognition module and a functional cytidine deaminase can be reconstituted.
- the two partial complexes may also be provided as separate molecules, or may be provided as a single fusion protein, either directly or by linking via a suitable linker.
- the position where citidine deaminase is inserted is not particularly limited, but for example, when the CRISPR-SpCas9 system is used as the nucleic acid sequence recognition module, SpCas9 (SEQ ID NO: 4) is located at positions 204 to 1054 (eg, positions 204 and 535).
- one of the amino acid residues at positions 127 to 848 (eg, positions 127, 538, 614, 690, 735, 848) of SaCas9 (SEQ ID NO: 5). It is preferable to divide SaCas9 between amino acid residues shifted by 1 amino acid residue on the C-terminal side (eg, positions 128, 539, 615, 691, 736, 849). ..
- the split site of cytidine deaminase is not particularly limited as long as the two fragmented fragments can be reconstituted into functional cytidine deaminase, and can be split at one site as an N-terminal fragment and a C-terminal fragment.
- three or more fragments generated by dividing at two or more locations can be appropriately connected to form two fragments.
- the three-dimensional structure of cytidine deaminase is known, and those skilled in the art can appropriately select the divided portion based on the information.
- miniaturized cytidine deaminase means cytidine deaminase having a reduced molecular weight as compared with wild-type cytidine deaminase by deleting a part of amino acid residues of wild-type cytidine deaminase.
- a miniaturized cytidine deaminase specifically, in one embodiment, (1) An amino acid sequence consisting of regions of amino acid residues at positions 30 to 150 in the amino acid sequence represented by SEQ ID NO: 1.
- the cytidine deaminase consisting of the amino acid sequence shown in SEQ ID NO: 1 is PmCDA1 (Petromyzon marinus cytosinedeaminase 1) derived from lamprey, and examples of such PmCDA1 orthologs include mammals (eg, humans, pigs, cows, horses, monkeys). Etc.)-derived AID (Activation-induced cytidine deaminase; AICDA) and the like.
- the nucleotide sequence and amino acid sequence of PmCDA1 cDNA can be referred to GenBank accession No. EF094822 and ABO15149
- the nucleotide sequence and amino acid sequence of human AID cDNA can be referred to GenBank accession No. NM_020661 and NP_065712, respectively.
- Amino acid sequence into which the amino acids of (iv) are inserted (iv) 1 to 50, preferably 1 to 20, and more preferably 1 to number (5, 4, 3 or 2) in the amino acid sequence shown in SEQ ID NO: 1.
- Examples thereof include citidine deaminase containing an amino acid sequence in which one of the amino acids is replaced with another amino acid, or (v) an amino acid sequence in which they are combined.
- amino acid residues at positions selected from the positions corresponding to these amino acid residues in citidine deaminase derived from animals other than Yttrium eel are listed (the alphabets in parentheses indicate amino acid residues). Of these, amino acid residues at positions selected from the group consisting of positions 122, 126 and 139 are preferable.
- hydrophilic amino acid residues include arginine, asparagine, aspartic acid, glutamic acid, glutamine, lysine, serine and threonine.
- the amino acid sequence of citidine deaminase is highly conserved among vertebrates, and by aligning the amino acid sequence of citidine deaminase from the desired animal with the amino acid sequence of PmCDA1, the corresponding deletion target site or corresponding The site of mutation can be identified. If the corresponding amino acid residue is a hydrophilic amino acid, it may not be replaced or may be replaced with another hydrophilic amino acid residue. For example, in the case of human AID, the amino acid corresponding to S30 of PmCDA1 is threonine at position 27, the amino acid corresponding to V150 of PmCDA1 is isoleucine at position 138, and the amino acid corresponding to W122 of PmCDA1 is position 109.
- Phenylalanine the amino acid corresponding to L126 of PmCDA1 is leucine at position 113, and the amino acid corresponding to W139 of PmCDA1 is arginine at position 127.
- W122 human AID F109
- W122 is glutamic acid residue or glutamic acid residue, taking PmCDA1 as an example.
- L126 L113 of human AID
- W139 R127 of human AID
- hydrophobic amino acid residues exposed to the outside of the above cytidine deaminase may be replaced with hydrophilic amino acid residues.
- positions 122, 126 and 139 Two or more amino acid residues at positions selected from the group consisting of (for example, positions 122 and 139) can be mentioned, and specifically, W122E / W139R, W122E / W139Q, W122Q / W139R, W122Q. Substitutions involving combinations of mutations such as / W139Q can be mentioned.
- similarity refers to the optimal alignment (preferably, the algorithm is optimal) when two amino acid sequences are aligned using a mathematical algorithm known in the art. It means the ratio (%) of the same amino acid and similar amino acid residues to all the overlapping amino acid residues in (which may consider the introduction of gaps in one or both of the sequences for alignment).
- similar amino acids means amino acids that are similar in physicochemical properties, such as aromatic amino acids (Phe, Trp, Tyr), aliphatic amino acids (Ala, Leu, Ile, Val), polar amino acids (Gln, Asn).
- modified DNA means that one nucleotide on the DNA strand (eg, dC) is converted or deleted to another nucleotide (eg, dT, dA or dG). This means that a nucleotide or nucleotide sequence is inserted between certain nucleotides on a DNA strand.
- the double-stranded DNA to be modified is not particularly limited as long as it is double-stranded DNA existing in the host cell, but is preferably genomic DNA (eg, chromosomal DNA, mitochondrial DNA, chloroplastome DNA, etc.). be.
- modification of a targeted site of double-stranded DNA may result in the conversion, deletion, or to the targeted site of one or more nucleotides of the targeted site with another one or more nucleotides.
- the "targeted site" of double-stranded DNA is all or part of the "target nucleotide sequence” specifically recognized and bound by the nucleic acid sequence recognition module, or in the vicinity of the target nucleotide sequence ( It means one or both of 5'upstream and 3'downstream), and the range thereof can be appropriately adjusted between 1 base and several hundred bases depending on the purpose.
- nucleic acid sequence recognition module means a molecule or molecular complex capable of specifically recognizing and binding to a specific nucleotide sequence (that is, a target nucleotide sequence) on a DNA chain.
- the binding of the nucleic acid sequence recognition module to the target nucleotide sequence allows the cytidine deaminase linked to the module to act specifically on the targeted site of the double-stranded DNA.
- the complex of the present disclosure (also referred to as “nucleic acid modifying enzyme complex”) is a complex in which the above-mentioned nucleic acid sequence recognition module and citidine deaminase are linked, and is deamination to which a specific nucleotide sequence recognition ability is imparted. It means a molecular complex having a deamination activity.
- the “complex” includes not only those composed of a plurality of molecules but also those having a nucleic acid sequence recognition module and cytidine deaminase in a single molecule, such as a fusion protein.
- the CRISPR-Cas system when used as the nucleic acid sequence recognition module, it may be a complex composed of a guide RNA, a Cas effector protein (also referred to as Cas protein or Cas nuclease), and citidine deaminase. It may also be a complex composed of a guide RNA and a fusion protein of Cas effector protein and citidine deaminase.
- the complex of the present disclosure can efficiently modify double-stranded DNA without containing an inhibitor of base excision repair.
- the use of miniaturized cytidine deaminase significantly reduces the off-target effect even when used in combination with an inhibitor of uracil DNA glycosylase, as compared with the case of using wild-type cytidine deaminase. It was proved that it could be done. It is speculated that the suppression of this off-target effect is due to the removal of the domain of cytidine deaminase, which has an affinity for unwanted DNA. Therefore, the complex of the present disclosure may be further ligated with an inhibitor of uracil DNA glycosylase.
- uracil DNA glycosylase inhibitor used in the present disclosure examples include a uracil DNA glycosylase inhibitor (UGI) derived from PBS1 which is a Bacillus subtilis bacterium, or a uracil DNA glycosylase inhibitor (UGI) derived from PBS2 which is a Bacillus subtilis bacterium. UGI), but not limited to (Wang, Z., and Mosbaugh, D. W. (1988) J. Bacteriol. 170, 1082-1091).
- PBS2-derived UGI is known to have the effect of making it difficult for mutations, cleavages, and recombination other than C to T on DNA to occur, so it is suitable to use PBS2-derived UGI.
- the target nucleotide sequence in the double-stranded DNA recognized by the nucleic acid sequence recognition module is not particularly limited as long as the module can specifically bind, and may be any sequence in the double-stranded DNA.
- the length of the target nucleotide sequence may be sufficient for the nucleic acid sequence recognition module to specifically bind, for example, depending on the size of the genome when introducing a mutation at a specific site in the genomic DNA of a mammal. It is 12 nucleotides or more, preferably 15 nucleotides or more, and more preferably 17 nucleotides or more.
- the upper limit of the length is not particularly limited, but is preferably 25 nucleotides or less.
- Examples of such a nucleic acid sequence recognition module include a CRISPR-Cas system in which at least one DNA cleavage ability of Cas is inactivated (hereinafter, also referred to as "CRISPR-mutant Cas system"), a zinc finger motif, a TAL effector, and a PPR motif.
- CRISPR-mutant Cas system in which at least one DNA cleavage ability of Cas is inactivated
- TAL effector a DNA cleavage ability of Cas is inactivated
- PPR motif fragments containing a DNA-binding domain of a protein that can specifically bind to DNA such as restriction enzymes, transcription factors, RNA polymerases, etc. and not having the ability to cleave DNA double strands
- Preferred include CRISPR-mutant Cas system, zinc finger motif, TAL effector, PPR motif and the like.
- the zinc finger motif is a combination of 3 to 6 zinc finger units of different Cys2His2 types (1 finger recognizes about 3 bases), and can recognize the target nucleotide sequence of 9 to 18 bases.
- Zinc finger motifs are Modular assembly method (Nat Biotechnol (2002) 20: 135-141), OPEN method (Mol Cell (2008) 31: 294-301), CoDA method (NatMethods (2011) 8: 67-69), E. coli. It can be produced by a known method such as the one-hybrid method (Nat Biotechnol (2008) 26: 695-701).
- Japanese Patent No. 4968498 can be referred to.
- the TAL effector has a repeating structure of modules in units of about 34 amino acids, and the 12th and 13th amino acid residues (called RVD) of one module determine the binding stability and base specificity. Ru. Since each module is highly independent, it is possible to create a TAL effector specific to the target nucleotide sequence simply by connecting the modules together.
- the TAL effector is a production method using open resources (REAL method (CurrProtoc Mol Biol (2012) Chapter 12: Unit 12.15), FLASH method (Nat Biotechnol (2012) 30: 460-465), Golden Gate method (Nucleic Acids Res). (2011) 39: e82), etc.) have been established, and it is possible to design a TAL effector for a target nucleotide sequence relatively easily.
- RRL method Current Retrimerase
- FLASH method Near Biotechnol (2012) 30: 460-465
- Golden Gate method Nucleic Acids Res. (2011) 39: e82
- the PPR motif is composed of 35 amino acids and is configured to recognize a specific nucleotide sequence by a series of PPR motifs that recognize one nucleobase, and the 1st, 4th, and ii (-2) amino acids of each motif. Only recognize the target base. Since there is no dependence on the motif composition and there is no interference from the motifs on both sides, it is possible to produce a PPR protein specific to the target nucleotide sequence simply by connecting the PPR motifs, as with the TAL effector. For details on the preparation of the PPR motif, Japanese Patent Application Laid-Open No. 2013-128413 can be referred to.
- fragments such as restriction enzymes, transcription factors, and RNA polymerases
- DNA binding domains of these proteins are well known, so that fragments containing the domains and having no DNA double-strand cleavage ability can be easily designed. And can be built.
- nucleic acid sequence recognition modules can also be provided as a fusion protein with citidine deaminase, or a protein binding domain such as SH3 domain, PDZ domain, GK domain, GB domain and their binding partners.
- the nucleic acid sequence recognition module and citidine deaminase may be fused to each other and provided as a protein complex through the interaction between the domain and its binding partner.
- intein can be fused with the nucleic acid sequence recognition module and cytidine deaminase, respectively, and both can be linked by ligation after each protein synthesis.
- Zinc finger motifs do not have high efficiency in producing zinc fingers that specifically bind to the target nucleotide sequence, and selection of zinc fingers with high binding specificity is complicated, so many zinc finger motifs that actually function are produced. Is not easy.
- the TAL effector and PPR motifs have a higher degree of freedom in recognizing the target nucleic acid sequence than the zinc finger motif, but there is a problem in terms of efficiency because it is necessary to design and construct a huge protein each time according to the target nucleotide sequence. Remains.
- the CRISPR-Cas system recognizes the sequence of target double-stranded DNA with a guide RNA complementary to the target nucleotide sequence, and thus synthesizes oligo DNA that can specifically hybridize with the target nucleotide sequence. Any sequence can be targeted simply by doing so. Therefore, in a more preferred embodiment of the present disclosure, a CRISPR-mutant Cas system in which only one or both of Cass have been deactivated is used as the nucleic acid sequence recognition module.
- the CRISPR-mutant Cas system requires CRISPR-RNA (crRNA) containing sequences complementary to the target nucleotide sequence and, optionally, trans-activating RNA (tracrRNA) and (tracrRNA) required for recruitment of the mutant Cas effector protein. If provided as a chimeric RNA with crRNA), it is provided as a complex with a mutant Cas effector protein.
- RNA molecules consisting of crRNA alone or a chimeric RNA of crRNA and tracrRNA that constitutes a nucleic acid sequence recognition module in combination with a mutant Cas effector protein are collectively referred to as "guide RNA".
- the “targeted strand” means the strand that hybridizes with crRNA, and the opposite strand is a strand that becomes a single strand by hybrid formation of the target strand and crRNA. It will be referred to as a "non-targeted strand".
- the target nucleotide sequence is represented by one strand (for example, when the PAM sequence is represented or when the positional relationship between the target nucleotide sequence and PAM is represented), it is represented by the sequence of the non-target strand.
- Cas effector protein used in the present disclosure is not particularly limited as long as it can form a complex with a guide RNA to recognize and bind to the target nucleotide sequence in the target gene and the adjacent protospaceradjacent motif (PAM).
- Cas9 or Cpf1 includes, for example, Cas9 (SpCas9; PAM sequence NGG (N is A, G, T or C; the same applies hereinafter)) derived from Streptococcus pyogenes, and Cas9 (StCas9; PAM) derived from Streptococcus thermophilus.
- Cas9 (NmCas9; PAM sequence NNNNGATT) from Neisseria meningitidis, Cas9 (SaCas9; PAM sequence: NNGRR (T)) from Staphylococcus aureus, Campiroc Examples include, but are not limited to, Cas9 (CjCas9; PAM sequence NNNVRYM (V is A, G or C; R is A or G; Y is T or C; M is A or C)) derived from jejuni). .. From the point of view of PAM constraints, SpCas9 is preferred (substantially 2 bases and can theoretically be targeted almost anywhere on the genome).
- Cpf1 examples include Cpf1 (FnCpf1; PAM sequence NTT) derived from Francisella novisida, Cpf1 (AsCpf1; PAM sequence NTTT) derived from Acidaminococcus sp., And Lachnospiraceae bacteria (AsCpf1; PAM sequence NTTT).
- Cpf1 LbCpf1; PAM sequence NTTT
- Lachnospiraceaebacterium examples include, but are not limited to, Cpf1 (LbCpf1; PAM sequence NTTT) derived from Lachnospiraceaebacterium).
- the mutant Cas effector proteins used in the present disclosure include those in which the ability to cleave both strands of the double-stranded DNA of the Cas effector protein is inactivated, and those having a nickase activity in which only the ability to cleave one strand is inactivated. , Both can be used.
- H840A variants that lack the ability to cleave the strand (and thus have nickase activity against the opposite strand of the strand that forms a complementary strand to the guide RNA), as well as its double variant (dCas9), can be used.
- the 10th Asp residue is converted to the Ala residue and / or the 556th Asp residue, the 557th His residue and / or the 580th Asn residue is converted to the Ala residue. It is possible to make a mutant of the above.
- a mutant in which the 8th Asp residue is converted to an Ala residue and / or the 559th His residue is converted to an Ala residue can also be used.
- a mutant lacking the ability to cleave both strands in which the Asp residue at position 917 was converted to Ala residue (D917A) or the Glu residue at position 1006 was converted to Ala residue (E1006A). Can be used.
- Other mutant Cas effector proteins can be used as well, as long as they lack the ability to cleave at least one strand of double-stranded DNA.
- the Cas effector protein may contain further deletions or mutations in addition to the above mutations.
- a mutant Cas effector protein having a different PAM recognition sequence from the wild type protein is also known, and such a protein is, for example, a mutant of SpCas9 of E108G / S217A / A262T / S409I / E480K / E543D / M694I / E1219V (xCas93).
- nucleic acid of the present disclosure The contact between the complex of the present disclosure, which comprises a complex in which a nucleic acid sequence recognition module and a deaminase (for example, citidine deaminase) is bound, and the double-stranded DNA is ,
- the nucleic acid encoding the complex (hereinafter, may be referred to as "nucleic acid of the present disclosure") may be introduced into a cell having the double-stranded DNA of interest.
- nucleic acid of the present disclosure can be used to produce the complex of the present disclosure and each constituent molecule of the complex by a molecular biological method.
- the nucleic acid sequence recognition module and citidine deaminase can form a complex in the host cell as a nucleic acid encoding their fusion protein or after being translated into a protein using a binding domain, intein, or the like. In various forms, they may be prepared as nucleic acids encoding each of them.
- the nucleic acid may be DNA or RNA.
- DNA it is preferably double-stranded DNA and is provided in the form of an expression vector placed under the control of a functional promoter in the host cell or in the form of an expression vector containing the DNA.
- RNA it is preferably single-stranded RNA.
- encoding a complex refers to encoding each of the molecules constituting the complex, and encoding a fusion protein having two or more constituent molecules in a single molecule. Both are included.
- DNA encoding a nucleic acid sequence recognition module such as a zinc finger motif, a TAL effector, and a PPR motif can be obtained for each module by any of the above-mentioned methods.
- the DNA encoding a sequence recognition module such as a limiting enzyme, a transcription factor, or an RNA polymerase covers a region encoding a desired portion (a portion containing a DNA binding domain) of the protein, for example, based on their cDNA sequence information. It can be cloned by synthesizing an oligo DNA primer as described above, using the total RNA or mRNA fraction prepared from the cell producing the protein as a template, and amplifying it by the RT-PCR method.
- an oligo DNA primer is synthesized so that deletion of a desired amino acid residue can be achieved based on the cDNA sequence information of the cytidine deaminase to be used, and the cytidine deaminase is produced. It can be cloned by using the total RNA or mRNA fraction prepared from cells as a template and amplifying it by the RT-PCR method. For example, the DNA encoding the miniaturized PmCDA1 of lamprey is based on the cDNA sequence (accessionNo.
- EF094822 registered in the NCBI database, and an appropriate primer is designed for the appropriate region of CDS, and the mRNA derived from lamprey is derived.
- the DNA encoding human AID can be cloned in the same manner based on the cDNA sequence (accessionNo. AB040431) registered in the NCBI database. Further, when the donor DNA is used for modifying the targeted site, the donor DNA can also be cloned in the same manner as described above based on the sequence information of the site and the like.
- the cloned DNA can be digested as is, or optionally with restriction enzymes, or appropriate linker and / or nuclear translocation signals (if the double-stranded DNA of interest is mitochondrial or blastomeal DNA, each organella translocation signal). ) Can be added and then ligated with the DNA encoding the nucleic acid sequence recognition module to prepare the DNA encoding the fusion protein. Alternatively, by fusing the DNA encoding the nucleic acid sequence recognition module and the DNA encoding citidine deaminase with the DNA encoding the binding domain or its binding partner, respectively, or by fusing both DNAs with the DNA encoding the separated intein.
- the nucleic acid sequence recognition conversion module and citidine deaminase may be capable of forming a complex after being translated in the host cell.
- the linker and / or nuclear localization signal can be ligated at appropriate positions in one or both DNAs, if desired.
- the donor DNAA may be prepared as a single DNA, or may contain a nucleic acid sequence recognition module and / or a nucleic acid encoding cytidine deaminase. It may be provided as one DNA.
- the DNA encoding the nucleic acid sequence recognition module the DNA encoding citidine deaminase, and the donor DNA
- the DNA strands are chemically synthesized, or the synthesized oligo DNA short strands that partially overlap are used by the PCR method or the Gibson Assembly method.
- the donor DNA is a single-stranded nucleic acid
- the plasmid DNA containing the DNA is digested with a restriction enzyme to form a single strand, and RNA is synthesized by an RNA polymerase.
- RNA chain can be prepared by synthesizing cDNA with reverse transcriptase and degrading the RNA chain with RNaseH.
- it can be prepared by digesting a plasmid containing donor DNA with a nickase-type restriction enzyme and separating and purifying it by electrophoresis.
- the advantage of constructing a full-length DNA in combination with chemical synthesis or the PCR method or the Gibson Assembly method is that the codons used can be designed over the entire length of the CDS according to the host into which the DNA is introduced. When expressing heterologous DNA, it is expected that the protein expression level will increase by converting the DNA sequence into codons that are frequently used in the host organism.
- the genetic code usage frequency database http://www.kazusa.or.jp/codon/index.html published on the homepage of the Kazusa DNA Research Institute.
- the literature describing the frequency of codon usage in each host may be referred to.
- An expression vector containing a nucleic acid sequence recognition module and / or a nucleic acid encoding a deaminase can be produced, for example, by linking the DNA downstream of a promoter in a suitable expression vector.
- Expression vectors include E. coli-derived plasmids (eg, pBR322, pBR325, pUC12, pUC13); Bacteriophage-derived plasmids (eg, pUB110, pTP5, pC194); Yeast-derived plasmids (eg, pSH19, pSH15); Insect cell expression.
- Plasmid eg pFast-Bac
- animal cell expression plasmid eg pA1-11, pXT1, pRc / CMV, pRc / RSV, pcDNAI / Neo
- bacteriophage such as ⁇ phage
- insect virus vector such as baculovirus (eg) Examples: BmNPV, AcNPV)
- animal virus vectors such as retrovirus, vaccinia virus, adenovirus, adeno-associated virus (AAV), etc. are used.
- the AAV vector is preferably used because the transgene can be expressed for a long period of time and because it is derived from a non-pathogenic virus and is safe.
- cytidine deaminase has a reduced molecular weight as compared with wild-type cytidine deaminase, and therefore, if necessary, a nucleic acid sequence recognition module having a lower molecular weight (for example,).
- a nucleic acid sequence recognition module having a lower molecular weight (for example,).
- the molecular weight can be reduced by deleting a part of the nucleobase module (for example, deleting the 1024th to 1054th positions of SpCas9). That is, when the CRISPR-Cas system is used as the nucleic acid sequence recognition module, the nucleic acid encoding the Cas effector protein, the nucleic acid encoding the guide RNA, and the nucleic acid encoding the cytidine deaminase are all integrated into a single AAV vector. It can also be installed. Further, in the present specification, the "nucleic acid sequence recognition module" includes not only the wild type but also a variant thereof having a nucleic acid sequence recognition ability (eg, the above-mentioned variant of SpCas9).
- a vector derived from a serotype suitable for infection of a target tissue or organ When using a viral vector as an expression vector, it is preferable to use a vector derived from a serotype suitable for infection of a target tissue or organ.
- a vector based on AAV1, 2, 3, 4, 5, 7, 8, 9 or 10 when targeting the heart.
- AAV1,3,4,6 or 9 based vector, AAV1,5,6,9 or 10 based vector when targeting lung, liver Uses AAV2,3,6,7,8,9-based vectors, and AAV1,2,6,7,8,9-based vectors when targeting skeletal muscle.
- AAV2 for cancer treatment.
- serotype of AAV for example, WO2005 / 0333321 A2 can be referred to.
- the promoter may be any promoter as long as it is suitable for the host used for gene expression. Although conventional methods with DSBs may significantly reduce host cell viability due to toxicity, it is desirable to use an inducible promoter to increase the number of cells before induction begins, but the complex of the present disclosure. Since sufficient cell proliferation can be obtained even if the promoter is expressed, the constituent promoter can be used without limitation. For example, if the host is an animal cell, SR ⁇ promoter, SV40 promoter, LTR promoter, CMV (cytomegalovirus) promoter, RSV (Laus sarcoma virus) promoter, MoMuLV (Morony mouse leukemia virus) LTR, HSV-TK (herpes simplex virus).
- Viral thymidine kinase promoter and the like are used. Of these, the CMV promoter, SR ⁇ promoter and the like are preferable.
- the trp promoter, lac promoter, recA promoter, ⁇ PL promoter, lpp promoter, T7 promoter and the like are preferable.
- the SPO1 promoter, SPO2 promoter, penP promoter and the like are preferable.
- the host is yeast, Gal1 / 10 promoter, PHO5 promoter, PGK promoter, GAP promoter, ADH promoter and the like are preferable.
- the host is an insect cell
- a polyhedrin promoter, a P10 promoter, or the like is preferable.
- the host is a plant cell
- the CaMV35S promoter, CaMV19S promoter, NOS promoter and the like are preferable.
- a vector containing an enhancer, a splicing signal, a terminator, a poly A addition signal, a drug resistance gene, a selection marker such as an auxotrophic complementary gene, an origin of replication, etc. shall be used, if desired. Can be done.
- RNA encoding the nucleic acid sequence recognition module and / or citidine deaminase can be obtained, for example, by transcribing the above-mentioned nucleic acid sequence recognition module and / or DNA encoding citidine deaminase into mRNA by a known in vitro transcription system. Can be prepared.
- the DNA encoding the guide RNA contains a crRNA sequence (eg, FnCpf1 as a Cas effector protein) containing a nucleotide sequence complementary to the target nucleotide sequence (also referred to herein as a "targeting sequence").
- a crRNA sequence eg, FnCpf1 as a Cas effector protein
- a nucleotide sequence complementary to the target nucleotide sequence also referred to herein as a "targeting sequence”
- the coding sequence of SEQ ID NO: 2; crRNA containing AAUU UCUAC UGUU GUAGA U can be used on the 5'side of the targeting sequence, and the underlined sequences form a base pair to form a stem-loop structure).
- the crRNA coding sequence and, if necessary, the known tracrRNA coding sequence for example, as the tracrRNA coding sequence when recruiting Cas9 as a Cas effector protein, gttttagagctagaaatagcaagttaaaataaggctagtccgttatcaacttgaaaaagtggcaccgagtcggtgcttttttttt;
- An oligo DNA sequence linked with in the case of SaCas9), etc.
- the length of the targeting sequence is not particularly limited as long as it can specifically bind to the target nucleotide sequence, but is, for example, 15 to 30 nucleotides, preferably 18 to 25 nucleotides.
- the targeting sequence for example, when Cas9 is used as the Cas effector protein, PAM (for example, in the case of SpCas9) is used from the CDS sequence of the target gene using a public guide RNA design website (CRISPRDesignTool, CRISPRdirect, etc.). , NGG) lists 20mer sequences adjacent to the 3'side, and when C within 7 nucleotides is converted to T in the 3'direction from the 5'end, amino acid changes occur in the protein encoded by the target gene. This can be done by selecting such an array. Further, when a targeting sequence length other than 20 mer is used, the sequence can be appropriately selected.
- a candidate sequence having a small number of off-target sites in the target host genome can be used as the targeting sequence. If the guide RNA design software used does not have the ability to search off-target sites in the host genome, for example, for 8-12 nucleotides on the 3'side of the candidate sequence (the seed sequence that is highly discriminating in the target nucleotide sequence), the host genome. You can search off-target sites by performing a Blast search on.
- the DNA encoding the guide RNA can also be inserted into the same expression vector as above, but the promoter includes pol III promoters (eg, SNR6, SNR52, SCR1, RPR1, U3, U6, H1 promoter, etc.). And a terminator (eg, poly T sequence (T 6 sequence, etc.)) is preferably used.
- pol III promoters eg, SNR6, SNR52, SCR1, RPR1, U3, U6, H1 promoter, etc.
- a terminator eg, poly T sequence (T 6 sequence, etc.) is preferably used.
- the targeted site of double-stranded DNA comprises contacting the complex of the present disclosure with double-stranded DNA of a host cell.
- the modification method of the present disclosure comprises a complex (including a fusion protein) in which a nucleic acid sequence recognition module and cytidine deaminase are bound.
- the complex of the present disclosure which comprises a complex (including a fusion protein) in which a nucleic acid sequence recognition module and cytidine deaminase are bound
- double-stranded DNA is performed as a cell-free enzymatic reaction.
- 1. And 2. It is desirable to carry out by introducing the complex or nucleic acid or vector of the present disclosure described in the above into a host and culturing the host.
- nucleic acid sequence recognition module and a complex with cytidine deaminase into cells resulted in deletion or insertion of one or more nucleotides as well as nucleotide conversion at the target site.
- modification of the targeted site is targeted not only to the conversion of one or more nucleotides of the targeted site to another one or more nucleotides, but also to the deletion of one or more nucleotides. It may be the insertion of one or more nucleotides into the site.
- the modification method of the present disclosure may include a step of introducing donor DNA into cells.
- a nucleic acid sequence recognition module and / or a nucleic acid encoding a deaminase (eg, citidine deaminase) or an expression vector containing the nucleic acid is introduced into a host cell, and the host cell is cultured to obtain a nucleic acid sequence recognition module and a deaminase (eg, citidine deaminase). ) Can also be expressed intracellularly. Since the modification method of the present disclosure does not involve DNA double-strand break (DSB), it is possible to edit the genome with low toxicity, and such a method can be applied to a wide range of biological materials.
- DSB DNA double-strand break
- the cells into which the nucleic acid sequence recognition module and / or the nucleic acid encoding citidine deaminase are introduced are from cells of microorganisms such as E. coli, which is a prokaryotic organism, and yeast, which is a lower eukaryote, to mammals such as humans. It can include cells of all species, including cells of higher eukaryotes such as vertebrates including animals, insects and plants.
- Escherichia spp. for example, Escherichia spp., Bacillus spp., Yeast, insect cells, insects, animal cells and the like are used.
- Escherichia spp. Are Escherichia coli K12 / DH1 [Proc. Natl. Acad. Sci.
- yeast examples include Saccharomyces cerevisiae AH22, AH22R-, NA87-11A, DKD - 5D, 20B-12, Schizosaccharomycespombe NCYC1913, NCYC2036, Pichia pastoris Etc. are used.
- insect cells examples include, when the virus is AcNPV, Spodoptera frugiperda cells (Sf cells), MG1 cells derived from the middle intestine of Trichoplusiani, and High Five TM cells derived from eggs of Trichoplusia ni. Cells derived from Mamestra brassicae, cells derived from Estigmena acrea, etc. are used.
- Sf cells silk moth-derived cell lines (Bombyx mori N cells; BmN cells) or the like are used as insect cells.
- Sf cells for example, Sf9 cells (ATCCCRL1711), Sf21 cells [above, In Vivo, 13, 213-217 (1977)] and the like are used.
- insects for example, silk moth larvae, Drosophila, crickets, etc. are used [Nature, 315, 592 (1985)].
- animal cells examples include monkey COS-7 cells, monkey Vero cells, Chinese hamster ovary (CHO) cells, dhfr gene-deficient CHO cells, mouse L cells, mouse AtT-20 cells, mouse myeloma cells, rat GH3 cells, and humans.
- Cell lines such as FL cells, pluripotent stem cells such as human and other mammalian iPS cells and ES cells, and primary cultured cells prepared from various tissues are used.
- zebrafish embryos, Xenopus oocytes and the like can also be used.
- the plant cells were prepared from various plants (for example, grains such as rice, wheat and corn, commercial crops such as tomato, cucumber and eggplant, garden plants such as carnation and Vietnamese ginkgo, experimental plants such as tobacco and white indigo plant). Suspended cultured cells, callus, protoplasts, leaf sections, root sections, etc. are used.
- the introduction of the expression vector depends on the type of host, known methods (eg, lysoteam method, competent method, PEG method, CaCl 2 coprecipitation method, electroporation method, microinjection method, particle gun method, lipofection method, etc. It can be carried out according to the Agrobacterium method, etc.).
- Donor DNA can also be introduced into cells by a similar method. When the expression vector and the donor DNA are introduced as different molecules, the expression vector and the donor DNA may be introduced at the same time or at different timings.
- E. coli can be transformed, for example, according to the methods described in Proc. Natl. Acad. Sci. USA, 69, 2110 (1972) and Gene, 17, 107 (1982).
- Bacillus can be vector-introduced, for example, according to the method described in Molecular & General Genetics, 168, 111 (1979).
- Yeast can be vector-introduced according to, for example, Methods in Enzymology, 194, 182-187 (1991), Proc. Natl. Acad. Sci. USA, 75, 1929 (1978).
- Insect cells and insects can be vector-introduced according to, for example, the methods described in Bio / Technology, 6, 47-55 (1988).
- Animal cells can be vector-introduced, for example, according to the method described in Cell Engineering Annex 8 New Cell Engineering Experiment Protocol, 263-267 (1995) (published by Shujunsha), Virology, 52, 456 (1973).
- Culturing of cells into which the nucleic acid of the present disclosure has been introduced can be carried out according to a known method depending on the type of host.
- a liquid medium is preferable as the medium used for culturing.
- the medium preferably contains a carbon source, a nitrogen source, an inorganic substance and the like necessary for the growth of the transformant.
- a carbon source for example, glucose, dextrin, soluble starch, sucrose, etc .
- a nitrogen source for example, ammonium salts, nitrates, corn steep liquor, peptone, casein, meat extract, soybean meal, etc.
- Inorganic or organic substances such as potato extracts; examples of the inorganic substances include calcium chloride, sodium dihydrogen phosphate, magnesium chloride and the like, respectively.
- yeast extract, vitamins, growth promoting factors and the like may be added to the medium.
- the pH of the medium is preferably about 5 to about 8.
- an M9 medium containing glucose and casamino acid is preferable.
- a drug such as 3 ⁇ -indrill acrylic acid may be added to the medium to allow the promoter to work efficiently. Culturing of E. coli is usually carried out at about 15 to about 43 ° C.
- Ventilation or stirring may be performed.
- Culture of Bacillus spp. Is usually carried out at about 30 to about 40 ° C. If necessary, ventilation or stirring may be performed.
- As a medium for culturing yeast for example, Burkholder's minimum medium [Proc. Natl. Acad. Sci. USA, 77, 4505 (1980)] and SD medium containing 0.5% casamino acid [Proc. Natl. Acad. Sci. USA, 81, 5330 (1984)] and the like.
- the pH of the medium is preferably about 5 to about 8.
- Culturing is usually carried out at about 20 ° C to about 35 ° C. If necessary, aeration or stirring may be performed.
- a medium for culturing insect cells or insects for example, Grace's Insect Medium [Nature, 195, 788 (1962)] to which an additive such as deactivated 10% bovine serum is appropriately added is used.
- the pH of the medium is preferably from about 6.2 to about 6.4.
- Culturing is usually carried out at about 27 ° C. Ventilation and agitation may be performed as needed.
- MEM minimum essential medium
- fetal bovine serum Science, 122, 501 (1952)]
- DMEM Dalveco modified Eagle's medium
- RPMI 1640 medium RPMI 1640 medium
- the pH of the medium is preferably about 6 to about 8.
- Culturing is usually carried out at about 30 ° C to about 40 ° C. Ventilation and agitation may be performed as needed.
- MS medium As a medium for culturing plant cells, MS medium, LS medium, B5 medium and the like are used.
- the pH of the medium is preferably about 5-8.
- Culturing is usually carried out at about 20 ° C to about 30 ° C. Ventilation and agitation may be performed as needed.
- the complex of the nucleic acid sequence recognition module and cytidine deaminase that is, the complex of the present disclosure can be expressed intracellularly.
- the nucleic acid sequence recognition module and / or the RNA encoding the miniaturized deaminase can be introduced into the host cell by a microinjection method, a lipofection method, or the like. RNA introduction can be repeated once or multiple times (eg, 2-5 times) at appropriate intervals.
- donor DNA means DNA containing a foreign insertion sequence
- the donor DNA usually has two regions upstream and downstream of the target site, which are adjacent to the target site (hereinafter, “adjacent”). It contains two types of sequences (hereinafter also referred to as “homology arms”) that are homologous to the sequences of "regions"). When distinguishing each homology arm, it may be distinguished by "5'homology arm” and “3'homology arm”.
- the "target site” of the double-stranded DNA means a region to be replaced by an insertion sequence contained in the donor DNA, or an interval between nucleotides into which the insertion sequence is to be inserted, and the target site. Does not include the adjacent sequence.
- sequence homologous to the adjacent region of the target site is not only the exact same sequence, but preferably 80% or more (eg, 85) of the completely identical sequence as long as homologous recombination can occur in the cell. It may be a sequence having the same identity of% or more, 90% or more, 95% or more, 96% or more, 97% or more, 98% or more, 99% or more).
- Inserted sequences include drug resistance genes (eg, canamycin resistance gene, ampicillin resistance gene, puromycin resistance gene, etc.), thymidine kinase gene, diphtheriatoxin gene and other selectable marker sequences, green fluorescent protein (GFP), as required. , Red fluorescent protein, ⁇ -glucuronidase (GUS), reporter gene sequences such as FLAG, etc. can be included.
- LoxP sequence, FRT sequence or transposon-specific terminal inversion sequence may be provided before and after these genes so that these genes can be excised after cell selection or the like is completed.
- Preferred transposons include, for example, piggyBac, which is a transposon derived from lepidopteran insects (Kaji, K. et al., Nature, 458: 771-775 (2009), Waltjen et al., Nature, 458: 766. -770 (2009), WO 2010/012077).
- piggyBac is a transposon derived from lepidopteran insects
- an expression vector containing the above drug resistance gene is co-introduced, and transient drug selection (for several days) is performed. May be done.
- Whether the inserted sequence is inserted into the target site or replaced with the target site is confirmed by decoding the sequence and screening the chromosomal DNA separated and extracted from the cells by Southern hybridization or PCR. If the drug resistance gene or the like is present in the donor DNA, its expression can be confirmed as an index.
- the donor DNA may be linear (eg, synthetic double-stranded DNA), circular (eg, plasmid DNA), or single-stranded DNA (eg, single-stranded oligodeoxynucleotide). (SsODN)) or double-stranded DNA.
- the donor DNA can be appropriately designed depending on the base length of the insertion sequence, the homologous recombination activity of the host cell, and the like. For example, if the insertion sequence is 100 bases or less in length, ssODN or synthetic double-stranded DNA is usually used, and if it is longer than that, synthetic double-stranded DNA or plasmid DNA is usually used.
- the length of the donor DNA is also not particularly limited, and can be appropriately designed depending on the length of the insertion sequence and the like.
- the length of the inserted sequence is not particularly limited, and is usually in the range of 1 base length to tens of thousands of bases (for example, in the case of ssODN, 100 bases or less (eg, 70 bases or less, 50 bases or less)). It can be appropriately designed according to the purpose.
- the length of each homology arm is also not particularly limited. When the donor DNA is ssODN, the one having a length of 10 to 150 bases is usually used, and when the donor DNA is a synthetic double-stranded DNA, it is usually 10 to.
- the donor DNA is a plasmid DNA
- a DNA having a length of 5000 bases is usually used, and a DNA having a length of 100 bases to 5000 bases, preferably 500 bases to 1000 bases is preferably used.
- These donor DNAs were designed with reference to publicly known literature (eg OchiaiH, Int J Mol Sci, 16: 21128-21137 (2015), Hockemeyer D et al., Nat Biotefchnol, 27: 851-857 (2009)). can do.
- the modification method of the present disclosure it is also possible to modify the targeted site by using a plurality of target nucleotide sequences at different positions. Therefore, in one embodiment of the present disclosure, two or more nucleic acid sequence recognition modules that specifically bind to different target nucleotide sequences can be used. In this case, each one of these nucleic acid sequence recognition modules and cytidine deaminase form a complex. Here, common cytidine deaminase can be used.
- a common complex of mutant Cas effector protein and citidine deaminase is used, and different target nucleotide sequences are used as guide RNA (crRNA or crRNA-tracrRNA chimera). It is possible to prepare and use two or more kinds of chimeric RNAs of two or more crRNAs or two or more crRNAs each forming a complementary strand with tracrRNA.
- nucleic acid sequence recognition module when a zinc finger motif, a TAL effector, or the like is used as the nucleic acid sequence recognition module, for example, cytidine deaminase can be bound to each nucleic acid sequence recognition module that specifically binds to a different target nucleotide.
- an expression vector containing the DNA encoding the complex is introduced into the host cell as described above, but in order to efficiently introduce the mutation, it is constant. It is desirable that the expression of the complex above a certain level is maintained for a period of time or longer. From this point of view, it is certain that the expression vector will be integrated into the host genome, but since sustained expression of the complex increases the risk of off-target cleavage, after successful target site modification has been achieved. , It is preferable to remove it promptly. Examples of means for removing the DNA integrated into the host genome include a method using a Cre-loxP system, a FLP-FRT system, a method using a transposon, and the like.
- transiently expressing the complex of the present disclosure in the host cell for a period of time required for the deamination reaction to occur at the desired time and the modification of the targeted site to be fixed. Editing of the host genome can be efficiently realized while avoiding the risk of off-target cleavage.
- Those skilled in the art can appropriately determine a suitable expression induction period based on the culture conditions and the like used. For example, when budding yeast is liquid-cultured in 0.02% galactose-inducing medium, an expression-inducing period of 20 to 40 hours is exemplified.
- a construct (expression vector) containing a nucleic acid encoding the complex in a form in which the expression period can be controlled is prepared.
- the expression period can be controlled includes those in which the nucleic acid of the present disclosure is placed under the control of an inducible regulatory region.
- the "inducible regulatory region” is not particularly limited, and examples thereof include an operon between a temperature-sensitive (ts) mutant repressor and an operator controlled by the repressor.
- ts mutant repressor examples include, but are not limited to, ts variants of the cI repressor derived from ⁇ phage.
- ⁇ phage cI repressor ts
- it binds to the operator at 30 ° C or lower (eg, 28 ° C) and suppresses downstream gene expression, but at high temperatures of 37 ° C or higher (eg, 42 ° C), the operator.
- Gene expression is induced to dissociate from. Therefore, the host cell into which the nucleic acid of the present disclosure has been introduced is usually cultured at 30 ° C. or lower, the temperature is raised to 37 ° C.
- the host cell is cultured for a certain period of time to carry out a deamination reaction, and the target gene is subjected to the deamination reaction.
- the temperature can be quickly returned to 30 ° C or lower to minimize the period during which the expression of the target gene is suppressed, and even when targeting an essential gene for the host cell, side effects can occur. You can edit efficiently while holding down.
- temperature-sensitive mutations for example, by mounting a temperature-sensitive mutant of a protein required for autonomous replication of a vector into a vector containing a DNA encoding the complex of the present disclosure, after expression of the complex, Autonomous replication is not possible rapidly, and the vector spontaneously sheds with cell division.
- temperature-sensitive mutant proteins include, but are not limited to, temperature-sensitive mutants of Rep101 ori required for replication of pSC101 ori.
- Rep101 ori acts on pSC101ori at 30 ° C or lower (eg, 28 ° C) to enable autonomous replication of the plasmid, but at 37 ° C or higher (eg, 42 ° C) it loses its function and the plasmid becomes autonomous. It cannot be duplicated. Therefore, when used in combination with the cI repressor (ts) of the above ⁇ phage, the transient expression of the complex of the present disclosure and the removal of the plasmid can be performed at the same time.
- the DNA encoding the complex of the present disclosure is subjected to host cells under the control of an inducible promoter (eg, lac promoter (induced by IPTG), cspA promoter (induced by cold shock), araBAD promoter (induced by arabinose), etc.).
- an inducible promoter eg, lac promoter (induced by IPTG), cspA promoter (induced by cold shock), araBAD promoter (induced by arabinose), etc.
- the promoter is introduced into the medium, and the inducer is added (or removed from the medium) to the medium at an appropriate time to induce the expression of the complex, and the complex is cultured for a certain period of time to undergo a nucleic acid modification reaction, and the target gene is mutated.
- Transient expression of the complex can be achieved after introduction.
- Saccharomyces cerevisiae BY4741 strain (leucine and uracil requirement) was used and cultured in a Dropout composition suitable for the auxotrophy of standard YPDA medium or SD medium.
- the culture was carried out between 25 ° C. and 30 ° C. by static culture on an agar plate or shaking culture on a liquid medium.
- the lithium acetate method was used, and selection was performed with an SD medium suitable for appropriate auxotrophy.
- the mutation rate was calculated and evaluated by using the number of surviving colonies on the SD plate as the total number of cells and the number of surviving colonies on the Canavanine plate as the number of resistant mutant strains.
- the cell suspension was appropriately diluted and applied to SD plate medium and SD + 100 mg / l S-aminoethyl-L-cysteine (Thialysine), and the number of colonies appearing after 3 days was determined. It was counted as the number of viable cells.
- the off-target mutation rate was calculated and evaluated by using the number of surviving colonies on the SD plate as the total number of cells and the number of surviving colonies on the Thirysine plate as the number of resistant mutants.
- DNA was processed and constructed by any of PCR method, restriction enzyme treatment, ligation, Gibson assembly method, and artificial chemical synthesis.
- plasmid pRS415 for leucine selection and pRS426 for uracil selection were used as the backbone for the yeast / E. coli shuttle vector.
- the plasmid was amplified with E. coli strain XL-10gold or DH5 ⁇ and introduced into yeast by the lithium acetate method.
- the vector 1251 used as a control in the following examples contains a sequence encoding the conventional dCas9-dSH3-CDA (UGI is not included), and the vector 1252 is the conventional nCas9-dSH3-CDA (UGI). Contains an array encoding (does not contain) (dSH3 is a linker).
- pAL008 contains a sequence encoding CDA-nCas9, and CDA is fused to the N-terminal side of Cas9 without a linker.
- HEK293T cells Human fetal kidney-derived cells
- DME-glutamax medium Thermo Fisher Scientific, USA
- penicillin-streptomycin Life Technologies, Carlsbad, CA, USA
- FBS fetal bovine serum
- the cells were cultured at 37 ° C and 5% CO 2 conditions.
- 5% trypsin was used for cell recovery.
- HEK293T cells stored in a deep freezer were lysed in a water bath at 37 ° C and seeded in 75T-flask to 5x10 6 cells.
- ⁇ Sequencing> For the analysis of mutation frequency and mutation site, cultured cells were collected 24 to 72 hours after transfection, DNA was extracted, the target region was amplified by PCR, and then amplicon analysis was performed using the next-generation sequencer Miniseq. Data processing was performed by CLC workbench. In some cases, construct-expressing cells were sorted by a cell sorter using the fluorescence of GFP or RFP as an index for cell enrichment.
- Example 1 Verification of miniaturization of cytidine deaminase due to deletion of the terminal region
- the nucleic acid modifying enzyme complex containing miniaturized cytidine deaminase disclosed in Non-Patent Document 4 also contains a uracil-DNA glycosylase inhibitor (UGI).
- UGI uracil-DNA glycosylase inhibitor
- the use of UGI is expected to enhance unwanted off-target effects.
- the mutagenesis efficiency is saturated in the presence of UGI in yeast cells, and the difference is difficult to see. Therefore, first, a complex obtained by removing UGI from the complex disclosed in Non-Patent Document 4 was prepared.
- Example 2 Verification of miniaturization of cytidine deaminase based on three-dimensional structure Therefore, instead of simply deleting the terminal region of cytidine deaminase, the deletion site of cytidine deaminase is determined based on the structure of cytidine deaminase. did.
- the structure of CDA1 ⁇ 161 was analyzed using three-dimensional structure analysis software, it was a distorted spherical shape (Fig. 6, left figure). Therefore, by extracting only the white part in the left figure of FIG. 6 (that is, the region at positions 30 to 150 of PmCDA1), the shape was made closer to a spherical shape.
- PmCDA1 (30-150) improved the modification efficiency of the target site as compared with CDA1 ⁇ 161, which has a high molecular weight of cytidine deaminase.
- cytidine deaminase sometimes referred to as PmCDA1 (30-150; W122E) in which tryptophan at position 122 was replaced with glutamic acid showed particularly high modifying activity.
- PmCDA1 (30-150; W122E) the modification efficiency of the target site was verified using a complex in which hydrophobic amino acids were further replaced with hydrophilic amino acids.
- the outline of the construct used in this experiment is shown in FIG. 12, the mutation-introduced portion (white portion) in the three-dimensional structure is shown in FIG. 13, and the results are shown in FIGS. 14 to 16.
- PmCDA1 (30-150; W122E) is particularly high in cytidine deaminase (sometimes referred to as PmCDA1 (30-150; W122E; W139R)) in which tryptophan at position 139 is replaced with arginine. Modification activity was observed.
- FIG. 17 The outline of the construct used in this experiment is shown in FIG. 17, and the results are shown in FIGS. 18 to 20. From FIGS. 18 to 20, PmCDA1 (30-150; W122E) is particularly high in cytidine deaminase (sometimes referred to as PmCDA1 (30-150; W122E; W139Q)) in which tryptophan at position 139 is replaced with glutamine. Modification activity was observed.
- Example 3 Verification of Modification Efficiency of Nucleic Acid-Modifying Enzyme Complex Using Split SpCas9
- the modification efficiency of the nucleic acid-modifying enzyme complex could be improved by a completely different approach.
- a cytidine deaminase fused to the end of the Cas protein is used, but by embedding the cytidine deaminase inside the Cas protein, the stability as a complex is improved and the substrate DNA is used.
- a proof-of-concept experiment was conducted under the hypothesis that access to the protein would be improved. The outline of the construct used in this experiment is shown in FIG. 21, and the results are shown in FIGS. 22 to 24. From FIGS. 22 to 24, high modification efficiency was observed in all the produced complexes.
- Example 4 Verification of modification efficiency of nucleic acid modifying enzyme complex having miniaturized cytidine deaminase and UGI
- UGI modified cytidine deaminase
- FIG. 33 The outline of the construct used in this experiment is shown in FIG. 33, and the results are shown in FIGS. 34 and 35. From FIGS. 34 and 35, high modification efficiency was observed in all the produced complexes.
- Example 5 Verification of Off-Target Effect of Nucleic Acid-Modifying Enzyme Complex The off-target effect was verified for the complex in which high modification efficiency was observed in the above Examples. The results are shown in FIGS. 36 to 41. From these results, the off-target effect was significantly suppressed in all the prepared complexes as compared with the control (using KN1252_UG1).
- Example 6 Verification of modification using small Cas9 (SaCas9) Next, using SaCas9 instead of SpCas9 in Example 3, it was verified whether or not modification was observed in the same manner.
- SaCas9 and miniaturized deaminase an expression cassette of a nucleic acid modifying enzyme complex containing a guide RNA expression cassette (consisting of a promoter, a gRNA coding sequence and a poly T sequence) can be loaded into an AAV vector (about 4.4 kb). ) It is now possible to: The outline of the construct used in this experiment to be encoded is shown in FIGS. 42 (nucleic acid modifying enzyme complex) and the upper figure (guide RNA) of FIG.
- FIGS. 44 and 45 the outline of the experimental procedure is shown in the lower figure of FIG. 43, and the results are shown in FIGS. 44 and 45. From FIGS. 44 and 45, high modification efficiency was observed in all the produced complexes. It is also expected to suppress the off-target effect. Furthermore, interestingly, it was shown that the insertion portion of deaminase into Cas9 changes the site where the mutation is likely to be introduced. Therefore, by adjusting the insertion site, it is possible to adjust the mutation introduction site.
- Example 7 Removal of DNA-binding region of PmCDA1 and restoration of deaminase activity DNA deaminase has a unique affinity for DNA and causes non-specific deamination. From the structure of hAID, which is a human homologue of PmCDA1, it has been clarified that a complex is formed with double-stranded DNA in a region different from the catalyst core (Fig. 46a). From the amino acid sequences of hAID and PmCDA1, the potential DNA binding sites of PmCDA1 were located at residues 21-27 and residues 172-192 of the total length of 208 amino acids of the protein (FIG. 46a).
- the nCas9 fusion structure may have a great influence on its base editing characteristics.
- deaminase can be embedded in the middle. Structurally, the position of the 1054 amino acid in the RuvC domain of Cas9 is on the flexible protein surface and is close to the non-target DNA strand to be deaminated.
- the tCDA1EQ fused to the N-terminus showed variability in editing efficiency among the target sites evaluated by the CAN1 assay, but the embedded one showed consistent editing efficiency equivalent to that of the original Target-AID (Fig.). 46d, FIG. 51).
- Example 8 Evaluation of AID-2S and AID-3S in mammalian cells
- the editing efficiency and window of AID-2S and AID-3S in human HEK293T cells were evaluated, and it was reported that the off-target effect was reduced. It was compared with the existing improved cytosine base editors YE1, YE2, R33A + K34A.
- Four well-studied on-target sites HEK2, HEK3, RNF2, VEGFA
- Target-AID, AID-2S, and YE1 consistently showed high efficiency for all four target sites tested.
- AID-3S and YE2 showed moderate to high efficiency depending on the target site.
- R33A + K34A was inefficient at the target site of HEK3 (Fig. 46e).
- the average editing window width of AID-2S was narrower than that of Target-AID and was about the same as that of YE1 and YE2 (FIG. 46f).
- gRNA-independent off-target effect was evaluated by the orthogonal SaCas9 R-loop assay using HEK293T cells (Fig. 47b).
- Sites 1-6 which are off-targets for SaCas9, were selected based on previous studies, and site 7 (VEGFA locus) is highly sensitive to CBE deamination due to its C-rich context. Selected because it may indicate.
- Target-AID showed off-target editing that was detectable at all seven sites (Fig. 47c), whereas AID2S had no antitargeting that was detectable at sites 1, 3, and at sites 2, 5, 6, and 7. Off-target editing was greatly reduced, and the results were equivalent to those of YE2 and R33A + K34A.
- YE1 showed slightly higher off-target editing at sites 6 and 7.
- AID-3S was the lowest at 7 sites and was barely detected. It is considered that this is because the affinity with DNA is lost and the access of the enzyme to other than the DNA strand to which Cas9 is bound is sterically restricted.
- AID-2S and -3S reduced R-loop off-target editing by an average of about 4.5 and 13.7 times compared to the original Target-AID, but on-target editing was nearly efficient. It was maintained (Fig. 47c, 47d). Together with the yeast LYP1 assay, these results consistently support that AID-2S and -3S have significantly reduced gRNA-independent off-target effects throughout the genome.
- Example 9 Minimized modified PmCDA1 (tCDA1EQ) of the cytosine base editing system is significantly smaller (121 amino acids) than the wild type (208 amino acids).
- the small molecular size as a genome editing component is particularly advantageous for in vivo delivery tools such as AAV vectors whose DNA length is limited to 4-5 kb. Even with a small SaCas9 system, the addition of a base editing component clearly exceeds the size limit (Fig. 46g).
- tCDA1EQ was added to the residues 615-616 of nSaCas9 in the HNH domain facing the polynucleotide-binding cleft. Incorporated in position.
- a small Scp1 promoter and SpA terminator were used to construct a gRNA expression cassette having a total length of 4036 bp and 332 bp.
- SaCas9 version of Target-AID has also been developed.
- This SaAID has a total length of 5220 bp by adding a linker, a UGI, a CMV promoter, and an SV40 terminator to the total length of PmCDA1, and no gRNA cassette is added.
- Cells transfected with the fluorescent signal of iRFP670 expressed from the vector backbone were selected to normalize transfection efficiencies that depended on the size of the vector. At the two target sites tested, both constructs showed comparable editing efficiencies, albeit with differences in mutation windows (FIG. 52).
- AID-3S has also been demonstrated in the SaCas9 ortholog, providing the smallest base editing system of the same size that can be mounted on a single AAV vector, facilitating its application to safer gene therapy.
- the present disclosure provides a complex for modifying double-stranded DNA, which is smaller in size than conventional ones, has high modification efficiency, and suppresses the off-target effect.
- the nucleic acid encoding such a complex is also mounted on an adeno-associated virus vector, and it becomes easy to deliver the complex to a target site, so that it can be particularly useful in an application phase such as gene therapy.
Landscapes
- Life Sciences & Earth Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Genetics & Genomics (AREA)
- Engineering & Computer Science (AREA)
- Organic Chemistry (AREA)
- Zoology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Wood Science & Technology (AREA)
- General Engineering & Computer Science (AREA)
- Biotechnology (AREA)
- Biomedical Technology (AREA)
- Molecular Biology (AREA)
- General Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- Microbiology (AREA)
- Medicinal Chemistry (AREA)
- Physics & Mathematics (AREA)
- Biophysics (AREA)
- Plant Pathology (AREA)
- Crystallography & Structural Chemistry (AREA)
- Virology (AREA)
- Enzymes And Modification Thereof (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Saccharide Compounds (AREA)
Abstract
Description
(項目1)
核酸配列認識モジュールと、デアミナーゼとが結合した複合体であって、
該核酸配列認識モジュールは、二本鎖DNA中の標的ヌクレオチド配列と特異的に結合し、
該デアミナーゼは、該デアミナーゼに対応する野生型デアミナーゼよりもサイズが小さく、かつ改変した結果露出する断面の面積または該面積を示す指数が所定値以下となるように改変されており、
該二本鎖DNAの標的化された部位を改変する能力を有する、複合体。
(項目2)
前記デアミナーゼは、前記デアミナーゼを改変した結果露出する断面に現れる疎水性アミノ酸残基の数が、所定値以下となるように改変されており、該改変は欠失を含む、上記項目に記載の複合体。
(項目3)
前記デアミナーゼは、改変させたアミノ酸残基の数に対する、改変した結果露出する断面に現れる疎水性残基の割合が、所定値以下となるように改変されており、該改変は欠失を含む、上記項目のいずれか一項に記載の複合体。
(項目4)
前記デアミナーゼは、前記デアミナーゼを改変した結果露出する断面に現れる疎水性アミノ酸残基の数が、最小化するように改変される、上記項目のいずれか一項に記載の複合体。
(項目5)
前記デアミナーゼは、改変させたアミノ酸残基の数に対する、改変した結果露出する断面に現れる疎水性残基の割合が、最小化するように改変される、上記項目のいずれか一項に記載の複合体。
(項目6)
前記デアミナーゼは、前記野生型デアミナーゼのN末端側およびC末端側が改変される、上記項目のいずれか一項に記載の複合体。
(項目7)
前記デアミナーゼにおける露出した疎水性の内部アミノ酸残基の少なくとも1つが、親水性のアミノ酸残基に置換される、上記項目のいずれか一項に記載の複合体。
(項目8)
前記デアミナーゼがシチジンデアミナーゼを含む、上記項目のいずれか一項に記載の複合体。
(項目9)
前記デアミナーゼが、
(1)配列番号1で示されるアミノ酸配列における30位~150位のアミノ酸残基の領域からなるアミノ酸配列、
(2)配列番号1で示されるアミノ酸配列からなるタンパク質のオルソログであって、(1)の領域に対応する領域からなるアミノ酸配列、
(3)(1)若しくは(2)のアミノ酸配列において、1若しくは数個のアミノ酸が欠失、置換、挿入及び/若しくは付加されたアミノ酸配列、又は
(4)(1)若しくは(2)のアミノ酸配列と90%以上の類似性又は同一性を有するアミノ酸配列、
からなる、上記項目のいずれか一項に記載の複合体。
(項目10)
前記(3)のアミノ酸配列が、配列番号1で示されるアミノ酸配列における122位、126位及び139位からなる群から選択される位置のアミノ酸残基又は該位置に対応するアミノ酸残基の親水性アミノ酸残基への1箇所以上の置換を含む、上記項目のいずれか一項に記載の複合体。
(項目11)
前記(3)のアミノ酸配列が、配列番号1で示されるアミノ酸配列における122位のアミノ酸残基及び139位のアミノ酸残基、又は該位置に対応するアミノ酸残基の親水性アミノ酸残基への2箇所以上の置換を含む、上記項目のいずれか一項に記載の複合体。
(項目12)
前記核酸配列認識モジュールが、Casタンパク質の少なくとも1つのDNA切断能が失活したCRISPR-Casシステム、ジンクフィンガーモチーフ、TALエフェクター及びPPRモチーフからなる群より選択される、上記項目のいずれか一項に記載の複合体。
(項目13)
前記核酸配列認識モジュールが、Casタンパク質の1つのDNA切断能が失活したCRISPR-Casシステムである、上記項目のいずれか一項に記載の複合体。
(項目14)
前記Casタンパク質がCas9タンパク質である、上記項目のいずれか一項に記載の複合体。
(項目15)
核酸配列認識モジュールのN末端断片と、デアミナーゼと、核酸配列認識モジュールのC末端断片とが結合した複合体であって、
該核酸配列認識モジュールのN末端断片とC末端断片とがリフォールディングした場合に、該核酸配列認識モジュールは、二本鎖DNA中の標的ヌクレオチド配列と特異的に結合し、該二本鎖DNAの標的化された部位を改変する能力を有する、複合体。
(項目16)
前記デアミナーゼは、前記デアミナーゼに対応する野生型デアミナーゼよりもサイズが小さく、かつ改変した結果露出する断面の面積または該面積を示す指数が所定値以下となるように改変されている、上記項目に記載の複合体。
(項目17)
前記デアミナーゼは、前記デアミナーゼを改変した結果露出する断面に現れる疎水性アミノ酸残基の数が、所定値以下となるように改変されており、該改変は欠失を含む、上記項目のいずれか一項に記載の複合体。
(項目18)
前記デアミナーゼは、改変させたアミノ酸残基の数に対する、改変した結果露出する断面に現れる疎水性残基の割合が、所定値以下となるように改変されており、該改変は欠失を含む、上記項目のいずれか一項に記載の複合体。
(項目19)
前記デアミナーゼは、前記デアミナーゼを改変した結果露出する断面に現れる疎水性アミノ酸残基の数が、最小化するように改変される、上記項目のいずれか一項に記載の複合体。
(項目20)
前記デアミナーゼは、改変させたアミノ酸残基の数に対する、改変した結果露出する断面に現れる疎水性残基の割合が、最小化するように改変される、上記項目のいずれか一項に記載の複合体。
(項目21)
前記デアミナーゼは、前記野生型デアミナーゼのN末端側およびC末端側が改変される、上記項目のいずれか一項に記載の複合体。
(項目22)
前記デアミナーゼにおける露出した疎水性の内部アミノ酸残基の少なくとも1つが、親水性のアミノ酸残基に置換される、上記項目のいずれか一項に記載の複合体。
(項目23)
前記デアミナーゼがシチジンデアミナーゼを含む、上記項目のいずれか一項に記載の複合体。
(項目24)
前記デアミナーゼが、
(1)配列番号1で示されるアミノ酸配列における30位~150位のアミノ酸残基の領域からなるアミノ酸配列、
(2)配列番号1で示されるアミノ酸配列からなるタンパク質のオルソログであって、(1)の領域に対応する領域からなるアミノ酸配列、
(3)(1)若しくは(2)のアミノ酸配列において、1若しくは数個のアミノ酸が欠失、置換、挿入及び/若しくは付加されたアミノ酸配列、又は
(4)(1)若しくは(2)のアミノ酸配列と90%以上の類似性又は同一性を有するアミノ酸配列、
からなる、上記項目のいずれか一項に記載の複合体。
(項目25)
前記核酸配列認識モジュールが、Casタンパク質の少なくとも1つのDNA切断能が失活したCRISPR-Casシステム、ジンクフィンガーモチーフ、TALエフェクター及びPPRモチーフからなる群より選択される、上記項目のいずれか一項に記載の複合体。
(項目26)
上記項目のいずれか一項に記載の複合体をコードする核酸。
(項目27)
上記項目に記載の核酸を含むベクター。
(項目28)
アデノ随伴ウイルスベクターである、上記項目に記載のベクター。
(項目29)
細胞の有する二本鎖DNAの標的化された部位を改変する方法であって、上記項目のいずれか一項に記載の複合体を該二本鎖DNAと接触させる工程を含む、方法。
(項目30)
二本鎖DNAと複合体との接触が、前記細胞への、上記項目のいずれか一項に記載の核酸またはベクターの導入により行われる、上記項目に記載の方法。
[1]
核酸配列認識モジュールと、シチジンデアミナーゼとが結合した複合体であって、
該核酸配列認識モジュールは、二本鎖DNA中の標的ヌクレオチド配列と特異的に結合し、
該シチジンデアミナーゼは、
(1)配列番号1で示されるアミノ酸配列における30位~150位のアミノ酸残基の領域からなるアミノ酸配列、
(2)配列番号1で示されるアミノ酸配列からなるタンパク質のオルソログであって、(1)の領域に対応する領域からなるアミノ酸配列、
(3)(1)若しくは(2)のアミノ酸配列において、1若しくは数個のアミノ酸が欠失、置換、挿入及び/若しくは付加されたアミノ酸配列、又は
(4)(1)若しくは(2)のアミノ酸配列と90%以上の類似性又は同一性を有するアミノ酸配列、
からなり、
該二本鎖DNAの標的化された部位を改変する、複合体。
[2]
前記(3)のアミノ酸配列が、配列番号1で示されるアミノ酸配列における122位、126位及び139位からなる群から選択される位置のアミノ酸残基又は該位置に対応するアミノ酸残基の親水性アミノ酸残基への1箇所以上の置換を含む、[1]に記載の複合体。
[3]
前記(3)のアミノ酸配列が、配列番号1で示されるアミノ酸配列における122位のアミノ酸残基及び139位のアミノ酸残基、又は該位置に対応するアミノ酸残基の親水性アミノ酸残基への2箇所以上の置換を含む、[1]又は[2]に記載の複合体。
[4]
前記核酸配列認識モジュールが、Casタンパク質の少なくとも1つのDNA切断能が失活したCRISPR-Casシステム、ジンクフィンガーモチーフ、TALエフェクター及びPPRモチーフからなる群より選択される、[1]~[3]のいずれかに記載の複合体。
[5]
前記核酸配列認識モジュールが、Casタンパク質の1つのDNA切断能が失活したCRISPR-Casシステムである、[1]~[3]のいずれかに記載の複合体。
[6]
前記Casタンパク質がCas9タンパク質である、[4]又は[5]に記載の複合体。
[7]
核酸配列認識モジュールのN末端断片と、シチジンデアミナーゼと、核酸配列認識モジュールのC末端断片とが結合した複合体であって、
該核酸配列認識モジュールのN末端断片とC末端断片がリフォールディングした場合に、該核酸配列認識モジュールは、該二本鎖DNA中の標的ヌクレオチド配列と特異的に結合することを特徴とする、該二本鎖DNAの標的化された部位を改変する、複合体。
[8]
前記シチジンデアミナーゼが、
(1)配列番号1で示されるアミノ酸配列における30位~150位のアミノ酸残基の領域からなるアミノ酸配列、
(2)配列番号1で示されるアミノ酸配列からなるタンパク質のオルソログであって、(1)の領域に対応する領域からなるアミノ酸配列、
(3)(1)若しくは(2)のアミノ酸配列において、1若しくは数個のアミノ酸が欠失、置換、挿入及び/若しくは付加されたアミノ酸配列、又は
(4)(1)若しくは(2)のアミノ酸配列と90%以上の類似性又は同一性を有するアミノ酸配列、
からなる、[7]に記載の複合体。
[9]
前記核酸配列認識モジュールが、Casタンパク質の少なくとも1つのDNA切断能が失活したCRISPR-Casシステム、ジンクフィンガーモチーフ、TALエフェクター及びPPRモチーフからなる群より選択される、[7]又は[8]に記載の複合体。
[10]
[1]~[9]のいずれかに記載の複合体をコードする核酸。
[11]
[10]に記載の核酸を含むベクター。
[12]
アデノ随伴ウイルスベクターである、[11]に記載のベクター。
[13]
細胞の有する二本鎖DNAの標的化された部位を改変する方法であって、[1]~[9]のいずれかに記載の複合体を該二本鎖DNAと接触させる工程を含む、方法。
[14]
二本鎖DNAと複合体との接触が、該細胞への、[10]~[12]のいずれかに記載の核酸又はベクターの導入により行われる、[13]に記載の方法。
本明細書の全体にわたり、単数形の表現は、特に言及しない限り、その複数形の概念をも含むことが理解されるべきである。従って、単数形の冠詞(例えば、英語の場合は「a」、「an」、「the」など)は、特に言及しない限り、その複数形の概念をも含むことが理解されるべきである。また、本明細書において使用される用語は、特に言及しない限り、当該分野で通常用いられる意味で用いられることが理解されるべきである。したがって、他に定義されない限り、本明細書中で使用される全ての専門用語および科学技術用語は、本開示の属する分野の当業者によって一般的に理解されるのと同じ意味を有する。矛盾する場合、本明細書(定義を含めて)が優先する。
本開示の一局面において、核酸配列認識モジュールと、デアミナーゼとが結合した複合体であって、該核酸配列認識モジュールは、二本鎖DNA中の標的ヌクレオチド配列と特異的に結合し、該デアミナーゼは、該デアミナーゼに対応する野生型デアミナーゼよりもサイズが小さく、かつ改変した結果露出する断面の面積または該面積を示す指数が所定値以下となるように改変されており、該二本鎖DNAの標的化された部位を改変する能力を有する、複合体が提供される。一実施形態において、改変した結果露出する断面の面積または該面積を示す指数は、デアミナーゼの種類によって適宜設定される所定値以下とすることができ、例えば、そのような指数として、後述の露出断面指数や疎水性アミノ酸残基の数を用いることができる。
(1)配列番号1で示されるアミノ酸配列における30位~150位のアミノ酸残基の領域からなるアミノ酸配列、
(2)配列番号1で示されるアミノ酸配列からなるタンパク質のオルソログであって、(1)の領域に対応する領域からなるアミノ酸配列、
(3)(1)若しくは(2)のアミノ酸配列において、1若しくは数個のアミノ酸が欠
失、置換、挿入及び/若しくは付加されたアミノ酸配列、又は
(4)(1)若しくは(2)のアミノ酸配列と90%以上の類似性又は同一性を有するアミノ酸配列
からなるシチジンデアミナーゼが挙げられる。ただし、上記(3)及び(4)のシチジンデアミナーゼからは、野生型のシチジンデアミナーゼや、配列番号1の28位~161位の領域(134アミノ酸配列からなる領域)を少なくとも含む、該シチジンデアミナーゼの断片は除かれるものとする。以下では、「小型化シチジンデアミナーゼ」と「野生型シチジンデアミナーゼ」の両方を包含する用語として、単に「シチジンデアミナーゼ」との用語を用いることがある。
これに対し、CRISPR-Casシステムは、標的ヌクレオチド配列に対して相補的なガイドRNAにより目的の二本鎖DNAの配列を認識するので、標的ヌクレオチド配列と特異的にハイブリッド形成し得るオリゴDNAを合成するだけで、任意の配列を標的化することができる。
従って、本開示のより好ましい実施態様においては、核酸配列認識モジュールとして、Casの1つのみ、又は両方のDNA切断能が失活したCRISPR-変異Casシステムが用いられる。
また、核酸配列認識モジュールとデアミナーゼ(例えばシチジンデアミナーゼ)とが結合した複合体を含んでなる本開示の複合体と、二本鎖DNAとの接触は、目的の二本鎖DNAを有する細胞に、該複合体をコードする核酸(以下では、「本開示の核酸」と称することがある。)を導入することにより、実施されてもよい。また、本開示の核酸を用いて、分子生物学的手法により、本開示の複合体や、該複合体の各構成分子を製造することもできる。従って、核酸配列認識モジュールと、シチジンデアミナーゼとは、それらの融合タンパク質をコードする核酸として、あるいは、結合ドメインやインテイン等を利用してタンパク質に翻訳後、宿主細胞内で複合体を形成し得るような形態で、それらをそれぞれコードする核酸として調製してもよい。ここで核酸は、DNAであってもRNAであってもよい。DNAの場合は、好ましくは二本鎖DNAであり、宿主細胞内で機能的なプロモーターの制御下に配置した発現ベクターの形態で、あるいは該DNAを含む発現ベクターの形態で提供される。RNAの場合は、好ましくは一本鎖RNAである。
シチジンデアミナーゼをコードするDNAも、同様に、使用するシチジンデアミナーゼのcDNA配列情報をもとに、所望のアミノ酸残基の欠失が達成できるようにオリゴDNAプライマーを合成し、当該シチジンデアミナーゼを産生する細胞より調製した全RNA若しくはmRNA画分を鋳型として用い、RT-PCR法によって増幅することにより、クローニングすることができる。例えば、ヤツメウナギの小型化PmCDA1をコードするDNAは、NCBIデータベースに登録されているcDNA配列(accessionNo. EF094822)をもとに、CDSの適切な領域に対して適当なプライマーを設計し、ヤツメウナギ由来mRNAからRT-PCR法によりクローニングできる。また、ヒトAIDをコードするDNAは、NCBIデータベースに登録されているcDNA配列(accessionNo. AB040431)をもとに、同様にクローニングできる。また、標的化された部位の改変にドナーDNAを用いる場合には、該ドナーDNAも、該部位の配列情報等に基づき、上記と同様にクローニングできる。
クローン化されたDNAは、そのまま、又は所望により制限酵素で消化するか、適当なリンカー及び/又は核移行シグナル(目的の二本鎖DNAがミトコンドリアや葉緑体DNAの場合は、各オルガネラ移行シグナル)を付加した後に、核酸配列認識モジュールをコードするDNAとライゲーションして、融合タンパク質をコードするDNAを調製することができる。あるいは、核酸配列認識モジュールをコードするDNAと、シチジンデアミナーゼをコードするDNAに、それぞれ結合ドメイン若しくはその結合パートナーをコードするDNAを融合させるか、両DNAに分離インテインをコードするDNAを融合させることにより、核酸配列認識変換モジュールとシチジンデアミナーゼとが宿主細胞内で翻訳された後に複合体を形成できるようにしてもよい。これらの場合も、所望により一方若しくは両方のDNAの適当な位置に、リンカー及び/又は核移行シグナルを連結することができる。また、標的化された部位の改変にドナーDNAを用いる場合には、該ドナーDNAAは、単一DNAとして作製してもよいし、核酸配列認識モジュール及び/又はシチジンデアミナーゼをコードする核酸を含む単一のDNAとして提供されてもよい。
発現ベクターとしては、大腸菌由来のプラスミド(例、pBR322,pBR325,pUC12,pUC13);枯草菌由来のプラスミド(例、pUB110,pTP5,pC194);酵母由来プラスミド(例、pSH19,pSH15);昆虫細胞発現プラスミド(例:pFast-Bac);動物細胞発現プラスミド(例:pA1-11、pXT1、pRc/CMV、pRc/RSV、pcDNAI/Neo);λファージなどのバクテリオファージ;バキュロウイルスなどの昆虫ウイルスベクター(例:BmNPV、AcNPV);レトロウイルス、ワクシニアウイルス、アデノウイルス、アデノ随伴ウイルス(AAV)などの動物ウイルスベクターなどが用いられる。遺伝子治療における利用を考慮すれば、導入遺伝子を長期にわたり発現させられる点や非病原性ウイルス由来で安全性の点からは、AAVベクターが好適に用いられる。
例えば、宿主が動物細胞である場合、SRαプロモーター、SV40プロモーター、LTRプロモーター、CMV(サイトメガロウイルス)プロモーター、RSV(ラウス肉腫ウイルス)プロモーター、MoMuLV(モロニーマウス白血病ウイルス)LTR、HSV-TK(単純ヘルペスウイルスチミジンキナーゼ)プロモーターなどが用いられる。なかでも、CMVプロモーター、SRαプロモーターなどが好ましい。
宿主が大腸菌である場合、trpプロモーター、lacプロモーター、recAプロモーター、λPLプロモーター、lppプロモーター、T7プロモーターなどが好ましい。
宿主がバチルス属菌である場合、SPO1プロモーター、SPO2プロモーター、penPプロモーターなどが好ましい。
宿主が酵母である場合、Gal1/10プロモーター、PHO5プロモーター、PGKプロモーター、GAPプロモーター、ADHプロモーターなどが好ましい。
宿主が昆虫細胞である場合、ポリヘドリンプロモーター、P10プロモーターなどが好ましい。
宿主が植物細胞である場合、CaMV35Sプロモーター、CaMV19Sプロモーター、NOSプロモーターなどが好ましい。
別の実施態様において、本開示の複合体を、宿主細胞の二本鎖DNAと接触させる工程を含む、二本鎖DNAの標的化された部位を改変する方法(以下では、「本開示の改変方法」と称することがある。)が提供される。核酸配列認識モジュールとシチジンデアミナーゼとが結合した複合体(融合タンパク質を含む。)を含んでなる本開示の複合体と、二本鎖DNAとの接触は、無細胞系の酵素反応として行われてもよいが、本開示の主たる目的に沿えば、1.及び2.に記載した本開示の複合体又は核酸若しくはベクターを、宿主に導入し、当該宿主を培養することにより実施されることが望ましい。
エシェリヒア属菌としては、例えば、エシェリヒア・コリ(Escherichia coli)K12・DH1〔Proc. Natl. Acad. Sci. USA,60,160(1968)〕,エシェリヒア・コリJM103〔Nucleic Acids Research,9,309 (1981)〕,エシェリヒア・コリJA221〔Journalof Molecular Biology,120,517 (1978)〕,エシェリヒア・コリHB101〔Journal of MolecularBiology,41,459 (1969)〕,エシェリヒア・コリC600〔Genetics,39,440 (1954)〕などが用いられる。
バチルス属菌としては、例えば、バチルス・サブチルス(Bacillus subtilis)MI114〔Gene,24,255 (1983)〕,バチルス・サブチルス207-21〔Journalof Biochemistry,95,87 (1984)〕などが用いられる。
酵母としては、例えば、サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)AH22,AH22R-,NA87-11A,DKD-5D,20B-12、シゾサッカロマイセス・ポンベ(Schizosaccharomycespombe)NCYC1913,NCYC2036,ピキア・パストリス(Pichia pastoris)KM71などが用いられる。
昆虫としては、例えば、カイコの幼虫、ショウジョウバエ、コオロギなどが用いられる〔Nature,315,592 (1985)〕。
大腸菌は、例えば、Proc. Natl. Acad. Sci. USA,69,2110 (1972)やGene,17,107 (1982)などに記載の方法に従って形質転換することができる。
バチルス属菌は、例えば、Molecular & General Genetics,168,111 (1979)などに記載の方法に従ってベクター導入することができる。
酵母は、例えば、Methods in Enzymology,194,182-187 (1991)、Proc. Natl. Acad. Sci. USA,75,1929(1978)などに記載の方法に従ってベクター導入することができる。
昆虫細胞及び昆虫は、例えば、Bio/Technology,6,47-55 (1988)などに記載の方法に従ってベクター導入することができる。
動物細胞は、例えば、細胞工学別冊8 新細胞工学実験プロトコール,263-267 (1995)(秀潤社発行)、Virology,52,456 (1973)に記載の方法に従ってベクター導入することができる。
例えば、大腸菌又はバチルス属菌を培養する場合、培養に使用される培地としては液体培地が好ましい。また、培地は、形質転換体の生育に必要な炭素源、窒素源、無機物などを含有することが好ましい。ここで、炭素源としては、例えば、グルコース、デキストリン、可溶性澱粉、ショ糖などが;窒素源としては、例えば、アンモニウム塩類、硝酸塩類、コーンスチープ・リカー、ペプトン、カゼイン、肉エキス、大豆粕、バレイショ抽出液などの無機又は有機物質が;無機物としては、例えば、塩化カルシウム、リン酸二水素ナトリウム、塩化マグネシウムなどがそれぞれ挙げられる。また、培地には、酵母エキス、ビタミン類、生長促進因子などを添加してもよい。培地のpHは、好ましくは約5~約8である。
大腸菌を培養する場合の培地としては、例えば、グルコース、カザミノ酸を含むM9培地〔Journal of Experiments in MolecularGenetics, 431-433, Cold Spring Harbor Laboratory, New York 1972〕が好ましい。必要により、プロモーターを効率よく働かせるために、例えば、3β-インドリルアクリル酸のような薬剤を培地に添加してもよい。大腸菌の培養は、通常約15~約43℃で行なわれる。必要により、通気や撹拌を行ってもよい。
バチルス属菌の培養は、通常約30~約40℃で行なわれる。必要により、通気や撹拌を行ってもよい。
酵母を培養する場合の培地としては、例えば、バークホールダー(Burkholder)最小培地〔Proc. Natl. Acad. Sci. USA,77,4505(1980)〕や0.5%カザミノ酸を含有するSD培地〔Proc. Natl. Acad. Sci. USA,81,5330 (1984)〕などが挙げられる。培地のpHは、好ましくは約5~約8である。培養は、通常約20℃~約35℃で行なわれる。必要に応じて、通気や撹拌を行ってもよい。
昆虫細胞又は昆虫を培養する場合の培地としては、例えばGrace's Insect Medium〔Nature,195,788 (1962)〕に非働化した10%ウシ血清等の添加物を適宜加えたものなどが用いられる。培地のpHは、好ましくは約6.2~約6.4である。培養は、通常約27℃で行なわれる。必要に応じて通気や撹拌を行ってもよい。
動物細胞を培養する場合の培地としては、例えば、約5~約20%の胎児ウシ血清を含む最小必須培地(MEM)〔Science,122,501 (1952)〕,ダルベッコ改変イーグル培地(DMEM)〔Virology,8,396(1959)〕,RPMI 1640培地〔The Journal of the American Medical Association,199,519(1967)〕,199培地〔Proceeding of the Society for the Biological Medicine,73,1 (1950)〕などが用いられる。培地のpHは、好ましくは約6~約8である。培養は、通常約30℃~約40℃で行なわれる。必要に応じて通気や撹拌を行ってもよい。
植物細胞を培養する培地としては、MS培地、LS培地、B5培地などが用いられる。培地のpHは好ましくは約5~約8である。培養は、通常約20℃~約30℃で行なわれる。必要に応じて通気や撹拌を行ってもよい。
以上のようにして、核酸配列認識モジュールとシチジンデアミナーゼとの複合体、即ち本開示の複合体を細胞内で発現させることができる。
温度感受性変異を利用する場合、例えば、ベクターの自律複製に必要なタンパク質の温度感受性変異体を、本開示の複合体をコードするDNAを含むベクターに搭載することにより、該複合体の発現後、速やかに自律複製が出来なくなり、細胞分裂に伴って該ベクターは自然に脱落する。このような温度感受性変異タンパク質としては、pSC101oriの複製に必要なRep101 oriの温度感受性変異体が挙げられるが、これに限定されない。Rep101 ori(ts)は30℃以下(例、28℃)では、pSC101oriに作用してプラスミドの自律複製を可能にするが、37℃以上(例、42℃)になると機能を失い、プラスミドは自律複製できなくなる。従って、上記λファージのcIリプレッサー(ts)と併用することで、本開示の複合体の一過的発現と、プラスミド除去とを、同時に行うことができる。
出芽酵母Saccharomyces cerevisiae BY4741株(ロイシン及びウラシル要求性)を用い、標準的なYPDA培地ないしSD培地の栄養要求性に合わせたDropout組成で培養した。培養は25℃から30℃の間で、寒天プレートでの静置培養又は液体培地での振とう培養を行った。形質転換は酢酸リチウム法を用い、適切な栄養要求性に合わせたSD培地で選抜を行った。ガラクトースによる発現誘導には、適切なSD培地で一晩予備培養した後、炭素源を2%グルコースから2%ラフィノースに代えたSR培地に植え継いで一晩培養、さらに炭素源を0.2%ガラクトースに代えたSGal培地に植え継いで3時間から二晩程度培養して発現誘導を行った。
生存細胞数及びCan1変異率の測定には、細胞懸濁液をSDプレート培地及びSD-Arg+60mg/l Canavanineプレート培地あるいはSD+300mg/lCanavanineプレート培地に適宜希釈して塗布し、3日後に出現するコロニー数を生存細胞数としてカウントした。SDプレートでの生存コロニー数を全細胞数とし、Canavanineプレートでの生存コロニー数を耐性変異株数として、変異率を算出・評価した。
オフターゲット効果の検証するため、細胞懸濁液をSDプレート培地及びSD+100 mg/lのS-aminoethyl-L-cysteine(Thialysine) に適宜希釈して塗布し、3日後に出現するコロニー数を生存細胞数としてカウントした。SDプレートでの生存コロニー数を全細胞数とし、Thialysineプレートでの生存コロニー数を耐性変異株数として、オフターゲット変異率を算出・評価した。
DNAは、PCR法、制限酵素処理、ライゲーション、Gibson Assembly法、人工化学合成のいずれかによって、加工・構築した。プラスミドは酵母・大腸菌シャトルベクターとしてロイシン選抜用のpRS415、及びウラシル選抜用のpRS426をバックボーンとして用いた。プラスミドは大腸菌株XL-10goldないしDH5αで増幅し、酢酸リチウム法で酵母に導入した。
非特許文献3に記載の手法及びplasmidに準じ、各ドメインの切断や入れ替え、変異の導入を行った。哺乳動物発現用のSaCas9含有ベクターとして、AddgeneよりSaABEmax(#119814)を入手して改変した。Scp1配列、polyAシグナルは人工化学合成を行った。KN1086、KN1150、KN1025及びKN1149のgRNAの各標的配列を、それぞれ配列番号8~11として示す。また、各コンストラクトの代表として、pAL008、pAL022、V5679、pAL047及びpAL050の全長配列を、それぞれ配列番号12~16として示す。なお、以下の実施例でコントロールとして用いたベクター1251は、従来型のdCas9-dSH3-CDA(UGIは含まない)をコードする配列を含み、ベクター1252は、従来型のnCas9-dSH3-CDA(UGIは含まない)をコードする配列を含む(dSH3はリンカーである)。また、pAL008は、CDA-nCas9をコードする配列を含み、Cas9のN末側にリンカーなしでCDAを融合している。
AID(id:5W1C)の立体構造はNCBIのMMDBより入手し、ソフトウェア(Cn3D)上で解析した。アラインメントはClustalWによって行った。
ヒト胎児腎臓由来細胞(HEK293T細胞)を用いた。細胞を、100μg/mL ペニシリン-ストレプトマイシン(Life Technologies,Carlsbad, CA, USA)及び10%胎仔ウシ血清(FBS)(Biosera, Nuaille, France)を添加したDME-glutamax培地(ThermoFisher Scientific, USA)を用いて、37℃、5% CO2条件で培養を行った。細胞の回収には5%トリプシンを用いた。 ディープフリーザーで保存したHEK293T細胞を37℃のウォーターバスにて溶解し、5x106 cellsになるように75T-flaskに播種した。1-3日間培養後に細胞を回収し、0.5x105 cells/wellになるように24ウェルプレートの各ウェルに播種した。1-3日培養後に60-80%コンフルエント状態の各ウェルの細胞に対して、約1μgの上記の各プラスミドDNAを3 μlのLipofectamine 2000(Life Technologies, Carlsbad, USA)を用いてトランスフェクションした。
変異頻度及び変異箇所の解析は、トランスフェクション24時間ないし72時間後に培養細胞を回収してDNAを抽出、標的領域をPCRにて増幅後、次世代シーケンサーMiniseqを用いてアンプリコン解析を行った。データ処理はCLCworkbenchにて行った。場合により、細胞濃縮のため、GFPないしRFPの蛍光を指標に、コンストラクト発現細胞をセルソーターにて分取を行った。
非特許文献4に開示された小型化シチジンデアミナーゼを含む核酸改変酵素複合体には、ウラシル-DNAグリコシラーゼ阻害剤(UGI)も含まれているが、UGIを用いると、望まれないオフターゲット効果が増強すると予想される。またデアミナーゼ活性を比較評価するにあたって、酵母細胞内ではUGI存在下では変異導入効率が飽和して差が見えにくい。そのため、まず、上記非特許文献4に開示された複合体から、UGIを除いた複合体を作製した。また、該複合体のN末端側領域をさらに欠失させた複合体も作製し、これらの複合体での標的部位の改変効率を検証した。本実験で用いたコンストラクトの概要を図2に、結果を図3~図5に示す。図3~図5より、予想外にも、非特許文献4において高い改変効率が認められた、CDA1Δ161を用いた複合体でさえ、UGIを用いない場合には、野生型CDA1を用いた複合体と比較して改変効率が3分の2以下となった。さらには、CDA1Δ161のN末端側を2アミノ酸残基以上欠失させたCDA1では、改変が顕著に低下した。上記結果から、本発明者らは、非特許文献4に開示の複合体が高い標的部位の改変効率を達成できたのは、酵母細胞内でのUGIによるDNA改変効率の向上効果によるところが大きく、UGIを用いない場合あるいは異なる生物種への適用の際には、単純にCDA1の末端領域を欠失するだけでは、所望の改変効率を達成できる複合体が得られないとの結論に達した。
そこで、単純にシチジンデアミナーゼの末端領域を欠失させるのではなく、シチジンデアミナーゼの構造に基づき、シチジンデアミナーゼの欠失部位を決定することとした。まず、立体構造解析ソフトを用いて、CDA1Δ161の構造を解析したところ、歪んだ球状の形状であった(図6左図)。そこで、図6左図の白色部分(即ち、PmCDA1の30-150位の領域)だけを取り出すことで、形状をより球状に近づけた。なお、以下では、アミノ酸残基の位置は、野生型シチジンデアミナーゼのアミノ酸配列(即ち、配列番号1で示される配列)に基づき示す。このようにして作製した小型化シチジンデアミナーゼ(PmCDA1(30-150)と称する場合がある。)について、さらに構造解析より、露出した内部アミノ酸残基(ここでは、91位、122位126位、128位、及び150位のアミノ酸残基)(図7右図の白色部分)を疎水性から親水性のアミノ酸残基に置換した複合体も作製した。本実験で用いたコンストラクトの概要を図8に、結果を図9~図11に示す。図9~図11より、PmCDA1(30-150)では、シチジンデアミナーゼの分子量が高いCDA1Δ161と比較して、標的部位の改変効率が向上することが示された。また、PmCDA1(30-150)について、122位のトリプトファンをグルタミン酸に置換したシチジンデアミナーゼ(PmCDA1(30-150;W122E)と称する場合がある。)では、特に高い改変活性が認められた。
また、全く異なるアプローチにより、核酸改変酵素複合体の改変効率が向上できるか否か検証した。従来の核酸改変酵素複合体では、Casタンパク質の末端にシチジンデアミナーゼを融合したものが用いられているが、シチジンデアミナーゼをCasタンパク質の内部に埋め込むことにより、複合体としての安定性の向上及び基質DNAへのアクセス向上がするのではないかとの仮説の下、実証実験を行った。本実験で用いたコンストラクトの概要を図21に、結果を図22~図24に示す。図22~図24より、作製した全ての複合体において、高い改変効率が認められた。
上記実施例2及び実施例3で高い改変効率が認められた複合体について、UGIをさらに併用することで、改変効率が向上するか否かを検証した。本実験で用いたコンストラクトの概要を図33に、結果を図34及び図35に示す。図34及び図35より、作製した全ての複合体において、高い改変効率が認められた。
上記実施例で高い改変効率が認められた複合体について、オフターゲット効果を検証した。結果を図36~図41に示す。かかる結果より、作製した全ての複合体において、コントロール(KN1252_UG1を用いたもの)と比較して、オフターゲット効果が顕著に抑制されていた。
次に、実施例3のSpCas9に代えてSaCas9を用いて、同様に改変が認められるか否かを検証した。SaCas9及び小型化デアミナーゼを用いることで、ガイドRNA発現カセット(プロモーター、gRNAコード配列及びポリT配列からなる)を含む核酸改変酵素複合体の発現カセットを、AAVベクターへ搭載可能なサイズ(約4.4kb)以下とすることが可能となった。コードする本実験で用いたコンストラクトの概要を図42(核酸改変酵素複合体)及び図43上図(ガイドRNA)を、実験手順の概要を図43下図に、結果を図44及び45に示す。図44及び図45より、作製した全ての複合体において、高い改変効率が認められた。また、オフターゲット効果の抑制も期待される。さらに、興味深いことに、デアミナーゼのCas9への挿入部分により、変異が導入される確率の高い部位が変動することが示された。そのため、上記挿入部位を調整することで、変異導入部位の調整も可能となる。
DNAデアミナーゼは、DNAに固有の親和性を持ち、非特異的な脱アミノ化を引き起こす。PmCDA1のヒトホモログであるhAIDの構造から、触媒コアとは異なる領域で二本鎖DNAと複合体を形成することが明らかになっている(図46a)。hAIDとPmCDA1のアミノ酸配列から、PmCDA1の潜在的なDNA結合部位は、タンパク質の全長208アミノ酸のうち、21-27残基と172-192残基に位置していた(図46a)。予測されるDNA結合領域を削除するために、まず、C末端から一連の切断体(1-201、1-197、1-190、1-183、1-179、1-176、1-161)を作り、酵母Saccharomyces cerevisiae(BY4741)細胞で塩基編集活性を試験した(図48)。これまでの報告では、47アミノ酸を切断したPmCDA1(1-161)は、酵母において完全長のPmCDA1(1-208)と同等の編集効率を示すことが報告されているが、nCas9のC末端に融合し、ウラシルDNAグリコシラーゼ阻害剤(UGI)を付加していないものでは、切断が進むにつれて活性が徐々に低下した。次に、nCas9のN末端に融合させることで、1-161残基のN末端から一連の切断を行った。CDA1(1-161)のN末端切断体では、まず活性がさらに低下したが、その後、21および28アミノ酸まで切断を進めると回復した(図49)。CDA1の予測構造から、N末端とC末端を同時に切断することで、断面積が最小化され、疎水性残基の露出が少ない滑らかなタンパク質表面が得られることがわかった(図49)。さらに、酵素コアドメインをそのまま残しつつ、疎水性表面の露出を最小限に抑えた最小のものと予測されるCDA1(30-150)に切り詰めたところ(図46b、図49)、活性が回復した(図49)。これらの結果は、その編集活性の変化が、タンパク質のコンフォメーションの安定性に起因することを示唆している。さらにその活性を向上させるために、切断後に露出した疎水性残基に一連の変異を導入した。まず6つの変異を試験したところ、W122EがCDA1(30-150)に対して有意に活性を獲得することがわかった(図50)。さらに7つの変異をW122Eと組み合わせて試験したところ、活性がさらに向上するW133R/Qが見つかった(図50)。以下、W122EとW133Qを含むCDA1(30-150)をtCDA1EQと呼ぶ。
次に,ヒトHEK293T細胞におけるAID-2SとAID-3Sの編集効率とウィンドウを評価し,オフターゲット効果が低減されていると報告されている既存の改良型シトシン塩基編集剤YE1,YE2,R33A+K34Aと比較した。よく研究されている4つのオンターゲット部位(HEK2、HEK3、RNF2、VEGFA)をプラスミドDNAベクターのトランスフェクションにより編集し、アンプリコンディープシークエンスにより解析した。Target-AID, AID-2S, YE1は、試験した4つのターゲットサイトすべてに対して一貫して高い効率を示した。AID-3SとYE2は、ターゲットサイトに依存して中程度から高程度の効率を示した。R33A+K34AはHEK3のターゲットサイトでは効率が悪かった(図46e)。AID-2Sの平均編集ウィンドウ幅はTarget-AIDよりも狭く、YE1やYE2と同程度であった(図46f)。
改変PmCDA1(tCDA1EQ)は、野生型(208アミノ酸)に比べてサイズが大幅に小さく(121アミノ酸)なっている。ゲノム編集コンポーネントとして分子サイズが小さいことは、特にDNAの長さが4~5kbに制限されているAAVベクターのようなin vivoデリバリーツールにとって有利である。小型のSaCas9システムを用いても、塩基編集コンポーネントを追加すると、明らかにサイズ制限を超えてしまう(図46g)。そこで、AAVベクターに搭載可能なサイズで、必要な塩基編集コンポーネントをすべて含むSaAID-3Sを開発するために、tCDA1EQを、ポリヌクレオチド結合クレフトに面したHNHドメイン内のnSaCas9の615-616残基の位置に組み込んだ。また、小型のScp1プロモーターとSpAターミネーターを用いて、全長4036bpと332bpのgRNA発現カセットを構成した。比較のために、従来型のSaCas9版Target-AID(SaAID)も開発した。このSaAIDは、全長のPmCDA1にリンカー、UGI、CMVプロモーター、SV40ターミネーターを加え、全長5220bpとし、gRNAカセットは加えていない。ベクターのサイズによって異なるトランスフェクション効率を正規化するために、ベクターバックボーンから発現させたiRFP670の蛍光シグナルでトランスフェクションした細胞を選別した。試験した2つの標的部位において、両コンストラクトは、変異ウィンドウに違いはあるものの(図52)、同等の編集効率を示した(図46h)。
以上のように、本開示の好ましい実施形態を用いて本開示を例示してきたが、本開示は、特許請求の範囲によってのみその範囲が解釈されるべきであることが理解される。本明細書において引用した特許、特許出願及び他の文献は、その内容自体が具体的に本明細書に記載されているのと同様にその内容が本明細書に対する参考として援用されるべきであることが理解される。本願は、日本国特許庁に2020年9月4日に出願された特願2020-149419に対して優先権主張をするものであり、その内容はその全体があたかも本願の内容を構成するのと同様に参考として援用される。
、アデノ随伴ウイルスベクターにも搭載し、標的部位に複合体をデリバリーすることも容易になるため、特に遺伝子治療などの応用局面で有用となり得る。
Claims (30)
- 核酸配列認識モジュールと、デアミナーゼとが結合した複合体であって、
該核酸配列認識モジュールは、二本鎖DNA中の標的ヌクレオチド配列と特異的に結合し、
該デアミナーゼは、該デアミナーゼに対応する野生型デアミナーゼよりもサイズが小さく、かつ改変した結果露出する断面の面積または該面積を示す指数が所定値以下となるように改変されており、
該二本鎖DNAの標的化された部位を改変する能力を有する、複合体。 - 前記デアミナーゼは、前記デアミナーゼを改変した結果露出する断面に現れる疎水性アミノ酸残基の数が、所定値以下となるように改変されており、該改変は欠失を含む、請求項1に記載の複合体。
- 前記デアミナーゼは、改変させたアミノ酸残基の数に対する、改変した結果露出する断面に現れる疎水性残基の割合が、所定値以下となるように改変されており、該改変は欠失を含む、請求項1または2に記載の複合体。
- 前記デアミナーゼは、前記デアミナーゼを改変した結果露出する断面に現れる疎水性アミノ酸残基の数が、最小化するように改変される、請求項1~3のいずれか一項に記載の複合体。
- 前記デアミナーゼは、改変させたアミノ酸残基の数に対する、改変した結果露出する断面に現れる疎水性残基の割合が、最小化するように改変される、請求項1~4のいずれか一項に記載の複合体。
- 前記デアミナーゼは、前記野生型デアミナーゼのN末端側およびC末端側が改変される、請求項1~5のいずれか一項に記載の複合体。
- 前記デアミナーゼにおける露出した疎水性の内部アミノ酸残基の少なくとも1つが、親水性のアミノ酸残基に置換される、請求項1~6のいずれか一項に記載の複合体。
- 前記デアミナーゼがシチジンデアミナーゼを含む、請求項1~7のいずれか一項に記載の複合体。
- 前記デアミナーゼが、
(1)配列番号1で示されるアミノ酸配列における30位~150位のアミノ酸残基の領域からなるアミノ酸配列、
(2)配列番号1で示されるアミノ酸配列からなるタンパク質のオルソログであって、(1)の領域に対応する領域からなるアミノ酸配列、
(3)(1)若しくは(2)のアミノ酸配列において、1若しくは数個のアミノ酸が欠失、置換、挿入及び/若しくは付加されたアミノ酸配列、又は
(4)(1)若しくは(2)のアミノ酸配列と90%以上の類似性又は同一性を有するアミノ酸配列、
からなる、請求項1~8のいずれか一項に記載の複合体。 - 前記(3)のアミノ酸配列が、配列番号1で示されるアミノ酸配列における122位、126位及び139位からなる群から選択される位置のアミノ酸残基又は該位置に対応するアミノ酸残基の親水性アミノ酸残基への1箇所以上の置換を含む、請求項9に記載の複合体。
- 前記(3)のアミノ酸配列が、配列番号1で示されるアミノ酸配列における122位のアミノ酸残基及び139位のアミノ酸残基、又は該位置に対応するアミノ酸残基の親水性アミノ酸残基への2箇所以上の置換を含む、請求項9または10に記載の複合体。
- 前記核酸配列認識モジュールが、Casタンパク質の少なくとも1つのDNA切断能が失活したCRISPR-Casシステム、ジンクフィンガーモチーフ、TALエフェクター及びPPRモチーフからなる群より選択される、請求項1~11のいずれか一項に記載の複合体。
- 前記核酸配列認識モジュールが、Casタンパク質の1つのDNA切断能が失活したCRISPR-Casシステムである、請求項1~12のいずれか一項に記載の複合体。
- 前記Casタンパク質がCas9タンパク質である、請求項12または13に記載の複合体。
- 核酸配列認識モジュールのN末端断片と、デアミナーゼと、核酸配列認識モジュールのC末端断片とが結合した複合体であって、
該核酸配列認識モジュールのN末端断片とC末端断片とがリフォールディングした場合に、該核酸配列認識モジュールは、二本鎖DNA中の標的ヌクレオチド配列と特異的に結合し、該二本鎖DNAの標的化された部位を改変する能力を有する、複合体。 - 前記デアミナーゼは、前記デアミナーゼに対応する野生型デアミナーゼよりもサイズが小さく、かつ改変した結果露出する断面の面積または該面積を示す指数が所定値以下となるように改変されている、請求項15に記載の複合体。
- 前記デアミナーゼは、前記デアミナーゼを改変した結果露出する断面に現れる疎水性アミノ酸残基の数が、所定値以下となるように改変されており、該改変は欠失を含む、請求項15または16に記載の複合体。
- 前記デアミナーゼは、改変させたアミノ酸残基の数に対する、改変した結果露出する断面に現れる疎水性残基の割合が、所定値以下となるように改変されており、該改変は欠失を含む、請求項15~17のいずれか一項に記載の複合体。
- 前記デアミナーゼは、前記デアミナーゼを改変した結果露出する断面に現れる疎水性アミノ酸残基の数が、最小化するように改変される、請求項15~18のいずれか一項に記載の複合体。
- 前記デアミナーゼは、改変させたアミノ酸残基の数に対する、改変した結果露出する断面に現れる疎水性残基の割合が、最小化するように改変される、請求項15~19のいずれか一項に記載の複合体。
- 前記デアミナーゼは、前記野生型デアミナーゼのN末端側およびC末端側が改変される、請求項15~20のいずれか一項に記載の複合体。
- 前記デアミナーゼにおける露出した疎水性の内部アミノ酸残基の少なくとも1つが、親水性のアミノ酸残基に置換される、請求項15~21のいずれか一項に記載の複合体。
- 前記デアミナーゼがシチジンデアミナーゼを含む、請求項15~22のいずれか一項に記載の複合体。
- 前記デアミナーゼが、
(1)配列番号1で示されるアミノ酸配列における30位~150位のアミノ酸残基の領域からなるアミノ酸配列、
(2)配列番号1で示されるアミノ酸配列からなるタンパク質のオルソログであって、(1)の領域に対応する領域からなるアミノ酸配列、
(3)(1)若しくは(2)のアミノ酸配列において、1若しくは数個のアミノ酸が欠失、置換、挿入及び/若しくは付加されたアミノ酸配列、又は
(4)(1)若しくは(2)のアミノ酸配列と90%以上の類似性又は同一性を有するアミノ酸配列、
からなる、請求項15~23のいずれか一項に記載の複合体。 - 前記核酸配列認識モジュールが、Casタンパク質の少なくとも1つのDNA切断能が失活したCRISPR-Casシステム、ジンクフィンガーモチーフ、TALエフェクター及びPPRモチーフからなる群より選択される、請求項15~24のいずれか一項に記載の複合体。
- 請求項1~25のいずれか一項に記載の複合体をコードする核酸。
- 請求項26に記載の核酸を含むベクター。
- アデノ随伴ウイルスベクターである、請求項27に記載のベクター。
- 細胞の有する二本鎖DNAの標的化された部位を改変する方法であって、請求項1~25のいずれか一項に記載の複合体を該二本鎖DNAと接触させる工程を含む、方法。
- 二本鎖DNAと複合体との接触が、前記細胞への、請求項26~28のいずれか一項に記載の核酸またはベクターの導入により行われる、請求項29に記載の方法。
Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP21864464.9A EP4209589A4 (en) | 2020-09-04 | 2021-09-06 | MINIATURIZED CYTIDINE INDEAMINASE-CONTAINING COMPLEX FOR MODIFYING DOUBLE-STRANDED DNA |
| CN202180054915.9A CN116134141A (zh) | 2020-09-04 | 2021-09-06 | 包含小型化胞苷脱氨酶的双链dna修饰用复合体 |
| BR112023003972A BR112023003972A2 (pt) | 2020-09-04 | 2021-09-06 | Complexos de um módulo de reconhecimento de sequência de ácido nucleico ligado a uma desaminase e de um fragmento amino-terminal de um módulo de reconhecimento de sequência de ácido nucleico, uma desaminase e um fragmento carboxi-terminal de um módulo de reconhecimento de sequência de ácido nucleico ligado ao mesmo, ácido nucleico, vetor, e, método para alterar um sítio alvo de um ácido desoxirribonucleico de fita dupla de uma célula |
| KR1020237011179A KR20230061474A (ko) | 2020-09-04 | 2021-09-06 | 소형화 시티딘 데아미나아제를 포함하는 이중쇄 dna의 개변용 복합체 |
| JP2022546998A JPWO2022050413A1 (ja) | 2020-09-04 | 2021-09-06 | |
| CA3194019A CA3194019A1 (en) | 2020-09-04 | 2021-09-06 | Miniaturized cytidine deaminase-containing complex for modifying double-stranded dna |
| US18/044,047 US20230323335A1 (en) | 2020-09-04 | 2021-09-06 | Miniaturized cytidine deaminase-containing complex for modifying double-stranded dna |
| AU2021336262A AU2021336262A1 (en) | 2020-09-04 | 2021-09-06 | Miniaturized cytidine deaminase-containing complex for modifying double-stranded dna |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2020149419 | 2020-09-04 | ||
| JP2020-149419 | 2020-09-04 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2022050413A1 true WO2022050413A1 (ja) | 2022-03-10 |
Family
ID=80492029
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2021/032689 WO2022050413A1 (ja) | 2020-09-04 | 2021-09-06 | 小型化シチジンデアミナーゼを含む二本鎖dnaの改変用複合体 |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US20230323335A1 (ja) |
| EP (1) | EP4209589A4 (ja) |
| JP (1) | JPWO2022050413A1 (ja) |
| KR (1) | KR20230061474A (ja) |
| CN (1) | CN116134141A (ja) |
| AU (1) | AU2021336262A1 (ja) |
| BR (1) | BR112023003972A2 (ja) |
| CA (1) | CA3194019A1 (ja) |
| WO (1) | WO2022050413A1 (ja) |
Citations (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003087341A2 (en) | 2002-01-23 | 2003-10-23 | The University Of Utah Research Foundation | Targeted chromosomal mutagenesis using zinc finger nucleases |
| WO2005033321A2 (en) | 2003-09-30 | 2005-04-14 | The Trustees Of The University Of Pennsylvania | Adeno-associated virus (aav) clades, sequences, vectors containing same, and uses therefor |
| WO2010012077A1 (en) | 2008-07-28 | 2010-02-04 | Mount Sinai Hospital | Compositions, methods and kits for reprogramming somatic cells |
| WO2011072246A2 (en) | 2009-12-10 | 2011-06-16 | Regents Of The University Of Minnesota | Tal effector-mediated dna modification |
| JP2013128413A (ja) | 2010-03-11 | 2013-07-04 | Kyushu Univ | Pprモチーフを利用したrna結合性蛋白質の改変方法 |
| WO2013176772A1 (en) | 2012-05-25 | 2013-11-28 | The Regents Of The University Of California | Methods and compositions for rna-directed target dna modification and for rna-directed modulation of transcription |
| WO2015133554A1 (ja) | 2014-03-05 | 2015-09-11 | 国立大学法人神戸大学 | 標的化したdna配列の核酸塩基を特異的に変換するゲノム配列の改変方法及びそれに用いる分子複合体 |
| JP2016520318A (ja) * | 2013-05-29 | 2016-07-14 | セレクティスCellectis | Ii型crisprシステムにおける新規のコンパクトなcas9足場 |
| WO2019189147A1 (ja) | 2018-03-26 | 2019-10-03 | 国立大学法人神戸大学 | 細胞の有する二本鎖dnaの標的部位を改変する方法 |
| JP2020511970A (ja) * | 2017-03-23 | 2020-04-23 | プレジデント アンド フェローズ オブ ハーバード カレッジ | 核酸によってプログラム可能なdna結合蛋白質を含む核酸塩基編集因子 |
| WO2020091069A1 (ja) * | 2018-11-01 | 2020-05-07 | 国立大学法人東京大学 | 2分割されたCpf1タンパク質 |
| JP2020521451A (ja) * | 2017-05-25 | 2020-07-27 | ザ ジェネラル ホスピタル コーポレイション | 望ましくないオフターゲット塩基エディター脱アミノ化を制限するためのスプリットデアミナーゼの使用 |
| JP2020149419A (ja) | 2019-03-14 | 2020-09-17 | 富士ゼロックス株式会社 | メッセージ通知装置、コンテンツ管理システム及びプログラム |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018165629A1 (en) * | 2017-03-10 | 2018-09-13 | President And Fellows Of Harvard College | Cytosine to guanine base editor |
| AU2020215730A1 (en) * | 2019-01-31 | 2021-07-29 | Beam Therapeutics Inc. | Nucleobase editors having reduced non-target deamination and assays for characterizing nucleobase editors |
| WO2020168132A1 (en) * | 2019-02-13 | 2020-08-20 | Beam Therapeutics Inc. | Adenosine deaminase base editors and methods of using same to modify a nucleobase in a target sequence |
-
2021
- 2021-09-06 US US18/044,047 patent/US20230323335A1/en active Pending
- 2021-09-06 CA CA3194019A patent/CA3194019A1/en active Pending
- 2021-09-06 BR BR112023003972A patent/BR112023003972A2/pt unknown
- 2021-09-06 AU AU2021336262A patent/AU2021336262A1/en active Pending
- 2021-09-06 CN CN202180054915.9A patent/CN116134141A/zh active Pending
- 2021-09-06 JP JP2022546998A patent/JPWO2022050413A1/ja active Pending
- 2021-09-06 WO PCT/JP2021/032689 patent/WO2022050413A1/ja unknown
- 2021-09-06 EP EP21864464.9A patent/EP4209589A4/en active Pending
- 2021-09-06 KR KR1020237011179A patent/KR20230061474A/ko active Search and Examination
Patent Citations (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003087341A2 (en) | 2002-01-23 | 2003-10-23 | The University Of Utah Research Foundation | Targeted chromosomal mutagenesis using zinc finger nucleases |
| JP4968498B2 (ja) | 2002-01-23 | 2012-07-04 | ユニバーシティ オブ ユタ リサーチ ファウンデーション | ジンクフィンガーヌクレアーゼを用いる、標的化された染色体変異誘発 |
| WO2005033321A2 (en) | 2003-09-30 | 2005-04-14 | The Trustees Of The University Of Pennsylvania | Adeno-associated virus (aav) clades, sequences, vectors containing same, and uses therefor |
| WO2010012077A1 (en) | 2008-07-28 | 2010-02-04 | Mount Sinai Hospital | Compositions, methods and kits for reprogramming somatic cells |
| WO2011072246A2 (en) | 2009-12-10 | 2011-06-16 | Regents Of The University Of Minnesota | Tal effector-mediated dna modification |
| JP2013128413A (ja) | 2010-03-11 | 2013-07-04 | Kyushu Univ | Pprモチーフを利用したrna結合性蛋白質の改変方法 |
| WO2013176772A1 (en) | 2012-05-25 | 2013-11-28 | The Regents Of The University Of California | Methods and compositions for rna-directed target dna modification and for rna-directed modulation of transcription |
| JP2016520318A (ja) * | 2013-05-29 | 2016-07-14 | セレクティスCellectis | Ii型crisprシステムにおける新規のコンパクトなcas9足場 |
| WO2015133554A1 (ja) | 2014-03-05 | 2015-09-11 | 国立大学法人神戸大学 | 標的化したdna配列の核酸塩基を特異的に変換するゲノム配列の改変方法及びそれに用いる分子複合体 |
| JP2020511970A (ja) * | 2017-03-23 | 2020-04-23 | プレジデント アンド フェローズ オブ ハーバード カレッジ | 核酸によってプログラム可能なdna結合蛋白質を含む核酸塩基編集因子 |
| JP2020521451A (ja) * | 2017-05-25 | 2020-07-27 | ザ ジェネラル ホスピタル コーポレイション | 望ましくないオフターゲット塩基エディター脱アミノ化を制限するためのスプリットデアミナーゼの使用 |
| JP2020521446A (ja) * | 2017-05-25 | 2020-07-27 | ザ ジェネラル ホスピタル コーポレイション | 二部塩基エディター(bbe)構造およびii型−c−cas9ジンクフィンガー編集 |
| WO2019189147A1 (ja) | 2018-03-26 | 2019-10-03 | 国立大学法人神戸大学 | 細胞の有する二本鎖dnaの標的部位を改変する方法 |
| WO2020091069A1 (ja) * | 2018-11-01 | 2020-05-07 | 国立大学法人東京大学 | 2分割されたCpf1タンパク質 |
| JP2020149419A (ja) | 2019-03-14 | 2020-09-17 | 富士ゼロックス株式会社 | メッセージ通知装置、コンテンツ管理システム及びプログラム |
Non-Patent Citations (48)
| Title |
|---|
| "Journal of Experiments in Molecular Genetics", 1972, COLD SPRING HARBOR LABORATORY, pages: 431 - 433 |
| "New Cell Engineering Experimental Protocol", 1995, SHUJUNSHA, pages: 263 - 267 |
| BIO/TECHNOLOGY, vol. 6, 1988, pages 47 - 55 |
| BOWIE ET AL., SCIENCE, vol. 247, 1990, pages 1306 - 1310 |
| CURRPROTOC MOL BIOL, vol. 12, 2012, pages 15 |
| GENE, vol. 17, 1982, pages 107 |
| GENE, vol. 24, 1983, pages 255 |
| GENETICS, vol. 39, 1954, pages 440 |
| HIDETAKA KAYA, KAZUHIRO ISHIBASHI, SEIICHI TOKI: "A Split Staphylococcus aureus Cas9 as a Compact Genome-Editing Tool in Plants", PLANT AND CELL PHSIOLOGY, OXFORD UNIVERSITY PRESS, UK, vol. 58, no. 4, 1 April 2017 (2017-04-01), UK , pages 643 - 649, XP055538981, ISSN: 0032-0781, DOI: 10.1093/pcp/pcx034 * |
| HOCKEMEYER D ET AL., NAT BIOTEFCHNOL, vol. 27, 2009, pages 851 - 857 |
| HU JH ET AL., NATURE, vol. 556, no. 7699, 2018, pages 57 - 63 |
| IN VIVO, vol. 13, 1977, pages 213 - 217 |
| JOURNAL OF BIOCHEMISTRY, vol. 95, 1984, pages 87 |
| JOURNAL OF MOLECULAR BIOLOGY, vol. 120, 1978, pages 517 |
| JOURNAL OF MOLECULAR BIOLOGY, vol. 41, 1969, pages 459 |
| JUNJIE TAN, FEI ZHANG, DANIEL KARCHER, RALPH BOCK: "Engineering of high-precision base editors for site-specific single nucleotide replacement", NATURE COMMUNICATIONS, vol. 10, no. 1, 1 December 2019 (2019-12-01), XP055677711, DOI: 10.1038/s41467-018-08034-8 * |
| KAJI, K. ET AL., NATURE, vol. 458, 2009, pages 766 - 770 |
| LEGUT M ET AL., CELL REP, vol. 30, no. 9, 2020, pages 2859 - 2868 |
| METHODS IN ENZYMOLOGY, vol. 194, 1991, pages 182 - 187 |
| MILLER SM ET AL., NAT BIOTECHNOL., vol. 38, no. 4, 2020, pages 471 - 481 |
| MOL CELL, vol. 31, 2008, pages 294 - 301 |
| MOLECULAR & GENERAL GENETICS, vol. 168, 1979, pages 111 |
| NAT BIOTECHNOL, vol. 20, 2002, pages 135 - 141 |
| NAT BIOTECHNOL, vol. 26, 2008, pages 695 - 701 |
| NAT BIOTECHNOL, vol. 30, 2012, pages 460 - 465 |
| NATMETHODS, vol. 8, 2011, pages 67 - 69 |
| NATURE, vol. 195, 1962, pages 788 |
| NATURE, vol. 315, 1985, pages 592 |
| NISHIDA, K. ET AL.: "353", SCIENCE, no. 6305, 2016, pages aaf8729 |
| NISHIMASU H ET AL., SCIENCE, vol. 361, no. 6408, 2018, pages 1259 - 1262 |
| NUCLEIC ACIDS RES, vol. 39, 2011, pages e82 |
| NUCLEIC ACIDS RESEARCH, vol. 9, 1981, pages 309 |
| OCHIAI H, INT J MOL SCI, vol. 16, 2015, pages 21128 - 21137 |
| OJI A ET AL., SCI REP, vol. 6, 2016, pages 31666 |
| PROC. NATL. ACAD. SCI. USA, vol. 60, 1968, pages 160 |
| PROC. NATL. ACAD. SCI. USA, vol. 69, 1972, pages 2110 |
| PROC. NATL. ACAD. SCI. USA, vol. 75, 1987, pages 1929 |
| PROC. NATL. ACAD. SCI. USA, vol. 77, 1980, pages 4505 |
| PROC. NATL. ACAD. SCI. USA, vol. 81, 1984, pages 5330 |
| PROCEEDING OF THE SOCIETY FOR THE BIOLOGICAL MEDICINE, vol. 73, 1950, pages 1 |
| SCIENCE, vol. 122, 1952, pages 501 |
| See also references of EP4209589A4 |
| TAN JUNJIE, ZHANG FEI, KARCHER DANIEL, BOCK RALPH: "Expanding the genome-targeting scope and the site selectivity of high-precision base editors", NATURE COMMUNICATIONS, vol. 11, no. 1, 1 December 2020 (2020-12-01), XP055906673, DOI: 10.1038/s41467-020-14465-z * |
| THE JOURNAL OF THE AMERICAN MEDICAL ASSOCIATION, vol. 199, 1967, pages 519 |
| VIROLOGY, vol. 52, 1973, pages 456 |
| VIROLOGY, vol. 8, 1959, pages 396 |
| WALTON RT ET AL., SCIENCE, vol. 368, no. 6488, 2020, pages 290 - 296 |
| WANG, ZMOSBAUGH, D. W, J. BACTERIOL, vol. 170, 1988, pages 1082 - 1091 |
Also Published As
| Publication number | Publication date |
|---|---|
| BR112023003972A2 (pt) | 2023-04-18 |
| US20230323335A1 (en) | 2023-10-12 |
| KR20230061474A (ko) | 2023-05-08 |
| AU2021336262A1 (en) | 2023-05-18 |
| EP4209589A1 (en) | 2023-07-12 |
| JPWO2022050413A1 (ja) | 2022-03-10 |
| EP4209589A4 (en) | 2024-09-25 |
| AU2021336262A9 (en) | 2024-09-05 |
| CN116134141A (zh) | 2023-05-16 |
| CA3194019A1 (en) | 2022-03-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20210171935A1 (en) | Method for modifying genome sequence to introduce specific mutation to targeted dna sequence by base-removal reaction, and molecular complex used therein | |
| JP6780860B2 (ja) | 標的化したdna配列の核酸塩基を特異的に変換するゲノム配列の改変方法及びそれに用いる分子複合体 | |
| EP3115457B1 (en) | Genomic sequence modification method for specifically converting nucleic acid bases of targeted dna sequence, and molecular complex for use in same | |
| JP2022520428A (ja) | Ruvcドメインを有する酵素 | |
| KR102626503B1 (ko) | 뉴클레오타이드 표적 인식을 이용한 표적 서열 특이적 개변 기술 | |
| EP3447139B1 (en) | Method for increasing mutation introduction efficiency in genome sequence modification technique, and molecular complex to be used therefor | |
| US20210095271A1 (en) | System and method for genome editing | |
| JP2023063448A (ja) | 細胞の有する二本鎖dnaの標的部位を改変する方法 | |
| KR20240055073A (ko) | 클래스 ii, v형 crispr 시스템 | |
| WO2022050413A1 (ja) | 小型化シチジンデアミナーゼを含む二本鎖dnaの改変用複合体 | |
| JP7454881B2 (ja) | Crisprタイプi-dシステムを利用した標的ヌクレオチド配列改変技術 | |
| JP2021533742A (ja) | 新規転写アクチベーター | |
| US20190218533A1 (en) | Genome-Scale Engineering of Cells with Single Nucleotide Precision | |
| WO2024206910A2 (en) | Components for genomic editing | |
| KR20230016751A (ko) | 염기 편집기 및 이의 용도 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 21864464 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2022546998 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 3194019 Country of ref document: CA |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112023003972 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 20237011179 Country of ref document: KR Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2021864464 Country of ref document: EP Effective date: 20230404 |
|
| ENP | Entry into the national phase |
Ref document number: 112023003972 Country of ref document: BR Kind code of ref document: A2 Effective date: 20230302 |
|
| ENP | Entry into the national phase |
Ref document number: 2021336262 Country of ref document: AU Date of ref document: 20210906 Kind code of ref document: A |