WO2019144889A1 - 中乌宁晶型及其制备方法 - Google Patents
中乌宁晶型及其制备方法 Download PDFInfo
- Publication number
- WO2019144889A1 WO2019144889A1 PCT/CN2019/072874 CN2019072874W WO2019144889A1 WO 2019144889 A1 WO2019144889 A1 WO 2019144889A1 CN 2019072874 W CN2019072874 W CN 2019072874W WO 2019144889 A1 WO2019144889 A1 WO 2019144889A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- crystal form
- zhongwuing
- medium
- crystal
- sample
- Prior art date
Links
Images












Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D221/00—Heterocyclic compounds containing six-membered rings having one nitrogen atom as the only ring hetero atom, not provided for by groups C07D211/00 - C07D219/00
- C07D221/02—Heterocyclic compounds containing six-membered rings having one nitrogen atom as the only ring hetero atom, not provided for by groups C07D211/00 - C07D219/00 condensed with carbocyclic rings or ring systems
- C07D221/22—Bridged ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/08—Bridged systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/439—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom the ring forming part of a bridged ring system, e.g. quinuclidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
Definitions
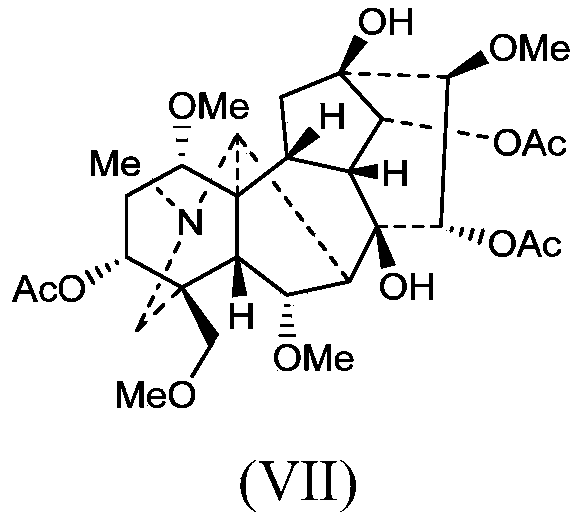
- the X-ray powder diffraction pattern of the Zhongwuing crystal form A of the present invention is still 2theta values of 11.6° ⁇ 0.2°, 19.8° ⁇ 0.2°, 24.2° ⁇ 0.2°, 26.9°. There is a characteristic peak at ⁇ 0.2°.
- the X-ray powder diffraction pattern of the Zhongwuing crystal form C of the present invention is still 2theta values of 19.9° ⁇ 0.2°, 21.0° ⁇ 0.2°, 23.8° ⁇ 0.2°, 26.6°. There is a characteristic peak at ⁇ 0.2°.
- High-humidity test The sample was placed in an open flat weighing bottle and sampled at a relative humidity of 90% for 10 days, and then sampled according to the stability test, and X-ray powder diffraction analysis was performed.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Cardiology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Life Sciences & Earth Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Heart & Thoracic Surgery (AREA)
- Hospice & Palliative Care (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Natural Medicines & Medicinal Plants (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Alternative & Traditional Medicine (AREA)
- Biotechnology (AREA)
- Botany (AREA)
- Medical Informatics (AREA)
- Microbiology (AREA)
- Mycology (AREA)
Abstract
Description







| 批号 | 熔点(℃) |
| 中乌宁晶型A样品 | 206.6~208.4℃ |
| 溶剂 | 溶解性 | 说明 |
| 水 | 易溶 | 1g溶质能在1~10ml溶剂中溶解 |



Claims (14)
- 一种中乌宁晶型A,其特征在于,使用Cu-Kα辐射,其X-射线粉末衍射图谱在2theta值为8.3°±0.2°、10.6°±0.2°、13.3°±0.2°、13.7°±0.2°、19.0°±0.2°处具有特征峰。
- 根据权利要求1所述的中乌宁晶型A,其特征在于,使用Cu-Kα辐射,其X-射线粉末衍射图谱还在2theta值为11.6°±0.2°、19.8°±0.2°、24.2°±0.2°、26.9°±0.2°处具有特征峰。
- 一种制备权利要求1或2所述的中乌宁晶型A的方法,其特征在于,所述方法包括以下步骤:(1)将中乌宁加入溶剂中,升温至50-90℃溶解,得到中乌宁的溶液;(2)将所述中乌宁的溶液降温至-10-30℃并搅拌析晶,过滤;(3)将步骤(2)过滤得到的固体真空干燥,得到中乌宁晶型A。
- 根据权利要求3所述的方法,其特征在于,所述溶剂选自水、甲醇、乙醇、异丙醇、丙酮、乙腈中的一种或多种。
- 一种中乌宁晶型B,其特征在于,使用Cu-Kα辐射,其X-射线粉末衍射图谱在2theta值为6.6°±0.2°、9.2°±0.2°、13.4°±0.2°、14.3°±0.2°、15.5°±0.2°处具有特征峰。
- 根据权利要求5所述的中乌宁晶型B,其特征在于,使用Cu-Kα辐射,其X-射线粉末衍射图谱还在2theta值为18.3°±0.2°、20.2°±0.2°、23.6°±0.2°、24.2°±0.2°处具有特征峰。
- 一种制备权利要求5或6所述的中乌宁晶型B的方法,其特征在于,所述方法包括以下步骤:(1)取根据权利要求1所述的中乌宁晶型A在40-60℃下在有机溶剂中悬浮搅拌,得到悬浮液;(2)将所述悬浮液降温至-10-30℃并搅拌析晶,过滤;(3)将步骤(2)过滤得到的固体真空干燥,得到中乌宁晶型B。
- 根据权利要求7所述的方法,其特征在于,所述有机溶剂为N-甲基吡咯烷酮。
- 一种中乌宁晶型C,其特征在于,使用Cu-Kα辐射,其X-射线粉末衍射图谱在2theta值为7.90°±0.2°、10.1°±0.2°、13.0°±0.2°、17.4°±0.2°、19.4°±0.2°处具有特征峰。
- 根据权利要求9所述的中乌宁晶型C,其特征在于,使用Cu-Kα辐射,其X-射线粉末衍射图谱还在2theta值为19.9°±0.2°、21.0°±0.2°、23.8°±0.2°、26.6°±0.2°处具有特征峰。
- 一种制备权利要求9或10所述的中乌宁晶型C的方法,其特征在于,所述方法包括以下步骤:将根据权利要求5所述的中乌宁晶型B真空加热,得到中乌宁晶型C。
- 根据权利要求11所述的方法,其特征在于,真空加热的温度为140℃-230℃,优选170℃-200℃。
- 一种药物组合物,其包含权利要求1或2所述的中乌宁晶型A,权利要求5或6所述的中乌宁晶型B或权利要求9或10所述的中乌宁晶型C,及药学上可接受的赋形剂。
- 权利要求1或2所述的中乌宁晶型A,权利要求5或6所述的中乌宁晶型B或权利要求9或10所述的中乌宁晶型C或权利要求13所述的药物组合物在制备强心剂和抗心衰剂中的用途。
Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP19743596.9A EP3744712B1 (en) | 2018-01-24 | 2019-01-23 | Crystalline forms of mesaconine and preparation method therefor |
| EA202091779A EA202091779A1 (ru) | 2018-01-24 | 2019-01-23 | Кристаллические формы мезаконина и способы их получения |
| JP2020562818A JP7072674B2 (ja) | 2018-01-24 | 2019-01-23 | メサコニンの結晶形およびその調製方法 |
| US16/963,026 US10941116B2 (en) | 2018-01-24 | 2019-01-23 | Crystalline forms of mesaconine and preparation methods therefor |
| AU2019212668A AU2019212668B2 (en) | 2018-01-24 | 2019-01-23 | Crystalline forms of mesaconine and preparation method therefor |
| CA3089268A CA3089268C (en) | 2018-01-24 | 2019-01-23 | Crystalline forms of mesaconine and preparation methods therefor |
| KR1020207022252A KR102528854B1 (ko) | 2018-01-24 | 2019-01-23 | 메사코닌의 결정형 및 그의 제조방법 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201810071927.2 | 2018-01-24 | ||
| CN201810071927 | 2018-01-24 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2019144889A1 true WO2019144889A1 (zh) | 2019-08-01 |
Family
ID=67365964
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2019/072873 WO2019144888A1 (zh) | 2018-01-24 | 2019-01-23 | 一种制备中乌宁的方法及其相关中间体 |
| PCT/CN2019/072874 WO2019144889A1 (zh) | 2018-01-24 | 2019-01-23 | 中乌宁晶型及其制备方法 |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2019/072873 WO2019144888A1 (zh) | 2018-01-24 | 2019-01-23 | 一种制备中乌宁的方法及其相关中间体 |
Country Status (9)
| Country | Link |
|---|---|
| US (2) | US11465971B2 (zh) |
| EP (2) | EP3744712B1 (zh) |
| JP (2) | JP7072674B2 (zh) |
| KR (2) | KR102528854B1 (zh) |
| CN (3) | CN110066248B (zh) |
| AU (1) | AU2019212668B2 (zh) |
| CA (1) | CA3089268C (zh) |
| EA (1) | EA202091779A1 (zh) |
| WO (2) | WO2019144888A1 (zh) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR102528854B1 (ko) * | 2018-01-24 | 2023-05-03 | 굿닥터 파머수티클 그룹 씨오., 엘티디. | 메사코닌의 결정형 및 그의 제조방법 |
| CN112250632B (zh) * | 2019-12-19 | 2023-01-24 | 好医生药业集团有限公司 | 一种3,14,15-三乙酰乌头宁碱的制备方法 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101759640A (zh) * | 2008-12-25 | 2010-06-30 | 上海中药制药技术有限公司 | 高纯度苯甲酰新乌头原碱的制备方法 |
| CN102146057A (zh) | 2011-02-18 | 2011-08-10 | 四川大学 | C19-二萜生物碱及其制备方法和以该化合物为活性成分的药物组合物及用途 |
| CN102977020A (zh) * | 2012-12-05 | 2013-03-20 | 四川大学 | 乌头碱型生物碱及其制备方法和以该化合物为强心和抗心衰活性成分的药物组合物及用途 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE19511235C2 (de) * | 1995-03-27 | 1999-09-23 | Klinge Co Chem Pharm Fab | Verwendung von Aconitinen und Aconinen aus der Eisenhutknolle zur Prophylaxe einer echten Migräne |
| CN102973679B (zh) * | 2012-12-21 | 2014-06-11 | 四川省中医药科学院 | 乌头属药材或其加工品中总生物碱提取物的制备方法 |
| CN107445893A (zh) * | 2017-08-31 | 2017-12-08 | 珠海润都制药股份有限公司 | 盐酸去甲乌药碱新晶型及其制备方法 |
| CN108409657B (zh) * | 2017-11-24 | 2021-08-20 | 孙益民 | 高纯高乌甲素及其制备方法 |
| KR102528854B1 (ko) * | 2018-01-24 | 2023-05-03 | 굿닥터 파머수티클 그룹 씨오., 엘티디. | 메사코닌의 결정형 및 그의 제조방법 |
-
2019
- 2019-01-23 KR KR1020207022252A patent/KR102528854B1/ko active IP Right Grant
- 2019-01-23 US US16/963,973 patent/US11465971B2/en active Active
- 2019-01-23 JP JP2020562818A patent/JP7072674B2/ja active Active
- 2019-01-23 KR KR1020207022930A patent/KR102518166B1/ko active IP Right Grant
- 2019-01-23 EA EA202091779A patent/EA202091779A1/ru unknown
- 2019-01-23 AU AU2019212668A patent/AU2019212668B2/en active Active
- 2019-01-23 WO PCT/CN2019/072873 patent/WO2019144888A1/zh unknown
- 2019-01-23 US US16/963,026 patent/US10941116B2/en active Active
- 2019-01-23 CN CN201910064717.5A patent/CN110066248B/zh active Active
- 2019-01-23 EP EP19743596.9A patent/EP3744712B1/en active Active
- 2019-01-23 WO PCT/CN2019/072874 patent/WO2019144889A1/zh unknown
- 2019-01-23 JP JP2020562817A patent/JP6952206B2/ja active Active
- 2019-01-23 CN CN202011307213.0A patent/CN112375080A/zh active Pending
- 2019-01-23 CN CN201910064716.0A patent/CN110066247B/zh active Active
- 2019-01-23 CA CA3089268A patent/CA3089268C/en active Active
- 2019-01-23 EP EP19743594.4A patent/EP3744711B1/en active Active
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101759640A (zh) * | 2008-12-25 | 2010-06-30 | 上海中药制药技术有限公司 | 高纯度苯甲酰新乌头原碱的制备方法 |
| CN102146057A (zh) | 2011-02-18 | 2011-08-10 | 四川大学 | C19-二萜生物碱及其制备方法和以该化合物为活性成分的药物组合物及用途 |
| CN102977020A (zh) * | 2012-12-05 | 2013-03-20 | 四川大学 | 乌头碱型生物碱及其制备方法和以该化合物为强心和抗心衰活性成分的药物组合物及用途 |
Non-Patent Citations (10)
| Title |
|---|
| "Chinese Pharmacopoeia", vol. 4, 2015 |
| "the Chinese Pharmacopoeia", vol. 4, 2015, article "determination method of the melting point" |
| CHEMICAL ABSTRACTS, Columbus, Ohio, US; abstract no. 6792-09-2 |
| CHINESE PHARMACOPOEIA, vol. 1, 2015 |
| MORIO, SHIN-ICHI: "Über Mesaconitin, ein zweites neues Aconitum‐Alkaloid. (VI. Mitteilung über Aconitum‐Alkaloide) = Aconitum Alkaloids. VI. Mesaconitine, a Second New Aconitum Alkaloid", JUSTUS LIEBIGS ANNALEN DER CHEMIE, vol. 476, no. 1, 31 December 1929 (1929-12-31), pages 189 - 190, XP009522472, ISSN: 0075-4617, DOI: 10.1002/jlac.19294760108 * |
| SAKAI, S. ET AL.: "Partial Synthesis of Isodelphinine and Penduline", YAKUGAKU ZASSHI, vol. 104, no. 7, 25 July 1984 (1984-07-25) - 31 December 1984 (1984-12-31), pages 747 - 748, XP055627143, ISSN: 0031-6903 * |
| See also references of EP3744712A4 |
| XIU-XIU LIU ET AL., CHEM. PHARM. BULL, vol. 60, no. 1, 2012, pages 144 - 149 |
| XI-XIAN JIAN ET AL., NAT. PROD. COMMUN, vol. 7, no. 6, 2012, pages 713 - 720 |
| ZHONG-TANG ZHANG ET AL., NAT. PROD. COMMUN, vol. 10, no. 12, 2015, pages 2075 - 2084 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN110066247A (zh) | 2019-07-30 |
| AU2019212668A1 (en) | 2020-08-13 |
| EP3744712A1 (en) | 2020-12-02 |
| KR20200110368A (ko) | 2020-09-23 |
| KR102528854B1 (ko) | 2023-05-03 |
| CN112375080A (zh) | 2021-02-19 |
| AU2019212668B2 (en) | 2021-04-15 |
| WO2019144888A1 (zh) | 2019-08-01 |
| US10941116B2 (en) | 2021-03-09 |
| CA3089268A1 (en) | 2019-08-01 |
| KR20200108004A (ko) | 2020-09-16 |
| US20200339514A1 (en) | 2020-10-29 |
| JP2021512941A (ja) | 2021-05-20 |
| EP3744712A4 (en) | 2021-07-07 |
| EP3744711A4 (en) | 2021-07-07 |
| JP6952206B2 (ja) | 2021-10-20 |
| JP7072674B2 (ja) | 2022-05-20 |
| EA202091779A1 (ru) | 2020-10-14 |
| US20210040042A1 (en) | 2021-02-11 |
| EP3744711B1 (en) | 2022-09-21 |
| CN110066247B (zh) | 2021-01-01 |
| EP3744712B1 (en) | 2023-04-12 |
| KR102518166B1 (ko) | 2023-04-05 |
| JP2021511390A (ja) | 2021-05-06 |
| CN110066248A (zh) | 2019-07-30 |
| US11465971B2 (en) | 2022-10-11 |
| EP3744711A1 (en) | 2020-12-02 |
| CA3089268C (en) | 2023-03-07 |
| CN110066248B (zh) | 2020-10-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5611846B2 (ja) | 置換ヘテロ環縮合ガンマ−カルボリン類固体 | |
| TW201039818A (en) | Polymorphs of eltrombopag and eltrombopag salts and processes for preparation thereof | |
| US20220235039A1 (en) | Crystalline salt forms of 6-(cyclopropanecarboxamido)-4-((2-methoxy-3-(1-methyl-1h-1,2,4-triazol-3-yl)phenyl)amino)-n-(methyld3) pyridazine-3-carboxamide | |
| WO2019228485A1 (zh) | 一种甲磺酸乐伐替尼新晶型及其制备方法 | |
| WO2019144889A1 (zh) | 中乌宁晶型及其制备方法 | |
| CN107043376A (zh) | 一种利格列汀新晶型及其制备方法 | |
| WO2020186960A1 (zh) | 一种草乌甲素c晶型及其制备方法与应用 | |
| WO2023125102A1 (zh) | 1H-呋喃并[3,2-b]咪唑并[4,5-d]吡啶化合物的合成方法 | |
| JP2022525125A (ja) | ブレイアコニチンaのe結晶形及びその製造方法と応用 | |
| EA043852B1 (ru) | Кристаллические формы мезаконина и способы их получения | |
| CN105777656B (zh) | 萘普替尼对甲苯磺酸盐的β晶型及制备方法和含有其的药物组合物 | |
| WO2016019867A1 (zh) | 一种6-芳基氨基吡啶酮甲酰胺化合物的结晶及其制备方法 | |
| EP3660031A1 (en) | Crystalline or amorphous form of steroid derivative fxr agonist, preparation method therefor and use thereof | |
| CN108659038B (zh) | 1-硬脂酰-2-丙戊酰-sn-甘油-3-磷脂酰胆碱的多晶型物及制备方法 | |
| CN112876530A (zh) | 一种地屈孕酮中间体的晶型及其制备方法 | |
| CN114630668B (zh) | 一种Aprocitentan晶型及其制备方法和用途 | |
| CN109651413B (zh) | 以溴代氧化荷苞牡丹碱为配体的稀土配合物及其合成方法和应用 | |
| TWI724651B (zh) | 貝前列素-314d單水合物晶體及其製備方法 | |
| WO2022262841A1 (zh) | 一种螺环化合物的盐型、晶型及其制备方法 | |
| CN109456335B (zh) | 氧化荷苞牡丹碱的合成方法 | |
| CN104710493B (zh) | 一种醋酸甲泼尼龙晶型 | |
| CN116041351A (zh) | 盐酸咪达唑仑新晶型及其制备方法 | |
| CN108264481A (zh) | 柳氮磺吡啶晶型及其制备方法 | |
| CN104628662A (zh) | 阿那曲唑及其一水合物的新晶型、制备和用途 | |
| CN104610176A (zh) | 阿那曲唑及其一水合物的新晶型、制备和用途 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 19743596 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 3089268 Country of ref document: CA Ref document number: 2020562818 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20207022252 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2019212668 Country of ref document: AU Date of ref document: 20190123 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2019743596 Country of ref document: EP Effective date: 20200824 |